Embed Size (px)
Citation preview

LETTERS
Defective tryptophan catabolism underliesinflammation in mouse chronic granulomatous diseaseLuigina Romani1, Francesca Fallarino1, Antonella De Luca1, Claudia Montagnoli1, Carmen D’Angelo1,Teresa Zelante1, Carmine Vacca1, Francesco Bistoni1, Maria C. Fioretti1, Ursula Grohmann1, Brahm H. Segal2
& Paolo Puccetti1
Half a century ago, chronic granulomatous disease (CGD) was firstdescribed as a disease fatally affecting the ability of children tosurvive infections. Various milestone discoveries have since beenmade, from an insufficient ability of patients’ leucocytes to killmicrobes to the underlying genetic abnormalities1. In this inhe-rited disorder, phagocytes lack NADPH oxidase activity and donot generate reactive oxygen species, most notably superoxideanion, causing recurrent bacterial and fungal infections. Patientswith CGD also suffer from chronic inflammatory conditions, mostprominently granuloma formation in hollow viscera. The precisemechanisms of the increased microbial pathogenicity have beenunclear2, and more so the reasons for the exaggerated inflamma-tory response3–6. Here we show that a superoxide-dependent stepin tryptophan metabolism along the kynurenine pathway isblocked in CGD mice with lethal pulmonary aspergillosis, leadingto unrestrained Vc11 cd T-cell reactivity, dominant production ofinterleukin (IL)-17, defective regulatory T-cell activity and acuteinflammatory lung injury. Although beneficial effects are inducedby IL-17 neutralization or cd T-cell contraction, complete cureand reversal of the hyperinflammatory phenotype are achievedby replacement therapy with a natural kynurenine distal to theblockade in the pathway. Effective therapy, which includes co-administration of recombinant interferon-c (IFN-c), restoresproduction of downstream immunoactive metabolites and enablesthe emergence of regulatory Vc41 cd and Foxp31 ab T cells.Therefore, paradoxically, the lack of reactive oxygen speciescontributes to the hyperinflammatory phenotype associatedwith NADPH oxidase deficiencies, through a dysfunctionalkynurenine pathway of tryptophan catabolism. Yet, this conditioncan be reverted by reactivating the pathway downstream of thesuperoxide-dependent step.
Indoleamine 2,3-dioxygenase (IDO), a ‘metabolic’ enzyme con-served through the past 600 million years of evolution, suppressesT-cell responses and promotes tolerance in mammalian pregnancy,tumour resistance, chronic infection, autoimmunity and allergicinflammation7–10. During inflammation, IDO is upregulated indendritic cells and phagocytes by proinflammatory stimuli—mostnotably, IFN-c—and the enzyme then uses superoxide as a ‘cofactor’for oxidative cleavage of the indole ring of tryptophan, yielding anintermediate that deformylates to L-kynurenine11,12 (SupplementaryInformation and Supplementary Fig. 2). As a result, several IDO-dependent mechanisms mitigate inflammation and preventautoimmunity13–15. By infecting p47phox2/2 mice with Aspergillusfumigatus—a frequent cause of life-threatening disease in patientswith CGD—we tested the hypothesis that defective IDO functionoccurs in experimental CGD, affecting antifungal resistance, inflam-mation and T-cell homeostasis. Owing to the absence of superoxide,
an impaired IDO activity might compromise microbial tryptophanstarvation and kynurenine production by phagocytes, resultingin reduced antimicrobial defence and an exaggerated inflammatoryresponse16.
We infected p47phox-deficient mice with A. fumigatus intratra-cheally and evaluated parameters of antifungal resistance and inflam-mation, with IDO message, protein expression and enzymaticactivity. Abnormal susceptibility to pulmonary infection wasobserved in the knockout mice, in terms of survival and histopatho-logy in the lungs (Fig. 1a), abundance of inflammatory cells inthe bronchoalveolar lavage (BAL) fluid (Fig. 1b) and productionof inflammatory markers (Fig. 1c). Lethal invasive pulmonaryaspergillosis developed in CGD mice within a context of acuteinflammatory lung injury, different from that associated withneutrophil-dependent oxidative stress. In sorted pulmonary CD31
cells and/or lung homogenates from knockout mice, infection pre-ponderantly induced IL-17 and IL-23, whereas IFN-c, IL-10, andtransforming growth factor-b (TGF-b) were defective (Fig. 1d). Inthoracic lymph node CD41 T cells from CGD mice, A. fumigatuscaused a greater than 15-fold increase in message expression of Rorc(encoding the TH17 cell transcription factor, RORct), and virtuallyno induction of TH1-associated Tbet and Foxp3 (regulatory T-cellspecification factor) (Supplementary Fig. 1). A detrimental effect ofIL-17 was demonstrated by IL-17 neutralization in vivo followed byin vitro assessment of immunological and antifungal parameters; abimodal effect of IL-17 was demonstrable in vitro on the conidiocidalactivity of polymorphonuclear cells (PMNs), with significant inhibi-tion at the level of nanograms per millilitre; improved survivaloccurred in CGD mice treated with the anti-IL-17 antibody (Supple-mentary Table 1). These data suggested that CGD mice manifest adefect in the initial lung defence against A. fumigatus infection that isassociated with a greater—but damaging—inflammatory response.
Despite their prompt mobilization after infection (Fig. 1a, b),PMNs from CGD mice were relatively inefficient at initiating tryp-tophan catabolism. Consistent with the hypothesis that IDO func-tional activity was blocked post-translationally in CGD mice, IFN-cinduced Indo transcript (Fig. 2a) and IDO protein (Fig. 2b) expres-sions in knockout PMNs that were comparable to those in wild-typecontrols. Yet, these cells were completely unable to mediate trypto-phan conversion to L-kynurenine (Fig. 2c). In vivo, infection resultedin detectable L-kynurenine in lung homogenates from wild-type butnot knockout mice (Fig. 2d). This effect could, at least in part, be dueto a defective expression of IDO protein in the knockout mice(Fig. 2e), likely as a result of the reduced potential of T cells to releaseIFN-c. We used real-time polymerase chain reaction (PCR) forquantitative and comparative assessment of Indo expression in lungmononuclear phagocytes from naive mice of both genotypes, and
1Department of Experimental Medicine, University of Perugia, 06126 Perugia, Italy. 2Division of Infectious Diseases, Roswell Park Cancer Institute, Buffalo, New York 14263, USA.
Vol 451 | 10 January 2008 | doi:10.1038/nature06471
211Nature ©2007 Publishing Group

extended the examination to a series of genes encoding enzymesdownstream of IDO17 (Supplementary Fig. 2). Like Indo, the expres-sions of Kmo, Kynu and Haao were all enhanced by IFN-c in bothgenotypes. However, IFN-c was unable to mediate conversion ofexternally added L-tryptophan to L-kynurenine by mononuclear pha-gocytes from p47phox2/2 mice, confirming that, in experimentalCGD, an inefficient IDO mechanism prevents metabolic steps thatare subsequent and consequent to IDO.
Among CD31 T lymphocytes, cells with the cd T-cell-typereceptors (cd T cells) constitute an ancient lineage that is presentin all vertebrates and responds to often-unprocessed microbialantigen and non-peptide metabolites18. cd T cells control innateresponses, including those mediated by PMNs, through their pro-duction of IL-17 (refs 19, 20), and they take part in the transitionalresponse (occurring temporally between the rapid innate and
EosLymTotal
30
20
250
125
0
500
250
0
1,500
750
0
1,500
750
0
1,500
750
0
1,500
750
0
500
250
0
500
250
0
100
50
0
100
50
0
10
0Mac Neu
BA
L ce
lls (×
105 )
*
*
***
WT (un.)WT (inf.)KO (un.)KO (inf.)
Lung homogenate
0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6
Lung CD3+ cells
IL-4
IL-17 IL-10 IL-12 IL-23 TNF-α
MP
O fo
ld c
hang
e
*WTKO
a
b
d
c
*
**
**
*
**
*
**
*
**
*
WT
86420
KOUn. Inf. Un. Inf.
(days)
TGF-βIFN-γIL-17 IL-10
*
******
*
***
WT (un.) WT (inf.) KO (inf.)KO (un.)
Figure 1 | Invasive pulmonary aspergillosis in the p47phox mouse knockoutmodel of CGD. Knockout (KO) and wild-type (WT) mice were infectedintratracheally with A. fumigatus conidia (5 3 106) on day 0, to be examinedfor course of infection. The median survival time in knockout mice was 7.5days (dead/total, 6/6), whereas it exceeded 60 days (dead/total, 0/6) in wild-type animals (P 5 0.004). a, Lung histology was performed on day 4, a timewhen the amount of pulmonary chitin (a major constituent of A. fumigatuscell wall) was fivefold higher in knockout than in wild-type mice.Histopathology revealed abundant pyogranulomatous lesions with centralneutrophilic infiltrates in knockout mice (periodic acid-Schiff; scale bar,100mm). Shown in insets are haematoxylin and eosin stains of magnifieddetails, with exemplary foci of neutrophilic inflammation (scale bar, 50 mm).Data are representative of several experiments, in which mortality andhistology data were highly consistent among similarly treated mice with thesame genotype. b, On day 4, the abundances of macrophages (Mac),lymphocytes (Lym), neutrophils (Neu), and eosinophils (Eos) were assessedin BAL fluid, revealing an exuberant neutrophilic response in the infectedknockout mice, in which eosinophils and increased lymphocyte numberswere also found. c, Day-3 metalloproteinase-9 (assessed as gelatinolyticactivity in upper blot) and myeloperoxidase (MPO; lower immunoblot, withb-tubulin normalization) were higher in lung neutrophils from knockoutmice. Myeloperoxidase blots (n 5 3) were quantified by scanningdensitometry and represented as fold change in infected (Inf) mice relativeto uninfected (Un) controls of the same genotype (in which foldchange 5 1). d, Secreted cytokines (in picograms per millilitre) from wild-type (open bars) and knockout (filled bars) mice were measured in lunghomogenates on days 2 and 6, whereas pulmonary CD31 T cells wereassayed for cytokine release in response to soluble anti-CD3 (1 mg ml21).Day 0 indicates uninfected mice, and comparisons are shown betweensimilarly treated groups of the two genotypes. In all panels, quantitative dataare means 6 s.d. from three experiments. *P , 0.05; **P , 0.005–0.001.
Kyn
uren
ine
(µM
)
WTKO
*
Kyn
uren
ine
(µM
)WT
10
4
3
2
1
00 2
Time (days)6
5
0
KO
Med
iumIF
N-γ
Med
ium
IFN-γ
Med
ium
IFN-γ
WT KO
Indo
Gapdh
a
HEK-M
HEK-I
IFN-γ
Med
ium
IFN-γ
Med
ium
WT KO
IDOβ-tubulin
Med
iumIF
N-γ
c
b
d
WT KO KOW
TKOW
T
IDO
β-tubulin
e
2 60
Figure 2 | Defective IDO-dependent conversion of tryptophan toL-kynurenine in p47phox knockout mice. a, PCR with reverse transcription(RT–PCR) of Indo (which encodes IDO) in PMNs from wild-type andknockout mice, either untreated or treated overnight with IFN-c(200 U ml21). One experiment is shown representative of three. b, IDOprotein expression was assessed by immunoblot analysis in similarly treatedPMNs by an IDO-specific antibody. The positive control consisted of IDO-expressing HEK-I transfectants; the negative control consisted of mock-transfected HEK-M cells. Loading controls consisted of samples re-probedwith b-tubulin-specific antibody. One experiment is shown representative ofthree. The mean fold changes (6 s.d.) in the three experiments (that is, day-6infection to no-infection ratios using normalized densitometry values) were5.4 6 1.2 and 6.3 6 1.5 for wild-type and knockout mice, respectively.c, Functional IDO (means 6 s.d. with n 5 3) in response to IFN-c wasmeasured in wild-type and knockout mice in terms of the ability of the PMNsto metabolize tryptophan to L-kynurenine, measured by high-performanceliquid chromatography. *P , 0.01 (IFN-c versus medium treatment).d, Detection of L-kynurenine occurred in the lungs of wild-type but notknockout mice challenged intratracheally with A. fumigatus. Wild-type andknockout mice, infected for 2 or 6 days, were assayed for the presence ofL-kynurenine in lung homogenates. Day 0 indicates uninfected controls.Error bars, means 6 s.d. from four experiments. e, IDO protein expressionwas assessed in the lungs of wild-type and knockout mice challengedintratracheally with A. fumigatus for 2 or 6 days. One experiment is shownrepresentative of three, demonstrating reduced IDO expression in theknockout mice.
LETTERS NATURE | Vol 451 | 10 January 2008
212Nature ©2007 Publishing Group
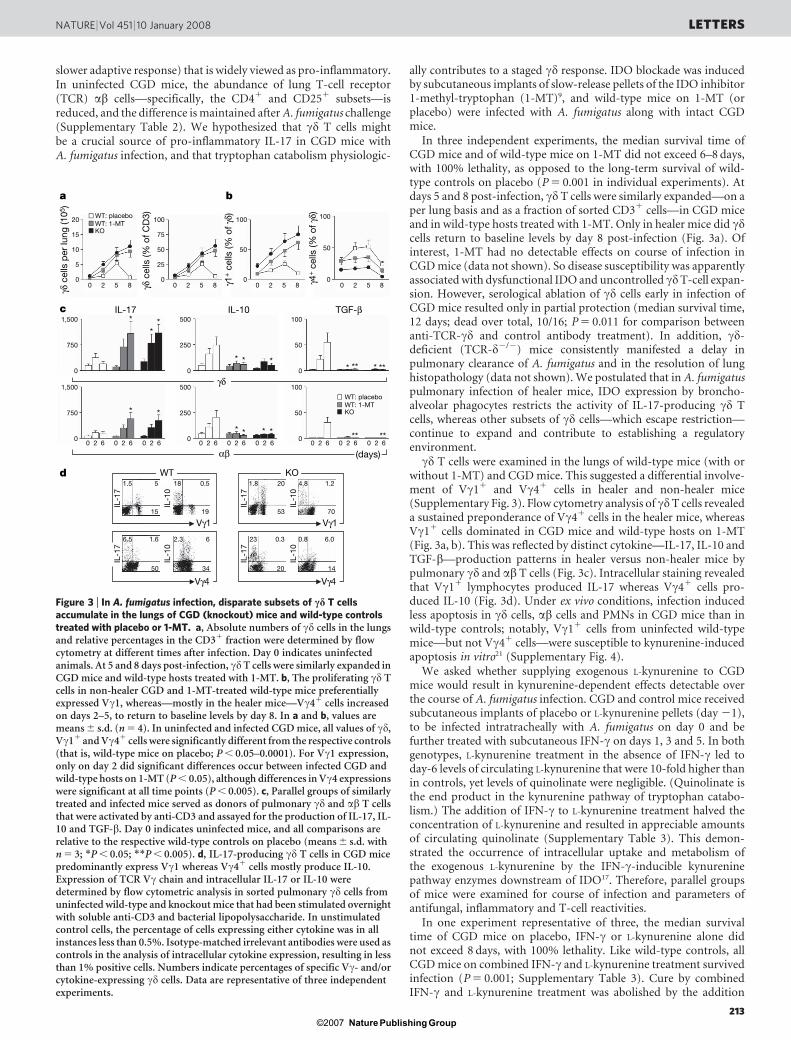
slower adaptive response) that is widely viewed as pro-inflammatory.In uninfected CGD mice, the abundance of lung T-cell receptor(TCR) ab cells—specifically, the CD41 and CD251 subsets—isreduced, and the difference is maintained after A. fumigatus challenge(Supplementary Table 2). We hypothesized that cd T cells mightbe a crucial source of pro-inflammatory IL-17 in CGD mice withA. fumigatus infection, and that tryptophan catabolism physiologic-
ally contributes to a staged cd response. IDO blockade was inducedby subcutaneous implants of slow-release pellets of the IDO inhibitor1-methyl-tryptophan (1-MT)9, and wild-type mice on 1-MT (orplacebo) were infected with A. fumigatus along with intact CGDmice.
In three independent experiments, the median survival time ofCGD mice and of wild-type mice on 1-MT did not exceed 6–8 days,with 100% lethality, as opposed to the long-term survival of wild-type controls on placebo (P 5 0.001 in individual experiments). Atdays 5 and 8 post-infection, cd T cells were similarly expanded—on aper lung basis and as a fraction of sorted CD31 cells—in CGD miceand in wild-type hosts treated with 1-MT. Only in healer mice did cdcells return to baseline levels by day 8 post-infection (Fig. 3a). Ofinterest, 1-MT had no detectable effects on course of infection inCGD mice (data not shown). So disease susceptibility was apparentlyassociated with dysfunctional IDO and uncontrolled cdT-cell expan-sion. However, serological ablation of cd cells early in infection ofCGD mice resulted only in partial protection (median survival time,12 days; dead over total, 10/16; P 5 0.011 for comparison betweenanti-TCR-cd and control antibody treatment). In addition, cd-deficient (TCR-d2/2) mice consistently manifested a delay inpulmonary clearance of A. fumigatus and in the resolution of lunghistopathology (data not shown). We postulated that in A. fumigatuspulmonary infection of healer mice, IDO expression by broncho-alveolar phagocytes restricts the activity of IL-17-producing cd Tcells, whereas other subsets of cd cells—which escape restriction—continue to expand and contribute to establishing a regulatoryenvironment.
cd T cells were examined in the lungs of wild-type mice (with orwithout 1-MT) and CGD mice. This suggested a differential involve-ment of Vc11 and Vc41 cells in healer and non-healer mice(Supplementary Fig. 3). Flow cytometry analysis of cdT cells revealeda sustained preponderance of Vc41 cells in the healer mice, whereasVc11 cells dominated in CGD mice and wild-type hosts on 1-MT(Fig. 3a, b). This was reflected by distinct cytokine—IL-17, IL-10 andTGF-b—production patterns in healer versus non-healer mice bypulmonary cd and ab T cells (Fig. 3c). Intracellular staining revealedthat Vc11 lymphocytes produced IL-17 whereas Vc41 cells pro-duced IL-10 (Fig. 3d). Under ex vivo conditions, infection inducedless apoptosis in cd cells, ab cells and PMNs in CGD mice than inwild-type controls; notably, Vc11 cells from uninfected wild-typemice—but not Vc41 cells—were susceptible to kynurenine-inducedapoptosis in vitro21 (Supplementary Fig. 4).
We asked whether supplying exogenous L-kynurenine to CGDmice would result in kynurenine-dependent effects detectable overthe course of A. fumigatus infection. CGD and control mice receivedsubcutaneous implants of placebo or L-kynurenine pellets (day 21),to be infected intratracheally with A. fumigatus on day 0 and befurther treated with subcutaneous IFN-c on days 1, 3 and 5. In bothgenotypes, L-kynurenine treatment in the absence of IFN-c led today-6 levels of circulating L-kynurenine that were 10-fold higher thanin controls, yet levels of quinolinate were negligible. (Quinolinate isthe end product in the kynurenine pathway of tryptophan catabo-lism.) The addition of IFN-c to L-kynurenine treatment halved theconcentration of L-kynurenine and resulted in appreciable amountsof circulating quinolinate (Supplementary Table 3). This demon-strated the occurrence of intracellular uptake and metabolism ofthe exogenous L-kynurenine by the IFN-c-inducible kynureninepathway enzymes downstream of IDO17. Therefore, parallel groupsof mice were examined for course of infection and parameters ofantifungal, inflammatory and T-cell reactivities.
In one experiment representative of three, the median survivaltime of CGD mice on placebo, IFN-c or L-kynurenine alone didnot exceed 8 days, with 100% lethality. Like wild-type controls, allCGD mice on combined IFN-c and L-kynurenine treatment survivedinfection (P 5 0.001; Supplementary Table 3). Cure by combinedIFN-c and L-kynurenine treatment was abolished by the addition
IL-1
7
IL-1
0
IL-1
7
IL-1
7IL
-17
IL-1
0
IL-1
0IL
-10
γδ c
ells
per
lung
(105
)
γδ c
ells
(% o
f CD
3)
γ1+ c
ells
(% o
f γδ)
γ4+ c
ells
(% o
f γδ)
c
γδ
αβ
**
* *
* * * * **
*********
**
*IL-17 IL-10 TGF-β
b
WT: placebo20
15
10
5
0
1,500
750
0
500
250
0
100
50
0
1,500
750
00 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6 0 2 6
500
250
0
100
50
0
100
75
50
25
0
100
50
0
100
50
00 2 5 8 0 2 5 8 0 2 5 8 0 2 5 8
WT: 1-MTKO
WT: placeboWT: 1-MTKO
(days)
d KOWT1.5 5
15
18 0.5
19
2.3 6
34
6.5 1.6
50
1.8 20
53
4.8 1.2
70
0.8 6.0
14
23 0.3
20
Vγ4Vγ4
Vγ1 Vγ1
a
Figure 3 | In A. fumigatus infection, disparate subsets of cd T cellsaccumulate in the lungs of CGD (knockout) mice and wild-type controlstreated with placebo or 1-MT. a, Absolute numbers of cd cells in the lungsand relative percentages in the CD31 fraction were determined by flowcytometry at different times after infection. Day 0 indicates uninfectedanimals. At 5 and 8 days post-infection, cdT cells were similarly expanded inCGD mice and wild-type hosts treated with 1-MT. b, The proliferating cd Tcells in non-healer CGD and 1-MT-treated wild-type mice preferentiallyexpressed Vc1, whereas—mostly in the healer mice—Vc41 cells increasedon days 2–5, to return to baseline levels by day 8. In a and b, values aremeans 6 s.d. (n 5 4). In uninfected and infected CGD mice, all values of cd,Vc11 and Vc41 cells were significantly different from the respective controls(that is, wild-type mice on placebo; P , 0.05–0.0001). For Vc1 expression,only on day 2 did significant differences occur between infected CGD andwild-type hosts on 1-MT (P , 0.05), although differences in Vc4 expressionswere significant at all time points (P , 0.005). c, Parallel groups of similarlytreated and infected mice served as donors of pulmonary cd and ab T cellsthat were activated by anti-CD3 and assayed for the production of IL-17, IL-10 and TGF-b. Day 0 indicates uninfected mice, and all comparisons arerelative to the respective wild-type controls on placebo (means 6 s.d. withn 5 3; *P , 0.05; **P , 0.005). d, IL-17-producing cd T cells in CGD micepredominantly express Vc1 whereas Vc41 cells mostly produce IL-10.Expression of TCR Vc chain and intracellular IL-17 or IL-10 weredetermined by flow cytometric analysis in sorted pulmonary cd cells fromuninfected wild-type and knockout mice that had been stimulated overnightwith soluble anti-CD3 and bacterial lipopolysaccharide. In unstimulatedcontrol cells, the percentage of cells expressing either cytokine was in allinstances less than 0.5%. Isotype-matched irrelevant antibodies were used ascontrols in the analysis of intracellular cytokine expression, resulting in lessthan 1% positive cells. Numbers indicate percentages of specific Vc- and/orcytokine-expressing cd cells. Data are representative of three independentexperiments.
NATURE | Vol 451 | 10 January 2008 LETTERS
213Nature ©2007 Publishing Group
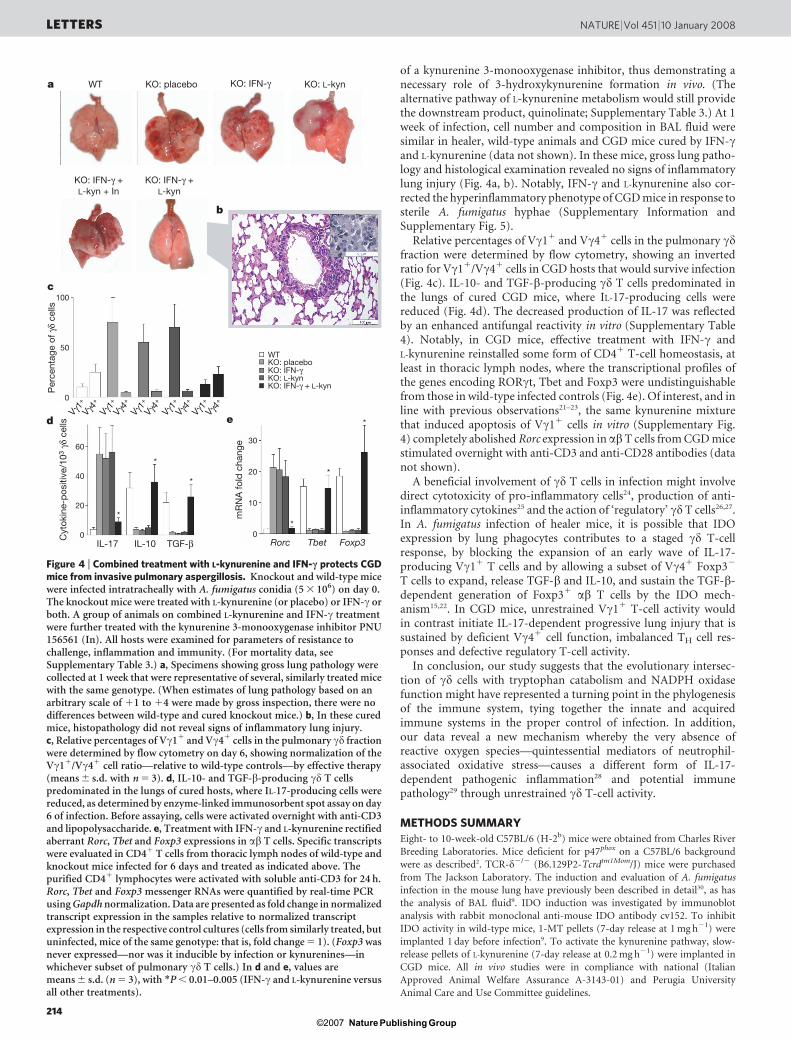
of a kynurenine 3-monooxygenase inhibitor, thus demonstrating anecessary role of 3-hydroxykynurenine formation in vivo. (Thealternative pathway of L-kynurenine metabolism would still providethe downstream product, quinolinate; Supplementary Table 3.) At 1week of infection, cell number and composition in BAL fluid weresimilar in healer, wild-type animals and CGD mice cured by IFN-cand L-kynurenine (data not shown). In these mice, gross lung patho-logy and histological examination revealed no signs of inflammatorylung injury (Fig. 4a, b). Notably, IFN-c and L-kynurenine also cor-rected the hyperinflammatory phenotype of CGD mice in response tosterile A. fumigatus hyphae (Supplementary Information andSupplementary Fig. 5).
Relative percentages of Vc11 and Vc41 cells in the pulmonary cdfraction were determined by flow cytometry, showing an invertedratio for Vc11/Vc41 cells in CGD hosts that would survive infection(Fig. 4c). IL-10- and TGF-b-producing cd T cells predominated inthe lungs of cured CGD mice, where IL-17-producing cells werereduced (Fig. 4d). The decreased production of IL-17 was reflectedby an enhanced antifungal reactivity in vitro (Supplementary Table4). Notably, in CGD mice, effective treatment with IFN-c andL-kynurenine reinstalled some form of CD41 T-cell homeostasis, atleast in thoracic lymph nodes, where the transcriptional profiles ofthe genes encoding RORct, Tbet and Foxp3 were undistinguishablefrom those in wild-type infected controls (Fig. 4e). Of interest, and inline with previous observations21–23, the same kynurenine mixturethat induced apoptosis of Vc11 cells in vitro (Supplementary Fig.4) completely abolished Rorc expression in abT cells from CGD micestimulated overnight with anti-CD3 and anti-CD28 antibodies (datanot shown).
A beneficial involvement of cd T cells in infection might involvedirect cytotoxicity of pro-inflammatory cells24, production of anti-inflammatory cytokines25 and the action of ‘regulatory’ cd T cells26,27.In A. fumigatus infection of healer mice, it is possible that IDOexpression by lung phagocytes contributes to a staged cd T-cellresponse, by blocking the expansion of an early wave of IL-17-producing Vc11 T cells and by allowing a subset of Vc41 Foxp32
T cells to expand, release TGF-b and IL-10, and sustain the TGF-b-dependent generation of Foxp31 ab T cells by the IDO mech-anism15,22. In CGD mice, unrestrained Vc11 T-cell activity wouldin contrast initiate IL-17-dependent progressive lung injury that issustained by deficient Vc41 cell function, imbalanced TH cell res-ponses and defective regulatory T-cell activity.
In conclusion, our study suggests that the evolutionary intersec-tion of cd cells with tryptophan catabolism and NADPH oxidasefunction might have represented a turning point in the phylogenesisof the immune system, tying together the innate and acquiredimmune systems in the proper control of infection. In addition,our data reveal a new mechanism whereby the very absence ofreactive oxygen species—quintessential mediators of neutrophil-associated oxidative stress—causes a different form of IL-17-dependent pathogenic inflammation28 and potential immunepathology29 through unrestrained cd T-cell activity.
METHODS SUMMARY
Eight- to 10-week-old C57BL/6 (H-2b) mice were obtained from Charles River
Breeding Laboratories. Mice deficient for p47phox on a C57BL/6 background
were as described2. TCR-d2/2 (B6.129P2-Tcrdtm1Mom/J) mice were purchased
from The Jackson Laboratory. The induction and evaluation of A. fumigatus
infection in the mouse lung have previously been described in detail30, as has
the analysis of BAL fluid9. IDO induction was investigated by immunoblot
analysis with rabbit monoclonal anti-mouse IDO antibody cv152. To inhibit
IDO activity in wild-type mice, 1-MT pellets (7-day release at 1 mg h21) were
implanted 1 day before infection9. To activate the kynurenine pathway, slow-
release pellets of L-kynurenine (7-day release at 0.2 mg h21) were implanted in
CGD mice. All in vivo studies were in compliance with national (Italian
Approved Animal Welfare Assurance A-3143-01) and Perugia University
Animal Care and Use Committee guidelines.
Cyt
okin
e-p
ositi
ve/1
03 γ
δ ce
lls
Rorc Tbet Foxp3
*
*
*
mR
NA
fold
cha
nge
IL-17
6030
20
10
0
40
20
0IL-10 TGF-β
d eVγ1
+
Vγ4+
Vγ1+
Vγ4+
Vγ1+
Vγ4+
Vγ1+
Vγ4+
Vγ1+
Vγ4+
b
a
Per
cent
age
of γ
δ ce
lls
WT
100
50
0
KO: placeboKO: IFN-γKO: L-kynKO: IFN-γ + L-kyn
*
*
*
c
KO: IFN-γ +L-kyn
KO: placebo KO: IFN-γ KO: L-kynWT
KO: IFN-γ +L-kyn + In
Figure 4 | Combined treatment with L-kynurenine and IFN-c protects CGDmice from invasive pulmonary aspergillosis. Knockout and wild-type micewere infected intratracheally with A. fumigatus conidia (5 3 106) on day 0.The knockout mice were treated with L-kynurenine (or placebo) or IFN-c orboth. A group of animals on combined L-kynurenine and IFN-c treatmentwere further treated with the kynurenine 3-monooxygenase inhibitor PNU156561 (In). All hosts were examined for parameters of resistance tochallenge, inflammation and immunity. (For mortality data, seeSupplementary Table 3.) a, Specimens showing gross lung pathology werecollected at 1 week that were representative of several, similarly treated micewith the same genotype. (When estimates of lung pathology based on anarbitrary scale of 11 to 14 were made by gross inspection, there were nodifferences between wild-type and cured knockout mice.) b, In these curedmice, histopathology did not reveal signs of inflammatory lung injury.c, Relative percentages of Vc11 and Vc41 cells in the pulmonary cd fractionwere determined by flow cytometry on day 6, showing normalization of theVc11/Vc41 cell ratio—relative to wild-type controls—by effective therapy(means 6 s.d. with n 5 3). d, IL-10- and TGF-b-producing cd T cellspredominated in the lungs of cured hosts, where IL-17-producing cells werereduced, as determined by enzyme-linked immunosorbent spot assay on day6 of infection. Before assaying, cells were activated overnight with anti-CD3and lipopolysaccharide. e, Treatment with IFN-c and L-kynurenine rectifiedaberrant Rorc, Tbet and Foxp3 expressions in ab T cells. Specific transcriptswere evaluated in CD41 T cells from thoracic lymph nodes of wild-type andknockout mice infected for 6 days and treated as indicated above. Thepurified CD41 lymphocytes were activated with soluble anti-CD3 for 24 h.Rorc, Tbet and Foxp3 messenger RNAs were quantified by real-time PCRusing Gapdh normalization. Data are presented as fold change in normalizedtranscript expression in the samples relative to normalized transcriptexpression in the respective control cultures (cells from similarly treated, butuninfected, mice of the same genotype: that is, fold change 5 1). (Foxp3 wasnever expressed—nor was it inducible by infection or kynurenines—inwhichever subset of pulmonary cd T cells.) In d and e, values aremeans 6 s.d. (n 5 3), with *P , 0.01–0.005 (IFN-c and L-kynurenine versusall other treatments).
LETTERS NATURE | Vol 451 | 10 January 2008
214Nature ©2007 Publishing Group

Full Methods and any associated references are available in the online version ofthe paper at www.nature.com/nature.
Received 19 August; accepted 13 November 2007.
1. Assari, T. Chronic granulomatous disease; fundamental stages in ourunderstanding of CGD. Med. Immunol. 5, 4 (2006).
2. Chang, Y. C., Segal, B. H., Holland, S. M., Miller, G. F. & Kwon-Chung, K. J.Virulence of catalase-deficient Aspergillus nidulans in p47phox2/2 mice.Implications for fungal pathogenicity and host defense in chronic granulomatousdisease. J. Clin. Invest. 101, 1843–1850 (1998).
3. Romani, L. Immunity to fungal infections. Nature Rev. Immunol. 4, 1–23 (2004).4. Marciano, B. E. et al. Long-term interferon-c therapy for patients with chronic
granulomatous disease. Clin. Infect. Dis. 39, 692–699 (2004).5. Ott, M. G. et al. Correction of X-linked chronic granulomatous disease by gene
therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1.Nature Med. 12, 401–409 (2006).
6. Bylund, J. et al. Enhanced inflammatory responses of chronic granulomatousdisease leukocytes involve ROS-independent activation of NF-kB. Eur. J. Immunol.37, 1087–1096 (2007).
7. Grohmann, U., Fallarino, F. & Puccetti, P. Tolerance, DCs and tryptophan: muchado about IDO. Trends Immunol. 24, 242–248 (2003).
8. Mellor, A. L. & Munn, D. H. IDO expression by dendritic cells: tolerance andtryptophan catabolism. Nature Rev. Immunol. 4, 762–774 (2004).
9. Grohmann, U. et al. Reverse signaling through GITR ligand enablesdexamethasone to activate IDO in allergy. Nature Med. 13, 579–586 (2007).
10. Sharma, M. D. et al. Plasmacytoid dendritic cells from mouse tumor-draininglymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase.J. Clin. Invest. 117, 2570–2582 (2007).
11. Schwarcz, R. & Pellicciari, R. Manipulation of brain kynurenines: glial targets,neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 303, 1–10 (2002).
12. Stone, T. W. & Darlington, L. G. Endogenous kynurenines as targets for drugdiscovery and development. Nature Rev. Drug Discov. 1, 609–620 (2002).
13. Munn, D. H. et al. GCN2 kinase in T cells mediates proliferative arrest and anergyinduction in response to indoleamine 2,3-dioxygenase. Immunity 22, 633–642(2005).
14. Platten, M. et al. Treatment of autoimmune neuroinflammation with a synthetictryptophan metabolite. Science 310, 850–855 (2005).
15. Puccetti, P. & Grohmann, U. IDO and regulatory T cells: a role for reverse signallingand non-canonical NF-kB activation. Nature Rev. Immunol. 7, 817–823 (2007).
16. Popov, A. et al. Indoleamine 2,3-dioxygenase-expressing dendritic cells formsuppurative granulomas following Listeria monocytogenes infection. J. Clin. Invest.116, 3160–3170 (2006).
17. Belladonna, M. L. et al. Kynurenine pathway enzymes in dendritic cells initiatetolerogenesis in the absence of functional IDO. J. Immunol. 177, 130–137 (2006).
18. Born, W. K., Reardon, C. L. & O’Brien, R. L. The function of cd T cells in innateimmunity. Curr. Opin. Immunol. 18, 31–38 (2006).
19. Lockhart, E., Green, A. M. & Flynn, J. L. IL-17 production is dominated by cd T cellsrather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol.177, 4662–4669 (2006).
20. Shibata, K., Yamada, H., Hara, H., Kishihara, K. & Yoshikai, Y. Resident Vd11 cd Tcells control early infiltration of neutrophils after Escherichia coli infection via IL-17production. J. Immunol. 178, 4466–4472 (2007).
21. Fallarino, F. et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 9,1069–1077 (2002).
22. Fallarino, F. et al. The combined effects of tryptophan starvation and tryptophancatabolites down-regulate T cell receptor f-chain and induce a regulatoryphenotype in naive T cells. J. Immunol. 176, 6752–6761 (2006).
23. De Luca, A. et al. Functional yet balanced reactivity to Candida albicans requiresTRIF, MyD88, and IDO-dependent inhibition of Rorc. J. Immunol. 179, 5999–6008(2007).
24. Kirby, A. C., Newton, D. J., Carding, S. R. & Kaye, P. M. Pulmonary dendritic cellsand alveolar macrophages are regulated by cd T cells during the resolution of S.pneumoniae-induced inflammation. J. Pathol. 212, 29–37 (2007).
25. Andrew, E. M. & Carding, S. R. Murine cd T cells in infections: beneficial ordeleterious? Microbes Infect. 7, 529–536 (2005).
26. Peterman, G. M., Spencer, C., Sperling, A. I. & Bluestone, J. A. Role of cd T cells inmurine collagen-induced arthritis. J. Immunol. 151, 6546–6558 (1993).
27. Andrew, E. M. et al. Delineation of the function of a major cd T cell subset duringinfection. J. Immunol. 175, 1741–1750 (2005).
28. Hizawa, N., Kawaguchi, M., Huang, S. K. & Nishimura, M. Role of interleukin-17F inchronic inflammatory and allergic lung disease. Clin. Exp. Allergy 36, 1109–1114(2006).
29. Hultqvist, M., Backlund, J., Bauer, K., Gelderman, K. A. & Holmdahl, R. Lack ofreactive oxygen species breaks T cell tolerance to collagen type II and allowsdevelopment of arthritis in mice. J. Immunol. 179, 1431–1437 (2007).
30. Montagnoli, C. et al. Immunity and tolerance to Aspergillus involve functionallydistinct regulatory T cells and tryptophan catabolism. J. Immunol. 176, 1712–1723(2006).
Supplementary Information is linked to the online version of the paper atwww.nature.com/nature.
Acknowledgements This work was supported by Specific Targeted ResearchProject ‘MANASP’ (L.R.), and funding from the Juvenile Diabetes ResearchFoundation (P.P.) We thank P. Mosci for maintaining the mutant strains of miceand performing histopathology; and G. Andrielli for digital art and image editing.
Author Information Reprints and permissions information is available atwww.nature.com/reprints. Correspondence and requests for materials should beaddressed to L.R. ([email protected]) or P.P. ([email protected]).
NATURE | Vol 451 | 10 January 2008 LETTERS
215Nature ©2007 Publishing Group

METHODSLung cell isolation and reagents. The p47phox mouse knockout model of CGD is
described in ref. 31. For isolation of lung cells, lungs were aseptically removed
and cut into small pieces in cold medium. The dissected tissue was then incu-
bated in medium containing collagenase XI (0.7 mg ml21; Sigma-Aldrich) and
type IV bovine pancreatic DNase (30mg ml21; Sigma-Aldrich) for 30–45 min at
37 uC. Adding 10 ml of medium stopped the action of the enzymes, and digested
lungs were further disrupted by gently pushing the tissue through a nylon screen.
The single-cell suspension was then washed and centrifuged at 200g.
Contaminating red blood cells were lysed, and cells were then washed withPBS containing 0.5% fetal bovine serum, counted and incubated with fluorescein
isothiocyanate (FITC)-labelled GL3 (hamster anti-cd T-cell receptor,
PharMingen) followed by anti-FITC MicroBeads (Miltenyi Biotech) or with
biotin-labelled anti-mouse Gr-1 followed by avidin-conjugated MicroBeads
(for isolation of PMNs32). In selected experiments, we used CD31 or TCR abT cells isolated by magnetic-activated sorting or the plastic-adherent fraction
(.95% mononuclear phagocytes) of lung cell suspensions. Thoracic lymph-
node CD41 cells were obtained as described9.
Pulmonary aspergillosis, collection and analysis of BAL fluid, and histopatho-logy. The induction and evaluation of A. fumigatus infection in the murine lung
have previously been described in detail30,33, as has the analysis of BAL fluid9.
Briefly, after the trachea was cannulated, the lungs were lavaged twice with 0.5 ml
PBS, and the fluid was pooled. Cells in BAL fluid were counted and analysed by
using slide preparations stained with Giemsa. Preparation and administration of
sterile A. fumigatus hyphal cell walls were conducted as described34. Cytokine
contents in BAL fluid were also determined as described9. For lung histopatho-
logy, paraffin-embedded sections (3–4 mm) were stained with periodic acid–
Schiff or haematoxylin and eosin. CD41 T cells from thoracic lymph nodes wereused for analysing Rorc, Tbet and Foxp3 transcript expression by real-time PCR
using Gapdh normalization (Supplementary Table 5)9,30,33.
IDO expression and functional analysis, and 1-MT and L-kynurenineimplants. IDO induction was investigated by immunoblot with a rabbit mono-
clonal anti-mouse IDO antibody (cv152). This antibody is produced in our
laboratory by a rabbit–rabbit hybridoma (consisting of spleen cells fused with
the 240E1 plasmacytoma partner35, obtained from K. L. Knight), and it was
raised in the same way as the previously characterized polyclonal reagent36, with
which the monoclonal antibody shares the pattern of reactivity. Indo-transfected
and mock-transfected human embryonic kidney (HEK) 293 cells were used as
the respective positive (HEK-I) and negative (HEK-M) controls. IDO functional
activity was measured in vitro in terms of the ability to metabolize tryptophan to
L-kynurenine whose concentrations were measured by high-performance liquid
chromatography36. Downstream metabolism of exogenous L-kynurenine was
also assessed by high-performance liquid chromatography, and expressed as
quinolinate production. To inhibit IDO activity in wild-type mice, 1-MT or
placebo pellets (7-day release at 1 mg h21; Innovative Research of America) were
implanted 1 day before infection9,37. Like 1-MT, yet to activate rather than blockthe kynurenine pathway, slow-release pellets of L-kynurenine (7-day release at
0.2 mg h21; Innovative Research of America) were implanted in CGD mice. The
kynurenine 3-monooxygenase inhibitor PNU 156561 (4,5-dichlorobenzoylala-
nine; synthesized in the Pharmacia and Upjohn Research Laboratories of
Nerviano, Italy; obtained from M. Varasi and R. Schwarcz) has previously been
described17, and it was administered in vivo at 40 mg kg21 by intraperitoneal
injections on days 1, 3 and 5 post-infection.
Flow cytometry. Flow cytometry analyses involving surface expression of TCR
ab, CD4, CD8 and CD25 molecules have previously been described9. cd T cells
from the lungs were isolated and enriched with FITC-labelled GL3, as described
above. For analysis of Vc use, cells were co-stained with FITC-conjugated 2.11
(anti-Vc1; a gift from P. Pereira) and UC3-10A6 (anti-Vc4; PharMingen). Stains
were pre-blocked with Fc Block (anti-mouse CD16/CD32; PharMingen). For
intracellular cytokine staining, cells were activated overnight with soluble anti-
CD3 (5mg ml21) and lipopolysaccharide (1mg ml21) and assayed by Cytoperm-
Cytofix Intracellular Staining kit (PharMingen).
PCR analyses. RT–PCR (for Indo) and real-time PCR (for Rorc, Tbet, Foxp3,
Indo, Kmo, Kynu and Haao) analyses were conducted as described using the
primers listed in Supplementary Table 5. Real-time PCR was also used for ana-
lysing Vc transcript expression in sorted cd T cells, as described38, using the
primers in Supplementary Table 5.
Miscellaneous procedures. Cytokines were measured by enzyme-linkedimmunosorbent assays using specific kits or previously described reagents and
procedures9,30. Pulmonary chitin was quantified as a measure of A. fumigatus
growth in the lungs32. Conidiocidal activity was determined as described32.
Gelatin zymography was used to measure metalloproteinase-9 activity, whereas
myeloperoxidase was measured by immunoblot analysis32. For depletion of cd T
cells, mice received five intraperitoneal injections of 200mg purified GL3 anti-
body (days 21 to 13, relative to infection on day 0), and control mice received
equal amounts of hamster IgG. Treatment with anti-TCR-cd antibody caused
durable (greater than 80%) reduction of cd T cells in wild-type and CGD mice.
Neutralization of IL-17 involved intraperitoneal injections of 50 mg rat anti-IL-
17 monoclonal antibody (MAB421; R&D Systems) on days 0 and 11, and
control mice received equal amounts of low-endotoxin, azide-free rat IgG
(SouthernBiotech). In selected experiments, rat IgG2A isotype control (R&D
Systems) was also used. Induction of apoptosis in T cells and PMNs by
kynurenines in vitro was determined by FITC- or phycoerythrin-labelled
annexin V staining, as described17,21. IFN-c treatment in vivo involved
administering the recombinant cytokine (Genentech) subcutaneously at
20,000 U per mouse on days 1, 3 and 5 post-infection39. Specimens showinggross lung pathology were photographed by using an OPMI 1FC surgical micro-
scope (Zeiss) and a magnification factor of 0.6.
Statistical analysis. In the in vivo experiments, mortality data were analysed by
Mann–Whitney U-test. Paired data were evaluated by Student’s t-test, and a one-
way analysis of variance was used for multiple comparisons. All in vitro deter-
minations are means 6 s.d. from at least three independent experiments. The
required numerosity of animals per group was computed by power analysis, so asto yield a power of at least 80% with an a-level of 0.05.
31. Jackson, S. H., Gallin, J. I. & Holland, S. M. The p47phox mouse knock-out model ofchronic granulomatous disease. J. Exp. Med. 182, 751–758 (1995).
32. Bellocchio, S. et al. TLRs govern neutrophil activity in aspergillosis. J. Immunol. 173,7406–7415 (2004).
33. Romani, L. et al. Thymosin a1 activates dendritic cell tryptophan catabolism andestablishes a regulatory environment for balance of inflammation and tolerance.Blood 108, 2265–2274 (2006).
34. Morgenstern, D. E., Gifford, M. A., Li, L. L., Doerschuk, C. M. & Dinauer, M. C.Absence of respiratory burst in X-linked chronic granulomatous disease miceleads to abnormalities in both host defense and inflammatory response toAspergillus fumigatus. J. Exp. Med. 185, 207–218 (1997).
35. Spieker-Polet, H., Sethupathi, P., Yam, P. C. & Knight, K. L. Rabbit monoclonalantibodies: generating a fusion partner to produce rabbit–rabbit hybridomas.Proc. Natl Acad. Sci. USA 92, 9348–9352 (1995).
36. Fallarino, F. et al. Modulation of tryptophan catabolism by regulatory T cells.Nature Immunol. 4, 1206–1212 (2003).
37. Grohmann, U. et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. NatureImmunol. 3, 1097–1101 (2002).
38. Tramonti, D., Andrew, E. M., Rhodes, K., Newton, D. J. & Carding, S. R. Evidence forthe opposing roles of different cd T cell subsets in macrophage homeostasis. Eur.J. Immunol. 36, 1729–1738 (2006).
39. Jackson, S. H. et al. IFN-c is effective in reducing infections in the mouse model ofchronic granulomatous disease (CGD). J. Interferon Cytokine Res. 21, 567–573(2001).
doi:10.1038/nature06471
Nature ©2007 Publishing Group





























