Embed Size (px)
Citation preview

1- Porphyrins are cyclic compounds formed by the linkage of 4 pyrrole rings 1- Porphyrins are cyclic compounds formed by the linkage of 4 pyrrole rings
through methenyl bridges (-CH=).through methenyl bridges (-CH=).
2- Porphyrins differ from each other in the type and arrangement of side chains 2- Porphyrins differ from each other in the type and arrangement of side chains
attached to numbered positions of pyrrole rings: 1,2,3,4,5,6,7, and 8.attached to numbered positions of pyrrole rings: 1,2,3,4,5,6,7, and 8.
3- 3- The side chains The side chains which stituted in numbered may be substituted in numbered which stituted in numbered may be substituted in numbered
positions are: positions are: A = Acetate = CHA = Acetate = CH22-COOH-COOH
P = Propionate = -CHP = Propionate = -CH22-CH-CH22-COOH-COOH
M = Methyl = -CHM = Methyl = -CH33
V = Vinyl = -CH=CHV = Vinyl = -CH=CH22
4- 4- Structure of heme:Structure of heme:
a- Heme is a complex of iron (in ferrous state, Fe ++) and one of porphyrins a- Heme is a complex of iron (in ferrous state, Fe ++) and one of porphyrins
called: proto porphyrin IX.called: proto porphyrin IX.
b- Iron is held in the center of heme molecule by bonds to the 4 nitrogen atoms b- Iron is held in the center of heme molecule by bonds to the 4 nitrogen atoms
of pyrrole rings of protoporphyrin.of pyrrole rings of protoporphyrin.

5- 5- Hemoproteins : Hemoproteins :
a- These are a group of specialized proteins that contain heme as a prosthetic a- These are a group of specialized proteins that contain heme as a prosthetic
group. They are:group. They are:1) Hemoglobin.1) Hemoglobin. 2) Myoglobin.2) Myoglobin.
3) Respiratory cytochromes. 3) Respiratory cytochromes. 4) Cytochrome p450.4) Cytochrome p450.
5) Catalase and peroxidase.5) Catalase and peroxidase. 6) Tryptophan 6) Tryptophan
oxygenase.oxygenase.b- b- The role of the heme group in each protein is: The role of the heme group in each protein is:
1) 1) In hemoglobin and myoglobin: In hemoglobin and myoglobin: Acts as oxygen carrier.Acts as oxygen carrier.
2) 2) In cytochromes : In cytochromes : Acts as an electron carrier.Acts as an electron carrier.
3) 3) In catalase and peroxidase:In catalase and peroxidase:Acts as a part of active site of the enzyme that catalyzes the breakdown of Acts as a part of active site of the enzyme that catalyzes the breakdown of
hydrogen peroxide (Hhydrogen peroxide (H22OO22) (For details, see iron metabolism page 125).) (For details, see iron metabolism page 125).
B- B- Heme Synthesis: Heme Synthesis: 1- The 2 starting materials are succinyl CoA (derived from citric acid cycle in 1- The 2 starting materials are succinyl CoA (derived from citric acid cycle in
mitochondria) and glycine. The reactions need ALA synthase enzyme and mitochondria) and glycine. The reactions need ALA synthase enzyme and
pyridoxal phosphate as activator for glycine. pyridoxal phosphate as activator for glycine.
2- Then ALA passes from mitochondria to cytoplasm where 2 molec cules are 2- Then ALA passes from mitochondria to cytoplasm where 2 molec cules are
condensed to form porphobilinogen :condensed to form porphobilinogen :
3- Porphobilinogen is a pyrrole ring to which a propiona (-CH3- Porphobilinogen is a pyrrole ring to which a propiona (-CH22-CH-CH22-COOH), and -COOH), and
acetate (-CHacetate (-CH22-COOH) groups are attached. 4 Porphobilinogens are condensa -COOH) groups are attached. 4 Porphobilinogens are condensa
ted to give protoporphyrin as follows:ted to give protoporphyrin as follows:

4- 4- Then heme formation is completed by incorporation (FeThen heme formation is completed by incorporation (Fe++++) into protoporphyrin ) into protoporphyrin
as follows: as follows:
Comments on heme synthesis: Comments on heme synthesis:
2 3 2CH COOH CH CO
2 2CO H OO2 3 2 2 2
Pr opionate Ethylalcohol Vinyl
CH2 CH2 COOH CH CH CH CH OH CH CH
5- Regulation of heme synthesis: 5- Regulation of heme synthesis:
Delta aminolevulinic acid synthase enzyme (ALA synthase)is the key Delta aminolevulinic acid synthase enzyme (ALA synthase)is the key
enzyme in heme synthesis.enzyme in heme synthesis.a- It is inhibited by :a- It is inhibited by :
Heme itself, by feedback inhibition.Heme itself, by feedback inhibition. Glucose and steroids.Glucose and steroids.
b- It is stimulated by :b- It is stimulated by : Certain drugs as phenobarbital and iron.Certain drugs as phenobarbital and iron.

C- Porphyrias : C- Porphyrias :
1- These are a group of diseases resulting from a deficiency of one of the 1- These are a group of diseases resulting from a deficiency of one of the
enzymes needed for heme synthesis.enzymes needed for heme synthesis.
2- 2- Porphyrias lead to disturbance. in heme synthesis and causes:Porphyrias lead to disturbance. in heme synthesis and causes:
a- a- Anaemia: Anaemia: due to decrease-production of heme.due to decrease-production of heme.
b- Abdominal pain and neuropsychatric symptoms due to toxic effect of b- Abdominal pain and neuropsychatric symptoms due to toxic effect of
the accumulated porphyrin intermediates. the accumulated porphyrin intermediates.
c- c- Photosensitivity: Photosensitivity: Some porphyrin derivatives when exposed to light Some porphyrin derivatives when exposed to light
react with molecular oxygen to form oxygen radi cals, which cause react with molecular oxygen to form oxygen radi cals, which cause
skin damage.skin damage.
3- Porphyrias are either 3- Porphyrias are either hereditary or aquired hereditary or aquired (caused by envi ronmental (caused by envi ronmental
poisons as lead).poisons as lead).
4- All hereditary porphyrias are 4- All hereditary porphyrias are autosomal dominant autosomal dominant except con genital except con genital
erythropoietic porphyria which is autosomal recessive.erythropoietic porphyria which is autosomal recessive.
5- 5- Classification:Classification:
a- Liver and bone-marrow are the organs where heme synthesis occurs.a- Liver and bone-marrow are the organs where heme synthesis occurs.
b- According to the site of enzyme deficiency, porphyrias can be b- According to the site of enzyme deficiency, porphyrias can be
classified into hepatic (liver), erythropoietic (bone marrow) and classified into hepatic (liver), erythropoietic (bone marrow) and
erythrohepatic (liver and bone marrow).erythrohepatic (liver and bone marrow).

c- c- The following table gives summary of the major findings of porphyrias :The following table gives summary of the major findings of porphyrias :

A- Hemoglobin is found only in the red blood cells.A- Hemoglobin is found only in the red blood cells.
B- Its main function is to transport oxygen from the lungs to the capillaries of the B- Its main function is to transport oxygen from the lungs to the capillaries of the
tissues and carbon dioxide (COtissues and carbon dioxide (CO22) from the tissues to the lungs.) from the tissues to the lungs.
C- C- Structure of hemoglobin:Structure of hemoglobin:
1- Hemoglobin is conjugated protein which consists of specialized protein called: 1- Hemoglobin is conjugated protein which consists of specialized protein called:
globin that is tightly bound to 4 heme molecules.globin that is tightly bound to 4 heme molecules.
2- Globin is a protein with four peptide chains joined together by noncovalent 2- Globin is a protein with four peptide chains joined together by noncovalent
bonds (tetramer).bonds (tetramer).
3- Several different kinds of hemoglobin are normally found in human. They vary 3- Several different kinds of hemoglobin are normally found in human. They vary
in the primary structure of the peptide chains of globin. These are HbA, HbAin the primary structure of the peptide chains of globin. These are HbA, HbA22, ,
HbF, HbAHbF, HbA11..a- Hemoglobin a- Hemoglobin
A :A :1) It is the major hemoglobin in adults (97%).1) It is the major hemoglobin in adults (97%).
2) Its globin comprises 4 polypeptide chains: 2) Its globin comprises 4 polypeptide chains:
Two α - chains (141 amino acids).Two α - chains (141 amino acids). Two β - chains (146 amino acids).Two β - chains (146 amino acids).
The globin is abbreviated αThe globin is abbreviated α22 β β22..

b- Hemoglobin Ab- Hemoglobin A2 2 : :
1) It accounts about 2% of adult human hemoglobin.1) It accounts about 2% of adult human hemoglobin.
2) Its globin consists of 2 α - chains and 2 delta chains (δ) : α2) Its globin consists of 2 α - chains and 2 delta chains (δ) : α22 δ δ2 2
c- Fetal hemoglobin (HbF) :c- Fetal hemoglobin (HbF) :
1) This is hemoglobin present in the fetus during intrautrine fetal life.1) This is hemoglobin present in the fetus during intrautrine fetal life.
2) It consists of 2 α - chains and 2 gamma chains (α2) It consists of 2 α - chains and 2 gamma chains (α22 γ γ22))
3 Hemoglobin F accounts about 1% of adult human hemoglobin. 3 Hemoglobin F accounts about 1% of adult human hemoglobin. d- Hemoglobin Al (Glycated hemoglobin) :d- Hemoglobin Al (Glycated hemoglobin) :
1) Hemoglobin A, reacts non enzymatically with glucose to form a derivative 1) Hemoglobin A, reacts non enzymatically with glucose to form a derivative
known as glycated hemoglobin or HbAknown as glycated hemoglobin or HbA1c1c..
2) Normally the concentration of HbA2) Normally the concentration of HbA1c1c is very low (5-8%) but in diabetes is very low (5-8%) but in diabetes
mellitus, where blood sugar levels may be high, the concentration of HbAmellitus, where blood sugar levels may be high, the concentration of HbA1c 1c
may reach 12% or more of the total hemoglobin.may reach 12% or more of the total hemoglobin.E- hemoglobinopathies: E- hemoglobinopathies:
1- These are a group of diseases caused by either abnormal globin formation or 1- These are a group of diseases caused by either abnormal globin formation or
synthesis of insufficient quantities of normal hemoglobin.synthesis of insufficient quantities of normal hemoglobin.
2- Many disorders are present, but here, two of them discussed:2- Many disorders are present, but here, two of them discussed:
a- Sickle cell anaemia:a- Sickle cell anaemia:
1) The blood cells of these patients contain abnormal hemoglobin call ed 1) The blood cells of these patients contain abnormal hemoglobin call ed
hemoglobin S (HbS).hemoglobin S (HbS).

2) A molecule of HbS contains 2 nor mal α - chains and 2 mutant β chains in 2) A molecule of HbS contains 2 nor mal α - chains and 2 mutant β chains in
which which glutamateglutamate at. po sition six has been replaced with valine. at. po sition six has been replaced with valine.
3) Glutamate is polar while valine is nonpolar. This single error makes 3) Glutamate is polar while valine is nonpolar. This single error makes
hemoglobin S less soluble especially in its deoxygenated form. This will hemoglobin S less soluble especially in its deoxygenated form. This will
lead to:lead to: The molecules of HbS aggregate to form fibers that deform red cells The molecules of HbS aggregate to form fibers that deform red cells
into a crescent or sickle shape. into a crescent or sickle shape. HemolysisHemolysis of RBSickling of cells will block the flow of blood in small of RBSickling of cells will block the flow of blood in small
capi llaries leading to capi llaries leading to hypoxia, pain and death of cells hypoxia, pain and death of cells supplied by these supplied by these
capillaries.capillaries. Cs. Cs. 4) 4) Two types of sickle cell anaemia are present:Two types of sickle cell anaemia are present: Homozygous recessive disorder:Homozygous recessive disorder: occurs in individuals who have two occurs in individuals who have two
mutant genes coding for synthesis of 1 chains (one gene from father and mutant genes coding for synthesis of 1 chains (one gene from father and
the other from mother). the other from mother). Heterozygous disorder:(sickle cell trait) Heterozygous disorder:(sickle cell trait) occurs in individuals having occurs in individuals having
one normal gene and one sickle cell gene. Usually patients with sickle one normal gene and one sickle cell gene. Usually patients with sickle
cell trait do not show clinical symptoms except if they exposed to very cell trait do not show clinical symptoms except if they exposed to very
low oxygen tension.low oxygen tension.b- Thalassemia: b- Thalassemia: 1) Are anaemias characterized by reduced synthesis of eith er alpha (α - 1) Are anaemias characterized by reduced synthesis of eith er alpha (α -
tha1assemias) or beta (β- thalassemia) chains of hemoglobin.tha1assemias) or beta (β- thalassemia) chains of hemoglobin.
2) The causes are most often due to 2) The causes are most often due to gene deletionsgene deletions..
3) Thalassemia may be either 3) Thalassemia may be either homozygoushomozygous with severe anaemia or with severe anaemia or
heterozygousheterozygous (thalassemia trait) with no clinical symptoms. (thalassemia trait) with no clinical symptoms.

F- Abnormal derivatives of hemoglobin: F- Abnormal derivatives of hemoglobin:
1- Methemoglobin (Met-Hb) :1- Methemoglobin (Met-Hb) :a- It is oxidized hemoglobin in which the ferrous ions (Fea- It is oxidized hemoglobin in which the ferrous ions (Fe++++) of hemoglobin has been ) of hemoglobin has been
oxidized to the ferric state (Feoxidized to the ferric state (Fe++++++).).
b- Oxidation is caused by some drugs,Hb- Oxidation is caused by some drugs,H22OO22 and number of free radicals. and number of free radicals.
C- Met-Hb binds oxygen irreversibly and is unable to act as an oxygen carrier. C- Met-Hb binds oxygen irreversibly and is unable to act as an oxygen carrier.
d- If present in high concentration, met-Hb will lead to hypoxia and cyanosis.d- If present in high concentration, met-Hb will lead to hypoxia and cyanosis.
2- Carboxyhemoglobin (COHb) :2- Carboxyhemoglobin (COHb) :a- It is hemoglobin combining with carbon monoxide (CO).a- It is hemoglobin combining with carbon monoxide (CO).
b- Carbon monoxide combines at the same position in the Hb molecule as Ob- Carbon monoxide combines at the same position in the Hb molecule as O22, with , with
affinity about 200 times greater than Oaffinity about 200 times greater than O22..
c- Concenirafion of COHb above 40% usually result in uncon -sciousness, and may be c- Concenirafion of COHb above 40% usually result in uncon -sciousness, and may be
fatal.fatal.3- Sulfhemoglobin (S-Hb) :3- Sulfhemoglobin (S-Hb) :
a- It is hemoglobin combining with sulfur.a- It is hemoglobin combining with sulfur.
b- It results from exposure of hemoglobin to the toxic effects of certain drugs as b- It results from exposure of hemoglobin to the toxic effects of certain drugs as
sulfonamides.sulfonamides.
c- S-Hb produces anoxia and cyanosis because it can not act as oxygen carrier.c- S-Hb produces anoxia and cyanosis because it can not act as oxygen carrier.
4- Hematin:4- Hematin:a- It is hemoglobin without iron (i.e. protoporphyrin combi ning,with globin).a- It is hemoglobin without iron (i.e. protoporphyrin combi ning,with globin).
b- It may be formed following intravascular hemolysis.b- It may be formed following intravascular hemolysis.

G- Hemoglobin catabolism:G- Hemoglobin catabolism:1- The average life span of the red blood cells is 120 days.1- The average life span of the red blood cells is 120 days.
2- At the end of that time, they are removed from circulation by the cells of 2- At the end of that time, they are removed from circulation by the cells of
reticuloendothelial (RE) system mostly present in liver, spleen and bone reticuloendothelial (RE) system mostly present in liver, spleen and bone
marrow, where they are hemolyzed (extravascular hemolysis) and marrow, where they are hemolyzed (extravascular hemolysis) and
hemoglobin comes out.hemoglobin comes out.
3- Globin. molecule is hydrolyzed into free amino acids.3- Globin. molecule is hydrolyzed into free amino acids.
4- 4- Formation of bilirubin:Formation of bilirubin: a- The heme ring is catabolized by the microsomal heme oxyge nase a- The heme ring is catabolized by the microsomal heme oxyge nase
enzymes of the RE cells.enzymes of the RE cells.
b- In this reaction (which needs Ob- In this reaction (which needs O22 and NADPH) ,iron (Fe and NADPH) ,iron (Fe++++) is removed for ) is removed for
re-use. The remaining of heme ring is cleaved between pyrrole rings re-use. The remaining of heme ring is cleaved between pyrrole rings
number I and II to form. Biliverdin (green pigment) and carbori number I and II to form. Biliverdin (green pigment) and carbori
monoxide (CO).monoxide (CO).
c- Biliverdin is then reducec into bilirubin (golden yellow) in a reaction c- Biliverdin is then reducec into bilirubin (golden yellow) in a reaction
requires biliverdin reductase enzyme.requires biliverdin reductase enzyme.5- 5- Transport of bilirubin in the plasma: Transport of bilirubin in the plasma:
Bilirubin is nonpolar, and is insoluble in plasma. There fore it binds Bilirubin is nonpolar, and is insoluble in plasma. There fore it binds
by noncovalent bonds to plasma albumin. This form is called: by noncovalent bonds to plasma albumin. This form is called:
unconjugated or indirect bilirubin.unconjugated or indirect bilirubin.

6- 6- Uptake of bilirubin by the liver: Uptake of bilirubin by the liver:
a- Bilirubin dissociates from the carrier albumin molecule and enters a a- Bilirubin dissociates from the carrier albumin molecule and enters a
hepatocytes.hepatocytes.
b- Bilirubin is conjugated with one or two molecules of gluc uronic acid b- Bilirubin is conjugated with one or two molecules of gluc uronic acid
(the acid form of glucose) to form bilirubin monoglucuronide and (the acid form of glucose) to form bilirubin monoglucuronide and
bilirubin diglucuronide.This form is called conjugated or direct bilirubin diglucuronide.This form is called conjugated or direct
bilirubin. This reaction needs UDP-lucuronyltrahsferase enzyme:bilirubin. This reaction needs UDP-lucuronyltrahsferase enzyme:
7- 7- Secretion of bilirubin into bile:Secretion of bilirubin into bile: Bilirubin diglucuronide is actively transported against concentration Bilirubin diglucuronide is actively transported against concentration
gradient into the bile canaliculi and then into the bile.gradient into the bile canaliculi and then into the bile.
8- 8- Van den Bergh reaction : Van den Bergh reaction :
a- This is a reaction between bilirubin and Ehrlich diazo reagent giving a- This is a reaction between bilirubin and Ehrlich diazo reagent giving
a reddish purple compound.a reddish purple compound.
b- Conjugated bilirubin reacts directly with the reagent. Thus it may b- Conjugated bilirubin reacts directly with the reagent. Thus it may
called: direct bilirubin.called: direct bilirubin.
c- Unconjugated bilirubin does not react with the reagent directly c- Unconjugated bilirubin does not react with the reagent directly
except after addition of alcohol. Thus it may be called: indirect except after addition of alcohol. Thus it may be called: indirect
bilirubin.bilirubin.
UDP Glucurony transferaseUDP Glucuronate UDP
Bilirubin Bilirubin glucuronide(s)

A- Normal plasma bilirubin level is up to 1 mg/dl (17.1 umol/L).A- Normal plasma bilirubin level is up to 1 mg/dl (17.1 umol/L).
B- Hyperbilirubinemia results when plasma bilirubin exceeds 1 mg/dl. B- Hyperbilirubinemia results when plasma bilirubin exceeds 1 mg/dl.
C- Hyperbilirubinemia may be due to increase conjugated and/or un conjugated C- Hyperbilirubinemia may be due to increase conjugated and/or un conjugated
bilirubin(s).bilirubin(s).

D- In hyperbilirubinemia, bilirubin accumulates in blood and when it reachs 3 mg/dl, it D- In hyperbilirubinemia, bilirubin accumulates in blood and when it reachs 3 mg/dl, it
diffuses into the tissues which become yellow. This condition is called: diffuses into the tissues which become yellow. This condition is called: jaundice or jaundice or
icterus.icterus.
E- Clinically, jaundice can be detected by yellow colouration of skin, sclera and mucous E- Clinically, jaundice can be detected by yellow colouration of skin, sclera and mucous
membranes.membranes.
F- F- Types of hyperbilirubinemia: Types of hyperbilirubinemia:
It can be classified into prehepatic, hepatic and post hepatic.It can be classified into prehepatic, hepatic and post hepatic.
1- Prehepatic: (Hemolytic jaundice) :1- Prehepatic: (Hemolytic jaundice) :
a- Hyperbilirubinemia, mostiy of unconjugated type occurs in all forms of a- Hyperbilirubinemia, mostiy of unconjugated type occurs in all forms of
hemolytic anaemias i.e. excessive destruction of RBCs inside blood vessels.hemolytic anaemias i.e. excessive destruction of RBCs inside blood vessels.
b- It is due to increase the amount of plasma unconjugated bilirubin more than the b- It is due to increase the amount of plasma unconjugated bilirubin more than the
capacity of the liver that can deal with.capacity of the liver that can deal with.
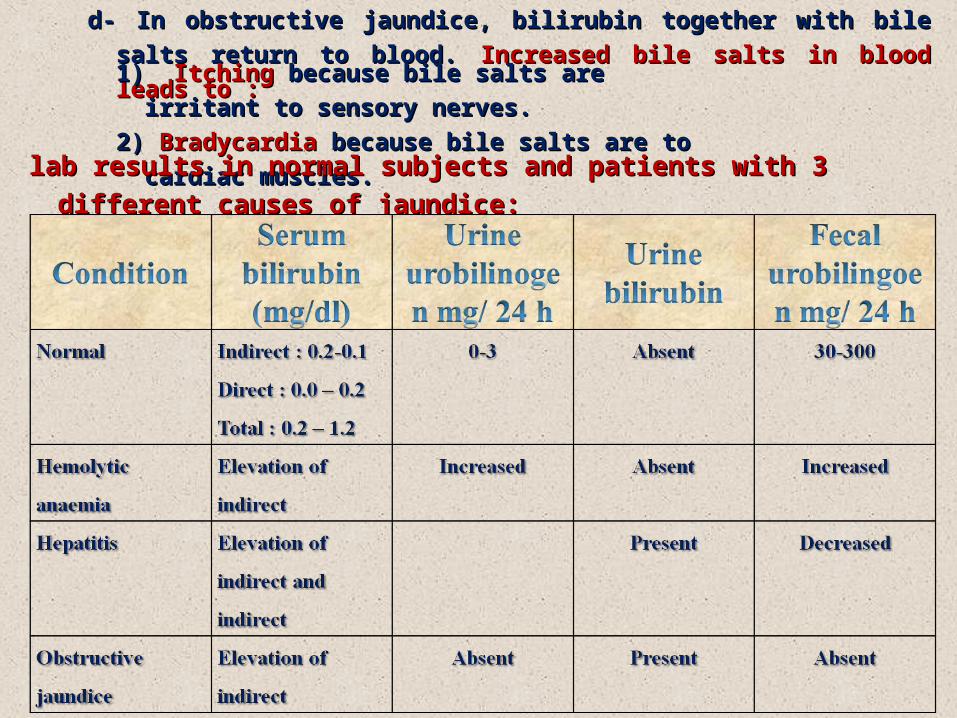
c- c- Biochemical changes: Biochemical changes:
1) Increased production of bilirubin leads to increased production of 1) Increased production of bilirubin leads to increased production of
urobilinogen which appears in urine in large amounts.urobilinogen which appears in urine in large amounts.
2) No bilirubin appears in urine. (So that the combination of increased 2) No bilirubin appears in urine. (So that the combination of increased
urobilinogen and absence of bilirubin in urine is suggestive of hemolytic urobilinogen and absence of bilirubin in urine is suggestive of hemolytic
jaundice).jaundice).

2- Hepatic: (hepatocellular jaundice) :2- Hepatic: (hepatocellular jaundice) :
a- It is due to liver cells damage by cirrhosis, infective hepatitis or toxins.a- It is due to liver cells damage by cirrhosis, infective hepatitis or toxins.
b- Usually there is an associated obstruction of some biliary /canaliculi.b- Usually there is an associated obstruction of some biliary /canaliculi.
c- Hyperbilirubinemia is thus a mixture of unconjugated and conjugated types.c- Hyperbilirubinemia is thus a mixture of unconjugated and conjugated types.
d- d- Biochemical changes:Biochemical changes:
1) Urobilinogen appears in normal trace amounts.1) Urobilinogen appears in normal trace amounts.
2) Bilirubin also appears in urine.2) Bilirubin also appears in urine.
e- Abnormalities at hepatpcellular level may be due to conge nital causes (discussed e- Abnormalities at hepatpcellular level may be due to conge nital causes (discussed
later) as defect of transport of bilirubin into the cell, defective conjugation or later) as defect of transport of bilirubin into the cell, defective conjugation or
defective excretion into the bile canalculi.defective excretion into the bile canalculi.
3- Posthepatic (cholestatic jaundice) : 3- Posthepatic (cholestatic jaundice) :
a- Cholestasis (stopagge of bile flow) may be due to mechanical obstruction of a- Cholestasis (stopagge of bile flow) may be due to mechanical obstruction of
biliary tree by gallstone in common bile duct or carcinoma of the head of the biliary tree by gallstone in common bile duct or carcinoma of the head of the
pancreas or carcinoma of the biliary tree.pancreas or carcinoma of the biliary tree.
b- Hyperbilirubinemia is mostly of conjugated type.b- Hyperbilirubinemia is mostly of conjugated type.
c- c- Biochemical changes:Biochemical changes:
1) Urobilinogen is absent in urine.1) Urobilinogen is absent in urine.
2) Urobilinogen is absent in stool giving clay colour stool. 2) Urobilinogen is absent in stool giving clay colour stool.
3) Bilirubin appears in urine.3) Bilirubin appears in urine.
d- In obstructive jaundice, bilirubin together with bile salts return to blood. d- In obstructive jaundice, bilirubin together with bile salts return to blood.
Increased bile salts in blood leads to :Increased bile salts in blood leads to :
1) 1) Itching Itching because bile salts are irritant to sensory nerves. because bile salts are irritant to sensory nerves.
2) 2) Bradycardia Bradycardia because bile salts are to cardiac muscles.because bile salts are to cardiac muscles.
lab results in normal subjects and patients with 3 different causes of jaundice:lab results in normal subjects and patients with 3 different causes of jaundice:

Physiologic jaundice of neonates: Physiologic jaundice of neonates:
1- This is a transient condition occurs in some newborn infants especially if they are 1- This is a transient condition occurs in some newborn infants especially if they are
premature.premature.
2- 2- It results from:It results from:
a) At birth, liver contains very little UDP-glucuronyltrans ferase enzyme, which is a) At birth, liver contains very little UDP-glucuronyltrans ferase enzyme, which is
important for conjugation of bilirubin.important for conjugation of bilirubin.
b) Accerelated hemolysis of RBCs.b) Accerelated hemolysis of RBCs.
3- 3- This leads to This leads to increased unconjugated bilirubin which lasts 2 - 3 days in full term increased unconjugated bilirubin which lasts 2 - 3 days in full term
infants and and jaundice about 6 days in premature infants.infants and and jaundice about 6 days in premature infants.
4- If unconjugated bilirubin exceeds the concentration which can be tightly bound to 4- If unconjugated bilirubin exceeds the concentration which can be tightly bound to
plasma albumin (20-25 mg/dl), free bili rubin can pass blood-brain barrier, causing plasma albumin (20-25 mg/dl), free bili rubin can pass blood-brain barrier, causing
kernicterus (toxic encephalopathy) which can cause mental retardation.kernicterus (toxic encephalopathy) which can cause mental retardation.
5- Kernicterus develops because the excess bilirubin is soluble in the lipid of the basal 5- Kernicterus develops because the excess bilirubin is soluble in the lipid of the basal
ganglia of the brain.ganglia of the brain.
6- 6- Treatment : Treatment :
Neonatal jaundice is treated by pheno-barbital and exposure of jaundiced Neonatal jaundice is treated by pheno-barbital and exposure of jaundiced
baby to visible light (phototherapy) as bili rubin is broken down in light.baby to visible light (phototherapy) as bili rubin is broken down in light.

H- Congenital hyperbilirubinemia:H- Congenital hyperbilirubinemia:
1- Gilbert's disease:1- Gilbert's disease:
a- It is asymptomatic a- It is asymptomatic unconjugated hyperbilirubinemia.unconjugated hyperbilirubinemia.
b- Bilirubin concentration is usually less than 3 mg/dl.b- Bilirubin concentration is usually less than 3 mg/dl.
c- It is due to a c- It is due to a defect in the uptake of bilirubin defect in the uptake of bilirubin by the liver parenchymal cells and a by the liver parenchymal cells and a
mild deficiency of mild deficiency of UDP-glucuronyltra- nsferase.UDP-glucuronyltra- nsferase.
2. Crigler - Najjar syndrome:2. Crigler - Najjar syndrome:
a- It is severe unconjugated hyperbilirubinemia.a- It is severe unconjugated hyperbilirubinemia.
b- It occurs rarely in neonates, leading to kernicterus and often to early death.b- It occurs rarely in neonates, leading to kernicterus and often to early death.
c- Bilirubin concentration is usually exceeds 20 mg/dl.c- Bilirubin concentration is usually exceeds 20 mg/dl.
d- It is due to marked reduction in the UDP-glucuronyltransferase.d- It is due to marked reduction in the UDP-glucuronyltransferase.
3. Dubin - Johnson syndrome: 3. Dubin - Johnson syndrome: a- It is a conjugated hyperbilirubinemia which occurs during adult life.a- It is a conjugated hyperbilirubinemia which occurs during adult life.
b- It is due to defect in the hepatic secretion of conjugated bilirubin into the bile.b- It is due to defect in the hepatic secretion of conjugated bilirubin into the bile.
I- Neonatal/jaundice: I- Neonatal/jaundice:
These are a group of diseases including phys iologic and congenintal jaundice These are a group of diseases including phys iologic and congenintal jaundice
and other hemolytic diseases as Rh incompatibility.and other hemolytic diseases as Rh incompatibility.





![The intricacies of the stacking interaction in a pyrrole ... · The intricacies of the stacking interaction in a pyrrole–pyrrole system Tomasz Sieran´ski1 Received: ... [32, 33]](https://img.dokumen.tips/doc/110x75/5ea942711f125e3c163555f9/the-intricacies-of-the-stacking-interaction-in-a-pyrrole-the-intricacies-of.jpg)

![Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel ...fulir.irb.hr/3840/1/BanZ_Nucleobase_Molecules-22_2017_2213.pdf · nucleobase cytosine [28,29], while the guanidiniocarbonyl-pyrrole](https://img.dokumen.tips/doc/110x75/5eadaa6c2f808b2f2c0bb939/nucleobaseaguanidiniocarbonyl-pyrrole-conjugates-as-novel-fulirirbhr38401banznucleobasemolecules-2220172213pdf.jpg)





















