Embed Size (px)
Citation preview

www.elsevier.com/locate/cplett
Chemical Physics Letters 404 (2005) 49–52
Direct observation of the k3P state of 12C18O
Jacob Baker a,*, Francoise Launay b
a Division of Environmental Health and Risk Management, School of Geography, Earth and Environmental Sciences,
University of Birmingham, Edgbaston, Birmingham B15 2TT, UKb Observatoire de Paris, Section de Meudon, LERMA, UMR 8112 du CNRS, 92195 Meudon Cedex, France
Received 27 October 2004; in final form 27 October 2004
Abstract
A weak rotationally resolved absorption band of 12C18O has been identified from photographic VUV spectra and assigned to the
k3P (v = 3) X1R+ (v = 0) forbidden transition. The experimentally determined band origin and upper state rotational constant are
in close agreement to that derived from isotopic scaling. A consideration of the intensity structure of the band suggests that the band
gains its intensity mainly through a k3P (v = 3)–E1P (v = 0) interaction. This is the first direct observation of the k3P state for12C18O.
� 2005 Elsevier B.V. All rights reserved.
1. Introduction
Carbon monoxide (CO) is the most abundant mole-
cule after molecular hydrogen in interstellar space and
is widely used as a tracer for mapping out the density
and flow of molecules in the interstellar medium [1].
Measurements of column densities of the different iso-
topomers of CO is also used to map isotopic ratios of
elemental carbon and oxygen [2]. These ratios are ulti-
mately linked to stellar origins and hence provide infor-mation on the stellar sources and evolution of the
interstellar medium [3]. To be used reliably as a tracer,
it is necessary to understand all the processes governing
the CO abundance, including processes giving rise to
isotopic fractionation.
Photodestruction of CO, which occurs in the 90–
112 nm VUV region, is one of the most important
processes governing the CO abundance and since thisoccurs mainly through absorption into rotationally
resolved bands followed by predissociation, radiation
0009-2614/$ - see front matter � 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.cplett.2004.11.138
* Corresponding author. Fax: +44 121 414 3078.
E-mail address: [email protected] (J. Baker).
shielding and isotope shifts are expected to lead to isoto-
pic fractionation [4–6]. The modelling studies of VanDishoek and Black [6] show that the E1P–X1R+ (1–0)
band is most effective in contributing to isotopic frac-
tionation. The E1P (v = 1) Rydberg state is in fact per-
turbed by the k3P valence state, which causes local
rotational line shifts and enhanced predissociation rates
[7–9]. The k3P state, which is believed to be strongly
predissociated by a 3P repulsive state, acts as a doorway
state to enhanced dissociation [7]. Forbidden transitionsdirectly to the k3P state from the X1R+ ground state
may also become a significant dissociation source in
the inner regions of dense interstellar clouds where
self-shielding effects diminish the importance of the
stronger absorption bands.
The k3P valence state has previously been directly
observed for 12C16O and 13C16O [8,10–13]. Berden
et al. [12] in 1997 showed that the vibrational labellingof this state in prior studies was incorrect and needed
to be incremented by one unit. In this article we report
on a weak band appearing in the VUV absorption spec-
trum of 12C18O which we assign to the k3P–X1R+ (3–0)
band. This represents the first direct observation of the
k3P state for 12C18O.

50 J. Baker, F. Launay / Chemical Physics Letters 404 (2005) 49–52
2. Experimental
This study made use of the 10.68 m VUV spectro-
graph at the Meudon observatory. Previously, we have
reported on absorption bands corresponding to forbid-
den transitions from the X1R+ (v = 0) ground state tothe v = 1, 3, 4 and 6 vibrational levels of the k3P state
of the normal isotopomer of carbon monoxide 12C16O,
recorded over the pressure range of 0.1–1.5 Torr [8,10].
The strongest of the reported absorption bands is the
k–X (3–0) band. This band has also been identified for
the 13C16O isotope recorded at a pressure of 0.02 Torr
[13].
The absorption spectrum of the rarer isotope 12C18Ohas previously been photographed at a maximum pres-
sure of 0.02 Torr using the spectrograph at Meudon,
as part of a study of singlet–singlet absorption transi-
tions [14,15]. A careful examination of a photographic
plate containing the absorption spectrum of 12C18O at
0.02 Torr revealed seven new faint lines to the lower en-
ergy side of the much stronger E1P–X1R+ (0–0) absorp-
tion band and occurring in the region of the expectedk–X (3–0) band at �107.8 nm.
The experimental details of the spectrograph have
been described in detail elsewhere [8,14]. The 12C18O
gas was purchased from the Commissariat a l�EnergieAtomique with a certified isotopic purity of 97.6%. Cal-
ibration was achieved by recording known atomic emis-
sion lines (Cu II, Ge II, Si II) from a windowless hollow
cathode lamp as well as known lines appearing in the ac-tual 12C18O absorption spectrum. The photographic
plates used to record the spectra have an approximately
logarithmic response to the transmitted light between
the threshold and saturation regions of the plate.
3. Results and discussion
Table 1 gives the line positions and assignments for
the faint absorption band of 12C18O observed at
�107.8 nm. Initial assignments were based on compari-
Table 1
Line positions for the k3P (v = 3)–X1R+ (v = 0) band of 12C18Oa
J QQ O � C RR O � C
0
1 92740.50(40)b 0.11 92744.92(40)b �0.142 92737.87(40) 0.13 92744.92(40)b 0.17
3 92733.57(40) �0.214 92728.15(40) �0.36 92740.50(40)b 0.30
5 92736.02(40) 0.05
6
7 92723.58(40) �0.05a All values are in units of cm�1. Values between parentheses are
estimated errors. O � C signifies observed � calculated value.b Overlapped lines.
sons with the corresponding bands of 12C16O and13C16O [8,13]. Only the two strongest rotational
branches QQ and RR were observed, which correspond
to transitions to the f and e parity levels, respectively,
of the f2 (3P1) spin–orbit component of the k3P state.
Although, the lines were clearly identified on the photo-graphic plate and measured with the Meudon photoelec-
tric comparator [16] they did not appear clearly on
positive prints and so we are unable to show the actual
photographic spectrum here.
Table 2 (last column) gives the results of a rota-
tional fit of the measured line positions. The 3P ex-
cited state rotational energy levels were fitted to the
f2 eigenvalue of the effective 3P Hamiltonian matrixof Brown and Merer [17] while the X1R+ (v = 0)
ground state rotational energy levels were fitted to
the expression F(J) = BJ(J + 1) � D(J(J + 1))2 with
B = 1.8309808 cm�1 and D = 5.554 · 10�6 cm�1 derived
from Ref. [18]. The band origin, T30, and upper state
rotational constant, B3, were varied in the fit, while
other molecular constant were fixed to the correspond-
ing values determined for 13C16O [13]. Table 2 alsopresents the molecular constants for the corresponding
bands of 12C16O and 13C16O for comparison.
To check this k–X (3–0) assignment for 12C18O, the
band origin T30 and upper state rotational constant B3
were estimated via isotopic scaling of the k state equilib-
rium molecular constants of 12C16O derived from previ-
ous VUV absorption studies (Table VI of Ref. [10])
but with the corrected vibrational assignment [12] (seeTable 3). Isotopically scaled molecular parameters can
then be estimated from [19]
T iv0¼ T e � ZPE ðX1RþÞi þ qxeðvþ 1=2Þ� q2xexeðvþ 1=2Þ2 þ q3xeyeðvþ 1=2Þ3;
Biv ¼ q2Be � q3aeðvþ 1=2Þ;
where ZPE (X1R+) is the zero point energy of the ground
electronic state and q ¼ffiffiffiffiffiffiffiffiffil=li
pwith l and li the reduced
Table 2
Molecular constantsa for the k3P (v = 3) state of 12C16O, 13C16O and12C18O
Molecular parameter 12C16O b 13C16O c 12C18O d
T30 92782.67(2) 92742.78(10) 92739.1(3)
B3 1.24405(26) 1.1917(6) 1.1860(36)
D · 106 9.86(80) 9.0 fixed 9.0 fixed
A 30.976(12) 30.95(4) 30.95 fixed
(o + p + q) 0.298(14) 0.31(4) 0.3 fixed
a All molecular constants are in units of cm�1. The numbers in
parenthesis are errors to one standard deviation, in the least significant
figure.b Ref. [8].c Ref. [13].d This work. The standard deviation of the fit was r = 0.22 cm�1.
The error in T30 includes an estimate for the calibration error.

Table 3
Equilibrium molecular constants for the k3P and B1R+ states of12C16O
Molecular parameter k3Pa B1R+b
Te 90968.34(160) 86951.32
xe 846.64 (91) 2084.8
xexe 5.46(11) �24.01xeye �15.60Be 1.29817(105)
ae 0.01531(31)
re (A) 1.3761
All values are in cm�1 except re.a Rederived from Ref. [10] using corrected vibrational numbering.
The vibrational constants were derived from v = 1, 3, 4, 6, levels which
do not show any obvious vibrational perturbation shifts, while the
rotational constants were derived from v = 1, 3, 4 (these level either do
not show any obvious rotational perturbations or have been deper-
turbed). The constants given reproduce the vibrational band origins of12C16O (v = 1, 3, 4, 6) to <0.5 cm�1. The numbers in parenthesis are
errors to one standard deviation and are relatively large due to the
limited degrees of freedom in the fit (one) and large correlations
between the fitted parameters.b Derived from v = 0 to 3 term values [14,21]. The negative values
for the anharmonicity constants xexe and xeye arise from the strong
perturbation of the B1R+ state with the D 01R+ state [21]. The degrees of
freedom in the fit is zero and so no fitting errors are given.
J. Baker, F. Launay / Chemical Physics Letters 404 (2005) 49–52 51
masses of 12C16O and the isotopomer, i, in question. For12C18O, q = 0.97584 and ZPE (X1R+)i = 1055.537 cm�1
(derived from Ref. [13]). The isotopically scaled values
of T30 and B3 calculated for 12C18O are 92740.8 and
1.1864 cm�1, respectively. These compare very closely
* *
* *
* *
05
1
9273092740927509276092770
Transition
* *
* *
* *
9273092740927509276092770
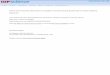
Fig. 1. A simulation of the forbidden k3P (v = 3)–X1R+ (v = 0) absorption b
represented by the dashed horizontal line were observed (see text for details
to the experimentally determined values of 92739.1(3)
and 1.1860(36) cm�1, respectively. Hence these isotopi-
cally scaling calculations confirm the 12C18O k–X (3–0)
band assignment.
Finally, we consider the intensity structure of the
band. Fig. 1 is a simulation of the absorption bandassuming a rotational temperature of T = 298 K and
a Gaussian linewidth of 0.6 cm�1 (FWHM). The line
positions were calculated from upper and lower state
term energy differences, where the term energies were
determined as described above using the upper state
molecular constants given in Table 2 and the known
ground state constants [18]. 3P–1R+ linestrength fac-
tors were taken from Kovacs [20], where the 3P stateis treated as an intermediate Hund�s coupling case (a/b).
In these linestrength formulae there is an adjustable
parameter r = D/E, where D and E correspond, in
the present case, to the effective transition moments
arising from mixing of 1R+ and 1P character into
the 3P state, respectively. For the corresponding band
of both 12C16O and 13C16O a value of r � �0.1 was
determined and is used here. This implies that the k–X (3–0) band gains it intensity mainly through a
k3P–1P interaction. The intensity is plotted on a log-
arithmic scale to simulate the response of the photo-
graphic plate above the threshold.
Only a few rotational lines were clearly visible on
the photographic plate and these are labelled with
asterisks in Fig. 1. It is clear that the absorption band
*
R
Q5
92690927009271092720
Energy (cm )-1
*
RR
10
92690927009271092720
and of 12C18O. Only the asterisked rotational lines above the threshold
).

52 J. Baker, F. Launay / Chemical Physics Letters 404 (2005) 49–52
was close to the detection threshold region and lines
below the dashed horizontal line were below threshold.
The spectrum recorded for 13C16O at 0.02 Torr was
similar but higher J lines were also observed [13].
The intensity simulation of Fig. 1 suggests that we
may have expected to observe a few higher J linesfor 12C18O. There are a few possibilities as to why
these higher J lines (appearing below 92723 cm�1)
were not observed. One possibility is that close to
threshold non-uniformities in the sensitivity of the
photographic plate may be significant. Another possi-
bility is an underlying diffuse band that helps to raise
the blue (higher transition energy) side of the band
above the threshold. The diffuse B1R+–X1R+ (3–0)band would be expected to occur in this energy region
and has been observed for 12C16O but at much higher
pressures [21]. An estimate of the B–X (3–0) band ori-
gin for 12C18O can be obtained via isotopic scaling
(vide supra) of the equilibrium molecular constants
of the B state of 12C16O derived from Refs. [14,21]
(see Table 3 (last column)). The B–X (3–0) band origin
so determined is 92675 ± 45 cm�1, which places it onthe red (lower transition energy) side rather than the
blue side of the k–X (3–0) band, so this diffuse band
does not explain our observation and is probably
too weak to have any effect. The uncertainty in the
given band origin is estimated from the uncertainty
in the experimental band origin of the diffuse B–X
(3–0) band of 12C16O [21].
One other possibility lies with the line strength fac-tors used. As mentioned above the forbidden transition
gains oscillator strength through a k3P–1P perturba-
tion and the line strength factors used assumes that
the perturbing states are widely separated from the
k3P state such that the rotational energy is insignifi-
cant compared to the energy separation of the states
[20]. If the k3P (v = 3) state is perturbed mainly by
the E1P (v = 0) state then this assumption is no longerstrictly valid. The molecular constants of the E1P(v = 0) state of 12C18O are well known and can be used
to determine rovibronic term energy values [15], while
the molecular constants of the k3P (v = 3) state are gi-
ven in the last column of Table 2. From this we deter-
mine an E1P (J = 1, v = 0)–k3P1 (J = 1, v = 3) energy
separation of 189 cm�1 and an E1P (J = 10, v = 0)–
k3P 1 (J = 10, v = 3) energy separation of 264 cm�1.This implies that the higher J line strengths will be
weaker relative to the lower J line strengths as a result
of this increasing energy separation with J. This to-
gether with non-uniformities in the sensitivity of the
plate close to threshold provides an explanation of
the observations.
4. Conclusion
A faint absorption band of 12C18O has been observed
and assigned to the forbidden k3P–X1R+ (3–0) transition
with ameasured band origin of 92739.1 ± 0.3 cm�1. Only
the two strongest branches QQ and RRwere observed cor-responding to transitions to the f- and e-parity levels,
respectively, of the f2 (3P1) spin–orbit component of the
k3P state. A consideration of the intensity structure of
this band indicates that it gains intensity through a k3P(v = 3)–E1P (v = 0) spin–orbit interaction.
Acknowledgements
We are indebted to Francois Rostas for supporting
and encouraging the work. We thank Maurice Benhar-
rous for technical assistance.
References
[1] N. Neininger, M. Guelin, H. Ungerechts, R. Lucas, R. Wielebin-
ski, Nature 395 (1998) 871.
[2] P. Harjunpaa, K. Lehtinen, L.K. Haikala, Astron. Astrophys.
421 (2004) 1087.
[3] A. Heikkila, L.E.B. Johansson, H. Olofsson, Astron. Astrophys.
332 (1998) 493.
[4] A.E. Glassgold, P.J. Huggins, W.D. Langer, Astrophys. J. 290
(1985) 615.
[5] Y.P Viala, C. Letzelter, M. Eidelsberg, F. Rostas, Astron.
Astrophys. 193 (1988) 265.
[6] E.F. Van Dishoeck, J.H. Black, Astrophys. J. 334 (1988) 771.
[7] J. Baker, J.L. Lemaire, S. Couris, A. Vient, D. Malmasson, F.
Rostas, Chem. Phys. 178 (1993) 569.
[8] J. Baker, F. Launay, J. Mol. Spectrosc. 165 (1994) 75.
[9] W. Ubachs, I. Velchev, P. Cacciani, J. Chem. Phys. 113 (2000) 547.
[10] J. Baker, J. Mol. Spectrosc. 167 (1994) 323.
[11] A. Mellinger, C.R. Vidal, J. Chem. Phys. 101 (1994) 104.
[12] G. Berden, R.T. Jongma, D. Van der Zande, G. Meijer, J. Chem.
Phys. 107 (1997) 8303.
[13] J. Baker, F. Launay, J. Mol. Spectrosc. 203 (2000) 196.
[14] M. Eidelsberg, J.-Y. Roncin, A. Le Floch, F. Launay, C.
Letzelter, J. Rostas, J. Mol. Spectrosc. 121 (1987) 309.
[15] M. Eidelsberg, F. Rostas, Astron. Astrophys. 235 (1990) 472.
[16] F. Launay, Proceedings International Conference on Image
Processing Techniques in Astronomy, Utrecht, Reidel, Dordrecht,
1975, p. 265.
[17] J.M. Brown, A.J. Merer, J. Mol. Spectrosc. 74 (1979) 488.
[18] G. Guelachvili, D. De Villeneuve, R. Farrenq, W. Urban, J.
Verges, J. Mol. Spectrosc. 98 (1983) 64.
[19] G. HerzbergSpectra of Diatomic Molecules, vol. 1, Van Nostrand,
New York, 1950.
[20] I. Kovacs, Rotational Structure in the Spectra of Diatomic
Molecules, Hilger, London, 1969.
[21] J. Baker, W.-U.L. Tchang-Brillet, P.S. Julienne, J. Chem. Phys.
102 (1995) 3956.