Embed Size (px)
Citation preview

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/12001499
TowardstheMHC-PeptideCombinatorics
ARTICLEinHUMANIMMUNOLOGY·JUNE2001
ImpactFactor:2.14·DOI:10.1016/S0198-8859(01)00219-1·Source:PubMed
CITATIONS
17
READS
13
4AUTHORS,INCLUDING:
PandjassarameKangueane
Bioinformation
112PUBLICATIONS756CITATIONS
SEEPROFILE
MeenaSakharkar
UniversityofTsukuba
120PUBLICATIONS1,222CITATIONS
SEEPROFILE
PrasannaRKolatkar
QatarBiomedicalResearchInstitute
79PUBLICATIONS1,956CITATIONS
SEEPROFILE
Availablefrom:PandjassarameKangueane
Retrievedon:05February2016

Towards the MHC-Peptide Combinatorics
Pandjassarame Kangueane, Meena K. Sakharkar,Prasanna R. Kolatkar, and Ee Chee Ren
ABSTRACT: The exponentially increased sequence in-formation on major histocompatibility complex (MHC)alleles points to the existence of a high degree of poly-morphism within them. To understand the functionalconsequences of MHC alleles, 36 nonredundant MHC-peptide complexes in the protein data bank (PDB) wereexamined. Induced fit molecular recognition patternssuch as those in MHC-peptide complexes are governed bynumerous rules. The 36 complexes were clustered into 19subgroups based on allele specificity and peptide length.The subgroups were further analyzed for identifying com-mon features in MHC-peptide binding pattern. The fourmajor observations made during the investigation were:(1) the positional preference of peptide residues defined bypercentage burial upon complex formation is shown for all
the 19 subgroups and the burial profiles within entries ina given subgroup are found to be similar; (2) in class Ispecific 8- and 9-mer peptides, the fourth residue isconsistently solvent exposed, however this observation isnot consistent in class I specific 10-mer peptides; (3) ananchor-shift in positional preference is observed towardsthe C terminal as the peptide length increases in class IIspecific peptides; and (4) peptide backbone atoms areproportionately dominant at the MHC-peptide interface.Human Immunology 62, 539–556 (2001). © AmericanSociety for Histocompatibility and Immunogenetics,2001. Published by Elsevier Science Inc.
KEYWORDS: MHC-peptide interface; residue prefer-ence; binding pattern; binding value
ABBREVIATIONSEBNA Epstein-Barr nuclear antigenEBV Epstein-Barr virusHBV hepatitis B virusHIV human immunodeficiency virusHMBP human myelin basic proteinHTLV human T lymphotropic virus
MHC major histocompatibility complexPDB protein data bankRT reverse transcriptaseSv Sendai virusVsv vesicular stomatitis virus
INTRODUCTIONT-cell mediated immune response involves the formationof major histocompatibility complex (MHC) peptidecomplex [1, 2]. Class I and class II molecules require thebinding of endogenously and exogenously processedshort antigenic peptides, respectively, to induce immuneresponse [3–5]. Peptide binding to MHC molecules oc-curs in several steps, mostly via the conformational re-arrangement of the peptide-binding groove [6–9].
Therefore the structure of the MHC-peptide complex atthe time point of peptide recognition may be differentfrom the equilibrium crystal structure [10].
Deciphering the structural principles that link MHCpolymorphism [11–13] with specific binding directlyleads to the development of tools for predicting antigenicpeptides from sequence information [14]. Availableknowledge on MHC has made these molecules interest-ing pharmaceutical targets for vaccination, immunosup-pressive therapy of autoimmune diseases, and cancerthrough the design of potential peptide and nonpeptideligands of MHC [15–17]. The complete sequence andgene map of the human MHC, a region on chromosome6 [18], provides impetus not only for studying thefunctions of MHC genes but also other undoubtedlyimportant immune related genes. Intellectual interdisci-plinary fusion of biomedicine and information technol-ogy has revolutionized T-cell immunology by providing
From the BioInformatics Centre (P.K., M.K.S., P.R.K), National Uni-versity of Singapore, and the Institute of Molecular & Cell Biology (P.R.K.),,and the Department of Microbiology (P.K., C.E.R.), National University ofSingapore, Singapore.
Address reprint requests to: Dr. Pandjassarame Kangueane, BioInfor-matics Centre, Medical Drive, MD7, #02-07, National University ofSingapore, Singapore 119 260; Tel: 165 774 7149; Fax: 165 7782466; E-mail: [email protected].
Received July 25, 2000; revised January 8, 2001; accepted January 10,2001.
Human Immunology 62, 539–556 (2001)0198-8859/01/$–see front matter© American Society for Histocompatibility and Immunogenetics, 2001
Published by Elsevier Science Inc. PII S0198-8859(01)00219-1

bioinformatics tools to explore the combinatorial solu-tion space in cell-mediated immune response [19–26].
The SCOP classification [27] of the MHC moleculesunder the immunoglobulin superfamily and the effortstaken by Steven E. Brenner, Derren Barken, and MichaelLevitt to supplement each protein data bank (PDB) entrywith experimental data under PRESAGE [28] are prom-ising steps towards a structure-based functional immu-nology paradigm. Extensive distillation of structural dataon multimeric protein complexes at the PDB to decodethe molecular principles of protein-protein recognition[29], and the recent analysis of a nonredundant MHC-peptide data set to improve the structure based MHC-peptide prediction procedure are encouraging [20]. Thecurrently available structural data on MHC-peptide com-plexes in PDB [30] is partially curated and somewhatredundant in nature. We created a highly curated dataset rich in MHC-peptide complexes and classified theminto different subsets by visual inspection of the struc-tural data. Given the current limitations of a purelyautomatic procedure, we believe this approach producesthe most accurate and useful information. The preparedMHC-peptide data set is further clustered based onMHC allele specificity and peptide length. Each of theclustered subgroup is analyzed for MHC-peptide bindingpattern and the results are discussed in this article.
MATERIALS AND METHODSData Set CreationStructural information on MHC-peptide complexes wasretrieved from the PDB as flat files with the “.pdb”extension [31, 32]. The retrieved data amounts to 43 innumber (Tables 1 and 2) and formed a MHC-peptidedata set for further analysis.
Data ClusteringThe MHC-peptide structural data set was primarily clus-tered into two groups based on MHC class. Further, theclass I complexes were clustered into 13 subgroups basedon allele specificity and peptide length. Similarly, theremaining eight class II complexes were clustered intosix subgroups. Therefore, the 36 nonredundant com-plexes were clustered into 19 subgroups for further anal-ysis.
PDB Chain Identifiers andMHC-Peptide ComplexesThe chain identifiers representing the a chain in MHCfor all class I entries except 1KBG (PDB code) is “A”(Table 1); “H” represents the chain identifier for the achain in 1KBG. Most of the class I specific peptides arerepresented by the chain identifier “C,” whereas the restare represented by the chain identifier “P” (Table 1). The
“a” and “b” chains in class II entries are represented bychain identifiers “A” and “B,” respectively (Table 2). Theclass II specific peptides are represented by either one ofthe following chain identifiers: “C,” “E,” or “B with somealphanumeric tags” (Table 2). Understanding the currentdata structure with reference to a specific biologicalfunction, such as MHC-peptide binding, will provideways to curate them to a more consistent format and thusaid in the development of an automated data retrievalsystem.
Information Redundancy in MHC-PeptideStructural DataWe considered the class I data set entries with PDB code1A9K, 1AKJ, 1AO7, 1BD2, 1B0R, 1A9B, and 1KBGredundant (within the limitation of this article), as theseentries are duplicates describing a particular sequenceinformation or they contain incomplete structural infor-mation. For example, the PDB entry 1A9k was classifiedas obsolete by PDB and hence is indicated by a “#” inTable 1. In class II data set the PDB entry 1AQDrepresents a DR1-peptide complex, where the peptidesequence is *VGSDWRFLRGYHQYA. The coordinatesfor the first residue valine (V) are not available in thePDB file. Similarly, the peptide sequence in 1BX2 (PDBcode) is ENPVVHFFKNIVTPR*, and the coordinatesfor the last residue arginine (R) are not available. Resi-dues for which the structural information is not availableare marked by an asterisk mark (*) solely to indicate thispoint (Table 2). In some entries, there are two complexesper asymmetric unit each composed of three polypeptidechains, describing identical MHC-peptide sequence data.In such cases, we take the data for one and leave theother.
MHC-Peptide Data AnalysisIn our attempt to study the principles of MHC-peptidebinding using the available structural data the followingparameters were concentrated on:
Interface area between the peptide and the MHC. Interfacearea for class I MHC-peptide complexes was defined asthe change in their solvent accessible surface area(‚ASA) when going from a monomeric MHC moleculeto a dimeric MHC-peptide complex state. Similarly, theinterface area for class II MHC-peptide complexes wasdefined as the change in their solvent accessible surfacearea (‚ASA) when going from a dimeric MHC moleculeto a trimeric MHC-peptide complex state. The ASAs ofthe complexes and the individual polypeptides were cal-culated using an algorithm described by Lee and Richard[61]. The mean ‚ASA upon complex formation (goingfrom a monomeric state to a dimeric state) was calculatedas half the sum of total ‚ASA for both the MHC
540 P. Kangueane et al.

molecule and the peptide. Calculating ‚ASA upon com-plex formation will provide an opportunity to comparestructural properties with experimental biochemical orimmunologic binding data.
Gap volume between the peptide and the MHC. The gapvolume between the MHC and the peptide in eachcomplex was calculated using the program SURFNET[62, 63].
Gap index. Complementing MHC-peptide interactingsurfaces were evaluated by defining a gap index [29],where gap index (Å) is defined as the ratio of gap volumebetween the MHC and the peptide (Å3) to the interfacearea (Å2) per MHC-peptide.
Number of intermolecular hydrogen bonds between the peptideand the MHC. The number of intermolecular hydrogenbonds between the peptide and the MHC was calculatedusing HBPLUS [64] in which hydrogen bonds are de-fined according to standard geometric criteria.
Predominant peptide atoms (backbone or side-chain) in theinteraction. An atom in each of the peptide residue wasconsidered to be involved in MHC-peptide interaction ifthe distance between the atom in the peptide and anyatom in the MHC is less than or equal to x (Å). Thedistribution of peptide backbone and side chain atomswas calculated within arbitrarily defined distances rang-ing from 0.0 to 5.0 (Å) at 0.1 (Å) increments.
TABLE 1 Class I MHC-peptide complexes in PDB
MHCsource Subgroup
PDBcode
MHCallele CIM
RedundantPeptide set CIP
Nonredundantpeptide set PL Peptide source
R[Å]
Releaseyear
Human Subset 1–group 1 1HHJ(33) A*0201 {a} ILKEPVHGV {c} ILKEPVHGV 9 Synthetic 2.50 1993Human 1AKJ(34) A*0201 {a} Ilkepvhgv {c} 9 HIV-1 RT 2.65 1997Human 1HHK(33) A*0201 {a} LLFGYPVYV {c} LLFGYPVYV 9 Synthetic 2.50 1993Human 1AO7(35) A*0201 {a} llfgypvyv {c} 9 HTLV-1 Tax 2.60 1997Human 1BD2(36) A*0201 {a} llfgypvyv {c} 9 HTLV-1 Tax 2.50 1998Human 1B0G(37) A*0201 {a} ALWGFFPVL {c} ALWGFFPVL 9 Human-Peptide
P10492.60 1998
Human #1A9K(–) A*0201 {a} alwgffpvl {c} 9 Human-PeptideP1049
2.50 1998
Human 1HHG(33)
A*0201 {a} TLTSCNTSV {c} TLTSCNTSV 9 HIV-1 gp 120 2.60 1993Human 1HHI(33) A*0201 {a} GILGFVFTL {c} GILGFVFTL 9 Synthetic 2.50 1993Human 1BOR(38) A*0201 {a} gilgfvftcde {c} 9 Influenza matrix 2.90 1998Human Subset 1–group 2 2CLR(39) A*0201 {a} MLLSVPLLLG {c} MLLSVPLLLG 10 Synthetic 2.00 1995Human 1HHH(33) A*0201 {a} FLPSDFFPSV {c} FLPSDFFPSV 10 HBV nucleocapsid 3.00 1993Human Subset 2–group 1 1TMC(40) A*6801 {a} EVAPPEYHRK {c} EVAPPEYHRK 10 Synthetic 2.30 1995Human Subset 3–group 1 1AGB(41) B*0801 {a} GGRKKYKL {c} GGRKKYKL 8 HIV-1 gag 2.20 1997Human 1AGC(41) B*0801 {a} GGKKKYQL {c} GGKKKYQL 8 HIV-1 gag 2.10 1997Human 1AGD(41) B*0801 {a} GGKKKYKL {c} GGKKKYKL 8 HIV-1 gag 2.05 1997Human 1AGE(41) B*0801 {a} GGRKKYRL {c} GGRKKYRL 8 HIV-1 gag 2.30 1997Human 1AGF(41) B*0801 {a} GGKKRYKL {c} GGKKRYKL 8 HIV-1 gag 2.20 1997Human Subset4–group 1 1HSA(42) B*2705 {a} ARAAAAAAA {c} ARAAAAAAA 9 — 2.10 1992Human Subset 5–group 1 1A1N(43) B*3501 {a} VPLRPMTY {c} VPLRPMTY 8 HIV-1 Nef 2.00 1998Human Subset 5–group 2 1A9E(44) B*3501 {a} LPPLDITPY {c} LPPLDITPY 9 EBV-Ebna3c 2.50 1998Human 1A9B(44) B*3501 {a} lpplditpy {c} 9 EBNA-3C 3.20 1998Human Subset 6–group 1 1A1M(45) B*5301 {a} TPYDINQML {c} TPYDINQML 9 HIV-2 Gag 2.30 1998Human 1A1O(45) B*5301 {a} KPIVQYDNF {c} KPIVQYDNF 9 HIV-1 Nef 2.30 1998Murine Subset 7–group 1 1OSZ(46) H-2KB {a} RGYLYQGL {c} RGYLYQGL 8 Vsv nucleoprotein 2.10 1999Murine 2VAB(47) H-2KB {a} RGYVYQGL {p} RGYVYQGL 8 SV nucleoprotein 2.50 1996Murine 1KBG(48) H-2K
B
{h} RGYVYuGL {p} 8 Synthetic 2.20 1999Murine 1VAC(49) H-2KB {a} SIINFEKL {p} SIINFEKL 8 Ovalbumin 2.50 1996Murine Subset 7–group 2 1VAD(49) H-KB {a} SRDHSRTPM {p} SRDHSRTPM 9 Yeast a-glucosidase 2.50 1996Murine 2VAA(47) H-2K
B
{a} FAPGNYPAL {p} FAPGNYPAL 9 Vsv nucleoprotein 2.30 1996Murine Subset 8–group 1 1BZ9(50) H-2DB {a} FAPGVFPYM {c} FAPGVFPYM 9 Peptide P1027 2.80 1998Murine 1CE6(51) H-2DB {a} FAPGNYPAL {c} FAPGNYPAL 9 SV nucleoprotein 2.90 1999Murine 1QLF(51) H-2DB {a} FAPSNYPAL {c} FAPSNYPAL 9 SV-nucleoprotein 2.65 1999Murine Subset 9–group 1 1BII(52) H-2DD {a} RGPGRAFVTI {p} RGPGRAFVTI 10 HIV-1 P18–110 2.40 1998Murine Subset 10–group 1 1LDP(53) H-2LD {a} APAAAAAAM {p} APAAAAAAM 9 Natural Peptide 3.10 1998
PDB 5 protein data bank, PL 5 peptide length, R 5 resolution, CIM 5 chain identifier for MHC, CIP 5 chain identifier for peptide. Release year 5 the yearin which the entry was released by PDB, Obsolete entries are marked by “superscripted #” at the left end of the PDB code. References are cited at the right endof each PDB code. Duplicate peptides were removed in the non-redundant peptide set.
541Towards the MHC-Peptide Combinatorics

RESULTS AND DISCUSSIONThe MHC-peptide project [15, 16] was explored withgreat momentum during the last decade by defininganchor residues in peptide binding motifs [65] and bythe subsequent utilization of those principles in compu-tational tools [22–26]. The speculated nightmare incollecting immunologic or biochemical binding data,after rigorous cloning, in vitro expression and purificationof every MHC allele to perform binding assay with acombinatorial library of peptides, sets the limitation todefine anchor residues in peptide motifs for a wide arrayof MHC alleles. Simultaneous progress in computationalstructural biology [66] provided a structure-based meth-odology for MHC-peptide prediction [19, 20]. Assign-ing quantitative prediction score for the derived three-dimensional models and the subsequent grouping [21] orranking [19, 20, 69] of the modeled complexes is ofparticular interest. Ranking of the modeled MHC-pep-tide complexes using pairwise potentials has been provedto preferentially select hydrophobic-hydrophobic inter-action [19]. Recent attempts were made to exploit theavailable structural data on MHC-peptide binding topredict the binding affinities for MHC peptide-ligands[67]. However, the analysis was restricted to very fewalleles and future cross validation requires more biophys-ical information on diversified MHC-peptide complexesto recalibrate interaction functions. Addressing the fun-damental question of what these complexes do and, mostimportantly, how they do it by taking into account thesubtle polymorphic changes in the MHC binding grooveis our interest. This will establish an understanding forthe consequences of multiple interactions involved in thecascade of events during cell-mediated immune response.Both class I and class II MHC molecules possess a pep-tide binding functional groove characterized by a similarstructural fold [10]. In order to establish a quantitativeunderstanding for MHC-peptide binding, the availableinformation have to be thoroughly analyzed. The task is
to identify a set of combination rules that will lead to avat with high concentration of knowledge rich interac-tion variables describing MHC-peptide binding.
Types of Rules Describing MHC-Peptide BindingBroadly, the types of rules may be biological, chemical,and physical, or a combination of the three. Biologically,two or more parameters describe MHC-peptide binding:(i) MHC diversity [11] and (ii) peptide diversity [16].Chemically, five or more parameters describe MHC-peptide binding: (i) chemical property of amino acids; (ii)chemical property of atoms constituting the amino acid;(iii) overall chemistry of the peptide [19, 20]; (iv) overallchemistry of MHC grooves [14]; and (v) chemistry of theenvironment. Physically, eight or more parameters de-scribe MHC-peptide binding: (i) peptide length; (ii)amino acid size; (iii) residue position [65]; (iv) peptideresidue conformation; (v) overall conformation of thebound peptide [20]; (vi) peptide complementation withthe MHC; (vii) peptide backbone contribution [20]; and(viii) peptide side-chain contribution [20]. Thus, morethan 15 types of parameters can be used to describeMHC-peptide binding either individually or in combi-nation. Some of these parameters were identified andtheir contribution towards MHC-peptide binding pre-diction has been reported [14, 19, 20, 67]. The sensitiv-ity and specificity of MHC-peptide binding predictionscan be further improved by identifying novel interactionparameters from MHC-peptide crystal data.
Information from MHC-Peptide DataWhile extracting knowledge from data repositories, weprepare data of highest quality that are quantitativelyrich in information content [74]. In our current investi-gation on MHC-peptide binding it is required to answerthe following questions. How much structural data iscurrently available on MHC-peptide binding? Is thisdata sufficient enough to address caveats in MHC-pep-
TABLE 2 Class II MHC-peptide complexes in PDB
MHCsource Subgroups
PDBcode
MHCallele CIM Peptide set CIP PL Peptide source
R[Å]
Releaseyear
Human Subset 1–group 1 1AQD(54) DR1 {a,b} *VGSDWRFLRGYHQYA {c} 15 Endogenous peptide 2.45 1998Human Subset 1–group 2 1DLH(55) DR1 {a,b} PKYVKQNTLKLAT {c} 13 Influenza virus 2.80 1994Human 1SEB(56) DR1 {a,b} AAAAAAAAAAAAA {c} 13 Endogenous peptide 2.70 1996Human Subset 2–group 1 1BX2(57) DR2 {a,b} ENPVVHFFKNIVTPR* {c} 15 HMBP 2.60 1998Human Subset 3–group 1 1A6A(58) DR3 {a,b} PVSKMRMATPLLMQA {c} 15 CLIP fragment 2.75 1998Human Subset 4–group 1 2SEB(59) DR4 {a,b} AYMRADAAAGGA {e} 12 Collagen II 2.50 1998Murine Subset 5–group 1 1IAO(60) I-AD {a,b} RGISQAVHAAHAEI {b} 14 Egg ovalbumin 2.60 1998Murine 2IAD(60) I-AD {a,b} GHATQGVTAASSHE {b} 14 Influenza
Hemagglutinin2.40 1998
PDB 5 protein data bank, PL 5 peptide length, R 5 resolution, CIM 5 chain identifier for MHC, CIP 5 chain identifier for peptide. Release year 5 the yearin which the entry was released by PDB, References are cited at the right end of each PDB code. An asterisk (*) marks those residues for which the structuralinformation is not available.
542 P. Kangueane et al.

tide binding? At the time of preparing this article, 36nonredundant MHC-peptide structural complexes wereidentified in PDB. Of the 36 complexes, 28 were class IMHC-peptide complexes and the remaining 8 complexeswere class II MHC-peptide complexes. Again, out of 28nonredundant class I entries listed in Table 1, 10 aremurine H-2 peptide complexes and the remaining 18 arehuman MHC-peptide complexes. Similarly, of the eightclass II entries listed in Table 2, two are murine com-plexes and the remaining six are human MHC-peptidemolecules. In 1998 18 more MHC-peptide complexeswere released by PDB (Figure 1a) and the availablestructural information has tremendously improved ourknowledge [14]. MHC-peptide complexes are availablefor HLA-A*0201, HLA-A*6801, HLA-B*0801, HLA-B*2701, HLA-B*3501, HLA-B*5301, H-2KB, H-2DB,and H-2LD (Tables 1 and 2). Among them, HLA-A*0201-peptide complexes are the most represented(Figure 1b). Therefore a representative structure-basedbinding information on allele specific MHC-peptidecomplexes will deduce the relationship that map thestructure function differences influenced by sequencevariation. The overwhelming advancements in r-DNAtechnology and high-throughput structural genomicsprojects [68] will speed up MHC-peptide research in thenear future.
Information on PDB code, MHC name, chain identi-fier for MHC and peptide, peptide length, peptide se-quence, peptide source, MHC source, resolution, PDBrelease year, binding value, gap volume, gap index,‚ASA, and intermolecular hydrogen bonds is provided(Tables 1–4). These tables summarize the current knowl-edge available on MHC alleles, their structures, and theirfunctions. If information on binding constants and bio-physical binding pattern is made available for MHC-peptide complexes, it is possible to relate functional datawith structural data and, therefore, with the sequencedata.
MHC Residues and Their Contribution toPeptide BindingThe residues forming the functional peptide bindingdomains in MHC alleles are highly polymorphic; there-fore, their peptide-binding pattern varies due to bothpeptide and MHC allele diversity. The specific bindingof antigenic peptides to a MHC residue is characterizedby the sequence position of the residue, chemical prop-erty, physical conformation in the groove, and biologicaldiversity across MHC alleles. The contribution made byMHC residue positional preference toward peptide bind-ing is shown as a quantitative measure of ‚ASA uponcomplex formation in both class I data set (Figure 2) andclass II data set (Figure 3). The results demonstrate thatnot all the MHC residues forming the binding groove are
involved in peptide binding. The MHC residue positionspreferentially involved in peptide binding are shown in alimited data set and this does not help to draw any clearconclusion on peptide binding (Figures 2 and 3). Butthere is a preferential selection among residues in the sixpockets (A–F) to bind specific peptides. In 1998, Zhangand coworkers [14] revealed that the class I MHC pock-ets could be classified into families distinguishable bytheir common physico-chemical properties and peptideside chain selectivities. Therefore it is possible to classifyall the pockets in known MHC alleles into distinguish-able pocket families. If a relationship is established be-tween these pocket families [14] and the residue posi-tions preferentially involved in peptide binding (Figures2 and 3), predicting the functional significance of MHCalleles from sequence information will become trivial.
Peptide Binding Pattern in SubgroupsThe binding of peptides to MHC alleles is studied ineach subgroup, as a measure of ‚ASA upon complexformation. Hence, the percentage burial of a peptideresidue is calculated. The peptide backbone conforma-tion in a given subgroup data is similar [20], peptideburial profiles described as a quantitative measure of‚ASA are similar among subgroups (Figures 4 and 5),and therefore the physical binding pattern is identical atleast at both the ends of the peptide. The middle part ofthe peptide may zigzag or bulge out of the bindinggroove using very different paths within the bindinggroove. Although a qualitative understanding of thisproperty was observed in specific cases [33], our analysisis comprehensive for an updated data set and comparisonamong the entries is graphically represented by a quan-titative measure for binding. Visual inspection of theseprofiles in this comparative analysis explains that thezigzag noise is relatively less within subgroups. Thepeptide binding pattern among entries in a subgroup aresimilar but not identical. This will also provide a meansto compare sequence anchors with structural anchors andhence relate to biochemical/immunologic binding val-ues. The binding values for most of the entries in ourdata set are not available as a quantitative measure ofbinding. However, literature survey helped us identifythe binding values for the structural and sequence infor-mation represented in PDB codes: 1HHJ, 1HHK,1HHI, 1HHH and 1VAC (Table 3).
Residue Sequence Anchor and Structural AnchorIt is important not to consider only the basic anchorresidues of a peptide motif for a given MHC moleculebecause the nonanchor positions of peptides also contrib-ute considerably to the interaction with MHC [65]. Forexample, in HLA-A*0201 bound 9-mer peptides clus-tered under subset 1, group 1, the primary anchor resi-
543Towards the MHC-Peptide Combinatorics

FIGURE 1 Distributionof MHC-peptide complexesat the Protein Data Bank.(a) Distribution based onPDB release year. (b) Dis-tribution based on MHCallele specificity. (c) Distri-bution based on peptidelength.
544 P. Kangueane et al.

TABLE 3 Some properties at the class I MHC peptide interface
PDBcode Subgroups
Bindingstrength
Interfacearea [Å2]
H-bonds[number]
Volume[Å3]
Gap index[Å]
1HHJ Subset 1–group 1 242(69) [IC50] 880.4 14 827.4 0.941HHK 2.5(70) [IC50] 885.0 10 1083.4 1.221B0G — 869.0 12 441.3 0.511HHG — 803.3 12 1039.9 1.291HHI 6(71) [IC50] 857.8 9 455.7 0.532CLR Subset 1–group 2 — 910.9 10 911.3 1.001HHH 2.5(72) [IC50] 940.5 11 655.9 0.701TMC Subset 2–group 1 — 955.5 14 926.2 0.971AGB Subset 3–group 1 — 844.4 15 881.7 1.041AGC — 830.5 18 688.1 0.831AGD — 846.0 16 816.1 0.961AGE — 832.8 15 920.6 1.101AGF — 883.1 14 765.4 0.871HSA Subset 4–group 1 — 727.8 14 1148.4 1.581A1N Subset 5–group 1 — 879.7 11 670.2 0.761A9E Subset 5–group 1 — 895.5 12 779.3 0.871A1M Subset 6–group 1 — 845.4 12 971.2 1.141A1O — 994.5 10 778.8 0.781OSZ Subset 7–group 1 — 946.9 18 756.2 0.802VAB — 881.7 12 1301.0 1.471VAC 5900(73) [M21 s21] 892.2 14 691.2 0.771VAD Subset 7–group 2 — 880.5 21 939.5 1.072VAA — 938.2 16 738.3 0.791BZ9 Subset 8–group 1 — 884.0 10 897.0 1.011CE6 — 867.7 15 787.7 0.911QLF — 893.7 13 567.3 0.631BII Subset 8–group 2 — 937.4 14 792.0 0.841LDP Subset 9–group 1 — 771.4 9 889.3 1.15
Interface area indicates a measure of the mean change in accessible area (mean DASA) for the peptide and the MHC molecules when going from a monomericMHC molecule to a dimeric MHC-peptide complex state. Solvent accessible surface area both for the MHC-peptide complexes as well as the individual peptidesand MHC molecules was calculated using the algorithm implemented by Lee and Richard [61]. The gap volume between the peptide and the MHC was calculatedusing SURFNET [63]. The number of intermolecular hydrogen bonds between the peptide and the MHC were calculated using HBPLUS [64] in which hydrogenbonds are defined according to standard geometric criteria. Gap index, defining the complementarity of the interacting surfaces in the MHC-peptide have beenevaluated by the formula as defined elsewhere [29]. The derived knowledge presented here is only for the non-redundant peptide set. Binding strength for thesepeptides was expressed in IC50 units, denoting the peptide’s concentration required to inhibit the binding of the standard peptide by 50%. The entries for whichthe relative binding strengths are not easily available are marked by “—”.
TABLE 4 Some properties at the class I MHC peptide interface
PDBcode Subgroups
Bindingstrength
Interfacearea [Å2]
H-bonds(number)(Total)
ChainA
ChainB
Volume[Å3]
Gap index[Å]
1AQD Subset 1–group 1 — 1211.2 18 10 8 1182.7 0.981DLH Subset 1–group 2 — 1168.4 17 10 7 1081.8 0.931SEB — 834.8 12 7 5 964.1 1.151BX2 Subset 2–group 1 — 1034.4 15 9 6 1308.6 1.271A6A Subset 3–group 1 — 1171.0 19 10 9 1204.7 1.032SEB Subset 4–group 1 — 960.5 14 5 9 836.1 0.871IAO Subset 5–group 1 — 1087.4 14 8 6 1449.5 1.332IAD Subset 5–group 2 — 963.5 13 7 6 1475.1 1.53
Interface area indicates a measure of the mean change in accessible area (mean DASA) for the peptide and the MHC molecules when going from a monomericMHC molecule to a dimeric MHC-peptide complex state. Solvent accessible surface area for the MHC-peptide complexes, monomeric peptides and monomericMHC molecules is calculated using the algorithm implemented by Lee and Richard [61]. The gap volume between the peptide and the MHC was calculated usingSURFNET [63]. The number of intermolecular hydrogen bonds between the peptide and the MHC were calculated using HBPLUS [64] in which hydrogen bondsare defined according to standard geometric criteria. Gap index, defining the complementarity of the interacting surfaces in the MHC-peptide have been evaluatedby the formula as defined elsewhere [29]. The entries for which the relative binding strengths are not easily available are marked by “—”.
545Towards the MHC-Peptide Combinatorics

dues at positions 2 [L (leucine)/I (isoleucine)] and 9 [V(valine)/L (leucine)] are buried (Figure 4a). Interestingly,residues at position 4 are consistently exposed irrespec-tive of the amino acid preference at that position and theauxiliary anchor for residues at position 6 is unpredict-ably influenced by residue property (Figure 4a). Residuesat positions 3 and 7 are relatively well buried comparedwith the residue at position 8. The percentage burial ofthe residue at position 5 is influenced by the residueproperty.
Similarly, in A*0201 specific 10-mer peptides clus-tered under subset 1, group 2, the primary anchor atposition 2 [L (leucine) and 9/10 (V (valine)/L (leucine)]are also buried (Figure 4b). The residue at position 4 isagain exposed in all entries, whereas the auxiliary anchorresidue at position 6 is influenced by the residue prop-erty. Residues at positions 3 and 7 are also well buried.But the percentage of burial for residues at positions 5and 8 is only 60%.
The molecular problem of position-specific peptideanchor is highly combinatorial in nature for each MHCallele. Interesting features of such anchor specificity haveto be identified and applied in the context of everysubgroup data discussed in this article (Figures 4 and 5).It should also be noted that the data in each subgroup
does not highlight all the properties of MHC peptidebinding. Hence, a representative MHC-peptide data setwill add value to our current knowledge on MHC-peptide binding in future.
Peptide Length and Binding PatternThe data set was also grouped into seven subgroups basedon peptide length. MHC allele specific peptide struc-tures are currently available for peptides of length 8, 9,10, 12, 13, 14, and 15 (Figure 1c). Out of the currentlyavailable MHC allele specific peptide structures, pep-tides of length 9 are the most represented. Peptides oflength 8, 9, and 10 are bound to class I MHC molecules(Table 1) and peptides of length 12, 13, 14, and 15 arebound to class II MHC molecules (Table 2).
The 8-mer peptides. In peptides of length 8 residues boundto class I molecules, all amino acid residues at positions1, 2, 3, 5, and 8 are buried whereas those at positions 4,6, and 7 are exposed from the groove (Figure 6a). Theavailable structural data describing 8-mer peptides isboth limited and less diverse at the sequence level (Table1). To deduce the complete nature of 8-mer peptidesbinding to MHC molecules, we need a wider pool ofallele specific structural data enfolding information onsequence diversity.
The 9-mer peptides. In 9-mer peptides all residues at po-sitions 1, 2, 3, 7, and 9 are buried, whereas the residueat position 4 is consistently exposed in all the complexes.
FIGURE 2 Class I MHC residue positions involved in pep-tide binding, illustrated as a measure of ‚ASA when goingfrom a monomeric MHC molecule to a dimeric MHC-peptidecomplex state.
546 P. Kangueane et al.
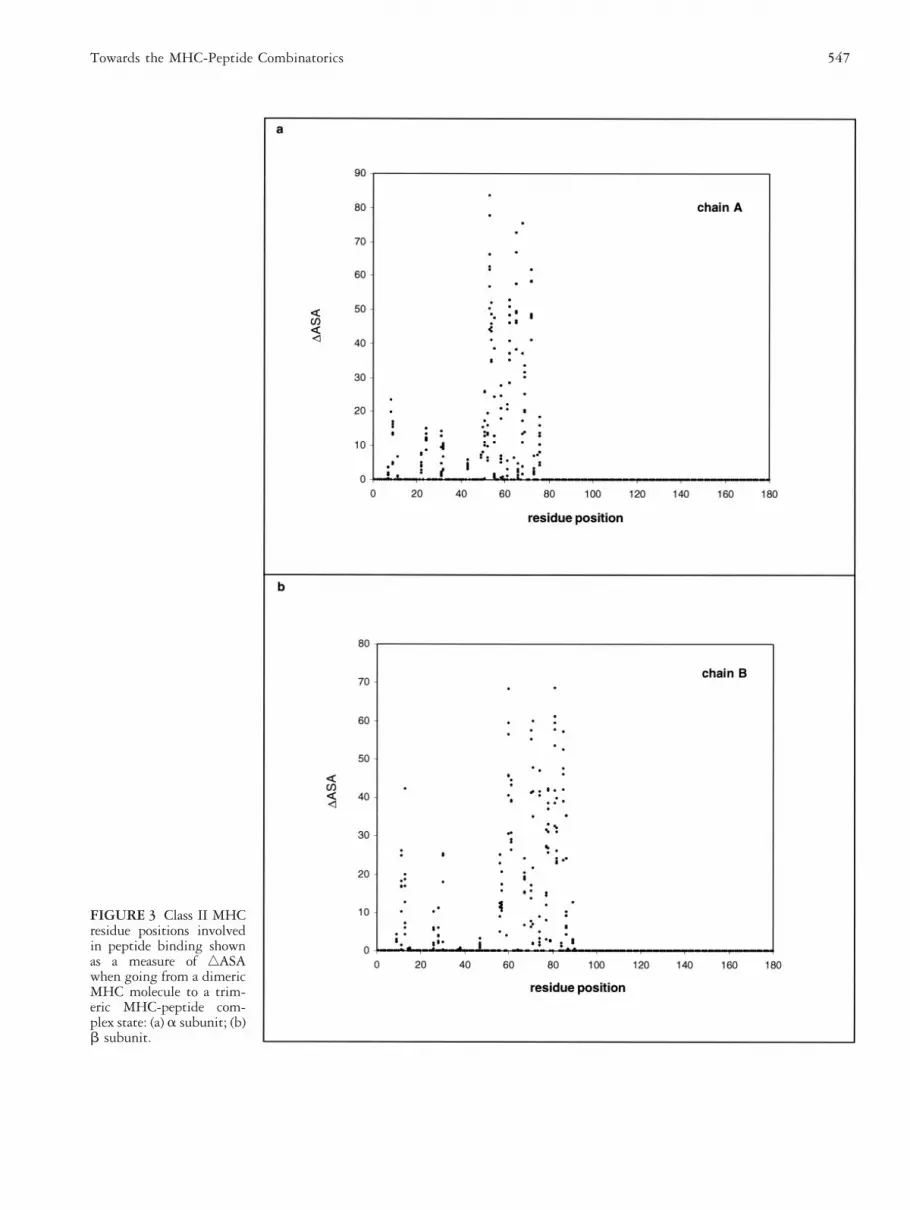
FIGURE 3 Class II MHCresidue positions involvedin peptide binding shownas a measure of ‚ASAwhen going from a dimericMHC molecule to a trim-eric MHC-peptide com-plex state: (a) a subunit; (b)b subunit.
547Towards the MHC-Peptide Combinatorics

FIGURE 4 MHC classI peptide positional an-chor defined by percent-age of burial (% burial)when going from a mono-meric peptide state toa dimeric MHC-peptidecomplex state is shown indifferent clustered sub-groups: (a) subset 1, group1; (b) subset 1, group 2;(c) subset 2, group 1; (d)subset 3, group 1; (e) sub-set 4, group 1; (f) subset 5,group 1; (g) subset 5,group 2; (h) subset 6,group 1; (i) subset 7,group 1; (j) subset 7,group 2; (k) subset 8,group 1; (l) subset 8,group 2; and (m) subset 9,group 1. The details aboutthe subgroups are given inTable 1.
548 P. Kangueane et al.

The residues at positions 5, 6, and 8 are unpredictablyexposed and buried (Figure 6b).
The 10-mer peptides. In 10-mer peptides all residues atpositions 1, 2, 3, and 10 are buried and those residues atposition 8 are consistently buried upon complex forma-tion (Figure 6c). Residues at positions 4, 5, 6, 7, and 9are again unpredictably buried and exposed.
The 12-mer peptides. In class II specific 12-mer peptides,residues at position 3, 6, 8, 9, and 11 are buried and therest are exposed (Figure 6d).
The 13-mer peptides. In 13-mer peptides, residues at po-sition 3, 6, 8, 9, 11, and 12 are buried and the rest areexposed (Figure 6e).
The 14-mer peptides. In 14-mer peptides, residues at po-sition 4, 7, 9, 10, and 12 are buried and the rest areexposed (Figure 6f).
The 15-mer peptides. In 15-mer peptides, residues at po-sition 5, 8, 10, 11, and 13 are buried and the rest areexposed (Figure 6g).
Anchor Shift in Positional PreferencePeptide anchor positions defined by the percentage ofburial upon complex formation illustrates that the firstanchor from the N terminal is at position 3, 3, 4, and 5for MHC class II specific peptides of length 12, 13, 14,and 15 residues, respectively. The second anchor is atposition 6, 6, 7, and 8 for peptides of length 12, 13, 14,and 15 residues, respectively. The third anchor is atpositions 8/9, 8/9, 9/10, and 10/11 for peptides of length12, 13, 14, and 15 residues, respectively. Lastly, thefourth anchor is at positions 11, 11/12, 12, and 13 forpeptides of length 12, 13, 14, and 15, respectively. Thereis an anchor shift in positional preference towards the Cterminal at all the four anchors in class II specific pep-tides, as the peptide length increases (Figures 6d–6g). Itshould also be noted that the number of residues exposed
FIGURE 5 MHC classII peptide positional an-chor defined by percent-age of burial (% burial)when going from a mono-meric peptide state to atrimeric MHC-peptidecomplex state is shown indifferent subgroups: (a)subset 1, group 1; (b) sub-set 1, group 2; (c) subset2, group 1; (d) subset 3,group 1; (e) subset 4,group 1; and (f) subset 5,group 1. The details aboutthe subgroups are given inTable 2.
549Towards the MHC-Peptide Combinatorics

FIGURE 6 Residue po-sitional anchor defined bypercentage of burial (%burial) is shown for MHCbound peptides. Class Ispecific peptides: (a) 8mer; (b) 9 mer; and (c) 10mer. Class II specific pep-tides: (d) 12 mer; (e) 3mer; (f) 14 mer; and (g)15 mer.
550 P. Kangueane et al.

between two consecutive anchor positions remains thesame (Figure 6d–6g). This information implies that thepeptide entry into the class II MHC groove follows asimilar path irrespective of the peptide length.
Intermolecular Hydrogen Bonds inMHC-Peptide ComplexesThe number of intermolecular hydrogen bonds formedbetween the peptide and the MHC varies from onecomplex to another and, even within complexes groupedunder a subgroup (Tables 3 and 4). In class I MHC-peptide complexes the number of intermolecular hydro-gen bonds inversely corelates with increasing peptidelength (Figure 7a). The implication is that the number ofintermolecular hydrogen bonds is more for peptides oflength 8 residues than peptides of length more than 8residues. However, the correlation is not very strong.Contrary to observation in class I MHC-peptide com-plexes, the number of intermolecular hydrogen bonds inclass II MHC-peptide complexes directly correlates withincreasing peptide length (Figure 7b). It will be reason-able to compare intermolecular hydrogen bonding withbinding constants to estimate the significance of inter-molecular hydrogen bonding in MHC peptide binding.
Interface Area in MHC-Peptide ComplexesThe mean interface area, in class I MHC-peptide com-plexes, is 871.81 6 62.59 (Å2) and in class II MHC-peptide complexes it is 1064.16 6 124.80 (Å2). Inter-face area increases with increase in peptide length in bothclass I complexes (Figure 7c) and class II complexes(Figure 7d). It is also shown that the number of inter-molecular hydrogen bonds increases with increasing in-terface area in both class I complexes (Figure 7e) and classII complexes (Figure 7f). No clear-cut conclusion can bemade toward epitope selection using this information.However, it is possible to understand peptide binding asa measure of interface area upon the availability of bind-ing constants for a representative set of MHC-peptidecrystal structures.
Gap Index in MHC-Peptide ComplexesThe Gap index (Å) in a MHC peptide complex is theratio of gap volume between the MHC and the peptide(Å3) to the interface area (Å2) per complex. Gap indexevaluates the peptide complementation with the MHCreceptors (Tables 3 and 4). In class I MHC peptidecomplexes, the mean gap index is 0.95 6 0.24 Å and thesame in class II MHC peptide complexes is 1.12 6 0.20Å. On average, the ratio of gap volume to interface areais more in class II complexes than in class I complexes.This suggests that the interface area is greater than thegap volume in class I MHC peptide complexes. On thecontrary, the mean gap volume is greater than the inter-
face area in class II MHC peptide complexes. Not muchdifference can be identified in gap index between com-plexes of different alleles either in class I or in class IIMHC-peptide complexes.
Backbone or Side Chain at theMHC-Peptide InterfaceThe dominance of peptide backbone and side chain at-oms at the interface is described for each MHC class. Inclass I MHC peptide complexes, within cut-off distancesranging from 2.7–3.4 Å, the proportion of backboneatoms dominates over side chain atoms (Figure 8a).However, at cut-off distances . 3.4 Å, side chain atomsare dominant over the backbone atoms. In class II MHCpeptide complexes scanned for interactions, at cut-offdistances Ä 2.4 Å the percent distribution of backboneatoms is more prominent than the side chain atoms(Figure 8b). The results suggests that in class II data set,backbone peptide atoms are proportionately dominant atthe MHC peptide interface, whereas in the class I dataset, peptide backbone atoms dominate within a windowof cut-off distances. A methodology was proposed bySchueler-Furman and coworkers [20] to select backbonestructures from a data set for the prediction of peptidestructures in the class I MHC groove. They revealed thatthe structure of a peptide in the groove is influenced byits interaction with the groove. The information pre-sented in this section suggests that the peptide backboneatoms play a crucial role in MHC-peptide binding in-teractions. This question has to be revisited in future foridentifying common features in a fairly homogenousMHC-peptide data. The accumulated structural knowl-edge on MHC molecules and the computational advance-ments in modeling procedures provide a methodology topredict the structure for each of the allelic variants. Butthe application of molecular modeling to estimate thefunctional consequences of MHC alleles in the context ofpeptide binding requires recalibration of interactionfunction. Hence, this study on MHC peptide binding isonly the beginning, and future advancement in compu-tational tools and database integration techniques willaid in understanding the principles of MHC-peptidebinding.
CONCLUSIONThe peptide-binding pattern in the MHC groove de-pends on the sequence of the peptide and the MHCallele. Our investigation suggests that the peptide-bind-ing patterns in MHC molecules are similar within com-plexes grouped under a subgroup based on allele speci-ficity and peptide length. The fourth residue in class Ispecific 8- and 9-mer peptides is consistently solventexposed irrespective of the chemical property of the
551Towards the MHC-Peptide Combinatorics

FIGURE 7 The corre-lation between peptidelength and intermolecularhydrogen bonding isshown: (a) class I MHC-peptide complexes; (b)class II MHC-peptidecomplexes. The correla-tion of interface area (Å2)to peptide length (c and d)and intermolecular hydro-gen bonding (e and f) isalso shown for class I andclass II. MHC-peptidecomplexes.
552 P. Kangueane et al.
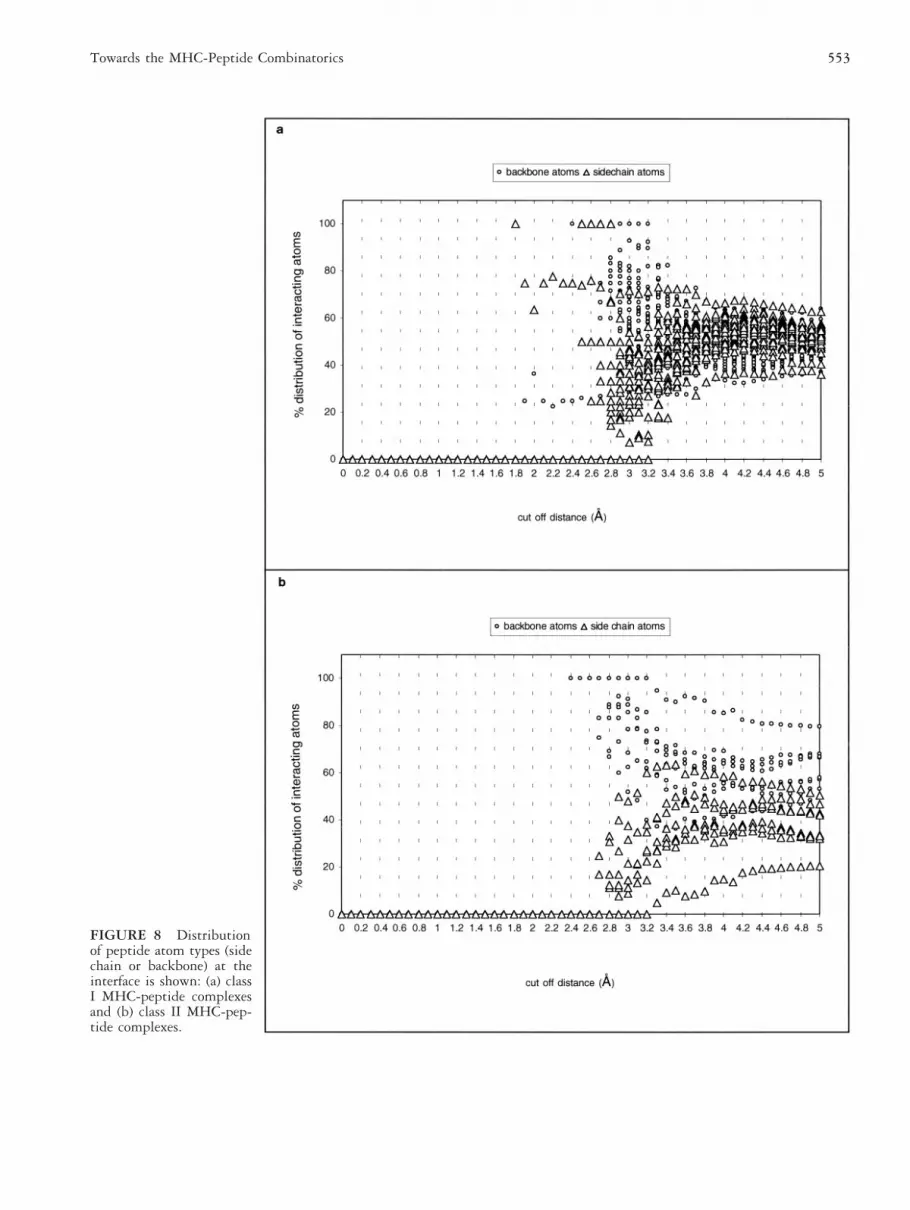
FIGURE 8 Distributionof peptide atom types (sidechain or backbone) at theinterface is shown: (a) classI MHC-peptide complexesand (b) class II MHC-pep-tide complexes.
553Towards the MHC-Peptide Combinatorics

residue at this position. However, this is not always truewith the class I specific 10-mer peptides. It should benoted that the conclusion made here is based on peptidesbound to HLA-A*0201, HLA-A*6801, HLA-B*0801,HLA-B*2701, HLA-B*3501, HLA-B*5301, H-2KB,H-2DB, and H-2LD molecules whose structures areknown in a complex state. In class II MHC peptidecomplexes there is a positional anchor shift towards theC terminal of the peptide as the length of the boundpeptide increases. At the MHC-peptide interface, pep-tide backbone atoms are more prominent than side chainatoms in both class I and class II MHC-peptide com-plexes. The information presented in this article providesinsight into peptide binding patterns in the MHCgrooves. Successful incorporation of these variables into aprediction algorithm will improve the efficiency ofMHC-peptide binding prediction. The importance ofvan der Waals contribution [20], MHC groove specificpeptide backbone conformation [20], evaluation of hy-drophobic-hydrophobic interactions in models [19], theuse of Fresno [67], and the effect of knowledge basedgrouping rules [21] in MHC-peptide binding predictionhave been reported already. However, all available pre-diction methodologies require sufficient cross validationand model refinement for application on a regular basis.
The successful prediction of short antigenic peptidesfrom a pool of viral/bacterial genome sequences usingMHC-peptide prediction models will be immunologi-cally and technologically advantageous in identifying theimmunogenic peptides. This could be further testedusing in vivo models for consideration as vaccines andtherapeutics. Apart from peptide MHC specificity otherundoubtedly important mechanisms, such as enzymemediated antigen processing, peptide transport, loadingof peptides to MHC molecules, and the phenomenon ofTCR repertoires, need to be understood in conjunctionwith each another. In the future, a knowledge-basedrelationship has to be established between sequence,structure, and biochemical or immunologic data describ-ing MHC-peptide binding. A comprehensive under-standing on MHC-peptide binding using knowledge-based combination rules will enlighten the roadmapsexplaining complex immunochemical events.
ACKNOWLEDGMENTS
We thank Dr. Roman Laskowski, Department of Biochemistryand Molecular Biology, University College, London, for mak-ing the SURFNET program available to us. We wish toexpress our sincere appreciation to all members of the researchcentre for many discussions on the subject of this article. Thiswork was supported by the research grants and facilities pro-vided by National Science and Technology Board of Singapore(NSTB), Institute for Molecular and Cell Biology (IMCB),
Singapore 117094 and National University of Singapore(NUS), Singapore 119260.
REFERENCES
1. Sette A, Nepom GT: Antigen recognition. Curr OpinImmunol 12:77, 2000.
2. Abbas AK, Janeway CA: Immunology: improving onnature in the twenty first century. Cell 100:129, 2000.
3. Abrams SI, Schlom J: Rational antigen modification as astrategy to upregulate or downregulate antigen recogni-tion. Curr Opin Immunol 12:85, 2000.
4. Pamer E, Cresswell P: Mechanisms of MHC class I re-stricted antigen processing. Annu. Rev. Immunol 16:323,1998.
5. Jensen PE: Peptide binding and antigen presentation byclass II histocompatibility glycoproteins. Biopolymers 43:303, 1997.
6. Joshi RV, Zarutslie JA, Stern LJ: A three step kineticmechanism for peptide binding to MHC class II proteins.Biochemistry 39:3751, 2000.
7. Kasson PM, Rabinowitz JD, Schmitt L, Davis MM, Mc-Connell HM: Kinetics of peptide binding to the class IIMHC protein I-Ek. Biochemistry 39:1048, 2000.
8. Natarajan SK, Assadi M, Nasseri SS: Stable peptide bind-ing to MHC class II molecule is rapid and is determinedby a receptive conformation shaped by prior associationwith low affinity peptides. J Immunol 162:4030, 1999.
9. Springer S, Doring K, Skipper JC, Townsend AR, Cerun-dolo V: Fast association rates suggest a conformationalchange in the MHC class I molecule H-2Db upon peptidebinding. Biochemistry 37:3001, 1998.
10. Batalia MA, Collins EJ: Peptide binding by class I andclass II MHC molecules. Biopolymers 43:281, 1997.
11. Bodmer JG, Marsh SGE, Albert ED, Bodmer WF, Bon-trop RE, Dupont B, Erlich HA, Hansen JA, Mach B,Mayr WR, Parham P, Petersdorf EW, Sasazuki T, Schre-uder GM, Strominger JL, Svejgaard A, Terasaki PI: No-menclature for factors of the HLA system, 1998. TissueAntigens 53:407, 1999.
12. http://www.anthonynolan.org.uk
13. http://www.ebi.ac.uk/imgt/hla
14. Zhang C, Anderson A, DeLisi C: Structural principles thatgovern the peptide binding motifs of class I MHC mol-ecules. J Mol Biol 281:929, 1998.
15. Buus S: Description and prediction of peptide-MHCbinding: the “human MHC project.” Curr Opin Immunol11:209, 1999.
16. Pinilla C, Martin R, Gran B, Appel JR, Boggiano C,Wilson DB, Houghten RA: Exploring immunologicalspecificity using synthetic peptide combinatorial libraries.Curr Opin Immunol 11:193, 1999.
17. Krebs S, Rognan D: From peptides to peptidomimetics:
554 P. Kangueane et al.

design of nonpeptide ligands for major histocompatibilityproteins. Pharm Acta Helv 73:173, 1998.
18. The MHC Sequencing Consortium: Complete sequenceand gene map of a human major histocompatibility com-plex. Nature 401:921, 1999.
19. Altuvia Y, Sette A, Sidney J, Southwood S, Margalit H: Astructure-based algorithm to predict potential bindingpeptides to MHC molecules with hydrophobic bindingpockets. Hum Immunol 58:1, 1997.
20. Schueler-Furman O, Elber R, Margalit H: Knowledge-based structure prediction of MHC class I bound peptides:a study of 23 complexes. Fold Des 3:549, 1998.
21. Kangueane P, Sakharkar MK, Lim KS, Hao H, Lin K,Chee RE, Kolatkar PR: Knowledge-based grouping ofmodeled HLA peptide complexes. Hum Immunol 6:460,2000.
22. Milik M, Sauer D, Brunmark AP, Yuan L, Vitiello A,Jackson MR. Peterson PA, Skolnick J, Glass CA: Appli-cation of an artificial neural network to predict specificclass I MHC binding peptide sequences. Nat Biotechnol16:753, 1998.
23. Honeyman MC, Brusic V, Stone NL, Harrison LC: Neuralnetwork-based prediction of candidate T-cell epitopes.Nat Biotechnol 16:966, 1998.
24. Mamitsuka H: Predicting peptides that bind to MHCmolecules using supervised learning of hidden Markovmodels. Proteins 33:460, 1998.
25. Parker KC, Shields M, DiBrino M, Brooks A, Coligan JE:Peptide binding to MHC class I molecules: implicationsfor antigenic peptide prediction. Immunol Res 14:34,1995.
26. Schafer JR, Jesdale BM, George JA, Kouttab NM, DeGroot AS: Prediction of well-conserved HIV-1 ligandsusing a matrix-based algorithm, EpiMatrix. Vaccine 16:1880, 1998.
27. Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, MurzinAG, Chothia C: SCOP: a structural classification of pro-teins database. Nucleic Acids Res 28:257, 2000.
28. Brenner SE, Barken D, Levitt M: The PRESAGE databasefor structural genomics. Nucleic Acids Res 27:251, 1999.
29. Jones S, Thornton JM: Principles of protein-protein in-teractions. Proc Natl Acad Sci USA 93:13, 1996.
30. Berman HM, Westbrook J, Feng Z, Gilliland G, BhatTN, Weissig H, Shindyalov IN, Bourne PE: The proteindata bank. Nucleic Acids Res 28:235, 2000.
31. http://www.rcsb.org/pdb
32. Bernstein FC, Koetzle TF, Williams GJ, Meyer EE, BriceMD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M:The protein data bank: a computer-based archival file formacromolecular structures. J Mol Biol 112:535, 1977.
33. Madden DR, Garboczi DN, Wiley DC: The antigenicidentity of peptide-MHC complexes: a comparison of theconformations of five viral peptides presented by HLA-A2. Cell 75:693, 1993.
34. Gao GF, Tormo J, Gerth UC, Wyer JR, McMichael AJ,Stuart D.I., Bell JI, Jones EY, Jakobsen BK: Crystalstructure of the complex between human CD8 alpha (al-pha) and HLA-A2. Nature 387:630, 1997.
35. Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE,Wiley DC: Structure of the complex between humanT-cell receptor, viral peptide and HLA-A2. Nature 384:134, 1996.
36. Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE,Wiley DC: Two human T cell receptors bind in a similardiagonal mode to the HLA-A2/Tax peptide complex us-ing different TCR amino acids. Immunity 8:403, 1998.
37. Zhao R, Loftus DJ, Appella E, Collins EJ: Structuralevidence of T cell xeno-reactivity in the absence of mo-lecular mimicry. J Exp Med 189:359, 1999.
38. Bouvier M, Guo HC, Smith KJ, Wiley DC: Crystal struc-tures of HLA-A*0201 complexed with antigenic peptideswith either the amino or carboxyl-terminal group substi-tuted by a methyl group. Proteins 33:97, 1998.
39. Collins EJ, Garboczi DN, Wiley DC: Three-dimensionalstructure of a peptide extending from one end of a class IMHC binding site. Nature 371:626, 1994.
40. Collins EJ, Garboczi DN, Karpusas MN, Wiley DC: Thethree-dimensional structure of a class I major histocom-patibility complex molecule missing the alpha 3 domainof the heavy chain. Proc Natl Acad Sci USA 92:1218,1995.
41. Reid SW, McAdam S, Smith KJ, Klenerman P,O’Callaghan CA, Harlos K, Jakobsen BK, McMichael AJ,Bell JI, Stuart DI, Jones EY: Antagonist HIV-1 Gagpeptides induce structural changes in HLA B8. J Exp Med184:2279, 1996.
42. Madden DR, Gorga JC, Strominger JL, Wiley DC: Thethree-dimensional structure of HLA-B27 at 2.1 A resolu-tion suggests a general mechanism for tight peptide bind-ing to MHC. Cell 70:1035, 1992.
43. Smith KJ, Reid SW, Stuart DI, McMichael AJ, Jones EY,Bell JI: An altered position of the alpha 2 helix of MHCclass I is revealed by the crystal structure of HLA-B*3501.Immunity 4:203, 1996.
44. Menssen R, Orth P, Ziegler A, Saenger W: Decamer likeconformation of a nona-peptide bound to HLA-B*3501due to non-standard positioning of the C terminus. J MolBiol 285:645, 1999.
45. Smith KJ, Reid SW, Harlos K, McMichael AJ, Stuart DI,Bell JI, Jones EY: Bound water structure and polymorphicamino acids act together to allow the binding of differentpeptides to MHC class I HLA-B53. Immunity 4:215,1996.
46. Ghendler Y, Teng MK, Liu JH, Witte T, Liu J, Kim KS,Kern P, Chang HC, Wang JH, Reinherz EL: Differentialthymic selection outcomes stimulated by focal structuralalteration in peptide/major histocompatibility complexligands. Proc Natl Acad Sci USA 95:10061, 1998.
47. Fremont DH, Matsumura M, Stura EA, Peterson PA,
555Towards the MHC-Peptide Combinatorics

Wilson IA: Crystal structures of two viral peptides incomplex with murine MHC class I H-2Kb. Science 257:919, 1992.
48. Speir JA, Abdel-Motal UM, Jondal M, Wilson IA: Crystalstructure of an MHC class I presented glycopeptide thatgenerates carbohydrate-specific CTL. Immunity 10:51,1999.
49. Fremont DH, Stura EA, Matsumura M, Peterson PA,Wilson IA: Crystal structure of an H-2Kb-ovalbuminpeptide complex reveals the interplay of primary andsecondary anchor positions in the major histocompatibil-ity complex binding groove. Proc Natl Acad Sci USA92:2479, 1995.
50. Zhao R, Loftus DJ, Appella E. Collins EJ: Structuralevidence of T cell xeno-reactivity in the absence of mo-lecular mimicry. J Exp Med 189:359, 1999.
51. Glithero A, Tormo J, Haurum JS, Arsequell G, ValenciaG, Edwards J, Springer S, Townsend A, Pao YL, WormaldM, Dwek RA, Jones EY. Elliott T: Crystal structures oftwo H-2Db/glycopeptide complexes suggest a molecularbasis for CTL cross-reactivity. Immunity 10:63, 1999.
52. Achour A, Persson K, Harris RA, Sundback J, SentmanCL, Lindqvist Y, Schneider G, Karre K: The crystal struc-ture of H-2Dd MHC class I complexed with the HIV-1-derived peptide P18-110 at 2.4 A resolution: implicationsfor T cell and NK cell recognition. Immunity 9:199,1998.
53. Speir JA, Garcia KC, Brunmark A, Degano M, PetersonPA, Teyton L, Wilson IA: Structural basis of 2C TCRallorecognition of H-2Ld peptide complexes. Immunity8:553, 1998.
54. Murthy VL, Stern LJ: The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implica-tions for the structural basis of the specificity of peptidebinding. Structure 5:1385, 1997.
55. Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG,Strominger JL, Wiley DC: Crystal structure of the humanclass II MHC protein HLA-DR1 complexed with an in-fluenza virus peptide. Nature 368:215, 1994.
56. Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG,Chi YI, Stauffacher C, Strominger JL, Wiley DC: Three-dimensional structure of a human class II histocompati-bility molecule complexed with superantigen. Nature368:711, 1994.
57. Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfen-nig KW: Crystal structure of HLA-DR2 (DRA*0101,DRB1*1501) complexed with a peptide from humanmyelin basic protein. J Exp Med 188:1511, 1998.
58. Ghosh P, Amaya M, Mellins E, Wiley DC: The structureof an intermediate in class II MHC maturation: CLIPbound to HLA-DR3. Nature 378:457, 1995.
59. Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC:X-ray crystal structure of HLA-DR4 (DRA*0101,DRB1*0401) complexed with a peptide from humancollagen II. Immunity 7:473, 1997.
60. Scott CA, Peterson PA, Teyton L, Wilson, IA: Crystalstructures of two I-Ad-peptide complexes reveal that highaffinity can be achieved without large anchor residues.Immunity 8:319, 1998.
61. Lee B, Richard FM: The interpretation of protein struc-tures: estimation of static accessibility. J Mol Biol 55:379,1971.
62. Laskowski RA: SURFNET computer-program. London:Department of Biochemistry and Molecular Biology, Uni-versity College, 1991.
63. Laskowski RA: SURFNET: a program for visualizingmolecular surfaces, cavities and intermolecular interac-tions. J Mol Graph 13:323, 1995.
64. McDonald IK, Thornton JM: Satisfying hydrogen bond-ing potential in proteins. J Mol Biol 238:777, 1994.
65. Rammensee HG, Friede T, Stevanoviic S: MHC ligandsand peptide motifs: first listing. Immunogenetics 41:178,1995.
66. Jones DT, Thornton JM: Potential energy functions forthreading. Curr Opin Struct Biol 6:210, 1996.
67. Rognan D, Lauemoller SL, Holm A, Buus S, Tschinke V:Predicting binding affinities of protein ligands fromthree-dimensional models: application to peptide bindingto class I major histocompatibility proteins. J Med Chem42:4650, 1999.
68. Service RF: Structural genomics offers high-speed look atproteins. Science 287:1954, 2000.
69. Sette A, Sidney J, del Guercio MF, Southwood S, RuppertJ, Dahlberg C, Grey HM, Kubo RT: Peptide binding tothe most frequent HLA-A class I alleles measured byquantitative molecular binding assays. Mol Immunol 31:813, 1994.
70. Lauvau G, Kakimi K, Niedermann G, Ostankovitch M,Yotnda P, Firat H, Chisari FV, van Endert PM: Humantransporters associated with antigen processing (TAPs)select epitope precursor peptides for processing in theendoplasmic reticulum and presentation to T cells. J ExpMed 190:1227, 1999.
71. Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A:Human memory CTL response specific for influenza Avirus is broad and multispecific. Hum Immunol 61:438,2000.
72. Livingston BD, Crimi C, Fikes J, Chesnut RW, Sidney J,Sette A: Immunization with the HBV core 18-27 epitopeelicits CTL responses in humans expressing differentHLA-A2 supertype molecules. Hum Immunol 60:1013,1999.
73. Chen W, Khilko S, Fecondo J, Margulies DH, McCluskeyJ: Determinant selection of major histocompatibility com-plex class I-restricted antigenic peptides is explained byclass I-peptide affinity and is strongly influenced by non-dominant anchor residues. J Exp Med 180:1471, 1994.
74. Rechenmann F: From data to knowledge. Bioinformatics16:411, 2000.
556 P. Kangueane et al.





























