Embed Size (px)
Citation preview

1
Computational Design of an
Enzyme-catalyzed Diels-Alder
reaction
Author: Max Pettersson
Supervisor: Prof. Tore Brinck
Institution of Applied Physical Chemistry, KTH
Date: 3/11-2016

2
Abstract The Diels-Alder is an important reaction that is one of the primary tools for synthesizing cyclic
carbon structures, while simultaneously introducing up to four stereocenters in the resulting
product. Not only is it a widely explored reaction in organic chemistry, but a vital tool in industry to
construct novel compounds for pharmacological applications. Still, a remaining concern is the fact
that upon the introduction of stereogenic carbons, the possibility of stereoselective control is greatly
diminished. A common solution to the problem of undesirable stereoisomers is to employ chiral
auxiliaries and ligands as means to increase the yield of a certain stereoisomer. However,
incorporating these types of compounds in order to obtain an enantiomerically pure product
increases the amount of synthetic steps to be regulated, implying that one or more purification steps
are necessary to obtain the desired result. An accompanying thought leans toward the
environmental aspect, as the principles of green chemistry are of great importance.
This thesis presents the attempts to explore the possibility of engineering an enzyme that can
catalyze an asymmetric Diels-Alder reaction through the use of molecular modeling. Based on
previous work, the catalytically proficient enzyme ketosteroid isomerase had been deemed a
probable candidate as a Diels-Alderase. To evaluate the enzyme thoroughly, a set of compounds was
scored against the active binding site where the best hits against the wild type were saved and
evaluated repeatedly after the introduction of rational mutations.
Although no conclusive indication of an optimal design could be obtained at the end of this work,
valuable insight was retrieved on plausible design strategies, which eventually could help lead to the
first catalytically proficient Diels-Alderase.

3
Sammanfattning Diels-Alder är en viktig reaktion då den är ett redskap för att syntetisera cykliska kolstrukturer,
samtidigt som uppemot fyra stereocentra introduceras i den resulterande produkten. Reaktionen
används inte enbart inom organisk kemi, utan är även ett viktigt redskap inom industriella
sammanhang för att ta fram nya preparat som direkt kan tillämpas inom farmakologi. En återstående
problematik är faktumet att introduktionen av nya stereogena kol bidrar till att drastiskt minska
möjligheten att bibehålla en stereoselektiv kontroll. En vanlig lösning för att undvika oönskade
stereoisomerer är att nyttja kirala hjälpmolekyler och ligander för att öka utbytet av en specifik
stereoisomer. Dock innebär införandet av dessa hjälpmolekyler i strävan att erhålla en
enantiomeriskt ren produkt ett ökat antal syntes-steg att hantera, vilket antyder att ett eller flera
reningssteg är nödvändiga för att uppnå önskat resultat. Ur en miljösynpunkt är detta värt att ha i
åtanke, då principerna för grön kemi är viktiga.
Detta arbete utforskar möjligheterna att konstruera ett enzym som kan katalysera en asymmetrisk
Diels-Alder-reaktion, med hjälp av molekylär modellering. Baserat på tidigare arbeten har enzymet
ketosteroid isomeras valts ut som en potential kandidat till ett Diels-Alderase. För att noggrant
evaluera enzymet så screenades ett set av substrat mot dess aktiva säte, där de bästa träffarna
gentemot vildtypen sparades och återevaluerades allteftersom rationella mutationer kontinuerligt
introducerades.
Trots avsaknaden av klara indikationer på att en optimal design har kunnat tas fram vid slutet av
detta arbete, så erhölls värdefull insikt på möjliga design-strategier, vilket skulle kunna bistå
sökandet av det första katalytiskt effektiva Diels-Alderase.

4
Acknowledgement I would like to thank my supervisor professor Tore Brinck for giving me the opportunity to work on
this project, for providing valid input on more difficult matters and for interesting, fun, and overall
helpful discussions. I appreciate the fact that I was given a lot of freedom in my working environment
and that I could ask for help regarding even the smallest of matters. I would also like to express a
heartfelt thank you to Camilla Gustafsson, Björn Dahlgren and Joakim Halldin Stenlid, whom all
welcomed me with open arms and served to nurture my scientific spirit by always assisting, helping
and encouraging me throughout my work. Thank you for all the laughter, wonderful discussions and
interesting topics we surveyed, both at the office desk and at the lunch table. Finally, a warm thank
you to all the people at Applied Physical Chemistry for creating a lovely environment to work, laugh
and dwell in.
You are all, truly, wonderful people.

5
Acronyms and abbreviations
DNE
Diene
DPH
Dienophile
DFT
Density Functional Theory
LGA
Lamarckian Genetic Algorithm
LS
Local Search
GA
Genetic Algorithm
KSI
Ketosteroid Isomerase
B3LYP
Becke’s 3-parameter Lee-Yang-Parr Hybrid functional
M06-2X
Minnesota 06 hybrid functional
FMO
Frontier Molecuar Orbital
HOMO
Highest Occupied Molecular Orbital
LUMO
Lowest Unoccupied Molecular Orbital
NED
Normal Electron Demand
IED
Inverse Demand
EDG
Electron Donating Group
EWG
Electron Withdrawing Group
ADT
AutoDock Tools
PDB
Protein Data Bank
TS
Transition state
LDA
Local Density Approximation
GGA General Gradient Approximation

6
Table of contents Computational Design of an Enzyme-catalyzed Diels-Alder reaction ..................................................... 1
Abstract ................................................................................................................................................... 2
Sammanfattning ...................................................................................................................................... 3
Acknowledgement ................................................................................................................................... 4
Acronyms and abbreviations ................................................................................................................... 5
1. Introduction ......................................................................................................................................... 8
1.1 Background .................................................................................................................................... 8
1.2 Ketosteroid Isomerase .................................................................................................................. 8
1.3 The Diels-Alder reaction ................................................................................................................ 9
1.4 Mechanism of D-A with KSI ......................................................................................................... 13
2. Theoretical Overview ........................................................................................................................ 16
2.1. Molecular Docking ...................................................................................................................... 16
2.1.1 Problems with Molecular Docking ....................................................................................... 16
2.1.2 AutoDock – A semi-empirical force field .............................................................................. 17
2.1.3 Autogrid ................................................................................................................................ 18
2.1.4 Lamarckian Genetic Algorithm ............................................................................................. 18
2.2 Molecular Dynamics .................................................................................................................... 19
2.2.1 Statistical Mechanics ............................................................................................................ 20
2.2.2 Molecular Dynamics simulation ........................................................................................... 21
2.2.3 Classical mechanics - Force fields ......................................................................................... 22
2.3 Quantum Chemistry .................................................................................................................... 25
2.3.1 The Schrödinger equation .................................................................................................... 25
2.3.2 The Born-Oppenheimer approximation ............................................................................... 26
2.3.3 Methods for solving the electronic Schrödinger equation................................................... 26
2.3.4 Density Functional Theory .................................................................................................... 27
3. Methodology ..................................................................................................................................... 29
3.1 Computational details ................................................................................................................. 29
3.1.1 Protein preparation .............................................................................................................. 29
3.1.2 Ligand preparation ............................................................................................................... 29
3.1.3 Molecular dynamics preparation ......................................................................................... 29
4. Results and Discussion ...................................................................................................................... 30
4.1 Molecular Docking with AutoDock 4.2 ........................................................................................ 30
4.1.1 Initial findings ....................................................................................................................... 30

7
4.1.2 Obtaining starting coordinates for MD simulation .............................................................. 34
4.2 Evaluation with Molecular Dynamics .......................................................................................... 44
4.2.1 MD simulation of MTE1 ........................................................................................................ 44
4.2.2 MD simulation of MTE2 ........................................................................................................ 48
4.2.3 MD simulation of MTE3 ........................................................................................................ 50
5. Conclusion ......................................................................................................................................... 56
References ............................................................................................................................................. 58
Appendix 1 ............................................................................................................................................. 61
Appendix 2 ............................................................................................................................................. 63
MTE1 – Cluster analysis of conformations ........................................................................................ 63
MTE2 – Cluster analysis of conformations ........................................................................................ 65
MTE3 – Cluster analysis of conformations ........................................................................................ 70

8
1. Introduction
1.1 Background Synthetic organic chemists are often interested in performing reactions that produce cyclic
compounds to be used in medical applications while simultaneously controlling the stereochemistry
of the reaction, as different stereoisomers usually demonstrate different biological properties, even
though they are made up of the same chemical structures [1]. A particularly useful reaction that
willingly allows for the synthesis of such structures is the Diels-Alder reaction, described by Diels and
Alder in 1928, a [4+2] cycloaddition reaction, which produces cyclohexane type structures and
forming two σC-C-bonds at the expense of two π-bonds [2]. However, the formation of two new σC-C-
bonds introduces up to four new stereogenic centers in the formed cyclic structure, a much desired
property overall in synthesis, but less so in the production of pharmaceuticals. As one particular
stereoisomer possesses the sought after properties that enables treatment of certain medical
conditions, the other may instead exhibit toxicological effects, which indicates that the reaction
needs to be put under rigid stereocontrol [1] While approaches has been taken to solve this by
employing chiral auxiliaries [3], this puts strain on the environment and a ‘greener’ approach is
preferable. An ingenious solution to this problem has been taken in the utilization of computational
design of enzymes, which are naturally chiral molecules that can generate products through
asymmetric reactions with high catalytic proficiency [4][5]. The use of computational methods
provides seemingly fast and accurate evaluation of attempted designs, as the toolbox of
computational chemists is undergoing steady evolution [4][5][6]. With high computational power
and improved molecular modeling programs at disposal, the in silico-generated designs allow for a
qualitative exploration of working systems. The enzyme 3-oxo-∆5-ketosteroid isomerase has been
described in earlier work as a potential Diels-Alderase and is considered one of the most efficient
catalytic machineries amongst enzymes [7]. This work explores the rational design of 3-oxo-∆5-
ketosteroid isomerase while employing a semirational design strategy developed by Brinck and co-
workers, thoroughly described elsewhere [8]. However, as a rough presentation is in order, the
protocol consists of three main stages; A) Static design with molecular docking, B) Dynamic design
with molecular dynamics and C) A quantum chemical evaluation. Appendix 1 presents the overall
flowchart, including the sub-steps of the main stages.
1.2 Ketosteroid Isomerase The enzyme 3-oxo-∆5-ketosteroid isomerase (KSI) from Pseudomonas testosteroni has been widely
considered as a relevant enzyme worth investigating due to its proficient catalytic machinery [9]. The
main reason as to why KSI is regarded as a plausible candidate for the catalysis of the Diels-Alder
reaction is due to its ability to abstract a proton from a simple carbon atom through a heterolytic C-H
bond cleavage [7][10], of which the details will be discussed later. The more prevalent situation is
that a proton abstraction occurs when an electronegative heteroatom (for example oxygen or
nitrogen) is the associated partner of said proton, such as the case in general acid-base reactions.
However, breaking of a C-H bond is commonly associated with a large activation barrier, and the fact
that this reaction proceeds rather efficiently is a testament to the catalytic proficiency of KSI [7].
KSI, obtained from the Protein Data Bank (PDB code: 1QJG), is complexed with the 3-oxo-∆5-
ketosteroid, equilenin, which will proceed via a catalytic isomerization to yield the 3-oxo-∆4-
ketosteroid isomer [11]. The isomerization follows the abstraction of the proton from the C4β

9
position on equilenin and the transfer of it to the C6β position. This is made possible by a catalytic
triad consisting of Asp99 (protonated), Asp38 (deprotonated) and Tyr14, which are buried in the active
site of KSI. The two amino acids Asp99 and Tyr14 will form hydrogen bonding interactions with the
carboxyl oxygen (C3-O), creating a Low Barrier Hydrogen Bond (LBHB). This has been disputed as one
of many other reasons as to why the catalytic machinery of KSI is so effective, alongside discussions
of electrostatic interactions and van der Waal’s forces [12]. While Asp99 and Tyr14 serve the purpose
of assisting in the stabilization of the proton transfer TS, the carboxyl oxygen of Asp38 is situated
roughly in the middle of the active site, within 2.8-3.6 Å of the C4β and C6β position of the steroid.
The close distances to the C4β proton as well as its low pKa of 4.57, allows Asp38 to act as the general
base for the proton transfer [7][9][11][12].
Inside the active site, these three amino acids make up an oxyanion hole, which serves to stabilize
the intermediary dienolate that is created upon isomerization of a 3-oxo-∆5-ketosteroid. From
Scheme 2 (section 1.4) it can be distinguished that following the deprotonation of the C4β proton, a
negative charge will accumulate on the ketosteroid C3-O, which in turn will be stabilized by hydrogen
bonding interactions from Asp99 and Tyr14, a stabilization of approximately 11 kcal/mol [12].
According to Pollack [12], 75 % of KSI:s catalytic ability is obtained from stabilization of the
intermediate, whereas 25 % is accounted for by the enolization.
The catalytic triad of amino acids in the active site was determined by mutation of these amino acids,
and the measurement of the loss of catalytic activity (kcat) in KSI as a result of these specific
mutations. The mutation of Tyr14 to Phe14 (Y14F) caused kcat to decrease by 104.7-fold, and the D38N
mutation decrease kcat by 105.6-fold. Mutation of Asp99 would also prove to demonstrate severe
impact on the catalytic rate. At pH 7, the mutagenesis of D99A and D99N would lower kcat 3000-fold
and 27-fold, respectively. From the investigation of the effect these mutations had on kcat, the
conclusion could be made that these three amino acids are vital to the enzymatic functions of KSI. It
was later determined that another amino acid, in close vicinity of the catalytic triad, also played an
important part in enhancing the catalytic rate. Directly behind Tyr14 lies another tyrosine residue,
Tyr55, which forms a hydrogen bond with the Tyr14 oxygen, which main purpose providing assistance
in catalytic activity. It has been shown that mutation of Tyr55 into other residues will lower the
catalytic activity of the active site [7][9][10][11][12][13].
After a brief overview of the KSI enzyme, a concluding remark can be made that KSI is an ingenious
example of evolution with a powerful potential as a catalyst. This does also provide hope that
eventual mutagenesis and rational design of the active site can improve the catalytic rate even
further.
1.3 The Diels-Alder reaction The overview of the D-A reaction presented in this section is based on the text book Organic
chemistry by Clayden et al [14].
Scheme 1. The mechanism of the D-A reaction.

10
The D-A reaction is a [4 + 2] cycloaddition (Scheme 1) where a conjugated diene interacts with a
dienophile, a species ready to interact with a diene that results in a cyclic species. The D-A reaction is
particularly useful for creating 6-membered cyclohexane rings while simultaneously introducing up
to four new stereogenic centers in the product. The common assumption is that the D-A reaction
occurs through a concerted mechanism. One of the criteria is that the diene needs to be in a cis-
conformation for the reaction to take place, where the reason can be explained with frontier
molecular orbital (FMO) theory.
Figure 1.The HOMO of 1,3-Butadiene.
Figure 1 depicts the highest occupied molecular orbital (HOMO) of a simple conjugated diene, 1,3-
Butadiene in its cis conformation, and figure 2 depicts the lowest unoccupied molecular orbital
(LUMO) of the dienophile, ethylene. These particular compounds serve an illustrative purpose in
regards to FMO theory, where the blue phase of the HOMO overlaps with the blue phase of the
LUMO and the red phase of the HOMO overlaps with the red phase of the LUMO.
Figure 2. The LUMO of ethylene.
The common situation observed is that the HOMO of the diene interacts with the LUMO of the
dienophile, usually ascribed as ‘normal electron demand’ (NED) Diels-Alder. A simpler rendering of
the FMO overlap between the two compounds can be seen in figure 3, where the electron rich diene
is seen overlapping the electron poor dienophile, in a rough representation of a D-A transition state
(TS).

11
Figure 3. A schematic rendition of the FMO overlap for an NED-DA reaction.
The D-A reaction is more prone to occur if the HOMO-LUMO energy gap is lowered and this very
situation can be promoted by introducing electron donating groups (EDG) onto the diene and
electron withdrawing groups (EWG) to the dienophile. An EDG on the diene will contribute with its
electrons through donation into the conjugated system of the diene, which will result in an increase
in the dienes HOMO energy, as opposed to the EWG and dienophile system which serves to
withdraw the electrons from the already electron poor dienophile, subsequently lowering the LUMO
energy. Therefore, substitution can have a drastic effect on the reaction rate.
Figure 4. The schematic version of a NED D-A.
Due to the nature of the HOMO-LUMO gap being overall close in energy; it is also possible to invert
the overlapping FMO, by exchanging the EDG on a diene with an EWG and vice versa for the
dienophile. This leads to the ‘inverted electron demand’ (IED) D-A, who’s FMO overlap is depicted
below. In the IED D-A reaction the HOMO of the dienophile is seen interacting with the LUMO of the
dienophile. The same reasoning as earlier is applied, as an EDG will raise the energy of the
dienophile’s HOMO, while the diene experiences a lower LUMO when substituted with an EWG.
Figure 5. FMO overlap for the IED-DA reaction.
12
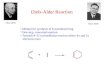
Figure 6. The schematic overview of IED D-A and their respective substituents.
Depending on the substitution of the diene and dienophile it can influence the regioselectivity in the
final product. Different substituents will affect the HOMO and LUMO of respective compound, as an
EDG and EWG will cause distortions of the HOMO/LUMO coefficients. The compounds investigated in
this work are tilted in their interaction due to this effect, resulting in an asymmetric overlap between
diene and dienophile.
Figure 7. The two TS-structures investigated in this work. A slight tilt in the overlap between these can be noticed, as a consequence of the carbonyl oxygens present in the molecules.
The substitution pattern will also have an effect on the stereoseletivity of the D-A product as the
reaction can pass through two different TS. These are known as the endo and exo TS, which will
result in the endo and exo products, respectively. The endo adduct is generally preferred over the exo
adduct. The usual accepted explanation is that the subtituent on the dienophile interacts with the π-
system of the diene and this is illustrated using the TS-structure presented to the right in figure 7, in
figure 8.
Figure 8. The demonstrated interactions giving rise to the endo adduct.
13
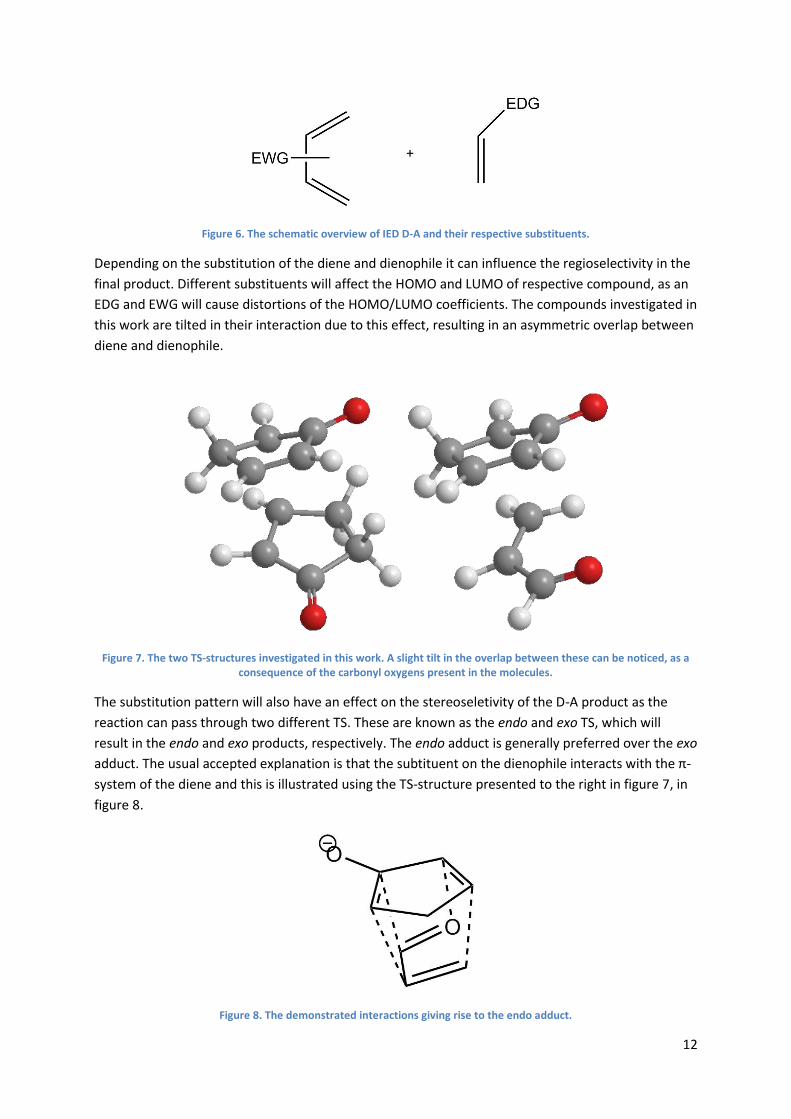
1.4 Mechanism of D-A with KSI After some review regarding KSI and its general function, now is the time to delve deeper into the
active mechanism of KSI and conveys the reason as to why this particular enzyme was chosen as a
candidate for Diels-Alderase. As previously mentioned, KSI is complexed with the steroid equilenin,
and even though the reaction mechanism for the isomerization of equilenin also has been pointed
out, it is worth going through it with accurate figures and explanation as this may serve an intuitive
purpose.
Starting with the transfer of the C4β proton from equilenin by Asp38, it can be distinguished from
Scheme 2 that the intermediate species formed in the active site is an anion, or more specifically an
enolate, with a negative charge accumulation on the C3 oxygen. This is the first step of the reaction
mechanism, enolization. As the reaction proceeds on to reketonization, the previously abstracted
C4β proton is now transferred from Asp38 to the C6β position on equilenin by a series of electron
rearrangements within the molecule. The isomerization is completed and the π-bond has clearly
changed locations.
Scheme 2. The natively complexed ligand of KSI, equilenin is shown undergoing the reaction with the catalytic triad.
With a little more background information about the mechanism of the catalytic reaction, a rather
ingenious idea was brought forward in regards to utilizing KSI as a Diels-Alderase. By making use of

14
an α,β-saturated ketone and the general acid/base mechanism in KSI, the abstraction of the ketones
α proton would generate a diene in situ, while the TS intermediate will attain stability through
interactions Asp99 and Tyr14. Earlier studies have shown that Asp38 is an accomplished base in
heterolytic cleavage of C-H bonds, which leaves for a broad possibility of substrate choices in regards
to pKa of the α-proton [7]. Although a diene can readily be generated within the active site of KSI
there is still the problem with the required s-cis conformation of the diene. A quite simple solution to
this problem would be to use cyclic α,β-saturated ketones as pro-dienes, seeing as the abstraction of
the α proton would result in an enolate species and thereby provide a diene in the correct s-cis
conformation.
The substrates that have been under main investigation in this thesis are the 2-cyclopenten-1-one (1)
and acrolein (2), with a varying arsenal of substituents positioned according to figure 9 (a figure of
attempted substitutions can be found in appendix 1).
Figure 9. The substitution pattern available to the investigated compounds.
It should be mentioned that (2) was only considered as a dienophile during this investigation as it
has no certainty of always remaining in an s-cis conformation. Although the energy barrier for
rotation along the σ-bond of acrolein is surmountable, the trans-conformation is lower in energy and
therefore the more stable and often encountered conformation.
Since the active site was constructed to house 3-oxo-∆5-ketosteroids and only one substrate at the
time, it is important to note that a Diels-Alderases active site would have to house two substrates at
the same time. Therefore the active site is required to stabilize the D-A TS in a likewise manner as it
stabilizes its steroids.
According to the theozyme model constructed by Brinck et al [7] the catalysis will occur as according
to the figures below in alphabetic order. It is worth noting that although only a presentation is given
for (1) below, (2) will undergo the same mechanism.

15
Figure 10. The pro-diene is seen interacting with the Asp99
and Tyr14
in a) where the α-proton is about to be abstracted by Asp
38, generating the diene in situ. In b) the proton has successfully been abstracted and can be observed on Asp
38.
Figure 11. In c) the dienophile (another molecule of (2) here) has approached the diene from below and formed the first TS. d) Presents the anionic product obtained as the reaction has been performed.
Figure 12. The re-protonation with the proton still present on Asp38
in e) which is seen returned to its original position in f). The resulting product is the endo adduct of the two interacting molecules.

16
In their work, (1) was used as a substrate with the intention to act as both diene and dienophile.
Initially, the first molecule of (1) will form hydrogen bonds to Asp99 and Tyr14 with its ketone oxygen
acting as hydrogen bond acceptor, while simultaneously positioning its α proton in close proximity of
the basic Asp38 residue. Of course there will be no guarantee for a perfect adaption of orientation
immediately as the pro-diene approaches the active site, but a reaction will only occur as the α
proton is facing Asp38. In a swift fashion the second molecule of (1), the dienophile, will approach the
diene from ‘slightly beneath’, according to figure
Following the formation of the diene a regular D-A reaction would take place and a newly formed
product is obtained. From figure 12.f) it is worth noting the transfer of the proton to its original
position. The Endo adduct is also the most probable product to end up with in accordance with the
Endo rule described earlier (section 1.3).
2. Theoretical Overview
2.1. Molecular Docking Since the methodology known as molecular docking was established in the 1980:s [15] it has grown
to be a valuable asset in drug discovery, providing a fast and effective means of detecting potential
ligands to be used in drug design [16]. The field of molecular docking has led to a spread of different
computational docking programs which aims to perform a specific screening process, both
concerning protein-protein docking and protein-ligand docking, where a few assorted programs are
described elsewhere [17]. The remainder of this section will consider protein-ligand docking with the
AutoDock software as well as some brief review of the relevant background and coverage of some
off the more important parameter files in AutoDock [18].
2.1.1 Problems with Molecular Docking
The study of molecular interactions can reveal a tremendous amount of information regarding
biological processes. What makes molecular docking a complicated matter is the fact that enzymes
are dynamic entities, where the backbone possesses certain degrees of freedom and the side chains
can adapt different conformations. Of course, the ligand can explore some conformational space, but
the enzyme is a bit more restricted due to entropic penalties, causing some dynamic restrictions,
associated with enzyme movement [19].
In the common biochemistry class it is not unusual to introduce students to the well-known “lock and
key” description of a molecular interaction between enzyme and ligand. However, this analogy does
not describe the entire situation, mainly due to the fact that there is still a lot we do not know about
enzymes. Another good description that has been used is the “induced fit”, and this is considerably
closer to reality than the “lock and key” description [19]. Another good way of looking at the
situations is considering the system as a “hand in a glove” situation, where the glove is adapting
(within limits) to the different conformations the hand can adopt [17][20]. But the central purpose of
molecular docking is to, as earlier mentioned, efficiently screen a large set (or library) of compounds
against a macromolecular target with satisfactory precision. This means that the ligand should be
able to explore an extensive conformational space and ligand orientations with accurate
determination of binding mode and affinity towards the target, while still remaining computationally
fast [17][18]. By introducing flexible side chains in an enzyme there is a possibility of increasing the

17
prediction of correct binding modes at the cost of the simulation being more computationally
expensive, while at the same time increasing the chance of false positives amongst the result due to
the larger conformational space [42]. Since only a few side chain residues are considered as flexible,
this aspect fails to treat backbone mobility of the enzyme [20].
Although molecular docking is a quite difficult problem in regards to optimization, it is still an
extensively useful method in quickly predicting binding modes and affinities, as well as acquiring
starting coordinates for further analysis with for example molecular dynamics. Molecular dynamics
(treated explicitly later) aims to thoroughly analyze a ligands conformational space while treating the
entire enzyme as being dynamic, including backbone flexibility along with the different
conformations of the side chains. However, as this approach requires additional computational
power, molecular dynamics is not recommended for screening of compounds to the same extent as
docking. But how does the evaluation of binding affinity and binding modes occur in AutoDock?
2.1.2 AutoDock – A semi-empirical force field
AutoDock 4.2 utilizes a conceptually simple semi-empirical force field to evaluate the free binding
energy in the formation of a ligand-protein complex that has been parameterized by using a training
set consisting of a large number of protein-inhibitor complexes, where all 3D structures and inhibitor
constants KI had previously been determined. The information relayed in this section is based on the
work by Huey et al [21]. The force field utilizes a set of pair-wise evaluations V and the term ∆Sconf
describing the conformational entropy, which is lost upon the binding of the ligand to the
macromolecular target,
∆𝐺 = (𝑉𝑏𝑜𝑢𝑛𝑑𝐿−𝐿 − 𝑉𝑢𝑛𝑏𝑜𝑢𝑛𝑑
𝐿−𝐿 ) + (𝑉𝑏𝑜𝑢𝑛𝑑𝑃−𝑃 − 𝑉𝑢𝑛𝑏𝑜𝑢𝑛𝑑
𝑃−𝑃 ) + (𝑉𝑏𝑜𝑢𝑛𝑑𝑃−𝐿 − 𝑉𝑢𝑛𝑏𝑜𝑢𝑛𝑑
𝑃−𝐿 + ∆𝑆𝑐𝑜𝑛𝑓) Eq.1
Where L and P refers to the ligand and protein (or macromolecule) respectively. For each docking
simulation the binding energy is estimated in a two-step process where the first course of action is i)
to determine the energy that arises intramolecularly as both molecules transition from unbound to
bound conformation, where the two terms in the first parenthesis of Eq.1 describes the ligands
intramolecular energy in the bound and unbound states. The two terms in the second parenthesis
describes the same type of intramolecular interactions for the macromolecule. ii) The second
estimation occurs in the last parenthesis where the ligand forms a complex with the macromolecule,
and the intermolecular energy is evaluated. It is important to note that 𝑉𝑢𝑛𝑏𝑜𝑢𝑛𝑑𝑃−𝐿 will be zero, as it is
assumed that the ligand and protein are at a great enough distance from each other that no
interactions will take place.
Each pair-wise evaluation in eq.1 consists of terms that aim to describe enthalpic as well as entropic
contributions to the free binding energy and is described as follows:
𝑉 = 𝑊𝑣𝑑𝑤∑(𝐴𝑖𝑗
𝑟𝑖𝑗12 −
𝐵𝑖𝑗
𝑟𝑖𝑗6 ) +𝑊ℎ𝑏𝑜𝑛𝑑∑𝐸(𝑡) (
𝐶𝑖𝑗
𝑟𝑖𝑗12 −
𝐷𝑖𝑗
𝑟𝑖𝑗10) +
𝑖,𝑗
𝑊𝑒𝑙𝑒𝑐∑𝑞𝑖𝑞𝑗
휀(𝑟𝑖𝑗)𝑟𝑖𝑗+𝑊𝑠𝑜𝑙∑(𝑆𝑖𝑉𝑗 + 𝑆𝑗𝑉𝑖)𝑒
(−𝑟𝑖𝑗2
2𝜎2)
𝑖,𝑗𝑖,𝑗𝑖,𝑗
Eq.2
Where the interactions considered are dispersion/respulsion, hydrogen bonding, electrostatics and
desolvation, respectively.
Each respective interaction parameter is preceded by the experimentally determined weighting
factors W. The A and B are parameters retrieved from the AMBER force field [22]. E(t) is defined as a

18
weighted directional, where t is the angle away from ideal bonding geometry. The parameters C and
D have been assigned to have a maximum well depth for hydrogen bonds, where the depth is 5
kcal/mol for O-H, N-H at 1.9 Å and 1 kcal/mol for S-H at 2.5 Å. The third term is the electrostatic
interactions evaluated by a screened Coulomb potential. The fourth and last term contains the
desolvation potential which is dependent on the volume of atoms V surrounding particular atom,
shielding that atom from solvent molecules. S is the solvation parameter and σ is a distance-
weighted factor set to 3.5 Å. The conformational entropy that is lost upon binding of the ligand,
∆Sconf, is proportional to the amount of bonds with ability to rotate, Ntors, where all torsional degrees
of freedom are included and is described accordingly
∆𝑆𝑐𝑜𝑛𝑓 = 𝑊𝑐𝑜𝑛𝑓𝑁𝑡𝑜𝑟𝑠 Eq.3
2.1.3 Autogrid
To attain a swift performance when executing a docking simulation, AutoDock 4.2 makes use of pre-
calculated grid maps that contain information about interaction energies for a set of atom types
present in the ligand that is to be docked. These grid maps are calculated with the program AutoGrid.
In the ADT GUI, the dimensions of a grid box can be defined over a selected partition of the
macromolecule and by specifying the grid point spacing, each of these points houses information on
the potential energy of the atoms in the ligand in relation to the macromolecule.
AutoGrid requires a grid parameter file (.gpf) that among other things holds the information on what
maps should be generated for significant atom types, the size of the grid box along with coordinates
declaring its location, the rigid receptor to be used in the docking simulation and more.
It is important to remember that this is simply a pre-calculation to improve calculation speed. The
AutoGrid program manages to reduce the complexity of the problem from N2 to N, where N is the
number of interacting atoms [18][21].
2.1.4 Lamarckian Genetic Algorithm
The French scientist Jean-Baptiste de Lamarck took an interest in evolution during the late 1700s and
proposed an evolutionary theory that could be summed up as, whatever traits an individual acquires
during its lifetime will affect the individuals traits that it passes on to its offspring. Even though this is
generally agreed upon to be an incorrect understanding of how evolution works, Lamarck is
accredited as being the first to present a truly coherent evolutionary theory [23].
During a molecular docking simulation all orientational, conformational and positional samplings
needs to be explored, turning docking into a difficult optimization problem [18]. Genetic algorithms
(GA) have previously been successfully employed for these types of problems as they are effective in
conducting a global search. A GA intends to discover solutions by means of procedures that are
inspired by evolutionary principles. During a docking, a ligand is situated in a particular state in
relation to the protein, where the translation, orientation and conformation of said ligand is
described by a set of values. If these values changes, the same is true for the ligands state. This is
therefore known as a ligands state variables and in a GA each state variable constitutes a particular
gene. This means that the entire ensemble of state variables makes up the ligands, or individuals,
genotype. The individual’s genotype is mapped by applying a developmental mapping function to its
corresponding phenotype, which composes the ligands atomic coordinates. As the phenotype has
been mapped the individual’s fitness is evaluated, where the fitness corresponds to the total energy

19
of interaction between a ligand and a protein. Similarly, this methodology is applied for the entirety
of the population.
The user is free to choose between a GA, an adaptive Local Search method (LS) or a hybrid GA-LS
method in AutoDock 4.2. The LS performs local energy minimizations and depending on the
previously registered energies, the step size is adjusted, where an increase in energy will double the
step size and a decrease in energy will result in the step size being halved. The adaptive LS is based
on work by Solis and Wets [24]. The combination of the GA and adaptive LS is what composes the
LGA. But the aspect of what makes this hybrid method “Lamarckian” has yet to be mentioned.
As the global search is performed, occasionally, a random mutation will arise, which might improve
the fitness of a certain individual. During the mating between two individuals it is possible that there
will be a crossover of genetic material that is passed on to the offspring, where the offspring can
plausibly be evaluated as being better fit than its parents. This is all in accordance with Darwinian
and Mendelian genetics [18]. As the LS progresses the phenotype can readily be altered, meaning
that the ligand performs some local movement, resulting in an energy decrease (increased fitness).
From the current phenotype there can be an inverse mapping to the genotype. This is analogue to
Lamarck’s claims about traits that are acquired during an individual’s lifetime can be effectively
transmitted to its offspring. The inverse mapping from the phenotype to the genotype is therefore by
definition “Lamarckian”, ultimately resulting in the LGA.
2.2 Molecular Dynamics There are several different biological systems that are seemingly interesting to study and that have
been studied on a macroscopic level. These systems commonly contain an overwhelming number of
particles and therefore also present a large number of conformations and unique interactions that
appear insurmountable in regards of detailed inspection. An investigation can however be performed
through the use of computer-aided simulations, where a small portion of these macroscopic systems
can be properly examined, but with a considerably less amount of particles included. Different
approaches have been taken to achieve manageable systems that can be carefully studied, where
one of the most famous methods employed is the Monte Carlo method. The general concept of the
Monte-Carlo method is to gather a lot of samplings by randomly generating a certain trial move and
then making a choice of whether to accept or reject the move. Although the Monte Carlo method
has proved useful in its application of randomness in studying for example fluid dynamics, it does not
consider changes over time. When contemplating various biological systems such as substrate
passage between transmembrane proteins, determination of binding free energies between ligand-
enzyme complexes and protein folding analysis (only to name a few), including time-dependency
serves to quite accurately describe the step-wise development of the system that one seeks to study.
At the same time, in order to obtain the sought after accuracy of these time-dependent biological
systems the employment of molecular dynamics (MD) are of utmost necessity. Additionally, in order
to describe these macroscopic systems at a suitable microscopic level, namely through expression of
atomic positions and their respective velocities, statistical mechanics are of essence. Since MD
simulations treat clearly large N-body problems, a means of rationalizing these computationally
demanding calculations is via utilization of mechanical force fields.
Following this introductory passage will be a brief overview of statistical mechanics, the fundamental
aspects of a typical MD simulation while also including some parts on force fields.

20
All theory in this section is taken from Molecular Modelling – Principles and Applications by Andrew
Leach [25].
2.2.1 Statistical Mechanics
The association between simulations at the microscopic level and macroscopic properties is made
with the help of statistical mechanics. The very purpose of statistical mechanics is to find a way to
study macroscopic properties through the use of the microscopic simulations, or more accurately
put, to describe macroscopic properties with the help of position and momentum for each of the N
particles present in the system. A useful approach in defining the state of the system comprised of N
particles is that the position can be described by 3N coordinates and the momenta can be described
by 3N components, resulting in 6N dimensions which define the system. More accurately, the
position and momenta of these particles are what define a microscopic state and the 6N dimension
made up from these particles is referred to as the phase space. As a single point in phase space
assists in the description of a systems current state, an entire collection of these single points makes
up what is known as an ensemble, and an ensemble is a collection of microscopic states. These
ensembles are used as expectation values in statistical mechanics, implying that a macroscopic
system could be viewed upon as a series of replications which are all considered at the same time.
This situation can be described by the following expression
⟨𝐴⟩𝑒𝑛𝑠 = ∫∫𝑑𝒑𝑁 𝑑𝒓𝑁𝐴(𝒑𝑁 , 𝒓𝑁) 𝜌(𝒑𝑁 , 𝒓𝑁) Eq.4
Where ⟨𝐴⟩ is the ensemble average, or the average value of the property A taken over all replications
of the system and
𝐴(𝒑𝑁 , 𝒓𝑁) Eq.5
Describes the property A as a function of the momenta p and position r in the system. Also, the
𝜌(𝒑𝑁 , 𝒓𝑁) Eq.6
Is the ensembles probability density, given by
𝜌(𝒑𝑁 , 𝒓𝑁) =1
𝑄exp(−
𝐸(𝒑𝑁 , 𝒓𝑁)
𝑘𝐵𝑇)
Eq.7
Where E is the energy, T is temperature and 𝑘𝐵 is the Boltzmann constant. Q is the partition function
and can be written as
𝑄 =∫∫𝑑𝒑𝑁 𝑑𝒓𝑁 exp(−Ĥ(𝒑𝑁, 𝒓𝑁)
𝑘𝐵𝑇)
Eq.8
Since this describes the overall procedure to estimate the properties of a macroscopic system by the
evaluation of the ensemble average of A, how does the approach look for a MD simulation where
time-dependency is introduced? Well, it is in fact very similar, except it has to be considered that a

21
macroscopic system usually contains numbers of atoms in the order of 1023 and solving this while
including time-dependency is unfeasible with modern computational power. The integral presented
in Eq.4 is complex in itself, implying that a different route has to be taken to establish a time-
dependent MD simulation with an acceptable computational time. For a system comprised of N
number of particles, the instantaneous value of the property A may be expressed as
𝐴(𝒑𝑁(𝑡), 𝒓𝑁(𝑡)) ≡ 𝐴(𝑝1𝑥, 𝑝1𝑦, 𝑝1𝑧, 𝑝2𝑥 , … , 𝑥1, 𝑦1, 𝑧1, 𝑥2, … , 𝑡) Eq.9
Where 𝑝1𝑥refers to the momentum of particle 1 in the x direction where 𝑥1 is the momentums x
coordinate and so on. As the instantaneous value is reliant on the changes occurring over time in the
system due to different interactions between particles taking place, it is appropriate to express the
property A as an average value even in the MD simulation. This average is based on the simulation
time, resulting in the time average of the property A, expressed by
⟨𝐴⟩𝑡𝑖𝑚𝑒 = lim𝜏→∞
1
𝜏∫ 𝐴(𝒑𝑁(𝑡), 𝒓𝑁(𝑡))𝑑𝑡
𝜏
𝑡=0
≈1
𝑀∑𝐴(𝒑𝑁, 𝒓𝑁)
𝑀
𝑡=1
Eq.10
Where t is the simulation time and M is the number of time steps exercised during simulation and
𝐴(𝒑𝑁 , 𝒓𝑁) is the instantaneous value of property A.
Hence, both microscopic and macroscopic properties can be described with statistical mechanics. But
a key component is still lacking as MD simulations averages over time and experimental performance
samples ensemble averages. By employing the ergodic hypothesis the microscopic and macroscopic
systems can be evaluated on the following assumption
⟨𝐴⟩𝑒𝑛𝑠 = ⟨𝐴⟩𝑡𝑖𝑚𝑒 Eq.11
The ergodic hypothesis states that a system may explore each and every possible state if it is allowed
to continue indefinitely through time. As such it is impeccable that a MD simulation manages to
sample enough of the phase space as a fixed time limit is specified for a simulation run. If enough
states are explored one can claim that the MD simulation will correspond to experimental accuracy.
There are ways to effectively perform MD simulations with fewer particles without penalizing the
‘real’ behavior of the system. The use of periodic boundary conditions (PBC) serves to limit the
number of particles; therefore successfully lowering the computation time as the number N will be
lower. A simulation may then be conducted for a satisfactory amount of time, producing a required
amount of conformations.
2.2.2 Molecular Dynamics simulation
The primary goal for an MD simulation is to evaluate the exerted force arising upon the interactions
of particles within a system. This information is thus obtained by solving the Newton’s equations of
motion,
𝑭 = 𝒎𝒂 Eq.12
This equation houses a differential equation as well which can be presented as

22
𝑑2𝒓𝑖𝑑𝑡2
=𝑭𝑖𝒎𝑖
Eq.13
Where 𝒓𝑖 is the position of the particle, 𝒎𝑖 its mass and 𝑭𝑖 the force applied on said particle in a
particular direction. To study the dynamics of the system, at a certain time t, the particles initial
position, its velocity and the acceleration should be known. These parameters have been
approximated with a Taylor series expansion while considering the position 𝒓(𝑡) and the time steps
before, 𝒓(𝑡 + 𝛿𝑡) and after, 𝒓(𝑡 − 𝛿𝑡). The Taylor expansions are presented accordingly
𝑖)𝒓(𝑡 + 𝛿𝑡) = 𝐫(t) + 𝐯(t)𝛿𝑡 +1
2𝒂(𝑡)𝛿𝑡2 +
1
6𝒃(𝑡)𝛿𝑡3 +⋯ Eq.14
𝑖𝑖)𝒓(𝑡 − 𝛿𝑡) = 𝐫(t) − 𝐯(t)𝛿𝑡 +1
2𝒂(𝑡)𝛿𝑡2 −
1
6𝒃(𝑡)𝛿𝑡3 +⋯ Eq.15
When i) and ii) are added it produces the following
𝒓(𝑡 + 𝛿𝑡) + 𝒓(𝑡 − 𝛿𝑡) = 2𝐫(t) + 𝒂(𝑡)𝛿𝑡2 + 𝑂(𝛿𝑡4) Eq.16
That can be rearranged to
𝒓(𝑡 + 𝛿𝑡) = 2𝐫(t) − 𝒓(𝑡 − 𝛿𝑡) + 𝒂(𝑡)𝛿𝑡2 Eq.17
Resulting in the original Verlet algorithm. From Eq.16 it can be seen that the Verlet algorithm will be
correct up to 4th order in positions. In the expression, 𝐫(t) represents the position and 𝒂(𝑡) is simply
the acceleration. The velocity 𝐯(t) is not explicitly included in the Verlet algorithm as the addition of
i) and ii) cancels the term out. One can however calculate the velocity from the information provided
by the positions
𝐯(t) =𝒓(𝑡 + 𝛿𝑡) − 𝒓(𝑡 − 𝛿𝑡)
2𝛿𝑡 Eq.18
Overall the calculation Verlet algorithm follows the following sequence:
1. Determine 𝒂(𝑡) from the force, 𝐹[𝒓(𝑡)}/𝑚.
2. Calculate 𝒓(𝑡 + 𝛿𝑡) from 𝒂(𝑡) and 𝒓(𝑡 − 𝛿𝑡).
3. If desired, 𝐯(t) can be determined as in Eq.18
Dynamic systems are reliant on choosing a fitting enough time step, which can be described as a
sequence of frames, similarly to when filming movies during the era of Buster Keaton. If a time step
is chosen and appears too small, the trajectory will only explore a narrow part of phase space. A too
large time step will cause the sought after event to be missed, as the simulation will produce a too
great of a separation, due to a cause of overestimated energy between particles.
2.2.3 Classical mechanics - Force fields
When studying biological systems one usually works with an amount of particles which cannot be
treated with methods from quantum chemistry, or even the most fitting of MD algorithms. As has

23
been discussed in the section of Statistical mechanics, MD simulations make use of the property A
and ultimately ensemble averages. Molecular mechanics are used in order to make calculations like
this feasible, where force fields are employed to describe the general interactions of atoms. A
potential energy surface (PES) is used to describe a molecule’s energy as a function of geometry and
a PES is helpful when parametrizing a force field.
The common force field is comprised of the following terms
𝐸𝑝𝑜𝑡 =𝐸𝑠𝑡𝑟𝑒𝑡𝑐ℎ + 𝐸𝑏𝑒𝑛𝑑 + 𝐸𝑡𝑜𝑟𝑠𝑖𝑜𝑛 + 𝐸𝑛𝑜𝑛−𝑏𝑜𝑛𝑑 Eq.19
Where the potential energy depends on different energy terms describing the stretching of bonds,
angle bending and torsion in bonds, respectively. The final term is a collection of non-bonded
interactions
𝐸𝑛𝑜𝑛−𝑏𝑜𝑛𝑑 = 𝐸𝑒𝑙𝑒𝑐𝑡𝑟𝑜 + 𝐸𝑣𝑑𝑊 + 𝐸𝐻−𝑏𝑜𝑛𝑑 Eq.20
The contributions to the non-bonded energy are obtained from the electrostatic, van der Waal’s and
hydrogen bond interactions, respectively.
The stretching and bending terms are usually expressed as variations of Hooke’s law
𝐸𝑠𝑡𝑟𝑒𝑡𝑐ℎ =𝑘
2(𝑙 − 𝑙0)
2 Eq.21
𝐸𝑏𝑒𝑛𝑑 =𝑘
2(𝜃 − 𝜃0)
2 Eq.22
Where it can be seen that k is a force constant, describing the compression of a bond and the angle
distortion. As it requires more energy to compress a bond than to distort an angle, the force
constants for bond stretching are considerably larger.
The bond and angle distortions can be described more accurately by applying a higher order
polynomial.
A similar expansion can be applied when defining the torsional term, stated below
𝐸𝑡𝑜𝑟𝑠𝑖𝑜𝑛 = ∑𝑉𝑛2
𝑁
𝑛=0
[1 + cos(𝑛𝜔 − 𝛾) Eq.23
And an expansion for the MM2 force field is defined accordingly
𝐸𝑡𝑜𝑟𝑠𝑖𝑜𝑛 =𝑉12(1 + cos(𝜔)) −
𝑉22(1 + cos(2𝜔)) +
𝑉32(1 + cos(3𝜔)) Eq.24
Each of the terms (in order) corresponds to bond dipole interactions, conjugation and
hyperconjugation and steric interactions. Another necessary component is also necessary to consider

24
regarding angular terms, namely the part treating the out-of-plane bending along with improper
torsion. Three of the common methods employed are the angle-to-plane
𝜐(𝜃) =𝑘
2𝜃2 Eq.25
The distance-to-plane
𝜐(ℎ) =𝑘
2ℎ2 Eq.26
And the third component considers the improper torsion
𝜐(𝜔) = 𝑘(1 − cos(2𝜔)) Eq.27
It is time to consider the expressions used for the non-bonding part of common force fields.
Electrostatic interactions in MD simulations cannot be accounted for by molecular orbital (MO)
calculations for the simple reason that the system is too big. The calculations would be far too
expensive, which leaves for a different approach to estimate the charges. It is common to assign
Coulombic interactions to describe the electrostatics with a dielectric constant that averages the
polarization effects
𝐸𝑒𝑙𝑒𝑐𝑡𝑟𝑜 =∑∑𝑞𝑖𝑞𝑗
4𝜋휀0𝑟𝑖𝑗
𝑁𝐵
𝑗=1
𝑁𝐴
𝑖=1
Eq.28
Where NA and NB are the number of point charges for two molecules,휀0 is the dielectric constant and
𝑟𝑖𝑗 is the distance between to charges.
The last two non-bonding terms employ different potentials. The van der Waal interactions use a
6/12 potential, more commonly known as the Lennard-Jones potential
𝐸𝑣𝑑𝑊 = 4휀𝑖𝑗 [(𝜎𝑖𝑗
𝑟𝑖𝑗)
12
− (𝜎𝑖𝑗
𝑟𝑖𝑗)
6
] Eq.29
The potential for hydrogen bonds is not always included but holds a similar appearance to the L-J
potential. However, it is necessary to know the identities of the hydrogen bonds before the
calculation is performed.
All parameters has now been the subject of a brief overview and the MD section closes with the full
expression for a basic MM force field
𝐸𝑝𝑜𝑡(𝒓𝑁) = ∑
𝑘𝑖2(𝑙𝑖 − 𝑙0,𝑖)
2
𝑏𝑜𝑛𝑑𝑠
+ ∑𝑘𝑖2(𝜃𝑖 − 𝜃0,𝑖)
2
𝑎𝑛𝑔𝑙𝑒𝑠
+ ∑𝑉𝑛2
𝑡𝑜𝑟𝑠𝑖𝑜𝑛𝑠
(1 + cos(𝑛𝜔 − 𝛾))
+∑ ∑ (4휀𝑖𝑗 [(𝜎𝑖𝑗
𝑟𝑖𝑗)
12
− (𝜎𝑖𝑗
𝑟𝑖𝑗)
6
] +𝑞𝑖𝑞𝑗
4𝜋휀0𝑟𝑖𝑗)
𝑁
𝑗=𝑖+1
𝑁
𝑖=1
Eq.30

25
2.3 Quantum Chemistry Quantum mechanics (QM) is agreed to be one of the most profoundly shocking theories to ever have
been established and despite being a particularly difficult subject, it has evolved rapidly during the
past hundred years while revolutionizing both scientific conduct and society. A long time has passed
since the early 1900s and QM has evolved into a pragmatic tool, not least within chemistry.
Computational chemists rely on Quantum chemical (QC) principles to investigate molecular systems,
reaction mechanisms, activation barriers etc. However, the equations describing these systems are
quite complex and striving for exact solutions with modern QC methods are simply not possible. The
sophisticated nature of molecular systems is notably obvious when evaluated by a QC approach and
one quickly finds that as the number of particles increases, so does the computational time and
power as well, sometimes drastically. Hence computational chemists are relying on good
approximations that can be made to simplify necessary calculations while maintaining satisfactory
enough solutions. Coincidentally, enhanced computational power share an equal amount of benefits
as good approximations, if not more.
This section aims to give a brief review of the fundamental aspects of QC and shortly mention some
of the methods, while finishing with an overview of the method used in this thesis, Density
Functional Theory (DFT).
All theory regarding the QC principles are taken from the text book Modern Quantum Chemistry:
Introduction to Advanced Electronic Structure Theory, by Szabo and Ostlund [26].
2.3.1 The Schrödinger equation
It is not an overstatement in that Edwin Schrödinger, by publishing his work on the widely known
partial differential equation bearing his own name, revolutionized physics [27].
Ĥ𝛹 = 𝐸𝛹 Eq.31 Where Ĥ is the Hamiltonian, Ψ is the wave function of the system and E is the energy obtained as an
eigenvalue to the Hamiltonian operator. The Hamiltonian operator is therefore a descriptor of the
systems total energy and is comprised of the quantum mechanical operators for kinetic and potential
energy. This includes terms that treats kinetic and potential energy for both nuclei and electrons and
as the particles number increases, so does the computational demands for that system.
The Hamiltonian is defined for N electrons and M nuclei as
Ĥ = −∑1
2∇𝑖2 −∑
1
2𝑀𝐴∇𝐴2 −∑∑
𝑍𝐴𝑟𝑖𝐴
+∑∑1
𝑟𝑖𝑗+∑ ∑
𝑍𝐴𝑍𝐵𝑅𝐴𝐵
𝑀
𝐵>𝐴
𝑀
𝐴=1
𝑁
𝑗>𝑖
𝑁
𝑖=1
𝑀
𝐴=1
𝑁
𝑖=1
𝑀
𝐴=1
𝑁
𝑖=1
Eq.32
Where MA is the ratio of the masses of nucleus A to an electron and ZA is nucleus A: s atomic number.
The operators ∇𝑖2 and ∇𝐴
2 are of Laplacian nature and handles differentiation regarding the
coordinates of the ith electron and the Ath nucleus. The terms in Eq. 32 corresponds to the kinetic
energy for electrons, kinetic energy for nuclei, coulombic interactions between the electrons and
nuclei, electron repulsion and nuclei repulsion respectively.
As a solution to the Schrödinger equation is sought after, the Hamiltonian should bring about some
cause for concern for a computational chemist, as the operator contains several terms to be
evaluated. Given that the system also consists of a lot of particles this calls for extensive

26
computational demand. It is still possible to withhold exact solutions of the wave function, assuming
the system is off an extremely simple nature, although most investigations will not be conducted on
simple systems. How can the computational demands be met for complicated systems while still
producing accurate enough results which reflect reality?
2.3.2 The Born-Oppenheimer approximation
The applied solution to this problem is known as the Born-Oppenheimer approximation and it makes
use of the fact that atomic nuclei are heavier by a considerable amount when compared to the
electron. As the nuclei have greater mass than electrons it is naturally assumed that electrons
outmaneuver nuclei fairly easily, in regards of speed. A fitting approximation is therefore that the
nuclei of a molecule can be considered as fixed and that the electrons move in a field relative to the
fixed nuclei. With all taken in consideration, the Hamiltonian can now be written as
Ĥ𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑛 = −∑1
2∇𝑖2 −
𝑁
𝑖=1
∑∑𝑍𝐴𝑟𝑖𝐴
+∑∑1
𝑟𝑖𝑗
𝑁
𝑗>𝑖
𝑁
𝑖=1
𝑀
𝐴=1
𝑁
𝑖=1
Eq.33
Since the nuclei are regarded as fixed the term associated with the nuclei kinetic energy can be
neglected. The nuclei repulsion can be treated as constant and is added to the operator eigenvalue.
The resulting Hamiltonian is known as the electronic Hamiltonian and this indicates that the
Schrödinger equation could be solved simply by regarding the motions of N electrons in the field of
M point charges. This abbreviates the Schrödinger equation into the following format
Ĥ𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑛𝛹𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑛 = 𝐸𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑛𝛹𝑒𝑙𝑒𝑐𝑡𝑟𝑜𝑛 Eq.34
The assumption resulting in the electronic Schrödinger constitutes a major simplification compared
to the many-particle Schrödinger equation. However, although the electronic Schrödinger results in a
drastic decrease of computational effort, the fact remains that producing a solution still requires
solving a problem consisting of multiple components. Still, this approximation is more than enough
and is a vital part in the field of quantum chemistry.
2.3.3 Methods for solving the electronic Schrödinger equation
One of the most widely recognized approaches in attempting to solve the electronic Schrödinger was
presented in 1930 by Hartree and Fock, and is attributed as the Hartree-Fock approximation.
According to the HF approximation a wave function can be expressed as a Slater determinant. The
relevant background will be quickly summarized below.
In the beginning of the era of quantum mechanics the concept of electron spin was established,
where functions describing if an electron possessed the property of spin up, alternatively spin down,
was presented. By incorporating a so called spin coordinate, electrons could be defined by 3 spatial
coordinates alongside the spin property, allowing for the possibility of two spin orbitals. Each spin
orbital follows the Pauli principle, meaning that each spin orbital can house a maximum of one
electron. The electrons should also be indistinguishable in which orbital houses them. Because of this
the interchange of electrons can follow a symmetrical or anti-symmetrical approach. If the criterion
of indistinguishable electrons is met, a wave function can be formulated, consisting of a linear
combination of two wave functions. The two respective wave functions contain information about
the electron spin, and these wave functions can be expressed as the Slater determinant, where the

27
spin orbitals and electrons are presented in the columns and rows, respectively, in a matrix. After
forming the Slater determinant, the variational principle is invoked stating that the energy solved for
the system will be either exactly that of the actual system, or larger according to
𝐸𝑒𝑥𝑎𝑐𝑡 ≤ 𝐸𝑣𝑎𝑟𝑖𝑎𝑡𝑖𝑜𝑛𝑎𝑙 =⟨𝛹∗|Ĥ|𝛹⟩
⟨𝛹∗|𝛹⟩ Eq.35
The energy is consecutively minimized with respect to the spin orbitals. The HF approximation aims
to simplify calculations by generating solutions for each individual electron. Each electron is
optimized in consideration to all other electrons, which produces a mean field.
However, the HF method fails to take into consideration that two electrons are unable to exist in the
same place at once at the same time, which means that the electron repulsion will not be taken into
consideration. The fault lies in that the stated mean field does not compensate for this correlation
energy. This will always lead to the computed energy being higher than its actual value during a HF
calculation. Because of these existing problematics a set of methods has been developed, known as
post Hartree-Fock methods that aim to compensate for faulty correlation energy. The post HF
methods will not be considered in this text as they were not used in this thesis work.
As the HF method and many others, such as Configuration Interaction (Full-CI), have earned their
rightful mark in history as pragmatic approaches to deliver a satisfactory description of molecular
systems, a major disadvantage is the fact that they are computationally expensive. Despite these
problematic traits there is another methodology that makes use of a systems electron density rather
than its wave functions. The method goes by the name of DFT and will be treated in the upcoming
section.
2.3.4 Density Functional Theory
The theory in this section was obtained from Molecular Modelling – Principles and Applications by
Andrew Leach [25] and review articles written by Burke [28] and Becke [29]
The foundation for DFT was laid out in 1964 with the publication of the Hohenberg-Kohn theorem
[30], stating that a system’s energy can be stated as a function of the atomic electron density
𝐸 = 𝐸(𝜌(𝒓)) Eq.36
And the total electronic energy can be expressed by a functional accordingly
𝐸(𝜌) = 𝐸𝐾𝐸(𝜌) + 𝐸𝐶(𝜌) + 𝐸𝐻(𝜌) + 𝐸𝑋𝐶(𝜌) Eq.37
The terms represent the kinetic energy, the electron-nuclei interaction, the electron-electron
coulombic interaction and the contributions from exchange correlation, respectively. Due to the
inclusion of the electron-electron repulsion in the above expression, the electrons are considered to
interact with a mean field of the electron density, with seeming similarity as in HF theory.
There is an important consequence arising from the Hohenberg-Kohn theorem in that all ground
state properties for a particular system can be determined exactly by the electron density, where an
incorrectly described density in Eq.37 will produce an energy higher than the systems true energy.

28
Should the density be exactly described, one would also know external potential (namely, the
electron-nuclei interaction) and therefore the unique wave function for that system, indicating that
in theory everything is known. But this knowledge is not enough and a DFT variational principle is
required to, as accurately as possible, describe the system. For DFT, this means that the lowest
energy determined will conform to the systems exact density.
In the most common formulation of DFT, presented by Kohn and Sham, the approach taken is that a
single Slater determinant, comprised of orthonormal and real molecular orbitals, will represent the
density. This will result in the so called ‘Kohn-Sham orbitals’, which aim to optimize the systems
energy by solving a set of one-electron equations. However, since DFT takes the mean field approach
it is also necessary to include the electronic correlation. With respect to the aforementioned criteria
the Kohn-Sham equations are presented
[−∇2
2+ 𝑉𝑛𝑢𝑐𝑙𝑒𝑎𝑟(𝒓) + ∫𝑑𝑣´
𝜌(𝒓)
|𝒓 − 𝒓´|+ 𝑉𝑋𝐶(𝒓)]𝛹𝑖(𝒓) = 휀𝑖𝛹𝑖(𝒓) Eq.38
Where the exchange-correlation functional, 𝑉𝑋𝐶(𝒓), can be obtained from analytical expressions for
the local density approximation (LDA), which assumes that there exists a uniform electron gas model
that claims the electron density is constant in all space. Given the assumption that the charge density
through a molecule varies slowly (i.e, behaving as a uniform electron gas), if the exchange-correlation
energy per particle in the uniform gas is given by 휀𝑋𝐶 , then the total exchange-correlation energy,
𝐸𝑋𝐶 , as a consequence of being integrated over all space is given by
𝐸𝑋𝐶[𝜌(𝒓)] ≅ ∫𝜌(𝒓)휀𝑋𝐶 [𝜌(𝒓)]𝑑𝒓 Eq.39
The functional describing the exchange-correlation can then be written as
𝑉𝑋𝐶(𝒓) =𝛿𝐸𝑋𝐶[𝜌(𝒓)]
𝛿𝜌(𝒓) Eq.40
As the LDA incorporates the mean field approximation the exchange correlation energy is penalized
upon evaluation, approaches have been taken to compensate for this. An early example of attempts
to minimize the error was the incorporation of generalized gradient approximations (GGA: s), which
will not be examined further here [31]. The GGA: s showed improved accuracy upon application in
calculations of chemical nature and in the 1990s, the German native Axel D. Becke introduced hybrid
functionals [32], where perhaps the most famous functional used in modern DFT is the B3LYP
functional, where a mixture of GGA and HF exchange was introduced.
The peculiar thing about hybrid functionals is that they all have to be selected with care for what
particular system one intends to investigate. In this thesis, the B3LYP functional was not incorporated
as it has been noted previously that this functional do not perform adequately in describing the D-A
reaction [7]. Instead, the M06-2X functional by Truhlar [33] has been the preferred method of
execution, as inspired by the findings of Brinck et al [7].

29
3. Methodology
3.1 Computational details
3.1.1 Protein preparation
The enzyme crystal structure for Pseudomonas testosterone Kestosteroid Isomerase was retrieved
from the Protein Data Bank with the PDB entry 1QJG. The PDB structure was minimized using the
AMBER14 set of programs with the AMBER FF14SB force field and the side chain Asp38 was
protonated using the mutagenesis tool in PYMOL. To conduct the docking experiment, a pdbqt-file
for the rigid part and the flexible parts of the enzyme was prepared with AutoDock Tools. The
catalytic triad consisting of Tyr14, Asp38 and Asp99 were selected as flexible residues and separated
into an enzyme_flex.pdbqt file. The remaining side chains were incorporated in an
enzyme_rigid.pdbqt file. ADT was then used to set the grid box for the docking simulation, where the
box dimensions used initially was x = 40, y = 40, z = 40, and the grid box was defined around the
active site, making sure to include all important side chains. These settings were saved in a .gpf file.
3.1.2 Ligand preparation
Starting coordinates for the TS structures of acrolein and cyclopentenone were obtained from Brinck
et al (artikel IX – envisioning diels alderase). Utilizing this scaffold, substituents were added to the
different sites presented in Figure… with Gaussview. The coordinates for the TS structures was locked
in place and the substituents was geometrically optimized, along with a computation of the atomic
charges with DFT, using the M06-2X functional at the 6-31+G(d) level. The ligands was then
converted from .pdb to .pdbqt format using ADT, with the DFT-computed charges added to the
.pdbqt file. The raccoon program was used to prepare multiple ligands for docking simultaneously
(raccoon reference). Using the Raccoon GUI, all ligands were added together with the rigid and
flexible part of the enzyme. The .gpf file containing the information on the grid box was incorporated
as well, and the .dpf file was generated in Raccoon. Following this step, AutoGrid was run in order to
generate grid maps. The LGA was used to create up to 100 conformations at a time, with 2 500 000
energy evaluations and 40 000 as a maximum number of generations.
3.1.3 Molecular dynamics preparation
The MD simulations were performed with the AMBER14 [34] set of programs, using the Amber force
field: FF14SB [35]. The general Amber force field (GAFF) [36] was employed to obtain force field
parameters for the ligands by using Antechamber [37] and Parmchk [38] in AmberTools. The partial
charges for the ligands were computed using RESP charges [39]. The enzyme was charge neutralized
using 2 Cl- ions and solvated with TIP3P water [40] (8.0 Å solvent shell) using XLEaP. The systems
were minimized two times using Sander, where the first minimization ran for 1000 iterations while
holding the protein fixed and the second minimization ran for 2500 iterations with no constriction to
the protein. For the minimization a steepest descent algorithm was performed for the first half of
both simulations and the other half of the minimization employed a conjugate gradient method.
Following the minimization the systems were heated to 300 K for duration of 20 ps, while putting
mild restraints on the protein. Unconstrained production was then performed, using pmemd.CUDA
[41], for 4 ns with a temperature of 300 K, pressure at 1 bar and a 2 fs time step. For hydrogen
atoms, the SHAKE algorithm was used. Lastly, trajectory analysis was performed with the program
CPPTRAJ in AmberTools.

30
4. Results and Discussion
4.1 Molecular Docking with AutoDock 4.2
Figure 13. An example of a docked structure, presented more thorough later in the text.
Earlier work, performed by Brinck et al, determined that KSI had the potential to house a D-A
reaction as it contained relevant catalytic components to generate an in-situ diene by proton
abstraction [7]. Although the catalytic mechanism was investigated, an evaluation of beneficial
substrate and enzyme design was not covered by the investigation at the time. A presentation of the
investigation conducted during this thesis will be presented below, dealing with the different aspects
of advantageous design, both in regards to the evaluated substrates and the attempts at a better
suited active site.
4.1.1 Initial findings
Figure 14. An extracted sample of some residues making up the active site of KSI.

31
Initial screenings using (1) (Figure 7 and 9) as diene and dienophile did not provide satisfactory poses
and demonstrated weak binding affinities in the wildtype KSI (hits with binding affinities higher than
-5.0 kcal/mol were discarded). As the substrates based on this scaffold would not yield a sought after
result the decision was made to solely focus on the 2-cyclopenten-1-one/Acrolein TS scaffold.
However, initial dockings with the monosubstituted (1) did indicate that a pyridine-based substituent
in the b2 position of the dienophile (Figure 7) presented some poses where the interactions between
substrate and the catalytic triad were upheld, although the planar positioning required in respect to
Asp38 was absent. It could also be determined that the binding-affinity slightly increased for all
attempted disubstituted substrates where pyridine was utilized as a substituent on the dienophile,
while retaining some interactions necessary for catalysis, yet still not to a satisfactory extent. Due to
these findings alone it was decided to further the investigation with pyridine as a plausible
substituent fitting for the dienophile, while shifting focus to the (2) compound, which was considered
as the dienophile throughout the rest of this work.
After obtaining a plausible dienophile the screening continued with attempted substitutions at the
b1 position on the diene. The reasoning was that a substituent placed in that position might obtain
some hydrogen-bonded interaction with Asp38 following deprotonation of the pro-diene and
therefore also retain the desired conformation such as presented in figure 13. The screening still did
not provide satisfactory results in reproducing the desired pose necessary for the deprotonation of
pro-diene without including several outliers in the docking clusters. Granted the failure in obtaining
desired binding poses at a high frequency, when the correct pose appeared in association with the
necessary interactions, they presented a fairly high binding affinity towards the active site, reaching
values around -6 kcal/mol.
Seeking to obtain a higher frequency of satisfactory poses and a good enough binding affinity,
rational mutagenesis of the active site was employed, resulting in a set of mutant versions of KSI. An
initial attempt explored the possibility of exchanging Phe80 and Phe82 with an alanine residue to
observe the effect on binding affinity. The trend throughout all screened compounds showed that
the exchange of Phe80and Phe82 with a smaller residue lowered the binding affinity significantly. After
ascertaining the effect of introducing a smaller residue to the deeper part of the active site, both
residues were mutated accordingly; F80N, F80T, F82N and F82T. While the binding energy showed
some increase it had little effect on the positioning of the docked structures, which was the desired
effect of said mutation. A similar exploration was conducted with Phe54 into an alanine, also resulting
in a lowering of the binding energy. Mutations of Phe54 into larger residues also provided similar
results with a decrease of the estimated binding energy. However, this was determined to depend on
different reasons altogether. While the F54A mutation decreased the binding affinity likely due to a
lack of steric interactions, the attempted F54N and F54T mutations resulted in that disubstituted
substrates adapted the wrong conformations caused by lack of compartmental space. For some
substitutions the substrate could not enter the active site completely as the opening of the entrance
became smaller, making the active site less accessible. Another overview of the residues in the
closest vicinity to the active site made for a discovery of Val65. With earlier attempts to obtain a
fitting conformation of the docked TS structures, mutation of Val65 provided interesting results as
both the binding affinity and desired conformation of substrates increased in frequency. The initial
assumption made was that since Val65 is located at the “bottom” of the active site, a mutation into a
larger residue would serve to decrease the size of the active size, forcing the docked structures to
adopt a more planar conformation. As mentioned earlier, this is a desired effect in order to make the

32
β-proton of the pro-diene more accessible to the acting base, Asp38 in terms of reach. The V65F,
V65N and V65Q mutations were incorporated and they all showed promising results, both in regards
to binding and conformation.
To further deduce eventual possibilities of enhancing the selection of desired conformations the
active site was examined with respect to the substituent in the dienes a1 position. Based on the
earlier work by Brinck et al it was clear that said substituent should point towards the entrance to the
active site and therefore, by exclusion, a fitting area to scout for plausible mutations would be the
entrance. An option presented itself in the form of Ser58 located to the “upper right” relative to Val65.
The obvious choice was to introduce a longer polar side chain with a large conformational freedom
compared to Ser58, subsequently being able to “steer” the substrate into a desired conformation. As
such, S58E, S58N and S58Q were the attempted mutations, although S58E was quickly discarded due
to the fact that it might pose a problem with the systems pH and the pKa of glutamic acid that could
possibly result in undesired deprotonation by the wrong residue. It was also discovered that
asparagine caused an increased sampling of conformations with the most probable cause being the
lack of one bond as compared to glutamine, redirecting the C3-O of the diene towards Gln58 instead
of the crucial interaction with Asp99 and Tyr14. However, as hypothesized, the Gln residue did in fact
benefit the ordering the compound into desired conformation.
As mentioned in section 1.5, the threshold for incorporated mutations was set to 3 in order to not
alter the active site to a wider extent which consequently might affect the backbone structure of KSI
overall. This in turn might have effect on KSI: s catalytic properties, inducing unsought lowering of
catalytic proficiency. The examination of the active site was thus directed towards the “bottom” of
the active site where Val84 was located. Once again the idea was to minimize the size of the active
site to favor affinity and conformation of the substrates, while simultaneously considering the size of
the final product. Attempted mutations were V84N and V84Q, both satisfying binding affinity and
conformational conditions.
In the end, four sets of mutations were produced, all conforming to the set mutation threshold:
V84N-S58Q-V65N, V84N-S58Q-V65Q, V84Q-S58Q-V65N, and V84Q-S58Q-V65Q. However, V84N-
S58Q-V65N and V84Q-S58Q-V65Q had to be excluded from further examination as they produced
variable results. The V84N-S58Q-V65N set did not provide sufficient enough binding energies while
the V84Q-S58Q-V65Q set caused a too sterically hindered active site. Due to time restrictions the
remaining investigation was limited to the V84N-S58Q-V65Q set, since this set provided the most
beneficial results overall.

33
Figure 15. The main residues under investigation when designing the active site. It can be seen that the Val65
and Val84
are located “below” the catalytic triad. Both residues proved great in order to structure the TS-structure and obtain the correct binding interactions. Asp
38 and Asp
99 are denoted as Ash
38 and Ash
99 due to a necessary renaming after
performing an energy minimization of the enzyme structure.
Figure 16. A view of the active site from a different angle. To the left and back in the figure, near the position of Ser58
, the entrance to the active site is located. Above Ser
58, Phe
54 is located, which demonstrated an importance for the access of
the substrates. The mutation F54Y proved to distort the conformation of the TS-structures that were docked and a mutation into smaller residues revealed a significant decrease in the binding energy.

34
4.1.2 Obtaining starting coordinates for MD simulation
A favorable docking result tends to produce several similar conformations with a root mean square
deviation (RMSD) tolerance of2.0 Å (the tolerance is set at 2.0 Å in this work, but can be altered out
of necessity) from the highest ranked conformation [18][21][42]. All these similar conformations are
collected in a cluster and the fewer amounts of clusters produced the better the result. If an
AutoDock simulation is run and is specified to generate 50 conformational poses and all are collected
within one cluster, this is an indication of an accurate prediction of the most probable interactions
between ligand and macromolecule. However, upon the introduction of flexible bonds in the ligand
as well as in the macromolecule (flexible side chains in the active site, more specifically) these
clusters might increase in numbers due to the increased possibility to explore the conformational
space. While the majority of the macromolecule is treated as a rigid structure this will of course not
lead to an accurate sampling of the possible conformations, providing an accuracy of approximately
70 % in the prediction of binding modes. As it stands, this raise a cause for concern as the clusters
may contain samplings of incorrectly presented binding interactions, as well as false positives [42].
The docking results presented here contain several clusters, although the expected accuracy error is
taken into consideration upon evaluation and will be discussed further alongside the presented
docked conformations.
The first docked TS-structure that gave a satisfactory verdict is presented below in figure 17.
Figure 17. The TS-structure and the schematic structure of the diene and dienophile, making up MTE1 are presented.
This compound, hereon denoted as MTE1, was docked against the mutated KSI (V84N-S58Q-V65Q)
with the GA run specified to generate 50 conformations, where the best generated pose presented
an estimated binding energy of -7.21 kcal/mol and a mean binding energy of -6.06 kcal/mol for
cluster rank 1. The rank 1 cluster presented 28 conformations within the specified RMSD tolerance of
2.0 Å and the other 22 conformations were presented in different clusters at a lower cluster rank.
Samples of the 6 best conformations are presented below in figure 18.

35
Figure 18. A cluster of successful binding poses for the MTE1 complex.
As can be distinguished from figure … the docked structure upholds the sought after binding
interactions where the C3-O of the diene interacts with Asp99 and Tyr14. Asp38 present to the left and
directly above the diene allowing for advantageous positioning of the re-protonation that would
follow the expected D-A reaction. The dienophile is located at the bottom of the active site where
the main interactions are likely ascribed to hydrophobic interactions. To the right is Gln58 that
appears to form some hydrogen interactions with the oxygen of the dienes hydroxyl group, serving
its purpose of “steering” the diene into its expected conformation. Below is a figure representing the
molecular surface of KSI, which indicates that the shape complementarity of the TS-structure is
consistent with the presented mutation of the enzyme. With the obtained conformations presenting
rather few outliers within the same cluster, the result is considered as a positive indication that the
observed binding modes are the dominant ones. Additional changes to the substrate design were
performed in hope of increasing eventual binding affinity, for which the results will be presented for
MTE2, MTE3 and MTE4 following the initial presentation of the MTE1 results.
Table 1. The assorted binding energies for the poses shown in figure 18. All binding energies and clusters can be viewed in Appendix 2.
Rank Sub-rank Run Binding Energy
Cluster RMSD Reference RMSD
1 1 42 -7.21 0.00 85.19 1 2 14 -6.91 0.70 85.59 1 3 47 -6.90 0.40 85.30 1 4 40 -6.67 0.26 85.30 1 5 44 -6.65 0.53 85.32 1 6 29 -6.54 0.23 85.27

36
Figure 19. All docking poses compared to the molecular surface of the enzyme, shown in white. It can be seen that the docking poses follows the shape of the active site quite well.
In the second investigated TS-structure, denoted MTE2, a methyl group was attached to the carbonyl
group of the dienophile (figure 20), intended to increase the hydrophobic interactions with the active
site, causing an increase in binding affinity.
Figure 20. The MTE2 complex. An additional methyl group is included on the b1 position of the dienophile.
For the MTE2 structure a total of 100 GA runs were performed in an attempt to obtain a better
overview of the conformations within each cluster. For this particular structure, the results were a bit
ambiguous seeing as out of 7 obtained clusters, the rank 1 cluster contained 73 of the conformations,

37
but the overall binding energy was lower. The best ranked conformation presented an estimated
binding energy of -6.45 kcal/mol and the mean binding energy was estimated to be -4.62 kcal/mol,
an increase of 1.44 kcal/mol as compared to MTE1. Below is a presentation of the top 4
conformations with correct binding interactions, followed by a depiction of 2 highly ranked outliers.
Figure 21. Some of the successful docking poses obtained from the docking of the MTE2 complex.
Similarly to the MTE1 the C3-O of the diene can be seen interacting with Asp99 and Tyr14, while the
dienophile is located directly under the diene, partially demonstrating hydrogen interactions with
Gln58. A cause for concern is the fact that Asp38 can be seen interacting with the hydrogen on the
dienes hydroxyl group. Due to earlier difficulties in obtaining desired conformations, the hydroxyl
group was introduced simply to allow for eventual interaction with the polar Gln58 residue. However,
as the Asp38 residue acts as a base on the pro-diene, a striking thought is that the deprotonation
might occur on the hydroxyl proton instead. The hydroxyl group was not initially planned to be used
as a substituent, as alternatives such as amines, imines, alkanolamines, etc, were attempted, with no
sufficient result reported. It was later decided that the hydroxyl group would remain as the
substituent of choice and used in a primary pursuit of investigation.
Table 2. The binding energy and ranking for the poses of MTE2. The unsought poses are presented as outliers.
Rank Sub-rank Run Binding energy
Cluster RMSD
Reference RMSD
1 1 51 -6.45 0.00 85.25 1 2 60 -6.36 0.68 85.53 1 3 67 -5.96 1.70 84.54 OUTLIER 1 4 95 -5.79 1.68 84.48 OUTLIER 1 5 52 -5.76 0.40 85.35 1 6 40 -5.64 0.18 85.27
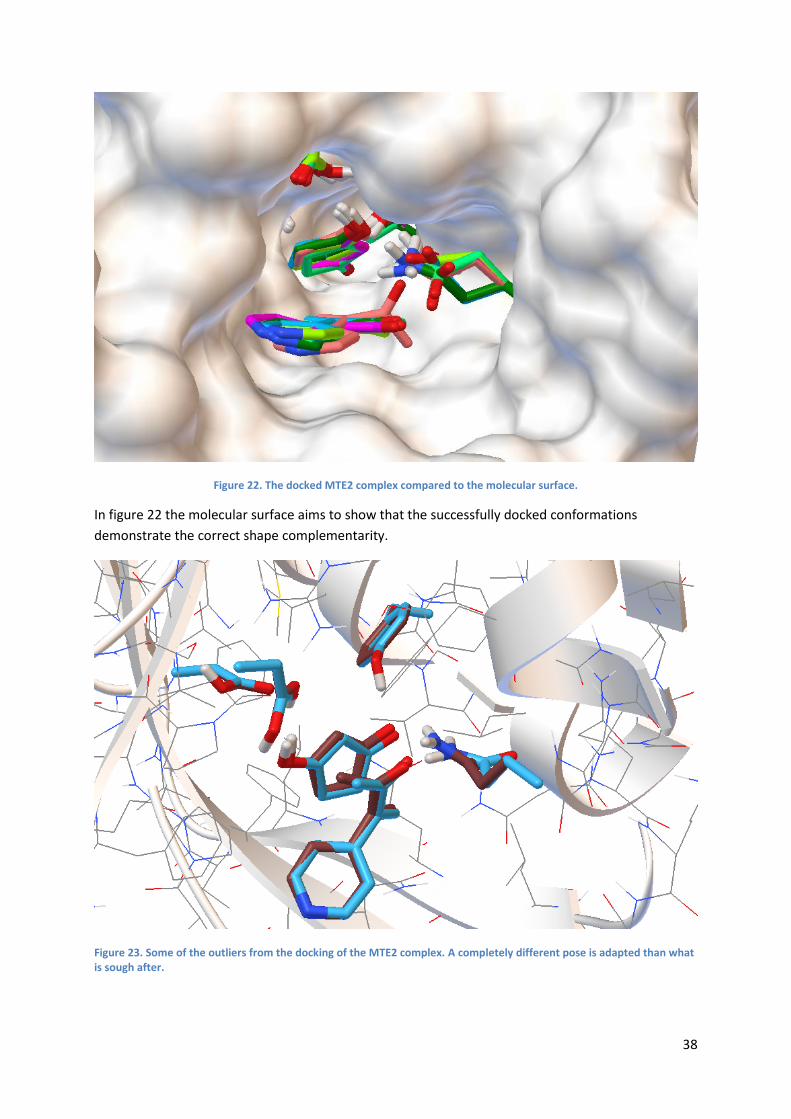
38
Figure 22. The docked MTE2 complex compared to the molecular surface.
In figure 22 the molecular surface aims to show that the successfully docked conformations
demonstrate the correct shape complementarity.
Figure 23. Some of the outliers from the docking of the MTE2 complex. A completely different pose is adapted than what is sough after.

39
Figure 23 and 24 presents some of the outliers obtained within the rank 1 cluster where the expected
interactions are not present. Likely this is due to the added methyl group on the carbonyl of the
diene causing the structure to explore other conformations, allowing more room for the compound.
The figure depicting the molecular surface illustrates that the rational design possibly could have
rendered the active site too small for a structure with an added methyl group.
Figure 24. The outliers of MTE2 complared to the molecular surface.
The next structure explored the option of adding a methyl group on the pyridine substituent, next to
the nitrogen. Remaining true to the earlier naming conventions, this structure is denoted as MTE3.
Figure 25. The structure of the MTE3 complex is presented. A methyl group has been added to the pyridine group.

40
For the MTE3 TS-structure, 100 GA runs were performed, (as for MTE2) generating an overall
increase in binding energy but fewer numbers of conformations present in the rank 1 cluster. The
best conformation demonstrated an estimated binding energy of -6.74 kcal/mol and the mean
binding energy was -4.64 kcal/mol (a poor -0.02 kcal/mol lower compared to MTE2). Out of 7
reported clusters, the rank 1 cluster contained 61 conformations, meaning that fewer conformations
were obtained. The figures below show the 8 best ranked conformations, energy-wise. However, 4
out of these top ranked conformations were deemed to be outliers.
Figure 26. The successfully obtained docking poses for MTE3.
Figure 26 demonstrates the obtained conformations that fulfilled the interactions criteria, but with a
slightly increased variation in positioning even for seemingly equal conformations and figure 27 show
the molecular surface. An estimated guess is that the methyl group added to the pyridine substituent
serves to increase the hydrophobic interactions, while the methyl group on the carbonyl group can
cause certain distortions as a result of steric clashes, which might explain the conformational
variation observed for MTE2.
Table 3. The binding energies and subsequent rankings of poses. Outliers are presented as well.
Rank Sub-rank Run Binding energy
Cluster RMSD
Reference RMSD
1 1 92 -6.74 0.00 85.21 1 2 32 -6.63 1.66 84.66 OUTLIER 1 3 13 -6.39 1.69 84.60 OUTLIER 1 4 18 -6.05 1.03 85.02 OUTLIER 1 5 5 -6.05 1.82 84.77 1 6 60 -6.04 0.74 84.89 1 7 33 -6.01 1.12 85.50 1 8 29 -5.74 1.69 84.30 OUTLIER

41
Figure 27. The successful dockings of MTE3 compared to the molecular surface.
In figure 28 and 29 below, the outliers for MTE3 are presented. Due to restricted access of space in
the active site the TS-structure attempts to explore different conformations by twisting itself into
more fitting poses. This early on led to the belief that further addition of substituents to the complex
would result in it being too large to even enter the site, or at least, not providing the sought after
binding interactions. The assumption would prove, as results will show, to be fairly correct.
Figure 28. Outliers obtained from the MTE3 docking.

42
Figure 29. The outliers for MTE3 compared to the molecular surface.
The last investigated TS-structure is presented below, where the main purpose was to determine if
the substrate design was pushing the limits in regards to steric clashes with the residues in the active
site. The complex is denoted as MTE4.
Figure 30. The representation for the MTE4 complex.
Like MTE2 and MTE3, MTE4 was subjected to 100 GA runs, generating a considerably lower binding
energy overall, but a variety of clusters with the rank 1 and rank 2 clusters, containing 35
conformations each. Some samples are presented in figure 31 to demonstrate the inability of the
designed complex to achieve the desired binding interactions. As no correct binding interactions
were detected out of all 100 generated conformations, there is no need for a presentation of the
binding energies. In figure 32 which presents the sampled conformation along with the molecular
surface, the conformation with bold bonds is shown as an interesting comparison in relation to the

43
others. A new conformation not previously detected is presented, where the pyridine group is
twisted to fit the complex into the active site. A quick conclusion can be drawn from simple visual
inspection that the MTE4 complex is too large for the active site, resulting in the absence of the
necessary interactions that leads to catalysis.
Figure 31. The MTE4 complex showed no favorable interactions whatsoever and was therefore removed from further consideration and evaluation as a plausible substrate design.
Figure 32. The MTE4 complex compared to the molecular surface. The bold representations demonstrate that the complex explored new conformations, yet to have been seen before.

44
The MTE1, MTE2 and MTE3 complexes were chosen to be evaluated with molecular dynamics.
Complete cluster analysis can be found in Appendix 2.
4.2 Evaluation with Molecular Dynamics Based on previous work conducted by Brinck et al [43] the focus in the MD simulations rests on
studying the specific binding interactions of the ligand towards the catalytic residues in the active
site. The results presented here can only be considered as the tip of the iceberg in regards to
determine if the substrate and active site design was sufficient enough. As described in their work,
the so called ‘near attack conformers’ (NAC) for a D-A reaction can be viewed as an overlap between
the diene and dienophile, where the free energy of activation is determined by the ability of the
ligand to go from the bound state to NAC and thereafter from NAC to TS. Basically, this means that
the structure should retain its interactions with the residues necessary for catalysis to take place,
while simultaneously maintaining the interacting relationship between a diene and dienophile. As no
MD simulations was performed for the diene and dienophile by themselves, but only for the poses
obtained from docking the TS-structure, the free energy of activation could not be determined.
Therefore, this section relied on visual inspection only, where a 4 ns simulation was performed for
MTE1, MTE2 and MTE3, respectively. A 10 ns simulation was conducted for MTE1 and MTE3 as well
to see if eventual change occurred later during the simulation, which might tell of eventual
interesting behavior. Seeing as this is the case, the only important conclusion that could be drawn
from this initial investigation was whether the active site design was sufficient enough to contain the
diene and dienophile in an organized state. Namely, could the active site preserve the poses
obtained from the docking stage within the limits off a TS-like configuration?
4.2.1 MD simulation of MTE1
The results obtained from the molecular docking of MTE1 showed good promise as candidates for
the MD simulation. However, during the MD simulation it became apparent that the complex worked
poorly for the active site design. Following just a few ps into the simulation the dienophile showed
signs of leaving the site, indicating that the design was not feasible. Following is a presentation of the
system at 0, 1 and 10 ns respectively. The simulation at 10 ns is presented to demonstrate the
instability of the design.

45
Figure 33. MTE1 at 0 ns. The NACs are still maintained and this type of positioning of the TS-complex was the sought after one.

46
Figure 34. The MTE1 simulation at 1 ns. It is obvious that the dienophile is not suitable for this particular active site design. It is difficult to distinguish the exact cause of error as it might potentially lie in the substrate design but also in the active site design. It is more probable that it is the active site that is too hydrophilic and therefore cannot sustain the TS-complex for various reasons. Whereas these reasons are due to protein backbone restructuring or other issues have yet to been determined for this work.

47
Figure 35. As 10 ns has passed, the diene has maximized its hydrogen bonding interactions, while the dienophile is no longer actively participating in the active site. Due to the instability of the complex toward the active site, the complex was subsequently discarded for this particular mutant.

48
4.2.2 MD simulation of MTE2
Presented below are two figures representing the conformations interacting with the active site at
the initial stage of the MD simulation (i.e. at 0 ns) and after some eloped time (1 ns), respectively.
From figure 33 it can be seen that the diene and dienophile exist in a TS-like state, such as that
described in section 4.1.2. However, quite quickly the dienophile seemingly left its location from
beneath the diene, veering away from the active site. This can be viewed in figure 34 and although
the simulation was performed up to 4 ns, the dienophile never reclaimed its position. This indicated
that the design of MTE2 was a failure which would probably not result in a satisfactory interaction
with the active site. As such, the work with MTE2 was abandoned.
Figure 36. The starting position for the MD simulation at 0 ns. The TS-structure is still retained and the diene can be seen interacting favorable with Asp
99 and Tyr
14.

49
Figure 37. At the 1 ns mark the dienophile had already began dislodging from the active site. As the simulation went on the dienophile traversed even farther from the active site, showing no favorable interactions. As such, this design proved to be a failure.

50
4.2.3 MD simulation of MTE3
While the MTE1 and MTE2 designs had failed to retain a TS-like configuration, a different observation
was made for the MTE3 design. The following five figures presents the development of the MTE3 MD
simulation at 0, 1, 2, 3 and 4 ns, respectively. At the start of the simulation the ligands can be seen
positioned quite close to the TS-structure, with the diene maintaining a steady interaction with both
Asp99 and Tyr14. As the simulation approaches 1 ns the hydroxyl proton initiates interaction with an
approaching water molecule, which appears to be tilting the diene a bit, although it still remains in a
steadfast interaction with Asp99 and Tyr14. The dienophile retains its position below the diene while
exploring different conformations, At 2 ns, both the diene and Asp38 are interacting with the water
molecule mentioned earlier and the overall system appears to be in a relatively stable state, except
for the dienophile which positions itself almost perpendicular to the diene, resulting in a temporary
dislodging from the more planar state it adopted earlier in relation to the diene. A positive aspect
however, is that the dienophile seems to prevail in positioning itself directly below the diene, as
opposed to in MTE2 where it left the active site almost immediately. Approaching the 3 ns mark the
water molecule has begun to move further away from its previous position. Asp38 is now interacting
with the hydroxyl oxygen on the diene and the dienophile reclaims a more planar conformation next
to the diene. However, at 4 ns the interaction between the hydroxyl group on the diene and the
Asp38 residue has ceased, with the likely reason being that the diene has tilted away from its original
position. Even though the complex was unable in maintaining the necessary overlap for the diene
and dienophile to adopt a TS-like configuration, the complex did not reposition itself entirely. This
indicates that the designed substrate might strive to adopt a proper relative positioning, if exposed
to slight modification. It is also unfortunate that no MD simulations were conducted for the diene
and dienophile individually, as this would have enabled calculation of the free energy of binding. A
value for the free energy of binding could distinguish between a decent result and wishful thinking,
as a visual inspection will not accurately reflect the obtained result. The observation that the
complex does not explore the active site in a completely random fashion serves as a strong indication
that the main interactions are preserved. This does not confirm to what extent they are preserved
since the results depend on a visual inspection of the trajectory.
When a longer simulation (10 ns) was performed, it became obvious that even MTE3 had failed.
Passing the 3-4 ns limit the dienophile became unstable in the active site and evacuated the active
site. This was a reoccuring phenomenon for each following simulation with a new random seed.

51
Figure 38. The starting coordinates obtained from the docking phase, at 0 ns during the start of the simulation.

52
Figure 39. The simulation shows stability in that the dienophile remains in its original starting position. However, the hydroxyl group on the diene can be seen interacting with a water molecule through hydrogen bonding, which would eventually lead to the compound becoming slightly tilted.

53
Figure 40. 2 ns into the simulation and both diene and dienophile remain within close border of the starting coordinates. As mentioned earlier in figure 36, the hydroxyl group demonstrates interactions with a single water molecule. This is also true for Asp
38, showing some interactions with the water molecule. As no further calculations was performed beyond
this point it is difficult to ascertain if the complex is of good of poor design, although it shows promise.

54
Figure 41. This figure shows the simulation at 3 ns and it is clear that the diene has tilted to the side a bit. The overlapping effect between diene and dienophile is not excellent, but both molecules are retaining important interactions within the active site.

55
Figure 42. At the 4 ns mark the diene is seen clearly tilted. The overlap between the diene and dienophile is fairly poor, but in contrast to what has been reported in previous work [5][43] regarding catalytic antibodies, designed to catalyze the D-A reaction, the molecules do not adapt random interactions within the active site. A possible conclusion to be drawn from this is that even though Siegel and co-workers managed catalysis with molecules adapting random conformations, with a quite low catalytic activity, the pre-organization of the enzyme for this particular substrate is not too poor. This indicates that with some minor design revision, the suggested mutant and substrate might produce a fair result. But since the dienophile showed instability after 4 ns one might suggest that the active site design is not optimal for this particular substrate, and that the pyridine group used as a substituent might be a fair guess if screened towards a different mutant.

56
5. Conclusion This work has demonstrated that by employing the protocol developed by Brinck et al [8][43], it is
possible to employ rational design in order to enhance the capabilities of enzymes. Although no
specific substrate or active site design could be accurately determined, some insight was gained of
which residues could be utilized for design purposes to enhance the binding energy, while
simultaneously providing an increased frequency of optimal conformations. By investigating different
substitution patterns on the diene and dienophile, a notable increase in binding affinity and
consistent binding interactions was observed in the wildtype KSI as well, as compared to the initial
trial dockings conducted with the scaffold TS-structures presented in figure 7. This serves as a good
indication that by generating an appropriate design of a substrate, a D-A reaction could readily be
performed in native KSI. The optimal increase in enzyme-substrate affinity is obtained by focusing on
designing the substrates in concert with one another. This increase is ascribed to the enzymes ability
to organize the substrates in a fashion that allows an easier access to the TS. This has been described
earlier and is enforced by the result of the early state MD (<4 ns) of the MTE3 complex.
The use of pyridine as a potential substituent cannot be excluded, as it provided overall positive
results, no matter what environment it was evaluated in. The pyridine substituent allows for
potential hydrogen bonding as well as pi-pi interactions with the prevalent aromatic side chains in
the active site. However, the redesign of the active site most likely has to take a different turn. A
longer evaluation of the MTE1 and MTE3 complexes (up to 10 ns), with a random seed, showed no
improvement whatsoever in regards to the stability of the complex. For the MTE1 complex, the
dienophile left the active site within the scope of 1 ns, and the MTE3 complex simulations managed a
stable enough positioning in the active site for up to 3-4 ns. Each mutant protein was relaxed before
molecular docking and molecular dynamics simulations were conducted, and the active site did not
demonstrate a large distortion in either case, compared to the wild type KSI. But since no actual
RMSD comparisons were conducted, and the backbone distortion was only visually inspected, it
poses a problem when one has to qualitatively discuss the problem of introduced mutations effect
on the protein backbone structure. The relaxation of the mutants was conducted mainly to
investigate that the active site did not collapse on itself upon introduction of new side chains, and to
obtain as thorough docking results as possible.
The introduction of more hydrophilic side chains may initially have provided an enhanced binding
affinity and a good pre-organization of the TS-complex. But the question remains to see if this
actually prevails. It can be argued that introducing hydrophilic residues into such a hydrophobic site,
such as in KSI, is counterproductive. It allows for the introduction of several possible hydrogen bonds,
but one of the characteristics for KSI is the pKa of the Asp99 and Asp38, which has shown to be
dependent on the hydrophobic environment [9][11]. While the introduction of hydrogen bonding
capabilities might affect the binding affinity positively, it is difficult to properly evaluate the exact
influence this will have on the reaction at a later stage. The eventual shift in pKa values might turn
out to decrease the catalytic efficiency, while the binding affinity is demonstrated to be potentially
quite favorable.
It can thus be concluded that the pyridine substituent might be a quite valuable choice, whereas the
hydroxyl group on the diene is a cause for concern due to eventual deprotonization. But with a
proper redesign of the active site, with introduction of several hydrophobic side chains, and also one
or more hydrophobic substituents on the diene, the hydrophobic interactions could plausibly be fully

57
utilized, while retaining a proper pre-organization of the TS-complex and avoiding a larger shift in the
pKa of the vitally important catalytic triad.
A concluding remark on how to speed up the screening process, which also may include more
frequent hits using the AutoDock set of programs, will follow. As the results suggest that some
particular structures are favorable to others, one might consider utilizing AutoDock Vina [44] for a
quicker initial virtual screening of compounds. Due to the difference in scoring tactics utilized in Vina
and AD4, Vina could screen large libraries of substituted TS-structures (as no incorporated charge
parameter is included Vina performs faster calculations) and focus on shape complementarity, while
AD4 could be employed when hits have been discovered with Vina, subsequently incorporating a
charge parameter. As the TS-structure is reliant on the emerging anionic charge, the structural fit
would allow for the organization of diene and dienophile, hopefully maximizing the overlap between
the two compounds. An additional property that is invaluable to the increased efficiency of the
virtual screening process lies in the speed of the virtual screening process. Not simply in regards to
the screening of ligands towards the wild type protein, but also when trying out different mutations.
This results in a simple workflow where the ligand library is screened towards the wild type protein,
the wild type is mutated and the most successful hits are screened against the different mutants.
Preferably the mutations will be performed with the same side chain each time, until one mutation is
deemed the best. From there, one can evaluate continued mutations swiftly with Vina, until a set of
mutants are obtained with several newly incorporated side chains. It would probably be wise to set a
binding affinity threshold for the virtually screened compounds as an initial guideline regarding the
mutations.

58
References
1. Nguyen, L.A., He, H. & Pham-Huy, C., 2006. Chiral drugs: an overview. International
journal of biomedical science : IJBS, 2(2), pp.85–100. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/17221858.
2. Diels, O. & Alder, K,.1928. Justus Liebigs Ann. Chem. 460, pp. 98-122.
3. Borman, S., 2001. ASYMMETRIC CATALYSIS WINS. Chemical & Engineering
News, 79(42), pp.5–6. Available at:
http://pubs.acs.org/cen/topstory/7942/7942notw1.html.
4. Röthlisberger, D. et al., 2008. Kemp elimination catalysts by computational enzyme
design. Nature, 453(7192), pp.190–195. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/18354394.
5. Siegel, J.B. et al., 2010. Computational Design of an Enzyme Catalyst for a
Stereoselective Bimolecular Diels-Alder Reaction. Science, 329(5989), pp.309–313.
Available at: http://www.sciencemag.org/cgi/doi/10.1126/science.1190239.
6. Zanghellini, A. et al., 2006. New algorithms and an in silico benchmark for
computational enzyme design. Protein Science, 15(12), pp.2785–2794. Available at:
http://doi.wiley.com/10.1110/ps.062353106.
7. Linder, M. et al., 2012. Envisioning an enzymatic Diels–Alder reaction by in situ
acid–base catalyzed diene generation. Chemical Communications, 48(45), p.5665.
Available at: http://www.ncbi.nlm.nih.gov/pubmed/22547054.
8. Linder, M. et al., 2011. Designing a New Diels–Alderase: A Combinatorial,
Semirational Approach Including Dynamic Optimization. Journal of Chemical
Information and Modeling, 51(8), pp.1906–1917. Available at:
http://pubs.acs.org/doi/abs/10.1021/ci200177d.
9. Ha, N.C. et al., 2001. Structure and enzymology of Delta5-3-ketosteroid isomerase.
Current opinion in structural biology, 11(6), pp.674–8. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/11751047.
10. Sharma, K., Vázquez-Ramírez, R. & Kubli-Garfias, C., 2006. A theoretical model of
the catalytic mechanism of the Δ5-3-ketosteroid isomerase reaction. Steroids, 71(7),
pp.549–557. Available at:
http://linkinghub.elsevier.com/retrieve/pii/S0039128X05002801.
11. Cho, H.-S. et al., 1999. Crystal Structure of 5-3-Ketosteroid Isomerase from
Pseudomonas testosteroni in Complex with Equilenin Settles the Correct Hydrogen
Bonding Scheme for Transition State Stabilization. Journal of Biological Chemistry,
274(46), pp.32863–32868. Available at:
http://www.jbc.org/cgi/doi/10.1074/jbc.274.46.32863.
12. Pollack, R.M., 2004. Enzymatic mechanisms for catalysis of enolization: ketosteroid
isomerase. Bioorganic Chemistry, 32(5), pp.341–353. Available at:
http://linkinghub.elsevier.com/retrieve/pii/S0045206804000550.
13. Schwans, J.P., Kraut, D.A. & Herschlag, D., 2009. Determining the catalytic role of
remote substrate binding interactions in ketosteroid isomerase. Proceedings of the
National Academy of Sciences, 106(34), pp.14271–14275. Available at:
http://www.pnas.org/content/106/34/14271.full.
14. Clayden, J. et al., 2001. Organic Chemistry, Available at:
http://books.google.com/books?id=Dy5-vEst37oC&pgis=1.
15. Kuntz, I. D., et al. (1982). A geometric approach to macromolecule-ligand
interactions. J Mol Biol 161(2), pp. 269-288.

59
16. Sousa, S.F., Fernandes, P.A. & Ramos, M.J., 2006. Protein-ligand docking: Current
status and future challenges. Proteins: Structure, Function, and Bioinformatics, 65(1),
pp.15–26. Available at: http://doi.wiley.com/10.1002/prot.21082.
17. Sousa, S.F. et al., 2013. Protein-Ligand Docking in the New Millennium – A
Retrospective of 10 Years in the Field. Current Medicinal Chemistry, 20(18),
pp.2296–2314. Available at: http://www.ncbi.nlm.nih.gov/pubmed/23531220.
18. Morris, G.M. et al., 1998. Automated docking using a Lamarckian genetic algorithm
and an empirical binding free energy function. Journal of Computational Chemistry,
19(14), pp.1639–1662. Available at: http://doi.wiley.com/10.1002/%28SICI%291096-
987X%2819981115%2919%3A14%3C1639%3A%3AAID-JCC10%3E3.0.CO%3B2-
B.
19. Berg, J.M., Tymoczko, J.L. & Stryer, L., 2012. Biochemistry.
20. Carlson, H.A., 2002. Protein flexibility is an important component of structure-based
drug discovery. Curr Pharm Des, 8(17), pp.1571–1578. Available at:
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Ci
tation&list_uids=12052201.
21. Huey, R. et al., 2007. Software news and update a semiempirical free energy force
field with charge-based desolvation. Journal of Computational Chemistry, 28(6),
pp.1145–1152.
22. Weiner, S. et al., 1984. A new force field for molecular mechanical simulation of
nucleic acids and proteins. Journal of the American Chemical Society. Available at:
http://pubs.acs.org/cgi-bin/abstract.cgi/jacsat/1984/106/i03/f-
pdf/f_ja00315a051.pdf?sessid=6006l3.
23. Appleman, P., 2000. Darwin.
24. Solis, F.J. & Wets, R.J.B., 1981. Minimization by Random Search Techniques.
Mathematics of Operations Research, 6, pp.19–30.
25. Leach, A.R., 2001. Molecular Modelling: Principles and Applications,
26. Szabo, A. & Ostlund, N.S., 1996. Modern Quantum Chemistry: Introduction to
Advanced Electronic Structure Theory, Available at:
http://www.amazon.com/Modern-Quantum-Chemistry-Introduction-
Electronic/dp/0486691861.
27. Schrödinger, E., 1926. Quantisierung als Eigenwertproblem. Annalen der Physik,
384(4), pp.489–527. Available at: http://doi.wiley.com/10.1002/andp.19263840404.
28. Burke, K., 2012. Perspective on density functional theory. The Journal of Chemical
Physics, 136(15), p.150901. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/22519306.
29. Becke, A.D., 2014. Perspective: Fifty years of density-functional theory in chemical
physics. The Journal of Chemical Physics, 140(18), p.18A301. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/24832308\nhttp://scitation.aip.org/content/aip/jo
urnal/jcp/140/18/10.1063/1.4869598.
30. Hohenberg, P.; Kohn, W., 1964. Hohenberg, P.; Kohn, W. Phys. Rev., 136, pp.B864–
B871.
31. Johnson, K.H., 1973. Scattered-Wave Theory of the Chemical Bond. Advances in
Quantum Chemistry, 7(C), pp.143–185.
32. Becke, A.D., 1993. Density-functional thermochemistry. III. The role of exact
exchange. The Journal of Chemical Physics, 98(7), p.5648. Available at:
http://link.aip.org/link/JCPSA6/v98/i7/p5648/s1&Agg=doi\nhttp://scitation.aip.org/co
ntent/aip/journal/jcp/98/7/10.1063/1.464913.
33. Zhao, Y. & Truhlar, D.G., 2008. The M06 suite of density functionals for main group
thermochemistry, thermochemical kinetics, noncovalent interactions, excited states,

60
and transition elements: two new functionals and systematic testing of four M06-class
functionals and 12 other function. Theoretical Chemistry Accounts, 120(1-3), pp.215–
241. Available at: http://link.springer.com/10.1007/s00214-007-0310-x.
34. Case, D.A. et al., 2014. Amber 14. University of California, San Francisco, (January).
Available at: http://ambermd.org/.
35. Maier, J.A. et al., 2015. ff14SB: Improving the Accuracy of Protein Side Chain and
Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation,
11, pp.3696–3713. Available at: http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00255.
36. Wang, J. et al., 2004. Development and testing of a general Amber force field. Journal
of Computational Chemistry, 25(9), pp.1157–1174.
37. Wang, J. et al., 2005. Antechamber, An Accessory Software Package For Molecular
Mechanical Calculations. Journal of computational chemistry, 25(2), pp.1157–1174.
38. Wang, J. et al., 2006. Automatic atom type and bond type perception in molecular
mechanical calculations. Journal of Molecular Graphics and Modelling, 25(2),
pp.247–260.
39. Cornell, W.D. et al., 1993. Application of RESP Charges To Calculate Conformational
Energies, Hydrogen Bond Energies, and Free Energies of Solvation. Journal of the
American Chemical Society, 115(7), pp.9620–9631.
40. Mark, P. & Nilsson, L., 2001. Structure and dynamics of the TIP3P, SPC, and SPC/E
water models at 298 K. Journal of Physical Chemistry A, 105(43), pp.9954–9960.
41. G??tz, A.W. et al., 2012. Routine microsecond molecular dynamics simulations with
AMBER on GPUs. 1. generalized born. Journal of Chemical Theory and
Computation, 8(5), pp.1542–1555.
42. Morris, G.M. et al., 2009. Software news and updates AutoDock4 and
AutoDockTools4: Automated docking with selective receptor flexibility. Journal of
Computational Chemistry, 30(16), pp.2785–2791.
43. Linder, M. et al., 2012. Computational design of a Diels–Alderase from a thermophilic
esterase: the importance of dynamics. Journal of Computer-Aided Molecular Design,
26(9), pp.1079–1095. Available at: http://link.springer.com/10.1007/s10822-012-
9601-y.
44. Trott, O. & Olson, A.J., 2010. AutoDock Vina. J. Comput. Chem., 31, pp.445–461.

61
Appendix 1
Figure A.1. The suggested flowchart developed by Brinck and co-workers, upon which this work has been based on.

62
Figure A.2. A sample of some of the attempted substitutions for the substrate design.

63
Appendix 2
MTE1 – Cluster analysis of conformations CLUSTER ANALYSIS OF CONFORMATIONS
_________________________________
Number of conformations = 50
RMSD cluster analysis will be performed using the ligand atoms only (18 / 48 total atoms).
Outputting structurally similar clusters, ranked in order of increasing energy.
________________________________________________________________________________
Number of distinct conformational clusters found = 6, out of 50 runs,
Using an rmsd-tolerance of 2.0 A
CLUSTERING HISTOGRAM
____________________
________________________________________________________________________________
Cluster rank
Lowest Binding Energy
Run
Mean Binding Energy
Number in Cluster
Histogram
_______________________________________________________________________________
1 | -7.21 | 42 | -6.06 | 28 |############################
2 | -6.27 | 20 | -5.51 | 2 |##
3 | -6.13 | 17 | -5.31 | 15 |###############
4 | -5.15 | 18 | -5.14 | 2 |##
5 | -4.69 | 27 | -4.62 | 2 |##
6 | -4.07 | 48 | -4.07 | 1 |#
_____|___________|_____|___________|_____|______________________________________
Number of multi-member conformational clusters found = 5, out of 50 runs.
RMSD TABLE
__________

64
Rank Sub-rank Run Binding energy
Cluster RMSD
Reference RMSD
Grep Pattern
1 1 42 -7.21 0.00 85.19 RANKING
1 2 14 -6.91 0.70 85.59 RANKING
1 3 47 -6.90 0.40 85.30 RANKING
1 4 40 -6.67 0.26 85.30 RANKING
1 5 44 -6.65 0.53 85.32 RANKING
1 6 29 -6.54 0.23 85.27 RANKING
1 7 43 -6.51 0.22 85.23 RANKING
1 8 46 -6.47 0.56 85.19 RANKING
1 9 23 -6.36 0.41 85.25 RANKING
1 10 3 -6.35 0.17 85.16 RANKING
1 11 38 -6.29 0.50 85.22 RANKING
1 12 12 -6.18 1.57 85.52 RANKING
1 13 49 -6.10 0.65 85.46 RANKING
1 14 8 -6.08 1.38 85.12 RANKING
1 15 37 -6.08 1.39 84.90 RANKING
1 16 35 -6.04 0.43 85.23 RANKING
1 17 16 -6.04 0.36 85.39 RANKING
1 18 34 -5.88 1.45 84.82 RANKING
1 19 30 -5.87 0.68 85.44 RANKING
1 20 7 -5.85 0.78 85.21 RANKING
1 21 33 -5.69 1.59 85.15 RANKING
1 22 1 -5.59 0.53 85.46 RANKING
1 23 2 -5.56 0.77 85.77 RANKING
1 24 39 -5.55 0.89 85.68 RANKING
1 25 21 -5.39 1.49 85.34 RANKING

65
1 26 10 -5.38 1.51 85.24 RANKING
1 27 25 -5.34 1.55 85.39 RANKING
1 28 4 -4.21 1.19 85.57 RANKING
2 1 20 -6.27 0.00 86.17 RANKING
2 2 15 -4.75 1.97 86.15 RANKING
3 1 17 -6.13 0.00 85.15 RANKING
3 2 36 -5.98 1.49 85.50 RANKING
3 3 6 -5.71 1.58 84.32 RANKING
3 4 28 -5.47 1.63 84.67 RANKING
3 5 5 -5.46 1.53 84.82 RANKING
3 6 32 -5.31 1.09 85.06 RANKING
3 7 24 -5.29 1.54 84.82 RANKING
3 8 19 -5.29 1.86 85.24 RANKING
3 9 31 -5.10 1.12 85.00 RANKING
3 10 22 -5.09 1.39 84.54 RANKING
3 11 13 -5.08 1.86 85.32 RANKING
3 12 9 -5.02 1.27 84.47 RANKING
3 13 26 -4.99 1.38 84.31 RANKING
3 14 41 -4.88 1.54 85.06 RANKING
3 15 50 -4.85 1.76 84.16 RANKING
4 1 18 -5.15 0.00 85.50 RANKING
4 2 11 -5.13 0.48 85.50 RANKING
5 1 27 -4.69 0.00 84.68 RANKING
5 2 45 -4.56 0.69 84.45 RANKING
6 1 48 -4.07 0.00 85.44 RANKING
MTE2 – Cluster analysis of conformations CLUSTER ANALYSIS OF CONFORMATIONS

66
_________________________________
Number of conformations = 100
RMSD cluster analysis will be performed using the ligand atoms only (19 / 49 total atoms).
Outputting structurally similar clusters, ranked in order of increasing energy.
________________________________________________________________________________
Number of distinct conformational clusters found = 7, out of 100 runs,
Using an rmsd-tolerance of 2.0 A
CLUSTERING HISTOGRAM
____________________
Cluster rank
Lowest Binding Energy
Run
Mean Binding Energy
Number in Cluster
Histogram
_______________________________________________________________________________
1 | -6.45 | 51 | -4.62 | 73 |#######################################
| | | | |##################################
2 | -4.94 | 47 | -4.84 | 3 |###
3 | -4.59 | 96 | -3.60 | 7 |#######
4 | -4.12 | 82 | -3.20 | 5 |#####
5 | -3.56 | 46 | -2.89 | 8 |########
6 | -3.14 | 39 | -3.14 | 1 |#
7 | -2.82 | 45 | -2.32 | 3 |###
_____|___________|_____|___________|_____|______________________________________
Number of multi-member conformational clusters found = 6, out of 100 runs.
RMSD TABLE
__________
Rank Sub-rank Run Binding energy
Cluster RMSD
Reference RMSD
Grep Pattern
_____________________________________________________________________
1 1 51 -6.45 0.00 85.25 RANKING

67
1 2 60 -6.36 0.68 85.53 RANKING
1 3 67 -5.96 1.70 84.54 RANKING
1 4 95 -5.79 1.68 84.48 RANKING
1 5 52 -5.76 0.40 85.35 RANKING
1 6 40 -5.64 0.18 85.27 RANKING
1 7 22 -5.55 0.46 85.29 RANKING
1 8 98 -5.52 0.26 85.28 RANKING
1 9 28 -5.47 1.81 84.35 RANKING
1 10 41 -5.47 1.49 84.95 RANKING
1 11 18 -5.45 0.47 85.33 RANKING
1 12 33 -5.38 0.38 85.37 RANKING
1 13 10 -5.38 0.32 85.33 RANKING
1 14 76 -5.33 1.40 85.40 RANKING
1 15 68 -5.28 1.78 84.30 RANKING
1 16 55 -5.27 0.44 85.33 RANKING
1 17 64 -5.26 0.39 85.24 RANKING
1 18 99 -5.22 1.74 84.75 RANKING
1 19 5 -5.19 1.80 84.50 RANKING
1 20 73 -5.18 1.83 84.33 RANKING
1 21 12 -5.12 1.38 85.16 RANKING
1 22 31 -5.12 1.77 84.41 RANKING
1 23 44 -5.10 1.83 84.31 RANKING
1 24 89 -5.03 1.42 84.92 RANKING
1 25 37 -5.03 1.77 84.38 RANKING
1 26 59 -5.02 1.00 85.26 RANKING
1 27 72 -4.98 1.70 84.42 RANKING
1 28 16 -4.89 1.39 85.18 RANKING

68
1 29 49 -4.89 1.68 85.48 RANKING
1 30 57 -4.85 0.57 85.56 RANKING
1 31 54 -4.84 1.15 85.13 RANKING
1 32 14 -4.80 0.79 85.04 RANKING
1 33 91 -4.79 1.72 84.72 RANKING
1 34 88 -4.79 1.40 85.06 RANKING
1 35 58 -4.78 0.87 84.37 RANKING
1 36 2 -4.77 1.75 84.39 RANKING
1 37 93 -4.77 0.54 85.41 RANKING
1 38 71 -4.75 1.84 84.80 RANKING
1 39 79 -4.69 1.60 84.55 RANKING
1 40 15 -4.62 1.75 84.38 RANKING
1 41 8 -4.51 1.87 84.35 RANKING
1 42 100 -4.49 1.45 85.14 RANKING
1 43 85 -4.48 0.68 85.35 RANKING
1 44 80 -4.43 1.54 85.89 RANKING
1 45 13 -4.42 0.79 85.75 RANKING
1 46 29 -4.42 1.48 84.81 RANKING
1 47 20 -4.38 0.84 85.63 RANKING
1 48 75 -4.37 0.46 85.23 RANKING
1 49 1 -4.35 1.53 86.09 RANKING
1 50 26 -4.35 1.47 84.81 RANKING
1 51 38 -4.27 1.45 84.80 RANKING
1 52 84 -4.20 1.47 84.82 RANKING
1 53 3 -4.12 1.62 85.86 RANKING
1 54 11 -4.07 1.53 84.82 RANKING
1 55 81 -3.94 1.34 85.64 RANKING

69
1 56 34 -3.87 1.98 85.74 RANKING
1 57 69 -3.82 0.50 85.34 RANKING
1 58 24 -3.81 0.52 85.26 RANKING
1 59 30 -3.79 1.42 85.87 RANKING
1 60 50 -3.77 0.67 85.36 RANKING
1 61 19 -3.68 0.51 85.27 RANKING
1 62 78 -3.64 0.59 85.29 RANKING
1 63 62 -3.63 1.47 86.09 RANKING
1 64 36 -3.62 0.57 85.35 RANKING
1 65 53 -3.61 1.14 85.36 RANKING
1 66 6 -3.60 0.57 85.27 RANKING
1 67 66 -3.58 1.59 86.07 RANKING
1 68 92 -3.54 1.16 85.72 RANKING
1 69 86 -3.45 1.28 85.53 RANKING
1 70 63 -3.35 1.64 86.12 RANKING
1 71 90 -3.30 0.71 85.55 RANKING
1 72 83 -2.99 1.55 85.74 RANKING
1 73 48 -2.99 1.65 84.58 RANKING
2 1 47 -4.94 0.00 85.99 RANKING
2 2 42 -4.86 0.35 85.89 RANKING
2 3 27 -4.72 0.24 86.00 RANKING
3 1 96 -4.59 0.00 85.12 RANKING
3 2 61 -3.84 1.33 84.56 RANKING
3 3 56 -3.42 1.08 84.70 RANKING
3 4 25 -3.40 1.57 84.23 RANKING
3 5 77 -3.36 1.37 85.30 RANKING
3 6 74 -3.34 1.82 84.81 RANKING

70
3 7 32 -3.24 1.90 84.33 RANKING
4 1 82 -4.12 0.00 85.41 RANKING
4 2 7 -3.84 0.19 85.31 RANKING
4 3 43 -2.96 1.82 85.08 RANKING
4 4 35 -2.80 1.49 84.91 RANKING
4 5 17 -2.27 0.83 85.11 RANKING
5 1 46 -3.56 0.00 85.66 RANKING
5 2 23 -3.13 0.50 85.61 RANKING
5 3 4 -2.99 1.52 85.56 RANKING
5 4 21 -2.97 0.97 85.51 RANKING
5 5 70 -2.86 1.67 84.97 RANKING
5 6 9 -2.80 1.25 85.36 RANKING
5 7 94 -2.70 1.88 84.28 RANKING
5 8 65 -2.13 1.85 84.60 RANKING
6 1 39 -3.14 0.00 86.31 RANKING
7 1 45 -2.82 0.00 84.15 RANKING
7 2 97 -2.08 1.91 85.11 RANKING
7 3 87 -2.04 1.06 83.69 RANKING
______________________________________________________________________
MTE3 – Cluster analysis of conformations CLUSTER ANALYSIS OF CONFORMATIONS
_________________________________
Number of conformations = 100
RMSD cluster analysis will be performed using the ligand atoms only (20 / 50 total atoms).
Outputting structurally similar clusters, ranked in order of increasing energy.
________________________________________________________________________________

71
Number of distinct conformational clusters found = 7, out of 100 runs,
Using an rmsd-tolerance of 2.0 A
CLUSTERING HISTOGRAM
____________________
Cluster rank
Lowest Binding Energy
Run
Mean Binding Energy
Number in Cluster
Histogram
1 | -6.74 | 92 | -4.64 | 61
#############################################################
2 | -5.23 | 43 | -4.26 | 16 |################
3 | -5.02 | 36 | -4.09 | 10 |##########
4 | -4.44 | 4 | -4.44 | 1 |#
5 | -4.00 | 97 | -3.39 | 8 |########
6 | -3.48 | 81 | -3.30 | 3 |###
7 | -2.83 | 17 | -2.83 | 1 |#
_____|___________|_____|___________|_____|______________________________________
Number of multi-member conformational clusters found = 5, out of 100 runs.
RMSD TABLE
__________
Rank Sub-rank Run Binding energy
Cluster RMSD
Reference RMSD
Grep Pattern
1 1 92 -6.74 0.00 85.21 RANKING
1 2 32 -6.63 1.66 84.66 RANKING
1 3 13 -6.39 1.69 84.60 RANKING
1 4 18 -6.05 1.03 85.02 RANKING
1 5 5 -6.05 1.82 84.77 RANKING
1 6 60 -6.04 0.74 84.89 RANKING
1 7 33 -6.01 1.12 85.50 RANKING
72
1 8 29 -5.74 1.69 84.30 RANKING
1 9 48 -5.74 1.53 85.44 RANKING
1 10 100 -5.68 1.64 85.34 RANKING
1 11 91 -5.67 1.51 84.96 RANKING
1 12 23 -5.64 1.31 85.84 RANKING
1 13 54 -5.59 1.13 85.01 RANKING
1 14 96 -5.56 1.31 85.00 RANKING
1 15 51 -5.53 1.53 84.91 RANKING
1 16 88 -5.53 1.03 84.96 RANKING
1 17 76 -5.37 1.55 84.95 RANKING
1 18 57 -5.31 1.54 84.89 RANKING
1 19 83 -5.29 1.54 85.21 RANKING
1 20 68 -5.25 1.57 85.37 RANKING
1 21 78 -5.21 0.99 84.89 RANKING
1 22 47 -5.18 1.18 84.98 RANKING
1 23 84 -5.12 1.34 85.84 RANKING
1 24 73 -4.99 1.95 85.03 RANKING
1 25 11 -4.97 1.74 84.54 RANKING
1 26 35 -4.96 1.33 84.91 RANKING
1 27 1 -4.86 1.46 85.24 RANKING
1 28 94 -4.76 1.23 85.02 RANKING
1 29 80 -4.55 0.86 85.09 RANKING
1 30 69 -4.50 1.27 85.23 RANKING
1 31 85 -4.47 1.24 85.19 RANKING
1 32 7 -4.45 1.25 85.21 RANKING
1 33 71 -4.41 1.97 85.46 RANKING
1 34 52 -4.38 1.46 84.95 RANKING

73
1 35 38 -4.25 1.47 85.20 RANKING
1 36 89 -4.24 1.62 85.62 RANKING
1 37 98 -4.22 1.62 84.93 RANKING
1 38 21 -4.19 1.60 85.70 RANKING
1 39 14 -4.19 1.34 85.31 RANKING
1 40 70 -4.16 1.54 84.74 RANKING
1 41 3 -4.15 1.68 85.77 RANKING
1 42 16 -4.02 1.03 85.16 RANKING
1 43 95 -3.96 1.91 85.97 RANKING
1 44 75 -3.93 1.39 85.60 RANKING
1 45 41 -3.92 1.43 85.50 RANKING
1 46 49 -3.80 1.46 85.69 RANKING
1 47 9 -3.78 1.48 85.49 RANKING
1 48 55 -3.70 1.31 84.94 RANKING
1 49 77 -3.67 1.63 85.85 RANKING
1 50 19 -3.65 1.41 85.35 RANKING
1 51 82 -3.60 1.56 85.49 RANKING
1 52 12 -3.56 1.28 84.94 RANKING
1 53 65 -3.50 1.88 85.86 RANKING
1 54 58 -3.49 1.45 85.60 RANKING
1 55 46 -3.40 1.39 85.42 RANKING
1 56 40 -3.37 1.67 85.77 RANKING
1 57 26 -3.37 1.60 85.87 RANKING
1 58 6 -3.36 1.48 85.24 RANKING
1 59 72 -3.35 1.59 85.41 RANKING
1 60 59 -3.24 1.58 85.31 RANKING
1 61 27 -2.60 1.98 85.01 RANKING

74
2 1 43 -5.23 0.00 84.66 RANKING
2 2 50 -5.20 0.40 84.66 RANKING
2 3 28 -5.16 1.00 84.81 RANKING
2 4 63 -5.01 0.43 84.62 RANKING
2 5 8 -4.58 1.00 84.48 RANKING
2 6 31 -4.57 0.86 84.81 RANKING
2 7 44 -4.50 1.04 84.47 RANKING
2 8 87 -4.41 0.97 84.44 RANKING
2 9 25 -4.41 1.00 84.41 RANKING
2 10 93 -4.17 1.97 84.72 RANKING
2 11 42 -3.74 1.93 84.34 RANKING
2 12 79 -3.73 1.92 84.34 RANKING
2 13 2 -3.71 1.28 84.59 RANKING
2 14 64 -3.66 1.35 84.93 RANKING
2 15 22 -3.06 1.98 84.38 RANKING
2 16 34 -2.94 1.96 84.43 RANKING
3 1 36 -5.02 0.00 84.57 RANKING
3 2 86 -4.87 1.09 84.59 RANKING
3 3 53 -4.53 1.21 84.66 RANKING
3 4 99 -4.33 1.15 84.58 RANKING
3 5 62 -4.29 1.19 84.69 RANKING
3 6 74 -4.27 1.20 84.37 RANKING
3 7 45 -3.61 1.21 84.67 RANKING
3 8 10 -3.49 1.14 84.54 RANKING
3 9 39 -3.33 1.14 84.21 RANKING
3 10 37 -3.21 1.64 83.87 RANKING
4 1 4 -4.44 0.00 83.87 RANKING

75
5 1 97 -4.00 0.00 85.28 RANKING
5 2 56 -3.88 1.34 85.30 RANKING
5 3 61 -3.59 1.78 85.27 RANKING
5 4 90 -3.51 1.55 85.41 RANKING
5 5 20 -3.46 0.72 85.00 RANKING
5 6 66 -3.36 1.59 85.29 RANKING
5 7 30 -2.67 1.95 84.79 RANKING
5 8 15 -2.62 1.76 85.26 RANKING
6 1 81 -3.48 0.00 84.96 RANKING
6 2 24 -3.27 0.25 84.90 RANKING
6 3 67 -3.15 0.22 84.90 RANKING
7 1 17 -2.83 0.00 84.59 RANKING
_______________________________________________________________________