Embed Size (px)
Citation preview

&get_box_var;ORIGINAL ARTICLE
Stimulation of Soluble Guanylate Cyclase Prevents CigaretteSmoke–induced Pulmonary Hypertension and EmphysemaNorbert Weissmann1*, Borja Lobo2*, Alexandra Pichl1*, Nirmal Parajuli1*, Michael Seimetz1, Raquel Puig-Pey2,Elisabet Ferrer2, Vıctor I. Peinado2,3, David Domınguez-Fandos2, Athanasios Fysikopoulos1, Johannes-Peter Stasch4,Hossein A. Ghofrani1, Nuria Coll-Bonfill2, Reiner Frey4, Ralph T. Schermuly1, Jessica Garcıa-Lucio2, Isabel Blanco2,3,Mariola Bednorz1, Olga Tura-Ceide2,3, Elsa Tadele1, Ralf P. Brandes5, Jan Grimminger1, Walter Klepetko6,Peter Jaksch6, Robert Rodriguez-Roisin2,3, Werner Seeger1,7, Friedrich Grimminger1, and Joan A. Barbera2,3
1Justus-Liebig University, Excellence Cluster Cardiopulmonary System, Universities of Giessen and Marburg Lung Center (UGMLC),DZL, Giessen, Marburg, Germany; 2Department of Pulmonary Medicine, Hospital Clınic, Institut d’Investigacions Biomediques August Pii Sunyer, University of Barcelona, Barcelona, Spain; 3Centro de Investigacion Biomedica en Red de Enfermedades Respiratorias,Valencia, Spain; 4Bayer HealthCare, Wuppertal, Germany; 5Department of Medicine, Institute for Cardiovascular Physiology, GoetheUniversity of Frankfurt, Frankfurt, Germany; 6Department of Cardiothoracic Surgery, University Hospital of Vienna, Vienna, Austria;and 7Max-Planck-Institute for Heart and Lung Research, Bad Nauheim, Germany
Abstract
Rationale: Chronic obstructive pulmonary disease (COPD) isa major cause of death worldwide. No therapy stopping progress ofthe disease is available.
Objectives: To investigate the role of the soluble guanylate cyclase(sGC)–cGMP axis in development of lung emphysema andpulmonary hypertension (PH) and to test whether the sGC–cGMPaxis is a treatment target for these conditions.
Methods: Investigations were performed in human lung tissue frompatients with COPD, healthy donors, mice, and guinea pigs. Micewere exposed to cigarette smoke (CS) for 6 hours per day, 5 daysper week for up to 6 months and treated with BAY 63-2521. Guineapigs were exposed to CS from six cigarettes per day for 3 months,5 days per week and treated with BAY 41-2272. Both BAYcompounds are sGC stimulators. Gene and protein expressionanalysis were performed by quantitative real-time polymerase chainreaction and Western blotting. Lung compliance, hemodynamics,
right ventricular heart mass alterations, and alveolar and vascularmorphometry were performed, as well as inflammatory cell infiltrateassessment. In vitro assays of cell adhesion, proliferation, andapoptosis have been done.
Measurements andMain Results: The functionally essential sGCb1-subunit was down-regulated in patients with COPD and inCS-exposedmice. sGC stimulators prevented the development of PHand emphysema in the two different CS-exposed animal models.sGC stimulation prevented peroxynitrite-induced apoptosis ofalveolar and endothelial cells, reduced CS-induced inflammatory cellinfiltrate in lung parenchyma, and inhibited adhesion of CS-stimulated neutrophils.
Conclusions: The sGC–cGMP axis is perturbed by chronicexposure to CS. Treatment of COPD animal models with sGCstimulators can prevent CS-induced PH and emphysema.
Keywords: chronic obstructive pulmonary disease; emphysema;pulmonary hypertension; soluble guanylate cyclase; pharmacology
(Received in original form November 19, 2013; accepted in final form April 7, 2014 )
*These authors contributed equally.
Supported by Bayer HealthCare; German Research Foundation, Excellence Cluster Cardiopulmonary System; the Hessian Government (LOEWE); grant PS09-0536 from the Fondo de Investigacion Sanitaria, Instituto de Salud Carlos III, Spanish Ministry of Economy and Competitiveness; and Fundacio La Marato TV3(#040430).
Author Contributions: N.W., J.A.B., conception, study design, analysis and interpretation of data, drafting and revising the manuscript. B.L., A.P., N.P., andM.S., design of the study, acquisition, analysis, and interpretation of data, drafting the manuscript. R.P.-P., E.F., V.I.P., D.D.-F., I.B., O.T.-C., and J.G.,acquisition, analysis, and interpretation of data, drafting the manuscript. A.F., J.-P.S., H.A.G., N.C.-B., R.F., R.T.S., M.B., E.T., R.P.B., W.K., P.J., W.S., andF.G., study design, interpretation of data, drafting the manuscript. J.G.-L., study design, acquisition and interpretation of data, drafting the manuscript.R.R.-R., acquisition, analysis, and interpretation of data.
Correspondence and requests for reprints should be addressed to Norbert Weissmann, Ph.D., Excellence Cluster Cardiopulmonary System, Aulweg 130,D-35392 Giessen, Germany. E-mail: [email protected]; or Joan Albert Barbera, M.D., Department of Pulmonary Medicine,Hospital Clınic, Villarroel, 170, 08036 Barcelona, Spain. E-mail: [email protected]
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Am J Respir Crit Care Med Vol 189, Iss 11, pp 1359–1373, Jun 1, 2014
Copyright © 2014 by the American Thoracic Society
Originally Published in Press as DOI: 10.1164/rccm.201311-2037OC on April 16, 2014
Internet address: www.atsjournals.org
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1359

Chronic obstructive pulmonary disease(COPD) is characterized by persistentairflow obstruction associated with anenhanced inflammatory response in theairways and the lung to noxious particles orgases, most commonly cigarette smoke (CS)(1). COPD is caused by the obstructionof small airways and emphysema, whichconsists of the enlargement of air spaces,destruction of lung parenchyma, loss ofelasticity, and closure of terminal airways(2, 3). The pathogenesis of COPDimplicates the recruitment of macrophages,neutrophils, and lymphocytes in the smallairways and the lung parenchyma, andthe induction of oxidative stress, allof which result in airway remodelingand parenchymal destruction (4, 5). Themechanism of disease development is stillnot fully understood (1), despite COPDbeing the fourth leading cause of deathworldwide, with a rising incidence. Todate there is still no therapy availablethat can either prevent or even cure theprogression of COPD. Currently, symptomsonly can be alleviated to slightly improve thequality of life of patients with COPD.
Recent findings have suggested thatendothelial dysfunction and pulmonaryhypertension (PH) play a key role inthe pathophysiology of COPD (6). Thishypothesis is supported by the observation
that a large proportion of patients withCOPD also have PH, with percentagesranging from 30 to 70% (7), and thatsmokers not suffering from COPD candevelop pulmonary vascular remodeling(8, 9). We recently showed that vascularremodeling and PH precede alveolardestruction in an emphysema model ofmice chronically exposed to CS, suggestingthat molecular alterations of the vasculaturemay trigger lung emphysema development(10, 11). The essential signaling moleculenitric oxide (NO), derived from inducibleNO synthase (iNOS), was identified asa possible mediator of alveolar destructionand vascular remodeling through thesubsequent formation of peroxynitrite(ONOO2) (10). However, the downstreamtargets of NO/ONOO2 in the pathogenesisof CS-induced PH and lung emphysemahave not yet been identified. Besides itspathophysiologic impact, NO is an effectiveand selective pulmonary vasodilator, whichactivates soluble guanylate cyclase (sGC).sGC normally exists as a heterodimer,consisting of a larger a-subunit anda smaller heme-binding b-subunit. Manystudies showed that the b1 N-terminusconstituted the heme-binding domain andit is known that NO binds to the heme ofsGC (12) (supporting the importance ofthis subunit for sGC function) (12–14).Although an a2- and b2-subunit havebeen identified (12, 15, 16) it is wellaccepted that the a1- and b1-subunitform the physiologically relevantheterodimer. Several investigationssuggested that dysfunction of theb1-subunit is key to development ofdiseases (14, 17, 18). The binding of NOto sGC results in the formation of thesecondary messenger cGMP (14, 19).cGMP has not only been shown to mediateNO-driven pulmonary vasodilatation,but also to inhibit smooth muscle cellproliferation, platelet aggregation, andinflammatory cell recruitment (20–22). Inaddition, ONOO2 has been suggested tooxidize and inactivate sGC (23–26).Stimulation of sGC has been shown toantagonize PH in experimental models(19, 27–30), and the novel sGC stimulatorriociguat recently met the primary endpointof improved exercise tolerance in a phaseIII study in patients with pulmonaryarterial hypertension and is now approvedfor the treatment of pulmonary arterialhypertension and chronic thromboembolicPH (28, 31).
Based on these observations, wehypothesized that sGC is involved in thedevelopment of CS-induced emphysemaand PH, and that the sGC–cGMP axis maytherefore be a potential target for thetreatment of COPD and COPD-associatedPH. To address this hypothesis, weinvestigated possible alterations of sGCexpression in patients with COPD. Inaddition, we targeted the sGC–cGMP axisin two experimental models of COPD: miceand guinea pigs chronically exposed toCS (32–34). We assessed the effects of thesGC stimulator compounds BAY 63-2521(riociguat) and BAY 41-2272, whichhave a dual mode of action, sensitizing sGCto endogenous NO and directly stimulatingsGC independently of NO, therebyrestoring the NO–sGC–cGMP pathway(14, 19, 35, 36). No relevant differenceswith respect to the biochemical andpharmacologic profile of both compoundsare known. We aimed to compare theeffect of two different but tightly relatedcompounds in two different species onsmoke-induced emphysema and PH asa strategy that, if the outcome was positive,would substantiate the relevance of theeffects of such drugs.
Some of the results of these studies havebeen previously reported in form ofabstracts (37–41).
Methods
Detailed descriptions of methods areprovided in the online supplement.
Patient CharacteristicsHuman lung tissues were obtained fromtransplanted patients with COPD withand without PH (42) and donor controlsubjects. Patient characteristics are given inTable E3 in the online supplement. Thestudies were approved by the EthicsCommittee of the Justus-Liebig-UniversitySchool of Medicine, Giessen, Germany andby the Ethics Committee of the HospitalClinic, Barcelona, Spain.
Exposure to CSMouse experiments were approved by thegovernmental ethics committee for animalwelfare (Regierungsprasidium Giessen,Germany). Male C57BL/6J mice (bodyweight, 19–20 g) were divided intofollowing groups (16 mice per group): (1)healthy control (no CS exposure), (2) CS
At a Glance Commentary
Scientific Knowledge on theSubject: The molecular mechanismsunderlying chronic obstructivepulmonary disease are not fullyunderstood. Currently no treatmentthat prevents the developmentof emphysema and pulmonaryhypertension in chronic obstructivepulmonary disease is available.
What This Study Adds to theField: We provide evidence that thesoluble guanylate cyclase (sGC)-cGMPaxis is affected by cigarette smokeexposure. Our data from investigationsconducted in humans, mice, andguinea pigs using sGC stimulatorsdemonstrate that maintenance of thesGC–cGMP pathway can preventcigarette smoke–induced pulmonaryhypertension and lung emphysema inanimal models.
ORIGINAL ARTICLE
1360 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014
exposure only, (3) placebo treatment (CSexposure), (4) sGC stimulator (BAY 63-2521) treatment (3 mg/kg body weight;CS exposure), and (5) BAY 63-2521treatment (10 mg/kg body weight; CSexposure).
Smoke-challenged animals wereexposed to the smoke of 3R4F cigarettes6 hours per day, 5 days per week for6 months. Nonexposed mice were treatedsimilarly, but without CS exposure. The200-ml solutions of BAY 63-2521 werefreshly prepared each day and suspendedin 4.0% methocel and 1.3% polyethyleneglycol. On CS exposure days, thesuspension was applied by gavage 1 hourbefore exposure. This time point of thelast application was chosen to allow
determination of peak plasma levels of BAY63-2521 on termination of the experiments.The solvent was used as placebo. After6 months, half of each group was randomlydivided into two subgroups. In Subgroup1 lung function tests were performed and inSubgroup 2 hemodynamics were quantified.From these subgroups, half of the lungswere fixed for alveolar morphometry andhalf were fixed for vascular morphometry.Portions of the expression studies weredone in a separate group of mice exposedfor 8 months to tobacco smoke, becauseit was previously shown that no majordifferences exist between 6 and 8 monthsof smoke exposure except the slightlyincreasing severity of lung emphysema andPH with time (10).
Procedures involving guinea pigs wereapproved by the Ethics Committee forAnimal Experimentation of the Universityof Barcelona, Spain. Thirty-nine maleDunkin Hartley guinea pigs (starting 300 g)were randomly distributed in four groups:(1) a nonexposed control group (Control,n = 7), (2) a nonexposed group receivingBAY 41-2272 (Control1 sGCS, n = 8), (3)a group exposed to CS receiving placebo(CS, n = 12), and (4) a group exposed to CSreceiving BAY 41-2272 (CS1 sGCS, n = 12).Animals from CS groups were exposed tothe smoke generated from six nonfiltered3R4F cigarettes per day for 3 months,5 days per week using a nose-only system(32). BAY 41-2272 was administered orallyby gavage at a dose of 3 mg/kg/day.
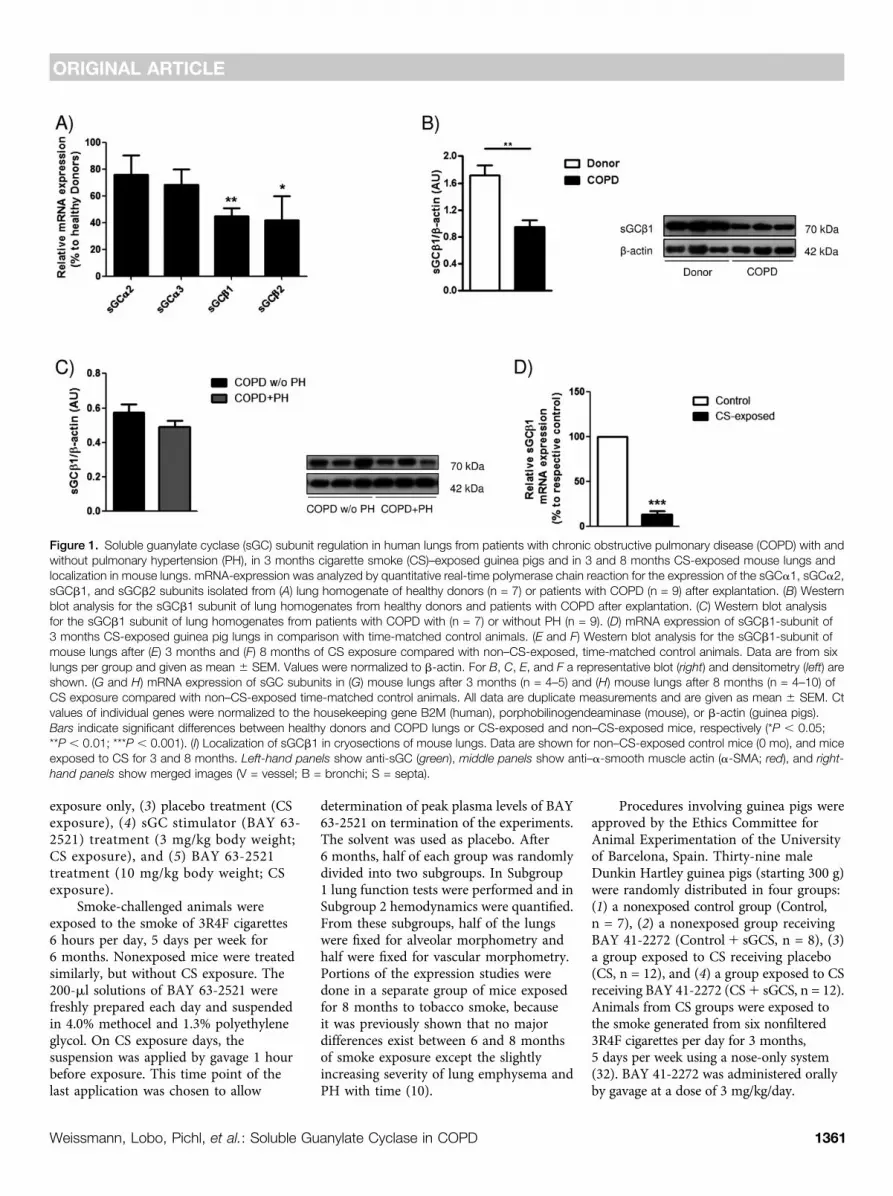
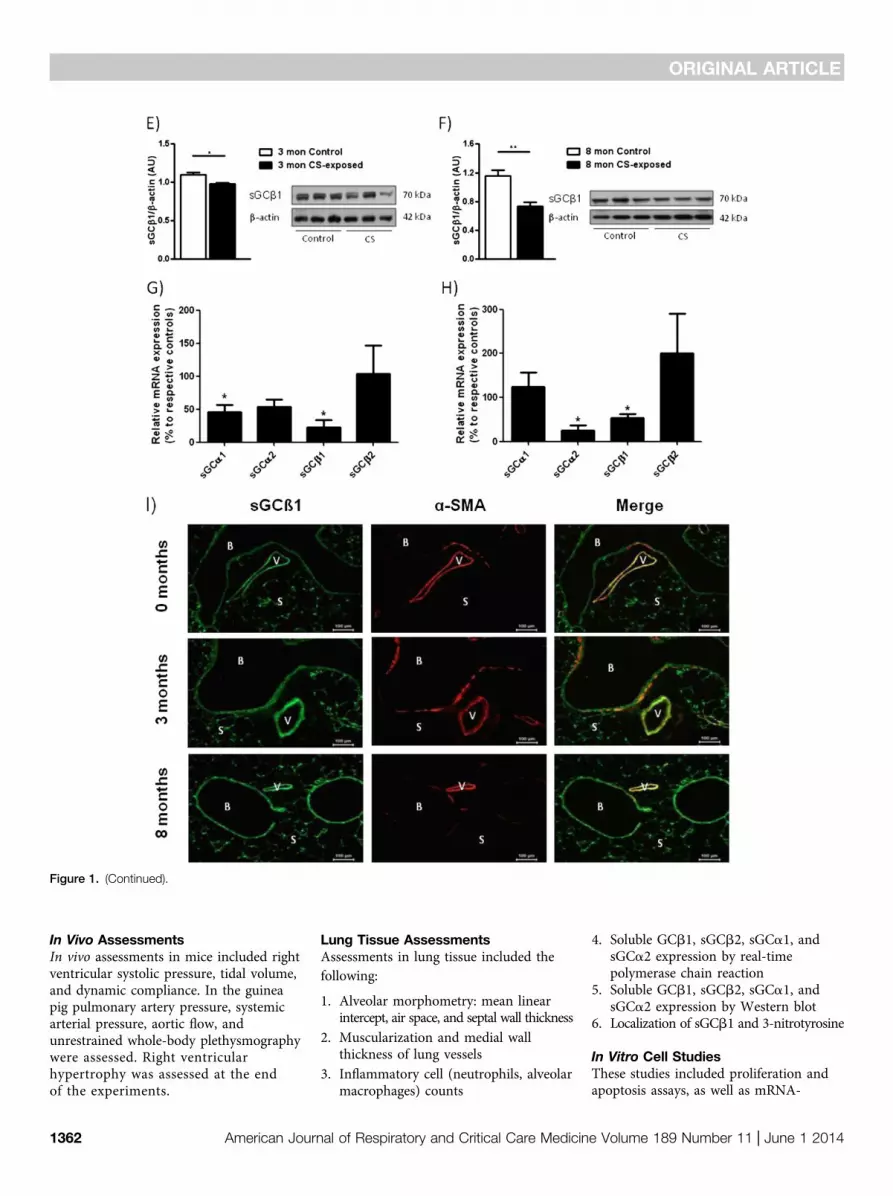
Figure 1. Soluble guanylate cyclase (sGC) subunit regulation in human lungs from patients with chronic obstructive pulmonary disease (COPD) with andwithout pulmonary hypertension (PH), in 3 months cigarette smoke (CS)–exposed guinea pigs and in 3 and 8 months CS-exposed mouse lungs andlocalization in mouse lungs. mRNA-expression was analyzed by quantitative real-time polymerase chain reaction for the expression of the sGCa1, sGCa2,sGCb1, and sGCb2 subunits isolated from (A) lung homogenate of healthy donors (n = 7) or patients with COPD (n = 9) after explantation. (B) Westernblot analysis for the sGCb1 subunit of lung homogenates from healthy donors and patients with COPD after explantation. (C) Western blot analysisfor the sGCb1 subunit of lung homogenates from patients with COPD with (n = 7) or without PH (n = 9). (D) mRNA expression of sGCb1-subunit of3 months CS-exposed guinea pig lungs in comparison with time-matched control animals. (E and F) Western blot analysis for the sGCb1-subunit ofmouse lungs after (E) 3 months and (F) 8 months of CS exposure compared with non–CS-exposed, time-matched control animals. Data are from sixlungs per group and given as mean 6 SEM. Values were normalized to b-actin. For B, C, E, and F a representative blot (right) and densitometry (left) areshown. (G and H) mRNA expression of sGC subunits in (G) mouse lungs after 3 months (n = 4–5) and (H) mouse lungs after 8 months (n = 4–10) ofCS exposure compared with non–CS-exposed time-matched control animals. All data are duplicate measurements and are given as mean 6 SEM. Ctvalues of individual genes were normalized to the housekeeping gene B2M (human), porphobilinogendeaminase (mouse), or b-actin (guinea pigs).Bars indicate significant differences between healthy donors and COPD lungs or CS-exposed and non–CS-exposed mice, respectively (*P , 0.05;**P , 0.01; ***P , 0.001). (I) Localization of sGCb1 in cryosections of mouse lungs. Data are shown for non–CS-exposed control mice (0 mo), and miceexposed to CS for 3 and 8 months. Left-hand panels show anti-sGC (green), middle panels show anti–a-smooth muscle actin (a-SMA; red), and right-
hand panels show merged images (V = vessel; B = bronchi; S = septa).
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1361
In Vivo AssessmentsIn vivo assessments in mice included rightventricular systolic pressure, tidal volume,and dynamic compliance. In the guineapig pulmonary artery pressure, systemicarterial pressure, aortic flow, andunrestrained whole-body plethysmographywere assessed. Right ventricularhypertrophy was assessed at the endof the experiments.
Lung Tissue AssessmentsAssessments in lung tissue included thefollowing:
1. Alveolar morphometry: mean linearintercept, air space, and septal wall thickness
2. Muscularization and medial wallthickness of lung vessels
3. Inflammatory cell (neutrophils, alveolarmacrophages) counts
4. Soluble GCb1, sGCb2, sGCa1, andsGCa2 expression by real-timepolymerase chain reaction
5. Soluble GCb1, sGCb2, sGCa1, andsGCa2 expression by Western blot
6. Localization of sGCb1 and 3-nitrotyrosine
In Vitro Cell StudiesThese studies included proliferation andapoptosis assays, as well as mRNA-
Figure 1. (Continued).
ORIGINAL ARTICLE
1362 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014
expression analyses in lung endothelialand alveolar type II epithelial cells (AECII);and adhesion assays to fibrinogen ofhuman polymorphonuclear leukocytes(hPMNL), treated and untreated with CSextract (CSE).
Data AnalysisAll data are given as mean 6 SEM.For comparison of more than twogroups analysis of variance followed byStudent-Newman-Keuls post hoc testwas performed. For comparison oftwo groups, a Student t test wasapplied. Correlations between variableswere done by Pearson analysis. P valuesless than 0.05 were consideredsignificant.
Results
sGC Regulation in Human COPD,CS-exposed Mouse, and GuineaPig LungsAnalysis of sGC subunit regulation in lungsfrom patients with COPD compared withhealthy donors revealed a down-regulationof sGCb1 and sGCb2 expression at themRNA level (Figure 1A) and a down-regulation of the sGCb1 subunit at theprotein level (Figure 1B). Comparing lungsfrom patients with COPD with and withoutPH we did not find significant differencesin the protein levels of the functionallyessential sGC subunit b1 (Figure 1C).Similar to human COPD the sGCb1
mRNA subunit was down-regulated in3 months CS-exposed guinea pigs(Figure 1D). Focusing on mice againa similar down-regulation of the sGCb1protein was found after 3 and 8 monthsof CS exposure (Figures 1E and 1F). Inaddition, as assessed on the mRNA levela down-regulation of sGCa1 and sGCb1was observed after 3 months, and a down-regulation of sGCa2 and sGCb1 wasobserved after 8 months of CS-exposurein this species (Figures 1G and 1H).Immunofluorescence staining suggestedthat the sGCb1 protein was locatedthroughout the lung in bronchi, vessels,and septa. Focusing on the pulmonaryvasculature it was evident that the sGCb1subunit was colocalized with a-smooth
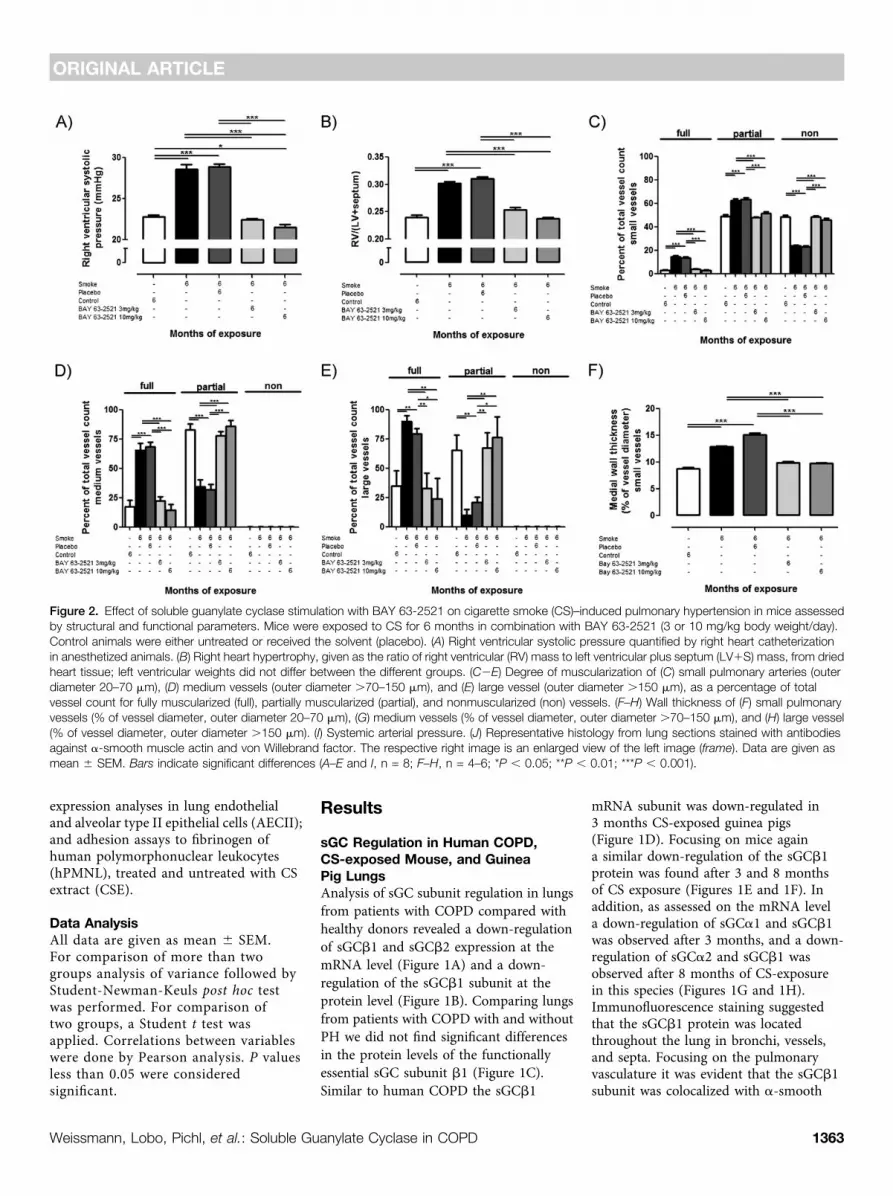
Figure 2. Effect of soluble guanylate cyclase stimulation with BAY 63-2521 on cigarette smoke (CS)–induced pulmonary hypertension in mice assessedby structural and functional parameters. Mice were exposed to CS for 6 months in combination with BAY 63-2521 (3 or 10 mg/kg body weight/day).Control animals were either untreated or received the solvent (placebo). (A) Right ventricular systolic pressure quantified by right heart catheterizationin anesthetized animals. (B) Right heart hypertrophy, given as the ratio of right ventricular (RV) mass to left ventricular plus septum (LV1S) mass, from driedheart tissue; left ventricular weights did not differ between the different groups. (C2E) Degree of muscularization of (C) small pulmonary arteries (outerdiameter 20–70 mm), (D) medium vessels (outer diameter .70–150 mm), and (E) large vessel (outer diameter .150 mm), as a percentage of totalvessel count for fully muscularized (full), partially muscularized (partial), and nonmuscularized (non) vessels. (F–H) Wall thickness of (F) small pulmonaryvessels (% of vessel diameter, outer diameter 20–70 mm), (G) medium vessels (% of vessel diameter, outer diameter .70–150 mm), and (H) large vessel(% of vessel diameter, outer diameter .150 mm). (I) Systemic arterial pressure. (J) Representative histology from lung sections stained with antibodiesagainst a-smooth muscle actin and von Willebrand factor. The respective right image is an enlarged view of the left image (frame). Data are given asmean 6 SEM. Bars indicate significant differences (A–E and I, n = 8; F–H, n = 4–6; *P , 0.05; **P , 0.01; ***P , 0.001).
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1363
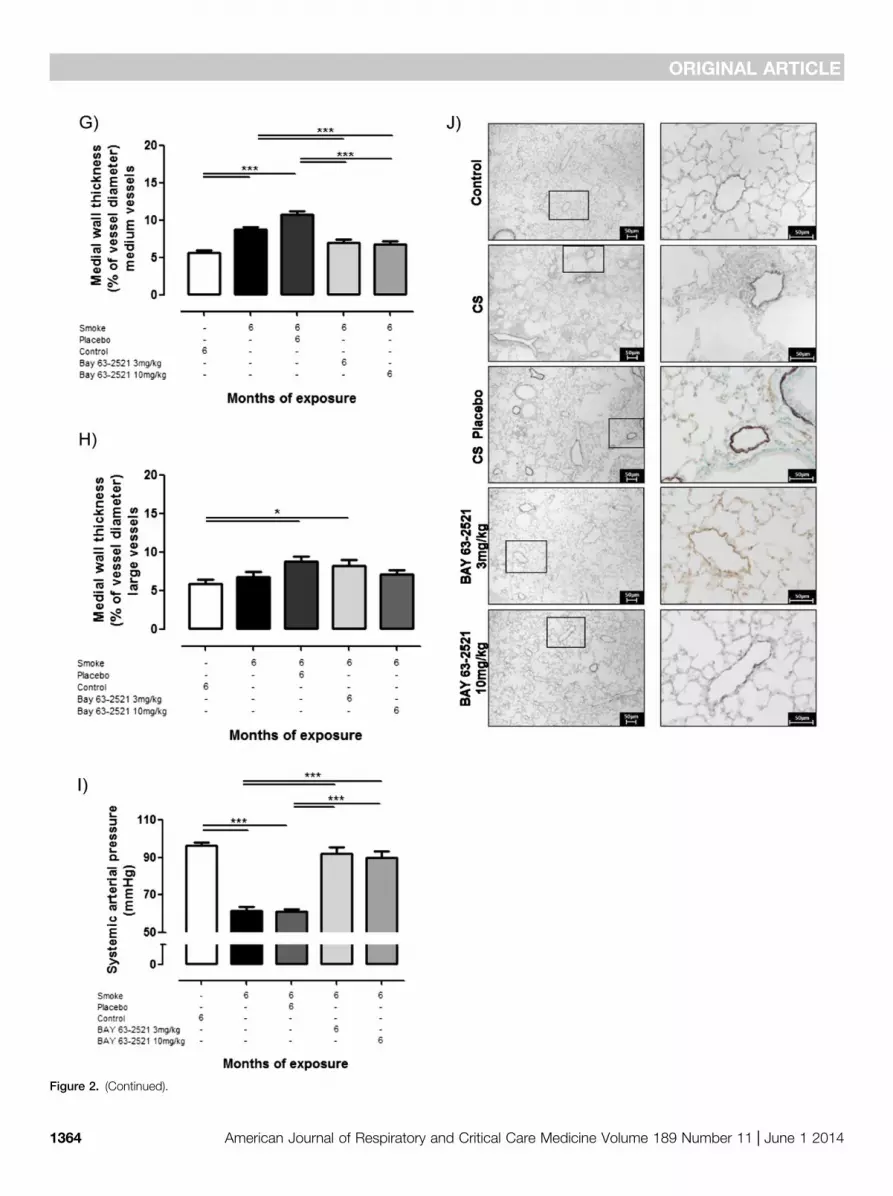
Figure 2. (Continued).
ORIGINAL ARTICLE
1364 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014

muscle actin–positive cells. Becausevascular localization was not restricted tothis cell type endothelial and adventitialcells may also express sGCb1 (Figure 1I).
Effect of sGC Stimulation on CS-induced PH and Pulmonary VascularRemodeling in Mice and Guinea PigsMice exposed to CS for 6 months developedPH and pulmonary vascular remodeling, asshown by quantification of right ventricularsystolic pressure (Figure 2A), right hearthypertrophy (Figure 2B), degree of vascularmuscularization (Figures 2C–2E), and themedial wall thickness (Figures 2F–2H).Treatment with BAY 63-2521, either atdoses of 3 or 10 mg/kg body weight/day,resulted in complete protection againstthe development of PH, based on allfour parameters of PH characterization.Systemic arterial pressure was reduced on
smoke exposure and was normalized bytreatment with BAY 63-2521 (Figure 2I).
Guinea pigs exposed to CS for3 months also showed a higher pulmonaryarterial pressure (Figure 3A) and totalpulmonary resistance (Figure 3B). Cardiacoutput (Figure 3C) and systemic arterialpressure (Figure 3D) did not differsignificantly from nonexposed animals.Treatment with BAY 41-2272 (3 mg/kgbody weight/day) during CS exposureresulted only in a tendency towarda decrease of pulmonary resistance, whichdid not differ from nonexposed guinea pigs,and clearly reduced pulmonary vascularremodeling induced by CS exposure(Figure 3E). The number of muscularizedintrapulmonary vessels with a diameter lessthan 50 mm and positive immune reactivityto a-smooth muscle actin was greater inCS-exposed animals than in control
animals (71 6 10% and 28 6 12%,respectively; P , 0.05). By contrast, inguinea pigs exposed to CS and treatedwith BAY 41-2272, the percentage ofmuscularized vessels was significantly lowerthan in CS-exposed only (P , 0.05)(Figure 3E). In addition, total pulmonaryresistance and intrapulmonary vesselmuscularization were significantly related(P , 0.001; r = 0.59).
Effect of sGC Stimulation Treatmenton CS-induced Lung EmphysemaDevelopmentFigure 4 shows that mice exposed to CSalso developed lung emphysema, quantifiedby determination of the airspace, the septalwall thickness, and the mean distancebetween alveolar septa (mean linearintercept) (Figures 4A24C). This wasreflected by an increase in compliance and
Figure 3. Effect of soluble guanylate cyclase (sGC) stimulation with BAY 41-2272 on cigarette smoke (CS)–induced pulmonary hypertension in guineapigs. Guinea pigs were exposed to cigarette smoke for 3 months in combination with BAY 41-2272. Control animals received the solvent (placebo). (A)Pulmonary arterial pressure, (B) total pulmonary resistance (pulmonary artery pressure/cardiac output), (C) cardiac output, and (D) systemic arterialpressure. (E) Percentage of muscularized intrapulmonary vessels less than 50 mm diameter. Control1sGCS = control and BAY 41-2272 3 mg/kg/day;CS = cigarette smoke; CS1sGCS = cigarette smoke and BAY 41-2272 3 mg/kg/day. Data are given from n = 7–12. Bars indicate significant differences(*P , 0.05; **P , 0.01; ***P , 0.001).
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1365
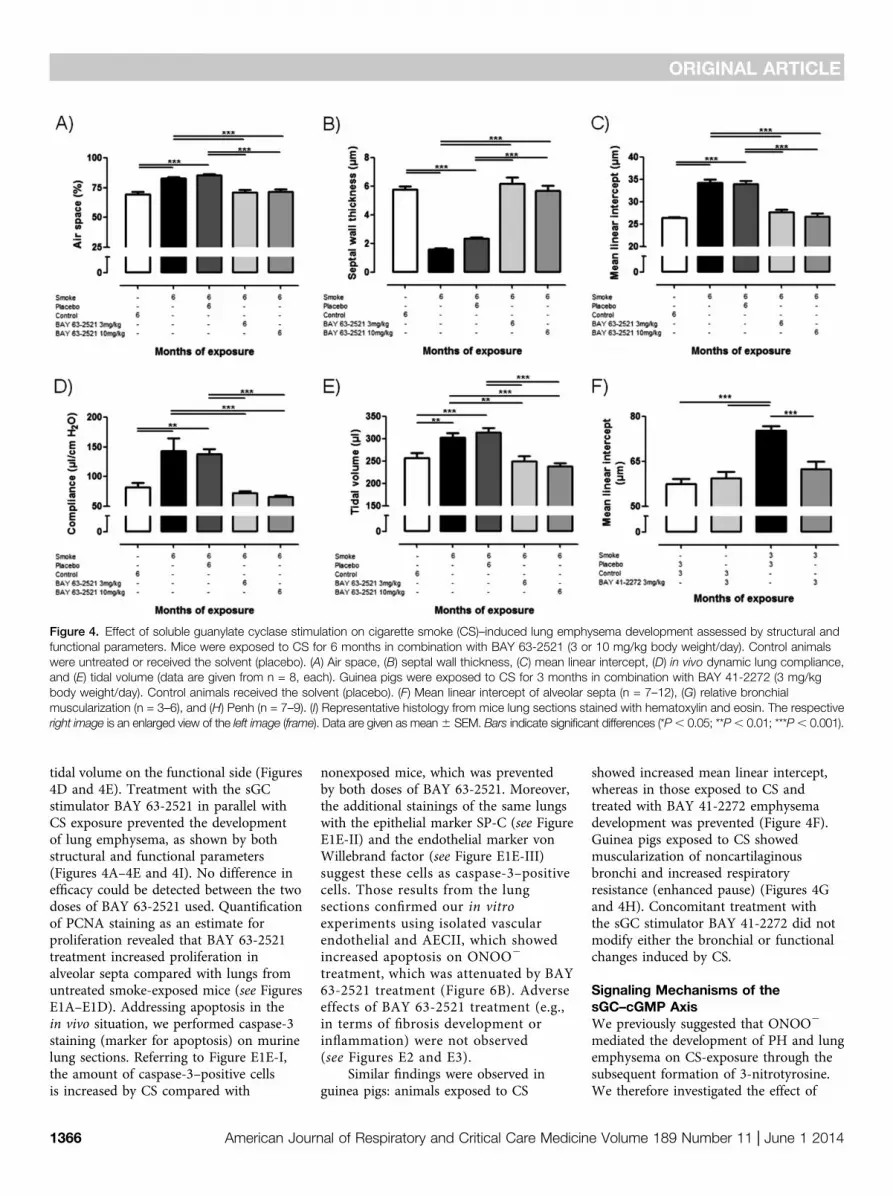
tidal volume on the functional side (Figures4D and 4E). Treatment with the sGCstimulator BAY 63-2521 in parallel withCS exposure prevented the developmentof lung emphysema, as shown by bothstructural and functional parameters(Figures 4A–4E and 4I). No difference inefficacy could be detected between the twodoses of BAY 63-2521 used. Quantificationof PCNA staining as an estimate forproliferation revealed that BAY 63-2521treatment increased proliferation inalveolar septa compared with lungs fromuntreated smoke-exposed mice (see FiguresE1A–E1D). Addressing apoptosis in thein vivo situation, we performed caspase-3staining (marker for apoptosis) on murinelung sections. Referring to Figure E1E-I,the amount of caspase-3–positive cellsis increased by CS compared with
nonexposed mice, which was preventedby both doses of BAY 63-2521. Moreover,the additional stainings of the same lungswith the epithelial marker SP-C (see FigureE1E-II) and the endothelial marker vonWillebrand factor (see Figure E1E-III)suggest these cells as caspase-3–positivecells. Those results from the lungsections confirmed our in vitroexperiments using isolated vascularendothelial and AECII, which showedincreased apoptosis on ONOO2
treatment, which was attenuated by BAY63-2521 treatment (Figure 6B). Adverseeffects of BAY 63-2521 treatment (e.g.,in terms of fibrosis development orinflammation) were not observed(see Figures E2 and E3).
Similar findings were observed inguinea pigs: animals exposed to CS
showed increased mean linear intercept,whereas in those exposed to CS andtreated with BAY 41-2272 emphysemadevelopment was prevented (Figure 4F).Guinea pigs exposed to CS showedmuscularization of noncartilaginousbronchi and increased respiratoryresistance (enhanced pause) (Figures 4Gand 4H). Concomitant treatment withthe sGC stimulator BAY 41-2272 did notmodify either the bronchial or functionalchanges induced by CS.
Signaling Mechanisms of thesGC–cGMP AxisWe previously suggested that ONOO2
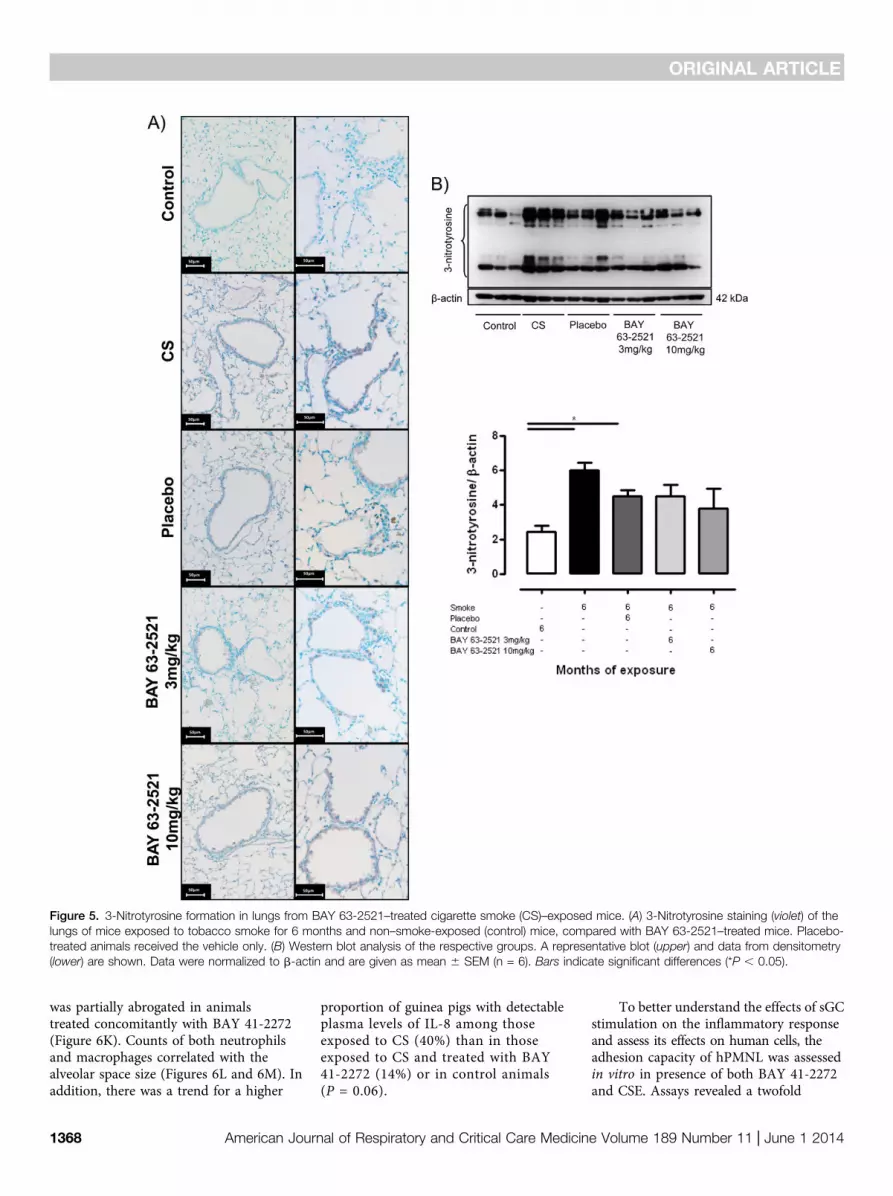
mediated the development of PH and lungemphysema on CS-exposure through thesubsequent formation of 3-nitrotyrosine.We therefore investigated the effect of
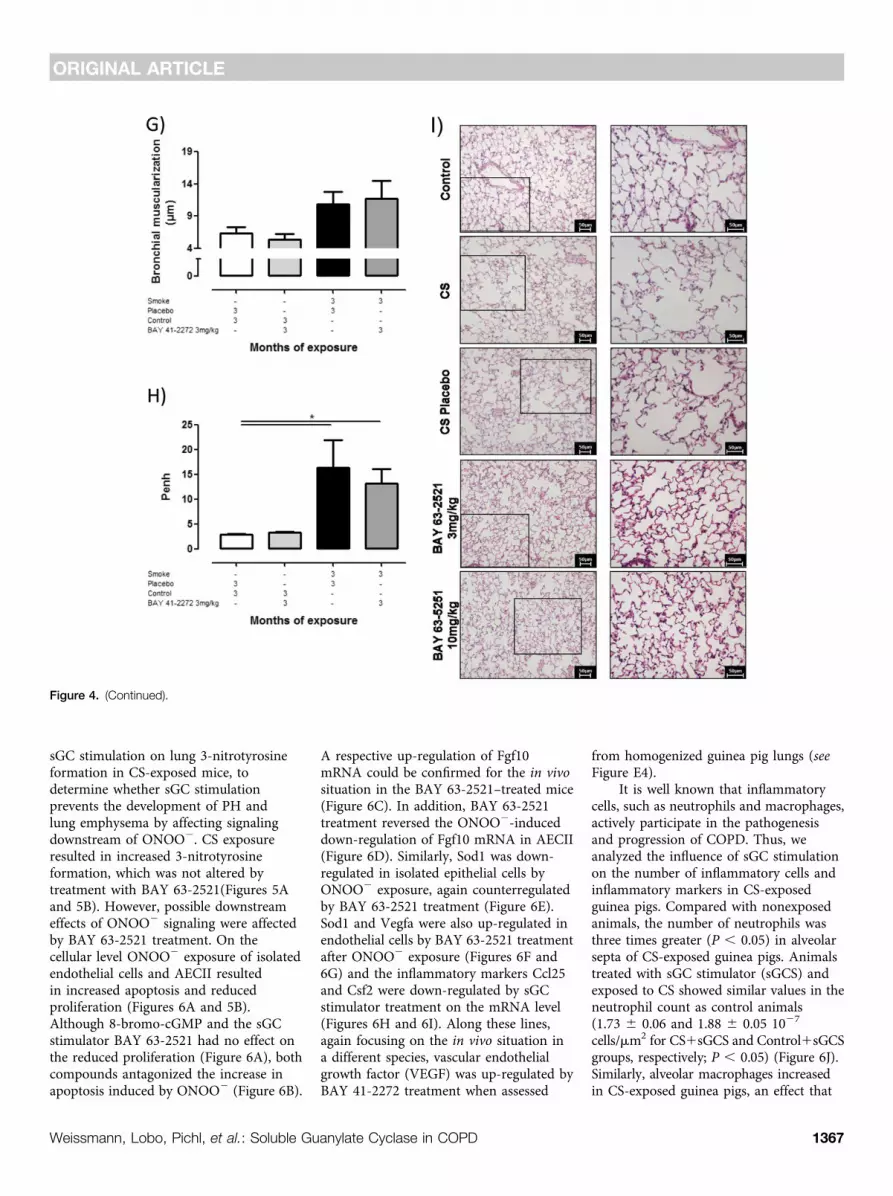
Figure 4. Effect of soluble guanylate cyclase stimulation on cigarette smoke (CS)–induced lung emphysema development assessed by structural andfunctional parameters. Mice were exposed to CS for 6 months in combination with BAY 63-2521 (3 or 10 mg/kg body weight/day). Control animalswere untreated or received the solvent (placebo). (A) Air space, (B) septal wall thickness, (C) mean linear intercept, (D) in vivo dynamic lung compliance,and (E) tidal volume (data are given from n = 8, each). Guinea pigs were exposed to CS for 3 months in combination with BAY 41-2272 (3 mg/kgbody weight/day). Control animals received the solvent (placebo). (F) Mean linear intercept of alveolar septa (n = 7–12), (G) relative bronchialmuscularization (n = 3–6), and (H) Penh (n = 7–9). (I) Representative histology from mice lung sections stained with hematoxylin and eosin. The respectiveright image is an enlarged view of the left image (frame). Data are given as mean6 SEM. Bars indicate significant differences (*P, 0.05; **P, 0.01; ***P, 0.001).
ORIGINAL ARTICLE
1366 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014
sGC stimulation on lung 3-nitrotyrosineformation in CS-exposed mice, todetermine whether sGC stimulationprevents the development of PH andlung emphysema by affecting signalingdownstream of ONOO2. CS exposureresulted in increased 3-nitrotyrosineformation, which was not altered bytreatment with BAY 63-2521(Figures 5Aand 5B). However, possible downstreameffects of ONOO2 signaling were affectedby BAY 63-2521 treatment. On thecellular level ONOO2 exposure of isolatedendothelial cells and AECII resultedin increased apoptosis and reducedproliferation (Figures 6A and 5B).Although 8-bromo-cGMP and the sGCstimulator BAY 63-2521 had no effect onthe reduced proliferation (Figure 6A), bothcompounds antagonized the increase inapoptosis induced by ONOO2 (Figure 6B).
A respective up-regulation of Fgf10mRNA could be confirmed for the in vivosituation in the BAY 63-2521–treated mice(Figure 6C). In addition, BAY 63-2521treatment reversed the ONOO2-induceddown-regulation of Fgf10 mRNA in AECII(Figure 6D). Similarly, Sod1 was down-regulated in isolated epithelial cells byONOO2 exposure, again counterregulatedby BAY 63-2521 treatment (Figure 6E).Sod1 and Vegfa were also up-regulated inendothelial cells by BAY 63-2521 treatmentafter ONOO2 exposure (Figures 6F and6G) and the inflammatory markers Ccl25and Csf2 were down-regulated by sGCstimulator treatment on the mRNA level(Figures 6H and 6I). Along these lines,again focusing on the in vivo situation ina different species, vascular endothelialgrowth factor (VEGF) was up-regulated byBAY 41-2272 treatment when assessed
from homogenized guinea pig lungs (seeFigure E4).
It is well known that inflammatorycells, such as neutrophils and macrophages,actively participate in the pathogenesisand progression of COPD. Thus, weanalyzed the influence of sGC stimulationon the number of inflammatory cells andinflammatory markers in CS-exposedguinea pigs. Compared with nonexposedanimals, the number of neutrophils wasthree times greater (P , 0.05) in alveolarsepta of CS-exposed guinea pigs. Animalstreated with sGC stimulator (sGCS) andexposed to CS showed similar values in theneutrophil count as control animals(1.73 6 0.06 and 1.88 6 0.05 1027
cells/mm2 for CS1sGCS and Control1sGCSgroups, respectively; P , 0.05) (Figure 6J).Similarly, alveolar macrophages increasedin CS-exposed guinea pigs, an effect that
Figure 4. (Continued).
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1367
was partially abrogated in animalstreated concomitantly with BAY 41-2272(Figure 6K). Counts of both neutrophilsand macrophages correlated with thealveolar space size (Figures 6L and 6M). Inaddition, there was a trend for a higher
proportion of guinea pigs with detectableplasma levels of IL-8 among thoseexposed to CS (40%) than in thoseexposed to CS and treated with BAY41-2272 (14%) or in control animals(P = 0.06).
To better understand the effects of sGCstimulation on the inflammatory responseand assess its effects on human cells, theadhesion capacity of hPMNL was assessedin vitro in presence of both BAY 41-2272and CSE. Assays revealed a twofold
Figure 5. 3-Nitrotyrosine formation in lungs from BAY 63-2521–treated cigarette smoke (CS)–exposed mice. (A) 3-Nitrotyrosine staining (violet) of thelungs of mice exposed to tobacco smoke for 6 months and non–smoke-exposed (control) mice, compared with BAY 63-2521–treated mice. Placebo-treated animals received the vehicle only. (B) Western blot analysis of the respective groups. A representative blot (upper) and data from densitometry(lower) are shown. Data were normalized to b-actin and are given as mean 6 SEM (n = 6). Bars indicate significant differences (*P , 0.05).
ORIGINAL ARTICLE
1368 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014
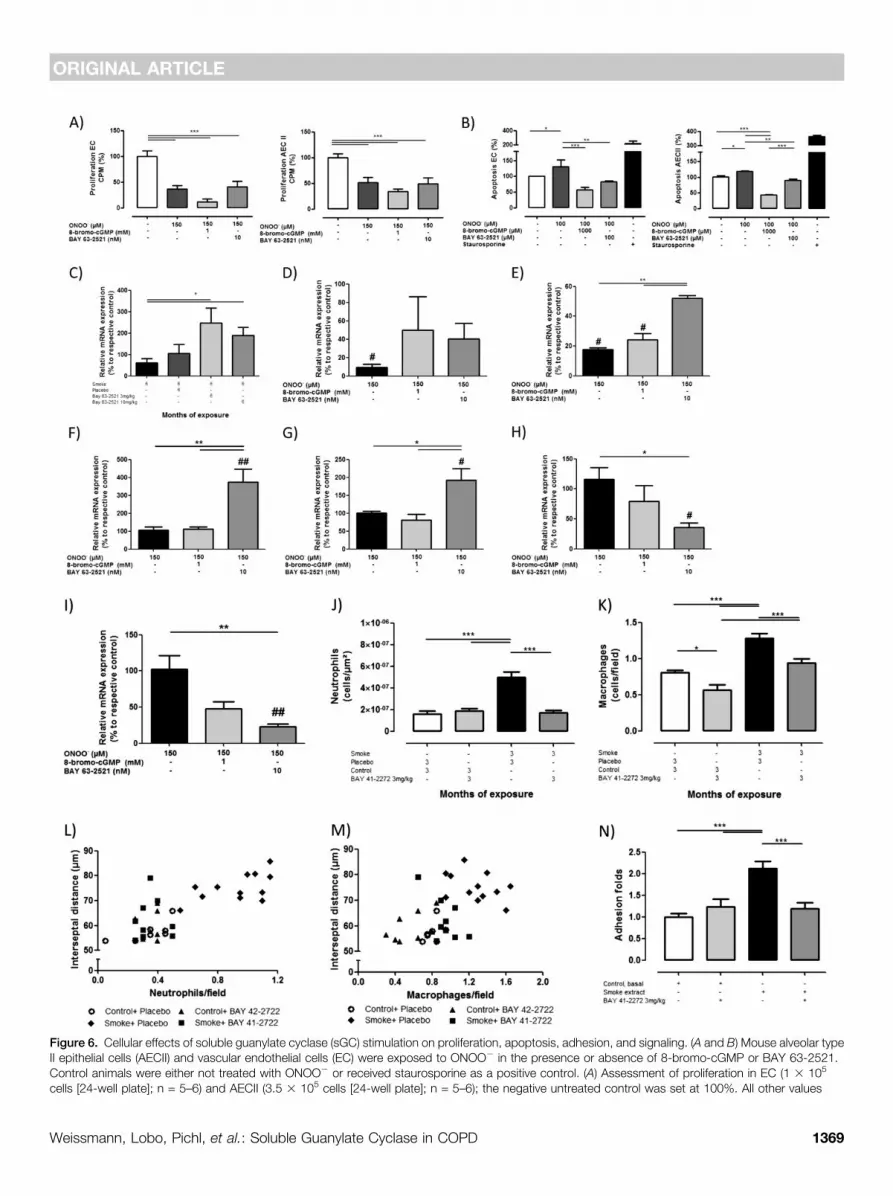
Figure 6. Cellular effects of soluble guanylate cyclase (sGC) stimulation on proliferation, apoptosis, adhesion, and signaling. (A and B) Mouse alveolar typeII epithelial cells (AECII) and vascular endothelial cells (EC) were exposed to ONOO2 in the presence or absence of 8-bromo-cGMP or BAY 63-2521.Control animals were either not treated with ONOO2 or received staurosporine as a positive control. (A) Assessment of proliferation in EC (1 3 105
cells [24-well plate]; n = 5–6) and AECII (3.5 3 105 cells [24-well plate]; n = 5–6); the negative untreated control was set at 100%. All other values
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1369

increased adhesion to a fibrinogen matrixcapacity of hPMNL when incubated withCSE with respect to baseline conditions.Addition of the sGC stimulator BAY41-2272 (1 mM) inhibited the adhesioncapacity of neutrophils in presence of CSE,keeping it similar to baseline conditions(1.19 6 0.45 and 1.23 6 0.46 folds,respectively) (Figure 6N). A screening ofmouse pulmonary vascular endothelialcells exposed to CSE showed a down-regulation of b2-integrin on sGCstimulator treatment and a trend tolower expression of intercellular adhesionmolecule-1 and P-selectin (see Figure E5).
Discussion
Our data suggest that the sGC–cGMP axisis critical for the development of lungemphysema and PH, and that thesGC–cGMP axis is a potential target fortreatment of CS-induced PH and alveolardestruction. We provide evidence that sGCis partially inactivated by CS, therebysuppressing the protective effect ofcGMP on the structural and functionaldeterioration of the lung in CS-inducedemphysema and PH. This conclusionis based on the following findings: (1)the sGCb1 subunit protein, which is thefunctionally essential sGC subunit (12), wassignificantly down-regulated in lungs frompatients with severe COPD (with andwithout PH), and in the mouse and in theguinea pig model of emphysema inducedby long-term exposure to CS; (2) treatmentwith the sGC stimulators BAY 63-2521 andBAY 41-2272 prevented the developmentof lung emphysema, vascular remodeling,and/or PH in two different rodent modelsof COPD, without adverse effects oftreatment; (3) BAY 41-2272 abrogatedinflammatory cell recruitment in lungparenchyma; (4) induction of apoptosis byONOO2 in both endothelial and AECII
could be prevented by the application ofeither a cGMP analog or a sGC stimulator;and (5) BAY 63-2521 or BAY 41-2272induced an up-regulation of Fgf10, thesuperoxide scavenger Sod1, and Vegfa aswell as a down-regulation of markers forinflammation or adhesion as assessedin vivo, in ONOO2-, or CSE-challengedpulmonary vascular endothelial.
The NO–sGC–cGMP signaling pathwayplays a critical role in the physiology andpathophysiology of the pulmonaryvasculature (30). A dysregulation of thispathway caused by reduced bioavailabilityof, or responsiveness to, endogenousNO may contribute to pulmonary diseasesand PH (17, 27, 43). NO generated by NOSbinds to sGC, which produces cGMPfrom GTP. The effects of cGMP can bemediated by cGMP-dependent proteinkinases (26) that modulate apoptosis,proliferation, migration, and extracellularmatrix protein expression (21, 44, 45).
Soluble GC consists of a large (a) andsmall heme-binding (b) subunit. For bothsubunits the isoforms a1 (synonym a3),a2, b1, and b2 have been described (12,13). No sGC heterodimer including theb2-subunit has yet been found (13); theb1-subunit therefore seems essential forsGC function (12). Besides the expressionof the sGC subunits the activity of theenzyme may also contribute to the observedsmoke effect. Although not determinedin our study it was suggested that sGCa1b1 are the essential subunits determiningsGC activity (12). Moreover, thesGC–cGMP system is essential for vascularhomeostasis and regulation of vasculartone, and can interfere with the pulmonaryvascular structure and remodeling (14, 19).In our study we show that this essentialb1-subunit is down-regulated in patientswith COPD, in mice, and in guineapigs chronically exposed to CS. In additionto such a down-regulation, sGC-dependentcGMP formation can also be suppressed
by oxidation (25). The redox state of sGCcan be altered by reactive oxygen andnitrogen species, such as superoxide (O2
.2)and ONOO2 (17, 26, 47) through theoxidation of the ferrous heme group of theb1-subunit, leading finally to a heme-freesGC. In this regard, it was previously shownthat CS-induced emphysema and PH inmice is affected by iNOS up-regulation,most likely caused by subsequent ONOO2
formation (10).Furthermore, it was previously shown
that ONOO2 induced apoptosis in AECIIand vascular endothelial cells, and reducedproliferation of AECII (10). Our data nowreveal that (1) the induction of apoptosis byONOO2 could be inhibited by both sGCstimulators and cGMP analogs; (2) sGCstimulators can increase mediators ofvascular integrity and lung maintenance,such as Vegfa (3) and Fgf10 (47, 48); (3)sGC stimulator treatment can increaseantioxidant enzymes, such as Sod1 (whichcan reduce sGC oxidation and thusinactivation); and (4) sGC stimulatortreatment can reduce inflammation underONOO2 exposure.
We speculate that a deactivation of sGCby oxidation, besides its down-regulation,can be antagonized by a cGMP-increasingtreatment. Therefore, either the down-regulation of cGMP can lead to emphysemaand PH development on CS exposure,and/or cGMP can antagonize the non–sGC-targeting effects of ONOO2.
It is also well accepted thatinflammatory cells, such as neutrophils andmacrophages, actively participate in thepathogenesis and progression of COPDand emphysema by releasing interleukins,elastase, and metalloproteinases (3, 10). Inour CS-exposed guinea pig model, airspace size was related to the number ofneutrophils and macrophages in lungtissue, suggesting that the abrogation ofthe development of emphysema could alsobe related to the attenuation of the
Figure 6. (Continued). are calculated in relation to the negative control. (B) Determination of apoptosis in EC (2 3 104 cells [96-well plate]; n = 8–9) andAECII (4 3 104 cells [96-well plate]; n = 4). Values were normalized to untreated cells (negative control, set at 100%) and are given as mean 6 SEM.Bars indicate significant differences (*P , 0.05; **P , 0.01; ***P , 0.001). Assessment of mRNA expression levels of (C) Fgf10 in lung homogenate,(D) Fgf10 in murine AECII, (E) Sod1 in murine AECII, (F) Sod1 in EC, (G) Vegfa in EC, (H) Ccl25 in EC, and (I) Csf2 in murine EC. In D–I #P, 0.05 and ##P,0.01 = significant to respective control (n = 5). For guinea pig experiments animals were exposed to cigarette smoke for 3 months in combinationwith BAY 41-2272 treatment. Control animals received the solvent (placebo). (J) Presence of intraseptal neutrophils (cells x1027/mm2), (K) alveolarmacrophages (cells per field), correlations between (L) neutrophils (r = 0.784, P , 0.001) and (M) macrophages (r = 0.535, P , 0.001) cell counts and themean interseptal distance. Control1sGCS = control and BAY 41-2272 3 mg/kg/day; CS = cigarette smoke; CS1sGCS = cigarette smoke and BAY41-2272 3 mg/kg/day. (N) In vitro human polymorphonuclear cells adhesion capacity to fibrinogen when incubated with basal medium (RPMI), cigarettesmoke extract (1/5), or BAY 41-2272 (1 mM) (sGCS) (n = 7–12).
ORIGINAL ARTICLE
1370 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014

inflammatory response induced by CSexposure. A prevention of emphysema wasproved by normal lung distensibility.However, there was no improvement inthe respiratory resistance, likely becauseof the concomitant effects of CS on thebronchial structure, which were notprevented by sGC stimulation. Accordingly,prevention of emphysema development inCS animals can be explained, at least inpart, by the antiinflammatory effects of sGCstimulation.
NO-cGMP signaling exertsantiinflammatory and antithromboticeffects by preventing the activation andadherence of circulating inflammatory cellsand platelets. Mechanisms underlying theseeffects have been suggested to involvechanges in the expression pattern ofadhesion molecules. P-selectin, togetherwith L-selectin, b2-integrin, andintercellular adhesion molecules 1 and 2 arenecessary for the inflammatory cells toroll and adhere to vascular endotheliumbefore their migration into lung structures.Reduced expression of P-selectin on theendothelial surface results in an impairedcapacity of inflammatory cells to infiltratetissues (22, 49, 50). Along these lines, sGCstimulator treatment down-regulated b2-integrin in pulmonary vascular endothelialcells exposed to CSE and a trend in thesame direction for intercellular adhesionmolecule-1 and P-selectin. In the presentstudy, sGC stimulation inhibited theadhesion of CS-stimulated neutrophils toa fibrinogen matrix indicating at leasta direct pharmacologic action oninflammatory cells (51, 52) additional tothe effects described on endothelial cells.Furthermore, it has been recently shownthat sGC mediates the angiogenic andpermeability-promoting activities of VEGF(53), pointing to the key role of sGC asa downstream effector of VEGF-triggeredresponses. Consistent with this concept, wecould show that BAY 41-2272 increasedVegf expression in comparison with smokein guinea pigs. Along these lines sGC
stimulators also increase Vegfa in isolatedmouse endothelial cells challenged withONOO2, indicating that sGC stimulatorscould exert partially their protective rolepreventing emphysema through thisgrowth factor (3). Similar results have beenshown with statin, which stimulates NOproduction, thereby reinforcing the criticalrole of the NO–cGMP pathway in lungalterations of COPD (54).
We also investigated the developmentof CS-induced PH in detail. There iscurrently discussion on whether in COPDchanges in pulmonary circulation emergesecondary to changes in the airways andlung parenchyma, or if they can antecedeor even trigger emphysema development.The latter concept has recently beensupported by investigations showing thatsmokers who have not developed COPD candisplay conspicuous abnormalities inpulmonary vessels (8, 9). Moreover, recentwork has shown that more than 50% ofpatients with COPD may suffer from PH(7–9, 29, 55). We have recently determinedin mice exposed to CS that (1) PH clearlyprecedes emphysema development, asshown for other species (32); (2) PHdevelopment is not driven by hypoxia; and(3) emphysema and PH are induced bydifferent pathways, with emphysemadevelopment being dependent on non-bone-marrow–derived iNOS-containingcells, and PH development being dependenton bone-marrow–derived iNOS-containingcells (10).
Based on these findings, it wasproposed that the up-regulation of iNOSand the subsequent production of ONOO2
in the vascular compartment can triggeremphysema development independentlyfrom vascular remodeling (10). We nowshow that maintenance of cGMP levelscan also prevent pulmonary vascularremodeling and the development of PHinduced by CS. These effects are in line withthe ability of sGC stimulators to antagonizePH induced by mechanisms other thanCS, including PH in hypoxic mice, PH
induced by monocrotaline in rats, PH inneonatal rats (14, 19, 27, 30), in lambs (56),and more recently in men (28, 31, 57, 58).Of note, in the guinea pig model, sGCstimulation prevented pulmonary vascularremodeling but did not modify significantlythe increase in pulmonary artery pressureinduced by CS exposure, presumablybecause of the lower dose used in thatexperiment (3 mg/kg). Similar observationswere made in a hypertensive rat modelin which a low dose of BAY 41-2272attenuated the structural changes withoutmodifying hemodynamic parameters (59).In support of our data and the importantrole of cGMP, it was shown thatsildenafil, a PDE5 inhibitor that preventsthe degradation of cGMP (60), preservedalveolar growth and lung angiogenesis,and decreased pulmonary vascularresistance, right ventricular hypertrophy,and medial wall thickness in oxygen-induced lung injury in newbornrats (61).
In conclusion, we provide evidencethat the sGC–cGMP axis is perturbed byCS exposure. Our data from investigationsconducted in humans, mice, and guineapigs using sGC stimulators demonstratethat maintenance of the sGC–cGMPpathway can prevent CS-induced lungemphysema and PH. If these data aretransferable to the human situation(with a compound that is alreadyapproved for treatment of other diseases inhumans), stopping the progress of thedisease would be a major advance.Further investigations should, however,focus on the potential of sGCstimulators to reverse establishedemphysema. n
Author disclosures are available with the textof this article at www.atsjournals.org.
Acknowledgment: The authors thank Xia Tian,Ingrid Breitenborn-Muller, Cristina Bonjoch,Lisa Frohlich, Carmen Homberger, ElisabethKappes, Miriam Wessendorf, and Karin Quanzfor technical assistance.
References
1. Vestbo J, Hurd SS, Agustı AG, Jones PW, Vogelmeier C, Anzueto A,Barnes PJ, Fabbri LM, Martinez FJ, Nishimura M, et al. Globalstrategy for the diagnosis, management, and prevention of chronicobstructive pulmonary disease: GOLD executive summary. Am JRespir Crit Care Med 2013;187:347–365.
2. Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 2000;343:269–280.
3. Yoshida T, Tuder RM. Pathobiology of cigarette smoke-inducedchronic obstructive pulmonary disease. Physiol Rev 2007;87:1047–1082.
4. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L,Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. Thenature of small-airway obstruction in chronic obstructive pulmonarydisease. N Engl J Med 2004;350:2645–2653.
5. Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lungparenchyma in COPD: role of T cells. Chest 2002;121(5, Suppl)160S–165S.
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1371

6. Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement inCOPD. Chest 2008;134:808–814.
7. Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD:epidemiology, significance, and management: pulmonary vasculardisease: the global perspective. Chest 2010;137(6, Suppl)39S–51S.
8. Barbera JA, Blanco I. Pulmonary hypertension in patients with chronicobstructive pulmonary disease: advances in pathophysiology andmanagement. Drugs 2009;69:1153–1171.
9. Santos S, Peinado VI, Ramirez J, Morales-Blanhir J, Bastos R, Roca J,Rodriguez-Roisin R, Barbera JA. Enhanced expression of vascularendothelial growth factor in pulmonary arteries of smokers andpatients with moderate chronic obstructive pulmonary disease. Am JRespir Crit Care Med 2003;167:1250–1256.
10. Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC,Milger K, Egemnazarov B, Turowska A, Fuchs B, et al. Inducible NOSinhibition reverses tobacco-smoke-induced emphysema andpulmonary hypertension in mice. Cell 2011;147:293–305.
11. Ferrer E, Peinado VI, Castañeda J, Prieto-Lloret J, Olea E, Gonzalez-Martın MC, Vega-Agapito MV, Dıez M, Domınguez-Fandos D, ObesoA, et al. Effects of cigarette smoke and hypoxia on pulmonarycirculation in the guinea pig. Eur Respir J 2011;38:617–627.
12. Derbyshire ER, Marletta MA. Structure and regulation of solubleguanylate cyclase. Annu Rev Biochem 2012;81:533–559.
13. Pyriochou A, Papapetropoulos A. Soluble guanylyl cyclase: moresecrets revealed. Cell Signal 2005;17:407–413.
14. Schermuly RT, Janssen W, Weissmann N, Stasch JP, Grimminger F,Ghofrani HA. Riociguat for the treatment of pulmonary hypertension.Expert Opin Investig Drugs 2011;20:567–576.
15. Harteneck C, Wedel B, Koesling D, Malkewitz J, Bohme E, Schultz G.Molecular cloning and expression of a new alpha-subunit of solubleguanylyl cyclase. Interchangeability of the alpha-subunits of theenzyme. FEBS Lett 1991;292:217–222.
16. Yuen PS, Potter LR, Garbers DL. A new form of guanylyl cyclase ispreferentially expressed in rat kidney. Biochemistry 1990;29:10872–10878.
17. Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP.NO-independent stimulators and activators of soluble guanylatecyclase: discovery and therapeutic potential. Nat Rev Drug Discov2006;5:755–768.
18. Lang M, Kojonazarov B, Tian X, Kalymbetov A, Weissmann N,Grimminger F, Kretschmer A, Stasch JP, Seeger W, Ghofrani HA,et al. The soluble guanylate cyclase stimulator riociguat amelioratespulmonary hypertension induced by hypoxia and SU5416 in rats.PLoS ONE 2012;7:e43433.
19. Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D,Schluter KD, Dingendorf A, Hackemack S, Kolosionek E, Kaulen C,et al. Expression and function of soluble guanylate cyclase inpulmonary arterial hypertension. Eur Respir J 2008;32:881–891.
20. Feil R, Kemp-Harper B. cGMP signalling: from bench to bedside.Conference on cGMP generators, effectors and therapeuticimplications. EMBO Rep 2006;7:149–153.
21. Joshi CN, Martin DN, Fox JC, Mendelev NN, Brown TA, Tulis DA. Thesoluble guanylate cyclase stimulator BAY 41-2272 inhibits vascularsmooth muscle growth through the cAMP-dependent proteinkinase and cGMP-dependent protein kinase pathways. J PharmacolExp Ther 2011;339:394–402.
22. Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perretti M,Moncada S, Hobbs AJ. Antiinflammatory activity of solubleguanylate cyclase: cGMP-dependent down-regulation of P-selectinexpression and leukocyte recruitment. Proc Natl Acad Sci USA 2004;101:1386–1391.
23. Meurer S, Pioch S, Pabst T, Opitz N, Schmidt PM, Beckhaus T, WagnerK, Matt S, Gegenbauer K, Geschka S, et al. Nitric oxide-independentvasodilator rescues heme-oxidized soluble guanylate cyclasefrom proteasomal degradation. Circ Res 2009;105:33–41.
24. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as anemerging therapeutic target in cardiopulmonary disease. Circulation2011;123:2263–2273.
25. Stasch JP, Schmidt PM, Nedvetsky PI, Nedvetskaya TY, H S AK,Meurer S, Deile M, Taye A, Knorr A, Lapp H, et al. Targeting the
heme-oxidized nitric oxide receptor for selective vasodilatation ofdiseased blood vessels. J Clin Invest 2006;116:2552–2561.
26. Tabima DM, Frizzell S, Gladwin MT. Reactive oxygen and nitrogenspecies in pulmonary hypertension. Free Radic Biol Med 2012;52:1970–1986.
27. Dumitrascu R, Weissmann N, Ghofrani HA, Dony E, Beuerlein K,Schmidt H, Stasch JP, Gnoth MJ, Seeger W, Grimminger F, et al.Activation of soluble guanylate cyclase reverses experimentalpulmonary hypertension and vascular remodeling. Circulation 2006;113:286–295.
28. Ghofrani HA, Hoeper MM, Halank M, Meyer FJ, Staehler G, Behr J,Ewert R, Weimann G, Grimminger F. Riociguat for chronicthromboembolic pulmonary hypertension and pulmonary arterialhypertension: a phase II study. Eur Respir J 2010;36:792–799.
29. Hoeper MM, Halank M, Wilkens H, Gunther A, Weimann G, Gebert I,Leuchte H, Behr J. Riociguat for interstitial lung disease andpulmonary hypertension: a pilot trial. Eur Respir J 2013;41:853–860.
30. Weissmann N, Hackemack S, Dahal BK, Pullamsetti SS, Savai R, MittalM, Fuchs B, Medebach T, Dumitrascu R, Eickels Mv, et al. Thesoluble guanylate cyclase activator HMR1766 reverses hypoxia-induced experimental pulmonary hypertension in mice. Am J PhysiolLung Cell Mol Physiol 2009;297:L658–L665.
31. Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC,Keogh AM, Langleben D, Kilama MO, Fritsch A, et al.; PATENT-1Study Group. Riociguat for the treatment of pulmonary arterialhypertension. N Engl J Med 2013;369:330–340.
32. Wright JLCA, Churg A. A model of tobacco smoke-induced airflowobstruction in the guinea pig. Chest 2002;121(5, Suppl)188S–191S.
33. Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-inducedCOPD: insights from animal models. Am J Physiol Lung Cell MolPhysiol 2008;294:L612–L631.
34. Padilla-Carlin DJ, McMurray DN, Hickey AJ. The guinea pig as a modelof infectious diseases. Comp Med 2008;58:324–340.
35. Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, FeurerA, Gerzer R, Minuth T, Perzborn E, Pleiss U, et al. NO-independentregulatory site on soluble guanylate cyclase. Nature 2001;410:212–215.
36. Stasch JP, Schmidt P, Alonso-Alija C, Apeler H, Dembowsky K, HaerterM, Heil M, Minuth T, Perzborn E, Pleiss U, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basisand cardiovascular implications of a new pharmacological principle.Br J Pharmacol 2002;136:773–783.
37. Pichl A, Parajuli N, Seimetz M, Stasch JP, Frey R, Schermuly RT, SeegerW, Grimminger F, Ghofrani HA, Weissmann N. Stimulation of solubleguanylate cyclase by riociguat prevents tobacco smoke-inducedpulmonary hypertension in mice. Thieme Pneumologie 2012;66:A310.
38. Lobo BP-PR, Ferrer E, Dominguez-Fandos D, Coll N, Garcia J, MusriMM, Peinado VI, Barbera JA. Stimulation of soluble guanylatecyclase in guinea pigs chronically exposed to cigarette smokereduces intrapulmonary vascular remodeling and preventsemphysema. Am J Respir Crit Care Med 2013;187:A4666.
39. Pichl A, Parajuli N, Seimetz M, Stasch JP, Frey R, Schermuly RT,Seeger W, Grimminger F, Ghofrani HA, Weissmann N. The solubleguanylate cylase stimulator riociguat prevents from tobacco smoke-induced pulmonary hypertension and emphysema. Presented atthe Pulmonary Vascular Research Institute Workshops & Debates.January 25, 2013, Istanbul, Turkey.
40. Seimetz M, Parajuli N, Pichl A, Stasch JP, Frey R, Schermuly RT,Ghofrani HA, Seeger W, Grimminger F, Weissmann N. Effects of thesoluble guanylate cyclase stimulator riociguat on emphysemadevelopment in tobacco-smoke exposed mice. Am J Respir CritCare Med 2011;183:A3107.
41. Seimetz M, Parajuli N, Pichl A, Stasch JP, Frey R, Schermuly RT,Ghofrani HA, Seeger W, Grimminger F, Weissmann N. Prevention ofcigarette smoke-induced pulmonary hypertension by the solubleguanylate cyclase stimulator riociguat. Am J Respir Crit Care Med2012;185;A3416.
42. Peinado VI, Gomez FP, Barbera JA, Roman A, Angels Montero M,Ramırez J, Roca J, Rodriguez-Roisin R. Pulmonary vascularabnormalities in chronic obstructive pulmonary disease undergoinglung transplant. J Heart Lung Transplant 2013;32:1262–1269.
ORIGINAL ARTICLE
1372 American Journal of Respiratory and Critical Care Medicine Volume 189 Number 11 | June 1 2014

43. Li H, Forstermann U. Nitric oxide in the pathogenesis of vasculardisease. J Pathol 2000;190:244–254.
44. Chiche JD, Schlutsmeyer SM, Bloch DB, de la Monte SM, RobertsJD Jr, Filippov G, Janssens SP, Rosenzweig A, Bloch KD.Adenovirus-mediated gene transfer of cGMP-dependent proteinkinase increases the sensitivity of cultured vascular smooth musclecells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J Biol Chem 1998;273:34263–34271.
45. Mendelev NN, Williams VS, Tulis DA. Antigrowth properties of BAY 41-2272 in vascular smooth muscle cells. J Cardiovasc Pharmacol 2009;53:121–131.
46. Wedgwood S, Steinhorn RH, Bunderson M, Wilham J,Lakshminrusimha S, Brennan LA, Black SM. Increased hydrogenperoxide downregulates soluble guanylate cyclase in the lungs oflambs with persistent pulmonary hypertension of the newborn. Am JPhysiol Lung Cell Mol Physiol 2005;289:L660–L666.
47. Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T,Yagishita N, Matsui D, Koga Y, Itoh N, et al. Fgf10 is essential forlimb and lung formation. Nat Genet 1999;21:138–141.
48. Ramasamy SK, Mailleux AA, Gupte VV, Mata F, Sala FG, Veltmaat JM,Del Moral PM, De Langhe S, Parsa S, Kelly LK, et al. Fgf10 dosageis critical for the amplification of epithelial cell progenitors and forthe formation of multiple mesenchymal lineages during lungdevelopment. Dev Biol 2007;307:237–247.
49. Liu PXB, Xu B, Hock CE, Nagele R, Sun FF, Wong PY. NO modulatesP-selectin and ICAM-1 mRNA expression and hemodynamicalterations in hepatic I/R. Am J Physiol 1998;275:H2191–H2198.
50. Overbeek SABS, Braber S, Henricks PA, Kleinjan M, Kamp VM,Georgiou NA, Garssen J, Kraneveld AD, Folkerts G. Cigarette smokeinduces b2-integrin-dependent neutrophil migration across humanendothelium. Respir Res 2011;12:75.
51. Canalli AA, Franco-Penteado CF, Saad ST, Conran N, Costa FF.Increased adhesive properties of neutrophils in sickle cell diseasemay be reversed by pharmacological nitric oxide donation.Haematologica 2008;93:605–609.
52. Dal-Secco DFA, Freitas A, Abreu MA, Garlet TP, Rossi MA, Ferreira SH,Silva JS, Alves-Filho JC, Cunha FQ. Reduction of ICAM-1 expressionby carbon monoxide via soluble guanylate cyclase activation
accounts for modulation of neutrophil migration. NaunynSchmiedebergs Arch Pharmacol 2010;381:483–493.
53. Morbidelli LPA, Pyriochou A, Filippi S, Vasileiadis I, Roussos C, Zhou Z,Loutrari H, Waltenberger J, Stossel A, Giannis A, et al. The solubleguanylyl cyclase inhibitor NS-2028 reduces vascular endothelialgrowth factor-induced angiogenesis and permeability. Am J PhysiolRegul Integr Comp Physiol 2010;298:R824–R832.
54. Wright JL, Zhou S, Preobrazhenska O, Marshall C, Sin DD, Laher I,Golbidi S, Churg AM. Statin reverses smoke-induced pulmonaryhypertension and prevents emphysema but not airway remodeling.Am J Respir Crit Care Med 2011;183:50–58.
55. Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension inCOPD. Eur Respir J 2008;32:1371–1385.
56. Evgenov OV, Ichinose F, Evgenov NV, Gnoth MJ, Falkowski GE, ChangY, Bloch KD, Zapol WM. Soluble guanylate cyclase activatorreverses acute pulmonary hypertension and augments thepulmonary vasodilator response to inhaled nitric oxide in awakelambs. Circulation 2004;110:2253–2259.
57. Ghofrani HA, Voswinckel R, Gall H, Schermuly R, Weissmann N,Seeger W, Grimminger F. Riociguat for pulmonary hypertension.Future Cardiol 2010;6:155–166.
58. Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, BolkowD, Weissmann N, Muck W, Unger S, Wensing G, et al. First acutehaemodynamic study of soluble guanylate cyclase stimulatorriociguat in pulmonary hypertension. Eur Respir J 2009;33:785–792.
59. Masuyama HTT, Tsuruda T, Kato J, Imamura T, Asada Y, Stasch JP,Kitamura K, Eto T. Soluble guanylate cyclase stimulation oncardiovascular remodeling in angiotensin II-induced hypertensiverats. Hypertension 2006;48:972–978.
60. Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z,Shapiro S, White RJ, Chan M, Beardsworth A, et al.; PulmonaryArterial Hypertension and Response to Tadalafil (PHIRST) StudyGroup. Tadalafil therapy for pulmonary arterial hypertension.Circulation 2009;119:2894–2903.
61. Ladha F, Bonnet S, Eaton F, Hashimoto K, Korbutt G, Thebaud B.Sildenafil improves alveolar growth and pulmonary hypertension inhyperoxia-induced lung injury. Am J Respir Crit Care Med 2005;172:750–756.
ORIGINAL ARTICLE
Weissmann, Lobo, Pichl, et al.: Soluble Guanylate Cyclase in COPD 1373





























