Embed Size (px)
Citation preview

Original Contribution
Intratracheal administration of mitochondrial DNA directly provokeslung inflammation through the TLR9–p38 MAPK pathway
Xiaoling Gu a, Guannan Wu a, Yanwen Yao a, Junli Zeng a, Donghong Shi b, Tangfeng Lv a,Liang Luo c, Yong Song a,n
a Department of Respiratory Medicine, Jinling Hospital, Nanjing University School of Medicine, Nanjing, Jiangsu Province 210002, People’s Republic of Chinab Department of Medical Imaging, Jinling Hospital, Nanjing University School of Medicine, Nanjing, Jiangsu Province 210002, People’s Republic of Chinac Intensive Care Unit, Wuxi Second Affiliated Hospital, Nanjing Medical University, Wuxi, Jiangsu Province 210004, People’s Republic of China
a r t i c l e i n f o
Article history:Received 18 November 2014Received in revised form14 February 2015Accepted 25 February 2015Available online 13 March 2015
Keywords:Mitochondrial DNALungInflammationTLR9–p38 MAPK signal transductionpathwayFree radicals
a b s t r a c t
An increasing number of studies have focused on the phenomenon that mitochondrial DNA (mtDNA)activates innate immunity responses. However, the specific role of mtDNA in inflammatory lung diseaseremains elusive. This study was designed to examine the proinflammatory effects of mtDNA in lungs andto investigate the putative mechanisms. C57BL/6 mice were challenged intratracheally with mtDNA withor without pretreatment with chloroquine. Changes in pulmonary histopathology, cytokine concentra-tions, and phosphorylation levels of p38 MAPK were assayed at four time points. In in vitro experiments,THP-1 macrophages were pretreated or not pretreated with chloroquine, TLR9 siRNA, p38 MAPK siRNA,or SB203580 and then incubated with mtDNA. The levels of cytokines and p-p38 MAPK were detected byELISA and Western blot, respectively. The intratracheal administration of mtDNA induced infiltration ofinflammatory cells, production of proinflammatory cytokines (including IL-1β, IL-6, and TNF-α), andactivation of p38 MAPK. The chloroquine pretreatment resulted in an abatement of mtDNA-induced locallung inflammation. In vitro experiments showed that the exposure of THP-1 macrophages to mtDNA alsoled to a significant upregulation of IL-1β, IL-6, and TNF-α and the activation of p38 MAPK. And theseresponses were inhibited either by chloroquine and TLR9 siRNA or by SB203580 and p38 MAPK siRNApretreatment. The intratracheal administration of mtDNA induced a local inflammatory response in themouse lung that depended on the interactions of mtDNA with TLR9 and may be correlated withinfiltrating macrophages that could be activated by mtDNA exposure via the TLR9–p38 MAPK signaltransduction pathway.
& 2015 Elsevier Inc. All rights reserved.
The etiological factors leading to pulmonary inflammation areoften complicated. Although it has been well established thatbacteria and specific environmental exposures are responsible forvarious lung diseases with profound inflammation, many cases ofinflammatory lung disease are idiopathic and do not present anyidentifiable etiology. For instance, most cases of acute respiratorydistress syndrome do not have a specific etiology and cannot bewell explained by existing mechanisms. There has recently beenincreasing interest in clarifying the role of endogenous dangersignals in the development of inflammatory lung injury and thesystemic inflammatory response. The results of our previous study
showed that the plasma concentration of mitochondrial DNA(mtDNA)1, a newly identified damage-associated molecular pat-tern, is positively correlated with the risk of systemic inflamma-tory response syndrome (SIRS) in acute traumatic patients [1].Additionally, a recently published study reported that acid aspira-tion results in a 120-fold increase in the mtDNA concentration incell-free bronchoalveolar lavage fluid in an animal model of acutegastric aspiration lung injury [2]. However, the specific role ofmtDNA in the development of inflammatory lung injury remainsto be further determined.
Mitochondria DNA is generally considered to be derived from thecircular genomes of bacteria that evolved into the mitochondria oftoday’s eukaryotic cells [3–7]. Similar to bacterial DNA, mtDNA iscircular and contains a higher frequency of unmethylated cytosine–phosphate–guanine (CpG) dinucleotides [8–10]. DNA that containsunmethylated CpG motifs can be recognized by the innate immunesystem and exerts powerful immunostimulatory effects [11–13].Previous studies conducted by Oka et al. [14] have demonstratedthat mtDNA that escapes from autophagy-mediated degradation can
Contents lists available at ScienceDirect
journal homepage: www.elsevier.com/locate/freeradbiomed
Free Radical Biology and Medicine
http://dx.doi.org/10.1016/j.freeradbiomed.2015.02.0340891-5849/& 2015 Elsevier Inc. All rights reserved.
Abbreviations: mtDNA, mitochondrial DNA; SIRS, systemic inflammatoryresponse syndrome; nDNA, nuclear DNA; CpG, cytosine–phosphate–guanine;MAPK, mitogen-activated protein kinase; TLR9, Toll-like receptor 9; DAPI, 40 ,6-diamidino-2-phenylindole; siRNA, small interfering RNA; CQ, chloroquine; SB,SB203580
n Corresponding author. Fax: þ86 25 80863591.E-mail address: [email protected] (Y. Song).
Free Radical Biology and Medicine 83 (2015) 149–158

trigger local inflammation in the heart. Additionally, Zhang et al. [15]reported that mtDNA released into the circulation by shock canactivate the neutrophil p38 mitogen-activated protein kinase (MAPK)signaling pathway via TLR9 and potentially contribute to the devel-opment of posttraumatic SIRS. The results of these recent studiesindicate that mtDNA can not only participate in the activation ofinnate immunity in the original cell but also initiate systemic innateimmunity.
It is well known that CpG-containing DNA activates inflamma-tory responses via an endosomally located pattern recognitionreceptor, namely TLR9 [16–18], which is constitutively expressedin endothelial cells and macrophages in the lung [19,20]. It hasbeen reported that the intravenous administration of mtDNA cancause SIRS and lung inflammation [21]. However, it is not clearwhether the lung inflammation observed in this animal model isdirectly induced by mtDNA or is just a secondary result of SIRS.Even if this published study has confirmed the immunostimula-tory potential of mtDNA in the lungs, the related molecularmechanisms remain unclear. Because DNA is readily endocytosedby macrophages [22], which act as the first line of host defense inthe lung, it is logical to hypothesize that mtDNA is capable ofcausing lung inflammation via active alveolar macrophages andmay be mediated by a signaling pathway similar to the TLR9–p38MAPK pathway in neutrophils. To determine the possible role ofmtDNA in the induction of lung inflammation, we intratracheallyadministered mtDNA to mice and simultaneously examined thein vitro effect of mtDNA on THP-1 macrophages, a human mono-cytic and macrophage cell line, to elucidate the specific signaltransduction mechanism that mediates the inflammatory activa-tion of THP-1 induced by mtDNA exposure.
Materials and methods
Main reagents
40,6-Diamidino-2-phenylindole (DAPI), phorbol 12-myristate13-acetate (PMA), chloroquine, and protease inhibitor cocktailwere purchased from Sigma. Mouse anti-β-actin antibody andrabbit anti-TLR9 antibody were obtained from Abcam. Antibodiesto p38 MAPK and phospho-p38 MAPK (Thr180/Tyr182) werepurchased from Cell Signaling. Alexa Fluor 594-conjugated goatanti-rabbit IgG was purchased from Invitrogen. Goat anti-rabbitIgG–horseradish peroxidase (HRP), goat anti-mouse IgG–HRP, andrabbit anti-CD68 antibody were obtained from Boster Biotechnol-ogy Co. Ltd. (Wuhan, China). The small interfering RNA (siRNA)targeting p38 MAPK, siRNA targeting TLR9, and negative controlsiRNA were purchased from GenePharma (Shanghai, China), andLipofectamine 2000 and Opti-MEM I reduced-serum mediumwere obtained from Invitrogen Corp. (Carlsbad, CA, USA). Heat-treated fetal bovine serum and RPMI 1640–Hepes medium wereobtained from Wisent, Inc.
Mice and ethics statement
Healthy male C57BL/6 mice weighing 20–22 g and age 7–8weeks were obtained from the Animal Feeding Center of YangzhouUniversity. The animal care and experimental procedures wereperformed in compliance with the Institutional Animal Care andUser Guidelines and were approved by the Model Animal ResearchCenter of Jinling Hospital.
Cell line, culture, transfection, and differentiation
THP-1 cells were obtained from the American Type CultureCollection and were cultured in complete medium (RPMI 1640–Hepes
containing 100 U/ml penicillin/streptomycin and 10% fetal bovineserum) according to the supplier’s guidelines. The transfection ofTHP-1 cells was performed according to the manufacturer’s recom-mended protocol (Invitrogen Corp.). Briefly, THP-1 cells were seeded at5�105 cells/ml in 10-cm dishes in 12 ml of growth medium withoutantibiotics. The siRNA solution and Lipofectamine 2000 solution wereprepared in 1.5 ml of Opti-MEM I reduced-serum medium. The siRNAand Lipofectamine solutions were then incubated together for 20 minat room temperature to form the siRNA–Lipofectamine complex, andthis complex was then added to the cells to a final siRNA concentrationof 50 nM. The cells were incubated at 37 1C in a CO2 incubator. Theknockdown efficiency was verified 48 to 72 h after transfection(Supplementary Fig. s1). For differentiation into macrophages,THP-1 cells were plated at 1�106 cells/well in six-well platesand differentiated by exposure to 100 ng/ml PMA for 24 h. Thedifferentiated, plastic-adherent cells were washed twice withfresh complete medium and allowed to rest for 6 h beforeexposure to various conditions.
Preparation of mtDNA and nuclear DNA (nDNA)
The mouse liver mitochondria were isolated using mito-chondrial isolation kits for tissues (Pierce Scientific, Rockford,IL, USA) according to the protocol supplied by the manufac-turer. Mitochondria isolation kits for cells (Pierce Scientific)were used to isolate the mitochondria from cultured THP-1 celllines. The mitochondrial isolation was performed under sterileconditions at 4 1C. The isolated mitochondrial pellets weresuspended in Hanks’ balanced salt solution buffer (Gibco LifeTechnologies, Gaithersburg, MD, USA). The nuclear fractions ofTHP-1 cells and hepatocytes were reserved for the subsequentpreparation of nDNA.
The mitochondrial DNA and nDNA were extracted from theisolated mitochondrial pellets and nuclear fractions, respectively,under sterile conditions with the DNeasy blood and tissue kits(Qiagen, Valencia, CA, USA), according to the protocol supplied bythe manufacturer. The DNA concentrations and purity wereexamined by spectrophotometry. The A260/280 ratio of both themtDNA and the nDNA samples was 1.8 to 2.0, confirming the lackof any significant protein contamination. The endotoxin levels inthe DNA samples were assessed using the Limulus amoebocytelysate assay, and no detectable levels were observed. To furtherexclude any significant nDNA contamination and ensure the purityof the mtDNA, we conducted real-time PCR to probe the samplesfor the presence of the nuclear genes GAPDH and 18S and for thepresence of the mitochondrial genes MT-ND2 and mtCOI, and wefound that the nDNA content was less than 0.1% in the isolatedmtDNA samples. Primers for the nDNA markers GAPDH (forward50-CCATGTTCGTCATGGGTGTGAAC-30 and reverse 50-GCCAGTAGA-GGCAGGGATGATGTTC-30) and 18S (forward 50-GTTCATCCTG-TTCCTGCTCC-30 and reverse 50-GTTCATCCTGTTCCTGCTCC-30) andfor the mtDNA markers MT-ND2 (forward 50-CACAGAAGCTGCCAT-CAAGTA-30 and reverse 50-CCGGAGAGTATATTGTTGAAGAG-30) andmtCOI (forward 50-GCCCCAGATATAGCATTCCC-30 and reverse 50-GTTCATCCTGTTCCTGCTCC-30) were all synthesized by Invitrogen.
Animal procedures
Seven- to eight-week-old mice were randomly divided intothree groups (groups A, B, and C) with 25 animals per group. Allof the mice were anesthetized with intraperitoneal pentobarbitalsodium (3 mg/kg) and placed in the supine position with the headtilted back. The intratracheal administration of nDNA (3 mg/kg,group A) or mtDNA (3 mg/kg, group B and group C) in a volume of60 ml was performed via a microsprayer (Penn-Century, Wyndmoor,PA, USA). The mice in group C were intraperitoneally pretreated
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158150

with chloroquine (30 mg/kg) for 2 h before mtDNA exposure. All ofthe mice were sacrificed 0, 3, 6, 12, or 24 h after administration, andtheir lungs were subsequently harvested. The large left lobe wasfixed in 4% paraformaldehyde for histological analysis, and theremaining lung lobes were stored at �80 1C before the subsequentexperimental assays. Here, the 0-h time point means that the micewere untreated shams and were immediately sacrificed aftertracheal intubation procedure.
THP-1 macrophage activation assays
Differentiated THP-1 macrophages were exposed to mtDNA(10 mg/ml) or nDNA (10 mg/ml). For the inhibitor studies, THP-1macrophages were pretreated with chloroquine (10 mg/ml, 30 min)or SB203580 (1 mM, 30 min), which is a specific inhibitor of thep38 MAPK signal transduction pathway, or were transfected withTLR9 siRNA or p38 MAPK siRNA before mtDNA exposure. The cell-conditioned medium was collected by centrifugation 0, 3, 6, 12, or24 h after exposure and then assayed for proinflammatory cyto-kines by ELISA as described below. THP-1 macrophages were lysedwith extraction buffer containing a protease inhibitor cocktail(Roche) 0, 30, 60, or 120 min after exposure to determine the totaland phosphorylated p38 MAPK levels by Western blot asdescribed below.
Histopathology and immunofluorescence of the lungs
The left lungs were fixed overnight at 4 1C in 4% paraformalde-hyde and processed by successive dehydration with an alcoholseries and xylene. The tissues were then embedded in paraffin andcut into 5-mm-thick sections for hematoxylin–eosin staining orimmunofluorescence assay.
Hematoxylin–eosin (H&E) staining was carried out according tothe instructions provided by the manufacturer to determine theseverity of the lung inflammation. All of the tissue sections wereevaluated by an experienced blinded pathologist and scoredaccording to criteria described previously [23]. Briefly, accordingto three criteria, i.e., vascular congestion and interstitial edema,alveolar structural disturbance, and atelectasis and inflammatorycell infiltration, and using a semiquantitative scale, the histologicalscore of the lung sections was calculated as the sum of the scores(0–3) given for each criterion. The cumulative histology scoreranged from 0 to 9.
To observe macrophages in the lung, we conducted an immu-nofluorescence assay as described below. Briefly, paraffin-embedded lung tissue sections were dewaxed in xylene andrehydrated in graded ethanol washes (100, 90, 70, and 50%). Thesamples were then washed twice for 5 min with phosphate-buffered saline (PBS) and subsequently incubated with 0.3% TritonX-100 for 8 min at room temperature. The samples were washedin PBS for 10 min, blocked with 3% bovine serum albumin (BSA) for1 h at 37 1C, and incubated overnight at 4 1C with a rabbit anti-CD68 antibody at a 1:200 dilution in 2% BSA. The samples werethen washed three times with PBS for 5 min each and subse-quently incubated with a goat anti-rabbit Alexa Fluor 594-conjugated secondary antibody (1:400) for 1 h at 37 1C in thedark. Finally, the nuclei were stained with DAPI (5 mg/ml). Thesamples were visualized with a laser scanning confocal micro-scope (FluoView FV10i, Japan) to detect the CD68þ macrophagesusing red fluorescence.
Enzyme-linked immunosorbent assay
The middle lobe of the right lung was homogenized and lysedwith extraction buffer containing a protease inhibitor cocktail(Roche), and we then collected the tissue supernatants via
centrifugation at 12,000 rpm for 30 min. The concentrations ofvarious cytokines in the mouse lung extracts and the supernatants(conditioned medium) of THP-1 macrophages were assessed usingappropriate ELISA kits as recommended by the manufacturer (4ABiotech Co., Ltd, Beijing, China). The lowest limits of detection formouse interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α),and IL-6 were 7, 7, and 4 pg/ml, respectively, and the lowestdetection limits for human IL-1β, IL-6, and TNF-α were 2, 1, and4 pg/ml, respectively.
Western blot analysis
The cell proteins were obtained from THP-1 macrophages, andthe tissue proteins were obtained from the right lower lungs of themice in each group. We performed a Western blot analysis of thecellular and tissue lysates as previously described [24]. Briefly, thecells or tissue homogenates were lysed in ice-cold protein extrac-tion buffer containing a protease inhibitor cocktail (Roche) for30 min. After the whole lysates were centrifuged at 12,000g for30 min, the supernatants were collected. The protein concentra-tions in the supernatants were determined by the Bradfordmethod. The proteins (20 mg) from the lung tissue and THP-1macrophages were separated in 12% SDS–PAGE gels and electro-phoretically transferred onto a polyvinylidene fluoride membraneusing an electroblotting apparatus (Bio-Rad). The membraneswere blocked in 5% BSA in PBS/Tween 20 (PBST) for 1 h at 37 1C,and the blocked membranes were then incubated overnight at 4 1Cwith anti-β-actin mouse monoclonal antibody (1:1000), anti-phospho-p38 MAPK (Thr180/Tyr182) rabbit monoclonal antibody(1:800), or anti-p38 MAPK rabbit polyclonal antibody (1:800) inPBST containing 2% BSA. After three washes with PBST buffer, themembranes were incubated with secondary HRP-conjugated anti-body for 1 h at 37 1C. The protein signals were enhanced bychemiluminescence detection kits (SuperSignal West Pico) anddetected using an Odyssey Scanning System (Li-Cor Biosciences,Lincoln, NE, USA).
Statistical analysis
The study data are expressed as the means 7 standarddeviation and assessed for statistical significance using Student’st test. A two-tailed p value less than 0.05 was consideredstatistically significant. All of the calculations and statisticalanalyses were performed using SPSS software for Windows(version 17.0).
Results
MtDNA induces histopathological changes in the lungs via TLR9
To examine the potential of mtDNA to induce lung inflamma-tion, mtDNA was intratracheally instilled into the lungs of normalmale C57BL/6 mice. Serial lung sections obtained from mice aftermtDNA exposure were stained with H&E and examined by lightmicroscopy. Representative microscopic images of these H&E-stained lung specimens are shown in Fig. 1. The mtDNA adminis-tration caused marked inflammatory cell infiltration accompaniedby thickening of the alveolar septa, which is suggestive of lunginflammation and acute lung injury. However, the mice in thenDNA control group showed no pathological changes in pulmon-ary inflammation. Additionally, as shown in Table 1, the histolo-gical score of the pulmonary tissues in the mtDNA exposure groupwas also markedly increased as early as 3 h after instillation,peaked at 6 h, and then gradually decreased, but the values afterthis decrease remained significantly higher compared with those
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158 151

found for the nDNA control group (p o 0.001 for all four selectedtime points). Considering that TLR9 is the only known Toll-likepattern recognition receptor for mtDNA, to further determinewhether TLR9 is a key upstream molecule that mediates theinflammatory response induced by mtDNA in the lung, mice wereintraperitoneally pretreated with chloroquine for 2 h and thensubjected to intratracheal mtDNA instillation. As shown in Fig. 1Cand Table 1, pretreatment with chloroquine (CQ) significantlyameliorated the mtDNA-induced inflammatory pathologicalchanges in the lungs and reduced the histological score of thepulmonary tissues after mtDNA exposure. These results suggestedthat TLR9 mediates the inflammatory histopathological changes inthe lungs stimulated by mtDNA.
Additionally, because alveolar macrophages act as the firstline of host defense in the lung, we conducted an immuno-fluorescence assay with the paraffin-embedded lung tissuesections to determine whether macrophages had infiltratedinto the mtDNA-treated pulmonary tissues. Representative
microscopic findings from these immunofluorescence-labeledlung specimens are shown in Fig. 2. The mice treated withcontrol nDNA presented no obvious CD68þ macrophage infil-tration at all selected time points after the intratrachealinstillation. However, exposure to mtDNA resulted in a pro-found increase in the amount of CD68þ macrophages in thelung tissues at all selected time points, which suggested thatthe mtDNA-induced lung inflammation was accompanied byCD68þ macrophage infiltration and that macrophages prob-ably play an important role in the initiation and developmentof mtDNA-induced inflammation.
MtDNA triggers cytokine responses in the lungs via TLR9
As further evidence of the mtDNA-induced inflammation, theconcentrations of proinflammatory cytokines in the lung homo-genate were assessed by ELISA. As shown in Fig. 3, in the mtDNAinstillation group at all four time points (3, 6, 12, and 24 h), all ofthe three examined proinflammatory cytokines (IL-1β, IL-6, andTNF-α) exhibited statistically significant increases compared withthe levels observed in the nDNA exposure control group. Theconcentrations of both IL-1β and TNF-α were highest 6 h after themtDNA challenge and then gradually decreased, but thesedecreased levels remained significantly higher compared withthose found in the respective nDNA controls. The levels of IL-6 inthe lung homogenate reached a peak as early as 3 h after mtDNAinstillation and then decreased, and the decreased levels remainedsignificantly higher than the levels observed in the control group.These data indicated that mtDNA triggers a significant increase inIL-1β, IL-6, and TNF-α production in the lungs and provideevidence for the potential ability of mtDNA to induce lunginflammation.
Fig. 1. Representative photomicrographs of lung histopathology stained with H&E. (A) Control group treated with nDNA (3 mg/kg). (B) mtDNA challenge group treated withmtDNA (3 mg/kg). (C) Chloroquine inhibitory group, which was pretreated with CQ (30 mg/kg, ip) before mtDNA (3 mg/kg) challenge. Original magnification, �400. Theseobservations are representative of results of experiments with five mice per time point per group.
Table 1Histological scores of lung tissue.
Time point nDNA mtDNA CQ þ mtDNA pa pb
Mean SD Mean SD Mean SD
0 h 0.3 0.67495 0.3 0.67495 0.3 0.48305 1.0 1.03 h 0.5 0.70711 3.6 0.69921 2.5 1.08012 o0.001 0.0156 h 0.5 0.70711 4.9 1.19722 3.5 1.08012 o0.001 0.01312 h 0.3 0.48305 4.8 1.47573 3.0 0.66667 o0.001 0.00224 h 0.3 0.67495 3.9 0.99443 3.0 0.8165 o0.001 0.04
a mtDNA vs nDNA.b CQ þ mtDNA vs mtDNA.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158152

Fig. 2. Representative photomicrographs of immunofluorescence-labeled CD68þ macrophages in lung specimens. The macrophages were labeled with antibodies to CD68(red), and the nuclei were stained with DAPI (blue). Scale bar, 30 mm. These observations are representative of results from experiments with five mice per time pointper group.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158 153

The proinflammatory cytokines (IL-1β, IL-6, and TNF-α) in themice of the CQ pretreatment group were also assessed andcompared with those of the mtDNA-exposure group. As shownin Fig. 3, pretreatment with CQ resulted in a significant reductionin the IL-6 concentration in the lung homogenates at all fourselected time points compared with the levels found in themtDNA-exposure group (p o 0.05, Fig. 3B). Although pretreat-ment with CQ led only to a decreasing trend in the IL-1β level,with a limbic p value of 0.063 at 12 h, the suppression effect of CQon IL-1β production assessed at the other three selected timepoints (3, 6, and 24 h) was statistically significant (p o 0.05,Fig. 3A). CQ pretreatment also significantly reduced the concen-trations of TNF-α in the lung homogenates at 3 and 6 h aftermtDNA instillation but did not significantly downregulate thelevels of TNF-α at 12 and 24 h compared with the levels foundin the mtDNA-exposure group (Fig. 3C). Taken together, these datasuggested that TLR9 mediates the inflammatory cytokine responsein the lungs after mtDNA exposure.
Intratracheal mtDNA activates p38 MAPK in the lungs via TLR9
Because p38 MAPK is a key early intermediary in the inflam-matory response and is one of the downstream targets of the TLR9signaling pathway, which is the only known Toll-like receptorpathway for mtDNA, we performed a Western blot assay todetermine the effect of mtDNA on the total and phosphorylatedp38 MAPK levels in the lung. As shown in Fig. 4, mtDNA instilla-tion induced a significant upregulation of phosphorylated p38MAPK at 6 h, and this level returned to the baseline 12 h after
mtDNA stimulation. However, the expression of total p38 MAPKwas not significantly changed within 24 h of mtDNA exposure.Conversely, nDNA administration had no effect on the expressionof total or phosphorylated p38 MAPK (Fig. 4). These observationssuggested that mtDNA induces p38 MAPK activation in vivo andthat this activation is probably involved in the mtDNA-inducedlung inflammation. Further more, pretreatment with CQ signifi-cantly suppressed p38 MAPK activation in lung tissue after mtDNAexposure (Fig. 4). It confirmed that mtDNA activates the p38MAPKpathway via TLR9 in the lung of model mice.
MtDNA activates p38 MAPK in cultured THP-1 macrophages via TLR9
Given that the above-presented evidence indicated that mtDNAexposure can trigger significant pulmonary inflammation via thepattern recognition receptor TLR9 and that this inflammation wasaccompanied by CD68þ macrophage infiltration, to clarifywhether macrophages play an important role in the developmentof mtDNA-induced inflammation, we performed further experi-ments to determine whether mtDNA exposure can induce cyto-kine production and activate p38 MAPK in cultured THP-1macrophages.
Differentiated THP-1 macrophages were incubated with eithernDNA or mtDNA (10 mg/ml), and a Western blot assay wasperformed to determine whether mtDNA stimulation activatesthe p38 MAPK pathway in THP-1 macrophages. As shown inFig. 5, mtDNA exposure induced significant upregulation ofphosphorylated p38 MAPK as early as 30 min after incubation,and the increased expression of phosphorylated p38 MAPK
Fig. 3. Cytokine responses in the lungs induced by control nDNA, mtDNA, and CQ þ mtDNA. (A) Concentration of IL-1β in the lung homogenate. (B) Concentration of IL-6 inthe lung homogenate. (C) Concentration of TNF-α in the lung homogenate. *p o 0.05; **p o 0.001. The results are from experiments with five mice per time point per group.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158154

remained 1 h after exposure and then weakened to the base-line level at 2 h after mtDNA stimulation. However, the expres-sion of total p38 MAPK was not significantly changed within2 h of mtDNA administration. Conversely, nDNA exposure hadno effect on the expression of total or phosphorylated p38MAPK (Fig. 5). These observations suggested that mtDNAinduces p38 MAPK activation in cultured THP-1 macrophages.
Furthermore, we attempted to investigate whether TLR9 is akey upstream molecule that mediates the p38 MAPK activationinduced by mtDNA in THP-1 macrophages. We pretreated THP-1cells with TLR9 siRNA before mtDNA exposure. Additionally,because endosomal acidification is a prerequisite for several TLRs(including TLR9) [25–27], THP-1 macrophages were preincubatedwith CQ, an inhibitor of endosomal acidification, before mtDNAadministration. As shown in Fig. 5, both TLR9 siRNA transfectionand CQ preincubation resulted in no upregulation of the expres-sion of phosphorylated p38 MAPK in THP-1 macrophages aftermtDNA exposure. This means that mtDNA activates p38 MAPK incultured THP-1 macrophages via TLR9.
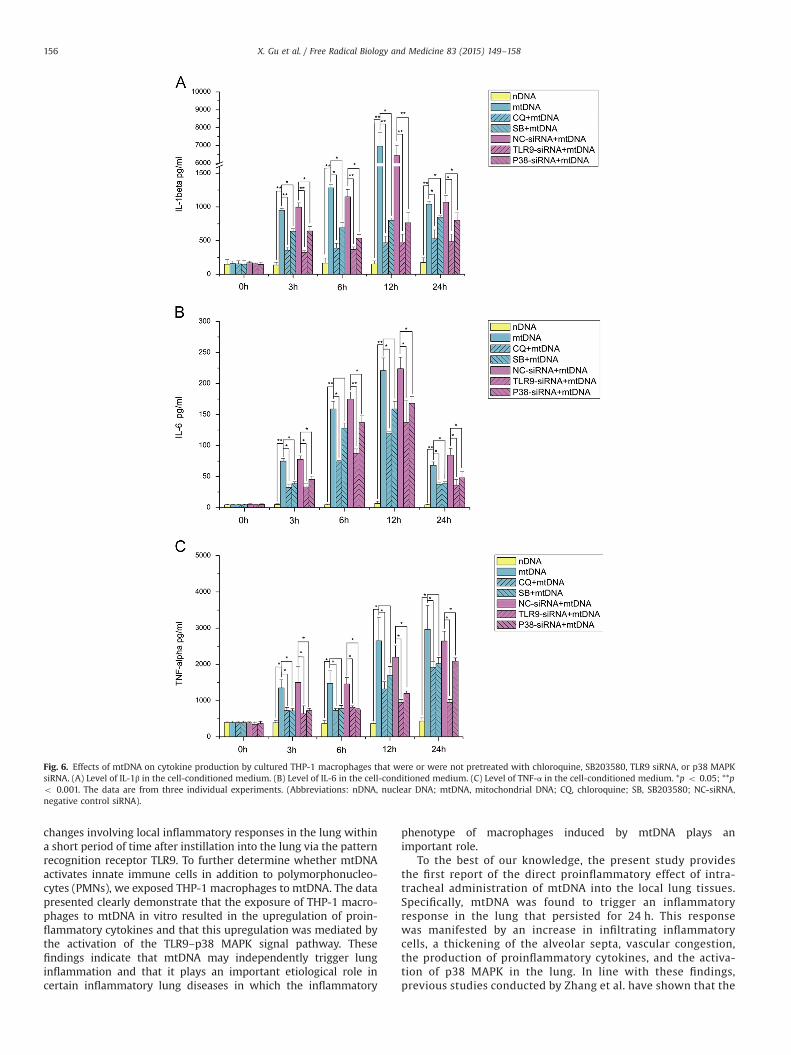
MtDNA induces cytokine production by THP-1 macrophages via theTLR9–p38 MAPK pathway
The concentrations of proinflammatory cytokines in the super-natants of the THP-1 macrophages after mtDNA exposure were
assessed by ELISA. As shown in Fig. 6, in the mtDNA stimulationgroup at all four time points (3, 6, 12, and 24 h), all three examinedcytokines (IL-1β, IL-6, and TNF-α) exhibited significant increasescompared with the levels found in the nDNA-exposure controlgroup. The concentrations of both IL-1β and IL-6 were highest 12 hafter the mtDNA challenge and then decreased at 24 h, but thedecreased levels remained significantly elevated compared withthose found in the nDNA controls. A persistent increase in theconcentrations of TNF-α in the THP-1 macrophage culture med-ium treated with mtDNA was observed within 24 h after admin-istration, and this increase was significantly higher than that foundin the nDNA-exposure control group. These data indicated thatmtDNA triggered a marked increase in IL-1β, IL-6, and TNF-αproduction in the cultured THP-1 macrophages.
Because the above-presented evidence indicated that mtDNAexposure activates the p38 MAPK pathway in cultured THP-1macrophages, we performed further experiments using the phar-macological probe SB203580 and p38 MAPK siRNA to clarify thecausality between the p38 MAPK signal and the cytokine produc-tion induced by mtDNA. As shown in Fig. 5, both preincubationwith SB203580 and pretreatment with p38 MAPK siRNA success-fully suppressed the p38 MAPK activation triggered by mtDNA. Inaddition, pretreatment with SB203580 resulted in a significantreduction in IL-1β production by mtDNA-stimulated THP-1 macro-phages at all four selected time points (p o 0.05, Fig. 6A).Although the suppression effect of SB203580 on TNF-α productionwas statistically significant only at 3 h after mtDNA administration,a trend toward TNF-α inhibition at 6, 12, and 24 h was alsoobserved, with p values of 0.061, 0.06, and 0.062, respectively(Fig. 6C). Analogously, although preincubation with SB203580 ledto a significant reduction in IL-6 production only at 3 and 24 hafter mtDNA exposure, a trend toward IL-6 inhibition at theremaining two selected time points (6 and 12 h) was alsoobserved, with p values of 0.093 and 0.066, respectively(Fig. 6B). Additionally, as shown in Fig. 6, pretreatment with p38MAPK siRNA significantly reduced the production of all threeinflammatory cytokines in mtDNA-stimulated THP-1 macrophagesat all four selected time points. These data therefore suggested animportant role for the p38 MAPK signal in mediating cytokineproduction in THP-1 macrophages stimulated with mtDNA.
Furthermore, we attempted to investigate whether TLR9 is akey upstream molecule that mediates the inflammatory cytokineresponse induced by mtDNA in THP-1 macrophages. We pre-treated THP-1 cells with TLR9 siRNA or with CQ before mtDNAexposure. As shown in Fig. 5, both TLR9 siRNA transfection and CQpreincubation suppressed the p38 MAPK activation induced bymtDNA. Additionally, the data shown in Fig. 6 demonstrate thatpretreatment with CQ or TLR9 siRNA transfection resulted in asignificant reduction in the production of all three examinedcytokines (IL-1β, IL-6, and TNF-α) triggered by mtDNA exposureat all four selected time points (3, 6, 12, and 24 h). Theseobservations suggested that TLR9 acts as a key upstream moleculein mtDNA-induced cytokine production and p38 MAPK activationin THP-1 macrophages.
Discussion
Although it is well known that both bacteria and specificenvironmental exposures can lead to various lung diseases withprofound inflammation, many cases of inflammatory lung diseaseare idiopathic, and the etiological factors leading to pulmonaryinflammation are always unknown. Therefore, we investigated thepotential of mtDNA, which is a newly identified danger-associatedmolecular pattern, for directly inducing lung inflammation in vivo.The present study showed that mtDNA instigated a series of
Fig. 4. Protein expression profiles of phosphorylated (p) p38 MAPK in the lungsinduced by control nDNA, mtDNA, and CQ þ mtDNA. The results are representativeof experiments with five mice per time point per group.
Fig. 5. Protein expression profiles of phosphorylated (p) p38 MAPK in THP-1macrophages that were or were not pretreated with chloroquine, SB203580, TLR9siRNA, or p38 MAPK siRNA and were then challenged with mtDNA. The results arerepresentative of three individual experiments.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158 155
changes involving local inflammatory responses in the lung withina short period of time after instillation into the lung via the patternrecognition receptor TLR9. To further determine whether mtDNAactivates innate immune cells in addition to polymorphonucleo-cytes (PMNs), we exposed THP-1 macrophages to mtDNA. The datapresented clearly demonstrate that the exposure of THP-1 macro-phages to mtDNA in vitro resulted in the upregulation of proin-flammatory cytokines and that this upregulation was mediated bythe activation of the TLR9–p38 MAPK signal pathway. Thesefindings indicate that mtDNA may independently trigger lunginflammation and that it plays an important etiological role incertain inflammatory lung diseases in which the inflammatory
phenotype of macrophages induced by mtDNA plays animportant role.
To the best of our knowledge, the present study providesthe first report of the direct proinflammatory effect of intra-tracheal administration of mtDNA into the local lung tissues.Specifically, mtDNA was found to trigger an inflammatoryresponse in the lung that persisted for 24 h. This responsewas manifested by an increase in infiltrating inflammatorycells, a thickening of the alveolar septa, vascular congestion,the production of proinflammatory cytokines, and the activa-tion of p38 MAPK in the lung. In line with these findings,previous studies conducted by Zhang et al. have shown that the
Fig. 6. Effects of mtDNA on cytokine production by cultured THP-1 macrophages that were or were not pretreated with chloroquine, SB203580, TLR9 siRNA, or p38 MAPKsiRNA. (A) Level of IL-1β in the cell-conditioned medium. (B) Level of IL-6 in the cell-conditioned medium. (C) Level of TNF-α in the cell-conditioned medium. *p o 0.05; **po 0.001. The data are from three individual experiments. (Abbreviations: nDNA, nuclear DNA; mtDNA, mitochondrial DNA; CQ, chloroquine; SB, SB203580; NC-siRNA,negative control siRNA).
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158156

intraperitoneal injection of mtDNA can induce systemicinflammation and acute lung injury [21]. However, that studydid not clarify whether mtDNA itself can induce an inflamma-tory response in the lung because there are t least two possiblemechanisms responsible for inflammation-associated lunginjury in the context of a systemic inflammatory response:the lung injury may be due to inflammatory mediators thatwere extensively released into the bloodstream, and thephenomenon of lung injury may also be caused directly bymtDNA that traveled through the endothelial–epithelial barrierand reached the lung via the blood circulation.
In addition, the time-course changes in the proinflammatorycytokines recorded in the present study first showed that theconcentrations of both IL-1β and TNF-α in the lung were elevatedas early as 3 h after mtDNA instillation, reached peaks at 6 h, andthen decreased. Consistent with this finding, the immu-nofluorescence-labeled lung specimens also showed that thenumber of CD68þ macrophages that had infiltrated into the lungwas increasing at 3 h, reached maximal levels at 6 h, and thengradually decreased at 12 and 24 h after mtDNA challenge. Thisobservation indirectly suggested that the macrophages were apossible source of the proinflammatory cytokines and that thesecells were involved in the pulmonary inflammation induced bymtDNA administration. However, the time course for IL-6 in thelung homogenate was slightly different from that found for theinfiltrated CD68þ macrophages in the lung: the IL-6 levels reacheda peak 3 h after mtDNA instillation and then gradually decreasedwithin 24 h after mtDNA exposure. This finding indicates that themacrophages may not be the only source of IL-6 and thatadditional innate immune cells may also participate in the initia-tion and development of mtDNA-induced lung inflammation.
Although the capacity of mtDNA to trigger innate immuneresponses through both the TLR9–p38 MAPK and the NLRP3–caspase-1 signaling pathways has been extensively investigated[15,28,29], the potential ability of cell-free mtDNA to induce aninflammatory phenotype in macrophages and the involvedmechanisms are not fully understood. The data presented hereclearly demonstrate that mtDNA exposure results in the upregula-tion of proinflammatory cytokines as well as the activation of p38MAPK in cultured THP-1 macrophages. In agreement with ourin vitro data, Zhang et al. have reported that mtDNA challengeinduces a robust inflammatory response in primary culturedperitoneal macrophages, as manifested by the production ofinflammatory mediators, including TNF-α, IL-6, and IL-10 [21].However, it is interesting that the time course of the examinedcytokines in the supernatants of THP-1 macrophages was notentirely consistent with that found in the in vivo experiments. Thisis probably because the cell lines do not fully reflect the responsesof primary cells. Therefore, the reality in the lung may be slightlydifferent from the in vitro results.
The present finding that mtDNA induces pneumonitis in themurine model raises intriguing questions regarding the involve-ment of self-mtDNA in inflammation. It can be envisaged thatcertain pathological states resulting in tissue injury and necrosiscould lead to the release of mtDNA from cells and that theseendogenous mtDNA could subsequently promote destructive pro-cesses in the host through the initiation of autoinflammation andthereby create a vicious circle of cell destruction and inflamma-tion. In fact, in a previous study [15], Zhang and co-workersshowed that hemorrhagic shock-induced mtDNA release contri-butes to the development of posttraumatic SIRS and organ injuryby activating PMNs through p38 MAPK. Additionally, in a recentreport, we demonstrated that the plasma mtDNA concentration ispositively correlated with the risk of SIRS in patients with acutetraumatic injury [1]. Therefore, it seems likely that therapiesincluding nuclease or suppressive oligodeoxynucleotides may
minimize or counteract the impact of extracellular mtDNA, parti-cularly in cases of mtDNA-induced sterile inflammation.
Although the present study indicated that mtDNA is clearlyinvolved in inflammation, there are several limitations that war-rant further discussion. The major limitation of this study is that itstill remains unclear which components of mtDNA contribute toits proinflammatory effect in the lung. Additionally, because theindividual preparation of mtDNA is moderately different and theisolated mtDNA may contain scanty proteins or nDNA, eachpreparation of mtDNA probably has a slightly different ratio ofthese components. Thus, unlike using commercial reagent pre-parations, the application of these extracted mtDNA preparationsat the same concentration does not always guarantee the sameresponses in different assays. Therefore, further studies applyingcommercial standardized mtDNA are required to determine theproinflammatory potential of mtDNA in the lung and to clarify themechanisms involved in this process.
Conclusions
To summarize, the present study demonstrated that the intra-tracheal administration of mtDNA induces a local inflammatoryresponse in the mouse lung that depends on the interactions ofmtDNA with TLR9 and may be correlated with infiltrated macro-phages. Because the exposure of cultured THP-1 macrophages tomtDNA led to the upregulation of several proinflammatory cyto-kines via the TLR9–p38 MAPK signal pathway, it is likely thatmacrophage activation by mtDNA may play an important role inthe mtDNA-induced lung inflammation. These events may parti-cipate in the genesis of certain idiopathic inflammatory lungdiseases and provide novel therapeutic targets for these diseases.
Acknowledgments
The present study was supported by the National NaturalScientific Foundation of China (Nos. 81170064 and 81370172).The National Natural Scientific Foundation of China grants didnot directly participate in the literature search, determination ofstudy eligibility criteria, data analysis or interpretation, or pre-paration, review, or approval of the manuscript for publication.Statements in this article should not be construed as an endorse-ment by the National Natural Scientific Foundation of China.
Appendix A. Supplementary material
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.freeradbiomed.2015.02.034.
References
[1] Gu, X.; Yao, Y.; Wu, G.; Lv, T.; Luo, L.; Song, Y. The plasma mitochondrial DNA isan independent predictor for post-traumatic systemic inflammatory responsesyndrome. PLoS One 8:e72834; 2013.
[2] Davidson, B. A.; Vethanayagam, R. R.; Grimm, M. J.; Mullan, B. A.; Raghaven-dran, K.; Blackwell, T. S.; Freeman, M. L.; Ayyasamy, V.; Singh, K. K.; Sporn, M.B.; et al. NADPH oxidase and Nrf2 regulate gastric aspiration-inducedinflammation and acute lung injury. J. Immunol. 190:1714–1724; 2013.
[3] Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 14:255–274; 1967.[4] Karlin, S.; Campbell, A. M. Which bacterium is the ancestor of the animal
mitochondrial genome? Proc. Natl. Acad. Sci. USA 91:12842–12846; 1994.[5] Cardon, L. R.; Burge, C.; Clayton, D. A.; Karlin, S. Pervasive CpG suppression in
animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 91:3799–3803; 1994.[6] Gray, M. W.; Burger, G.; Lang, B. F. Mitochondrial evolution. Science
283:1476–1481; 1999.[7] Gray, M. W.; Burger, G.; Lang, B. F. The origin and early evolution of
mitochondria. Genome Biol. 2; 2001. REVIEWS1018.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158 157

[8] Hochhauser, D. Relevance of mitochondrial DNA in cancer. Lancet 356:181–-182; 2000.
[9] Pollack, Y.; Kasir, J.; Shemer, R.; Metzger, S.; Szyf, M. Methylation pattern ofmouse mitochondrial DNA. Nucleic Acids Res. 12:4811–4824; 1984.
[10] Taanman, J. W. The mitochondrial genome: structure, transcription, transla-tion and replication. Biochim. Biophys. Acta 1410:103–123; 1999.
[11] Sparwasser, T.; Miethke, T.; Lipford, G.; Borschert, K.; Hacker, H.; Heeg, K.;Wagner, H. Bacterial DNA causes septic shock. Nature 386:336–337; 1997.
[12] Klinman, D. M.; Kamstrup, S.; Verthelyi, D.; Gursel, I.; Ishii, K. J.; Takeshita, F.;Gursel, M. Activation of the innate immune system by CpG oligodeoxynucleo-tides: immunoprotective activity and safety. Springer Semin. Immunopathol.22:173–183; 2000.
[13] Krieg, A. M. The role of CpG motifs in innate immunity. Curr. Opin. Immunol.12:35–43; 2000.
[14] Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.;Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA thatescapes from autophagy causes inflammation and heart failure. Nature485:251–255; 2012.
[15] Zhang, Q.; Itagaki, K.; Hauser, C. J. Mitochondrial DNA is released by shock andactivates neutrophils via p38 MAP kinase. Shock 34:55–59; 2010.
[16] Wagner, H. Interactions between bacterial CpG-DNA and TLR9 bridge innateand adaptive immunity. Curr. Opin. Microbiol. 5:62–69; 2002.
[17] Ashkar, A. A.; Rosenthal, K. L. Toll-like receptor 9, CpG DNA and innateimmunity. Curr. Mol. Med. 2:545–556; 2002.
[18] Chuang, T. H.; Lee, J.; Kline, L.; Mathison, J. C.; Ulevitch, R. J. Toll-like receptor9 mediates CpG-DNA signaling. J. Leukocyte Biol. 71:538–544; 2002.
[19] Li, J.; Ma, Z.; Tang, Z. L.; Stevens, T.; Pitt, B.; Li, S. CpG DNA-mediated immuneresponse in pulmonary endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol.287:L552–L558; 2004.
[20] Guo, Z.; Garg, S.; Hill, K. M.; Jayashankar, L.; Mooney, M. R.; Hoelscher, M.;Katz, J. M.; Boss, J. M.; Sambhara, S. A distal regulatory region is required forconstitutive and IFN-beta-induced expression of murine TLR9 gene. J. Immu-nol. 175:7407–7418; 2005.
[21] Zhang, J. Z.; Liu, Z.; Liu, J.; Ren, J. X.; Sun, T. S.; Mitochondrial, D. N. A. inducesinflammation and increases TLR9/NF-kappaB expression in lung tissue. Int. J.Mol. Med. 33:817–824; 2014.
[22] Yi, A. K.; Tuetken, R.; Redford, T.; Waldschmidt, M.; Kirsch, J.; Krieg, A. M. CpGmotifs in bacterial DNA activate leukocytes through the pH-dependentgeneration of reactive oxygen species. J. Immunol. 160:4755–4761; 1998.
[23] Sun, T.; Wang, X.; Liu, Z.; Liu, S.; Zhang, J. Patterns of cytokine release andevolution of remote organs from proximal femur fracture in COPD rats. Injury42:825–832; 2011.
[24] Sun, Z.; Wang, C.; Shi, C.; Sun, F.; Xu, X.; Qian, W.; Nie, S.; Han, X. ActivatedWnt signaling induces myofibroblast differentiation of mesenchymal stemcells, contributing to pulmonary fibrosis. Int. J. Mol. Med. 33:1097–1109; 2014.
[25] Hong, Z.; Jiang, Z.; Liangxi, W.; Guofu, D.; Ping, L.; Yongling, L.; Wendong, P.;Minghai, W. Chloroquine protects mice from challenge with CpG ODN and LPSby decreasing proinflammatory cytokine release. Int. Immunopharmacol.4:223–234; 2004.
[26] Yasuda, H.; Leelahavanichkul, A.; Tsunoda, S.; Dear, J. W.; Takahashi, Y.; Ito, S.;Hu, X.; Zhou, H.; Doi, K.; Childs, R.; et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am. J. Physiol.Renal Physiol. 294:F1050–F1058; 2008.
[27] Sacre, S. M.; Lo, A.; Gregory, B.; Simmonds, R. E.; Williams, L.; Feldmann, M.;Brennan, F. M.; Foxwell, B. M. Inhibitors of TLR8 reduce TNF production fromhuman rheumatoid synovial membrane cultures. J. Immunol. 181:8002–8009;2008.
[28] Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki,K.; Hauser, C. J. Circulating mitochondrial DAMPs cause inflammatoryresponses to injury. Nature 464:104–107; 2010.
[29] Shimada, K.; Crother, T. R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.;Ramanujan, V. K.; Wolf, A. J.; Vergnes, L.; Ojcius, D. M.; et al. Oxidizedmitochondrial DNA activates the NLRP3 inflammasome during apoptosis.Immunity 36:401–414; 2012.
X. Gu et al. / Free Radical Biology and Medicine 83 (2015) 149–158158




























![09 Lecture [호환 모드] - Konkukhome.konkuk.ac.kr/~parkyong/Classes/chapter 9.pdf · and we would expect the smallest one ... A Substitution Reaction A tertiary alkyl halide and](https://img.dokumen.tips/doc/110x75/5ad7ae727f8b9a3e578c9016/09-lecture-parkyongclasseschapter-9pdfand-we-would-expect-the.jpg)
