Embed Size (px)
Citation preview

135
J. Electrounul. Chem., 314 (1991) 135-152 Elsevier Sequoia S.A., Lausanne
JEC 01639
Redox site loading, electrolyte concentration and temperature effects on charge transport and electrode kinetics of electrodes modified with osmium containing poly( 4-vinylpyridine) films in sulphuric acid
Robert J. Forster and Johannes G. Vos *
School of Chemical Sciences, Dublin City University, Dublin 9 (Ireland)
(Received 18 March 1991; in revised form 2 May 1991)
Abstract
The kinetic parameters for homogeneous charge transport and heterogeneous electron-transfer, DO and k o respectively, have been obtained as a function of the redox site loading and the concentration of sulphuric acid as the supporting electrolyte for electrodes modified with [Os(bipy)2(PVP),,Cl]Cl; 25 2 n > 5, bipy = 2,2’-bipyridyl, PVP = poly(4-vinylpyridine). Both short timescale potential step and cyclic voltammetry have been used to examine homogeneous charge transport, while sampled current voltam- metry has been used to examine electrode kinetics. A linear correlation between the homogeneous charge transport rate measured by potential step methods and /co is observed. The relevant activation parameters for homogeneous charge percolation, E,, AS* and AG* as well as the relevant enthalpy
terms ((A&” &I and (AHt”)iacal) together with the entropy terms ((AS;), and (ASt)ideal) for heterogeneous electron transfer have been evaluated. The temperature dependence of the Os(II/III) formal potential has been used to calculate (AS,) in order to examine the effect of osmium loading and electrolyte concentration on the local microenvironment of the immobilised redox couple.
INTRODUCTION
An important feature determinin g the practical applicability of polymer modified electrodes concerns the rate of homogeneous charge transport within the film [l]. In this contribution we report on the effects of redox site and electrolyte concentration, as well as temperature on the homogeneous charge transport rate Dcr, for [Os(bipy)z(PVP),Cl]Cl films in sulphuric acid (bipy = 2,2’-bipyridyl; PVP = poly(C vinylpyridine); 25 > n > 5). The physico-chemical properties of the poly(4+inyl- pyridine) backbone, to which the osmium centres are coordinatively bound, are
* To whom correspondence should be addressed.
0022-0728/91/$03.50 0 1991 - Blsevier Sequoia S.A. All rights reserved

136
expected to be dependent on both the osmium loading and sulphuric acid con- centration. Changes in the polymer morphology or structure are likely to affect processes, such as internal polymer fluidity or ion diffusion rates, which may limit the rate at which charge can be transported through the film [2]. By examining the sensitivity of DCT to osmium loading and sulphuric acid concentration, in conjunc- tion with thermodynamic parameters, the rate determining step of the homogeneous charge transport process for each redox site loading has been established. Charge transport rates have been evaluated at different timescales, allowing information about the nature of initial charge injection and percolation rates, as well as charge propagation through the bulk material to be obtained.
Heterogeneous electron transfer involving surface attached layers may represent better models for examining electron transfer theories than outer sphere electro- chemical reactions since the reactant and product can be identified with precursor and successor states [3]. Despite this amenability to both thermodynamic and kinetic investigation, the examination of heterogeneous electron transfer reactions of polymer modified electrodes containing immobile redox centres has received little attention [4]. In order to probe the kinetics of heterogeneous electron transfer and the nature of its coupling to homogeneous charge propagation, the rate of heteroge- neous electron transfer, k O, as well as the relevant enthalpic and entropic parame- ters have been evaluated as a function of redox site loading and sulphuric acid concentration.
EXPERIMENTAL
Materials The modifying metallopolymers were prepared by refluxing [Os(bipy),Cl,] with
the appropriate amount of PVP in ethanol. A detailed description of the synthesis, spectroscopic and thermal properties of the materials is reported elsewhere [5].
Apparatus and procedures Electrochemical measurements were performed using an EG&G Model 273
potentiostat/ galvanostat. Where necessary, IR compensation was achieved via positive feedback circuitry. Transient amperometric/coulometric measurements were made using a Philips 3311 digital storage oscilloscope interfaced to a BBC microcomputer for data interrogation and allowing signal averaged results to be obtained. Sampled current voltammetry was performed using samples times of 1,2, 4 and 10 ms, a pulse width of 200 ms and an interval of 20 s between successive pulses. The current was sampled before application of the potential step and confirmed to be zero, which ensures that the depletion layer of the oxidised species due to the preceding step has been completely re-reduced. Adherent electrode coatings were obtained by evaporation of the required volume of a 1% solution of the metallopolymer in methanol on the electrode surface in a solvent saturated chamber followed by air drying. All potentials are referenced with respect to the potassium chloride saturated calomel electrode (SCE) without regard for liquid junction potentials. A non isothermal electrochemical cell, where the reference

137
electrode was isolated from the main compartment and held at a constant tempera- ture of 25 o C, was used for the investigation of temperature effects on k ’ and E O. This was connected via a salt bridge to the main electrochemical cell in which the temperature could be controlled to within f 0.5 o C.
The formal potentials of the Os(II/III) oxidation were obtained using slow sweep rate cyclic voltammetry (1 mV/s) where the anodic and cathodic branches con- verged. Surface coverages were estimated by graphical integration of the back- ground corrected slow sweep rate cyclic voltammograms (1 mV/s), and were typically 2 X lo-* mol cmm2. The quantity of osmium immobilised on the electrode surface was kept constant as the loading was varied. This means that the layer thickness for the n = 25 loading is approximately 3.5 times greater than the n = 5 loading. However, the same results within experimental error are obtained for homogeneous and heterogeneous charge transfer when layer thickness is kept constant. Where the layer thickness is varied more extremely DC- typically decreases with increasing film thickness. The advantage of keeping the quantity of osmium on the electrode constant is that measurable signals are obtained where the electrolyte, redox site concentrations and temperature are low. Layer thickness was estimated from the individual densities of the dry metallopolymers as measured by flotation in non swelling solvents; n = 25, 1.07 g/cm3, n = 20, 1.08 g/cm3; n = 15, 1.09 g/cm3; n = 10, 1.20 g/cm3 and n = 5 1.40 g/cm3. These gave a value for the maximum concentration of osmium centres within the film as follows: n = 25, 0.33 mol/l; n = 20, 0.38 mol/l; n = 15, 0.46 mol/l; n = 10, 0.70 mol/l and n = 5, 1.17 mol/l. These values have been used to calculate Dcr from the experimentally determined
In studies where the electrolyte concentration was varied, all the experiments were carried out with fresh coatings. This avoids possible problems from “memory effects” where the rate of charge transport through a film would be dependent on the electrolytes to which it had previously been exposed. The values for DcT and k” as well as the activation parameters are reproducible to within +2% on a single coating and to f 10% between coatings. Transfer coefficients are accurate to + 0.05.
RESULTS
Homogeneous charge transport
General In potential step experiments linear Cottrell plots [6] showing zero current
intercepts were obtained for all active site/ electrolyte concentration combinations for times up to at least 20 ms, indicating a semi-infinite diffusion/migration response [7]. In order to calculate DcT from these plots the response must be diffusional in character and migration must be absent. A migrational contribution can be diagnosed in several ways, such as a non-zero current intercept in the Cottrell plot [8] or a peak in the It’j2 vs t”* plot [9], neither of which are observed . in this investigation. Further, in the present study DcT, evaluated using potential
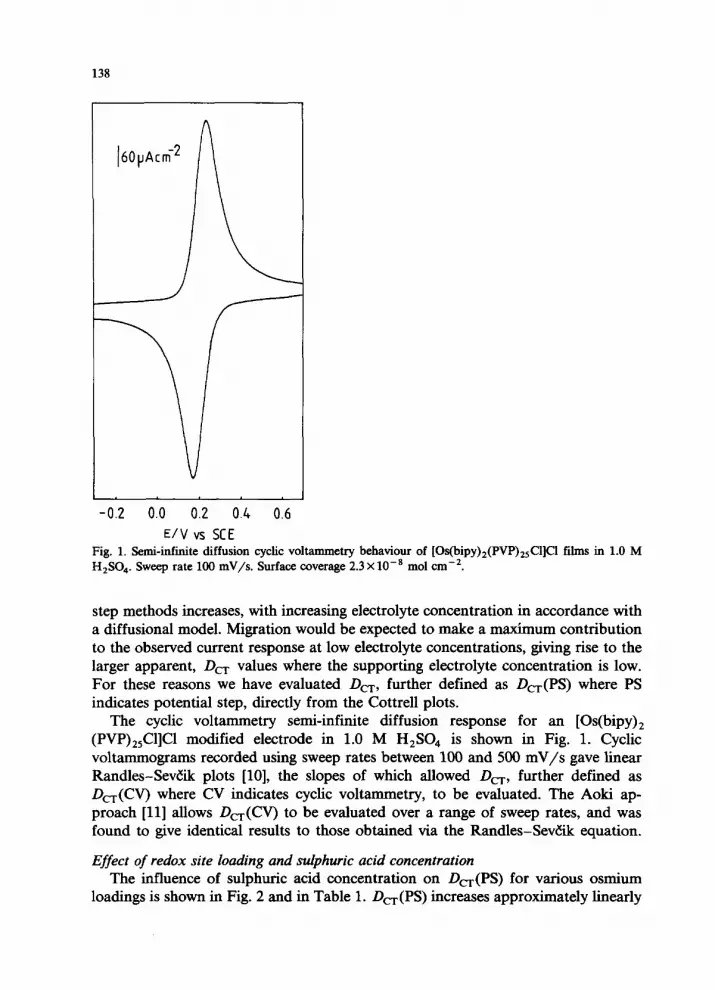
138
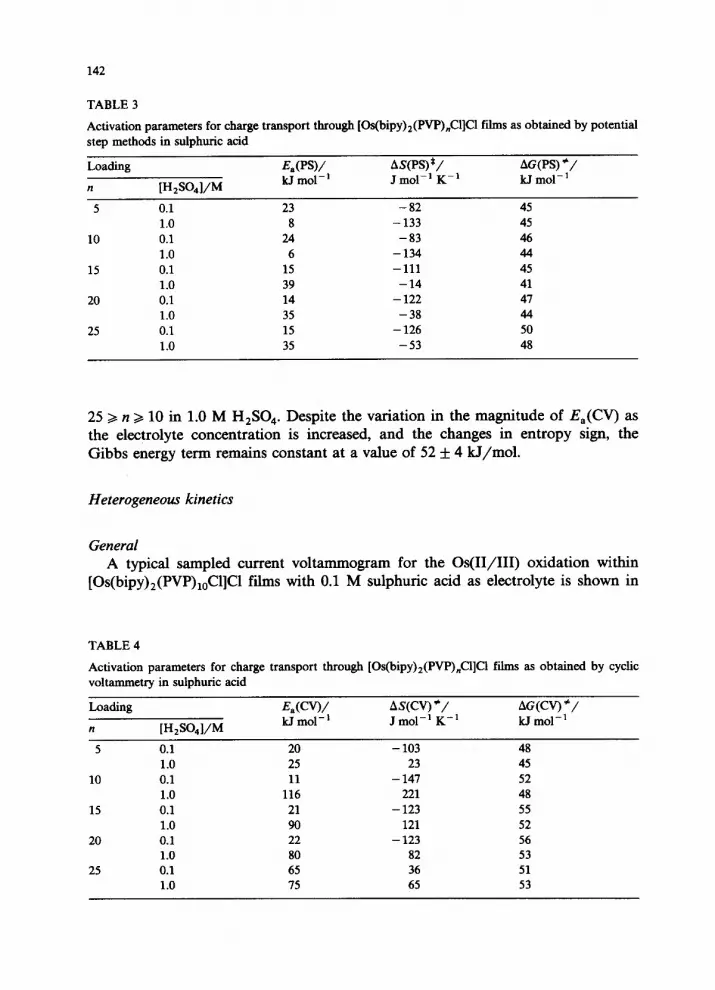
-0.2 0.0 0.2 0.4 0.6
E/V vs SCE Fig. 1. Semi-infinite diffusion cyclic voltammetry behaviour of [Os(bipy)2(PVP),,Cl]cl films in 1.0 M H,SO.,. Sweep rate 100 mV/s. Surface coverage 2.3 X10-s mol cm-‘.
step methods increases, with increasing electrolyte concentration in accordance with a diffusional model. Migration would be expected to make a maximum contribution to the observed current response at low electrolyte concentrations, giving rise to the larger apparent, Dcr values where the supporting electrolyte concentration is low. For these reasons we have evaluated Dcr, further defined as D,(PS) where PS indicates potential step, directly from the Cottrell plots.
The cyclic voltammetry semi-infinite diffusion response for an [Os(bipy), (PVP),,Cl]Cl modified electrode in 1.0 M H,SO, is shown in Fig. 1. Cyclic voltammograms recorded using sweep rates between 100 and 500 mV/s gave linear Randles-SevZik plots [lo], the slopes of which allowed DC-, further defined as DC-(W) where CV indicates cyclic voltammetry, to be evaluated. The Aoki ap- proach [ll] allows D,(O) to be evaluated over a range of sweep rates, and was found to give identical results to those obtained via the Randles-SevZik equation.
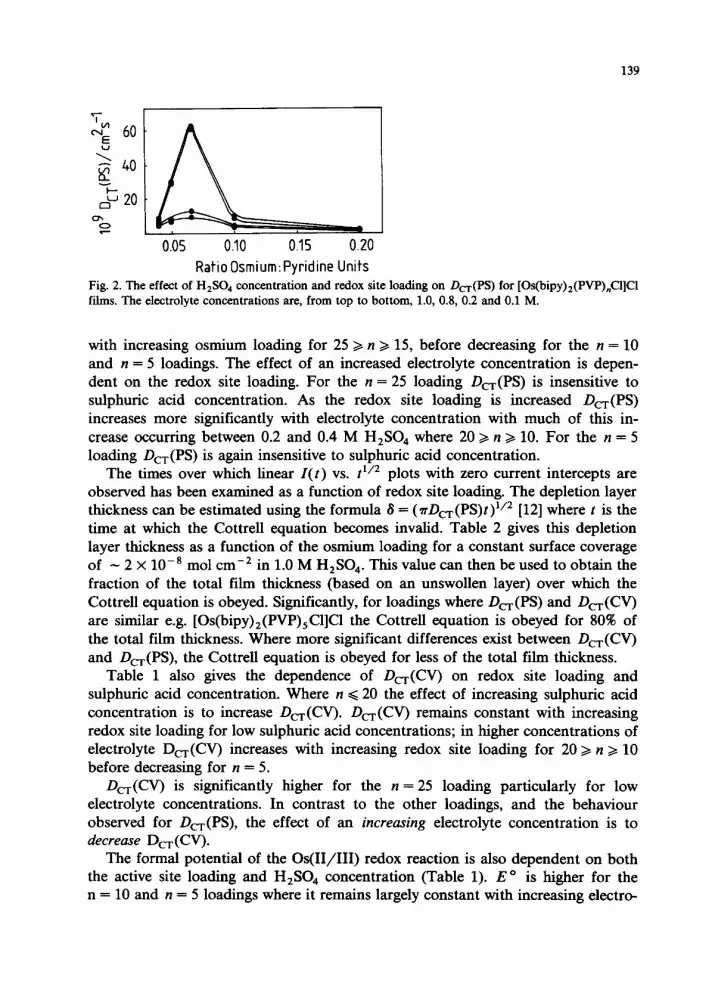
Effect of redox site loading and sulphuric acid concentration The influence of sulphuric acid concentration on D,(PS) for various osmium
loadings is shown in Fig. 2 and in Table 1. D,(PS) increases approximately linearly
139
A; 60
2 40
-i-- 0" 20 m 0
0.05 0.10 0.15 0.20
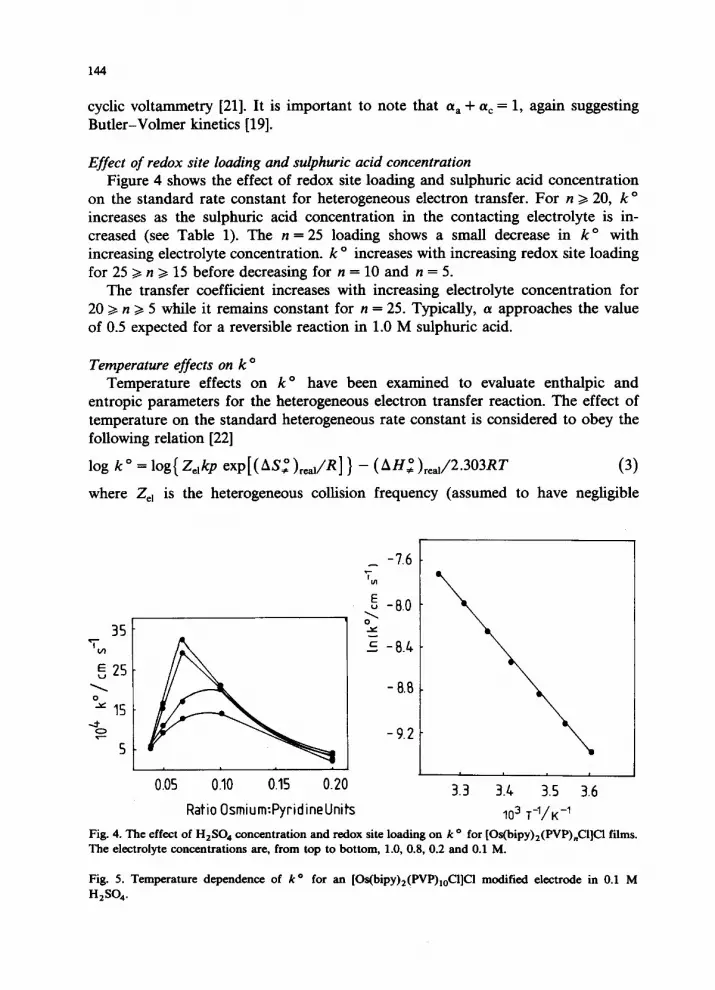
RatioOsmium:Pyridine Units Fig. 2. The effect of H2S04 concentration and redox site loading on D,(PS) for [Os(bipy)z(PVP),Cl]C1 films. The electrolyte concentrations are, from top to bottom, 1.0, 0.8, 0.2 and 0.1 M.
with increasing osmium loading for 25 >, n z 15, before decreasing for the n = 10 and n = 5 loadings. The effect of an increased electrolyte concentration is depen- dent on the redox site loading. For the n = 25 loading Dc,(PS) is insensitive to sulphuric acid concentration. As the redox site loading is increased D,(PS) increases more significantly with electrolyte concentration with much of this in- crease occurring between 0.2 and 0.4 M H,SO, where 20 z n 2 10. For the n = 5 loading Dc,(PS) is again insensitive to sulphuric acid concentration.
The times over which linear I(t) vs. t’/* plots with zero current intercepts are observed has been examined as a function of redox site loading. The depletion layer thickness can be estimated using the formula 6 = ( sDCT(PS)t)l/* [12] where t is the time at which the Cottrell equation becomes invalid. Table 2 gives this depletion layer thickness as a function of the osmium loading for a constant surface coverage of - 2 x lop8 mol cm-* in 1.0 M H,SO,. This value can then be used to obtain the fraction of the total film thickness (based on an unswollen layer) over which the Cottrell equation is obeyed. Significantly, for loadings where &(PS) and D,(CV) are similar e.g. [Os(bipy),(PVP),Cl]Cl the Cottrell equation is obeyed for 80% of the total film thickness. Where more significant differences exist between Dcr(CV) and D,(PS), the Cottrell equation is obeyed for less of the total film thickness.
Table 1 also gives the dependence of D,(CV) on redox site loading and sulphuric acid concentration. Where n Q 20 the effect of increasing sulphuric acid concentration is to increase Dcr(CV). D,(CV) remains constant with increasing redox site loading for low sulphuric acid concentrations; in higher concentrations of electrolyte D&X) increases with increasing redox site loading for 20 2 n 2 10 before decreasing for n = 5.
D&CV) is significantly higher for the n = 25 loading particularly for low electrolyte concentrations. In contrast to the other loadings, and the behaviour observed for D,(PS), the effect of an increasing electrolyte concentration is to decrease DcT (0’).
The formal potential of the Os(II/III) redox reaction is also dependent on both the active site loading and H2S04 concentration (Table 1). E o is higher for the n = 10 and n = 5 loadings where it remains largely constant with increasing electro-
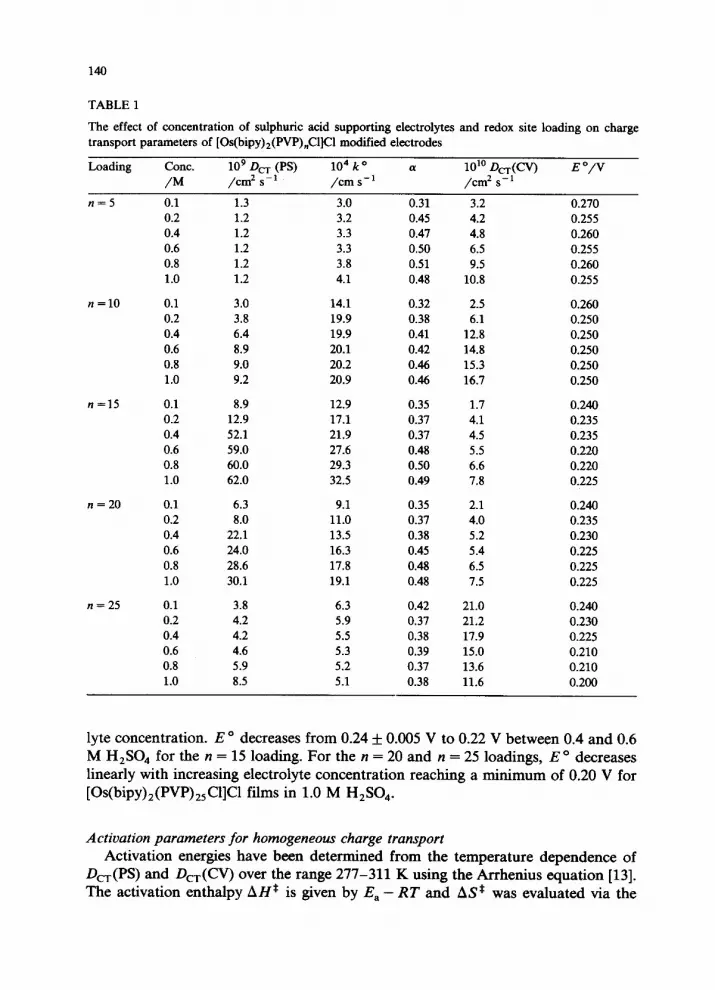
140
TABLE 1
The effect of concentration of sulphuric acid supporting electrolytes and redox site loading on charge
transport parameters of [Os(bipy),(PVP),Cl]Cl modified electrodes
Loading Cont.
/M
lo9 D, (PS) /cm2 s-’
104k0 (I 10” D,(W) E “/V /cm s-l /cm* s-l
n=5
n=lO
n=15
” = 20
n = 25
0.1 1.3 3.0 0.31 3.2 0.270 0.2 1.2 3.2 0.45 4.2 0.255 0.4 1.2 3.3 0.47 4.8 0.260 0.6 1.2 3.3 0.50 6.5 0.255 0.8 1.2 3.8 0.51 9.5 0.260 1.0 1.2 4.1 0.48 10.8 0.255
0.1 3.0 14.1 0.32 2.5 0.260 0.2 3.8 19.9 0.38 6.1 0.250 0.4 6.4 19.9 0.41 12.8 0.250 0.6 8.9 20.1 0.42 14.8 0.250 0.8 9.0 20.2 0.46 15.3 0.250 1.0 9.2 20.9 0.46 16.7 0.250
0.1 8.9 12.9 0.35 1.7 0.240 0.2 12.9 17.1 0.37 4.1 0.235 0.4 52.1 21.9 0.37 4.5 0.235 0.6 59.0 27.6 0.48 5.5 0.220 0.8 60.0 29.3 0.50 6.6 0.220 1.0 62.0 32.5 0.49 7.8 0.225
0.1 6.3 9.1 0.35 2.1 0.240 0.2 8.0 11.0 0.37 4.0 0.235 0.4 22.1 13.5 0.38 5.2 0.230 0.6 24.0 16.3 0.45 5.4 0.225 0.8 28.6 17.8 0.48 6.5 0.225 1.0 30.1 19.1 0.48 7.5 0.225
0.1 3.8 6.3 0.42 21.0 0.240 0.2 4.2 5.9 0.37 21.2 0.230 0.4 4.2 5.5 0.38 17.9 0.225 0.6 4.6 5.3 0.39 15.0 0.210 0.8 5.9 5.2 0.37 13.6 0.210 1.0 8.5 5.1 0.38 11.6 0.200
lyte concentration. E o decreases from 0.24 + 0.005 V to 0.22 V between 0.4 and 0.6 M H,SO, for the n = 15 loading. For the n = 20 and n = 25 loadings, E o decreases linearly with increasing electrolyte concentration reaching a minimum of 0.20 V for [Os(bipy),(PVP),,Cl]Cl films in 1.0 M H,SO,.
Activation parameters for homogeneous charge transport Activation energies have been determined from the temperature dependence of
D,(PS) and Q-r(W) over the range 277-311 K using the Arrhenius equation [13]. The activation enthalpy AH* is given by E, - RT and AS* was evaluated via the
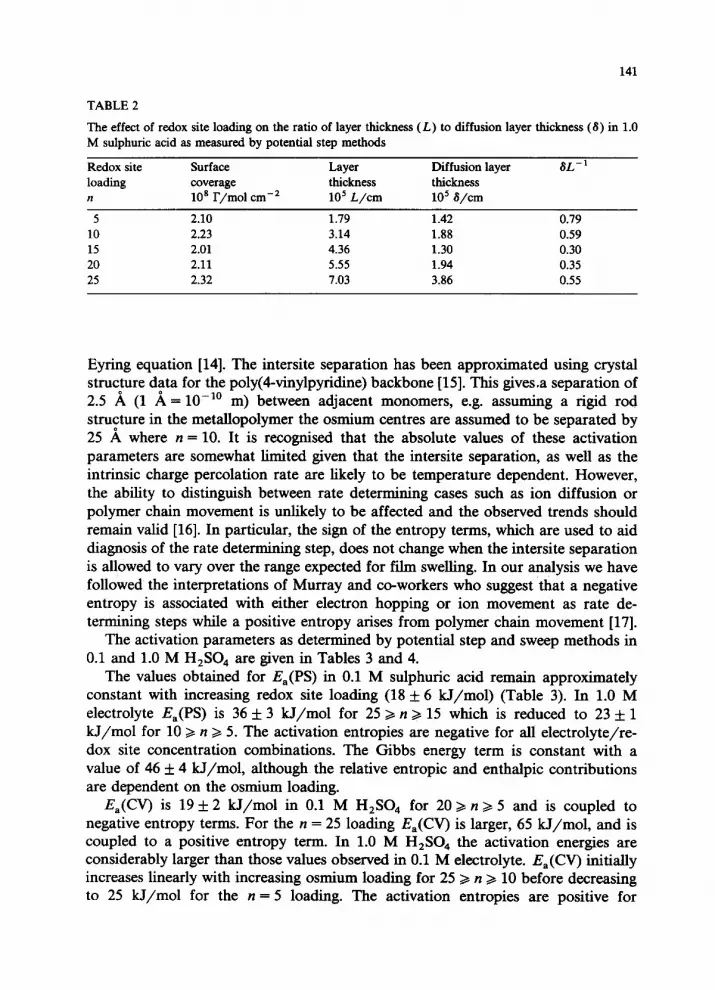
141
TABLE 2
The effect of redox site loading on the ratio of layer thickness (L) to diffusion layer thickness (8) in 1.0
M sulphuric acid as measured by potential step methods
Redox site Surface Layer Diffusion layer
loading coverage thickness thickness
n 10’ r/m01 cm-’ lo5 L/cm lo5 S/cm
5 2.10 1.79 1.42
10 2.23 3.14 1.88
15 2.01 4.36 1.30
20 2.11 5.55 1.94
25 2.32 7.03 3.86
SL-’
0.79
0.59 0.30
0.35
0.55
Eyring equation [14]. The intersite separation has been approximated using crystal structure data for the poly(4-vinylpyridine) backbone [15]. This givesa separation of 2.5 A (1 A = lo-” m) between adjacent monomers, e.g. assuming a rigid rod strufture in the metallopolymer the osmium centres are assumed to be separated by 25 A where n = 10. It is recognised that the absolute values of these activation parameters are somewhat limited given that the intersite separation, as well as the intrinsic charge percolation rate are likely to be temperature dependent. However, the ability to distinguish between rate determining cases such as ion diffusion or polymer chain movement is unlikely to be affected and the observed trends should remain valid [16]. In particular, the sign of the entropy terms, which are used to aid diagnosis of the rate determinin g step, does not change when the intersite separation is allowed to vary over the range expected for fihn swelling. In our analysis we have followed the interpretations of Murray and co-workers who suggest that a negative entropy is associated with either electron hopping or ion movement as rate de- termining steps while a positive entropy arises from polymer chain movement [17].
The activation parameters as determined by potential step and sweep methods in 0.1 and 1.0 M H,SO, are given in Tables 3 and 4.
The values obtained for E,(PS) in 0.1 M sulphuric acid remain approximately constant with increasing redox site loading (18 + 6 kJ/mol) (Table 3). In 1.0 M electrolyte E,(PS) is 36 f 3 kJ/mol for 25 > n > 15 which is reduced to 23 f 1 kJ/mol for 10 > n > 5. The activation entropies are negative for all electrolyte/re- dox site concentration combinations. The Gibbs energy term is constant with a value of 46 f 4 kJ/mol, although the relative entropic and enthalpic contributions are dependent on the osmium loading.
E,(0) is 19 + 2 kJ/mol in 0.1 M H,SO, for 20 > n z 5 and is coupled to negative entropy terms. For the n = 25 loading E,(W) is larger, 65 kJ/mol, and is coupled to a positive entropy term. In 1.0 M H,SO, the activation energies are considerably larger than those values observed in 0.1 M electrolyte. E,(CV) initially increases linearly with increasing osmium loading for 25 2 n > 10 before decreasing to 25 kJ/mol for the n = 5 loading. The activation entropies are positive for
142
TABLE 3
Activation parameters for charge transport through [Os(bipy),(PVP),Cl]Cl films as obtained by potential step methods in sulphuric acid
Loading &(PS)/ AS(PS)*/ AG(PS) ‘/
n F-WWM kJ mol-’ J mol-’ K-’ kJ mol-’
5 0.1 23 -82 45 1.0 8 - 133 45
10 0.1 24 -83 46 1.0 6 -134 44
15 0.1 15 -111 45 1.0 39 -14 41
20 0.1 14 - 122 47 1.0 35 -38 44
25 0.1 15 -126 50 1.0 35 -53 48
25 > n > 10 in 1.0 M H,SO,. Despite the variation in the magnitude of E,(CV) as the electrolyte concentration is increased, and the changes in entropy sign, the Gibbs energy term remains constant at a value of 52 f 4 kJ/mol.
Heterogeneous kinetics
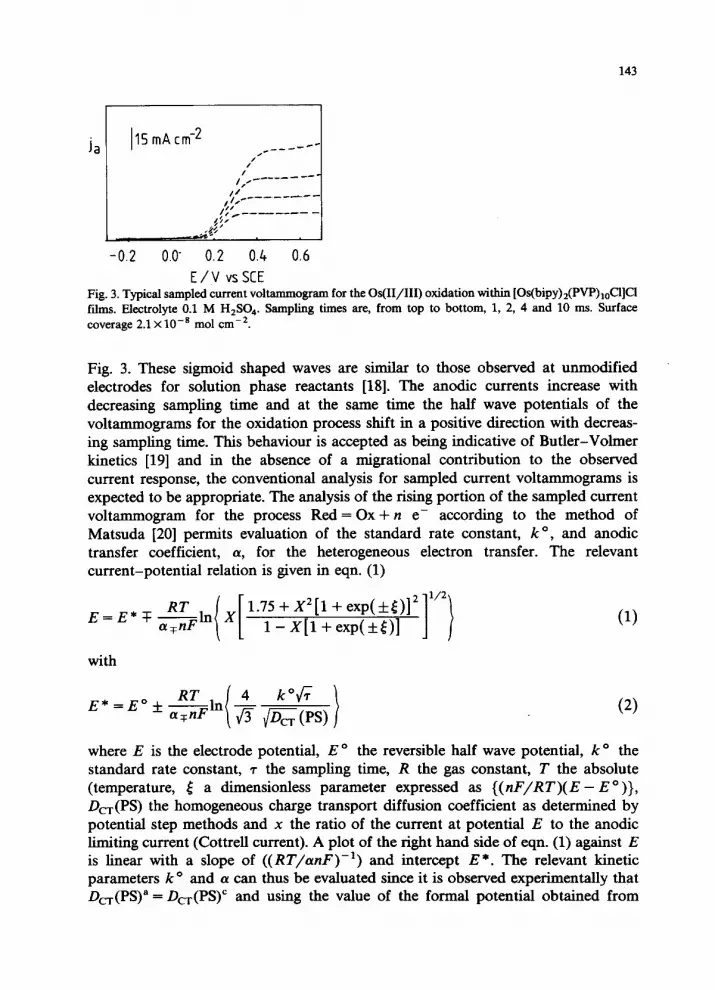
General A typical sampled current voltammogram for the Os(II/III) oxidation within
[Os(bipy)2(PVP),,C1]C1 films with 0.1 M sulphuric acid as electrolyte is shown in
TABLE 4
Activation parameters for charge transport through [Os(bipy)2(PVP)$l]C films as obtained by cyclic voltammetry in sulphuric acid
Loading &l(Cv)/ AS(Cv) l / AG(Cv) +/
Ff,SO.,I/M kJ mol-’ J mol-’ K-’ kJ mol-’ ”
5 0.1 20 - 103 48 1.0 25 23 45
10 0.1 11 - 147 52 1.0 116 221 48
15 0.1 21 - 123 55 1.0 90 121 52
20 0.1 22 - 123 56 1.0 80 82 53
25 0.1 65 36 51 1.0 75 65 53
143
la 15 mA cm-*
,/--- __--
I’ I/------ --
/l l,---------- 1, 5, - $2, c-------
.*g - -0.2 0.0’ 0.2 0.4 0.6
E/V vs SCE Fig. 3. Typical sampled current voltammogram for the Os(II/III) oxidation within [Os(bipy)z(PVP),,CI]Cl films. Electrolyte 0.1 M H,S04. Sampling times are, from top to bottom, 1, 2, 4 and 10 ms. Surface coverage 2.1 X lo-* mol cm-*.
Fig. 3. These sigmoid shaped waves are similar to those observed at unmodified electrodes for solution phase reactants [18]. The anodic currents increase with decreasing sampling time and at the same time the half wave potentials of the voltammograms for the oxidation process shift in a positive direction with decreas- ing sampling time. This behaviour is accepted as being indicative of Butler-Volmer kinetics [19] and in the absence of a migrational contribution to the observed current response, the conventional analysis for sampled current voltammograms is expected to be appropriate. The analysis of the rising portion of the sampled current voltammogram for the process Red = Ox + n e- according to the method of Matsuda [20] permits evaluation of the standard rate constant, k O, and anodic transfer coefficient, (Y, for the heterogeneous electron transfer. The relevant current-potential relation is given in eqn. (1)
E=E*T 1.75 + X2[1 + exp(+t)]* “*)
1 - X[l + exp(+t)l 1 j with
E*=E”f
1 (1)
(2)
where E is the electrode potential, E o the reversible half wave potential, k” the standard rate constant, r the sampling time, R the gas constant, T the absolute (temperature, 5 a dimensionless parameter expressed as {( nF/RT)( E - E o )}, Dc,(PS) the homogeneous charge transport diffusion coefficient as determined by potential step methods and x the ratio of the current at potential E to the anodic limiting current (Cottrell current). A plot of the right hand side of eqn. (1) against E is linear with a slope of ((RT/anP)-‘) and intercept E *. The relevant kinetic parameters k” and (Y can thus be evaluated since it is observed experimentally that Dcr(PS)‘= Dcr(PS)’ and using the value of the formal potential obtained from
144
cyclic voltammetry [21]. It is important to note that (Y, + (Y, = 1, again suggesting Butler-Volmer kinetics [19].
Effect of redox site loading and sulphuric acid concentration Figure 4 shows the effect of redox site loading and sulphuric acid concentration
on the standard rate constant for heterogeneous electron transfer. For n > 20, k o increases as the sulphuric acid concentration in the contacting electrolyte is in- creased (see Table 1). The n = 25 loading shows a small decrease in k o with increasing electrolyte concentration. k o increases with increasing redox site loading for 25 z n >, 15 before decreasing for n = 10 and n = 5.
The transfer coefficient increases with increasing electrolyte concentration for 20 z n z 5 while it remains constant for n = 25. Typically, (Y approaches the value of 0.5 expected for a reversible reaction in 1.0 M sulphuric acid.
Temperature effects on k O Temperature effects on k” have been examined to evaluate enthalpic and
entropic parameters for the heterogeneous electron transfer reaction. The effect of temperature on the standard heterogeneous rate constant is considered to obey the following relation [22]
log k” = log{ Z,,kp exp[ (AS," ),,,/R] } - (AH,“),,/2.303RT (3)
where Z,, is the heterogeneous collision frequency (assumed to have negligible
35 7*
5 25 \
Ox 15 % -5
0.05 0.10 0.15 0.20
Ratio 0smium:PyridineUnits
_ -7.6
L
< -8.0 0 Y
+ -8.4
- 8.8
- 9.2
3.3 3.4 3.5 3.6
IO3 T-‘/K-’
Fig. 4. The effect of H,SO, concentration and redox site loading on k o for [Os(bipy)2(PVP),Cl]Cl films. The electrolyte concentrations are, from top to bottom, 1.0, 0.8, 0.2 and 0.1 M.
Fig. 5. Temperature dependence of /co for an [Os(bipy)2(PVP),,-$l]Cl modified electrode in 0.1 M H,SO,.

145
temperature dependence), k and p are constants (assumed equal to unity), (AS,“),,,
and (AH,O),,, ep r resent the entropic and enthalpic barriers at the standard poten- tial after correction for the entropic and enthalpic driving forces, AH,” ( = T A$: ) and A&E respectively. AS,0 can be calculated from the temperature dependence of the formal potential of the redox couple [23,24]:
AS; = F(d E “/dT) ‘(4)
This reaction entropy difference between the reduced and oxidised forms of the redox couple can also be evaluated using the Born electrostatic model [25]:
(As: horn = 40.38( z&,/~~, - z&/u,) J mol-’ K-’ (5)
where tOx and zaed are the net charges, and aox and aRed are the equivalent radii in angstroms of the oxidised and reduced species respectively.
The temperature dependence of k o can also be used to calculate (AH o ) ideal and
(AS”)ideat, which give the enthalpic and entropic barriers to electron transfer at a particular potential where they are evaluated, from eqns. (6) and (7) [26]
(AH” )ideat = (AHO),, + aTAS; (6)
(7) Figure 5 shows the temperature dependence of k o for an [Os(bipy)z(PVP),,,Cl]Cl
modified electrode in 0.1 M H,SO,. This behaviour is typical of all redox site/ electrolyte concentration combinations and shows that log k” varies linearly with T-’ indicating the applicability of eqn. (3). (AS:),,, and (AH:),ea, can thus be calculated from the slope and intercept respectively assuming Z,, = - low4 cm s-l.
Table 5 gives the dependence of these thermodynamic parameters on the redox site loading in 0.1 and 1.0 M H,SO,. In 0.1 M H,SO, the enthalpy and entropy of
TABLE 5
Thermodynamic parameters for the heterogeneous electron transfer reaction from [Os@ipy)2(PVP),C1]C1 modified electrodes in sulphuric acid
Loading (AH: )d (AS:),,/ AW (AH,O )idd IH,SO,],M /kJ mol-’ J mol-’ K-’ J mol-’ K-’ /lcJ mol-’
(ASi )idd/
n J mol-’ K-’
5 0.1 40 -9 20 42 -3 1.0 22 -61 18 25 -58
10 0.1 25 -55 19 21 -49 1.0 34 -21 20 36 -18
15 0.1 20 -71 21 22 -64 1.0 38 -13 19 40 -4
20 0.1 18 -80 20 20 -72 1.0 39 -8 20 42 11
25 0.1 16 -82 19 19 -75 1.0 41 -1 20 44 7
146
: 0.217 -
9 $ 0.215 - w
0.213 .
0.211 -
10 20 30 40 T /"C
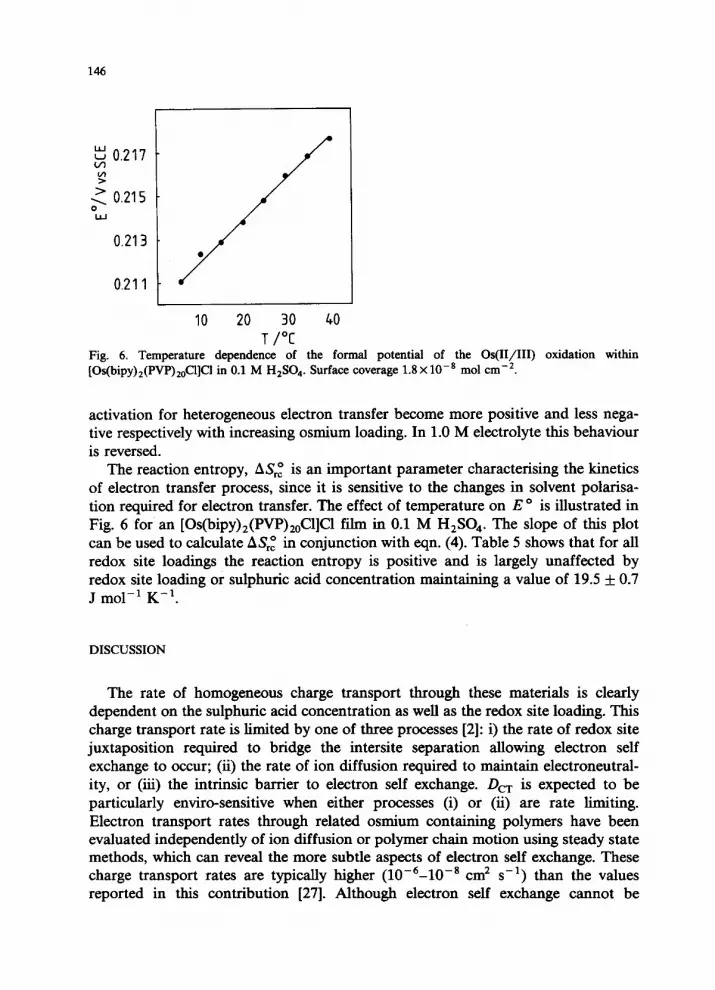
Fig. 6. Temperature dependence. of the formal potential of the Os(II/III) oxidation within [Os(bipy),(PVP)&l]Cl in 0.1 M H,SO.,. Surface coverage 1.8 x lo-* mol cm-*.
activation for heterogeneous electron transfer become more positive and less nega- tive respectively with increasing osmium loading. In 1.0 M electrolyte this behaviour is reversed.
The reaction entropy, ASrz is an important parameter characterising the kinetics of electron transfer process, since it is sensitive to the changes in solvent polarisa- tion required for electron transfer. The effect of temperature on E o is illustrated in Fig. 6 for an [Os(bipy)z(PVP),,C1]C1 film in 0.1 M H,SO,. The slope of this plot can be used to calculate A&z in conjunction with eqn. (4). Table 5 shows that for all redox site loadings the reaction entropy is positive and is largely unaffected by redox site loading or sulphuric acid concentration maintaining a value of 19.5 + 0.7 J mol-’ K-‘.
DISCUSSION
The rate of homogeneous charge transport through these materials is clearly dependent on the sulphuric acid concentration as well as the redox site loading. This charge transport rate is limited by one of three processes [2]: i) the rate of redox site juxtaposition required to bridge the intersite separation allowing electron self exchange to occur; (ii) the rate of ion diffusion required to maintain electroneutral- ity, or (iii) the intrinsic barrier to electron self exchange. DcT is expected to be particularly em&-sensitive when either processes (i) or (ii) are rate limiting. Electron transport rates through related osmium containing polymers have been evaluated independently of ion diffusion or polymer chain motion using steady state methods, which can reveal the more subtle aspects of electron self exchange. These charge transport rates are typically higher (1O-6-1O-8 cm2 s-l) than the values reported in this contribution [27]. Although electron self exchange cannot be

147
excluded formally, these observations, in conjunction with the thermodynamic data, where the lowest activation energies are consistent with ion diffusion through polymer matrices [28], suggest that electron self exchange does not represent the rate determining step of the charge transport process for these [Os(bipy),(PVP),Cl]Cl films.
Differences between D,,(PS) and D&CV) The differences in the charge transport rate as evaluated by potential step and
sweep methods suggest that DCT depends either on the experimental timescale or on the region of the film in which it is evaluated. The low E,(PS) and negative ASt(PS) terms (see below) for all redox site/electrolyte concentration combinations suggest D,(PS) is limited by ion diffusion. Under the longer timescales of the CV experiment either polymer chain motion (low loading/high electrolyte concentra- tions) or ion diffusion (high loadings/low electrolyte concentrations) limit DcT(CV). These observations suggest that the mass transport which is coupled to the initial charge injection and percolation, measured by D&I’S), occurs via highly mobile species e.g. counterions already resident within the film, while only at longer times do counterions, solvent and polymer chains move as dictated by the Os(II/III) concentration gradient established by electron transport. The extent of film oxida- tion over which the Cottrell equation is valid is linked closely to the relative difference between D,(PS) and D&W). Where D&PS) and D&W) are of similar magnitude e.g. [Os(bipy)2(PVP),Cl]Cl in 1.0 M H,SO, the Cottrell equation is valid for up to 80% of the calculated film thickness. Deviations from Cottrell behaviour under near exhaustive oxidation conditions are expected due to finite diffusion or uneven film thickness [29,30]. For other loadings, larger differences are observed between the charge transport rates evaluated using the potential step and sweep techniques. The fact that the variation between D,(PS) and DC&V) depends on both the redox site and electrolyte concentration suggests that the physico-chemical properties of the polymer phase influence the mobility of the species whose transport is coupled to charge propagation.
This time dependence of DcT is not entirely unexpected, given, for example, that the nature of the ion movement required is distinctly different for the two tech- niques. In the CV experiment, extensive oxidation of the fihn occurs requiring counterion diffusion to occur over greater spatial regions of the layer than does the potential step technique. As well as this spatial resolution the quantity of counter- ions which must diffuse is also considerably larger in the case of cyclic voltammetry experiments. In the extreme case, it is conceivable that on the recorded, short timescale of the potential step experiment counterion diffusion may be localised within the film.
Alternatively, it is possible that the osmium concentration or activity is different through the film thickness. This could arise if film swelling is greater at the film/electrolyte interface. However, in order for such a process to be fully responsi- ble for the differences observed between D&I’S) and D&W), then differential film swelling of over 300% would be required.

148
Effect of osmium loading sulphuric acid concentration and temperature on DCT D,(PS) is sensitive to both redox site loading and electrolyte concentration,
suggesting that either ion diffusion or polymer chain motion is rate limiting. These possible rate determining cases can be distinguished ‘from the temperature depen- dence of D,(PS). The values of E,(PS) are less than 40 kJ/mol and are coupled to negative entropy terms for all redox site loadings and sulphuric acid concentrations. These activation energies are consistent with ion diffusion through polymer matrices [28]. Negative entropy terms are also consistent with an ion diffusion limitation requiring an ordering deformation of the polymer matrix due to film expansion and reduced chain mobility [17]. In 1.0 M sulphuric acid the electrolyte concentration is above the osmium concentration for the 25 >, n > 15 loadings; this coupled to protonation of the large number of unbound pyridine units is likely to cause extensive film swelling [31]. This allows ion diffusion to proceed at a rate such that overall charge transport can increase with increasing osmium concentration and decreasing intersite separation. This increase in D,(PS) with increasing osmium concentration is not sustained indefinitely and Dc,(PS) decreases for the n = 10 and n = 5 loadings. This is considered to be associated with decreased fihn swelling or an ion exclusion process. Ion exclusion for high loadings, such as n = 5, is likely since the fixed site concentration is always greater than the electrolyte concentra- tion. The reduced value of E,(PS) for n = 5, which probably represents the barrier to ion diffusion within the film, agrees with the interpretation of impeded ion permeation rather than restricted ion diffusion within the film.
In 0.1 M electrolyte, the fixed site concentration is above the electrolyte con- centration for all loadings. Both D,(PS) and E,(PS) remain largely unchanged as the osmium loading is increased. These observations suggest that the rate of ion diffusion or availability is such that D,(PS) cannot increase with decreasing intersite separation. The fact that D&PS) is independent of redox site loading in 0.1 M H,S04 suggests strongly that electron self exchange does not represent the rate determinin g step of the charge transport process.
In high electrolyte concentrations, D&W) increases with increasing osmium loading for 20 > n > 10 before decreasing for n = 5. This suggests that the rate determining step of the charge transport process changes between the n = 10 and n = 5 loadings. This interpretation is supported by the thermodynamic data. In 1.0 M H,SO, E,(CV) is large and coupled to positive entropy terms for 20 2 n z 10. In contrast, E,(W) is reduced to 25 kJ/mol which is coupled to a negative entropy term for the n = 5 loading. This suggests that, for low redox site loadings, the films are extensively swollen and polymer chain motion is required to allow electron self exchange to occur. As the redox site loading is increased film swelling appears to be reduced, decreasing matrix fluidity and thus reducing the requirement of polymer chain motion and allowing D,(W) to increase. A further increase in osmium loading to n = 5 reduces film swelling resulting in an ion diffusion limitation.
In 0.1 M electrolyte both Dcr(CV) and E,(W) are insensitive to active site loading for 20 > n > 5. The low activation energies and negative entropy terms suggest that ion diffusion limits Dcr(CV) for these loadings. It is to be noted that

149
little variation is observed in G # (CV). This shows that entropy effects, imposed mostly by the polymer matrix, are primarily responsible for the large variations observed in the activation energies upon changes in electrolyte composition or redox site loading, and that the fundamental electron transfer process is largely unaf- fected.
The n = 25 loading exhibits unusual behaviour in that &&CV) decreases with increasing stdphuric acid concentration. The activation energy is 70 f 5 kJ/mol in both 0.1 and 1.0 M H,SO, and is coupled to a positive entropy term. This suggests that polymer chain motion limits D,(W) for this loading in both electrolyte concentrations. It appears therefore that the low metal content of the film together with electrostatic repulsion of the protonated pyridine monomer units results in extensive film swelling. Such swelling is likely to increase with increasing electrolyte concentration thus increasing the intersite separation. This acts to decrease D&CV) since polymer chain motion is rate limiting.
The physico-chemical properties of the metallopolymer films as dictated by the redox site loading and the electrolyte concentration also appear to be reflected in the formal potentials of the Os(II/III) reaction. Thus E o is reduced in high electrolyte concentrations or low loadings where electrolyte is more freely available within the layer. It is significant to note that E o is largest for the n = 5 loading where counterion diffusion represents the rate determining step in all electrolyte concentrations.
Effect of osmium loading, sulphuric acid concentration and temperature on k” and (Y The rate of heterogeneous electron transfer is considerably less than those
obtained for model monomeric species such as [Os(bipy),(Pic)Cl]Cl, Pit = 4- methylpyridine. This behaviour has been observed for previous systems [32], and interpreted in terms of a reduced area of the electrode surface which is active for electron exchange [33,34]. Such a process appears reasonable since the polymer backbone is non-conducting, and electron transfer between the film and underlying electrode can occur only at the immobilised osmium centres. However, it is to be expected that an increased osmium loading would increase the number of these active sites resulting in an increased k ‘. The fact that k” increases only for 25 > n 2 15 and 25 > n > 10 for high and low electrolyte concentrations respec- tively, suggests that this model cannot fully explain these results.
In 0.1 M electrolytes both (AH:),, and (AS,“), increase as the redox site loading is increased, the enthalpy becoming more positive and the entropy becom- ing less negative (see Table 5). However, in 1.0 M H,SO, the trend is reversed with both the enthalpy and entropy terms decreasing with increasing redox site loading. This observation suggests that the driving forces may be different for the two electrolyte concentrations. Despite the opposing effects of redox site loading on the enthalpy and entropy terms in 0.1 and 1.0 M electrolyte, (AH; )real and (AS,” )real follow a linear relation. Negative entropy terms such as those reported here are unusual for solution species but have been observed previously for polymer mod- ified electrodes [35]. The negative terms may arise from the assumption in eqn. (3)

150
that kp is unity. If the entropies are to have positive values, then it is likely that kp is less than unity, in which case the electrode kinetics would be non-adiabatic [3].
Such non-adiabaticity may also provide an explanation of the low k ’ values and the dependence of k” on both redox site and electrolyte concentration. The results presented in the homogeneous charge transport section clearly demonstrated the influence of redox site loading on ion diffusion within the film. Processes, such as ion diffusion and availability will directly influence the double layer thickness and the position of the outer Helmholtz plane. Given that heterogeneous electron transfer is assumed to take place at the outer Helmholtz plane, ion availability will determine the distance over which heterogeneous electron transfer will take place. In the case of homogeneous electron transfer reactions e.g. between transition metal complexes, non-adiabaticity has been shown to result in a electron transfer rate which decreases exponentially with increasing separation of the reactants [36]. We suggest that heterogeneous electron transfer at modified electrodes exhibits a similar dependence on reacting site separation, which, in the case of heterogeneous electron transfer is the double layer thickness. This may explain the low values of k” observed compared to solution species. This model would also predict the increase of k o with increasing electrolyte concentration and the variation of k O with redox site loading. The interpretation of homogeneous and heterogeneous electron transfer being coupled via ion availability and diffusion, is supported by the observation of a linear correlation between Dcr(PS) and k O, which suggests that the effect of changing the redox site and electrolyte concentration, affects similarly both the heterogeneous electron-transfer reaction and the homogeneous charge transport process occurring within the film [35].
The reaction entropy (AS,“) is considered to be sensitive to changes in the solvent polarisation required to allow electron transfer [23,24]. For all electrolytes examined here, a positive reaction entropy term of similar magnitude for all redox site loadings was obtained. Such positive. entropy terms have been previously reported [35] and attributed to more extensive solvent ordering in the oxidised state. For the present case we suggest that increased polarisation of water molecules adjacent to the aromatic rings of the osmium complex occurs in the higher oxidation state. The values obtained for the reaction entropy and the fact that they are relatively constant is in qualitative agreement with the Born model (eqn. 5). This equation gives a value of 16 J mol-’ K-’ for AS,0 using the charges on the reduced and oxidised forms of the metal complex and the metal complex radius, a, = aRed = 7.5 A.
The transfer coefficient for a model monomeric compound ([Os@ipy),(Pic) Cl]PF,) at a bare glassy carbon electrode is 0.5 f 0.05. Given that the transfer coefficient can yield information as to the position of the reaction site with respect to the outer Helmholtz plane, the similarity of the transfer coefficient observed for high electrolyte concentrations and the solution species tends to suggest that the position of the reacting transition state with respect to the electrode is similar in both cases. This agrees with the Dcr and k” results discussed previously which

151
indicated the importance of ion availability in both homogeneous and heteroge- neous charge transfer.
CONCLUSIONS
The homogeneous charge transport results presented are significant iu terms of the nature of the mobile species which maintain el~~oneutr~ty at short and long times. The results also indicate that the charge transport behaviour is strongly dependent on the physico-chemical properties of the polymer matrix. By exploring the effect of redox site loading and electrolyte concentration, the requirements of high electron transport rates and rapid ion diffusion are seen to be mutually somewhat exclusive. Where the redox site loading is high, and electron self exchange is most likely to be favoured, an ion diffusion station is observed. This means that the highest rates of charge transport, as measured by potential step, are observed for intermediate loadings where film swelling allows a higher ion diffusion rate. In the case of charge transport measured under longer timescale cyclic voltammetry conditions, the highest charge transport rate is observed for low loadings where polymer chain motion limits D&X).
Electrode kinetics and the rate of homogeneous charge transport as measured by potential step are seen to be strongly coupled suggesting that processes such as ion diffusion similarly influence both processes. The fact that the reaction entropy remains constant when the electrolyte is changed or the redox site loading increased suggests that the local chemical microenvironment of the osmium centre remains largely unchanged as the redox site loading is varied. It also appears, that an increase in electrolyte concentration, which is expected to increase the ion popnla- tion within the film does not alter the solvation of the redox centre significantly. This emphasises the importance of considering not only the redox properties of the dectroactive centre, but also the physi~che~~ properties of the Polymeric matrix. The observation that k” and D&PS) are sensitive to both redox site and electrolyte ~n~ntration while (A$?) is not, suggests that it is largely changes in the polymer phase which are responsible for the ion availability within the film and hence, the observed variations of k O.
ACKNOWLEDGEMENT
The financial support of EOLAS the Irish Science and Technology Agency is gratefully acknowledged.
1 L.R. Faulkner, Filectro&im. Acta, 34 (1989) 1699. 2 A.R. Hillman, in R.G. Linford (Ed.), Electrochmical Science and Technology of Polymers, Vol. 1,
Elseviw, Amsterdam, 1987, Ch. 5. 3 J.T. Hupp and M.J. Weaver, J. Electroanal. Chem., 145 (1983) 43.

152
4 N. Gyama, T. Ohsaka, H. Yamamoto and M. Kaneko, J. Phys. Chem., 90 (1986) 3850. 5 R.J. Forster and J.G. Vos, Macromolecules, 23 (1990) 4372. 6 F.G. Cottrell, Z. Phys. Chem., 42 (1902) 385. 7 C.P. Andrieux and J.M. Sav&tnt, J. Phys. Chem., 92 (1988) 6761. 8 W.T. Yap and R.A. Durst, J. Electroanal. Chem., 216 (1987) 11. 9 W.T. Yap, R.A. Durst, E.A. Blubaugh and D.D. Blubaugh, J. Electroanal. Chem., 144 (1983) 69.
10 A. Se&i, Collect. Czech. Chem. Commtm., 44 (1948) 327. 11 K. Aoki, K. Tokuda and H. Matsuda, J. Electroanal. Chem., 146 (1983) 417. 12 A.J. Bard and L.R. Faulkner, Electrochemical Methods - Fundamentals and Applications, Wiley,
New York, 1980. 13 S.M. Oh and L.R. Faulkner, J. Electroanal. Chem., 269 (1989) 77. 14 R.C. Bowers and R.W. Murray, Anal. Chem., 38 (1966) 461. 15 D. Ghesquiere, B. Ban and C. Chachaty, Macromolecules, 10 (1977) 743. 16 R. Lange and K. Doblhofer, J. Electroanal. Chem. 237 (1987) 13. 17 P. Daum, J.R. Lenhard, D.R. Rohson and R.W. Murray, J. Am. Chem. Sot., 102 (1980) 4649. 18 T. Ohsaka, N. Oyama, S. Yamaguchi, and H. Matsuda, Bull. Chem. Sot. Jpn., 54 (1981) 2475. 19 K.J. Vetter, Electrochemical Kinetics, Academic Press, New York and London, 1987, p. 107. 20 H. Matsuda, Bull. Chem. Sot. Jpn., 53 (1980) 3439. 21 R.J. Forster and J.G. Vos, unpublished results, 1990. 22 M.J. Weaver, J. Phys. Chem., 83 (1976) 2645. 23 E.L. Yee, R.J. Cave, K.L. Guyer, P.D. Tyma and M.J. Weaver, J. Am. Chem. Sot., 101 (1979) 1131. 24 J.T. Hupp and M.J. Weaver, J. Electroanal. Chem., 131 (1984) 619. 25 E.L. Yee and M.J. Weaver, Inorg. Chem., 19 (1980) 1077. 26 N. Sutin, M.J. Weaver and E.L. Yee, Inorg. Chem., 19 (1980) 1096. 27 C.E.D. Chidsey and R.W. Murray, Science, 231 (1986) 231. 28 P. Daum and R.W. Murray, J. Phys. Chem., 85 (1981) 389. 29 W.G. Albery, M.G. Boutelle, P.J. Colby and A.R. HiBman, J. Electroanal. Chem., 133 (1982) 135. 30 K. Aoki, K. Tokuda, H. Matsuda and N. Oyama, J. Electroanal. Chem., 176 (1984) 139. 31 P. Ferruti and R. Barbucci, Adv. Polym. Sci., 58 (1984) 55. 32 T. Ohsaka, T. Okajima and N. Gyama, J. Electroanal. Chem., 215 (1986) 191. 33 C. Amatore, J.M. Saw&ant and D. Tessier, J. Electroanal. Chem., 146 (1983) 37. 34 C. Amatore, J.M. Saveant and D. Tessier, J. Electroanal. Chem., 147 (1983) 39. 35 T. Oh&a, H. Yamamoto and N. Gyama, J. Phys. Chem., 91 (1987) 3775. 36 M.D. Newton and N. Sutin, Amm. Rev. Phys. Chem., 35 (1984) 437.





























