Embed Size (px)
Citation preview

Hyperventilation and CerebralBlood FlowBY MARCUS E. RAICHLE, M.D.,* AND F. PLUM, M.D.f
Abstract:Hyperventila-tion andCerebralBlood Flow
• Hypocapnic-hyperventilation has a profound, but probably temporary, effecton CBF, producing approximately a 2% decline in CBF for each 1 torr declinein Pcov Th's effect appears to be mediated through changes in perivascular pHof the cerebral resistance vessels acting directly on the vessel wall. At low PC0;(the vasoconstrictor effect of short-term hypocapnic-hyperventilation is attenu-ated by resultant cerebral hypoxia. During prolonged hyperventilation CBFreturns toward normal as the pH in the CSF is restored.
Short-term hypocapnic-hyperventilation can be lifesaving in the treatmentof acute intracranial hypertension. On the other hand, prolonged hyperventila-tion has not been convincingly shown to benefit patients, whether with severehead injury or cerebral infarction, or during carotid endarterectomy withoutbypass.
Additional Key Wordscerebral infarction
intracranial hypertension head injurycarotid endarterectomy
Introduction• Hypocapnic-hyperventilation (HV) has aprofound effect on cerebral blood flow inhealthy man and animals. Because of thiseffect, it has played a central role in thedevelopment of our current understanding ofthe regulatory mechanism of the cerebralcirculation. In addition, it has been employedin a variety of clinical situations wheremanipulation of cerebral circulation and brainacid-base metabolism has been sought. In thisreview we will consider first the relationshipbetween hypocapnic-hyperventilation and ce-rebral blood flow (CBF), and second theclinical application of this tool.
Influence of Hyperventilationon Cerebral Blood FlowAcute hypocapnic-hyperventilation (HV) inhealthy animals and man causes an immediatecerebral vasoconstriction with a consequentrise in cerebral vascular resistance (CVR) and
#The Edward Mallinckrodt Institute of Radiology, 510South Kingshighway, St. Louis, Missouri 63110.
tDepartment of Neurology, Cornell UniversityMedical College, New York, New York 10021.
566
a fall in cerebral blood flow (CBF), changesthat parallel the fall in arterial carbon dioxidetension (Pco-,)- It is generally accepted thatthe effect of HV is mediated by this change inPoo.,, for it is the most potent cerebralvasoconstrictive agent known. Acute changesin Pco., between 20 and 60 torr have beenshown to change CBF 1 to 2 ml/min/100 gmof brain per 1 torr change in Poo.,-1
MECHANISM OF ACTIONThe specific mechanism for the effect of Pco2
on CBF has been the subject of considerablecontroversy. It is well established that an acuterise in Poo2 causes a decrease in CVR whichincreases the CBF, and a fall in PCon has theopposite effect. However, during sustainedalteration in POo.. the CBF and absolute carbondioxide tension often fail to correlate closely.This has led to the hypothesis that alteration ofthe pH of the brain extracellular spacemediates the cerebral vascular response tocarbon dioxide and hence HV, and that braininterstitial fluid pH is a major regulator ofCBF.2
One can marshal considerable indirectevidence that the pH of brain extracellular fluidis an important factor governing the response
Stroke, Vol. 3, Sopttmber-October 1972
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

HYPERVENTILATION AND CBF
of the cerebral vessels to Pco.,- Underlying thepremise is the fact that carbon dioxide diffusesreadily and almost immediately across theblood-brain barrier, creating simultaneous pHchanges in both blood and extracellular fluid(ECF), whereas charged ions are impeded intheir passage across the barrier, creatingconsiderable pH differences between the twocompartments in systemic metabolic acidosisand alkalosis. If one alters the brain ECF pHby changing the bicarbonate concentration ofthe cortex as a whole" or the CSF surroundingarterioles,4 vasodilatation and contraction arereported to occur in response to lowering andraising the ECF pH while the PCo2 remainsconstant.2
By contrast, if the intravascular pH isacutely raised or lowered and the Pco., is keptconstant, the ECF pH stays the same, and CBFdoes not change.5 With more sustained acid-base disorders or changes in the blood carbondioxide levels, brain ECF pH does shift, thechange being mediated by more slowly chang-ing influences than the quick respiratoryadjustments that alter PCo.,.° Under thesecircumstances, the relation between CBF andECF pH sometimes appears particularlystrong. The examples are many: at highaltitude the P00., is low but both the CSF pHand the CBF are normal.7 In severe diabeticacidosis, a condition in which an acidotic CSFhas sometimes been found,8 CBF is increasedabove normal in spite of a low PCo.,-° Agnoli10
found that in chronic normoxic respiratoryacidosis, CSF, pH, and CBF returned towardnormal while Pco2 remained high, andSkinh0jn reported that CBF was normal inpatients with either hypocapnia or hypercap-nia, so long as the pH in the CSF was normal.Most telling in this regard was the study ofFencl et al.12 in which steady-state metabolicacidosis or alkalosis was successively inducedfor several days in healthy men: CBF,estimated from cerebral arteriovenous differ-ences, appeared to be a linear function of thelumbar CSF pH and not of the Pco-.-According to Betz and Heuser18 cortical pH islow and CBF is high in the reactive cerebralhyperemia that follows transient hypoxia, eventhough the cortical Pc0., is low.
Conflicting with the interpretation of theabove studies are the results of previousexperiments using HV of several hours'
Stroke, Vol. 3, September-October 1972
duration. HV in man and animals causes animmediate rise in blood and CSF pH and a fallin CBF. With sustained HV the Pco. remainslow and blood pH high, but the brain ECF pHand bicarbonate as measured in the CSF14 andon the cortex16 progressively fall, becauselactic acid accumulates in the brain and CSF.14
This accumulation of lactate with its accom-panying reduction in CSF buffering capacityhas been invoked to explain why, if the PCo2
1S
abruptly restored to normal after a period ofHV, the CSF pH overshoots and transientlybecomes relatively acidotic. Such a situationcreates a model to test whether the brain ECFpH or the PCo-, most affects CBF. If it is thePCo.,, CBF should drop when hypocapniabegins, remain low until it ends, and return tonormal as PCo2 is restored. If it is the pH ofthe brain ECF, CBF should drop whenhypocapnia begins, rise during hypocapnia asthe pH of the CSF and brain falls, and exceedcontrol when the Pco.. is restored.
Different workers have used parts of thismodel but with discrepant results. At sea levelSeveringhaus and Lassen10 noted no return ofCBF (estimated by cerebral arteriovenousblood gases) toward control in awake humansafter 90 minutes of active HV; CSF pH wasnot recorded. With several hours of passivehyperventilation under general anesthesia,Wollman et al.17 using humans, Plum et al.18
using dogs, and Betz et al.10 using cats allfound that CBF underwent no return towardnormal during several hours of constanthypocapnia. McDowell and Harper,20 usinganesthetized, paralyzed baboons, noted asignificant increase in CBF over control afterthree hours of HV in association with alowered pH of the CSF. However, the level ofanesthesia had changed, making the datadifficult to interpret, especially since CBFremained low during the antecedent period ofHV despite a significant decline in CSF pH.Alexander and his associates21 observed thatCBF in goats rose during continued HV onlywhen the animals were hypoxic, and suggestedthat hypoxia had influenced the studies per-formed in man at high altitude.
In an attempt to resolve these discrepan-cies, Raichle, Posner, and Plum22 re-examinedthe effect of prolonged HV on CBF. Westudied both awake men and anesthetized andawake animals with results that support the pH
567
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from
RAICHLE, PLUM
hypothesis and offer an explanation for theconflicting earlier experimental results. Theresults of our animal HV experiments aresimilar to many that one can find in theliterature, and conflict with both our humandata and the hypothesis that ECF pH controlsCVR. The pattern of changes in PCO;.predictably and significantly differed fromobserved changes in the CSF pH during andafter HV, but CBF appeared to follow thePCo., rather than the CSF pH. However, therewas also a significant decline in CBF of ourcontrol animals during five hours of generalanesthesia, paralysis, and passive ventilation.McDowell and Harper23 also noted a progres-sive and significant decline in CBF over severalhours of observation in paralyzed dogs underhalothane anesthesia. Although they did notcomment on it, Michenfelder and colleagues24
reported a similar decline in CBF in paralyzeddogs receiving halothane anesthesia measuredboth directly by collecting the cerebral venouseffluent from an isolated sagittal sinus, andindirectly using 8BKr.
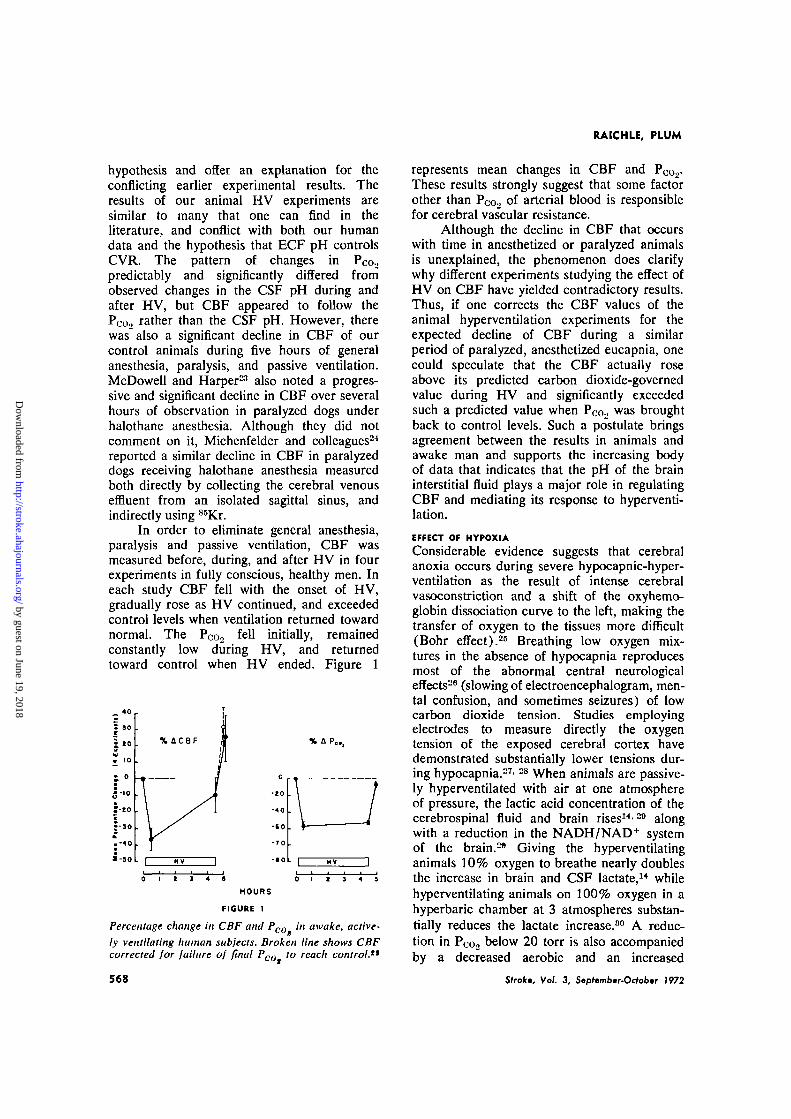
In order to eliminate general anesthesia,paralysis and passive ventilation, CBF wasmeasured before, during, and after HV in fourexperiments in fully conscious, healthy men. Ineach study CBF fell with the onset of HV,gradually rose as HV continued, and exceededcontrol levels when ventilation returned towardnormal. The PCo2 fell initially, remainedconstantly low during HV, and returnedtoward control when HV ended. Figure 1
a-so
HflCBF P..,
o
-to
-40
-to
-TO
- • 0 L L
HOURS
FIGURE 1
Percentage change in CBF and Pco in awake, active-ly ventilating human subjects. Broken line shows CBFcorrected for failure of final Poo to reach control."
568
represents mean changes in CBF and P0o2-These results strongly suggest that some factorother than PCo., of arterial blood is responsiblefor cerebral vascular resistance.
Although the decline in CBF that occurswith time in anesthetized or paralyzed animalsis unexplained, the phenomenon does clarifywhy different experiments studying the effect ofHV on CBF have yielded contradictory results.Thus, if one corrects the CBF values of theanimal hyperventilation experiments for theexpected decline of CBF during a similarperiod of paralyzed, anesthetized eucapnia, onecould speculate that the CBF actually roseabove its predicted carbon dioxide-governedvalue during HV and significantly exceededsuch a predicted value when Pco., was broughtback to control levels. Such a postulate bringsagreement between the results in animals andawake man and supports the increasing bodyof data that indicates that the pH of the braininterstitial fluid plays a major role in regulatingCBF and mediating its response to hyperventi-lation.
EFFECT OF HYPOXIA
Considerable evidence suggests that cerebralanoxia occurs during severe hypocapnic-hyper-ventilation as the result of intense cerebralvasoconstriction and a shift of the oxyhemo-globin dissociation curve to the left, making thetransfer of oxygen to the tissues more difficult(Bohr effect).28 Breathing low oxygen mix-tures in the absence of hypocapnia reproducesmost of the abnormal central neurologicaleffects28 (slowing of electroencephalogram, men-tal confusion, and sometimes seizures) of lowcarbon dioxide tension. Studies employingelectrodes to measure directly the oxygentension of the exposed cerebral cortex havedemonstrated substantially lower tensions dur-ing hypocapnia.27'28 When animals are passive-ly hyperventilated with air at one atmosphereof pressure, the lactic acid concentration of thecerebrospinal fluid and brain rises14-M alongwith a reduction in the NADH/NAD+ systemof the brain.28 Giving the hyperventilatinganimals 10% oxygen to breathe nearly doublesthe increase in brain and CSF lactate,14 whilehyperventilating animals on 100% oxygen in ahyperbaric chamber at 3 atmospheres substan-tially reduces the lactate increase.80 A reduc-tion in Pc0., below 20 torr is also accompaniedby a decreased aerobic and an increased
Strokt, Vol. 3, Sepfemb.r-Octot.r 1972
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

HYPERVENTILATION AND CBF
anaerobic utilization of glucose81 which isreversed during hypocapnic-hyperventilationon 100% oxygen at 3 atmospheres abso-lute.0
Several experiments demonstrate that theinduced brain tissue hypoxia modifies thevasomotor response to HV at low PCo2.Reivich and his colleageus82 demonstrated inhumans that CBF was significantly lower at thesame PCo2 during HV on 100% oxygen at 2.0atmospheres than at sea level pressures.Wollman et al.ss induced metabolic alkalosis inanesthetized subjects by sodium bicarbonateinfusion and controlled P 0 0 2 at 19 torr. Thealkalosis intensified the degree of cerebralhypoxia through the Bohr effect, and resultedin a 17% increase in CBF. Haggendahl andWinso made similar observations.34
Therapeutic ApplicationsThe profound effect of HV on CBF has led toits use in several clinical situations. Fourdeserve particular mention: cerebral infarction,head injury, acute intracranial hypertension,and the operative setting of carotid endarterec-tomy. Before considering each of these situa-tions separately, two general points about theuse of HV as a clinical tool should be made.
First, in evaluating any proposal for theuse of HV as a therapeutic tool, one mustconstantly bear in mind that if properlyperformed, it adds considerably to the com-plexity and expense of patient care. To giveprolonged HV one must control the patient'sairway and constantly monitor the assistivebreathing device. This requires intubation ortracheostomy and meticulous tracheobronchialtoilet. If the patient is paralyzed, as is oftennecessary, even more skilled personnel arerequired.
Second, patients with severe brain injuryfrequently hyperventilate spontaneously.88 Thishas been correlated with a poor prognosis,"5"87
possibly the result of the severity of theneurological injury itself. However, in suchpatients one almost universally finds a subnor-mal or moderately low arterial oxygen tensionin addition to the low PCo2 and elevated pH.Detailed examination of the lungs and airwaysusually reveals serious disease even when thishas not been immediately obvious.88 Such asituation calls for treatment of the underlyinghypoxia. If such treatment is delayed or never
Stroke, Vol. 3, September-October 1972
initiated, one cannot hope to improve theprognosis in these patients.
CEREBRAL INFARCTION
Strokes can severely disrupt the normalregulation of the cerebral circulation inman,88-41 as can experimentally occluding themiddle cerebral artery in animals.42-(B Inareas of infarction the CBF is usually reducedbut occasionally it may be increased in or nearthe area of ischemia, particularly during theearly stages of the lesion. This focal cerebralhyperemia, which may be relative or abso-lute,45 has been termed "luxury perfusion" byLassen38 because of a presumably overabun-dant CBF relative to the metabolic needs of thetissue.
Normally, mechanisms intrinsic to thebrain's vascular bed maintain the CBF inde-pendent of the blood pressure within fairlywide limits—so-called autoregulation. Auto-regulation frequently disappears in areas ofinfarction along with the normal response toacute changes in the arterial Pco.>- "Luxuryperfusion" and loss of vascular autoregulationare postulated to result from an acute,metabolic acidosis localized within the brain,88
although the association is by no meansproved. Whatever the cause, the focal loss ofnormal vasomotor responses, so-called vaso-paralysis, is thought to be the basis ofparadoxical responses that vasodilator andvasoconstrictor agents occasionally induce,e.g., during vasodilatation produced by theinhalation of carbon dioxide CBF in theaffected area decreases.40'40 Conversely, whengeneralized vasoconstriction is produced byhyperventilation, CBF in the affected regioncan increase at a time when flow elsewheredecreases.40 These paradoxical responses pre-sumably occur because the vessels in thediseased region, being maximally dilated, areunable to respond to changes in PCo,- As aresult, with hypercapnia the resistance innormal vessels falls and blood is shunted awayfrom the ischemic focus. With HV the reverseoccurs.40 In addition to these focal changes,widespread loss of CBF autoregulation hasbeen described in patients with stroke,47
occasionally affecting the opposite hemis-phere.
Based on the above features of experi-mental and clinical cerebral infarction, hyper-ventilation has been proposed as a therapeutic
569
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

RAICHLE, PLUM
tool to improve blood flow to the ischemicregion and counteract the tissue acidosis, thehope being to restore normal CBF autoregula-tion, improve cell function, and prevent orreduce cerebral edema.38' *°-41
Unfortunately, the above predictions havenot been borne out by either controlled animalexperiments or observations of patients. Solo-way and his colleagues,47 commencing hyper-ventilation before arterial occlusion andcontinuing after occlusion for three hours, ob-served a significant reduction in the size of theinfarct when compared to controls. Obviouslythe experiment has no applicability to themedical problem of stroke. No effect wasobserved by the same authors when hyperventi-lation was started one hour after occlusion.48
Similar negative results were obtained byYamaguchi et al.40 when they initiated hyper-ventilation four to six hours after arterialocclusion. Brock et aI.B0 found that CBF andneurological signs actually worsened in apatient who received hyperventilation for thefirst time 36 hours after onset of symptoms.Meyer et al.51 observed a reduction in CBF inthe infarcted hemisphere in all patients duringhyperventilation, and on no occasion did theyencounter a paradoxical increase in CBF. Theauthors49-51 concluded that hyperventilationin cerebral infarction was potentially dangerousbecause collateral vessels feeding the ischemicarea, although naturally dilated, retained theresponsiveness to Pco2 and constricted duringhyperventilation. This view is supported by thework of Yamamoto et al.B2 and Yamaguchi40 inexperimental animals.
In contrast to the above studies, Battistiniet al.B8 reported that in cats the area of anexperimental infarction was reduced to aboutone-third of control when they initiatedhyperventilation one-half hour after occlusionand continued for four to six hours. The sameauthors were able to demonstrate an increasein tissue pH to near normal values and asignificant reduction in the water content of thedamaged hemisphere. However, the blood gasvalues reported for their experiment reveal thatthey called Pco., 25 to 30 torr hyperventilation.Since this is the normal PCOo for the cat, itmeans that their control group at Pcoo 40 torrwas actually hypercapnic and they never reallytested feline hypocapnia. In a single patientwith cerebral infarction, Paulson54 was able to
570
restore autoregulation with hyperventilation,but the patient's neurological condition did notchange. In the only controlled study ofpatients, conducted by Christensen,5" artificialhyperventilation to 25 torr for 72 hours did notappear to influence the clinical neurologicaldeficit of the patients. There was no significantdifference in mortality in the hyperventilatedgroup, which was high in both the paralyzedand passively ventilated subjects and con-trols.
Several theoretical reasons argue againsthyperventilating patients with stroke. First,during hyperventilation of even a few hours'duration there is a return of CSF pH and CBFtoward prehyperventilation values,7'22 limitingthe duration of the intended therapeutic effects.Furthermore, upon discontinuation of hyper-ventilation, particularly if abrupt, one has asudden state of hypercapnia,21 which couldtheoretically negate the therapeutic advantagesgained in the antecedent period of hyperventi-lation. As has already been mentioned, there isevidence that the hyperemia surrounding areasof infarction include dilated collateral ves-sels49' B2 supplying the infarcted region. Thesevessels appear to retain their responsiveness tocarbon dioxide and may constrict during HV,reducing blood flow to the ischemic region.Finally, hyperventilation, when properly carriedout for a prolonged period of time, iscomplicated and enhances the risk of pulmo-nary infection.55
HEAD INJURY
Both experimental and clinical observationshave demonstrated that head injury can abolishcerebral autoregulation.B(V-59 Often one can alsofind brain swelling with increased intracranialpressure,80"82 regional increases in CBF,luxury perfusion66-B8' ^ and a CSF lacticacidosis.58 The overall CBF is variably affect-ed63 and may be normal in the face of thepreceding abnormalities.157 As yet, a goodcorrelation has not been established betweenthe quantity of trauma and the development ofthese abnormalities, or between the depen-dence of one abnormality on another. How-ever, it has been proposed that the loss of CBFautoregulation exposes the brain capillariesand veins to an increased intraluminal pres-sure, forcing fluid into the extracellular spaceof the brain.00'81 Increases in systemic arterialpressure, transmitted directly to the capillaries
Strok; Vol. 3, Sepftmber-Octobir 1972
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

HYPERVENTILATION AND CBF
because of the loss of autoregulation, arepostulated to cause an immediate worsening ofthe brain edema.00
Hyperventilation has been advocated inthe treatment of severe head injuries in order toreduce tissue acidosis, restore autoregulation,and reduce increased intracranial pressure,presumably by reducing cerebral blood volumeand decreasing CSF production.154- *-• °*-ns
The effectiveness of short-term hyperven-tilation along with withdrawal of ventricularCSF for the relief of transient increases inintracranial pressure in head-injured patientshas been clearly demonstrated by Johnston andhis co-workers.04 They point out, however, thatonly by continuously monitoring intracranialpressure can this form of therapy be effectivelyused.
Gordon and Rossanda01"'"07 report the onlylarge series of patients treated with prolongedhyperventilation. Their work is a retrospectiveevaluation of 251 patients with severe headtrauma, 51 of whom were hyperventilated(Pcoo 25 to 30 torr) from between six hoursto 41 days (mean ten days). Few details oftheir case selection are given, but they found areduction in mortality from 32.8% to 9.8%.The number of surviving patients with severeneurological sequelae increased substantially inthe treated group, but the percentage recover-ing completely did not differ in the two groups.One must question both the social utility andspecific effectiveness of hyperventilation in thisstudy, particularly since most patients withsevere head injury hyperventilate spontaneous-ly anyway.™
Respiratory abnormalities frequentlycomplicate severe head injuries, often contrib-uting to increased morbidity and mortali-ty «n, 08, no Conventional measures aimed atimproving pulmonary toilet, airway, and oxy-genation reduced mortality from 90% to 40%in one series.70
ACUTE INTRACRANIAL HYPERTENSION
There is much evidence, recently summarizedby Langfitt,71 that when acute intracranialhypertension is caused by a space-occupyinglesion or brain swelling, short-term hyperventi-lation can be lifesaving. Langfitt's concept isthat within finite limits the brain can accom-modate a gradually expanding mass lesion orswelling by a reduction of its CSF compart-ment. When these limits have been reached or
Stroke, Vol. 3, September-October 1972
when sudden increases in volume occur, slightchanges in the size of the mass, the brain, orthe blood volume can cause a marked increasein intracranial pressure, accompanied by trans-tentorial herniation and sudden neurologicaldeterioration. Hypocapnic-hyperventilation, byreducing the cerebral blood volume throughcerebral vasoconstriction, can promptly reducethis pressure below a critical level, allowing onetime to initiate more permanent correctiveaction. However, hyperventilation can be atwo-edged sword under these circumstances.Kitahata et al.,72 using positive pressurebreathing, observed that the intracranial pres-sure declined until the PCo2 reached 23 torr.Beyond this point the intracranial pressureincreased because of an elevated airwaypressure; applying negative expiratory airwaypressure further reduced the intracranial pres-sure.
CAROTID ENDARTERECTOMY
To perform carotid endarterectomy withoutbypass necessitates the temporary occlusion ofthe internal carotid artery. Most patientstolerate this uneventfully, but signs of focalcerebral ischemia and infarction develop in afew. Various techniques have been employedto determine which patients are intolerant tocross clamping. The one receiving widestattention has been to measure the pressure inthe internal carotid artery distal to the clamp(so-called "stump pressure" or carotid arteryback pressure), on the premise that it directlyreflected the perfusion pressure to the affectedhemisphere. Moore and Hall78 found a goodcorrelation between such pressures and clinicaltolerance: all patients who had ischemicsymptoms had pressures less than 25 torr,while all those who tolerated the procedure hadpressures greater than 25 torr.
Based on "stump pressure" measurementsat the time of endarterectomy, Fourcade etal.74 proposed the use of hyperventilationduring this procedure. Working on the premisethat occlusion would lead to cerebral ischemia,vasomotor paralysis, and paradoxical respon-ses, they employed hyperventilation and ob-served a significant increase in stump pressureover control. They concluded that this repre-sented an increased perfusion pressure, andhence shunting of blood to the affectedhemisphere. They did not present data on the
571
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

RAICHLE, PLUM
tolerance of their hyperventilated patientscompared with controls.
Boysen et al.75 and Ladegaard-Pedersen76
combined measurements of "stump pressure"with regional CBF measurements and de-scribed a more complex, although useful,relationship between "stump pressure" andCBF. They found that if stump pressures werehigh during hypocapnia (above 70 torr),hypercapnia induced a significant increase inmean rCBF and a moderate fall in stumppressure. If the hypocapnic occlusion pressurewas about 50 torr, hypercapnia induced asimilar decrease in occlusion pressure but onlya moderate increase in mean rCBF. Finally, ifthe hypocapnic stump pressure was 30 torr orless, regional impaired flow responses wereencountered. It was concluded that patientsmaintaining high stump pressures (greater than50 torr with hypercapnia or 65 torr withhypocapnia) could tolerate any level of Poo.,-Patients whose pressure was below these levelsmight benefit from moderate hypocapnia andcertainly should not be subjected to hypercap-nia.
Agnoli et al.77 and Pistolese et al.7S
observed a 42% reduction in mean hemispher-al CBF with carotid clamping at normocapnia.An additional 18% reduction in flow occurredwhen clamping occurred during hypocapnia,partially due to a fall in mean systemic bloodpressure. They saw no evidence of an improvedblood flow induced by hyperventilation, andconcluded that although hyperventilation wasnot harmful in terms of CBF, it did not offerclear benefit.
As already mentioned, Soloway's exper-imental study47 indicated that hyperventilationcounteracts cerebral tissue hypoxia duringreduced cerebral perfusion: hyperventilationinitiated before occlusion of the middlecerebral artery of the cat reduced the incidenceand size of the consequent infarct comparedwith normocapnic controls.
The above observations leave unclear therole of hyperventilation in carotid endarterec-tomy. When the internal carotid artery stumppressure is below a critical level (about 30torr), blood flow to the affected hemisphere islikely to be marginal and steps must be takento ensure adequate CBF. A shunt or bypass hasproved its effectiveness in this regard (seeDiscussion, ref. 74). Whether hyperventilation
572
is a dependable substitute remains to beseen.
References1. Reivich M: Arterial pCO., and cerebral
hemodynamics. Amer J Physiol 206: 25-35,1964
2. Lessen NA: Brain extracellular fluid pH: Themain factor controlling cerebral blood flow.ScandJ Lab Clin Invest 2: 247-251, 1968
3. Elliott KAC, Jasper HH: Physiological saltsolution for brain surgery: Studies of local pHand pial vessel reaction to buffered andunbuffered isotonic solution. J Neurosurg 6:140-152, 1949
4. Lessen NA, Wahl M, Deet|en P, et a l :Regulation of cerebral arteriolar diameter byextracellular pH variation in the perivascularfluid. In Russell RWR (ed) : Brain and BloodFlow. Pitman Co, Ltd, London, p 174-177, 1971
5. Harper AM, Bell RA: The effect of metabolicacidosis and alkalosis on the blood flowthrough the cerebral cortex. J Neurol Neuro-surg Psychiat 26: 341-344, 1963
6. Siesjo BK, Kjallquist A: A new theory forregulation of extracellular pH in the brain.Scond J Lab Clin Invest 24: 1-9, 1969
7. Severinghaus JW, Chiodi H, Eger El, et a l :Cerebral blood flow in man at high altitude:Role of cerebrospinal fluid pH in normaliza-tion of flow in chronic hypocapnia. CirculationResearch 19: 274-282, 1966
8. Posner JB, Plum F: Spinal fluid pH andneurologic symptoms in systemic acidosis.New Eng J Med 277: 605-613, 1967
9. Kety SS, Polis BP, Nadler CS, et a l : The bloodflow and oxygen consumption of the humanbrain in diabetic acidosis and coma. J ClinInvest 27: 500-510, 1948
10. Agnoli A: Adaptation of CBF during inducedchronic normoxic respiratory acidosis. Scand JLab Clin Invest 22 (Suppl 102) : 8 D, 1968
11. Skinh0j E: CBF adaptation in man to chronichypo- and hypercapnia and Its relation to CSFpH. Scand J Lab Clin Invest 22 (Suppl 102):8:A, 1968
12. Fend V, Vale JR, Broch JA: Respiration andcerebral blood flow in metabolic acidosis andalkalosis in humans. J Appl Physiol 27: 67-76,1969
13. Betz E, Heuser D: Cerebral cortical blood flowduring changes in acid-base equilibrium of thebrain. J Appl Physiol 23: 726-733, 1967
14. Plum F, Posner JB: Blood and cerebrospinalfluid lactate during hyperventilation. Amer JPhysiol 212: 864-870, 1967
15. Severinghaus JW: Regulation of pH on thecerebral cortex. J Physiol 181: 35P, 1965
Stroke, Vol. 3, Stpfembar-Ocfobsr 1972
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

HYPERVENTILATION AND CBF
16. Severinghaus JW, Lassen N: Step hypocapniato separate arterial from tissue pCO2 in theregulation of cerebral blood flow. CirculationResearch 20: 272-278, 1967
17. Wollman H, Smith TC, Stephen GW, et a l :Effects of extremes of respiratory and meta-bolic alkalosis on cerebral blood flow in man. JAppl Physiol 24: 60-65, 1968
18. Plum F, Posner JB, Zee D: The relationship ofcerebral blood flow to CO2 tension in theblood and pH in the cerebrospinal fluidrespectively. Scand J Lab Clin Invest 22 (Suppl102): 8:F, 1968
19. Betz E, Pickerodt V, Weidner A: Respiratoryalkalosis: Effect on CBF, PO2 and acid-baserelation in cerebral cortex with a note on watercontent. Scand J Lab Clin Invest 22 (Suppl102) : IV:D, 1968
20. McDowell DG, Harper A M : CBF and CSF pH inthe monkey during prolonged hypocapnia.Scand J Lab Clin Invest 22 (Suppl 102):VII I :E, 1968
2 1 . Alexander SC, Marshall BE, Agnoli A: Cere-bral blood flow in the goat with sustainedhypocarbia. Scand J Lab Clin Invest 22 (Suppl102): VIII:C, 1968
22. Raichle ME, Posner JB, Plum F: Cerebralblood flow during and after hyperventilation.Arch Neurol 23: 394-403, 1970
23. McDowell DG, Harper A M : Blood flow andoxygen uptake of the cerebral cortex of thedog during anesthesia with different volatileagents. Acta Neurol Scand 14 (Suppl 14) :146-151, 1965
24. Michenfelder JD, Messick JM, Theye RA:Simultaneous cerebral blood flow measured bydirect and indirect methods. J Surg Res 8:475-481, 1968
25. Gote J: The Influence of oxygen affinity ofblood and cerebral blood flow on cerebraloxygen supply. In Brock M, et al (eds) :Cerebral Blood Flow. Springer-Verlag, NewYork, p 75-78, 1969
26. Davis A, Wallace W M : Factors affectingchanges produced in the electroencephalogramby standardized hyperventilation. Arch NeurolPsychiat47: 606-625, 1942
27. Malette WG: Cerebral anoxia resulting fromhyperventilation. Surg Forum 9: 208-215,1959
28. Sugioka K, Davis DA: Hyperventilation withoxygen—a possible cause of cerebral hypoxia.Anesthesiology 2 1 : 135-143, 1960
29. Granholm L, Lukjanova L, Siesjo BK: Theeffect of marked hyperventilation upon tissuelevels of NADH, lactate, pyruvote, phospho-creatine, and adenosine phosphates of rat
brain. Acta Physiol Scand 77: 179-190,1969
30. Plum F, Posner JB, Smith WW: Effect ofhyperbaric-hyperoxic hyperventilation on blood,brain, and CSF lactate. Amer J Physiol 215:1240-1244, 1968
31. Alexander SC, Smith TC, Strobel G, et a l :Cerebral carbohydrate metabolism of manduring respiratory and metabolic alkalosis. JAppl Physiol 24: 66-72, 1968
32. Reivich M, Dickson J, Clark J, et a l : Role ofhypoxia in cerebral circulatory and metabolicchanges during hypocarbia in man: Studies inhyperbaric milieu. Scand J Lab Clin Invest 22(Suppl 102): IV: B, 1968
33. Wollman H, Smith TC, Strobel G, et a l : Effectof extremes of respiratory and metabolicalkalosis on cerebral blood flow in man. J ApplPhysiol 24: 60-65, 1968
34. Haggendahl E, Winso I: Influence of hypoxiaon the response of CBF to hypocapnia. InBrock M, et al (eds) : Cerebral Blood Flow.Springer-Verlag, New York, p 70-74, 1969
35. Rout MW, Lane DJ, Wollner L: Prognosis inacute cerebrovascular accidents in relation torespiratory pattern and blood gas tensions.Brit Med J 3: 7-9, 1971
36. Huang CT, Cook AW, Lyons HA: Severecraniocerebral trauma and respiratory abnor-malities. Arch Neurol 9: 545-554, 1963
37. Vapalahti M, Troupp H: Prognosis for patientswith severe brain injuries. Brit Med J 3: 404-407, 1971
38. Lassen NA: The luxury-perfusion syndromeand its possible relation to acute metabolicacidosis localized within the brain. Lancet 2:1113-1116, 1966
39. Hoedt-Rasmussen K, Skinh0j E, Paulson O, eta l : Regional cerebral blood flow in acuteapoplexy. Arch Neurol 17: 271-281, 1967
40. Paulson OB: Regional cerebral blood flow inapoplexy due to occlusion of the middlecerebral artery. Neurology 20: 63-77, 1970
4 1 . Paulson OB, Olesen J, Christensen MS:Restoration of autoregulation of cerebralblood flow by hypocapnia. Neurology 22: 286-293, 1972
42. Waltz AG, Sundt TM, Owen CA: Effect ofmiddle cerebral artery occlusion on corticalblood flow in animals. Neurology 16: 1185-1 190, 1966
43. Waltz AG: Effect of blood pressure on bloodflow in ischemic cerebral cortex: The phenom-enon of autoregulation and luxury perfusion.Neurology 18: 613-621, 1968
44. Blair RDG, Waltz AG: Regional cerebral bloodflow during acute ischemia: Correlation of
Sfrokt, Vol. 3, Stpf»mbor-Ortober 1972 573
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

RAICHLE, PLUM
autoradiographic measurements with observa-tion of cortical microcirculation. Neurology20: 802-808, 1970
45. Yamaguchl T, Waltz AG, Okazaki H: Hyper-emia and ischemia in experimental cerebralinfarction. Neurology 2 1 : 565-578, 1971
46. Brawley BW, Strandness DE, Kelly WA: Thephysiologic response to therapy in experimen-tal cerebral ischemia. Arch Neurol 17: 180-187, 1967
47. Soloway M, Nadel W, Albin MS, et a l : Theeffect of hyperventilation on subsequent cere-bral infarction. Anesthesiology 29: 975-980,1968
48. Soloway M, Moriarty G, Fraser JG, et a l : Theeffect of delayed hyperventilation on experi-mental middle cerebral artery occlusion.In Russell RWR (ed) : Brain and Blood Flow.Pitman Co, Ltd, London, p 98-101, 1971
49. Yamaguchi T, Regli F, Waltz AG: Effect ofPaCO2 on hyperaemia and ischaemia inexperimental cerebral infarction. In RussellRWR (ed) : Brain and Blood Flow. Pitman Co,Ltd, London, p 191-194, 1971
50. Brock M, Hadjldimos AA, Schurman K:Possible adverse effect of hyperventilation onrCBF during the acute phase of total proximalocclusion of a main cerebral artery. In BrockM, et al (eds) : Cerebral Blood Flow. Springer-Verlag, New York, p 254-257, 1969
51. Meyer JS, Fukuuchi Y, Kanda T, et a l :Interaction between cerebral metabolism andblood flow. In Russell RWR (ed) : Brain andBlood Flow. Pitman Co, Ltd, London, p 156-164, 1971
52. Yamamoto YL, Phillips KM, Hodge CP, et a l :Effect of alteration in arterial carbon dioxideon experimental focal cerebral ischaemia. InRussell RWR (ed) : Brain and Blood Flow.Pitman Co, Ltd, London, p 205-208, 1971
53. Battistinl N, Casacchia P, Fieschl C, et a l :Treatment of experimental cerebral infarctionwith passive hyperventilation. In Russell RWR(ed) : Brain and Blood Flow. Pitman Co, Ltd,London, p 102-106, 1971
54. Paulson OB: Restoration of autoregulation byhypocapnia. In Russell RWR (ed) : Brain andBlood Flow. Pitman Co, Ltd, London, p 313-321, 1971
55. Christensen MS: Stroke treated with prolongedhyperventilation. In Russell RWR (ed) : Brainand Blood Flow. Pitman Co, Ltd, London, p358-364, 1971
56. Taylor AR: Disturbance of autoregulationfollowing head injury—acute and chronicobservation. In Brock M, et al (eds) : CerebralBlood Flow. Springer-Verlag, New York, p 202-204 1969
57. Reivich M, Marshall WJS, Kassell N: Loss ofautoregulation produced by cerebral trau-ma. In Brock M, et al (eds) : Cerebral BloodFlow. Springer-Verlag, New York, p 205-208,1969.
58. Baldy-Moulmier M, Frerebeau C: Cerebralblood flow in cases of coma following severehead injury. In Brock M, et al (eds) : CerebralBlood Flow. Springer-Verlag, New York, p216-218, 1969
59. Zupping R: Cerebral acid-base and gas metab-olism in brain injury. J Neurosurg 33: 498-505, 1970
60. Langfitt TW, Marshall WJS, Kassell NF, et al :The pathophysiology of brain swelling pro-duced by mechanical trauma and hypertension.Scand J Lab Clin Invest 22 (Suppl 102):XIV:B, 1968
61 . Schurta HS, Langfitt TW, Kassell NF: Brainswelling produced by injury and aggravated byarterial hypertension. Brain 9 1 : 281-294,1968
62. Johnston IH, Johnston JA, Jennett B: Intro-cranial pressure changes following head in-jury. Lancet 2: 433-436, 1970
63. Meyer S, Kondo A, Nomura F, et a l : Cerebralhemodynamics and metabolism followingbrain trauma. Demonstration of luxury perfu-sion following brain stem laceration. In BrockM, et al (ed) : Cerebral Blood Flow. Springer-Verlag, New York, p 199-201, 1969
64. Oppelt WW, Maren TH, Owens ES, et al :Effect of acid-base alteration on cerebrospinalfluid production. Proc Soc Exp Biol Med 114:86-89, 1963
65. Rossanda M: Prolonged hyperventilation Intreatment of unconscious patients with severebrain injuries. Scand J Lab Clin Invest 22(Suppl 102): XIII:E, 1968
66. Gordon E, Rossanda M: Artificial hyperventi-lation in the treatment of patients with severebrain lesion. In Brock M, et al (eds) : CerebralBlood Flow. Springer-Verlag, New York, p258-259, 1969
67. Gordon E: Controlled respiration in themanagement of patients with traumatic brainInjuries. Acra Anaesth Scand 15: 193-208,1971
68. Valpalahti M, Troupp H, Heiskanen O:Extremely severe brain injuries treated withhyperventilation and ventricular drainage. InBrock M, et al (eds) : Cerebral Blood Flow.Springer-Verlag, New York, p 266-267,1969
69. Naerra N: Blood-gas analysis in unconsciousneurosurgical patients on admission to thehospital. Acta Anaesth Scand 7: 191-199,1963
574 Stroke, Vol. 3, Sep/omber-October 7972
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

HYPERVENTILATION AND CBF
70. Maciver IN, Frew IJC, Matheson JG: The roleof respiratory insufficiency in the mortality ofsevere head injuries. Lancet I: 390-393,1958
7 1 . Langfirt TW: Increased intracranial pressure.Clin Neurosurg 16: 436-471, 1969
72. Kitahata LM, Galicich JH, Sato I: The effect ofpassive hyperventilation on intraventricularpressure in the dog. J Neurosurg 34: 185-193,1971
73. Moore WS, Hall AD: Carotid artery backpressure. A test of tolerance to temporarycarotid occlusion. Arch Surg 99: 702-710,1969
74. Fourcade HE, et a l : The effect of CO2 andsystemic hypertension on cerebral perfusionpressure during carotid endarterectomy. Anes-thesiology 33: 383-390, 1970
75. Boysen G, Ladegaard-Pedersen HJ, HenriksenH, et a l : The effects of PaCO2 on regionalcerebral blood flow and internal carotidarterial pressure during carotid clamping.Anesthesiology 35: 286-300, 1971
76. Ladegaard HJ, Boysen G, Henriksen H, et a l :Relation of regional cerebral blood flow todistal internal carotid artery pressure duringclamping of the carotid artery. In Russell RWR(ed): Brain and Blood Flow. Pitman Co, Ltd,London, p 336-341, 1971
77. Agnoli A, Fieschi C, Prencipe M, et a l : rCBFstudies during carotid surgery. In RussellRWR (ed) : Brain and Blood Flow. Pitman Co,Ltd, London, p 346-350, 1971
78. Pistolese GR, Citone G, Faraglia V, et a l : Effectof hypercapnia on cerebral blood flow duringthe clamping of the carotid arteries in surgicalmanagement of cerebrovascular insufficiency.Neurology 21:95-102, 1971
Stroke, Vol. 3, September-Odober 7972 575
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from

Marcus E. Raichle and F. PLUMHyperventilation and Cerebral Blood Flow
Print ISSN: 0039-2499. Online ISSN: 1524-4628 Copyright © 1972 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Stroke doi: 10.1161/01.STR.3.5.566
1972;3:566-575Stroke.
http://stroke.ahajournals.org/content/3/5/566World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://stroke.ahajournals.org//subscriptions/
is online at: Stroke Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer available in the
Permissions in the middle column of the Web page under Services. Further information about this process isOnce the online version of the published article for which permission is being requested is located, click Request
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office.Stroke Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on June 19, 2018http://stroke.ahajournals.org/
Dow
nloaded from





























