Embed Size (px)
Citation preview

POLIMORFISME NRAS, RUNX1, NPM1, FLT3, DAN DELESI KROMOSOM 5 del(5q) SEBAGAI PREDIKTOR MYELODYSPLASTIC SYNDROME
RELATED ACUTE MYELOID LEUKEMIA (MDS-RELATED AML) DI SURABAYA
DISERTASI
Untuk Memenuhi Persyaratan
Memperoleh Gelar Doktor
Oleh
MULYA DINATA
NIM. 117070100011029
PROGRAM STUDI DOKTOR ILMU KEDOKTERAN
KEKHUSUSAN BIOMEDIK
FAKULTAS KEDOKTERAN
UNIVERSITAS BRAWIJAYA
MALANG
2019

i i
POLIMORFISME NRAS, RUNX1, NPM1, FLT3, DAN DELESI KROMOSOM 5 del(5q) SEBAGAI PREDIKTOR MYELODYSPLASTIC SYNDROME
RELATED ACUTE MYELOID LEUKEMIA (MDS-RELATED AML) DI SURABAYA
DISERTASI
Nama Mahasiswa : Mulya Dinata
NIM : 117070100011029
Program Studi : Doktor Ilmu Kedokteran
Minat : Biomedik
Menyetujui
KOMISI PEMBIMBING
Ketua,
(Prof.Dr.dr. Edi Widjajanto, MS.,SpPK.(K))
Anggota 1 Anggota 2
(Dr.dr. Pudji Rahadju, SpTHT-KL.(K)) (Dr.dr. S. Ugroseno, SpPD-KHOM.)
Mengetahui, Ketua Program Doktor Ilmu Kedokteran
(Prof.Dr.dr. Kusworini, M.Kes.,SpPK.) NIP. 194804081979031001

i i i
IDENTITAS TIM PENGUJI DISERTASI
JUDUL DISERTASI: POLIMORFISME NRAS, RUNX1, NPM1, FLT3, DAN
DELESI KROMOSOM 5 del(5q) SEBAGAI PREDIKTOR MYELODYSPLASTIC
SYNDROME RELATED ACUTE MYELOID LEUKEMIA (MDS-related AML)
DI SURABAYA.
NAMA : Mulya Dinata
NIM : 117070100011029
Program Studi : Doktor Ilmu Kedokteran
Minat : Biomedik
KOMISI PEMBIMBING
Promotor : Prof.Dr.dr. Edi Widjajanto, MS.,SpPK.(K).
Ko-Promotor 1 : Dr.dr. Pudji Rahaju, SpTHT-KL.(K)
Ko-Promotor 2 : Dr.dr. S. Ugroseno, SpPD-KHOM.
TIM PENGUJI
Penguji 1 : Prof.Dr.dr. Kusworini, M.Kes.,SpPK(K).
Penguji 2 : Dr.dr. Karyono Mintaroem, SpPA(K).
Penguji Luar : Prof.dr. Soebandiri, SpPD.,K-HOM.
Ujian Proposal : 26 Juli 2016
Seminar Hasil : 29 Mei 2019
Ujian Tertutup : 31 Juli 2019
Ujian Terbuka :

i v
Lembar originalitas

v
KATA PENGANTAR
Pertama saya haturkan puji syukur kepada Tuhan Yang Maha Esa,
Karena kemurahan, anugrahNya dan kebaikanNya, saya dapat menyelesaikan
penulisan disertasi untuk Program Doktor Ilmu Kedokteran ini yang berjudul :
Polimorfisme NRAS, RUNX1, NPM1, FLT3, dan delesi kromosom 5 del(5q)
sebagai prediktor Myelodysplastic syndrome related acute myeloid
leukemia (MDS-related AML) di Surabaya.
Di dalam tulisan ini, disajikan pokok-pokok bahasan yang meliputi
abnomalitas molekuler dan sitogenetik progresivitas berpengaruh terhadap MDS-
related AML yang melibatkan polimorfisme NRAS, RUNX1, NPM1, FLT3 dan
delesi kromosom 5 del(5q). Delesi kromosom 5 del(5q) dengan mutasi N-RAS,
FLT3, NPM1 lebih dari satu gen (60%) dapat meningkatkan kemungkinan
transformasi menjadi AML, sedangkan RUNX (RUNX1) (15%) dapat
menghambat deferensiasi dan meningkatkan proliferasi.
Dengan selesainya disertasi ini penulis menyampaikan ucapan terima
kasih yang sebesar-besarnya kepada :
1. Rektor Universitas Brawijaya Malang Prof. Dr. Ir. Nuhfil Hanani AR, MS
atas ijin dan kesempatan yang diberikan untuk mengikuti pendidikan
Program Studi Doktor Ilmu Kedokteran di Universitas Brawijaya.
2. Dekan Universitas Brawijaya Malang Dr. dr. Wisnu Barlianto, M.Si., Med.,
SpA(K) atas kesempatan, dukungan dan fasilitas yang diberikan dalam
penyelesaian pendidikan Program Studi Doktor Ilmu Kedokteran di
Universitas Brawijaya.
3. Prof. Dr. dr. Kusworini, SpPK(K).,M.Kes, selaku ketua Program Studi
Doktor Ilmu Kedokteran Universitas Brawijaya Malang, atas pemberian ijin

v i
untuk mengikuti pendidikan di Fakultas kedokteran, Universitas Brawijaya
dan selaku Tim penguji disertasi, dan membantu saya selama studi hingga
selesainya disertasi saya sebagai tugas akhir Program Studi Doktor Ilmu
Kedokteran Universitas Brawijaya.
4. Rektor Universitas Katolik Widya Mandala Surabaya Drs. Kuncoro Foe,
G.Dip.Sc., Ph.D., Apt. yang telah memberikan ijin dan dana kepada penulis
untuk tugas belajar pada jenjang doktor di Program Studi Doktor Ilmu
Kedokteran di Universitas Brawijaya.
5. Dekan Fakultas Kedokteran Universitas Katolik Widya Mandala Surabaya
Prof. Dr. Dr.Med. Paul L. Tahalele, Sp.BTKV (K)., FICS yang telah
memberikan ijin dan dana kepada penulis untuk tugas belajar pada jenjang
doktor di Program Studi Doktor Ilmu Kedokteran di Universitas Brawijaya.
6. Prof dr. W.F. Maramis SpKJ(K), beliau yang pertama kali memberikan
dorongan untuk tugas belajar pada jenjang doktor di Program Studi Doktor
Ilmu Kedokteran di Universitas Brawijaya.
7. Prof Dr. dr. Edi Widjajanto, MS, SpPK(K), selaku promotor disertasi saya,
sejak ujian kualifikasi, penelitian dan penyusunan disertasi telah berkenan
membimbing dan memberikan masukan dan saran hingga saya bisa
menyelesaikan disertasi ini.
8. Dr. dr. Pudji Rahadju, sp THT-KL.(k), selaku dosen pembimbing 6 karya
ilmiah, yang dapat saya selesai dengan baik berkat bantuan, bimbingan
beliau, sehingga saya dapat menyelesaikan disertasi ini.
9. Dr. dr. S. Ugroseno Y.B., SpPD-KHOM, Promotor dari Departemen
Penyakit Dalam devisi Hematologi dan Onkologi, Rumah Sakit Pendidikan

v i i
dr. Soetomo, Surabaya, yang membimbing penelitian ini terutama terutama
sampel penelitian, bagian laboratorium, diagnosis dan klinik.
10. Dr. dr. Windhu purnomo, MS, pembimbing statistik dari Fakultas Public
Health universitas Airlangga departemen studi Biostatistik dan populasi,
selaku pembimbing sejak Makalah Ilmiah sampai selesainya disertasi.
11. Dr. dr. Karyono Mintaroem, SpPA(K), selaku dosen penguji 6 karya ilmiah
dan tim penguji ujian tertutup dan terbuka.
12. Prof. dr. Soebandiri, SpPD-KHOM, selaku Tim penguji dari devisi
Hematologi dan Onkologi, Rumah Sakit Pendidikan dr. Soetomo, Surabaya
selaku Tim penguji proposal sampai disertasi.
13. Prof. Drs. Sutiman B. sumitro, SU.,D.Sc, selaku dosen penguji 6 karya
ilmiah dan beliau banyak memberikan masukkan untuk memperbaiki
penulisan 6 karya ilmiah.
14. Prof. Dr. dr. Handono Kalim, Sp.PD.KR, selaku tim penguji kualifikasi yang
telah memberikan wawasan untuk membuat penelitian pendahuluan dan
sebagai dosen MKPD yang memberikan masukkan yang menunjang
disertasi.
15. Dra. Diana Lyrawati, Apt., MSc., PhD, selaku pembimbing MKPD yang
banyak memberikan bimbingaan terutama tentang journal internasional,
sehingga 2 journal saya dapat accepted
16. Agustina Tri Endharti S.Si., PhD selaku pembimbing untuk mencari
journal, verifikasi dan memasukkan journal internasional.
17. Dr. dr. Yetty Hernaningsih ,SpPK(K), kepala bagian Departemen Patologi
Klinik Rumah Sakit pendidikan dr Soetomo, Surabaya yang membimbing,
memberikan ijin, untuk menggunakan laboratorium .

v i i i
18. Prof. Dr. dr. Ariyati, MS,SpPK(K), bagian Kimia Klinik Departemen Patologi
Klinik Rumah Sakit pendidikan dr Soetomo, Surabaya, memberikan ijin
kepada saya untuk mengambil sampel penelitian pendahuluan.
19. Dr. dr. Hartono Kahar, SpPK(K), MQIH, bagian Hematologi, Departemen
Patologi Klinik Rumah Sakit pendidikan dr Soetomo, Surabaya,
memberikan support untuk menggambil sampel penelitian dari data pasien
yang mengambil darah di bagian Patologi Klinik.
20. dr. Arifoel Hajat, SpPK(K), bagian Hematologi Departemen Patologi Klinik
Rumah Sakit pendidikan dr Soetomo, Surabaya, yang membimbing dan
memberikan konsultasi diagnosis laboratorium hapusan sumsum Tulang.
21. Prof. Dr. dr. jusak Nugraha, SpPK(K), bagian Immunologi Departemen
Patologi Klinik Rumah Sakit pendidikan dr Soetomo, Surabaya,
memberikan support dan bimbingan penyusunan disertasi ini.
22. dr. Endang Retnowati Kusumowidagdo, MS, SpPK(K) (Alm), bagian
Immunologi Departemen Patologi Klinik Rumah Sakit pendidikan dr
Soetomo, Surabaya, memberikan support dan bimbingan penyusunan
desertasi ini.
23. Prof. Dr. dr. Tjipto Suwandi, SpOK (Alm), Fakultas Public Health universitas
Airlangga, Surabaya, memberikan support dan bimbingannya pada awal sy
mendaftar sebagai mahasisiwa S3.
24. Prof. Dr. dr. Irwan Setiabudi, SpPK(K) (Alm), bagian Departemen Patologi
Klinik Fakultas kedokteran Universitas Hang Tuah, Surabaya, memberikan
support pada awal sy mendaftar sebagai mahasisiwa S3.
25. Semua Guru Besar dan dosen serta seluruh staf dan pegawai di Program
Studi Doktor Universitas Brawijaya Malang, yang dengan sabar membantu

i x
dan memberikan semangat kepada saya selama menempuh pendidikan S3
ini.
26. Wibi Irawan SSi MBiomed, stab.laboratorium Biokimia Biomolekuler, FK
Universitas Brawijaya Malang, membantu pemeriksaan laboratorium
Desertasi ini.
27. Semua Guru Besar dan dosen serta seluruh staf dan pegawai Patologi
Klinik, Fakultas kedokteran Universitas Airlangga Surabaya, yang dengan
sabar membantu pemeriksaan laboratorium dan memberikan semangat
kepada saya selama menempuh pendidikan S3 ini.
28. Semua Guru Besar dan dosen serta seluruh staf dan pegawai bagian
penyakit Dalam divisi Hematologi-Onkologi, Fakultas kedokteran
Universitas Airlangga Surabaya, yang dengan sabar membantu dan
memberikan semangat kepada saya selama menempuh pendidikan S3 ini.
29. Semua sejawat Dokter, seluruh teman dosen dan pegawai serta staf di
Fakultas Kedokteran Universitas Katolik Widya Mandala Surabaya, yang
mendampingi, menyemangati dan membantu saya dalam menjalani studi
S3 ini hingga selesai.
30. Semua sejawat Dokter, seluruh teman serta staf di klinik Sentra Medika
Surabaya, antara lain Windi, Imam, menyemangati dan membantu saya
dalam menyelesaikan studi S3 ini.
31. Semua sejawat Dokter Alumni Fakultas Kedokteran Universitas Airlangga,
Surabaya tahun 1978, Teman SMA Santo Louis Surabaya, menyemangati
dan membantu saya dalam menyelesaikan studi S3 ini.
32. Keluarga besar saya ibu, Adik dan keluarga, khususnya istri saya drg
Trijuliati SpOrt., anak saya drg Floretta Charlene, ARAD, Alfonsis

x
Claresta,S.AK.,ADV.Dipl,GMA, Michael Rodney yang selalu memberikan
semangat, mendampingi dan memotivasi saya untuk menyelesaikan studi
S3 ini.
33. Guru guru saya sejak TK, SD, SMP, SMA hingga pendidikan saat ini, saya
ucapkan terima kasih yang tak terhingga. Berkat Bapak dan ibu guru, saya
bisa menempuh pendidikan S3 ini.
34. Kepada semua saudara, sahabat dan kerabat yang tidak dapat saya
sebutkan satu per-satu, yang telah berperan dan turut serta dalam studi S3
saya ini
Sangat disadari bahwa dengan kekurangan dan keterbatasan yang
dimiliki penulis, masih dirasakan banyak kekurangtepatan, oleh karena itu penulis
mengharapkan saran yang membangun agar tulisan ini bermanfaat bagi yang
membutuhkan.
Surabaya, 29 Oktober 2019
Penulis

x i
RINGKASAN
Mulya Dinata. NIM. 117070100011029. Fakultas Kedokteran Universitas
Brawijaya Malang, 29 Mei 2019. Polimorfisme NRAS, RUNX1, NPM1, FLT3, Dan
Delesi Kromosom 5 del(5q) Sebagai Prediktor Myelodysplastic Syndrome
Related Acute Myeloid Leukemia (MDS-Related AML) di Surabaya.
Myelodysplastic syndrome (MDS) adalah sekelompok kelainan haematopoeitic stem cells (HSC) yang ditunjukkan dengan kegagalan sumsum tulang (displastik) yang meningkat. Penyakit yang didominasi orang tua antara 70 tahun dan dapat dikatakan kira-kira 1 orang menderita MDS pada 500 populasi diatas 60 tahun. Jumlah kasus MDS lebih kurang 20-30% berkembang menjadi Acute Myelocytic Leukemia (AML), prevalensi risiko menjadi AML cukup tinggi, diperlukan diagnosis sedini mungkin sebelum transformasi menjadi AML.
Etiologi kasus Leukemia sampai saat ini belum diketahui penyebabnya dengan pasti. Banyak penelitian telah dilakukan untuk mengetahui faktor risiko. Berbagai faktor risiko yaitu penggunaan pestisida, medan listrik, bahan kimia (Benzena), virus, abnormalitas sitogenetik (contoh: Down syndrome), usia ibu yang relatif tua saat melahirkan, ibu yang merokok saat hamil, konsumsi alkohol saat hamil, medan magnet, pekerjaan orang tua, radiasi prenatal dan postnatal.
Data penelitian pendahuluan didapatkan bagian Patologi Klinik RSUD dr Soetomo atau Fakultas Kedokteran Universitas Airlangga Surabaya, Jawa Timur, jumlah pasien 228 orang selama 10 bulan pada tahun 2014, ikut serta penelitian 181 orang, kasus AML 37,28%, rentang umur terbanyak 46 - 61 tahun 11,40% sedangkan Kasus MDS 3,95%.
Hasil penelitian pasien rawat jalan atau inap di instalasi Ilmu Penyakit Dalam Hematologi-Onkologi FK Universitas Airlangga, RSUD dr Soetomo, Surabaya, Indonesia, tahun 2017 - 2018 (satu tahun), sebanyak 36 subyek sampel, 31 sampel dengan diagnosis AML sedangkan 5 sampel diagnosis MDS dikeluarkan dari penelitian. Usia 47- 57 tahun dan 58 – 68 tahun masing-masing sebanyak 19,35% dan 22,58%, sedangkan usia tua yaitu 80-90 tahun (3,24%). Jenis kelamin laki-laki lebih banyak dari perempuan yaitu laki-laki (70,97%), perempuan (29,03%). Hasil pemeriksaan immunophenotyping yaitu myeloid lineage 96,76%.
Tujuan penelitian ini adalah membuktikan pengaruh polimorfisme NRAS, RUNX1, NPM1, FLT3, dan delesi kromosom 5 del(5q) sebagai prediktor Myelodisplastic Syndrome related Acute Myeloid Leukemia (MDS-related AML) pada penderita di Surabaya.
Diagnosis pasien (klasifikasi Leukemia FAB, WHO 2008) berdasarkan pemeriksaan klinik, immunophenotyping, hapusan aspirasi sumsum tulang (BMA) yaitu pertama AML (86,11%), dan Non AML (MDS) sebanyak 13,89%, abnormalitas genetik (PCR), abnormalitas kromosom (CISH).
Metode penelitian ini cross sectional analytic, uji analisis statistik yang dipergunakan Logistic Regression (Regresi Logistik), Receiver operating Characteristics (ROC), 24 sampel adalah delesi kromosom 5 del(5q) (positif) dan 7 sampel adalah kromosom 5q normal (negatif) dengan berbagai variasi mutasi gen. ROC didapatkan area dibawah kurva (AUC) 76,5%, confidence interval 95%, dan signifikan (P = 0,036), menunjukkan bahwa delesi kromosom 5 del(5q) atau kromosom 5 normal dengan berbagai variasi mutasi atau tidak ada mutasi NRAS, RUNX1, NPM1, FLT3 variasi tersebut sebanyak 16 macam variasi, sebagai prediktor MDS-related AML adalah valid dan bisa didapatkan cut-off

x i i
value (0,7825584010). Delesi kromosom 5 del(5q) dengan variasi gen cut-off probbilitas ≥ 0,7825584010 prediksi kuat MDS-related AML, sebaliknya kromosom 5 normal dengan variasi NRAS, RUNX1, NPM1, FLT3 ≤ probabilitas 0,7825584010 prediktor lemah MDS-related AML. Mutasi NRAS mempunyai β tertinggi 2,446, PR(Prealence Risk) 11,543 adalah PR tertinggi berarti meningkatkan risiko MDS-related AML sebesar 11,543 kali; mutasi RUNX1 β (-) 1,694, PR 0,184 adalah PR terendah, berarti menghambat risiko MDS-related AML sebesar 0,184 kali sedangkan Mutasi NPM1 mempunyai β 1,618, PR 5,043; mutasi FLT3 β 0,147; PR 1,158 meningkatkan risiko MDS-related AML.
Hasil penelitian didapatkan prediktor kuat MDS-related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen cut-off probbilitas > 0,7825584010, sedangkan Prediktor lemah untuk MDS-related AML yaitu kromosom q normal dengan variasi gen cut-off < probabilitas 0,7825584010. Mutasi gen RUNX1 mempunyai aktivitas menghambat 0,184 kali terjadinya MDS–related AML, sebaliknya mutasi gen NRAS, NPM1, FLT3 mempunyai aktivitas meningkat risiko MDS-related AML tertinggi adalah NRAS 11,543 kali.
Kesimpulan: variasi RUNX1, NPM1, FLT3, N-RAS dan delesi kromosom 5 del(5q) sebagai prediktor kuat MDS–related AML di Surabaya. Progresivitas MDS-related AML sebagai prediktor lemah ditunjukan oleh sitogenatik normal yaitu kromosom 5, mutasi gen RUNX1 bersifat menghambat progresivitas sedangkan mutasi gen NRAS, NPM1, FLT3 meningkatkan progresivitas MDS-related AML. Masing-masing prediktor lemah atau kuat besarnya risiko prediktor ditentukan oleh masing-masing gen yang mutasi, setiap mutasi gen NRAS, RUNX1, NPM1, FLT3, sesuai urutan sebagai berikut NRAS (1), NPM1 (2), FLT3 (3), RUNX1 (4).

x i i i
SUMMARY Mulya Dinata. NIM. 117070100011029. Medical Faculty of Brawijaya University Malang, 29th Mei 2019. NRAS, RUNX1, NPM1, FLT3 Polimorfism And Chromosome 5 del(5q) Delation As Myelodysplastic Syndrome-Related Acute Myeloid Leukemia (MDS-Related AML) Predictor In Surabaya.
Myelodysplastic syndrome (MDS) is a group of haematopoeitic stem cells (HSC) abnormality which is showed by the increase fail in bone marrow (dysplastic). A disease dominated by 70 years old people can be one in 500 population over 60 years. The number of MDS case grows more less 20-30% into Acute Myelocytic Leukemia (AML), the risk prevalency to be AML is high enough. It needs earlier diagnosis before the transformation into AML.
The previous research data of Clinical Patology of dr Soetomo hospital or Medical faculty of Airlangga University, Surabaya, East Java states that there are 228 patients during 10 months in 2014 with 37,28% of AML case, 11,40% mostly with age range between 46 – 61 years old. Meanwhile, the MDS case is about 3.95%.
Based on the research result from in or outpatients in Hematology and Medical oncology department of Internal Medicine, Airlangga University, School of Medical Dr. Soetomo Hospital, Surabaya, Indonesia, there are 36 subject of sample, 31 sample with AML diagnosis and 5 sample of MDS diagnosis that is issued by the researcher. There are 19,35% and 22,58% of Each of age 47-57 years old and 58-68 years old, besides 3,24% of age 80-90 years old. The number of man (70,97%) is more than the woman (29,03%). The immunophenotyping checkup result and myeloid lineage 96,76%.
The patient diagnosis based on the clinical checkup, immunophenotyping, Bone marrow aspiration is first AML(86,11%), and there are 13,89% Non AML (MDS), genetic abnormality (PCR), chromosome abnormality (CISH). This research is cross sectional analytic using statistic Logistic Regression, Receiver operating Characteristics (ROC), 24 samples are deletion of 5 del(5q) chromosome as possitive and 7 samples are normal 5 chromosome as negative with many kinds of genetic mutation. ROC that is found in the area of under curve (AUC) is 76.5%, confidence interval 95%, and the significance is (P = 0.036), shows that deletion of 5 del(5q) chromosome or normal 5 chromosome with various mutation or no NRAS, RUNX1, NPM1, FLT3 mutation. There are 16 kinds of mutation which influence to MDS-related AML in which it is valid and can be found cut-off value (0.7825584010). Deletion of 5 del(5q) chromosome with variety of gen cut-off probability is > 0.7825584010 will be strongly predicted to be MDS-related AML, otherwise normal 5 chromosome with various mutation of RUNX1, FLT3, NRAS, NPM1 which are < 0.7825584010 probability in which the weak predictor becomes MDS-related AML. NRAS mutation has highest β 2.446, PR 11.543. The highest PR means increasing 11.543 times of MDS-related AML; the mutation of RUNX1 β (-) 1.694, PR 0.184 is the lowest PR. it means that it increases 0.184 times of MDS-related AML. Whereas, NPM1 mutation has β 1.618, PR 5.043; FLT3 β 0.147 mutation; PR 1.158. The variable of β (-)1.694, inhibits the risk of MDS-related AML. Otherwise, the mutation of FLT3 β variable: 0.147, NRAS β 2.446 and NPM1 β 1.618 increases the risk of MDS-related AML.
The research result found that strong predictor will be MDS-related AML , that is deletion of del(5q) chromosome 5 with genetic variation of cut-off probability > 0.7825584010. Meanwhile, the weak predictor for MDS-related AML

x iv
is normal 5 chromosome with genetic variation of cut-off that is < 0.7825584010 probability for being MDS-related AML. RUNX1 gene mutation has activity to inhibit the occurrence of MDS-related AML as much as 0.184 times. On the contrary, NRAS gene mutation, NPM1 and FLT3 have activities to increase higher than others such as NRAS 11.543 times.
The conclusion: the variation of N-RAS, RUNX1, NPM1, FLT3, and chromosome 5 del(5q) deletion are strong predictors MDS–related AML patients in Surabaya. RUNX1 has activity to inhibit the process of MDS-related AML; while NRAS, NPM1 and FLT3 have activity to increase the probability of MDS-related AML occurrence. Each of weak or strong predictors risk is determined by each of mutated genes. Every gene mutation of NRAS, RUNX1, NPM1, FLT3 is based on its order as follow NRAS (1), NPM1 (2), FLT3 (3), RUNX1 (4).

xv
DAFTAR ISI
LEMBAR PERSETUJUAN ............................................................................... ii
IDENTITAS TIM PENGUJI DISERTASI .......................................................... iii
LEMBAR ORIGINALITAS ................................................................................ iv
SURAT KETERANGAN BEBAS PLAGIASI .................................................... v
RINGKASAN .................................................................................................... vi
SUMMARY ....................................................................................................... viii
DAFTAR ISI...................................................................................................... ix
DAFTAR TABEL .............................................................................................. xv
DAFTAR GAMBAR .......................................................................................... xvii
DAFTAR SINGKATAN ..................................................................................... xix
BAB 1 PENDAHULUAN ................................................................................... 1
1.1 Latar Belakang .............................................................................. 1
1.2 Rumusan Masalah ........................................................................ 6
1.3 Tujuan Penelitian........................................................................... 7
1.3.1 Tujuan umum ....................................................................... 7
1.3.2 Tujuan Khusus ..................................................................... 7
1.4 Manfaat Penelitian ........................................................................ 8
BAB 2 TINJAUAN PUSTAKA .......................................................................... 9
2.1 Acute Myeloid Leukemia (AML) .................................................... 9
2.1.1 Patogenesis AML ................................................................. 11
2.2 Myelodysplastic syndrome (MDS) ................................................ 19
2.2.1 Patogenesis MDS ................................................................ 21
2.3 Patomekanisme AML .................................................................... 25

xv i
2.3.1 Mutasi kategori I (kelainan yang mengaktifkan jalur
transduksi sinyal) ................................................................. 28
2.3.1.1 Mutasi gen Fms-Related Tyrosine Kinase 3
(FLT3) ...................................................................... 28
2.3.1.2 Mutasi gen RAS ....................................................... 31
2.3.2 Mutasi kategori II (Mutasi faktor transkripsi) ....................... 35
2.3.2.1 Core-binding factor (CBF) leukemia ....................... 36
2.3.2.2 Mutasi gen PU.1 ...................................................... 37
2.3.2.3 Mutasi gen NPM1 .................................................... 38
2.4 Patomekanisme MDS ................................................................... 40
2.4.1 Abnormalitas sitogenetik ..................................................... 40
2.4.2 Abnormalitas molekuler MDS .............................................. 41
2.4.2.1 Mutasi Gen RAS ...................................................... 41
2.4.2.2 Mutasi Gen yang lain ............................................... 42
2.5 Peran gen RUNX1, NPM1, N-RAS pada delesi kromosom 5
del(5q) pada MDS ......................................................................... 43
BAB 3 KERANGKA KONSEP DAN HIPOTESIS PENELITIAN ...................... 54
3.1 Kerangka Konsep Penelitian ......................................................... 54
3.2 Hipotesis Penelitian ....................................................................... 55
BAB 4 METODE PENELITIAN ........................................................................ 56
4.1 Jenis dan Desain Penelitian .......................................................... 56
4.2 Waktu dan Tempat Penelitian ....................................................... 56
4.3 Pengambilan sampel penelitian .................................................... 56
4.3.1 Kriteria inklusi ....................................................................... 57
4.3.2 Kriteria Eksklusi ................................................................... 57

xv i i
4.3.3 Besar sampel ....................................................................... 57
4.3.4 Cara Pengambilan Sampel .................................................. 58
4.4 Variabel penelitian ......................................................................... 58
4.5 Definisi Operasional ...................................................................... 58
4.5.1 MDS-related AML ................................................................ 58
4.5.2 MDS (Myelodysplastic Syndrome) ...................................... 59
4.5.3 NRAS (Rat sarcoma) ........................................................... 59
4.5.4 RUNX1 atau CBF (runt-related transcription factor 1) ........ 59
4.5.5 NPM1 (Nucleophosmin) ...................................................... 60
4.5.6 FLT3 (Fms-like tyrosine kinase 3) ....................................... 60
4.5.7 Delesi kromosom 5q ............................................................ 60
4.6 Bahan dan metode pemeriksaan .................................................. 61
4.6.1 Hapusan darah tepi ............................................................. 61
4.6.2 Aspirasi Sumsum Tulang (Bonemarrow Aspiration) ........... 61
4.6.3 Pemeriksaan fenotipe AML ................................................. 61
4.6.4 Polymerase Chain Reaction (PCR) dipergunakan untuk
pemeriksaan Gen NRAS, RUNX1, NPM1, FLT3 ................ 63
4.6.5 Analisis kromosom dan CISH (Chromic In Situ
Hybridization) ....................................................................... 67
4.7 Kerangka Operasional .................................................................. 70
4.8 Rancangan analisa data ............................................................... 71
BAB 5 HASIL PENELITIAN ............................................................................ 72
5.1 Karakteristik dasar pasien AML .................................................... 72
5.1.1 Hasil data karakteristik dasar .............................................. 72

xv i i i
5.1.2 Polimorfisme NRAS sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid
Leukemia (MDS-related AML). ............................................ 73
5.1.3 Menentukan polimorfisme RUNX1 sebagai prediktor
perubahan Myelodisplastic Syndrome menjadi Acute
Myeloid Leukemia (MDS-related AML) ............................... 73
5.1.4 Menentukan polimorfisme NPM1 sebagai prediktor
perubahan Myelodisplastic Syndrome menjadi Acute
Myeloid Leukemia (MDS-related AML) ............................... 74
5.1.5 Menentukan polimorfisme FLT3 sebagai prediktor
perubahan Myelodisplastic Syndrome menjadi Acute
Myeloid Leukemia (MDS-related AML) ............................... 75
5.1.6 Menentukan polimorfisme delesi kromosom 5 del(5q) dan
Kromosom 5 normal sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid
Leukemia (MDS-related AML) ............................................. 76
5.1.6,7 Poliforfisme delesi kromosom ............................................ 88
5.1.8 Analisis statistik .................................................................. 88
5.1.8.1 Pemeriksaan morfologi ........................................... 88
5.1.8.2 Metode statistik ROC .............................................. 89
5.1.8.3 Hasil analisis ROC .................................................. 89
5.1.8.4 Sensitivitas dan spesifisitas .................................... 91
5.1.8.5 Progresivitas gen NRAS ......................................... 93
5.1.8.6 Rumus probabilitas ................................................. 93
5.1.8.7 Variasi statistic ........................................................ 93

x ix
5.2 Hasil penelitian diskriptif dan statistic ROC, Regresi Logistik ..... 94
BAB 6 PEMBAHASAN UMUM ....................................................................... 96
6.1 Data dasar karakteristik subyek penelitian ................................... 96
6.2 Polimorfisme NRAS sebagai prediktor MDS-related AML ........... 99
6.3 Polimorfisme RUNX1 sebagai prediktor MDS-related AML ......... 102
6.4 Polimorfisme NPM1 sebagai prediktor MDS-related AML ........... 104
6.5 Polimorfisme FLT3N sebagai prediktor MDS-related AML .......... 105
6.6 Polimorfisme delesi kromosom 5 del(5q) sebagai predictor
MDS-related AML .......................................................................... 109
6.7 Polimorfisme kromosom 5 normal sebagai prediktor MDS-
related AML ................................................................................... 110
6.8 Hasil penelitian dan diskusi ........................................................... 113
6.9 Keterbatasan ................................................................................. 115
BAB 7 KESIMPULAN DAN SARAN ................................................................ 116
7.1 Kesimpulan .................................................................................... 117
7.2 Saran ............................................................................................. 117
DAFTAR PUSTAKA ......................................................................................... 119
LAMPIRAN ....................................................................................................... 127

xx
DAFTAR TABEL
Tabel 2.1 Risiko mutasi kromosom berkembang menjadi AML ................... 18
Tabel 2.2 Gambaran klinik mutasi gen AML ................................................. 19
Tabel 2.3 Membandingkan antara abnormalitas genetik dan sitogenetik
yang ditemukan pada MDS dan AML ......................................... 49
Tabel 2.4 Flourescence In Situ Hybridization dapat memperbaiki
pemeriksaan delesi 5q31 pada kasus MDS tanpa membuktikan
kelainan 5q- ................................................................................... 51
Tabel 4.1 Informasi primer PCR (NRAS, RUNX1, NPM1, FLT3) ................. 73
Tabel 5.1 Diskripsi data Dasar Karakteristik 36 pasien AML, MDS-related
AML ............................................................................................... 84
Tabel 5.2 Hasil pemeriksaan Polimorfisme NRAS berdasarkan umur, jenis
kelamin, gen NRAS mutasi/tiddak mutasi .................................... 85
Tabel 5.3 Hasil pemeriksaan Polimorfisme RUNX1 berdasarkan umur,
jenis kelamin, gen RUNX1 mutasi/tidak mutasi ............................ 85
Tabel 5.4 Hasil pemeriksaan Polimorfisme NPM1 berdasarkan umur, jenis
kelamin, gen NPM1 mutasi/tidak mutasi ...................................... 86
Tabel 5.5 Hasil pemeriksaan Polimorfisme FLT3 berdasarkan umur, jenis
kelamin, gen FLT3 mutasi/tidak mutasi ........................................ 87
Tabel 5.6 Hasil pemeriksaan Polimorfisme delesi Kromosom 5 del(5q)
dan kromosom 5 normal berdasarkan umur, jenis kelamin ......... 88
Tabel 5.7 Ringkasan Pengolahan Kasus ...................................................... 89
Tabel 5.8 Kasus dinyatakan valid ................................................................. 89
Tabel 5.9 Area Dibawah Kurva ..................................................................... 90
Tabel 5.10 Hasil Variabel : probabilitas MDS-related AML ............................ 92

xx i
Tabel 5.11 Nilai validitas kombinasi NRAS, RUNX1, NPM1, FLT3 dan
Delesi kromosom 5 del(5q) sebagai prediktor MDS - related
AML ............................................................................................... 92
Tabel 5.12 Hasil Regresi Logistik .................................................................... 93
Tabel 5.13 Variasi gen NRAS, RUNX1, NPM1, FLT3 terhadap Probabilitas
MDS-related AML .......................................................................... 94

xx i i
DAFTAR GAMBAR
Gambar 2.1 Hematopoisis, defernsiasi sel dan faktor transkripsi .................. 15
Gambar 2.2 Patogenesis AML, MDS dan MDS-related AML ........................ 17
Gambar 2.3 Mekanisme karsinogenesis akibat faktor lingkungan dan diet .. 22
Gambar 2.4 Proses epigenetic transformasi menjadi AML ............................ 23
Gambar 2.5 Apoptosis, antioksidan dan aktivasi NF-kB ................................ 24
Gambar 2.6 Perkembangan sel Th1 dan Th2 dan sitokin yang diproduksi
masing-masing ............................................................................ 25
Gambar 2.7 Frekwensi dan distribusi mutasi NPM1, CEPBA, RUNX1,
MLL-PTD, dan FLT3-ITD pada karyotype normal AML ............. 32
Gambar 2.8 Siklus GDP-GTP dari RAS ......................................................... 35
Gambar 2.9 Faktor transkripsi AML-1 dan leukemogenesis .......................... 39
Gambar 2.10 Radikal bebas mempunyai pengaruh terhadap peningkatan
risiko terjadinya MDS yang berhubungan dengan AML, MDS
dan AML ................................................................................... 53
Gambar 2.11 Anemia mempunyai pengaruh terhadap peningkatan risiko
terjadinya MDS yang berhubungan dengan AML, MDS dan
AML ........................................................................................... 54
Gambar 2.12 Kerangka teori .......................................................................... 56
Gambar 3.1 Kerangka konsep penelitian ....................................................... 60
Gambar 4.1 Kerangka operasional ................................................................. 79
Gambar 5.1 Kurva ROC .................................................................................. 90
Gambar 5.2 Probabilitas dibawah 0,7825584010 (low risk), diatas
0,7825584010 (high risk), Sensitivitas 0,708, spesifisitas 0,714 79
Gambar 5.3 Hasil PCR.................................................................................... 82

xx i i i
Gambar 5.4 Pemeriksaan sitogenetik delesi kromosom 5 del (5q) (CISH) ... 82
Gambar 5.5 Hasil pemeriksaan Immunophenotyping .................................... 83
Gambar 5.6 Morfologi sel hapusan darah tepi dan sumsum tulang .............. 84

xx iv
DAFTAR SINGKATAN
AKT : AK virus dengan AK8 (retrovirus) T (tymoma)
ALAS : 5 Aminolevulinic (acid) Syntetase
ALL : acute lymphocytic leukemia
AML : acute myeloid leukemia
Apaf-1 : apoptotic protease activating factor-1
APL : Acute Promyelocytic Leukemia
Apo2L : Apo2 ligand
Bcl-2 : B-cell lymphoma protein 2
BFU-E : Bursa form unit-eritroid
Bim-1 : Bcl2 interacting protein BIM
C KIT : Proto-oncogen C-KIT/tyrosine-protein kinase KIT
CAD : caspase-activated DNAse
CBF : core binding factor
CBFβ : Core binding Factor β
Cbl : Cobalamin
CD : Cluster of Differentiation
CDR : comman deleted region
CEBPA : enchener-binding protein α
CFU-E : coloni forming unit-eritroid
CML : chronic myeloid leukemia
CMML : chronic myelomomocytic leukemia
CN : Cytogenetic normal
CN-AML : cytogenetic normal – AML
CR : complete remission

xxv
CSC : Cancer Stem Cell
CTL : cytotoxic T lymphocyte
DAMPs : damage associated molecular pattern
DBA : Diamond – black anemia
DC : dendritic cell
DED : death effector domain
Del(5q) : delesi (5q)
DFS : Disease-Free Survival
DIC : disseminated intravascular coagulation
DISC : death inducing signaling complex
DNA : Deoxyribonucleic acid
DR3 : death receptor 3 Duplication
EFS : Even-Free Survival
ELN : European Leukemia Net .
ERK(MAPK) : Mitogen-activated protein kinase
ETO : Eight-Twenty One
FAB : Frans, American and British
FADD : Fas associated death domain
Fas : fatty acid synthetase
FasL : fatty acid synthetase ligand
FasR : fatty acid synthetase receptor
FDA : Food and Durg Administration
FGFR : Fibroblast growth factor receptor
FISH : fluorescence in situ hybridization
FLT3-ITDs : Fms-related tyrosine kinase 3 gene internal tandem

xxv i
FPD : Familial Platelet disorder
FTIs : Farmesyl transferase inhibitors
GAPs : GTPase-activating proteins
GATA 1 : GATA-binding factor 1
G-CSF : granulocyte-colony stimulating factor
GEFs : guanine nucleotide exchange factors
GPD : guanine di phosphate
GPx : Gluthatione Peroksidase
GRB2 : Growth factor receptor-bounds
GTP : guanine tri phosphate
HOX : Homeobox
HSCs : Hematopoetic stem Cells
IF : Intrinsic factor
IFN-ɤ : interferon ɤ
IL- 2 : Interleukin – 2
IL- 4 : Interleukin – 4
IL-10 : interleukin – 10
inv(16) : inversi (16)
IRE : Iron Responsive Element
IRP : Iron Regulation Protein
ITP : Idiopatic Thrombositopenia Purpura
JAK2 : Janus-associated kinase 2
JNK : C-Jun N-terminal kinase
Leukemia
LSCs : Leukemia Stem Cells

xxv i i
MAP : Microtubule-associated protein
MCV : mean corpuscular volume
MDS : Myelodysplasic syndrome
MDS-related AML : Myelodysplastic syndrome-related acute myeloblastic
MEK(MAPKK) : Mitogen-activated protein kinase kinase
MLLT3-MLL : mixed lineage leukemia T3
MMA : Methyl Malonic Acid
MPDs : Myeloproliferative disorders
MRD : Minimal Residual Disease
mRNA : messenger RNA
m-TORC : mammatian target of rapamycin complex 2
MyD88 : Myeloid differentiation factor
MYH11 : Myosin Heavy Chain 11
NCCN : National comprehensive Cancer network
NPM1 : Nucleophosmin member 1
NPM1c+ : Nucleophosmin 1 Cytoplamic Localization
OS : Over all Survival
PAMPs : pathogen associated molecular patterns
PCR : polymerase chain reaction
PDGFR : Platelet-derived growth factor
PI3K : phosphatidy Iinositol 3-Kinase
PKB : Protein kinase B
PLP : Pyridoxal 5’- phosphate
PML : Promyelocytic leukemia
PML/RARA : promyelocytic leukemia/retinoic acid receptor α gene

xxv i i i
PRRS : pattern regocnation receptor
PSA : Pure sideroblastic anemia
PTKRS : Protein-tyrosine kinase
PTPN11 : 3 phosphoinosite phosphatase
PU.1 : Purine.1- rich DNA
RA : refractory anemia
RAEB : Refractory anemia excess blasts
RAEB-t : Refractory anemia excess blasts in transformation
RAF(MAPKKK) : Mitogen-activated protein kinase kinase kinase
RAF-1 : Sorafenib-1mechanism
RARS : refractory anemia withring sideroblastic
RARα : retinoic acid reseptor
RAS : Rat sarcoma
RBC : Red Blood Cell
RNA : Ribonucleic acid
RNA : ribonucleic acid
ROS : Reactive Oxygen Species
RUNX1 : runt-related transcription factor 1
SA : Sideroblastic anemia
SAM : S-adenosyl methionine
SEER : Surveilence Epidemiology and End Result
SH2 : Src homology region 2
SH3 : Src homology region 3
Smac/DIABLO : second mitochondrial activator of caspase/Direct IAP
SNP array : single-nucleotide polymorphism

xx ix
SOD : Superoksida Dismutase
SOS : Son of sevenless
STAT 5 : signal transducer and activator of transcription
t(15;17) : translocation (15;17)
TC II : Transcobalamin II
TCR : T cell receptor
TEL : translocation ets. Leukemia
Th1 : T helper 1
THF : Tetrahidrofolate
TIBC : Total Iron binding capacity
TILs : tumor infiltrating lymphocytes
TKD : Tyrosine Kinase Domain
TLR : Toll like receptor
t-MDS/AML : therapy-related MDS/AML
TNFα : Tumor necrosis factor alpha
TNR : tumor necrotic reseptor
TP53 : tumor protein p53
TRADD : TNF receptor associated death domain
T-reg : T – regulatory cells
WHO : world Health Organization
WT1 : wilm’s tumor

1
BAB 1
PENDAHULUAN
1.1 Latar belakang
Myelodysplastic Syndrome (MDS) adalah sekelompok kelainan
haematopoeitic stem cells (HSC) yang ditunjukkan dengan kegagalan sumsum
tulang, dikorelasikan dengan abnormalitas kuantitatif dan kualitatif sel pada darah
perifer (Hoffbrand & Moss, 2012).Pada MDS terjadi hematopoisis yang semula
mempunyai organisasi teratur (apoptosis mengeliminasi sel ganas) menjadi
displastik dan tidak efektif (anti apoptosis) pada sumsum tulang tampak sel muda
(blast) sedikit meningkat, selanjutnya dapat terjadi transformasi MDS menjadi
Acute Myeloid Leukemia (AML).Hal tersebut mungkin disebabkan kombinasi dari
faktor genetik, epigenetik dan sinyal reseptor yang abnormal serta faktor
lingkungan mikro (gambar 2.1; 2.2) (Ceesay,et al.,2012, Gilliland&
Gribben,2012).
Kerusakan HSC premalignant klonal yang sitopenia refrakter,
hematopoisis yang tidak efektif, hematopoitik displastik, deferensiasi selmultiple
lineages yang terhambat akanmeningkatkan risiko berkembangnya menjadi AML
(Pirruccello et al., 2006). Konsekuensi dari sel darah yang mengalami regulasi
abnormal dan diferensiasi yang terhambat pada tingkat
progenitormembuktikanproliferasi tidak terkendali sistem prekursor hematopoisis.
Keseimbangan homeostatik terganggu karena proliferasi yang meningkat, dan
apoptosis mulai meningkat pada stadium awal proses penyakit. Penderita yang
mempunyai risiko tinggi menjadi MDS, terjadi proliferasi meningkat sedikit demi
sedikit dan juga terjadi hambatan pada apoptosis (anti apoptosis) (Tefferi &
Vardman, 2009).

2
Data penelitian pendahuluan yang diambil peneliti di bagian Patologi
Klinik RSUD dr Soetomo atau Fakultas Kedokteran Universitas Airlangga
Surabaya, Jawa Timur periode tanggal 2 Januari sampai dengan 30 Oktober
tahun 2014 didapatkan jumlah penderita leukemia 228 orang; 181 orang masuk
sebagai subyek penelitian umur 14 – 77 tahun. Hasil penelitian berdasarkan
morfologi sel hapusan darah tepi (HDT) dengan klasifikasi FAB didapatkan
sebagai berikut: AML 70 orang (38,66%), Myelodisplastic Syndrome (MDS) 9
orang (4,98%). Diagnosis AML risiko pada perempuan 40 orang (57%) lebih
tinggi dari pada laki-laki 30 orang (43%).
Data penelitian berdasarkan usia sebagai berikut penderita dengan AML:
risiko tertinggi AML pada usia 46 – 61 tahun sebanyak 70 orang (37,14%).
Penderita dengan MDS pada usia 14 – 29 tahun mempunyai risiko tertinggi
dibandingkan dengan kelompok usia lain sebanyak 4 orang (44,4%) dan pada
usia tua antara 62 – 77 tahun sebanyak 1 orang (11,1%).
MDS adalah sindroma kelainan hematologi yang didominasi orang tua
antara 70 tahun dan diprediksi 1 orang menderita MDS dari 500 populasi umur
diatas 60 tahun diduga karena terpapar radiasi dan atau kemoterapi. Menurut
Besa, 2011 bahwa insiden MDS meningkat sesuai umur yaitu 20,8 – 36,3 per
100.000 populasi umur diatas 70 tahun, jarang terjadi pada usia dibawah 50
tahun (Tefferi & Vardman, 2009; Natelson & Pyatt, 2013;Besa, 2011). MDS
jarang terjadi pada anak, pria mempunyai risiko MDS lebih tinggi dari pada
wanita (Ceesayet al., 2012; Chapoval, 2010). MDS primer diperkirakan 85% dari
seluruh MDS, 45 – 50% terjadi abnormalitas sitogenetik misal del(5q), del(7q),
del(20q) dan trisomy 8. MDS sekunder diperkirakan 7 – 12% dari seluruh MDS,
dan terdapat lebih dari 80% dikorelasikan dengan kemoterapi dan radiasi.
Abnormalitas kromosom lebih dari 90% termasuk abnormalitas kromosom 5 dan

3
7. Lingkungan kerja (paparan kimia: Benzene) menyebabkan penyakit, insiden
kurang 1% dari seluruh MDS (Natelson& Pyatt, 2013;Rahim et al., 2015).
Van Den Berghemenyatakan bahwa Delesi kromosom 5 del(5q) sebagai
abnormalitas sitogenetikmerupakan bagian dan mewakili penentuan kerusakan
molekuler yang mendasari pathogenesis MDS.Myelodysplastic Syndrome (MDS)
yang mempunyai prevalensi 15% dari penderita MDS mempunyai semua kasus
dengan gambaran klinik tersebut diatas. Dalam perbandingan dengan subtipe
MDS yang lain, dapat dikarakterisasiadanya del(5q), hapusan sumsum tulang <
5%, insiden perempuan lebih besar dan prognosis baik karena mempunyai risiko
yang rendah untuk transformasi menjadi leukemia (Berghe et.al., 1974;
Mohamedali & Mufti, 2008). Klasifikasi WHO pada MDS mengidentifikasi
sindrom-5q sebagai kesatuan klinis yang berbeda yang memiliki gambaran
klinik, dan laboratorium klinis (Mohamedali & Mufti, 2008).
Prevalensi diagnosis MDS yang berkembang menjadi Acute Myeloid
Leukemia (AML) (MDS-related AML) sekitar 20 - 30% (Walter et al., 2012). MDS-
related AML membutuhkan waktu antara 5-6 tahun, diikuti paparan lingkungan
makro dan mikro sampai terjadi leukemogenesis, faktor risiko tergantung
besarnya paparan. Pada saat terjadi MDS-related AML antara 12 – 130 bulan,
insiden tersebut dihubungkan dengan mutasi gen dan kromosom, pemeriksaan
aspirasi sumsum tulang pada kasus AML dijumpai sedikit fase displastik (Ceesay
et al., 2012).
AML (Acute Myeloid Leukemia) adalah penyakit heterogen yang terdiri
dari berbagai macam gambaran klinik dan kelainan genetik, termasuk mutasi
sitogenetik, mutasi gen dan perubahan ekspresi gen (Owen & Fitzgibbon, 2012).
Pada tahun 2006, American Cancer Society memprediksi bahwa 11.930
pria dan wanita (6350 pria dan 5580 wanita) di Amerika Serikat telah ditetapkan
dengan diagnosis AML (Deschler & Lübbert, 2006). Pada kelompok median

4
umur 67 tahun, insiden lebih tinggi pada pria dibandingkan wanita yaitu 4,3 :
2,9.Insiden AML meningkat sesuai umur yaitu 1,7 kali pada populasi umur kurang
dari 65 tahun, dan 15,6 kali pada populasi yang sama umur lebih dari 65 tahun
(Wetzler et al., 2012; Deschler and Lübbert, 2006). Menurut penelitian Aulyaet
al., 2014 insiden Leukemia Akut di Indonesia diprediksi sekitar 3,4 kasus setiap
100.000 populasi per tahun.
AML penyakit yang kompleks dan bermacam-macam dasar
patomekanisme: genetik, genotip molekuler yang mana belum dapat dikenali
secara klinis dan morfologi fenotifnya. Patogenesis AML melibatkan mutasi dua
tipe abnormalitas atau mutasi genetik yaitu mutasi faktor jalur transkripsi dan
transduksi sinyal. Tipe mutasi gen yang paling banyak pada AML adalah
mekanisme faktor jalur transkripsi, berperan sebagai regulator utama pada
deferensiasi berbagai jenis perkembangan sel hematopoitik. Konsekuensi
langsung dari mutasi tersebut adalah menghambat proses deferensiasi sel
mieloid. Tipe mutasi gen kedua jalur transduksi sinyal, proliferasi meningkat dan
pertumbuhan sel yang berlebihan (Hoffmanet al., 2013). Mutasi faktor jalur
transduksi sinyal melibatkan mutasi SCFR (Stem cell factor reseptor) atau
tyrosine-protein kinase (Kit) dan gen fms-related tyrosine kinase 3 (FLT3), RAS
(rat sarcoma) dalam jalur transduksi yang sama (Hoffman et al., 2013;
Takahashi, 2011). Dengan adanya point mutation NRAS akan resisten terhadap
GAP (GTPase-activating proteins) sehingga hidrolisis GTP terhambat, sehingga
terjadi akumulasi RAS dalam bentuk aktif. Jumlah protein RAS aktif dapat
meningkatkan kemungkinan transformasi, point mutation pada RAS banyak
ditemukan pada berbagai tumor termasuk kanker darah (Yusup, 2008).
Mutasi faktor transkripsi melibatkan translokasi core binding factor (CBF)
yang berikatan dengan RARαakan menyusun gen MLL(mixed lineage leukemia).
Mutasi yang melibatkan fusi CBF-α dan CBF-β, pasien dengan CBF-AML

5
terdeteksi kurang lebih 15% pada kasus AML. CBF adalah faktor transkripsi
heterodimerik, terdiri dari DNA yang mengikat α-subunit, disandi oleh satu dari
tiga anggota keluarga RUNX (RUNX1 atau AML1), dan β-subunit disandi oleh
gen CBFβ. Ikatan AML1 dan CBFβ dengan gen lain menghasilkan protein
chimeric mengubah kompleks CBF dan menghambat aktivasi transkripsi
(Andrew, et al.,2018).
Pada kasus AML diidentifikasi adanya variasi dari translokasi kromosom
yang melibatkan AML1 maupun CBFβ. 1. Variasi kromosom CBF pada AML
berupa inv(16)t(16;16), pada CBFβ yang membuat fusi dengan gen MYH11. 2.
Translokasi kromosom t(8;21), yang dihubungkan dengan fusi AML1 dan eight-
twenty-one (ETO) (Hoffmanet al., 2013). Osato, 2004 menyatakan bahwa
translokasi menghasilkan gabungan protein yang berperan penting pada
leukemia yaitu menghambat deferensiasi dan proliferasi meningkat. 3. Gen AML1
dapat ditemukan pada turunan fusi yang lain, fusi yang melibatkan gen ecotropic
viral integration 1(EVI1), terjadi translokasi t(3;21). 4. Gen translocation ets
leukemia (TEL) pada AML dimana terjadi translokasi t(12;21). Pada kasus fusi
gen tersebut diatas diperkirakan 25 % dari seluruh kasus AML. Inaktivasi CBFα/β
akibat fusi AML-1/ETO menghasilkan gangguan hematopoisis yang dapat
berakhir dengan leukemogenesis.
Mutasi gen NPM1 termasuk mutasi faktor jalur transkripsi, akan
menimbulkan berbagai gambaran biologis, klinis dan membentuk fusi dengan
gen yang lain dalam leukemogenesis (Takahashi, 2011). Beberapa penelitian
menyatakan bahwa genotip mutan NPM1 tanpa mutasi FLT3-ITD menunjukkan
prognosis lebih baik dibandingkan genotip mutan NPM1 disertai mutasi FLT3-ITD
(terdapat 60% mutasi NPM1 dengan FLT3-ITD) (Hoffmanet al., 2013; Dinata
et.al., 2019).

6
Kasus Leukemia sampai saat ini belum diketahui penyebabnya dengan
pasti. Banyak penelitian telah dilakukan untuk mengetahui faktor risiko. Berbagai
faktor risiko yaitu penggunaan pestisida, medan listrik, bahan kimia (Benzene),
virus, abnormalitas sitogenetik (contoh: Down syndrome), usia ibu yang relatif tua
saat melahirkan, ibu yang merokok saat hamil, konsumsi alkohol saat hamil,
medan magnet, pekerjaan orang tua, radiasi prenatal dan postnatal
(Simanjoranget al., 2010; Snyder, 2012).
Pada penelitian ini kami menggunakan klasifikasi diagnosis MDS dan
AML menurut klasifikasi WHO 2008 dan FAB(French-American-
british).Pemeriksaan MDS dan AML, morfologi hapusan darah tepi, aspirasi
sumsum tulang yang mana MDS: myeloblast kurang dari 20%, AML: myeloblast
lebih dari 20%. Pemeriksaan penunjang lain yaitu darah lengkap,
immunophenotyping(Pirruccello et.al., 2006), abnormalitas sitogenetik
menggunakan CISH (chromogen In Situ Hybridryzation) dan molekuler
menggunakan PCR (Polimerase Chain Reaction) (Hoffmanet al., 2013;Do̎hner et
al.,2010). Penatalaksana di klinik MDS mempunyai pengobatan yang berbeda
dengan AML, oleh karena itu diagnosis yang tepat sangat diperlukan, khususnya
MDS atau AML (Owen & Fitzgibbon, 2012; Gililand& Gribben, 2012; Takahashi,
2011).
Penjelasan tersebut diatas, maka peneliti mempunyai pendapat bahwa
abnomalitas molekuler dan sitogenetik progresivitasberpengaruh terhadap MDS-
related AML yang melibatkan polimorfisme NRAS, RUNX1, NPM1, FLT3 dan
delesi kromosom 5 del(5q) atau kromosom 5 normal. Delesi kromosom 5 del(5q)
dengan mutasi NRAS, FLT3, NPM1 lebih dari satu gen (60%) dapat
meningkatkan kemungkinan transformasi menjadi AML. Keluarga RUNX
(RUNX1) mempunyai risiko 15% menghambat deferensiasi (differentiation arrest)
dan NRAS, FLT3 meningkatkan proliferasi (proliferation drive).

7
1.2 Rumusan masalah
Apakah polimorfisme RUNX1, NPM1, FLT3, N-RAS dan delesi kromosom
5 del(5q) dapat menjadi prediktor terhadap perubahanMyelodisplastic Syndrome
menjadi Acute Myeloid Leukemia(MDS-related AML) pada penderitadi Surabaya.
1.3 Tujuan penelitian
1.3.1 Tujuan umum
Membuktikan polimorfisme NRAS, RUNX1, NPM1, FLT3, dan delesi
kromosom 5 del(5q) sebagai prediktor perubahan Myelodisplastic Syndrome
menjadi Acute Myeloid Leukemia (MDS-related AML) pada penderita di
Surabaya.
1.3.2 Tujuan khusus
1. Menentukan polimorfisme NRAS sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-related
AML).
2. Menentukan polimorfisme RUNX1 sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-related
AML).
3. Menentukan polimorfisme NPM1 sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-related
AML).
4. Menentukan polimorfisme FLT3 sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia(MDS-related
AML).

8
5. Menentukan polimorfisme delesi kromosom 5 del(5q) sebagai prediktor
perubahan Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia
(MDS-related AML).
6. Menentukan kromosom 5q normal sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-related
AML).
1.4 Manfaat penelitian
1.4.1 Manfaat Keilmuan
Salah satu sumber informasi polimorfisme NRAS, RUNX1, NPM1, FLT3
dan delesi kromosom 5 del(5q) sebagai prediktor perubahan Myelodisplastic
Syndrome (MDS) menjadi Acute Myeloid Leukemia (AML) pada penderita di
Surabaya, serta memperkaya informasi ilmu pengetahuan dan dapat
dipergunakan untuk acuan karya ilmiah selanjutnya
1.4.2 Manfaat Praktis
Manfaatdiagnosis dan klinis, polimorfisme NRAS, RUNX1, NPM1, FLT3
dan delesi kromosom 5 del(5q) atau kromosom 5 normal dapat dipergunakan
untuk diagnosis penunjang MDS dalam menentukan progresivitas menjadi AML
atau MDS-related AML. Diagnosis penunjang tersebut diatas dapat dipergunakan
untuk penatalaksana terapi yang tepat.

10
BAB 2
TINJAUAN PUSTAKA
2.1 Acute Myeloid Leukemia (AML)
AML adalah penyakit keganasan yang menghambat deferensiasi dan
proliferasi secara tidak terkendali (pada AML tubuh memproduksi myeloblast
yang berlebihan) (Fitriani, 2009). Peneliti lain mendifinisikan AML yaitu penyakit
heterogen yang terdiri dari berbagai macam gambaran klinik dan kelainan
genetik, termasuk mutasi sitogenetik, mutasi gen dan perubahan ekspresi gen
(Owen& Fitzgibbon, 2012).
Menurut WHO (World Health organization) insiden leukemia terjadi
hampir di seluruh dunia, kasus kanker yang tercatat sekitar 250 kasus baru per
tahun dengan Case Fatality Rate (CFR) 76%. Kasus baru kanker sebanyak
100.000 terdapat penderita AML dijumpai sekitar 2,5%, dan ALL dijumpai sekitar
1,3%. Di Indonesia insiden Leukemia Akut diprediksi sekitar 3,4 kasus setiap
100.000 populasi per tahun (Aulya et al., 2014 ). Menurut data yang diambil oleh
peneliti di bagian Patologi Klinik RSUD dr Soetomo atau Fakultas Kedokteran
Universitas Airlangga Surabaya, Jawa Timur periode tanggal 2 Januari sampai
dengan 30 Oktober tahun 2014 didapatkan jumlah penderita 181 orang, dengan
rentang umur 14 – 77 tahun. Hasil penelitian berdasarkan morfologi sel hapusan
darah tepi (HDT) yaitu: AML 70 orang (38,66%), Myelodisplastic Syndrome
(MDS) 9 orang (4,98%). Perempuan dengan diagnosis Leukemia mempunyai
risiko lebih tinggi dari pada laki-laki yaitu perempuan 107 orang (59,12%),
sedangkan laki-laki 74 orang (40,88%). Diagnosis MDS perempuan mempunyai
risiko lebih tinggi dari pada laki-laki yaitu perempuan 6 orang (66,67%),
sedangkan pada lak-laki-laki 3 orang (33,33%), demikian juga diagnosis AML

11
risiko perempuan lebih tinggi dari pada laki-laki yaitu perempuan 40 orang (40%),
dan laki-laki 30 orang (30%) (accepted, Journal: Drug Invention today).
Data penelitian berdasarkan usia sebagai berikut diagnosis Leukemia
risiko tertinggi pada usia 30 – 45 tahun (37,02%) dari 181 orang. Penderita
dengan diagnosis AML: risiko tertinggi AML pada usia 46 – 61 tahun (37,14%)
dari 70 orang. Data AML dengan klasifikasi FAB didapatkan AML-M5 (40%),
AML-M3 (25,72%), AMl-M4 (15,71%). Penderita dengan diagnosis MDS Usia 14
– 29 tahun yang mempunyai risiko tinggi sebanyak 4 orang (44,4%), pada usia
tua 62 – 77 tahun 1 orang (11,1%) (accepted,journal: Drug InventionToday).
Etiologi leukemia sampai saat ini belum diketahui dengan pasti, beberapa
penelitian telah dilakukan untuk mengetahui faktor risiko. Berbagai faktor risiko
yaitu penggunaan pestisida, medan listrik, bahan kimia (Benzene), obat
(chloramphenicol) virus, abnormalitas sitogenetik (contoh: Down syndrome), usia
ibu yang relatif tua saat melahirkan, ibu yang merokok saat hamil, konsumsi
alkohol saat hamil, medan magnet, pekerjaan orang tua, radiasi prenatal dan
postnatal (Simanjorang et al.,2010, Snyder, 2012, Wetzler et al., 2012).
Klasifikasi AML menurut WHO 2008 dan FAB, klasifikasi WHO termasuk
gambaran klinik, abnormalitas sitogenetik dan molekuler, ditambahkan juga
morfologi sel. Klasifikasi FAB (French-American-British classification)
menggunakan deskripsi morfologi blast misalnya seri myeloid, diagnosis AML
pada pemeriksaan aspirasi sumsum tulang ditemukan myeloblast lebih dari 20%,
pemeriksaan penunjang lain. Klasifikasi FAB digunakan untuk diagnosis AML
dengan diskripsi M0 – M7, (lampiran 7) (Wetzler et al., 2012;Killick et al.,
2014).Menurut Weinberg and Arber, 2010, kalsifikasi WHO 2008 memperbaiki
tentang kategori AML dengan myelodysplasiarelated changes (AML-MRC) yaitu
myeloblast darah tepi atau sumsum tulang kurang 20%, mempunyai riwayat
penyakit MDS, abnormalitas sitogenetik yang berhubungan dengan MDS,

12
morfologi dysplasia (multilineage),50% sel mengalami kriteria dysplasia, AML-
MRC adalah leukemia yang agresif dan prognosis buruk (Weinberg et al.,2010;
Do̎hner et al., 2010).
2.1.1 Patogenesis AML
AML penyakit yang kompleks dan bermacam-macam dasar
patomekanisme: genetik, genotip molekuler sehingga belum dapat dikenali
secara klinis dan morfologi fenotifnya. Gagasan patogenesis AML dapat diterima
dengan melibatkan mutasi dua tipe abnormalitas atau mutasi genetik yaitu mutasi
faktor jalur transduksi sinyal dan transkripsi. Insiden mutasi gen yang terbanyak
pada AML adalah mekanisme faktor jalur transkripsi, berperan sebagai regulator
utama pada deferensiasi berbagai jenis perkembangan sel hematopoitik.
Konsekuensi langsung dari mutasi tersebut adalah menghambat proses
deferensiasi sel mieloid. Tipe mutasi berikutnya menyebabkan proliferasi dan
pertumbuhan sel yang berlebihan (Hoffman et al., 2013). Contoh mutasi faktor
transkripsi melibatkan translokasi core binding factor (CBF) yang berikatan
dengan RARαakan menyusun gen MLL(mixed lineage leukemia). Contoh yang
lain yaitu mutasi faktor jalur transduksi sinyal melibatkan mutasi SCFR (Stem cell
factor reseptor) atau tyrosine-protein kinase (KIT) dan gen fms-related tyrosine
kinase 3 (FLT3) dalam jalur transduksi yang sama (tabel 2.2.). Pada penderita
AML yang diperiksa dengan PCR didapatkan abnormalitas yang menunjukkan
adanya mutasi beberapa gen (FLT3, N-RAS) yang menyebabkan proliferasi dan
pertumbuhan sel yang berlebihan (Hoffman et al., 2013; Takahashi, 2011).
Dua grup terdiri dari faktor transkripsi diamati aktivitas transkripsi yang
menghasilkan protein yang berperan penting dalam menentukan progenitor
hematopoitik. Grup pertama terdiri dari regulator utama faktor transkripsi, seperti
AML1 terlibat dalam perkembangan seluruh haematopoitic lineages. Grup kedua

13
terdiri dari faktor transkripsi yang menghambat secara spesifik perkembangan
setiap haematopoietic lineages. Contoh: faktor transkripsi tipe GATA1, yang
mendukung perkembangan progenitor hematopoitik menghasilkan erythroid
lineages atau C/EBPα, yang mana mendukung diferensiasi granulocytic (AML)
(Takahashi, 2011; Hoffman et al.,2013).
Mutasi yang melibatkan fusi CBF-α dan CBF-β, pasien dengan CBF-
AMLterdeteksi kurang lebih 15% dari kasus AML. Pada usia pertengahan
mempunyai signifikan yang lebih rendah dan prognosis lebih baik dibandingkan
penderita dengan kariotipe yang normal atau abnormalitas kromosom. Hasil
pengobatan rata-rata CR (complete remission) lebih tinggi dan insiden relapse
lebih rendah (Boissel et al., 2006). CBF adalah faktor transkripsi heterodimerik,
terdiri dari DNA yang mengikat α-subunit, disandi oleh satu dari tiga anggota
keluarga gen RUNX (RUNX1 atau AML1), dan β-subunit disandi oleh gen CBFβ.
Ikatan gen AML1 dan CBFβ dengan gen lain menghasilkan protein chimeric
mengubah kompleks CBF dan menghambat aktivasi transkripsi.
Pada kasus AML diidentifikasi adanya variasi dari translokasi kromosom
yang melibatkan AML1 maupun CBFβ. Variasi kromosom CBF pada AML berupa
inv(16)t(16;16), pada CBFβ terjadi fusi dengan gen smooth muscles myosin
heavy chain (SMMHC), disamping itu terjadi translokasi kromosom t(8;21), yang
dihubungkan dengan fusi gen AML1 dan gen eight-twenty-one (ETO) (Hoffman et
al., 2013).
Menurut Ley et al., 2013 bahwa jumlah penderita AML de novo sebesar 7
- 12% yang menunjukkan adanya translokasi t(8;21), fusi gen RUNX1 dengan
RUNX1T1 (eight-twenty-one atau ETO). Variasi kromosom inv(16)t(16;16) dan
fusi gen CBFβ dengan SMMHC ditemukan pada 10 – 12 % kasus (gambar 2.1.).
Osato, 2004 menyatakan bahwa translokasi menghasilkan gabungan protein
yang berperan penting pada leukemia yang menghambat deferensiasi, proliferasi

14
meningkat. Gen AML1 dapat ditemukan pada turunan fusi yang lain. Keadaan
tersebut dapat dideteksi pada AML, seperti fusi yang melibatkan gen ecotropic
viral integration 1(EVI1), atau gen translocation ets leukemia (TEL) pada AML
dimana terjadi translokasi secara berturutan t(3;21) dan t(12;21). AML melibatkan
CBF yang mengalami translokasi menjadi AML1-ETO, bertindak sebagai
dominant-negative inhibitorAML1, seperti ditunjukkan pada transcriptional
activation assays (gambar 2.1.)(Owen & Fitzgibbon, 2012). Fusi CBF sering
terjadi pada penderita AML dan umumnya terjadi keseimbangan translokasi
resiprok (reciprocal translocation).Pada seluruh kasus AML terdapat kasus
tersebut diatas diperkirakan 25%. Ceesay et al., 2012; Juniarka, 2010
menyatakan AML-1-related translocations atau haploinsufficiency,tidak langsung
mengakibatkan leukemia pada binatang coba. Keadaan ini menunjukkan bahwa
diperlukan perubahan genetik lain untuk menjadi fenotif leukemia.
Kresno, 2012 menyatakan tentang fusi protein, bahwa translokasi tunggal
tidak dapat menyebabkan terjadinya fenotif leukemia. Fusi gen RUNX1-
RUNX1T1 mengekspresikan progenitor hematopoisis pada orang dewasa tidak
menginduksi AML, tetapi dapat menyebabkan myeloproliferative phenotype.
Pada penelitian binatang coba yang diberi perlakuan dengan bahan kimia
sebagai mutagen akan memicu terjadinya AML (Ceesay et al., 2012, Agrawalet
al., 2007).

15
Gambar 2.1 Hematopoeisis, deferensiasi sel dan faktor transkripsi(Dikutip: Kresno, 2012).
Keterangan: Mutasi gen AML1 disertai fusi dengan gen lain (ETO, Evi-1, TEL) akan menghambat aktivitas deferensiasi (deferensiasi “arrest”). Bentuk keganasan darah, faktor jalur mutasi: transkripsi selanjutnya AML, CML, meningkatkan hematopoiesis proliferasi.
Menurut penelitian binatang coba tersebut diatas, Fusi gen RUNX1-
RUNX1T1, dan paparan bahan kimia akan memicu terjadinya AML. Mutasi gen
tertentu (RUNX1) pada pasien AML, MDS maupun orang normal mempunyai
prosentase yang berbeda. Hal tersebut didukung oleh hasil penelitian Osato,
2004, yang menyatakan bahwa frekwensi mutasi pada gen RUNX1/AML1
didapatkan berbagai variasi pada penderita kanker maupun orang normal.
Pertama AML(subtipe M0), 185 kasus, mutasi 39 kasus, prosentase mutasi 21%.
AML dengan bermacam subtipe menurut kriteria FAB didapatkan total 619 kasus,
mutasi 53 kasus, prosentase mutasi 8,6%; kedua MDS (subtipe RAEB/RAEB-t),
138 kasus, mutasi 22 kasus, prosentase mutasi 15,9%; MDS (subtipe MDS
leukemia), 53 kasus, mutasi 8 kasus, prosentase mutasi 15,0%; MDS dengan
bermacam subtipe menurut FAB/WHO, total 362 kasus, mutasi 33 kasus,
prosentase mutasi 9,1%; ketiga tipe Orang sehat (healthy) terdiri dari orang
Jepang, Inggris, Prancis, Belanda, total 338 kasus, mutasi 9 kasus, prosentase
2,6% (Osato, 2004; Rocquain et al., 2010).

16
Therapy-related (t)-MDS/AML ditemukan pada sporadik atau kasus yang
didapat antara 10 – 20%. Kasus (t)-MDS/AML dapat dibagi menjadi dua grup
yaitu alkylating agents atau radiation related,topoisomerase II inhibitor related
dan Grup MDS/AML (merupakan grup kasus keturunan atau Familial MDS/AML)
akan berkembang dalam 5 - 6 tahun tergantung paparan menjadi proses
leukemogenik dan faktor risiko yang berhubungan dengan dosis paparan.
Beberapa grup membutuhkan waktu berkembang lebih pendek yaitu 12 – 130
bulan, kejadian ini dihubungkan dengan keseimbangan antara translokasi
kromosom dan Frank AML (tanda klinik misalnya hematuria) yang mendahului
fase displastik.Mutasi t-MDS/AML ditunjukkan dalam tiga kelas yaitu : kelas 1,
faktor mutasi mengaktifkan jalur transduksi sinyal (FLT3, RAS); kelas 2 faktor
mutasi yang melibatkan transkripsi (RUNX1, NPM1), dan kelas 3 mutasi yang
melibatkan tumor-suppressor gene p53. Data statistik ditemukan bahwa
frekwensi mutasi satu gen (point mutation) 20 – 30% dari semua kasus t-
MDS/AML (gambar 2.2.) (Ceesayet al., 2012).
MDS berkembang pada keadaan lebih dari satu mutasi abnormalitas
sitogenetik contoh -7/del(7q), -5/del(5q), +8 (gambar 2.2, tabel 2.1). Beberapa
penelitian yang menyatakan bahwa myelodysplastic stem cells yang mengalami
perubahan genetik dan epigenetik didapatkan pada sepertiga kasus yang mana
akan berkembang menjadi AML (MDS-related AML) (Gambar 2.2) (Walteret al.,
2012).
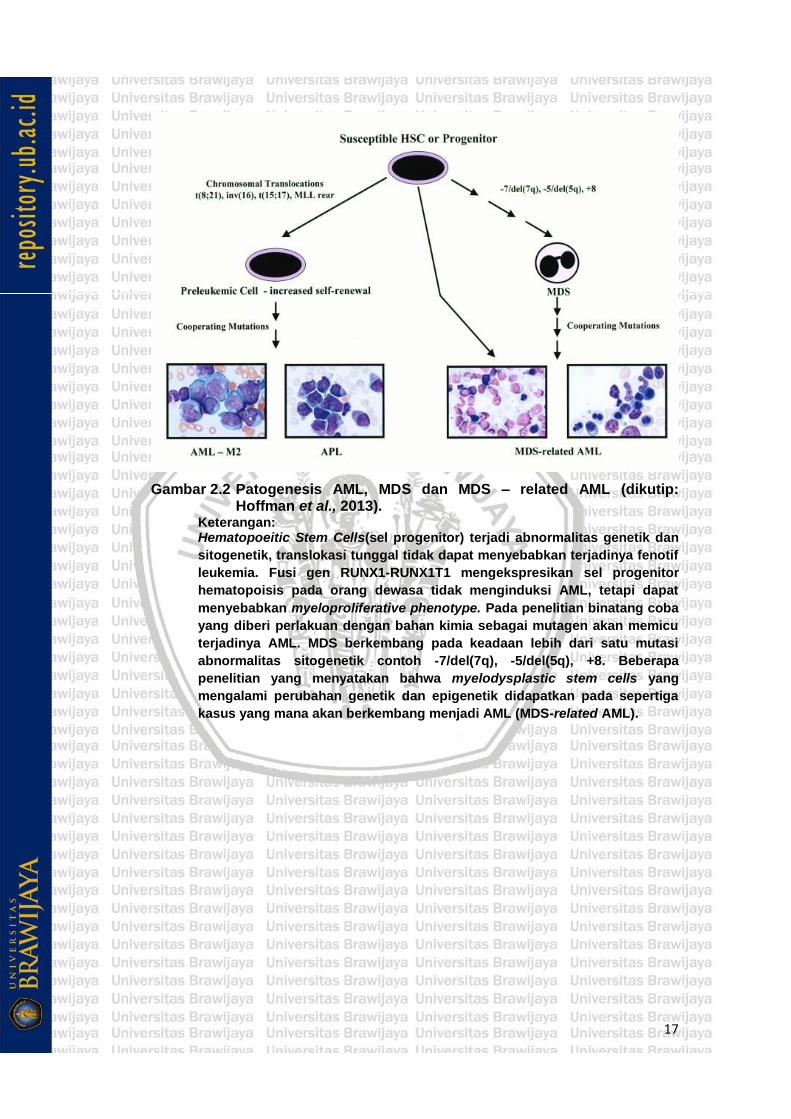
17
Gambar 2.2 Patogenesis AML, MDS dan MDS – related AML (dikutip: Hoffman et al., 2013).
Keterangan: Hematopoeitic Stem Cells(sel progenitor) terjadi abnormalitas genetik dan
sitogenetik, translokasi tunggal tidak dapat menyebabkan terjadinya fenotif
leukemia. Fusi gen RUNX1-RUNX1T1 mengekspresikan sel progenitor
hematopoisis pada orang dewasa tidak menginduksi AML, tetapi dapat
menyebabkan myeloproliferative phenotype. Pada penelitian binatang coba
yang diberi perlakuan dengan bahan kimia sebagai mutagen akan memicu
terjadinya AML. MDS berkembang pada keadaan lebih dari satu mutasi
abnormalitas sitogenetik contoh -7/del(7q), -5/del(5q), +8. Beberapa
penelitian yang menyatakan bahwa myelodysplastic stem cells yang
mengalami perubahan genetik dan epigenetik didapatkan pada sepertiga
kasus yang mana akan berkembang menjadi AML (MDS-related AML).

18
Tabel 2.1 Risiko mutasi kromosom berkembang menjadi AML
(dikutip dari:Takahashi, 2011)
TRANSLOKASI/
INVERSI
GEN MORFOLOGI
INSIDEN*
t(8;21)(q22;q22) RUNX1;RUNX1T1 M2 with Auer rods 6%
inv(16)(p13q22) or
t(16;16)(p13;q22)
CBFB;MYH11 M4E0 7%
t(15;17)(q22;q11-21) PML;RARA M3/M3v 7%
t(9;11)(p22;q23) MLL;AF9 M5 2%
t(6;11)(q27;q23) MLL;AF6 M4 and M5 ~1%
inv(3)(q21q26) or
t(3;3)(q21;q26)
EVI;RPN1 M1, M4, M6, M7? ~1%
t(6;9)(p23;qQ34) DEK;NUP214 M2, M4 ~1%
ChromosalImbalances+8 .. M2, M4 and M5 9%
-7/7q- .. No FAB preference 7%
-5/5q- .. No FAB preference 7%
-17/17p- TP53 No FAB preference 5%
-20/20q- .. No FAB preference 3%
9q- .. No FAB preference 3%
+22 .. M4, M4E0 3%
+21 .. No FAB preference 2%
+13 .. M0, M1 2%
+11 MLL1+ M1,M2 2%
Complex karyotype* 10%
Normal karyotype 44%
Keterangan : AML (Acute Myeloid Leukemia). *Ditentukan diantara 1,311 pasien dengan diikuti oleh studi 8461 kanker dan
Leukemia Group B. + Turunan dua sebagiandari MLL1 ++Tiga atau lebih penyimpangan kromosom dalam ketiadaan t(8;21),
inv(16)t(16;16), t(15;17), or t(9;11). Sumber: diadaptasi dari Dohner K, Mrozek K, Dohner H, Bloomfield CD. Leukemia Diagnosis and Classifications. Amsterdam: Elsevier Inc., (in press).
19
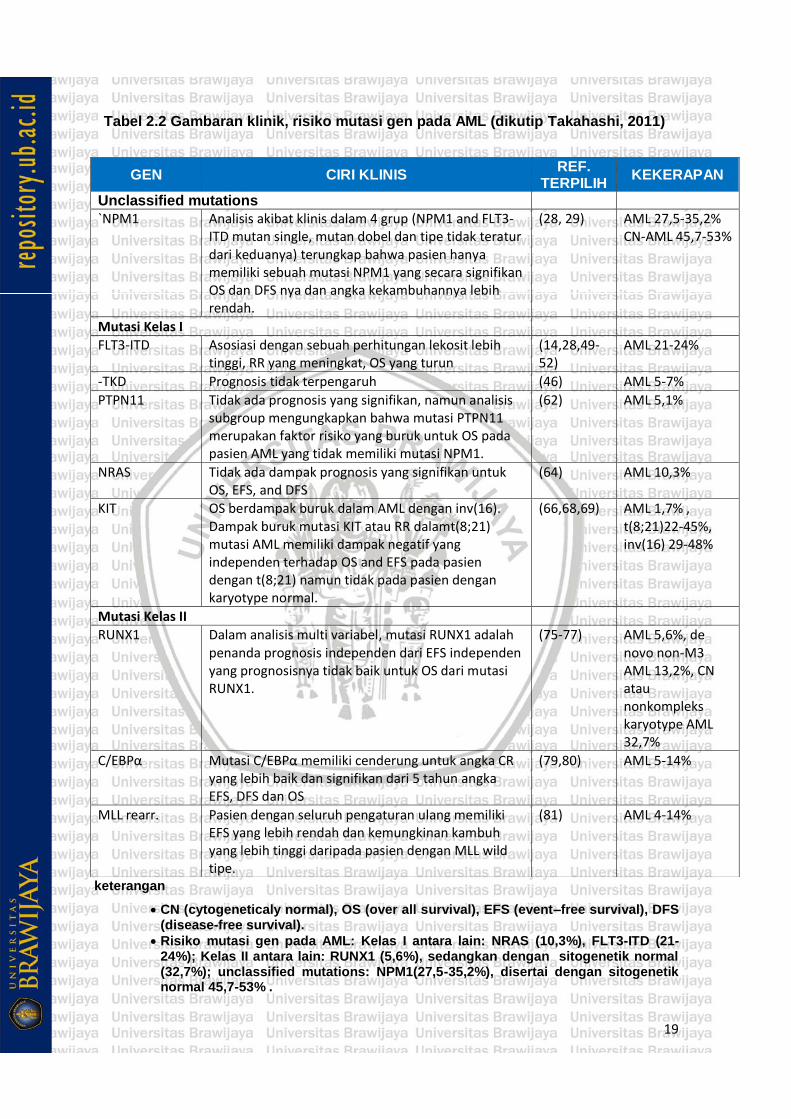
Tabel 2.2 Gambaran klinik, risiko mutasi gen pada AML (dikutip Takahashi, 2011)
keterangan
• CN (cytogeneticaly normal), OS (over all survival), EFS (event–free survival), DFS (disease-free survival).
• Risiko mutasi gen pada AML: Kelas I antara lain: NRAS (10,3%), FLT3-ITD (21-24%); Kelas II antara lain: RUNX1 (5,6%), sedangkan dengan sitogenetik normal (32,7%); unclassified mutations: NPM1(27,5-35,2%), disertai dengan sitogenetik normal 45,7-53% .
GEN CIRI KLINIS REF.
TERPILIH KEKERAPAN
Unclassified mutations
`NPM1 Analisis akibat klinis dalam 4 grup (NPM1 and FLT3-ITD mutan single, mutan dobel dan tipe tidak teratur dari keduanya) terungkap bahwa pasien hanya memiliki sebuah mutasi NPM1 yang secara signifikan OS dan DFS nya dan angka kekambuhannya lebih rendah.
(28, 29) AML 27,5-35,2% CN-AML 45,7-53%
Mutasi Kelas I
FLT3-ITD Asosiasi dengan sebuah perhitungan lekosit lebih tinggi, RR yang meningkat, OS yang turun
(14,28,49-52)
AML 21-24%
-TKD Prognosis tidak terpengaruh (46) AML 5-7%
PTPN11 Tidak ada prognosis yang signifikan, namun analisis subgroup mengungkapkan bahwa mutasi PTPN11 merupakan faktor risiko yang buruk untuk OS pada pasien AML yang tidak memiliki mutasi NPM1.
(62) AML 5,1%
NRAS Tidak ada dampak prognosis yang signifikan untuk OS, EFS, and DFS
(64) AML 10,3%
KIT OS berdampak buruk dalam AML dengan inv(16). Dampak buruk mutasi KIT atau RR dalamt(8;21) mutasi AML memiliki dampak negatif yang independen terhadap OS and EFS pada pasien dengan t(8;21) namun tidak pada pasien dengan karyotype normal.
(66,68,69) AML 1,7% , t(8;21)22-45%, inv(16) 29-48%
Mutasi Kelas II
RUNX1 Dalam analisis multi variabel, mutasi RUNX1 adalah penanda prognosis independen dari EFS independen yang prognosisnya tidak baik untuk OS dari mutasi RUNX1.
(75-77) AML 5,6%, de novo non-M3 AML 13,2%, CN atau nonkompleks karyotype AML 32,7%
C/EBPα Mutasi C/EBPα memiliki cenderung untuk angka CR yang lebih baik dan signifikan dari 5 tahun angka EFS, DFS dan OS
(79,80) AML 5-14%
MLL rearr. Pasien dengan seluruh pengaturan ulang memiliki EFS yang lebih rendah dan kemungkinan kambuh yang lebih tinggi daripada pasien dengan MLL wild tipe.
(81) AML 4-14%

20
2.2 Myelodysplastic syndrome (MDS)
Definisi MDS adalah sekelompok kelainan haematopoeitic stem cells
(HSC)yang ditunjukkan dengan kegagalan sumsum tulang (displastik) yang
meningkat, dikorelasikan dengan abnormalitas kuantitatif dan kualitatif sel pada
darah perifer (Hoffbrand& Moss, 2012).
Insiden MDS dihubungkan dengan umur, penderita MDS mempunyai
tanda dan gejala yaitu umur antara 60 - 75 tahun, beberapa penderita lebih muda
dari 50 tahun, MDS jarang terjadi pada anak, pria mempunyai risiko MDS lebih
tinggi dari pada wanita (Ceesayet al., 2012; Chapoval, 2010; Sangle, 2012;
Kubasch & Platzbecker, 2018). MDS primer diperkirakan 85% dari seluruh MDS,
45 – 50% terjadi abnormalitas sitogenetik misal del(5q), del(7q), del(20q) dan
trisomy 8. MDS sekunder diperkirakan 7 – 12% dari seluruh MDS, dan terdapat
lebih dari 80% dihubungkan dengan kemoterapi dan radiasi. Abnormalitas
kromosom lebih dari 90% termasuk abnormalitas kromosom 5 dan 7. Lingkungan
kerja (paparan kimia: Benzene) menyebabkan penyakit, insiden kurang dari 1%
dari seluruh MDS (Natelson& Pyatt, 2013;Klepin et al., 2014).Etiologi MDS tidak
diketahui dengan pasti tetapi faktor abnormalitas sitogenetik, molekuler,
epigenetik dapat berkembang menjadi MDS. Abnormalitas kromosom: delesi
kromosom 5 del (5q) mempunyai prosentase berbeda antara kasus MDS dengan
kasus AML, demikian juga abnormalitas molekuler (RUNX1, NPM1, FLT3,
NRAS). Beberapa translokasi t(15;17), i(16), t(8;21) tidak didapatkan pada MDS,
keadaan ini dapat dipergunakan untuk membantu mengetahui perubahan MDS
menjadi AML (MDS-related AML) (Walteret al., 2012; Bejaret al., 2011;
Takahashi, 2011; Luzzatto& Kadardimitris, 2010; Alcindor& Bridges, 2001).
MDS sekunder karena pengobatan kemoterapi keganasan lain,
mempunyai risiko terjadi abnormalitas sitogenetik diperkirakan 80% (Ceesayet
al., 2010; Young, 2012). Prevalensi MDS yang berkembang menjadi Acute

21
Myeloid Leukemia (AML) (MDS-related AML) sekitar 20 - 30% (Walteret al.,
2012, Montalban-Bravo G. et al., 2014;Bejar et al., 2011).
Penelitian Walter et al., pada tahun 2012, menyatakan bahwa sampel
secondary-AML(sAML) 11 sampel terdiri dari gen yang mengalami mutasi
berulang, pemeriksaan hapusan sumsum tulang, mutasi gen PCR antara lain :
NPM1, RUNX1, TP53, WT1, abnormalitas kromosom antara lain del(5), del(20),
del(17). 7 sampel dalam proses MDS menjadi sAML, termasuk 4 kasus
mengalami proses yang progresivitas cepat untuk menjadi sAML yaitu < 6 bulan
(6,7% dari semua mutasi yang spesifik menjadi sAML) dan 3 kasus proses
progresivitas lambat yaitu > 20 bulan (37,8% dari mutasi yang spesifik menjadi
sAML). Pada MDS terjadi mutasi transition dan transversion pada sebanyak 7
sampel, sedangkan 2 sampel telah diberi terapi Decitabine selama 4-11 bulan,
setelah diagnosis MDS, sebelum terjadi proses sAML, kemudian Sampel
tersebut mengalami progresivitas menjadi sAML (mengalami transversion).
Peneliti tidak mengetahui hasilnya seandainya ke 2 sampel tidak diberikan terapi
Decitabine (Jabbour & Kantarjian. 2011).Progresivitas menjadi sAML ditandai
dengan meningkatnya myeloblast antara 7% - 66%, yang lain antara 13% - 43%,
tidak mengalami perubahan klon dan sedikit peningkatan mutasi titik (point
mutation) < 2% selama progresivitas MDS menjadi sAML. Terapi pada awal
mutasi adalah suatu strategi eliminasi sel ganas dan akan memperbaiki respon
kemoterpi konvensional pasien sAML. Peneltian ini sebagai biomarker dan
pengertian yang lebih baik tentang patogenesis MDS (Walter et al., 2012).
Klasifikasi MDS menurut WHO 2008 dan FAB (lampiran 7), klasifikasi
WHO termasuk gambaran klinik, abnormalitas sitogenetik dan molekuler,
ditambahkan juga morfologi sel. Klasifikasi FAB (French-American-British
classification) menggunakan deskripsi morfologi Anemia. Diagnosis klinik MDS
dilengkapi dengan pemeriksaan morfologi hapusan darah tepi, aspirasi sumsum

22
tulang, imunositokimia berupa displastik hematopoitik dan anemia, myeloblast
kurang dari 20%(Besa, 2011).Menurut Weinberg and Arber, kalsifikasi WHO
2008 diperbaiki termasuk kategori AML with myelodysplasia related changes
(AML-MRC) yaitu myeloblast darah tepi atau bonemarrow kurang 20%,
mempunyai riwayat penyakit MDS, abnormalitas sitogenetik yang berhubungan
dengan MDS, morfologi dysplasia (multilineage), 50% sel mengalami kriteria
dysplasia, AML-MRC adalah leukemia yang agresif dan prognosis buruk
(Weinberg & Arber,2010; Besa, 2011).
Gambar 2.3 Mekanisme karsinogenesis akibat faktor lingkungan dan
diet(Dikutip dari: kresno, 2012).
2.2.1 Patogenesis MDS
Pada MDS terjadi hematopoisis yang semula mempunyai organisasi yang
teratur dan efektif (contoh: apoptosis mengeliminasi sel ganas) menjadi displastik
dan tidak efektif (contoh: antiaptosis, pada sumsum tulang tampak sedikit
displastik), selanjutnya terjadi transformasi MDS menjadi AML (Gutierrez &
Romerro-Olivia, 2013). Keadaan tersebut mungkin merupakan kombinasi dari
faktor genetik, epigenetik dan sinyal reseptor yang abnormal serta faktor
lingkungan mikro (gambar 2.1; 2.2) (Ceesay et al., 2012, Gilliland& Griffin, 2012).

23
Gambar 2.4 Progresivitas MDS-related AML(Dikutip dari: Saunthararajahet
al., 2011).
HSC: Hematopoietic stem cells
Keterangan:
A. Hematopoisis normal: HSC memperbarui sel dengan sendirinya (self-renewal)
untuk lineage – committed daughter cells (sel progenitor). Sel progenitor
melakukanaktivitas fungsideferensiasi, proliferasi, maturitas sel normal
B. Pada awal abnormalitas mempengaruhi HSC sehingga terjadi evolusi menuju
ganas. Selanjutnya supresi epigenetik mengganggu peran sel progenitormaka
terjadi deferensiasi lambat. Pada awal sakit deferensiasi terhambat ringan,
precursor meningkat, terjadi maturitas sel menurundi sumsum tulang.
C. Supresi epigenetik progresif sehingga jumlah maturitas sel menurunprogrsif
di sumsum tulang.Selanjutnya diferensiasi terhambat, dan proliferasi
meningkat.
Kerusakan kelompok HSC premalignant yang bersifat sitopenia refrakter ,
hematopoetik displastik, deferensiasi sel multiple lineages yang terhambat dan
meningkatkan risiko berkembangnya menjadi AML (Pirruccello et al., 2006).
Konsekuensi dari sel darah yang mengalami regulasi abnormal dan diferensiasi
yang terhambat pada tingkat progenitor diatas, menyebabkan proliferasi yang
tidak terkendali pada sistem prekursor hematopoisis.
Keseimbangan homeostatik akan terganggu pada proliferasi dan
apoptosis yang meningkat pada awal penyakit karena adanya proses progresif.
Penderita yang mempunyai risiko tinggi menjadi MDS, proses proliferasi
meningkat sedikit demi sedikit juga terjadi hambatan pada mekanisme apoptosis
(anti apoptosis) (Tefferi& Vardman, 2009). Molekuler yang tidak dipengaruhi
regulasi sinyal yang meningkat seperti NF-kB, phosphor-Akt, Bim-1, Bcl-2, Bcl-
XL, dan heat shock protein chaperones menghambat apoptosis, penyebaran dan

24
evolusi menjadi ganas (AML) (gambar 2.3.)(Banerjeeet al., 2008). Menurut
penelitian Aulya et al., 2014 menunjukkan peran NFkB sebagai suatu faktor
transkripsi yang membuat suatu sinyal mampu membawa informasi gen
apoptosis. Contoh P53 (oksidan)dapat merangsang jumlah sel mononuklear
(makrofag) selain itu melalui TNF akan mengaktifkan procaspase, caspase
meningkatkan apoptosis sehingga dapat dipertimbangkan sebagai pengobatan
leukemia akut melalui jalur penghambatan NFkB.
Identifikasi abnormalitas sitogenetik atau molekuler berulang (recurrent
mutation) dan perubahan epigenetik adalah mekanisme dasar terjadinya MDS.
Perubahan tersebut tidak spesifik untuk MDS oleh karena beberapa perubahan
dapat ditemukan juga pada AML dan myeloproliferative disorders (MPDs). Status
non-hematopoitik (kemoterpi, radiasi, bahan kimia) yang meningkatkan proses
MDS menjadi AML telah menjadi subyek penelitian yang intensif (Gilliland&
gribben, 2012; Banerjeeet al., 2008;Klepin et al., 2014).
Gambar 2.5 Apoptosis, Aktivasi NF-kB (dikutip dari: Banerjeeet al., 2008) Keterangan gambar: Peran NFkB sebagai suatu faktor transkripsi yang mempunyai sinyal mampu
membawa informasi gen apoptosis Contoh P53 (oksidan), TNF akan
mengaktifkan procaspase, caspase meningkatkan apoptosis sehingga dapat
dipertimbangkan sebagai pengobatan leukemia akut melalui jalur
penghambatan NFkB. Antioksidan menghambat NF-kB, dapat membantu dalam

25
menghadapi progresivitas sel kanker. Penelitian lain menyatakan bahwa NF-kB
mempunyai peran penting dalam pertumbuhan kanker dan perkembangannya.
Target dari NF-kB adalah progresi tumor, inflamasi, imortalitas sel, promosi
tumor dan metasase.Korelasi antara inflamasi dan kanker sebagai faktor utama
yang mengendalikan kemampuan sel ganas untuk melawan
immunosurveillanceberdasarkan apoptosis dan bertindak sebagai faktor
pertumbuhan (kresno, 2012).
Faktor imunostimulasi dan imunosupresi dalam lingkungan mikro, yaitu
stimulasi dan supresi mempengaruhi perkembangan tumor(lihat gambar
2.6)(Alpdogan, 2013).
Gambar 2.6 Perkembangan sel Th1, Th2 dan sitokin yang diproduksi masing-masing (Dikutip dari Kresno S.B., 2010).
Keterangan gambar :
Antigen tumor dan produk tumor menarik sel dendrit (DC) ke lokasi tumor,
kemudian sel DC menangkap antigen tumor, berubah menjadi DC matur yang
menghasilkan IL-12 dan merangsang sel CD4+/Th1 untuk memproduksi IFN-ϒ. Sel
ini membantu ekspansi CD8+ menghancurkan sel tumor melalui jalur apoptosis
(granzym B dan perforin). Antigen dan produk tumor lain mempromosikan
maturasi DC yang menghasilkan sitokin pro-inflamasi IL-6 dan TNF-α. DC ini
membantu pembentukan sel CD4+/Th2 yang memproduksi IL-4 dan IL-3 yang akan
menyingkirkan tumor menjadi tidak efektif (Alpdogan, 2013).
Sel punca hematopoetik (HSCs) memiliki molekul yang khas pada
permukaan selnya, yaitu molekul glikoprotein CD34 (cluster of differentiation).Sel
induk hematopoietik berfungsi sebagai sel induk semua sel darah termasuk

26
eritrosit, leukosit dan trombosit. Sel induk (stem cell) tersebut adalah sel-sel
langka yang menyusun kurang dari 0,01% sel sumsum tulang. Isolasi dan
pengukuran kuantitatif sel ini sulit dilakukan karena kelangkaan dan kemiripan
bentuknya dengan sel-sel lainnya. Alasan tersebut diperlukan
penandaan/marker. Sel induk CD34+ adalah sel yang mengekspresikan sel induk
hematopoitik dan sel progenitor lainnya.Dalam sistem hemopoesis, sel induk
mengalami proses komitmen untuk menjadi sel progenitor multipotensial yang
msing-masing selanjutnya menjadi sel hematopoetik normal. Proses tersebut
diatas diatur secara ketat oleh gen tertentu yang menyandi faktor
transkripsi.Misalnya: Penanda molekul CD (CD4 dan CD8) yang mana
merupakan penanda sel Thelper dan sitotoksik secara berturut-turut(Provan &
Gribben, 2012; Kresno, 2012; Takahashi, 2011; Chen et al., 2004).
Perubahan ekpresi maupun struktur gen yang menyandi faktor transkripsi
dapat menyebabkan fungsi faktor transkripsi terganggu dan mengakibatkan
transformasi sel (Kresno, 2012). Leukemia adalah penyakit yang didapat yang
disebabkan akumulasi kelainan kromosom dan mutasi genetic yang
mengubahsifat biokimiawi atau kadar ekspresi protein. Gen yang direkomendasi
pada translokasi kromosom seringkali menyandi faktor trasnkripsi, maka faktor
trasnkripsi mempunyai peranan yang cukup penting pda leukemogenesis (faktor
Transkripsi PU-1 adalah regulator utama gen myeloid (gambar 2.9) (Kresno,
2012).Kasus MDS terdeteksi adanya sel CD34+, yang dapat mempengaruhi
sistem hematopoitik dan mempunyai korelasi dengan AML, mungkin karena
beberapa regulator penting untuk gen myeloid.PU.1 adalah pengatur pusat
seluruh garis keturunan hematopoietik dan aktivitas sel induk. Sementara itu,
C/EBP α(CCAAT/enhancer binding protein alpha) merupakan salah satu faktor
transkripsi spesifik dari garis keturunan hematopoietik. Gen myeloid tersebut
merupakan regulator perkembangan granulosit, dan PU.1 diekspresikan pada sel

27
induk CD34+. Ekspresi PU.1 mengalami supresi maka akan terjadi supresi
diferensiasi (differentiation arrest) dan meningkatkan proliferasi (proliferation
drive), cenderung tidak terkendali (Provan& Gribben, 2012; Kresno, 2012;
Takahashi, 2011; Chenet al., 2004).
2.3 Patomekanisme AML
Pada tahun 1998, Rowley mengemukakan peran translokasi kromosom
pada leukemogenesis. Abnormalitas sitogenetik dianggap sebagai parameter
yang lebih tepat untuk menentukan Remisi Komplit (CR) dan harapan hidup
jangka panjang pada penderita leukemia dibandingkan dengan penentuan
menggunakan respon sumsum tulang dan proporsi sel blast. Sejak tahun 1996
perkembangan ilmu dan teknologi telah memungkinkan untuk mengidentifikasi
kelainan atau mutasi gen yang terlibat dalam leukemogenesis di tingkat
molekuler. Menurut Leis, 1996 bahwa studi tentang kelainan kromosom atau
molekuler pada leukemia, baik pada orang dewasa maupun pada anak-anak.
Setelah tahun 1996 menunjukkan bahwa kelainan genetik berhubungan erat
dengan prognosis, sehingga evaluasi sitogenetik maupun molekuler saat ini
diperlukan untuk penatalaksana leukemia secara optimal (Kresno, 2012; Leis,
1996).
Beberapa bukti tersebut diatas menunjukkan bahwa berbagai perubahan
genetik yang berbeda, membentuk mutasi fusi gen dalam leukemogenesis.
Menurut Kresno, 2012 bahwa pada binatang coba menunjukkan mutasi tunggal
tidak cukup untuk menyebabkan AML. Pada mutasi fusi gen RUNX-RUNX1T1
dan CBFB-MYH11, masing-masing disebabkan t(8;21) dan inv(16) t(16;16),
berhasil mengganggu diferensiasi sel (Kresno, 2012;Rowley, 1998). Kresno,
2012; Owen&Fitzgibbon, 2012, menyatakan bahwa mutasi berikutnya dalam sel
progenitor (multistep leukemogenesis) diperlukan untuk menjadi fenotif

28
leukemik.Perkembangan leukemia adalah refleksi dari faktor lingkungan dan
genetik. Keganasan ini berkembang dari sel yang memiliki kemampuan
memperbarui diri sendiri (self renewal), baik sel induk (stem cell), sel myeloid
maupun sel limfoid, berproliferasi secara tidak terkendali. Kerentanan sel untuk
transformasi tergantung akumulasi kelainan genetik, kemampuan self renewal
dan kecepatan relatif sel tersebut untuk berdeferensiasi. Berbagai kelainan dan
mutasi genetik terlibat dalam perkembangan leukemia, dapat dijumpai pada
semua atau sebagian jenis leukemia, ada juga yang spesifik untuk leukemia
tertentu (Owen&Fitzgibbon, 2012; Kresno, 2012, Hoffmanet al., 2013).
Menurut Owen&Fitzgibbon, 2012 perkembangan leukemogenesis,
mengenai hipotesis sel punca kanker (cancer stem cell hypothesis/CSC), dalam
hal ini leukemia stem cells (LSCs). Sel sasaran mengalami transformasi
leukemik, pada saat ini ada 3 hipotesis yaitu pertama, berbagai jenis sel
dalam hirarki sel punca (stem cell) dan sel progenitor menunjukkan kerentanan
untuk transformasi. Keadaan ini berarti mutasi akan mengubah pola deferensiasi
normal dan mempromosikan ekspansi klonal sel leukemik dengan status
diferensiasi spesifik. Kedua, mutasi yang bertanggung jawab atas transformasi
dan progresivitas menjadi leukemia terjadi pada sel punca primitif multipoten dan
mengakibatkan terbentuknya LSC. Heterogenitas tumor terjadi karena
kemampuan LSC untuk berdeferensiasi menjadi lineage tertentu dengan fenotip
spesifik. Ketiga, leukemia akut memerlukan serangkaian perubahan genetik
yang progresif mulai dari ekspansi klonal LSC yang mengalami transformasi
(Owen&Fitzgibbon, 2012).
Takahashi, 2011 membuat model “two-hit leukemogenesis” yaitu sel
punca pre-leukemik mengalami transformasi awal tetapi belum mengalami
mutasi berikutnya yang diperlukan untuk berkembang menjadi leukemia.
Disregulasi gen yang terlibat dalam self-renewal dan diferensiasi sel punca

29
leukemia menggunakan jalur regulasi yang sama antara sel normal dengan jalur
yang digunakan oleh sel ganas. Mutasi dapat terjadi pada setiap gen dan setiap
lokus untaian DNA, menimbulkan berbagai onkogen yang jumlahnya hampir tidak
terbatas (Kresno, 2012, Takahashi, 2011).
Model klasik leukemogenesis mempunyai 2 jenis mutasi yang
dikelompokkan dalam kelas yang berbeda tetapi saling bekerjasama dalam
leukemogenesis (Gilliland &Gribben, 2012, Takahashi, 2011).Kategori I adalah
mutasi yang mengakifkan jalur transduksi sinyal meningkatkan proliferasi dan
atau survival sel progenitor leukemia. Sel progenitor leukemia sebagai contoh:
mutasi yang mengakibatkan aktivasi reseptor tyrosine kinase FLT3 atau aktivasi
jalur sinyal NRAS dan C-KIT. Jenis mutasi kategori ini pada umumnya adalah
point mutation (Owen&Fitzgibbon, 2012, Bejaret al., 2011).Kategori II yaitu
mutasi yang menggunakan jalur transkripsi atau komponen kompleks ko-aktivasi
transkripsi mengakibatkan gangguan diferensiasi dan atau perubahan sifat self-
renewal dari committed progenitor hematopoietic. Kelompok ini yaitu mutasi CBF,
PML- RARA, CEBPA, MLL (tabel 2.2) (Owen&Fitzgibbon, 2012, Takahashi,
2011). Pada umumnya mutasi kategori ini disebabkan translokasi kromosom.
Kresno, 2012 memprediksi bahwa kategori III pada leukemogenesis yaitu mutasi
gen yang terlibat dalam regulasi siklus sel atau apoptosis (tabel 2.2).
Konsep diagnosis molekuler leukemia yaitu mengidentifikasi kelainan gen
mempunyai peran penting dalam mengendalikan berbagai fase pertumbuhan sel
dengan cara mendeteksi perubahan kode gen RNA/DNA yang mempunyai
kelainan. Abnormalitas molekuler pada leukemia dapat dipergunakan untuk
memprediksi prognosis, respon terhadap pengobatan dan mendeteksi sisa sel
leukemik atau MRD (minimal residual disease). Selain dipergunakan untuk
diagnosis, klasifikasi atau subklasifikasi dan penentuan prognosis. Pemeriksaan
abnormalitas sitogenetik pada leukemia dapat digunakan untuk

30
memprediksi keberhasilan pengobatan, transplantasi stem sel, target
terapi, dan pemantauan penyakit.
2.3.1 Mutasi Kategori I (Kelainan yang mengaktifkan jalur transduksi
sinyal)
2.3.1.1 Mutasi Gen Fms-Related Tyrosine kinase 3 (FLT3)
Gen FLT3 adalah reseptor tirosin kinase memainkan peran penting dalam
mengontrol kelangsungan hidup sel, proliferasi dan diferensiasi sel
hematopoitik.Ligan FLT3 dieskpresikan oleh sel stroma pada sumsum tulang, sel
lain, dan faktor pertumbuhan (growth factors) untuk menstimulasi sel punca
proliferasi, sel progenitor, sel dendrit, sel NK. Mutasi gen FLT3 telah dideteksi
sekitar 30% pada AML dan pada pasienALL atau MDS dalam prosentase yang
kecil (Gilliland& Griffin, 2002).
Lokasi gen FLT3 disebut juga FLK2 (fetal Liver Kinase-2), hSTK1 (human
stem Cell Kinase-1) pada pita kromosom 13q12, struktur ikatan membran
berhubungan dengan reseptor tyrosine kinase kelas III seperti KIT, Fms, dan
PDGFR (Platelet-derived growth factor). Menurut penelitian Owen&Fitzgibbon,
2012 peran sinyal FLT3 pada leukemogenesis, gen FLT3 Internal tandem
duplication (ITD), terbentuk dengan FLT3 domain juxtamembrane (exon 14 dan
15), diprediksi mempunyai mutasi terbanyak, diperkirakan 30% dari kasus AML
(Bain,et al., 2011). Pada kasus AML mutasi titik (point mutation) tipe missense
lebih dari 7% mempunyai efek mengaktivasi struktur dari domain tyrosine kinase
dari FLT3 yang berkode exon 20.Mutasi FLT3-ITD dan FLT3 domain kinase
meningkatkan peran aktif protein FLT3, keadaan tersebut didukung ligand-
independent untuk proliferasi dan kelangsungan hidup sel leukemia (Gililand&
Gribben, 2010; Brunetet al., 2008).

31
Mutasi aktivitas onkogen FLT3-ITD merupakan sinyal abnormal termasuk
STAT5 dan menghambat faktor transkripsi myeloid seperti PU.1 dan C/EBPα
(kerangka teori).
Menurut penelitian Shihet al., 2004 insiden terjadinya mutasi melalui jalur
transduksi sinyal (FLT3-ITD dan N-RAS), jumlah kasus MDS yang mengalami
proses transformasi menjadi AML sebanyak 82 kasus; 70 kasus diagnosis MDS-
related AML, 5 kasus mempunyai mutasi FLT3-ITD. 70 kasus tersebut diatas
ditemukan 7 kasus FLT3-ITD dalam proses menjadi AML. Insiden kasus mutasi
FLT3-ITD pada diagnosis MDS secara signifikan lebih kecil dibandingkan dengan
kasus MDS-related AML.Pada kasus yang ditemukan mutasi FLT3-ITD
mempunyai progresivitas menjadi AML dalam waktu lebih cepat dibandingkan
kasus yang FLT3-ITD normal.Pada kasus yang ditemukan mutasi FLT3-ITD
secara signifikan mempunyai harapan hidup lebih pendek dari pada kasus
dengan FLT3-ITD normal. Diagnosis AML dengan mutasi FLT3 adalah bagian
penting pada kasus AML (tabel 2.1, 2.2), karena mempunyai korelasi dengan
prognosis. Menurut Shihet al., 2004 studi pada orang dewasa dan anak
menyatakan bahwa diagnosis AML dengan mutasi FLT3-ITD mempunyai
prognosis buruk (Badar et al., 2014;Klepin et al., 2014).
Implikasi klinik AML dengan mutasi FLT3-TKD masih kontroversi risiko 5 -
7% (tabel 2.3) dan dibutuhkan lebih banyak studi untuk menjelaskan mutasi gen
FLT3-TKD yang memberikan prognosis buruk (Takahashi, 2011). Frekwensi
mutasi FLT3 berbeda dalam subgrup sitogenetik dari AML, frekwensi terbanyak
terutama pada kariotipe normal yaitu 30 – 35% pada kasus AML dengan t(15;17)
(Owen&Fitzgibbon, 2012).
Pada mutasi gen FLT3-ITD dengan kariotipe normal mempunyai
prognosis yang buruk yaitu kekambuhan (relapse) AML yang meningkat,
keadaan tersebut menyebabkan penurunan harapan hidup bebas dari sakit

32
(event-free survival atau EFS) dan hidup sehat (overall survival atau OS)
(gambar 2.7.) Complate Remission (CR) tidak dipengaruhi tetapi menurut
OwendanFitzgibbon pada studi tahun 2012, menunjukkan bahwa durasi waktu
CR secara signifikan menurun pada kasus tersebut.
Pada pemeriksaan aspirasi sumsum tulang hewan coba yang
menggunakan model mutasi FLT3-ITD menyebabkan myeloproliferative disease,
tetapi tidak menyebabkan AML. Menurut penelitian Owen and Fitzgibbon tahun
2012 menyatakan bahwa FLT3-ITDs mempengaruhi EFS (prognosis yang buruk)
(gambar 2.7.).
Gambar 2.7 Frekuensi dan distribusi mutasi NPM1, CEPBA, RUNX1, MLL-PTD, dan FLT3-ITD pada AML dengan karyotipenormal (Owen&Fitzgibbon, 2012).
Menurut penelitian Agersborget al., 2015 bahwa mutasi FLT3, NPM1, dan
WT1 adalah abnormalitas molekuler yang dapat dipergunakan sebagai kriteria
diagnosis pasien AML yang obyektif lebih baik dari pada aspirasi sumsum tulang.
Mutasi tersebut telah dideteksi sebanyak 49% pada pasien AML. Peneliti ini
menunjukkan bahwa diperkirakan 50% dari pasien AML dapat dibuat diagnosis
dengan mendeteksi abnormalitas melekuler, tidak memperhatikan morfologi
sumsum tulang.

33
2.3.1.2 Mutasi Gen RAS(rat sarcoma)
Mutasi pada semua gen RAS kromosom homologus, N-RAS, KRAS dan
HRAS, menempati codon 12, 13 dan 61. Semua protein RAS (p21ras), NRAS
adalah anggota keluarga RAS superfamily dari GTPase. Aktivasi mutasi exon 2
(codon 12/13)(1p13.2), exon 3 (codon 61), and exon 4 (codon 146) antara lain
ditemukan pada Myeloid leukemia (14%) (Colicelli, 2004).
Pengikatan RAS berinteraksi dengan protein sasaran (RAF-1,
Phosphatidylinositol-3 kinase), SOS mengaktivasi RAS sebagai respon terhadap
faktor pertumbuhan. Pengikatan GTP pada protein RAS membentuk kompleks
GTP-RAS melalui interaksi protein lain yaitu GTPase-activating protein (GAP).
Ikatan GTP-RAS ini akan segera inaktif menjadi bentuk GDP-RAS. Aktivitas
GTPase dari RAS meningkat kalau ada interaksi dengan protein GAP. Dengan
adanya point mutation pada gen RAS akan menurunkan aktivitas GTPase,
akibatnya ikatan GTP-RAS akan inaktif secara perlahan-lahan sehingga akan
menimbulkan respon seluler yang berlebihan terhadap sinyal dari
reseptor.Jumlah protein RAS aktif yang dapat meningkatkan kemungkinan
transformasi, Kompleks GTP-RAS akan mentransmisikan sinyal di dalam sel.
Point mutation pada gen RAS banyak ditemukan pada berbagai tumor termasuk
kanker darah (tabel 2.8) (Yusup, 2008).
Mutasi NRAS adalah mutasi RAS yang paling sering pada AML,
terdeteksi antara 10% - 30% dari kasus (Owen&Fitzgibbon, 2012; Hoffman et al.,
2013). Pada tahun 2011 Takahashi menyatakan bahwa gambaran klinik mutasi
gen NRAS tidak secara signifikan mempengaruhi OS (over all survival), EFS
(event-free survival), dan DFS (disease-free survival), mempunyai insiden 10,3%
pada kasus AML (tabel 2.2). Shihet al., 2004, menyatakan bahwa mutasi FLT3
atau NRAS menunjukkan progresivitas MDS untuk menjadi AML yaitu 30% dari
semua kasus MDS.

34
Fungsi jalur RAS-RAF-MAP-kinaseadalah meneruskan sinyal dari
lingkungan ekstraseluler ke dalam nukleus dimana gen spesifik diaktifkan. Faktor
pertumbuhan (Epidermal Growth Factor) akan menyebabkan dimerisasi reseptor,
kemudian akan terjadi autofosforilasi tyrosine. Selanjutnya tyrosine yang
terfosforilasi akan bertindak sebagai tempat ikatan berafinitas tinggi bagi suatu
protein adaptor bernama Growth factor receptor-bounds2 (GRB2). GRB2 yang
mengandung domain SH2 dan SH3, protein ini tidak bertindak sebagai enzim
tetapi berfungsi menfasilitasi interaksi protein, termasuk interaksi yang
menghasilkan aktivitas RAS oleh GEF (guanine nucleotide exchange factors).
Varian onkogenik RAS mempunyai sifat resisten terhadap GAP (GTPase-
activating proteins) sehingga hidrolisis GTP terhambat, sehingga terjadi
akumulasi RAS dalam bentuk aktif. Jumlah protein RAS aktif yang dapat
meningkatkan kemungkinan transformasi. RAS berada dalam keadaan aktif
berinteraksi dengan protein sasaran misalnya RAF-1 protein kinase dan
Phosphatidylinositol-3 kinase (PI3K). GEF merupakan regulator positif bagi RAS.
SOS dalam hal ini mempunyai peran penting dalam aktivasi RAS sebagai respon
terhadap stimulasi faktor pertumbuhan protein-tyrosine kinasereseptors(PTKRs).
RAF diaktifkan oleh RAS, kemudian mengalami fosforilasi dan mengaktivasi
kinase-kinase lain disebut Mitogen-activated protein kinase kinase
(MEK/MAPKK). MEK memfosforilasi dan mengaktifkan extracellular signal-
regulated kinase (Erk1) dan Erk2 (MAPKs), Mitogen-activated protein kinase
(MAPK) mem-fosforilasi kinase dalam sitoplasma yang mengatur faktor translasi
maupun faktor transkripsi dalam nukleus (gambar 2.8.) (Owen&Fitzgibbon, 2012;
Ahearnet al., 2012).
RAF(sorafenib) dan MAPK bukan satu-satunya sasaran dari RAS, banyak
efektor RAS yang lain, di antaranya jalur survival PI3K (Phosphatidylinositol 3-

35
kinase), dan jalur c-Jun N-terminal kinase (JNK) (Owen&Fitzgibbon, 2012,
Budiasih, 2012).
RAS diprediksi adalah target potensial untuk pengobatan pada AML.
Farnesyl transferase inhibitors (FTIs) adalah obat yang bekerja pada target
farnesyl dari protein RAS. Pada modifikasi protein post translasi penting sebagai
mediator transformasi sel. Menurut laporan studi Owen and Fitzgibbon tahun
2012 menyatakan bahwa nonpeptidometric FTI tipifarnib digunakan sebagai
ketentuan kemoterapi standar yaitu kombinasi idarubicin dan cytarabine, dapat
memperbaiki hasil pengobatan dibandingkan dengan kontrol yang konvensional
(Owen&Fitzgibbon, 2012).
Gambar 2.8 Siklus GDP – GTP dari RAS (dikutip dari: Ahearn, 2012),
Keterangan gambar: a. SOS mengaktifkan RAS dengan meningkatkan pengikatan RAS pada GTP
dengan bantuan GEFs (guanine nucleotide exchange factors). Ikatan RAS
dengan GTPase dari RAS ditingkatkan melalui interaksi dengan protein lain
yang dikenal sebagai GTPase-activiting protein (GAP). Interaksi ini
mengakibatkan hidrolisis GTP menjadi GDP.
b. RAS berada dalam keadaan aktif, RAS dapat berinteraksi dengan protein
sasaran misalnya RAF-1 protein kinase dan Phosphatidylinositol-3 kinase
(PI3K). GEFs merupakan regulator positif bagi RAS. SOS dalam hal ini
mempunyai peran penting mengaktivasi RAS sebagai respon terhadap
stimulasi faktor pertumbuhan protein-tyrosine kinase (PTKRs). Dalam
perannya ini SOS bergabung dengan protein yang mengandung

36
phosphotyrosine melalui molekul adaptor Growth factor receptor-bound 2
(GRB2) yang mengandung domain SH2 dan SH3. Protein ini tidak bertindak
sebagai enzim tetapi berfungsi membantu interaksi protein, termasuk
interaksi yang menghasilkan aktivitas RAS oleh GEF. Varian onkogenik RAS
mempunyai sifat resisten terhadap GAP sehingga hidrolisis GTP terhambat,
sehingga terjadi akumulasi RAS dalam bentuk aktif. Jumlah protein RAS aktif
yang dapat meningkatkan kemungkinan transformasi.
c. Multiplex regulasi dan sinyal RAS. Famili terdiri dari GEFs, GAPs dan
effectors dilaporkan meregulasi RAS atau transmisi dari RAS-GTP. CAPRI
(Calcium Promoted RAS Inactivator); GAP1(IP44BP); GAP1(InsP4)-pengikat
protein; MEK( MAPK/ERK kinase); NF1(Neurofibromin1); PLCƐ
(phospholipase CƐ); PI3K (phosphoinositide 3-kinase); RALGDS (RAL
guanine – nucleotide dissociation stimulator); RASAL (RASGAP-activating-
like); RASGRF (RAS-specific guanine-nucleotide-releasing factor); RASGRP
(RAS-specific guanine-nucleotide-relesing protein); RASSF (RAS association
domain-conteining family); RIN1 ( RAS and RAB interactor1); SYNGAP
(synaptic RASGAP); TIAM1 (T lymphoma invasion and metastasis-inducing
1) (kresno, 2012, Ahearnet al., 2012) .
Pada sel kanker, migrasi dari sel juga dipengaruhi oleh beberapa gen
yang terlibat. Salah satu dari gen tersebut adalah Ras superfamili dari small
GTP- binding protein merupakan gen yang paling banyak dipelajari. Ras
superfamili ini terdiri dari 30 jenis antara lain: Rho, Ras, Arf/sar 1 dan Rab/Ran
subfamily (Lumongga, 2008; Colicelli, 2004).
AML menghambat maturitas sel sehingga proses diferensiasi seri myeloid
berhenti pada sel-sel muda (blast) sehingga terjadi akumulasi blast di sumsum
tulang. Keadaan ini akan menyebabkan gangguan hematopoisis normal dan
selanjutnya akan mengakibatkan sindrom kegagalan sumsum tulang yang
ditandai dengan adanya sitopenia. Selain itu sel-sel bast yang terbentuk juga
punya kemampuan untuk migrasi keluar sumsum tulang dan berinfiltrasi ke
organ-organ lain seperti kulit, tulang, jaringan lunak, dan sistem saraf pusat serta
merusak organ-organ tersebut dengan segala akibatnya.
2.3.2 Mutasi Kategori II (Mutasi faktor transkripsi)
Faktor transkripsi memegang peranan utama pada diferensiasi berbagai
jenis sel termasuk sel hematopoitik. Kresno, 2012 menyatakan dalam studinya

37
bahwa faktor transkripsi hematopoitik dapat terlibat dalam interaksi berbagai
protein yang kompleks dan spesifik dapat menjadi petanda jenis sel dan stadium
diferensiasi. Dalam sistem hematopoisis, sel induk (stem cell) mengalami proses
menjadi sel progenitor multipotensial yang selanjutnya menjadi sel hematopoitik
normal. Proses tersebut diatas diatur oleh gen tertentu yang menyandi faktor
transkripsi. Perubahan struktur gen ekspresi dan yang menyandi faktor
transkripsi, dapat mengakibatkan fungsi faktor transkripsi terganggu dan terjadi
transformasi sel (Kresno, 2012;Takahashi, 2011).
2.3.2.1 Core-binding factor (CBF) leukemia
Core-binding factor (CBF) adalah regulator utama pada hematopoisis dan
sering menjadi target translokasi kromosom dihubungkan dengan leukemia
(Owen and Fitzgibbon, 2012). Faktor transkripsi ini tersusun dari dua subunit,
subunit α dikode RUNX1 disebut juga runt-related transcription factor 1, AML1
(acute myeloid leukemia 1), dan subunit β dikode CBFB (core binding factor B).
Gen RUNX1 terdiri dari 9 exons dan spans lebih dari 150 kb. Kode Runt domain
sebagai bagian dari exon 3, exon 4, dan exon 5.Lokasi kromosom dari AML1
adalah pada kromosom 21q22, sedangkan AML2 dan AML3 berada pada
kromosom regio 1p36 dan 6p21 (Juniarka, 2011, Taniuchi et al., 2012).Hilangnya
homozigot dari fungsi RUNX1 atau CBFB pada binatang percobaan (tikus yang
telah direkayasa genetika) tidak mempunyai sistem hematopoisis yang definitif,
keadaan tersebut menerangkan bahwa kedua komponen dari CBF diperlukan
untuk mengembangkan sistem hematopoitik normal.CBF AML adalah subtipe
yang relatif sering pada AML dewasa, diklasifikasikan dalam kategori risiko baik.
Kasus AML de novo antara 7% dan 12%, mempunyai t(8;21) dimana gen RUNX1
fusi menjadi RUNX1T1 (ETO), sedangkan 10-12% kasus lain mempunyai
inv(16)/t(16;16) dimana CBFB fusi menjadi MYH11. Gen partner CBF dalam

38
setiap kasus mempunyai dasar aktivitas menuju ekspresi tetap RUNX1-
RUNX1T1 (AML-ETO) atau protein melebur menjadi CBFβ-MYH11,
menyebabkan anggota dari kompleks membantu menekan dan gen ekspresi
rendah mengatur kompleks CBF (Owen&Fitzgibbon, 2012; Ley et al., 2013).
Leukemia ditemukan rata-rata jauh lebih tinggi pada keluarga dengan
dominan kuat mutasi K83E (57%) dibanding dengan delesi lengkap RUNX1
(24%), Oleh karena itu, aktivitas rendah RUNX1 sepertinya berhubungan dengan
tingginya leukemogenisitas.Hipotesa diperluas ke arah leukemogenesis yang
diinduksi oleh chimeric genes.Penelitian terbaru kebanyakan mengindikasikan
bahwa fungsi utama dari chimeric genes adalah inhibisi transdominan dari wild-
type RUNX1, menyebabkan turunnya aktivitas RUNX1 secara drastis di sel yang
terpengaruh. Sekarang lebih dari 10 partner gen yang ber fusi dengan RUNX1
telah diidentifikasi di dalam translokasi kromosom. Setiap chimeric gene mungkin
mempunyai mekanisme leukemogenesis nya sendiri, kekurangan RUNX1
mungkin menjadi mekanisme yang sama pada leukemia dengan mutasi RUNX1.
Diterimanya trisomy 21 menggarisbawahi gagasan ini.RUNX1 +/-
mutant alleles seperti sering diduplikasi. Penemuan ini bisa menjadi bukti
tambahan terhadap hipotesa bahwa berkurangnya aktivitas RUNX1
meningkatkan status leukemogenic(Osato, 2004).
Data terbaru dari studi in vitro dan model hewan transgenic
menunjukkan alur dominant-negative untuk fusi gen. RUNX1-RUNX1T1 berperan
sebagai dominant-negativeinhibitor dari wild type CBF dengan transkripsi fusi
yang mempertahankan DNA dari RUNX1 dan domain heterodimerization tetapi
kurangnya aktivasi domain transkripsi C-terminal. Mekanisme dari aktivitas
dominant-negative pada fusi CBFβ-MYH11 tidak jelas, tetapi semua mencegah
transaksi dari target CBF. Namun, sudah jelas bahwa translokasi saja tidaklah
cukup untuk menghasilkan fenotif leukemia. Contohnya, kondisi alel untuk

39
RUNX1-RUNX1T1 ditunjukkan dalam hematopoitik progenitor dewasa yang tidak
cukup untuk menyebabkan AML, tetapi hasil menunjukkan Myeloproliferative
phenotype.Pada kasus tikus percobaan memberikan binatang tersebut perlakuan
dengan paparan chemical muntagens untuk menjadi fenotif AML
(Owen&Fitzgibbon, 2012; Kresno, 2012; Aggrawalet al., 2007).
2.3.2.2 Mutasi gen PU.1
Faktor transkripsi PU.1 kode dari gen Sfpi1 (spleen focus-forming virus
proviral integration/Purine-rich DNA) diferensiasi myelomonocytic selama
hematopoisis normal dan sebagai regulator yang mempunyai komitmen pada
progenitor multipotent hematopoiesis (gambar 2.9.) (Hoffman et al.,
2013).Menurut Kresno, 2012 menyatakan pengaturan ekspresi mRNA PU.1
memegang peran penting dalam komitmen sel progenitor multipotensial menjadi
sel myeloid, maupun diferensiasi dan maturasi sel selanjutnya (gambar
2.9.)(Provan& Gribben, 2012; Kresno 2012; Takahashi, 2011). Abnormalitas
pada level ekspresi PU.1 adalah leukemogenik. Pada penelitian Hoffamanet al.,
2013 menyatakan bahwa tikus ekspresi gen Sfpi1 sebagai regulasi kontrol
transkripsi pada bagian distal upsteam regulatory element (URE) itu sebagai
penyelamat yang efektif, tetapi penurunan URE dapat menurunkan ekspresi
PU.1 sampai 80% pada sumsum tulang dan dapat berkembang menjadi AML.
Peneliti tersebut diatas menyatakan bahwa menurunnya level PU.1 masih cukup
untuk mendukung kelangsungan progenitor myeloid, sehingga mengganggu
regulasi dari ekspresi beberapa reseptor sitokin. Pada kasus AML telah
dilaporkan penekanan transkripsi PU.1 yang mengekspresikan fusi gen PML-
RARα atau mutasi internal tandem duplication FLT3 (FLT3-ITD), dan AML1-ETO,
fungsi PU.1 tidak aktif oleh karena ditempat itu tidak ada ko-aktivasi JUN
(Hoffmanet al., 2013; Provan& Gribben, 2012; Kresno 2012).

40
Gambar 2.9 Faktor transkripsi AML-1 dan leukemogenesis(Dikutip: Kresno,
2012).
Keterangan:
PU.1 menurun atau tidak aktif maka deferensiasi terhambat pada sel
mielosit dan monosit.
2.3.2.3 Mutasi gen NPM1
Faliniet al., 2005, mutasi gen Nucleophosmin member 1 (NPM1) pertama
kali di laporkan, mutasi NPM1 terjadi pada exon 12, dengan prosentase lebih dari
95% terdiri dari 4-bp insersi di posisi 960. NPM1 lokasi pada kromosom pita
5q35, nucleus-sitoplasma dikode gen yang mana mempengaruhi protein terdiri
dari agregasi nucleus protein, regulasi dari berkumpulnya protein ribosomal,
inisiasi duplikasi centrosome dan regulasi dari p53 (onco-suppressors) dari
nucleus ke sitoplasma mungkin akan menjadi transformasi ke ganas. Mutasi
NPM1 ditemukan pada kira-kira 35% penderita dewasa dengan AML (tidak
termasuk hasil mutasi NPM1, pada bagian protein NPM1 dalam sitoplasma) dan
sering terjadi abnormalitas genetik ditemukan pada AML dengan kariotipe normal
(CN). Pemikiran bahwa NPM1 mempunyai fungsi supresor tumor, kerusakan
yang berada di lokasi subseluler dan dikorelasikan dengan rata-rata CR
(complete remission) yang signifikan setelah dilakukan kemoterapi. Jumlah kasus
AML dengan kariotipe normal antara 60 – 80% ditemukan gen mutasi NPM1
dapat membantu prognosis kasus AML dengan kariotipe normal (CN) (Hoffmanet
al., 2013;Falini & Bolli, 2009).

41
Owen and Fitzgibbon, 2012 menyatakan bahwa kasus AML dengan
kariotipe normal sering terjadi abnormalitas gen NPM1 antara 45 – 55%.Mutasi
NPM1 adalah tipe heterogenus dan dibatasi pada exon 12. Pada saat NPM1
disitoplasma dideteksi menggunakan imunohistokimia untuk prediksi mutasi
NPM1 pada AML (Tanet al., 2008) dan keadaan mutasi NPM1 dihubungkan
dengan prognosis yang lebih baik, deteksi mutasi NPM1 mungkin menggunakan
alat yang lebih baik sehingga dapat memprediksi meskipun minimal monitoring of
minimal residual (MRD) (Tanet al., 2008; Hoffmanet al., 2013; Owen& Fitzgibbon,
2012).
Hoffmanet al., 2013 menyatakan bahwa mutasi NPM1 berhubungan
dengan mutasi gen FLT3-ITD, dan FLT3-TKD, tetapi pada penelitian
ditemukanmutasi gen NPM1 dengan FLT3-ITD insiden 60%. Menurut Owen&
Fitzgibbon, 2012 bahwa Insiden mutasi NPM1 dengan mutasi FLT3-ITD
diperkirakan 40%. Mutasi NPM1 sebagai petanda prognostik, hal tersebut telah
dikonfirmasi oleh beberapa peneliti Kejadian yang tidak perlu diragukan yaitu
kasus dengan mutasi NPM1c+/wild-type FLT3 mempunyai prognosis lebih baik
dibandingkan dengan kasus AML dengan kariotipe normal.Pada kasus tersebut
mendapatkan keadaan EFS dan OS dengan pengobatan kemoterapi
konvensional, keadaan itu hampir sama dengan leukemia CBF. Owen and
Fitzgibbon, 2012 menyatakan bahwa mutasi NPM1 dibatasi hanya pada
penderita yang tidak mempunyai mutasi FLT3-ITD.
Pada awal penelitian menunjukkan bahwa mutasi NPM1 sangat stabil
yaitu pada diagnosis dan relapse, dengan demikian memungkinkan menegakkan
kejadian primer pada leukemogenesis. Selanjutnya FLT3 atau mutasi sekunder
yang lain memerlukan pemicu untuk berkembang menjadi AML
(Owen&Fitzgibbon, 2012; Tanet al., 2008). Mutasi NPM1 akan menimbulkan
berbagai gambaran biologis dan klinis membentuk fusi dengan gen yang lain

42
dalam leukemogenesis. Beberapa penelitian menyatakan bahwa genotip mutan
NPM1 tanpa mutasi FLT3-ITD menunjukkan prognosis lebih baik dibandingkan
genotip mutan NPM1 disertai mutasi FLT3-ITD.Pada saat ini beberapa jenis
inhibitor FLT3 sedang dilakukan uji klinik, mungkin dikemudian hari dapat
dipergunakan untuk terapi (Owen&Fitzgibbon, 2012, Young, 2012, Tanet al.,
2008).
2.4 Patomekanisme MDS
2.4.1 Abnormalitas sitogenetik
Abnormalitas molekuler dan sitogenetik yang terjadi pada MDS adalah
penting, terutama memahami sitogenetik untuk mendeteksi kromosom yang
abnormal.Abnormalitas sitogenetik dapat terjadi single atau complex yang
abnormal (lebih dari tiga kromosom abnormal).abnormalitas tersebut membentuk
fusi gen dan mutasi kromosom yang tidak seimbang.
Pada MDS abnormalitaskromosom dapat sebagian atau keseluruhan yang
hilang, pada umumnya - 5, 5q -, - 7, 7q -, 11q -, 13q -, 20q - dan -Y (gambar
2.1.).Jarang material kromosom bertambah dan maksimal terbesar +8, contoh:
trisomy 8 (+8) adalah penambahan kromosom yang sering terjadi pada MDS.
Keadaan tersebut mempunyai nilai prognostik dan kontribusi untuk patogenesis
MDS, kromosom yang hilang akan menjadi pertimbangan karena menyebabkan
tumor-suppressor genesatau haploinsufficiensyofgenesdiperlukanuntuk
hematopoisis yang normal(Montalban-Bravo & Gracia-Manero, 2018).
Van Den Berghe menetapkan sindrom-5q sebagai kesatuan klinis yang
terdapat pada MDS (1974), pada saat dia menemukan pasien dengan anemia
makrositik, thrombositosis, diseritropoisis, megakariositik yang hipolobulasi dan
delesi kromosom 5 del(5q). Delesi kromosom 5 del(5q) sebagai abnormalitas
sitogenetik merupakan bagian dan mewakili penentuan kerusakan molekuler

43
yang mendasari pathogenesis MDS. Myelodysplastic Syndrome (MDS) yang
mempunyai prevalensi 15% dari penderita MDS mempunyai semua kasus
dengan gambaran klinik tersebut diatas. Dalam perbandingan dengan subtipe
MDS yang lain, dapat dikarakterisasi adanya del(5q), hapusan sumsum tulang <
5%, insiden perempuan lebih besar dan prognosis baik karena mempunyai risiko
yang rendah untuk transformasi menjadi leukemia (Berghe et.al., 1974;
Mohamedali & Mufti, 2008).
Abnormalitas sitogenetik diperkirakan 30 – 50% dari MDS primer, dan
MDS sekunder mempunyai prevalensi 80%disebabkan pengobatan antara lain:
kemoterapi keganasan penyakit yang lain. MDS primer adalah penyakit yang
belum diketahui faktor predisposisi dan terjadi sporadik, sedangkan MDS-related
AML jarang didapatkan yang bersifat keturunan. Kasus MDS mempunyai faktor
risiko terjadi mutasi (contoh : fusi dua gen, RUNX1 dan CEBPα). MDS sekunder
terjadi seperti keracunan kronik karena pengobatan kanker, kombinasi dari
radiasi dan radiometricalkylating agent (busulfan, nitrosourea) (Ceesayet al.,
2012; Young, 2012).
MDS dalam perjalanannya menjadi AML, maka semua abnormalitas
sitogenetik yang ditemukan pada MDS juga ditemukan pada AML, walaupun
keberadaannya mempunyai Insiden yang berbeda-beda. Bagaimana memastikan
translokasi yang seimbang seperti yang ditemukan pada AML, contoh : t(15;17),
inv(16) dan t(8;21), kelainan tersebut diatas sangat jarang ditemukan pada MDS
(gambar 2.2.) (Ceesayet al., 2012).
2.4.2 Abnormalitas molekuler MDS
2.4.2.1 Gen mutasi RAS
Semua protein RAS memiliki ikatan dengan guanosine triphosphate
(GTP) bahwa ikatan tersebut adalah sinyal utama intraseluler. Protein RAS

44
berada antara GTP dan ikatan guanosine diphosphate (GDP), ikatan GDP RAS
tidak dapat mengaktivasi jalur transduksi sinyal (Owen&Fitzgibbon,
2012).Keadaan inaktif, GDP-berikatan RAS diaktifkan oleh GEF, menginduksi
pelepasan GDP kemudian RAS berikatan dengan GTP. Gen RAS menjadi aktif,
aktivitas GTPase dari RAS ditingkatkan melalui interaksi dengan protein yang
dikenal dengan GTPase-activiting protein (GAP). Interaksi ini mengakibatkan
hidrolisis GTP menjadi GDP. RAS menjadi inaktif, GDP dalam keadaan bound
state(Ahearnet al., 2012, Kresno, 2012).
RAS berada dalam keadaan aktif, RAS dapat berinteraksi dengan protein
lain misal RAF-1 protein kinase. SOS mempunyai peran penting mengaktivasi
RAS sebagai respon terhadap simulasi faktor pertumbuhan (Growth factor) pada
protein-tyrosine kinase (PTKRs). Dalam perannya SOS bergabung dengan
protein yang mengandung phosphotyrosine melalui molekul adaptor GBR2 yang
mengandung domain SH2 dan SH3. Protein ini berfungsi sebagai fasilitas
interaksi protein antara lain GEF aktivasi RAS. Jalur RAS adalah yang terbaik
dan menjadi disregulasi pada keganasan (Ahearnet al., 2012;Gelsi-Boyer et al.,
2008).Insiden mutasi NRASpada MDS dilaporkan bervariasi (diperkirakan 10%
pada kasus MDS), sedangkan kasus CMML mempunyai prosentase relative lebih
tinggi.Mutasi NRAS sering ditemukan saat menegakkan diagnosis, selain itu
NRAS didapatkan pada perjalanan penyakit (Ceesayet al., 2012;Montalban-
Bravo &.Gracia-monero, 2018).
2.4.2.2 Gen mutasi yang lain
Pada kasus MDS-related AML adalah secondaryAMLyang faktor
penyebanyadihubungkan dengan MDS dimana terjadi paparan kimia, radiasi,
kemoterapi pada kasus tersebut dibandingkan dengan herediter. Pola bentuk
keturunan merupakan mutasi gen tunggal yang bersifat autosomal dominan.

45
Pada mutasi seringkali dihubungkan dengan sindroma genetik seperti: Diamond-
blackfan anemia (DBA), severe congenital neutropenia. MDS-related AML dapat
juga terjadi melalui mekanisme DNA repair seperti: Fanconi anemia, diperkirakan
35 – 50% kasus usia 40 tahun berkembang menjadi MDS-related AML. Keadaan
tersebut diatas dapat menunjukkan bahwa mutasi genetik pada MDS-related
AML tidak dapat diidentifikasi dari kasus yang sporadik, diduga kedua kasus
mempunyai latar belakang patofisiologi yang berbeda (gambar 2.2.)(Ceesayet
al., 2012).
Internal tandem duplication (FLT3-ITD) adalah mutasi yang jarang pada
MDS, angka kejadian tersebut kira-kira 5% dari kasus dengan stadium lanjut atau
mempunyai risiko yang tinggi untuk berkembang menjadi AML. Gen mutasi AML-
1 jarang didapatkan pada MDS primer (Ceesayet al., 2012). Bains et al., 2011
menyatakan bahwa insiden mutasi FLT3 dan NPM1 pada kasus MDS yaitu 2,0%
dan 4,0%, secara berurutan, dan mutasi dibatasi pada kasus yang mempunyai
risiko intermediate dan tinggi terhadap MDS.
Mutasi JAK2 V617F adalah bagian penting untuk diagnostik MPDs,
karena mutasi gen tersebut mempunyai risiko 90% kasus polisitemia vera,
myelofibrosis primer 50% dan trombositopenia. Overlapping diantara MPD dan
MDS dapat dibedakan dengan baik. Beberapa kasus kromosom 5 adalah
komponen proliferatif, sehingga jumlah trombosit dan leukosit meningkat.Pada
penderita MPDs dapat diprediksi lebih kurang 10% didapatkan mutasi JAK2
V617F. Pada RARS dengan trombositosis (RARS – T) mempunyai prevalensi
kurang lebih 66% didapatkan mutasi JAK2 V617F. Mutasi JAK2 adalah subgrup
penderita MDS yang proliferatif pada sumsum tulang, keadaan tersebut
mengeluarkan sinyal proliferatifmenuju ke dysplastic Haematopoeitic(Ceesayet
al., 2012).

46
2.5 Peran gen RUNX1, NPM1, N-RAS, dan delesi kromosom 5
del(5q)pada MDS
Myelodysplastic Syndrome (MDS) adalah grup penyakit yang ditandai
hematopoitik tidak efektif, pada umumnya karena kegagalan yang terjadi di
sumsum tulang (Walteret al.,2012).Perkembangan kasus MDS lebih kurang 30%
menjadi secondary AML. Secara klinik membedakan antara MDS dan AML
dengan memeriksa morfologi aspirasi sumsum tulang sejak pasien di diagnosis
MDS, yaitu dysplastic hematopoietic dan didapatkan myeloblast kurang dari 20%,
sedangkan diagnosis AML mempunyai myeloblast lebih dari 20%. Keadaan
tersebut perlu dipertimbangkan adanya dua kelainan berbeda yaitu abnormalitas
sitogenetik dan molekuler, tetapi kelainan ini tidak dapat memastikan atau
mempredikasi MDS secara menyakinkan berkembang menjadi AML (Walteret al.,
2012).
Latar belakang progresivitas dari cellular dysplasia menjadi kanker
menunjukkan adanya mutasi genetik dan telah banyak dipelajari dengan intensif
pada epitel dan jaringan (contoh: colon dan oropharynx). Pada perkembangan
MDS-related AML sampai sekarang tidak dimengerti kapan dan keadaan mutasi
gen tertentu berkembang dari MDS menjadi secondary AML.Candidate-gene
resequencing diidentifikasi ada beberapa gen yang kemudian menjadi recurrent
mutations selama perkembangan dari MDS menjadi secondary AML antara lain :
Fms-related tyrosine kinase 3 gene (FLT3) risiko mutasi FLT3-ITD 21-
24%;mutasi Nucleophosmin 1 (NPM1) 27,5-35,2%; mutasi runt-related
transcription factor 1 (RUNX1) 5,6%, sitogenetiknormal(CN) or non kompleks
karyotipe AML 32,7%; N-RAS 10,3% dan Tumor protein p53 (TP53) , tetapi
prediksi tentang berapa kali mutasi dan distribusi gen mutasi pada penyakit
sangat terbatas(Walteret al., 2012; Bejaret al., 2011; Takahashi, 2011).

47
Therapy-related (t)-MDS/AML didapatkan sebanyak 10-20% dari kasus
yang didapat. Dua grup besar dikenal sebagai alkylating agentsatau dikaitkan
dengan radiasi dan topoisomerase II inhibitor. Pada grup MDS atau AML
berkembang dalam 5-6 tahun diikuti dari paparan sampai terjadi leukemogenesis
dan risiko akan meningkat dikaitkan dengan total dosis kumulatif paparan. Pada
grup tersebut waktu tercepat periode laten 12-130 bulan, prevalensi dihubungkan
dengan keseimbangan translokasi dan AML dengan sedikit fase dysplastic
(Ceesayet al., 2012).
Mutasi dibagi dalam tiga kelas digambarkan pada (t)-MDS/AML faktor
mutasi kelas satu yaitu melibatkan tyrosine kinase gen RAS atau BRAF pada
jalur transduksi sinyal seperti FLT3, C-KIT, c-FMS atau JAK2, atau downstream
pada RAS-BRAF-MEK, Erk; jalur transduksi sinyal seperti N-RAS, K-RAS, BRAF
atau protein-tyrosine phosphatase (PTPN11/Shp2), terjadi proliferasi tidak
terkendali (Michaudet al., 2008). Mutasi kelas dua yaitu melibatkan inaktivasi
mutasi gen faktor transkripsi hematopoitik seperti RUNX1, NPM1 atau RARα
mengganggu diferensiasi(deferensiasi terhambat) (Takahashi, 2011). Kelas tiga
yaitu mutasi yang melibatkan tumor-supressor gen p53 dilakukan studi yang
serius untuk solid tumors, MDS-related AML.
Pada mutasi genetik tunggal didapatkan insiden t-MDS/AML 20-30%,
pada umumnya kasus dengan mutasi p53 diikuti abnormalitas sitogenetik yang
kompleks, mempunyai prognosis buruk.Fusi bersama terjadi pada mutasi kelas
satu dan kelas dua, tetapi tidak dengan setiap kelas yang berbeda (Ceesayet al.,
2012; Takahashi, 2011; Michaudet al., 2008).
Perubahan keseimbangan homeostatik karena faktor lingkungan makro
(paparan radiasi, kimia) dan mikro (faktor imunitas), dapat mempengaruhi
abnormalitas sitogenetik dan molekuler. Abnormalitas tersebut akan menjadi
gangguan pada siklus hematopoitik yaitu transkripsi PU.1. PU.1 memegang

48
peran penting pada sel progenitor multipotensial, jika terhambat pada tingkat
precursor, hambatan PU.1 menyebabkan proliferation drive karena abnormalitas
molekuler faktor transduksi sinyal FLT3, N-RAS, dan deferentiation arrestterjadi
padaabnormalitas molekuler faktor transkripsi RUNX1, NPM1(Hoffmanet
al.,2013). Deferentiation arrest meningkatkan jumlah myeloblast, jika 3 (tiga) atau
lebih gen terjadi mutasi akan berkembang menjadi AML. Anemia (anemia
megaloblastik, sideroblastik) dan radikal bebas mempengaruhi Apoptosis
(proapoptotik), selanjutnya mempengaruhi sel CD34+ displastik berkembang
menjadi MDS dan mengakibatkan hambatan apoptosis akan menginduksi
transformasi ganas, akhirnya berkembang menjadi AML(gambar 2.10).
Abnormalitas sitogenetik ditemukan pada MDS yaitu delesi kromosom 5 del(5q),
kelainan tersebut didapatkan pada AML dalam insiden (prosentase) yang
berbeda (tabel 2.3), tetapi beberapa translokasi t(15;17), i(16), t(8;21) tidak
didapatkan pada MDS, keadaan ini dapat dipergunakan untuk membantu
mengetahui perubahan MDS-related AML.
Menurut Horriganet al., 1996 (tabel 2.3) menyatakan bahwa pada kasus
MDS dalam proses AML (antara lain t-MDS, RAEB-t) insiden delesi kromosom 5
del(5q) 65-100%, sedangkan kasus MDS yang tidak dalam proses menjadi AML
(antara lain: RAEB) 6-7%. AML (kriteria FAB M0 – M5) tidak didapatkan delesi
kromosom 5 del(5q), (tabel 2.3; 2.4)(Jiang et al.,2009;Horrigan et al., 1996).
Malloet al.,2008,menyatakan bahwa MDS dengan abnormalitas
sitogenetik delesi kromosom 5 del(5q) menggunakan pemeriksaan
FISH(Flourecene In Situ Hybridization) didapatkan 6% kasus del(5q) yang tidak
jelas (tabel 2.4) dibandingkan dengan imunokaryotyping yang mempunyai
akurasi lebih rendah.

49
Pemeriksaan abnormalitas molekuler tunggal yaitu FLT3, NPM1, RUNX1,
CEBPA, N-RAS dan delesi kromosom 5q del(5q)mempunyai risiko 20 – 30%
mutasi tersebut berkembang menjadi MDS-related AML(Walteret al., 2012;
Bejaret al., 2011; Takahashi, 2011;Luzzatto& Kadardimitris, 2010;Alcindor&
Bridges, 2001; Malloet al., 2008).
Tabel 2.2 Membandingkan antara abnormalitas genetik dan sitogenetik
yang ditemukan pada MDS dan AML (dikutip dari Horriganet al.,
1996)

50
Pasien No. Umur/ jenis kelamin
Diagnosis Partial Karyotype* %dengan † del(5)
Hasil PCR ‡
Cases presenting with MDS
1 68/M RAEB – T inv(9)p11q13)c,2-138
dm
-- Tidak hilang
2 28/F RAS del(5)(q13q33), others 6% Tidak hilang
3 54/F RAEBS 46 XX -- Tidak hilang
4 62/M RAEBS 46 XY -- Tidak hilang
5 77/F MDSS del(s)(q15q35) 100% Hilang
6 55/M t-MDSS§ dic(5;17)(q11;p11) 95% Hilang
7 61/M RAEBS 46 XY -- Tidak hilang
8 68/M RAEB del(s)(q13q33),-7,
others
7% Tidak hilang
9 86/M tMDS del(11)(q14,q24) -- Tidak hilang
10 57/F RAEB-T§ -7;+21 -- Tidak hilang
11 74/M MDSS inv(3)(q21q26,
i(17)(q10))
-- Tidak hilang
12 69/F t-MDS§ del(5)(q13q33),
del(12)(q22q24.1)
100% Hilang
13 79/M MDS§ del(5)(q13q35) 65% Hilang
14 65/M RAEB-T§ 46 XY -- Tidak hilang
15 65/F MDSS t(11;19)(q23;p13.3) -- Tidak hilang
16 85/M RAEB del(20)(q11q13) -- Tidak hilang
17 77/M RAEB-T 46 XY -- Tidak hilang
Cases presenting with AML
18 25/M M2 del(9)(q12q22) -- Tidak hilang
19 45/F M6 46 XX -- Tidak hilang
20 47/F M0 -5, -7, others 91% Loss
21 58/F M1 del(7)p(10) -- Tidak hilang
22 47/M M3 t(15;17)(q22q11.2) -- Tidak hilang

51
23 38/F M4Eo inv(16)(p13q22) -- Tidak hilang
24 48/M M5 del(7)(q31q36, t(6;11)) -- Hilang
25 66/M M6 del(5)(q15q33),-7,
others
100% Hilang
26 55/M AML inv(3)(q21q26) -- Tidak hilang
27 64/M M1 46 XX -- Tidak hilang
28 69/M M4 46 XY -- Tidak hilang
29 80/M MO t(14;14)(q11q32),+10 -- Tidak hilang
Keterangan:
Singkatan: MDS(Myelodysplasia); t-MDS, therapy-related-Myelodysplasia (muncul setelah kemoterapi cytotoxic); AML (acute myeloid leukemia); t-AML(acute myeloid leukemia muncul setelah kemoterapi cytotoxic chemotherapy untuk kanker lain); RA refrakter anemia dengan excess blasts; RAEB-t,refrakter anemia with excess blasts dalam transformasi *complete karyotypetidak ditampilkan; hanya yang ditemukan abnormal pada tabel
diatas † prosentase total metaphase yang membawa abnormal chromosome 5 -5, del(5q),
atau dic(5) ketika terdeteksi ‡ hilang: kehilangan heterozygotsity paling sedikit satu penanda informatif, tidak
ada kehilangan: heterozygosity terdeteksi pada penanda informatif. § MDS berkembang menjadi AML
Tabel 2.3 Flourescence In Situ Hybridization dapat memperbaiki pemeriksaan delesi 5q31 pada kasus myelodysplastic Syndrome tanpa membuktikan kelainan 5q- (dikutip dari Mar Mello, et al., 2008)
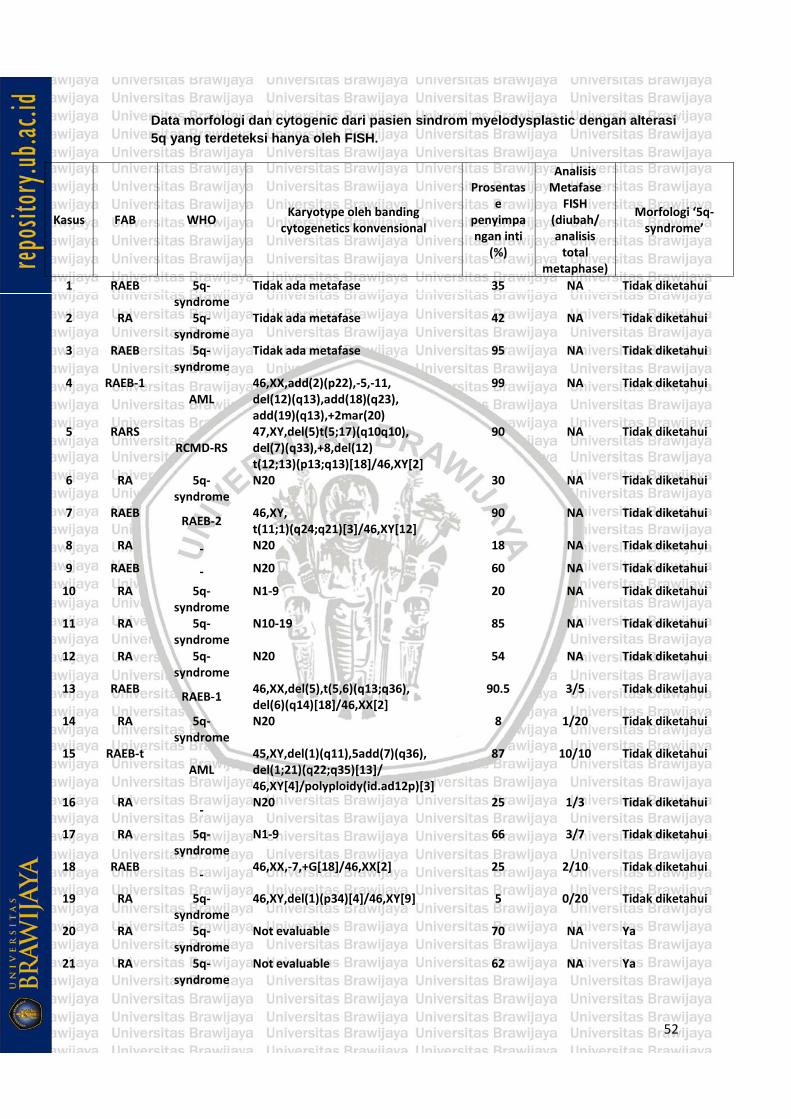
52
Data morfologi dan cytogenic dari pasien sindrom myelodysplastic dengan alterasi
5q yang terdeteksi hanya oleh FISH.
Kasus FAB WHO Karyotype oleh banding
cytogenetics konvensional
Prosentase
penyimpangan inti
(%)
Analisis Metafase
FISH (diubah/ analisis
total metaphase)
Morfologi ‘5q-syndrome’
1 RAEB 5q- syndrome
Tidak ada metafase 35 NA Tidak diketahui
2 RA 5q- syndrome
Tidak ada metafase 42 NA Tidak diketahui
3 RAEB 5q- syndrome
Tidak ada metafase 95 NA Tidak diketahui
4 RAEB-1 AML
46,XX,add(2)(p22),-5,-11, del(12)(q13),add(18)(q23), add(19)(q13),+2mar(20)
99 NA Tidak diketahui
5 RARS RCMD-RS
47,XY,del(5)t(5;17)(q10q10), del(7)(q33),+8,del(12) t(12;13)(p13;q13)[18]/46,XY[2]
90 NA Tidak diketahui
6 RA 5q- syndrome
N20 30 NA Tidak diketahui
7 RAEB RAEB-2
46,XY, t(11;1)(q24;q21)[3]/46,XY[12]
90 NA Tidak diketahui
8 RA - N20 18 NA Tidak diketahui
9 RAEB - N20 60 NA Tidak diketahui
10 RA 5q- syndrome
N1-9 20 NA Tidak diketahui
11 RA 5q- syndrome
N10-19 85 NA Tidak diketahui
12 RA 5q- syndrome
N20 54 NA Tidak diketahui
13 RAEB RAEB-1
46,XX,del(5),t(5,6)(q13;q36), del(6)(q14)[18]/46,XX[2]
90.5 3/5 Tidak diketahui
14 RA 5q- syndrome
N20 8 1/20 Tidak diketahui
15 RAEB-t AML
45,XY,del(1)(q11),5add(7)(q36), del(1;21)(q22;q35)[13]/ 46,XY[4]/polyploidy(id.ad12p)[3]
87 10/10 Tidak diketahui
16 RA -
N20 25 1/3 Tidak diketahui
17 RA 5q- syndrome
N1-9 66 3/7 Tidak diketahui
18 RAEB -
46,XX,-7,+G[18]/46,XX[2] 25 2/10 Tidak diketahui
19 RA 5q- syndrome
46,XY,del(1)(p34)[4]/46,XY[9] 5 0/20 Tidak diketahui
20 RA 5q- syndrome
Not evaluable 70 NA Ya
21 RA 5q- syndrome
Not evaluable 62 NA Ya

53
22 RA 5q- syndrome
Not evaluable 52 NA Ya
23 RAEB RAEB-2
50,XX,+8,+11,add(11),(p13), +mar[15]/46,XX[7]
53.5 NA Tidak
24 RAEB -
49,XY,+2,+3,-7,+12 +mar[12]/46,XY[6]
72.5 NA Tidak
25 RAEB RAEB-2
No metaphases 44.5 NA Ya
26 RA 5q- syndrome
N20 50 3/3 Ya
27 RARS 5q- syndrome
No metaphases 21 NA Ya
28 RAEB RAEB-2
N20 14 1/1 Ya
29 RA 5q- syndrome
N10-19 18 6/7 Ya
30 RA RA
No metaphases 7 NA Ya
31 RA CRDM
No metaphases 30 NA Ya
32 CMML MDS/MPD CMML
46,XY,T(3;5)(p21;q14)[11] 50 NA Ya
33 Unkown -
45,X,-Y[21]/42,XY,5,-7,-8, t(15;?)(p13;?),-16,-17, add(17)(p13),+mar[21]/46,XY[8]
53.5 10/10 Tidak diketahui
34 Unkown -
45,xy,-5,-17,-21, +2mar[21]/46XY[9]
68 5/7 Tidak diketahui
35 Unkown -
47X,add(Y)(q21),-5,-15,-17 ,18,-21, +22,+5mar[22]/46,XY[8]
35 4/10 Tidak diketahui
36 Unkown -
45,XX,-4,-5,-12,-17, +3mar[4]/46,XX,[26]
38.5 3/10 Tidak diketahui
37 Unkown - No metaphases 40 NA Tidak diketahui
38 Unkown N20 6 2/11 Tidak diketahui
39 RAEB+MM
46,XX,add(11)(q25),del(16),del(22) +19,-21,del(22)(q11)(15)/47, XX,der(7),add(11)(q25), del(16)(q22),+19,-21,del(22)(q11), +t(3)/92,xxx,id,+r,+r,+r[1]/46,XX(6)
18 0/8 Tidak diketahui
40 RARS+MM
RCMD- RAS+MM
N20 49 0/2 Tidak diketahui
Keterangan N20: karyotype normal dalam 20 metafase; N10-19: karyotype normal dalam 10-19 metafase; N1-9: karyotype normal dalam 1-9 metafase; NA: tidak tersedia; MM: myeloma multipel. Pasien iniditunjukan olehsitogenetik konvensional, kromosom 5 monosomy kecuali FISH menunjukan delesi 5q. ‘kasus dengan monosomy 5.’ Kasus dengan trisomy 5.
Hubungan antara radikal bebas dengan progresivitas MDS-related AML
(lihat gambar 2.10)

54
Gambar 2.10 Radikal bebas mempunyai pengaruh terhadap peningkatan risiko terjadinya MDS yang berhubungan dengan AML, MDS-related AML.
Keterangan gambar Hubungan antara Radikal bebas dengan apoptosis
a. Stimulus dapat mengawali fase inisiasi melalui aktivitas berbagai reseptor transmembran; Contoh khas dari stimulasi ini adalah pengikatan Fas(CD95) yang merupakan protein homotrimerik dengan FasL, tumor necrotic factor alfa (TNF-α) dengan TNFR dan beberapa yang lain. Pada pengikatan fatty acid synthetase ligand (Fas/FasL) terjadi oligomerisasi dari reseptor yang mengakibatkan bagian intraseluler dari CD95 menggumpal dan dikenal dengan sebutan “death domain”. Protein lain yang kemudian diambil dari sitoplasma dan berfungsi “death domain” adalah Fas associated death domain (FADD) berfungsi mengaktifkan caspase.
b. Untuk mempermudah proses ini molekul FADD mengandung molekul pengikat disebut death effector domain (DED) yang juga dimiliki oleh procaspase-8, dimana keduanya dapat saling berikatan.
c. TNFR tidak mengandung death domain tetapi menggunakan protein TRAF sebagai adaptor sinyal untuk merekurt molekul-molekul transduksi seperti TNF receptor associated death domain (TRADD). Fas mengandung death domain pada bagian intrasitoplasmik dan berinteraksi dengan molekul adaptor sinyal yang mengandung death domain (FADD) dan dengan demikian merekurt molekul transduksi sinyal misalnya FLICE (proapoptosis).
d. Berbagai bukti bahwa pengendalian apoptosis dihubungkan dengan gen yang mengatur siklus, termasuk di antaranya gen p53. Di lain pihak berbagai jenis gen berfungsi anti apoptosis, di antaranya keluarga bcl2 dan beberapa jenis onkogen virus yang dikenal memiliki potensi untuk mengakibatkan transformasi sel menjadi ganas. Gen ini termasuk keluarga gen yang anggota keluarganya makin lama makin bertambah; beberapa anggota keluarga gen ini

55
bersifat anti apoptosis (Bcl2, Bcl-x1, Mc11), tetapi beberapa anggota keluarga yang lain ternyata bersifat pro apotosis (Bax, Bcl-xs, Bid, Bak).
Anemia mempunyai pengaruh terhadap insiden MDS-related AML,
gambar 2.11, defisiensi asam Folat, cycobalamin akan menyebabkan gangguan
terhadap pembentukan DNA, dimana terjadi megaloblatik anemia, transformasi
menjadi AML. Defiensi Fe (zat besi), anemia tersebut menyebabkan transfuse
berulang, sehingga kumulasi zat Besi, trasnformasi menjadi AML.
Gambar 2.11 Anemia mempunyai pengaruh terhadap peningkatan
risiko terjadinya MDS yang berhubungan dengan AML. Keterangan gambar
A. Defisiensi folat dan atau Cobalamin (Cbl) (anemia megaloblastik) 1. Asam Folat diabsorbsi dari usus halus bagian proksimal, 2. Sementara itu ileum Asam Folat masuk dalam sel dalam bentuk
methyltetrahydrofolate (methyl-THF). 3. Cbl ditransfer dan masuk dalam sel bersama dengan transcobalamin II
(TC II). Didalam sitoplasma, reaksi Cbl memerlukan suatu katalis untuk sintesis metionin, grup CH3 dari methyl-THF ditransfer ke homositein, THF dan metionin diproduksi hasilnya berurutan, bentuk Polyglutamated dari THF dikonversi menjadi 5,10-methylene THF, sebagai donor single-carbon grup CH2 yang mana reaksinya memerlukan katalis Thymidylate synthetase

56
4. Pada saat bersamaan dUMP dikonversi menjadi dTMP memerlukan katalis Thymidylate synthetase, selanjutnya dTMP digunakan dalam sintesis DNA.
5. Pada kondisi Cbl tersedia cukup banyak konversi 5,10-methylene–THF menjadi methyl-THF dihambat oleh S-adenosylmethionine (SAM).
6. Pada keadaan difisiensi folat dan atau Cbl, SAM mendapatkan suplai dalam jumlah kecil. Konsekuensinya hambatan SAM menurun, maka perubahan formasi 5,10-methylene-THF menjadi methyl-THF menurun.
7. Keadaan tersebut pada satu pihak menyebabkan proses sintesa dTMP turun secara drastis sehingga tidak tersedia untuk sintesis DNA, dan pada pihak lain dUMP tersedia berlebihan.
8. Para ahli berpendapat bahwa dUMP diubah menjadi dUTP melebihi kapasitas kerja enzim dUTP dalam sel, sehingga melalui konversi kembali menjadi dUMP, akibatnya terjadi penumpukan dUTP di dalam sel, sehingga terjadi kelambatan dalam sintesis DNA, menunjukkan perubahan karakteristik megaloblastik.
B. Anemia sideroblastik 1. Pada keadaan normal kira-kira 90% iron(Fe) tersedia setiap hari dari diet
diambilkan dari eritroblast, dimana biosintesis heme membutuhkan Fe pada proses akhir sintesis, di dalam mitokondria Fe ²⁺( iron ) masuk ring protoporphyrin IX dengan bantuan ferrochelatase.
2. Tahap pertama sampai dengan tahap ketiga lokasinya berada di sitoplasma, keseluruhan tahap biosintesis Heme di mitokondria. Tahap pertama dan tahap selanjutnya terdiri dari kondensasi glycine dan succinyl-CoA menjadi 5-aminolevulinic acid. Reaksi tersebut dikatalisis 5-aminolevulinate syntetase (ALAS) dan reaksi tersebut membutuhkan pyridoxal 5’-phosphate (PLP) sebagai kofaktor. Ekspresi ALAS2 diregulasi oleh dua faktor setinggi transkripsi dan translasi.
3. Kontrol transkripsi yang mengatur program deferensiasi dari erythroid lineage, kemudian mempengaruhi pada eritropoitin menginduksi faktor transkripsi termasuk juga GATA-1. Berbeda dari contoh tipe gene ekspresi pada level kontrol translasi, ALAS2 termasuk mengatur sintesa
Heme yang mana bahan utamanya Fe²⁺. 4. Pusat dari mekanisme regulasi adalah iron protein regulator (IRP)1,
trans-acting protein membutuhkan empat atom Fe untuk berfungsi dan juga dapat berikatan dengan mRNA disebut Iron-responsive element (IRE). Pada saat iron dalam suplai yang minimal, iron kosong pada IRP1 berikatan dengan IRE dan ALAS2 mRNA translasi dalam keadaan menurun maka sintesa heme menurun dan dampaknya hemoglobin mature, tapi menunjukkan produksi sel darah merah hipokromik.
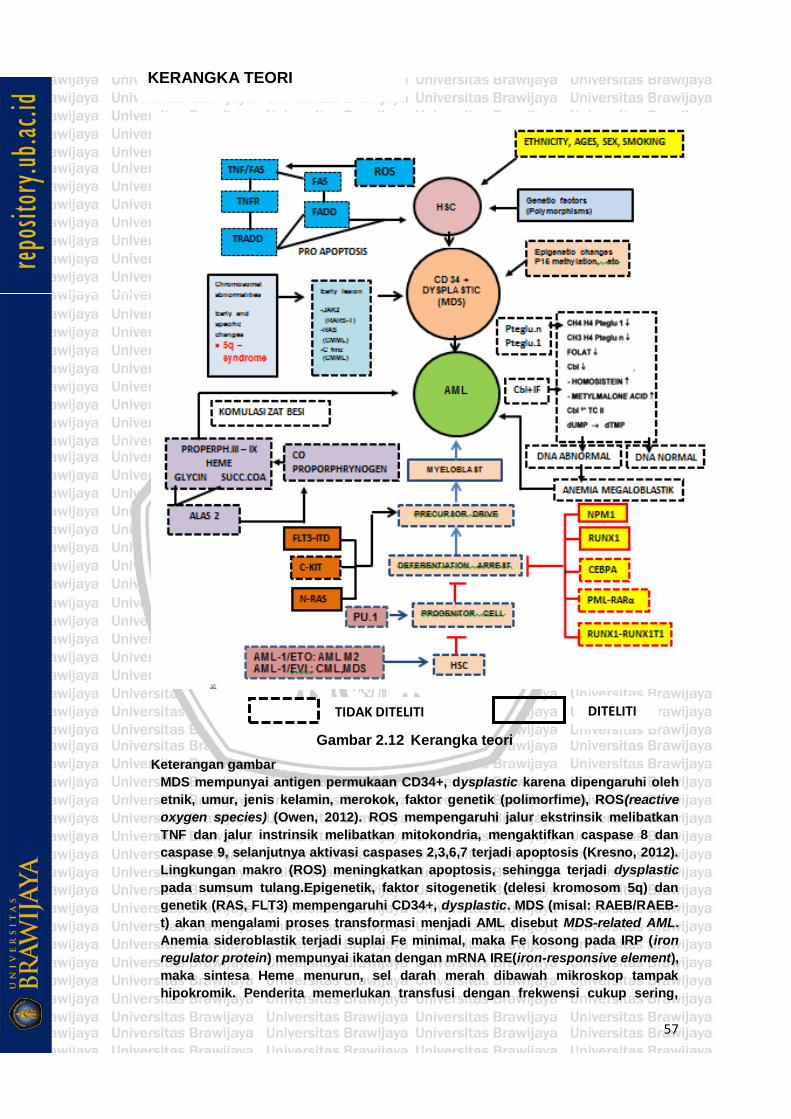
57
Gambar 2.12 Kerangka teori
Keterangan gambar
MDS mempunyai antigen permukaan CD34+, dysplastic karena dipengaruhi oleh
etnik, umur, jenis kelamin, merokok, faktor genetik (polimorfime), ROS(reactive
oxygen species) (Owen, 2012). ROS mempengaruhi jalur ekstrinsik melibatkan
TNF dan jalur instrinsik melibatkan mitokondria, mengaktifkan caspase 8 dan
caspase 9, selanjutnya aktivasi caspases 2,3,6,7 terjadi apoptosis (Kresno, 2012).
Lingkungan makro (ROS) meningkatkan apoptosis, sehingga terjadi dysplastic
pada sumsum tulang.Epigenetik, faktor sitogenetik (delesi kromosom 5q) dan
genetik (RAS, FLT3) mempengaruhi CD34+, dysplastic. MDS (misal: RAEB/RAEB-
t) akan mengalami proses transformasi menjadi AML disebut MDS-related AML.
Anemia sideroblastik terjadi suplai Fe minimal, maka Fe kosong pada IRP (iron
regulator protein) mempunyai ikatan dengan mRNA IRE(iron-responsive element),
maka sintesa Heme menurun, sel darah merah dibawah mikroskop tampak
hipokromik. Penderita memerlukan transfusi dengan frekwensi cukup sering,
KERANGKA TEORI
TIDAK DITELITI DITELITI

58
yang mana akan terjadi Iron overload. Akumulasi besi dalam tubuh akan merusak
fungsi jantung dan cedera organ hepar. Beberapa penelitian pada penderita
anemia akansurvive dan bahkan berhasil hidup beberapa tahun, tetapi 15%
mempunyai risiko berkembang menjadi akut leukemia (Luzzatto and
Kadardimiris, 2010; Alcindor and Bridges, 2001). Demikian juga anemia
(defisiensi Folat dan Cobalamin), defisiensi Folat meningkatkan homosistein,
methylmalonyl-CoA (Methylmalonic acid) dan difisiensi Cobalamin menurunkan
kemampuan dUMP menjadi dTMP, dUTP meningkat.DNA normal menurun, dTMP
menurun karena defisiensi Folat dan Cobalamin (anemia megaloblastik). Anemia
megaloblastik adalah tanda langsung dari proses hematopoisis yang inefektif
dari sumsum tulang. Hematopoitic stem cell (HSC) dipengaruhi mutasi gen
tunggal atau kompleks (AML-1/ETO, AML-1/EVI) menghambat fungsi sel
progenitor, PU.1 mempengaruhi sel progenitor yaitu menghambat fungsi
prekursor maka terjadi proliferasi meningkat dan deferensiasi menjadi lambat.
Prekursor dihambat oleh mutasi gen tunggal misal: N-RAS, FLT3, dan NPM1,
RUNX1 deferensiasi menjadi lambat, proliferasi meningkat (jumlah myeloblast
meningkat) (Owenet al., 2012).

60
BAB 3
KERANGKA KONSEP DAN HIPOTESIS PENELITIAN
3.1 Kerangka Konsep
Gambar 3.1 Kerangka Konsep

61
Keterangan kerangka konsep MDS mempunyai antigen permukaan sel CD34+, dysplastic karena
dipengaruhi oleh etnik, umur, jenis kelamin, merokok, faktor genetik (polimorfime), reactive oxygen species (ROS), kimia(benzene) (Owen et al., 2012). Lingkungan makro (ROS) meningkatkan apoptosis (proapoptosis) maka sel yang diprogram mati cukup banyak sehingga terjadi dysplastic pada sumsum tulang. Lingkungan makro yang lain adalah metabolit benzena (C6H6) yang paling poten menghambat erythropoiesis. Epigenetik, abnormalitas faktor sitogenetik: delesi kromosom 5 del(5q) dan genetic: RAS, FLT3 menyebabkan hematopoisis yang mempunyai organisasi yang baik dan efektif menjadi dysplastic dan tidak efektif mempengaruhi sel CD34+, dysplastic mendorong peran transformasi menuju proses AML (Ceesay et al., 2012; Gilliland & Gribben, 2012). MDS (RAEB/RAEB-t, klasifikasi FAB) akan mengalami proses transformasi menjadi AML disebut MDS-related AML. Mutasi gen tunggal (FLT3-ITD, NRAS) atau kompleks mempengaruhi Hematopoietic stem cell (HSC) menghambat fungsi sel progenitor, maka deferensiasi menjadi lambat sedangkan mengahambat precursor akan meningkatkan proliferasi. Mutasi gen tunggal NRAS, RUNX1, NPM1, FLT3 menyebabkan deferensiasi sel melambat dan proliferasi meningkat, karena supresi apoptosis (antiapoptosis) jumlah myeloblast meningkat evolusi menjadi keganasan (AML) (Owen et al., 2012).
3.2 Hipotesis penelitian
Polimorfisme gen NRAS, RUNX1, NPM1, FLT3, delesi kromosom 5
del(5q) dan kromosom 5 (normal) dapat sebagai prediktor perubahan
Myelodisplastic Syndrome related Acute Myeloid Leukemia (MDS-related
AML) di Surabaya.

62
BAB 4
METODE PENELITIAN
4.1 Jenis dan Desain Penelitian
Rancangan penelitian ini adalah cross sectional analytic, gen RUNX1,
NPM1, FLT3, N-RAS, dan delesi kromosom 5 del(5q) sebagai prediktor
Myelodisplastic SyndromerelatedAcute Myeloid Leukemia (MDS-related AML) di
Surabaya.
4.2 Waktu dan tempat Penelitian
Penelitian dilaksanakan dalam waktu 11 (sebelas) bulan, dari bulan Mei
2017 sampai dengan bulan April 2018 (lampiran 1).Pengambilan sampel
penelitian dan penentuan MDS dan AML dilaksanakan di LaboratoriumInstalasi
Ilmu penyakit Dalam bagian Hematologi-onkologi Fakultas Kedokteran
Universitas Airlangga, RSUD dr Soetomo, Surabaya, Indonesia.Pemeriksaan
Immunophenotyping dilaksanakan di laboratorium Instalasi Patologi Klinik
Fakultas Kedokteran Universitas Airlangga, RSUD dr Soetomo, Surabaya,
Indonesia. Pemeriksaan gen dan kromosom dilaksanakan di laboratorium
Instalasi Biomedik Fakultas Kedokteran Universitas Brawijaya, Malang,
Indonesia.
4.3 Populasi dan sampel penelitian diduga AML
Populasi dalam penelitian adalah semua pasien yang diduga AML yang
datang memeriksakan darah di Laboratorium InstalasiPatologi Klinik Fakultas
Kedokteran Universitas Airlangga, RSUD dr Soetomo, Surabaya, Indonesia,
rawat Inap di Ilmu Penyakit Dalam bagian Hematologi-Onkologi FK Universitas

63
Airlangga, RSUD Dr. Soetomo, Surabaya. Sampel penelitian adalah penderita
AML yang memenuhi kriteria penelitian.
4.3.1 Kriteria inklusi:
4.3.1.aPasien baru AML umur (14 - 77) tahun .
4.3.1.b Bersedia mengikuti penelitian dengan memberikan persetujuan
inform concern.
4.3.2 Kriteria Eksklusi :
4.3.2.aSampel pasien yang mengalami kerusakan data selama penelitian
berlangsung dan tidak mungkin lagi pengambilan data ulang.
4.3.2.bMempunyai penyakit yang mempengaruhi sampel antara lain:
keganasan, anemia makrositer, HIV positif.
4.3.3 Besar sampel
Besar sampel pada penelitian ini dihitung berdasarkan rumus karena
pertimbangan:
4.3.3.aKomparasi
4.3.3.bData kuantitatif
Pilihan rumus besar sampel uji hipotesis komparatif antar
mean/rerata(Windhu & Taufan, 2018).

64
4.3.4 Cara Pengambilan Sampel
4.3.4.1 Sampel diambil yaitu darah perifer, hapusan, pewarnaan Wright,
diperiksa dengan mikroskop cahaya Olympus CX-41, di di Instalasi
penyakit Dalam divisi Hematologi-onkologi RSUD Dr Soetomo
Surabaya, bila pada hapusan darah tepi ditemukan blast lebih dari
20% dilakukan aspirasi sumsum tulang.
4.3.4.2 Aspirasi sumsum tulang, morfologi, pewarnaan Wright, menggunakan
mikroskop cahaya Olympus CX-41 di Instalasi penyakit Dalam divisi
Hematologi-onkologi RSUD Dr Soetomo Surabaya.
4.3.4.3 Sampel yang diambil yaitu aspirasi sumsum tulang, isolasi DNA untuk
pemeriksaan gen NRAS ,RUNX1, NPM1, FLT3 mengunakan PCR
(Polymerase Chain Reactions), di departemen biokimia dan biologi
Molekuler, Fakultas Kedokteran Universitas Brawijaya, Malang.
4.3.4.4 Sampel yang diambil yaitu aspirasi sumsum tulang, isolasi DNA untuk
pemeriksaan kromosom, yaitu Delesi kromosom 5 del(5q)
menggunakan CISH (Chromogen In Situ Hibridization)di departemen
biokimia dan biologi Molekuler, Fakultas Kedokteran Universitas
Brawijaya, Malang.

65
4.4 Variabel penelitian
Variabel yang digunakan dalam penelitian ini adalah:
4.4.1 Variabel tergantung: MDS-related AML {delesi kromosom 5 del(5q).
4.4.2 Variabel bebas: N-RAS, RUNX1, NPM1, FLT3.
4.5 Definisi Operasional
4.5.1 MDS-related AML
MDS-related AML adalah AML yang ada hubungannya dengan MDS
yang mengalami transformasi.AML adalah penyakit heterogen yang terdiri
dari berbagai macam gambaran klinik dan kelainan genetik, termasuk
mutasi sitogenetik, mutasi gen dan perubahan ekspresi gen, diagnosis
klinik, Immunophenotyping, morfologi sel sumsum tulang (myeloblast >
20%), menggunakan klasifikasi FAB.MDS-related AML adalah kasus
AML yang sebelumnya menderita MDS.
Cara perolehan : pasiendidapatkan dari Rawat Inap Instalasi Ilmu
penyakit Dalam bagian Hematologi-onkologi Fakultas Kedokteran
Universitas Airlangga, RSUD dr Soetomo, Surabaya dengan cara
mengmbil aspirasi sumsum tulang pemeriksaan hapusan, mikroskop
cahaya Olympus CX-41, diperiksa oleh 3 pembaca yang berkompeten,
pembesaran 400 kali, interpretasi blast (sel muda) yaitu lebih dari 20%.
Pemeriksaan yang lain yaitu delesi kromosom 5 del(5q) menggunakan
CISH (chromogen In Situ Hybridization), cara ukur menggunakan
mikroskop cahaya E-100 NIKON, positif yaitu tampak warna “Brownish”.
Satuan/Unit Hapusan aspirasi sumsum tulang blast lebih dari 20%, hasil
pemeriksaan Immunophenotyping yaitu Myeloid leanage, hasil CISH:
positif berarti terjadi delesi kromosom 5 del(5q), negatif berarti kromosom
5 normal (Hoffbrand & Moss, 2012).

66
4.5.2 MDS (Myelodysplastic Syndrome)
MDS adalahsekelompok kelainan haematopoeitic stem cells
(HSC) yang ditunjukkan dengan kegagalan sumsum tulang (displastik)
yang meningkat, dikorelasikan dengan abnormalitas kuantitatif dan
kualitatif sel pada darah perifer, sumsum tulang, diagnosis klinik.
Menggunakan klasifikasi French American British (FAB), WHO 2008 yaitu
Refrakter anemia, morfologi blast ditemukan < 20%, abnormalitas
kromosom: delesi kromosom 5 del(5q).
Cara perolehan: gejala klinik, aspirasi sumsum tulang, hapusan
sumsum tulang, pemeriksaan morfologi menggunakan mikroskop cahaya
Olympus CX-41, yaitublast kurang dari 20%, pemeriksaan lain
Immunophenotyping hasil interpretasi non myeloid lineage, pemeriksaan
CISH: positif pada hapusan tampak warna “brownish”, berarti terjadi
delesi kromosom 5 del(5q), negatif berarti kromosom 5 normal (Hoffbrand
& Moss, 2012).
4.5.3 NRAS (Rat sarcoma)
NRAS adalah Mutasi pada semua gen RAS kromosom
homologus, N-RAS, KRAS dan HRAS, menempati codon 12, 13 dan
61.Semua protein RAS (p21ras), NRAS adalah anggota keluarga RAS
superfamily dari GTPase. Aktivasi mutasi exon 2 (codon 12/13)(1p13.2),
exon 3 (codon 61), and exon 4 (codon 146) antara lain ditemukan pada
Myeloid leukemia (14%) (Colicelli, 2004).
Mutasi NRAS adalah mutasi RAS yang paling sering pada AML,
terdeteksi antara 10% - 30% dari kasus (Owen & Fitzgibbon, 2012;
Hoffman et al., 2013).

67
Cara perolehan: pengambilan Aspirasi Sumsum Tulang. Cara
ukur isolasi DNA, pemeriksaan dengan menggunakan PCR (Polymeraase
Chain Reactions) yaitu Denaturasi, Annelling, ekstensi. Satuan/unit:
tampak band (pita), Positif berarti mutasi gen NRAS.
4.5.4 RUNX1
RUNX1 adalah regulator utama pada hematopoisis dan sering
menjadi target translokasi kromosom dihubungkan dengan leukemia
(Owen and Fitzgibbon, 2012).
Cara perolehan: pengambilan Aspirasi Sumsum tulang. Cara ukur
isolasi DNA, penguji menggunakan PCR yaitu Denaturasi, Annelling,
ekstensi. Satuan/unit: tampak band (pita): Positif berarti mutasi gen
RUNX1.
4.5.5 NPM1
NPM1 (Nucleophosmin member 1) adalah mutasi gen terjadi
pada exon 12, dengan prosentase lebih dari 95% terdiri dari 4-bp insersi
di posisi 960. NPM1 lokasi pada kromosom pita 5q35.
Mutasi NPM1 ditemukan pada kira-kira 35% penderita dewasa
dengan AML dan sering terjadi abnormalitas genetik ditemukan pada
AML dengan kariotipe normal (CN), Jumlah kasus AML dengan kariotipe
normal antara 60 – 80% (Hoffman et al., 2013; Falini & Bolli, 2009).Mutasi
FLT3-ITD menunjukkan prognosis lebih baik dibandingkan genotip mutan
NPM1 disertai mutasi FLT3-ITD (Owen & Fitzgibbon, 2012, Young, 2012,
Tan et al., 2008).
Cara perolehan: pengambilan Aspirasi Sumsum tulang. Cara ukur
isolasi DNA, penguji menggunakan PCR yaitu Denaturasi, Annelling,

68
ekstensi. Satuan/unit: tampak band (pita): Positif berarti mutasi gen
NPM1.
4.5.6 FLT3
FLT3(Fms-like tyrosine kinase 3) adalah reseptor tirosin kinase
yang mempunyai peran penting dalam mengontrol kelangsungan hidup
sel, proliferasi dan diferensiasi sel hematopoitik.Gen FLT3 mempunyai
fungsi untuk meningkatkan proliferasi sedangkan pada mutasi gen FLT3
proliferasi meningkat tidak terkendali.Lokasi gen FLT3 disebut juga FLK2
(fetal Liver Kinase-2), hSTK1 (human stem Cell Kinase-1) pada pita
kromosom 13q12, struktur ikatan membran berhubungan dengan reseptor
tyrosine kinase kelas III seperti KIT, Fms, dan PDGFR (Platelet-derived
growth factor). Ligan FLT3 dieskpresikan oleh sel stroma pada sumsum
tulang, dan faktor pertumbuhan (growth factors) untuk menstimulasi
proliferasi sel punca, sel progenitor, sel dendrit, sel NK. Gen FLT3
Internal tandem duplication (ITD), terbentuk dengan FLT3 domain
juxtamembrane (exon 14 dan 15), diprediksi merupakan mutasi
terbanyak, diperkirakan 30% dari kasus AML (Bain, et al., 2011; Owen &
Fitzgibbon, 2012).
Insiden kasus mutasi FLT3-ITD pada diagnosis MDS secara
signifikan lebih kecil dibandingkan dengan kasus MDS-related AML.Pada
kasus yang ditemukan mutasi FLT3-ITD mempunyai progresivitas
menjadi AML dalam waktu lebih cepat dibandingkan kasus yang FLT3-
ITD normal.
Menurut penelitian Agersborg et al., 2015 bahwa mutasi FLT3,
NPM1, dan WT1 adalah abnormalitas molekuler yang dapat dipergunakan
sebagai kriteria diagnosis pasien AML yang obyektif lebih baik dari pada
aspirasi sumsum tulang.

69
Cara perolehan: pengambilan Aspirasi Sumsum Tulang. Cara
ukur isolasi DNA, pemeriksaan dengan menggunakan PCR (Polymeraase
Chain Reactions) yaitu Denaturasi, Annelling, ekstensi. Satuan/unit:
tampak band(pita): Positif berarti mutasi gen FLT3-ITD.
4.5.7 Delesi kromosom 5q atau kromosom 5 normal
Delesi kromosom 5 del(5q) adalah delesi pada kromosom 5
mempunyai segmen lengan panjang del(5q). Pada MDS abnormalitas
kromosom dapat sebagian atau keseluruhan yang hilang, pada umumnya
- 5, 5q -, - 7, 7q -, 11q -, 13q -, 20q - dan -Y . Jarang material kromosom
bertambah dan maksimal terbesar +8, contoh: trisomy 8 (+8) adalah
penambahan kromosom yang sering terjadi pada MDS. Keadaan tersebut
mempunyai nilai prognostik dan kontribusi untuk patogenesis MDS,
kromosom yang hilang akan menjadi pertimbangan karena menyebabkan
tumor-suppressor genes atau haploinsufficiensyofgenes diperlukanuntuk
hematopoisis yang normal (Montalban-Bravo & Gracia-Manero, 2018).
Cara perolehan: pengambilan Aspirasi Sumsum tulang. Cara Ukur:
Pemeriksaan yang lain yaitu delesi kromosom 5 del(5q) menggunakan
CISH (chromogen In Situ Hybridization), cara ukur menggunakan
mikroskop cahaya E-100 NIKON. Satuan/Unit: Positif: Delesi kromosom 5
del(5q), hapusan tampak warna “Brownish” , negatif: kromosom 5
normal.
Bahan dan metode pemeriksaan

70
4.6.1 Hapusan darah tepi
4.6.1.aPengambilan darah vena, menggunakan antikoagulan EDTA atau
langsung dibuat hapusan pada deck gelas.
4.6.1.bDikeringkan, kemudian diberikan pewarnaan Wright, merata sampai
menutup hapusan darah, selama 5 menit, diberikan buffer 20 menit
kemudian dicuci dengan air mengalir.
4.6.1.cDibaca dengan mikroskop Olympus seri CX-41 (Bainet al., 2012)
4.6.1.dHasil hapusan darah perifer atau aspirasi sumsum tulang dibaca 3(tiga)
orang spesialis Patologi Klinik dan Penyakit dalam konsultan
Hematologi.
4.6.2 Aspirasi Sumsum Tulang (Bonemarrow Aspiration)
4.6.2.aPengambilan sampel aspirasi sumsum tulang menggunakan jarum tipe
SALAH dengan GUARD (kontrol kedalaman) (Bainet al., 2012).
4.6.2.bDesinfeksi (betadine dan alkohol), premedikasi, anastesi lokal, jarum
dan mandrin, spuit, desinfektan dan verband, deck glass, pewarnaan.
4.6.2.cDilihat dibawah mikroskop Olympus seri CX-41.
4.6.2.d Hasil hapusan darah perifer atau aspirasi sumsum tulang dibaca
3(tiga) orang spesialis Patologi Klinik dan Penyakit dalam konsultan
Hematologi.
4.6.3 Pemeriksaan fenotipe AML (Bain et al., 2012)
4.6.3.aFlow Immunophenotyping metode yang dipergunakan untuk penentuan
immunophenotype sel menggunakan teknik flow cytometry. Hal ini
sangat berguna untuk mendiagnosis berbagai jenis leukemia
(Matutes et al., 2012).

71
4.6.3.b Pemeriksaan fenotipe dengan menggunakan campuran antibody
monoclonal CD7 FITC CD33 PE/CD7/CD33 BD, HLA-DR
FITC(isothocyanate)/CD13 PE, CD45 (2D1) Per /CP BD, CD34 (8G12)
PE BD, ANTI MPO (myeloperoxidase) (5B8) FITC BD. Mesin
flowsitometer yang dipergunakan: BD Facs Calibur, BD FACSort, atau BD
FACScan Flow Cytometer.Tabung satu berisi FITC CD7/CD33, tabung
dua berisi FITC HLA-DR/CD13, tabung tiga berisi PE CD34/ANTI-
MPO.Masing-masing tabung (satu, dua, tiga) ditambahkan PE CD45 (Kit
Manual BD test). Prosedur pemeriksaan sebagai berikut:
1. CD 45 sebanyak 20 µl dimasukkan ke dalam tabung falcon 5 ml :
satu, dua, tiga, kemudian ditambahkan sampel sebanyak 50 µl,
selanjutnya diinkubasi suhu kamar pada ruang gelap selama 30
menit.
2. Ditambahkan lysing solution Cat.No.349202 LOT: 6204937 sebanyak
1 ml, vortex, kemudian dilanjutkan dengan inkubasi suhu kamar pada
ruang gelap selama 10 menit.
3. Campuran tersebut diatas, dilakukan sentrifugasi dengan kecepatan
250 g (1.800 rpm) selama 5 menit, selanjutnya supernatan dibuang
dengan menuangnya secara cepat pada posisi 180º. Pada pellet
didasar tabung ditambahkan PBS (Phosphate Buffered saline
solution)/Facs Flow sebanyak 500 µl, dicampur kemudian sentrifugasi
250 g selama 5 menit. Tabung satu dan dua dibaca pada
flowsitometer.
4. Tabung tiga ditambahkan cytofix/cytoperm (Kit Cytofix/Cytoperm
buffer dari BD) sebanyak 500 µl dan diinkubasi pada tempat gelap
pada suhu kamar selama 20 menit.

72
5. Selanjutnya pencucian dengan menambahkan Perm/wash buffer dari
BD: FBS (Fetus Bovine Serum) sebanyak 1 ml dan sel dicampur
merata dan dilakukan sentrifugasi 250 g (1800 rpm) selama 5 menit.
Setelah sentrifugasi, supernatan dibuang dengan menuangnya secara
cepat pada posisi 180º.
6. Selanjutnya ditambahkan ANTI-MPO sebanyak 20 µl, kemudian
diinkubasi pada tempat gelap dalam suhu kamar selama 30 menit.
7. Campuran tersebut diatas ditambahkan Perm/Wash buffer dari BD
sebanyak 1ml, kemudian dilakukan sentrifugasi 250 g selama 5 menit.
Selanjutnya ditambahkan Formaldehid 1% dengan pengenceran PBS
(Phosphat Buffered Saline Solution) sebanyak 500 µl.
Kemudian dibaca dengan alat flowsitometer dengan software Cellquest
“Pro”, dilakukan gating dari populasi blast pada CD45.
Hasil: bila CD34+/MPO
+/CD13+/CD33
+ berarti fenotipe AML.
4.6.4 Polymerase Chain Reaction (PCR) dipergunakan untuk pemeriksaan
Gen NRAS, RUNX1, NPM1, FLT3.
4.6.4.a Primer: (Lampiran 7)
Informasi primer PCR (NRAS, RUNX1, NPM1, FLT3), peneliti
mendapatkan bahan material dan prosedur pemerikasaan (Genetika
Science Indonesia, jalan Lingkaran luar Barat, Kembangan, Jakarta
Barat, 11610, Email [email protected]).

73
Tabel 4.1 Informasi primer PCR (NRAS, RUNX1, NPM1, FLT3) yang
dipergunakan dalam penelitian ini
GEN SUSUNAN ASAM AMINO
1 N_RAS_F N_RAS_R
31-mer oligo (25 nmole scale, PCR Grade, 3 OD) GAC TGA ATA TAA ACT TGT GGT AGT TGG ACC T 28-mer oligo (25 nmole scale, PCR Grade, 3 OD) ACC AAG ATT TAC CTC TAT TGT TGG ATC A
2 RUNX1_F
RUNX1_R
22-mer oligo (25 nmole scale, PCR Grade, 3 OD)
TAC AGC CAA TCT GCA CTG TGC T
22-mer oligo (25 nmole scale, PCR Grade, 3 OD)
CAC CTG TGT GGA ACA GAT CTC C.
3 NPM1_F NPM1_R
27-mer oligo (25 nmole scale, PCR grade, 3 OD) CTG ATG TCT ATG AAG TGT TGT TCC
26-mer oligo (25 nmole scale, PCR Grade, 3 OD) CTC TGC ATT ATA AAA AGG ACA GCC AG
4
FLT3-ITD_F FLT3-ITD_R
16-mer oligo (25 nmole scale, PCR grade, 3 OD) AGG AGG GCA ACT ACT T
21-mer oligo (25 nmole scale, PCR Grade, 3 OD) CTT CAC TTG AAT TGG TAG CAT
Keterangan: F : forward, R : reverse
4.6.4.bPrinsip: PolymeraseChain Reaction (PCR) adalah proses replikasi
bahan genetik dengan menggunakan enzim DNA polymerase. Enzim ini
digunakan untuk memulai sintesis DNA dari DNA primer pada
template.DNA primer terdiri dari 9 – 20 basa dan menentukan sisi dimana
replikasi DNA dimulai. Dengan PCR tiap rangkaian bahan genetik khusus
dapat ditunjukkan dan direplikasi beberapa kali secara mudah dengan
memilih sepasang primer yang akan menjadi pangkal dari rangkaian DNA
yang diinginkan. PCR diprediksi pada proses "anneling” dari kedua
oligonukleotida (primer) untuk mengetahui komposisi dari rangkaian suatu
target oligonukleotida yang diinginkan dan diperpanjang dengan bantuan
enzim DNA polymerase. Tiap-tiap reaksi diulang-ulang mulai dari tahap
denaturasi sampai tahap ekstensi, sehingga akan bertambah secara
eksponential (Bain et al., 2012, Loegito, 2007).

74
4.6.4.cAlat atau reagen
CI0L c-PAGE® 10% 12 wells 10 plates/box, Ex Run (Electrophoresis
Bufferfor SDS PAGE), Agarose-Biotechnology Grade – 100 g, 10X Tris-
Acetate-EDTA (tae) buffer-pH 8,0 Ultra pure Grade – 1L, Genezol
Reagen 100 ml, KODFX Neo, BstNI, 3000 unit, Proteo Silver Silver Stain
Kit, Gen NRAS, RUNX1, NPM1, FLT3
4.6.4.dProsedur pemeriksaan PCR
4.6.4.d.1Isolasi/ekstraksi DNA
1.1 Proses isolasi/ekstraksi DNA dilakukan dengan penambahan buffer
AL yang dilakukan setelah penambahan proteinase K dimana
bersama-sama dengan proteinase K berfungsi untuk melisiskan sel
sehingga dinding sel rusak. Supernatan yang dihasilkan dari proses
sentrifugasi suspensi tersebut dimasukkan ke dalam kolom mini
berfilter sehingga DNA tersangkut/terikat di dalamnya.
1.2 Penambahan buffer AW1 dan buffer AW2 yang dilakukan kedalam
kolom mini berfilter yang berfungsi untuk pencucian dimana proses
pencucian tersebut dapat meningkatkan kemurnian DNA nantinya
serta meyakinkan penghilangan kontaminan secara keseluruhan
dari proses tersebut tanpa mempengaruhi pengikatan DNA pada
filter.
1.3 Penambahan buffer AE ke dalam kolom mini dan dilakukan proses
sentrifugasi setelah proses pencucian berfungsi untuk mengelusi
DNA dari filter dan juga berfungsi dalam penyimpanan purifikasi
DNA. Supernatan yang dihasilkan merupakan isolat DNA yang
kemudian disimpan di dalam freezer suhu -20º C.

75
1.4 Teknis pengisolasian DNA dengan kit komersial pertama-tama
adalah satu ml suspensi dimasukkan ke dalam tabung
mikrosentrifus. Kemudian ditambahkan dengan 1 ml
CTAB(cetyltrimetylammonium bromide) sebagai suatu modifikasi
metode dari prosedur yang ditunjukkan pada handbook produsen
kit, kemudian divortex hingga homogen.Suspensi tersebut
disentrifus selama 1 menit dengan kecepatan 13000 rpm dan
supernatan yang dihasilkan dibuang sehingga menyisakan pelet
pada tabung mikrosentrifus.
1.5 Pelet tidak dihasilkan, maka suspensi disisakan sebanyak 200 µl
supernatan pada tabung mikrosentrifus. Kemudian ditambahkan
proteinase K sebanyak 30 µl ke dalam tabung mikrosentrifus, dan
dihomogenkan dengan vortex kemudian diinkubasi selama 20 menit
pada suhu 65º C.
1.6 Setelah itu, ditambahkan buffer AL sebanyak 300 µl dan
dihomogenkan dengan vortex. Suspensi diinkubasi selama 10 menit
di dalam water bath pada suhu 65º C. Setelah itu, ditambahkan 500
µl etanol (96-100%), dihomogenkan dengan vortex dan disentrifus
selama 2 menit dengan kecepatan 10000 rpm.
1.7 Supernatan yang dihasilkan dipipet dan dimasukkan ke dalam
kolom mini yang sudah dipasang pada collection tube, kemudian
disentrifus 8000 RPM selama 1 menit.
1.8 Sebanyak 500 µl buffer AW1 ditambahkan ke dalam kolom mini
yang masih terpasang pada collection tube dan disentrifus 8000
rpm selama 1 menit.

76
1.9 Tahap selanjutnya adalah kolom mini dipindahkan ke dalam
collection tube yang baru dan ditambahkan dengan 500 µl buffer
AW2 ke dalam kolom mini, kemudian disentrifus 13000 rpm selama
3 menit.
1.10 Kolom mini dipindahkan kembali ke dalam collection tube dan
disentrifus kembali selama 1 menit 13000 rpm. Kolom mini
dipindahkan ke dalam tabung mikrosentrifus steril dan ditambahkan
ke dalamnya sebanyak 80 µl buffer AE yang kemudian disentrifus
8000 rpm selama 1 menit.
1.11 Supernatan yang diperoleh pada tabung mikrosentrifus
merupakan isolat DNA yang akan digunakan pada tahap
selanjutnya dan isolat tersebut disimpan pada freezer dengan suhu
20º C (QIAamp® DNA Blood Mini Kit Handbook dengan modifikasi
Bioteknologi PROM).
4.6.4.d.2. Pengujian dilakukan dengan PCR.
Konsentrasi primer yang sesuai/tepat adalah konsentrasi primer
yang menghasilkan nilai threshold cycle (Ct) yang paling rendah dan tanpa
menghasilkan atau seminimal mungkin menghasilkan primer-dimer pada
kurva puncak pelelehan (Pestana, 2010).
1. PCR (Polymerase Chain Reaction) diambil 2 μL RT reaction product
pada PCR tube tambahkan 12,5 μL green Go-Taq, 1nM primer
forward dan reverse (Primer dalam lampiran)
2. Tambahkan nuclease free water sampai volume menjadi 25 μL.
3. PCR reaksi dilakukan :
3.1 Tahap I dengan 1 siklus pada 94⁰C selama 5 menit

77
3.2 Tahap II dengan 30 siklus, terdiri dari 94⁰C selama 30 detik, 58⁰C
selama 30 datik dan 72⁰C selama 1 menit.
3.3 Tahap III dengan 1 siklus pada 72⁰C selama 10 menit.
4.6.5 Analisis kromosom dan CISH (Chromic In Situ
Hybridization)Pemeriksaan dengan CISH dapat dilakukan pada sel atau
jaringan tanpa harus membuat sel berada dalam stadium metafase (sukartini,
2013, Bain et al., 2012).
Pemeriksaan sitogenetik yaitu Delesi kromosom 5 del(5q) pada
dasarnya melakukan analisis kromosom.
Reagen CISH :del(5q) Deletion Probe
Metode CISH (Chromogenic in-situ Hybridization) delesi kromosom 5 del
(5q)
1. Slide sel dicuci dengan HEPES buffer dan keringkan,
2. tambahkan DNase dalam 2x SSC buffer dan inkubasi pada 37oC selama
1 jam.
3. Cuci dengan dH2O steril dan bilas dengan 2X SSC buffer selama 10
menit.
4. Refiksasi dengan 4% paraformaldehide 10 menit pada suhu ruang,
5. selanjutnya bilas dengan dH2O steril selama 10 menit dan dehidrasi
dengan alcohol berseri dari 70% sampai 100%, masing-masing selama
10 menit.
6. Keringkan pada 37°C selama 10 menit.
7. Masing-masing reaksikan dengan 10uL cDNA (probe 5delq) yang telah
berlabel biotin.

78
8. Slide didenaturasi pada 85oC selama 10 menit, dan diinkubasi pada 37oC
semalam untuk proses hibridisasi.
9. Cuci slide dengan washing solution pada 55oC selama 45 menit dan
diinkubasi pada washing solution selama tiga kali 60 menit.
10. Inkubasi pada 2 X SSC buffer 15 menit pada suhu ruang.
11. Inkubasi pada 0,1 X SSC buffer pada 50oC selama 15 menit dan
12. lanjutkan inkubasi pada buffer yang sama selama 30 menit pada suhu
ruang.
13. Dehidrasi kembali slide menggunakan etanol seri dari 30% sampai 95%,
yang masing-masing mengandung 0,3 M ammonium asetat, dan etanol
absolut, masing-masing selama 1 menit.
14. Cuci dengan TNT buffer, ulangi selama tiga kali, masing-masing selama 5
menit.
15. Kemudian diblocking menggunakan TNB buffer semalam.
16. Reaksikan dengan SA-HRP (1:500 dalam TNB buffer), selama 30 menit
dalam suhu ruang.
17. Cuci dengan TNT buffer, tiga kali, masing-masing selama 15 menit.
Kemudian
18. Inkubasi dalam chromogen – DAB 10 menit, dan bilas dengan TNT buffer,
tiga kali, masing-masing selama 15 menit.
19. Serap dengan kertas tissue sisa TNT buffer.
20. Conterstaning dengan mayer hematoxilen selama 10 menit dan
21. bilas dengan tap water.
22. Kering-anginkan slide dan teteskan entelan dan tutup dengan cover
glass. Amati pada mikroskop cahaya Olympus CX-41 dengan
pembesaran 400X (sukartini, 2013, Bain et al., 2012).

79
4.6 Kerangka Operasional
Gambar 4.1 Kerangka operasional
Keterangan gambar Pasien diduga menderita AML, MDS-related AML diberikan penjelasan dan
memberikan tanda tangan pada inform concent. Pasien diambil darah tepi dan
aspirasi sumsum tulang. Hasil pemeriksaan hapusan darah tepi, hapusan aspirasi
sumsum tulang (1), hasil pemeriksaan morfologi blast lebih dari 20% dan aspirasi
sumsum tulang (2) digunakan untuk pemeriksaan Immunophenotyping interpretasi
Myeloid lineage, memberikan diagnosis AML, MDS-related AML. Non AML
dilakukan eksklusi, sedangkan AML, MDS-related AML dimasukkan inklusi, Umur
14 – 77 tahun. Aspirasi Sumsum Tulang (2) dilakukan pemeriksaan PCR dan CISH,
hasil pemeriksaan mutasi gen NRAS, RUNX1, NPM1, FLT3 menggunakan PCR dan
pemeriksaan kromosom yaitu delesi kromosom 5 del(5q) menggunakan CISH.
Pengumpulan data yang telah masuk dalam kriteria inklusi dimasukkan tabel untuk
analisa.
MAKROSITER
RRER

80
4.7 Rancangan analisa data
Analisa data dilakukan setelah semua pemeriksaan selesai, peneliti
memasukkan data sebagai berikut
4.8.1 Analisa deskriptif
Sampel sebanyak 31 sampel: berdasarkan umur, jenis kelamin,
pendidikan.
4.8.2 Analisa Regresi Logistik
4.8.2.1 Variable dependen mempunyai skala pengukuran nominal yang terdiri
dari dua kategori positif (0) atau negatif (1) sedangkan variable bebas
(independen) dapat mempunyai skala pengukuran nominal, ordinal,
interval maupun rasio.
4.8.2.2 Model regresi logistik : MDS related AML = del(5q)
4.8.2.3 Probabilitas:
{𝐝𝐞𝐥(5q)=1}=1/1+e-(β0 +β1.RUNX1+β2.FLT3+β3.NRAS+β4.NPM1)
4.8.3 Analisa ROC (Receiver Operating characteristics)
4.8.3.1 Analisis ROC peringkat dan hasil tes diagnostik berkelanjutan versus
gold standar.
4.8.3.2 Area di bawah kurva (AUC) memiliki interpretasi yang berarti untuk
klasifikasi penyakit dari subjek yang sehat.Metode estimasi AUC dan
pengujiannya dalam uji diagnostik tunggal dan juga studi komparatif.
4.8.3.3 Keuntungan kurva ROC untuk menentukan nilai cut-off optimal
(Hajian -Tilaki, 2013 Hulley & Cummings, 1988).

84
BAB 5
HASIL PENELITIAN
5.1 Karakteristik dasar pasien AML
5.1.1 Hasil data karakteristik dasar
Hasil penelitian dasar karakteristik yang mana peneliti mengambil di
Rawat Inap di Laboratorium/instalasi Ilmu Penyakit Dalam Hematologi-Onkologi
Fakultas Kedokteran Universitas Airlangga, RSUD dr Soetomo, Surabaya,
Indonesia, jumlah seluruh sampel 36 orang, 5 sampel di ekslusi, yang
dipergunakan untuk penelitian 31 sampel pada tahun 2017 - 2018, sebagai
berikut:
Tabel 5.1 Deskripsi Data Dasar karakteristik 36 pasien AML, MDS-related
AML
Karakteristik Jumlah
(31 sampel) Prosentase (%)
Usia (tahun)
14 – 24 25 – 35 36 – 46 47 – 57 58 – 68 69 – 79 80 – 90
4 4 9 6 7 0 1
12,90 12,90 29,03 19,35 22,58
0 3,24
Jenis kelamin Laki-laki
Perempuan 22 9
70,97 29,03
Pekerjaan
Pegawai swasta PNS/TNI
Wiraswasta Ibu rumah tangga
Petani Pelajar
15 6 3 2 4 1
48,39 19,35 9,67 6,45 12,90 3,24
Pendidikan
SD SMP SMA
Perguruan Tinggi
2 11 12 6
6,45 35,49 38,71 19,35
Immunophenotyping
Myeloid Lineage
30
96,76
Data yang didapatkan sebanyak 36 subyek sampel, 31 sampel dengan
diagnosis AML sedangkan 5 sampel dengan diagnosis klinik, morfologi sebagai

85
MDS, maka total sampel yang dilakukan analisis 31 sampel, eksklusi 5 sampel
dengan diagnosis MDS (lampiran 2).
Data dasar karakteristik subyek penelitian ini sebagai berikut pada usia
terbanyak yang mengikuti penelitian ini adalah kelompok usia 36 – 46 tahun
tertinggi, usia tua yaitu 80 – 90 tahun (3,24%). Jenis kelamin laki-laki insiden
lebih tinggi dari pada perempuan yaitu laki-laki lebih tinggi perempuan.
5.1.2 Polimorfisme NRAS sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
MDS-related AML dapat diprediksi dengan beberapa gen mutasi, antara
lain: NRAS, sebagai berikut:
Tabel 5.2 Hasil pemeriksaan Polimorfisme NRAS berdasarkan umur, jenis
kelamin, gen NRAS mutasi/tiddak mutasi
Umur (Tahun) Jenis Kelamin NRAS Mutasi
Keterangan L P (+) (-)
14 – 24 3 1 2 2
Jumlah sampel yang dilakukan
analisis 31 sampel
25 – 35 2 2 3 1 36 – 46 7 2 8 1 47 – 57 3 3 4 2 58 – 68 6 1 5 2 69 – 79 - - - - 80 – 90 1 - 1 -
JUMLAH 22 9 23 8
Keterangan: NRAS mutasi : terdapat mutasi (+), tidak ada mutasi (-) Jenis kelamin : L : laki-laki, P : perempuan
Hasil pemeriksaan NRAS didapatkan 23 sampel dari 31 sampel mutasi
NRAS (74,19%). Mutasi NRAS terbanyak pada umur 36 – 46 tahun selanjutnya
Umur lanjut antara 80 – 90 tahun 1 orang (3,22%) (Tabel 5.2). Sampel Laki-laki
lebih banyak dari pada perempuan.
5.1.3 Polimorfisme RUNX1 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).

86
MDS-related AML dapat diprediksi dengan beberapa gen mutasi, antara
lain: RUNX1, sebagai berikut:
Tabel 5.3 Hasil pemeriksaan Polimorfisme RUNX1 berdasarkan umur, jenis
kelamin, gen RUNX1 mutasi/tidak mutasi
Umur (tahun) Jenis Kelamin RUNX1 Mutasi
Keterangan L P (+) (-)
14 - 24 3 1 2 2
Jumlah sampel yang dilakukan analisis 31
sampel
25 – 35 1 3 3 1 36 – 46 6 3 6 3 47 – 57 5 1 3 3 58 – 68 6 1 3 4 69 - 79 - - - - 80 - 90 1 - 1 -
JUMLAH 22 9 18 13
Keterangan:
RUNX1 mutasi: terdapat mutasi (+), tidak ada mutasi (-) Jenis kelamin: L : laki-laki, P : perempuan
Hasil pemeriksaan RUNX1 didapatkan 18 sampel mutasi RUNX1
(58,06%), sedangkan mutasi gen RUNX1 terbanyak pada umur 36 – 46 tahun,
Umur lanjut antara 80 – 90 tahun 1 orang (3,22%) (Tabel 5.3). Sampel Laki-laki
lebih banyak dari pada perempuan.
5.1.4 Polimorfisme NPM1 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
MDS-related AML dapat diprediksi dengan beberapa gen mutasi, antara
lain: NPM1, sebagai berikut:
Tabel 5.4 Hasil pemeriksaan Polimorfisme NPM1 berdasarkan umur, jenis
kelamin, gen NPM1 mutasi/tidak mutasi
Umur (Tahun) Jenis Kelamin NPM1 Mutasi
Keterangan L P (+) (-)
14 - 24 3 1 4 -
Jumlah sampel yang dilakukan analisis 31
sampel
25 – 35 1 3 2 2 36 – 46 6 3 8 1 47 – 57 5 1 4 2 58 – 68 6 1 7 - 69 - 79 - - - - 80 - 90 1 - 1 -
JUMLAH 22 9 26 5
Keterangan: NPM1 mutasi: terdapat mutasi (+), tidak ada mutasi (-)

87
Jenis kelamin: L : laki-laki, P : perempuan
Hasil pemeriksaan NPM1 didapatkan 26 sampel mutasi NPM1
(83,87%), sedangkan Mutasi NPM1 terbanyak pada umur 36 – 46 tahun
sebanyak 8 sampel (25,80%), umur lanjut antara 80 – 90 tahun 1 orang (3,22%)
(Tabel 5.4). Sampel Laki-laki lebih banyak dari pada perempuan.
5.1.5 Polimorfisme FLT3 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
MDS-related AML dapat diprediksi dengan beberapa gen mutasi, antara
lain: FLT3, sebagai berikut:
Tabel 5.5 Hasil pemeriksaan Polimorfisme FLT3 berdasarkan umur, jenis
kelamin, gen FLT3 mutasi/tidak mutasi
Umur (Tahun) Jenis Kelamin FLT3 Mutasi
Keterangan L P (+) (-)
14 - 24 3 1 3 1
Jumlah sampel yang dilakukan analisis 31
sampel
25 – 35 1 3 4 - 36 – 46 5 3 6 3 47 – 57 6 1 5 1 58 – 68 6 1 6 1 69 - 79 - - - - 80 - 90 1 - 1 -
JUMLAH 22 9 25 6
Keterangan: FLT3 mutasi: terdapat mutasi (+), tidak ada mutasi (-) Jenis kelamin: L : laki-laki, P : perempuan
Hasil pemeriksaan FLT3 didapatkan 25 sampel mutasi FLT3 (80,64%).
Mutasi FLT3 terbanyak pada umur 36 – 46 tahun, umur lanjut antara 80 – 90
tahun 1 orang (3,22%) (Tabel 5.5). Insiden sampel Laki-laki lebih banyak dari
pada perempuan.

88
5.1.6,7 Polimorfisme delesi kromosom 5 del(5q) dan Kromosom 5 normal
sebagai prediktor perubahan Myelodisplastic Syndrome menjadi
Acute Myeloid Leukemia (MDS-related AML).
MDS-related AML dapat diprediksi dengan beberapa gen mutasi, dan
delesi kromosom 5 del(5q) dan kromosom 5 normal sebagai berikut:
Tabel 5.6 Hasil pemeriksaan Polimorfisme delesi Kromosom 5 del(5q) dan
kromosom 5 normal berdasarkan umur, jenis kelamin
Umur (Tahun)
Jenis Kelamin Delesi Kromosom 5 Del(5q) Keterangan
L P (+) (-)
14 – 24 3 1 4 - Jumlah sampel
yang dilakukan analisis 31
sampel
25 – 35 1 3 2 2 36 – 46 6 3 7 2 47 – 57 5 2 5 2 58 – 68 6 - 5 1 69 – 79 - - - - 80 – 90 1 - 1 -
JUMLAH 22 9 24 7
Keterangan: Delesi Kromosom 5 del(5q): delesi kromosom 5 del(5q) (+), kromosom 5 normal (-) Jenis kelamin: L : laki-laki, P : perempuan
Hasil pemeriksaan delesi kromosom 5 del(5q) didapatkan 24 sampel
(77,41%), Delesi kromosom 5 del(5q) terbanyak pada umur 36 – 46 tahun
sebanyak 7 sampel (22,58%), insiden umur terbanyak antara umur 25-35 tahun,
36-46 tahun, 47-57 tahun yang mana rata-rata 6,45% (Tabel 5.6). Sampel Laki-
laki lebih banyak dari pada perempuan.
5.1.8. Analisis statistik polimorfisme gen NRAS, RUNX, NPM1, FLT3 dan
delesi keomosom 5 del(5q) menjadi MDS-related AML.
Analisis statistik menggunakan ROC (Receiver Operating
Characteristics) dan Regresi Logistik sebagai berikut:
5.1.8.1. Pemeriksaan morfologi, gen dan kromosom, analisis statistik
Pada penelitian ini terdapat 31 sampel yang mana akan dilakukan
analisis dengan Regresi Logistik dan ROC.

89
Tabel 5.7 Ringkasan Pengolahan Kasus
Kasus N
(besar sampel) Prosentase
Kasus yang diseleksi
Kasus di analisis 31 100,0
Kasus tidak di analisis 0 0 Total 31 100,0 Kasus yang tidak masuk seleksi
0 .0
Total 31 100,0
5.1.8.2. Metode statistik ROC (Receiver operating Characteristics)
Diskripsi ROC curve analysis
Analisa kurva ROC memungkinkan kita untuk membuat kurva ROC dan
laporan sensitivitas yang lengkap. Kurva ROC adalah alat yang fundamental
untuk evaluasi test diagnostik. Dalam kurva ROC true positive rate di-plot dalam
fungsi false positive rate (1- Specificity) untuk cut-off point yang berbeda dari
setiap parameter. Setiap point dalam kurva ROC menunjukkan pasangan
sensitif/spesifik yang berhubungan dengan setiap keputusan. Area di bawah
kurva ROC adalah pengukuran dari seberapa baik sebuah parameter dapat
membedakan Antara dua grup diagnostik (abnormal/normal).
5.1.8.3 Hasil analisis ROC
Hasil pemeriksaan kurva ROC menunjukkan hasil dibawah kurva
76,5% (tabel 5.9), faktor gen (RUNX1, FLT3, NRAS, NPM1) mempengaruhi
MDS-related AML.
Tabel 5.8 Hasil pemeriksaan delesi kromosom 5 del(5q)
Delesi kromosom 5 del(5q) (MDS-related AML) Valid ( N )
Positif 24 Negatif 7
Keterangan: Nilai yang lebih besar pada hasil tes variabel menunjukkan bukti yang lebih kuat untuk keadaan aktual yang positif. Keadaan aktual yang positif ditunjukkan dengan (+), Negatif ditunjukkan dengan (-)

90
Tabel 5.8. menyatakan bahwa sampel penelitian sebanyak 31 terdapat
24 (dua puluh empat) delesi kromosom 5 del(5q) (positif), 7(tujuh) kromosom 5
normal (negatif).
Tabel 5.9 Area dibawah kurva (AUC)
Area 95% confidence interval P
0,765(76,5%) Batas bawah Batas atas 0,036
0,574(57,4%) 0,956(95,6%)
Tabel 5.9, area dibawah kurva (AUC) 76,5%, confidence interval 95%,
dan signifikan (P = 0,036), menunjukkan bahwa faktor (RUNX1, FLT3, NRAS,
NPM1) yang mempengaruhi MDS-related AML adalah valid dan bisa didapatkan
cut-off value (gambar 5.1).
Gambar 5.1 Kurva ROC dari perubahan MDS menjadi AML.
Kurva ROC Probabilitas dibawah 0,7825584010 (low risk), diatas
0,7825584010 (high risk), sebagai berikut (lihat gambar):
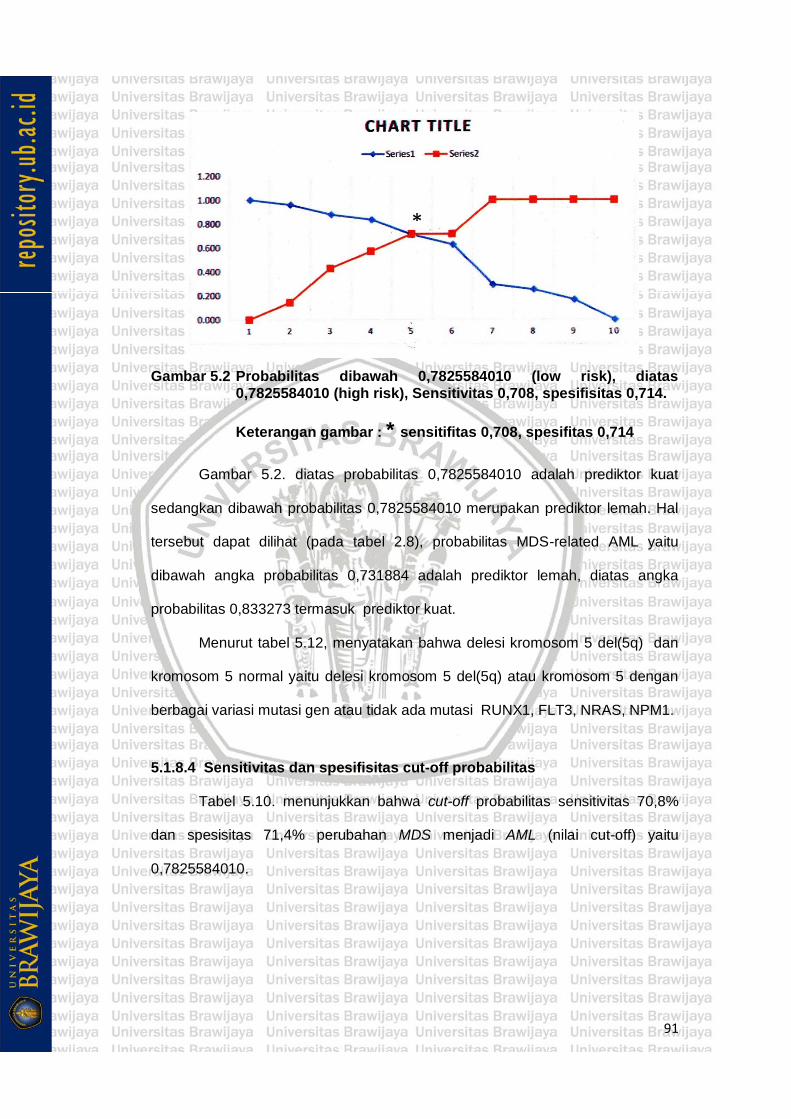
91
Gambar 5.2 Probabilitas dibawah 0,7825584010 (low risk), diatas 0,7825584010 (high risk), Sensitivitas 0,708, spesifisitas 0,714.
Keterangan gambar : * sensitifitas 0,708, spesifitas 0,714
Gambar 5.2. diatas probabilitas 0,7825584010 adalah prediktor kuat
sedangkan dibawah probabilitas 0,7825584010 merupakan prediktor lemah. Hal
tersebut dapat dilihat (pada tabel 2.8), probabilitas MDS-related AML yaitu
dibawah angka probabilitas 0,731884 adalah prediktor lemah, diatas angka
probabilitas 0,833273 termasuk prediktor kuat.
Menurut tabel 5.12, menyatakan bahwa delesi kromosom 5 del(5q) dan
kromosom 5 normal yaitu delesi kromosom 5 del(5q) atau kromosom 5 dengan
berbagai variasi mutasi gen atau tidak ada mutasi RUNX1, FLT3, NRAS, NPM1.
5.1.8.4 Sensitivitas dan spesifisitas cut-off probabilitas
Tabel 5.10. menunjukkan bahwa cut-off probabilitas sensitivitas 70,8%
dan spesisitas 71,4% perubahan MDS menjadi AML (nilai cut-off) yaitu
0,7825584010.
*

92
Tabel 5.10 Hasil Variabel : probabilitas MDS-related AML
Positif jika ≥ a No. Sensitifitas 1 – spesifisitas spesifisitas
0.0000000000 1 1.000 1.000 0.000 .4342392130 2 .958 .857 0.143 .6182394205 3 .875 .571 0.429 .7169388935 4 .833 .429 0.571 .7825584010 5 .708 .286 0.714 .8429902415 6 .625 .286 0.714 .8573538955 7 .292 0.000 1.000 .9132656980 8 .250 0.000 1.000 .9668813630 9 .167 0.000 1.000
1.0000000000 10 .000 0.000 1.000
Keterangan: Tes hasil variabel hasil: probabilitas MDS-related AML mempunyai paling sedikit satu ikatan antara kelompok keadaan positif dan negatif yg sesungguhnya. a. nilai cut-off paling kecil adalah nilai tes observasi minimum minus 1, dan nilai
cut-off paling besar adalah hasil tes observasi maksimum plus 1, semua nilai cut-off lainnya adalah rata-rata dari 2 nilai tes observasi secara berurutan.
Kombinasi pemeriksaan genetik dan sitogenetik NRAS, RUNX1,
NPM1, FLT3 dan Delesi kromosom 5 del(5q) sebagai parameter untuk prediktor
MDS-related AML, tabel 5.11 menunjukkan bahwa pemeriksaan parameter
tersebut diatas sebagai prediksi adanya mutasi lebih baik (PPV 90,9%) dari pada
merupakan prediksi tidak terjadi mutasi (NPV 42,9 %). Tabel 5.11 menunjukkan
spesifisitas maupun sensitivitas diatas 70%, maka pemeriksaan genetic dan
sitogenetik dapat dipergunakan sebagai sekrening maupun diagnosis.
Tabel 5.11, Nilai validitas kombinasi NRAS, RUNX1, NPM1, FLT3 dan
Delesi kromosom 5 del(5q) sebagai prediktor MDS-related AML

93
5.1.8.5 Progresivitas gen NRAS, RUNX1, NPM1, FLT3 terjadi MDS-related AML
Area dibawah kurva (AUC) 76,5%, confidence interval 95%, dan
signifikan (P = 0,036), menunjukkan bahwa faktor gen RUNX1, FLT3, NRAS,
NPM1 sebagai prediktor perubahan MDS-related AML adalah valid dan bisa
didapatkan cut-off value (Tabel 5.9; Gambar 5.1).
Tabel 5.12 Hasil Regresi Logistik (hubungan prediktor gen dengan MDS-
related AML)
Variables Β Wald Df PR
NRAS 2,446 3,236 1 11,543 RUNX1 (-)1,694 1,617 1 0,184 NPM1 1,618 1,688 1 5,043 FLT3 0,147 0,012 1 1,158
Constant del(5q) -,761 ,160 1 0,467
PR: Prevalence Risk Keterangan: Mutasi NRAS mempunyai β tertinggi 2,446, PR 11,543 adalah PR tertinggi berarti meningkatkan risiko MDS-related AML sebesar 11.543 kali; mutasi RUNX1 β (-) 1,694, PR 0,184 adalah PR terendah, berarti menurunkan risiko MDS-related AML sebesar 0,184 kali sedangkan Mutasi NPM1 mempunyai β 1,618, PR 5,043; mutasi FLT3 β 0,147; PR 1,158 meningkatkan risiko MDS-related AML.
5.1.8.6 Rumus Probabilitas
{𝐝𝐞𝐥(5q)=1}=1/1+e-(β0 +β1.RUNX1+β2.FLT3+β3.NRAS+β4.NPM1)
5.1.8.7 Variasi statistik 16 variasi gen mutasi/tidak mutasi dengan delesi
kromosom 5 del(5q)/kromosom 5 normal
Hasil penelitian stud statistik didapatkan 16 variasi dari Gen NRAS,
RUNX1, NPM1, FLT3 terhadap Probabilitas MDS-related AML (delesi
kromosom 5 del(5q), variasi tersebut dapat dilihat di (tabel 5.12)

94
Tabel 5.13 Variasi Gen NRAS, RUNX1, NPM1, FLT3 terhadap Probabilitas
MDS-related AML
NRAS RUNX1 NPM1 FLT3
Prob. MDS related AML
MDS related AML [Delesi kromosom 5
del(5q)]
1 - + - - 0.079074 -
2 - + - + 0.090463 -
3 - + + - 0.302167 -
4 - - - - 0.318429 -
5 - + + + 0.334033 -
6 - - - + 0.351147 -
7 + + - - 0.497750 -
8 + + - + 0.534445 -
9 - - + - 0.702033 -
10 - - + + 0.731844 -
11 + + + - 0.833273 +
12 + - - - 0.843565 +
13 + + + + 0.852708 +
14 + - - + 0.862000 +
15 + - + - 0.964532 +
16 + - + + 0.969231 + Keterangan: Abnormalitas gen NRAS, RUNX1, NPM1, FLT3 dan abnormalitas delesi kromosom 5 del(5q) sebagai prediktor MDS-related AML: delesi kromosom 5 del(5q) negatif (-) sebagai prediktor lemah (0,731884) dibawah cut-off 0,7825584010, sedangkan delesi kromosom 5 del(5q) positif (+) sebagai prediktor kuat (0,833273) diatas cut-off 0,7825584010. Gen RUNX1 mempunyai aktivitas menghambat MDS menjadi AML (tabel 5.1) (-) 1,694, dapat ditunjukkan pada Mutasi NRAS, RUNX1, NPM1, FLT3 (deret variasi no. 13) probabilitas MDS-related AML 0.852708, sedangkan mutasi NRAS, NPM1, FLT3 (deret variasi no. 16) probabilitas MDS-related AML 0.969231 dan keduanya mempunyai delesi kromosom 5 del(5q) positif (+).
5.2. Hasil penelitian diskriptif dan statistic ROC, Regresi Logistik
Hasil penelitian didapatkan prediktor kuat menjadi MDS-related AML
yaitu delesi kromosom 5 del(5q) dengan variasi mutasi gen > cut-off
0,7825584010. Prediktor lemah untuk terjadinya MDS-related AML yaitu
kromosom 5 normal dengan variasi mutasi NRAS, RUNX1, NPM1, FLT3 < cut-off
0,7825584010 (tabel 5.10, 5.11, 5.12). Pada kromosom 5 normal dengan variasi
mutasi gen NRAS, RUNX1, NPM1, FLT3 sebagai berikut: mutasi gen RUNX1
dengan berbagai variasi terdapat 6 dari 10 variasi (60%), mutasi gen RUNX1 β (–

95
) 1,694 dan PR 0,184, termasuk mutasi gen RUNX1 tunggal dengan probabilitas
paling rendah (0,079074) untuk menjadi MDS-related AML. NRAS terdapat 2 dari
10 variasi (20%), setiap mutasi NRAS β 2,446 dan PR 11,543 (tabel 5.11). NPM1
terdapat 2 dari 10 variasi (20%), setiap mutasi NPM1 β 1,618 dan PR 5,043
(tabel 5.11). FLT3 terdapat 5 dari 10 variasi (50%), setiap mutasi FLT3 β 0,147
dan PR 1,158 (tabel 5.11). Mutasi gen NRAS, RUNX1, FLT3, mempunyai
probabilitas (0,534445) lebih rendah dibandingkan probabilitas mutasi gen
NPM1, FLT3 (0,731844) untuk menjadi MDS-related AML (tabel 5.12). Prediktor
kuat untuk terjadinya MDS-related AML yaitu delesi kromosom 5 del(5q) dengan
variasi mutasi gen NRAS, RUNX1, NPM1, FLT3 > probabilitas cut-off
0,7825584010 (tabel 5.10, 5.11, 5.12). Mutasi gen NRAS dengan berbagai
variasi terdapat 6 dari 6 variasi (100%), termasuk mutasi gen NRAS tunggal
dengan probabilitas (0,843565), dibandingkan dengan probabilitas mutasi gen
NRAS, RUNX1, NPM1 (0,833273), sedangkan mutasi gen NRAS, RUNX1,
NPM1, FLT3 (0,852708). Mutasi gen NRAS, NPM1, FLT3 mempunyai
probabilitas menjadi MDS-related AML tertinggi yaitu 0,969231. Hasil
pemeriksaan Immunotyping : myeloid lineage, morfologi dari sel darah tepi dan
sumsum tulang, pemeriksaan molekuler: mutasi gen menggunakan PCR,
pemeriksaan sitogenetik menggunakan CISH: delesi kromosom 5 del(5q) dan
kromosom 5 normal (lampiran 6)

96
BAB 6
PEMBAHASAN
6.1 Data dasar karakteristik subyek penelitian
Pada penelitian ini sampel sebanyak 31 sampel sebagai berikut pada
usia terbanyak yang mengikuti penelitian ini adalah 36 – 46 tahun (29,03%); 58 –
68 tahun (22,58%), sedangkan usia tua yaitu 80 - 90 tahun (3,24%). Jenis
kelamin laki-laki lebih banyak dari perempuan. Hasil pemeriksaan
immunophenotyping yaitu myeloid lineage 96,76%, diagnosis pasien
berdasarkan pemeriksaan klinik, immunophenotyping, hapusan aspirasi sumsum
tulang (BMA).
Subyek penelitian ini sebanyak 36 sampel, 5 sampel dilakukan eksklusi
karena diagnosis klinik MDS, Leukemia non myelocytic, setelah dilakukan
pemeriksaan Immunophenotyping (gambar 5.5), pemeriksaan hapusan sumsum
tulang (gambar 5.6), sehingga subyek yang dipergunakan dengan diagnosis AML
sebanyak 31 sampel, pemeriksaan abnormalitas gen menggunakan PCR
(gambar 5.3), dan pemeriksaan sitogenetik (CISH) (gambar 5.4).
Menurut data yang diambil oleh peneliti di bagian Patologi Klinik RSUD dr
Soetomo Fakultas Kedokteran Universitas Airlangga Surabaya, Jawa Timur
periode tanggal 2 Januari sampai dengan 30 Oktober tahun 2014 didapatkan
jumlah penderita 181 orang, dengan rentang umur 14 – 77 tahun. Hasil penelitian
pendahuluan, berdasarkan morfologi sel hapusan darah tepi (HDT) yaitu: AML
tertinggi 38,66 % (70 orang), CML 37,57%, ALL 14,92%, Myelodisplastic
Syndrome (MDS) 4,98% (9 orang). Diagnosis MDS perempuan mempunyai risiko
lebih tinggi dari pada laki-laki demikian juga diagnosis AML risiko perempuan
lebih tinggi dari pada laki-laki yaitu perempuan 40 orang (40%), dan laki-laki 30
orang (30%). Data penelitian ini, penderita dengan diagnosis AML sebanyak 70

97
orang: risiko tertinggi AML pada usia 46 – 61 tahun (37,14%). Penderita dengan
diagnosis MDS Usia 14 – 29 tahun yang mempunyai risiko tinggi sebanyak 4
orang (44,4%), pada usia tua 62 – 77 tahun 1 orang (11,1%) (accepted, akan
publikasi di Journal Drug Invension Today).
Menurut WHO (World Health organization) insiden leukemia terjadi
hampir di seluruh dunia, kasus kanker yang tercatat sekitar 250 kasus baru per
tahun dengan Case Fatality Rate (CFR) 76%. Kasus baru kanker sebanyak
100.000 didapatkan penderita AML sekitar 2,5%, dan ALL sekitar 1,3%. Pada
tahun 2006, American Cancer Society memprediksi bahwa 11.930 pria dan
wanita (6350 pria dan 5580 wanita) di Amerika Serikat telah didiagnosis
menderita AML (Deschler & Lübbert, 2006). Pada kelompok median umur 67
tahun, insiden lebih tinggi pada pria dibandingkan wanita yaitu 4,3 : 2,9. Di
Indonesia insiden Leukemia Akut diprediksi sekitar 3,4 kasus setiap 100.000
populasi per tahun (Aulya et al., 2014 ). Hasil penelitian, data dasar karakteristik
dibandingkan dengan hasil peneliti di luar negeri yaitu berdasarkan jenis kelamin
laki-laki lebih banyak dari pada perempuan, rentang umur lebih muda yaitu 46-61
tahun, 58-68 tahun, sedangkan usia lanjut 80 – 90 tahun (3,24%). Keadaan
tersebut mungkin terjadi karena jumlah sampel yang kecil, fasilitas kesehatan
kualitas masih kurang memadai.
Analisis statistik sampel penelitian 31 terdapat 24 (dua puluh empat)
delesi kromosom 5 del(5q) (positif), dengan berbagai variasi mutasi NRAS,
RUNX1, NPM1, FLT3. 7(tujuh) kromosom 5 (normal) dengan berbagai variasi
mutasi NRAS, RUNX1, NPM1, FLT3. Tabel 5.7, 5.8 area dibawah kurva (AUC)
76,5%, confidence interval 95%, dan signifikan (P = 0,036), menunjukkan bahwa
faktor mutasi gen NRAS, RUNX1, NPM1, FLT3 dan delesi kromosom 5 del(5q)
sebagai prediktor MDS-related AML adalah valid dan bisa didapatkan cut-off
value (gambar 5.1, 5.2; tabel 5.9).

98
Hasil penelitian tersebut diatas, didukung oleh penelitian dari 1) Shiozawa
et al., 2017, ekspresi gen dan risiko MDS transformasi menjadi leukemik,
memeriksa mutasi gen antara lain NRAS, RUNX1, FLT3, abnormalitas
sitogenetik antara lain: delesi kromosom 5 del(5q), sel imatur (blast) dan gejala
klinik. Pada kurva ROC didapatkan AUC 86%, confidence interval 95%,
menunjukkan bahwa ekspresi gen mendukung prognostik, diagnostik MDS
transformasi leukemik (Shiozawa et al., 2017). Selain penelitian tersebut diatas,
2) penelitian Osato, 2004 menyatakan bahwa frekwensi mutasi pada gen
RUNX1/AML1 didapatkan berbagai variasi pada penderita kanker maupun orang
normal, yaitu AML (M0) prosentase mutasi 21%, MDS (RAEB/RAEB-t)
prosentase mutasi 15,9%, MDS-related AML prosentase mutasi 15,0%,
sedangkan orang normal prosentase mutasi 2,6% (Osato, 2004; Rocquain et al.,
2010). 3) Etiologi MDS tidak diketahui dengan pasti tetapi faktor abnormalitas
sitogenetik, molekuler, epigenetik dapat berkembang menjadi MDS.
Menurut penelitian Agersborg et al., 2015 bahwa mutasi FLT3, NPM1,
dan WT1 adalah abnormalitas molekuler yang dapat dipergunakan sebagai
kriteria diagnosis pasien AML yang obyektif lebih baik dari pada aspirasi sumsum
tulang (Walter et al., 2012). Mutasi gen tersebut dideteksi sebanyak 49% pada
penderita AML dan 50% kasus AML dapat dibuat diagnosis dengan mendeteksi
abnormalitas molekuler, tidak memperhatikan morfologi sumsum tulang.
Delesi kromosom 5 del(5q) atau kromosom 5 normal dengan berbagai
variasi mutasi gen atau tidak mutasi (normal) NRAS, RUNX1, NPM1, FLT3,
variasi tersebut sebanyak 16 macam variasi (tabel 5.12).

99
6.2 NRAS sebagai prediktor MDS-related AML
Area dibawah kurva (AUC) 76,5%, confidence interval 95%, dan
signifikan (P = 0,036), menunjukkan bahwa faktor gen RUNX1, FLT3, NRAS,
NPM1 sebagai prediktor perubahan MDS-related AML adalah valid dan bisa
didapatkan cut-off value (Tabel 5.9; Gambar 5.1).
Pada penelitian ini dapat sebagai prediktor kuat untuk menjadi MDS-
related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen (NRAS), cut-off
probbilitas > 0,7825584010 (tabel 5.12), sebagai berikut:
1. Mutasi gen NRAS, RUNX1, dan NPM1 dengan probabilitas 0,833273,
mempunyai risiko tinggi menjadi MDS-related AML.
2. Mutasi gen NRAS dengan probabilitas 0.843565, mempunyai risiko lebih
tinggi menjadi MDS-related AML dibandingkan dengan ad.1.
3. Mutasi gen NRAS, RUNX1, NPM1, FLT3 dengan probabilitas 0,852708,
mempunyai risiko lebih tinggi menjadi MDS-related AML dibandingkan
dengan ad.2.
4. Mutasi gen NRAS, FLT3 dengan probabilitas 0,862000, mempunyai risiko
lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.3.
5. Mutasi gen NRAS, NPM1 dengan probabilitas 0,964532, mempunyai
risiko lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.4.
6. Mutasi gen NRAS, NPM1, F;T3 dengan probabilitas 0,969231,
mempunyai risiko tertinggi untuk menjadi MDS-related AML dibandingkan
variasi yang lain.
Variasi stastistik NRAS 100%, artinya didaptkan 6 mutasi gen NRAS dari
6 variasi sebagai prediktor kuat MDS-related AML, variasi NRAS, RUNX1, NPM1
sebagai prediktor lemah, sedangkan variasi sebagai prediktor paling kuat adalah
NRAS, NPM1, FLT3. Mutasi NRAS ditemukan 23 sampel dari 31 sampel
(74,19%) pada diagnosis AML. Mutasi gen NRAS β 2,446 dan PR 11,543 berarti

100
setiap peningkatan 1 satuan NRAS, maka akan ada peningkatan 11,543 kali
kemungkinan terjadi MDS-related AML (lihat tabel 5.11, 5.2).
Prediktor lemah untuk menjadi MDS-related AML yaitu kromosom 5
normal dengan variasi gen NRAS mempunyai probabilitas < 0,7825584010 (tabel
5.12), sebagai berikut:
1. Mutasi gen NRAS, RUNX1, dengan probabilitas 0,497750 sebagai
prediktor lemah untuk menjadi MDS-related AML.
2. Mutasi gen NRAS, RUNX1, FLT3 dengan probabilitas 0,534445, sebagai
prediktor lemah untuk menjadi MDS-related AML.
Mutasi NRAS ditemukan 6 mutasi NRAS dari 6 variasi termasuk mutasi
NRAS dengan probabilitas: 0,843565, dibandingkan dengan probabilitas mutasi
RUNX1, NRAS, NPM1: 0,833273. Mutasi NRAS, NPM1, FLT3 sebagai prediktor
paling kuat menjadi MDS-related AML dengan probabilitas 0,969231. Hal
tersebut diatas terjadi karena mutasi NRAS β 2,446 dan PR 11,543 berarti setiap
peningkatan 1 satuan NRAS, maka terjadi peningkatan 11,543 kali kemungkinan
MDS-related AML.
Menurut peneliti Takshashi, Hoffman et al., bahwa mutasi faktor jalur
transduksi sinyal melibatkan mutasi SCFR (Stem cell factor reseptor) atau
tyrosine-protein kinase (KIT) dan gen fms-related tyrosine kinase 3 (FLT3) dalam
jalur transduksi yang sama. Pada penderita AML yang diperiksa dengan PCR
didapatkan abnormalitas genetik yang menunjukkan mutasi jalur transduksi
sinyal gen NRAS, FLT3 yang menyebabkan proliferasi atau pertumbuhan sel
yang tidak terkendali (Hoffman et al., 2013; Takahashi, 2011).
Menurut penelitian Walter et al., 2012, menyatakan bahwa MDS menjadi
Secondary-AML (sAML) atau MDS-related AML mengalami proses yang
progresivitas cepat untuk menjadi sAML yaitu < 6 bulan (6,7% dari mutasi yang
spesifik menjadi sAML) dan progresivitas lambat yaitu > 20 bulan (37,8% dari

101
mutasi yang spesifik menjadi sAML) (Walter et al., 2012). Pada penelitian
terdahulu dapat dihubungkan dengan waktu (Walter et al., 2012) sebagai
prediktor kuat untuk menjadi MDS-related AML yaitu delesi kromosom 5 del(5q)
dengan variasi gen cut-off > 0,7825584010, korelasi dengan waktu < 6 bulan
untuk menjadi sAML (Walter et al., 2012), sedangkan prediktor lemah untuk
menjadi MDS-related AML yaitu kromosom 5 normal dengan variasi NRAS,
RUNX1, NPM1, FLT3 < probabilitas 0,7825584010, waktu > 20 bulan untuk
menjadi sAML (MDS-related AML) (Walter et al., 2012). Keadaan tersebut diatas
didukung oleh penelitian Ceesay et al., 2012, MDS transformasi menjadi AML
membutuhkan waktu antara 5-6 tahun, diikuti paparan lingkungan makro dan
mikro sampai terjadi leukemogenesis, faktor risiko tergantung besarnya paparan.
Pada saat terjadi MDS-related AML antara 12 – 130 bulan, insiden tersebut
dihubungkan dengan mutasi gen dan kromosom, pemeriksaan aspirasi sumsum
tulang pada kasus AML dijumpai dalam jumlah sedikit fase displastik (lihat tabel
5.2 – 5.6) (Ceesay et al., 2012).
Hasil penelitian tersebut diatas dapat dinyatakan bahwa Mutasi NRAS,
RUNX1, NPM1, FLT3 adalah prediktor terjadinya MDS-related AML dengan
delesi kromosom 5 del(5q) (p = 0,036). Cut-off probabilitas sensitifitas 70,8% dan
spesifisitas 71,4% perubahan MDS-related AML yaitu 0,7825584010. Diskusi
tersebut diatas, pemeriksaan NRAS, RUNX1, NPM1, FLT3 dan sitogenetik delesi
kromosom 5 del(5q) dapat dipergunakan untuk memperbaiki prediksi MDS-
related AML, dan terapi sedini mungkin, sedangkan untuk dihubungkan dengan
waktu memerlukan penelitian tersendiri.

102
6.3 RUNX1 sebagai prediktor MDS-related AML
Pada penelitian ini dapat sebagai prediktor kuat untuk menjadi MDS-
related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen RUNX1, cut-off
probbilitas > 0,7825584010 (tabel 5.12), sebagai berikut:
1. Mutasi gen NRAS, RUNX1, dan NPM1 dengan probabilitas 0,833273,
sebagai prediktor kuat menjadi MDS-related AML.
2. Mutasi gen NRAS, RUNX1, NPM1, FLT3 dengan probabilitas 0,852708,
sebagai prediktor kuat menjadi MDS-related AML dari ad.1.
Variasi stastistik RUNX1 33,33%, artinya terdapat 2 mutasi gen RUNX1
dari 6 variasi sebagai prediktor kuat, sedangkan variasi paling kuat adalah
NRAS, RUNX1, NPM1, FLT3. Mutasi RUNX1 terdapat 18 dari 31 sampel
(58.06%) diagnosis AML.
Prediktor lemah untuk menjadi MDS-related AML yaitu kromosom 5
normal dengan variasi gen RUNX1 < probabilitas 0,7825584010 (tabel 5.12),
sebagai berikut:
1. Mutasi gen RUNX1, dengan probabilitas 0,079074 mempunyai risiko
rendah menjadi MDS-related AML.
2. Mutasi gen RUNX1, FLT3, dengan probabilitas 0,090463 mempunyai
risiko rendah menjadi MDS-related AML tetapi potensi menjadi MDS-
related AML lebih tinggi dibandingkan dengan ad.1.
3. Mutasi gen RUNX1, NPM1, dengan probabilitas 0,302167, mempunyai
risiko rendah menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad.2.
4. Mutasi gen RUNX1, NPM1, FLT3 dengan probabilitas 0,334033,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 3.

103
5. Mutasi gen NRAS, RUNX1 dengan probabilitas 0,497750, mempunyai
risiko rendah untuk menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad.4
6. Mutasi gen NRAS, RUNX1, FLT3 dengan probabilitas 0,534445,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 1-5.
Mutasi gen RUNX1 dengan berbagai variasi terdapat 6 dari 10 variasi
termasuk mutasi gen RUNX1 dengan probabilitas 0,079074, dibandingkan
dengan probabilitas mutasi NRAS, RUNX1 (0,497750). Mutasi NRAS, RUNX1,
FLT3 mempunyai probabilitas MDS-related AML tertinggi 0,534445, Kromosom 5
normal. Hal tersebut diatas terjadi mutasi RUNX1 dengan berbagai variasi
terdapat 18 dari 31 sampel (58,06%), pada umur 58-86 tahun (22,58%), insiden
laki-laki lebih banyak dari perempuan. Mutasi gen RUNX1 β (-) 1,694 dan PR
0,184 berarti setiap peningkatan 1 satuan RUNX1, maka akan ada penurunan
0,184 kali kemungkinan MDS-related AML (lihat tabel 5.11, 5.3)
Menurut Hoffman et al., insiden faktor mutasi gen yang terbanyak pada
AML adalah mekanisme faktor jalur transkripsi yang berperan sebagai regulator
utama pada deferensiasi berbagai jenis perkembangan sel hematopoitik, mutasi
tersebut menghambat proses deferensiasi sel myeloid, contoh: Core Binding
Factor (CBF) adalah faktor transkripsi (RUNX1 atau AML1) (Hoffman et al.,
2013).
Hasil penelitian tersebut diatas dapat dinyatakan bahwa Mutasi NRAS,
RUNX1, NPM1, FLT3 adalah prediktor terjadinya MDS-related AML dengan
delesi kromosom 5 del(5q) (p = 0,036). Cut-off probabilitas sensitifitas 70,8% dan
spesifisitas 71,4% perubahan MDS-related AML yaitu 0,7825584010. Diskusi
tersebut diatas, pemeriksaan NRAS, RUNX1, NPM1, FLT3 dan delesi kromosom
5 del(5q) dapat dipergunakan sebagai prediktor MDS-related AML

104
6.4 NPM1 sebagai prediktor MDS-related AML
Pada penelitian ini dapat sebagai prediktor kuat untuk menjadi MDS-
related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen NPM1, cut-off
probbilitas > 0,7825584010 (tabel 5.12), sebagai berikut:
1. Mutasi gen NRAS, RUNX1, dan NPM1 dengan probabilitas 0,833273,
mempunyai risiko tinggi menjadi MDS-related AML.
2. Mutasi gen NRAS, RUNX1, NPM1, FLT3 dengan probabilitas 0,852708,
mempunyai risiko lebih tinggi menjadi MDS-related AML dibandingkan
dengan ad.1.
3. Mutasi gen NRAS, NPM1 dengan probabilitas 0,964532, mempunyai
risiko lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.2.
4. Mutasi gen NRAS, NPM1, FLT3 dengan probabilitas 0,969231,
mempunyai risiko tertinggi untuk menjadi MDS-related AML dibandingkan
variasi yang lain.
Variasi stastistik NPM1 4 (66,66%), artinya 4 dari 6 variasi sebagai
prediktor kuat didapatkan insiden mutasi gen NPM1, variasi paling lemah adalah
NRAS, RUNX1, NPM1, sedangkan variasi paling kuat adalah NRAS, NPM1,
FLT3. Mutasi NPM1 terdapat 26 dari 31 sampel (83,87%) diagnosis AML.
Prediktor lemah untuk menjadi MDS-related AML yaitu kromosom 5
normal dengan variasi gen RUNX1 < probabilitas 0,7825584010 (tabel 5.12),
sebagai berikut:
1. Mutasi gen RUNX1, NPM1, dengan probabilitas 0,302167, mempunyai
risiko rendah menjadi MDS-related AML.
2. Mutasi gen RUNX1, NPM1, FLT3 dengan probabilitas 0,334033,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 1.

105
3. Mutasi gen NPM1 dengan probabilitas 0,702033, mempunyai risiko
rendah untuk menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad 2.
4. Mutasi gen NPM1, FLT3 dengan probabilitas 0,731844, sebagai prediktor
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad. 1-3
Mutasi gen NPM1 dengan berbagai variasi terdapat 4 dari 10 variasi
termasuk mutasi gen NPM1 dengan probabilitas 0,302167, dibandingkan dengan
probabilitas mutasi NPM1, FLT3: 0,731844. Mutasi NRAS, NPM1, FLT3
mempunyai probabilitas MDS-related AML tertinggi 0,969231, deleasi Kromosom
5 del(5q). Hal tersebut diatas terjadi mutas gen NPM1 dengan berbagai variasi
terdapat 26 dari 31 sampel (83,87%), pada umur 58-86 tahun (22,58%), insiden
laki-laki lebih banyak dari perempuan. Mutasi gen NPM1 β 1,618 dan PR 5,043
berarti setiap peningkatan 1 satuan NPM1, maka akan ada peningkatan 5,043
kali kemungkinan menjadi MDS-related AML (lihat tabel 5.11, 5.3).
Hasil penelitian tersebut diatas dapat dinyatakan bahwa Mutasi NRAS,
RUNX1, NPM1, FLT3 adalah prediktor terjadinya MDS-related AML dengan
delesi kromosom 5 del(5q) (p = 0,036). Cut-off probabilitas sensitifitas 70,8% dan
spesifisitas 71,4% perubahan MDS-related AML yaitu 0,7825584010. Diskusi
tersebut diatas, pemeriksaan NRAS, RUNX1, NPM1, FLT3 dan delesi kromosom
5 del(5q) dapat dipergunakan untuk memperbaiki prediksi MDS-related AML, dan
terapi sedini mungkin.
6.5 FLT3 sebagai prediktor MDS-related AML.
Area dibawah kurva (AUC) 76,5%, confidence interval 95%, dan
signifikan (P = 0,036), menunjukkan bahwa faktor gen RUNX1, FLT3, NRAS,
NPM1 yang mempengaruhi MDS-related AML adalah valid dan bisa didapatkan
cut-off value (Tabel 5.9; Gambar 5.1).

106
Pada penelitian ini dapat sebagai prediktor kuat untuk menjadi MDS-
related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen FLT3, cut-off
probbilitas > 0,7825584010 (tabel 5.12), sebagai berikut
1. Mutasi gen NRAS, RUNX1, NPM1, FLT3 dengan probabilitas 0,852708,
mempunyai risiko lebih tinggi menjadi MDS-related AML.
2. Mutasi gen NRAS, FLT3 dengan probabilitas 0,862000, mempunyai risiko
lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.1.
3. Mutasi gen NRAS, NPM1, FLT3 dengan probabilitas 0,969231,
mempunyai risiko tertinggi untuk menjadi MDS-related AML dibandingkan
variasi yang lain.
Variasi stastistik FLT3 (50%) yaitu 3 dari 6 variasi sebagai prediktor kuat
didapatkan insiden mutasi gen FLT3, variasi paling lemah adalah NRAS,
RUNX1, NPM1, FLT3, sedangkan yang variasi paling kuat adalah NRAS, NPM1,
FLT3. Mutasi FLT3 terdapat 25 (80,64%) dari 31 sampel dengan diagnosis AML,
mutasi gen NPM1 β 0,147 dan PR 1,158 berarti setiap peningkatan 1 satuan
NPM1, maka akan meningkatkan 1,158 kali kemungkinan terjadi MDS-related
AML (lihat tabel 5.11, 5.2).
Prediktor lemah untuk menjadi MDS-related AML yaitu kromosom 5
normal dengan variasi gen NRAS < probabilitas 0,7825584010 (tabel 5.12),
sebagai berikut:
1. Mutasi gen RUNX1, FLT3 dengan probabilitas 0,090463, mempunyai
risiko rendah untuk menjadi MDS-related AML.
2. Mutasi gen NRAS, RUNX1, FLT3 dengan probabilitas 0,534445,
mempunyai risiko rendah untuk menjadi MDS-related AML.
3. Mutasi gen FLT3 dengan probabilitas 0,351147, mempunyai risiko rendah
untuk menjadi MDS-related AML.

107
4. Mutasi gen NRAS, RUNX1, FLT3 dengan probabilitas 0,534445,
mempunyai risiko rendah untuk menjadi MDS-related AML.
5. Mutasi gen NPM1, FLT3 dengan probabilitas 0,731844, mempunyai risiko
rendah untuk menjadi MDS-related AML.
Mutasi FLT3 dengan berbagai variasi terdapat 5 (50%) dari 10 variasi
termasuk mutasi RUNX1, FLT3 dengan probabilitas 0,090463, dibandingkan
dengan probabilitas mutasi FLT3: 0,351147. Mutasi gen NRAS, NPM1, FLT3 dan
delesi kromosom 5 del(5q, mempunyai probabilitas MDS-related AML tertinggi
0,969231.. Keadaan tersebut diatas terjadi karena mutasi FLT3 β 0,147 dan PR
1,158 berarti setiap peningkatan 1 satuan FLT3, maka terjadi peningkatan 1,158
kali kemungkinan terjadi MDS-related AML. RUNX1, FLT3 mempunyai
probabilitas lebih kecil dari pada FLT3 diduga RUNX1 menghambat terjadinya
MDS-related AML.
Menurut Shih et al., 2004 studi pada orang dewasa dan anak menyatakan
bahwa diagnosis AML dengan mutasi FLT3 mempunyai prognosis buruk.
Keadaan tersebut menurut penelitian Owen and Firtzgibbon, 2012 peran sinyal
FLT3 pada leukemogenesis, gen FLT3-(ITD) Internal tandem duplication,
diprediksi mempunyai mutasi terbanyak, diperkirakan 30% dari kasus AML. Pada
kasus AML mutasi titik (point mutation) tipe missense, lebih dari 7% mempunyai
efek mengaktivasi struktur domain tyrosine kinase dari FLT3 yang berkode exon
20. Mutasi FLT3-ITD dan FLT3 domain kinase meningkatkan peran aktivitas
protein FLT3, keadaan tersebut didukung ligand-independent untuk proliferasi
dan kelangsungan hidup sel leukemia (Gililand and Griffin, 2002, Brunet et al.,
2008). Mutasi aktivitas gen FLT3-ITD merupakan sinyal abnormal termasuk
STAT5 dan menghambat faktor transkripsi myeloid seperti PU.1 dan C/EBPα.
Diskusi hasil penelitian ini yaitu mutasi gen NRAS (74,19%), RUNX1
(58,06%), NPM1 (83,87%), FLT3 (80,64%), delesi kromosom 5 del(5q) (77,81%)

108
pada kasus AML dibandingkan dengan hasil peneliti terdahulu mempunyai
prosentase insiden mutasi gen yang lebih tinggi, kecuali delesi Kromosom 5
del(5q) pada penelitian terdahulu menyatakan bahwa delesi kromosom 5 del(5q)
sebagai prediktor MDS-related AML (65-100%). Pada penelitian ini didapatkan
RUNX1 mempunyai aktivitas menghambat MDS-related AML, penelitian yang
terdahulu menyebutkan bahwa jika terdapat mutasi gen RUNX1 mempunyai
prognosis baik.
Menurut peneliti Takshashi, Hoffman et.al. bahwa mutasi faktor jalur
transduksi sinyal melibatkan mutasi SCFR (Stem cell factor reseptor) atau
tyrosine-protein kinase (KIT) dan gen fms-related tyrosine kinase 3 (FLT3) dalam
jalur transduksi yang sama. Pada penderita AML yang diperiksa dengan PCR
didapatkan delesi kromosom 5 del(5q) yang menunjukkan mutasi jalur transduksi
sinyal yaitu gen NRAS, FLT3 yang menyebabkan proliferasi atau pertumbuhan
sel tidak terkendali (Hoffman et al., 2013; Takahashi, 2011).
Hasil penelitian tersebut diatas dapat dinyatakan bahwa Mutasi NRAS,
RUNX1, NPM1, FLT3 adalah prediktor terjadinya MDS-related AML dengan
delesi kromosom 5 del(5q) (p = 0,036). Cut-off probabilitas sensitifitas 70,8% dan
spesifisitas 71,4% perubahan MDS-related AML yaitu 0,7825584010. Diskusi
tersebut diatas, pemeriksaan NRAS, RUNX1, NPM1, FLT3 dan delesi kromosom
5 del(5q) dapat dipergunakan untuk memperbaiki prediksi MDS-related AML, dan
terapi sedini mungkin. Mutasi gen RUNX1 mempunyai prognosis baik, disertai
pernyataan bahwa RUNX1 mempynyai aktivitas menghambat terjadinya MDS-
related AML.

109
6.6 Delesi kromosom 5 del(5q) sebagai prediktor MDS-related AML.
Kromosom 5: delesi kromosom 5 del(5q) mempunyai prosentase berbeda
antara kasus MDS dengan kasus AML (Walter et al., 2012; Bejar et al., 2011;
Takahashi, 2011).
Pada penelitian ini dapat sebagai prediktor kuat untuk menjadi MDS-
related AML yaitu delesi kromosom 5 del(5q) dengan variasi gen NRAS, RUNX1,
NPM1, FLT3 cut-off probbilitas > 0,7825584010 (tabel 5.12), sebagai berikut
1. Mutasi gen NRAS, RUNX1, dan NPM1 dengan probabilitas 0,833273,
mempunyai risiko tinggi menjadi MDS-related AML.
2. Mutasi gen NRAS dengan probabilitas 0.843565, mempunyai risiko lebih
tinggi menjadi MDS-related AML dibandingkan dengan ad.1.
3. Mutasi gen NRAS, RUNX1, NPM1, FLT3 dengan probabilitas 0,852708,
mempunyai risiko lebih tinggi menjadi MDS-related AML dibandingkan
dengan ad.2.
4. Mutasi gen NRAS, FLT3 dengan probabilitas 0,862000, mempunyai risiko
lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.3.
5. Mutasi gen NRAS, NPM1 dengan probabilitas 0,964532, mempunyai
risiko lebih tinggi menjadi MDS-related AML dibandingkan dengan ad.4.
6. Mutasi gen NRAS, NPM1, FLT3 dengan probabilitas 0,969231,
mempunyai risiko tertinggi untuk menjadi MDS-related AML dibandingkan
variasi yang lain.
Variasi stastistik delesi kromosom 5 del(5q) dan variasi NRAS, RUNX1,
NPM1, FLT3, kombinasi NRAS, RUNX1, NPM1 sebagai prediktor kuat untuk
progresvitas MDS-related AML, tetapi dibandingkan dengan kobinasi NRAS,
NPM1, FLT3 adalah prediktor paling kuat. Mutasi delesi kromosom 5del(5q)
terdapat 24 dari 31 sampel (74,41%) diagnosis AML, mutasi gen NRAS β 2,446

110
dan PR 11,543 berarti setiap peningkatan 1 satuan NRAS, maka akan ada
peningkatan 11,543 kali kemungkinan terjadi MDS-related AML. Mutasi gen
RUNX1 β (-) 1,694 dan PR 0,184 berarti setiap peningkatan 1 satuan RUNX1,
maka akan ada penurunan 0,184 kali kemungkinan MDS-related AML (lihat tabel
5.11, 5.2, 5.3).
Diskusi tersebut diatas, pemeriksaan NRAS, RUNX1, NPM1, FLT3 dan
delesi kromosom 5 del(5q) sebagai prediktor menjadi MDS-related AML, dan
terapi sedini mungkin.
6.7 Kromosom 5 normal sebagai prediktor MDS-related AML.
Menurut Berghe, et al., 1974 pertama kali menemukan abnormalitas
sitogenetik pada kasus MDS. Abnormalitas kromosom: delesi kromosom 5 del
(5q) mempunyai prosentase berbeda antara kasus MDS dengan kasus AML,
demikian juga pada mutasi gen NRAS, RUNX1, NPM1, FLT3
Prediktor lemah untuk menjadi MDS-related AML yaitu kromosom 5
normal dengan variasi gen NRAS, RUNX1, NPM1, FLT3 < probabilitas cut-off
0,7825584010 (tabel 5.12), sebagai berikut:
1. Mutasi gen RUNX1, dengan probabilitas 0,079074 mempunyai risiko
rendah menjadi MDS-related AML.
2. Mutasi gen RUNX1, FLT3, dengan probabilitas 0,090463 mempunyai
risiko rendah menjadi MDS-related AML tetapi potensi menjadi MDS-
related AML lebih tinggi dibandingkan dengan ad.1.
3. Mutasi gen RUNX1, NPM1, dengan probabilitas 0,302167, mempunyai
risiko rendah menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad.2.

111
4. Gen NRAS, RUNX1, NPM1, FLT3 normal dengan probabilitas 0,318429,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad.3.
5. Mutasi gen RUNX1, NPM1, FLT3 dengan probabilitas 0,334033,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 4.
6. Mutasi gen FLT3 dengan probabilitas 0,351147, mempunyai risiko rendah
untuk menjadi MDS-related AML tetapi potensi lebih tinggi dibandingkan
dengan ad 5.
7. Mutasi gen NRAS, RUNX1 dengan probabilitas 0,497750, mempunyai
risiko rendah untuk menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad.6
8. Mutasi gen NRAS, RUNX1, FLT3 dengan probabilitas 0,534445,
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 7.
9. Mutasi gen NPM1 dengan probabilitas 0,702033, mempunyai risiko
rendah untuk menjadi MDS-related AML tetapi potensi lebih tinggi
dibandingkan dengan ad 8.
10. Mutasi gen NPM1, FLT3 dengan probabilitas 0,731844, sebagai prediktor
mempunyai risiko rendah untuk menjadi MDS-related AML tetapi potensi
lebih tinggi dibandingkan dengan ad 1- 9.
Variasi stastistik delesi kromosom 5 normal dan variasi NRAS, RUNX1,
NPM1, FLT3 , mutasi tunggal gen RUNX1 sebagai prediktor paling lemah untuk
progresvitas MDS-related AML sedangkan mutasi gen NPM1, FLT3 adalah
prediktor lemah tetapi lebih kuat terjadinya MDS-related AML dibandingkan
mutasi gen RUNX1. Kromosom 5 normal terdapat 7 dari 31 sampel (22,59%)
diagnosis AML. Mutasi gen RUNX1 β (-) 1,694 dan PR 0,184, berarti setiap

112
peningkatan 1 satuan RUNX1, maka akan ada penurunan 0,184 kali
kemungkinan MDS-related AML. mutasi NPM1 β 1,618 dan PR 5,043, berarti
setiap peningkatan 1 satuan NPM1, maka akan ada peningkatan 5,043 kali
kemungkinan terjadinya MDS-related AML (lihat tabel 5.11, 5.4). Mutasi FLT3
terdapat 25 dari 31 sampel (80,64%). Mutasi FLT3 β 0,147 dan PR 1,158 berarti
setiap peningkatan 1 satuan FLT3, maka akan ada peningkatan 1,158 kali
kemungkinan MDS-related AML
Menurut penelitian Owen & Firtzgibbon, 2012 bahwa frekwensi dan
distribusi FLT3 30-33%, kromosom 5 normal. Menurut Takahashi, 2011
memberikan gambaran risiko mutasi gen pada AML, sebagai berikut : frekwensi
insiden mutasi NPM1 pada penderita AML 27,5 – 35,2%, sedangkan dengan
kromosom 5 normal didapatkan 45,7 – 53%. RUNX1 pada penderita AML de
novo (13,2%), RUNX1 pada penderita AML dengan kromosom normal (32,7%)
(Takahashi, 2011).
Menurut Falini et al., 2005, Mutasi gen NPM1 pada kasus AML dewasa
ditemukan 35% dan ditemukan mutasi gen NPM1 pada AML dengan kromosom
normal. Jumlah kasus AML dengan kromosom 5 normal antara 60 – 80%
ditemukan mutasi gen NPM1, dapat membantu prognosis kasus AML (Hoffman
et al., 2013).
Owen & Firtzgibbon, 2012 menyatakan bahwa kasus AML dengan
kromosom 5 normal terjadi mutasi gen NPM1 dengan insiden antara 45 – 55%.
Pada diskusi tersebut diatas kromosom 5 normal dan variasi mutasi gen
NRAS, RUNX1, NPM1, FLT3 digunakan sebagai prediktor lemah terjadinya
MDS-related AML.

113
6.8 Hasil penelitian dan diskusi
Didapatkan MDS-related AML mempunyai delesi kromosom 5 del(5q) : 24
sampel (77,41%), kromosom 5 (normal): 4 (22,59%), diduga AML kriteria FAB
M0 – M5 didapatkan kromosom 5 normal, mungkin AML de novo. Menurut
Horrigan et al., 1996 bahwa pada kasus MDS masih dalam proses MDS-related
AML (antara lain t-MDS, RAEB-t) insiden delesi kromosom 5 del(5q) 65-100%,
sedangkan kasus MDS yang tidak dalam proses MDS-related AML (antara lain :
RAEB) 6-7%. Penelitian Weinberg & Arber, 2010 menyatakan bahwa AML de
novo dengan dysplasia hanya 10-15% dari seluruh kasus AML (Weinberg &
Arber, 2010).
NRAS adalah anggota keluarga RAS superfamily dari GTPase. Aktivasi
mutasi exon 2 (codon 12/13)(1p13.2), exon 3 (codon 61), and exon 4 (codon
146) antara lain ditemukan pada Myeloid leukemia (14%) (Colicelli, 2004). Point
mutation NRAS adalah mutasi RAS yang paling sering pada AML, terdeteksi
antara 10% - 30% dari kasus (owen & Firtzgibbon, 2012; Hoffman et al., 2013).
Menurut Takahashi, 2011 menyatakan bahwa gambaran klinik mutasi gen NRAS
tidak secara signifikan mempengaruhi OS (over all survival), EFS (event-free
survival), dan DFS (disease-free survival), mempunyai insiden 10,3% pada kasus
AML. Fungsi jalur RAS-RAF-MAP-kinase adalah meneruskan sinyal dari
lingkungan ekstraseluler ke dalam nukleus dimana gen spesifik diaktifkan. Faktor
pertumbuhan (Epidermal Growth Factor) akan menyebabkan dimerisasi reseptor,
autofosforilasi tyrosine. Tyrosine yang terfosforilasi sebagai tempat ikatan
berafinitas tinggi bagi protein adaptor bernama Growth factor receptor-bounds2
(GRB2). GRB2 (domain SH2 dan SH3), protein ini tidak bertindak sebagai enzim
tetapi berfungsi menfasilitasi interaksi protein, termasuk interaksi yang
menghasilkan aktivitas RAS oleh GEF (guanine nucleotide exchange factors).
Varian onkogenik RAS mempunyai sifat resisten terhadap GAP (GTPase-

114
activating proteins) sehingga hidrolisis GTP terhambat, sehingga terjadi
akumulasi RAS dalam bentuk aktif. Jumlah protein RAS aktif yang dapat
meningkatkan kemungkinan transformasi (Owen & Firtzgibbon,, 2012; Ahearn et
al., 2012).
Hasil pembahasan tersebut diatas: Mutasi NRAS, RUNX1, NPM1, FLT3
dan delesi kromosome 5 del(5q) sebagai prediktor myelodysplastic syndrome-
related acute myeloid leukemia (MDS-related AML) di Surabaya. Progresivitas
MDS-related AML sebagai prediktor lemah ditunjukan oleh sitogenatik normal
yaitu kromosom 5, mutasi gen RUNX1 bersifat menghambat progresivitas
sedangkan mutasi gen NRAS, NPM1, FLT3 meningkatkan progresivitas MDS-
related AML. Prediktor kuat pada progresivitas MDS-related AML ditunjukkan
abormalitas sitogenetik yaitu delesi kromosom 5 del(5q) dan mutasi gen NRAS,
RUNX1, NPM1, FLT3. Masing-masing prediktor lemah atau kuat besarnya risiko
prediktor ditentukan oleh masing-masing gen yang mutasi, setiap mutasi gen
NRAS, RUNX1, NPM1, FLT3, sesuai urutan sebagai berikut NRAS (1), NPM1
(2), FLT3 (3), RUNX1 (4).
Patomekanisme terjadinya keganasan dan dihubungkan dengan faktor
jalur mutasi : 1) Fungsi jalur RAS-RAF-MAP-kinase adalah meneruskan sinyal
dari lingkungan ekstraseluler ke dalam nukleus dimana gen spesifik diaktifkan
(faktor mutasi jalur Transduksi sinyal). Varian onkogenik RAS (NRAS)
mempunyai sifat resisten terhadap GAP (GTPase-activating proteins) sehingga
hidrolisis GTP terhambat, sehingga terjadi akumulasi RAS dalam bentuk aktif.
Jumlah protein RAS aktif yang dapat meningkatkan kemungkinan transformasi.
2) Faktor jalur transkripsi yang berperan sebagai regulator utama pada
deferensiasi berbagai jenis perkembangan sel hematopoitik, mutasi tersebut
menghambat proses deferensiasi sel myeloid, contoh: Core Binding Factor (CBF)
adalah faktor transkripsi (RUNX1 atau AML1).

115
6.9 Kebaruan Penelitian (Novelty)
Mutasi atau normal RUNX1, NPM1, FLT3, NRAS dan delesi kromosom
5 del(5q) atau Kromosom 5 normal adalah prediktor MDS-related AML pada
penderita di Surabaya, Indonesia.
Mutasi NRAS B 2,446 dan PR 11,543 berarti setiap peningkatan
NRAS, maka akan ada peningkatan terjadinya MDS-related AML. Meknisme
terjadinya Point mutation pada gen RAS banyak ditemukan pada berbagai
tumor termasuk kanker darah yaitu pengikatan RAS berinteraksi dengan
protein sasaran (RAF-1, Phosphatidylinositol-3 kinase), SOS mengaktivasi
RAS sebagai respon terhadap faktor pertumbuhan. Pengikatan GTP pada
protein RAS membentuk kompleks GTP-RAS melalui interaksi protein lain
yaitu GTPase-activating protein (GAP). Ikatan GTP-RAS ini akan segera
inaktif menjadi bentuk GDP-RAS. Aktivitas GTPase dari RAS meningkat kalau
ada interaksi dengan protein GAP. Dengan adanya point mutation pada gen
RAS akan menurunkan aktivitas GTPase, akibatnya ikatan GTP-RAS akan
inaktif secara perlahan-lahan sehingga akan menimbulkan respon seluler
yang berlebihan terhadap sinyal dari reseptor. Jumlah protein RAS aktif yang
dapat meningkatkan kemungkinan transformasi, Kompleks GTP-RAS akan
mentransmisikan sinyal di dalam sel.
Mutasi gen RUNX1 B - 1,694 dan PR 0,184, berarti setiap peningkatan
RUNX1, maka akan terjadi penurunan kemungkinan terjadinya MDS-related
AML. Menurut referensi, mekanisme terjadinya hipotesis menurunnya aktivitas
RUNX1 dapat meningkatkan leukemogenesis, yaitu Leukemia ditemukan rata-
rata jauh lebih tinggi pada keluarga dengan dominan kuat mutasi K83E (57%)
dibandingkan dengan delesi lengkap RUNX1 (24%), Oleh karena itu, aktivitas
rendah RUNX1 diduga berhubungan dengan tingginya leukemogenisitas.
Penelitian terbaru kebanyakan mengindikasikan bahwa fungsi utama dari

116
chimeric genes adalah inhibisi transdominan dari wild-type RUNX1,
menyebabkan turunnya aktivitas RUNX1 secara drastis di sel yang
terpengaruh. Pada akhir-akhir ini lebih dari 10 partner gen yang ber fusi
dengan RUNX1 telah diidentifikasi di dalam translokasi kromosom. Setiap
chimeric gene mungkin mempunyai mekanisme leukemogenesis nya sendiri,
kekurangan RUNX1 mungkin menjadi mekanisme yang sama pada leukemia
dengan mutasi RUNX1. Semakin rendah aktivitas RUNX1, semakin tinggi
leukemogenisitas

116
BAB 7
KESIMPULAN DAN SARAN
7.1 Kesimpulan
a. Polimorfisme NRAS sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
b. Polimorfisme RUNX1 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
c. Polimorfisme NPM1 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
d. Polimorfisme FLT3 sebagai prediktor perubahan Myelodisplastic
Syndrome menjadi Acute Myeloid Leukemia (MDS-related AML).
e. Polimorfisme delesi kromosom 5 del(5q) sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-
related AML).
f. Polimorfisme kromosom 5 normal sebagai prediktor perubahan
Myelodisplastic Syndrome menjadi Acute Myeloid Leukemia (MDS-
related AML).
g. Mutasi NRAS, RUNX1, NPM1, FLT3 dan delesi kromosome 5 del(5q)
sebagai prediktor myelodysplastic syndrome-related acute myeloid
leukemia (MDS-related AML) di Surabaya. Progresivitas MDS-related
AML sebagai prediktor lemah ditunjukan oleh sitogenatik normal yaitu
kromosom 5, mutasi gen RUNX1 bersifat menghambat progresivitas
sedangkan mutasi gen NRAS, NPM1, FLT3 meningkatkan
progresivitas MDS-related AML. Prediktor kuat pada progresivitas
MDS-related AML ditunjukkan abormalitas sitogenetik yaitu delesi
kromosom 5 del(5q) dan variasi mutasi gen NRAS, RUNX1, NPM1,

117
FLT3. Masing-masing prediktor lemah atau kuat besarnya risiko
prediktor ditentukan oleh masing-masing gen yang mutasi, setiap
mutasi gen NRAS, RUNX1, NPM1, FLT3, sesuai urutan sebagai
berikut NRAS (1), NPM1 (2), FLT3 (3), RUNX1 (4).
7.2 Keterbatasan
a. Pada penelitian ini tidak menggunakan sistem kwantitatif, untuk
mendapatkan hasil penelitian yang lebih akurat menggunakan
kwantitatif RT-PCR, dan kwantitatif FISH.
b. Faktor etiologi tidak tampak pada hasil penelitian ini, oleh karena itu
perlu dilakukan penelitian dengan menggunakan sampel yang besar
dan memperhitungkan lokasi tempat tinggal, pekerjaan, gaya hidup,
seandainya memungkinkan pada pekerjaan yang mempunyai faktor
kuat terjadinya Leukemia (zat kimia Benzena C6H6).
c. Sampel dengan diagnosis MDS dengan karakteristik sampel dan
metode seperti tersebut diatas, diikuti untuk menentukan prediktor
MDS-related AML dengan menggunakan gen NRAS, RUNX1, NPM1,
FLT3, delesi kromosom 5 del(5q) dan waktu dalam bulan atau tahun.
7.3 Saran
1. Penelitian selanjutnya menggunakan jumlah sampel lebih besar,
pemeriksaan sumsum tulang (berdasarkan morfologi),
Immunophenotyping, pemeriksaan macam variasi mutasi gen yang
terlibat dalam komponen MDS-related AML
2. Abnormalitas sitogenetik menggunakan macam kromosom lebih banyak.
Pemeriksaan gen menggunakan PCR, pemeriksaan kromosom
menggunakan FISH (Flourecene In Situ Hybridization).

118
3. Penelitian yang menghubungkan antara waktu terjadinya transformasi
(MDS-related AML) dengan variasi genetik dan kromosom.

119
DAFTAR PUSTAKA
Agersborg S., Thangavelu M., Ma W., et al., Classification of Acute Myeloid
leukemia and Myelodysplastic Syndrome based on Molecular Profiling,
blood 2015; 126: 3836: P. 1 – 5.
Agrawal S., Koschmieds S., Baumer N. Pim2 complements FLT3 wild-type
receptor in hematopoitic progenitor cell transformation, Leukemia, 2008;
22: 78 – 86.
Ahearn I.M., Haigis k., sagi D.B. Regulating the regulator: post-translational
modification of RAS, Nature review molecular cell biology, 2012; 10 : 1-4.
Alcindor T., Bridges KR., Sideroblastic Anemias, Revused, Havard, 2001; 1 – 10
Alpdogan O., Advances in immune regulator in transplantation, Dep.Medical
oncology, 2013 :1-33.
Andrew k., Duployez N., BoisselN., et al., Acute Myeloid Leukemia: The good,
the bad, and the Ugly, American Society of Clinical Oncology, Asco
Educational Book, 2018; P.555 - 570
Annu Rev Genet. 1998;32:495-519
Aulya Z.V., Arthamin M.Z., Chilmi S., Widodo M.A., Sujuti H. Kombinasi
Elektroporasi dan Apirin Menghambat Aktivasi Nuclear Factor Kappa B
(NFkB) pada kultur Sel Mononuklear Darah Tepi Pasien leukemia Akut.
Majalah Kesehatan Fakultas kedokteran Universitas Brawijaya; 2014: 1(1)
P. 10 – 15.
Badar T., Patel K.P., Thompson P.A., et al., detectable FLT3 or RAS mutation at
the time of transformation from MDS to AML predicts for very poor
outcomes, Departemen of leukemia, The university of Texas MD
Anderson Cancer Center, Houston, Texas, USA 2014.
Bain BJ., Bates I., laffan MA., lewis S.M., practical haematology in: Molecular and
cytogenetic analysis, 11th Ed. Churchill livingstone, 2012 P. 139 – 174.
Bains A., Luthra R, Medeiros LJ. FLT3 AND NPM1 Mutations in Myelodysplastic
Syndromes: Frequency and Potential Value for Predicting Progression to
Myeloid Leukemia. Am J Clin Pathol. 2011; 135(1): 62 – 69.

120
Bajaj R., Fang Xu, Bixia Xiang, Wilox K., et al., Evidence-based genomic
diagnosis characterized chromosomal and cryptic imbalances in 30
elderly patients with myelodysplastic syndrome and acute myeloid
leukemia, 2011; 4: 1-10
Banerjee A., Grumont R., gugasyan R., et al., NF-kB and C-Rel cooperate to
promote the survival of TRL4-activated B cells by neutralizing BIM via
distinct mechanisms, Blood, 2008; 112: 5063 – 5073.
Bejar R., Stevenson K., abdel-Wahab O., Galili N. Clinical Effect of Point
Mutations in Myelodysplastic syndrome, N Engl J Med 2011; 363: 1-5.
Besa E.C., Myelodysplastic syndrome, Medscape reference, 2011 : 1-7
Boissel N., Leroy H, Brethon B., et al., Incidence and prognostic impact of c-Kit,
FLT3, and Ras gene mutations in core binding factor acute myeloid
leukemia. Leukemia 2006; 20: P. 965 - 970.
Brunet S., Esteve J., Nomdedeu J., Ribera J.M. Prognostic significance of
mutations of nucleophosmin (NPM1) gene and interal tandem duplication
of FLT3 gene (FLT3-ITD) in acute myeloid leukemia (AML) with normal
karyotype. J Clin. Oncol 2008 Maay 20; 26 : 7024.
Cazzola M., Myelodysplastic syndrome with isolated 5q delation (5q-syndrome).
A clonal stem cell disorder characterized by defective ribosome
biogenesis, haematologica, 2008, 13377(93): P.967 – 972.
Ceesay MM, Wendy I, Mufti G. Molecular Hematology. In: Provan D, Gribben J
G, editors. Myelodysplastic Syndrome. Third Edition,London: Wiley-
Blackwell; 2012.P.89 - 103.
Chen G., Zeng W., Myazato A. Distinctive gene expression profile of CD34 cells
from patients with myelodysplastic syndrome characterized by specific
chromosomal abnormalities. Blood 2004 Dec.15;104(13):4210 – 4218.
Chopoval S. Regulation of the T helper Cell type 2 (Th2)/T regulatory cell (Treg.)
balance by Il-4 and STAT6, Journal of leukocyte biology; 87., 2010,P.
1011 – 1017.
Colicelli J., Human RAS superfamily Proteins and Related GTPases, Cell
Signaling Technology. 2004; 250: P.1-32.

121
Deschler B, Lϋbbert M. Acute Myeloid Leukemia: Epidemiology and etiology
Cancer 2006; 107 (9): 2099-2107.
Dinata M., Rahaju P., Ugroseno S.Y.B., Widjjanto E. NPM1, FLT3 and deletion of
chromosome 5 del(5q) as predictor of myelodysplastic syndrome (MDS)
to be acute myelocytic leukemia (AML) in Surabaya, Durg Invention
Today (Dit). 2019, P.1841-1845
Do̎hner H., Estey E.H., Amadori S., et al., Diagnosis and management of acute
myeloid leukemia in adults: recommendations from an international expert
panel, on behalf of the European LeukemiaNet; Blood: 115; 3 : 2010.
Falini B., Bolli N. Altered Nucleophosmin transport in acute myeloid leukemia with
mutated NPM1: molecular basis and clinical implications. Leukemia 2009
october; 23: 1731 – 1743.
Falini B., Mecccci C., Tiacci E. Cytoplasmic Nucleophosmin in Acute
Myelogenous Leukemia with a Normal Karyotype.N Engl J Med 2005;
352:254 – 266.
Farr C.J., Saiki R.K., Erlich H.A., et al., Analysis of RAS gene mutations in acute
myeloid leukemia y polymerase chain reaction and oligonucleotide
probes, Medical Sciences, 1988; 85: P. 1629 – 1633.
Fitriani G.P. Laporan Kasus Acute Myeloblastic Leukemia (AML); bagian Ilmu
Kesehatan Anak, Fakultas kedokteran UNLAM-RSUD Uli, Banjarmasin;
2009; P.1- - 49.
Gelsi-Boyer V., Trouplin V., Adelaide J., Aceto N. Genome profiling of chronic
myelomonocytic leukemia: frequent alterations of RAS and RUNXI genes,
BMC Cancer 2008; 8 (299): P.1-12.
Gilliland D.G., Griffin J.D. the roles of FLT3 in hematopoiesis and leukemia,
Blood 2002; 100(5): P. 1532-1542
Gilliland DG, Gribben JG. Molecular Hematology. In: Provan D, Gribben J G,.
Secondary myelodysplasia/acute myelogenous leukemia: assessment of
risk. Third Edition,London: Wiley-Blackwell; 2012.P.51-56.
Gutierrez S.E., Romero-Oliva F.A., Epigenetic changes: a common theme in
acute myelogenous leukemogenesis, Journal of Hematology & Oncology
2013; 6:57; P. 1-14.

122
Hajian-Tilaki K., Receiver operating Characteristic (ROC) Curve Analysis for
Medical Diagnostic Test Evaluation, Caspian J intern med 4(2), 2013
P.627 – 635.
Havard health Publication, Acute Myeloid Leukemia (AML), medically reviewed ;
2017
Hoffbrand A.V., Moss P.A.H., Essential Haematology. 6th Edition, Wiley-
Blackwell; 2011 P. 1150 – 222.
Hoffman R., Benz E.J.Jr, Silberstein L.E. Hematology Basic Principles and
practice In: Quinitas-Cadarma A., Cortes JE., Kantarjian H. Biology of
chronic and Acute Myeloid Leukemia. 6th Edition, Elservier Saunders;
2013 p. 853 – 862.
Horrigan SK., Westbrook CA., Kim AH., Banerjee M., et al., Polymerase chain
reaction-based diagnosis of del(5q) in acute myeloid leukemia and
myelodysplastic syndrome identifies a minimal deletion interval, Blood,
1996; P: 2665-2670.
Hulley S.B, Cummings S.T. designing clinical research an epidemiologic
approach. In: Newman T.B., Browner W.S., CummingsS.T., Hulley S.B.
Designing a new study: II. Cross-sectional and case-control studies.
United State of America United State of America; 1988 P.75 – 97.
Jabbour E., Kantarjian H. Hand Book of Cancer Chemotherapy. In: Skeel R.T.,
Khleif S.N., Myeloproliferative Neoplasms and Myelodysplastic
Syndromes. 8th Edition, Lippincott Williams & Wilkins; 2011. P. 418-436.
Jiang Y, Dunbar A, Gondek LP, et.al Aberrant DNA methylation is a dominant
mechanism in MDS progression to AML. Blood. 2009;113(6): P. 1315–
1325.
Juniarka I.G.A. RUNX1, Program Pasca sarjana Ilmu Farmasi Universitas Gajah
Mada, 2011. P. 1 - 11
Killick SB., Carter C., Culligan D., Guidelines for the Diagnosis and Management
of Adult Myelodysplastic Syndromes. Br.J.Haematology 2014; 164(4):
P.503 – 25.
Klepin H.D., Rao A.V., Pardee T.S., Acute myeloid leukemia and Myelodysplastic
Syndromes in Older Adults, J clin Oncol 32: 2014: P.2541-2552.

123
Kresno S.B. Ilmu Dasar Onkologi, Edisi Kedua, Badan Penerbit Fakultas
Kedokteran universitas Indonesia; 2012: 112-371
Kubasch A.S., Platzbecker U., Beyond the Edge of Hypomethylating Agents:
Novel Combination Strategies for Older Adults with Advanced MDS and
AML, Cancers; 2018.
Ley C.M., Miller C., Ding L., Genomic and Epigenomic landscapes of Adult De
Novo Acute myeloid Leukemia, N Eng J Med 2013; 368: 2059 – 2074.
Loegito H.M., Dasar-dasar Biologi sel dan Biologi Molekuler, 2010. P. 198 – 242.
Lumongga F. Invasi Sel Kanker, Departemen Patologi Anatomi Fakultas
Kedokteran Universitas Sumatera utara, Medan. USU Repository 2008.
P. 1 – 11.
Luzzatto L, kadardimitris A., myelodysplastic syndrome, Molecular Hematology,
third Ed. Wiley-Blackwell, 2010, P: 89 – 103.
Mallo M., Arenillas L., Espinet B., et al., Fluorescene in situ hybridization
improves the detection of 5q31 deletion in myelodysplastic syndrome
without cytogenetic evidence of 5q-, Haematologica, 2008; p: 1001-1008.
Matutes E, Morilla R, Morilla AM. Practical Haematology. In: Bain JB, Bates I,
Laffan MA, Lewis SM. Immunophenotyping. Eleventh Edition, London:
Churchill Livingstone; 2012.P.353 – 371.
Michaud J., Simpson K.M., Escher R. Intergrative analysis of RUNX1
downstream pathways and target genes. BMC Genomics 2008 July 31, 9;
363 : 1-17.
Mohamedali A., Mufti G.J., Van-den Berghe’s 5q-syndome in 2008. Bjh. 2008;
144 : 157-168.
Montalban-Bravo G., Garcia-Manero G., Myelodysplastic Syndromes: 2018
update On diagnosis, risk-stratification and management, Annual clinical
Updates in Hematological Malignancies; American Journal of
Hematology; 93, 2018.
Montalban-Bravo G., Lee E., Merchan B., et al., Integrating Genetics and
Epigenetics In MDS: Advances in Pathogenesis and Disease Evolution.
Br J Haematol. 2014 ; 166(5): P. 646–659.

124
Natelson E.A., Pyatt D. acquired Myelodysplasia or Myelodysplastic Syndrome:
Clearing the Fog, Advances in Hematology 2013; 2013(11): P. 1-11.
Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX
leukemia. Oncogene 2004; 23: 4284 – 4296.
Ottone T., Zaza S., Divona M., et al., Identification of emerging FLT3 ITD-positive
clones during clinical remission and kinetics of disease relapse in acute
myeloid leukemia with mutated nucleophosmin, British Journal of
Haematology(bjh), 2013; 161: P.533 – 540.
OwenC.J., Fitzgibbon J. Molecular Hematology. In: Provan D, Gribben J G,
editors. the genetic of acute myeloid leukemias. Third Edition,London:
Wiley-Blackwell; 2012.P. 42 - 49.
Pirruccello J.S., Young K.H., Aoun P. Myeloblast phenotypic changes in
Myelodysplasia. Am J Clin Pathol 2006; 125: 884 – 984.
Provan D, Gribben J.G., molecular hematology. 3th, London, UK: Wiley-Blackwell;
2012. P. 42 – 163.
Rahim A., Suaniti N.M., Rita W.S. analisis Fenol dalam Urine Pekerja salah satu
stasiun Pengisian Bahan Bakar Umum di kota Denpasar, Kimia FMIPA
Universitas Udayana, Bukit Jimbaran, Bali.Jurnal Kimia 2015; 9(1): P. 105
– 108
Rocquain J., Carbuccia N., Trouplin V., Raynaud S., et al., Combined mutations
of ASXL1, CBL, FLT3, IDH1, IDH1, JAK2, KRAS, NPM1, NRAS, RUNX1,
TET2 AND WT1 genes in myelodysplastic syndromes and acute myeloid
leukemias, BMC cancer 2010; 10;401:P. 1-7.
Rowley J.D., The critical role of chromosome translocations in human leukemias
1998. Annu. Rev. Genet.; 32: P. 495-519.
Saunthararajah Y., Triozzi P., Rini B., et al., P53-Independent, normal stem cell
sparing epigenetic-differentiation therapy for myeloid and other
malignancies. 2012 ; 39(1): 97–108.
Shih L-Y, Huang C-F., Wang P-N., Wu J-H, et al., Acuquisition of FLT3 or N-ras
mutations is frequently associated with progression of myelodysplastic
syndrome to acute myeloid leukemia, Leukemia; 2004; 18;466-475.

125
Shiozawa Y., Malcovati L., Galli A. et al., Gene expression and risk of leukemic
transformation in myelodysplasia; Blood; 2017
Simanjorang C., Adisasmita A.C., Tehuteru E.S. Gambaran epidemiologi Kasus
Leukemia Anak di Rumah Sakit Kanker “Dharmais”, 2004-2008,
Indonesian Journal of Cancer 2010; 4 (1) : 15-21.
Snyder R., Leukemia and Benzene, international Journal of environmental
Reserch and public health 2012; 9(8): P.2875 – 2893.
Sukartini N., Aspek Laboratorium Sitogenetik dan Molekuler Genetik pada
leukemia, Pertemuan Ilmiah Tahunan XII Perhimpunan Dokter Spesialis
Patologi Klinik Indonesia, 26-28 September 2013. P. 25 – 30.
Takahashi S. Current findings for recurring mutations in acute myeloid
leukemia, Journal of Hematology & Onkology 2011; 4 (36): 1-8.
Tan A.YC., Westerman D.A., Carney D.A. Detection of NPM1 exon 12 mutations
and FLT3 – internal tandem duplications by high resolution melting
analysis in normal karyotype acute myeloid leukemia. Journal of
Hematology&Oncology 2008 July 29 ; 1: 10.
Taniuchi I., Osato M., Ito Y. Runx1: no longer just for Leukemia, The EMBO
Journal 2012; 31: P.4098 – 4099.
Tefferi A., Vardman J.M.D., Myelodysplastic syndromes, N Engl. J.Med., 2009;
361: P. 1872 – 1885.
Van den Berghe H., Cassiman J.J., David G., et al., Distinct haematological
disorder with deletion of long arm of no 5 chromosome. Nature.
1974;251:437–438.
Vardiman J.W., Harris N.L., Brunning R.D. The world Health organization (WHO)
classification of the myeloid neoplasms. Blood 2002; 100: 2292 – 2302.
Walter M.J., dong S., Li D., et al., Clonal Architecture of Secondary Acute
myeloid Leukemia, N Engl J Med 2012; 366(12): P.1090-1093.
Weinberg O.K., Arber D.A. Acute Myeloid Leukemia with myelodysplasia related
changes: a new definition. Surgical Pathology 3; 2010; P.1153–1164

126
Wetzler M, Marcucci G, bloomfield CD. Harrison’s principles of Internal Medicine.
In: Longo DL, Kasper Dl, Jameson, et al., Acute and chronic Myeloid
leukemia. 18th Edition, United State of America; 2012.P. 905 – 914.
Windhu Purnomo., Taufan Bramantoro. Pengantar Metodologi Penelitian bidang
kesehatan. Airlangga University Press, 2018 P.1-34.
Young NS. Harrison’s principles of Internal Medicine. In: Longo DL, Kasper Dl,
Jameson, et al., Apalstic anemia Myelodysplasia, and releted bone
marrow failure syndrome. 18th Edition, United State of America;
2012.P.887- 897.
Yusuf A.H. Aspek Genetika Kanker, Bagian Histologi, Fakultas kedokteran
Universitas Indonesia, Jakarta; 2008 P.1 – 9.

DATA SAMPEL PENELITIAN S3 2017/2018
No Nama Jenis
Kelamin Umur
(Tahun) Immunophe
notyping
Sitogenetik Molekuler/Kromosom Diagnosis
PK/peneliti/hom Sampel Diagnosis
NRAS RUNX1 NPM1 FLT3 DEL(5q) 1 NI 2 28 1 1 1 0 1 0 AML/AML/AML BMA 1
2 NA 1 48 1 1 0 0 1 1 AML/AML/AML Darah/BMA (Meninggal)
1
3 MO 1 74 1 1 0 1 1 1 AML/AML/MDS BMA
(Meninggal) 2
4 MI 1 81 1 1 1 1 1 1 AML M1/AML/AML BMA 1 5 SU 2 71 1 1 1 1 1 1 AML/AML /MDS BMA/Darah 2 6 ABR 1 36 1 1 0 1 1 1 AML M7/AML/AML M1 BMA 1
7 BUN 2 30 1 0 0 1 1 0 AML M3
VARIAN/AML/AML BMA 1
8 BUM 1 42 1 1 1 1 0 1 AML M3/AML/AML M3 BMA 1 9 ROS 1 40 1 1 1 0 1 0 AML M7/AML/AML BMA 1
10 ENR 2 26 1 1 1 0 1 1 AML M3
VARIAN/AML/AML Darah/BMA 1
11 AFA 1 18 1 0 0 1 1 1 AML/AML/AML M5 Darah/BMA 1 12 BEN 1 53 1 1 0 1 0 1 AML/AML/AML BMA I/II 1 13 PWA 1 24 1 1 0 1 1 1 AML M5/AML/AML M4 BMA 1 14 SAM 1 67 1 1 0 1 1 1 AML M7/AML/AML M4 BMA 1
15 TNA 2 75 1 0 1 1 1 1 AML/AML/MDS Perifer/Diagnosis dari Singapore
Post Kemoterapi 2
16 ARI F. 1 60 1 1 0 1 1 1 AML/AML/AML BMA 1 17 MHA 1 55 1 0 0 1 1 1 AML/AML/AML BMA 1 18 RIF 2 50 1 1 1 1 1 1 AML/AML/AML BMA 1
19 RIS 2 37 1 1 1 1 1 1 Tidak
Representative(I) AML/AML/ AML(II)
Darah/BMA 1
20 SUKA 1 61 1 0 0 1 0 1 AML M2/AML/AML BMA 1 21 NRH 2 65 1 1 1 1 1 0 AML/AML/AML BMA 1 22 ARI 1 40 1 1 1 1 1 1 AML/AML/AML BMA 1
23 NRD 1 44 1 1 1 1 1 1 AML M3/AML/AML BMA/Darah
Perifer 1
24 YLI 2 42 1 1 1 1 1 1 AML M5/AML/AML BMA 1
25 MFS 1 21 1 1 1 1 1 1 AML/AML/MDS BMA/Darah
Perifer 2
26 SJR 1 45 1 1 0 1 0 1 AML/AML M7/AML(I) Post Kemoterapi(II)
BMA 1
27 SPD 1 58 1 1 1 1 1 0 AML/AML M2/AML BMA 1 28 MAS 1 53 1 0 1 1 1 0 AML/AML/AML BMA 1

29 AJR 2 17 1 0 1 1 1 1 AML M7/AML/AML M7 BMA/Darah
Perifer. (Meninggal)
1
30 HAF 1 59 1 0 0 0 1 1 AML/AML/AML M2 BMA 1
31 MIS 1 57 1 1 1 0 1 1
AML/AML/AML BMA 1
32 ANS 1 35 1 1 1 1 1 1 AML/AML M7/AML BMA 1 33 IMM 1 18 1 1 1 1 0 1 AML/AML M7/AML Darah/BMA 1 34 SFR 2 50 1 1 1 1 1 0 AML/AML/MDS Darah/BMA 2 35 SHO 1 64 1 1 1 1 1 1 AML/AML/AML M3 BMA 1 36 ADH 2 42 1 0 0 1 0 0 AML/AML/AML M3 BMA 1
KETERANGAN :
• Jenis kelamin : Laki (1), Perempuan (2)
• Immunophenotyping : Myeloid Lineage (AML) (1), Non Myeloid Leneage (0)
• Pemeriksaan sitogenetik: o RUNX1 : RUNX1 ABNORMAL (1), RUNX1 NORMAL (0) o FLT3 : FLT3 ABNORMAL (1), FLT3 NORMAL (0) o NRAS : NRAS ABNORMAL (1), NRAS NORMAL (0) o NPM1 : NPM1 ABNORMAL (1), NPM1 NORMAL (0) o Del(5q) : KROMOSOM Del(5q) (1), KROMOSOM (5q) NORMAL (0)
• Diagnosis : AML (1), MDS (2)
• Nomor: 3, 5, 15, 25, 34 : EKSKLUSI


127
Lampiran 1 Surat Keterangan Bebas Plagiasi

128
Lampiran 2 Etichal clearance

129
Logistic Regression
VARIABLES del(5q) MDS.related.AML
METHOD=ENTER RUNX, FLT3, NRAS, NPM1
CRITERIA=PIN (.05) POUT (.10) ITERATE (20) CUT (.5).
Logistic Regression
Data Analysis\Mulya Dinata
Case Processing Summary
Unweighted Casesa N percent
Selected Cases Included in Analysis 31 100.0
Missing Cases 0 .0
Total 31 100.0
Unselected Cases 0 .0
Total 31 100.0
a. If weight is in effect, see classification table for the total number of cases
Dependent Variable Encoding
Original value Internal Value (-) 0 (+) 1
Block 1 : Method = Enter
Omnibus Tests of Model Coefficients
Chi-square df Sig.
Step 1 Step 5.281 4 .260
Block 5.281 4 .260
Model 5.281 4 .260
Model Summary
Step 1 -2 log likelihood Cox & Snell R square Nagelkerke R square
1 27.837a .157 .239 a. Estimation terminated at iteration number 5 because parameter estimates
changed by less than .001
Classification Tableᵃ
Predicted
Del(5q) MDS related AML
Precentage Corrected
Observed (-) (+)
Step 1 Del(5q) MDS related AML (- ) 1 6 14.3
(+) 1 23 95,8
Overall Percentage 77.4 a. The cut value is .500

130
Variables in the Equation
B(β) S.E Wald df Sig. Exp(B)
Step 1a RUNX1 -1.694 1.332 1.617 1 .204 .184
FLT3 .147 1.343 .012 1 .913 1.158
NRAS 2.446 1.360 3.236 1 .072 11.543
NPM1 1.618 1.245 1.688 1 .194 5.043
CONSTANT -.761 1.901 .160 1 .689 .467
a. Variable(s) entered on step 1 : RUNX1, FLT3, NRAS, NPM1 Probabilitas
{𝐝𝐞𝐥(5q)=1}=1/1+e-(β0 +β1.RUNX1+β2.FLT3+β3.NRAS+β4.NPM1)
ROC Curve Data Analysis\Mulya Dinata Case Processing Summary
Del(5q) MDS related AML Valid N (listwise)
Positvea 24
Negative 7
Larger values of the test result variable(s) Indicate stronger evidence for a positive Actual state a. The positive actual state is (+).
ROC CURVE

131
Area Under the Curve
Test Result Variable(s) : a
Prob. MDS related AML
Asymptotic
Sig.b
Asymptotic 95% Confidence Interval
Area Std.Error Lower bound Upper bound
.765 .097 .036 .574 .956
The test result variable(s): Prob.MDS.related.AML has at least one tie between the positive actual state group and the negative actual state group. Statistic may be biased a. Under the nonparametric assumption b. Null hypothesis : true area = 0.5
Coordinates of the curve
The result Variable(s): Prob.MDS related AML
Positive if Greater Than or Equal
To a
Sensitivity 1 – specificity
0.0000000000 1.000 1.000
.4342392130 .958 .857
.6182394205 .875 .571
.7169388935 .833 .429
.7825584010 .708 .286
.8429902415 .625 .286
.8573538955 .292 0.000
.9132656980 .250 0.000
.9668813630 .167 0.000
1.0000000000 .000 0.000
The test result variable(s): Prob.MDS.related.AML has at least one tie between the positive actual state group and the negative actual state group a. The smallest cutoff value is the minimum observed test value minus 1, and the largest cutoff value is the maximum observed test value plus 1. All the other cutoff values are the averages of two consecutive ordered observed test values.
RECODE Prob. MDS related AML (0.7825584010 thru Highest=1) (ELSE=0) INTO MDS.related.AML.
Sensitivity Specificity 1 – specificity
0.0000000000 1 1.000 0.000 1.000
.4342392130 2 .958 0.143 .857
.6182394205 3 .875 0.429 .571
.7169388935 4 .833 0.571 .429
.7825584010 5 .708 0.714 .286
.8429902415 6 .625 0.714 .286
.8573538955 7 .292 1.000 0.000
.9132656980 8 .250 1.000 0.000
.9668813630 9 .167 1.000 0.000
1.0000000000 10 .000 1.000 0.000

132
NO RUNX1 FLT3 NRAS NPM1 Prob
MDS related AML
MDS related AML
1 1 0 0 0 0.079074 0
2 1 1 0 0 0.090463 0
3 1 0 0 1 0.302167 0
4 1 0 0 1 0.302167 0
5 0 0 0 0 0.318429 0
6 1 1 0 1 0.334033 0
7 0 1 0 0 0.351147 0
8 1 0 1 0 0.497750 0
9 1 1 1 0 0.534445 0
10 0 0 0 1 0.702033 0
11 0 1 0 1 0.731844 0
12 1 0 1 1 0.833273 1
13 0 0 1 0 0.843565 1
14 1 1 1 1 0.852708 1
15 0 1 1 0 0.862000 1
16 0 0 1 1 0.964532 1
17 0 0 1 1 0.964532 1
18 0 1 1 1 0.969231 1

133
Probabilitas Variasi Gen RUNX1, FLT3, NRAS, NPM1Terhadap MDS-related AML
NO RUNX1 FLT3 NRAS NPM1 PROB
1 1 1 1 0 0.534445353
2 0 1 1 0 0.861999811
3 0 1 1 1 0.969231141
4 1 1 1 1 0.85270798
5 1 1 1 1 0.85270798
6 0 1 1 1 0.969231141
7 0 1 0 1 0.731844299
8 1 0 1 1 0.833272503
9 1 1 1 0 0.534445353
10 1 1 1 0 0.534445353
11 0 1 0 1 0.731844299
12 0 0 1 1 0.964531585
13 0 1 1 1 0.969231141
14 0 1 1 1 0.969231141
15 1 1 0 1 0.334033073
16 0 1 1 1 0.969231141
17 0 1 0 1 0.731844299
18 1 1 1 1 0.85270798
19 1 1 1 1 0.85270798
20 0 0 0 1 0.702033488
21 1 1 1 1 0.85270798
22 1 1 1 1 0.85270798
23 1 1 1 1 0.85270798
24 1 1 1 1 0.85270798
25 1 1 1 1 0.85270798
26 0 0 1 1 0.964531585
27 1 1 1 1 0.85270798
28 1 1 0 1 0.334033073
29 1 1 0 1 0.334033073
30 0 1 0 1 0.731844299
31 1 1 1 0 0.534445353
32 1 1 1 1 0.85270798
33 1 0 1 1 0.833272503
34 1 1 1 0 0.534445353
35 1 1 1 1 0.85270798
36 0 0 0 1 0.702033488

134
DATA SAMPEL PENELITIAN S3 2017/2018
No Nama Jenis
Kelamin Umur
(Tahun) Immunophe
notyping
Sitogenetik Molekuler/Kromosom Diagnosis
PK/peneliti/hom Sampel Diagnosis
NRAS RUNX1 NPM1 FLT3 DEL(5q) 1 NI 2 28 1 1 1 0 1 0 AML/AML/AML BMA 1
2 NA 1 48 1 1 0 0 1 1 AML/AML/AML Darah/BMA (Meninggal)
1
3 MO 1 74 1 1 0 1 1 1 AML/AML/MDS BMA
(Meninggal) 2
4 MI 1 81 1 1 1 1 1 1 AML M1/AML/AML BMA 1 5 SU 2 71 1 1 1 1 1 1 AML/AML /MDS BMA/Darah 2 6 ABR 1 36 1 1 0 1 1 1 AML M7/AML/AML M1 BMA 1
7 BUN 2 30 1 0 0 1 1 0 AML M3
VARIAN/AML/AML BMA 1
8 BUM 1 42 1 1 1 1 0 1 AML M3/AML/AML M3 BMA 1 9 ROS 1 40 1 1 1 0 1 0 AML M7/AML/AML BMA 1
10 ENR 2 26 1 1 1 0 1 1 AML M3
VARIAN/AML/AML Darah/BMA 1
11 AFA 1 18 1 0 0 1 1 1 AML/AML/AML M5 Darah/BMA 1 12 BEN 1 53 1 1 0 1 0 1 AML/AML/AML BMA I/II 1 13 PWA 1 24 1 1 0 1 1 1 AML M5/AML/AML M4 BMA 1 14 SAM 1 67 1 1 0 1 1 1 AML M7/AML/AML M4 BMA 1
15 TNA 2 75 1 0 1 1 1 1 AML/AML/MDS Perifer/Diagnosis dari Singapore
Post Kemoterapi 2
16 ARI F. 1 60 1 1 0 1 1 1 AML/AML/AML BMA 1 17 MHA 1 55 1 0 0 1 1 1 AML/AML/AML BMA 1 18 RIF 2 50 1 1 1 1 1 1 AML/AML/AML BMA 1
19 RIS 2 37 1 1 1 1 1 1 Tidak
Representative(I) AML/AML/ AML(II)
Darah/BMA 1
20 SUKA 1 61 1 0 0 1 0 1 AML M2/AML/AML BMA 1 21 NRH 2 65 1 1 1 1 1 0 AML/AML/AML BMA 1 22 ARI 1 40 1 1 1 1 1 1 AML/AML/AML BMA 1
23 NRD 1 44 1 1 1 1 1 1 AML M3/AML/AML BMA/Darah
Perifer 1

135
24 YLI 2 42 1 1 1 1 1 1 AML M5/AML/AML BMA 1
25 MFS 1 21 1 1 1 1 1 1 AML/AML/MDS BMA/Darah
Perifer 2
26 SJR 1 45 1 1 0 1 0 1 AML/AML M7/AML(I) Post Kemoterapi(II)
BMA 1
27 SPD 1 58 1 1 1 1 1 0 AML/AML M2/AML BMA 1 28 MAS 1 53 1 0 1 1 1 0 AML/AML/AML BMA 1
29 AJR 2 17 1 0 1 1 1 1 AML M7/AML/AML M7 BMA/Darah
Perifer. (Meninggal)
1
30 HAF 1 59 1 0 0 0 1 1 AML/AML/AML M2 BMA 1
31 MIS 1 57 1 1 1 0 1 1
AML/AML/AML BMA 1
32 ANS 1 35 1 1 1 1 1 1 AML/AML M7/AML BMA 1 33 IMM 1 18 1 1 1 1 0 1 AML/AML M7/AML Darah/BMA 1 34 SFR 2 50 1 1 1 1 1 0 AML/AML/MDS Darah/BMA 2 35 SHO 1 64 1 1 1 1 1 1 AML/AML/AML M3 BMA 1 36 ADH 2 42 1 0 0 1 0 0 AML/AML/AML M3 BMA 1
KETERANGAN :
• Jenis kelamin : Laki (1), Perempuan (2)
• Immunophenotyping : Myeloid Lineage (AML) (1), Non Myeloid Leneage (0)
• Pemeriksaan sitogenetik: o RUNX1 : RUNX1 ABNORMAL (1), RUNX1 NORMAL (0) o FLT3 : FLT3 ABNORMAL (1), FLT3 NORMAL (0) o NRAS : NRAS ABNORMAL (1), NRAS NORMAL (0) o NPM1 : NPM1 ABNORMAL (1), NPM1 NORMAL (0) o Del(5q) : KROMOSOM Del(5q) (1), KROMOSOM (5q) NORMAL (0)
• Diagnosis : AML (1), MDS (2)
• Nomor: 3, 5, 15, 25, 34 : EKSKLUSI

136
Lampiran 3 IDENTITAS PESERTA PENELITIAN
N A M A (L/P) :
U M U R :
A L A M A T/Hp/TELP. :
PENDIDIKAN :
PEKERJAAN / MASA KERJA I :
PEKERJAAN / MASA KERJA II :
PEKERJAAN LAIN : (BERHUBUNGAN DENGAN KIMIA BENZENE, SOLVENT, RADIASI, BAHAN
TAMBANG)
RIWAYAT PENYAKIT PESERTA :
RIWAYAT PENYAKIT KELUARGA :
KEBIASAAN:
I. MEROKOK/BATANG PER HARI
II. MINUMAN BERALKOHOL
III. OBAT-OBATAN (PENENANG)
IV. OLAH-RAGA HARI/MINGGU/JAM PER KALI

137
Lampiran 4 Informed consent

138

139

140

141

142
LEMBAR PENGUNDURAN DIRI
Saya yang bertanda tangan dibawah ini :
Nama
:……………………………………………………………………
Umur
:……………………………………………………………………
Alamat
:……………………………………………………………………
Tlp / Email
:……………………………………………………………………
Fakultas / Instansi
:……………………………………………………………………
Dengan ini menyatakan MENGUNDURKAN DIRI sebagai subjek penelitian
Dengan judul penelitian:
“POLIMORFISME GEN NPM1, RUNX1, FLT3, N-RAS, DAN DELESI KROMOSOM 5
del(5q) MEMPENGARUHI MYELODYSPLASTIC SYNDROME (MDS) MENJADI
ACUTE MYELOID LEUKEMIA (AML) PADA PENDERITA DI SURABAYA ” .
Demikian lembar pengunduran diri ini saya buat dengan penuh kesadaran dan
tanpa paksaan.
Surabaya, …………………………
Yang Membuat Pernyataan
(……………………………)
Saksi 1 Saksi 2
(……………………………) (………..……………………)

143
Lampiran 5 KLASIFIKASI DIAGNOSIS AML
KLASIFIKASI AML, FAB (Desai., 2000)
M0 AML with minimal differentiation
M1 AML without maturation
M2 AML with maturation
M3 Promyelocytic leukemia
M4 Myelomonocytic leukemia
M5 Monocytic leukemia
M6 Erythroleukemia
M7 Megakaryoblastic leukemia
KLASIFIKASI AML, WHO 2008 (Hoffbrand., 2011) Acute myeloid leukaemia (with recurrent genetic abnormalities)
AML with t(8;21)
AML with inv(16)
AML with t(15;17) (q22;q12); PML-RARA
Acute myeloid leukaemia with myelodysplasia-related changes
Therapy-related myeloid neoplasms (t-AML)
Acute myeloid leukaemia, not otherwise specified
AML with minimal differentiation
AML without defferentiation
AML with maturation
Acute Myelomonocytic leukemia
Acute Monoblastic/Monocytic leukemia
Acute erythroid leukemia
Acute Megakaryoblastic leukemia

144
Acute Basophilic leukaemia
Acute panmyelosis with myelofibrosis
Myeloid sarcoma
Myeloid proliferations related to down syndrome
Transient abnormal myelopoiesis
Myeloid leukaemia
KLASIFIKASI DIAGNOSIS MDS
KLASIFIKASI MDS, FAB (Desai., 2000)
Refractory anemia (RA)
Refractory anemia with ring sideroblast (RARS)
Refractory Anemia with excess Blast (RAEB)
Refractory Anemia with excess blast in Transformasi (RAEB-T).
Chronic myelomonocytic leukemia (CMML)

145
Tabel 7., KLASIFIKASI MDS, WHO 2008 (Hoffbrand., 2011).

146
Lampiran 6 Informasi primer PCR (RUNX1, NPM1, N-RAS, FLT3)
(Genetika Science Indonesia, jalan Lingkaran luar Barat,
Kembangan, Jakarta Barat, 11610,
Email [email protected])
GEN SUSUNAN ASAM AMINO
1 N_RAS_F N_RAS_R
31-mer oligo (25 nmole scale, PCR Grade, 3 OD) GAC TGA ATA TAA ACT TGT GGT AGT TGG ACC T 28-mer oligo (25 nmole scale, PCR Grade, 3 OD) ACC AAG ATT TAC CTC TAT TGT TGG ATC A
2 RUNX1_F
RUNX1_R
22-mer oligo (25 nmole scale, PCR Grade, 3 OD)
TAC AGC CAA TCT GCA CTG TGC T
22-mer oligo (25 nmole scale, PCR Grade, 3 OD)
CAC CTG TGT GGA ACA GAT CTC C.
3 NPM1_F NPM1_R
27-mer oligo (25 nmole scale, PCR grade, 3 OD) CTG ATG TCT ATG AAG TGT TGT TCC
26-mer oligo (25 nmole scale, PCR Grade, 3 OD) CTC TGC ATT ATA AAA AGG ACA GCC AG
4
FLT3-ITD_F FLT3-ITD_R
16-mer oligo (25 nmole scale, PCR grade, 3 OD) AGG AGG GCA ACT ACT T
21-mer oligo (25 nmole scale, PCR Grade, 3 OD) CTT CAC TTG AAT TGG TAG CAT
Keterangan: F : forward, R : reverse

147
Lampiran 6
Hasil pemeriksaan Immunotyping : myeloid lineage, morfologi dari sel
darah tepi dan sumsum tulang, pemeriksaan molekuler menggunakan PCR,
pemeriksaan sitogenetik menggunakan CISH : delesi kromosom 5 del(5q)
dan kromosom 5 normal.
1. Hasil Pemeriksaan Immunophenotyping
Pemeriksaan Immunophenotyping dengan menggunakan Fasc Calibur,
peneliti mengharapkan dapat sebagi penunjang diagnosis yaitu Myeloid Leneage
atau Non Myeloid Leneage, sebagai berikut (gambar 5.5)

148
Gambar 5.1 Hasil pemeriksaan Immunophenotyping
Keterangan ganbar CD13, CD33, CD34, MPO positif Immunophenotyping: Myeloid Lineage
2. Hasil pemeriksaan morfologi sel darah tepi dan aspirasi sumsum tulang
Hasil pemeriksaan morfologi sel darah tepi dan aspirasi sumsum tulang,
kami mengharapkan diagnosis AML, MDS-related AML dengan kriteria blast lebih
dari 20%, auer Rod (+), dapat dilihat sebagai berikut:

149
Gambar 5.2 Morfologi sel hapusan darah tepi dan sumsum tulang
Keterangan gambar: A. Hapusan darah tepi (AML), PEMBESARAN 10X B. Hapusan darah tepi (AML), PEMBESARAN 100X (ujung petunjuk auer rod) C. Hapusan sumsum tulang (AML), PEMBESARAN 10X D. Hapusan sumsum tulang (AML), PEMBESARAN 100X (ujung petunjuk myeloblast)
3. Hasil Pemeriksaan PCR
Hasil pemeriksaan PCR dalam bentuk band dimana terlihat band berarti
terjadi mutasi pada gen NRAS, RUNX1, NPM1, FLT3 dapat dilihat pada gambar
dibawah ini (gambar 5.3)
Gambar 5.3 Hasil PCR Agarose-Biotechnology gen NRAS, RUNX1, NPM1,
FLT3
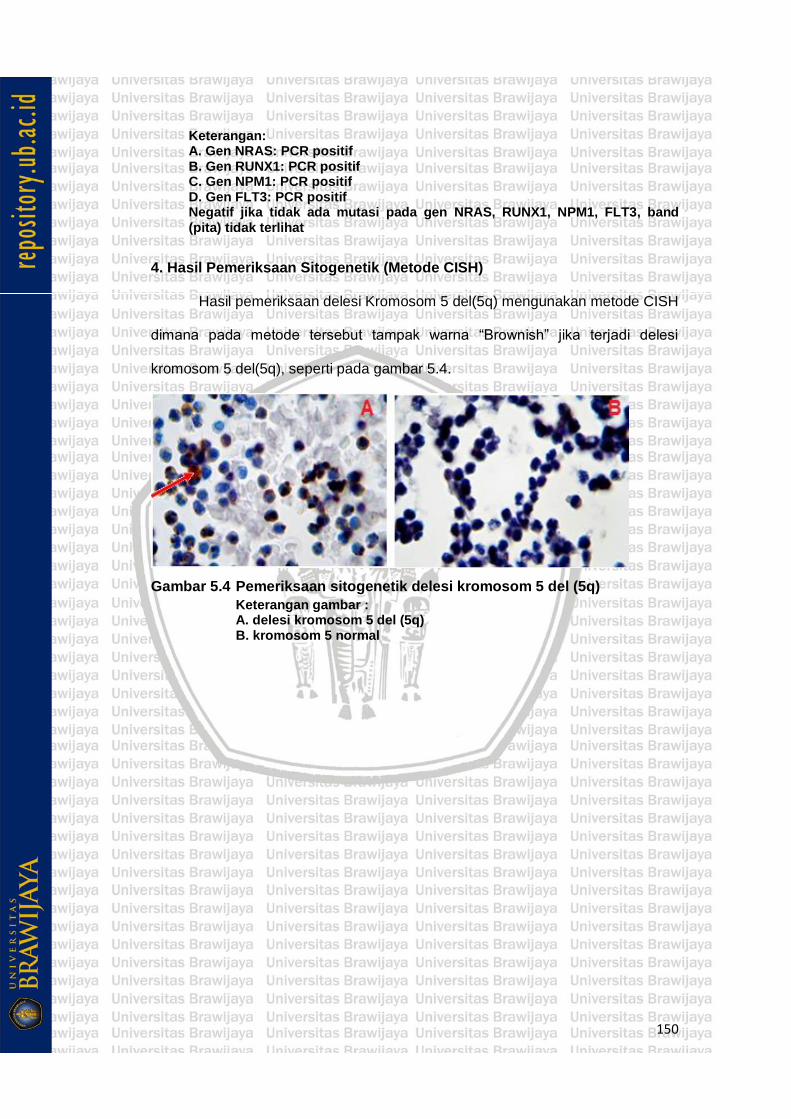
150
Keterangan: A. Gen NRAS: PCR positif B. Gen RUNX1: PCR positif C. Gen NPM1: PCR positif D. Gen FLT3: PCR positif Negatif jika tidak ada mutasi pada gen NRAS, RUNX1, NPM1, FLT3, band (pita) tidak terlihat
4. Hasil Pemeriksaan Sitogenetik (Metode CISH)
Hasil pemeriksaan delesi Kromosom 5 del(5q) mengunakan metode CISH
dimana pada metode tersebut tampak warna “Brownish” jika terjadi delesi
kromosom 5 del(5q), seperti pada gambar 5.4.
Gambar 5.4 Pemeriksaan sitogenetik delesi kromosom 5 del (5q)
Keterangan gambar : A. delesi kromosom 5 del (5q) B. kromosom 5 normal

151

152

153

154














