Embed Size (px)
Citation preview

Theoretical Study of Hydrogenation ofThiouracils and Their Base Pairswith Adenine
LEIF A. ERIKSSON,1 EUGENE S. KRYACHKO,2 MINH T. NGUYEN2
1Department of Biochemistry, Uppsala University, Box 576, S-751 23 Uppsala, Sweden2Department of Chemistry, University of Leuven, Celestijnenlaan 200 F, B-3001 Leuven, Belgium
Received 11 December 2002; accepted 10 October 2003Published online 29 January 2004 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/qua.10856
ABSTRACT: The purpose of the present work is to employ theoretical chemicaltools to explore the subtle features of the hydrogenation of uracil and its relatedthioderivatives. The reactions of hydrogenation are followed for the free uracil, 2-thiouracil, 4-thiouracil, and 2,4-dithiouracil as well as for adenine–thiouracil base pairs.It is shown for the first time that, in contrast to the hydrogenation pathways to thecarbon atoms C5 and C6 of the studied molecules, characterized by the transition states,the hydrogenations at the sulphur atoms for thiouracils are barrierless and thus the latterare the kinetically most favorable pathways for hydrogenation. As also shown, thesefeatures are retained under the hydrogenation of the adenine–thiouracil base pairs. Thepresent study clearly demonstrates that the base pairing destruction is the keyradiation-induced lesion in the adenine–4-thiouracil and adenine–2,4-dithiouracil basepairs when the hydrogen radical H�, as one of the primary radiation products, resides atthe sulphur atom S4. © 2004 Wiley Periodicals, Inc. Int J Quantum Chem 99: 841–853, 2004
Key words: thiouracil; adenine–thiouracil base pairs; hydrogenation; base pairingdestruction
Introduction
I n the Watson–Crick architecture of DNA [1],guanine is paired to cytosine via three hydrogen
bonds and adenine to thymine by means of only
two, excluding a weak COH . . . O bond. In RNA,thymine is replaced by uracil, which, geometricallyspeaking, is more compact due to the absence of thethymine’s methyl group [1b–1f]. Its thiated deriva-tive(s), with the exocyclic oxo group(s) being sub-stituted by the heavier thio group(s), are corre-spondingly referred to as 2-thiouracil (2TU),4-thiouracil (4TU), or 2,4-dithiouracil (2,4DTU).Thiouracils have been identified as minor constitu-ents of tRNA and of peptide nucleic acids (PNAs)and as potent anticancer drugs [1b, 1c, 2, 3]. Crys-tallographic [2f] and NMR [3a] data demonstrate
E. S. Kryachko’s present address is Department de Chimie,Bat B6C, Universite de Liege, Allee de la Chimie 3, Sart-Tilman,B-4000 Liege, Belgium. He is on leave of absence from Bogoli-ubov Institute for Theoretical Physics, Kiev, 03143 Ukraine.
Correspondence to: E. S. Kryachko; e-mail: [email protected]
International Journal of Quantum Chemistry, Vol 99, 841–853 (2004)© 2004 Wiley Periodicals, Inc.

that 4TU forms the hydrogen (H) bond with ade-nine in the position 14 that actually corresponds tothe reversed Hoogsteen pair where the sulphuratom S4 plays the role of H-bond acceptor [2a, 2b].2TU is paired to adenine and diaminopurine inPNAs [2c].
Thiobases have certain unique features that arealso largely retained in partly thiated DNA [3k].Thiouracils have been thoroughly studied experi-mentally [4] due to their significant importance inbiophysics, biochemistry, and pharmacology (seeRefs. [5, 6] and references therein). It is well knownthat while 2TU [7d] acts as a cancerogenic agent [6a,6b] 4TU behaves as a cross-linked agent in RNAtranscriptional regulation [6d]. The substitution ofuracil by 2TU can be responsible for misrecognitionin mRNA [3b] and also reduces its interaction en-ergy with adenine [4g]. Adenine is also known toform a rather strong H bond with 2TU in thecodon–anticodon complex although without in-volving the thio group [5d].
Theoretical aspects of thiouracils and the hydro-gen-bonded base pairs with thiouracils have re-cently become of considerable interest [7, 8]. Inparticular it was shown [7e, 8b] that the hydrogen-bonded guanine–thiouracil base pair is only 2 kcal/mol less stable than the guanine–uracil one (seealso Ref. [9]). On the other hand, the Watson–Crickadenine–thiouracil pair is slightly stronger than theunmodified adenine–uracil base (AU) in the case of2TU (by � 0.4 kcal/mol; cf. Ref. [4g]) and lessstronger for 4TU (ca. 0.6 kcal/mol) and 2,4DTU (ca.4 kcal/mol) [8f].
Despite the aforementioned importance of thio-uracil-containing base pairs, less is known, how-ever, about their damage under radiation and thefunction of their damaged analogs [3g, 10], in par-ticular whether there is any specificity in the radi-ation damage of the base pairs containing thioura-cils compared with the normal Watson–Crick pairs[11, 12] and, if so, whether and how these damagedbase pairs affect the structural motifs of DNA(PNAS) and RNA. These questions will in part beaddressed in the present work by performing atheoretical study on how the radical H�, as one ofthe most reactive primary products of radiation,modifies the A–thiouracil (ATU) base pairs. Thestructures and lower-energy pathways of the reac-tions of thiouracils and ATU base pairs with H� (thehydrogenation reactions) will be compared withthe original unmodified analogs. The computa-
tional framework of the present work is outlined inRefs. [13–15].*
Hydrogenated pyrimidines can originate eitherthrough protonation of radical anions formed fromelectron ejection and uptake in irradiated samplesor by H-atom additions from decomposition of ra-diolyzed water in the near surrounding. The effectsof hydrogenation (and hydrogen abstraction) havebeen explored in earlier work [16–19], although theemphasis has then been on the relative stabilities ofthe various radicals formed, the computation ofhyperfine coupling constants (HFCCs), and the ef-fects of vibrational averaging on the HFCCs. Thesestudies have been aimed at providing a theoreticalunderstanding of the observations made in experi-mental studies, primarily performed in low-tem-perature electron spin resonance (ESR) measure-ments of irradiated nucleobase crystals.
From experimental data it is well established thathydrogenation primarily occurs at the C5AC6 dou-ble bonds in pyrimidines, although also hydroge-nation at O4 has been observed for thymine anduracil. The relative stabilities of the C5OH andC6OH radical adducts are highly similar; C6OHbeing the most stable (by ca. 3 kcal/mol) in thymineand 1-methyl thymine but C5OH being 2 kcal/molmore stable than C6OH in uracil [16]. In cytosine,
*All computations performed in the present work were con-ducted at the hybrid Hartree–Fock density functional theoryB3LYP level in conjunction with the 6-31�(d,p) basis set, usingthe Gaussian 98 package [13]. Such computational level wasoften employed in the early theoretical studies of the DNA basesand base pairs, in particular the thiated ones (see Refs. [8c–f3]and references therein) and provided an accurate description oftheir geometric, energetic, and spectroscopic features. Harmonicvibrational frequencies and zero-point vibrational energies(ZPVEs) were also calculated at the current level to verifywhether the structure in question was a local minimum or atransition structure on the potential energy surface and to obtainthe thermodynamic quantities reported. The expectation valueof the S2 operator was for all the radicals studied equal or closeto the ideal value of 0.75. Being aware of the fundamentalshortcoming of the computational density functional theory inan inaccurate description of the self-interaction correction (SIC)term [14], some selected structures were further refined viaperforming single-point (sp) calculations at the second-, third-and partial fourth-order Møller–Plesset perturbation method(MP2, MP3, MP4SDQ) within a frozen-core approximation inconjunction with the 6-311��G(d,p) basis set and at coupledcluster method including single excitations (CCSs). It is also wellknown that the SIC error influences the shape of the potentialenergy surface around the transition state of the hydrogen ab-straction reaction [15]. For example, B3LYP consistently lowersthe barrier heights by more than 1 kcal/mol, which can besuccessfully “cured” by performing single-point calculations atthe coupled cluster computational level [15].
ERIKSSON, KRYACHKO, AND NGUYEN
842 VOL. 99, NO. 5

the N3OH adducts is the most stable form in vacuo(7–8 kcal/mol more stable than C5OH and C6OHin this case), whereas in solution the relative ener-gies of the three main adducts are similar [19].
We earlier explored hydrogenation reactions to(unsaturated) hydrocarbon guests in radiolyzedzeolites [20], which has provided valuable insightinto the energetics and pathways for hydrogenationacross double bonds in related systems. It was con-cluded that initial van der Waals complexes arehighly difficult to localize when performing com-putations of these model systems in vacuo and thatsuch initial complexes actually provide little infor-mation about the reactions occurring in real sys-tems. Similar observations have been made in thecase of hydroxylation reactions (HO� radical addi-tions) to pyrimidines [21], a reaction type also oc-curring due to radiolytic decomposition of water.For the hydrogenation reactions, it was found thatthe reactions were exothermic by between 20–55kcal/mol, depending on the stability of the adductradical formed, and that they proceed via a transi-tion barrier (the transition states located in all caseshave an imaginary frequency corresponding to themode of attack) lying between 0 and 3.7 kcal/molabove the initial reactants. Similar conclusionscould be also drawn for the hydroxylation of pyri-midines.
Hydrogenation of Thiouracils
BASIC FEATURES OF HYDROGENATION OFURACIL
We begin with considering some of the key fea-tures of the hydrogenated uracil [12, 20]. Uracilfavors hydrogenation at its C5 and C6 carbons (seeFig. 1 for the standard enumeration of atoms inuracil) with a slight preference to the former,amounting to � 2 kcal/mol. The energy gain uponformation of the C5-hydrogenated uracil (5H-U, forshort), relative to the isolated reactants, is 37.3 kcal/mol (31.6 kcal/mol after ZPVE), whereas for 6H-Uit is 35.2 kcal/mol (29.6 kcal/mol including ZPVE).The exothermicities for hydrogenation of uracil arehence highly similar to those of, e.g., 1,2-dimethyl-cyclopentene (31.2 kcal/mol) or cyclohexene (33.6kcal/mol) [20]. Correcting for thermal energy (cf.Table I), we obtain the Gibbs free energy differencefor the two systems being �G298K � 1.3 kcal/mol.The main contribution to the Gibbs energy differ-
ence comes from the entropy effect, �S(5H�6H)�U ��2.6 cal/mol � T.
In Table I we list some of the key geometricfeatures of free uracil, the transition states to hydro-genation and the hydrogenated species 5H-U and6H-U. The latter two species display a rather dif-ferent structural distortions upon hydrogenationthat somewhat disagree with the previous findings[16]. The 5H-U adduct attains a distorted ring sys-tem due to the localization of the unpaired electron(and thus due to a partial change in the hybridiza-tion from sp2 to sp3) at C6, with a dihedral anglemeasured over C4OC5OC6ON1 of �30.2°. In6H-U, on the other hand, the unpaired spin be-comes more delocalized within the ring and thusthe ground-state ring structure is planar. This dif-ference is also clearly visible in the computedHFCCs of the protons listed in Table I. The rationalefor the distortion in 5H-U, which was not observedwhen optimizating at the B3LYP/6-31G(d,p) level[16], is due to the effect of the additional diffusefunctions employed in the present work. Similareffects have been noted for the nucleobase anions[23].
Experimentally, the two C5 hydrogens in 5H-Ushould be equal or nearly equal (2 � 35.5 G [24]; 2 �32.1 G [25], 35.3/35.7 G [26]), as also obtained at theB3LYP/6-311G(2df,p) level (2 � 34.5 G [16]). Thecurrent, slightly distorted, structure gives the val-ues 14.3/45.4 G. The large negative isotropic cou-pling at C6 (�18.5 G [24]; �18.8 G [25]; �19.4 [26];�20.0 G [16]) is well reproduced at the currentlevel, �18.6 G. One reason for the equal C5OHHFCCs, observed experimentally, could be a low-temperature vibrational averaging. A similar effecthas been observed in the case of the C6-hydroge-nated cytosine radical, where the vibrational aver-aging of the asymmetrically located hydrogens(HFCCs 14.6/48.0 G) leads to a closer agreementwith experimental data (2 � 37.7 G; experimentalvalue is 2 � 40 G [27]) [19]. This avenue was,however, not explored in the present work. For the6H-U radical, on the other hand, the computedHFCCs agree well already from the beginning (ex-perimental values: C6OH, 45/51 G; C5OH, �18.0 G[24]; previous study: C6OH, 2 � 40.5 G; C5OH,�17.9 G [16]). The current data are the following:C6OH, 2 � 41.2 G; C5OH, �20.6 G.
Forming the two radicals, the systems will passover transition states, found to lie 1.8 kcal/molabove the isolated reactants for 5H-U and 2.9 kcal/mol for 6H-U (including ZPVE corrections). Theobserved barriers are highly similar to those found
HYDROGENATION OF THIOURACILS AND THEIR BASE PAIRS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 843

for hydrogenation of unsaturated hydrocarbons(e.g., 2.3 kcal/mol in 1,2,-dimethylcyclopentene; 1.9kcal/mol in cyclohexene [20]). In the transitionstates, the attacking hydrogen atom is positionedca. 2 Å above the ring and forms almost straightangles with the hydrogen already bound to thecarbon under attack. Apart from a slight lengthen-ing of the C5OC6 bond in the transition structures,the remaining geometry of the uracil is essentiallyunaffected by the incoming hydrogen, indicative ofthe transition states being early in the reaction path-way. The difference in barrier height of ca. 1 kcal/mol, which is retained also when comparing entha-plies and Gibbs energies, provides an explanationfor the observed preference for the 5H-U productover 6H-U. Assuming a purely kinetics controlledreaction, and using Boltzmann statistics to evaluatethe relative distribution in product formation, a1-kcal/mol barrier difference yields a 5H-U:6H-Ureaction ratio of approximately 5:1.
MECHANISM OF HYDROGENATION OFTHIOURACILS
The mechanism of hydrogenation of thiouracilsis somewhat distinct from that of uracil. The mainreason for this difference is the modified chemistryresulting from exchange of one or both of the oxy-gen atoms by sulphur [8f]. The latter exhibit(s) astrong attraction with the incoming hydrogen andhence compete(s) with C5 and C6 as being the pre-ferred sites of the H� attack. In unsubstituted uracil,the hydrogenation adducts at O4 and O2 are placed
13.6 and 27.9 kcal/mol above the most stable ad-duct (5H-U), respectively [16].
As for uracil, the C5-hydrogenated form is themost stable product of 2TU, 33.7 kcal/mol below
FIGURE 1. Most favorable structures of the hydroge-nated uracil. Bond lengths are given in Å and bond an-gles in degrees. The indicated numbering of atoms isvalid throughout the present work.
FIGURE 2. Hydrogenated 2TU, 4TU, and 2,4DTU.Bond lengths are given in Šand bond angles in de-grees. For the selected structures, the energies corre-spond to the B3LYP/6-31�G(d,p) and single-pointMP2, MP3, MP4SDQ, and CCS computational levels inconjunction with the 6-311��G(d,p) basis set (readingfrom the top down, respectively).
ERIKSSON, KRYACHKO, AND NGUYEN
844 VOL. 99, NO. 5
the isolated reactants. This is 4.1 kcal/mol morestable than 6H-2TU and 14.1 kcal/mol below the2H-2TU product (cf. Table II). The former magni-tude slightly increases by the single-point calcula-tions performed at the MP2, MP3, and MP4SDQlevels in conjunction with the 6-311��G(d,p) basisset while CCS(sp)/6-311��G(d,p) leaves it un-changed. Also, the barriers to addition at theC5AC6 double bond are similar to those of uracil,0.8 kcal/mol for 5H-2TU and 3.1 kcal/mol for6H-2TU (see Table II and Fig. 2). As shown inFigure 2, the right well of the barrier of the H� ad-dition to C5 becomes deeper by 1.2, 8.1, 7.9, and 6.5kcal/mol at the MP2(sp)/6-311��G(d,p), MP3(sp)/6-311��G(d,p), MP4SDQ(sp)/6-311��G(d,p), andCCS(sp)/6-311��G(d,p) levels, respectively. Thelatter affects the 6H-2TU left well of the barrier ofthe H� addition to C6 as well: The difference be-
tween the MP2(sp)/6-311��G(d,p) and B3LYP/6-31�G(d,p) values nearly vanishes whereas theMP3(sp)/6-311��G(d,p), MP4SDQ(sp)/6-311��G(d,p),and CCS(sp)/6-311��G(d,p) change it to 8.2, 7.3,and 6.4 kcal/mol, respectively, keeping thus theoffset between the two aforementioned barriersnearly the same (see Fig. 2). Note that the CCS(sp)/6-311��G(d,p) estimations (both fall in the range of5–7 kcal/mol) are in fair agreement with the gen-eral trend of the underestimation of the transition-state barrier heights by B3LYP (see Ref. [15] andreferences therein). The difference compared to theunsubstituted uracil arises from the sulphur atomat S2. Hydrogenation at this atom proceeds withoutbarrier and will hence be the main adduct formedfor this system.
The effect of hydrogenation at the C5 and C6positions also resembles the corresponding nonthi-
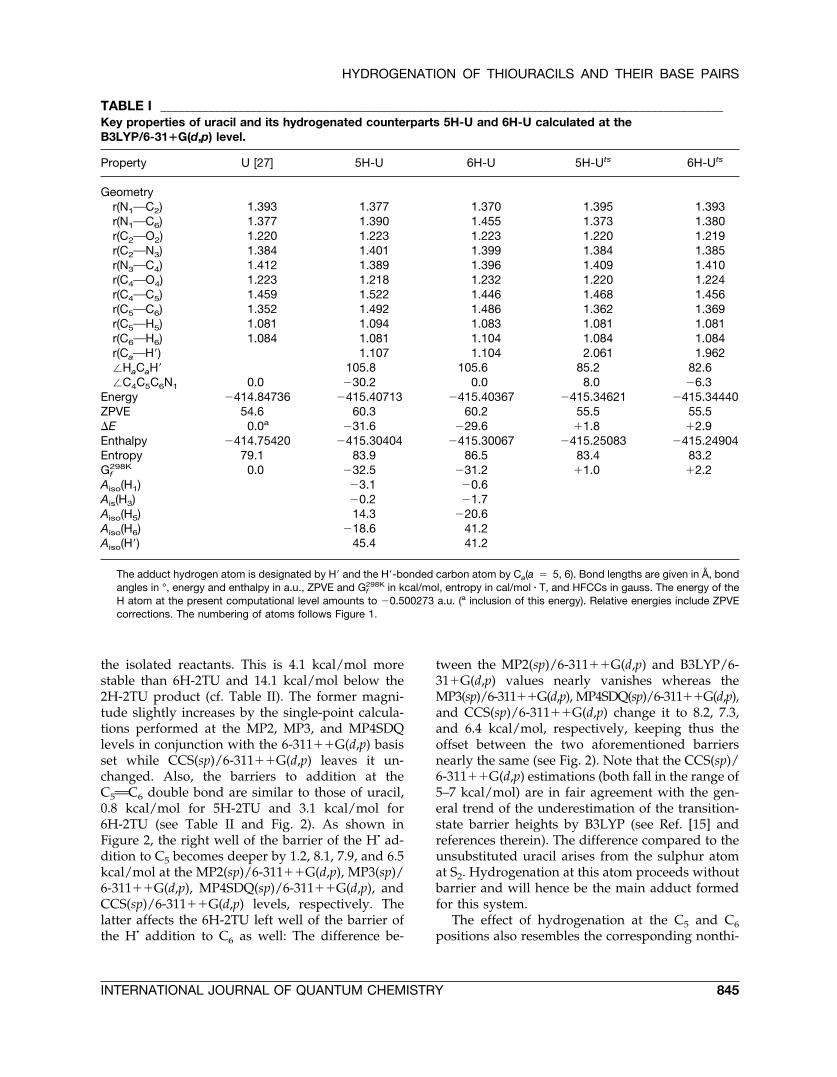
TABLE I ______________________________________________________________________________________________Key properties of uracil and its hydrogenated counterparts 5H-U and 6H-U calculated at theB3LYP/6-31�G(d,p) level.
Property U [27] 5H-U 6H-U 5H-Uts 6H-Uts
Geometryr(N1OC2) 1.393 1.377 1.370 1.395 1.393r(N1OC6) 1.377 1.390 1.455 1.373 1.380r(C2OO2) 1.220 1.223 1.223 1.220 1.219r(C2ON3) 1.384 1.401 1.399 1.384 1.385r(N3OC4) 1.412 1.389 1.396 1.409 1.410r(C4OO4) 1.223 1.218 1.232 1.220 1.224r(C4OC5) 1.459 1.522 1.446 1.468 1.456r(C5OC6) 1.352 1.492 1.486 1.362 1.369r(C5OH5) 1.081 1.094 1.083 1.081 1.081r(C6OH6) 1.084 1.081 1.104 1.084 1.084r(CaOH�) 1.107 1.104 2.061 1.962�HaCaH� 105.8 105.6 85.2 82.6�C4C5C6N1 0.0 �30.2 0.0 8.0 �6.3
Energy �414.84736 �415.40713 �415.40367 �415.34621 �415.34440ZPVE 54.6 60.3 60.2 55.5 55.5�E 0.0a �31.6 �29.6 �1.8 �2.9Enthalpy �414.75420 �415.30404 �415.30067 �415.25083 �415.24904Entropy 79.1 83.9 86.5 83.4 83.2Gf
298K 0.0 �32.5 �31.2 �1.0 �2.2Aiso(H1) �3.1 �0.6Ais(H3) �0.2 �1.7Aiso(H5) 14.3 �20.6Aiso(H6) �18.6 41.2Aiso(H�) 45.4 41.2
The adduct hydrogen atom is designated by H� and the H�-bonded carbon atom by Ca(a � 5, 6). Bond lengths are given in Å, bondangles in °, energy and enthalpy in a.u., ZPVE and Gf
298K in kcal/mol, entropy in cal/mol � T, and HFCCs in gauss. The energy of theH atom at the present computational level amounts to �0.500273 a.u. (a inclusion of this energy). Relative energies include ZPVEcorrections. The numbering of atoms follows Figure 1.
HYDROGENATION OF THIOURACILS AND THEIR BASE PAIRS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 845
ated uracils in terms of geometries and hyperfinecouplings. In the two transition states, the hydro-gen atom is located � 2 Å above the ring and formsthe H�CH angle between 82 and 85°. In the prod-ucts, the 5H-2TU ring system becomes distorted(C4OC5OC6ON1 angle is ca. 21°) whereas in 6H-2TU it is planar. The distorted ring structure in5H-2TU has as effect that the two C5 hydrogens arenonequivalent and have HFCCs 19.2/42.5 G,whereas the C5 alpha-proton is �19.5 G. For 6H-2TU, the HFCCs are 2 � 40.4 G at C6OH and �20.6at C5OH.
In 2H-2TU, the C2OS2 bond is elongated by 0.1 Åas the effect of hydrogenation, whereas the C5AC6bond is essentially unaltered compared to the 2TUsystem. The spin in 2H-2TU is localized to S2 (0.21)and C2 (0.67). The hydrogen is oriented perpendic-ularly to the ring and forms a straight angleH�OS2OC2. The ring becomes slightly distorteddue to the spin component at C2 and the mainHFCC in this system is at S2OH, 47.7 G (Table II).
The calculated geometric, energetic, and mag-netic properties for the three main hydrogenationadducts of 4TU are gathered in Table III, and in
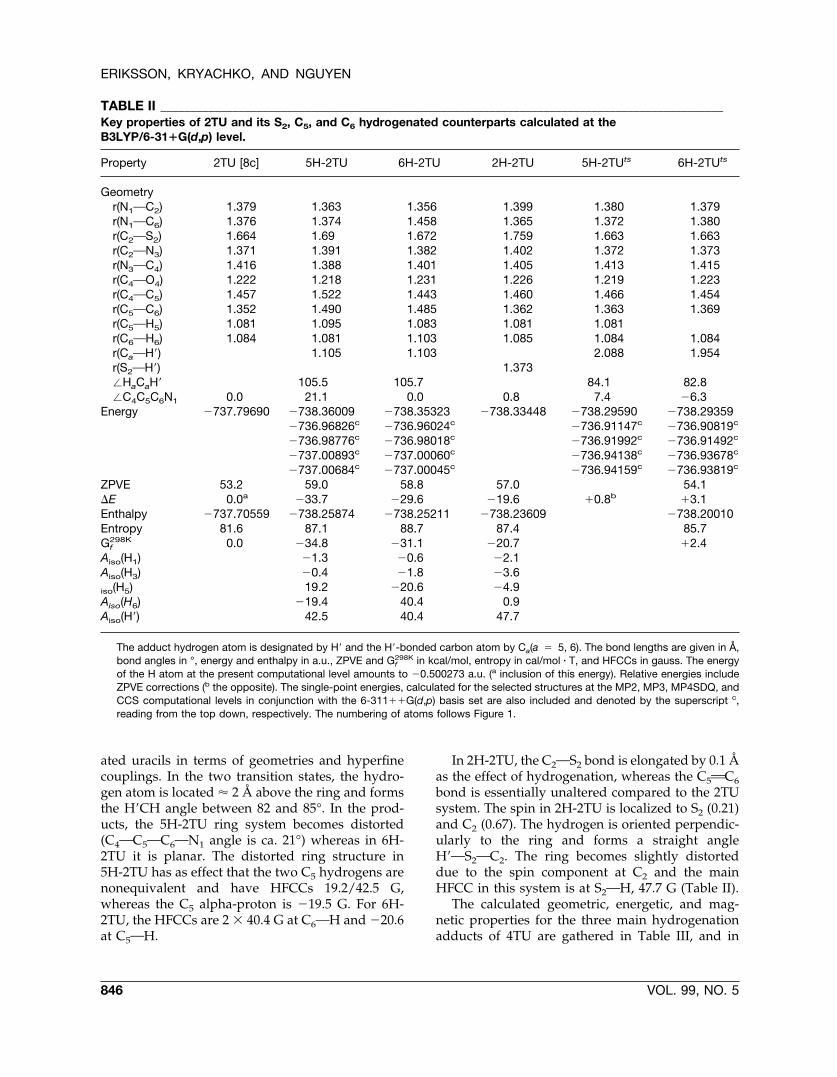
TABLE II ______________________________________________________________________________________________Key properties of 2TU and its S2, C5, and C6 hydrogenated counterparts calculated at theB3LYP/6-31�G(d,p) level.
Property 2TU [8c] 5H-2TU 6H-2TU 2H-2TU 5H-2TUts 6H-2TUts
Geometryr(N1OC2) 1.379 1.363 1.356 1.399 1.380 1.379r(N1OC6) 1.376 1.374 1.458 1.365 1.372 1.380r(C2OS2) 1.664 1.69 1.672 1.759 1.663 1.663r(C2ON3) 1.371 1.391 1.382 1.402 1.372 1.373r(N3OC4) 1.416 1.388 1.401 1.405 1.413 1.415r(C4OO4) 1.222 1.218 1.231 1.226 1.219 1.223r(C4OC5) 1.457 1.522 1.443 1.460 1.466 1.454r(C5OC6) 1.352 1.490 1.485 1.362 1.363 1.369r(C5OH5) 1.081 1.095 1.083 1.081 1.081r(C6OH6) 1.084 1.081 1.103 1.085 1.084 1.084r(CaOH�) 1.105 1.103 2.088 1.954r(S2OH�) 1.373�HaCaH� 105.5 105.7 84.1 82.8�C4C5C6N1 0.0 21.1 0.0 0.8 7.4 �6.3
Energy �737.79690 �738.36009 �738.35323 �738.33448 �738.29590 �738.29359�736.96826c �736.96024c �736.91147c �736.90819c
�736.98776c �736.98018c �736.91992c �736.91492c
�737.00893c �737.00060c �736.94138c �736.93678c
�737.00684c �737.00045c �736.94159c �736.93819c
ZPVE 53.2 59.0 58.8 57.0 54.1�E 0.0a �33.7 �29.6 �19.6 �0.8b �3.1Enthalpy �737.70559 �738.25874 �738.25211 �738.23609 �738.20010Entropy 81.6 87.1 88.7 87.4 85.7Gf
298K 0.0 �34.8 �31.1 �20.7 �2.4Aiso(H1) �1.3 �0.6 �2.1Aiso(H3) �0.4 �1.8 �3.6iso(H5) 19.2 �20.6 �4.9Aiso(H6) �19.4 40.4 0.9Aiso(H�) 42.5 40.4 47.7
The adduct hydrogen atom is designated by H� and the H�-bonded carbon atom by Ca(a � 5, 6). The bond lengths are given in Å,bond angles in °, energy and enthalpy in a.u., ZPVE and Gf
298K in kcal/mol, entropy in cal/mol � T, and HFCCs in gauss. The energyof the H atom at the present computational level amounts to �0.500273 a.u. (a inclusion of this energy). Relative energies includeZPVE corrections (b the opposite). The single-point energies, calculated for the selected structures at the MP2, MP3, MP4SDQ, andCCS computational levels in conjunction with the 6-311��G(d,p) basis set are also included and denoted by the superscript c,reading from the top down, respectively. The numbering of atoms follows Figure 1.
ERIKSSON, KRYACHKO, AND NGUYEN
846 VOL. 99, NO. 5
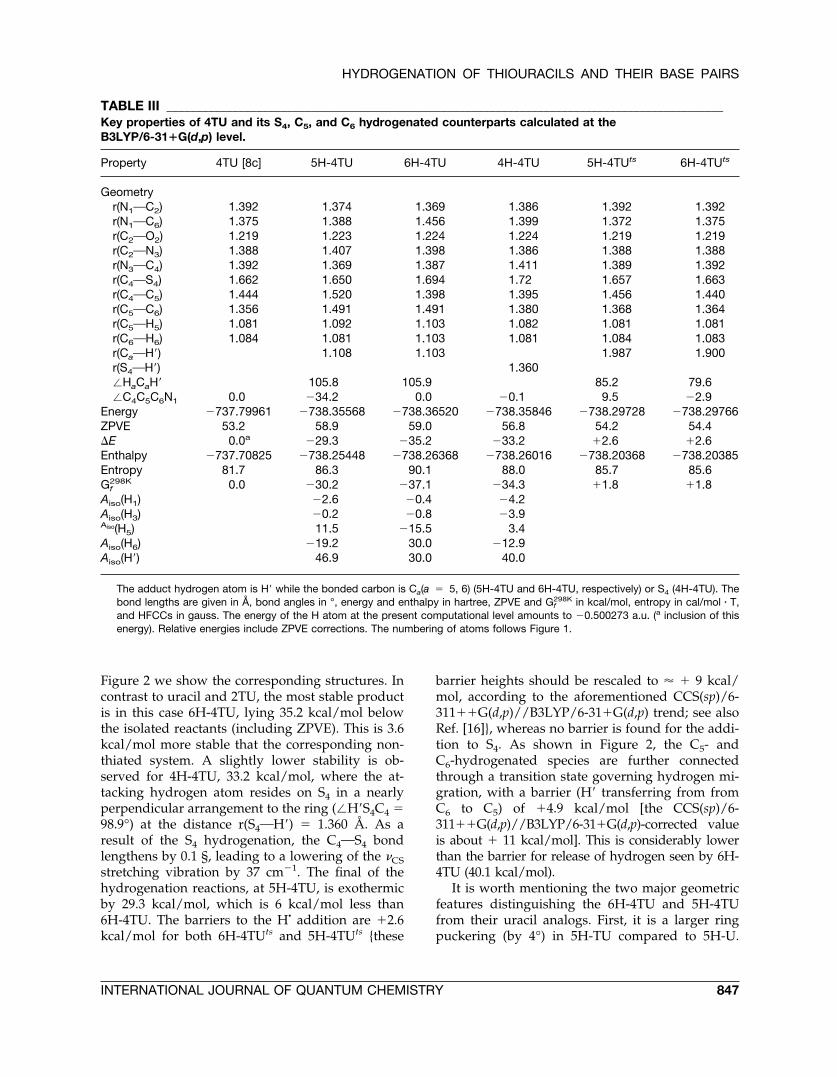
Figure 2 we show the corresponding structures. Incontrast to uracil and 2TU, the most stable productis in this case 6H-4TU, lying 35.2 kcal/mol belowthe isolated reactants (including ZPVE). This is 3.6kcal/mol more stable that the corresponding non-thiated system. A slightly lower stability is ob-served for 4H-4TU, 33.2 kcal/mol, where the at-tacking hydrogen atom resides on S4 in a nearlyperpendicular arrangement to the ring (�H�S4C4 �98.9°) at the distance r(S4OH�) � 1.360 Å. As aresult of the S4 hydrogenation, the C4OS4 bondlengthens by 0.1 §, leading to a lowering of the �CSstretching vibration by 37 cm�1. The final of thehydrogenation reactions, at 5H-4TU, is exothermicby 29.3 kcal/mol, which is 6 kcal/mol less than6H-4TU. The barriers to the H� addition are �2.6kcal/mol for both 6H-4TUts and 5H-4TUts {these
barrier heights should be rescaled to � � 9 kcal/mol, according to the aforementioned CCS(sp)/6-311��G(d,p)//B3LYP/6-31�G(d,p) trend; see alsoRef. [16]}, whereas no barrier is found for the addi-tion to S4. As shown in Figure 2, the C5- andC6-hydrogenated species are further connectedthrough a transition state governing hydrogen mi-gration, with a barrier (H� transferring from fromC6 to C5) of �4.9 kcal/mol [the CCS(sp)/6-311��G(d,p)//B3LYP/6-31�G(d,p)-corrected valueis about � 11 kcal/mol]. This is considerably lowerthan the barrier for release of hydrogen seen by 6H-4TU (40.1 kcal/mol).
It is worth mentioning the two major geometricfeatures distinguishing the 6H-4TU and 5H-4TUfrom their uracil analogs. First, it is a larger ringpuckering (by 4°) in 5H-TU compared to 5H-U.
TABLE III _____________________________________________________________________________________________Key properties of 4TU and its S4, C5, and C6 hydrogenated counterparts calculated at theB3LYP/6-31�G(d,p) level.
Property 4TU [8c] 5H-4TU 6H-4TU 4H-4TU 5H-4TUts 6H-4TUts
Geometryr(N1OC2) 1.392 1.374 1.369 1.386 1.392 1.392r(N1OC6) 1.375 1.388 1.456 1.399 1.372 1.375r(C2OO2) 1.219 1.223 1.224 1.224 1.219 1.219r(C2ON3) 1.388 1.407 1.398 1.386 1.388 1.388r(N3OC4) 1.392 1.369 1.387 1.411 1.389 1.392r(C4OS4) 1.662 1.650 1.694 1.72 1.657 1.663r(C4OC5) 1.444 1.520 1.398 1.395 1.456 1.440r(C5OC6) 1.356 1.491 1.491 1.380 1.368 1.364r(C5OH5) 1.081 1.092 1.103 1.082 1.081 1.081r(C6OH6) 1.084 1.081 1.103 1.081 1.084 1.083r(CaOH�) 1.108 1.103 1.987 1.900r(S4OH�) 1.360�HaCaH� 105.8 105.9 85.2 79.6�C4C5C6N1 0.0 �34.2 0.0 �0.1 9.5 �2.9
Energy �737.79961 �738.35568 �738.36520 �738.35846 �738.29728 �738.29766ZPVE 53.2 58.9 59.0 56.8 54.2 54.4�E 0.0a �29.3 �35.2 �33.2 �2.6 �2.6Enthalpy �737.70825 �738.25448 �738.26368 �738.26016 �738.20368 �738.20385Entropy 81.7 86.3 90.1 88.0 85.7 85.6Gf
298K 0.0 �30.2 �37.1 �34.3 �1.8 �1.8Aiso(H1) �2.6 �0.4 �4.2Aiso(H3) �0.2 �0.8 �3.9Aiso(H5) 11.5 �15.5 3.4Aiso(H6) �19.2 30.0 �12.9Aiso(H�) 46.9 30.0 40.0
The adduct hydrogen atom is H� while the bonded carbon is Ca(a � 5, 6) (5H-4TU and 6H-4TU, respectively) or S4 (4H-4TU). Thebond lengths are given in Å, bond angles in °, energy and enthalpy in hartree, ZPVE and Gf
298K in kcal/mol, entropy in cal/mol � T,and HFCCs in gauss. The energy of the H atom at the present computational level amounts to �0.500273 a.u. (a inclusion of thisenergy). Relative energies include ZPVE corrections. The numbering of atoms follows Figure 1.
HYDROGENATION OF THIOURACILS AND THEIR BASE PAIRS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 847

Second, the C4AS4 bond length is shortened by 0.01Å in 5H-4TU and elongated by 0.03 Å in 6H-4TU(cf. with 0.005 Å in 5H-U and 0.009 Å in 6H-U). Theformer effect is due to a weakening of the C4OC5
bond in 5H-4TU (its bond length varies from 1.444Å in 4TU to 1.520 Å) caused by the H� addition tocarbon C5. The latter is obviously due to a strength-ening of the C4OC5 in 6H-4TU (its bond lengthdecreases from 1.444 Å to 1.398 Å). Both effectslikely have their origin in a more spatial extensionof the valence orbitals of the sulphur atom com-pared to the oxygen resulting in larger overlap withthe valence orbitals of the carbon C5.
The present calculations show that the spin den-sity, obtained from Mulliken population analysis,in 6H-4TU resides mainly on C5 (0.67) and S4 (0.49),giving thus rise to the main isotropic proton hyper-fine couplings Aiso equal to 30.0 G on C6OH and�15.5 G on C5OH. In 5H-4TU, the atomic spindensity is mainly localized on C6 (0.92), for themain HFCCs Aiso being 46.9 G (C5H). The largepositive proton coupling is also originated in thiscase due to the nonplanar distortion of the ring. In4H-4TU, finally, the spin density is nearly equallydistributed between C4 and C6 (0.52 on each), withthe largest Aiso values of 40.0 G and �12.9 G foundon S4OH and C6OH, respectively.
The situation for the doubly thiated system, 2,4-DTU, is highly similar to that of 4TU (see Fig. 2 andTable IV for structures and properties). The hydro-genation product at C6 is 35.6 kcal/mol more stablethan the free reactants, whereas for the C5 hydro-genation we gain 31.9 kcal/mol. 4H-2,4DTU isagain of intermediate stability relative to the two(�E � �33.9 kcal/mol), whereas 2H-2,4DT shows amuch weaker bonding (�E � �20.5 kcal/mol). Byanalogy with the situations in 2H-2TU and 4H-4TU,the COS bond length in 4H-2,4DTU increases uponhydrogenation by ca. 0.1 Å. The added hydrogenresides in precisely the same arrangement as in4H-4TU (see above), and the sulphur atom is dis-placed out of the ring plane by ca. 6°.
As in the cases above, the barriers to the H-atomaddition at C5 and C6 are small (2.1 and 2.0 kcal/mol after ZPVE correction, respectively). As shownin both Figure 2 and Table IV, the CCS(sp)/6-311��G(d,p)//B3LYP/6-31�G(d,p) correction tothe 6H-2,4DTU barrier height amounts to � � 8kcal/mol. Note that the interbarrier offset is thencorrected to only 0.5 kcal/mol. The addition toeither of the two sulphur atoms is, however, barrierfree at the present level of theory.
Concluding this section, one explanation for thedifferent mechanisms of hydrogenation of the thio-uracils relative to the parent uracil may be sought inthe highest occupied molecular orbitals (HOMOand HOMO-1) of the systems. Let us assume that,under its approach to a given molecule, H� primar-ily interacts with its HOMO and HOMO-1. Figure 3depicts the HOMOs and HOMO-1s of U, 2TU, 4TU,and 2,4DTU. In uracil, the highest occupancy (0.72)of the HOMO is found on the �z orbital, placedperpendicular to the ring, and localized on the car-bon C5, which may explain its role as being themost favorable site of attack. In the thiouracils, onthe other hand, the largest occupancies are ob-served on the �x and �y orbitals localized on the S4atom in 4TU and 2,4DTU whose HOMOs havelower energies compared to that in uracil.
Hydrogenation of Adenine–ThiouracilBase Pairs
Let us start this section with analyzing the effectof hydrogenation at C5 in uracil, for it being thepreferred site of H� addition in the normal AU basepair (cf. Fig. 4). The structural effects on the overallbase pairing upon C5 hydrogenation of uracil areminor. This is also manifested in the interactionenergy only being 2.1 kcal/mol weaker in A–5H-Ucompared to AU. For the energetically most stableH� adduct in 4TU (6H-4TU), the effects on stabilityand H-bonding pattern in the base pairing withadenine are also small (�E � �9.5 kcal/mol with-out ZPVE and �8.3 kcal/mol with ZPVE correc-tions, compared to the nonhydrogenated A–4TUbase pair whose interaction energy amounts to�12.3 and �11.0 kcal/mol, respectively [8f]).
If, on the other hand, the hydrogenation occursat S4 of 4TU, for it being the kinetically most favor-able pathway, the effects on the base pairing withadenine become more pronounced. The (N)H . . . Sbond between A and 4TU stretches by 0.18 Å andbends by about 10° (see Fig. 4). The binding energyis reduced by 4 kcal/mol relative to the A–4TUbase pair. The weakened interaction is also seenspectroscopically, for instance, in that the symmet-rical and asymmetrical modes of the NH2 group inadenine are blue-shifted by 96 and 19 cm�1, respec-tively. The IR activity of the former is reduced by afactor of 2. The preopening of the A–4H-4TU basepair on the major groove side might have furtherconsequences on the possibility of intruding watermolecules in the interbase region and on interbase
ERIKSSON, KRYACHKO, AND NGUYEN
848 VOL. 99, NO. 5
flipping [29] (see also Ref. [1i] for a recent review).In addition, such hydrogenation results in a sub-stantial interbase twist of 12.5°, which most likelyinfluences its stacking capabilities.
The behavior of the A–2,4DTU base pair underhydrogenation is different from the above cases. Ifthe H� addition occurs at C6, as being the thermo-dynamically most stable adduct (although not be-ing the kinetical most favored reaction pathway),the geometric effects in terms of the hydrogenbonding are small. On the other hand, we observe adecrease of the interbase twist angle, from 24.0°,
inherent to the original A–2,4DTU base pair [8f], to18.9° (Fig. 4). Energetically, the binding energy isreduced by 2.9 kcal/mol and amounts to �6.2 kcal/mol (including ZPVE).
Hydrogenation of the A–2,4DTU base pair at C5and S4 displays several common features. The in-teraction energies of the resulting complexes aresimilar, viz., �8.5 and �7.1 kcal/mol for A–5H-2,4DTU and A–4H-2,4DTU, respectively. In bothcases, the (N)H . . . S hydrogen bond between Aand 2,4DTU is slightly elongated. For instance, inthe S4-hydrogenated case it is elongated by 0.1 Å,
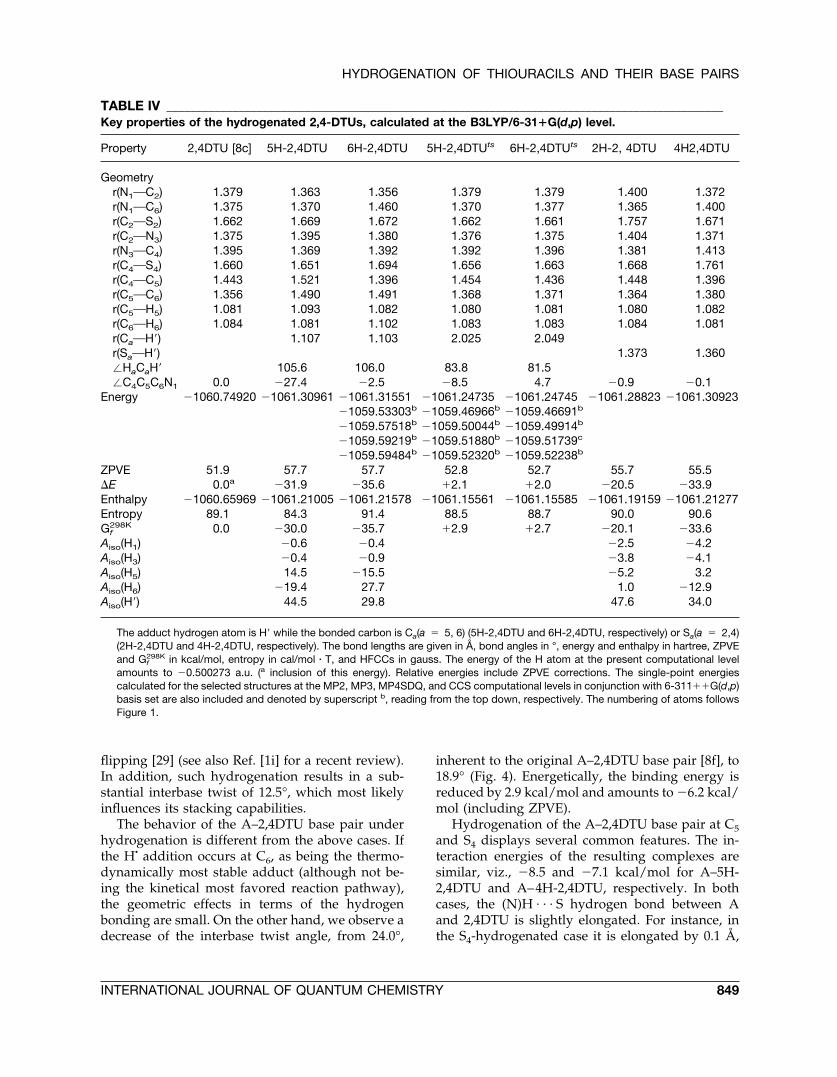
TABLE IV _____________________________________________________________________________________________Key properties of the hydrogenated 2,4-DTUs, calculated at the B3LYP/6-31�G(d,p) level.
Property 2,4DTU [8c] 5H-2,4DTU 6H-2,4DTU 5H-2,4DTUts 6H-2,4DTUts 2H-2, 4DTU 4H2,4DTU
Geometryr(N1OC2) 1.379 1.363 1.356 1.379 1.379 1.400 1.372r(N1OC6) 1.375 1.370 1.460 1.370 1.377 1.365 1.400r(C2OS2) 1.662 1.669 1.672 1.662 1.661 1.757 1.671r(C2ON3) 1.375 1.395 1.380 1.376 1.375 1.404 1.371r(N3OC4) 1.395 1.369 1.392 1.392 1.396 1.381 1.413r(C4OS4) 1.660 1.651 1.694 1.656 1.663 1.668 1.761r(C4OC5) 1.443 1.521 1.396 1.454 1.436 1.448 1.396r(C5OC6) 1.356 1.490 1.491 1.368 1.371 1.364 1.380r(C5OH5) 1.081 1.093 1.082 1.080 1.081 1.080 1.082r(C6OH6) 1.084 1.081 1.102 1.083 1.083 1.084 1.081r(CaOH�) 1.107 1.103 2.025 2.049r(SaOH�) 1.373 1.360�HaCaH� 105.6 106.0 83.8 81.5�C4C5C6N1 0.0 �27.4 �2.5 �8.5 4.7 �0.9 �0.1
Energy �1060.74920 �1061.30961 �1061.31551 �1061.24735 �1061.24745 �1061.28823 �1061.30923�1059.53303b �1059.46966b �1059.46691b
�1059.57518b �1059.50044b �1059.49914b
�1059.59219b �1059.51880b �1059.51739c
�1059.59484b �1059.52320b �1059.52238b
ZPVE 51.9 57.7 57.7 52.8 52.7 55.7 55.5�E 0.0a �31.9 �35.6 �2.1 �2.0 �20.5 �33.9Enthalpy �1060.65969 �1061.21005 �1061.21578 �1061.15561 �1061.15585 �1061.19159 �1061.21277Entropy 89.1 84.3 91.4 88.5 88.7 90.0 90.6Gf
298K 0.0 �30.0 �35.7 �2.9 �2.7 �20.1 �33.6Aiso(H1) �0.6 �0.4 �2.5 �4.2Aiso(H3) �0.4 �0.9 �3.8 �4.1Aiso(H5) 14.5 �15.5 �5.2 3.2Aiso(H6) �19.4 27.7 1.0 �12.9Aiso(H�) 44.5 29.8 47.6 34.0
The adduct hydrogen atom is H� while the bonded carbon is Ca(a � 5, 6) (5H-2,4DTU and 6H-2,4DTU, respectively) or Sa(a � 2,4)(2H-2,4DTU and 4H-2,4DTU, respectively). The bond lengths are given in Å, bond angles in °, energy and enthalpy in hartree, ZPVEand Gf
298K in kcal/mol, entropy in cal/mol � T, and HFCCs in gauss. The energy of the H atom at the present computational levelamounts to �0.500273 a.u. (a inclusion of this energy). Relative energies include ZPVE corrections. The single-point energiescalculated for the selected structures at the MP2, MP3, MP4SDQ, and CCS computational levels in conjunction with 6-311��G(d,p)basis set are also included and denoted by superscript b, reading from the top down, respectively. The numbering of atoms followsFigure 1.
HYDROGENATION OF THIOURACILS AND THEIR BASE PAIRS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 849

which is accompanied by an 11° deviation fromlinearity of the bond in question (Fig. 4). In addi-tion, the (C)H . . . S bond becomes weakened: ForA–4H-2,4DTU, the bond length increases by asmuch as 0.2 Å and the interbase twisting increasesfrom 24.0° in the original base pair to 30.8° (A–5H-2,4DTU) and 35.4° (A–4H-2,4DTU), respectively.The A–2,4DTU base pair, hydrogenated at S2, liesca. 10.8 kcal/mol above the energy of the S4-hydro-genated species.
The most remarkable changes in the HFCCs forthe hydrogenated bases under the base pair forma-tion with adenine occur in A–4H-4TU, A–4H-2,4TU, and A–6H-2,4DTU (see Fig. 4). In the formertwo cases Aiso (H�), with H� residing at S4, is re-duced by 5.4 and 2.6 G, respectively, while in thelatter Aiso (H�) increases by 3.8 G.
Summary and Conclusions
A key purpose of the present work is to establishthe subtle features of the hydrogenation pathwaysfor uracil, its thiated derivatives, and their basepairs with adenine. It has been theoretically re-vealed that the reactions of hydrogenation of U,2TU, 4TU, and 2,4DTU at C5,6 and S2,4 are exother-mic, varying between 19–45 kcal/mol, dependingon the stability of the adduct radical formed. Thelargest and nearly equal exothermicity is predictedfor the 6H hydrogenation of 4TU and 2,4DTU. True,there exist two different types of hydrogenationpathways: One, finalized at C5 or C6, proceeds via atransition barrier lying between 6 and 8 kcal/mol,taking the CCS(sp)/6-311��G(d,p)//B3LYP/6-31�G(d,p) corrections into account, above the initialreactants. The other, ending at the hydrogenatedsulphur atoms, is barrierless and therefore is actu-ally the kinetically most stable reaction pathway.We have shown that such reaction features are re-tained under the Watson–Crick pairings of thesebases with adenine.
We demonstrated that the base pairing destruc-tion is the key radiation-induced lesion in theA–thiouracil base pairs when the hydrogen radicalH�, as one of the primary radiation products, re-sides on the sulphur atom S4. Such base pairingdestruction may certainly lead to their preopening,
FIGURE 3. Shapes of the HOMOs and HOMO-1s ofuracil, 2TU, 4TU, and 2,4DTU. �, orbital energy.
ERIKSSON, KRYACHKO, AND NGUYEN
850 VOL. 99, NO. 5
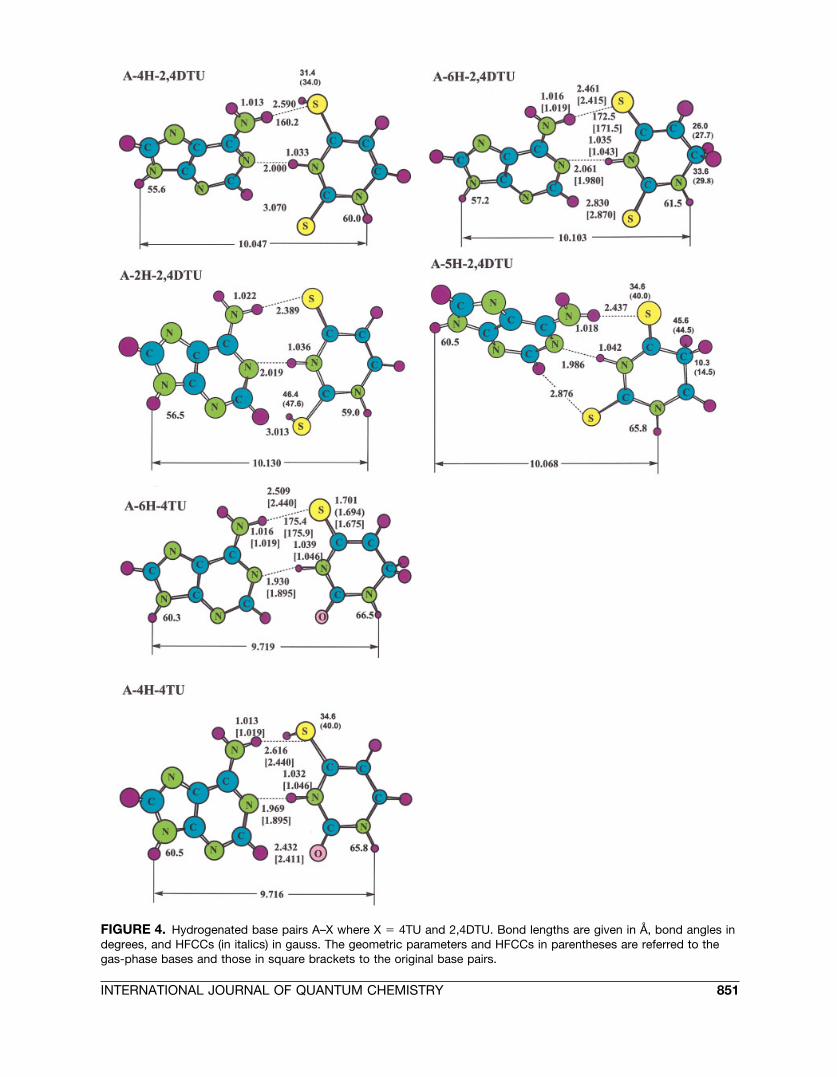
FIGURE 4. Hydrogenated base pairs A–X where X � 4TU and 2,4DTU. Bond lengths are given in Å, bond angles indegrees, and HFCCs (in italics) in gauss. The geometric parameters and HFCCs in parentheses are referred to thegas-phase bases and those in square brackets to the original base pairs.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 851

interbase flipping, and intruding of water mole-cule(s) into the interbase region on the majorgroove side that may indeed affect their stackingcapabilities as well. Finally, we suggest that ourfindings are also applicable to the A–4TU–thyminebase of DNA [2]. Altogether with our recent studyon the thiated bases and base pairs [8f], mainlyfocused on the structural and energetical aspects ofbonding of thiouracils with water and adenine,their tautomerisms, protonation, and deprotona-tion, the present work wealfares our understandingof the key functional mechanism of the radiation-induced lesion in the A–TU base pairs and providesa sufficient answer on the questions of what makesthe thiated nucleobases so markedly different fromthe normal ones and what is the key effect of thethio substitution that causes a variety of bizarreeffects having significant importance in biophysics,biochemistry, and pharmacology.
ACKNOWLEDGMENT
L. A. E. gratefully acknowledges grants of com-puting time at the national supercomputing facili-ties in Linkoping and Stockholm as well as researchgrants from the National Sciences Research Council(VR). E. S. K. and M. T. N. acknowledge researchgrant of the Catholic University of Leuven. Theauthors also thank the referees for valuable sugges-tions and useful comments.
References
1. (a) Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737(reprinted in Ann NY Acad Sci 1995, 758, 737); (b) Saenger,W. Principles of Nucleic Acid Structure; Springer: Berlin,1984; (c) Jeffrey, G. A.; Saenger, W. Hydrogen bonding inbiological Structures; Springer: New York, 1991; (d) Sinden,R. R. DNA Structure and Function; Academic: San Diego,1994; (e) Blackburn, G. M.; Gait, M. J., eds. Nucleic Acids inChemistry and Biology; Oxford University Press: Oxford,UK, 1996; p 21; (f) Leontis, N. B.; Westhof, E. Q Rev Biophys1998, 31, 399. For recent works see (g) Kryachko, E. S. Int JQuantum Chem 2002, 90, 910; (h) Kryachko, E. S.; Sabin, J. R.Int J Quantum Chem 2003, 91, 695; (i) Kryachko, E. S. InFundamental World of Quantum Chemistry: A Tribute Vol-ume to the Memory of Per-Olov Lowdin; Brandas, E. J.;Kryachko, E. S., Eds.; vol. 2; Kluwer: Dordrecht, The Neth-erlands, 2003; Chapter 22 and references therein.
2. (a) Saenger, W.; Suck, D. J Mol Biol 1970, 50, 153; (b)Gotschalk, E. M.; Kopp, E.; Lezius, A. G. Eur J Biochem 1971,24, 168; (c) Nielsen, P. E. Acc Chem Res 199, 32, 624; (d)Thewald, U.; Bugg, C. E. J Am Chem Soc 1972, 94, 8892; (e)Scheit, K. H.; Gartner, E. Biochim Biophys Acta 1969, 182, 10;(f) Kim, S. H.; Sussman, J. L.; Suddath, F. L.; Quigley, G. J.;
McPherson, A.; Wang, A. H.; Seeman, N. C.; Rich, A. ProcNatl Acad Sci USA 1974, 71, 4970; (g) Martin, R. P.; Scheller,J. M.; Stahl, A. J. C.; Dirheimer, G. Biochem Biophys ResCommun 1976, 70, 997; (h) Altweg, M.; Kubli, E. NucleicAcid Res 1980, 8, 215; (i) Kim, M. J.; Zhing, W. D.; Hong, Z.;Kao, C. C. J Virol 2000, 74, 10312; (j) Kadokura, M.; Wada, T.;Seio, K.; Sekine, M. J Org Chem 2000, 65, 5104.
3. (a) Reid, B. R.; Ribeiro, N. S.; Gould, G.; Robillard, G.; Hil-bers, C. W.; Shulman, R. G. Proc Natl Acad Sci USA 1975, 72,2049; (b) Beck, C. F.; Howlett, G. J Mol Biol 1977, 111, 1; (c)Vormbrock, R.; Morawetz, R.; Gassen, H. G. Biochim Bio-phys Acta 1974, 340, 348; (d) Lin, G. H.; Sundaralingham, M.Acta Crystallogr B 1971, 27, 961; (e) Lin, G. H.; Sundaraling-ham, M. J Am Chem Soc 1971, 93, 1235; (f) Lohse, J.; Dahl, O.;Nielsen, P. E. Proc Natl Acad Sci USA 1999, 96, 11804; (g)Lopez-Torres, M.; Romero, M.; Barja, G. Mol Cell Endocrinol2000, 168, 127; (h) Palumbo, A.; d’Ischia, M.; Cioffi, F. A.FEBS Lett 2000, 485, 109; (i) Palumbo, A.; d’Ischia, M. Bio-chem Biophys Res Commun 2001, 282, 793; (j) Joyce, G. F.Nature 2002, 418, 214; (k) Xu, Y. Z. Progr Natural Sci 2000,10, 401.
4. (a) Katritzky, A. R.; Lagowski, J. M. In Advances in Hetero-cyclic Chemistry, vol. 1; Katritzky, A. R.; Boulton, A. J., Eds.;Academic: New York, 1963; p 400; (b) Kwiatkowski, J. S.;Pullman, B. In Advances in Heterocyclic Chemistry, vol. 8;Katritzky, A. R.; Boulton, A. J., Eds.; Academic: New York,1975; p 256; (c) Katritzky, A. R.; Baykut, G.; Rachwal, S.;Szafran, M.; Caster, K. C. J Chem Soc Perkin Trans 1989, 2,1499; (d) Winckelmann, I.; Larsen, E. H. Synthesis 1986, 1041;(e) Rubin, Y. V.; Savin, F. A.; Blagoi, Y. P. Stud Biophys 1988,123, 203; (f) Clivio, P.; Favre, A.; Fontaine, C.; Fourrey, J. L.;Gasche, J.; Guittet, E.; Laugaa, P. Tetrahedron 1992, 48, 1605;(g) Dossantos, D. V.; Vianna, A. L.; Fourrey, J. L.; Favre, A.Nucleic Acids Res 1993, 21, 201; (h) Saintome, C.; Clivio, P.;Fourrey, J. L.; Woisard, A.; Laugaa, P.; Favre, A. Tetrahedron2000, 56, 1197; (i) Liu, J. Q.; Taylor, J. S. Nucleic Acids Res1998, 26, 3300; (j) Favre, A.; Fourrey, J. L. Acc Chem Res 1995,28, 375; (k) Fourrey, J. L.; Gasche, J.; Fontaine, C.; Guittet, E.;Favre, A. Chem Comm 1989, 1334; (l) Favre, A.; Dubreuil,Y. L.; Fourrey, J. L. New J Chem 1991, 15, 593; (m) Blazek,E. R.; Alderfer, J. L.; Tabaczynski, W. A.; Stamoudis, V. C.;Churchill, M. E.; Peak, J. G.; Peak, M. J Photochem Photobiol1993, 57, 255; (n) Favre, A.; Moreno, G.; Salet, C.; Vinzens, F.J Photochem Photobiol 1993, 58, 689; (o) Favre, A.; Saintome,C.; Fourrey, J. L.; Clivio, P.; Laugaa, P.; J Photochem Photo-biol B 1998, 42, 109; (p) Kamalakannan, P.; Vankappayya, D.;Balasubramanian, T. J Chem Soc Dalton Trans 2002, 3381; (q)Fathalla, O. A.; Zaghary, W. A.; Radwan, H. H.; Awad, S. M.;Mohamed, M. S. Arch Pharmacal Res 2002, 25, 258.
5. (a) Miller, W. H.; Roblin, R. O.; Astwood, E. B. J Am ChemSoc 1945, 67, 2201; (b) Jeemer, R.; Rossels, J. Biochim BiophysActa 1953, 11, 438; (c) Hammers, R. Biochim Biophys Acta1956, 21, 170; (d) Carbon, J. A.; Hung, L.; Jones, D. S. ProcNatl Acad Sci USA 1965, 53, 979; (e) Lipsett, M. N.; Doctor,B. P. J Biol Chem 1967, 242, 4072.
6. (a) Yoshida, M.; Takeishi, T.; Ukida, T. Biochim Biophys Acta1971, 228, 15; (b) Beck, C. F.; Howlett, G. J. J Mol Biol 1977,111, 1; (c) Saxand, N. I.; Lewis, R. J. Dangerous Properties ofIndustrial Materials; Van Nostrand Reinhold: New York,1989; (d) McEvoy, G. K., Ed. AHFS Drug Information; Amer-ican Society of Hospital Pharmacists: Bethesda, MD, 1989; (e)Wang, Z.; Rana, T. Biochemistry 1996, 35, 6491.
ERIKSSON, KRYACHKO, AND NGUYEN
852 VOL. 99, NO. 5

7. (a) Les, A.; Adamowicz, L. J Am Chem Soc 1990, 112, 1504;(b) Leszczynski, J. Int J Quantum Chem 1992, 18, 9; (c)Lapinski, L. Rostkowska, H.; Nowak, M. J.; Kwiatkowski,J. S.; Leszczynski, J. Vibrat Spectrosc 1996, 13, 23; (d) Leszc-zynski, J.; Sponer, J. J Mol Struct Theochem 1996, 388, 237; (e)Hueso Urena, F.; Moreno Carretero, M. N.; Low, J. N.; Mas-terton, A. G. J Mol Struct Theochem 1997, 435, 133; (f)Sponer, J.; Leszczynski, J.; Hobza, P. J Phys Chem A 1997,101, 9489; (g) Rubin, Y. V.; Morozov, Y.; Venkateswarlu, D.;Leszczynski, J. J Phys Chem A 1998, 102, 2194; (h) Rubin, Y.;Leszczynski, J. Nucleosides Nucleotides 1999, 18, 1119.
8. (a) Lamsabhi, M.; Alcami, M.; Mo, O.; Bouab, W.; Esseffar,M.; Abboud, J. L. M.; Yanez, M. J Phys Chem A 2000, 104,5122; (b) Kawahara, S. I.; Uchimaru, T.; Sekine, M. J MolStruct Theochem 2000, 530, 109; (c) Kryachko, E. S.; Nguyen,M. T.; Zegers-Huyskens, T. J Phys Chem A 2001, 105, 3379;(d) Kryachko, E. S.; Nguyen, M. T.; Zeegers-Huyskens, T.Chem Phys 2001, 264, 21; (e) Marino, T.; Russo, N.; Sicilia, E.;Toscano, M. Int J Quantum Chem 2001, 82, 44; (f) Kryachko,E. S.; Nguyen, M. T. Adv Quantum Chem 2001, 40, 79; (g)Civcir, P. U. J Phys Org Chem 2001, 14, 171; (h) Moustafa, H.;El-Taher, S.; Shibl, M. F.; Hilal, R. Int J Quantum Chem 2002,87, 378.
9. Crnugelj, M.; Dukhan, D.; Barascut, J. L.; Imbach, J. L.;Plavec, J. J Chem Soc Perkin Trans 2, 2000, 255.
10. (a) Peak, J. G.; Peak, M. J.; Foote, C. S. Photochem Photobiol1986, 44, 111; (b) Peak, M. J.; Atsushti, I.; Foote, C. S.; Peak,J. G. Photochem Photobiol 1988, 47, 809; (c) Ito, A.; Robb,F. T.; Peak, J. G.; Peak, M. J. Photochem Photobiol 1988, 47,231; (d) Churchill, M. E.; Schmitz, A. M.; Peak, J. G.; Peak,M. J. Photochem Photobiol 1990, 52, 1017; (e) Komeda, K.;Iwamoto, S.; Kominami, S.; Hashimoto, S.; Ihara, M.;Ohnishi, T. Mutat Res 1992, 272, 268; (f) Komeda, K.;Iwamoto, S.; Kominami, S.; Ohnishi, T. Photochem Photobiol1997, 65, 115; (g) Mehta, P.; Mehta, S. C. Asian J Chem 1999,11, 528; (h) Gredilla, R.; Barja, G.; Lopez-Torres, M. FreeRadical Res 2001, 35, 417.
11. (a) Sevilla, M. D. In Excited States in Organic Chemistry;Pullman, B.; Goldblum, N., Eds.; Reidel: Boston, 1977; p 15;(b) Colson, A. O.; Sevilla, M. D. Int J Radiat Biol 1995, 67, 627.
12. Wetmore, S. D.; Eriksson, L. A.; Boyd, R. J. In TheoreticalBiochemistry: Processes and Properties of Biological Sys-tems; Eriksson, L. A., Ed.; Elsevier: Amsterdam, 2001; Chap-ter 11 and references therein.
13. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgom-ery, J. A. Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.;Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.;Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.;Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski,J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.;Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman,J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.;Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Mar-tin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.;Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill,
P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.;Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98,revision A.5; Gaussian, Inc.: Pittsburgh, PA, 1998.
14. See, e. g., (a) Kryachko, E. S.; Ludena, E. V. Energy DensityFunctional Theory of Many-Electron Systems; Kluwer; Dor-drecht, The Netherlands, 1990; (b) Csonka, G. I.; Johnson,B. G. Theor Chem Acc 1998, 99, 158; (c) Polo, V.; Kraka, E.;Cremer, D. Theor Chem Acc 2002, 107, 291; (d) Polo, V.;Kraka, E.; Cremer, D. Mol Phys 2002, 100, 1771; (e) Polo, V.;Grafenstein, J.; Kraka, E.; Cremer, D. Theor Chem Acc 2003,109, 22, and references therein.
15. (a) Johnson, B. G.; Gonzales, C. A.; Gill, P. M. W.; Pople, J. A.Chem Phys Lett 1994, 221, 100; (b) Durant, J. L. Chem PhysLett 1996, 256, 595; (c) Ref. [14b]; (d) Truhlar, D. G. FaradayDiscuss 1998, 110, 362; (e) Lynch, B. J.; Truhlar, D. G. J PhysChem A 2001, 105, 2936; (f) Patchkovskii, S.; Ziegler, T.J Chem Phys 2002, 116, 7806, and references therein.
16. Wetmore, S. D.; Boyd, R. J.; Reiksson, L. A. J Phys Chem B1998, 102, 5369.
17. Wetmore, S. D.; Himo, F.; Boyd, R. J.; Eriksson, L. A. J PhysChem B 1998, 102, 7484.
18. Wetmore, S. D.; Boyd, R. J.; Himo, F.; Eriksson, L. A J PhysChem B 1999, 103, 3051.
19. Adamo, C., Heitzmann, M.; Meilleur, F.; Rega, N.; Scalmani,G.; Grand, A.; Cadet, J.; Barone, V. J Am Chem Soc 2001, 123,7113.
20. Werst, D. W.; Han, P.; Choure, S. C.; Vinokur, E. I.; Xu, L.;Trifunac, A. D.; Eriksson, L. A. J Phys Chem B 1999, 103,9219.
21. Wetmore, S. D.; Boyd, R. J.; Llano, J.; Lundqvist, M. J.;Eriksson, L. A. In Recent Advances in Density FunctionalMethods, vol. 3; Barone, V.; Bencini, A.; Fantucci, P., Eds.;World Scientific: Singapore, 2002; Chapter 10.
22. (a) Padva, A.; O’Donnell, T. J.; LeBreton, P. R. Chem PhysLett 1976, 41, 278; (b) Deeble, D.; Das, C.; v. Sonntag, C. JPhys Chem 1985, 89, 5784; (c) Urano, S.; Yang, X.; LeBreton,P. R. J Mol Struct 1989, 214, 315; (d) Krauss, M.; Osman, R. JPhys Chem 1993, 97, 13515; (e) Miaskiewicz, K.; Miller, J.;Osman, R. Biochim Biophys Acta 1994, 1218, 283.
23. Wetmore, S. D.; Boyd, R. J.; Eriksson, L. A. Chem Phys Lett2000, 322, 129.
24. Zehner, H.; Flossmann, W.; Westhof, E.; Muller, A. Mol Phys1976, 32, 869.
25. Novais, H. M.; Steenken, S. J Phys Chem 1987, 91, 426.26. Heark, J. N.; McDowell, C. A. J Chem Phys 1974, 61, 1129.27. Westhof, E.; Lion, Y.; van der Vorst, A. Int J Radiat Biol 1977,
32, 499.28. Kryachko, E. S.; Nguyen, M. T.; Zeegers-Huyskens, T. J Phys
Chem A 2001, 105, 1288.29. See, e. g., (a) Mol, C. D.; Parikh, S. S.; Putnam, C. D.; Lo, T. P.;
Tainer, J. A. Annu Rev Biophys Biomol Struct 1999, 28, 101;(b) Stofer, E.; Chipot, C.; Lavery, R. J Am Chem Soc 1999,121, 9503; (c) Kryachko, E. S.; Volkov, S. N. Int J QuantumChem 2001, 82, 193.
HYDROGENATION OF THIOURACILS AND THEIR BASE PAIRS
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 853





























