Embed Size (px)
Citation preview

UN
CO
RR
EC
TED
PR
OO
FRole of nodal signaling and the microenvironmentunderlying melanoma plasticity
Lynne-Marie Postovit1,2,*, Naira V. Margaryan1,
Elisabeth A. Seftor1 and Mary J. C. Hendrix1
1Children’s Memorial Research Center, Cancer Biology and
Epigenomics Program, Robert H. Lurie Comprehensive Cancer
Center, Northwestern University’s Feinberg School of Medicine,
Chicago, IL, USA2Department of Anatomy and Cell Biology, Schulich School of
Medicine and Dentistry, University of Western Ontario, London,
ON, Canada
*Address correspondence to Lynne-Marie Postovit,
e-mail: [email protected]
Abstract
The incidence of melanoma has increased dramati-
cally over the last 50 yr, and although melanoma
accounts for only 10% of all skin cancers, it is respon-
sible for over 80% of skin cancer deaths. Recent stud-
ies have uncovered critical molecular events
underlying melanocytic transformation and mel-
anomagenesis. Among these noteworthy observa-
tions are the acquisition of stem cell-associated
proteins, such as the Notch receptors and Nodal,
which have also been implicated in melanoma pro-
gression. For example, we have demonstrated that
Nodal expression is limited to invasive vertical
growth phase and metastatic melanoma lesions, and
that inhibition of Nodal signaling promotes the rever-
sion of metastatic melanoma cells toward a more
differentiated, less invasive non-tumorigenic pheno-
type. In addition, molecular cross-talk exists between
the Notch and Nodal signaling pathways. Interest-
ingly, the acquisition of stem cell-associated plastic-
ity is often acquired via epigenetic mechanisms, and
is therefore receptive to reprogramming in response
to embryonic microenvironments. Here, we review
the concept of melanoma plasticity, with an empha-
sis on the emerging role of Nodal as a regulator of
melanoma tumorigenesis and progression, and pres-
ent findings related to epigenetic reprogramming.
Key words XXXX ⁄ XXXX ⁄ XXXX2
Received 24 January 2008, revised and accepted for
publication 19 March 2008
doi: 10.1111/j.1755-148X.2008.00463.x
Melanoma defined
Melanoma arises from the transformation of neural
crest derived melanocytes that reside in the basal layer
of the epidermis. Normally, melanocytes are evenly dis-
persed into epidermal-melanin units, consisting of
approximately 36 keratinocytes per melanocyte. Each
melanocyte transfers pigment-containing melanosomes
to the keratinocytes in its unit via dendritic processes.
The melanin contained in these melanosomes absorbs
and scatters ultraviolet radiation, thereby shielding the
nucleic acids in the skin from damage. Interestingly,
in vitro skin models have demonstrated that the kerati-
nocytes dynamically regulate this process by controlling
dendrite growth, melanocyte proliferation and melanin
production (Chin, 2003; Hsu et al., 2002).
During melanoma progression there is a general dys-
function of this complex epidermal-melanin unit. Initially,
the melanocyte:keratinocyte ratio increases, resulting in
the formation of common nevi (moles) which may lead
to dysplastic nevi with structural atypia. Dysplastic nevi
can subsequently progress into a radial growth phase
(RGP) melanoma, characterized by lateral growth that is
largely confined to the epidermis. Radial growth phase
tumors may then acquire the ability to invade into the
dermis and subcutaneous tissue, to form a vertical
growth phase (VGP) melanoma. Histologically, VGP mel-
anomas are best characterized as expansive nodules of
malignant cells that have penetrated the epidermal
basement membrane. Unlike RGP melanomas, which
remain dependent on keratinocyte-derived growth
factors and cannot undergo anchorage-indepen-
dent growth, VGP melanomas have escaped keratino-
cyte control, can undergo anchorage-independent
growth and have acquired metastatic competency (Ben-
nett, 2008; Chin, 2003; Hsu et al., 2002). Metastatic
melanoma is characterized by a high mortality rate of
over 80% and a median survival of only 7.5 months. In
the last 40 yr, the incidence of melanoma in the USA
has increased by 15-fold and cutaneous melanoma has
become the most common cancer afflicting young
adults. When diagnosed prior to the onset of VGP
disease, melanoma is generally curable with surgery
(Chin et al., 2006; Chudnovsky et al., 2005b). However,
patients with metastatic melanoma have few clinical
options, because of a high resistance to therapy,
exacerbated by a very rapid disease progression. It is
P C R 4 6 3 B Dispatch: 11.4.08 Journal: PCR CE: Ulagammal
Journal Name Manuscript No. Author Received: No. of pages: 10 PE: Aswini
ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard 1
Pigment Cell Melanoma Res. INVITED REVIEW
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
therefore imperative that the molecular events that
characterize the melanocytic neoplasia be determined,
so that targets for early detection and intervention can
be developed.
Multipotent melanoma cells
There is a notable body of literature detailing the molec-
ular signature of aggressive melanoma cells and embry-
onic stem cells (ESCs), which reveals an intriguing
similarity in the pluripotent gene expression patterns
that characterize these cell types (Hendrix et al., 2007).
In the field of melanoma research, studies have utilized
comparative global gene analyses to decipher some of
the major gene expression patterns that arise as a con-
sequence of genomic and epigenetic transforming
events and that characterize the transition of melano-
cytes to poorly and then highly metastatic melanoma
cells (Bittner et al., 2000; Hendrix et al., 2003; Hoek,
2007; Smith et al., 2005). With this approach, we and
others have shown that aggressive melanoma cells
manifest a functional plasticity characterized by the
simultaneous expression of genes from a variety of cell
types, including stem cells, concomitant with a reduc-
tion in the expression of genes specific to their parental
cell lineage (Table 1). For example, aggressive mela-
noma cells aberrantly express genes (and proteins),
such as Vascular Endothelial Cadherin (VE-Cadherin),
which are normally associated with endothelial cells,
and also express Keratins, which are intermediate fila-
ments characteristically associated with epithelial cells
(Hendrix et al., 2003). Furthermore, the expression of
melanocyte-specific markers is dramatically reduced,
and sometimes absent, in aggressive melanoma cells:
Melan A is reduced by more than fivefold and Tyrosi-
nase, which catalyses the conversion of tyrosine to the
pigment melanin, is reduced by more than 35-fold in
aggressive melanomas relative to their poorly aggres-
sive counterparts (Hendrix et al., 2003). In addition,
reduced Tyrosinase levels are associated with immune
evasion (Takeuchi et al., 2003). Collectively, this gene
expression pattern confers a functional plasticity upon
aggressive melanoma cells that enables them to thrive
and metastasize. For example, VE-Cadherin expression
by melanoma cells is essential for the formation of
tumor-derived vascular networks, thought to provide the
tumor with a paravascular perfusion pathway, while the
expression of Keratins is associated with enhanced inva-
sion and metastasis (Hendrix et al., 1992, 2001).
Aggressive melanomas also express stem cell-associ-
ated proteins (including the Notch receptors, CD133,
Wnt-5a, and Nodal) which have been shown to play a
role in the maintenance of pluripotency (Balint et al.,
2005; Frank et al., 2005; Hendrix et al., 2003; Hoek
et al., 2004; Weeraratna et al., 2002). These intriguing
findings support the premise that aggressive melanoma
cells acquire a multipotent, plastic phenotype, a concept
that challenges our current thinking of how to target
tumor cells with stem cell-like properties. Indeed, while
previous therapeutic strategies have focused on elimi-
nating a homogeneous tumor expressing traditional bio-
markers, new treatment modalities should attempt to
target a heterogeneous population of cancer cells
whose stem cell-like phenotype facilitates adaptation
and consequently survival (Hendrix et al., 2007). There-
fore, pluripotency-promoting pathways, which maintain
tumor cell plasticity, would be ideal targets for early
diagnosis and therapeutic intervention.
Nodal as a melanoma plasticity biomarker
The recent studies in our laboratory have revealed a
new regulator of melanoma plasticity and tumorigenic-
ity, called Nodal (Topczewska et al., 2006). Nodal is a
member of the Transforming Growth Factor Beta (TGF-
b) superfamily and is a pivotal inhibitor of hESC differen-
tiation (James et al., 2005; Mesnard et al., 2006; Vallier
et al., 2005). Indeed, Nodal has been shown to maintain
the pluripotency of ESCs and is one of the first genes
to be down-regulated as totipotent hESCs differentiate
during embryoid body formation. Moreover, inhibition of
the Nodal signaling pathway, through pharmacological
inhibition of its receptor, results in hESC differentiation
(Vallier et al., 2004). We recently discovered that Nodal
expression is positively associated with melanoma
tumor progression: As indicated by Western blot analy-
sis, tumorigenic melanoma cells lines (C8161, WM793,
and 1205Lu) express high levels of Nodal, whereas
Nodal is absent in normal melanocytes and in non-
tumorigenic melanoma cells (C81-61) (Figure 1A). Nodal
expression is also positively correlated with melanoma
progression clinically (Figure 1B–E). Indeed, immunohis-
Table 1 Molecular profile of aggressive melanoma cells
expressing multiple cellular phenotypesa
Gene Function Ratiob
ESM-1 Endothelial surface molecule 44
VE-Cadherin Endothelial adhesion molecule 10.7
TIE-1 Endothelial protein receptor >100
EphA2 (Eck) Epithelial cell kinase 77
Keratins 7,8,18 Epithelial intermediate filaments 20–80
Mart-1 ⁄ Melan A Melanocyte antigen 5.1flLSP1 Lymphocyte specific protein 4.8
HCLS1 Hematopoietic lineage protein 10
KIT Stem cell factor receptor 2.5
Nodal Embryonic stem cell marker
and morphogen
20
Notch Stem cell marker and receptor 5.3
aAltered gene expression in human melanoma cells was identified
by cDNA microarray analysis and confirmed by Western blot.bSelected genes are reported as a ratio for highly aggressive C8161
human cutaneous melanoma cells compared to poorly aggressive,
isogenically matched C81-61 cells.
Postovit et al.
2 ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
Ftochemical analysis has shown that Nodal protein is
absent in normal skin and rare in poorly invasive RGP
melanomas. This is in contrast to invasive VGP melano-
mas and melanoma metastases where Nodal expres-
sion is detectable in up to 60% of cases. We have also
demonstrated that Nodal plays an instrumental role in
the maintenance of melanoma cell plasticity and tumori-
genicity (Topczewska et al., 2006). Metastatic C8161
melanoma cells re-expressed Tyrosinase, a melanocyte
marker, and down-regulated VE-Cadherin and Keratin
8 ⁄ 18, markers of endothelial and epithelial lineages
respectively, in response to Nodal inhibition with a
Nodal specific Morpholino (MONodal) (Table 2). As a
complement to these findings, we utilized an orthotopic
mouse model to examine the effect of Nodal inhibition
on melanoma tumor formation (Figure 2). Palpable
subcutaneous tumors arose within 7 days following the
injection of only 250 000 control C8161 cells. In
contrast, knocking down Nodal expression resulted in a
significant reduction in C8161 tumorigenicity when the
same number of cells was injected (Figure 2A). Indeed,
a 30% diminution of tumor incidence in addition to a
decrease in tumor growth occurred when Nodal
expression was inhibited (Topczewska et al., 2006). Pre-
A
B C
D E
Figure 1.11 Nodal expression correlates
with melanoma progression. (A) Western
blot analysis of Nodal in: C8161, human
metastatic cutaneous melanoma cells;
normal human melanocytes; C81-61, non-
tumorigenic melanoma cells (isogenically
matched to C8161 cells); WM793 human
vertical growth phase cutaneous
melanoma cells; 1205Lu, melanoma cells
derived from an experimental metastasis
of WM793; and A375P, a tumorigenic
human cutaneous melanoma cell line.
Actin is used as a loading control. Nodal
expression is exclusive to the tumorigenic
melanoma cells lines. (B–E)
Immunohistochemical analysis of Nodal
staining (red color) in (B,C) primary
cutaneous melanomas and (D,E)
cutaneous melanoma metastases.
Tumor-associated mast cells are also
immunoreactive for Nodal protein. Isotype
controls are pictured in the insets. Brown
areas are melanotic cells, and bars equal
50 lm.
Table 2 Summary of biomarker
expression in metastatic melanoma cells
treated with a morpholino designed to
inhibit nodal expression (MONodal)
or exposed to a hESC-derived
microenvironment (hESC CMTX) as
compared to control cellsa
Nodal (stem cell)
Tyrosinase
(Melanocyte)
Melan A
(Melanocyte)
VE-Cadherin
(Endothelial)
Keratin 8 ⁄ 18
(Epithelial)
Control +++ – – +++ +++
MONodal – ››› flflfl flflflhESC CMTX flflfl ››› flflfl
aRelative expression of biomarkers indicative of melanoma plasticity (Nodal, VE-Cadherin and
Keratin 8 ⁄ 18) or a differentiated phenotype (Tyrosinase and Melan A) were assessed using
RT-PCR analysis and confirmed by Western blot.
LO
WR
ES
OL
UT
ION
CO
LO
UR
FIG
Nodal and melanoma plasticity
ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard 3
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
UN
CO
RR
EC
TED
PR
OO
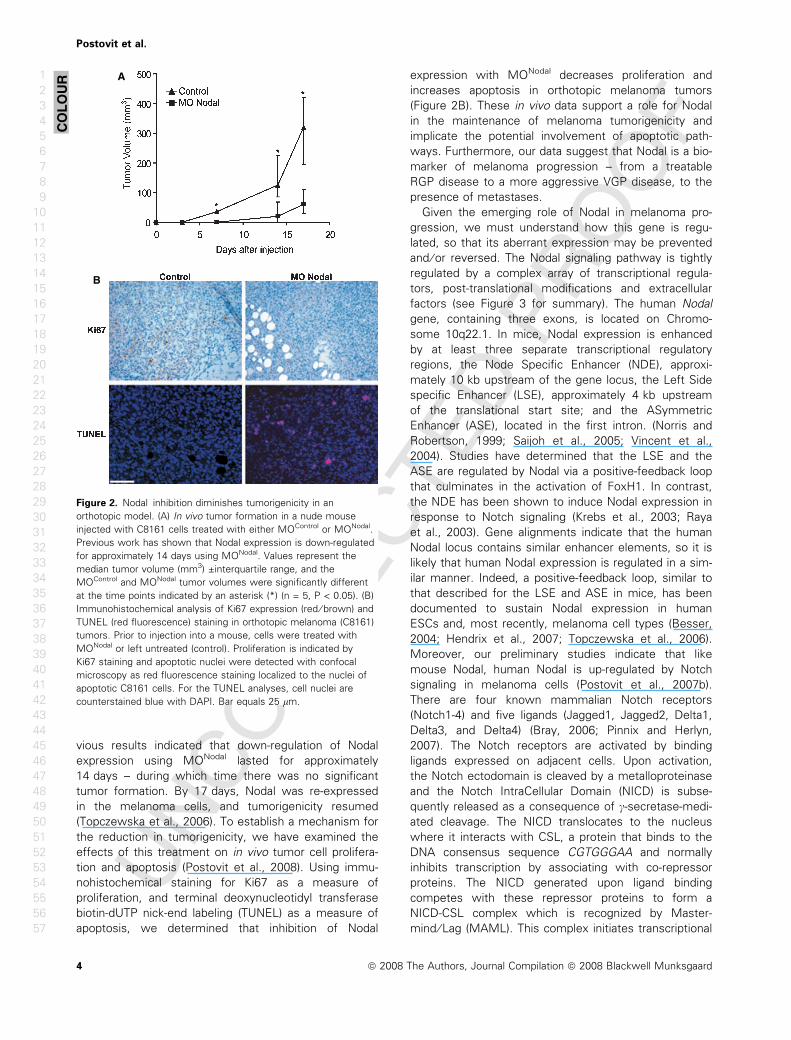
Fvious results indicated that down-regulation of Nodal
expression using MONodal lasted for approximately
14 days – during which time there was no significant
tumor formation. By 17 days, Nodal was re-expressed
in the melanoma cells, and tumorigenicity resumed
(Topczewska et al., 2006). To establish a mechanism for
the reduction in tumorigenicity, we have examined the
effects of this treatment on in vivo tumor cell prolifera-
tion and apoptosis (Postovit et al., 2008). Using immu-
nohistochemical staining for Ki67 as a measure of
proliferation, and terminal deoxynucleotidyl transferase
biotin-dUTP nick-end labeling (TUNEL) as a measure of
apoptosis, we determined that inhibition of Nodal
expression with MONodal decreases proliferation and
increases apoptosis in orthotopic melanoma tumors
(Figure 2B). These in vivo data support a role for Nodal
in the maintenance of melanoma tumorigenicity and
implicate the potential involvement of apoptotic path-
ways. Furthermore, our data suggest that Nodal is a bio-
marker of melanoma progression – from a treatable
RGP disease to a more aggressive VGP disease, to the
presence of metastases.
Given the emerging role of Nodal in melanoma pro-
gression, we must understand how this gene is regu-
lated, so that its aberrant expression may be prevented
and ⁄ or reversed. The Nodal signaling pathway is tightly
regulated by a complex array of transcriptional regula-
tors, post-translational modifications and extracellular
factors (see Figure 3 for summary). The human Nodal
gene, containing three exons, is located on Chromo-
some 10q22.1. In mice, Nodal expression is enhanced
by at least three separate transcriptional regulatory
regions, the Node Specific Enhancer (NDE), approxi-
mately 10 kb upstream of the gene locus, the Left Side
specific Enhancer (LSE), approximately 4 kb upstream
of the translational start site; and the ASymmetric
Enhancer (ASE), located in the first intron. (Norris and
Robertson, 1999; Saijoh et al., 2005; Vincent et al.,
2004). Studies have determined that the LSE and the
ASE are regulated by Nodal via a positive-feedback loop
that culminates in the activation of FoxH1. In contrast,
the NDE has been shown to induce Nodal expression in
response to Notch signaling (Krebs et al., 2003; Raya
et al., 2003). Gene alignments indicate that the human
Nodal locus contains similar enhancer elements, so it is
likely that human Nodal expression is regulated in a sim-
ilar manner. Indeed, a positive-feedback loop, similar to
that described for the LSE and ASE in mice, has been
documented to sustain Nodal expression in human
ESCs and, most recently, melanoma cell types (Besser,
2004; Hendrix et al., 2007; Topczewska et al., 2006).
Moreover, our preliminary studies indicate that like
mouse Nodal, human Nodal is up-regulated by Notch
signaling in melanoma cells (Postovit et al., 2007b).
There are four known mammalian Notch receptors
(Notch1-4) and five ligands (Jagged1, Jagged2, Delta1,
Delta3, and Delta4) (Bray, 2006; Pinnix and Herlyn,
2007). The Notch receptors are activated by binding
ligands expressed on adjacent cells. Upon activation,
the Notch ectodomain is cleaved by a metalloproteinase
and the Notch IntraCellular Domain (NICD) is subse-
quently released as a consequence of c-secretase-medi-
ated cleavage. The NICD translocates to the nucleus
where it interacts with CSL, a protein that binds to the
DNA consensus sequence CGTGGGAA and normally
inhibits transcription by associating with co-repressor
proteins. The NICD generated upon ligand binding
competes with these repressor proteins to form a
NICD-CSL complex which is recognized by Master-
mind ⁄ Lag (MAML). This complex initiates transcriptional
A
B
Figure 2. Nodal inhibition diminishes tumorigenicity in an
orthotopic model. (A) In vivo tumor formation in a nude mouse
injected with C8161 cells treated with either MOControl or MONodal.
Previous work has shown that Nodal expression is down-regulated
for approximately 14 days using MONodal. Values represent the
median tumor volume (mm3) ±interquartile range, and the
MOControl and MONodal tumor volumes were significantly different
at the time points indicated by an asterisk (*) (n = 5, P < 0.05). (B)
Immunohistochemical analysis of Ki67 expression (red ⁄ brown) and
TUNEL (red fluorescence) staining in orthotopic melanoma (C8161)
tumors. Prior to injection into a mouse, cells were treated with
MONodal or left untreated (control). Proliferation is indicated by
Ki67 staining and apoptotic nuclei were detected with confocal
microscopy as red fluorescence staining localized to the nuclei of
apoptotic C8161 cells. For the TUNEL analyses, cell nuclei are
counterstained blue with DAPI. Bar equals 25 lm.
CO
LO
UR
Postovit et al.
4 ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
Factivation of target genes such as c-myc (Bray, 2006;
Krebs et al., 2003; Raya et al., 2003). Of note, the NDE
of the Nodal gene contains two CSL binding sites, and
this region has been shown to respond to Notch signal-
ing (Krebs et al., 2003; Raya et al., 2003). We have
recently determined that inhibiting Notch in metastatic
melanoma cells with a c-secretase inhibitor (DAPT)
results in decreased Nodal expression. Moreover, using
specific siRNAs, we have found that Notch-4 may pref-
erentially regulate Nodal expression in these cells
(Postovit et al., 2007b).
As a complement to canonical regulators of transcrip-
tion, Nodal expression is also governed by gene methyl-
ation and miRNA-directed degradation. For example, we
have determined that there is a sizable CpG island
(>1300 base pairs) near the transcription start site (TSS)
of the Nodal gene, and that this site may regulate Nodal
expression (Postovit et al., 2007a). Moreover, a novel
miRNA (miR-430) has been shown to block the transla-
tion of a Nodal homolog, squint, in zebrafish (Choi et al.,
2007). MiR-430 target sites are also present in the
mammalian Nodal gene; and so it is likely that Nodal
expression is similarly affected by miRNA-mediated
degradation in humans.
Nodal is also regulated post-translationally by subtili-
sin-like pro-protein convertases, including PACE-4 and
Furin (Beck et al., 2002), and by glycosylation. In a
manner akin to most TGF-b family members, Nodal is
synthesized as a pro-protein that is activated following
proteolytic processing by covertases at R-X-(K ⁄ R)-R and
R-X-X-R consensus sequences (Schier, 2003). Removal
of the pro-domain potentiates autocrine signaling but
reduces Nodal stability and signaling range, thereby
promoting autocrine signaling (Le Good et al., 2005).
Conversely, glycosylation of mature Nodal affords the
protein with increased stability so that it can potentiate
paracrine signaling events (Le Good et al., 2005). Hence,
post-translational modifications of Nodal are important
mediators of Nodal signaling outcomes.
Nodal propagates its signal by binding to hetero-
dimeric complexes between type I (ALK 4 ⁄ 7) and type II
(ActRIIB) activin-like kinase receptors. Assembly of this
complex results in the phosphorylation and activation of
ALK 4 ⁄ 7 by ActRIIB, followed by the ALK 4 ⁄ 7-mediated
Figure 3.11 Regulation of the nodal signaling pathway. Nodal is secreted from the cell where it can act as an autocrine or paracrine factor. The
Nodal precursor is cleaved and activated by the pro-protein convertases (SPC), Pace-4, and Furin. Nodal propagates its signal by binding to
heterodimeric complexes between type I (ALK 4 ⁄ 7) and type II (ActRIIB) activin-like kinase receptors, resulting in the phosphorylation and
activation of ALK 4 ⁄ 7 by ActRIIB, followed by the ALK 4 ⁄ 7-mediated phosphorylation of Smad-2 and possibly Smad-3. The epidermal
growth factor-coreceptor (EGF-CFC), Cripto-1 (Cripto), is often a component of this receptor complex and can enhance Nodal signaling.
Phosphorylated Smad-2 ⁄ 3 associates with Smad-4 and translocates to the nucleus where it regulates gene expression through the
association with transcription factors such as FoxH1. Nodal up-regulates its own expression by stimulating transcriptional activation at the
Left Side specific and Asymmetric Enhancers (LSE and ASE). Extracellular Nodal inhibitors, most notably Lefty A and B (Lefty), spatially and
temporally restrict Nodal signaling levels through antagonism of Nodal and ⁄ or Cripto-1. Notch receptors become activated following binding to
ligands, such as Delta and Jagged, expressed on adjacent cells. Once activated, the intracellular component of the Notch receptor (NICD) is
released following a c-secretase-dependent cleavage. The NICD translocates to the nucleus where it interacts with CSL, a protein that binds
to the DNA and inhibits transcription by associating with co-repressor proteins. The NICD competes with these repressor proteins to form
a NICD-CSL complex enabling the transcriptional activation of target genes, including Nodal, which has CSL binding domains in its Node
Specific Enhancer. Nodal levels may also be regulated by miRNA and possibly DNA methylation of a CpG island.
LO
WR
ES
OL
UT
ION
CO
LO
UR
FIG
Nodal and melanoma plasticity
ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard 5
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
phosphorylation of Smad-2 and possibly Smad-3
(outlined in Figure 3). Phosphorylated Smad 2 ⁄ 3subsequently associates with Smad-4 and then
translocates to the nucleus where it regulates gene
expression through an association with transcription
factors such as FoxH1 and Mixer (Schier, 2003). Genetic
studies in zebrafish and mice have defined an
essential role for Cripto-1, an Epidermal Growth Factor–
Cripto-1 ⁄ FRL1 ⁄ cryptic (EGF–CFC) family member, in
Nodal function. Indeed, embryological studies have
determined that Cripto-1 directly associates with ALK 4
(with its CFC domain) and Nodal (with its EGF domain)
and that these associations may be required for Nodal
to propagate its signal (Bianco et al., 2002; Yeo and
Whitman, 2001). This prerequisite is perhaps best
exemplified in Cripto-1 null mice which die at day 7.5 as
a result of the inability to gastrulate (a Nodal-dependent
phenomenon) (Ding et al., 1998; Liguori et al., 1996).
Studies have determined that Nodal may also signal in a
Cripto-1-independent fashion. For example, Reissmann
et al. (2001) revealed that Nodal can bind to activate
ALK 7 in the absence of Cripto-1, but that Cripto-1 mark-
edly enhances this process. Another study determined
that the Nodal precursor can bind to ALK 4 in the extra-
embryonic ectoderm of the developing mouse embryo
in a Cripto-1-independent manner, and that this binding
results in the expression of Nodal-responsive genes
(Ben-Haim et al., 2006). Finally, using murine knock out
models, Liguori et al. (2007) recently demonstrated that
Nodal can signal extensively and control axis specifica-
tion in the absence of Cripto, if its inhibitor Cerberus is
also knocked out.
Nodal up-regulates its own transcription via a positive-
feedback loop. Hence, to control the levels of this
potent morphogen, hESCs also secrete Nodal inhibitors
such as Lefty A, Lefty B, Cerberus and Tomoregulin-1
(Schier, 2003; Tabibzadeh and Hemmati-Brivanlou,
2006). Of these factors, the Lefty molecules, highly
divergent members of the TGFb superfamily, are
expressed to the greatest extent. In fact, studies have
demonstrated that in conjunction with Nodal and Oct
3 ⁄ 4, Lefty A and B are among the most enriched genes
expressed in hESCs (Sato et al., 2003; Tabibzadeh and
Hemmati-Brivanlou, 2006). Extracellular Nodal inhibitors
control Nodal signaling by spatially and temporally
restricting the Nodal-mediated activation of ALK 4 ⁄ 7.
For example, Lefty A and B specifically antagonize the
Nodal signaling pathway by binding to and interacting
with Nodal and ⁄ or with Cripto-1 in a manner that blocks
ALK activation (Schier, 2003; Shen, 2007). This restric-
tion of Nodal signaling can occur in the extracellular
microenvironment, where Nodal and sometimes Cripto-
1 are present, as well as at the cell surface. Of note,
the Lefty proteins have not been found to bind ALK4 or
ActRIIB; hence these Lefty proteins are not competitive
inhibitors of the ALK receptor complex. Furthermore, in
embryological systems, the Lefty genes are often
downstream targets of Nodal signaling, thereby provid-
ing a powerful negative-feedback loop for this pathway
(Schier, 2003; Shen, 2007). In contrast, we have deter-
mined that Nodal-expressing melanoma cells do not
express Lefty, thereby allowing Nodal signaling to go
unchecked in this tumor-associated system (Postovit
et al., 2007a, 2008).
The myriad of regulatory mechanisms characterizing
the Nodal signaling pathway likely underlies its propen-
sity for aberrant expression in melanoma. However, this
complexity also affords a number of putative strategies
for the inhibition of Nodal signaling and the circumven-
tion of melanoma progression. One such approach
involves the epigenetic silencing of Nodal expression.
Epigenetic reprogramming of multipotentmelanoma cells
Although the role of genetic mutations in oncogenic
transformation is indisputable, a great deal of evidence
suggests that the tumorigenic potential of a transformed
cell is also attributable to epigenetic modifications.
Unlike genetic changes, epigenetic adjustments do not
affect the primary DNA sequence. Rather, they involve
interactions among cells and cell products, which lead
to alterations in reversible phenomena such as cell sig-
naling and DNA modifications (Postovit et al., 2007a;
Rothhammer and Bosserhoff, 2007). Exemplifying the
importance of epigenetics in melanomagenesis is a
recent study by Jaenisch et al. in which nuclear trans-
plantation of a melanoma nucleus into an oocyte gave
rise to ESCs with the capacity to differentiate into non-
tumorigenic cell types such as melanocytes and fibro-
blasts (Hochedlinger et al., 2004). Nuclear transplanta-
tion into an oocyte induces a dramatic hypomethylation
of the donor DNA, exposing promoters and enabling
transcription (Lotem and Sachs, 2006). These phenom-
ena confer the broad developmental spectrum observed
when normal or neoplastic somatic nuclei are trans-
planted. As differentiation and cell fate specification
ensue, there is a marked increase in DNA methylation
leading to the down-regulation of most genes and a
consequential specialization in gene expression (Lotem
and Sachs, 2006). In Jaenisch’s study, the tumorigenic
potential of the melanoma nuclei was temporarily
reversed in response to nuclear transplantation, even
though genetic mutations persisted (Hochedlinger et al.,
2004). Hence, the mutations which characterized the
melanoma genome worked in concert with epigenetic
factors to sustain transformation in the donor melanoma
cells.
Epigenetic modifications, initiated via microenviron-
mental factors, have also emerged as major players
in melanocyte transformation and transdifferentiation
(Bedogni et al., 2005; Carreira et al., 2006)3 . For
example, Goding et al. recently determined that the
microphthalmia-associated transcription factor, Mitf, epi-
Postovit et al.
6 ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
genetically regulates diaphanous-related formin (Dia1)
expression. Dia1, which promotes actin polymerization
and coordinates the actin cytoskeleton and microtubule
networks at the cell periphery, inhibits invasion and
induces cell-cycle arrest. Hence, by regulating Dai1 tran-
scription, alterations in Mitf, which occur in response to
microenvironmental factors, influence melanoma cell
proliferation and invasion (Carreira et al., 2006). In
another study, Bedogni et al. (2005) demonstrated that
hypoxia, which characterizes the microenvironment of
many solid tumors and has been shown to promote
melanoma cell invasion and metastasis, also contributes
to melanocyte transformation (Bedogni et al., 2005).
This study revealed that constitutively active Akt, which
is observed in a high percentage of melanomas, can
transform melanocytes exclusively when oxygen levels
are low (Bedogni et al., 2005). Furthermore, the skin’s
distance from superficial blood vessels renders it is
mildly hypoxic with oxygen levels between 1 and 5%.
This microenvironmental milieu permits melanocytes to
stabilize the transcriptional co-factor hypoxia inducible
factor 1 alpha (HIF-1a), which promotes hypoxia-associ-
ated gene expression. It was discovered that this
up-regulation of HIF-1a enhances melanocyte transfor-
mation by synergizing with constitutively active Akt to
promote anchorage-independent growth in vitro, and
tumor formation in vivo (Bedogni et al., 2005). This
finding exemplifies how the microenvironment can
complement aberrant genetic changes to promote
melanomagenesis. Moreover, these findings highlight
the importance of epigenetic phenomena (and resultant
gene expression patterns) in melanocyte transformation
and melanoma progression.
Epigenetic phenomena are theoretically reversible.
Hence, the plastic, stem cell-like phenotype of aggres-
sive tumor cells should be receptive to reprogramming
(i.e. redifferentiation) (Gerschenson et al., 1986). In sup-
port of this concept, embryonic microenvironments
have been shown to inhibit the tumorigenicity of a vari-
ety of cancer cell lines (Gerschenson et al., 1986; Pierce
et al., 1982; Podesta et al., 1984). For example, B16
murine melanoma cells were unable to form tumors
and appeared to differentiate toward a neuronal pheno-
type following exposure to microenvironmental factors
derived from the embryonic skin of a developing mouse
(Gerschenson et al., 1986). In another set of experi-
ments, Bissell and colleagues documented that Rous
sarcoma virus, which causes a rapidly growing tumor
when injected into hatched chicks, is non-tumorigenic
when injected into 4-day-old chick embryos, despite viral
replication and v-src oncogene activation (Dolberg and
Bissell, 1984).
More recently, we employed an in vitro 3D model
to examine whether the microenvironment of human
embryonic stem cells (hESCs) could similarly reprogram
the metastatic melanoma cell phenotype (Postovit et al.,
2006;4 Postovit et al., 2008). In this model, hESCs were
allowed to ‘condition’ a 3D matrix (CMTX), which would
subsequently receive multipotent metastatic melanoma
cells. Because the hESCs were removed prior to the
addition of melanoma cells, the melanoma cells were
exposed only to the extracellular microenvironment of
the hESCs, thereby removing the complexity of cell–cell
interactions from the vast array of mechanisms that
may be working to epigenetically modulate cell behav-
ior. Utilizing this approach we determined that, similar
to Nodal inhibition, exposure of melanoma cells to a
hESC microenvironment results in the re-expression of
Melan-A, a melanocyte specific marker, as well as a
reduction in the expression of VE-Cadherin (Table 2)
(Abbott et al., 2008; Hendrix et al., 2007; Topczewska
et al., 2006). Aggressive melanoma cells exposed to
hESC microenvironments also experienced an 87%
reduction in Nodal expression concomitant to a signifi-
cant decrease in tumorigenicity (Postovit et al., 2008).
Indeed, exposure to the hESC microenvironment signifi-
cantly diminished the ability of human metastatic mela-
noma cells to undergo anchorage-independent growth,
a phenomenon that was rescued by the inclusion of
rNodal (100 ng ⁄ ml). Moreover, exposure of these cells
to hESC-derived CMTX resulted in an inhibition of tumor
growth in an orthotopic mouse model (Hendrix et al.,
2007; Postovit et al., 2008). In a manner similar to Nodal
inhibition, we found that exposure to hESC CMTX
decreased proliferation and increased apoptosis in the
orthotopic tumors (Postovit et al., 2008), implicating the
potential involvement of apoptotic pathways in the
tumor suppressive effects of the hESC microenviron-
ment. Collectively, these findings illuminate the remark-
able ability of hESC-derived factors to inhibit melanoma
tumorigenicity and suggest that this tumor-suppressive
phenomenon is mediated via an inhibition of Nodal
expression and signaling.
The ability of hESCs to reprogram aggressive mela-
noma cells is reversible over time (Abbott et al., 2008;
Postovit et al., 2007a, 2008). As such, this phenomenon
is likely because of epigenetic alterations, such as DNA
methylation. Given the location of a sizable CpG island
near the TSS of the Nodal gene, together with the
marked down-regulation in Nodal expression observed
in melanoma cells exposed to hESC microenvironments,
we hypothesized that the Nodal CpG island is differen-
tially methylated in cells exposed to a hESC micro-
environment. Using bisufite-sequencing technology, we
determined that exposure of aggressive melanoma cells
to matrices conditioned by hESCs resulted in a marked
increase in site-specific methylation in the Nodal CpG
island. Although hESC microenvironments did not drasti-
cally affect global methylation, we observed specific
areas, in the first half of the CpG island, where a 32%
increase in DNA methylation occurred (Postovit et al.,
2007a). Sequence analyses determined that these areas
contain putative consensus sequences for transcription
factors including Sp1, Egr-1, and GATA-4. It is therefore
Nodal and melanoma plasticity
ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard 7
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
plausible that hESC-derived microenvironments can alter
Nodal expression in melanoma cells by epigenetically
methylating transcription factor binding sites. These
modifications may canonically decrease the accessibility
of the Nodal promoter for transcriptional activators,
thereby decreasing Nodal expression commensurate
with differentiating melanoma cells and abrogating their
tumorigenicity. Our ongoing studies will further explore
this possibility to decipher some of the epigenetic
mechanisms by which embryonic microenvironments
reprogram aggressive cancer cells.
Conclusions and future perspectives
There is consensus in the melanoma research commu-
nity and affiliated patient advocacy groups that new
therapeutic strategies are needed to treat advanced
stages of this disease. However, one of the greatest
obstacles to achieving success has been a lack of
understanding of the basic biology underlying the onco-
genic transformation of melanocytes, the aggressive
plasticity of aggressive melanoma, and the possible epi-
genetic regulators of the metastatic phenotype. What
we have come to appreciate is that aggressive mela-
noma cells share many characteristics with embryonic
progenitor cells. In fact, our work has illuminated a new
pathway in melanoma which is directly related to tumor
cell plasticity, and represents the convergence of
embryonic and tumorigenic signaling pathways – via
Nodal, a member of the TGF-b superfamily responsible
for the pluripotency of hESCs.
Protein and immunohistochemical analyses of Nodal
demonstrate that this embryonic morphogen is aber-
rantly expressed in melanoma cells with tumorigenic
potential and in VGP and metastatic lesions. These obser-
vations suggest that Nodal expression may be associated
with the acquisition of an aggressive phenotype in mela-
noma. Additional studies are needed to determine the
prognostic value of Nodal as a new biomarker for disease
progression. Compelling evidence supporting a direct
relationship between Nodal expression, tumorigenicity
and plasticity is provided in Morpholino experiments
showing that when Nodal was down-regulated in aggres-
sive melanoma cells, tumor formation was significantly
diminished and apoptosis was induced. However, tumori-
genicity resumed when Nodal was re-expressed in these
same melanoma cells. Equally noteworthy is the finding
linking down-regulation of Nodal expression in melanoma
to loss of plasticity markers, such as VE-Cadherin and
Keratin 8 ⁄ 19, and the re-expression of melanocyte differ-
entiation pathway specific genes such as Tyrosinase and
Melan-A. Future studies will focus on new strategies to
down-regulate Nodal expression for a longer duration. In
addition, the molecular cross-talk revealed between
Notch-4 and Nodal may provide novel approaches to tar-
geting a broader signaling pathway underlying melanoma
aggressiveness and plasticity.
Based on the plastic phenotype of aggressive mela-
noma cells and their similar characteristics to stem
cells, we tested the possibility that the microenviron-
ment of hESCs could reprogram the multipotent pheno-
type. This unique experimental approach generated
important clues related to the epigenetic reprogram-
ming of plastic melanoma cells, including an 87%
reduction in Nodal expression and a 32% increase in
DNA methylation near the transcriptional start site of
the Nodal gene. These exciting results have stimulated
additional studies related to the identification of factor(s)
in the hESC microenvironment that might contribute to
this important reprogramming. As a consequence of
exposure to the normal hESC microenvironment, the
melanoma cells lost plasticity markers, regained mela-
noma differentiation markers, and underwent apoptosis;
similar to that described for Nodal down-regulation. Col-
lectively, these observations begin to elucidate a new
pathway in melanoma progression that deserves addi-
tional scientific scrutiny. Targeting Nodal and associated
pathways with molecular cross-talk may provide valu-
able new insights into managing the plastic melanoma
phenotype.
Acknowledgements
This work was supported by grants from the Illinois Regenerative
Medicine Institute, U.S. National Institutes of Health (CA50702 and
CA121205) and Charlotte Geyer Foundation to M.J.C.H., and a
Canadian Institutes of Health Research Post-doctoral Fellowship to
L.M.P. The authors wish to thank Drs Brian Nickoloff and Bento
Soares for helpful scientific discussions. We apologize to those col-
leagues whose studies were not cited in this review because of
space limitations.
References
Abbott, D.E., Bailey, C.M., Postovit, L.M., Seftor, E.A., Margaryan,
N.V., and Hendrix, M.J. (2008). The epigenetic influence of
tumor and embryonic microenvironments: how different are
they? Cancer Microenvironment. In press.5
Balint, K., Xiao, M., Pinnix, C.C., Soma, A., Veres, I., Juhasz, I.,
Brown, E.J., Capobianco, A.J., Herlyn, M., and Liu, Z.J. (2005).
Activation of Notch1 signaling is required for beta-catenin-medi-
ated human primary melanoma progression. J. Clin. Invest. 115,
3166–3176.
Beck, S., Le Good, J.A., Guzman, M., Ben, H.N., Roy, K., Beermann,
F., and Constam, D.B. (2002). Extraembryonic proteases regulate
Nodal signalling during gastrulation. Nat. Cell Biol. 4, 981–985.
Bedogni, B., Welford, S.M., Cassarino, D.S., Nickoloff, B.J., Giac-
cia, A.J., and Powell, M.B. (2005). The hypoxic microenvironment
of the skin contributes to Akt-mediated melanocyte transforma-
tion. Cancer Cell 8, 443–454.
Ben-Haim, N., Lu, C., Guzman-Ayala, M., Pescatore, L., Mesnard,
D., Bischofberger, M., Naef, F., Robertson, E.J., and Constam,
D.B. (2006). The nodal precursor acting via activin receptors
induces mesoderm by maintaining a source of its convertases
and BMP4. Dev. Cell 11, 313–323.
Bennett, D.C. (2008). How to make a melanoma: what do we
know of the primary clonal envents? Pigment Cell Melanoma
Res. 21, 27–38.
Postovit et al.
8 ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
Besser, D. (2004). Expression of nodal, lefty-a, and lefty-B in undif-
ferentiated human embryonic stem cells requires activation of
Smad2 ⁄ 3. J. Biol. Chem. 279, 45076–45084.
Bianco, C. et al. (2002). Cripto-1 activates nodal- and ALK4-depen-
dent and -independent signaling pathways in mammary epithelial
Cells. Mol. Cell. Biol. 22, 2586–2597.6
Bittner, M. et al. (2000). Molecular classification of cutaneous
malignant melanoma by gene expression profiling. Nature 406,
536–540.
Bray, S.J. (2006). Notch signalling: a simple pathway becomes
complex. Nat. Rev. Mol. Cell Biol. 7, 678–689.
Carreira, S., Goodall, J., Denat, L., Rodriguez, M., Nuciforo, P.,
Hoek, K.S., Testori, A., Larue, L., and Goding, C.R. (2006). Mitf
regulation of Dia1 controls melanoma proliferation and invasive-
ness. Genes Dev. 20, 3426–3439.
Chin, L. (2003). The genetics of malignant melanoma: lessons from
mouse and man. Nat. Rev. Cancer 3, 559–570.
Chin, L., Garraway, L.A., and Fisher, D.E. (2006). Malignant mela-
noma: genetics and therapeutics in the genomic era. Genes Dev.
20, 2149–2182.
Choi, W.Y., Giraldez, A.J., and Schier, A.F. (2007). Target protectors
reveal dampening and balancing of Nodal agonist and antagonist
by miR-430. Science 318, 271–274.
Chudnovsky, Y., Adams, A.E., Robbins, P.B., Lin, Q., and Khavari,
P.A. (2005a). Use of human tissue to assess the oncogenic activity
of melanoma-associated mutations. Nat. Genet. 37, 745–749.7
Chudnovsky, Y., Khavari, P.A., and Adams, A.E. (2005b). Melanoma
genetics and the development of rational therapeutics. J. Clin.
Invest. 115, 813–824.
Ding, J., Yang, L., Yan, Y.T., Chen, A., Desai, N., Wynshaw-Boris,
A., and Shen, M.M. (1998). Cripto is required for correct orienta-
tion of the anterior–posterior axis in the mouse embryo. Nature
395, 702–707.
Dolberg, D.S., and Bissell, M.J. (1984). Inability of Rous sarcoma
virus to cause sarcomas in the avian embryo. Nature 309, 552–
556.
Frank, N.Y., Margaryan, A., Huang, Y., Schatton, T., Waaga-Gasser,
A.M., Gasser, M., Sayegh, M.H., Sadee, W., and Frank, M.H.
(2005). ABCB5-mediated doxorubicin transport and chemoresis-
tance in human malignant melanoma. Cancer Res. 65, 4320–
4333.
Gerschenson, M., Graves, K., Carson, S.D., Wells, R.S., and Pierce,
G.B. (1986). Regulation of melanoma by the embryonic skin.
Proc. Natl Acad. Sci. USA 83, 7307–7310.
Hendrix, M.J. et al. (1992). Coexpression of vimentin and keratins
by human melanoma tumor cells: correlation with invasive and
metastatic potential. J. Natl Cancer Inst. 84, 165–174.
Hendrix, M.J., Seftor, E.A., Meltzer, P.S., Gardner, L.M., Hess,
A.R., Kirschmann, D.A., Schatteman, G.C., and Seftor, R.E.
(2001). Expression and functional significance of VE-cadherin in
aggressive human melanoma cells: role in vasculogenic mimicry.
Proc. Natl Acad. Sci. USA 98, 8018–8023.
Hendrix, M.J., Seftor, E.A., Hess, A.R., and Seftor, R.E. (2003).
Vasculogenic mimicry and tumour-cell plasticity: lessons from
melanoma. Nat. Rev. Cancer 3, 411–421.
Hendrix, M.J., Seftor, E.A., Seftor, R.E., Kasemeier-Kulesa, J.,
Kulesa, P.M., and Postovit, L.M. (2007). Reprogramming meta-
static tumour cells with embryonic microenvironments. Nat. Rev.
Cancer 7, 246–255.
Hochedlinger, K., Blelloch, R., Brennan, C., Yamada, Y., Kim, M.,
Chin, L., and Jaenisch, R. (2004). Reprogramming of a melanoma
genome by nuclear transplantation. Genes Dev. 18, 1875–1885.
Hoek, K.S. (2007). DNA microarray analyses of melanoma gene
expression: a decade in the mines. Pigment Cell Res. 20, 644–
484.8
Hoek, K. et al. (2004). Expression profiling reveals novel pathways
in the transformation of melanocytes to melanomas. Cancer Res.
64, 5270–5282.
Hsu, M.Y., Meier, F., and Herlyn, M. (2002). Melanoma develop-
ment and progression: a conspiracy between tumor and host.
Differentiation 70, 522–536.
James, D., Levine, A.J., Besser, D., and Hemmati-Brivanlou, A.
(2005). TGFbeta ⁄ activin ⁄ nodal signaling is necessary for the
maintenance of pluripotency in human embryonic stem cells.
Development 132, 1273–1282.
Krebs, L.T. et al. (2003). Notch signaling regulates left-right asym-
metry determination by inducing Nodal expression. Genes Dev.
17, 1207–1212.
Le Good, J.A., Joubin, K., Giraldez, A.J., Ben-Haim, N., Beck, S.,
Chen, Y., Schier, A.F., and Constam, D.B. (2005). Nodal stability
determines signaling range. Curr. Biol. 15, 31–36.
Liguori, G., Tucci, M., Montuori, N., Dono, R., Lago, C.T., Pacifico,
F., Armenante, F., and Persico, M.G. (1996). Characterization of
the mouse Tdgf1 gene and Tdgf pseudogenes. Mamm. Genome
7, 344–348.
Liguori, G.L., Borges, A.C., D’Andrea, D., Liguoro, A., Goncalves,
L., Salgueiro, A.M., Persico, M.G., and Belo, J.A. (2007). Cripto-
independent Nodal signaling promotes positioning of the A–P
axis in the early mouse embryo. Dev. Biol. Epub ahead of print.9
Lotem, J., and Sachs, L. (2006). Epigenetics and the plasticity of
differentiation in normal and cancer stem cells. Oncogene 14,
7663–72.
Mesnard, D., Guzman-Ayala, M., and Constam, D.B. (2006). Nodal
specifies embryonic visceral endoderm and sustains pluripotent
cells in the epiblast before overt axial patterning. Development
133, 2497–2505.
Norris, D.P., and Robertson, E.J. (1999). Asymmetric and node-
specific nodal expression patterns are controlled by two distinct
cis-acting regulatory elements. Genes Dev. 13, 1575–1588.
Pierce, G.B., Pantazis, C.G., Caldwell, J.E., and Wells, R.S. (1982).
Specificity of the control of tumor formation by the blastocyst.
Cancer Res. 42, 1082–1087.
Pinnix, C.C., and Herlyn, M. (2007). The many faces of Notch
signaling in skin-derived cells. Pigment Cell Res. 20, 458–465.
Podesta, A.H., Mullins, J., Pierce, G.B., and Wells, R.S. (1984). The
neurula stage mouse embryo in control of neuroblastoma. Proc.
Natl Acad. Sci. USA 81, 7608–7611.
Postovit, L.M., Seftor, E.A., Seftor, R.E., and Hendrix, M.J. (2006).
A 3-D model to study the epigenetic effects induced by the
microenvironment of human embryonic stem cells. Stem Cells
24, 501–505.
Postovit, L.M., Costa, F.F., Bischof, J.M., Seftor, E.A., Wen, B.,
Seftor, R.E., Feinberg, A.P., Soares, M.B., and Hendrix, M.J.
(2007a). The commonality of plasticity underlying multipotent
tumor cells and embryonic stem cells. J. Cell. Biochem. 101,
908–917.
Postovit, L.M., Seftor, E.A., Seftor, R.E., and Hendrix, M.J.
(2007b). Targeting Nodal in malignant melanoma cells. Expert.
Opin. Ther. Targets 11, 497–505.
Postovit, L.M., Margaryan, N.V., Seftor, E.A., Kirschmann, D.A.,
Lipavsky, A., Wheaton, W.W., Abbott, D.E., Seftor, R.E.B., and
Hendrix, M.J. (2008). Human embryonic stem cell microenviron-
ment suppresses the tumorigenic phenotype of aggressive can-
cer cells. Proc. Natl Acad. Sci. USA. In press.10
Raya, A. et al. (2003). Notch activity induces Nodal expression and
mediates the establishment of left-right asymmetry in vertebrate
embryos. Genes Dev. 17, 1213–1218.
Reissmann, E., Jornvall, H., Blokzijl, A., Andersson, O., Chang, C.,
Minchiotti, G., Persico, M.G., Ibanez, C.F., and Brivanlou, A.H.
(2001). The orphan receptor ALK7 and the Activin receptor ALK4
Nodal and melanoma plasticity
ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard 9
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57

UN
CO
RR
EC
TED
PR
OO
F
mediate signaling by Nodal proteins during vertebrate develop-
ment. Genes Dev. 15, 2010–2022.
Rothhammer, T., and Bosserhoff, A.K. (2007). Epigenetic events in
malignant melanoma. Pigment Cell Res. 20, 92–111.
Saijoh, Y., Oki, S., Tanaka, C., Nakamura, T., Adachi, H., Yan, Y.T.,
Shen, M.M., and Hamada, H. (2005). Two nodal-responsive enh-
ancers control left-right asymmetric expression of Nodal. Dev.
Dyn. 232, 1031–1036.
Sato, N., Sanjuan, I.M., Heke, M., Uchida, M., Naef, F., and Brivan-
lou, A.H. (2003). Molecular signature of human embryonic stem
cells and its comparison with the mouse. Dev. Biol. 260, 404–413.
Schier, A.F. (2003). Nodal signaling in vertebrate development.
Annu. Rev. Cell Dev. Biol. 19, 589–621.
Shen, M.M. (2007). Nodal signaling: developmental roles and regu-
lation. Development 134, 1023–1034.
Smith, A.P., Hoek, K., and Becker, D. (2005). Whole-genome expres-
sion profiling of the melanoma progression pathway reveals
marked molecular differences between nevi ⁄ melanoma in situ
and advanced-stage melanomas. Cancer Biol. Ther. 4, 1018–1029.
Tabibzadeh, S., and Hemmati-Brivanlou, A. (2006). Lefty at the
crossroads of ‘stemness’ and differentiative events. Stem Cells
24, 1998–2006.
Takeuchi, H., Kuo, C., Morton, D.L., Wang, H.J., and Hoon, D.S.
(2003). Expression of differentiation melanoma-associated
antigen genes is associated with favorable disease out-
come in advanced-stage melanomas. Cancer Res. 63, 441–448.
Topczewska, J.M., Postovit, L.M., Margaryan, N.V., Sam, A., Hess,
A.R., Wheaton, W.W., Nickoloff, B.J., Topczewski, J., and Hen-
drix, M.J. (2006). Embryonic and tumorigenic pathways converge
via Nodal signaling: role in melanoma aggressiveness. Nat. Med.
12, 925–932.
Vallier, L., Reynolds, D., and Pedersen, R.A. (2004). Nodal inhibits
differentiation of human embryonic stem cells along the neuroec-
todermal default pathway. Dev. Biol. 275, 403–421.
Vallier, L., Alexander, M., and Pedersen, R.A. (2005). Activin ⁄ Nodal
and FGF pathways cooperate to maintain pluripotency of human
embryonic stem cells. J. Cell Sci. 118, 4495–4509.
Vincent, S.D., Norris, D.P., Le Good, J.A., Constam, D.B., and Rob-
ertson, E.J. (2004). Asymmetric Nodal expression in the mouse
is governed by the combinatorial activities of two distinct regula-
tory elements. Mech. Dev. 121, 1403–1415.
Weeraratna, A.T., Jiang, Y., Hostetter, G., Rosenblatt, K., Duray, P.,
Bittner, M., and Trent, J.M. (2002). Wnt5a signaling directly
affects cell motility and invasion of metastatic melanoma. Cancer
Cell 1, 279–288.
Yeo, C., and Whitman, M. (2001). Nodal signals to Smads through
Cripto-dependent and Cripto-independent mechanisms. Mol. Cell
7, 949–957.
Postovit et al.
10 ª 2008 The Authors, Journal Compilation ª 2008 Blackwell Munksgaard
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
Author Query Form
Journal: PCR
Article: 463
Dear Author,
During the copy-editing of your paper, the following queries arose. Please respond to these by marking
up your proofs with the necessary changes/additions. Please write your answers on the query sheet if
there is insufficient space on the page proofs. Please write clearly and follow the conventions shown on
the attached corrections sheet. If returning the proof by fax do not write too close to the paper’s edge.
Please remember that illegible mark-ups may delay publication.
Many thanks for your assistance.
Query reference Query Remarks
1 Au: The first author has been retained as corresponding author. Please
check.
2 Au: Please supply up to six keywords for indexing.
3 Au: Bedgoni et al., 2005 has been changed to Bedogni et al., 2005 so that
this citation matches the list.
4 Au: Postovit et al., 2005 has been changed to Postovit et al., 2006 so that
this citation matches the list.
5 Au: Please update volume number, page detail in reference Abbott et al.
(2008).
6 Au: The journal style is to list the names of first 3 authors followed by
et al. for 11 or more authors. Please provide author names according to
journal style for all references in the list.
7 Au: Chudnovsky et al. (2005a) not cited. Please cite reference in text or
delete from the list.
8 Au: Please check the page detail in Hoek (2007).
9 Au: Please update volume number, page detail in reference Liguori et al.
(2007).
10 Au: Please update volume number, page detail in reference Postovit et al.
(2008).
11 Au: This figure has been supplied as a low resolution file. Please supply
high quality, high resolution figure files to the Production Editor along
with all other proof corrections. Please go to the following link to access
the Blackwell Publishing guidelines on supplying electronic artwork:
http://www.blackwellpublishing.com/bauthor/illustration.asp




















![EE -304 Electrical Network Theory (Nodal Analysis) [2016]](https://img.dokumen.tips/doc/110x75/6362d19c40b666b8ec0e8895/ee-304-electrical-network-theory-nodal-analysis-2016.jpg)








