Embed Size (px)
Citation preview

1
Biologie für Mediziner
Mikrobiologisch/Genetischer Teil
Sommersemester 2016
Prof. Dr. Eckhard Boles Institut für Molekulare Biowissenschaften Goethe-Universität Frankfurt Max-von-Laue-Str. 9 60438 Frankfurt am Main Tel.: 069-798-29513 Email: [email protected] Homepage: http://www.bio.uni-frankfurt.de/boles

2
Programm Sommersemester 2016 - Mikrobiologie und Genetik
KURS I 1 Organisatorisches; Einführung in den mikrobiologischen Teil des Praktikums 2 Sterilisationstechniken: Erhitzen, Autoklavieren
Herstellen von Nährmedien, Agarplatten 3 Mikroskopische Untersuchung von Mikroorganismen: Lebende Bäckerhefe; Bakterienfärbung mit
Methylenblau, Gram-Färbung KURS II 1 Mikroben in unserer Umwelt: Luftkeime, Abklatschplatten, Fingerkeime vor und nach Desinfektion,
Überprüfung der Sterilität mit Nährbouillon, Tröpfcheninfektion 2 Reinkulturen: Verdünnungsausstrich, Verdünnungsreihe KURS III 1 Auswertung der Experimente von Kurs II 2 Bestimmen einer Antibiotikaresistenz 3 Antibiogramm 4 Replica plating (Samtstempelmethode) mit Hefekolonien 5 Genetischer Fingerabdruck 6 Sporenfärbung KURS IV 1 Auswertung der Experimente von Kurs III 2 Phagentiter-Bestimmung 3 Mutationsauslösung durch UV-Strahlen KURS V 1 Auswertung der Experimente von Kurs IV 2 Regulation der Enzymsynthese (β-Galaktosidase/lacZ von E. coli) 3 Stoffwechselleistungen von Mikroorganismen Termine:
Tag1 Fr So Tag der Arbeit Mi 8. Woche Fr Gruppe 7 (G) +8 (H) Versuch 52 Sa Mo 18. KW Do Gruppe 5 (E) + 6 (F) Versuch 3 Sa3 So Di Fr Gruppe 7 (G) +8 (H) Versuch 3 So4 Mo Mi 4. Woche Sa Mo 27. KW5 Di Do Christi Himmelfahrt So Di6 Mi Fr Mo 23. KW Mi 13. Woche
7 Do Sa Di Do8 Fr So Mi 9. Woche Fr9 Sa Mo 19. KW Do Gruppe 1 (A) + 2 (B) Versuch 4 Sa10 So Di Fr Gruppe 3 (C) + 4 (D) Versuch 4 So11 Mo 15. KW Mi 5. Woche Sa Mo 28. KW12 Di Do Gruppe 1 (A) + 2 (B) Versuch 3 So Di13 Mi 1. Woche Fr Gruppe 3 (C) + 4 (D) Versuch 3 Mo 24. KW Mi 14. Woche
14 Do Gruppe 1 (A) + 2 (B) Versuch 1 Sa Di Do15 Fr Gruppe 3 (C) + 4 (D) Versuch 1 So Mi 10. Woche Fr16 Sa Mo 20. KW Pfingsmontag Do Gruppe 5 (E) + 6 (F) Versuch 4 Sa17 So Di Fr Gruppe 7 (G) +8 (H) Versuch 4 So18 Mo 16. KW Mi 6. Woche Sa Mo 29. KW19 Di Do Gruppe 5 (E) + 6 (F) Versuch 2 So Di20 Mi 2. Woche Fr Gruppe 7 (G) +8 (H) Versuch 2 Mo 25. KW Mi21 Do Gruppe 5 (E) + 6 (F) Versuch 1 Sa Di Do22 Fr Gruppe 7 (G) +8 (H) Versuch 1 So Mi 11. Woche Fr23 Sa Mo 21. KW Do Gruppe 1 (A) + 2 (B) Versuch 5 Sa24 So Di Fr Gruppe 3 (C) + 4 (D) Versuch 5 So25 Mo 17. KW Mi 7. Woche Sa Mo26 Di Do Fronleichnam So Di27 Mi 3. Woche Fr Mo 26. KW Mi28 Do Gruppe 1 (A) + 2 (B) Versuch 2 Sa Di Do29 Fr Gruppe 3 (C) + 4 (D) Versuch 2 So Mi 12. Woche Fr30 Sa Mo 22. KW Do Gruppe 5 (E) + 6 (F) Versuch 5 Sa31 Di So
Praktikumsplan Biologie Sommersemester 2016
April Mai Juni Juli12:45 -16:00 Uhr bzw. 16:15 - 19:30 Uhr

3
Kriterien für die Scheinvergabe: Das Praktikum Biologie für Mediziner besteht aus 2 Teilen: Teil I - Zellbiologie, Zoologie, Humangenetik (WS) Teil II - Mikrobiologie, Molekulargenetik (SS) Für beide Teile zusammen wird ein gemeinsamer Schein vergeben. Voraussetzungen hierfür sind: - Regelmäßige Teilnahme, d.h. es dürfen nicht mehr als 10 % der Praktikumszeit versäumt
werden. Dies entspricht einem Fehltermin für das gesamte Praktikum (Teil I + Teil II). Wer später als 30 Minuten nach Veranstaltungsbeginn zum Praktikum erscheint, hat einen
Fehltermin. - Aktive Mitarbeit. - Testat-Gespräche, die jeweils am Ende der Kurse abgehalten werden (s. u.). Im SS2016
werden mit allen Studierenden jeweils zwei solcher Gespräche geführt. Für jedes bestandene Testat erhält man einen Bonuspunkt, der zum Ergebnis der Klausur addiert wird.
- Bestehen der beiden Abschlussklausuren. Die regelmäßige Teilnahme an der
Lehrveranstaltung ist die Voraussetzung für die Teilnahme an den Erfolgskontrollen (vgl. § 15 Abs. 2 der Studienordnung). Die Erfolgskontrolle erfolgt durch Klausuren mit 20 Fragen im Antwort-Wahl-Verfahren, für deren Bearbeitung 30 Minuten zur Verfügung stehen. Es gelten die Regelungen der §§ 18 und 20 der Studienordnung. Die Klausur umfasst Fragen zu Inhalten von Praktikum und begleitender Vorlesung. Für jede richtig beantwortete Frage erhält man einen Punkt. Die Klausur ist bestanden, wenn mindestens 60 % der maximalen Gesamtpunktzahl (= 12 Punkte) erreicht werden. Eine nicht bestandene Klausur kann einmal wiederholt werden.
Testat-Gespräche Ab dem zweiten Kurstag werden am Ende jedes Kurses Testat-Gespräche abgehalten. Hierbei handelt es sich um ca. 15 - 20minütige Gruppengespräche, die von den HiWis und evtl. dem Kursleiter mit in der Regel jeweils 3 - 5 Studierenden geführt werden. Im Rahmen dieser Gespräche wird theoretisches Wissen zum Praktikum überprüft. Ein Testat wird mit "bestanden" oder "nicht bestanden" bewertet. Für jedes bestandene Testat erhält man einen Bonuspunkt, der zum Ergebnis der Klausur addiert wird. (Beispiel: Die Klausur gilt als bestanden, wenn man mindestens 60 % davon, d. h. 12 Punkte, erreicht hat. Bei zwei bestandenen Testatgesprächen muss man in diesem Fall also nur 10 Klausurfragen richtig beantworten, um zu bestehen.) Die Testat-Punkte behalten auch für eine eventuelle Nachklausur ihre Gültigkeit. Die Gesprächstermine und die Zuordnung der Studierenden zu den HiWis bzw. dem Kursleiter werden nach dem Zufallsprinzip festgelegt und im Kurs für den jeweiligen Tag bekannt gegeben.

Versuchstag 1
4
1. Grundlagen steriler Arbeitsweisen - Mikroskopieren Versuch 1.1: Agarplatten - feste Nährmedien Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten Agarplatten Wie erhält man keimfreie Nährmedien? Das einzige zuverlässige Sterilisationsverfahren mit feuchter Hitze und zugleich die sicherste Sterilisationsmethode überhaupt ist das Autoklavieren, d.h. die Sterilisation mit gespanntem (= unter Druck stehendem), gesättigten Wasserdampf. Bei diesem Verfahren erhitzt man das Nährmedium in einem geschlossenen Druckbehälter, dem Autoklav, bei 121°C für 20 min. Dabei werden selbst die äußerst hitzeresistenten Endosporen einiger Bakterienarten abgetötet. Als Verfestigungsmittel für mikrobiologische Nährböden verwendet man meist Agar (eigentlich: Agar-Agar). Agar ist ein gelbildendes, komplex zusammengesetztes Naturprodukt aus Polysacchariden, das aus marinen Rotalgen durch Extraktion mit heißem Wasser gewonnen wird und nach Reinigung und Trocknung als feines Pulver oder als Granulat in den Handel kommt. Agar wird von den allermeisten Mikroorganismen nicht angegriffen. Sein Schmelz- und Erstarrungsverhalten in wäßrigem Milieu machen den Agar zum idealen Geliermittel für die Mikrobiologie. Eine 1,5 – 2 %ige wäßrige Agarsuspension schmilzt bei 80-90°C (also auch während des Autoklavierens) und erstarrt beim Abkühlen bei 38-32°C zu einem stabilen Gel. Die Kultivierung von Mikroorganismen auf und in festen Nährböden geschieht im Labor in der Regel in Petrischalen, runden, flachen Doppelschalen mit übergreifendem, lose aufliegendem Deckel. In diese wird das mit Agar versetzte Nährmedium nach dem Autoklavieren hineingegossen und bei Zimmertemperatur abkühlen gelassen. Als Agarplatte bezeichnet man eine dünne Scheibe aus agarhaltigem, verfestigten Nährboden in einer Petrischale. Agarplatten dürfen nur zur Beimpfung des Nähragars geöffnet werden. Sie werden ausschließlich in geschlossenem Zustand transportiert, beschriftet oder ausgewertet. Bitte nie den Deckel, sondern stets den Boden der Schale beschriften! Hierdurch wird eine Verwechslung durch (ungewollten) Deckeltausch vermieden. Im 2. Kurs sollen verschiedene Versuche mit Hefen durchgeführt werden, die man sehr einfach auf sogenannten YEPD-Agarplatten kultivieren kann. Dafür sollen die notwendigen Agarplatten vorbereitet werden. Versuchsdurchführung: Pro Gruppe werden 5 Agarplatten benötigt. 1 Agarplatte fasst ca. 30-40 ml Medium. Jede Gruppe bereitet 200 ml Nährmedium vor: 2 g Hefeextrakt (Yeast extract) 4 g Pepton 2 g Glucose 3,5 g Agar - abwiegen (weiter auf nächster Seite)

Versuchstag 1
5
- in Flasche geben - auf 200 ml mit Wasser auffüllen - schütteln - autoklavieren (121°C, 20 min) - abkühlen auf 80 - 50°C - luftblasenfrei mischen (Schwenken oder Rollen) - in Petrischalen gießen - abkühlen und aushärten lassen - lagern bei 4°C Versuch 1.2: Abtötung von vegetativen Zellen und Bakteriensporen Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten
Sterilisation Wie erhält man keimfreie Nährmedien und Glaswaren? Vergleich der Keimtötung durch einfaches Erhitzen (80°C, 15 min) und Autoklavieren (121°C, 20 min) inclusive Kontrollinkubation (Zimmertemperatur). Beschriftungen der Reagenzröhrchen und Nähragarplatten mit Gruppennummer und Temperaturen nicht vergessen!! Sie erhalten je 4 ml einer Hefekultur (Saccharomyces cerevisiae) und einer Bakterienkultur (Bacillus subtilis) und 4 leere, sterile Reagenzröhrchen mit Deckel sowie zwei Nähragarplatten, eine mit LB-Medium und eine mit YEPD-Medium. (LB-Medium: 0,5% NaCl, 0,5% Hefeextrakt, 1% Trypton, pH 7,5). Pipettieren Sie steril (i) in 2 dieser Röhrchen jeweils ca. 1 ml der Hefekultur und (ii) in die zwei anderen jeweils ca. 1 ml der Bakterienkultur. Den Rest der Kulturen verwahren Sie bitte auf (=Inkubation bei Zimmertemperatur). Stellen Sie jeweils eins der Röhrchen mit den Hefen und eins mit den Bakterien für 15 min in ein Wasserbad bei 80°C, nehmen es dann heraus und lassen es zunächst am Arbeitsplatz stehen. Stellen Sie die jeweils anderen beiden Röhrchen mit den Hefe- und Bakterienkulturen in den Autoklaven und autoklavieren diese Kulturen zusammen mit dem unter Versuch 1.1 hergestellten Nährmedium (121°C, 20 min). Am Ende des Versuchstages unterteilen Sie die beiden Nähragarplatten jeweils in 3 Felder und plattieren je einen Tropfen der Hefe- und Bakterienkulturen darauf aus, S. cerevisiae auf YEPD und B. subtilis auf LB. Dazu beschriften Sie zunächst die Felder mit ZT (für die Restkultur = Kontrolle, die bei Zimmertemperatur aufbewahrt wurde), 80° (für die Kultur, die im Wasserbad bei 80°C inkubiert wurde) und A (für die Kultur, die autoklaviert wurde). Drehen Sie die Agarplatten nicht eher um, bis die Ausstriche getrocknet sind, damit sie nicht ineinander verlaufen. Die Agarplatten werden dann für 1-2 Tage bei 30°C inkubiert, damit die überlebenden Mikroorganismen zu Kolonien heranwachsen können. Damit kann dann nachgewiesen

Versuchstag 1
6
werden, unter welchen Bedingungen die Hefen und die Bakterien abgetötet wurden und unter welchen sie überlebten. Auswertung erfolgt am 2. Versuchstag. Versuch 1.3: Mikroskopische Untersuchung von Mikroorganismen Die Mikroskopie ist eine Standard-Technik zur Untersuchung von Form, Größe und Beweglichkeit von Mikroorganismen. Für die Beobachtung lebender Bakterien ist die Phasenkontrast-Mikroskopie besonders geeignet, bei der eine Kontraststeigerung gegenüber der Hellfeld-Mikroskopie durch Veränderung der Lichtführung erreicht wird. Präparate mit gefärbten Bakterien werden dagegen im Hellfeld angeschaut. Die Mikroorganismen werden bei 400facher oder 1000facher Vergrößerung betrachtet. Bei 1000facher Vergrößerung ist die Ölimmersion einzusetzen. Dabei werden das Immersionsobjektiv und der Objektträger mit Immersionsöl verbunden, das gegenüber Luft einen höheren Brechungsindex aufweist. Dadurch wird das Auflösungsvermögen erhöht. Aufgaben dieses Versuches: A: Sich mit der Bedienung des Mikroskops zur Betrachtung von Mikroorganismen in groben Zügen vertraut machen. B: Betrachten von lebenden Hefezellen aus vorgegebenen Reinkulturen (Präparation nach Punkten 1-3. Bitte kein Immersionsöl verwenden, da es sich um ein Feuchtpräparat handelt. Bei Vergrößerung 40 (Objektiv) x 10 (Okular) betrachten. Objektiv hinterher gründlich säubern! Achtung: das Objekt darf nicht hitzefixiert werden!!!! C1: Bakterienfärbung mit Methylenblau Untersuchungsobjekt ist: Bacillus megaterium (mit Hitzefixierung!! siehe unten) C2: Gramfärbung Untersuchungsobjekte sind: Bacillus subtilis (Gram-positiv) und Escherichia coli (Gram-negativ) (mit Hitzefixierung!! siehe unten) Alle mikroskopischen Bilder sind zu zeichnen (mehrere Zellen) und zu beschriften: • Name des präparierten Organismus • Färbemethode • Vergrößerung A) Zum Gebrauch des Mikroskops Während des Semesters wechseln täglich die Benutzer der Mikroskope. Da wir für Schäden und Verlust einzelner Teile den letzten Benutzer haftbar machen müssen, beachten Sie bitte folgendes: 1. Prüfen Sie zu Beginn des Kurses, ob alle Teile vorhanden und unbeschädigt sind.
Melden Sie Beanstandungen sofort einem Assistenten oder dem Kursleiter. 2. Heben Sie das Mikroskop nie am Einblicktubus in die Höhe, sondern fassen Sie es
stets am Stativ an. 3. Stoßen Sie nie die Frontlinse der Objektive auf den Objektträger. 4. Lassen Sie keine Farb- und Salzlösungen vom Objektträger auf den Kondensor
fließen.

Versuchstag 1
7
5. Lassen Sie bitte am Ende des Kurses das Mikroskop auf dem Arbeitstisch stehen, damit es überprüft werden kann.
Zur Beruhigung: In den letzten 13 Jahren mußte kein Student/keine Studentin für Schäden haftbar gemacht werden. Anleitung zum Mikroskopieren 1. Bringen Sie den Objekttisch an den oberen Anschlag. 2. Lichtquelle einschalten. (Die Marke am Drehschalter der Leuchte kennzeichnet die
zum Mikroskopieren geeignete Stellung.) 3. Objekt auf den Objekttisch legen. Blende an der Leuchte öffnen. 4. Schwaches Objektiv (3,2x) einrasten. Bild mit dem groben Einstelltrieb scharf stellen
(Feintrieb erst bei den starken Vergrößerungen verwenden!). Beginnen Sie nie mit den großen Objektiven. Gehen Sie schrittweise an die stärkste Vergrößerung heran.
5. Da das Sehfeld bei den schwachen Objektiven durch die Hilfslinse (Lichtsammellinse) eingeengt wird, klappt man sie zunächst heraus.
6. Mit der Kondensorblende können Sie den Bildkontrast beeinflussen. 7. Das 100x -Objektiv (und nur dieses!) ist mit Immersionsöl zu benutzen. Reinigen Sie
die Frontlinse dieses Objektivs nach Gebrauch mit einem weichen, mit Spüli-Lösung benetzten Tuch (nie mit Alkohol reinigen!).
Bleibt das Sehfeld dunkel oder schlecht ausgeleuchtet, dann senken Sie den Kondensor und beobachten beim Heben, wohin die Ausleuchtung im Sehfeld zieht. Holen Sie mit den Zentrierschrauben die Helligkeit in die Sehfeldmitte zurück. Zum Zentrieren schließen Sie weitgehend die Leuchtfeldblende, bilden diese durch Senken oder Heben des Kondensors scharf ab und rücken mit Hilfe der Zentrierschrauben das Bild der Blende in die Sehfeldmitte. Zum Mikroskopieren öffnen Sie die Leuchtfeldblende wieder, bis ihr Rand aus dem Sehfeld gerade eben verschwindet. Bei Problemen fragen Sie bitte umgehend Ihre Assistentin / Ihren Assistenten oder den Kursleiter. Herstellung eines mikroskopischen Präparates B) Feuchtpräparat/Lebendbetrachtung 1. Auf einen sauberen Objektträger einen kleinen Tropfen Wasser aufbringen. 2. Impföse ausglühen und abkühlen lassen. 3. Eine kleine (!!) Menge Mikroben-Material mit der Impföse aufnehmen und im
Wassertropfen suspendieren. Dabei den Wassertropfen großflächig auf dem Glasplättchen verstreichen.
C) Hitzefixierung 1. Auf einen sauberen Objektträger einen kleinen Tropfen Wasser aufbringen. 2. Impföse ausglühen und abkühlen lassen. 3. Eine kleine Menge Mikroben-Material mit der Impföse aufnehmen und im
Wassertropfen suspendieren. Dabei den Wassertropfen großflächig auf dem Glasplättchen verstreichen.
4. Ausstrich an der Luft trocknen lassen. 5. Objektträger mit der beschichteten Seite nach oben dreimal durch die Flamme des
Spiritusbrenners ziehen. 6. Färbung (siehe unten).

Versuchstag 1
8
C1) Färbung mit Methylenblau 1. Den hitzefixierten Ausstrich mit Methylenblau-Lösung überschichten und ca. eine
Minute einwirken lassen. 2. Mit Wasser abspülen (in Photowanne). 3. Objektträger vorsichtig mit Filtrierpapier trocknen. 4. Mikroskopieren (ohne Deckglas). Zunächst mit kleinster Vergrößerung, zuletzt mit der
größten (Objektiv 100x) und mit Immersionsöl. C2) Bakterienfärbung nach Gram 1. Den hitzefixierten Bakterienausstrich 1 Minute mit Kristallviolett-Lösung
überschichten. 2. Farblösung abgießen, nicht nachspülen. 3. Mehrmals Lugol‘sche Lösung für 1 Minute einwirken lassen und jeweils abgießen. 4. Mit Wasser abspülen. 5. Mit Aceton / Ethanol mehrmals (ca. 3x) abspülen (jeweils 5 Sekunden lang). 6. Mit Wasser abspülen. 7. Safranin-Lösung 1 Minute einwirken lassen. 8. Mit Wasser abspülen. 9. Mit Filtrierpapier vorsichtig trocknen und mikroskopieren.
Beurteilung Gram-positive Bakterien sind dunkelblau-violett, Gram-negative rot bis rötlich.
Fragen zum Kurstag I: 1. Durch welchen Trick lassen sich lebende Keime nachweisen? 2. Was bedeutet „autoklavieren“? 3. Wie lassen sich Bakterien identifizieren? 4. Was ist Agar-Agar? 5. Was ist eine Bakterienkolonie? 6. Welche Bakterienformen gibt es? 7. Was müssen Sie beim Betrachten von Bakterien beachten und warum? 8. Beschreiben Sie das Prinzip der Gram-Färbung! 9. Wie unterscheiden sich verschiedene Bakterien (Prokaryonten) und Hefezellen (Eukaryonten) hinsichtlich Zellgröße und Zellform?

Versuchstag 2
9
2. Mikroben in der Umwelt - Isolierung von Reinkulturen Vorbereitung: - morphologische Grundformen der Bakterien - Pathogenitätsfaktoren und Pathogenese - Endosporen (Klostridien/Bacillus) - Autoklavieren/Sterilisation/Desinfektion - Vereinzelung von Bakterienzellen - chemische Wachstumskontrolle Versuch 2.1: Mikroben in unserer unmittelbaren Umwelt Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten Hinweise für den Umgang mit Nähragar-Platten Im ersten Versuch wollen wir mit Hilfe von Nähragarplatten feststellen, ob und wenn ja, wie viele Bakterien oder Pilzsporen sich in unserer unmittelbaren Umgebung befinden. Die Agarplatten dürfen nur zur Beimpfung des Nähragars geöffnet werden. Sie werden ausschließlich in geschlossenem Zustand transportiert, beschriftet oder ausgewertet. Bitte nie den Deckel, sondern stets den Boden der Schale beschriften! Hierdurch wird eine Verwechslung durch (ungewollten) Deckeltausch vermieden. Zur Beschriftung gehört immer die Angabe des Wochentages sowie die Arbeitsplatznummer(n). Nach Beimpfung der Platten mit Mikroben-Suspension muß diese erst in den Agar eintrocknen, ehe die Platten umgedreht werden dürfen. • Luftkeime Pro Gruppe werden 3 sterile LB-Agarplatten 15, 30 bzw. 60 Min. lang offen stehen gelassen, dann geschlossen und anschließend 6 Tage bebrütet (Auswertung am 3. Versuchstag). Aus jeder Mikrobenzelle, die sich aus der Luft auf der Agarplatte niedergelassen hat, entsteht eine Kolonie. Wie hoch ist die Anzahl der Keime in Abhängigkeit von der Zeit? Wie unterscheiden sich die Kolonien? • Abklatschplatten Pro Gruppe werden 4 Platten mit LB-Medium verwendet, um Keime z.B. auf Möbeln, Geldscheinen, Fußböden, an Schuhsohlen, Fenstern, Wänden oder anderen Objekten Ihrer Wahl nachzuweisen. Vermerken Sie den Abklatschort auf dem Plattenboden. • Keime an den Händen Zwei LB-Agarplatten werden (durch Markierungen auf dem Plattenboden) in je 4-6 Felder unterteilt. Jedes Gruppenmitglied berührt dann mit dem Zeigefinger den Nähragar in einem Feld. Anschließend wird die Platte sofort geschlossen. Nach intensiver Händedesinfektion (5 ml Sterillium 1-2 Min. (!) in den Händen und auf den Fingern gut verreiben) wird der Abdruck auf der zweiten Platte wiederholt. Vergleichen Sie den Bewuchs der Platte nach einwöchiger Bebrütungszeit! War Ihr Finger nach der Desinfektion keimfrei?

Versuchstag 2
10
• Verteilung von Keimen an den Händen durch Händeschütteln Eine YEPD-Agarplatte wird (durch Markierungen auf dem Plattenboden) in 4- 6 Felder unterteilt. Zunächst desinfizieren sich wieder wie oben alle Gruppenmitglieder die Hände mit Sterillium. Dann fasst ein Gruppenmitglied mit einer Hand in ein Gefäß, dass in Wasser aufgelöste Backhefe enthält. Er/Sie schüttelt dem 2. Gruppenmitglied die Hand (bitte beachten, dass sich auch die Finger berühren), dieses schüttelt dem dritten die Hand usw. Zum Schluß berühren wieder alle Gruppenmitglieder mit dem Zeigefinger die Agarplatte auf einem der Felder (bitte beschriften). Am 3. Versuchstag vergleichen Sie den Bewuchs der Platte nach zweitägiger Bebrütungszeit! Gemeinsames Arbeiten von alle zusammen • Verteilung von Keimen in der Luft durch Husten Es werden 10 YEPD-Agarplatten mit 1-10 beschriftet und in einer Reihe hintereinander im Raum offen aufgestellt. Ein/e Kursteilnehmer/in gurgelt kurz mit einer Backhefesuspension. Danach hustet er/sie kräftig mehrmals in Richtung der Agarplatten. Nach 5-10 min werden diese geschlossen und anschließend 2 Tage bei 30°C bebrütet. Am 3. Versuchstag wird anhand der Anzahl der Hefekolonien ausgewertet, wie weit sich die Hefezellen im Raum verteilt hatten. Versuch 2.2: Herstellung einer Reinkultur Gemeinsames Arbeiten von Gruppen zu je 2 Studenten Von einer mit einem Gemisch verschiedener Hefestämme beimpften YEPD-Platte sollen Reinkulturen jedes vertretenen Stammes angelegt werden. Wir bedienen uns zweier Methoden: A: Vereinzelung mittels Ausstrich (mit einer sterilen Impföse) Diese simple Technik wird Ihnen von Ihren Betreuern gezeigt. B: Vereinzelung mittels Verdünnungsreihe Mit einer sterilen Impföse wird ein wenig Zellmaterial (nicht zu viel nehmen!) in steriles Wasser überführt und durch leichtes Schütteln des Röhrchens suspendiert (die Flüssigkeit soll leicht trübe sein). Die Anzahl der Hefezellen im Wasser beträgt nun ca. 106 Zellen pro ml. Wir verdünnen dann diese Suspension in mehreren Schritten bis auf etwa 102 Zellen pro ml nach dem folgenden Schema:
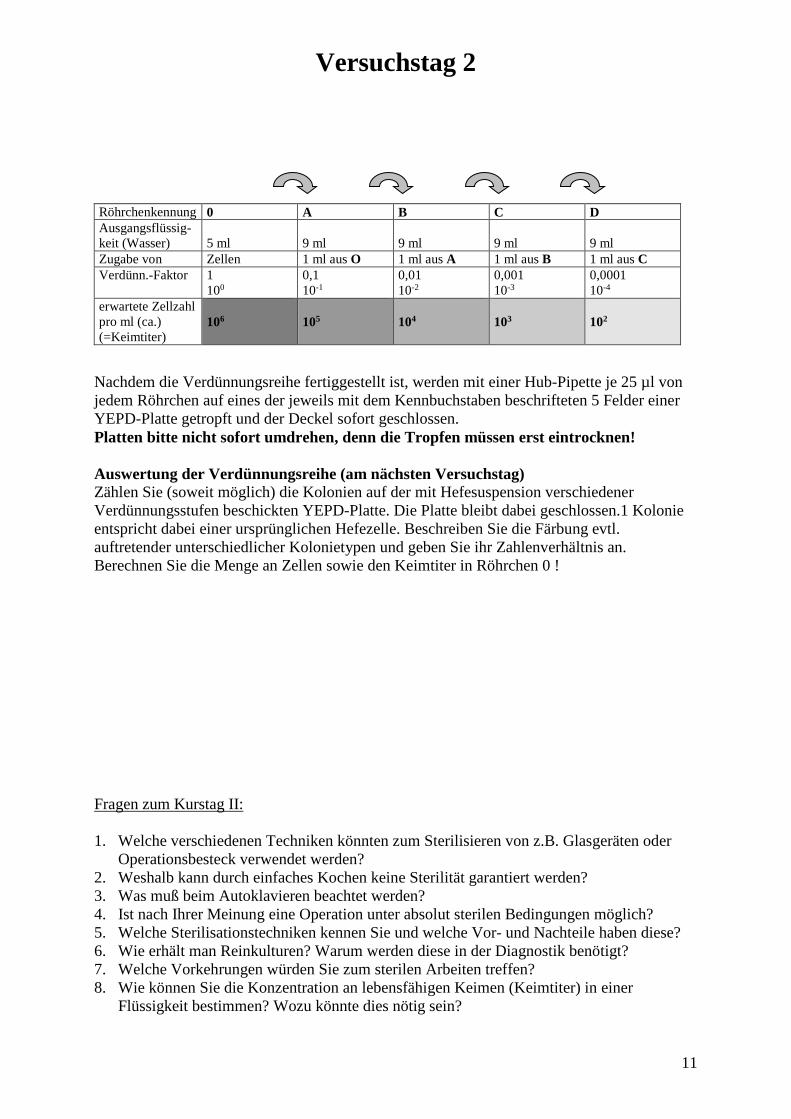
Versuchstag 2
11
Röhrchenkennung 0 A B C D Ausgangsflüssig-keit (Wasser)
5 ml
9 ml
9 ml
9 ml
9 ml
Zugabe von Zellen 1 ml aus O 1 ml aus A 1 ml aus B 1 ml aus C Verdünn.-Faktor 1
100 0,1 10-1
0,01 10-2
0,001 10-3
0,0001 10-4
erwartete Zellzahl pro ml (ca.) (=Keimtiter)
106
105
104
103
102
Nachdem die Verdünnungsreihe fertiggestellt ist, werden mit einer Hub-Pipette je 25 µl von jedem Röhrchen auf eines der jeweils mit dem Kennbuchstaben beschrifteten 5 Felder einer YEPD-Platte getropft und der Deckel sofort geschlossen. Platten bitte nicht sofort umdrehen, denn die Tropfen müssen erst eintrocknen! Auswertung der Verdünnungsreihe (am nächsten Versuchstag) Zählen Sie (soweit möglich) die Kolonien auf der mit Hefesuspension verschiedener Verdünnungsstufen beschickten YEPD-Platte. Die Platte bleibt dabei geschlossen.1 Kolonie entspricht dabei einer ursprünglichen Hefezelle. Beschreiben Sie die Färbung evtl. auftretender unterschiedlicher Kolonietypen und geben Sie ihr Zahlenverhältnis an. Berechnen Sie die Menge an Zellen sowie den Keimtiter in Röhrchen 0 ! Fragen zum Kurstag II: 1. Welche verschiedenen Techniken könnten zum Sterilisieren von z.B. Glasgeräten oder Operationsbesteck verwendet werden? 2. Weshalb kann durch einfaches Kochen keine Sterilität garantiert werden? 3. Was muß beim Autoklavieren beachtet werden? 4. Ist nach Ihrer Meinung eine Operation unter absolut sterilen Bedingungen möglich? 5. Welche Sterilisationstechniken kennen Sie und welche Vor- und Nachteile haben diese? 6. Wie erhält man Reinkulturen? Warum werden diese in der Diagnostik benötigt? 7. Welche Vorkehrungen würden Sie zum sterilen Arbeiten treffen? 8. Wie können Sie die Konzentration an lebensfähigen Keimen (Keimtiter) in einer Flüssigkeit bestimmen? Wozu könnte dies nötig sein?

Versuchstag 3
12
3. Antibiotika – Antibiogramm - Replikaplattierung – Genetischer Fingerabdruck - Endosporen
Vorbereitung: - Antibiotika - Wirkorte/Wirkweise von Antibiotika - Antibiotikaresistenzen - Antibiogramm - Replikaplattierung - Genetischer Fingerabdruck - Endosporen Versuch 3.1: einfaches Bestimmen von Antibiotikaresistenzen durch
Ausstrich auf Nährmediums-Agarplatten mit und ohne Antibiotikum
Gemeinsames Arbeiten von Gruppen zu je 2 Studenten Bei 2 Bakterienisolaten (Reinkulturen) soll bestimmt werden, ob eine Kanamycin-Resistenz vorliegt. Wir verwenden hierfür einen Nähragar, der reichlich Nährstoffe für die Bakterien enthält und dem Kanamycin (50 mg/l) zugegeben worden ist. Durch Vergleich des Bakterienwachstums auf der Nähragarplatte ohne Kanamycin mit dem auf der Platte mit Kanamycin wird die Sensibilität/Resistenz gegenüber dem Antibiotikum deutlich. Versuch Sie erhalten 2 Bakterienstämme (E. coli D, E. coli S) auf einer LB-Stammplatte, die Sie 1.) auf eine LB-Kan-Platte und danach 2.) mit der gleichen Impföse auf eine frische LB-Platte ausstreichen. Die Platten werden dazu jeweils in zwei Hälften unterteilt. Die beiden Platten werden 2 Tage bei 30°C bebrütet und dann bis zur Auswertung am nächsten Kurstag gekühlt aufbewahrt. Auswertung (am nächsten Versuchstag) Beschreiben Sie die Wachstumserscheinungen bei den 2 Ausstrichen auf der LB- und LB-Kan-Platte. Gab es Unterschiede zwischen den beiden Bakterienstämmen? Versuch 3.2: Antibiogramm (Plättchentest, Agardiffusionstest) Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten Jede Gruppe erhält eine LB-Agarplatte, ein Röhrchen mit 0,5 ml einer E. coli Übernachtkultur (37°) sowie ein Röhrchen mit 15 ml LB-Weichagar (0,5%) (50-60°C). Beschriften Sie bitte die LB-Agarplatte mit Ihrer Gruppennummer sowie dem Buchstaben der E. coli Kultur. Die 15 ml LB-Weichagar werden rasch zu den 0,5 ml der E. coli Übernachtkultur hinzugegossen,

Versuchstag 3
13
kurz gemischt und dann auf die LB-Agarplatte gegossen (Achtung: rasch arbeiten, damit der Weichagar nicht fest wird). Nachdem der Weichagar auf der LB-Agarplatte fest geworden ist (5-10 min) werden mit Stempelgeräten auf jede Platte 5 vorgefertigte, verschiedene Antibiotika-enthaltende Filterplättchen aufgebracht. Die Platten werden 24-48 Stunden bei 37°C inkubiert.
Die Konzentration der verwendeten Antibiotika ist der untenstehenden Tabelle zu entnehmen. Die Sensitivität eines Keims gegen das getestete Antikbiotikum ist an einer Hemmzone im Diffusionsbereich des jeweiligen Antibiotikums um die Filterplättchen zu erkennen. Es werden insgesamt 4 verschiedene E. coli Isolate zur Verfügung gestellt (E. coli G, S, B, D), die verschiedene Antibiotika-Resistenzen aufweisen. Auswertung (am nächsten Versuchstag): Bitte werten Sie sowohl Ihre eigene Platte sowie auch die Platten Ihrer Nachbargruppen aus und tragen Sie die Ergebnisse in die Tabelle ein. Bei der Auswertung soll vermerkt werden:
- ist der Keim resistent oder sensitiv gegen das jeweilige Antibiotikum - möglicher Grund einer beobachteten Resistenz - Durchmesser der Hemmzone - ist die Hemmzone trüb oder klar
Nr. Antibiotikum
(Konzentration) E. coli G E. coli S E. coli B E. coli D Wirkort des
Antibiot. 1 Ampicillin
(10 µg/Plättchen)
2 Vancomycin (30µg/Plättchen)
3 Tetracyclin (30µg/Plättchen)
4 Ciprofloxacin (5 µg/Plättchen)
5 Kanamycin (30 µg/Plättchen)

Versuchstag 3
14
Versuch 3.3: Replikaplattierung Gemeinsames Arbeiten von Gruppen zu je 2 Studenten Durch Übertragung eines Teils der Zellen einer Kolonie (die aus mindestens 106 bis maximal 1010 Zellen bestehen kann) auf andere Nähragar-Platten kann man z.B: A: Kolonien erneut wachsen lassen (vegetative, klonale Vermehrung). B: Die Zellen dieser Kolonien auf ihre Antibiotikaresistenz testen C: Die Zellen dieser Kolonien auf ihre Nährstoffabhängigkeit prüfen. Man überträgt dabei das "Koloniemuster" auf Nähragarplatten anderer Zusammensetzungen. Diese Übertragung von Zellen jeder Kolonie (in plattengetreuem Muster) geschieht am einfachsten unter Anwendung der sog. Samtstempel-Methode (replica plating). Sie erlaubt die schnelle Übertragung des Koloniemusters der Ausgangsplatte auf bis zu 20 neue Platten mit verschiedenen Selektivnährmedien. In unserem Versuch übertragen wir Teile der Hefe-Kolonien auf 3 verschiedene Medien. Die Medien enthalten: 1.) Selektivmedium ohne Adenin, Glucose als Energiequelle (= G-ade) 2.) Vollmedium mit Adenin, Glycerin statt Glucose als Energiequelle (= Gly+Ade) 3.) Vollmedium mit Adenin, Glucose als Energiequelle (= G+Ade) Die Agarplatten werden in dieser Reihenfolge gestempelt!!
Nach Bebrütung für mindestens 2-3 Tage (bei 30oC) wird das Wachstum auf den Selektivmedien untersucht. Versuch 3.4: Genetischer Fingerabdruck Als genetischer Fingerabdruck wird ein DNA-Profil eines Individuums bezeichnet, das für dieses in hohem Maße charakteristisch ist. Die DNA wird aus Zellen gewonnen, die aus Gewebeteilen oder Sekreten, zum Beispiel Sperma, Hautzellen oder Speichel stammen. Das Verfahren wird in der Molekularbiologie auch als Genetic Fingerprinting oder DNA Fingerprinting bezeichnet. Für den genetischen Fingerabdruck werden derzeit zwischen 8 und 15 spezifische Abschnitte aus der DNA mit Hilfe der PCR-Methode (Polymerasekettenreaktion) vervielfältigt und ihre Größe/Länge per Gelelektrophorese sichtbar gemacht. Bei dieser Methode werden repetitive DNA-Sequenzen untersucht, dabei handelt es sich meist um sogenannte polymorphe Mikrosatelliten. Mikrosatelliten-DNA besteht aus 10-50 Kopien von Folgen aus 1-6 Basenpaaren, die direkt hintereinander vorkommen. Beispiele für solche Wiederholungsfolgen sind A, AC, AAT oder AAC. Für den genetischen Fingerabdruck ist interessant, dass die Zahl solcher Wiederholungsfolgen in einem gegebenen Mikrosatelliten hoch polymorph ist. In einem Allel eines Menschen können es z.B. 30 sein (also z.B. AC(AC)28AC), in dem eines anderen Menschen 25. Die Ursachen dafür sind Verrutschungen in DNA-Sequenzwiederholungen während der DNA-Replikation. Beim genetischen Fingerabdruck werden nun 8-15 solcher polymorpher Mikrosatelliten in hochautomatisierten Verfahren auf die Anzahl ihrer Wiederholungsfolgen, also ihre Länge,

Versuchstag 3
15
untersucht. Damit lässt sich dann statistisch eine unter mehreren 100 Millionen Personen eindeutig identifizieren. Die Identifizierung erfolgt dabei nach dem Ausschlussprinzip. Bei der kriminalistischen Aufklärung einer Vergewaltigung wird z.B. ein Vaginalabstrich mit Spermaresten des Täters untersucht. Das Ergebnis der PCR-Analyse wird mit DNA-Proben von Tatverdächtigen verglichen. Weicht nur in einem einzigen der 8-15 untersuchten Mikrosatelliten die Anzahl der Wiederholungsfolgen ab, so ist der Tatverdächtige unschuldig. Stimmen alle Wiederholungsfolgen überein, dann liegt die Wahrscheinlich bei weit über 99%, dass der Verdächtige auch der Täter ist. Aufgabe: In der Abbildung ist das Ergebnis einer kriminalistischen Aufklärung einer Vergewaltigung zu sehen. Es wurden in diesem Fall beispielhaft nur zwei Mikrosatelliten per PCR amplifiziert und per Gelelektrophorese aufgetrennt. In Spur 1 wurden Speichelzellen des Opfers und in Spur 2 ein Vaginalabstrich des Opfers analysiert. (Frage: warum finden sich beim Opfer zwei Banden, beim Vaginalabstrich vier bzw. nur drei Banden?) In den Spuren 3-7 wurden Speichelzellen von 5 potentiellen Tätern analysiert. Wer ist am wahrscheinlichsten der Täter? Versuch 3.5: Sporenfärbung mit einer stationären und einer exponentiell wachsenden Bakterienkultur Untersuchungsobjekt ist: Bacillus subtilis, vegetative Zellen und Sporen Sie bekommen zwei Reagenzröhrchen mit 1ml Kulturen von B. subtilis. In der einen Kultur befinden sich die Bakterien in Nährmedium in der exponentiellen Wachstumsphase. In der zweiten Kultur befinden sie sich in Wasser. Streichen Sie jeweils einen Tropfen der Bakterienkulturen mit der Impföse auf einem Objektträger aus und führen Sie nach Hitzefixierung eine Sporenfärbung durch. Fertigen Sie eine Zeichnung an! Wie unterscheiden sich die beiden Kulturen? Hitzefixierung 1. Auf einen sauberen Objektträger einen kleinen Tropfen Bakterienkultur aufbringen. 2. Impföse ausglühen und abkühlen lassen. 3. Den Tropfen Bakterienkultur mit der ausgeglühten und abgekühlten Impföse
großflächig auf dem Objektträger ausstreichen.
Mikrosatellit 1 Mikrosatellit 2

Versuchstag 3
16
4. Ausstrich an der Luft trocknen lassen. 5. Objektträger mit der beschichteten Seite nach oben dreimal durch die Flamme des
Spiritusbrenners ziehen. 6. Färbung (siehe unten). Sporenfärbung 1. Auf den hitzefixierten Ausstrich Malachitgrün-Lösung entweder 10 Minuten bei
Zimmertemperatur einwirken lassen oder durch Erhitzen (Flüssigkeit dampft!) die Einwirkzeit auf ca. 1 Minute reduzieren.
2. Mit Wasser abspülen. 3. Safranin-Lösung 20 Sekunden einwirken lassen. 4. Mit Wasser abspülen. 5. Mit Filtrierpapier vorsichtig trocknen und mikroskopieren. Beurteilung Sporen sind grün, vegetative Zellen rot. Fragen zum Kurstag III: 1. Welchen Wirkort hat das Kanamycin bei Bakterien? 2. Was ist ein Antibiogramm und wozu benutzt man es? 3. Vervollständigen Sie die Tabelle zum Antibiogramm! 4. Warum töten ß-Lactam-Antibiotika wachsende grampositive Bakterienkulturen besser ab als gramnegative? 5. Warum sollen Antibiotika nur unter ärztlicher Kontrolle eingesetzt werden und nicht in Selbstmedikation? 6. Welches ist der häufigste Mechanismus für die Resistenz gegen Kanamycin und gegen ß-Lactam-Antibiotika? 7. Warum zeigen ß-Lactam-Antibiotika bei Mycoplasma Species keine bakterizide (bakterientötende) Wirkung? 8. Wie wirken die verschiedenen anderen Antibiotika? 9. Wozu verwendet man die Samtstempeltechnik? 10. Wie funktioniert die PCR (Polymerasekettenreaktion)? 11. Wie funktioniert ein genetischer Fingerabdruck? 12. Wie entstehen Endosporen und wodurch zeichnen sie sich aus?

Versuchstag 4
17
4. Viren/Phagen - Mutagenitätsnachweis Vorbereitung: - Viren - Bakteriophagen - Transduktion - Konjugation - Transformation - Mutationen - Mutationsreparaturmechanismen Versuch 4.1: Phagentiter-Bestimmung Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten Einleitung Phagen (Bakterienviren) sind subzelluläre Partikel von 50 - 100 nm Größe, die aus DNA (alternativ auch RNA) und Proteinen bestehen. DNA bzw. RNA beinhalten die genetische Information für die intrazelluläre Vermehrung des Phagen und außerdem mindestens ein für das Hüllprotein (Kapsid) kodierendes Gen. Große Phagen, wie z.B. die T-Phagen von E. coli oder der Phage KAPPA von Serratia marcescens, haben ein differenziertes Aussehen, ihre Proteine sind in Hülle und Infektionsfortsatz ("Kopf und Schwanz") organisiert. Sie können Bakterienzellen infizieren und lassen sich in ihnen vermehren. Bei der Infektion verbleiben die Proteinteile außen an der Zellwand, nur die DNA (bei RNA-Phagen: RNA) gelangt in das Zellinnere. Die injizierte Phagen-DNA (RNA) wird von den für diese Zwecke vorhandenen Einrichtungen der Wirtszelle vermehrt. Sie veranlaßt diese auch, neue Proteinhüllen mit Schwänzen zu produzieren und schließlich die Phagen-DNA darin zu verpacken, so dass viele neue infektionsfähige Phagen (Virions) entstehen. Diese werden durch Aufplatzen der Zelle (Lysis) freigesetzt, woran die Zelle zugrunde geht. Versuchsbeschreibung Der Phage Lambda ist ein temperenter Phage und kann nach der Infektion einer E. coli-Zelle entweder den lytischen Vermehrungszustand oder den lysogenen Zustand einschlagen. Der größte Teil der Phagen zerstört jedoch die E. coli-Zellen. Phagen vermehren sich also nicht auf Nährmedien sondern vermehren sich in Bakterien, die wiederum auf Nährmedien wachsen. Um also Phagen sichtbar zu machen oder ihre Anzahl zu bestimmen, lässt man ihre Wirtsbakterien auf Nährmedium wachsen und bestimmt dann die Zerstörung der Bakterien durch die Phagen. Ähnlich wie man Bakterien als Kolonien auf Nähragarplatten sichtbar machen kann, kann man Phagen als sogenannte Plaques sichtbar machen. Dazu lässt man eine Nähragarplatte mit einem Bakterienrasen bewachsen, wobei man die Bakterien zuvor mit den Phagen vermischt. Während der Inkubation bei 42°C zerstören die Phagen die Bakterien, vermehren sich und werden nachher als Löcher (=Plaques) im Bakterienrasen sichtbar. Jeder Phage bildet dann entsprechend einen Plaque.

Versuchstag 4
18
Durchführung :
Sie erhalten ein Phagenlysat des Phagen Lambda, dessen Titer Sie bestimmen sollen (Phagentiter = Anzahl der Phagen/ml). Dazu bekommen Sie eine Übernachtkultur von E. coli-Bakterien, mit denen Sie einen Bakterienrasen mittels der Weichagartechnik herstellen sollen. - Verdünnungsreihe für das Phagenlysat (bereits 10-6 vorverdünnt): bis 10-9 in Phagenpuffer (1 ml Phagen + 9 ml Puffer) - von den Verdünnungsstufen 10-7 bis 10-9 jeweils 300µl mit 300µl Übernachtkultur der E. coli (ca. OD600nm = 2) mischen - Inkubation: 10 min, 37°C-Wasserbad ohne Schütteln - mit je 10 ml LB-Weichagar ( 50°C- Wasserbad) versetzen, mischen und auf vorgewärmte LB-Platten ausgießen (nicht ausstreichen) - Inkubation: über Nacht bei 42°C, Plattenboden nach unten (nicht zu lange inkubieren, da E. coli die Plaques überwachsen können) Versuch 4.2: Mutagenitätsnachweis
Einleitung Mutationen, d.h. Änderungen der Basensequenz in der DNA und somit der genetischen Information, können durch energiereiche Strahlung, (UV-Licht, Röntgenstrahlen, Radioaktivität) oder durch recht verschiedene chemische Substanzen (wie salpetrige Säure, Epoxide, Alkylsulfonsäure-Ester, Hydroxylamin, Alkimine, Acridine, Loste, Aflatoxine etc.) ausgelöst werden. Solche Mutagene erhöhen die Häufigkeit der Mutationen über die spontane Mutationsrate oft um mehrere Zehnerpotenzen. Meist (besonders bei höheren Konzentrationen oder Dosen) sind solche genotoxischen Stoffe oder Strahlen nicht nur mutagen, sondern unmittelbar giftig, d.h. sie töten Zellen ab oder hemmen Lebensprozesse wie z.B. das Wachstum. Versuchsprinzip Sie sollen heute die mutagene Wirkung von UV-Strahlen bestimmen. Es werden Hefezellen, die durch Mutation, z.B. Punktmutation, in einem Gen nicht mehr in der Lage sind, eine bestimmte Aminosäure, nämlich Tryptophan, zu synthetisieren (sogenannte Mangelmutanten, siehe Auxotrophie), auf einen diese Aminosäure nicht enthaltenden Nährboden (Agar) aufgebracht. Da diese Hefen zur Vermehrung auf diese Aminosäure angewiesen sind, würden sie absterben bzw. könnten sich nicht auf diesem Mangelmedium vermehren. Nun setzt man die Hefen der UV-Strahlung aus. Bilden sich nach dem anschließenden Bebrüten Hefe-Kolonien, so sind einzelne Hefen gewachsen und haben die Fähigkeit zur Synthese der entsprechenden Aminosäure zurückerlangt. Es handelt sich hierbei um sogenannte Revertanten, bei denen die zur Auxotrophie führende Punktmutation in einem Gen rückgängig gemacht wurde - sie wurden wieder prototroph. Man geht davon aus, dass diese Rückmutation sehr wahrscheinlich der Wirkung des UV-Lichtes zuzuschreiben ist und es sich somit um eine mutagene Strahlung handelt, welche eine Punktmutation in einem Gen bewirkt.

Versuchstag 4
19
Durchführung - Je 0,5 ml Übernachtkultur Hefe mit 9,5 ml SCD/-Trp (0,5% Weichagar) mischen und auf eine SCD/-Trp-Platte giessen. - Agar fest werden lassen - Bestrahlung mit UV-Lampe : 0 sec (Negativkontrolle) 5 sec 10 sec 1 min 2 min - Platten sofort in Alu-Box (dunkel) stellen und 2 Tage bei 30°C inkubieren - pro Gruppe 2 Platten:
Negativkontrolle und eine Bestrahlungszeit (Zeiten auf die Bankreihen aufteilen , damit ein großes Bestrahlungsspektrum entsteht)
Fragen zu Kurstag IV:
1. Wie könnte man nachweisen, dass bei der Injektion nur Phagen-DNA und nicht Hüllprotein in die Zelle gelangt?
2. Was ist Lysogenie? Was ist ein temperenter Phage?
3. Was ist ein Plaque?
4. Nennen Sie mindestens fünf durch Viren ausgelöste Krankheiten des Menschen.
5. Beschreiben Sie das Prinzip des AMES-Tests.
6. Wie werden Mutationen ausgelöst?
7. Wie können DNA-Schädigungen repariert werden?
8. Nennen Sie die wichtigsten DNA-Reparatursysteme?
9. Warum verpackt man die UV-bestrahlten Hefen in einer dunklen Alu-Box und lässt sie nicht im Licht stehen?

Versuchstag 5
5. Genregulation - Stoffwechsel Vorbereitung: - Wachstum von Bakterien - Bakteriengenetik - Operon (Lactose-Operon von E. coli) - Genregulation - Enzymaktivität - Stoffwechselwege - Mikrobielle Gärungen In diesem Versuch sollen Sie erkennen, dass Zellen ökonomisch arbeiten (müssen). Bakterienzellen benötigen zu ihrem Wachstum eine Kohlenstoffquelle. In einem Vollmedium ist Kohlenstoff reichlich enthalten. In unserem Fall z.B. im Hefeextrakt und im Glycerin. Bei den früheren Versuchen haben wir Pepton bzw. Trypton - also partiell hydrolysiertes Eiweiß - verwendet. In allen Fällen erhält man ein gutes Zellwachstum. Die Bakterienzellen bilden eine große Population. Bei gegebenen Umweltbedingungen werden sich solche Individuen durchsetzen, die sich am schnellsten vermehren. Langsam wachsende Bakterien werden im Laufe der Zeit aus einer Population einfach herausverdünnt. Im Laufe der Jahrmillionen der Evolution sind dabei Unterschiede von millionstel von Sekunden in der Generationszeit entscheidend gewesen. Es ist daher nicht verwunderlich, dass Bakterien (und andere Organismen) vielerlei Regelmechanismen entwickelt haben, die es ihnen ermöglichen, mit der zur Verfügung stehenden Energie ausgesprochen wirtschaftlich umzugehen. Eine Bakterienzelle ist in der Lage, für die unterschiedlichen Reaktionen des Stoffwechsels eine Vielzahl spezifischer Enzyme zu synthetisieren. Für alle diese Enzyme gibt es je einen Genabschnitt, der die Information zur Bildung dieses Enzyms von einer Generation auf die nächste weitergibt. Aber selbst wenn eine Information vorliegt, darf nicht außer acht gelassen werden, dass zu ihrer Realisation Zeit und Energie benötigt werden. D.h. würde eine Bakterienzelle alle Enzyme bilden, die es potentiell bilden könnte, so wäre seine Vermehrungsrate sehr gering, es würde unnötig viel Energie verschwenden und somit sparsameren Zellen gegenüber ins Hintertreffen geraten. Bakterien der Art Escherichia coli können Milchzucker (Lactose) abbauen. Das geschieht durch das Enzym β-Galactosidase. Es gibt ein weiteres Enzym, die Galactosidtransferase, welches die Aufnahme der Lactose durch die Zellmembran hindurch beschleunigt. Da wir die Bakterien in der Regel in einem lactosefreien Medium kultivieren, wäre die Bildung dieser Enzyme unökonomisch. Andererseits müssten die Bakterien in der Lage sein, die beiden Enzyme umgehend zu bilden, sobald sie in ein lactosehaltiges Medium geraten. Durch einen heute recht gut verstandenen Regelmechanismus ist E. coli tatsächlich in der Lage, die Synthese von Galactosidtransferase (LacY), β-Galactosidase (LacZ) und einem dritten Enzym (LacA) anzukurbeln oder zu drosseln. Es handelt sich hierbei um eines der einfachsten und experimentell bestuntersuchten Systeme einer Genregulation, das lac-Operon. Das Genprodukt von lacI, der "Repressor", reagiert mit dem Operator o des lac-Operons, wodurch die Synthese der Genprodukte der Gene lacZ, lacY und lacA blockiert wird. Lactose inaktiviert den Repressor, so dass nunmehr das lac-Operon transkribiert wird und β-

Versuchstag 5
21
Galactosidase und Galactosidtransferase gebildet werden können. Andererseits funktioniert das nur in der Abwesenheit von Glucose, da Glucose mit Hilfe eines anderen Mechanismus (cAMP, CAP) die Transkription des lac-Operons blockiert. Wenn dieses Modell, bekannt als JACOB- MONOD-Modell, richtig ist, müsste unser heutiger Versuch auch klappen. Wir wollen heute zeigen, dass 1. die Bakterien in einem Medium, welches Lactose enthält, das Enzym β-Galactosidase bilden können. 2. bei Zellen, denen Glucose angeboten wird, dieses Enzym nicht nachweisbar ist. 3. bei Zellen, denen sowohl Lactose als auch Glucose angeboten wird, dieses Enzym auch nicht gebildet wird Wie weisen wir das Enzym nun nach? Der direkte experimentelle Nachweis der Lactosespaltung ist recht umständlich. Wir bedienen uns deshalb eines besonderen Testsystems. Man weiß nämlich, dass β-Galactosidase nicht nur Lactose, sondern auch andere Verbindungen spaltet, die eine beta -glykosidische Bindung enthalten. So wird z. B. o - Nitrophenyl -beta - D - Galactosid (ONPG), eine unphysiologische, in der Natur nicht vorkommende Substanz, gespalten. Dabei entsteht o - Nitrophenol. ONPG ist farblos, o- Nitrophenol ist gelb. Damit kann der Ablauf der Hydrolyse und somit die spezifische Enzymaktivität einfach an der zunehmenden Gelbfärbung der Reaktionsflüssigkeit verfolgt werden. Bevor wir jedoch eine solche Reaktion verfolgen können, müssen wir das Enzym aus den Zellen herausholen, damit es mit dem Substrat reagieren kann. Wir behandeln deshalb die Bakterien mit dem Detergenz Natriumdodecylsulfat (SDS). SDS schädigt (durchlöchert) die Zellmembran, so dass die β-Galactosidase ausläuft bzw. das ONPG in die Zellen eindringen kann. Versuch: Wir lassen E. coli Bakterien in 3 verschiedenen Medien (mit Lactose oder Glucose oder Glucose/Lactose) wachsen. Nach bestimmten Zeiten (0 - 60 min) entnehmen wir aus den Kulturröhrchen jeweils Proben: a ) mit Lactose (nach 0, 20, 40, 60 min) b ) mit Glucose (0 und 60 min) c) mit Glucose und Lactose (0 und 60 min) Man behandelt diese Proben jeweils bei 37°C mit SDS und ONPG solange bis eine leichte Gelbfärbung eintritt, längstens jedoch für 20 min, stoppt dann durch Zugabe von Natriumcarbonat ab und misst dann im Photometer die Extinktion E (=Absorption, Optische Dichte) bei 420 nm Wellenlänge. Die Stärke der Gelbfärbung bzw. die Extinktion bei 420 nm ist ein Maß für die Menge an β-Galactosidase und damit der Expression des lac-Operons. Um eine quantitative Beurteilung zu ermöglichen, berechnen wir aus den ermittelten Werten von E(420nm) und der Bakterienmenge, die im Photometer als Optische Dichte (OD) bei 550 nm Wellenlänge bestimmt wird, die jeweiligen spezifischen Enzymaktivitäten (U/OD550nm) und vergleichen, inwieweit diese durch die unterschiedlichen Zucker induziert werden.

Versuchstag 5
22
Gemeinsames Arbeiten von Gruppen zu je 4-6 Studenten Experimentelle Durchführung Reagenzgläser wie folgt beschicken Ansatz A: 4,5 ml Nährlösung 1,5 ml Bakteriensuspension 1,5 ml Lactoselösung Ansatz B: 3 ml Nährlösung 1 ml Bakteriensuspension 1 ml Glucoselösung Ansatz C: 3 ml Nährlösung 1 ml Bakteriensuspension 1 ml Glucose-/Lactose Mischung - Ansätze gut mischen Die Ansätze werden dann bei 37°C im Rollschüttler oder Wasserbad inkubiert. Zu folgenden Zeiten werden jeweils 2 Proben entnommen, und zwar je 1 ml und 0,5 ml: Ansatz A (0, 20, 40, 60 min) Ansatz B und C (0 und 60 min)
1. Nachweis der Menge an Bakterien und ihrer Vermehrung Zur Bestimmung der Menge an Bakterien werden die 0,5 ml Proben direkt in Küvetten pipettiert und die Trübung der Proben (=Optische Dichte, OD) (unverdünnt) im Photometer bei lambda = 550 nm gemessen. (Die Nährlösung dient als Leerwert/Referenzwert).

Versuchstag 5
23
2. Test auf enzymatische Aktivität: Die 1 ml Proben werden zum Nachweis der enzymatischen Aktivität eingesetzt. Als Referenzwert/Leerwert wird statt Probe 1 ml Nährlösung eingesetzt: Probe 1 ml Enzymtestpuffer (Phosphatpuffer/SDS/ONPG) 1 ml Dann Stoppuhr direkt starten Inkubationszeit : 1-20 min. bei 37°C (im Wasserbad) (bis leichte Geldfärbung zu sehen ist) Dann: die Reaktion wird durch Zugabe von 1 M Na2CO3 1 ml abgestoppt Stoppuhr stoppen (Na2CO3 macht den Reaktionsansatz alkalisch und im alkalischen Bereich wird die β-Galactosidase inaktiviert) Messung der Extinktion bei 420 nm Tragen Sie Ihre Messergebnisse in die folgende Tabelle ein und berechnen Sie die spezifischen Enzymaktivitäten: Bakterienmenge Aktivität Inkubationszeit spez.Enzymaktivität Lactose min OD550nm E420nm min 0 20 40 60 Glucose min OD550nm E420nm min 0 60 Lactose/Glucose min OD550nm E420nm min 0 60

Versuchstag 5
24
Die Berechnung der spezifischen Enzymaktivitäten erfolgt mit der Formel:
E = ε x c x d
die nach c aufgelöst wird und durch die Zeit der Inkubation in Minuten (t) und die OD550nm dividiert wird. Außerdem wird die Probenverdünnung im Reaktionsansatz berücksichtigt (1 ml Probe wird durch den Puffer und das Natriumcarbonat auf 3 ml verdünnt):
spezifische Enzymaktivität (mU/OD550) = E420nm x (V/v) / (t x ε x d x OD550nm)
E = zu messende Extinktion (420 nm) der Probe, V/v = Gesamtvolumen/Probevolumen = 3,
t = Inkubationszeit Enzymtest
ε420nm(ONP) = 4,5 x 10-3 mL x nmol-1 x cm-1, d = Schichtdicke der Küvetten (1 cm),
1mU = 1 nmol/(mL x min) (U = Unit = Enzymeinheit)
Skizzieren Sie im Diagramm die jeweilige Veränderung der spezifischen Enzymaktivitäten bei Wachstum auf den verschiedenen Zuckern, die ein Maß für die Expression des lac-Operons ist:
Zeit
spe
z. E
nzy
ma
kti
vit
ät





























