Embed Size (px)
Citation preview

Scope and limitations of the designer proline-richantibacterial peptide dimer, A3-APO, alone or insynergy with conventional antibiotics
Marco Cassone a, Paraskevi Vogiatzi a, Raffaele La Montagna a, Vanessa De Olivier Inacio b,Predrag Cudic b, John D. Wade c, Laszlo Otvos Jr.a,*aSbarro Institute for Cancer Research and Molecular Medicine, Temple University, Philadelphia, PA 19122, United StatesbDepartment of Chemistry and Biochemistry, Florida Atlantic University, Boca Raton, FL 33431, United StatescHoward Florey Institute and Department of Chemistry, University of Melbourne, Victoria 3010, Australia
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 6
a r t i c l e i n f o
Article history:
Received 30 June 2008
Received in revised form
22 July 2008
Accepted 22 July 2008
Published on line 5 August 2008
Keywords:
A3-APO
Synergy
Antimicrobial peptide
DnaK
a b s t r a c t
The proline-rich antimicrobial peptide dimer, A3-APO, was designed based on a statistical
analysis of native antibacterial peptide and protein sequences. Analysis of a series of
structural analogs failed to identify any single or multiple amino acid modification or
architectural changes that would significantly improve its potential as a clinical therapeutic.
However, a single chain Chex1-Arg20 version, a natural in vivo metabolite, showed a 2 to 8-
fold increase in activity against test Enterobacteriaceae strains. In addition to bacterial species
close to Escherichia coli in phylogeny, A3-APO analogs were able to effectively kill Pseudomonas
aeruginosa and Staphylococcus saprophyticus. Antibacterial efficacy analysis together with
biochemical experiments provided further evidence for a multiple mode of action of A3-
APO that includes binding and inhibition of the bacterial heat shock protein DnaK. Through
inactivating of resistance enzymes, A3-APO was able to recover the lost activity of con-
ventional antibiotics including chloramphenicol, b-lactams, sulfonamides or trimethoprim
against multidrug resistant strains with partial or full synergy. However, the synergy
appeared to be individual strain and small molecule drug combination-dependent.
# 2008 Elsevier Inc. All rights reserved.
avai lable at www.sc iencedi rec t .com
journal homepage: www.e lsev ier .com/ locate /pept ides
1. Introduction
The antimicrobial drug industry is not keeping in pace with
the overwhelming appearance and circulation of pathogens
causing known or novel infectious syndromes. Perhaps the
most worrisome event is the worldwide spread of antibiotic
resistance in hospitals and the community alike [17,28].
Without discounting the efficacy of preventive measures,
there is an impelling need to develop new antimicrobial
molecules, and to use antimicrobial combinations capable of
exerting synergistic activities. Antimicrobial peptides, which
* Corresponding author at: BioLife Sciences Building, 1900 North 12th Sfax: +1 215 204 4021.
E-mail address: [email protected] (L. Otvos Jr.).
0196-9781/$ – see front matter # 2008 Elsevier Inc. All rights reserveddoi:10.1016/j.peptides.2008.07.016
are largely diffused in nature, are a promising emerging class
of anti-infective drugs. Among those, proline-rich peptides are
unique since they have a very specific mechanism of action,
allowing structure–activity relationships (SAR) studies [21]. In
general, antimicrobial peptides carry little potential to induce
resistance [11], however their systemic use is frequently
hampered by unacceptable pharmacokinetic parameters and
low safety margins [5].
Peptide A3-APO is a flagship representative of a new class of
synthetic peptide antimicrobials derived from natural insect
products [22]. A3-APO selectively binds to the multihelical lid
treet, Philadelphia, PA 19122, United States. Tel.: +1 215 204 4020;
.

p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 6 1879
region of the 70-kDa bacterial heat shock protein DnaK [21] and
inhibits DnaK functions including protein refolding [15]. A
combination of DnaK binding and bacterial membrane disin-
tegration leads to effective killing of genetically related
Enterobacteriaceae such as Escherichia coli, Klebsiella pneumoniae,
Salmonella typhimurium and Haemophilus influenzae [14]. The
membrane-onlyactivitiesof theproline-rich dimersare enough
to combat Pseudomonas aeruginosa strains, albeit at higher
concentrations than needed to kill bacteria with complemen-
tary DnaK sequences [4]. The specific role of several specific
residues as well as some structural aspects of this peptide
warrant further investigation, and this was the first aim of the
current study.
The Asp2-Lys3 dipeptide and Tyr6-Leu-Pro-Arg-Pro10
pentapeptide fragments are crucial for antibacterial activity
[17]. A combination of protein folding and membrane stability
assays indicate that while the role of the positively charged
Lys3 is likely to initiate attachment to the negatively charged
bacterial membrane surface, the prolines serve as structural
constrains, and Asp2, Tyr6, Leu7 and Arg9 are involved in
binding to the DnaK protein of E. coli and related bacteria [4].
These seven residues are conserved in the designer dimer A3-
APO and are positioned as Asp4-Lys5 and Tyr9-Leu-Pro-Arg-
Pro13. The two extra N-terminal residues and the C-terminal
half of the peptide are thought to partially destroy bacterial
membranes and drive the DnaK-inactivating pharmacophore
into cells [22]. SAR of the designer dimer family indicate that in
two of three inactive analogs, the Tyr-Leu-Pro-Arg-Pro
pentapeptide is altered; in one Ile is substituted for Leu, and
in another Lys is substituted for Arg [23]. This suggests that
killing of susceptible strains requires the intact DnaK-binding
domain. However, one of the inactive analogs featuring an
Arg! Lys change and additional modifications at the carboxy-
terminus, does bind a fluorescein-labeled E. coliDnaK D-E helix
preparation suggesting that perhaps alterations in the C-
terminal delivery unit (compared to A3-APO) are responsible
for the lack of antibacterial activity. We wanted to test this
hypothesis experimentally. Examination of the C-terminal
delivery halves indicate that the active peptides are compact
and lack residues that do not promote entry into cells.
Positively charged residues, especially Arg [25] together with
Pro are known to facilitate peptide entry into cells [9]. Yet, the
frequent appearance of His (a residue both positively charged
and hydrophobic) in native proline-rich antibacterial
sequences such as apidaecins, warrant the synthesis and
testing of a few His-analogs in the place of Arg. While the
function of residues N-terminal to the YLPRP active site other
than the Asp-Lys dipeptide fragment is unclear [4], it is
tempting to speculate that the improved antibacterial activity
of A3-APO compared to some analogs is due to its cell-entry
focused N-terminal heptapeptide fragment. In support,
another pyrrhocoricin-apidaecin construct, based on apidae-
cin 1a without the N-terminal Lys-Val dipeptide fragment fails
to kill the test bacterial strains. The argument presented above
questions the need for the near-C-terminal Val, the only
residue remaining in A3-APO without DnaK-binding or cell-
penetrating functions. Likewise, we are not fully convinced
that the N-terminal Chex residue or the C-terminal Dab are the
best amino acid mimics, although they show favorable
efficacy and toxicity properties. Additionally, we not pre-
viously considered reversing the N-terminal DnaK-binding—
C-terminal delivery architecture or using single chain (non-
dimeric) peptides but with the sequence optimizations
included.
The second aim of this study was to test the potential of A3-
APO as a synergic agent in combination with antimicrobials
currently on the market and to which resistance is increas-
ingly observed. In a preliminary report we documented how
preincubation of b-lactamase-expressing E. coli strains with
the designer antimicrobial peptide dimer, A3-APO, is able to
reinstall the lost activity of amoxicillin against these b-lactam
resistant bacteria [20]. The inhibition of protein refolding
mediated by A3-APO through DnaK binding is the basis for a
possible restoration of the efficacy of antibiotics that are
inactivated by specific enzymes. Since inhibition of DnaK
impairs the function of many enzymes, A3-APO could be the
first molecule able to be synergic in a broad range of
combinations, and also to be effective just when it is most
needed and other combinations fail, i.e. in the presence of
highly active, antibiotic-cleaving or diverting enzymes
responsible for high-level resistance. Encouraged by the
success of our preliminary results with E. coli and b-lactam
antibiotics, in the current study we expanded the investiga-
tions to other Enterobacteriaceae and different small molecule
antibiotics that are impaired by increasingly diffuse resis-
tance-causing enzymes.
2. Materials and methods
2.1. Peptide synthesis
The peptides were synthesized by solid-phase methods.
Peptide chain assembly was carried out on a CEM Liberty
microwave-assisted automated synthesizer using TentaGel S-
Ram-Fmoc resin with an initial load of 0.3 mmol/g (Advanced
ChemTech). Standard Fmoc-chemistry was used throughout
[10] with a 4-fold molar excess of the acylating amino acids.
Non-natural amino acids were coupled manually to ensure
completion. The peptides were cleaved from the solid support
with trifluoroacetic acid (TFA) in the presence of thioanisole
(5%), and water (5%) as scavengers. After cleavage, the
peptides were purified by reversed-phase high performance
liquid chromatography (RP-HPLC). The final products were
characterized by RP-HPLC and matrix-assisted laser deso-
rption/ionization mass spectroscopy (MALDI-MS). Mass spec-
tra (PerSeptive Biosystems, Voyager DE instrument) identified
correct and highly purified samples.
2.2. Bacterial strains
For the study we used bacteria belonging to the species E. coli
(HK101, HK179, HK131, BF1023, SOTE40, 5770, S5081, S4362,
SEQ102, and 045-849), K. pneumoniae (HK123, HK186, HK127,
012-3132, MH4, RP1 and K6), S. typhimurium (ATCC 14028, S5
and G10215), P. aeruginosa (ATCC 39329 and 10), H. influenzae
(ATCC 49247 and R387), Haemophilus ducreyi 51620, Bulkholderia
cepacia 8Q, Proteus vulgaris ATCC 6896, Proteus mirabilis ATCC
7002, Staphylococcus aureus 27660, Staphylococcus saprophyticus
ATCC 15305, and Enterococcus faecalis JWL. Each strain was

p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 61880
grown to mid-exponential phase in an incubator at 37 8C and
5% CO2, and in the case of E. coli, the flasks were agitated at
200 rpm. For Haemophilus strains, hemin and nicotinamide
adenine dinucleotide (NAD) were added at the concentration
of 15 mg/L each. All assays involving live bacteria were
performed in a BSL2 environment.
2.3. Minimal inhibitory concentration (MIC)determination assays
MICs were determined in duplicate by a liquid growth
inhibition microdilution assay in sterile polypropylene 96-
well plates (Nunc F96 microtiter plates), with a final volume
of 100 mL [1]. The cell concentrations were estimated by
measuring the ultraviolet absorbance at 595 nm and apply-
ing the formula CFU/mL = A595 (3.8 � 108), where CFU is the
number of colony-forming units. Briefly, 50 mL of a suspen-
sion of midlogarithmic phase bacterial cultures diluted to
5 � 105 cfu/mL in 1/4 strength Mueller–Hinton broth was
added to 50 mL of 2-fold serially diluted peptides, dissolved in
the same medium. For S. saprophyticus, additional MICs were
acquired in full strength broth. The final peptide concentra-
tions ranged between 0.12 and 128 mg/mL, (approximately
0.03–32 mM for the full-length peptides and 0.06–64 mM for
the two single chain peptides (A3 single chain and A3 Inverse
single chain; Table 1). For each strain a well with no peptide
was included as growth control, and for each test a row of
medium-only wells was included as a sterility control. The
plates were then incubated at 37 8C, 5% CO2 for 16–20 h
without shaking, and growth inhibition was measured by
recording the absorbance at 595 nm using a microplate
reader. MICs were identified as the lowest antimicrobial
concentrations where the 595 nm absorbance value did not
exceed that of the medium only.
2.4. Synergism assays
Two different assays were employed to study the presence of
synergism between A3-APO and conventional antibiotics in
Table 1 – Sequences of synthetic peptides used in this study
A3-APO (H-Chex-Arg-Pro-Asp-Lys-Pro-Arg
Glu4A3 (H-Chex-Arg-Pro-Glu-Lys-Pro-Arg-
Arg5A3 (H-Chex-Arg-Pro-Asp-Arg-Pro-Arg
Gly11A3 (H-Chex-Arg-Pro-Asp-Lys-Pro-Arg
Lys12A3 (H-Chex-Arg-Pro-Asp-Lys-Pro-Arg
A3-desVal (H-Chex-Arg-Pro-Asp-Lys-Pro-Arg
Arg2A4 (H-Pip-Arg-Pro-Glu-Arg-Pro-Arg-Pr
His2A4 (H-Pip-His-Pro-Glu-Arg-Pro-Arg-Pro
A3 Inverse (H-Chex-Arg-Pro-Arg-Pro-Pro-Arg-
A3 Inverse single chain H-Chex-Arg-Pro-Arg-Pro-Pro-Arg-P
A3 Single chain H-Chex-Arg-Pro-Asp-Lys-Pro-Arg-
Chex-pyrr-Dap dimer (H-Chex-Asp-Lys-Gly-Ser-Tyr-Leu
Residues in italics indicate the amino acid changes compared to the seq
bacteria harboring antibiotic-disrupting or antibiotic-insensi-
tive enzymes. With the first method, bacteria were preincu-
bated with A3-APO in full-strength Mueller–Hinton broth at
concentrations of 1/2 or 1/4 of its MIC and the conventional
antibiotics were serially diluted in the entire concentration
range [11]. Preincubated bacteria where then added and the
OD at 600 nm was measured after 16–20 h. The second method
we used was the conventional checkerboard assay [8]. Bacteria
grown to mid-logarithmic phase in 1/4 strength Mueller–
Hinton broth were preincubated with serially diluted con-
centrations of A3-APO and the conventional antibiotics were
added in a similar 2-fold dilution pattern. The occurrence and
extent of synergy were calculated based on the sum of the MIC
ratios for each compound alone and in the presence of the
other at half MIC (FIC score). Synergy was defined by a FIC
score below 0.5. The small molecule antibiotics included
amoxicillin, chloramphenicol, sulfamethoxazole or the
trimethoprim/sulfamethoxazole (TMP/SXT) combination as
resistance to all these is manifested through the action of
specific enzymes. Those E. coli, S. typhimurium and K.
pneumoniae strains were selected for which the involvement
of enzymatic activity was known or suggested by the
resistance profile. A few tests were also undertaken on
bacteria lacking specific antibiotic-resistance enzymes, in
which synergy was not expected.
2.5. b-galactosidase assay
The inhibition of the enzyme-refolding functions of DnaK by
peptide A3-APO was confirmed by measuring the b-
galactosidase activity of live cells. To this end, a modified
ortho-nitrophenol (ONP)-substrate method was employed
according to manufacturer’s instructions (b-galactosidase
assay, Stratagene, Cedar Creek, TX). The test strain here
was K. pneumoniae K6 harvested after 1 h of exposure to
A3-APO alone, chloramphenicol or TMP/SXT alone or the
small molecule antibiotics together with peptide A3-APO.
All the antibiotics were used at a concentration 1/4 of
their MIC.
-Pro-Tyr-Leu-Pro-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg)2-Dab
Pro-Tyr-Leu-Pro-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg)2-Dab-NH2
-Pro-Tyr-Leu-Pro-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg)2-Dab-NH2
-Pro-Tyr-Leu-Gly-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg)2-Dab-NH2
-Pro-Tyr-Leu-Pro-Lys-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg)2-Dab-NH2
-Pro-Tyr-Leu-Pro-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Arg)2-Dab
o-Tyr-Leu-Pro-Lys-Pro-Arg-Pro-Pro-Arg-Pro-Arg)2-Dab-NH2
-Tyr-Leu-Pro-Lys-Pro-Arg-Pro-Pro-Arg-Pro-Arg)2-Dab-NH2
Pro-Val-Arg)2-Dab-Asp-Lys-Pro-Arg-Pro-Tyr-Leu-Pro-Arg-Pro-NH2
ro-Val-Arg-Dab-Asp-Lys-Pro-Arg-Pro-Tyr-Leu-Pro-Arg-Pro-NH2
Pro-Tyr-Leu-Pro-Arg-Pro-Arg-Pro-Pro-Arg-Pro-Val-Arg-NH2
-Pro-Arg-Pro-Thr-Pro-Pro-Arg-Pro-Ile-Tyr-Asn-Arg)2-Dap(DapAc)-NH2
uence of A3-APO in the non-architecture-modified analogs.
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 6 1881
2.6. DnaK binding assay
To identify which A3-APO analogs bind DnaK, a dot-blot assay
was used. Ten micrograms of each peptide was immobilized
on a nitrocellulose membrane, which was blocked by incuba-
tion in phosphate-buffered saline (PBS) - 0.1% Tween20
mixture with 5% bovine serum albumin (BSA) for 2 h at room
temperature. Fifty micrograms of DnaK (Stressgen, Victoria,
British Columbia) were added in 5 mL PBS - 0.1% Tween20 for
1 h. Bound DnaK protein was detected after overnight
incubation with a bacterial DnaK-specific mouse monoclonal
antibody diluted 1/10,000 (clone 8E2/2, Stressgen) at 4 8C
overnight, followed by development of the membrane with
horseradish peroxidase (HRP)-conjugated anti-mouse IgG
antibody (dilution 1:20,000, Pierce, Rockford, Illinois).
3. Results
3.1. Analog and assay design
The new analogs (Table 1) include single residue conservative
changes in the active sites (Glu4A3, Arg5A3, Gly11A3,
Lys12A3), elimination of the valine (A3-desVal), multiple
amino acid substitutions including pipecolic acid at the amino
terminus (Arg2A4, His2A4) and a single chain analog (A3 Single
chain). In the last set of new derivatives we included a reverse
design with a single or dual N-terminal membrane penetrating
and a single C-terminal pharmacophore orientation (A3
Inverse single chain, A3 Inverse). This design places Pro to
the C-terminus. The antimicrobial efficacies of these analogs
were compared with the parent peptide A3-APO and an earlier
dimer that shows less attractive antibacterial properties [6].
A3-APO kills bacteria in full-strength Mueller–Hinton broth,
Table 2 – Minimal inhibitory concentrations of peptide A3-APOthe structural analogues A3 single chain and A3-desVal againinhibition assay in quarter strength Mueller–Hinton broth at 3
Bacterial strain
A3-APO Arg5A3 Glu4A3
E. coli HK101 2 2 2
E. coli HK179 2 2 2
E. coli SOTE40 8 8 8
E. coli 5770 2 2 2
E. coli HK131 4 4 8
E. coli SEQ102 2 2 2
E. coli 045-849 4 2 4
K. pneumoniae HK 123 4 4 8
K. pneumoniae HK 186 8 4 8
K. pneumoniae HK 127 8 8 8
K. pneumoniae 012-3132 2 2 2
K. pneumoniae K6 4 4 8
S. typhimurium ATCC 14028 8 4 8
S. typhimurium S5 8 8 8
S. typhimurium G10215 4 4 4
P. aeruginosa ATCC 39329 8 4 8
P. aeruginosa 10 8 8 8
S. saprophyticus ATCC 15305 2 2 2
but earlier analogs do not [22]. To be able to discriminate
between the efficacies of the new peptides and draw
conclusions on the relative efficacy of analogs, in this report
all antimicrobial assays were conducted in 1/4 strength
Mueller–Hinton broth.
3.2. Antibacterial activity and DnaK binding of peptidescontaining single residue conservative changes
Peptide Arg5A3 has a conservative mutation at the hypothe-
tical membrane-active residue Lys, Gly11A3 features a non-
charged substitution at the DnaK-binding domain and Glu4A3
as well as Lys12A3 involve conservative and charged sub-
stitutions in the fragments interacting with DnaK (Table 1). As
Table 2 shows, A3-APO is indeed a close-to-optimal dimer in
killing Enterobacteriaceae. All but one (Gly11A3) of the new
derivatives with charged substitutions were about equally
potent as the parent designer peptide. Glu4A3 and Lys12A3
showed a slightly decreased activity in three strains each, but
still within the expected experimental variability range.
Arg5A3 exhibited marginally improved activity against 3 of
the 15 E. coli, K. pneumoniae, and S. typhimurium strains studied.
Although this minor improvement can be expected due to the
stronger positive charge of Arg compared to Lys and the
generally observable superiority of Arg in cell and nuclear
penetration-enhancement [2], the MIC differences were in the
experimental error range, so no clear-cut conclusion can be
drawn. Since the Glu4A3 and Lys12A3 analogs showed no
major loss in the antibacterial activity, it is likely that Asp4 and
Arg12 in A3-APO interacts with DnaK through ionic forces. In
contrast, a major drop in the antibacterial activity was
observed for Gly11A3 that features a Pro! Gly substitution,
in spite of both residues being reverse-turn formers [13]. This
derivative reproducibly killed all 15 E. coli, K. pneumoniae, and S.
, of its conservative amino acid replacement analogues andst selected strains measured in an overnight liquid growth7 8C
MIC (mg/mL)
Lys12A3 Gly11A3 A3 single chain A3-desVal
2 4 0.5 2
2 4 1 2
8 32 2 2
2 4 0.5 1
8 32 1 4
2 4 1 2
4 8 2 2
8 32 2 16
8 32 0.25 4
8 32 0.25 2
2 8 0.25 2
4 16 2 4
8 16 1 4
8 16 0.5 4
4 8 0.25 2
4 8 32 8
4 8 32 4
2 2 4 2

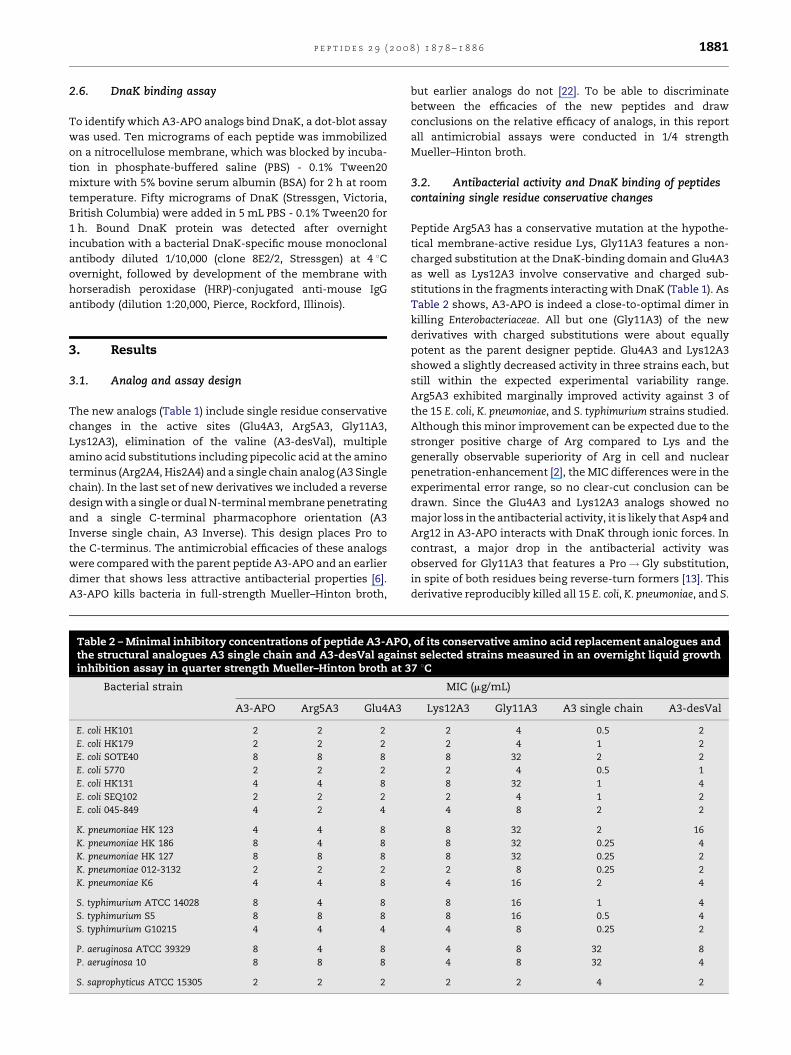
Fig. 1 – Binding of A3-APO, conservative change analogs
and control peptides to recombinant Escherichia coli DnaK.
The peptides were dried on a nitrocellulose sheet and
bound DnaK was developed with an N-terminal specific
antibody. The peptides used are from top to bottom—left
column: G11A3, K12A3, R5A3, E4A3; right column: A3-
APO, magainin II, scrambled Rb2 fragment, blank.
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 61882
typhimurium strains 2 to 8-fold less efficiently than the other 4
analogs.
To confirm the specificity of the above mentioned
substitutions to DnaK interaction, we investigated whether
the conservative changes affect efficacy against bacteria
whose DnaK is not a known target of native pyrrhocoricin
or other proline-rich peptides. Here the test strains included P.
aeruginosa, B. cepacia, Proteus vulgaris, P. mirabilis, H. influenzae
and H. ducreyi as Gram-negatives as well as S. aureus, S.
saprophyticus and E. faecalis as Gram-positives. Since in these
experiments DnaK-binding did not play a role, all five
peptides, including the Enterobacteriaceae less active Gly11A3,
showed almost identical antibacterial efficacies. Among the
named species, P. aeruginosa and S. saprophiticus were
susceptible to the peptides (Table 2), while the others were
not (MIC > 32 mg/mL) (data not shown). We documented
earlier that modified proline-rich peptides kill P. aeruginosa
through membrane disintegration [4], while a study is
undergoing to characterize the mechanism of action towards
S. saprophyticus.
To correlate the retention or loss of antibacterial activity
with DnaK interactions, we studied the binding of A3-APO and
the single residue conservative change derivatives to DnaK
protein on dot-blot. The negative control peptides were the
only-membrane active antibacterial peptide magainin II and a
scrambled Rb2 protein fragment that similarly to the
antibacterial peptides of this study is positively charged and
of the same overall length [2]. The results fully supported our
hypothesis: peptides K12A3, R5A3, G11A3 and E4A3 bound to
DnaK just like A3-APO did. Magainin II and the scrambled Rb2
fragment did not bind at all (Fig. 1). Although dot-blot is a
rather qualitative assay, the less active analog G11A3
exhibited significantly weaker DnaK binding. These assays
provided further proof for the notion that DnaK inhibition is
one of the mechanisms by which the proline-rich native
peptides and designer analogs kill bacteria [22].
3.3. Antimicrobial activity of A3-APO derivativesfeaturing structural modifications
This group of analogs included the valine-eliminated deriva-
tive, the single-chain Chex1-Arg20 analog, the reversed DnaK-
binding—delivery unit architecture in single chain and
dimeric version and two multiply modified A3-APO analogs.
These latter contained the Glu4, Arg5, Lys12 alterations that
were previously shown not to negatively influence the
antibacterial efficacy, and pipecolic acid instead of 1-amino-
cyclohexane carboxylic acid at the N-terminus. One of them
also contained a histidine for an N-terminal arginine. Chex is a
cyclic valine mimic and was introduced into the early proline-
rich dimers to replace the native Val and stabilize the peptides
from premature aminopeptidase cleavage in body fluids [6]. In
pipecolic acid the free amino group required for maximum
activity [14] is placed inside the hydrocarbon ring, endowing
the secondary amine with a stronger positive charge than the
primary amine in Chex. Clearly, the lack of a primary amine
deteriorated the antimicrobial activity. The Pip-containing
peptides were also inactive (MIC > 32 mg/mL) against both E.
coli SEQ102 and S. typhimurium S5, strains that are efficiently
killed by A3-APO and the other primary amine-containing
analogs (Table 2). Likewise, the N-terminal DnaK-binding and
C-terminal membrane penetrating-unit architecture cannot
be changed. The inverse derivatives A3 inverse and A3 inverse
single chain remained completely inactive (MIC > 32) against
both E. coli and K. pneumoniae, the two most sensitive bacterial
species to native proline-rich peptides and their designer
analogs. Arg2A4 and His2A4 exhibited MIC values of 32 and
>64 against E. coli SEQ102, and >64 for K. pneumoniae K6 and S.
typhimurium S5.
Against expectations, the elimination of the C-terminal
valine did not influence the antimicrobial efficacy in most of
the cases (Table 2). A3-desVal showed a comparable, and in
several cases slightly better activity in all but one the tested
strains. This observation confirms that Arg and Pro are indeed
the responsible residues for cellular penetration. A3-desVal
and A3-APO were 2–4 times more active than the first
generation Chex-pyrr-Dap dimer against the strains sensitive
to this control peptide; the Chex-pyrr-Dap dimer control was
inactive against representative E. coli, K. pneumoniae and S.
typhimurium strains that are killed by A3-APO by a median MIC
of 4 mg/mL.
Even more surprisingly, the most active peptide of this
study was the Chex1-Arg20 A3 single chain analog. This
peptide consistently exhibited lower MIC values compared to
A3-APO against the test phylogenetically related E. coli, K.
pneumoniae and S. typhimurium strains. The A3 Single chain
peptide is the most active antimicrobial peptide against these
Enterobacteriaceae we developed during our antibacterial
peptide drug development program. Interestingly, the oppo-
site was true for the Pseudomonas and S. saprophyticus strains,
which are killed by A3-APO but display a DnaK sequence that
is significantly different from that of E. coli, Klebsiella and
Salmonella. Since staphylococci are halophiles, for S. saprophy-
ticus we also run the MICs of A3-APO and the A3 Single chain
peptide in full-strength broth, to have a comparison under
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 6 1883
conditions allowing the fastest possible growth of the bacteria.
In this case, the difference between the MIC for A3-APO (8 mg/
mL) and the A3 Single chain (>64 mg/mL) was even more
pronounced. The data obtained for Pseudomonas and S.
saprophyticus show that while the dimeric and presumably
increasingly membrane-penetrating A3-APO retains activity
on some bacteria independently of DnaK specificity, this is not
the case for the otherwise more active, but less membrane
destroying A3 Single chain derivative.
3.4. Synergy with small molecule antibiotics
We tested the synergistic activity of A3-APO in combination
with amoxicillin, chloramphenicol, sulfamethoxazole or TMP/
SXT on various E. coli, S. typhimurium, K. pneumoniae and H.
influenzae strains. The culprit resistance-carrying enzymes for
these bacteria are b-lactamase (penicillins and cephalospor-
ins), chloramphenicol acetyltransferase, dihydrofolate reduc-
tase (TMP), and tetrahydropteroic synthetase (sulfonamides).
We performed two sets of assay involving the same strains
and antibiotics but using two different techniques.
In the first set of assays, we expanded the same assay design
as previously described [20] that is we used A3-APO in fixed
concentration at 1/2 or 1/4 of its MIC, and the small molecule
antibiotics in the full concentration range from 0.5 to 64 mg/mL.
During these experiments, preincubation of bacteria with A3-
APO lowered the MIC of amoxicillin against H. influenzae R387,
but did not help the b-lactam againstK. pneumoniaeK6 or RP1, or
S. typhimurium G10215. Likewise, preincubation with A3-APO
could not restore any efficacy of sulfonamide against E. coli
S5081 or TMP against E. coli S4362. Partial synergy was observed
for chloramphenicolagainstH. influenzaeR387. In this series, the
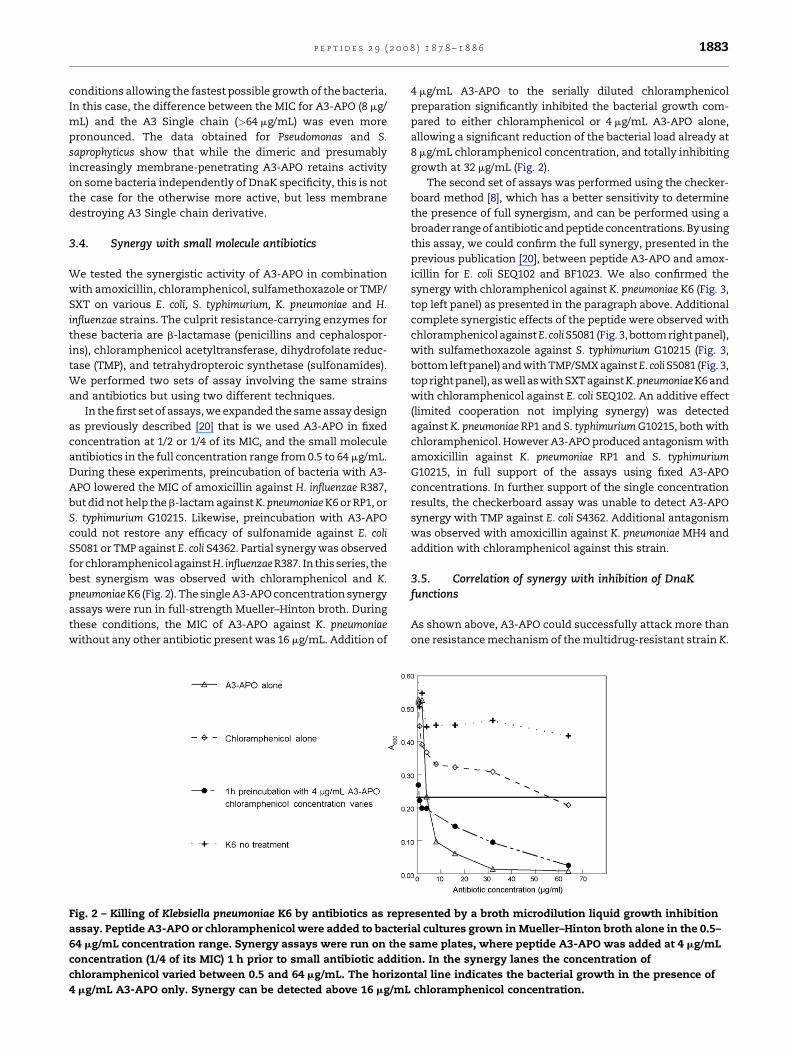
best synergism was observed with chloramphenicol and K.
pneumoniaeK6 (Fig. 2). The single A3-APO concentration synergy
assays were run in full-strength Mueller–Hinton broth. During
these conditions, the MIC of A3-APO against K. pneumoniae
without any other antibiotic present was 16 mg/mL. Addition of
Fig. 2 – Killing of Klebsiella pneumoniae K6 by antibiotics as repr
assay. Peptide A3-APO or chloramphenicol were added to bacter
64 mg/mL concentration range. Synergy assays were run on the
concentration (1/4 of its MIC) 1 h prior to small antibiotic additi
chloramphenicol varied between 0.5 and 64 mg/mL. The horizon
4 mg/mL A3-APO only. Synergy can be detected above 16 mg/mL
4 mg/mL A3-APO to the serially diluted chloramphenicol
preparation significantly inhibited the bacterial growth com-
pared to either chloramphenicol or 4 mg/mL A3-APO alone,
allowing a significant reduction of the bacterial load already at
8 mg/mL chloramphenicol concentration, and totally inhibiting
growth at 32 mg/mL (Fig. 2).
The second set of assays was performed using the checker-
board method [8], which has a better sensitivity to determine
the presence of full synergism, and can be performed using a
broader range of antibiotic and peptide concentrations. Byusing
this assay, we could confirm the full synergy, presented in the
previous publication [20], between peptide A3-APO and amox-
icillin for E. coli SEQ102 and BF1023. We also confirmed the
synergy with chloramphenicol against K. pneumoniae K6 (Fig. 3,
top left panel) as presented in the paragraph above. Additional
complete synergistic effects of the peptide were observed with
chloramphenicol againstE. coliS5081 (Fig. 3, bottom right panel),
with sulfamethoxazole against S. typhimurium G10215 (Fig. 3,
bottom left panel) and with TMP/SMX againstE. coliS5081 (Fig. 3,
top right panel), aswell aswith SXT againstK.pneumoniaeK6and
with chloramphenicol against E. coli SEQ102. An additive effect
(limited cooperation not implying synergy) was detected
against K. pneumoniae RP1 and S. typhimurium G10215, both with
chloramphenicol. However A3-APO produced antagonism with
amoxicillin against K. pneumoniae RP1 and S. typhimurium
G10215, in full support of the assays using fixed A3-APO
concentrations. In further support of the single concentration
results, the checkerboard assay was unable to detect A3-APO
synergy with TMP against E. coli S4362. Additional antagonism
was observed with amoxicillin against K. pneumoniae MH4 and
addition with chloramphenicol against this strain.
3.5. Correlation of synergy with inhibition of DnaKfunctions
As shown above, A3-APO could successfully attack more than
one resistance mechanism of the multidrug-resistant strain K.
esented by a broth microdilution liquid growth inhibition
ial cultures grown in Mueller–Hinton broth alone in the 0.5–
same plates, where peptide A3-APO was added at 4 mg/mL
on. In the synergy lanes the concentration of
tal line indicates the bacterial growth in the presence of
chloramphenicol concentration.
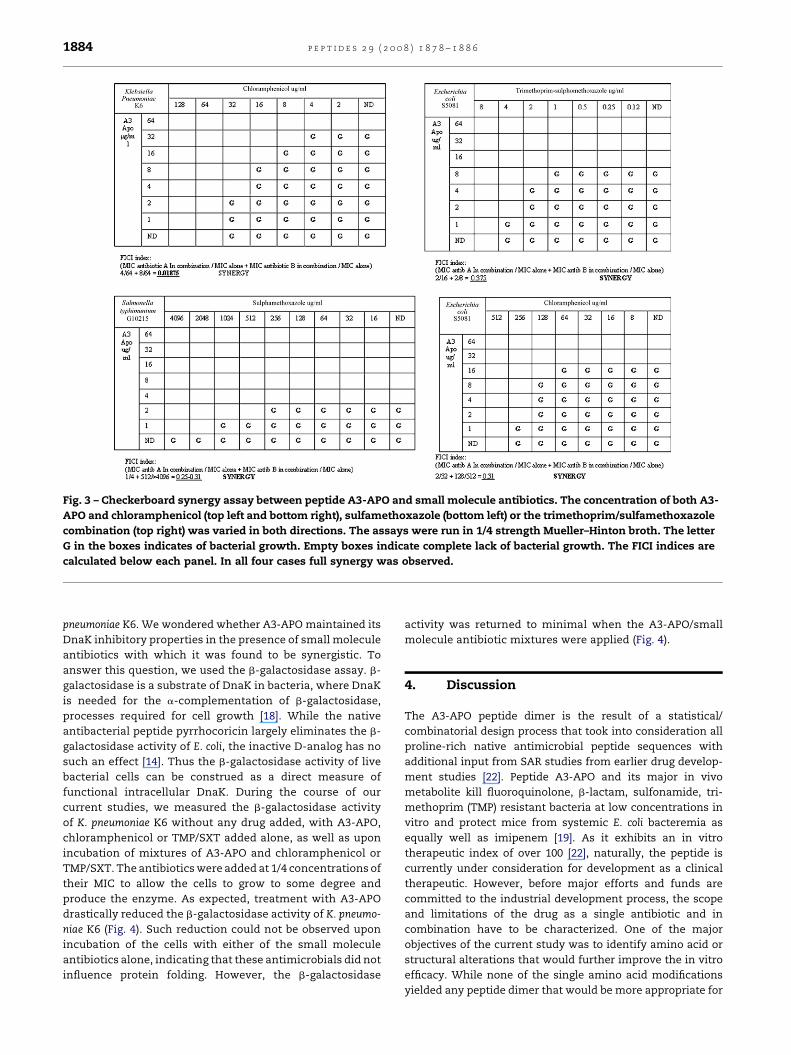
Fig. 3 – Checkerboard synergy assay between peptide A3-APO and small molecule antibiotics. The concentration of both A3-
APO and chloramphenicol (top left and bottom right), sulfamethoxazole (bottom left) or the trimethoprim/sulfamethoxazole
combination (top right) was varied in both directions. The assays were run in 1/4 strength Mueller–Hinton broth. The letter
G in the boxes indicates of bacterial growth. Empty boxes indicate complete lack of bacterial growth. The FICI indices are
calculated below each panel. In all four cases full synergy was observed.
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 61884
pneumoniae K6. We wondered whether A3-APO maintained its
DnaK inhibitory properties in the presence of small molecule
antibiotics with which it was found to be synergistic. To
answer this question, we used the b-galactosidase assay. b-
galactosidase is a substrate of DnaK in bacteria, where DnaK
is needed for the a-complementation of b-galactosidase,
processes required for cell growth [18]. While the native
antibacterial peptide pyrrhocoricin largely eliminates the b-
galactosidase activity of E. coli, the inactive D-analog has no
such an effect [14]. Thus the b-galactosidase activity of live
bacterial cells can be construed as a direct measure of
functional intracellular DnaK. During the course of our
current studies, we measured the b-galactosidase activity
of K. pneumoniae K6 without any drug added, with A3-APO,
chloramphenicol or TMP/SXT added alone, as well as upon
incubation of mixtures of A3-APO and chloramphenicol or
TMP/SXT. The antibiotics were added at 1/4 concentrations of
their MIC to allow the cells to grow to some degree and
produce the enzyme. As expected, treatment with A3-APO
drastically reduced the b-galactosidase activity of K. pneumo-
niae K6 (Fig. 4). Such reduction could not be observed upon
incubation of the cells with either of the small molecule
antibiotics alone, indicating that these antimicrobials did not
influence protein folding. However, the b-galactosidase
activity was returned to minimal when the A3-APO/small
molecule antibiotic mixtures were applied (Fig. 4).
4. Discussion
The A3-APO peptide dimer is the result of a statistical/
combinatorial design process that took into consideration all
proline-rich native antimicrobial peptide sequences with
additional input from SAR studies from earlier drug develop-
ment studies [22]. Peptide A3-APO and its major in vivo
metabolite kill fluoroquinolone, b-lactam, sulfonamide, tri-
methoprim (TMP) resistant bacteria at low concentrations in
vitro and protect mice from systemic E. coli bacteremia as
equally well as imipenem [19]. As it exhibits an in vitro
therapeutic index of over 100 [22], naturally, the peptide is
currently under consideration for development as a clinical
therapeutic. However, before major efforts and funds are
committed to the industrial development process, the scope
and limitations of the drug as a single antibiotic and in
combination have to be characterized. One of the major
objectives of the current study was to identify amino acid or
structural alterations that would further improve the in vitro
efficacy. While none of the single amino acid modifications
yielded any peptide dimer that would be more appropriate for

Fig. 4 – Inhibition of b-galactosidase activity of live K.
pneumoniae K6 cells. The enzymatic activity of the culture
was measured as described in Section 2. While A3-APO
eliminates the enzymatic activity either alone or in
combination with small molecule antibiotics, neither
chloramphenicol (CHF) nor the trimethoprim/
sulfamethoxazole combination (TMP/SXT) alone
influences the cells’ ability to produce active enzyme.
p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 6 1885
clinical development than A3-APO, some interesting data
emerged from the panel of structurally modified analogues.
Quite surprisingly, one half of A3-APO, the Chex1-Arg20 A3
Single chain derivative showed an overall improved activity
spectrum against test Enterobacteriaceae. The dimeric structure
was earlier identified as necessary in native antimicrobial
peptides for maximum interaction with bacterial membranes,
with the non-natural diaminobutyric acid scaffold providing
stability against carboxypeptidase cleavage [6]. Apparently,
after primary sequence optimization from native peptides to
the designer A3-APO, the dimeric construct is no longer
necessary, at least for efficacy. Notably, we recently deter-
mined that Chex1-Arg20 fragment is one of the major in vivo
metabolites of peptide A3-APO [19]. In addition to the
microbiological improvements, the shorter version is more
stable in blood ex vivo than the prodrug and is easier to mass-
produce.
It is an intriguing question why peptide A3-APO and its
conservative change analogs killed S. saprophyticus. The
antibacterial activity spectrum of native proline-rich peptides
such as drosocin, correlate with the E. coli similarity of the
DnaK D-E helix sequences of sensitive bacteria [3]. The
sequence of S. saprophyticus DnaK D-E helix region [14] has
low similarity to that of E. coli (48%), just like the non-sensitive
S. aureus [21]. Nevertheless, a few Gram-positive strains are
killed by the native peptides pyrrhocoricin and drosocin at
moderate concentrations, including Bacillus megaterium (44%
homology to E. coli DnaK D-E helix) and Micrococcus luteus [12].
A3-APO showed a broader spectrum of activity compared to
the A3 Single chain peptide, which seems to be restricted to
the specific DnaK sequence of E. coli and other Enterobacter-
iaceae whose DnaKs display a high similarity to E. coli. As
shown in Table 2, while the A3 Single chain peptide was
superior to A3-APO in E. coli, Klebsiella and Salmonella, the
opposite was true for Pseudomonas and S. saprophyticus. From
our data it can be thus hypothesized that while the A3 Single
chain Chex1-Arg20 derivative is able to penetrate the bacterial
membrane and specifically bind DnaK, the dimer A3-APO is
able not only to penetrate, but also to damage the membrane
to such an extent that it retains killing activity even towards
some strains in which the DnaK binding does not play a role.
On the other hand, the inhibition of DnaK may be more
effective in the A3 Single chain peptide, potentially because of
steric restrictions towards the dimer.
S. saprophyticus is the leading pathogen in cystitis in young
women [24]. Regardless of the mode of action, the known
efficacy of proline-rich peptide dimers in animal models of
urinary tract infections (UTI) [7], offers A3-APO or its dimeric
analogs as viable prospective alternatives to current anti-
microbials for the treatment of UTI in a clinical setting.
In view of A3-APO’s potential in inhibiting antibiotic-
inactivating enzymes such as b-lactamases [11], we also
studied the synergistic activity of A3-APO and a series of
antibiotics against which the resistance mechanism involves
production of drug-inactivating enzymes. Synergy between
two antibiotics is defined by a 10-fold decrease of viable
bacterial count using the antibiotics in combination as
opposed to each drug alone [16]. This effect provides an
additional therapeutic choice by allowing the use of conven-
tional antibiotics against multi-drug resistant bacteria, for
which there may currently be no other drug available [27]. The
classical synergic combinations, such as aminoglycoside
antibiotics with b-lactams [26], are however ineffective when
resistance is due to the presence of a modified target or
antibiotic-cleaving enzymes.
We could show A3-APO synergy with each of the tested
antibiotics and against all four bacterial species when the
bacteria were preincubated with the antibacterial peptide
prior to addition of the small molecules to inhibit protein
folding. Importantly, in some multi-resistant strains we were
able to retrieve multiple synergic effects. The synergy with
small molecule antibiotics was a generally observable phe-
nomenon, although the effect is individual strain and small
molecule drug combination-dependent. A possible explana-
tion may reside in the different structural stability of different
enzymes, so that the protein refolding activity of DnaK may be
less important for some enzymes. Also, resistance factors
other than properly folded enzymes or compositional differ-
ences among different strains of the same species may be
responsible for maintaining the resistant phenotype. To verify
our results, we also evaluated some antibiotic combinations
for which synergy was not implied due to the absence of a
functional antibiotic-inactivating enzyme. No synergism was
detected in any of those cases.
Acknowledgement
This work was supported in part by the Sbarro Health Research
Organization.
r e f e r e n c e s
[1] Amsterdam D. Susceptibility testing of antimicrobials inliquid media. In: Lorian V, editor. Antibiotics in laboratory

p e p t i d e s 2 9 ( 2 0 0 8 ) 1 8 7 8 – 1 8 8 61886
medicine. Philadelphia, PA: Lippincott Williams andWilkins; 1996. p. 52–111.
[2] Bagella L, Sun A, Tonini T, Abbadessa G, Cottone G, PaggiGM, et al. A small molecule based on the pRb2/p130 spacerdomain leads to inhibition of cdk2 activity, cell cycle arrestand tumor growth reduction in vivo. Oncogene2007;26:1829–39.
[3] Bikker FJ, Kaman-van Zanten WE, de Vries-van de Ruit AM,Voskamp-Visser I, van Hooft PA, Mars-Groenendijk RH,et al. Evaluation of the antibacterial spectrum of drosocinanalogues. Chem Biol Drug Des 2006;68:148–53.
[4] Bower MA, Cudic M, Campbell W, Wade JD, Otvos Jr L.Walking the fine line between intracellular and membraneactivities of antibacterial peptides. Lett Pept Sci2004;10:463–73.
[5] Bush K, Macielag M, Weidner-Wells M. Taking inventory:antibacterial agents currently at or beyond Phase I. CurrOpin Microbiol 2004;7:466–76.
[6] Cudic M, Condie BA, Weiner DJ, Lysenko ES, Xiang ZQ,Insug O, et al. Development of novel antibacterial peptidesthat kill resistant isolates. Peptides 2002;23:2071–83.
[7] Cudic M, Lockatell CV, Johnson DE, Otvos Jr L. In vitro and invivo activity of a designed antibacterial peptide analogagainst uropathogens. Peptides 2003;24:807–20.
[8] Eliopoulos GM, Moellering RC. Antimicrobial combinations.In: Lorian V, editor. Antibiotics in laboratory medicine. 3rded., Baltimore, MD: Lippincott Williams & Wilkins; 1991. p.432–92.
[9] Fernandez-Carneado J, Kogan MJ, Castel S, Giralt E.Potential peptide carriers: amphipathic proline-richpeptides derived from the N-terminal domain of gamma-zein. Angew Chem Int Ed Engl 2004;43:1811–4.
[10] Fields GB, Noble RL. Solid-phase peptide synthesis using 9-fluorenylmethoxycarbonyl amino acids. Int J Pept ProteinRes 1990;35:161–214.
[11] Ge Y, MacDonald DL, Holroyd KJ, Thornsberry C, Wexler H,Zasloff M. In vitro properties of pexiganan, an analogof magainin. Antimicrob Agents Chemother 1999;43:782–8.
[12] Hoffmann R, Bulet P, Urge L, Otvos Jr L. Range of activityand metabolic stability of synthetic antibacterialglycopeptides from insects. Biochim Biophys Acta1999;1426:459–67.
[13] Hollosi M, Kawai M, Fasman GD. Studies on proline-containing tetrapeptide models of beta-turns. Biopolymers1985;24:211–42.
[14] Kragol G, Hoffmann R, Chattergoon MA, Lovas S, Cudic M,Bulet P, et al. Identification of crucial residues for theantibacterial activity of the proline-rich peptide,pyrrhocoricin. Eur J Biochem 2002;269:4226–37.
[15] Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R, OtvosJr L. The antibacterial peptide pyrrhocoricin inhibits theATPase actions of DnaK and prevents chaperone-assistedprotein folding. Biochemistry 2001;40:3016–26.
[16] Leng B, Meyers BR, Hirschman SZ, Keusch GT.Susceptibilities of gram-negative bacteria to combinationsof antimicrobial agents in vitro. Antimicrob AgentsChemother 1975;8:164–71.
[17] Levy SB, Marshall B. Antibacterial resistance worldwide:causes, challenges and responses. Nat Med 2004;10:S122–129.
[18] Lopez Ferreira N, Alix JH. The DnaK chaperone is necessaryfor a-complementation of b-galactosidase in Escherichia coli.J Bacteriol 2002;184:7047–54.
[19] Noto PB, Abbadessa G, Cassone M, Mateo GD, Agelan A,Wade JD, et al. Alternative stabilities of a proline-richantibacterial peptide in vitro and in vivo. Protein Sci2008;17:1249–55.
[20] Otvos Jr L, de Olivier Inacio V, Wade JD, Cudic P. Priorantibacterial peptide-mediated inhibition of protein foldingin bacteria mutes resistance enzymes. Antimicrob AgentsChemother 2006;50:3146–9.
[21] Otvos Jr L, de Olivier Inacio V, Rogers ME, Consolvo PJ,Condie BA, Lovas S, et al. Interaction between heat shockproteins and antimicrobial peptides. Biochemistry2000;39:14150–9.
[22] Otvos Jr L, Snyder C, Condie BA, Bulet P, Wade JD. Chimericantimicrobial peptides exhibit multiple modes of action. IntJ Pept Res Ther 2005;11:29–42.
[23] Otvos Jr L, Wade JD, Lin F, Condie BA, Hanrieder J,Hoffmann R. Designer antibacterial peptides killfluoroquinolone-resistant clinical isolates. J Med Chem2005;48:5349–59.
[24] Raz R, Colodner R, Kunin CM. Who are you, Staphylococcussaprophyticus? Clin Infect Dis 2005;40:896–8.
[25] Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL,Wender PA. Arginine-rich molecular transporters for drugdelivery: role of backbone spacing in cellular uptake. J MedChem 2002;45:3612–8.
[26] Serra P, Brandimarte C, Martino P, Carlone S, Giunchi G.Synergistic treatment of enterococcal endocarditis: in vitroand in vivo studies. Arch Intern Med 1977;137:1562–7.
[27] Tarasi A, Cassone M, Monaco M, Tarasi D, Pompeo ME,Venditti M. Activity of moxifloxacin in combination withvancomycin or teicoplanin against Staphylococcus aureusisolated from device-associated infections unresponsive toglycopeptide therapy. J Chemother 2003;15:239–43.
[28] Weber JT, Courvalin P. An emptying quiver: antimicrobialdrugs and resistance. Emerg Infect Dis 2005;11:791–3.





























