Embed Size (px)
Citation preview

NeuroToxicology 31 (2010) 454–460
Oxidative stress-mediated inhibition of brain creatine kinase activity bymethylmercury
Viviane Glaser a, Guilhian Leipnitz b, Marcos Raniel Straliotto a, Jade Oliveira a,Vanessa Valgas dos Santos a, Clovis Milton Duval Wannmacher b, Andreza Fabro de Bem a,Joao Batista Teixeira Rocha c, Marcelo Farina d, Alexandra Latini a,*a Laboratorio de Bioenergetica e Estresse Oxidativo, Departamento de Bioquımica, Centro de Ciencias Biologicas, Universidade Federal de Santa Catarina,
Campus Universitario – Trindade, Florianopolis, SC, Brazilb Instituto de Ciencias Basicas da Saude, Universidade de Rio Grande do Sul, Porto Alegre, RS, Brazilc Departamento de Quımica, Centro de Ciencias Naturais e Exatas, Universidade Federal de Santa Maria, Santa Maria, RS, Brazild Laboratorio de Neurotoxicidade de Metais, Departamento de Bioquımica, Centro de Ciencias Biologicas, Universidade Federal de Santa Catarina, Florianopolis, SC, Brazil
A R T I C L E I N F O
Article history:
Received 8 December 2009
Received in revised form 7 May 2010
Accepted 28 May 2010
Available online 4 June 2010
Keywords:
Creatine kinase
Methylmercury
Neurotoxicity
A B S T R A C T
Methylmercury (MeHg), a potent neurotoxicant, easily passes through the blood–brain barrier and
accumulates in brain causing severe irreversible damage. However, the underlying neurotoxic
mechanisms elicited by MeHg are still not completed defined. In this study, we aimed to investigate
the in vitro toxic effects elicited by crescent concentrations (0–1500 mM) of MeHg on creatine kinase
(CK) activity, thiol content (NPSH) and protein carbonyl content (PCC) in mouse brain preparations. In
addition, CK activity, MTT reduction and DCFH-DA oxidation (reactive oxygen species (ROS) formation)
were also measured in C6 glioma cell linage. CK activity was severely reduced by MeHg treatment in
mouse brain preparations. This inhibitory effect was positively correlated to the MeHg-induced
reduction of NPSH levels and increment in PCC. Moreover, the positive correlation between brain CK
activity and NPSH levels was observed at either 15 or 60 min of MeHg pre-incubation. In addition, MeHg-
treated C6 cells showed also a significant inhibition of CK activity at MeHg concentrations, as low as,
50 mM in parallel to reduced mitochondrial function and increased ROS production. Taking together,
these data demonstrate that MeHg severely affects CK activity, an essential enzyme for brain energy
buffering to maintain cellular energy homeostasis. This effect appears to be mediated by oxidation of
thiol groups that might cause subsequent oxidative stress.
� 2010 Elsevier Inc. All rights reserved.
Contents lists available at ScienceDirect
NeuroToxicology
1. Introduction
Creatine kinases (CKs, EC 2.7.3.2), a family of enzymescatalyzing the reversible transfer of a phosphoryl group betweenATP and creatine, plays a key role in the energy metabolism oftissues that have intermittently high and fluctuating energyrequirements, such as skeletal and cardiac muscle, and nervoustissue (Wallimann and Hemmer, 1994). There are distinct CKisoenzymes, which are compartmentalized specifically in theplaces where energy is produced (mitochondria) or utilized(cytosol). The cytosolic CK isoforms (Cy-CK) are expressed in atissue-specific manner, the brain-specific (BB-CK), the skeletalmuscle-specific (MM-CK) and the cardiac muscle-specific (MB-CK)isoenzymes (Wallimann et al., 1992). The mitochondrial forms ofCK (Mi-CK) consist of the muscle-specific sarcomeric isoform Mib-
* Corresponding author. Tel.: +55 48 37219589; fax: +55 48 37219672.
E-mail address: [email protected] (A. Latini).
0161-813X/$ – see front matter � 2010 Elsevier Inc. All rights reserved.
doi:10.1016/j.neuro.2010.05.012
CK and the ubiquitous isoform Mia-CK, which is mainly found inbrain tissue mitochondria (Wallimann and Hemmer, 1994; Wall-imann et al., 1992).
Cy-CK, which in part is associated with specific subcellularcompartments or structures (Wallimann et al., 1992; Wegmannet al., 1992), exists as homo- and heterodimers in the cytosol, andtheir function is to prevent fluctuations of ATP during periods ofhigh energy demand, such as in cardiac and skeletal musclecontraction, Ca2+-pump activity, photoreceptor-mediated lighttransduction, and neuronal excitation.
Mi-CK, located in cristae and intermembrane space (Adamset al., 1989), uses ATP supplied by the ANT (adenine nucleotidetranslocase) to form PCr (phosphocreatine) (Dolder et al., 2003),which is then delivered via the outer membrane VDAC (voltage-dependent anion channel) to the cytosol (Adams et al., 1989).These contact site complexes (CK/ANT/VDAC) has been pointed outas a functional a structural element of the permeability transitionpore (PTP) (Beutner et al., 1998). The opening of the PTP, possiblyregulated by Mi-CK oligomers (Andrienko et al., 2003), is

V. Glaser et al. / NeuroToxicology 31 (2010) 454–460 455
considered a key event in the mitochondrial pathways leading tocellular apoptosis (Vyssokikh and Brdiczka, 2003).
Due to the specific localization of CK isofoms, CK/phosphocrea-tine-system could in principle provide a spatial ‘‘energy shuttle’’ or‘‘energy circuit’’ bridging sites of energy generation with sites ofenergy consumption (Wallimann et al., 1992; Saks et al., 2004).
The brain, like other tissues with high and variable rates of ATPmetabolism, presents high PCr concentration and CK activity. Theimportance of creatine and the CK system for normal cell functionhas been elucidated in transgenic mice lacking the expression of CK(Crozatier et al., 2002; Janssen et al., 2003; Steeghs et al., 1997).These animals showed muscular and neurological dysfunctionsand phenotypes that have some similarities with the clinicalsymptoms of humans suffering from the so-called ‘‘creatinedeficiency syndrome’’ (Leuzzi, 2002). It is well described thatinhibition of CK activity is implicated in the pathogenesis of anumber of diseases, especially in the brain (Aksenov et al., 2000;Beal, 2002, 2000), because of the central role of the PCr/CK systemin the regulation of brain ATP concentrations. Therefore, altera-tions in CK functioning have been proposed in CNS diseases withaltered energy metabolism and may represent an important step ofa neurodegenerative pathway that leads to neuronal loss in thebrain (Tomimoto et al., 1993; Hemmer and Wallimann, 1993).
CK isoenzymes are extremely susceptible to damage by reactivespecies (Aksenov et al., 2000; Konorev et al., 1998), and thisappears to be mediated by oxidation of a cysteinyl residue(cysteine282) that is critical for substrate binding (Kenyon, 1996). Ithas been demonstrated that the substitution of this cysteine282
with a serine results in a 500-fold decrease in enzyme activity(Kenyon, 1996). Consequently, CK is highly susceptible toinactivation by oxidative reactions (Wang et al., 2001). In thisscenario, Mi-CK appears to be more vulnerable than Cy-CK, due toits mitochondrial localization (Koufen and Stark, 2000). Most of thereactive species originate directly or indirectly from the activity ofthe mitochondrial respiratory chain, in particular under conditionsof increased oxidative stress like ischemia/reperfusion injury,aging (Beal, 2002; Raha and Robinson, 2000), as well as, in certainneurodegenerative diseases such as amyotrophic lateral sclerosis,Huntington’s disease, and Alzheimer’s disease (Beal, 2000). In linewith this, it has been demonstrated a compromised CK system incommon neurodegenerative diseases (Aksenov et al., 2000;Ferrante et al., 2000; Wendt et al., 2002).
On the other hand, environmental pollutants, including theorganic form of mercury, methylmercury (MeHg), have beenshown to cause severe and irreversible neurobehavioral andneuropsychological disorders in both humans and animals (Choi,1989; Clarkson et al., 2003). Even though, MeHg-inducedneurotoxicity is a widely reported phenomenon, the molecularmechanisms related to its toxicity are not completely understood.The current mechanisms involved in the MeHg-induced neurotox-icity are mainly related to intracellular calcium impairment (Siroisand Atchison, 2000), alteration of glutamate homeostasis andoxidative stress (Aschner et al., 2007). Indeed, the antioxidantglutathione (GSH) system appears to be an important moleculartarget of MeHg-induced neurotoxicity (Stringari et al., 2008),corroborated by decreased GSH levels and activities of GSH-relatedenzymes in the brain of MeHg-exposed animals. Considering thatMeHg is a potent electrophilic molecule that compromise thecellular antioxidant system (oxidizes thiol groups), in the presentinvestigation we study the in vitro effect of MeHg on CK activity, asensitive thiol-containing enzyme. In addition, we also study the in
vitro effect of MeHg on the neurochemical parameters, namelynon-protein thiol group (NPSH) levels, protein carbonyl content(PCC), DCFH-DA oxidation (reactive oxygen species (ROS) forma-tion) and MTT reduction in mouse brain preparations and in C6glioma cell linage homogenates.
2. Experimental procedures
2.1. Animals and reagents
Male Swiss albino mice of 60 days of life obtained from the CentralAnimal House of the Centre for Biological Sciences, UniversidadeFederal de Santa Catarina, Florianopolis, SC, Brazil, were used. Theanimals were maintained on a 12-h light/dark cycle (lights on07:00–19:00 h) in a constant temperature (22 � 1 8C) colony room,with free access to water and protein commercial chow (Nuvital-PR,Brazil). The experimental protocol was approved by the EthicsCommittee for Animal Research (PP00084/CEUA) of the UniversidadeFederal de Santa Catarina, Florianopolis, SC, Brazil, and followed theGuiding Principles in the Use of Animals in Toxicology, adopted by theSociety of Toxicology in July 1989. All efforts were made to minimizethe number of animals used and their suffering.
All chemicals were of analytical grade and purchased from Sigma(St. Louis, MO, USA) except methylmercury (II) chloride which wasobtained from Aldrich Chemical Co. (Milwaukee, WI). The CKactivity, NPSH content, and cell viability assay were performed in aVarian Cary 50 spectrophotometer (Varian Inc., Palo Alto, CA, USA)with temperature control. The rate of oxidation of 20–70-dichloro-fluorescein (DCFH) was quantified by using a Tecan Austria GmbHM200 (Tecan, Grodig/Salzburg, Austria) fluorescence spectropho-tometer. For brain tissue preparations, an Eppendorf 5415 R(Eppendorf, Hamburg, Germany) centrifuge was used. The oxidationof DCFH was also assessed by using a Nikon inverted microscopeusing the TE-FM Epi-Fluorescence accessory.
2.2. Cerebral cortex supernatant preparation
Animals were killed by decapitation without anesthesia, thebrain was rapidly excised on a Petri dish placed on ice and thecerebral cortex was dissected, weighed and kept chilled untilhomogenization which was performed using a ground glass typePotter-Elvejhem homogenizer. Homogenates were further cen-trifuged at 1000 � g for 10 min at 4 8C, the pellet was discarded andthe supernatants obtained were incubated at 37 8C for 15 min or1 h with MeHg (0–1500 mM). Immediately after incubation,aliquots were taken to determine the biochemical parameters.
By using this large range of MeHg concentration, we deter-mined the necessary MeHg concentration to reduce 50% (IC50) ofthe CK activity and NPSH content (Dixon and Webb, 1964; Latiniet al., 2005).
2.3. Maintenance and treatment of cell line
The C6 astroglioma cell line was obtained from the AmericanType Culture Collection (Rockville, Maryland, USA) and wasmaintained essentially according to the procedure previouslydescribed (dos Santos et al., 2006). The cells were seeded in flasksand cultured in DMEM (pH 7.4) containing 5% fetal bovine serum,2.5 mg/mL Fungizone1 and 100 U/L gentamicin. Cells were kept at37 8C in an atmosphere of 5% CO2/95% air. Exponentially growingcells were detached from the culture flasks using 0.05% trypsin/ethylene–diaminetetracetic acid and seeded in 24-well plates(10 � 103 cells/well). After cells reached confluence, the culturemedium was removed by suction and cells were pre-incubated inthe presence of MeHg (0–1500 mM) for 15 min or 1 h, in serum-free DMEM (pH 7.4), at 37 8C in an atmosphere of 5% CO2/95% air.
2.4. Measurement of creatine kinase (CK) activity
CK activity was measured in a 60 mM Tris–HCl buffer, pH 7.5,containing 7 mM phosphocreatine, 9 mM MgSO4, and approxi-mately 1 mg protein in a final volume of 0.13 mL. After 20 min pre-

V. Glaser et al. / NeuroToxicology 31 (2010) 454–460456
incubation at 37 8C, the reaction was started by the addition of0.42 mmol ADP (2.8 mM final concentration). The reaction wasstopped after the incubation for 15 min by the addition of 1 mmolp-hydroxymercuribenzoic acid (6.25 mM final concentration).The reagent concentrations and the incubation time were chosento assure linearity of the enzymatic reaction. Appropriate controlswere carried out to measure the spontaneous hydrolysis ofphosphocreatine. The creatine formed was estimated accordingby colorimetric measurement (Hughes, 1962). The color wasdeveloped by the addition of 0.1 mL 2% a-naphtol and 0.1 mL0.05% diacetyl in a final volume of 1 mL and read after 20 min at540 nm. Results were expressed as mmol creatine formed/min/mgprotein.
2.5. Non-protein thiol groups (NPSH) measurement
NPSH groups, whose levels are mainly represented byglutathione (around 90%; Cooper and Kristal, 1997), weredetermined as described previously (Ellman, 1959) in a fractionobtained after treating supernatants with 1 volume of 10%trichloroacetic acid. After centrifugation, an aliquot of supernatantwas diluted in 800 mM sodium phosphate buffer, pH 7.4, and500 mM DTNB (5,50-dithiobis-2-nitrobenzoic acid) were added.Color development resulting from the reaction between DTNB andthiols reaches a maximum in 5 min and is stable for more than30 min. Absorbance was determined at 412 nm after 10 min.Results were calculated as mmol NPSH/mg protein.
2.6. Protein carbonyl content (PCC)
The oxidative damage to protein was measured by thedetermination of protein carbonyl groups content (PCC), basedon the reaction with dinitrophenylhydrazine (DNPH) (Reznick andPacker, 1994). MeHg-exposed cortical supernatants were treatedwith 4 mmol DNPH dissolved in 2.5N HCl or with 2.5N HCl (blankcontrol) and left in the dark for 1 h. Samples were then precipitatedwith 1 volume 20% TCA and centrifuged for 5 min at 10,000 � g.The pellet was then washed with 1 mL ethanol:ethyl acetate (1:1,v/v) and re-dissolved in 550 mL 6 M guanidine prepared in 2.5NHCl. Then, the tubes were incubated at 37 8C for 5 min to assurecomplete dissolution of the pellet and the resulting sample wasdetermined at 365 nm. The difference between the DNPH-treatedand HCl-treated samples was used to calculate the PCC. The resultswere calculated as nmol of carbonyls groups/mg protein, using theextinction coefficient of 22,000 � 106 mM�1 cm�1 for aliphatichydrazones.
2.7. Measurement of mitochondrial function
Mitochondrial function of C6 glioma cells was assessed byfollowing the MTT (3-[4,5dimethylthiazol-2-yl]-2,5-diphenylte-trazolic bromide) reduction. Active mitochondrial dehydrogenasescleavage and reduce the soluble yellow MTT dye into the insolublepurple formazan (Mosmann, 1983). Brain slices or cells wereincubated for 1 h with MeHg (0–1500 mM). At the end of theincubation period, MTT test were performed. The formazanformation was spectrophotometrically assayed at 570 and630 nm, and the net DA(570–630) was taken as an index ofmitochondrial function. Results were compared to control samplesto which 100% activity was attributed.
2.8. ROS production measurement through the DCFH-DA oxidation
Intracellular ROS production was detected using the non-fluorescent cell permeating compound, 20–70-dichlorofluoresceindiacetate (DCFH-DA). DCFH-DA is hydrolyzed by intracellular
esterases to DCFH, which is trapped within the cell. This non-fluorescent molecule is then oxidized to fluorescent dichloro-fluorescin (DCF) by action of cellular oxidants. MeHg-exposed C6cells were treated with DCFH-DA (10 mM) for 30 min at 37 8C.Afterwards, the cells were photographed or scraped into PBS with0.2% Triton X-100. The fluorescence was measured with excitationat 485 nm and emission at 520 nm. Calibration curve wasperformed with standard DCF (0–1 mM) and the level of ROSproduction was calculated as nmol DCF formed/mg protein (Vieirade Almeida et al., 2008).
2.9. Protein determination
Brain and cell homogenate protein content was determined bythe method of Lowry et al. (1951) using bovine serum albumin asthe standard.
2.10. Statistical analysis
Results are presented as mean � standard deviation. Assays wereperformed in triplicate and data were analyzed using one-wayanalysis of variance (ANOVA) followed by the post hoc Duncanmultiple range test when F was significant. For analysis of dose-dependent effects, linear regression was used. Pearson correlation, r,was analyzed between CK activity and NPSH levels. Differencesbetween the groups were rated significant at P � 0.05. All analyseswere carried out in an IBM-compatible PC computer using theStatistical Package for the Social Sciences (SPSS) software.
3. Results
3.1. MeHg treatment strongly inhibited CK activity and induced
oxidative stress in mouse cerebral cortex homogenates
MeHg in vitro effect was first investigated on CK activity in mousecortical homogenates. Fig. 1A shows that 15 or 60 min of pre-incubation with the mercurial elicited a strong inhibition (up to 85%)of CK activity. In addition, the MeHg-inhibitory effect was in aconcentration-dependent fashion (15 min pre-incubation =[F(6,28) = 22.41, P < 0.001, b(linear regression) =�0.64, P < 0.001, R2
(best
fit non-linear regression) = 0.81]; 60 min pre-incubation = [F(6,28) = 20.06,P < 0.001, b(linear regression) = �0.76, P < 0.001, R2
(best fit non-linear
regression) = 0.81]).In parallel, the effect of MeHg on NPSH levels in cortical
supernatants was also investigated. Fig. 1B shows that 15 or 60 minMeHg exposure significantly decreased NPSH content (up to 95%)also in a concentration-dependent manner (15 min pre-incubation:[F(6,28) = 23.17, P < 0.001; b(linear regression) = �0.84, P < 0.001, R2
(best
fit non-linear regression) = �0.89]; 60 min pre-incubation: [F(6,28) = 9.09,P < 0.001, b(linear regression) = 0.80, P < 0.001, R2
(best fit non-linear
regression) = 0.86]).The IC50 (MeHg concentration necessary to reduce 50% of the CK
activity) was determined as previously described (Dixon andWebb, 1964; Latini et al., 2005). The IC50 values obtained for the CKinhibition induced by MeHg exposition was 189.6 � 1.18 and87.0 � 1.15 mM for 15 and 60 min of pre-incubation, respectively.
Fig. 1A and B also shows the high sensitivity of this enzyme andof NPSH levels to the pre-incubation conditions, depicted by thereduction in CK activity and thiol content in control samples at15 min or 1 h pre-incubation.
Fig. 2A and B shows that MeHg-induced inhibition of CK activitywas positively correlated with NPSH levels, either with 15 or60 min pre-incubation (15 min pre-incubation: r = 0.85, P < 0.001;60 min pre-incubation: r = 0.87, P < 0.001). By using GraphPadsoftware, it is also possible to extrapolate and calculate that atMeHg IC50 for CK inhibition, NPSH levels are slightly reduced
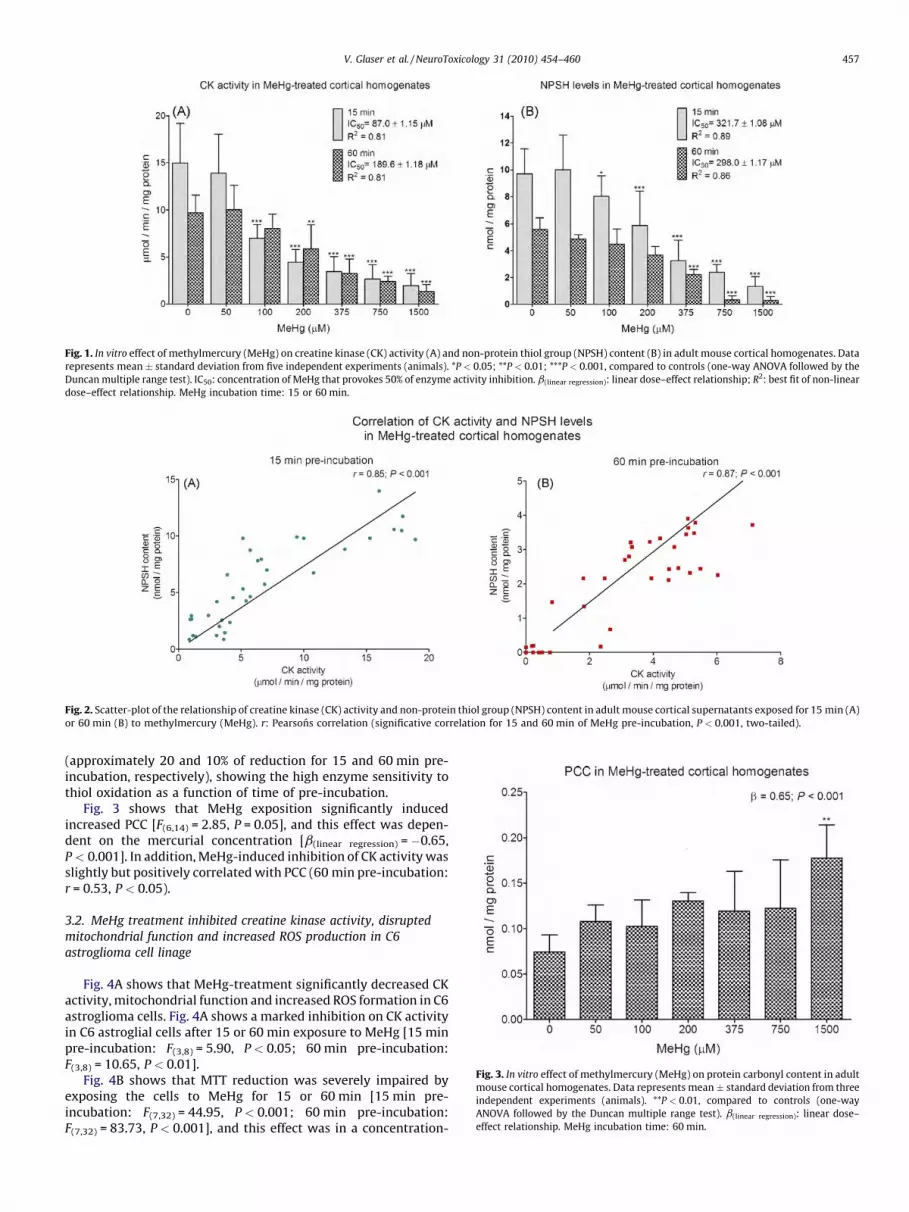
[(Fig._1)TD$FIG]
Fig. 1. In vitro effect of methylmercury (MeHg) on creatine kinase (CK) activity (A) and non-protein thiol group (NPSH) content (B) in adult mouse cortical homogenates. Data
represents mean � standard deviation from five independent experiments (animals). *P < 0.05; **P < 0.01; ***P < 0.001, compared to controls (one-way ANOVA followed by the
Duncan multiple range test). IC50: concentration of MeHg that provokes 50% of enzyme activity inhibition. b(linear regression): linear dose–effect relationship; R2: best fit of non-linear
dose–effect relationship. MeHg incubation time: 15 or 60 min.[(Fig._2)TD$FIG]
Fig. 2. Scatter-plot of the relationship of creatine kinase (CK) activity and non-protein thiol group (NPSH) content in adult mouse cortical supernatants exposed for 15 min (A)
or 60 min (B) to methylmercury (MeHg). r: Pearsons correlation (significative correlation for 15 and 60 min of MeHg pre-incubation, P < 0.001, two-tailed).
[(Fig._3)TD$FIG]
Fig. 3. In vitro effect of methylmercury (MeHg) on protein carbonyl content in adult
mouse cortical homogenates. Data represents mean � standard deviation from three
independent experiments (animals). **P < 0.01, compared to controls (one-way
ANOVA followed by the Duncan multiple range test). b(linear regression): linear dose–
effect relationship. MeHg incubation time: 60 min.
V. Glaser et al. / NeuroToxicology 31 (2010) 454–460 457
(approximately 20 and 10% of reduction for 15 and 60 min pre-incubation, respectively), showing the high enzyme sensitivity tothiol oxidation as a function of time of pre-incubation.
Fig. 3 shows that MeHg exposition significantly inducedincreased PCC [F(6,14) = 2.85, P = 0.05], and this effect was depen-dent on the mercurial concentration [b(linear regression) = �0.65,P < 0.001]. In addition, MeHg-induced inhibition of CK activity wasslightly but positively correlated with PCC (60 min pre-incubation:r = 0.53, P < 0.05).
3.2. MeHg treatment inhibited creatine kinase activity, disrupted
mitochondrial function and increased ROS production in C6
astroglioma cell linage
Fig. 4A shows that MeHg-treatment significantly decreased CKactivity, mitochondrial function and increased ROS formation in C6astroglioma cells. Fig. 4A shows a marked inhibition on CK activityin C6 astroglial cells after 15 or 60 min exposure to MeHg [15 minpre-incubation: F(3,8) = 5.90, P < 0.05; 60 min pre-incubation:F(3,8) = 10.65, P < 0.01].
Fig. 4B shows that MTT reduction was severely impaired byexposing the cells to MeHg for 15 or 60 min [15 min pre-incubation: F(7,32) = 44.95, P < 0.001; 60 min pre-incubation:F(7,32) = 83.73, P < 0.001], and this effect was in a concentration-
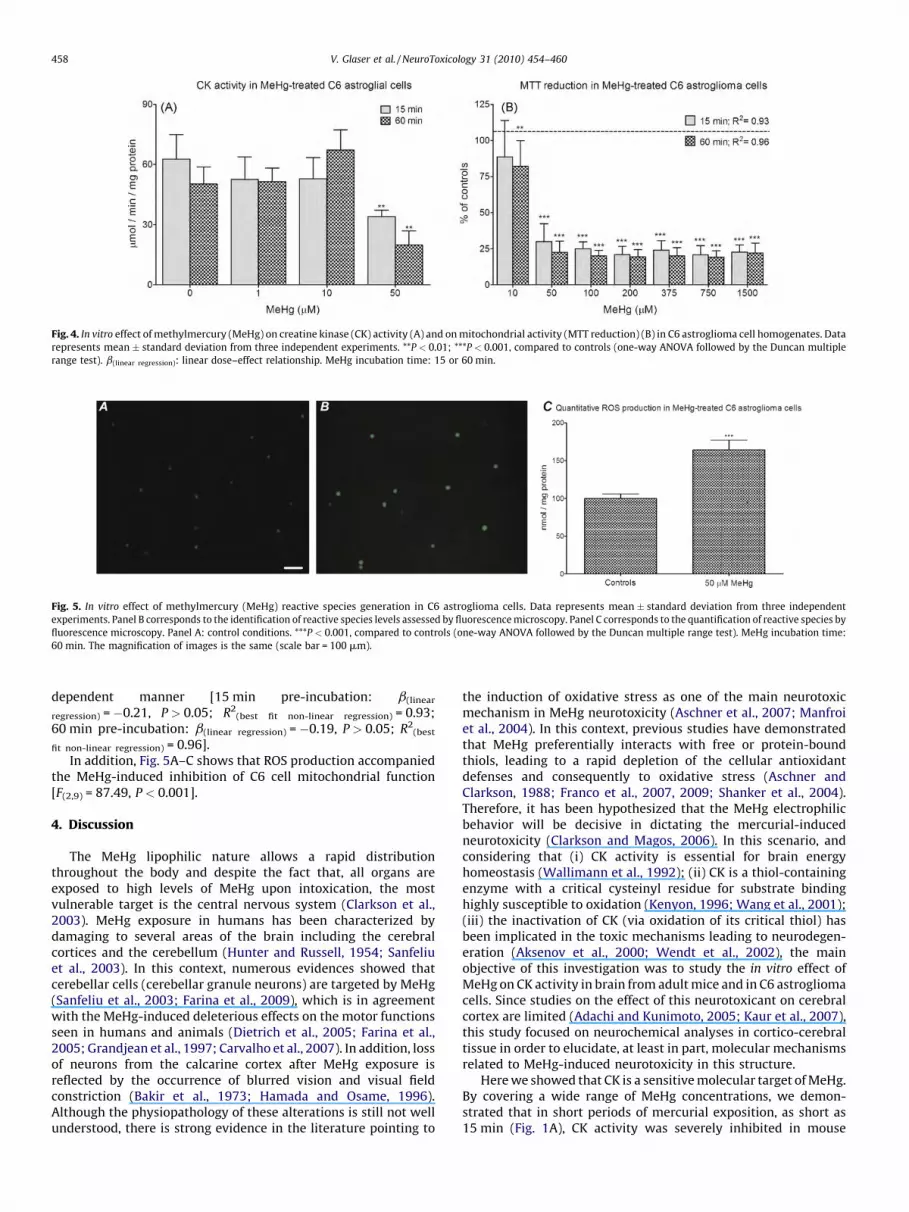
[(Fig._5)TD$FIG]
Fig. 5. In vitro effect of methylmercury (MeHg) reactive species generation in C6 astroglioma cells. Data represents mean � standard deviation from three independent
experiments. Panel B corresponds to the identification of reactive species levels assessed by fluorescence microscopy. Panel C corresponds to the quantification of reactive species by
fluorescence microscopy. Panel A: control conditions. ***P < 0.001, compared to controls (one-way ANOVA followed by the Duncan multiple range test). MeHg incubation time:
60 min. The magnification of images is the same (scale bar = 100 mm).
[(Fig._4)TD$FIG]
Fig. 4. In vitro effect of methylmercury (MeHg) on creatine kinase (CK) activity (A) and on mitochondrial activity (MTT reduction) (B) in C6 astroglioma cell homogenates. Data
represents mean � standard deviation from three independent experiments. **P < 0.01; ***P < 0.001, compared to controls (one-way ANOVA followed by the Duncan multiple
range test). b(linear regression): linear dose–effect relationship. MeHg incubation time: 15 or 60 min.
V. Glaser et al. / NeuroToxicology 31 (2010) 454–460458
dependent manner [15 min pre-incubation: b(linear
regression) = �0.21, P > 0.05; R2(best fit non-linear regression) = 0.93;
60 min pre-incubation: b(linear regression) = �0.19, P > 0.05; R2(best
fit non-linear regression) = 0.96].In addition, Fig. 5A–C shows that ROS production accompanied
the MeHg-induced inhibition of C6 cell mitochondrial function[F(2,9) = 87.49, P < 0.001].
4. Discussion
The MeHg lipophilic nature allows a rapid distributionthroughout the body and despite the fact that, all organs areexposed to high levels of MeHg upon intoxication, the mostvulnerable target is the central nervous system (Clarkson et al.,2003). MeHg exposure in humans has been characterized bydamaging to several areas of the brain including the cerebralcortices and the cerebellum (Hunter and Russell, 1954; Sanfeliuet al., 2003). In this context, numerous evidences showed thatcerebellar cells (cerebellar granule neurons) are targeted by MeHg(Sanfeliu et al., 2003; Farina et al., 2009), which is in agreementwith the MeHg-induced deleterious effects on the motor functionsseen in humans and animals (Dietrich et al., 2005; Farina et al.,2005; Grandjean et al., 1997; Carvalho et al., 2007). In addition, lossof neurons from the calcarine cortex after MeHg exposure isreflected by the occurrence of blurred vision and visual fieldconstriction (Bakir et al., 1973; Hamada and Osame, 1996).Although the physiopathology of these alterations is still not wellunderstood, there is strong evidence in the literature pointing to
the induction of oxidative stress as one of the main neurotoxicmechanism in MeHg neurotoxicity (Aschner et al., 2007; Manfroiet al., 2004). In this context, previous studies have demonstratedthat MeHg preferentially interacts with free or protein-boundthiols, leading to a rapid depletion of the cellular antioxidantdefenses and consequently to oxidative stress (Aschner andClarkson, 1988; Franco et al., 2007, 2009; Shanker et al., 2004).Therefore, it has been hypothesized that the MeHg electrophilicbehavior will be decisive in dictating the mercurial-inducedneurotoxicity (Clarkson and Magos, 2006). In this scenario, andconsidering that (i) CK activity is essential for brain energyhomeostasis (Wallimann et al., 1992); (ii) CK is a thiol-containingenzyme with a critical cysteinyl residue for substrate bindinghighly susceptible to oxidation (Kenyon, 1996; Wang et al., 2001);(iii) the inactivation of CK (via oxidation of its critical thiol) hasbeen implicated in the toxic mechanisms leading to neurodegen-eration (Aksenov et al., 2000; Wendt et al., 2002), the mainobjective of this investigation was to study the in vitro effect ofMeHg on CK activity in brain from adult mice and in C6 astrogliomacells. Since studies on the effect of this neurotoxicant on cerebralcortex are limited (Adachi and Kunimoto, 2005; Kaur et al., 2007),this study focused on neurochemical analyses in cortico-cerebraltissue in order to elucidate, at least in part, molecular mechanismsrelated to MeHg-induced neurotoxicity in this structure.
Here we showed that CK is a sensitive molecular target of MeHg.By covering a wide range of MeHg concentrations, we demon-strated that in short periods of mercurial exposition, as short as15 min (Fig. 1A), CK activity was severely inhibited in mouse

V. Glaser et al. / NeuroToxicology 31 (2010) 454–460 459
cortical homogenates. Although, a linear concentration–effect wasobserved in 15 or 60 min of MeHg pre-incubation (b = 0.64;P < 0.001), a stronger concentration–effect relationship wasdemonstrated when applying the polynomial (non-linear) regres-sion, pointing to the high susceptibility of the single critical thiol(cysteine282; pKa = 5.4; Wang et al., 2001) of CK towards theelectrophilic activity of MeHg. This inhibitory MeHg-induced effecton CK is in agreement to the MeHg affinity constant for the SHgroups, which is approximately 1010–16 (Onyido et al., 2004).Therefore, it could be assumed that any thiol-containing enzyme atphysiological pH would be a molecular target of MeHg toxicity,including that of CK. Moreover, we should also consider that ahigher selectivity of MeHg toward specific nucleophilic moleculescould also be determined by the pKa value. Therefore, the CK thiolgroup, because of its low pKa 5.4 would be potentially morevulnerable to oxidation than the thiol group of glutathione (GSH;pKa = 8.7; Srinivasan et al., 1997) at pH 7.4 and at equimolarconcentrations. This is in line with our present results demon-strating that at MeHg concentrations that provoked 50% of CKinhibition (IC50 values), NPSH levels were slightly reduced(approximately 20 and 10% of reduction for 15 and 60 min pre-incubation; Fig. 1B). It could be also considered that the high GSHlevels (up to 12 mM; Cooper and Kristal, 1997), main contributor tothe cellular NPSH content, could initially protect the critical thiolgroup of CK from MeHg oxidation (by a mass low effect). However,when NPSH concentrations are slightly reduced, CK thiol groupbecame a sensitive target of MeHg to oxidative modification, andthis is in line, with the positive relationship observed between CKactivity and NPSH levels (r > 0.85; Fig. 2A and B). In this scenario, itshould valuable to measure the IC50 on commercial purified CK andcomparing it with the values observed in the present investigation.
In addition, it has been demonstrated by our group and others,that MeHg-induced NPSH oxidation is associated with ROSgeneration, mitochondrial dysfunction and consequently proteinoxidation (Franco et al., 2007, 2009; Shenker et al., 1999). Indeed,the mitochondrial electron transfer chain, where reactive speciesare mainly produced has been described as the preferential MeHgaccumulation site in the cell, and would contribute to furtherbiochemical and ultra-structural changes in the organelle leadingto neurotoxicity (Atchison and Hare, 1994; Mori et al., 2007).Therefore, our next step was to assess whether the MeHgexposition enhances the oxidation of biomolecules throughenhanced ROS generation. As shown in Fig. 3, we observedsignificant protein oxidation (increased PCC; Fig. 3A) in corticalMeHg-treated homogenates, indicating that apart from CKinhibition and depletion of NPSH, the oxidation of cytosolicproteins (brain homogenates) contribute to perpetuate the MeHg-initiated oxidative stress.
On the other side, CK in conjunction with its tight functionalcoupling to oxidative phosphorylation (OXPHOS) is able tomodulate the mitochondrial function, and it has been demonstrat-ed that the lack of equilibrium between these energy systems(OXPHOS and CK) might potentiate the energy deficit and favoursreactive species formation in the mitochondria. This is in line, withour present data and those from Franco et al. (2009) and Wagneret al. (2009) demonstrating that MeHg-induced oxidative stresscaused a severe mitochondrial dysfunction, as seen by theinhibition of MTT reduction and increased reactive species contentin C6 astroglioma cells (Fig. 4A–C). In parallel to these alterations,CK activity was markedly inhibited (up to 46 and 60% for 15 and60 min pre-treatment, respectively) at lower MeHg concentrations(50 mM) than those observed in cortical homogenates, reinforcingthe idea that the cytosolic components including proteins andNPSH could initially protect CK activity from the toxicity of MeHg.
Finally, it should be taken into consideration the differentialsusceptibility of astrocytes and neurons to the toxic effects elicited
by MeHg, as well as the interaction between both cell types. In thisregard, it is important to mention the role of astrocytes as mediatorof MeHg-induced neuronal toxicity, which is linked, at least in part,to the capability of glial cells to remove glutamate from theextracellular milieu, avoiding excitotoxic events (Dave et al., 1994).Moreover, it is noteworthy the role of astrocytes in offeringglutathione precursors to neurons (Dringen and Hirrlinger, 2003),which seems to be related to the fact that astrocytes increaseneuronal resistance against MeHg neurotoxicity (Morken et al.,2005). With particular emphasis to the present study, the NPSHlevels in both cell types could be responsible for dissimilar effectsof MeHg toward astrocytes and neurons. This could be also relatedto the characteristic aerobic oxidative metabolism of neurons,where the reduction of mitochondrial thiol content will directlyimpact on the OXPHOS system and on CK activity and induce aprogressive decrease in ATP levels. Differentially, astrocytes areable to survive under these conditions by increasing the anaerobicmetabolism in order to prevent further ATP depletion (Almeidaet al., 2001). However, additional studies should be performed tocompletely answer this question.
Summarizing, the data presented here clearly demonstrate thatMeHg severely affects CK activity, an essential enzyme for brainenergy buffering to maintain cellular energy homeostasis, and thiseffect appears to be mediated by oxidation of thiol groups andconsequently by inducing oxidative stress.
Conflict of interest
None declared.
Acknowledgements
This work was supported by grants from FAPESC (Fundacao deApoio a Pesquisa Cientıfica e Tecnologica do Estado de SantaCatarina), CNPq (Conselho Nacional de Desenvolvimento Cientıficoe Tecnologico), INCT for Excitotoxicity and Neuroprotection/MCTand CAPES (Coordenacao de Aperfeicoamento de Pessoal de NıvelSuperior). Farina M, Rocha JBT, Wannmacher CMD and Latini A areCNPq fellows.
References
Adachi T, Kunimoto M. Acute cytotoxic effects of mercuric compounds in culturedastrocytes prepared from cerebral hemisphere and cerebellum of newborn rats.Biol Pharm Bull 2005;28:2308–11.
Adams V, Bosch W, Schlegel J, Wallimann T, Brdiczka D. Further characterization ofcontact sites from mitochondria of different tissues: topology of peripheralkinases. Biochim Biophys Acta 1989;981:213–25.
Aksenov M, Aksenova M, Butterfield DA, Markesbery WR. Oxidative modification ofcreatine kinase BB in Alzheimer’s disease brain. J Neurochem 2000;74:2520–7.
Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes andneurons to nitric oxide: the role of glycolytically generated ATP in astrocyteprotection. Proc Natl Acad Sci USA 2001;98:15294–9.
Andrienko T, Kuznetsov AV, Kaambre T, Usson Y, Orosco A, Appaix F, et al. Metabolicconsequences of functional complexes of mitochondria, myofibrils and sarcoplas-mic reticulum in muscle cells. J Exp Biol 2003;206:2059–72.
Aschner M, Clarkson TW. Uptake of methylmercury in the rat brain: effects of aminoacids. Brain Res 1988;462:31–9.
Aschner M, Syversen T, Souza DO, Rocha JB, Farina M. Involvement of glutamate andreactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res2007;40:285–91.
Atchison WD, Hare MF. Mechanisms of methylmercury-induced neurotoxicity. FASEB J1994;8:622–9.
Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, et al. Methyl-mercury poisoning in Iraq. Science 1973;181:230–41.
Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neu-rosci 2000;23:298–304.
Beal MF. Coenzyme Q10 as a possible treatment for neurodegenerative diseases. FreeRadical Res 2002;36:455–60.
Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase,mitochondrial creatine kinase and adenylate translocator display properties ofthe permeability transition pore. Implication for regulation of permeability tran-sition by the kinases. Biochim Biophys Acta 1998;1368:7–18.

V. Glaser et al. / NeuroToxicology 31 (2010) 454–460460
Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari EM, Rocha JB, et al. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomo-tor deficits and cerebellar toxicity in mice. Toxicology 2007;239:195–203.
Choi BH. The effects of methylmercury on the developing brain. Prog Neurobiol1989;32:447–70.
Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit RevToxicol 2006;36:609–62.
Clarkson TW, Magos L, Myers GJ. The toxicology of mercury–current exposures andclinical manifestations. N Engl J Med 2003;349:1731–7.
Cooper AJ, Kristal BS. Multiple roles of glutathione in the central nervous system. BiolChem 1997;378:793–802.
Crozatier B, Badoual T, Boehm E, Ennezat PV, Guenoun T, Su J, et al. Role of creatinekinase in cardiac excitation–contraction coupling: studies in creatine kinase-deficient mice. FASEB J 2002;16:653–60.
Dave V, Mullaney KJ, Goderie S, Kimelberg HK, Aschner M. Astrocytes as mediators ofmethylmercury neurotoxicity: effects on D-aspartate and serotonin uptake. DevNeurosci 1994;16:222–31.
Dietrich MO, Mantese CE, dos Anjos G, Souza DO, Farina M. Motor impairment inducedby oral exposure to methylmercury in adult mice. Environ Toxicol Pharmacol2005;19:169–75.
Dixon M, Webb EC. Enzymes. London: Longmans; 1964.Dolder M, Walzel B, Speer O, Schlattner U, Wallimann T. Inhibition of the mitochondrial
permeability transition by creatine kinase substrates. Requirement for microcom-partmentation. J Biol Chem 2003;278:17760–6.
dos Santos AQ, Nardin P, Funchal C, de Almeida LM, Jacques-Silva MC, Wofchuk ST, etal. Resveratrol increases glutamate uptake and glutamine synthetase activity in C6glioma cells. Arch Biochem Biophys 2006;453:161–7.
Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem 2003;384:505–16.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys 1959;82:70–7.Farina M, Franco JL, Ribas CM, Meotti FC, Missau FC, Pizzolatti MG, et al. Protective
effects of Polygala paniculata extract against methylmercury-induced neurotox-icity in mice. J Pharm Pharmacol 2005;57:1503–8.
Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, et al. Probucol increasesglutathione peroxidase-1 activity and displays long-lasting protection againstmethylmercury toxicity in cerebellar granule cells. Toxicol Sci 2009;112:416–26.
Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, et al.Neuroprotective effects of creatine in a transgenic mouse model of Huntington’sdisease. J Neurosci 2000;20:4389–97.
Franco JL, Braga Hde C, Nunes AK, Ribas CM, Stringari J, Silva AP, et al. Lactationalexposure to inorganic mercury: evidence of neurotoxic effects. NeurotoxicolTeratol 2007;29:360–7.
Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, et al. Methylmercuryneurotoxicity is associated with inhibition of the antioxidant enzyme glutathioneperoxidase. Free Radical Biol Med 2009;47:449–57.
Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, et al. Cognitive deficit in7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Ter-atol 1997;19:417–28.
Hamada R, Osame M. Minamata disease and other mercury syndromes. In: Chang LW,editor. Toxicology of metals. Boca Raton: CRC Press; 1996337–51.
Hemmer W, Wallimann T. Functional aspects of creatine kinase in brain. Dev Neurosci1993;15:249–60.
Hughes BP. A method for the estimation of serum creatine kinase and its use incomparing creatine kinase and aldolase activity in normal and pathological sera.Clin Chim Acta 1962;7:597–603.
Hunter D, Russell DS. Focal cerebellar and cerebellar atrophy in a human subject due toorganic mercury compounds. J Neurol Neurosurg Psychiatry 1954;17:235–41.
Janssen E, Terzic A, Wieringa B, Dzeja PP. Impaired intracellular energetic communi-cation in muscles from creatine kinase and adenylate kinase (M-CK/AK1) doubleknock-out mice. J Biol Chem 2003;278:30441–9.
Kaur P, Aschner M, Syversen T. Role of glutathione in determining the differentialsensitivity between the cortical and cerebellar regions towards mercury-inducedoxidative stress. Toxicology 2007;230:164–77.
Kenyon GL. Energy metabolism. Creatine kinase shapes up. Nature 1996;381:281–2.Konorev EA, Hogg N, Kalyanaraman B. Rapid and irreversible inhibition of creatine
kinase by peroxynitrite. FEBS Lett 1998;427:171–4.Koufen P, Stark G. Free radical induced inactivation of creatine kinase: sites of
interaction, protection, and recovery. Biochim Biophys Acta 2000;1501:44–50.Latini A, da Silva CG, Ferreira GC, Schuck PF, Scussiato K, Sarkis JJ, et al. Mitochondrial
energy metabolism is markedly impaired by D-2-hydroxyglutaric acid in rattissues. Mol Genet Metab 2005;86:188–99.
Leuzzi V. Inborn errors of creatine metabolism and epilepsy: clinical features, diagno-sis, and treatment. J Child Neurol 2002;17: 3S89–97, discussion 83S97.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folinphenol reagent. J Biol Chem 1951;193:265–75.
Manfroi CB, Schwalm FD, Cereser V, Abreu F, Oliveira A, Bizarro L, et al. Maternal milkas methylmercury source for suckling mice: neurotoxic effects involved with thecerebellar glutamatergic system. Toxicol Sci 2004;81:172–8.
Mori N, Yasutake A, Hirayama K. Comparative study of activities in reactive oxygenspecies production/defense system in mitochondria of rat brain and liver, and theirsusceptibility to methylmercury toxicity. Arch Toxicol 2007;81:769–76.
Morken TS, Sonnewald U, Aschner M, Syversen T. Effects of methylmercury on primarybrain cells in mono- and co-culture. Toxicol Sci 2005;87:169–75.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application toproliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
Onyido I, Norris AR, Buncel E. Biomolecule–mercury interactions: modalities of DNAbase–mercury binding mechanisms. Remediation strategies. Chem Rev 2004;104:5911–29.
Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. TrendsBiochem Sci 2000;25:502–8.
Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method forcarbonyl assay. Methods Enzymol 1994;233:357–63.
Saks VA, Kuznetsov AV, Vendelin M, Guerrero K, Kay L, Seppet EK. Functional couplingas a basic mechanism of feedback regulation of cardiac energy metabolism. MolCell Biochem 2004;256/257:185–99.
Sanfeliu C, Sebastia J, Cristofol R, Rodriguez-Farre E. Neurotoxicity of organomercurialcompounds. Neurotox Res 2003;5:283–305.
Shanker G, Aschner JL, Syversen T, Aschner M. Free radical formation in cerebralcortical astrocytes in culture induced by methylmercury. Brain Res Mol BrainRes 2004;128:48–57.
Shenker BJ, Guo TL, Onyido I, Shapiro IM. Induction of apoptosis in human T-cells bymethyl mercury: temporal relationship between mitochondrial dysfunction andloss of reductive reserve. Toxicol Appl Pharmacol 1999;157:23–35.
Sirois JE, Atchison WD. Methylmercury affects multiple subtypes of calcium channelsin rat cerebellar granule cells. Toxicol Appl Pharmacol 2000;167:1–11.
Srinivasan U, Mieyal PA, Mieyal JJ. pH profiles indicative of rate-limiting nucleophilicdisplacement in thioltransferase catalysis. Biochemistry 1997;36:3199–206.
Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschap A, Ruitenbeek W, et al.Altered Ca2+ responses in muscles with combined mitochondrial and cytosoliccreatine kinase deficiencies. Cell 1997;89:93–103.
Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, et al. Prenatalmethylmercury exposure hampers glutathione antioxidant system ontogenesisand causes long-lasting oxidative stress in the mouse brain. Toxicol Appl Phar-macol 2008;227:147–54.
Tomimoto H, Yamamoto K, Homburger HA, Yanagihara T. Immunoelectron microscop-ic investigation of creatine kinase BB-isoenzyme after cerebral ischemia in gerbils.Acta Neuropathol 1993;86:447–55.
Vieira de Almeida LM, Pineiro CC, Leite MC, Brolese G, Leal RB, Gottfried C, et al.Protective effects of resveratrol on hydrogen peroxide induced toxicity in primarycortical astrocyte cultures. Neurochem Res 2008;33:8–15.
Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrialmembrane pore (VDAC) and the adenine nucleotide translocase in regulation ofenergy metabolism and apoptosis. Acta Biochim Pol 2003;50:389–404.
Wagner C, Vargas AP, Roos DH, Morel AF, Farina M, Nogueira CW, et al. Comparativestudy of quercetin and its two glycoside derivatives quercitrin and rutin againstmethylmercury (MeHg)-induced ROS production in rat brain slices. Arch Toxicol2009.
Wallimann T, Hemmer W. Creatine kinase in non-muscle tissues and cells. Mol CellBiochem 1994;133/134:193–220.
Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compart-mentation, structure and function of creatine kinase isoenzymes in tissues withhigh and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellularenergy homeostasis. Biochem J 1992;281:21–40.
Wang PF, McLeish MJ, Kneen MM, Lee G, Kenyon GL. An unusually low pK(a) for Cys282 inthe active site of human muscle creatine kinase. Biochemistry 2001;40:11698–705.
Wegmann G, Zanolla E, Eppenberger HM, Wallimann T. In situ compartmentation ofcreatine kinase in intact sarcomeric muscle: the acto-myosin overlap zone as amolecular sieve. J Muscle Res Cell Motil 1992;13:420–35.
Wendt S, Dedeoglu A, Speer O, Wallimann T, Beal MF, Andreassen OA. Reduced creatinekinase activity in transgenic amyotrophic lateral sclerosis mice. Free Radical BiolMed 2002;32:920–6.





























