Embed Size (px)
Citation preview

Molecular Psychiatry (2021) 26:2685–2706https://doi.org/10.1038/s41380-020-00999-7
REVIEW ARTICLE
Current and future applications of induced pluripotent stemcell-based models to study pathological proteins inneurodegenerative disorders
Aurélie de Rus Jacquet1,2 ● Hélèna L. Denis1,2 ● Francesca Cicchetti 1,2● Melanie Alpaugh1,2
Received: 25 September 2020 / Revised: 2 December 2020 / Accepted: 9 December 2020 / Published online: 25 January 2021© The Author(s) 2020, corrected publication 2021
AbstractNeurodegenerative disorders emerge from the failure of intricate cellular mechanisms, which ultimately lead to the loss ofvulnerable neuronal populations. Research conducted across several laboratories has now provided compelling evidence thatpathogenic proteins can also contribute to non-cell autonomous toxicity in several neurodegenerative contexts, includingAlzheimer’s, Parkinson’s, and Huntington’s diseases as well as Amyotrophic Lateral Sclerosis. Given the nearly ubiquitousnature of abnormal protein accumulation in such disorders, elucidating the mechanisms and routes underlying theseprocesses is essential to the development of effective treatments. To this end, physiologically relevant human in vitro modelsare critical to understand the processes surrounding uptake, release and nucleation under physiological or pathologicalconditions. This review explores the use of human-induced pluripotent stem cells (iPSCs) to study prion-like proteinpropagation in neurodegenerative diseases, discusses advantages and limitations of this model, and presents emergingtechnologies that, combined with the use of iPSC-based models, will provide powerful model systems to propel fundamentalresearch forward.
The origin of the prion hypothesis forneurodegenerative diseases
The prion disorder scrapie was first described in animals asearly as the 18th century, but the disease-causing agent, theprion protein (PrP), was only identified in the 1980s [1, 2]. Inthe intervening time, a number of disorders involving the sameprotein were reported in mammals, including chronic wastingdisease in cervids, bovine spongiform encephalopathy in cowsand Creutzfeldt–Jakob’s disease (CJD) in humans [1]. Thesedisorders, known as transmissible spongiform encephalo-pathies, first came to the public eye when the bovine
spongiform encephalopathy epidemic made front page newsin the early 2000s. Research into PrP gained momentum whenevidence of a novel variant of CJD was linked to ingestion ofinfected meat [3]. Unlike other disease-causing agents, such asviruses and bacteria, the key information leading to patho-genicity of prion and prion-like proteins is not encoded in thegenetic material but instead, in the structure and biophysicalproperties of the misfolded protein. In general, the formationof dimers and oligomers is the rate-limiting-step in the proteinaggregation process [4]. The presence of the misfolded proteincan alter this by acting as a catalyst for the conformationalchange of the endogenous protein [4]. While these mechan-isms of protein infectivity were first described for the PrPprotein, the majority, if not all proteins associated with neu-rodegenerative diseases such as Alzheimer’s disease (AD),Parkinson’s disease (PD), Huntington’s disease (HD) andAmyotrophic Lateral Sclerosis (ALS), are now recognized tohave prion-like properties (Fig. 1a).
Evidence of prion-like properties for proteins such as α-Synuclein (α-Syn), tau, amyloid-β (Aβ), huntingtin (HTT),superoxide dismutase (SOD1) and TAR DNA-binding 43protein (TDP-43) have slowly been accruing over decades ofresearch through the use of diverse techniques such asin vitro protein conversion assays and immunohistochemical
* Francesca [email protected]
* Melanie [email protected]
1 Centre de Recherche du CHU de Québec - Université Laval, AxeNeurosciences, Québec, QC G1V 4G2, Canada
2 Département de Psychiatrie & Neurosciences, Université Laval,Québec, QC G1V 0A6, Canada
1234
5678
90();,:
1234567890();,:

assessment of post-mortem tissue. Some of the earliestindications of prion-like properties came from the study ofbrains of PD patients who had received fetal tissue trans-plants. Detailed analysis of the grafts unveiled the presenceof aggregated α-Syn within this previously healthy fetaltissue [5–8]. Seminal work conducted by Braak and
colleagues on the characterization of the spread of α-Synacross disease stages provided an additional indication thatproteins may indeed propagate between cells. From a sys-tematic study of post-mortem tissue, Braak et al. hypothe-sized that α-Syn pathology follows a specific pattern ofpropagation, originating in the medulla oblangata and
2686 A. de Rus Jacquet et al.

migrating through synaptically connected regions, includinginto the substantia nigra pars compacta [9]. These findingsoffered a strong conceptual framework for subsequent stu-dies of the prion-like behavior of α-Syn. For example, it wasthereafter reported that injections of either PD brain homo-genates or α-Syn fibrils into the brains of mice and non-human primates were sufficient to induce the loss of dopa-minergic neurons and the development of motor impair-ments [10–12]. More recent experimental paradigms haveexpanded on this notion by demonstrating that injection ofα-Syn into the peripheral nervous system is sufficient tocause central disease in rodents [13, 14]. Meanwhile,detection of pathogenic α-Syn seeds in the brains or cerebralspinal fluid of PD patients has been shown to be a selectiveand sensitive diagnostic tool [15, 16], further supporting therelationship between misfolded species and diseaseprogression.
The tracking of tau spreading throughout the progressionof AD was also performed by Braak et al. several yearsprior to the description of α-Syn pathology in PD [17].More recently, other proteins associated with commonneurodegenerative disorders have been administered viainjection of brain homogenates derived from post-mortemtissue of AD and frontotemporal lobe dementia (FTD)patients and were reported to lead to Aβ, tau, and TDP-43pathology [18–21]. Furthermore, tau [22], Aβ [23], α-Syn[10, 24], mutant HTT (mHTT) [25], and SOD1 [26] oli-gomers/fibrils have been used in a wide variety of in vitroand in vivo experiments to demonstrate their neurotoxicity.There is now ample evidence that prion-like proteotoxicityis sufficient to provoke the manifestation of common neu-rodegenerative diseases in a non-cell autonomous manner.However, despite extensive research, much remains to be
learned about the mechanisms of prion-like properties aswell as the degree to which they contribute to diseasemanifestation and/or progression. Disease-relevant modelsare needed to unravel the molecular mechanisms of prionpathogenesis and thereby design novel protein-targetedtherapies. One powerful model system with the potential togreatly enhance our understanding of the contributions ofpathological proteins to neurodegenerative disordersinvolves the use of human-induced pluripotent stem cells(iPSCs). In this review, we discuss molecular and species-specific advantages of iPSCs, studies that have previouslybeen performed in iPSCs to model prion-like protein pro-pagation, current limitations that need to be considered, aswell as recently developed technologies that are likely toadvance the field.
Considerations for modeling propertiesof pathological proteins
Species-specific cellular differences
Mouse models have been developed and widely used tostudy environmental and genetic determinants of neurode-generative diseases [27, 28]. However, these genetic modelsfail to reproduce all pathological features of human neuro-degenerative disorders. For example, rodent models of AD,PD and HD rarely display overt neuronal cell death or theprogressive neurodegeneration which characterizes thesehuman conditions [27, 29, 30]. In addition, evidencedemonstrating intrinsic functional differences betweenhuman and mouse cells has begun to emerge, includingdifferences in developmental stages, the rate of proteindegradation during development, RNA processing, glial cellbiology and global transcriptomes [28, 31–34]. Thesespecies-specific differences likely account for some of thechallenges faced by mouse-based systems in modelinghuman diseases.
Species-specific protein differences
While all of these differences have important consequencesfor the understanding and interpretation of studies usingmice, one particularly salient difference, in the context ofprion-like disorders, is variations in the peptide sequences,isoform expression, or post-translational modification ofproteins associated with common neurodegenerative dis-eases (Fig. 1b). The details of the most frequent modifiers ofprotein toxicity and/or aggregation, as well as differencesbetween mice and humans, are summarized in Table 1.While such difficulties can be overcome using humanizedmice, purified fibrils, or by adding high doses of seed-competent proteins into a culture system, the inherent
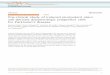
Fig. 1 Prion-like proteins in human diseases and iPSC models.Protein aggregation is associated with multiple neurodegenerativedisorders, involves disease-specific proteins, and affects uniquepopulations of neurons (a). Variability in the propensity to aggregateand inefficient cross-species seeding between human and mouse pro-tein homologs have been observed (b). Ex vivo/test tube and in vitroassays including primary cultures, immortalized cells and iPSC-derived cells are commonly used to study protein propagation.Advantages and disadvantages of each model are listed (c). iPSC-based models enable the production of human neuronal populations ofinterest that better recapitulate physiological features of human dis-eases compared to rodent-based models. This unprecedented access tohuman cells opens new avenues for the study of mechanisms asso-ciated with propagation, seeding, and toxicity of pathological proteins(d). As with any disease model, a number of advantages and dis-advantages need to be carefully considered to inform and guideexperimental choices (d). α-Syn alpha-synuclein, Aβ amyloid beta,AD Alzheimer’s disease, ALS Amyotrophic lateral sclerosis, Cas9CRISPR associated protein 9, CRISPR Clustered Regularly Inter-spaced Short Palindromic Repeats, GABA gamma aminobutyric acid,HD Huntington’s disease, LB Lewy body, mHTT mutant huntingtin,PD Parkinson’s disease, SOD1 superoxide dismutase 1, TDP-43 TARDNA-binding 43 protein.
IPSCs to study prion-like mechanisms 2687

structural differences in the proteins can be efficaciouslycircumvented through the use of human cell models(Fig. 1c). For these reasons, neurons and glia derived fromiPSCs are attractive alternatives to rodent primary culturesas they produce human-specific cell types.
PrP
Prion diseases are rare amongst neurodegenerative protei-nopathies in that they are not human-specific conditions,as other mammalian species also spontaneously develop
Table 1 Characteristics and species-specific differences in common [219–272] prion-like proteins.
α-Syn alpha-synuclein, Aβ amyloid beta, AD Alzheimer’s disease, ALS Amyotrophic lateral sclerosis, AP amyloid plaques, APP amyloid precursorprotein gene, CAA cerebral amyloid angiopathy, CBD corticobasal degeneration, CHR chromosome, CJD Creutzfeldt–Jakob’s disease, CP coredplaques, COP cored-only plaques, CTE chronic traumatic encephalopathy, DLB dementia with Lewy bodies, DN dystrophic neurites, DNP/CNP denseneuritic plaques/cored neuritic plaques, DP diffuse plaques, DS Down’s syndrome, FI fatal insomnia, FTLD frontotemporal lobar degeneration, FTDP-17 frontotemporal dementia with parkinsonism linked to chromosome 17, GFT glial fibrillary tangles,GGI globular glial inclusions,GI glial inclusions,GSS Gerstmann–Sträussler–Scheinker syndrome, HD Huntington’s disease, HpScl hippocampal sclerosis, HTT huntingtin protein, Htt huntingtin gene,MAPT microtubule-associated protein tau gene, mHTTmutant huntingtin,MSA multi-system atrophy, Namino-terminal inserts, NCI neural cytoplasmicinclusions, NFL neurofibrillary lesions, NFT neurofibrillary tangles, NII neuronal intranuclear inclusions, NP neuritic plaques, NT neuropil threads, PiDPick’s disease, PD Parkinson’s disease, PDD Parkinson’s disease dementia, PHF paired helical filaments, PMA progressive muscular atrophy, PolyQpolyglutamine, PSP supranuclear palsy, PRNP gene encoding for PrP, PrP prion protein, PrP-CAA pure prion protein cerebral amyloid angiopathy,PrP-SA PrP‐systemic amyloidosis, SF straight filaments, SNCA gene encoding for α-Syn, SOD1 superoxide dismutase protein, SOD1 superoxidedismutase gene, S serine, SORT1 transmembrane receptor sortilin, T threonine, TARDP TDP-43 gene, TDP-43 TAR-binding domain protein-43, TsAsthorn-shaped astrocytes, VPSPr variably protease-sensitive prionopathy, Y tyrosine, 3R 3 repeats, 4R 4 repeats.
2688 A. de Rus Jacquet et al.

transmissible spongiform encephalopathies (e.g. cervids)[35]. Most human neurodegenerative disorders do not havea naturally occurring mammalian equivalent, thus in vivostudies generally involve investigation of the humanpathological protein expressed in the model organism ofinterest. However, the presence of bona fide transmissiblespongiform encephalopathies (e.g. CJD, kuru and scrapie)in multiple species has diversified the sources of misfoldedproteins available in the prion field. PrP scrapie (PrPSc), theinfectious form of PrP, contained in homogenates fromsheep, cows and humans are all routinely tested in multiplemammalian species to better understand the factors essentialto interspecies disease transmission. Such studies haveidentified the amino acid sequence, folding conformationand glycosylation of the prion protein as factors that con-tribute to the presence or absence of species barriers[36–38]. One recent study has even suggested that singleamino acid residues in horse and dog PrP contribute to therelative resistance of these animals to experimentally-induced disease [39]. While the gene encoding for PrP(PRNP) is highly conserved in mammals, substitutions haveoccurred across evolution. Mice and humans differ by only20 amino acids [40] (Table 1), but this is sufficient to limittransmission of disease from humans to mice. To overcomethis, transgenic animals have been generated to express achimeric form of PrP which facilitates human to mousetransmission [41].
Tau
While the tau protein shares similarities between mice andhumans, not all splice variants are equally represented ineach species and the murine N-terminal peptide sequencelacks 11 residues present in the canonical human homolog[42, 43] (Table 1). In the human adult central nervoussystem (CNS), tau has 6 common isoforms that derive fromalternative splicing of Exons 2, 3 and 10. The tau variantthat includes Exon 10 has four microtubule-binding repeatsand is referred to as 4R tau, while exclusion of Exon 10results in 3R tau which contains only three microtubule-binding domains [42]. Inclusion or exclusion of Exon 10 isdevelopmentally regulated in humans, with 3R taudemonstrating dominant expression in the juvenile nervoussystem, while 3R and 4R tau are found in similar propor-tions in the adult CNS. In mice, this change is more dra-matic with 3R tau being entirely absent from the adultmouse brain [42] (Table 1). In humans, the balance between3R and 4R is very important. In fact, mutations in MAPTaffecting alternative splicing of Exon 10 lead to the devel-opment of tauopathies with preferential accumulation ofeither 3R or 4R, depending on the mutation [44–47]. Thepresence or absence of the extra microtubule bindingdomain also impacts spreading and seeding of tau, with
analysis of cross-seeding between 4R and 3R tau, sug-gesting that 3R tau can recruit 4R tau, but that the reversedoes not occur [48]. Furthermore, both 3R and 4R tau havethe capacity to trigger aggregation, but injected 4R tau canspread to more remote areas in vivo [49]. Given the com-plex relationship between 3R and 4R tau in human disease,the absence of 3R tau in the adult mouse CNS may haveimportant ramifications. Recent studies have additionallydescribed discrepancies in the N-terminal portion of mouseand human tau. These changes are suspected to alter inter-actions between tau and other proteins, including end-binding proteins, which may impact the release of tau aswell as the formation of aggregates [43, 50].
Aβ
Unlike tau, no differences in Aβ isoform expression havebeen reported between humans and mice. Structurally,mouse and human Aβ are very similar, differing by only 3amino acid substitutions in the N-terminal domain [51, 52](Table 1). Despite the similarity in sequence, cell cultureanalyses with human and mouse Aβ show both differentialseeding capacity and toxicity [51]. After 48 h of exposure tohuman Aβ, congo red positive amyloid fibrils are visible incultured SH-SY5Y cells, while no aggregates are detected72 h following treatment with rodent Aβ [51]. Animal stu-dies show that overexpression of mouse Aβ is insufficient toinduce pathological changes or to exacerbate plaque for-mation when mouse and human Aβ are simultaneouslyoverexpressed [52]. A transgenic background is alsorequired for Aβ-containing homogenates from AD patientsto induce behavioral and neuropathological phenotypes[53].
α-Syn
Comparison of α-Syn between species has revealed thatonly 7 amino acids differ between canonical human andmouse homologs (Table 1). However, these changes impactthe electrophoretic mobility, conformation of fibrils, clea-vage by proteases and cross-seeding [54, 55]. Both humanand mouse α-Syn can efficiently seed proteins from thesame species, suggesting no inherent differences in thekinetics of aggregation. Instead, the alterations in the aminoacid sequence seem to only influence cross-species seeding[55]. Importantly, the rate of cross-seeding can be modifiedby adjusting the amino acid sequence [54, 55]. More recentwork has shown that mouse and human α-Syn producedistinct fibrillar structures with human protein formingtwisted fibrils and the mouse protein generating straightfibrils [54]. These structures could explain why mouse α-Syn can inhibit aggregation of the human protein (Table 1)[56]. This inhibitory effect was clearly demonstrated in one
IPSCs to study prion-like mechanisms 2689

series of experiments where human α-Syn was expressed inmice in the presence or absence of mouse α-Syn within thegenetic background. Primary neurons expressing onlyhuman α-Syn formed Lewy-body like inclusions, whichwere absent when mouse α-Syn was also expressed [56].Similar results were observed in the brains of transgenicanimals, where the lack of mouse α-Syn increased thetendency of human α-Syn to aggregate [56].
HTT
Multiple changes in the N-terminal structure have beendetected between canonical mouse and human homologs ofHTT, including differences in the average number ofpolyglutamine repeats and in the organization of the poly-proline region [57] (Table 1). The potential consequences ofthe fluctuations in the polyglutamine number are inferredfrom clinical data indicating that the age of onset is inver-sely correlated with the number of CAG repeats [58]. Theconsequences of variations in the polyproline region are lessapparent, but they result in an altered structure of the mouseprotein [57] which may in turn change HTT’s capacity tomisfold. Further evidence of the relevance of this polypro-line stretch comes from the heightened phenotype of chi-meric knock-in mouse models expressing the pathogenichuman HTT exon fragment 1 (HTTExon 1) in the normalmouse gene [59], as opposed to the milder phenotypedetected in a non-chimeric knock-in mouse model withequivalent CAG expansions [60].
SOD1
Between mice and humans, 25 amino acids differ in thecanonical SOD1 protein [61]. Similar to what has beenreported for Aβ, these differences result in a mouse proteinwhich is less prone to aggregate than its human homolog[62]. Furthermore, when human SOD1 is added into thebrains of SOD1 transgenic mice, the mouse form of theprotein is absent from the aggregates, while the endogenoushuman protein is present [61].
TDP-43
TDP-43 has very high homology between mice andhumans, with only 14 of 414 amino acids differing betweenthe canonical sequences of the two species [63]. Despite thisoverlap, there are numerous physiological differences. Forexample, human and mouse TDP-43 are transcribed dif-ferently such that human TDP-43 has nine less mRNAtranscript variants and one fewer protein isoform thanmouse TDP-43 [64]. These features have important func-tional consequences, which are highlighted by differencesin progranulin release in mouse and human cells.
Progranulin release is largely dependent on the receptorprotein sortilin 1, whose alternative splicing is influenced byTDP-43. In mouse cells, elimination of TDP-43 results inincreased expression of Sort1ex17b, while in human cellsthe SORT1Δex17b isoform is increased [65]. This alteredisoform ratio in the two species leads to a difference in thenumber of receptors detectable within the membrane,with murine cells displaying constant expression levels,while human cells show an increase in the number ofreceptors [65].
Current in vitro systems modeling thebehavior of prion-like proteins
Non-iPSC-based models
Studies evaluating the capacity of different proteins to seedpathology have frequently used test tube-based measuressuch as the amyloid seeding assay (ASA) [66], real-timequaking-induced conversion (RT-QuiC) [67] and proteinmisfolding cyclic amplification (PMCA) [68]. Test-tubeassays are valuable methods for determining whether seed-competent forms of proteins are present within biologicalsamples, as well as measuring the kinetics of the seedingreaction [69, 70] (Fig. 1c). However, such assays are spe-cialized towards assessment of seeding under controlledconditions and do not represent the rate of aggregateformation within cellular systems. As this review isfocused on cellular models, test-tube assays will not bediscussed further.
Assessment of spreading, toxicity and seeding, underphysiological conditions, has traditionally been performedin primary and immortalized cells (Fig. 1c). Primary cellsare isolated directly from humans or model organisms, andgenerally retain characteristics of their tissue of origin.Challenges with these cells include the presence of multiplecell types and low yields. Cell number is further limited bythe lack of cell division of some primary cells in culture[71]. While this is representative of physiological condi-tions, it does prevent expansion of cells after isolation.Alternatively, several groups use immortalized cell lines[72]. These cells are homogenous and easy to grow, as theyhave the capacity to expand over extended periods of time,although the presence of genetic mutations may not repre-sent normal physiology (Fig. 1c).
Despite these drawbacks, work with cell culture modelshas identified multiple routes by which pathogenic proteinscan spread, including through the release and uptake ofextracellular vesicles or soluble proteins, synapses, glialphagocytosis and tunneling nanotubes [73–75]. While thebasic uptake and release of specific proteins seem to be wellconserved, the rate of uptake and response differs between
2690 A. de Rus Jacquet et al.

models depending on the tissue source and the metabolic stateof the cells, with more active non-terminally differentiatedprecursor cells demonstrating greater uptake than matureneurons [76]. Some toxicity experiments have further showndifferential susceptibility of cell lines and primary cells totoxicity induced by aggregates [77]. In genomic studies, pri-mary cells have been compared to their immortalizedequivalents and, when cell type-specific genes were con-sidered, the R2 value approached 0.5, indicating a poor con-cordance between the two cell types [78]. Genes related tocell division and apoptosis were particularly affected, with astrong enrichment in immortalized cells. A similar phenom-enon has been described for the human neuroblastoma cellline SH-SY5Y. SH-SY5Y cells have elevated N-Myc levels,which has been suggested to modify their response to apop-totic stimuli [79]. Such changes render toxicity studies inimmortalized cells challenging. Primary cells are furtherproblematic as availability of human primary brain cells islimited and cells from animal models can differ from theirhuman counterparts (Fig. 1c).
Human iPSC-based systems
Pluripotent stem cells were successfully isolated and cul-tured from mice embryos for the first time in the early 1980s[80]. Their capacity to maintain pluripotency in cell culture,almost indefinitely, has made them an important researchtool. The potential of these cells to contribute to both basicand clinical research was enhanced when a landmark studyidentified c-Myc, Klf4, Sox2 and Oct3/4 as the fouressential transcription factors for inducing pluripotency inadult mouse somatic cells [81]. Subsequent work confirmedthat these same factors induced pluripotency in adult humansomatic cells [82, 83]. This discovery has led to thedevelopment of numerous protocols for the differentiationof various cell types, including cells of the CNS such asastrocytes [84], neurons [85], oligodendrocytes [86] andmicroglia [87].
The feasibility and utility of iPSC models to study prion-like proteins have been demonstrated in studies assessingthe specific toxicity profiles of different pools of Aβ derivedfrom control or AD patient brains [88], as well as thespontaneous aggregation of tau harboring P301L andV337M disease-associated mutations [89]. In addition to theadvantages of having a model with human proteins, iPSCscan mimic multiple pathological features observed inpatients, including selective vulnerability of neuronal sub-populations [90, 91], mitochondrial dysfunction [92, 93] orimpairment of protein degradation pathways [94, 95].Modeling neurodegenerative diseases with iPSC-derivedcells is a powerful approach that offers a human-basedsystem to elucidate mechanisms of disease onset and
progression, including pathogenic prion-like seed formationand trans-cellular protein propagation (Fig. 1c).
Protein release, uptake and propagation iniPSC-based systems
A significant number of studies related to protein spreadinghave focused on characterizing the release, subsequentuptake and response of cells to pathogenic proteins; majorevents that contribute to protein propagation. After uptakeby naive cells, pathological proteins act as seeds by inter-acting with homologous proteins to increase the likelihoodof a shift from a normal to a pathological conformation [96].To date, iPSCs have been used to study how disease-associated proteins such as tau [74, 97, 98], Aβ [99], α-Syn[100], mHTT [101] and SOD1 [26] are released and takenup by neighboring cells (Table 2). Unless otherwise stated,the studies reviewed in the following sections refer tohuman iPSCs.
Release of pathogenic proteins
Studies assessing protein release from iPSCs have largelyfocused on tau with only one study addressing Aβ. Studiesrelated to tau have demonstrated that healthy iPSC-derivedneurons release both soluble and aggregated forms of theprotein. The secretion of soluble forms of tau occurs inde-pendently of the classic endoplasmic reticulum and Golgiapparatus secretory pathway, and appears to be temperature-dependent [97, 98, 102]. AD and control iPSC lines havebeen used for these studies and both models demonstratedthat release occurred independently of cell death. However,AD-related lines indicate that different forms of tau may bereleased under pathological conditions. Expression of anAD-relevant mutation in the Presenilin 1 (PSEN1) gene wasassociated with a change in the lengths of the tau fragmentsreleased into the media [98]. Similarly, iPSC lines withgenetic mutations in the AD-related genes Aβ precursorprotein (APP) or PSEN1 have been found to release moreAβ42 than Aβ40 [99]. While these two studies involvedPSEN1, mutations in the tau repeat domain (tau-P301L-V337M) have also been shown to alter release of patholo-gical proteins. Specifically, such cells were described tospontaneously form and secrete aggregated tau proteins[89], which is in contrast to healthy cells where the majorityof released tau was non-aggregated and free-floating[98, 102, 103]. Collectively, these studies indicate thatprotein release is a physiological mechanism that is notspecific to pathogenic processes, but that the form of theprotein released can be influenced by disease-causingmutations.
IPSCs to study prion-like mechanisms 2691

Uptake of pathogenic proteins
The biology of tau uptake has also been a focus of researchrelated to protein propagation. In particular, it has beenreported that misfolded and native forms of tau are inter-nalized to similar levels in control iPSC-derived corticalneurons, although there are some differences in the cellularmachinery involved in uptake [74]. Indeed, healthy iPSC-derived excitatory cortical neurons internalize P301S taumonomers via dynamin-dependent endocytosis or macro-pinocytosis, while P301S tau aggregates are primarily takenup by micropinocytosis [74]. Additional studies on the
mechanisms of prion-like propagation of tau have shown thatexogenous tau oligomers or preformed fibrils can increase thelevel of pathological phosphorylated tau within healthy orMAPT P301L-expressing iPSC-derived cortical neurons,trigger a conformational change, and recruit endogenous tauto promote aggregation [104, 105] (Table 2).
Disease models using iPSCs have also contributed to theelucidation of mechanisms of uptake of SOD1 aggregates.For example, iPSC-derived motor neurons exposed toSOD1-preformed aggregates undergo Rac1-mediatedmembrane ruffling and blebbing characteristic of macro-pinocytosis, and subsequent internalization of these protein
Table 2 Summary of iPSC and prion-like mechanistic studies.
α-SYN alpha-synuclein, Aβ amyloid beta, AD Alzheimer’s disease, CJD Creutzfeldt-Jakob’s disease, E glutamic acid, GSSGerstmann–Sträussler–Scheinker syndrome, GABAergic gamma aminobutyric acid expressing neurons, iPSC induced pluripotent stem cells, Klysine, mHTT mutant huntingtin, PRNP gene encoding for PrP, PrPC cellular non-pathogenic prion protein, PrPSC pathogenic prion protein, ROSreactive oxygen species, S serine, SNCA gene encoding for α-SYN, SOD1 superoxide dismutase 1, WT HTT wild-type huntingtin.
2692 A. de Rus Jacquet et al.

aggregates [26]. Finally, the propagation of mHTT aggre-gates was highlighted using an ex vivo chimeric model ofHD, where organotypic brain slices were prepared from asevere transgenic mouse model of HD (R6/2) and injectedwith control iPSC-derived human neurons. mHTT aggre-gates could be transferred from mouse tissue to healthyhuman neurons and affect their morphology, suggesting thattrans-cellular spread of mHTT from mouse to human neu-rons mediated neuronal impairment [101].
Propagation and associated toxicity of pathogenicproteins
While a significant amount of work has been conducted tounderstand the factors that influence prion-like protein release,uptake and the mechanisms mediating these processes, fewstudies have addressed the functional consequences of toxicproteins on iPSC survival and function. Two different methodshave been utilized for these experiments; genetic mutation/overexpression and addition of exogenous pathological pro-teins. Studies using genetic manipulation demonstrated thatSNCA duplication, E46K and E57K α-Syn, as well as mutatedforms of tau, tau-A152T and tau-P301L-V337M, result inspontaneous α-Syn oligomerization or tau aggregation[89, 106, 107]. Impairments in the anterograde axonal trans-port of iPSC-derived cortical neurons along with synapticdegeneration accompanied α-Syn oligomerization, recapitu-lating elements of early neurodegeneration in synucleino-pathies [106]. Similarly, iPSC-derived cortical neuronsexpressing tau-A152T to model FTD showed that accumula-tion of intracellular insoluble tau and phospho-tau (S396)correlated with neuronal vulnerability to cellular stressors,including exogenous Aβ (1–42) [107]. While the mutation oftau facilitated accumulation of pathological proteins in theaforementioned publication, not all genetic modifications stu-died have shown similar results. For example, iPSC-derivedneurons harboring the rare Y218N PRNP mutation associatedwith Gerstmann-Sträussler-Scheinker syndrome internalized,but failed to replicate and propagate PrPSC [108].
Exogenous administration of tau, Aβ, α-Syn and mHTThave all been tested experimentally. Specifically, addition oftau increased aggregation/phosphorylation of the endogen-ous tau protein and triggered conformational changes withinneurons [104, 105]. Addition of Aβ oligomers promoted theproduction of harmful reactive oxygen species in controlcortical neurons [109]. Addition of mHTT fibrils toGABAergic cells induced neuronal atrophy and neur-ite shortening [25] while addition of α-Syn ribbons andfibrils caused changes in spontaneous calcium oscillationsand mitochondrial morphology [110]. One very recentstudy further evaluated possible interactions between PrPand other prion-like proteins. iPSC-derived cortical neuronsgenetically edited to suppress PrP expression and their
isogenic controls were exposed to α-Syn, Aβ or tau solubleaggregates, as well as pathogenic human brain lysates fromAD, Pick’s disease or dementia with Lewy bodies. Cellslacking PrP had no neurodegenerative phenotypes, whileisogenic controls exhibited functional and morphologicalalterations [111]. These intriguing results suggest that PrPmediates, at least in part, the toxicity triggered by otherprion-like proteins and could serve as a therapeutic targetfor a number of neurodegenerative diseases. Together, thesestudies provide strong support for the use of iPSC models tostudy the effects of pathological proteins on human neuronsby mirroring phenotypes observed in disease settings.
Glia and pathological proteins
Neuron-to-neuron spread of prion-like proteins is anessential aspect of pathology, but evidence suggests thatmicroglia and astrocytes may also play a role, either throughattempted degradation of proteins or via glia-to-neuron orglia-to-glia transfer.
Glia-mediated protein clearance
Phagocytic abilities of microglia and astrocytes are essentialto maintain tissue homeostasis and several studies using non-iPSC models, as well as in vivo models, have demonstratedtheir ability to recognize and uptake α-Syn [112–115] tau[116–118], Aβ [119, 120] or mHTT [121] aggregates.Recently, studies using iPSC-based systems have also con-tributed to this body of work by demonstrating that expressionof APOE4 [122], the PSEN1ΔE9 mutation [123], a homo-zygous KM670/671NL mutation in APP [124] or knockout ofAPP [124] is sufficient to disrupt uptake of Aβ1-42 byastrocytes. Furthermore, astrocytes expressing the PSEN1ΔE9mutation not only lost their ability to uptake the Aβ1–42fragment, but also increased the number of Aβ fragmentsthey released into the media [123].
Few studies on the functional consequences of disease-related mutations on iPSC-derived microglia are available.However, two recent reports have shown that microgliadifferentiated from iPSCs expressing either the APOE4variant or containing a mutated C9orf72 gene, a known riskfactor for ALS and FTD, have a reduced ability to degradeinternalized Aβ1-40 fragments, leading to pathological Aβ1-40 accumulation [122, 125]. When internalized by a reci-pient glial cell, pathological proteins can accumulate, forminclusion bodies and subsequently trigger a series of cellulardysfunctions that further drive the propagation process.
Protein spread
In addition to their role in protein clearance, microglia andastrocytes may also exacerbate pathological protein spread
IPSCs to study prion-like mechanisms 2693

by participating in cell-to-cell transfer. Embryonic stemcell-derived astrocytes exposed to aggregated α-Syn pro-duce tunneling nanotubes to communicate with neighboringhealthy astrocytes and recover healthy mitochondria fromthese cells. This process results in the passage of accumu-lated α-Syn from the damaged cell to the healthy neighbor[126]. Exposure of iPSC-derived astrocytes with differentPRNP variants to prions originating from the brain of CJDpatients expressing the same PRNP variant also results inthe internalization, self-propagating replication and accu-mulation of PrPsc [127]. Lysates prepared from PrPsc-posi-tive astrocytes infect and propagate the PrPsc pathology tonaive astrocytes. In addition to passing pathological pro-teins to other astrocytes, iPSC-derived astrocytes with thePD-related mutation LRRK2 G2019S convey endogenousα-Syn to iPSC-derived dopaminergic neurons [91]. Whilestudies on transfer between glia and other cells is limited,the evidence to date suggests that they may be importantcontributors and more models incorporating multiple celltypes could provide valuable information.
Idiopathic vs. familial etiology
A unique application of iPSC-based technologies is tocompare the role and behavior of pathological proteins inidiopathic vs. familial forms of neurodegenerative diseases.For example, studies of sporadic and familial AD haveshown differences in the levels of phospho tau (Thr 231),secreted Aβ40 and Aβ42/Aβ40 ratios [128, 129]. While fewchanges were reported in familial vs. sporadic neuronalcultures, a greater Aβ42/Aβ40 ratio in familial AD wasobserved. For example, motor neurons differentiated from32 sporadic ALS-derived iPSCs (e.g., SOD1 and TDP-43mutants) displayed shorter neurites, higher levels of lactatedehydrogenase and abnormal production of protein aggre-gates as compared to control neurons [130]. However, theseverity of these phenotypes varied between differentsporadic patients, and overall the cells did not recapitulatethe SOD1 protein aggregation observed in SOD1 familialALS, but instead demonstrated formation of cytosolic TDP-43 aggregates. This study therefore indicates that sporadic
Fig. 2 Challenges and perspectives of iPSCs for modeling protei-nopathies. Neurons differentiated from iPSCs tend to have an immaturephenotype, which can impact cellular functions critical to the successfulmodeling of prion-like protein propagation and seeding (a, b). Newtechniques such as brain organoids (c), microfluidic devices (d) andCRISPR/Cas9 gene editing (e) are important tools to overcome these
limitations and further establish iPSCs as a model of choice to study theproperties and role of prion-like proteins in neurodegenerative diseases.Cas9 CRISPR associated protein 9, CRISPR Clustered RegularlyInterspaced Short Palindromic Repeats, Pop. 1 population 1, Pop. 2population 2, Pop. 3 population 3, 3D three-dimensional.
2694 A. de Rus Jacquet et al.

ALS can be modeled using iPSC-based technologies andthat these systems do not necessarily match phenotypesfrom familial forms of disease. Such methods have thecapacity to not only increase our understanding of diseasemechanisms, but also to screen for potential drug candidatesthat mitigate neurodegeneration across all forms of ALS.
While such investigations are still nascent, they showgreat promise towards improving our understanding of thegenetic determinants of protein spreading and seeding andoffer unique models to design personalized medicine.Genetic-based models may not be representative of themore common idiopathic forms of neurodegenerative dis-eases and iPSC systems could provide a platform to modelidiopathic disease as well. However, further validation ofiPSC models generated from idiopathic cases will berequired to confirm that they accurately recapitulatepathology across the spectrum of etiologies and neurode-generative diseases.
Challenges and perspectives of iPSC-derivedmodels
Biochemical maturation and aging of iPSC-derivedneurons
Functional maturity and age-related cellular changes areparamount considerations when studying prion-like proteins(Fig. 2), as some proteins, such as tau (Table 1), are char-acterized by changes in splicing or expression throughoutdevelopment [42]. Since iPSC-derived neurons show animmature phenotype, they tend to express more juvenileforms of proteins including predominant expression of 3Rtau [131]. Although iPSCs are generally derived from adultcells, the process of reprogramming resets markers of cel-lular age such that the cells effectively return to anembryonic state [132]. In addition to altering the pattern ofexpression of proteins such as tau, age is the greatest riskfactor for developing neurodegenerative disorders. While themechanisms are still unknown, age has been associated withincreased protein spreading [133]. To some extent, co-culture with glia, 3D-culturing and increased time in culturecan overcome these limitations but these may not be suffi-cient to fully replicate processes that take decades[131, 134]. An alternative emerging technology involves theexpression of progerin, a mutated form of lamin A respon-sible for the premature aging phenotype observed in patientssuffering from the Hutchinson-Gilford progeria syndrome.When progerin-related aging is induced in dopaminergicneurons derived from PD patients, the cells acquire char-acteristic disease phenotypes, including altered morphologyand protein inclusions [135]. Another interesting approachinvolves the direct conversion of adult cells into neurons,
thereby avoiding a pluripotent intermediate stage [136–139].This method has been shown, in the case of HD, to generateinduced neurons which retain markers of cellular age anddisplay stronger disease phenotypes, including overt neuro-nal degeneration and protein aggregation [140]. While thistechnology is still quite novel, there are indications that itmay more faithfully mimic age-dependent molecularmechanisms of degeneration and could therefore providenew insights into the processes of protein release, uptake andseeding.
As the field moves forward, models that consistentlydisplay spontaneous formation of protein aggregates willbecome invaluable resources to mimic disease phenotypesand elucidate early events responsible for protein misfold-ing and formation of pathogenic seeds. Spontaneous for-mation of intracellular misfolded/aggregated proteins hasbeen observed in both sporadic and familial forms of dis-ease [89, 106, 107, 141], however, these results have beenvariable. In one study, sixteen lines of iPSC with five cloneseach were generated from sporadic ALS and only three ofthem displayed TDP-43 protein aggregation, while none ofthe eight lines with SOD1 mutations generated detectableaggregates [141]. Even in the lines that displayed aggre-gation, this was not detected in every clonal line from thesame patient [141]. In AD, a review citing fourteen differentiPSC studies, the majority of which used familial forms ofthe disease, reported oligomers or aggregates in only four ofthe studies [142]. Together, the available literature suggeststhat detection of protein aggregation does not readily occurin early iPSC-derived neurons and currently requires largenumbers of patients and clones, which is beyond the cap-abilities of many groups. These challenges could be at leastpartially overcome by implementing the maturation strate-gies delineated above.
Functional maturation of iPSC-derived cell types
Neuronal activity is an important parameter that regulatesthe propagation of pathogenic prion-like proteins such astau [143, 144], Aβ [145] and α-Syn [146, 147]. Forexample, activation of AMPA receptors, release of pre-synaptic glutamate, active synapses and synaptic contactsare important mechanisms regulating the trans-synaptic andtrans-cellular propagation of tau [144, 148, 149]. To modelprion-like protein propagation using iPSC-derived neurons,it is critical to ensure that the cells produced displayappropriate physiological electrical properties (Fig. 2).Methods commonly used to determine the functionalmaturity of neurons differentiated from iPSCs includewhole-cell patch-clamp recordings, electrode arrays andassessment of functional receptors (NMDA, AMPA,GABAA) by calcium imaging [150, 151]. At early stages ofdifferentiation, iPSC-derived neurons have an immature
IPSCs to study prion-like mechanisms 2695

electrophysiological phenotype, but in vitro maturation andformation of functional neural networks can be achieved byextending the time in culture [150, 151]. The method usedto produce neurons has important consequences on elec-trophysiological maturity. An alternative approach usingNGN2-mediated neuronal induction of iPSCs coupled withglia co-culture demonstrated significant electrical maturityas early as 14 days post-induction [152, 153]. In addition,direct conversion of fibroblasts to neurons results in popu-lations that tend to lack spontaneous and evoked neuro-transmission and have passive membrane properties similarto immature neurons after 3–4 weeks in culture [154].However, electrically active induced neurons can beobtained by increasing the time in culture to 85–100 daysand co-culturing them with glia [140, 155, 156]. Therefore,ensuring the functional maturity of neurons is critical toaccurately recapitulate disease progression and proteinpropagation.
Astrocyte maturity is an equally important aspect ofdisease modeling, as an increasing number of studiesimplicate astrocytes in the progression of neurodegenerativedisorders [91, 126, 157, 158]. Several astrocyte differ-entiation protocols have been established, but they differ inthe timing (4-weeks to several months) and nature of thedifferentiation cocktail [159]. Overall, these protocols pro-duce cells that express known markers of astrocytes (e.g.GFAP, S100β), respond to inflammatory stimuli, supportneuronal growth and enhance neuron survival [159–163].However, they fail to recapitulate the complex stellatemorphology composed of branches and fine processestypical of astrocytes in vivo. In contrast to neurons, themolecular and functional characteristics of mature astrocyteand the impact of aging on their neuroprotective or neuro-toxic properties have not been extensively studied. Giventhe essential role of astrocytes in the maintenance ofappropriate neuronal functions and immune processes of thebrain, maturity may have important ramifications in regardsto the spreading of pathological proteins.
Genomic variability
Disease modeling using iPSCs is often intended to leveragea patient’s genetic background to induce a disease pheno-type. However, while highly penetrant disease-associatedmutations may drive strong in vitro phenotypic alterations,genetic variants with a low penetrance or small allelic foldchange could result in smaller effects that may be maskedby the inter-individual or reprogramming-induced varia-bility intrinsic to iPSC-based models [164]. For example, ithas been estimated that the comparison of iPSC-derivedcells prepared from 20 to 80 unrelated individuals would benecessary to accurately recapitulate phenotypes induced byvariants with small allelic fold changes [165]. However,
implementing studies with large cohorts of patient-derivedcells is costly and technically challenging, which has con-sequently limited most reports to a small number ofiPSC lines.
Microenvironment and connectivity
In their native brain tissue, neurons and non-neuronal cellsevolve in a complex 3D microenvironment where cell-extracellular matrix interactions, direct cell-cell contacts andparacrine signals mediate tissue homeostasis, cellular func-tions, and survival [166–168]. While organoids provide atechnological advance in 3D modeling, they more accuratelyrepresent a developing rather than a mature CNS tissue [169]and lack functional and structural brain connectivity, twofeatures associated with clinical phenotypes in neurodegen-erative diseases [170]. Furthermore, organoids do not containvasculature that could promote long-term survival andmaturation via oxygen and nutrient distribution, as well asthrough direct contributions of endothelial cells to organo-genesis [171]. A number of groups have recently reported thesuccessful integration of vascular-like networks in culturedorganoids [172–175], which hold great promises for the futuredevelopment of increasingly complex 3D models of the CNS.In the following section, we will discuss in more details thepotential of organoids as a new tool to model prion-likeprotein propagation.
Modeling the behavior of prion-like proteinsin complex systems
iPSC-derived organoids
iPSCs self-organize and form 3D human-like cerebral tissuein vitro (Fig. 2). These spheroid structures, or organoids,can be patterned to produce regionalized in vitro humanbrain tissue including the midbrain [176], neocortex [177]and even a complex cerebral-like circuit composed ofinterconnected, but distinct brain regions [178]. The uniqueorganization of the human brain has made the modeling ofhuman diseases in mice difficult, but these new in vitroculture systems have the potential to overcome such lim-itations. For example, 3D cerebral-like organoids canrecapitulate features of human brain development with thepresence of cell layers of the subventricular zone unique tothe human brain and absent in mice [178]. Microcephalyhas traditionally been difficult to study in mice, possiblybecause of the different behaviors of human and miceneural progenitor cells. However, cerebral-like organoidssuccessfully modeled this human-specific disorder [178],highlighting the powerful potential of 3D tissue models tostudy neurological disorders. Human cerebral-like
2696 A. de Rus Jacquet et al.

organoids are multicellular structures comprising progeni-tors, neurons, astrocytes and oligodendrocytes [179–181].Microglia originate from a different developmental lineagethan neuronal cells and are consequently, often absent inorganoids. Interestingly, recent studies reported the pre-sence of microglia that developed within organoids, sug-gesting that adaptations of existing protocols can beimplemented to obtain organoids comprising various cellpopulations [180, 182]. Furthermore, microglia can be dif-ferentiated separately and added exogenously to the cul-tured organoids. These microglia penetrate organoids andacquire in vivo-like functions, such as the ability to clear Aβaggregates [122]. While organoids are a powerful new tool,it should be noted that the complexity of the model tends tolead to significant variability in the response to experimentalconditions, which generally requires the analysis of anincreased number of samples to ensure the reproducibilityand validity of the results.
The potential of organoids can be harnessed to studyprion-like proteins and mechanisms of propagation. Forexample, control cerebral organoids can be inoculated withbrain homogenates prepared from CJD patients and displayfeatures indicative of PrP infectivity, self-seeding and pro-pagation [183]. Future studies could also fuse differentbrain organoids to produce fully connected structures thatsupport cell migration and material exchange from oneorganoid to another [184, 185]. Such systems could be usedto monitor the propagation of prion-like proteins from asource to a naive organoid to determine the cell typesinvolved, the resulting modifications in 3D morphology andany potential tissue reorganization. Changes occurringwithin an organoid can be monitored by live-cell imagingeither in a full organoid [186] or in organoid slices [187](Fig. 2). Live-cell imaging in whole organoids requires theuse of bright fluorescent reporter proteins to compensatefrom the loss of signal intensity due to the considerable gapbetween the objective lens and the structure of interest andthe presence of a gel-like extracellular matrix surroundingthe organoid [186]. Several non-invasive optical sectioningmicroscopes such as spinning disk confocal [186], two-photon [188] and light-sheet microscopes [189, 190] havebeen tested and ensured low phototoxicity, rapid imageacquisition and sufficient sensitivity to enable appropriatespatial and temporal resolution. End-point 3D imaging offixed whole organoids is also possible and can be improvedby optical clearing techniques [191]. Whole organoid 3Dimaging is a powerful approach to visualize tissue archi-tecture and cell-cell interactions that are lost in 2D culturesand sectioned tissues. A combination of organoid-basedmodels and advanced microscopy techniques are powerful,yet unexplored tools, to study prion-like protein propagationand associated cellular dysfunction in a unique, human-based multicellular 3D model.
Microfluidic technologies
Axon isolation chambers are ideal devices to studymechanisms of prion-like protein propagation and uptakeby isolating neuronal somas, axons and synaptic compart-ments in separated, but interconnected chambers (Fig. 2).These devices can be designed to accommodate severalpopulations of neurons in bi- and tripartite chambers that areindependent but synaptically interconnected throughmicrogroove arrays [143, 149] (Fig. 2). Pathogenic seeds ofprion-like proteins can be added to a fluidically isolatedchamber without diffusing to the other compartments.Microfluidic devices can be used to address a unique rangeof biological questions such as elucidating the effects ofprion-like proteins in distinct compartments of the neurons,the mechanisms and direction of their axonal transport[192], as well as to discover antibodies or screen for inhi-bitors of trans-neuronal protein propagation [193–195].These devices have been extensively used to demonstratein vitro trans-cellular propagation of proteins including α-Syn [192, 196] and tau [143, 149, 197] in primary mouseneuronal cultures. For example, it was found that α-Syn(1–120) preformed fibrils propagate to distal parts of theneuronal somas and compartmentalized neurites. Whensomas are treated with preformed fibrils, phosphorylated α-Syn (p-α-Syn) aggregates are found in the soma followedby propagation to the neurites. When neurites are treatedwith the preformed fibrils, p-α-Syn pathology is identifiedin the neurites followed by propagation back to the somawhere aggregates are formed [192]. Other studies showedthat tau seeds can be transferred from a donor to a recipientneuron via anterograde transport from the soma [198], andin a tripartite chamber device, tau pathology propagatesfrom the first population of neurons to a second and thirdpopulation in a trans-synaptic fashion [143]. Overall, theuse of microfluidic axon isolation devices has revealedimportant information on the propagation potential of dif-ferent species of prion-like proteins and their modes oftransport. Future experiments with human iPSC-derivedneurons will hopefully confirm these findings in a humanmodel as well as provide an opportunity to study how cellsderived from patients with neurodegenerative diseasesimpact the findings. Further progress can also be made viathe coupling of microfluidics devices with advancedmicroscopy techniques to resolve spatial and temporalevents leading to pathology. One example of this is a recentstudy that cultured motor neurons derived from ALS patientiPSCs in a microfluidic device to understand how organelletrafficking rate and direction was changed in these cells[199]. The coupling of this system with live-cell micro-scopy permitted isolation of distal and proximal regions ofthe axons [199]. To further determine the mechanisms ofprotein propagation, axon isolation chambers partnered with
IPSCs to study prion-like mechanisms 2697

microelectrode arrays and iPSC-derived neurons will bepowerful systems that could be used to study the propaga-tion of prion-like proteins in response to neuronal stimula-tion [200].
Combining gene editing with iPSC modeling
Gene editing technologies are powerful methods tomanipulate a gene of interest (e.g. gene knockdown) or tovisualize a protein (e.g. tagged endogenous protein).Among the multiple gene editing technologies available,such as Zinc Finger Nuclease (ZFN), TranscriptionActivator-Like Effector Nuclease (TALEN) and ClusteredRegularly Interspaced Short Palindromic Repeats(CRISPR), CRISPR offers distinct advantages such asbroad applicability, low cost, relative ease of use and rapidimplementation [201–203]. The CRISPR system is wellsuited to the study of prion proteins and has been adopted asthe gene-editing method of choice in studies that address adiversity of biological questions [91, 204, 205] (Fig. 2). Itenables the insertion of fluorescent protein tags to endo-genous proteins of interest and alleviates the need to over-express tagged proteins that could result in proteinmislocalization or misfolding, protein imbalance and pos-sibly, dysregulation of cellular processes [201, 206].Fluorescent labeling of prion-like proteins is a widely usedapproach to study their behavior, functions and modes ofpropagation [207]. However, methods using viral vectors orplasmid transfection to transiently or stably express theexogenously tagged protein of interest lead to proteinoverexpression. To overcome limitations associated withoverexpression, CRISPR/Cas9 was used to introduce aFLAG tag into the endogenous SNCA locus, leading to theproduction of cells that expresses FLAG-tagged α-Syn atendogenous levels [91]. The CRISPR system can also beused to identify proteins involved in a pathway of interestby modulating their endogenous expression levels usingCRISPR interference (CRISPRi) and CRISPR activation(CRISPRa) of single genes, or as a screening strategy [208].Greater silencing efficiencies can be obtained by CRISPRicompared to RNA interference (RNAi), as demonstrated byan incomplete 40% knockdown of mHTT by RNAi [209]and a complete knockdown with CRISPRi [210]. Further-more, CRISPRi can be used as a screening strategy touncover pathways essential to the prion-like propagation ofproteins. For example, a CRISPRi screen in an immorta-lized cell line successfully identified key proteins and cel-lular pathways that mediate tau propagation [205]. Otherprion-like proteins, such as α-Syn, have been successfullydeleted [211] or tagged [91] in iPSC lines using CRISPR-based technologies to characterize their function and iden-tify drug candidates.
Another important advantage of gene-editing technolo-gies in the study of prion-like proteins in iPSC-based modelis the generation of isogenic lines, i.e. the correction ofdisease-causing mutations to produce mutant and controllines that differ only at the specific mutation of interest. Theimplementation of isogenic controls ensures geneticallydefined conditions, particularly relevant to the study ofgenetic forms of neurodegenerative diseases. Gene correc-tion has been applied successfully to repair mutations inprion-like proteins, including the N276K mutation in tau[212], abnormal CAG repeats in mHTT [210] and SNCAtriplication [213]. Alternatively, an iPSC line originatingfrom a healthy donor can be mutated at a specific locus toproduce a disease iPSC line on a control background[214, 215]. However, it should be noted that gene editing ofiPSCs has also proven somewhat challenging, with limiteduptake, low editing efficiency and significant cell deathassociated with transfection of the plasmid DNA elementsrequired for CRISPR/Cas9-mediated gene correction. Fur-thermore, a majority of the corrections tend to be mono-allelic and require a second round of manipulations toachieve biallelic gene editing [216, 217]. In the past fewyears, new strategies have emerged to improve iPSC sur-vival, reduce off-target mutations and increase the fre-quency of biallelic gene corrections. Recent examples usingCRISPR/Cas9 gRNA ribonucleoprotein complexes showedthat directly delivering precomplexed Cas9-gRNA, insteadof plasmid DNA, has the potential to solve specific tech-nical challenges associated with the editing of iPSCs[216, 218].
Conclusions
To date, iPSCs have frequently been used to validate thephysiological relevance of findings from cell lines and toinvestigate the different mechanisms involved in variousaspects of prion-like protein spreading and seeding. Whilethe resulting findings have been informative, challengesrelated to the immaturity of neurons and the absence ofsupportive non-neuronal cells remain. New technologicaladvances such as 3D modeling and induced neurons willlikely contribute to overcome these challenges and advanceour understanding of prion-like mechanisms of neurode-generation. Furthermore, human iPSC-based models pro-vide unlimited access to vulnerable neuronal populationsthat were traditionally unavailable and offer a human-specific background to study the properties of proteins ofinterest. In light of the current literature and despite lim-itations such as neuronal immaturity and the absence of anative microenvironment, iPSC-based models provideexciting perspectives and unique advantages to elucidate therole of pathological prion-like proteins in neurodegenerative
2698 A. de Rus Jacquet et al.

diseases and the molecular mechanisms regulating propa-gation and neurotoxicity.
Acknowledgements FC is a recipient of a Researcher Chair from theFonds de Recherche du Québec en Santé (FRQS) providing salarysupport and operating funds and receives funding from the CanadianInstitutes of Health Research (CIHR) to conduct her HD-relatedresearch. MA is supported by post-doctoral fellowships from bothCIHR and FRQS and HLD is supported by an FRQS doctoralresearch award.
Author contributions ARJ and MA researched data for the article andwrote the manuscript. HLD designed the figures and tables. FC con-tributed to discussions and helped edit the manuscript. All authorsreviewed the content before publication.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict ofinterest.
Publisher’s note Springer Nature remains neutral with regard tojurisdictional claims in published maps and institutional affiliations.
References
1. Das AS, Zou WQ. Prions: beyond a single protein. ClinMicrobiol Rev. 2016;29:633–58.
2. Prusiner SB, Groth DF, Cochran SP, Masiarz FR, McKinley MP,Martinez HM. Molecular properties, partial purification, andassay by incubation period measurements of the hamster scrapieagent. Biochem. 1980;19:4883–91.
3. Ironside JW. Neuropathological findings in new variant CJD andexperimental transmission of BSE. FEMS Immunol Med Mic.1998;21:91–95.
4. Lee CC, Nayak A, Sethuraman A, Belfort G, McRae GJ. Athree-stage kinetic model of amyloid fibrillation. Biophys J.2007;92:3448–58.
5. Ahn TB, Langston JW, Aachi VR, Dickson DW. Relationship ofneighboring tissue and gliosis to alpha-synuclein pathology in afetal transplant for Parkinson’s disease. Am J Neurodegen Dis.2012;1:49–59.
6. Chu Y, Kordower JH. Lewy body pathology in fetal grafts. AnnNY Acad Sci. 2010;1184:55–67.
7. Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW.Lewy body-like pathology in long-term embryonic nigral trans-plants in Parkinson’s disease. Nat Med. 2008;14:504–6.
8. Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al.Lewy bodies in grafted neurons in subjects with Parkinson’sdisease suggest host-to-graft disease propagation. Nat Med.2008;14:501–3.
9. Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN,Braak E. Staging of brain pathology related to sporadic Parkin-son’s disease. Neurobiol Aging. 2003;24:197–211.
10. Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, LeeVM. Intracerebral inoculation of pathological alpha-synucleininitiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–86.
11. Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S,Perez-Villalba A, et al. Lewy body extracts from Parkinsondisease brains trigger alpha-synuclein pathology and neurode-generation in mice and monkeys. Ann Neurol. 2014;75:351–62.
12. Dehay B, Bezard E. Intrastriatal injection of alpha-synucleinfibrils induces Parkinson-like pathology in macaques. Brain.2019;142:3321–2.
13. Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS,Lee S, et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease.Neuron. 2019;103:627–41 e627.
14. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, HamiltonAM, et al. Gut-seeded alpha-synuclein fibrils promote gut dys-function and brain pathology specifically in aged mice. NatNeurosci. 2020;23:327–36.
15. Groveman BR, Orru CD, Hughson AG, Raymond LD, ZanussoG, Ghetti B, et al. Rapid and ultra-sensitive quantitation ofdisease-associated alpha-synuclein seeds in brain and cere-brospinal fluid by alphaSyn RT-QuIC. Acta Neuropathol Com-mun. 2018;6:7.
16. Singh S, DeMarco ML. In vitro conversion assays diagnostic forneurodegenerative proteinopathies. JALM. 2020;5:142–157.
17. Braak H, Braak E, Bohl J. Staging of Alzheimer-related corticaldestruction. Eur Neurol. 1993;33:403–8.
18. Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S,Schaefer C, Kilger E, et al. Exogenous induction of cerebralbeta-amyloidogenesis is governed by agent and host. Science.2006;313:1781–4.
19. Gary C, Lam S, Herard AS, Koch JE, Petit F, Gipchtein P, et al.Encephalopathy induced by Alzheimer brain inoculation in anon-human primate. Acta Neuropathol Commun. 2019;7:126.
20. Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S,Hench J, et al. Brain homogenates from human tauopathiesinduce tau inclusions in mouse brain. PNAS. 2013;110:9535–40.
21. Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ,et al. Patient-derived frontotemporal lobar degeneration brainextracts induce formation and spreading of TDP-43 pathologyin vivo. Nat Commun. 2018;9:4220.
22. Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, ClosAL, Jackson GR, Kayed R. Tau oligomers impair memory andinduce synaptic and mitochondrial dysfunction in wild-typemice. Mol Neurodegen. 2011;6:39.
23. Mulder CK, Dong Y, Brugghe HF, Timmermans HA, TilstraW, Westdijk J, et al. Immunization with small amyloid-beta-derived cyclopeptide conjugates diminishes amyloid-beta-induced neurodegeneration in mice. J Alzheimer’s Dis.2016;52:1111–23.
24. Polinski NK, Volpicelli-Daley LA, Sortwell CE, Luk KC, Cre-mades N, Gottler LM, et al. Best practices for generating andusing Alpha-synuclein pre-formed fibrils to model Parkinson’sdisease in rodents. J Parkinson’s Dis. 2018;8:303–22.
25. Masnata M, Sciacca G, Maxan A, Bousset L, Denis HL, LauruolF, et al. Demonstration of prion-like properties of mutant hun-tingtin fibrils in both in vitro and in vivo paradigms. ActaNeuropathol. 2019;137:981–1001.
26. Zeineddine R, Pundavela JF, Corcoran L, Stewart EM, Do-Ha D,Bax M, et al. SOD1 protein aggregates stimulate macro-pinocytosis in neurons to facilitate their propagation. Mol Neu-rodegen. 2015;10:57.
27. Blesa J, Przedborski S. Parkinson’s disease: animal models anddopaminergic cell vulnerability. Front Neuroanat. 2014;8:155.
28. Dawson TM, Golde TE, Lagier-Tourenne C. Animal models ofneurodegenerative diseases. Nat Neurosci. 2018;21:1370–9.
29. Holley SM, Kamdjou T, Reidling JC, Fury B, Coleal-Bergum D,Bauer G, et al. Therapeutic effects of stem cells in rodent modelsof Huntington’s disease: Review and electrophysiological find-ings. CNS Neurosci Ther. 2018;24:329–42.
30. Jankowsky JL, Zheng H. Practical considerations for choosing amouse model of Alzheimer’s disease. Mol Neurodegen.2017;12:89.
IPSCs to study prion-like mechanisms 2699

31. Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, et al.Uniquely hominid features of adult human astrocytes. J Neu-rosci. 2009;29:3276–87.
32. Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, et al.Forebrain engraftment by human glial progenitor cells enhancessynaptic plasticity and learning in adult mice. Cell Stem Cell.2013;12:342–53.
33. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blu-menthal PD, et al. Purification and characterization of progenitorand mature human astrocytes reveals transcriptional and func-tional differences with mouse. Neuron. 2016;89:37–53.
34. Rayon T, Stamataki D, Perez-Carrasco R, Garcia-Perez L, Bar-rington C, Melchionda M, et al. Species-specific pace of devel-opment is associated with differences in protein stability.Science. 2020;369:eaba7667.
35. Prusiner SB. Prions. PNAS. 1998;95:13363–83.36. Barria MA, Balachandran A, Morita M, Kitamoto T, Barron R,
Manson J, et al. Molecular barriers to zoonotic transmission ofprions. Emerg Infect Dis. 2014;20:88–97.
37. Wiseman FK, Cancellotti E, Piccardo P, Iremonger K, Boyle A,Brown D, et al. The glycosylation status of PrPC is a key factorin determining transmissible spongiform encephalopathy trans-mission between species. J Virol. 2015;89:4738–47.
38. Hasegawa K, Mohri S, Yokoyama T. Comparison of the localstructural stabilities of mammalian prion protein (PrP) by frag-ment molecular orbital calculations. Prion. 2013;7:185–91.
39. Sanchez-Garcia J, Fernandez-Funez P. D159 and S167 are pro-tective residues in the prion protein from dog and horse, twoprion-resistant animals. Neurobiol Dis. 2018;119:1–12.
40. Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J,Will R, et al. Changing a single amino acid in the N-terminus ofmurine PrP alters TSE incubation time across three speciesbarriers. EMBO J. 2001;20:5070–8.
41. Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J,et al. Abbreviated incubation times for human prions in miceexpressing a chimeric mouse-human prion protein transgene.PNAS. 2003;100:4784–9.
42. Hernandez F, Merchan-Rubira J, Valles-Saiz L, Rodriguez-Matellan A, Avila J. Differences between human and murine tauat the N-terminal end. Front Aging Neurosci. 2020;12:11.
43. Hernandez F, Cuadros R, Olla I, Garcia C, Ferrer I, Perry G,et al. Differences in structure and function between human andmurine tau. Biochim Biophys Acta Mol Basis Dis.2019;1865:2024–30.
44. Stanford PM, Shepherd CE, Halliday GM, Brooks WS, SchofieldPW, Brodaty H, et al. Mutations in the tau gene that cause anincrease in three repeat tau and frontotemporal dementia. Brain.2003;126(Pt 4):814–26.
45. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H,et al. Association of missense and 5′-splice-site mutations in tauwith the inherited dementia FTDP-17. Nature. 1998;393:702–5.
46. Metrick MA II, Ferreira NDC, Saijo E, Kraus A, Newell K,Zanusso G, et al. A single ultrasensitive assay for detection anddiscrimination of tau aggregates of Alzheimer and Pick diseases.Acta Neuropathol Commun. 2020;8:22.
47. Hu WT, Parisi JE, Knopman DS, Boeve BF, Dickson DW,Ahlskog JE, et al. Clinical features and survival of 3R and 4Rtauopathies presenting as behavioral variant frontotemporaldementia. Alzheimer Dis Assoc Disord. 2007;21:S39–43.
48. Dinkel PD, Siddiqua A, Huynh H, Shah M, Margittai M. Var-iations in filament conformation dictate seeding barrier betweenthree- and four-repeat tau. Biochemistry. 2011;50:4330–6.
49. Levarska L, Zilka N, Jadhav S, Neradil P, Novak M. Of rodentsand men: the mysterious interneuronal pilgrimage of misfoldedprotein tau in Alzheimer’s disease. J Alzheimer’s Dis.2013;37:569–77.
50. Sayas CL, Medina M, Cuadros R, Olla I, Garcia E, Perez M,et al. Role of tau N-terminal motif in the secretion of human tauby End Binding proteins. PLoS ONE. 2019;14:e0210864.
51. Ueno H, Yamaguchi T, Fukunaga S, Okada Y, Yano Y, HoshinoM, et al. Comparison between the aggregation of human androdent amyloid beta-proteins in GM1 ganglioside clusters. Bio-chemistry. 2014;53:7523–30.
52. Jankowsky JL, Younkin LH, Gonzales V, Fadale DJ, Slunt HH,Lester HA, et al. Rodent A beta modulates the solubility anddistribution of amyloid deposits in transgenic mice. J Biol Chem.2007;282:22707–20.
53. Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA,Schwarz RD, et al. Evidence for seeding of beta -amyloid byintracerebral infusion of Alzheimer brain extracts in beta -amyloidprecursor protein-transgenic mice. J Neurosci. 2000;20:3606–11.
54. Tanaka G, Yamanaka T, Furukawa Y, Kajimura N, Mitsuoka K,Nukina N. Sequence- and seed-structure-dependent polymorphicfibrils of alpha-synuclein. Biochim Biophys Acta Mol Basis Dis.2019;1865:1410–20.
55. Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD,et al. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep.2016;16:3373–87.
56. Fares MB, Maco B, Oueslati A, Rockenstein E, Ninkina N,Buchman VL, et al. Induction of de novo alpha-synucleinfibrillization in a neuronal model for Parkinson’s disease. PNAS.2016;113:E912–921.
57. Tartari M, Gissi C, Lo Sardo V, Zuccato C, Picardi E, Pesole G,et al. Phylogenetic comparison of huntingtin homologues revealsthe appearance of a primitive polyQ in sea urchin. Mol BiolEvol. 2008;25:330–8.
58. Walker FO. Huntington’s disease. Lancet. 2007;369:218–28.59. Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF.
Time course of early motor and neuropathological anomalies in aknock-in mouse model of Huntington’s disease with 140 CAGrepeats. J Comp Neurol. 2003;465:11–26.
60. Heng MY, Detloff PJ, Albin RL. Rodent genetic models ofHuntington disease. Neurobiol Dis. 2008;32:1–9.
61. Seetharaman SV, Taylor AB, Holloway S, Hart PJ. Structures ofmouse SOD1 and human/mouse SOD1 chimeras. Arch BiochemBiophys. 2010;503:183–90.
62. Karch CM, Borchelt DR. Aggregation modulating elements inmutant human superoxide dismutase 1. Arch Biochem Biophys.2010;503:175–82.
63. Wang HY, Wang IF, Bose J, Shen CK. Structural diversity andfunctional implications of the eukaryotic TDP gene family.Genomics. 2004;83:130–9.
64. D’Alton S, Altshuler M, Lewis J. Studies of alternative isoformsprovide insight into TDP-43 autoregulation and pathogenesis.RNA. 2015;21:1419–32.
65. Gumina V, Onesto E, Colombrita C, Maraschi A, Silani V, RattiA. Inter-species differences in regulation of the progranulin-Sortilin axis in TDP-43 Cell models of neurodegeneration. Int JMol Sci. 2019;20:5866.
66. Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D,et al. Prion detection by an amyloid seeding assay. PNAS.2007;104:20914–9.
67. Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B,et al. Rapid end-point quantitation of prion seeding activity withsensitivity comparable to bioassays. PLoS Pathog. 2010;6:e1001217.
68. Saborio GP, Permanne B, Soto C. Sensitive detection of patho-logical prion protein by cyclic amplification of protein misfold-ing. Nature. 2001;411:810–3.
69. Raymond GJ, Hope J, Kocisko DA, Priola SA, Raymond LD,Bossers A, et al. Molecular assessment of the potential
2700 A. de Rus Jacquet et al.

transmissibilities of BSE and scrapie to humans. Nature.1997;388:285–8.
70. Caughey B, Orru CD, Groveman BR, Hughson AG, Manca M,Raymond LD, et al. Amplified detection of prions and otheramyloids by RT-QuIC in diagnostics and the evaluation oftherapeutics and disinfectants. Prog Mol Biol Transl Sci.2017;150:375–88.
71. Soto-Gamez A, Quax WJ, Demaria M. Regulation of survivalnetworks in senescent cells: from mechanisms to interventions. JMol Biol. 2019;431:2629–43.
72. Kaur G, Dufour JM. Cell lines: valuable tools or useless artifacts.Spermatogenesis. 2012;2:1–5.
73. Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, YanamandraK, et al. Heparan sulfate proteoglycans mediate internalizationand propagation of specific proteopathic seeds. PNAS. 2013;110:E3138–3147.
74. Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, BillintonA, et al. Extracellular monomeric and aggregated tau efficientlyenter human neurons through overlapping but distinct pathways.Cell Rep. 2018;22:3612–24.
75. Jansen AH, Batenburg KL, Pecho-Vrieseling E, Reits EA.Visualization of prion-like transfer in Huntington’s diseasemodels. Biochim Biophys Acta Mol Basis Dis.2017;1863:793–800.
76. Jurgielewicz BJ, Yao Y, Stice SL. Kinetics and specificity ofHEK293T extracellular vesicle uptake using imaging flowcytometry. Nanoscale Res Lett. 2020;15:170.
77. Heravi M, Dargahi L, Parsafar S, Tayaranian Marvian A,Aliakbari F, Morshedi D. The primary neuronal cells are moreresistant than PC12 cells to alpha-synuclein toxic aggregates.Neurosci Lett. 2019;701:38–47.
78. Deng L, Pollmeier L, Zhou Q, Bergemann S, Bode C, Hein L,et al. Gene expression in immortalized versus primary isolatedcardiac endothelial cells. Sci Rep. 2020;10:2241.
79. Kang JH, Rychahou PG, Ishola TA, Qiao J, Evers BM, ChungDH. MYCN silencing induces differentiation and apoptosis inhuman neuroblastoma cells. Biochem Biophys Res Commun.2006;351:192–7.
80. Evans M. Discovering pluripotency: 30 years of mouseembryonic stem cells. Nat Rev Mol Cell Biol. 2011;12:680–6.
81. Takahashi K, Yamanaka S. Induction of pluripotent stem cellsfrom mouse embryonic and adult fibroblast cultures by definedfactors. Cell. 2006;126:663–76.
82. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al.Reprogramming of human somatic cells to pluripotency withdefined factors. Nature. 2008;451:141–6.
83. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J,Frane JL, Tian S, et al. Induced pluripotent stem cell linesderived from human somatic cells. Science. 2007;318:1917–20.
84. Suga M, Kondo T, Inoue H. Modeling neurological disorderswith human pluripotent stem cell-derived astrocytes. Int J MolSci. 2019;20:3862.
85. Chang CY, Ting HC, Liu CA, Su HL, Chiou TW, Lin SZ, et al.Induced pluripotent stem cell (iPSC)-based neurodegenerativedisease models for phenotype recapitulation and drug screening.Molecules. 2020;25:2000.
86. Chanoumidou K, Mozafari S, Baron-Van Evercooren A, Kuhl-mann T. Stem cell derived oligodendrocytes to study myelindiseases. Glia. 2020;68:705–20.
87. Hasselmann J, Blurton-Jones M. Human iPSC-derived micro-glia: a growing toolset to study the brain’s innate immune cells.Glia. 2020;68:721–39.
88. Hong W, Wang Z, Liu W, O’Malley TT, Jin M, Willem M, et al.Diffusible, highly bioactive oligomers represent a critical min-ority of soluble Abeta in Alzheimer’s disease brain. Acta Neu-ropathol. 2018;136:19–40.
89. Reilly P, Winston CN, Baron KR, Trejo M, Rockenstein EM,Akers JC, et al. Novel human neuronal tau model exhibitingneurofibrillary tangles and transcellular propagation. NeurobiolDis. 2017;106:222–34.
90. Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM,Wichterle H, et al. Astrocytes expressing ALS-linked mutatedSOD1 release factors selectively toxic to motor neurons. NatNeurosci. 2007;10:615–22.
91. di Domenico A, Carola G, Calatayud C, Pons-Espinal M, MunozJP, Richaud-Patin Y, et al. Patient-specific iPSC-derived astro-cytes contribute to non-cell-autonomous neurodegeneration inparkinson’s disease. Stem Cell Rep. 2019;12:213–29.
92. Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J,Sundberg M, et al. Pharmacological rescue of mitochondrialdeficits in iPSC-derived neural cells from patients with familialParkinson’s disease. Sci Transl Med. 2012;4:141ra190.
93. Li L, Roh JH, Chang EH, Lee Y, Lee S, Kim M, et al. iPSCmodeling of presenilin1 mutation in Alzheimer’s disease withcerebellar ataxia. Exp Neurobiol. 2018;27:350–64.
94. Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, et al.Acid sphingomyelinase modulates the autophagic process bycontrolling lysosomal biogenesis in Alzheimer’s disease. J ExpMed. 2014;211:1551–70.
95. Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM,Ferraiuolo L, et al. The C9orf72 protein interacts with Rab1a andthe ULK1 complex to regulate initiation of autophagy. EMBO.2016;35:1656–76.
96. Scialo C, De Cecco E, Manganotti P, Legname G. Prion andprion-like protein strains: deciphering the molecular basis ofheterogeneity in neurodegeneration. Viruses. 2019;11:261.
97. Chai X, Dage JL, Citron M. Constitutive secretion of tau protein byan unconventional mechanism. Neurobiol Dis. 2012;48:356–66.
98. Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, et al.Human secreted tau increases amyloid-beta production. Neuro-biol Aging. 2015;36:693–709.
99. Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB,et al. APP metabolism regulates tau proteostasis in human cer-ebral cortex neurons. Cell Rep. 2015;11:689–96.
100. Reyes JF, Olsson TT, Lamberts JT, Devine MJ, Kunath T,Brundin P. A cell culture model for monitoring alpha-synucleincell-to-cell transfer. Neurobiol Dis. 2015;77:266–75.
101. Pecho-Vrieseling E, Rieker C, Fuchs S, Bleckmann D, EspositoMS, Botta P, et al. Transneuronal propagation of mutant hun-tingtin contributes to non-cell autonomous pathology in neurons.Nat Neurosci. 2014;17:1064–72.
102. Kanmert D, Cantlon A, Muratore CR, Jin M, O’Malley TT, LeeG, et al. C-terminally truncated forms of Tau, but not full-lengthTau or its C-terminal fragments, are released from neuronsindependently of cell death. J Neurosci. 2015;35:10851–65.
103. Guix FX, Corbett GT, Cha DJ, Mustapic M, Liu W, Mengel D,et al. Detection of aggregation-competent tau in neuron-derivedextracellular vesicles. Int J Mol Sci. 2018;19:663.
104. Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC,Hatcher NG, et al. Internalized Tau oligomers cause neurode-generation by inducing accumulation of pathogenic tau in humanneurons derived from induced pluripotent stem cells. J Neurosci.2015;35:14234–50.
105. Verheyen A, Diels A, Dijkmans J, Oyelami T, Meneghello G,Mertens L, et al. Using human iPSC-derived neurons to modelTAU aggregation. PLoS ONE. 2015;10:e0146127.
106. Prots I, Grosch J, Brazdis RM, Simmnacher K, Veber V, Hav-licek S, et al. Alpha-Synuclein oligomers induce early axonaldysfunction in human iPSC-based models of synucleinopathies.PNAS. 2018;115:7813–8.
107. Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU,et al. Human iPSC-derived neuronal model of Tau-A152T
IPSCs to study prion-like mechanisms 2701

frontotemporal dementia reveals tau-mediated mechanisms ofneuronal vulnerability. Stem Cell Rep. 2016;7:325–40.
108. Matamoros-Angles A, Gayosso LM, Richaud-Patin Y, diDomenico A, Vergara C, Hervera A, et al. iPS cell cultures froma Gerstmann-Straussler-Scheinker patient with the Y218N PRNPmutation recapitulate tau pathology. Mol Neurobiol.2018;55:3033–48.
109. Jarosz-Griffiths HH, Corbett NJ, Rowland HA, Fisher K, JonesAC, Baron J, et al. Proteolytic shedding of the prion protein viaactivation of metallopeptidase ADAM10 reduces cellular bind-ing and toxicity of amyloid-beta oligomers. J Biol Chem.2019;294:7085–97.
110. Gribaudo S, Tixador P, Bousset L, Fenyi A, Lino P, Melki R,et al. Propagation of alpha-Synuclein Strains within humanreconstructed neuronal network. Stem Cell Rep.2019;12:230–44.
111. Corbett GT, Wang Z, Hong W, Colom-Cadena M, Rose J, LiaoM, et al. PrP is a central player in toxicity mediated by solubleaggregates of neurodegeneration-causing proteins. Acta Neuro-pathol. 2020;139:503–26.
112. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, et al. Directtransfer of alpha-synuclein from neuron to astroglia causesinflammatory responses in synucleinopathies. J Biol Chem.2010;285:9262–72.
113. Lindstrom V, Gustafsson G, Sanders LH, Howlett EH, Sig-vardson J, Kasrayan A, et al. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracel-lular deposits and mitochondrial damage. Mol Cell Neuro.2017;82:143–56.
114. Braidy N, Gai WP, Xu YH, Sachdev P, Guillemin GJ, Jiang XM,et al. Uptake and mitochondrial dysfunction of alpha-synucleinin human astrocytes, cortical neurons and fibroblasts. TranslNeuro. 2013;2:20.
115. Cavaliere F, Cerf L, Dehay B, Ramos-Gonzalez P, De Giorgi F,Bourdenx M, et al. In vitro alpha-synuclein neurotoxicity andspreading among neurons and astrocytes using Lewy bodyextracts from Parkinson disease brains. Neurobiol Dis.2017;103:101–12.
116. Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D,et al. The role of microglia in processing and spreading ofbioactive tau seeds in Alzheimer’s disease. J Neuroinflam.2018;15:269.
117. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM.Microglial internalization and degradation of pathological tau isenhanced by an anti-tau monoclonal antibody. Sci Rep.2015;5:11161.
118. Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW,Bajaj L, et al. TFEB enhances astroglial uptake of extracellulartau species and reduces tau spreading. J Exp Med.2018;215:2355–77.
119. Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F,et al. Adult mouse astrocytes degrade amyloid-beta in vitro andin situ. Nat Med. 2003;9:453–7.
120. Paresce DM, Ghosh RN, Maxfield FR. Microglial cells inter-nalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–65.
121. Pearce MMP, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-liketransmission of neuronal huntingtin aggregates to phagocytic gliain the Drosophila brain. Nat Commun. 2015;6:6768.
122. Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, et al.APOE4 causes widespread molecular and cellular alterationsassociated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–54 e1147.
123. Oksanen M, Petersen AJ, Naumenko N, Puttonen K, Lehtonen S,Gubert Olive M, et al. PSEN1 mutant iPSC-derived model
reveals severe astrocyte pathology in Alzheimer’s disease. StemCell Rep. 2017;9:1885–97.
124. Fong LK, Yang MM, Dos Santos Chaves R, Reyna SM, Lang-ness VF, Woodruff G, et al. Full-length amyloid precursor pro-tein regulates lipoprotein metabolism and amyloid-beta clearancein human astrocytes. J Biol Chem. 2018;293:11341–57.
125. Lorenzini I, Alsop E, Levy J, Gittings LM, Rabichow BE, lall d,et al. Activated iPSC-microglia from C9orf72 ALS/FTD patientsexhibit endosomal-lysosomal dysfunction. bioRxiv. 2020:277459.
126. Rostami J, Holmqvist S, Lindstrom V, Sigvardson J, WestermarkGT, Ingelsson M, et al. Human astrocytes transfer aggregatedAlpha-Synuclein via tunneling nanotubes. J Neurosci.2017;37:11835–53.
127. Krejciova Z, Alibhai J, Zhao C, Krencik R, Rzechorzek NM,Ullian EM, et al. Human stem cell-derived astrocytes replicatehuman prions in a PRNP genotype-dependent manner. J ExpMed. 2017;214:3481–95.
128. Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C,et al. Probing sporadic and familial Alzheimer’s disease usinginduced pluripotent stem cells. Nature. 2012;482:216–20.
129. Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Teglasi A,Bock I, et al. Neurons derived from sporadic Alzheimer’s diseaseiPSCs reveal elevated TAU hyperphosphorylation, increasedamyloid levels, and GSK3B activation. Alzheimer’s Res Ther.2017;9:90.
130. Fujimori K, Ishikawa M, Otomo A, Atsuta N, Nakamura R,Akiyama T, et al. Modeling sporadic ALS in iPSC-derived motorneurons identifies a potential therapeutic agent. Nat Med.2018;24:1579–89.
131. Miguel L, Rovelet-Lecrux A, Feyeux M, Frebourg T, Nassoy P,Campion D, et al. Detection of all adult Tau isoforms in a 3Dculture model of iPSC-derived neurons. Stem Cell Res.2019;40:101541.
132. Studer L, Vera E, Cornacchia D. Programming and reprogram-ming cellular age in the era of induced pluripotency. Cell StemCell. 2015;16:591–600.
133. Wegmann S, Bennett RE, Delorme L, Robbins AB, Hu M,McKenzie D, et al. Experimental evidence for the age depen-dence of tau protein spread in the brain. Sci Adv. 2019;5:eaaw6404.
134. Nekrasov ED, Vigont VA, Klyushnikov SA, Lebedeva OS,Vassina EM, Bogomazova AN, et al. Manifestation of Hun-tington’s disease pathology in human induced pluripotent stemcell-derived neurons. Mol Neurodegen. 2016;11:27.
135. Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, TuEY, et al. Human iPSC-based modeling of late-onset disease viaprogerin-induced aging. Cell Stem Cell. 2013;13:691–705.
136. Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC,Wernig M. Direct conversion of fibroblasts to functional neuronsby defined factors. Nature. 2010;463:1035–41.
137. Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A,et al. Direct conversion of human fibroblasts to dopaminergicneurons. PNAS. 2011;108:10343–8.
138. Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D,Taverna S, Leo D, et al. Direct generation of functional dopa-minergic neurons from mouse and human fibroblasts. Nature.2011;476:224–7.
139. Abernathy DG, Kim WK, McCoy MJ, Lake AM, Ouwenga R,Lee SW, et al. MicroRNAs induce a permissive chromatinenvironment that enables neuronal subtype-specific reprogram-ming of adult human fibroblasts. Cell Stem Cell.2017;21:332–48 e339.
140. Victor MB, Richner M, Olsen HE, Lee SW, Monteys AM, Ma C,et al. Striatal neurons directly converted from Huntington’s
2702 A. de Rus Jacquet et al.

disease patient fibroblasts recapitulate age-associated diseasephenotypes. Nat Neurosci. 2018;21:341–52.
141. Burkhardt MF, Martinez FJ, Wright S, Ramos C, Volfson D,Mason M, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci.2013;56:355–64.
142. Arber C, Lovejoy C, Wray S. Stem cell models of Alzheimer’sdisease: progress and challenges. Alzheimer’s Res Ther.2017;9:42.
143. Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A,Rilett K, et al. Neuronal activity enhances tau propagation andtau pathology in vivo. Nat Neurosci. 2016;19:1085–92.
144. Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H,et al. Neuronal activity regulates extracellular tau in vivo. J ExpMed. 2014;211:387–93.
145. Pignataro A, Middei S. Trans-synaptic spread of amyloid-beta inAlzheimer’s disease: paths to beta-amyloidosis. Neural Plast.2017;2017:5281829.
146. Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, RyanTA, Edwards RH. Neural activity controls the synaptic accu-mulation of alpha-synuclein. J Neurosci. 2005;25:10913–21.
147. Paillusson S, Clairembault T, Biraud M, Neunlist M, Derkin-deren P. Activity-dependent secretion of alpha-synuclein byenteric neurons. J Neurochem. 2013;125:512–7.
148. Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Phy-siological release of endogenous tau is stimulated by neuronalactivity. EMBO Rep. 2013;14:389–94.
149. Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, deStrooper B, et al. Synaptic contacts enhance cell-to-cell Taupathology propagation. Cell Rep. 2015;11:1176–83.
150. Pruunsild P, Bengtson CP, Bading H. Networks of culturediPSC-derived neurons reveal the human synaptic activity-regulated adaptive gene program. Cell Rep. 2017;18:122–35.
151. Amin H, Maccione A, Marinaro F, Zordan S, Nieus T, Ber-dondini L. Electrical responses and spontaneous activity ofhuman iPS-derived neuronal networks characterized for 3-monthculture with 4096-electrode arrays. Front Neurosci. 2016;10:121.
152. Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al.Rapid single-step induction of functional neurons from humanpluripotent stem cells. Neuron. 2013;78:785–98.
153. Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, PietilainenO, et al. Combining NGN2 programming with developmentalpatterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 2018;23:2509–23.
154. Koppensteiner P, Boehm S, Arancio O. Electrophysiologicalprofiles of induced neurons converted directly from adult humanfibroblasts indicate incomplete neuronal conversion. CellReprogram. 2014;16:439–46.
155. Drouin-Ouellet J, Lau S, Brattas PL, Rylander Ottosson D, PircsK, Grassi DA, et al. REST suppression mediates neural con-version of adult human fibroblasts via microRNA-dependent and-independent pathways. EMBO Mol Med. 2017;9:1117–31.
156. Xue Y, Qian H, Hu J, Zhou B, Zhou Y, Hu X, et al. Sequentialregulatory loops as key gatekeepers for neuronal reprogrammingin human cells. Nat Neurosci. 2016;19:807–15.
157. de Rus Jacquet A, Tancredi JL, Lemire AL, DeSantis MC, Li W-P, O’Shea EK. The LRRK2 G2019S mutation alters astrocyte-to-neuron communication via extracellular vesicles and inducesneuron atrophy in a human iPSC-derived model of Parkinson’sdisease. bioRxiv. 2020:178574.
158. Birger A, Ben-Dor I, Ottolenghi M, Turetsky T, Gil Y, SweetatS, et al. Human iPSC-derived astrocytes from ALS patients withmutated C9ORF72 show increased oxidative stress and neuro-toxicity. EBioMedicine. 2019;50:274–89.
159. Chandrasekaran A, Avci HX, Leist M, Kobolak J, Dinnyes A.Astrocyte differentiation of human pluripotent stem cells: new
tools for neurological disorder research. Front Cell Neurosci.2016;10:215.
160. de Rus Jacquet A. Preparation and co-culture of ipsc-deriveddopaminergic neurons and astrocytes. Curr Protoc Cell Biol.2019;85:e98.
161. Tcw J, Wang M, Pimenova AA, Bowles KR, Hartley BJ, LacinE, et al. An efficient platform for astrocyte differentiation fromhuman induced pluripotent stem cells. Stem Cell Rep.2017;9:600–14.
162. Li X, Tao Y, Bradley R, Du Z, Tao Y, Kong L, et al. Fastgeneration of functional subtype astrocytes from human plur-ipotent stem cells. Stem Cell Rep. 2018;11:998–1008.
163. Hedegaard A, Monzon-Sandoval J, Newey SE, Whiteley ES,Webber C, Akerman CJ. Pro-maturational effects of HumaniPSC-derived cortical astrocytes upon iPSC-derived corticalneurons. Stem Cell Rep. 2020;15:38–51.
164. Volpato V, Webber C. Addressing variability in iPSC-derivedmodels of human disease: guidelines to promote reproducibility.Dis Model Mech. 2020;13:dmm042317.
165. Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Ala-soo K, Knights AJ, et al. Molecular and functional variation iniPSC-derived sensory neurons. Nat Genet. 2018;50:54–61.
166. Barros CS, Franco SJ, Muller U. Extracellular matrix: functionsin the nervous system. Cold Spring Harb Perspect Biol. 2011;3:a005108.
167. Spampinato SF, Bortolotto V, Canonico PL, Sortino MA, GrilliM. Astrocyte-derived paracrine signals: relevance for neurogenicniche regulation and blood-brain barrier integrity. Front Pharm.2019;10:1346.
168. Kornberg TB, Roy S. Communicating by touch–neurons are notalone. Trends Cell Biol. 2014;24:370–6.
169. Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, EckerJR. Cerebral organoids recapitulate epigenomic signatures of thehuman fetal brain. Cell Rep. 2016;17:3369–84.
170. Pievani M, Filippini N, van den Heuvel MP, Cappa SF, FrisoniGB. Brain connectivity in neurodegenerative diseases–fromphenotype to proteinopathy. Nat Rev Neurol. 2014;10:620–33.
171. Vargas-Valderrama A, Messina A, Mitjavila-Garcia MT, Gue-nou H. The endothelium, a key actor in organ development andhPSC-derived organoid vascularization. J Biomed Sci.2020;27:67.
172. Pham MT, Pollock KM, Rose MD, Cary WA, Stewart HR, ZhouP, et al. Generation of human vascularized brain organoids.Neuroreport. 2018;29:588–93.
173. Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ,et al. Engineering of human brain organoids with a functionalvascular-like system. Nat Methods. 2019;16:1169–75.
174. Worsdorfer P, Dalda N, Kern A, Kruger S, Wagner N, KwokCK, et al. Generation of complex human organoid modelsincluding vascular networks by incorporation of mesodermalprogenitor cells. Sci Rep. 2019;9:15663.
175. Grebenyuk S, Ranga A. Engineering organoid vascularization.Front Bioeng Biotechnol. 2019;7:39.
176. Monzel AS, Smits LM, Hemmer K, Hachi S, Moreno EL, vanWuellen T, et al. Derivation of human midbrain-specific orga-noids from neuroepithelial stem cells. Stem Cell Rep.2017;8:1144–54.
177. Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Brauninger M, et al. Human cerebral organoids recapitulate geneexpression programs of fetal neocortex development. PNAS.2015;112:15672–7.
178. Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS,Hurles ME, et al. Cerebral organoids model human braindevelopment and microcephaly. Nature. 2013;501:373–9.
179. Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, HuberN, et al. Functional cortical neurons and astrocytes from human
IPSCs to study prion-like mechanisms 2703

pluripotent stem cells in 3D culture. Nat Methods.2015;12:671–8.
180. Marton RM, Miura Y, Sloan SA, Li Q, Revah O, Levy RJ, et al.Differentiation and maturation of oligodendrocytes in humanthree-dimensional neural cultures. Nat Neurosci.2019;22:484–91.
181. Qian X, Song H, Ming GL. Brain organoids: advances, appli-cations and challenges. Development. 2019;146:dev166074.
182. Ormel PR, Vieira de Sa R, van Bodegraven EJ, Karst H,Harschnitz O, Sneeboer MAM, et al. Microglia innately developwithin cerebral organoids. Nat Commun. 2018;9:4167.
183. Groveman BR, Foliaki ST, Orru CD, Zanusso G, Carroll JA,Race B, et al. Sporadic Creutzfeldt-Jakob disease prion infectionof human cerebral organoids. Acta Neuropathol Commun.2019;7:90.
184. Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G,Roselaar N, et al. Fusion of regionally specified hPSC-derivedorganoids models human brain development and interneuronmigration. Cell Stem Cell. 2017;21:383–98 e387.
185. Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA.Fused cerebral organoids model interactions between brainregions. Nat Methods. 2017;14:743–51.
186. Bolhaqueiro ACF, van Jaarsveld RH, Ponsioen B, OvermeerRM, Snippert HJ, Kops G. Live imaging of cell division in 3Dstem-cell organoid cultures. Methods Cell Biol.2018;145:91–106.
187. Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A,Wynshaw-Boris A, et al. Human iPSC-derived cerebral orga-noids model cellular features of lissencephaly and reveal pro-longed mitosis of outer radial glia. Cell Stem Cell.2017;20:435–49 e434.
188. Browne AW, Arnesano C, Harutyunyan N, Khuu T, MartinezJC, Pollack HA, et al. Structural and functional characterizationof human stem-cell-derived retinal organoids by live imaging.Invest Ophthalmol Vis Sci. 2017;58:3311–8.
189. Held M, Santeramo I, Wilm B, Murray P, Levy R. Ex vivo livecell tracking in kidney organoids using light sheet fluorescencemicroscopy. PLoS ONE. 2018;13:e0199918.
190. Andilla J, Jorand R, Olarte OE, Dufour AC, Cazales M, Mon-tagner YL, et al. Imaging tissue-mimic with light sheet micro-scopy: a comparative guideline. Sci Rep. 2017;7:44939.
191. Dekkers JF, Alieva M, Wellens LM, Ariese HCR, Jamieson PR,Vonk AM, et al. High-resolution 3D imaging of fixed andcleared organoids. Nat Protoc. 2019;14:1756–71.
192. Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM,Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewybody pathology leading to synaptic dysfunction and neurondeath. Neuron. 2011;72:57–71.
193. Nobuhara CK, DeVos SL, Commins C, Wegmann S, Moore BD,Roe AD, et al. Tau antibody targeting pathological speciesblocks neuronal uptake and interneuron propagation of Tauin vitro. Am J Pathol. 2017;187:1399–412.
194. Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC,et al. Alpha-synuclein immunotherapy blocks uptake and tem-plated propagation of misfolded alpha-synuclein and neurode-generation. Cell Rep. 2014;7:2054–65.
195. Cheong R, Paliwal S, Levchenko A. High-content screening inmicrofluidic devices. Expert Opin Drug Discov. 2010;5:715–20.
196. Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sour-igues Y, et al. Neuron-to-neuron transmission of alpha-synucleinfibrils through axonal transport. Ann Neurol. 2012;72:517–24.
197. Takeda S, Wegmann S, Cho H, DeVos SL, Commins C, RoeAD, et al. Neuronal uptake and propagation of a rare phos-phorylated high-molecular-weight tau derived from Alzheimer’sdisease brain. Nat Commun. 2015;6:8490.
198. Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N,Lachaud C, et al. Neuron-to-neuron wild-type Tau proteintransfer through a trans-synaptic mechanism: relevance tosporadic tauopathies. Acta Neuropathol Commun. 2014;2:14.
199. Pal A, Glass H, Naumann M, Kreiter N, Japtok J, Sczech R, et al.High content organelle trafficking enables disease state profilingas powerful tool for disease modelling. Sci Data. 2018;5:180241.
200. Lewandowska MK, Radivojevic M, Jackel D, Muller J, Hierle-mann AR. Cortical axons, isolated in channels, display activity-dependent signal modulation as a result of targeted stimulation.Front Neurosci. 2016;10:83.
201. Bukhari H, Muller T. Endogenous fluorescence tagging byCRISPR. Trends Cell Biol. 2019;29:912–28.
202. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol.2019;20:490–507.
203. Adli M. The CRISPR tool kit for genome editing and beyond.Nat Commun. 2018;9:1911.
204. Kaczmarczyk L, Mende Y, Zevnik B, Jackson WS. Manipulatingthe prion protein gene sequence and expression levels withCRISPR/Cas9. PLoS ONE. 2016;11:e0154604.
205. Chen JJ, Nathaniel DL, Raghavan P, Nelson M, Tian R, Tse E,et al. Compromised function of the ESCRT pathway promotesendolysosomal escape of tau seeds and propagation of tauaggregation. J Biol Chem. 2019;294:18952–66.
206. Rice AM, McLysaght A. Dosage-sensitive genes in evolutionand disease. BMC Biol. 2017;15:78.
207. Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, et al.Glucocerebrosidase depletion enhances cell-to-cell transmissionof alpha-synuclein. Nat Commun. 2014;5:4755.
208. Kampmann M. CRISPRi and CRISPRa screens in mammaliancells for precision biology and medicine. ACS Chem Biol.2018;13:406–16.
209. Stanek LM, Sardi SP, Mastis B, Richards AR, Treleaven CM,Taksir T, et al. Silencing mutant huntingtin by adeno-associatedvirus-mediated RNA interference ameliorates disease manifes-tations in the YAC128 mouse model of Huntington’s disease.Hum Gene Ther. 2014;25:461–74.
210. Xu X, Tay Y, Sim B, Yoon SI, Huang Y, Ooi J, et al. Reversal ofphenotypic abnormalities by CRISPR/Cas9-mediated gene cor-rection in huntington disease patient-derived induced pluripotentstem cells. Stem Cell Rep. 2017;8:619–33.
211. Chen Y, Dolt KS, Kriek M, Baker T, Downey P, Drummond NJ,et al. Engineering synucleinopathy-resistant human dopaminer-gic neurons by CRISPR-mediated deletion of the SNCA gene.Eur J Neurosci. 2019;49:510–24.
212. Hallmann AL, Arauzo-Bravo MJ, Mavrommatis L, Ehrlich M,Ropke A, Brockhaus J, et al. Astrocyte pathology in a humanneural stem cell model of frontotemporal dementia caused bymutant TAU protein. Sci Rep. 2017;7:42991.
213. Heman-Ackah SM, Manzano R, Hoozemans JJM, Scheper W,Flynn R, Haerty W, et al. Alpha-synuclein induces the unfoldedprotein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum Mol Genet. 2017;26:4441–50.
214. Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q,Alagappan R, et al. Generation of isogenic pluripotent stem cellsdiffering exclusively at two early onset Parkinson point muta-tions. Cell. 2011;146:318–31.
215. Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L,Glatza M, et al. Genetic correction of a LRRK2 mutation inhuman iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell.2013;12:354–67.
216. Jackow J, Guo Z, Hansen C, Abaci HE, Doucet YS, Shin JU,et al. CRISPR/Cas9-based targeted genome editing for correction
2704 A. de Rus Jacquet et al.

of recessive dystrophic epidermolysis bullosa using iPS cells.PNAS. 2019;116:26846–52.
217. Li XL, Li GH, Fu J, Fu YW, Zhang L, Chen W, et al. Highlyefficient genome editing via CRISPR-Cas9 in human pluripotentstem cells is achieved by transient BCL-XL overexpression.Nucleic Acids Res. 2018;46:10195–215.
218. Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM,Collingwood MA, et al. A high-fidelity Cas9 mutant delivered asa ribonucleoprotein complex enables efficient gene editing inhuman hematopoietic stem and progenitor cells. Nat Med.2018;24:1216–24.
219. Prusiner SB. Molecular biology of prion diseases. Science.1991;252:1515–22.
220. Makrinou E, Collinge J, Antoniou M. Genomic characterizationof the human prion protein (PrP) gene locus. Mamm Genome.2002;13:696–703.
221. Schmitz M, Lullmann K, Zafar S, Ebert E, Wohlhage M,Oikonomou P, et al. Association of prion protein genotype andscrapie prion protein type with cellular prion protein chargeisoform profiles in cerebrospinal fluid of humans with sporadicor familial prion diseases. Neurobiol Aging. 2014;35:1177–88.
222. Giannopoulos PN, Robertson C, Jodoin J, Paudel H, Booth SA,LeBlanc AC. Phosphorylation of prion protein at serine 43induces prion protein conformational change. J Neurosci.2009;29:8743–51.
223. Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H,et al. Different patterns of truncated prion protein fragmentscorrelate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. PNAS. 1998;95:8322–7.
224. Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in thehisto-molecular pathology of human prion disease. Brain Pathol.2019;29:278–300.
225. Zhong Q, Congdon EE, Nagaraja HN, Kuret J. Tau isoformcomposition influences rate and extent of filament formation. JBiol Chem. 2012;287:20711–9.
226. Hefti MM, Farrell K, Kim S, Bowles KR, Fowkes ME, Raj T,et al. High-resolution temporal and regional mapping of MAPTexpression and splicing in human brain development. PLoSONE. 2018;13:e0195771.
227. Lacovich V, Espindola SL, Alloatti M, Pozo Devoto V, Crom-berg LE, Carna ME, et al. Tau Isoforms Imbalance Impairs theAxonal Transport of the Amyloid Precursor Protein in HumanNeurons. J Neurosci. 2017;37:58–69.
228. Liu C, Gotz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinctsubcellular localization, with the 1N isoform being enriched inthe nucleus. PLoS ONE. 2013;8:e84849.
229. Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C,Loeffler T, et al. Phosphorylation of different tau sites duringprogression of Alzheimer’s disease. Acta Neuropathol Commun.2018;6:52.
230. Ercan-Herbst E, Ehrig J, Schondorf DC, Behrendt A, Klaus B,Gomez Ramos B, et al. A post-translational modification sig-nature defines changes in soluble tau correlating with oligo-merization in early stage Alzheimer’s disease brain. ActaNeuropathol Commun. 2019;7:192.
231. Katsumoto A, Takeuchi H, Tanaka F. Tau pathology in chronictraumatic encephalopathy and Alzheimer’s disease: similaritiesand differences. Front Neurol. 2019;10:980.
232. Barthelemy NR, Bateman RJ, Hirtz C, Marin P, Becher F, SatoC, et al. Cerebrospinal fluid phospho-tau T217 outperforms T181as a biomarker for the differential diagnosis of Alzheimer’sdisease and PET amyloid-positive patient identification. Alz-heimer’s Res Ther. 2020;12:26.
233. Barthelemy NR, Mallipeddi N, Moiseyev P, Sato C, BatemanRJ. Tau phosphorylation rates measured by mass spectrometry
differ in the intracellular brain vs. extracellular cerebrospinalfluid compartments and are differentially affected by Alzhei-mer’s disease. Front Aging Neurosci. 2019;11:121.
234. Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, Network RIotEHsD. et al. The role of tau in thepathological process and clinical expression of Huntington’sdisease. Brain. 2015;138(Pt 7):1907–18.
235. Bamburg JR, Bernstein BW, Davis RC, Flynn KC, Goldsbury C,Jensen JR, et al. ADF/Cofilin-actin rods in neurodegenerativediseases. Curr Alzheimer Res. 2010;7:241–50.
236. Spillantini MG, Goedert M, Crowther RA, Murrell JR, FarlowMR, Ghetti B. Familial multiple system tauopathy with preseniledementia: a disease with abundant neuronal and glial tau fila-ments. PNAS. 1997;94:4113–8.
237. Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E,Torrejon-Escribano B, et al. Glial and neuronal tau pathology intauopathies: characterization of disease-specific phenotypes and taupathology progression. J Neuropathol Exp Neurol. 2014;73:81–97.
238. Strang KH, Golde TE, Giasson BI. MAPT mutations, tauopathy,and mechanisms of neurodegeneration. Lab Invest.2019;99:912–28.
239. Leong YQ, Ng KY, Chye SM, Ling APK, Koh RY. Mechanismsof action of amyloid-beta and its precursor protein in neuronalcell death. Metab Brain Dis. 2020;35:11–30.
240. Rezaei-Ghaleh N, Amininasab M, Kumar S, Walter J, Zweck-stetter M. Phosphorylation modifies the molecular stability ofbeta-amyloid deposits. Nat Commun. 2016;7:11359.
241. Kumar S, Wirths O, Stuber K, Wunderlich P, Koch P, Theil S,et al. Phosphorylation of the amyloid beta-peptide atSer26 stabilizes oligomeric assembly and increases neurotoxi-city. Acta Neuropathol. 2016;131:525–37.
242. Thal DR, Walter J, Saido TC, Fandrich M. Neuropathology andbiochemistry of Abeta and its aggregates in Alzheimer’s disease.Acta Neuropathol. 2015;129:167–82.
243. Takahashi RH, Nagao T, Gouras GK. Plaque formation and theintraneuronal accumulation of beta-amyloid in Alzheimer’s dis-ease. Pathol Int. 2017;67:185–93.
244. Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, MaghamesCM, Meymand ES, et al. Amyloid-Beta (Abeta) plaques promoteseeding and spreading of alpha-synuclein and Tau in a mousemodel of lewy body disorders with abeta pathology. Neuron.2020;105:260–75 e266.
245. Batarseh YS, Duong QV, Mousa YM, Al Rihani SB, Elfakhri K,Kaddoumi A. Amyloid-beta and astrocytes interplay in amyloid-beta related disorders. Int J Mol Sci. 2016;17:338.
246. Irwin DJ, Hurtig HI. The Contribution of Tau, Amyloid-Beta andAlpha-Synuclein Pathology to Dementia in Lewy Body Dis-orders. JADP. 2018;8:444.
247. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A,Dutra A, et al. Mutation in the alpha-synuclein gene identified infamilies with Parkinson’s disease. Science. 1997;276:2045–7.
248. Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, BergD, et al. Genome-wide association study reveals genetic riskunderlying Parkinson’s disease. Nat Genet. 2009;41:1308–12.
249. Beyer K, Ariza A. Alpha-Synuclein posttranslational modifica-tion and alternative splicing as a trigger for neurodegeneration.Mol Neurobiol. 2013;47:509–24.
250. Burre J, Sharma M, Sudhof TC. Cell biology and pathophy-siology of alpha-Synuclein. Cold Spring Harb Perspect Med.2018;8:a024091.
251. Villar-Pique A, Lopes da Fonseca T, Outeiro TF. Structure,function and toxicity of alpha-synuclein: the Bermuda triangle insynucleinopathies. J Neurochem. 2016;139(Suppl 1):240–55.
252. Calo L, Wegrzynowicz M, Santivanez-Perez J, Grazia, Spillan-tini M. Synaptic failure and alpha-synuclein. Mov Disord.2016;31:169–77.
IPSCs to study prion-like mechanisms 2705

253. Peng C, Gathagan RJ, Lee VM. Distinct alpha-Synuclein strainsand implications for heterogeneity among alpha-Synucleinopathies. Neurobiol Dis. 2018;109(Pt B):209–18.
254. A novel gene containing a trinucleotide repeat that is expandedand unstable on Huntington’s disease chromosomes. The Hun-tington’s Disease Collaborative Research Group. Cell.1993;72:971–83.
255. Ruzo A, Ismailoglu I, Popowski M, Haremaki T, Croft GF,Deglincerti A, et al. Discovery of novel isoforms of huntingtinreveals a new hominid-specific exon. PLoS ONE. 2015;10:e0127687.
256. Mort M, Carlisle FA, Waite AJ, Elliston L, Allen ND, Jones L,et al. Huntingtin exists as multiple splice forms in human brain. JHuntington’s Dis. 2015;4:161–71.
257. Neueder A, Landles C, Ghosh R, Howland D, Myers RH, FaullRLM, et al. The pathogenic exon 1 HTT protein is produced byincomplete splicing in Huntington’s disease patients. Sci Rep.2017;7:1307.
258. Romo L, Mohn ES, Aronin N. A fresh look at huntingtin mRNAprocessing in Huntington’s disease. J Huntington’s Dis.2018;7:101–8.
259. Ehrnhoefer DE, Sutton L, Hayden MR. Small changes, bigimpact: posttranslational modifications and function of hunting-tin in Huntington disease. Neuroscientist. 2011;17:475–92.
260. Cicchetti F, Lacroix S, Cisbani G, Vallieres N, Saint-Pierre M,St-Amour I, et al. Mutant huntingtin is present in neuronalgrafts in Huntington disease patients. Ann Neurol.2014;76:31–42.
261. Hartlage-Rubsamen M, Ratz V, Zeitschel U, Finzel L, Mach-ner L, Koppen J, et al. Endogenous mouse huntingtin is highlyabundant in cranial nerve nuclei, co-aggregates to Abeta pla-ques and is induced in reactive astrocytes in a transgenicmouse model of Alzheimer’s disease. Acta NeuropatholCommun. 2019;7:79.
262. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P,Hentati A, et al. Mutations in Cu/Zn superoxide dismutase geneare associated with familial amyotrophic lateral sclerosis. Nature.1993;362:59–62.
263. Groner Y, Lieman-Hurwitz J, Dafni N, Sherman L, Levanon D,Bernstein Y, et al. Molecular structure and expression of the genelocus on chromosome 21 encoding the Cu/Zn superoxide dis-mutase and its relevance to Down syndrome. Ann NY Acad Sci.1985;450:133–56.
264. Zinman L, Liu HN, Sato C, Wakutani Y, Marvelle AF, MorenoD, et al. A mechanism for low penetrance in an ALS family witha novel SOD1 deletion. Neurology. 2009;72:1153–9.
265. Fay JM, Zhu C, Proctor EA, Tao Y, Cui W, Ke H, et al. Aphosphomimetic mutation stabilizes SOD1 and rescues cellviability in the context of an ALS-associated mutation. Structure.2016;24:1898–906.
266. Jiang H, Shimizu H, Shiga A, Tanaka M, Onodera O, Kakita A,et al. Familial amyotrophic lateral sclerosis with an I104Fmutation in the SOD1 gene: Multisystem degeneration withneurofilamentous aggregates and SOD1 inclusions. Neuro-pathology. 2017;37:69–77.
267. Milani P, Gagliardi S, Cova E, Cereda C. SOD1 transcriptionaland posttranscriptional regulation and its potential implicationsin ALS. Neurol Res Int. 2011;2011:458427.
268. Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK.Molecular mechanisms of TDP-43 misfolding and pathology inamyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:25.
269. Xiao S, Sanelli T, Chiang H, Sun Y, Chakrabartty A, Keith J,et al. Low molecular weight species of TDP-43 generated byabnormal splicing form inclusions in amyotrophic lateralsclerosis and result in motor neuron death. Acta Neuropathol.2015;130:49–61.
270. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M,Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporallobar degeneration and amyotrophic lateral sclerosis. Ann Neu-rol. 2008;64:60–70.
271. Robinson JL, Geser F, Stieber A, Umoh M, Kwong LK, VanDeerlin VM, et al. TDP-43 skeins show properties of amyloid ina subset of ALS cases. Acta Neuropathol. 2013;125:121–31.
272. Lin WL, Dickson DW. Ultrastructural localization of TDP-43 infilamentous neuronal inclusions in various neurodegenerativediseases. Acta Neuropathol. 2008;116:205–13.
2706 A. de Rus Jacquet et al.