Embed Size (px)
Citation preview

ACADEMIC DISSERTATION
To be presented for public examination with the permission of the Medical Faculty of theUniversity of Helsinki in the Richard Faltin Auditorium of the Surgical Hospital on
December 1st, 2000, at 12 noon.
Helsinki 2000
Department of MedicineDivision of NephrologyUniversity of Helsinki
Finland
FAMILIAL FACTORS AND DIABETIC
NEPHROPATHY IN TYPE 1 DIABETES
Johan Fagerudd

ISBN 952-91-2849-5ISBN 952-91-2850-9 (pdf)Helsinki 2000Yliopistopaino

Felix qui potuit rerum cognoscere causas
To Pia, Viktor and Blanca

Johan Fagerudd
4
Contents
List of original publications ..................................................................................... 6
Abbreviations .......................................................................................................... 7
Introduction ............................................................................................................ 8
Review of the literature ........................................................................................... 9History and classification of diabetes .................................................................. 9The natural history of diabetic nephropathy ....................................................... 9
The changing natural history of diabetic nephropathy ................................. 10Renal structural changes in diabetic nephropathy............................................. 11
Structural changes in relation to albuminuria .............................................. 12Structural changes in relation to kidney function ........................................ 12
Pathogenesis of diabetic nephropathy ............................................................... 12Hyperglycemia ........................................................................................... 12Abnormalities in extracellular matrix .......................................................... 13Hemodynamic factors ................................................................................. 14Growth factors ............................................................................................ 14Genetic factors ............................................................................................ 15Genetics...................................................................................................... 16Other factors ............................................................................................... 16
Aims of the study .................................................................................................. 18
Study design and subjects ..................................................................................... 19Familial predisposition to hypertension (I) ....................................................... 19Familial predisposition to diabetes (II) ............................................................. 19Familial abnormalities in glucose metabolism (III) ........................................... 20Familial abnormalities in urinary albumin excretion rate (IV–V) ...................... 20
Methods ................................................................................................................ 22Assessment of medical history .......................................................................... 22Assessment of blood pressure and hypertension ................................................ 22Assessment of diabetic complications ............................................................... 23Assessment of glucose metabolism ................................................................... 23Assessment of albuminuria in relatives ............................................................. 23Assessment of smoking and anthropometric measurements .............................. 24Assays .............................................................................................................. 24Statistical analysis ............................................................................................ 24

5
Diabetic nephropathy
Results .................................................................................................................. 25Familial predisposition to hypertension (I) ....................................................... 25Familial predisposition to diabetes (II) ............................................................. 26Familial abnormalities in glucose metabolism (III) ........................................... 27Familial abnormalities in urinary albumin excretion rate (IV–V) ...................... 28
Discussion ............................................................................................................. 31Subjects and methods ....................................................................................... 31Parental mortality ............................................................................................ 32Familial predisposition to hypertension ............................................................ 33Familial predisposition to diabetes ................................................................... 34Familial abnormalities in urinary albumin excretion rate .................................. 37Familial clustering – genes or environment? .................................................... 39
Summary and conclusions ..................................................................................... 40
Acknowledgments ................................................................................................ 41
References ............................................................................................................. 43
Original publications ............................................................................................ 51

Johan Fagerudd
6
List of original publications
This thesis is based on the following original publications, which will be referred to in thetext by their Roman numerals:
I. Fagerudd JA, Tarnow L, Jacobsen P, Stenman S, Nielsen FS, Pettersson-Fernholm KJ,Grönhagen-Riska C, Parving HH, Groop PH: Predisposition to essential hypertensionand development of diabetic nephropathy in IDDM patients. Diabetes 47: 439–444,1998.
II. Fagerudd JA, Pettersson-Fernholm KJ, Grönhagen-Riska C, Groop PH: The impact ofa family history of Type II (non-insulin-dependent) diabetes mellitus on the risk of dia-betic nephropathy in patients with Type I (insulin-dependent) diabetes mellitus.Diabetologia 42: 519–526, 1999.
III. Fagerudd JA, Pettersson-Fernholm KJ, Grönhagen-Riska C, Groop PH: Glucose me-tabolism in relatives of type 1 diabetic patients with albuminuria. (submitted).
IV. Fagerudd JA, Pettersson-Fernholm KJ, Riska MK, Grönhagen-Riska C, Groop PH:Albuminuria in non-diabetic relatives of IDDM patients with and without diabetic neph-ropathy. Kidney International 58: 959–965, 2000.
V. Fagerudd JA, Riska MK, Pettersson-Fernholm KJ, Groop PH: No evidence of an exag-gerated albuminuric response to physical exercise in non-diabetic siblings of type 1 dia-betic patients with diabetic nephropathy. The Scandinavian Journal of Clinical & Labo-ratory Investigation 60: 449–456, 2000.

7
Diabetic nephropathy
Abbreviations
ACE angiotensin-converting enzymeAGEs advanced glycosylation end-productsAUC area under curveBMI body mass indexDN+ diabetic nephropathy (micro- or macroalbuminuria)DN– normal urinary albumin excretion rateELISA enzyme-linked immunosorbent assayESRD end-stage renal diseaseGBM glomerular basement membraneGFR glomerular filtration rateHbA
1cglycosylated hemoglobin A
1cIGT impaired glucose toleranceITT insulin tolerance testNS not significantOGTT oral glucose tolerance testOR odds ratioPKC protein kinase CRIA radioimmunoassaySEM standard error of meanTGF-β transforming growth factor-βUAER urinary albumin excretion rateW wattWHR waist/hip ratio24 h ABPM 24 hour ambulatory blood pressure monitoring95% CI 95% confidence interval

Johan Fagerudd
8
Introduction
The discovery of insulin by Banting and Bestin the early 1920’s was one of modernmedicine’s most important achievements. Thepatients with diabetes due to an incapabilityof their pancreatic beta-cells to produce insu-lin, subsequently termed insulin-dependentor type 1 diabetic patients, could be rescuedfrom otherwise inescapable death. However,although exogenous insulin prevented acutedeath from diabetic ketoacidosis, it was un-able to fully normalize glucose metabolism,thus leading to higher blood glucose levels ininsulin-treated diabetic patients than inhealthy subjects. Later, long-lasting diabeteswas found to be associated with secondarycomplications involving the heart, blood ves-sels, eyes, nerves, and the kidneys.
In 1936, Kimmelstiel and Wilson [1] de-scribed the specific structural changes in dia-betic kidney disease (diabetic nephropathy)in combination with the clinical features ofelevated blood pressure, grossly enhanced ex-cretion of protein in the urine, edema, andrenal failure. In addition, it became evidentthat the condition was not solely a disease ofthe kidneys but was associated also with se-
vere forms of retinal changes leading to vi-sual impairment, with dysfunction of the ner-vous system, and with a massively increasedrisk for cardiovascular morbidity and earlydeath.
Epidemiological studies have demonstratedthat diabetic nephropathy occurs in approxi-mately one-third to one half of all patientswith type 1 diabetes [2] and, today, diabetesis the most important cause of renal failure inthe industrialized world [3, 4]. Although el-evated blood glucose is of crucial importance[5], it is not the only determinant of diabeticnephropathy. Only a subgroup of patientsseems to be susceptible to this long-term com-plication of diabetes, and, since the conditionhas been found to cluster in families [6], thedevelopment of diabetic nephropathy is likelyto be governed also by genetic factors.
In order to elucidate the nature of such ge-netic determinants, the present studies wereundertaken to examine whether any associa-tion exists between diabetic nephropathy andthe familial clustering of traits such as elevatedblood pressure, diabetes, and elevated excre-tion of protein in the urine.

9
Diabetic nephropathy
Review of the literature
History and classification of diabetes
A wonderful but not very frequent affection amongmen, being a melting down of the flesh and limbsinto urine … life is short, offensive and distress-ing, thirst unquenchable, death inevitable.
This description of the diabetic state byAretaeus of Cappadocia is approximately 2000years old [7]. Although he was the first knownto use the term ‘diabetes’ (Greek: dia[through] and bainein [go]), he had no knowl-edge of the underlying abnormalities of thedisease. However, the typical symptoms andfindings of the diabetic patient, with sweeturine, intense thirst, profuse urination, weightloss, vomiting, drowsiness, coma, and death,are described in the old Hindu literature prob-ably originating from the period between2500 and 600 BC [8]. In European medicine,attention was drawn to the sweet taste of theurine in some patients by Willis in Englandin the 17th century [8], but it was not untilthe work by Claude Bernard in the middle ofthe 19th century that the basic principles ofglucose metabolism were understood [9].
Hindu medicine recognized two types ofdiabetes, one affecting strong and corpulentpersons, the other weak and lean ones, andstated that the two subtypes should be treateddifferently [8]. Investigations in the era ofmodern medicine confirmed this observation,and in 1936, Himsworth proposed at least twoclinical types of diabetes, one insulin-sensi-tive due to insulin deficiency, the other insu-lin-insensitive [10]. Later, the terms ‘juvenileonset’ and ‘maturity onset’ diabetes werewidely used, but the lack of a general consen-sus regarding their definition caused confu-sion. Therefore, at the end of the 1970s and
beginning of 1980s, two separate expert com-mittees [11, 12] agreed on the classificationof diabetes into type 1 or insulin-dependentdiabetes mellitus and type 2 or non-insulin-dependent diabetes mellitus in addition to aspectrum of other types of diabetes. The di-agnostic criteria for diabetes have recentlybeen revised [13, 14] as the knowledge of theconsequences of minor elevations of bloodglucose levels has increased.
The natural history of diabeticnephropathy
Although proteinuria had long been recog-nized as associated with diabetes, observationsby Kimmelstiel and Wilson in 1936 becamethe foundation of the work leading to the cur-rent understanding of diabetes-specific lesionsof the kidney, also called diabetic nephropa-thy [1]; they described the specific renal his-tology of diabetic nephropathy in associationwith the clinical features of hypertension, al-buminuria, edema, and renal failure in anautopsy study on a series of eight patients ofwhom seven had diabetes.
All patients with type 1 diabetes do notdevelop nephropathy. According to epidemio-logical studies performed to describe the natu-ral history of the disease [2, 15], diabeticnephropathy develops in slightly less than half(35–45%) the patients during the 40 yearsfollowing diagnosis of diabetes. A strikingpeak in annual incidence rate occurs after 10to 20 years of diabetes, after which the riskfor nephropathy diminishes. Furthermore,male gender increases risk.
The stages in development and progressionof diabetic nephropathy in type 1 diabetes are

Johan Fagerudd
10
depicted in Table 1 (modified from Mogensen[16]). The initial stage is characterized by re-nal hyperfunction that is normalized by theinitiation of insulin therapy [17]. The kid-ney structure is normal at the onset of diabe-tes, but already during the first, clinically si-lent phase of normoalbuminuria, renal struc-tural changes become discernible [18, 19].The first clinical sign of renal microvascularcomplications is a slight increase in urinaryalbumin excretion rate (UAER). This condi-tion, referred to as microalbuminuria or in-cipient diabetic nephropathy, is a powerfulpredictor of subsequent overt nephropathy[20–22], even though its predictive valueseems to be dependent on the duration of thediabetes [23]. Blood pressure rises togetherwith the increase in albuminuria and is clearlyelevated early at the stage of overt diabeticnephropathy [24, 25]. At the time proteinuriadevelops, kidney function is usually normal,but soon, glomerular filtration rate (GFR)relentlessly deteriorates. The rate of declinein GFR averages approximately 10 ml/min/year, with a considerable interindividual vari-ability [26, 27]. The process culminates in acomplete loss of kidney function.
Without intervention other than insulin,the prognosis of diabetic nephropathy is poor.Half the patients are dead within seven yearsafter the development of persistent proteinuria[2], predominantly due to renal failure, butalso due to a massively increased risk for car-diovascular death [2, 28, 29]. Life-expectancy
in type 1 diabetes is highly dependent on thedevelopment of nephropathy. In comparisonto the background population, there is a two-fold increase in relative mortality even in type1 diabetic patients who do not develop pro-teinuria. However, in type 1 diabetic patientswith nephropathy, relative mortality is ex-tremely high, increasing to a maximum of100-fold at age 35 [30]. In addition, qualityof life is severely impaired in patients withnephropathy due to the clustering of othermicro- and macrovascular complications [28,31–34].
The changing natural historyof diabetic nephropathy
The natural history of diabetic nephropathyhas changed dramatically through progress inthe care of diabetic patients during recentdecades. Intensified insulin therapy has a ben-eficial effect on development of diabetic renalcomplications. Shortly after onset of diabe-tes, initiation of insulin pump therapy nor-malizes the initial glomerular hyperfiltration[35, 36]. Later, as demonstrated in The Dia-betes Control and Complications Trial, inten-sified insulin therapy, achieved either by in-sulin pump treatment or multiple insulin in-jections, resulting in a lowering of meanglycosylated hemoglobin A
1c (HbA
1c) from
9% to 7% over six years, diminishes risk formicroalbuminuria by 39% and that for albu-minuria by 54% [5]. Furthermore, improve-
Stage
I. Acute hypertrophy-hyperfunction
II. Normoalbuminuria
III. Microalbuminuria(incipient diabetic nephropathy)
IV. Proteinuria(overt diabetic nephropathy)
V. End stage renal disease
Table 1. Stages in the development of diabetic nephropathy in type 1 diabetes
Chronology
At diagnosis
1–5 years
6–15 years
15–25 years
Over 25 years
UAER
Normala
Normal(<20 µg/min)↑(20–200 µg/min)↑↑↑(>200 µg/min)Not measurable
Kidney structure
Kidney size ↑glomerular size ↑GBM thickness ↑mesangial expansion ↑GBM thickness ↑↑mesangial expansion ↑↑PronouncedabnormalitiesAdvanced abnormalities
Bloodpressure
Normalb
Normalb
↑
↑↑↑
↑↑↑
aMay be transiently elevated. bAs in the background population.
GFR
↑↑
↑↑
↑↑
↑–↓↓
↓↓↓
Review of the literature
11
Diabetic nephropathy
ment in glycemic control (change in HbA1c
from 10% to 8.5%) by means of continuoussubcutaneous insulin infusion retards the pro-gression of morphological renal changes [37].Whether intensified insulin therapy has asimilar beneficial effect on the rate of declinein GFR is unclear [38–40], although such aneffect was demonstrated in a study includingpatients with microalbuminuria at baseline[40]. Thus, intensive glycemic control hasgreat potential, especially in the primary pre-vention of diabetic nephropathy, and shouldalways be an aim.
Effective antihypertensive treatment reducesthe rate of decline of GFR [41–43]. Inhibi-tors of the angiotensin-converting enzyme(ACE) have been proposed to have a specificrenal protective effect beyond their blood pres-sure-lowering ability in patients with overtnephropathy [44, 45], although recent evi-dence suggests that other agents, for instancecalcium-channel blockers, may be equally ca-pable of preserving kidney function [46].However, in the primary prevention of dia-betic nephropathy, ACE inhibitors are indis-putably efficient [47–50] and may also pre-vent progression of diabetic retinopathy [51].
Prior to the 1970´s, renal replacement therapywas generally not offered to a patient withuremia which was due to diabetic nephropa-thy, since the prognosis was considered as toopoor [4, 52]. Today, diabetes is the most com-mon reason for actively treated uremia in theWestern world [3, 4]. Renal-transplant recipi-ents seem to have a favourable prognosis com-pared to patients treated with dialysis [53],and promising results have emerged fromcombined pancreas–kidney transplantation[54].
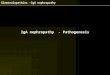
These improvements in the care of the dia-betic patient have indeed had an impact onprognosis. In one cohort with excellent gly-cemic control, a pronounced decrease hasoccured in the cumulative incidence of neph-ropathy [55]. Furthermore, recent evidencesuggests that median survival time from on-set of proteinuria has doubled to 14 years [2,56], most likely an effect of intensified anti-hypertensive treatment (Figure 1). Finally,
according to the Finnish Registry for KidneyDiseases, median survival time in type 1 dia-betic patients with diabetic nephropathy whodeveloped end-stage renal disease (ESRD) inthe time period 1985–1998, was approxi-mately 5.5 years from onset of uremia [4]. Inother words, the prognosis of a patient withESRD today is quite similar to that of a pa-tient with the first sign of diabetic renal dis-ease – dipstick-positive proteinuria – two orthree decades ago. However, despite these re-cent improvements, diabetic nephropathy isstill an important cause of increased morbid-ity and premature death among patients withdiabetes, and constitutes a heavy and grow-ing burden on the health care systemsthroughout the world.
Renal structural changesin diabetic nephropathy
Kimmelstiel and Wilson, the first investiga-tors to describe the characteristic “intercapil-lary”, nodular thickening of the mesangium,related these findings to the clinical syndrome
100
80
60
40
20
0
Cumulative death rate (%)
Duration of nephropathy (years)0 4 8 12 16
Before 1953
1970’s and 1980’s
Figure 1. Cumulative death rate after onset of diabeticnephropathy in patients diagnosed with diabetes before1953 according to Andersen et al [2] and in patientsdiagnosed with diabetes mainly in 1970s and 1980saccording to Rossing et al [56]. The prognosis ofdiabetic nephropathy has improved (median survival7 vs 14 years after onset of proteinuria).
Review of the literature

Johan Fagerudd
12
Review of the literature
of diabetic nephropathy [1]. Characteristicsof the glomerular and interstitial lesions inadvanced diabetic nephropathy are listed inTable 2.
Structural changes in relation to albuminuria
As a group, type 1 diabetic patients with long-duration diabetes and a UAER within therange of normoalbuminuria display mildstructural lesions in their kidneys, such asthickening of the glomerular basement mem-brane (GBM) and mesangial expansion [57].There is, however, within the group a consid-erable overlap, with kidney structure rang-ing from normal to rather advanced lesions[57]. At the stage of microalbuminuria, thereis a further thickening of the GBM, togetherwith more pronounced expansion of themesangium [57–60]. In overt nephropathy,these structural changes become more ad-vanced, although a considerable heterogene-ity exists in lesions between patients and evenfrom one glomerulus to another within thesame patient [59].
As can be expected, UAER correlates witha variety of glomerular lesions such as GBMthickening and degree of mesangial expan-sion [58]. However, in one follow-up studywith two kidney biopsies performed at a five-year interval in type 1 diabetic patients with
UAER varying from normo- to macroalbu-minuria, the structural variable most consis-tently correlating with increase in UAER wasthe mesangial volume fraction [61].
Structural changes in relation to kidney function
Even the initial glomerular hyperfiltration,early in the course of type 1 diabetes, is asso-ciated with structural changes in the kidneysuch as increase in kidney size [17] and in thesize of the glomeruli [62]. The glomerularfiltration surface area, i.e., the part of the cap-illary wall that is directed towards the uri-nary space and is not in contact with themesangium, is similarly increased [63]. Inovert diabetic nephropathy, decline in GFRcorrelates strongly with the decreasing glom-erular filtration surface area [64, 65]. Latestages of diabetic nephropathy are character-ized by a high percentage of occluded glom-eruli in combination with compensatory hy-pertrophy and increased filtration surface areain the non-occluded glomeruli [66].
Pathogenesis of diabetic nephropathy
Hyperglycemia
Hyperglycemia is the key player in the devel-opment of diabetic nephropathy. The charac-teristic structural lesions of diabetic nephr-opathy are absent at the onset of type 1 dia-betes. However, after two years of diabetes,GBM thickening and mesangial expansion arealready distinguishable, and at five years, thesechanges are advanced [18, 19]. Furthermore,when normal kidneys are transplanted into adiabetic milieu, lesions typical of diabeticnephropathy develop [67–71]. In type 1 dia-betic patients with microalbuminuria, im-proved glycemic control by means of intensi-fied insulin therapy retards the progressionof morphological changes [37]. Furthermore,as demonstrated in a group of type 1 diabeticpatients with mild to advanced glomerularlesions at the time of pancreas transplanta-tion, ten years of normoglycemia has inducedreversal of renal structural lesions [72]. Fi-
Table 2. Structural lesions in diabetic nephropathy(adapted from Mauer et al [244])
GBM thickeninga
Mesangial expansion (diffuse glomerulosclerosis)a
Intense immunofluorescence staining for albumin inGBM, tubular basement membrane and Bowman’scapsulea
Kimmelstiel-Wilson nodules (nodularglomerulosclerosis)b
Afferent and efferent glomerular arteriolar hyalinizationb
Tubular basement membrane thickeningSubendothelial hyaline exudative lesions (fibrinoid cap)Parietal Bowman’s capsular surface “capsular drop”b
aAlways present. bIf present, highly characteristic ofdiabetic nephropathy.

13
Diabetic nephropathyReview of the literature
nally, intervention studies have demonstratedin diabetic patients with intensive glycemiccontrol a decreased risk for progression ofUAER [5, 73].
Which are the biochemical mechanisms re-sponsible for the harmful effects of high bloodglucose on the kidney? Chronic exposure tohyperglycemia causes formation of advancedglycosylation end-products (AGEs) via non-en-zymatic glycosylation of extracellular macro-molecules [74]. AGEs are formed from earlyglycosylation products in a chemical reactionthat is irreversible. Tissue accumulation ofAGEs is associated with cross-linking of long-lived extracellular proteins and induces ab-normalities in critical matrix protein functionssuch as basement membrane self-assembly andthe binding of heparan sulfate proteoglycan.AGEs also induce increased formation of ex-tracellular matrix via stimulation of produc-tion of growth-promoting cytokines [74].High levels of AGEs have been found circu-lating in the serum and are found incorpo-rated into the arterial walls of diabetic pa-tients who have nephropathy [75]. Further-more, administration of advanced glycosylatedalbumin to non-diabetic rats induces pro-teinuria and morphological changes similarto those seen in diabetic nephropathy [76].The important role of AGEs in developmentof diabetic nephropathy is highlighted whenadministration to diabetic rats of aminogua-nidine, an inhibitor of formation of AGEs,prevents the expected rise in albuminuria[77].
Through the polyol pathway, glucose istransformed into sorbitol, a reaction in whichaldose reductase is the rate-limiting enzyme.When sorbitol and other polyols accumulateintracellularly, disturbances in the cellularosmoregulation and a decrease in the intrac-ellular myoinositol follow, with tissue dam-age as the consequence [78]. After treatmentof diabetic rats with an aldose reductase in-hibitor, diminished proteinuria was evident[79, 80]. Unfortunately, findings in humanbeings have been conflicting [81, 82].
The protein kinase C (PKC) family includesat least eleven isoenzymes that act as intrac-
ellular serine/threonine kinases and are in-volved in various cellular signal transductions.Glucose-induced activation of PKC is associ-ated with increased permeability, increasedproduction of cytokines and of extracellularmatrix, with cell proliferation, and with an-giogenesis in vascular cells [83]. A specificinhibitor of the PKC-β isoform has beenshown in diabetic rats to normalize glomeru-lar hyperfiltration and decrease UAER [84].
Abnormalities in extracellular matrix
In the healthy glomerulus, the barrier betweenthe capillary and the urinary space can bethought of as a membrane perforated by poresand coated with an inner layer of negativelycharged molecules, mainly heparan sulfateproteoglycan and sialic acid [85]. The trans-glomerular passage is therefore dependent onboth the size and the charge of a molecule inaddition to hemodynamic forces. The heparansulfate proteoglycan content is decreased inthe capillary wall of type 1 diabetic patientswith nephropathy [86]. In addition, theundersulfation of heparan sulfate moleculesdemonstrated in experimental diabetes [87],results in a further reduction in the anionicsites of the GBM. One consequence is an in-creased ability of the negatively charged al-bumin to pass through the glomerular filter.Since this increased permeability is not lim-ited to the glomerulus but is present through-out the vascular bed, a decrease in the anioniccontent of the lining of the endothelium hasbeen proposed as a common denominator forthe deleterious triumvirate of long-term dia-betic complications: nephropathy, retinopa-thy, and macrovascular disease [88]. Interest-ingly, treatment of patients with type 1 dia-betes with low molecular weight heparins,assumed to restore the heparan sulfateproteoglycan content, has been found to re-duce albuminuria [89] and to improve reti-nal hard exudates [90]. Abnormalities in theextracellular matrix may thus be importantin the cascade leading in diabetes both tonephropathy and to associated micro- andmacrovascular complications.

Johan Fagerudd
14
Review of the literature
Hemodynamic factors
Autopsy findings in a patient with long-standing diabetes and unilateral renal arterystenosis revealed only mild ischemic changeson the stenotic side, whereas advanced dia-betic nephropathy was evident on the con-tralateral side exposed to both hyperglycemiaand hypertension [91]. Surgical induction ofunilateral renal artery stenosis in rats hasshown a similar effect [92]. Hemodynamicfactors thus seem to influence the develop-ment of diabetic nephropathy. As put forwardby Hostetter, Rennke, and Brenner [93],intraglomerular hypertension and single-nephron hyperfiltration induced by the dia-betic state may lead to increased transglom-erular passage of proteins, resulting in theiraccumulation in the mesangium. This mayact as a stimulus for proliferation of mesangialcells, leading to sclerosis of the glomeruli.Compensatory hyperfiltration in the surviv-ing glomeruli then completes the vicious cycleand induces progressive loss of renal function.
This hypothesis gains support from severalobservations. Glomerular hyperfiltration isfrequently present in both type 1 and type 2diabetes, in particular during poor metaboliccontrol [17, 94]. Patients who later developmicroalbuminuria or proteinuria have showna higher GFR early in the course of their dia-betes [95]. In an 8-year follow-up study inpatients with type 1 diabetes [96], initialhyperfiltration predicted development of mi-cro- or macroalbuminuria, although anotherstudy found the role of early hyperfiltrationto be less pronounced [97]. Furthermore, re-duction in number of nephrons is associatedwith a reduced filtration surface area andsingle nephron glomerular hyperfiltration[98]. Indeed, several conditions associatedwith a reduced number of nephrons, such aslow birth weight [99, 100] and short stature[101] and also loss of one kidney [102, 103],have been linked in diabetes to an increasedrisk of elevated UAER. Hemodynamic fac-tors thus seem to play an important role inthe development and progression of diabeticglomerulopathy.
The issue as to whether the rise in sys-temic blood pressure usual in diabetic neph-ropathy precedes or follows the rise in UAER,has been subject to debate [104–106]. How-ever, results from a longitudinal study ap-plying 24 h ambulatory blood pressure moni-toring (24 h ABPM) indicate a parallel risein UAER and in systemic blood pressure inthe transition from normo- to microalbumin-uria [24]. In that study, base-line systemicblood pressure did not differ betweenprogressors and non-progressors, but the risein progressors’ UAER was closely correlatedwith their rise in 24 h ambulatory blood pres-sure during follow-up.
Growth factors
The term ‘growth factor’ is used for any sub-stance capable of inducing cellular differen-tiation or proliferation and embraces an in-creasing number of peptides. In the initiationand progression of diabetic nephropathy,growth factors such as growth hormone, in-sulin-like growth factor, epidermal growthfactor, transforming growth factor, platelet-derived growth factor, tumor necrosis factor-α, and fibroblastic growth factors have all beensuggested to play a role [107]. Of these, trans-forming growth factor-β (TGF-β) is of par-ticular interest due to its wide effects on ex-tracellular matrix [108]. In the normal situa-tion, TGF-β induces deposition of extracel-lular matrix after tissue injury. However, inthe diabetic state, a sustained secretion ofTGF-β resulting in an inappropriate produc-tion of collagen IV, fibronectin, and proteo-glycans among others, may be caused by sev-eral stimuli. First, high glucose concentrationinduces messenger RNA expression of TGF-β in glomerular cells [109] and the increasedproduction of collagen by mesangial cells ex-posed to high glucose is at least in part medi-ated by TGF-β [110]. Second, activation ofthe renin-angiotensin system with enhancedformation of angiotensin II may influenceextracellular matrix protein synthesis throughTGF-β [111]. Third, AGEs stimulate collagenproduction by mesangial cells via pathways

15
Diabetic nephropathyReview of the literature
that involve TGF-β [112]. The importanceof TGF-β is further underlined by the dem-onstration of a rapid development of glom-erulosclerosis after in vivo transfection of theTGF-β1 gene into the kidneys of non-diabeticrats [113].
Genetic factors
Renal disease, regardless of underlying cause,has been found to aggregate in families [114,115]. In addition, substantial racial differencesexist in the occurrence of diabetic nephropa-thy – for instance, non-Ashkenazi Jewish type1 diabetic patients are at higher risk than areAshkenazi patients [116]; moreover, PimaIndian, African-American, and Mexican-American type 2 diabetic patients have ahigher risk than do Caucasian patients [117–119]. A genetic predisposition both to dia-betic and non-diabetic nephropathy thusseems likely.
Seaquist et al [6] were the first to describea familial aggregation of albuminuria in type 1diabetes. Elevated UAER was found among83% of diabetic siblings of patients withnephropathy, but in only 17% of the siblingsof patients without nephropathy. Three sub-sequent studies [120–122] confirmed thisfinding, although their degree of familial clus-tering was less pronounced. A recent study ofmainly normoalbuminuric type 1 diabetic sib-ling pairs demonstrated a familial effect notonly on UAER, but also on the severity andpattern of glomerular structural lesions [123].Familial aggregation of nephropathy has alsobeen demonstrated in type 2 diabetes [124–127]. Interestingly, minor abnormalities inUAER seem to be present in individuals witha potential genetic susceptibility to diabeticnephropathy even in the absence of diabetes.This suggestion is based on the finding of anelevated UAER in ordinary urine collections[126, 128–130] as well as of an exaggeratedalbuminuric response to physical exercise[129] in non-diabetic offspring and siblingsof type 2 diabetic patients with micro- or mac-roalbuminuria. Whether similar abnormali-ties in UAER are present also in the non-dia-
betic relatives of type 1 diabetic patients withnephropathy remains unknown.
Susceptibility to diabetic nephropathy maybe linked to familial predisposition to hyperten-sion, since parents of type 1 diabetic patientswith proteinuria have been found to havehigher arterial blood pressure than do parentsof non-proteinuric patients, a finding origi-nally described by Viberti and co-workers[131]. Although some subsequent studieshave confirmed this finding, others haveyielded conflicting results [132–137, 201],making the role of familial predisposition toessential hypertension in development of dia-betic nephropathy in type 1 diabetes stillunclear.
Furthermore, activity of the sodium–lithium countertransporter in red blood cells,a potential indicator of a genetic predisposi-tion to hypertension [139, 140], has beenfound to be increased in type 1 diabetic pa-tients with nephropathy [132, 141] and intheir parents [135]. Abnormalities have alsobeen reported in the closely related sodium–hydrogen countertransporter [142]. However,conflicting results have also appeared [134]and the exact role of these phenotypic mark-ers of essential hypertension in the develop-ment of diabetic nephropathy remains to beestablished.
Earle and co-workers [136] demonstratedthat a parental history of cardiovascular diseasewas associated with increased risk for diabeticnephropathy. A Danish case-control study[143] did not, however, find any differencein prevalence of cardiovascular disease be-tween parents of patients with and withoutnephropathy. The prevalence of cardiovascu-lar disease was much lower in the latter study,probably due to lower age (parental mean age58 vs 64), which could explain some of thedifferences between the two studies.
In non-diabetic subjects with a family his-tory of diabetes, a spectrum of metabolic de-rangements such as insulin resistance, ab-dominal obesity, elevated blood pressure, andlipid abnormalities has been described [144,145]. Interestingly, similar metabolic andhemodynamic derangements are also found at

Johan Fagerudd
16
Review of the literature
the early stages of diabetic nephropathy [26,146–151]. It could therefore be hypothesizedthat a family history of diabetes increases therisk for diabetic nephropathy, and indeed, inthe Pima Indians, a link was demonstratedbetween familial diabetes and diabetic neph-ropathy [152]. Whether a family history ofdiabetes increases the risk for nephropathy intype 1 diabetes is unknown. However, familymembers of patients with microalbuminuriahave shown metabolic derangements such ashyperinsulinemia, elevated cholesterol andapolipoprotein B levels, and an increased LDL/HDL cholesterol ratio [153].
Genetics
Because the evidence for genetic factors in thedevelopment of diabetic nephropathy is con-vincing, efforts have been made to identifyone or several genes increasing susceptibilityto diabetic nephropathy. Recent results fromsegregation analyses in pedigrees of type 2diabetic patients speak in favor of a major geneeffect in the development of albuminuria [154,155]. Similar analyses are very difficult toperform in type 1 diabetes due to the scarcityof large pedigrees with many affected indi-viduals. However, in the study on type 1 dia-betic sibling pairs by Quinn et al [121], thecumulative incidence rate for diabetic nephr-opathy after 25 years of diabetes was muchhigher in siblings of patients with nephropa-thy than in siblings of patients without neph-ropathy, 71% vs 25%, respectively. This find-ing has been interpreted as indicative of amajor gene effect on diabetic nephropathy alsoin type 1 diabetes. Therefore, in the light ofpresent understanding, diabetic nephropathyseems to be the result of one or several majoror minor genes in combination with environ-mental factors, of which the most importantis hyperglycemia [156].
Table 3 presents some of the genes sug-gested to be involved in the development ofdiabetic nephropathy. Thus far, the study de-sign most widely used to identify such geneshas been the candidate gene approach in case-control study settings. As an example, the
candidate gene most thoroughly studied hasbeen the insertion(I)/deletion(D) polymor-phism in the ACE gene. This polymorphismis especially attractive, since it accounts forhalf of the variance in serum ACE level [157].Numerous studies have addressed the issue ofa role for the ACE I/D-polymorphism in thedevelopment of diabetic nephropathy with di-verging results [158]. Some evidence suggeststhat the D-allele may increase the risk for dia-betic nephropathy in Japanese type 2 diabeticpatients, while the effect in Caucasian type 1diabetic patients, if present at all, seems to beonly minor [158, 159]. However, it shouldbe noted that case-control studies are espe-cially prone to biases such as selective survivaland population stratification. At present, noneof the candidate genes in Table 3 qualifies asa gene with a major impact on the develop-ment of diabetic nephropathy.
Linkage analysis in type 1 diabetic siblingpairs discordant for diabetic nephropathy hasidentified a region on chromosome 3 in link-age with diabetic nephropathy [160]. Themost obvious candidate gene in this region isthe angiotensin II type 1 receptor gene, butsubsequent analysis has found no evidence ofDNA sequence differences in this gene withany impact on the development of nephropa-thy. However, this region may harbor a yet-unidentified gene with a major effect on de-velopment of diabetic nephropathy.
Other factors
Restriction of dietary protein intake retards inanimal models the progression of renal dis-ease [161]. Suggested mediators of this ben-eficial effect include a reduction in glomeru-lar hyperfiltration/hypertension, a preserva-tion of proper glomerular permeability, andactions mediated via lipid metabolism. Inhumans, dietary protein restriction has beenthoroughly studied in non-diabetic kidneydiseases, and, according to a meta-analysis[162], a low protein diet has a beneficial ef-fect on the rate of loss of kidney function. Indiabetic nephropathy, the studies conductedthus far have been small, and in some cases,

17
Diabetic nephropathyReview of the literature
uncontrolled [163–167], and although thecombined results indicate that protein restric-tion slows the progression of diabetic nephr-opathy [162], larger studies are needed todefine the role of protein restriction in dia-betic renal disease.
Type 1 diabetic patients with diabeticnephropathy display abnormalities of lipid me-tabolism including slightly elevated levels oftotal and LDL cholesterol, triglycerides, andapolipoprotein B [148, 168]. Such an athero-genic profile may contribute to the massivelyincreased risk for cardiovascular complicationsin patients with nephropathy [30]. In type 2diabetes, high cholesterol levels increase therisk for development of elevated UAER [169].In type 1 diabetes, multiple abnormalities oflipid metabolism are already evident at themicroalbuminuria stage [148, 168], but littleinformation exists on the role of lipid abnor-malities in the progression from normal toelevated UAER. In overt nephropathy, hyper-cholesterolemia has been associated with anaccelerated decline in renal function [170–
172] and with cardiovascular death [170].However, although a recent animal study re-ported positive effects on renal function oflipid-lowering therapy with lovastatin [173],treatment with statins in hypercholester-olemic patients with elevated UAER failedto show any benefit regarding progression ofUAER or rate of decline of GFR [174]. How-ever, no data are available from long-term,prospective, randomised trials.
Among other factors, smoking has beenfound to increase the risk for development andprogression of diabetic nephropathy [175–177] and thereby constitutes an importantmodifiable risk factor in diabetic nephropa-thy.
In addition, some evidence suggests a ben-eficial effect of the c-peptide molecule on kid-ney function in type 1 diabetes [178], andpatients with persisting c-peptide secretiondue to only partial destruction of pancreaticβ-cells may be protected against diabetic mi-crovascular late complications [179].
Table 3. Examples of candidate genes for diabetic nephropathy
Mechanism
1. Renin angiotensin system
2. Blood pressure regulation, cardiovascular disease
3. Other
Candidate gene
Angiotensin converting enzymeAngiotensinogenAngiotensin II type 1 receptorKallikrein
α-adducinApolipoprotein ENitric oxide synthase (eNOS)Glycogen synthase
Aldose reductaseTransforming growth factor-βHeparan sulphate proteoglycan (Perlecan gene)Methylenetetrahydrofolate reductase

Johan Fagerudd
18
Aims of the study
One-third of all patients with type 1 diabetesdevelop diabetic nephropathy. In addition tobeing the most important cause of chronicrenal failure in the industrialized world, dia-betic nephropathy is also associated with amassively increased risk for cardiovascularcomplications. At present, growing evidenceexists of a role for genetic factors in the de-velopment of diabetic nephropathy. Identifi-cation of such predisposing factors would al-low targeting of high-risk individuals and apossibility for intervention even at diagnosisof diabetes. Little is known, however, aboutthe nature of the genetic factors increasingthe risk for nephropathy. The main objectivesof the present study were therefore to answerthe following questions:
1. Is diabetic nephropathy in type 1 diabe-tes associated with familial predispositionto hypertension? (I)
2. Is diabetic nephropathy in type 1 diabe-tes associated with familial predispositionto diabetes? (II)
3. If so, which abnormalities in glucose me-tabolism are responsible for the excessprevalence of diabetes seen in relatives oftype 1 diabetic patients with diabeticnephropathy? (III)
4. Are abnormalities in UAER present innon-diabetic first-degree relatives of type1 diabetic patients with diabetic nephr-opathy? (IV, V)

19
Diabetic nephropathy
All studies were cross-sectional case-control stud-ies. Characteristics of the type 1 diabetic pa-tients are depicted in Tables 4 and 5. The stud-ies were conducted in accordance with theDeclaration of Helsinki [180], and the studyprotocols were approved by the local ethicscommittee. All patients and relatives gavetheir written informed consent prior to theirparticipation.
A total of 318 Caucasian type 1 diabeticpatients took part. Type 1 diabetes was de-fined as onset of diabetes before the age of 35,initiation of insulin therapy within a year af-ter diagnosis, and present treatment with atleast two daily insulin injections without theuse of any other antidiabetic drug. The sub-jects were recruited from a random sample ofall patients with diabetic nephropathy attend-ing the renal outpatient clinic or the dialysisunit of the Helsinki University Central Hos-pital between November, 1995, and Decem-ber, 1997 (n = 137), and from a consecutivesample of all patients attending the diabeticoutpatient clinic of the same hospital fromSeptember, 1990, to February, 1992 (n = 73),and via an advertisement in the newsletter ofthe Helsinki Diabetes Association (n = 20).Consequently, a total number of 174 patientswith signs of diabetic nephropathy and 56 pa-tients with normal UAER were studied inHelsinki. In addition, 88 Danish patients at-tending the outpatient clinic at the Steno Dia-betes Center, Gentofte, Denmark, participatedin Study I. Of these, 44 patients were ran-domly selected from among all type 1 diabeticpatients with diabetic nephropathy who hadtheir GFR measured in 1993, together with44 patients from an age-, duration- and sex-matched control group with normal UAER.
Study design and subjects
Familial predispositionto hypertension (I)
Study aim. To evaluate whether diabetic neph-ropathy is associated with a familial predis-position to hypertension.
Subjects. The parents (n = 109) of 73 patientswith overt diabetic nephropathy and those (n= 112) of 73 patients with normal UAER anda duration of diabetes exceeding 15 years(Tables 4 and 5). In each group, 44 of the pa-tients were recruited from the Steno DiabetesCenter in Gentofte, Denmark, while the restcame from the outpatient clinics of theHelsinki University Central Hospital.
Study design. Any antihypertensive medica-tion taken by the parents was recorded, andblood pressure was measured auscultatorilyin all parents not on antihypertensive medi-cation (n = 162). Furthermore, 131 of the162 parents taking no antihypertensivemedication volunteered for a 24 h ABPM.The prevalence and cumulative incidenceof hypertension in the two groups of par-ents were calculated after exclusion of thoseparents with clear evidence of hypertensionsecondary to renal disease. The impact ofparental hypertension on the developmentof hypertension in the diabetic offspring wasalso assessed.
Familial predispositionto diabetes (II)
Study aim. To elucidate whether diabetic neph-ropathy is associated with a familial predis-position to type 2 diabetes.
Johan Fagerudd
20
Study design and subjects
Subjects. Parents of 137 patients with overtdiabetic nephropathy and of 54 patients withnormal UAER (Tables 4 and 5) recruited fromthe outpatient clinics of the Helsinki Uni-versity Central Hospital and via the HelsinkiDiabetes Association.
Study design. Prevalence of known diabetes andhypertension as well as overall and cardiovas-cular death rate in the parents were assessedin the two groups of patients. Furthermore,an oral glucose tolerance test (OGTT) andanthropometric measurements were carriedout in 95 and 55 living, non-diabetic parentsof patients with and without diabetic nephr-opathy, respectively.
Familial abnormalities inglucose metabolism (III)
Study aim. In order to explore the mechanismsbehind a possible association between diabeticnephropathy and familial type 2 diabetes, glu-cose metabolism was assessed in first-degreerelatives of type 1 diabetic patients with andwithout elevated UAER.
Subjects. First-degree relatives (n = 114) of 43patients with elevated UAER (microalbumin-uria, n = 18; overt diabetic nephropathy, n =25), and 93 relatives of 39 patients with nor-mal UAER. The patients were recruited fromthe outpatient clinics of the Helsinki Uni-versity Central Hospital and via the HelsinkiDiabetes Association.
Study design. After exclusion of relatives withtreatment for diabetes or with fasting hyper-glycemia, an OGTT was performed on all rela-tives, with measurement of plasma glucoseand serum insulin responses. On a second oc-casion, insulin sensitivity was measured bythe short insulin tolerance test (ITT) in 106(89%) and 84 (89%) relatives of patients withand without elevated UAER.
Familial abnormalities in urinaryalbumin excretion rate (IV–V)
Study aim. Non-diabetic relatives of type 2diabetic patients with elevated UAER havebeen found to display abnormalities in UAER.The aim was to discover whether this was also
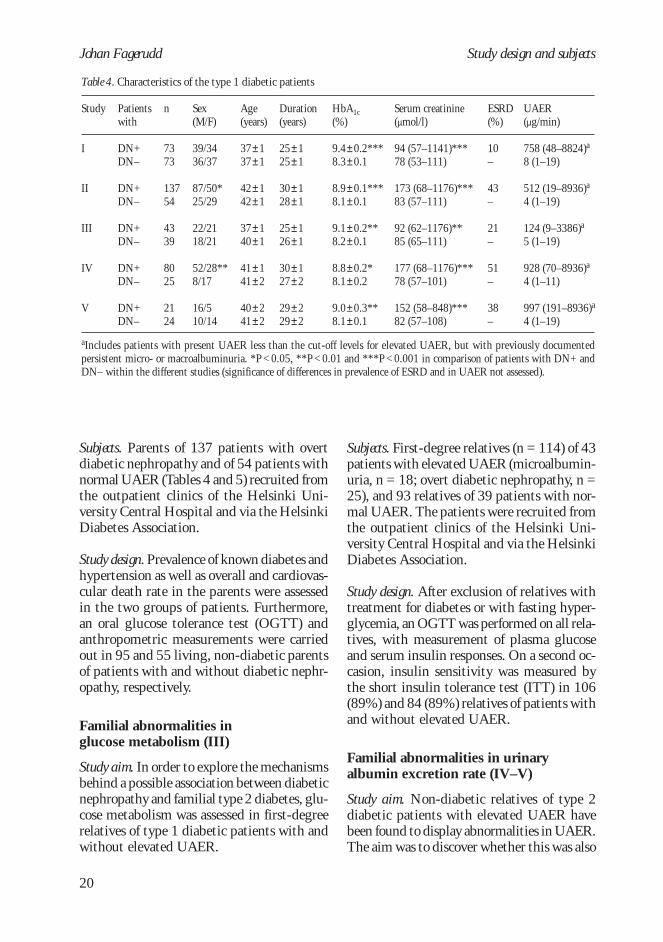
Table 4. Characteristics of the type 1 diabetic patients
Patientswith
DN+DN–
DN+DN–
DN+DN–
DN+DN–
DN+DN–
Study
I
II
III
IV
V
n
7373
13754
4339
8025
2124
Sex(M/F)
39/3436/37
87/50*25/29
22/2118/21
52/28**8/17
16/510/14
Age(years)
37±137±1
42±142±1
37±140±1
41±141±2
40±241±2
Duration(years)
25±125±1
30±128±1
25±126±1
30±127±2
29±229±2
HbA1c(%)
9.4±0.2***8.3±0.1
8.9±0.1***8.1±0.1
9.1±0.2**8.2±0.1
8.8±0.2*8.1±0.2
9.0±0.3**8.1±0.1
ESRD(%)
10–
43–
21–
51–
38–
UAER(µg/min)
758 (48–8824)a
8 (1–19)
512 (19–8936)a
4 (1–19)
124 (9–3386)a
5 (1–19)
928 (70–8936)a
4 (1–11)
997 (191–8936)a
4 (1–19)
aIncludes patients with present UAER less than the cut-off levels for elevated UAER, but with previously documentedpersistent micro- or macroalbuminuria. *P < 0.05, **P < 0.01 and ***P < 0.001 in comparison of patients with DN+ andDN– within the different studies (significance of differences in prevalence of ESRD and in UAER not assessed).
Serum creatinine(µmol/l)
94 (57–1141)***78 (53–111)
173 (68–1176)***83 (57–111)
92 (62–1176)**85 (65–111)
177 (68–1176)***78 (57–101)
152 (58–848)***82 (57–108)

21
Diabetic nephropathyStudy design and subjects
Table 5. Characteristics of the relatives
Relatives ofpatients with
DN+DN–
DN+DN–
DN+DN–
DN+DN–
DN+DN–
Study
I
II
III
IV
V
n
109112
9555
11493
18652
2124
Parents/siblings
109/–112/–
95/–55/–
56/5844/49
77/109*30/22
–/21–/24
Sex(M/F)
42/6742/70
36/5922/33
49/6543/50
85/10122/30
12/914/10
Age(years)
66±168±1
65±165±1
51±153±2
52±154±2
43±243±2
BMI(kg/m2)
27.1±0.5*25.9±0.3
28.2±0.427.2±0.6
26.1±0.4*25.0±0.4
26.4±0.326.0±0.6
25.1±0.624.5±0.6
Smokers(%)
4736
1924
2530
2725
4333
*P<0.05 in comparison of relatives of patients with DN+ and DN– within the studies.
the case in non-diabetic relatives of type 1diabetic patients with diabetic nephropathy.
Subjects. In Study IV, UAER was measured in186 first-degree relatives of 80 patients withovert diabetic nephropathy and in 52 relativesof 25 patients with normal UAER (Tables 4and 5). In Study V, basal and exercise-inducedUAER was measured in three urine collec-tions from 21 and 24 age-, sex- and body massindex (BMI)-matched siblings of patients withand without diabetic nephropathy, respec-tively. These patients were recruited from the
outpatient clinics of the Helsinki UniversityCentral Hospital and via the Helsinki Diabe-tes Association.
Study design. In Study IV, UAER was mea-sured in one overnight urine collection fromrelatives, in whom diabetes was excluded bymedical history. In Study V, an OGTT wasperformed to exclude abnormalities in glu-cose metabolism. UAER was measured fromurine collections which the relatives per-formed overnight, during the OGTT, andduring a submaximal bicycle ergometer test.

Johan Fagerudd
22
Assessment of medical history
A careful medical history regarding the pres-ence of hypertension, diabetes, cardiovascu-lar disease, smoking habits and regular medi-cation was taken from all participating rela-tives in Studies I to V by use of a standard-ized questionnaire. Hypertension was consid-ered present if the subject was on medicationprescribed for elevated blood pressure. Dia-betes was defined as a diagnosis of diabetesmade by a physician. Diabetes in the relativeswas classified as type 1 diabetes if age at on-set of the disease was less than or equal to 40years, and since diagnosis it had been treatedwith no other antidiabetic drug than insulin.All other cases of diabetes were classified astype 2 diabetes. A history of cardiovasculardisease was defined as a history of acute myo-cardial infarction or stroke. In cases with in-complete information, medical records werereviewed.
In Studies II and III, the participating type 1diabetic patients were all interviewed with astandardized questionnaire regarding the healthstatus of their first-degree relatives. The ques-tionnaire assessed whether the relatives werealive, the age of the relatives at the time of thestudy and presence of diabetes or antihyperten-sive treatment in the relatives. If a relative wasdeceased, cause of death and age at death wererequested. Parental cardiovascular death wasconsidered to have taken place if the probandreported death from cardiac causes, stroke, orrupture of an aortic aneurysm. In Study II, thereliability of the information obtained from thepatients was tested by interviewing living par-ents and by reviewing medical records and deathcertificates in those deceased. Confirmation ofdata was carried out for all parents classified asdiabetic by their diabetic offspring (n = 66; 50
Methods
parents of patients with and 16 of patients with-out nephropathy) and furthermore, in a sampleof 221 (71%) of all 311 presumed non-diabeticparents (217 parents of patients with and 94parents of patients without nephropathy). Thesample of 221 presumed non-diabetic parents,comprising 138 (64%) of patients with and 83(88%) of patients without nephropathy, was se-lected as follows: first, their medical history wasobtained from those parents subsequently in-terviewed at the outpatient clinic when they hadtheir assessment of oral glucose tolerance (n =95 and 55). Second, in order to test the reliabil-ity of data on those presumed non-diabetic par-ents not attending the outpatient clinic, themedical record and death certificate validationprocedure was then performed on a subgroup of71 parents (n = 43 and 28) of whom 18 werealive (n = 14 and 4) and 53 dead (n = 29 and24).
Assessment of blood pressureand hypertension
Systolic (Korotkoff I) and diastolic (KorotkoffV) office blood pressure was measured ausculta-torily on the right arm with an ordinary cali-brated mercury sphygmomanometer (StudiesI to IV) or a Hawksley random zero sphyg-momanometer (Study I) with a properly sizedcuff after at least 5 min of rest. The mean valueof at least two recordings was used in theanalysis. In Study V, an aneroid sphygmoma-nometer was employed to measure ausculta-tory blood pressure after the subject had rested10 min supine.
In Study I, 24 h ambulatory blood pressurewas measured oscillometrically with aSpaceLabs 90207 device (SpaceLabs Inc.,Redmond, WA, USA). The monitor waschecked before, after, and once each month
Methods

23
Diabetic nephropathy
during the study by comparison with a cali-brated mercury sphygmomanometer accord-ing to the instructions of the manufacturer.Blood pressure was measured every 15 minfrom 07:00 to 22:00 and every 30 min from22:00 to 07:00 with a properly sized cuff dur-ing 24 h of normal daily activities. Blood-pressure monitoring was accepted if there wasat least one successful blood-pressure record-ing per hour during at least 20 of the 24 h ofmonitoring. Day- and night-time blood pres-sures were calculated based on individuallyrecorded awake and sleeping hours.
Hypertension was defined as current use ofantihypertensive medication, an office bloodpressure exceeding or equal to 140/90 (Stud-ies I, III) or 160/95 mmHg (Study II), or a 24h ABPM exceeding or equal to 135/85 mmHg[181, 182].
Assessment of diabetic complications
The degree of renal involvement in the dia-betic patients was based on at least three urinecollections [183]. Overt diabetic nephropathy wasdefined as an UAER exceeding either 200 µg/min (overnight urine collections) or 300 mg/24 h (24 h urine collections), and microalbu-minuria as UAER 20 to 200 µg/min or 30 to300 mg/24 h in two out of three consecutiveurine collections in the absence of any clini-cal or laboratory evidence of other renal dis-ease. Normal UAER was defined as UAERpersistently below 20 µg/min or 30 mg/24 h.In some patients, UAER had decreased, forinstance after initiation of antihypertensivetherapy. In these cases, classification of mi-croalbuminuria and overt diabetic nephropa-thy was based on past UAER recordings.ESRD was considered present after initiationof renal replacement therapy (dialysis or kid-ney transplantation). A history of retinal pho-tocoagulation served as an indicator of severeretinopathy.
Assessment of glucose metabolism
An OGTT was performed in Studies II, III,and V. Plasma glucose and serum insulin were
measured in the morning after a 10 to 12 hovernight fast and at 30, 60, and 120 minafter ingestion of 75 g of glucose in a volumeof 200 ml of water. Diabetes and impairedglucose tolerance (IGT) in the OGTT weredefined according to 1985 World HealthOrganization criteria [184]. Abnormal glu-cose tolerance was defined as IGT or diabe-tes. Incremental area under the curve (AUC)was calculated according to the trapezoidalrule.
In order to measure insulin sensitivity, theshort ITT [185] was applied in Study III. Twointravenous cannulas were inserted, one in adeep cubital vein and a second in retrogradeposition in a dorsal vein of the contralateralhand. The hand was kept in a heated (+55°C) box in order to achieve arterialization ofvenous blood. After a baseline period of 20 to30 min, fasting plasma glucose was measuredtwice, and an intravenous bolus of short-act-ing insulin (0.1 IU/kg body weight) wasgiven. Arterialized venous blood samples formeasurement of plasma glucose level weredrawn every min from 3 to 15 min after theinsulin bolus. After the 15 min test-period, aglucose infusion was initiated, and the sub-ject received a meal. The percentage declinein logarithmically transformed plasma glu-cose per min during 3 to 15 min after admin-istration of the insulin bolus was calculatedby least square analysis and expressed as theK
ITT-value (%/min).
Assessment of albuminuria in relatives
In Study IV, UAER was measured in the non-diabetic relatives from a timed overnight urinecollection. In Study V, UAER was measuredfrom three timed urine collections: (I) overnight,(II) during an OGTT, and (III) during asubmaximal bicycle ergometer test. In orderto maintain sufficient diuresis, the subjectswere given 600 ml of water to drink duringthe OGTT, and approximately 10 ml of wa-ter per kilogram body weight before the ex-ercise test was initiated. Prior to the exercisetest, each subject lay for 10 min supine. Thetest began at a level of 40 W in male and 30
Methods

Johan Fagerudd
24
W in female subjects, and the work-load wasincreased by 40 W in male and by 30 W infemale subjects every 4 min. The target was90% of the age-adjusted maximal heart rate(205 minus age in years divided by 2). Bloodpressure was measured at rest and during thelast min at every work-load level, while heartrate was being recorded with a pulse-sensordevice at rest and at 1-min intervals duringthe test. The test was followed by a 10-minrest in the supine position, after which thesubject voided the final urine sample.
Assessment of smokingand anthropometric measurements
Smoking was defined as present smoking of atleast one daily cigarette, cigar, or pipe duringthe year prior to participation in the studies.Body weight (to the closest 0.1 kg) and height(to the closest cm) were measured in lightclothing. BMI was calculated as weight (kg) /(height (m)2). Waist circumference was measuredmidway between the iliac crest and the low-est rib and hip circumference at the widest partof the gluteal region. Waist/hip ratio (WHR)was calculated as waist (cm)/hip (cm).
Assays
Plasma glucose was measured in duplicate by aglucose oxidase method (Beckman GlucoseAnalyser II, Beckman, Fullerton, CA, USA)with a coefficient of variation of 1.0%. Seruminsulin was measured by enzyme-linkedimmunosorbent assay (ELISA; Dako Diagnos-tics Ltd, Cambridgeshire, UK) in Study III andby radioimmunoassay (RIA; Insulin-RIA,Pharmacia-Upjohn, Uppsala, Sweden) in StudyV with coefficients of variation of 9% and 8%,respectively. In Studies II to V and in the Finn-ish subjects in Study I, urinary albumin wasmeasured by use of RIA (Albumin-RIA,Pharmacia-Upjohn, Uppsala, Sweden) with acoefficient of variation of 4%. In the Danishsubjects in Study I, urinary albumin was mea-
sured by ELISA [186]. HbA1c
was measured byhigh-pressure liquid chromatography with anormal range of 4.0 to 6.0% (Finland) and 4.1to 6.1% (Denmark). Serum creatinine was as-sayed by a kinetic Jaffé method (normal rangein Finland: women 50–110, men 55–115µmol/l; normal range in Denmark: women 40–110, men 60–130 µmol/l). Serum cholesterol(normal range: 3.6–7.0 mmol/l), HDL-choles-terol (normal range: women 1.10–2.35, men0.95–2.00 mmol/l) and triglycerides (normalrange: 0.4–1.7 mmol/l) were all measured ona Hitachi 917 automated analyzer with enzy-matic colorimetric tests.
Statistical analysis
The significance of difference in categoricalvariables between the groups was tested withthe Chi squared test. The significance of dif-ference in normally distributed continuous vari-ables was tested with Student’s t-test, whiledifferences in non-normally distributed vari-ables were assessed with the Mann-WhitneyU-test or Student’s t-test after logarithmictransformation. Adjustment for confoundingfactors was performed by analysis of covari-ance. The cumulative incidence of parentalhypertension and overall and cardiovascularparental death rate was calculated with a life-table method, which takes into account the vari-ability in length of follow-up. The significanceof the difference in cumulative incidence andsurvival between the two groups was deter-mined with the logrank test. In order to evalu-ate the independent association between fa-milial factors and diabetic nephropathy inStudy II, a multiple forward stepwise logistic re-gression analysis was performed and the ad-justed odds ratio (OR) and the 95% confi-dence interval (95% CI) calculated. A two-tailed P-value less than 0.05 was consideredstatistically significant. Normally distributedcontinuous variables are presented as mean±standard error of mean (SEM) and non-nor-mally distributed variables as median (range).
Methods
25
Diabetic nephropathy
Results
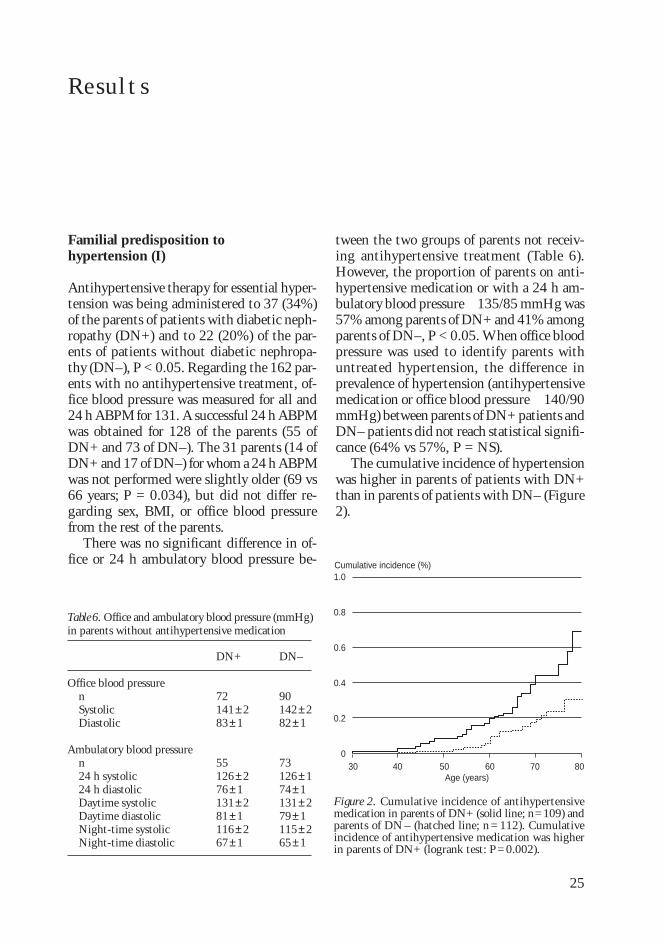
Familial predisposition tohypertension (I)
Antihypertensive therapy for essential hyper-tension was being administered to 37 (34%)of the parents of patients with diabetic neph-ropathy (DN+) and to 22 (20%) of the par-ents of patients without diabetic nephropa-thy (DN–), P < 0.05. Regarding the 162 par-ents with no antihypertensive treatment, of-fice blood pressure was measured for all and24 h ABPM for 131. A successful 24 h ABPMwas obtained for 128 of the parents (55 ofDN+ and 73 of DN–). The 31 parents (14 ofDN+ and 17 of DN–) for whom a 24 h ABPMwas not performed were slightly older (69 vs66 years; P = 0.034), but did not differ re-garding sex, BMI, or office blood pressurefrom the rest of the parents.
There was no significant difference in of-fice or 24 h ambulatory blood pressure be-
tween the two groups of parents not receiv-ing antihypertensive treatment (Table 6).However, the proportion of parents on anti-hypertensive medication or with a 24 h am-bulatory blood pressure 135/85 mmHg was57% among parents of DN+ and 41% amongparents of DN–, P < 0.05. When office bloodpressure was used to identify parents withuntreated hypertension, the difference inprevalence of hypertension (antihypertensivemedication or office blood pressure 140/90mmHg) between parents of DN+ patients andDN– patients did not reach statistical signifi-cance (64% vs 57%, P = NS).
The cumulative incidence of hypertensionwas higher in parents of patients with DN+than in parents of patients with DN– (Figure2).
1.0
0.8
0.6
0.4
0.2
0
Cumulative incidence (%)
Figure 2. Cumulative incidence of antihypertensivemedication in parents of DN+ (solid line; n=109) andparents of DN – (hatched line; n = 112). Cumulativeincidence of antihypertensive medication was higherin parents of DN+ (logrank test: P=0.002).
40 50 60 70 8030Age (years)
Table 6. Office and ambulatory blood pressure (mmHg)in parents without antihypertensive medication
Office blood pressurenSystolicDiastolic
Ambulatory blood pressuren24 h systolic24 h diastolicDaytime systolicDaytime diastolicNight-time systolicNight-time diastolic
DN–
90142±282±1
73126±174±1131±279±1115±265±1
DN+
72141±283±1
55126±276±1131±281±1116±267±1
Results
Johan Fagerudd
26
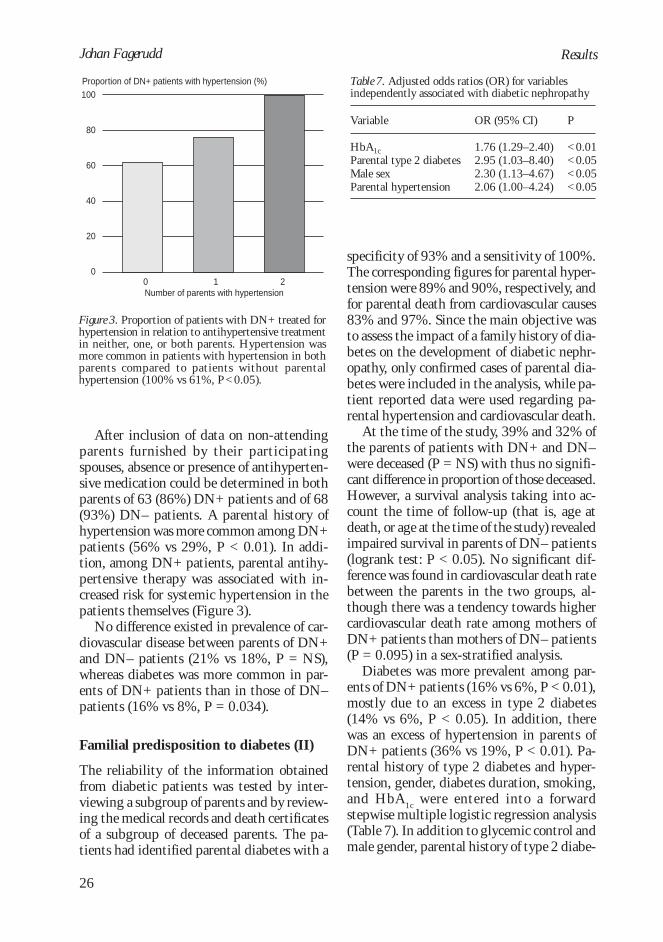
After inclusion of data on non-attendingparents furnished by their participatingspouses, absence or presence of antihyperten-sive medication could be determined in bothparents of 63 (86%) DN+ patients and of 68(93%) DN– patients. A parental history ofhypertension was more common among DN+patients (56% vs 29%, P < 0.01). In addi-tion, among DN+ patients, parental antihy-pertensive therapy was associated with in-creased risk for systemic hypertension in thepatients themselves (Figure 3).
No difference existed in prevalence of car-diovascular disease between parents of DN+and DN– patients (21% vs 18%, P = NS),whereas diabetes was more common in par-ents of DN+ patients than in those of DN–patients (16% vs 8%, P = 0.034).
Familial predisposition to diabetes (II)
The reliability of the information obtainedfrom diabetic patients was tested by inter-viewing a subgroup of parents and by review-ing the medical records and death certificatesof a subgroup of deceased parents. The pa-tients had identified parental diabetes with a
specificity of 93% and a sensitivity of 100%.The corresponding figures for parental hyper-tension were 89% and 90%, respectively, andfor parental death from cardiovascular causes83% and 97%. Since the main objective wasto assess the impact of a family history of dia-betes on the development of diabetic nephr-opathy, only confirmed cases of parental dia-betes were included in the analysis, while pa-tient reported data were used regarding pa-rental hypertension and cardiovascular death.
At the time of the study, 39% and 32% ofthe parents of patients with DN+ and DN–were deceased (P = NS) with thus no signifi-cant difference in proportion of those deceased.However, a survival analysis taking into ac-count the time of follow-up (that is, age atdeath, or age at the time of the study) revealedimpaired survival in parents of DN– patients(logrank test: P < 0.05). No significant dif-ference was found in cardiovascular death ratebetween the parents in the two groups, al-though there was a tendency towards highercardiovascular death rate among mothers ofDN+ patients than mothers of DN– patients(P = 0.095) in a sex-stratified analysis.
Diabetes was more prevalent among par-ents of DN+ patients (16% vs 6%, P < 0.01),mostly due to an excess in type 2 diabetes(14% vs 6%, P < 0.05). In addition, therewas an excess of hypertension in parents ofDN+ patients (36% vs 19%, P < 0.01). Pa-rental history of type 2 diabetes and hyper-tension, gender, diabetes duration, smoking,and HbA
1c were entered into a forward
stepwise multiple logistic regression analysis(Table 7). In addition to glycemic control andmale gender, parental history of type 2 diabe-
100
80
60
40
20
0
Proportion of DN+ patients with hypertension (%)
0 1 2Number of parents with hypertension
Figure 3. Proportion of patients with DN+ treated forhypertension in relation to antihypertensive treatmentin neither, one, or both parents. Hypertension wasmore common in patients with hypertension in bothparents compared to patients without parentalhypertension (100% vs 61%, P<0.05).
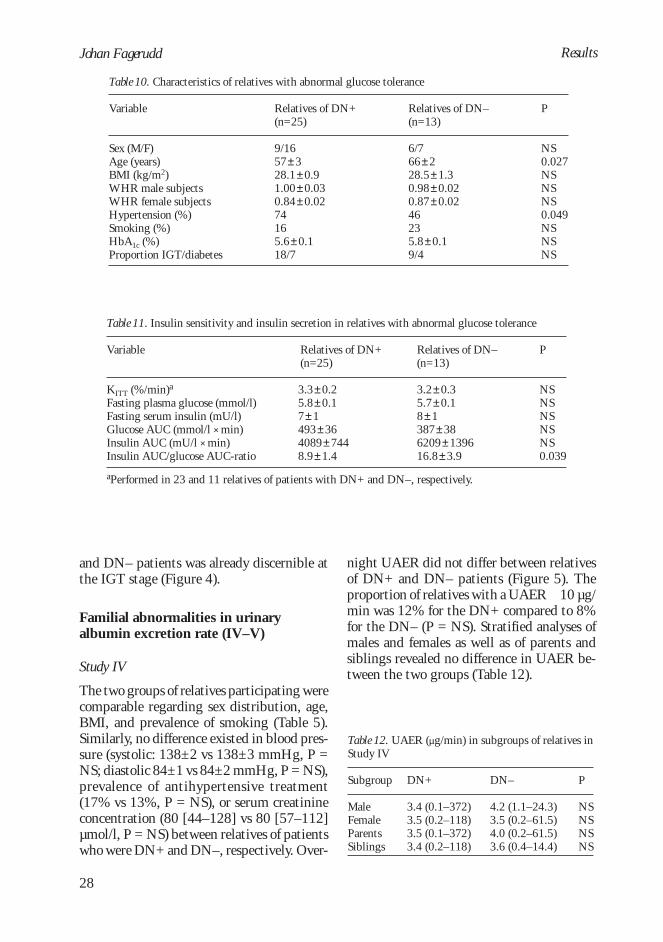
Table 7. Adjusted odds ratios (OR) for variablesindependently associated with diabetic nephropathy
Variable
HbA1cParental type 2 diabetesMale sexParental hypertension
OR (95% CI)
1.76 (1.29–2.40)2.95 (1.03–8.40)2.30 (1.13–4.67)2.06 (1.00–4.24)
P
<0.01<0.05<0.05<0.05
Results

27
Diabetic nephropathy
tes and hypertension were both independentlyassociated with diabetic nephropathy.
An OGTT was performed in parents withno history of diabetes (Tables 5 and 8). Par-ents of patients with nephropathy had higherfasting plasma glucose levels and had moreoften been treated for hypertension. Fathersof DN+ patients also had higher WHR thanfathers of DN– patients.
Familial abnormalities in glucosemetabolism (III)
No differences in insulin sensitivity or in in-sulin secretion were discernible between rela-tives of DN+ and DN– patients (Table 9).The relatives of the DN+ showed an excess ofhypertension (46% vs 29%, P < 0.05) andmore frequently a family history of type 2
diabetes (34% vs 20%, P < 0.05) than didrelatives of the DN–. Selection of one relativeat random for each diabetic patient did notinfluence the results (DN+ vs DN–: K
ITT4.0±0.2 vs 3.9±0.1 %/min, P = NS; ratio ofinsulin AUC/glucose AUC 21.1±4.0 vs22.1±4.0, P = NS).
Twenty-five relatives of patients with DN+and 13 relatives of patients with DN– hadabnormal glucose tolerance (IGT or diabetes;Table 10). Relatives of DN+ patients wereyounger. As shown in Table 11, no differenceexisted in insulin sensitivity between the twogroups of glucose-intolerant relatives. How-ever, their insulin response to a rise in plasmaglucose (insulin AUC/glucose AUC-ratio) inthe OGTT was impaired among relatives ofDN+ patients (Table 11). This difference ininsulin secretion between relatives of DN+
Table 8. Characteristics of parents with no history of diabetes
Variable
Fathers/mothersAge (years)WHR, fathersWHR, mothersBMI, fathers (kg/m2)BMI, mothers (kg/m2)Fasting plasma glucose (mmol/l)Glucose AUC (mmol/l × min)Abnormal glucose tolerancea (%)Antihypertensive therapy (%)
DN+n=95
36/5965±10.97±0.010.85±0.0128.0±0.728.4±0.65.7±0.1326±183334
DN–n=55
22/3365±10.94±0.010.83±0.0126.1±0.927.9±0.75.4±0.1300±212418
P
NSNS<0.05NS<0.05NS<0.05NSNS<0.05
aIGT or diabetes
Table 9. Plasma glucose and serum insulin response to oral glucose load and insulin sensitivity measured in first-degree relatives of DN+ and DN–
Variable
Fasting plasma glucose (mmol/l)Fasting serum insulin (mU/l)Glucose AUC (mmol/l × min)Insulin AUC (mU/l × min)Insulin AUC/glucose AUC-ratioKITT (%/min)a
Relatives of DN+(n=114)
5.3±0.15±1276±163349±24817.9±1.94.0±0.1
Relatives of DN–(n=93)
5.3±0.15±1225±123380±28017.8±1.94.2±0.1
P
NSNSNSNSNSNS
aMeasured in 101 and 84 relatives of DN+ and DN–, respectively.
Results
Johan Fagerudd
28
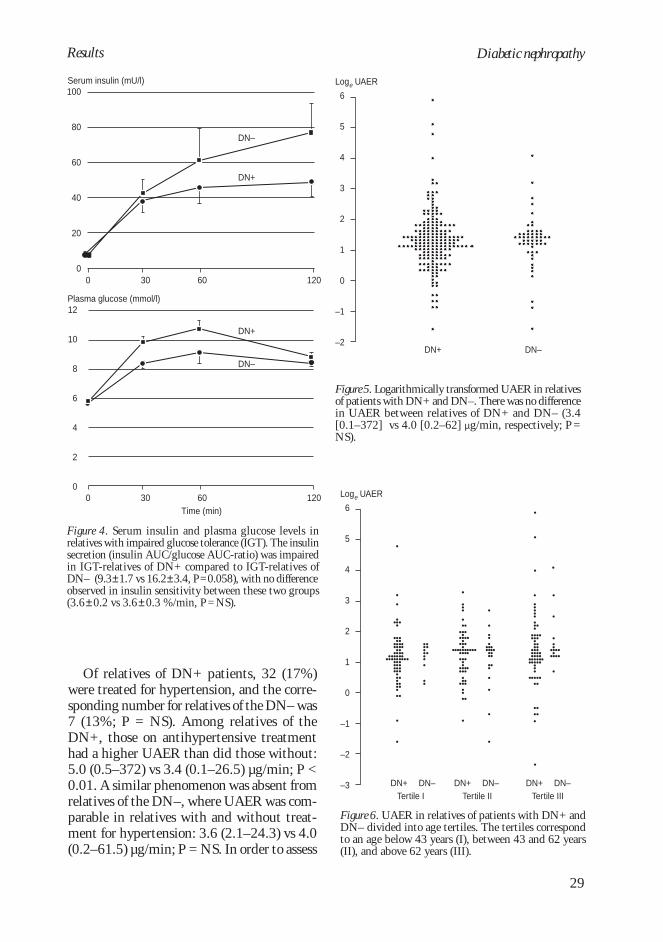
and DN– patients was already discernible atthe IGT stage (Figure 4).
Familial abnormalities in urinaryalbumin excretion rate (IV–V)
Study IV
The two groups of relatives participating werecomparable regarding sex distribution, age,BMI, and prevalence of smoking (Table 5).Similarly, no difference existed in blood pres-sure (systolic: 138±2 vs 138±3 mmHg, P =NS; diastolic 84±1 vs 84±2 mmHg, P = NS),prevalence of antihypertensive treatment(17% vs 13%, P = NS), or serum creatinineconcentration (80 [44–128] vs 80 [57–112]µmol/l, P = NS) between relatives of patientswho were DN+ and DN–, respectively. Over-
night UAER did not differ between relativesof DN+ and DN– patients (Figure 5). Theproportion of relatives with a UAER 10 µg/min was 12% for the DN+ compared to 8%for the DN– (P = NS). Stratified analyses ofmales and females as well as of parents andsiblings revealed no difference in UAER be-tween the two groups (Table 12).
Table 10. Characteristics of relatives with abnormal glucose tolerance
Variable
Sex (M/F)Age (years)BMI (kg/m2)WHR male subjectsWHR female subjectsHypertension (%)Smoking (%)HbA1c (%)Proportion IGT/diabetes
Relatives of DN+(n=25)
9/1657±328.1±0.91.00±0.030.84±0.0274165.6±0.118/7
Relatives of DN–(n=13)
6/766±228.5±1.30.98±0.020.87±0.0246235.8±0.19/4
P
NS0.027NSNSNS0.049NSNSNS
Table 11. Insulin sensitivity and insulin secretion in relatives with abnormal glucose tolerance
Variable
KITT (%/min)a
Fasting plasma glucose (mmol/l)Fasting serum insulin (mU/l)Glucose AUC (mmol/l × min)Insulin AUC (mU/l × min)Insulin AUC/glucose AUC-ratio
Relatives of DN+(n=25)
3.3±0.25.8±0.17±1493±364089±7448.9±1.4
Relatives of DN–(n=13)
3.2±0.35.7±0.18±1387±386209±139616.8±3.9
P
NSNSNSNSNS0.039
aPerformed in 23 and 11 relatives of patients with DN+ and DN–, respectively.
Table 12. UAER (µg/min) in subgroups of relatives inStudy IV
Subgroup
MaleFemaleParentsSiblings
DN+
3.4 (0.1–372)3.5 (0.2–118)3.5 (0.1–372)3.4 (0.2–118)
DN–
4.2 (1.1–24.3)3.5 (0.2–61.5)4.0 (0.2–61.5)3.6 (0.4–14.4)
P
NSNSNSNS
Results
29
Diabetic nephropathy
Of relatives of DN+ patients, 32 (17%)were treated for hypertension, and the corre-sponding number for relatives of the DN– was7 (13%; P = NS). Among relatives of theDN+, those on antihypertensive treatmenthad a higher UAER than did those without:5.0 (0.5–372) vs 3.4 (0.1–26.5) µg/min; P <0.01. A similar phenomenon was absent fromrelatives of the DN–, where UAER was com-parable in relatives with and without treat-ment for hypertension: 3.6 (2.1–24.3) vs 4.0(0.2–61.5) µg/min; P = NS. In order to assess
Figure 4. Serum insulin and plasma glucose levels inrelatives with impaired glucose tolerance (IGT). The insulinsecretion (insulin AUC/glucose AUC-ratio) was impairedin IGT-relatives of DN+ compared to IGT-relatives ofDN– (9.3±1.7 vs 16.2±3.4, P=0.058), with no differenceobserved in insulin sensitivity between these two groups(3.6±0.2 vs 3.6±0.3 %/min, P=NS).
DN+
DN–
DN+
DN–
12
10
8
6
4
2
0
Plasma glucose (mmol/l)
100
80
60
40
20
0
Serum insulin (mU/l)
Time (min)0 30 60 120
0 30 60 120
Results
6
5
4
3
2
1
0
–1
–2
–3
Loge UAER
Figure 6. UAER in relatives of patients with DN+ andDN– divided into age tertiles. The tertiles correspondto an age below 43 years (I), between 43 and 62 years(II), and above 62 years (III).
Tertile I Tertile II Tertile IIIDN+ DN– DN+ DN– DN+ DN–
DN+ DN–
6
5
4
3
2
1
0
–1
–2
Loge UAER
Figure 5. Logarithmically transformed UAER in relativesof patients with DN+ and DN–. There was no differencein UAER between relatives of DN+ and DN– (3.4[0.1–372] vs 4.0 [0.2–62] µg/min, respectively; P =NS).

Johan Fagerudd
30
was observable in any of the tertiles. In orderto control for the range in number of rela-tives studied per diabetic patient, one rela-tive per diabetic patient was randomly se-lected. In this analysis, no difference in UAERwas observed between the two groups: DN+(n = 80) vs DN– (n = 25): 3.6 (0.1–168) vs3.6 (0.2–61.5) µg/min; P = NS.
Study V
As a result of the matching procedure, thetwo groups of siblings participating in StudyV were similar regarding age, gender, BMI,and smoking habits (Table 5). Furthermore,no significant differences were apparent inserum creatinine, blood pressure levels, orplasma glucose or serum insulin levels in theOGTT (Table 13). All siblings had normalglucose tolerance except for one sister of aDN+ patient with IGT. UAER measuredfrom timed urine collections performed over-night, during the OGTT, and during thesubmaximal exercise test did not differ be-tween the two groups of relatives (Figure 7).Furthermore, the exercise-induced propor-tional increase in UAER was approximatelyeight-fold in both groups of siblings; DN+vs DN–: 8.3 (2.1–193) vs 7.9 (1.7–119)-foldincrease; P = NS.
any effect of impaired survival among rela-tives of patients with nephropathy, the rela-tives were further divided into tertiles accord-ing to age (Figure 6). No difference in UAER
8
7
6
5
4
3
2
1
0
Loge UAER
DN+ DN– DN+ DN– DN+ DN–
Overnight OGTT Exercise
Figure 7. Logarithmically transformed UAER in siblingsof patients with DN+ and with DN–. There was nosignificant difference between siblings of patients withDN+ and DN– in UAER measured overnight: median(range): 3.8 (1.3–24.1) vs 3.5 (2.0–21.0) µg/min; P=NS, during the OGTT 6.3 (3.2–26.0)] vs 4.8 (1.9–15.7)µg/min; P = NS or during the exercise test 44.8(7.0–535) vs 30.0 (3.4–1614) µg/min; P=NS.
Table 13. Renal function, blood pressure, and glucose metabolism in siblings of patients with DN+ and DN– in Study V
Variable
Serum creatinine (µmol/l)Systolic blood pressure (mmHg)Diastolic blood pressure (mmHg)Fasting plasma glucose (mmol/l)Glucose AUC (mmol/l × min)Fasting serum insulin (mU/l)Insulin AUC (mU/l × min)
DN+(n=21)
78±2125±276±25.1±0.1125±277±14282±690
DN–(n=24)
82±3124±376±35.0±0.1135±266±13644±801
P
NSNSNSNSNSNSNS
Results

31
Diabetic nephropathyDiscussion
Discussion
Subjects and methods
The Finnish patients were recruited from thenephrological unit and the diabetic outpatientclinic of the Helsinki University Central Hos-pital as well as from among members of theHelsinki Diabetes Association, and the Dan-ish patients in Study I from the outpatientclinic of the Steno Diabetes Center inGentofte, but in both countries the health caresystems as well as the epidemiology of type 1diabetes [187] and its long-term complica-tions [2, 188] are very similar. It is thereforeunlikely that the inclusion of patients fromtwo different source populations in Study Iwould cause problems in terms of validity ofthe results. Most of the patients with type 1diabetes residing in the Copenhagen area aretreated at the Steno Diabetes Center. Simi-larly, Helsinki University Central Hospitalruns the only nephrological unit in theHelsinki area. Patients from these sources canbe considered representative of type 1 diabeticpatients with long-duration diabetes with andwithout diabetic nephropathy. In addition,patients came from the diabetic outpatientclinic of the Helsinki University Central Hos-pital and via an advertisement in the news-letter of the Helsinki Diabetes Association.These patients could have been affected bysome selection bias: first, complicated casesare more easily referred to a university hospi-tal; second, members of a diabetes associationcan be assumed to be more actively involvedin gaining knowledge of their disease thanthe average diabetic patient. However, as theaim of the present studies was to assess theeffect of familial traits on the development ofdiabetic nephropathy, it is highly unlikely thatany selection bias would have had an effecton occurrence of familial traits in patients with
and without diabetic nephropathy. In termsof familial factors, the patients included inthis study are therefore most likely represen-tative of Caucasian type 1 diabetic patientswith and without diabetic nephropathy.
In Study I, 24 h ABPM was used to mea-sure parental blood pressure in addition tooffice blood pressure. The SpaceLabs 9020724 h ABPM device achieved a B-rating [189]when tested according to the British Hyper-tension Society Protocol [190] and was con-sequently recommended by the authors formeasurement of 24 h ambulatory blood pres-sure. Day- and night-time blood pressureswere calculated by use of individually recordedwaking and sleeping hours [191]. Most stud-ies have reported a better correlation with end-organ damage [192–194] and a better pre-dictive value for future cardiovascular events[195] and for treatment-induced regressionof left ventricular hypertrophy [196] with 24h ABPM than with conventional office bloodpressure. Thus, 24 h ABPM is considered su-perior to conventional blood pressure in iden-tifying subjects with clinically relevant hy-pertension. This superiority is even more pro-nounced if office blood pressure is measuredon a single occasion, since high screening val-ues tend to overestimate and low screeningvalues underestimate true blood pressure, aphenomenon referred to as regression dilutionbias [197]. At present, little information isavailable from prospective studies on the cut-off level for hypertension in 24 h ABPM, butin Study I we applied the operational thresh-old for hypertension ( 135/85 mmHg) pro-posed by Staessen et al [181] and the Ameri-can Society of Hypertension [182].
In Study III, whole-body insulin sensitiv-ity was measured with the short ITT [185].When glucose disappearance rate is measured
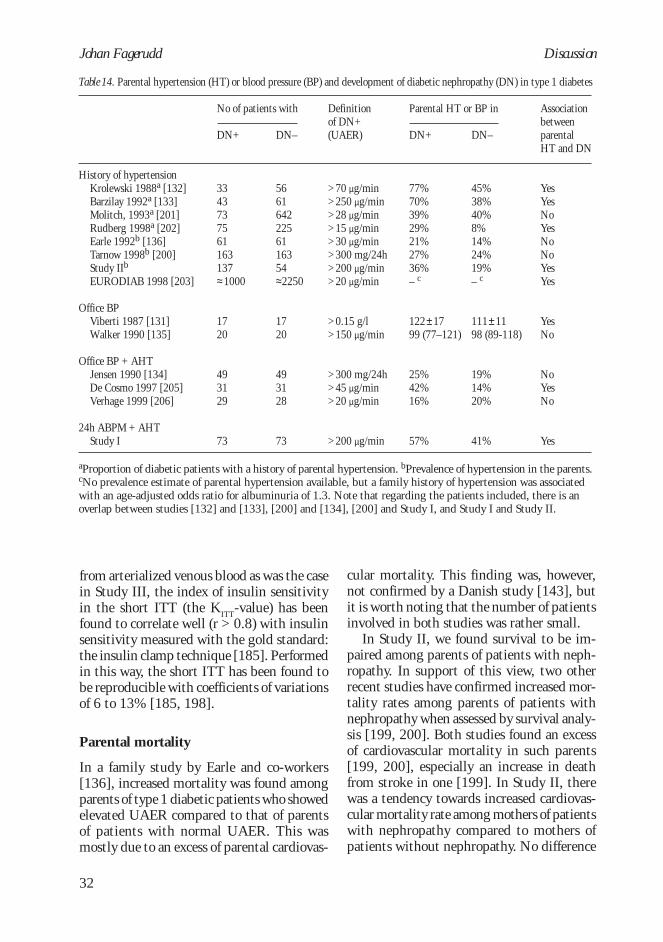
Johan Fagerudd
32
from arterialized venous blood as was the casein Study III, the index of insulin sensitivityin the short ITT (the K
ITT-value) has been
found to correlate well (r > 0.8) with insulinsensitivity measured with the gold standard:the insulin clamp technique [185]. Performedin this way, the short ITT has been found tobe reproducible with coefficients of variationsof 6 to 13% [185, 198].
Parental mortality
In a family study by Earle and co-workers[136], increased mortality was found amongparents of type 1 diabetic patients who showedelevated UAER compared to that of parentsof patients with normal UAER. This wasmostly due to an excess of parental cardiovas-
cular mortality. This finding was, however,not confirmed by a Danish study [143], butit is worth noting that the number of patientsinvolved in both studies was rather small.
In Study II, we found survival to be im-paired among parents of patients with neph-ropathy. In support of this view, two otherrecent studies have confirmed increased mor-tality rates among parents of patients withnephropathy when assessed by survival analy-sis [199, 200]. Both studies found an excessof cardiovascular mortality in such parents[199, 200], especially an increase in deathfrom stroke in one [199]. In Study II, therewas a tendency towards increased cardiovas-cular mortality rate among mothers of patientswith nephropathy compared to mothers ofpatients without nephropathy. No difference
Discussion
Table 14. Parental hypertension (HT) or blood pressure (BP) and development of diabetic nephropathy (DN) in type 1 diabetes
History of hypertensionKrolewski 1988a [132]Barzilay 1992a [133]Molitch, 1993a [201]Rudberg 1998a [202]Earle 1992b [136]Tarnow 1998b [200]Study IIb
EURODIAB 1998 [203]
Office BPViberti 1987 [131]Walker 1990 [135]
Office BP + AHTJensen 1990 [134]De Cosmo 1997 [205]Verhage 1999 [206]
24h ABPM + AHTStudy I
No of patients with
DN+
3343737561163137≈1000
1720
493129
73
DN–
56616422256116354≈2250
1720
493128
73
Parental HT or BP in
DN+
77%70%39%29%21%27%36%– c
122±1799 (77–121)
25%42%16%
57%
Definitionof DN+(UAER)
>70 µg/min>250 µg/min>28 µg/min>15 µg/min>30 µg/min>300 mg/24h>200 µg/min>20 µg/min
>0.15 g/l>150 µg/min
>300 mg/24h>45 µg/min>20 µg/min
>200 µg/min
DN–
45%38%40%8%14%24%19%– c
111±1198 (89-118)
19%14%20%
41%
AssociationbetweenparentalHT and DN
YesYesNoYesNoNoYesYes
YesNo
NoYesNo
Yes
aProportion of diabetic patients with a history of parental hypertension. bPrevalence of hypertension in the parents.cNo prevalence estimate of parental hypertension available, but a family history of hypertension was associatedwith an age-adjusted odds ratio for albuminuria of 1.3. Note that regarding the patients included, there is anoverlap between studies [132] and [133], [200] and [134], [200] and Study I, and Study I and Study II.

33
Diabetic nephropathy
was evident in cardiovascular death rate infathers alone or in all parents, and we couldconfirm no particular increase in deaths fromstroke among parents of patients with nephr-opathy (data not shown). However, Study IIdid not have the same power as the other re-cent studies [199, 200]. Thus, diabetic neph-ropathy in type 1 diabetic patients is associ-ated with impaired survival in the parents, aphenomenon that seems to be due to an ex-cess of cardiovascular disease.
Familial predisposition to hypertension
Viberti and co-workers [131] were the firstto report an association between high bloodpressure in non-diabetic parents and pro-teinuria in their offspring with type 1 diabe-tes. Retrospective analysis of a family studyconducted in the 50’s revealed a higher arte-rial blood pressure in 26 parents of 17 pa-tients with a urinary protein concentrationexceeding 0.15 g/l than in 26 parents of 17non-proteinuric patients. This led the authorsto put forward the hypothesis that suscepti-bility to diabetic nephropathy is linked to afamilial predisposition to hypertension. How-ever, at the time Study I was initiated, a con-troversy existed regarding this subject, withsome studies supporting the original obser-vation [132, 133], and others unable to findhigher blood pressure [134, 135] or an excessof hypertension [136, 201] in parents of pa-tients with signs of diabetic nephropathy.
Table 14 presents an update of studies pub-lished thus far dealing with the subject. Theseexhibit huge variation in sample size, andtheir definitions of diabetic nephropathy rangefrom the upper level of normoalbuminuria ina Swedish study on pediatric patients [202]to ESRD because of diabetic nephropathy, asin Study II. The methods of determining fa-milial hypertension vary from assessing pa-rental history of hypertension [132, 133, 136,200–204], to measuring parental office bloodpressure [131, 135], to combining antihyper-tensive medication and office blood pressure[134, 205, 206] or 24 h ABPM [207] in or-der to calculate total prevalence of parental
hypertension. There is an overlap betweensome of the studies.
At first glance, the results of the studiesseem to diverge substantially, but a more de-tailed analysis enables some conclusions to bedrawn. First, although the difference in pa-rental history of hypertension between pa-tients with and without nephropathy is notstatistically significant in all studies, there isindeed an evident overall tendency toward anexcess of hypertension among parents of pa-tients with nephropathy. The results of thelarge EURODIAB study [203] speak in fa-vor of such a view. Second, studies relying onoffice blood pressure may be hampered byobvious methodological shortcomings, as pre-viously discussed. Since no routine treatmentfor hypertension was available when bloodpressure was measured by Viberti et al [131],this may explain why the same group detectedno difference in a subsequent study authoredby Walker et al [135]. Regarding hyperten-sion defined as office blood pressure or as an-tihypertensive treatment, only one of fourstudies [134, 205–207] demonstrates a sig-nificant difference between parents of patientswith and without nephropathy. The relativelyhigh prevalence of hypertension in Study Ican be explained by a lower cut-off level (140/90 mmHg) for untreated hypertension thanin two of the other studies (160/95 mmHg)[134, 206] and to the fact that the parents inStudy I were older than in the other studies.Study I is the only study having used 24 hABPM to identify parents with untreatedhypertension. In conclusion, an excess of fa-milial hypertension appears in type 1 diabeticpatients with diabetic nephropathy, but inorder to detect this excess, proper evaluationof parental blood pressure status and sufficientstatistical power are required.
Thus far, Study I is also the only studymaking any attempt to evaluate severity ofhypertension by taking into account the ageof onset of parental hypertension. Parents ofpatients with diabetic nephropathy showedan increased cumulative incidence rate of hy-pertension, and a pronounced excess of hy-pertension with a rather young age at onset
Discussion

Johan Fagerudd
34
(Figure 2). Two studies on adolescent [202]and pediatric patients [137] support thesefindings. In the study by Rudberg et al [202],a parental history of hypertension was associ-ated with a four-fold risk for micro- or mac-roalbuminuria in the offspring. Likewise,Freire and co-workers [137] found that amongnormoalbuminuric patients with an UAERin the highest tertile, 79% had a family his-tory of hypertension compared to only 29%of patients with UAER in the lowest tertile.Based on the patients’ age, the mean age ofthe parents in these studies must have beenlow, and both report strong associations be-tween elevated UAER and parental hyperten-sion. Thus, a familial predisposition to a phe-notypically more severe hypertension, mani-fested in the parents at a relatively young age,may be of particular importance as a deter-minant of renal outcome in patients with type1 diabetes. However, before drawing any fur-ther conclusions in terms of identifying high-risk patients, our hypothesis needs to be testedin large-scale prospective study settings.
Hypertension was more often present inthose patients suffering from nephropathywho had a parental history including antihy-pertensive therapy. Although this findingmust be regarded as preliminary because com-parisons were made between relatively smallgroups, it suggests that for patients with dia-betic nephropathy, a familial predispositionto hypertension is associated not only withtheir increased risk for diabetic nephropathy,but also with their increased risk for systemichypertension.
Why would a predisposition to hyperten-sion increase risk for nephropathy in type 1diabetes? The importance of hemodynamicfactors in the genesis of diabetic nephropathyhas been discussed above. It is of interest thata family history of hypertension has been as-sociated both with elevated systemic bloodpressure and with increased glomerular fil-tration in recent-onset type 1 diabetic patientsas well as in non-diabetic subjects [208, 209].Of these abnormalities, glomerular hyperfil-tration may be more important in the initia-tion of renal damage, since such hyperfiltra-
tion [96] is a more powerful predictor of sub-sequent microalbuminuria or overt diabeticnephropathy than is the level of systemicblood pressure [24, 96, 104, 105]. However,at a later stage, the level of systemic bloodpressure is strongly positively correlated withthe rate of decline in renal function.
Familial predisposition to diabetes
In Pima Indians, diabetic nephropathy in theparents, more than parental diabetes alone,has been associated with an increased risk fordiabetes in the offspring [152], suggesting alink between familial diabetes and develop-ment of diabetic nephropathy. Studies in type1 diabetes evaluating parental hypertension[131, 134, 135, 202] and prevalence [136,143, 200] or mechanisms [205, 206] of pa-rental cardiovascular disease, have not re-ported any excess of parental diabetes in type1 diabetic patients with nephropathy. How-ever, these studies have in general been small.Support of our observation in Study II comesfrom two recent large-scale studies. An asso-ciation between parental diabetes and albu-minuria in the type 1 diabetic offspring wasobserved in the EURODIAB Study [203]. Inthe Pittsburgh Epidemiology of DiabetesComplications Study [210], the prevalence ofnephropathy was significantly higher inunivariate analysis among type 1 diabetic pa-tients with familial type 2 diabetes than inthose without.
In Study II, parental type 2 diabetes wasassociated with an almost three-fold risk fornephropathy in the offspring, which exceedsthe 95% CI of the EURODIAB Study [203].Additionally, in the EURODIAB Study, thefinding was limited to female patients alone,whereas the association in male patients, af-ter adjustment for glycemic control, failed toreach statistical significance. However, theclassification of patients into albuminuric andnormoalbuminuric in the EURODIAB Studywas based on a single urine collection, andthe control group had had a relatively shortdiabetes duration and could therefore haveincluded some proportion of patients at con-
Discussion

35
Diabetic nephropathy
siderable risk for subsequent development ofnephropathy. Misclassification of cases andcontrols in a study with a case-control designwill lead to an underestimation of any associ-ated phenomenon. Therefore, the strongerassociation between diabetic nephropathy andparental diabetes found in our Study II is mostlikely a result of the more robust classifica-tion of cases and controls we used.
In the insulin resistance syndrome, hyper-tension and abnormalities of glucose metabo-lism are closely intertwined [14], so that anassociation of diabetic nephropathy with pa-rental diabetes may be just another reflectionof its association with parental hypertension.However, the association of diabetic nephr-opathy with a family history of diabetes wasindependent of that of a family history of hy-pertension in Study II, which was also the casein the EURODIAB Study [203]. Therefore,a family history of diabetes and familial pre-disposition to hypertension seem to influencethe risk of nephropathy via different mecha-nisms.
In order to identify the mechanisms link-ing a family history of diabetes to diabeticnephropathy in type 1 diabetes, a further char-acterization of the mechanisms behind thefamilial clustering of diabetes is needed. Thetwo key variables involved in the regulationof glucose metabolism are insulin sensitivityand insulin secretion [211]. Study II found aparticular excess of parental type 2 diabetes,defined as all cases of diabetes other than thosewith their onset before age 40 and that hadbeen treated with insulin alone, but it is evi-dent that this definition will include cases ofdiabetes with a wide range of different etio-logic mechanisms [212]; it is thus impossibleto further characterize the pathogeneticmechanisms based on this observation.
Impaired insulin sensitivity has been foundpresent in the early stages of diabetic nephr-opathy in both type 1 [146, 147, 213] andtype 2 diabetes [150, 214]. This impairmentin insulin sensitivity has in part been attrib-uted to genetic factors, since insulin resistance,either based on fasting hyperinsulinemia[153] or measured with the short ITT [205],
has been found present in relatives of type 1diabetic patients with micro- or macroalbu-minuria. Furthermore, assessment of the non-diabetic parents participating in Study II re-vealed, in parents of patients with diabeticnephropathy, an excess of abdominal obesity,higher fasting plasma glucose, and more hy-pertension – all factors associated with theinsulin resistance syndrome [14]. However,in Study III, we found no significant impair-ment of insulin sensitivity in non-diabeticrelatives of patients with nephropathy com-pared to that in relatives of patients withoutnephropathy, despite Study III’s being thelargest one thus far that has addressed thisquestion.
Differences in study design may explainsome of this discrepancy. Study III focusedonly on relatives with normal fasting bloodglucose, and this differs from the strategyapplied by De Cosmo and coworkers, who alsoincluded diabetic parents [205]. In Study III,there was an excess of diabetes among rela-tives of patients with nephropathy, and ex-clusion of these diabetic individuals mostlikely resulted in an underestimation of thetrue abnormalities in glucose metabolism inthis group. However, chronic hyperglycemiahas well-known effects on glucose metabo-lism, including an impairment in both insu-lin sensitivity and insulin secretion [215]. Ifwe had included diabetic relatives in StudyIII, we would have been unable to excludethe fact that any impairment observed in, forinstance, insulin sensitivity was merely a con-sequence of their chronic hyperglycemia. Fo-cusing on those relatives without severe hy-perglycemia offered us an opportunity to iden-tify the underlying mechanisms of the excessof diabetes in relatives of patients with neph-ropathy.
Moreover, De Cosmo’s group [205] ob-served parents alone, not, as in the presentstudy, all first-degree relatives. Indeed, inStudy III, insulin sensitivity tended to beimpaired, especially in fathers of patients withnephropathy (K
ITT 2.9 vs 3.7%/min, respec-
tively). We measured insulin sensitivity withthe short ITT, a method found to be both re-
Discussion

Johan Fagerudd
36
liable and reproducible in estimating whole-body insulin sensitivity [185, 198]. However,one cannot ignore the fact that the use of moresophisticated methods, for instance the insu-lin clamp technique, could have detected adifference in insulin sensitivity between thetwo groups. A role for impaired insulin sen-sitivity cannot, therefore, be excluded in thefamilial clustering of diabetes in patients withnephropathy, but if impaired insulin sensi-tivity does play a major role, one would haveexpected it to be detected with our samplesize, despite these various shortcomings. An-other recent study found, in parents of pa-tients with nephropathy, no clustering of fac-tors associated with the metabolic syndrome[206].
The new finding in Study III was impairedinsulin secretion in relatives of patients withnephropathy, a finding not evident when as-sessed in all relatives, but isolated in relativeswith signs of abnormal glucose tolerance. Sincethis was the result of a post-hoc analysis, thefinding should be interpreted with caution.It is, however, rational to focus on those rela-tives with evidence of mild abnormalities inglucose metabolism, since only a minority ofall the relatives in this study will ever developdiabetes, and the mechanisms responsible foran excess of familial diabetes in patients withnephropathy will most likely be found in thatgroup. In this respect, it is noteworthy thatinsulin response to a glucose load is related todegree of derangement of glucose metabolism,with a more marked hypoinsulinemia as thedegree of hyperglycemia increases [216]. Thequestion arises, whether the impairment ofinsulin secretion observed is a primary phe-nomenon, or whether it is just a consequenceof subclinical hyperglycemia. Although thelatter is possible, it should be emphasized thatall the relatives studied had only mild abnor-malities in glucose metabolism – in fact, theywould all have been classified as non-diabeticbased on their fasting plasma glucose level.Furthermore, no differences existed betweenthe groups in the proportions of IGT and dia-betes, or in the level of HbA
1c. Finally, al-
though based on comparison between very
small groups, the impairment in insulin se-cretion in relatives of patients with nephropa-thy was already present at the stage of IGT.Therefore, even though our results are prelimi-nary and need to be confirmed, they suggestthat the excess of diabetes in the family his-tory for type 1 diabetic patients with nephr-opathy at least in part may be due to a pri-mary, possibly inherited, impairment in insu-lin secretion.
By what mechanism could such familialabnormalities increase the risk for diabeticnephropathy in the type 1 diabetic patient?First, patients with an inherited tendency to-ward impaired insulin sensitivity may be atincreased risk for nephropathy since goodmetabolic control is likely to be more diffi-cult to achieve in such patients. They may alsorequire higher doses of exogenous insulin, andthe resultant hyperinsulinemia may increaseUAER via increased endothelial permeability[217] or via effects on glomerular hemody-namics [218, 219]. Factors associated withimpaired insulin sensitivity, such as hyperten-sion [220] and dyslipidemia [221], may alsoexert deleterious effects on the glomerulus.
Second, type 1 diabetic patients with re-sidual endogenous insulin secretion are atlower risk of diabetic microvascular compli-cations than are patients with complete lossof beta-cell function [179]. An inherited in-sulin-secretion defect may increase risk fornephropathy by causing a more complete lossof beta-cell function at the time of onset ofdiabetes. This could simply result in worsemetabolic control, with harmful effects on thekidneys. However, beneficial effects of c-pep-tide on renal function have also been described[222], and the absence of circulating c-pep-tide could thereby increase risk for diabeticnephropathy. Furthermore, gestational diabe-tes develops when the beta cells are unable torespond to the increased need for insulin dueto the insulin resistance induced by pregnancy[223]. The recent observation in Pima Indi-ans that offspring of mothers who have dia-betes during their pregnancy are at increasedrisk for elevated UAER [224] may providean additional mechanism for an increased risk
Discussion

37
Diabetic nephropathy
for nephropathy in patients with familial de-fects in insulin secretion, because intrauter-ine hyperglycemia exerts detrimental effectson fetal kidney function [225].
In the light of our findings, several ques-tions need to be answered in large-scale studysettings for type 1 diabetic patients. First, isit possible to confirm that preserved beta-cellfunction protects against development of dia-betic nephropathy? If so, is this just an effectof enhanced metabolic control, or is there per-haps some protective effect from the c-pep-tide molecule? Second, what is the relationbetween familial diabetes and residual beta-cell function in the type 1 diabetic patient?Third, is there any association between ma-ternal gestational diabetes and developmentof diabetic nephropathy in the offspring? Theanswers to these questions will determine theimportance of our finding of impaired insu-lin secretion in glucose-intolerant relatives ofpatients with elevated UAER.
Familial abnormalities in urinaryalbumin excretion rate
Studies in both type 1 [6, 120, 121, 123] andtype 2 diabetes [124, 126, 127] have reportedfamilial clustering of microalbuminuria and/or proteinuria in diabetic siblings. Thus, inthe presence of diabetes, genetic factors seemto influence UAER. Several family studieshave reported an elevated UAER also in non-diabetic relatives of those type 2 diabetic pa-tients showing micro- or macroalbuminuriawhen compared with that in relatives ofnormoalbuminuric patients [126, 128–130].Therefore, at least for relatives of type 2 dia-betic patients, abnormalities leading to an el-evated UAER seem to be present even in theabsence of diabetes in subjects with a poten-tial genetic susceptibility to diabetic nephr-opathy. Studies IV and V thus far offer theonly such data available in type 1 diabetes andindicate that similar abnormalities in UAERare absent from non-diabetic relatives of pa-tients with type 1 diabetes and nephropathy.
Since there was no difference in UAERbetween relatives of patients with and with-
out nephropathy, the power to detect such adifference is of crucial importance. Assum-ing that the distributions of UAER in bothgroups were similar to those observed, StudyIV would have detected a relatively small dif-ference in median value between the twogroups (5.1 µg/min in relatives of DN+ vs4.0 µg/min in relatives of DN– patients).Studies performed in type 2 diabetes [126,128, 129] have reported clearly elevated (ap-proximately 2-fold) UAER in non-diabeticrelatives of patients with nephropathy com-pared to that in relatives of patients withoutnephropathy. A recent study of relatives ofFinnish type 2 diabetic patients found thisdifference to be somewhat smaller [130]. Al-though our study cannot totally exclude aminimal elevation in UAER in non-diabeticrelatives of type 1 diabetic patients with neph-ropathy, it seems justified to question the rel-evance of an elevation in UAER in such rela-tives not detected in a sample of our size.
Features associated with elevated albumin-uria such as cardiovascular morbidity andmortality [226, 227], diabetes [228], insulinresistance [229], and hypertension [230] haveall been found to cluster in family membersof type 1 diabetic patients with diabetic neph-ropathy [131, 136, 153, 203, 204, 207].Therefore, since we studied UAER in surviv-ing non-diabetic relatives in Study IV and innon-diabetic siblings with no antihyperten-sive therapy in Study V, an underestimationof the “true” UAER in relatives of type 1 dia-betic patients with nephropathy may havetaken place. However, also in type 2 diabetes,relatives of patients with nephropathy are atincreased risk for hypertension [231] and dia-betes [152] compared to relatives of patientswithout nephropathy. Most importantly, theoriginal observation by Gruden et al [128]was made in a case-control study with 20 off-spring in each group, all of whom were nor-motensive and had normal glucose tolerance.The confirming observation by Strojek et al[129] was also based on offspring selected tobe normotensive and to have normal oral glu-cose tolerance. The studies on siblings of type2 diabetic patients [126, 130] may readily
Discussion

Johan Fagerudd
38
have been affected by selective mortality. Inother words, a similar selection bias is likelyto affect all the studies in type 2 diabetes.Therefore, the most likely explanation for thediscrepancy between ours and the previousfindings is that a true difference exists betweenmechanisms regulating UAER in non-dia-betic relatives of type 1 and type 2 diabeticpatients.
In Study V, the relatives selected were thosenot receiving antihypertensive therapy, norwas any difference observed in the prevalenceof hypertension between the two groups ofrelatives in Study IV. The latter fact was some-what surprising, since there was an excess offamilial hypertension in the whole studypopulation, as demonstrated in Study II. Arecent study has concluded that common ge-netic determinants of UAER and blood pres-sure exist in families with type 2 diabetes[232]. Therefore, the absence of a differencein blood pressure level in Studies IV and Vmay have contributed to the lack of differ-ence in UAER. However, it should be notedthat all [126, 128, 130] but one [129] of thefour studies in type 2 diabetes found no dif-ference in blood pressure between relatives ofpatients with and without nephropathy de-spite the difference observed in UAER.
Exercise increases albuminuria in both dia-betic and non-diabetic subjects [233, 234],but more pronouncedly in diabetic patientswith early signs of diabetic nephropathy[233]. Furthermore, a prominent rise in ex-ercise-induced UAER has been predictive ofsubsequent microalbuminuria [235]. Thestudy by Strojek et al [129] found an exag-gerated albuminuric response to physical ex-ercise in relatives of albuminuric comparedto normoalbuminuric type 2 diabetic patients,a 16-fold vs a 6-fold increase, respectively[129]. No such tendency was evident in StudyV, which further supports the theory of dif-ferences in mode of inheritance of UAER be-tween type 1 and type 2 diabetes.
How can such a difference between type 1and type 2 diabetes be explained? One possi-bility may be that a common familial sus-ceptibility to elevated UAER exists in both
types of diabetes, but that an additional trig-ger, such as diabetes, dyslipidemia, or hyper-tension, is needed for the susceptibility tomanifest itself as increased UAER. Non-dia-betic individuals with a family history of type2 diabetes display various metabolic and he-modynamic alterations [144, 145] notpresent in non-diabetic subjects with a fam-ily history of type 1 diabetes [236]. It maybe hypothesized that such an unmasking trig-ger is present in the non-diabetic relatives oftype 2 diabetic patients, but not in those oftype 1 diabetic patients. It is therefore of in-terest that in Study IV, hypertensive relativeshad higher UAER than normotensive rela-tives when it was specifically assessed in therelatives of patients with nephropathy. Nosuch effect appeared among relatives of pa-tients without nephropathy. The comparisonwas, however, made between small groupsand does not allow any further conclusionsto be drawn, but if the albuminuric responseto hypertension indeed differs between rela-tives of patients with and without nephropa-thy, it would favor this hypothesis. Moreover,the finding that persistently elevated UAERis often present at diagnosis [237] and evenprecedes development of type 2 diabetes[238] but is usually absent at diagnosis oftype 1 diabetes [239] is compatible with thishypothesis.
On the other hand, the discrepancy mayalso reflect differences in susceptibility to el-evated UAER between type 1 and type 2 dia-betes. In support of this, only 30% of type 2diabetic patients with microalbuminuria(comparable to the cases included in the stud-ies in type 2 diabetes [126, 128–130]) havebeen found to have structural lesions typicalof diabetic nephropathy [240]. Type 1 dia-betic patients with overt proteinuria (compa-rable to our cases) have the morphologicallyrather monotonous pattern typical of diabeticnephropathy [59]. Microalbuminuria is, fur-thermore, a strong predictor of overt diabeticnephropathy in type 1 diabetes [20–22], whilein type 2 diabetes, microalbuminuria is, in-stead, prognostic of cardiovascular events[241]. Therefore, differences regarding
Discussion

39
Diabetic nephropathyDiscussion
mechanisms behind elevated UAER, and per-haps also in genetic susceptibility to albumin-uria, may exist between type 1 and type 2diabetes.
Familial clustering – genes orenvironment?
The basis for genetic susceptibility to diabeticnephropathy is mostly based upon evidenceof familial clustering, not only of diabeticnephropathy [6, 120–123], but also of traitsclustering in the family members of patientswith nephropathy, as in the present studies.However, familial clustering can be demon-strated for almost any disease [242], and it isdifficult to differentiate to what extent this isdue to shared genes on the one hand and tosimilar known and unknown environmentalfactors on the other. A simulation study hasprovided some insight into this problem[243]. By assuming no genetic susceptibilityat all, it found that familial clustering of en-vironmental factors with a relative risk fordisease of less than 10 lead to only a minorincrease in familial clustering of disease, evenin the case of complete correlation of expo-sure. Therefore, genetic factors, acting eitheron their own or in concert with the environ-
ment, ought to be important in the familialclustering of a disease. This view is supportedby recent segregation analyses in type 2 dia-betes [154, 155].
During the last decade, many attemptshave been made to identify one or severalgenes that increase susceptibility to diabeticnephropathy, but thus far, no gene has beenidentified with any major impact on the de-velopment of nephropathy [156]. What arethe implications of the present studies in thesearch for diabetic nephropathy susceptibil-ity genes? First, the association of diabeticnephropathy with familial hypertension isconfirmed (Study I). If identified, genes asso-ciated with hypertension of early onset are ofparticular interest. Second, genes related todiabetes, to the metabolic syndrome, or espe-cially to impaired insulin secretion qualify ascandidate genes for diabetic nephropathy(Studies II and III). Finally, and most impor-tantly, differences may exist between suscep-tibility to elevated UAER in type 1 and type2 diabetes (Studies IV and V). Distinctsubphenotyping of the type of diabetes in thepatients studied may therefore prove worth-while, and results from studies with mixedtype 1 and type 2 diabetic patients should beinterpreted with caution.

Johan Fagerudd
40
1. Diabetic nephropathy was associated witha familial predisposition to hypertension.Genetic or environmental factors relatedto hypertension, especially to a more se-vere form of hypertension manifested rela-tively early in life, may be important inthe development of diabetic nephropathyin type 1 diabetes.
2. Diabetic nephropathy was associated witha familial predisposition to diabetes. Fac-tors related to familial clustering of dia-betes may predispose to diabetic nephr-opathy in type 1 diabetes.
3. In relatives of patients with diabetic neph-ropathy, diminished insulin secretion
Summary and conclusions
rather than impaired insulin sensitivity,characterized early abnormalities in glu-cose metabolism. This may explain theexcess of diabetes found in relatives of pa-tients with nephropathy. In the develop-ment of diabetic nephropathy, factors re-lated to impaired insulin secretion needto be further evaluated.
4. No abnormalities of UAER were presentin non-diabetic first-degree relatives of type1 diabetic patients with diabetic nephropa-thy. This differs from what has been foundin type 2 diabetes and may suggest differ-ences in susceptibility to albuminuria be-tween type 1 and type 2 diabetes.

41
Diabetic nephropathy
This work was carried out at the Departmentof Medicine at the Helsinki University Cen-tral Hospital. I wish to express my deep grati-tude to Professor Frej Fyhrqvist, Head of theDivision of Internal Medicine, and to DocentCarola Grönhagen-Riska, Head of the Divi-sion of Nephrology, for creating a inspiringatmosphere and providing me with excellentresearch facilities.
I am greatly indebted to my supervisor, Do-cent Per-Henrik Groop, for introducing meto the exciting field of diabetic nephropathy.Your energy, enthusiasm, devotion, encour-aging attitude, and friendly approach createdthe foundation onto which this work wasbuilt. I cannot think of one single occasionduring the years of this thesis when you didnot respond immediately to my concerns. Iam glad to have you as my friend.
The careful review and the constructivecriticism of the two reviewers, Docent LeenaMykkänen and Docent Kaj Metsärinne, hada great impact on the final version of themanuscript. Thank you for many valuablecomments.
I had the opportunity to work for a coupleof months at the Steno Diabetes Center as aresearch fellow under the supervision of Pro-fessor Hans-Henrik Parving. Thank you,Hans-Henrik, for sharing with me your im-mense knowledge in the field and for givingme the opportunity to learn from your insight-ful way of conducting science. I have alsotaken great pleasure in many stimulating dis-cussions during the years with Professor LeifGroop, who was the first to open my eyes tothe fascinating complexities of diabetes.
As a member of our diabetic nephropathyresearch group, I have had the pleasure to work
with many outstanding individuals. I wouldespecially like to thank Kim Pettersson-Fernholm, who has made a substantial con-tribution to this thesis. Mikael Riska is ac-knowledged for sharing with me moments ofjoy and of hardship.
My stay at Steno gave my family and memany moments of delight to remember. LiseTarnow never hesitated to help us – we hadsome unforgettable days in your beautifulhome in the countryside. I also value highlythe time spent with Fleming Nielsen, HenrikPost Hansen, Peder Jacobsen, Per Christian-sen, and all the other friendly Danes, not onlyat Steno, but also at different meetingsthroughout the world.
My friend Carol Forsblom has contributedprofoundly to this work. I thank you for allthe hours you have unselfishly put into help-ing me to solve various problems, for yourknowledge of the literature and for your ad-mirably uncompromising attitude towardsscience. The third of the Musketeers from ouryears at the Fourth Department of Medicine,Mikko Lehtovirta, is another extraordinaryperson whose company I have always verymuch enjoyed. Svante Stenman has, regard-less of adverse circumstances, never failed toreach out a helping hand and has always pro-vided me with numerous solutions to prob-lems related to computers and statistics. Thelayout of this book is the result of Svante’swork. In addition, Carola Saloranta has beena source of encouragement whenever needed.
During the many hours at the outpatientclinic of our research unit, I have had thepleasure to be assisted by efficient labora-tory technicians who have created a friendlyand warm atmosphere not only for me, but
Acknowledgments

Johan Fagerudd
42
Acknowledgments
also for the patients. Riina Laine assistedme during most of these studies, but I havealso enjoyed working with Tuula Riihimäkiand Tarja Vesisenaho. I highly appreciatethe volunteer work of Arja Kehusmaa. Theother members of the research laboratory,Anna-Maija Teppo, Seija Heikkinen, andArja Tapio among others, are also warmlyacknowledged for their assistance.
Nothing could have been achieved with-out the volunteer efforts of the diabetic pa-tients and their relatives. We had no difficul-ties in recruiting family members for exten-sive and time-consuming experiments. Theireagerness to contribute to an increase inknowledge about the disease that had causedso much harm and disability to a close rela-tive was often evident.
The linguistic revision of this book wasskillfully done by Carol Norris. I had never
expected such a dull process to be accompa-nied with so much laughter.
The support of my parents, Karl-Johan andLisbeth, has helped me through many timesof trouble not only during this thesis butthroughout my whole life. You have given methe values on which to build my life. Kaunoand Elina, your help with taking care of ourchildren has given me not only time to carryout my work, but also peace of mind, know-ing the warm atmosphere you have alwayscreated for them.
I dedicate this book to my family – mywife Pia, our son Viktor, and our daughterBlanca. You are the jewels of my crown andthe treasures of my kingdom – without you,I have nothing. I thank you for your patience,love, and support.
Helsinki, November 2000.

43
Diabetic nephropathy
1. Kimmelstiel P, Wilson C: Intercapillary lesions in theglomeruli of the kidney. Am J Pathol 12: 83–105, 1936.
2. Andersen AR, Christiansen JS, Andersen JK, Kreiner S,Deckert T: Diabetic nephropathy in Type 1 (insulin-de-pendent) diabetes: an epidemiological study. Diabetologia25: 496–501, 1983.
3. Markell MS, Friedman EA: Diabetic nephropathy. Man-agement of the end-stage patient. Diabetes Care 15:1226–38, 1992.
4. Finnish Registry for Kidney Diseases: Report. 1998.5. Anonymous: The effect of intensive treatment of diabe-
tes on the development and progression of long-termcomplications in insulin-dependent diabetes mellitus.The Diabetes Control and Complications Trial ResearchGroup. N Engl J Med 329: 977–86, 1993.
6. Seaquist ER, Goetz FC, Rich S, Barbosa J: Familial clus-tering of diabetic kidney disease. Evidence for geneticsusceptibility to diabetic nephropathy. N Engl J Med320: 1161–5, 1989.
7. Keen H: Diabetes diagnosis, International textbook ofdiabetes mellitus. Edited by Alberti KGMM, DeFronzoRA, Keen H, Zimmet P. Oxford, Great Britain, JohnWiley & Sons Ltd, 1992, pp 3–18.
8. Frank LL: Diabetes mellitus in the text of old Hindumedicine. Am J Gastroenterol 27: 76–81, 1957.
9. Young FG: Claude Bernard and the discovery of glyco-gen. A century of retrospect. BMJ 1: 1431–7, 1957.
10. Himsworth HP: Diabetes mellitus: its differentiation intoinsulin-sensitive and insulin-insensitive types. Lancet i:117–9, 1936.
11. Anonymous: Classification and diagnosis of diabetes mel-litus and other categories of glucose intolerance. NationalDiabetes Data Group. Diabetes 28: 1039–57, 1979.
12. Anonymous: WHO Expert Committee on Diabetes Mel-litus: second report. World Health Organ Tech Rep Ser646: 1–80, 1980.
13. Anonymous: Report of the Expert Committee on the Di-agnosis and Classification of Diabetes Mellitus. DiabetesCare 20: 1183–97, 1997.
14. Alberti KG, Zimmet PZ: Definition, diagnosis and clas-sification of diabetes mellitus and its complications. Part1: diagnosis and classification of diabetes mellitus provi-sional report of a WHO consultation. Diabet Med 15:539–53, 1998.
15. Krolewski AS, Warram JH, Christlieb AR, Busick EJ,Kahn CR: The changing natural history of nephropathyin type I diabetes. Am J Med 78: 785–94, 1985.
16. Mogensen CE: Microalbuminuria, blood pressure and dia-betic renal disease: origin and development of ideas.Diabetologia 42: 263–85, 1999.
17. Mogensen CE, Andersen MJ: Increased kidney size and
glomerular filtration rate in untreated juvenile diabetes:normalization by insulin-treatment. Diabetologia 11:221–4, 1975.
18. Østerby R: Morphometric studies of the peripheral glom-erular basement membrane in early juvenile diabetes. I.Development of initial basement membrane thickening.Diabetologia 8: 84–92, 1972.
19. Østerby R: A quantitative electron microscopic study ofmesangial regions in glomeruli from patients with shortterm juvenile diabetes mellitus. Lab Invest 29: 99–110,1973.
20. Viberti GC, Hill RD, Jarrett RJ, Argyropoulos A,Mahmud U, Keen H: Microalbuminuria as a predictorof clinical nephropathy in insulin-dependent diabetesmellitus. Lancet 1: 1430–2, 1982.
21. Parving HH, Oxenbøll B, Svendsen PA, Christiansen JS,Andersen AR: Early detection of patients at risk of de-veloping diabetic nephropathy. A longitudinal study ofurinary albumin excretion. Acta Endocrinol (Copenh)100: 550–5, 1982.
22. Mogensen CE, Christensen CK: Predicting diabetic neph-ropathy in insulin-dependent patients. N Engl J Med311: 89–93, 1984.
23. Forsblom CM, Groop PH, Ekstrand A, Groop LC: Pre-dictive value of microalbuminuria in patients with insu-lin-dependent diabetes of long duration. BMJ 305: 1051–3, 1992.
24. Poulsen PL, Hansen KW, Mogensen CE: Ambulatoryblood pressure in the transition from normo- to microal-buminuria. A longitudinal study in IDDM patients.Diabetes 43: 1248–53, 1994.
25. Parving HH, Andersen AR, Smidt UM, Oxenboll B,Edsberg B, Christiansen JS: Diabetic nephropathy andarterial hypertension. Diabetologia 24: 10–2, 1983.
26. Parving HH, Smidt UM, Friisberg B, Bonnevie-NielsenV, Andersen AR: A prospective study of glomerular fil-tration rate and arterial blood pressure in insulin-depen-dent diabetics with diabetic nephropathy. Diabetologia20: 457–61, 1981.
27. Viberti GC, Bilous RW, Mackintosh D, Keen H: Moni-toring glomerular function in diabetic nephropathy. Aprospective study. Am J Med 74: 256–64, 1983.
28. Kussman MJ, Goldstein H, Gleason RE: The clinicalcourse of diabetic nephropathy. JAMA 236: 1861–3,1976.
29. Borch-Johnsen K, Kreiner S: Proteinuria: value as pre-dictor of cardiovascular mortality in insulin dependentdiabetes mellitus. BMJ 294: 1651–4, 1987.
30. Borch-Johnsen K, Andersen PK, Deckert T: The effect ofproteinuria on relative mortality in type 1 (insulin-de-pendent) diabetes mellitus. Diabetologia 28: 590–6, 1985.
References

Johan Fagerudd
44
31. Vigstrup J, Mogensen CE: Proliferative diabetic retin-opathy: at risk patients identified by early detection ofmicroalbuminuria. Acta Ophthalmol (Copenh) 63: 530–4, 1985.
32. Klein R, Klein BE, Moss SE, Davis MD, DeMets DL:The Wisconsin epidemiologic study of diabetic retinopa-thy. II. Prevalence and risk of diabetic retinopathy whenage at diagnosis is less than 30 years. Arch Ophthalmol102: 520–6, 1984.
33. Winocour PH, Durrington PN, Ishola M, Anderson DC,Cohen H: Influence of proteinuria on vascular disease,blood pressure, and lipoproteins in insulin dependent dia-betes mellitus. BMJ 294: 1648–51, 1987.
34. Tuomilehto J, Borch-Johnsen K, Molarius A, Forsen T,Rastenyte D, Sarti C, Reunanen A: Incidence of cardio-vascular disease in Type 1 (insulin-dependent) diabeticsubjects with and without diabetic nephropathy in Fin-land. Diabetologia 41: 784–90, 1998.
35. Beck-Nielsen H, Richelsen B, Mogensen CE, Olsen T,Ehlers N, Nielsen CB, Charles P: Effect of insulin pumptreatment for one year on renal function and retinal mor-phology in patients with IDDM. Diabetes Care 8: 585–9, 1985.
36. Wiseman MJ, Saunders AJ, Keen H, Viberti G: Effect ofblood glucose control on increased glomerular filtrationrate and kidney size in insulin-dependent diabetes. NEngl J Med 312: 617–21, 1985.
37. Bangstad HJ, Osterby R, Dahl-Jorgensen K, Berg KJ,Hartmann A, Hanssen KF: Improvement of blood glu-cose control in IDDM patients retards the progression ofmorphological changes in early diabetic nephropathy.Diabetologia 37: 483–90, 1994.
38. Reichard P, Nilsson BY, Rosenqvist U: The effect of long-term intensified insulin treatment on the developmentof microvascular complications of diabetes mellitus. NEngl J Med 329: 304–9, 1993.
39. Viberti GC, Bilous RW, Mackintosh D, Bending JJ, KeenH: Long term correction of hyperglycaemia and progres-sion of renal failure in insulin dependent diabetes. BMJ286: 598–602, 1983.
40. Feldt-Rasmussen B, Mathiesen ER, Jensen T, LauritzenT, Deckert T: Effect of improved metabolic control onloss of kidney function in type 1 (insulin-dependent) dia-betic patients: an update of the Steno studies. Diabeto-logia 34: 164–70, 1991.
41. Mogensen CE: Long-term antihypertensive treatmentinhibiting progression of diabetic nephropathy. BMJ 285:685–8, 1982.
42. Parving HH, Andersen AR, Smidt UM, Svendsen PA:Early aggressive antihypertensive treatment reduces rateof decline in kidney function in diabetic nephropathy.Lancet 1: 1175–9, 1983.
43. Parving HH, Hommel E, Smidt UM: Protection of kid-ney function and decrease in albuminuria by captopril ininsulin dependent diabetics with nephropathy. BMJ 297:1086–91, 1988.
44. Björck S, Mulec H, Johnsen SA, Norden G, Aurell M:Renal protective effect of enalapril in diabetic nephropa-thy. BMJ 304: 339–43, 1992.
45. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD: The ef-fect of angiotensin-converting-enzyme inhibition on dia-betic nephropathy. The Collaborative Study Group. NEngl J Med 329: 1456–62, 1993.
46. Tarnow L, Rossing P, Jensen C, Parving HH: Long-termrenoprotective effect of nisoldipine and lisinopril in type
1 diabetic patients with diabetic nephropathy. J Am SocNephrol 11: A0690 (abstract), 1999.
47. Marre M, Chatellier G, Leblanc H, Guyene TT, MenardJ, Passa P: Prevention of diabetic nephropathy withenalapril in normotensive diabetics with microalbumin-uria. BMJ 297: 1092–5, 1988.
48. Viberti G, Mogensen CE, Groop LC, Pauls JF: Effect ofcaptopril on progression to clinical proteinuria in patientswith insulin-dependent diabetes mellitus and microal-buminuria. European Microalbuminuria Captopril StudyGroup. JAMA 271: 275–9, 1994.
49. Mathiesen ER, Hommel E, Hansen HP, Smidt UM,Parving HH: Randomised controlled trial of long termefficacy of captopril on preservation of kidney functionin normotensive patients with insulin dependent diabe-tes and microalbuminuria. BMJ 319: 24–5, 1999.
50. Anonymous: Randomised placebo-controlled trial oflisinopril in normotensive patients with insulin-dependentdiabetes and normoalbuminuria or microalbuminuria. TheEUCLID Study Group. Lancet 349: 1787–92, 1997.
51. Chaturvedi N, Sjolie AK, Stephenson JM, AbrahamianH, Keipes M, Castellarin A, Rogulja-Pepeonik Z, FullerJH: Effect of lisinopril on progression of retinopathy innormotensive people with type 1 diabetes. The EUCLIDStudy Group. EURODIAB Controlled Trial of Lisinoprilin Insulin-Dependent Diabetes Mellitus. Lancet 351: 28–31, 1998.
52. Raine AEG: Evolution worldwide of the treatment ofpatients with advanced diabetic nephropathy by renalreplacement therapy, The kidney and hypertension indiabetes mellitus, 2nd edition Edition. Edited byMogensen CE. Boston, Kluwer Academic Publishers,1994, pp 449–58.
53. Port FK, Wolfe RA, Mauger EA, Berling DP, Jiang K:Comparison of survival probabilities for dialysis patientsvs cadaveric renal transplant recipients. JAMA 270:1339–43, 1993.
54. Smets YF, Westendorp RG, van der Pijl JW, de CharroFT, Ringers J, de Fijter JW, Lemkes HH: Effect of si-multaneous pancreas-kidney transplantation on mortal-ity of patients with type-1 diabetes mellitus and end-stage renal failure. Lancet 353: 1915–9, 1999.
55. Bojestig M, Arnqvist HJ, Hermansson G, Karlberg BE,Ludvigsson J: Declining incidence of nephropathy ininsulin-dependent diabetes mellitus. N Engl J Med 330:15–8, 1994.
56. Rossing P, Hougaard P, Borch-Johnsen K, Parving HH:Predictors of mortality in insulin dependent diabetes:10 year observational follow up study. BMJ 313: 779–84, 1996.
57. Fioretto P, Steffes MW, Mauer M: Glomerular structurein nonproteinuric IDDM patients with various levels ofalbuminuria. Diabetes 43: 1358–64, 1994.
58. Walker JD, Close CF, Jones SL, Rafftery M, Keen H,Viberti G, Østerby R: Glomerular structure in type-1(insulin-dependent) diabetic patients with normo- andmicroalbuminuria. Kidney Int 41: 741–8, 1992.
59. Østerby R: Glomerular structural changes in type 1 (in-sulin-dependent) diabetes mellitus: causes, consequences,and prevention. Diabetologia 35: 803–12, 1992.
60. Bangstad HJ, Østerby R, Dahl-Jorgensen K, Berg KJ,Hartmann A, Nyberg G, Frahm Bjorn S, Hanssen KF:Early glomerulopathy is present in young, type 1 (insu-lin-dependent) diabetic patients with microalbuminuria.Diabetologia 36: 523–9, 1993.
References

45
Diabetic nephropathyReferences
61. Fioretto P, Steffes MW, Sutherland DE, Mauer M: Se-quential renal biopsies in insulin-dependent diabeticpatients: structural factors associated with clinical pro-gression. Kidney Int 48: 1929–35, 1995.
62. Østerby R, Gundersen HJ: Glomerular size and struc-ture in diabetes mellitus. I. Early abnormalities.Diabetologia 11: 225–9, 1975.
63. Kroustrup JP, Gundersen HJ, Østerby R: Glomerularsize and structure in diabetes mellitus. III. Early en-largement of the capillary surface. Diabetologia 13: 207–10, 1977.
64. Ellis EN, Steffes MW, Goetz FC, Sutherland DE, MauerSM: Glomerular filtration surface in type I diabetes mel-litus. Kidney Int 29: 889–94, 1986.
65. Østerby R, Parving HH, Nyberg G, Hommel E,Jorgensen HE, Lokkegaard H, Svalander C: A strongcorrelation between glomerular filtration rate and fil-tration surface in diabetic nephropathy. Diabetologia31: 265–70, 1988.
66. Gundersen HJ, Østerby R: Glomerular size and struc-ture in diabetes mellitus. II. Late abnormalities.Diabetologia 13: 43–8, 1977.
67. Lee CS, Mauer SM, Brown DM, Sutherland DE, MichaelAF, Najarian JS: Renal transplantation in diabetes mel-litus in rats. J Exp Med 139: 793–800, 1974.
68. Mauer SM, Barbosa J, Vernier RL, Kjellstrand CM,Buselmeier TJ, Simmons RL, Najarian JS, Goetz FC:Development of diabetic vascular lesions in normal kid-neys transplanted into patients with diabetes mellitus.N Engl J Med 295: 916–20, 1976.
69. Mauer SM, Miller K, Goetz FC, Barbosa J, Simmons RL,Najarian JS, Michael AF: Immunopathology of renal ex-tracellular membranes in kidneys transplanted into pa-tients with diabetes mellitus. Diabetes 25: 709–12, 1976.
70. Mauer SM, Steffes MW, Connett J, Najarian JS,Sutherland DE, Barbosa J: The development of lesionsin the glomerular basement membrane and mesangiumafter transplantation of normal kidneys to diabetic pa-tients. Diabetes 32: 948–52, 1983.
71. Østerby R, Nyberg G, Hedman L, Karlberg I, PerssonH, Svalander C: Kidney transplantation in type 1 (in-sulin-dependent) diabetic patients. Early glomerulopa-thy. Diabetologia 34: 668–74, 1991.
72. Fioretto P, Steffes MW, Sutherland DE, Goetz FC, MauerM: Reversal of lesions of diabetic nephropathy after pan-creas transplantation. N Engl J Med 339: 69–75, 1998.
73. Wang PH, Lau J, Chalmers TC: Meta-analysis of ef-fects of intensive blood-glucose control on late compli-cations of type I diabetes. Lancet 341: 1306–9, 1993.
74. Brownlee M: Glycation of macromolecules, Interna-tional Textbook of Diabetes Mellitus, First Edition.Edited by Alberti KGMM, DeFronzo RA, Keen H,Zimmet P. Chichester, UK, John Wiley & Sons Ltd,1992, pp 669–82.
75. Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E,Delaney V, Friedman EA, Cerami A, Vlassara H: Ad-vanced glycosylation end products in patients with dia-betic nephropathy. N Engl J Med 325: 836–42, 1991.
76. Vlassara H, Striker LJ, Teichberg S, Fuh H, Li YM,Steffes M: Advanced glycation end products induceglomerular sclerosis and albuminuria in normal rats.Proc Natl Acad Sci U S A 91: 11704–8, 1994.
77. Edelstein D, Brownlee M: Aminoguanidine amelioratesalbuminuria in diabetic hypertensive rats. Diabetolo-gia 35: 96–7, 1992.
78. Beyer TA, Hutson NJ: Introduction: evidence for the roleof the polyol pathway in the pathophysiology of diabeticcomplications. Metabolism 35: 1–3, 1986.
79. Beyer-Mears A, Cruz E, Edelist T, Varagiannis E: Di-minished proteinuria in diabetes mellitus by sorbinil, analdose reductase inhibitor. Pharmacology 32: 52–60,1986.
80. Tilton RG, Chang K, Pugliese G, Eades DM, ProvinceMA, Sherman WR, Kilo C, Williamson JR: Preventionof hemodynamic and vascular albumin filtration changesin diabetic rats by aldose reductase inhibitors. Diabetes38: 1258–70, 1989.
81. Ranganathan S, Krempf M, Feraille E, Charbonnel B:Short term effect of an aldose reductase inhibitor on uri-nary albumin excretion rate (UAER) and glomerular fil-tration rate (GFR) in type 1 diabetic patients with in-cipient nephropathy. Diabetes Metab 19: 257–61, 1993.
82. Passariello N, Sepe J, Marrazzo G, De Cicco A, PelusoA, Pisano MC, Sgambato S, Tesauro P, D'Onofrio F: Ef-fect of aldose reductase inhibitor (tolrestat) on urinaryalbumin excretion rate and glomerular filtration rate inIDDM subjects with nephropathy. Diabetes Care 16:789–95, 1993.
83. Koya D, King GL: Protein kinase C activation and thedevelopment of diabetic complications. Diabetes 47:859–66, 1998.
84. Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, ClermontA, Bursell SE, Kern TS, Ballas LM, Heath WF, StrammLE, Feener EP, King GL: Amelioration of vascular dys-functions in diabetic rats by an oral PKC beta inhibitor.Science 272: 728–31, 1996.
85. Viberti GC, Walker JD, Pinto J: Diabetic nephropathy,International textbook of diabetes mellitus. Edited byAlberti KGMM, DeFronzo RA, Keen H, Zimmet P.Oxford, Great Britain, John Wiley & Sons Ltd, 1992, pp1267–328.
86. Vernier RL, Steffes MW, Sisson-Ross S, Mauer SM:Heparan sulfate proteoglycan in the glomerular basementmembrane in type 1 diabetes mellitus. Kidney Int 41:1070–80, 1992.
87. Cohen MP, Klepser H, Wu VY: Undersulfation of glom-erular basement membrane heparan sulfate in experimen-tal diabetes and lack of correction with aldose reductaseinhibition. Diabetes 37: 1324–7, 1988.
88. Deckert T, Feldt-Rasmussen B, Borch-Johnsen K, JensenT, Kofoed-Enevoldsen A: Albuminuria reflects wide-spread vascular damage. The Steno hypothesis.Diabetologia 32: 219–26, 1989.
89. Myrup B, Hansen PM, Jensen T, Kofoed-Enevoldsen A,Feldt-Rasmussen B, Gram J, Kluft C, Jespersen J,Deckert T: Effect of low-dose heparin on urinary albu-min excretion in insulin-dependent diabetes mellitus.Lancet 345: 421–2, 1995.
90. van der Pijl JW, van der Woude FJ, Swart W, van Es LA,Lemkes HH: Effect of danaparoid sodium on hard exu-dates in diabetic retinopathy. Lancet 350: 1743–5, 1997.
91. Berkman J, Rifkin H: Unilateral nodular diabetic glom-erulosclerosis (Kimmelstiel-Wilson): report of a case.Metabolism 22: 715–22, 1973.
92. Mauer SM, Steffes MW, Azar S, Sandberg SK, BrownDM: The effects of Goldblatt hypertension on develop-ment of the glomerular lesions of diabetes mellitus inthe rat. Diabetes 27: 738–44, 1978.
93. Hostetter TH, Rennke HG, Brenner BM: The case forintrarenal hypertension in the initiation and progression

Johan Fagerudd
46
of diabetic and other glomerulopathies. Am J Med 72:375–80, 1982.
94. Schmitz A, Hansen HH, Christensen T: Kidney func-tion in newly diagnosed type 2 (non-insulin-dependent)diabetic patients, before and during treatment.Diabetologia 32: 434–9, 1989.
95. Mogensen CE: Early glomerular hyperfiltration in insu-lin-dependent diabetics and late nephropathy. Scand JClin Lab Invest 46: 201–6, 1986.
96. Rudberg S, Persson B, Dahlquist G: Increased glomerularfiltration rate as a predictor of diabetic nephropathy – an8-year prospective study. Kidney Int 41: 822–8, 1992.
97. Yip JW, Jones SL, Wiseman MJ, Hill C, Viberti G: Glom-erular hyperfiltration in the prediction of nephropathyin IDDM: a 10-year follow-up study. Diabetes 45: 1729–33, 1996.
98. Brenner BM, Garcia DL, Anderson S: Glomeruli andblood pressure. Less of one, more the other? Am JHypertens 1: 335–47, 1988.
99. Rossing P, Tarnow L, Nielsen FS, Hansen BV, BrennerBM, Parving HH: Low birth weight. A risk factor fordevelopment of diabetic nephropathy? Diabetes 44:1405–7, 1995.
100. Nelson RG, Morgenstern H, Bennett PH: Birth weightand renal disease in Pima Indians with type 2 diabetesmellitus. Am J Epidemiol 148: 650–6, 1998.
101. Rossing P, Tarnow L, Nielsen FS, Boelskifte S, BrennerBM, Parving HH: Short stature and diabetic nephropa-thy. BMJ 310: 296–7, 1995.
102. Nielsen FS, Gall MA, Parving HH: Acquired oligoneph-ropathy and diabetic nephropathy. Am J Kidney Dis 26:898–903, 1995.
103. Silveiro SP, da Costa LA, Beck MO, Gross JL: Urinaryalbumin excretion rate and glomerular filtration rate insingle-kidney type 2 diabetic patients. Diabetes Care 21:1521–4, 1998.
104. Mathiesen ER, Ronn B, Jensen T, Storm B, Deckert T:Relationship between blood pressure and urinary albu-min excretion in development of microalbuminuria. Dia-betes 39: 245–9, 1990.
105. Anonymous: Risk factors for development of microalbu-minuria in insulin dependent diabetic patients: a cohortstudy. Microalbuminuria Collaborative Study Group,United Kingdom. BMJ 306: 1235–9, 1993.
106. Nørgaard K, Feldt-Rasmussen B, Borch-Johnsen K,Saelan H, Deckert T: Prevalence of hypertension in type1 (insulin-dependent) diabetes mellitus. Diabetologia 33:407–10, 1990.
107. Flyvbjerg A, Gronbaek H, Bak M, Nielsen B, Christian-sen T, Hill C, Logan A, Orskov H: Diabetic kidney dis-ease: the role of growth factors. Nephrol Dial Transplant13: 1104–7, 1998.
108. Yokoyama H, Deckert T: Central role of TGF-beta inthe pathogenesis of diabetic nephropathy and macrovas-cular complications: a hypothesis. Diabet Med 13: 313–20, 1996.
109. Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I,Tomino Y, Koide H: mRNA expression of growth fac-tors in glomeruli from diabetic rats. Diabetes 42: 450–6, 1993.
110. Ziyadeh FN, Sharma K, Ericksen M, Wolf G: Stimula-tion of collagen gene expression and protein synthesis inmurine mesangial cells by high glucose is mediated byautocrine activation of transforming growth factor-beta.J Clin Invest 93: 536–42, 1994.
111. Kagami S, Border WA, Miller DE, Noble NA: Angio-tensin II stimulates extracellular matrix protein synthe-sis through induction of transforming growth factor-betaexpression in rat glomerular mesangial cells. J Clin In-vest 93: 2431–7, 1994.
112. Throckmorton DC, Brogden AP, Min B, Rasmussen H,Kashgarian M: PDGF and TGF-beta mediate collagenproduction by mesangial cells exposed to advancedglycosylation end products. Kidney Int 48: 111–7, 1995.
113. Isaka Y, Fujiwara Y, Ueda N, Kaneda Y, Kamada T, ImaiE: Glomerulosclerosis induced by in vivo transfection oftransforming growth factor-beta or platelet-derivedgrowth factor gene into the rat kidney. J Clin Invest 92:2597–601, 1993.
114. Lei HH, Perneger TV, Klag MJ, Whelton PK, Coresh J:Familial aggregation of renal disease in a population-based case-control study. J Am Soc Nephrol 9: 1270–6,1998.
115. Freedman BI, Spray BJ, Tuttle AB, Buckalew VM, Jr.:The familial risk of end-stage renal disease in AfricanAmericans. Am J Kidney Dis 21: 387–93, 1993.
116. Kalter-Leibovici O, Van Dyk DJ, Leibovici L, Loya N,Erman A, Kremer I, Boner G, Rosenfeld JB, Karp M,Laron Z: Risk factors for development of diabetic neph-ropathy and retinopathy in Jewish IDDM patients. Dia-betes 40: 204–10, 1991.
117. Nelson RG, Kunzelman CL, Pettitt DJ, Saad MF, BennettPH, Knowler WC: Albuminuria in type 2 (non-insulin-dependent) diabetes mellitus and impaired glucose tol-erance in Pima Indians. Diabetologia 32: 870–6, 1989.
118. Cowie CC, Port FK, Wolfe RA, Savage PJ, Moll PP,Hawthorne VM: Disparities in incidence of diabetic end-stage renal disease according to race and type of diabetes.N Engl J Med 321: 1074–9, 1989.
119. Haffner SM, Mitchell BD, Pugh JA, Stern MP, KozlowskiMK, Hazuda HP, Patterson JK, Klein R: Proteinuria inMexican Americans and non-Hispanic whites withNIDDM. Diabetes Care 12: 530–6, 1989.
120. Borch-Johnsen K, Norgaard K, Hommel E, MathiesenER, Jensen JS, Deckert T, Parving HH: Is diabetic neph-ropathy an inherited complication? Kidney Int 41: 719–22, 1992.
121. Quinn M, Angelico MC, Warram JH, Krolewski AS:Familial factors determine the development of diabeticnephropathy in patients with IDDM. Diabetologia 39:940–5, 1996.
122. Anonymous: Clustering of long-term complications infamilies with diabetes in the diabetes control and com-plications trial. The Diabetes Control and ComplicationsTrial Research Group. Diabetes 46: 1829–39, 1997.
123. Fioretto P, Steffes MW, Barbosa J, Rich SS, Miller ME,Mauer M: Is diabetic nephropathy inherited? Studies ofglomerular structure in type 1 diabetic sibling pairs.Diabetes 48: 865–9, 1999.
124. Pettitt DJ, Saad MF, Bennett PH, Nelson RG, KnowlerWC: Familial predisposition to renal disease in two gen-erations of Pima Indians with type 2 (non-insulin-de-pendent) diabetes mellitus. Diabetologia 33: 438–43,1990.
125. Freedman BI, Tuttle AB, Spray BJ: Familial predisposi-tion to nephropathy in African-Americans with non-in-sulin-dependent diabetes mellitus. Am J Kidney Dis 25:710–3, 1995.
126. Faronato PP, Maioli M, Tonolo G, Brocco E, Noventa F,Piarulli F, Abaterusso C, Modena F, de Bigontina G, Velussi
References

47
Diabetic nephropathy
M, Inchiostro S, Santeusanio F, Bueti A, Nosadini R: Clus-tering of albumin excretion rate abnormalities in Cauca-sian patients with NIDDM. The Italian NIDDM Nephr-opathy Study Group. Diabetologia 40: 816–23, 1997.
127. Canani LH, Gerchman F, Gross JL: Familial clusteringof diabetic nephropathy in Brazilian type 2 diabetic pa-tients. Diabetes 48: 909–13, 1999.
128. Gruden G, Cavallo-Perin P, Olivetti C, Repetti E, SivieriR, Bruno A, Pagano G: Albumin excretion rate levels innon-diabetic offspring of NIDDM patients with andwithout nephropathy. Diabetologia 38: 1218–22, 1995.
129. Strojek K, Grzeszczak W, Morawin E, Adamski M, LackaB, Rudzki H, Schmidt S, Keller C, Ritz E: Nephropathyof type II diabetes: evidence for hereditary factors? Kid-ney Int 51: 1602–7, 1997.
130. Forsblom CM, Kanninen T, Lehtovirta M, Saloranta C,Groop LC: Heritability of albumin excretion rate in fami-lies of patients with Type II diabetes. Diabetologia 42:1359–66, 1999.
131. Viberti GC, Keen H, Wiseman MJ: Raised arterial pres-sure in parents of proteinuric insulin dependent diabet-ics. BMJ 295: 515–7, 1987.
132. Krolewski AS, Canessa M, Warram JH, Laffel LM,Christlieb AR, Knowler WC, Rand LI: Predispositionto hypertension and susceptibility to renal disease in in-sulin-dependent diabetes mellitus. N Engl J Med 318:140–5, 1988.
133. Barzilay J, Warram JH, Bak M, Laffel LM, Canessa M,Krolewski AS: Predisposition to hypertension: risk fac-tor for nephropathy and hypertension in IDDM. KidneyInt 41: 723–30, 1992.
134. Jensen JS, Mathiesen ER, Norgaard K, Hommel E,Borch-Johnsen K, Funder J, Brahm J, Parving HH,Deckert T: Increased blood pressure and erythrocyte so-dium/lithium countertransport activity are not inheritedin diabetic nephropathy. Diabetologia 33: 619–24, 1990.
135. Walker JD, Tariq T, Viberti G: Sodium-lithiumcountertransport activity in red cells of patients withinsulin dependent diabetes and nephropathy and theirparents. BMJ 301: 635–8, 1990.
136. Earle K, Walker J, Hill C, Viberti G: Familial clusteringof cardiovascular disease in patients with insulin-depen-dent diabetes and nephropathy. N Engl J Med 326: 673–7, 1992.
137. Freire MB, Ferreira SR, Vivolo MA, Oliveira JM, ZanellaMT: Familial hypertension and albuminuria in normo-tensive type I diabetic patients. Hypertension 23: 1256–8, 1994.
138. Anonymous: Effect of intensive therapy on the develop-ment and progression of diabetic nephropathy in theDiabetes Control and Complications Trial. The DiabetesControl and Complications (DCCT) Research Group.Kidney Int 47: 1703–20, 1995.
139. Canessa M, Adragna N, Solomon HS, Connolly TM,Tosteson DC: Increased sodium-lithium countertransportin red cells of patients with essential hypertension. NEngl J Med 302: 772–6, 1980.
140. Woods JW, Falk RJ, Pittman AW, Klemmer PJ, WatsonBS, Namboodiri K: Increased red-cell sodium-lithiumcountertransport in normotensive sons of hypertensiveparents. N Engl J Med 306: 593–5, 1982.
141. Mangili R, Bending JJ, Scott G, Li LK, Gupta A, VibertiG: Increased sodium-lithium countertransport activityin red cells of patients with insulin-dependent diabetesand nephropathy. N Engl J Med 318: 146–50, 1988.
142. Trevisan R, Li LK, Messent J, Tariq T, Earle K, WalkerJD, Viberti G: Na+/H+ antiport activity and cell growthin cultured skin fibroblasts of IDDM patients with neph-ropathy. Diabetes 41: 1239–46, 1992.
143. Nørgaard K, Mathiesen ER, Hommel E, Jensen JS,Parving HH: Lack of familial predisposition to cardio-vascular disease in type 1 (insulin-dependent) diabeticpatients with nephropathy. Diabetologia 34: 370–2,1991.
144. Groop L, Forsblom C, Lehtovirta M, Tuomi T, KarankoS, Nissen M, Ehrnstrom BO, Forsen B, Isomaa B, SnickarsB, Taskinen MR: Metabolic consequences of a family his-tory of NIDDM (the Botnia study): evidence for sex-spe-cific parental effects. Diabetes 45: 1585–93, 1996.
145. Haffner SM, Stern MP, Hazuda HP, Mitchell BD,Patterson JK, Ferrannini E: Parental history of diabetesis associated with increased cardiovascular risk factors.Arteriosclerosis 9: 928–33, 1989.
146. Yip J, Mattock MB, Morocutti A, Sethi M, Trevisan R,Viberti G: Insulin resistance in insulin-dependent dia-betic patients with microalbuminuria. Lancet 342: 883–7, 1993.
147. Mäkimattila S, Virkamäki A, Groop PH, Cockcroft J,Utriainen T, Fagerudd J, Yki-Järvinen H: Chronic hy-perglycemia impairs endothelial function and insulinsensitivity via different mechanisms in insulin-dependentdiabetes mellitus. Circulation 94: 1276–82, 1996.
148. Groop PH, Elliott T, Ekstrand A, Franssila-Kallunki A,Friedman R, Viberti GC, Taskinen MR: Multiple lipo-protein abnormalities in type I diabetic patients withrenal disease. Diabetes 45: 974–9, 1996.
149. Fliser D, Pacini G, Engelleiter R, Kautzky-Willer A,Prager R, Franek E, Ritz E: Insulin resistance andhyperinsulinemia are already present in patients withincipient renal disease. Kidney Int 53: 1343–7, 1998.
150. Groop L, Ekstrand A, Forsblom C, Widen E, Groop PH,Teppo AM, Eriksson J: Insulin resistance, hypertensionand microalbuminuria in patients with type 2 (non-in-sulin-dependent) diabetes mellitus. Diabetologia 36:642–7, 1993.
151. Stuhldreher WL, Becker DJ, Drash AL, Ellis D, KullerLH, Wolfson SK, Orchard TJ: The association of waist/hip ratio with diabetes complications in an adult IDDMpopulation. J Clin Epidemiol 47: 447–56, 1994.
152. McCance DR, Hanson RL, Pettitt DJ, Jacobsson LT,Bennett PH, Bishop DT, Knowler WC: Diabetic nephr-opathy: a risk factor for diabetes mellitus in offspring.Diabetologia 38: 221–6, 1995.
153. Yip J, Mattock M, Sethi M, Morocutti A, Viberti G: In-sulin resistance in family members of insulin-dependentdiabetic patients with microalbuminuria [letter]. Lancet341: 369–70, 1993.
154. Fogarty DG, Hanna LS, Wantman M, Warram JH,Krolewski AS, Rich SS: Segregation analysis of urinaryalbumin excretion in families with type 2 diabetes. Dia-betes 49: 1057–63, 2000.
155. Imperatore G, Knowler WC, Pettitt DJ, Kobes S, BennettPH, Hanson RL: Segregation analysis of diabetic nephr-opathy in Pima Indians. Diabetes 49: 1049–56, 2000.
156. Krolewski AS: Genetics of diabetic nephropathy: evidencefor major and minor gene effects. Kidney Int 55: 1582–96, 1999.
157. Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, CorvolP, Soubrier F: An insertion/deletion polymorphism in theangiotensin I-converting enzyme gene accounting for half
References

Johan Fagerudd
48
the variance of serum enzyme levels. J Clin Invest 86:1343–6, 1990.
158. Fujisawa T, Ikegami H, Kawaguchi Y, Hamada Y, UedaH, Shintani M, Fukuda M, Ogihara T: Meta-analysis ofassociation of insertion/deletion polymorphism of angio-tensin I-converting enzyme gene with diabetic nephr-opathy and retinopathy. Diabetologia 41: 47–53, 1998.
159. Tarnow L, Gluud C, Parving HH: Diabetic nephropathyand the insertion/deletion polymorphism of the angio-tensin-converting enzyme gene. Nephrol Dial Transplant13: 1125–30, 1998.
160. Moczulski DK, Rogus JJ, Antonellis A, Warram JH,Krolewski AS: Major susceptibility locus for nephropa-thy in type 1 diabetes on chromosome 3q: results of noveldiscordant sib-pair analysis. Diabetes 47: 1164–9, 1998.
161. Jacobson HR: Chronic renal failure: pathophysiology.Lancet 338: 419–23, 1991.
162. Pedrini MT, Levey AS, Lau J, Chalmers TC, Wang PH:The effect of dietary protein restriction on the progres-sion of diabetic and nondiabetic renal diseases: a meta-analysis. Ann Intern Med 124: 627–32, 1996.
163. Ciavarella A, Di Mizio G, Stefoni S, Borgnino LC, VanniniP: Reduced albuminuria after dietary protein restrictionin insulin-dependent diabetic patients with clinical neph-ropathy. Diabetes Care 10: 407–13, 1987.
164. Zeller K, Whittaker E, Sullivan L, Raskin P, JacobsonHR: Effect of restricting dietary protein on the progres-sion of renal failure in patients with insulin-dependentdiabetes mellitus. N Engl J Med 324: 78–84, 1991.
165. Dullaart RP, Beusekamp BJ, Meijer S, van Doormaal JJ,Sluiter WJ: Long-term effects of protein-restricted dieton albuminuria and renal function in IDDM patientswithout clinical nephropathy and hypertension. Diabe-tes Care 16: 483–92, 1993.
166. Barsotti G, Ciardella F, Morelli E, Cupisti A,Mantovanelli A, Giovannetti S: Nutritional treatmentof renal failure in type 1 diabetic nephropathy. ClinNephrol 29: 280–7, 1988.
167. Walker JD, Bending JJ, Dodds RA, Mattock MB,Murrells TJ, Keen H, Viberti GC: Restriction of dietaryprotein and progression of renal failure in diabetic neph-ropathy. Lancet 2: 1411–5, 1989.
168. Groop PH: Lipidaemia and diabetic renal disease, Thekidney and hypertension in diabetes mellitus, 3rd edi-tion Edition. Edited by Mogensen CE. Boston, KluwerAcademic Publishers, 1996, pp 307–20.
169. Ravid M, Brosh D, Ravid-Safran D, Levy Z, RachmaniR: Main risk factors for nephropathy in type 2 diabetesmellitus are plasma cholesterol levels, mean blood pres-sure, and hyperglycemia. Arch Intern Med 158: 998–1004, 1998.
170. Krolewski AS, Warram JH, Christlieb AR: Hypercho-lesterolemia – a determinant of renal function loss anddeaths in IDDM patients with nephropathy. Kidney IntSuppl 45: S125–31, 1994.
171. Breyer JA, Bain RP, Evans JK, Nahman NS, Jr., LewisEJ, Cooper M, McGill J, Berl T: Predictors of the pro-gression of renal insufficiency in patients with insulin-dependent diabetes and overt diabetic nephropathy. TheCollaborative Study Group. Kidney Int 50: 1651–8,1996.
172. Mulec H, Johnsen SA, Wiklund O, Bjorck S: Choles-terol: a renal risk factor in diabetic nephropathy? Am JKidney Dis 22: 196–201, 1993.
173. Inman SR, Stowe NT, Cressman MD, Brouhard BH,
Nally JV, Jr., Satoh S, Satodate R, Vidt DG: Lovastatinpreserves renal function in experimental diabetes. Am JMed Sci 317: 215–21, 1999.
174. Hommel E, Andersen P, Gall MA, Nielsen F, Jensen B,Rossing P, Dyerberg J, Parving HH: Plasma lipoproteinsand renal function during simvastatin treatment in dia-betic nephropathy. Diabetologia 35: 447–51, 1992.
175. Chase HP, Garg SK, Marshall G, Berg CL, Harris S, Jack-son WE, Hamman RE: Cigarette smoking increases therisk of albuminuria among subjects with type I diabetes.JAMA 265: 614–7, 1991.
176. Sawicki PT, Didjurgeit U, Muhlhauser I, Bender R,Heinemann L, Berger M: Smoking is associated withprogression of diabetic nephropathy. Diabetes Care 17:126–31, 1994.
177. Orth SR, Ritz E, Schrier RW: The renal risks of smok-ing. Kidney Int 51: 1669–77, 1997.
178. Wahren J, Johansson BL, Wallberg-Henriksson H: DoesC-peptide have a physiological role? Diabetologia 37Suppl 2: S99–107, 1994.
179. Eff C, Faber O, Deckert T: Persistent insulin secretion,assessed by plasma C-peptide estimation in long-termjuvenile diabetics with a low insulin requirement. Dia-betologia 15: 169–72, 1978.
180. Anonymous: Human experimentation. Code of ethics ofthe World Medical Association. Declaration of Helsinki.BMJ 2: 177, 1964.
181. Staessen JA, Bieniaszewski L, O’Brien ET, Fagard R:Special feature: what is a normal blood pressure in am-bulatory monitoring? Nephrol Dial Transplant 11: 241–5, 1996.
182. Pickering T: Recommendations for the use of home (self)and ambulatory blood pressure monitoring. AmericanSociety of Hypertension Ad Hoc Panel. Am J Hypertens9: 1–11, 1996.
183. Mogensen CE, Chachati A, Christensen CK, Close CF,Deckert T, Hommel E, Kastrup J, Lefebvre P, MathiesenER, Feldt-Rasmussen B: Microalbuminuria: an earlymarker of renal involvement in diabetes. Uremia Invest9: 85–95, 1985.
184. Anonymous: Diabetes mellitus. Report of a WHO StudyGroup. World Health Organ Tech Rep Ser 727: 1–113,1985.
185. Akinmokun A, Selby PL, Ramaiya K, Alberti KG: Theshort insulin tolerance test for determination of insulinsensitivity: a comparison with the euglycaemic clamp.Diabet Med 9: 432–7, 1992.
186. Feldt-Rasmussen B, Dinesen B, Deckert M: Enzymeimmunoassay: an improved determination of urinary al-bumin in diabetics with incipient nephropathy. Scand JClin Lab Invest 45: 539–44, 1985.
187. Tuomilehto J, Karvonen M, Padaiga Z, Tuomilehto-WolfE, Kohtamaki K: Epidemiology of insulin-dependentdiabetes mellitus around the Baltic Sea. The DIABALTStudy Group. Horm Metab Res 28: 340–3, 1996.
188. Tuomilehto J, Borch-Johnsen K, Molarius A,Jormanainen V, Lounamaa R, Gronhagen-Riska C,Reunanen A, Sarti C: The unchanging incidence of hos-pitalization for diabetic nephropathy in a population-based cohort of IDDM patients in Finland. Diabetes Care20: 1081–6, 1997.
189. O’Brien E, Mee F, Atkins N, O’Malley K: Accuracy ofthe SpaceLabs 90207 determined by the British Hyper-tension Society protocol. J Hypertens 9: 573–4, 1991.
190. O’Brien E, Petrie J, Littler W, de Swiet M, Padfield PL,
References

49
Diabetic nephropathy
O’Malley K, Jamieson M, Altman D, Bland M, AtkinsN: The British Hypertension Society protocol for theevaluation of automated and semi-automated blood pres-sure measuring devices with special reference to ambula-tory systems. J Hypertens 8: 607–19, 1990.
191. Gosse P, Ansoborlo P, Lemetayer P, Clementy J: Day-time and nighttime ambulatory blood pressures shouldbe calculated over the true sleep/waking cycle and notover arbitrary periods. Am J Hypertens 9: 269–72, 1996.
192. Sokolow M, Werdegar D, Kain HK, Hinman AT: Rela-tionship between level of blood pressure measured casu-ally and by portable recorders and severity of complica-tions in essential hypertension. Circulation 34: 279–98,1966.
193. Shimada K, Kawamoto A, Matsubayashi K, Ozawa T:Silent cerebrovascular disease in the elderly. Correlationwith ambulatory pressure. Hypertension 16: 692–9,1990.
194. Giaconi S, Levanti C, Fommei E, Innocenti F, SeghieriG, Palla L, Palombo C, Ghione S: Microalbuminuria andcasual and ambulatory blood pressure monitoring in nor-motensives and in patients with borderline and mild es-sential hypertension. Am J Hypertens 2: 259–61, 1989.
195. Verdecchia P, Porcellati C, Schillaci G, Borgioni C, CiucciA, Battistelli M, Guerrieri M, Gatteschi C, Zampi I,Santucci A: Ambulatory blood pressure. An independentpredictor of prognosis in essential hypertension. Hyper-tension 24: 793–801, 1994.
196. Mancia G, Zanchetti A, Agabiti-Rosei E, Benemio G,De Cesaris R, Fogari R, Pessina A, Porcellati C, RappelliA, Salvetti A, Trimarco B: Ambulatory blood pressure issuperior to clinic blood pressure in predicting treatment-induced regression of left ventricular hypertrophy.SAMPLE Study Group. Study on Ambulatory Monitor-ing of Blood Pressure and Lisinopril Evaluation. Circu-lation 95: 1464–70, 1997.
197. MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, NeatonJ, Abbott R, Godwin J, Dyer A, Stamler J: Blood pres-sure, stroke, and coronary heart disease. Part 1, Prolongeddifferences in blood pressure: prospective observationalstudies corrected for the regression dilution bias. Lancet335: 765–74, 1990.
198. Hirst S, Phillips DI, Vines SK, Clark PM, Hales CN:Reproducibility of the short insulin tolerance test. DiabetMed 10: 839–42, 1993.
199. Lindsay RS, Little J, Jaap AJ, Padfield PL, Walker JD,Hardy KJ: Diabetic nephropathy is associated with anincreased familial risk of stroke. Diabetes Care 22: 422–5, 1999.
200. Tarnow L, Rossing P, Nielsen FS, Fagerudd JA, PoirierO, Parving HH: Cardiovascular morbidity and earlymortality cluster in parents of type 1 diabetic patientswith diabetic nephropathy. Diabetes Care 23: 30–3, 2000.
201. Molitch ME, Steffes MW, Cleary PA, Nathan DM:Baseline analysis of renal function in the Diabetes Con-trol and Complications Trial. The Diabetes Control andComplications Trial Research Group. Kidney Int 43:668–74, 1993.
202. Rudberg S, Stattin EL, Dahlquist G: Familial and peri-natal risk factors for micro- and macroalbuminuria inyoung IDDM patients. Diabetes 47: 1121–6, 1998.
203. Roglic G, Colhoun HM, Stevens LK, Lemkes HH, ManesC, Fuller JH: Parental history of hypertension and pa-rental history of diabetes and microvascular complica-tions in insulin-dependent diabetes mellitus: the
EURODIAB IDDM Complications Study. Diabet Med15: 418–26, 1998.
204. Fagerudd JA, Pettersson-Fernholm KJ, Grönhagen-RiskaC, Groop PH: The impact of a family history of Type II(non-insulin-dependent) diabetes mellitus on the risk ofdiabetic nephropathy in patients with Type I (insulin-dependent) diabetes mellitus. Diabetologia 42: 519–26,1999.
205. De Cosmo S, Bacci S, Piras GP, Cignarelli M, PlacentinoG, Margaglione M, Colaizzo D, Di Minno G, GiorginoR, Liuzzi A, Viberti GC: High prevalence of risk factorsfor cardiovascular disease in parents of IDDM patientswith albuminuria. Diabetologia 40: 1191–6, 1997.
206. Verhage B, Vervoort G, Wolkotte C, Elving LD, WetzelsJF, Willems H, Smits P, Verbeek AL, Berden JH: Preva-lence of "syndrome X" features in parents of type 1 dia-betic patients with or without nephropathy. Diabetes Care22: 1048–52, 1999.
207. Fagerudd JA, Tarnow L, Jacobsen P, Stenman S, NielsenFS, Pettersson-Fernholm KJ, Grönhagen-Riska C,Parving HH, Groop PH: Predisposition to essential hy-pertension and development of diabetic nephropathy inIDDM patients. Diabetes 47: 439–44, 1998.
208. Hannedouche TP, Marques LP, Natov S, Delgado AG,Boitard C, Lacour B, Grunfeld JP: Renal abnormalitiesin normotensive insulin-dependent diabetic offspring ofhypertensive parents. Hypertension 19: 378–84, 1992.
209. Bianchi G, Cusi D, Barlassina C, Lupi GP, Ferrari P,Picotti GB, Gatti M, Polli E: Renal dysfunction as a pos-sible cause of essential hypertension in predisposed sub-jects. Kidney Int 23: 870–5, 1983.
210. Erbey JR, Kuller LH, Becker DJ, Orchard TJ: The asso-ciation between a family history of type 2 diabetes andcoronary artery disease in a type 1 diabetes population.Diabetes Care 21: 610–4, 1998.
211. DeFronzo RA: Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM.Diabetes 37: 667–87, 1988.
212. Groop LC, Tuomi T: Non-insulin-dependent diabetesmellitus – a collision between thrifty genes and an afflu-ent society. Ann Med 29: 37–53, 1997.
213. Ekstrand AV, Groop PH, Grönhagen-Riska C: Insulinresistance precedes microalbuminuria in patients withinsulin-dependent diabetes mellitus. Nephrol Dial Trans-plant 13: 3079–83, 1998.
214. Nosadini R, Solini A, Velussi M, Muollo B, Frigato F,Sambataro M, Cipollina MR, De Riva F, Brocco E,Crepaldi G: Impaired insulin-induced glucose uptake byextrahepatic tissue is hallmark of NIDDM patients whohave or will develop hypertension and microalbumin-uria. Diabetes 43: 491–9, 1994.
215. Yki-Järvinen H: Acute and chronic effects ofhyperglycaemia on glucose metabolism. Diabetologia 33:579–85, 1990.
216. Zimmet P, Whitehouse S, Alford F, Chisholm D: Therelationship of insulin response to a glucose stimulus overa wide range of glucose tolerance. Diabetologia 15: 23–7, 1978.
217. Nestler JE, Barlascini CO, Tetrault GA, Fratkin MJ, CloreJN, Blackard WG: Increased transcapillary escape rateof albumin in nondiabetic men in response tohyperinsulinemia. Diabetes 39: 1212–7, 1990.
218. Scholey JW, Meyer TW: Control of glomerular hyper-tension by insulin administration in diabetic rats. J ClinInvest 83: 1384–9, 1989.
References

Johan Fagerudd
50
219. Tucker BJ, Anderson CM, Thies RS, Collins RC, BlantzRC: Glomerular hemodynamic alterations during acutehyperinsulinemia in normal and diabetic rats. KidneyInt 42: 1160–8, 1992.
220. Ferrannini E, Buzzigoli G, Bonadonna R, Giorico MA,Oleggini M, Graziadei L, Pedrinelli R, Brandi L,Bevilacqua S: Insulin resistance in essential hypertension.N Engl J Med 317: 350–7, 1987.
221. Reaven GM, Lerner RL, Stern MP, Farquhar JW: Role ofinsulin in endogenous hypertriglyceridemia. J Clin In-vest 46: 1756–67, 1967.
222. Johansson BL, Kernell A, Sjoberg S, Wahren J: Influ-ence of combined C-peptide and insulin administrationon renal function and metabolic control in diabetes type1. J Clin Endocrinol Metab 77: 976–81, 1993.
223. Kuhl C: Etiology and pathogenesis of gestational diabe-tes. Diabetes Care 21 Suppl 2: B19–26, 1998.
224. Nelson RG, Morgenstern H, Bennett PH: Intrauterinediabetes exposure and the risk of renal disease in diabeticPima Indians. Diabetes 47: 1489–93, 1998.
225. Smith FG, Lumbers ER: Effects of maternal hyperglyce-mia on fetal renal function in sheep. Am J Physiol 255:F11–4, 1988.
226. Damsgaard EM, Froland A, Jorgensen OD, MogensenCE: Microalbuminuria as predictor of increased mortal-ity in elderly people. BMJ 300: 297–300, 1990.
227. Yudkin JS, Forrest RD, Jackson CA: Microalbuminuriaas predictor of vascular disease in non-diabetic subjects.Islington Diabetes Survey. Lancet 2: 530–3, 1988.
228. Metcalf PA, Baker JR, Scragg RK, Dryson E, Scott AJ,Wild CJ: Microalbuminuria in a middle-aged workforce.Effect of hyperglycemia and ethnicity. Diabetes Care 16:1485–93, 1993.
229. Forsblom CM, Eriksson JG, Ekstrand AV, Teppo AM,Taskinen MR, Groop LC: Insulin resistance and abnor-mal albumin excretion in non-diabetic first-degree rela-tives of patients with NIDDM. Diabetologia 38: 363–9,1995.
230. Gerber LM, Shmukler C, Alderman MH: Differences inurinary albumin excretion rate between normotensive andhypertensive, white and nonwhite subjects. Arch InternMed 152: 373–7, 1992.
231. Nelson RG, Pettitt DJ, de Courten MP, Hanson RL,Knowler WC, Bennett PH: Parental hypertension andproteinuria in Pima Indians with NIDDM. Diabetologia39: 433–8, 1996.
232. Fogarty DG, Rich SS, Hanna L, Warram JH, KrolewskiAS: Urinary albumin excretion in families with type 2
diabetes is heritable and genetically correlated to bloodpressure. Kidney Int 57: 250–7, 2000.
233. Feldt-Rasmussen B, Baker L, Deckert T: Exercise as aprovocative test in early renal disease in type 1 (insulin-dependent) diabetes: albuminuric, systemic and renalhaemodynamic responses. Diabetologia 28: 389–96,1985.
234. Torffvit O, Castenfors J, Agardh CD: A study of exer-cise-induced microalbuminuria in type I (insulin-depen-dent) diabetes mellitus. Scand J Urol Nephrol 25: 39–43, 1991.
235. O’Brien SF, Watts GF, Powrie JK, Shaw KM: Exercisetesting as a long-term predictor of the development ofmicroalbuminuria in normoalbuminuric IDDM patients.Diabetes Care 18: 1602–5, 1995.
236. Degnbol B, Green A: Diabetes mellitus among first- andsecond-degree relatives of early onset diabetics. Ann HumGenet 42: 25–47, 1978.
237. Anonymous: UK Prospective Diabetes Study (UKPDS).X. Urinary albumin excretion over 3 years in diet-treatedtype 2, (non-insulin-dependent) diabetic patients, andassociation with hypertension, hyperglycaemia andhypertriglyceridaemia. Diabetologia 36: 1021–9, 1993.
238. Mykkänen L, Haffner SM, Kuusisto J, Pyörälä K, LaaksoM: Microalbuminuria precedes the development ofNIDDM. Diabetes 43: 552–7, 1994.
239. Anonymous: Microalbuminuria in type I diabetic pa-tients. Prevalence and clinical characteristics. Microal-buminuria Collaborative Study Group. Diabetes Care 15:495–501, 1992.
240. Fioretto P, Mauer M, Brocco E, Velussi M, Frigato F,Muollo B, Sambataro M, Abaterusso C, Baggio B,Crepaldi G, Nosadini R: Patterns of renal injury inNIDDM patients with microalbuminuria. Diabetologia39: 1569–76, 1996.
241. Schmitz A, Vaeth M: Microalbuminuria: a major riskfactor in non-insulin-dependent diabetes. A 10-year fol-low-up study of 503 patients. Diabet Med 5: 126–34,1988.
242. MacMahon B: Epidemiologic approaches to family re-semblance, Genetic Epidemiology. Edited by Morton NE,Chung CS. New York, Academic Press, 1978, pp 3–11.
243. Khoury MJ, Beaty TH, Liang KY: Can familial aggrega-tion of disease be explained by familial aggregation ofenvironmental risk factors? Am J Epidemiol 127: 674–83, 1988.
244. Mauer SM, Steffes MW, Brown DM: The kidney in dia-betes. Am J Med 70: 603–12, 1981.
References