Embed Size (px)
Citation preview

Nucleoside phosphorylases from thermophiles
Recombinant expression and biocatalytic use for modified nucleosides
Kathleen Szeker, Berlin 2012


Nucleoside phosphorylases from thermophiles
Recombinant expression and biocatalytic use for modified nucleosides
vorgelegt von
Diplom-Ingenieurin Kathleen Szeker
aus Potsdam
von der Fakultät III – Prozesswissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Ingenieurwissenschaften
- Dr.-Ing. –
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender Prof. Dr. Roland Lauster
Gutachter Prof. Dr. Peter Neubauer
Gutachterin Prof. Dr. Marion Ansorge-Schumacher
Gutachter Prof. Dr. Igor A. Mikhailopulo
Tag der wissenschaftlichen Aussprache: 11.06.2012
Berlin 2012
D83


Abstract
Modified nucleosides are valuable pharmaceutical agents used in the treatment of cancer and viral
infections. Moreover, they serve as building blocks in the synthesis of therapeutic oligonucleotides
with advanced properties.
While the chemical modification of pyrimidine nucleosides is generally well established, the synthesis
of modified purine nucleosides is often rather challenging, resulting in multistage processes with low
yield. Alternative synthetic routes include the chemo-enzymatic synthesis of purine nucleosides from
a pyrimidine nucleoside serving as pentofuranosyl donor and a purine base functioning as
pentofuranosyl acceptor. As biocatalysts, nucleoside phosphorylases (NPs) are used to catalyze the
regio- and stereoselective transfer reaction, whereby natural or chemically prepared artificial
precursors can be applied as substrate. Unfortunately, a number of highly interesting nucleoside
analogues are hardly recognized as substrate by NPs that are currently in use. Moreover, high
temperatures are desirable to increase the concentration of poorly soluble purine bases, but many
enzymes are rapidly deactivated by heat. Both factors limit the scope and the efficiency of NP
mediated syntheses of modified nucleosides and prompted us to study novel, thermostable
nucleoside phosphorylase variants as potential biocatalysts.
Therefore a set of 5 NPs from 4 different thermophilic microorganisms (Deinococcus geothermalis,
Geobacillus thermoglucosidasius, Thermus thermophilus, Aeropyrum pernix) has been overexpressed
in E. coli. The recombinant proteins were characterized in order to assess their potential application
as biocatalysts. Thermal properties (temperature optima, stability) varied significantly and were
dependent on the source microorganism and the type of enzyme. Investigations of the substrate
specificities revealed striking differences in the ability to tolerate modified nucleosides as substrate.
The data allowed us to select and test the most promising combinations of enzymes for enzymatic
transglycosylation reactions. In focus of the present work was thereby the synthesis of 2′-fluorinated
purine nucleosides as well as 2,6-dihalogenated purine nucleosides. 2′-Fluorinated nucleosides were
found to have valuable pharmaceutical properties and impart favourable characteristics to synthetic
oligonucleotides. On the other hand, 2,6-dihalogenated purine nucleosides are versatile precursors
for a variety of purine modified nucleosides. In comparison to E. coli enzymes that are described in
literature as biocatalysts for the synthesis of 2′-fluorinated purine nucleosides, the application of the
novel, thermostable enzymes permits the operation at higher temperature, and appears to be more
efficient in the synthesis 2′-fluorinated purine nucleosides. Furthermore, 2,6-dihalogenated purines
were readily accepted as substrates and the respective (deoxy-)ribosides were rapidly produced by
the novel enzyme preparations.
The results corroborate the general potential of thermostable NPs in the synthesis of modified
nucleosides and specifically pave the way towards improved, environmentally friendly synthetic
procedures affording valuable 2′-fluorinated and 2,6-dihalogenated purine nucleoside analogues.

The present work was performed from April 2009 – April 2012 in the research group of Prof. Dr.
Peter Neubauer (Laboratory of Bioprocess Engineering) at the Department of Biotechnology,
Technische Universität Berlin.

Publications
Szeker, K., Niemitalo, O., Casteleijn, M.G., Juffer, A.H., and Neubauer, P., 2011.
“High-temperature cultivation and 5' mRNA optimization are key factors for the efficient overexpression of thermostable Deinococcus geothermalis purine nucleoside phosphorylase in Escherichia coli”
Journal of Biotechnology, 156(4), 268-274
Szeker, K., Zhou, X., Schwab, T., Casanueva, A., Cowan, D., Mikhailopulo, I.A., and Neubauer, P., 2012.
“Comparative investigations on thermostable pyrimidine nucleoside phosphorylases from Geobacillus thermoglucosidasius and Thermus thermophilus.”
Journal of Molecular Catalysis B: Enzymatic, in press
Conference contributions
K. Szeker, X. Zhou, A. Scholz, M. Ansorge-Schumacher, I. A. Mikhailopulo, and P. Neubauer
“Thermostable nucleoside phosphorylases for the synthesis of purine nucleoside analogues”
X. Zhou, K. Szeker, I. A. Mikhailopulo, and P. Neubauer
“Thermostable biocatalysts with purine nucleoside activity”
Catalyzing Bio-Economy – Biocatalysts for industrial biotechnology, Annual meeting of the DECHEMA-VAAM-Section Biotransformations, Frankfurt, Germany, April 2012
K. Szeker, X. Zhou, I. A. Mikhailopulo, and P. Neubauer
“Characterizing thermostable nucleoside phosphorylases for their use as biocatalysts”
Biotrans 2011, Giardini Naxos, Italy, October 2011
K. Szeker, X. Zhou, I. A. Mikhailopulo, and P. Neubauer
“Overexpression and biocatalytic characterization of thermostable nucleoside phosphorylases in Escherichia coli”
1st European Congress of Applied Biotechnology, Berlin, Germany, September 2011
K. Szeker, O. Niemitalo, and P. Neubauer
“Increasing the expression level of purine nucleoside phosphorylase from thermophilic origin in Escherichia coli”
6th Conference on recombinant protein production – A comparative view on host physiology, Vienna, Austria, February 2011
K. Szeker, M. Casteleijn, and P. Neubauer
“Optimization of soluble expression of recombinant thermophilic nucleoside phosphorylases”
14th International Biotechnology Symposium and Exhibition, Rimini, Italy, September 2010
K. Szeker, and P. Neubauer
“Novel biocatalysts for the preparation of modified nucleosides”
Talk at BIG-NSE / UniCat Mini Symposium – Protein Engineering, Berlin, Germany, May 2010
K. Szeker, and P. Neubauer
“Biocatalysts in nucleoside chemistry”
Talk at German-Hungarian biotechnology seminar, Miskolc, Hungary, July 2009

List of abbreviations
AdoP Adenosine phosphorylase
A. hydrophila Aeromonas hydrophila
A. pernix Aeropyrum pernix
AhPNP Purine nucleoside phosphorylase from A. hydrophila
Anhydro-Urd O2,2′-Anhydro-1-(-D-arabinofuranosyl)uracil
ApMTAP 5′-Methylthioadenosine phosphorylase from A. pernix
ApUP Uridine phosphorylase of A. pernix
ara-A 9-(-D-Arabinofuranosyl)adenine
ara-U 1-(-D-Arabinofuranosyl)uracil
ASOs Antisense oligonucleotides
AZT Azidothymidine
B. cereus Bacillus cereus
B. subtilis Bacillus subtilis
CAI Codon adaption index
D. geothermalis Deinococcus geothermalis
DgPNP Purine nucleoside phosphorylase from D. geothermalis
dAdo2′F 9-(2-Deoxy-2-fluoro-β-D-arabinofuranosyl)adenine
dAdo2′F 2′-Deoxy-2′-fluoroadenosine
dUrd2′F 1-(2-Deoxy-2-fluoro-β-D-arabinofuranosyl)uracil
dUrd2′F 2′-Deoxy-2′-fluorouridine
DTT Dithiothreitol
EcPNP Purine nucleoside phosphorylase of E. coli
EcTP Thymidine phosphorylase of E. coli
EcUP Uridine phosphorylase of E. coli
GsPyNP Pyrimidine nucleoside phosphorylase from G. stearothermophilus
G. stearothermophilus Geobacillus stearothermophilus
G. thermoglucosidasius Geobacillus thermoglucosidasius
GtPNP Purine nucleoside phosphorylase from G. thermoglucosidasius
GtPyNP Pyrimidine nucleoside phosphorylase from G. thermoglucosidasius

HPLC High-performance liquid chromatography
IPTG Isopropyl β-D-1-thiogalactopyranoside
KP buffer Potassium phosphate buffer
LB medium Lysogeny broth medium
MTAP 5′-Methylthioadenosine phosphorylase
NdRT N-deoxyribosyltransferase
NP Nucleoside phosphorylase
NP buffer Sodium phosphate buffer
PNP Purine nucleoside phosphorylase
PyNP Pyrimidine nucleoside phosphorylase
P. furiosus Pyrococcus furiosus
S. solfataricus Sulfolobus solfataricus
SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis
TB medium Terrific broth medium
T. thermophilus Thermus thermophilus
TP Thymidine phosphorylase
TtPyNP Thymidine phosphorylase from T. thermophilus
UP Uridine phosphorylase
XanoP Xanthosine phosphorylase


Table of contents
1. Introduction ............................................................................................. 5
1.1. Modified nucleosides in pharmacy and life sciences ............................................. 5
1.1.1. Natural nucleosides .............................................................................................. 5
1.1.2. Modified nucleosides in clinical use ..................................................................... 5
1.1.3. Modified nucleosides as building blocks for synthetic nucleic acids ................... 9
1.2. Synthetic routes towards nucleoside analogues .................................................. 11
1.2.1. Chemical synthesis ............................................................................................. 11
1.2.2. Enzymes for nucleoside synthesis ...................................................................... 12
1.2.3. The NP-catalyzed transglycosylation of nucleosides ......................................... 14
1.2.4. Formulation of NPs for technical use ................................................................. 15
1.3. Nucleoside phosphorylases ................................................................................. 16
1.3.1. Physiological role ................................................................................................ 16
1.3.2. Basic nature of the catalytic mechanism ........................................................... 17
1.3.3. Classification of NPs ........................................................................................... 20
1.3.4. From natural to recombinant production of nucleoside phosphorylases ......... 22
1.4. Motivation and structure of the present work .................................................... 24
1.4.1. Enzyme-assisted synthesis of modified purine nucleosides .............................. 24
1.4.2. Taking advantage of thermostable nucleoside phosphorylases ........................ 25
1.4.3. Recombinant expression of thermostable NPs in E. coli ................................... 25
1.4.4. Experimental outline and key objectives ........................................................... 25
2. Experimental part ................................................................................... 27
2.1. Generation of expression plasmids ..................................................................... 27
2.1.1. Gene isolation .................................................................................................... 27
2.1.2. Plasmids .............................................................................................................. 27
2.1.3. Recombinational cloning .................................................................................... 28
2.1.4. Cloning via restriction and digestion .................................................................. 29
2.1.5. Verification of the cloning steps and propagation of vector constructs ........... 30
2.1.6. Site-directed mutagenesis .................................................................................. 30

2 Introduction
2.2. Bioinformatics ..................................................................................................... 32
2.2.1. Amino acid sequence analysis and homology modelling .................................. 32
2.2.2. Secondary mRNA prediction and sequence optimization ................................. 32
2.3. Bacterial growth and recombinant protein expression ........................................ 33
2.3.1. Preparation of recombinant E. coli cell banks ................................................... 33
2.3.2. Recombinant protein expression ....................................................................... 33
2.4. Preparation of protein samples ........................................................................... 34
2.4.1. Cell disruption .................................................................................................... 34
2.4.2. Protein purification ............................................................................................ 34
2.5. Protein analytics ................................................................................................. 35
2.5.1. SDS-PAGE analysis .............................................................................................. 35
2.5.2. Determination of the protein concentration ..................................................... 35
2.5.3. Protein unfolding studies ................................................................................... 36
2.6. Activity assays ..................................................................................................... 36
2.6.1. Spectroscopic assay for PNP activity .................................................................. 36
2.6.2. Standard assay with purified proteins ............................................................... 37
2.6.3. Thermal properties of the enzymes ................................................................... 37
2.6.4. Kinetic parameters ............................................................................................. 38
2.6.5. Substrate screenings .......................................................................................... 38
2.6.6. Synthetic reactions ............................................................................................. 38
2.7. HPLC analysis ...................................................................................................... 38
3. Recombinant expression of nucleoside phosphorylases .......................... 41
3.1. Introduction ........................................................................................................ 41
3.1.1. Recombinant expression of thermostable proteins in E. coli ............................ 41
3.1.2. Target enzymes of this study ............................................................................. 42
3.2. Sequence analysis and theoretical predictions .................................................... 44
3.3. Expression of DgPNP ........................................................................................... 45
3.3.1. Towards the functional expression of DgPNP .................................................... 45
3.3.2. DgPNP expression optimization by reducing secondary 5′mRNA stability ........ 48

Introduction 3
3.3.3. Functional expression of DgPNP with N-terminal hexahistidine tag ................. 53
3.3.4. DgPNP expression - summary and conclusions ................................................. 55
3.4. Expression of ApMTAP ........................................................................................ 56
3.4.1. Expression of the wild type ApMTAP gene without tag .................................... 56
3.4.2. ApMTAP expression with N-terminal hexahistidine tag .................................... 57
3.4.3. ApMTAP - summary and conclusions ................................................................. 62
3.5. Expression of GtPNP ........................................................................................... 62
3.5.1. GtPNP expression with C-terminal hexahistidine tag ........................................ 63
3.5.2. GtPNP expression with N-terminal hexahistidine tag ........................................ 63
3.5.3. GtPNP expression - summary and conclusion ................................................... 63
3.6. Expression of GtPyNP .......................................................................................... 64
3.6.1. Chemical lysis buffer decreases apparent thermal stability of GtPyNP ............. 64
3.6.2. GtPyNP expression with N-terminal hexahistidine tag ...................................... 65
3.6.3. GtPyNP expression - summary and conclusions ................................................ 66
3.7. Expression of TtPyNP .......................................................................................... 66
3.7.1. TtPyNP expression with N-terminal hexahistidine tag ...................................... 66
3.7.2. TtPyNP expression - summary and conclusions ................................................. 68
3.8. Expression of ApUP ............................................................................................. 68
3.8.1. Expression of ApUP without tag ......................................................................... 69
3.8.2. Expression of ApUP with N-terminal hexahistidine tag ..................................... 71
3.8.3. ApUP expression - summary and discussion ...................................................... 71
3.9. Recombinant expression of NPs - summary and conclusions ............................... 72
4. Characterization of thermostable NPs ..................................................... 75
4.1. Thermostable PyNPs ........................................................................................... 75
4.1.1. Homology modelling .......................................................................................... 76
4.1.2. Thermal characteristics ...................................................................................... 76
4.1.3. Kinetic parameters ............................................................................................. 78
4.1.4. Phosphorolysis of 2′-fluorosubstituted pyrimidine nucleosides ........................ 80
4.1.5. PNP activity of PyNPs ......................................................................................... 83

4 Introduction
4.1.6. Characterization of thermostable PyNPs - summary and conclusions .............. 84
4.2. Thermostable enzymes with PNP activity ........................................................... 85
4.2.1. Sequence analysis and homology modelling ..................................................... 86
4.2.2. Thermal characteristics ...................................................................................... 88
4.2.3. Substrate specificities ......................................................................................... 91
4.2.4. Characterization of thermostable PNP enzymes - summary and conclusions .. 95
5. Enzymatic transglycosylations with thermostable NPs ............................ 97
5.1. Introduction ........................................................................................................ 97
5.1.1. Chemical synthesis of 2′-fluorinated nucleosides .............................................. 97
5.1.2. The chemo-enzymatic synthesis of 2′-fluorinated purine nucleosides ............. 99
5.1.3. 2,6-Dihalogenated purine nucleosides ............................................................ 100
5.2. Synthesis of 2′-fluorosubstituted purine nucleosides ......................................... 101
5.2.1. Synthesis of 2′-deoxy-2′-fluoroadenosine ........................................................ 101
5.2.2. Synthesis of 9-(2-deoxy-2-fluoro--D-arabinofuranosyl)adenine ................... 104
5.3. Synthesis of 2,6-dihalogenated purine nucleosides ............................................ 107
5.3.1. Synthesis of 2,6-dihalogenated purine ribosides ............................................. 108
5.3.2. Synthesis of 2,6-dihalogenated purine deoxyribosides ................................... 110
5.4. Enzymatic transglycosylations– summary and conclusions ................................ 112
5.4.1. 2′-Fluorinated purine nucleosides .................................................................... 112
5.4.2. 2,6-Dihalogenated purine nucleosides ............................................................ 113
6. Final conclusions ................................................................................... 115
References .................................................................................................. 119
Appendix ..................................................................................................... 135
Zusammenfassung ...................................................................................... 137
Acknowledgements ..................................................................................... 138

Introduction 5
1. Introduction
Nucleoside phosphorylases (NPs) are enzymes that catalyse the reversible phosphorolysis of
nucleosides. The purpose of the present study is to utilize thermostable variants for the chemo-
enzymatic synthesis of modified nucleosides. The major focus of this chapter is to give an overview
about the underlying motivation. First, the pharmaceutical and scientific value of nucleoside
analogues will be demonstrated. Next, chemical and chemo-enzymatic routes towards their
preparation will be discussed, followed by a closer glance on NPs. Finally the approach of this work
will be explained and the experimental strategy presented.
1.1. Modified nucleosides in pharmacy and life sciences
Natural nucleosides are major constituents of nucleic acids. Since they play a key role for the cellular
life, reflected by the storage of genetic information or metabolic regulation, it is obvious that
modifications can turn them into highly bioactive compounds. Therefore modified nucleosides find
broad application in both pharmaceutical industry and as molecular biological tools.
1.1.1. Natural nucleosides
Nucleosides consist out of a nucleobase that is connected to the anomeric centre (C1′) of a sugar via
a β-glycosidic linkage. In natural nucleosides this sugar represents either a D-ribose or a
D-2-deoxyribose. Depending on the heterocyclic base, nucleosides are classified as purine or
pyrimidine nucleosides. While purine bases are linked via the N-9 atom, pyrimidine bases are linked
via the N1 atom to the sugar moiety. In Figure 1 the four natural deoxynucleosides that constitute
the building blocks of our DNA are shown.
O
OH
OH
NH
N
O
O
Thymidine
O
OH
OH
N
NN
N
NH2
Deoxyadenosine
O
OH
OH
NH
NN
N
O
Deoxyguanosine
NH2
CH3
O
OH
OH
N
N
NH2
O
Deoxycytidine
1'2'3'
4'
5'
1 2
345
6
1'2'3'
4'
5'
12
34
567
89
Pyrimidine nucleosides Purine nucleosides
Figure 1: Natural deoxynucleosides
1.1.2. Modified nucleosides in clinical use
As mimics of natural nucleic acid constituents, modified nucleosides can have a high impact on
fundamental processes involving the replication and transcription of our genetic material. This
characteristic was the basis for the development of therapies against hyperproliferative diseases –

6 Introduction
including viral infections and cancer - that are particularly dependent on high levels of nucleic acid
synthesis.
Nucleosides with antiviral activity
A number of nucleoside analogues were found to exert antiviral activity by inhibiting viral DNA
polymerases, e.g. the DNA and RNA dependent reverse transcriptase. The discovery of zidovudine
(azidothymidine, AZT) (Figure 2) as anti HIV compound in 1985 (Mitsuya et al. 1985) marked a
breakthrough in the therapy of HIV infections. Indeed AZT, later marketed as “Retrovir” by
GlaxoSmithKline, was the first drug approved by the FDA for the treatment of HIV infection. The
disclosure of other nucleoside analogues with anti-HIV activity followed (Figure 2). The common
feature is the absence of the 3′-hydroxyl group that is responsible for the chain terminator effect
within reverse transcription.
O
N3
HO
NH
N
O
O
Zidovudine Zalcitabine Didanosine Stavudine
Lamivudine Abacavir Emtricitabine
H3C
OHO
N
N
NH2
OO
HO
NH
NN
N
O
OHO
NH
N
O
O
H3C
S
O
HO
N
N
NH2
O
OHO
N
NN
N
NH
NH2S
O
HO
N
N
NH2
O
F
Figure 2: Nucleoside reverse-transcriptase inhibitors in current clinical use for the treatment of HIV infections. While zidovudine marks the oldest anti-HIV drug (launched in 1987), emtricitabine represents the last drug approved by the FDA in (2003). For further details on trade names and companies see (Flexner 2007).
Today, nucleoside reverse transcriptase inhibitors play a crucial role as components of the so-called
highly active antiretroviral therapy (HAART) that has dramatically improved the quality of life and
prognosis of patients infected by HIV. The medication includes a combination of two or more
nucleoside reverse transcriptase inhibitors and protease inhibitors (Murphy et al. 2001). Other
modified nucleoside analogues are in clinical use for treatment of infections with Hepatitis B, Herpes
Simplex, Varicella-Zoster and Hepatitis C virus (De Clercq 2004).
The viral polymerase is one of the most common targets of nucleoside reverse transcriptase
inhibitors in antiviral therapy (Berdis 2008). After the incorporation into a nascent DNA chain,
artificial nucleosides prevent the formation of the 5′ to 3′ phosphodiester linkages that are essential
for DNA chain elongations. Noteworthy nucleoside reverse transcriptase inhibitors require

Introduction 7
intracellular phosphorylation to their triphosphate forms for activity – a process that is dependent on
deoxynucleosides kinases. In case of AZT the phosphorylation was found non-selective. Conversely
the azidothymidine triphosphate competed about 100-fold better for the HIV reverse transcriptase
than for the cellular DNA polymerase (Furman et al. 1986).
A less common strategy in antiviral therapies constitutes the induction of lethal mutagenesis by the
administration of promutagenic nucleoside analogues. The rationale behind is to take advantage of
the error-prone viral DNA synthesis, while the cellular DNA polymerase is characterized by higher
fidelity and proofreading capacity. By a careful selection of the dose, the promutagenic nucleosides
would thus have only little impact on the integrity of the cellular genome, while the viability of the
virus is negatively affected. An example of such an antiviral nucleoside drug is Ribavirin that is used
for the treatment of Hepatitis C (Berdis 2008).
Despite the impressive progress in antiviral therapy, the emergence of drug-resistant mutants and
adverse side effects of many nucleoside analogues calls for the development of new drugs that may
complement the currently used ones. Examples of potential future nucleoside drugs are 2′-deoxy-4′-
C-ethynyl-2-fluoroadenosine and 2′-deoxy-4′-C-ethynyl-2-chloroadenosine. Both were reported to be
highly active against multi-drug resistant HIV and exert favourable toxicity profiles (Ohrui 2011).
Modified nucleosides as anticancer agents
Similar to viral infections, cancer is characterized by uncontrolled DNA synthesis and is therefore
conceived as hyperproliferative disease. Nucleoside reverse transcriptase inhibitors exert their
antiviral activity predominantly directly by chain termination. By contrast, nucleoside anticancer
agents typically inhibit additionally also other enzymes involved in the metabolism of nucleic acid
constituents. A prominent additional target for example is ribonucleotide reductase. DNA damage,
provoked by different factors, will eventually lead to the activation of signalling pathways that
initiate apoptotic processes. Hence apoptosis is considered as the final outcome of the treatment
with nucleoside anticancer agents (Sampath et al. 2003).
Examples of nucleoside analogues with anticancer activity currently in clinical use are shown in
Figure 3. A remarkable case study represents the discovery of Clofarabine that was approved in 2004
by the FDA for the treatment of acute lymphoblastic leukaemia in paediatrics and is marketed as
“Clolar” by Genzyme. Clofarabine can be considered as second-generation nucleoside anticancer
agent and is closely related to Cladribine and Fludarabine - clinical drugs for the treatment of
leukaemia. Both compounds are adenosine analogues and hence quite similar from the structural
point of view. Nevertheless, there are distinct advantages and disadvantages of both drugs resulting
from the nature of the specific substituents. The advantage of Cladribine lies in the 2-chloro
substitution of the purine ring, that confers improved stability against adenosine deaminase (Carson
et al. 1980).

8 Introduction
Cytarabine Gemcitabine
Cladribine Fludarabinephosphate Clofarabine
O
HO
HO
N
N
NH2
O
OH
O
HO
HO
N
N
NH2
O
F
F
Acute myeloid leukemia (Pfizer) Pancreatic and lung cancer (Eli Lilly)
O
N
NN
N
NH2
HO
HO Cl
Hairy cell leukemia (Pfizer)
O
N
NN
N
NH2
HO
OH
O FP
O
HO
OH
Chronic lymphocytic leukemia (Sandoz)
O
N
NN
N
NH2
HO
F
HO Cl
Refractory acute lymphoblastic leukemiain pediatrics (Genzyme)
Figure 3: Modified nucleosides in anticancer therapy
Even though the 2-fluoro substitution present in Fludarabine has a similar effect, the 2-chloro
substitution is preferred due to the severe toxic effect of the hydrolysis product 2-fluoroadenine that
may evolve from Fludarabine in the cell (Bonate et al. 2006). The disadvantage of both drugs is the
susceptibility to glycosyl bond cleavage. In case of Fludarabine the instability is caused by nucleoside
phosphorylase activity while in case of Cladribine both hydrolytic and enzymatic activity is
responsible. Owing to the arabino configuration of the C2′ hydroxyl group, the situation in
Fludarabine is better than in Cladribine. Nevertheless, it was later shown that the stability of the
glycosyl bond is even further improved by the presence of a fluorine atom in the C2′ arabino position,
which conferred resistance against nucleoside phosphorylase degradation (Montgomery et al. 1986).
Finally Montgomery and co-workers synthesized a number of 2′-fluoro-2-halo derivatives of 9-β-D-
arabinofuranosyladenine and found that 2-fluoro, 2-bromo and 2-chloro substituted derivatives of 9-
(2-deoxy-2-fluoro--D-arabinofuranosyl)adenine showed anti-leukaemic activity in the mouse model
(Montgomery et al. 1992). Following studies showed that the 2-chloro congener, later assigned as
Clofarabine, exerted the best activity (Bonate et al. 2006).
Despite the discovery of numerous anticancer agents, there is still the need for novel and advanced
drugs. A driving force for future developments lies in the poor selectivity of many topical
chemotherapeutic drugs that leads to the damage of healthy cells and organs. Moreover, multidrug
resistance emerges after prolonged incubation, caused for example by the activation of
transmembrane proteins effluxing active agents from the cell (Lowenthal and Eaton 1996,
Stavrovskaya 2000). A recently developed nucleoside drug with anticancer activity is Sapacitabine
that is now in clinical trial. Sapacitabine was demonstrated to be expedient for the treatment of solid

Introduction 9
tumours and haematological malignancies and was reported to have the potential of overcoming
resistance to some of the currently used drugs (Liu et al. 2012, Serova et al. 2007). Another objective
is to target nucleoside-transporter deficient cells that are highly resistant to nucleoside analogues.
Such challenges might be tackled by encapsulation or conjugation of target compounds to
nanoparticles (Hajdo et al. 2010). The development of suicide gene therapies represents an attempt
towards the selective killing of tumour cells. Parker and co-workers have demonstrated the feasibility
of a system employing modified nucleosides as prodrugs that are selectively cleaved in E. coli purine
nucleoside phosphorylase (PNP) transfected cancer cells. With this method cytotoxic purine bases as
for example 2-fluoroadenine could be locally liberated (Parker et al. 1997, Parker et al. 2003).
Other fields of pharmaceutical application
The potential of modified nucleoside drugs is not restricted to the treatment of viral infections and
cancer. In fact, modified nucleosides that act as purine nucleoside inhibitors are known for their
therapeutic properties. A recent example in this field constitutes an immucillin purine nucleoside
phosphorylase inhibitor that proved to be potent in a primate animal model for therapy against the
protozoan parasite Plasmodium falciparum. The parasite is responsible for most of the malarial
deaths each year (Cassera et al. 2011).
Moreover, insights into the biological activities on adenosine receptors have further broadened the
scope of potential applications of adenosine analogues. A number of adenosine analogues acting as
adenosine receptor agonists are now in clinical trials and may later be used for the treatment of
inflammation, type 2 diabetes, and arrhythmia (Samsel and Dzierzbicka 2011).
1.1.3. Modified nucleosides as building blocks for synthetic nucleic acids
The modulation of gene expression through the use of synthetic nucleic acids is an exciting research
field in both fundamental and clinical science. Antisense oligonucleotides (ASOs) and small
interfering RNAs (siRNAs) are the most widely used strategies. The profound impact on clinical drug
development is reflected by the numerous oligonucleotides that are currently in clinical trials (Watts
and Corey 2010, Watts and Corey 2012). Fomivirsen is the first ASO approved by the FDA and is used
for the treatment of cytomegalovirus retinitis (Grillone and Lanz 2001).
Pioneering studies on antisense ASOs have been reported in 1978 by Paul Zamecnik (the “Father of
Antisense”(Agrawal 2010)) and Mary Stephenson. The idea was to administer an artificial
oligonucleotide complementary to a target RNA into cells and thereby inhibiting the expression of a
target gene. The work published by Zamecnik and Stephenson demonstrated the feasibility of the
approach for the inhibition of Rous sarcoma virus replication in fibroblast cultures (Stephenson and
Zamecnik 1978, Zamecnik and Stephenson 1978).
A more recent development is the use of siRNAs that are double stranded RNA oligonucleotides
capable of entering the RNA interference pathway naturally occurring in the cell. The manipulation of
gene expression in the nematode Caenorhabditis elegans by taking advantage of RNA interference
was reported in 1998 (Fire et al. 1998). In 2006 Andrew Fire and Craig C. Mello were rewarded for

10 Introduction
their groundbreaking contribution with the Nobel Prize in Physiology or Medicine. RNA interference
(RNAi) is characterised by the cleavage of double stranded RNAs into small RNAs that afterwards
associates with proteins to form a RNA induced silencing complex. In the subsequent process the
sense strand is released, while the antisense siRNA is used to identify and destroy the homologous
mRNA target (Hammond et al. 2000, Martinez et al. 2002).
Modified nucleosides in antisense oligonucleotides and siRNA
In both antisense and RNA interference technologies, early optimism was significantly diminished
after a multitude of severe hurdles were discovered (Gura 1995). In particular, the following
obstacles and requirements have been identified as bottlenecks: i) instability of the oligonucleotides
due to nuclease-mediated degradation, ii) poor selective and stable binding to the target RNA, iii) un-
wanted triggered immune responses and iv) limitations on the level of delivery and binding to other
proteins.
Soon it was found that chemical modifications of the nucleotides could convey favourable properties
to tackle some of the aforementioned limitations. Thus, in first-generation oligonucleotides,
phosphodiester linkages were replaced by phosphorothioate linkages in order to prevent the
degradation by nucleases. Among other modified building blocks, 2′-fluorinated nucleosides proved
to have unique, favourable properties for antisense and RNA interference.
O
HO F
HO
NH
N
O
O
O
HO
F
HO
NH
N
O
O
Figure 4: Uridine substituted with fluorine in the 2′-ribo position (left) and 2′-arabino position (right)
The substitution of the 2′-ribo position with a fluorine atom locks the sugar moiety predominantly in
a C3′-endo conformation that is characteristic for the sugars in RNA helices. This feature was
exploited for the development of ASOs that better bind to the RNA target molecule due to increased
thermodynamic stability of the generated DNA/RNA duplex (Kawasaki et al. 1993). Likewise many
studies have demonstrated the favourable properties of 2′-fluorinated nucleotides in RNA
interference (Allerson et al. 2005, Deleavey et al. 2010, Manoharan et al. 2011, Morrissey et al.
2005). Particularly it was reported that the 2′-fluoro substitution leads, in comparison to non-
modified oligonucleotides, to constructs with enhanced serum and thermal stability of the duplex
with reduced immunogenicity. Furthermore, higher in vitro and in vivo potency, also in comparison
with some other modified oligonucleotides (including locked nucleic acids) was reported (Manoharan
et al. 2011).

Introduction 11
Replacement of the 2′-arabino position of the sugar with a fluorine atom leads to equally favourable
properties for RNA technology. The recruitment of RNase H that cleaves RNA in RNA/DNA duplexes
improves the efficacy of many ASOs (Watts and Corey 2012). However, chemical modifications that
are conventionally used to increase the stability of the RNA/DNA duplex or confer nuclease
resistance do not support RNase activity. In order to achieve nevertheless RNase susceptibility,
typically “gapmers” are used in which unmodified nucleotides are introduced in the middle of
oligonucleotides containing also modified constituents (Monia et al. 1993, Watts and Corey 2012).
Also, it was shown that 2′-fluoroarabinonucleic acid constituents supported RNase H activity while
retaining a high binding affinity to the RNA target (Damha et al. 1998, Watts and Damha 2008).
Hence 2′-fluoroarabinonucleosides are attractive components of oligonucleotides used for gene
silencing (Kalota et al. 2006). Recently, the expedient properties of oligonucleotides with N3′-P5′
phosphoramidate linkages (Gryaznov et al. 1995, Gryaznov 1999) were combined with the
advantages of the fluorination of the 2′-arabino position of the sugar residue. The synthesis and use
of the resulting 2′-arabino-fluorooligonucleotide N3′-P5′ phosphoramidates was patented (Gryaznov
and Schultz 2011).
Modified nucleosides for the stabilization of aptamers
The binding of oligonucleotides to other proteins in addition to the intended hybridization with the
target RNA is generally considered as unwanted off-target effect in antisense and RNA interference
strategies. However, the high affinity binding of single-stranded nucleic acids, to distinct molecular
targets has been found to be of interest also for other therapeutic approaches (Thiel et al. 2009). The
specificity of these “aptamers” is conferred by their three-dimensional structure. The incorporation
of 2′-fluoro pyrimidine nucleotides has been shown to increase the resistance against nucleases and
leads to equal or higher binding affinities to the target ligand (Adler et al. 2008, Khati et al. 2003). In
2004 the first aptamer therapeutic (Pegaptanib) has gained FDA approval and is now marketed by
Pfizer for the treatment of exudative (wet) age-related macular degeneration. The oligonucleotide
that is substituted with 2′-fluorinated pyrimidine nucleotides, is selectively directed against a
vascular endothelial growth factor (Gragoudas et al. 2004, Ng and Adamis 2006).
Noteworthy, current applications of 2′-fluorinated nucleosides often concentrate on 2′-fluro
substituted pyrimidine nucleosides - a phenomenon that might reflect the fact that the chemical
synthesis of the according purine nucleosides is significantly more challenging and therefore related
to higher costs. This aspect will be further discussed throughout the present study.
1.2. Synthetic routes towards nucleoside analogues
1.2.1. Chemical synthesis
For the chemical synthesis of nucleosides, typically a convergent approach is followed, which means
that heterocyclic base and ribose moiety are independently prepared and afterwards coupled. A
commonly used method is the silyl-Hilbert-Johnson (or Vorbrüggen) reaction, in which a silylated

12 Introduction
(nucleophilic) heterocyclic base reacts with a protected (electrophilic) sugar acetate in the presence
of a Lewis acid (Vorbrüggen and Ruh-Pohlenz 2001).
Even though the Vorbrüggen reaction and similar methods have been widely applied for the
synthesis of nucleosides, the chemical synthesis is often challenged by regio- and stereospecific
requirements that are prerequisites for the biological activity of nucleosides. In natural nucleosides
the carbohydrate moiety is connected to the N9 atom of purine bases and to the N1 atom of
pyrimidine bases, respectively. However, the presence of multiple nucleophilic sites of the
heterocyclic base poses difficulties for the regioselective formation of the glycosyl bond during
chemical synthesis. Moreover, natural nucleosides can be exclusively found in the β-anomeric
configuration in which the base is oriented above the plane of the sugar. Indeed, this requirement
can be readily achieved in the synthesis of ribonucleosides through the formation of a cyclic cation
intermediate of the ribose moiety prior to the formation of the glycosyl bond. For the synthesis of
deoxyribonucleosides the situation is more complicate due to the absence of the 2′ hydroxyl group
involved in the neighbouring group participation described above. More details on the specific
challenges encountered in the synthesis of 2′-fluorinated and 2,6-dihalogenated nucleosides will be
discussed in section 5.1.
In summary, the chemical synthesis is typically a multistep process requiring protection and
deprotection of functional groups and sophisticated procedures to achieve regio- and
stereoselectivity. Furthermore, the chemical preparation usually involves reagents that are harmful
to health and environment.
1.2.2. Enzymes for nucleoside synthesis
Enzymes are able to efficiently catalyze reactions with strict regio- and stereoselectivity under mild
reaction conditions. Protection and deprotection steps, as well as the employment of hazardous
chemical reagents are unnecessary. If utilized as biocatalysts, enzymes have thus the potential to
replace complicate chemical reactions routes. A number of enzymes interacting with nucleic acid
constituents have been investigated with respect to their ability to aid in nucleoside synthesis. It was
found that enzyme catalyzed reactions can be exploited in two major fields: i) the selective
modification of nucleosides, and ii) the formation of the glycosyl bond connecting heterocyclic base
and pentofuranose moiety.
The hydrolytic deamination of 6-aminopurine, catalysed by adenosine deaminase, represents an
example for the first category of enzymes and has been widely applied for the synthesis of modified
purine nucleosides (Santaniello et al. 2005). Moreover, enzymes have been used for the selective
modification of the sugar moiety of nucleosides. Examples include lipases, used for the regioselective
acylation of hydroxyl groups (Moris and Gotor 1993), and nucleoside oxidases that oxidize the
CH2OH group of the sugar moiety of nucleosides, which can be used for the synthesis of carboxylic
nucleoside derivatives (Mahmoudian et al. 1998).

Introduction 13
The second major application field of enzymes in nucleoside chemistry concerns the formation of the
glycosyl bond between the heterocyclic base and the pentofuranose moiety. In practice this strategy
involves the transfer of a glycosyl residue from a nucleoside donor to an acceptor base and is
catalysed by NPs or N-deoxyribosyltransferases (NdRT). Although the overall reaction – the
interchange of the base of a nucleoside - is essentially the same (Figure 5), the catalytic mechanism
of the transglycosylation reaction mediated by NPs and NdRTs is different. NdRTs (EC 2.4.2.6)
catalyze the direct transfer of a deoxyribofuranosyl moiety, while with NPs the intermediate product
α-D-pentofuranosyl-1-phosphate is formed. For this reason the NP catalyzed reaction requires the
presence of inorganic phosphate. Furthermore, NPs and NdRTs differ in their substrate specificities.
Both ribo- and deoxyribonucleosides are natural substrates of nucleoside phosphorylases. Contrarily
NdRTs are specific for 2′-deoxyribonucleosides. Regarding the specificity toward the heterocyclic
bases, NdRTs are classified in two categories. Type I is specific for the interchange of purine bases
(purine <-> purine) whereas type II NdRTs catalyze the transfer of purine and pyrimidine bases
(purine <-> purine, pyrimidine <-> pyrimidine, purine <-> pyrimidine) (Holguin and Cardinaud 1975).
In particular NdRTs from Lactobacilli have been extensively studied and used for the synthesis of
natural and artificial nucleosides (Carson and Wasson 1988, Fernandez-Lucas et al. 2010, Huang et al.
1983, Kaminski 2002, Okuyama et al. 2003).
Remarkably, metabolic enzymes of purine and pyrimidine nucleotide metabolism have also been
used to completely reshape the synthesis of nucleosides in vitro. Even such an approach appears
tedious for production of bulk chemicals, it is an important tool for the synthesis of isotope labelled
nucleic acid constituent (Schultheisz et al. 2008, Schultheisz et al. 2010).
O
HO OH
HO
NH
N
O
O
Pyrimidine nucleoside(Pentofuranosyl donor)
Pyrimidine basePurine base (pentofuranosyl acceptor)
Purine nucleoside
N
NNH
N
NH2
NH
NH
O
O
O
HO OH
HO
N
NN
N
NH2
Figure 5: General scheme of a transglycosylation reaction with a pyrimidine nucleoside as pentofuranosyl donor and a purine base as pentofuranosyl acceptor. The overall reaction can be catalyzed either by a single type II N-deoxyribosyltransferase or by two NPs in the presence of inorganic phosphate. As example the synthesis of adenosine from uridine as pentofuranosyl donor and adenine as pentofuranosyl acceptor is shown.

14 Introduction
1.2.3. The NP-catalyzed transglycosylation of nucleosides
The transglycosylation of nucleosides, mediated by nucleoside phosphorylases, proceeds in two
consecutive steps (Figure 6). In the first step a nucleoside that serves as pentofuranosyl donor is
phosphorolytically cleaved into the corresponding heterocyclic base and α-D-pentofuranosyl-1-
phosphate. In a second step this activated carbohydrate moiety is coupled to the heterocyclic base
that is used as pentofuranosyl acceptor. Hence, in the first reaction inorganic phosphate is
consumed, while in the second reaction phosphate is released.
Both reactions are catalyzed by NPs. Depending on the type of NPs used different modes of
transglycosylations are possible. If a purine nucleoside phosphorolyzing enzyme is exclusively
employed as biocatalyst, interchange of purine bases can be achieved. For this purpose
7-methylguanosine and 7-methylinosine have been reported as effective ribofuranosyl donors
(Hennen and Wong 1989, Ubiali et al. 2012). Likewise the interchange of pyrimidine bases can be
accomplished by utilizing a pyrimidine phosphorolyzing enzyme in a transglycosylation with a
pyrimidine nucleoside serving as pentofuranosyl donor and a different pyrimidine base serving as
pentofuranosyl acceptor. Following this methodology Serra and co-workers have synthesized 5-
fluoro-2′-deoxythymidine with immobilized E. coli thymidine phosphorylase (Serra et al. 2011).
Pi
+
Uridine
Phosphate
Pi
Uracil α-D-pentofuranose-1-phosphate
Adenine
Phosphate
PyNP
PyNP : Pyrimidine nucleoside phosphorylase
PNP : Purine nucleoside phosphorylase
O
HO OH
HO
NH
N
O
O NH
NH
O
O
O
HO OH
HO
OPO32-
N
NNH
N
NH2
O
HO OH
HO
N
NN
N
NH2
PNP
Figure 6: Purine nucleoside synthesis at the example of uridine as pentofuranosyl donor and adenine, functioning as pentofuranosyl acceptor
The spectrum of possible transglycosylation reactions is expanded through the combined use of
pyrimidine and purine nucleoside phosphorolyzing enzymes. With this strategy a pyrimidine base can
be substituted by a purine base or vice versa. An example is the synthesis of 5′-methyluridine by
employing guanosine as pentofuranosyl donor and thymine as acceptor (Gordon et al. 2011, Ishii et
al. 1989). The advantage is that guanine, the phosphorolysis product of guanosine precipitates from
the reaction and thereby shifts the equilibrium of the first reaction in the favourable direction.
However, of particular interest for the present study is the synthesis of modified purine nucleosides

Introduction 15
accomplished by a transglycosylation reaction with pyrimidine nucleosides acting as pentofuranosyl
donor and purine bases acting as pentofuranosyl acceptor (Figure 6). The rationale behind this
approach is that a number of modifications on the carbohydrate moiety of pyrimidine nucleosides
can be relatively easily introduced by chemical transformations. The procedure involves the
intermediate formation of an intramolecular anhydrous bond between the O-2 atom of the
pyrimidine base and the 2′, 3′, or 5′ position of the carbohydrate moiety and subsequent opening of
the oxygen containing bridge by the treatment with nucleophilic agents (illustrated in (Mikhailopulo
and Miroshnikov 2011)). By contrast, comparably simple methods for the modification of the
carbohydrate moiety of purine nucleosides do not exist. The coupling of sugar modified moieties,
donated by pyrimidine nucleosides, to natural or chemically prepared purine bases is hence an
attractive approach for the synthesis of purine nucleoside analogues (Krenitsky et al. 1981,
Lewkowicz et al. 2000, Mikhailopulo 2007, Tuttle et al. 1993, Utagawa et al. 1985b, Utagawa 1999).
1.2.4. Formulation of NPs for technical use
The least laborious way to exploit the enzymatic activities of NPs in enzymatic transglycosylations of
nucleosides is the utilization of whole cells displaying the desired activities. With this strategy
downstream processing is virtually not required which leads to a cost-effective biocatalyst
preparation. Moreover, the whole cell represents a kind of natural immobilization vehicle which
simplifies the handling and conveys stability to the biocatalysts. On the downside, whole cell
biocatalysts are complex systems exhibiting not only NP activities but various other catalytic
reactions. The overall process can hence become complicate. An example represents the synthesis of
adenosine from uridine and adenine catalyzed by E. coli BL21, as described by Lewkowicz and co-
workers (Lewkowicz et al. 2000). Even though the authors reported a 94 % yield of adenosine after
only one hour, they also observed that after prolonged incubation times the second (PNP catalyzed)
reaction was reversed and after 24 h only adenine and uracil were present. A possible explanation is
that α-D-pentofuranose-1-phosphate served as energy source for the cells and has been consumed.
Adenosine deaminase activity is another by-activity of whole cells that is often encountered as
obstacle. Comparative investigations of E. coli “adenosine nucleoside phosphorylase” (today referred
to as EcPNP) and adenosine deaminase, have shown that the latter is more sensitive to heat (Koch
and Vallee 1958). Investigations of Utagawa and co-workers revealed later that conducting the
enzymatic synthesis of ara-A at high temperature (60 °C instead of 37 °C) was a good strategy to
avoid adenosine deaminase activity of the Enterobacter aerogenes cells employed as biocatalyst
(Utagawa et al. 1980).
In other cases, by-activities of whole cells are rationally used to pursue the desired synthetic
direction. For example, cytidine deaminase activity of selected E. coli cells was used to transform 1-β-
D-arabinofuranosyl cytidine and 2′-deoxy-2′-fluorocytidine into the respective uridine derivatives,
prior to the transglycosylation reactions affording 1-β-D-arabinofuranosyl guanine and 2′-deoxy-2′-
fluoroguanosine (Mikhailopulo 2007, Zaitseva et al. 1999).

16 Introduction
The use of purified enzyme preparations offers the possibility to apply very high enzyme loadings
which is in some cases required due to poor substrate activities of chemically modified precursors
(Tuttle and Krenitsky 1992, Tuttle et al. 1993). Furthermore, purified enzymes can be selectively
applied and side-reactions as described above are not of concern.
In view of industrial applications however, it is highly desirable to make use of immobilized enzyme
preparations instead of directly employing purified enzyme solutions. Immobilized biocatalysts can
be easily recovered from the reaction mixture which simplifies the downstream processing and
facilitates biocatalyst recycling. Furthermore, immobilization is a useful tool to increase the stability
of enzymes to withstand harsh reaction conditions. In this regard the preservation of the multimeric
nature of NPs deserves special attention.
Diverse methods of immobilization have, therefore, been exploited for the use of nucleoside
phosphorylases as biocatalysts. Earlier studies report on the successful co-immobilization of NPs on
anion exchange resins (DEAE) (Hori et al. 1991, Mahmoudian 2000). To further increase chemical
stability, Zuffi and co-workers have co-immobilized E. coli UP and PNP by covalent linkage on epoxy
activated Sepabead®s resins (Zuffi et al. 2004). The higher stability allowed the use of the biocatalyst
at relatively high temperature (60 °C) in the presence of organic solvents (40 % DMSO). Rocchietti
and co-workers found the covalent linkage on glyoxyl-agarose as most suitable for B. subtilis PNP
immobilization (Rocchietti et al. 2004). Likewise PNP and PyNP from G. stearothermophilus have
been successfully covalently immobilized on aminopropylated macroporous glass (Taran et al. 2009).
On the other hand, there seems to be evidence that ionic adsorption is advantageous over covalent
immobilization for certain NPs. This appears to apply especially for members of the NPII family,
which are characterized by a homodimeric structure and a domain movement within catalysis
(Pugmire and Ealick 2002). Thus for both the immobilization of B. subtilis PyNP and E. coli TP, ionic
adsorption was found to be a suitable method while covalent immobilization was generally
detrimental (Rocchietti et al. 2004, Serra et al. 2011). A completely different strategy was followed
by Visser and co-workers. They applied spherezyme selfimmobilization (Brady et al. 2008) to further
increase the stability of an E. coli UP derivative that gained improved thermal stability through
directed evolution (Visser et al. 2011).
1.3. Nucleoside phosphorylases
The reversible phosphorolysis of nucleosides by nucleoside phosphorylases was first described by
Kalckar in 1947. Since then these remarkable enzymes have been in focus of numerous research
endeavours. The following sections will give a brief overview of some of the lessons learned.
1.3.1. Physiological role
Nucleoside phosphorylases catalyze the reversible phosphorolysis of ribo- and deoxyribonucleosides
in the presence of inorganic phosphate. The products of the reaction are α-D-pentofuranose-1-
phosphate and the nucleobase of the nucleoside substrate. In vitro studies revealed that the

Introduction 17
equilibrium of the phosphorolysis reaction is shifted towards the reverse (synthetic) reaction,
whereby this effect is significantly more pronounced in PNPs than in pyrimidine nucleoside
phosphorolyzing enzymes. Thus the equilibrium constant of the phosphorolysis reaction determined
for E. coli uridine phosphorylase with uridine as substrate was in the range of 0.54 – 0.61 (Vita et al.
1983), while the equilibrium constant obtained with E. coli PNP with inosine as substrate was 0.0175
(Jensen and Nygaard 1975). Due to these findings, PNPs have long been considered as enzymes
involved in the salvage of purine bases. However, in vivo the phosphorolysis reaction is highly
preferred over the synthetic reaction, since the products are further metabolized. Purine bases are
salvaged by hypoxanthine-guanine phosphoribosyltransferease or oxidized by xanthine oxidase to
uric acid (Bzowska et al. 2000), whereas the liberated pentose-1-phosphate is used in the catabolism
as energy source (Sgarrella et al. 1997). The catabolic role of nucleoside phosphorylases is also
reflected by the fact that they often belong to the same regulon as other nucleoside catabolising
enzymes (Hammer-Jespersen and Munch-Ptersen 1975, Tozzi et al. 1981).
However, in some specific cases NPs are used for the salvage of nucleobases, and thus permit an
alternative to the de novo synthesis of nucleosides. For example many protozoan parasites lack the
de novo purine nucleoside synthesis and rely on NPs instead to obtain building blocks for nucleic acid
synthesis (Hammond and Gutteridge 1984). This phenomenon makes the PNP of the malaria parasite
Plasmodium falciparum a promising target for the development of anti malaria drugs (Silva et al.
2007).
In humans, PNP deficiency was found to coincide with defected T-cell immunity, while B-cell function
was unaffected (Giblett et al. 1975). This discovery was the driving force for extensive efforts
towards the development of PNP inhibitors that were now recognized as potential
immunosuppressive agents for organ transplantation, as well as for the treatment of T-cell leukaemia
and T-cell related autoimmune diseases (Bzowska et al. 2000, Silva et al. 2007). Noteworthy, human
thymidine phosphorylase was found to be an angiogenic factor and proved to be identical to the
platelet-derived endothelial cell growth factor (Akiyama et al. 2004, Furukawa et al. 1992).
1.3.2. Basic nature of the catalytic mechanism
The basic principle of the catalytic mechanism is presumably similar in all nucleoside phosphorylases
(Pugmire and Ealick 2002). Studies on human PNP revealed that the nucleoside is bound in a high
energy conformation, producing steric strain that is favourable for the glycosidic bond cleavage.
Through electron flow from O4′ of the pentose moiety to the purine ring, an oxocarbenium ion is
formed, stabilized by the negative charge of the phosphate ion. Finally, the phosphate ion
participates in a nucleophilic attack at the C1 position. Active site residues interactions at the N-7
position are likely to support the flow of electrons from the glycosidic bond to the purine ring.
Active sites in E. coli and human PNP
As it will be discussed in more detail in the following section (1.3.3) nucleoside phosphorylases of the
NP-I fold encompass bacterial and eukaryotic type PNPs that are characterized by distinct substrate

18 Introduction
specificities. In both type of enzymes a ternary complex is formed which involves the enzyme and
both substrates (Bzowska et al. 2000). Moreover the geometric arrangement of purine, ribose and
phosphate binding site is similar (Mao et al. 1997). However, the mode of substrate binding differs
significantly and may account for the different substrate specificities of eukaryotic PNPs (specific for
6-oxopurines) and bacterial PNPs (accepting both 6-oxopurines and 6-aminopurines). The following
paragraphs are mostly based on (Bzowska et al. 2000) and will shortly list some differences with the
example of EcPNP and human PNP.
Figure 7: Proposed catalytic mechanism of hPNP. Figure adopted from (Pugmire and Ealick 2002)
A basic difference of the active site of both type of enzymes, is that in hexameric (bacterial type) PNP
the active site is composed out of amino acids belonging to two subunits: In EcPNP His4 and Arg43
are donated by the neighbouring subunit and interact directly via hydrogen bonds with the ligands
(Figure 8). By contrast, in hPNP Phe159 is the only residue from the neighbouring subunit and does
not directly interact with ligands. Its role is rather to complete the hydrophobic environment near
the pentose group.
In EcPNP the base binding site is more exposed and accessible than in hPNP. The N1-H of the purine
base is involved in a hydrogen bond with Glu201 in hPNP, whereas in EcPNP N1-H is linked to a water
molecule and has no contact to a protein residue. In 6-Aminopurines the aminogroup interacts with
Asp204 in EcPNP, which plays obviously a key role, since it is conserved in enzymes homologous to
EcPNP. In hPNP Glu201 and Asn243 belong to the essential residues involved in base binding. The
role of Asn243 may be attributed to the recognition of 6-oxo purines as substrates: The replacement
of Asn243 by Asp changed specificity of trimeric PNPs from 6-oxopurines to that of bacterial type
PNPs.
The phosphate binding site is more positively charged in EcPNP than hPNP, which is the result of
three Arg residues interacting with phosphate (Arg24, Arg87, Arg43), whereas in human PNP there is
only one Arg residue (Arg84) that interacts with phosphate.
Both PNP types use hydrophobic interactions for the hydrophobic site of pentose ring, while
hydrophilic site of the pentose ring, with hydroxyl groups, faces the phosphate binding site.

Introduction 19
Figure 8: Key active site residues of EcPNP and their interactions with inosine. W = water molecule. Figure taken from (Bennett et al. 2003)
Overview of different nucleoside phosphorylases
All nucleoside phosphorylases are pentosyltransferases that catalyze the β-glycosidic bond cleavage
of nucleosides, whereby a pentufranosyl-1-phosphate is formed under inversion of the configuration.
Based on the specific substrates, enzyme names and enzyme commission number (EC) were
assigned. Table 1 lists enzymes and acronyms that will be of relevance in the present report.
Table 1. Nucleoside phosphorylases
Enzyme name Acronym EC number
Purine nucleoside phosphorylase PNP 2.4.2.1
Pyrimidine nucleoside phosphorylase PyNP 2.4.2.2
Uridine phosphorylase UP 2.4.2.3
Thymidine phosphorylase TP 2.4.2.4
5′ -Methylthioadenosine phosphorylase MTAP 2.4.2.28

20 Introduction
1.3.3. Classification of NPs
In 2002 a comprehensive classification of nucleoside phosphorylases was established by Pugmire and
Ealick. It was found that NPs can be assigned to one of two groups that are distinct in the structural
fold of their subunits (Figure 9). The nucleoside phosphorylase-I family is characterized by a common
single domain α/β subunit fold with trimeric or hexameric quaternary structure and includes NPs
accepting purine nucleosides (viz. PNP and MTAP) as well as uridine phosphorylase (UP). Members of
the NP-II family are homodimers, in which each subunit consists out of two domains: a large mixed
α/β domain, separated by a cleft from a smaller α-helical domain. NPs assigned to this family share a
high degree of sequence identity and are currently known to exclusively accept pyrimidine bases and
corresponding nucleosides as substrate but not purine bases or purine nucleosides.
NP-I family
• Subunits with common α/β-fold (single-domain)
• Substrates: Purine nucleosides and uridine
Hexamers Trimers
NP-II family
• Homodimeres, two-domain subunits
• High degree of sequence identity
• Significant domain movement needed for catalysis
• PyNP: phoyphorolyses of thymidine and uridine
• TP: specific for thymidineBacterial PNP UP
Specific for:
6-aminopurines
6-oxopurine
Mammalian PNP MTAP
Specific for:
6-oxopurines
Figure 9: Classification of NPs based on Pugmire and Ealick (Pugmire and Ealick 2002)
NP-I family
On the basis of substrate specificity, molecular mass, quaternary structures and amino acid
sequences, members of the NP-I family can be further categorized. Hence Pugmire and Ealick have
defined a trimeric and a hexameric subgroup of the NP-I family. PNPs with trimeric quaternary
structure, described before by Bzowska as low-molecular-mass PNP (Bzowska et al. 2000), are
specific for 6-oxopurines and their nucleosides, but do not accept 6-aminopurines or nucleosides
thereof (e.g. adenosine) as substrate. By contrast, high-molecular-mass PNPs (with hexameric
quaternary structure) accept both 6-aminopurines as well as 6-oxopurines as substrate. However, in
some cases high-molecular-mass PNPs were found to have a significant higher specificity towards
adenosine in comparison to 6-oxopurine (nucleosides) (Mcelwain et al. 1988, Sgarrella et al. 2007,
Trembacz and Jezewska 1993). Therefore, respective enzymes are also referred to as adenosine

Introduction 21
phosphorylases, as it is the case for the high-molecular-mass PNP from B. cereus (Sgarrella et al.
2007).
While trimeric PNPs are found in eukaryotes, the hexameric form is prevalent in bacteria. However,
in a number of bacteria both, a PNP with substrate specificity of low-molecular-mass as well as a PNP
with substrate specificity for high-molecular-mass can be found. Examples include the PNPs from
E. coli, G. stearothermophilus, B. subtilis and B. cereus (Bzowska et al. 2000).
Sequence analysis has shown that the trimeric subgroup of the NP-I family also encompasses
5′-methylthioadenosine phosphorylase (2.4.2.28) and the hexameric NP-I subgroup includes uridine
phosphorylase (2.4.2.3).
NP-II family
TP and PyNP form the nucleoside phosphorylase-II family, which share a common two-domain
subunit fold and a high level of sequence identity (Pugmire and Ealick 2002). Despite the similarity of
the reaction catalyzed, uridine phosphorylase (UP; EC 2.4.2.3) belongs to the phosphorylase-I family
with distinct structural characteristics. From the catalytic point of view, TP is distinguished from UP
due to its high specificity for the 2′-deoxy-D-ribofuranose moiety of pyrimidine nucleosides (Pugmire
and Ealick 2002). By contrast, PyNP does not discriminate between uridine and thymidine and
accepts both compounds as natural substrates (Saunders et al. 1969).
Other relevant aspects concerning the classification of NPs
Ten years later, the classification of nucleoside phosphorylases presented by Pugmire and Ealick in
2002 is still helpful for the classification of NPs or for assumptions regarding quaternary structures or
substrate specificities of unknown NP gene products. However, not all relevant aspects have been
covered by the theory. Some critical aspects will be shortly stated in this section.
A number of PNPs with dimeric or tetrameric quaternary structures were reported (reviewed in
Bzowska et al. 2000), while in the classification in 2002 exclusively trimeric and hexameric quaternary
PNP structures are considered. However, many of the earlier studies have relied on gel filtration or
electrophoresis in order to determine quaternary structures. Such methods may lead to incorrect
conclusions, as illustrated by Bzowska and co-workers (Bzowska et al. 2000): By gel filtration they
found that PNP from Cellulomonas has a tetrameric quaternary structure, even though a trimeric
structure was expected (Tebbe et al. 1997). However, in the crystal structure the enzyme was later
found to be a trimer (Tebbe et al. 1999), as it was originally expected from the PNP subgroup
classification.
In other cases it appears to be obvious that quaternary structures are not consistent with the
theoretically expected structures. For example human UP was found to be a homodimer (Roosild et
al. 2009), even though the classification implies that UPs are hexamers. Likewise the crystal structure
of trypasonomal UP revealed a homodimeric enzyme (Larson et al. 2010). Moreover, a number of
hexameric PNPs were found to display substrate specificities of the low-molecular-mass PNPs that
were previously associated by Pugmire and Ealick with a trimeric quaternary structure. Examples of

22 Introduction
these hexameric enzymes are the PNPs from T. thermophilus (Tahirov et al. 2004),
Plasmodium falciparum (Daddona et al. 1986, Schnick et al. 2005, Shi et al. 2004), and from the
hyperthermophilic archaeon P. furiosus (Cacciapuoti et al. 2007).
Other deviations from the general perspective on NPs concern the substrate spectra. For example
the NP from Klebsiella was shown to accept both pyrimidine nucleosides as well as purine
nucleosides as substrates. The relative activity was highest for uridine (368 %), but the enzyme also
had high activity towards deoxyinosine (254 %) and other purine nucleosides, while thymidine
showed significant less substrate activity (29 %) (Ling et al. 1990). Other studies revealed that purine
nucleoside phosphorylases with cytidine activity exist (Mikhailopulo and Miroshnikov 2011). And
finally, a novel NP enzyme specificity was recently discovered. The putative MTAP from
Pseudomonas aeruginosa was found to be a NP with specificity towards 5′-deoxy-5′-methylinosine,
whereas 5′-deoxy-5′-methyladenosine was not accepted as substrate (Guan et al. 2011). 5′-Deoxy-5′-
methylinosine specific enzymes have not been described before.
New insights have been also gathered by comprehensive studies on arachaeal PNPs and MTAPs.
Examples include the MTAPI (Appleby et al. 2001, Cacciapuoti et al. 1994) and MTAPII from
Sulfolobus solfataricus (Cacciapuoti et al. 2005), as well as PNP (Cacciapuoti et al. 2007) and MTAP
(Cacciapuoti et al. 2004) from P. furiosus. The research in this area also opens the way for new
conclusions regarding the structural and functional differences between MTAP and PNP (Cacciapuoti
et al. 2011).
1.3.4. From natural to recombinant production of nucleoside phosphorylases
The phosphorolytic cleavage of nucleosides by NPs has been described for a multitude of living
organisms, spanning the three domains of life (Pugmire and Ealick 2002). Microorganisms have been
exploited in particular as natural nucleoside phosphorylase (NP) producers for biocatalytic
applications. In this regard, comprehensive screening studies have led to the identification of
microorganisms that are especially suitable for the enzymatic synthesis of nucleosides and analogues
thereof. Thus, Utagawa and co-workers have tested more than 240 microorganisms representing 26
bacterial genera towards their ability to produce ara-A (9-β-D-arabinofuranosyladenine) from ara-U
(9-β-D-arabinofuranosyluracil) and adenine, and selected an Enterobacter aerogenes strain as best
producer (Utagawa et al. 1980). Since the reaction did not proceed without inorganic phosphate, it
was assumed that the transglycosylation reaction was catalyzed by nucleoside phosphorylases.
B. stearothermophilus, possessing one PyNP and two PNPs was found to be the best producer of a
number of 6-modified nucleosides among 100 microorganisms screened (Trelles et al. 2005).
Likewise E. coli strains with specific catalytic properties including nucleoside phosphorylase activity
have been selected and successfully applied for the synthesis of modified nucleosides (Barai et al.
2002, Zaitseva et al. 1999, Zinchenko et al. 1990). Recently T. thermophilus strains were screened as
whole cell biocatalysts for their productivity in the synthesis of natural purine nucleosides
(Almendros et al. 2009). The T. thermophilus strains were found to have beneficial properties for this
application due to high productivity values, the absence of adenosine deaminase activity and the

Introduction 23
thermostability, which permitted to conduct the screening at 65 °C. However, based on the results it
was not clear whether NdRTs or NPs are responsible for the transglycosylation reactions.
It is obvious that such screening approaches offer a great potential for the identification of
biocatalysts with desirable substrate specificities or other favourable properties as enhanced thermal
stability. However, with the aim in view to design efficient industrial processes, the volumetric yield
of NPs reached with such strategies is often rather unsatisfactory. The reasons can be found in
difficulties to cultivate NP producer strains to high cell densities or in the low amount of NPs
naturally present in the cells. In fact, even screening approaches are often restricted to
microorganisms that are easy to cultivate in standard culture media (Condezo et al. 2006). On the
other hand, strategies to increase the expression level of NPs per cell have been developed. Thus, the
addition of nucleosides and related compounds to the culture medium was used in order to
maximize the induction of NPs in Enterobacter aerogenes (Wei et al. 2008). The extent of culture
growth, that is the growth phase of the cells at the time of harvest, constitutes another influential
parameter and was investigated to maximize the specific activity of E. coli BL21 whole cell
biocatalysts (Rogert et al. 2002).
On the other hand, the advances in recombinant DNA technology opened new dimensions of the
high-level production of proteins: By changing the genetic context of a gene coding sequence,
unnaturally high expression levels can be reached. If thereby the source species of the gene
sequence coincides with the host used to produce the target protein, the methodology is referred to
as homologous expression. With this strategy overproducing strains of E. coli UP, TP, and PNP have
been generated (Esipov et al. 2002). In contrast, heterologous expression is referred to the
methodology where a gene is derived from a species that does not coincide with the expression host,
into which the gene is inserted for overexpression. The availability of such a technology has greatly
expanded the scope of NPs that can be efficiently produced and studied. This includes NPs from
microorganisms that are difficult to cultivate, currently not available, or even not cultivable at all. The
mere gene sequence information is sufficient to have a template for the chemical synthesis of the
corresponding DNA that can be subsequently inserted into an appropriate expression system.
Heterologous expression has thus lead to the expression of highly interesting NPs from a variety of
remarkable microorganisms that thrive under extreme conditions or are otherwise difficult to
cultivate. Examples include: purine nucleoside phosphorylase from the cold- adapted marine
bacterium Pseudoalteromonas sp. Bsi590 (Li et al. 2008), uridine phosphorylase from the pathogenic
protozoan Trypanosoma brucei (Larson et al. 2010), purine nucleoside phosphorylase from the
hyperthermophilic archaeon P. furiosus (Cacciapuoti et al. 2007), 5′-deoxy-5′-methylthioadenosine
phosphorylase from S. solfataricus (Cacciapuoti et al. 2005), as well as the purine nucleoside
phosphorylase from the alkaliphilic microorganism Bacillus halodurans (Visser et al. 2010). In future,
metagenomic approaches that have been recognized as powerful strategies for the discovery of new
genes within the search for “ideal” biocatalysts (Cowan et al. 2004), might also become important
tools for the discovery of novel NP variants. Recently, Cieslinski and co-workers have found a MTAP
that resembles the MTAP from Psychrobacter arcticus 273-4 by screening a metagenomic library that
was generated from environmental DNA isolated from Antarctic topsoil. Remarkably the scientists

24 Introduction
were actually screening for lipolytic enzymes; and the clone expressing the MTAP has just captured
their interest because it showed unusual degree of pink fluorescence on the screening-agar
containing rhodamine B (Cieslinski et al. 2009).
1.4. Motivation and structure of the present work
1.4.1. Enzyme-assisted synthesis of modified purine nucleosides
The ultimate goal of the present study is to improve the chemo-enzymatic route towards modified
purine nucleosides by taking advantage of novel nucleoside phosphorylases, derived from
thermophilic microorganisms. In focus is thereby the synthesis of purine nucleoside analogues with
modifications on the carbohydrate moiety (2′-fluorinated purine nucleosides) or with modifications
on the purine base (2,6-dihalogenated purine nucleosides). 2′-Fluorinated purine nucleosides display
interesting pharmaceutical activities (Bonate et al. 2006, Tuttle and Krenitsky 1992, Tuttle et al.
1993) and are furthermore valuable precursors for the synthesis of synthetic oligonucleotides with
advanced properties (Adler et al. 2008, Allerson et al. 2005, Damha et al. 1998, Deleavey et al. 2010,
Gragoudas et al. 2004, Kalota et al. 2006, Kawasaki et al. 1993, Khati et al. 2003, Manoharan et al.
2011, Morrissey et al. 2005, Ng and Adamis 2006, Watts and Damha 2008), whereas
2,6-dihalogenated purine nucleosides are convenient precursors in the preparation of various
therapeutic purine nucleoside analogues (Kazimierczuk et al. 1984, Montgomery et al. 1986, Tennilä
et al. 2000).
While the chemical synthesis of numerous pyrimidine nucleoside analogues is well established, the
synthesis of equivalent purine nucleoside analogues is often rather complicate and is typically a
multistage process that is hampered by the formation of regio- and stereoisomers. The utilization of
NPs as biocatalysts offers the possibility to transfer pentofuranosyl moieties from a pyrimidine
nucleoside pentofuranosyl donor to a purine base serving as pentofuranosyl acceptor. Such
biocatalytic applications of enzymes that operate under strict stereo- and regioselectivity is generally
a promising approach towards the development of efficient and high yielding synthetic routes, and
offers, furthermore, the possibility to design environmentally friendly processes.
By employing chemically prepared, artificial precursors as substrates, novel, modified nucleosides
can be synthesized with this strategy. Unfortunately, a number of highly interesting nucleoside
analogues are hardly recognized as substrate by NPs that are currently in use. For example the
catalytic rates of E. coli NPs in reactions involving 2′-fluorinated nucleosides are dramatically reduced
in comparison to the activities towards natural nucleosides. Therefore, high amounts of enzyme
loading and prolonged incubation times are needed to obtain reasonable yields (see e.g. Tuttle and
Krenitsky 1992). It is therefore appealing to study novel NP variants that possibly display more
favourable substrate specificities.

Introduction 25
1.4.2. Taking advantage of thermostable nucleoside phosphorylases
A major limitation towards the wide application of enzymes in chemical synthesis concerns the
instability under harsh reaction conditions, for example at high temperature. However, the operation
at higher temperature has many advantages: higher reaction rates can be achieved, due to a
decrease in viscosity and an increase in diffusion coefficients of substrates. The final yield may be
increased due to higher solubility of substrates that are only poorly soluble at ambient temperature,
as it is also the case for many purine bases. And finally there is a lower risk of contamination by
common mesophiles.
For these reasons inherently thermostable enzymes derived from thermophilic microorganisms have
been recognized as powerful biocatalysts for industrial processes (Haki and Rakshit 2003). It has been
shown, that thermal stability is not the result of a unique phenomenon. Instead, nature has found
diverse strategies towards enzyme stabilization: increased electrostatic interactions, greater
hydrophobicity and better atom packing, deletion or shortening of loops and disulfide bonds are just
some examples. By taking advantage of enzymes from thermophiles the intrinsic stability of these
proteins, that is the product of evolution, can be exploited. Noteworthy thermal stability is often
accompanied by a higher resistance against denaturants and other harsh reaction conditions that
may be encountered in industry.
1.4.3. Recombinant expression of thermostable NPs in E. coli
Since thermophilic microorganisms are generally difficult to cultivate to high cell densities (Krahe et
al. 1996), the target enzymes of this study will be expressed in E. coli. This approach offers the
possibility to produce the enzymes in high yields and simplifies the downstream processing: Major
parts of the mesophilic endogenous E. coli enzymes can be simply removed by heat precipitation.
Although recombinant expression can be understood primarily just as a tool here, it should be noted
that the outcome is pivotal for the entire study. In fact, the efficient expression of thermostable
enzymes is challenged by various factors. Knowledge gained in this field of research may further
support the use of thermostable enzymes in industrial processes.
1.4.4. Experimental outline and key objectives
For the realization of the aims outlined above, three work packages have been defined. The results of
each work package will be presented in each one of the following chapters.
The first work package represents the Recombinant expression of thermostable NPs. This work
package includes the selection of target enzymes, cloning and optimization of the expression in
E. coli. The second work package is the Characterization of recombinant NPs. Here the aim is to
determine characteristics of the enzymes that will be important for the final application. In focus will
be temperature optima, thermal stabilities and substrate spectra of the enzymes. The final work
package is the Application in the synthesis of modified nucleosides. This work package will show

26 Introduction
whether the generated thermostable enzymes are indeed beneficial for the synthesis of modified
purine nucleosides that have been defined as target compounds.

2. Experimental part
2.1. Generation of expression plasmids
2.1.1. Gene isolation
G. thermoglucosidasius 11955 was grown for 1.5 days at 52 °C in LB (10 g l-1 tryptone, 5 g l-1 yeast
extract, 10 g l-1 NaCl, pH 7.0). The genomic DNA was isolated using an adapted standard protocol
(Moore and Dowhan 2002). Shortly the procedure involved the treatment with proteinase K in the
presence of SDS, ethanol precipitation of nucleic acids, a washing step (with 70 % ethanol) and re-
suspension of the air-dried pellet in a solution containing RNase A. Genomic DNA of T. thermophilus
HB27 was obtained from Thomas Schwab from the Institute of Biophysics and Physical Biochemistry,
University of Regensburg. Genomic DNA of D. geothermalis and A. pernix were isolated before by
Marco Casteleijn in the Bioprocess Engineering Laboratory of the University of Oulu, Finland. The
target gene sequences were amplified from genomic DNA using Pfu DNA polymerase (Fermentas,
Lithuania; now Thermo Scientific).
2.1.2. Plasmids
The pCTUT7 vector was chosen as basis for the generation of NP expression vectors. This vector is
characterized by an IPTG inducible lac promoter derivate and a pBR322 origin of replication. In
addition this vector contains attR recombination sites through which a gene coding sequence can be
conveniently introduced by recombinational cloning. For more details see (Šiurkus et al. 2010).
For some expression studies pET21a (Novagen) was used that is based on the T7 expression system.
The T7 promoter located upstream of the target sequence on the plasmid, is recognized by the RNA
polymerase of the T7 bacteriophage. The corresponding expression cassette is usually genomically
integrated in the expression strains, indicated by the genotype annotation (DE3). The T7 expression
system is considered as very efficient.
Table 2. Chaperone plasmid set (Takara)
Plasmid Chaperone Promoter Inducer
pG-KJE8 dnaK-dnaJ-grpE
groES-groEL
araB
Pzt1
L-Arabinose
Tetracyclin
pGro7 groES-groEL araB L-Arabinose
pKJE7 dnaK-dnaJ-grpE araB L-Arabinose
pG-Tf2 groES-groEL-tig Pzt1 Tetracyclin
pTf16 Tig araB L-Arabinose

28 Experimental part
For co-expression experiments a set of chaperone plasmids (Takara Bio Inc., Otsu, Japan) was used.
Chaperone expression, that has been shown to promote proper folding of recombinantly expressed
proteins (Baneyx and Mujacic 2004, Nishihara et al. 1998, Schlieker et al. 2002), is here induced by
addition of L-arabinose or tetracycline. The plasmids have a chloramphenicol resistance marker and a
p15A origin of replication. For more details see Table 2.
2.1.3. Recombinational cloning
a) Construction of the DgPNP entry vector (pEntrDgeo1497)
The following primer pair was used to amplify the gene coding for DgPNP: forward -
5′ GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAAACCTGTATTTTCAGGGCATGGTGGTGGCGCGTGTAC
CGG 3′, reverse - 5′ GGGGACCACTTTGTACAAGAAAGCTGGGTTCACATGCTGTTGGAAGGTACT 3′ (the
underlined portion represents the gene specific template, the 5′ overhang includes attB1
recombination site and an engineered TEV cleavage site in the forward primer and the attB2
recombination site in the reverse primer). The purified PCR fragment (PeqGold gel extraction kit C-
line, Peqlab Biotechnologie GmbH, Erlangen, Germany) was inserted into the pDONR201™ vector
(Invitrogen) by the site specific “BP” recombination reaction with BP Clonase II enzyme mix
(Invitrogen) according to the manufacturer’s manual.
b) Construction of DgPNP expression vectors (pCTUT7_DgPNP and pCTUT7A_DgPNP)
Following Invitrogen’s manual, the DgPNP gene was transferred from the entry vector to the
cytoplasmic expression vector pCTUT7 (Šiurkus et al. 2010) that confers a N-terminal hexahistidine
tag, by the site specific “LR” recombination reaction (LR clonase II enzyme mix, Invitrogen). The
resulting expression vector was assigned as pCTUT7_DgPNP. The DgPNP gene was also transferred
into the pCTUT7A vector, a derivative of the pCTUT7 vector, resulting in the plasmid
pCTUT7A_DgPNP. Construction of the pCTUT7A destination vector is described in the following
section.
A) B)
Figure 10: Vector maps of destination vectors pCTUT7 (A) and pCTUT7A (B) in which the chloramphenicol resistance cassette (Cmr) was replaced by an ampicillin resistance cassette (Ampr) and the plasmid stabilizing parB locus.

Experimental part 29
2.1.4. Cloning via restriction and digestion
PCR-amplified fragments containing the gene coding sequences (see Appendix) were cloned into the
vector backbones through the treatment with FastDigest restriction endonucleases (Fermentas,
Lithuania) and subsequent ligation (T4 DNA Ligase, Roche). The oligonucleotide sequences
(synthesized by TIB Molbiol, Berlin Germany) that were used for the amplification of the inserts,
restriction enzymes and vector backbones used for the construction of each expression vector are
summarized in Table 3.The Vector NTI software (Invitrogen) was used to plan and analyze cloning
procedures.
The plasmid pCTUT7 was modified by replacing the chloramphenicol with an ampicillin resistance
cassette, and introducing the plasmid stabilizing parB locus [22]. Therefore the ampicillin resistance
cassette was amplified with the first primer pair shown Table 3 in the first line. The 580 bp long parB
region of plasmid R1 (GenBank A20060.1) was amplified with the second primer pair from the
plasmid pKG1022, kindly provided by Prof. Thomas Schweder. The resulting vector was assigned as
pCTUT7A, and was used as destination vector for recombinational coning, as well as vector backbone
for the construction of expression vectors via restriction and ligation (see Table 3).
For the construction of the plasmid pKS2_ApUP (Figure 11) the primer extensions for the insert
amplification were designed in such a way, that a hexahistidine tag is fused to the N-terminus of the
target protein. A BamHI restriction site was engineered between hexahistidine and gene coding
sequence; hence the pKS2_ApUP vector could be conveniently used for cloning of the other target
genes to be expressed with N-terminal hexahistidine tag (Table 3).
AGGAGATATACATATGATGAGAGGATCGCATCACCATCACCATCACGGATCCGGAGACGAGAGTCTA
NdeI BamHI6 x his ApUP
Figure 11: Vector map of pKS2_ApUP. Native gene sequences can be inserted in this vector by NdeI/HindIII digestion. Alternatively inserts can be cloned by BamHI/HindIII digestion; in this case a hexahistidine tag will be fused to the N-terminus of the target protein.

30 Experimental part
2.1.5. Verification of the cloning steps and propagation of vector constructs
The progress of cloning was followed by the visualization of amplified DNA fragments and digested
vectors on agarose gels. After the ligation step, plasmids were transformed by electroporation
(electroporator 2510, Eppendorf) into E. coli TOP10 (for details see Table 5) following a standard
protocol (Sambrook and Russell 2001). For the propagation of pCTUT7 and pCTUT7A an E. coli strain
with resistance to the ccdB gene product (E. coli ccdB+, Table 5), encoded on both vectors, was used.
Single colonies obtained after overnight cultivation on agar plates were screened for the presence of
the target gene sequence via PCR. Positive clones were cultivated and the plasmids were extracted
and purified (Invisorb® Spin Plasmid kit, Invitek, Berlin, Germany). The plasmid concentration was
determined by a spectroscopic measurement at 260 nm (Nanodrop, Thermo Scientific). In a next step
the plasmids were digested with restriction enzymes. If the restriction pattern was in accordance
with the theoretically expected pattern, plasmids were sent for sequencing to confirm that the target
sequence is correct.
2.1.6. Site-directed mutagenesis
Site-directed sequence optimizations were normally introduced by the primers that served for insert
amplification. With this method the following expression plasmids were generated: pKS1_DgPNP1,
pKS1_DgPNP2, pKS1_ApMTAP1, pKS1_ApMTAP2, pKS1_ApUP1 (see Table 3 for corresponding primer
pairs). Another approach involved the amplification of an entire plasmid that served as template.
With this method the GtPyNP sequence was modified to match the corresponding data bank amino
acid sequence. Kapa Hifi DNA polymerase (Kapa Biosystems, Woburn, United States) that is especially
suitable for long range amplification with high fidelity was used for this purpose. The template
sequence was afterwards removed by DpnI digestion. The freshly, in vitro synthesized plasmids
containing the modified sequence are not recognized by DpnI since they lack methylation patterns
generated in vivo by dam positive E. coli strains. The mutagenesis primers used for this purpose
were:
Forward primer: 5′ GCAGGAGCGAAGCGGCTCGCAACAGCGATG 3′
Reverse primer: 5′ CATCGCTGTTGCGAGCCGCTTCGCTCCTGC 3′

Experimental part 31
Table 3. Construction of expression vectors via restriction and digestion
Vector Primer pair for insert amplification (5′ – 3 ′) Vector backbone
Restrict. enzymes
pCTUT7A F1: GAAATGTGCGCGGAACC
R1: TACTAGCCATGGCAATCTAAAGTATATATGAGTAAAC
F2: GGTTCCGCGCACATTTCAACAAACTCCGGGAGG
R2: TGCATGAAGCTTACAACATCAGCAAGGAGA
pCTUT7 NcoI
HindIII
pKS1_DgPNP F: TACTAGCATATGGTGGTGGCGCGTGTAC
R: CAGCATAAGCTTTCACATGCTGTTGGAAGG
pCTUT7A NdeI
HIndIII
pKS1_DgPNP1 F: TACTAGCATATGATTGCGCGTGTACCGGCAA
R: same as for pKS1_DgPNP
pCTUT7A NdeI
HIndIII
pKS1_DgPNP2 F: TACTAGCATATGATTGCCCGAGTACCCGCACGTCCTTTCGCTTCCCCGC
R: same as for pKS1_DgPNP
pCTUT7A NdeI
HIndIII
pKS2_DgPNP F: TACTAGGGATCCGTGGCGCGTGTACCGG
R: same as for pKS1_DgPNP
pKS2_ApUP BamHI
HindIII
pKS1_ApMTAP F: ACTAGCATATGAGGAAGCCGGTTCACCTCG
R: AGCATAAGCTTCTAGACTCCTCCTGTGAGG
pCTUT7A NdeI
HIndIII
pKS1_ApMTAP1 F: ACTAGCATATGAGGAAGCCAGTTCACCTAGAGGCAGGGCCCGGC
R: same as for pKS1_ApMTAP
pCTUT7A NdeI
HIndIII
pKS1_ApMTAP2 F: ATATACATATGAGGAAACCAGTACACCTAGAGGCAGGGCC
R: same as for pKS1_ApMTAP
pCTUT7A NdeI
HIndIII
pKS2_ApMTAP F: ACTAGGGATCCAGGAAGCCGGTTCACCTCG
R: same as for pKS1_ApMTAP
pKS2_ApUP BamHI
HindIII
pET21a_GtPNP F: ACTAGCATATGAGCATCCATATCGAAGCAA
R: AGCATGAATTCTTATTCTACGCGAATCGCC
pET21a NdeI
EcoRI
pET21a_GtPNPh F: same as for pET21a_GtPNP
R: CAGCATAAGCTTTTCTACGCGAATCGCCG
pET21a NdeI HindIII
pKS2_GtPNP F: ACTAGGGATCCTTGAGCATCCATATCGAAG
R: AGCATAAGCTTTTATTCTACGCGAATCGCC
pKS2_ApUP BamHI HindIII
pET21a_GtPyNP F: ACTAGCATATGGTCGATTTAATTGCGAAAA
R: AGCATGAATTCTTATGAAATGGTTTCGTAT
pET21a NdeI EcoRI
pKS2_GtPyNP F: ACTAGGGATCCATGGTCGATTTAATTGCGA
R: AGCATGCGGCCGCTTATGAAATGGTTTCGTATATA
pKS2_ApUP BamHI
NotI
pKS2_TtPyNP F: ACTAGGGATCCAACCCCGTGGTCTTCATC
R: AGCATGCGGCCGCCTAGATGGCCTCCAGGA
pKS2_ApUP BamHI
NotI
pKS1_ApUP F: TACTAGCATATGGGAGACGAGAGTCTAAGG
R: CAGCATAAGCTTCTATGTGCGTCTGCACGC
pCTUT7A NdeI HindIII
pKS1_ApUP1 F1: same as for pKS1_ApUP
R1: GGGCCACATCCCCACGCCGGACCCTCAGATGG
F2: CCATCTGAGGGTCCGGCGTGGGGATGTGGCCC
R2: same as fpr pKS1_ApUP
pCTUT7A NdeI HindIII
pKS2_ApUP F1: CATCACCATCACCATCACGGATCCGGAGACGAGAGTCTAAGG
F2: TACTAGCATATGATGAGAGGATCGCATCACCATCACCATCACGG
R: same as for pKS1_ApUP
pCTUT7A NdeI HindIII
pKS2_ApUPsh F: ACTAGGGATCCGTGGCCCGCTACGTTCTCC
R: same as for pKS1_ApUP
pKS2_ApUP BamHI HindIII
Bold letters indicate restrictions sites and the underlined portion represents the insert-specific sequence. F = forward, R = reverse.

32 Experimental part
2.2. Bioinformatics
Table 4. Bioinformatic tools
Program Reference Application
Vector NTI Invitrogen Vector design,
molecular weight prediction
CYSPRED (Fariselli and Casadio 1999) Cysteine oxidation state
prediction
GenScript Rare Codon
Analysis Tool
GenScript
(https://www.genscript.com)
Codon adaption index
determination
Rare codon caltor http://people.mbi.ucla.edu/sumchan/
caltor.html
Rare codon analysis
Mfold (Zuker 2003) mRNA secondary structure
prediction
CCP4mg (McNicholas et al. 2011). Visualization of 3D protein models
2.2.1. Amino acid sequence analysis and homology modelling
Amino acid sequence identities were assessed with the protein basic local alignment tool (BLAST)
(Altschul et al. 1990) of the National Center for Biotechnology Information web server
(http://blast.ncbi.nlm.nih.gov). For multiple sequence alignments and phylogenetic analysis the
ClustalW2 - multiple sequence alignment or the ClustalW2 - phylogeny tool (Larkin et al. 2007),
respectively, have been used, both accessible through the European Bioinformatics Institute web
server (http://www.ebi.ac.uk). Three-dimensional models of the protein structures of the target
proteins were built by homology modelling using the Swiss-model workspace (Bordoli et al. 2009).
2.2.2. Secondary mRNA prediction and sequence optimization
Secondary mRNA prediction and optimization of the 5′ mRNA of the DgPNP constructs are described
in detail in (Szeker et al. 2011). For the prediction of the stability of 5′ mRNA regions of other
constructs a sequence comprising the Shine-Dalgarno sequence (AGGAGA) and the first 37
nucleotides of the coding region was used for secondary structure prediction (Kudla and co-worker
have shown, that after these 37 coding nucleotides, the correlation between mRNA folding energy
and protein expression drops significantly (Kudla et al. 2009)). For retrieving the minimum free
energy of secondary structure formation in this region, the mfold web server was used (Zuker 2003).
Optimized sequences were generated by synonymous codon substitutions.

Experimental part 33
2.3. Bacterial growth and recombinant protein expression
Table 5. Genotype of the used E. coli strains.
Strain Genotype Company
E. coli TOP10 F–mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacΧ74 recA1
araD139 Δ(ara-leu) 7697 galU galK rpsL (StrR) endA1 nupG λ-
Invitrogen
E. coli ccdB+ F– mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1
araΔ139 Δ(ara-leu)7697 galU galK rpsL (StrR) endA1 nupG fhuA::IS2
Invitrogen
E. coli BL21 F– ompT hsdSB(rB– mB
–) gal dcm Novagen
E. coli Origami Δ( ara–leu)7697 ΔlacX74 ΔphoA PvuII phoR araD139 ahpC galE galK rpsL F'[lac+ lacI )pro] gor522::Tn10 trxB (KanR, StrR, TetR)
Novagen
E. coli Rosetta2 F–ompT hsdSB(rB– mB
–) gal dcm pRARE2 (CamR) Novagen
E. coli Rosetta (DE3) F–ompT hsdSB(rB– mB
–) gal dcm (DE3) pRARE2 (CamR) Novagen
2.3.1. Preparation of recombinant E. coli cell banks
The generated expression vectors (Table 3) were transformed into appropriate E. coli expression
strains (Table 5). Single colonies were picked and cultivated overnight in LB medium. Glycerol was
added to the culture broth to a final concentration of 25 % and aliquots were frozen and stored
at -80 °C.
2.3.2. Recombinant protein expression
Cultivations were performed in a shaking incubator with 2.5 cm shaking orbit (Lab-Therm LT-X,
Kühner, Basel, Schweiz), at 250 rpm for expression in 24-squarewell Deep well plates (HJ-Bioanalytik,
Mönchengladbach, Germany) with 10 ml total volume and a liquid volume of 3 ml, or 200 rpm for the
expression in Ultra Yield Flasks™ that were covered with AirOTop™ seals (Thomson Instrument
Company, USA).
Different media were used for expression studies. Initial expression studies with DgPNP constructs at
different temperatures were performed in LB medium. Therefore cells from LB agar plates grown
overnight were used to inoculate LB medium to an initial OD600 value of 0.2. Protein expression was
induced by addition of 100 µM or 1 mM IPTG after 2 h (cultivations performed at 42 °C), 2 1/2 h
(37 °C) and 3 h (30 °C). Cells were harvested 3 h after induction. For expression in TB medium
(Sambrook and Russell 2001) cells from LB agar plates grown overnight were used to inoculate the
main culture to an initial OD600 value of 0.1. Furthermore medium that is based on enzyme controlled
substrate delivery (EnPresso®, BioSilta, Finland) was used. Therefore the main culture was inoculated
to a final OD600 of 0.15 with a fresh preculture that was cultivated before on LB agar plates (8 – 10 h
at 37 °C). After overnight cultivation protein expression was induced by addition of IPTG, and at the
same time a “booster tablet” was added, according to the instructions of the manufacturer. Finally,
cells were harvested by centrifugation (16,000 ×g, 5 min, 4 °C) and the pellets were stored at –20 °C.
Chaperone co-expression experiments were performed as outlined in the manufacturer’s manual
(Takara Bio Inc., Otsu, Japan). Shortly, E. coli BL21, co-transformed with the expression plasmid and a

34 Experimental part
chaperone plasmid (Table 2), was cultivated in LB medium as described above; however, the medium
additionally contained 20 µg ml-1 chloramphenicol for chaperone plasmid selection, and
0.5 - 4 mg ml-1 L-arabinose or 1 – 10 mg ml-1 tetracycline for the induction of the respective
chaperones (Table 2). Three hours after incubation at 30 °C, target protein expression was induced
by the addition of 100 µM IPTG.
2.4. Preparation of protein samples
2.4.1. Cell disruption
In order to analyze protein expression by SDS-PAGE, cell disruption was performed in small-scale.
Therefore cell pellets that equalled the amount of 1 ml of OD600 = 5 were resuspended in 300 µl
BugBuster™ Protein Extraction Reagent (Novagen), containing 1 µl ml-1 Benzonase® (Novagen). The
suspension was afterwards incubated for 20 – 30 min on a shaking platform. Alternatively cells were
disrupted by sonication. For this purpose a cell pellet corresponding to the amount of cells in 1 ml of
OD600 = 5 was resuspended in 500 µl potassium phosphate buffer (10 mM , pH 7.0). This cell
suspension was sonified on ice using a UP200S sonicator (Hielscher Ultrasonics GmbH, Teltow,
Germany). The sonication was performed twice with 30 % power input for 3 min in 30 s intervals
using a sonotrode of 2 mm in diameter. After cell disruption soluble and insoluble protein fractions
were separated by centrifugation (20,000 × g; 15 min; 4 °C).
In order to prepare cell lysates for protein purification, cell pellets were re-suspended in NPI-10
binding buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) at 5 ml per gram wet
weight in a falcon tube. This cell suspension was sonified on ice using the UP200S sonicator. The
sonication was performed twice with 30 % power input for 3 min in 30 s intervals using a sonotrode
of 7 mm in diameter.
2.4.2. Protein purification
Cell lysates were centrifuged (20,000 × g, 15 min, 4 °C) to separate soluble from insoluble fractions.
The soluble portion of the cell lysate was heated for 15 min at 50 °C (DgPNP), 60 °C (GtPyNP), 65 °C
(GtPNP), 80 °C (TtPyNP), or 85 °C (ApMTAP). Coagulated proteins were removed by centrifugation
(20,000 × g, 15 min, 4 °C). Additionally the cell lysate was filtered using Rotilab®-sytinge filters, CME,
sterile, 0.22 µm pore size from Carl Roth (Karlsruhe Germany). The target proteins were further
purified with a Fast Protein Liquid Chromatography system (Äkta™ avant, GE healthcare) via
immobilized metal ion affinity chromatography using a 5 ml Ni-NTA Superflow cartridge (Qiagen),
following the instructions given by the manual. Shortly, the column was equilibrated with 10 column
volumes of NPI-10 buffer (see 2.4.1) and the cell lysate was applied using the system pump. Then,
the column was washed with NPI-20 buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH
8.0) until the absorption signal at 280 nm returned to the baseline value. Finally the hexahistidine
tagged target protein was eluted with elution buffer NPI-250 (50 mM NaH2PO4, 300 mM NaCl,
250 mM imidazole, pH 8.0). Fractions containing the purified protein were pooled and subsequently

Experimental part 35
the excess of salt and imidazole was removed by the use of a HiPrep 26/10 Desalting column (GE
Healthcare). Therefore the column was equilibrated before with at least 5 column volumes desalting
buffer (50 mM KH2PO4, 0.15 M NaCl, pH 7.0). For more details see the instructions given by the
manufacturer. Finally, the purified protein solution was aliquoted, rapidly frozen in liquid nitrogen
and stored at -80 °C.
2.5. Protein analytics
2.5.1. SDS-PAGE analysis
Soluble, insoluble, and total protein fractions were analyzed by SDS-PAGE. For this purpose either
precast gels (NuPage® 4-12 % Bis-Tris gel, Invitrogen) or 15 % Tris-glycine SDS-polyacrylamide gels
with 5 % stacking gels were prepared (Sambrook and Russell 2001). Culture samples were normalized
based on their optical densities at 600 nm prior to cell lysis and subsequently prepared for SDS-PAGE
analysis as described in standard protocols (Sambrook and Russell 2001). Shortly, soluble or total
protein fractions were combined with 1 volume of 2 x SDS gel-loading buffer (100 mM Tris·Cl pH 6.8,
200 mM DTT, 4 % SDS, 0.2 % bromphenol blue, 20 % glycerol). For analysis of insoluble protein
fractions the pellet was resuspended in an adequate amount of 1 x SDS gel-loading buffer containing
8 M urea. Afterwards samples were incubated for 5 min at 99 °C, cooled to room temperature and 5
– 10 µl were applied for SDS-PAGE analysis. The protein molecular weight marker (#Sm0431) was
purchased from Fermentas (Vilnius, Lithuania; now Thermo Scientific). After gel electrophoresis
(approx. 90 min at 150 V), gels were washed with water and stained for approximately 1 h with a
solution containing 80 mg l-1 Coomassie Brialliant Blue G-250 and 35 mM HCl. The procedure does
not require extensive destaining, but in order to increase the contrast gels were washed overnight in
water.
2.5.2. Determination of the protein concentration
The protein concentration of the purified protein solution was determined by measuring the
absorption at 280 nm (Nanodrop, Thermo Scientific). The absorption coefficients were theoretically
calculated from the amino acid sequence (Vector NTI software, Invitrogen) and are listed in Table 6,
whereby the amino acid sequence that results from the expression in the pKS2 vector served as input
sequence. The theoretical prediction of the absorption coefficient at 280 nm of folded proteins in
water has been reported to be fairly reliable for proteins containing tryptophan residues (Pace et al.
1995). All target proteins of this study contain at least one tryptophan residue per subunit.

36 Experimental part
Table 6. Theoretically calculated absorption coefficients.
Protein A[280] of 1 mg ml-1
DgPNP 0.63
ApMTAP 0.88
GtPNP 0.88
GtPyNP 0.42
TtPyNP 0.56
2.5.3. Protein unfolding studies
Thermal denaturation of 10 µM purified TtPyNP protein dissolved in potassium phosphate (50 mM,
pH 7.5) was monitored with a Jasco J-815 circular dichroism (CD) spectrometer in a 0.1 cm cuvette by
following the loss of ellipticity at 220 nm. Unfolding was induced by raising the temperature in 0.1 °C
increments at a ramp rate of 1 °C min-1 with a Peltier-effect temperature controller. The measured
ellipticity was normalized, and the apparent melting temperature (TappM) was determined. DSC
experiments were performed with 43 µM TtPyNP protein dissolved in potassium phosphate (50 mM,
pH 7.5) by heating the samples in a CSC 5100 Nano differential scanning calorimeter with a scan rate
of 1 °C min-1. The DSC data were analyzed with the program CpCalc (version 2.1, Calorimetry Sciences
Corporation, 1995) to determine TappM at which half of the protein is unfolded. The irreversibility of
thermal denaturation precluded thermodynamic analysis of the CD and DSC unfolding traces.
2.6. Activity assays
2.6.1. Spectroscopic assay for PNP activity
In initial experiments, soluble fractions of DgPNP cell lysates were analyzed with respect to PNP
activity by a spectroscopic assay. Therefore the spectroscopic assay developed by Kalckar (Kalckar
1947) was adapted for use in 96-microwell plates. The final reaction mixture (200 µl) contained:
50 mM potassium phosphate buffer pH 7.5, 0.5 mM inosine (Carl Roth) and 0.4 U ml-1 microbial
xanthine oxidase (Sigma-Aldrich). After addition of 20 µl diluted soluble cell extract, the UV
compatible microplate containing the reaction mixtures was incubated for 1 min at 25 °C in a
temperature-controllable microplate reader (Synergy™ Mx, Biotek,Winoosky, United States), and
subsequently the change of absorption at 293 nm was recorded. Quantification of uric acid (Sigma-
Aldrich) was performed at the same temperature and in the same buffer, to correlate the change of
absorption to the liberation of uric acids during the enzymatic assy. One unit of PNP is defined as the
amount of PNP which liberates 1 µmol of uric acid from inosine, in the presence of excess of xanthine
oxidase.

Experimental part 37
2.6.2. Standard assay with purified proteins
Standard activity assays were performed in 50 mM potassium phosphate buffer, pH 7.0 containing
1 mM of uridine (GtPyNP, TtPyNP) or 1 mM inosine (DgPNP, GtPNP, ApMTAP) as substrate. After
2 min of pre-heating, 1-2 µl of diluted enzyme was added per 100 µl of reaction mixture. The
following standard reaction temperatures were defined for each protein: 55 °C (DgPNP), 60 °C
(GtPyNP), 70 °C (GtPNP), 80 °C (ApMTAP, TtPyNP). These temperatures either coincide with the
temperature optimum of the respective protein (DgPNP, GtPyNP, GtPNP) or were set to 80 °C due to
practical reasons (TtPyNP, ApMTAP). Samples were withdrawn after defined time intervals and the
reaction was immediately stopped by adding 1 vol of reaction mixture to ½ vol of 10 % trichloroacetic
acid (TCA). Precipitated proteins were removed by centrifugation (20,000 ×g, 15 min, 10 °C) and the
samples were stored at – 20 °C for later analysis by HPLC.
From the HPLC results the concentration of residual nucleoside (substrate) and liberated nucleobase
(product of the phosphorolytic cleavage) was retrieved. The substrate conversion was calculated
with the following formula:
%100(%)
nucleosidebase
baseConversion
Only conversion rates that were linear with respect to time and amount of enzyme added, were
considered for further analysis. This was usually the case if not more than 10 % of the substrate was
converted.
2.6.3. Thermal properties of the enzymes
Thermal stability
Enzyme preparations were diluted in 50 mM potassium phosphate buffer (pH 7.0). The final enzyme
concentrations were 13 µg ml-1 (GtPNP), 26 µg ml-1 (DgPNP), 46 µg ml-1 (GtPyNP, TtPyNP), and
55 µg ml-1 (ApMTAP). It is expected that at higher enzyme concentrations as eventually used in
synthetic applications later (typically 100 µg ml-1) enzymes will be even more stable, as it was also
reported for E. coli TP and E. coli PNP (Krenitsky et al. 1981).
Aliquots were incubated in 0.2 ml tubes in a thermocycler at the respective temperatures. After
defined time intervals, tubes were withdrawn and the residual activity of the incubated enzyme
solution was determined under the standard conditions outlined in section 2.6and plotted over the
reaction time. In order to determine the half life, the resulting curve was fitted (Sigma Plot)to the
decay function
tik
evv
0 and subsequently the half life was calculated 1
2/1 5.0ln
ikt.
Temperature optimum
Standard reaction mixtures were pre-heated for 2 min at the respective temperatures. Diluted
enzyme solutions were added and the reaction was stopped after 3 min.

38 Experimental part
2.6.4. Kinetic parameters
Activity tests were performed in triplicates for at least 5 different substrate concentrations spanning
0.25 to 5 times of the Michaelis-Menton constant. The initial reaction rates were plotted over the
substrate concentrations. The resulting substrate saturation curve was fitted to the hyperbolic
Michaelis-Menton function (Sigma Plot). From the resulting equation Km and Vmax were directly
retrieved, and kcat could be easily calculated. For details see(Copeland 2000).
2.6.5. Substrate screenings
Phosphorolysis rates for natural and modified nucleosides were determined for 1 mM nucleoside
substrate concentrations. 10 mM sodium phosphate buffer pH 6.5 was used for dUrd2′F and dUrd2′F,
reactions with other nucleosides were investigated in 50 mM potassium phosphate buffer. A typical
enzyme loading for the phosphorolysis assays with artificial nucleosides was 0.1 mg ml-1. Nucleosides
and manufacturers are listed in Table 7.
2.6.6. Synthetic reactions
Reactions were performed employing 2 mM pyrimidine nucleosides as pentofuranosyl donor and
1 mM purine base as pentofuranosyl acceptor. The buffer (pH 6.5 ) was either 10 mM NP buffer or
2 mM NP buffer, if dihalogenated purine bases were substrates of the reaction. The concentration of
the NPs was 0.1 mg ml-1 each, if not otherwise stated. Reactions were typically performed in 200 –
1000 µl scale in Eppendorf tubes that were incubated on a thermoshaker (Eppendorf) at 300 rpm.
After defined time intervals samples were withdrawn. Reactions were stopped by the addition of TCA
as outlined in section 2.6. Alternatively samples were instantly diluted in ice-cold buffer. The latter
strategy was used to investigate the synthesis of (acid labile) purine deoxyribosides.
2.7. HPLC analysis
The concentration of nucleosides and nucleobases was determined by following the absorption at
260 nm during HPLC analysis using a reversed phase C18 column (Gemini-Nx 5u, 150 × 4.60 mm,
Phenomenex, Torrance, United States) with the following gradient: from 97 % 20 mM ammonium
acetate and 3 % acetonitrile to 60 % 20 mM ammonium acetate and 40 % acetonitrile in 10 min.
Retention times and calibration factors that were determined under these conditions are listed in
(Table 7).
Authentic samples of the ribo- and deoxyribosides of dihalogenated purines were not available in the
course of this study. The retention times indicated in Table 7 in fact just represent the retention
times of the new product peaks observed in HPLC. In these cases the calibration factors are only
estimates derived indirectly from the decrease of substrate concentrations.

Experimental part 39
Table 7. Properties of nucleosides and nucleobases in HPLC analysis.
Compound Obtained from Retention
time [min]
Calibration factor
(area [AU] / [mM])
Uridine Sigma-Aldrich 3.2 5372
Uracil Sigma-Aldrich 2.4 4257
Thymidine Sigma-Aldrich 4.7 4610
Thymine Sigma-Aldrich 4.0 4365
1-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)uracil (dUrd2′F)
Metkinen Chemistry
(Kuusisto, Finland) 4.6 5449
2′-Deoxy-2′-fluorouridine (dUrd2′F) TCI Deutschland (Eschborn,
Germany) 4.4 5185
O2,2′-anhydro-1-(β-D-
arabinofuranosyl)uracil (anhydro-Urd) Prof. Igor A. Mikhailopulo 2.3 3388
1-(β-D-arabinofuranosyl)uracil (ara-U) Sigma-Aldrich 3.8 5324
Adenosine Carl Roth (Karlsruhe,
Germany) 5.0 7836
Adenine Carl Roth (Karlsruhe,
Germany) 4.2 6958
Inosine Carl Roth (Karlsruhe,
Germany) 4.0 4436
Hypoxanthine Sigma-Aldrich 3.0 4736
2′-Deoxy-2′-fluoroadenosine (dAdo2′F) Metkinen Chemistry
(Kuusisto, Finland) 5.8 15162
9-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)adenine (dAdo2′F)
Metkinen Chemistry
(Kuusisto, Finland) 5.7 11336
Cytidine Sigma-Aldrich 2.6 4224
Cytosine
2,6-Dichloropurine (26DCP)
Sigma-Aldrich
TCI Deutschland (Eschborn,
Germany)
2.0
7.6
3220
2386
6-Chloro-2-fluoropurine (6C2FP) TCI Deutschland (Eschborn,
Germany) 7.0 3676
2,6-Dichloropurine ribosides - 8.2 3610
2,6-Dichloropurine deoxyribosides - 8.8 3165
6-Chloro-2-fluoropurine ribosides - 7.5 3924
6-Chloro-2-fluoropurine deoxyriboside - 8.2 4038
(-) not purchased, but presumably synthesized.


3. Recombinant expression of nucleoside phosphorylases
3.1. Introduction
3.1.1. Recombinant expression of thermostable proteins in E. coli
For many objectives in recombinant protein expression E. coli is the first choice as expression host.
Extensive investigations within the last decades made this rod-shaped bacterium to a microbial cell
factory that is easy to cultivate and to genetically manipulate in laboratory-scale (Baneyx 1999). For
industrial-scale, methods of high cell-density cultivations have been developed that enable rapid and
cost-efficient production of recombinant proteins (Shiloach and Fass 2005). In the present study
E. coli will thus serve as expression host for the production of target enzymes derived from
thermophilic microorganisms. This strategy offers an additional advantage for later protein
purification, since the majority of E. coli enzymes can be easily removed by heat precipitation.
Despite impressive progresses, the high-level production of recombinant proteins in E. coli can be
challenging. A common problem is the aggregation of misfolded protein to insoluble inclusion bodies
or a poor product yield and a multitude of strategies to overcome such problems have been
developed. Generally these approaches address the expression system (expression vector,
expression strain, fusion tag, co-expression of molecular chaperones) (Esposito and Chatterjee 2006,
Sørensen and Mortensen 2005) or the expression conditions (cultivation temperature, aeration, level
of induction, media composition) (Berrow et al. 2006, Donovan et al. 1996, Krause et al. 2010, Schein
and Noteborn 1988). Moreover it is possible to use the degeneracy of the genetic code to re-design
coding sequences according to the requirements for high-level protein expression (Welch et al.
2009). The advances in synthetic biology in recent years make this approach a more and more
reasonable strategy.
Some factors appear to be especially relevant to be considered for the successful expression of
thermostable proteins in the mesophilic host E. coli. For example poor expression resulting from
codon bias between thermophilic donor and E. coli has been tackled by co-expression of rare tRNAs
or codon optimization (Wang and Zhang 2009). Disulfide bond formation, being a widespread feature
stabilizing intracellular proteins from thermophiles, can pose another obstacle (Beeby et al. 2005,
Cacciapuoti et al. 1999). Formation of disulfide bonds can be achieved in E. coli by translocating the
nascent protein to the oxidizing periplasm or by using mutant strains with a mild oxidative cytoplasm
(de Marco 2009). Co-expression of sulfhydryl oxidase that catalyses de novo disulfide bond formation
represents a novel technology and allows the expression of disulfide bond containing proteins in the
cytoplasm with intact reducing pathways (Hatahet et al. 2010, Nguyen et al. 2011). Other factors
including optimal folding temperature and the need of specific activation factors may play a pivotal
role for the functional expression of genes from thermophiles: Hence, in some cases the expression
of thermophilic enzymes has been successful in the thermophilic host T. thermophilus, while the
functional expression of the same enzymes in E. coli failed (Angelov et al. 2009, Hidalgo et al. 2004).

42 Recombinant expression of NPs
The potential requirement of a higher cultivation temperature for the soluble expression of genes
from thermophiles was also addressed by Koma and co-workers (Koma et al. 2006). By cultivation at
43 °C or even 46 °C they successfully expressed genes from thermophiles in E. coli that were only
hardly expressed in soluble form at 37 °C.
Another critical factor that might be of special relevance for thermostable protein expression is the
formation of secondary mRNA structures in the translation initiation region of mRNA. This
phenomenon has been recognized as an important determinant for poor recombinant protein
expression levels in general (de Smit and van Duin 1990, Griswold et al. 2003, Kudla et al. 2009) and
likewise the optimization of the 5′ mRNA sequence has been objective for a number of scientific
endeavours (Care et al. 2008, Cèbe and Geiser 2006, Griswold et al. 2003, Jung et al. 2010, Khan et al.
2007, Na et al. 2010, Niemitalo et al. 2005, Sadaf et al. 2008). Recent findings make us believe that
this factor has to be particularly addressed for the recombinant expression of genes from
thermophiles in mesophilic hosts: A computational study of Gu and co-workers (Gu et al. 2010) gives
insight into the natural variation of secondary mRNA structure stability within genes, genomes and
among different species. By analyzing the genomes of 340 species the authors showed that stability
of secondary mRNA structures within the coding sequence of genes is generally reduced near the
start codon. Moreover they found that among prokaryotes this effect becomes weaker with higher
optimal growth temperature. Since secondary mRNA structures are destabilized at higher
temperature there seems to be less selection pressure on the reduction of secondary 5′ mRNA
structure stability in thermophiles than in mesophiles. In practice this may implicate that the
expression of genes from thermophiles in mesophilic hosts, at “unnatural” low temperature, is in
particular hampered by translation inhibiting 5′ mRNA structures. For these reasons mRNA stability
will also be addressed in the present study in order to optimize the expression level of the
thermostable target enzymes.
3.1.2. Target enzymes of this study
Two main criteria were decisive for the selection of the thermostable nucleoside phosphorylases
studied in this work. On the one hand the final set of enzymes should comprise biocatalysts suitable
for the reversible phosphorolysis of both pyrimidine and purine nucleosides. The motivation lies in
the combined use of both categories of enzymes in order to carry out transglycosylation reactions as
outlined in section 1.2.3. Hence, half of the selected enzymes (Figure 12) are expected to show
substrate specificities for pyrimidine nucleosides (UP, PyNP), while the other half are expected to
show substrate specificity for purine nucleosides (PNP, MTAP).
On the other hand, the selection should represent a preferably high diversity of biocatalytical
properties in order to cover broad future application fields. In this respect the expected temperature
optima of the biocatalysts are of special interest. As mentioned before thermal stability is generally
considered as advantageous for bio-catalyzed synthesis of nucleosides and therefore in focus of this
study. However, the degree of thermal stability that is appropriate may substantially depend on the
specific process. If, for example, the temperature is restricted due to thermal lability of reactants, a

Recombinant expression of NPs 43
biocatalyst with respective moderate temperature optimum might be a better choice than a
biocatalyst exhibiting an unusually high degree of thermal stability but showing at the same time
only poor activity at the process temperature.
In order to meet these requirements, the six nucleoside phosphorylases in focus of this study
originate from different thermophilic microorganisms with distinct temperature optima (Figure 12).
To some extent, this strategy also allows studying enzymes that are formally equal but evolved in a
distinct phylogenetic context. Possibly resulting differences in substrate specificity are of high
interest for biocatalytical applications for the synthesis of modified nucleosides.
40
50
60
70
80
90
100
Tem
pop
to
f sou
rce
mic
roo
rgan
ism
[C
]
DgPNP
GtPNP
ApUP
TtPyNP
Deinococcus geothermalis
• Isolated from hot springs in Naples, Italy
• Extremely gamma-radiation resistant
• Gram-positive
Geobacillus thermoglucosidasius
• Isolated in 1983 by Suzuki and co-workers in Japan, assigned as Bacillus thermoglucosidasius
• Re-classified as Geobacillus thermoglucosidasius in 2001
• Gram-positive
Thermus thermophilus
• Isolated in the 70’s from hot springs in Japan
• Genome sequenced in 2004
• Gram-negative
Aeropyrum pernix
• Isolated from coastal solfataric thermal vent in Kodakara-Jima Island in Japan
• Areobic hyperthermophilic crenarchaeon
• Gram-negative
• Complete genome sequence published in 1999
• Re-annotation of the genome in 2006
ApMTAP
GtPyNP
°
Figure 12: Overview of target enzymes and microorganisms that served as source for gene isolation. The assigned protein names consist out of two letters representing the source microorganism followed by the functional name of the enzyme. For further details regarding the source microorganisms see references (Ferreira et al. 1997, Henne et al. 2004, Kawarabayasi et al. 1999, Nazina et al. 2001, Oshima and Imahori 1974, Sako et al. 1996, Suzuki et al. 1983, Yamazaki et al. 2006).

44 Recombinant expression of NPs
3.2. Sequence analysis and theoretical predictions
In order to anticipate potential difficulties of the recombinant expression of the target proteins, the
coding sequences were analyzed beforehand in silico. Therefore coding sequences were retrieved
from the National Centre of Biotechnology Information (GenBank accession numbers are listed in
(Table 8) and analyzed with respect to rare codon usage and probability of disulfide bond formation.
Rare codon usage is an important determinant for the efficient expression of heterologous proteins
in E. coli. Since the genetic code is degenerate, a number of different codons can code for the same
amino acid. In E. coli the usage of such synonymous codons is especially biased in genes highly and
continuously expressed during exponential growth (Hénaut and Danchin 1996), and correlates with
the abundance of corresponding tRNAs (Dong et al. 1996, Ikemura 1981). The difference of codon
usage of heterologous target gene sequences can lead to insufficient tRNA pools which can decrease
efficiency (translational stalling, premature translation termination) and accuracy (translational
frameshifting, amino acid misincorporation) during the translation of a mRNA sequence into a
protein (Akashi 1994, Kane 1995, Kurland and Gallant 1996). In Table 8 the frequency of individual
codons rarely used in E. coli (coded less than 2 % usage for the particular amino acid in genes highly
and continuously expressed during exponential growth) in the target gene sequences of the present
study are listed. In addition, the codon adaption index (CAI), as a measure for the codon usage bias
between the heterologous target sequences and genes highly expressed in E. coli was calculated for
each gene. A value of 1.0 is considered as ideal, whereas low numbers indicate that the recombinant
genes may be only poorly expressed in the desired expression host (for details see (Sharp and Li
1987)). Both, the frequency of rare codons as well as the deduced CAI indicate a general trend:
Codon bias appears to be more critical for the expression of the NP genes derived from
microorganisms with high optimal growth temperature (> 60 °C) in comparison to that of
microorganisms with lower optimal growth temperature (< 60 °C). Adequate measures to avoid the
lack of rare tRNA pools have thus been considered and will be discussed in the following sections.
The heterologous expression of thermostable proteins in the cytoplasm of E. coli may also be
challenged by the requirement of disulfide bond formation that is in some cases needed for the
stabilization of intracellular proteins from thermophiles (Beeby et al. 2005, Cacciapuoti et al. 1999).
In order to assess the likelihood of the presence of disulfide bonds in the native proteins, the
oxidation state of the cysteine residues of the target sequences was predicted. It has been previously
shown that the sequence environment of free cysteine residues and cysteines involved in disulfide
bonds differ (Fiser et al. 1992). The online tool used here for prediction of the cysteine bonding state
(CYSPRED) discriminates disulfide bonds from free cysteines by exploiting this information through
the training of a neural network. The source data are the amino acid sequences flanking cysteines of
proteins with resolved three-dimensional structure as well as evolutionary information (Fariselli and
Casadio 1999). According to the predictions a single cysteine residue (Cys112) of ApMTAP is in a
bonding state, as well as two cysteines of ApUP (Cys227 and Cys279). In case of ApMTAP it appears
hence likely that Cys112 is involved in an intermolecular disulfide bond, while for ApUP both
intramolecular and intermolecular stabilization through disulfide bonds is possible.

Recombinant expression of NPs 45
Finally the molecular weight of the monomeric subunits was theoretically predicted and is shown in
Table 8 to aid the interpretation of SDS-PAGE gels of cultivation samples shown in the following
sections.
Table 8. Properties of the gene coding regions and predictions for the gene products
Assigned abbreviation DgPNP GtPNP GtPyNP TtPyNP ApMTAP ApUP
GenBank accession
number ABF45792 EFG53380 ZP_06809030 AAS81754.1 NP_147653 NP_148386
Rar
e co
do
n a
bu
nd
ancy
AGG (Arg) 1 - - 6 17 19
CTA (Leu) - 1 2 1 3 3
AGA (Arg) - - - - - 2
ATA (Ile) - - 1 3 7 9
CGG (Arg) 6 3 5 23 1 3
GGA (Gly) 3 8 12 2 3 4
CCC (Pro) 4 - - 14 8 6
CGA (Arg) 1 1 2 - - -
CAI 0.74 0.69 0.70 0.56 0.50 0.54
Cys
tein
e
resi
du
e -
oxi
dat
ion
stat
e
101-NB 38-NB 270-NB 103-NB 112-B 225-NB
129- NB 91-NB 129-NB 227-B
279-B
Pre
dic
ted
siz
e
of
gen
e p
rod
uct
(kD
a)
Native
protein 28.5 26.1 46.0 45.4 25.6 30.3
Expressed
from pKS2 30.0 27.6 47.6 46.9 27.0 31.7
NB = non-bonding state, B = bonding state, number of the cysteine residue applies to the native amino acid sequence, CAI= Codon adaption index
3.3. Expression of DgPNP
Purine nucleoside phosphorylase from D. geothermalis (DgPNP) was chosen as first target enzyme to
be overexpressed in E. coli. Since the natural host shows optimal growth at temperature between
45 °C – 50 °C it is expected that the correctly folded recombinant protein would be temporarily
stable in this temperature range.
3.3.1. Towards the functional expression of DgPNP
For pilot expression experiments the DgPNP encoding sequence was cloned via recombinational
cloning into the pCTUT7 expression vector (Šiurkus et al. 2010). This vector is characterized by a
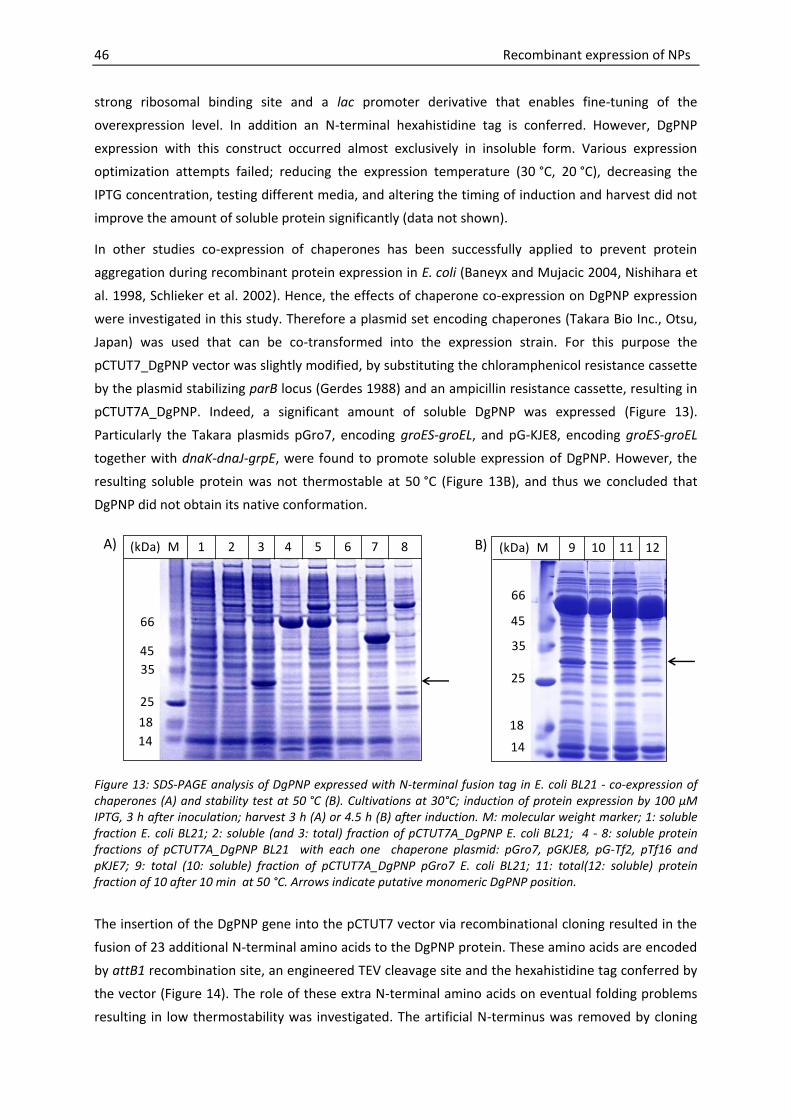
46 Recombinant expression of NPs
strong ribosomal binding site and a lac promoter derivative that enables fine-tuning of the
overexpression level. In addition an N-terminal hexahistidine tag is conferred. However, DgPNP
expression with this construct occurred almost exclusively in insoluble form. Various expression
optimization attempts failed; reducing the expression temperature (30 °C, 20 °C), decreasing the
IPTG concentration, testing different media, and altering the timing of induction and harvest did not
improve the amount of soluble protein significantly (data not shown).
In other studies co-expression of chaperones has been successfully applied to prevent protein
aggregation during recombinant protein expression in E. coli (Baneyx and Mujacic 2004, Nishihara et
al. 1998, Schlieker et al. 2002). Hence, the effects of chaperone co-expression on DgPNP expression
were investigated in this study. Therefore a plasmid set encoding chaperones (Takara Bio Inc., Otsu,
Japan) was used that can be co-transformed into the expression strain. For this purpose the
pCTUT7_DgPNP vector was slightly modified, by substituting the chloramphenicol resistance cassette
by the plasmid stabilizing parB locus (Gerdes 1988) and an ampicillin resistance cassette, resulting in
pCTUT7A_DgPNP. Indeed, a significant amount of soluble DgPNP was expressed (Figure 13).
Particularly the Takara plasmids pGro7, encoding groES-groEL, and pG-KJE8, encoding groES-groEL
together with dnaK-dnaJ-grpE, were found to promote soluble expression of DgPNP. However, the
resulting soluble protein was not thermostable at 50 °C (Figure 13B), and thus we concluded that
DgPNP did not obtain its native conformation.
14
66
45
35
25
1814
66
45
35
25
18
A) (kDa) M 1 2 3 4 5 6 7 8 (kDa) M 9 10 11 12B)
Figure 13: SDS-PAGE analysis of DgPNP expressed with N-terminal fusion tag in E. coli BL21 - co-expression of chaperones (A) and stability test at 50 °C (B). Cultivations at 30°C; induction of protein expression by 100 μM IPTG, 3 h after inoculation; harvest 3 h (A) or 4.5 h (B) after induction. M: molecular weight marker; 1: soluble fraction E. coli BL21; 2: soluble (and 3: total) fraction of pCTUT7A_DgPNP E. coli BL21; 4 - 8: soluble protein fractions of pCTUT7A_DgPNP BL21 with each one chaperone plasmid: pGro7, pGKJE8, pG-Tf2, pTf16 and pKJE7; 9: total (10: soluble) fraction of pCTUT7A_DgPNP pGro7 E. coli BL21; 11: total(12: soluble) protein fraction of 10 after 10 min at 50 °C. Arrows indicate putative monomeric DgPNP position.
The insertion of the DgPNP gene into the pCTUT7 vector via recombinational cloning resulted in the
fusion of 23 additional N-terminal amino acids to the DgPNP protein. These amino acids are encoded
by attB1 recombination site, an engineered TEV cleavage site and the hexahistidine tag conferred by
the vector (Figure 14). The role of these extra N-terminal amino acids on eventual folding problems
resulting in low thermostability was investigated. The artificial N-terminus was removed by cloning

Recombinant expression of NPs 47
the DgPNP gene in the same expression vector used before via restriction digestion and ligation
(pKS1_DgPNP). The theoretically determined molecular size of the monomeric unit of the new gene
product was 28.5 kDa, which is 3.1 kDa smaller than the protein expressed by the previous constructs
pCTUT7_DgPNP and pCTUT7A_DgPNP. Subsequent expression at 30 °C resulted in completely soluble
DgPNP, albeit in a low amount. Raising the cultivation temperature to 37 °C or even 42 °C, resulted in
considerably higher expression levels (Figure 15A). The new gene product was stable at 50 °C for at
least 20 min (Figure 15B), which suggested that DgPNP was folded correctly now.
His tag attB1 Tev cleavage site DgPNP
ATGCATCACCATCACCATCACGCTAGCACAAGTTTGTACAAAAAAGCAGGCTTCGAAAACCTGTATTTTCAGGGCATGGTGGTG…
Figure 14: Vector map of pCTUT7_DgPNP. Upstream of the DgPNP coding sequence, additional amino acids are encoded by attB1 recombination site, an engineered TEV cleavage site and the hexahistidine tag. .
14
66
45
35
25
18
25
14
66
45
35
18
(kDa) M30 °C 37 °C 42 °C
T S T S T S(kDa) M
0 min 10 min 20 min
T S T S T S
A) B)
Figure 15: SDS-PAGE analysis of DgPNP expressed without fusion tag at different temperatures (A), and stability test at 50 °C (B). The cultivation temperatures varied between 30 °C and 42 °C; cultivation in LB medium; 100 µM IPTG. For the stability test crude cell extract of the expression trial at 42 °C was incubated for indicated time periods at 50 °C. The arrows indicate the bands corresponding to the molecular weight theoretically calculated for the native monomeric DgPNP subunit (28.5 kDa). T = total protein fraction; S = soluble protein fraction, M = molecular weight marker.

48 Recombinant expression of NPs
3.3.2. DgPNP expression optimization by reducing secondary 5′mRNA stability
The previous section describes how a suitable expression vector was found to express DgPNP in
soluble and presumably correctly folded form. However, the poor yield of recombinant protein
prompted us to investigate strategies to increase the expression level. This section deals with the
expression optimization through the reduction of the stability of secondary mRNA structures in the
5′ region. The results presented in this section were previously published (Szeker et al. 2011). The
theoretical studies on mRNA secondary structure prediction and optimization were provided by Olli
Niemitalo and André H. Juffer (Biocenter Oulu and Department of Biochemistry, University of Oulu,
Finland).
Among other factors, the formation of secondary mRNA structures in the translation initiation region
of mRNA has been recognized as an important determinant for poor recombinant protein expression
levels in general (de Smit and van Duin 1990, Griswold et al. 2003, Kudla et al. 2009). If stable
secondary structures are formed in this (5′ mRNA) region, an efficient translation initiation is
impeded. Accordingly, the optimization of 5′ mRNA sequences to tackle this problem has been
objective for a number of scientific endeavours (Care et al. 2008, Cèbe and Geiser 2006, Griswold et
al. 2003, Jung et al. 2010, Khan et al. 2007, Na et al. 2010, Niemitalo et al. 2005, Sadaf et al. 2008).
We assumed that secondary 5′ mRNA structures are also the reason for the low expression of DgPNP,
since these structures are destabilized at higher temperature – which would explain, why the
expression level increased with higher growth temperature as shown in (Figure 15A) in the previous
section. In order to test this hypothesis the secondary-structural properties of pKS1_DgPNP mRNA
were predicted theoretically (Table 9). The free energy of formation of secondary structures in the
translation initiation region surpassed the threshold for inhibition of translation initiation of about
-6 kcal mol-1 (de Smit and van Duin 1990, de Smit and van Duin 1994) by -12 kcal mol-1 or more
depending on temperature. The Shine-Dalgarno sequence and the initiation codon were involved in
base pairs in the minimum free energy substructure encompassing the translation initiation region
(Figure 16A).
A) B) C) D)
Figure 16: Substructures of predicted free energy secondary mRNA structures.Taken from (Szeker et al. 2011).

Recombinant expression of NPs 49
With these findings the hypothesis that the formation of stable 5′ mRNA structure is causing poor
expression levels in the pKS1_DgPNP construct was further supported. Hence, our aim was to
attenuate the stability of these structures. Our strategy to follow this purpose was to combine the
following two approaches:
(i) Optimization of the 5′ mRNA sequence
(ii) High-temperature cultivation
Design of optimized 5′ mRNA sequences
The gene sequence was optimized in silico to reduce secondary structures. Using the pKS1_DgPNP
mRNA as template, the second codon, GTG, was removed, as its function as a start codon had been
superseded by the artificially introduced ATG start codon, and because in the second codon position
the originally N-formylmethionine-encoding GTG encoded for a nonnative valine. It was also thought
that elimination of the G-rich codon might reduce formation of secondary structures (Kudla et al.
2009). However, no silent codon substitutions could be found by automated optimization that would
have reduced the stability of secondary structures to a satisfactory level. This was the case both
before and after removal of the GTG. Amino-acid-changing codon substitutions were thus
considered, using as template the sequence from which the GTG at the second codon position had
been deleted. The new second codon was also GTG, encoding for valine. To avoid affecting the
protein structure too much, only codons encoding for physico-chemically similar amino acids were
considered in place of the valine, and ATT, encoding for isoleucine, the most similar (Wei et al. 1997)
amino acid, was chosen. The substitution was implemented along with the earlier removal of GTG,
forming DgPNP1. For pKS1_DgPNP1, the free energy of secondary structure formation in the
translation initiation region was still at least -3.7 kcal mol-1 lower than the threshold for inhibition. On
the other hand, the secondary structures were more dynamical (Table 9), not adequately described
by the minimum free energy substructure (Figure 16B).
With DgPNP1 as the template three additional silent substitutions, Ala3: GCGGCC, Arg4:
CGTCGA, Pro6: CCGCCC were found by an automated optimization. Those together with the
silent substitution Arg8: AGGCGT resulted in DgPNP2. The free energy of secondary structure
formation at the translation initiation region of pKS1_ DgPNP2 was at 42 °C approximately at the
threshold for uninhibited translation initiation. The minimum free energy substructure (Figure 16C,D)
had little value in describing the largely unordered secondary structures (Table 9). For all three gene
variants, the free energy of secondary structure formation was lower for lower temperatures, with a
difference of at least 3 kcal mol-1 between 30 °C and 42 °C and at least 1.3 kcal mol-1 between 37 °C
and 42 °C.
Protein expression and activity tests of derived constructs
The effects of the 5′ mRNA optimizations and high-temperature cultivation on the expression of
functional DgPNP were analyzed in parallel. Therefore samples of E. coli strains expressing the

50 Recombinant expression of NPs
different DgPNP constructs at temperatures between 30 °C and 42 °C were analyzed by denaturing
gel electrophoresis and an activity assay.
The presence of E. coli intrinsic PNP was a critical issue for the determination of DgPNP activity from
crude soluble E. coli extracts. However, activity tests of E. coli BL21 cells alone and E. coli BL21 cells
expressing the recombinant enzyme revealed a sufficiently low E. coli PNP background (Figure 17A).
In the following experiments PNP activities of E. coli BL21, cultivated under same conditions as the
recombinant cultures, were determined and served as blank values.
Table 9. Secondary-structural properties of mRNA substructures of DgPNP gene variants at the translation initiation region. Adapted from (Szeker et al. 2011).
Variant 5′codons T (°C) ΔGF
(kcal mol-1
)
Substructure Base pairs
(bp)
Mean identity
(bp)
DgPNP ATG-GTG-GTG-GCG-CGT-GTA-CCG-GCA-AGG 30 -22.6 Figure 16A 17 16.1
37 -19.9 15.7
42 -17.9 15.3
DgPNP1 ATG-ATT-GCG-CGT-GTA-CCG-GCA-AGG 30 -13.4 Figure 16B 12 6.2
37 -11.3 6.8
42 -9.7 7.0
DgPNP2 ATG-ATT-GCC- CGA-GTA-CCC-GCA-CGT 30 -9.6 Figure 16C 8 3.5
37 -7.7 3.1
42 -6.4 Figure 16D 14 5.7
Compared to the original sequence, variants DgPNP1 and DgPNP2 lacked the second codon and contained a number of nucleotide substitutions (bold). The free energy of secondary structure formation (ΔGF) at the translation initiation region was predicted. The substructure of the minimum free energy secondary structure concerning the translation initiation region was found to be identical at different temperatures for DgPNP and DgPNP1, but differed between 30/37 °C and 42 °C for DgPNP2. To see how well each substructure described the dynamical secondary structures at the translation initiation region, the number of base pairs in the substructure was calculated to be compared against the Boltzmann-weighted mean number of shared base pairs between it and ensemble structures (mean identity). The presence of competing structures drops mean identity below the number of base pairs in the structure.
Optimization of the 5′ mRNA enhances total DgPNP expression
The level of total protein expression correlates well with the predicted free energies associated with
5′ mRNA folding (Table 9 and Figure 17B). The highest free energy, for example, was predicted for the
5′ mRNA folding of the DgPNP2 construct. The almost complete elimination of translation initiation
inhibition by secondary 5′ mRNA structure formation was thus expected. In agreement with this
prediction a very high amount of total DgPNP2 expression was detected under all conditions tested.
The soluble expression of DgPNP and the exerted PNP activity were significantly increased in the
constructs with the optimized 5′ sequence (Figure 17C), too. Nevertheless, SDS-PAGE analysis (Figure
17B) clearly shows that the level of total DgPNP expression was more enhanced than the level of
soluble DgPNP expression. This implies that the 5′ mRNA optimization provoked to some extent the
formation of insoluble protein. The only exception is the expression trial of DgPNP1 at 30 °C, here
5′ mRNA optimization resulted in equal higher levels of soluble and total DgPNP expression.

Recombinant expression of NPs 51
Various strategies of 5′ mRNA optimization for the enhancement of recombinant protein expression
have been reported before (Care et al. 2008, Cèbe and Geiser 2006, Griswold et al. 2003, Jung et al.
2010, Khan et al. 2007, Na et al. 2010, Niemitalo et al. 2005, Sadaf et al. 2008). The strategy
presented here, demonstrates the benefits of a computational approach.
Higher cultivation temperature increases the yield of soluble and active DgPNP
Raising the cultivation temperature from 30 °C to 37 °C or even 42 °C resulted in considerably higher
yields of soluble and active recombinant DgPNP. This applied both to the original pKS1_DgPNP
construct as well as to the new pKS1_DgPNP1 and pKS1_DgPNP2 constructs (Figure 17B, C). For
example the DgPNP activity per cell harbouring pKS1_DgPNP resulted in an over 10-fold increase by
increasing the cultivation temperature from 30 °C to 42 °C.
This phenomenon can be well explained by the temperature dependence of secondary structure
stability. If sufficient heat is supplied to compensate for the base bonding energies, secondary RNA
structures are disassembled. Likewise translation inhibiting 5′ mRNA structures (de Smit and van Duin
1990, Griswold et al. 2003) are destabilized or completely eliminated at elevated temperatures and
translation initiation efficiency is increased. The same concept of translational control by heat is
known from so-called RNA thermometers. Through secondary structure formation of mRNA,
translation is inhibited at lower temperatures. At higher temperature the structure melts and
translation initiation can occur. Such RNA-based genetic control systems can be found in heat- and
cold-shock responses or in virulence gene expression (Narberhaus et al. 2006). Neupert and co-
workers exploited this principle for temperature-controlled recombinant protein expression in E. coli
and designed synthetic RNA thermometers (Neupert et al. 2008).
High-temperature cultivation versus 5′ mRNA optimization
The experimental design allows a separate analysis of the effects of high-temperature cultivation and
5′ mRNA optimization. SDS-PAGE analysis of total protein fractions revealed that the 5′ sequence
optimization had a stronger impact on total protein expression than the high-temperature approach
(Figure 17B). Regarding the increase of exhibited PNP activity per cell, however, the high-
temperature cultivation approach seems to be more effective than the optimization of the 5′ mRNA
(Figure 17C). Hence, other factors than the stability of 5′ mRNA structures and their curtailing effect
on total protein expression have to be considered for the interpretation of the results.
A possible cause for the described phenomenon lies in the amino acid change that was introduced in
the course of 5′ mRNA optimization. One could argue that the replacement of valine by isoleucine
might lead to misfolding and aggregation, or impeded activity of the enzyme in another way. We
consider this explanation as rather unlikely for two reasons: Firstly, samples represented by a similar
intensity of soluble band also exhibited PNP activities of similar range. For example the DgPNP
construct cultivated at 42 °C displays a soluble band intensity similar to that of DgPNP1 cultivated at
37 °C. At the same time the determined activities are in a similar range (Figure 17B,C). Secondly, the
formation of insoluble protein does not seem to correlate to the new derived sequences themselves

52 Recombinant expression of NPs
but rather to the overall expression level: At 30 °C no aggregation can be observed for DgPNP1,
whereas at 37 °C and 42 °C a considerable amount of protein becomes insoluble.
T S T S T S T S T S T S T S T S T S T S T S T S T S T S T S T S T S T S
DgPNP
IPTG [mM]
Cultivation temperature
0.00
0.02
0.04
0.06
0.08
0.10
0 1 2 3 4 5 6 7
ΔA
29
3 [m
in-1
]
Soluble cell extract [%]
BL21-DgPNP2
BL21
A)
B)
C)
0.00
0.05
0.10
0.15
0.20
0.25
0.1 1 0.1 1 0.1 1 0.1 1 0.1 1 0.1 1 0.1 1 0.1 1 0.1 1
DgPNP DgPNP1 DgPNP2 DgPNP DgPNP1 DgPNP2 DgPNP DgPNP1 DgPNP2
30 C 37 C 42 C
DgP
NP
act
ivit
y [U
OD
600-1
]
° ° °
Figure 17: Expression analysis of cultures expressing the original and the optimized DgPNP constructs at different temperatures: E. coli BL21 PNP background activity (A), SDS-PAGE analysis of protein fractions from OD600-normalized samples (B), and determined recombinant PNP activities (C). The PNP activities of soluble protein fractions from E. coli BL21 alone (BL21) and E. coli BL21 expressing DgPNP2 (both at 42 °C) were analyzed with respect to the percentage of crude cell extract in the sample (A). The same cultivation samples from DgPNP, DgPNP1 and DgPNP2, expressed at 30 °C, 37 °C, or 42 °C and induced with 100 μM or 1 mM IPTG served for SDS-PAGE analysis (B) and for the determination of DgPNP activity (C). T= total; S= soluble protein fractions. Error bars: standard deviation from 3 independent enzymatic reactions. Figure adapted from (Szeker et al. 2011).
Other interpretations of the beneficial effects of the high-temperature cultivation beyond the
destabilization of translation inhibiting 5′ mRNA might be found in the scientific literature. Hence, the
transient exposure of E. coli to 42 °C, known as heat shock treatment, has been shown to enhance

Recombinant expression of NPs 53
the soluble expression of some recombinant proteins (Chen et al. 2002, Oganesyan et al. 2007). The
authors discuss the potential role of molecular chaperones, which are part of the heat shock
response and can assist folding or prevent protein aggregation. Another possible explanation for the
beneficial effect of the high-temperature cultivation on enzyme activity may be found in the
thermophilic nature of the purine nucleoside phosphorylase expressed here. Thermophilic proteins
were found to be characterized by slow folding rates (Kaushik et al. 2002, Ogasahara et al. 1998)
which should increase with higher temperature. Indeed it was shown that the solubility of a number
of thermophilic proteins was enhanced when cultivating the recombinant E. coli strains at high
temperatures (Koma et al. 2006). In the same study chaperone co-expression experiments were also
performed and did not improve the expression, making it unlikely that increased levels of molecular
chaperons are responsible for the positive result.
Combination of both methods yields best result
The combination of sequence optimization and high-temperature cultivation resulted in the highest
PNP activity per cell (Figure 17C). Compared to the original sequence expressed at 30 °C the activity
per cell was increased over 18-fold by expressing optimized PNP sequences at 42 °C.
3.3.3. Functional expression of DgPNP with N-terminal hexahistidine tag
The preceding two subsections have shown that i) DgPNP is not correctly folded if expressed with
“long” N-terminal tag and that ii) secondary structures within the 5′ mRNA impede an efficient
protein expression if this tag is removed and only the wild type DgPNP gene is expressed. Although
we successfully overcame this problem by high-temperature cultivation and optimization of the
5′ mRNA sequence, the expression of hexahistidine-tagged DgPNP would be desirable for certain
reasons. Firstly, a hexahistidine tag permits a simple and fast purification of the target protein via
immobilized metal ion affinity chromatography. Secondly, cloning a hexahistidine coding tag
upstream of the target gene provides a defined 5′ mRNA sequence in the region relevant for an
efficient translation initiation. If one time shown that the expression works well with such a
construct, a variety of target genes could be cloned downstream of this sequence without the need
to bother about secondary 5′ mRNA structure formation and the destabilization thereof for protein
expression. Having in mind that the aim is to clone and express 5 more target enzymes (for details
see section3.1.2), it would be hence of great value to make use of such straight-forward cloning
approach. This motivated us to investigate, whether the expression of correctly folded DgPNP with
N-terminal his tag is possible, despite the negative results shown for the pCTUT7_DgPNP construct in
section 3.3.1. Since we assumed that the multitude of additional amino acids at the N-terminus of
DgPNP lead to folding problems in this original vector construct, the aim was now to remove the
superfluous codons between N-terminal histidine tag and the start of the target gene sequence. The
resulting vector will be assigned as pKS2_DgPNP.
Initial expression experiments with pKS2_DgPNP were performed in TB medium at 37 °C. As in the
previous experiments, the E. coli BL21 strain was used. Protein expression was induced by adding
IPTG to a final concentration of 50 µM. Under these conditions, DgPNP was mostly expressed in

54 Recombinant expression of NPs
insoluble form (Figure 18). However, in contrast to the previous experiments with pCTUT7_DgPNP
(Figure 13), a significant amount of soluble DgPNP can be found, too. In addition, the amount of
soluble DgPNP does not decrease after thermo-treatment at 50 °C which indicates that DgPNP is
correctly folded.
14
18
25
35
45
66
0
4
8
12
16
20
0 1 2 3 4 5 6O
pti
cal d
ensi
ty (
60
0 n
m)
Cultivation time [h]
Induction
(kDa) M
Before 50 °C incubation
After 50 °C incubation
S IN S IN
A) B)
Figure 18: Expression of DgPNP with N-terminal hexahistidine tag from E. coli BL21 pKS2_DgPNP. (A) SDS-PAGE analysis of soluble (S) and insoluble (IN) protein fractions before and after a 5 min incubation step at 50 °C. (B) Optical density at 600 nm over cultivation time. Cells were cultivated in TB medium at 37 °C; 50 µM IPTG 2.5 h after inoculation. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric DgPNP subunit including N-terminal hexahistidine tag (30 kDa). M= molecular weight marker.
Since it has been shown that media with enzyme based glucose delivery have beneficial effects on
protein expression and cell density (Krause et al. 2010, Panula-Perälä et al. 2008, Ukkonen et al.
2011), EnPresso medium was used in the following experiments aiming at the optimization of the
yield of soluble DgPNP. Culture samples were analyzed by SDS-PAGE with respect to the recombinant
expression of DgPNP. Initial screening experiments showed that in samples taken 24 h after induction
significantly higher levels of recombinant DgPNP were found than in samples taken 7 h after
induction (data not shown). In the following experiments DgPNP was thus expressed for 24 h, which
is also in agreement with the instructions given by the EnPresso’s manual. Analysis of different IPTG
concentrations at 30 °C showed that 20 µM was sufficient to ensure an efficient expression of
DgPNP, while increasing the IPTG concentration to 100 µM leads mainly to a higher level of insoluble
aggregates (Figure 19A). In a next experiment the IPTG concentration was kept at 20 µM while three
different cultivation temperatures were tested (30 °C, 37 °C, 42 °C). At 42 °C significantly less soluble
protein was found than at 30 ° and 37 °C, while the amount of soluble DgPNP appeared to be slightly
higher at 37 °C than at 30 °C (Figure 19B). According to these results the optimal conditions for
DgPNP expression were defined (37 °C, 20 µM IPTG, 24 h expression) for future production of DgPNP.
Under these conditions we also obtained a reasonable cell density of OD600 = 25 (Figure 19C).

Recombinant expression of NPs 55
18
25
35
45
66
116
(kDa) M0 µM 20 µM 100 µM
IN S IN S IN S
30 °C 37 °C 42 °C
S S S
A) B)
0
5
10
15
20
25
0 10 20 30 40
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
C)
Induction
Figure 19: Expression study with E. coli BL21 pKS2 _DgPNP using EnPresso medium. SDS-PAGE analysis of expression study with (A) variable IPTG concentration at 30 °C; and (B) variable cultivation temperatures where cultures were induced with 20 µM IPTG. Cells were harvested 24 h after induction. (C) Growth curve of the cultivation performed at 37 °C with 20 µM IPTG. Arrows indicate the bands corresponding to the molecular weight theoretically calculated for the monomeric DgPNP subunit including a hexahistidine tag (30 kDa). M=molecular weight marker; IN=insoluble protein fraction; S=soluble protein fraction.
3.3.4. DgPNP expression - summary and conclusions
The experiments described in this section demonstrate how DgPNP can be efficiently expressed in
correctly folded form. For initial experiments the target sequence was cloned into a vector that
confers a hexahistidine tag at the N-terminus. The artificial N-terminal tag that was generated
contained not only this hexahistidine tag, but also a number of other amino acids, that were the
result of the recombinational cloning approach used. In this vector construct, DgPNP could only be
expressed in insoluble form. Chaperone co-expression appeared to aid soluble expression of DgPNP,
but thermo-treatment at 50 °C indicated that the protein had not obtained its natural conformation.
The removal of the N-terminal tag allowed the expression of soluble and active enzyme, but in
extremely low amount. The formation of translation inhibiting secondary 5′ mRNA structures was
found to be the reason for the poor expression. We tackled this problem by sequence optimization of
the 5′ mRNA and a high-temperature cultivation approach. Both methods were very effective to
reduce the stability of the 5′ mRNA and increase the yield of active DgPNP. The combination of both
methods gave the best result. This strategy might be also useful for the expression of other wild type
genes (lacking artificial N-terminal tags), especially for those derived from thermophiles. Two reasons
strongly support this assumption: The first reason is that secondary structures in the 5′ mRNA were
found to be less reduced in thermophiles than in mesophiles (Gu et al. 2010), suggesting that the
expression in mesophilic hosts is frequently impeded by 5′ mRNA structure formation. The second
reason is that the high-temperature cultivation was reported to have a positive effect on the soluble
expression of thermophilic proteins (Koma et al. 2006), which makes these proteins, in contrast to
their mesophilic counterparts, especially amenable to a high-temperature cultivation approach. In
section 3.4.1 the successful application of this strategy for the expression optimization of another

56 Recombinant expression of NPs
thermostable nucleoside phosphorylase (5′ methythioadenosine phosphorylase from the
hyperthermophilic archaeon A. pernix) will be presented.
This section has furthermore revealed that eliminating the N-terminal tag completely is not
necessary to obtain functional DgPNP. Instead, it was sufficient to reduce the N-terminal tag to the
hexahistidine sequence by cloning via restriction and digestion. This methodology opens the door for
simple purification methods based on the IMAC technology. A positive side effect is that the mRNA in
the region relevant for translation initiation consists now out of the hexahistidine coding sequence
instead of the DgPNP insert. This makes 5′ mRNA optimization and high-temperature cultivation
unnecessary for the efficient expression of DgPNP. For these reasons the resulting vector backbone
(pKS2) was also used for the expression of the other target sequences of this study.
Finally we have shown here that the yield of soluble DgPNP (when expressed from pKS2 in
E. coli BL21) could be substantially increased - mainly by minimizing the formation of insoluble
DgPNP aggregates through reduced protein expression rates. This effect was achieved by controlling
the substrate feed (EnPresso medium) and reducing the IPTG concentration.
3.4. Expression of ApMTAP
The gene coding for 5′-methylthioadenosine phosphorylase was isolated from the hyperthermophilic
archaeon A. pernix that shows optimal growth between 90 °C and 95 °C (Sako et al. 1996). It was
hence anticipated that the resulting recombinant protein (ApMTAP) would also be temporarily stable
in this temperature range.
3.4.1. Expression of the wild type ApMTAP gene without tag
The initial aim was to express the natural amino acid sequence of ApMTAP without any artificial
fusion tag. Therefore the pKS1 vector (see section3.3.1 for details) and the natural ApMTAP gene
sequence were used, the latter only modified in its start codon that was changed from TTG to ATG.
Both start codons encode N-formylmethionine, but ATG is more efficiently recognized in E. coli.
Before starting with the cloning procedure, the secondary structural properties of the resulting
pKS1_ApMTAPmRNA were predicted theoretically (Table 10). The result showed that a stable
substructure in the 5′ region is formed, that was expected to disturb efficient expression of ApMTAP
in a similar manner as discussed for DgPNP. Since the combination of 5′ mRNA sequence optimization
and high-temperature cultivation improved the expression level of DgPNP significantly, we
considered the same approach right from the start of ApMTAP expression studies. On first sight,
sequence optimization appeared to be easier in this case: two silent substitutions were found by
automated optimization (Pro4: CCGCCA and Leu7: CTCCTA). In the resulting ApMTAP1 construct
the fee energy of 5′ mRNA secondary structure formation was increased by approx. 3.6 kcal mol-1
(Table 10). SDS-PAGE analysis of culture samples showed that these substitutions alone were not
enough to achieve a considerable positive effect on the ApMTAP expression level (Figure 20A). On
the other hand, the expression level increased significantly with the expression temperature, and we

Recombinant expression of NPs 57
concluded that a better result could be obtained if secondary structures could be further destabilized
by sequence optimization. For this purpose two additional silent substitutions were introduced (Lys3:
AAGAAA, Val5: GTTGTA), resulting in ApMTAP2 (Table 10). With this optimized construct
considerably higher total and soluble expression levels could be achieved than with the original
ApMTAP sequence (Figure 20B). The same expression study was also repeated with the E. coli
Rosetta strain (Novagen) to compensate for the abundance of rare codons in the ApMTAP gene
sequence (Table 8) by overexpression of rare tRNAs. However, the resulting ApMTAP expression
patterns (Figure 20C) did not change significantly compared to the patterns obtained
withE. coli BL21.
The conclusions that can be drawn on the level of SDS-PAGE analysis are the same as described for
DgPNP expression: both sequence optimization and high-temperature cultivation lead to increased
total and soluble expression levels of ApMTAP and the best result was obtained when both methods
were combined, i.e. expression of the optimized sequence at 42 °C.
Table 10. Secondary-structural properties of ApMTAP 5′ gene variants
Variant 5′ codons ΔGF(kcal mol-1)
ApMTAP ATG-AGG-AAG-CCG-GTT-CAC-CTC -16.0
ApMTAP1 ATG-AGG-AAG-CCA-GTT-CAC-CTA -12.4
ApMTAP2 ATG-AGG-AAA-CCA-GTA-CAC-CTA -10.7
Silent nucleotide substitutions (bold) lead to variants ApMTAP1 and ApMTAP2.
3.4.2. ApMTAP expression with N-terminal hexahistidine tag
Despite the positive results presented in the previous section, the fusion of a hexahistidine tag to
ApMTAP would be of advantage, too. As discussed before, this strategy permits a simple, straight-
forward purification procedure that is a prerequisite to rapidly move forward to the characterization
of the recombinant protein. The experiments on DgPNP expression have shown that functional
expression with hexahistidine tag is possible, if the tag does not contain too many other additional
amino acids. Hence, the same vector backbone that was suitable for DgPNP expression with N-
terminal hexahistidine tag was used for the following studies with ApMTAP, resulting in the vector
pKS2_ApMTAP.
Initial screening in TB medium
The first aim was to quickly decide which expression strain is beneficial for ApMTAP expression.
Therefore both E. coli BL21 and E. coli Rosetta were investigated as expressions strains. The latter
was considered to be of advantage for ApMTAP expression, since the native gene contains numerous
codons that are only rarely used in E. coli (Table 8).

58 Recombinant expression of NPs
(kDa) M
ApMTAP ApMTAP1
30 °C 37 °C 42 °C 30 °C 37 °C 42 °C
T S T S T S T S T S T S
116
66
45
35
25
18
14
(kDa) M
30 °C 37 °C 42 °C
ApMTAP ApMTAP2 ApMTAP ApMTAP2 ApMTAP ApMTAP2
T S T S T S T S T S T S
116
66
45
35
25
18
14
A)
30 °C 37 °C 42 °C
ApMTAP ApMTAP2 ApMTAP ApMTAP2 ApMTAP ApMTAP2
T S T S T S T S T S T S
116
66
45
35
25
18
14
(kDa) M
B)
C)
Figure 20: SDS-PAGE analysis of expression studies with original (ApMTAP) and optimized (AMTAP1 and ApMTAP2) ApMTAP variants expressed in E. coli BL21 (A,B) or E. coli Rosetta (C). Cultivations were performed at 30 °C, 37 °C, or 42 °C. The cell lysates were incubated for 10 min at 90 °C prior to separation of soluble from insoluble protein fractions. Arrows indicate the bands corresponding to the molecular weight theoretically calculated for the monomeric ApMTAP subunit (25.6 kDa). T= total, S= soluble protein fractions; M= molecular weight marker.

Recombinant expression of NPs 59
The expression was performed in TB medium at 37 °C with an induction of 20 µM IPTG. Since the
gene product was expected to be highly thermostable, the culture samples were thermo-treated
at85 °C prior to SDS-PAGE analysis. The results show that a thermostable protein was expressed –
with molecular weight according to the theoretically calculated value for the monomeric ApMTAP
subunit including the hexahistidine tag (Figure 21A). This indicates that correctly folded ApMTAP was
expressed. The amount of ApMTAP, however, did not considerably depend on the choice of the
expression strain. On the other hand the cell density that was obtained with BL21 was significantly
higher than that obtained with the Rosetta strain (Figure 21B). Hence, only E. coli BL21 was used for
the following expression optimization.
Expression in EnPresso medium
The aim was to investigate whether the expression level of soluble ApMTAP can be further increased
by making use of EnPresso medium. Therefore the expression with E. coli BL21 at 30 and 37 °C with
three different levels of induction was analyzed. No strong effects of these parameters on the
amount of ApMTAP could be seen (Figure 22). However, the results show, that 20 µM IPTG is enough
for an efficient induction, and ApMTAP band intensity seemed to be higher in comparison to other
bands at 30 °C than at 37 °C.
(kDa) MBL21 Rosetta
S IN S IN
116
66
45
35
25
18
14
0
2
4
6
8
10
12
0 1 2 3 4 5 6
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
BL21
Rosetta
Induction
A) B)
Figure 21: ApMTAP expression in TB medium. (A) SDS-PAGE analysis of ApMTAP expressed with N-terminal hexahistidine tag (pKS2_ApMTAP) in E. coli BL21 or E. coli Rosetta and (B) growth curves of the cultivations. ApMTAP expression was induced with 20 µM IPTG at 37 °C 2.5 h after inoculation;cells were harvested 3 h after induction. The cell lysate was incubated at 85 °C prior to separation of soluble from insoluble protein fractions. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric ApMTAP subunit including hexahistidine tag (27 kDa); S = soluble, IN= insoluble protein fractions; M= molecular weight marker.
Another experiment was performed to check the influence of the time period of induction at 37 °C.
Therefore samples taken 6 h and 22 h after induction were analyzed. The results clearly show that
the expression level after the longer time period of induction, i.e. 22 h, lead to a higher expression
level of ApMTAP (Figure 23).

60 Recombinant expression of NPs
In contrast to DgPNP expression, where the amount of soluble protein could be significantly
increased in favour of insoluble protein with the help of EnPresso medium, the amount of soluble
ApMTAP per cell could not be increased with EnPresso medium in comparison to TB medium. On the
other hand, the volumetric yield of soluble ApMTAP could be considerably increased due to the
higher cell density reached: The optical density reached with EnPresso medium was more than two-
fold higher than that with TB medium (Figure 21B, Figure 23B).
(kDa) M
30 °C 37 °C
0 20 50 100 0 20 50 100
S S IN S IN S IN S S IN S IN S IN
116
66
45
35
25
18
14
IPTG (µM)
Figure 22: SDS-PAGE analysis of ApMTAP expression in EnPresso medium at 30 and 37 °C with 0 - 100 µM IPTG. E. coli BL21 pKS2_ApMTAP culture samples were harvested 25 h after induction; cell lysates were incubated at 85 °C prior to separation of soluble (S) from insoluble (IN) protein fractions. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric ApMTAP subunit including hexahistidine tag (27 kDa). M= molecular weight marker.
(kDa) M22 h 6 h
IN S IN S
116
66
45
35
25
18
14
0
5
10
15
20
25
30
0 10 20 30 40
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
Induction
A) B)
Figure 23: ApMTAP expression in EnPresso medium over time. (A) SDS-PAGE analysis of samples harvested 6 h and 22 h after induction and (B) corresponding growth curve. E. coli BL21 pKS2_ApMTAP was cultivated at 37 °C with 20 µM IPTG. The cell lysate was incubated at 85 °C prior to separation of soluble (S) from insoluble (IN) protein fractions. The arrow indicates bands corresponding to the molecular weight theoretically calculated for the monomeric ApMTAP subunit including hexahistidine tag (27 kDa); M= molecular weight marker.

Recombinant expression of NPs 61
Evidence for intersubunit disulfide bonds in ApMTAP
According to the theoretical predictions presented in Table 8, the cysteine in position 112 of ApMTAP
forms a disulfide bond. Therefore it seems likely that ApMTAP is stabilized by intersubunit disulfide
linkages. As it will be shown later (in section 4.2.1), based on amino acid sequence alignments
ApMTAP shows high similarity to MTAPI from S. solfataricus. The characterization of the
S. solfataricus enzyme in turn revealed a hexameric structures, in which three intersubunit disulfide
bonds link dimers to each other to form a hexamer (Appleby et al. 2001, Cacciapuoti et al. 1994). The
close evolutionary relationship of ApMTAP to MTAP from S. solfataricus hence corroborates the
presence of stabilizing intersubunit disulfide linkages, even though Cys129 instead of the predicted
Cys112 would be involved in the disulfide bond according to the amino acid sequence alignment.
Within the purification of ApMTAP, SDS-PAGE analysis revealed the presence of two bands. One
band corresponded to the molecular weight of the monomeric subunit (27 kDa), while the other
band represented a molecular weight of more than 116 kDa. It was confirmed by mass spectrometric
analysis (by Knut Büttner, Division of microbial physiology and molecular biology, Ernst-Moritz-Arndt-
University of Greifswald, Germany) that this second band also represents ApMTAP (data not shown).
For the reasons outlined in the previous paragraph we assumed that this second band represents
some oligomerization state of the monomeric subunit that is possibly stabilized by intersubunit
disulfide bonds. Further analysis indeed revealed that this second band was the predominant band
when ApMTAP samples were not treated with DTT prior to SDS-PAGE analysis. Conversely, the band
representing the molecular weight of the monomeric subunit was predominant after default sample
preparation that includes an incubation step at 95 °C in the presence of 200 µM DTT. Noteworthy
this phenomenon was not observed with the other proteins that were analyzed (Figure 24).
116
66
45
35
25
18
(kDa) M
- DTT treatment
TtP
yNP
Ap
MTA
P
DgP
NP
GtP
NP
GtP
yNP
TtP
yNP
Ap
MTA
P
DgP
NP
GtP
NP
GtP
yNP
Figure 24: Influence of DTT treatment on SDS-PAGE analysis of ApMTAP. Samples on the right side were prepared according to the standard protocol that includes DTT treatment. Samples on the left side were not treated with DTT. Arrows highlight the different positions of the bands representing ApMTAP in dependence of DTT treatment.

62 Recombinant expression of NPs
3.4.3. ApMTAP - summary and conclusions
Both the native ApMTAP sequence and an ApMTAP construct comprising a hexahistidine tag at the
N-terminus were cloned and overexpressed. Stable secondary 5′ mRNA structures impaired the
efficient expression of the native ApMTAP amino acid sequence initially. In analogy to DgPNP
expression optimization the yield of soluble ApMTAP was increased by a combination of 5′ mRNA
sequence optimization and high-temperature cultivation. The results further support the general
applicability of this strategy that is discussed in sections 3.3.2 and 3.3.4 in detail. Cloning ApMTAP
into a vector conferring N-terminally a hexahistidine tag (assigned here as pKS2_ApMTAP) proved to
be a good alternative to this approach for the purpose of this study: The ApMTAP gene product is still
thermostable, the expression is efficient and the hexahistidine tag enables the application of a simple
purification procedure that is favourable for following characterization studies (presented in
chapter 4 and 5).
Expression studies with pKS2_ApMTAP in E. coli BL21 or E. coli Rosetta as host yielded similar
amounts of recombinant protein, despite the presence of numerous rare codons. The volumetric
yield of ApMTAP expressed in BL21 was significantly higher in EnPresso medium than in TB medium,
due to the higher cell density reached. SDS-PAGE analysis and theoretical predictions strongly
support the presence of at least one intersubunit disulfide bond. However, the positive results
obtained here on the level of SDS-PAGE (and in the following sections on the basis of enzyme
activity) with E. coli BL21 or E. coli Rosetta suggest that it is not absolutely necessary to make use of
specialized expression systems. An example of such a specialized expression system is E. coli Origami,
a mutant strain with a mild oxidative cytoplasm that is offered by Novagen for the expression of
proteins containing disulfide bonds, but is generally more difficult to grow to high cell densities.
Noteworthy the successful cytoplasmic expression of ApMTAP in E. coli BL21 was recently also
reported by others (Zhu et al. 2012). The authors refer to a PNP, but comparison of the primers used
for the isolation reveals that the gene coding region coincides with the ApMTAP sequence here
under investigation.
The conclusions that can be drawn regarding the expression vector are basically the same as for
DgPNP expression. In both cases, expression of the target gene with hexahistidine tag in the pKS2
vector lead to presumably correctly folded protein in relatively high yield. Hence, the expression of
the following proteins will be directly started with this vector.
3.5. Expression of GtPNP
The gene coding for purine nucleoside phosphorylase (GenBank accession number EFG53380) was
isolated from G. thermoglucosidasius 11955. Since this microorganism shows optimal growth at 55 °C
(Suzuki et al. 1983), it was expected that the resulting protein is also temporarily stable at this
temperature. GtPNP was expressed in two different constructs. In the first vector, a hexahistidine tag
was fused to the C-terminus, in the second vector a hexahistidine tag was fused to the N-terminus.

Recombinant expression of NPs 63
3.5.1. GtPNP expression with C-terminal hexahistidine tag
Initially the GtPNP gene was cloned into the vector pET21a in such a way, that only the native gene is
expressed without any fusion partner. Expression resulted in high amount of both soluble GtPNP that
was retained in the soluble protein fraction after incubating the cell lysate at temperatures up to
65 °C (data not shown). To simplify the purification the vector was modified in such a way that a
hexahistidine tag is cloned to the C-terminus of the protein. Surprisingly, expression under similar
conditions as before (TB medium, 37 °C) resulted in almost exclusively insoluble recombinant GtPNP
(Figure 25).
3.5.2. GtPNP expression with N-terminal hexahistidine tag
Cloning and expression studies of DgPNP and ApMTAP have meanwhile shown that both proteins
were well expressed from the vector pKS2 that confers an N-terminal hexahistidine tag. The same
vector was used now for GtPNP expression. E. coli BL21 was transformed with pKS2_GtPNP,
cultivated at 37 °C in TB medium and induced with 100 µM IPTG. Under these conditions, GtPNP was
expressed in very high yield in predominantly soluble form. In addition the gene product could be still
found in the soluble protein fraction after incubation at 65 °C for 10 min. These positive results made
any further expression optimization unnecessary.
(kDa) M S IN
116
66
45
35
25
18
14
Induction
0
2
4
6
8
10
12
14
0 2 4 6
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
A) B)
Figure 25: Expression of GtPNP with C-terminal hexahistidine tag. (A) SDS-PAGE analysis of E. coli Rosetta2 (DE3) pET21a_GtPNP culture sample (B) growth curve of the cultivation. Cells cultivated in TB medium at 37 °C; induction of GtPNP expression with 100 µM IPTG; harvest 3 h after induction. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric GtPNP subunit including hexahistidine tag (27.6 kDa). S= soluble, IN= insoluble protein fraction; M= molecular weight marker.
3.5.3. GtPNP expression - summary and conclusion
The expression studies on GtPNP have further highlighted the critical role of fusion tags on the
functional expression of nucleoside phosphorylases: Apparently not only the length (see
section3.3.1), but also the position of the fusion tag is decisive for proper folding. While the C-

64 Recombinant expression of NPs
terminal hexahistidine tag lead to aggregation, GtPNP was very efficiently expressed in soluble form
from pKS2 with N-terminal fusion of the hexahistidine tag. The high yield of functional GtPNP made
further expression optimization superfluous.
(kDa) M S IN
116
66
45
35
25
18
14
0
4
8
12
16
0 2 4 6 8O
pti
cal d
ensi
ty (
60
0 n
m)
Cultivation time [h]
Induction
A) B)
Figure 26: Expression of GtPNP with N-terminal hexahistidine tag. (A) SDS-PAGE analysis of E. coli BL21 pKS2_GtPNP samples (B) growth curve of the cultivation. GtPNP expression in TB medium was induced with 100 µM IPTG at 37 °C. Samples were harvested 3.5 h after induction; the cell lysate was incubated at 65 °C prior to separation of soluble (S) from insoluble (IN) protein fractions. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric GtPNP subunit including hexahistidine tag (27.6 kDa); M= molecular weight marker.
3.6. Expression of GtPyNP
Next to the purine nucleoside phosphorylase the pyrimidine nucleoside phosphorylase of
G. thermoglucosidasius 11955 was studied. It was likewise expected that the recombinant protein is
stable at least at 55 °C. GtPyNP was expressed with and without N-terminal hexahistidine tag.
Parts of the expression study on GtPyNP have been previously published (Szeker et al. 2012).
3.6.1. Chemical lysis buffer decreases apparent thermal stability of GtPyNP
In analogy to GtPNP, GtPyNP was first expressed in the vector pET21a in such a way that no tag is
fused to the protein. Expression in TB medium under standard conditions (37 °C, 100 µM IPTG)
resulted in a high yield of recombinant GtPyNP that was almost exclusively produced in soluble form
(Figure 27). However, unexpectedly and in contrast to GtPNP, the gene product appeared to be not
thermostable. When the cell lysate was incubated at temperatures exceeding 45 °C, GtPyNP was not
completely retained in the soluble protein fraction.
With the assumption that GtPyNP did not obtain its native conformation and hence lacked
thermostability, two measures were undertaken to overcome this problem. Firstly, the same plasmid
(pET21a_GtPyNP) was expressed in E. coli Origami cells. The idea behind was that disulfide bond
formation could have been necessary to obtain thermostable GtPyNP. In contrast to usual E. coli

Recombinant expression of NPs 65
expression strains, Origami promotes disulfide bond formation of recombinant proteins expressed in
the cytoplasm of E. coli. The second measure undertaken applied to the amino acid sequence.
Compared to the data bank sequence, the gene cloned here encoded glutamine (side chain not
charged at physiological pH) on position 214 instead of arginine (positively charged side chain). This
mutation could have been the result of errors that occurred during the cloning procedure, for
example during the amplification of the gene via PCR. Hence, the coding region was modified by site-
directed mutagenesis, to match the respective databank entry (gene bank accession number
ZP_06809030). However, both measures did not change the apparent lack of thermostability as
shown in Figure 27A.
Finally the solution for the problem was found in the nature of the buffer, in which the cell lysate was
incubated to estimate the stability at high temperatures. So far, cells were disrupted through
treatment with a chemical lysis buffer, namely Bugbuster® protein extraction reagent (Novagen).
However, in following experiments cell pellets were resuspended in a phosphate buffer (50 mM,
pH 8) and disrupted by sonication. Investigation of cell lysates obtained with this method showed
that GtPyNP appeared to be stable at temperatures up to 60 °C now (Figure 27B).
3.6.2. GtPyNP expression with N-terminal hexahistidine tag
The positive results obtained with the pKS2 vector prompted us to use the same vector for GtPyNP
expression. E. coli BL21 pKS2_GtPyNP was cultured under standard conditions (TB medium, 37 °C)
and recombinant protein expression was induced by the addition of 100 µM IPTG. GtPyNP was
expressed in very high yield in predominantly soluble form and was retained in the soluble protein
fraction after incubation of the cell lysate at 60 °C. Further optimization was considered as
unnecessary.
(kDa) M- 45 °C 50 °C 55 °C
S IN S IN S IN S IN
116
66
45
35
25
18
14
(kDa) M55 °C 60 °C 65 °C 70 °C 75 °C
S IN S IN S IN S IN S IN
116
66
45
35
25
18
14
A) B)
Figure 27: SDS-PAGE analysis of thermo-treated E. coli BL21 pKS2_GtPyNP culture samples. Cell disruption was performed by chemical means with lysis buffer (A) or by sonication of cells resuspended in phosphate buffer (B). Soluble (S) and insoluble (IN) fractions of culture samples that were treated at the indicated temperatures for 10 min are shown. Arrows indicate the bands corresponding to the molecular weight theoretically calculated for the monomeric GtPyNP subunit (46 kDa); M= molecular weight marke.

66 Recombinant expression of NPs
A) (kDa) M S IN
116
66
45
35
25
18
14
0
2
4
6
8
10
12
0 2 4 6 8
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
B)
Induction
Figure 28: Expression of GtPyNP with N-terminal hexahistidine tag. (A) SDS-PAGE analysis of E. coli BL21 pKS2_GtPyNP culture sample, (B) growth curve of the cultivation. GtPyNP expression in TB medium was induced with 100 µM IPTG at 37 °C. Samples were harvested 3.5 h after induction. The cell lysate was incubated at 60 °C prior to the separation of the soluble (S) from the insoluble (IN) protein fraction. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric GtPyNP subunit including hexahistidine tag (47.6 kDa). M= molecular weight marker
3.6.3. GtPyNP expression - summary and conclusions
So far, incubation of the cell lysates of expression cultures at high temperatures and subsequent
SDS-PAGE analysis has served as quick tool to evaluate whether the target protein is properly folded.
However, this section has demonstrated the limitations of this procedure, since it was shown that
the buffer composition dramatically influenced the apparent stability of GtPyNP. In chapter 4- that
deals with the characterization of the target enzymes, stability issues will be investigated in more
detail.
As observed for all other target proteins investigated so far, the pKS2 vector was found to be a good
expression vector for GtPyNP, too. Indeed, the expression efficiency under standard conditions was
so high, that optimization was considered as unnecessary and investigations on GtPyNP could
proceed directly with the characterization of the recombinant protein.
3.7. Expression of TtPyNP
The gene coding for pyrimidine nucleoside phosphorylase was isolated from
T. thermophilus HB27.Since this microorganism shows optimal growth at around 68 °C (Oshima and
Imahori 1974), it was expected that the resulting recombinant protein (TtPyNP) is also temporarily
stable at this temperature.
Parts of the results presented in this section have been previously published (Szeker et al. 2012).

Recombinant expression of NPs 67
3.7.1. TtPyNP expression with N-terminal hexahistidine tag
TtPyNP was cloned in vector pKS2. The resulting expression plasmid (pKS2_TtPyNP) was transformed
in E. coli BL21 and E. coli Rosetta. Initial studies showed that TtPyNP was predominantly expressed in
soluble form and was completely retained in the soluble fraction after incubation at 80 °C (data not
shown).
The expression level of TtPyNP was generally poor. In order to optimize the yield of soluble TtPyNP
different cultivation media were studied. Experiments within the course of this study revealed that
the most reproducible result was obtained through the use of media with enzyme based glucose
delivery (EnBase Flo, EnPresso) in contrast to conventional complex media (LB, TB) (data not shown).
In Figure 29 an exemplary expression study in the different media is shown in which the best result
was obtained with EnPresso medium. The figure also indicates that despite the abundance of rare
codons in the TtPyNP gene, the expression level per cell could not be significantly increased by the
use of the E. coli Rosetta strain in which rare tRNAs are overexpressed.
(kDa) MRosetta2 BL21
LB TB EP LB TB EP
116
66
45
35
25
18
Figure 29: SDS-PAGE analysis of pKS2_TtPyNP expression in E. coli Rosetta2 or BL21 in different media. The cultivations were performed at 37 °C in LB, TB, or EnPresso (EP) medium; induction of TtPyNP expression with 1 mM IPTG. Soluble fractions of samples were treated for 5 min at 80 °C after cell disruption. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric TtPyNP subunit including hexahistidine tag (approx. 47 kDa); M= molecular weight marke.
Further investigation on TtPyNP expression in E. coli BL21 cultivated in EnBase Flo medium showed,
that the expression level increased only moderately with the expression time. Nevertheless, a higher
volumetric yield could be obtained by extending the time period after induction (24 h), due to the
higher optical density reached: The final OD600 obtained with EnBase Flo was 24 (Figure 30); instead,
with TB medium only OD600 =11 was reached (data not shown).

68 Recombinant expression of NPs
0
4
8
12
16
20
24
0 10 20 30 40
Op
tica
l den
sity
(6
00
nm
)
Cultivation time [h]
(kDa) M
S IN
Thermo -treatment
-Thermo-
treatment-
0 8 24 0 8 24 0 8 24 0 8 24
116
66
45
35
25
18
14
A) B)
Induction
Figure 30: Expression pKS2_TtPyNP E. coli BL21 in dependence of expression time. (A) SDS-PAGE analysis, (B) growth curve of the cultivation. The cultivation was performed at 30 °C in EnBase Flo medium, induction with 1 mM IPTG. Cells were harvested 0, 8, and 24 h after induction. Soluble (S) and insoluble (IN) protein fractions were analyzed before and after thermo-treatment for 5 min at 80 °C. The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric TtPyNP subunit including hexahistidine tag (47 kDa); M= molecular weight marke.
3.7.2. TtPyNP expression - summary and conclusions
TtPyNP was successfully expressed with N-terminal hexahistidine tag. The gene product was retained
in the soluble protein fraction after heat-treatment at 80 °C, which indicates that TtPyNP was
correctly folded. The expression level was generally poor. Expression optimization resulted in slightly
higher expression levels, but the final yield of TtPyNP was significantly lower compared to that of the
other proteins investigated in this study, for example GtPyNP. Nevertheless, suitable expression
conditions were found for production of a sufficient amount of TtPyNP for characterization studies
and synthetic reactions as described in chapters 4 and 5.
3.8. Expression of ApUP
The gene coding for uridine phosphorylase was the second gene isolated from the hyperthermophilic
archaeon A. pernix in this study. Due to its origin, the resulting recombinant protein (ApUP) was
hence expected to be also temporarily stable at 90 - 95 °C, where A. pernix shows optimal growth.
The ApUP sequence is characterized by a number of peculiarities that might pose a challenge for the
successful recombinant expression. According to the theoretical predictions listed in Table 8, the
ApUP gene sequence contains numerous rare codons. Moreover, 2 cysteine residues per subunit
were predicted to be in the bonding state, which suggests that inter- or intrasubunit disulfide bonds
play a role for stabilization. One of the cysteines predicted to be in the bonding state (Cys225) is part
of a CXC motif that was reported to be required for folding in the disulfide bond containing proteins
PfPNP (PNP from P. furiosus) and SsMTAPII (MTAPII from S. solfataricus) (Cacciapuoti et al. 2009).
Furthermore, the gene sequence contains two tandemly repeated AGG triplets. This AGGAGG
sequence motif is part of the natural consensus Shine-Dalgarno sequence and was reported to inhibit

Recombinant expression of NPs 69
recombinant protein expression by competing with the “real” Shine-Dalgarno sequence for ribosome
binding (Ivanov et al. 1992, Jin et al. 2006). Noteworthy the gene coding sequence, as it was
annotated originally started with the initiating codon located downstream of this internal ribosomal
binding site. Just after the re-annotation of the A. pernix genome in 2006 the full length ApUP gene
starting before the AGGAGG motif was assigned (Yamazaki et al. 2006). Finally, when the native gene
is cloned in the expression vector used here (resulting in pKS1_ApUP) the free energy of the 5′ mRNA
folding is similarly low (-17.0 kcal mol-1) as reported before for ApMTAP (Table 10), suggesting that
secondary structures might impair the efficient expression.
3.8.1. Expression of ApUP without tag
For the first series of experiments the native ApUP gene (without fusion tag) was overexpressed in
three different E. coli expression strains. These strains include E. coli BL21 as standard strain, E. coli
Origami – that allows disulfide bond formation in the cytoplasm and E. coli Rosetta-gami in which
additionally rare tRNAs are overexpressed to compensate for the abundance of codons in the ApUP
gene that are only rarely used in E. coli.
No thermo-treatment Thermo-treatment at 90 °C
Origami Rosetta-gami BL21 Origami Rosetta-gami BL21
+P +P +P +P +P +P
(kDa) M S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN
116
66
45
35
25
18
14
Figure 31: SDS-PAGE analysis of ApUP expression in E. coli Origami, Rosetta-gami, and BL21. Expression strains harbouring pKS1_ApUP (+P) and empty strains without expression plasmid were cultivated at 37 °C in TB medium, induction of ApUP expression with 20 µM IPTG. Soluble (S) and insoluble (IN) protein fractions were analyzed before (left side) and after thermo-treatment at 90 °C (right side). The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric ApUP subunit (30.3 kDa); M= molecular weight marker
Expression experiments were performed in TB medium at 37 °C, Protein expression was induced by
adding IPTG to a final concentration of 20 µM. The same expression strains but without recombinant
plasmids were cultivated in parallel as a reference. Soluble and insoluble protein fractions were
analyzed by SDS-PAGE before and after heat treatment at 90 °C (Figure 31). A protein with a
molecular size according to the theoretically calculated value for monomeric ApUP was found in the
soluble protein fraction before and after the heat treatment. It was not totally clear, whether this
protein really represents ApUP since a thermostable protein with similar size was also found in

70 Recombinant expression of NPs
“empty” Origami and Rosetta-gami strains. In BL21, however, the expression was more pronounced;
a clear band was observed in BL21 with the recombinant plasmid exclusively. On the other hand,
SDS-PAGE analysis also indicates that the gene product is not totally stable at 90 °C, since the amount
of soluble ApUP seems to decrease after the thermo-treatment. It remains to be interesting whether
the reason can be found in the conditions of the thermo-treatment that might be too harsh (too high
temperature, improper buffer), or in the recombinant protein itself that might have not obtained its
native conformation.
ApUP ApUP1
No thermo-treatment Thermo-treatment No thermo-treatment Thermo-treatment
30 °C 37 °C 42 °C 30 °C 37 °C 42 °C 30 °C 37 °C 42 °C 30 °C 37 °C 42 °C
(kDa) M S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN S IN
116
66
45
35
25
18
14
Figure 32: SDS-PAGE analysis of ApUP expression in E. coli BL21 with the original gene sequence (ApUP) and the optimized sequence, lacking an internal ribosomal binding site (ApUP1). Cultivations were performed at 30, 37, and 42 °C in TB medium; induction with 20 µM IPTG. Soluble (S) and insoluble (IN) protein fractions were analyzed before and after thermo-treatment which was initially performed at 85 °C (ApUP) or 90 °C (ApUP1). The arrow indicates the bands corresponding to the molecular weight theoretically calculated for the monomeric ApUP subunit (30.3 kDa); M= molecular weight marker
A closer look at the ApUP gene revealed the presence of a sequence motif that could serve as
internal ribosomal binding site. In this case, a second, smaller protein would be produced from the
same mRNA molecule. It is possible that in the expression experiment with BL21 and pKS1_ApUP
shown in Figure 31 a mixture of the thermostable full length protein and a slightly shorter,
thermolabile fragment was produced. This would also be an explanation for the apparent loss of
soluble ApUP after thermo-treatment. In order to investigate this hypothesis, the potential internal
ribosomal binding site was removed by silent mutations (AGGAGG CGGCGT); the resulting
expression vector was assigned as pKS1_ApUP1. In addition, sequence analysis of pKS1_ApUP mRNA
has also revealed, that stable secondary structures (free energy of formation of -17 kcal mol-1 at
37 °C) are likely to occur in the 5′ mRNA region. The following expression experiments were hence
performed at different expression temperatures, to investigate whether the yield of recombinant
protein can be increased by high-temperature cultivation, as done before (sections 3.3.2 and 3.4.1).
SDS-PAGE analysis of the recombinant cultures (Figure 32) indeed showed that the yield of soluble

Recombinant expression of NPs 71
ApUP was slightly better at 42 °C. However, the removal of the potential internal ribosomal binding
site did not have a visible positive effect on ApUP expression.
3.8.2. Expression of ApUP with N-terminal hexahistidine tag
In order to produce high amounts of ApUP that can be easily purified for characterization studies,
ApUP1 was also expressed with an N-terminal hexahistidine tag (pKS2_ApUP1). Next to the full
length protein, also the shorter fragment starting after the internal ribosomal binding site was cloned
in this way, resulting in a vector assigned as pKS2_ApUPsh. The respective open reading frame that
starts after the internal ribosomal binding site and coincides with the coding region as it was
annotated for the ApUP gene in the past. Since both the older and the topical annotation are based
on theoretical predictions only, both open reading frames were considered for the final investigation
on ApUP expression of this study. The expression conditions were chosen as before for the native
gene. However, SDS-PAGE analysis of the culture samples revealed that both ApUP constructs with
the N-terminal hexahistidine tag resulted exclusively in the formation of insoluble protein,
independent of the length of the coding region cloned (ApUP1, ApUPsh) and the cultivation
temperature.
30 °C 37 °C 42 °C
ApUP1 ApUPsh ApUP1 ApUPsh ApUP1 ApUPsh
(kDa) M T S T S T S T S T S T S
116
66
45
35
25
18
14
Figure 33: SDS-PAGE analysis of ApUP expression with N-terminal hexahistidine tag in E. coli Rosetta. Both the expression of the full length protein (ApUP1) and the shorter fragment that represents the former ApUP annotation (ApUPsh) was investigated. Soluble (S) and total (T) protein fractions were analyzed after thermo-treatment at 90 °C. The arrows indicate the vertical position of bands presumably representing the full length protein and the shorter fragment.M= molecular weight marker
3.8.3. ApUP expression - summary and discussion
ApUP expression proved to be very challenging. Even at very low expression levels, a high amount of
recombinant protein was expressed in insoluble form. Various aspects were considered for
expression optimization: Different expression strains were tested, an apparent internal ribosomal
binding site was removed and finally also a shorter version of the ApUP gene that represents the old

72 Recombinant expression of NPs
ApUP annotation was considered. The best result, i.e. the highest amount of soluble and
thermostable ApUP, was obtained by the expression of the full-length ApUP gene without any tag.
Fusion of an N-terminal hexahistidine tag - that was a successful strategy to obtain high yields of all
the other proteins studied here – resulted in the synthesis of ApUP in exclusively insoluble form. It
might be of interest for future experiments to study i) whether it is possible to express ApUP with N-
terminal hexahistidine tag by further reducing the expression level and making use of media with
enzyme controlled glucose delivery, ii) whether other lysis buffers would lead to a different result
(having in mind the experiences with GtPyNP expression analysis) and iii) to establish another
purification method and purify ApUP expressed without hexahistidine tag. However, since in the
meantime ApUP was successfully recombinantly expressed by others (Montilla Arevalo et al. 2011)
and investigated with respect to its application as biocatalyst for the synthesis of nucleosides, the
focus of this study shifted more to the other 5 target enzymes, already successfully expressed, and
ApUP expression was not further investigated. It is interesting to note, that the authors of the
invention claim to have successfully expressed uridine phosphorylase from A. pernix by making use of
an expression vector in which thioredoxin is fused to the N-terminal end and a hexahistidine tag to
the C-terminal end, and that the ApUP gene sequence that was cloned represents the older
annotation of the gene, that is the shorter fragment starting after the internal ribosomal binding site.
In another very recent study ApUP expression from plasmid pET30a in E. coli BL21 was reported.
According to the primer sequences used, here the full length gene was cloned (Zhu et al. 2012).
Apparently a hexahistidine tag was fused to the C-terminus, since the reverse primer lacks a stop
codon and a hexahistidine tag is indicated downstream of the restriction site used in pET30a.
3.9. Recombinant expression of NPs - summary and conclusions
Six nucleoside phosphorylases were selected from four different thermophilic microorganisms that
show optimal growth between 50 °C and 95 °C. With the aim in view to generate biocatalysts for
transglycosylation of nucleosides from pyrimidine nucleoside donor to purine base acceptor, the
selection comprises enzymes with specificity towards purine and pyrimidine nucleosides. Five of the
six target proteins were successfully overexpressed in E. coli in moderate or high yield. Based on
thermostability studies with SDS-PAGE analysis these enzymes obtained the correctly folded form.
Overexpression of ApUP proved to be challenging. A variety of attempts to produce thermostable
ApUP failed and finally investigations on expression optimization was discontinued for the sake of
time and owing to the fact that the successful expression and biocatalytic application of ApUP was
already reported in the meantime by others (Montilla Arevalo et al. 2011, Zhu et al. 2012). A number
of obstacles were encountered and had to be overcome also for the five proteins that were
successfully overexpressed. The initially selected expression system proved to be not suitable, most
likely because too many additional amino acids were conferred to the N-terminus of the target
sequence, which possibly precluded the correct folding of the monomeric subunits or impaired the
oligomerization step, respectively. After the elimination of additional N-terminal amino acids, the
formation of stable secondary 5′ mRNA structures impaired the efficient expression of wild type
sequences without N-terminal fusion tag. This hurdle was tackled by an approach involving sequence

Recombinant expression of NPs 73
optimization and high-temperature cultivation. This strategy might also be useful for the expression
of other native gene sequences without N-terminal fusion, especially when derived from
thermophilic microorganisms. The choice of an expression vector that confers N-terminal a
hexahistidine tag proved to be a good alternative and was eventually used for all the five target
proteins. For the expression of a single protein that was not hampered by the formation of stable
secondary 5′ mRNA (GtPNP), also the C-terminal fusion of a hexahistidine tag was investigated, but
this strategy lead to aggregation of the protein product. One of the candidates (GtPyNP) was readily
expressed in soluble form but appeared to lack thermal stability. Closer investigations revealed that
the chemical lysis buffer used had an adverse effect on the thermostability of this protein.
Remarkably an adverse effect of the same lysis buffer was not observed for any other target protein.
Some proteins were readily expressed in soluble form and in very high yield (GtPNP, GtPyNP) while
the expression levels of other proteins was poor (TtPyNP), leading to a low volumetric yield or the
ratio of soluble (presumably correctly folded) to insoluble (presumably not correctly folded) protein
was unfavourable (DgPNP, ApMTAP) under standard conditions (TB medium, 37 °C, 100 µM IPTG).
Both the volumetric yield and the ratio of soluble to insoluble protein could be improved by fine-
tuning the IPTG concentration and by making use of media in which the growth rate is restricted by
enzyme based glucose delivery. Despite the abundance of rare codons in some of the target
sequences, the use of an E. coli strain overproducing rare tRNAs (Rosetta) had either no positive
effect or yielded only moderate improvement that was offset by the decreased final cell density
reached in comparison to the standard expression strain used (BL21).
In the final expression system all target proteins are hence expressed in E. coli BL21 from the same
vector backbone that confers an N-terminal hexahistidine tag. This strategy also paves the way for a
simple purification strategy for all target proteins, a prerequisite for rapidly progressing to the next
work package, the characterization of the biocatalytical properties of the purified enzymes.


4. Characterization of thermostable NPs
The final aim of this study was the synthesis of modified nucleosides by an enzymatic
transglycosylation reaction employing thermostable nucleoside phosphorylases. In order to transfer
a pentofuranosyl moiety from pyrimidine nucleoside to purine base two types of enzymes are
required. In a first step an enzyme with PyNP activity phosphorolytically cleaves the donor
nucleoside; in the second step an enzyme with PNP activity catalyzes the glycosyl bond formation
between the intermediate product (pentofuranosyl-1-phosphate) and the purine base.
This chapter is devoted to the characterization of thermostable NPs that could be employed in this
reaction scheme described. In focus are thermal properties (temperature optimum, stability) and
substrate specificities towards natural and artificial nucleosides. The results help to evaluate which
enzymes are most suitable for a specific catalytic reaction. Moreover, the data help to define the
process operating windows for transglycosylations employing the studied thermostable biocatalysts.
4.1. Thermostable PyNPs
PyNPs have been hardly studied in detail. Only few examples can be found in the scientific literature:
PyNP from B. subtilis (Gao et al. 2006), G. stearothermophilus (Hamamoto et al. 1996, Hori et al.
1990, Saunders et al. 1969) and from T. thermophilus (Shimizu and Kunishima 2007). With respect to
practical use as biocatalyst, only the G. stearothermophilus enzyme has been described (Taran et al.
2009).
The following sections deal with the characterization of recombinantly expressed PyNPs derived from
the thermophilic microorganisms G. thermoglucosidasius 11955 (GtPyNP) and T. thermophilus HB27
(TtPyNP). Albeit the expression and crystallization of PyNP from T. thermophilus HB8 has been
reported (Shimizu and Kunishima 2007), information about the biocatalytic characterization is not
available. Here, data reporting on thermal properties and steady state kinetics for the natural
substrates uridine and thymidine is presented. Of special interest is furthermore the ability of the
thermostable PyNPs to phosphorolyze unnatural pyrimidine nucleosides. Specifically the two 2′-
fluorosubstituted pyrimidine nucleosides 2′-deoxy-2′-fluorouridine (dUrd2′F) and 1-(2-deoxy-2-fluoro-
-D-arabinofuranosyl)uracil (dUrd2′F) are tested as substrates. Both compounds are highly interesting
pentofuranosyl donors in enzymatic transglycosylation reactions aiming at the synthesis of the
respective sugar-modified purine nucleosides. A precondition is however, the availability of PyNPs
that can efficiently catalyze the phosphorolysis of these donor nucleosides.
Major parts of the results presented in this chapter, have been published previously (Szeker et al.
2012). Thermal unfolding of TtPyNP (Figure 37) was studied by Thomas Schwab from the laboratory
of Reinhard Sterner (Institute of Biophysics and Physical Biochemistry, University of Regensburg,
Germany).

76 Characterization of thermostable NPs
4.1.1. Homology modelling
Three entries of solved crystal structures of PyNPs can be found in the Protein Database Bank
(http://www.rcsb.org/pdb/) (Bernstein et al. 1977). Both target enzyme sequences were blasted
against the amino acid sequences belonging to these PDB entries. GtPyNP aligned best with the PyNP
from G. stearothermophilus ATCC 12980 (assigned here as GsPyNP, PDB ID: 1BRW) with 78 %
sequence identity. TtPyNP aligned best with the PyNP from T. thermophilus HB8 (PDB ID: 2DSJ), to
which it is almost identical (approx. 98 % sequence identity) but showed also a high degree of
sequence identity (approx. 50 %) to GsPyNP. The amino acid sequence alignments of GsPyNP,
GtPyNP, TtPyNP, and E. coli thymidine phosphorylase (EcTP) are shown in Figure 34. Amino acids that
have been described to be involved in substrate binding or in the catalytic mechanism, respectively,
are indicated (Mendieta et al. 2004, Pugmire and Ealick 1998).
The chain B of GsPyNP (PDB ID: 1BRW) was used as template to model both TtPyNP and GtPyNP. The
structure of GsPyNP was elucidated in its closed conformation (Pugmire and Ealick 1998), uracil and
the phosphate ion can be seen in the active site pocket. The structural folds of the models of GtPyNP
and TtPyNP are almost identical to the template structure GsPyNP, which can be seen in the
superposition of the secondary structure elements Figure 35.
4.1.2. Thermal characteristics
In order to assess the optimal temperature range at which the PyNPs can be used in potential bio-
synthetic applications, the optimal temperatures and thermal stabilities of the enzymes were
studied.
GtPyNP showed a temperature optimum of 60 °C, while the relative activity of TtPyNP increased with
the reaction temperature up to the highest temperature tested (99 °C, Figure 36A). An apparent
melting temperature of ≥ 102 °C and 103 °C was determined by circular dichroism and differential
scanning calorimetry, respectively (Figure 37). Hence, we assume that the temperature optimum of
TtPyNP is in the range of 95 °C – 103 °C.
The stability half life of GtPyNP was determined to be 1.6 h at 70 °C; while at 60 °C no significant loss
of activity could be seen within 16 h of incubation. The stability half life of TtPyNP exceeds 23 h at
80 °C (Figure 36B). At 90 °C TtPyNP was almost completely deactivated within 6 hours (data not
shown).
Compared to other reported enzymes with pyrimidine nucleoside phosphorylase activity, including
UPs and TPs, the thermal stability and the temperature optimum of TtPyNP is extremely high (Table
11). It seems therefore appealing to further investigate whether this highly thermostable biocatalyst
can be expediently used for enzymatic transglycosylations reactions aiming at the synthesis of
modified nucleosides. Based on the results obtained here, further characterization of GtPyNP and
TtPyNP will be performed at 60 °C and 80 °C, respectively. These temperatures seem adequate since
both enzymes show sufficiently high activity and at the same time stay stable for prolonged
incubation times.

Characterization of thermostable NPs 77
Figure 34: Multiple sequence alignment of PyNP from G. stearothermophilus (GsPyNP), G. thermoglucosidasius (GtPyNP), T. thermophilus (TtPyNP), and TP from E. coli (EcTP). Shading represents the degree of sequence identity. Residues of the active site pocket are highlighted. Figure taken from (Szeker et al. 2012).
Figure 35: Three-dimensional structure models. The superposition of GtPyNP (purple), TtPyNP (yellow), and GsPyNP (grey) 3D structures are shown. Structural models of GtPyNP and TtPyNP were built based on homology modelling using the structure of GsPyNP as template. Figure taken from (Szeker et al. 2012).

78 Characterization of thermostable NPs
Figure 36: Thermal characteristics. Relative activity of GtPyNP and TtPyNP over the reaction temperature (A) where the highest reaction rate determined was set to 100 % for each enzyme. Thermostability (B) was investigated by incubating protein samples for defined time intervals and subsequently determining the residual activity, where the activity of enzyme samples that were not thermo-treated was set to 100 %. Figure adapted from (Szeker et al. 2012).
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2
20 30 40 50 60 70 80 90 100 110
No
rmal
ized
elli
pti
city
Temperature [°C]
-40
-30
-20
-10
0
10
20
30
40
75 80 85 90 95 100 105 110 115 120
c p[k
cal K
-1m
ol-1
]
Temperature [°C]
A) B)
Figure 37: Thermal unfolding of TtPyNP. Thermal unfolding trace monitored by loss of the far-UV circular dichroism signal at 220 nm provided an apparent melting temperature of at least 102 °C (A). The apparent melting temperature determined by differential scanning calorimetry (B) was 103 °C. Figure adapted from (Szeker et al. 2012).
4.1.3. Kinetic parameters
In order to compare the thermostable PyNPs prepared in this study with PyNPs described by others,
parameters describing substrate specificity and catalytic efficiency were studied (Table 12).
Michaelis-Menten kinetics for the natural substrates uridine and thymidine were investigated by
determining the reaction rates at different substrate concentration. The amount of phosphate, that

Characterization of thermostable NPs 79
in fact represents the second substrate, was kept constant whereby the concentration (50 mM)
considerably exceeded the concentration of the nucleoside substrate. Enzyme reactions with GtPyNP
were performed at 60 °C and with TtPyNP at 80 °C. The previous section has shown that both
enzymes are stable over extended time periods at these temperatures.
Table 11. Thermal characteristics of reported PyNP, UP, TP. Table adapted from (Szeker et al. 2012).
Enzyme Organism Thermal stability (t1/2) Temp. optimum Reference
TP E. coli <10 min (55 °C) (Krenitsky and Tuttle
1982)
UP E. coli 9.9 h (60 °C) 40 °C (Visser et al. 2011)
PyNP G. thermoglucosidasius 1.6 h (70 °C) 60 °C This study
UP* E. coli 3.3 h (70 °C) 60 °C (Visser et al. 2011)
UP Enterobacter aerogenes 1 week (60 °C) 65 °C (Utagawa et al. 1985a)
PyNP G. stearothermophilus 25 min (70 °C) 70 °C (Hamamoto et al. 1996,
Hori et al. 1990)
UP Erwinia carotovora - 70 °C (Shirae and Yokozeki
1991)
PyNP T. thermophilus >23 h (80 °C) > 95 °C This study
* UP from E. coli was engineered for enhanced stability, for details see (Visser et al. 2011)
The Michaelis-Menten constant (Km) is defined as the substrate concentration at which the reaction
rate is half of the maximal velocity. It is a common parameter used to describe the affinity of an
enzyme towards a specific substrate. The estimated Km values of TtPyNP for the natural substrates
uridine and thymidine are slightly lower than the Km values determined by Hori and co-workers(Hori
et al. 1990) for GsPyNP. In contrast to TtPyNP, GtPyNP is characterized by extremely low substrate
affinities (high Km values) towards both natural substrates.
The catalytic efficiency of an enzyme can be described by the ratio of kcat/Km (Copeland 2000). These
ratios are 15 times (substrate uridine) and 25 times (substrate thymidine) higher for TtPyNP than for
GtPyNP. The kcat/Km ratio is also a measure to compare an enzyme’s specificity towards different
substrates (Copeland 2000). The results of the present study show that both enzymes are more
specific for uridine than for thymidine, but the difference in specificity is more pronounced for
GtPyNP: the kcat/Km ratio is 2fold higher for uridine than for thymidine. In contrast, the kcat/Km ratio of
TtPyNP is only 1.23 fold higher for uridine vs that of thymidine.
In applications, where high substrate concentrations are used (cs>> Km) the kcat value alone, also
referred to as turnover number, may be the most appropriate parameter describing the efficiency of
the biocatalyst. It describes on how many substrate molecules a single enzyme molecule is acting in
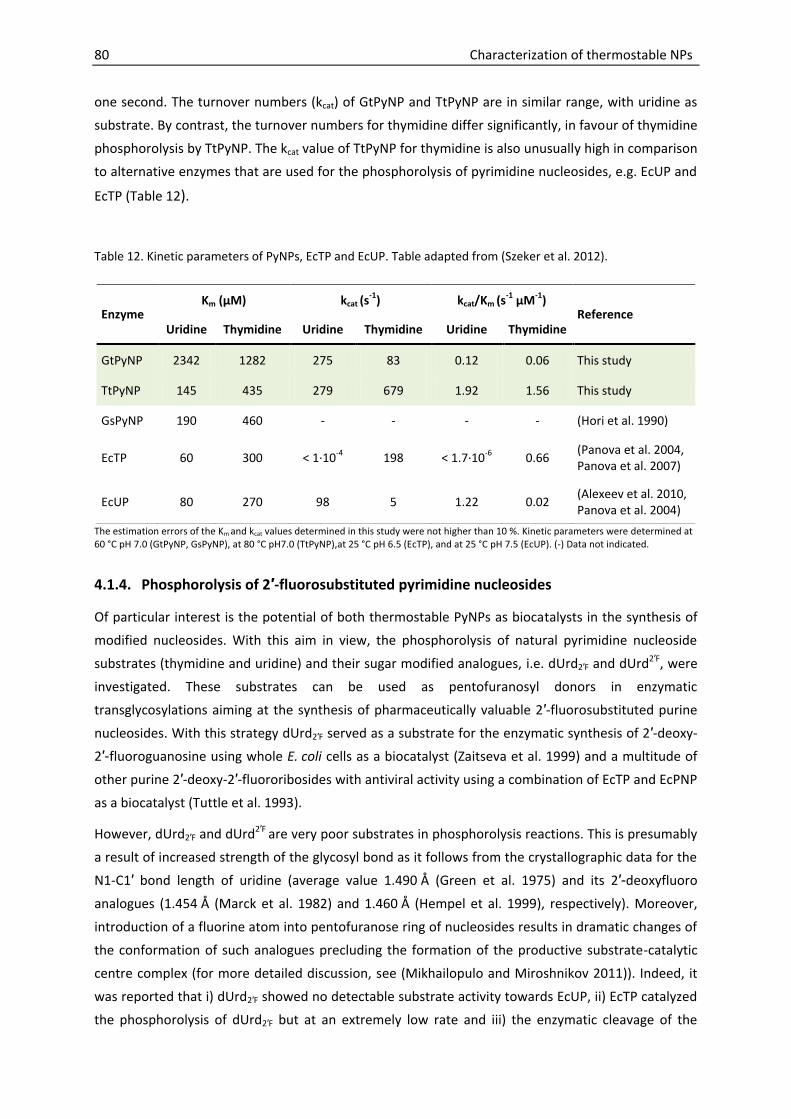
80 Characterization of thermostable NPs
one second. The turnover numbers (kcat) of GtPyNP and TtPyNP are in similar range, with uridine as
substrate. By contrast, the turnover numbers for thymidine differ significantly, in favour of thymidine
phosphorolysis by TtPyNP. The kcat value of TtPyNP for thymidine is also unusually high in comparison
to alternative enzymes that are used for the phosphorolysis of pyrimidine nucleosides, e.g. EcUP and
EcTP (Table 12).
Table 12. Kinetic parameters of PyNPs, EcTP and EcUP. Table adapted from (Szeker et al. 2012).
The estimation errors of the Km and kcat values determined in this study were not higher than 10 %. Kinetic parameters were determined at 60 °C pH 7.0 (GtPyNP, GsPyNP), at 80 °C pH7.0 (TtPyNP),at 25 °C pH 6.5 (EcTP), and at 25 °C pH 7.5 (EcUP). (-) Data not indicated.
4.1.4. Phosphorolysis of 2′-fluorosubstituted pyrimidine nucleosides
Of particular interest is the potential of both thermostable PyNPs as biocatalysts in the synthesis of
modified nucleosides. With this aim in view, the phosphorolysis of natural pyrimidine nucleoside
substrates (thymidine and uridine) and their sugar modified analogues, i.e. dUrd2′F and dUrd2′F, were
investigated. These substrates can be used as pentofuranosyl donors in enzymatic
transglycosylations aiming at the synthesis of pharmaceutically valuable 2′-fluorosubstituted purine
nucleosides. With this strategy dUrd2′F served as a substrate for the enzymatic synthesis of 2′-deoxy-
2′-fluoroguanosine using whole E. coli cells as a biocatalyst (Zaitseva et al. 1999) and a multitude of
other purine 2′-deoxy-2′-fluororibosides with antiviral activity using a combination of EcTP and EcPNP
as a biocatalyst (Tuttle et al. 1993).
However, dUrd2′F and dUrd2′F are very poor substrates in phosphorolysis reactions. This is presumably
a result of increased strength of the glycosyl bond as it follows from the crystallographic data for the
N1-C1′ bond length of uridine (average value 1.490 Å (Green et al. 1975) and its 2′-deoxyfluoro
analogues (1.454 Å (Marck et al. 1982) and 1.460 Å (Hempel et al. 1999), respectively). Moreover,
introduction of a fluorine atom into pentofuranose ring of nucleosides results in dramatic changes of
the conformation of such analogues precluding the formation of the productive substrate-catalytic
centre complex (for more detailed discussion, see (Mikhailopulo and Miroshnikov 2011)). Indeed, it
was reported that i) dUrd2′F showed no detectable substrate activity towards EcUP, ii) EcTP catalyzed
the phosphorolysis of dUrd2′F but at an extremely low rate and iii) the enzymatic cleavage of the
Enzyme Km (µM) kcat (s
-1) kcat/Km (s-1 µM-1)
Reference Uridine Thymidine Uridine Thymidine Uridine Thymidine
GtPyNP 2342 1282 275 83 0.12 0.06 This study
TtPyNP 145 435 279 679 1.92 1.56 This study
GsPyNP 190 460 - - - - (Hori et al. 1990)
EcTP 60 300 < 1·10-4 198 < 1.7·10-6 0.66 (Panova et al. 2004, Panova et al. 2007)
EcUP 80 270 98 5 1.22 0.02 (Alexeev et al. 2010, Panova et al. 2004)

Characterization of thermostable NPs 81
glycosidic bond of dUrd2′F equally afforded a high amount of enzyme and prolonged reaction time (6
days) (Tuttle and Krenitsky 1992, Tuttle et al. 1993).
In this study we have investigated the phosphorolysis of these challenging substrates by GtPyNP and
TtPyNP (Figure 38). Our results indicate that TtPyNP might be a good alternative to the use of E. coli
enzymes, but the use of GtPyNP is apparently not suitable for the applications discussed above: no
activity towards dUrd2′F and only poor activity towards dUrd2′F (0.44 % substrate conversion) was
detected with GtPyNP as biocatalyst after 30 min reaction time.
By contrast, the TtPyNP catalyzed reaction under the same conditions resulted in the phosphorolytic
cleavage of 0.65 % of dUrd2′F and 7.0 % of dUrd2′F. Since the optimal reaction temperature of TtPyNP
is significantly higher than 60 °C (section 4.1.2), we repeated the same reaction also at 80 °C. Now,
the TtPyNP catalyzed reaction resulted in 1.4 % phosphorolyzed dUrd2′Fand 15.6 % of dUrd2′F.
However, under these conditions the formation of two new peaks was observed by HPLC analysis of
the reaction mixture that contained dUrd2′F. The retention times coincide with those of authentic
samples of O2,2′-anhydro-1-(-D-arabinofuranosyl)uracil (anhydro-Urd) (2.3 min) and 1-(-D-
arabinofuranosyl)uracil (ara-U) (3.8 min). Hence, the formation of anhydro-Urd resulting from HF
release from the dUrd2′F molecule and the subsequent hydrolysis of anhydro-Urd resulting in ara-U
appear to be a reasonable explanation (Scheme 1). Phosphorolysis of both 2′-fluorosubstituted
pyrimidine nucleosides catalyzed by TtPyNP was also monitored over prolonged reaction times
(Figure 39A). The results show that the conversion of dUrd2′F at 80 °C could be increased to 46 % after
17 h; side-product formation was not observed. The final amount of phosphorolyzed dUrd2′F after
17 h at 80 °C (65 %) was in similar range as the amount obtained at 60 °C (60 %). By contrast, side
product formation, as discussed above, at 80 °C was significantly higher than at 60 °C (Figure 39B):
After 17 h 8.3 % of dUrd2′F reacted to anhydro-Urd at 80 °C, while the same value is decreased to
1.2 % at 60 °C.
O
HO
HO
F
HN
N
O
O
O
HO
HO
N
N
O
OO
NH
N
O
O
HO
HO
OH
-HF
Hydrolysis
O2,2'-Anhydro-(-D-ara-
binofuranosyl)uracil
1-(-D-arabinofuranosyl)-
uracil (ara-U)
dUrd2'F
Scheme 1
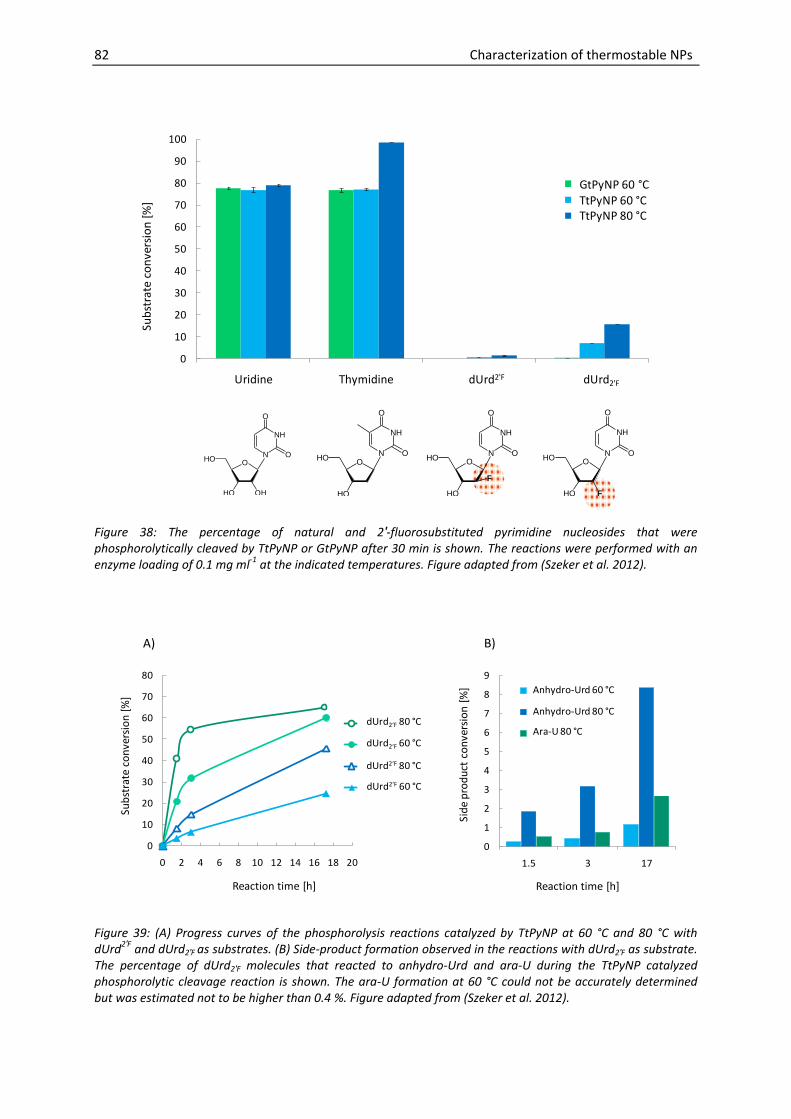
82 Characterization of thermostable NPs
0
10
20
30
40
50
60
70
80
90
100
Uridine Thymidine 2’Fana-U Urd2'F
Sub
stra
te c
on
vers
ion
[%
]
GtPyNP 60 C
TtPyNP 60 C
TtPyNP 80 C
GtPyNP 60 °CTtPyNP 60 °CTtPyNP 80 °C
O
HO OH
HO
NH
N
O
OO
HO
HO
NH
N
O
OO
HO
HO
NH
N
O
O
F
O
HO F
HO
NH
N
O
O
dUrd2'F dUrd2'F
Figure 38: The percentage of natural and 2′-fluorosubstituted pyrimidine nucleosides that were phosphorolytically cleaved by TtPyNP or GtPyNP after 30 min is shown. The reactions were performed with an enzyme loading of 0.1 mg ml-1 at the indicated temperatures. Figure adapted from (Szeker et al. 2012).
0
10
20
30
40
50
60
70
80
0 2 4 6 8 10 12 14 16 18 20
Sub
stra
te c
on
vers
ion
[%
]
Reaction time [h]
FU 80 °C
FU 60 °C
FanaU 80 °C
FanaU 60 °C
dUrd2‘F 80 °C
dUrd2‘F 60 °C
dUrd2‘F 80 °C
dUrd2‘F 60 °C
0
1
2
3
4
5
6
7
8
9
1.5 3 17
Sid
e p
rod
uct
co
nve
rsio
n [
%]
Reaction time [h]
Anhydro-Urd 60 °C
Anhydro-Urd 80 °C
Ara-U 80 °C
A) B)
Anhydro-Urd 60 °C
Anhydro-Urd 80 °C
Ara-U 80 °C
Figure 39: (A) Progress curves of the phosphorolysis reactions catalyzed by TtPyNP at 60 °C and 80 °C with dUrd2′F and dUrd2′F as substrates. (B) Side-product formation observed in the reactions with dUrd2′F as substrate. The percentage of dUrd2′F molecules that reacted to anhydro-Urd and ara-U during the TtPyNP catalyzed phosphorolytic cleavage reaction is shown. The ara-U formation at 60 °C could not be accurately determined but was estimated not to be higher than 0.4 %. Figure adapted from (Szeker et al. 2012).

Characterization of thermostable NPs 83
4.1.5. PNP activity of PyNPs
The previous section has shown that both PyNPs efficiently catalyze the phosphorolysis of the natural
pyrimidine nucleoside substrates uridine and thymidine. Considerably weaker activities were also
found for the 2′-fluorosubstituted uridine analogues.
In the further course of this study it was also recognized that not only pyrimidine nucleoside
analogues, but, unexpectedly also purine nucleosides are phosphorolyzed by both PyNPs. Initially we
considered it equivocal, whether this PNP activity indeed originates from the PyNPs themselves or
whether it could be the result of E. coli PNP contamination of the purified enzyme preparations. In
order to investigate this issue, the temperature optima of the apparent PNP activities of both PyNPs
were determined with inosine as substrates (Figure 40). The dependence of the reaction rate over
time is in accordance with the temperature dependence observed with the natural substrate uridine
(Figure 36A). These results clearly indicate that the PNP activity is inherent to the PyNPs and not the
result of a possible contamination with the PNP of the host (EcPNP).
0
20
40
60
80
100
20 30 40 50 60 70 80 90 100
Rel
ativ
e ac
tivi
ty [%
]
Temperature [°C]
GtPyNP
TtPyNP
Figure 40: Temperature dependence of the PNP activity of GtPyNP and TtPyNP. The highest reaction rate observed for each enzyme was set to 100 %. The purine nucleoside substrate was inosine.
To further characterize the PNP “side” activity of the PyNPs, estimated specific activities for different
purine and pyrimidine nucleoside substrates were determined (Table 13). The results show that
GtPyNP seems to have a preference for adenosine over inosine, while for TtPyNP no clear preference
can be seen. Even though the reaction rates for the purine nucleosides are extremely low in
comparison to the reaction rates observed with natural enzymes, they are significantly higher than
the reaction rates observed with the 2′-fluorosubstituted pyrimidine nucleosides. A simple conclusion
may be that the enzymes readily accept modifications on the heterocyclic base, but have stricter
requirements in respect to the sugar residue. These results prompted us to also investigate enzyme
activity towards cytidine, which only differs from uridine by one oxo-group replaced by an amino-

84 Characterization of thermostable NPs
group on the pyrimidine base. However, no phosphorolytic activity of GtPyNP and only poor activity
of TtPyNP were determined for cytidine as substrate. It appears quite remarkable that a single
substitution on the pyrimidine ring has such a pronounced impact on activity, while the substitution
of uracil by the purine bases adenine or hypoxanthine, results in still reasonable reaction rates.
Table 13. Estimated specific activities of GtPyNP and TtPyNP acting on pyrimidine and purine nucleosides.
Substrate Estimated specific activity [U mg-1 ]
GtPyNP (60 °C) TtPyNP (80 °C)
Uridine 51.51 148.80
Thymidine 22.44 286.00
Adenosine 4.57 × 10-1 8.09 × 10-1
Inosine 3.08 × 10-1 9.00 × 10-1
dUrd2′F 1.58× 10-3 2.62 × 10-2
dUrd2′F UDL 2.42 × 10-3
Cytidine UDL 8.7 × 10-4
Experiments performed with 1 mM substrate, at 60 °C (GtPyNP) or 80 °C (TtPyNP). UDL = under detection level
4.1.6. Characterization of thermostable PyNPs - summary and conclusions
PyNPs isolated from thermophilic microorganisms are promising biocatalysts for the efficient
synthesis of modified nucleosides (Taran et al. 2009). Up to now, the biocatalytic characterization of
PyNPs from thermophilic microbes was restricted to PyNPs from G. stearothermophilus strains
(Hamamoto et al. 1996, Hori et al. 1990, Saunders et al. 1969). We have studied here the
biocatalytical properties of two additional thermostable PyNP, originating from
G. thermoglucosidasius and T. thermophilus.
Our results indicate that both enzymes show excellent biocatalytical properties for applications with
natural pyrimidine nucleosides as substrates (thymidine, uridine) and a reaction temperature of
60 °C. In addition, the unusually high thermal stability of TtPyNP makes this biocatalyst also suitable
for reactions requiring an even higher reaction temperature of 80 °C.
We have further tested both thermostable PyNPs towards their ability to phosphorolyze
2′-fluorosubstituted pyrimidine nucleosides that have been shown to be very poor substrates in
phosphorolysis reactions employing EcUP or EcTP as biocatalyst (Tuttle and Krenitsky 1992, Tuttle et
al. 1993). Our results reveal striking differences of the substrate specificities of GtPyNP and TtPyNP,
in favour of the latter. The yield of phosphorolyzed 2′-fluorosubstituted pyrimidine nucleosides could
be enhanced by extending the reaction time and increasing the reaction temperature from 60 °C to
80 °C. However, the higher reaction temperature was not suitable for dUrd2′F since this substrate was

Characterization of thermostable NPs 85
not stable. By contrast, the higher reaction rate was a useful measure to increase the rate of the
phosphorolysis reaction of the more stable epimeric counterpart dUrd2′F.These findings make TtPyNP
a candidate as powerful biocatalyst in the transglycosylation reactions aiming at the synthesis of
2′-fluoro substituted purine nucleosides.
It was further discovered that both PyNPs studied here are able to accept also the purine nucleosides
inosine and adenosine as substrate. To our best knowledge PNP activity was so far only observed for
enzymes with the NP-I fold. PyNP however, belongs to the structurally unrelated NP-II family that is
likely to have evolved independently (Pugmire and Ealick 2002). The fact that purine nucleosides are
weakly accepted as substrates of PyNPs may rise the question whether PyNPs have diverged from
ancient enzymes with broader substrate specificity, also involved in the metabolism of purine
nucleosides.
The PNP “side activity” of the PyNPs could be of interest for synthetic reactions, because it could
allow to perform transglycosylation from a pyrimidine nucleoside donor to a purine base acceptor
with a single enzyme. In a similar way, NdRTs can be used for transglycosylations (see section 1.2.2).
However, these enzymes are specific for deoxyribosides and are therefore not suitable for the
synthesis of ribosides.
4.2. Thermostable enzymes with PNP activity
PNPs are thoroughly investigated enzymes. In fact, BRENDA (BRaunschweig ENzyme Database,
http://www.brenda-enzymes.org (Scheer et al. 2011)) currently lists PNPs from 73 different
organisms. Particularly the recognition of PNP as therapeutic target (for more details see section
1.3.1) has triggered a multitude of scientific studies devoted to the elucidation of structural
properties and catalytic mechanism (reviewed in (Bzowska et al. 2000, Silva et al. 2007)). Also, the
PNP-catalyzed reversible phosphorolysis of purine nucleosides has been recognized as an important
reaction that can be used for the synthesis of modified nucleosides (see section 1.2.3 for more
details). Therefore a number of PNPs have been investigated and subsequently exploited as
biocatalysts in transglycosylation reactions of nucleosides. Examples include the PNPs from E. coli
(Krenitsky et al. 1981), G. stearothermophilus strains (Hamamoto et al. 1996, Hamamoto et al. 1997a,
Hori et al. 1989b, Hori et al. 1991, Taran et al. 2009), Bacillus halodurans (Gordon et al. 2011, Visser
et al. 2010), and from A. hydrophila (Ubiali et al. 2012).
The use of NPs as biocatalysts in enzymatic transglycosylation reactions is also in focus of this work.
While in the last section the characterization of biocatalytical properties of thermostable PyNPs has
been presented, this section is devoted to thermostable enzymes with PNP activity. Specifically two
novel PNPs derived from the thermophilic microorganisms D. geothermalis and
G. thermoglucosidasius were investigated. In both microorganisms each two genes are annotated as
PNPs. As it will be discussed in more detail in the following section, high-molecular-mass-type PNPs
(hexameric form) were chosen to be studied here. In contrast to the low-molecular-mass (trimeric)
species, these PNPs are known not to be restricted to the phosphorolysis of 6-oxopurine nucleosides
and are therefore generally considered to have broader substrate specificity (see section 1.3.3).

86 Characterization of thermostable NPs
Moreover we investigated MTAP from the hyperthermophilic archaeon A. pernix. MTAPs from
hyperthermophilic sources have been successfully recombinantly expressed and structural features
have been thoroughly studied (Appleby et al. 2001, Cacciapuoti et al. 2005, Cacciapuoti et al. 2011).
Investigations of substrate specificities has shown that some MTAPs are able to accept both 6-oxo
and 6-aminopurine nucleosides as substrates (Cacciapuoti et al. 1994, Cacciapuoti et al. 2003), which
makes them interesting candidates as versatile catalysts for NP synthesis in our opinion. Indeed, a
recent study reports on the application of UP and PNP from A. pernix for the production of
5′-methyluridine (Zhu et al. 2012), whereby the coding region of the primers used for the isolation of
the thermostable PNP coincides with the coding sequence for ApMTAP. Hence the enzyme assigned
by the authors as PNP is most likely identical to ApMTAP here under investigation. However, in this
report (Zhu et al. 2012) no data concerning the biocatalytical properties of ApMTAP are disclosed. In
order to gain further knowledge concerning the potential application fields of ApMTAP as biocatalyst,
we therefore investigated the biocatalytical properties and substrate specificities toward natural and
artificial nucleosides.
4.2.1. Sequence analysis and homology modelling
The PNPs recombinantly expressed and characterized here are: PNP from D. geothermalis (GenBank
accession numbers ABF45792, YP_604961.1), PNP from G. thermoglucosidasius 11955 (sequence in
accordance with GenBank accession numbersEFG53380, AEH47728.1), and MTAP from A. pernix
(GenBank accession number NP_147653).
Phylogenetic analysis
A phylogenetic analysis with PNPs and MTAPs that are already reported in literature (Figure 41)
reveals that the PNPs from D. geothermalis and G. thermoglucosidasius studied here, show close
evolutionary relationships to other PNPs displaying the characteristics of hexameric, high molecular
mass PNPs (see section 1.3.3). These bacterial type PNPs are generally known to accept both,
6-oxopurine nucleosides (inosine, guanosine) and 6-aminopurine nucleosides (e.g. adenosine) as
substrate. Representatives are the 234 amino acid long PNP from G. stearothermophilus (Hamamoto
et al. 1997a), the 239 amino acid long PNP from E. coli (Jensen and Nygaard 1975) and the 238 amino
acid long PNP from A. hydrophila (Ubiali et al. 2012). Also the 235 amino acid long PNP from Bacillus
anthracis belongs to this group. This protein is identical to B. cereus adenosine phosphorylase that
was reported to prefer adenosine over 6-oxopurine nucleosides as substrate (Dessanti et al. 2012,
Sgarrella et al. 2007). The same applies to the 233 amino acid long PNP from B. subtilis (Jensen 1978),
although the preference for adenosine seems not so clear from the data of a more recent study (Xie
et al. 2011).
The phylogenetic analysis also illustrates that the second PNP of G. thermoglucosidasius (274 amino
acids) that was not examined in this study belongs to a group of PNPs found in Geobacilli and Bacilli
species, representing the eukaryotic (low molecular mass) type PNPs, implying specificity towards
6-oxopurine nucleosides, whereas 6-aminopurine nucleosides are not accepted as substrates.
Representatives of this group of PNPs are the 274 amino acid long PNP from G. stearothermophilus

Characterization of thermostable NPs 87
(Hamamoto et al. 1997b), the 272 amino acid long PNP from Bacillus halodurans (Visser et al. 2010),
and the 271 amino acid long PNP from B. subtilis(Jensen 1978, Xie et al. 2011). The second PNP from
D. geothermalis (238 amino acids) shows high sequence identity (61 % sequence identity) with the
235 amino acid long PNP from T. thermophilus which is in fact a hexameric protein but shows
substrate specificity as typical for trimeric (eukaryotic type) PNPs (Tahirov et al. 2004).
The MTAP from A. pernix cloned here (244 amino acids) appears to have an evolutionary relationship
to MTAPI from S. solfataricus (236 amino acids). The latter, has been shown to accept MTA and
adenosine, as well as 6-oxopurine nucleosides. By contrast, the MTAPII from S. solfataricus (270
amino acids), is specific for MTA and adenosine (Cacciapuoti et al. 2005) and appears closely related
to the second MTAP from A. pernix (275 amino acids) that is therefore catalytically less interesting
for this study and not investigated.
Figure 41: Phylogentic analysis of some PNPs and MTAPs described in literature. Enzymes are indicated by: Two-letter code for the source microorganism, enzyme name (PNP, MTAP), number of amino acids and the GenBank accession number. In the case of TtPNP the PDB code is given instead. Species abbreviations used: Gt = G. thermoglucosidasius, Gs = G. stearothermophilus, Bh = Bacillus halodurans, Ba = Bacillus anthracis, Bs = B. subtilis, Hs= Homo sapiens, Ec = E. coli, Ap = A. pernix, Ss = S. solfataricus, Pf = P. furiosus, Ah = A. hydrophila, Tt = T. thermophilus.
Homology modelling
The amino acid sequences of the PNPs investigated here (DgPNP, GtPNP) were blasted against the
amino acid sequences belonging to PNPs with resolved crystal structures (PDB database). Both
enzymes aligned best with the PNP from Bacillus anthracis (assigned here as BaPNP, PDB ID: 1XE3)
with 61 % (DgPNP) and 77 % (GtPNP) sequence identity, respectively. The PNP from Bacillus anthracis
(causative microorganism for anthrax) is identical to the B. cereus adenosine phosphorylase, for

88 Characterization of thermostable NPs
which the crystal structure was equally resolved (PDB ID: 2AC7, 3UAW) and investigated with respect
to the substrate specificity of the enzyme (Dessanti et al. 2012, Sgarrella et al. 2007). The crystal
structure of BaPNP was used as template to model DgPNP and GtPNP. The structural fold of the
models of DgPNP and GtPNP is almost identical to the template structure. As an example the
predicted structure of a monomer of GtPNP is shown, superimposed on the template BaPNP
hexamer (Figure 42A).
ApMTAP aligned best to the MTAP of S. solfataricus (PDB ID: 1JDS) with 45 % sequence identity and
an ApMTAP 3-dimensional model was built with SsMTAP as template. The S. solfataricus enzyme was
shown to be a hexamer with a broad substrate spectrum (Appleby et al. 2001, Cacciapuoti et al.
1994). Three intersubunit disulfide bonds link the dimers to each other to form a hexamer. In
SsMTAP the cysteine residues on position 125 are involved in this disulfide linkage. Since both in the
amino acid sequence alignment and the three dimensional superposition of SsMTAP and ApMTAP
Cys125 of SsMTAP corresponds to Cys129 of ApMTAP, it appears likely that in ApMTAP this cysteine
residue (on position 129) is equally involved in a disulfide linkage. This finding is in contradiction to
the predicted oxidation state of the cysteine residues summarized in Table 8, where Cys112 instead
of Cys129 was predicted to be in the bonding state. The crystal structure of SsMTAP together with
the superimposed monomeric model of ApMTAP is shown in (Figure 42B). However, a closer
investigation on the oligomerization state is necessary to determine the validity of this model, since
SDS-PAGE analysis of ApMTAP samples not treated with DTT may give evidence for a tetrameric
configuration (Figure 24).
GtPNP ApMTAP
Figure 42: GtPNP model based on BaPNP; ApMTAP model (with disulfide bridges) based on SsMTAP. The overall hexameric template structures as well as the superimposed monomeric models are shown. In the MTAP structure the disulfide bridges connecting each two dimers are highlighted (red).
4.2.2. Thermal characteristics
In order to assess the optimal temperature range at which the enzymes with PNP activity studied
here can be used in biocatalytic reactions, the optimal temperatures and thermal stabilities were
studied. The results are summarized in Table 14.

Characterization of thermostable NPs 89
Analysis of the reaction rates in dependence of the reaction temperature revealed that DgPNP shows
optimal activity at around 55 °C (Figure 43) which is slightly higher than the optimal growth
temperature of the source microorganism D. geothermalis (Ferreira et al. 1997). At 60 °C DgPNP
rapidly loses activity (stability half life approx. 1.6 h), while at 55 °C DgPNP is relatively stable over
prolonged incubation times (Figure 44). In comparison to the PNP from E. coli, DgPNP appears thus to
have a slightly lower temperature optimum, but is superior in regard to a higher stability at elevated
temperatures (Table 14).
Table 14. Thermal characteristics of enzymes with PNP activity from thermophilic microorganisms and E. coli.
Enzyme Organism Thermal stability (t1/2) Temp. optimum Reference
PNP* D. geothermalis > 8 h (55 °C)
1.7 h (60 °C) 55 °C This study
PNP* G. thermoglucosidasius > 8 h (70 °C)
6.3 h (75 °C) 70 °C This study
MTAP A. pernix > 27 h (90 °C) > 90 °C This study
PNP Bacillus halodurans 20.8 h (60 °C) 70 °C (Visser et al. 2010)
PNP I G. stearothermophilus - 70 °C (Hamamoto et al. 1996)
PNP II* G. stearothermophilus > 24 h ( 70 °C) 70 °C (Hamamoto et al. 1997a,
Taran et al. 2009)
PNP P. furiosus > 4 h (100 °C) 120 °C (Cacciapuoti et al. 2007)
MTAP P. furiosus > 5 h (100 °C)
43 min (130 °C) 125 °C (Cacciapuoti et al. 2003)
MTAP S. solfataricus > 2 h (100 °C)
15 min (130 °C) 120 °C (Cacciapuoti et al. 1994)
MTAPII S. solfataricus > 5 min (120 °C) 120 °C (Cacciapuoti et al. 2005)
PNP* E. coli > 30 min (50 °C)
< 30 min (55 °C) 60 °C (Li et al. 2008)
* bacterial, hexameric, high molecular mass type PNP
GtPNP showed optimal activity at 70 °C (Figure 43), which is about 10 °C higher than the temperature
optimum of GtPyNP (Table 11). Moreover, GtPNP is significantly more stable at higher temperature
in comparison to GtPyNP. At 70 °C no significant loss of activity can be seen within 8 h incubation
time (Figure 44), while stability half life of GtPyNP at 70 °C was only 1.6 h (Table 11). Similarly, the PNP
of E. coli was shown to be more thermoactive than E. coli thymidine phosphorylase, which can be
seen as the equivalent of PyNP in E. coli (Krenitsky et al. 1981). Likewise the PNPII was reported to

90 Characterization of thermostable NPs
be more thermostable than the PyNP in G. stearothermophilus (Taran et al. 2009). The PNP from
G. thermoglucosidasius here under investigation displays similar thermal properties as the PNPII from
G. stearothermophilus (Table 14). In fact, best to our knowledge, both enzymes represent the most
thermostable reported bacterial type PNPs that share a broader substrate specificity by accepting
both 6-oxopurine and 6-aminopurine nucleosides.
The reaction rates of ApMTAP increased with the reaction temperature up to the highest
temperature tested (99 °C, Figure 43). Furthermore, the enzyme appears to be extremely
thermostable with a stability half life exceeding 27 h at 90 °C (Figure 44). The high degree of
thermostability is conceivable, having in mind that the source microorganism is actually the
hyperthermophilic archaeon A. pernix, that shows optimal growth between 90 °C and 95 °C (Sako et
al. 1996). As the following section will show, ApMTAP together with the MTAPs of P. furiosus
(Cacciapuoti et al. 2007) and S. solfataricus (Cacciapuoti et al. 2005) thus belongs to the most
thermostable enzymes described to phosphorolyze adenosine as well as 6-oxopurine nucleosides
(Table 14). Both other enzymes have, however, not been investigated towards potential applications.
0
20
40
60
80
100
20 30 40 50 60 70 80 90 100
Rel
ativ
e ac
tivi
ty [%
]
Temperature [°C]
DgPNP
GtPNP
ApMTAP
Figure 43: Temperature optima of reaction rates. Relative activity of DgPNP, GtPNP,and ApMTAP over the reaction temperature assessed by the phosphorolysis of inosine. The highest reaction rate determined was set to 100 % for each enzyme.
As a conclusion for their application, it is not recommended to use DgPNP and GtPNP at
temperatures exceeding 55 °C and 70 °C, respectively, because both enzymes are rapidly inactivated
above these temperatures. By contrast, ApMTAP is highly thermostable and can be used even at
90 °C. The following chapter will show, that the reaction temperature of biocatalytic reactions
involving ApMTAP as catalyst will therefore not be restricted by the stability of the enzyme but
rather by the stability of substrates, intermediates, and products, or by the stability of the second
enzyme needed for the phosphorolysis of pyrimidine nucleosides.

Characterization of thermostable NPs 91
0
20
40
60
80
100
120
0 2 4 6 8
Res
idu
al a
ctiv
ity
[%]
Incubation time [h]
GtPNP 70 C
GtPNP 75 C
0
20
40
60
80
100
120
0 2 4 6 8
Res
idu
al a
ctiv
ity
[%]
Incubation time [h]
DgPNP 55 C
DgPNP 60 C
0
20
40
60
80
100
120
0 5 10 15 20 25 30
Res
idu
al a
ctiv
ity
[%]
Incubation time [h]
ApMTAP 90 C
°
°
°
°
°
Figure 44: Thermal stabilities of purine nucleoside phosphorolyzing enzymes. Protein samples were incubated for defined time intervals and subsequently the residual activity was determined. The activity of enzyme preparations that were not thermo-treated was set to 100 %.
4.2.3. Substrate specificities
The first aim was to determine which nucleosides are recognized as substrates by the thermostable
enzymes with PNP activity here under investigation. Therefore a number of natural and artificial
purine nucleoside analogues were screened towards their substrate activity. Enzyme concentrations
and sampling times were adjusted to ensure that the phosphorolysis reaction proceeded in linear
dependence of time and amount of NP added. The resulting apparent specific activities are
summarized in Table 15.
Phosphorolysis of natural purine nucleosides
All three enzymes recognize both inosine and adenosine as substrate. To compare the catalytical
properties with respect to recognition of adenine and hypoxanthine nucleosides, the relative
reaction rates were calculated from the data shown in Table 15 and listed together with the
corresponding rates of other enzymes reported in literature in Table 17. As pointed out before, PNPs
can be grouped in two categories: PNPs belonging to the low-molecular mass eukaryotic type
enzymes are specific for 6-oxopurine nucleosides (e.g. xanthosine phosphorylase from E. coli, PNPI
from G. stearothermophilus, PNP from B. subtilis and Mycobacterium smegmatis). PNPs categorized
as high-molecular mass, bacterial-type enzymes are generally characterized by a broader substrate
specificity and accept both 6-oxopurine nucleosides and 6-aminopurine nucleosides (e.g. the PNP of
E. coli encoded by the deoD gene, PNP encoded by the deoD gene of A. hydrophila, Ado-PNP from
Mycobacterium smegmatis, PNPII from G. stearothermophilus). Some members of the high-
molecular mass PNPs appear to have, however, a clear preference for adenosine over inosine as
substrate and are therefore also referred to as adenosine phosphorylases (examples listed in Table
17 are AdoP from B. cereus and from B. subtilis). The phylogenetic analysis illustrated in section 4.2.1
has shown that DgPNP and GtPNP are closely related to both the “normal” high-molecular mass PNPs
(e.g. PNPII from G. stearothermophilus, encoded by punB, E. coli PNP encoded by deoD, A. hydrophila
PNP encoded by deoD) as well as to the adenosine-specific nucleoside phosphorylases from Bacillus

92 Characterization of thermostable NPs
anthracis (enzyme is identical with AdoP from B. cereus) and B. subtilis (Figure 41). However, the
data summarized in Table 17 clearly indicate that from the catalytical properties both target PNPs
studied here behave similar to normal high-molecular mass PNPs that recognize both inosine and
adenosine efficiently as substrates.
Among the enzymes considered for the phylogenetic study, ApMTAP showed the closest
evolutionary relationship to the MTAPI from S. solfataricus (Figure 41). The similarity between both
amino acid sequences is also reflected by their catalytic properties. Both enzymes recognize
adenosine and inosine as substrate with a preference for the latter (Table 17).
Comparing the absolute activities for inosine determined in the present study with Vmax determined
by others for EcPNP (deoD) at 55 °C (211 U mg-1 (Li et al. 2008)) shows that the thermostable
enzymes investigated here appear to be less active. However, the results presented at this point are
possibly gained under nonsaturating substrate concentrations (1 mM) and therefore not necessarily
represent Vmax values. Moreover, it should be noted, that the PNPs investigated here are superior
with respect to thermostability. The data in Table 14 show that the stability half life of E. coli PNP at
55 °C is actually less than 30 min, while all enzymes recombinantly expressed in the present study are
stable at this temperature over prolonged incubation times. In comparison to AhPNP specific
activities (determined with 5 mM substrate concentration) (Ubiali et al. 2012), the activities
determined here are partially significantly higher, even though the substrate concentration was only
1 mM (Table 15). For PNPII of G. stearothermophilus a specific activity of 683 U mg-1 was reported
with adenosine as substrate at 70 °C (Hamamoto et al. 1997a). This value clearly exceeds the specific
activity determined here for GtPNP and the other enzymes. However, as stated above, the Vmaxvalues
(determined with saturating substrate concentrations) are possibly higher than the enzyme activities
obtained with 1 mM nucleoside concentration and listed for now in Table 15.
Table 15. Apparent specific activities obtained with a nucleoside substrate concentration of 1 mM.
Substrate Apparent specific activity [U mg-1]
DgPNP (55 °C) GtPNP (70 °C) ApMTAP (80 °C) AhPNP1 GsPNPII (70 °C)
2
Adenosine 151.88 379.16 41.27 35 683
Inosine 43.69 192.49 45.42 75 nd
dAdo2′F 0.023 0.009 0.013 nd nd
dAdo2′F 0.029 0.021 0.015 nd nd
Cytidine 0.0111 0.0147 0.0368 nd nd
1 = data from (Ubiali et al. 2012), were determined with 5 mM substrate concentration. 2 = data from (Hamamoto et al. 1997a). nd = not determined/ data not available; (*) 5 mM substrate concentration
In summary, the data presented here appear to be useful to categorize the target enzymes according
to their substrate specificity. Obviously all the enzymes show substrate specificities of the high
molecular mass (bacterial type) PNPs. The broader substrate specificity of these group of PNPs in

Characterization of thermostable NPs 93
comparison to the low molecular mass PNP is an important factor for possible future applications.
For example a halogen substitution on the C-2 position of the purine ring is tolerated only by high
molecular mass PNPs (Bzowska et al. 2000). On the other hand, kinetic studies will be needed to
investigate whether the differences in reaction rates observed here are due to substrate binding or
catalytic efficiency of the biocatalysts. Moreover, the maximal velocities and turnover number that
can be extracted from such a study will be more suitable determinants to compare the efficiency of
the thermostable PNPs among each other and with other reported enzymes.
Phosphorolysis of 2′-fluorosubstituted purine nucleoside analogues
Of particular interest for the present study is the synthesis of 2′-fluorosubstituted purine ribosides
from the respective pyrimidine nucleosides serving as pentofuranosyl donor and a purine base
serving as pentofuranosyl acceptor. In this transglycosylation reaction two types of NPs are
employed. The first enzyme, a PyNP phosphorolytically cleaves the donor nucleosides, while the
second enzyme with PNP activity is intended to catalyze the glycosyl bond formation between
pentofuranosyl-1-phosphate and the purine base (illustrated previously in Figure 6).The focus of this
section is to find out, which of the herein reported enzymes with PNP activity might be particularly
suitable for this application. For this purpose we have investigated the phosphorolysis of the two 2′-
fluorosubstituted purine nucleosides, 2′-deoxy-2′-fluoroadenosine (dAdo2′F) and 9-(2-deoxy-2-fluoro-
-D-arabinofuranosyl)adenine (dAdo2′F). The idea is that the enzyme that catalyzes most efficiently
the phosphorolysis of these substrates will also be the most suitable candidate for the reverse
reaction, required for the synthesis of the 2′-fluorosubstituted purine nucleosides.
The specific activities obtained with each 1 mM substrate for natural purine nucleosides and
2′-fluorosubstituted purine nucleosides are summarized in Table 15. The data show that the highest
absolute phosphorolysis rates for both compounds were obtained with DgPNP. By contrast, GtPNP
that actually appears to be the most efficient catalyst for natural purine nucleosides (inosine,
adenosine) shows less activity than DgPNP for dAdo2′F and is the weakest catalyst for dAdo2′F.
Table 16. Relative reaction rates (% of adenosine phosphorolysis).
dAdo2′F dAdo2′F
DgPNP (55 °C) 0.015 0.019
GtPNP (70 °C) 0.002 0.001
ApMTAP (80 °C) 0.032 0.036
1 mM, 50 mM KP, pH 7.0
This narrow specificity of GtPNP is also reflected by the extremely low relative reaction rates towards
the fluoro-substituted purine nucleosides shown in Table 16. Here the data of Table 15 describing the
phosphorolysis of fluoro-substituted purine nucleosides are expressed as relative reaction rates
whereby the reaction rate observed for adenosine phosphorolysis is set to 100 %. From these

94 Characterization of thermostable NPs
relative reaction rates one could also speculate that ApMTAP shows the broadest specificity. With
this enzyme dAdo2′F and dAdo2′F are phosphorolyzed with 0.032 % and 0.036 %of the reaction rate
observed with adenosine. By comparison, the reaction rate reported for EcPNP with 2′-deoxy-2′-
fluoroguanosine as substrate (1 mM) was 0.0031 % of the reaction rate with deoxyguanosine as
substrate (Tuttle et al. 1993).
Table 17. Relative reaction rates of enzymes with PNP activity for purine nucleosides.
Enzyme Source MO Relative reaction rates [%] Reference
Ino dIno Ado dAdo
PNP D. geothermalis 29 nd 100 nd This study
PNP G. thermoglucosidasius 51 nd 100 nd This study
AdoP/PNP B. cereus 2 2 100 46.7 (Sgarrella et al. 2007)1
AdoP/PNP B. subtilis 1 < 1 24 100 (Jensen 1978)2
PNPII (punB) G. stearothermophilus 46 40 100 90 (Hamamoto et al. 1997a)3
Ado-PNP Mycobacterium smegmatis 43 nd 100 58 (Buckoreelall et al. 2011)4
PNP (DeoD) E. coli 46 100 61 61 (Jensen and Nygaard 1975)
PNP (DeoD) A. hydrophila 55 100 26 20 (Ubiali et al. 2012)5
XanoP (XapA) E. coli 100 82 < 0.02 < 0.02 (Koszalka et al. 1988)6
PNPI (punA) G. stearothermophilus 100 100 0 nd (Hamamoto et al. 1996)
PNP B. subtilis 83 63 < 1 < 1 (Jensen 1978)6
PNP Mycobacterium smegmatis 100 nd - - (Buckoreelall et al. 2011)4
MTAP A. pernix 100 nd 91 nd This study
MTAPI S. solfataricus 100 nd 33 nd (Cacciapuoti et al. 2005)7
MTAP P. furiosus 36 nd 92 nd (Cacciapuoti et al. 2007)7
MTAPII S. solfataricus - nd 100 nd (Cacciapuoti et al. 2005)7
The reaction rates determined in this study where obtained with 1 mM nucleoside substrate concentration, 50 mM potassium phosphate buffer, pH 7.0 at 55 °C (DgPNP), 70 °C (GtPNP), and 80 °C (ApMTAP). 1) 150 µM nucleoside, 25 °C, 8.5 mM Na2HPO4, pH 7.5. 2) 1 mM nucleoside, 20 mM potassium arsenate, pH 7.1, 37 °C. For the PNP 100 % activity was assigned for the phosphorolysis of guanosine. 3) 20 mM nucleoside, 100 mM KP buffer, pH8, 70 °C. 4) 100 µM nucleoside, Ado-PNP had 137 % activity for 2-fluoroadenosine. 5) calculated from specific activities determined with 5 mM nucleoside substrate, 50 mM KP buffer, pH 7.5. 6) Numbers represent relative Vmax. 7) Numbers represent relative kcat values that were determined at 80 °C (P. furiosus enzymes) and 70 °C (S. solfataricus). For the MTAP from P. furiosus 100 % activity was assigned for the phosphorolysis of MTA. XanoP = abbreviation used here for xanthosine phosphorylase, the second PNP in E. coli encoded by the xapA gene that shows substrate specificity as eukaryoatic PNPs (specificity for 6-oxopurine nucleosides). (-) No activity was detected. (nd) = not determined/ data not available.
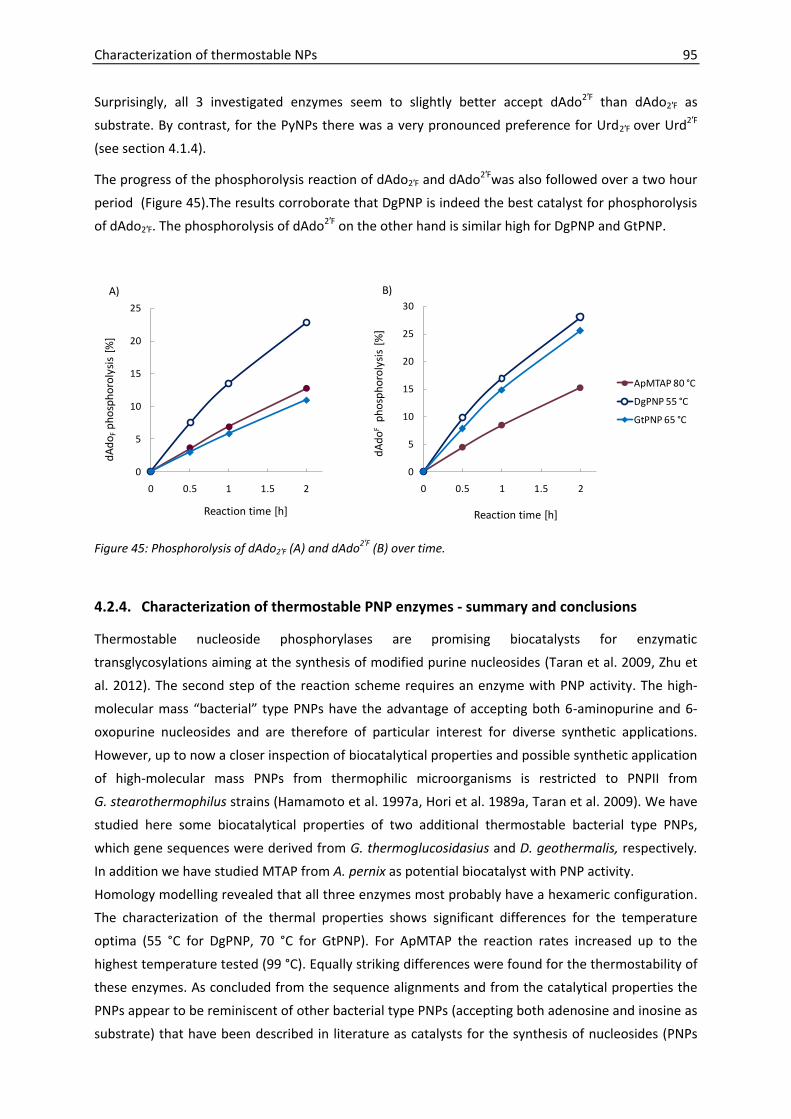
Characterization of thermostable NPs 95
Surprisingly, all 3 investigated enzymes seem to slightly better accept dAdo2′F than dAdo2′F as
substrate. By contrast, for the PyNPs there was a very pronounced preference for Urd2′F over Urd2′F
(see section 4.1.4).
The progress of the phosphorolysis reaction of dAdo2′F and dAdo2′Fwas also followed over a two hour
period (Figure 45).The results corroborate that DgPNP is indeed the best catalyst for phosphorolysis
of dAdo2′F. The phosphorolysis of dAdo2′F on the other hand is similar high for DgPNP and GtPNP.
0
5
10
15
20
25
0 0.5 1 1.5 2
dA
do
Fp
ho
sph
oro
lysi
s [%
]
Reaction time [h]
0
5
10
15
20
25
30
0 0.5 1 1.5 2
dA
do
Fp
ho
sph
oro
lysi
s [%
]
Reaction time [h]
ApMTAP 80 °C
DgPNP 55 °C
GtPNP 65 °C
A) B)
Figure 45: Phosphorolysis of dAdo2′F (A) and dAdo2′F (B) over time.
4.2.4. Characterization of thermostable PNP enzymes - summary and conclusions
Thermostable nucleoside phosphorylases are promising biocatalysts for enzymatic
transglycosylations aiming at the synthesis of modified purine nucleosides (Taran et al. 2009, Zhu et
al. 2012). The second step of the reaction scheme requires an enzyme with PNP activity. The high-
molecular mass “bacterial” type PNPs have the advantage of accepting both 6-aminopurine and 6-
oxopurine nucleosides and are therefore of particular interest for diverse synthetic applications.
However, up to now a closer inspection of biocatalytical properties and possible synthetic application
of high-molecular mass PNPs from thermophilic microorganisms is restricted to PNPII from
G. stearothermophilus strains (Hamamoto et al. 1997a, Hori et al. 1989a, Taran et al. 2009). We have
studied here some biocatalytical properties of two additional thermostable bacterial type PNPs,
which gene sequences were derived from G. thermoglucosidasius and D. geothermalis, respectively.
In addition we have studied MTAP from A. pernix as potential biocatalyst with PNP activity.
Homology modelling revealed that all three enzymes most probably have a hexameric configuration.
The characterization of the thermal properties shows significant differences for the temperature
optima (55 °C for DgPNP, 70 °C for GtPNP). For ApMTAP the reaction rates increased up to the
highest temperature tested (99 °C). Equally striking differences were found for the thermostability of
these enzymes. As concluded from the sequence alignments and from the catalytical properties the
PNPs appear to be reminiscent of other bacterial type PNPs (accepting both adenosine and inosine as
substrate) that have been described in literature as catalysts for the synthesis of nucleosides (PNPs

96 Characterization of thermostable NPs
from A. hydrophila (Ubiali et al. 2012), from G. stearothermophilus (Taran et al. 2009), and from
E. coli (Tuttle et al. 1993)). The advantage of all three biocatalysts in comparison to AhPNP and EcPNP
lies in the higher thermostability. From this point of view also ApMTAP is superior to GsPNP. We have
further investigated substrate specificities towards 2′-fluorosubstituted nucleosides. The idea was
that the enzyme most suitable for the phosphorolysis of 2′-fluorosubstituted purine nucleosides will
also be most suitable one for the reverse (synthetic) reaction. For dAdo2′F phosphorolysis, DgPNP
proved to be the best catalyst due to two criteria. Firstly, the highest reaction rate was observed with
DgPNP. Secondly, in the final application with dUrd2′F as donor the temperature should not exceed
60°C, since the donor is thermosensitive (see section 4.1.4). The phosphorolysis of dAdo2′F, however,
is similar high for DgPNP and GtPNP. Taking into account that the donor dUrd2′F is quite stable and
significant higher phosphorolysis rates were achieved with TtPyNP at 80°C than at 60 °C, GtPNP
appears to be superior over DgPNP as second enzyme in the transglycosylation reaction. However,
for this application also ApMTAP is possibly a good alternative due to its high stability at 80 °C.
These findings render all three enzymes promising for biocatalytic applications aiming at the
synthesis of 2′-fluorosubtituted nucleosides. The next chapter will focus on transglycosylations
employing PyNPs and enzymes with PNP activity at the same time and show, whether the combined
use of the generated thermostable enzymes will translate in high yield of synthetic purine
nucleosides. Moreover, the combination of both enzymes in transglycosylation reactions will also
allow to further characterize the substrate specificities of the PNPs towards artificial heterocyclic
bases, for which corresponding nucleosides were not readily available to investigate the reverse,
phosphorolytic reaction here.

5. Enzymatic transglycosylations with thermostable NPs
In this chapter the use of the generated thermostable biocatalysts for the synthesis of modified
purine nucleosides will be investigated. Our interest concerns the synthesis of sugar modified purine
nucleosides (2′-fluorinated nucleosides) and the synthesis of purine nucleosides with modified
nucleobases (2,6-dihalogenated purine nucleoside).
5.1. Introduction
5.1.1. Chemical synthesis of 2′-fluorinated nucleosides
The small size and high electronegativity renders fluorine a remarkable element. The C-F bond is one
of the strongest known bonds and organofluorine compounds are hence often characterized by high
chemical stability. On the other hand the bioisosteric replacements with fluorine have lead to the
disclosure of drugs with enhanced bioavailability, metabolic stability, and biological activity
(Hagmann 2008, Müller et al. 2007). Fluorination is therefore considered as an important tool in drug
discovery (Rentmeister et al. 2009).
The 2′ position of the carbohydrate moiety is the distinguishing feature between ribo- and
deoxyribonucleosides and is an attractive target for the selective introduction of a fluorine atom.
Here, fluorine can function as isosteric replacement of the hydrogen atom or as an isopolar mimic of
the hydroxyl group, respectively, whereby the C-F bond length (1.41 Å) is much closer to the C-O
bond length (1.35 Å) than to the C-H bond length (1.09 Å) (Liu et al. 2008, Müller et al. 2007).
Fluorine substitutions at the 2′ position of nucleosides have been shown to contribute to chemical
stability, in particular in acidic environment and lead to increased metabolic stability ((Liu et al. 2008)
and references therein).
Generally two strategies towards the synthesis of 2′-fluorinated nucleosides have been disclosed: i)
the direct fluorination of nucleosides and ii) the convergent synthesis that involves the condensation
of a fluorine-substituted ribose with a heterocyclic base. While the first approach may represent a
very efficient synthetic route for specific cases, the second approach is more versatile.
2′-Deoxy-2′-fluoro ribonucleosides
The efficient synthesis of 2′-deoxy-2′-fluorouridine starting from the natural ribonucleoside uridine
was already reported by Codington et al. in 1964. The approach that became the standard method
for fluorination of pyrimidine nucleosides involves the synthesis of O2,2′-anhydro-1-(-D-
arabinofuranosyl)uracil as intermediate product followed by a nucleophilic fluorination through
treatment with hydrogen fluoride. To avoid hazardous hydrogen fluoride, Olah’s reagent (mixture of
hydrogen fluoride and pyridine) has later been successfully employed as alternative fluorinating
reagent (Liu et al. 2008, Shi et al. 2005).
An analogous reaction route for the synthesis of 2′-deoxy-2′-fluoro ribofuranosyl purines does not
exist due to a lack of a suitable oxo-group on the heterocyclic base that could participate with the C-

98 Enzymatic transglycosylations with thermostable NPs
2′ atom in an anhydro bridge formation. Alternatively the synthesis of 2′-deoxy-2′-fluoroadenosine
and 2′-deoxy-2′-fluoroguanosine has been described by a nucleophilic displacement of a
corresponding triflate at the 2′-position (Kawasaki et al. 1993, Ranganathan 1977). However, due to
the number of different steps, the overall yield is limited. Furthermore it should be noted, that the
synthesis of the starting material (e.g. ara-A) also entails a number synthetic steps or chemo-
enzymatic approaches (Roshevskaia et al. 1986).
Another disadvantage of the described method is that it is not easily adaptable to the synthesis of
other 2′-deoxy-2′-fluoro ribofuranosyl purines. Therefore Thomas et al. investigated the convergent
synthesis by coupling 3,5-di-O-benzoyl-2-deoxy-2-fluoro-D-ribofuranosyl bromide to
2,6-dichloropurine (Thomas et al. 1994). The product could be readily converted to other analogues
resulting in the 2-fluoroadenine, 2-chloroadenine, 2,6 diaminopurine and guanine congeners by
standard procedures. The drawback of this approach is the formation of stereoisomers (α- and β-
anomers) and the need to separate both forms. Conversely, a chemo-enzymatic approach as it is
followed in the present work exclusively results in the biological active β-anomers and reduces the
number of required catalytic steps.
2′-Deoxy-2′-fluoro arabinonucleosides
While protocols for the efficient synthesis of 2′-deoxy-2′-fluoro ribonucleosides via direct fluorination
of preformed pyrimidine nucleosides have been developed, the synthesis of the according 2′-deoxy-
2′-fluoro arabinofuranosyl pyrimidine nucleosides is significantly more challenging. In fact the
susceptibility of pyrimidine nucleosides to form O2,2′-anhydro-bonds that is actually exploited for the
synthesis of pyrimidine 2′-deoxy-2′-fluoro ribonucleosides, is somehow precluding the efficient
synthesis of the respective arabinofuranosyl analogues via a direct fluorination approach. The
problem is that prior to the fluorination at the 2′-arabino position, the C2 carbonyl group of the
pyrimidine base will displace the leaving group at the C2-ribo activated function and O2,2′-anhydro-1-
(-D-arabinofuranosyl)pyrimidines is formed (Watts and Damha 2008). A subsequent SN2 type
reaction that entails the inversion of the configuration will then lead to the 2′-substituted
ribofuranosyl pyrimidine.
Therefore, the convergent synthetic approach towards 2′-deoxy-2′-fluoro arabinofuranosyl
pyrimidines has been in focus of research, and efficient protocols have been established. In 1979
Watanabe and co-workers described the synthesis of a series of 2′-deoxy-2′-fluoro arabinofuranosyl
pyrimidines with antiviral activity (Watanabe et al. 1979). Key steps of the synthetic procedure are
the fluorination and subsequent bromination of a protected ribofuranose derivative, and the
coupling with silylated pyrimidine. The protocol was further improved by Tann and co-workers in
1985 by proposing an efficient method for the selective synthesis of the respective 1-α-bromo sugar
(Tann et al. 1985). The advantage is that the 1-α-bromo sugar, in contrast to the stereoisomeric 1-β-
bromo sugar leads to preferred synthesis of (natural) β-nucleosides. Hence, 2′-deoxy-2′-fluoro
arabinofuranosyl pyrimidine can be prepared stereoselectively in high overall yield (Howell et al.
1988).

Enzymatic transglycosylations with thermostable NPs 99
On the other hand, the synthesis of the purine nucleoside congeners is significantly more complicate.
The major obstacle is the formation of stereo- and regioisomers (e.g. 7- or 9-substituted α- and
β-anomers) within the condensation of the ribose and purine base moiety, that entails the need of
tedious purification steps and eventually decreases the overall yield (Montgomery et al. 1986,
Tennilä et al. 2000, Wright et al. 1969). Despite impressive achievements in the development of
synthetic procedures for specific compounds, as for example for clofarabine (Bauta et al. 2004), the
synthesis of 2′-deoxy-2′-fluoro arabinofuranosyl purines remains in general a complicate endeavour.
Owing to these difficulties, alternative synthetic routes towards 2′-deoxy-2′-fluoro arabinofuranosyl
purines have been investigated. Thus, dAdo2′F and 9-(2-deoxy-2-fluoro--D-
arabinofuranosyl)hypoxanthine have been synthesized by direct fluorination of preformed
nucleosides (Krzeminski et al. 1991, Maruyama et al. 1999, Sivets et al. 2006). The fluorination step
itself was reported as fairly efficient (approx. 30 %). However, the overall synthesis is an advanced
multistep procedure involving the need of selective protection steps and is not comparable to the
facile and efficient synthesis of 2′-deoxy-2′-fluoro ribofuranosyl pyrimidines via direct fluorination
(described before).
Noteworthy, despite the aforementioned difficulties arising from the C2 carbonyl group, the direct
fluorination at the 2′-arabino position of pyrimidine nucleosides has been recently proposed
(Turkman et al. 2010). The procedure was applied for the synthesis of radiolabelled 2′-deoxy-2′-
fluoro-5-methyl-1-β-D-arabinofuranosyluracil ([18F]FMAU).
5.1.2. The chemo-enzymatic synthesis of 2′-fluorinated purine nucleosides
Motivated by the difficulties arising within the chemical synthesis of purine nucleoside analogues,
enzyme-assisted synthetic routes towards 2′-fluorinated purine nucleosides have been investigated.
Hereby, the enzymatic transfer of a 2′-fluorinated pentofuranose moiety from a pyrimidine
nucleoside donor to a purine base acceptor represents an attractive approach. With this strategy
Tuttle and co-workers synthesized 2′-deoxy-2′-fluoro ribofuranosyl purines employing 2′-deoxy-2′-
fluorouridine as pentofuranosyl donor (Tuttle et al. 1993). The enzymatic transglycosylation was
mediated by large amounts of EcTP and EcPNP and required long reaction times (up to 57 days). In
order to improve the availability of poorly soluble purine bases and to increase the efficiency of the
reaction, the temperature was increased from 37 °C to 50 °C. To retain the activity of the E. coli NPs,
enzymes were immobilized. Moreover, the first 24 h were run at 37 °C, in order to accumulate
pentose-1-phosphate that is known to further stabilize the NPs. The results demonstrate that this
“high” temperature approach was a clear advantage with respect to reduced reaction times and
increased yields.
The same strategy was patented for the synthesis of a number of 9-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)purines (Tuttle and Krenitsky 1992), whereby 1-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)thymine served as pentofuranosyl donor. Although the high-temperature approach
(at 50 °C with immobilized E. coli enzymes) was used, enzyme loadings were high and the reaction
time fairly long (e.g. 6 days for 2,6-Diamino-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-9H-purine).

100 Enzymatic transglycosylations with thermostable NPs
The enzymatic synthesis of 2′-deoxy-2′-fluoroguanosine with 2′-deoxy-2′-fluorouridine or -cytidine as
pentofuranosyl donors was also accomplished by the use of whole cells of E. coli BMT-4D/lA as
biocatalyst (Zaitseva et al. 1999).
Recently the possibility to use NdRTs for the transfer of 2′-deoxy-2′-fluoro ribofuranosyl moieties was
disclosed (Fernandez-Lucas et al. 2010). With dUrd2′F as donor and adenine as acceptor, dAdo2′F was
synthesized employing Lactobacillus reuteri NdRT
A modified approach was followed by Yamada and co-workers (Yamada et al. 2009). Here the
pentofuranosyl moiety was not provided by the phosphorolysis of a corresponding pyrimidine
nucleoside. Instead, the authors successfully synthesized 2-deoxy-2-fluoro arabinofuranosyl-α-1-
phosphate that was afterwards enzymatically linked to a purine base affording the desired 9-(2-
deoxy-2-fluoro-β-D-arabinofuranosyl)purine. In order to perform the reaction at 50 °C, thermostable
PNP from G. stearothermophilus was used. The approach is motivated by the fact that the
phosphorolysis reaction of the pyrimidine donor, that proceeds very slowly if the 2′ position is
substituted with a fluorine atom in the arabino position, can be avoided.
5.1.3. 2,6-Dihalogenated purine nucleosides
Purine nucleosides with variable substitutions on position 2 and 6 of the heterocyclic base are of
pharmaceutical interest due to antimicrobial and anticancer activities (Bellezza et al. 2008, Bonate et
al. 2006, Cappellacci et al. 2011, Rodenko et al. 2007). Insights into the biological activities of
adenosine receptors have further expanded potential therapeutic application fields (Nair et al. 1995,
Poulsen and Quinn 1998, Samsel and Dzierzbicka 2011, Vittori et al. 2000).
2,6-Dichloropurine nucleosides are valuable precursors for this class of nucleoside derivatives (e.g.
(Kazimierczuk et al. 1984, Montgomery et al. 1986, Tennilä et al. 2000). The chlorine atoms display
electron withdrawing centres on the purine ring, making these positions amenable for nucleophilic
substitutions. Hereby the chlorine atom on ring position 6 is much more reactive than the chlorine
atom at position 2 (Dobak et al. 2008, Schaeffer and Thomas 1958). This difference in reactivity
enables the synthesis of 2,6-substituted derivatives either by selective monosubstitution of the C-6
chlorine atom or by consecutive substitutions of the C-6 and C-2 chlorine atoms by diverse functional
groups (Salvatori et al. 2002, Schaeffer and Thomas 1958, Tennilä et al. 2000, Vittori et al. 2000,
Wright et al. 1987). Likewise 2,6-dichloropurine nucleosides can serve as starting material for cross-
coupling reactions leading to the efficient synthesis of 2-substituted 6-methylpurine nucleosides
(Hocek and Dvorakova 2003).
Simple and efficient methods for the preparation of 2,6-dichloropurine have been developed, for
example the facile synthesis from xanthine has been described (Zeng et al. 2004). By contrast, the
coupling reaction to carbohydrate moieties gave rise to regioisomers and, moreover, mixtures of the
α- and β-anomers in the case of 2′-deoxynucleosides (Vorbrüggen and Ruh-Pohlenz 2001). By strictly
obeying to regio- and stereoselective requirements, enzymatically catalyzed coupling reaction of 2,6-
dichloropurine to pentofuranosyl moieties could therefore present an attractive alternative.

Enzymatic transglycosylations with thermostable NPs 101
5.2. Synthesis of 2′-fluorosubstituted purine nucleosides
The aim was to synthesize 2′-deox-2′-fluoroadenosine (dAdo2′F) and 9-(2-deoxy-2-fluoro--D-
arabinofuranosyl)adenine (dAdo2′F) by enzymatic transglycosylations employing 2′-deoxy-2′-
fluorouridine (dUrd2′F) or 1-(2-deoxy-2-fluoro--D-arabinofuranosyl)uracil (dUrd2′F) as pentofuranosyl
donor and adenine as pentofuranosyl acceptor.
+ Pi+ Pi
Uracil
PyNPPNP/MTAP
NH
NH
O
O
O
HO F
HO
OPO32-
O
HO F
HO
N
NN
N
NH2
O
HO F
HO
NH
N
O
O
O
HO
F
HO
NH
N
O
O O
HO
F
HO
N
NN
N
NH2
O
HO
F
HO
OPO32-
dUrd2‘F
dUrd2‘F
Adenine
N
NNH
N
NH2
dAdo2‘F
dAdo2‘F
Figure 46: Enzymatic synthesis of dAdo2′F and dAdo2′F from respectively sugar-modified uridine analogues (dUrd2′F, dUrd2′F) and adenine, mediated by PyNP and PNP or MTAP.
5.2.1. Synthesis of 2′-deoxy-2′-fluoroadenosine
The experiments presented in section 4.1.4 indicated that TtPyNP compared to GtPyNP is clearly the
more efficient catalysts for the phosphorolysis of dUrd2′F. Even at 60 °C - where GtPyNP shows
optimal activity, but TtPyNP only about 12 % of the activity displayed at 80 °C (Figure 36)- the TtPyNP
catalyzed phosphorolysis of dUrd2′F is about 16 times more efficient than the GtPyNP catalyzed
reaction. On the other hand, in section 4.2.3 it was shown that among the tested enzymes with PNP
activity DgPNP is most efficiently catalyzing the phosphorolysis of dAdo2′F (determined at 55 °C),
followed by ApMTAP (at 80 °C). However, due to the themolability of the donor nucleoside (dUrd2′F)
discussed in section 4.1.4, the final reaction should not be conducted at a temperature considerably
exceeding 60 °C. This restriction makes the use of ApMTAP rather unattractive since at 60 °C this
highly thermoactive biocatalyst shows only about 22 % of the activity displayed at 80 °C (Figure 43).
For these reasons we tested the following two combinations of enzymes for the synthesis of dAdo2′F:
i) TtPyNP + DgPNP at 55 °C and ii) TtPyNP + GtPNP at 65 °C. Reactions were stopped after 2 and 18 h,
respectively. The formation of dAdo2′F was confirmed by comparing the retention time and UV
spectrum of the newly evolved peak with that of authentic sample of dAdo2′F. From the peak areas
obtained by HPLC measurements, the fraction of phosphorolytically cleaved donor (dUrd2′F)
molecules and the fraction of the acceptor (adenine) molecules that were transformed into the

102 Enzymatic transglycosylations with thermostable NPs
product (dAdo2′F ) were calculated (Figure 47). The second value can also be understood as the yield
with respect to the applied acceptor molecule.
0
10
20
30
40
18 h 2 h 18 h 2 h 18 h
TtPyNP 65 °C TtPyNP+GtPNP 65 °C TtPyNP+DgPNP 55 °C
Sub
stra
te c
on
vers
ion
%
phosphorylized FU
FA formation
Uracil/(dUrd2‘F + Uracil) × 100 %
dAdo2‘F/(Adenine + dAdo2‘F) × 100 %
Figure 47: Synthesis of 2′-deoxy-2′-fluoroadenosine (dAdo2′F) from 2′-deoxy-2′-fluorouridine serving as donor and adenine serving as pentofuranosyl acceptor.
The results show that the yield was in fact in similar range for both enzyme combinations tested and
that prolonged incubation times are needed to obtain a reasonable outcome (after 18 h the yield was
less than 20 %). When only TtPyNP was added to the reaction mixture the phosphorolysis reaction of
the donor molecule proceeded as expected while product formation was negligible (Figure 47).
When only PNPs were added, no substrate conversion was observed. In the reaction employing
TtPyNP and GtPNP at 65 °C, after 18 h 3.4 % of the donor molecules reacted to the unwanted side-
product O2,2′-anhydro-1-(-D-arabinofuranosyl)uracil (anhydro-Urd). In the TtPyNP-DgPNP catalyzed
reaction at 55 °C anhydro-Urd formation was significantly lower (2 %). Therefore the TtPyNP-DgPNP
catalyzed reaction was considered as more suitable and was further investigated with respect to the
reaction time (Figure 48). The formation of the product (dAdo2′F) increased in linear relation to the
reaction time, while the phosphorolysis of dUrd2′F appeared to follow a hyperbolic function over the
reaction time. Therefore it seems likely that the second reaction represents the rate-limiting step.
The final yield obtained after 24 h was 24 %. At this final point 44 % of the donor molecules were
phosphorolytically cleaved to uracil and the pentose-1-phosphate intermediate. Taking into account
that the initial donor nucleoside concentration was 2 mM and the initial acceptor concentration was
1 mM, these data show that in the 1 ml reaction vessel used, about 0.88 µmol pentose-1-phosphate
intermediate was formed, and 0.24 µmol of the intermediate further reacted to the final product.
Overall 2.3 % of the donor molecules reacted to the unwanted side-product anhydro-Urd. 1-(-D-
Arabinofuranosyl)uracil (ara-U) was not detected.
In summary, the combined use of TtPyNP and DgPNP at 55 °C was found to be a good approach for
the synthesis of dAdo2′F. The data suggest that the current product yield (23 % with respect to the
pentofuranosyl acceptor) is restricted by the reaction rates of the biocatalysts. Hence, the yield could
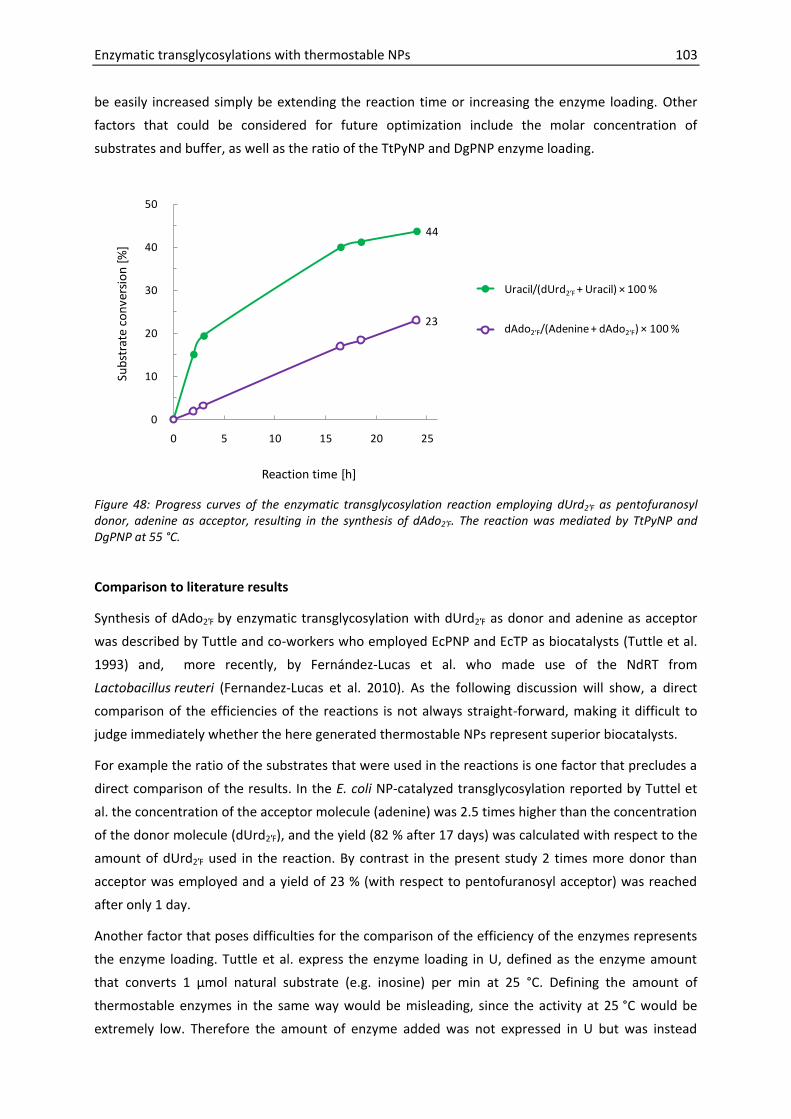
Enzymatic transglycosylations with thermostable NPs 103
be easily increased simply be extending the reaction time or increasing the enzyme loading. Other
factors that could be considered for future optimization include the molar concentration of
substrates and buffer, as well as the ratio of the TtPyNP and DgPNP enzyme loading.
44
23
0
10
20
30
40
50
0 5 10 15 20 25
Sub
stra
te c
on
vers
ion
[%
]
Reaction time [h]
phosphorylized FU
FA formation
Uracil/(dUrd2‘F + Uracil) × 100 %
dAdo2‘F/(Adenine + dAdo2‘F) × 100 %
Figure 48: Progress curves of the enzymatic transglycosylation reaction employing dUrd2′F as pentofuranosyl donor, adenine as acceptor, resulting in the synthesis of dAdo2′F. The reaction was mediated by TtPyNP and DgPNP at 55 °C.
Comparison to literature results
Synthesis of dAdo2′F by enzymatic transglycosylation with dUrd2′F as donor and adenine as acceptor
was described by Tuttle and co-workers who employed EcPNP and EcTP as biocatalysts (Tuttle et al.
1993) and, more recently, by Fernández-Lucas et al. who made use of the NdRT from
Lactobacillus reuteri (Fernandez-Lucas et al. 2010). As the following discussion will show, a direct
comparison of the efficiencies of the reactions is not always straight-forward, making it difficult to
judge immediately whether the here generated thermostable NPs represent superior biocatalysts.
For example the ratio of the substrates that were used in the reactions is one factor that precludes a
direct comparison of the results. In the E. coli NP-catalyzed transglycosylation reported by Tuttel et
al. the concentration of the acceptor molecule (adenine) was 2.5 times higher than the concentration
of the donor molecule (dUrd2′F), and the yield (82 % after 17 days) was calculated with respect to the
amount of dUrd2′F used in the reaction. By contrast in the present study 2 times more donor than
acceptor was employed and a yield of 23 % (with respect to pentofuranosyl acceptor) was reached
after only 1 day.
Another factor that poses difficulties for the comparison of the efficiency of the enzymes represents
the enzyme loading. Tuttle et al. express the enzyme loading in U, defined as the enzyme amount
that converts 1 µmol natural substrate (e.g. inosine) per min at 25 °C. Defining the amount of
thermostable enzymes in the same way would be misleading, since the activity at 25 °C would be
extremely low. Therefore the amount of enzyme added was not expressed in U but was instead

104 Enzymatic transglycosylations with thermostable NPs
simply set to 0.1 mg ml-1 in the present study. However, if the specific activities of the NPs are
known, it is obviously possible to calculate how much enzyme (in mg ml-1) was used in the study of
Tuttle and co-workers. For example it was stated that 3900 U EcPNP was employed in a reaction
volume of 20 ml (at least in the first 6 days). A maximal velocity of 90.5 U mg-1 (Li et al. 2008) and
230 U mg-1 (Jensen and Nygaard 1975) was reported for EcPNP at 37 °C. Since the temperature
optimum of EcPNP is 60 °C (Li et al. 2008) it can hence be speculated that the specific activity at 25 °C
is significantly lower than 230 U mg-1. Therefore it can be concluded that the enzyme loading of
EcPNP was considerably higher than 0.85 mg ml-1 within the first 6 days, whereas in the present
study only 0.1 mg ml-1 DgPNP was used.
As stated above, a direct comparison of the efficiencies of the enzymes is not possible with the
current data. However, taking the information on yield, reaction time and enzyme loading together,
there seems to be strong evidence that the efficiency of DgPNP and TtPyNP could considerably
exceed the efficiency of the E. coli NPs employed previously (Tuttle et al. 1993) for the synthesis of
dAdo2′F.
Furthermore, the here generated thermostable enzymes appear to be also superior to NdRT from
Lactobacillus reuteri, that was recently described as biocatalyst for the synthesis of dAdo2′F. The
reaction was performed at 40 °C with each 1 mM substrate concentration and an enzyme loading of
0.34 µg per 40 µl, which corresponds to 0.0085 mg ml-1. The specific activity assessed for dAdo2′F
synthesis after 24 h was 0.3 x 10-3 U mg-1. Note that there is a typo in the exponent in the publication
(Fernandez-Lucas et al. 2010). According to our calculations, if these data are transformed into yield
with respect to the amount of adenine this would correspond to approximately 3.8 x 10-4 %. By
contrast, in the present study DgPNP and TtPyNP (enzyme loading each 0.1 mg ml-1) were employed
at 55 °C with 2 mM dUrd2′F and 1 mM adenine, resulting in a yield of 23 % with respect to adenine.
These data indicate that, even if both experiments would have been performed with the same
enzyme loading, the final yield of dAdo2′F obtained with DgPNP and TtPyNP as biocatalyst would
considerably exceed the yield that would have been obtained with Lactobacillus reuteri NdRT.
5.2.2. Synthesis of 9-(2-deoxy-2-fluoro--D-arabinofuranosyl)adenine
After the successful synthesis of dAdo2′F the aim was now to synthesize the epimeric counterpart 9-
(2-deoxy-2-fluoro--D-arabinofuranosyl)adenine (dAdo2′F). Therefore dUrd2′F should serve as
pentofuranosyl donor and, again, adenine should serve as pentofuranosyl acceptor.
TtPyNP was again used as PyNP, because only this enzyme recognized dUrd2′F as a substrate, while
for GtPyNP no substrate activity could be seen (for further details see 4.1.4). Nevertheless, it should
be noted that also for TtPyNP the reaction rate observed with dUrd2′F as substrate was about 10
times lower than that recorded for dUrd2′F.
According to their phosphorolytic activity on dAdo2′F, DgPNP (at 55 °C) and GtPNP (at 70 °C) appear to
be the most efficient catalysts that could be used for dAdo2′F synthesis. Despite these findings we
decided to test the following two combinations first: i) TtPyNP + GtPNP at 70 °C (temperature is
limited by GtPNP stability) and ii) TtPyNP + ApMTAP at 80 °C (temperature is limited by TtPyNP

Enzymatic transglycosylations with thermostable NPs 105
stability). The use of DgPNP was not tested because this would limit the reaction temperature to
55 °C where the first, TtPyNP-catalyzed phosphorolysis of dUrd2′F would presumably proceed
unreasonably slow. While for dAdo2′F synthesis the reaction temperature was also limited by the
instability of the donor nucleoside dUrd2′F at temperatures above 60 °C, dUrd2′Fappeared to be stable
at even 80 °C and is therefore not limiting the reaction temperature.
0.37 0.48 0.650
5
10
15
20
25
30
18 h 2 h 2 h
TtPyNP 65 °C TtPyNP + GtPNP 70 °C TtPyNP + ApMTAP 80 °C
Sub
stra
te c
on
vers
ion
%
Series1
Series2
Uracil/(dUrd2‘F + Uracil) × 100 %
dAdo2‘F/(Adenine + dAdo2‘F) × 100 %
Figure 49: Biocatalyst screening for the synthesis of dAdo2′F by an enzymatic transglycosylation reaction employing dUrd2′F as donor and adenine as pentofuranosyl acceptor.
The procedure of the experiments was essentially the same as described for the synthesis of dAdo2′F
in the previous section. In a first step the enzymatic transglycosylation was investigated with both
the aforementioned enzyme combinations after a reaction time of 2 h (Figure 49). The yield of
dAdo2′F with respect to adenine was in a similar range (0.48 % and 0.65 %). If TtPyNP was employed
as sole biocatalyst the yield was only 0.37 % after 18 h. Hence TtPyNP alone was not further
investigated as biocatalyst for dAdo2′F synthesis. If PNPs were employed as sole biocatalysts no
substrate conversion could be seen (data not shown). In a next step the enzymatic transglycosylation
reaction affording dAdo2′F was monitored over time (Figure 50). The results indicate that the
combination of TtPyNP and ApMTAP at 80 °C was clearly superior over the combination of TtPyNP
and GtPNP at 70 °C, resulting in 24 % yield of dAdo2′F with respect to adenine after a reaction period
of 24 h.
Comparison to literature results
Tuttle and Krenitsky have patented the enzymatic synthesis of a number of 9-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)purines, whereby similarly as described for the synthesis of purine 2′-deoxy-2′-
fluororibosides the procedure involved the use of EcPNP and EcTP as biocatalysts (Tuttle and
Krenitsky 1992). The enzymatic transglycosylation reaction described in the present study followed
the same principle, with the exception that dUrd2′F instead of 1-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)thymine was used as pentofuranosyl donor.

106 Enzymatic transglycosylations with thermostable NPs
37
24
0
10
20
30
40
50
0 5 10 15 20 25
Sub
stra
te c
on
vers
ion
[%
]Reaction time [h]
Phosphorylized FanaU
FanaA formation
32
14
0
10
20
30
40
50
0 5 10 15 20 25
Sub
stra
te c
on
vers
ion
[%
]
Reaction time [h]
Uracil/(dUrd2‘F + Uracil) × 100 %
dAdo2‘F/(Adenine + dAdo2‘F) × 100 %
TtPyNP + GtPNP 70 °C TtPyNP + ApMTAP 80 °C
Figure 50: Progress curves for the enzymatic synthesis of dAdo2′F mediated by TtPyNP + GtPNP at 70 °C or by TtPyNP + ApMTAP at 80 °C. Pentofuranosyl donor was dUrd2′F and acceptor was adenine.
The synthesis of dAdo2′F is not included in the patent, but the example of 2,6-diamino-9-(2-deoxy-2-
fluoro-β-D-arabinofuranosyl)-9H-purine likewise illustrates the synthetic challenges encountered
with EcPNP and EcTP as biocatalyst. The enzyme loading was extremely high, extending even the
load that was used by the same group for the synthesis of dAdo2′F (Tuttle et al. 1993). For example
2900 U ml-1 of EcPNP were used by Tuttle and Krenitsky for 2,6-diamino-9-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)-9H-purine synthesis, while in the equivalent reaction for dAdo2′F synthesis only
195 U ml-1of EcPNP were used. With the assumptions and calculations outlined in the previous
section, an enzyme loading of 2900 U ml-1 would correspond to more than 12.6 mg ml-1.
Furthermore, the enzymes were not used in soluble form but as immobilized catalysts – presumably
because the reaction was conducted at 50 °C in order to increase the reaction rates as described in
(Tuttle et al. 1993). Nevertheless, the reaction required a relatively long incubation time of 6 days.
From the amount of isolated dAdo2′F (0.2 g) one can calculate the isolated yield with respect to 1-(2-
deoxy-2-fluoro-β-D-arabinofuranosyl)thymine, which is approx. 62 %. In the present study the yield
of dAdo2′F from adenine was 24 % after only 1 day, whereby the loading with each enzyme was only
0.1 mg ml-1. Hence, it appears that the high thermal stability and the favourable substrate specificity
of TtPyNP and ApMTAP indeed translate into considerably improved biocatalytical efficiencies.
Yamada and co-workers have described the synthesis of dAdo2′F from chemically prepared 2-deoxy-2-
fluoroarabinofuranosyl-α-1-phosphate and adenine as substrates, whereby the purine base was
applied in excess in the reaction mixture (5 mM versus 2.4 mM). The formation of the gycosidic bond
was catalyzed by PNP from G. stearothermophilus that allowed the operation at 50 °C without the
need of enzyme stabilization measures. With this strategy the yield of dAdo2′F with respect to 2-
deoxy-2-fluoroarabinofuranosyl-α-1-phosphate was about 50 %, reached after only 2.5 days. Even
though this process seems significantly more efficient than the enzymatic transglycosylation
described by Tuttle et al. with E. coli enzymes, the disadvantage lies in the complexity of the chemical
synthesis of 2-deoxy-2-fluoroarabinofuranosyl-α-1-phosphate. In fact, the authors motivated their
work with the reason that the rate of the phosphorolysis reaction in the enzymatic transglycosylation

Enzymatic transglycosylations with thermostable NPs 107
is very low. In this respect, the enzymatic transglycosylation employing the thermostable biocatalysts
(ApMTAP and TtPyNP) described in the present work represents a promising alternative.
5.3. Synthesis of 2,6-dihalogenated purine nucleosides
After having investigated the synthesis of carbohydrate-modified adenosine analogues, the next aim
was to test the applicability of the thermostable NPs for the synthesis of base-modified purine
nucleosides, namely 2,6-dichloropurine and 6-chloro-2-fluoropurine ribosides and deoxyribosides,
respectively. Since these target compounds were hardly available, the suitability of the enzymes with
PNP activity was investigated directly in the synthetic reaction, employing pentose-1-phosphate and
the modified purine base. Pentose-1-phosphates were thereby provided by the PyNP-catalyzed
phosphorolysis of uridine or thymidine. The reactions scheme for the synthesis of 2,6-dihalogenated
purine ribosides is illustrated in Figure 51. The reaction scheme for the respective deoxyribosides is
basically the same, with the exception that thymidine serves as pentofuranosyl donor.
To our best knowledge, enzymatic transglycosylations affording the 2,6-dihalogenated ribosides and
deoxribosides that are subject of this section have not been previously disclosed in the scientific
literature.
O
HO OH
HO
NH
N
O
O
+ Pi+ Pi
Uracil
PyNPPNP/MTAP
NH
NH
O
O
O
HO OH
HO
OPO32-
O
HO OH
HO
N
NN
N
Cl
Cl
Uridine
2,6-Dichloropurine
N
NNH
N
Cl
Cl
N
NNH
N
Cl
F
6-Chloro-2-fluoropurine
O
HO OH
HO
N
NN
N
Cl
F
2,6-Dichloropurine riboside
6-Chloro-2-fluoropurine riboside
Figure 51: Reaction scheme for the enzymatic synthesis of 2,6-dihalogenated ribosides. Uridine serves as pentofuranosyl donor, while either 2,6-dichloropurine or 6-chloro-2-fluoropurine functions as pentofuranosyl acceptor. The enzymatic transfer reaction can be catalyzed by the concurrent activity of PyNP and PNP, or MTAP respectively.

108 Enzymatic transglycosylations with thermostable NPs
5.3.1. Synthesis of 2,6-dihalogenated purine ribosides
Different combinations of enzymes were tested for their ability to catalyze the enzymatic synthesis of
2,6-dichloropurine riboside and 6-chloro-2-fluoropurine riboside. Uridine (2 mM) was used as ribose-
1-phosphate donor; the purine bases (2,6-dichloropurine, 6-chloro-2-fluoropurine) were employed in
1 mM concentration. In order to avoid spontaneous reactions of the halogenated purine bases, the
reaction temperature was restricted to a maximum of 65 °C. For screening the efficiency of different
combinations of biocatalysts, the reaction mixtures were analyzed after an incubation time of
30 min.
Indeed with all combinations of enzymes tested new peaks emerged, indicating the formation of 2,6-
dichloropurine riboside (retention time 8.2 min) and 6-chloro-2-fluoropurine riboside (7.6 min).
Authentic samples of the products were not available, but product formation as shown in (Figure 52)
could be indirectly quantified from the decrease of the concentration of the heterocyclic base. In
control experiments employing the natural purine base adenine as pentofuranosyl acceptor,
adenosine was obtained in very high yield with respect to the base (approx. 90 %). If the
dihalogenated purine were applied as pentofuranosyl acceptor, the yield with respect to the base
was between 49 % and 65 % for all the different combinations of enzymes tested. The highest
substrate conversion was obtained in reactions involving GtPNP, but a striking difference between
the different enzymes with PNP activity was not disclosed. The formation of adenosine was also
efficient when a PyNP was employed as sole biocatalyst. At 65 °C for example the same fraction of
adenine molecules (90 %) were converted to adenosine after 30 min, whether or not a second
enzyme with PNP activity was added to the reaction mixture (Figure 52B). These results further
corroborate the PNP by-activity of the PyNPs as discussed in section 4.1.5. However, if the artificial
halogenated purine bases were employed as pentofuranosyl acceptors the product formation was
dramatically lower in reactions with only PyNP as biocatalyst compared to the 2-enzyme reactions
(Figure 52). The use of a PNP enzyme (DgPNP, GtPNP or ApMTAP) as sole catalyst did not afford any
substrate conversion.
The transglycosylation reactions employing GtPNP and GtPyNP at 65 °C and artificial purine bases as
pentofuranosyl acceptors were further investigated by monitoring the progress of the reactions over
time. In addition transglycosylations with TtPyNP and ApMTAP were monitored at 80 °C over a 2 h
period, since the stability test had meanwhile shown that the dihalogenated purine bases did not
react to side products under these conditions. The results show that an equilibrium of the reaction
was relatively rapidly established, within a 1 – 2 h period (Figure 53). Further incubation did not
increase the product concentration. In the reactions where 2,6-dichloropurine was used applied as
pentofuranosyl acceptor, about 56 mol % of the purine base molecules were converted to the
corresponding riboside. Apparently, the only difference between the reactions employing
GtPNP/GtPyNP at 65 °C and TtPyNP/ApMTAP at 80 °C is that the equilibrium is faster reached in the
latter case (Figure 53A,B). Basically the same phenomenon was observed with 6-chloro-2-
fluoropurine, although the final outcome was significantly better: 63 – 67 mol % of base molecules
were transformed into the respective ribonucleoside (Figure 53C,D).

Enzymatic transglycosylations with thermostable NPs 109
0
20
40
60
80
100
GtPyNP GtPyNP + DgPNP
GtPyNP GtPyNP + DgPNP
GtPyNP GtPyNP + DgPNP
Adenine 2,6DCP 6C2FP
Sub
stra
te c
on
vers
ion
[m
ol
%]
Uridine Phosphorolysis
Glycosylated base
Uracil/(Uridine+ Uracil) × 100 %
Product/(Purine base + Product) × 100 %
0
20
40
60
80
100
GtPyNP GtPyNP + GtPNP
GtPyNP + ApMTAP
GtPyNP GtPyNP + GtPNP
GtPyNP + ApMTAP
GtPyNP GtPyNP + GtPNP
GtPyNP + ApMTAP
Adenine 2,6DCP 6C2FP
Sub
stra
te c
on
vers
ion
[m
ol
%]
0
20
40
60
80
100
TtPyNP TtPyNP + GtPNP
TtPyNP TtPyNP + GtPNP
TtPyNP TtPyNP + GtPNP
Adenine 2,6DCP 6C2FP
Sub
stra
te c
on
vers
ion
[m
ol
%]
A)
B)
C)
Figure 52. Synthesis of 2,6-dihalogentated purine ribosides with (A) DgPNP and GtPyNP at 55 °C, (B) GtPNP or ApMTAP in combination with GtPyNP at 65 °C and (C) GtPNP and TtPyNP at 65 °C . Uridine (2 mM) served as pentofuranosyl donor; adenine, 2,6-dichloropurine (2,6DCP) and 6-chloro-2-fluoropurine (6C2FP) were investigated as acceptors (1 mM). Transglycosylations were performed in 2 mM sodium phosphate buffer, pH 6.5. Reactions were stopped after 30 min.

110 Enzymatic transglycosylations with thermostable NPs
56
0
20
40
60
80
0 1 2 3 4 5 6 7
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
GtPyNP + GtPNP 65°C
56
0
20
40
60
80
0 1 2
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
TtPyNP + ApMTAP 80°C
Uracil
26DCP riboside
Uracil/(Uridine+ Uracil) × 100 %
2,6DCPriboside/(2,6DCP + 2,6 DCPriboside) × 100 %
54
63
0
20
40
60
80
0 1 2
Sub
stra
te c
on
vers
ion
mo
l [%
]
Reaction time [h]
TtPyNP + ApMTAP 80°C
Uracil
6C2FP riboside
54
67
0
20
40
60
80
0 1 2 3 4 5 6 7
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
GtPyNP + GtPNP 65°C
Uracil/(Uridine+ Uracil) × 100 %
6C2FPriboside/(6C2FP + 6C2FPriboside) × 100 %
A) B)
C) D)
Figure 53: Synthesis of dihalogenated ribosides over time. Formation of 2,6-dichloropurine riboside employing GtPNP + GtPyNP at 65 °C (A) or TtPyNP + ApMTAP at 80 °C (B); formation of 6-chloro-2-fluoropurine riboside employing GtPNP + GtPyNP at 65 °C (C) or TtPyNP + ApMTAP at 80 °C (D). Uridine (2 mM) served as pentofuranosyl donor; adenine, 2,6-dichloropurine (2,6DCP) and 6-chloro-2-fluoropurine (6C2FP) were investigated as acceptors (1 mM). Transglycosylations were performed in 2 mM sodium phosphate buffer, pH 6.5.
5.3.2. Synthesis of 2,6-dihalogenated purine deoxyribosides
In analogy to the 2,6 dihalogenated ribosides the synthesis of the deoxyriboside congeners was
investigated. Hereby thymidine served as pentofuranosyl donor. Initial tests revealed that the
synthesized purine deoxyribosides were not stable under acidic conditions and hydrolyzed to
pentose and purine base moieties (data not shown). For these reasons stopping the reactions
through addition of TCA proved not to be appropriate. Instead samples were now 3-fold diluted with
ice-cold buffer. Two different enzyme combinations were tested: TtPyNP + GtPNP at 65 °C and
TtPyNP + ApMTAP at 80 °C and the reaction mixture was analyzed after 30 min. With both
combinations new peaks emerged in the HPLC chromatogram of respective reaction mixture
samples, suggesting the formation of 2,6-dichloropurine deoxyriboside (retention time 8.8 min) and
6-chloro-2-fluoropurine deoxyriboside (8.2 min). Authentic samples of the products were not
available; product formation was thus again just indirectly quantified from the decrease of the
concentration of the heterocyclic base.
Monitoring of the substrate conversion over time reveals an interesting development in all reaction
mixtures studied here (Figure 54): Instead of increasing or constant product concentration, a

Enzymatic transglycosylations with thermostable NPs 111
decrease of the product concentrations over time was observed. At the same time the ratio of
thymine/thymidine increased. These results indicate that either the product or the intermediate
product (deoxyribose-1-phosphate) is instable and possible hydrolyzed. If for example the phosphate
would be cleaved form the deoxyribose-1-phosphate, this intermediate product would not be
anymore substrate for both the PyNP and the PNP catalyzed reactions. As a consequence both
enzymes would irreversibly catalyze the phosphorolysis reactions. Presumably it is indeed the
instability of the pentose-1-phosphate that is responsible for the observed effect. This would also be
an explanation for a previously observed effect. In Figure 38 it is shown that almost 100 % thymidine
molecules were phosphorolyzed at 80 °C with TtPyNP, while at 60 °C or with GtPyNP as biocatalyst
apparently an equilibrium with about 75 % phosphorolyzed molecules was reached. Also in the
transglycosylation reactions investigated now, the (intermediate) product instability appears to be
more pronounced at higher temperature; in both cases the apparent product decay was significantly
faster at 80 °C than at 65 °C (Figure 54).
0
20
40
60
80
100
0 1 2
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
TtPyNP + ApMTAP 80°CThymidine
26DCPdeoxyriboside
0
20
40
60
80
100
0 2 4 6
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
GtPyNP + GtPNP 65°C Thymine/(Thymidine+ Thymine) × 100 %
Product/(2,6DCP + Product) × 100 %
0
20
40
60
80
100
0 1 2
Sub
stra
te c
on
vers
ion
[m
ol
%]
Reaction time [h]
TtPyNP + ApMTAP 80°CThymidine
6C2FP deoxyriboside
0
20
40
60
80
100
0 2 4 6
Sub
stra
te c
on
vers
ion
mo
l [%
]
Reaction time [h]
GtPyNP+GtPNP 65°CThymine/(Thymidine+ Thymine) × 100 %
Product/(6C2FP + Product) × 100 %
A) B)
C) D)
Figure 54: Synthesis of dihalogenated deoxyribosides. Formation of 2,6-dichloropurine deoxyriboside employing GtPyNP + GtPNP at 65 °C (A) or TtPyNP + ApMTAP at 80 °C (B) and formation of 6-chloro-2-fluoropurine deoxyriboside employing GtPyNP + GtPNP at 65 °C (C) or TtPyNP + ApMTAP at 80 °C (D). Thymidine (2 mM) served as pentofuranosyl donor and 2,6-dichloropurine (2,6DCP) as acceptors (1 mM). Transglycosylations were performed in 2 mM sodium phosphate buffer, pH 6.5. Reactions were stopped after defined time intervals through the addition of 2 volumes of ice-cold buffer to 1 volume reaction mixture and further subsequent cooling.

112 Enzymatic transglycosylations with thermostable NPs
Future starting points for increasing the yield of 2,6-dihalogenated purine deoxyribosides
Due to the apparent lability of the intermediate product (2-deoxyribofuranosyl -α-1-phosphate) a
high temperature approach appears not to be suitable for the synthesis of the purine deoxyribosides
here under investigation and the reactions should be repeated employing DgPNP/GtPyNP at 55 °C.
For further optimization other factors that might have a positive effect on the stability of the
(intermediate) product might helpful to study (buffer composition, pH). Another option would be to
precisely determine when it would be best to stop the reaction. In the examples shown with GtPyNP
and GtPNP at 65 °C, it would thus appear appealing to stop the reaction after only 30 min, which
would result in approximately 70 % yield of nucleoside from the respective base. The use of
immobilized enzyme preparations could facilitate such an approach. A simple technical solution
could thus represent the batch-wise operation in a stirred tank reactor, in which the immobilized
enzymes are retained after product recovery, e.g. by a bottom stainless steel screen (Illanes and
Altamirano 2008). By the instant removal of the biocatalysts the presumably labile intermediate
product (deoxyribose-α-1-phosphate) would no longer be formed and the product concentration
should not decrease. A further advantage of this strategy would be that the biocatalyst could be
repeatedly used and product contamination with the biocatalysts would be avoided. It is also
conceivable to use immobilized biocatalysts in a continuous approach, for example in a stirred tank
reactor or in a packed column through which the substrate stream passes (Illanes and Altamirano
2008). However, continuous processes are usually selected for rather large scale productions and
hence the target production scale should be determined and considered for choosing the most
adequate reactor configuration.
5.4. Enzymatic transglycosylations– summary and conclusions
The results presented in this chapter have shown that the thermostable NPs were successfully
employed as biocatalysts for the enzymatic synthesis of both i) sugar modified purine nucleosides
(dAdo2′F, dAdo2′F)and for the synthesis of ii) purine nucleosides with modified purine base
(2,6-dihalogenated ribosides or deoxyribosides).
5.4.1. 2′-Fluorinated purine nucleosides
2′-Fluorinated adenine nucleosides (dAdo2′F, dAdo2′F) were synthesized via enzymatic
transglycosylation reactions employing 2′-fluorinated deoxyuridines (dUrd2′F, dUrd2′F) as
pentofuranosyl donors and adenine as pentofuranosyl acceptor. Different combinations of the
thermostable NPs that have been generated were investigated towards their suitability to catalyze
the reactions.
Synthesis of dAdo2′F
The combination of TtPyNP and DgPNP employed at 55 °C was found as most suitable fordAdo2′F
synthesis. Apart from the final yield, the increasing side product formation from dUrd2′F at higher
temperature was considered for the evaluation of enzyme combinations. With TtPyNP and DgPNP at

Enzymatic transglycosylations with thermostable NPs 113
55 °C, the final yield of dAdo2′F from adenine was 23 % after 1 day. The fact that the product
concentration increased in quasi linear relation to time indicates that the final outcome was limited
by the reaction rates of the enzymes. Therefore, higher enzyme loadings and prolonged reaction
times would presumably easily lead to higher yields. Moreover the ratio of the substrates and
enzymes in the reaction mixture could be optimized in future experiments.
The presented results should therefore be regarded as preliminary results, indicating the feasibility of
the approach to use thermostable enzymes for dAdo2′F synthesis. However, already at this stage the
results show that the efficiency of the TtPyNP/DgPNP enzyme preparation in the synthesis of dAdo2′F
is presumably higher than the efficiency of Lactobacillus reuteri NdRT that was recently described as
biocatalyst for the same reaction (Fernandez-Lucas et al. 2010).
Comparing the results to the data reported by Tuttle et al. (82 % yield with respect to dUrd2′F after 17
days), who have accomplished the transglycosylation reaction employing EcPNP and EcTP at
37 °C(Tuttle et al. 1993),it is difficult owing to the different reaction conditions that were used (molar
ratio of the substrates, different enzyme loading). In fact, the final yield reported by Tuttle is higher
than that obtained in the present study. However, taking into account that reaction time and enzyme
loading were both significantly higher, we can speculate that the combination of TtPyNP and DgPNP
at 55 °C has the potential to catalyze the same reaction with significantly higher efficiency. Future
investigations should include experiments with molar substrate ratios as used by Tuttle and co-
workers, higher enzyme loadings and longer reaction times. The results will then show whether the
use of TtPyNP/DgPNP is advantageous over the use of the E. coli enzymes.
Synthesis of dAdo2′F
The best conditions for dAdo2′F synthesis were found with TtPyNP/ApMTAP at 80 °C, which lead to
24 % yield after 1 day. Again the product concentration increased quasi linear in relation to time.
Hence, same measures as proposed for dAdo2′F synthesis (higher enzyme loading, prolonged reaction
times) would presumably also lead here to and improved final outcome.
However, already the preliminary results presented here indicate that the thermostable enzyme
preparation (TtPyNP+ ApMTAP) applied, seems to be more efficient than the combination of EcPNP
and EcTP used by Tuttle and Krenitsky in 1992 for the synthesis of 9-(2-deoxy-2-fluoro-β-D-
arabinofuranosyl)purines via enzymatic transglycosylation.
On the other hand, the advantage compared to the method developed by Yamada et al. (Yamada et
al. 2009), who have used GsPNP to catalyze the synthesis of dAdo2′F from 2-deoxy-2-
fluoroarabinofuranosyl-α-1-phosphate and adenine, is that the substrate used in the present study
(dUrd2′F) is presumably easier chemically prepared than 2-deoxy-2-fluoroarabinofuranosyl-α-1-
phosphate.
5.4.2. 2,6-Dihalogenated purine nucleosides
2,6-Dihalogenated purine nucleosides were synthesized via enzymatic transglycosylation reactions
employing either thymidine or uridine as pentofuranosyl donor and 2,6-dichloropurine or 6-chloro-2-

114 Enzymatic transglycosylations with thermostable NPs
fluoropurine as pentofuranosyl acceptor. Note that authentic samples of the products were not
available as reference and the identity of the products should hence be confirmed by adequate
analytical investigations in future experiments.
2,6-Dihalogenated purine ribosides
Different combinations of the thermostable NPs were investigated towards their suitability to
catalyze the synthesis of 2,6-dichloropurine riboside and 6-chloro-2-fluoropurine riboside via
enzymatic transglycosylations. The results indicate that the situation is very different from that
observed concerning the synthesis of 2′-fluorinated purine nucleosides. Obviously, the equilibrium of
the transglycosylation reaction was rather rapidly reached, with the consequence that the final
outcome evaluated within a 6 h period seems not to depend significantly on the choice of
temperatures and enzyme combination. Instead, rather the ratio of product and substrate
concentrations at equilibrium conditions seems to limit the final yield (56 % 2,6-DCPriboside,
63 - 67 % 6C2FP riboside with respect to purine base). Future optimization studies should therefore
target the equilibrium conditions rather than the enzyme loading and extended reaction times as
proposed for the synthesis of 2′-fluorinated nucleosides. Possible starting points include the pH and
the concentration of the phosphate buffer, as well as the ratio of pentofuranosyl donor and
acceptor. Furthermore, other ribofuranosyl donors that are essentially irreversibly phosphorolyzed
could be considered in order to favourably shift the equilibrium conditions.
2,6-Dihalogenated purine deoxyribosides
Within the experiments aiming at the synthesis of 2,6-dihalogenated purine deoxyribosides a totally
different obstacle was encountered. Apparently, the intermediate compound 2-deoxyribofuranosyl -
α-1-phosphate was not stable, leading to the exclusive formation of thymine and dihalogenated
purine after prolonged reaction times. Further optimizations should therefore aim on conditions that
are favourable for the stability of 2-deoxyribofuranosyl-α-1-phosphate, for example reduced reaction
temperatures, or different buffer compositions. Additionally, reactions should not proceed to long
but rather stopped after a defined period. This could be realized by making use of immobilized
enzymes. An advantage of this approach would be that the recovered biocatalysts could be re-used
and that a number of reactor configurations and operation modes would be conceivable.

6. Final conclusions
Key objectives of the present work have been accomplished and concern: i) the recombinant
expression of thermostable nucleoside phosphorylases (NPs) in E. coli, ii) the characterization of the
generated NPs with respect to potential biocatalytical applications and finally iii) the synthesis of
modified purine nucleosides (2′-fluorinated and 2,6-dihalogenated purine nucleosides) that are
difficult to access by chemical methods. In the following sections, the main conclusions that can be
drawn from the course of this study will be listed.
Recombinant expression of thermostable NPs in E. coli
Six NPs from thermophilic microorganisms have been selected for the recombinant
expression in E. coli: purine nucleoside phosphorylase (PNP) from Deinococcus geothermalis
and from Geobacillus thermoglucosidasius, 5'-Methylthioadenosine phosphorylase (MTAP)
and uridine phosphorylase (UP) from Aeropyrum pernix, and pyrimidine nucleoside
phosphorylase (PyNP) from Geobacillus thermoglucosidasius and Thermus thermophilus.
Except ApUP all enzymes have been successfully overexpressed in biologically active form in
E. coli. The expression vector confers a hexahistidine tag that is fused to the N-terminus and
contains an IPTG inducible lac promoter.
Soluble expression levels were very different, strongly depending on the target enzyme. The
expression of GtPNP and GtPyNP was very robust, resulting in high yields of soluble
recombinant enzyme and was relatively independent of the specific expression conditions
chosen. By contrast, expression of DgPNP and ApMTAP was slightly less efficient and more
prone to aggregation. Here, the expression and/or final cell density were optimized by fine-
tuning the inducer concentration and controlling the growth rate by employing medium with
enzyme-based glucose delivery. Expression of TtPyNP was more challenging and could not be
considerably improved. Most reproducible conditions were found by expression with
enzyme-based glucose delivery.
Initial expression studies have demonstrated the sensitivity of DgPNP towards N-terminal
fusion tags. We reasoned that excessive additional amino acids conferred by recombinational
cloning into a “destination” vector rendered PNP prone to aggregation. To avoid similar
problems with other NPs the respective expression construct was not further used and genes
were rather cloned by the conventional strategy involving restriction and ligation.
When native sequences of DgPNP and ApMTAP without any fusion tag were expressed, the
formation of stable secondary 5′ mRNA structures was found to impair the efficient
expression. The strategy to overcome the problem studied here, involved sequence
optimization and high-temperature cultivation (up to 42 °C) of the E. coli expression strains.
It seems likely that this strategy would also translate into better yields in the expression of
other thermostable proteins expressed without artificial N-terminal tag.

116 References
Characterization of thermostable NPs
Thermal properties of NPs varied according to the source microorganism and type of
enzyme. The following temperature optima were determined: 55 °C (DgPNP), 60 °C (GtPyNP)
and 70 °C (GtPNP). In case of TtPyNP and ApMTAP the reaction rate increased up to the
highest temperature tested. The degree of thermal stability correlated with the temperature
optima. DgPNP represents the least thermostable enzyme (t1/2 = 1.7 h at 60 °C) whereas
ApMTAP is the most thermostable NP here under investigation (t1/2 > 27 h at 90 °C).
As it is expected from the gene annotations the PyNPs readily catalyze the phosphorolytic
cleavage of uridine and thymidine, whereas the PNPs recognize both inosine and adenosine
as substrate, which is typical for bacterial type PNPs. The PNP substrate specificities are also
in agreement with the phylogenetic analysis that was based on amino acid alignments to
studied PNPs. ApMTAP is phylogenetically closely related to MTAPI from
Sulfolobus solfataricus and shows likewise similar substrate activities (accepting both inosine
and adenosine).
Unexpectedly, purine nucleosides (inosine, adenosine) were also recognized as substrates by
the PyNPs, even there was a clear preference for uridine and thymidine. This “PNP” activity
of enzymes with the NP-II fold is best to our knowledge first time described.
Cytidine was weakly recognized as substrate by PNPs and ApMTAP, whereas the cytidine
phosphorolysis rate determined for TtPyNP was significantly lower. For GtPyNP no substrate
activity of cytidine was seen.
The fluorination of the 2′-position of uridine leads to a dramatic decrease of substrate activity
for PyNPs, but is significantly better tolerated by TtPyNP than by GtPyNP. Fluorination of the
arabino position (dUrd2′F) is more critical, than the fluorination of the ribo position (dUrd2′F).
In fact, 2′-arabino fluorinated uridine is not recognized at all by GtPyNP as substrate.
In analogy, the phosphorolysis of adenosine analogues, fluorinated on the 2′ position, by NPs
with PNP activity (DgPNP, GtPNP, ApMTAP) was investigated. The data show that the highest
absolute phosphorolysis rates for both compounds (dAdo2′F and dAdo2′F) were obtained with
DgPNP. By contrast, GtPNP, that actually appears to be the most efficient catalyst for natural
purine nucleosides (inosine, adenosine) shows less activity than DgPNP for dAdo2′F and is the
weakest catalyst for dAdo2′F.
Kinetic parameters of PyNPs were determined for the natural substrates uridine and
thymidine at 60 °C (GtPyNP) and 80 °C (TtPyNP). In contrast to TtPyNP, GtPyNP is
characterized by extremely low substrate affinities (high Km values) towards both natural
substrates. The turnover numbers (kcat) of GtPyNP and TtPyNP are in similar range, with
uridine as substrate. By contrast, the turnover numbers for thymidine differ significantly, in
favour of thymidine phosphorolysis by TtPyNP. The kcat value of TtPyNP for thymidine is also
unusually high in comparison to EcTP, which demonstrates the potential for biocatalytic
applications.

Enzymatic transglycosylations with thermostable NPs 117
Enzymatic transglycosylations with thermostable NPs
The generated thermostable NPs were successfully applied in transglycosylation reactions
affording purine nucleoside analogues.
dAdo2′F was synthesized from dUrd2′F (pentofuranosyl donor) and adenine (pentofuranosyl
acceptor). The combination of TtPyNP and DgPNP applied at 55 °C was found to be the best
strategy for dAdo2′F synthesis. The operation at considerably higher temperatures proved to
be not feasible in reactions employing dUrd2'Fas substrate since dUrd2'F is relative labile,
reacting to unwanted side products at high temperature.
The synthesis of dAdo2'F was most efficiently catalyzed by ApMTAP + TtPyNP at 80 °C (from
dUrd2'F as donor and adenine as pentofuranosyl acceptor.
The efficient synthesis of sugar-fluorinated nucleosides was generally limited by the reaction
rates of the enzymes. Therefore it is expected that higher enzyme loadings and prolonged
incubation times will lead to higher yield. However, already the preliminary results presented
here, indicate that the use of the thermostable enzyme preparations instead of
conventionally used E. coli enzymes permits the operation at higher temperature and
translates into increased efficiencies of the respective transglycosylation reactions.
Both, 2,6-dichloropurine and 6-chloro-2-fluoropurine are well accepted as substrates by NPs
with PNP activity here under investigation (DgPNP, GtPNP, ApMTAP). The respective
ribosides can hence be readily prepared by employing uridine as pentofuranosyl donor and a
combination of PyNP and PNP (ApMTAP). In contrast to the synthesis of sugar-fluorinated
purine nucleosides, the final outcome (56 % to 67 % yield with respect to the purine base) is
not limited by the reaction rate of the enzymes, but rather by the conditions of the
equilibrium, that is rapidly established.
The synthesis of 2,6-dihalogenated deoxyribosides from thymidine as pentofuranosyl donor
and dihalogenated purine bases as pentofuranosyl acceptor is likewise efficiently catalyzed
by the thermostable NPs investigated in the present study. However, prolonged reaction
times lead to the exclusive formation of thymine and dihalogenated purine. This
phenomenon is accelerated at higher temperature and is presumably due to the instability of
the intermediate product deoxyribofuranosyl -α-1-phosphate.
In control experiments transglycosylation reactions were also performed by employing either
PyNP or PNP (ApMTAP) as sole biocatalyst. With the exclusive use of PNPs or ApMTAP as
biocatalyst, no product formation was observed. By contrast, owing to the PNP side activity
of the PyNPs, natural purine nucleosides (inosine, adenosine) were efficiently synthesized, by
applying PyNP as sole biocatalyst. However, this catalytic activity of the PyNP was not of
practical use for the synthesis of purine nucleoside analogues, since substrate activities of
modified purine nucleosides were very poor for PyNPs.

118 References
Outlook
In summary, the data demonstrate the promising potential of the investigated thermostable NPs for
applications aiming at the synthesis of unnatural nucleosides from chemically modified precursors.
Future experiments may be envisioned to be devoted to i) the optimization of the synthetic reactions
presented in this work and to ii) the use of the repertoire of thermostable NPs for the disclosure of
novel reactions.
A number of variables appear to be promising to study in regard to the optimization of the
transglycosylation reactions. Examples include the variation of substrate ratios (pentofuranosyl
donor versus acceptor), the buffer composition (concentration of phosphate ions, pH) and the
enzyme loading ratios. In order to better understand the nature of the reactions, it might
furthermore be of advantage if the reactions would be mathematically modelled. Such an approach
could also pave the way for in silico optimizations. Since purine bases are in general only poorly
soluble in aqueous solutions, and thermostability is often correlated to higher stability of enzymes in
the presence of organic solvents, it would furthermore be appealing to investigate enzymatic
transglycosylations in organic solvents. Finally the use of immobilized enzyme preparations would be
advantageous in regard to industrial applications.
Eventually it would be interesting to expand the repertoire of nucleoside products that can be
synthesized through the use of the generated NPs. After having shown here that both sugar-modified
and base modified purine nucleosides can be synthesized it would be conceivable to investigate the
synthesis of purine nucleosides that are modified on both moieties at the same time. Furthermore,
extensive substrate activity studies with chemically prepared precursors could reveal which
substrates are readily recognized and clarify the scope of feasible future applications.

References
Adler, A., Forster, N., Homann, M., and Goringer, H.U., 2008. "Post-SELEX chemical optimization of a trypanosome-specific RNA aptamer". Comb Chem High Throughput Screen, 11(1), 16-23
Agrawal, S., 2010. "Remembering Paul C. Zamecnik, M.D., "Father of Antisense" (1912-2009)". Oligonucleotides, 20(1), 47-50
Akashi, H., 1994. "Synonymous Codon Usage in Drosophila-Melanogaster - Natural-Selection and Translational Accuracy". Genetics, 136(3), 927-935
Akiyama, S., Furukawa, T., Sumizawa, T., Takebayashi, Y., Nakajima, Y., Shimaoka, S., and Haraguchi, M., 2004. "The role of thymidine phosphorylase, an angiogenic enzyme, in tumor progression". Cancer Sci, 95(11), 851-857
Alexeev, C.S., Panova, N.G., Polyakov, K.M., and Mikhailov, S.N., 2010. "Substrate specificity of E. coli uridine phosphorylase". Abstracts of XIX International Round Table on Nucleosides, Nucleotides and Nucleic Acids, IRT 2010(Lyon 2010), p. 94
Allerson, C.R., Sioufi, N., Jarres, R., Prakash, T.P., Naik, N., Berdeja, A., Wanders, L., Griffey, R.H., Swayze, E.E., and Bhat, B., 2005. "Fully 2'-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA". J Med Chem, 48(4), 901-904
Almendros, M., Gago, J.V.S., and Carlos, J.B., 2009. "Thermus thermophilus Strains Active in Purine Nucleoside Synthesis". Molecules, 14(3), 1279-1287
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J., 1990. "Basic local alignment search tool". J Mol Biol, 215(3), 403-10
Angelov, A., Mientus, M., Liebl, S., and Liebl, W., 2009. "A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles". Syst Appl Microbiol, 32(3), 177-85
Appleby, T.C., Mathews, I.I., Porcelli, M., Cacciapuoti, G., and Ealick, S.E., 2001. "Three-dimensional structure of a hyperthermophilic 5'-deoxy-5'-methylthioadenosine phosphorylase from Sulfolobus solfataricus". J Biol Chem, 276(42), 39232-39242
Baneyx, F., 1999. "Recombinant protein expression in Escherichia coli". Curr Opin Biotechnol, 10(5), 411-421
Baneyx, F. and Mujacic, M., 2004. "Recombinant protein folding and misfolding in Escherichia coli". Nat Biotechnol, 22(11), 1399-1408
Barai, V.N., Zinchenko, A.I., Eroshevskaya, L.A., Kalinichenko, E.N., Kulak, T.I., and Mikhailopulo, I.A., 2002. "A universal biocatalyst for the preparation of base- and sugar-modified nucleosides via an enzymatic transglycosylation". Helv Chim Acta, 85(7), 1901-1908
Bauta, W.E., Schulmeier, B.E., Burke, B., Puente, J.F., Cantrell, W.R., Lovett, D., Goebel, J., Anderson, B., Ionescu, D., and Guo, R.C., 2004. "A new process for antineoplastic agent clofarabine". Org Process Res Dev, 8(6), 889-896
Beeby, M., O'Connor, B.D., Ryttersgaard, C., Boutz, D.R., Perry, L.J., and Yeates, T.O., 2005. "The genomics of disulfide bonding and protein stabilization in thermophiles". PLoS Biol, 3(9), 1549-1558
Bellezza, I., Tucci, A., and Minelli, A., 2008. "2-Chloroadenosine and Human Prostate Cancer Cells". Anti-Cancer Agents Med Chem, 8(7), 783-789
Bennett, E.M., Li, C., Allan, P.W., Parker, W.B., and Ealick, S.E., 2003. "Structural basis for substrate specificity of Escherichia coli purine nucleoside phosphorylase". J Biol Chem, 278(47), 47110-47118
Berdis, A.J., 2008. "DNA polymerases as therapeutic targets". Biochemistry-Us, 47(32), 8253-8260 Bernstein, F.C., Koetzle, T.F., Williams, G.J., Meyer, E.F., Jr., Brice, M.D., Rodgers, J.R., Kennard, O.,
Shimanouchi, T., and Tasumi, M., 1977. "The Protein Data Bank: a computer-based archival file for macromolecular structures". J Mol Biol, 112(3), 535-542

120 References
Berrow, N.S., Bussow, K., Coutard, B., Diprose, J., Ekberg, M., Folkers, G.E., Levy, N., Lieu, V., Owens, R.J., Peleg, Y., Pinaglia, C., Quevillon-Cheruel, S., Salim, L., Scheich, C., Vincentelli, R., and Busso, D., 2006. "Recombinant protein expression and solubility screening in Escherichia coli: a comparative study". Acta Crystallogr D, 62(10), 1218-1226
Bonate, P.L., Arthaud, L., Cantrell, W.R., Jr., Stephenson, K., Secrist, J.A., 3rd, and Weitman, S., 2006. "Discovery and development of clofarabine: a nucleoside analogue for treating cancer". Nat Rev Drug Discov, 5(10), 855-863
Bordoli, L., Kiefer, F., Arnold, K., Benkert, P., Battey, J., and Schwede, T., 2009. "Protein structure homology modeling using SWISS-MODEL workspace". Nat Protoc, 4(1), 1-13
Brady, D., Jordaan, J., Simpson, C., Chetty, A., Arumugam, C., and Moolman, F.S., 2008. "Spherezymes: a novel structured self-immobilisation enzyme technology". BMC Biotechnol, 8(8),
Buckoreelall, K., Wilson, L., and Parker, W.B., 2011. "Identification and Characterization of Two Adenosine Phosphorylase Activities in Mycobacterium smegmatis". J Bacteriol, 193(20), 5668-5674
Bzowska, A., Kulikowska, E., and Shugar, D., 2000. "Purine nucleoside phosphorylases: properties, functions, and clinical aspects". Pharmacol Therapeut, 88(3), 349-425
Cacciapuoti, G., Porcelli, M., Bertoldo, C., Derosa, M., and Zappia, V., 1994. "Purification and Characterization of Extremely Thermophilic and Thermostable 5'-Methylthioadenosine Phosphorylase from the Archaeon Sulfolobus-Solfataricus - Purine Nucleoside Phosphorylase-Activity and Evidence for Intersubunit Disulfide Bonds". J Biol Chem, 269(40), 24762-24769
Cacciapuoti, G., Fusco, S., Caiazzo, N., Zappia, V., and Porcelli, M., 1999. "Heterologous expression of 5'-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus: characterization of the recombinant protein and involvement of disulfide bonds in thermophilicity and thermostability". Protein Expr Purif, 16(1), 125-135
Cacciapuoti, G., Bertoldo, C., Brio, A., Zappia, V., and Porcelli, M., 2003. "Purification and characterization of 5'-methylthioadenosine phosphorylase from the hyperthermophilic archaeon Pyrococcus furiosus - Substrate specificity and primary structure analysis". Extremophiles, 7(2), 159-168
Cacciapuoti, G., Moretti, M.A., Forte, S., Brio, A., Camardella, L., Zappia, V., and Porcelli, M., 2004. "Methylthioadenosine phosphorylase from the archaeon Pyrococcus furiosus - Mechanism of the reaction and assignment of disulfide bonds". Eur J Biochem, 271(23-24), 4834-4844
Cacciapuoti, G., Forte, S., Moretti, M.A., Brio, A., Zappia, V., and Porcelli, M., 2005. "A novel hyperthermostable 5'-deoxy-5'-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus". FEBS J, 272(8), 1886-1899
Cacciapuoti, G., Gorassini, S., Mazzeo, M.F., Siciliano, R.A., Carbone, V., Zappia, V., and Porcelli, M., 2007. "Biochemical and structural characterization of mammalian-like purine nucleoside phosphorylase from the Archaeon Pyrococcus furiosus". FEBS J, 274(10), 2482-2495
Cacciapuoti, G., Peluso, I., Fuccio, F., and Porcelli, M., 2009. "Purine nucleoside phosphorylases from hyperthermophilic Archaea require a CXC motif for stability and folding". FEBS J, 276(20), 5799-5805
Cacciapuoti, G., Marabotti, A., Fuccio, F., and Porcelli, M., 2011. "Unraveling the structural and functional differences between purine nucleoside phosphorylase and 5'-deoxy-5'-methylthioadenosine phosphorylase from the archaeon Pyrococcus furiosus". Bba-Proteins Proteom, 1814(10), 1358-1366
Cappellacci, L., Petrelli, R., Franchetti, P., Vita, P., Kusumanchi, P., Kumar, M., Jayaram, H.N., Zhou, B., Yen, Y., and Grifantini, M., 2011. "Synthesis and biological activity of novel N6-substituted and 2,N6-disubstituted adenine ribo- and 3'-C-methyl-ribonucleosides as antitumor agents". Eur J Med Chem, 46(5), 1499-1504
Care, S., Bignon, C., Pelissier, M.C., Blanc, E., Canard, B., and Coutard, B., 2008. "The translation of recombinant proteins in E. coli can be improved by in silico generating and screening random

References 121
libraries of a -70/+96 mRNA region with respect to the translation initiation codon". Nucleic Acids Res, 36(1), 1-6
Carson, D.A., Wasson, D.B., Kaye, J., Ullman, B., Martin, D.W., Jr., Robins, R.K., and Montgomery, J.A., 1980. "Deoxycytidine kinase-mediated toxicity of deoxyadenosine analogs toward malignant human lymphoblasts in vitro and toward murine L1210 leukemia in vivo". P Natl Acad Sci USA, 77(11), 6865-6869
Carson, D.A. and Wasson, D.B., 1988. "Synthesis of 2',3'-Dideoxynucleosides by Enzymatic Trans-Glycosylation". Biochem Bioph Res Co, 155(2), 829-834
Cassera, M.B., Hazleton, K.Z., Merino, E.F., Obaldia, N., 3rd, Ho, M.C., Murkin, A.S., DePinto, R., Gutierrez, J.A., Almo, S.C., Evans, G.B., Babu, Y.S., and Schramm, V.L., 2011. "Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model". PLoS One, 6(11), e26916
Cèbe, R. and Geiser, M., 2006. "Rapid and easy thermodynamic optimization of the 5'-end of mRNA dramatically increases the level of wild type protein expression in Escherichia coli". Protein Expres Purif, 45(2), 374-380
Chen, J., Acton, T.B., Basu, S.K., Montelione, G.T., and Inouye, M., 2002. "Enhancement of the solubility of proteins overexpressed in Escherichia coli by heat shock". J Mol Microbiol Biotechnol, 4(6), 519-524
Cieslinski, H., Dlugolecka, A., Kur, J., and Turkiewicz, M., 2009. "An MTA phosphorylase gene discovered in the metagenomic library derived from Antarctic top soil during screening for lipolytic active clones confers strong pink fluorescence in the presence of rhodamine B". FEMS Microbiol Lett, 299(2), 232-240
Codington, J.F., Fox, J.J., and Doerr, I.L., 1964. "Nucleosides. XVIII. Synthesis of 2'-Fluorothymidine, 2'-Fluorodeoxyuridine, and Other 2'-Halogeno-2'-Deoxy Nucleosides". J Org Chem, 29(3), 558-564
Condezo, L.A., Fernández-Lucas, J., García-Burgos, C.A., Alcántara, A.R., and Sinisterra, J.V., "Enzymatic Synthesis of Modified Nucleosides", in "Biocatalysts in the Pharmaceutical and Biotechnology Industries", R.N. Patel, Editor. 2006, Crc Press, Boca Raton. p. 403-426.
Copeland, R.A., "Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis". Second ed. 2000, New York: Wiley-VCH.
Cowan, D.A., Arslanoglu, A., Burton, S.G., Baker, G.C., Cameron, R.A., Smith, J.J., and Meyer, Q., 2004. "Metagenomics, gene discovery and the ideal biocatalyst". Biochem Soc T, 32(Pt 2), 298-302
Daddona, P.E., Wiesmann, W.P., Milhouse, W., Chern, J.W., Townsend, L.B., Hershfield, M.S., and Webster, H.K., 1986. "Expression of Human Malaria Parasite Purine Nucleoside Phosphorylase in Host Enzyme-Deficient Erythrocyte Culture - Enzyme Characterization and Identification of Novel Inhibitors". J Biol Chem, 261(25), 1667-1673
Damha, M.J., Wilds, C.J., Noronha, A., Brukner, I., Borkow, G., Arion, D., and Parniak, M.A., 1998. "Hybrids of RNA and arabinonucleic acids (ANA and 2'F-ANA) are substrates of ribonuclease H". J Am Chem Soc, 120(49), 12976-12977
De Clercq, E., 2004. "Antiviral drugs in current clinical use". J Clin Virol, 30(2), 115-133 de Marco, A., 2009. "Strategies for successful recombinant expression of disulfide bond-dependent
proteins in Escherichia coli". Microb Cell Fact, 8(26), de Smit, M.H. and van Duin, J., 1990. "Secondary structure of the ribosome binding site determines
translational efficiency: A quantitative analysis". P Natl Acad Sci 87(19), 7668-7672 de Smit, M.H. and van Duin, J., 1994. "Control of translation by mRNA secondary structure in
Escherichia coli. A quantitative analysis of literature data". J Mol Biol, 244(2), 144-150 Deleavey, G.F., Watts, J.K., Alain, T., Robert, F., Kalota, A., Aishwarya, V., Pelletier, J., Gewirtz, A.M.,
Sonenberg, N., and Damha, M.J., 2010. "Synergistic effects between analogs of DNA and RNA improve the potency of siRNA-mediated gene silencing". Nucleic Acids Res, 38(13), 4547-4557
Dessanti, P., Zhang, Y., Allegrini, S., Tozzi, M.G., Sgarrella, F., and Ealick, S.E., 2012. "Structural basis of the substrate specificity of Bacillus cereus adenosine phosphorylase". Acta Crystallogr D, 68(Pt 3), 239-248

122 References
Dobak, I., Ostafin, M., Poleshchuk, O.K., Milecki, J., and Nogaj, B., 2008. "Reactivity of 2,6-Dichloropurine Ribonucleoside studied by 35Cl NQR Spectroscopy". Appl Magn Reson 34(1-2), 47-53
Dong, H.J., Nilsson, L., and Kurland, C.G., 1996. "Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates". J Mol Biol, 260(5), 649-663
Donovan, R.S., Robinson, C.W., and Glick, B.R., 1996. "Review: Optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter". J Ind Microbiol, 16(3), 145-154
Esipov, R.S., Gurevich, A.I., Chuvikovsky, D.V., Chupova, L.A., Muravyova, T.I., and Miroshnikov, A.I., 2002. "Overexpression of Escherichia coli genes encoding nucleoside phosphorylases in the pET/Bl21 (DE3) system yields active recombinant enzymes". Protein Expres Purif, 24(1), 56-60
Esposito, D. and Chatterjee, D.K., 2006. "Enhancement of soluble protein expression through the use of fusion tags". Curr Opin Biotech, 17(4), 353-358
Fariselli, P. and Casadio, R., 1999. "A neural network based predictor of residue contacts in proteins". Protein Eng, 12(1), 15-21
Fernandez-Lucas, J., Acebal, C., Sinisterra, J.V., Arroyo, M., and de la Mata, I., 2010. "Lactobacillus reuteri 2'-Deoxyribosyltransferase, a Novel Biocatalyst for Tailoring of Nucleosides". Appl Environ Microb, 76(5), 1462-1470
Ferreira, A.C., Nobre, M.F., Rainey, F.A., Silva, M.T., Wait, R., Burghardt, J., Chung, A.P., and da Costa, M.S., 1997. "Deinococcus geothermalis sp. nov. and Deinococcus murrayi sp. nov., two extremely radiation-resistant and slightly thermophilic species from hot springs". Int J Syst Bacteriol, 47(4), 939-947
Fire, A., Xu, S.Q., Montgomery, M.K., Kostas, S.A., Driver, S.E., and Mello, C.C., 1998. "Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans". Nature, 391(6669), 806-811
Fiser, A., Cserzo, M., Tudos, E., and Simon, I., 1992. "Different Sequence Environments of Cysteines and Half Cystines in Proteins Application to Predict Disulfide Forming Residues". FEBS Lett, 302(2), 117-120
Flexner, C., 2007. "HIV drug development: the next 25 years". Nat Rev Drug Discov, 6(12), 959-966 Furman, P.A., Fyfe, J.A., Stclair, M.H., Weinhold, K., Rideout, J.L., Freeman, G.A., Lehrman, S.N.,
Bolognesi, D.P., Broder, S., Mitsuya, H., and Barry, D.W., 1986. "Phosphorylation of 3'-Azido-3'-Deoxythymidine and Selective Interaction of the 5'-Triphosphate with Human-Immunodeficiency-Virus Reverse-Transcriptase". P Natl Acad Sci USA, 83(21), 8333-8337
Furukawa, T., Yoshimura, A., Sumizawa, T., Haraguchi, M., Akiyama, S., Fukui, K., Ishizawa, M., and Yamada, Y., 1992. "Angiogenic Factor". Nature, 356(6371), 668-668
Gao, X.F., Huang, X.R., and Sun, C.C., 2006. "Role of each residue in catalysis in the active site of pyrimidine nucleoside phosphorylase from Bacillus subtilis: A hybrid QM/MM study". J Struct Biol, 154(1), 20-26
Gerdes, K., 1988. "The ParB (hok sok) Locus of Plasmid R1: a General Purpose Plasmid Stabilization System". Bio-Technol, 6(12), 1402-1405
Giblett, E.R., Ammann, A.J., Sandman, R., Wara, D.W., and Diamond, L.K., 1975. "Nucleoside-Phosphorylase Deficiency in a Child with Severely Defective T-Cell Immunity and Normal B-Cell Immunity". Lancet, 1(7914), 1010-1013
Gordon, G.E.R., Visser, D.F., Brady, D., Raseroka, N., and Bode, M.L., 2011. "Defining a process operating window for the synthesis of 5-methyluridine by transglycosylation of guanosine and thymine". J Biotechnol, 151(1), 108-113
Gragoudas, E.S., Adamis, A.P., Cunningham, E.T., Feinsod, M., Guyer, D.R., and Neova, V.I.S.O., 2004. "Pegaptanib for neovascular age-related macular degeneration". New Engl J Med, 351(27), 2805-2816
Green, E.A., Rosenstein, R.D., Shiono, R., Abraham, D.J., Trus, B.L., and Marsh, R.E., 1975. "Crystal-Structure of Uridine". Acta Crystallogr B, 31(Jan15), 102-107
Grillone, L.R. and Lanz, R., 2001. "Fomivirsen". Drugs Today, 37(4), 245-255

References 123
Griswold, K.E., Mahmood, N.A., Iverson, B.L., and Georgiou, G., 2003. "Effects of codon usage versus putative 5'-mRNA structure on the expression of Fusarium solani cutinase in the Escherichia coli cytoplasm". Protein Expr Purif, 27(1), 134-142
Gryaznov, S.M., Lloyd, D.H., Chen, J.K., Schulz, R.G., Dedionisio, L.A., Ratmeyer, L., and Wilson, W.D., 1995. "Oligonucleotide N3'-->P5' Phosphoramidates". P Natl Acad Sci USA, 92(13), 5798-5802
Gryaznov, S.M., 1999. "Oligonucleotide N3'-->P5' phosphoramidates as potential therapeutic agents". Biochim Biophys Acta, 1489(1), 131-140
Gryaznov, S.M. and Schultz, R.G., 2011. "2'-Arabino-Fluorooligonucleotide N3'-P5' Phosphoramidates: Their synthesis and use", US 2011/0294213 A1 (US) C12N 5/09, C07H 19/20, G01N 33/53, C070 21/02, C070 21/04, C070 19/04, C070 19/10
Gu, W., Zhou, T., and Wilke, C.O., 2010. "A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes". PLoS Comput Biol, 6(2), e1000664
Guan, R., Ho, M.C., Almo, S.C., and Schramm, V.L., 2011. "Methylthioinosine Phosphorylase from Pseudomonas aeruginosa. Structure and Annotation of a Novel Enzyme in Quorum Sensing". Biochemistry-Us, 50(7), 1247-1254
Gura, T., 1995. "Antisense Has Growing Pains". Science, 270(5236), 575-577 Hagmann, W.K., 2008. "The many roles for fluorine in medicinal chemistry". J Med Chem, 51(15),
4359-4369 Hajdo, L., Szulc, A.B., Klajnert, B., and Bryszewska, M., 2010. "Metabolic Limitations of the Use of
Nucleoside Analogs in Cancer Therapy May Be Overcome by Application of Nanoparticles as Drug Carriers: A Review". Drug Develop Res, 71(7), 383-394
Haki, G.D. and Rakshit, S.K., 2003. "Developments in industrially important thermostable enzymes: a review". Bioresource Technol, 89(1), 17-34
Hamamoto, T., Noguchi, T., and Midorikawa, Y., 1996. "Purification and characterization of purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus TH 6-2". Biosci Biotech Bioch, 60(7), 1179-1180
Hamamoto, T., Noguchi, T., and Midorikawa, Y., 1997a. "Cloning of purine nucleoside phosphorylase II gene from Bacillus stearothermophilus TH 6-2 and characterization of its gene product". Biosci Biotech Bioch, 61(2), 276-280
Hamamoto, T., Okuyama, K., Noguchi, T., and Midorikawa, Y., 1997b. "Cloning and expression of purine nucleoside phosphorylase I gene from Bacillus stearothermophilus TH 6-2". Biosci Biotech Bioch, 61(2), 272-275
Hammer-Jespersen, K. and Munch-Ptersen, A., 1975. "Multiple regulation of nucleoside catabolizing enzymes: regulation of the deo operon by the cytR and deoR gene products". Mol Gen Genet, 137(4), 327-335
Hammond, D.J. and Gutteridge, W.E., 1984. "Purine and Pyrimidine Metabolism in the Trypanosomatidae". Mol Biochem Parasit, 13(3), 243-261
Hammond, S.M., Bernstein, E., Beach, D., and Hannon, G.J., 2000. "An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells". Nature, 404(6775), 293-296
Hatahet, F., Nguyen, V.D., Salo, K.E., and Ruddock, L.W., 2010. "Disruption of reducing pathways is not essential for efficient disulfide bond formation in the cytoplasm of E. coli". Microb Cell Fact, 967
Hempel, A., Camerman, N., Grierson, J., Mastropaolo, D., and Camerman, A., 1999. "FF-β-D-Arabinofuranosyluracil". Acta Crystallogr C, 55632-633
Hénaut, A. and Danchin, A., "Analysis and Predictions from Escherichia coli sequences", in "Escherichia coli and Salmonella typhimurium cellular and molecular biology", F. Neidhardt, et al., Editors. 1996, ASM press, Washington, DC. p. 2047 -2066.
Henne, A., Bruggemann, H., Raasch, C., Wiezer, A., Hartsch, T., Liesegang, H., Johann, A., Lienard, T., Gohl, O., Martinez-Arias, R., Jacobi, C., Starkuviene, V., Schlenczeck, S., Dencker, S., Huber, R., Klenk, H.P., Kramer, W., Merkl, R., Gottschalk, G., and Fritz, H.J., 2004. "The genome sequence of the extreme thermophile Thermus thermophilus". Nat Biotechnol, 22(5), 547-553

124 References
Hennen, W.J. and Wong, C.H., 1989. "A New Method for the Enzymatic-Synthesis of Nucleosides Using Purine Nucleoside Phosphorylase". J Org Chem, 54(19), 4692-4695
Hidalgo, A., Betancor, L., Moreno, R., Zafra, O., Cava, F., Fernandez-Lafuente, R., Guisan, J.M., and Berenguer, J., 2004. "Thermus thermophilus as a cell factory for the production of a thermophilic Mn-dependent catalase which fails to be synthesized in an active form in Escherichia coli". Appl Environ Microbiol, 70(7), 3839-3844
Hocek, M. and Dvorakova, H., 2003. "An efficient synthesis of 2-substituted 6-methylpurine bases and nucleosides by Fe- or Pd-catalyzed cross-coupling reactions of 2,6-dichloropurines". J Org Chem, 68(14), 5773-5776
Holguin, J. and Cardinaud, R., 1975. "Trans-N-Deoxyribosylase - Purification by Affinity Chromatography and Characterization". Eur J Biochem, 54(2), 505-514
Hori, N., Watanabe, M., Yamazaki, Y., and Mikami, Y., 1989a. "Purification and Characterization of 2nd Thermostable Purine Nucleoside Phosphorylase in Bacillus-Stearothermophilus Jts-859". Agr Biol Chem Tokyo, 53(12), 3219-3224
Hori, N., Watanabe, M., Yamazaki, Y., and Mikami, Y., 1989b. "Purification and Characterization of Thermostable Purine Nucleoside Phosphorylase of Bacillus-Stearothermophilus Jts-859". Agr Biol Chem Tokyo, 53(8), 2205-2210
Hori, N., Watanabe, M., Yamazaki, Y., and Mikami, Y., 1990. "Purification and Characterization of Thermostable Pyrimidine Nucleoside Phosphorylase from Bacillus-Stearothermophilus Jts-859". Agr Biol Chem Tokyo, 54(3), 763-768
Hori, N., Watanabe, M., Sunagawa, K., Uehara, K., and Mikami, Y., 1991. "Production of 5-methyluridine by immobilized thermostable purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus JTS 859". J Biotechnol, 17(2), 121-131
Howell, H.G., Brodfuehrer, P.R., Brundidge, S.P., Benigni, D.A., and Sapino, C., 1988. "Antiviral Nucleosides - a Stereospecific, Total Synthesis of 2'-Fluoro-2'-Deoxy-β-D-Arabinofuranosyl Nucleosides". J Org Chem, 53(1), 85-88
Huang, M.C., Montgomery, J.A., Thorpe, M.C., Stewart, E.L., Secrist, J.A., and Blakley, R.L., 1983. "Formation of 3-(2'-Deoxyribofuranosyl) and 9-(2'-Deoxyribofuranosyl) Nucleosides of 8-Substituted Purines by Nucleoside Deoxyribosyltransferase". Arch Biochem Biophys, 222(1), 133-144
Ikemura, T., 1981. "Correlation between the Abundance of Escherichia coli Transfer-RNAs and the Occurrence of the Respective Codons in Its Protein Genes - a Proposal for a Synonymous Codon Choice That Is Optimal for the Escherichia coli Translational System". J Mol Biol, 151(3), 389-409
Ishii, M., Shirae, H., and Yokozeki, K., 1989. "Enzymatic Production of 5-Methyluridine from Purine Nucleosides and Thymine by Erwinia carotovora AJ 2992". Agr Biol Chem Tokyo, 53(12), 3209-3218
Ivanov, I., Alexandrova, R., Dragulev, B., Saraffova, A., and AbouHaidar, M.G., 1992. "Effect of tandemly repeated AGG triplets on the translation of CAT-mRNA in E. coli". FEBS Lett, 307(2), 173-176
Jensen, K.F. and Nygaard, P., 1975. "Purine Nucleoside Phosphorylase from Escherichia coli and Salmonella typhimurium - Purification and Some Properties". Eur J Biochem, 51(1), 253-265
Jensen, K.F., 1978. "Two purine nucleoside posphorylases in Bacillus subtilis. Purification and some properties of adenosine-specific phosphorylase". BBA-Enzymol, 525(2), 346-356
Jin, H., Zhao, Q., Gonzalez de Valdivia, E.I., Ardell, D.H., Stenstrom, M., and Isaksson, L.A., 2006. "Influences on gene expression in vivo by a Shine-Dalgarno sequence". Mol Microbiol, 60(2), 480-492
Jung, S.T., Kang, T.H., and Georgiou, G., 2010. "Efficient expression and purification of human aglycosylated Fcgamma receptors in Escherichia coli". Biotechnol Bioeng, 107(1), 21-30
Kalckar, H.M., 1947. "Differential spectrophotometry of purine compounds by means of specific enzymes; studies of the enzymes of purine metabolism". J Biol Chem, 167(2), 461-475

References 125
Kalota, A., Karabon, L., Swider, C.R., Viazovkina, E., Elzagheid, M., Damha, M.J., and Gewirtz, A.M., 2006. "2'-Deoxy-2'-fluoro-β-D-arabinonucleic acid (2'F-ANA) modified oligonucleotides (ON) effect highly efficient, and persistent, gene silencing". Nucleic Acids Res, 34(2), 451-461
Kaminski, P.A., 2002. "Functional cloning, heterologous expression, and purification of two different N-deoxyribosyltransferases from Lactobacillus helveticus". J Biol Chem, 277(17), 14400-14407
Kane, J.F., 1995. "Effects of Rare Codon Clusters on High-Level Expression of Heterologous Proteins in Escherichia coli". Curr Opin Biotech, 6(5), 494-500
Kaushik, J.K., Ogasahara, K., and Yutani, K., 2002. "The Unusually Slow Relaxation Kinetics of the Folding-unfolding of Pyrrolidone Carboxyl Peptidase from a Hyperthermophile, Pyrococcus furiosus". J Mol Biol, 316(4), 991-1003
Kawarabayasi, Y., Hino, Y., Horikawa, H., Yamazaki, S., Haikawa, Y., Jin-no, K., Takahashi, M., Sekine, M., Baba, S., Ankai, A., Kosugi, H., Hosoyama, A., Fukui, S., Nagai, Y., Nishijima, K., Nakazawa, H., Takamiya, M., Masuda, S., Funahashi, T., Tanaka, T., Kudoh, Y., Yamazaki, J., Kushida, N., Oguchi, A., Kikuchi, H., and et al., 1999. "Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1". DNA Res, 6(2), 83-101, 145-52
Kawasaki, A.M., Casper, M.D., Freier, S.M., Lesnik, E.A., Zounes, M.C., Cummins, L.L., Gonzalez, C., and Cook, P.D., 1993. "Uniformly Modified 2'-Deoxy-2'-Fluoro Phosphorothioate Oligonucleotides as Nuclease-Resistant Antisense Compounds with High-Affinity and Specificity for RNA Targets". J Med Chem, 36(7), 831-841
Kazimierczuk, Z., Cottam, H.B., Revankar, G.R., and Robins, R.K., 1984. "Synthesis of 2'-Deoxytubercidin, 2'-Deoxyadenosine, and Related 2'-Deoxynucleosides Via a Novel Direct Stereospecific Sodium-Salt Glycosylation Procedure". J Am Chem Soc, 106(21), 6379-6382
Khan, M.A., Sadaf, S., and Akhtar, M.W., 2007. "Role of Silent Gene Mutations in the Expression of Caprine Growth Hormone in Escherchia coli". Biotechnol Progr, 23(5), 1049-1052
Khati, M., Schuman, M., Ibrahim, J., Sattentau, Q., Gordon, S., and James, W., 2003. "Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2'F-RNA aptamers". J Virol, 77(23), 12692-12698
Koch, A.L. and Vallee, G., 1958. "The Properties of Adenosine Deaminase and Adenosine Nucleoside Phosphorylase in Extracts of Escherichia coli". J Biol Chem, 234(5), 1213-1218
Koma, D., Sawai, T., Harayama, S., and Kino, K., 2006. "Overexpression of the genes from thermophiles in Escherichia coli by high-temperature cultivation". Appl Microbiol Biotechnol, 73(1), 172-180
Koszalka, G.W., Vanhooke, J., Short, S.A., and Hall, W.W., 1988. "Purification and Properties of Inosine-Guanosine Phosphorylase from Escherichia Coli K12". J Bacteriol, 170(8), 3493-3498
Krahe, M., Antranikian, G., and Märkl, H., 1996. "Fermentation of extremophilic microorganisms". FEMS Microbiol Rev, 18271-285
Krause, M., Ukkonen, K., Haataja, T., Ruottinen, M., Glumoff, T., Neubauer, A., Neubauer, P., and Vasala, A., 2010. "A novel fed-batch based cultivation method provides high cell-density and improves yield of soluble recombinant proteins in shaken cultures". Microb Cell Fact, 9
Krenitsky, T.A., Koszalka, G.W., and Tuttle, J.V., 1981. "Purine nucleoside synthesis, an efficient method employing nucleoside phosphorylases". Biochemistry-Us, 20(12), 3615-3621
Krenitsky, T.A. and Tuttle, J.V., 1982. "Correlation of Substrate-Stabilization Patterns with Proposed Mechanisms for 3 Nucleoside Phosphorylases". BBA-Protein Struct M, 703(2), 247-249
Krzeminski, J., Nawrot, B., Pankiewicz, K.W., and Watanabe, K.A., 1991. "Nucleosides .157. Synthesis of 9-(2-Deoxy-2-Fluoro-β-D-Arabinofuranosyl)Hypoxanthine - the first Direct Introduction of a 2'-β-Fluoro Substituent in Preformed Purine Nucleosides - Studies Directed toward the Synthesis of 2'-Deoxy-2'-Substituted Arabinonucleosides ". Nucleos Nucleot, 10(4), 781-798
Kudla, G., Murray, A.W., Tollervey, D., and Plotkin, J.B., 2009. "Coding-sequence determinants of gene expression in Escherichia coli". Science, 324(5924), 255-258
Kurland, C. and Gallant, J., 1996. "Errors of heterologous protein expression". Curr Opin Biotech, 7(5), 489-493

126 References
Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J., and Higgins, D.G., 2007. "Clustal W and Clustal X version 2.0". Bioinformatics, 23(21), 2947-2948
Larson, E.T., Mudeppa, D.G., Gillespie, J.R., Mueller, N., Napuli, A.J., Arif, J.A., Ross, J., Arakaki, T.L., Lauricella, A., Detitta, G., Luft, J., Zucker, F., Verlinde, C.L., Fan, E., Van Voorhis, W.C., Buckner, F.S., Rathod, P.K., Hol, W.G., and Merritt, E.A., 2010. "The crystal structure and activity of a putative trypanosomal nucleoside phosphorylase reveal it to be a homodimeric uridine phosphorylase". J Mol Biol, 396(5), 1244-59
Lewkowicz, E.S., Martínez, N., Rogert, M.C., Porro, S., and Iribarren, A.M., 2000. "An improved microbial synthesis of purine nucleosides". Biotechnol Lett, 221277-1280
Li, X., Jiang, X., Li, H., and Ren, D., 2008. "Purine nucleoside phosphorylase from Pseudoalteromonas sp. Bsi590: molecular cloning, gene expression and characterization of the recombinant protein". Extremophiles, 12(3), 325-333
Ling, F., Inoue, Y., and Kimura, A., 1990. "Purification and Characterization of a Novel Nucleoside Phosphorylase from a Klebsiella Sp and Its Use in the Enzymatic Production of Adenine-Arabinoside". Appl Environ Microb, 56(12), 3830-3834
Liu, P., Sharon, A., and Chu, C.K., 2008. "Fluorinated Nucleosides: Synthesis and Biological Implication". J Fluor Chem, 129(9), 743-766
Liu, X., Kantarjian, H., and Plunkett, W., 2012. "Sapacitabine for cancer". Expert Opin Investig Drugs, 21(4), 541-555
Lowenthal, R.M. and Eaton, K., 1996. "Toxicity of chemotherapy". Hematol Oncol Clin North Am, 10(4), 967-990
Mahmoudian, M., Rudd, B.A.M., Cox, B., Drake, C.S., Hall, R.M., Stead, P., Dawson, M.J., Chandler, M., Livermore, D.G., Turner, N.J., and Jenkins, G., 1998. "A versatile procedure for the generation of nucleoside 5'-carboxylic acids using nucleoside oxidase". Tetrahedron, 54(28), 8171-8182
Mahmoudian, M., 2000. "Biocatalytic production of chiral pharmaceutical intermediates". Biocatal Biotransfor, 18105-118
Manoharan, M., Akinc, A., Pandey, R.K., Qin, J., Hadwiger, P., John, M., Mills, K., Charisse, K., Maier, M.A., Nechev, L., Greene, E.M., Pallan, P.S., Rozners, E., Rajeev, K.G., and Egli, M., 2011. "Unique Gene-Silencing and Structural Properties of 2'-Fluoro-Modified siRNAs". Angew Chem Int Edit, 50(10), 2284-2288
Mao, C., Cook, W.J., Zhou, M., Koszalka, G.W., Krenitsky, T.A., and Ealick, S.E., 1997. "The crystal structure of Escherichia coli purine nucleoside phosphorylase: a comparison with the human enzyme reveals a conserved topology". Structure, 5(10), 1373-1383
Marck, C., Lesyng, B., and Saenger, W., 1982. "The Crystal-Structures of 2'-Deoxy-2'-Fluorocytidine and 2'-Deoxy-2'-Fluorouridine". J Mol Struct, 82(1-2), 77-94
Martinez, J., Patkaniowska, A., Urlaub, H., Luhrmann, R., and Tuschl, T., 2002. "Single-stranded antisense siRNAs guide target RNA cleavage in RNAi". Cell, 110(5), 563-574
Maruyama, T., Takamatsu, S., Kozai, S., Satoh, Y., and Izawa, K., 1999. "Synthesis of 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)adenine bearing a selectively removable protecting group". Chem Pharm Bull, 47(7), 966-970
Mcelwain, M.C., Williams, M.V., and Pollack, J.D., 1988. "Acholeplasma-Laidlawii B-Pg9 Adenine-Specific Purine Nucleoside Phosphorylase That Accepts Ribose-1-Phosphate, Deoxyribose-1-Phosphate, and Xylose-1-Phosphate". J Bacteriol, 170(2), 564-567
McNicholas, S., Potterton, E., Wilson, K.S., and Noble, M.E., 2011. "Presenting your structures: the CCP4mg molecular-graphics software". Acta Crystallogr D, 67(Pt 4), 386-94
Mendieta, J., Martin-Santamaria, S., Priego, E.M., Balzarini, J., Camarasa, M.J., Perez-Perez, M.J., and Gago, F., 2004. "Role of histidine-85 in the catalytic mechanism of thymidine phosphorylase as assessed by targeted molecular dynamics simulations and quantum mechanical calculations". Biochemistry-Us, 43(2), 405-414
Mikhailopulo, I.A., 2007. "Biotechnology of Nucleic Acid Constituents - State of the Art and Perspectives". Curr Org Chem, 11(4), 317-335

References 127
Mikhailopulo, I.A. and Miroshnikov, A.I., 2011. "Biologically important nucleosides: modern trends in biotechnology and application". Mendeleev Commun, 21(2), 57-68
Mitsuya, H., Weinhold, K.J., Furman, P.A., Stclair, M.H., Lehrman, S.N., Gallo, R.C., Bolognesi, D., Barry, D.W., and Broder, S., 1985. "3'-Azido-3'-Deoxythymidine (Bw A509u) - an Antiviral Agent That Inhibits the Infectivity and Cytopathic Effect of Human Lymphotropic-T Virus Type-Iii Lymphadenopathy-Associated Virus Invitro". P Natl Acad Sci USA, 82(20), 7096-7100
Monia, B.P., Lesnik, E.A., Gonzalez, C., Lima, W.F., Mcgee, D., Guinosso, C.J., Kawasaki, A.M., Cook, P.D., and Freier, S.M., 1993. "Evaluation of 2'-Modified Oligonucleotides Containing 2'-Deoxy Gaps as Antisense Inhibitors of Gene-Expression". J Biol Chem, 268(19), 14514-14522
Montgomery, J.A., Shortnacy, A.T., Carson, D.A., and Secrist, J.A., 1986. "9-(2-Deoxy-2-Fluoro-FF-β-D-Arabinofuranosyluracil-D-Arabinofuranosyl)Guanine - a Metabolically Stable Cytotoxic Analog of 2'-Deoxyguanosine". J Med Chem, 29(11), 2389-2392
Montgomery, J.A., Shortnacy-Fowler, A.T., Clayton, S.D., Riordan, J.M., and Secrist, J.A., 3rd, 1992. "Synthesis and biologic activity of 2'-fluoro-2-halo derivatives of 9-β-D-arabinofuranosyladenine". J Med Chem, 35(2), 397-401
Montilla Arevalo, R., Deroncelé Thomas, V.M., López Gómez, C., Pascual Gilabert, M., Estévez Company, C., and Castells Boliart, J., 2011. "Thermostable biocatalyst combination for nucleoside synthesis", EP 2 338 985 A1 (Institut Univ. de Ciència i Tecnologia, Barcelona, ES) C12N 9/10, C12P 19/38
Moore, D. and Dowhan, D., "Purification and concentration of DNA from aqueous solutions", in "Current protocols in molecular biology", F.M. Ausubel, et al., Editors. 2002, John Wiley & Sons, Houston. p. Unit 2.1A.
Moris, F. and Gotor, V., 1993. "A Useful and Versatile Procedure for the Acylation of Nucleosides through an Enzymatic-Reaction". J Org Chem, 58(3), 653-660
Morrissey, D.V., Lockridge, J.A., Shaw, L., Blanchard, K., Jensen, K., Breen, W., Hartsough, K., Machemer, L., Radka, S., Jadhav, V., Vaish, N., Zinnen, S., Vargeese, C., Bowman, K., Shaffer, C.S., Jeffs, L.B., Judge, A., MacLachlan, I., and Polisky, B., 2005. "Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs". Nat Biotechnol, 23(8), 1002-1007
Müller, K., Faeh, C., and Diederich, F., 2007. "Fluorine in pharmaceuticals: looking beyond intuition". Science, 317(5846), 1881-1886
Murphy, E.L., Collier, A.C., Kalish, L.A., Assmann, S.F., Para, M.F., Flanigan, T.P., Kumar, P.N., Mintz, L., Wallach, F.R., Nemo, G.J., and Study, V.A.T., 2001. "Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease". Ann Intern Med, 135(1), 17-26
Na, D., Lee, S., and Lee, D., 2010. "Mathematical modeling of translation initiation for the estimation of its efficiency to computationally design mRNA sequences with desired expression levels in prokaryotes". BMC Systems Biology, 4(1), 71
Nair, V., Adah, S.A., and Ha, S.B., 1995. "Strategically Functionalized Adenosines: Agonists for Adenosine Receptors". Nucleos Nucleot Nucl, 14(3-5), 537-553
Narberhaus, F., Waldminghaus, T., and Chowdhury, S., 2006. "RNA thermometers". FEMS Microbiology Reviews, 30(1), 3-16
Nazina, T.N., Tourova, T.P., Poltaraus, A.B., Novikova, E.V., Grigoryan, A.A., Ivanova, A.E., Lysenko, A.M., Petrunyaka, V.V., Osipov, G.A., Belyaev, S.S., and Ivanov, M.V., 2001. "Taxonomic study of aerobic thermophilic bacilli: descriptions of Geobacillus subterraneus gen. nov., sp nov and Geobacillus uzenensis sp nov from petroleum reservoirs and transfer of Bacillus stearothermophilus Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermoglucosidasius and Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. thermocatenulatus, G. thermoleovorans, G. kaustophilus, G. thermoglucosidasius and G. thermodenitrificans". Int J Syst Evol Micr, 51(Pt 2), 433-446
Neupert, J., Karcher, D., and Bock, R., 2008. "Design of simple synthetic RNA thermometers for temperature-controlled gene expression in Escherichia coli". Nucleic Acids Res, 36(19), e124

128 References
Ng, E.W.M. and Adamis, A.P., 2006. "Anti-VEGF aptamer (pegaptanib) therapy for ocular vascular diseases". Ann Ny Acad Sci, 1082151-171
Nguyen, V.D., Hatahet, F., Salo, K.E., Enlund, E., Zhang, C., and Ruddock, L.W., 2011. "Pre-expression of a sulfhydryl oxidase significantly increases the yields of eukaryotic disulfide bond containing proteins expressed in the cytoplasm of E.coli". Microb Cell Fact, 10(1), 1
Niemitalo, O., Neubauer, A., Liebal, U., Myllyharju, J., Juffer, A.H., and Neubauer, P., 2005. "Modelling of translation of human protein disulfide isomerase in Escherichia coli - A case study of gene optimisation". J Biotechnol, 120(1), 11-24
Nishihara, K., Kanemori, M., Kitagawa, M., Yanagi, H., and Yura, T., 1998. "Chaperone coexpression plasmids: Differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2 in Escherichia coli". Appl Environ Microb, 64(5), 1694-1699
Oganesyan, N., Ankoudinova, I., Kim, S.H., and Kim, R., 2007. "Effect of osmotic stress and heat shock in recombinant protein overexpression and crystallization". Protein Expr Purif, 52(2), 280-285
Ogasahara, K., Nakamura, M., Nakura, S., Tsunasawa, S., Kato, I., Yoshimoto, T., and Yutani, K., 1998. "The Unusually Slow Unfolding Rate Causes the High Stability of Pyrrolidone Carboxyl Peptidase from a Hyperthermophile, Pyrococcus furiosus: Equilibrium and Kinetic Studies of Guanidine Hydrochloride-Induced Unfolding and Refolding". Biochemistry-Us, 37(50), 17537-17544
Ohrui, H., 2011. "Development of modified nucleosides that have supremely high anti-HIV activity and low toxicity and prevent the emergence of resistant HIV mutants". P Jpn Acad B-Phys, 87(3), 53-65
Okuyama, K., Shibuya, S., Hamamoto, T., and Noguchi, T., 2003. "Enzymatic synthesis of 2'-deoxyguanosine with nucleoside deoxyribosyltransferase-II". Biosci Biotech Bioch, 67(5), 989-995
Oshima, T. and Imahori, K., 1974. "Description of Thermus Thermophilus (Yoshida and Oshima) comb. nov, a Nonsporulating Thermophilic Bacterium from a Japanese Thermal Spa". Int J Syst Bacteriol, 24(1), 102-112
Pace, C.N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T., 1995. "How to Measure and Predict the Molar Absorption-Coefficient of a Protein". Protein Sci, 4(11), 2411-2423
Panova, N.G., Shcheveleva, E.V., Alexeev, C.S., Mukhortov, V.G., Zuev, A.N., Mikhailov, S.N., Esipov, R.S., Chuvikovsky, D.V., and Miroshnikov, A.I., 2004. "Use of 4-thiouridine and 4-thiothymidine in studies on pyrimidine nucleoside phosphorylases". Mol Biol+, 38(5), 770-776
Panova, N.G., Alexeev, C.S., Kuzmichov, A.S., Shcheveleva, E.V., Gavryushov, S.A., Polyakov, K.M., Kritzyn, A.M., Mikhailov, S.N., Esipov, R.S., and Miroshnikov, A.I., 2007. "Substrate specificity of Escherichia coli thymidine phosphorylase". Biochemistry (Mosc), 72(1), 21-28
Panula-Perälä, J., Šiurkus, J., Vasala, A., Wilmanowski, R., Casteleijn, M.G., and Neubauer, P., 2008. "Enzyme controlled glucose auto-delivery for high cell density cultivations in microplates and shake flasks". Microb Cell Fact, 7(31),
Parker, W.B., King, S.A., Allan, P.W., Bennett, L.L., Secrist, J.A., Montgomery, J.A., Gilberg, K.S., Waud, W.R., Wells, A.H., Gillespie, G.Y., and Sorscher, E.J., 1997. "In vivo gene therapy of cancer with E. coli purine nucleoside phosphorylase". Hum Gene Ther, 8(14), 1637-1644
Parker, W.B., Allan, P.W., Hassan, A.E., Secrist, J.A., 3rd, Sorscher, E.J., and Waud, W.R., 2003. "Antitumor activity of 2-fluoro-2'-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase". Cancer Gene Ther, 10(1), 23-9
Poulsen, S.A. and Quinn, R.J., 1998. "Adenosine receptors: new opportunities for future drugs". Bioorg Med Chem, 6(6), 619-641
Pugmire, M.J. and Ealick, S.E., 1998. "The crystal structure of pyrimidine nucleoside phosphorylase in a closed conformation". Structure, 6(11), 1467-1479
Pugmire, M.J. and Ealick, S.E., 2002. "Structural analyses reveal two distinct families of nucleoside phosphorylases". Biochemical Journal, 3611-25

References 129
Ranganathan, R., 1977. "Modification of 2'-Position of Purine Nucleosides - Syntheses of 2'-α-Substituted-2'-Deoxyadenosine Analogs". Tetrahedron Lett, (15), 1291-1294
Rentmeister, A., Arnold, F.H., and Fasan, R., 2009. "Chemo-enzymatic fluorination of unactivated organic compounds". Nat Chem Biol, 5(1), 26-28
Rocchietti, S., Ubiali, D., Terreni, M., Albertini, A.M., Fernandez-Lafuente, R., Guisan, J.M., and Pregnolato, M., 2004. "Immobilization and stabilization of recombinant multimeric uridine and purine nucleoside phosphorylases from Bacillus subtilis". Biomacromolecules, 5(6), 2195-2200
Rodenko, B., van der Burg, A.M., Wanner, M.J., Kaiser, M., Brun, R., Gould, M., de Koning, H.P., and Koomen, G.J., 2007. "2,N6-disubstituted adenosine analogs with antitrypanosomal and antimalarial activities". Antimicrob Agents Chemother, 51(11), 3796-3802
Rogert, M.C., Trelles, J.A., Porro, S., Lewkowicz, E.S., and Iribarren, A.M., 2002. "Microbial synthesis of antiviral nucleosides using Escherichia coli BL21 as biocatalyst". Biocatal Biotransfor, 20(5), 347-351
Roosild, T.P., Castronovo, S., Fabbiani, M., and Pizzorno, G., 2009. "Implications of the structure of human uridine phosphorylase 1 on the development of novel inhibitors for improving the therapeutic window of fluoropyrimidine chemotherapy". Bmc Struct Biol, 9
Roshevskaia, L.A., Barai, V.N., Zinchenko, A.I., Kvasiuk, E.I., and Mikhailopulo, I.A., 1986. "[Preparative synthesis of the antiviral nucleoside 9-β-D-arabinofuranosyladenine by using bacterial cells]". Antibiot Med Biotekhnol, 31(3), 174-178
Sadaf, S., Khan, M.A., and Akhtar, M.W., 2008. "Expression enhancement of bubaline somatotropin in E. coli through gene modifications in the 5'-end coding region". J Biotechnol, 135(2), 134-139
Sako, Y., Nomura, N., Uchida, A., Ishida, Y., Morii, H., Koga, Y., Hoaki, T., and Maruyama, T., 1996. "Aeropyrum pernix gen. nov., sp. nov., a novel aerobic hyperthermophilic archaeon growing at temperatures up to 100 °C". Int J Syst Bacteriol, 46(4), 1070-1077
Salvatori, D., Volpini, R., Vincenzetti, S., Vita, A., Costanzi, S., Lambertucci, C., Cristalli, G., and Vittori, S., 2002. "Adenine and deazaadenine nucleoside and deoxynucleoside analogues: inhibition of viral replication of sheep MVV (in vitro model for HIV) and bovine BHV-1". Bioorg Med Chem, 10(9), 2973-80
Sambrook, J. and Russell, D.W., "Molecular cloning : a laboratory manual". 3rd ed. 2001, New York: Cold Spring Harbor Laboratory Press.
Sampath, D., Rao, V.A., and Plunkett, W., 2003. "Mechanisms of apoptosis induction by nucleoside analogs". Oncogene, 22(56), 9063-9074
Samsel, M. and Dzierzbicka, K., 2011. "Therapeutic potential of adenosine analogues and conjugates". Pharmacol Rep, 63(3), 601-617
Santaniello, E., Ciuffreda, P., and Alessandrini, L., 2005. "Synthesis of modified purine nucleosides and related compounds mediated by adenosine deaminase (ADA) and adenylate deaminase (AMPDA)". Synthesis-Stuttgart, (4), 509-526
Saunders, P.P., Wilson, B.A., and Saunders, G.F., 1969. "Purification and comparative properties of a pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus". J Biol Chem, 244(13), 3691-3697
Schaeffer, H.J. and Thomas, H.J., 1958. "Synthesis of Potential Anticancer Agents. XIV. Ribosides of 2,6-Disubstituted Purines". J Am Chem Soc, 80(14), 3738-3742
Scheer, M., Grote, A., Chang, A., Schomburg, I., Munaretto, C., Rother, M., Sohngen, C., Stelzer, M., Thiele, J., and Schomburg, D., 2011. "BRENDA, the enzyme information system in 2011". Nucleic Acids Res, 39D670-D676
Schein, C.H. and Noteborn, M.H.M., 1988. "Formation of Soluble Recombinant Proteins in Escherichia coli Is Favored by Lower Growth Temperature". Bio-Technol, 6(3), 291-294
Schlieker, C., Bukau, B., and Mogk, A., 2002. "Prevention and reversion of protein aggregation by molecular chaperones in the E. coli cytosol: implications for their applicability in biotechnology". J Biotechnol, 96(1), 13-21
Schnick, C., Robien, M.A., Brzozowski, A.M., Dodson, E.J., Murshudov, G.N., Anderson, L., Luft, J.R., Mehlin, C., Hol, W.G.J., Brannigan, J.A., and Wilkinson, A.J., 2005. "Structures of Plasmodium

130 References
falciparum purine nucleoside phosphorylase complexed with sulfate and its natural substrate inosine". Acta Crystallogr D, 61(Pt 9), 1245-1254
Schultheisz, H.L., Szymczyna, B.R., Scott, L.G., and Williamson, J.R., 2008. "Pathway engineered enzymatic de novo purine nucleotide synthesis". ACS Chem Biol, 3(8), 499-511
Schultheisz, H.L., Szymczyna, B.R., Scott, L.G., and Williamson, J.R., 2010. "Enzymatic De Novo Pyrimidine Nucleotide Synthesis". J Am Chem Soc, 133(2), 297-304
Serova, M., Galmarini, C.M., Ghoul, A., Benhadji, K., Green, S.R., Chiao, J., Faivre, S., Cvitkovic, E., Le Tourneau, C., Calvo, F., and Raymond, E., 2007. "Antiproliferative effects of sapacitabine (CYC682), a novel 2'-deoxycytidine-derivative, in human cancer cells". Brit J Cancer, 97(5), 628-636
Serra, I., Serra, C.D., Rocchietti, S., Ubiali, D., and Terreni, M., 2011. "Stabilization of thymidine phosphorylase from Escherichia coli by immobilization and post immobilization techniques". Enzyme Microb Tech, 49(1), 52-58
Sgarrella, F., Poddie, F.P.A., Meloni, M.A., Sciola, L., Pippia, P., and Tozzi, M.G., 1997. "Channelling of deoxyribose moiety of exogenous DNA into carbohydrate metabolism: Role of deoxyriboaldolase". Comp Biochem Phys B, 117(2), 253-257
Sgarrella, F., Frassetto, L., Allegnini, S., Camici, M., Carta, M.C., Fadda, P., Tozzi, M.G., and Ipata, P.L., 2007. "Characterization of the adenine nucleoside specific phosphorylase of Bacillus cereus". Bba-Gen Subjects, 1770(10), 1498-1505
Sharp, P.M. and Li, W.H., 1987. "The codon Adaptation Index -a measure of directional synonymous codon usage bias, and its potential applications". Nucleic Acids Res, 15(3), 1281-1295
Shi, J.X., Du, J.F., Ma, T.W., Pankiewicz, K.W., Patterson, S.E., Tharnish, P.M., McBrayer, T.R., Stuyver, L.J., Otto, M.J., Chu, C.K., Schinazi, R.F., and Watanabe, K.A., 2005. "Synthesis and anti-viral activity of a series of D- and L-2'-deoxy-2'-fluororibonucleosides in the subgenomic HCV replicon system". Bioorgan Med Chem, 13(5), 1641-1652
Shi, W.X., Ting, L.M., Kicska, G.A., Lewandowicz, A., Tyler, P.C., Evans, G.B., Furneaux, R.H., Kim, K., Almo, S.C., and Schramm, V.L., 2004. "Plasmodium falciparum purine nucleoside phosphorylase - Crystal structures, immucillin inhibitors, and dual catalytic function". J Biol Chem, 279(18), 18103-18106
Shiloach, J. and Fass, R., 2005. "Growing E. coli to high cell density - A historical perspective on method development". Biotechnol Adv, 23(5), 345-357
Shimizu, K. and Kunishima, N., 2007. "Purification, crystallization and preliminary X-ray diffraction study on pyrimidine nucleoside phosphorylase TTHA1771 from Thermus thermophilus HB8". Acta Crystallogr F, 63308-310
Shirae, H. and Yokozeki, K., 1991. "Purifications and Properties of Orotidine-Phosphorolyzing Enzyme and Purine Nucleoside Phosphorylase from Erwinia carotovora AJ 2992". Agric Biol Chem 55(7), 1849-1857
Silva, R.G., Nunes, J.E.S., Canduri, F., Borges, J.C., Gava, L.M., Moreno, F.B., Basso, L.A., and Santos, D.S., 2007. "Purine nucleoside phosphorylase: A potential target for the development of drugs to treat T-cell- and apicomplexan parasite-mediated diseases". Curr Drug Targets, 8(3), 413-422
Šiurkus, J., Panula-Perälä, J., Horn, U., Kraft, M., Rimseliene, R., and Neubauer, P., 2010. "Novel approach of high cell density recombinant bioprocess development: Optimisation and scale-up from microlitre to pilot scales while maintaining the fed-batch cultivation mode of E. coli cultures". Microb Cell Fact, 9(1), 35
Sivets, G.G., Kalinichenko, E.N., and Mikhailopulo, I.A., 2006. "Synthesis of C2'-ß-fluoro-substituted adenine nucleosides via pivaloyl derivatives of adenosine and 3'-deoxyadenosine". Lett Org Chem, 3(5), 402-408
Sørensen, H.P. and Mortensen, K.K., 2005. "Advanced genetic strategies for recombinant protein expression in Escherichia coli". J Biotechnol, 115(2), 113-128
Stavrovskaya, A.A., 2000. "Cellular mechanisms of multidrug resistance of tumor cells". Biochemistry (Mosc), 65(1), 95-106

References 131
Stephenson, M.L. and Zamecnik, P.C., 1978. "Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide". P Natl Acad Sci USA, 75(1), 285-288
Suzuki, Y., Kishigami, T., Inoue, K., Mizoguchi, Y., Eto, N., Takagi, M., and Abe, S., 1983. "Bacillus thermoglucosidasius sp. nov., a New Species of Obligately Thermophilic Bacilli". Syst Appl Microbiol, 4(4), 487-495
Szeker, K., Niemitalo, O., Casteleijn, M.G., Juffer, A.H., and Neubauer, P., 2011. "High-temperature cultivation and 5'mRNA optimization are key factors for the efficient overexpression of thermostable Deinococcus geothermalis purine nucleoside phosphorylase in Escherichia coli". J Biotechnol, 156(4), 268-274
Szeker, K., Zhou, X., Schwab, T., Casanueva, A., Cowan, D., Mikhailopulo, I.A., and Neubauer, P., 2012. "Comparative investigations on thermostable pyrimidine nucleoside phosphorylases from Geobacillus thermoglucosidasius and Thermus thermophilus". J Mol Catal B-Enzym, in press(doi:10.1016/j.molcatb.2012.02.006),
Tahirov, T.H., Inagaki, E., Ohshima, N., Kitao, T., Kuroishi, C., Ukita, Y., Takio, K., Kobayashi, M., Kuramitsu, S., Yokoyama, S., and Miyano, M., 2004. "Crystal structure of purine nucleoside phosphorylase from Thermus thermophilus". J Mol Biol, 337(5), 1149-1160
Tann, C.H., Brodfuehrer, P.R., Brundidge, S.P., Sapino, C., and Howell, H.G., 1985. "Fluorocarbohydrates in Synthesis. An Efficient Synthesis of 1-(2-Deoxy-2-Fluoro-β-D-Arabinofuranosyl)-5-Iodouracil (β-FIAU) and 1-(2-Deoxy-2-Fluoro-β-D-Arabinofuranosyl)Thymine (β-FMAU)". J Org Chem, 50(19), 3644-3647
Taran, S.A., Verevkina, K.N., Feofanov, S.A., and Miroshnikov, A.I., 2009. "Enzymatic Transglycosylation of Natural and Modified Nucleosides by Immobilized Thermostable Nucleoside Phosphorylases from Geobacillus stearothermophilus". Russ J Bioorg Chem+, 35(6), 739-745
Tebbe, J., Wielgus-Kutrowska, B., Schroder, W., Luic, M., Shugar, D., Saenger, W., Koellner, G., and Bzowska, A., 1997. "Purine nucleoside phosphorylase (PNP) from Cellulomonas sp., a third class of PNP different from both ''low-molecular weight'' mammalian and ''high-molecular weight'' bacterial PNPs". Protein Eng, 1090
Tebbe, J., Bzowska, A., Wielgus-Kutrowska, B., Schroder, W., Kazimierczuk, Z., Shugar, D., Saenger, W., and Koellner, G., 1999. "Crystal structure of the purine nucleoside phosphorylase (PNP) from Cellulomonas sp and its implication for the mechanism of trimeric PNPs". J Mol Biol, 294(5), 1239-1255
Tennilä, T., Azhayeva, E., Vepsalainen, J., Laatikainen, R., Azhayev, A., and Mikhailopulo, I.A., 2000. "Oligonucleotides containing 9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-adenine and -guanine: Synthesis, hybridization and antisense properties". Nucleos Nucleot Nucl, 19(10-12), 1861-1884
Thiel, A., Pries, R., Jeske, S., Trenkle, T., and Wollenberg, B., 2009. "Effect of Head and Neck Cancer Supernatant and CpG-Oligonucleotides on Migration and IFN-alpha Production of Plasmacytoid Dendritic Cells". Anticancer Res, 29(8), 3019-3025
Thomas, H.J., Tiwari, K.N., Clayton, S.J., Secrist, J.A., and Montgomery, J.A., 1994. "Synthesis and Biologic Activity of Purine 2’-Deoxy-2’-fluoro-ribonucleosides". Nucleos Nucleot, 3(1-3), 309-323
Tozzi, M.G., Sgarrella, F., and Ipata, P.L., 1981. "Induction and Repression of Enzymes Involved in Exogenous Purine Compound Utilization in Bacillus cereus". Bba-Gen Subjects, 678(3), 460-466
Trelles, J.A., Valino, A.L., Runza, V., Lewkowicz, E.S., and Iribarren, A.M., 2005. "Screening of catalytically active microorganisms for the synthesis of 6-modified purine nucleosides". Biotechnol Lett, 27(11), 759-763
Trembacz, H. and Jezewska, M.M., 1993. "Specific Adenosine Phosphorylase from Hepatopancreas of Gastropod Helix pomatia". Comp Biochem Phys B, 104(3), 481-487
Turkman, N., Gelovani, J.G., and Alauddin, M.M., 2010. "A novel method for stereospecific fluorination at the 2'-arabino-position of pyrimidine nucleoside: synthesis of [(18)F]-FMAU". J Labelled Compd Rad, 53(13-14), 782-786

132 References
Tuttle, J.V. and Krenitsky, T.A., 1992. "Therapeutic nucleosides ", EP 0 285 432 B1 (Wellcome Found, GB) C07H 19/16
Tuttle, J.V., Tisdale, M., and Krenitsky, T.A., 1993. "Purine 2'-deoxy-2'-fluororibosides as antiinfluenza virus agents". J Med Chem, 36(1), 119-125
Ubiali, D., Serra, C.D., Serra, I., Morelli, C.F., Terreni, M., Albertini, A.M., Manitto, P., and Speranza, G., 2012. "Production, Characterization and Synthetic Application of a Purine Nucleoside Phosphorylase from Aeromonas hydrophila". Adv Synth Catal, 354(1), 96-104
Ukkonen, K., Vasala, A., Ojamo, H., and Neubauer, P., 2011. "High-yield production of biologically active recombinant protein in shake flask culture by combination of enzyme-based glucose delivery and increased oxygen transfer". Microb Cell Fact, 10(107),
Utagawa, T., Morisawa, H., Miyoshi, T., Yoshinaga, F., Yamazaki, A., and Mitsugi, K., 1980. "A novel and simple method for the preparation of adenine arabinoside by bacterial transglycosylation reaction". FEBS Lett, 109(2), 261-263
Utagawa, T., Morisawa, H., Shigeru, Y., Yamazaki, A., Yoshinaga, F., and Hirose, Y., 1985a. "Properties of Nucleoside Phosphorylase from Enterobacter aerogenes". Agric Biol Chem, 49(11), 3239-3246
Utagawa, T., Morisawa, H., Yoshinaga, F., Yamazaki, A., Mitsugi, K., and Hirose, Y., 1985b. "Enzymatic-Synthesis of Nucleoside Antibiotics .1. Microbiological Synthesis of Adenine-Arabinoside". Agric Biol Chem, 49(4), 1053-1058
Utagawa, T., 1999. "Enzymatic preparation of nucleoside antibiotics". J Mol Catal B-Enzym, 6(3), 215-222
Visser, D., Hennessy, F., Rashamuse, K., Louw, M., and Brady, D., 2010. "Cloning, purification and characterisation of a recombinant purine nucleoside phosphorylase from Bacillus halodurans Alk36". Extremophiles, 14(2), 185-192
Visser, D.F., Hennessy, F., Rashamuse, J., B., P., and Brady, D., 2011. "Stabilization of Escherichia coli uridine phosphorylase by evolution and immobilization". J Mol Catal B-Enzym, 68279-285
Vita, A., Huang, C.Y., and Magni, G., 1983. "Uridine Phosphorylase from Escherichia coli B - Kinetic Studies on the Mechanism of Catalysis". Arch Biochem Biophys, 226(2), 687-692
Vittori, S., Lorenzen, A., Stannek, C., Costanzi, S., Volpini, R., AP, I.J., Kunzel, J.K., and Cristalli, G., 2000. "N-cycloalkyl derivatives of adenosine and 1-deazaadenosine as agonists and partial agonists of the A(1) adenosine receptor". J Med Chem, 43(2), 250-260
Vorbrüggen, H. and Ruh-Pohlenz, C., "Handbook of Nucleoside Synthesis". 1 ed. 2001: Wiley-Interscience.
Wang, Y. and Zhang, Y.H., 2009. "Overexpression and simple purification of the Thermotoga maritima 6-phosphogluconate dehydrogenase in Escherichia coli and its application for NADPH regeneration". Microb Cell Fact, 8(30),
Watanabe, K.A., Reichman, U., Hirota, K., Lopez, C., and Fox, J.J., 1979. "Nucleosides. 110. Synthesis and antiherpes virus activity of some 2'-fluoro-2'-deoxyarabinofuranosylpyrimidine nucleosides". J Med Chem, 22(1), 21-24
Watts, J.K. and Damha, M.J., 2008. "2'F-arabinonucleic acids (2'F-ANA) - History, properties, and new frontiers". Can J Chem, 86(7), 641-656
Watts, J.K. and Corey, D.R., 2010. "Clinical status of duplex RNA". Bioorg Med Chem Lett, 20(11), 3203-3207
Watts, J.K. and Corey, D.R., 2012. "Silencing disease genes in the laboratory and the clinic". J Pathol, 226(2), 365-379
Wei, L., Altman, R.B., and Chang, J.T., 1997. "Using the radial distributions of physical features to compare amino acid environments and align amino acid sequences". Pac Symp Biocomput, 465-476
Wei, X.K., Ding, Q.B., Zhang, L., Guo, Y.L., Ou, L., and Wang, C.L., 2008. "Induction of nucleoside phosphorylase in Enterobacter aerogenes and enzymatic synthesis of adenine arabinoside". J Zhejiang Univ Sci B, 9(7), 520-526
Welch, M., Villalobos, A., Gustafsson, C., and Minshull, J., 2009. "You're one in a googol: optimizing genes for protein expression". J R Soc Interface, 6

References 133
Wright, G.E., Hildebrand, C., Freese, S., Dudycz, L.W., and Kazimierczuk, Z., 1987. "Convenient Synthesis of 2-Halo-2'-Deoxyadenosines". J Org Chem, 52(20), 4617-4618
Wright, J.A., Taylor, N.F., and Fox, J.J., 1969. "Nucleosides .60. Fluorocarbohydrates .22. Synthesis of 2-Deoxy-2-Fluoro-D-Arabinose and 9-(2-Deoxy-2-Fluoro-α- and -β-D-Arabinofuranosyl)Adenines". J Org Chem, 34(9), 2632-2636
Xie, X.X., Xia, J.G., He, K.F., Lu, L.N., Xu, Q.Y., and Chen, N., 2011. "Low-molecular-mass purine nucleoside phosphorylase: characterization and application in enzymatic synthesis of nucleoside antiviral drugs". Biotechnol Lett, 33(6), 1107-1112
Yamada, K., Matsumoto, N., and Hayakawa, H., 2009. "Stereoselective Synthesis of 2-Deoxy-2-Fluroarabinofuranosyl-α-1-phosphate and its Application to the Synthesis of 2'-Deoxy-2'-Fluoroarabinofuranosyl Purine Nucleosides by a chemo-enzymatic method". Nucleos Nucleot Nucl, 28(11-12), 1117-1130
Yamazaki, S., Yamazaki, J., Nishijima, K., Otsuka, R., Mise, M., Ishikawa, H., Sasaki, K., Tago, S., and Isono, K., 2006. "Proteome analysis of an aerobic hyperthermophilic crenarchaeon, Aeropyrum pernix K1". Mol Cell Proteomics, 5(5), 811-823
Zaitseva, G.V., Zinchenko, A.I., Barai, V.N., Pavlova, N.I., Boreko, E.I., and Mikhailopulo, I.A., 1999. "Chemical and enzymatic synthesis and antiviral properties of 2'-deoxy-2'-fluoroguanosine". Nucleos Nucleot, 18(4-5), 687-688
Zamecnik, P.C. and Stephenson, M.L., 1978. "Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide". P Natl Acad Sci USA, 75(1), 280-284
Zeng, Q., Huang, B., Danielsen, K., Shukla, R., and Nagy, T., 2004. "Facile and Practical Synthesis of 2,6-Dichloropurine". Org Process Res Dev, 8(6), 962-963
Zhu, S.Z., Ren, L., Wang, J.J., Zheng, G.J., and Tang, P.W., 2012. "Two-step efficient synthesis of 5-methyluridine via two thermostable nucleoside phosphorylase from Aeropyrum pernix". Bioorg Med Chem Lett, 22(5), 2102-2104
Zinchenko, A.I., Barai, V.N., Bokut, S.B., Kvasyuk, E.I., and Mikhailopulo, I.A., 1990. "Synthesis of 9-(β-D-arabinofuranosyl)guanine using whole cells of Escherichia coli". Appl Microbiol Biotechnol, 32(6), 658-661
Zuffi, G., Ghisotti, D., Oliva, I., Capra, E., Frascotti, G., Tonon, G., and Orsini, G., 2004. "Immobilized biocatalysts for the production of nucleosides and nucleoside analogues by enzymatic transglycosylation reactions". Biocatal Biotransfor, 22(1), 25-33
Zuker, M., 2003. "Mfold web server for nucleic acid folding and hybridization prediction". Nucleic Acids Res, 31(13), 3406-3415


Appendix
DgPNP gene sequence (Dgeo1497, 786 bp)
gtggtggcgcgtgtaccggcaaggcctttcgcttccccgcctgctaccctggaccgcgtgagtgtccacctgaatgcccggcccggcgaaattgcc
gaaactgtcctgcttcccggcgaccccctgcgcgcgcagcacatcgcggagacgttcttcgagaacccggtgcagcataacagcgtgcgcggcat
gctgggctttaccggcacgtaccggggcgtccccgtcagcgtgcagggcactggtatgggaatcgcctcatccatgatctacgtcaacgagctgat
ccgagactacggttgccagaccctgattcgcgtcggaacggcaggcagctatcagccggacgtgcacgtgcgcgacctcgtcctggcacaggcc
gcctgcactgacagcaatatcaacaacatccgcttcggtctgcggaacttcgccccgattgcggactttgagctgctgctgcgcgcctaccaaatg
gcccgggaccgtggcttcgcgacccatgtcggcaacatcctgtcctcggacaccttctatcaggacgatccggagagctacaaactctgggcgca
gtacggcgtgctggcggtggagatggaagccgccgggctgtataccctcgccgccaagtacggcgtgcgcgcgctcaccatcctgaccatctccg
accacctcgtcacacgggaggaaaccaccgctgaggagcggcagacgacatttaacggcatgattgaggtcgcgctggacgccgcactgggac
tggcagtaccttccaacagcatgtga
ApMTAP gene sequence (APE0993.1, 735 bp)
ttgaggaagccggttcacctcgaggcagggcccggcgacgtggcaccactcgtggtggcagtcggcgacccggggagggctgagaggctggcc
acaggcctcctcgaggacgctaggctggtgtcctccgccaggggtctgaaggtatacacgggcagcttcaacggctcggaggtaacgatagcca
cccacggcataggaggcccctcggcggctgtagtcttcgaggagctgaggatgcttggggctgaggttctggtgaggctcggcacctcgggcggc
ctatccaaggacctcaggctgggggacgtggtggtcgccgcgggggcgggctgctactggggctccgggggtagcatccagtacgcgggcgag
aggcccatgtgcctcccggcctcccccgaccccatattgacggcggggatatacaggggcctctcctccaggctcggggatagggtggttctagc
ccccgtcatgtcgagcgacgccttctacgccgagacgcccgaggctgccgggaggtggaggagcctcggcatggcggctgttgagatggagctc
cacaccctcttcagcatatcctggatacggggcttccgctcggccggggtgctcatagtctccgacctcctcctacccgagggtttcaaacgcatca
caccaggggagcttgcaaggagggaggttgaggttggcagggctctcctcgaggtcctcacaggaggagtctag
GtPNP gene sequence (ADNQ01000003.1, 708 bp)
ttgagcatccatatcgaagcaaagcaacaagaaattgctgagaaaattttgctgccgggtgatccgctccgcgcccaatatatcgcggaaacatt
tttagaaggagcgacatgctacaatcgcgtgcgcggcatgcttggatttacaggtacatacaaagggcaccgcatttctgtgcaagggacagga
atgggagttccttcgatttcgatttatgtcaatgaattaatccaaagctatcatgtacaaacactcattcgcgttggaacgtgcggagcgattcaaa
aagatgtcaacgttcgcgacgttattttggcgatgagcgcatccacggattcgaacatgaaccgtttgactttccgtgggcgggattatgcgccga
cagcaaattttgctttgttgcggaccgcttatgaagtcggcgccgaaaaagggcttccgctaaaggtcggaagtgtctttactgctgacatgtttta
taatgacgaaccggactgggagacgtgggcccgctacggtgttttagctgttgagatggaaacagcggcgctgtatacgctggcggcaaaatttg
gccgaaaagcgctttctgtgctgacggtaagcgaccatattttaacaggagaagaaacaacggcacaagaacggcaaacaacgtttaacgata
tgattgaagtggcgctggaaacggcgattcgcgtagaataa
GtPyNP gene sequence (NZ_ADNQ01000001.1, 1296 bp)
atggtcgatttaattgcgaaaaaacgggatggttatgagctttcaaaagaagaaattgattttatcatccgcggttacacgaacggcgacattcct
gattatcaaatgagcgcgttcgcgatggcggtgtttttccgcggcatgacagaagaagaaacggcggcgctaacgatggcgatggtccgctccg
gagatgtcatcgatttatcgaaaatcgaaggaatgaaagtcgacaagcattcaacgggtggcgtcggcgatacgacgacgcttgtgttagggcc
gcttgttgcgtctgtcggcgtgcctgtcgcaaaaatgtcggggcgagggcttggacataccggcgggaccattgataaattagagtccgttccagg
gtttcatgtggaaatcgataacgagcaatttattgagcttgtgaataaaaacaaaatcgcgattatcggccagacaggcaatttaacgccagccg
ataaaaagctgtatgcgctccgtgacgttacggcgacggtggacagcattccactgatcgcttcgtcgattatgagcaaaaaaattgccgctggc

136 Appendix
gctgacgcgattgtgttggatgtgaaaacgggagccggcgcgtttatgaaagattttgcaggagcgaagcggctcgcaacagcgatggtggaaa
tcggcaagcgcgtcggccggaaaacgatggcggttatttccgacatgagccagccgctcggatacgctgttggaaacgcgctcgaagtgaaaga
agcgattgatacgcttaaaggaaaagggccagaagatttacaagaactatgtttgacgcttggaagctatatggtatatttggcggaaaaagcct
cttcattagaggaagcgcgcgcgctgttagaagcgtcgattcgggaaggaaaagcgttagaaacgttcaaagtgtttctcagcgcgcaaggcgg
cgacgcatcggttgtcgatgatccaacgaaactgccgcaagcgaaataccgatgggagcttgaagccccggaagatgggtatgtcgcggaaatt
gtcgctgacgaagtcggaacggctgcgatgctgcttggagccgggcgggcgacaaaagaagcaacgatcgatctttctgtcggcctcgtcttgca
caaaaaggtcggcgatgcggtgaaaaaaggcgaatcgcttgtgacgatttacagcaatacggaaaatattgaagaagtcaaacaaaagcttgc
caaaagcattcgcctctcctccattcctgttgccaagccgacgcttatatacgaaaccatttcataa
TtPyNP gene sequence (AE017221.1, 1272 bp)
atgaaccccgtggtcttcatccgggagaagcgggaagggaaaaagcaccgccgggaggacctcgaggccttcctcctcggctacctgcgggac
gaggtgccggactaccaggtggccgcctggctcatggccgccttcctaaggggcctggacgccgaggagaccctctggctcaccgaaaccatgg
cccgctcggggaaggttctggacctctccggccttccccaccccgtggacaagcactcctcggggggcgtgggggacaaggtgagcctggtggtg
gggccgatcctcgccgcaagcgggtgcaccttcgccaagatgtcgggccggggcctggcccacaccggggggaccatagacaagctggagtcg
gtgccgggctggcggggggagatgacggaggcggagtttttggagagggcccggagggtgggcctcgtcatcgccgcccaaagcccggacctc
gcccccctggatgggaagctttacgccctccgcgacgtgaccgccacggtggagagcgtgcctttgatcgcgagctccatcatgagcaagaagct
cgccgccggggcgcggagcattgtcttggacgtgaaggtgggccggggggccttcatgaagaccctggaggaggcccggcttttggccaagacc
atggtggccatcggccagggggcgggaaggcgggtgagggccctcctcacctccatggaggcccccctggggcgggcggtgggcaacgccata
gaggtgcgggaggcgataggggccctcaagggggagggccccgaggatcttctggaggtggccctggccctggcggaggaggccttaaagctt
gaggggctggaccccgccctcgcccggaaggccctggagggcggggcggccttggagaagttccgggccttcctcgaggcccaggggggagac
ccccgggcggtggaggacttctcccttttgcccctcgccgaggagcaccccctccgtgccgagcgggagggcgtggtgcaggaggtggacgccta
caaggtgggcctcgccgtcctcgccctgggcggggggcggaagcggaagggggagcccattgaccacggggtgggggtctacctgctcaagaa
gcccggggaccgggtggagcggggggaggccttggccctggtctaccaccggaggcggggcctggaggaggcccttgggcacctgcgcgaggc
ctacgccctgggggaggaagcgcaccccgcccccttggtcctggaggccatctag
ApUP (full length) gene sequence (APE2105.1, 849 bp)
ttgggagacgagagtctaaggagcgccgcccgtcccgagggggagggagggctgcagtaccatctgagggtcaggaggggggatgtggcccg
ctacgttctcctcccgggagaccccgagaggacagaccttatagcccgcctctgggatgaagcgaggcttgtagcgcaccaccgggagtacagg
acgtggaccggcttctacaaggggacatcgataagtgtaacaagcaccgggataggctctcccagcacggcgatagccgttgaggagctgctga
gggttggagccgagactttcataagagtaggcactatgggcggtataagggaggatctgcggcccggcaccctggttatagggagtgcggcggt
taggatggaggggacgagcggccagtacgctccccgggggttcccagcggccgccagctatgacgttgtggcggcgctggtggaggctgctgag
gcgctcggggttaggtatgaggttggcgttgttgccagcacggacagcttctacctgggccaggggaggccggggtacggggggtatatgacgc
cggaggcttcggaagtcatacccctcctcaggtcagccggcgtcctcggcttcgagatggaggcctccgccctcttcaccctatcccagctctacg
gcgccagggcagggtgcgtgtgcgcggtagtggcaaacagggttagcggggagtttgtggtaaacgcgggggttgaagacgctgctagggttgc
ctccgaggcggtagccatactagcaggctgggacagggagagggagaagaggggtaagaaatggttttacccgagcctggcgtgcagacgca
catag
(The underlined bps represent the putative internal ribosome binding site and the bold letter mark the related potential
start codon)

Zusammenfassung
Modifizierte Nukleoside sind wertvolle Pharmazeutika, die für die Behandlung von Krebs und viralen
Erkrankungen eingesetzt werden. Außerdem dienen sie als Bausteine für die Synthese
therapeutischer Oligonukleotide mit besonderen Eigenschaften.
Während es für die Herstellung chemisch modifizierter Pyrimidinnukleoside einfache und bewährte
Verfahren gibt, stellt die organische Synthese modifizierter Purinnukleoside oft eine Herausforderung
dar, was zu mehrstufigen Verfahren mit niedriger Ausbeute führt. Die chemisch-enzymatische
Herstellung, bei der ein Pyrimidinnukleosid als Pentofuranosyl-Donor und eine Purinbase als
Pentufuranosyl-Akzeptor dient, kann daher eine attraktive Alternative sein. Für den regio- und
stereospezifischen Transfer der Zuckereinheit kommen Nukleosidphosphorylasen (NPs) als
Biokatalysatoren zum Einsatz, wobei als Substrate sowohl natürlich vorkommende, als auch
chemisch modifizierte, künstliche Vorstufen verwendet werden können. Leider ist die
Substrataktivität einer Vielzahl von hochinteressanten Nukleosid-Analoga jedoch bei den
gegenwärtig genutzten NPs sehr gering. Darüber hinaus ist es von Vorteil den Syntheseprozess bei
hohen Temperaturen durchzuführen um die Konzentration schlecht löslicher Purinbasen zu erhöhen,
doch dies führt bei vielen Enzymen zum schnellen Aktivitätsverlust. Beide Faktoren schränken den
Anwendungsbereich und die Effizienz der Synthese modifizierter Nukleoside durch NPs ein. Ziel
dieser Arbeit ist es, neue, thermostabile Varianten von NPs und ihren potentiellen Einsatz als
Biokatalysatoren zu untersuchen.
Hierfür wurden 5 NPs von 4 verschiedenen thermophilen Mikroorganismen (Deinococcus
geothermalis, Geobacillus thermoglucosidasius, Thermus thermophilus, Aeropyrum pernix) in E. coli
überexprimiert. Die Temperaturoptima und Thermostabilität der rekombinant hergestellten Enzyme
unterscheiden sich signifikant, vor allem in Abhängigkeit von Ursprungsmikroorganismus und
Enzymtyp. Die Untersuchung der Substratspezifität zeigt, dass modifizierte Nukleoside in sehr
unterschiedlichem Ausmaß als Substrate erkannt werden. Der Einsatz der vielversprechendsten
Enzym-Kombination in enzymatischen Transglykosylierungsreaktionen wurde untersucht. Hierbei
stand die Synthese 2′-fluorinierter Purinnukleoside sowie 2,6-dihalogenierte Purinnukleoside im
Fokus dieser Arbeit. 2′-Fluorinierte Nukleoside haben wertvolle pharmazeutische Eigenschaften und
verleihen synthetischen Oligonukleotiden günstige Eigenschaften, wohingegen 2,6-dihalogenierte
Purinnukleoside hervorragende Vorstufen für die Herstellung modifizierter Purinnukleoside sind. Im
Vergleich zu E. coli Enzymen, die als Biokatalysatoren für die Synthese 2′-fluorinierter
Purinnukleoside bereits in der Literatur beschrieben sind, ermöglichen die hier generierten Enzyme
die Durchführung der Transglykosylierung bei höherer Temperatur und scheinen dabei effizienter zu
sein. Gleichzeitig werden 2,6-dihalogenierte Purine sehr gut als Substrate erkannt und die Synthese
der entsprechenden (Deoxy )riboside verläuft dementsprechend schnell.
Die Ergebnisse dieser Arbeit verdeutlichen generell das Potential thermostablier Enzyme als
Biokatalysatoren und ebnen insbesondere den Weg für verbesserte und umweltfreundlichere
Verfahren für die Synthese wertvoller 2′-fluorinierter und 2,6-dihalogenierter Purinnkleosid-Analoga.

Acknowledgements
I would like to use the opportunity to thank all the people who have contributed to the
accomplishment of this work.
First of all I want to thank my supervisor Prof. Peter Neubauer for giving me the interesting research
topic, for his continuous support and for the inspiring ideas, discussions and positive view of life.
Many thanks also to Prof. Igor A. Mikhailopulo, for the intense scientific discussions, the numerous
emails from Minsk, and for sharing enthusiasm on nucleoside analogues and pictures of Murka with
me. Furthermore I would like to thank Prof. Marion Ansorge-Schumacher for helpful discussions on
enzyme technology.
Progress of this work has substantially benefitted from viable scientific collaborations and some of
the data shown have thus been obtained by other scientists in external institutions. In particular I
want to thank Olli Niemitalo and Prof. André H. Juffer for the fruitful collaboration towards the
expression optimization of DgPNP for which they have contributed the theoretical predictions of 5’
mRNA secondary structure stability and in silico sequence optimization. I am furthermore grateful for
the helpful suggestions of Thomas Schwab from the group of Prof. Reinhard Sterner, who has also
performed thermal unfolding studies with TtPyNP presented in this work. Additionally I want to
thank Knut Büttner for promptly helping out with a mass spectrometric measurement of ApMTAP
and Leif Garbe for the discussion of analytical problems. Thanks also to Marco Casteleijn for his
ongoing scientific and personal support. I want to express my gratitude to Prof. Don Cowan for giving
me the opportunity of an inspiring research stay in his lab at the other end of the world - at the
Institute for Microbial Biotechnology and Metagenomics (IMBM) of the University of the Western
Cape in Cape Town.
I am deeply thankful for the BIG-NSE scholarship of the cluster of excellence “Unifying concepts in
Catalysis” coordinated by the Technische Universität Berlin that gave me the opportunity to focus on
my research project. Special thanks to Dr. Jean-Philippe Lonjaret for his dedication to make the BIG-
NSE graduate program a success. Furthermore I would like to thank Alex Azhayev from Metkinen
Chemistry for kindly providing us with modified nucleosides for substrate screening studies.
I am grateful for the support by students with who I had the chance to work together on the
nucleoside phosphorylase project. Many thanks to Julia Geyer, who joined the project in the early
stage during her study work and with who it was always pleasant to work. Thanks to Dr. Thomas
Böhme (self proclaimed „Goldfinger“) and Dr. Bernd Janocha, whose entertaining protocols were
always enjoyable to read: Both have dared to extend their chemical expertise to the field of
biotechnology in the program „Campus Biotech“. During their 6-month practical training in our lab
they have gathered important results on ApMTAP and DgPNP expression and characterization that
are also included in this thesis.
Especially I would like to express my gratitude to Xinrui Zhou, who has joined our group in 2011,
working together with me on nucleoside phosphorylases. Her scrupulous work, her commitment and

her patience has greatly accelerated the progress of this project and the majority of data presented
in this work are the result of our productive teamwork. It has been a great pleasure to work with you.
I would like to thank the team of the department of enzyme technology, especially Andy Maraite
who helped with the first protein purification and Alexander Scholz for his invaluable work on
immobilization that is opening up future industrial perspectives.
I want to thank all the members of the IMBM in Cape Town for cordially welcoming me in- and
outside of the lab. Especially I would like to thank Heide Goodman, Ana Casanueva, Munaka Schnaka,
Randall and Layla.
Many thanks to the present and fromer co-workers at the bioprocess engineering laboratory.
Margitta Seidenstücker, Irmgard Maue-Mohn, Dirk Itzeck, and Brigtitte Burckhardt for their
unshakable drive towards a cleaner lab, their helpful advises and practical support. Herta Klein-
Leuendorf for help with bureaucracy. Stefan Junne for being ready to give competent help even the
time schedule is actually already full. My dear „kolezhanka“ Julia Glazyrina, who helped not only in
scientific matters but also with her encouraging words and pleasant coffee breaks. Jennifer Jaitzig for
sharing rooms with me, her invaluable hints and advises and for many discussions concerning science
and life. Friederike Hillig for waiting with the lunch break, Mihaela Paella and Maciek Pullarek for
their entertaining discussions, Jian Li for all the gold coins he spent. Erich for his exciting stories,
Divine for his self-propagating smile, Mirja, Eva, Kathrin and all the other co-workers that contributed
to a great working atmosphere. And the Biosilta team for sponsoring yummy sweets.
Finally I want to thank my friends, who made it easier to deal with frustrating experiments, my
parents who have supported me in every phase of my life and my brother who dragged me to do
some sports even after long working days.
Ermin – for your enormous support, your confidence and the wonderful life you share with me.
































