Embed Size (px)
Citation preview

1
HRB is Essential for Influenza A Virus Replication and Promotes Genome Trafficking in 1
Late-Stage Infection 2
3
Amie J. Eisfeld1,*, Gabriele Neumann1, and Yoshihiro Kawaoka1,2,3,4,5,* 4
5
1University of Wisconsin-Madison, School of Veterinary Medicine, Department of 6
Pathobiological Sciences, Influenza Research Institute, Madison, Wisconsin, USA 7
8
2University of Tokyo, Institute of Medical Science, Division of Virology, Department of 9
Microbiology and Immunology, Tokyo, Japan 10
11
3Division of Virology, Department of Microbiology and Immunology, Institute of Medical 12
Science, University of Tokyo, Tokyo 108-8639, Japan 13
14
4Department of Special Pathogens, International Research Center for Infectious Diseases, 15
Institute of Medical Science, University of Tokyo, 108-8639, Japan 16
17
5ERATO Infection-Induced Host Responses Project, Saitama 332-0012, Japan 18
19
*Corresponding author20
Copyright © 2011, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved.J. Virol. doi:10.1128/JVI.05064-11 JVI Accepts, published online ahead of print on 13 July 2011
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

2
Correspondence should be addressed to: 21
Amie J. Eisfeld, Ph.D. 22
Influenza Research Institute 23
University of Wisconsin-Madison 24
575 Science Drive 25
Madison, WI 53711 26
Phone: (608) 890-2908 27
Fax: (608) 890-2912 28
Email: [email protected]
30
or31
32
Yoshihiro Kawaoka, D.V.M., Ph.D. 33
Influenza Research Institute 34
University of Wisconsin-Madison 35
575 Science Drive 36
Madison, WI 53711 37
Phone: (608) 265-4925 38
Fax: (608) 262-9641 39
Email: [email protected]
41
Running title: HRB promotes influenza vRNP trafficking42
Abstract word count: 23443
Text word count: 514044
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

3
ABSTRACT 45
Influenza A virus uses cellular protein transport systems (e.g., CRM1-mediated nuclear 46
export and Rab11-dependent recycling endosomes) for genome trafficking from the nucleus to 47
the plasma membrane, where new virions are assembled. However, the detailed mechanisms of 48
these events have not been completely resolved and additional cellular factors are probably 49
required. Here, we investigated the role of the cellular human immunodeficiency virus (HIV) 50
rev-binding protein (HRB), which interacts with influenza virus nuclear export protein (NEP), 51
during the influenza virus life cycle. By using siRNAs and overexpression of a dominant-52
negative HRB protein fragment, we show that cells lacking functional HRB have significantly 53
reduced production of influenza virus progeny and that this defect results from impaired viral 54
ribonucleoprotein (vRNP) delivery to the plasma membrane in late-stage infection. Since HRB 55
co-localizes with influenza vRNPs early after their delivery to the cytoplasm, it may mediate a 56
connection between the nucleocytoplasmic transport machinery and the endosomal system, thus 57
facilitating the transfer of vRNPs from nuclear export to cytoplasmic trafficking complexes. We 58
also found an association between NEP and HRB in the perinuclear region, suggesting that NEP 59
may contribute to this process. Our results identify HRB as a second endosomal factor with a 60
crucial role in influenza virus genome trafficking, suggest cooperation between unique 61
endosomal compartments in the late steps of the influenza virus life cycle, and provide a 62
common link between the cytoplasmic trafficking mechanisms of influenza virus and HIV. 63
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

4
INTRODUCTION64
Influenza A virus is a highly contagious respiratory pathogen responsible for up to 0.5 65
million deaths annually (43). Seasonal epidemics are punctuated by rare but recurring 66
pandemics, and recently, highly pathogenic H5N1 avian influenza viruses have caused human 67
infections with high fatality (~60%) (42). These threats, along with the potential emergence of an 68
influenza virus strain with both high transmissibility and high pathogenicity, emphasize the 69
importance of controlling influenza viruses to promote global health. To achieve this, more 70
detailed knowledge of how viruses replicate and interact with their hosts is needed. Any essential 71
replication mechanism or host molecule interaction could be exploited for use in the 72
development of novel prevention or intervention strategies. Recent studies have revealed that 73
host molecules are required for influenza genome transport to newly forming virions (2, 10). 74
Here, we aimed to clarify the mechanisms of influenza genome trafficking further.75
Viral ribonucleoprotein (vRNP) complexes are the genetic elements of influenza virus, 76
and their incorporation into budding virions is a prerequisite for the formation of infectious 77
viruses. vRNPs are composed of individual negative-sense viral RNAs (vRNA) associated with 78
viral nucleoprotein (NP) and the heterotrimeric polymerase complex (PB2, PB1 and PA). A set 79
of eight vRNPs representing the eight unique genome segments is required for an influenza A 80
virus to be infectious. Upon infection, vRNPs within virions are released from endosomes and 81
transported to the nucleus where they serve as templates for the production of viral protein-82
encoding mRNAs and positive-sense complementary RNAs (cRNA). Subsequently, the cRNAs 83
serve as templates for vRNA synthesis. In late-stage infection, new vRNPs are assembled in the 84
nucleus and must undergo both nuclear export and transport across the cytoplasm to gain access 85
to the viral budding sites at the plasma membrane. 86
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

5
Influenza virus utilizes cellular protein trafficking systems to facilitate the journey of 87
vRNPs from the nucleus to the plasma membrane. vRNP nuclear export ensues through the 88
coordinated actions of the M1 matrix protein—which associates with vRNP—the cellular CRM1 89
nuclear export receptor, and the viral nuclear export protein (NEP) (6, 11, 22-24, 27, 31, 35, 38, 90
40). NEP encodes a nuclear export signal, binds CRM1 and M1, and is thought to bridge the 91
complex between M1-vRNP and the cellular nuclear export machinery (1, 27, 31). After nuclear 92
export, vRNPs accumulate at the microtubule organizing center (MTOC) (2, 24), where they 93
associate with the cellular Rab11 GTPase, a major component of recycling endosomes (2, 10). 94
vRNPs are later observed in punctate foci that co-localize with Rab11 in the peripheral 95
cytoplasm, and are transported to the cell surface in a manner dependent on Rab11 GTPase 96
activity (2, 10); microtubules also may be involved in this process (2, 24). Near the plasma 97
membrane, the vRNP foci coalesce and dissociate from Rab11 for their presumed incorporation 98
into budding virions (10). While it is clear that cellular protein trafficking systems are necessary 99
for influenza virus genome transport, additional unknown cellular factors likely contribute to this 100
process.101
Previously, the influenza virus NEP protein was shown to interact with the cellular 102
human immunodeficiency virus (HIV) rev-binding protein (HRB) in a yeast two-hybrid system 103
(31), but its role in the influenza virus life cycle was not examined. HRB contains a putative Arf 104
GTPase activating protein (GAP) domain at its N-terminus (7, 33), regulates cellular endocytic 105
processes (7, 8, 17, 33, 37), and may be a component of the nuclear pore complex (NPC) (12, 13, 106
29). We hypothesized that HRB contributes to influenza vRNP trafficking through effects on 107
either nucleocytoplasmic transport or vesicular transport systems. By using a combination of 108
siRNA-mediated protein knockdown and co-immunofluorescence analyses, we found that HRB 109
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

6
is critical for the efficient production of influenza virus progeny and is involved in mediating 110
vRNP transport to the plasma membrane. Our results highlight the complex nature of influenza 111
vRNP trafficking and suggest interplay between multiple endocytic compartments in vRNP 112
delivery to cell surface sites of influenza virus formation. 113
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

7
MATERIALS AND METHODS 114
Cells, viruses, and infections. Transformed human embryonic kidney cells (293), 115
human lung carcinoma cells (A549), Madin-Darby canine kidney cells (MDCK), human cervical 116
adenocarcinoma cells (HeLa), baby hamster kidney cells (BHK), and Cercopithecus aethiops117
kidney fibroblast cells (CV-1) were cultured at 37°C in 5% CO2 in the following media: 118
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum 119
(FBS) (293 and CV-1), DMEM containing 5% FBS (BHK), a 1:1 mixture of DMEM and Ham’s 120
F12 nutrient mixture supplemented with 10% FBS (A549), minimum essential medium (MEM) 121
supplemented with 5% newborn calf serum (MDCK), or MEM supplemented with 10% FBS 122
(HeLa). All media contained 50 units/ml of penicillin and 50 μg/ml of streptomycin (Invitrogen, 123
Carlsbad, CA). 124
Influenza A/WSN/33 (H1N1; WSN) was generated by using plasmid-based reverse 125
genetics as described previously (28), and virus stock amplification and plaque assays were 126
performed in MDCK cells. Vesicular stomatitis virus (VSV, strain Indiana), vaccinia virus (VV, 127
strain Ankara; a kind gift from Dr. Paul Ahlquist, University of Wisconsin-Madison), and 128
adenovirus 5 (Ad5, strain McEwen; a kind gift from Dr. Paul Kinchington, University of 129
Pittsburgh) stock virus amplifications and plaque assays were performed in BHK, CV-1, or A549 130
cells, respectively. For multi-cycle growth experiments after siRNA treatment, cells grown in 24-131
well plates were infected with 100 plaque-forming units (PFU) of the indicated virus by direct 132
inoculation of the culture medium. Supernatants were collected from influenza virus- and VSV-133
infected cells at 48 h and 24 h, respectively, and virus titrations were performed in MDCK or 134
BHK cells. To harvest VV or Ad5, infected monolayers were scraped into the overlying media at 135
48 h, and the mixtures were subjected to three consecutive freeze-thaw cycles followed by 136
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

8
centrifugation to clear insoluble debris. The resultant supernatants were subjected to virus 137
titration in CV-1 or A549 cells for VV and Ad5, respectively. For similar experiments after 138
plasmid transfection, cells in 12-well plates were inoculated with 200 PFU of WSN and 139
supernatants were harvested at 72 h for plaque assays in MDCK cells. For immunofluorescence 140
and immunoblot experiments, cells were infected with WSN as described in the Figure Legends. 141
siRNAs, plasmids and transfections. siRNA transfections were performed as 142
previously described (10). The final concentration of all siRNAs in the culture medium was 20 143
nM, and siRNA transfections were allowed to proceed for 48 h before subsequent plasmid 144
transfections or virus infections. The following siRNAs were used: an extensively validated non-145
targeting siRNA (AllStars Neg; Qiagen, Valencia, CA; cat. no. 1027281); a previously described 146
siRNA targeting influenza NP mRNA (NP-1496; synthesized by Qiagen) (14); a previously 147
described siRNA targeting the human AGFG1 (HRB) gene (synthesized by Qiagen) (36, 45); 148
and a validated mixture of siRNAs targeting essential host survival genes (Death Control, Qiagen 149
cat. no. SI04381048). 150
To generate the HRB dominant-negative mutant, total RNA was isolated from A549 cells 151
using the RNeasy Mini Kit (Qiagen), and poly-A RNA was reverse transcribed using SuperScript 152
II reverse transcriptase and an oligo-dT primer (Invitrogen) according to the manufacturers’ 153
instructions. A segment of the AGFG1/HRB gene corresponding to amino acids 361–562 154
( N360) was amplified from the resultant cDNA using gene-specific primers with 5’ Xho I and 155
3’ Hind III overhangs and was inserted in-frame with enhanced green fluorescent protein (GFP) 156
in the pEGFP-N1 vector (Clontech, Mountain View, CA), producing the p N360-GFP plasmid. 157
The plasmid expressing NEP fused to enhanced yellow fluorescent protein (YFP) was created by 158
first PCR amplifying YFP (Clontech) with 5’ Not I and 3’ Bgl II overhangs and then inserting 159
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

9
the product into the pCAGGS/MCS (18, 30) backbone. The WSN NEP open reading frame 160
lacking the stop codon was then amplified by using 5’ Eco RI and 3’ Not I overhangs and 161
inserted upstream and in-frame with YFP in pCAGGS/MCS to produce pNEP-YFP. All 162
plasmids were sequenced to confirm proper fragment insertion. Primer sequences are available 163
upon request. For viral replication studies, 293 cells were transfected with plasmid DNAs by the 164
calcium phosphate precipitation method using a calcium phosphate transfection kit (Invitrogen) 165
according to the manufacturer’s recommendations. All other plasmid transfections were 166
performed with the TransIT LT1 transfection reagent (Mirus Bio, Madison, WI). 167
Cell viability assay. Cell viability was determined by assaying total intracellular ATP 168
with the CellTiter-Glo kit (Promega, Madison, WI) according to the manufacturer’s 169
recommendations, with some modifications. Briefly, at the times indicated in the Figure, 170
triplicate cultures of siRNA-treated 293 cells in 24-well dishes were lysed directly with 100 μl of 171
Glo Lysis Buffer (Promega) for 15 minutes at room temperature. Subsequently, 50 μl of the 172
resultant lysate was mixed with an equal volume of CellTiter-Glo reagent and luminescence was 173
read directly using a Tecan microplate reader (Tecan Group Ltd, Switzerland). 174
Mini-replicon assay. In vitro viral polymerase activity in siRNA or plasmid-transfected 175
293 cells was compared using a dual luciferase reporter assay system (Promega) according to the 176
manufacturer’s instructions. Briefly, 48 h after siRNA transfection, triplicate cultures in 24-well 177
plates were transfected with a plasmid expressing a firefly luciferase reporter under the control of 178
the influenza WSN NA terminal genome sequences (pPolWSNNA F-Luc; 0.025 μg), together 179
with the protein expression plasmids pCAGGS-PB2, pCAGGS-PB1, pCAGGS-PA, and 180
pCAGGS-NP, which express the WSN polymerase complex proteins (0.25 μg each) (20, 28). 181
Cells were also co-transfected with an internal control plasmid (0.025 μg) expressing Renilla182
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

10
luciferase regulated by the herpes simplex virus type 1 thymidine kinase promoter (pGL.74; 183
Promega), which exhibits basal transcriptional activation in mammalian cells. Firefly and Renilla184
luciferase activities were measured 48 h after plasmid transfection by using the Tecon microplate 185
reader. The level of viral gene expression was determined after normalization to the level of 186
cellular gene expression from the same sample (firefly luciferase light units/Renilla luciferase 187
light units). This produced a ratio indicative of specific effects on viral polymerase activity; the 188
average ratio for triplicate reads is reported.189
Immunological assays. Co-immunofluorescence and immunoblotting were performed 190
as described previously (10). The following primary antibodies were used: monoclonal mouse 191
anti-HRB (Santa Cruz Biotechnology, Santa Cruz, CA; catalog no. sc-166651; used at 1:100 for 192
immunofluorescence and 1:500 for immunoblotting); polyclonal rabbit anti-calnexin (Santa Cruz 193
Biotechnology cat. no. sc-11397, used at 1:5000 for immunoblotting); mouse anti-GFP antibody 194
(Clontech cat. no. 632375; used at 1:20,000 for immunoblotting); a previously described 195
polyclonal rabbit anti-serum against influenza vRNPs (R528; used at 1:5000 for immunoblotting 196
and 1:2500 for immunofluorescence) (26); polyclonal rabbit anti-actin (Santa Cruz 197
Biotechnology cat. no. sc-10731, used at 1:1000 for immunoblotting); a previously described 198
rabbit anti-serum against influenza NEP (R5023; used at 1:200 for immunofluorescence) (27); 199
and a previously described monoclonal mouse antibody that recognizes influenza NP in the form 200
of vRNP (MAb 3/1; used at 1:1000 for immunofluorescence) (10). For secondary detection in 201
immunoblot analysis, we used horseradish peroxidase-conjugated antibodies, including goat anti-202
rabbit (Invitrogen; 1:2000) and goat anti-mouse (Thermo Scientific, Rockford, IL; 1:2000). 203
Proteins were detected with the Lumi-Light PLUS western blotting substrate (Roche, 204
Indianapolis, IN), Kodak Biomax XAR film, and a Konica Minolta SRX-101A X-ray film 205
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

11
developer. Alexa Fluor (AF) 546-conjugated goat anti-mouse and AF 488-conjugated goat anti-206
rabbit antibodies were used for secondary detection in immunofluorescence analyses (Invitrogen; 207
1:1000). Where indicated in the Figure Legends, nuclei were stained with 0.4 μg/ml Hoechst 208
33258 (Invitrogen), or cells were treated with 20 ng/ml of leptomycin B (LMB) (Sigma-Aldrich, 209
St. Louis, MO) or an equivalent volume of dimethyl sulfoxide (DMSO). All fluorescence 210
images were captured with a Zeiss LSM 510 Meta point-scan laser microscope system, as 211
previously described (10). Images were exported as TIFF files and cropped using Adobe CS4 212
software, but were otherwise unaltered. 213
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

12
RESULTS 214
HRB expression and function are essential for efficient influenza virus production. 215
HRB is a critical host factor in the HIV life cycle, and both siRNA-mediated knockdown of HRB 216
expression and overexpression of a C-terminal fragment of the HRB protein ( N360) strongly 217
impair HIV replication (45). To determine whether HRB is involved in the influenza virus life 218
cycle, we examined the effect of reducing HRB expression or overexpressing the HRB N360 219
fragment on influenza A/WSN/1933 (H1N1; WSN) multi-cycle growth in 293 cells. HRB-220
specific siRNA treatment resulted in efficient protein knockdown without affecting a non-221
targeted cellular protein (calnexin; CANX). Parallel transfections with non-targeting control 222
siRNA (AllStars Neg) or siRNA targeting influenza NP mRNA did not result in knockdown of 223
either HRB or CANX (Figure 1A). Further, cells treated with siRNA targeting HRB exhibited 224
no major differences in cellular morphology (data not shown) or viability (Fig. 1B) compared to 225
cells treated with non-targeting or NP-specific siRNA controls over a 96-h time course. This was 226
in contrast to cells treated with death-inducing siRNAs, in which clear morphological changes 227
consistent with cytotoxicity (data not shown) and a sharp reduction in viability at the 96-h time 228
point (Fig. 1B) were observed. Because these data indicated that prolonged reduction in HRB 229
expression was not detrimental to cell viability, we next tested the effects of the HRB siRNA on 230
multi-cycle influenza virus growth. HRB siRNA induced a statistically significant (P = 0.0062), 231
287-fold reduction in WSN titer relative to cells treated with the AllStars negative control siRNA 232
(Fig. 1C). This result was highly reproducible and observed in three independent experiments. 233
Moreover, similar treatments did not cause a reduction in vesicular stomatitis virus (VSV) or 234
adenovirus type 5 (Ad5) replication and led to only a minor reduction in vaccinia virus 235
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

13
replication (Fig. 1D). Therefore, while cells exhibiting significantly reduced HRB expression 236
remained viable, they did not support the efficient production of progeny influenza viruses. 237
To corroborate these findings, we assessed multi-cycle growth in cells overexpressing the 238
HRB N360 protein fragment fused to green fluorescent protein (GFP), which was previously 239
shown to act in a dominant-negative manner (36). We used GFP alone as a negative control and 240
influenza NEP fused to yellow fluorescent protein (YFP) as a positive control, as our earlier 241
work indicated that NEP overexpression impairs influenza replication (data not shown). We 242
observed abundant levels of all plasmid-expressed proteins (Fig. 1E) and efficient transfection 243
efficiency, with fluorescence in at least 75% of cells under all conditions by 48 h post-244
transfection (data not shown). Plasmid-transfected cells were infected with influenza WSN at 245
this time, and at 72 h post-infection (hpi) cells expressing either NEP-YFP or N360-GFP246
exhibited greater than 200-fold reductions in WSN titers relative to cells expressing only GFP 247
(Fig. 1F). Taken together, our results show a critical dependence of influenza virus on HRB 248
expression and function for the generation of infectious virus particles. 249
HRB knockdown does not impair influenza virus entry or viral gene expression.250
HRB interacts with adaptor-related protein complex 2 (AP2), which is involved in clathrin-251
mediated endocytosis (37), and EPS15, a mediator of EGFR endocytosis (8) and is a regulator of 252
clathrin-dependent endocytosis and endocytic sorting (7, 17, 33). Because influenza virus enters 253
cells through a clathrin-dependent endocytosis mechanism (19) and virus entry depends on 254
EGFR signaling (9), we wondered whether HRB knockdown might impair influenza virus entry. 255
To test this, we assessed viral gene expression in WSN-infected cells after HRB or AllStars Neg 256
control siRNA treatments. Cells treated with either siRNA and subjected to immunofluorescence 257
staining with an NP-specific antibody exhibited a comparable number of infected cells and a 258
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

14
similar fluorescence signal at 6 hpi (data not shown). Consistent with this finding, we saw no 259
differences in influenza virus NP expression at 8 or 11 hpi (Fig. 2A). These results suggest that 260
HRB knockdown does not impair influenza virus entry or gene expression and that HRB 261
functions at a later stage in the influenza virus life cycle. 262
To further determine whether the disruption of HRB expression or function interferes 263
with viral polymerase activity, we evaluated the effects of HRB siRNA treatment or 264
overexpression of the N360-GFP fragment in a mini-replicon system, which provides an 265
overall representation of the efficiency of vRNA, cRNA and mRNA production. Cells were 266
treated with siRNAs or plasmids expressing fluorescently tagged proteins, as described above, 267
and were transfected with plasmids encoding the WSN polymerase complex proteins (PB2, PB1, 268
PA and NP) and a viral segment encoding a reporter firefly luciferase gene. Using this assay, we 269
detected a minimal effect on viral reporter gene expression with HRB-specific siRNA and no 270
effect with overexpression of N360-GFP (Fig. 2B). This was in contrast with strong reductions 271
in viral reporter gene expression observed with either NP-specific siRNA or NEP-YFP protein 272
expression. Together, these data imply that HRB functions in the late stage of influenza virus 273
infection and is not involved in regulating influenza virus entry, polymerase activity or protein 274
expression.275
HRB and NEP partially co-distribute in the cytoplasm during influenza virus 276
infection. Although HRB interacts with NEP in a yeast two-hybrid system, an association in the 277
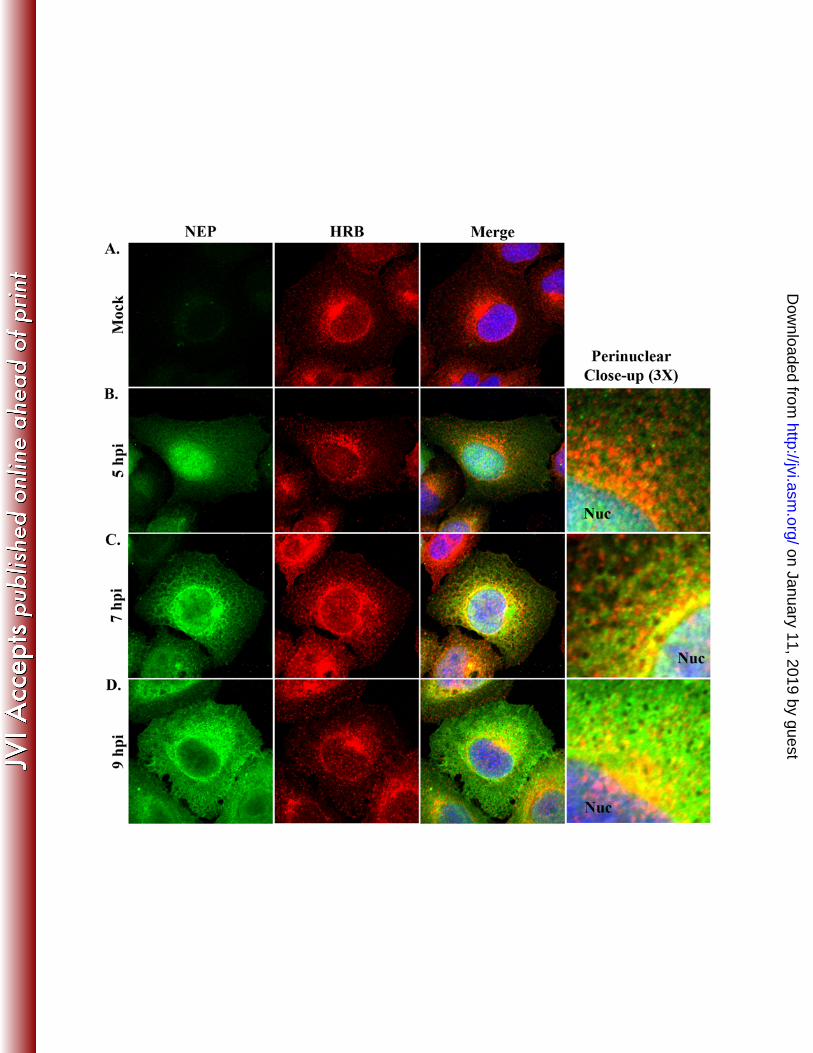
context of influenza virus infection has not been demonstrated. In uninfected cells, HRB exhibits 278
juxtanuclear and punctate cytoplasmic localization, with occasional low-level accumulation in 279
the nucleus (7, 33). This distribution pattern is distinct from the generally diffuse nuclear and 280
cytoplasmic profile of NEP (6, 11, 22, 40). To determine whether influenza virus infection 281
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

15
induces changes in the HRB staining pattern, and to specifically address whether HRB and NEP 282
associate in influenza virus-infected cells, we performed co-immunofluorescence analysis of 283
WSN-infected A549 cells.284
As in previous reports (7, 33), HRB in mock-infected cells was observed in peripheral 285
cytoplasmic foci and accumulated in the perinuclear region, with occasional diffuse localization 286
in the nucleus (Fig. 3A). The overall HRB distribution pattern in WSN-infected cells was 287
comparable to that of mock-infected cells at all time points (Fig. 3B-D). At 5 hpi, NEP exhibited 288
prominent nuclear localization, with some diffuse staining in the cytoplasm and no co-289
localization with HRB (Fig. 3B). By 7 hpi, NEP began to accumulate in the perinuclear region of 290
the cytoplasm and partially co-localized with HRB immediately adjacent to the nucleus (Fig. 291
3C). Of note, NEP also exhibited a mesh-like cytoplasmic staining pattern that was reminiscent 292
of cellular cytoskeletal filaments (Fig. 3C); this localization pattern has not been described 293
previously. By 9 hpi, NEP nuclear levels were consistently reduced, perinuclear levels were 294
increased, and partial co-distribution with HRB occurred near the nuclear membrane (Fig. 3D). 295
Thus, our data reveal novel details about the localization of NEP in late-stage infected cells and 296
suggest that NEP and HRB may interact in the perinuclear region.297
HRB and vRNP co-localize at the MTOC in influenza virus-infected cells. We298
previously demonstrated that cytoplasmic influenza vRNPs undergo multi-stage trafficking 299
following nuclear export (10). Specifically, vRNPs initially accumulate at the MTOC, are co-300
transported with Rab11 through the cytoplasm in punctate foci, and accumulate at the plasma 301
membrane in late-stage infected cells. Since the HRB localization pattern in infected cells 302
mirrors that of vRNPs (Fig. 3), we hypothesized that HRB co-localizes with vRNPs to promote 303
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

16
trafficking. To test this hypothesis, we examined the HRB spatio-temporal distribution pattern in 304
relation to influenza vRNPs in WSN-infected A549 cells.305
To detect vRNPs by immunofluorescence analysis in influenza virus-infected cells, we 306
previously used a mouse monoclonal antibody (MAb 3/1) against influenza NP that exhibits a 307
punctate cytoplasmic staining pattern and nearly completely co-localizes with cytoplasmic 308
vRNA (10). Because we could not use MAb 3/1 with mouse anti-HRB (also a MAb) for indirect 309
co-immunofluorescence analyses, we needed a compatible antibody that could identify vRNPs. 310
The R528 rabbit polyclonal anti-serum (used in Fig. 2A) was generated against purified 311
influenza vRNPs and has been used to identify NP by immunofluorescence (26). Therefore, we 312
tested whether R528 could detect influenza vRNPs by performing co-immunofluorescence with 313
MAb 3/1. Both antibodies exhibited nearly identical staining patterns, recognizing NP in the 314
nucleus, accumulating at the MTOC and in punctate foci in the peripheral cytoplasm (Fig. 4). 315
Because MAb 3/1 is known to recognize vRNPs, we interpret these results to indicate that R528 316
also identifies trafficking vRNPs in the cytoplasm of influenza virus-infected cells.317
To assess the relationship between HRB and vRNP during infection, we next used R528 318
with mouse anti-HRB in co-immunofluorescence analysis. Similar to previous results (Fig. 3), 319
HRB was primarily juxtanuclear at 5 and 7 hpi (Fig. 5A and B). At 5 hpi, vRNPs were restricted 320
to the nucleus and, as such, did not co-localize with the predominantly cytoplasmic HRB (Fig. 321
4A). Upon export from the nucleus at 7 hpi, vRNPs co-localized with HRB in the MTOC region 322
(Fig. 5B, panel i). Some vRNPs could also be seen en route to the plasma membrane in close 323
proximity to HRB (Fig. 5B, panel vi). At 9 hpi, vRNPs were distributed throughout the 324
peripheral cytoplasm and near the plasma membrane, and no longer prominently co-localized 325
with HRB at the MTOC (Fig. 5C). By 11 hpi, vRNPs were observed in abundance near the 326
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

17
plasma membrane, and although some HRB was also dispersed in this region, no specific co-327
localization with vRNP was observed (Fig. 5D, panels ii and vi). These data suggest a potential 328
interaction between HRB and vRNP in the MTOC region and indicate that HRB is not a 329
component of transport complexes that deliver vRNPs to the plasma membrane. 330
HRB does not directly mediate vRNP nuclear export. HRB associates with the 331
cellular nuclear export factor CRM1 (29) and influenza NEP (31), both of which are required for 332
influenza vRNP nuclear export (27, 31). Indeed, the NEP nuclear export signal is required for its 333
association with HRB in the yeast two-hybrid assay (31). To clarify whether HRB participates in 334
vRNP nuclear export by facilitating an interaction between vRNP-M1-NEP complexes and 335
CRM1, we examined HRB, NEP, and vRNP distribution in cells treated with leptomycin B 336
(LMB), a specific inhibitor of CRM1. We reasoned that if HRB is required to bridge the 337
interaction between vRNP-M1-NEP and CRM1, then LMB treatment may result in HRB nuclear 338
accumulation and co-localization with vRNP or NEP.339
In mock-infected cells treated with LMB, we observed little to no accumulation of HRB 340
in the nucleus, and the HRB distribution pattern was similar to that observed in DMSO-treated 341
control cells, except that LMB treatment frequently resulted in more evenly distributed HRB 342
around the periphery of the nucleus (Fig. 6A). This indicates that HRB does not shuttle between 343
the nucleus and the cytoplasm in a CRM1-dependent manner. In influenza virus-infected cells, 344
LMB treatment induced nearly complete vRNP nuclear retention and significantly increased the 345
nuclear levels of NEP in most cells, consistent with previous findings (11, 22, 24, 40), but it did 346
not cause HRB nuclear accumulation (Fig. 6B and C). As a consequence of vRNP nuclear 347
accumulation and HRB cytoplasmic localization, vRNPs and HRB did not co-localize in the 348
presence of LMB (Fig. 6B). LMB treatment also reduced co-localization between HRB and 349
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

18
NEP in the perinuclear region, although some cytoplasmic forms of NEP persisted and low 350
levels of co-localization could be observed at the MTOC (Fig. 6D). These data argue against a 351
role for HRB in mediating interactions that promote influenza vRNP nuclear export and imply 352
that the HRB promotes cytoplasmic, rather than nuclear, activities of influenza virus.353
HRB knockdown provokes aberrant vRNP cytoplasmic trafficking in late-stage 354
infection. Our results excluded a role for HRB in influenza virus entry and polymerase activity, 355
and suggested that HRB may function in vRNP cytoplasmic transport but not vRNP nuclear 356
export. To directly test this possibility, we assessed vRNP trafficking in HRB siRNA-treated 357
cells, by using co-immunofluorescence of HRB and vRNPs. 293 cells were not suitable for these 358
studies because of their low cytoplasmic volume and weak adherence properties, and HRB 359
siRNA treatments in A549 cells induced insufficient knockdown of HRB protein expression to 360
impair influenza virus growth (data not shown). However, we observed efficient and specific 361
reduction in HRB protein expression in HeLa cells (Fig. 7A). Although influenza virus 362
undergoes abortive infection in this cell-type, the blocks occur principally at the levels of virus 363
entry and viral budding (15). Therefore, we used HeLa cells as an alternative model system to 364
examine influenza vRNP trafficking in the absence of abundant HRB protein expression. Of 365
note, non-targeting siRNA-treated mock-infected HeLa cells exhibited HRB distribution similar 366
to that observed in untreated A549 cells, with juxta- or perinuclear accumulation accompanied 367
by punctate foci in the peripheral cytoplasm (Fig. 7B). 368
By 6 h after influenza virus infection, cells in both siRNA treatments exhibited prominent 369
nuclear vRNP staining, despite appreciable differences in the level of HRB protein expression 370
(Fig. 7C). This result confirms our previous finding that the lack of HRB does not impair 371
influenza virus uptake or inhibit viral gene expression. At 9 hpi, HeLa cells treated with non-372
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

19
targeting siRNAs exhibited vRNPs in the perinuclear region and scattered throughout the 373
cytoplasm, with some vRNP accumulation near the plasma membrane (Fig. 7D). At the same 374
time point in HRB siRNA-treated cells, vRNPs accumulated in the perinuclear region, exhibiting 375
tight association with the nuclear membrane, and were only minimally observed in the peripheral 376
cytoplasm or at the plasma membrane. The effect of siRNA-mediated HRB knockdown was 377
even more pronounced at 15 hpi (Fig. 7E). No obvious differences in vRNP nuclear export were 378
observed for either siRNA transfection condition at any time point (Fig, 7C–E). These data 379
suggest that cells with minimal HRB expression remain competent for vRNP nuclear export, but 380
are impaired in their ability to transport vRNP complexes from the perinuclear region to the 381
plasma membrane. 382
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

20
DISCUSSION 383
Previous studies have implicated the cellular CRM1 nuclear export and Rab11 recycling 384
endosome pathways in influenza vRNP transport from the nucleus to plasma membrane sites of 385
virion formation (2, 10, 11, 22, 27, 40). However, the detailed mechanisms of vRNP trafficking 386
and the requirement for additional cellular co-factors remain to be illuminated. Here, we 387
identified a second cellular endosomal factor integral to vRNP intracellular transport, the HRB 388
Arf GAP protein. Our results clearly indicate that HRB expression and function are essential for 389
efficient production of influenza virus progeny and implicate HRB in regulating vRNP 390
trafficking from the perinuclear region to the plasma membrane. Since HRB has a role in both 391
nucleocytoplasmic and endosomal trafficking, we suggest that HRB operates as a linker between 392
these systems in influenza virus-infected cells, to facilitate vRNP cell surface delivery and 393
formation of infectious virus particles. 394
Upon its discovery, HRB was hypothesized to contribute to nucleocytoplasmic 395
trafficking due to its ability to bind HIV rev and facilitate rev function (3, 8, 13), its interaction 396
with the nuclear export receptor CRM1 (12, 29), and the presence of multiple nucleoporin-like 397
phenylalanine-glycine (FG) repeats in its C-terminus (13). Given these observations and the 398
identification of HRB as an interaction partner for influenza NEP (31)—a known associate of 399
CRM1 and an integral mediator of influenza vRNP nuclear export—we considered the potential 400
for HRB to affect vRNP nuclear export to be important. However, several lines of evidence 401
argue against this possibility. First, HRB exhibited exclusively cytoplasmic distribution in most 402
cells (infected and mock-infected), and did not consistently accumulate in the nucleus after 403
influenza virus infection. Second, LMB treatment did not increase HRB nuclear accumulation in 404
either mock-infected or infected cells, indicating that HRB does not traffic through the CRM1-405
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

21
dependent nuclear export pathway, which is required for influenza vRNP nuclear export (27). 406
Most importantly, siRNA-mediated knockdown of HRB protein expression had no affect on 407
vRNP nuclear export, and instead induced accumulation of vRNPs in the perinuclear region. 408
These observations strongly imply that HRB does not regulate vRNP nuclear export, but rather 409
participates in an early event in the vRNP cytoplasmic trafficking mechanism. 410
The tight association of vRNPs with the outer periphery of the nucleus in HRB siRNA-411
treated cells suggests that HRB may be involved in the release of vRNPs from CRM1-RanGTP 412
nuclear export complexes. CRM1 associates with cargo in the nucleus in a RanGTP-dependent 413
manner, and cargo-CRM1-RanGTP complexes are transported to the cytoplasm through the 414
nuclear pore complex (NPC) (32). On the cytoplasmic face of the NPC, RANBP1 and RANBP2 415
cooperate to facilitate RanGTP hydrolysis to RanGDP, thereby inducing the disassociation of 416
RanGDP and CRM1 and the release of cargo into the cytoplasm (16). Importantly, expression of 417
a mutant Ran protein deficient in GTP hydrolysis results in CRM1 accumulation at the nuclear 418
periphery (16). HRB preferentially accumulates at the periphery of the nucleus, contains a 419
putative GTPase activating protein (GAP) domain in its N-terminus (33) and interacts with 420
CRM1 (29). In influenza virus-infected cells, it is therefore conceivable for HRB to interact with 421
CRM1 and RanGTP-associated vRNP nuclear export complexes, possibly assisted by 422
interactions with NEP, to promote RanGTP hydrolysis following their transit across the NPC. A 423
lack of HRB expression would then result in the accumulation of unhydrolyzed RanGTP at the 424
nuclear periphery and the failure of CRM1 and vRNP to dissociate from the perinuclear region in 425
late-stage infection. This concept is consistent with our observations for HRB-specific siRNA 426
treated, influenza virus-infected cells, where vRNPs were abundantly retained at the periphery of 427
the nucleus. The HRB GAP domain is predicted to affect Arf GTPases, and to our knowledge no 428
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

22
Arf GAP protein is known to influence RanGTP. However, other GAP proteins exhibit broad 429
specificity against multiple small GTPases (4, 21, 39, 44). The verification of authentic HRB 430
GAP activity and identification of HRB-targeted small GTPases will be necessary to clarify the 431
potential role of HRB in modulating RanGTP at the nuclear membrane. 432
HRB may also facilitate other protein-protein interactions that promote vRNP recruitment 433
to cytoplasmic transport complexes. In addition to its N-terminal putative Arf GAP domain, 434
HRB contains multiple linear protein interaction motifs, including an established VAMP7 435
binding domain; a consensus clathrin binding motif and three consensus AP2 appendage binding 436
motifs; multiple FG repeats typical of nucleoporins; and four C-terminal asparagine-proline-437
phenylalanine (NPF) motifs, which mediate interactions with proteins containing an EPS15 438
homology (EH) domain (8, 33, 36). Several EH domain-containing proteins (e.g., EHD1, EHD3, 439
and EHD4) associate with endocytic recycling compartments containing Rab11 (25). Therefore, 440
HRB could recruit vRNPs to Rab11 cytoplasmic transport complexes through coordinate 441
interactions with NEP-associated vRNPs and an EH domain-containing protein. In support of 442
this idea, we observed co-localization between HRB and both vRNPs and NEP at the MTOC, the 443
site of initial vRNP-Rab11 association, immediately after vRNP nuclear export. Additional 444
studies are required to define the specific components of influenza vRNP nuclear export and 445
cytoplasmic trafficking complex intermediates. 446
In summary, we identified HRB as a novel host factor involved in an early step in 447
influenza virus cytoplasmic genome transport. HRB was previously implicated in the HIV life 448
cycle, facilitating the cytoplasmic trafficking of rev-directed viral RNAs by promoting their 449
release from the perinuclear region. Influenza virus and HIV genomes both use the CRM1 450
pathway for nuclear export (27, 29, 31); however, this is the first study to implicate a common 451
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

23
cytoplasmic trafficking strategy for the genomes of these two viruses. Since influenza virus, 452
unlike HIV, does not use the endosomal sorting complex required for transport (ESCRT) for new 453
virion production (5, 34, 41), it will be interesting to delineate the point of divergence between 454
the influenza virus and HIV genome trafficking mechanisms. 455
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

24
ACKNOWLEDGEMENTS 456
We thank Susan Watson for editing the manuscript. This work was supported by National 457
Institute of Allergy and Infectious Disease Public Health Service research grants; by a grant-in-458
aid for Specially Promoted Research from the Ministries of Education, Culture, Sports, Science, 459
and Technology; and by grants-in-aid from the Ministry of Health and by ERATO (Japan 460
Science and Technology Agency). 461
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

25
REFERENCES 462
1. Akarsu, H., W. P. Burmeister, C. Petosa, I. Petit, C. W. Muller, R. W. Ruigrok, and 463
F. Baudin. 2003. Crystal structure of the M1 protein-binding domain of the influenza A 464
virus nuclear export protein (NEP/NS2). EMBO J 22:4646-55.465
2. Amorim, M. J., E. A. Bruce, E. K. Read, A. Foeglein, R. Mahen, A. D. Stuart, and P. 466
Digard. 2011. A Rab11 and microtubule dependent mechanism for cytoplasmic transport 467
of influenza A virus vRNA. J Virol 85:4143-56.468
3. Bogerd, H. P., R. A. Fridell, S. Madore, and B. R. Cullen. 1995. Identification of a 469
novel cellular cofactor for the Rev/Rex class of retroviral regulatory proteins. Cell 470
82:485-94.471
4. Bowzard, J. B., D. Cheng, J. Peng, and R. A. Kahn. 2007. ELMOD2 is an Arl2 472
GTPase-activating protein that also acts on Arfs. J Biol Chem 282:17568-80.473
5. Bruce, E. A., L. Medcalf, C. M. Crump, S. L. Noton, A. D. Stuart, H. M. Wise, D. 474
Elton, K. Bowers, and P. Digard. 2009. Budding of filamentous and non-filamentous 475
influenza A virus occurs via a VPS4 and VPS28-independent pathway. Virology 476
390:268-78.477
6. Bui, M., E. G. Wills, A. Helenius, and G. R. Whittaker. 2000. Role of the influenza 478
virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol 74:1781-6.479
7. Chaineau, M., L. Danglot, V. Proux-Gillardeaux, and T. Galli. 2008. Role of HRB in 480
clathrin-dependent endocytosis. J Biol Chem 283:34365-73.481
8. Doria, M., A. E. Salcini, E. Colombo, T. G. Parslow, P. G. Pelicci, and P. P. Di Fiore.482
1999. The eps15 homology (EH) domain-based interaction between eps15 and hrb 483
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

26
connects the molecular machinery of endocytosis to that of nucleocytosolic transport. J 484
Cell Biol 147:1379-84.485
9. Eierhoff, T., E. R. Hrincius, U. Rescher, S. Ludwig, and C. Ehrhardt. 2010. The 486
epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) 487
into host cells. PLoS Pathog 6:e1001099.488
10. Eisfeld, A. J., E. Kawakami, T. Watanabe, G. Neumann, and Y. Kawaoka. 2011. 489
RAB11A is Essential for Influenza Genome Transport to the Plasma Membrane. J Virol 490
85:6117-26.491
11. Elton, D., M. Simpson-Holley, K. Archer, L. Medcalf, R. Hallam, J. McCauley, and 492
P. Digard. 2001. Interaction of the influenza virus nucleoprotein with the cellular 493
CRM1-mediated nuclear export pathway. J Virol 75:408-19.494
12. Floer, M., and G. Blobel. 1999. Putative reaction intermediates in Crm1-mediated 495
nuclear protein export. J Biol Chem 274:16279-86.496
13. Fritz, C. C., M. L. Zapp, and M. R. Green. 1995. A human nucleoporin-like protein 497
that specifically interacts with HIV Rev. Nature 376:530-3.498
14. Ge, Q., M. T. McManus, T. Nguyen, C. H. Shen, P. A. Sharp, H. N. Eisen, and J. 499
Chen. 2003. RNA interference of influenza virus production by directly targeting mRNA 500
for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci 501
U S A 100:2718-23.502
15. Gujuluva, C. N., A. Kundu, K. G. Murti, and D. P. Nayak. 1994. Abortive replication 503
of influenza virus A/WSN/33 in HeLa229 cells: defective viral entry and budding 504
processes. Virology 204:491-505.505
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

27
16. Kehlenbach, R. H., A. Dickmanns, A. Kehlenbach, T. Guan, and L. Gerace. 1999. A 506
role for RanBP1 in the release of CRM1 from the nuclear pore complex in a terminal step 507
of nuclear export. J Cell Biol 145:645-57.508
17. Khwaja, S. S., H. Liu, C. Tong, F. Jin, W. S. Pear, J. van Deursen, and R. J. Bram.509
2010. HIV-1 Rev-binding protein accelerates cellular uptake of iron to drive Notch-510
induced T cell leukemogenesis in mice. J Clin Invest 120:2537-48.511
18. Kobasa, D., M. E. Rodgers, K. Wells, and Y. Kawaoka. 1997. Neuraminidase 512
hemadsorption activity, conserved in avian influenza A viruses, does not influence viral 513
replication in ducks. J Virol 71:6706-13.514
19. Lakadamyali, M., M. J. Rust, and X. Zhuang. 2004. Endocytosis of influenza viruses. 515
Microbes Infect 6:929-36.516
20. Li, C., M. Hatta, C. A. Nidom, Y. Muramoto, S. Watanabe, G. Neumann, and Y. 517
Kawaoka. 2010. Reassortment between avian H5N1 and human H3N2 influenza viruses 518
creates hybrid viruses with substantial virulence. Proc Natl Acad Sci U S A 107:4687-92.519
21. Liu, K., and G. Li. 1998. Catalytic domain of the p120 Ras GAP binds to RAb5 and 520
stimulates its GTPase activity. J Biol Chem 273:10087-90.521
22. Ma, K., A. M. Roy, and G. R. Whittaker. 2001. Nuclear export of influenza virus 522
ribonucleoproteins: identification of an export intermediate at the nuclear periphery. 523
Virology 282:215-20.524
23. Martin, K., and A. Helenius. 1991. Nuclear transport of influenza virus 525
ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. 526
Cell 67:117-30.527
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

28
24. Momose, F., Y. Kikuchi, K. Komase, and Y. Morikawa. 2007. Visualization of 528
microtubule-mediated transport of influenza viral progeny ribonucleoprotein. Microbes 529
Infect 9:1422-33.530
25. Naslavsky, N., and S. Caplan. 2011. EHD proteins: key conductors of endocytic 531
transport. Trends Cell Biol 21:122-31.532
26. Neumann, G., M. R. Castrucci, and Y. Kawaoka. 1997. Nuclear import and export of 533
influenza virus nucleoprotein. J Virol 71:9690-700.534
27. Neumann, G., M. T. Hughes, and Y. Kawaoka. 2000. Influenza A virus NS2 protein 535
mediates vRNP nuclear export through NES-independent interaction with hCRM1. 536
EMBO J 19:6751-8.537
28. Neumann, G., T. Watanabe, H. Ito, S. Watanabe, H. Goto, P. Gao, M. Hughes, D. R. 538
Perez, R. Donis, E. Hoffmann, G. Hobom, and Y. Kawaoka. 1999. Generation of 539
influenza A viruses entirely from cloned cDNAs. Proc Natl Acad Sci U S A 96:9345-50.540
29. Neville, M., F. Stutz, L. Lee, L. I. Davis, and M. Rosbash. 1997. The importin-beta 541
family member Crm1p bridges the interaction between Rev and the nuclear pore complex 542
during nuclear export. Curr Biol 7:767-75.543
30. Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-544
expression transfectants with a novel eukaryotic vector. Gene 108:193-9.545
31. O'Neill, R. E., J. Talon, and P. Palese. 1998. The influenza virus NEP (NS2 protein) 546
mediates the nuclear export of viral ribonucleoproteins. EMBO J 17:288-96.547
32. Pemberton, L. F., and B. M. Paschal. 2005. Mechanisms of receptor-mediated nuclear 548
import and nuclear export. Traffic 6:187-98.549
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

29
33. Pryor, P. R., L. Jackson, S. R. Gray, M. A. Edeling, A. Thompson, C. M. Sanderson, 550
P. R. Evans, D. J. Owen, and J. P. Luzio. 2008. Molecular basis for the sorting of the 551
SNARE VAMP7 into endocytic clathrin-coated vesicles by the ArfGAP Hrb. Cell 552
134:817-27.553
34. Rossman, J. S., X. Jing, G. P. Leser, and R. A. Lamb. 2010. Influenza virus M2 554
protein mediates ESCRT-independent membrane scission. Cell 142:902-13.555
35. Sakaguchi, A., E. Hirayama, A. Hiraki, Y. Ishida, and J. Kim. 2003. Nuclear export 556
of influenza viral ribonucleoprotein is temperature-dependently inhibited by dissociation 557
of viral matrix protein. Virology 306:244-53.558
36. Sanchez-Velar, N., E. B. Udofia, Z. Yu, and M. L. Zapp. 2004. hRIP, a cellular 559
cofactor for Rev function, promotes release of HIV RNAs from the perinuclear region. 560
Genes Dev 18:23-34.561
37. Schmid, E. M., M. G. Ford, A. Burtey, G. J. Praefcke, S. Y. Peak-Chew, I. G. Mills, 562
A. Benmerah, and H. T. McMahon. 2006. Role of the AP2 beta-appendage hub in 563
recruiting partners for clathrin-coated vesicle assembly. PLoS Biol 4:e262.564
38. Shimizu, T., N. Takizawa, K. Watanabe, K. Nagata, and N. Kobayashi. 2011. Crucial 565
role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear 566
export of vRNP. FEBS Lett 585:41-6.567
39. Tomoda, T., J. H. Kim, C. Zhan, and M. E. Hatten. 2004. Role of Unc51.1 and its 568
binding partners in CNS axon outgrowth. Genes Dev 18:541-58.569
40. Watanabe, K., N. Takizawa, M. Katoh, K. Hoshida, N. Kobayashi, and K. Nagata.570
2001. Inhibition of nuclear export of ribonucleoprotein complexes of influenza virus by 571
leptomycin B. Virus Res 77:31-42.572
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

30
41. Watanabe, R., and R. A. Lamb. 2010. Influenza virus budding does not require a 573
functional AAA+ ATPase, VPS4. Virus Res 153:58-63.574
42. World Health Organization (W.H.O.). April 21, 2011, posting date. Global Alert and 575
Response; Cumulative Number of Confirmed Human Cases of Avian Influenza 576
A/(H5N1) Reported to World Health Organization (WHO). 577
http://www.who.int/csr/disease/avian_influenza/country/cases_table_2011_04_21/en/inde578
x.html 579
43. World Health Organization (W.H.O). April 2009, posting date. Fact Sheet N°211. 580
http://www.who.int/mediacentre/factsheets/fs211/en/ 581
44. Xiao, G. H., F. Shoarinejad, F. Jin, E. A. Golemis, and R. S. Yeung. 1997. The 582
tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein 583
(GAP) in modulating endocytosis. J Biol Chem 272:6097-100.584
45. Yu, Z., N. Sanchez-Velar, I. E. Catrina, E. L. Kittler, E. B. Udofia, and M. L. Zapp.585
2005. The cellular HIV-1 Rev cofactor hRIP is required for viral replication. Proc Natl 586
Acad Sci U S A 102:4027-32.587
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

31
FIGURE LEGENDS 588
Figure 1. HRB perturbation interferes with influenza virus growth. (A) 293 cells were 589
transfected with a validated, non-targeting commercial negative control siRNA (AllStars Neg, 590
referred to as ‘Neg’ on the figure), siRNA targeting influenza NP mRNA (NP) or siRNA 591
targeting HRB, and total cell lysates were subjected to immunoblot analysis with mouse anti-592
HRB or rabbit anti-calnexin (CANX; loading control). (B) Cell viability was measured using 593
the CellTiter-Glo assay. 293 cells were transfected with siRNAs as described for panel A, 594
except that a cell death-inducing siRNA mixture (Death) was also included. The data are 595
represented as an average luciferase reading ± standard deviation (SD) for triplicate 596
transfections. (C) Cells were transfected with siRNAs as described for panel A and were super-597
infected with influenza virus A/WSN/33 (WSN). At 48 h post-infection, supernatants were 598
assayed for infectious virus by plaque assay in MDCK cells. The data shown are a compilation 599
of three independent experiments, in which triplicate transfections were performed for each 600
siRNA, and are represented as an average ± SD. A paired Student’s t-test was used to compare 601
replication in AllStars Neg siRNA-treated cells versus either NP or HRB siRNA treatments, and 602
the P-value is indicated above the graph. (D) 293 cells were transfected with AllStars Neg or 603
HRB siRNA as described, and infected with VSV, Ad5 or vaccinia virus. Infectious viruses 604
from each condition were quantified as described in the Materials and Methods. Paired 605
Student’s t-tests were performed to compare replication between the siRNA treatment conditions 606
for each virus, and significant P-values are indicated above the graph. Data are represented as 607
means of triplicate transfections from two independent experiments ± standard error of the mean. 608
(E) 293 cells were transfected with plasmids expressing GFP, NEP-YFP or a N360-GFP and 609
expression levels were determined by immunoblot analysis of whole cell lysates with anti-GFP 610
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

32
antibodies at 48 h post-transfection. (F) At 48 h post-transfection, plasmid-transfected cells 611
were super-infected with influenza virus WSN, and the level of infectious virus in supernatants 612
was assayed by plaque assay in MDCK cells after 72 h. Data are represented as an average of 613
triplicate infections performed for each transfection condition ± SD. 614
615
Figure 2. HRB is not required for early events in the influenza virus life cycle. (A) siRNA-616
treated 293 cells were mock-infected or infected with influenza virus WSN at a multiplicity of 617
infection (MOI) of 3 PFU per cell, and total cell lysates were subjected to immunoblot analysis 618
with rabbit polyclonal antibody R528 (against influenza vRNP) or rabbit anti-actin (loading 619
control) at different times after infection. siRNA treatments are indicated to the left, time points 620
are shown at the top and antibodies are to the right. Duplicate samples were prepared for each 621
infection condition at each time point. (B) 293 cells were transfected with AllStars Neg 622
(referred to as ‘Neg’ on figure), NP or HRB-specific siRNAs; or plasmids expressing GFP, NEP-623
YFP or N360-GFP; and were subsequently transfected with plasmids for the influenza WSN 624
mini-replicon assay. Viral gene expression levels were quantified as described in the Materials 625
and Methods. Data are represented as average ratios of firefly (viral)/Renilla (cellular) gene 626
expression from triplicate transfections ± SD. 627
628
Figure 3. HRB and NEP distribution in influenza virus-infected cells. A549 cells were 629
mock-infected (A) or infected with influenza WSN (MOI 3) and fixed with 4% 630
paraformaldehyde at 5 (B), 7 (C) and 9 (D) hpi. Permeabilized cells were stained with rabbit 631
polyclonal anti-serum against influenza NEP (R5023) and a mouse monoclonal antibody against 632
HRB, combined with AF 488 goat anti-rabbit and AF 546 goat anti-mouse, and Hoechst 33258. 633
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

33
Individual NEP (green) and HRB (red) staining patterns are shown for each time point, along 634
with a merged panel including Hoechst nuclear staining. A 3X zoom highlighting the 635
perinuclear region from the merged panels is shown in (B-D). Nuc, nucleus 636
637
Figure 4. Polyclonal rabbit anti-vRNP (R528) identifies influenza vRNPs with similar 638
specificity to that of monoclonal antibody 3/1. A549 cells were infected with WSN at an MOI 639
of 3 PFU per cell and fixed with 4% paraformaldehyde at 9 hpi. Permeabilized cells were 640
stained with R528 and monoclonal antibody 3/1 (MAb 3/1), combined with AF 488 goat anti-641
rabbit (green) and AF 546 goat anti-mouse (red). Individual and merged staining patterns are 642
shown.643
644
Figure 5. vRNP and HRB spatio-temporal dynamics in influenza virus-infected cells. A549645
cells were infected with influenza WSN (MOI 3) and harvested at 5 (A), 7 (B), 9 (C) or 11 (D) 646
hpi. Cells were stained with rabbit anti-vRNP (R528) and mouse anti-HRB, followed by AF 488 647
goat anti-rabbit (green) and AF 546 goat anti-mouse (red). Representative staining profiles are 648
shown, with the specific time points indicated at the top. For each panel, (i) shows a merged 649
image of HRB and vRNP, and individual staining profiles are shown in (ii) and (iii), 650
respectively. In (ii), white traces indicate the plasma membrane boundaries. Individual and 651
merged staining is also shown for enlargements (3X) of boxed regions from (i): (iv, HRB), (v, 652
vRNP) and (vi, Merge). A staining key is shown in the lower right corner of each panel.653
654
Figure 6. Leptomycin B (LMB) does not induce HRB nuclear accumulation. A549 cells 655
were mock-infected or infected with influenza WSN (MOI 3), and at 4 hpi they were treated with 656
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from

34
LMB and further incubated with LMB for 5 h, followed by fixation in paraformaldehyde. (A) 657
DMSO (control) and LMB-treated mock-infected cells were stained with monoclonal mouse 658
anti-HRB and AF 546 goat anti-mouse secondary antibodies. Infected, DMSO or LMB-treated 659
cells were stained as described in Fig. 5 (B) or Fig. 3 (C), respectively. All cells were 660
counterstained with Hoechst 33258. Individual HRB (red) and vRNP or NEP (green) panels are 661
shown, along with a merged panel including Hoechst. For (A-C), drug treatments are shown at 662
the top and specific stains are indicated to the left. (D) Enlarged images of the merged panels in 663
(C), highlighting HRB and NEP distribution around the periphery of the nucleus. A color key is 664
shown at the left and drug treatments are shown at the right. 665
666
Figure 7. HRB knockdown causes retention of vRNP in the peri-nuclear region. (A) Total 667
cell lysates from AllStars Neg (referred to as ‘Neg’ on the figure), NP and HRB siRNA-treated 668
HeLa cells at 48 h post-transfection were subjected to immunoblot analysis with mouse anti-669
HRB or rabbit anti-CANX (loading control). (B) Negative control siRNA-treated HeLa cells 670
were mock-infected and subjected to staining with mouse anti-HRB and rabbit anti-vRNP 671
(R528), followed by AF 546 goat anti-mouse and AF 488 goat anti-rabbit secondary antibodies. 672
(C-E) HeLa cells treated with either negative control (AllStars Neg) or HRB siRNA were 673
infected with influenza WSN (MOI 5) and fixed at 6, 9 and 15 hpi. Cells were stained as 674
described for panel (B). Individual HRB (red) and vRNP (green) staining profiles, as well as 675
merged images are shown for each condition. siRNA treatments are shown to the left and time 676
points and stains are indicated at the top of each panel. 677
on January 11, 2019 by guesthttp://jvi.asm
.org/D
ownloaded from























![Hrb [1].No Te Rindas](https://img.dokumen.tips/doc/110x75/55919f111a28abc50a8b462f/hrb-1no-te-rindas.jpg)











