Embed Size (px)
Citation preview

AMPA Receptor Antibodies in LimbicEncephalitis Alter Synaptic
Receptor LocationMeizan Lai, MD,1 Ethan G. Hughes, BS,2 Xiaoyu Peng, BS,2 Lei Zhou, BS,1 Amy J. Gleichman, BS,2
Huidy Shu, MD, PhD,3 Sabrina Mata, MD,4 Daniel Kremens, MD, JD,5 Roberta Vitaliani, MD,6
Michael D. Geschwind, MD,3 Luis Bataller, MD,7 Robert G. Kalb, MD,8 Rebecca Davis, BA,1
Francesc Graus, MD,9 David R. Lynch, MD, PhD,1,8 Rita Balice-Gordon, PhD,2
and Josep Dalmau, MD, PhD1
Objective: To report the clinical and immunological features of a novel autoantigen related to limbic encephalitis (LE) and theeffect of patients’ antibodies on neuronal cultures.Methods: We conducted clinical analyses of 10 patients with LE. Immunoprecipitation and mass spectrometry were used toidentify the antigens. Human embryonic kidney 293 cells expressing the antigens were used in immunocytochemistry andenzyme-linked immunoabsorption assay. The effect of patients’ antibodies on cultures of live rat hippocampal neurons wasdetermined with confocal microscopy.Results: Median age was 60 (38–87) years; 9 were women. Seven had tumors of the lung, breast, or thymus. Nine patientsresponded to immunotherapy or oncological therapy, but neurological relapses, without tumor recurrence, were frequent andinfluenced the long-term outcome. One untreated patient died of LE. All patients had antibodies against neuronal cell surfaceantigens that by immunoprecipitation were found to be the glutamate receptor 1 (GluR1) and GluR2 subunits of the �-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). Human embryonic kidney 293 cells expressing GluR1/2 re-acted with all patients’ sera or cerebrospinal fluid, providing a diagnostic test for the disorder. Application of antibodies tocultures of neurons significantly decreased the number of GluR2-containing AMPAR clusters at synapses with a smaller decreasein overall AMPAR cluster density; these effects were reversed after antibody removal.Interpretation: Antibodies to GluR1/2 associate with LE that is often paraneoplastic, treatment responsive, and has a tendencyto relapse. Our findings support an antibody-mediated pathogenesis in which patients’ antibodies alter the synaptic localizationand number of AMPARs.
Ann Neurol 2009;65:424–434
Limbic encephalitis (LE) is an inflammatory disorderthat predominantly affects the gray matter of the me-dial temporal lobes, amygdala, and orbitofrontal cor-tex.1 As a result, patients experience short-term mem-ory deficits, emotional and behavioral disturbances,seizures, and sometimes dementia. Until recently, mostcases of LE were thought to be paraneoplastic, medi-ated by immune responses against intracellular anti-
gens, and poorly responsive to treatment.2 However,the clinical and immunological spectra of LE are farmore extensive than initially considered, and recentstudies demonstrated a larger category of allied disor-ders in which the antigens are on the cell surface.3,4
For example, a series of 45 patients with LE demon-strated that 64% had antibodies against neuronal cellsurface antigens; of these, 45% were the voltage-gated
From the 1Department of Neurology, University of Pennsylvania;2Department of Neuroscience, University of Pennsylvania, School ofMedicine, Philadelphia, PA; 3Department of Neurology, Memory& Aging Center, University of California, San Francisco MedicalCenter, San Francisco, CA; 4Department of Neurological and Psy-chiatric Sciences, University of Florence, Florence, Italy; 5Depart-ment of Neurology, Jefferson Medical College of Thomas JeffersonUniversity, Philadelphia, PA; 6Department of Neurology, TrevisoRegional Hospital, Treviso, Italy; 7University Hospital La Fe, Va-lencia, Spain; 8Department of Pediatrics, Division of NeurologyChildren’s Hospital of Philadelphia, Philadelphia, PA; and 9Serviceof Neurology, Institut d’Investigacio Biomedica August Pi i Sunyer,Hospital Clinic, Barcelona, Spain.
Address correspondence to Dr Dalmau, Division Neuro-Oncology,Department of Neurology, 3 W. Gates, University of Pennsylvania,
3400 Spruce Street, Philadelphia, PA 19104. E-mail:[email protected]
M.L., E.G.H., and X.P. have contributed equally to this work.
Potential conflict of interest: Nothing to report.
Received Sep 5, 2008, and in revised form Oct 14. Accepted forpublication Oct 31, 2008.
Published in Wiley InterScience (www.interscience.wiley.com).DOI: 10.1002/ana.21589
Additional Supporting Information may be found in the online ver-sion of this article.
424 © 2009 American Neurological Association

potassium channels and 55% other antigens, most un-known.5 Given that disorders with antibodies to cellsurface autoantigens are often treatment responsive,characterization of the antigens is important to facili-tate a prompt and specific diagnosis.3,5 In addition, theimmune response provides a link between the functionof the antigens and neuronal events involved in mem-ory, learning, cognition, and seizures. For example, an-tibodies to NR1/NR2 heteromers of the N-methyl-D-aspartate (NMDA) receptor associate with a treatableencephalitis characterized by psychosis, memory defi-cits, seizures, dyskinesias, and autonomic instability.6
This study describes a novel cell surface autoantigenin 10 patients with LE. Our findings are relevant forthree reasons; (1) the antigen is the �-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor(AMPAR), a subtype of glutamate receptor that under-lies mechanisms of memory, learning, and seizures; (2)application of patients’ antibodies into cultures of liverat hippocampal neurons altered the number of AM-PAR clusters at synapses, supporting an antibody-mediated pathogenesis of the LE; and (3) we describethe associated syndrome and provide a diagnostic testfor this disorder.
Patients and MethodsThe 10 patients of this study were identified among 109cases who fulfilled strict criteria of LE2 seen at the Hospitalof the University of Pennsylvania or whose sera, cerebrospi-nal fluid (CSF), or tissues were referred for immunologicalstudies from July 2004 to July 2008. Thirty-nine cases hadantibodies to known antigens (16 voltage-gated potassiumchannels, 7 NMDA receptor, 6 glutamic acid decarboxylase[GAD], 4 Hu, 4 Ma2, 1 amphiphysin, and 1 CV2/collaps-ing response mediator protein-5 [CRMP5]), 43 had antibod-ies to unknown antigens, and 27 had no antibodies. Amongthe 43 patients with antibodies to unknown antigens, 10were selected for having antibodies with a similar pattern ofreactivity with the neuropil of brain and cerebellum. Nine ofthese 10 patients were seen by the authors (4 by J.D., 5 byother authors), and the information of 1 was provided by thereferring physician. The clinical features of four patients havebeen partially reported.4,5 In addition to the above cases, se-rum or CSF of 110 individuals served as control samples.These included 20 patients with anti-NMDA receptor en-cephalitis, 20 LE and voltage-gated potassium channel anti-bodies, 20 paraneoplastic encephalitis and antibodies to in-tracellular antigens (Hu, Ma2, CV2/CRMP5, oramphiphysin), 10 patients with Rasmussen’s encephalitis, 10patients with lupus erythematosus, 10 patients with small-cell lung cancer without paraneoplastic syndromes, 10 pa-tients with thymoma without LE, and 10 blood donors.
Information on clinical antibody testing, animal tissueprocessing, IgG biotinylation, immunohistochemistry on tis-sue and cultures of live rat hippocampal neurons, immuno-cytochemistry on human embryonic kidney 293 (HEK293)cells, and enzyme-linked immunoabsorption assay is pro-vided in the Supplemental Materials and Methods. Studies
were approved by the University of Pennsylvania Institu-tional Review Board.
Immunoprecipitation and ImmunoblotRat hippocampal neuronal cultures were prepared as reportedpreviously.7 Live neurons grown in 100mm wells (density106 neurons/well) were incubated at 37°C with filtered pa-tient’s serum (diluted 1:500) for 1 hour. Neurons were thenwashed with phosphate-buffered saline, lysed with buffer(NaCl 150mM, EDTA 1mM, tris(hydroxymethyl)amin-omethane [Tris]-HCl 100mM, deoxycholate acid 0.5%, 1%Triton X-100 [Sigma Labs, St. Louis, MO], pH 7.5) con-taining protease inhibitors (P8340; Sigma Labs), and centri-fuged at 16.1 � 103 g for 20 minutes at 4°C. The superna-tant was retained and incubated with protein A/G agarosebeads (20423; Pierce, Rockford, IL) overnight at 4°C, cen-trifuged, and the pellet containing the beads with patients’antibodies bound to the target cell surface antigens was thenwashed with phosphate-buffered saline, aliquoted, and keptat �80°C. An aliquot of this pellet was resuspended in Lae-mmli buffer, boiled for 10 minutes, separated in a 4 to 15%sodium dodecyl sulfate polyacrylamide gel electrophoresis,and the proteins visualized with EZBlue gel staining (G1041;Sigma Labs). Distinctive protein bands precipitated by pa-tients’ sera were excised from the gel and analyzed usingmass spectrometry at the proteomic facility at the Universityof Pennsylvania.
After characterization of the antigens, frozen aliquots ofthe indicated pellets were separated in a sodium dodecyl sul-fate polyacrylamide gel electrophoresis as described earlier,transferred to nitrocellulose (162-0115; Bio-Rad, Hercules,CA), and blotted with the indicated GluR1 (1:1,000;Chemicon, Temecula, CA) or GluR2/3 (1:200; Chemicon)antibodies.
Quantitative Analysis of AMPA Receptor ClustersUsing Confocal MicroscopyTo determine the degree of immunolabeling of AMPAR bypatients’ antibodies, we exposed 14-day in vitro (div) live rathippocampal neurons to patient’s CSF and a rabbit poly-clonal antibody against GluR1 or GluR2/3, washed, fixed,and incubated with the appropriate fluorescent-conjugatedsecondary antibodies (see Supplemental Methods section Im-munocytochemistry Using Live Rat Hippocampal Neurons).Images were obtained using a laser-scanning confocal micro-scope (Leica TCS SP2; Leica, Deerfield, IL). For each image,laser light levels and detector gain and offset were adjusted sothat no pixel values were saturated. Images were thresholded,and the number of individual clusters along neuronal den-drites was determined using interactive software (Meta-Morph; Universal Imaging, West Chester, PA; or ImageJ).8
To determine the effects of patients’ antibodies on thenumber and localization of AMPAR clusters, we treated neu-rons with patient or control CSF (1:15 dilution in Neuro-Basal � B27 medium; GIBCO, Carlsbad, CA) from 11 to17 div. Every 3 days, 20 of the 300�l medium in each cul-ture well was removed and replaced with 20�l fresh patientor control CSF. In another series of experiments, neuronswere treated with patient CSF from 11 to 14 div followed bytreatment with control CSF from 14 to 17 div. On 14 or 17
Lai et al: Anti-AMPA Receptor Encephalitis 425

div, neurons were fixed in freshly made paraformaldehyde(4% paraformaldehyde, 4% sucrose in phosphate-bufferedsaline) for 5 minutes, permeabilized in 0.25% Triton X-100for 10 minutes, and blocked in 5% normal goat serum for 1hour. Neurons were then incubated with patient’s CSF (1:15dilution in Triton X-100), a rabbit polyclonal antibodyagainst GluR1 (1:1,000; Chemicon) or GluR2/3 (1:1,200;Chemicon), a mouse monoclonal antibody against PSD-95(1:500; Affinity BioReagents, Golden, CO), or a guinea pigpolyclonal antibody against VGlut (1:1,000, polyclonal;Chemicon) for 2 hours, followed by the appropriatefluorescent-conjugated secondary antibodies (Jackson Immu-nologicals, West Grove, PA). Images were obtained and an-alyzed as described earlier.
StatisticsDifferences in antibody titers among groups were analyzedusing the Mann–Whitney U test. The effects of IgG andCSF on neuronal cultures were analyzed using the Kruskal–Wallis nonparametric analysis of variance followed byDunn’s pairwise comparison.
ResultsClinical FeaturesDemographic information, symptoms, diagnostic tests,treatment, and outcome are shown in Tables 1 and 2.Patient median age was 60 years (range, 38–87 years);nine were women. Nine patients presented with sub-acute (�8 weeks) confusion, disorientation, and mem-ory loss, classic of LE; one patient presented with a4-month history of progressive memory loss, behavioralchange, and agitation that initially suggested a rapidlyprogressive dementia. Four patients had seizures, oneof them at the relapse of the disorder 16 months afterthe first episode of encephalitis. The neurological ex-amination was always consistent with an encephalopa-thy predominantly involving the limbic system.
The CSF showed lymphocytic pleocytosis in ninepatients (median white blood cell count, 24/�l; range,6–75/�l;). The brain magnetic resonance imagingdemonstrated increased fluid-attenuated inversion re-covery signal involving the medial temporal lobes ineight of nine patients examined. Two patients had ad-ditional abnormalities in the anterior septal nuclei, onewith several cortical areas of transiently increased signalon fluid-attenuated inversion recovery imaging, and theother with transiently increased signal in the cerebel-lum.
Seven patients had an underlying neoplasm, 5 ofthem diagnosed by the time of the initial episode of LEand 2 at first relapse of LE. The other three patientshad extensive studies (two combined body computedtomography/fluorodeoxyglucose positron emission to-mography, and one computed tomography of thechest, abdomen, and pelvis, along with serologic tumormarkers, and ultrasound of the breast) without show-ing a neoplasm (median follow-up, 16 months; range,
8–50 months). Five patients had a history or concur-rent findings of systemic autoimmunity (see Table 2;see Supplemental Results section Concurrent Autoim-mune Findings).
Nine patients received immunotherapy; six of themhad an underlying tumor and also received oncologicaltherapy. All nine patients responded to the indicatedtreatments at the first episode of encephalitis. Five pa-tients had one to three neurological relapses between 2and 101 months (median, 16 months) after the firstepisode of encephalitis. None of the seven patientswith tumors experienced development of tumor recur-rences.
At relapse of LE, four of five patients responded totreatment, but the response was always partial, and onepatient died after a prolonged episode of status epilep-ticus. Among the four patients with a single episode ofencephalitis, two returned to their baseline activities,one had partial improvement, and one returned tomost of her baseline activities but had residual hyper-somnia and depression for several months. This patienthad small-cell lung cancer and died 7 months afterneurological recovery; the cause of death was a cardiacdysrhythmia caused by severe coronary atherosclerosis(see Supplemental Results: Autopsy Findings, Case 9).
One patient was not treated and died of LE. She wasa 44-year-old woman who experienced development ofa rapidly progressive encephalitis resulting in suddendeath. The final diagnosis was LE with thymoma andoverlapping GluR1/2 and CV2/CRMP5 antibodies(see Supplemental Results: Autopsy Findings, Case 5).
Identification of Antibodies to Cell Surface AntigensUsing immunohistochemistry of rat brain, we foundthat all 10 patients’ serum and CSF showed intensereactivity with the neuropil of the hippocampus, sub-iculum, caudate-striatum, and molecular layer of thecerebellum; there was substantially less reactivity withother areas of the brain, cerebellum, and brainstem(Fig 1). In cultures of live, nonpermeabilized, rat hip-pocampal neurons, all patients’ serum and CSF pro-duced intense immunolabeling of the cell surface andneuronal processes (Fig 2A). Further analysis of this re-activity using confocal microscopy demonstrated thatthe target antigens were distributed in a punctuatemanner along dendrites (see Fig 2B).
Three patients had additional antibodies to previ-ously characterized antigens (see Table 2): one hadGAD antibodies, another had CV2/CRMP5 antibod-ies, and another had SOX1 and voltage-gated calciumchannel antibodies without evidence of Lambert–Eatonmyasthenic syndrome. In the latter patient, SOX1 an-tibodies predicted the presence of a small-cell lung can-cer.
426 Annals of Neurology Vol 65 No 4 April 2009

Table 1. Clinical Features, Cerebrospinal Fluid, Electroencephalographic, and Magnetic Resonance Imaging Findings
CaseNo.a
Sex/Age(yr)
Symptom Presentation CSF Initial EEG Initial Brain MRI(FLAIR)
GluR1/2Antibodies
(Main Antigen)
14 F/65 Initial episode and relapses: short-term memory loss, confusion,agitation, aggressive behavior,confabulation. Transientdownbeat nystagmus
30 WBC,protein 97,positive OBand ISAb
Normal,repeatedtwice
Mild increased signalin medial temporallobes
GluR1/2-positive (GluR1)
24 F/44 Initial episode and relapses: short-term memory loss; confusion,combativeness; focal motorseizures; right beating nystagmus
44 WBC,protein 91,no OB,positive ISAb
Diffuse thetaactivity;episodes ofepilepticactivity in lefttemporal lobe
Initial MRI normalFollow-up 5 dayslater: mild temporallobe increased signal(right�left)
GluR1/2-positive (GluR2)
34 M/38 Initial episode and relapse: short-term memory loss, confusion,agitation, perseverationgeneralized tonic-clonic seizures
7 WBC,protein 50,positive OBand ISAb
NA Increased signal in theright medial andlateral temporal lobe,right frontal, leftinsular and leftoccipital regions
GluR1/2-positive (GluR2)
4 F/64 Increased seizures, confusion,disorientation, lethargy, short-term memory loss
75 WBC,protein 79
Slow activityin the righttemporalregion; noepilepticactivity
Postoperative changesin the right temporallobe; new increasedsignal in the leftmedial temporal lobe
GluR1/2-positive (GluR2)
55 F/44 Confusion, behavioral change,hypersomnia, progressiveunresponsiveness; mild gaitunsteadiness, low-grade fever
15 WBC,normalprotein
NA NAb CT normal GluR1/2-positive(GluR2�R1)
6 F/38 Short-term memory loss,confabulation
6 WBC,normalprotein
Normal Increased signal inmedial temporal lobes,left septal nucleus, leftcerebellum
GluR1/2-positive (GluR2)
7 F/87 Initial episode: short-termmemory loss, disorientation Atrelapse: memory loss, generalizedtonic-clonic seizures
Initiallynormal;protein 50 atrelapse
Diffuse slowactivity (7-8c/sec), deltaactivity inanteriorfrontotemporalareas
Increased signal inmedial temporal lobes;mild transient contrastenhancement in theleft hippocampus
GluR1/2-positive (GluR1)
8 F/61 Initial episode and relapse: short-term memory loss, decreased levelof consciousness
24 WBC,protein 420
Theta activityin posteriortemporalregions
Normal MRI GluR1/2-positive (GluR2)
9 F/59 Progressive memory loss,behavioral change, agitation,confabulation (4 months); milddysdiadochokinesia
17 WBC,protein 51,positive OB
Bilateralsharp wavesin temporallobes; noseizures
Increased signal inmedial temporal lobes,and medialorbitofrontal region
GluR1/2-positive (GluR1)
10 F/67 Confusion, combativeness,insomnia, hallucinations, short-term memory loss
32 WBC,normalprotein, noOBs
Sharp wavesin temporallobes
Mild temporal lobeincreased signal
GluR1/2-positive (GluR2)
Cerebrospinal fluid (CSF) reference values: white blood cell count (WBC) � 4/�l; protein level: 16-46 mg/dl. All patients had normalglucose levels in the CSF.aThe initial clinical features of Patients 1, 2, and 3, were described in detail elsewhere4 (Cases 2, 5, and 6); the long-term follow-up isprovided here. Patient 5 corresponds to Case 4 of Tables 1 and 2 in Graus and colleagues.5bLimbic encephalitis confirmed at autopsy.EEG � electroencephalogram; MRI � magnetic resonance imaging; FLAIR � fluid-attenuated inversion recovery; GluR1/2 �glutamate receptors 1 and 2; OB � oligoclonal bands; ISAb � intrathecal synthesis of antibodies; NA � not available.
Lai et al: Anti-AMPA Receptor Encephalitis 427

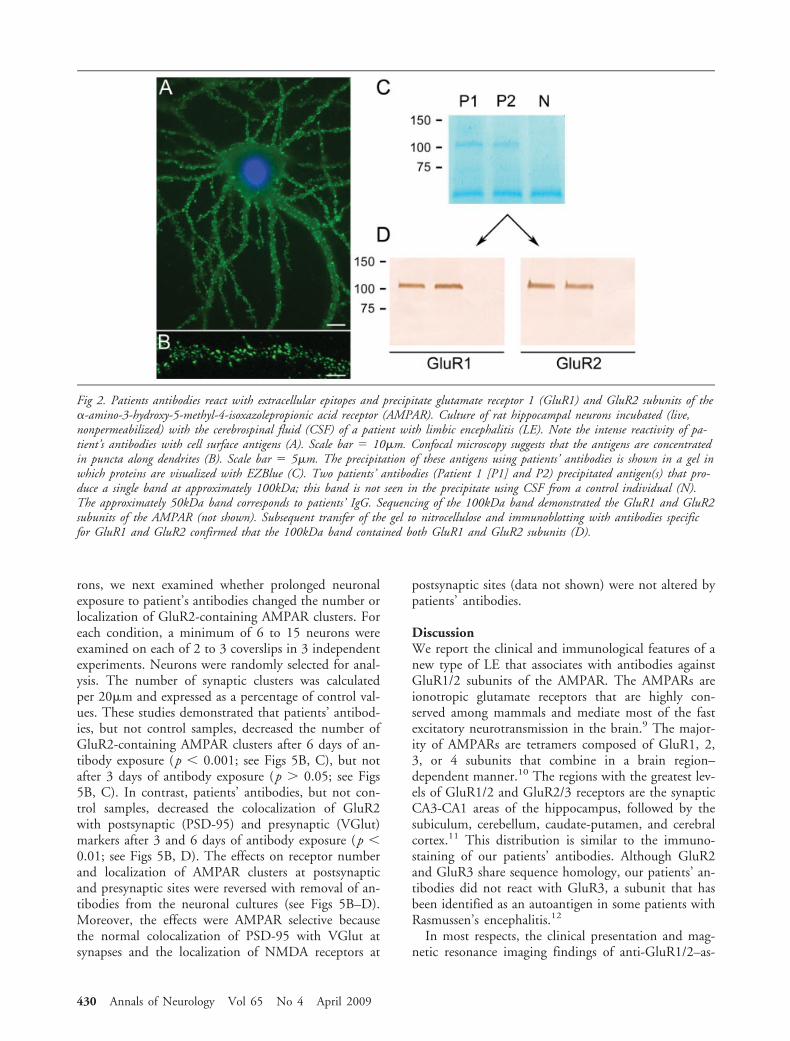
Cell Surface Synaptic Antigens Are the GlutamateReceptors 1 and 2 Subunits of the AMPA ReceptorTo identify the cell surface antigens, we incubated liverat hippocampal neurons with patients’ sera and theantigens immunoprecipitated. These studies produceda distinctive protein band of approximately 100kDa(see Fig 2C) that when analyzed by mass spectrometrycontained sequences derived from the GluR1 andGluR2 subunits of the AMPAR. Because GluR1 andGluR2 have similar molecular weight and share se-quence homology, we further examined whether theprecipitated band contained GluR1, GluR2, or bothsubunits, using Western blot with antibodies specificfor each subunit. These studies confirmed that the
band precipitated by patients’ antibodies containedboth GluR1 and GluR2 subunits (see Fig 2D).
Because GluR1 and GluR2 coassembled in neuronsand immunoprecipitated together, the primary epitopescould be located on either or both subunits. Thus, weexamined which of the two subunits contained the mainepitopes recognized by patients’ antibodies usingHEK293 cells transfected with single subunits (GluR1or GluR2). Nontransfected HEK cells and cells trans-fected with GluR3 were used as controls. Six patientshad antibodies against GluR2, three against GluR1, andone against both GluR1 and GluR2. None of the pa-tients’ antibodies reacted with GluR3 (see Supplemen-tary Fig S1). All patients’ antibodies reacted with cells
Table 2. Tumor Association, Other Autoimmune Features, Treatment, and Outcome
CaseNo.
Tumor(antigen)
Time fromSymptomsof LE toTumor
Diagnosis
Other AutoimmuneDisorders orAntibodies
Treatment Number ofRelapses/Intervalbetween
Presentationand LastRelapse
Outcome (follow-up in months)
14 — — — At presentation: plasmaexchange, corticosteroidsAt relapse: IVIg,corticosteroids; chronictreatment withazathioprine
3/7 months First episode: returned tobaseline; subsequent relapsing-remitting behavioral problem andmemory deficit; residual stabledeficits after third relapse (50)
24 Thymiccarcinoma(GluR1/2)
Concurrentwith firstepisode ofencephalitis
ANA, dsDNA,cardiolipin antibodies
Tumor removal Atpresentation and relapses:IVIg, corticosteroids;chronic treatment withazathioprine
3/101 months First episode: returned tobaseline; subsequent relapsing-remitting memory deficit Residualshort-term memory deficit afterthird relapse (120)
34 Malignantthymoma(GluR2)
Concurrentwith relapseofencephalitis
Stiff-person syndrome,diabetes mellitus, GADantibodies
Tumor removal, radiationtherapy; corticosteroids,plasma exchange, IVIg
1/60 months First episode: returned tobaseline; mild residual memorydeficit after relapse; steroid-dependant muscle spasms andrigidity (36)
4 Non-SCLC(N/A)
Concurrentwith firstepisode ofencephalitis
Chronic seizures causedby cortical dysplasia(confirmed by surgery)
Tumor removal;corticosteroids
— Returned to baseline (8)
55 Thymoma(N/A)
Concurrentwith firstepisode ofencephalitis
CV2/CRMP5antibodies
— (1 atypical)a; 24months
Unexpected dead,cardiorespiratory arrest (0.5);autopsy results included insupplemental material
6 — — — IVIg, corticosteroids — Returned to baseline (8)
7 — — ANA, hypothyroidism, Corticosteroids 1/16 months First episode: partial improvementfollowed by progressivedeterioration; died at relapse afterstatus epilepticus (16)
8 Breastcancer(GluR1/2)
Concurrentwith relapseofencephalitis
Hypothyroidism At presentation:corticosteroids At relapse:tumor removal, plasmaexchange andcorticosteroids
1/9 months First episode and relapseresponded to corticosteroids andplasma exchange; last follow-up:residual short-term memory lossand behavioral problems (28)
9 SCLC(GluR1)
6 months Raynaud’s syndrome,�ANA speckledpattern (1:160); VGCCand SOX1 antibodies
Tumor removal,chemotherapy;corticosteroids, IVIg
— Returned to baseline; died ofmyocardial infarction (15)Autopsy results in includedsupplemental material
10 Breastcancer(N/A)
Concurrentwith firstepisode ofencephalitis
— Tumor removal, radiationtherapy, corticosteroidschemotherapy (includingcyclophosphamide), IVIg
— Rapid recovery of memory; mildpersistent depression, apathy, andreduced verbal fluency (3)
aAtypical: episode of confusion, hallucinations, of unclear cause, attributed to a “psychotic break”; resolved spontaneously 2 years beforethe diagnosis of glutamate receptors 1 and 2 (GluR1/2)–associated limbic encephalitis (LE).IVIg � intravenous immunoglobulin; SCLC � small-cell lung cancer; VGCC � voltage-gated calcium channel; ANA � antinuclearantibody; dsDNA � double stranded DNA; GAD � glutamic acid decarboxylase; CRMP5 � collapsin response mediator protein-5.
428 Annals of Neurology Vol 65 No 4 April 2009

cotransfected with both GluR1 and GluR2 (GluR1/2)(Figs 3A–C; see Table 1). None of the 110 control pa-tients’ samples produced the neuropil reactivity shownin Figure 1 or reacted with cells transfected with GluR1or GluR2 (see Supplementary Fig S1).
Taken together, these findings establish the presenceof antibodies to GluR1/2 in this subgroup of patientswith LE, and provide a diagnostic test for this disorder.We next developed an enzyme-linked immunoabsorp-tion assay–based on lysates of HEK293 cells expressingGluR1/2, and this proved to be useful for the quanti-fication of antibodies (see Figs 3D–G). Analysis ofGluR1/2 antibodies using normalized amounts of IgGfrom paired serum and CSF samples indicated that alltested patients had intrathecal synthesis of antibodies(see Fig 3F).
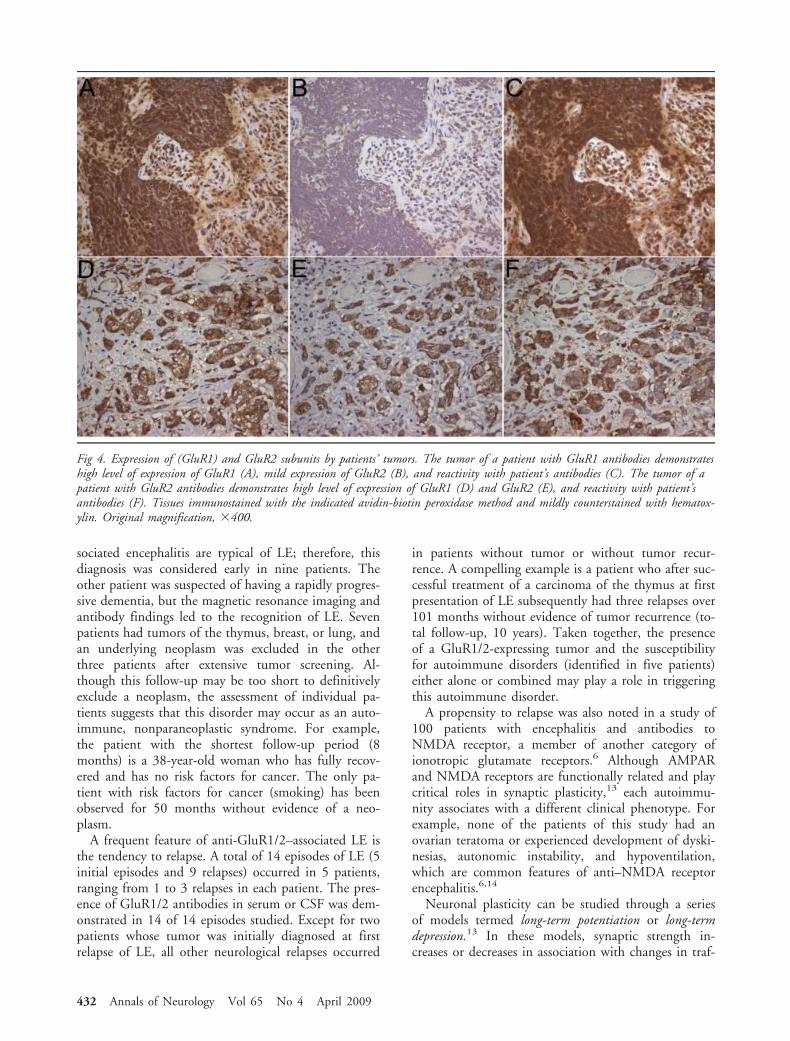
After identifying the GluR1/2 subunits as the targetantigens, we next examined whether these subunitswere present in patients’ tumors (see SupplementalMaterials and Methods: Immunohistochemistry onTissue). All tumors examined (4/4) expressed GluR1/2subunits. Two tumors predominantly expressed onesubunit, which correlated with the patients’ antibody
specificity. The other two tumors expressed similar lev-els of GluR1/2, and both patients had GluR2 antibod-ies (Fig 4; see Table 2).
Patients’ Antibodies Decrease the Number of AMPAReceptor Clusters and Alter Receptor Localizationat SynapsesGiven that the GluR2 subunit was the most frequenttarget of patients’ antibodies and that most hippocam-pal AMPARs are composed of GluR1/2 heteromers, wenext investigated whether patients’ GluR2 antibodiesaffected the levels of GluR2 subunits in cultures of livehippocampal neurons. For these studies, we first se-lected a representative patient’s CSF containing anti-bodies that competed with the reactivity of other pa-tients’ GluR2 antibodies, indicating they target thesame epitopes (data not shown). Then, the extent ofspecific GluR2 immunolabeling was quantified by con-focal microscopy. These studies demonstrated that91% of the clusters labeled by patient’s antibodies cor-responded to GluR2/3 (Fig 5A).
Having shown the high specificity of patients’ anti-bodies for GluR2 in cultures of live hippocampal neu-
Fig 1. Immunolabeling of rat brain by patients’ antibodies. Sagittal section of rat brain incubated with the cerebrospinal fluid(CSF) of a patient with limbic encephalitis and novel antibodies. Note the intense reactivity of the patient’s antibodies with theneuropil of hippocampus (Hip), subiculum (S), molecular layer of the cerebellum and Purkinje cells (CB), caudate-putamen (CPu),and cerebral cortex (Ctx). Other regions of the brain (eg, corpus callosum [cc] and brainstem [B]) do not show significant immu-nolabeling. Boxed areas in (A) correspond to hippocampus and cerebellum, and are shown at high magnification in (B) and (D).The box in (B) is located at the dentate gyrus and is shown amplified in (C). (B–D) The nuclei of the cells are demonstrated with4,6-diamidino-2-phenylindole (DAPI). Immunofluorescent technique, �2.5 (A); �200 (B, D); �400 (C).
Lai et al: Anti-AMPA Receptor Encephalitis 429
rons, we next examined whether prolonged neuronalexposure to patient’s antibodies changed the number orlocalization of GluR2-containing AMPAR clusters. Foreach condition, a minimum of 6 to 15 neurons wereexamined on each of 2 to 3 coverslips in 3 independentexperiments. Neurons were randomly selected for anal-ysis. The number of synaptic clusters was calculatedper 20�m and expressed as a percentage of control val-ues. These studies demonstrated that patients’ antibod-ies, but not control samples, decreased the number ofGluR2-containing AMPAR clusters after 6 days of an-tibody exposure (p � 0.001; see Figs 5B, C), but notafter 3 days of antibody exposure (p � 0.05; see Figs5B, C). In contrast, patients’ antibodies, but not con-trol samples, decreased the colocalization of GluR2with postsynaptic (PSD-95) and presynaptic (VGlut)markers after 3 and 6 days of antibody exposure (p �0.01; see Figs 5B, D). The effects on receptor numberand localization of AMPAR clusters at postsynapticand presynaptic sites were reversed with removal of an-tibodies from the neuronal cultures (see Figs 5B–D).Moreover, the effects were AMPAR selective becausethe normal colocalization of PSD-95 with VGlut atsynapses and the localization of NMDA receptors at
postsynaptic sites (data not shown) were not altered bypatients’ antibodies.
DiscussionWe report the clinical and immunological features of anew type of LE that associates with antibodies againstGluR1/2 subunits of the AMPAR. The AMPARs areionotropic glutamate receptors that are highly con-served among mammals and mediate most of the fastexcitatory neurotransmission in the brain.9 The major-ity of AMPARs are tetramers composed of GluR1, 2,3, or 4 subunits that combine in a brain region–dependent manner.10 The regions with the greatest lev-els of GluR1/2 and GluR2/3 receptors are the synapticCA3-CA1 areas of the hippocampus, followed by thesubiculum, cerebellum, caudate-putamen, and cerebralcortex.11 This distribution is similar to the immuno-staining of our patients’ antibodies. Although GluR2and GluR3 share sequence homology, our patients’ an-tibodies did not react with GluR3, a subunit that hasbeen identified as an autoantigen in some patients withRasmussen’s encephalitis.12
In most respects, the clinical presentation and mag-netic resonance imaging findings of anti-GluR1/2–as-
Fig 2. Patients antibodies react with extracellular epitopes and precipitate glutamate receptor 1 (GluR1) and GluR2 subunits of the�-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). Culture of rat hippocampal neurons incubated (live,nonpermeabilized) with the cerebrospinal fluid (CSF) of a patient with limbic encephalitis (LE). Note the intense reactivity of pa-tient’s antibodies with cell surface antigens (A). Scale bar � 10�m. Confocal microscopy suggests that the antigens are concentratedin puncta along dendrites (B). Scale bar � 5�m. The precipitation of these antigens using patients’ antibodies is shown in a gel inwhich proteins are visualized with EZBlue (C). Two patients’ antibodies (Patient 1 [P1] and P2) precipitated antigen(s) that pro-duce a single band at approximately 100kDa; this band is not seen in the precipitate using CSF from a control individual (N).The approximately 50kDa band corresponds to patients’ IgG. Sequencing of the 100kDa band demonstrated the GluR1 and GluR2subunits of the AMPAR (not shown). Subsequent transfer of the gel to nitrocellulose and immunoblotting with antibodies specificfor GluR1 and GluR2 confirmed that the 100kDa band contained both GluR1 and GluR2 subunits (D).
430 Annals of Neurology Vol 65 No 4 April 2009
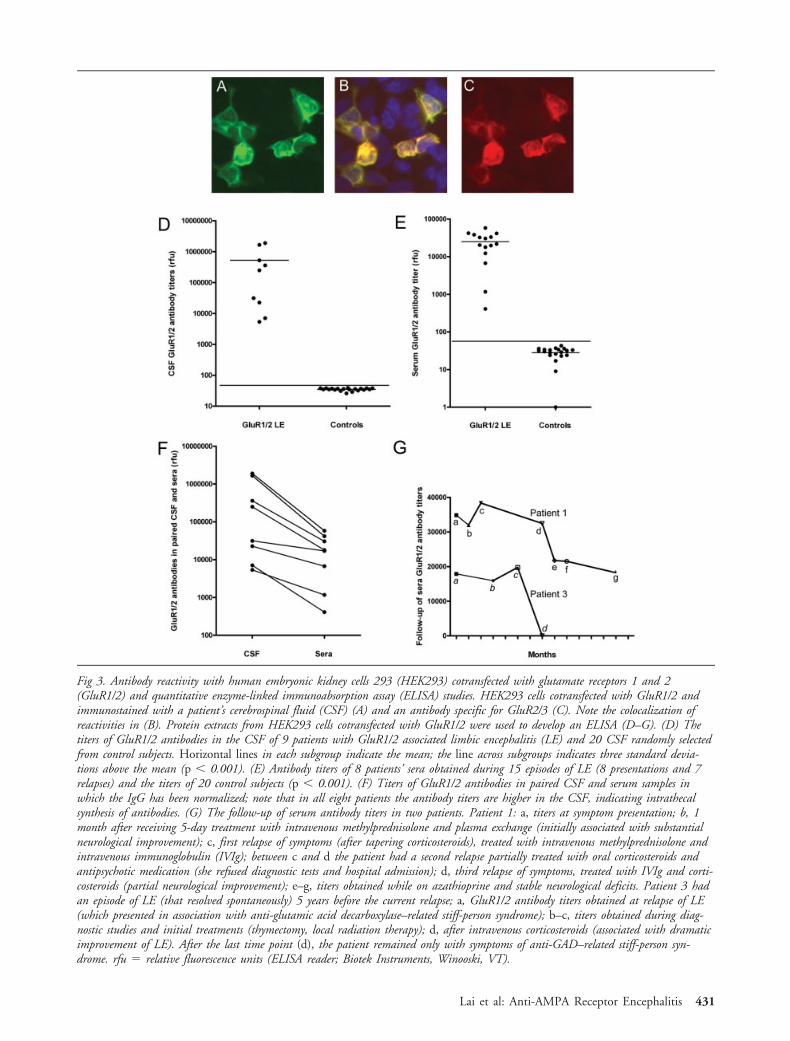
Fig 3. Antibody reactivity with human embryonic kidney cells 293 (HEK293) cotransfected with glutamate receptors 1 and 2(GluR1/2) and quantitative enzyme-linked immunoabsorption assay (ELISA) studies. HEK293 cells cotransfected with GluR1/2 andimmunostained with a patient’s cerebrospinal fluid (CSF) (A) and an antibody specific for GluR2/3 (C). Note the colocalization ofreactivities in (B). Protein extracts from HEK293 cells cotransfected with GluR1/2 were used to develop an ELISA (D–G). (D) Thetiters of GluR1/2 antibodies in the CSF of 9 patients with GluR1/2 associated limbic encephalitis (LE) and 20 CSF randomly selectedfrom control subjects. Horizontal lines in each subgroup indicate the mean; the line across subgroups indicates three standard devia-tions above the mean (p � 0.001). (E) Antibody titers of 8 patients’ sera obtained during 15 episodes of LE (8 presentations and 7relapses) and the titers of 20 control subjects (p � 0.001). (F) Titers of GluR1/2 antibodies in paired CSF and serum samples inwhich the IgG has been normalized; note that in all eight patients the antibody titers are higher in the CSF, indicating intrathecalsynthesis of antibodies. (G) The follow-up of serum antibody titers in two patients. Patient 1: a, titers at symptom presentation; b, 1month after receiving 5-day treatment with intravenous methylprednisolone and plasma exchange (initially associated with substantialneurological improvement); c, first relapse of symptoms (after tapering corticosteroids), treated with intravenous methylprednisolone andintravenous immunoglobulin (IVIg); between c and d the patient had a second relapse partially treated with oral corticosteroids andantipsychotic medication (she refused diagnostic tests and hospital admission); d, third relapse of symptoms, treated with IVIg and corti-costeroids (partial neurological improvement); e–g, titers obtained while on azathioprine and stable neurological deficits. Patient 3 hadan episode of LE (that resolved spontaneously) 5 years before the current relapse; a, GluR1/2 antibody titers obtained at relapse of LE(which presented in association with anti-glutamic acid decarboxylase–related stiff-person syndrome); b–c, titers obtained during diag-nostic studies and initial treatments (thymectomy, local radiation therapy); d, after intravenous corticosteroids (associated with dramaticimprovement of LE). After the last time point (d), the patient remained only with symptoms of anti-GAD–related stiff-person syn-drome. rfu � relative fluorescence units (ELISA reader; Biotek Instruments, Winooski, VT).
Lai et al: Anti-AMPA Receptor Encephalitis 431
sociated encephalitis are typical of LE; therefore, thisdiagnosis was considered early in nine patients. Theother patient was suspected of having a rapidly progres-sive dementia, but the magnetic resonance imaging andantibody findings led to the recognition of LE. Sevenpatients had tumors of the thymus, breast, or lung, andan underlying neoplasm was excluded in the otherthree patients after extensive tumor screening. Al-though this follow-up may be too short to definitivelyexclude a neoplasm, the assessment of individual pa-tients suggests that this disorder may occur as an auto-immune, nonparaneoplastic syndrome. For example,the patient with the shortest follow-up period (8months) is a 38-year-old woman who has fully recov-ered and has no risk factors for cancer. The only pa-tient with risk factors for cancer (smoking) has beenobserved for 50 months without evidence of a neo-plasm.
A frequent feature of anti-GluR1/2–associated LE isthe tendency to relapse. A total of 14 episodes of LE (5initial episodes and 9 relapses) occurred in 5 patients,ranging from 1 to 3 relapses in each patient. The pres-ence of GluR1/2 antibodies in serum or CSF was dem-onstrated in 14 of 14 episodes studied. Except for twopatients whose tumor was initially diagnosed at firstrelapse of LE, all other neurological relapses occurred
in patients without tumor or without tumor recur-rence. A compelling example is a patient who after suc-cessful treatment of a carcinoma of the thymus at firstpresentation of LE subsequently had three relapses over101 months without evidence of tumor recurrence (to-tal follow-up, 10 years). Taken together, the presenceof a GluR1/2-expressing tumor and the susceptibilityfor autoimmune disorders (identified in five patients)either alone or combined may play a role in triggeringthis autoimmune disorder.
A propensity to relapse was also noted in a study of100 patients with encephalitis and antibodies toNMDA receptor, a member of another category ofionotropic glutamate receptors.6 Although AMPARand NMDA receptors are functionally related and playcritical roles in synaptic plasticity,13 each autoimmu-nity associates with a different clinical phenotype. Forexample, none of the patients of this study had anovarian teratoma or experienced development of dyski-nesias, autonomic instability, and hypoventilation,which are common features of anti–NMDA receptorencephalitis.6,14
Neuronal plasticity can be studied through a seriesof models termed long-term potentiation or long-termdepression.13 In these models, synaptic strength in-creases or decreases in association with changes in traf-
Fig 4. Expression of (GluR1) and GluR2 subunits by patients’ tumors. The tumor of a patient with GluR1 antibodies demonstrateshigh level of expression of GluR1 (A), mild expression of GluR2 (B), and reactivity with patient’s antibodies (C). The tumor of apatient with GluR2 antibodies demonstrates high level of expression of GluR1 (D) and GluR2 (E), and reactivity with patient’santibodies (F). Tissues immunostained with the indicated avidin-biotin peroxidase method and mildly counterstained with hematox-ylin. Original magnification, �400.
432 Annals of Neurology Vol 65 No 4 April 2009

ficking of AMPAR.11 The results in our patients areanalogous to those seen in some long-term depressionparadigms.9 Our study shows that patients’ antibodiesspecifically bind to AMPAR clusters causing a decreasein the number of the receptors at synapses and, to alesser degree, the total number of receptor clustersalong dendrites. Because the major effect was on thesynaptic location of the receptors, this finding suggestsa mechanism whereby the antibodies disrupt receptortrafficking/turnover, relocating them from synaptic toextrasynaptic sites/intracellular pool. The reversibilityof these effects provides an explanation for the im-provement of patients’ symptoms with plasma ex-change, intravenous immunoglobulin, or corticoste-roids.
Nine patients dramatically improved after the firstepisode of LE. All nine patients received immunother-apy and, when appropriate, oncological therapy. How-ever, the long-term outcome depended on the appro-priate management of the relapses. In some patients,treatment was challenging because in each episode theybecame agitated, belligerent, and unmanageable athome, yet refusing hospital admission and medication.In two patients, the recovery from each relapse was in-complete, resulting in cumulative residual memory orbehavioral deficits. After the third relapse, both pa-tients stabilized with prolonged use of azathioprine.One was left with moderate short-term memory defi-
cits; the other required institutionalization in a skillednursing facility where she has been living for 3 years.Another patient, an 87-year old woman, died at thesecond relapse shortly after a prolonged episode of sta-tus epilepticus.
The neurological outcome was not influenced by thepresence of a tumor (as long as this was well con-trolled) but was adversely influenced by the presence ofoverlapping immune responses. For example, after re-covering from anti-GluR1/2–associated LE, one patientsuffered from prolonged residual symptoms of anti-GAD–associated stiff-person syndrome; the GluR1/2antibodies had disappeared after treatment, but theGAD antibody levels remained increased (data notshown). Another patient, with GluR1/2 and CRMP5antibodies, had rapid neurological deterioration that re-sulted in death; the autopsy confirmed prominent cy-totoxic T-cell infiltrates in the limbic system. A possi-
Š Fig 5. Patient’s antibodies selectively bind to glutamate recep-tor 2 (GluR2) and alter the number and localization of�-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor(AMPAR) in live neurons. (A) Seventeen days in vitro (div)hippocampal neurons immunostained with patient’s cerebrospi-nal fluid (CSF) (b�w, red) and with a commercial antibodyspecific for GluR2/3 (b�w, green). White column indicatesthe number of GluR2/3-containing clusters labeled by patient’sCSF; black column indicates the number of GluR2/3-containing clusters that are not labeled by patient’s CSF. Be-cause patient’s antibodies react with GluR2, but not GluR3(see text), the findings indicate that nearly all clusters labeledwith patient’s CSF correspond to GluR2 (91%, yellow punctain overlay). Scale bar � 5�m. (B–D) Hippocampal neuronscultured with control CSF or patient’s CSF and subsequentlyimmunostained for GluR2/3 (b�w, red), the postsynapticmarker PSD-95 (green), and the presynaptic marker VGlut(blue). White columns indicate the number of GluR2/3-containing clusters in neurons cultured for 6 days with controlCSF; light gray columns indicate the number of clusters inneurons cultured for 3 days with patient’s CSF; dark graycolumns indicate the number of clusters in neurons culturedfor 6 days with patient’s CSF; black columns indicate thenumber of clusters in neurons cultured for 3 days with pa-tient’s CSF and subsequently cultured for 3 days with controlCSF (3-day recovery). Note that patient’s CSF, applied from11 to 17 div (6-day treatment), reduces the number ofGluR2/3-labeled puncta compared with cultures exposed tocontrol CSF (B: “GluR2/3”; C: p � 0.001). Moreover, thepatient’s CSF applied for 3 or 6 days, but not the controlCSF, reduces the number of GluR2/3 clusters that colocalizewith PSD-95 (yellow puncta) and the number of GluR2/3clusters that colocalize with VGlut (white puncta) (B:“GluR2/3 and PSD-95 and VGlut”; D: p � 0.01). Theseeffects were reversed after removing the antibodies from thecultures and allowing the neurons to recover for 3 days (C,D). Scale bar � 5�m. Results were analyzed using theKruskal–Wallis nonparametric analysis of variance followed byDunn’s pairwise comparison.
Lai et al: Anti-AMPA Receptor Encephalitis 433

ble explanation for these outcomes is that theaccompanying immune responses, particularly if associ-ated with cytotoxic T-cell mechanisms, are more diffi-cult to treat, or that the neuronal dysfunction is lessreversible than that caused by the anti-GluR1/2 im-mune response, which appears to be directly mediatedby antibodies.15,16
Anti-GluR1/2–associated LE represents a new cate-gory of immune-mediated encephalitis that may occurwith or without systemic tumors and shows a propen-sity to relapse. The disorder is treatable and can nowbe diagnosed serologically. Potential cases are patientswho are currently categorized as “antibody-negativeLE” or “steroid-responsive LE.” Future studies shoulddetermine the extent of tumor association and overlap-ping autoimmunities, the role of chronic immune sup-pression in preventing relapses, and the molecularmechanisms whereby antibodies alter the synaptic lo-calization of AMPAR.
This work was supported in part by the National Cancer Institute,2R56CA089054 and RO1CA107192 (J.D.); National Institute ofHealth, R21 MH057683 (R.B-G.); National Institute of Health,NSR56-45986, NSR01-45986 and Foederer Foundation of theChildren’s Hospital of Philadelphia (D.L.); National Institute ofMental Health, F31MH083395 (A.G.); The National Institute onAging, AG023481, and the McBean Family Foundation, and LarryL. Hillblom Foundation (H.S.).
We thank Drs A. Voloschin, S. Zanini, and G. C.Muscas for providing clinical information.
References1. Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis”
and its association with carcinoma. Brain 1968;91:481–496.2. Gultekin SH, Rosenfeld MR, Voltz R, et al. Paraneoplastic lim-
bic encephalitis: neurological symptoms, immunological find-ings and tumour association in 50 patients. Brain 2000;123(pt7):1481–1494.
3. Bataller L, Kleopa KA, Wu GF, et al. Autoimmune limbic en-cephalitis in 39 patients: immunophenotypes and outcomes.J Neurol Neurosurg Psychiatry 2007;78:381–385.
4. Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsivelimbic encephalitis identified by neuropil antibodies: MRI andPET correlates. Brain 2005;128:1764–1777.
5. Graus F, Saiz A, Lai M, et al. Neuronal surface antigen anti-bodies in limbic encephalitis: clinical-immunologic associations.Neurology 2008;71:930–936.
6. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA re-ceptor encephalitis: case series and analysis of the effects of an-tibodies. Lancet Neurol 2008;7:1091–1098.
7. Buchhalter JR, Dichter MA. Electrophysiological comparisonof pyramidal and stellate nonpyramidal neurons in dissociatedcell culture of rat hippocampus. Brain Res Bull 1991;26:333–338.
8. Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astro-cytes regulate inhibitory synapse formation via Trk-mediatedmodulation of postsynaptic GABAA receptors. J Neurosci2005;25:3638–3650.
9. Shepherd JD, Huganir RL. The cell biology of synapticplasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol2007;23:613–643.
10. Palmer CL, Cotton L, Henley JM. The molecular pharmacol-ogy and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev 2005;57:253–277.
11. Sprengel R. Role of AMPA receptors in synaptic plasticity. CellTissue Res 2006;326:447–455.
12. Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies toglutamate receptor GluR3 in Rasmussen’s encephalitis. Science1994;265:648–651.
13. Genoux D, Montgomery JM. Glutamate receptor plasticity atexcitatory synapses in the brain. Clin Exp Pharmacol Physiol2007;34:1058–1063.
14. Iizuka T, Sakai F, Ide T, et al. Anti-NMDA receptor enceph-alitis in Japan: long-term outcome without tumor removal.Neurology 2008;70:504–511.
15. Bernal F, Graus F, Pifarre A, et al. Immunohistochemical anal-ysis of anti-Hu-associated paraneoplastic encephalomyelitis.Acta Neuropathol (Berl) 2002;103:509–515.
16. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of theCNS. Lancet Neurol 2008;7:327–340.
434 Annals of Neurology Vol 65 No 4 April 2009





























