Embed Size (px)
Citation preview

Regioselective Functionalization of Polyols via
Organoboron Catalysis
by
Lina Chan
A thesis submitted in conformity with the requirements for the degree of Masters of Science Graduate Department of Chemistry
University of Toronto
© Copyright by Lina Chan, 2011

ii
Regioselective Functionalization of Polyols via Organoboron
Catalysis
Lina Chan
Masters of Science
Department of Chemistry
University of Toronto
2011
Abstract
With the increasing realization of their involvement in numerous biological processes, synthetic
oligosaccharides present promising potential in drug and vaccine discovery. Selective
functionalization of hydroxy groups in polyols represent a long-standing goal in chemistry since
the chemical synthesis of O-glycosides often requires extensive protecting group manipulation.
Organoboron catalysis is a recent strategy for regioselective activation of the equatorial hydroxy
group of cis-vicinal diols. Following the initial findings that diarylborinic acid catalyzes the
regioselective acylation of carbohydrate derivatives, kinetic studies were conducted to obtain
better insight on the mechanism. Thereafter, the ability of diarylborinic acid to catalyze the
regioselective alkylation of carbohydrates was demonstrated. Finally, investigations in the
capability of diarylborinic acid to influence regiochemical outcome of glycosylation reactions
were explored. Similarly, kinetic experiments were devised to shed light on the mechanism of
the reaction.

iii
Dedication:
This dissertation is dedicated to Keith Fagnou.
Being the best is nothing. Being the best and being modest is everything.

iv
Acknowledgments
First and foremost, I am thankful to my family. Without their encouragement and belief in me, I
would have not been able to come this far. While everyone else’s kid went to medical or law
school, my parents didn’t force that on me. They let me pick my own path and supported it no
matter what, and that is something I greatly admire about them.
I am eternally grateful to Dr. Keith Fagnou. He has changed my life and everyone who has been
part of the Fagnou lab understands where I am coming from when I say he was a great mentor
and friend. I am glad I got the chance to join his lab and if it weren’t for him, all the
opportunities I’ve been given since then would have never happened.
I can’t say that without mentioning Malcolm Huestis and David Stuart. Most of the success I
have achieved was guided by their chemistry advice, and now I am especially thankful to
Malcolm as he is always looking out for me. I would like to thank Ho-yan Sun and David
Lapointe for always having time to help me out again and again. It was awesome being able to
work with such a great people and I’m definitely proud to be a Fagnou girl.
Over my time at the University of Toronto, Dr. Mark S. Taylor has given me guidance, support,
and the opportunity of a lifetime, and I truly thank him. Working in his lab, I experienced a lot
and did some ‘sweet’ chemistry! I hope to take all that I have learned and continue to grow.
A big thank you to the Taylor gang! I am especially thankful to Corey McClary and Alice Wei.
When I first joined the Taylor lab, Corey and Alice were the people who made me feel truly
welcome. I also want to thank Christina Gouliaras, as she has been with me through all the harsh
times in Ottawa and Toronto.
A special thank you goes to Kevin Kou for helping me waste the evenings on endless
conversations, and for truly believing in me. I hope we’ll be friends for a lifetime.
Lastly, I want to thank my very awesome supervisor at Genentech, Mike Siu. Thanks Mike! For
giving me a chance that literally changed my life.

v
Table of Contents
Abstract ................................................................................................................... ii
Acknowledgments ...................................................................................................vi
Table of Contents.....................................................................................................v
List of Abbreviations ........................................................................................... viii
List of Tables ......................................................................................................... xii
List of Figures .......................................................................................................xiv
List of Schemes.................................................................................................... xvii
Chapter 1: Mechanistic Studies on the Monofunctionalization of Polyols by
Boron Catalysis ...................................................................................................... 1 1.0 Introduction...................................................................................................................... 1
1.1 Mechanistic Proposal of Borinic Acid-Catalyzed Functionalization of Diols ............ 6
1.2 Kinetic Studies on the Mechanism ................................................................................. 7 1.2.1 Pseudo First-order Kinetics in cis-1,2-Cyclohexanediol .........................................................9 1.2.2 Dependence of the Initial Rate on the Concentration of 4-Toluenesulfonyl Chloride ..........10 1.2.3 Dependence of the Initial Rate on the Concentration of N,N-Diisopropylethylamine ..........12 1.2.4 Dependence of the Initial Rate on the Concentration of 2-Aminoethyl Diphenylborinate
…………………...………………………………………………………………….……...…………13 1.2.5 Effect of 2-Aminoethanol ......................................................................................................15 1.2.6 Electronic and Steric Effects on Reactivity ...........................................................................18
1.3 Conclusion ...................................................................................................................... 24
Chapter 2: Regioselective Alkylation of Carbohydrate Derivatives Catalyzed
by a Diarylborinic Acid Derivative ..................................................................... 26 2.0 Introduction.................................................................................................................... 26
2.1 Reactivity of the Hydroxy Group................................................................................. 27
2.2 Reductive Cleavage of Benzylidene Acetals ................................................................ 29

vi
2.3 Organotin-Mediated Regioselective Alkylation .......................................................... 31 2.3.1 Stannyl Ethers ........................................................................................................................32 2.3.2 Stannylene Acetals .................................................................................................................36
2.4 Other Transition Metal Promoted Alkylations........................................................... 39
2.5 Boron Activation of Hydroxy Groups for Regioselective Alkylation ....................... 41
2.6 Diarylborinic Acid Catalyzed Regioselective Alkylation ........................................... 43 2.6.1 Synthesis of the Carbohydrate Substrates ..............................................................................44 2.6.2 Reaction Development and Extension of Scope ....................................................................45
2.7 Monoalkylation via Halide Catalysis ........................................................................... 54
2.8 DFT Calculations ........................................................................................................... 58
2.9 Conclusion ...................................................................................................................... 59
Chapter 3: Regioselective Glycosylation by a Diarylborinic Acid Derivative 61 3.0 Introduction.................................................................................................................... 61
3.1 Inherent Reactivity of Hydroxy Groups in Glycosylations........................................ 62
3.2 Regioselective Glycosylations of Carbohydrate Derivatives via Activating Agents 64 3.2.1 Activation via Organotin Reagents ........................................................................................65 3.2.2 Activation via Arylboronic Acids ..........................................................................................69
3.3 Regioselective Activation of Glycosyl Acceptors by a Diarylborinic Acid Catalyst 72 3.3.1 Synthesis of Glucosamine Derived Donors ...........................................................................74 3.3.2 Regioselective Glycosylations with Nitrogen-Containing Glycosyl Donors.........................76 3.3.3 Regioselective Glycosylations to Form β-Mannoside Linkages............................................80 3.3.4 Regioselective Glycosylations with Halide Ion Catalysis .....................................................83 3.3.5 Regioselective Glycosylations with Stoichiometric Boronic Acid ........................................86
3.4 Kinetic Studies: Reagent Order ................................................................................... 88 3.4.1 Pseudo First-order Kinetics in Glycosyl Donor .....................................................................88 3.4.2 Dependence of the Initial Rate on the Concentration of Glycosyl Donor .............................89 3.4.3 Dependence of the Initial Rate on the Concentration of Glycosyl Acceptor .........................91 3.4.4 Dependence of the Initial Rate on the Concentration of Borinic Acid-Catalyst....................92 3.4.5 Dependence of the Initial Rate on the Concentration of Promoter ........................................94 3.4.6 Dependence of Rate on the Nature of the Catalyst Used .......................................................95 3.4.7 Effect of 2-Aminoethanol ......................................................................................................96
3.5 Conclusion ...................................................................................................................... 97

vii
Chapter 4: Final Conclusion................................................................................ 98
Chapter 5: Experimental Procedures ............................................................... 101 5.0 General Information.................................................................................................... 101
5.1 Experimental and Characterization Data ................................................................. 102 5.1.1 General Procedure A: Tosylation Kinetic Experiments.......................................................102 5.1.2 General Procedure B: Hydrolysis of Diarylboronic Esters ..................................................103 5.1.3 General Procedure C: Borinic Acid-Catalyzed Alkylation ..................................................103 5.1.4 General Procedure D: Borinic Acid-Catalyzed Alkylation with Halide Salts .....................103 5.1.5 General Procedure E: Borinic Acid-Catalyzed Glycosylation.............................................131 5.1.6 General Procedure F: Borinic Acid-Catalyzed Synthesis of Sugar Substrates Bearing a
Carbonate at O-2 and O-3 ....................................................................................................131 5.1.7 General Procedure G: Borinic Acid-Catalyzed Glycosylation with Halide Catalysis .........134 5.1.8 General Procedure H: Borinic Acid-Mediated Glycosylation .............................................135 5.1.9 General Procedure I: Glycosylation Kinetic Experiments ...................................................136
Appendix A: NMR Spectra................................................................................ 137
Appendix B: DFT Calculations ......................................................................... 195

viii
List of Abbreviations
1H proton NMR
13C carbon 13 NMR
°C degrees Celcius
aq. aqueous
Ac acetyl
Bn benzyl
BOM benzyloxymethyl
Bu butyl
Bz benzoyl
CDI N,N’-carbonyldiimidazole
d doublet
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
DCE dichloroethane
DCM dichloromethane
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
EI electron impact
Et ethyl

ix
equiv equivalents
ESI electron spray ionization
fk Fukui index
FTIR fourier-transform infrared spectrometry
HMPA hexamethylphosphoramide
HPLC high-performance liquid chromatography
HRMS high-resolution mass spectrometry
hr hour
IR infrared spectrometry
kobs observed rate constant
M molar
m multiplet
M+ parent molecular ion
Me methyl
mg milligram
MHz megahertz
mL milliliters
mmol millimoles
MS molecular sieves
Nap naphthyl

x
NIS N-iodosuccinimide
NMR nuclear magnetic resonance
NOESY nuclear overhauser enhancement spectroscopy
n.r. no reaction
Oct octyl
OH hydroxy
Ph phenyl
Piv pivaloyl
PMB p-methoxybenzyl
ppm parts per million
rpm revolutions per minute
rt room temperature
s singlet
t tert
t triplet
TBAB tetrabutylammonium bromide
TBAF tetrabutylammonium fluoride
TBAI tetrabutylammonium iodide
TBMDSOTf tert-butyldimethylsilyl trifluoromethane sulfonate
TBSCl tert-butyldimethylsilyl chloride

xi
TCP tetrachlorophthaloyl
TCT 2,4,6-trichloro[1,3,5]triazine
TESCl triethylchlorosilane
THF tetrahydrofuran
TLC thin layer chromatography
Tr trityl
Troc 2,2,2-trichloroethyl chloroformate
µL microliter

xii
List of Tables
Table 1.1 – Synthesis of Diarylborinate Catalysts ....................................................................... 19
Table 1.2 – Synthesis of Borinate Esters Containing Other Ligands........................................... 20
Table 1.3 – Synthesis of 8-Hydroxyquinoline Diarylborinate Ester Catalysts ............................ 21
Table 1.4 – Sulfonylation of cis-1,2-Cyclohexanediol Using 8-Hydroxyquinoline Diarylborinate
Esters as Catalysts......................................................................................................................... 22
Table 2.1 – Solvent Screen for Alkylation of Carbohydrate Derivatives .................................... 46
Table 2.2 – Optimization for Alkylation of Carbohydrate Derivatives ....................................... 47
Table 2.3 – Further Optimization for Alkylation of Carbohydrate Derivatives........................... 48
Table 2.4 – Further Optimization for Alkylation of Carbohydrate Derivatives........................... 49
Table 2.5 – Optimization of Silver(I) Salt.................................................................................... 50
Table 2.6 – Scope of Regioselective Alkylation of Monosaccharides......................................... 51
Table 2.7 – Scope of Regioselective Allylation of Monosaccharides.......................................... 52
Table 2.8 – Scope of Regioselective p-Methoxybenzylation of Monosaccharides ..................... 53
Table 2.9 – Alkylation of Using Sodium Iodide and Brønsted bases .......................................... 55
Table 2.10 – Optimization of Iodide Salts ................................................................................... 55
Table 2.11 – Glycosylation of Thiogalactosides Using TBAI in Halide Catalysis...................... 57
Table 2.12 – Glycosylation of Thiogalactosides Using KI in Halide Catalysis........................... 58

xiii
Table 3.1 – Scope of Optimized Reaction Conditions for Glycosylation.................................... 73
Table 3.2 – Glycosylation Screen with 2-Phthalimido-β-D-Glucopyranosyl Bromide ............... 76
Table 3.3 – Optimization of Reaction Conditions........................................................................ 77
Table 3.4 – Glycosylation Screen with 2-[(2,2,2-Trichloroethoxy)carbonylamino]α-D-
Glucopyranosyl Bromide .............................................................................................................. 78
Table 3.5 – Glycosylation Screen with 2-Azido-α-D-Galactopyranosyl Chloride ...................... 79
Table 3.6 – Synthesis of 2,3-O-Carbonyl-α-L-Rhamnopyranoside ............................................. 81
Table 3.7 – Screening Conditions for Desilylation ...................................................................... 82
Table 3.8 – Glycosylation with 4-Benzoyl-2,3-O-Carbonyl-α-L-Rhamnopyranosyl Bromide... 83
Table 3.9 – Glycosylation of Methyl-6-(tert-Butyldimethylsilyloxy)-α-D-Mannopyranoside via
Organoboron and Halide Ion Catalysis......................................................................................... 84
Table 3.10 – Glycosylation of Methyl-6-(tert-Butyldimethylsilyloxy)-α-D-Galactopyranoside
via Organoboron and Halide Ion Catalysis ................................................................................... 85
Table 3.11 – Glycosylation Using 2,3,4,6-Tetra-O-Benzyl-α-D-Mannopyranosyl Chloride ...... 86
Table 3.12 – Boronic Acid-Activated Glycosylation of Rhamnose Derivative........................... 87

xiv
List of Figures
Figure 1.1 – D-Glucose Selective Molecular Fluorescence Sensor............................................... 4
Figure 1.2 – Boronic Acid-Based Chiral Saccharide Sensor ......................................................... 4
Figure 1.3 – Proposed Catalytic Cycle for Regioselective Monofunctionalization of Diols......... 7
Figure 1.4 – Formation of 2-Hydroxycyclohexyl 4-Methylbenzenesulfonate Over Time ............ 8
Figure 1.5 – Plot of cis-1,2-Cyclohexanediol Concentration Versus Time Under Pseudo First-
order Reaction Conditions ............................................................................................................ 10
Figure 1.6 – Formation of Product Over Time With Variation in the Concentration of 4-
Toluenesulfonyl Chloride ............................................................................................................. 11
Figure 1.7 – Initial Rate Dependence on the Concentration of 4-Toluenesulfonyl Chloride ...... 11
Figure 1.8 – Formation of Product Over Time With Variation in the Concentration of N,N-
Diisopropylethylamine.................................................................................................................. 12
Figure 1.9 – Initial Rate Dependence on the Concentration of N,N-Diisopropylethylamine ...... 13
Figure 1.10 – Formation of Product Over Time With Variation in the Concentration of 2-
Aminoethyl Diphenylborinate ...................................................................................................... 14
Figure 1.11 – Initial Rate Dependence on the Concentration of 2-Aminoethyl Diphenylborinate
....................................................................................................................................................... 14
Figure 1.12 – Consumption of cis-1,2-Cyclohexanediol Over Time Using 2-Aminoethyl
Diphenylborinate (A) or Diphenylborinic Acid (B) as the Catalyst ............................................. 16
Figure 1.13 – Rate of Consumption of cis-1,2-Cyclohexanediol Using 2-Aminoethyl
Diphenylborinate (A) or Diphenylborinic Acid (B) as the Catalyst ............................................. 16

xv
Figure 1.14 – Consumption of cis-1,2-Cyclohexanediol Over Time in the Absence (A) and
Presence (B) of Excess 2-Aminoethanol ...................................................................................... 17
Figure 1.15 – Rate of Consumption of cis-1,2-Cyclohexanediol Over Time in the Absence (A)
and Presence (B) of Excess 2-Aminoethanol................................................................................ 18
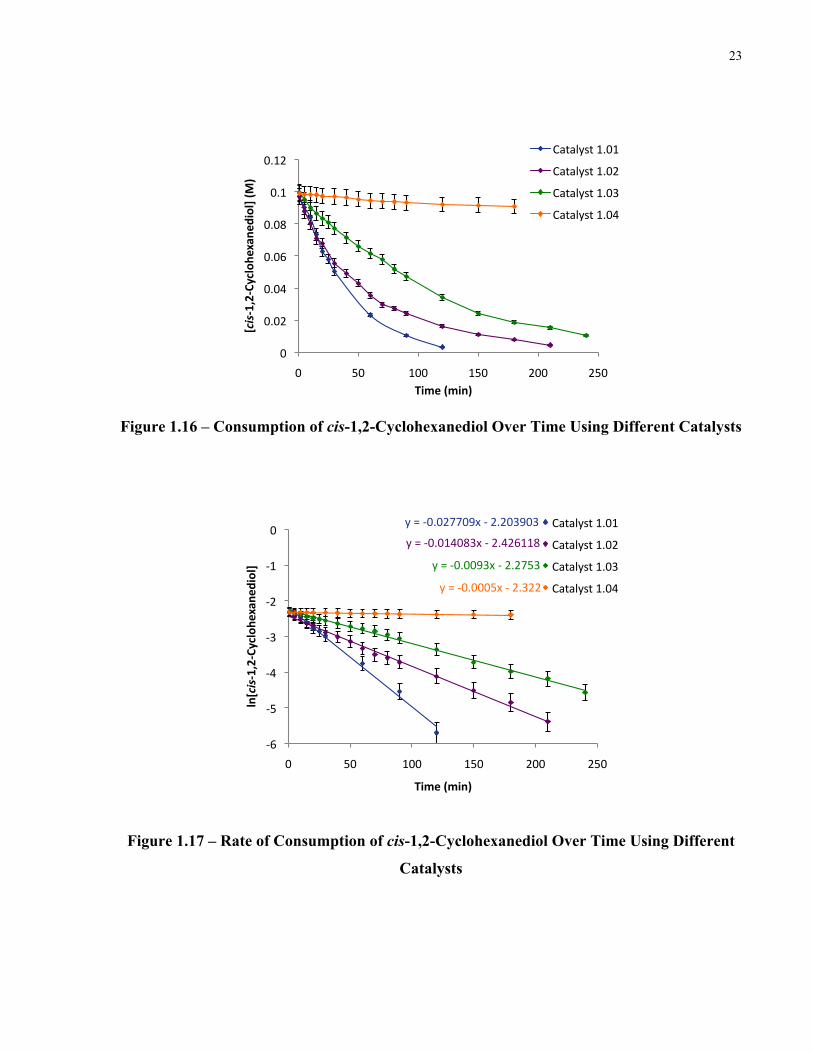
Figure 1.16 – Consumption of cis-1,2-Cyclohexanediol Over Time Using Different Catalysts. 23
Figure 1.17 – Rate of Consumption of cis-1,2-Cyclohexanediol Over Time Using Different
Catalysts ........................................................................................................................................ 23
Figure 2.1 – Incompatible Monosaccharide Derivatives ............................................................. 54
Figure 2.2 – Calculated Structures and Condensed Fukui Indices (B3LYP/6-311+G(d,p)) ....... 60
Figure 3.1 – Plot of 2,3,4,6-Tetra-O-Acetyl-α-D-Glucopyranosyl Bromide Concentration Versus
Time Under Pseudo First-order Reaction Conditions................................................................... 89
Figure 3.2 – Formation of Product Over Time With Variation in the Concentration of Glycosyl
Donor ............................................................................................................................................ 90
Figure 3.3 – Initial Rate Dependence on the Concentration of Glycosyl Donor ......................... 90
Figure 3.4 – Formation of Product Over Time With Variation in the Concentration of Glycosyl
Acceptor ........................................................................................................................................ 91
Figure 3.5 – Initial Rate Dependence on the Concentration of Glycosyl Acceptor..................... 92
Figure 3.6 – Formation of Product Over Time With Variation in the Concentration of 2-
Aminoethyl Diphenylborinate ...................................................................................................... 93
Figure 3.7 – Initial Rate Dependence on the Concentration of 2-Aminoethyl Diphenylborinate 93
Figure 3.8 – Formation of Product Over Time With Variation in the Concentration of Silver
Oxide............................................................................................................................................. 94

xvi
Figure 3.9 – Initial Rate Dependence on the Concentration of Silver Oxide .............................. 95
Figure 3.10 – Formation of Product Over Time Using Diphenylborinic Acid (A) or 2-
Aminoethyl Diphenylborinate (B) as the Catalyst........................................................................ 96
Figure 3.11 – Formation of Product Over Time in the Absence (A) and Presence (B) of Excess
2-Aminoethanol ............................................................................................................................ 97

xvii
List of Schemes
Scheme 1.1 – Boronic Acid–Diol Complexation Equilibria .......................................................... 3
Scheme 1.2 – Boron Containing Fluorescent Chemosensors for Polyols ...................................... 3
Scheme 1.3 – Diphenylborinic Acid-Catalyzed Aldol Reaction of Pyruvic Acids........................ 5
Scheme 1.4 – Borinic Ester-Catalyzed Regioselective Acylation of Carbohydrate Derivatives ... 6
Scheme 1.5 – Regioselective Sulfonylation of Carbohydrate Derivatives..................................... 6
Scheme 1.6 – Regioselective Tosylation of cis-1,2-Cyclohexanediol ........................................... 8
Scheme 1.7 – Tosylation of cis-1,2-Cyclohexanediol Under Pseudo First-order Reaction
Conditions ....................................................................................................................................... 9
Scheme 1.8 – Reaction Order in 4-Toluenesulfonyl Chloride ..................................................... 10
Scheme 1.9 – Reaction Order in N,N-Diisopropylethylamine ..................................................... 12
Scheme 1.10 – Reaction Order in 2-Aminoethyl Diphenylborinate ............................................ 13
Scheme 1.11 – Effect of 2-Aminoethanol .................................................................................... 15
Scheme 1.12 – Cleavage of 2-Aminoethanol by Acid Catalysis.................................................. 15
Scheme 1.13 – Effect of Excess 2-Aminoethanol ........................................................................ 17
Scheme 1.14 – Schaab’s Synthesis of Borinic Acids ................................................................... 19
Scheme 1.15 – Tavassoli’s Synthesis of Borinic Acids ............................................................... 19
Scheme 1.16 – Electronic and Steric Effects on Reactivity ......................................................... 22
Scheme 2.1 – Selective Alkylation of O-2 Over O-3 Using Phase Transfer Catalysis................ 28

xviii
Scheme 2.2 – Selective Alkylation of O-6 Over O-4 Using Phase Transfer Catalysis................ 28
Scheme 2.3 – Regioselective Alkylation of the Anomeric Position by Alkyl Triflates............... 29
Scheme 2.4 – Preferential Reactivity of Hydroxy Groups in D-Glucal ....................................... 29
Scheme 2.5 – Bhattacharjee and Gorin’s Reductive Cleavage of Benzylidene Acetals .............. 30
Scheme 2.6 – Nánási and Lipták’s Reductive Cleavage of Benzylidene Acetals........................ 30
Scheme 2.7 – Garegg’s Reductive Cleavage of Benzylidene Acetals ......................................... 30
Scheme 2.8 – Solvent Effect on Reductive Cleavage of Benzylidene Acetals ............................ 31
Scheme 2.9 – Stannyl Ethers and Stannylene Acetals ................................................................. 31
Scheme 2.10 – Organotin-Mediated Sulfamoylation of Adenosine Derivative........................... 32
Scheme 2.11 – Regioselective O-Stannylation ............................................................................ 33
Scheme 2.12 – Ogawa and Matsui’s Benzoylation of Polyols..................................................... 33
Scheme 2.13 – Ogawa’s Benzylation of Alkyl β-D-Glucopyranoside Derivatives ..................... 34
Scheme 2.14 – Ogawa’s Benzylation of Methyl β-D-Galactopyranoside ................................... 34
Scheme 2.15 – Ogawa’s Benzylation of Methyl α-D-Glucopyranoside ...................................... 35
Scheme 2.16 – Veyrières’ Benzylation of α-D-Mannopyranoside Derivatives ........................... 35
Scheme 2.17 – Effect of Halide Salts on Trialkyltin Species....................................................... 35
Scheme 2.18 – Veyrières’ Benzylation of α-D-Glucopyranoside Derivatives ............................ 36
Scheme 2.19 – Moffatt’s Benzylation of β-D-Ribofuranosyl Nucleosides.................................. 36
Scheme 2.20 – David’s Benzylation of α-D-Galactopyranoside Derivatives .............................. 37
Scheme 2.21 – Nashed and Anderson’s Benzylation of α-D-Galactopyranoside Derivatives .... 37

xix
Scheme 2.22 – David and Thieffry’s Benzylation of D-Galactopyranoside Derivatives ............ 38
Scheme 2.23 – Ley’s Selective Alkylation of Diols Using Dibutyltin Dimethoxide................... 38
Scheme 2.24 – Tin(II) Catalyzed Benzylation of α-L-Rhamnopyranoside Derivatives .............. 39
Scheme 2.25 – Schuerch’s Alkylation of α-D-Mannopyranoside Derivatives ............................ 40
Scheme 2.26 – Gridley’s Methylation of β-D-Glucopyranoside Derivatives .............................. 40
Scheme 2.27 – Demchenko’s Alkylation of Carbohydrate Derivatives....................................... 40
Scheme 2.28 – Kartha’s Alkylation of Carbohydrate Derivatives ............................................... 41
Scheme 2.29 – Aoyama’s Alkylation of Fucose and Arabinose Derivatives............................... 42
Scheme 2.30 – Onomura’s Monoalkylation of 1,2-Diols............................................................. 42
Scheme 2.31 – Evtushenko’s Methylation of Methyl Glucopyranosides .................................... 42
Scheme 2.32 – Borinic Acid-Catalyzed Monoacylation of cis-1,2-Diols .................................... 43
Scheme 2.33 – Proposal for Regioselective Alkylation of Polyols.............................................. 43
Scheme 2.34 – TBS-Protection of the Primary Hydroxy Group.................................................. 44
Scheme 2.35 – Synthesis of 3,4,6-Tri-O-Benzyl-D-Mannopyranoside ....................................... 45
Scheme 2.36 – Extension of Halide Catalysis Method to Other Substrates................................. 56
Scheme 3.1 – Selective Glycosylation Due to Steric Effects ....................................................... 62
Scheme 3.2 – Regiodifferentiation in Glycosylation Reactions................................................... 63
Scheme 3.3 – Glycosylations of n-Pentenyl Glycosides and n-Pentenyl Ortho Esters................ 64
Scheme 3.4 – Ogawa’s Regioselective Glycosylation of Carbohydrate Derivatives................... 65

xx
Scheme 3.5 – Augé and Veyrières’ Regioselective Glycosylation Method................................. 66
Scheme 3.6 – Regioselective Glycosylation of 1,6-Anhydro-β-D-Galactopyranose................... 67
Scheme 3.7 – Regioselective Glycosylation of Methyl β-Lactoside............................................ 67
Scheme 3.8 – Regioselective Glycosylation of Methyl β-D-Galactopyranoside ......................... 68
Scheme 3.9 – Regioselective Glycosylation of Methyl β-D-Galactopyranoside With Silver Silica
Alumina as a Promoter.................................................................................................................. 68
Scheme 3.10 – Protection of Carbohydrate Derivatives by Boronic Acids ................................. 69
Scheme 3.11 – Regioselective Glycosylation Using Boronic Acids as a Protecting Group........ 69
Scheme 3.12 – Boronic Acids as a Transient Masking Agent ..................................................... 70
Scheme 3.13 – Tetracoordinated Arylboronate Complex ............................................................ 71
Scheme 3.14 – Regioselective Glycosylation Activated by Arylboronic Acids .......................... 71
Scheme 3.15 – Synthesis of 2-Phthalimido-β-D-Glucopyranosyl Bromide ................................ 74
Scheme 3.16 – Synthesis of 2-Phthalimido-α-D-Glucopyranosyl Chloride ................................ 74
Scheme 3.17 – Synthesis of 2-[(2,2,2-Trichloroethoxy)carbonylamino]-α-D-Glucopyranosyl
Bromide......................................................................................................................................... 75
Scheme 3.18 – Synthesis of 2-Azido-α-D-Galactopyranosyl Chloride ....................................... 75
Scheme 3.19 – Unsuccessful Glycosylation of 2-Phthalimido-β-D-Glucopyranosyl Chloride ... 78
Scheme 3.20 – Formation of β-Mannoside Linkages................................................................... 80
Scheme 3.21 – Synthesis of 4-Benzoyl-2,3-O-Carbonyl-α-L-Rhamnopyranosyl Bromide ........ 82
Scheme 3.22 – Synthesis of Methyl-6-(tert-Butyldimethylsilyloxy)-2,3-O-Carbonyl-α-D-
Mannopyranoside.......................................................................................................................... 82

xxi
Scheme 3.23 – Glycosylation Under Pseudo First-order Reaction Conditions............................ 89
Scheme 3.24 – Reaction Order in Glycosyl Donor ...................................................................... 90
Scheme 3.25 – Reaction Order in Glycosyl Acceptor.................................................................. 91
Scheme 3.26 – Reaction Order in 2-Aminoethyl Diphenylborinate ............................................ 92
Scheme 3.27 – Reaction Order in Silver Oxide ........................................................................... 94
Scheme 3.28 – Effect of the Nature of Catalyst Used.................................................................. 95
Scheme 3.29 – Effect of 2-Aminoethanol .................................................................................... 96

1
1 Mechanistic Studies on the Monofunctionalization of Polyols
by Boron Catalysis
1.0 Introduction
Direct, regioselective protection of hydroxy groups in diols and polyols represents an important
and long-standing goal in chemistry. The selective functionalization of carbohydrates is an
increasingly active area of research due to its broad potential in synthesis. In addition, this
strategy enables readily available starting materials such as sugar feedstocks to be converted into
value-added intermediates that are otherwise difficult to prepare in short reaction sequences. The
development of regioselective methods has undergone significant advances in recent years with
efforts focused towards strategies based on catalysis, which include enzyme-catalyzed1 methods,
organocatalytic methods2, Lewis acid-promoted processes3, and tandem catalytic reactions of
1 (a) Therisod, M.; Klibanov, A. M. J. Am. Chem. Soc. 1987, 109, 3977–3981. (b) Wang, Y.-F.; Lalonde, J. J.; Momongan, M.; Bergbreiter, D. E.; Wong, C.-H. J. Am. Chem. Soc. 1988, 110, 7200–7205. 2 (a) Griswold, K. S.; Miller, S. J. Tetrahedron 2003, 59, 8869–8875. (b) Kawabata, T.; Muramatsu, W.; Nishio, T.; Shibata, T.; Schedel, H. J. Am. Chem. Soc.2007, 129, 12890–12895.

2
persilyated sugar derivatives4. In light of these studies, organoboron reagents are emerging as an
attractive alternative in comparison to organometallic reagents. The ability for organoboron
compounds to form reversible covalent interactions with diols has been employed extensively in
the design of receptors and sensors for carbohydrates in aqueous media.5
In aqueous solutions, an equilibria boronic acid–diol complexation exists between the boronic
acid and boronate ester (Scheme 1.1).5b These species also undergo acid-base equilibria to form
anionic tetracoordinated species, where the acidity of the boronate ester is generally higher than
the boronic acid. It is postulated that this effect is due to the relative lower angle strain present in
the tetracoordinate boronate ester in comparison to the tricoordinate conjugate acid. Lorand and
Edwards exploited this phenomenon by measuring the pH depression to determine the
association constants between phenylboronic acid and various polyols (including carbohydrate
derivatives) in water.6 For more than four decades, these studies remained the dominant source
of quantitative data in regards to phenylboronic acid–diol complex.
More recently, Wang and co-workers reported quantitative studies of arylboronic acid–diol
equilibria using indicator–displacement assays to investigate pH and substitution effects on
binding affinity.7
3 Sn(IV) derivatives: (a) Iwasaki, F.; Maki, T.; Onomura, O.; Nakashima, W.; Matsumura, Y. J. Org. Chem. 2000, 65, 996–1002. (b) Martinelli, M. J.; Vaidyanathan, R.; Pawlak, J. M.; Nayyar, N. K.; Dhokte, U. P.; Doecke, C. W.; Zollars, L. M. H.; Moher, E. D.; Van Khau, V.; Kosmrjl, B. J. Am. Chem. Soc. 2002, 124, 3578–3585. (c) Demizu, Y.; Kubo, Y.; Miyoshi, H.; Maki, T.; Matsumura, Y.; Moriyama, N.; Onomura, O. Org. Lett. 2008, 10, 5075–5077. La(III) salts: Dhiman, R. S.; Kluger, R. Org. Biomol. Chem. 2010, 8, 2006–2008. 4 (a) Wang, C.-C.; Lee, J.-C.; Luo, S.-Y.; Kulkami, S. S.; Huang, Y.-W.; Lee, C.-C.; Chang, K.-L.; Hung, S.-C. Nature 2007, 446, 896–899. (b) Français, A.; Urban, D.; Beau, J.-M. Angew. Chem., Int. Ed. 2007, 46, 8662–8665. 5 (a) Nishiyabu, R.; Kubo, Y.; James, T. D.; Fossey, J. S. Chem. Commun. 2011, 47, 1106–1123. (b) James, T. D. Boronic Acids (Ed. D. G. Hall) 2005, pp. 441–479. 6 Lorand, J. P.; Edwards, J. O. J. Org. Chem. 1959, 24, 769–774. 7 (a) Springsteen, G.; Wang, B. Tetrahedron 2002, 58, 5291–5300; (b) Yan, J.; Springsteen, G.; Deeter, S.; Wang, B. Tetrahedron 2004, 60, 11205–11209.

3
Scheme 1.1 – Boronic Acid–Diol Complexation Equilibria
In the intervening years, several reports demonstrated the ability of boronic acid-based receptors
to relay the reversible, covalent binding of polyols into colorimetric or fluorescence-based
signals.
In a report by Czarnik, it was found that pKa modulation of anthrylboronic acid serves as a
useful fluorescent chemosensor for carbohydrates (Scheme 1.2).8
Scheme 1.2 – Boron Containing Fluorescent Chemosensors for Polyols
8 Yoon, J.; Czarnik, A. W. J. Am. Chem. Soc. 1992, 114, 5874–5875.
HO OH
R1 R2
HO OH
R1 R2
-2 H2O
Keq-trig
Keq-tet
-2 H2O
+ H2O- H+
+ H2O- H+
Ka2Ka1
BHO OH
BOO
R2R1
B O
OR2
R1
HOB OHOH
HO
B(OH)2
OOH
HOOH
OH
OHOH+
H2O
OOH
OO
OH
OH
BOH

4
Later, James and Shinkai reported a boronic acid based photoinduced electron transfer sensor
designed to selectively bind D-glucose (Figure 1.1).9 This was followed up by a chiral variant
that could detect one enantiomer in the presence of the other (Figure 1.2).10
Figure 1.1 – D-Glucose Selective Molecular Fluorescence Sensor
Figure 1.2 – Boronic Acid-Based Chiral Saccharide Sensor
More recent developments in this field have led to the synthesis of sensors that incorporate
boronic acid functional groups into three-dimensional scaffolds with spacings and orientations
complementary to the carbohydrate of interest.11
In contrast to the vast literature on the properties and applications boronic acids as receptors and
sensors, only one quantitative study on the application of borinic acids has been reported. In the
study conducted by Smith and co-workers, diphenylborinic acid and phenylboronic acid were
investigated as a glycoside transporter for aqueous/organic interfaces.12 It was revealed that the
two boron acids differ in their extraction efficiency and transport rate.
9 James, T. D.; Sandanayake, K. R. A. S.; Shinkai, S. Angew. Chem., Int. Ed. 1994, 33, 2207–2209. 10 James, T. D.; Sandanayake, K. R. A. S.; Shinkai, S. Nature 1995, 374, 345–347. 11 Shan, J.; Cheng, Y.; Reid, S.; Li, M.; Wang, B. Med. Res. Rev. 2010, 30, 171–257. 12 Morin, G. T.; Hughes, M. P.; Paugam, M.-F.; Smith, B. D. J. Am. Chem. Soc. 1994, 116, 8895–8901.
N
N
B BHO
OHOH
HO
OMe
N
OMe
N
B
B
OHHO
OHHO
Me
Me

5
Several studies on related compounds bearing two hydroxy groups such as catechols, α-
hydroxycarboxylic acids13, and the enol tautomers of pyruvic acids14, have also demonstrated the
ability to interact with boronic acids in a two-point fashion. They have also been exploited in
organoboron-based recognition assays.
Previously, our group has demonstrated the ability of catalytic boron to direct aldol reactions of
pyruvic acids in aqueous conditions where stabilization of the enol tautomer of pyruvic acids
underlies the process (Scheme 1.3).15 The studies revealed that borinic acids displayed far
superior catalytic activity and selectivity in comparison to boronic acids.
Scheme 1.3 – Diphenylborinic Acid-Catalyzed Aldol Reaction of Pyruvic Acids
Such interactions may become an area of interest for reaction development due to key features;
these include their tolerance of aqueous medium, favourable kinetics, and selectivity for cis-
vicinal diol moieties.16 Inspired by this reactivity, our group has developed methods for selective
acylation17 and sulfonylation18 of vicinal cis-diol moieties in carbohydrates catalyzed by 2-
aminoethyl diphenylborinate ester 1.01 (Scheme 1.4 and 1.5). These methods have been shown
to provide reliable selectivity for a given regiochemical outcome and generality for a wide range
of sugar substrate and protecting group combinations.
13 Wang, W.; Gao, X. Curr. Org. Chem. 2002, 6, 1285–1317. 14 Zhu, L.; Zhong, Z.; Anslyn, E. V. J. Am. Chem. Soc. 2005, 127, 4260–4269. 15 Lee, D.; Newman, S. G.; Taylor, M. S. Org. Lett. 2009, 11, 5486–5489. 16 (a) James, T. D.; Sandanayake, K. R. A. S.; Shinkai, S. Angew. Chem., Int. Ed. 1996, 35, 1910–1922.(b) Davis, A. P.; James, T. D. In Functional Synthetic Receptors; Schrader, T., Hamilton, A. D., Eds.; Wiley: Weinheim, 2005; pp 45-109. 17 Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. 18 Lee, D.; Williamson, C.; Chan, L.; Taylor, M. S. Unpublished results.
R OH
O
O
R' H
O
O
O
HO
R R'
(1.0–5.0 equiv)
Ph2BOH (0.5–20 mol %)H2O, rt
56–90% (13 examples)

6
Scheme 1.4 – Borinic Ester-Catalyzed Regioselective Acylation of Carbohydrate
Derivatives
While these methods are synthetically useful, the development of novel catalysts that show
increased reactivity and that can be applied to asymmetric catalysis still remains a challenge. In
order to develop new catalysts, a better mechanistic understanding of the reaction is crucial since
this mode of catalytic reactivity is not well precedented. As well, such studies would allow for
improvements to be made on existing reaction conditions and be useful in developing new
reactions. Herein a detailed investigation on the mechanism of the diarylborinic acid-catalyzed
regioselective monosulfonylation of cis-1,2-cyclohexanediol is described.
Scheme 1.5 – Regioselective Sulfonylation of Carbohydrate Derivatives
1.1 Mechanistic Proposal of Borinic Acid-Catalyzed Functionalization
of Diols
A proposed mechanism for the diarylborinic acid-catalyzed monofunctionalization reaction is
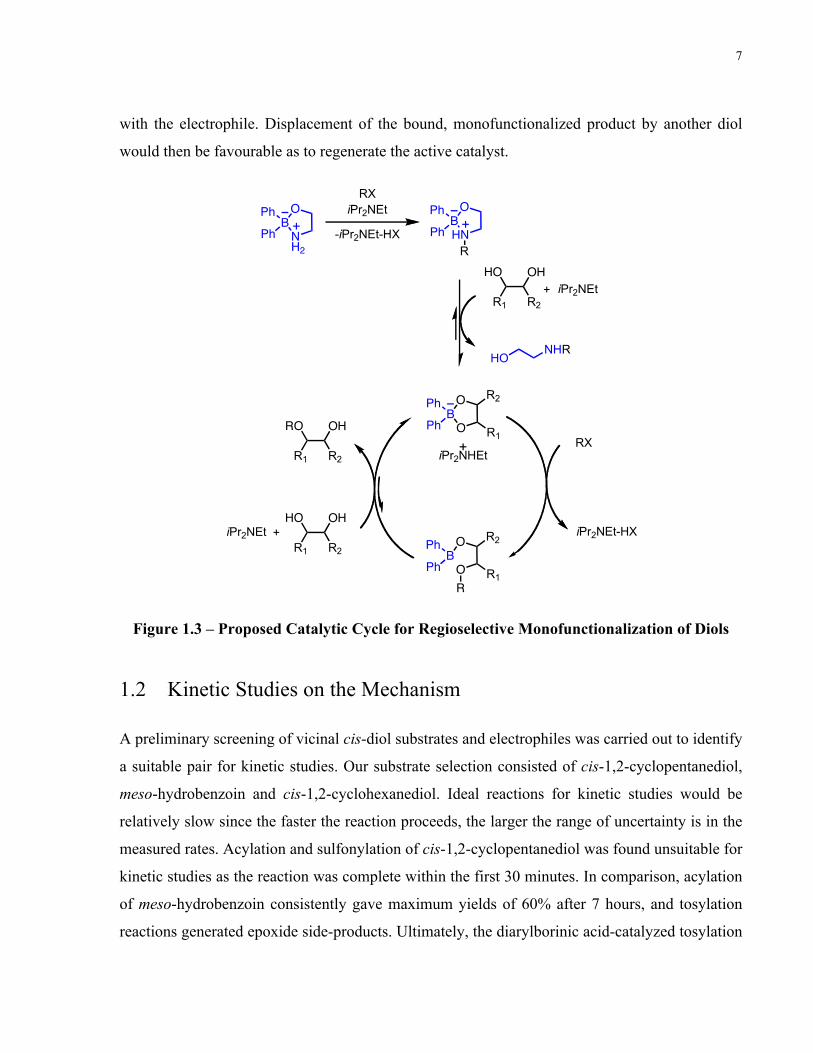
schematically depicted in the diagram below (Figure 1.3). In the sequence of steps postulated to
occur, 2-aminoethyl diphenylborinate 1.01 serves as a precatalyst from which under the reaction
conditions, the ethanolamine ligand is functionalized prior to displacement by the diol substrate.
Upon substrate binding, the organoboron catalyst and diol generate a cyclic ‘ate’ complex. Such
substrate-catalyst binding selectively activates one B-O bond over the other towards reactivity
PhB
Ph NH2
O
(5-10 mol %)
RCl (1.2–2.0 equiv)iPr2NEt (1.2–2.0 equiv)
MeCN, rt
R2R1
HO
HO R2R1
HO
RO
17 examples69–99%
1.01
OOTBSHO
HOOH
SiPr
PhB
Ph NH2
O
(10 mol %)
TsCl (1.2 equiv)iPr2NEt (1.2 equiv)
MeCN, rt
OOTBSHO
TsOOH
SiPr
97%
1.01
7
with the electrophile. Displacement of the bound, monofunctionalized product by another diol
would then be favourable as to regenerate the active catalyst.
Figure 1.3 – Proposed Catalytic Cycle for Regioselective Monofunctionalization of Diols
1.2 Kinetic Studies on the Mechanism
A preliminary screening of vicinal cis-diol substrates and electrophiles was carried out to identify
a suitable pair for kinetic studies. Our substrate selection consisted of cis-1,2-cyclopentanediol,
meso-hydrobenzoin and cis-1,2-cyclohexanediol. Ideal reactions for kinetic studies would be
relatively slow since the faster the reaction proceeds, the larger the range of uncertainty is in the
measured rates. Acylation and sulfonylation of cis-1,2-cyclopentanediol was found unsuitable for
kinetic studies as the reaction was complete within the first 30 minutes. In comparison, acylation
of meso-hydrobenzoin consistently gave maximum yields of 60% after 7 hours, and tosylation
reactions generated epoxide side-products. Ultimately, the diarylborinic acid-catalyzed tosylation
PhB
Ph NH2
O
HO OH
PhB
Ph O
O
PhB
Ph O
O
R
RXiPr2NEt
-iPr2NEt-HX
PhB
Ph HN
O
R
R1 R2iPr2NEt
HONHR
R1
R2
iPr2NHEt
R2
R1
RX
iPr2NEt-HX
RO OH
R1 R2
HO OH
R1 R2iPr2NEt +
+

8
reaction of cis-1,2-cyclohexanediol afforded the desired product in >99% yield after 4 hours
(Scheme 1.6, Figure 1.4).
Scheme 1.6 – Regioselective Tosylation of cis-1,2-Cyclohexanediol
Figure 1.4 – Formation of 2-Hydroxycyclohexyl 4-Methylbenzenesulfonate Over Time
With the goal of obtaining a more detailed understanding of the catalytic cycle, and more
specifically the intimate role of each of the reaction components, the order for each reagent in the
sulfonylation of cis-1,2-cyclohexanediol was obtained using the method of initial rates.
Employing 2-aminoethyl diphenylborinate 1.01 as the catalyst, the concentration of each reaction
component was varied and the progression of the reaction over 4 hours at room temperature was
monitored. The initial time of each kinetic run corresponded to the time at which N,N-
diisopropylethylamine was added to the reaction vial. During the course of the reaction, aliquots
of the reaction mixture were removed and quenched with methanol, thus stopping the reaction.
OH
OH
OTs
OHTsCl (1.1 equiv)iPr2NEt (1.1 equiv)MeCN, 0.2 M4 hr, rt
PhB
Ph NH2
O
(1 mol %)
100%
1.01
!"
#!"
$!"
%!"
&!"
'!!"
!" (!" %!" )!" '#!" '*!" '&!" #'!" #$!"
!"#$
%&'()
*+,$(-.
/(
0*1+(-1*2/(

9
The solvent was then removed and the resulting samples were analyzed by 1H-NMR
spectroscopy for the formation of product with mesitylene as an internal standard. Integrations
for the internal standard peak and an isolated proton peak of the product were used to calculate
moles of product formed and therefore % conversion in each of the aliquots. For each of the
reagents, the concentration of product formed was calculated and values not exceeding 10%
conversion were plotted against time. The slope was extrapolated to determine the initial rate of
the reaction, which was then plotted against concentration of the reagent to establish its order in
the reaction.
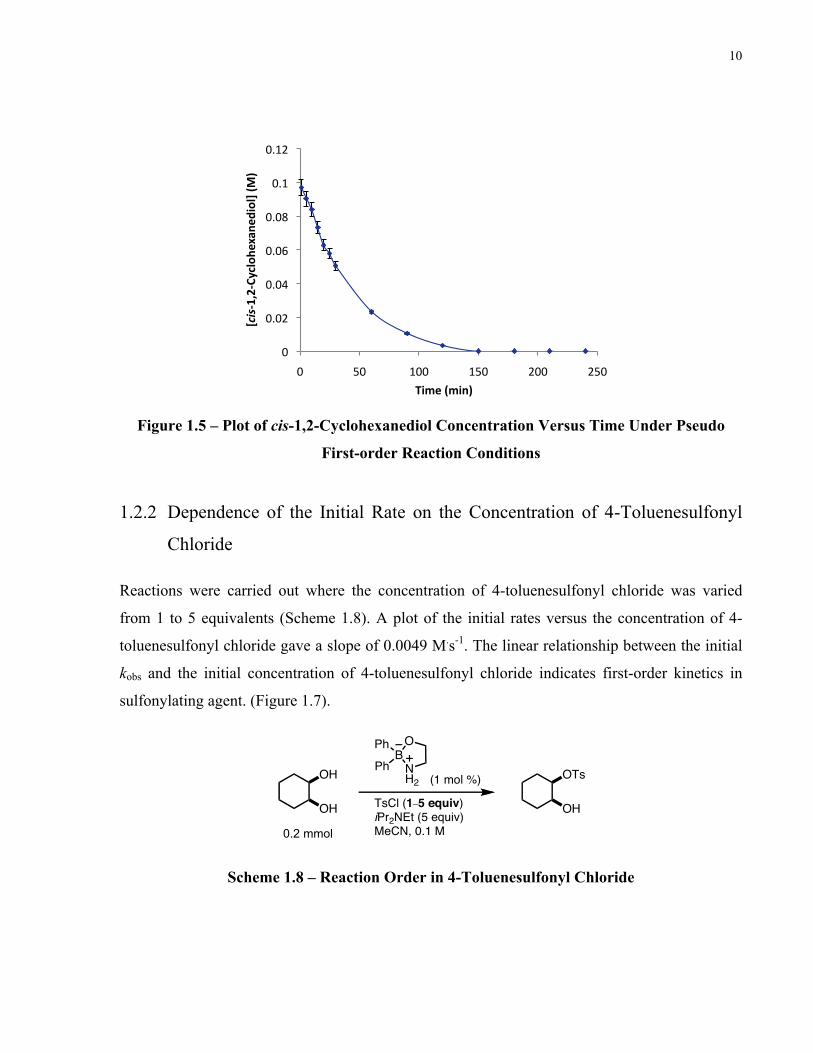
1.2.1 Pseudo First-order Kinetics in cis-1,2-Cyclohexanediol
The reaction was carried out under pseudo first-order conditions where cis-1,2-cyclohexanediol
was a limiting reagent and all other reagents were in excess in order to maintain a constant
concentration (Scheme 1.7). A plot of cis-1,2-cyclohexanediol concentration over time fit to the
exponential function:
[cis-1,2-cyclohexanediol](t) = [cis-1,2-cyclohexanediol]0e–kt
indicating the reaction is first-order in substrate (Figure 1.5).
Scheme 1.7 – Tosylation of cis-1,2-Cyclohexanediol Under Pseudo First-order Reaction
Conditions
OH
OH
OTs
OHTsCl (5 equiv)iPr2NEt (5 equiv)MeCN, 0.1 M
PhB
Ph NH2
O
(1 mol %)
0.2 mmol
1.01
10
Figure 1.5 – Plot of cis-1,2-Cyclohexanediol Concentration Versus Time Under Pseudo
First-order Reaction Conditions
1.2.2 Dependence of the Initial Rate on the Concentration of 4-Toluenesulfonyl
Chloride
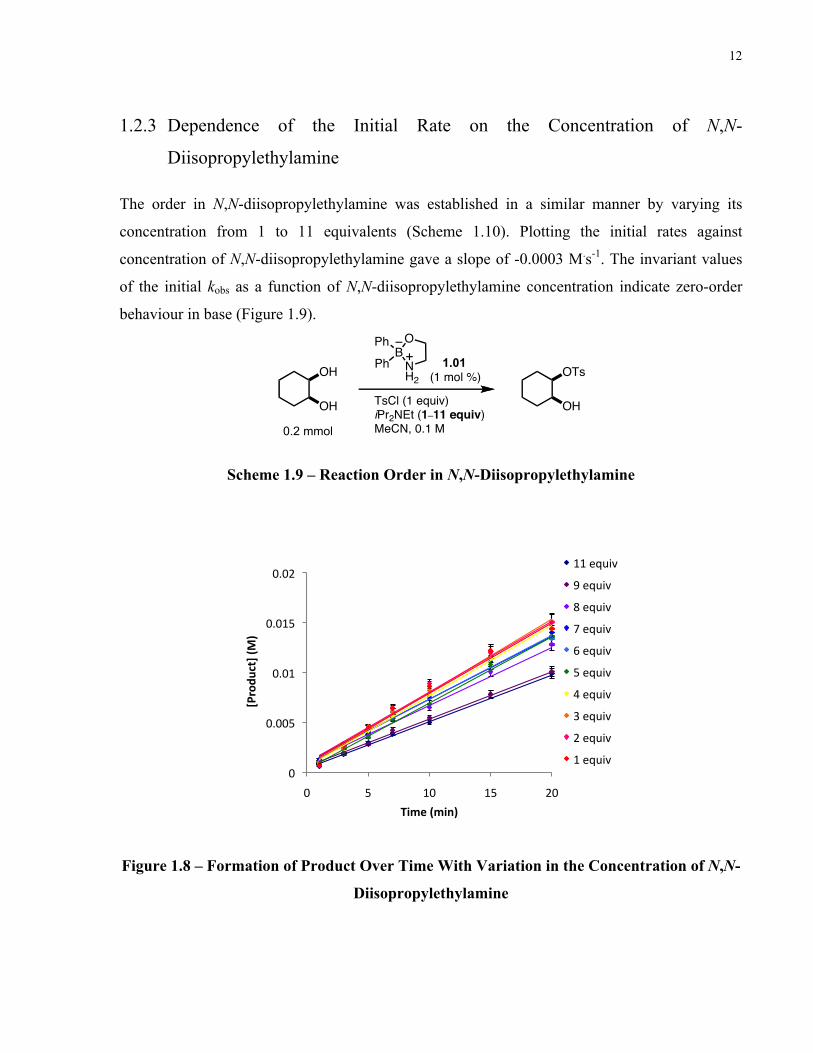
Reactions were carried out where the concentration of 4-toluenesulfonyl chloride was varied
from 1 to 5 equivalents (Scheme 1.8). A plot of the initial rates versus the concentration of 4-
toluenesulfonyl chloride gave a slope of 0.0049 M.s-1. The linear relationship between the initial
kobs and the initial concentration of 4-toluenesulfonyl chloride indicates first-order kinetics in
sulfonylating agent. (Figure 1.7).
Scheme 1.8 – Reaction Order in 4-Toluenesulfonyl Chloride
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#("
!#($"
!" )!" (!!" ()!" $!!" $)!"
!!"#"#$%"&'()*+,
-./,
01*)234563
718,3481/63
OH
OH
OTs
OHTsCl (1–5 equiv)iPr2NEt (5 equiv)MeCN, 0.1 M
PhB
Ph NH2
O
(1 mol %)
0.2 mmol

11
Figure 1.6 – Formation of Product Over Time With Variation in the Concentration of 4-
Toluenesulfonyl Chloride
Figure 1.7 – Initial Rate Dependence on the Concentration of 4-Toluenesulfonyl Chloride
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#!("
!#!)"
!" (" $!" $(" %!"
!"#$%&
'()*+,-*
./01*+0/2-*
(#!"*+,-."
'#!"*+,-."
&#!"*+,-."
%#!"*+,-."
$#!"*+,-."
!"#"$%$$&'(")"$%$$$*"
$"
$%$$$+"
$%$$*"
$%$$*+"
$%$$,"
$%$$,+"
$%$$-"
$%$$-+"
$" $%*" $%," $%-" $%&" $%+" $%."
! !"#$%#
&'($
)*&+!,-./.#-,0!/1,$23,!456.7$%8($
12
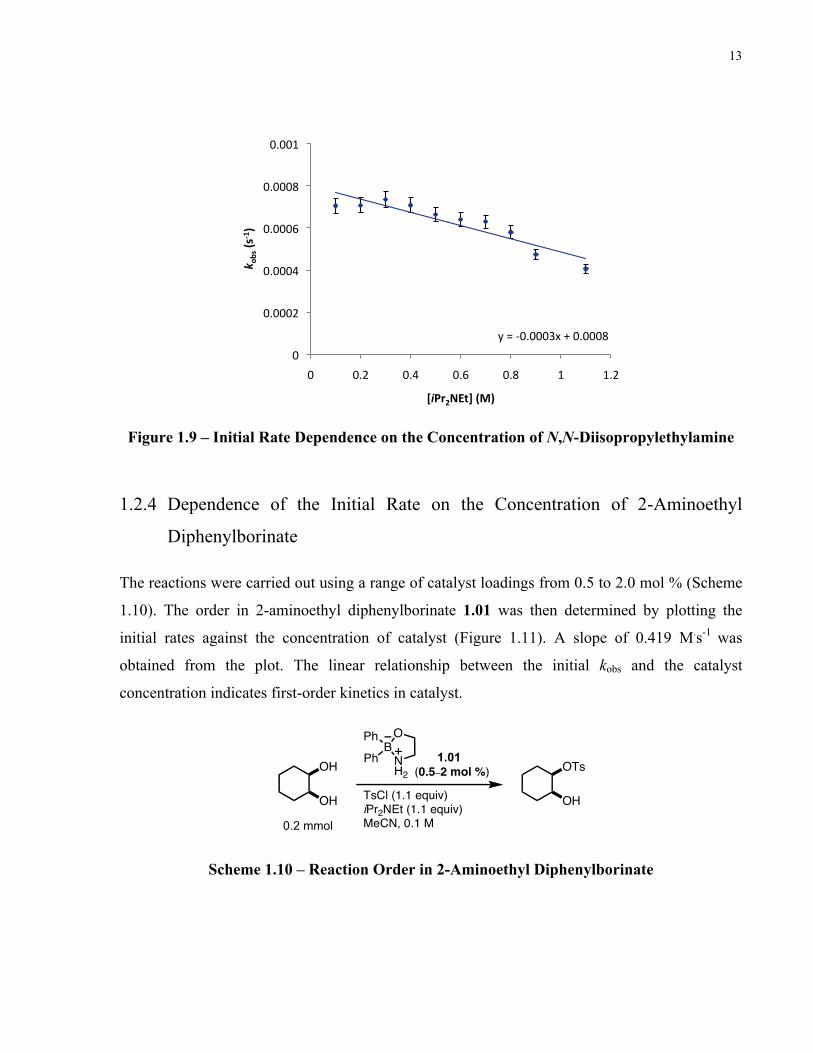
1.2.3 Dependence of the Initial Rate on the Concentration of N,N-
Diisopropylethylamine
The order in N,N-diisopropylethylamine was established in a similar manner by varying its
concentration from 1 to 11 equivalents (Scheme 1.10). Plotting the initial rates against
concentration of N,N-diisopropylethylamine gave a slope of -0.0003 M.s-1. The invariant values
of the initial kobs as a function of N,N-diisopropylethylamine concentration indicate zero-order
behaviour in base (Figure 1.9).
Scheme 1.9 – Reaction Order in N,N-Diisopropylethylamine
Figure 1.8 – Formation of Product Over Time With Variation in the Concentration of N,N-
Diisopropylethylamine
OH
OH
OTs
OHTsCl (1 equiv)iPr2NEt (1–11 equiv)MeCN, 0.1 M
PhB
Ph NH2
O
(1 mol %)
0.2 mmol
1.01
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!" $" %!" %$" &!"
!"#$%&
'()*+,-*
./01*+0/2-*
%%"'()*+"
,"'()*+"
-"'()*+"
."'()*+"
/"'()*+"
$"'()*+"
0"'()*+"
1"'()*+"
&"'()*+"
%"'()*+"
13
Figure 1.9 – Initial Rate Dependence on the Concentration of N,N-Diisopropylethylamine
1.2.4 Dependence of the Initial Rate on the Concentration of 2-Aminoethyl
Diphenylborinate
The reactions were carried out using a range of catalyst loadings from 0.5 to 2.0 mol % (Scheme
1.10). The order in 2-aminoethyl diphenylborinate 1.01 was then determined by plotting the
initial rates against the concentration of catalyst (Figure 1.11). A slope of 0.419 M.s-1 was
obtained from the plot. The linear relationship between the initial kobs and the catalyst
concentration indicates first-order kinetics in catalyst.
Scheme 1.10 – Reaction Order in 2-Aminoethyl Diphenylborinate
!"#"$%&%%%'(")"%&%%%*"
%"
%&%%%+"
%&%%%,"
%&%%%-"
%&%%%*"
%&%%."
%" %&+" %&," %&-" %&*" ." .&+"
! !"#$%#
&'($
)"*+,-./0$%1($
OH
OH
OTs
OHTsCl (1.1 equiv)iPr2NEt (1.1 equiv)MeCN, 0.1 M
PhB
Ph NH2
O
(0.5–2 mol %)
0.2 mmol
1.01

14
Figure 1.10 – Formation of Product Over Time With Variation in the Concentration of 2-
Aminoethyl Diphenylborinate
Figure 1.11 – Initial Rate Dependence on the Concentration of 2-Aminoethyl
Diphenylborinate
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!#!&$"
!#!'"
!" $" %!" %$" &!"
!"#$%&
'()*+,-*
./01*+0/2-*
&#!"()*"+"
%#,$"()*"+"
%#$"()*"+"
%#&$"()*"+"
%#!"()*"+"
!#,$"()*"+"
!#$"()*"+"
!"#"$%&'(&)(*"+"$%$$$,$-"
$"
$%$$$)"
$%$$$&"
$%$$$-"
$%$$$."
$%$$'"
$%$$')"
$%$$'&"
$" $%$$$&" $%$$$." $%$$')" $%$$'-" $%$$)"
! !"#$%#
&'($
)*&+,-.!/0123$4-51/.23"!6-.70/8$%9($

15
1.2.5 Effect of 2-Aminoethanol
Experiments were performed to examine the effect the nature of the borinic acid catalyst had on
the reaction rate. Reactions were carried out using 1 mol % of either 2-aminoethyl
diphenylborinate 1.01 (Condition A) or diphenylborinic acid (Condition B) as the catalyst
(Scheme 1.12). Diphenylborinic acid 1.02 can be furnished by treatment of 2-aminoethyl
diphenylborinate 1.01 with 1 M hydrochloric acid in a 1:1 mixture of methanol:acetone (Scheme
1.13).15 The results showed that the rate of the reaction with diphenylborinic acid was faster in
comparison to 2-aminoethyl diphenylborinate 1.01 (Figure 1.12 and 1.13). This outcome is
consistent with our hypothesis that diphenylborinic acid 1.02 serves as a catalyst and that 2-
aminoethyl diphenylborinate 1.01 is a precatalyst for the reaction.
• Condition A: 2-Aminoethyl Diphenylborinate 1.01
• Condition B: Diphenylborinic Acid 1.02
Scheme 1.11 – Effect of 2-Aminoethanol
Scheme 1.12 – Cleavage of 2-Aminoethanol by Acid Catalysis
OH
OH
OTs
OHTsCl (5 equiv)iPr2NEt (5 equiv)
MeCN, 0.1 M0.2 mmol
Catalyst (1 mol %)
BNH2
O HCl, 1M
MeOH:Acetone (1:1)B OH
1.01 1.02, 96%

16
Figure 1.12 – Consumption of cis-1,2-Cyclohexanediol Over Time Using 2-Aminoethyl
Diphenylborinate (A) or Diphenylborinic Acid (B) as the Catalyst
Figure 1.13 – Rate of Consumption of cis-1,2-Cyclohexanediol Using 2-Aminoethyl
Diphenylborinate (A) or Diphenylborinic Acid (B) as the Catalyst
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#("
!#($"
!" $)" )!" *)" (!!" ($)" ()!"
!!"#"#$%"&'()*+,
-./,
01*)234563
718,3481/63
+,-./0,-"1"
+,-./0,-"2"
!"#"$%&%'(()"$"'&'%*+"
!"#"$%&%,*-)"$"'&.''+"
$/"
$,"
$-"
$*"
$'"
$."
%"
%" '%" -%" /%" 0%" .%%" .'%" .-%"
!"#!"#$%&'$()*!+,-
./"-
01+!23
415-3651"73
12345623"7"
12345623"8"
17
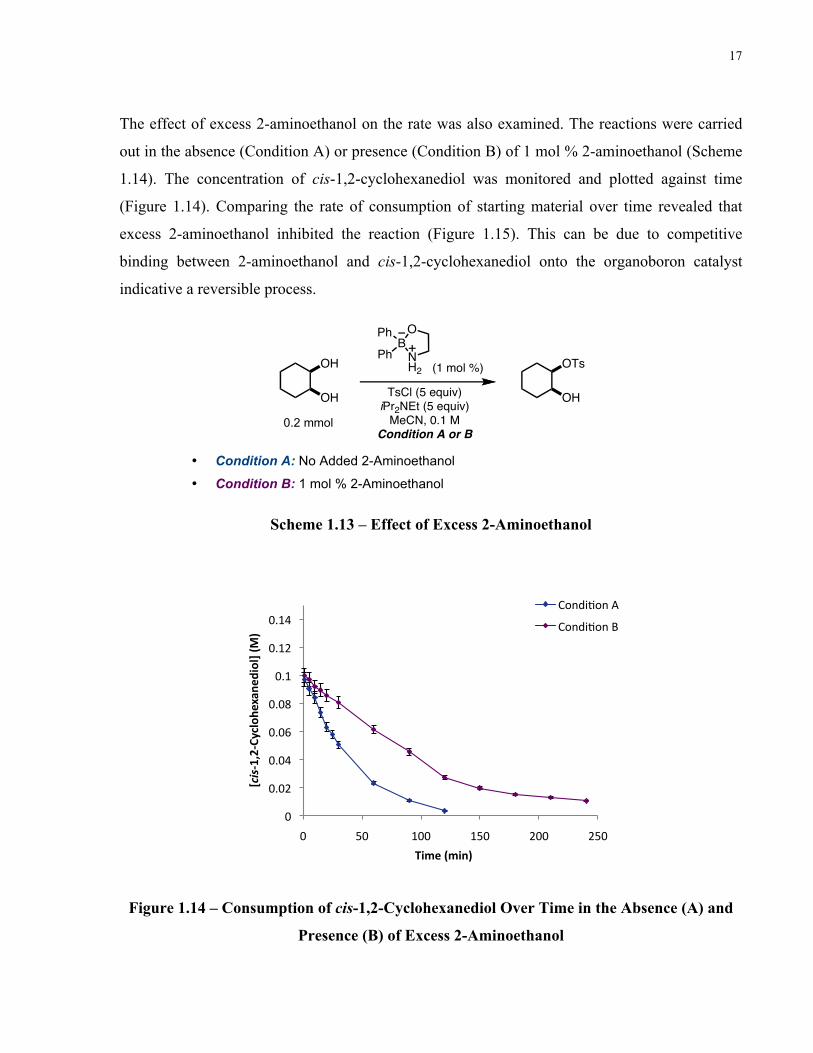
The effect of excess 2-aminoethanol on the rate was also examined. The reactions were carried
out in the absence (Condition A) or presence (Condition B) of 1 mol % 2-aminoethanol (Scheme
1.14). The concentration of cis-1,2-cyclohexanediol was monitored and plotted against time
(Figure 1.14). Comparing the rate of consumption of starting material over time revealed that
excess 2-aminoethanol inhibited the reaction (Figure 1.15). This can be due to competitive
binding between 2-aminoethanol and cis-1,2-cyclohexanediol onto the organoboron catalyst
indicative a reversible process.
• Condition A: No Added 2-Aminoethanol
• Condition B: 1 mol % 2-Aminoethanol
Scheme 1.13 – Effect of Excess 2-Aminoethanol
Figure 1.14 – Consumption of cis-1,2-Cyclohexanediol Over Time in the Absence (A) and
Presence (B) of Excess 2-Aminoethanol
OH
OH
OTs
OHTsCl (5 equiv)iPr2NEt (5 equiv)
MeCN, 0.1 MCondition A or B
PhB
Ph NH2
O
(1 mol %)
0.2 mmol
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#("
!#($"
!#(%"
!" )!" (!!" ()!" $!!" $)!"
!!"#"#$%"&'()*+,
-./,
01*)234563
718,3481/63
*+,-./+,"0"
*+,-./+,"1"
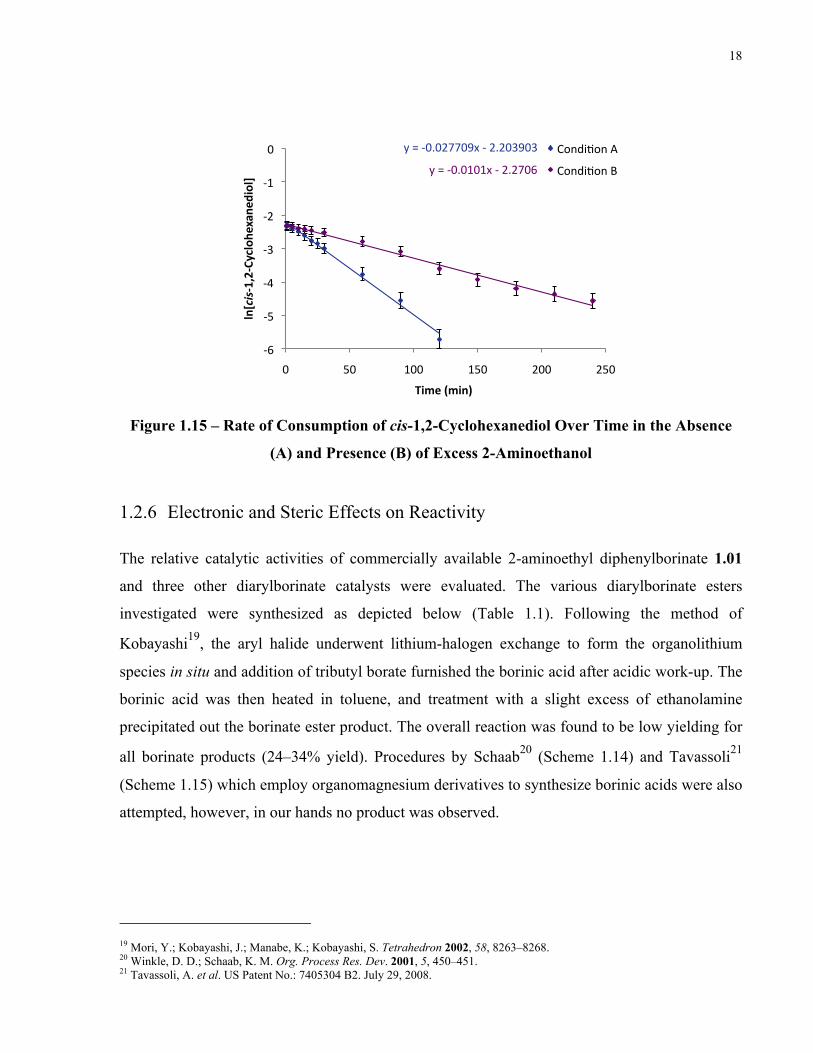
18
Figure 1.15 – Rate of Consumption of cis-1,2-Cyclohexanediol Over Time in the Absence
(A) and Presence (B) of Excess 2-Aminoethanol
1.2.6 Electronic and Steric Effects on Reactivity
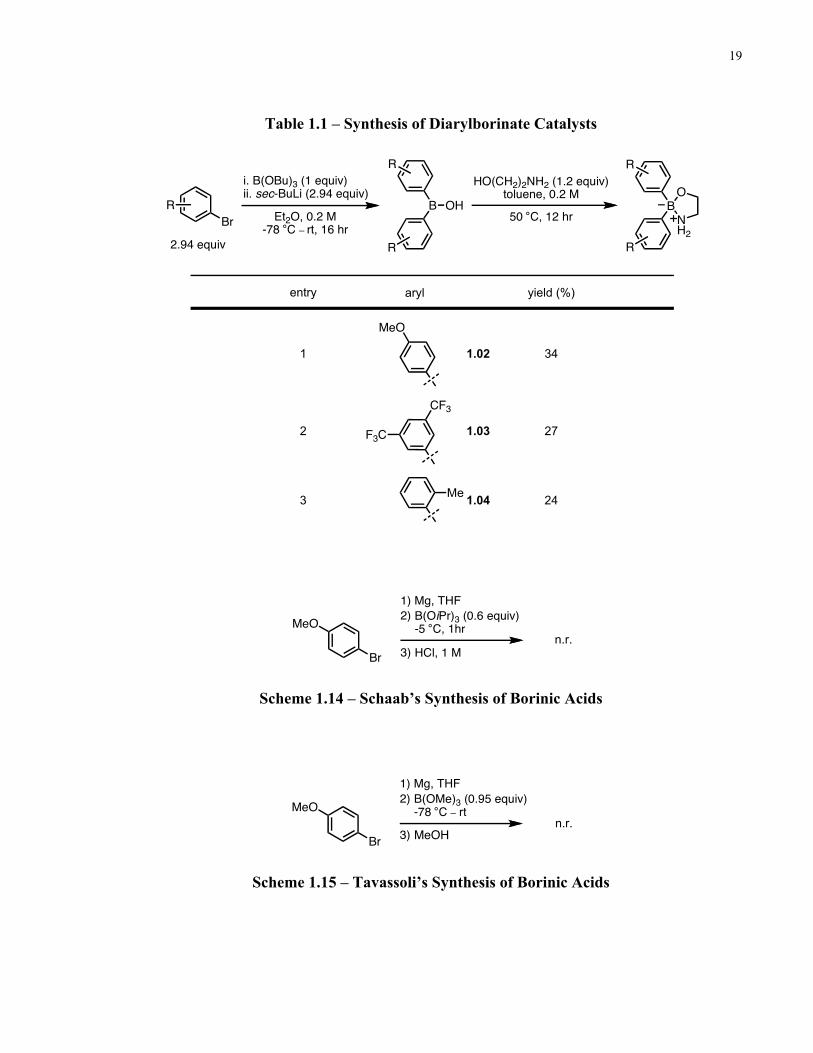
The relative catalytic activities of commercially available 2-aminoethyl diphenylborinate 1.01
and three other diarylborinate catalysts were evaluated. The various diarylborinate esters
investigated were synthesized as depicted below (Table 1.1). Following the method of
Kobayashi19, the aryl halide underwent lithium-halogen exchange to form the organolithium
species in situ and addition of tributyl borate furnished the borinic acid after acidic work-up. The
borinic acid was then heated in toluene, and treatment with a slight excess of ethanolamine
precipitated out the borinate ester product. The overall reaction was found to be low yielding for
all borinate products (24–34% yield). Procedures by Schaab20 (Scheme 1.14) and Tavassoli21
(Scheme 1.15) which employ organomagnesium derivatives to synthesize borinic acids were also
attempted, however, in our hands no product was observed.
19 Mori, Y.; Kobayashi, J.; Manabe, K.; Kobayashi, S. Tetrahedron 2002, 58, 8263–8268. 20 Winkle, D. D.; Schaab, K. M. Org. Process Res. Dev. 2001, 5, 450–451. 21 Tavassoli, A. et al. US Patent No.: 7405304 B2. July 29, 2008.
!"#"$%&%'((%)*"$"'&'%+)%+"
!"#"$%&%,%,*"$"'&'(%-"
$-"
$."
$/"
$+"
$'"
$,"
%"
%" .%" ,%%" ,.%" '%%" '.%"
!"#!"#$%&'$()*!+,-
./"-
01+!23
415-3651"73
01234512"6"
01234512"7"
19
Table 1.1 – Synthesis of Diarylborinate Catalysts
Scheme 1.14 – Schaab’s Synthesis of Borinic Acids
Scheme 1.15 – Tavassoli’s Synthesis of Borinic Acids
MeO
CF3
F3C
Me
R
i. B(OBu)3 (1 equiv)ii. sec-BuLi (2.94 equiv)
2.94 equiv
Br
R
B OH
R
Et2O, 0.2 M-78 °C – rt, 16 hr
HO(CH2)2NH2 (1.2 equiv)toluene, 0.2 M
50 °C, 12 hr
R
B
R
NH2
O
entry yield (%)aryl
1
2
3
34
27
24
1.02
1.03
1.04
B(OiPr)3 (0.6 equiv)-5 °C, 1hr
Br HCl, 1 M
MeO
1) Mg, THF2)
3)n.r.
B(OMe)3 (0.95 equiv)-78 °C – rt
Br
MeO
1) Mg, THF2)
3) MeOHn.r.

20
Switching the 2-aminoethanol group on the borinate ester to other ligands was also investigated
to evaluate whether it would facilitate isolation of the product (Table 1.2). No product was
observed using L-glycine or L-proline, however, 8-hydroxyquinoline afforded the target borinate
ester 1.05 in 89% yield.
Table 1.2 – Synthesis of Borinate Esters Containing Other Ligands
Electron rich and electron deficient diarylborinate esters containing 8-hydroxyquinoline as a
ligand were also synthesized in a similar manner (Table 1.3). Although reaction with 8-
hydroxyquinoline gave cleaner diarylborinate products by 1H-NMR, the yields were lower than
those with 2-aminoethanol.
entry yield (%)conditions
1
2
3
n.r.
n.r.
1.05, 89
ligand
B OH
NH O
H2NOH
O
NOH
BNR
O R'
OH
EtOH:H2O (1:1), rt, 16 hr
toluene, 50 °C, 16 hr
1 equiv
EtOH:H2O (1:1), rt, 16 hr
RHN R'
OH (1 equiv)
BN
O
BNH2
O O
BHN
O O
product
Conditions

21
Table 1.3 – Synthesis of 8-Hydroxyquinoline Diarylborinate Ester Catalysts
Subsequently, the catalysts were tested out in sulfonylation reactions with cis-1,2-
cyclohexanediol (Table 1.4). The 8-hydroxyquinoline diarylborinate derivatives were observed
to give the desired product in comparable yields to their corresponding 2-aminoethyl
diarylborinate catalysts. For ease of synthesis, kinetic studies were then carried out using the 2-
aminoethyl diarylborinate esters as catalysts.
The reactions were carried out with 1 mol % of a diarylborinate ester catalyst and the
disappearance of cis-1,2-cyclohexanediol was monitored over time (Scheme 1.16). A plot of the
initial rates revealed that 2-aminoethyl diphenylborinate 1.01 was the fastest, followed by the
bis(4-methoxyphenyl) catalyst 1.02 and the bis(3,5-bis(trifluoromethyl)phenyl) catalyst 1.03
(Figure 1.16). The more sterically demanding di-o-tolyl catalyst 1.04 gave the slowest rate. After
4 hours, the reaction showed less than 10% product formation.
MeO
CF3
F3C
R
i. B(OBu)3 (1 equiv)ii. sec-BuLi (2.94 equiv)
2.94 equiv
Br
R
B OH
R
Et2O, 0.2 M-78 °C – rt, 16 hr
entry yield (%)aryl
1
2
22
19
1.06
1.07
NOH
BN
O
toluene50 °C, 16 hr
(1 equiv)R
R
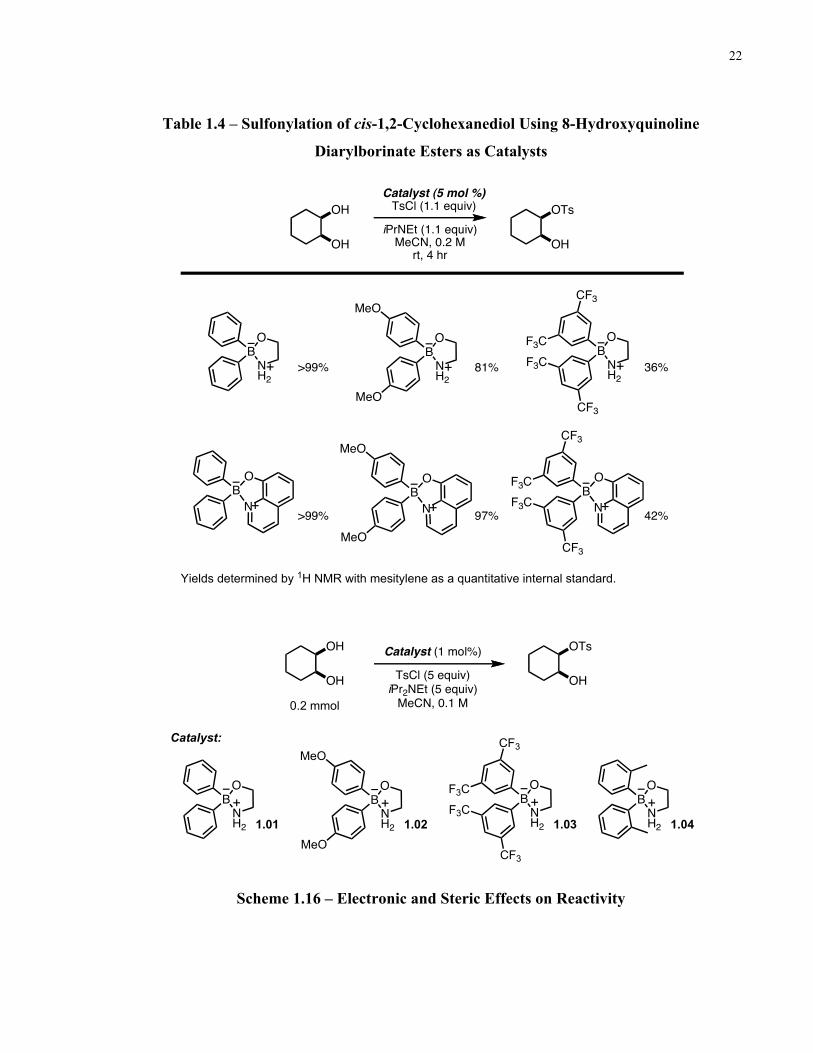
22
Table 1.4 – Sulfonylation of cis-1,2-Cyclohexanediol Using 8-Hydroxyquinoline
Diarylborinate Esters as Catalysts
Scheme 1.16 – Electronic and Steric Effects on Reactivity
Catalyst (5 mol %)TsCl (1.1 equiv)
iPrNEt (1.1 equiv)MeCN, 0.2 M
rt, 4 hr
OH
OH
OTs
OH
>99%
>99% 97%
81%
BN
O
BNH2
O
42%
36%
CF3
F3CF3C
CF3
CF3
F3CF3C
CF3
BN
OB
N
O
MeO
MeO
MeO
B
MeO
NH2
OB
NH2
O
Yields determined by 1H NMR with mesitylene as a quantitative internal standard.
OH
OH
OTs
OHTsCl (5 equiv)iPr2NEt (5 equiv)
MeCN, 0.1 M
Catalyst (1 mol%)
0.2 mmol
BNH2
O
1.01 1.02 1.03 1.04
BNH2
OB
NH2
OB
NH2
O
MeO
MeO
CF3
F3C
F3C
CF3
Catalyst:
23
Figure 1.16 – Consumption of cis-1,2-Cyclohexanediol Over Time Using Different Catalysts
Figure 1.17 – Rate of Consumption of cis-1,2-Cyclohexanediol Over Time Using Different
Catalysts
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#("
!#($"
!" )!" (!!" ()!" $!!" $)!"
!!"#"#$%"&'()*+,
-./,
01*)234563
718,3481/63
*+,+-./,"(#!("
*+,+-./,"(#!$"
*+,+-./,"(#!0"
*+,+-./,"(#!%"
!"#"$%&%'((%)*"$"'&'%+)%+"
!"#"$%&%,-%.+*"$"'&-'/,,."
!"#"$%&%%)+*"$"'&'(0+"
!"#"$%&%%%0*"$"'&+''"
$/"
$0"
$-"
$+"
$'"
$,"
%"
%" 0%" ,%%" ,0%" '%%" '0%"
!"#!"#$%&'$()*!+,-
./"-
01+!23
415-3651"73
12324!53",&%,"
12324!53",&%'"
12324!53",&%+"
12324!53",&%-"

24
The fact that both electron-rich and electron-deficient diarylborinic acids displayed lower
catalyst activity than diphenylborinic acid was unexpected. It can be reasoned that the electron
rich bis(4-methoxyphenyl) catalyst 1.02 decreases the Lewis acidity of boron causing the B-O
bonds to be weaker. This would then negatively affect diol binding, however, should speed up
reactivity with the electrophile. In the case of the electron deficient bis(3,5-
bis(trifluoromethyl)phenyl) catalyst 1.03, the Lewis acidity of boron is increased which
strengthens the B-O bonds. A tighter binding between the catalyst and substrate would then
inhibit the release of product or decrease reactivity of the bound diol. The reactivity of di-o-tolyl
catalyst 1.04 can be attributed to the bulky methyl group on the arenes. The binding of the diol is
an unfavourable process as boron is more sterically congested, thus very low reactivity is
observed. We have observed steric effects on equilibrium constants for boric acid-diol
interactions.22
1.3 Conclusion
The mechanistic insight we obtained from the results of the various experiments performed have
been consistent with the proposed catalytic cycle. The initial rate kinetic studies showed:
• first-order kinetics in substrate, electrophile, and catalyst
• zero-order kinetics in base
The zero-order kinetics in base signifies that this component is not involved in the rate-
determining step. The first-order dependence on the substrate, electrophile and catalyst may
reflect that attack of the electrophile by the bound cyclic ‘ate’ complex is the rate-limiting step.
The fact that we observe higher initial rates for the reaction catalyzed by the diphenylborinic acid
in comparison to its ethanolamine derivative is consistent with our proposal that the latter acts as
a precatalyst under the reaction conditions. Inhibition by 2-aminoethanol indicates that the
disassociation of the ligand is a reversible process and is also consistent with the proposal that 2-
aminoethyl diphenylborinate is a precatalyst. Variation in the electronic and steric properties of
22 Chudzinski, M. G.; Chi, Y.; Taylor, M. S. Manuscript submitted (July 15, 2011).

25
the borinic acid catalyst did not show any improvements on the reaction rate, however, it was
revealed that electron rich catalysts undergo faster reactions than electron poor catalysts.

26
2 Regioselective Alkylation of Carbohydrate Derivatives
Catalyzed by a Diarylborinic Acid Derivative23
2.0 Introduction
Complex oligosaccharides and glycoconjugates display prevalent roles in a diverse range of
biological processes including cell-cell signaling, immune response, and the development of
human diseases and cancer.24 With the increasing realization of their potential in drug and
vaccine discovery, chemical methods for their preparation have been sought after.25 However,
the synthesis of carbohydrate derivatives is a synthetic challenge due to their complex structures
requiring extensive protecting group manipulations.26 For decades, selective protection of
carbohydrates for applications in glycosylations has been pursued intensively. Monoalkylation,
in particular benzylation, represents one of the main strategies used in the preparation of
23 A significant portion of the work described in this chapter has been published: Chan, L.; Taylor, M. S. Org. Lett. 2011, 13, 3090–3093. 24 Boltje, T. J.; Buskas, T.; Boons, G.-J. Nature Chem. 2009, 1, 611–622. 25 Seeberger, P. H.; Werz, D. B. Nature 2007, 446, 1046–1051. 26 Wuts, P. G. M.; Greene, T. W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2007.

27
carbohydrates as alkyl ether groups are relatively stable and have low propensities to migrate.27
In recent years, transition metal-mediated approaches for the monofunctionalization of
carbohydrates have introduced new possibilities, with tin playing a central role. This section will
present a survey of the developments in this area of research with an emphasis on methods
involving regioselective installation of benzyl and related groups. Later, the development of a
regioselective diarylborinic acid-catalyzed preparation of monoalkylated carbohydrates will be
described.
2.1 Reactivity of the Hydroxy Group
In certain cases, selective protection of the most acidic hydroxy group, usually C-2, may be
achieved with varying degrees of efficiency.28 Generally in a 6-membered ring, it is well known
that an equatorial hydroxy group is functionalized preferentially in the presence of an axial
hydroxy group. In a study by Williams and Richardson, it was found that acylation of mannose,
glucose and galactose derivatives all showed that the reactivity of the equatorial hydroxy groups
were greater than the axial hydroxy groups.29 Likewise, a primary hydroxy group may be
protected selectively with a bulky substituent in the presence of several other secondary hydroxy
groups.30 For example, the primary hydroxy group can be selectively silylated in the presence of
free secondary hydroxy groups using tert-butyldimethylsilyl chloride.26 However, applications of
this method generally are not efficient as they may require several steps to occur prior to the
desired transformation. Thus, a more direct approach would avoid the tedious and costly steps.
In 1976, Garegg and co-workers reported the use of phase transfer catalysis to carry out
monobenzylation of carbohydrate derivatives.31 In phase transfer catalysis, the reaction is
heterogeneous where at least one of the reagents exist in a different phase. Reactions are able to
occur due to the use of a catalyst to increase the concentration of the insoluble reagent in the
reaction phase. It was observed that methyl 4,6-O-benzylidene-α-D-glucopyranoside could be
27 Gómez, A. M. In Glycoscience; Fraser-Reid, B., Tatsuka, K., Thiem, J., Eds.; Springer-Verlag: Berlin, 2008; pp 103–177. 28 Garegg, P. J. Pure Appl. Chem. 1984, 56, 845–858. 29 Williams, J. M.; Richardson, A. C. Tetrahedron 1967, 23, 1369–1378. 30 David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. 31 Garegg, P. J.; Iversen, T.; Oscarson, S. Carbohydr. Res. 1976, 50, C12–C14.

28
selectively alkylated at the O-2 position over O-3 using benzyl bromide, tetrabutylammonium
hydrogen sulfate, aqueous sodium hydroxide in dichloromethane under refluxing conditions
(Scheme 2.1).
Scheme 2.1 – Selective Alkylation of O-2 Over O-3 Using Phase Transfer Catalysis
With methyl 2,3-O-benzyl-α-D-glucopyranoside, base catalyzed phase transfer conditions
selectively benzylated the O-6 position over the O-4 position to give 68% and 18% yields
respectively (Scheme 2.2).28
Scheme 2.2 – Selective Alkylation of O-6 Over O-4 Using Phase Transfer Catalysis
In a report by Schmidt, it was demonstrated that the anomeric position of partially unprotected
carbohydrate derivatives could undergo regioselective alkylation using sodium hydride and alkyl
triflates (Scheme 2.3).32 The regioselectivity arises from the higher acidity of the anomeric
32 Schmidt, R. R.; Klotz, W. Synlett 1991, 168–169.
OOHO
OMe
O
HO
PhBnBr (1.7 equiv)
TBAHS (0.2 equiv)
R1 = H, R2 = Bn, 54%R1 = Bn, R2 = H, 20%R1 = Bn, R2 = Bn, 6%
OOR1O
OMe
O
R2O
Ph
5% NaOHCH2Cl2, 0.06 Mreflux, 2 days
OOHO OMeO
HO
PhBnBr (1.7 equiv)
TBAHS (0.2 equiv)
R1 = H, R2 = Bn, 50%R1 = Bn, R2 = H, 20%R1 = Bn, R2 = Bn, 7%
OOR1O OMe
O
R2O
Ph
5% NaOHCH2Cl2, 0.06 Mreflux, 2 days
OOH
BnO
OMe
HO
BnO
BnBr (1.7 equiv)TBAHS (0.2 equiv)
R1 = H, R2 = Bn, 68%R1 = Bn, R2 = H, 18%
OOR2
BnO
OMe
R1O
BnO5% NaOHCH2Cl2, 0.06 Mreflux, 2 days

29
hydroxy group in comparison to other unprotected hydroxy groups that results from indirect
stabilization by the ring oxygen atom.
Scheme 2.3 – Regioselective Alkylation of the Anomeric Position by Alkyl Triflates
In the total synthesis the KH-1 adenocarcinoma antigen by Danishefsky, the preferential
reactivity of the hydroxy groups in D-glucal, C-6 > C-3 > C-4, were exploited to generate the
desired product for glycosylation (Scheme 2.4).33
Scheme 2.4 – Preferential Reactivity of Hydroxy Groups in D-Glucal
2.2 Reductive Cleavage of Benzylidene Acetals
In 1968, Bhattacharjee and Gorin introduced an efficient method for regioselective ring-opening
of cyclic benzylidene acetals on hexafuranose and hexopyranose compounds (Scheme 2.5).34 An
aluminum dihydride chloride reactive species generated from an equimolar mixture of lithium
aluminum hydride and aluminum chloride complexes to the acetal, promoting ring-opening to
the corresponding ether. Reductive cleavage of the methyl 4,6-O-benzylidene derivative of α-D-
galactopyranoside gave preferentially the 6-O-benzyl derivative, whereas preferential formation
of the 4-O-benzyl derivative was observed for α-D-glucopyranoside and α-D-mannopyranoside.
33 Deshpande, P. P.; Danishefsky, S. J. Nature 1997, 387, 164–166. 34 Bhattacharjee, S. S.; Gorin, P. A. J. Can. J. Chem. 1968, 47, 1195–1206.
OOH
BnOOH
BnO
BnO
OOH
BnO OdecylBnO
BnOCH2Cl2, NaH
rt, 2hr
decyl triflate
64%
OOH
HOHO
NaH, BnBr
TESCl, pyrii.i.
OOBn
HOTESO

30
Scheme 2.5 – Bhattacharjee and Gorin’s Reductive Cleavage of Benzylidene Acetals
Shortly thereafter, Nánási, Lipták and coworkers reported that under similar reaction conditions,
dibenzylidene isomers of α-D-mannopyranosides and α-L-rhamnopyranosides could be
selectively cleaved (Scheme 2.6).35
Scheme 2.6 – Nánási and Lipták’s Reductive Cleavage of Benzylidene Acetals
A related strategy for reductive ring opening of 4,6-O-benzylidene acetals was introduced by
Garegg (Scheme 2.7).28 Previously, Horne and Jordan had demonstrated that the use of sodium
cyanoborohydride and hydrogen chloride in methanol could reductively open the
phenylethylidene acetal of ethylene glycol.36 Under slightly modified reaction conditions, excess
sodium cyanoborohydride and hydrogen chloride in tetrahydrofuran delivered the major product
with the benzyl group at the O-6 position in good to high yields.
Scheme 2.7 – Garegg’s Reductive Cleavage of Benzylidene Acetals
35 Lipták, A.; Imre, J.; Harangi, J.; Nánási, P. Tetrahedron 1982, 38, 3721–3727. 36 Horne, D. A.; Jordan, A. Tetrahedron Lett. 1978, 16, 1357–1358.
OO
BnO OBn
O
OBn
Ph
OOBn
BnO OBn
HO
OBn
AlCl3, LiAlH4
29%
CH2Cl2:Et2O (1:3)40 °C
OOO OBn
OOPh
AlCl3, LiAlH4
81%
CH2Cl2:Et2O (1:1)
PhH
OOBnO OBn
OOHPh
OO
BnOOMe
O
BnO
Ph
OOBn
BnOOMe
HO
BnO93%
HCl, THFNaCNBH3

31
It was also found that when borane-trimethylamine was employed as a reducing agent with
aluminum chloride as the acid, the regioselectivity of the opening of 4,6-O-benzylidene acetals
could be controlled by the choice of the solvent (Scheme 2.8).37 Using tetrahydrofuran as the
solvent, the benzyl group was installed at the O-6 position, whereas toluene led to the benzyl
group at the O-4 position of the pyranoside.
Scheme 2.8 – Solvent Effect on Reductive Cleavage of Benzylidene Acetals
2.3 Organotin-Mediated Regioselective Alkylation
Since their introduction in the 1970s, trialkylstannyl ethers and dialkylstannylene acetals have
become widely applied in the synthesis of carbohydrate derivatives (Scheme 2.9).38 Although
stoichiometric amounts of potentially toxic di- or trialkyltin (IV) byproducts are generated, these
intermediates have been extensively used since they provide a reliable, high-yielding route to
generate monosubstituted products with high regioselectivity.
Scheme 2.9 – Stannyl Ethers and Stannylene Acetals
37 Ek, M.; Garegg, P. J.; Hultberg, H.; Oscarson, S. J. Carbohydrate Chem. 1983, 2, 305–311. 38 Grindley, T. B. Adv. Carbohydr. Chem. Biochem. 1998, 53, 17–142.
OOBnO OBn
O
OBn
Ph Me3N.BH3, AlCl3
toluene: R = Bn, R' = H, 52%THF: R = H, R' = Bn, 72%
solventO
OR'
BnO OBnRO
OBn
OOH
HO OMe
OH
OH
OOH
BnO OMe
OH
OH
BnBr, DMF
95%
OOH
O OMe
O
OH
Bu2SnBu2SnO
100 °CMeOH
OOHO
OMe
O
HO
Ph OOHO
OMe
O
BnO
Ph
89%
(Bu3Sn)2O
MeOH
OOHO
OMe
O
O
Ph
Bu3Sn
BnBr, DMF
90 °C

32
These methods involve a two-step procedure whereby the sugar substrate is initially heated with
the tin reagent to form one or two Sn-O bonds. In the case of stannyl ethers, the tin forms a
coordination bond with a neighbouring oxygen atom.39 The formation of such intermediates
enhances the nucleophilicity of the oxygen atoms in the stannyl ether or stannylene acetal.
Subsequent treatment with the appropriate electrophile yields the corresponding
monofunctionalized product. The outcome of the reaction is dependant on several parameters
such as substrate, electrophile, additives, etc., however, generally the primary hydroxy group and
the equatorial hydroxy group in a vicinal cis-diol configuration are functionalized preferentially.
2.3.1 Stannyl Ethers
In 1971, Moffatt reported one of the first examples of the utility of alkoxy trialkyltin ethers in
carbohydrate chemistry (Scheme 2.10)40. The 5’-hydroxy group of 4’-fluoro-2’,3’-O-
isopropylidene adenosine was converted to the tributylstannyl ether using bis(tributyltin) oxide.
Subsequent sulfamoylation with sulfamoyl chloride yielded the desired sulfamate product in high
yield.
Scheme 2.10 – Organotin-Mediated Sulfamoylation of Adenosine Derivative
Investigations by Smith revealed that the hydroxy groups at the 1, 4 and 6 positions of
unsubstituted carbohydrates preferentially undergo O-stannylation with bis(tributyltin) oxide to
form the corresponding ethers (Scheme 2.11).41
39 Smith, P. J.; White, R. F. M.; Smith, L. J. Organomet. Chem. 1972, 40, 341–343. 40 Jenkins, I. D.; Verheyden, J. P. H.; Moffatt, J. G. J. Am. Chem. Soc. 1971, 93, 4323–4324. 41 Crowe, A. J.; Smith, P. J. J. Organomet. Chem. 1976, 110, C57–C59.
N
NN
N
NH2
O
OO
HO
Me Me
F
N
NN
N
NH2
O
OO
H2NO2S
Me Me
F
i. (Bu3Sn)2O, benzene
ii. ClSO2NH2, 5 °C
87%

33
Scheme 2.11 – Regioselective O-Stannylation
Ogawa and Matsui subsequently made several contributions to this area.42 In 1977, they reported
a novel approach to regioselectively acylate polyols using trialkylstannyl alkoxides (Scheme
2.12). Treatment of methyl α-D-glucopyranoside with bis(tributylstannyl)oxide in refluxing
toluene followed by the addition of benzoyl chloride at room temperature afforded methyl 2,6-di-
O-benzoyl-α-D-glucopyranoside and methyl 2,3,6-tri-O-benzoyl-α-D-glucopyranoside in 82%
and 15% yields respectively. Likewise, reactivity with methyl α-D-mannopyranoside and methyl
β-D-galactopyranoside were also shown to give high yields (90% and 92% respectively). The
results of the study also revealed that the stereochemistry of the diols in the five-membered
coordination ring is crucial for regioselectivity as cis-vicinal diols are expected to be much
favoured than trans-vicinal diols.
Scheme 2.12 – Ogawa and Matsui’s Benzoylation of Polyols
Building on the preceding results, Ogawa and co-workers reported a stannylation-alkylation
sequence to obtain monoalkylated carbohydrate derivatives (Scheme 2.13).43 Compared to
acylation, however, alkylation presented to be far more challenging. Stannylation of 2,2,2-
trichloroethyl 2-deoxyl-2-phthalimido-β-D-glucopyranoside using bis(tributylstannyl)oxide
followed by reaction with benzyl bromide for 4 days at 75 to 80 °C afforded the monobenzylated
product at the primary position in 60% yield. However, leaving the reaction to proceed for 8.5
42 (a) Ogawa, T.; Matsui, M. Carbohydr. Res. 1977, 56, C1–C6. (b) Ogawa, T.; Matsui, M. Tetrahedron 1981, 37, 2363–2369. 43 Ogawa, T.; Nakabayashi, S.; Sasajima, K. Carbohydr. Res. 1981, 96, 29–39.
OOH
HOOH
HO
OHtoluene
OOSnBu3
HO OSnBu3Bu3SnO
OH
(Bu3Sn)2O
OOH
HO
OMe
HO
HO
OOBz
HO
OMe
R'O
RO
R = Bz, R' = H, 82%R = R' = Bz, 15%
i. (Bu3Sn)2O, toluene 140 °C
ii. BzCl

34
days at 100 to 105 °C gave the 3,6-dibenzyl ether, 4,6-dibenzyl ether and monobenzyl ether in
9%, 4% and 14% yields respectively.
Scheme 2.13 – Ogawa’s Benzylation of Alkyl β-D-Glucopyranoside Derivatives
Shortly thereafter, a regioselective approach to the alkylation of alkyl β-D-galactopyranosides
using trialkylstannyl ethers was disclosed (Scheme 2.14).44 Reaction of methyl β-D-
galactopyranoside with bis(tributylstannyl)oxide, followed by alkylation with benzyl bromide
for 3 days at 85 °C resulted in the 3,6-dibenzyl ether in 48% yield and the 6-monobenzyl ether in
24% yield.
Scheme 2.14 – Ogawa’s Benzylation of Methyl β-D-Galactopyranoside
Extension of the stannylation-alkylation method to regioselective alkylation of methyl α-D-
glucopyranoside was demonstrated thereafter (Scheme 2.15).45 Methyl α-D-glucopyranoside was
condensed with bis(tributylstannyl)oxide and subsequent reaction with benzyl bromide at 80 to
90 °C for 2 days gave the 3,6-dibenzyl ether, 2,6-dibenzyl ether, 4,6-dibenzyl ether and 6-
monobenzyl ether in 4%, 30%, 6% and 48% yields respectively.
44 Ogawa, T.; Nukada, T.; Matsui, M. Carbohydr. Res. 1982, 101, 263–270. 45 Ogawa, T.; Takahashi, Y.; Matsui, M. Carbohydr. Res. 1982, 102, 207–215.
OOH
HO OCH2CCl3HO
NPhth
OOBn
HO OCH2CCl3HO
NPhth60%
i. (Bu3Sn)2O, benzene
ii. BnBr, 4 days 75–80 °C
OOBn
RO OCH2CCl3R'O
NPhth
i. (Bu3Sn)2O, benzene
ii. BnBr, 8.5 days 100–105 °C
R = R' = H, 14%R = Bn, R' = H, 9%R = H, R' = Bn, 4%
OOH
HO OMe
HO
OH
i. (Bu3Sn)2O, toluene 130 °C
ii. BnBr, 3 days 85 °C
R = H, 24%R = Bn, 48%
OOBn
RO OMe
HO
OH

35
Scheme 2.15 – Ogawa’s Benzylation of Methyl α-D-Glucopyranoside
In a report by Veyrières, the addition of tetrabutylammonium bromide as an additive was found
to strongly accelerate the alkylation step (Scheme 2.16).46 The reaction of allyl bromide and the
stannylated derivative of benzyl 6-O-trityl-α-D-mannopyranoside with tetrabutylammonium
bromide at 80 °C for 2 days was found to give 62% of the 3-O-allyl ether and 15% of the 2-O-
allyl ether.
Scheme 2.16 – Veyrières’ Benzylation of α-D-Mannopyranoside Derivatives
It is postulated that the addition of halide salts forms an anionic pentacoordinate tin species that
reversibly dissociates into a highly reactive quaternary ammonium alkoxides and tributyltin
halide adduct (Scheme 2.17).47 The large separation in the cation-anion pair of the newly formed
quaternary ammonium alkoxide attributes to the enhanced reactivity of the alkoxides.
Scheme 2.17 – Effect of Halide Salts on Trialkyltin Species
46 Alais, J.; Veyrières, A. J. Chem. Soc., Perkin Trans. I 1981, 377–381. 47 Tagliavini, G.; Zanella, P. Anal. Chim. Acta. 1968, 40, 33–39.
OOH
HOOMe
HO
HO
i. (Bu3Sn)2O, toluene 130 °C
ii. BnBr, 2 days 80–90 °C
R = R" = H, R' = Bn, 4%R = Bn, R' = R" = H, 30%R = R' = H, R" = Bn, 6%R = R' = R" = H, 48%
OOBn
R'OOMe
R''O
RO
OOTr
HOOBn
HOOH
i. (Bu3Sn)2O, toluene 130 °C
ii. AllylBr, TBAB 2 days, 80 °C
R = H, R' = Allyl, 62%R = Allyl, R' = H, 15%
OOTr
R'OOBn
HOOR
Bu3SnORBr
Bu3SnBrOR ROBu3SnBr +

36
Similarily, it was shown that the benzyl 2-acetamido-3-O-benzyl-2-deoxy-α-D-glucopyranoside
stannyl derivative could be benzylated at the 6-position in 86% yield (Scheme 2.18).48
Scheme 2.18 – Veyrières’ Benzylation of α-D-Glucopyranoside Derivatives
2.3.2 Stannylene Acetals
After demonstrating the viability of using stannyl ether intermediates to carry out regioselective
stannylation and subsequent alkylation reactions, Moffatt and co-workers disclosed the utility of
O-stannylene acetals in regioselective acylation and alkylation of nucleosides (Scheme 2.19).49
Upon treatment of β-D-ribofuranosyl uridine with equimolar equivalents of dibutyltin oxide to
afford the corresponding 2’,3’-O-stannylene acetal, reaction with benzyl bromide at 100 °C for 1
hour gave a 1:1 mixture of the 2’-O-benzyluridine and 3’-O-benzyluridine products in a
combined yield of 65% with no indication of other benzylated products.
Scheme 2.19 – Moffatt’s Benzylation of β-D-Ribofuranosyl Nucleosides
Later, David and co-workers reported the utility of stannylene acetals for regioselective
monobenzylation of cis-vicinal diols of carbohydrate derivatives (Scheme 2.20).50 Reaction of
48 Veyrières, A. J. Chem. Soc., Perkin Trans. I 1981, 1626–1629. 49 Wagner, D.; Verheyden, J. P. H.; Moffatt, J. G. J. Org. Chem. 1974, 39, 24–30. 50 Augé, C.; David, S.; Veyrières, A. J. Chem. Soc. Chem. Comm. 1976, 375–376.
OOH
BnOOBn
HO
AcHN
i. (Bu3Sn)2O, toluene reflux
ii. BnBr, TBAB 3 days, 80 °C
86%
OOBn
BnOOBn
HO
AcHN
NO
OHHO
HOi. (Bu2SnO)n, MeOH
ii. BnBr, DMF, 100 °C
NH
O
O NO
ORR'O
HO
NH
O
O
R = Bn, R' = HR = H, R' = Bn

37
the 3,4-O-stannylene derived from benzyl α-D-galactopyranoside with benzyl bromide at 100 °C
for 2 hours resulted in exclusive benzylation at the O-3 position with 66% yield. It is noteworthy
that benzylation of the same carbohydrate substrate using sodium hydride was found to give a
mixture of four products, and in other cases, preferential attack occurs at the O-4 position51.
Scheme 2.20 – David’s Benzylation of α-D-Galactopyranoside Derivatives
Likewise, independent studies by Nashed and Anderson reported the use of dibutyltin oxide to
mediate regioselective alkylation of allyl 2,6-di-O-benzyl-α-D-galactopyranoside to afford the
corresponding 3-O-allylated product in 79% yield (Scheme 2.21).52 The practicality of this
method was subsequently demonstrated in a series of reports for the syntheses of
oligosaccharides.53 Furthermore, extension of the regioselective alkylation to benzylation of
1,2:5,6-di-O-isopropylidene-L-inositol was found to give 95% yield of the 3-O-benzylated
derivative using benzyl bromide in a 1:1 mixture of benzene and dimethylformamide.30
Scheme 2.21 – Nashed and Anderson’s Benzylation of α-D-Galactopyranoside Derivatives
David and Thieffry subsequently developed milder reaction conditions in which the alkylation
step is carried out with an equimolar quantity of tetrabutylammonium iodide (Scheme 2.22).54
An improvement of this method was developed where the reaction takes place in non-polar
solvents such as toluene or benzene rather than the traditional polar solvents. Treatment of the
51 Flowers, H. M. Carbohydr. Res. 1975, 39, 245–251. 52 Nashed, M. A.; Anderson, L. Tetrahedron Lett. 1976, 39, 3503–3506. 53 (a) Nashed, M. A.; Anderson, L. Carbohydr. Res. 1977, 56, 419–422. (b) Slife, C. W.; Nashed, M. A.; Anderson, L. Carbohydr. Res. 1981, 93, 219–230. (c) Nashed, M. A.; Chowdhary, M. S.; Anderson, L. Carbohydr. Res. 1982, 102, 99–110. 54 David, S.; Thieffry, A.; Veyrières, A. J. Chem. Soc., Perkin Trans 1 1981, 1796–1801.
OOAllyl
HOOBn
HO
BnO
i. (Bu2SnO)n, benzene
ii. BnBr, DMF 2 hr, 100 °C
66%
OOAllyl
BnO
OBn
HO
BnO
OOBn
HOOAllyl
HO
BnO
i. (Bu2SnO)n, MeOH
ii. AllylI, DMF 1 hr, 100 °C
79%
OOBn
AllylO
OAllyl
HO
BnO

38
benzyl 2,3-di-O-benzyl-α-D-glucopyranoside stannylene derivative with benzyl bromide under
the reaction conditions was found to give the 6-O-benzylated product in 80% yield. Application
of the reaction conditions to carbohydrate derivatives bearing three free hydroxy groups was also
successful. Benzyl 2-O-benzyl-β-D-galactopyranoside afforded the 3-O-benzyl ether product in
68% yield, whereas benzyl 3-O-benzyl-β-D-galactopyranoside gave preferentially the 6-O-benzyl
ether product in 72% yield. Interestingly, reaction using benzyl β-D-galactopyranoside resulted in
exclusively the 3-O-benzylated product in 67% yield.
Scheme 2.22 – David and Thieffry’s Benzylation of D-Galactopyranoside Derivatives
A much more recent report by Ley and co-workers revealed the use of dibutyltin dimethoxide to
effect selective alkylation of varying diols in benzene or toluene (Scheme 2.23).55 Under Dean-
Stark conditions, the corresponding stannylene acetal is generated in situ and treated with the
alkylating reagent to afford the selectively substituted product. This contrasts with the dibutyltin
oxide method as it is not necessary to remove and replace the solvent prior to the addition of the
alkylating agent.
Scheme 2.23 – Ley’s Selective Alkylation of Diols Using Dibutyltin Dimethoxide
55 Boons, G.-J.; Castle, G. H.; Clase, J. A.; Grice, P.; Ley, S. V.; Pinel, C. Synlett 1993, 913–914.
OOH
HO OBn
HO
OBn
i. (Bu2SnO)n, benzene
ii. BnBr, TBAI, benzene
68%
OOH
BnO OBn
HO
OBn
OOH
HO OBn
HO
OH
i. (Bu2SnO)n, benzene
ii. BnBr, TBAI, benzene
67%
OOH
BnO OBn
HO
OH
OOHO OBnO
OH
PhBu2Sn(OMe)2BenzeneDean-Stark OO
BnO OBnO
OH
Ph
BnBr
i.
ii.
83%

39
Selective monobenzylation of alkyl α-L-rhamnopyranoside derivatives in the presence of
catalytic tin(II) chloride has also been reported by Toman et al. (Scheme 2.24).56 Treatment of
methyl α-L-rhamnopyranoside with excess benzyl bromide and triethylamine in acetonitrile
afforded a mixture of the 3-O-benzylated and 2-O-benzylated products in 52% and 5% yields
respectively.
Scheme 2.24 – Tin(II) Catalyzed Benzylation of α-L-Rhamnopyranoside Derivatives
2.4 Other Transition Metal Promoted Alkylations
Further developments in the area of carbohydrate chemistry have revealed a variety of other
transition metals capable of promoting regioselective alkylation. Several examples have
demonstrated the capability of metal chelation to impart regioselective alkylation of diols. Earlier
work by Avela showed that copper chelates of the dianions of methy 4,6-O-benzylidene-α- and -
β-D-glucopyranoside react with methyl iodide to give preferential substitution at the 3-O
position57. It is postulated that the formation of the copper(II) salts deactivate the carbohydrate
dianions, and as a result invokes the reaction to occur at the C-3 hydroxy group. Dialkylation is
also suppressed under these conditions.58 With methyl 2,3-di-O-benzyl-D-glucopyranoside,
differences in the reaction stoichiometry were also found to affect the selectivity of the reaction
either at the 4-O versus 6-O position.
An extension of this work was reported by Schuerch and co-workers, in which conditions to
form benzyl and allyl ethers regioselectively on carbohydrate derivatives were developed
56 Toman, R.; Rosík, J.; Zikmund, M. Carbohydr. Res. 1982, 103, 165–169. 57 Gridley, J. J.; Osborn, H. M. I.; Suthers, W. G. Tetrahedron Lett. 1999, 40, 6991–6994. 58 Osborn, H. M. I.; Brome, V. A.; Harwood, L. M.; Suthers, W. G. Carbohydr. Res. 2001, 332, 157–166.
O
OMeMe
HOHO OH
O
OMeMe
HOR'O OR
SnCl2 (10 mol %)
BnBr, NEt3MeCN, 110 °C
R = H, R' = Bn, 52%R = Bn, R' = H, 5%

40
(Scheme 2.25).59 Reaction of methyl 2,3-O-isopropylidene-α-D-mannopyranoside and benzyl
iodide with copper(II) chloride gave only the 4-O-benzylated product in >99% yield.
Scheme 2.25 – Schuerch’s Alkylation of α-D-Mannopyranoside Derivatives
In addition, Gridley et al. have developed a method for regioselective alkylation of various alkyl
1-O/S/Se-4,6-benzylidene-β-D-glucopyranoside using copper(II) chloride activation (Scheme
2.26).57 Treatment of the phenyl 4,6-O-benzylidene-β-D-glucopyranoside dianion, generated
from two equivalents of sodium hydride, with copper(II) chloride and methyl iodide afforded the
corresponding 3-O-methyl ether product in 64% yield.
Scheme 2.26 – Gridley’s Methylation of β-D-Glucopyranoside Derivatives
Demchenko and co-workers have also reported the use of nickel chelates to mediate
regioselective alkylation of carbohydrate derivatives (Scheme 2.27).60 Several 4,6-O-
benzylidene derivatives of D-glucopyranosides and D-galacopyranosides have been shown to
undergo regioselective alkylation using nickel(II) chloride in the presence of NaH in good yields.
Scheme 2.27 – Demchenko’s Alkylation of Carbohydrate Derivatives
59 Eby, R.; Webster, K. T.; Schuerch, C. Carbohydr. Res. 1984, 129, 111–120. 60 Gangadharmath, U. B.; Demchenko, A. V. Synlett 2004, 12, 2191–2193.
OOH
O
OMe
HOO
>99%
i.ii.
NaH, THFCuCl2BnIiii.
OOH
O
OMe
BnOO
OOHO OPhO
OH
Ph OOMeO OPh
O
OH
Phi.ii.
64%
NaH, THFCuCl2MeIiii.
OOHO
OMe
O
HO
Ph OOHO
OMe
O
BnO
Phi.ii.
61%
NaH, THF/DMFNiCl2BnBr, 16 hriii.

41
Very recently, a mild and regioselective method for benzylation of carbohydrate derivatives has
appeared where silver carbonate acts as a promoter (Scheme 2.28).61 Kartha et al. reported that
using p-methoxybenzyl chloride, octyl β-D-galactosidepyranoside could be selectively alkylated
at the 6-O position in 91% yield. Silver carbonate has also been proven to be highly efficient in
regioselective glycosylation of ‘naked’ galactosides using glycosyl halides.62
Scheme 2.28 – Kartha’s Alkylation of Carbohydrate Derivatives
2.5 Boron Activation of Hydroxy Groups for Regioselective
Alkylation
The selective formation of cyclic boronates between arylboronic acids and vicinal 1,2-diols or
1,3-diols with the exocyclic CH2OH moiety of carbohydrate derivatives has been well known in
the area of carbohydrate chemistry.63 Although the majority of these studies disclose the use of
boronate esters as protecting agents, several studies have emerged demonstrating their ability as
activating agents for diols.
Aoyama and co-workers reported that 3,4-boronate esters generated from phenylboronic acid and
either methyl α-L-fucopyranoside or methyl α-L-arabinopyranoside could be alkylated selectively
at the O-3 position by 1-iodobutane in the presence of triethylamine and silver oxide (Scheme
2.29).64 They proposed that the ‘ate’ complex formed from the amine and boronate ester promote
regioselective alkylation at the equatorial B-O bond.
61 Malik, S.; Dixit, V. A.; Bharatam, P. V.; Kartha, K. P. R. Carbohydr. Res. 2010, 345, 559–564. 62 Kartha, K. P. R.; Kiso, M.; Hasegawa, A.; Jennings, H. J. J. Chem. Soc., Perkin Trans. 1 1995, 3023–3026. 63 Duggan, P. J.; Tyndall, E. M. J. Chem. Soc., Perkin Trans. 1 2002, 1325–1339. 64 Oshima, K.; Kitazono, E. -i.; Aoyama, Y. Tetrahedron Lett. 1997, 38, 5001–5004.
OOH
HO OOct
HO
OH
pMBnCl, Ag2CO3
toluene, dark, 60 °C
91%
OOpMBn
HO OOct
HO
OH

42
Scheme 2.29 – Aoyama’s Alkylation of Fucose and Arabinose Derivatives
More recently, Onomura demonstrated an approach to monoalkylate 1,2-diols using catalytic
quantities of boronic acids in the presence of potassium carbonate (Scheme 2.30).65 They
reported that the efficiency of the reaction was controlled by the acidity of the diol and catalyst.
Less acidic diols required strong Lewis acids, whereas more acidic diols needed weak Lewis
acids.
Scheme 2.30 – Onomura’s Monoalkylation of 1,2-Diols
Studies by Evtushenko have also found that boric acid in catalytic quantities can help influence
regioselective methylation of methyl glycopyranosides with diazomethane (Scheme 2.31).66 The
reaction, however, results in variable yields and in some cases a mixture of products may be
observed.
Scheme 2.31 – Evtushenko’s Methylation of Methyl Glucopyranosides
65 Maki, T.; Ushijima, N.; Matsumura, Y.; Onomura, O. Tetrahedron Lett. 2009, 50, 1466–1468. 66 Evtushenko, E. V. Carbohydr. Res. 1999, 316, 187–200.
O
OMeMe
OHOH
OHPhB(OH)2
O
OMeMe
OO
OH
B
Ph
O
OMeMe
OHOBu
OH
O
OMeMe
OO
OH
B
Ph
NEt3
NEt3
nBuIAg2O
50%
OH
OH
B(OH)2
FBnBr, K2CO3, DMF
OBn
OH
99%
(0.1 equiv)
O
OMeMe
HOHO
OH
O
OMeMe
R''OR'O
OR
B(OH)3 (0.04 equiv)
R = R" = H, R' = Me, 48%R = R' = H, R" = Me, 47%R' = H, R = R" = Me, 2%
MeOH, CH2N2

43
2.6 Diarylborinic Acid Catalyzed Regioselective Alkylation
As described above, the use of organotin reagents is the most current state-of-the-art method for
regioselective monoalkylation of carbohydrates. However, these processes generate
stoichiometric quantities of tin byproducts. The report of Aoyama indicates that organoboron
reagents are capable of regioselective monoalkylations, although the process requires the use of
equimolar quantities of the organoboron reagent. Development of a catalytic variant of these
reactions would thus be an attractive improvement. Very recently our group reported that
derivatives of diphenylborinic acid are capable of catalyzing the selective monoacylation of the
equatorial hydroxy groups of cis-vicinal diol motifs in a wide range of carbohydrate derivatives
(Scheme 2.32).17
Scheme 2.32 – Borinic Acid-Catalyzed Monoacylation of cis-1,2-Diols
Building on our previous results, we sought to determine whether a similar mode of reactivity
could be employed to achieve catalyst-controlled, regioselective monoalkylation of carbohydrate
derivatives bearing multiple secondary hydroxy groups (Scheme 2.33). Described herein are the
studies that led to the successful realization of such an approach. These studies point to new
opportunities for the regioselective functionalization of carbohydrate derivatives and may find
application in the preparation of complex oligosaccharide compounds.
Scheme 2.33 – Proposal for Regioselective Alkylation of Polyols
O
OMeMe
HOHO
OH
PhB
Ph NH2
O
(10 mol %)PhCOCl, iPr2NEt
MeCN
O
OMeMe
HOPhOCO
OH94%
HO OH
R1 R2
RO OH
R1 R2
RXLnBOH (cat.)
LnBOH
LnB
O O
R1 R2

44
2.6.1 Synthesis of the Carbohydrate Substrates
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 was synthesized for method
development since it was readily prepared on gram-scale from methyl α-D-mannopyranoside.
Following a procedure reported by our group17, commercially avaliable methyl-α-D-
mannopyranoside was silylated using tert-butyldimethyl silyl chloride under basic conditions to
deliver the substrate in 89% yield (Scheme 2.34). Subsequently, methyl α-D-galactopyranoside
and methyl β-D-galactopyranoside were silylated in a similar fashion to afford 2.05 and 2.06 in
46% and 91% yields respectively.
Scheme 2.34 – TBS-Protection of the Primary Hydroxy Group
The 3,4,6-tri-O-benzyl-D-mannopyranoside 2.03 was synthesized in six steps using a literature
procedure reported by Namme (Scheme 2.35).67 Commercial 1,2,3,4,6-penta-O-acetyl-D-
galactopyranose was first converted to the orthoester product via bromination at the anomeric
position, followed by cyclization upon treatment with 2,6-lutidine in methanol. Deacetylation
was then carried out followed by alkylation with benzyl bromide to afford a tribenzylated
galactopyranoside. Treatment with acetic acid cleaved the orthoester and subsequent
deacetylation delivered the target sugar in 29% yield overall.
67 Namme, R.; Mitsugi, T.; Takahashi, H.; Shiro, M.; Ikegami, S. Tetrahedron 2006, 62, 9183–9192.
OOH
HOHO
OH
OMe
TBSCl (1.2 equiv)pyridine, 0.7 M
OOTBS
HOHO
OH
OMe2.01, 89%

45
Scheme 2.35 – Synthesis of 3,4,6-Tri-O-Benzyl-D-Mannopyranoside
2.6.2 Reaction Development and Extension of Scope
Preliminary studies were conducted with methyl-6-(tert-butyldimethylsilyloxy)-α-D-
mannopyranoside 2.01, benzyl bromide, silver oxide and the commercially available
diphenylborinate ester as a catalyst. Protection of the 6-hydroxy group in α-D-mannopyranoside
was necessary to achieve efficient catalyst-controlled alkylation as a complex mixture of
products were formed when unprotected α-D-mannopyranoside was employed. This is
presumably due to competitive binding of the boron catalyst to the 4,6- and 3,4-diol groups.
The results of a solvent screen are summarized in Table 2.1. It was found that acetonitrile
afforded the product with the highest yield of 35%.
K2CO3 (2.2 equiv)
O
OAc
AcOAcO
OAc
OAc
i. 33% HBr in AcOHi. K2CO3 (2.1 equiv) MeOH, 0.2 M
34% over 4 steps
O
OBn
BnOBnO
OAc
OAc
O
OAc
AcOAcO
OO
OMeO
OBn
BnOBnO
OO
OMe
ii. 2,6-lutidine (2 equiv) MeOH, 0.4 M
ii. BnBr (3.2 equiv) NaH (3.2 equiv) DMF, 0.3 M 0 °C – rt
AcOH, 0.3 M
MeOH, 0.5 M0 °C – rt
O
OBn
BnOBnO
OH
OH2.03, 95% 89%
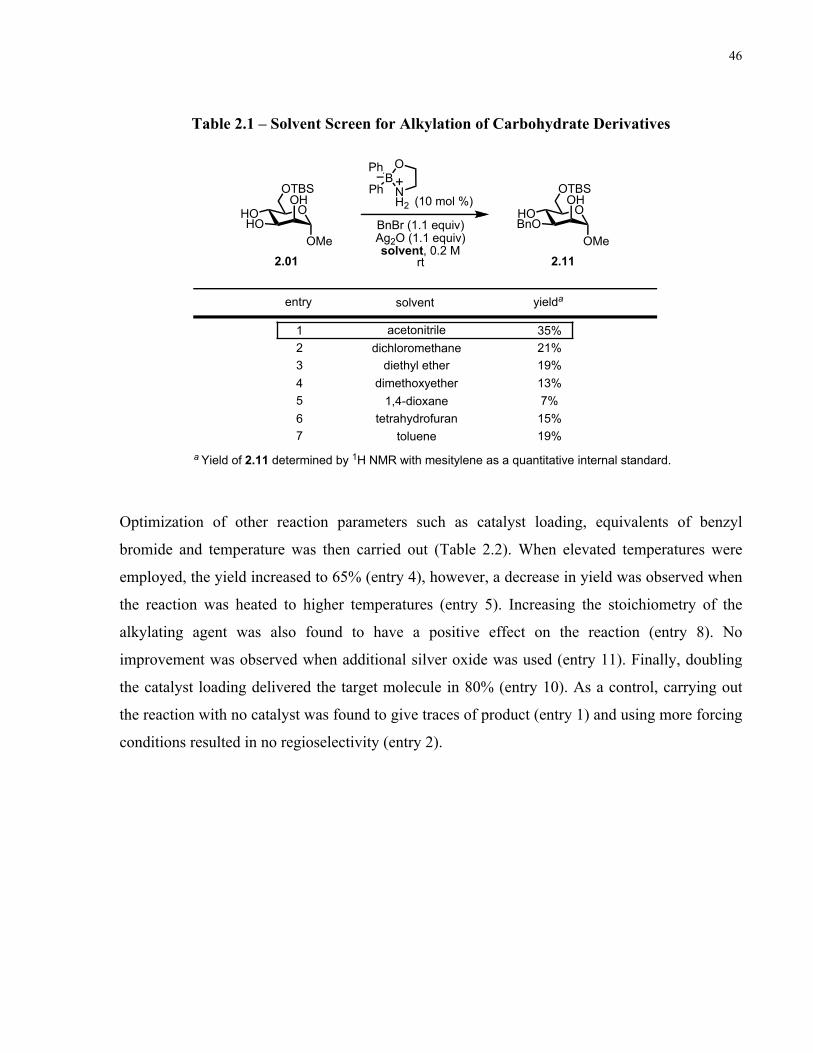
46
Table 2.1 – Solvent Screen for Alkylation of Carbohydrate Derivatives
Optimization of other reaction parameters such as catalyst loading, equivalents of benzyl
bromide and temperature was then carried out (Table 2.2). When elevated temperatures were
employed, the yield increased to 65% (entry 4), however, a decrease in yield was observed when
the reaction was heated to higher temperatures (entry 5). Increasing the stoichiometry of the
alkylating agent was also found to have a positive effect on the reaction (entry 8). No
improvement was observed when additional silver oxide was used (entry 11). Finally, doubling
the catalyst loading delivered the target molecule in 80% (entry 10). As a control, carrying out
the reaction with no catalyst was found to give traces of product (entry 1) and using more forcing
conditions resulted in no regioselectivity (entry 2).
entry yielda
15%19%
a Yield of 2.11 determined by 1H NMR with mesitylene as a quantitative internal standard.
solvent
123456
35%21%19%13%
7
7%
O
OTBS
HOHO
OH
OMeBnBr (1.1 equiv)Ag2O (1.1 equiv)solvent, 0.2 M
rt
PhB
Ph NH2
O
(10 mol %)O
OTBS
HOBnO
OH
OMe
dichloromethane
1,4-dioxane
diethyl etherdimethoxyether
toluene
acetonitrile
tetrahydrofuran
2.01 2.11
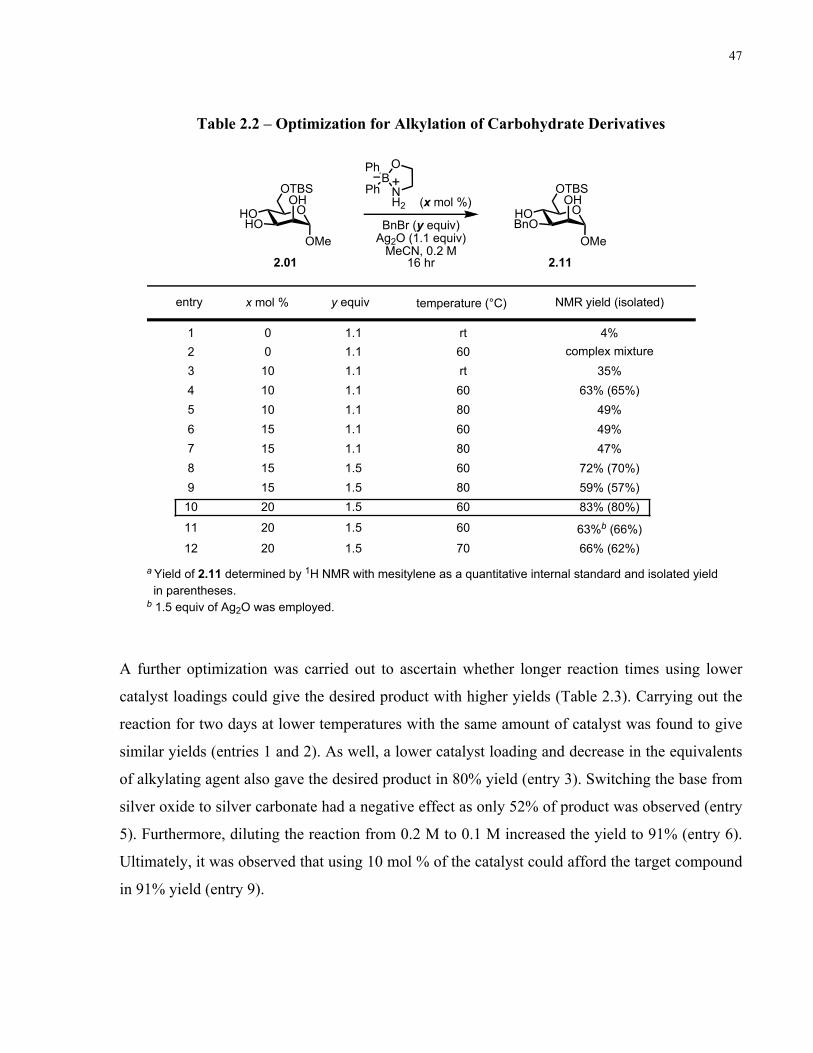
47
Table 2.2 – Optimization for Alkylation of Carbohydrate Derivatives
A further optimization was carried out to ascertain whether longer reaction times using lower
catalyst loadings could give the desired product with higher yields (Table 2.3). Carrying out the
reaction for two days at lower temperatures with the same amount of catalyst was found to give
similar yields (entries 1 and 2). As well, a lower catalyst loading and decrease in the equivalents
of alkylating agent also gave the desired product in 80% yield (entry 3). Switching the base from
silver oxide to silver carbonate had a negative effect as only 52% of product was observed (entry
5). Furthermore, diluting the reaction from 0.2 M to 0.1 M increased the yield to 91% (entry 6).
Ultimately, it was observed that using 10 mol % of the catalyst could afford the target compound
in 91% yield (entry 9).
entry NMR yield (isolated)
8910
11
12
49%
72% (70%)47%
59% (57%)
a Yield of 2.11 determined by 1H NMR with mesitylene as a quantitative internal standard and isolated yield in parentheses.b 1.5 equiv of Ag2O was employed.
15
x mol % y equiv
1.1
1.51.1
1.5
temperature (°C)
60
6080
8083% (80%)20 1.5 60
123456
4%complex mixture
35%63% (65%)
0
10
1.1
1.1
rt60rt60
7
49%80
O
OTBS
HOHO
OH
OMeBnBr (y equiv)
Ag2O (1.1 equiv)MeCN, 0.2 M
16 hr
PhB
Ph NH2
O
(x mol %) O
OTBS
HOBnO
OH
OMe
63%b (66%)60
66% (62%)70
0
1010
1.1
1.11.1
151515
20
20
1.5
1.5
2.01 2.11
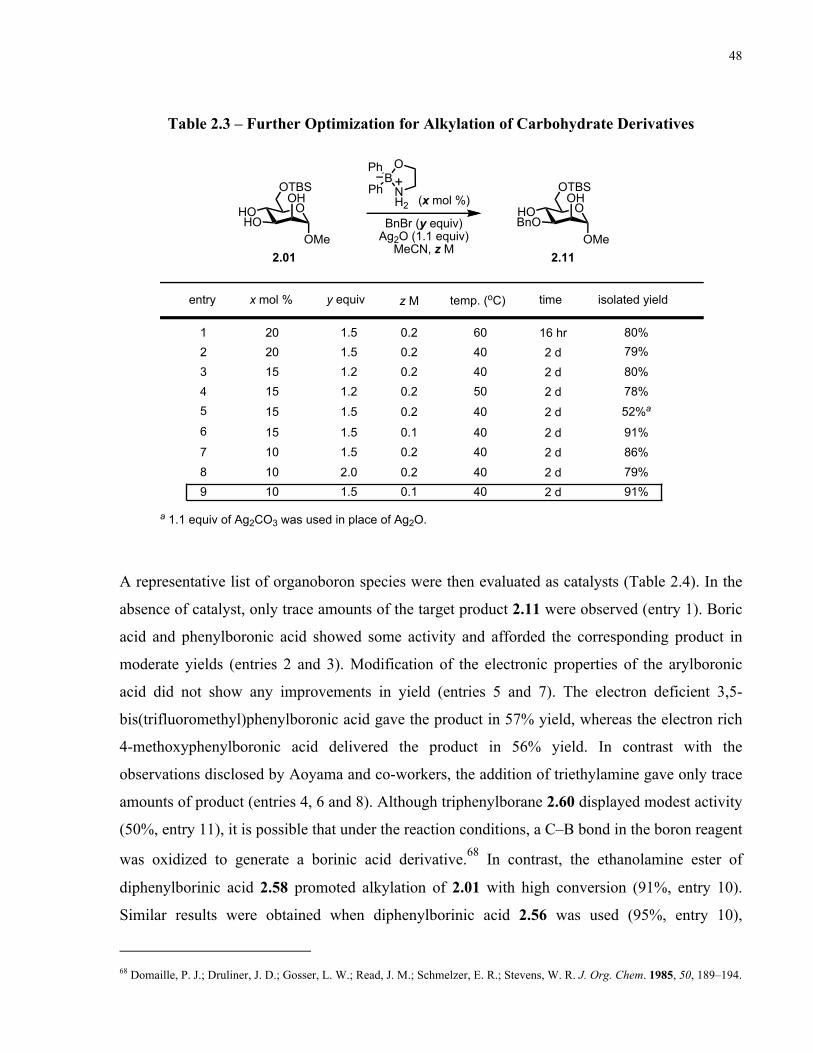
48
Table 2.3 – Further Optimization for Alkylation of Carbohydrate Derivatives
A representative list of organoboron species were then evaluated as catalysts (Table 2.4). In the
absence of catalyst, only trace amounts of the target product 2.11 were observed (entry 1). Boric
acid and phenylboronic acid showed some activity and afforded the corresponding product in
moderate yields (entries 2 and 3). Modification of the electronic properties of the arylboronic
acid did not show any improvements in yield (entries 5 and 7). The electron deficient 3,5-
bis(trifluoromethyl)phenylboronic acid gave the product in 57% yield, whereas the electron rich
4-methoxyphenylboronic acid delivered the product in 56% yield. In contrast with the
observations disclosed by Aoyama and co-workers, the addition of triethylamine gave only trace
amounts of product (entries 4, 6 and 8). Although triphenylborane 2.60 displayed modest activity
(50%, entry 11), it is possible that under the reaction conditions, a C–B bond in the boron reagent
was oxidized to generate a borinic acid derivative.68 In contrast, the ethanolamine ester of
diphenylborinic acid 2.58 promoted alkylation of 2.01 with high conversion (91%, entry 10).
Similar results were obtained when diphenylborinic acid 2.56 was used (95%, entry 10),
68 Domaille, P. J.; Druliner, J. D.; Gosser, L. W.; Read, J. M.; Schmelzer, E. R.; Stevens, W. R. J. Org. Chem. 1985, 50, 189–194.
entry isolated yieldy equiv
80%1.5
x mol %
20
temp. (oC)
60
time
16 hr
52%a
86%
91%
1.5
1.5
1.5
15
10
10
40
40
40
2 d
2 d
2 d
z M
0.2
0.2
0.2
0.1
O
OTBS
HOHO
OH
OMeBnBr (y equiv)
Ag2O (1.1 equiv)MeCN, z M
PhB
Ph NH2
O
(x mol %) O
OTBS
HOBnO
OH
OMe
78%1.215 50 2 d0.280%1.215 40 2 d0.2
91%1.515 40 2 d0.1
79%2.010 40 2 d0.2
a 1.1 equiv of Ag2CO3 was used in place of Ag2O.
79%1.520 40 2 d0.2
9
21
43
5
6
78
2.01 2.11

49
suggesting that 2.55 serves as a precatalyst from which the ethanolamine ligand is alkylated prior
to displacement by the carbohydrate substrate.
Table 2.4 – Further Optimization for Alkylation of Carbohydrate Derivatives
Experimentation with silver(I) salts was also conducted, however, all provided inferior results in
comparison to silver oxide (Table 2.5). Silver acetate gave the lowest yield with 37% product
(entry 1), whereas silver carbonate and silver triflate, with 1.1 equivalents of N,N-
diisopropylethylamine to sequester generated triflic acid, provided the product with moderate
yields of 42% and 57% respectively (entries 2 and 3). The addition of stoichiometric or catalytic
(20 mol %) amounts of tetrabutylammonium iodide or tetrabutylammonium bromide salts to the
reaction conditions led to the formation of complex mixtures.
entry NMR yielda
89
10
11
<5%
<5%56%
91%
a Yield of 2.11 determined by 1H NMR with mesitylene as a quantitative internal standard and isolated yield in parentheses.b Reaction carried out in the presence of 1.1 equiv NEt3
2.56b
catalyst
95%2.59
123456
<5%52%54%<5%
None
7
57%
O
OTBS
HOHO
OH
OMecatalyst (10 mol %)
Ag2O (1.1 equiv)MeCN, 0.2 M40 °C, 2 days
O
OTBS
HOBnO
OH
OMe
50%
PhB(OH)2b
2.56
2.572.57b
2.58
2.60
2.01 2.11
B(OH)3
PhB(OH)2
BnBr (1.5 equiv)
Catalysts:F3C
B(OH)2
CF3
BX
OMe
B(OH)2
2.56 2.572.58: X = OCH2CH2NH22.59: X = OH2.60: X = Ph

50
Table 2.5 – Optimization of Silver(I) Salt
With the optimal conditions in hand, we then proceeded to evaluate the scope of the reaction
using a variety of carbohydrate derivatives and alkylating agents. Illustrative examples of the
scope for selective installation of benzyl, naphthylmethyl, 4-bromobenzyl and benzyloxymethyl
protecting groups on ten carbohydrate substrates derived from mannose 2.01–2.03, rhamnose
2.04, galactose 2.05–2.07, 2.10, fucose 2.08 and arabinose 2.09 are included in Table 2.6. As
demonstrated therein, the reaction conditions enables reliable and high-yielding alkylation of the
equatorial hydroxy group of cis-vicinal diol pairs. The transformation is equally compatible with
variation in the stereochemistry of the anomeric position (2.05 vs 2.06). In the galacto series, a
sterically hindered substituent at C-5 was found to be not essential for obtaining high
regioselectivity (galactose 2.05 vs fucose 2.08 and arabinose 2.09). As exemplified by the 1,6-
anhydro derivatives of mannose 2.02 and galactose 2.07, where the pyranose ring adopts a 1C4
conformation instead of a 4C1, complementary regiochemical outcomes to those obtained for
mannose 2.01 and galactose 2.05 was obtained respectively. Selective alkylation of tribenzylated
mannose 2.03 delivered the benzyl ether at the O-1 position to afford the challenging β-mannosyl
stereochemical outcome at the anomeric position (entry 9). Using a slight excess of 4-
bromobenzyl bromide and silver oxide, D-Galactal 2.10 was found to undergo selective
dialkylation at the O-3 and O-6 positions (entry 34).
entry isolated yield
1234
42%37%57%91%
OOTBS
HOHO
OH
OMeBnBr (1.5 equiv)Ag(I) (1.1 equiv)
MeCN, 0.2 M40 °C, 2 days
PhB
Ph NH2
O
(10 mol %) OOTBS
HOBnO
OH
OMe
AgOTf + i-Pr2NEt (1.1 equiv)
Ag2CO3
Ag(I)
AgOAc
Ag2O
2.01 2.11

51
Table 2.6 – Scope of Regioselective Alkylation of Monosaccharides
R2R1 R!X (1.5 equiv)
Ag2O (1.1 equiv)MeCN, 40 oC, 2 days
PhB
Ph NH2
O
(10 mol %)OH
HO R2R1
OH
RO
entry yielda
8
9
101112
2.15
2.172.16
2.18
substrate
2.19
1234
56
2.112.122.13
7
2.14
2.202.21
O
OTBS
HO
OMe
OH
HO
O
O
ROOH
OH
O
OOH
OH
OH
O
OTBS
HO OMe
OH
OH
O
OMeMe
OHOH
OH
O
OMeMe
HOHO
OH
O
OTBS
RO
OMe
OH
HO
O
O
HOOH
OH
O
OOH
OH
OR
O
OMeMe
OHOR
OH
O
OMeMe
HORO
OH
O
OBn
BnOBnO
OH
OH
O
OBn
BnOBnO
OH
OBn
O
OTBS
HOHO
OH
OMe
O
OTBS
HORO
OH
OMe
O
OTBS
RO OMe
OH
OH
2.01
2.02
2.03
2.04
2.05
2.06
2.07
2.08
91%(R = Bn)(R = 4-BrBn) 99%
2.26
2.28
2.27
2.292.30
2.222.23
2.242.25
2.31
2.322.332.342.35
2.362.372.382.39
2021
222324
13
14151617
1819
25
26272829
(R = Nap)(R = BOM)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = Bn)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
63%
72%
83%73%99%
89%84%
73%73%71%
99%88%
76%72%74%
83%
77%95%82%90%
74%91%
66%
94%89%86%98%
product

52
Selective protection using allyl bromide with mannose 2.01, galactose 2.05 and fucose 2.08 was
also carried out, however, under the optimized reaction conditions poor yields were obtained
(Table 2.7). It can be rationalized that the low yields arise from the ability of allyl bromide to
undergo SN2 and SN2’reactions, and is less sterically hindered.
Table 2.7 – Scope of Regioselective Allylation of Monosaccharides
a Isolated yield on 0.2-1.0 mmol scale. X = Br for the installation of the Bn, 4-BrBn, and Nap groups; X = Cl for installation of the BOM group.b 2.5 equiv of 4-BrBnBr and 2.1 equiv of Ag2O were employed.
O
OHHO
HOO
ORHO
RO
OHO OMe
OH
OH
ORO OMe
OH
OH2.09
2.10
2.402.412.42
2.44
2.433233
34
3031
(R = Bn)(R = 4-BrBn)(R = Nap)(R = BOM)
(R = 4-BrBn)
71%82%
77%b
74%84%
R2R1 AllylBr (1.5 equiv)
Ag2O (1.1 equiv)MeCN, 40 oC, 2 days
PhB
Ph NH2
O
(10 mol %)OH
HO R2R1
OH
RO
entry isolated yieldsubstrate
1
2
3
53%
17%
56%
product
O
OMeMe
OHOH
OH
2.08
O
OMeMe
OHOAllyl
OH
O
OTBS
HO
OMe
OH
HO
O
OTBS
AllylO
OMe
OH
HO2.05
O
OTBS
HOHO
OH
OMe
O
OTBS
HOAllylO
OH
OMe2.01 2.45
2.46
2.47

53
Alkylation using p-methoxybenzyl chloride was found to be less selective in comparison as
mixtures of regioisomers were obtained (Table 2.8). In all cases, functionalization at the other
boron-bound hydroxy group was observed. The poor selectivity is presumably due to the weaker
chloride leaving group or possibly p-methoxybenzyl chloride is more prone to SN1 reactions.
Table 2.8 – Scope of Regioselective p-Methoxybenzylation of Monosaccharides
R2R1 PMBCl (1.5 equiv)
Ag2O (1.1 equiv)MeCN, 40 oC, 2 days
PhB
Ph NH2
O
(10 mol %)OH
HO R2R1
OH
RO
entry isolated yieldasubstrate
1
2
3
42% (33%)
61% (33%)
37% (15%)
product
O
OTBS
HOHO
OH
OMe
O
OTBS
HOPMBO
OH
OMe2.01 2.48
4
5
6
59% (20%)
62% (7%)
42% (29%)
O
OTBS
HO OMe
OH
OH
O
OTBS
PMBO OMe
OH
OH2.06
O
OMeMe
OHOH
OH O
OMeMe
OHOPMB
OH
2.08
O
OOH
OH
OH O
OOH
OH
OPMB
2.02
O
O
PMBOOH
OH
O
O
HOOH
OH2.07
OHO OMe
OH
OH
OPMBO OMe
OH
OH2.09
2.49
2.50
2.51
2.52
2.53a Yield in parentheses represent the isolated regioisomer.

54
Current limitations of the reaction condition include sugars that do not contain a cis-vicinal diol
motif such as glucose or xylose derivatives, thiogalactose derivatives and mannose derivatives
presented in Figure 2.1. Neither iodomethane or bromobutane was found to afford regioselective
alkylation under the optimized conditions indicating that it may be essential to use activated
(benzylic or alkoxymethyl) halides for high reactivity.
Figure 2.1 – Incompatible Monosaccharide Derivatives
2.7 Monoalkylation via Halide Catalysis
Several reports have described the use of tetrabutylammonium iodide as a key additive for
promoting increased product yields in alkylation reactions.69 It is postulated that in these
reactions, halide exchange occurs to generate the more reactive alkyl halide.
In an extension of this work, it was found that halide catalysis is also compatible with the
diphenylborinate ester catalyst to carry out regioselective alkylation. Activation of the alkyl
halide by the addition of iodide salts in conjunction with Brønsted bases rather than silver(I) salts
was evaluated. With sodium iodide as the halide salt, benzylation of 2.01 was carried out using
the optimized reaction conditions for silver oxide with potassium carbonate, N,N-
diisopropylethylamine or 1,2,2,6,6-pentamethylpiperidine as the Brønsted base (Table 2.9). N,N-
Diisopropylethylamine and 1,2,2,6,6-pentamethylpiperidine both gave only starting material
(entries 2 and 3), whereas potassium carbonate afforded the target sugar in 14% isolated yield
(entry 1).
The reaction outcome was significantly influenced by further experimentation as shown in Table
2.10. Elevating the reaction temperature to 60 °C and a shorter reaction time improved the yield
to 85% (entry 1). This yield was further increased to 89% when potassium iodide was employed
69 (a) Fox, D. L.; Whitely, N. R.; Cohen, R. J.; Salvatore, R. N. Synlett 2003, 13, 2037–2041; (b) Salvalore, R. N.; Smith, R. A.; Nischwitz, A. K.; Gavin, T. Tetrahedron Lett. 2005, 46, 8931–8935.
OOO
MeHO
OHHOO
OTBSHO
HOOH
SiPrO
OH
HO
OMe
HO
HO
OHO
OMe
HO
HO O
OH
HOHO
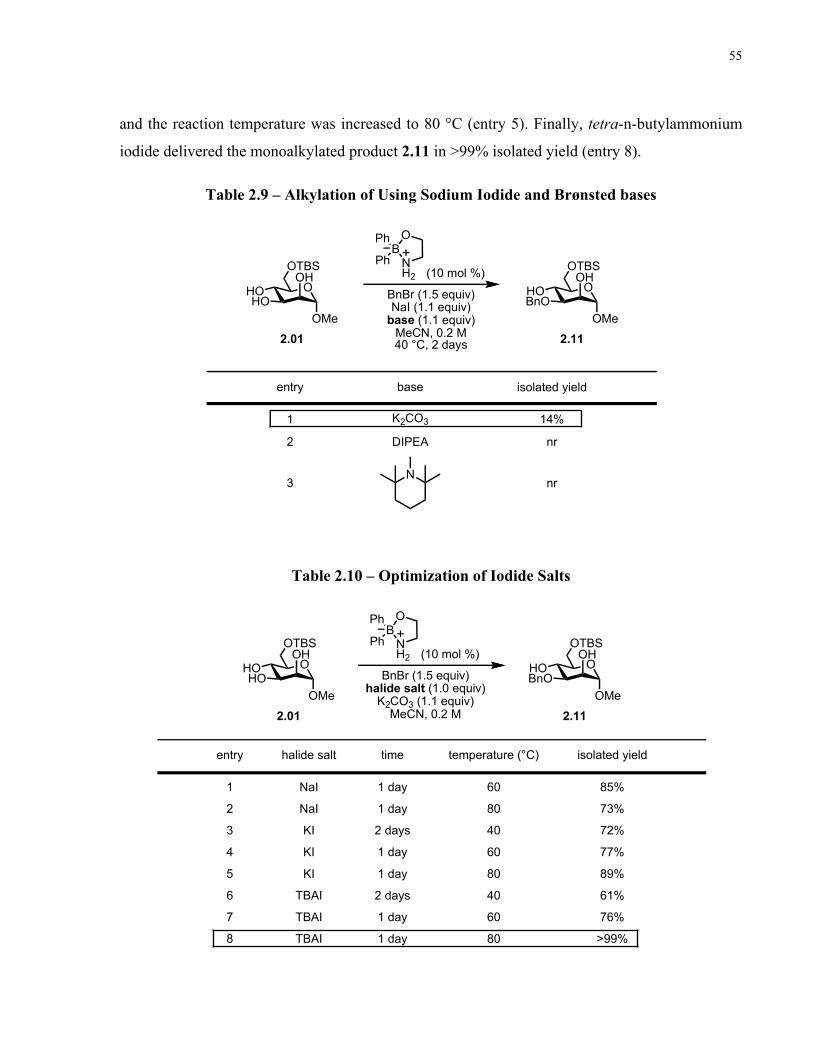
55
and the reaction temperature was increased to 80 °C (entry 5). Finally, tetra-n-butylammonium
iodide delivered the monoalkylated product 2.11 in >99% isolated yield (entry 8).
Table 2.9 – Alkylation of Using Sodium Iodide and Brønsted bases
Table 2.10 – Optimization of Iodide Salts
entry isolated yieldbase
BnBr (1.5 equiv)NaI (1.1 equiv)base (1.1 equiv)
MeCN, 0.2 M40 °C, 2 days
PhB
Ph NH2
O
(10 mol %)
1
2
3
K2CO3
DIPEA
N
nr
nr
14%
O
OTBS
HOHO
OH
OMe
O
OTBS
HOBnO
OH
OMe
2.01 2.11
BnBr (1.5 equiv)halide salt (1.0 equiv)
K2CO3 (1.1 equiv)MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %)
entry isolated yieldhalide salt time temperature (°C)
3
4
5
6
7
8
KI
KI
KI
TBAI
TBAI
TBAI
NaI
NaI
1
2
40
60
80
40
60
80
60
80
2 days
1 day
2 days
1 day
1 day
1 day
1 day
1 day
72%
61%
77%
76%
85%
73%
89%
>99%
O
OTBS
HOHO
OH
OMe
O
OTBS
HOBnO
OH
OMe
2.01 2.11

56
Further studies on the compatibility of the reaction conditions with other carbohydrate substrates
were tested (Scheme 2.31). Using 1,6-anhydro-β-D-galactopyranoside 2.07, the alkylation with
benzyl bromide and tetra-n-butylammonium iodide as an additive gave 78% yield of the
corresponding product 2.32. Switching the halide salt to potassium iodide surprisingly gave
>99% yield which is significantly greater than the amount furnished when silver oxide was
employed (76% yield, Table 2.6, entry 22). Alkylation of methyl-β-L-arabinopyranoside 2.09
using benzyl bromide and tetra-n-butylammonium iodide gave 52% yield of 2.40. Similar results
were obtained when potassium iodide was utilized (50% yield). In comparison, silver oxide
afforded benzylated arabinose 2.40 in 74% yield (Table 2.6, entry 30).
Scheme 2.36 – Extension of Halide Catalysis Method to Other Substrates
In efforts towards transferring this reactivity to thiogalactose derivatives, a screen of different
conditions with changes in stoichiometry of halide salt, temperature and reaction time was
carried out in the presence of tetra-n-butylammonium iodide (Table 2.11). Although no
reactivity was observed after one day even with heat (entry 1 to 3), low conversion to 2.55 was
observed after two days at room temperature (20% yield, entry 4). Increasing the temperature to
40 °C improved the yield to 32% (entry 5), however, excess halide salt resulted in no reactivity
(entry 6).
2.40, 52%
OO
HOOH
OH
OO
BnOOH
OH
O
OMeHOBnO
HOO
OMeHOHO
HO
BnBr (1.5 equiv)KI (1.0 equiv)
K2CO3 (1.1 equiv)MeCN, 0.2 M1 day, 80 °C
PhB
Ph NH2
O
(10 mol %)
2.32, >99%
BnBr (1.5 equiv)TBAI (1.0 equiv)
K2CO3 (1.1 equiv)MeCN, 0.2 M1 day, 80 °C
PhB
Ph NH2
O
(10 mol %)
2.07
2.09
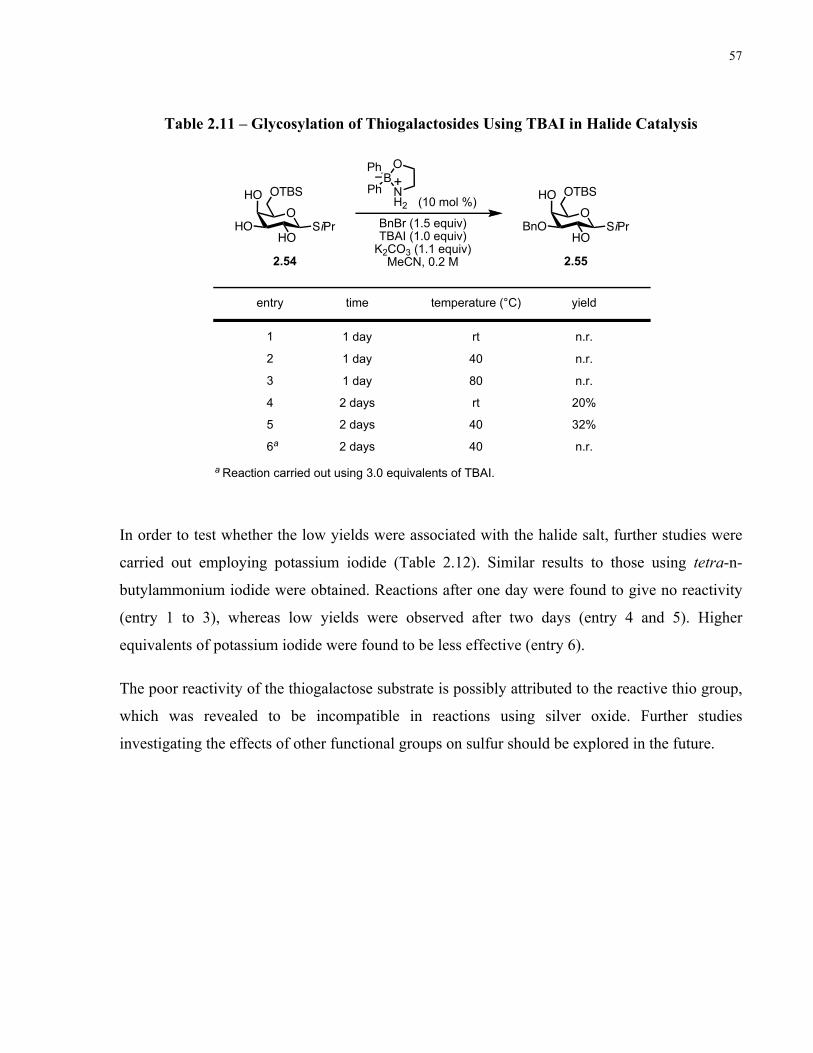
57
Table 2.11 – Glycosylation of Thiogalactosides Using TBAI in Halide Catalysis
In order to test whether the low yields were associated with the halide salt, further studies were
carried out employing potassium iodide (Table 2.12). Similar results to those using tetra-n-
butylammonium iodide were obtained. Reactions after one day were found to give no reactivity
(entry 1 to 3), whereas low yields were observed after two days (entry 4 and 5). Higher
equivalents of potassium iodide were found to be less effective (entry 6).
The poor reactivity of the thiogalactose substrate is possibly attributed to the reactive thio group,
which was revealed to be incompatible in reactions using silver oxide. Further studies
investigating the effects of other functional groups on sulfur should be explored in the future.
OOTBSHO
HOHO
SiPrO
OTBSHO
BnOHO
SiPrBnBr (1.5 equiv)TBAI (1.0 equiv)
K2CO3 (1.1 equiv)MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %)
entry yieldtime temperature (°C)
3
4
5
6a
1
2
rt
40
1 day
1 day
n.r.
n.r.
rt
40
40
80
2 days
1 day
2 days
2 days
20%
32%
n.r.
n.r.
2.54 2.55
a Reaction carried out using 3.0 equivalents of TBAI.

58
Table 2.12 – Glycosylation of Thiogalactosides Using KI in Halide Catalysis
2.8 DFT Calculations
Our mechanistic model for the regioselective alkylation reaction involves the formation of a
tetracoordinate borinate complex with a cis-vicinal diol motif (Figure 2.2). The B-O bonds of the
tetracoordinate adduct reacts preferentially with electrophiles to deliver the product of mono-
alkylation or acylation. Computational studies were performed to calculate Fukui indices. The
Fukui index takes into account the change in electron density of valence electrons when forming
molecules and relatively predicts which atom will have a higher tendency to either lose or accept
an electron for nucleophilic attack or electrophilic attack respectively.70 Previously, Fukui index
calculations of borinate esters derived from rhamnose 2.04 and fucose 2.08 demonstrated that
nucleophilic activity was found to be the highest at O-3, which was consistent with the observed
reactivity and also suggests an electronic basis for the observed selectivity.17 The boron-bound
oxygen atoms in the tetracoordinate complex have increased electron density in comparison to
non-bonded oxygen groups and the relative reactivity seems to reflect dipole–dipole interactions.
70 Yang, W.; Mortier, W. J. J. Am. Chem. Soc. 1986, 108, 5708–5711.
OOTBSHO
HOHO
SiPrO
OTBSHO
BnOHO
SiPrBnBr (1.5 equiv)KI (1.0 equiv)
K2CO3 (1.1 equiv)MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %)
entry yieldtime temperature (°C)
3
4
5
6a
1
2
80
rt
40
40
rt
40
1 day
2 days
1 day
1 day
2 days
2 days
n.r.
16%
17%
24%
n.r.
n.r.
2.54 2.55
a Reaction carried out using 3.0 equivalents of KI.

59
For rhamnose 2.04 and fucose 2.08, the C3–O bond is in opposition of the C4–O and C5–O
bonds, and of the C2–O and C1–O bonds respectively.
Similar calculations modeling the adducts of anhydro[1,6]mannose 2.02 and
anhydro[1,6]galactose 2.07 provided additional support for this proposal. The Fukui indices were
found to give the highest nucleophilicity at O-2 for anhydro[1,6]mannose 2.02 and at O-4 for
anhydro[1,6]galactose 2.07, which is consistent with the observed data. For the tribenzylated
mannose 2.03, the Fukui index indicated the highest nucleophilicity at the O-2 position since the
C1 position in comparison is more electron deficient. However, selective alkylation at the O-1
position is observed under the reaction conditions. This may be indicative of a proton transfer
equilibria playing a role in the regiochemical outcome.32
2.9 Conclusion
In conclusion, we have demonstrated that borinic acid catalysts represent an efficient and broadly
applicable method for the regioselective alkylation of carbohydrate derivatives bearing multiple
secondary hydroxy groups. Attractive features of this new mode of catalytic reactivity include its
operational simplicity and avoidance of using stoichiometric quantities of organotin-based
reagents.
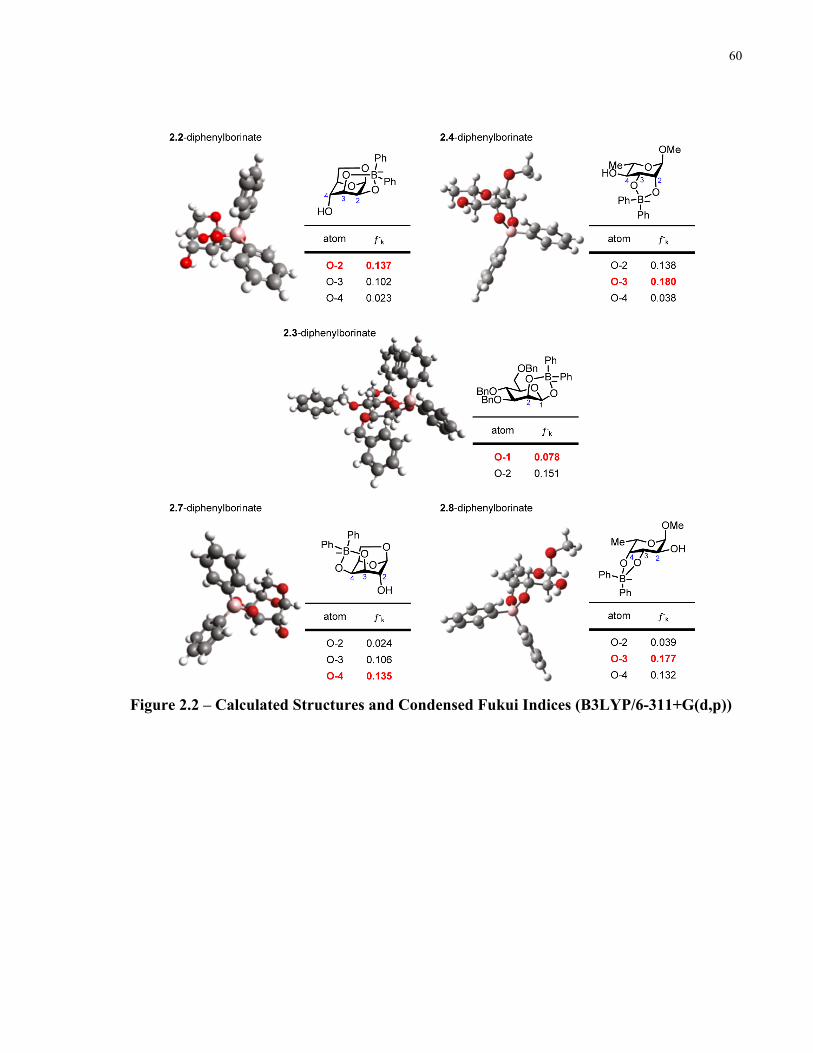
60
Figure 2.2 – Calculated Structures and Condensed Fukui Indices (B3LYP/6-311+G(d,p))

61
3 Regioselective Glycosylation by a Diarylborinic Acid
Derivative
3.0 Introduction
The stereo and regioselective formation of O-glycosidic bonds represents the key process in
oligosaccharide syntheses.71 Since the first introduction of glycoside synthesis by Michael72 and
Fischer73, and the seminal studies of Koenigs and Knorr74, the development of selective
glycosidation methods using readily available carbohydrate derivatives have been pursued for
more than a century. Regioselectivity in oligosaccharide synthesis, however, is a fundamental
challenge that arises from the fact that the glycosyl acceptor bears multiple, potentially reactive
hydroxy groups. Generally, this problem has been addressed through the use of protecting groups
to suppress glycosylations at undesired sites. As addressed earlier, the disadvantage of this
71 Zhu, X.; Schmidt, R. R. Angew. Chem. Int. Ed. 2009, 48, 1900–1934. 72 Michael, A. Am. Chem. J. 1879, 1, 305–312. 73 Fischer, E. Ber. Dtsch. Chem. Ges. 1893, 26, 2400–2412. 74 Koenigs, W.; Knorr, E. Ber. Dtsch. Chem. Ges. 1901, 34, 957–981.

62
strategy is that multiple inefficient steps are required to install and remove protecting groups,
generating chemical waste in the process.
Among other approaches towards addressing this issue, several are outlined below including
exploiting the inherent reactivity differences of the hydroxy groups and employing activating
agents such as organotin and organoboron reagents. Although there have been numerous
developments, none of these strategies rival the capabilities of enzymes.75
3.1 Inherent Reactivity of Hydroxy Groups in Glycosylations
Several reports of regioselective glycosylations have been achieved by exploiting inherent
differences in the steric and electronic properties of hydroxy groups for a limited group of
glycosyl substrates. These reactions, however, are generally optimized on a case-by-case basis
depending on the glycosyl donor and acceptor substrates employed, as outlined below. As well,
the regiochemical outcome of these processes may depend on the structure of the glycosyl donor
substrate. Various studies have reported the regioselective β-galactosylation of N-protected
glucosamine derivatives. In the synthesis of Lewis X pentasaccharide by Schmidt, glycosylation
of a 3-O,4-O-unprotected acceptor derived from N-TCP protected glucosamine using boron
trifluoride diethyl etherate as an activator was found to take place selectively at the 4-hydroxy
over the 3-hydroxy group (Scheme 3.1).76 It was proposed that the steric demand of the TCP
group suppressed reaction at the 3-hydroxy group.
Scheme 3.1 – Selective Glycosylation Due to Steric Effects
75 (a) Bennett, C. S.; Wong, C.-H. Chem. Soc. Rev. 2007, 36, 1227–1238; Hancock, S. M.; Vaughan, M. D.; Withers, S. G. Curr. Opin. Chem. Biol. 2006, 10, 509–519; (c) Gamblin, D. P.; Scanlan, E. M.; Davis, B. G. Chem. Rev. 2009, 109, 131–163. 76 Lay, L.; Manzoni, L.; Schmidt, R. R. Carbohydr. Res. 1998, 310, 157–171.
OO SPhHONTCP
OBn
O
OAc
OAcAcO
AcOOHO SPhHONTCP
OBnO
OAcAcO
AcOAcO
O NH
CCl3
BF3.OEt2 (0.05 equiv)
87%
–30 °C+

63
Similarily, work by the group of Magnusson reported that a N-TCP protected glucosamine
derivative could be selectively glycosylated to afford the 1→4 linked lactosamine product in the
presence of a free 3-hydroxy group using methylsulfenyl bromide and silver
trifluoromethanesulfonate as activators (Scheme 3.2).77 Glycosylation using a perbenzylated
fucosyl donor under the same reaction conditions was found to give a 3.6:1 mixture of the 1→3
and 1→4 linked disaccharides in 84% yield. In comparison, glycosylation using the
peracetylated fucosyl donor gave a 2.2:1 β:α mixture of the 4-O linked disaccharide in 58%
yield. The differences in the observed regioselectivities were attributed to several factors,
including the nature of the various protecting groups, and the nature and mode of activation of
the donor.
Scheme 3.2 – Regiodifferentiation in Glycosylation Reactions
77 Ellervik, U.; Magnusson, G. J. Org. Chem. 1998, 63, 9314–9322.
OO OPMBHONTCP
OBn
O
OAc
OBnAcO
AcOOHO OPMBHONTCP
OBnO
OBnAcO
AcOAcO
SPh
i. AgOTf, MeCN
84%
+ii. MeSBr, DCE –70 °C
OHO OPMBHONTCP
OBni. AgOTf, MeCN
+ii. MeSBr, DCE –70 °C
SEtO
OBnBnO
MeOBn
OO OPMBHONTCP
OBn
OOBnBnO
MeOBn
3.6:1, 84%
OHO OPMBHONTCP
OBni. AgOTf, MeCN
+ii. MeSBr, DCE –70 °C
SEtO
OAcAcO
MeOAc
OO OPMBHONTCP
OBn
O
OAcAcO
MeOAc
OO OPMBHONTCP
OBn
OOAcAcO
MeOAc
2.2:1, 58%
OHO OPMBONTCP
OBn
OOBnBnO
MeOBn
+
+

64
In a report by Fraser-Reid, glycosylations employing n-pentenyl ortho esters or n-pentenyl
glycosides as glycosyl donors were achieved with selectivity for the equatorial hydroxy group
and predominantly the axial hydroxy group respectively (Scheme 3.3).78 Reaction of the
mannose derived n-pentenyl glycoside with a mannoside diol afforded the disaccharide product
glycosylated at the axial hydroxy group as the major product, whereas with the mannose derived
n-pentenyl ortho ester only the equatorial glycosylated disaccharide was isolated. They
rationalize that the selectivity may arise from the distinct nature of the electrophilic species
involved (a trioxolenium ion versus oxocarbenium ion).
Scheme 3.3 – Glycosylations of n-Pentenyl Glycosides and n-Pentenyl Ortho Esters
3.2 Regioselective Glycosylations of Carbohydrate Derivatives via
Activating Agents
Regioselective reactions of carbohydrate derivatives have also been achieved by utilizing
activating agents to enhance the reactivity of specific hydroxy groups towards glycosyl donors.
In particular, organotin reagents have been widely applied; however, these protocols generally
require an additional synthetic step to incorporate the activating agent. As well, stoichiometric
quantities of toxic, lipophilic organotin species are generated in the process. More recently, an
78 Anilkumar, G.; Nair, L. G.; Fraser-Reid, B. Org. Lett. 2000, 2, 2587–2589.
NIS (1.3 equiv)
TBDMSOTf (cat.)CH2Cl2
OHO
OMe
AllylO
OHAllylOOBnO
O
BnO
OBnOBn
OBnO OBnO
OOBn
PhO
+
+NIS (1.3 equiv)
TBDMSOTf (cat.)CH2Cl2
OBnOBnO
OBnOBn
OHO
OMe
AllylO
OHAllylO
OHO
OMe
AllylO
OAllylO
OBnOBnO
OHOBn
OOAllylO
OHOAllyl
OMe69%
66%

65
alternative approach has been developed using arylboronic acids, in which a tetracoordinate
boronate complex of a cis-1,2-diol group undergoes selective reactions with electrophiles.
3.2.1 Activation via Organotin Reagents
The use of organotin reagents to control the activation of a specific hydroxy group for
glycosylation was first reported by Ogawa (Scheme 3.4).79 In these processes, participating
groups in the 2-position generated orthoesters which were then transformed with Lewis acids
into the corresponding disaccharides in moderate yields. Treatment of 2-O-acetyl-3,4,6-tri-O-
benzyl-α-D-mannopyranosyl chloride with stannylated methyl α-D-mannopyranoside, prepared
from bis(tributyltin) oxide, afforded a trimer derived from orthoester formation at the 3-O and 6-
O positions of methyl α-D-mannopyranoside in 34% yield. A minor amount (2% yield) of a
regioisomer in which orthoesters were generated at the 4-O and 6-O positions of methyl α-D-
mannopyranoside was also observed. The formation of the orthoesters is due to the participation
of the acetyl group at the 2-position of the glycosyl donor, generating a trioxolenium ion.
Scheme 3.4 – Ogawa’s Regioselective Glycosylation of Carbohydrate Derivatives
Augé and Veyrières demonstrated that glycosylation of the dibutylstannylene derivative of a
galactose-derived 3,4-cis-diol with glycosyl halides bearing no participating groups at the 2-
position gave good regio- and stereoselectivity when the reaction was carried out in a dipolar
aprotic solvent with lithium bromide or iodide salts as additives.80 Reaction of 2,3,4,6-tetra-O-
benzyl-α-D-galactopyranosyl chloride with an α-D-galactopyranoside stannylene derivative with
79 Ogawa, T.; Katano, K.; Matsui, M. Carbohydr. Res. 1978, 64, C3–C9. 80 Augé, C.; Veyrières, A. J. Chem. Soc. Perkin Trans. I 1979, 1825–1832.
OOBn
BnOBnO
OAc
Cl
OOH
HOHO
OH
OMe
i. (Bu3Sn)2O (1.5 equiv)
ii.O
O
HOO
OH
OMe
OOBn
BnOBnO
OO
Me
OOBn
BnOBnO
OO
Me
34%

66
lithium iodide in dry hexamethylphosphoroamide gave the desired α–linked disaccharide in 74%
yield and minor amounts of the β-linked isomer in 8% yield (Scheme 3.5).
The addition of lithium halide salts was inspired by a report by Gent and Gigg where they
demonstrated that tetrabutylammonium bromide helps avoid problems associated with the
instability of benzylated glycosyl bromides by converting 1,2-cis-glycosyl chloride into a 1,2-
trans-glycosyl bromide in 1,2-cis-glycoside synthesis.81 In later reports, it was found that the
same regioselectivity was observed in the absence of stannylene activation.80,82
Scheme 3.5 – Augé and Veyrières’ Regioselective Glycosylation Method
Martin-Lomas later reported the tributyltin ether-mediated regioselective glycosylations of 1,6-
anhydro-β-D-galactopyranose and methyl β-lactoside.83 Reaction of the stannylated 1,6-anhydro-
β-D-galactopyranose derivative with 2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl bromide in the
presence of tetraethylammonium bromide in toluene gave a 4:1 mixture of the 1,6-anhydro-4-O-
(2,3,4,6-tetra-O-benzyl-β- and -α-D-galactopyranosyl)-β-D-galactopyranose in 79% yield with
1,6-anhydro-3-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-β-D-galactopyranose as a minor
product in 14% yield (Scheme 3.6). Glycosylation of methyl β-lactoside occurred
regioselectively at the primary positions with complete α-stereoselectivity (Scheme 3.7). This
result contrasts with their previous findings of tributyltin ether-mediated benzylation of methyl
β-lactoside, in which the 3’-O position was the most reactive hydroxy group.83
81 Gent, P. A.; Gigg, R.; J. Chem. Soc. Perkin Trans. I 1975, 1521–1524. 82 Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062. 83 Cruzado, C.; Bernabe, M.; Martin-Lomas, M. Carbohydr. Res. 1990, 203, 296–301.
OBnO
BnOBnO
OBn
Cl
OBnO
BnOBnO
OBn
OHO
OAllylO
OBn
OBn
OHO
AllylO
OBn
OBn
HOi. (Bu2SnO)n, benzene
ii.
LiI (5 equiv)HMPA, 0.35 M
(2 equiv)
OBnO
BnOBnO
OBn
OHO
OAllylO
OBn
OBn
+
74%:8%

67
Scheme 3.6 – Regioselective Glycosylation of 1,6-Anhydro-β-D-Galactopyranose
Scheme 3.7 – Regioselective Glycosylation of Methyl β-Lactoside
More recently, Oscarson and co-workers reported the regioselective glycosidation at the primary
position of galactopyranosides bearing no protecting groups at the 2-,3-,4-,6-positions.84 The
dibutylstannylene derivative of β-D-galactopyranoside underwent regioselective glycosylation
with 2,3,4,6-tetra-O-benzyl-α-D-glucopyranoside in the presence of tetrabutylammonium iodide
to afford the α-linked disaccharide in 78% yield (Scheme 3.8).
84 Garegg, P. J.; Maloisel, J.-L.; Oscarson, S. Synthesis 1995, 409–414.
OBzO
BzOBzO
OBz
Br
i. (Bu3Sn)2O, toluene
ii.
Et4NBr (6 equiv)CH2Cl2, 0.04 M
(2 equiv)
OO
HOOH
OH
OO
OOH
OH
OBzO
BzOBzO
OBz
OO
OOH
OH
OBzO
BzOBzO
OBz
OO
HOO
OH
OBzO
BzOBzO
OBz
4:1, 79%+
14%
HO OMeOO
HO
HOHO
OH
OH
OH
HO OMeOO
HO
HOHO
OH
O
OH
OBzO
BzOBzO
OBz
Br
i. (Bu3Sn)2O, toluene
ii.
Et4NBr (6 equiv)CH2Cl2, 0.04 M
(2 equiv)
OBzO
BzOBzO
OBz
HO OMeOO
HO
HOHO
O
OH
OH
OBzO
BzOBzO
OBz 16%
58%
+

68
Scheme 3.8 – Regioselective Glycosylation of Methyl β-D-Galactopyranoside
In 2000, Kaji reported an alternative stannylene acetal-mediated regioselective glycosylation
strategy for methyl β-D-galactopyranoside and methyl α-L-rhamnopyranoside using silver silica
alumina as an effective promoter to access β(1→6)- and β(1→3)-linked disaccharides.
Treatment of the stannylene-acetal derivative of methyl β-D-galactopyranoside with per-O-
pivaloylglucosyl bromide afforded the disaccharide in 72% yield as the sole product (Scheme
3.9).
Scheme 3.9 – Regioselective Glycosylation of Methyl β-D-Galactopyranoside With Silver
Silica Alumina as a Promoter
OMeO
HO
HOHO
OH
OBnOBnO
BnO
OBn
Br
i. (Bu3Sn)2O, MeOH
ii.
Bu4NI (1.2 equiv)CH2Cl2
(1.2 equiv) OMeO
HO
HOHO
O
OBnOBnO
BnO
OBn
78%
OMeO
HO
HOHO
OH
OPivOPivO
PivO
OPiv
Br
i. (Bu2SnO)n, MeOH
ii.
Ag-silica aluminaCH2Cl2
(2 equiv)OMe
OHO
HOHO
OOBnOBnO
BnO
OBn
72%

69
3.2.2 Activation via Arylboronic Acids
Generally, the formation of boronic esters with cis-vicinal diols or 1,3-diols involving an
exocyclic 6-hydroxy group of carbohydrate derivatives has been employed as a strategy for OH
protection. A series of reports by Fréchet and co-workers demonstrated the ability of
polystyrylboronic acids to act as protective agents for cis-diols. In their studies, protection of
methyl α-D-xylopyranoside with polystyrylboronic acid, followed by treatment with acetic
anhydride afforded the 3-O monoacetylated product in 87% yield (Scheme 3.10).85
Scheme 3.10 – Protection of Carbohydrate Derivatives by Boronic Acids
Boons and co-workers reported an extension of this method where they demonstrated the use of
polystyrylboronic acid in the synthesis of di- and trisaccharides.86 Methyl 3-O-benzyl-
galactoside was immobilized on polystyrylboronic acid, and treated with an N-phthalimido
thioglucopyranoside derivative, N-iodosuccinimide and trimethylsilyl trifluoromethanesulfonate
in dichloromethane to obtain the β-disaccharide in 95% yield (Scheme 3.11).
Scheme 3.11 – Regioselective Glycosylation Using Boronic Acids as a Protecting Group
85 Fréchet, J. M. J.; Seymour, E. Tetrahedron Lett. 1976, 41, 3669–3672. 86 (a) Belogi, G.; Zhu, T.; Boons, G.-J. Tetrahedron Lett. 2000, 41, 6965–6968; (b) Belogi, G.; Zhu, T.; Boons, G.-J. Tetrahedron Lett. 2000, 41, 6969–6972.
OHO
OMeOH
HOi. pyridine, !
ii. Ac2O, pyridine
B(OH)2
O
OAc
OO B
OMe
87%
OMeO
HO
BnOHO
OH
pyridine, !
B(OH)2
OMeO
O
BnOHO
OB SEt
OBnOBnO
NPhth
OBni.
NIS, TMSOTf, CH2Cl2ii. acetone-H2O 60 °C
OMeO
HO
BnOO
OH
OBnOBnO
NPhth
OBn
95%

70
More recently, the group of Kaji has reported an alternative procedure where boronic acids act as
transient masking agents for hydroxy groups to carry out regioselective glycosylation of fully
unprotected methyl hexopyranosides in one-pot.87 In these reactions, the boronic acid forms
cyclic boronates with the 1,2-cis-diol or 4,6-diols of carbohydrate derivatives, thus allowing the
glycosyl donor to attack a free hydroxy group other than the masked hydroxy groups. Exposure
to aqueous sodium perborate after the glycosylation step easily cleaves the boronic acid and
affords the desired product. Treatment of methyl β-D-galactopyranoside with per-O-
pivaloylglucosyl bromide, phenylboronic acid, and silver silica alumina in dichloroethane
afforded the β(1→3)- and β(1→2)-linked disaccharides in a 92:8 mixture in 78% yield (Scheme
3.12).
Scheme 3.12 – Boronic Acids as a Transient Masking Agent
In contrast, studies by Aoyama and Oshima have demonstrated the ability of boronate esters to
activate hydroxy groups towards regioselective glycosylation. Previously, it was demonstrated
that triethylamine activated boronic esters generated from phenylboronic acid and a fucose
derivative undergo highly regioselective alkylations.88 Initial attempts to extend such studies to
the synthesis of di- and trisaccharides using the same conditions were futile, however, it was
found that intramolecular oxygen coordination to activate the boronate oxygen atom towards
glycosylation allowed such reactions to occur regioselectively with unprotected sugars (Scheme
87 Kaji, E.; Nishino, T.; Ishige, K.; Ohya, Y.; Shirai, Y. Tetrahedron Lett. 2010, 51, 1570–1573. 88 Oshima, K.; Kitazono, E.-I.; Aoyama, Y. Tetrahedron Lett. 1997, 38, 5001–5004.
OMeO
HO
HOOH
OH
Br
OPivOPivO
PivO
OPiv
1,2-DCE, 0.05 M3Å MS
Ag-silica alumina
+
BHO OH
(1 equiv)
(1 equiv)
(2 equiv)
OMeO
HO
OOH
OHOPivO
PivOPivO
OPiv
OMeO
HO
HOO
OH
OPivOPivO
PivO
OPiv
+
92:8, 78%

71
3.13).89 The two vicinal methyl groups on the boronate ester were proposed to block the benzylic
oxygen atom, preventing undesired glycosylation reactions at that site.
Scheme 3.13 – Tetracoordinated Arylboronate Complex
Treatment of the methyl-α-L-fucoside 3,4-boronate ester with 2,3,4,6-tetra-O-acetyl-α-D-
glucopyranosyl bromide, tetraethylammonium iodide, silver carbonate and molecular sieves in
tetrahydrofuran gave glycosylation exclusively at the 3-O position to afford a disaccharide in
74% yield. Increasing the equivalents of the glycosyl donor to 3.5 equivalents improved the yield
to 93% (Scheme 3.14). Following a similar procedure, other carbohydrate substrates bearing four
hydroxy groups were converted to trisaccharides with yields up to 84%. The regioselectivity for
glycosylation was rationalized as occurring at the least hindered oxygen atom of the boronate
ester.
Scheme 3.14 – Regioselective Glycosylation Activated by Arylboronic Acids
89 Oshima, K.; Aoyama, Y. J. Am. Chem. Soc., 1999, 121, 2315–2316.
O
OH
OHOH
OMeMe
O B O
MeMe
HMe
Me
Ag2CO3 O B O
MeMe
MeMe
Et4NI4Å MS
O
O
OHO
OMeMe
B O
Me
MeEt4N
OAcOAcO
AcO
OAcO
OH
OHO
OMeMeO
OH
OHOH
OMeMe
i. promoter, Ag2CO3 Et4NI, 4Å MS
ii.
Ag2CO3, THF
OAcOAcO
AcO
OAc
Br74%
O B O
MeMe
MeMe
promoter:

72
3.3 Regioselective Activation of Glycosyl Acceptors by a
Diarylborinic Acid Catalyst
Following our investigations of the diarylborinic acid-catalyzed regioselective functionalization
of carbohydrate derivatives containing cis-vicinal diol moieties, we sought to determine whether
we could achieve regioselective activation of glycosyl acceptors for glycosylation reactions.
Extensive investigations by other members of our laboratory90 resulted in the discovery of
reaction conditions suitable for the glycosylation of several derivatives of mannose, galactose,
fucose and arabinose to access 1,2-trans linkages from donors of gluco or galacto
configuration.91 In contrast to other protocols, this method is operationally simple and avoids the
use of stoichiometric quantities of organotin or organoboron reagents. In addition, this new mode
of reactivity displayed remarkable compatibility towards the synthesis of disaccharides and
trisaccharides.
Reaction development was carried out on methyl-6-(tert-butyldimethylsilyloxy)-α-D-
mannopyranoside and the optimal conditions were found to utilize the diphenylborinic acid
derivative as a catalyst, silver oxide as a promoter and base, and acetonitrile as a solvent. The
versatility of this method was illustrated by the variety of glycosyl donors and acceptors that are
tolerated (Table 3.1).
90 This work was done by Christina Gouliaras and Doris Lee. 91 Gouliaras, C.; Lee, D.; Chan, L.; Taylor, M. S. J. Am. Chem. Soc. DOI: 10.1021/ja2062715.

73
Table 3.1 – Scope of Optimized Reaction Conditions for Glycosylation
Given the success of the aforementioned reaction, subsequent investigations were carried out to
further expand the scope of the organo-boron catalyzed glycosylations. These include
glycosylations using glucosamine derived donors, the formation of β-mannoside linkages,
reactivity in conjunction with halide ion catalysis, and boronic acid-mediated glycosylations.
OOAc
AcOAcO
AcO OOOH
OOH
O
OAc
AcOAcO
AcO OO
OTBS
HO
HO
OMe
OOPiv
PivOPivO
PivOOMe
O
OMe
OH
OHO
OAc
AcOAcO
AcO OOTBS
HOO
OH
OMe
OOAc
AcOAcO
AcOO
OHO
O
OH
OMe
OOAc
AcOAcO
AcO
OAcO
OAc
AcO
OAc
OAcO
OAc
OO
OTBSOH
HO
OMeOAc
AcO
OAcO
OBn
OO
OTBSOH
HO
OMeOAc
AcO
OAcO O
O
OTBSOH
HO
OMeOAc
AcO
OMe
OAcOAc
OAcO
OHOMe
OH
OMe
OBnO
OBn
OO
OTBSOH
HO
OCH3OBn
BnO
OOAc
AcOAcO
AcO OO
OTBS
HO
HO
OMe
R3R4 Ag2O (1.1 equiv)
MeCN, 23–60 °C
PhB
Ph NH2
O
(10 mol %)HO
HO R3R4
HO
O
OR1R2
X
OR1R2
+
OO
HO
HOOMe
99%
94%
73%
81%
73%
68%a
86%a
71%a
80%
75%88%
74%
a Reaction was carried out using the glycosyl chloride

74
3.3.1 Synthesis of Glucosamine Derived Donors
Various glucosamine donors investigated for use in the glycosylation reactions were synthesized
by literature methods as depicted below.
3,4,6-Tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl bromide 3.01 was synthesized in
73% yield by treating commercially available 1,3,4,6-tetra-O-acetyl-2-deoxy-2-phthalimido-β-D-
glucopyranoside with hydrogen bromide (33% in acetic acid) (Scheme 3.15).92
Scheme 3.15 – Synthesis of 2-Phthalimido-β-D-Glucopyranosyl Bromide
The α-D-glucopyranosyl chloride version was also synthesized in order to compare their
reactivity. 1,3,4,6-Tetra-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranoside was deacetylated
at the anomeric position using imidazole and methanol to afford 3.02 in 72% yield.93
Chlorination was carried out following a protocol by Mong94 using trichlorotriazine,
dimethylformamide and diazabicyclo-[5.4.0]-undec-7-ene to afford the product in a 3:1 α:β
mixture in 46% yield. Recrystallization in cold (–20 °C) diethyl ether gave pure 3,4,6-tri-O-
acetyl-2-deoxy-2-phthalimido-α-D-glucopyranosyl chloride 3.03 in 39% yield (Scheme 3.16).
Scheme 3.16 – Synthesis of 2-Phthalimido-α-D-Glucopyranosyl Chloride
92 Ruttens, B.; Blom, P.; Van Hoof, S.; Hubrecht, I.; Van der Eycken, J. V. J. Org. Chem. 2007, 72, 5514–5522. 93 Brain n’ Beyond. Biotech. Pvt. Ltd. 2007. Patent W02007/52308 A2. 94 Chang, C.-W.; Chang, S.-S.; Chao, C.-S.; Mong, K.-K. T. Tetrahedron Lett. 2009, 50, 4536–4540.
OOAc
AcOAcO
PhthNOAc
i. 33% HBr in AcOH
ii. recrystallization in Et2O
OOAc
AcOAcO
PhthNBr
3.01, 73%
OOAc
AcOAcO
PhthNOAc
OOAc
AcOAcO
PhthN OHMeOH, 0.2 M
40 °C
3.02, 72%
Imidazole (1 equiv)
DBU (1.1 equiv)DCM, 0.3 M
60 °C
TCT (1.1 equiv)DMF (4 equiv)
OOAc
AcOAcO
PhthNCl
3.03, 39%

75
The synthesis of 3,4,6-tetra-O-acetyl-2-deoxy-2-[(2,2,2-trichloroethoxy)carbonylamino]α-D-
glucopyranosyl bromide 3.06 was carried out following the procedure by Herdewijn (Scheme
3.17).95 Beginning with D-glucosamine hydrochloride, protection of the amine with
(trichloroethoxy)carbonyl chloride, followed by peracetylation of the hydroxy groups and
bromination of the anomeric position furnished the desired α-product in 45% yield overall.
Scheme 3.17 – Synthesis of 2-[(2,2,2-Trichloroethoxy)carbonylamino]-α-D-Glucopyranosyl
Bromide
Following the procedure by Sewald96, 3,4,6-tri-O-acetyl-D-glucal 3.07 was treated with ferric
chloride hexahydrate, sodium azide and hydrogen peroxide in acetonitrile at -20 °C for 7 hours to
afford 3,4,6-tri-O-acetyl-2-deoxy-2-azido-α-D-galactopyranosyl chloride 3.08 in 76% yield as a
6:1 mixture of α:β products (Scheme 3.18). Attempts to obtain only the α-anomer through
recrystallization were unsuccessful.
Scheme 3.18 – Synthesis of 2-Azido-α-D-Galactopyranosyl Chloride
95 Lioux, T.; Busson, R.; Rozenski, J.; Nguyen-Distèche, M.; Frére, J.-M. Herdewijn, P. Collect. Czech. Chem. Commun. 2005, 70, 1615–1641. 96 Plattner, C.; Hofener, M.; Sewald, N. Org. Lett. 2011, 13, 545–547.
OOH
HOHO
NH2 OH
HCl
NaHCO3 (1.7 equiv)TrocCl (1.5 equiv)
H2O, 1.2 MO
OH
HOHO
TrocHN OH
3.04, 53%
OOAc
AcOAcO
TrocHN OAc
3.05, >99%
Ac2O (4.5 equiv)
pyridine, 0.5 M
33% HBrin AcOH
OOAc
AcOAcO
TrocHNBr3.06, 86%
OOAc
AcOAcO
OOAc
AcOCl
AcO
N3
NaN3 (1.1 equiv)FeCl3.6H2O (0.8 equiv)
H2O2 (1.1 equiv)MeCN, 0.12 M–20 °C, 7 hr 6:1 alpha:beta
3.08, 76%
OOH
HOHO
Ac2O (3.5 equiv)
pyridine, 0.5 M
3.07, 92%

76
3.3.2 Regioselective Glycosylations with Nitrogen-Containing Glycosyl Donors
The optimal reaction conditions for glycosylations using the diarylborinic acid were tested with
the glucosamine derived donors. Initial screening of 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-
D-glucopyranosyl bromide 3.01 with mannose 3.09, galactose 3.10, fucose 3.11 or arabinose 3.12
was carried out to determine ideal coupling partners for optimization studies (Table 3.2). Results
showed that only galactose 3.10 delivered the desired disaccharide in 42% isolated yield.
Reaction with mannose 3.09 and arabinose 3.12 gave no reactivity, whereas fucose 3.11 gave no
selectivity and resulted in a complex mixture of products.
Table 3.2 – Glycosylation Screen with 2-Phthalimido-β-D-Glucopyranosyl Bromide
OOAc
AcOAcO
PhthNBr
3.01
entry yieldacceptor
1
2
3
4
O
OTBS
HO OMe
HO
OH
O
OTBS
HOHO
OH
OMe
n.r.
3.13, 42%
n.r.
complex mixture
product
O
OMeMe
OHOH
OH
OHO OMe
HO
OH
OOAc
AcOAcO
PhthN
O
OTBS
HOO
OH
OMe
OOAc
AcOAcO
PhthN
OOAc
AcOAcO
PhthN
OOAc
AcOAcO
PhthN
O
OTBS
O OMe
HO
OH
O
OMeMe
OHO
OH
OO OMe
HO
OH
Ag2O (1.1 equiv)MeCN, 0.13 M
16 hr, rt
PhB
Ph NH2
O
(10 mol %)
R2R1
HO
O+ O
OAc
AcOAcO
PhthNR2R1
HO
HO
3.09
3.10
3.11
3.12
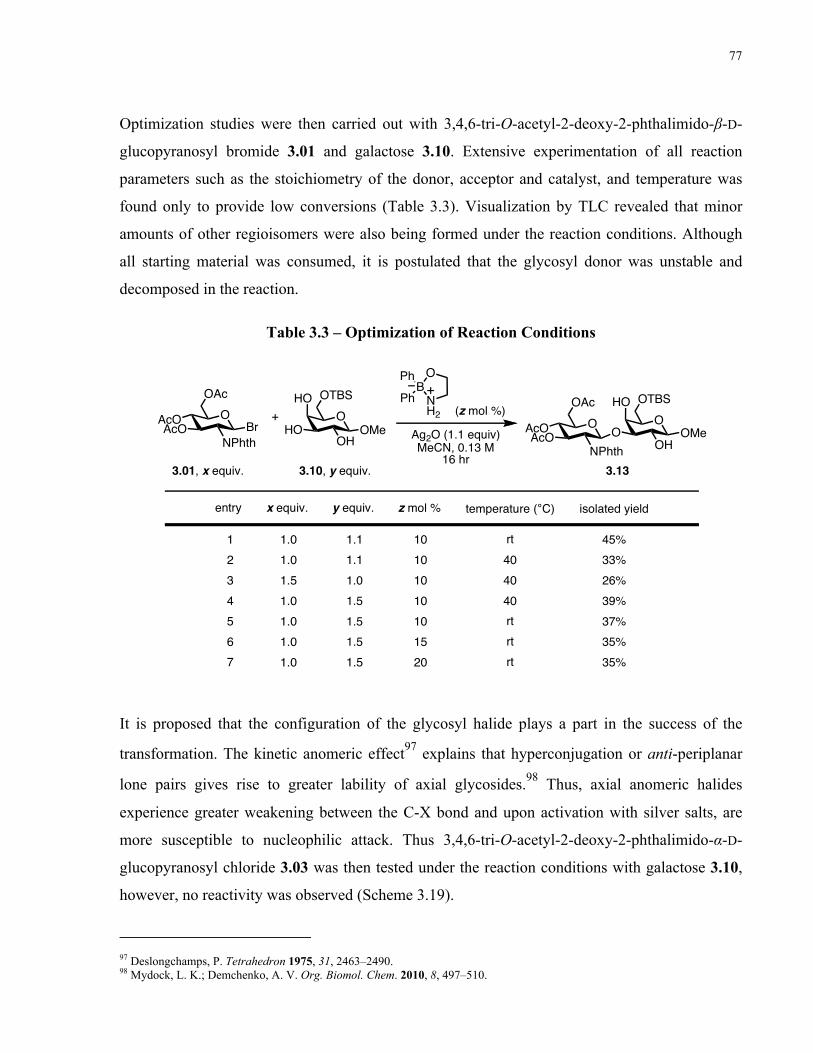
77
Optimization studies were then carried out with 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-
glucopyranosyl bromide 3.01 and galactose 3.10. Extensive experimentation of all reaction
parameters such as the stoichiometry of the donor, acceptor and catalyst, and temperature was
found only to provide low conversions (Table 3.3). Visualization by TLC revealed that minor
amounts of other regioisomers were also being formed under the reaction conditions. Although
all starting material was consumed, it is postulated that the glycosyl donor was unstable and
decomposed in the reaction.
Table 3.3 – Optimization of Reaction Conditions
It is proposed that the configuration of the glycosyl halide plays a part in the success of the
transformation. The kinetic anomeric effect97 explains that hyperconjugation or anti-periplanar
lone pairs gives rise to greater lability of axial glycosides.98 Thus, axial anomeric halides
experience greater weakening between the C-X bond and upon activation with silver salts, are
more susceptible to nucleophilic attack. Thus 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-α-D-
glucopyranosyl chloride 3.03 was then tested under the reaction conditions with galactose 3.10,
however, no reactivity was observed (Scheme 3.19).
97 Deslongchamps, P. Tetrahedron 1975, 31, 2463–2490. 98 Mydock, L. K.; Demchenko, A. V. Org. Biomol. Chem. 2010, 8, 497–510.
OOAc
AcOAcO
NPhthBr Ag2O (1.1 equiv)
MeCN, 0.13 M16 hr
PhB
Ph NH2
O
(z mol %)OOTBSHO
HOOH
OMe+ O
OAc
AcOAcO
NPhth
OOTBSHO
OOH
OMe
3.10, y equiv.3.01, x equiv.
temperature (°C) isolated yield
26%
35%
39%37%
35%
33%
y equiv.
1.0
1.5
1.51.5
1.1
1.5
404040rt
rtrt
x equiv.
1.01.51.01.0
1.01.0
z mol %
10101010
2015
entry
2345
76
45%1.1 rt1.0 101
3.13

78
Scheme 3.19 – Unsuccessful Glycosylation of 2-Phthalimido-β-D-Glucopyranosyl Chloride
To test whether the glycosylation was unsuccessful due to incompatibility with the phthalimido
protecting group, 3,4,6-tetra-O-acetyl-2-deoxy-2-[(2,2,2-trichloroethoxy)carbonylamino]α-D-
glucopyranosyl bromide 3.06 was evaluated under the reaction conditions (Table 3.4). The
reaction gave higher conversions for mannose 3.09 (entry 2) in comparison to that of galactose
3.10 (entry 1), however, the yield was still modest. Furthermore, heating the reaction had a
negative effect (entry 3) and increasing stoichiometry of acceptor (entry 4) or donor (entry 5) still
gave no significant improvements.
Table 3.4 – Glycosylation Screen with 2-[(2,2,2-Trichloroethoxy)carbonylamino]α-D-
Glucopyranosyl Bromide
Ag2O (1.1 equiv)MeCN, 0.13 M
16 hr, rt
PhB
Ph NH2
O
(10 mol%)O
OTBSHO
HOHO
OMen.r.+O
OAc
AcOAcO
PhthNCl
3.03 3.10
OOAc
AcOAcO
TrocHNBr
3.06
entry yieldacceptor
1
234
29%
45%
49%a38%
product
OOAc
AcOAcO
TrocHN
O
OTBS
HOO
OH
OMe
OOAc
AcOAcO
TrocHN
O
OTBS
O OMe
HO
OH
Ag2O (1.1 equiv)MeCN, 0.13 M
16 hr
PhB
Ph NH2
O
(10 mol %)
R2R1
OH
O+ O
OAc
AcOAcO
TrocHNR2R1
OH
HO
5 46%b
O
OTBS
HO OMe
HO
OH
O
OTBS
HOHO
OH
OMe3.09
3.10
temperature (°C)
rt
rt40rtrt
3.14
3.15
a Reaction carried out with 1.5 equivalent of glycosyl acceptorb Reaction carried out with 1.5 equivalent of glycosyl donor

79
The final nitrogen-containing glycosyl donor studied was the 3,4,6-tri-O-acetyl-2-deoxy-2-azido-
α-D-galactopyranosyl chloride 3.08 (Table 3.5). The glycosyl donor was screened with mannose
3.09 and galactose 3.10, and reaction parameters such as stoichiometry of reagents, temperature
and time were carried. However, conditions providing successful glycosylations could not be
identified.
Overall, the attempted glycosylation reactions with nitrogen-containing glycosyl donors were
unsuccessful as only up to modest yields were obtained.
Table 3.5 – Glycosylation Screen with 2-Azido-α-D-Galactopyranosyl Chloride
entry yieldacceptor
123a
4b
n.r.n.r.
n.r.n.r.
product
OOAc
AcOAcO
N3
O
OTBS
HOO
OH
OMe
OOAc
AcOAcO
N3
O
OTBS
O OMe
HO
OH
Ag2O (1.1 equiv)MeCN, 0.13 M
16 hr
PhB
Ph NH2
O
(10 mol %)
R2R1
OH
O
+ OOAc
AcOAcO
N3R2
R1
OH
HO
O
OTBS
HO OMe
HO
OH
O
OTBS
HOHO
OH
OMe3.09
3.10
temperature (°C)
rt40rtrt
OOAc
AcOCl
AcO
N36:1 alpha:beta
3.08
567a
8b
n.r.n.r.
n.r.n.r.
rt40rtrt
a Reaction carried out with 1.5 equivalent of glycosyl acceptorb Reaction carried out with 1.5 equivalent of glycosyl donorc Reaction was left stirring for 2 days

80
3.3.3 Regioselective Glycosylations to Form β-Mannoside Linkages
Of the glycosidic linkages possible, the β-mannoside link is one of the most difficult to form due
to both anomeric effect and neighbouring group participation effect favouring the formation of α-
mannosides. A strategy developed by Gorin and co-workers exploited the use of a cyclic
carbonate at the 2-O and 3-O positions of mannose to block neighbouring group participation
allowing the formation of β-linkages (Scheme 3.20).99
Scheme 3.20 – Formation of β-Mannoside Linkages
We envisioned that a diarylborinic acid variant of that reaction would offer a major improvement
since minimally protected carbohydrate derivatives bearing cis-1,2-diols could potentially act as
glycosyl acceptors (the β-mannosylation often works best when the nucleophile is unhindered).
To test the hypothesis, the mannose and rhamnose derivatives bearing a cyclic carbonate at the 2-
O and 3-O positions were required to be synthesized.
In initial efforts to form the cyclic carbonate on rhamnose, methyl α-L-rhamnopyranoside was
treated with phenyl chloroformate and N,N-diisopropylethylamine in dichloromethane, however,
no reactivity was observed (Table 3.6, entry 1). It was proposed that perhaps a diarylborinic acid
could help activate the hydroxy groups and impart regioselectivity. With the addition of 10 mol
% of diarylborinic acid derivative and the use of acetonitrile as the solvent, only starting material
was observed (entry 2). Switching the electrophile to bis(4-nitrophenyl) carbonate afforded a
complex mixture of products (entry 3). Ultimately, it was found that using
N,N’carbonyldiimidazole as the electrophile gave the desired cyclic carbonate 3.16 in 72% yield
(entry 4). Protection of the free hydroxy group with benzoyl chloride followed by treatment with
99 (a) Gorin, P. A. J.; Perlin, A. S. Can. J. Chem. 1961, 39, 2474–2485; (b) Bebault, G. M.; Dutton, G. G. S. Carbohydr. Res. 1974, 37, 309–319; (c) Backinowsky, L. V.; Balan, N. F.; Shashkov, A. S.; Kochetkov, N. K. Carbohydr. Res. 1980, 84, 225–235.
O
OAc
AcOOO
Br
O
O
OHOMe
O
OMe
MeMe
Ag2O, CHCl3 O
OAc
AcOOO
O
O
OO
Me
O
OMe
MeMe
82%
+

81
hydrogen bromide (33% in acetic acid) afforded the desired glycosyl bromide 3.18 in 30% yield
(Scheme 3.21).
Synthesis of the mannose donor was attempted under a similar protocol. Treatment of methyl-6-
(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 3.09 with catalytic 2-aminoethyl
diphenylborinic ester, N,N’carbonyldiimidazole and N,N-diisopropylethylamine in acetonitrile
delivered the desired cyclic carbonate 3.19 in >99% yield (Scheme 3.22). In order to install the
bromide at the anomeric position, steps for desilylation followed by peracetylation were
necessary. However, attempts to desilylate 3.19 were futile as either only starting material or a
complex mixture of products were obtained (Table 3.7). Thus, glycosylation reactions were
investigated using the rhamnose donor 3.18.
Table 3.6 – Synthesis of 2,3-O-Carbonyl-α-L-Rhamnopyranoside
entry yieldelectrophile
1a
2
3
4
n.r.
n.r.
72%
complex mixture
electrophileiPr2NEt (1.1 equiv)
MeCN, 0.2 M40 °C, 16 hr
PhB
Ph NH2
O
(10 mol %)O
OMeMe
HOHO
OH
O
OMeMe
HOO O
O3.16
O Cl
O
O O
OO2N NO2
O Cl
O
N N
O
NN
a Reaction without catalyst in dichloromethane
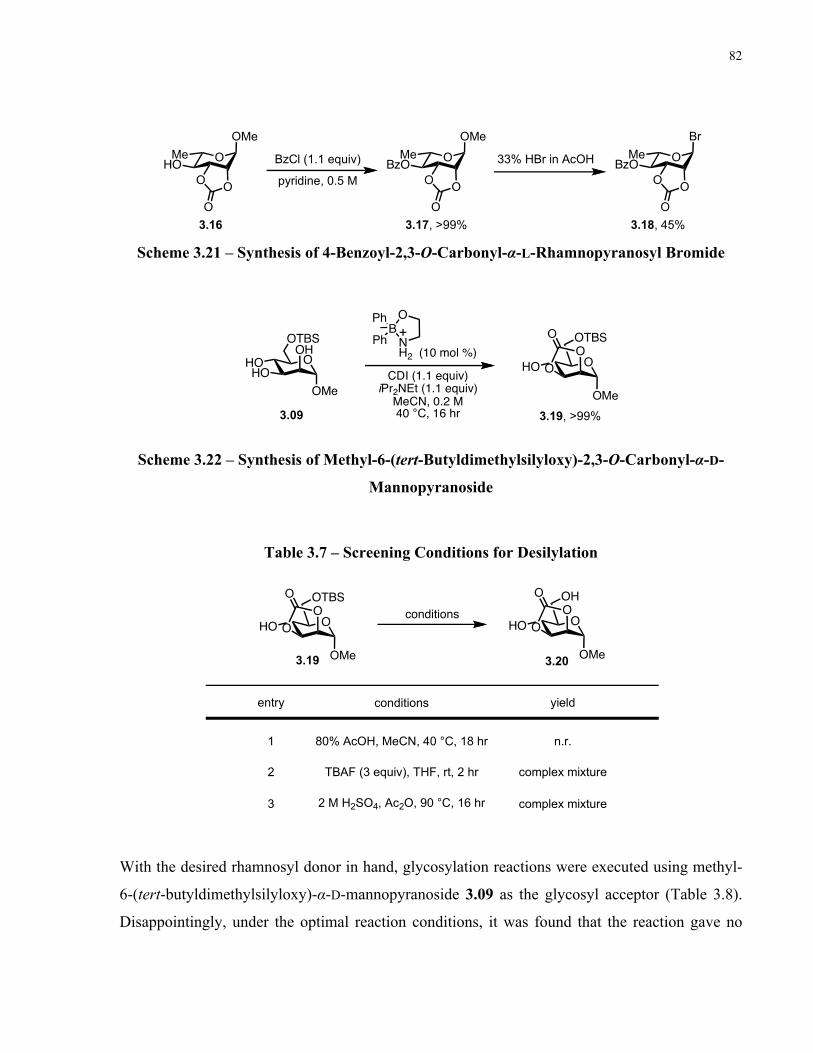
82
Scheme 3.21 – Synthesis of 4-Benzoyl-2,3-O-Carbonyl-α-L-Rhamnopyranosyl Bromide
Scheme 3.22 – Synthesis of Methyl-6-(tert-Butyldimethylsilyloxy)-2,3-O-Carbonyl-α-D-
Mannopyranoside
Table 3.7 – Screening Conditions for Desilylation
With the desired rhamnosyl donor in hand, glycosylation reactions were executed using methyl-
6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 3.09 as the glycosyl acceptor (Table 3.8).
Disappointingly, under the optimal reaction conditions, it was found that the reaction gave no
O
OMeMe
HOO O
O
BzCl (1.1 equiv) O
OMeMe
BzOO O
O
33% HBr in AcOH O
BrMe
BzOO O
O3.18, 45%3.17, >99%
pyridine, 0.5 M
3.16
O
OTBS
HO OO
OMe
O
OOTBS
HOHO
OH
OMeCDI (1.1 equiv)
iPr2NEt (1.1 equiv)MeCN, 0.2 M40 °C, 16 hr
PhB
Ph NH2
O
(10 mol %)
3.19, >99%3.09
entry yieldconditions
1
2
3
n.r.
complex mixture
complex mixture
O
OH
HO OO
OMe
O
conditions
3.203.19
O
OTBS
HO OO
OMe
O
80% AcOH, MeCN, 40 °C, 18 hr
TBAF (3 equiv), THF, rt, 2 hr
2 M H2SO4, Ac2O, 90 °C, 16 hr

83
desired product, and only starting material was isolated (entry 1). Additionally, longer reaction
times and higher temperatures lead to complex mixtures of product (entry 2 and 3).
Due to time constraints, further experimentation could not be carried out. Future work would
involve extensive optimization of all reaction parameters such as stoichiometry, reagents,
temperature and time. Furthermore, we also envision employing such donors for glycosylation at
the 6-O position.
Table 3.8 – Glycosylation with 4-Benzoyl-2,3-O-Carbonyl-α-L-Rhamnopyranosyl Bromide
3.3.4 Regioselective Glycosylations with Halide Ion Catalysis
Halide ion-catalyzed glycosylation reactions established by Lemieux and co-workers
demonstrate that halide salts such as tetraethylammonium chloride can be exploited to rapidly
anomerize glycosyl halides to form the less thermodynamically stable anomer in low
concentrations and which react readily.100 Studies were then carried out to examine whether the
halide ion-catalyzed glycosylation method would be compatible with borinic acid catalysis.
Preliminary tests to screen the glycosylation reaction between 2,3,4,6-tetra-O-acetyl-α-D-
glucopyranosyl bromide and methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 3.09
were carried out with 2-aminoethyl diphenylborinic ester, potassium carbonate and a halide salt
100 Lemieux, R. U.; Hendriks, K. B.; Stick, R. V.; James, K. J. Am. Chem. Soc. 1975, 97, 4056–4062.
Ag2O (1.1 equiv)MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %) OOTBS
HOOH
OMeO+ OMe
BzOO O
O
O
BrMe
BzOO O
O3.18
OOTBS
HOHO
OH
OMe
3.09
entry yieldtime (hr)
1
2
n.r.
complex mixture
temperature (°C)
16
48
rt
40
3 complex mixture48 80
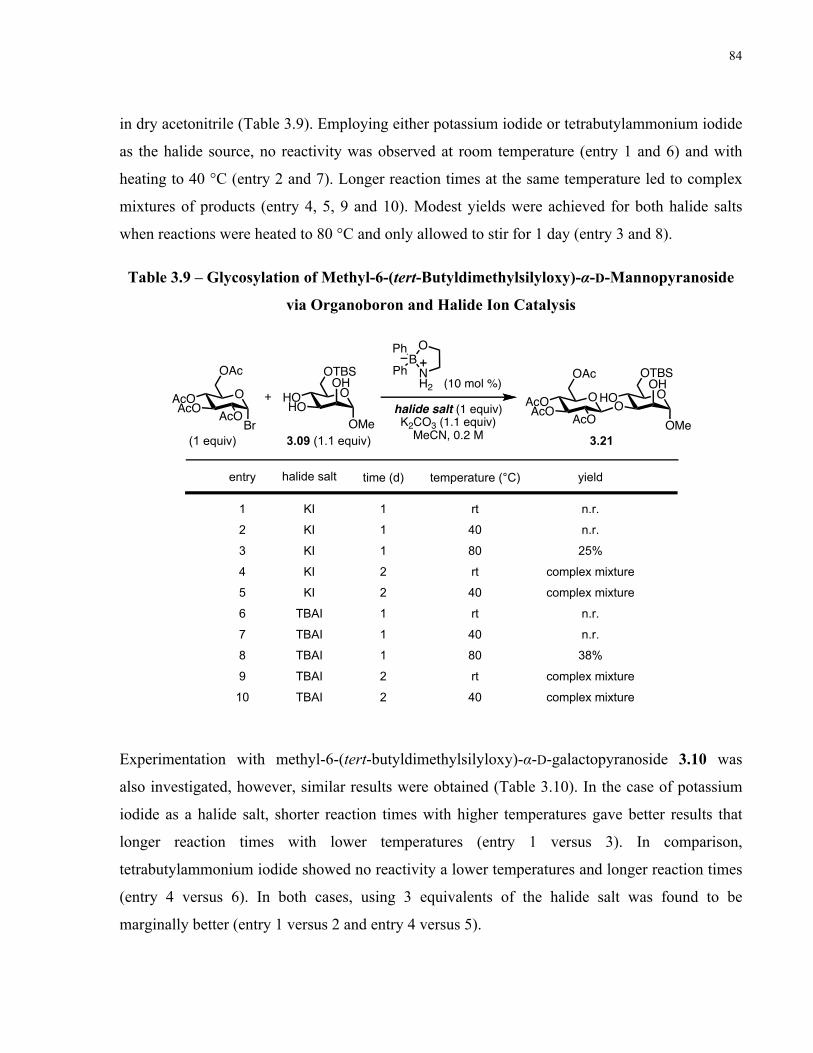
84
in dry acetonitrile (Table 3.9). Employing either potassium iodide or tetrabutylammonium iodide
as the halide source, no reactivity was observed at room temperature (entry 1 and 6) and with
heating to 40 °C (entry 2 and 7). Longer reaction times at the same temperature led to complex
mixtures of products (entry 4, 5, 9 and 10). Modest yields were achieved for both halide salts
when reactions were heated to 80 °C and only allowed to stir for 1 day (entry 3 and 8).
Table 3.9 – Glycosylation of Methyl-6-(tert-Butyldimethylsilyloxy)-α-D-Mannopyranoside
via Organoboron and Halide Ion Catalysis
Experimentation with methyl-6-(tert-butyldimethylsilyloxy)-α-D-galactopyranoside 3.10 was
also investigated, however, similar results were obtained (Table 3.10). In the case of potassium
iodide as a halide salt, shorter reaction times with higher temperatures gave better results that
longer reaction times with lower temperatures (entry 1 versus 3). In comparison,
tetrabutylammonium iodide showed no reactivity a lower temperatures and longer reaction times
(entry 4 versus 6). In both cases, using 3 equivalents of the halide salt was found to be
marginally better (entry 1 versus 2 and entry 4 versus 5).
halide salt (1 equiv)K2CO3 (1.1 equiv)
MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %)O
OTBS
HOHO
OH
OMe
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
3.09 (1.1 equiv)(1 equiv)
rt
40
80
rt
40
rt
40
80
rt
40
n.r.
n.r.
25%
complex mixture
complex mixture
n.r.
n.r.
38%
complex mixture
complex mixture
entry yieldtemperature (°C)
1
2
3
4
5
6
7
8
9
10
1
1
1
2
2
1
1
1
2
2
time (d)
KI
KI
KI
KI
KI
TBAI
TBAI
TBAI
TBAI
TBAI
halide salt
3.21
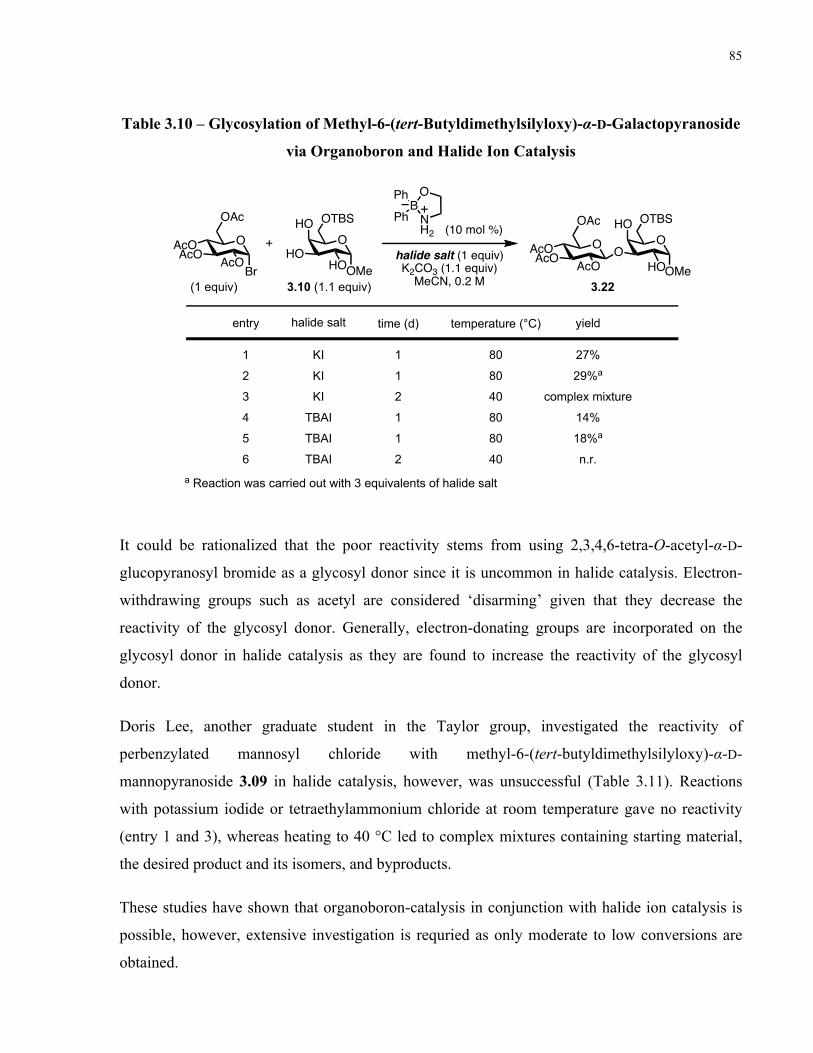
85
Table 3.10 – Glycosylation of Methyl-6-(tert-Butyldimethylsilyloxy)-α-D-Galactopyranoside
via Organoboron and Halide Ion Catalysis
It could be rationalized that the poor reactivity stems from using 2,3,4,6-tetra-O-acetyl-α-D-
glucopyranosyl bromide as a glycosyl donor since it is uncommon in halide catalysis. Electron-
withdrawing groups such as acetyl are considered ‘disarming’ given that they decrease the
reactivity of the glycosyl donor. Generally, electron-donating groups are incorporated on the
glycosyl donor in halide catalysis as they are found to increase the reactivity of the glycosyl
donor.
Doris Lee, another graduate student in the Taylor group, investigated the reactivity of
perbenzylated mannosyl chloride with methyl-6-(tert-butyldimethylsilyloxy)-α-D-
mannopyranoside 3.09 in halide catalysis, however, was unsuccessful (Table 3.11). Reactions
with potassium iodide or tetraethylammonium chloride at room temperature gave no reactivity
(entry 1 and 3), whereas heating to 40 °C led to complex mixtures containing starting material,
the desired product and its isomers, and byproducts.
These studies have shown that organoboron-catalysis in conjunction with halide ion catalysis is
possible, however, extensive investigation is requried as only moderate to low conversions are
obtained.
OOTBSHO
HOOMe
O
OAc
AcOAcO AcO
O
a Reaction was carried out with 3 equivalents of halide salt
halide salt (1 equiv)K2CO3 (1.1 equiv)
MeCN, 0.2 M
PhB
Ph NH2
O
(10 mol %)O
OAc
AcOAcO
BrAcO
+
3.10 (1.1 equiv)(1 equiv)
80
80
40
80
80
40
27%
29%a
complex mixture
14%
18%a
n.r.
entry yieldtemperature (°C)
1
2
3
4
5
6
1
1
2
1
1
2
time (d)
KI
KI
KI
TBAI
TBAI
TBAI
halide salt
3.22
OOTBSHO
HOHOOMe

86
Table 3.11 – Glycosylation Using 2,3,4,6-Tetra-O-Benzyl-α-D-Mannopyranosyl Chloride
3.3.5 Regioselective Glycosylations with Stoichiometric Boronic Acid
Previous studies by Aoyama and co-workers demonstrated the ability of boronic acids bearing 2-
hydroxy-2-propyl groups at the ortho position capable of regioselective glycosylation of
unprotected sugars. Further work in this area has not been reported which inspired us to expand
on this methodology especially since the organoboron reagent is not commercially available and
the scope is narrow in terms of glycosyl donors. Extensive investigation on the ability of
stoichiometric quantities of organoboron reagents to activate the cis-1,2-diol in rhamnose and
galactose derivatives towards regioselective glycosylation were carried out.
Starting with methyl α-L-rhamnopyranoside, several different boronic esters were synthesized by
condensation with arylboronic acids in refluxing toluene. The boronic esters were then exposed
to glycosylation conditions by treatment with 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide
in the presence of silver oxide and an activator in dry acetonitrile. Six different boronic acids
were tested with four different activators (Table 3.12). In the screen, it was found that all
reactions with quinuclidine as a promoter gave no reactivity, whereas tributylphosphine oxide
and dimethyl sulfoxide generally gave complex mixtures of product. Triethylamine was
identified to be the optimal promoter as it afforded only the desired disaccharide product. In
terms of the boronic acid, the more sterically demanding 8-quinolinyl-boronic acid delivered the
product in 82% yield (entry 21). p-Methoxyboronic acid, biphenyl-2-boronic acid and
conditions
PhB
Ph NH2
O
(10 mol %)O
OTBS
HOHO
OH
OMe
OOTBS
HOOH
OMe
O
OBn
BnOBnO
Cl
OBn+ O
OBn
BnOBnO
OBnO
3.09 (1.1 equiv)(1 equiv)
rt
40
rt
40
n.r.
complex mixture
n.r.
complex mixture
entry yieldtemperature (°C)
1
2
3
4
conditions
KI (1 equiv), K2CO3 (1.1 equiv)MeCN, 0.2 M
Et4NCl (1 equiv), iPr2NEt (1 equiv)DCM, 0.33 M
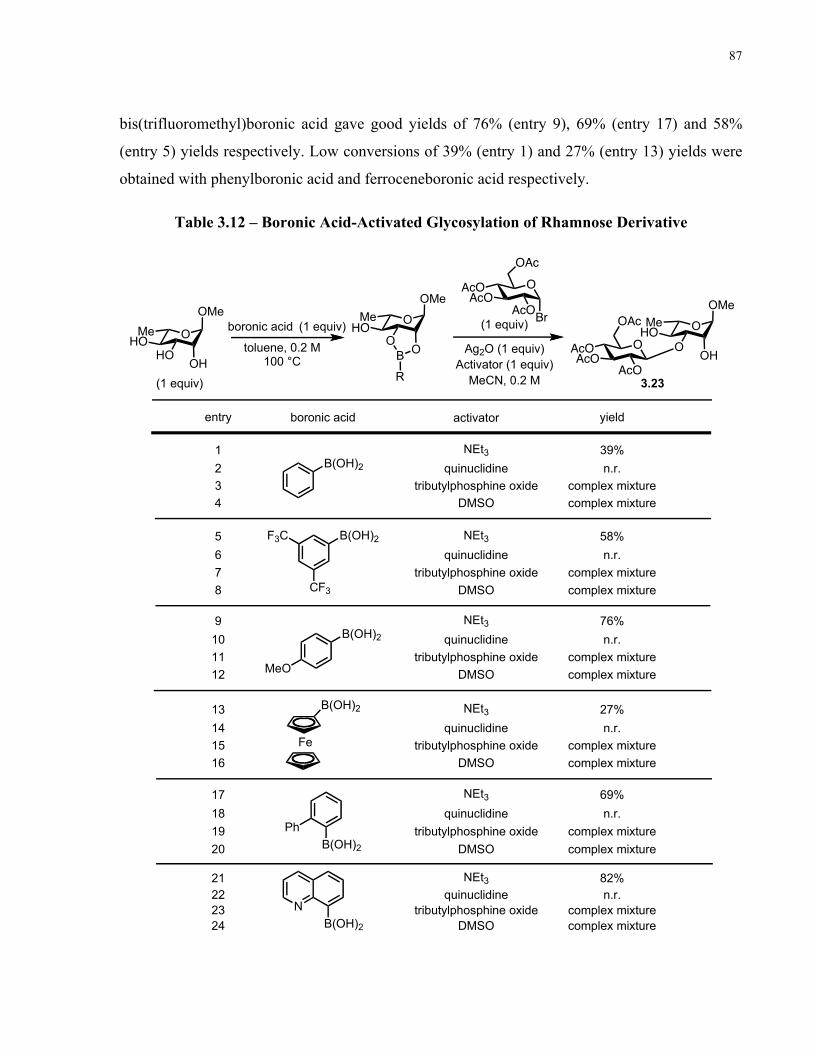
87
bis(trifluoromethyl)boronic acid gave good yields of 76% (entry 9), 69% (entry 17) and 58%
(entry 5) yields respectively. Low conversions of 39% (entry 1) and 27% (entry 13) yields were
obtained with phenylboronic acid and ferroceneboronic acid respectively.
Table 3.12 – Boronic Acid-Activated Glycosylation of Rhamnose Derivative
O
OMeMe
HOHO
OH
boronic acid (1 equiv)
toluene, 0.2 M100 °C
O
OMeMe
HOO
OB
R
OOAc
AcOAcO
BrAcO
Ag2O (1 equiv)Activator (1 equiv)
MeCN, 0.2 M
O
OAc
AcOAcO
AcO
O
OMeMeHO
OOH
(1 equiv)
(1 equiv)
3.23
NEt3quinuclidine
tributylphosphine oxideDMSO
NEt3quinuclidine
tributylphosphine oxideDMSO
NEt3quinuclidine
tributylphosphine oxideDMSO
NEt3quinuclidine
tributylphosphine oxideDMSO
NEt3quinuclidine
tributylphosphine oxideDMSO
NEt3quinuclidine
tributylphosphine oxideDMSO
complex mixture
39%n.r.
complex mixturecomplex mixture
58%n.r.
complex mixturecomplex mixture
76%n.r.
complex mixturecomplex mixture
69%n.r.
complex mixturecomplex mixture
82%n.r.
complex mixturecomplex mixture
entry yieldboronic acid activator
1234
5678
9101112
13141516
17181920
21222324
27%n.r.
complex mixture
F3C
CF3
B(OH)2
B(OH)2
MeO
B(OH)2
PhB(OH)2
NB(OH)2
Fe
B(OH)2

88
This is a promising initial result as it employs commercially available boronic acids and occurs
under operationally simple reaction conditions. With the optimized reaction conditions, future
work would require extension of this method to other unprotected carbohydrate substrates such
as thiogalactose derivatives.
3.4 Kinetic Studies: Reagent Order
Efforts in future reaction and catalyst design are best focused on accelerating the rate limiting
step of a reaction. To establish this, initial rate kinetic studies were performed to determine the
order in each component of the reaction of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide
and methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside following the methods
described in Chapter 1. Initial rates were determined for reactions where the concentration in
glycosyl donor, glycosyl acceptor, borinic acid-catalyst, and silver oxide were modified
independently.
3.4.1 Pseudo First-order Kinetics in Glycosyl Donor
Under pseudo first-order conditions, the conversion of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl
bromide to product was monitored over a period of 10 hours (Scheme 3.23). A plot of the
concentration of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide over time was found to fit
the exponential function:
[glycosyl donor](t) = [glycosyl donor]0e–kt
indicating that the reaction follows pseudo-first-order kinetics under the reaction conditions
(Figure 3.1).
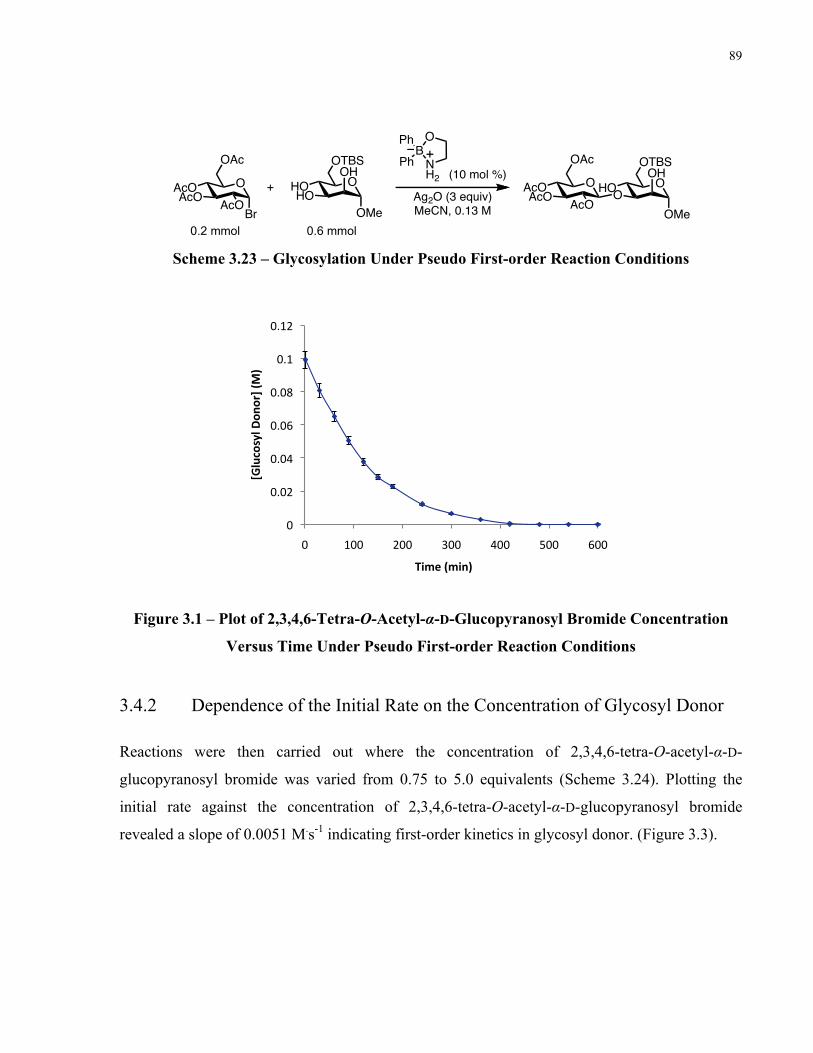
89
Scheme 3.23 – Glycosylation Under Pseudo First-order Reaction Conditions
Figure 3.1 – Plot of 2,3,4,6-Tetra-O-Acetyl-α-D-Glucopyranosyl Bromide Concentration
Versus Time Under Pseudo First-order Reaction Conditions
3.4.2 Dependence of the Initial Rate on the Concentration of Glycosyl Donor
Reactions were then carried out where the concentration of 2,3,4,6-tetra-O-acetyl-α-D-
glucopyranosyl bromide was varied from 0.75 to 5.0 equivalents (Scheme 3.24). Plotting the
initial rate against the concentration of 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide
revealed a slope of 0.0051 M.s-1 indicating first-order kinetics in glycosyl donor. (Figure 3.3).
OOTBS
HOHO
OH
OMeAg2O (3 equiv)MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.6 mmol0.2 mmol
PhB
Ph NH2
O
(10 mol %)
!"
!#!$"
!#!%"
!#!&"
!#!'"
!#("
!#($"
!" (!!" $!!" )!!" %!!" *!!" &!!"
!"#$%&'(#)*
&+&,-)./0)
1234).32+0)
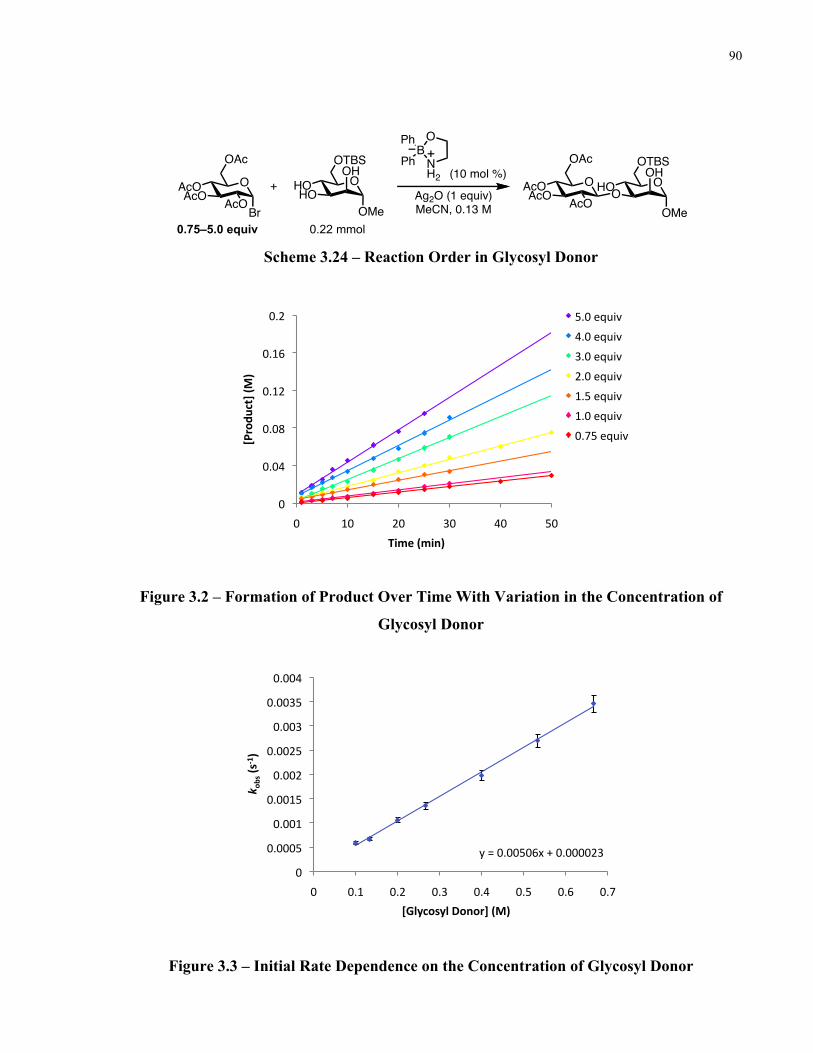
90
Scheme 3.24 – Reaction Order in Glycosyl Donor
Figure 3.2 – Formation of Product Over Time With Variation in the Concentration of
Glycosyl Donor
Figure 3.3 – Initial Rate Dependence on the Concentration of Glycosyl Donor
OOTBS
HOHO
OH
OMeAg2O (1 equiv)MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.22 mmol0.75–5.0 equiv
PhB
Ph NH2
O
(10 mol %)
!"
!#!$"
!#!%"
!#&'"
!#&("
!#'"
!" &!" '!" )!" $!" *!"
!"#$%&
'()*+,-*
./01*+0/2-*
*#!"+,-./"
$#!"+,-./"
)#!"+,-./"
'#!"+,-./"
&#*"+,-./"
&#!"+,-./"
!#0*"+,-./"
!"#"$%$$&$'(")"$%$$$$*+"
$"
$%$$$&"
$%$$,"
$%$$,&"
$%$$*"
$%$$*&"
$%$$+"
$%$$+&"
$%$$-"
$" $%," $%*" $%+" $%-" $%&" $%'" $%."
! !"#$%#
&'($
)*+,-!#,+$.!/!01$%2($$
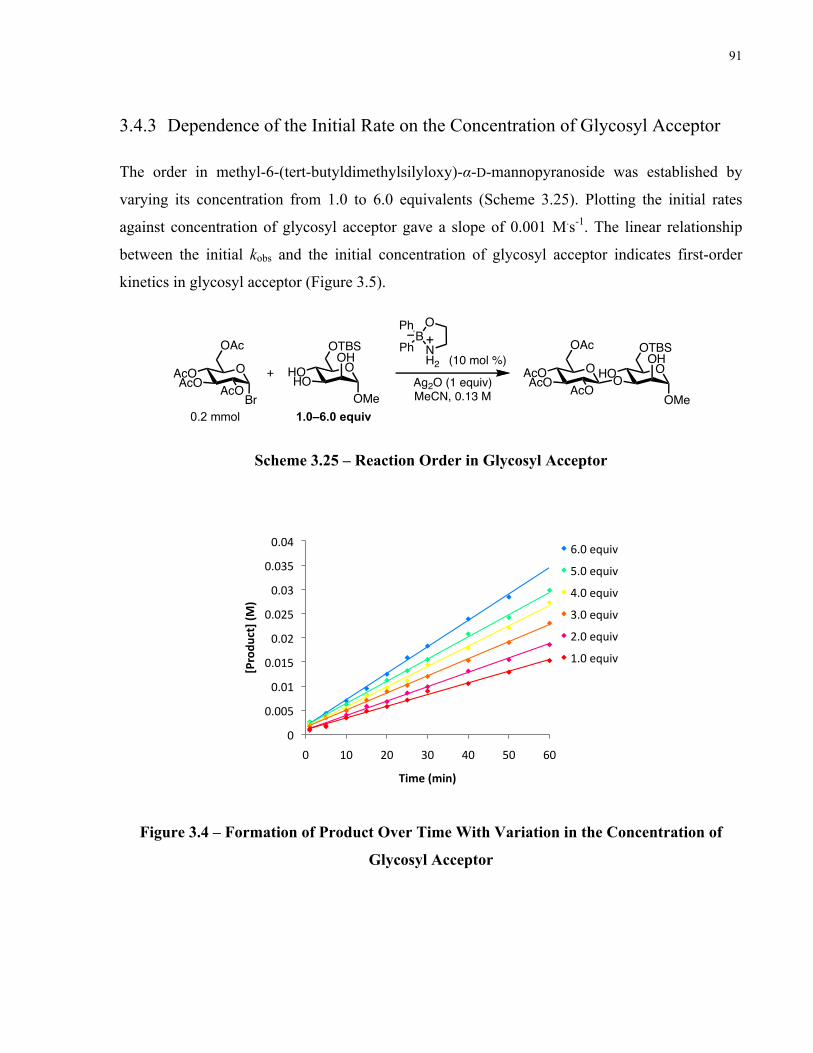
91
3.4.3 Dependence of the Initial Rate on the Concentration of Glycosyl Acceptor
The order in methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside was established by
varying its concentration from 1.0 to 6.0 equivalents (Scheme 3.25). Plotting the initial rates
against concentration of glycosyl acceptor gave a slope of 0.001 M.s-1. The linear relationship
between the initial kobs and the initial concentration of glycosyl acceptor indicates first-order
kinetics in glycosyl acceptor (Figure 3.5).
Scheme 3.25 – Reaction Order in Glycosyl Acceptor
Figure 3.4 – Formation of Product Over Time With Variation in the Concentration of
Glycosyl Acceptor
OOTBS
HOHO
OH
OMeAg2O (1 equiv)MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
1.0–6.0 equiv0.2 mmol
PhB
Ph NH2
O
(10 mol %)
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!#!&$"
!#!'"
!#!'$"
!#!("
!" %!" &!" '!" (!" $!" )!"
!"#$%&
'()*+,-*
./01*+0/2-*
)#!"*+,-."
$#!"*+,-."
(#!"*+,-."
'#!"*+,-."
&#!"*+,-."
%#!"*+,-."
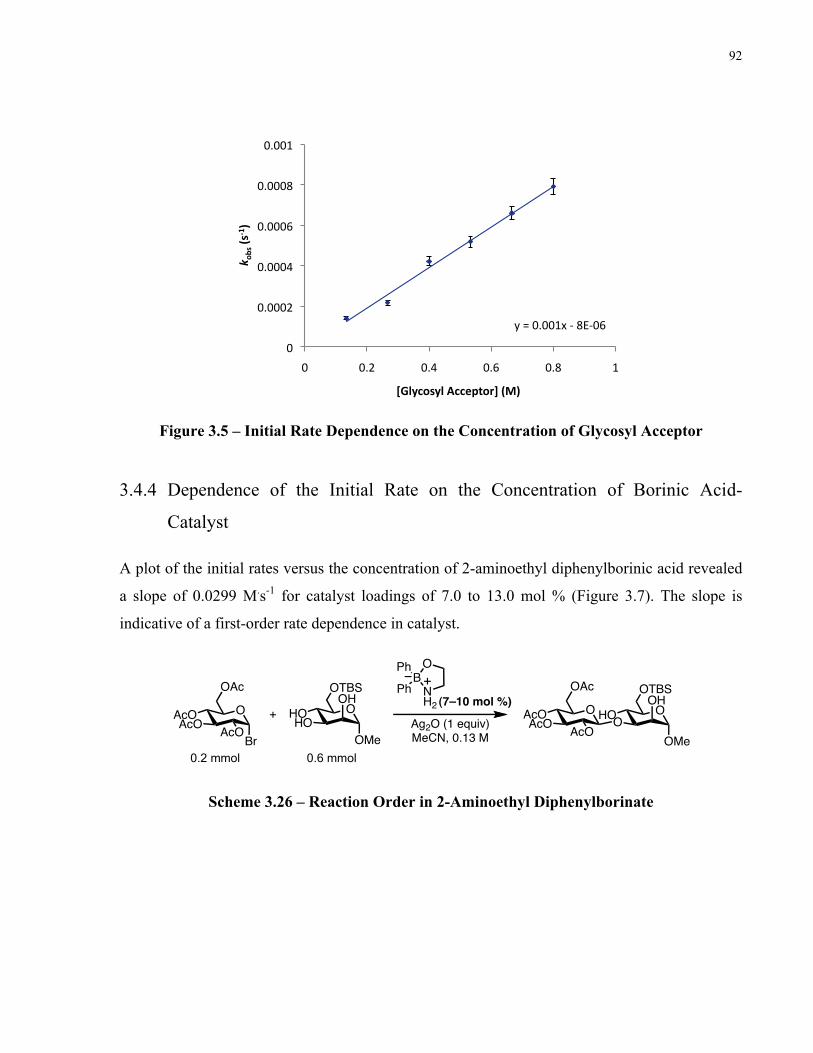
92
Figure 3.5 – Initial Rate Dependence on the Concentration of Glycosyl Acceptor
3.4.4 Dependence of the Initial Rate on the Concentration of Borinic Acid-
Catalyst
A plot of the initial rates versus the concentration of 2-aminoethyl diphenylborinic acid revealed
a slope of 0.0299 M.s-1 for catalyst loadings of 7.0 to 13.0 mol % (Figure 3.7). The slope is
indicative of a first-order rate dependence in catalyst.
Scheme 3.26 – Reaction Order in 2-Aminoethyl Diphenylborinate
!"#"$%$$&'"(")*($+"
$"
$%$$$,"
$%$$$-"
$%$$$+"
$%$$$)"
$%$$&"
$" $%," $%-" $%+" $%)" &"
! !"#$%#
&'($
)*+,-!#,+$.--/01!23$%4($
OOTBS
HOHO
OH
OMeAg2O (1 equiv)MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.6 mmol0.2 mmol
PhB
Ph NH2
O
(7–10 mol %)

93
Figure 3.6 – Formation of Product Over Time With Variation in the Concentration of 2-
Aminoethyl Diphenylborinate
Figure 3.7 – Initial Rate Dependence on the Concentration of 2-Aminoethyl
Diphenylborinate
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!#!&$"
!#!'"
!#!'$"
!" %!" &!" '!" (!" $!" )!"
!"#$%&
'()*+,-**
./01*+0/2-*
%'#!"*+,"-"
%&#!"*+,"-"
%%#!"*+,"-"
%!#!"*+,"-"
.#!"*+,"-"
/#!"*+,"-"
0#!"*+,"-"
!"#"$%$&''(")"&*+$,"
$"
$%$$$-"
$%$$$&"
$%$$$."
$%$$$/"
$%$$$,"
$%$$$0"
$%$$$1"
$%$$0" $%$$2" $%$-" $%$-&" $%$-/" $%$-0" $%$-2" $%$&"
! !"#$%#
&'($
)*&+,-.!/0123$4-51/.23"!6-.70/8$%9($
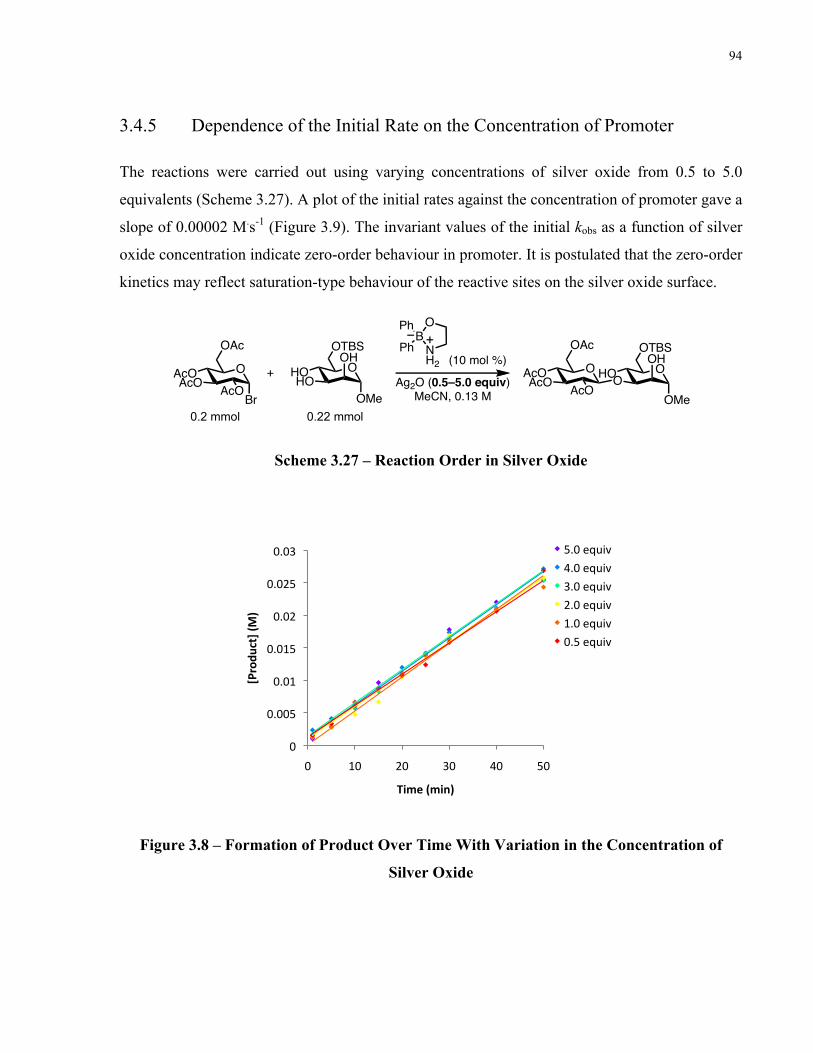
94
3.4.5 Dependence of the Initial Rate on the Concentration of Promoter
The reactions were carried out using varying concentrations of silver oxide from 0.5 to 5.0
equivalents (Scheme 3.27). A plot of the initial rates against the concentration of promoter gave a
slope of 0.00002 M.s-1 (Figure 3.9). The invariant values of the initial kobs as a function of silver
oxide concentration indicate zero-order behaviour in promoter. It is postulated that the zero-order
kinetics may reflect saturation-type behaviour of the reactive sites on the silver oxide surface.
Scheme 3.27 – Reaction Order in Silver Oxide
Figure 3.8 – Formation of Product Over Time With Variation in the Concentration of
Silver Oxide
OOTBS
HOHO
OH
OMeAg2O (0.5–5.0 equiv)
MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.22 mmol0.2 mmol
PhB
Ph NH2
O
(10 mol %)
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!#!&$"
!#!'"
!" %!" &!" '!" (!" $!"
!"#$%&
'()*+,-*
./01*+0/2-*
$#!")*+,-"
(#!")*+,-"
'#!")*+,-"
&#!")*+,-"
%#!")*+,-"
!#$")*+,-"
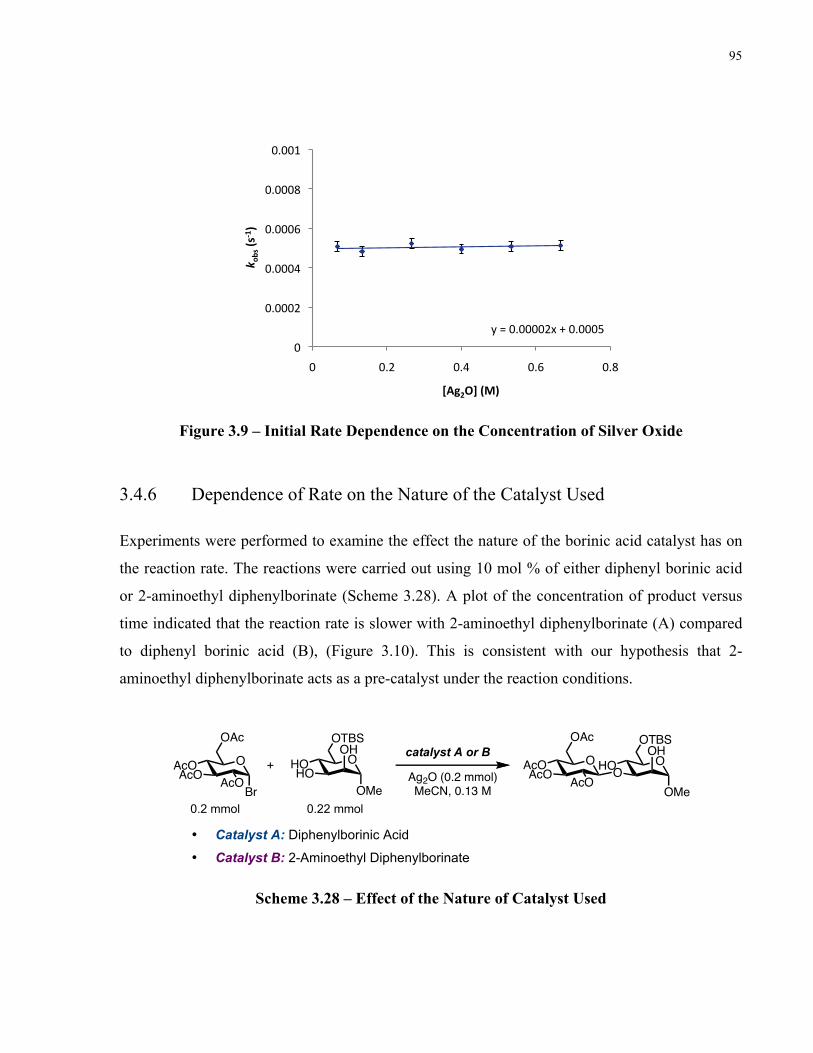
95
Figure 3.9 – Initial Rate Dependence on the Concentration of Silver Oxide
3.4.6 Dependence of Rate on the Nature of the Catalyst Used
Experiments were performed to examine the effect the nature of the borinic acid catalyst has on
the reaction rate. The reactions were carried out using 10 mol % of either diphenyl borinic acid
or 2-aminoethyl diphenylborinate (Scheme 3.28). A plot of the concentration of product versus
time indicated that the reaction rate is slower with 2-aminoethyl diphenylborinate (A) compared
to diphenyl borinic acid (B), (Figure 3.10). This is consistent with our hypothesis that 2-
aminoethyl diphenylborinate acts as a pre-catalyst under the reaction conditions.
• Catalyst A: Diphenylborinic Acid
• Catalyst B: 2-Aminoethyl Diphenylborinate
Scheme 3.28 – Effect of the Nature of Catalyst Used
!"#"$%$$$$&'"("$%$$$)"
$"
$%$$$&"
$%$$$*"
$%$$$+"
$%$$$,"
$%$$-"
$" $%&" $%*" $%+" $%,"
! !"#$%#
&'($
)*+,-.$%/($
OOTBS
HOHO
OH
OMeAg2O (0.2 mmol)MeCN, 0.13 M
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.22 mmol0.2 mmol
catalyst A or B

96
Figure 3.10 – Formation of Product Over Time Using Diphenylborinic Acid (A) or 2-
Aminoethyl Diphenylborinate (B) as the Catalyst
3.4.7 Effect of 2-Aminoethanol
Further experiments were performed to examine the effect of excess 2-aminoethanol on the rate.
The reactions were carried out in the absence (Condition A) or presence (Condition B) of 1 mol
% 2-aminoethanol (Scheme 3.29). Comparing the rate of formation of product over time
indicated that excess 2-aminoethanol did not impede the reaction (Figure 3.11). This suggests
that although 2-aminoethanol can bind back onto the borinic acid, it is displaced at a rate faster
than the limiting step.
• Condition A: No Added 2-Aminoethanol
• Condition B: 1 mol % 2-Aminoethanol
Scheme 3.29 – Effect of 2-Aminoethanol
!"#"$%$$$&'"("$%$$))"
!"#"$%$$$)'"("$%$$*+"
$"
$%$*"
$%$,"
$%$+"
$%$-"
$%$)"
$%$."
$%$/"
$" *$" ,$" +$" -$" )$" .$"
!"#$%&
'()*+,-*
./01*+0/2-*
01213!42"5"
01213!42"6"
OOTBS
HOHO
OH
OMeAg2O (0.2 mmol)MeCN, 0.13 M
Condition A or B
OOTBS
HOOH
OMe
O
OAc
AcOAcO
BrAcO
+ O
OAc
AcOAcO AcO
O
0.22 mmol0.2 mmol
PhB
Ph NH2
O
(10 mol %)
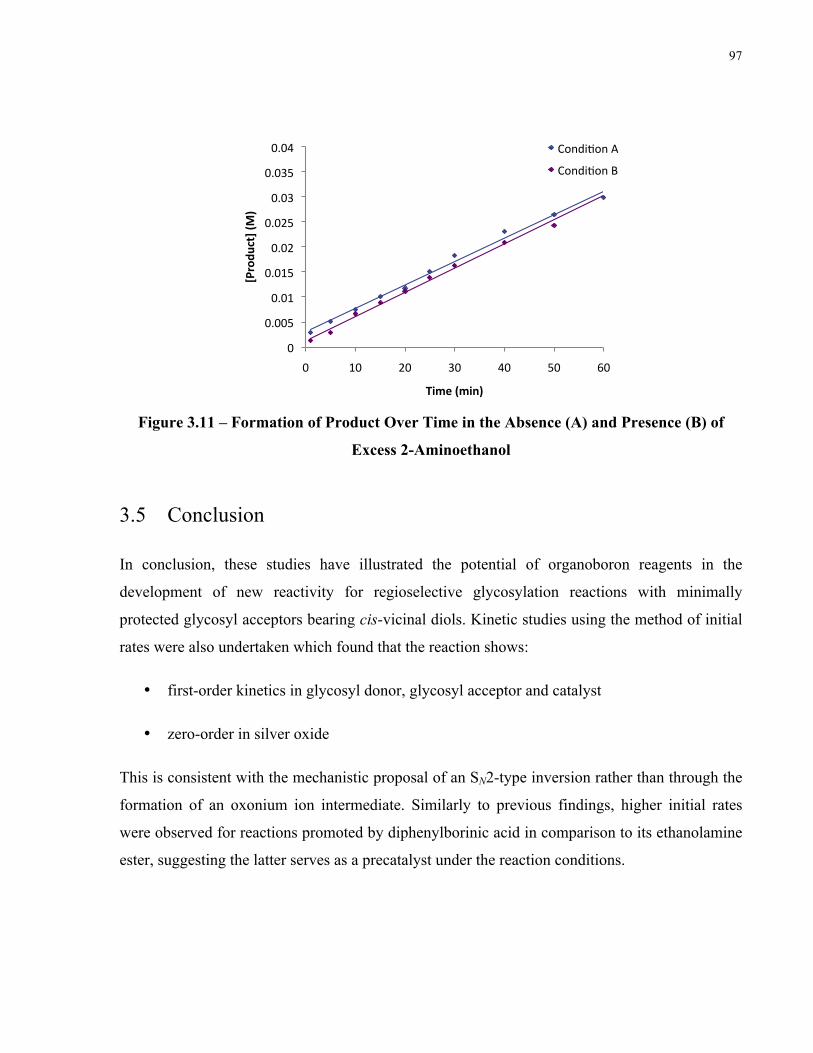
97
Figure 3.11 – Formation of Product Over Time in the Absence (A) and Presence (B) of
Excess 2-Aminoethanol
3.5 Conclusion
In conclusion, these studies have illustrated the potential of organoboron reagents in the
development of new reactivity for regioselective glycosylation reactions with minimally
protected glycosyl acceptors bearing cis-vicinal diols. Kinetic studies using the method of initial
rates were also undertaken which found that the reaction shows:
• first-order kinetics in glycosyl donor, glycosyl acceptor and catalyst
• zero-order in silver oxide
This is consistent with the mechanistic proposal of an SN2-type inversion rather than through the
formation of an oxonium ion intermediate. Similarly to previous findings, higher initial rates
were observed for reactions promoted by diphenylborinic acid in comparison to its ethanolamine
ester, suggesting the latter serves as a precatalyst under the reaction conditions.
!"
!#!!$"
!#!%"
!#!%$"
!#!&"
!#!&$"
!#!'"
!#!'$"
!#!("
!" %!" &!" '!" (!" $!" )!"
!"#$%&
'()*+,-*
./01*+0/2-*
*+,-./+,"0"
*+,-./+,"1"

98
4
Final Conclusion
The ability to selectively functionalize a hydroxy group on carbohydrate derivatives is
challenging due to their complex structure, which require extensive protecting group
manipulation. With the inspiration of using boron–diol interactions that are widely known in the
chemical sensing literature, our group has demonstrated the catalytic ability of organoboron
reagents to carry out acylation and sulfonylation reactions of carbohydrate derivatives
regioselectively. This led us to further explore and expand on the capability of the organoboron
catalyst.
In the first chapter, the mechanism for the sulfonylation of cis-1,2-cyclohexanediol was studied
by carrying out kinetic studies by the method of initial rates. The proposed mechanism follows
that the 2-aminoethyl diphenylborinate is converted to the active catalyst by functionalization of
the ethanolamine ligand. The diol substrate then binds to the catalyst forming a cyclic ‘ate’
complex which activates one of the two B–O bonds towards nucleophilic attack. Displacement of
the product is then achieved by attack of another diol substrate.

99
The various experiments performed revealed that the reaction follows first-order kinetics in
substrate, electrophile and catalyst, and zero-order kinetics in base. This reveals that attack of the
electrophile by the bound cyclic ‘ate’ complex is possibly part of the rate-determining step. As
well, the zero-order in base is indicative that the base does not have a significant part in the rate-
determining step. The observation that the 2-aminoethyl diphenylborinate catalyst shows a
slower reaction rate in comparison to the diphenylborinic acid suggests that the former is a
precatalyst under the reaction conditions. Slower rates were also observed upon the addition of 2-
aminoethanol revealing that the dissociation of the ligand is a reversible process. All these were
found to be consistent with our proposed catalytic cycle.
In the second chapter, the development of a regioselective method for the monoalkylation of
carbohydrate derivatives was explored. With having recently developed a method for
regioselective acylation of cis-diols by boron-catalysis, we sought to determine whether a similar
mode of reactivity could be employed. With extensive optimization of all reaction parameters
such as catalyst, base, solvent, concentration, stoichiometry and temperature, a set of conditions
were discovered which gave high-yielding results. This method was carried out on ten
carbohydrate substrates employing four alkylating agents to generate a scope table with thirty-
four entries. Thereafter, further studies revealed that the alkylation by diphenylborinic acid-
catalysis could also be achieved in combination with halide catalysis. Current limitations of this
protocol include the use of thiogalactosides and carbohydrate derivatives not bearing the cis-1,2-
diol moiety.
In the third chapter, we explore the ability of the diphenylborinic acid to catalyze regioselective
glycosylation reactions. Previously, extensive investigations carried out by other members in our
group have discovered suitable reaction conditions that were compatible for glycosylation
reactions using several derivatives of mannose, galactose, fucose and arabinose to access 1,2-
trans linkages from donors of glucose or galactose configuration. Building on these results,
further investigations were carried out to expand the scope of this method which included
glycosylations with glucosamine derived donors, the formation of β-mannoside linkages,
reactivity in halide ion catalysis, and boronic acid-mediated glycosylations.

100
In the study of glucosamine derived donors, several substrates were synthesized and tested,
however, all attempts to optimize the reaction conditions were found to only give modest yields.
In examining the ability to form β-mannoside linkages, a rhamnose derivative containing a cyclic
carbonate at the 2-O and 3-O positions was synthesized and investigated. Disappointingly, it was
found that either no reactivity or complex mixtures were obtained under tested reaction
conditions. Glycosylation using halide catalysis was also found to be futile as optimization of the
reaction conditions using a variety of iodide salts only gave low conversions. When exploring the
use of stoichiometric amounts of organoboron catalyst to carry out glycosylations, it was found
that 8-quinolinyl-boronic acid with triethylamine as an activator promoted the coupling of a
rhamnose acceptor and glucose derived donor with high yields.
Kinetic studies on the glycosylation reaction were then carried out which revealed the reaction is
first-order in glycosyl donor, glycosyl acceptor and catalyst, and zero-order in silver oxide. It
was also found that the reaction carried out using diphenylborinic acid was faster in comparison
to 2-aminoethyl diphenylborinate which is consistent with the proposal that the latter is a
precatalyst in the reaction. In contrast to previous findings, the presence of excess 2-
aminoethanol does not inhibit the reaction.

101
5
Experimental Procedures
5.0 General Information
General: Reactions were carried out without effort to exclude air or moisture, unless otherwise
indicated. Stainless steel syringes were used to transfer air- and moisture-sensitive liquids. Flash
chromatography was carried out using silica gel (Silicycle).
Materials: HPLC grade acetonitrile was dried and purified using a solvent purification system
equipped with columns of activated alumina, under argon (Innovative Technology, Inc.).
Deionized water was obtained from an in-house supply. All other reagents and solvents were
purchased from Sigma-Aldrich, Caledon, Carbosynth or Alfa Aesar, and used without further
purification. Diphenylborinic acid was prepared from 2-aminoethyl diphenylborinate according
to a reported procedure.Error! Bookmark not defined.
Instrumentation: 1H and 13C NMR spectra were recorded in CDCl3 using a Bruker Avance III
400 MHz or Varian Mercury 400 MHz spectrometer, referenced to residual protium in the

102
solvent. Spectral features are tabulated in the following order: chemical shift (δ, ppm);
multiplicity (s-singlet, d-doublet, t-triplet, q-quartet, m-complex multiplet); coupling constants
(J, Hz); number of protons; assignment. Assignments are based on analysis of coupling constants
and COSY spectra. In cases of uncertain assignments, structural confirmation was secured
through NOESY experiments. High-resolution mass spectra (HRMS) were obtained on a VS 70-
250S (double focusing) mass spectrometer at 70 eV. Infrared (IR) spectra were obtained on a
Perkin-Elmer Spectrum 100 instrument equipped with a single-reflection diamond / ZnSe ATR
accessory, either in the solid state or as neat liquids, as indicated. Spectral features are tabulated
as follows: wavenumber (cm–1); intensity (s-strong, m-medium, w-weak, br-broad).
5.1 Experimental and Characterization Data
5.1.1 General Procedure A: Tosylation Kinetic Experiments
Into an oven-dried 1 dram vial were added cis-1,2-cyclohexanediol, 4-toluenesulfonyl chloride,
and 2-aminoethyl diphenylborinate. The reaction vial was capped with a septum and purged with
argon. Dry acetonitrile (0.1 M) was added followed by N,N-diisopropylethylamine and the
resulting mixture was stirred vigorously (750 rpm) at room temperature. During the course of the
reaction, aliquots of the reaction mixture were removed and quenched with methanol. The
solvent was then removed and the resulting samples were analyzed by 1H-NMR spectroscopy for
the formation of product with mesitylene as an internal standard. Integrations for the internal
standard peak and an isolated proton peak of the product were used to calculate moles of product
formed and therefore % conversion.
OH
OH
OTs
OHTsCl, iPr2NEtMeCN
PhB
Ph NH2
O

103
5.1.2 General Procedure B: Hydrolysis of Diarylboronic Esters
In a 2-dram vial equipped with a stir bar was added diethylamino diphenylborinate ester (0.8
mmol, 180 mg), acetone (0.4 mL) and methanol (0.4 mL). To the resulting solution was added
HCl(aq) (1 M, 1 mL). The vial was capped in ambient atmosphere and stirred at 500 rpm at room
temperature. After 1 hour, the mixture was diluted with diethyl ether, washed with water, and
extracted several times with diethyl ether. The combined organic extracts were dried over
MgSO4, filtered, and concentrated in vacuo to afford 128 mg of 2a as a colourless oil (88%
yield). Spectral data were in agreement with previous reports.101
5.1.3 General Procedure C: Borinic Acid-Catalyzed Alkylation
The carbohydrate substrate (1 equiv), 2-aminoethyl diphenylborinate (10 mol %) and Ag2O (1.1
equiv) were weighed into a 1-dram vial, and dissolved in dry acetonitrile (0.1–0.2 M, see below).
The alkyl halide (1.5 equiv) was then added, and the reaction vessel was capped with a septum
and purged with argon. The mixture was stirred vigorously (750-1000 rpm) for 48 hr at 40 °C.
The resulting mixture was diluted with CH2Cl2, filtered through Celite® and concentrated to
dryness. The resulting crude material was purified by flash chromatography on silica gel using
the stated eluent system.
5.1.4 General Procedure D: Borinic Acid-Catalyzed Alkylation with Halide Salts
The carbohydrate substrate (1 equiv), 2-aminoethyl diphenylborinate (10 mol %), halide salt (1.0
equiv), potassium carbonate (1.1 equiv) were weighed into a 1-dram vial, and dissolved in dry
acetonitrile (0.1–0.2 M, see below). The alkyl halide (1.5 equiv) was then added, and the reaction
vessel was capped with a septum and purged with argon. The mixture was stirred vigorously
(750-1000 rpm) for 48 hr at 40 °C. The resulting mixture was diluted with MeOH, filtered
101 White, D. A. J. Inorg. Nucl. Chem. 1971, 33, 691–696.
PhB
Ph NH2
O HCl(aq), 1M
MeOH:Acetone (1:1)
PhB
PhOH

104
through Celite® and concentrated to dryness. The resulting crude material was purified by flash
chromatography on silica gel using the stated eluent system.
Carbohydrate substrates 2.01102, 2.03103, 2.05102, 2.06102 and 2.54102 were prepared following
literature methods. Spectral data were consistent with those presented in the literature.102,103
Substrates 2.02, 2.04 and 2.07–2.10 were purchased (Carbosynth, Alfa Aesar) and used as
received.
Benzyloxymethyl chloride was purchased at ~60% purity (technical grade) from Sigma-Aldrich
and was purified by flash chromatography prior to use.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyl-α-D-mannopyranoside (2.11)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol),
benzyl bromide (35.6 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and
Ag2O (50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere
at 40 °C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:25) afforded the title
compound as a colourless oil (72.4 mg, 91%); Rf = 0.32 (pentanes/EtOAc, 70:30); FTIR (νmax)
3482 (br), 2927 (m), 1462 (w), 1361 (w), 1251 (m), 1106 (s), 1052 (s), 967 (m), 835 (s), 777 (m)
cm-1; 1H NMR (400 MHz, CDCl3): δ 7.40–7.29 (m, 5H, ArH), 4.73 (d, J = 1.6 Hz, 1H, H-1),
4.71 (s, 2H, ArCH2), 3.97 (dd, J = 3.2, 1.6 Hz, 1H, H-2), 3.91–3.84 (m, 3H, H-4, H-6a and H-
6b), 3.68 (dd, J = 9.2, 3.6 Hz, 1H, H-3), 3.60 (apparent dt, J = 9.2, 4.8 Hz, 1H, H-5), 3.36 (s, 3H,
OCH3), 2.97 (d, J = 1.6 Hz, 1H, C4-OH), 2.40 (d, J = 2.4 Hz, 1H, C2-OH), 0.91 (s, 9H,
Si(C(CH3)3)(CH3)2), 0.10 (s, 6H, Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 138.0,
128.6, 128.0, 127.9, 100.4, 79.5, 72.2, 70.9, 69.2, 67.9, 64.8, 54.9, 25.9, 18.3, –5.4; HRMS m/z
calcd for C20H35O6Si [M+H]+: 399.2203. Found 399.2215.
102 Lee, D.; Taylor, M. S. J. Am. Chem. Soc. 2011, 133, 3724–3727. 103 Namme, R.; Mitsugi, T.; Takahashi, H.; Shiro, M.; Ikegami, S. Tetrahedron. 2006, 62, 9183–9192.
O
OMe
OTBSOH
BnOHO

105
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(4-bromobenzyl)-α-D-mannopyranoside (2.12)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol), 4-
bromobenzyl bromide (75.0 mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol
%) and Ag2O (50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon
atmosphere at 40 °C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:15)
afforded the title compound as a colourless oil (95.2 mg, >99%); Rf = 0.49 (pentanes/EtOAc,
70:30); FTIR (νmax) 3509 (br), 2927 (m), 1593 (w), 1488 (m), 1389 (m), 1251 (m), 1108 (s), 968
(s), 836 (s), 741 (s) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.46 (apparent dt, J = 9.2, 2.8 Hz, 2H,
ArH), 7.25 (apparent dt, J = 9.2, 2.8 Hz, 2H, ArH), 4.72 (d, J = 1.6 Hz, 1H, H-1), 4.66 (s, 2H,
ArCH2), 3.95–3.93 (m, 1H, H-2), 3.92–3.82 (m, 3H, H-4, H-6a and H-6b), 3.65 (dd, J = 8.8, 3.6
Hz, 1H, H-3), 3.58 (apparent dt, J = 9.6, 5.6 Hz, 1H, H-5), 3.35 (s, 3H, OCH3), 3.12 (d, J = 1.6
Hz, 1H, C4-OH), 2.41 (d, J = 2.8 Hz, 1H, C2-OH), 0.90 (s, 9H, Si(C(CH3)3)(CH3)2), 0.09 (s, 6H,
Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 137.0, 131.6, 129.5, 121.8, 100.3, 79.2,
71.3, 70.5, 69.6, 67.9, 64.9, 54.9, 25.9, 18.2, –5.4; HRMS m/z calcd for C20H34BrO6Si
[M+H]+: 477.1308. Found 477.1299.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(2-methylnaphthalenyl)-α-D-mannopyranoside
(2.13)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol), 2-
(bromomethyl)naphthalene (66.3 mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10
mol %) and Ag2O (50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon
atmosphere at 40 °C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:15)
afforded the title compound as a colourless oil (79.9 mg, 89.1%); Rf = 0.54 (pentanes/EtOAc,
O
OMe
OTBSOH
OHOBr
O
OMe
OTBSOH
OHO

106
70:30); FTIR (νmax) 3511 (br), 2927 (m), 1462 (m), 1334 (w), 1250 (m), 1106 (s), 1051 (s), 967
(m), 868 (w), 834 (s) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.86–7.81 (m, 4H, ArH), 7.52–7.45
(m, 3H, ArH), 4.87 (s, 2H, ArCH2), 4.74 (d, J = 1.6 Hz, 1H, H-1), 4.01–3.99 (m, 1H, H-2), 3.96–
3.88 (m, 3H, H-4, H-6a and H-6b), 3.74 (dd, J = 9.2, 3.6 Hz, 1H, H-3), 3.61 (apparent dt,
J = 10.0, 5.2 Hz, 1H, H-5), 3.36 (s, 3H, OCH3), 3.04 (d, J = 1.6 Hz, 1H, C4-OH), 2.46 (d, J = 2.4
Hz, 1H, C2-OH), 0.90 (s, 9H, Si(C(CH3)3)(CH3)2), 0.09 (s, 6H, Si(C(CH3)3)(CH3)2); 13C NMR
(100 MHz, CDCl3): δ 135.5, 133.3, 133.1, 128.5, 127.9, 127.8, 126.7, 126.2, 126.0, 125.8,
100.4, 79.4, 72.2, 70.9, 69.4, 67.9, 64.8, 54.8, 25.9, 18.3, –5.4; HRMS m/z calcd for
C24H37O6Si [M+H]+: 449.2359. Found 449.2350.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyloxymethyl-α-D-mannopyranoside (2.14)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol),
benzyloxymethyl chloride (41.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10
mol %) and Ag2O (50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon
atmosphere at 40 °C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:15)
afforded the title compound as a pale yellow oil (71.9 mg, 84%); Rf = 0.40 (pentanes/EtOAc,
70:30); FTIR (νmax) 3510 (br), 2928 (m), 1388 (m), 1251 (m), 1106 (s), 1024 (s), 968 (s), 834
(s), 801 (m), 734 (s) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.38–7.28 (m, 5H, ArH), 4.96 (d, J =
6.8 Hz, 1H, OCH2O), 4.91 (d, J = 6.8 Hz, 1H, OCH2O), 4.74 (d, J =11.6 Hz, 1H, ArCH2O), 4.72
(d, J = 2.4 Hz, 1H, H-1), 4.67 (d, J =11.6 Hz, 1H, ArCH2O), 3.90–3.86 (m, 1H, H-2), 3.94–3.79
(m, 4H, H-3, H-4, H-6a and H-6b), 3.60 (apparent dt, J = 9.2, 4.8 Hz, 1H, H-5), 2.89 (d, J = 1.2
Hz, 1H, C4-OH), 3.37 (s, 3H, OCH3), 2.33 (d, J = 2.8 Hz, 1H, C2-OH), 0.90 (s, 9H,
Si(C(CH3)3)(CH3)2), 0.09 (s, 6H, Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 137.2,
128.6, 128.0, 127.9, 100.4, 95.3, 80.1, 71.2, 70.4, 69.8, 68.4, 64.4, 54.8, 25.9, 18.3, –5.4; HRMS
m/z calcd for C21H37O7Si [M+H]+: 429.2308. Found 429.2297.
O
OMe
OTBSOH
BOMOHO

107
1,6-Anhydro-2-O-benzyl-β-D-mannopyranoside (2.15)
1,6-Anhydro-β-D-mannopyranoside 2.02 (32.4 mg, 0.200 mmol), benzyl bromide (35.6 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 30:70) afforded the title compound as a
pale yellow oil (36.8 mg, 73%); Rf = 0.22 (pentanes/EtOAc, 30:70); FTIR (νmax) 3414 (br), 2901
(w), 1640 (w), 1496 (w), 1454 (w), 1395 (w), 1209 (w), 1076 (s), 1047 (s), 900 (s), 678 (s) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.31–7.22 (m, 5H, ArH), 5.33 (apparent t, J = 1.6 Hz, 1H, H-1),
4.64 (d, J = 12.0 Hz, 1H, ArCH2), 4.52 (d, J = 12.0 Hz, 1H, ArCH2), 4.42–4.40 (m, 1H, H-5),
4.22 (dd, J = 7.2, 0.8 Hz, 1H, H-6a), 3.99–3.96 (m, 1H, H-3), 3.85–3.82 (m, 1H, H-4), 3.67 (dd,
J = 7.2, 6.0 Hz, 1H, H-6b), 3.48 (dd, J = 4.8, 2.0 Hz, 1H, H-2), 3.02 (d, J = 2.4 Hz, 1H, C3-OH),
2.28 (d, J = 8.8 Hz, 1H, C4-OH); 13C NMR (100 MHz, CDCl3): δ 137.0, 128.6, 128.3, 128.0,
100.3, 76.4, 72.6, 71.4, 71.1, 69.5, 64.9; HRMS m/z calcd for C13H20NO5 [M+NH4]+:
270.1342. Found 270.1344.
1,6-Anhydro-2-O-(4-bromobenzyl)-β-D-mannopyranoside (2.16)
1,6-Anhydro-β-D-mannopyranoside 2.02 (162.1 mg, 1.000 mmol), 4-bromobenzyl bromide
(374.9 mg, 1.500 mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol %) and Ag2O (254.9
mg, 1.100 mmol) were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for
48 hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound
as a colourless oil (252.8 mg, 96 % pure by 1H NMR: 73% yield). The contaminant is the 3-O-
(4-bromobenzyl) regioisomer. Rf = 0.25 (pentanes/EtOAc, 30:70); FTIR (νmax) 3479 (br), 2902
(m), 1488 (m), 1340 (m), 1101 (s), 1068 (s), 1049 (s), 969 (m), 817 (s), 681 (m) cm-1; 1H NMR
OOOH
OH
OBn
OOOH
OH
OBr

108
(400 MHz, CDCl3): δ 7.52 (apparent dt, J = 9.2, 2.4 Hz, 2H, ArH), 7.26 (apparent dt, J = 9.2, 2.4
Hz, 2H, ArH), 5.44 (apparent t, J = 1.6 Hz, 1H, H-1), 4.68 (d, J = 12.0 Hz, 1H, ArCH2), 4.56 (d,
J = 12.0 Hz, 1H, ArCH2), 4.54–4.50 (m, 1H, H-5), 4.31 (dd, J = 7.6, 1.2 Hz, 1H, H-6a), 4.09–
4.06 (m, 1H, H-3), 3.94–3.92 (m, 1H, H-4), 3.77 (dd, J = 7.2, 5.6 Hz, 1H, H-6b), 3.56 (dd,
J = 5.2, 2.0 Hz, 1H, H-2), 3.06 (d, J = 2.8 Hz, 1H, C3-OH), 2.35 (d, J = 8.0 Hz, 1H, C4-OH); 13C
NMR (100 MHz, CDCl3): δ 136.1, 131.7, 129.4, 122.2, 100.2, 76.5, 73.2, 71.6, 70.3, 69.7, 64.9;
HRMS m/z calcd for C13H19BrNO5 [M+NH4]+: 348.0454. Found 348.0447.
Major peaks from 3-O-(4-bromobenzyl) regioisomer: 5.37–5.35 (m, 1H, H-1), 4.68 (d,
J = 12.0 Hz, 1H, ArCH2), 4.56 (d, J = 12.0 Hz, 1H, ArCH2), 4.83–4.49 (m, 1H, H-5), 4.18 (dd,
J = 7.2, 0.8 Hz, 1H, H-6a), 2.44 (d, J = 9.6 Hz, 1H, C2-OH).
1,6-Anhydro-2-O-(2-methylnaphthalenyl)-β-D-mannopyranoside (2.17)
1,6-Anhydro-β-D-mannopyranoside 2.02 (32.4 mg, 0.200 mmol), 2-(bromomethyl)naphthalene
(66.3 mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 50:50) afforded the title compound as a
colourless oil (42.9 mg, 71%). Rf = 0.26 (pentanes/EtOAc, 30:70); FTIR (νmax) 3464 (br), 2941
(m), 1508 (w), 1396 (m), 1251 (m), 1123 (s), 1048 (s), 992 (s), 896 (s), 802 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.86–7.76 (m, 4H, ArH), 7.52–7.47 (m, 3H, ArH), 5.42 (apparent t,
J = 2.0 Hz, 1H, H-1), 4.87 (d, J = 12.0 Hz, 1H, ArCH2), 4.74 (d, J = 12.0 Hz, 1H, ArCH2), 4.48–
4.44 (m, 1H, H-5), 4.30 (dd, J = 7.2, 0.8 Hz, 1H, H-6a), 4.10–4.06 (m, 1H, H-3), 3.92–3.89 (m,
1H, H-4), 3.73 (dd, J = 7.2, 6.0 Hz, 1H, H-6b), 3.58 (dd, J = 5.2, 2.0 Hz, 1H, H-2), 3.12 (d, J =
2.4 Hz, 1H, C3-OH), 2.26 (d, J = 8.8 Hz, 1H, C4-OH); 13C NMR (100 MHz, CDCl3): δ 134.5,
133.3, 128.5, 128.0, 127.9, 127.7, 126.9, 126.3, 126.1, 125.5, 100.3, 76.5, 72.9, 71.6, 71.2, 69.7,
64.9; HRMS m/z calcd for C17H22NO5 [M+NH4]+: 320.1498. Found 320.1487.
OOOH
OH
O

109
1.6-Anhydro-2-O-benzyloxymethyl-β-D-mannopyranoside (2.18)
1,6-Anhydro-β-D-mannopyranoside 2.02 (32.4 mg, 0.200 mmol), benzyloxymethyl chloride
(41.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
pale yellow oil (37.1 mg, 97% pure by 1H NMR: 63% yield. The contaminant is the 3-O-
benzyloxymethyl regioisomer. Rf = 0.23 (pentanes/EtOAc, 30:70); FTIR (νmax) 3447 (br), 2898
(m), 1656 (w), 1406 (w), 1105 (s), 1047 (s), 973 (s), 902 (m), 820 (m), 699 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.31–7.21 (m, 5H, ArH), 5.36 (apparent t, J = 1.6 Hz, 1H, H-1), 4.82 (s,
2H, OCH2O), 4.64 (d, J =12.0 Hz, 1H, ArCH2O), 4.57 (d, J =12.0 Hz, 1H, ArCH2O), 4.45–4.40
(m, 1H, H-5), 4.25 (dd, J = 7.6, 0.8 Hz, 1H, H-6a), 3.99–3.96 (m, 1H, H-3), 3.85–3.80 (m, 1H,
H-4), 3.73–3.67 (m, 2H, H-2 and H-6b), 2.88 (d, J = 2.0 Hz, 1H, C3-OH), 2.36 (d, J = 8.8 Hz,
1H, C4-OH); 13C NMR (100 MHz, CDCl3): δ 137.1, 128.6, 128.1, 128.0, 100.7, 93.9, 76.3, 72.1,
71.5, 70.4, 70.3, 65.0; HRMS m/z calcd for C14H19O6 [M+H]+: 283.1182. Found 283.1195.
Major peaks from 3-O-benzyloxymethyl regioisomer: 5.28–5.30 (m, 1H, H-1), 4.83 (d, J =12.0
Hz, 1H, ArCH2O), 4.75 (d, J =12.0 Hz, 1H, ArCH2O), 4.42–4.38 (m, 1H, H-5), 4.11 (dd, J = 7.6,
1.2 Hz, 1H, H-6a), 2.98 (d, J = 2.4 Hz, 1H, C2-OH).
1,3,4,6-Tetra-O-benzyl-β-D-mannopyranose (2.19)
3,4,6-Tri-O-benzyl-D-mannopyranoside 2.03 (81.0 mg, 0.200 mmol), benzyl bromide (35.6 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
OOOH
OH
OBOM
OOBn
OBnOH
BnOBnO

110
Flash chromatographic purification (pentanes/EtOAc, 85:25) afforded the title compound as a
colourless oil (77.8 mg, 72%); Rf = 0.42 (pentanes/EtOAc, 70:30). Spectral data were in accord
with those presented in the literature.104
Methyl-3-O-benzyl-α-L-rhamnopyranoside (2.20)
Methyl-α-L-rhamnopyranoside 2.04 (35.6 mg, 0.200 mmol), benzyl bromide (35.6 µL, 0.300
mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220 mmol)
were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
colourless oil (44.6 mg, 83%); Rf = 0.56 (pentanes/EtOAc, 30:70); FTIR (νmax) 3430 (br), 2909
(m), 1497 (w), 1453 (m), 1197 (w), 1104 (s), 1053 (s), 985 (s), 905 (m), 740 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.39–7.29 (m, 5H, ArH), 4.71 (d, J = 1.6 Hz, 1H, H-1), 4.70 (d,
J = 11.6 Hz, 1H, ArCH2), 4.55 (d, J = 11.6 Hz, 1H, ArCH2), 4.02–4.00 (m, 1H, H-2), 3.68–3.59
(m, 2H, H-3 and H-5), 3.54 (apparent td, J = 9.2, 2.4 Hz, 1H, H-4), 3.36 (s, 3H, OCH3), 2.40 (d,
J = 2.4 Hz, 1H, C2-OH), 2.22 (d, J = 2.8 Hz, 1H, C4-OH), 1.31 (d, J = 6.4 Hz, 3H, CHCH3); 13C
NMR (100 MHz, CDCl3): δ 137.7, 128.7, 128.2, 127.9, 100.4, 79.8, 71.6, 71.5, 67.7, 67.5, 54.8,
17.6; HRMS m/z calcd for C14H24NO5 [M+NH4]+: 286.1654. Found 286.1650.
Methyl-3-O-(4-bromobenzyl)-α-L-rhamnopyranoside (2.21)
104 Cumpstey, I.; Chayajarus, K.; Fairbanks, A. J.; Redgrave, A. J.; Seward, C. M. P. Tetrahedron: Asymmetry. 2004, 15, 3207–3222
O
OMeMeHOBnO
OH
O
OMeMeHO
OOH
Br

111
Methyl-α-L-rhamnopyranoside 2.04 (35.6 mg, 0.200 mmol), 4-bromobenzyl bromide (75.0 mg,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
colourless oil (69.1 mg, >99%); Rf = 0.54 (pentanes/EtOAc, 30:70); FTIR (νmax) 3447 (br), 2910
(m), 1593 (w), 1488 (m), 1366 (w), 1265 (w), 1053 (s), 985 (s), 905 (w), 737 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.48 (apparent dt, J = 8.8, 2.4 Hz, 2H, ArH), 7.22 (dt, J = 8.8, 2.4 Hz, 2H,
ArH), 4.68 (d, J = 2.0 Hz, 1H, H-1), 4.64 (d, J = 12.0 Hz, 1H, ArCH2), 4.52 (d, J = 12.0 Hz, 1H,
ArCH2), 3.99–3.97 (m, 1H, H-2), 3.66–3.51 (m, 3H, H-3, H-4 and H-5), 3.35 (s, 3H, OCH3),
2.37 (d, J = 2.8 Hz, 1H, C2-OH), 2.24 (d, J = 2.8 Hz, 1H, C4-OH), 1.31 (d, J = 6.0 Hz, 3H,
CHCH3); 13C NMR (100 MHz, CDCl3): δ 136.7, 131.8, 129.5, 122.1, 100.3, 79.8, 71.7, 70.8,
67.8, 67.5, 54.9, 17.6; HRMS m/z calcd for C14H23BrNO5 [M+NH4]+: 364.0750. Found
364.0759.
Methyl-3-O-(2-naphthyl)methyl-α-L-rhamnopyranoside (2.22)
Methyl-α-L-rhamnopyranoside 2.04 (35.6 mg, 0.200 mmol), 2-(bromomethyl)naphthalene (66.3
mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
pale yellow oil (52.7 mg, 83%); Rf = 0.55 (pentanes/EtOAc, 30:70). Spectral data were in accord
with those presented in the literature.105
105 Borbás, A.; Szabó, Z. B.; Szilágyi, L.; Bényei, A.; Lipták, A. Carbohydr. Res. 2002, 337, 1941–1951.
O
OMeMeHO
OOH

112
Methyl-3-O-benzyloxymethyl-α-L-rhamnopyranoside (2.23)
Methyl-α-L-rhamnopyranoside 2.04 (35.6 mg, 0.200 mmol), benzyloxymethyl chloride (41.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
pale yellow oil (43.1 mg, 72%); Rf = 0.54 (pentanes/EtOAc, 30:70); FTIR (νmax) 3443 (br), 2892
(m), 1497 (w), 1454 (m), 1381 (m), 1053 (s), 982 (s), 905 (m), 802 (m), 740 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.38–7.28 (m, 5H, ArH), 4.92 (d, J = 6.8 Hz, 1H, OCH2O), 4.86 (d, J =
6.8 Hz, 1H, OCH2O), 4.75 (d, J = 12.0 Hz, 1H, ArCH2O), 4.67 (d, J = 1.6 Hz, 1H, H-1), 4.65 (d,
J = 12.0 Hz, 1H, ArCH2O), 3.99–3.97 (m, 1H, H-2), 3.69–3.61 (m, 2H, H-3 and H-5), 3.52
(apparent td, J = 9.2, 2.0 Hz, 1H, H-4), 3.36 (s, 3H, OCH3), 3.24 (d, J = 2.4 Hz, 1H, C4-OH),
2.35 (d, J = 3.2 Hz, 1H, C2-OH), 1.34 (d, J = 6.4 Hz, 3H, CHCH3); 13C NMR (100 MHz,
CDCl3): δ 136.8, 128.6, 128.1, 127.9, 100.3, 95.3, 81.4, 71.4, 70.5, 70.1, 67.6, 54.8, 17.7;
HRMS m/z calcd for C15H26NO6 [M+NH4]+: 316.1760. Found 316.1753.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyl-α-D-galactopyranoside (2.24)
Methyl-α-D-galactopyranoside 2.05 (61.7 mg, 0.200 mmol), benzyl bromide (35.6 µL, 0.300
mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220 mmol)
were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 80:20) afforded the title compound as a
O
OMeMeHOBOMO
OH
O
OMe
OTBS
HOBnO
HO

113
colourless oil (61.1 mg, 77%); Rf = 0.32 (pentanes/EtOAc, 70:30). Spectral data were in accord
with those presented in the literature.106
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(4-bromobenzyl)-α-D-galactopyranoside (2.25)
Methyl-α-D-galactopyranoside 2.05 (61.7 mg, 0.200 mmol), 4-bromobenzyl bromide (75.0 mg,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 70:30) afforded the title compound as a
colourless oil (90.7 mg, 95%); Rf = 0.29 (pentanes/EtOAc, 70:30); FTIR (νmax) 3508 (br), 2856
(m), 1517 (w), 1487 (m), 1389 (m), 1250 (m), 1148 (m), 1083 (s), 963 (m), 836 (s) cm-1; 1H
NMR (400 MHz, CDCl3): δ 7.49 (apparent dt, J = 8.8, 2.4 Hz, 2H, ArH), 7.25 (apparent dt,
J = 8.8, 2.4 Hz, 2H, ArH), 4.81 (d, J = 4.0 Hz, 1H, H-1), 4.69 (s, 2H, ArCH2), 4.07–4.06 (m, 1H,
H-4), 4.01 (ddd, J = 9.6, 8.4, 4.0 Hz, 1H, H-2), 3.87 (dd, J = 10.4, 6.0 Hz, 1H, H-6a), 3.80 (dd,
J = 10.4, 5.6 Hz, 1H, H-6b), 3.69 (apparent t, J = 5.6 Hz, 1H, H-5), 3.56 (dd, J = 9.6, 3.2 Hz, 1H,
H-3), 3.40 (s, 3H, OCH3), 2.67 (s, 1H, C4-OH), 2.10 (d, J = 8.4 Hz, 1H, C2-OH), 0.89 (s, 9H,
Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 137.0,
131.6, 129.4, 121.8, 99.5, 78.6, 71.2, 69.9, 68.7, 67.1, 62.7, 55.2, 25.8, 18.3, –5.4; HRMS m/z
calcd for C20H34BrO6Si [M+H]+: 477.1308. Found 477.1292.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(2-methylnaphthalenyl)-α-D-galactopyranoside
(2.26)
106 Aurrecoechea, J. M.; Gil, J. H.; López, B. Tetrahedron. 2003, 59, 7111–7122.
O
OMe
OTBS
HOO
HO
Br
O
OMe
OTBS
HOO
HO

114
Methyl-α-D-galactopyranoside 2.05 (61.7 mg, 0.200 mmol), 2-(bromomethyl)naphthalene (66.3
mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 70:30) afforded the title compound as a
colourless oil (73.3 mg, 82%); Rf = 0.24 (pentanes/EtOAc, 70:30); FTIR (νmax) 3509 (br), 2928
(m), 1462 (m), 1360 (w), 1250 (s), 1084 (s), 1051 (s), 963 (m), 836 (s), 773 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.85–7.82 (m, 4H, ArH), 7.54–7.45 (m, 3H, ArH), 4.90 (s, 2H, ArCH2),
4.81 (d, J = 4.0 Hz, 1H, H-1), 4.10–4.07 (m, 1H, H-4), 4.06–4.02 (m, 1H, H-2), 3.87 (dd,
J = 10.4, 6.0 Hz, 1H, H-6a), 3.79 (dd, J = 10.4, 5.6 Hz, 1H, H-6b), 3.77 (apparent t, J = 5.6 Hz,
1H, H-5), 3.64 (dd, J = 9.6, 3.2 Hz, 1H, H-3), 3.40 (s, 3H, OCH3), 2.70 (s, 1H, C4-OH), 2.23 (d,
J = 6.0 Hz, 1H, C2-OH), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2); 13C
NMR (100 MHz, CDCl3): δ 135.4, 133.2, 133.1, 128.5, 127.9, 127.7, 126.7, 126.2, 126.1, 125.7,
99.5, 78.5, 72.1, 70.7, 68.7, 67.0, 62.7, 55.2, 25.8, 18.3, –5.4; HRMS m/z calcd for C24H37O6Si
[M+H]+: 449.2359. Found 449.2341.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyloxymethyl-α-D-galactopyranoside (2.27)
Methyl-α-D-galactopyranoside 2.05 (61.7 mg, 0.200 mmol), benzyloxymethyl chloride (41.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 70:30) afforded the title compound as a
pale yellow oil (77.1 mg, 90%); Rf = 0.20 (pentanes/EtOAc, 70:30); FTIR (νmax) 3506 (br), 2928
(m), 1518 (w), 1462 (m), 1388 (m), 1251 (m), 1083 (s), 1036 (s), 963 (m), 836 (s) cm-1; 1H
NMR (400 MHz, CDCl3): δ 7.36–7.28 (m, ArH), 4.96 (d, J = 7.2 Hz, 1H, OCH2O), 4.91 (d, J =
7.2 Hz, 1H, OCH2O), 4.83 (d, J = 4.0 Hz, 1H, H-1), 4.72 (d, J =12.0 Hz, 1H, ArCH2O), 4.70 (d,
J =12.0 Hz, 1H, ArCH2O), 4.14–4.12 (m, 1H, H-4), 4.00 (ddd, J = 9.6, 8.0, 4.0 Hz, 1H, H-2),
3.89 (dd, J = 10.4, 5.6 Hz, 1H, H-6a), 3.82 (dd, J = 10.4, 5.2 Hz, 1H, H-6b), 3.78 (dd, J = 10.0,
3.2 Hz, 1H, H-3), 3.74 (apparent t, J = 5.2 Hz, 1H, H-5), 3.42 (s, 3H, OCH3), 2.80 (s, 1H, C4-
O
OMe
OTBS
HOBOMO
HO

115
OH), 2.51 (d, J = 8.0 Hz, 1H, C2-OH), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H,
Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 137.3, 128.5, 127.9, 127.8, 99.6, 94.7,
78.2, 77.4, 77.1, 76.8, 70.1, 63.0, 55.2, 25.8, 18.3, –5.4; HRMS m/z calcd for C21H37O7Si
[M+H]+: 429.2308. Found 429.2290.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyl-β-D-galactopyranoside (2.28)
Methyl-β-D-galactopyranoside 2.06 (308.4 mg, 1.000 mmol), benzyl bromide (178.2 µL, 1.500
mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol %) and Ag2O (254.9 mg, 1.100 mmol)
were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 70:30) afforded the title compound as a
colourless oil (305.7 mg, 77%); Rf = 0.27 (pentanes/EtOAc, 70:30). Spectral data were in accord
with those presented in the literature.107
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(4-bromobenzyl)-β-D-galactopyranoside (2.29)
Methyl-β-D-galactopyranoside 2.06 (308.4 mg, 1.000 mmol), 4-bromobenzyl bromide (374.9
mg, 1.500 mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol%) and Ag2O (247.6 mg,
1.100 mmol) were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 75:25) afforded the title compound as a
colourless oil (280.1 mg, 91%); Rf = 0.29 (pentanes/EtOAc, 70:30); FTIR (νmax) 3462 (br), 2929
(m), 2856 (m), 1592 (w), 1462 (m), 1254 (s), 1070 (s), 1011 (m), 837 (s), 778 (m) cm-1; 1H
NMR (400 MHz, CDCl3): δ 7.45 (apparent dt, J = 9.2, 2.4 Hz, 2H, ArH), 7.26 (apparent dt,
107 Villalobos, A.; Danishefsky, S. J. J. Org. Chem. 1990, 55, 2776–2786.
OOMe
OTBS
HOBnO
HO
OOMe
OTBS
HOO
HO
Br

116
J = 9.2, 2.4 Hz, 2H, ArH), 4.69 (s, 2H, ArCH2), 4.14 (d, J = 7.6 Hz, 1H, H-1), 4.03–3.99 (m, 1H,
H-4), 3.90 (dd, J = 10.0, 6.4 Hz, 1H, H-6a), 3.81 (dd, J = 10.0, 5.6 Hz, 1H, H-6b), 3.77 (dd,
J = 9.6, 8.0 Hz, 1H, H-2), 3.52 (s, 3H, OCH3), 3.42–3.40 (m, 1H, H-5), 3.37 (dd, J = 9.2, 3.2 Hz,
1H, H-3), 2.55–2.58 (m, 2H, C2-OH and C4-OH), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H,
Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 136.9, 131.6, 129.5, 121.8, 104.0, 80.6,
74.6, 71.2, 71.1, 66.2, 62.1, 56.9, 25.8, 18.3, –5.4; HRMS m/z calcd for C20H34BrO6Si
[M+H]+: 477.1309. Found 477.1294.
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(2-methylnaphthalenyl)-β-D-galactopyranoside
(2.30)
Methyl-β-D-galactopyranoside 2.06 (308.4 mg, 1.000 mmol), 2-(bromomethyl)naphthalene
(331.6 mg, 1.500 mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol %) and Ag2O (247.6
mg, 1.100 mmol) were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for
48 hr. Flash chromatographic purification (pentanes/EtOAc, 75:25) afforded the title compound
as a pale yellow oil (443.7 mg, >99%); Rf = 0.28 (pentanes/EtOAc, 70:30); FTIR (νmax) 3557
(br), 2928 (m), 1462 (w), 1389 (w), 1252 (m), 1154 (m), 1072 (s), 836 (s), 733 (s), 702 (m) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.84–7.80 (m, 4H, ArH), 7.54–7.45 (m, 3H, ArH), 4.91 (s, 2H,
ArCH2), 4.14 (d, J = 8.0 Hz, 1H, H-1), 4.02–4.05 (m, 1H, H-4), 3.92 (dd, J = 10.4, 6.8 Hz, 1H,
H-6a), 3.87–3.81 (m, 2H, H-2 and H-6b), 3.54 (s, 3H, OCH3), 3.45 (dd, J = 9.6, 3.2 Hz, 1H, H-
3), 3.40 (apparent t, J = 6.0 Hz, 1H, H-5), 2.78 (s, 1H, C4-OH), 2.66 (d, J = 2.0 Hz, 1H, C2-OH),
0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz,
CDCl3): δ 135.3, 133.3, 133.1, 128.5, 127.9, 127.7, 126.8, 126.2, 126.1, 125.7, 104.0, 80.5, 74.7,
72.1, 71.1, 66.2, 62.1, 56.8, 25.8, 18.3, –5.4; HRMS m/z calcd for C24H37O6Si [M+H]+:
449.2359. Found 449.2346.
OOMe
OTBS
HOO
HO

117
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-benzyloxymethyl-β-D-galactopyranoside (2.31)
Methyl-β-D-galactopyranoside 2.06 (61.7 mg, 0.200 mmol), benzyloxymethyl chloride (41.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 70:30) afforded the title compound as a
pale yellow oil (75.1 mg, 88%); Rf = 0.23 (pentanes/EtOAc, 70:30); FTIR (νmax) 3469 (br), 2929
(m), 1471 (w), 1387 (w), 1254 (m), 1206 (w), 1102 (s), 1041 (s), 837 (s), 736 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.35–7.27 (m, 5H, ArH), 4.97 (d, J = 7.2 Hz, 1H, OCH2O), 4.94 (d, J =
7.2 Hz, 1H, OCH2O), 4.73 (d, J =11.6 Hz, 1H, ArCH2O), 4.68 (d, J =11.6 Hz, 1H, ArCH2O),
4.20 (d, J = 7.6 Hz, 1H, H-1), 4.10–4.08 (m, 1H, H-4), 3.92 (dd, J = 10.4, 6.4 Hz, 1H, H-6a),
3.85 (dd, J = 10.4, 5.2 Hz, 1H, H-6b), 3.76 (apparent dt, J = 9.2, 1.2 Hz, 1H, H-2), 3.58 (dd, J =
9.2, 3.2 Hz, 1H, H-3), 3.55 (s, 3H, OCH3), 3.47 (apparent t, J = 5.6 Hz, 1H, H-5), 2.89 (d, J = 1.2
Hz, 1H, C2-OH), 2.66 (d, J = 3.2 Hz, 1H, C4-OH), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.08 (s, 6H,
Si(C(CH3)3)(CH3)2); 13C NMR (100 MHz, CDCl3): δ 137.1, 128.5, 128.1, 128.0, 104.1, 94.7,
80.6, 74.4, 70.5, 70.2, 67.7, 62.4, 56.9, 25.8, 18.3, –5.4; HRMS m/z calcd for C21H37O7Si
[M+H]+: 429.2308. Found 429.2294.
1,6-Anhydro-4-O-benzyl-β-D-galactopyranoside (2.32)
1,6-Anhydro-β-D-galactopyranoside 2.07 (32.4 mg, 0.200 mmol), benzyl bromide (35.6 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
yellow oil (38.4 mg, 96% pure by 1H NMR: 73% yield). The contaminant is the 3-O-benzyl
OOMe
OTBS
HOBOMO
HO
OOOH
BnO
OH

118
regioisomer. Rf = 0.24 (pentanes/EtOAc, 30:70); FTIR (νmax) 3428 (br), 2902 (m), 1454 (m),
1333 (m), 1210 (m), 1133 (s), 1050 (s), 932 (s), 852 (m), 738 (m) cm-1; 1H NMR (400 MHz,
CDCl3): δ 7.39–7.30 (m, 5H, ArH), 5.35–5.38 (m, 1H, H-1), 4.69 (d, J = 12.0 Hz, 1H, ArCH2),
4.64 (d, J = 12.0 Hz, 1H, ArCH2), 4.40 (apparent t, J = 4.4 Hz, 1H, H-5), 4.29 (d, J = 7.6 Hz, 1H,
H-6a), 4.42–3.91 (m, 1H, H-3), 3.84–3.77 (m, 2H, H-2 and H-4), 3.63 (dd, J = 7.2, 5.6 Hz, 1H,
H-6b), 2.76 (d, J = 2.4 Hz, 1H, C3-OH), 2.34 (d, J = 8.0 Hz, 1H, C2-OH); 13C NMR (100 MHz,
CDCl3): δ 137.3, 128.6, 128.3, 127.8, 101.6, 72.7, 71.8, 71.7, 71.6, 69.9, 64.1; HRMS m/z calcd
for C13H20NO5 [M+NH4]+: 270.1341. Found 270.1338. Major peaks from 3-O-benzyl
regioisomer: 4.78 (d, J = 11.6 Hz, 1H, ArCH2), 4.46 (d, J = 11.6 Hz, 1H, ArCH2), 4.19 (d,
J = 7.6 Hz, 1H, H-6a), 3.12 (d, J = 6.0 Hz, 1H, C4-OH).
1,6-Anhydro-4-O-(4-bromobenzyl)-β-D-galactopyranoside (2.33)
1,6-Anhydro-β-D-galactopyranoside 2.07 (162.1 mg, 1.000 mmol), 4-bromobenzyl bromide
(374.9 mg, 1.500 mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol %) and Ag2O (254.9
mg, 1.100 mmol) were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for
48 hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound
as a yellow oil (126.7 mg, 92% pure by 1H NMR: 72% yield). The contaminant is the 3-O-(4-
bromobenzyl) regioisomer. Rf = 0.23 (pentanes/EtOAc, 30:70); FTIR (νmax) 3424 (br), 2900
(m), 1592 (w), 1487 (m) 1333 (m), 1247 (m), 1132 (s), 1050 (s), 1011 (s), 931 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.47 (apparent dt, J = 9.2, 2.4 Hz, 2H, ArH), 7.20 (apparent dt, J = 9.2, 2.4
Hz, 2H, ArH), 5.35–5.33 (m, 1H, H-1), 4.61 (d, J = 12.0 Hz, 1H, ArCH2), 4.54 (d, J = 12.0 Hz,
1H, ArCH2), 4.39 (apparent t, J = 4.4 Hz, 1H, H-5), 4.26 (d, J = 7.6 Hz, 1H, H-6a), 4.02–3.98
(m, 1H, H-3), 3.81–3.77 (m, 2H, H-2 and H-4), 3.61 (dd, J = 7.2, 5.6 Hz, 1H, H-6b), 2.76 (d, J =
2.8 Hz, 1H, C3-OH), 2.67 (d, J = 8.0 Hz, 1H, C2-OH); 13C NMR (100 MHz, CDCl3): δ 136.3,
131.8, 129.3, 122.3, 101.6, 72.7, 71.9, 71.6, 70.8, 69.8, 64.2; HRMS m/z calcd for
C13H19BrNO5 [M+NH4]+: 348.0446. Found 348.0456. Major peaks from 3-O-(4-bromobenzyl)
OOOH
O
OH
Br

119
regioisomer: 4.71 (d, J = 11.6 Hz, 1H, ArCH2), 4.16 (d, J = 7.6 Hz, 1H, H-6a), 3.03 (d, J = 9.6
Hz, 1H, C4-OH).
1,6-Anhydro-4-O-(2-methylnaphthalenyl)-β-D-galactopyranoside (2.34)
1,6-Anhydro-β-D-galactopyranoside 2.07 (32.4 mg, 0.200 mmol), 2-(bromomethyl)naphthalene
(66.3 mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (1 mL, 0.2 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
yellow oil (46.1 mg, 97% pure by 1H NMR: 74% yield). The contaminant is the 3-O-(2-
methylnaphthalenyl) regioisomer. Rf = 0.23 (pentanes/EtOAc, 30:70); FTIR (νmax) 3433 (br),
2960 (w), 1509 (w), 1341 (m), 1238 (m), 1127 (s), 1018 (s), 923 (m), 824 (s), 751 (m) cm-1; 1H
NMR (400 MHz, CDCl3): δ 7.87–7.77 (m, 4H, ArH), 7.54–7.45 (m, 3H, ArH), 5.37 (apparent t,
J = 1.2 Hz, 1H, H-1), 4.86 (d, J = 11.6 Hz, 1H, ArCH2), 4.78 (d, J = 11.6 Hz, 1H, ArCH2), 4.43
(apparent t, J = 4.4 Hz, 1H, H-5), 4.34 (d, J = 7.6 Hz, 1H, H-6a), 4.06–4.04 (m, 1H, H-3), 3.88–
3.82 (m, 2H, H-2 and H-4), 3.67 (dd, J = 7.2, 5.2 Hz, 1H, H-6b), 2.70 (d, J = 0.8 Hz, 1H, C3-
OH), 2.25 (d, J = 7.6 Hz, 1H, C2-OH); 13C NMR (100 MHz, CDCl3): δ 134.6, 133.2, 133.1,
128.7, 127.9, 127.8, 127.0, 126.5, 126.4, 125.5, 101.5, 72.7, 71.9, 71.5, 71.4, 69.8, 64.3; HRMS
m/z calcd for C17H22NO5 [M+NH4]+: 320.1498. Found 320.1486.
Major peaks from 3-O-(2-methylnaphthalenyl) regioisomer: 4.93 (d, J = 12.0 Hz, 1H, ArCH2),
4.64 (d, J = 12.0 Hz, 1H, ArCH2), 4.23 (d, J = 8.0 Hz, 1H, H-6a), 3.12 (d, J = 9.6 Hz, 1H, C4-
OH).
1.6-Anhydro-4-O-benzyloxymethyl-β-D-galactopyranoside (2.35)
OOOH
O
OH
OOOH
BOMO
OH

120
1,6-Anhydro-β-D-galactopyranoside 2.07 (32.4 mg, 0.200 mmol), benzyloxymethyl chloride
(41.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
yellow oil (37.0 mg, 96% pure by 1H NMR: 63% yield). The contaminant is the 3-O-
benzyloxymethyl regioisomer. Rf = 0.22 (pentanes/EtOAc, 30:70); FTIR (νmax) 3436 (br), 2895
(m), 1454 (w), 1266 (w), 1132 (s), 1107 (m), 1025 (s), 930 (s), 850 (m), 734 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.39–7.30 (m, 5H, ArH), 5.39–5.40 (m, 1H, H-1), 4.91 (d, J = 10.8 Hz,
1H, OCH2O), 4.80 (d, J = 10.8 Hz, 1H, OCH2O), 4.64 (s, 2H, ArCH2O), 4.48 (apparent t,
J = 4.8 Hz, 1H, H-5), 4.32 (d, J = 7.2 Hz, 1H, H-6a), 4.05–3.99 (m, 2H, H-3 and H-4), 3.83 (d,
J = 9.6 Hz, 1H, H-2), 3.67 (dd, J = 6.8, 6.0 Hz, 1H, H-6b), 2.64 (d, J = 2.8 Hz, 1H, C3-OH), 1.95
(d, J = 9.6 Hz, 1H, C2-OH); 13C NMR (100 MHz, CDCl3): δ 137.1, 128.5, 128.0, 127.7, 101.5,
94.6, 73.4, 71.7, 71.6, 70.7, 70.6, 64.2; HRMS m/z calcd for C14H22NO6 [M+NH4]+: 300.1447.
Found 300.1459.
Major peaks from 3-O-benzyloxymethyl regioisomer: 5.38–5.36 (m, 1H, H-1), 4.22 (d,
J = 7.6 Hz, 1H, H-6a), 2.89 (d, J = 2.8 Hz, 1H, C4-OH).
Methyl-3-O-benzyl-α-L-fucopyranoside (2.36)
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), benzyl bromide (35.6 µL, 0.300 mmol),
2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220 mmol) were
stirred in acetonitrile (1 mL, 0.2 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a pale
yellow oil (50.4 mg, 94%); Rf = 0.26 (pentanes/EtOAc, 60:40); FTIR (νmax) 3445 (br), 2898 (m),
1497 (w), 1360 (m), 1191 (m), 1079 (s), 1049 (s), 956 (s), 875 (w), 749 (m) cm-1; 1H NMR (400
MHz, CDCl3): δ 7.39–7.28 (m, 5H, ArH), 4.76 (d, J = 4.0 Hz, 1H, H-1), 4.72 (s, 2H, ArCH2),
3.45 (ddd, J = 9.6, 8.0, 4.0 Hz, 1H, H-2), 3.86 (apparent q, J = 6.8 Hz, 1H, H-5), 3.82–3.80 (m,
1H, H-4), 3.61 (dd, J = 9.6, 3.2 Hz, 1H, H-3), 3.40 (s, 3H, OCH3), 2.40 (s, 1H, C4-OH), 2.17 (d,
O
OMeMe
HOOBnOH

121
J = 8.0 Hz, 1H, C2-OH), 1.29 (d, J = 6.8 Hz, 3H, CHCH3); 13C NMR (100 MHz, CDCl3): δ
137.9, 128.6, 128.0, 127.8, 99.5, 78.7, 72.0, 69.5, 68.4, 65.4, 55.3, 16.2; HRMS m/z calcd for
C14H24NO5 [M+NH4]+: 286.1654. Found 286.1658.
Methyl-3-O-(4-bromobenzyl)-α-L-fucopyranoside (2.37)
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), 4-bromobenzyl bromide (75.0 mg,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 55:45) afforded the title compound as a
pale yellow oil (61.9 mg, 89%); Rf = 0.31 (pentanes/EtOAc, 30:70); FTIR (νmax) 3534 (m), 3463
(m), 2906 (m), 1593 (w), 1491 (m), 1135 (s), 1067 (s), 1023 (s), 805 (s) cm-1; 1H NMR (400
MHz, CDCl3): δ 7.44 (apparent dt, J = 8.8, 2.4 Hz, 2H, ArH), 7.22 (apparent dt, J = 8.8, 2.4 Hz,
2H, ArH), 4.72 (d, J = 4.0 Hz, 1H, H-1), 4.64 (s, 2H, ArCH2), 3.92 (ddd, J = 9.6, 8.0, 4.0 Hz, 1H,
H-2), 3.82 (apparent q, J = 6.4 Hz, 1H, H-5), 3.78–3.76 (m, 1H, H-4), 3.54 (dd, J = 9.6, 3.2 Hz,
1H, H-3), 3.37 (s, 3H, OCH3), 2.46 (s, 1H, C4-OH), 2.28 (d, J = 8.4 Hz, 1H, C2-OH), 1.26 (d, J =
6.4 Hz, 3H, CHCH3); 13C NMR (100 MHz, CDCl3): δ 137.0, 131.6, 129.4, 121.8, 99.6, 78.7,
71.2, 69.6, 68.4, 65.5, 55.3, 16.2; HRMS m/z calcd for C14H23BrNO5 [M+NH4]+: 364.0760.
Found 364.0764.
Methyl-3-O-(2-naphthyl)methyl-α-L-fucopyranoside (2.38)
O
OMeMe
HOOOH
Br
O
OMeMe
HOOOH

122
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), 2-(bromomethyl)naphthalene (66.3 mg,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
pale yellow oil (51.1 mg, 86%); Rf = 0.24 (pentanes/EtOAc, 30:70); FTIR (νmax) 3428 (br), 2904
(m), 1602 (w), 1447 (w), 1265 (m), 1124 (m), 1083 (s), 1049 (s), 857 (m), 735 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.84–7.46 (m, 4H, ArH), 7.52–7.46 (m, 3H, ArH), 4.87 (s, 2H, ArCH2),
4.92–4.77 (d, J = 3.6 Hz, 1H, H-1), 4.00 (ddd, J = 9.6, 8.0, 4.0 Hz, 1H, H-2), 3.86–3.81 (m, 2H,
H-5 and H-4), 3.64 (dd, J = 9.6, 3.2 Hz, 1H, H-3), 3.38 (s, 3H, OCH3), 2.55 (s, 1H, C4-OH), 2.32
(d, J = 8.4 Hz, 1H, C2-OH), 1.29 (d, J = 6.8 Hz, 3H, CHCH3); 13C NMR (100 MHz, CDCl3): δ
135.4, 133.2, 133.1, 128.4, 127.9, 127.7, 126.7, 126.2, 126.1, 125.7, 99.6, 78.6, 72.1, 69.6, 68.4,
65.5, 55.3, 16.2; HRMS m/z calcd for C18H26NO5 [M+NH4]+: 336.1810. Found 336.1811.
Methyl-3-O-benzyloxymethyl-α-L-fucopyranoside (2.39)
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), benzyloxymethyl chloride (41.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
pale yellow oil (58.6 mg, 98%); Rf = 0.24 (pentanes/EtOAc, 30:70); FTIR (νmax) 3447 (br), 2896
(m), 1454 (w), 1361 (w), 1266 (m), 1191 (m), 1065 (s), 958 (s), 801 (w), 735 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.38–7.28 (m, 5H, ArH), 4.96 (d, J = 7.2 Hz, 1H, OCH2O), 4.92 (d, J =
7.2 Hz, 1H, OCH2O), 4.78 (d, J = 3.8 Hz, 1H, H-1), 4.72 (d, J = 12.0 Hz, 1H, ArCH2O), 4.66 (d,
J = 12.0 Hz, 1H, ArCH2O), 3.96–3.88 (m, 2H, H-2 and H-5), 3.81–3.78 (m, 2H, H-3 and H-4),
3.41 (s, 3H, OCH3), 2.49 (d, J = 8.0 Hz, 1H, C2-OH), 2.34 (s, 1H, C4-OH), 1.29 (d, J = 6.8 Hz,
3H, CHCH3); 13C NMR (100 MHz, CDCl3): δ 137.3, 128.5, 128.0, 127.9, 99.6, 94.7, 78.3, 71.0,
70.2, 68.0, 65.4, 55.3, 16.1; HRMS m/z calcd for C15H26NO6 [M+NH4]+: 316.1760. Found
316.1752.
O
OMeMe
HOOBOMOH

123
Methyl-3-O-benzyl-β-L-arabinopyranoside (2.40)
Methyl-β-L-arabinopyranoside 2.09 (164.1 mg, 1.000 mmol), benzyl bromide (178.2 µL, 1.500
mmol), 2-aminoethyl diphenylborinate (22.5 mg, 10 mol %) and Ag2O (247.6 mg, 1.100 mmol)
were stirred in acetonitrile (5 mL, 0.2 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (188.8 mg, 74%); Rf = 0.19 (pentanes/EtOAc, 30:70); FTIR (νmax) 3463 (m), 2922
(w), 2835 (w), 1450 (w), 1350 (m), 1136 (s), 1060 (s), 998 (s), 925 (w), 855 (m), 747 (s) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.39–7.28 (m, 5H, ArH), 4.76 (d, J = 3.6 Hz, 1H, H-1), 4.72 (d,
J = 11.6 Hz, 1H, ArCH2), 4.68 (d, J = 11.6 Hz, 1H, ArCH2), 4.01–3.93 (m, 2H, H-2 and H-4),
3.73–3.68 (m, 2H, H-6a and H-6b), 3.65 (dd, J = 9.6, 3.2 Hz, 1H, H-3), 3.39 (s, 3H, OCH3), 2.84
(br s, 1H, C4-OH), 2.61 (d, J = 7.6 Hz, 1H, C2-OH); 13C NMR (100 MHz, CDCl3): δ 137.8,
128.6, 128.1, 127.8, 99.9, 77.9, 72.2, 68.6, 66.9, 61.7, 55.5; HRMS m/z calcd for C13H22NO5
[M+NH4]+: 272.1152. Found 272.1154.
Methyl-3-O-(4-bromobenzyl)-β-L-arabinopyranoside (2.41)
Methyl-β-L-arabinopyranoside 2.09 (32.8 mg, 0.200 mmol), 4-bromobenzyl bromide (75.0 mg,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (1 mL, 0.2 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (56.1 mg, 84%); Rf = 0.20 (pentanes/EtOAc, 70:30); FTIR (νmax) 3445 (br), 2918
(br), 1592 (w), 1487 (m), 1340 (m), 1265 (m), 1140 (s), 1062 (s), 891 (m), 735 (s) cm-1; 1H
NMR (400 MHz, CDCl3): δ 7.41 (apparent dt, J = 8.8, 2.4 Hz, 2H, ArH), 7.18 (apparent dt, J =
8.8, 2.4 Hz, 2H, ArH), 4.70 (d, J = 3.6 Hz, 1H, H-1), 4.61 (d, J = 1.2 Hz, 1H, ArCH2), 3.92–3.86
O
OMeHOBnO
HO
O
OMeHOO
HO
Br

124
(m, 2H, H-2 and H-4), 3.67–3.60 (m, 2H, H-5a and H-5b), 3.53 (dd, J = 9.2, 3.2 Hz, 1H, H-3),
3.33 (s, 3H, OCH3), 2.54 (s, 1H, C4-OH), 2.22 (d, J = 8.0 Hz, 1H, C2-OH); 13C NMR (100 MHz,
CDCl3): δ 136.9, 131.7, 129.4, 121.9, 99.9, 77.9, 71.4, 68.7, 67.0, 61.8, 55.5; HRMS m/z calcd
for C13H21BrNO5 [M+NH4]+: 350.0603. Found 350.0586.
Methyl-3-O-(2-naphthyl)methyl-β-L-arabinopyranoside (2.42)
Methyl-β-L-arabinopyranoside 2.09 (32.8 mg, 0.200 mmol), 2-(bromomethyl)naphthalene (66.3
mg, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (42.9 mg, 71%); Rf = 0.21 (pentanes/EtOAc, 30:70); FTIR (νmax) 3447 (br), 2918
(m), 1604 (w), 1444 (w), 1343 (m), 1135 (s), 1060 (s), 924 (m), 859 (s), 699 (m) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.87–7.82 (m, 4H, ArH), 7.47–7.54 (m, 3H, ArH), 4.90 (d, J = 12.0 Hz,
1H, ArCH2), 4.86 (d, J = 12.0 Hz, 1H, ArCH2), 4.81 (d, J = 3.6 Hz, 1H, H-1), 4.05 (ddd, J = 9.2,
8.0, 3.6 Hz, 1H, H-2), 4.04–3.97 (m, 1H, H-4), 3.79–3.68 (m, 3H, H-3, H-5a and H-5b), 3.41 (s,
3H, OCH3), 2.79 (s, 1H, C4-OH), 2.45 (d, J = 8.0 Hz, 1H, C2-OH); 13C NMR (100 MHz,
CDCl3): δ 135.3, 133.2, 133.1, 128.5, 127.9, 127.7, 126.7, 126.3, 126.1, 125.7, 100.0, 77.7, 72.3,
68.7, 67.0, 61.8, 55.5; HRMS m/z calcd for C17H24NO5 [M+NH4]+: 322.1654. Found 322.1642.
Methyl-3-O-benzyloxymethyl-β-L-arabinopyranoside (2.43)
Methyl-β-L-arabinopyranoside 2.09 (32.8 mg, 0.200 mmol), benzyloxymethyl chloride (41.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
O
OMeHOO
HO
O
OMeHOBOMO
HO

125
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
pale yellow oil (46.6 mg, 82%); Rf = 0.23 (pentanes/EtOAc, 30:70); FTIR (νmax) 3431 (br), 2896
(m), 1454 (w), 1342 (w), 1192 (w), 1115 (m), 1139 (s), 1040 (s), 844 (w), 734 (s) cm-1; 1H NMR
(400 MHz, CDCl3): δ 7.39–7.29 (m, 5H, ArH), 4.96 (d, J = 6.8 Hz, 1H, OCH2O), 4.93 (d, J =
6.8 Hz, 1H, OCH2O), 4.80 (d, J = 3.6 Hz, 1H, H-1), 4.72 (d, J = 11.6 Hz, 1H, ArCH2O), 4.67 (d,
J = 11.6 Hz, 1H, ArCH2O), 4.02–3.98 (m, 1H, H-4), 3.98–3.91 (m, 1H, H-2), 3.81 (dd, J = 9.6,
3.6 Hz, 1H, H-3), 3.78–3.69 (m, 2H, H-5a and H-5b), 3.42 (s, 3H, OCH3), 2.57–2.53 (m, 2H, C4-
OH and C2-OH); 13C NMR (100 MHz, CDCl3): δ 137.2, 128.6, 128.0, 127.9, 99.9, 94.8, 77.6,
70.3, 68.3, 61.8, 55.5; HRMS m/z calcd for C14H24NO6 [M+NH4]+: 302.1603. Found 302.1592.
3,6-Di-O-(4-bromobenzyl)-D-galactal (2.44)
D-Galactal 2.10 (29.2 mg, 0.200 mmol), 4-bromobenzyl bromide (124.9 mg, 0.500 mmol), 2-
aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (97.3 mg, 0.420 mmol) were stirred
in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr. Flash chromatographic
purification (pentanes/EtOAc, 85:15) afforded the title compound as a colourless oil (76.1 mg,
79%); Rf = 0.46 (pentanes/EtOAc, 70:30); FTIR (νmax) 3558 (br), 3073 (br), 2859 (w), 1648 (m),
1400 (m), 1233 (s), 1160 (m), 1011 (s), 871 (s), 744 (m) cm-1; 1H NMR (400 MHz, CDCl3): δ
7.49–7.46 (m, 4H, ArH), 7.24–7.20 (m, 4H, ArH), 6.42 (dd, J = 6.4, 1.6 Hz, 1H, H-1), 4.70 (dd,
J = 6.4, 2.0 Hz, 1H, H-2), 4.61–4.50 (m, 4H, ArCH2), 4.20–4.18 (m, 1H, H-3), 4.09–4.07 (m,
1H, H-4), 4.02 (apparent t, J = 6.0 Hz, 1H, H-5), 3.78 (d, J = 6.0 Hz, 2H, H-6), 2.50 (d, J = 3.6
Hz, 1H, C4-OH); 13C NMR (100 MHz, CDCl3): δ 145.1, 136.8, 136.6, 131.7, 131.6, 129.4,
129.3, 121.9, 121.7, 99.2, 75.3, 72.9, 70.8, 69.7, 69.3, 63.0; HRMS m/z calcd for C20H21Br2O4
[M+H]+: 482.9806. Found 482.9816.
OOHO
O
Br
Br

126
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-allyl-α-D-mannopyranoside (2.45)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol), allyl
bromide (25.9 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O
(50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40
°C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:15) afforded the title
compound as a colourless oil (36.9 mg, 53%); Rf = 0.43 (pentanes/EtOAc, 70:30); 1H NMR (400
MHz, CDCl3): δ 5.98 (dddd, J = 16.0, 11.6, 10.4, 5.6 Hz, 1H, CH2CHCH2), 5.32 (dq, J = 16.0,
1.2 Hz, 1H, CH2CHCH2), 5.21 (dq, J = 10.4, 1.2 Hz, 1H, CH2CHCH2), 4.73 (d, J = 1.6 Hz, 1H,
H-1), 4.11–4.22 (m, 2H, CH2CHCH2), 3.98–3.95 (m, 1H, H-2), 3.91–3.81 (m, 3H, H-3, H-4 and
H-5), 3.63–3.57 (m, 2H, H-6a and H-6b), 3.36 (s, 3H, OCH3), 3.03 (d, J = 1.6 Hz, 1H, C4-OH),
2.40 (d, J = 2.8 Hz, 1H, C2-OH), 0.90 (s, 9H, Si(C(CH3)3)(CH3)2), 0.09 (s, 6H,
Si(C(CH3)3)(CH3)2).
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-allyl-α-D-galactopyranoside (2.46)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-galactopyranoside 2.05 (61.7 mg, 0.200 mmol), allyl
bromide (25.9 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O
(50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40
°C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 85:15) afforded the title
compound as a colourless oil (11.8 mg, 17%); Rf = 0.43 (pentanes/EtOAc, 70:30); 1H NMR (400
MHz, CDCl3): δ 5.94 (dddd, J = 16.0, 11.6, 10.4, 5.6 Hz, 1H, CH2CHCH2), 5.28 (dq, J = 16.0,
1.2 Hz, 1H, CH2CHCH2), 5.18 (dq, J = 10.4, 1.2 Hz, 1H, CH2CHCH2), 4.41 (ddt, J =12.8, 5.6,
1.2 Hz, 1H, CH2CHCH2), 4.73 (d, J = 7.6 Hz, 1H, H-1), 4.16 (ddt, J =12.8, 5.6, 1.2 Hz, 1H,
CH2CHCH2), 4.03–4.05 (m, 1H, H-4), 3.93 (dd, J = 10.4, 6.4 Hz, 1H, H-6a), 3.85 (dd, J = 10.4,
O
OTBS
HOAllylO
OH
OMe
O
OTBS
AllylO
OMe
OH
HO

127
5.2 Hz, 1H, H-6b), 3.60–3.55 (m, 1H, H-2), 3.53 (s, 3H, OCH3), 3.47–3.44 (m, 1H, H-5), 3.44–
3.37 (m, 1H, H-3), 2.68 (d, J = 1.6 Hz, 1H, C4-OH), 2.59 (d, J = 2.8 Hz, 1H, C2-OH), 0.90 (s,
9H, Si(C(CH3)3)(CH3)2), 0.09 (s, 6H, Si(C(CH3)3)(CH3)2).
Methyl-3-O-allyl-α-L-fucopyranoside (2.47)
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), allyl bromide (25.9 µL, 0.300 mmol),
2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220 mmol) were
stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr. Flash
chromatographic purification (pentanes/EtOAc, 60:40) afforded the title compound as a
colourless oil (24.4 mg, 56%); Rf = 0.23 (pentanes/EtOAc, 60:40); 1H NMR (400 MHz,
CDCl3): δ 5.94 (dddd, J = 16.0, 11.6, 10.4, 5.6 Hz, 1H, CH2CHCH2), 5.32 (dq, J = 16.0, 1.2 Hz,
1H, CH2CHCH2), 5.19 (dq, J = 10.4, 1.2 Hz, 1H, CH2CHCH2), 4.76 (d, J = 7.6 Hz, 1H, H-1),
4.20–4.15 (m, 2H, CH2CHCH2), 4.03–3.81 (m, 3H, H-2, H-4 and H-5), 3.54 (dd, J = 10.0, 3.6
Hz, H-3), 3.41 (s, 3H, OCH3), 2.49 (d, J = 8.0 Hz, 1H, C2-OH), 2.34 (s, 1H, C4-OH), 1.29 (d, J =
6.8 Hz, 3H, CHCH3).
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(4-methoxybenzyl)-α-D-mannopyranoside (2.48)
Methyl-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 2.01 (61.7 mg, 0.200 mmol), 4-
methoxybenzyl bromide (40.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol
%) and Ag2O (50.9 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon
atmosphere at 40 °C for 48 hr. Flash chromatographic purification (pentanes/EtOAc, 80:20)
afforded the title compound as a colourless oil (36.0 mg, 42%); Rf = 0.34 (pentanes/EtOAc,
70:30); 1H NMR (400 MHz, CDCl3): δ 7.24–7.16 (m, 2H, ArH), 6.82–6.77 (m, 2H, ArH), 4.63
O
OMeMe
OHOAllyl
OH
O
OTBS
HOPMBO
OH
OMe

128
(d, J = 1.2 Hz, 1H, H-1), 4.53 (s, 2H, ArCH2), 3.87–3.83 (m, 1H, H-2), 3.79–3.73 (m, 3H, H-4,
H-6a and H-6b), 3.71 (s, 3H, ArOCH3), 3.57 (dd, J = 9.2, 3.2 Hz, 1H, H-3), 3.50 (apparent dt,
J = 10.0, 5.2 Hz, 1H, H-5), 3.27 (s, 3H, OCH3), 2.84 (d, J = 1.6 Hz, 1H, C4-OH), 2.32 (d, J = 2.8
Hz, 1H, C2-OH), 0.90 (s, 9H, Si(C(CH3)3)(CH3)2), 0.09 (s, 6H, Si(C(CH3)3)(CH3)2).
1,6-Anhydro-2-O-(4-methoxybenzyl)-β-D-mannopyranoside (2.49)
1,6-Anhydro-β-D-mannopyranoside 2.02 (32.4 mg, 0.200 mmol), 4-methoxybenzyl bromide
(40.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (34.4 mg, 61% yield); Rf = 0.21 (pentanes/EtOAc, 40:60); 1H NMR (400 MHz,
CDCl3): δ 7.20–7.16 (m, 2H, ArH), 6.85–6.80 (m, 2H, ArH), 5.27–5.24 (m, 1H, H-1), 4.54 (d,
J = 11.2 Hz, 1H, ArCH2), 4.46 (d, J = 11.2 Hz, 1H, ArCH2), 4.36–4.39 (m, 1H, H-5), 4.11 (dd,
J = 7.2, 0.8 Hz, 1H, H-6a), 3.75 (s, 3H, OCH3), 3.74–3.73 (m, 1H, H-3), 3.70–3.65 (m, 2H, H-4
and H-6b), 3.60 (ddd, J = 11.2, 5.6, 2.0 Hz, 1H, H-2), 2.96 (d, J = 11.6 Hz, 1H, C3-OH), 2.27 (d,
J = 10.0 Hz, 1H, C4-OH).
Methyl-6-(tert-butyldimethylsilyloxy)-3-O-(4-methoxybenzyl)-β-D-galactopyranoside (2.50)
Methyl-β-D-galactopyranoside 2.06 (61.7 mg, 0.200 mmol), 4-methoxybenzyl bromide (40.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 75:25) afforded the title compound as a
colourless oil (31.7 mg, 37%); Rf = 0.28 (pentanes/EtOAc, 70:30); 1H NMR (400 MHz,
O
OOH
OH
OPMB
O
OTBS
PMBO OMe
OH
OH

129
CDCl3): δ 7.32–7.27 (m, 2H, ArH), 6.91–6.86 (m, 2H, ArH), 4.86 (d, J = 11.2 Hz, 1H, ArCH2),
4.59 (d, J = 11.2 Hz, 1H, ArCH2), 4.26 (d, J = 7.6 Hz, 1H, H-1), 4.01 (apparent t, J = 2.8 Hz, 1H,
H-4), 3.92 (dd, J = 10.4, 6.0 Hz, 1H, H-6a), 3.84 (dd, J = 10.4, 5.6 Hz, 1H, H-6b), 3.81 (s, 3H,
ArOCH3), 3.59–3.51 (m, 1H, H-2), 3.57 (s, 3H, OCH3), 3.52–3.43 (m, 2H, H-3 and H-5), 2.61
(d, J = 3.2 Hz, 1H, C4-OH), 2.32 (d, J = 4.0 Hz, 1H, C2-OH), 0.90 (s, 9H, Si(C(CH3)3)(CH3)2),
0.09 (s, 6H, Si(C(CH3)3)(CH3)2).
1,6-Anhydro-4-O-(4-methoxybenzyl)-β-D-galactopyranoside (2.51)
1,6-Anhydro-β-D-galactopyranoside 2.07 (32.4 mg, 0.200 mmol), 4-methoxybenzyl bromide
(40.7 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48
hr. Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (33.3 mg, 59% yield); Rf = 0.23 (pentanes/EtOAc, 40:60); 1H NMR (400 MHz,
CDCl3): δ 7.29–7.24 (m, 2H, ArH), 6.92–6.88 (m, 2H, ArH), 5.38–5.36 (m, 1H, H-1), 4.62 (d,
J = 11.6 Hz, 1H, ArCH2), 4.56 (d, J = 11.6 Hz, 1H, ArCH2), 4.37 (apparent t, J = 5.6 Hz, 1H, H-
5), 4.28 (d, J = 7.2 Hz, 1H, H-6a), 4.01–3.96 (m, 1H, H-3), 3.81 (s, 3H, OCH3), 3.81–3.78 (m,
2H, H-2 and H-4), 3.66–3.61 (m, 1H, H-6b), 2.68 (d, J = 2.0 Hz, 1H, C3-OH), 2.06 (d, J = 9.6
Hz, 1H, C2-OH).
Methyl-3-O-(4-methoxybenzyl)-α-L-fucopyranoside (2.52)
Methyl-α-L-fucopyranoside 2.08 (35.6 mg, 0.200 mmol), 4-methoxybenzyl bromide (40.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
O
O
PMBOOH
OH
O
OMeMe
OHOPMB
OH
130
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (37.0 mg, 62% yield); Rf = 0.25 (pentanes/EtOAc, 40:60); 1H NMR (400 MHz,
CDCl3): δ 7.35–7.29 (m, 2H, ArH), 6.94–6.88 (m, 2H, ArH), 4.79 (d, J = 4.0 Hz, 1H, H-1), 4.67
(s, 2H, ArCH2), 3.98–3.93 (m, 1H, H-2), 3.92–3.86 (m, 1H, H-5), 3.82 (s, 3H, ArOCH3), 3.84–
3.80 (m, 1H, H-4), 3.63 (dd, J = 9.6, 3.2 Hz, 1H, H-3), 3.43 (s, 3H, OCH3), 2.41–2.39 (m, 1H,
C4-OH), 2.16 (d, J = 8.0 Hz, 1H, C2-OH), 1.31 (d, J = 6.4 Hz, 3H, CHCH3).
Methyl-3-O-(4-methoxybenzyl)-β-L-arabinopyranoside (2.53)
Methyl-β-L-arabinopyranoside 2.09 (32.8 mg, 0.200 mmol), 4-methoxybenzyl bromide (40.7 µL,
0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %) and Ag2O (50.9 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 40:60) afforded the title compound as a
colourless oil (23.9 mg, 42% yield); 1H NMR (400 MHz, CDCl3): δ 7.32–7.27 (m, 2H, ArH),
6.91–6.87 (m, 2H, ArH), 4.79 (d, J = 3.6 Hz, 1H, H-1), 4.87 (d, J = 11.2 Hz, 1H, ArCH2), 4.63
(d, J = 11.2 Hz, 1H, ArCH2), 3.99–3.92 (m, 2H, H-2 and H-4), 3.81 (s, 3H, ArOCH3), 3.73–3.71
(m, 2H, H-5a and H-5b), 3.63 (dd, J = 9.2, 3.2 Hz, 1H, H-3), 3.42 (s, 3H, OCH3), 2.49 (s, 1H,
C4-OH), 2.11 (d, J = 7.6 Hz, 1H, C2-OH).
Isopropyl-6-(tert-butyldimethylsilyloxy)-β-D-thiogalactopyranoside (2.55)
Isopropyl-6-(tert-butyldimethylsilyloxy)-β-D-thiogalactopyranoside 2.54 (69.6 mg, 0.200 mmol),
benzyl bromide (35.6 µL, 0.300 mmol), 2-aminoethyl diphenylborinate (4.5 mg, 10 mol %),
tetrabutylammonium iodide (73.9 mg, 0.200 mmol) and potassium carbonate (30.4 mg, 0.220
mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 40 °C for 48 hr.
Flash chromatographic purification (pentanes/EtOAc, 85:25) afforded the title compound as a
OPMBO OMe
OH
OH
OOTBSHO
BnOHO
SiPr

131
colourless oil (28.0 mg, 32%); Rf = 0.34 (pentanes/EtOAc, 70:30); 1H NMR (400 MHz,
CDCl3): δ 7.35–7.15 (m, 5H, ArH), 4.72 (d, J = 11.6 Hz, 1H, ArCH2), 4.68 (d, J = 11.6 Hz, 1H,
ArCH2), 4.30 (d, J = 9.6 Hz, 1H, H-1), 4.00–3.97 (m, 1H, H-4), 3.80 (dd, J = 10.4, 6.4 Hz, 1H,
H-6a), 3.76–3.72 (m, 2H, H-2 and H-6b), 3.51 (apparent t, J = 5.6 Hz, 1H, H-5), 3.39–3.33 (m,
2H, H-3 and C2-OH), 3.20–3.09 (m, 1H, C4-OH), 2.59 (t, J = 5.6 Hz, 1H, SCH(CH3)2), 1.26 (d, J
= 6.8, 1.2 Hz, 6H, SCH(CH3)2), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H,
Si(C(CH3)3)(CH3)2).
5.1.5 General Procedure E: Borinic Acid-Catalyzed Glycosylation
The glycosyl donor (1.0 equiv), glycosyl acceptor (1.1 equiv), Ag2O (1.0 equiv) and 2-
aminoethyl diphenylborinate (10 mol %) were added to an oven-dried round bottom flask, under
an argon atmosphere. Dry acetonitrile (0.13 M) was added and the resulting mixture was stirred
vigorously (750-1000 rpm). The reaction was then diluted with dichloromethane and filtered
through a plug of Celite®. The resulting crude material was purified by flash chromatography on
silica gel.
Carbohydrate substrates 3.01108, 3.0293, 3.0394, 3.04109, 3.0595, 3.0695, 3.0796, 3.0896 were
prepared following literature methods. Spectral data were consistent with those presented in the
literature.
5.1.6 General Procedure F: Borinic Acid-Catalyzed Synthesis of Sugar Substrates
Bearing a Carbonate at O-2 and O-3
The sugar substrate (1 equiv), 1,1’-carbonyl diimidazole (1.1 equiv), 2-aminoethyl
diphenylborinate (10 mol %) and diisopropylethylamine (1.1 equiv) were stirred in acetonitrile
(0.2 M) under argon atmosphere at 40 °C for 16 hr. The reaction was subsequently diluted with
methanol and washed with water twice. The organic layer was dried (MgSO4), concentrated and
purified by silica gel chromatography.
108 Bianchi, A.; Bernardi, A. J. Org. Chem. 2006, 71, 4565–4577. 109 Mitchell, S. A.; Pratt, M. R.; Hruby, V. J.; Polt, R. J. Org. Chem. 2001, 66, 2327–2342.

132
Methyl-2,3-O-carbonate-α-L-rhamnopyranoside (3.16)
Methyl-2,3-O-carbonate-α-L-rhamnopyranoside was synthesized following General Procedure F
using methyl-α-L-rhamnopyranoside. Flash chromatographic purification (pentanes/EtOAc,
80:20) afforded the title compound as a colourless oil (72%); Rf = 0.22 (pentanes/EtOAc, 80:20); 1H NMR (400 MHz, CDCl3): δ 4.85 (s, 1H, H-1), 4.61 (d, J = 6.4 Hz, 1H, H-3), 4.56 (dd, J =
7.2, 0.9 Hz, 1H, H-2), 3.75–3.66 (m, 1H, H-5), 3.49 (dd, J = 8.4, 6.4 Hz, 1H, H-4), 3.36 (s, 3H,
OCH3), 2.55 (br s, 1H, C4-OH), 1.29 (d, J = 6.4 Hz, 3H, CHCH3).
Methyl-2,3-O-carbonate-4-O-benzoyl-α-L-rhamnopyranoside (3.17)
To a solution of methyl-2,3-O-carbonate-α-L-rhamnopyranoside 3.16 (355.9 mg, 1.0 mmol) in
pyridine (0.5 M) was added benzoyl chloride (127.8 µL, 1.1 mmol). The mixture was stirred
overnight at room temperature and was then diluted in dichloromethane. After washing twice
with water, the organic layer was dried over MgSO4, filtered, and concentrated in vacuo. Toluene
was added and the solution was concentrated in vacuo to azeotrope pyridine. The resulting crude
material was purified by silica gel chromatography (pentane/EtOAc, 90:10) to yield the desired
product as a pale yellow solid in quantitative yield; Rf = 0.29 (pentanes/EtOAc, 85:15); 1H NMR
(400 MHz, CDCl3): δ 7.99–7.96 (m, 2H, ArH), 7.57–7.52 (m, 1H, ArH), 7.43–7.37 (m, 2H,
ArH), 5.08 (dd, J = 9.6, 6.8 Hz, 1H, H-4), 4.95 (s, 1H, H-1), 4.83 (apparent t, J = 7.2 Hz, 1H, H-
3), 4.62 (d, J = 7.2 Hz, 1H, H-2), 3.95–3.86 (m, 1H, H-5), 3.49 (s, 3H, OCH3), 1.24 (d, J = 6.4
Hz, 3H, CHCH3).
O
OMeMe
HOO OO
O
OMeMe
BzOO O
O

133
Methyl-2,3-O-carbonate-4-O-benzoyl-α-L-rhamnosyl bromide (3.18)
To a solution of methyl-2,3-O-carbonate-4-O-benzoyl-α-L-rhamnopyranoside 3.17 (308.1mg, 1.0
mmol) was added hydrogen bromide (33% solution in acetic acid, 2.1 mL). The mixture was
stirred overnight hours at room temperature. The crude mixture was diluted in dichloromethane
and washed twice with ice-cold water to pH neutral. The organic layer was dried over MgSO4,
filtered, and concentrated in vacuo. The resulting crude material was purified over a plug of
silica (10 cm, pentanes/EtOAc, 80:20) to yield the desired product as a yellow solid in 45%
yield; Rf = 0.27 (pentanes/EtOAc, 80:20); 1H NMR (400 MHz, CDCl3): δ 8.10–8.04 (m, 2H,
ArH), 7.65–7.60 (m, 1H, ArH), 7.51–7.45 (m, 2H, ArH), 6.65 (s, 1H, H-1), 5.25 (dd, J = 9.6, 6.8
Hz, 1H, H-4), 5.14–5.06 (m, 2H, H-2 and H-3), 4.31–4.22 (m, 1H, H-5), 1.35 (d, J = 6.4 Hz, 3H,
CHCH3).
Methyl-2,3-O-carbonate-α-D-mannopyranoside (3.19)
Methyl-2,3-O-carbonate-6-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside was synthesized
following General Procedure F using methyl-6-(tert-butyldimethylsilyloxy)-α-D-
mannopyranoside. Flash chromatographic purification (pentanes/EtOAc, 80:20) afforded the title
compound as a colourless oil (>99%); Rf = 0.26 (pentanes/EtOAc, 80:20); 1H NMR (400 MHz,
CDCl3): δ 4.59 (d, J = 1.6 Hz, 1H, H-1), 3.81–3.75 (m, 2H, H-2 and H-3), 3.73–3.75 (m, 3H, H-
5, H-6a and H-6b), 3.46 (dt, J = 9.2, 5.2 Hz, 1H, H-4), 3.26 (s, 3H, OCH3), 3.23 (br s, 1H, C4-
OH), 0.89 (s, 9H, Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2).
O
BrMe
BzOO O
O
O
OTBS
HO OO
OMe
O

134
5.1.7 General Procedure G: Borinic Acid-Catalyzed Glycosylation with Halide
Catalysis
The glycosyl donor (1.0 equiv), glycosyl acceptor (1.1 equiv), 2-aminoethyl diphenylborinate (10
mol %), halide salt (1.0 equiv), potassium carbonate (1.1 equiv) were weighed into a 1-dram vial,
and dissolved in dry acetonitrile (0.2 M). The reaction vessel was capped with a septum and
purged with argon. The mixture was stirred vigorously (750-1000 rpm) and for the allotted time.
The resulting mixture was diluted with dichloromethane, filtered through Celite® and
concentrated to dryness. The resulting crude material was purified by flash chromatography on
silica gel.
Methyl-6-O-tert-butyldimethylsilyl-3-O-(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-α-D-
mannopyranoside (3.21)
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (82.0 mg, 0.200 mmol), methyl-6-O-tert-
butyldimethysilyl-α-D-mannopyranose (67.8 mg, 0.220 mmol), 2-aminoethyl diphenylborinate
(4.5 mg, 10 mol %), tetrabutylammonium iodide (73.9 mg, 0.200 mmol) and potassium
carbonate (30.4 mg, 0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon
atmosphere at 80 °C for 24 hr. Flash chromatographic purification (toluene/iPrOH, 95:5)
afforded the title compound as a white solid (48.5 mg, 38%); Rf = 0.40 (toluene/iPrOH, 90:10); 1H NMR (400 MHz, CDCl3): δ 5.23 (dd, J = 9.6, 9.6 Hz, 1H, H-3'), 5.06–5.01 (m, 2H, H-4' and
H-2'), 4.73 (d, J = 1.6 Hz, 1H, H-1), 4.61 (d, J = 8.0 Hz, 1H, H-1'), 4.21 (dd, J = 12.4, 2.4 Hz,
1H, H-6a'), 4.15 (dd, J = 12.4, 6.0 Hz, 1H, H-6b'), 3.96 (dd, J = 11.2, 2.4 Hz, 1H, H-6a), 3.83–
3.69 (m, 5H, H-6b, H-2, H-3, H-5' and H-4), 3.54 (ddd, J = 8.8, 6.0, 2.4 Hz, 1H, H-5), 3.37 (s,
3H, OCH3), 3.36 (d, J = 1.6 Hz, 1H, C4-OH), 2.21 (d, J = 3.6 Hz, 1H, C2-OH), 2.08 (s, 3H,
OCOCH3), 2.06 (s, 3H, OCOCH3), 2.03 (s, 3H, OCOCH3), 2.01 (s, 3H, OCOCH3), 0.89 (s, 9H,
Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2).
OOTBSHO
HOOMe
O
OAc
AcOAcO AcO
O

135
Methyl-6-O-tert-butyldimethylsilyl-3-O-(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-α-D-
galactopyranoside (3.22)
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (82.0 mg, 0.200 mmol), methyl-6-O-tert-
butyldimethysilyl-α-D-galactopyranose (67.8 mg, 0.220 mmol), 2-aminoethyl diphenylborinate
(4.5 mg, 10 mol %), potassium iodide (33.2 mg, 0.200 mmol) and potassium carbonate (30.4 mg,
0.220 mmol) were stirred in acetonitrile (2 mL, 0.1 M) under argon atmosphere at 80 °C for 24
hr. Flash chromatographic purification (toluene/iPrOH, 95:5) afforded the title compound as a
white solid (37.0 mg, 29%); Rf = 0.39 (toluene/iPrOH, 90:10); 1H NMR (400 MHz, CDCl3): δ
5.23 (dd, J = 9.6, 9.6 Hz, 1H, H-3'), 5.07 (dd, J = 10, 9.6 Hz, 1H, H-4'), 5.02 (dd, J = 10, 8 Hz,
1H, H-2'), 4.81 (d, J = 8 Hz, 1H, H-1'), 4.79 (d, J = 3.6 Hz, 1H, H-1), 4.23 (dd, J = 12.4, 4.8 Hz,
1H, H-6a'), 4.12 (dd, J = 12.4, 2.4 Hz, 1H, H-6b'), 4.06 (d, J = 3.2 Hz, 1H, H-4), 3.97 (dd, J =
9.2, 3.6 Hz, 1H, H-2), 3.85 (ddd, J = 8.8, 4.8, 4.8 Hz, 1H, H-5), 3.80−3.74 (m, 3H, H-6a, H-6b,
and H-3), 3.71 (ddd, J = 10, 4.8, 2.4 Hz, 1H, H-5'), 3.42 (s, 3H, OCH3), 2.08 (s, 3H, OCOCH3),
2.05 (s, 3H, OCOCH3), 2.02 (s, 3H, OCOCH3), 2.01 (s, 3H, OCOCH3), 0.88 (s, 9H,
Si(C(CH3)3)(CH3)2), 0.07 (s, 6H, Si(C(CH3)3)(CH3)2).
5.1.8 General Procedure H: Borinic Acid-Mediated Glycosylation
In a large test tube with a screw cap, the glycosyl acceptor (1.0 equiv) and boronic acid (1.0
equiv) was refluxed in toluene (0.2 M) for 16 hrs after which the solvent was then removed. To
the crude was then added the glycosyl donor (1.0 equiv), Ag2O (1.0 equiv), triethylamine (1.0
equiv) and dry acetonitrile (0.2 M). The reaction vessel was capped with a septum and purged
with argon. The mixture was stirred vigorously (750-1000 rpm) at room temperature for 16 hrs.
The resulting mixture was quenched with MeOH and extracted with water twice. The organic
layer was dried over MgSO4, filtered, and concentrated in vacuo. The resulting crude material
was purified by flash chromatography on silica gel.
OOTBSHO
HOOMe
O
OAc
AcOAcO AcO
O

136
Methyl-3-O-(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-α-L-rhamnopyranoside (3.23)
Methyl-3-O-(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-α-L-rhamnopyranoside was
synthesized following General Procedure H using 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl
bromide (1.0 equiv), methyl-α-L-rhamnopyranoside (1.0 equiv) and 8-quinolinyl borinic acid
(1.0 equiv). Flash chromatographic purification (pentanes/EtOAc, 60:40) afforded the title
compound as a white solid (82%); Rf = 0.26 (pentanes/EtOAc, 60:40); 1H NMR (400 MHz,
CDCl3): δ 5.38 (dd, J = 1.2, 1.2 Hz, 1H, H-1), 5.26 (dd, J = 3.4, 0.7 Hz, 1H, H-4'), 5.20 (dd,
J = 10.5, 7.9 Hz, 1H, H-2'), 5.03 (dd, J = 10.5, 3.4 Hz, 1H, H-3'), 4.54 (apparent t, J = 5.2 Hz,
1H, H-5), 4.50 (d, J = 7.9 Hz, 1H, H-1'), 4.29 (d, J = 7.6 Hz, 1H, H-6a), 4.04–4.00 (m, 2H, H-4
and H-3), 3.89 (qd, J = 6.4, 0.7 Hz, 1H, H-5'), 3.83 (br s, 1H, H-2), 3.67 (dd, J = 7.2, 5.2 Hz, 1H,
H-6b), 2.28 (d, J = 1.9 Hz, 1H, C2-OH), 2.09 (s, 3H, OCOCH3), 2.05 (s, 3H, OCOCH3), 2.03 (s,
3H, OCOCH3), 2.01 (s, 3H, OCOCH3), 1.24 (d, J = 6.4 Hz, 3H, C6'-CH3).
5.1.9 General Procedure I: Glycosylation Kinetic Experiments
Into an oven-dried 1 dram vial were added 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide,
methyl-6-O-tert-butyldimethysilyl-α-D-mannopyranose, 2-aminoethyl diphenylborinate and
Ag2O. The reaction vial was capped with a septum and purged with argon. Dry acetonitrile (0.13
M) was added and the resulting mixture was stirred vigorously (750 rpm) at room temperature.
The initial time of each kinetic run corresponds to the time at which dry acetonitrile was added to
the reaction vial. During the course of the reaction, aliquots of the reaction mixture were
removed and filtered over Celite® to remove the silver oxide, thus stopping the reaction. The
solvent was then removed and the resulting samples were analyzed by 1H-NMR spectroscopy for
the formation of product with mesitylene as an internal standard. Integrations for the internal
standard peak and the anomeric peak of the product were used to calculate moles of product
formed and therefore % conversion.
O
OAc
AcOAcO
AcO
O
OMeMeHO
OOH
3.23

137
Appendix A: NMR Spectra

195
Appendix B: DFT Calculations
Calculations were carried out with the Gaussian ’09 software package,110 on a Linux workstation
equipped with two quad-core AMD Shanghai processors built by HardData, Inc. (Edmonton,
Alberta, Canada). Geometry optimizations of the diphenylborinate esters of 1,6-anhydro-β-D-
mannopyranoside, 3,4,6-tri-O-benzyl-D-mannopyranoside and 1,6-anhydro-β-D-
galactopyranoside were carried out using density functional theory (B3LYP functional), with the
6-311+G** basis set. Condensed Fukui functions were calculated according to the method of
Yang and Mortier:111 a single-point energy calculation of the one-electron-oxidized form was
carried out, and the difference in Mulliken charges between the diphenylborinate adduct and its
oxidized form was calculated for each oxygen atom.
1,6-Anhydro-β-D-mannopyranoside 2,3-diphenylborinate
Atom Mulliken charge f-k
O-2 –0.360 0.137
O-3 –0.288 0.102
O-4 –0.240 0.023
110 Gaussian 09, Revision B.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2009. 111 Yang, W.; Mortier, W. J. J. Am. Chem. Soc. 1986, 108, 5708–5711.
OOO
OH
OB Ph
Ph
123

196
3,4,6-Tri-O-benzyl-β-D-mannopyranoside 2,3-diphenylborinate
Atom Mulliken charge f-k
O-1 –0.264 0.078
O-2 –0.182 0.151
1,6-Anhydro-β-D-galactopyranoside 3,4-diphenylborinate
Atom Mulliken charge f-k
O-2 –0.233 0.024
O-3 –0.263 0.106
O-4 –0.324 0.135
O
OBn
BnOBnO
OO
B PhPh
12
OO
OO
OH
BPh
Ph
123
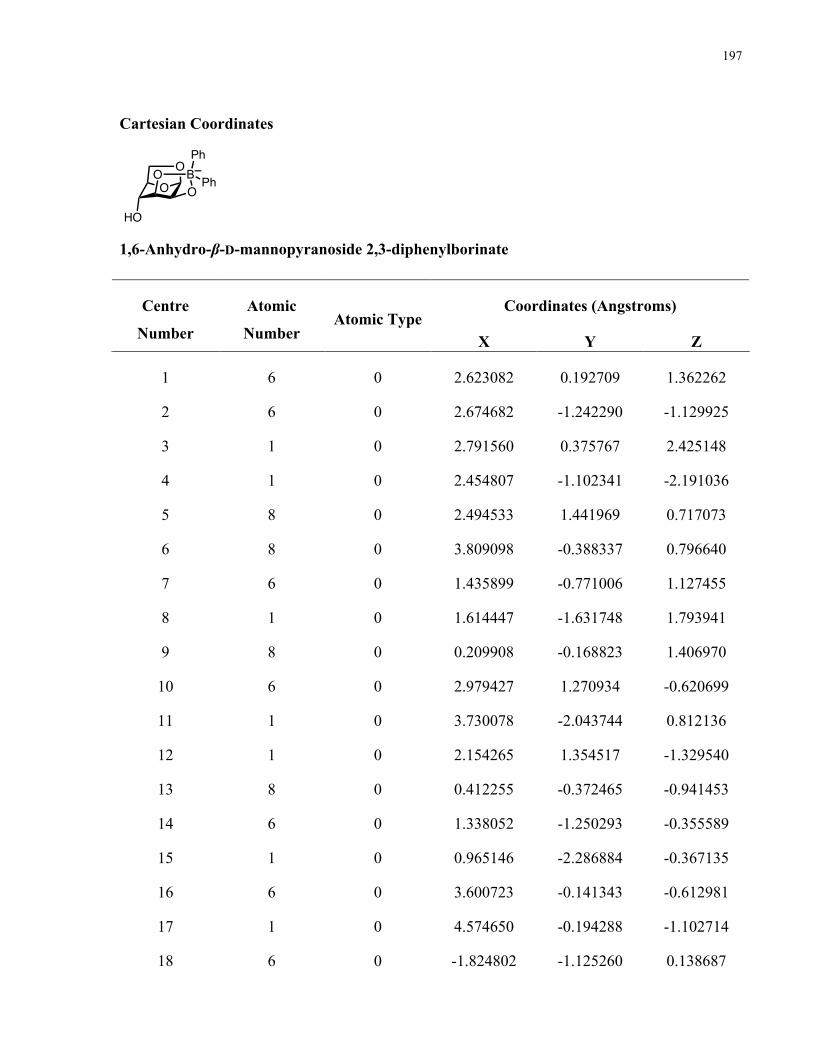
197
Cartesian Coordinates
1,6-Anhydro-β-D-mannopyranoside 2,3-diphenylborinate
Coordinates (Angstroms) Centre
Number
Atomic
Number Atomic Type
X Y Z
1 6 0 2.623082 0.192709 1.362262
2 6 0 2.674682 -1.242290 -1.129925
3 1 0 2.791560 0.375767 2.425148
4 1 0 2.454807 -1.102341 -2.191036
5 8 0 2.494533 1.441969 0.717073
6 8 0 3.809098 -0.388337 0.796640
7 6 0 1.435899 -0.771006 1.127455
8 1 0 1.614447 -1.631748 1.793941
9 8 0 0.209908 -0.168823 1.406970
10 6 0 2.979427 1.270934 -0.620699
11 1 0 3.730078 -2.043744 0.812136
12 1 0 2.154265 1.354517 -1.329540
13 8 0 0.412255 -0.372465 -0.941453
14 6 0 1.338052 -1.250293 -0.355589
15 1 0 0.965146 -2.286884 -0.367135
16 6 0 3.600723 -0.141343 -0.612981
17 1 0 4.574650 -0.194288 -1.102714
18 6 0 -1.824802 -1.125260 0.138687
OOO
HO
OB Ph
Ph
198
19 6 0 -2.582281 -1.366527 1.297989
20 6 0 -2.190448 -1.847969 -1.008421
21 6 0 -3.646329 -2.268281 1.314563
22 1 0 -2.318997 -0.837892 2.209852
23 6 0 -3.252671 -2.755987 -1.007790
24 1 0 -1.614413 -1.700736 -1.917949
25 6 0 -3.990173 -2.969515 0.156462
26 1 0 -4.209062 -2.429180 2.230822
27 1 0 -3.503887 -3.300078 -1.914979
28 1 0 -4.817643 -3.673140 0.163696
29 6 0 -1.148814 1.479399 -0.079671
30 6 0 -0.949524 2.477909 0.885961
31 6 0 -1.851091 1.855789 -1.237463
32 6 0 -1.422135 3.780474 0.711164
33 1 0 -0.396631 2.220056 1.782726
34 6 0 -2.319608 3.155743 -1.430978
35 1 0 -2.037780 1.112247 -2.007683
36 6 0 -2.109822 4.128330 -0.451515
37 1 0 -1.248212 4.528390 1.480813
38 1 0 -2.853832 3.411230 -2.342654
39 1 0 -2.476796 5.140888 -0.593581
40 5 0 -0.591322 -0.037827 0.129333
41 8 0 3.359342 -2.500024 -1.028102
42 1 0 3.682824 -2.556880 -0.120348

199
3,4,6-Tri-O-benzyl-β-D-mannopyranoside 2,3-diphenylborinate
Coordinates (Angstroms) Centre
Number
Atomic
Number Atomic Type
X Y Z
1 6 0 -0.027994 1.622977 -0.669089
2 6 0 1.674403 -0.070443 -1.363629
3 1 0 0.125192 2.164825 -1.615002
4 1 0 1.745600 0.422142 -2.349280
5 8 0 0.675369 -1.074450 -1.374379
6 8 0 -1.557662 -0.028035 0.287644
7 6 0 -1.117062 0.569014 -0.897708
8 1 0 -1.952906 1.050291 -1.436993
9 6 0 1.315877 0.993534 -0.305557
10 1 0 1.243364 0.508232 0.673597
11 8 0 2.344142 1.994952 -0.293238
12 8 0 -1.540368 -1.626577 -1.462904
13 6 0 3.015273 -0.739618 -1.068239
14 1 0 3.756425 0.035913 -0.873024
15 1 0 2.912643 -1.362233 -0.171382
16 8 0 3.542222 -1.496028 -2.156587
17 6 0 2.972330 -2.800999 -2.364433
18 1 0 3.439403 -3.146566 -3.290502
19 1 0 1.894833 -2.721626 -2.516798
O
OBn
BnOBnO
OOB PhPh

200
20 6 0 -0.657735 -0.633055 -1.756168
21 1 0 -0.634453 -0.416438 -2.836892
22 6 0 2.668702 2.497000 0.990619
23 1 0 3.026288 1.674818 1.631600
24 1 0 1.777307 2.917885 1.468625
25 8 0 -0.367566 2.577152 0.345710
26 6 0 -0.919248 3.809190 -0.072247
27 1 0 -0.546082 4.560190 0.635402
28 1 0 -0.526185 4.084773 -1.061720
29 6 0 -2.438914 3.877867 -0.092267
30 6 0 -3.234615 2.839541 0.393552
31 6 0 -3.053985 5.032978 -0.590507
32 6 0 -4.626205 2.952618 0.366159
33 1 0 -2.769656 1.932890 0.765242
34 6 0 -4.441414 5.150700 -0.604792
35 1 0 -2.442341 5.845609 -0.974926
36 6 0 -5.233501 4.105460 -0.126471
37 1 0 -5.228880 2.121789 0.715789
38 1 0 -4.903801 6.050770 -0.997486
39 1 0 -6.315191 4.186262 -0.149151
40 6 0 3.747622 3.550766 0.888869
41 6 0 4.424006 3.806305 -0.305631
42 6 0 4.091658 4.287195 2.028818
43 6 0 5.426440 4.776328 -0.356666
44 1 0 4.154432 3.243853 -1.189939
201
45 6 0 5.093384 5.252567 1.979692
46 1 0 3.568059 4.102561 2.962479
47 6 0 5.766702 5.501659 0.782744
48 1 0 5.941941 4.964148 -1.292781
49 1 0 5.346119 5.813333 2.873454
50 1 0 6.545918 6.255073 0.740773
51 6 0 3.265730 -3.772505 -1.241167
52 6 0 2.268399 -4.124562 -0.326297
53 6 0 4.547052 -4.316633 -1.087728
54 6 0 2.545390 -5.004816 0.721088
55 1 0 1.275887 -3.697606 -0.424727
56 6 0 4.825918 -5.197776 -0.046154
57 1 0 5.328401 -4.041500 -1.789838
58 6 0 3.822598 -5.543578 0.861763
59 1 0 1.759290 -5.253406 1.425051
60 1 0 5.822339 -5.615227 0.058751
61 1 0 4.038613 -6.227911 1.675844
62 6 0 -1.653832 -2.497481 1.004234
63 6 0 -1.804944 -3.870037 0.742598
64 6 0 -1.105924 -2.151007 2.249144
65 6 0 -1.432504 -4.845427 1.667652
66 1 0 -2.224120 -4.180259 -0.210665
67 6 0 -0.735242 -3.116493 3.188721
68 1 0 -0.964388 -1.098962 2.477043
69 6 0 -0.897893 -4.471781 2.902824
202
70 1 0 -1.562055 -5.897894 1.429414
71 1 0 -0.317214 -2.811874 4.144678
72 1 0 -0.614219 -5.226267 3.630879
73 6 0 -3.763421 -1.321736 -0.193018
74 6 0 -4.561123 -1.076962 0.939283
75 6 0 -4.441554 -1.493940 -1.409678
76 6 0 -5.951211 -0.994573 0.864821
77 1 0 -4.079383 -0.953631 1.905816
78 6 0 -5.834427 -1.419151 -1.499678
79 1 0 -3.855621 -1.690985 -2.301832
80 6 0 -6.598052 -1.165771 -0.361379
81 1 0 -6.533777 -0.804497 1.762589
82 1 0 -6.324695 -1.558005 -2.459847
83 1 0 -7.680614 -1.107168 -0.425332
84 5 0 -2.127933 -1.376707 -0.076668
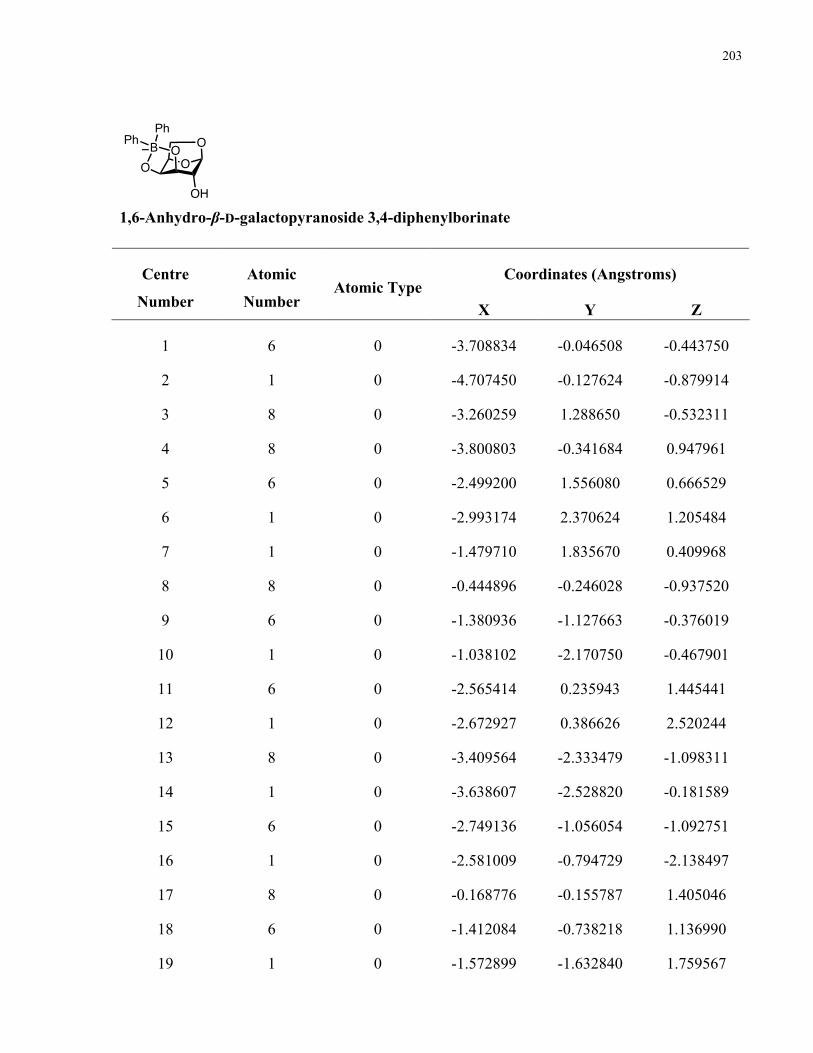
203
1,6-Anhydro-β-D-galactopyranoside 3,4-diphenylborinate
Coordinates (Angstroms) Centre
Number
Atomic
Number Atomic Type
X Y Z
1 6 0 -3.708834 -0.046508 -0.443750
2 1 0 -4.707450 -0.127624 -0.879914
3 8 0 -3.260259 1.288650 -0.532311
4 8 0 -3.800803 -0.341684 0.947961
5 6 0 -2.499200 1.556080 0.666529
6 1 0 -2.993174 2.370624 1.205484
7 1 0 -1.479710 1.835670 0.409968
8 8 0 -0.444896 -0.246028 -0.937520
9 6 0 -1.380936 -1.127663 -0.376019
10 1 0 -1.038102 -2.170750 -0.467901
11 6 0 -2.565414 0.235943 1.445441
12 1 0 -2.672927 0.386626 2.520244
13 8 0 -3.409564 -2.333479 -1.098311
14 1 0 -3.638607 -2.528820 -0.181589
15 6 0 -2.749136 -1.056054 -1.092751
16 1 0 -2.581009 -0.794729 -2.138497
17 8 0 -0.168776 -0.155787 1.405046
18 6 0 -1.412084 -0.738218 1.136990
19 1 0 -1.572899 -1.632840 1.759567
OO
OO
OH
BPh
Ph
204
20 6 0 1.769549 -1.189826 0.035696
21 6 0 2.341995 -1.577621 -1.188092
22 6 0 2.253662 -1.834003 1.185519
23 6 0 3.340491 -2.548797 -1.265091
24 1 0 1.986667 -1.108852 -2.101775
25 6 0 3.257302 -2.804426 1.125629
26 1 0 1.817932 -1.570018 2.144816
27 6 0 3.807624 -3.167534 -0.103435
28 1 0 3.756794 -2.826160 -2.230236
29 1 0 3.607227 -3.282392 2.037366
30 1 0 4.586195 -3.923001 -0.157187
31 6 0 1.255282 1.457527 -0.048719
32 6 0 2.071421 2.009861 0.953469
33 6 0 1.028225 2.252019 -1.183885
34 6 0 2.626466 3.284112 0.837345
35 1 0 2.272665 1.427608 1.848463
36 6 0 1.584558 3.526895 -1.317277
37 1 0 0.389000 1.860758 -1.969061
38 6 0 2.387633 4.051177 -0.304996
39 1 0 3.248142 3.681073 1.635751
40 1 0 1.386993 4.114340 -2.210294
41 1 0 2.819946 5.042856 -0.402190
42 5 0 0.607013 -0.029105 0.104490
138
5.925.92
9.199.19
0.9740.974
0.9740.974
33
1.031.03
1.021.02
3.033.03
11
22
1.011.01
4.634.63
-1-100112233445566778899ppm
O
OMe
OTBSOH
BnOHO
4a
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBSOH
BnOHO
4a
400 MHz, CDCl3
100 MHz, CDCl3
2.11
2.11
139
0 0112233445566778899ppmppm
0
1
2
3
4
5
6
7
8
9
O
OMe
OTBSOH
BnOHO
4a
5.875.87
9.369.36
0.9780.978
0.970.97
3.013.01
1.011.01
11
4.074.07
2.042.04
0.9780.978
1.981.98
1.831.83
-1-100112233445566778899ppm
O
OMe
OTBSOH
OHOBr
5a
400 MHz, CDCl3
400 MHz, CDCl3
2.11
2.12
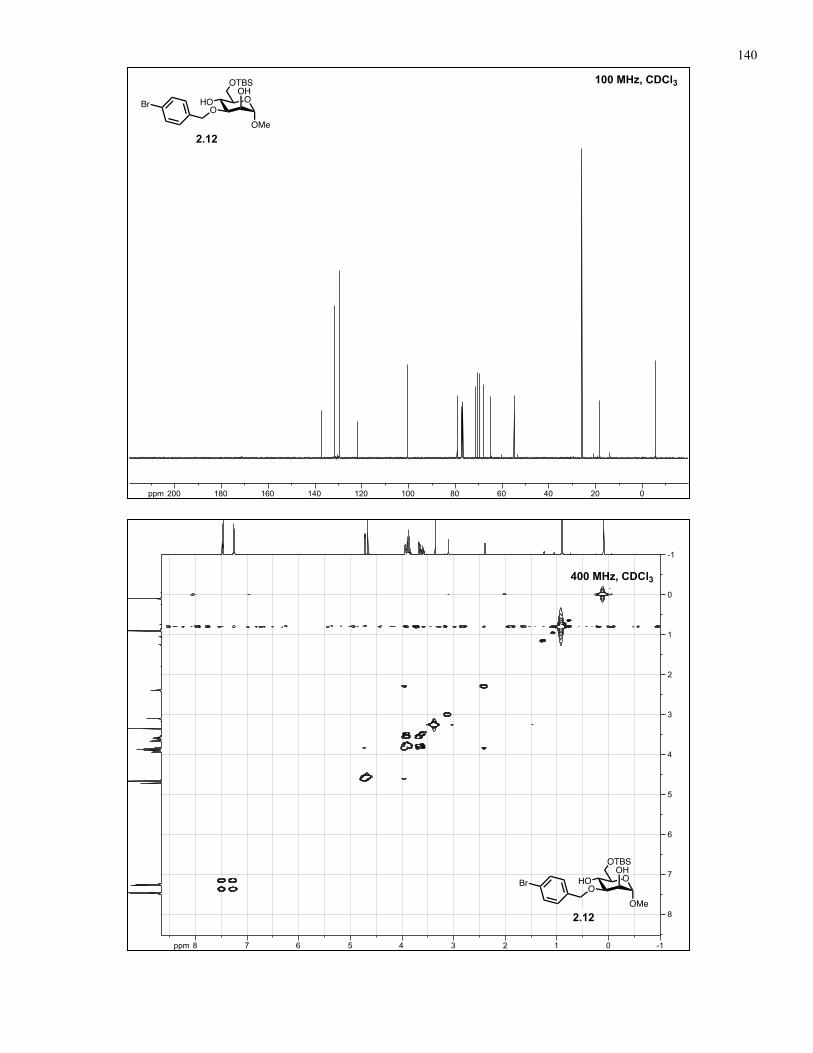
140
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBSOH
OHOBr
5a
-1 -1001122334455667788ppmppm
-1
0
1
2
3
4
5
6
7
8
O
OMe
OTBSOH
OHOBr
5a
400 MHz, CDCl3
100 MHz, CDCl3
2.12
2.12

141
5.825.82
9.19.1
0.9680.968
0.9620.962
2.992.99
11
11
33
11
0.9820.982
22
2.862.86
3.873.87
-1-100112233445566778899ppm
O
OMe
OTBSOH
OHO
6a
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBSOH
OHO
6a
400 MHz, CDCl3
100 MHz, CDCl3
2.13
2.13
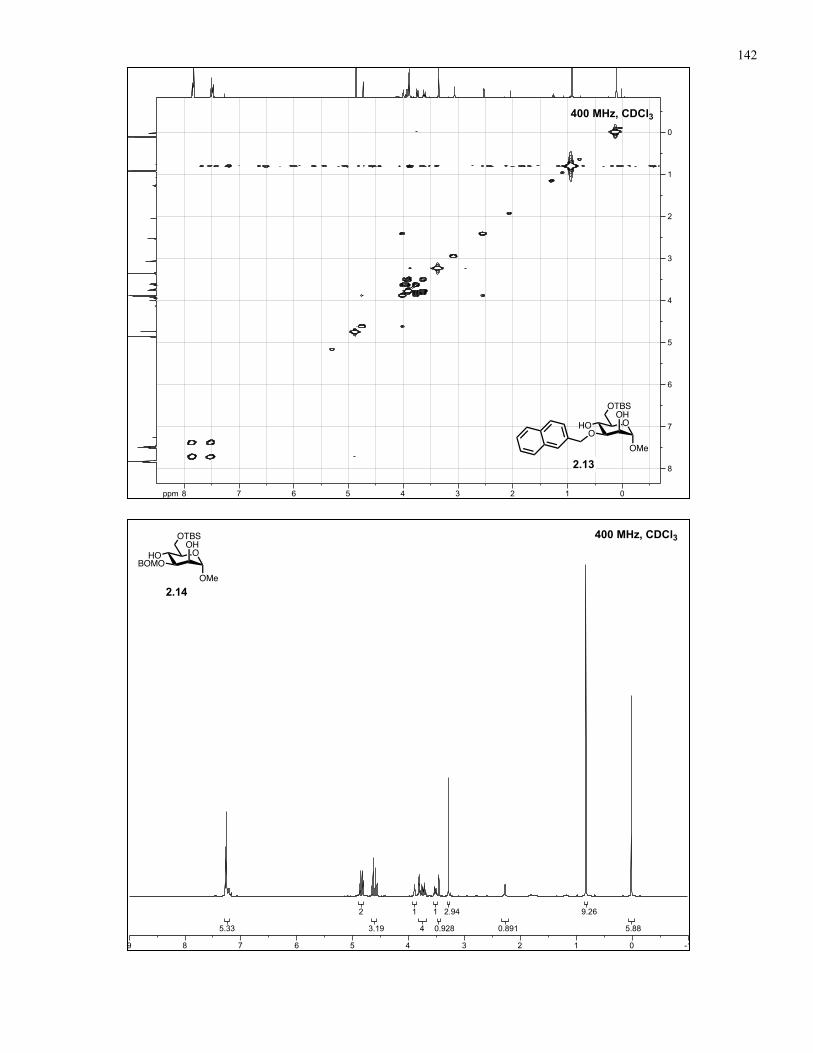
142
0 01122334455667788ppmppm
0
1
2
3
4
5
6
7
8
O
OMe
OTBSOH
OHO
6a
5.885.88
9.269.26
0.8910.891
2.942.94
0.9280.928
11
44
11
3.193.19
22
5.335.33
-1-100112233445566778899ppm
O
OMe
OTBSOH
BOMOHO
7a
400 MHz, CDCl3
400 MHz, CDCl3
2.13
2.14

143
0 02020404060608080100100120120140140160160180180200200ppmppm
LC-IV-2-F2-3/CDCl3/400 MHz
O
OMe
OTBSOH
BOMOHO
O
OMe
OTBSOH
BOMOHO
7a
10
0112233445566778899ppmppm
0
1
2
3
4
5
6
7
8
9
O
OMe
OTBSOH
BOMOHO
7a
400 MHz, CDCl3
100 MHz, CDCl3
2.14
2.14
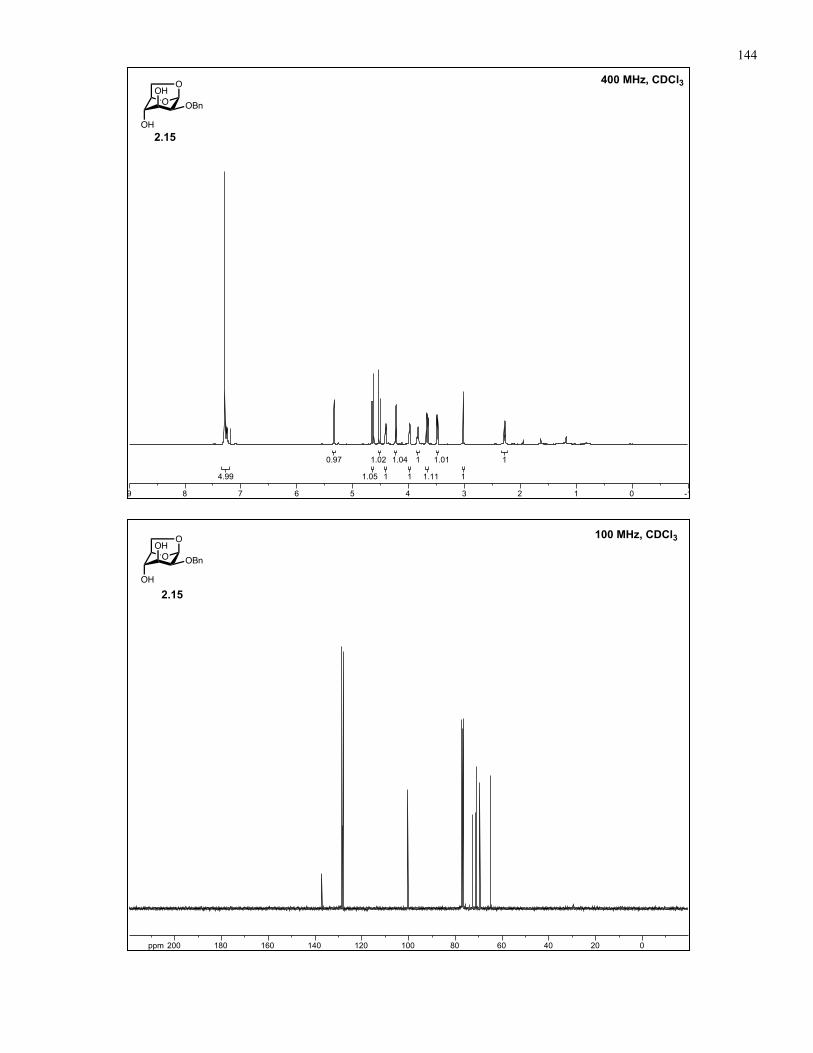
144
11
11
1.011.01
1.111.11
11
11
1.041.04
11
1.021.02
1.051.05
0.970.97
4.994.99
-1-100112233445566778899ppm
O
OOH
OH
OBn
4b
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OOH
OH
OBn
4b
400 MHz, CDCl3
100 MHz, CDCl3
2.15
2.15
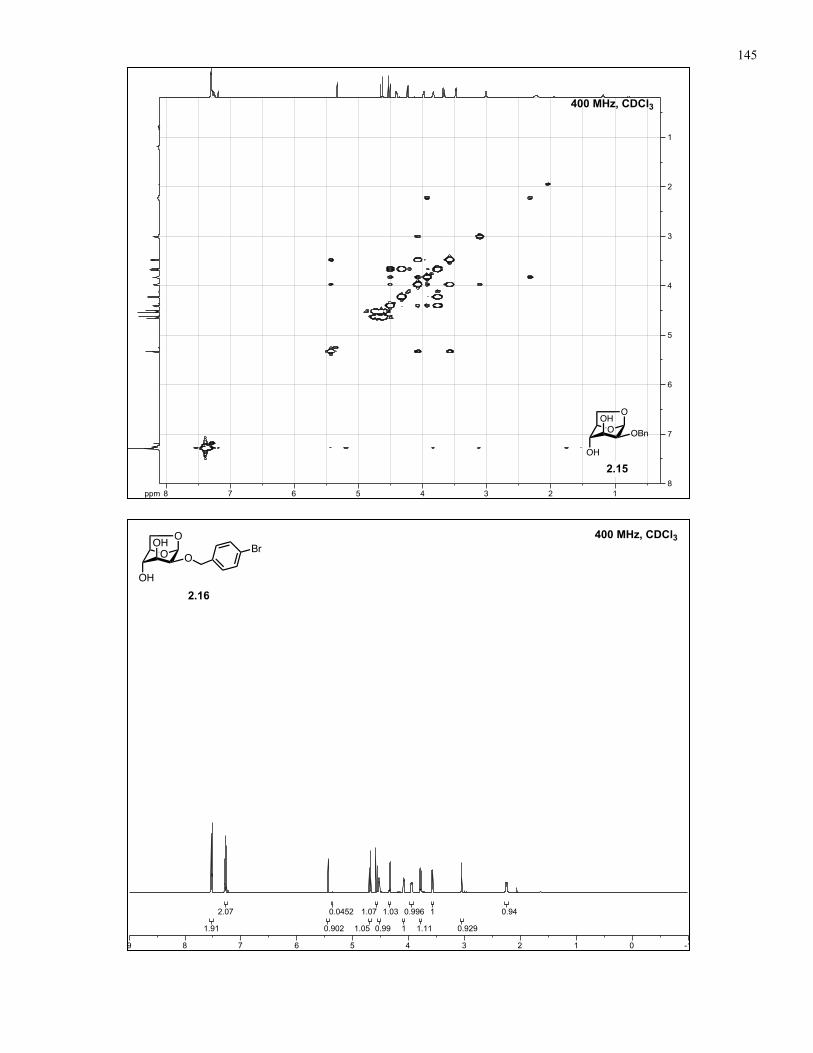
145
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OOH
OH
OBn
4b
0.940.94
0.9290.929
11
1.111.11
0.9960.996
11
1.031.03
0.990.99
1.071.07
1.051.05
0.04520.0452
0.9020.902
2.072.07
1.911.91
-1-100112233445566778899ppm
OOOH
OH
OBr
5b
400 MHz, CDCl3
400 MHz, CDCl3
2.15
2.16
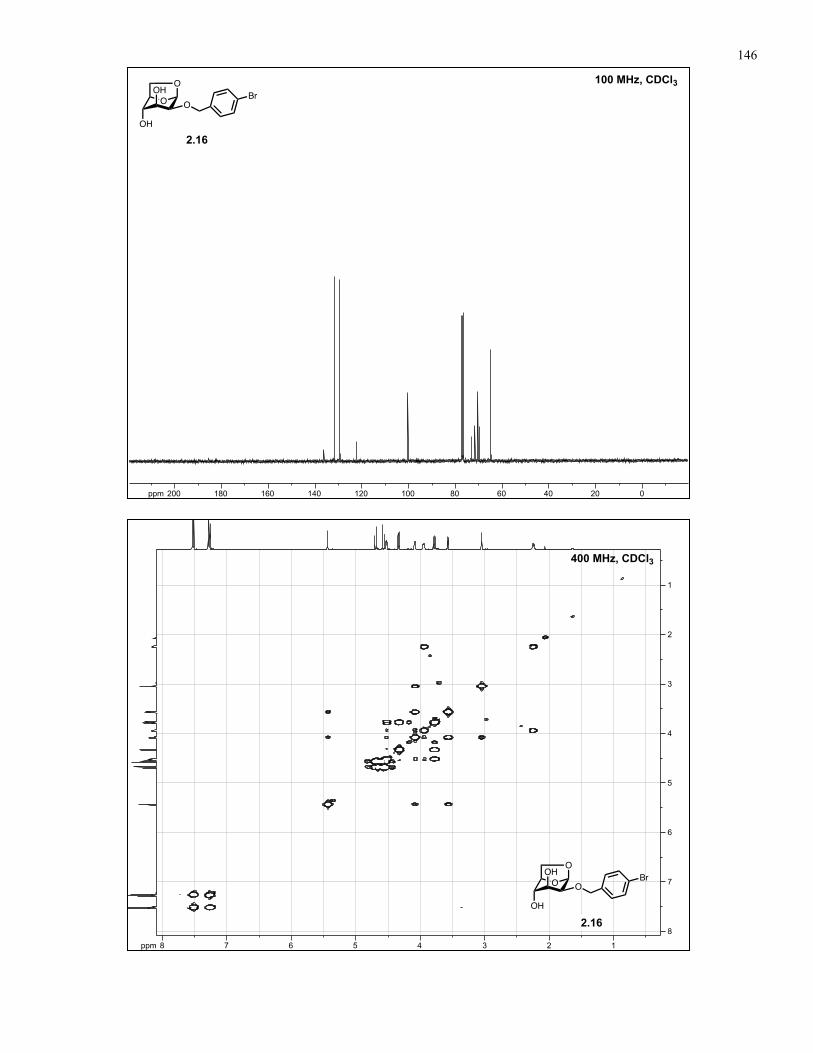
146
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OOH
OH
OBr
5b
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OOH
OH
OBr
5b
100 MHz, CDCl3
400 MHz, CDCl3
2.16
2.16

147
0.9490.949
11
1.031.03
1.231.23
1.031.03
1.081.08
1.11.1
1.031.03
1.161.16
1.111.11
0.9390.939
3.083.08
4.284.28
00112233445566778899ppm
OOOH
OH
O
6b
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OOH
OH
O
6b
100 MHz, CDCl3
400 MHz, CDCl3
2.17
2.17

148
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OOH
OH
O
6b
0.9310.931
0.9050.905
2.012.01
1.031.03
11
11
11
1.061.06
11
2.092.09
0.8950.895
4.94.9
-1-100112233445566778899ppm
OOOH
OH
OBOM
7b
400 MHz, CDCl3
400 MHz, CDCl3
2.17
2.18
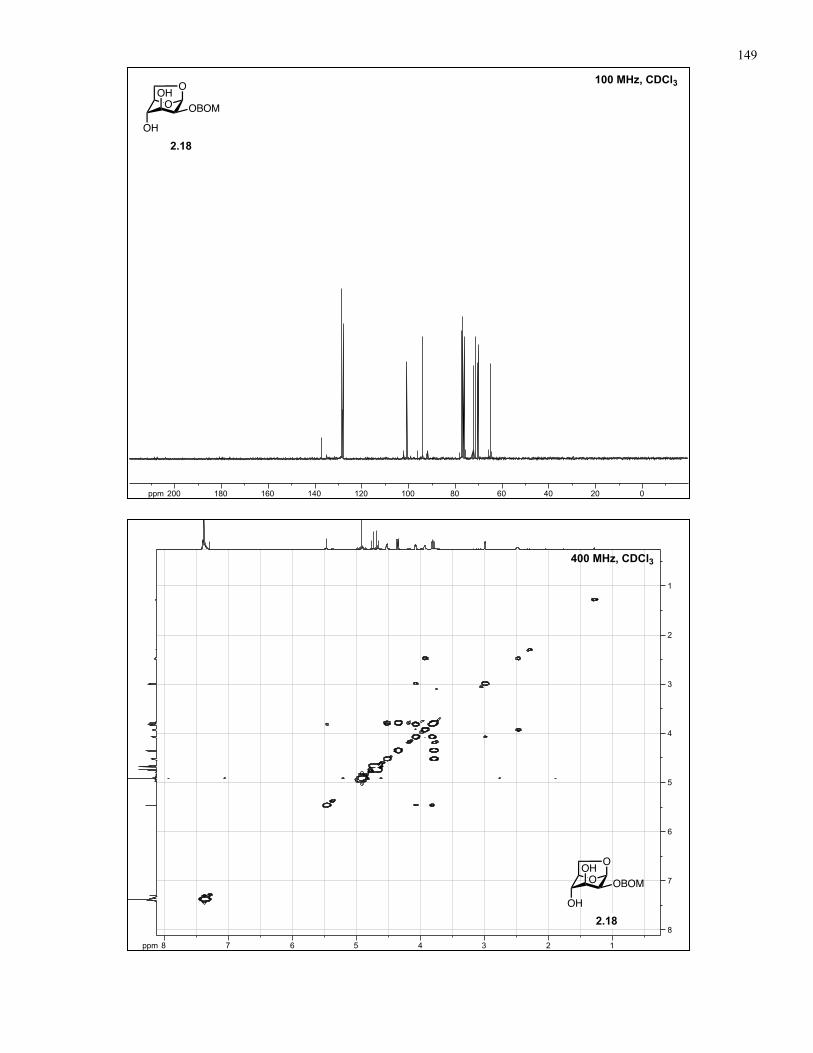
149
0 02020404060608080100100120120140140160160180180200200ppmppm
OOOH
OH
OBOM
7b
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
OOOH
OH
OBOM
7b
100 MHz, CDCl3
400 MHz, CDCl3
2.18
2.18
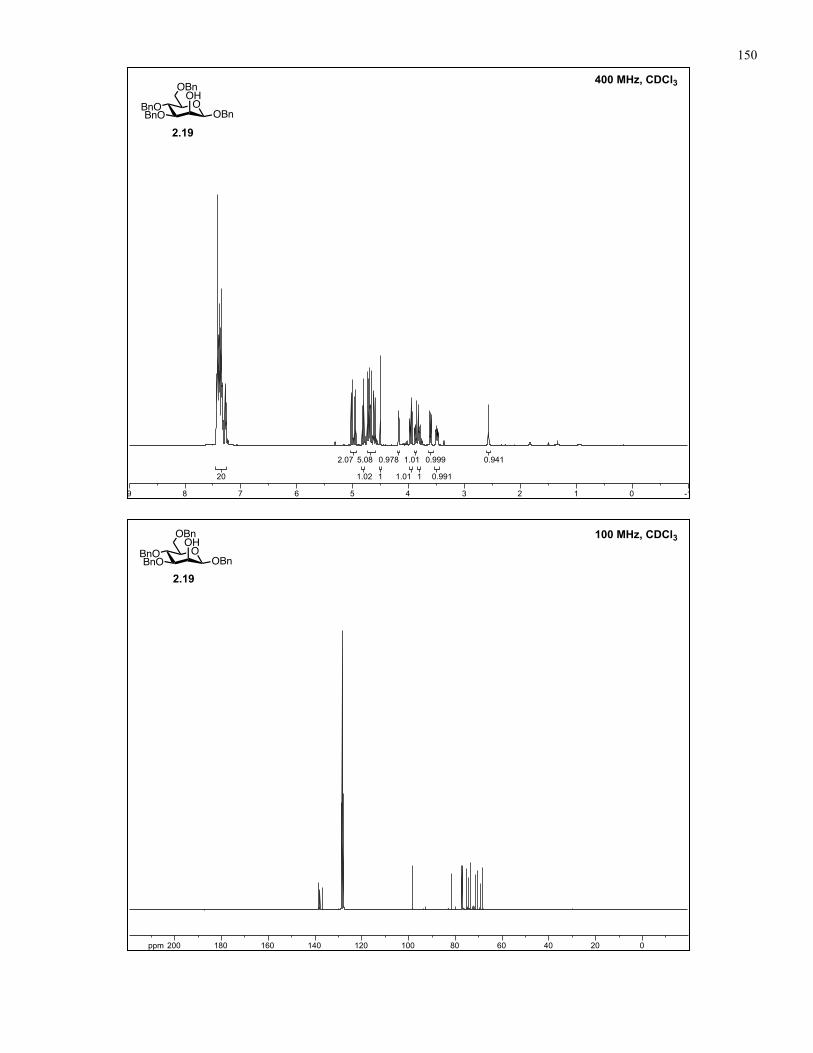
150
0.9410.941
0.9910.991
0.9990.999
11
1.011.01
1.011.01
0.9780.978
11
5.085.08
1.021.02
2.072.07
2020
-1-100112233445566778899ppm
OOBn
OBnOH
BnOBnO
4c
0 02020404060608080100100120120140140160160180180200200ppmppm
OOBn
OBnOH
BnOBnO
4c
100 MHz, CDCl3
400 MHz, CDCl3
2.19
2.19
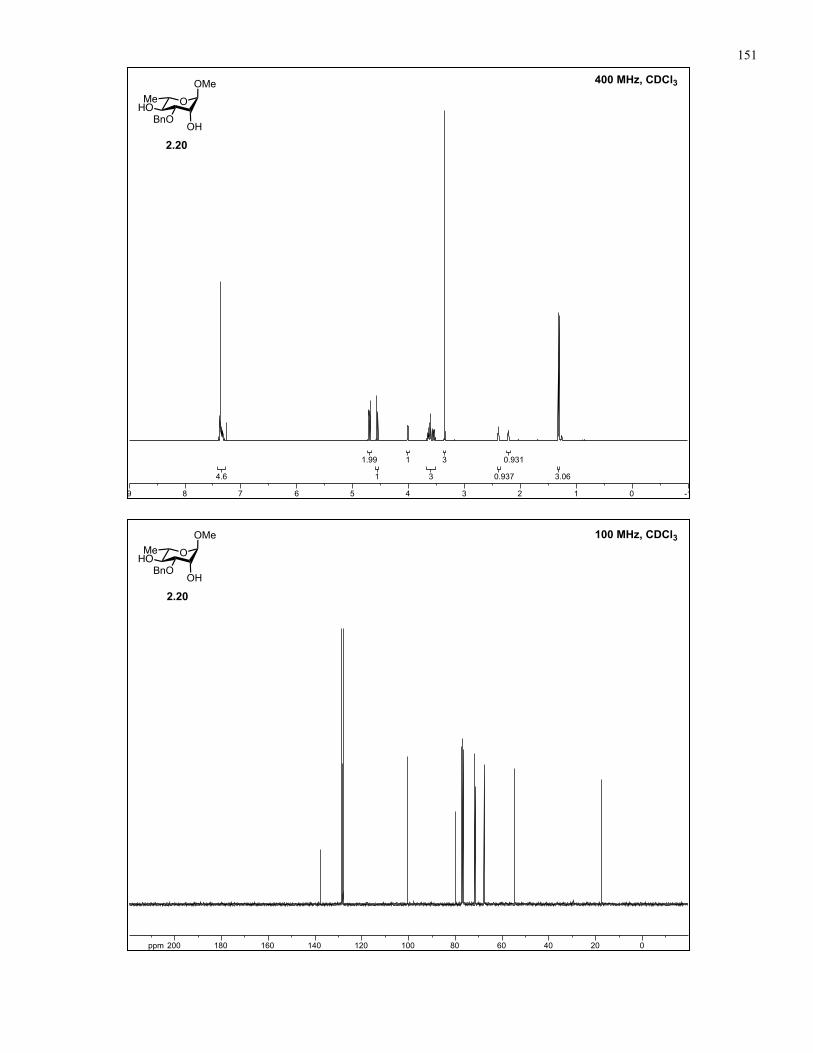
151
3.063.06
0.9310.931
0.9370.937
33
33
11
11
1.991.99
4.64.6
-1-100112233445566778899ppm
O
OMeMeHOBnO
OH
4d
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMeHOBnO
OH
4d
100 MHz, CDCl3
400 MHz, CDCl3
2.20
2.20

152
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OMeMeHOBnO
OH
4d
33
0.9040.904
0.9320.932
2.952.95
33
0.9770.977
11
11
0.9430.943
1.871.87
1.761.76
-1-100112233445566778899ppm
O
OMeMeHO
OOH
Br5d
400 MHz, CDCl3
400 MHz, CDCl3
2.20
2.21
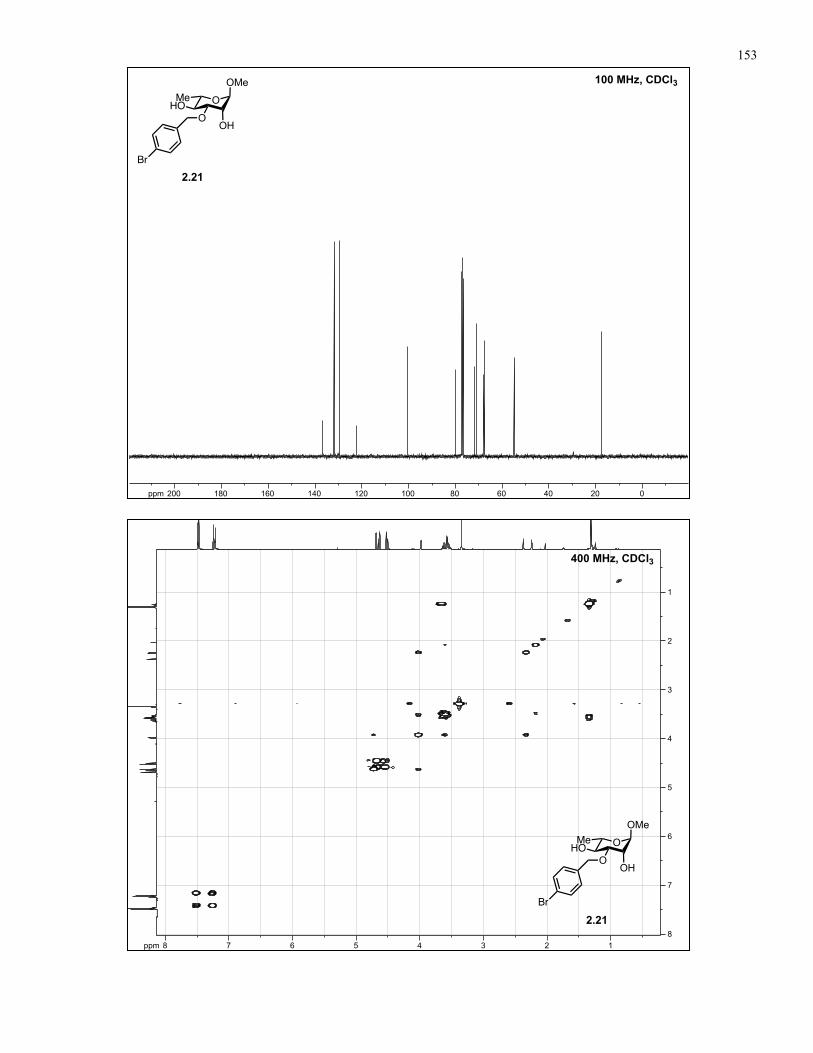
153
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMeHO
OOH
Br5d
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OMeMeHO
OOH
Br5d
100 MHz, CDCl3
400 MHz, CDCl3
2.21
2.21

154
3.023.02
0.9420.942
0.9540.954
33
33
0.9980.998
22
11
2.822.82
3.833.83
-1-100112233445566778899ppm
O
OMeMeHO
OOH
6d
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMeHO
OOH
6d
100 MHz, CDCl3
400 MHz, CDCl3
2.22
2.22
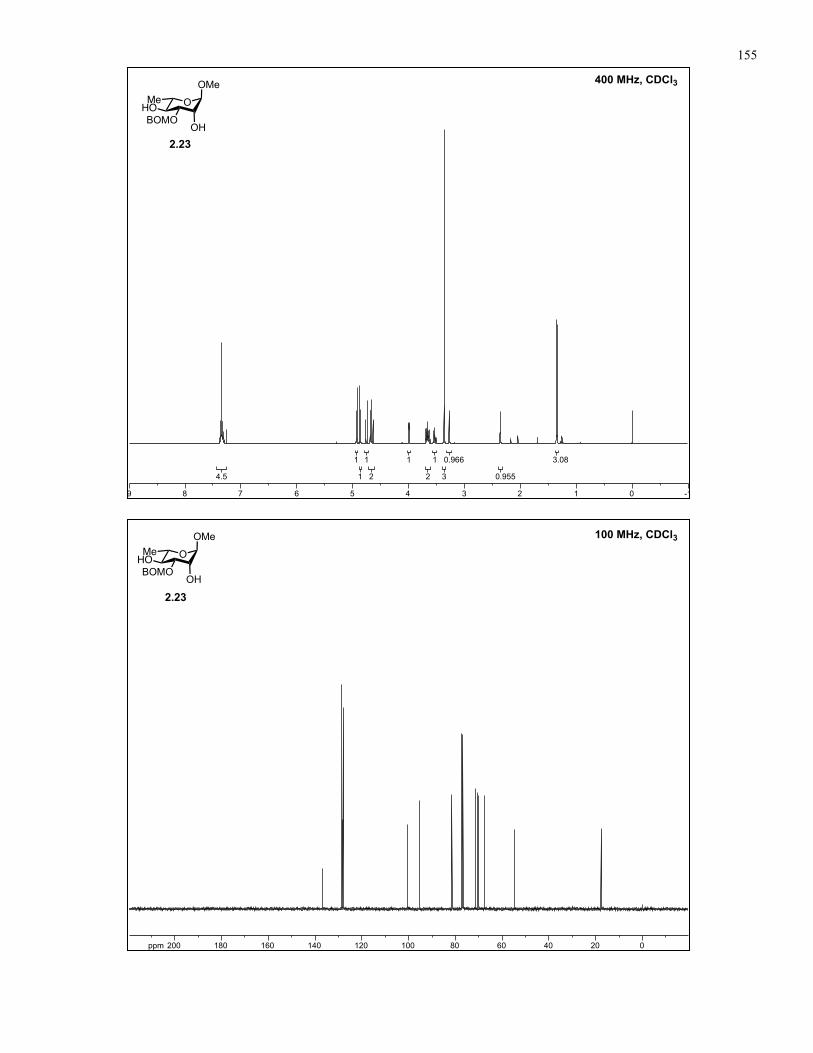
155
3.083.08
0.9550.955
0.9660.966
33
11
22
11
22
11
11
11
4.54.5
-1-100112233445566778899ppm
O
OMeMeHOBOMO
OH
7d
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMeHOBOMO
OH
7d
100 MHz, CDCl3
400 MHz, CDCl3
2.23
2.23
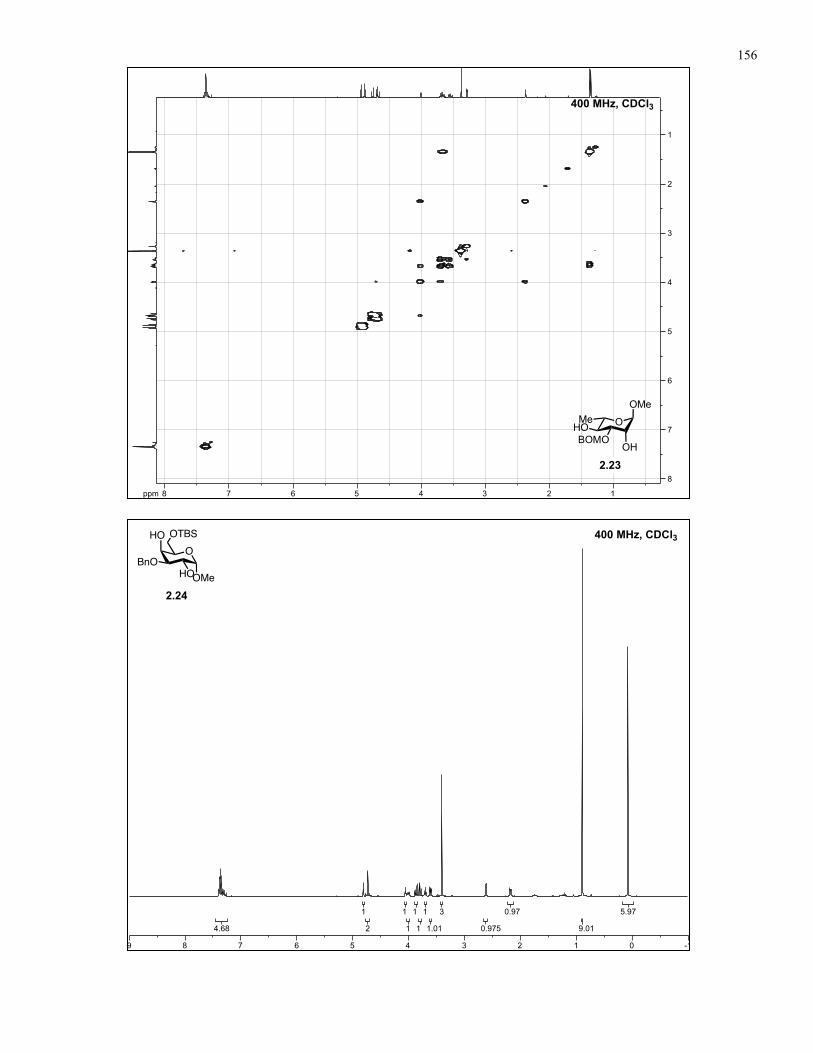
156
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OMeMeHOBOMO
OH
7d
5.975.97
9.019.01
0.970.97
0.9750.975
33
1.011.01
11
11
11
11
11
22
11
4.684.68
-1-100112233445566778899ppm
O
OMe
OTBS
HOBnO
HO
4e
400 MHz, CDCl3
400 MHz, CDCl3
2.23
2.24
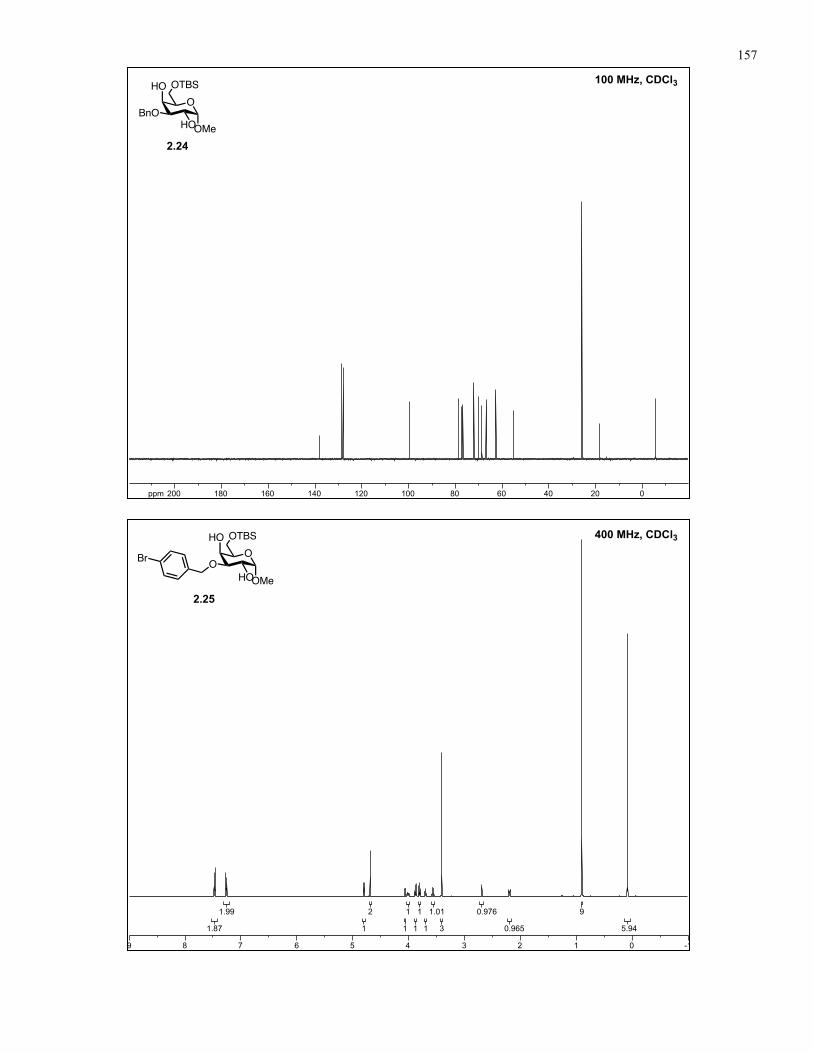
157
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBS
HOBnO
HO
4e
5.945.94
99
0.9650.965
0.9760.976
33
1.011.01
11
11
11
11
11
22
11
1.991.99
1.871.87
-1-100112233445566778899ppm
O
OMe
OTBS
HOO
HO
Br
5e
100 MHz, CDCl3
400 MHz, CDCl3
2.24
2.25
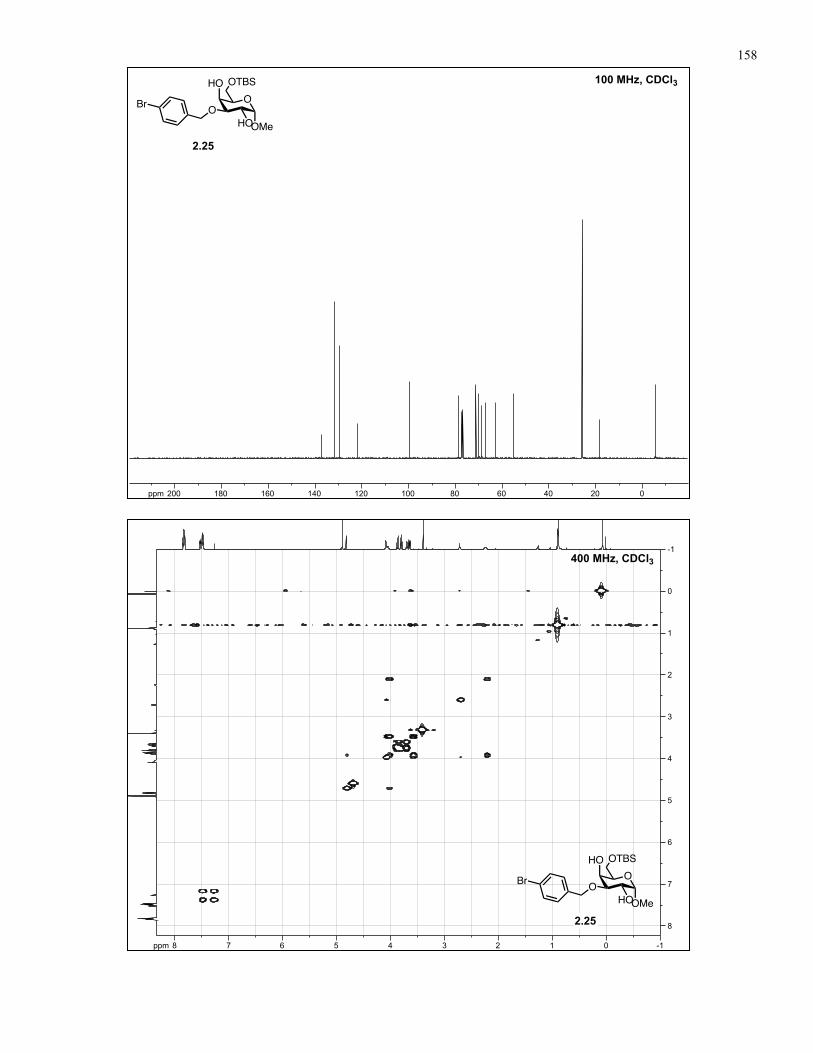
158
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBS
HOO
HO
Br
5e
-1 -1001122334455667788ppmppm
-1
0
1
2
3
4
5
6
7
8
O
OMe
OTBS
HOO
HO
Br
5e
100 MHz, CDCl3
400 MHz, CDCl3
2.25
2.25

159
5.855.85
9.029.02
0.9370.937
0.9480.948
2.972.97
11
11
11
11
22
11
22
2.912.91
3.933.93
-1-100112233445566778899ppm
O
OMe
OTBS
HOO
HO
6e
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBS
HOO
HO
6e
100 MHz, CDCl3
400 MHz, CDCl3
2.26
2.26

160
-1 -100112233445566778899ppmppm
-1
0
1
2
3
4
5
6
7
8
9
O
OMe
OTBS
HOO
HO
6e
5.955.95
99
0.9890.989
0.9310.931
33
3.013.01
11
11
11
22
11
22
4.74.7
-1-100112233445566778899ppm
O
OMe
OTBS
HOBOMO
HO
7e
400 MHz, CDCl3
400 MHz, CDCl3
2.26
2.27
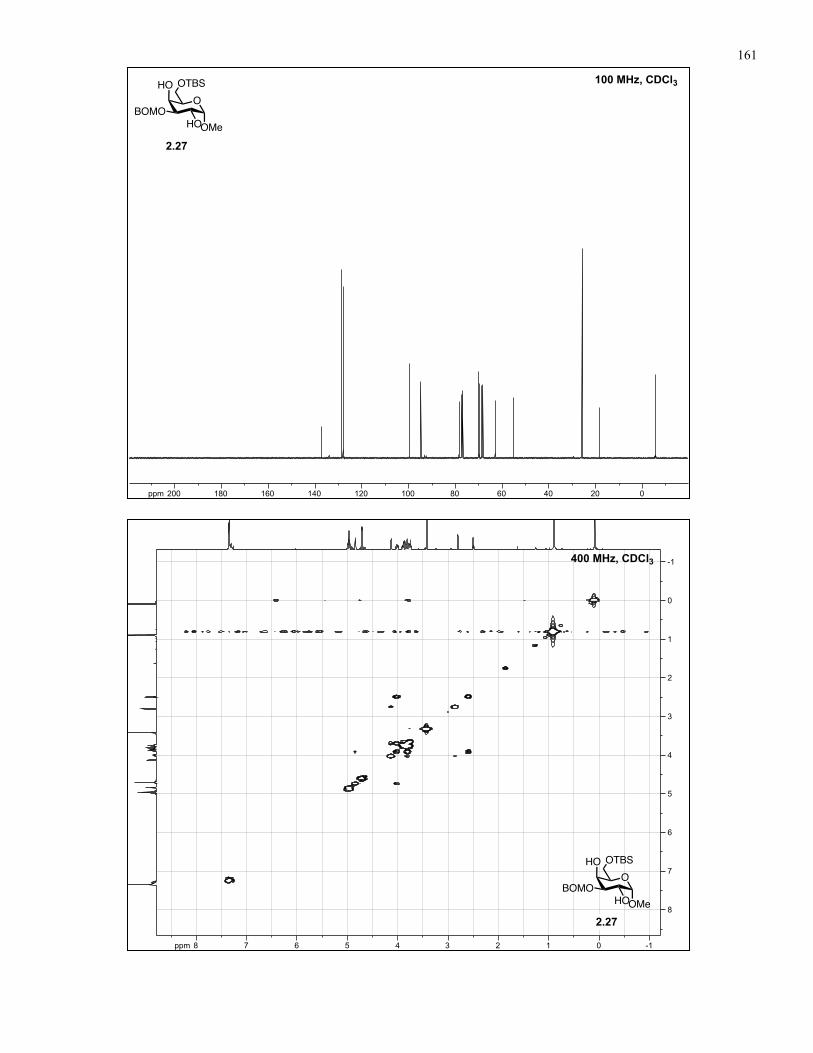
161
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMe
OTBS
HOBOMO
HO
7e
-1 -1001122334455667788ppmppm
-1
0
1
2
3
4
5
6
7
8
O
OMe
OTBS
HOBOMO
HO
7e
100 MHz, CDCl3
400 MHz, CDCl3
2.27
2.27
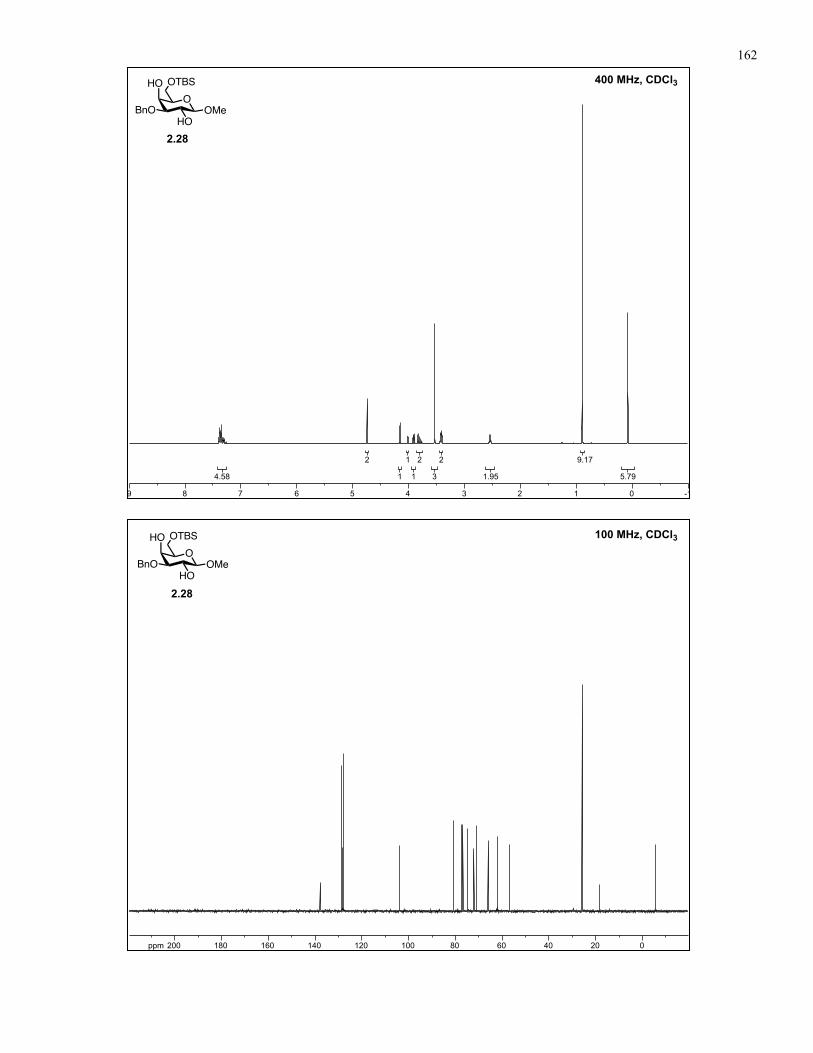
162
5.795.79
9.179.17
1.951.95
22
33
22
11
11
11
22
4.584.58
-1-100112233445566778899ppm
OOMe
OTBS
HOBnO
HO
4f
0 02020404060608080100100120120140140160160180180200200ppmppm
OOMe
OTBS
HOBnO
HO
4f
100 MHz, CDCl3
400 MHz, CDCl3
2.28
2.28
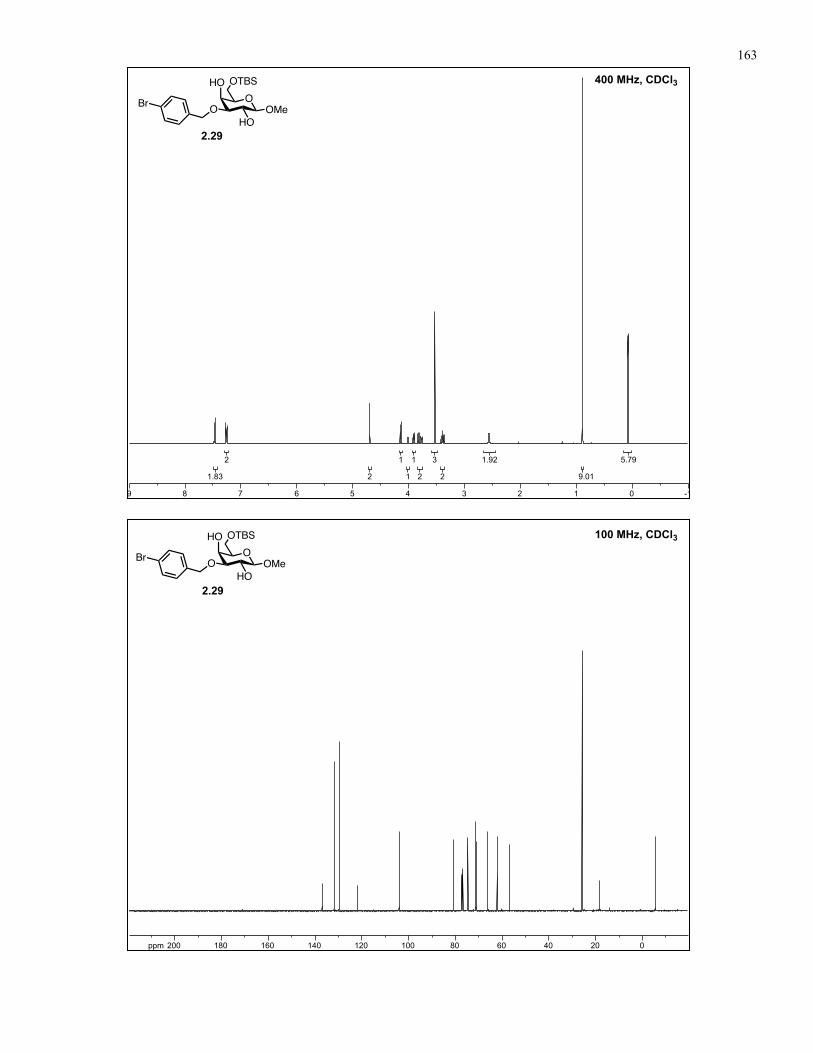
163
5.795.79
9.019.01
1.921.92
22
33
22
11
11
11
22
22
1.831.83
-1-100112233445566778899ppm
OOMe
OTBS
HOO
HO
Br
5f
0 02020404060608080100100120120140140160160180180200200ppmppm
OOMe
OTBS
HOO
HO
Br
5f
100 MHz, CDCl3
400 MHz, CDCl3
2.29
2.29
164
0 01122334455667788ppmppm
-1
0
1
2
3
4
5
6
7
8
OOMe
OTBS
HOO
HO
Br
5f
5.835.83
99
0.9680.968
0.9330.933
11
11
2.992.99
22
11
11
11
22
2.882.88
3.883.88
-1-100112233445566778899ppm
OOMe
OTBS
HOO
HO
6f
400 MHz, CDCl3
400 MHz, CDCl3
2.29
2.30

165
0 02020404060608080100100120120140140160160180180200200ppmppm
OOMe
OTBS
HOO
HO
6f
-1 -1001122334455667788ppmppm
-1
0
1
2
3
4
5
6
7
8
OOMe
OTBS
HOO
HO
6f
100 MHz, CDCl3
400 MHz, CDCl3
2.30
2.30
166
5.995.99
9.289.28
0.9420.942
0.9930.993
11
4.014.01
11
11
11
11
11
22
22
4.684.68
-1-100112233445566778899ppm
OOMe
OTBS
HOBOMO
HO
7f
0 02020404060608080100100120120140140160160180180200200ppmppm
OOMe
OTBS
HOBOMO
HO
7f
100 MHz, CDCl3
400 MHz, CDCl3
2.31
2.31
167
0 011223344556677ppmppm
0
1
2
3
4
5
6
7
OOMe
OTBS
HOBOMO
HO
7f
0.9960.996
0.9560.956
11
22
11
11
11
11
11
0.9620.962
4.714.71
-1-100112233445566778899ppm
OOOH
BnO
OH4g
400 MHz, CDCl3
400 MHz, CDCl3
2.31
2.32
168
0 02020404060608080100100120120140140160160180180200200ppmppm
OOOH
BnO
OH4g
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
OOOH
BnO
OH4g
100 MHz, CDCl3
400 MHz, CDCl3
2.32
2.32

169
11
0.930.93
1.111.11
2.022.02
1.021.02
11
11
11
11
11
2.062.06
2.012.01
-1-100112233445566778899ppm
OOOH
O
OH
Br
5g
0 02020404060608080100100120120140140160160180180200200ppmppm
OOOH
O
OH
Br
5g
400 MHz, CDCl3
100 MHz, CDCl3
2.33
2.33

170
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
OOOH
O
OH
Br
5g
0.9610.961
0.9210.921
1.031.03
2.012.01
11
11
1.011.01
1.011.01
11
0.9290.929
3.013.01
4.014.01
-1-100112233445566778899ppm
OOOH
O
OH6g
400 MHz, CDCl3
400 MHz, CDCl3
2.33
2.34
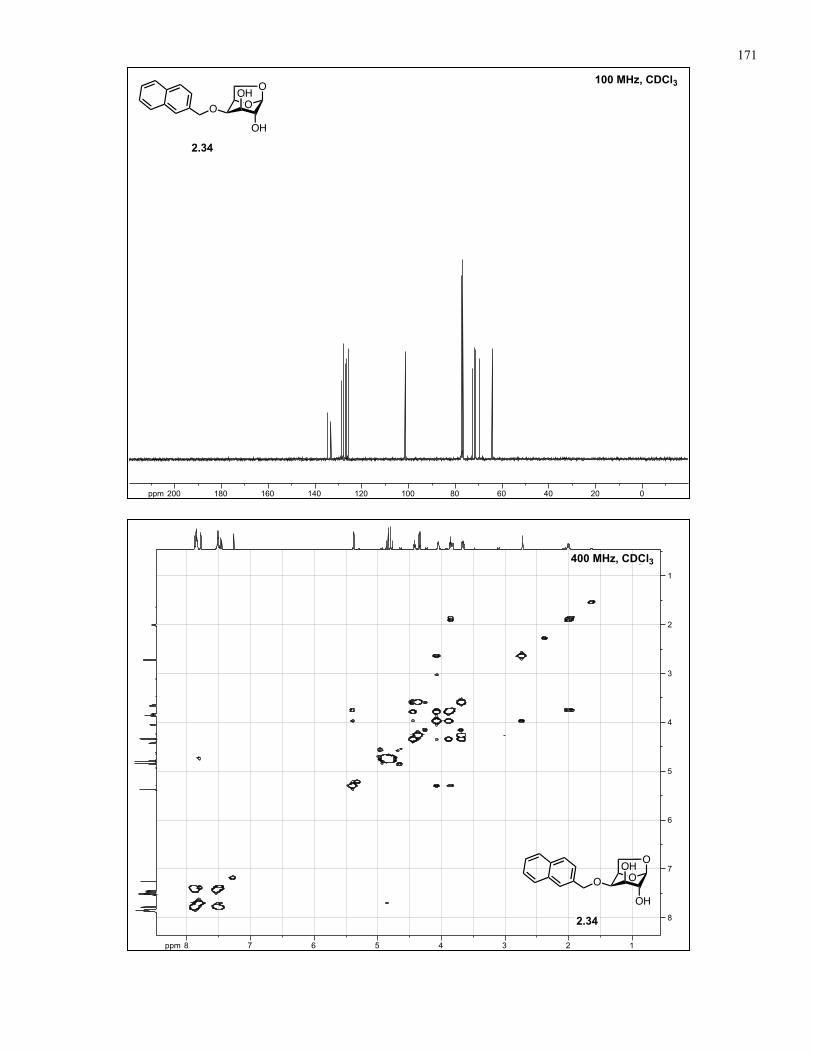
171
0 02020404060608080100100120120140140160160180180200200ppmppm
OOOH
O
OH6g
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
OOOH
O
OH6g
400 MHz, CDCl3
100 MHz, CDCl3
2.34
2.34
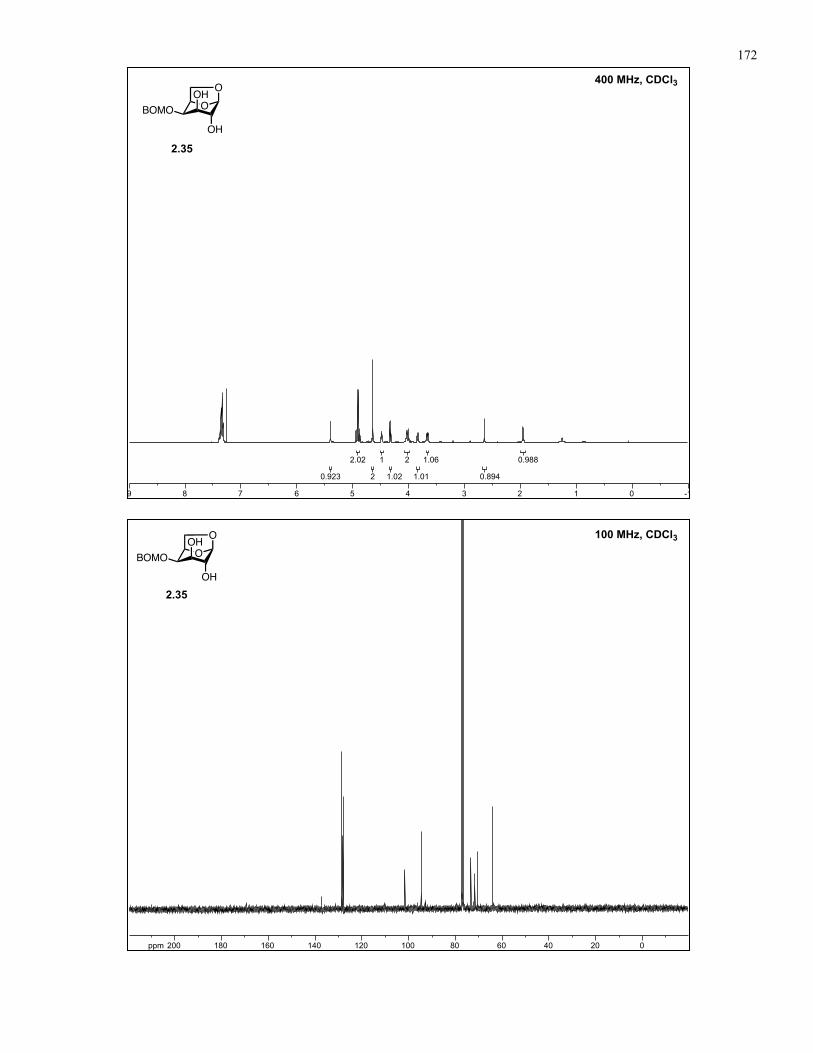
172
0.9880.988
0.8940.894
1.061.06
1.011.01
22
1.021.02
11
22
2.022.02
0.9230.923
-1-100112233445566778899ppm
OOOH
BOMO
OH7g
0 02020404060608080100100120120140140160160180180200200ppmppm
OOOH
BOMO
OH7g
400 MHz, CDCl3
100 MHz, CDCl3
2.35
2.35
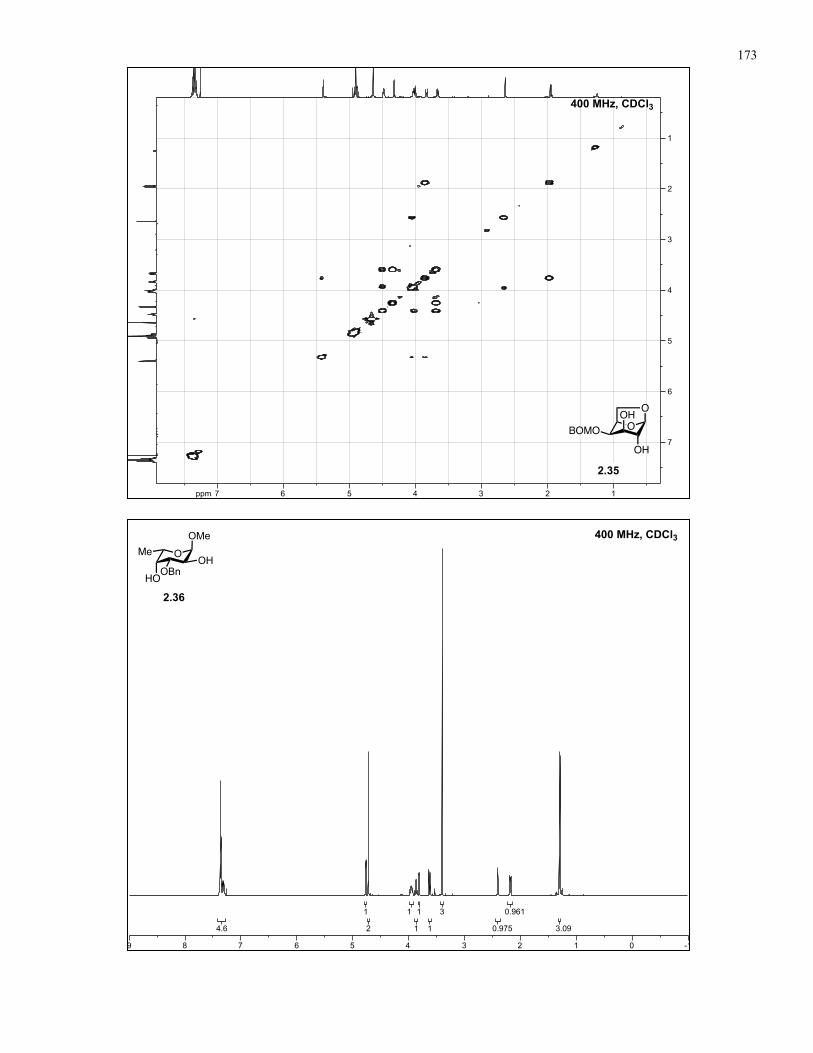
173
1 1223344556677ppmppm
1
2
3
4
5
6
7
OOOH
BOMO
OH7g
3.093.09
0.9610.961
0.9750.975
33
11
11
11
11
22
11
4.64.6
-1-100112233445566778899ppm
O
OMeMe
HOOBnOH
4h
400 MHz, CDCl3
400 MHz, CDCl3
2.35
2.36
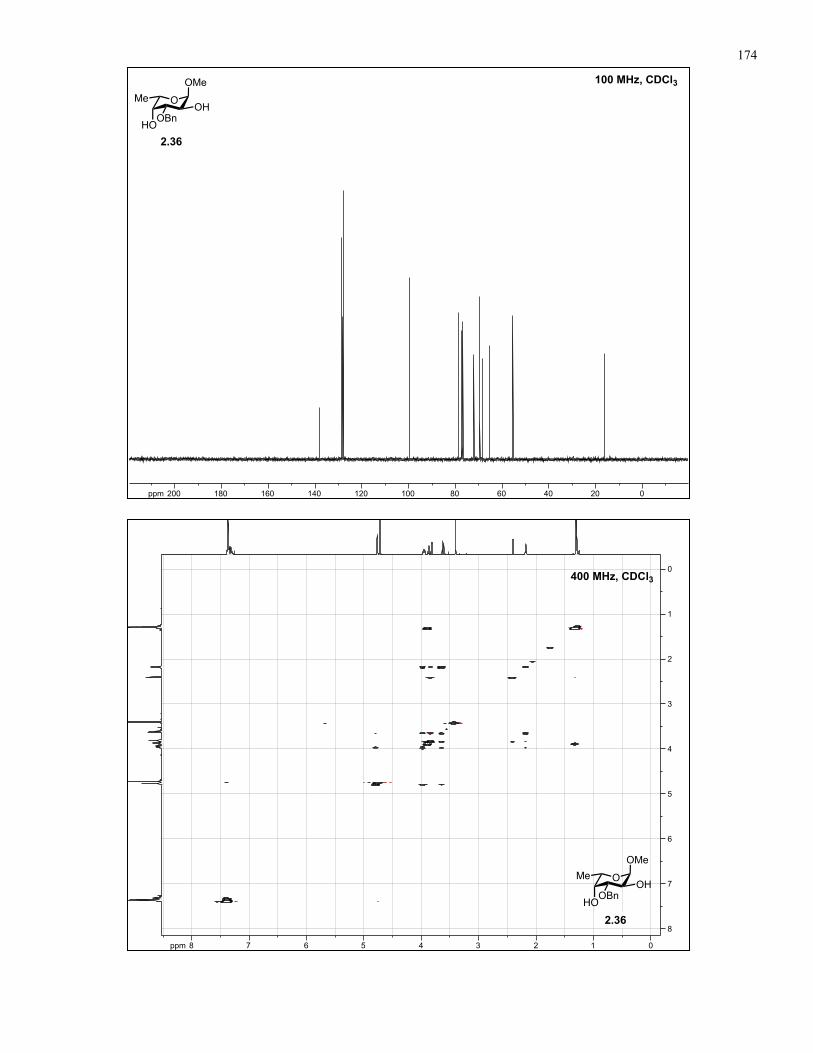
174
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMe
HOOBnOH
4h
0 01122334455667788ppmppm
0
1
2
3
4
5
6
7
8
O
OMeMe
HOOBnOH
4h
400 MHz, CDCl3
100 MHz, CDCl3
2.36
2.36
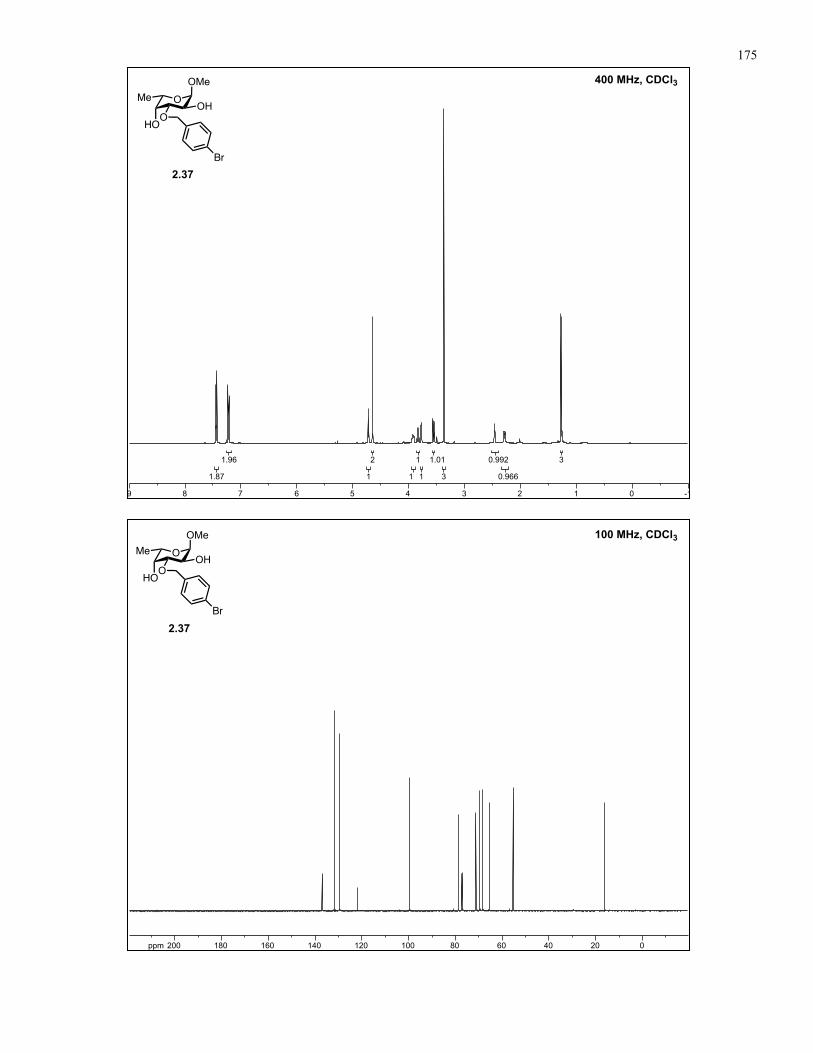
175
33
0.9660.966
0.9920.992
33
1.011.01
11
11
11
22
11
1.961.96
1.871.87
-1-100112233445566778899ppm
O
OMeMe
HOOOH
Br5h
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMe
HOOOH
Br5h
400 MHz, CDCl3
100 MHz, CDCl3
2.37
2.37
2.37
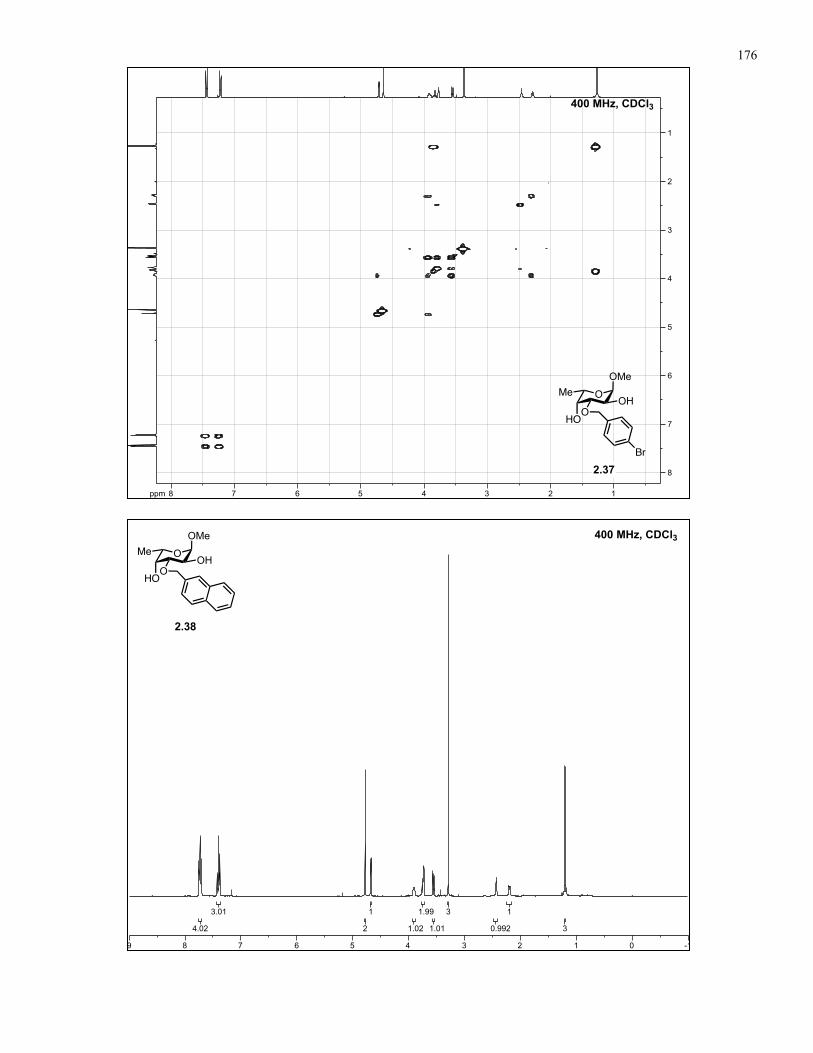
176
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OMeMe
HOOOH
Br5h
33
11
0.9920.992
33
1.011.01
1.991.99
1.021.02
11
22
3.013.01
4.024.02
-1-100112233445566778899ppm
O
OMeMe
HOOOH
6h
400 MHz, CDCl3
400 MHz, CDCl3
2.37
2.38
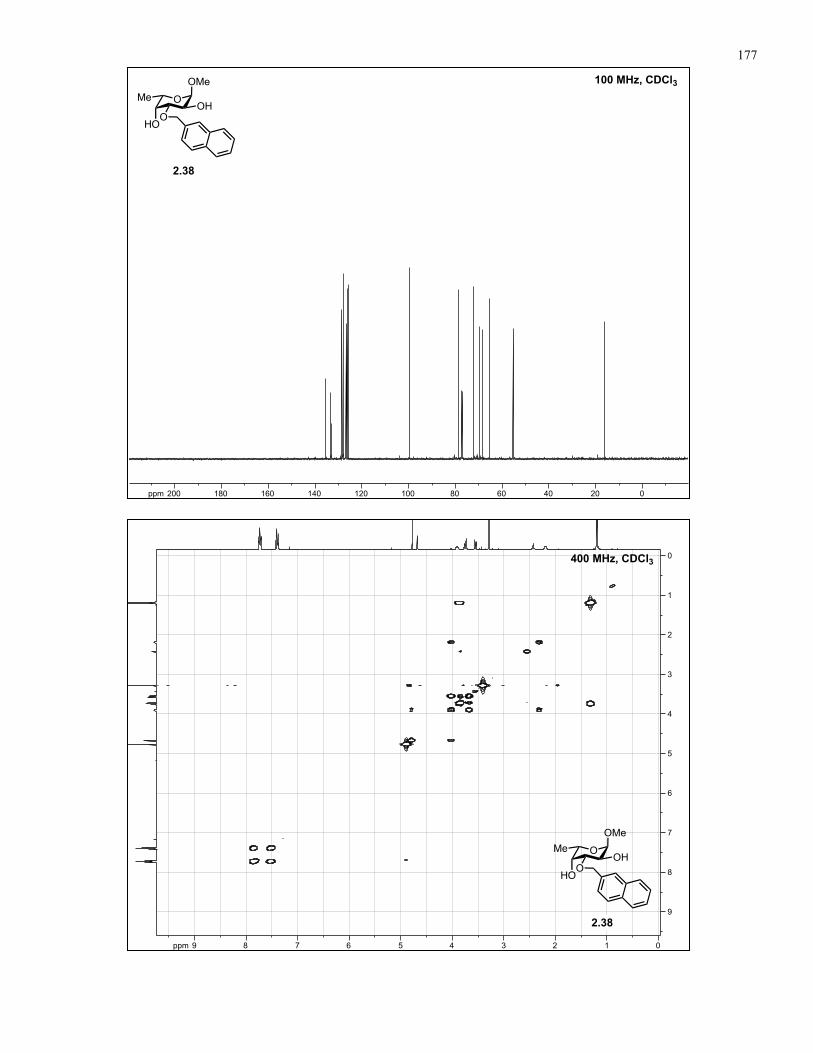
177
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMe
HOOOH
6h
0 0112233445566778899ppmppm
0
1
2
3
4
5
6
7
8
9
O
OMeMe
HOOOH
6h
400 MHz, CDCl3
100 MHz, CDCl3
2.38
2.38
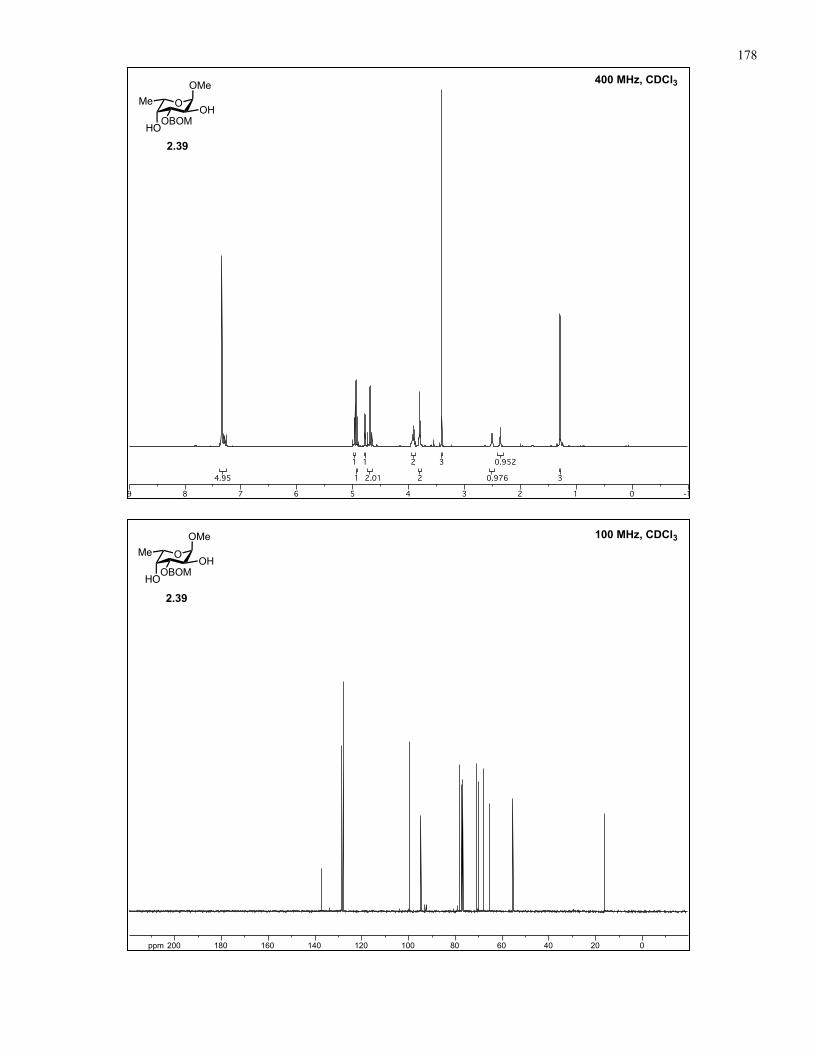
178
33
0.9520.952
0.9760.976
33
22
22
2.012.01
11
11
11
4.954.95
-1-100112233445566778899ppm
O
OMeMe
HOOBOMOH
7h
0 02020404060608080100100120120140140160160180180200200ppmppm
O
OMeMe
HOOBOMOH
7h
400 MHz, CDCl3
100 MHz, CDCl3
2.39
2.39
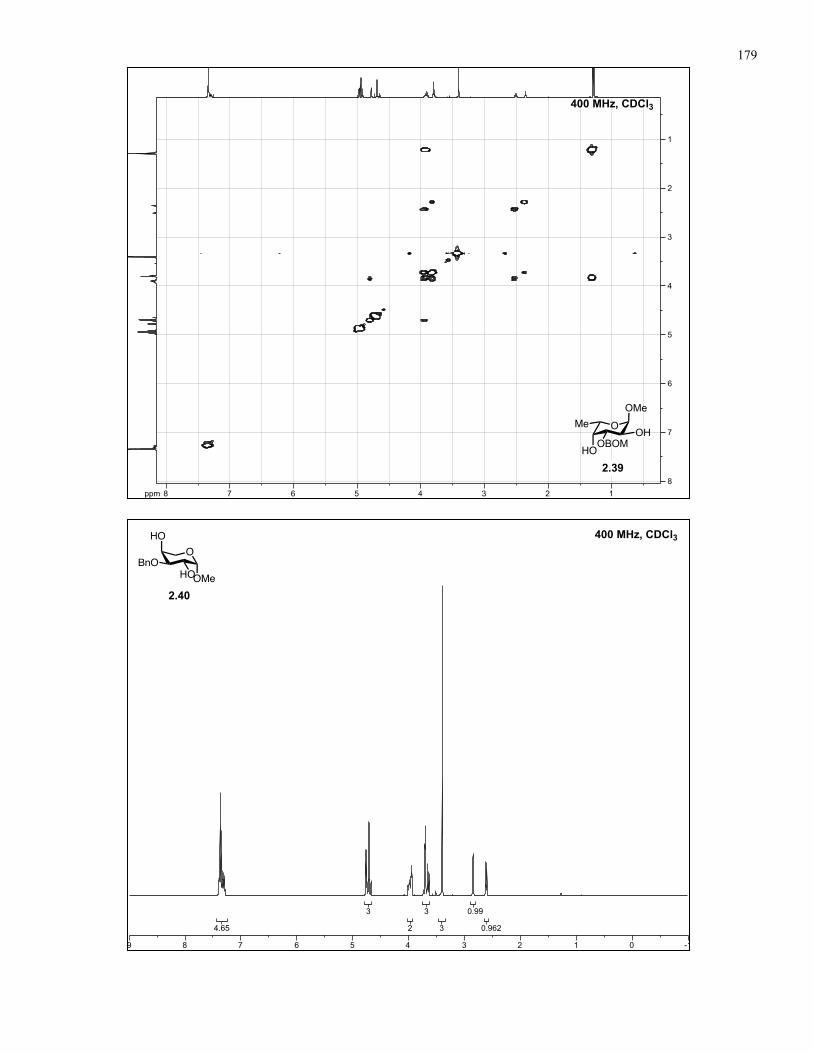
179
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
O
OMeMe
HOOBOMOH
7h
0.9620.962
0.990.99
33
33
22
33
4.654.65
-1-100112233445566778899ppm
4i
O
OMeHOBnO
HO 400 MHz, CDCl3
400 MHz, CDCl3
2.39
2.40

180
0 02020404060608080100100120120140140160160180180200200ppmppm
4i
O
OMeHOBnO
HO
1 122334455667788ppmppm
1
2
3
4
5
6
7
84i
O
OMeHOBnO
HO
400 MHz, CDCl3
100 MHz, CDCl3
2.40
2.40
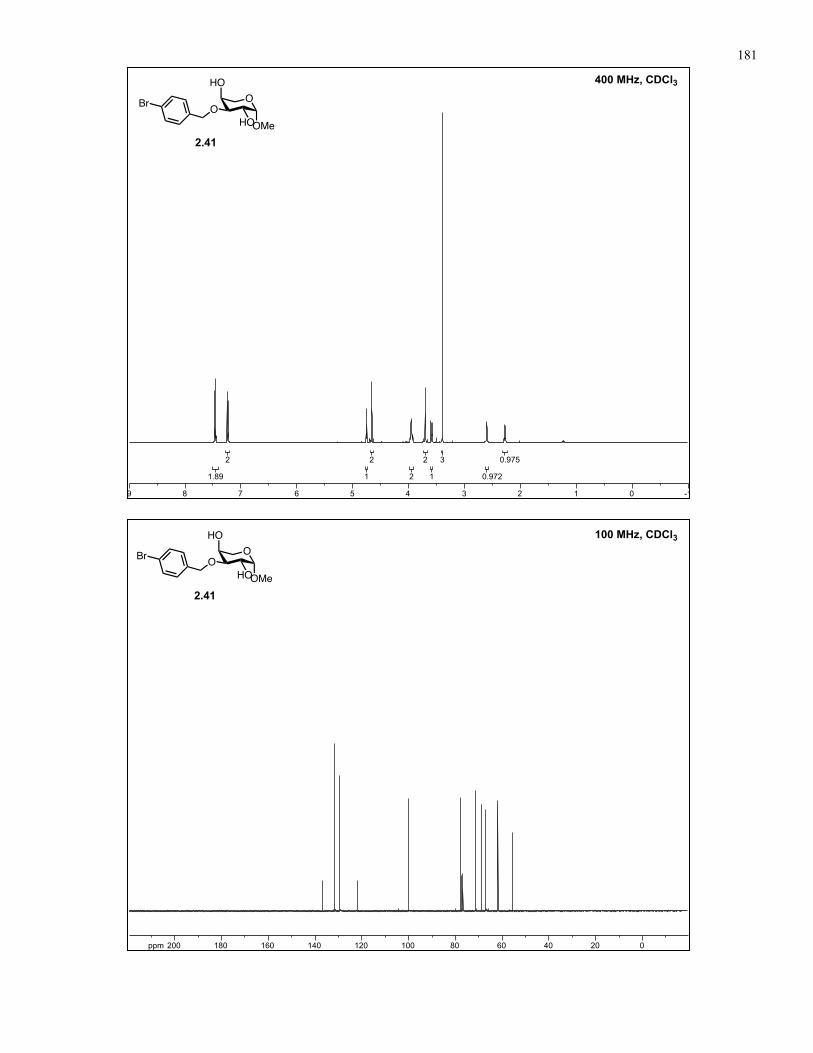
181
0.9750.975
0.9720.972
33
11
22
22
22
11
22
1.891.89
-1-100112233445566778899ppm
5i
O
OMeHOO
HO
Br
0 02020404060608080100100120120140140160160180180200200ppmppm
5i
O
OMeHOO
HO
Br
400 MHz, CDCl3
100 MHz, CDCl3
2.41
2.41
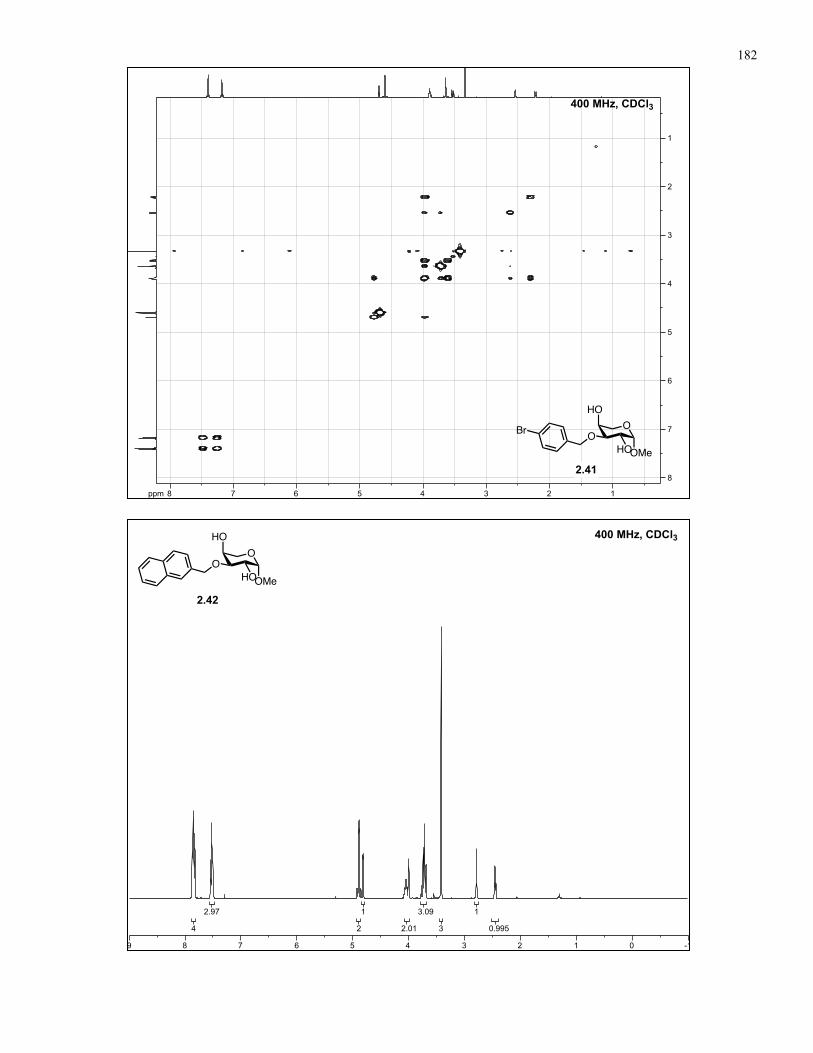
182
1 122334455667788ppmppm
1
2
3
4
5
6
7
85i
O
OMeHOO
HO
Br
0.9950.995
11
33
3.093.09
2.012.01
11
22
2.972.97
44
-1-100112233445566778899ppm
6i
O
OMeHOO
HO 400 MHz, CDCl3
400 MHz, CDCl3
2.41
2.42
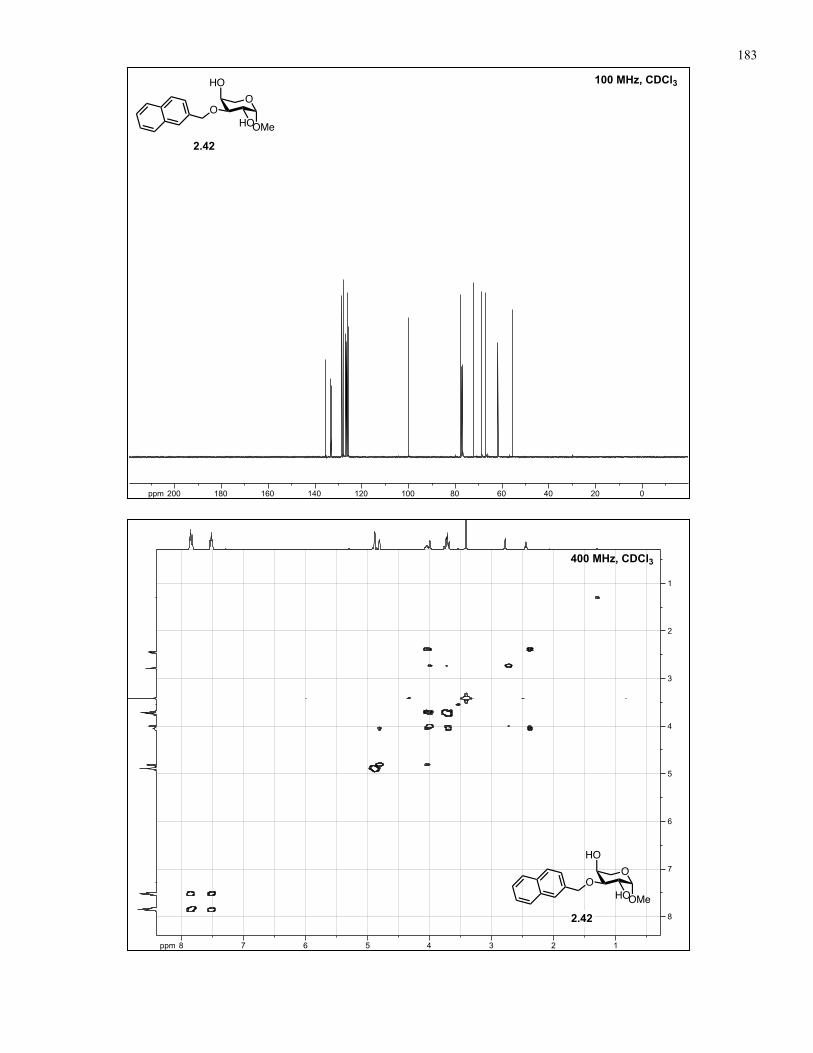
183
0 02020404060608080100100120120140140160160180180200200ppmppm
6i
O
OMeHOO
HO
1 122334455667788ppmppm
1
2
3
4
5
6
7
86i
O
OMeHOO
HO
400 MHz, CDCl3
100 MHz, CDCl3
2.42
2.42
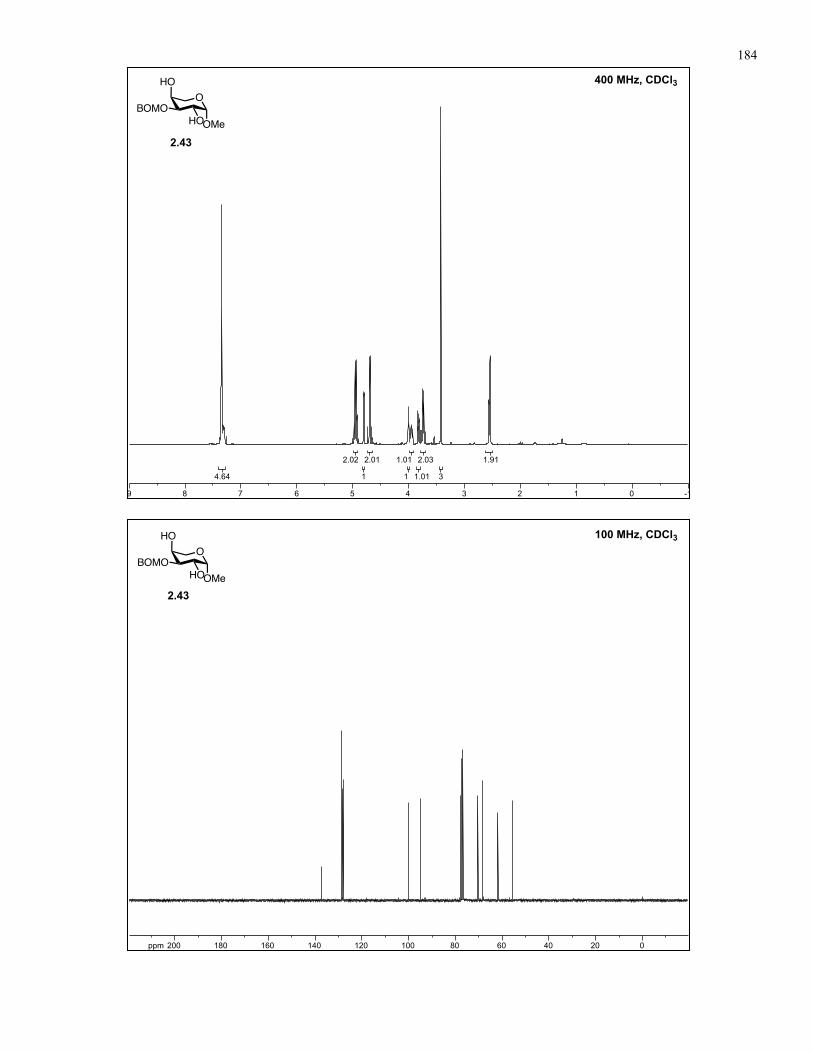
184
1.911.91
33
2.032.03
1.011.01
1.011.01
11
2.012.01
11
2.022.02
4.644.64
-1-100112233445566778899ppm
7i
O
OMeHOBOMO
HO
0 02020404060608080100100120120140140160160180180200200ppmppm
7i
O
OMeHOBOMO
HO
400 MHz, CDCl3
100 MHz, CDCl3
2.43
2.43
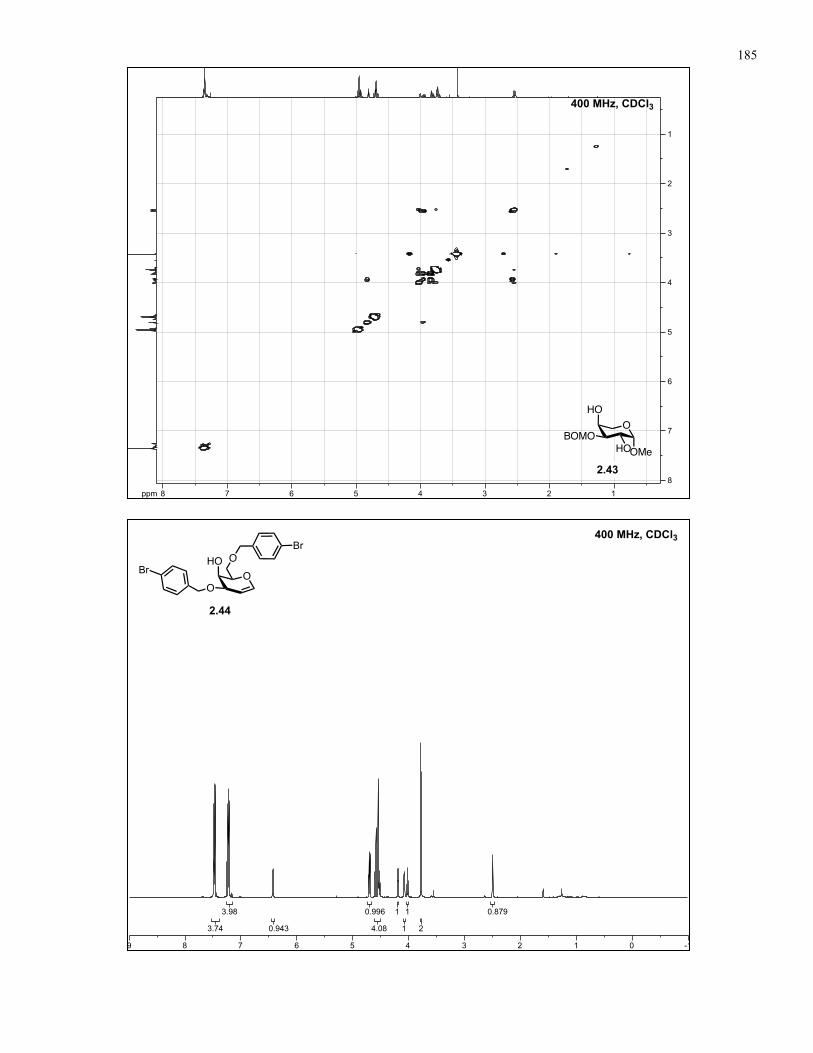
185
1 122334455667788ppmppm
1
2
3
4
5
6
7
87i
O
OMeHOBOMO
HO
0.8790.879
22
11
11
11
4.084.08
0.9960.996
0.9430.943
3.983.98
3.743.74
-1-100112233445566778899ppm
OOHO
O
Br
Br
5j
400 MHz, CDCl3
400 MHz, CDCl3
2.43
2.44
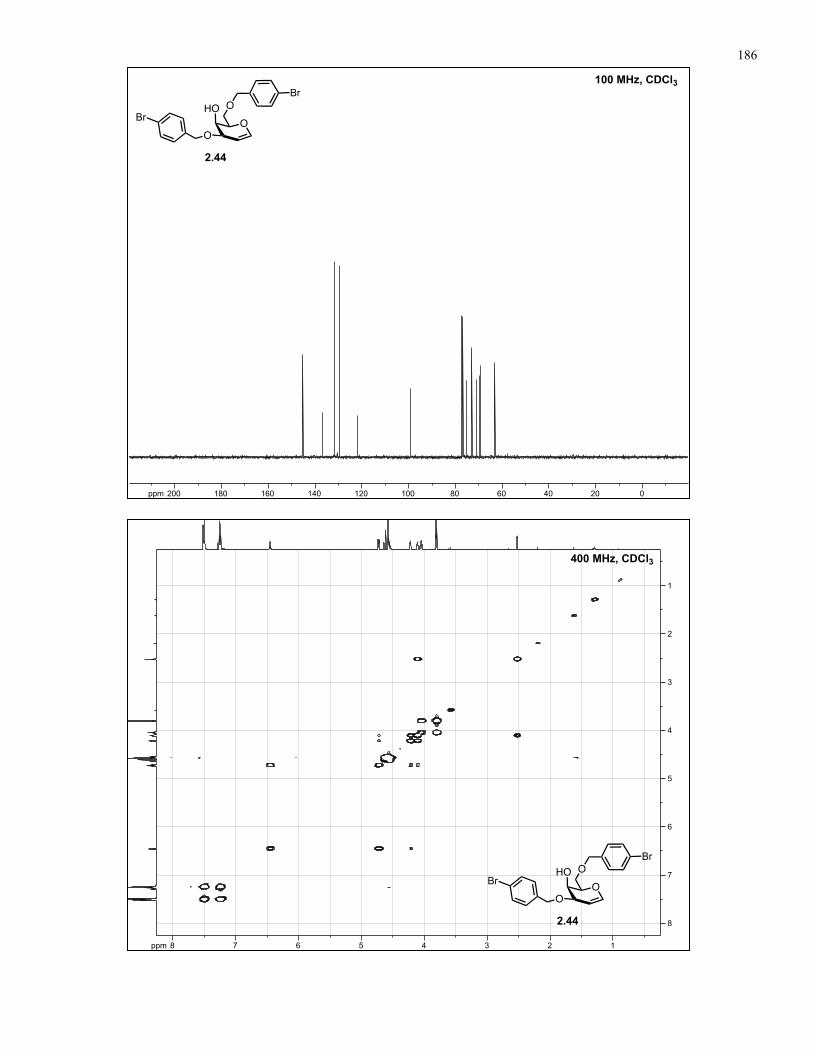
186
0 02020404060608080100100120120140140160160180180200200ppmppm
OOHO
O
Br
Br
5j
1 122334455667788ppmppm
1
2
3
4
5
6
7
8
OOHO
O
Br
Br
5j
400 MHz, CDCl3
100 MHz, CDCl3
2.44
2.44
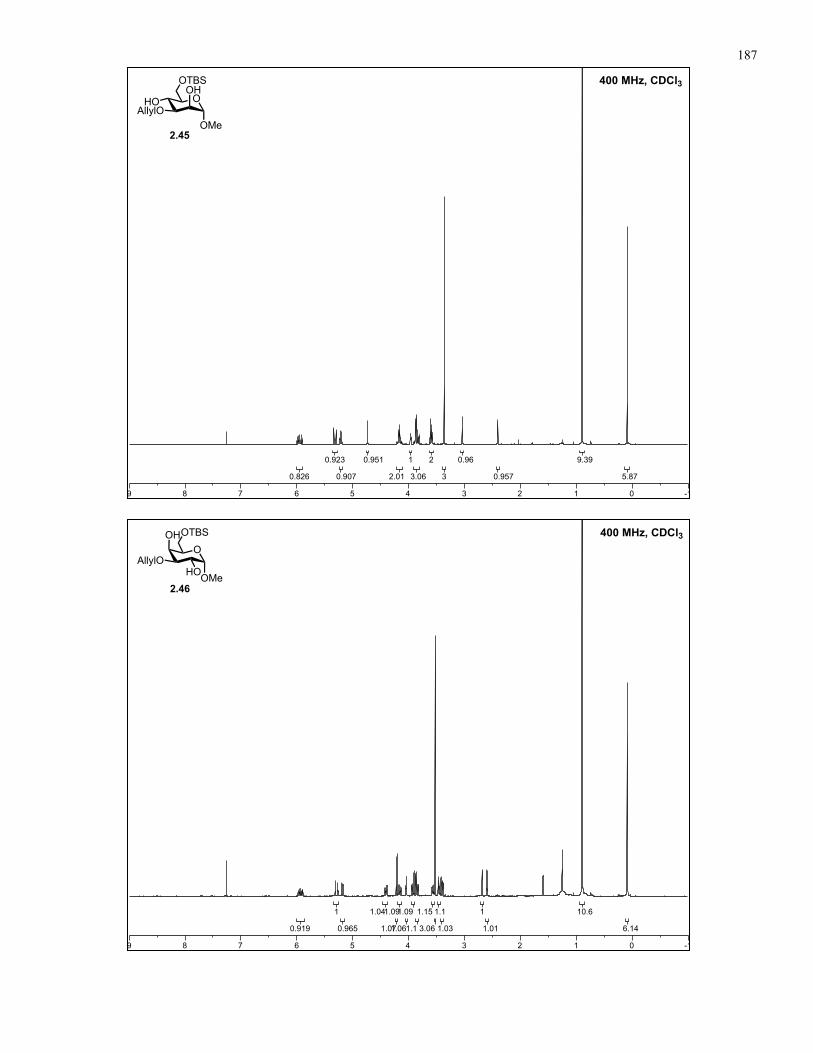
187
5.875.87
9.399.39
0.9570.957
0.960.96
33
22
3.063.06
11
2.012.01
0.9510.951
0.9070.907
0.9230.923
0.8260.826
-1-100112233445566778899ppm
O
OTBS
HOAllylO
OH
OMe2.45
6.146.14
10.610.6
1.011.01
11
1.031.03
1.11.1
3.063.06
1.151.15
1.11.1
1.091.09
1.061.06
1.091.09
1.071.07
1.041.04
0.9650.965
11
0.9190.919
-1-100112233445566778899ppm
O
OTBS
AllylO
OMe
OH
HO2.46
400 MHz, CDCl3
400 MHz, CDCl3
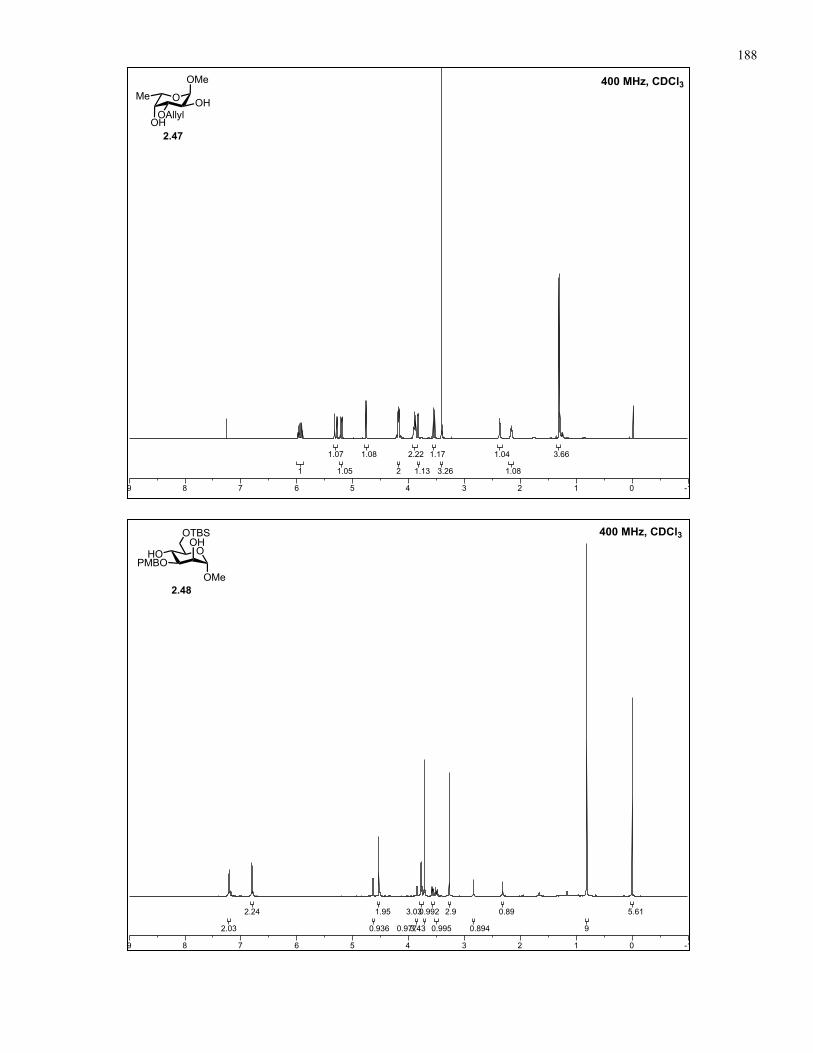
188
3.663.66
1.081.08
1.041.04
3.263.26
1.171.17
1.131.13
2.222.22
22
1.081.08
1.051.05
1.071.07
11
-1
-100112233445566778899ppm
O
OMeMe
OHOAllyl
OH
2.47
5.615.61
99
0.890.89
0.8940.894
2.92.9
0.9950.995
0.9920.992
3.433.43
3.033.03
0.9770.977
1.951.95
0.9360.936
2.242.24
2.032.03
-1-100112233445566778899ppm
O
OTBS
HOPMBO
OH
OMe2.48
400 MHz, CDCl3
400 MHz, CDCl3
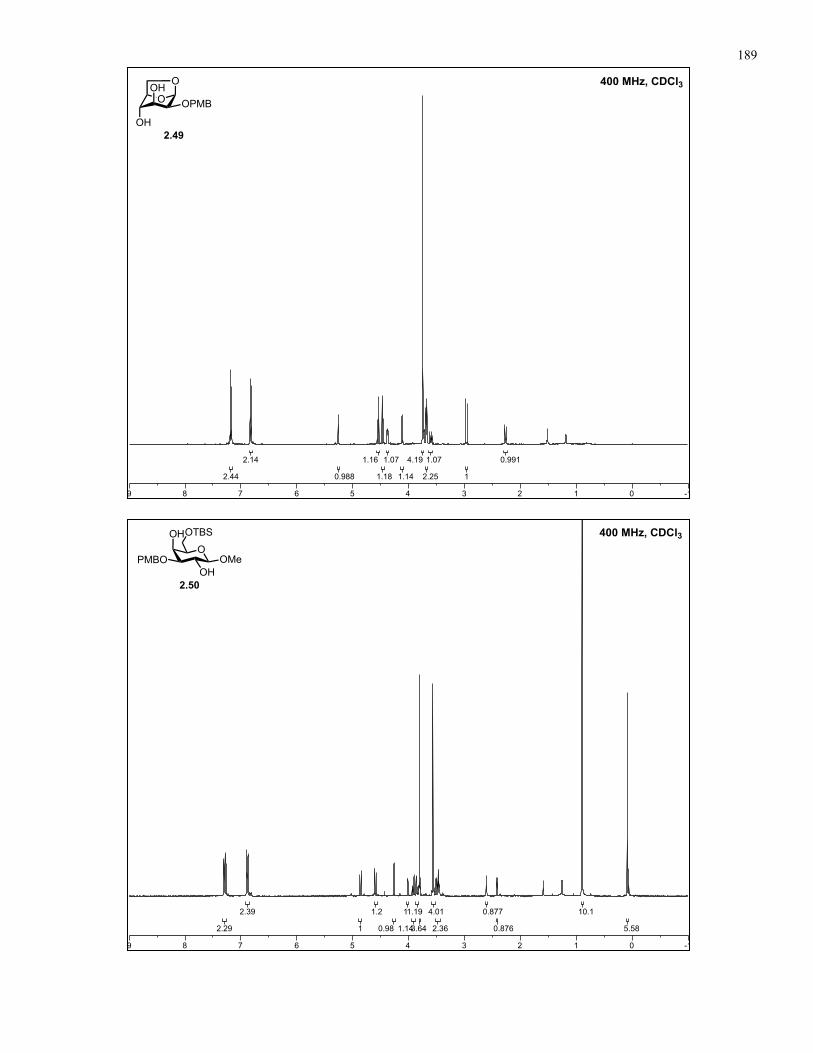
189
0.9910.991
11
1.071.07
2.252.25
4.194.19
1.141.14
1.071.07
1.181.18
1.161.16
0.9880.988
2.142.14
2.442.44
-1-100112233445566778899ppm
O
OOH
OH
OPMB
2.49
5.585.58
10.110.1
0.8760.876
0.8770.877
2.362.36
4.014.01
3.643.64
1.191.19
1.141.14
11
0.980.98
1.21.2
11
2.392.39
2.292.29
-1-100112233445566778899ppm
O
OTBS
PMBO OMe
OH
OH2.50
400 MHz, CDCl3
400 MHz, CDCl3
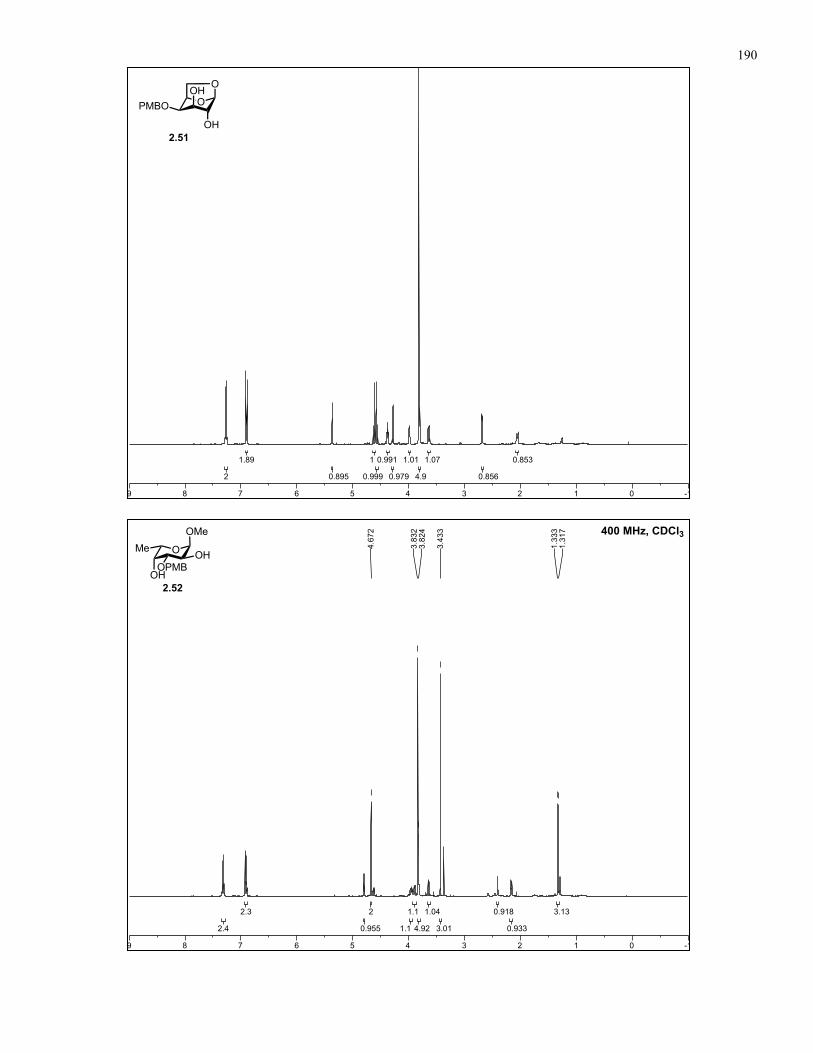
190
0.8530.853
0.8560.856
1.071.07
4.94.9
1.011.01
0.9790.979
0.9910.991
0.9990.999
11
0.8950.895
1.891.89
22
-1-100112233445566778899ppm
O
O
PMBOOH
OH2.51
3.133.13
0.9330.933
0.9180.918
3.013.01
1.041.04
4.924.92
1.11.1
1.11.1
22
0.9550.955
2.32.3
2.42.4
-1-100112233445566778899ppm-16.017
1.317
1.333
3.433
3.824
3.832
4.672
O
OMeMe
OHOPMB
OH
2.52
400 MHz, CDCl3
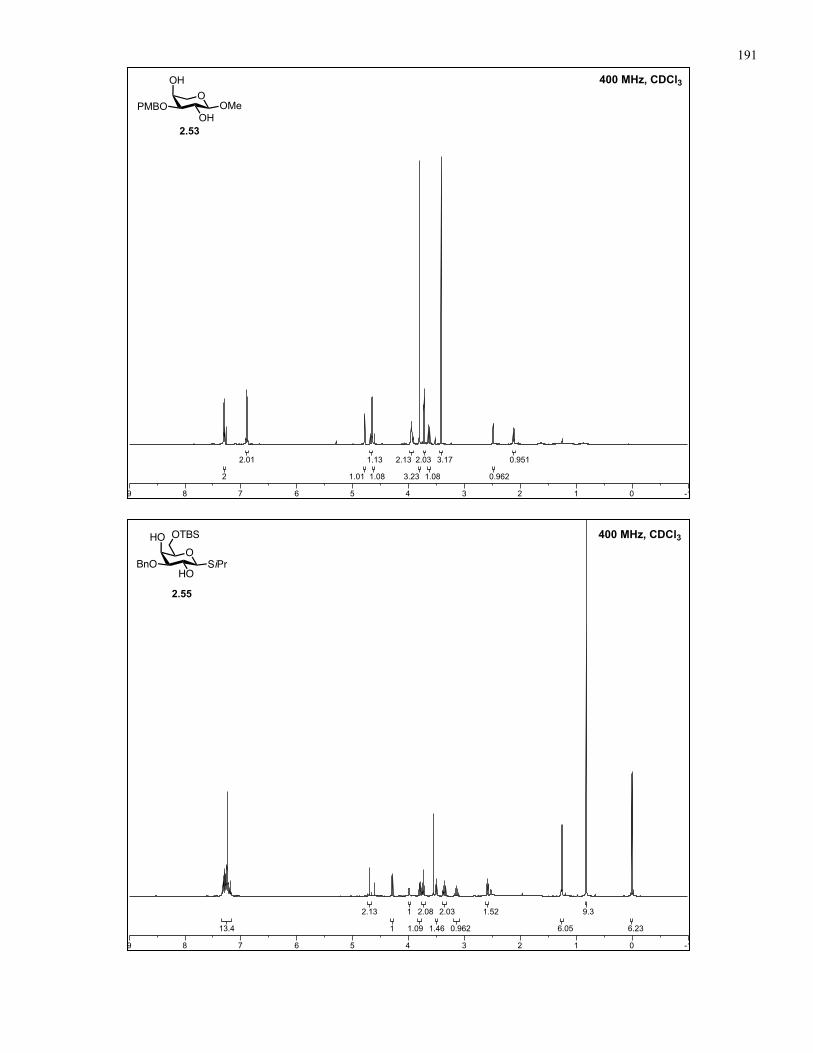
191
0.9510.951
0.9620.962
3.173.17
1.081.08
2.032.03
3.233.23
2.132.13
1.081.08
1.131.13
1.011.01
2.012.01
22
-1-100112233445566778899ppm
OPMBO OMe
OH
OH2.53
6.236.23
9.39.3
6.056.05
1.521.52
0.9620.962
2.032.03
1.461.46
2.082.08
1.091.09
11
11
2.132.13
13.413.4
-1-100112233445566778899ppm
OOTBSHO
BnOHO
SiPr
2.55
400 MHz, CDCl3
400 MHz, CDCl3
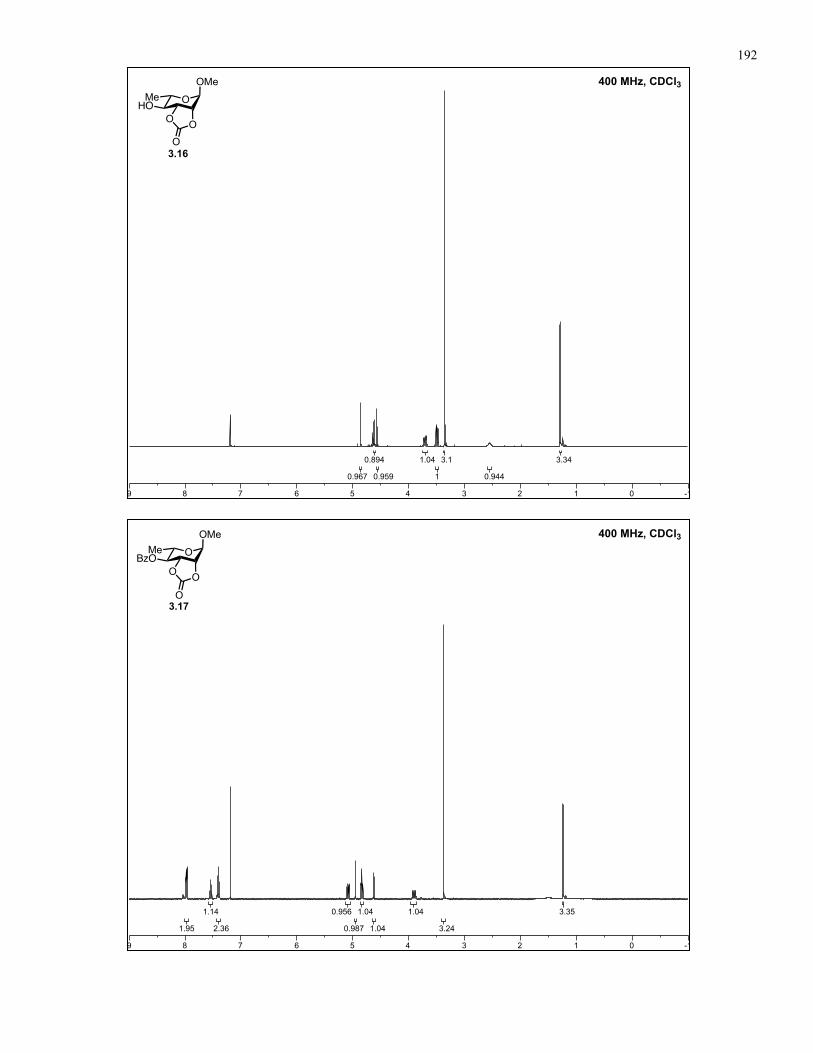
192
3.343.34
0.9440.944
3.13.1
11
1.041.04
0.9590.959
0.8940.894
0.9670.967
-1-100112233445566778899ppm
O
OMeMe
HOO O
O3.16
3.353.35
3.243.24
1.041.04
1.041.04
1.041.04
0.9870.987
0.9560.956
2.362.36
1.141.14
1.951.95
-1-100112233445566778899ppm
O
OMeMe
BzOO O
O3.17
400 MHz, CDCl3
400 MHz, CDCl3
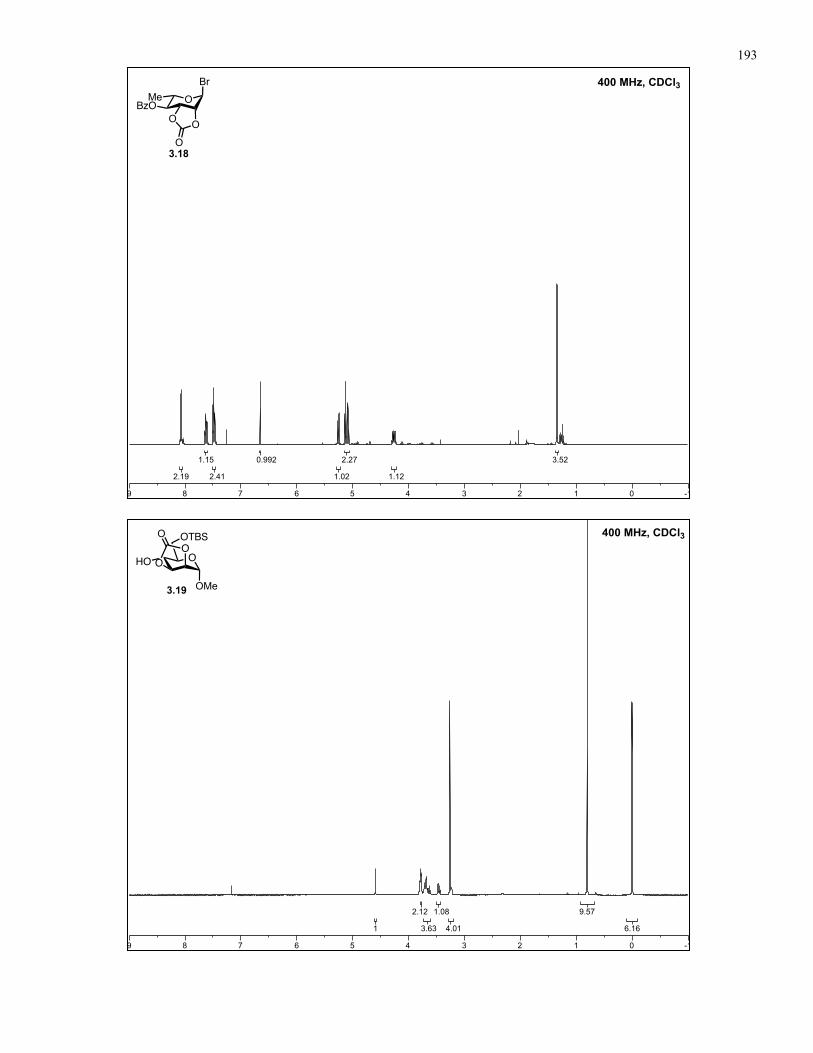
193
3.523.52
1.121.12
2.272.27
1.021.02
0.9920.992
2.412.41
1.151.15
2.192.19
-1-100112233445566778899ppm
O
BrMe
BzOO O
O3.18
6.166.16
9.579.57
4.014.01
1.081.08
3.633.63
2.122.12
11
-1-100112233445566778899ppm
3.19
O
OTBS
HO OO
OMe
O
400 MHz, CDCl3
400 MHz, CDCl3
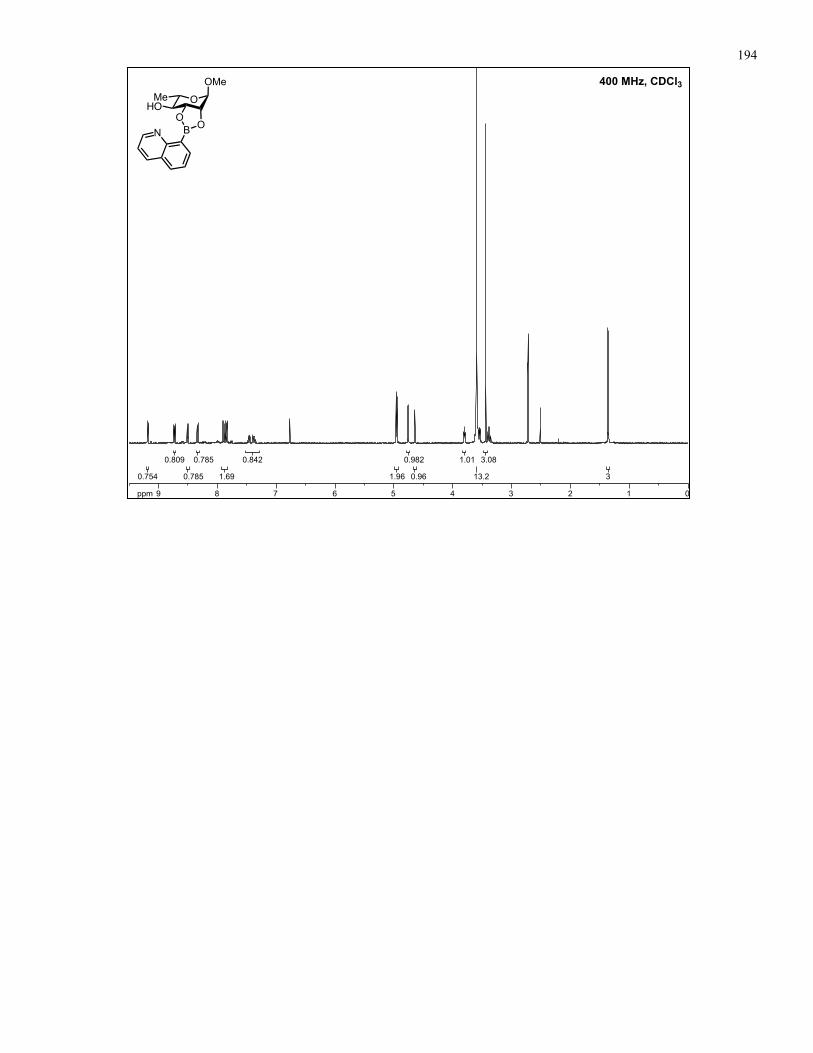
194
33
3.083.08
13.213.2
1.011.01
0.960.96
0.9820.982
1.961.96
0.8420.842
1.691.69
0.7850.785
0.7850.785
0.8090.809
0.7540.754
00112233445566778899ppmppm
O
OMeMe
HOO
OBN
400 MHz, CDCl3





























