Embed Size (px)
Citation preview

Chapter 1
PORPHYRINS AND METALLOPORPHYRINS
1.1. Introduction
Porphyrins are one of the vital chemical units essential for several life
processes on the earth. Many biological molecules function with prosthetic
groups essentially made of these units. Chlorophylls of chloroplasts which
drive photosynthesis, heme as a component of hemoglobin that transports
oxygen to animal tissues and as the central unit of myoglobin ensures the
storage of oxygen - all these have active sites essentially made of porphyrin
core'". Over the years, a great deal of concerted efforts have brought to light
substantial understanding of the structure-function relationship in these natural
porphyrins"'O
A large variety of synthetic porphyrins and their metalloderivatives were
made over the years to study the porphyrin based natural systems. The search
for anti-cancer drugs, useful catalysts, semiconductors and superconductors,
electronic materials with novel properties has also made this synthetic
porphyrin chemistry a very actively probed one by chemists, biologists and
physicists alike. The synthetic meso-substituted porphyrins offer a great
advantage to study the physical and chemical properties of the porphyrin
nucleus quantitatively by a judicious choice of the substituents that may be
attached on the periphery. Metalloporphyrins are widely and intensely
investigated in the area of catalysis and also as models and mimics of enzymes
l i e catalase, peroxidases, P450 cytochromes or as transmembrane electron 11-13 transport agents . They have also been used as NMR image enhancement
agents14, Nonlinear optical materials" and DNA-binding or cleavage agent1" . 17 . Currently there is interest in using chelated radioactiv
diagnostic imaging and therapeutic agents. In that con
excellent compounds because of their extremely high s
many metal ions.

1.2. The Porphyrin System
Porphyrins are basically cyclic tetrapyrrole derivatives with a highly
delocalised planar ?r-framework having a core structure 1. They can exist in
varied forms by having different peripheral substituents at all the eight pyrrole
P-carbon atom and the four meso-carbon centres and also by undergoing
certain structural variations.
Porphyrin is an 18-n: electron system and hence exhibits aromaticity18.
The simplest porphyrin is known as porphine which is the H-analogue (R1 -
R12 = H) of 1. Besides many synthetic and naturally occurring ones with the
core structure mentioned above, there are many biologically active systems
which also have porphyrin-like structures which are given in 2-7.
Porphin Chlorin Phorbin
2 3 4

Bacteriochlorin Porphyrazine Phthalocyanine
5 6 7
The prophyrin ring provides a vacant site at its center, ideally suited for
metal incorporation. The NH protons inside the ring of porphyrins possess
acidic character and hence can get deprotonated to give porphyrinato ions.
These dianion species with their electronically sensitive planar n-framework
and central cavity with more or less rigid size exhibit remarkable ligation
characteristic towards metal ions. Thus derivatives of porphyrins with almost
all metals and semimetals have been synthesisedl. A crucial factor to form
stable metalloporphyrins seems to be the compatibility of porphyrin ring size
with the ionic radii of the metal cations". Hence stable complexes generally
result when these two sizes match while their instability tends to increase when
the size of the cation is too big or too small with very few exceptions. The
porphyrinato dianion is ideally suited to act as a tetradenate ligand with metal
ions2'. Thus the minimum coordination number of the metal ion possible in a
metalloporphyrin is four. A size matching divalent metal ion would give
neutral complex, while a higher valent cation would carry with it balancing
anion@) mostly covalently bound with the metal, in addition to the
porphyrinato ion. f i e normal coordination geometry around the metal ion in
the former species would be square planar, while in the latter case the
coordination geometry would be square pyramidal. Coordination number
greater than four is also possible. The two ligands of the six coordinate
metalloporphyrins are found on the opposite sides of the porphyrinato plane
yielding complexes with tetragonal or octahedral geometries20. Ability to
exhibit variable oxidation states of metals in their metalloporphyrins is another

important feature in this class of compounds. They are also capable of
stabilizing metal ions in their unusual oxidation states, which have resulted in
extensive studies revealing interesting chemistry.
The nature of bonding between a central metal and the porphyrin ligand
is found to be originating essentially from the following two types of primary
interactions: a-coordination of nitrogen lone pairs directed towards the central
metal atoms and n-interaction of metal pn or dn orbitals with nitrogen-based n
orbitals2'. The appropriate symmetry-adapted linear combinations of
prophyrin-ligand orbitals involved in the bonding with metal orbitals are shown
in 8.
(b)
8
The symmetry adapted linear combi~tion ofpotphyin-Iigand orbitals involved in the
bonding with metal obitals (a) me suitable for einteraclonsand (6) for z-interactions
In the a-system the prophyrin is clearly a donor to the metal while in the
x-system porphyrin has the appropriate orbitals to act both as a n-donor and as
a n-acceptor.

The versatile characteristics of the ubiquitous porphyrin molecules can
be attributed largely to the extensively delocalised 7r-system which is
electronically very sensitive and tunable. A knowledge on all such crucial
factors is often necessary before one tries to design and develop the ideal
molecular system for any specific purpose. The studies on porphyrins so far
indicate the following factors to be very significant.
(i) The nature of peripheral substituents has great ability to tune the
electronic levels of porphyrin and their metalloderivatives.
(ii) The type of central metal ion has a very pronounced effect on the
electronic property of the porphyrins. The nature of interaction between
the metal ion and the porphyrinato moiety is such that both the species
mutually influence their electronic levels.
(iii) There is generally a thermodynamic drive for square planar complexes
to add on axial ligands, if available. Most of the metal(II) porphyrin
complexes thus tend to take octahedral geometry when exposed to
coordinating species. Such a change in geometry affects both the metal
electronic levels and porphyrinato orbital energies. Five coordinated
species for metal(1I) porphyrins with one neutral axial ligand are
generally unstable but have been suggested to occur as an intermediate
in solution state. Electronically, five coordinate species are different
from six coordinate complexes and are more reactive due to
coordinative unsaturation.
(iv) In most of the biological systems, the porphyrin moiety is often covered
and buried at specific sites by the long chain of the protein residues.
This steric crowding of protein chain around it can cause some tilt or
puckering in the planar n-framework of the metalloporphyrin. Such a
distortion then would cause a noticeable decrement in the extent of
overlap of certain n-molecular orbitals of the macrocycle with symmetry

matching metal orbitals. The net effect of this would be an enforced
strain on the molecule. The tendency of these entatic species would be to
release the strain that is supposed to be the driving force for these
species to behave as biological catalysts.
1.3. Electronic Properties of Porphyrins and Metalloporphyrins
The most useful spectroscopic technique for the study of porphyrin and
their metalloderivatives is the electronic absorption spectroscopy. As the
spectral absorptions are found to be sensitive to the nature of porphyrins and its
surroundings, vital information could be obtained on the nature of chemical
environment in which they exist and on the role these molecules play in key
biological functions they take part, all by just monitoring the electronic spectra
in respective conditions. Since the present study deals with some aspects of
aggregation characteristics of porphyrins and environment effects on their
electronic spectra provide a vital tool to study them. A brief description on the
origin of the spectra of the porphyrins and their metalloderivatives are given
below.
The electronic heart of a porphyrin is the inner 16-membered ring with
18-x electrons. The ring is structured with basic fourfold symmetry, including
four nitrogen atoms directed towards the center. This electronic heart is
responsible for the unique porphyrin-type optical spectra, which are then
perturbed to a greater or lesser extent by various chemical modifications to the
basic structure.
Porphyrin and its metal derivatives are of considerable spectroscopic
interest because of their simplicity and uniqueness. The optical absorptions of
porphyrin are determined essentially by the nelectrons on the porphyrin ring,
with only minor perturbation from the electrons of the central substituents.
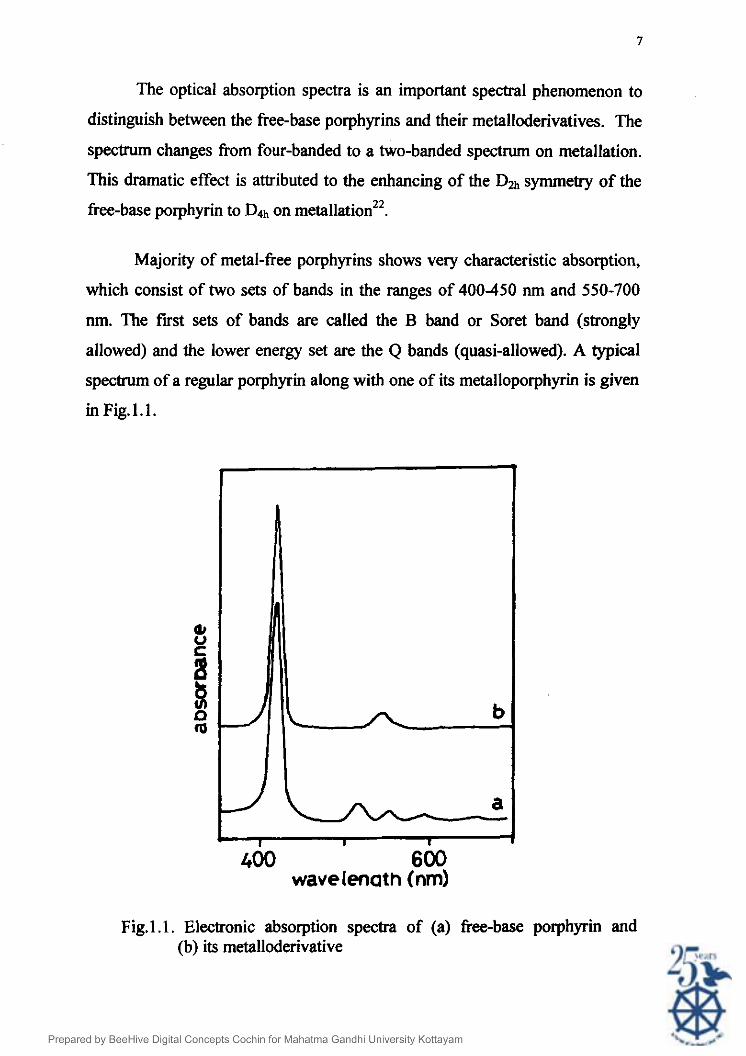
The optical absorption spectra is ati important spectral phenomenon to
distinguish between the free-base porphyrins and their metalloderivatives. The
spectrum changes from four-banded to a two-banded spectrum on metallation.
This dramatic effect is attributed to the enhancing of the DZh symmetry of the
free-base porphyrin to D4h on metal~ation~~.
Majority of metal-free porphyrins shows very characteristic absorption,
which consist of two sets of bands in the ranges of 400-450 nm and 550-700
nm. The fust sets of bands are called the B band or Soret band (strongly
allowed) and the lower energy set are the Q bands (quasi-allowed). A typical
spectrum of a regular porphyrin along with one of its metalloporphyrin is given
in Fig.l.1.
400 600 wavelenath (nm)
Fig.1 .l . Electronic absorption spectra of (a) free-base porphyrin and (b) its metalloderivative

The Q bands of free-base porphyrins are a set of four absorptions arising
from HOMO to n* transition. Of these, the first set of two lines is X-
component of Q while the second set is its y-component. Both these Q, and Q,
components are composed of two types of vibrational excitations too, the lower
energy one being Q(0,O) and the higher energy one Q(1,O). Thus the four lines
in the set are Q,(0,0), Q,(1,0), QJ0,O) and Q,(1,0) in the increasing order of
energy.
On metallation, the spectrum shows an intense B (Soret) band at -420
nm and two weaker Q bands at - 550-600 nmlsP. These spectral absorptions
arise from n-z* transitions of the aromatic porphyrin ligand.
The widely accepted model to fit this spectrum, the four-orbital model,
treats the porphyrin as a cyclic polyene and emphasizes the transition between
the two highest filled bonding molecular orbital levels, a,,, azU and the lowest
empty doubly degenerate antibonding molecular orbital levels eg*. The
allowed transitions, a,, -+ eg* and azU -+ eg* are assumed to be degenerate in
energy. As a consequence, the states undergo configuration interaction and
give rise to new states. The resulting spectrum shows a highenergy band B in
which the transition dipoles add and a low-energy band Q in which the
transition dipoles cancel. The two Q bands are vibronic components of the
same tran~ition*~.
1.4. Types of Porphyrins
There are a variety of porphyrins that are of special significance.
Described below in brief are some of them.
(a) Protoporphyrin
Protoporphyrin (9) contains four methyl groups, two vinyl groups and
two propionic acid groups on its periphery. Fifteen different isomeric
protoporphyrins differing in the sequence of substitution of the above groups in

eight available positions are possible. Hemin is the prosthetic group of
hemoglobin, myoglobin, peroxidase, P-type cytochromes, catalase, tryptophan
pyrrolase, cytochrome P450 and many other proteins. Synthesis of porphyrins
derived from those found in animal materials invariably involves hemin or
protoporphyrin as starting material.
Protoporphyrin dimethyl ester is most easily obtained from hemin by
iron removal and esterification. The original Grinstein procedure that
accomplishes both operations simultaneously involves passage of gaseous HCI
through a methanolic solution of heminz5.
0) De@teropo~vrin Deuterohemin (10) was a key intermediate in Fischer's total synthesis of
hemin. A Friedel-Crafts acylation of deuterohemin folowed by reduction and 26.27 dehydration gave protohemin . Because they have two free P-positions,
deuteroporphyrin and deuterohemin have been used as starting materials in the
synthesis of several porphyrins in which the vinyl groups of protoporphyrin are
replaced by other functionalities. The 2,4dibromo, diacetyl, dipropionyl and
dihydroxymethyl derivatives are all synthesised directly from
deuteroporphyrin. Purification of deuterohemin was done using
deuteroporphyrin in Caughey's methodz8. The iron chelate is obtained by
reinsertion of iron into deuteroporphyrin dimethylester. Deuteroporphyrin-free
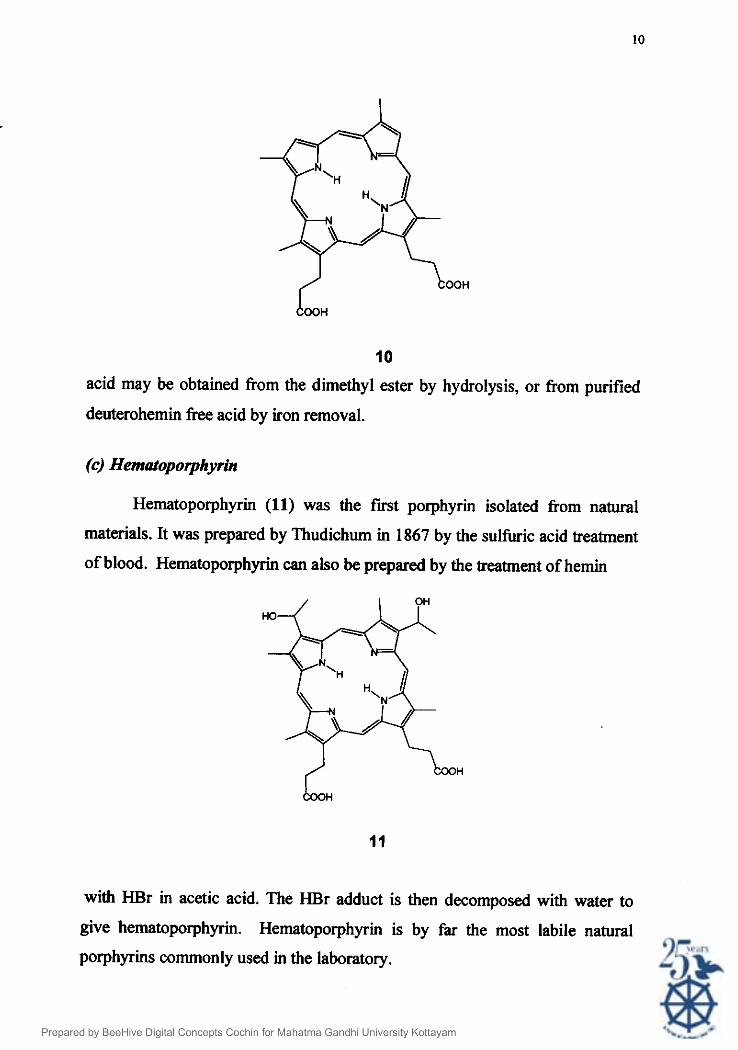
10
acid may be obtained from the dimethyl ester by hydrolysis, or from purified
deuterohemin free acid by iron removal.
(c) Hematoporphyrin
Hematoporphyrin (11) was the fmt porphyrin isolated from natural
materials. It was prepared by Thudichum in 1867 by the sulfuric acid treatment
of blood. Hematoporphyrin can also be prepared by the treatment of hemin
with HBr in acetic acid. The HBr adduct is then decomposed with water to
give hematoporphyrin. Hematoporphyrin is by far the most labile natural
porphyrins commonly used in the laboratory.

(d) Mesoporphyrin
Mesoporphyrin (12) is used in biological and chemical studies especially
where the liability of the vinyl groups of protoporphyrin is a factor. The
classical method of mesoporphyrin synthesis was HI reduction of protohemin29. -++& \ / ce) K 7 - - y . w .
\ 7 y //-, h!, OOH <.: .- - - - -4-
Better result was obtained by the catalytic reduction over PdO of protohemin,
protoporphyrin or protoporphyrin dimethyl ester in formic acid at elevated
temperature. The most convenient method for obtaining large amount of
mesoporphyrin was developed by caughey3'. In this method hydrogen was
bubbled through a formic acid solution of hemin over PdO.
(e) Diace@ldtwteroporphyrin
This prophyrin (13) possesses electron withdrawing acetyl groups in the
2- and 4-positions. Fischer's classical method is now used for the synthesis of

diacetyldeuteroporphyrin. Crude deuterohemin was dissolved in acetic
anhydride at 0°C followed by the addition of anhydrous SnCb under constant
stirring. Longer reaction times result in substantial decomposition, and shorter
times give substantial mount of the monoacetylated product. The solution is
further acidified using 0.1N HCI and thoroughly stirred to avoid acetic
anhydride formation. The crude diacetyldeuterohemin is isolated by suction
filtration.
Diformyldeuteroporphyrin (14) with two strongly electron-withdrawing
formyl groups has been used in the studies of the relationship of heme structure
to heme protein function3'. Fischer's method, alkylation of deuterohemin with
dichloromethyl methyl ether in the presence of SnBr4 followed by hydrolysis,
gives a mixture of products including the monoformyl compounds.
C-type cytochromes (15) contains a prosthetic group, a covalent bound
hemin, that can be regarded as been formed by the addition of two cystane
sulfhydryls across the double bonds of protoheme. Treatment of cytochrome C

with silver or mercury salts result in the formation of optically active
hematoporphyrin. Acid hydrolysis of horse heart cytochrome C results in the
formation of a homogenous amphoteric porphyrin fraction, which has been 32-34 shown to be porphyrin C .
1.5. Porphyrins in Life Chemistry
Metal complexes of porphyrins and related compounds are important
prosthetic groups that assemble to form a wide variety of proteins and enzymes
working as redox and rearrangement catalysts3'. The nature being a master
designer, has chosen and retained the best choice of molecular assemblies in
living systems to cany out very specifically the functions they are intended for
and also to achieve them with the maximum efficiency.
A large number of naturally occurring porphyrin have been isolated and
characterized. Of these protoporphyrins are the most abundant and widely
characterized ones. It is found in hemoglobin, myoglobin, heme enzymes and
most of the cytochromes5.

The biological and chemical importance of metalloporphyrins has
brought to focus intense interest in the nature of the metal ligand linkages in
such complexes as well as all the physicochemical properties of the
macrocycles. The macrocycle has the ability to function as a reservoir of
electrons and control the reactivity at the axial position of metal, which usually
serves as a catalytic site in heme enzyme. Because of their ubiquitousness and
the variety of their natural functions, heme proteins have been investigated on
multi- and interdisciplinary levels. These proteins all containing an iron
porphyrin as the prosthetic group, are responsible for oxygen transport and
storage (hemoglobin and myoglobin)36, electron transport (cytochrome~)~~,
oxygen reduction (cytochrome o x i d a ~ e ) ~ ~ , hydrogen peroxide utilization and
destruction (peroxidases and cata~ases)~', and hydrocarbon oxidation
(cytochrome, p.450)~~.
Also the various functions of heme proteins in the transport, storage and
reactions with dioxygen are made possible by different and selective
interactions of diverse proteins with the heme groups'0. These differences are
brought about largely by the axial ligands provided by ancillary groups of the
protein and from the nature of the pockets on either side of the porphyrin.
Important porphyrin based natural systems are the hemoglobin, myoglobin,
chlorophyll, heme enzymes and the cytochromes.
1.5.1. Hemoglobin and myoglobin
Hemoglobin and myoglobin are high molecular weight protein systems
containing iron(I1) protoporphyrin IX units. They are responsible for oxygen
transport and storage in higher animals9. Hemoglobin transport dioxygen from
its source to the site of use inside the muscle cells. There the oxygen is
transferred to myoglobin for use in respiration. Myoglobin which is responsible
for storing oxygen in cells, has high affmity for the dioxygen even at low
partial pressure but in lungs, hemoglobin takes up high amount of oxygen at
high partial pressure. This special property of hemoglobin is attributed tothe

'cooperative effect" caused by the decrement in size of ~ e ~ ' in central hole due 40,41
to spin change (from high spin to low spin) on dioxygen complexation .
The iron in hemoglobin and myoglobin is in the ferrous state and has to
have an N-base (histidine) coordinated to the metal from one of the sides of the
plane to have dioxygen bound at the vacant sixth coordination site. The
oxidized form of iron, ie. ferric state, called metmyoglobin and methemoglobin
will not bind oxygen. The free heme is immediately oxidized in the presence of
oxygen and water and thus renders useless for Oz transport.
Myoglobin exhibits greater affimity of O2 than hemoglobin and it is
largely converted to oxyrnyoglobin even at low O2 concentration in order to
effect the transport of O2 at the cell9. Upon the oxygenation of hemoglobin, two
of the heme groups move about 100 pm towards each other while two others
separate by about 700 pm due to the action of protein envelope. The net result
of this combined movement is that hemoglobin can exhibit relatively low
affmity for binding the f i t one or two oxygen molecules. But once they are
bound, the binding of subsequent O2 molecule is greatly enhanced. Conversely
the loss of one oxygen molecule from fully oxygenated hemoglobin causes the
rest to dissociate more readily when the oxygen pressure is decreased42.
1.5.2. Chlorophyll
Chlorophyll is the green colouring matter of leaves and green stems, and
its presence is essential for photosynthesis. In green plants it is the chlorophyll
which absorbs the light energy. Chlorophyll is basically magnesium derivative
of porphyrins and some slight structural changes in porphyrin moiety results in
different classes of chlorophyll with slight difference in photocatalytic
/ - properties5. All of the chlorophylls absorb light very intensely, particularly at
relatively long wavelength regions43. The light energy absorbed by a
chlorophyll molecule become delocalised and spread throughout the entire
electronic structure of the excited molecule.

The photosynthetic pigments in the chloroplasts of plants consists of two
functional units namely photosystem I and photosystem 1 1 ~ ~ . Photosystem I
contains chlorophyll, p-carotene and a single molecule of P-700, a specialised
chlorophyll a which serves as an energy trap. Photosystem II has a
characteristic reactive centre namely Pa80 a specialized chlorophyll-protein
complex. Photosystem I absorbs light at longer wavelength. Photosystem I1 is
activated by shorter wavelength, ie. 670 nm and below and it is responsible for
oxygen evolution.
Both the photosystems contain chlorophyll a and chlorophyll b. In
photosystem I, the ratio of chlorophyll a to chlorophyll b is higher than in
photosystem 11'. These two photosystems must cooperate to yield maximum
result in photosynthesis.
Enzymes are biological catalysts that govern, initiate and control
biological reactivity important for the life processes. They are produced by the
living organism and are usually present in only very small amounts in the
various cells. All known enzymes are proteins and some contain non-protein
moieties termed prosthetic groups that are essential for the manifestation of
catalytic a c t i ~ i t i e s ~ ' ~ ~ . In several natural enzymes, metalloporphyrins constitute
these prosthetic groups, some of which are discussed in brief here.
There are various enzymatic reactions in which one or both atoms of 4
are directly inserted into the organic substrate molecule to yield hydroxyl
groups. Enzymes catalyzing such reactions are called oxygenase, of which
there are two classes - the dioxygenase catalyzes insertion of both atoms of the
0 2 molecules into the organic substrate, whereas the monooxygenase inserts
only one4.

The most important monooxygenase is cytochrome P-450 found in the
microsomes of liver cells4'. Cytochrome P-450 contains protoheme. The CO
derivative of its reduced form absorbs maximally at 450 nm, hence the name
cytochrom P-450. During the enzyme reaction the ferric P-450 first combines
with a substrate followed by a oneelectron reduction to form a ferrous P-450
substrate complex. The reduced Fe(II) form of P-450 reacts with molecular O2
in such a way that one of the 0-atoms is reduced to water and the other is
introduced into the organic substrate.
Cytochrome P-450 enzymes catalyze hydroxylation of many different
k i d s of substrates including steroids, fatty acids, certain amino acids etc and
thus making them more water-soluble. They also promote hydroxylation of
various drugs.
1.5.3.2. Peroxidases and Catalase
Peroxidases are enzymes catalyzing the oxidation of a variety of organic
and inorganic compounds. Peroxidases obtained from plants contain hemine
groups. The different types of peroxidases are horseradish peroxidase, mylo
peroxidase, chloroperoxidase etcB.
Catalase is also a heme enzyme which catalyze the dismutation of H202
generated during various life processes. Catalase is made up of four identical
sub-units each containing one heme group. The axial metal sites appear to be
occupied by water and an amino acid residue.
1.5.4. The Cyt@chromes
The cytochromes are electron transferring proteins, containing iron
porphyrin, found in aerobic cells. Some cytochromes found in endoplasmic
reticulam, play a role in specialized hydroxylation reactions36. All cytochromes
undergo reversible Few-Fe(II) valency changes during their catalytic cycles.
In almost all the cytochromes both the fifth and sixth positions of the iron are
occupied by the R groups of specific amino acid residue of the proteins46.

Therefore, these cytochromes cannot bind with ligands like 4, CO or C N . An
important exception is cytochrome oxidase that normally binds O2 in its
biological function.
The iron protoporphyrin group of cytochrome c is covalently linked to
the protein by thioether bridges between the prophyrin ring and two cysteine
residue in the peptide chain whereas in other cytochromes the porphyrin ring is
non-covalently bound. Cytochrome c is the only common heme moiety in
which the heme is bound to the protein by a covalent linkage.
1.6 Applications of porphyrins and metalloporphyrins
Porphyrins and related macrocycles provide an extremely versatile
synthetic base for a variety of material applications. The broadly defmed
porphyrin research area is one of the most exciting, stimulating and rewarding
for scientists in the filed of chemistry, physics, biology and medicine. The
beautifully constructed porphyrinoid ligand, perfected over the course of
evolution, provides the chromophore for a multitude of iron, magnesium, cobalt
and nickel complexes which are primary metabolites and without which life
itself could not be maintained. The field is spreading rapidly in every direction
across the whole spectrum4'.
Diverse applications of porphyrins and metalloporphyrins to materials
chemistry have been developed over the past decade, both for their optical
properties and their applications as sensors. Notably, porphyrins and
metalloporphyrins have found applications as field-responsive materials,
particularly for optoelectronic applications, including mesomorphic materials
- and optical-limiting coatings. For example, the facile substitution of the
periphery of various porphyrins has generated a series of unusual liquid
crystalline materials. The porphyrin ligand serves as a platform on which one
can erect desirable molecular and materials properties. The nonlinear optical

properties of these materials are of special interest, in part for energy transfer
with molecular control, and in part for potential application in optical
communications, data storage and electrooptical signal processing. The
stability of mono- and di-cation porphyrin x-radicals makes these systems
especially interesting for photoionization processes.
Porphyrins and metalloporphyrins can also be used as nonlinear optical
materials. They have desirable properties for use in optoelectronics. They have
greater thermal stability and their extended lrconjugated macrocycle ring give
large nonlinear optical effects and subtle variation in their physical properties
can be made easily through chemical modification of their periphery48-i4. They
also play key roles in adsorbing light energy over a wide spectral range and
converting it into the highly directional transfer of electrons5*". It is a
marvelous but highly complex process that has inspired considerable interest in
the synthesis of porphyrh arrays. A biomimetic approach to the photosynthetic
apparatus may also lead to applications of similar systems as optoelectronic
devices.
Because of their inherent stability, unique optical properties, and
synthetic versatility, porphyrins and metalloporphyrins are excellent candidates
for a variety of sensing-materials applications. Research in this area has
focussed on incorporation of synthetic porphyrins and metalloporphyrins into a
variety of material matrices, such as polymers, glasses and films6'. The unique
spectral characteristics and synthetic versatility of porphyrins allow a variety of
sensing applications. They are also used in the detection of organic vapours and
ionic species in solution.
Photochemical reduction of water utilizes only a limited portion of sun's
ray. Porphyrins, whose absorption spectra wver an appreciable portion of the
spectrum of sunlight, are of great interest in this respect. Also
metalloporphyrins exhibit very high photochemical stability6'. Large attempts

are also made to utilize the photochemical properties of metalloporphyrins in
organic synthesis. For eg., it was found that irradiation by a W-visible light to
a solution of Fe(TDCPP)OH in 02-satuntted cyclohexane led to progressive
formation of cyclohexane6'. The author proposed a mechanism for this reaction
in which the fmt step is a photodissociation of the Fe(II1)-OH bond of the
catalyst leading to (TDCPP)Fe(II) and OH followed by H-atom abstraction of
cyclohexane by OH as represented in Scheme 1.
Scheme 1 Mechanism for the oxidation of alkanes to ketones by O2 catalyed by photoactivated Fe(TDCPP)OH
Metalloporphyrins of aluminium, zinc, manganese, cobalt and rhodium
complexes have been demonstrated to serve as excellent initiators for
controlled anionic and free radical polymerizations6466. The discovery of these
metalloporphyrins-based initiators has led to significant contributions to the
progress of precision macromolecular synthesis via "living polymerization"
(ie., growth pattern of the macromolecule can be viewed as analogous to the 67-69 growth of a biological organism) .
The worldwide approval of Photofib [a purified version of
Hernatoporphyrin derivative (HpD); a complex mixture of dimers and
oligomers in which porphyrin units are joined by ether, ester and carbon-carbon
bonds] for the treatment of various types of cancers has created enormous

70-71 interest among physicians, chemists, biologists and physicists . During the
early and mid 1970s, several groups including Diamond et. aL7', Kelly and
~ n e l l ~ ~ , and Dougherty et. a~. '~ , realized that together HpD and light (Photo
dynamic therapy) had a, potential capability for tumor destruction. This is now
one of the accepted modalities for the treatment of ~ a n c e r ' ~ . ~ ~ . In addition to its
use for cancer treatment, Photo dynamic therapy has also shown a potential for
applications in other areas: treatment of aged-related macular degeneration,
Psoriasis, bone marrow purging, arthritis and purification of blood infected
with various viruses inchding HIv4'. At present, all the photosensitizers in
clinical trials are based on tetrapyrroles (porphyrins, chlorins, bacteriochlorins
and pthalocynanines), and it seems that porphyrin in general will show
continued interest in the exiting area of photodynamic therapy.
1.7 References
1. J.W. Buchler, "The Porphyrins", D. Dolphin (Ed.), Academic, New York, Vol. 1, Part A, Structure and Synthesis (1978).
2. D. Ostfeld, Tsutsui, Acc. Chem. Res., 7, 52 (1974).
3. 3. O.A. Golubchikov and B.D. Berezin, Russ. Chem. Rev., 55(8), 1361 (1986).
4. 0. Hayashi, "The Enzymes", P. Boyer, H. Lardy, K. Myrback (Eds.), Academic Press, New York, Vo1.8 (1963).
5. A.L. Lehinger, "Biochemistry", Kalyani Publishers, New Delhi (1978).
6. E.L. Smith, R.L. Hill, I.R. Lehman, R.J. Lef Kowitz, P. Handler, A. White. "Principles of Biochemistry- General Aspects", McGraw-Hill Inc., Singapore (1983).
t - 7. T.S. Mashiko, "Comprehensive Coordination Chemistry", Geoffery and Wilkinson (Eds.), Vo1.2, Pergamon Press, Oxford (1987).
8. G.I. Likhtenshtein, "Chemical Physics of Redox Metalloenzyme Catalysis", Springer-Verlag, Berlin, (1988).

9. J.E. Huheey, E.A. Keiter, R.L. Keiter, "Inorganic Chemistry - Principles of Structure and Reactivity", Harper Collins College Publishers, New York
b (1993).
10. J.P. Collman, Inorg. C:hem., 36,5145 (1997).
1l.O.A. Golubchikov, B.D. Berezin, Russ. Chem., Rev., 55(8), 768 (1986).
12. B. Meunier, Chem. Rev., 92, 141 1 (1992).
13. V.V. Borovkov, R.P. Evstigneeva, L.N. Strekova, E.I. Fillippovich, Russ. Chem. Rev., 58(6), 602 (1989).
14. K.E. Keller, N. Foster, Inorg. Chem., 31, 1353 (1992).
15. K.S. Suslick, C.T. Chen, G.R. Meredith, L.T. Cheng, J. Am. Chem. Soc., 114,6928 (1992).
16.L.G. Marzilli, New J. Chem., 14,409 (1990).
17.N.E. Mukundan, G. Petho, D.W. Dixon, L.G. Marzilli, Inorg. Chem., 34, 3677 (1995).
18.M. Gouterman, "The Porphyrins", D. Dolphin (Ed.) Academic New York, Vol.111, Part A, Physical Chemistry (1978).
19. J.W. Buchler, "Porphyrins and Metalloporphyrins", K.M. Smith (Ed.), Elsevier (1 975).
20. W.R Scheidt, Accounts of Chemical Research, 10,339 (1977).
21.K. Tatsurni, R. Hoffinann, J. Am. Chem. Soc., 103,3328 (1981).
22.M. Gouterman, J. C'hem. Phys., 30,1139 (1959).
23.L.J. Boucher, Coord. Chem. Rev., 7,289 (1972).
24.M. Zerner, M. Goutennan, Theoret. Chim. Acta, 4,44 (1966).
25. M. Grinstein, J. Biol. Chem., 167,515 (1947).
26.H. Fischer, K. Zeile,, Justus Liebigs Ann. Chem., 468,98 (1929).
27.H. Fischer and R. Muller, Hoppe-Scylers, Z. Physiol. Chem., 142, 155 (1925).

28. W.S. Caughey, J.O.Alben, W.Y.Fujimoto, J.L. York, J. Org. Chem., 31, 2631 (1966).
29. S. Schwartz, M.H. Berg, I. Bossenmaier, H. Dinsmore, Methods Biochem. Anal., 8,221 (1960).
30. W.S.Caughey, W.Y. FujimotoJ. Bearden, T.H. Moss, Biochemistry, 5, 1255 (1966).
31.T.Asakura, M. Sono, J. Biol. Chem., 249,7089 (1974).
32.R Hill, D. Keilin, Prw. R. Soc. London, Ser. B., 107,286 (1930).
33.H. Theorell, Enzymologia, 6,88 (1939).
34.H. Theorell, K. Akeson, J. Am. Chem. Soc., 63, 1804 (1941).
35. R.A. Pasternack, A. Ghetto, P. Pagano, E.J. Gibbs, J. Am. Chem. Soc. 113,7799 (1991).
36.F.S. Mathews, Prog. Biophys. Mol. Biol., 45, 1 (1985).
37. Y. Hatefi, Ann. Rev. Biochem., 54,1015 (1985).
38. J.E. Frew, P. Jones, Adv. Inorg. Bioinorg. Mech., 3, 175 (1984).
39.R.T. Murray, M.T. Fischer, P.G. Debrunner, S.G. Sligar, Top. Mol. Struct. Biol., 6 (Metalloproteins, Pt.l), 157 (1985).
40. J.L. Hoard, "Hemes and Hemoproteins", B.Chance (Ed.), Academic Press, New York (1966).
41. W.R. Scheidt, C.A. Reed, Chem. Rev., 81,543 (1981).
42.M.S. Perutz, Nature, 228,726 (1970).
43.H.H. Seliger, W.D. McElory, Light; Physical and Biological Action, Academic Press, New York, 1965.
44.K. Saner, Annu. Rev. Phy. Chem., 30, 155 (1979).
45. K.E. White, M.J. Coon, Annu. Rev. Biochem., 49,356 (1980).
46.T. Takano, B.L. Trus, N. Mandel, G. Mandel, O.B. Kallai, R Swanson, R.F. Dickerson, J. Biol. Chem., 252,776 (1977).

47. J.-H. Chou, M.E. Kosal, H.S. Nalwa, N.A. Rakow, K.S. Suslick, "The Porphyrin Handbook", K.M. Kadish, K.M. Smith, R. Guillard (Eds.) Vol. 3, Academic Press, New York (2000).
48. H.S. Nalwa, "Nonlinear Optics in Organic Molecular and Polymeric Materials", H.S. Nalwa, S. Miyata (Eds.) CRC Press, Boca Raton, FL, 61 1- 797 (1997).
49. H.S. Nalwa, T. Watanabe, S. Miyata, "Nonlinear Optics in Organic Molecular and Polymeric Materials", H.S. Nalwa, S. Miyata (Eds.) CRC Press, Boca Raton, FL, 61 1-797 (1997).
50. H.S. Nalwa, "Handbook of Organic Conductive Molecules and Polymers", H.S. Nalwa (Ed.) John Wiley & Sons, Chichester, 261-363 & Vol. 1-4 (1997).
51. H.S. Nalwa, J.S. Shirk, "Pthalocyanines: Properties and Applications", C.C. Leznoff, A.B.P. Lever (Eds.)Vol. 4,79-118 (1996).
52.H.S. Nalwa, Adv. Mater., 5,341 (1993).
53.H.S. Nalwa, Appl. Organometal. Chem., 5,349 (1991).
54. D.S. Chemla, J. Zyss, "Nonlinear Optical Properties of Organic Molecules and Crystals", Academic Press, Orlando (1987).
55.G.W. Robison, Brookhaven Symp. Biol., 19, 16 (1967).
56. R.H. Pearlstein, New Compr. Biochem., 15,299 (1987).
57.G.R. Fleming, J.-L. Martin, J. Breton, Nature, 333, 190 (1988).
58.M.L. Paddock, S.H. Rongey, G. Feher, M.Y. Okamura, Proc. Natl. Acad. Sci., USA, 86 (1989).
59.G. Feher, J.P. Allen, M.Y. Okamura, D.C. Rees, Nature, 339, 11 1 (1989).
60.D. Holten, C. Kirmarier, Photosynth. 13,225 (1987).
61.A.E. Baron, J.D.S. Danielson, M. Gouterrnan, J.R. Wan, J.B. Callis, Rev. Sci. Instrum., 64, 3394 (1993).
62.0.A. Golubchikov, B.D. Berezin, Russ. Chem. Rev., 55,768 (1986).
63.D. Mansuy, Coord. Chem. Rev., 125, 129 (1993).

64.T. Aida, S. Inoue, Acc:. Chem. Res., 29.39 (1996).
65.S. Inoue, T. Aida, Chemtech, 24,28 (1994).
66. B.B. Wayland, S. Mukerjee, G. Posmik, D.C. Woska, L. Basickes, A.A.M. Gridnev, S.D. Ittel, ,4CS Sympo. Ser. 686,305 (1998).
67.M. Szwarc, M. Levy, K. Milkfish, J. Am. Chem. Soc., 78,2656 (1956).
68.T. Aida, Prog. Polym. Sci., 18,469 (1994).
69. S. Inoue, T. Aida, "New Method for Polymer Synthesis", W.J. Mijs (Ed.), Plenum, New York, 33 (1992)
70. J.S. McCaughan Jr., J. Clin. Laser Med. Surg., 14,223 (1996).
71.H. Kato, T. Okunaka, H. Shimatani, J. Clin. Laser Med. Surg., 14,235 (1996).
72.1. Diamond, S.G. Graneli, A.F. McDonagh, C.B. Wilson, S.L. Nielsen, Lancet, 2, 1 175 (1972).,
73.J.F. Kelly, M.E. Snell, J. Urol., 155,150 (1976).
74.T.J. Dougherty, G.B. Grindley, R. Fiel, K.R Weishaupt, D.G Boyle, J. Natl. Cancer. Inst., 55, 115 ( 1975).
75. T.J. Dougherty, J. Clin. Laser Med. Surg., 14,219 (1996).
76.FDA Approval for Photofiin for Early NSCLC, Oncology Times (1998).





























