Embed Size (px)
Citation preview

PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [Moutsatsou, P.]On: 28 July 2009Access details: Access Details: [subscription number 913459393]Publisher Informa HealthcareInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Cancer InvestigationPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713597231
Ursolic Acid Triggers Apoptosis and Bcl-2 Downregulation in MCF-7 BreastCancer CellsE. Kassi a; T. G. Sourlingas b; M. Spiliotaki a; Z. Papoutsi a; H. Pratsinis b; N. Aligiannis c; P. Moutsatsou a
a Laboratory of Biological Chemistry, Medical School, University of Athens, Athens, Greece b NCSRDemokritos, Institute of Biology, Athens, Greece c Laboratory of Pharmacognosy, Department of Pharmacy,University of Athens, Panepistimiopolis, Zografou-Athens, Greece
First Published:August2009
To cite this Article Kassi, E., Sourlingas, T. G., Spiliotaki, M., Papoutsi, Z., Pratsinis, H., Aligiannis, N. and Moutsatsou,P.(2009)'Ursolic Acid Triggers Apoptosis and Bcl-2 Downregulation in MCF-7 Breast Cancer Cells',Cancer Investigation,27:7,723 —733
To link to this Article: DOI: 10.1080/07357900802672712
URL: http://dx.doi.org/10.1080/07357900802672712
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial orsystematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply ordistribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug dosesshould be independently verified with primary sources. The publisher shall not be liable for any loss,actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directlyor indirectly in connection with or arising out of the use of this material.

Cancer Investigation, 27:723–733, 2009ISSN: 0735-7907 print / 1532-4192 onlineCopyright c© Informa Healthcare USA, Inc.DOI: 10.1080/07357900802672712
ORIGINAL ARTICLECellular and Molecular Biology
Ursolic Acid Triggers Apoptosis and Bcl-2Downregulation in MCF-7 Breast Cancer Cells
E. Kassi,1 T. G. Sourlingas,2 M. Spiliotaki,1 Z. Papoutsi,1 H. Pratsinis,2 N. Aligiannis,3 and P. Moutsatsou1
Laboratory of Biological Chemistry, Medical School, University of Athens, 75 M. Asias, GR-115 27 Athens, Greece,1 Institute of Biology, NCSRDemokritos, 153 10 Athens, Greece,2 Laboratory of Pharmacognosy, Department of Pharmacy, University of Athens, Panepistimiopolis, GR-157
71 Zografou-Athens, Greece3
ABSTRACT
In this report we determine the ability of ursolic acid (UA) to induce apoptosis and to modulateglucocorticoid receptor (GR) and Activator Protein-1 (AP-1) in MCF-7 cells. The UA-inducedapoptosis (53 µM), the PARP cleavage, and the decrease in Bcl-2 protein (53 µM) support thenotion that UA induces apoptosis through the intrinsic mitochondrial pathway. UA binds GR(relative binding affinity: 2.57) and translocates GR into nucleus, suggesting its potential as aGR modulator. UA had no effect on GRE- or TRE-driven gene expression. In summary, UA is aGR modulator and may be considered as a potential anticancer agent in breast cancer.
INTRODUCTION
Glucocorticoids (GCs) inhibit cellular growth and causeapoptosis in lymphocytes (1); however, they show heteroge-neous effects on the growth and chemosensitivity of varioustypes of carcinoma cells (2, 3). Dexamethasone treatment andglucocorticoid receptor (GR) activation in breast cancer cellshave resulted in the regulation of specific genes known toplay key roles in cellular functions, including growth, apop-tosis, differentiation, metastasis, and survival genes associatedwith inhibition of chemotherapy-induced apoptosis (4–6). Inclinical practice, GCs are effective in inducing apoptosis inmany hematological malignancies; however, they are ineffec-tive when coadministered with chemotherapy in the treatmentof nonhematological solid tumors, including breast cancer (7, 8).
GCs mediate their effects by binding to intracellular GR, anuclear transcription factor that regulates the transcription of
Keywords: Apoptosis, Bcl-2 protein, Breast cancer, MCF-7 cells,Ursolic acidCorrespondence to:Dr. P. MoutsatsouDepartment of Biological ChemistryMedical School, Athens University75 Mikras Asias Str.Goudi 11527, Athens, Greeceemail: [email protected]
GC responsive genes (9). Apart from the classical GR (knowntoday as GRα) another GR isoform, the GRβ, does not bind GCand antagonizes the GRα-mediated activity (10). Upon bind-ing to GCs, GRα changes its conformational shape, dissociatesfrom heat-shock proteins (HSPs), homodimerizes, translocatesinto nucleus where it interacts directly with its target DNAsequences, the glucocorticoid response elements (GREs), thusresulting in the stimulation of genes (known as transactiva-tion effect) (11). Moreover, GRα as a monomer may interact,via protein–protein interactions, with other transcription factorssuch as AP-1 and NF-kB, hence influencing the activity of theirtarget genes (known as transrepression effect). AP-1 (Activa-tor Protein-1), constituted by the c-fos and c-jun proteins (ashomodimers or heterodimers), regulates gene expression via itsbinding on a specific DNA site, the phorbol 12-o-tetradecanoate-13-acetate (TPA) responsive element (TRE). It is activated byvarious stimuli, including mitogens and oncoproteins, and reg-ulates many processes such as cellular proliferation, survival,and apoptosis (12–15); therefore it has a role in many types ofhuman cancer, including breast cancer.
Due to the ineffectiveness of GCs as adjuncts to chemother-apy in breast cancer the discovery of new agents is highly de-sirable. Ursolic acid (UA), a pentacyclic triterpenoid acid ofsimilar structure to dexamethasone, has shown antitumorigenicand chemopreventive activity via modulation of cancer-relatedpathways such as apoptosis, invasion, and metastasis (16–18).However, its effects in breast cancer remain largely unknown.
723
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

New strategies for anticancer therapies target apoptosis, theantiapoptotic Bcl-2 protein being an attractive molecular tar-get (19, 20). Poly(ADP-ribose) polymerase (PARP) cleavageis also a major hallmark and a sensitive parameter of apoptosis(21, 22), while caspase-3 and caspase-7 are the main effectors ofapoptotic PARP cleavage (22). More important, the final stagesof apoptosis involve the DNA fragmentation factor (DFF). Inits inactive form, DFF exists as a heterodimer, DFF45/40. Pro-teolysis of DFF45 by activated caspase-3, and to a lesser extentactivated caspase-7, releases DFF40 in its activated form (23,24). In view of the above and because of the crucial role of GRand AP-1 signaling in breast cancer (6, 14, 25), we decided toinvestigate whether UA (i) inhibits MCF-7 cellular growth, (ii)induces apoptosis in MCF-7 cells, (iii) modulates the antiapop-totic Bcl-2 protein, (iv) induces PARP cleavage and changes incaspase-3, caspase-7, and DFF45 levels, and (v) modulates GR-and AP-1-mediated signaling pathways. Our present data sup-port that UA induces apoptosis through the intrinsic mitochon-drial pathway, binds GR, and translocates it into the nucleus,suggesting its potential as a GR modulator. UA had no effect onGRE or TRE-driven gene expression.
MATERIALS AND METHODS
Isolation of ursolic acid
Ursolic acid was isolated from aerial parts of Salvia officinalis(Labiatae).
Air-dried, powdered plant material (1 kg) was extracted atroom temperature with CH2Cl2 (3L X 3) and then MeOH (3L X3). The CH2Cl2 soluble extract was concentrated under reducedpressure to give a residue (25 g), which was chromatographedover a silica gel 60H column and eluted with CH2Cl2–MeOH(100:0 →0:100) (0.5–1.5 L) to give 14 fractions. The sixth frac-tion (0.92 g), eluted with CH2Cl2–MeOH (97:3), was rechro-matographed over silica gel and eluted with CH2Cl2–MeOH(99.5:0.5 →98:2) to afford UA (18 mg).
Cell culture conditions
A cervical adenocarcinoma cell line (HeLa, ATCC CellBank) and a breast carcinoma cell line (MCF-7, ATCC CellBank) were used. All cell lines were grown in Dulbecco’s Mod-ified Eagle Media (DMEM) (Gibco BRL) supplemented with10% fetal bovine serum (FBS) (Gibco BRL), 1% penicillin–streptomycin (50 units/mL penicillin and 50 µg/mL strepto-mycin) (Gibco BRL) at 37◦C, 85% humidity, and 5% CO2
atmosphere. Stock cultures were subcultured every 4–5 daysusing a trypsin 0.25%/EDTA 0.02% solution (Gibco BRL).
Cell viability assay (MTT)
MCF-7 cell viability was estimated by a mod-ification of the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (26), which determines themetabolically active mitochondria of cells. Briefly, cells wereincubated in the absence and presence of UA (0.016–100 µM) in
DMEM phenol red free [PR (−)]. After 48 hr of incubation, themedium was replaced with MTT (Sigma) [final concentrationof 1 mg/mL in serum-free, PR (−)], and a further 4-hr incu-bation followed. The MTT formazan product was solubilizedthoroughly in isopropanol and the optical density was measuredat a test wavelength of 550 nm and a reference wavelength of690 nm (27).
Assessment of apoptosis
Flow cytometry
MCF-7 cells were plated at a density of 50,000/well in24-well plates and grown with DMEM containing 10% FBS.UA at final concentration 53 µM was added and incubationfollowed for 24, 48, and 72 hr. Apoptosis was assessed byflow cytometry in Annexin V-FITC and propidium iodide (PI)-stained cells (TACS Annexin V-FITC Apoptosis Detection Kit,Cat#TA4638, R&D Systems) with a FACS Calibur (BectonDickinson) cytometry.
Western blot analysis for Bcl-2
MCF-7 cells were incubated for 24, 48, and 72 hr in theabsence (vehicle control) and presence of UA (at final concen-tration 53 µM), washed twice in cold PBS, and lysed with 1 mLlysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mMEGTA, 1 mM EDTA, 1% NP-40 with aprotinin 2 µg/mL, leu-peptine 2 µg/mL, PMSF 100 µg/mL) at 4◦C for 30 min withoccasional vortexing and brief sonication. Centrifugation fol-lowed at 14,000 rpm for 10 min and each supernatant (60 µg)was subjected to 12% SDS-polyacrylamide gel electrophoresisand transferred to nitrocellulose membrane. Incubation with ananti-Bcl-2 antibody (N-19, Santa-Cruz) at 1:500 dilution and anantiactin antibody (MAB 1501, Chemicon) at 1:2000 dilution,washing with TBST, and incubation with secondary antibody,antirabbit (Pierce) at 1:7000 dilution and antimouse (Santa Cruz)at 1:2500 dilution, respectively, followed. Chemiluminescencedetection kit was used for visualization of the signal. Densit-ometric quantification of the bands, normalization with actin,and calculation of the ratio Bcl-2 without UA/Bcl-2 with UA(24 hr), Bcl-2 without UA/Bcl-2 with UA (48 hr), and Bcl-2without UA/Bcl-2 with UA (72 hr) were also carried out.
Western blot analysis for caspase-3,caspase-7, PARP, and DFF45
MCF-7 cells were incubated with either 30 or 53 µM UA for24 and 48 hr, respectively, for each concentration used. Cellswere processed as described above and 60 µg of protein fromeach sample were subjected to SDS-polyacrylamide gel elec-trophoresis. After transfer and blocking, the following primaryantibodies were used: 1/1000 dilution of monoclonal anti-caspase-3 (full length, Ab-2, Oncogene/Calbiochem), 1/200dilution of monoclonal anti-caspase-7 (full length, 9494, CellSignalling), 1/500 dilution of polyclonal anti-PARP-1 (9532,Cell Signalling), 1/200 dilution of polyclonal anti-DFF45(sc-9066, Santa Cruz), and 1/5000 dilution of monoclonal
724 E. Kassi et al.
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

anti-α-tubulin (T5168, Sigma). Incubations of all primaryantibodies, except for anti-α-tubulin (1-hr incubation), werecarried out overnight. The secondary antibodies used, theirdilutions, and all other processing were according to the man-ufacturers’ instructions. Immunodetection was accomplishedusing horseradish peroxidase-conjugated secondary antibodiesand enhanced chemiluminescence (ECL Western BlottingDetection Kit, RPN2209, Amersham). Stripping of the blottedmembranes for reprobing was carried out by incubating themembranes in stripping buffer (100 mM 2-mercaptoethanol,2% SDS, 62.5 mM Tris-HCl, pH 6.7) for 30 min at 50◦C withoccasional agitation.
Assessment of GR and AP-1 signalingpathways
Relative binding affinity assay
Cells and cytosol preparation: Monolayer culture of HeLacells – a cell line rich in GR which is widely used for GR stud-ies – were treated with trypsin 0.25%-EDTA 0.02% solution,centrifuged at 3,000 rpm for 20 min, and cell pellets were dis-rupted in buffer (50 mM Tris-base, 5 mM EDTA, 50 mM NaCl,10 mM NaMoO4, 10% glycine, 10−4 M PMSF, 10−3 M leu-peptin, 1 µg/µL trypsin inhibitor, 10−3 M DTT) using ultrasonicdisintegrator. Cytosolic extracts (30 µL) were incubated with10 µL 3H-triamcinolone acetonide (final concentration 20 ×10−9 M) in the absence and presence of DEX or UA (5 × 10−6
M−1 × 10−3 M) at 0◦C for 18 hr. Dextran-coated charcoal(DCC) (100 µL) slurry (0.05% Dextran, 0.5% charcoal) wasadded, and incubation for 15 min at 0◦C and centrifugation at5,000 rpm for 10 min followed. This procedure (DCC slurry)was carried out twice. In the supernatant (120 µL) the radioac-tivity was determined by liquid-scintillation spectrometry afteraddition of 2.4 mL of Lumagel plus. The relative binding affinityassay (RBA) was calculated as the ratio of the moles of DEX andUA required to decrease the amount of bound radioactivity by50%, multiplied by 100. IC50 values were calculated as the con-centration necessary for 50% displacement of 3H-triamcinoloneacetonide binding to the GR.
GR nuclear translocation
Subfractionation and Western blot analysis: We studied thepotential of UA to modulate GR signaling at a concentrationcomparable to that of dexamethasone. MCF-7 cells were treatedwith DEX (0.1 µM), RU486 (0.4 µM) or UA (0.1 µM) for 1 hr or3 hr in DMEM PR (−) medium and centrifuged at 4,000×g for5 min at 4◦C to obtain the cell pellet. Nuclear extracts were pre-pared according to a mini-extraction protocol with slight mod-ifications (28). Briefly, the cell pellet was suspended in 800 µLice-cold buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mMEDTA, 0.1 mM EGTA) containing 5 mM DFP, 1 mM PMSF,1 mM AEBSF, 1 M DTT, 2 mM aprotinin, 20 mM leupeptin,0.01 mM pepstatin, 0.01 mM bestatin, 0.035 mM TPCK, 0.04mM TLCK, and 0.01 mM ALLnL protein inhibitors. Cells wereallowed to swell on ice for 15 min, addition of 75 µL of a 10%
solution of TWEEN 20, vigorous vortex for 20 s, and centrifuga-tion at 10,000 × g for 7 min at 4◦C followed. The nuclear pelletwas suspended in 100 µL ice-cold buffer C (20 mM HEPESpH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA) contain-ing protein inhibitors (as described previously in buffer A), thenuclear homogenate was vigorously rocked at 4◦C for 15 min,centrifuged at 10,000 × g for 10 min at 4◦C, and the supernatantwas frozen in aliquots at −80◦C. Nuclear protein extracts (5 µg)were further submitted to 7.5% SDS PAGE electrophoresis andWestern blot analysis as described above. Blots were then in-cubated for 1 hr at room temperature in TBST (Tris bufferedsaline Tween 20), pH 7.6 containing 5% nonfat milk (blockingbuffer), washed with TBST, and then incubated at 4◦C overnightin blocking buffer with rabbit polyclonal antibody against theGR (E-20, Santa Cruz, dilution 1:1000). Incubation with the sec-ond antibody, horseradish peroxidase linked antirabbit (Pierce,dilution 1:10000) in blocking buffer for 1 hr at room temperaturefollowed and immunoreactivity was detected with the Westernblot detection system (West Pico, Pierce). In order to confirm thepurity of the isolated nuclei, 50 µg of cytosolic, mitochondrial,as well as nuclear extract were submitted to Western blot analy-sis using anti-histone H1 (sc-8030, Santa Cruz dilution 1:100).
Electrophoretic mobility shift assay
Consensus sequences of GR (5′-AGC TTG GTA CAC TGTGTC CTG AAT TCA-3′) and AP-1 (5′-CGC TTG ATG AGTCAG CCG GAA-3′) were used for gel shift assays. Double-stranded oligonucleotides were 5′ end-labeled with [γ−32P]ATP by T4 polynucleotide kinase. The labeled DNA was incu-bated with 5× binding buffer (50 mM HEPES pH 8.0, 500 mMNaCl, 1 mM EDTA pH 8.0, 0.5 mg/mL BSA, 0.5 mM PMSF,20% glycerol), 1 µg poly(dI-dC), 5 mM DTT, and 10 µg ofnuclear extract protein (obtained as previously described (28))for 30 min at room temperature. Electrophoresis followed at150 V for 3 hr on a nondenaturing 6% acrylamide gel, 0.5 XTris-borate EDTA buffer, pH 8.0, followed by gel drying andautoradiography. Specificity was ensured by competition witha 100-fold excess amount of unlabeled GR, or AP-1 specificoligonucleotides. Quantification was performed by densitomet-ric scanning of the autoradiograms using an image analysissystem (Quantity one, Biorad).
Plasmids and transfections
Before each transfection experiment MCF-7 cells were main-tained for 2 days in DMEM PR (−) containing 10% FBSDCC-treated. For each transfection experiment 2 × 105 cellswere plated per well in six-well dishes in DMEM PR (−) with10% FBS DCC-treated. After 24 hr, MCF-7 cells were trans-fected with 0.75 µg MMTV-Luc or 0.75 µg 5XColl-Luc and0.75 µg β-Gal expression vectors by using Polyfect Transfec-tion Reagent (Qiagen) according to manufacturer’s guidelines.After 24 hr, cells were washed once with PBS and 2 mL DMEMPR (−) 10% FBS DCC-treated was added, containing variousfinal concentrations of DEX (10−8–10−6 M), or RU486 (10−8–10−6 M), or UA (10−6–10−5 M). Co-incubation of RU486
Ursolic Acid and Breast Cancer 725
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009
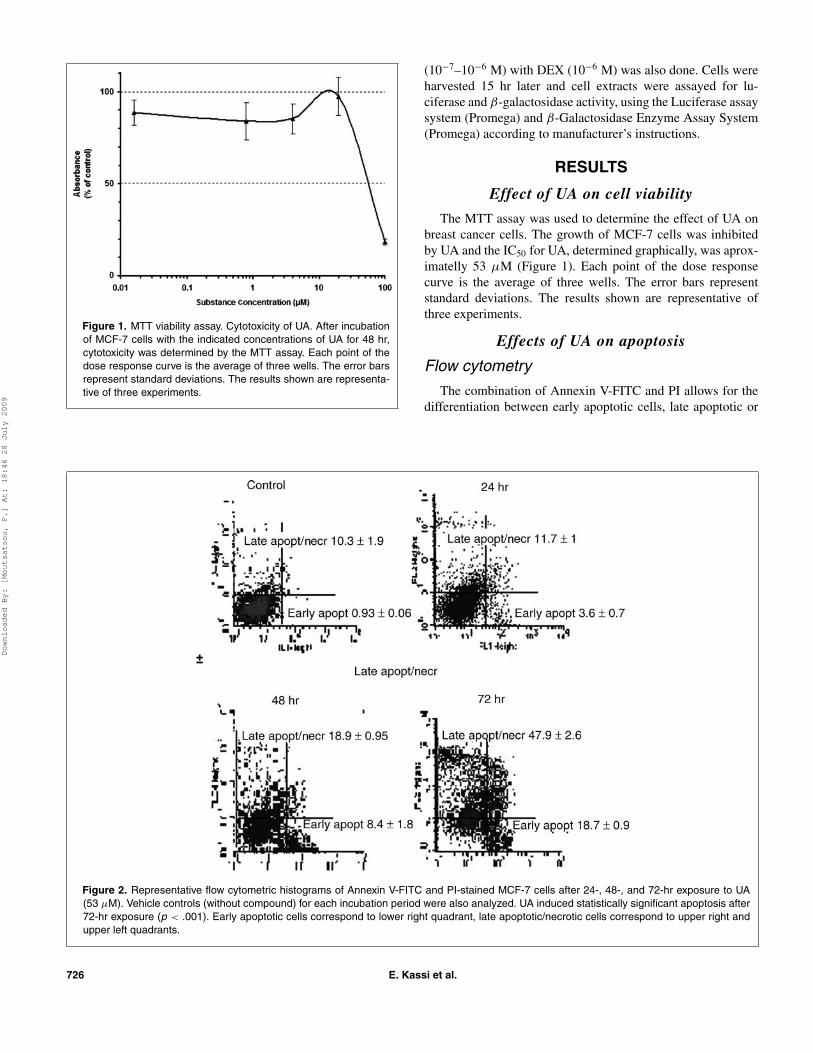
Figure 1. MTT viability assay. Cytotoxicity of UA. After incubationof MCF-7 cells with the indicated concentrations of UA for 48 hr,cytotoxicity was determined by the MTT assay. Each point of thedose response curve is the average of three wells. The error barsrepresent standard deviations. The results shown are representa-tive of three experiments.
(10−7–10−6 M) with DEX (10−6 M) was also done. Cells wereharvested 15 hr later and cell extracts were assayed for lu-ciferase and β-galactosidase activity, using the Luciferase assaysystem (Promega) and β-Galactosidase Enzyme Assay System(Promega) according to manufacturer’s instructions.
RESULTS
Effect of UA on cell viability
The MTT assay was used to determine the effect of UA onbreast cancer cells. The growth of MCF-7 cells was inhibitedby UA and the IC50 for UA, determined graphically, was aprox-imatelly 53 µM (Figure 1). Each point of the dose responsecurve is the average of three wells. The error bars representstandard deviations. The results shown are representative ofthree experiments.
Effects of UA on apoptosis
Flow cytometry
The combination of Annexin V-FITC and PI allows for thedifferentiation between early apoptotic cells, late apoptotic or
Figure 2. Representative flow cytometric histograms of Annexin V-FITC and PI-stained MCF-7 cells after 24-, 48-, and 72-hr exposure to UA(53 µM). Vehicle controls (without compound) for each incubation period were also analyzed. UA induced statistically significant apoptosis after72-hr exposure (p < .001). Early apoptotic cells correspond to lower right quadrant, late apoptotic/necrotic cells correspond to upper right andupper left quadrants.
726 E. Kassi et al.
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

necrotic cells, and viable cells. Figure 2 shows the analysis ofAnnexin V-FITC and PI-stained MCF-7 cells after 24-, 48-, and72-hr exposure to UA (53 µM). For each incubation period avehicle control (without UA) was used. UA at a concentration of53 µM induced significantly (p < .001) apoptosis (viable cells∼ 33.36% of total cells) in MCF-7 cells after 72-hr incubation.More specifically, the percentage of early apoptotic (% of totalcells) was as follows: Control = 0.93 ± 0.06; UA 24 hr = 3.6 ±0.67; UA 48 hr = 8.4 ± 1.86; and UA 72 hr = 18.74 ± 0.98 (av-erage ± SD; triplicate determinations) while the percentage oflate apoptotic/necrotic (% of total cells) was as follows: Control= 10.36 ± 1.94; UA 24 hr = 11.7 ± 1.03; UA 48 hr = 18.9 ±0.95; and UA 72 hr = 47.89 ± 2.59 (average ± SD; triplicatedeterminations).
PARP cleavage during UA-induced apoptosis
PARP cleavage, resulting in the 89-kDa fragment, is an ir-reversible event and a major hallmark and sensitive parameterof apoptosis (21, 22). In Figure 3(A), PARP cleavage is appar-ent after 48 hr of incubation with 30 µM UA. At 53 µM UA,cleavage is observed after 24 hr and by 48 hr of incubation, allof the 116 kDa full-length PARP has been cleaved. It has beenwell established that the main effector of apoptotic PARP cleav-age is caspase-3 (22). However, on using an antibody specificfor procaspase-3, which detects the full-length 32-kDa form aswell as the 17-kDa fragment, not only did we not observe frag-mentation, but we also could not detect the noncleaved form.Instead, we found unrelated bands above 45 kDa, which accord-ing to the manufacturer’s instructions may also be detected usingthis antibody (Figure 3(A)). Caspase-7 also cleaves PARP (22).But, procaspase-7 levels do not change in any of the treatedsamples (Figure 3(A)). In order to verify the noninvolvementof caspase-3 and caspase-7, we immunoblotted for DFF45. Itis known that one of the pathways during the final stages ofapoptosis involves the DFF. In its nonactive form, DFF existsas a heterodimer, DFF45/40. Proteolysis of DFF45 by activatedcaspase-3, and to a lesser extent activated caspase-7, releasesDFF40 in its activated form (23, 24). Since caspase-3 is notpresent in this cell line and procaspase-7 was not found to becleaved, the unchanged levels of DFF45 found in all treatedsamples, and shown in Figure 3(A), were to be expected.
Effect of UA on Bcl-2 protein levels
UA decreased the Bcl-2 protein levels in MCF-7 cells (Figure3(B)). The downregulation of Bcl-2 expression (as percentage ofcontrol), after normalization with actin, was statistically signifi-cant (p < .01). There was a downregulation of Bcl-2 expressioncompared to control after incubation with UA (53 µM) for 24,48, and 72 hr (Figure 3(B)).
Effect of UA on GR and AP-1 signalingpathways
In the following studies dexamethasone, a known GR-agonist, and RU486, a known GR-antagonist, were included.
Figure 3. A) Western blot analysis for PARP cleavage,procaspase-3, procaspase-7, and DFF45. MCF-7 cells were in-cubated with 30 and 53 µM UA for 24 and 48 hr, respectively,for each concentration used. Lanes: 1 = control untreated cells;2 = 30 µM UA, 24 hr; 3 = 30 µM, 48 hr; 4 = 53 µM UA, 24hr; and 5 = 53 µM, 48 hr. Equal protein loading was confirmedby anti-α-tubulin. (B) Effect of UA (53 µM) on Bcl-2 expression inMCF-7 cells. Western blot analysis shows less intense bands inMCF-7 cells treated with UA for 24, 48, and especially for 72 hrin comparison to untreated cells (control).
Relative binding affinity assay of UA
A plot of bound 3H-triamcinolone vs. log competitor (DEX,UA) moles was prepared, and the number of moles requiredto decrease the total bound radioactivity (disintegrations perminute of radioactive triamcinolone acetonide, DPM) by 50%was determined for DEX and UA (midpoint of sigmoidal curve).The ratio of DEX to UA moles, multiple by 100, is defined asthe relative binding affinity (RBA) of the competitor. The RBAvalues of DEX and UA were calculated to be 100 and 2.75,respectively. The IC50 values for DEX and UA were calculatedto be approximately 1 × 10−5M and 4 × 10−4 M, respectively(Figure 4).
Ursolic Acid and Breast Cancer 727
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009
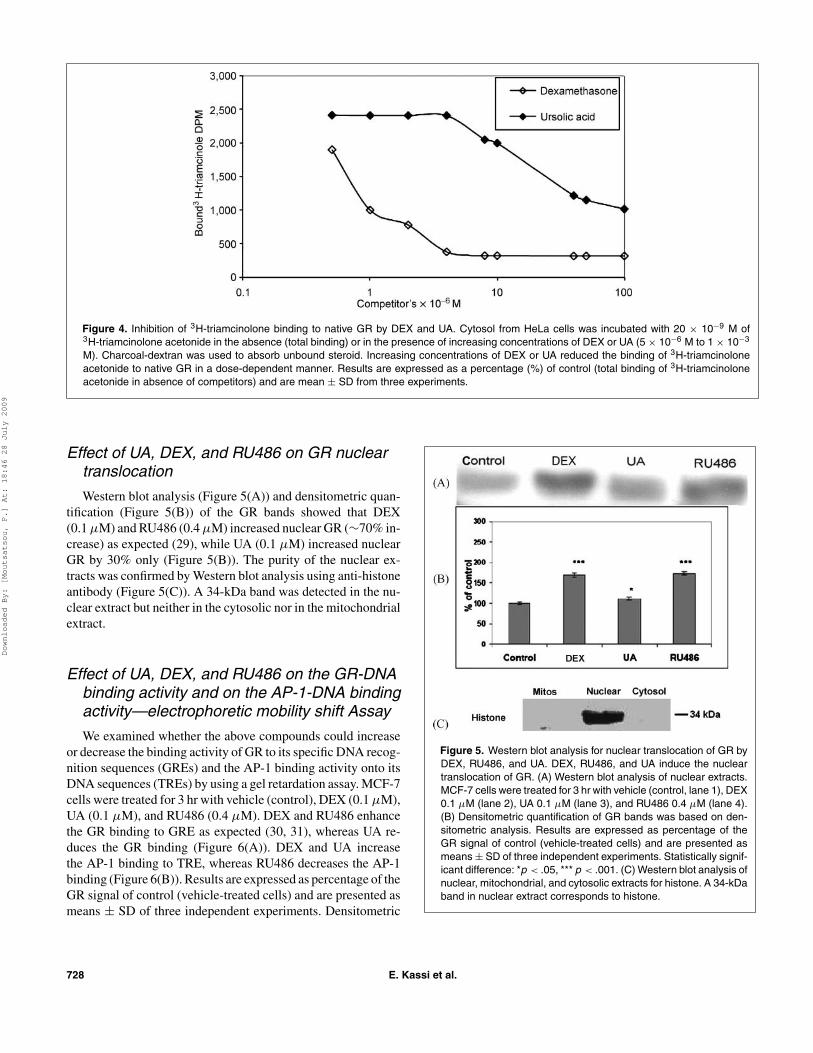
Figure 4. Inhibition of 3H-triamcinolone binding to native GR by DEX and UA. Cytosol from HeLa cells was incubated with 20 × 10−9 M of3H-triamcinolone acetonide in the absence (total binding) or in the presence of increasing concentrations of DEX or UA (5 × 10−6 M to 1 × 10−3
M). Charcoal-dextran was used to absorb unbound steroid. Increasing concentrations of DEX or UA reduced the binding of 3H-triamcinoloneacetonide to native GR in a dose-dependent manner. Results are expressed as a percentage (%) of control (total binding of 3H-triamcinoloneacetonide in absence of competitors) and are mean ± SD from three experiments.
Effect of UA, DEX, and RU486 on GR nucleartranslocation
Western blot analysis (Figure 5(A)) and densitometric quan-tification (Figure 5(B)) of the GR bands showed that DEX(0.1 µM) and RU486 (0.4 µM) increased nuclear GR (∼70% in-crease) as expected (29), while UA (0.1 µM) increased nuclearGR by 30% only (Figure 5(B)). The purity of the nuclear ex-tracts was confirmed by Western blot analysis using anti-histoneantibody (Figure 5(C)). A 34-kDa band was detected in the nu-clear extract but neither in the cytosolic nor in the mitochondrialextract.
Effect of UA, DEX, and RU486 on the GR-DNAbinding activity and on the AP-1-DNA bindingactivity—electrophoretic mobility shift Assay
We examined whether the above compounds could increaseor decrease the binding activity of GR to its specific DNA recog-nition sequences (GREs) and the AP-1 binding activity onto itsDNA sequences (TREs) by using a gel retardation assay. MCF-7cells were treated for 3 hr with vehicle (control), DEX (0.1 µM),UA (0.1 µM), and RU486 (0.4 µM). DEX and RU486 enhancethe GR binding to GRE as expected (30, 31), whereas UA re-duces the GR binding (Figure 6(A)). DEX and UA increasethe AP-1 binding to TRE, whereas RU486 decreases the AP-1binding (Figure 6(B)). Results are expressed as percentage of theGR signal of control (vehicle-treated cells) and are presented asmeans ± SD of three independent experiments. Densitometric
Figure 5. Western blot analysis for nuclear translocation of GR byDEX, RU486, and UA. DEX, RU486, and UA induce the nucleartranslocation of GR. (A) Western blot analysis of nuclear extracts.MCF-7 cells were treated for 3 hr with vehicle (control, lane 1), DEX0.1 µM (lane 2), UA 0.1 µM (lane 3), and RU486 0.4 µM (lane 4).(B) Densitometric quantification of GR bands was based on den-sitometric analysis. Results are expressed as percentage of theGR signal of control (vehicle-treated cells) and are presented asmeans ± SD of three independent experiments. Statistically signif-icant difference: *p < .05, *** p < .001. (C) Western blot analysis ofnuclear, mitochondrial, and cytosolic extracts for histone. A 34-kDaband in nuclear extract corresponds to histone.
728 E. Kassi et al.
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

Figure 6. Effects of DEX, RU486, and UA on the binding of GR and AP-1 to DNA. Gel shift assays of nuclear extracts and densitometricquantification of bands. MCF-7 cells were treated for 3 hr with vehicle (control), DEX (0.1 µM), UA (0.1 µM), and RU486 (0.4 µM). (A) DEX andRU486 enhance the GR binding to GRE, whereas UA reduces the GR binding. (B) DEX and UA increase the AP-1 binding to TRE, whereasRU486 decreases the AP-1 binding. Results are expressed as a percentage of the GR signal of control (vehicle-treated cells) and are presentedas means ± SD of three independent experiments. Statistically significant difference: ∗p < .05, ∗∗∗p < .001.
analysis showed that DEX and RU486 induced an increase by∼115% and ∼150%, respectively, in GR-DNA binding activ-ity, whereas UA decreased (∼70%) the binding of GR to GRE(Figure 6(A)). DEX and UA induced an increase (∼15%) onthe binding of AP-1 to TRE, whereas RU486 caused a decrease(∼50%) in AP-1-DNA binding activity (Figure 6(B)). Com-petition with specific cold competitor oligos (cold GREs) andnonspecific oligos was carried out and confirmed that indicatedband is GR (data not shown).
Effect of UA on GR-dependent transcriptionalregulation
Figures 7(A) and (B) demonstrate the effect of vehicle con-trol (in the absence of compounds), DEX, RU486, and UA onluciferase activity on MCF-7 cells transfected with MMTV-Luc (GREs) and on luciferase activity on MCF-7 cells trans-fected with 5X-Coll (TREs), respectively. To correct variationsin transfection efficiencies, values of the luciferase assay werenormalized using β-galactosidase activity. An effect of the testedcompounds on the β-galactosidase measurement was not ob-served. DEX and RU486 were used as positive controls. InFigure 7(A), DEX (10−8 M–10−6 M) increased significantly(p < .001) luciferase activity compared to vehicle control asexpected (32), while RU486 increased significantly luciferaseactivity at concentration 10−7 M–10−6 M (p < .001) and 10−8
M (p < .05). Coincubation of RU486 (10−7–10−6 M) with DEX
(10−6 M) lowered significantly (p < .001) the increasing effectof DEX. UA did not alter basal levels of luciferase activity atthe concentrations tested (10−6–10−5 M).
In Figure 7(B), incubation with DEX decreased significantly(p < .001) luciferase activity at a concentration range of 10−8
M–10−6 M compared to vehicle control as expected (33), whileRU486 decreased significantly luciferase activity at concentra-tion 10−6 M (p < .01) and 10−8–10−7 M (p < .001). Coincu-bation of RU486 (10−7–10−6 M) with DEX (10−6 M) loweredsignificanlty (p < .01) the effect of DEX. UA did not alter basalluciferase activity levels at the concentrations tested (10−6–10−5 M).
DISCUSSION
Many regulatory circuits are disrupted in cancerous cells,the defects in cell proliferation and/or apoptosis being amongthe main alterations that dictate malignant growth in almost alltypes of cancer, including breast cancer (19). GCs are currentlycoadministered with chemotherapy in breast cancer; however,data support that their use may be ineffective or even hazardousin the outcome of disease. Hence, the development of new agentsis a challenging issue.
UA is a dexamethasone-like natural compound, which hasbeen shown to be nontoxic even at high concentrations (up to80 mg/Kg b.w.) when injected intraperitoneally in rats (34, 35).Therefore, we examined the antigrowth and apoptotic effects
Ursolic Acid and Breast Cancer 729
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009
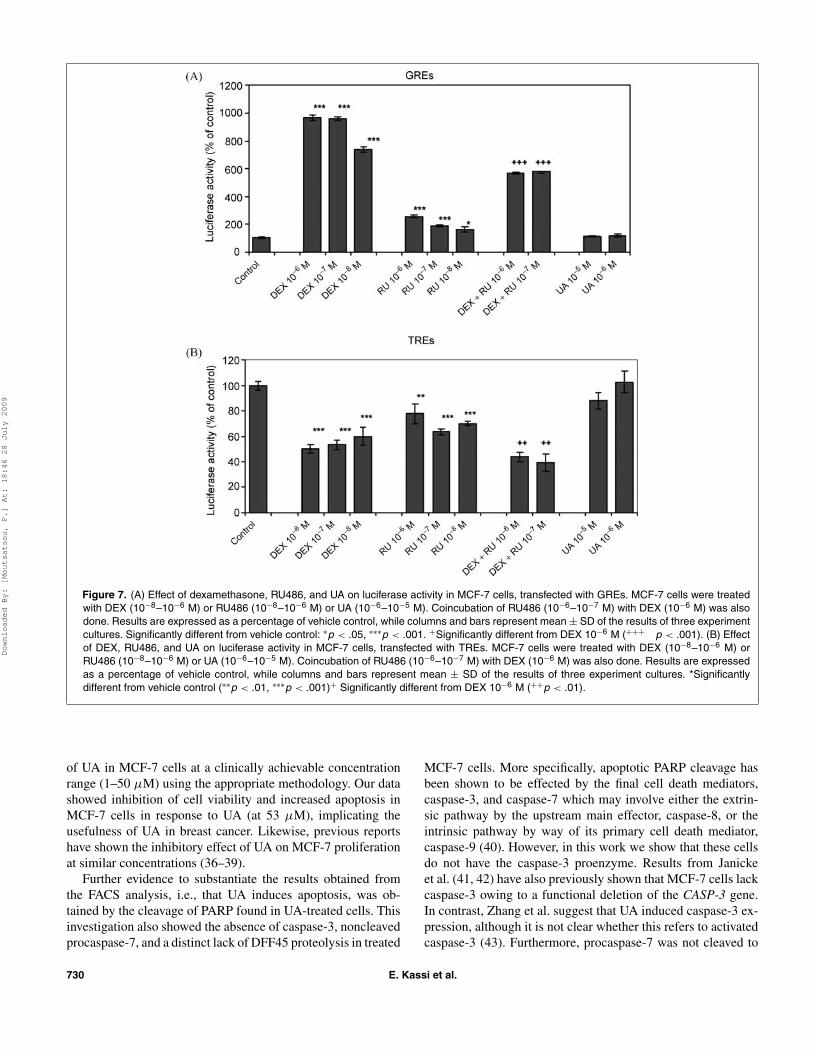
Figure 7. (A) Effect of dexamethasone, RU486, and UA on luciferase activity in MCF-7 cells, transfected with GREs. MCF-7 cells were treatedwith DEX (10−8–10−6 M) or RU486 (10−8–10−6 M) or UA (10−6–10−5 M). Coincubation of RU486 (10−6–10−7 M) with DEX (10−6 M) was alsodone. Results are expressed as a percentage of vehicle control, while columns and bars represent mean ± SD of the results of three experimentcultures. Significantly different from vehicle control: ∗p < .05, ∗∗∗p < .001. +Significantly different from DEX 10−6 M (+++ p < .001). (B) Effectof DEX, RU486, and UA on luciferase activity in MCF-7 cells, transfected with TREs. MCF-7 cells were treated with DEX (10−8–10−6 M) orRU486 (10−8–10−6 M) or UA (10−6–10−5 M). Coincubation of RU486 (10−6–10−7 M) with DEX (10−6 M) was also done. Results are expressedas a percentage of vehicle control, while columns and bars represent mean ± SD of the results of three experiment cultures. *Significantlydifferent from vehicle control (∗∗p < .01, ∗∗∗p < .001)+ Significantly different from DEX 10−6 M (++p < .01).
of UA in MCF-7 cells at a clinically achievable concentrationrange (1–50 µM) using the appropriate methodology. Our datashowed inhibition of cell viability and increased apoptosis inMCF-7 cells in response to UA (at 53 µM), implicating theusefulness of UA in breast cancer. Likewise, previous reportshave shown the inhibitory effect of UA on MCF-7 proliferationat similar concentrations (36–39).
Further evidence to substantiate the results obtained fromthe FACS analysis, i.e., that UA induces apoptosis, was ob-tained by the cleavage of PARP found in UA-treated cells. Thisinvestigation also showed the absence of caspase-3, noncleavedprocaspase-7, and a distinct lack of DFF45 proteolysis in treated
MCF-7 cells. More specifically, apoptotic PARP cleavage hasbeen shown to be effected by the final cell death mediators,caspase-3, and caspase-7 which may involve either the extrin-sic pathway by the upstream main effector, caspase-8, or theintrinsic pathway by way of its primary cell death mediator,caspase-9 (40). However, in this work we show that these cellsdo not have the caspase-3 proenzyme. Results from Janickeet al. (41, 42) have also previously shown that MCF-7 cells lackcaspase-3 owing to a functional deletion of the CASP-3 gene.In contrast, Zhang et al. suggest that UA induced caspase-3 ex-pression, although it is not clear whether this refers to activatedcaspase-3 (43). Furthermore, procaspase-7 was not cleaved to
730 E. Kassi et al.
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

its active form. In light of the fact that we distinctly found apop-totic cleavage of PARP in UA-treated MCF-7 cells, we furthersubstantiated these findings by immuno-analyzing for DFF45proteolytic cleavage. To date, experimental evidence has shownthat in vivo, these two caspases are the only effectors of theactivation of the DNA endonuclease, DFF40, in one of the finalapoptotic pathways leading to DNA fragmentation (23, 24). Ac-tivation of DFF40 is accomplished by the proteolysis of DFF45by caspase-3 and/or caspase-7. This results in the release andactivation of DFF40 and the execution of the final stages ofapoptosis. The lack of DFF45 proteolysis found in this studyis therefore convincing evidence of the absence of activatedcaspase-3 and caspase-7.
The downregulation of Bcl-2 protein levels by UA treatmentfound in this study supports the contention that UA-inducedapoptosis is mediated by the intrinsic mitochrondrial pathway.This downregulation leads to release of cytochrome c, acti-vation of caspase-9, and usually to the subsequent activationof caspase-3 by caspase-9. However, it has been shown thatcells lacking caspase-3 activity can compensate for this loss.Rabi et al. (44), using another triterpenoid, found that apoptoticMCF-7 cells lacking caspase-3 and caspase-7 activity, nonethe-less, showed PARP cleavage. Other investigators also found thatPARP cleavage can be independent of caspase-3 (45, 46). Inthese investigations, Bcl-2 protein levels were also found to bedownregulated (45, 46). The conclusion drawn from these find-ings was that, surprisingly, PARP can also be a direct substrateof caspase-9. Thus, our results showing the lack of caspase-3and caspase-7, PARP cleavage, and the decrease in Bcl-2 proteinlevels, along with the findings of others cited herein, support theconclusion that the induction of apoptosis by UA is through theintrinsic mitochondrial pathway and that caspase-9 may com-pensate for the caspase-3 and caspase-7 deficiency found in thisinvestigation, by directly cleaving PARP.
It is important to notice that UA has been shown to accom-plish apoptosis in different cell types by various pathways, suchas caspase-3 and caspase-9 activation, downregulation of c-IAPsfamily proteins, alteration of the Bax protein or changes in p53protein levels, increased intracellular Ca2+ levels (Ca2+influx),or even by changes in mitochondrial permeability transition(MPT) (17, 47, 48).
GC-induced apoptosis has been associated with various sig-naling cascades including Ca2+ mobilization, changes in ki-nases, transcription factors, caspases and Bcl-family proteins,disruption of mitochondrial membrane potential (��m), andrelease of cytochrome c to the cytosol (25). The role of GRin mediated GC-induced apoptosis is under current researchinterest. Evidence indicates that the transactivation and transre-pression effects of GR as well as the phosphorylation status ofGR are associated with GC-induced apoptosis (49–51). Moreimportant, accumulating evidence demonstrates the importantrole of GR-signaling in breast cancer (6). In light of the above,we addressed the question whether UA, a dexamethasone-likecompound, could modulate GR signaling in MCF-7 cells. Weused a panel of assays investigating GR-subcellular localizationand function such as (a) ligand binding to GR to stimulate re-
ceptor activation, (b) quantitative Western immunoblotting ofGR protein in nuclear fraction, (c) DNA-binding activity of GRto its cognate sequences (GREs), and (d) GR-mediated geneexpression in cells transfected with MMTV-Luc reporter gene.
The ability of UA to exert competitive inhibition on 3H-triamcinolone binding to GR, in a way similar to that of dexam-ethasone (IC501 × 10−5 M for DEX and 4 × 10−4 M for UA),suggests that UA might influence GR-dependent signaling via amodification of GR conformation (allosteric regulator). To elu-cidate further the ability of UA to induce the right conformationof GR able to translocate GR into nucleus and to bind on itsDNA elements (GREs), we conducted a Western blot analysisand a gel retardation assay. In these experiments we tested UA ata lower concentration (0.1 µM) than in the ligand binding assay,since evidence indicates that the biological potencies of com-pounds determined by different assays may not be comparable,the cell-free binding assays being the less sensitive (52).
UA resulted in increased nuclear GR content, a finding com-patible with previous reports (18). However, despite the fact thatUA triggered the nuclear translocation of GR in MCF-7 cells,the UA–GR complex did not favor the binding of GR on its GREelement (it rather reduced the basal GR-DNA binding activity).
To verify further the role of UA in GRE-mediated activ-ity, we examined the potential of UA to induce luciferase geneactivation in the transfected MCF-7 cells. UA did not triggerGRE-mediated activity. Of note, Wu et al. (4) reported thatdexamethasome treatment and GR transactivation to its GREelements in breast cancer cells regulate survival signaling, as-sociated with inhibition of chemotherapy-induced apoptosis. Tothis effect, our findings demonstrating the inability of UA toinduce GRE-dependent genes suggest that it may be a mech-anism by which, at least in part, MCF-7 cells show increasedsusceptibility to apoptosis in presence of UA. UA may induceapoptosis in MCF-7 cells by a yet unknown GR-mediated path-way. For example, Sionov et al. (53) have shown that some ofthe apoptotic effects of GR in T cells occur independently ofits nuclear translocation, but depend solely on its translocationto the mitochondria. Given that NFkB, a known target of GR-transrepression activity, is an important molecule for mammarycancer therapy, the role of UA in the crosstalk between GR andNFkB signaling in breast cancer warrants investigation (54–56).
Due to the crucial role of AP-1 in regulating the function ofbreast cancer cells, we hypothesized that it may be a relevantcontributor to UA-mediated effects in MCF-7 cells. In line withprevious reports, UA increased significantly the binding of AP-1protein to its specific DNA elements (18, 57).
GCs have been reported to be potent inhibitors of AP-1 re-sponsive genes, a mechanism mediated by the GR (11). It hasbeen suggested that the GC does not affect the binding ac-tivity of AP-1 but it rather downmodulates the transactivatingfunction of AP-1 through the interaction of GR with either thepreexisting unbound or DNA-bound AP-1 (57). In the presentstudy, UA neither transrepressed nor transactivated basal AP-1activity, implicating that UA may not respond through the AP-1signaling.
Ursolic Acid and Breast Cancer 731
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

Altogether, our results showing the PARP cleavage and thedecrease in Bcl-2 protein levels, along with the findings of otherscited herein, support the conclusion that the induction of apop-tosis by UA is through the intrinsic mitochondrial pathway. Inaddition, UA, a GR modulator, does not induce transactivation ofGRE-dependent survival genes in MCF-7 cells (4), which may,in part, contribute to its apoptotic effects in this cell system.
ACKNOWLEDGMENTS
We would like to thank Dr. Tomoshige Kino, Pediatric andReproductive Endocrinology Branch, National Institute of ChildHealth, USA, for providing us the MMTV-Luc expression vectorand Dr. V. Zoumbourlis, National Hellenic Research Founda-tion, Greece, for providing us the TRE expression vector. Wewould also like to thank Bodossaki Foundation and the GeneralSecretariat of Research and Technology, Ministry of Develop-ment, Greece (grant EPAN in cooperation with the companiesGiotis, Aktina, Attiki-Pittas, Pierre Fabre, and Gaia, and grantPAVE in cooperation with ELPEN Company) for the financialsupport.
REFERENCES1. Planey, S.L.; Litwack, G. Glucocorticoid-induced apoptosis in lym-
phocytes. Biochem Biophys Res Commun 2000, 279, 307–312.2. Zhang, C.; Beckermann, B.; Kallifatidis, G.; Liu, Z.; Rittgen, W.;
Edler, L.; Buchler, P.; Debatin, K.M.; Buchler, M.W.; Friess, H.; Herr,I. Corticosteroids induce chemotherapy resistance in the majorityof tumour cells from bone, brain, breast, cervix, melanoma andneuroblastoma. Int J Oncol 2006, 29, 1295–1301.
3. Lu, Y.S.; Lien, H.C.; Yeh, P.Y.; Yeh, K.H.; Kuo, M.L.; Kuo,S.H.; Cheng, A.L. Effects of glucocorticoids on the growth andchemosensitivity of carcinoma cells are heterogeneous and re-quire high concentration of functional glucocorticoid receptors.World J Gastroenterol 2005, 11, 6373–6380.
4. Wu, W.; Chaudhuri, S.; Brickley, D.R.; Pang, D.; Karrison, T.;Conzen, S.D. Microarray analysis reveals glucocorticoid-regulatedsurvival genes that are associated with inhibition of apoptosis inbreast epithelial cells. Cancer Res 2004, 64, 1757–1764.
5. Wu, W.; Pew, T.; Zou, M.; Pang, D.; Conzen, S.D. Glucocorticoidreceptor-induced MAPK phosphatase-1 (MPK-1) expression in-hibits paclitaxel-associated MAPK activation and contributes tobreast cancer cell survival. J Biol Chem 2005, 280, 4117–4124.
6. Moutsatsou, P.; Papavassiliou, A.G. The glucocorticoid receptorsignaling in breast cancer. J Cell Mol Med 2007.
7. Kris, M.G.; Hesketh, P.J.; Somerfield, M.R.; Feyer, P.; Clark-Snow,R.; Koeller, J.M.; Morrow, G.R.; Chinnery, L.W.; Chesney, M.J.;Gralla, R.J.; Grunberg, S.M. American Society of Clinical Oncologyguideline for antiemetics in oncology: update. J Clin Oncol 2006,24, 2932–2947.
8. Herr, I.; Pfitzenmaier, J. Glucocorticoid use in prostate cancerand other solid tumours: implications for effectiveness of cytotoxictreatment and metastases. Lancet Oncol 2006, 7, 425–430.
9. Beato, M.; Herrlich, P.; Schutz. G. Steroid hormone receptors:many actors in search of a plot. Cell 1995, 83, 851–857.
10. Bamberger, C.M.; Bamberger, A.M.; de Castro, M.; Chrousos, G.P.Glucocorticoid receptor beta, a potential endogenous inhibitor ofglucocorticoid action in humans. J Clin Invest 1995, 95, 2435–2441.
11. De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. The interplaybetween the glucocorticoid receptor and nuclear factor-kappaB or
activator protein-1: molecular mechanisms for gene repression.Endocr Rev 2003, 24, 488–522.
12. Eferl, R.; Wagner, E.F. AP-1: a double-edged sword in tumorigen-esis. Nat Rev Cancer 2003, 3, 859–868.
13. Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death,Nat Cell Biol 2002, 4, E131–E136.
14. Liu, Y.; Ludes-Meyers, J.; Zhang, Y.; et al. Inhibition of AP-1 tran-scription factor causes blockade of multiple signal transductionpathways and inhibits breast cancer growth. Oncogene 2002, 21,7680–689.
15. Milde-Langosch, K.; Kappes, H.; Riethdorf, S.; Loning, T.; Bam-berger, A.M. FosB is highly expressed in normal mammary epithe-lia, but down-regulated in poorly differentiated breast carcinomas.Breast Cancer Res Treat 2003, 77, 265–275.
16. Novotny, L.; Vachalkova, A.; Biggs, D. Ursolic acid: an anti-tumorigenic and chemopreventive activity. Minireview. Neoplasma2001, 48, 241–246.
17. Achiwa, Y.; Hasegawa, K.; Komiya, T.; Udagawa, Y. Ursolic acidinduces Bax-dependent apoptosis through the caspase-3 pathwayin endometrial cancer SNG-II cells. Oncol Rep 2005, 13, 51–57.
18. Cha, H.J.; Park, M.T.; Chung, H.Y.; Kim, N.D.; Sato, H.; Seiki, M.;Kim, K.W. Ursolic acid-induced down-regulation of MMP-9 geneis mediated through the nuclear translocation of glucocorticoidreceptor in HT1080 human fibrosarcoma cells. Oncogene 1998,16, 771–778.
19. Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000,100, 57–70.
20. Papadopoulos, K. Targeting the Bcl-2 family in cancer therapy.Semin Oncol 2006, 33, 449–456.
21. Duriez, P.J.; Shah, G.M. Cleavage of poly(ADP-ribose) poly-merase: a sensitive parameter to study cell death. Biochem CellBiol 1997, 75, 337–349.
22. Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleav-age during apoptosis: an update. Apoptosis 2002, 7, 321–328.
23. Liu, X.; Zou, H.; Widlak, P.; Garrard, W.; Wang, X. Activation ofthe apoptotic endonuclease, DFF40 (caspase-activated DNase ornuclease). J Biol Chem 1999, 274, 13836–13840.
24. Widlak, P.; Garrard, W. Discovery, regulation and action of themajor apoptotic nucleases DFF40/CAD and endonuclease G. JCell Biochem 2005, 94, 1078–1087.
25. Herr, I.; Gassler, N.; Friess, H.; Buchler, M.W. Regulation of differ-ential pro- and anti-apoptotic signaling by glucocorticoids. Apop-tosis 2007, 12, 271–291.
26. Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth andsurvival. Modifications to the tetrazolium dye procedure giving im-proved sensitivity and reliability. J Immunol Methods 1986, 89,271–277.
27. Kassi, E.; Papoutsi, Z.; Pratsinis, H.; Aligiannis, N.; Manoussakis,M.; Moutsatsou, P. Ursolic acid, a naturally occurring triterpenoid,demonstrates anticancer activity on human prostate cancer cells.J Cancer Res Clin Oncol 2007, 133, 493–500.
28. Schreiber, E.; Matthias, P.; Muller, M.M.; Schaffner, W. Rapid de-tection of octamer binding proteins with ‘mini-extracts,’ preparedfrom a small number of cells. Nucleic Acids Res 1989, 17, 6419.
29. Htun, H.; Barsony, J.; Renyi, I.; Gould, D.L.; Hager, G.L. Visualiza-tion of glucocorticoid receptor translocation and intranuclear or-ganization in living cells with a green fluorescent protein chimera.Proc Natl Acad Sci USA 1996, 93, 4845–4850.
30. Wagner, B.L.; Pollio, G.; Giangrande, P.; Webster, J.C.; Breslin, M.;Mais, D.E.; Cook, C.E.; Vedeckis, W.V.; Cidlowski, J.A.; McDonnell,D.P. The novel progesterone receptor antagonists RTI 3021-012and RTI 3021-022 exhibit complex glucocorticoid receptor antag-onist activities: implications for the development of dissociatedantiprogestins. Endocrinology 1999, 140, 1449–1458.
31. Bourgeois, S.; Pfahl, M.; Baulieu, E.E. DNA binding propertiesof glucocorticosteroid receptors bound to the steroid antagonistRU-486. Embo J 1984, 3, 751–755.
732 E. Kassi et al.
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009

32. Pariante, C.M.; Pearce, B.D.; Pisell, T.L.; Su, C.; Miller, A.H. Thesteroid receptor antagonists RU40555 and RU486 activate gluco-corticoid receptor translocation and are not excreted by the steroidhormones transporter in L929 cells. J Endocrinol 2001, 169, 309–320.
33. Vayssiere, B.M.; Dupont, S.; Choquart, A.; Petit, F.; Garcia, T.;Marchandeau, C.; Gronemeyer, H.; Resche-Rigon, M. Syntheticglucocorticoids that dissociate transactivation and AP-1 transre-pression exhibit antiinflammatory activity in vivo. Mol Endocrinol1997, 11, 1245–1155.
34. Saravanan, R.; Viswanathan, P.; Pugalendi, K.V. Protective effectof ursolic acid on ethanol-mediated experimental liver damage inrats. Life Sci 2006, 78, 713–718.
35. Somova, L.I.; Shode, F.O.; Ramnanan, P.; Nadar, A. Antihy-pertensive, antiatherosclerotic and antioxidant activity of triter-penoids isolated from Olea europaea, subspecies africana leaves.J Ethnopharmacol 2003, 84, 299–305.
36. Es-Saady, D.; Simon, A.; Jayat-Vignoles, C.; Chulia, A.J.; Delage,C. MCF-7 cell cycle arrested at G1 through ursolic acid, and in-creased reduction of tetrazolium salts. Anticancer Res 1996, 16,481–486.
37. He, X.; Liu R.H. Cranberry phytochemicals: Isolation, structureelucidation, and their antiproliferative and antioxidant activities. JAgric Food Chem 2006, 54, 7069–7074.
38. Zhang, W.; Li, Y.; Zhang, G.; Lu, J.; Ou, H. Experimental studyon MCF-7 cell apoptosis induced by ursolic acid. Zhong Yao Cai2005, 28, 297–301.
39. He, X.; Liu, R.H. Triterpenoids isolated from apple peels havepotent antiproliferative activity and may be partially responsible forapple’s anticancer activity. J Agric Food Chem 2007, 55, 4366–4370.
40. Chowdhury, I.; Tharakan, B.; Bhat, G.K. Current concepts in apop-tosis: the physiological suicide program revisited. Cell Mol Biol Lett2006, 11, 506–525.
41. Janicke, R.U.; Sprengart, M.I.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changesassociated with apoptosis. J Biol Chem 1998, 273, 9357–9360.
42. Janicke, R.U.; Ng, P.; Sprengart, M.I.; Porter, A.G. Caspase-3 isrequired for α-fodrin cleavage but dispensable for cleavage of otherdeath substrates in apoptosis. J Biol Chem 1998, 273, 15540–15545.
43. Zhang, G.P.; Lu, Y.Y.; Lv, J.C.; Ou, H.J. Effect of ursolic acid oncaspase-3 and PARP expression of human MCF-7 cells. Zhong-guo Zhong Yao Za Zhi 2006, 31, 141–144.
44. Rabi, T.; Wang, L.; Bannerjee, S.; Novel triterpenoid 25-hydroxy-3-oxoolean-12-en-28-oic acid induces growth arrest and apoptosisin breast cancer cells, Breast Cancer Res Treat 2007, 101, 27–36.
45. Cui, Q.; Yu, J.H.; Wu, J.N.; Tashiro, S.; Onodera, S.; Minami, M.;Ikejima, T. p53-mediated cell cycle arrest and apoptosis through
a caspase-3-independent, but caspase-9-dependent pathway inoridonin-treated MCF-7 human breast cancer cells. Acta Pharma-col Sin 2007, 28, 1057–1066.
46. Huang, J.; Wu, L.; Tashiro, S.; Onodera, S.; Ikejima, T. A compari-son of the signal pathways between TNFα - and oridonin-inducedmurine L929 fibrosarcoma cell death. Acta Med Okayama 2005,59, 261–270.
47. Lauthier, F.; Taillet, L.; Trouillas, P.; Delage, C.; Simon, A. Ursolicacid triggers calcium-dependent apoptosis in human Daudi cells.Anticancer Drugs 2000, 11, 737–745.
48. Choi, Y.H.; Baek, J.H.; Yoo, M.A.; Chung, H.Y.; Kim, N.D.; Kim,K.W. Induction of apoptosis by ursolic acid through activation ofcaspases and down-regulation of c-IAPs in human prostate ep-ithelial cells. Int J Oncol 2000, 17, 565–571.
49. Frankfurt, O.; Rosen, S.T. Mechanisms of glucocorticoid-inducedapoptosis in hematologic malignancies: updates. Curr Opin Oncol2004, 16, 553–563.
50. Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.;Kumar, R.; Thompson, E.B. p38 Mitogen-activated protein kinase(MAPK) is a key mediator in glucocorticoid-induced apoptosis oflymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor atserine 211. Mol Endocrinol 2005, 19, 1569–1583.
51. Amsterdam, A.; Tajima, K.; Sasson, R. Cell-specific regulation ofapoptosis by glucocorticoids: implication to their anti-inflammatoryaction. Biochem Pharmacol 2002, 64, 843–850.
52. Gutendorf, B.; Westendorf, J. Comparison of an array of in vitroassays for the assessment of the estrogenic potential and syntheticestrogens, phytoestrogens and xenoestrogens. Toxicology 2001,166, 79–89.
53. Sionov, R.V.; Cohen, O.; Kfir, S.; Zilberman, Y.; Yefenof, E. Roleof mitochondrial glucocorticoid receptor in glucocorticoid-inducedapoptosis. J Exp Med 2006, 203, 189–201.
54. Gonzalez, M.V.; Jimenez, B.; Berciano, M.T.; Gonzalez-Sancho,J.M.; Caelles, C.; Lafarga, M.; Munoz, A. Glucocorticoids antag-onize AP-1 by inhibiting the Activation/phosphorylation of JNKwithout affecting its subcellular distribution. J Cell Biol 2000, 150,1199–1208.
55. Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.;Wessely, O; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel,P.; Schutz, G. DNA binding of the glucocorticoid receptor is notessential for survival. Cell 1998, 93, 531–541.
56. Haffner, M.C.; Berlato, C.; Doppler, W. Exploiting our knowledgeof NF-kappaB signaling for the treatment of mammary cancer. JMammary Gland Biol Neoplasia 2006, 11, 63–73.
57. Jonat, C.; Rahmsdorf, H.J.; Park, K.K.; Cato, A.C.; Gebel, S.;Ponta, H.; Herrlich, P. Antitumor promotion and antiinflammation:down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hor-mone. Cell 1990, 62, 1189–1204.
Ursolic Acid and Breast Cancer 733
Downloaded By: [Moutsatsou, P.] At: 18:46 28 July 2009





























