Embed Size (px)
Citation preview

INFECTION AND IMMUNITY,0019-9567/00/$04.0010
Feb. 2000, p. 492–501 Vol. 68, No. 2
Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Host Cellular Immune Response to Pneumococcal LungInfection in Mice
ARAS KADIOGLU,1 NEILL A. GINGLES,1 KATE GRATTAN,1 ALISON KERR,2 TIM J. MITCHELL,2
AND PETER W. ANDREW1*
Department of Microbiology & Immunology, University of Leicester, Leicester,1 and Division of Infection & Immunity,University of Glasgow, Glasgow,2 United Kingdom
Received 17 June 1999/Returned for modification 9 September 1999/Accepted 5 November 1999
Although there is substantial evidence that pneumolysin is an important virulence factor in pneumococcalpneumonia, relatively little is known about how it influences cellular infiltration into the lungs. We investigatedhow the inability of mutant pneumococci to produce pneumolysin altered the pattern of inflammation andcellular infiltration into the lungs. The effect on bacterial growth in the lungs also was assessed. There werethree phases of growth of wild-type bacteria in the lungs: a decline followed by a rapid increase and then stasisor decline. The absence of pneumolysin was associated with a more rapid early decline and then a much slowerincrease in numbers. The pattern of inflammatory-cell accumulation also had distinct stages, and the timingof these stages was influenced by the presence of pneumolysin. Neutrophils began to accumulate about 12 to16 h after infection with wild-type pneumococci. This accumulation occurred after the early decline inpneumococcal numbers but coincided with the period of rapid growth. Following infection with pneumococciunable to make pneumolysin, neutrophil influx was slower and less intense. Coincident with the third stage ofpneumococcal growth was an accumulation of T and B lymphocytes at the sites of inflammation, but theaccumulation was not associated with an increase in the total number of lymphocytes in the lungs. Lymphocyteaccumulation in the absence of pneumolysin occurred but was delayed.
Streptococcus pneumoniae is an important respiratory patho-gen of humans, causing pneumonia (lobar and bronchopneu-monia), septicemia, otitis media, and meningitis.
The pneumococcus produces several factors that may beimportant in the development of disease. One such factor isthe pneumococcal toxin pneumolysin. We have shown thatpneumolysin is a multifunctional toxin that exhibits cytolyticactivity (hemolysis), and at sublytic concentrations it is knownto alter the functioning of immune cells (1, 15). This modula-tion of cells and thus the activity of the immune system in-cludes the inhibition of ciliary beat on human respiratory ep-ithelium (8, 9), the stimulation of tumor necrosis factor alphaand interleukin-1b release from human monocytes (12), theactivation of phospholipase A2 in pulmonary cells (20), and theinhibition of the neutrophil respiratory burst (18). The toxinalso activates the classical complement pathway in the absenceof antipneumolysin antibody (16).
Pneumolysin plays an important but as yet not completelydefined role in the development of bronchopneumonia. It hasbeen previously shown that pneumococci not expressing pneu-molysin have reduced virulence in the mouse compared to thewild-type organism, with slower pneumococcal growth in thelungs and delayed development of associated septicemia, cul-minating in a general reduction in the severity of the inflam-matory response (6). It has also been shown that immunizationwith a genetically engineered toxoid version of pneumolysinprotects mice from bronchopneumonia (2). It is also worthnoting that pneumolysin alone can reproduce the symptoms ofpneumococcal disease in the lungs (9). Hence, either by directdamage to the host or by stimulation of host inflammatorymediators leading to bronchopneumonia, pneumolysin may
play an important role in the induction and further mainte-nance of the inflammatory response.
Despite a considerable amount of work illustrating the re-quirement for pneumolysin in bronchopneumonia, studies ex-amining host tissue pathogenesis and the interactions betweenbacterial and host factors in bronchopneumonia are rare. Inthis paper, we discuss how bronchopneumonia develops in amurine model of bronchopneumonia and septicemia due toboth wild-type Streptococcus pneumoniae and a pneumolysin-negative mutant. We monitored for the first time in this par-ticular animal model the early events involved in the initialprogression of bronchopneumonia and its subsequent devel-opment after intranasal infection. We have focused not only onbacterial growth kinetics but also on the host tissue response interms of the onset of inflammation, the timing of inflammatorycell infiltrate into lungs, the nature and type of host immunecells involved in these processes, and the development of tissuehistopathology.
Our hypothesis is that in pneumococcal bronchopneumonia,pneumolysin is the major trigger of inflammation and toxemiaand that it acts by activating a cascade of host factors respon-sible for the recruitment of inflammatory cells. We will belooking to see how the pattern of bacterial growth and dissem-ination, host morbidity and mortality, and the progress of in-flammation and production of host factors develop by usingour model of bronchopneumonia and septicemia due to pa-rental wild-type and pneumolysin-negative mutant pneumo-cocci. By combining these results with data on bacterial growthin the lungs and blood, we intend to develop a picture ofcertain aspects of the host inflammatory response to pneumo-coccal infection.
MATERIALS AND METHODS
Pneumococcal strains. The wild-type S. pneumoniae strain used was serotype2 strain D39, NCTC 7466 (National Collection of Type Cultures, London,United Kingdom). The pneumolysin-negative mutant used, PLN-A, was made by
* Corresponding author. Mailing address: Department of Microbi-ology & Immunology, Medical Sciences Building, University Rd., Le-icester LE1 9HN, United Kingdom. Phone: (116) 2523018. Fax: (116)2525030. E-mail: [email protected].
492
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

insertion duplication mutagenesis (5). Pneumococci were cultured on blood agarbase containing 5% (vol/vol) horse blood or in brain heart infusion broth (Oxoid,Basingstoke, United Kingdom) containing 20% (vol/vol) fetal bovine serum(FBS; Gibco, Paisley, United Kingdom) supplemented with 1 mg of erythromy-cin (Sigma, Poole, United Kingdom) per ml for PLN-A.
Preparation of the challenge dose. S. pneumoniae was passaged through miceas described previously (6), and aliquots were stored at 270°C. Pneumococci canbe stored for at least 3 months at 270°C with no significant loss of viability. Whenrequired, the suspension was thawed at room temperature and bacteria wereharvested by centrifugation before being resuspended in sterile phosphate-buff-ered saline (PBS).
Intranasal challenge of mice. Female MF1 outbred mice weighing 30 to 35 g(Harlan Olac, Bicester, United Kingdom) were lightly anesthetized with 2.5%(vol/vol) fluothane (Zeneca, Macclesfield, United Kingdom) over oxygen (1.5 to2 liters/min). A 50-ml volume of PBS containing 106 CFU of S. pneumoniae wildtype or PLN-A was administered to the nostrils of mice held vertical. Mice weremonitored for symptoms of disease for 48 h (longer for those infected withPLN-A) or until they became moribund, at which point the experiment wasended.
Experiments were done to determine the growth of bacteria in vivo. At pre-selected time intervals following infection, groups of mice were deeply anesthe-tized with 5% (vol/vol) fluothane and blood was collected by cardiac puncture.Following this procedure, the mice were killed immediately by cervical disloca-tion. The lung were removed into 10 ml of sterile distilled water, weighed, andthen homogenized in a Stomacher-Lab blender (Seward Medical, London,United Kingdom). Viable counts in homogenates and in blood were determinedas described previously (6). The presence of a type 2 polysaccharide capsule wasconfirmed by the Quellung reaction.
Enumeration and differential analysis of lung leukocyte counts. At the sameprechosen intervals following infections as above, lungs from preselected groupsof mice were removed, as above, and leukocytes were prepared by a modificationof a previously published method (7, 13). After removal of the lungs fromsacrificed animals, the tissue was placed into 10 ml of Hanks balanced saltsolution. The lungs were then cut into small pieces and homogenized in 5 ml ofdigestion buffer (5% [vol/vol] FBS in RPMI 1640 with collagenase [Sigma] at 0.5mg/ml [207 collagen digestion units] and DNase I from bovine pancreas [Sigma]at 30 mg/ml [87 units]) through a tea strainer a total of three times. Afterhomogenization, lung samples were incubated at 37°C for 30 min. Subsequently,digested tissue samples were pipetted to break up tissue fragments and passedthrough a column containing approximately 1 cm of nonabsorbent cotton wool ina glass Pasteur pipette to remove large pieces of debris. Cells collected in Falcon2052 tubes (Becton Dickinson) were centrifuged at 322 3 g for 5 min at 4°C. Thesupernatant was removed, and the cells resuspended in 1 ml of 13 lysis solution(Pharmingen, San Diego, Calif.). After 5 min at room temperature to lyse theerythrocytes, the remaining cells were brought to isotonicity by adding an excessvolume of ice-cold 13 PBS. Following centrifugation at 322 3 g for 5 min at 4°C,the cells were washed with 1 ml of 13 PBS before a final resuspension in 1 ml of5% FBS in RPMI 1640. The cells were then enumerated with a hemocytometer(Improved Neubauer; Weber Scientific International Ltd.) with the addition oftrypan blue in 13 PBS at a 1:1 volume ratio of stain to cell suspension. The totalnumber of cells used from each lung sample was diluted with 5% (vol/vol) FBSin RPMI 1640 to give a total of 7 3 104 to 10 3 105 cells per 50 ml.
For differential analysis, cytocentrifugation of these cells was performed with50 ml of cell suspension centrifuged onto cytospin slides (Shandon) in a cyto-centrifuge (Cytospin 2; Shandon) at 108 3 g for 3 min. Following centrifugation,the slides were air dried briefly and then fixed in 100% methanol for 10 min.After fixation, differential staining was performed with Giemsa stain (BDH,Poole, United Kingdom). The slides were quantified independently by two ob-servers at 3500 magnification with a graticule-equipped eyepiece, and mononu-clear leukocytes, lymphocytes, and polymorphonuclear leukocytes were identi-fied. At least 200 cells were counted on each slide in total. By using thepercentage of each type of leukocyte obtained from each slide, cell numbers ofeach leukocyte population were calculated from the total number of cellscounted per milliliter. All slides were read by investigators blinded to theiridentity, and the coefficient of intraobserver variation was 3.4%.
Histology. At prechosen intervals, whole-lung samples from infected micewere excised, embedded in Tissue Tek OCT, and frozen in liquid nitrogen withan isopentane heat buffer to prevent snap freezing and tissue damage. Oncefrozen, the samples were stored at 270°C until required. At 1 day before sec-tioning, the samples were moved to 220°C. On the day of sectioning, 15-mmsections were taken at 218 to 225°C on a Bright microtome. The sections wereallowed to dry at room temperature for 20 min, and the embedding compoundsurrounding the tissue was peeled away and discarded. Once dried, the sectionswere stained with hematoxylin and eosin. After being stained, the sections werefixed with DPX mountant (BDH) for permanent storage.
Immunohistochemistry. Leukocyte recruitment into lung tissue was analyzedby an alkaline phosphatase anti-alkaline phosphatase (APAAP) staining methodas described previously (14). Rat anti-mouse monoclonal antibodies to T cells(CD3), B cells (CD19), macrophages (F/480), and neutrophils (7/4) (Serotec,Oxford, United Kingdom) were used. Four sections from each lung collected atchosen time points were used for each antibody to be tested, along with threesections for negative controls: (i) an isotype-matched control antibody; (ii) ex-
clusion of the primary antibody (or the secondary enzyme-conjugated antibody);and (iii) a sample not incubated with the substrate-chromogen solution. Oncestained, the sections were analyzed by one observer (A.K.) and positively stainedcells within the vicinity of inflamed bronchioles were enumerated. The distribu-tion patterns of positively stained leukocytes within the lungs were also observed.
Statistical analysis. Data were analyzed by a one-tailed Mann-Whitney U test,Student’s t test, and one-way analysis of variance. Statistical significance wasassumed at P , 0.05.
RESULTS
Symptoms following infection. All 75 mice challenged intra-nasally with 106 CFU of wild-type S. pneumoniae showed signsof illness (starry coat and hunched appearance) by 24 h postin-fection. By 48 h postinfection, all the mice had become mori-bund. In contrast, the most extreme symptom in mice infectedwith PLN-A was starry coat, seen in all 30 mice at 48 h.Thereafter, none of the 20 mice observed showed symptomswithin the remaining 9 days of the experiment.
Growth of wild-type and PLN-A pneumococci in lung tissueand blood. The growth of the pneumolysin-negative strain,PLN-A, in lung tissue and blood was different from that of theparental wild-type strain. In the lungs, over the first 16 h for thewild type and 8 to 10 h for PLN-A, bacterial numbers declinedsharply; they began to increase again 12 to 16 h postinfection(Fig. 1a). PLN-A growth was significantly slower (P , 0.05)(maximum doubling time, 178 min) than wild-type growth(maximum doubling time, 120 min), although both showedidentical growth patterns when grown in vitro in brain heartinfusion medium (data not shown). Both wild-type and PLN-Agrowth showed no further increase after 24 h postinfection.There was, however, a statistically significant difference be-tween wild-type and PLN-A levels at 2, 4, 6, 20, 24, and 48 h(P , 0.05 for all time points). There was no significant differ-ence at other time points. Levels of PLN-A in lung tissuedecreased by 72 h and significantly so by 96 h compared to the48-h levels (P , 0.05), but the bacterium still persisted 264 hpostinfection (Fig. 1b).
Neither the wild type nor PLN-A was detected in the blooduntil 12 h postinfection, at which time bacteria were isolatedfrom all mice (five mice for each infection) (Fig. 2a). The twoorganisms showed comparable rates of increase until 16 h, atwhich time wild-type numbers increased rapidly (maximumdoubling time, 145 min), reaching a peak by 24 to 48 h, whereasthe numbers of PLN-A organisms did not increase after 20 to24 h postinfection (maximum doubling time, 307 min). Thesedifferences between the wild type and PLN-A at 24 and 48 hwere statistically significant (P , 0.01 for both). Levels ofPLN-A organisms began to decrease after 48 h, but organismsstill persisted in the blood for at least 11 days (264 h) postin-fection (Fig. 2b), with no signs of illness in infected mice attimes after 48 h. Production of a type 2 capsule was confirmedby Quellung reactions at 24 and 48 h postinfection for wild-type and PLN-A organisms recovered from both lungs andblood.
Histological examination of lung tissue. Histological analy-sis of lung tissue sections from mice infected with wild-typepneumococci showed inflammation and cellular infiltrationcentered around bronchioles and perivascular areas. The fociof inflammation were restricted to certain bronchioles andperivascular areas close to these bronchioles at 24 h postinfec-tion. Inflammation presented itself as hypertrophy of bronchi-ole walls, heavy cellular infiltration around such bronchioles,and some edema. Bacteria were detected within alveoli andaround inflamed bronchioles.
By 48 h postinfection, bronchiole wall thickening had in-creased, and solid fibrous tissue and exudate filling the bron-
VOL. 68, 2000 ROLE OF PNEUMOLYSIN IN BRONCHOPNEUMONIA 493
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

chioles and alveolar spaces had appeared. Additionally, cellu-lar infiltration had increased, with extension of inflammatorycells from bronchioles and perivascular areas into the sur-rounding lung parenchyma and with several focal areas ofconsolidation becoming larger and more diffuse. The presenceof alveoli in lung sections at this time point was hardly distin-guishable due to intensive tissue edema. Overall, during thisperiod of infection, inflammation and tissue injury had encom-passed nearly all of the lung surface (Fig. 3a and b).
The histological changes seen in the lungs of mice infectedwith PLN-A were generally delayed compared to those in thelungs of mice infected with the wild type and were less severe,exhibiting considerably less tissue inflammation and cellularinfiltration into perivascular areas between infected bronchi-oles at 24, 48, 72, and 96 h. However, despite this lower severityand the lower levels of pathologic tissue damage, some bron-chioles did exhibit signs of inflammation by 48 h, with moder-
ate levels of cellular infiltration and hyperplasia. Compared towild-type-infected mice, however, the cellular infiltrationaround such bronchioles appeared to be less intense and didnot extend into the perivascular areas. Loss of alveolar struc-ture, parenchymal involvement, and interstitial edema weregreatly reduced, and no focal areas of tissue consolidation werepresent. General tissue edema was mild, and tissue fibrosis wasabsent (Fig. 4).
Total and differential leukocyte analysis of cytocentrifugedlung homogenates. Total leukocyte numbers and individuallymphocyte, macrophage, and polymorphonuclear cell num-bers were enumerated over the time course of infection inwild-type- and PLN-A–infected mice.
Total leukocyte levels in wild-type-infected lung tissue ho-mogenates increased by 12 h and reached significantly in-creased values (P , 0.05) by 24 h postinfection (Fig. 5) com-pared to the time zero levels. In contrast, total leukocyte levelsin PLN-A–infected lung tissue homogenates showed no signif-
FIG. 1. (a) Time course of the change in numbers of S. pneumoniae wild type(h) and PLN-A ({) in lungs of MF1 mice infected intranasally with 106 CFU(n 5 5 for each time point; error bars indicate standard error of the mean[SEM]). P , 0.05 for wild-type values at 20, 24 and 48 h compared to PLN-A. (b)Time course of the change in numbers of S. pneumoniae PLN-A in lungs of MF1mice infected intranasally with 106 CFU (n 5 5 for each time point; error barsindicate SEM).
FIG. 2. (a) Time course of the change in numbers of S. pneumoniae wild typeand PLN-A in blood of MF1 mice infected intranasally with 106 CFU (n 5 5 foreach time point; error bars indicate SEM). P , 0.01 for wild-type values at 24 and48 h compared to PLN-A values. Time course of the change in numbers of S.pneumoniae PLN-A in blood of MF1 mice infected intranasally with 106 CFU(n 5 5 for each time point; error bars indicate SEM).
494 KADIOGLU ET AL. INFECT. IMMUN.
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

icant change until 48 h postinfection, when they were signifi-cantly greater than those at time zero (P , 0.01). Interestingly,the total leukocyte levels in PLN-A–infected mice were lowerat 72 h postinfection than at 48 h. The total leukocyte levelsalso appeared to be lower in PLN-A–infected than in wild-type-infected tissue at each equivalent time point; however,between 12 and 48 h the differences did not reach statisticalsignificance.
When individual cell types were analyzed in wild-type-in-fected total-lung homogenates, polymorphonuclear cell num-bers in the lungs showed significant increases by 12, 24, and48 h postinfection (Fig. 6a) compared to time zero (P , 0.05,0.01, and 0.05 respectively). Macrophage levels decreased by24 h (P , 0.01), whereas lymphocyte levels showed no signif-icant change throughout the 48-h time course (Fig. 6a). Whenthe same analysis was carried out for PLN-A–infected total-lung homogenates, a different picture emerged. Although poly-
morphonuclear cell levels were again significantly higher at 24and 48 h postinfection (P , 0.01 and 0.05, respectively) than attime zero (Fig. 6b), there was no increase at 12 h and thenumber of cells was significantly smaller than the number ob-served for wild-type-infected tissue at each equivalent timepoint (P , 0.01). Additionally, as was the case for total leuko-cyte levels, the levels of polymorphonuclear cells in PLN-A–infected tissue also decreased substantially by 72 h comparedto the 24- and 48-h levels. Macrophage levels at 24 h postin-fection were significantly higher than in wild-type-infected tis-sue at the equivalent time point (P , 0.05), but a significantdecrease in macrophage levels in PLN-A–infected tissue oc-curred 72 h postinfection compared to time zero (P , 0.01). Asimilar decrease had occurred at 24 h in wild-type-infectedtissue. As in wild-type-infected lungs, lymphocyte levels inPLN-A–infected lungs showed no significant changes through-out the time course (Fig. 6b). The levels of each cell type were
FIG. 3. Light microscopy of lung tissue from mice infected with 106 CFU of wild-type S. pneumoniae, sacrificed at 24 h (a) (the large single arrow indicates an areaof cellular infiltration, the small single arrow indicates edema, and the large double arrow indicates bronchiole wall thickening) and 48 h (b) (the large arrows indicateareas of fibrosis, the small arrows indicate edema, and the open arrowhead indicates heavy cellular infiltration) postinfection. Magnifications, 3400.
VOL. 68, 2000 ROLE OF PNEUMOLYSIN IN BRONCHOPNEUMONIA 495
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

the same (P . 0.05) at time zero for both wild-type- andPLN-A–infected mice, and when data were analyzed with eachcell population as a percentage instead of total numbers ofcells, the same patterns were obtained (data not shown).
Immunohistochemical analysis of inflammatory cell infil-trates. To further analyze leukocyte infiltration into lung tis-sue, in situ analysis of leukocyte numbers, distribution pat-terns, and their anatomical localization in lung tissue over thetime course of infection with the wild type and PLN-A wasperformed by immunohistochemistry. Positively stained cellswere enumerated in inflamed areas of sectioned lung tissueonly.
In inflamed areas of wild-type-infected lung, the numbers ofneutrophils showed the greatest increase, reaching a statisti-cally significant peak (P , 0.05) at 24 h compared to time zero(Fig. 7). This also reflected the equivalent increase seen intotal-lung homogenate counts of neutrophils. Neutrophils
were observed within inflamed bronchioles and in bronchiolewalls but also to a much greater extent in the perivascular areassurrounding inflamed bronchioles and in alveolar spaces.These areas of lung tissue were heavily infiltrated with neutro-phils at 24 h. However, the numbers of neutrophils in tissue,especially in and around inflamed bronchioles, decreased sig-nificantly by 48 h postinfection compared to 24 h (P , 0.05).The numbers of neutrophils were still larger than those ofmacrophages or lymphocytes at equivalent time points, as wasalso the case for the numbers of total-lung homogenate.
T lymphocytes showed interesting patterns of distribution,with the numbers of cells in inflamed areas increasing signifi-cantly (P , 0.05) by 24 and 48 h postinfection compared totime zero. The numbers of T cells were large in tissue sur-rounding inflamed bronchioles and somewhat smaller in closeproximity to the bronchiole walls themselves by 24 h (Fig. 8a).By 48 h, however, the numbers of T cells around the bronchiole
FIG. 4. Light microscopy of lung tissue from mice infected with 106 CFU of S. pneumoniae PLN-A, sacrificed at 24 h (a) (the large arrow indicates a bronchiole,and the small arrow indicates slight cellular infiltration) and 48 h (b) (the large arrow indicates a bronchiole, and the small arrow indicates cellular infiltration)postinfection. Magnifications, 3300 (a) and 3400 (b).
496 KADIOGLU ET AL. INFECT. IMMUN.
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

walls had decreased but the numbers in perivascular tissue andaround inflamed bronchioles had increased, as had the totalnumber of T cells dispersed throughout the tissue as a whole(Fig. 8b). Thus, there is a shift in distribution patterns frombronchioles to tissue spaces between the inflamed bronchioles.
The total numbers of macrophages in inflamed areas re-mained constant at 0, 24, and 48 h postinfection. However, thenumbers of macrophages localized inside inflamed bronchiolesand in perivascular tissue areas surrounding such bronchiolesincreased by 24 h compared to time zero. Macrophages werealso seen in alveolar spaces by this time point. The number ofB lymphocytes in lung tissue increased steadily by 24 and 48 hpostinfection. This increase was observed in tissue in closeproximity to inflamed bronchioles and to a lesser extent withinalveolar spaces. By 48 h, B lymphocytes were also observedwithin inflamed bronchioles.
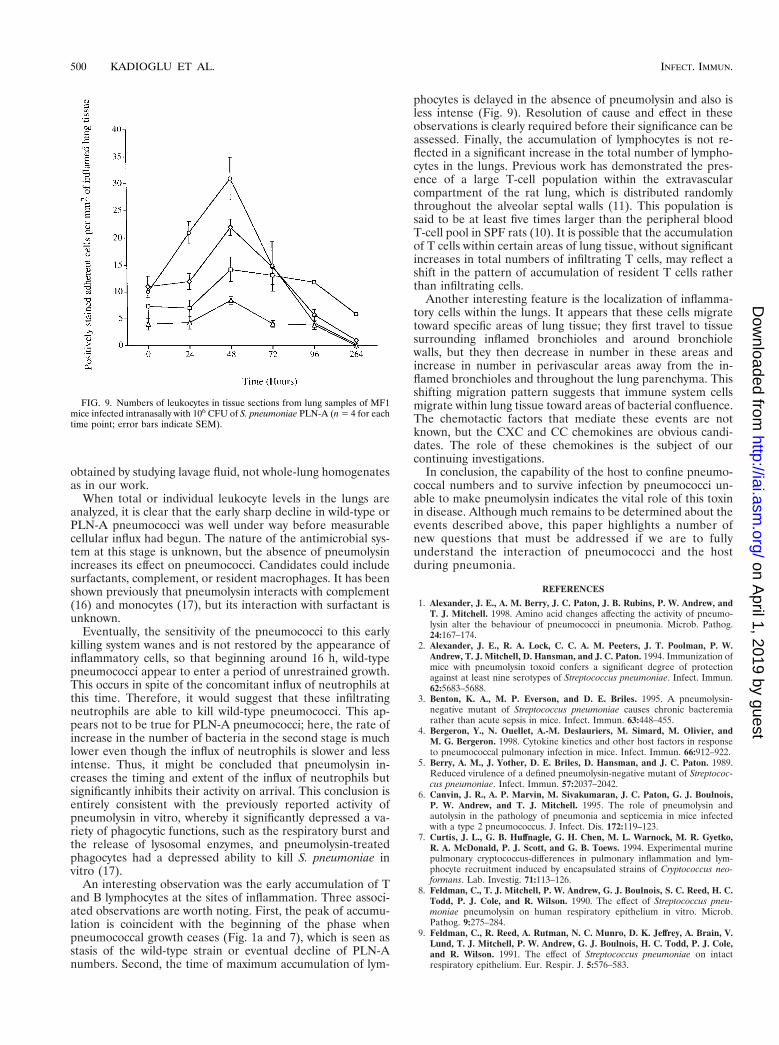
In inflamed areas of PLN-A–infected lung tissue, neutrophilnumbers increased by 24 h and continued to do so until reach-ing a peak significantly greater than the initial value (P , 0.05compared to time zero levels) by 48 h (Fig. 9), again in keepingwith equivalent increases in neutrophil counts in total-lunghomogenate. Neutrophil numbers in perivascular areas aroundinflamed bronchioles increased somewhat by 24 h and to agreater extent by 48 h, although the numbers within bronchi-oles did not increase. Neutrophil numbers in PLN-A–infectedtissue were significantly smaller at 24 h postinfection (P ,0.05) and smaller again at 48 h postinfection compared to thenumbers found at the equivalent time points in wild-type-infected tissue. However, neutrophils exhibited a similar ana-tomical localization pattern in both types of infected lungs.
No changes were seen in the numbers of T lymphocytes, Blymphocytes, and macrophages in PLN-A–infected tissue from0 to 24 h postinfection, unlike in wild-type-infected tissue (Fig.9). By 48 h postinfection, small increases in the numbers ofsuch cells were observed. Both macrophages and T lympho-cytes showed increased numbers by 48 h, with macrophagenumbers increasing within inflamed bronchioles and especiallyaround inflamed bronchiole tissue. T-cell numbers, on theother hand, showed small increases in and around inflamedbronchioles (Fig. 8c) but considerably less than for wild-type-
infected tissue. A similar pattern was also observed for Blymphocytes. The total number of cells was much smaller thanthat observed in wild-type-infected tissue sections, however.
The main differences between cell infiltrates into wild-type-and PLN-A–infected lung tissue were the changing distributionpatterns for T lymphocytes and neutrophils, the greater num-bers of infiltrating cells in wild-type-infected lung sections, anda general decrease in numbers of infiltrating cells by 48 hpostinfection in wild-type-infected tissue compared to 72 hpostinfection in PLN-A–infected tissue.
DISCUSSION
We have previously shown that pneumolysin is crucially in-volved in the pathogenesis of pneumococcal pneumonia (6).
FIG. 5. Total leukocyte counts from whole-lung homogenate cytospins fromMF1 mice infected intranasally with 106 CFU of S. pneumoniae wild type andPLN-A (n 5 5 for each time point; error bars indicate SEM). P , 0.05 forwild-type leukocyte levels at 24 h compared to time zero; P , 0.01 for PLN-A at48 h compared to time zero; P , 0.05 for PLN-A at 72 h compared to 48 h.
FIG. 6. (a) Differential leukocyte counts from whole-lung homogenate cyto-spins from MF1 mice infected intranasally with 106 CFU of wild-type S. pneu-moniae (n 5 5 for each time point; error bars indicate SEM). P , 0.05 forpolymorphonuclear cell levels at 12 and 48 h and P , 0.01 for levels at 24 hcompared to time zero. P , 0.01 for macrophage levels at 24 h compared to timezero. (b) Differential leukocyte counts from whole-lung homogenate cytospinsfrom MF1 mice infected intranasally with 106 CFU of S. pneumoniae PLN-A(n 5 5 for each time point; error bars indicate SEM). P , 0.01 for polymorpho-nuclear cell levels at 24 h and P , 0.05 for cell levels at 48 h compared to timezero. P , 0.01 for macrophage levels at 72 h compared to time zero.
VOL. 68, 2000 ROLE OF PNEUMOLYSIN IN BRONCHOPNEUMONIA 497
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

We found that a pneumolysin-negative mutant grew moreslowly in the lungs and induced much less inflammation thandid compared to the parent wild-type organism (6). In thispaper, we have analyzed pneumococcal growth in the lungsand blood of mice in more detail than before and have char-acterized the pattern of inflammatory-cell influx. These datarevealed that the pattern of survival and cell influx of wild-typeand PLN-A pneumococci is more complex than we previouslysaw.
When the growth kinetics of both wild-type and PLN-Aorganisms in lungs was examined, three phases were seen inthe first 48 h postinfection: (i) an early and sharp decline innumbers of pneumococci, (ii) an increase in numbers, and (iii)a stage where pneumococcal numbers remained constant ordeclined. Although the two strains showed similar patterns, themost obvious difference between the wild type and PLN-A wasthe extent of the change in two of the phases. The early, sharpdecline of pneumococcal numbers was much more evident forPLN-A and the increase in numbers after 16 h was muchsharper with wild-type pneumococci. Thus, pneumolysin ap-pears to be crucial to the survival of the pneumococcus at twodistinct stages of the infection.
The occurrence of these three phases of pneumococcal de-cline or growth at different times after infection implies thatdifferent antipneumococcal systems emerged over time or thattheir effectiveness changed with time. This view is reinforcedby the second period of decline in numbers of bacteria in thelungs seen with PLN-A after 48 h postinfection. The pattern ofinflux of inflammatory cells was consistent with this idea.
When pneumococcal growth in blood was examined, distinctstages were seen again: a rapid increase in the numbers of
pneumococci and the subsequent stabilization of these num-bers. A notable feature here was the much lower plateau of thePLN-A level reached in the blood compared with the level ofwild-type organisms. Another feature was the asymptomaticpersistence of PLN-A in the blood. In these experiments,pneumolysin did not influence the time of appearance of pneu-mococcal bacteremia or the early rate of increase in bacterialnumbers. However, it did influence the sensitivity of pneumo-cocci to the mechanism that eventually limits their numbers.Whatever the nature of this mechanism, it is one of stasisrather than cidal action. Previous investigation of bacteremiawith the same strain of pneumococci also showed the persis-tence of PLN-A in blood up to 7 days postinfection, with 50%survival of infected animals (3). The authors suggested that theabsence of pneumolysin during the early hours of infectionprevents the development of sepsis and delays death by at leastseveral days. Our previous finding that a delay in the appear-ance of pneumococci in the blood is associated with a delay inthe time of death would also suggest that the onset of bacte-remia is an important determinant of the time of death (6).
Previous work with a model of endotracheal instillation (21)has shown that PLN-A has a reduced capacity to injure thealveolar-capillary barrier and hence a reduced capacity to mul-tiply within lung tissue. It has also been shown that PLN-Afailed to cause the separation of tight junctions between epi-thelial cells. It was suggested that as a consequence, adherenceto separated epithelial cell edges and invasion of lung tissuewere decreased (19). PLN-A is also known to be less successfulin penetrating the interstitium of the lung from the alveoli andinvading the bloodstream than is the wild type. When purifiedpneumolysin is coinstilled with PLN-A, however, the pattern ofmultiplication is similar to that of the wild type (21). Althoughusing a different model, the authors showed that pneumolysinfacilitated the intra-alveolar replication of pneumococci, aswell as the penetration of these bacteria from alveoli intodeeper lung tissue, eventually resulting in the presence ofpneumococci in the bloodstream (21).
This previous work, combined with the observations de-scribed in this paper, suggests that the cytotoxic properties ofpneumolysin are essential for bacterial multiplication in thealveoli. Pneumolysin appears to play an important role espe-cially during the first 6 to 8 h of infection, when bacterialcolonization and growth within host tissue crucially occur toform the basis of future inflammatory reactions and systemicinfection.
To begin to explain the host mechanisms underlying thesepatterns of pneumococcal behavior, we analyzed the influx ofinflammatory cells into infected lungs. We found a sequentialinfiltration pattern of inflammatory cells which failed to elim-inate wild-type pneumococci from lungs and resulted in bac-terial proliferation, bacteremia, and eventual host death. Re-cently published work with CD1 Swiss mice intranasallychallenged with S. pneumoniae (4) also showed a sequentialmovement of different inflammatory cells into the lungs. Aswith our observations, it was reported that an early neutrophilinflux was followed by an increase in the number of lympho-cytes, but in contrast to our data, a large increase in macro-phages was seen. However, the data of Bergeron et al. (4) were
FIG. 7. Numbers of leukocytes in tissue sections from lung samples of MF1mice infected intranasally with 106 CFU of wild-type S. pneumoniae (n 5 4 foreach time point; error bars indicate SEM).
FIG. 8. (a) Light microscopy of APAAP-stained T lymphocytes (darkly stained cells) surrounding a bronchiole in lung tissue infected with 106 CFU of wild-typeS. pneumoniae 24 h postinfection. Arrows indicate positively stained T cells. (b) Light microscopy of APAAP-stained T lymphocytes (darkly stained cells) surroundinga bronchiole in lung tissue infected with 106 CFU of wild-type S. pneumoniae 48 h postinfection. Large arrows indicate positively stained T cells, and the small arrowindicates the bronchiole. (c) Light microscopy of APAAP-stained T lymphocytes (darkly stained cells) surrounding a bronchiole in lung tissue infected with 106 CFUof S. pneumoniae PLN-A 24 h postinfection. Large arrows indicate the small number of T cells. The small arrow indicates the bronchiole. Magnifications, 3400 (a) and3320 (b and c).
498 KADIOGLU ET AL. INFECT. IMMUN.
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from
obtained by studying lavage fluid, not whole-lung homogenatesas in our work.
When total or individual leukocyte levels in the lungs areanalyzed, it is clear that the early sharp decline in wild-type orPLN-A pneumococci was well under way before measurablecellular influx had begun. The nature of the antimicrobial sys-tem at this stage is unknown, but the absence of pneumolysinincreases its effect on pneumococci. Candidates could includesurfactants, complement, or resident macrophages. It has beenshown previously that pneumolysin interacts with complement(16) and monocytes (17), but its interaction with surfactant isunknown.
Eventually, the sensitivity of the pneumococci to this earlykilling system wanes and is not restored by the appearance ofinflammatory cells, so that beginning around 16 h, wild-typepneumococci appear to enter a period of unrestrained growth.This occurs in spite of the concomitant influx of neutrophils atthis time. Therefore, it would suggest that these infiltratingneutrophils are able to kill wild-type pneumococci. This ap-pears not to be true for PLN-A pneumococci; here, the rate ofincrease in the number of bacteria in the second stage is muchlower even though the influx of neutrophils is slower and lessintense. Thus, it might be concluded that pneumolysin in-creases the timing and extent of the influx of neutrophils butsignificantly inhibits their activity on arrival. This conclusion isentirely consistent with the previously reported activity ofpneumolysin in vitro, whereby it significantly depressed a va-riety of phagocytic functions, such as the respiratory burst andthe release of lysosomal enzymes, and pneumolysin-treatedphagocytes had a depressed ability to kill S. pneumoniae invitro (17).
An interesting observation was the early accumulation of Tand B lymphocytes at the sites of inflammation. Three associ-ated observations are worth noting. First, the peak of accumu-lation is coincident with the beginning of the phase whenpneumococcal growth ceases (Fig. 1a and 7), which is seen asstasis of the wild-type strain or eventual decline of PLN-Anumbers. Second, the time of maximum accumulation of lym-
phocytes is delayed in the absence of pneumolysin and also isless intense (Fig. 9). Resolution of cause and effect in theseobservations is clearly required before their significance can beassessed. Finally, the accumulation of lymphocytes is not re-flected in a significant increase in the total number of lympho-cytes in the lungs. Previous work has demonstrated the pres-ence of a large T-cell population within the extravascularcompartment of the rat lung, which is distributed randomlythroughout the alveolar septal walls (11). This population issaid to be at least five times larger than the peripheral bloodT-cell pool in SPF rats (10). It is possible that the accumulationof T cells within certain areas of lung tissue, without significantincreases in total numbers of infiltrating T cells, may reflect ashift in the pattern of accumulation of resident T cells ratherthan infiltrating cells.
Another interesting feature is the localization of inflamma-tory cells within the lungs. It appears that these cells migratetoward specific areas of lung tissue; they first travel to tissuesurrounding inflamed bronchioles and around bronchiolewalls, but they then decrease in number in these areas andincrease in number in perivascular areas away from the in-flamed bronchioles and throughout the lung parenchyma. Thisshifting migration pattern suggests that immune system cellsmigrate within lung tissue toward areas of bacterial confluence.The chemotactic factors that mediate these events are notknown, but the CXC and CC chemokines are obvious candi-dates. The role of these chemokines is the subject of ourcontinuing investigations.
In conclusion, the capability of the host to confine pneumo-coccal numbers and to survive infection by pneumococci un-able to make pneumolysin indicates the vital role of this toxinin disease. Although much remains to be determined about theevents described above, this paper highlights a number ofnew questions that must be addressed if we are to fullyunderstand the interaction of pneumococci and the hostduring pneumonia.
REFERENCES
1. Alexander, J. E., A. M. Berry, J. C. Paton, J. B. Rubins, P. W. Andrew, andT. J. Mitchell. 1998. Amino acid changes affecting the activity of pneumo-lysin alter the behaviour of pneumococci in pneumonia. Microb. Pathog.24:167–174.
2. Alexander, J. E., R. A. Lock, C. C. A. M. Peeters, J. T. Poolman, P. W.Andrew, T. J. Mitchell, D. Hansman, and J. C. Paton. 1994. Immunization ofmice with pneumolysin toxoid confers a significant degree of protectionagainst at least nine serotypes of Streptococcus pneumoniae. Infect. Immun.62:5683–5688.
3. Benton, K. A., M. P. Everson, and D. E. Briles. 1995. A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremiarather than acute sepsis in mice. Infect. Immun. 63:448–455.
4. Bergeron, Y., N. Ouellet, A.-M. Deslauriers, M. Simard, M. Olivier, andM. G. Bergeron. 1998. Cytokine kinetics and other host factors in responseto pneumococcal pulmonary infection in mice. Infect. Immun. 66:912–922.
5. Berry, A. M., J. Yother, D. E. Briles, D. Hansman, and J. C. Paton. 1989.Reduced virulence of a defined pneumolysin-negative mutant of Streptococ-cus pneumoniae. Infect. Immun. 57:2037–2042.
6. Canvin, J. R., A. P. Marvin, M. Sivakumaran, J. C. Paton, G. J. Boulnois,P. W. Andrew, and T. J. Mitchell. 1995. The role of pneumolysin andautolysin in the pathology of pneumonia and septicemia in mice infectedwith a type 2 pneumococcus. J. Infect. Dis. 172:119–123.
7. Curtis, J. L., G. B. Huffnagle, G. H. Chen, M. L. Warnock, M. R. Gyetko,R. A. McDonald, P. J. Scott, and G. B. Toews. 1994. Experimental murinepulmonary cryptococcus-differences in pulmonary inflammation and lym-phocyte recruitment induced by encapsulated strains of Cryptococcus neo-formans. Lab. Investig. 71:113–126.
8. Feldman, C., T. J. Mitchell, P. W. Andrew, G. J. Boulnois, S. C. Reed, H. C.Todd, P. J. Cole, and R. Wilson. 1990. The effect of Streptococcus pneu-moniae pneumolysin on human respiratory epithelium in vitro. Microb.Pathog. 9:275–284.
9. Feldman, C., R. Reed, A. Rutman, N. C. Munro, D. K. Jeffrey, A. Brain, V.Lund, T. J. Mitchell, P. W. Andrew, G. J. Boulnois, H. C. Todd, P. J. Cole,and R. Wilson. 1991. The effect of Streptococcus pneumoniae on intactrespiratory epithelium. Eur. Respir. J. 5:576–583.
FIG. 9. Numbers of leukocytes in tissue sections from lung samples of MF1mice infected intranasally with 106 CFU of S. pneumoniae PLN-A (n 5 4 for eachtime point; error bars indicate SEM).
500 KADIOGLU ET AL. INFECT. IMMUN.
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

10. Holt, P. G., A. Degebrodt, and T. Venaille. 1985. Preparation of interstitiallung cells by digestion of tissue slices: preliminary characterisation by mor-phology and performance in functional assays. Immunity 54:139–147.
11. Holt, P. G., and M. A. Schon-Hegard. 1987. Localisation of T cells, macro-phages and dendritic cells in rat respiratory tract tissue: implications forimmune function studies. Immunity 62:349–356.
12. Houldsworth, S., P. W. Andrew, and T. J. Mitchell. 1994. Pneumolysinstimulates production of tumor necrosis factor alpha and interleukin-1b byhuman mononuclear phagocytes. Infect. Immun. 62:1501–1503.
13. Huffnagle, G. B., R. M. Strieter, T. J. Standiford, R. A. McDonald, M. D.Burdick, S. L. Kunkel, and G. B. Toews. 1995. The role of monocyte che-motactic protein-1 (MCP-1) in the recruitment of monocytes and CD41
T-cells during a pulmonary Cryptococcus neoformans infection. J. Immunol.155:4790–4797.
14. Kadioglu, A., and P. Sheldon. 1998. Steroid pulse therapy for rheumatoidarthritis: effect on lymphocyte subsets and mononuclear adhesion. Br. J.Rheumatol. 37:282–286.
15. Mitchell, T. J., P. W. Andrew, G. J. Boulnois, C. J. Lee, R. A. Lock, and J. C.Paton. 1992. Molecular studies of pneumolysin as an aid to vaccine design.Zentbl. Bakteriol. 23:429–438.
16. Mitchell, T. J., P. W. Andrew, F. K. Saunders, A. N. Smith, and G. J.
Boulnois. 1991. Complement activation and antibody binding by pneumoly-sin via a region homologous to a human acute phase protein. Mol. Microbiol.5:1883–1888.
17. Nandoskar, M., A. Ferrante, E. J. Bates, N. Hurst, and J. C. Paton. 1986.Inhibition of human monocyte respiratory burst, degranulation, phospho-lipid methylation and bactericidal activity by pneumolysin. Immunity 59:515–520.
18. Paton, J. C., and A. Ferrante. 1983. Inhibition of human polymorphonuclearleukocyte respiratory burst, bactericidal activity, and migration by pneumo-lysin. Infect. Immun. 41:1212–1216.
19. Rayner, C. F. J., A. D. Jackson, A. Rutman, A. Dewar, T. J. Mitchell, P. W.Andrew, P. J. Cole, and R. Wilson. 1995. Interaction of pneumolysin-suffi-cient and -deficient isogenic variants of Streptococcus pneumoniae with hu-man respiratory mucosa. Infect. Immun. 63:442–447.
20. Rubins, J. B., P. W. Andrew, T. J. Mitchell, and D. E. Niewoehner. 1994.Pneumolysin activates phospholipase A2 in pulmonary artery endothelialcells. Infect. Immun. 62:3829–3836.
21. Rubins, J. B., D. Charboneau, J. C. Paton, T. J. Mitchell, and P. W. Andrew.1995. Dual function of pneumolysin in the early pathogenesis of murinepneumococcal pneumonia. J. Clin. Investig. 95:142–150.
Editor: E. I. Tuomanen
VOL. 68, 2000 ROLE OF PNEUMOLYSIN IN BRONCHOPNEUMONIA 501
on April 1, 2019 by guest
http://iai.asm.org/
Dow
nloaded from

ERRATUM
Host Cellular Immune Response to Pneumococcal LungInfection in Mice
ARAS KADIOGLU, NEILL A. GINGLES, KATE GRATTAN, ALISON KERR, TIM J. MITCHELL,AND PETER W. ANDREW
Department of Microbiology & Immunology, University of Leicester, Leicester, and Division of Infection & Immunity,University of Glasgow, Glasgow, United Kingdom
Volume 68, no. 2, p. 492–501, 2000. The symbols in Fig. 2, 5, 6, 7, and 9 should have been defined as follows.Page 494, Fig. 2: e, PLN-A; h, wild type.Page 497, Fig. 5: e, PLN-A; h, wild type. Figure 6: h, lymphocytes; e, macrophages; E, polymorph.Page 498, Fig. 7: h, CD3; e, macrophages; E, neutrophils; ‚, CD19.Page 500, Fig. 9: h, CD3; e, macrophages; E, neutrophils; ‚, CD19.
2390






























