Embed Size (px)
Citation preview

doi:10.1006/jmbi.2000.4224 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 304, 371±383
Solution Structure of a DNA Three-way JunctionContaining Two Unpaired Thymidine Bases.Identification of Sequence Features that DecideConformer Selection
Bernd N. M. van Buuren1, Franc J. J. Overmars2, Johannes H. Ippel1
Cornelis Altona2 and Sybren S. Wijmenga1*
1Department of MedicalBiosciences, Medical BiophysicsUmeaÊ University, S-90187UmeaÊ, Sweden2Leiden Institute of ChemistryGorlaeus Laboratories, PO Box9502, NL-2300 RA Leiden, TheNetherlands
Present address: F.J.J. Overmars,Box 1, NL-2600 MA Delft, The Neth
E-mail address of the [email protected]
0022-2836/00/030371±13 $35.00/0
The solution structure of a DNA three-way junction (3H) containing twounpaired thymidine bases at the branch site (3HT2), was determined byNMR. Arms A and B of the 3HT2 form a quasi-continuous stacked helix,which is underwound at the junction and has an increased helical rise.The unstacked arm C forms an acute angle of approximately 55 � withthe unique arm A. The stacking of the unpaired thymidine bases on armC resembles the folding of hairpin loops. From this data, combined withthe reported stacking behavior of 23 other 3HS2 s, two rules are derivedthat together correctly reproduce their stacking preference. These rulespredict, from the sequence of any 3HS2, its stacking preference. Thestructure also suggests a plausible mechanism for structure-speci®c recog-nition of branched nucleic acids by proteins.
# 2000 Academic Press
Keywords: DNA three-way junction; nucleic acid structure; HMG-boxproteins; Holliday junction; Hammerhead ribozyme*Corresponding author
Introduction
Branched nucleic acids play important roles inthe living cell and in its parasites; they can also beused as building blocks in nanoscale assemblies(Seeman,1996). In the cell they are involved innucleic acid rearrangements, such as splicing andgenetic recombination (reviewed in Lilley & Clegg,1993). The majority of these events involve aninterplay between nucleic acid structures and pro-teins. The latter are noteworthy in that they arestructure speci®c, i.e. they recognize their DNAsubstrate at the level of tertiary structure, ratherthan by sequence speci®city. The best-knownexample of a branched nucleic acid structure isthat of the Holliday, or four-way junction (4H(Lilley et al., 1995) in DNA, which comprises thecentral step in homologous and site-speci®c recom-bination (Altona et al., 1996; Lilley & Clegg, 1993).Three-way junctions (3Hs) are a recurring motifin RNA, where they play key roles in important
Gist-brocades, POerlands.
ing author:
cellular processes, such as translation (Noller,1991), splicing (Guthrie & Patterson, 1988), andprogrammed frameshifting (Rettberg et al., 1999).In DNA, 3Hs arise during recombination involvingphages (Minagawa et al., 1983; Jensch & Kemper,1986) and in the lagging strand during replication(Rosche et al., 1995). Furthermore, a 3H structure isobserved in the inverted terminal repeats of theAdeno-associated virus (Ren et al., 1999), where itis involved in viral DNA replication and site-speci®c integration of the virus DNA into the hostgenome. This feature of site-speci®c integration hasmade this virus the focus of efforts to develop agene therapy vector (Kotin et al., 1992).
Because of this broad variety of functional activi-ties, there is a wide interest in functional and struc-tural studies of 3Hs. A considerable amount ofstructural knowledge has been derived by severalbiophysical methods. It has been shown (Leontiset al., 1991; StuÈ hmeier et al., 1997a) that the intro-duction of formally unpaired bases at the branchsite, generating bulged 3Hs (3HSm), greatlyincreases 3H stability, the effect being most notice-able with two unpaired bases (3HS2). Most natu-rally occurring 3Hs indeed have evolutionaryconserved unpaired bases at the branch site(Noller, 1984; Ren et al., 1999). In solution, 3HS2s
# 2000 Academic Press

372 DNA Three-way Junction
exist as an equilibrium of three conformations(Altona et al., 1996; Welch et al., 1995; Lilley &Clegg, 1993). The unfolded form (Figure 1(a), cen-ter) is evident under conditions of low ionicstrength and in the absence of multivalent cations(Welch et al., 1995). Junctions that tend to adoptthe A/C-stacked conformation (Figure 1(a), right)appear to require multivalent cations for their fold-ing (Leontis et al., 1993; Welch et al., 1995). In con-trast, the junctions that adopt the A/B-stackedconformation (Figure 1(a), left) appear to fold atlower concentrations of multivalent cations, oreven in their absence (Overmars et al., 1996; Welchet al., 1995; Rosen & Patel, 1993a,b; Overmars,1997). Often a clear preference for one or the other
Figure 1. (a) Schematic of the conformations that can beat the branch point (3HSm). The notation proposed is used(continuous lines) are numbered clockwise with Arabic num30 termini; the helical arms are indicated by capital letterdenotes a sequence of formally unpaired bases. In the preeven in the presence of Na� only) the unfolded form (centearm A stacks on arm B (A/B stack), or into conformer I(Overmars et al., 1996; Leontis et al., 1993; Welch et al., 199polarity of the unpaired bases, going from 50 to 30 into theEach conformer can be either parallel or anti-parallel (seetation, the unstacked arm makes an acute angle with the uis obtuse. Y indicates the pyrimidine positions crucial for cof TWJ1 represented in the experimentally observed A/B-sttal letters, strand numbers are indicated with arabic num(see Table 3). (c) Schematic overview of the unambiguouslytra) for the eight central residues at the junction.
conformer is observed. This preference dependshighly on the sequence at or near the branch point.However, the exact manner in which sequencein¯uences the equilibrium is not yet understood. Adetailed 3D structure of a DNA 3H can giveinsight into the basic principles of this folding pro-cess. For RNA a number of high-resolution 3Hcrystal structures have been published (Cate et al.,1996; Scott et al., 1996; Pley et al., 1994), in additiona number of 3H structures are present in the largeribosomal subunit (Ban et al., 2000). For DNA nohigh-resolution structure is available, so far onlytwo low-resolution models have been proposedbased on NMR studies (Leontis et al., 1993; Rosen& Patel, 1993b).
adopted by three-way junctions that have unpaired bases(Altona, 1996; Altona et al., 1996). The backbone strands
erals with the arrowheads pointing in the direction of thes; base-pairing is symbolized by the ladder motifs. N-Nsence of polyvalent cations (or for some base sequencesr) folds into either conformer I (left), in which the uniqueI (right) in which arm A stacks on arm C (A/C stack)5; Rosen & Patel, 1993a,b; Overmars, 1997). Note that theunstacked arm in conformer I, is reversed in conformer II.also Lilley & Norman, (1999)). In an anti-parallel orien-
nique arm A, whereas in a parallel orientation, this angleonformer selection (see THE text). (b) Secondary structureacked conformation. Helical arms are indicated with capi-erals. Underlined residues indicate junction classi®cation
assigned NOEs (taken from 30 and 50 ms NOESY spec-

DNA Three-way Junction 373
Here we present the high-resolution NMR sol-ution structure of a DNA 3HS2 (TWJ1, Figure 1(b)),showing for the ®rst time in molecular detail thecoaxial stacking of the arms, the conformation ofthe strand crossing at the junction, as well as thepositioning of the unpaired residues at the branchsite. From this data combined with the reportedstacking behavior of 23 other 3HS2 s two rules arederived that together correctly account for theirstacking preference. These rules predict from thesequence of any 3HS2 its stacking preference. Fur-thermore, the structural data suggest a plausiblemechanism to explain the preference of structure-speci®c proteins, e.g. HMG box proteins, for bind-ing to branched DNA structures.
Results and Discussion
Overall structural features and NOE contactson the junction
The overall structural features of TWJ1 followdirectly from the 1D and 2D NOESY spectra inH2O and 2H2O and the phosphorus NMR spectra,as well as from characteristic chemical shift valuesas described by Overmars et al. (1996). It wasshown that TWJ1 folds into a stable 3H with astrong preference for an A/B-stacking. Here, wefocus on the NOEs at and near the junction whichare crucial for the determination of its 3D structure.The high quality of the 2H2O NOESY spectra facili-tated for a near-complete assignment of the NOEs.An overview of all observed NOEs in the 30 and50 ms NOESY spectra between the eight residuesaround the junction is given in Figure 1(c). Thebase-pair step determining the coaxial stacking ofthe A and B arms (G17 �C8/C18 �G27) is wellde®ned by 16 characteristic B-DNA type inter-resi-due NOEs. Ten of these de®ne the base stacking inthe continuous strand (G17-C18), and six de®nethe base stacking in the crossover region (C8-G27).No NOEs were detected that could indicate A/C-stacking. Although sequential B-DNA type NOEsare present for all base steps in the A/B-stackedhelix, some are less intense than expected for aB-DNA type helix, indicating deviations from idealB-DNA (vide infra). The unpaired bases and theclosing base-pair of arm C are restrained by 25NOEs between base-pair C7 �G30 and the twounpaired thymidine bases. Residue T28 has 13NOEs with C7, which indicates that the base ofT28 is stacked on the base of C7. The otherunpaired base, T29, has one base-base and eightsugar-base NOEs with T28, implying that T29 isstacked on T28. Only one sequential NOE(H10 $ H40) is observed for the step T29-G30, indi-cating that the base of T29 is pointing away fromG30. This NOE and the observed up®eld chemicalshift of T29H40 are characteristic for a sharp back-bone turn between these two residues (vanDongen et al., 1996; Orbons et al., 1986; Hilberset al., 1994). The orientation of arm C with respectto the A/B-stacked arms is restrained by 12 NOEs.
Ten of these are sequential, from G27 to T28 andfrom C7 to C8. The remaining two are long-rangecross-strand contacts, from C7 to G27, imposingadditional restraints on the angle between arm Cand the A/B-stacked arms. Additional long-rangeNOEs from G27 to T29 are observed in the longermixing time NOESYs, but were conservatively notincluded (vide infra).
Structure elucidation
Despite the complexity of the spectra, a largenumber of experimental constraints could beextracted for structure calculations (Table 1). Weemployed torsion angle molecular dynamics (Steinet al., 1997) for the structure elucidation (seeMaterials and Methods). Restraints were used con-servatively (Wijmenga & van Buuren, 1998) (errorbounds of 30 %, see Materials and Methods). Start-ing from 20 structures with random conformationsfor the 14 central residues (6-9, 16-19, 26-31), weobtained an unbiased set of 100 structures, ofwhich 60 converged. Final re®nements were per-formed for the 26 lowest-energy structures. As inthe structure calculation only experimentally veri®-able restraints were used in this re®nement (seeMaterials and Methods). Figure 2 gives stereoviews of this ®nal set and shows that TWJ1 has awell-de®ned 3D structure, which can also bejudged from the structural statistics (Table 1). Theheavy atom RMSDs for the whole molecule andfor the central 14 residues are 1.47 AÊ and 0.84 AÊ ,respectively. Locally, the arms around the junctionare even better de®ned, with a heavy atom RMSDof 0.59 and 0.32 AÊ for the A/B-stacked arms(Figure 2(c)) and dangling arm C (Figure 2(d)),respectively (Table 1). The high resolution is alsoevident from the small standard deviations in thehelix parameters and the inter-helix angle (Table 2).
For a given DNA structure proton chemicalshifts can be calculated with a good level of accu-racy (0.17 ppm chemical shift RMSD) (Wijmengaet al.,1997; Wijmenga & van Buuren, 1998). Thedeviation of back-calculated proton chemical shiftsfrom the experimentally observed shifts is there-fore an independent indicator for the accuracy ofNMR-derived DNA structures. For the ®nal set of26 structures the RMSD of the back-calculatedchemical shifts was 0.20 ppm, indicating that theyfaithfully re¯ect the experimental chemical shifts.Back-calculation of NOE intensities by CORMA(Keepers & James, 1984) showed a low R(1/6)-factor (Gonzalez et al., 1991) (9.7 %), also con®rm-ing the consistency of the structure with theexperimental constraints.
Description of the structure
The overall features of the TWJ1 structure areevident from the set of re®ned structures(Figure 2(a)) and the ribbon presentation(Figure 3(a)). Arms A and B are coaxially stacked.Arm C has an anti-parallel orientation with respect

Table 1. Structural statistics of the ®nal ensemble of 26 structures
A. Structural restraintsDistance restraintsa Intranucleotide 130
Internucleotide 107Hydrogen bonding 17
Repulsive 2Subtotal 256
Dihedral angle restraintsa Glycosidic 14Sugar pucker (d) 14Backbone (a,g,e,z) 47
Subtotal 75B. Violations of experimental restraintsb
Distance restraints Number of violations > 0.2 AÊ 6.8 � 0.9Maximum violation (AÊ ) 0.57
Dihedral angle restraints Number of violations > 5.0 � 9.7 � 1.4Maximum violation (deg.) 19.5
C. Mean residual violationsb Bonds (AÊ ) 0.01273 � 0.00008Angles (deg.) 1.857 � 0.007
Impropers (deg.) 0.76 � 0.01Distance restraints (AÊ ) 0.064 � 0.002
Dihedral restraints (deg.) 4.8 � 0.1D. Atomic R.M.S.D. (AÊ )c All residues 1.47 � 0.73
Central 14 residues (6-9.16-19,26-31) 0.84 � 0.44A/B-stack (8,9,16-19,26,27) 0.59 � 0.29
TT-bulge (6,7,28-31) 0.32 � 0.19F. Chemical shift RMSD (ppm)b Central 14 residues (6-9,16-19,26-31) 0.20 � 0.01Helix angle (deg.)b 53 � 11
a Restraints incorporated in the structure calculation for the central 14 residues (see methods).b Average values and standard deviations for the ®nal set of 26 structures.c Average pairwise RMSD of heavy atoms for the listed residues of the set of 26 structures.
374 DNA Three-way Junction
to the A/B-stacked helix and makes an acute angleof 53(�11)� with arm A for the ®nal set of 26 struc-tures (Table 2). This value for the inter-helix angleis in agreement with the ®ndings reported from arecent FRET study on another DNA three-wayjunction (StuÈ hmeier et al., 1997a). The precision inthe inter-helix angle follows from the relativelylarge set of 12 long-range and sequential NOEsfound in short mixing time NOESY spectra (30 msand 50 ms) that constrain the inter-helical angle(see above). As pointed out, the NOE restraintswere used with conservative error bounds andback-calculated with good-level accuracy from the®nal set of structures (R is 9.7 %). Also, the chemi-cal shifts, which were not used as restraints, wereback-calculated with high accuracy. In addition,long-range NOEs that should be present from thestructures according to back-calculated NOESYspectra (from G27 to T29) were indeed observed inlonger mixing time NOESY spectra (200 ms). Theywere, however, conservatively not used in thestructure determination. The fact that these NOEsare not present in the low mixing times might indi-cate some inter-helical dynamics, for example viaa hinge-like motion of limited degree betweenthe A/B-stacked arms and arm C (see Figure 2(c)and (d)).
A/B continuous stacked helices deviate fromB-DNA geometry
Although the A/B-stacked helices in the derivedstructures show overall B-DNA geometry, devi-ations do occur (Table 2). A reduced twist is
observed for the base-pair step at the junction(C8 �G17-C18 �G27), which has a value ca 10 �lower than found in B-DNA. For the other stepsthe twist value lies within the range usually foundin a regular B-DNA helix. The lower twist (20-25 �)for the base-step towards the hairpin loop in armsA and B corresponds to what is usually seen forCNNG-tetraloops (Ippel et al., 1998). The reducedtwist for the base-pair step at the junction was alsoobserved in a recent X-ray structure of a DNA 4H,although here non-canonical G �A base-pairs werepresent at the junction (Ortiz-LombardõÂa et al.,1999).
All base steps in the A/B-stacked arms show thenormal B-DNA type sequential NOEs. However,for some of these, the intensities are relativelyweak, leading to abnormally long sequential dis-tances. In particular, the sequential H2'' to H6/8distances for the base steps in the continuousstrand penultimate from the junction (G17-G16,G19-C18) and one further away (G16-C15) are sig-ni®cantly longer than normal (>3.3 AÊ ). Therefore,an increase in helical rise is deduced for these steps(Table 2). This ®nding is important, as it mayexplain the sequence dependence of stacking pre-ference, as well as the structure-speci®c recognition(vide infra).
To establish further the increased rise, additionaltests were carried out. The distance restraints,although already taken from short mixing timeNOESY spectra, underwent an additional correc-tion for spin diffusion in two ways and for eachnew structure re®nements were performed (seeMaterials and Methods). The resulting sets of
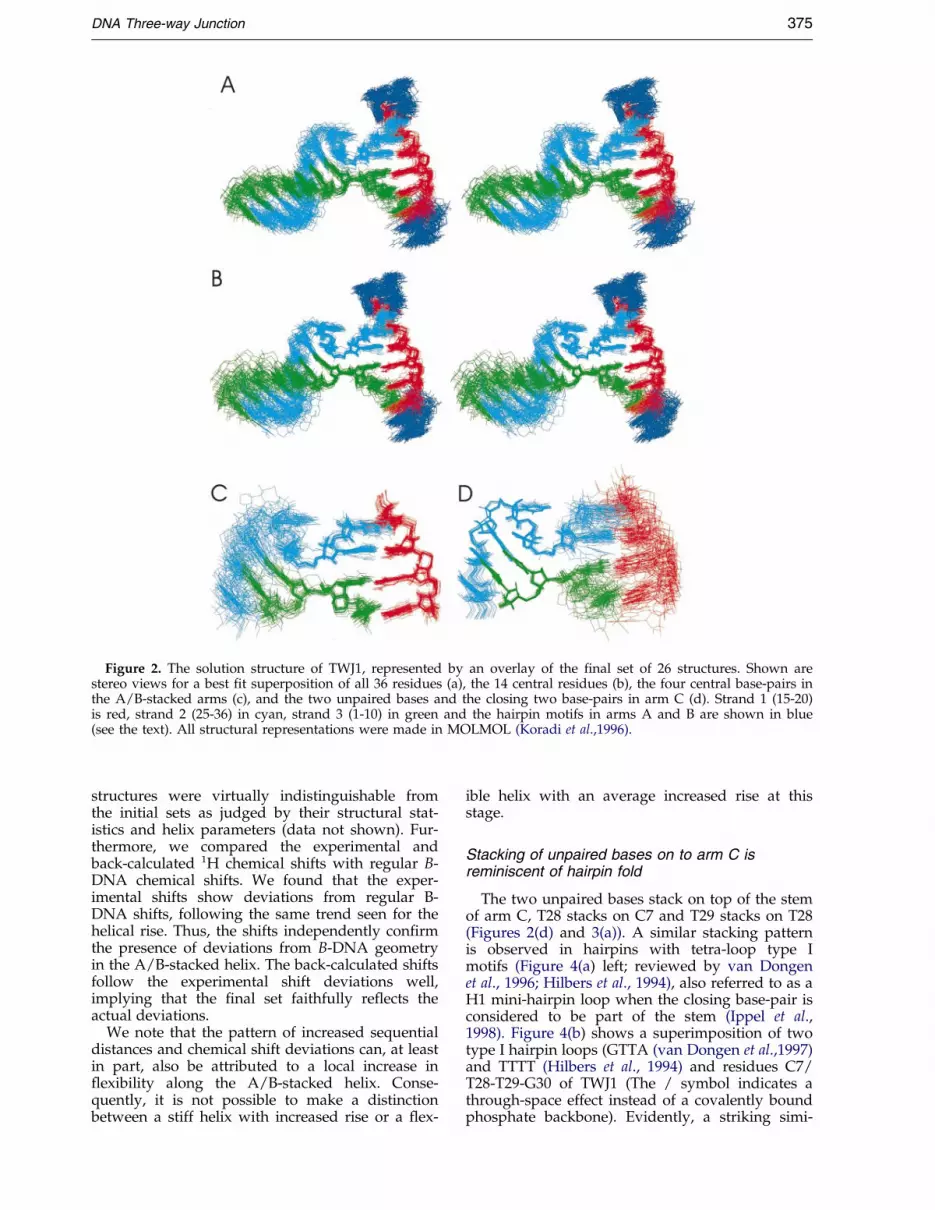
Figure 2. The solution structure of TWJ1, represented by an overlay of the ®nal set of 26 structures. Shown arestereo views for a best ®t superposition of all 36 residues (a), the 14 central residues (b), the four central base-pairs inthe A/B-stacked arms (c), and the two unpaired bases and the closing two base-pairs in arm C (d). Strand 1 (15-20)is red, strand 2 (25-36) in cyan, strand 3 (1-10) in green and the hairpin motifs in arms A and B are shown in blue(see the text). All structural representations were made in MOLMOL (Koradi et al.,1996).
DNA Three-way Junction 375
structures were virtually indistinguishable fromthe initial sets as judged by their structural stat-istics and helix parameters (data not shown). Fur-thermore, we compared the experimental andback-calculated 1H chemical shifts with regular B-DNA chemical shifts. We found that the exper-imental shifts show deviations from regular B-DNA shifts, following the same trend seen for thehelical rise. Thus, the shifts independently con®rmthe presence of deviations from B-DNA geometryin the A/B-stacked helix. The back-calculated shiftsfollow the experimental shift deviations well,implying that the ®nal set faithfully re¯ects theactual deviations.
We note that the pattern of increased sequentialdistances and chemical shift deviations can, at leastin part, also be attributed to a local increase in¯exibility along the A/B-stacked helix. Conse-quently, it is not possible to make a distinctionbetween a stiff helix with increased rise or a ¯ex-
ible helix with an average increased rise at thisstage.
Stacking of unpaired bases on to arm C isreminiscent of hairpin fold
The two unpaired bases stack on top of the stemof arm C, T28 stacks on C7 and T29 stacks on T28(Figures 2(d) and 3(a)). A similar stacking patternis observed in hairpins with tetra-loop type Imotifs (Figure 4(a) left; reviewed by van Dongenet al., 1996; Hilbers et al., 1994), also referred to as aH1 mini-hairpin loop when the closing base-pair isconsidered to be part of the stem (Ippel et al.,1998). Figure 4(b) shows a superimposition of twotype I hairpin loops (GTTA (van Dongen et al.,1997)and TTTT (Hilbers et al., 1994) and residues C7/T28-T29-G30 of TWJ1 (The / symbol indicates athrough-space effect instead of a covalently boundphosphate backbone). Evidently, a striking simi-

Figure 3. Comparison of the global fold of TWJ1 (a) and the Hammerhead ribozyme (Scott et al.,1996) (b). Thehelix arms are indicated with capital A, B, and C using the notation and de®nitions as described in the legend toFigure 1(a). In this notation, the hammerhead ribozyme has an A/B-stacked conformation with the arms in a parallelorientation. By contrast, TWJ1 has an A/B-stacked conformation with an anti-parallel orientation of the arms.
376 DNA Three-way Junction
larity exists between the two hairpin structuresand the fold of the unpaired bases on top of thelast base-pair of arm C. Even the characteristicsharp turn found in the backbone of DNA hairpinloops between L3 and L4 is present here (betweenT29 and G30 in TWJ1). Even though there is abreak in the 3H backbone between L1 (C7) and L2(T28), L2 is still virtually in the same position as inthe true hairpin situation. The reduced twist in theultimate base-pair step usually observed in CNNGhairpins is also found here (C7 �G30/A6 �T31,Table 2). On average, the helical rise and twistover the seven base-pairs in arm C fall within therange observed for standard B-DNA (Table 2), asdo the other helix parameters. Only the base-pairstep penultimate from the junction shows a slightly
Table 2. Helix inter-base-pair parametersa
Rise Twist
B-DNA 3.4 36armCb 3.7 � 0.3 34 � 4
C1 �G36-G2 �C35 3.5 � 0.1 36.9 � 0.3G2 �C35-T3 �A34 3.7 � 0.1 33.4 � 0.3T3 �A34-G4 �C33 3.5 � 0.1 37.2 � 0.5G4 �C33-C5 �G32 3.6 � 0.1 32.5 � 0.5C5 �G32-A6 �T31 4.2 � 0.2 37.4 � 0.8A6 �T31-C7 �G30 3.6 � 0.2 25.6 � 1.7
A/B stacked armsb 4.5 � 0.6 30 � 6G24 �C21-T25 �A20 3.6 � 0.1 22.2 � 0.7T25 �A20-C26 �G19 4.6 � 0.2 30.2 � 1.0C26 �G19-G27 �C18 4.6 � 0.2 36.3 � 1.3G27 �C18-C8 �G17 4.9 � 0.3 27.7 � 3.6C8 �G17-C9 �G16 4.9 � 0.2 39.1 � 2.0C9 �G16-G10 �C15 5.3 � 0.4 31.5 � 1.2G10 �C15-C11 �G14 3.5 � 0.1 23.0 � 0.6
a Average values and standard deviations for the ®nal set of26 structures.
b Average values and standard deviations for all base-pairsin the region.
increased rise. Thus, the stacking of unpaired basesonto arm C is reminiscent of the type I hairpinfold. This quasi-hairpin loop folding, together withthe well-known thermodynamic stabilities of truehairpin folds, has direct consequences for under-standing 3HS2 conformer stability (vide infra).
The conformation of the exchanging strands atthe junction
As with a 4H, a folded 3HS2 has two points ofstrand exchange. In TWJ1, the ®rst is found instrand 3 between C7 and C8 and the other betweenG27 and T28 in strand 2 (Figures 1(a), 2(c) and2(c)). At these points the two strands run anti-par-allel, and undergo a U-turn, i.e. the orientation ofthe ribose ring ¯ips 180 � going from C7 to C8 andfrom G27 to T28. The phosphate group betweenC7 and C8 points outwards and away from thephosphate group in the opposing strand, thusminimizing electrostatic repulsion. Between C7and C8 the turn is brought about by an increase ofeC7 to an average of 240 �, accompanied by a shiftof zC7 to the trans domain. In addition, in some ofthe structures gC8 is turned to the trans domain,although it mostly stays gauche(�). The turnbetween G27 and T28 is mostly affected by eG27,which is divided into distinct groups (gauche(-) andtrans), with no occurrence of gT28 in the transdomain. This is in accordance with the experimen-tal data, i.e. no unusual 31P chemical shifts occurand intra-nucleotide H6/8-H5'/5`` NOEs areabsent (see Materials and Methods). In the 4H crys-tal structure by (Ortiz-LombardõÂa et al., 1999) thecorresponding e angles are strictly con®ned to thegauche(-) domain. The phosphate groups at thepoints of strand exchange also point away fromeach other, similar to but more outspoken than inthe set of TWJ1 structures. The angle distributions

Figure 4. Comparison of theTWJ1 quasi-hairpin loop and hair-pin tetraloops. (a) Schematic of thethree distinct classes of foldingfound for tetraloops: type I (left),type II (middle) and type III (right)(Figure adapted from van Dongenet al. (1996); the tetra-loop is rep-resented as 50-L1-L2-L3-L4-30). InDNA, hairpin type I and II motifshave been found, whereas in RNAmotifs II and III are observed. Theclosing base pair (L1 �L4) has oftenbeen found to deviate from canoni-cal Watson-Crick (van Dongen et al.,1996; Hilbers et al., 1994). In type IIthe base of L2 folds back, eitherpartially or completely, into theminor groove. Type I and type IIIshow continuous stacking of thebases from the 50-side and the 30-side, respectively. (b) Superimposi-tion of the TWJ1 quasi-hairpin loop(C7/T28-T29-G30) onto two type Itetra-loops. GTTA hairpin loop ingreen (van Dongen et al.,1997),TTTT hairpin loop in blue (Hilberset al.,1994), and the TWJ1 loop inred. Note the close resemblancebetween the TWJ1 quasi-hairpinloop and the type I hairpin loops.
DNA Three-way Junction 377
found for TWJ1 may re¯ect the higher motionalfreedom of the backbone in solution. We ®nallynote that the inter-helix angle in TWJ1 (53 �) is lesssharp than in the 4H crystal structure (40 �). Thus,the turning phosphate (PC8), although still pointingto C9N4, is too far away (ca 6 to 7 AÊ ) to form thedirect hydrogen bond observed in the 4H crystalstructure.
Comparison with available 3H data
The 3HS2 (J3CC) reported by Rosen & Patel(1993b) prefers, like TWJ1, A/B-stacking. In J3CCthe fold of the 50-C/CCG-30 loop is reminiscent of atype II hairpin loop (Figure 4(a)). The only differ-ence between the fold of the loop sequence in J3CCand a true type II hairpin fold is that residue L2 isfound in the minor groove of arm B, as opposed tothe minor groove of arm C. In TWJ1 the loopresembles a type I hairpin fold and it is residue L3,which points towards the minor groove of arm B.
Leontis and coworkers reported a 3HS2 (TWJ-TT) that prefers the A/C stack (Leontis et al.,1993),despite the fact that it has the exact same basesequence at the junction as TWJ1. This indicatesthat the identity of the penultimate base-pair, andpossibly one further away, has a strong in¯uenceon stacking preference. In their latest re®nement
(Thiviyanathan et al., 1999), the last potentialWatson-Crick base-pair near the unpaired bases inthe unstacked arm B appears not to be formed. Inview of the observation of hairpin folds in TWJ1and J3CC, this is not surprising. The loop sequencein A/C-stacked TWJ-TT is 50-GTT/C-30 and it hasalready been shown that this leads to an unstablehairpin in which the closing G �C base-pair is dis-rupted (Ippel et al., 1995).
The best studied example of a RNA 3H is theHammerhead ribozyme, for which several X-raystructures have been published (Scott et al., 1996;Pley et al., 1994). Although it falls into another cat-egory of 3Hs, being a HS1HS4H (not consideringthe non-canonical base-pairs as bulge residues),comparison with the 3HS2 s is tempting. In thehammerhead, a tandem GA base-pair is incorpor-ated into the A/B-stacked helix. The unstackedarm B is topped by a large compact loop consistingof the one-residue bulge S1 (the cleaved cytidineresidue in the semi-continuous strand) and thefour-residue bulge (S4) of the CUGA sequence. Themost striking difference between TWJ1 and thehammerhead is the orientation of the unstackedarm with respect to the unique arm A (Figure 3).In TWJ1 this is anti-parallel, whereas it is parallelin the hammerhead. A magnesium-induced tran-sition from anti-parallel into parallel was found by

378 DNA Three-way Junction
Bassi et al. (1997) and was attributed from a chan-geover from an unde®ned loop to the well-struc-tured loop as observed in the X-ray structures.Geometric considerations suggest that the largecompact loop seen in the X-ray structure at highMg2� content forces the unstacked arm into theparallel orientation.
Sequence features governingconformer selection
The structural data of TWJ1, combined with thereported stacking behavior of 23 other 3HS2 s mol-ecules (Table 3), allow us to propose two newrules, which together grosso modo reproduce theexperimentally known conformational preferences:
the ``pyrimidine rule''the ``loop rule''The pyrimidine rule states that in a stacked con-
formation it is energetically advantageous when, inarm A, a pyrimidine (C or T) is located in thecrossover strand at the penultimate position. Inother words, when this pyrimidine is located instrand 3, A/B-stacking is preferred (Figure 1(a),left), whereas A/C-stacking is preferred when itresides in strand 1 (Figure 1(a), right). In terms ofthe classi®cation of 3HS2s (Altona, 1996), extendedto include the penultimate base-pairs, the notationfor the pyrimidine rule becomes simply (Table 3):second-row class RNN prefers A/B-stacking,whereas class YNN prefers A/C-stacking. All3HS2s of class RNN (examples 1-8) show the pre-dicted A/B-stacking, whereas those of class YNNyield either an outspoken major A/C-stacked form(examples 9-14) or a mixture of A/C and A/Bstacks (examples 15-22). This modulation of thepreference between pure and mixed forms can beexplained by the loop rule.
The loop rule states that a thermodynamicallystable (low free energy) quasi-hairpin loop in theA/B-stacking mode (50-L1/L2-L3-L4-30; Figure 4)will favor A/B-stacking, whereas a thermodyna-mically unstable one will not. A close resem-blance is seen between the quasi-hairpin foldand true hairpin folds (Figure 4). It seems there-fore reasonable to assume that the stability, or atleast the order of stabilities, of the quasi-hairpinloops in A/B-stacked 3HS2 s is the same as thatof true hairpin loops. The latter are well known(Hilbers et al., 1994; Ippel et al., 1998), i.e. hair-pin loops with sequence 50-Y1-N2-N3-R4-30 aremore stable than those with sequence 50-R1-N2-N3-Y4-30; in addition, loops with pyrimidines atpositions N2 and N3 are more stable than thecorresponding purine loops. This means, forexample, that the A/B-stacked conformation isstabilized when its quasi-hairpin loop is of type50-Y/N-N-R-30, whereas it is relatively destabi-lized when its quasi-hairpin loop is of the type50-R/N-N-Y-30. It seems likely that also in theA/C-stacked mode a quasi-hairpin loop occurs(see below). Note that for the A/C-stackedmode the break in the phosphate backbone
occurs on the 30-side of the quasi-hairpin loop(50-L1-L2-L3/L4-30), which may affect its stabilityrelative to that of loops of type (50-L1/L2-L3-L4-30). Nevertheless, we tentatively assume that theorder of stabilities of the former parallels that oftrue hairpins. However, the qualitative predic-tions on conformer selection made possible bythe new rules do not depend on the existence ornon-existence of a loop in the A/C-stackedforms. Below, we therefore consider for simpli-city only the presence of a loop in the A/B-stacking mode.
The two free energy terms postulated by the pyr-imidine and the loop rule can either enhance orcounteract each other. In the cases of A/B-stacking(Table 3, examples 1-8), both rules favor A/B-stacking, i.e. in the second row all are of classRNN and the quasi-hairpin loop in the A/B-stack-ing mode is most stable (type 50-Y/N-N-R-30). Thiswell explains the outspoken conformational purityof these A/B-stacked forms. For the next six3HS2 s (examples 9-14) the pyrimidine rule favorsA/C-stacking (class YNN) and the loop rule disfa-vors A/B-stacking (destabilizing loop of type 50-R/N-N-Y-30). Indeed, the A/C-stacking mode is pre-ferred in the presence of suf®cient Mg2�. Theremaining 3HS2 s (examples 15-22) are character-ized by con¯icting features: the pyrimidine rulefavors A/C-stacking (class YNN), whereas theloop rule favors A/B-stacking (A/B loop of type50-Y/N-N-R-30). All these 3HS2 s display, to amore or lesser degree, the predicted mixture ofconformers.
Could there be other factors that contribute toconformer selection or ®ne-tuning the free-energydifference between the A/B and A/C-stackedforms? As mentioned above, the presence orabsence of a loop in the A/C-stacking mode doesnot affect the qualitative predictions on conformerselection. However, it may well explain the moresubtle variations in stability among some of the3HS2 s. For example, the series of A/C-stackedjunctions (Table 3, junctions 9-14) exist as a mixtureof unfolded and predominantly A/C-stackedforms as shown by Welch et al. (1995). These junc-tions show a variation in stability as monitored bythe amount of Mg2� required for folding. This vari-ation is predicted when an A/C quasi-hairpin loopis assumed to be present (compare J1C2 versus theothers of this series). Variation in loop stability alsoexplains a subtle variation in stability in the A/B-stacked series; RPA2 with a loop 50-C/A-A-G-30requires more Mg2� to fold than its parent 3HS2(RPC2) with a more stable loop (50-C/C-C-G-30)(Table 3). Another factor expected to contribute isthe base-base stacking across the junction. How-ever, no clear correlation has been detected basedon stacking alone, see e.g. Welch et al. (1995) andOvermars et al. (1996). This is not surprising whenthe free energy of base stacking across the junction(maximum possible difference between A/B andA/C-stacked forms ca ÿ0.8 kcal/mol) is comparedwith that of loop folding (maximum difference
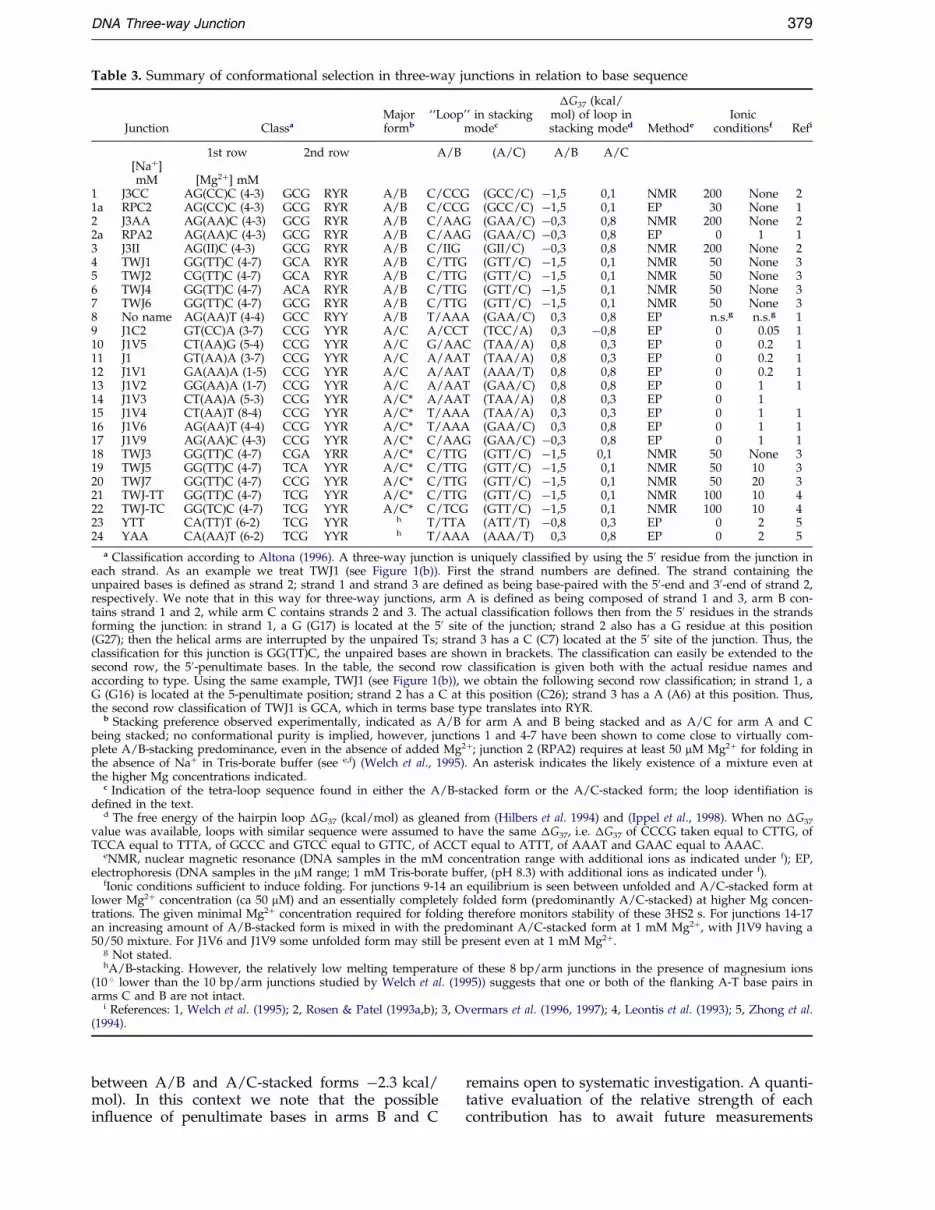
Table 3. Summary of conformational selection in three-way junctions in relation to base sequence
Junction ClassaMajorformb
``Loop'' in stackingmodec
�G37 (kcal/mol) of loop instacking moded Methode
Ionicconditionsf Refi
1st row 2nd row A/B (A/C) A/B A/C[Na�]mM [Mg2�] mM
1 J3CC AG(CC)C (4-3) GCG RYR A/B C/CCG (GCC/C) ÿ1,5 0,1 NMR 200 None 21a RPC2 AG(CC)C (4-3) GCG RYR A/B C/CCG (GCC/C) ÿ1,5 0,1 EP 30 None 12 J3AA AG(AA)C (4-3) GCG RYR A/B C/AAG (GAA/C) ÿ0,3 0,8 NMR 200 None 22a RPA2 AG(AA)C (4-3) GCG RYR A/B C/AAG (GAA/C) ÿ0,3 0,8 EP 0 1 13 J3II AG(II)C (4-3) GCG RYR A/B C/IIG (GII/C) ÿ0,3 0,8 NMR 200 None 24 TWJ1 GG(TT)C (4-7) GCA RYR A/B C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 None 35 TWJ2 CG(TT)C (4-7) GCA RYR A/B C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 None 36 TWJ4 GG(TT)C (4-7) ACA RYR A/B C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 None 37 TWJ6 GG(TT)C (4-7) GCG RYR A/B C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 None 38 No name AG(AA)T (4-4) GCC RYY A/B T/AAA (GAA/C) 0,3 0,8 EP n.s.g n.s.g 19 J1C2 GT(CC)A (3-7) CCG YYR A/C A/CCT (TCC/A) 0,3 ÿ0,8 EP 0 0.05 110 J1V5 CT(AA)G (5-4) CCG YYR A/C G/AAC (TAA/A) 0,8 0,3 EP 0 0.2 111 J1 GT(AA)A (3-7) CCG YYR A/C A/AAT (TAA/A) 0,8 0,3 EP 0 0.2 112 J1V1 GA(AA)A (1-5) CCG YYR A/C A/AAT (AAA/T) 0,8 0,8 EP 0 0.2 113 J1V2 GG(AA)A (1-7) CCG YYR A/C A/AAT (GAA/C) 0,8 0,8 EP 0 1 114 J1V3 CT(AA)A (5-3) CCG YYR A/C* A/AAT (TAA/A) 0,8 0,3 EP 0 115 J1V4 CT(AA)T (8-4) CCG YYR A/C* T/AAA (TAA/A) 0,3 0,3 EP 0 1 116 J1V6 AG(AA)T (4-4) CCG YYR A/C* T/AAA (GAA/C) 0,3 0,8 EP 0 1 117 J1V9 AG(AA)C (4-3) CCG YYR A/C* C/AAG (GAA/C) ÿ0,3 0,8 EP 0 1 118 TWJ3 GG(TT)C (4-7) CGA YRR A/C* C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 None 319 TWJ5 GG(TT)C (4-7) TCA YYR A/C* C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 10 320 TWJ7 GG(TT)C (4-7) CCG YYR A/C* C/TTG (GTT/C) ÿ1,5 0,1 NMR 50 20 321 TWJ-TT GG(TT)C (4-7) TCG YYR A/C* C/TTG (GTT/C) ÿ1,5 0,1 NMR 100 10 422 TWJ-TC GG(TC)C (4-7) TCG YYR A/C* C/TCG (GTT/C) ÿ1,5 0,1 NMR 100 10 423 YTT CA(TT)T (6-2) TCG YYR h T/TTA (ATT/T) ÿ0,8 0,3 EP 0 2 524 YAA CA(AA)T (6-2) TCG YYR h T/AAA (AAA/T) 0,3 0,8 EP 0 2 5
a Classi®cation according to Altona (1996). A three-way junction is uniquely classi®ed by using the 50 residue from the junction ineach strand. As an example we treat TWJ1 (see Figure 1(b)). First the strand numbers are de®ned. The strand containing theunpaired bases is de®ned as strand 2; strand 1 and strand 3 are de®ned as being base-paired with the 50-end and 30-end of strand 2,respectively. We note that in this way for three-way junctions, arm A is de®ned as being composed of strand 1 and 3, arm B con-tains strand 1 and 2, while arm C contains strands 2 and 3. The actual classi®cation follows then from the 50 residues in the strandsforming the junction: in strand 1, a G (G17) is located at the 50 site of the junction; strand 2 also has a G residue at this position(G27); then the helical arms are interrupted by the unpaired Ts; strand 3 has a C (C7) located at the 50 site of the junction. Thus, theclassi®cation for this junction is GG(TT)C, the unpaired bases are shown in brackets. The classi®cation can easily be extended to thesecond row, the 50-penultimate bases. In the table, the second row classi®cation is given both with the actual residue names andaccording to type. Using the same example, TWJ1 (see Figure 1(b)), we obtain the following second row classi®cation; in strand 1, aG (G16) is located at the 5-penultimate position; strand 2 has a C at this position (C26); strand 3 has a A (A6) at this position. Thus,the second row classi®cation of TWJ1 is GCA, which in terms base type translates into RYR.
b Stacking preference observed experimentally, indicated as A/B for arm A and B being stacked and as A/C for arm A and Cbeing stacked; no conformational purity is implied, however, junctions 1 and 4-7 have been shown to come close to virtually com-plete A/B-stacking predominance, even in the absence of added Mg2�; junction 2 (RPA2) requires at least 50 mM Mg2� for folding inthe absence of Na� in Tris-borate buffer (see e,f) (Welch et al., 1995). An asterisk indicates the likely existence of a mixture even atthe higher Mg concentrations indicated.
c Indication of the tetra-loop sequence found in either the A/B-stacked form or the A/C-stacked form; the loop identi®ation isde®ned in the text.
d The free energy of the hairpin loop �G37 (kcal/mol) as gleaned from (Hilbers et al. 1994) and (Ippel et al., 1998). When no �G37
value was available, loops with similar sequence were assumed to have the same �G37, i.e. �G37 of CCCG taken equal to CTTG, ofTCCA equal to TTTA, of GCCC and GTCC equal to GTTC, of ACCT equal to ATTT, of AAAT and GAAC equal to AAAC.
eNMR, nuclear magnetic resonance (DNA samples in the mM concentration range with additional ions as indicated under f); EP,electrophoresis (DNA samples in the mM range; 1 mM Tris-borate buffer, (pH 8.3) with additional ions as indicated under f).
fIonic conditions suf®cient to induce folding. For junctions 9-14 an equilibrium is seen between unfolded and A/C-stacked form atlower Mg2� concentration (ca 50 mM) and an essentially completely folded form (predominantly A/C-stacked) at higher Mg concen-trations. The given minimal Mg2� concentration required for folding therefore monitors stability of these 3HS2 s. For junctions 14-17an increasing amount of A/B-stacked form is mixed in with the predominant A/C-stacked form at 1 mM Mg2�, with J1V9 having a50/50 mixture. For J1V6 and J1V9 some unfolded form may still be present even at 1 mM Mg2�.
g Not stated.hA/B-stacking. However, the relatively low melting temperature of these 8 bp/arm junctions in the presence of magnesium ions
(10 � lower than the 10 bp/arm junctions studied by Welch et al. (1995)) suggests that one or both of the ¯anking A-T base pairs inarms C and B are not intact.
i References: 1, Welch et al. (1995); 2, Rosen & Patel (1993a,b); 3, Overmars et al. (1996, 1997); 4, Leontis et al. (1993); 5, Zhong et al.(1994).
DNA Three-way Junction 379
between A/B and A/C-stacked forms ÿ2.3 kcal/mol). In this context we note that the possiblein¯uence of penultimate bases in arms B and C
remains open to systematic investigation. A quanti-tative evaluation of the relative strength of eachcontribution has to await future measurements

380 DNA Three-way Junction
on the thermodynamics of selected three-wayjunctions.
What is the physical basis of the two rules for-mulated above? The loop rule has a ®rm foun-dation in the known relative stabilities of varioushairpin loops (Table 3); in general: 50-Y-Y-Y-R-30450-Y-R-R-R-30450-R-Y-Y-Y-30450-R-R-R-Y-30.This order in stabilities is explained well by stericstrain induced by the loop in the closing base-pair(van Dongen et al., 1996; Hilbers et al., 1994; Ippelet al., 1998). The physical origin of the pyrimidinerule seems less clear. In both known 4H crystalstructures (Ortiz-LombardõÂa et al., 1999; Eichmanet al., 2000) the turn in the crossing strand at thejunction is stabilized by a hydrogen bond betweenthe oxygen of the turning phosphate and the N4group of cytosine in the 30-penultimate position(the pyrimidine of the pyrimidine rule in the A/B-stacked mode). As already mentioned, this hydro-gen bond is at best only temporarily formed in thesolution structure of TWJ1. Moreover, Ortiz-Lom-bardia et al. (1999) point out that this interaction isbase speci®c and can also occur with adenine,which makes, it in the light of the pyrimidine rule(C or T at this position versus G or A), even moreunlikely that in aqueous solution this hydrogenbond plays a role in conformer selection. Theobservation of deviations in the helical rise in theA/B-stacked stem around the penultimate base-pair appears to indicate the presence of steric stressspread out over this region of the molecule. Wesuggest that the pyrimidine rule like the loop ruleoriginates from steric stress, such as could arisebetween the crossing phosphate and the down-stream penultimate base and which is (partly) alle-viated by the smaller steric size of a pyrimidinecompared to a purine base.
In conclusion, the present analysis shows thatthe two new rules (pyrimidine rule and loop rule),taken together, predict the conformer selection of3HS2 on a qualitative level, with the pyrimidinerule being more important. A more complete quan-titative analysis including other factors, such asbase stacking across the junction, will be publishedelsewhere.
Structure-specific recognition
HMG-box proteins bind to branched nucleic acidstructures with a higher level of af®nity than to thecorresponding ds-DNA sequences (PoÈhleret al.,1998; Bianchi, 1995). Comparison between thestructure of TWJ1 and that of the ds-DNA sub-strates in different HMG-DNA complexes gives arationale for this structure-speci®c recognition. Forexample, in the LEF-HMG complex (Love et al.,1995) the DNA is substantially underwound andhas an increased helical rise at the area of inter-action with the LEF-HMG. In TWJ1 we observe thesame characteristics, which makes it a ready-madetarget for HMG-box proteins and modeling showsthat the LEF-HMG directly ®ts on TWJ1. The 3HS2structure thus provides a plausible mechanism for
the structure-speci®c recognition of branched DNAspecies by HMG-domain proteins.
Materials and Methods
DNA synthesis, sample preparation, NMR spec-troscopy and resonance assignment were carried out asdescribed (Overmars et al.,1996). An additional 2D 2H2
NOESY build-up series was recorded on a BrukerDMX600 spectrometer with mixing times of 30, 50, 80,120 and 200 ms. These spectra were recorded in a sol-ution containing 50 mM NaCl buffered at pH 6.5, with aTWJ1 concentration of ca 3 mM as described (Overmarset al., 1996). The 2D NOESY data sets were acquiredwith a 4808 Hz spectral width in both dimensions, 4 Kcomplex data points in the t2 dimension and 400 incre-ments in the t1 dimension. Spectra were analyzed usingTriad NMR software from Tripos Inc., running on aSilicon Graphics workstation.
Structure restraints
Residues not belonging to the 14-residue central coreof TWJ1 were restrained for standard B-helix geometryor CTTG hairpin geometry (Ippel et al., 1998). For thecentral 14 residues (6-9, 16-19, 26-31, Figure1(b)) exper-imental restraints were employed. The distance restraintswere derived from the 30 ms and/or 50 ms 600MHzNOESY spectra in 2H2O using the Isolated Spin PairApproach (ISPA) by calibrating against all known dis-tances (Wijmenga & van Buuren, 1998). This calibrationshowed that distances could be determined with a highdegree of accuracy of 5 %, 8 %, 17 % for the intra-sugar,intra-nucleotide, inter-nucleotide distances, respectively.Nevertheless, conservative error bounds (Wijmenga &van Buuren, 1998) were used in the calculations. Atintermediate stages of the structure calculation, NOEsabsent in the NOESY spectra were converted into dis-tance restraints with a lower bound of 7 AÊ (non-NOEs),to improve success rate and precision. The w angles wereall restrained on the basis of diagnostic intra-residueH10/H20/H20 0-base distances (Wijmenga & van Buuren,1998). The ribose sugar rings were all found to be essen-tially S-type (70 % to 100 %) as follows from the charac-teristic intra-residue sugar-sugar and sugar-basedistances (Wijmenga et al.,1993; Wijmenga & vanBuuren, 1998). They were restrained via the d angle andthe endocylic torsion angles. The e angles for residues ina helical environment were restrained to 202(�30)�,because no unusual phosphorus shifts were detectedexcept in the CTTG loops (Gorenstein, 1984; Wijmenga &van Buuren, 1998). The remaining e angles wererestrained to 225(�60)� on the basis of steric factors(Wijmenga & van Buuren, 1998). All other torsion angleswere left unrestrained. Base-pairing was preserved byexplicit restraints on the length of the hydrogen bonds(Wijmenga & van Buuren, 1998), and by weak planarityrestraints (weight factor 2.0 kcal molÿ1 AÊ ÿ2) involving allbase atoms. In subsequent further re®nements on the 26lowest energy structures (vide infra) additional restraintswere imposed. The a and z were restrained to ÿ47(�50)�and ÿ95(�50)�, respectively, for residue in helicalenvironment because of the normal phosphorus chemicalshifts. Torsion angles g were restrained to 36(�50)� forthese residues because they showed no strong intra-residue NOE H50/H50 0 to H6/H8 contacts. Although thedistance estimates had conservative error bounds andwere obtained from NOESY spectra with short mixing

DNA Three-way Junction 381
times (30 and 50 ms), additional controls for spin diffu-sion were performed by deriving distances either viaMardigras (Borgias & James,1990) or via spin-diffusioncorrection on ISPA (Wijmenga & van Buuren, 1998).These distances were used in the re®nements again withconservative error bounds (�30 %). A summary of theused restraints is given in Table 1.
Structure calculation
A set of 20 starting structures was generated with ran-dom conformation for the central 14 nucleotides andstandard geometries for the helical stems and CTTGloops. From this set, 100 structures were generated viaX-PLOR 3.851 by means of a torsion angle dynamics(TAD) protocol designed for nucleic acids (Stein et al.,1997). The structural parts not belonging to the core of14 central residues were treated as ®xed groups in theTAD protocol. Only holonomic restraints (bond anglesetc.), and the usual simpli®ed force-®eld containing onlysoft-sphere repulse van der Waals constraints were used,together with the experimental restraints. This preventsbias from uncertain force-®eld terms, for example termsrepresenting electrostatic interactions. The accuracy ofthe structures was assessed by back-calculation of timedomain NOESY spectra, using in-house software andCORMA (Keepers & James, 1984), and by comparison ofback-calculated 1H chemical shifts (Wijmenga et al., 1997;Wijmenga & van Buuren, 1998) with the experimentalshifts. Of the set of 100 structures 60 % converged to oneenergy minimum, as judged from a statistical analysisbelow. The structures were ordered according to theiroverall energy. The lowest energy structures form anapproximate plateau. The mean overall energy and stan-dard deviation of the complete set was then calculatedand structures with an energy higher than two times thestandard deviation from the mean were removed. Thisprocess was repeated until a self-consistent set of struc-tures with a statistically similar lowest energy. Of thisset of 60 structures, the 26 lowest energy structures wereselected on the basis of overall energy, and analyzed forhelix parameters and inconsistencies with the experimen-tal spectra. Unusual angles for a, z and g were found forwhich no experimental evidence was present. Inaddition, the helical rise in the A/B-stacked helix wasfound to deviate from normal B-DNA. These deviationscorrelated with the obtained ISPA distance restraintsobtained from NOESY spectra with short mixing times.To remove any possible effect of spin diffusion, twodifferent sets of spin-diffusion corrected distances werederived (structure restraints). The set of 26 structureswent then through ®nal re®nements with the additionaltorsional restraints and with either the ISPA distance set,the modi®ed ISPA distance set, or the Mardigras dis-tance set. The sets of structures derived this way wereindistinguishable from one another by their structuralstatistics. The 26 structures re®ned with the Mardigrasderived restraints were selected for presentation andtheir statistics are listed in Table 1. Helical parametersfor this set of structures were calculated using CURVES5.1 (Lavery & Sklenar, 1988), and are listed in Table 2.
Coordinates
The coordinates for the set of ®nal 26 structurestogether with a full list of constraints in Xplor have beendeposited in the RCSB Data Bank, PDB-ID code is 1EZN.
Acknowledgments
This work was supported by grants from the SwedishNational Research Council, the Bioteknik Medel UmeaÊUniversity (S.W.). We thank Janny Hof and Anje Valken-burg for critical reading of the manuscript.
References
Altona, C. (1996). Classi®cation of nucleic acid junctions.J. Mol. Biol. 263, 568-581.
Altona, C., Pikkemaat, J. A. & Overmars, F. J. J. (1996).Three-way and four-way junctions in DNA: A con-formational viewpoint. Curr. Opin. Struct. Biol. 6,305-316.
Ban, N., Nissen, P., Hansen, J., Moore, P. B. & Steitz,T. A. (2000). The complete atomic structure of thelarge ribosomal subunit at 2.4 AÊ resolution. Science,289, 905-920.
Bassi, G. S., Murchie, A. I. H., Walter, F., Clegg, R. M. &Lilley, D. M. J. (1997). Ion-induced folding of thehammerhead ribozyme: a ¯uorescence resonanceenergy transfer study. EMBO J. 16, 7481-7489.
Bianchi, M. E. (1995). The HMG-box domain. In DNA-Protein: Structural Interactions (Lilley, D. M. J., ed.),p. 177, IRL Press, Oxford.
Borgias, B. A. & James, T. L. (1990). MARDIGRAS.J. Magn. Reson. 87, 475-487.
Cate, J. H., Gooding, A. R., Podell, E., Zhou, K., Golden,B. L., Kundrot, C. E., Cech, T. R. & Doudna, J. A.(1996). Crystal structure of a group I ribozymedomain: principles of RNA packing. Science, 273,1678-1685.
Eichman, B. N., Vargason, J. M., Mooers, B. H. M. &Ho, P. S. (2000). The Holliday junction in aninverted repeat DNA sequence: sequence effects onthe structure of four-way junctions. Proc. Natl Acad.Sci. USA, 97, 3971-3976.
Gonzalez, C., Rullmann, J. A. C., Bonvin, A. M. J. J.,Boelens, R. & Kaptein, R. (1991). Toward an NMRR factor. J. Magn. Reson. 91, 659-664.
Gorenstein, D. G. (1984). Phosphorus-31 NMR: Principlesand Applications, Academic Press, New York.
Guthrie, C. & Patterson, B. (1988). SpliceosomalsnRNAs. Annu. Rev. Genet. 22, 387-419.
Hilbers, C. W., Heus, H. A., van Dongen, M. J. P. &Wijmenga, S. S. (1994). The hairpin elements ofnucleic acid structure: DNA and RNA folding. InNucleic Acids and Molecular Biology, pp. 56-104,Springer, Berlin.
Ippel, J. H., Lanzotti, V., Galeone, A., Mayol, L., vanden Boogaart, J. E., Pikkemaat, J. A. & Altona, C.(1995). Thermodynamics of melting of the circulardumbbell d < pCGC-TT-GCG-TT>. Biopolymers, 36,701-710.
Ippel, J. H., Van den Elst, H., Van der Marel, G. A., VanBoom, J. H. & Altona, C. (1998). Structural simi-larities and differences between H1- and H2- familyDNA minihairpin loops: NMR studies of octamericminihairpins. Biopolymers, 46, 375-393.
Jensch, F. & Kemper, B. (1986). Endonuclease VIIresolves Y-junctions in branched DNAs in vitro.EMBO J. 5, 181-189.
Keepers, J. W. & James, T. L. (1984). A theoretical studyof distance determinations from NMR. Two-dimen-sional nuclear Overhauser effect spectra. J. Magn.Reson. 57, 404-426.

382 DNA Three-way Junction
Koradi, R., Billeter, M. & WuÈ thrich, K. (1996).MOLMOL: a program for display and analysis ofmacromolecular structures. J. Mol. Graph. 14, 51-55.
Kotin, R. M., Linden, R. M. & Berns, K. I. (1992).Characterization of a preferred site on humanchromosome 19q for integration of adeno-associatedvirus DNA by non-homologous recombination.EMBO J. 11, 5071-5078.
Lavery, R. & Sklenar, V. (1988). The de®nition of gener-alized helicoidal parameters and of axis curvaturefor irregular nucleic acids. J. Biomol. Struct. Dynam.6, 63-91.
Leontis, N. B., Kwok, W. & Newman, J. S. (1991). Stab-ility and structure of three-way DNA junctions con-taining unpaired nucleotides. Nucl. Acids Res. 19,759-766.
Leontis, N. B., Hills, M. T., Piotto, M., Malhotra, A.,Nussbaum, J. & Gorenstein, D. G. (1993). A modelfor the solution structure of a branched, three-strand DNA complex. J. Biomol. Struct. Dynam. 11,215-223.
Lilley, D. M. J. & Clegg, R. M. (1993). The structure ofbranched DNA species. Quart. Rev. Biophys. 22, 299-328.
Lilley, D. M. J. & Norman, D. G. (1999). The Hollidayjunction is ®nally seen with crystal clarity. NatureStruct. Biol. 6, 897-899.
Lilley, D. M. J., Clegg, R. M., Diekmann, S., Seeman,N. C., von Kitzing, E. & Hagerman, P. J. (1995). Anomenclature of junctions and branchpoints innucleic acids. Nucleic Acids Res. 23, 3363-3364.
Love, J. J., Li, X., Case, D. A., Giese, K., Grosschedl, R.& Wright, P. E. (1995). Structural basis for DNAbending by the architectural transcription factorLEF-1. Nature, 376, 791-795.
Minagawa, T., Murakami, A., Ryo, Y. & Yamagishi, H.(1983). Structural features of very fast sedimentingDNA formed by gene 49 defective T4. Virology, 126,183-189.
Noller, H. F. (1984). Structure of ribosomal RNA. Annu.Rev. Biochem. 53, 119-162.
Noller, H. F. (1991). Ribosomal RNA and translation.Annu. Rev. Biochem. 60, 191-227.
Orbons, L. P., Van der Marel, G. A., Van Boom, J. H. &Altona, C. (1986). Hairpin and duplex formation ofthe DNA octamer d(m5C-G-m5C-G-T-G-m5C-G) insolution. An NMR study. Nucleic. Acids. Res. 14,4187-4196.
Ortiz-LombardõÂa, M., GonzaÂlez, A., Eritja, R., AymamõÂ,J., AzorõÂn, F. & Coll, M. (1999). Crystal structure ofa DNA Holliday junction. Nature Struct. Biol. 6, 913-917.
Overmars, F. J. J. (1997). Conformational Aspects ofBranched DNA; NMR Studies of Three-way and Four-way Junctions, Leiden University, The Netherlands.
Overmars, F. J. J., Pikkemaat, J. A., Van den Elst, H.,Van Boom, J. H. & Altona, C. (1996). NMR studiesof DNA three-way junctions containing twounpaired thymidine bases: the in¯uence of thesequence at the junction on the stability of thestacking conformers. J. Mol. Biol. 255, 702-713.
Pley, H. W., Flaherty, K. M. & McKay, D. B. (1994).Three-dimensional structure of a hammerhead ribo-zyme. Nature, 372, 68-74.
PoÈhler, J. R. G., Norman, D. G., Bramham, J., Bianchi,M. E. & Lilley, D. M. J. (1998). HMG box proteinsbind to four-way DNA junctions in their open con-formation. EMBO J. 17, 817-826.
Ren, J., Qu, X., Chaires, J. B., Trempe, J. P., Dignam, S. S.& Dignam, J. D. (1999). Spectral and physicalcharacterization of the inverted repeat DNA struc-ture from adenoassociated virus 2. Nucleic AcidsRes. 27, 1985-1990.
Rettberg, C. C., Prere, M. F., Gesteland, R. F., Atkins,J. F. & Fayet, O. (1999). A three-way junction andconstituent stem-loops as the stimulator for pro-grammed -1 frameshifting in bacterial insertionsequence IS911. J. Mol. Biol. 286, 1365-1378.
Rosche, W. A., Trinh, T. Q. & Sinden, R. R. (1995).Differential DNA secondary structure-mediated del-etion mutation in the leading and the laggingstrands. J. Bacteriol. 177, 4385-4391.
Rosen, M. A. & Patel, D. J. (1993a). Conformationaldifferences between bulged pyrimidines (C-C) andpurines (A-A, I-I) at the branch point of three-stranded DNA junctions. Biochemistry, 32, 6563-6575.
Rosen, M. A. & Patel, D. J. (1993b). Structural featuresof a three-stranded DNA junction containing a C-Cjunctional bulge. Biochemistry, 32, 6576-6587.
Scott, W. G., Murray, J. B., Arnold, J. R. P., Stoddard,B. L. & Klug, A. (1996). Capturing the structure of acatalytic RNA intermediate: the hammerhead ribo-zyme. Science, 274, 2065-2069.
Seeman, N. C. (1996). The design and engineering ofnucleic acid nanoscale assemblies. Curr. Opin.Struct. Biol. 6, 519-526.
Stein, E. G., Rice, L. M. & Brunger, A. T. (1997).Torsion-angle molecular dynamics as a new ef®-cient tool for NMR structure calculation. J. Magn.Reson. 124, 154-164.
StuÈ hmeier, F., Welch, J. B., Murchie, A. I. H., Lilley,D. M. J. & Clegg, R. M. (1997a). Global structure ofthree-way DNA junctions with and withoutadditional unpaired bases: a ¯uorescence resonanceenergy transfer analysis. Biochemistry, 36, 13530-13538.
StuÈ hmeier, F., Lilley, D. M. J. & Clegg, R. M. (1997b).Effect of additional unpaired bases on the stabilityof three- way DNA junctions studied by ¯uor-escence techniques. Biochemistry, 36, 13539-13551.
Thiviyanathan, V., Luxon, B. A., Leontis, N. B.,Illangasekare, N., Donne, D. G. & Gorenstein, D. G.(1999). Hybrid-hybrid matrix structural re®nementof a DNA three-way junction from 3D NOESY-NOESY. J. Biomol. NMR, 14, 209-221.
van Dongen, M. J. P., Wijmenga, S. S., Van der Marel,G. A., Van Boom, J. H. & Hilbers, C. W. (1996). Thetransition from a neutral-pH double helix to a low-pH triple helix induces a conformational switch inthe CCCG tetraloop closing a Watson-Crick stem.J. Mol. Biol. 263, 715-729.
van Dongen, M. J. P., Mooren, M. M. W., Willems,E. F. A., Van der Marel, G. A., Van Boom, J. H.,Wijmenga, S. S. & Hilbers, C. W. (1997). Structuralfeatures of the DNA hairpin d(ATCCTA-GTTA-TAGGAT): formation of a G-A base pair in theloop. Nucl. Acids Res. 25, 1537-1547.
Welch, J. B., Walter, F. & Lilley, D. M. J. (1995). Twoinequivalent folding isomers of the three-way DNAjunction with unpaired bases: sequence dependenceof the folded conformation. J. Mol. Biol. 251, 507-519.
Wijmenga, S. S. & van Buuren, B. N. M. (1998). The useof NMR methods for conformational studies ofnucleic acids. Prog. NMR. Spec. 32, 287-387.

DNA Three-way Junction 383
Wijmenga, S. S., Mooren, M. M. & Hilbers, C. W. (1993).NMR of nucleic acids; from spectrum to structure.In NMR of Macromolecules, a Practical Approach(Roberts, G. C. K., ed.), p. 217, Oxford UniversityPress, New York.
Wijmenga, S. S., Kruithof, M. & Hilbers, C. W. (1997).Analysis of 1H chemical shifts in DNA: assessment
of the reliability of 1H chemical shift calculations foruse in structure re®nement. J. Biomol. NMR, 10, 337-350.
Zhong, M., Rashes, M. S., Leontis, N. B. & Kallenbach,N. R. (1994). Effects of unpaired bases on the con-formation and stability of three-arm DNA junctions.Biochemistry, 33, 3660-3667.
Edited by I. Timoco
(Received 24 July 2000; received in revised form 5 October 2000; accepted 10 October 2000)






![Sequence dependence of benzo[a]pyrene diol epoxide-DNA adduct conformer distribution: A study by laser-induced fluorescence/polyacrylamide gel electrophoresis](https://img.dokumen.tips/doc/110x75/6333eb0de9e768a27a0fce1d/sequence-dependence-of-benzoapyrene-diol-epoxide-dna-adduct-conformer-distribution.jpg)






















