Embed Size (px)
Citation preview

Quantification of Mitochondrial Acetylation DynamicsHighlights Prominent Sites of Metabolic Regulation*!S
Received for publication, May 7, 2013, and in revised form, June 30, 2013 Published, JBC Papers in Press, July 17, 2013, DOI 10.1074/jbc.M113.483396
Amelia J. Still‡1, Brendan J. Floyd‡1, Alexander S. Hebert§¶1, Craig A. Bingman‡, Joshua J. Carson‡,Drew R. Gunderson‡, Brendan K. Dolan‡, Paul A. Grimsrud‡, Kristin E. Dittenhafer-Reed!, Donald S. Stapleton‡,Mark P. Keller‡, Michael S. Westphall¶, John M. Denu!, Alan D. Attie‡, Joshua J. Coon§¶!, and David J. Pagliarini‡2
From the Departments of ‡Biochemistry, §Chemistry, and !Biomolecular Chemistry and ¶Genome Center of Wisconsin, University ofWisconsin–Madison, Madison, Wisconsin 53706
Background: Lysine acetylation, a prevalent post-translational modification, alters mitochondrial metabolism in responseto nutrient changes.Results: Quantitative proteomics distinguishes dynamic and static acetylation sites, highlighting 48 likely regulatory sites ofthousands identified.Conclusion: Acetylation of Acat1 lysine 260, a highly dynamic site, reversibly inhibits enzyme activity.Significance:Quantitative, state-specific proteomic analyses accelerate the functional characterization of acetylation in mito-chondrial remodeling.
Lysine acetylation is rapidly becoming established as a keypost-translational modification for regulating mitochondrialmetabolism. Nonetheless, distinguishing regulatory sites fromamong the thousands identified by mass spectrometry and elu-cidating how these modifications alter enzyme function remainprimary challenges. Here, we performed multiplexed quantita-tive mass spectrometry to measure changes in the mouse livermitochondrial acetylproteome in response to acute and chronicalterations in nutritional status, and integrated these data setswith our compendium of predicted Sirt3 targets. These analyseshighlight a subset of mitochondrial proteins with dynamicacetylation sites, including acetyl-CoA acetyltransferase 1(Acat1), an enzyme central tomultiplemetabolic pathways.Weperformed in vitro biochemistry and molecular modeling todemonstrate that acetylation of Acat1 decreases its activity bydisrupting the binding of coenzyme A. Collectively, our datareveal an important new target of regulatory acetylation andprovide a foundation for investigating the role of select mito-chondrial protein acetylation sites in mediating acute andchronic metabolic transitions.
Protein acetylation was first identified 50 years ago (1, 2) andhas since become established as a key regulator of gene tran-
scription through its role in chromatin remodeling. In recentyears, fueled by technological advances in mass spectrometry-based proteomics, lysine acetylation has become recognizedas a widespread post-translational modification (PTM)3 thatrivals phosphorylation and ubiquitination in its prevalence(3–5). Metabolic enzymes, including many in mitochondria,are among the most heavily acetylated proteins (6–8). Mecha-nistic and physiological studies have begun to reveal that acety-lation of these proteins plays a key role in regulating metabolicflux, and to suggest that aberrant levels of this PTMmight con-tribute to disorders associated with metabolic inflexibility,including obesity, type 2 diabetes, and cancer (9–15).
Despite mounting evidence that acetylation is a prevalentprotein modification, much work remains to establish it as apervasive mitochondrial regulatory mechanism. This includesthe need to distinguish bona fide functional acetylation sitesfrom what may be widespread spurious or adventitious modi-fications (16). This is highlighted by the considerable gapbetween the large number of identified mitochondrial lysineacetylation sites (!2,200) and the few with a validated regula-tory function (!12 proteins) (17). The reasons for this disparitystem in part from both the relative youth of the field and fromtechnical limitations of most previous mass spectrometry-driven investigations, which were largely unable to produceaccurate quantitative information on how acetylation levelschange between contrasting biological states.
Here, we applied our newly developed quantitative acetyl-proteomics methods to measure changes in the mouse livermitochondrial acetylproteome between two pairs of contrast-ing nutritional states (18). First, because PTMs are a primarymechanism of regulation at acute time scales, we assessedchanges in the mitochondrial acetylproteome between fastedand refed mice. Second, because chronic dysregulation of
* This work was supported by a Searle Scholars Award, a Shaw ScientistAward, a Glenn Foundation Award, and National Institutes of Health GrantsRC1DK086410, U01GM094622, and R01DK098672 (to D. J. P.); NationalScience Foundation Graduate Fellowship DGE-1256259 and National Insti-tutes of Health Training Grant 5T32GM007215-37 (to A. J. S.); NationalInstitutes of Health Grants R01GM080148 (to J. J. Coon), DK066369and DK058037 (to A. D. A.), AG038679 and GM065386 (to J. M. D.),T32DK007665 and T32GM008692 MSTP (to B. J. F.), and F32DK091049 (toP. A. G.); American Heart Association Grant 12PRE839 (to J. J. Carson); and aNational Science Foundation graduate fellowship and National Institutesof Health Training Grant 5T32GM08349 (to K. E. D.).
!S This article contains supplemental “Experimental Procedures,” Figs. S1–S4,and additional references.
1 These authors contributed equally to this work.2 To whom correspondence should be addressed. E-mail: pagliarini@
wisc.edu.
3 The abbreviations used are: PTM, post-translational modification; Acat1,acetyl-CoA acetyltransferase 1; FAO, fatty acid oxidation; OxPhos, oxida-tive phosphorylation.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 288, NO. 36, pp. 26209 –26219, September 6, 2013© 2013 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26209 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

PTMs can be an important factor in disease, we compared themitochondrial acetylproteome of obese (Leptin-deficient, Lep-tinob/ob) and lean (Leptin"/") C57/Bl6 (B6) mice. Our analysesreveal that, among a background of largely static acetylationsites, 237mitochondrial proteins have a significant change (q!0.1, Welch’s t test with Storey correction) in the relative occu-pancy of one or more acetylation sites during these transitionsin nutritional status. These include a range of enzymes fromcore mitochondrial metabolic processes, such as oxidativephosphorylation, fatty acid "-oxidation, branched chain aminoacid metabolism, and ketogenesis.
To further prioritize mitochondrial lysine acetylation sitesthat are themost likely to be important formetabolic flexibility,we integrated these large-scale data sets with our compendiumof predicted Sirt3 targets (18). Together, these analyses high-lighted a select set of mitochondrial proteins as being likelytargets for regulatory acetylation, including acetyl-CoA acetyl-transferase 1 (Acat1), which possessed several sites of highlydynamic acetylation. By performing site-specific acetyllysineincorporation and in vitro biochemical enzyme activity assays,we discovered that Sirt3-driven deacetylation of K260ac andK265ac significantly enhances Acat1 activity. Molecular mod-eling revealed that the inhibitory effects of acetylation onAcat1activity are likely due to decreased affinity for CoA caused by aloss of a favorable electrostatic interaction between one ormore positively charged lysines with the negatively charged3#-phosphate of CoA. Collectively, ourwork reveals Acat1 as animportant new metabolic target of reversible acetylation andprovides a resource of prioritized acetylation sites for futureinvestigations into the role of this PTM in regulatingmitochon-drial protein function (19).
EXPERIMENTAL PROCEDURES
Animal Models—Breeding, sacrificing, and tissue harvestingof mice were described previously for the fasting versus refeed-ing study (19) and the obese versus lean study (20). Briefly, malemice were bred and housed in an environmentally controlledfacility with a 12-h light-dark cycle (6 am–6 pm light cycle) andprovided water and standard rodent chow (Purina number5008). For the fasting versus refeeding study, chow wasremoved for 16 h (5 pm–9 am), after which half of the animalswere sacrificed, whereas the other half were allowed to feed adlibitum an additional 2 h before being sacrificed. Liver tissuewas dissected and immediately enriched for mitochondriausing differential centrifugation (19). For the obese versus leanstudy, chowwas removed for 4 h (8 am–12 pm) before sacrifice,and the liver was flash-frozen in liquid nitrogen before beingthawed and enriched for mitochondria.QuantitativeAcetylproteomeAnalysis—Crudemitochondria
were subjected to quantitative proteomics/acetylomics usingrecently developedmethods (18). Briefly, mitochondrial pelletswere resolubilized, and the proteinswere reduced and alkylatedand digested enzymatically and labeled with isobaric tags(6-plex TMT or 8-plex iTRAQ). Samples were mixed and sub-jected to strong cation exchange chromatography, followed byimmunoprecipitation with pan-acetyllysine antibody (Immu-nechem), and non-bound peptides were further fractionated byhigh pH reverse phase chromatography. All samples were sub-
jected to data-dependent Nano-LC-MS/MS analysis on anETD-enabled LTQ Orbitrap Velos (Thermo Fisher Scientific)using our previously described QuantMode instrumentationmethod (21) with higher energy collision-activated dissociationfragmentation. Our custom software COMPASS (22) was uti-lized to search the MS/MS spectra against a concatenated tar-get-decoymouse database (UniProt), to trim the identified pep-tides (1% false discovery rate), to normalize the intensities ofisobaric tag reporter ions, to group all peptides to parsimoniousprotein groups (1% false discovery rate), to localize acetylationsites to specific residues (95% probability), and to sum reporterion intensities for spectra identifying the same protein oracetyl-isoform. For more details, see the supplemental “Exper-imental Procedures”.Measurement of Oxygen Consumption Rate—The oxygen
consumption rate was measured using an XF-96 ExtracellularFlux Analyzer (Seahorse Bioscience) with methods based onprevious work (23). Briefly, isolatedmitochondria were quanti-fied by wet weight, and 40 #g were loaded per well and supple-mented with 10 mM glutamate and 2 mM malate. Basal respira-tion was measured for 3 min, followed by ADP injection to afinal concentration of 2mM.ADP-stimulated oxygen consump-tionwas calculated by subtracting basal respiration, after whichthe wells were averaged. Data analysis was performed using theXF96 software (version 1.4.1.4) and the “Level(Direct)Akos”algorithm. See the supplemental “Experimental Procedures”for further details.Biochemical Assessment of Acat1 Activity—Recombinant
Acat1 (amino acids 31–424) with a C-terminal hexahistidinetag was overexpressed in Escherichia coli and purified usingmetal affinity resin based on previous methods (24, 25). Forsite-specific acetyllysine incorporation, an acetyl-lysyl-tRNAsynthetase/tRNACUA pair that recognizes an amber codon wasco-expressed with Acat1 in the presence of 10 mM acetyllysineand 20 mM nicotinamide (26–28). Briefly, a plasmid encodingAcat1 was cotransformed with pBK AcKRS (acetyllysine tRNAsynthase) and pCDF PyIT (tRNACUA) into BL21(DE3) E. coli.Cells were grown to anA600 of 0.5 to 0.7 and then supplementedwith 10 mM acetyllysine and 20 mM nicotinamide for 30 minbefore the addition of 0.3 mM isopropyl 1-thio-"-D-galactopy-ranoside. Cells were purified using metal affinity resin as abovewith the inclusion of 20 mM nicotinamide in the lysis buffer.Incorporationwas verified bymass spectrometry and immuno-blots for Acat1 and acetyllysine. Sirt3 was purified as describedpreviously (29).Acat1 Activity Assays—Activity was assessed as described
previously (24, 30). Briefly, the reaction mixture contained 50mM Tris-HCl (pH 8.1), 20 mM MgCl2, 10 #M acetoacetyl-CoA,40 mM KCl, and 8 ng of purified Acat1 enzyme (diluted into abuffer of 50 mM HEPES (pH 6.6), 9.5% glycerol, and 0.5 mg/mlgelatin). The reaction was initiated with the addition of CoA.The final volume of the reaction was 300 #l. We measured thechange in absorbance at 303 nm over 30 min at room temper-ature (!25 °C). For the determinations ofKm andVmax forCoA,the above procedure was followed, and the CoA concentrationwas varied between 15 and 300 #M. All absorbance measureswere obtained using the BioTek Synergy 2 microplate spectro-photometer with Gen5 software. We analyzed the mean veloc-
Quantitative Mitochondrial Acetylation Dynamics
26210 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 36 • SEPTEMBER 6, 2013 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

ity over the first 5 min, whereas the reaction velocity curveremained linear over the first 10min.Km andVmax calculationswere obtained using the Enzkin package in Matlab and Sigma-Plot (version 12).Sirt3 Treatments—Sirt3 treatments were performed at 37 °C
for 30 min and contained 16 ng/#l Acat1, 0.375 #M Sirt3, 1 mMNAD", 0.5mMDTT in dilution buffer (50mMHEPES, pH 8.23,0.5mg/ml gelatin, 9% glycerol). Deacetylation was verified withimmunoblots for Acat1 and acetyllysine, and activity wasmeas-ured as above.Electrostatic Modeling—To assess the changes in charge
complementarity for CoA in the active site of Acat1 caused bylysine acetylation, the tetrameric coordinates of Protein DataBank code 2IBW (24) were prepared for electrostatic calcula-tions. Water and other ligands were removed from the model.Lysine residues were either fully charged in the calculation orneutralized to simulate the effect of acetylation. An electro-static map was calculated using the Adaptive Poisson-Boltz-mann Solver (31) plugin in PyMOL (The PyMOL MolecularGraphics System, Version 1.5.0.4, Schodinger, LLC) usingdefault parameters. Local electrostatic environment was visu-alized as a continuous red-blue gradient running from $4 to"4 kT/e on the solvent accessible surface of the protein, or asan isosurface extending through space at "1 kT/e.
RESULTS
Quantitative Analysis of Mitochondrial Acetylation Dynam-ics during Fasting and Refeeding—To quantify the dynamics ofmitochondrial acetylation with high resolution, we leveragedour recently developed strategy for efficient acetyl peptideenrichment (18) and our QuantMode method (21) for highlyaccurate quantitative proteomics with isobaric tags to profilemouse liver mitochondrial protein abundance and acetylationlevels across the transition between fasting and refeeding. Eightmice were fasted overnight for 16 h, after which half wereallowed to feed ad libitum for 2 h (Fig. 1A), a time point featur-ing marked PTM changes with limited protein abundancechanges (19). We identified 1,915 unique acetylation isoforms,of which 1,796 were quantified (Fig. 1B and supplemental Fig.
S1). Unsupervised average linkage hierarchical clustering of thedata based on relative acetyl isoform occupancy (site-specificacetyl fold change divided by protein abundance fold change)grouped the refed and fasted mice separately (Fig. 2A), indicat-ing that acetylation changes are a reproducible and distinguish-ing feature of the transition between fasting and refeeding (32,33). Importantly, our quantitative approach reveals that !10%(188) of identified acetyl isoforms exhibited a statistically sig-nificant change (q! 0.1) in acetyl occupancy in response to thisacute perturbation of metabolic status.
We hypothesize that the subset of mitochondrial proteinacetylation sites with changing occupancy upon refeeding rep-resent those most likely to have a regulatory function duringthis transition. For example, we detect acetylation on all fourmitochondrialmembers of themalate-aspartate shuttle (Mdh2,Got2, Slc25a11, and Slc25a12), which is responsible for relayingthe reducing potential of NADH generated from glycolysis intothe mitochondrial matrix (Fig. 2B) (34). Rat liver Mdh2 activityis increased upon feeding (35), and previous studies havereported that Mdh2 activity can be increased either by acetyla-tion (on Lys-185, Lys-301, Lys-307, and Lys-314) (36) or bySirt3-dependent deacetylation (of Lys-239) (18). Here, wedetect a large decrease in acetylation occupancy of Mdh2 Lys-239 with little or no changes to the other sites. This indicatesthat the Sirt3-controlled acetylation site (Lys-239) is likely thedominant regulator of Mdh2 during this acute metabolic per-turbation, consistent with an increased flux of reducing equiv-alents from the cytosol into the mitochondria for energy pro-duction upon feeding.
Quantifying the magnitude of acetylation changes across anacute time scale can provide insights into key acetyl sites with-out the confounding changes in protein levels that accompanylonger perturbations (Fig. 2C). For example, despite unchang-ing protein levels among almost all enzymes involved in mito-chondrial fatty acid oxidation (FAO), more than 90 distinctacetyl isoforms within this pathway are altered significantlyupon refeeding (Fig. 2D). These include four acetylation siteson carnitineO-palmitoyltransferase 2 (Cpt2; Lys-104, Lys-544,
FIGURE 1. Schematic of workflow for quantitative proteomic and acetylomic analyses of mouse liver mitochondria. A, in experiment one, eight lean micewere fasted for 16 h and either sacrificed immediately or refed for 2 h before being sacrificed. In experiment two, three obese mice and three lean mice werefasted for 4 h before being sacrificed. In each case, liver mitochondria were enriched and analyzed by LC-MS/MS (see “Experimental Procedures”). SCX Frac.,strong cation exchange fractionation. B, summary of protein and acetylation data. IDs, unique identifications at 1% false discovery rate; Quant, measurementsquantified with isotopic reporter ions in all mice assessed; %, measurements significantly changing with q ! 0.1. See also supplemental Fig. S1.
Quantitative Mitochondrial Acetylation Dynamics
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26211 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

Lys-537, and Lys-453), an enzyme involved in the transport offatty acids from the cytosol into the mitochondrial matrix for"-oxidation (37). Similarly, energy production from amino aciddegradation is robustly induced in the postprandial state (38),and several enzymes in amino acid catabolic processes exhibitlarge changes in relative acetylation occupancy without altera-tion to the underlying protein abundance levels. As one exam-ple, acetylation on Lys-329 of glutaminase (Gls2) decreases&2-fold following refeeding, which is consistent with this PTMserving an inhibitory function on this key enzyme of amino acidbreakdown. Overall, the vast majority of dynamic acetylationsites decreases in relative acetyl occupancy upon refeeding,consistent with previous reports that fasting can lead to hyper-acetylation of liver mitochondrial proteins (23).Quantitative Analysis of Mitochondrial Acetylation Dynam-
ics due toChronicObesity—Previous studies have noted generalchanges to the acetylproteome during the onset of obesity (13,39); however, these studies were also limited by the semi-quan-titative nature of their applied mass spectrometry methods. Toevaluate andmeasure the key obesity-induced changes in acety-lation of mitochondrial proteins with high accuracy and depthof coverage, we performed a large-scale quantitative compari-son of the liver mitochondrial acetylproteome from three lean(Leptin"/") and three obese (Leptinob/ob) mice at 10 weeks ofage, each fed standard chow diet ad libitum (Fig. 1A). This
approach provides a comparison that is similar in nature to thefirst perturbation (contrasting nutrient abundance) but mark-edly different in time scale (hours versus weeks). Leptin defi-ciency in B6 mice is known to cause hyperphagia-inducedobesity, which is accompanied by a dramatic response to over-nutrition (e.g. hyperinsulinemia, hepatic steatosis) by 10 weeks(40). These obese mice, however, have only a mild, controlledelevation in blood glucose andmodel the prediabetic state, pro-viding a model of obesity that is not confounded by diabetes.Here, we identified 1,723 unique acetyl isoforms, 1,601 ofwhichwere quantified, and 826 that were significantly changing (q !0.1; Fig. 1B, and supplemental Fig. S1).
Not surprisingly, given the relative magnitudes and dura-tions of the perturbations involved, the changes in proteinabundance and acetyl occupancy due to chronic obesityencompass a greater dynamic range than those seen following amere 2-h refeeding (Fig. 1B and Fig. 3, A and B). This compar-ison could reveal key acetylation events potentially importantfor regulatingmitochondrial function in states of chronic over-nutrition. For instance, it is known that the protein abundanceand the activity of complexes in the oxidative phosphorylation(OxPhos) pathway are increased in certain models of obesity(19, 41), and acetylation has been implicated in inhibiting allfive complexes of OxPhos (39, 42–45); however, the specificregulatory sites are as yet unclear. To confirm the increase inOxPhos flux in our model of obesity, we isolated liver mito-chondria from obese and lean livers andmeasured oxygen con-sumption using a Seahorse extracellular flux analyzer. Asexpected, mitochondria from obese mice exhibit an elevatedstate 3 oxygen consumption rate compared with their leancounterparts (Fig. 3D and supplemental Fig. S1). In our obeseversus lean acetylome comparison, we identified 156 acetyl iso-forms onOxPhos proteins (Fig. 3C). Ninety-seven of these siteschange significantly (q ! 0.1) in acetyl occupancy, 91 of whichdecrease in the obese state.We therefore predict that a subset ofthese 91 sites (which we further prioritize below) plays animportant role in the stimulation of OxPhos activity in obesity,irrespective of any protein abundance changes.
As with our fasting and refeeding study, we find that cata-bolic enzymes, including those of FAO and amino acid degra-dation, appear particularly susceptible to alterations in acetyla-tion levels. For example, acetylation of Lys-42 on long chainacyl-CoAdehydrogenase (Acadl) is significantly decreasedwithobesity. This PTMhas previously been shown to decrease enzy-matic activity (46), consistent with elevated mitochondrial FAOin obese animals. Aminoadipate aminotransferase (Aadat), a pro-tein involved in tryptophan degradation to kynurenine acid (47),has increased acetylation on Lys-263, which is involved in bind-ing the pyridoxal phosphate cofactor (48, 49). We predict thatacetylation of Lys-263 reduces aminoadipate aminotransferaseactivity by preventing this enzyme-cofactor interaction. Thisdecrease in activity could shift tryptophan degradation fromthe kynurenine-kynurenic acid pathway toward the kynu-renine-NAD" pathway, which has previously been linked tohypertension, diabetes, atherosclerosis, obesity, and immu-nodeficiency (50).
Notably, for our proteomics analyses we purified mitochon-dria only to the extent required to achieve near comprehensive
B D
A
-1.0
-0.5
0.0
0.5
1.0
Acetyl OccupancyFasted Refed
Fold
Δ (l
og2(R
/F))
C 2
1
-3
0
-1
-2
Fold
Δ (l
og2(R
/F))
0 200 600MitoCarta proteins
400
Acetyl isoformsProteins
-2.50
-2.75
-3.00
Fold
Δ (l
og2(R
/F))
0.25
-0.50
-0.25
0.00
Slc2
5a11
Got
2
Slc2
5a12
Mdh
2
Known e!ect of acetylationInhibitActivateNone
K239
Mitochondrialfatty acid oxidation
Fold
Δ (l
og2(R
/F))
-3
0
-1
-2Acetyl isoforms (q"0.1)Proteins
Pccb
Mut
Acad
sAc
advl
Acad
sbPc
caD
ecr1
Acad
lG
cdh
Had
hCp
t2H
adhb
Acaa
2H
adha
Eci1
Echs
1Ac
adm
Mce
eFIGURE 2. Global analysis of acute protein and acetylation changes dur-ing the transition from fasting to refeeding. A, heat map showing unsu-pervised average linkage hierarchical clustering of four fasted and four refedmice groups them according to nutrient status. Values are colored based onrelative acetyl occupancy, normalized to the average of all eight mice, on alog2 scale from less than $1 to greater than 1. R/F, refed/fasted. B, relativeacetyl occupancy fold change for proteins in the malate-aspartate shuttle.The acetylation site previously shown to inhibit Mdh2 activity is shown in red,whereas those previously shown to activate Mdh2 are shown in black. Othersites we detected as acetylated but that have no prior functional data areshown in gray. C, changes in acetylation are greater than changes in proteinabundance. All quantified MitoCarta proteins are ranked on the x axis byprotein abundance fold change, refed/fasted (black dots). Relative acetyl iso-form changes are plotted in the same position on the x axis as the correspond-ing protein measurement (gray dots). D, acetyl and protein abundancechanges in mitochondrial fatty acid oxidation. Proteins are marked in black,and significantly changing (q ! 0.1) acetyl isoforms are marked in gray.
Quantitative Mitochondrial Acetylation Dynamics
26212 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 36 • SEPTEMBER 6, 2013 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from
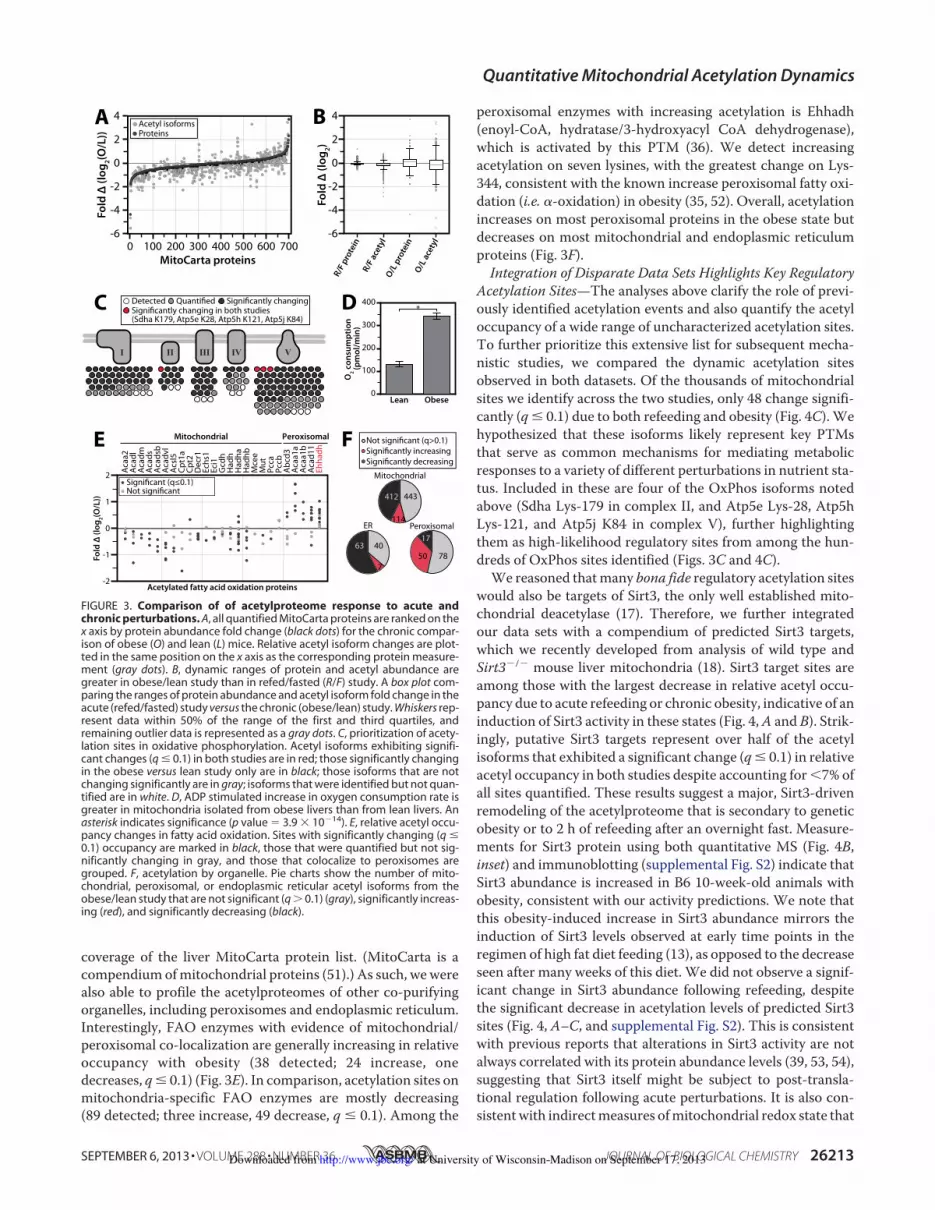
coverage of the liver MitoCarta protein list. (MitoCarta is acompendium ofmitochondrial proteins (51).) As such, wewerealso able to profile the acetylproteomes of other co-purifyingorganelles, including peroxisomes and endoplasmic reticulum.Interestingly, FAO enzymes with evidence of mitochondrial/peroxisomal co-localization are generally increasing in relativeoccupancy with obesity (38 detected; 24 increase, onedecreases, q! 0.1) (Fig. 3E). In comparison, acetylation sites onmitochondria-specific FAO enzymes are mostly decreasing(89 detected; three increase, 49 decrease, q ! 0.1). Among the
peroxisomal enzymes with increasing acetylation is Ehhadh(enoyl-CoA, hydratase/3-hydroxyacyl CoA dehydrogenase),which is activated by this PTM (36). We detect increasingacetylation on seven lysines, with the greatest change on Lys-344, consistent with the known increase peroxisomal fatty oxi-dation (i.e. $-oxidation) in obesity (35, 52). Overall, acetylationincreases on most peroxisomal proteins in the obese state butdecreases on most mitochondrial and endoplasmic reticulumproteins (Fig. 3F).Integration of Disparate Data Sets Highlights Key Regulatory
Acetylation Sites—The analyses above clarify the role of previ-ously identified acetylation events and also quantify the acetyloccupancy of a wide range of uncharacterized acetylation sites.To further prioritize this extensive list for subsequent mecha-nistic studies, we compared the dynamic acetylation sitesobserved in both datasets. Of the thousands of mitochondrialsites we identify across the two studies, only 48 change signifi-cantly (q ! 0.1) due to both refeeding and obesity (Fig. 4C). Wehypothesized that these isoforms likely represent key PTMsthat serve as common mechanisms for mediating metabolicresponses to a variety of different perturbations in nutrient sta-tus. Included in these are four of the OxPhos isoforms notedabove (Sdha Lys-179 in complex II, and Atp5e Lys-28, Atp5hLys-121, and Atp5j K84 in complex V), further highlightingthem as high-likelihood regulatory sites from among the hun-dreds of OxPhos sites identified (Figs. 3C and 4C).We reasoned thatmany bona fide regulatory acetylation sites
would also be targets of Sirt3, the only well established mito-chondrial deacetylase (17). Therefore, we further integratedour data sets with a compendium of predicted Sirt3 targets,which we recently developed from analysis of wild type andSirt3$/$ mouse liver mitochondria (18). Sirt3 target sites areamong those with the largest decrease in relative acetyl occu-pancy due to acute refeeding or chronic obesity, indicative of aninduction of Sirt3 activity in these states (Fig. 4,A and B). Strik-ingly, putative Sirt3 targets represent over half of the acetylisoforms that exhibited a significant change (q! 0.1) in relativeacetyl occupancy in both studies despite accounting for'7% ofall sites quantified. These results suggest a major, Sirt3-drivenremodeling of the acetylproteome that is secondary to geneticobesity or to 2 h of refeeding after an overnight fast. Measure-ments for Sirt3 protein using both quantitative MS (Fig. 4B,inset) and immunoblotting (supplemental Fig. S2) indicate thatSirt3 abundance is increased in B6 10-week-old animals withobesity, consistent with our activity predictions. We note thatthis obesity-induced increase in Sirt3 abundance mirrors theinduction of Sirt3 levels observed at early time points in theregimen of high fat diet feeding (13), as opposed to the decreaseseen after many weeks of this diet. We did not observe a signif-icant change in Sirt3 abundance following refeeding, despitethe significant decrease in acetylation levels of predicted Sirt3sites (Fig. 4, A–C, and supplemental Fig. S2). This is consistentwith previous reports that alterations in Sirt3 activity are notalways correlated with its protein abundance levels (39, 53, 54),suggesting that Sirt3 itself might be subject to post-transla-tional regulation following acute perturbations. It is also con-sistentwith indirectmeasures ofmitochondrial redox state that
4
2
-6
0
-2
-4
0 200 600MitoCarta proteins
400100 500 700300
AFo
ld Δ
(log
2(O/L
))
Signi#cantly changing in both studies (Sdha K179, Atp5e K28, Atp5h K121, Atp5j K84)
Signi#cantly changingQuanti#edDetected
VIIII IVII
4
2
0
-2
-4
-6
O/L p
rote
inO/L
acet
yl
R/F p
rote
inR/
F ace
tyl
Fold
Δ (l
og2)
BAcetyl isoformsProteins
Not signi#cant (q>0.1)Signi#cantly increasingSigni#cantly decreasing
Mitochondrial
443
114
412
Peroxisomal
7850
17ER
4063
7
E
D
PeroxisomalMitochondrial
Acetylated fatty acid oxidation proteins
Fold
Δ (l
og2(O
/L))
-2
1
0
-1
Signi#cant (q"0.1)Not signi#cant
2 Acaa
2Ac
adl
Acad
sAc
adsb
Acad
vlAc
sl5
Cpt1
aCp
t2D
ecr1
Echs
1Ec
i1G
cdh
Had
hH
adha
Had
hbM
cee
Mut
Pcca
Pccb
Abcd
3Ac
aa1a
Acaa
1bAc
ad11
Ehha
dh
Acad
m
O2 c
onsu
mpt
ion
(pm
ol/m
in)
ObeseLean0
100
200
300
400*
F
C
FIGURE 3. Comparison of of acetylproteome response to acute andchronic perturbations. A, all quantified MitoCarta proteins are ranked on thex axis by protein abundance fold change (black dots) for the chronic compar-ison of obese (O) and lean (L) mice. Relative acetyl isoform changes are plot-ted in the same position on the x axis as the corresponding protein measure-ment (gray dots). B, dynamic ranges of protein and acetyl abundance aregreater in obese/lean study than in refed/fasted (R/F) study. A box plot com-paring the ranges of protein abundance and acetyl isoform fold change in theacute (refed/fasted) study versus the chronic (obese/lean) study. Whiskers rep-resent data within 50% of the range of the first and third quartiles, andremaining outlier data is represented as a gray dots. C, prioritization of acety-lation sites in oxidative phosphorylation. Acetyl isoforms exhibiting signifi-cant changes (q ! 0.1) in both studies are in red; those significantly changingin the obese versus lean study only are in black; those isoforms that are notchanging significantly are in gray; isoforms that were identified but not quan-tified are in white. D, ADP stimulated increase in oxygen consumption rate isgreater in mitochondria isolated from obese livers than from lean livers. Anasterisk indicates significance (p value ( 3.9 ) 10$14). E, relative acetyl occu-pancy changes in fatty acid oxidation. Sites with significantly changing (q !0.1) occupancy are marked in black, those that were quantified but not sig-nificantly changing in gray, and those that colocalize to peroxisomes aregrouped. F, acetylation by organelle. Pie charts show the number of mito-chondrial, peroxisomal, or endoplasmic reticular acetyl isoforms from theobese/lean study that are not significant (q & 0.1) (gray), significantly increas-ing (red), and significantly decreasing (black).
Quantitative Mitochondrial Acetylation Dynamics
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26213 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

suggest an increase in theNAD"/NADH ratio in the refed state(55).
Among the dynamic Sirt3 targets observed in both large-scale analyses are multiple sites on enzymes from core meta-bolic pathways involved in responding to changing nutrient sta-tus. Three of these processes, branched chain amino acidmetabolism, fatty acid catabolism, and ketogenesis, exhibitacetylation changes in many pathway members and have incommon a single enzyme: acetyl-coenzyme A acetyltransferase1 (Acat1, NCBI gene ID 110446) (56, 57). Acat1 catalyzes twomajor reversible reactions: the cleavage of acetoacetyl-CoAinto two acetyl-CoA, and the cleavage of 2-methyl acetoacetyl-CoA into propionyl-CoA and acetyl-CoA. We identified 18total acetylation sites on this protein, nine of which are chang-ing significantly (q ! 0.1) in at least one comparison. Relativeacetyl occupancy at three of these sites changed significantly inboth analyses (Lys-187, Lys-260, and Lys-265) (Fig. 5A), thelatter two of which are predicted Sirt3 targets. Another recentSirt3 knock-out study also identifies Lys-260 and Lys-265, butnot Lys-187, as Sirt3 targets (15). In the present study, the acety-lation levels of both putative Sirt3-target sites on Acat1decrease markedly with obesity (relative to lean mice) and withrefeeding (relative to fasted mice), whereas acetylation on Lys-187 increases due to obesity. These data led us to hypothesizethat acetylation on Lys-187, Lys-260, and/or Lys-265 alters theactivity of Acat1. Moreover, as Sirt3 most often activates itstarget enzymes (17), we further hypothesized that deacetylationof Lys-260 and Lys-265 increases Acat1 activity in response tonutrient excess.
Acetylation of Lysine 260 and 265 on Acat1 Inhibits Enzy-matic Activity—Our analyses suggest that acetylation of selectAcat1 lysines is important for regulating enzyme activity. Tobegin testing this possibility, we individually mutated fiveacetylated lysine residues (Lys-84, Lys-187, Lys-260, Lys-265,and Lys-340) to glutamine to mimic the charge state of anacetylated lysine ormutated to arginine tomimic a deacetylatedstate (58). These sites were selected based on the structure ofhuman ACAT1 (24) and on the dynamic changes seen in bothdata sets. Acat1 variants and wild type protein were purifiedfrom E. coli and tested by an in vitro activity assay. Mutation oflysines 260 and 265 to arginine (K260R, K265R) resulted inincreased activity compared with the respective glutaminemutant (K260Q, K265Q), suggesting that acetylation woulddecrease activity (Fig. 5B). In contrast, K187R led to a loss ofactivity compared with K187Q. Interestingly, mutation of theother acetylation sites did not significantly change the activityof Acat1, further suggesting that Lys-187, Lys-260, and Lys-265may be the most important acetylation sites for regulating theactivity of this enzyme.
Although mutation of lysine to glutamine or arginine canapproximate the effect of acetylation on protein function, theseare imperfect analogs. To validate the ability of acetylation toregulate Acat1 enzymatic activity, we used site-specific acetyl-lysine incorporation, a technique that allows for the productionof a protein with nearly full acetylation occupancy on a specificlysine (26, 27). Acat1 variants were generated and purified thateach possessed a single acetylated lysine at position 187, 260, or265 (K187ac, K260ac, K265ac). Acetyllysine incorporation was
B
A C
Fold Δ (log2(O/L))-2 0 2 4
Sirt3 targetsMitoCarta
All isoforms
Hsd17b10 K104
BckdhaK357
Acat1 K260Acat1 K265
Hadha K644Bdh1 K97
-2
1
-1
0
Fold
Δ (l
og2(R
/F))
Acetyl Site R/F O/LAass K286Acaa2 K13Acat1 K187Acsf2 K570Acsm1 K534Ak3 K29Aldh2 K453Aldh4a1 K54Atp5e K28Atp5h K121Cps1 K1291Dld K66Dmgdh K757Etfdh K222Hadha K129Hmgcs2 K354Hspe1 K66Keg1 K124Lrpprc K291Mut K210Oat K392Suclg2 K280
Acetyl Site R/F O/LAcadl K322Acadvl K240Acadvl K277Acadvl K373Acat1 K260Acat1 K265Aldh2 K370Atp5j K84Auh K119Bdh1 K97Cs K52Eci2 K60Grpel1 K100Gstk1 K93Hadh K75Hadha K406Hadha K644Hadhb K189Hadhb K202Hmgcs2 K310Hmgcs2 K342Hspa9 K567Sdha K179Sucla2 K139Suclg1 K81Tst K175
PutativeSirt3-targets
Not PutativeSirt3-targets
-Log
10 (p
-val
ue) 1.0
0.01
0.1
10.0
Fold Δ (log2(R/F))-3 -2 -1 0 1
Sirt3 targetsMitoCarta
All isoforms
-1.0 -0.5 0.0 0.5 1.0Fold Δ (log2)
Eci2 K60Hmgcs2 K83
-3 -2 -1 10 32Fold Δ (log2(O/L))
0.0
0.4
0.8
R/F
Sirt3 protein abundance
O/L
Fold
Δ (l
og2) *
FIGURE 4. Integration of different metabolic comparisons identifies candidate regulatory acetylation events. A, volcano plots of fold change in relativeacetyl occupancy versus $log10 (p value). B and C, comparison of the relative acetyl occupancy fold change across the refed/fasted versus obese/leancomparisons. Only isoforms that were quantified in both studies are represented. B, putative Sirt3 targets are shown in red, other MitoCarta proteins are inblack, and non-MitoCarta proteins are colored gray. Inset, Sirt3 protein abundance fold change by mass spectrometry. An asterisk indicates significance (p !0.05). C, acetylation sites that are significantly changing in both studies (q ! 0.1). Fold changes (log2) are shown in red (positive) and blue (negative) forrefed/fasted (R/F) and obese/lean (O/L).
Quantitative Mitochondrial Acetylation Dynamics
26214 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 36 • SEPTEMBER 6, 2013 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

verified by immunoblot andMSanalyses, and in vitro treatmentof these proteins with recombinant Sirt3 effectively removedthis acetylation in an NAD"-dependent manner (Fig. 5C andsupplemental Fig. S3). Interestingly, in vitro Sirt3 treatmentmore readily removed the acetyl groups on Lys-260 and Lys-265 than that on Lys-187, consistent with the prediction thatK260ac and K265ac are Sirt3 targets in vivo.In vitro Acat1 activity assays of each isoform revealed that
Sirt3-mediated deacetylation of K260ac and K265ac dramati-cally increased enzyme activity in an NAD"-dependent man-ner (Fig. 5, D–F and supplemental Fig. S4). In contrast, weobserved minimal change in activity with identically treatedwild type or K187ac proteins. We further analyzed the kineticproperties of the Acat1 K260 in its acetylated and deacetylatedforms across several concentrations of substrate and deter-mined that acetylation decreases the Vmax of the enzyme whileincreasing its Km for CoA (Fig. 5F). As physiological levels ofCoA in liver are markedly higher than the Acat1 Km (59), thesealterations in Vmax will likely alter metabolic flux in vivo.
Consistent with the hypothesis that regulatory residues arelikely to be conserved throughout evolution (16, 60), Acat1 Lys-260 and Lys-265 are highly invariant and correspond to lysines263 and 268 in the human ACAT1 ortholog (K263_Hs andK268_Hs). Recently, Haapalainen and colleagues (24) solvedthe crystal structure of humanACAT1 in complex with its CoAsubstrate. This structure reveals that positively chargedK263_Hs is in close proximity to the negatively charged3#-phosphate of CoA, which suggests that charge complemen-tarity for CoA in the active site is important for substrate bind-ing. Using this structure (Protein Data Bank code 2IBW) (Fig.6A), we modeled a neutralized lysine residue at K263_Hs tosimulate the effect of acetylation (Fig. 6B). We then calculatedelectrostatic isosurfaces extending through space at "1 kT/ewith all lysines fully charged (Fig. 6C), or with K263_Hs neu-tralized (Fig. 6D). This analysis clearly reveals that the nega-tively charged 3#-phosphate of CoA is present within the posi-tively charged space enclosed by this electrostatic isosurfacein the fully charged enzyme, but not when K263_Hs is neutral-ized. These data suggest that acetylation of Acat1 Lys-260(ACAT1 K263_Hs) likely reduces the favorable electrostaticinteractions between CoA and the Acat1 binding pocket. Thisis similar to reports of the Sirt3-dependent regulation of otherenzymes by an electrostatic repulsion mechanism, includingMn-SOD (61, 62). Together, in conjunction with our in vitroactivity data, we propose that Sirt3 activates Acat1 by removinglysine acetylation that reduces the affinity of Acat1 for CoA.
DISCUSSION
Reversible lysine acetylation is rapidly becoming recognizedas a pervasive PTM that regulates cellular processes rangingfrom gene transcription to intermediary metabolism. Theimpact of this modification extends to mitochondria, where astrikingly high percentage of proteins are acetylated on one ormore lysines. A few of these sites are now known to modify
1
0
-2
-1
Fold
Δ (l
og2)
A K
63 K
75 K80
K171
K178
K187
K199
K220
Acat1 acetylation sitesK2
27K2
42K2
48K2
54K2
60K2
65K3
04K3
35K3
40
Refed/FastedObese/Lean
WT
K260
ac
K265
ac
K187
ac
220
200
140
180
120
80
100
160
Activ
ity +
Sirt
3 (%
of m
ock)
DSirt3targetNot Sirt3target
F
C
WT
K187
ac
K260
ac
K265
ac
anti-Acat1:anti-AcK:
Acat1:Sirt3:
B
Activ
ity o
f R m
utan
t (%
of Q
)
180160
120140
100
40
60
80
Sirt3targetNot Sirt3target
K84
K187
K260
K265
K340
Isoform
WT
K187ac
K260ac
K265ac
Treatment
Mock+Sirt3, NAD+
Mock+Sirt3, NAD+
Mock+Sirt3, NAD+
Mock+Sirt3, NAD+
Vmax(μM/min)0.62 ± 0.060.63 ± 0.050.37 ± 0.030.44 ± 0.050.44 ± 0.070.68 ± 0.060.22 ± 0.060.38 ± 0.02
KM e"ective(μM CoA)
17.75 ± 1.6916.74 ± 1.9422.20 ± 2.4221.56 ± 2.2224.44 ± 2.0017.68 ± 1.8922.06 ± 2.0230.35 ± 2.63
E
Activ
ity (µ
M A
cAcC
oA/m
in)
0.0
0.2
0.4
0.6
0 100 200 300[CoA] (µM)
+ Sirt3, NAD+untreated
n.s.
WTK260ac
FIGURE 5. Reversible, Sirt3-regulated acetylation of Acat1 Lys-260 inhib-its activity. A, relative acetyl occupancy fold change for 17 quantified sites onAcat1. An asterisk indicates q ! 0.1. B, in vitro enzyme activity assays of Acat1mutants generated by mutating lysine to glutamine (Q, acetyl mimic) or argi-nine (R, deacetyl mimic). Data are expressed as the average activity of the Argmutant as a percentage of that for the Gln mutant, at 300 #M CoA (* S.E., n %2). C, immunoblot for Acat1 and acetyllysine on samples of purified Acat1protein used in kinetic assays. Proteins were treated with or without NAD"
and Sirt3. AcK, acetyllysine. C, representative of samples used in D. D–F, in vitroenzyme activity assays of Acat1 with site-specific incorporation of acetylly-sine at the indicated residues, with or without deacetylase (Sirt3) treatment.D, data are expressed as the average activity after Sirt3 treatment as a percentof mock-treated enzyme, at 300 #M CoA (* S.E., n % 3; an asterisk indicates asignificant difference from mock treatment, p ' 0.05). E, aggregated enzymekinetic analysis (n ( 4; an asterisk indicates a significant difference from allother treatments, p ' 0.05; n.s. indicates no significant difference between
the highlighted treatments). F, table of kinetic parameters of Acat1 iso-forms * Sirt3 treatment (* S.E., n % 3; an asterisk indicates a significant dif-ference from mock treatment, p ' 0.05).
Quantitative Mitochondrial Acetylation Dynamics
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26215 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

metabolic flux by altering enzymatic activity or substrate avail-ability, adding to the growing notion that PTMs are key to reg-ulating mitochondrial function.
However, although thousands of mitochondrial acetylationsites have now been identified, little is known about theenzymes that control acetylation inmitochondria. There is onlya single well established mitochondrial protein deacetylase,Sirt3 (17). Likewise, there is no known acetyltransferase withinthis organelle, although emerging evidence suggests thatGCN5-L1 may be a regulator of a yet to be characterized pro-tein acetyltransferase complex (63). Further confounding thisissue is the fact that even at low concentrations of acetyl-CoA,acetylation can occur nonenzymatically (64, 65). Coupled withthe knowledge that mitochondrial enzymes tend to becomeacetylated following nutritional changes that produce anincrease in acetyl-CoA (66) and that there are a very limitednumber of validated regulatory sites, this has led to the specu-lation that many of the numerous mitochondrial acetylationeventsmay be nonenzymatic and, thus, likely nonregulatory. Assuch, rigorous efforts are needed to spotlight and validate bonafide regulatory acetylation sites from a potentially noisy back-ground of non-regulatory PTM “decorations” (67).
To prioritize candidate acetyl-regulated sites on mitochon-drial proteins, we performed large-scale quantitative acetyl-proteomics across multiple contrasting biological states. Ourdata reveal that only a select subset of sites significantly changein occupancy during drastic acute and chronic alterations innutritional status. From our recent study (18), we predict thatmany of these dynamic acetylation sites are targets of Sirt3,consistent with the role of this enzyme as a primary regulator ofmitochondrial acetylation. Together, our integrated analyses
highlight a collection of proteins with high-confidence candi-date regulatory sites (Fig. 4C). Although other acetylationsites may prove to be regulatory under distinct metabolictransitions, our data nonetheless caution that many othersmight represent unregulated and likely non-regulatory tar-gets of acetylation.
A second priority in the field is to rigorously validate anddefine the functions of this modification in regulating proteinactivity (16, 68). In this study, we investigated the importance ofacetylation on Acat1. Our quantitative analyses nominatedthree of the 18 acetylated lysines on this protein as likely regu-latory sites. Using site-specific acetyllysine incorporation, invitro enzymatic analyses and molecular modeling, we demon-strated that acetylation of Lys-260 and Lys-265 inhibits Acat1activity by disrupting CoA binding.
Acat1 is found at the cross-roads of branched chain aminoacid degradation, ketogenesis, and fatty acid catabolism. Inter-estingly, more than one-third (19/48) of our high-confidencecandidate regulatory sites are on proteins involved in thesethree pathways. As such, we hypothesize that the role of regu-latory acetylation is not limited to isolated control points, butrather that it impacts multiple enzymes of a pathway to collec-tively alter net flux, often alongside other PTMs. Indeed, otherkey regulators are known for each of these pathways (e.g. Bck-dha phosphorylation (69), Hmgcs2 acetylation (70) and phos-phorylation (19), and Acadl acetylation (46)). Interestingly,Acat1 Lys-265was also recently identified as a prominent site ofreversible succinylation, further suggesting that this is anunusually important site of post-translational regulation (71).Based on our work here, we predict that this modificationwould likewise dampen Acat1 activity.
Although each of the three pathways mentioned aboveoccurs in the liver, they are not simultaneously induced. Forexample, branched chain amino acid degradation is robustlyinduced by refeeding, as assessed by Bckdha phosphorylationstatus (19, 38) but fatty acid oxidation and ketogenesis areinhibited in that same state (56). Conversely, fatty acid oxida-tion and ketogenesis are highly active in the obese state (72).Thus, it seems reasonable to speculate that the overabundanceof PTMs on ketogenesis, FAO, and branched chain amino acidenzymes noted above, including those on Acat1, operate com-binatorially to help properly calibrate the individual fluxesthrough these interrelated but distinct pathways. Furthermore,although our biochemical data support a role for deacetylationof Acat1 in enhancing flux through any these pathways, itsacetylation alone would likely not be sufficient for completeinactivation of any one of them. Further integration of our datasets with other protein PTM data and quantitative metabolicflux analyses will be necessary to better define the physiologicalramifications of these modifications in collectively modifyingmetabolic output.
The results of our analyses both mirror current trends in theacetylation field and also raise new issues. Consistent with pre-vious work, we find that acetylation is widespread inmitochon-dria and that its levels generally tend to track with predictedacetyl-CoA abundance (66). Our data also suggest that Sirt3 ismore active in the refed state and early during the onset ofobesity and that it tends to enhance the activity of its target
FIGURE 6. Electrostatic modeling of acetylation effects using the crystalstructure of Homo sapiens ACAT1 (Protein Data Bank code 2IBW). Localelectrostatic environment of ACAT1 active site surrounding K263_Hs(K260_Mm, identified by the white arrow). A and C represent wild-type ACAT1,whereas B and D mimic acetylation on K263_Hs by neutralizing its charge. Theelectrostatic environment is visualized as a continuous red-blue gradientfrom $4 to "4 kT/e on the solvent accessible surface of the protein. In C andD, it is overlaid with an electrostatic isosurface extending through space at "1kT/e. The negatively charged 3#-phosphate of CoA is located within the spaceenclosed by the positive isosurface when K263_Hs is fully charged, but it islocated outside when this lysine is neutralized.
Quantitative Mitochondrial Acetylation Dynamics
26216 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 36 • SEPTEMBER 6, 2013 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

enzymes. However, our data reveal that acetyl occupancy atseveral putative Sirt3-regulated sites (e.g. Eci2 Lys-60, Gstk1Lys-93, Cs Lys-52, and Tst Lys-175) was significantly decreaseddue to refeeding but significantly increased due to obesity. Thissuggests that Sirt3 may possess an adjustable target preferenceunder different nutritional states. Moreover, our data indicatethat Sirt3 activity cannot necessarily be predicted from itsabundance, implying that other regulatory post-transcriptionalor post-translational features might be important for titratingits activity. Last, our data reveal that many probable non-Sirt3sites also change considerably in acetylation occupancy, sup-porting the notion that other mitochondrial regulatory acety-lation machinery awaits discovery.
Collectively, our work provides a quantitative map of thechanging mitochondrial protein acetylation landscape duringacute and chronic metabolic transitions, defines the role ofdeacetylation in stimulating Acat1 activity, and highlightsmany other high-priority acetylation sites for mechanistic fol-low-up work (19).
Acknowledgment—We thank A. J. Bureta for assistance with figureillustrations.
REFERENCES1. Allfrey, V. G., Faulkner, R., andMirsky, A. E. (1964) Acetylation andmeth-
ylation of histones and their possible role in the regulation of RNA syn-thesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794
2. Phillips, D. M. (1963) The presence of acetyl groups of histones. Biochem.J. 87, 258–263
3. Gnad, F., Forner, F., Zielinska, D. F., Birney, E., Gunawardena, J., andMann, M. (2010) Evolutionary constraints of phosphorylation in eu-karyotes, prokaryotes, and mitochondria. Mol. Cell. Proteomics 9,2642–2653
4. Norvell, A., and McMahon, S. B. (2010) Cell biology. Rise of the rival.Science 327, 964–965
5. Guan, K. L., and Xiong, Y. (2011) Regulation of intermediary metabolismby protein acetylation. Trends Biochem. Sci. 36, 108–116
6. Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., Cheng, T., Kho, Y.,Xiao, H., and Xiao, L. (2006) Substrate and functional diversity of lysineacetylation revealed by a proteomics survey.Mol. Cell 23, 607–618
7. Lundby, A., Lage, K., Weinert, B. T., Bekker-Jensen, D. B., Secher, A.,Skovgaard, T., Kelstrup, C. D., Dmytriyev, A., Choudhary, C., Lundby, C.,and Olsen, J. V. (2012) Proteomic analysis of lysine acetylation sites in rattissues reveals organ specificity and subcellular patterns. Cell Rep. 2,419–431
8. Anderson, K. A., andHirschey,M. D. (2012)Mitochondrial protein acety-lation regulates metabolism. Essays Biochem. 52, 23–35
9. Vadvalkar, S. S., Baily, C. N., Matsuzaki, S., West, M., Tesiram, Y. A., andHumphries, K. M. (2013) Metabolic inflexibility and protein lysine acety-lation in heart mitochondria of a chronic model of Type 1 diabetes.Biochem. J. 449, 253–261
10. Lombard, D. B., Tishkoff, D. X., and Bao, J. (2011) Mitochondrial sirtuinsin the regulation of mitochondrial activity and metabolic adaptation.Handb. Exp. Pharmacol. 206, 163–188
11. Vaitheesvaran, B., Yang, L., Hartil, K., Glaser, S., Yazulla, S., Bruce, J. E.,and Kurland, I. J. (2012) Peripheral effects of FAAH deficiency on fuel andenergy homeostasis: role of dysregulated lysine acetylation. PLoS One 7,e33717
12. Kim, H. S., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N.,Pennington, J. D., van der Meer, R., Nguyen, P., Savage, J., Owens, K. M.,Vassilopoulos, A., Ozden, O., Park, S. H., Singh, K. K., Abdulkadir, S. A.,Spitz, D. R., Deng, C. X., and Gius, D. (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial
integrity and metabolism during stress. Cancer Cell 17, 41–5213. Hirschey,M.D., Shimazu, T., Jing, E., Grueter, C.A., Collins, A.M., Aouiz-
erat, B., Stancáková, A., Goetzman, E., Lam, M. M., Schwer, B., Stevens,R. D., Muehlbauer, M. J., Kakar, S., Bass, N. M., Kuusisto, J., Laakso, M.,Alt, F. W., Newgard, C. B., Farese, R. V., Jr., Kahn, C. R., and Verdin, E.(2011) SIRT3 deficiency and mitochondrial protein hyperacetylation ac-celerate the development of the metabolic syndrome. Mol. Cell 44,177–190
14. Pereira, C. V., Lebiedzinska, M., Wieckowski, M. R., and Oliveira, P. J.(2012) Regulation and protection of mitochondrial physiology by sirtuins.Mitochondrion 12, 66–76
15. Rardin, M. J., Newman, J. C., Held, J. M., Cusack, M. P., Sorensen, D. J., Li,B., Schilling, B., Mooney, S. D., Kahn, C. R., Verdin, E., and Gibson, B. W.(2013) Label-free quantitative proteomics of the lysine acetylome in mi-tochondria identifies substrates of SIRT3 in metabolic pathways. Proc.Natl. Acad. Sci. U.S.A. 110, 6601–6606
16. Beltrao, P., Albanèse, V., Kenner, L. R., Swaney, D. L., Burlingame, A.,Villén, J., Lim, W. A., Fraser, J. S., Frydman, J., and Krogan, N. J. (2012)Systematic functional prioritization of protein posttranslationalmodifica-tions. Cell 150, 413–425
17. He, W., Newman, J. C., Wang, M. Z., Ho, L., and Verdin, E. (2012) Mito-chondrial sirtuins: regulators of protein acylation andmetabolism.TrendsEndocrinol. Metab. 23, 467–476
18. Hebert, A. S., Dittenhafer-Reed, K. E., Yu, W., Bailey, D. J., Selen, E. S.,Boersma, M. D., Carson, J. J., Tonelli, M., Balloon, A. J., Higbee, A. J.,Westphall, M. S., Pagliarini, D. J., Prolla, T. A., Assadi-Porter, F., Roy, S.,Denu, J. M., and Coon, J. J. (2013) Calorie restriction and SIRT3 triggerglobal reprogramming of the mitochondrial protein acetylome.Mol. Cell49, 186–199
19. Grimsrud, P. A., Carson, J. J., Hebert, A. S., Hubler, S. L., Niemi, N. M.,Bailey, D. J., Jochem, A., Stapleton, D. S., Keller, M. P., Westphall, M. S.,Yandell, B. S., Attie, A. D., Coon, J. J., and Pagliarini, D. J. (2012) A quan-titative map of the liver mitochondrial phosphoproteome reveals post-translational control of ketogenesis. Cell Metab. 16, 672–683
20. Zhao, E., Keller, M. P., Rabaglia, M. E., Oler, A. T., Stapleton, D. S.,Schueler, K. L., Neto, E. C., Moon, J. Y., Wang, P., Wang, I. M., Lum, P. Y.,Ivanovska, I., Cleary, M., Greenawalt, D., Tsang, J., Choi, Y. J., Kleinhanz,R., Shang, J., Zhou, Y. P., Howard, A. D., Zhang, B. B., Kendziorski, C.,Thornberry, N. A., Yandell, B. S., Schadt, E. E., and Attie, A. D. (2009)Obesity and genetics regulate microRNAs in islets, liver, and adipose ofdiabetic mice.Mamm. Genome 20, 476–485
21. Wenger, C. D., Lee,M. V., Hebert, A. S., McAlister, G. C., Phanstiel, D. H.,Westphall, M. S., and Coon, J. J. (2011) Gas-phase purification enablesaccurate, multiplexed proteome quantification with isobaric tagging.Nat.Methods 8, 933–935
22. Wenger, C. D., Phanstiel, D. H., Lee, M. V., Bailey, D. J., and Coon, J. J.(2011) COMPASS: A suite of pre- and post-search proteomics softwaretools for OMSSA. Proteomics 11, 1064–1074
23. Rogers, G. W., Brand, M. D., Petrosyan, S., Ashok, D., Elorza, A. A., Fer-rick, D. A., and Murphy, A. N. (2011) High throughput microplate respi-ratory measurements using minimal quantities of isolated mitochondria.PLoS One 6, e21746
24. Haapalainen, A. M., Meriläinen, G., Pirilä, P. L., Kondo, N., Fukao, T., andWierenga, R. K. (2007) Crystallographic and kinetic studies of humanmitochondrial acetoacetyl-CoA thiolase: the importance of potassiumand chloride ions for its structure and function. Biochemistry 46,4305–4321
25. Blommel, P. G., Becker, K. J., Duvnjak, P., and Fox, B. G. (2007) Enhancedbacterial protein expression during auto-induction obtained by alterationof lac repressor dosage and medium composition. Biotechnol. Prog. 23,585–598
26. Neumann, H., Peak-Chew, S. Y., and Chin, J. W. (2008) Genetically en-coding N&-acetyllysine in recombinant proteins. Nat. Chem. Biol. 4,232–234
27. Neumann, H., Hancock, S. M., Buning, R., Routh, A., Chapman, L., Som-ers, J., Owen-Hughes, T., van Noort, J., Rhodes, D., and Chin, J. W. (2009)A method for genetically installing site-Specific acetylation in recombi-nant histones defines the effects of H3 K56 acetylation. Mol. Cell 36,
Quantitative Mitochondrial Acetylation Dynamics
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26217 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

153–16328. Yu, W., Dittenhafer-Reed, K. E., and Denu, J. M. (2012) SIRT3 protein
deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochon-drial redox status. J. Biol. Chem. 287, 14078–14086
29. Hallows, W. C., Lee, S., and Denu, J. M. (2006) Sirtuins deacetylate andactivate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A.103, 10230–10235
30. Middleton, B. (1973) The acetoacetyl-coenzyme A thiolases of rat brainand their relative activities during postnatal development.Biochem. J. 132,731–737
31. Baker, N. A., Sept, D., Joseph, S., Holst,M. J., andMcCammon, J. A. (2001)Electrostatics of nanosystems: application to microtubules and the ribo-some. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041
32. de Hoon, M. J., Imoto, S., Nolan, J., and Miyano, S. (2004) Open sourceclustering software. Bioinformatics 20, 1453–1454
33. Saldanha, A. J. (2004) Java Treeview–extensible visualization of microar-ray data. Bioinformatics 20, 3246–3248
34. Locasale, J.W., andCantley, L. C. (2011)Metabolic flux and the regulationof mammalian cell growth. Cell Metab. 14, 443–451
35. Báez-Ruiz, A., Escobar, C., Aguilar-Roblero, R., Vázquez-Martínez, O.,and Díaz-Muñoz, M. (2005) Metabolic adaptations of liver mitochondriaduring restricted feeding schedules. Am. J. Physiol. Gastrointest. LiverPhysiol. 289, G1015–G1023
36. Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., Yao, J., Zhou, L.,Zeng, Y., Li, H., Li, Y., Shi, J., An,W., Hancock, S. M., He, F., Qin, L., Chin,J., Yang, P., Chen, X., Lei, Q., Xiong, Y., and Guan, K. L. (2010) Regulationof cellular metabolism by protein lysine acetylation. Science 327,1000–1004
37. Longo, N., Amat di San Filippo, C., and Pasquali, M. (2006) Disorders ofcarnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin.Med. Genet. 142C, 77–85
38. Adeva, M. M., Calviño, J., Souto, G., and Donapetry, C. (2012) Insulinresistance and themetabolism of branched-chain amino acids in humans.Amino Acids 43, 171–181
39. Kendrick, A. A., Choudhury, M., Rahman, S. M., McCurdy, C. E., Fried-erich,M., VanHove, J. L.,Watson, P. A., Birdsey, N., Bao, J., Gius, D., Sack,M. N., Jing, E., Kahn, C. R., Friedman, J. E., and Jonscher, K. R. (2011) Fattyliver is associated with reduced SIRT3 activity and mitochondrial proteinhyperacetylation. Biochem. J. 433, 505–514
40. Keller, M. P., Choi, Y., Wang, P., Davis, D. B., Rabaglia, M. E., Oler, A. T.,Stapleton, D. S., Argmann, C., Schueler, K. L., Edwards, S., Steinberg,H. A., Chaibub Neto, E., Kleinhanz, R., Turner, S., Hellerstein, M. K.,Schadt, E. E., Yandell, B. S., Kendziorski, C., and Attie, A. D. (2008) A geneexpression network model of type 2 diabetes links cell cycle regulation inislets with diabetes susceptibility. Genome Res. 18, 706–716
41. Buchner, D. A., Yazbek, S. N., Solinas, P., Burrage, L. C., Morgan, M. G.,Hoppel, C. L., and Nadeau, J. H. (2011) Increased mitochondrial oxidativephosphorylation in the liver is associated with obesity and insulin resist-ance. Obesity 19, 917–924
42. Finley, L. W., Haas, W., Desquiret-Dumas, V., Wallace, D. C., Procaccio,V., Gygi, S. P., andHaigis,M. C. (2011) Succinate dehydrogenase is a directtarget of sirtuin 3 deacetylase activity. PLoS One 6, e23295
43. Ahn, B. H., Kim, H. S., Song, S., Lee, I. H., Liu, J., Vassilopoulos, A., Deng,C. X., and Finkel, T. (2008) A role for the mitochondrial deacetylase Sirt3in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105,14447–14452
44. Bao, J., Scott, I., Lu, Z., Pang, L., Dimond, C. C., Gius, D., and Sack, M. N.(2010) SIRT3 is regulated by nutrient excess and modulates hepatic sus-ceptibility to lipotoxicity. Free Rad. Biol. Med. 49, 1230–1237
45. Cimen, H., Han, M. J., Yang, Y., Tong, Q., Koc, H., and Koc, E. C. (2010)Regulation of succinate dehydrogenase activity by SIRT3 in mammalianmitochondria. Biochemistry 49, 304–311
46. Hirschey,M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard,D. B., Grueter, C.A.,Harris, C., Biddinger, S., Ilkayeva,O. R., Stevens, R.D.,Li, Y., Saha, A. K., Ruderman,N. B., Bain, J. R., Newgard, C. B., Farese, R. V.,Jr., Alt, F. W., Kahn, C. R., and Verdin, E. (2010) SIRT3 regulates mito-chondrial fatty-acid oxidation by reversible enzyme deacetylation.Nature464, 121–125
47. Han, Q., Cai, T., Tagle, D. A., Robinson, H., and Li, J. (2008) Substratespecificity and structure of human aminoadipate aminotransferase/kynu-renine aminotransferase II. Biosci. Rep. 28, 205–215
48. Rossi, F., Garavaglia, S., Montalbano, V., Walsh, M. A., and Rizzi, M.(2008) Crystal structure of human kynurenine aminotransferase II, a drugtarget for the treatment of schizophrenia. J. Biol. Chem. 283, 3559–3566
49. Han, Q., Robinson, H., and Li, J. (2008) Crystal structure of human kynu-renine aminotransferase II. J. Biol. Chem. 283, 3567–3573
50. Oxenkrug, G. F. (2010) Metabolic syndrome, age-associated neuroendo-crine disorders, and dysregulation of tryptophan-kynureninemetabolism.Ann. N.Y. Acad. Sci. 1199, 1–14
51. Pagliarini, D. J., Calvo, S. E., Chang, B., Sheth, S. A., Vafai, S. B., Ong, S. E.,Walford, G. A., Sugiana, C., Boneh, A., Chen, W. K., Hill, D. E., Vidal, M.,Evans, J. G., Thorburn, D. R., Carr, S. A., and Mootha, V. K. (2008) Amitochondrial protein compendium elucidates complex I disease biology.Cell 134, 112–123
52. Noland, R. C.,Woodlief, T. L.,Whitfield, B. R.,Manning, S.M., Evans, J. R.,Dudek, R. W., Lust, R. M., and Cortright, R. N. (2007) Peroxisomal-mito-chondrial oxidation in a rodent model of obesity-associated insulin resist-ance. Am. J. Physiol. Endocrinol. Metab. 293, E986–E1001
53. Stein, L. R., and Imai, S. (2012) The dynamic regulation of NAD metabo-lism in mitochondria. Trends Endocrinol. Metab. 23, 420–428
54. Feldman, J. L., Dittenhafer-Reed, K. E., and Denu, J. M. (2012) SirtuinCatalysis and Regulation. J. Biol. Chem. 287, 42419–42427
55. Williamson,D.H., Lund, P., andKrebs,H.A. (1967)The redox state of freenicotinamide-adenine dinucleotide in the cytoplasm andmitochondria ofrat liver. Biochem. J. 103, 514–527
56. McGarry, J. D., and Foster, D. W. (1980) Regulation of hepatic fatty acidoxidation and ketone body production. Ann. Rev. Biochem. 49, 395–420
57. Korman, S. H. (2006) Inborn errors of isoleucine degradation: a review.Mol. Genet. Metab. 89, 289–299
58. Megee, P. C., Morgan, B. A., Mittman, B. A., and Smith, M. M. (1990)Genetic analysis of histone H4: essential role of lysines subject to reversi-ble acetylation. Science (New York, NY) 247, 841–845
59. Herrera, E., and Freinkel, N. (1967) Internal standards in the estimation ofacetyl-CoA in liver extracts. J. Lipid Res. 8, 515–518
60. Boekhorst, J., van Breukelen, B., Heck, A., Jr., and Snel, B. (2008) Compar-ative phosphoproteomics reveals evolutionary and functional conserva-tion of phosphorylation across eukaryotes. Genome Biol. 9, R144
61. Benovic, J., Tillman, T., Cudd, A., and Fridovich, I. (1983) Electrostatic facil-itation of the reaction catalyzed by the manganese-containing and the iron-containing superoxide dismutases.Arch. Biochem. Biophys. 221, 329–332
62. Tao, R., Coleman,M.C., Pennington, J. D., Ozden,O., Park, S.H., Jiang,H.,Kim, H. S., Flynn, C. R., Hill, S., HayesMcDonald,W., Olivier, A. K., Spitz,D. R., and Gius, D. (2010) Sirt3-mediated deacetylation of evolutionarilyconserved lysine 122 regulatesMnSOD activity in response to stress.Mol.Cell 40, 893–904
63. Scott, I., Webster, B. R., Li, J. H., and Sack, M. N. (2012) Identification of amolecular component of themitochondrial acetyltransferase programme:a novel role for GCN5L1. Biochem. J. 443, 655–661
64. Paik, W. K., Pearson, D., Lee, H. W., and Kim, S. (1970) Nonenzymaticacetylation of histones with acetyl-CoA. Biochim. Biophys. Acta 213,513–522
65. Tanner, K. G., Trievel, R. C., Kuo,M.H., Howard, R.M., Berger, S. L., Allis,C. D., Marmorstein, R., and Denu, J. M. (1999) Catalytic mechanism andfunction of invariant glutamic acid 173 from the histone acetyltransferaseGCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160
66. Newman, J. C., He, W., and Verdin, E. (2012) Mitochondrial protein acy-lation and intermediary metabolism: regulation by sirtuins and implica-tions for metabolic disease. J. Biol. Chem. 287, 42436–42443
67. Covian, R., and Balaban, R. S. (2012) Cardiac mitochondrial matrix andrespiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ.Physiol. 303, H940–H966
68. Xiong, Y., and Guan, K. L. (2012) Mechanistic insights into the regulationof metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164
69. Popov, K. M., Zhao, Y., Shimomura, Y., Kuntz, M. J., and Harris, R. A.(1992) Branched-chain $-ketoacid dehydrogenase kinase. Molecularcloning, expression, and sequence similarity with histidine protein ki-
Quantitative Mitochondrial Acetylation Dynamics
26218 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 288 • NUMBER 36 • SEPTEMBER 6, 2013 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from

nases. J. Biol. Chem. 267, 13127–1313070. Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B.,
Lombard, D. B., Li, Y., Bunkenborg, J., Alt, F. W., Denu, J. M., Jacobson,M. P., and Verdin, E. (2010) SIRT3 deacetylatesmitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production.Cell Metab. 12, 654–661
71. Park, J., Chen, Y., Tishkoff, D. X., Peng, C., Tan,M., Dai, L., Xie, Z., Zhang,Y., Zwaans, B. M., Skinner, M. E., Lombard, D. B., and Zhao, Y. (2013)SIRT5-mediated lysine desuccinylation impacts diverse metabolic path-ways.Mol. Cell 50, 919–930
72. Newgard, C. B. (2012) Interplay between lipids and branched-chain aminoacids in development of insulin resistance. Cell Metab. 15, 606–614
Quantitative Mitochondrial Acetylation Dynamics
SEPTEMBER 6, 2013 • VOLUME 288 • NUMBER 36 JOURNAL OF BIOLOGICAL CHEMISTRY 26219 at University of Wisconsin-Madison on September 17, 2013http://www.jbc.org/Downloaded from





























