Embed Size (px)
Citation preview

Multivalent bioconjugates for the inhibition of anthrax toxin, influenza virus, and HIV
by
Jacob T. Martin
A Thesis Submitted to the Graduate
Faculty of Rensselaer Polytechnic Institute
in Partial Fulfillment of the
Requirements for the degree of
DOCTOR OF PHILOSOPHY
Major Subject: Chemical and Biological Engineering
Approved by the Examining Committee:
_________________________________________ Dr. Ravi Kane, Thesis Adviser
_________________________________________ Dr. Peter Tessier, Member
_________________________________________ Dr. Steven Cramer, Member
_________________________________________ Dr. Shekhar Garde, Member
_________________________________________ Dr. Marlene Belfort, Member
Rensselaer Polytechnic Institute Troy, New York
April 2014 (For Graduation May 2014)

ii
© Copyright 2014
by
Jacob T. Martin
All Rights Reserved

iii
CONTENTS
CONTENTS .......................................................................................................................... iii
LIST OF TABLES ................................................................................................................. viii
LIST OF FIGURES ................................................................................................................. ix
ACKNOWLEDGMENT ....................................................................................................... xiii
ABSTRACT ......................................................................................................................... xv
1. Introduction ................................................................................................................. 1
1.1 Anthrax ............................................................................................................... 1
1.1.1 Overview ................................................................................................ 1
1.1.2 Intoxication pathway ............................................................................. 1
1.2 Influenza ............................................................................................................. 3
1.2.1 Overview ................................................................................................ 3
1.2.2 Virion morphology and infection pathway ............................................ 4
1.2.3 Antigenic variation ................................................................................. 5
1.2.4 Infection pathway .................................................................................. 5
1.3 Human Immunodeficiency Virus (HIV) ............................................................... 6
1.3.1 Overview ................................................................................................ 6
1.3.2 Infection pathway .................................................................................. 6
1.3.3 Current treatment strategies ................................................................. 7
1.4 Multivalent therapeutics .................................................................................... 8
1.4.1 Multivalency ........................................................................................... 8
1.4.2 Scaffolds for multivalent therapeutics ................................................... 9
1.4.3 A multivalent anthrax toxin inhibitor with in vivo efficacy .................. 10
1.4.4 Multivalent anthrax toxin inhibitors with control over average ligand spacing .................................................................................................. 11
1.4.5 Multivalent anthrax toxin inhibitors with predefined ligand spacing . 13

iv
1.5 Motivation ........................................................................................................ 18
2. Preclinical development of polyvalent inhibitors of anthrax toxin ........................... 19
2.1 Therapeutic suitability ..................................................................................... 19
2.1.1 Biocompatibility ................................................................................... 19
2.1.2 PGA inhibitor synthesis ........................................................................ 20
2.1.3 PGA inhibitor efficacy in vitro .............................................................. 20
2.1.4 Storage stability.................................................................................... 22
2.2 Efficacy in vivo .................................................................................................. 22
2.2.1 Toxin challenge .................................................................................... 22
2.2.2 Spore challenge .................................................................................... 23
2.3 Pharmacokinetics ............................................................................................. 25
2.3.1 Serum stability ..................................................................................... 25
2.3.2 PEGylation of PGA-based inhibitors to improve blood residence time26
2.4 Conclusions ...................................................................................................... 28
3. Radial heptavalent inhibitors of anthrax toxin .......................................................... 30
3.1 Abstract ............................................................................................................ 30
3.2 Introduction ..................................................................................................... 30
3.3 Materials and methods .................................................................................... 32
3.3.1 Preparation of toxin components ........................................................ 32
3.3.2 Cytotoxicity assay ................................................................................. 32
3.3.3 Serum stability ..................................................................................... 33
3.3.4 Rat intoxication .................................................................................... 33
3.4 Results and discussion...................................................................................... 33
3.4.1 Structure-based design of heptavalent inhibitors ............................... 33
3.4.2 Characterization of heptavalent inhibitors .......................................... 35
3.5 Conclusions ...................................................................................................... 38

v
4. Multivalent inhibitors of influenza virus.................................................................... 39
4.1 Rationale for the design of multivalent inhibitors of influenza virus entry ..... 39
4.1.1 Targeting a conserved epitope on the hemagglutinin spike ............... 39
4.1.2 De novo design of protein ligands that target a conserved epitope on the hemagglutinin spike ....................................................................... 41
4.1.3 Design of divalent inhibitors that target a conserved epitope on the hemagglutinin spike ............................................................................. 43
4.1.4 Design of polyvalent inhibitors that target a conserved epitope on the hemagglutinin spike ............................................................................. 46
4.2 Production of multivalent bioconjugates of hemagglutinin-binding proteins 46
4.2.1 Modification of hemagglutinin-binding proteins from previous studies .............................................................................................................. 46
4.2.2 Cloning and bacterial production of modified hemagglutinin-binding proteins ................................................................................................ 49
4.2.3 Preparation of hemagglutinin-binding proteins for thioether bioconjugation reactions ..................................................................... 53
4.2.4 Synthesis of divalent hemagglutinin-binding protein bioconjugates .. 54
4.2.5 Synthesis of polyvalent hemagglutinin-binding protein bioconjugates .............................................................................................................. 55
4.2.6 Purification of multivalent hemagglutinin-binding protein bioconjugates ....................................................................................... 59
4.3 Characterization of multivalent bioconjugates of hemagglutinin-binding proteins ............................................................................................................ 61
4.3.1 In vitro microneutralization assay of influenza virus infection inhibition .............................................................................................................. 61
4.3.2 Inhibition of influenza infection with multivalent hemagglutinin-binding protein conjugates ............................................................................... 62
4.4 Conclusions ...................................................................................................... 64
5. Multivalent HIV entry inhibitors ................................................................................ 65
5.1 Rationale for the design of multivalent HIV entry inhibitors ........................... 65
5.1.1 Targeting the cellular receptor CCR5 ................................................... 65

vi
5.1.2 Leukotoxin E is a protein ligand of CCR5 .............................................. 67
5.1.3 Design of multivalent inhibitors that target CCR5 ............................... 68
5.2 Production of polyvalent bioconjugates of leukotoxin E ................................. 69
5.2.1 Cloning and bacterial production of leukotoxin E variants and fusion proteins ................................................................................................ 69
5.2.2 Synthesis of polyvalent leukotoxin E bioconjugates ............................ 73
5.2.3 Purification of polyvalent leukotoxin E bioconjugates ........................ 74
5.3 Characterization of polyvalent conjugates of leukotoxin E ............................. 75
5.3.1 Flow cytometry assay of CCR5-binding inhibition ............................... 75
5.3.2 Inhibition of CCR5-binding with polyvalent bioconjugates of leukotoxin E ............................................................................................................ 76
5.4 Conclusions ...................................................................................................... 78
6. Multivalent oligonucleotide aptamer bioconjugates ................................................ 80
6.1 Rationale for the use of oligonucleotide aptamers as ligands ........................ 80
6.2 Production of polyvalent bioconjugates of the ssDNA aptamer sgc8c ........... 81
6.2.1 Synthesis of polyvalent bioconjugates of the ssDNA aptamer sgc8c .. 81
6.2.2 Purification of polyvalent bioconjugates of the ssDNA aptamer sgc8c84
6.3 Characterization of polyvalent bioconjugates of the ssDNA aptamer sgc8c ... 85
6.4 Conclusions ...................................................................................................... 86
7. Suggestions for future work ...................................................................................... 87
7.1 Identification of oligonucleotide aptamers that target cell receptors ............ 87
7.1.1 Rationale for the search for oligonucleotide aptamers that target cell receptors .............................................................................................. 87
7.1.2 Screening libraries of oligonucleotide aptamers ................................. 87
7.1.3 Characterization of screened oligonucleotide aptamers ..................... 92
7.2 Alternative designs for multivalent HIV entry inhibitors ................................. 92
7.2.1 Peptide ligand derivatives of CCL5 ....................................................... 92

vii
7.2.2 Rationale for the design of divalent CCR5-targeting HIV inhibitors .... 95
7.2.3 Synthesis of homodivalent bioconjugates of aptamers that bind CCR5 .............................................................................................................. 97
7.2.4 Synthesis of heterodivalent inhibitors that bind CCR5 ........................ 98
7.3 Bioengineered protein polymer scaffolds ........................................................ 99
7.3.1 Using proteins as monodisperse polymer scaffolds ............................ 99
7.3.2 Bioengineering scaffolds for precise control over multivalent architecture ........................................................................................ 100
8. Conclusions .............................................................................................................. 102
REFERENCES ................................................................................................................... 103
APPENDIX A – List of abbreviations ............................................................................... 118
APPENDIX B – Additional contributions ........................................................................ 120

viii
LIST OF TABLES
Table 1.1: Polymerization of NAS at various [M]/[CTA] ratios.a ...................................... 14
Table 3.1: Inhibition of anthrax toxin action in a rat intoxication model........................ 37
Table 4.1: Homobifunctional PEG linkers purchased for homodivalent conjugation of
hemagglutinin-binding proteins. ............................................................................. 46

ix
LIST OF FIGURES
Figure 1.1: Representation of the anthrax intoxication pathway. .................................... 2
Figure 1.2: Synthesis of a polyvalent anthrax toxin inhibitor of controlled molecular
weight. ..................................................................................................................... 12
Figure 1.3: Influence of peptide density on the potency of a polyvalent anthrax toxin
inhibitor. .................................................................................................................. 13
Figure 1.4: Design of polyvalent inhibitors with control over molecular weight and ligand
spacing. .................................................................................................................... 15
Figure 1.5: Inhibitory activity of polyvalent inhibitors of anthrax toxin derived from a
controlled molecular weight homopolymer, pNAS. ................................................ 16
Figure 1.6: Characterization of polyvalent inhibitors based on poly(AAm-co-NAS)
copolymers. ............................................................................................................. 17
Figure 2.1: Synthesis of a PGA-based polyvalent anthrax toxin inhibitor. ...................... 20
Figure 2.2: Inhibition of cytotoxicity in vitro by PGA-based polyvalent inhibitors. ......... 21
Figure 2.3: Inhibition of cytotoxicity in vitro by PGA-based polyvalent inhibitors
synthesized in bulk for preclinical studies. .............................................................. 21
Figure 2.4: Inhibitory activity of PGA-based anthrax toxin inhibitors after long-term
storage under various conditions. ........................................................................... 22
Figure 2.5: Inhibitory activity of a PGA-based polyvalent inhibitor against anthrax lethal
toxin in vivo. ............................................................................................................. 23
Figure 2.6: Inhibitory activity of a PGA-based polyvalent inhibitor against anthrax toxin in
vivo generated by infection with B. anthracis spores. ............................................ 24
Figure 2.7: Inhibitory activity of PGA-based inhibitors after incubation in serum. ........ 25
Figure 2.8: ELISA assay of PGA-based anthrax toxin inhibitor blood residence time. .... 26
Figure 2.9: Silver-stain (left) followed by barium iodide-stain (right) of a 4-12% acrylamide
SDS-PAGE of PGA-based anthrax toxin inhibitors conjugated with monofunctional
PEG20k or PEG40k. ...................................................................................................... 28
Figure 3.1: Structure-based design of heptavalent anthrax toxin inhibitors. ................. 32
Figure 3.2: Synthesis scheme of heptavalent anthrax toxin inhibitor. ............................ 34

x
Figure 3.3: Characterization of a well-defined heptavalent anthrax toxin inhibitor. ..... 36
Figure 3.4: Influence of linker length on the activity of heptavalent inhibitors. ............ 37
Figure 4.1: Combined image of HA from influenza A/South Carolina/1/1918 (SC1918/H1)
with structures of several strain-specific antibodies and the broadly-neutralizing
antibody CR6261. ..................................................................................................... 40
Figure 4.2: Comparison of crystal structure of the broadly-neutralizing Fab CR6261 in
complex with HA to the crystal structures of the designed hemagglutinin-binding
proteins in complex with HA. .................................................................................. 42
Figure 4.3: Estimated distance between HB protein binding sites on the HA trimer stalk.
................................................................................................................................. 45
Figure 4.4: SDS-PAGE of IMAC fractions from FLAG-HB36.5 purification revealing non-
FLAG-tagged HB36.5 co-production. ....................................................................... 48
Figure 4.5: Predicted net protein charge at various pH values of three HB36.5 variants.
................................................................................................................................. 49
Figure 4.6: SDS-PAGE of IMAC fractions from DDDG-FLAG-HB36.5_A341C_ΔHM
purification. .............................................................................................................. 52
Figure 4.7: UV chromatogram of DDDG-FLAG-HB36.5_ΔHM on a GE HiLoad 16/600
Superdex 200 SEC column. ...................................................................................... 52
Figure 4.8: Synthesis scheme for homodivalent PEG-protein bioconjugation. ............... 55
Figure 4.9: Synthesis schemes for polyvalent polymer-maleimide-protein bioconjugation.
................................................................................................................................. 56
Figure 4.10: NMR spectra of three different PGA samples functionalized with increasing
percentages of maleimide. ...................................................................................... 57
Figure 4.11: Silver-stained non-reducing SDS-PAGE of conjugation reactions of HBA (lanes
1-12) and HBB (lanes 13-25) before purification, 5.0 µg total HB protein per lane. 59
Figure 4.12: Influenza virus infection inhibition efficacy of homodivalent bioconjugates
of HBB. ...................................................................................................................... 62
Figure 4.13: Influenza virus infection inhibition efficacy of polyvalent bioconjugates of
HBA. .......................................................................................................................... 63

xi
Figure 5.1: Silver-stained reducing SDS-PAGE of IMAC fractions from LukE-H6C
purification. .............................................................................................................. 72
Figure 5.2: Silver-stained reducing SDS-PAGE of IMAC fractions from CH6-GFP-LukE
purification. .............................................................................................................. 72
Figure 5.3: Silver-stained non-reducing SDS-PAGE of LukE-H6C conjugation reaction
products before purification, 5.0 µg total LukE protein per lane. .......................... 73
Figure 5.4: Silver-stained non-reducing SDS-PAGE of LukE-H6C conjugation reaction
products after SEC purification on a Superdex200 column. ................................... 75
Figure 5.5: CCR5-binding inhibition efficacy of polyvalent PGA-LukE bioconjugates. .... 77
Figure 5.6: CCR5-binding inhibition efficacy of polyvalent HyA-LukE bioconjugates. .... 77
Figure 6.1: SYBR Gold-stained 15% acrylamide TBE-Urea PAGE of ssDNA model aptamer
polyvalent conjugation reactions with PGA-maleimide after 88 hours at ambient
temperature. ............................................................................................................ 83
Figure 6.2: Heat map of SYBR Gold-stained 4-20% acrylamide TBE-PAGE of ssDNA
aptamer sgc8c conjugation reactions with PGA-maleimide. .................................. 84
Figure 6.3: SEC multi-chromatogram of the PGA35k-sgc8c reaction product. ............... 85
Figure 6.4: Enhanced binding of polyvalent PGA-sgc8c conjugates to lymphocyte cell
receptors, assayed by flow cytometry. .................................................................... 86
Figure 7.1: General SELEX procedure for screening RNA aptamers. ............................... 88
Figure 7.2: The sequence of the initial DNA library and a representative mfold prediction.
................................................................................................................................. 89
Figure 7.3: Schematic representation of the new library design for screening of short
oligonucleotide aptamers. ....................................................................................... 90
Figure 7.4: Structure of CCL5 monomer (PDB ID 1HRJ), with regions important for binding
CCR5 highlighted. ..................................................................................................... 93
Figure 7.5: Inhibition of HIV-1Ba-L infection by Ac-CFAYIARPLPRA-Am-functionalized
liposomes. ................................................................................................................ 95
Figure 7.6: Divalent conjugates for targeting CCR5. ........................................................ 96
Figure 7.7: Synthesis scheme for homodivalent PEG-aptamer bioconjugates. .............. 97

xii
Figure 7.8: SYBR Gold-stained 15% acrylamide TBE-Urea PAGE of ssDNA model aptamer
homodivalent PEG conjugation reactions after 88 hours at ambient temperature.
................................................................................................................................. 98
Figure 7.9: Synthesis scheme for heterodivalent PEG-aptamer bioconjugates. ............. 99

xiii
ACKNOWLEDGMENT
This dissertation was made possible with the support of several people whom I would
like to acknowledge here.
First and foremost, I would like to thank Prof. Ravi Kane, my Ph.D. advisor. Ravi has
not only provided the majority of funding for my studies, but he has almost always been
available for feedback at any time of day, including late at night and even sometimes
while on personal vacations. He has also taught by example, especially with his patience
and careful approach to problems. Furthermore, I have been happy to find that we have
many shared interests, both academic and unrelated to our research. Ravi has always
treated me with respect, and I have greatly appreciated that throughout the years.
I would also like to thank my other doctoral committee members, especially Prof.
Pete Tessier, who has been very kind to me throughout the years. Despite not being my
primary advisor, Pete has almost always been willing to discuss my research and my
career. He has also inspired me with his teaching style and with the productivity of his
research group; I aspire to someday be as successful as he has been both as a primary
investigator and as a teacher in the classroom. Prof. Steve Cramer and Prof. Shekhar
Garde have also been substantial influences on me. Steve has been very supportive of
me and I really admire his consistent involvement with the students in the department.
Shekhar was a really great instructor to have for Thermodynamics, and he also always
provided very astute guidance to the department’s graduate student association during
the years that I participated. Shekhar has been a great model of a leader for me.
My colleagues in the Kane lab deserve special recognition for helping me with much
of the research that is presented herein. I am grateful to Dr. Dhananjoy Mondal for
teaching me organic synthesis techniques, Dr. Manish Arha and Dr. Isil Severcan for
helping me with cloning and aptamer screening, and Dr. Sunit Srivastava and Dr. Marc
Douaisi for testing countless samples for me in a variety of assays. I am also glad to have
spent time in lab with Dr. Indrani Banerjee, Dr. Sanket Patke, Dr. Mohan Boggara, and
Dr. Jeff Litt, as they were fun and friendly colleagues to work with. I also appreciate the

xiv
help of the RPI staff, some of whom I got to know very well, especially Lee Vilardi and
Rose Primett. Dorota, Nancy, and Sharon were also very friendly and helpful for me.
I would like to express my gratitude for the support of my family and friends as well.
My parents have been especially generous in helping me financially whenever I needed
it, and have always given me good advice in times when I doubted myself, so I am
extremely grateful for that. My friends from Delaware (especially Ricky Komdat, Matt
O’Brian, Steve Newth, Pat Kerrane, Dan Kerrigan, Steve Smith, and Tiffany Garber) and
Connecticut (especially Harrison Paine, Ray Naclerio, Aman Kidwai, Andrew Harris, James
Halperin, Phil Nizzardo, Steve Papen, Steve Ferketic, Ryan Notti, Kevin Duffy, and Jim
Warren) have made more trips to visit me in the past six years than I could keep track of,
and I am so glad that those friendships have persisted despite me living a drive of several
hours away for so many years. I hope that the new friends that I have made locally will
also remain friends for many years to come. Some of these friends (especially Dr. Mike
Riley, Erin Eldeen, Hannah White, Erin Keys, Ana Hoyos, Ashlee Vilardi, Ashley Cress,
Krunal Mehta, James Woo, Jeremy Sauer, Corey Lemley, Dr. Eric Sterner, Clay Albracht,
Panos Karampourniotis, Jasmine Shong, and Emily Gnacik) have really helped me to stay
sane and socially balanced, and others (especially Brady Cress, Dr. Sayaka Masuko, and
Dr. Leyla Gasimli) have not only been great friends but have also been genuine
inspirations to me with their dedication to their research. I am thankful for all of these
friendships; they definitely helped me to stay motivated throughout the years.
Finally, I would also like to acknowledge the funding I received from the Chauncey
and Doris Starr Fellowship during my first two years at RPI, as well as the NIH grants
which enabled my advisor, Prof. Kane, to continue to support me during my studies.

xv
ABSTRACT
Here I describe the structure-based design of macromolecular bioconjugates for the
effective inhibition of three different diseases with broad public interest: anthrax,
influenza, and HIV/AIDS. To that end, I have synthesized multivalent arrangements of
bioactive macromolecules (i.e., peptides, proteins, and oligonucleotides) on
biocompatible scaffolds in order to enhance the inhibitory efficacy of those
biomolecules. Multivalency is the simultaneous interaction of multiple binding elements
with multiple target receptors, and this phenomenon underlies a powerful strategy for
controlling the potency of bioactive molecules by manipulating their context. For
example, some multivalent ligand-receptor interactions are known to exhibit binding
avidities that are several orders of magnitude stronger than the corresponding
monovalent receptor-ligand binding affinities. An important aspect of this research was
the identification of appropriate targets from the etiology of each of these diseases. Once
suitable targets were identified, I used strategically-chosen scaffolds to control the
multivalent arrangements of targeting ligands. Specifically, the anthrax toxin protective
antigen heptamer was the target of multivalent anti-toxin inhibitors that display peptide
ligands on either linear or radial scaffolds. Similarly, a conserved region of the influenza
hemagglutinin glycoprotein was targeted by a series of linear divalent and polyvalent cell
entry inhibitors. Finally, a recently-identified protein ligand for CCR5, a co-receptor for
HIV cell attachment and entry, was used for polyvalent display on linear polymer
scaffolds, and the ability to obstruct CCR5 binding was shown. The results support the
potential for multivalent bioconjugates such as these to lead to very promising
therapeutic applications.

1
1. Introduction1
1.1 Anthrax1
1.1.1 Overview
The disease anthrax is caused by infection with the bacterial species Bacillus anthracis,
which can occur through the inhalation of bacterial spores.2 For this reason, B. anthracis
is well known to the general public as a potential bioterrorism and/or biowarfare agent.2,3
There are two major virulence factors which contribute to the pathogenicity of B.
anthracis. First, the bacteria produces a poly-D-glutamic acid capsule which protects
against macrophage phagocytosis4 and allows the bacteria to spread systemically.
Second, B. anthracis produces anthrax toxin, which attacks the cells of the immune
system and causes death of the host.5,6 Ciprofloxacin is an antibiotic that has been
approved by the Food and Drug Administration (FDA) in the United States for post-
inhalational treatment of anthrax spores.7 Antibiotics such as ciprofloxacin may help to
reduce the proliferation of the pathogen within the body, but they do not prevent the
damage caused by the release of bacterial toxins. Thus there is a need for an antitoxin
treatment.
1.1.2 Intoxication pathway
Anthrax toxin is comprised of individually nontoxic monomeric proteins known as
protective antigen (PA), lethal factor (LF), and edema factor (EF).5,6,8 The combination of
PA and LF is called lethal toxin (LeTx) and the combination of PA and EF is called edema
toxin (EdTx). The individual components assemble into toxic complexes upon interacting
with receptors on the surfaces of target cells, as represented in Figure 1.1. The detailed
mechanisms governing the entry of LF and EF into target cell cytosol has been reported
in the literature.9–12
Portions of this chapter previously appeared as: Martin, J. T.; Kane, R. S. Design of Polyvalent Polymer Therapeutics. In Functional Polymers by Post-Polymerization Modification; Theato, P.; Klok, H.-A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 267–290.
2
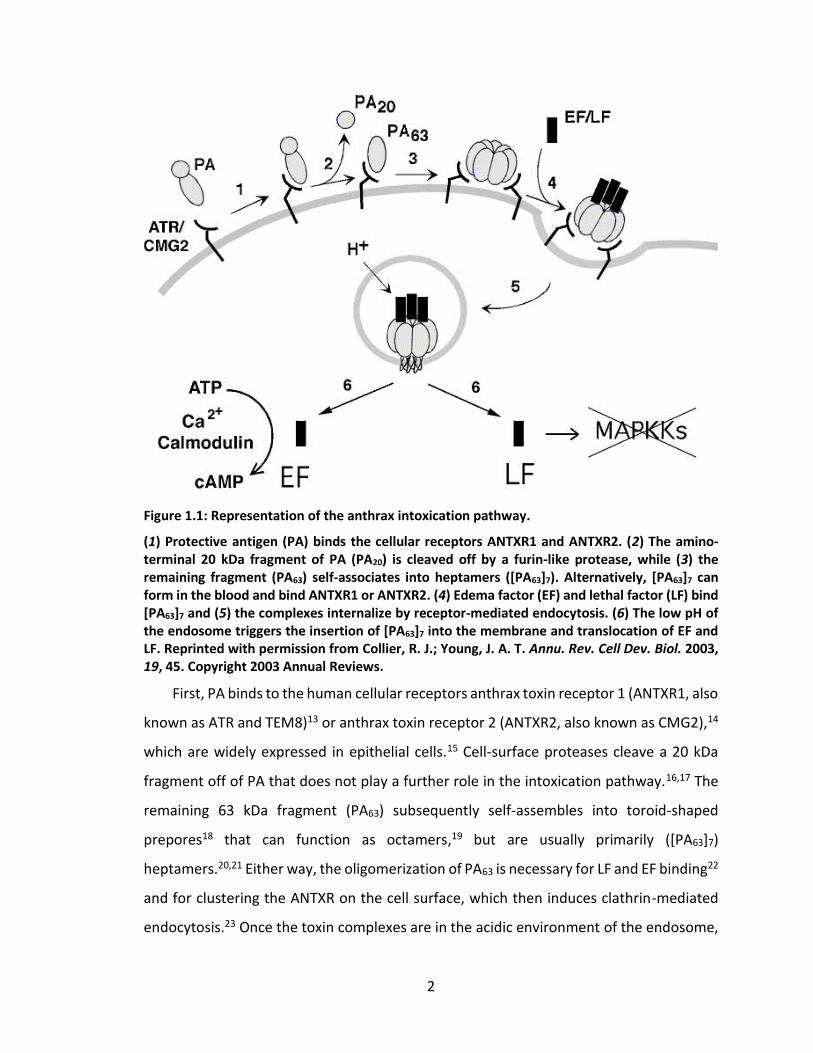
Figure 1.1: Representation of the anthrax intoxication pathway.
(1) Protective antigen (PA) binds the cellular receptors ANTXR1 and ANTXR2. (2) The amino-terminal 20 kDa fragment of PA (PA20) is cleaved off by a furin-like protease, while (3) the remaining fragment (PA63) self-associates into heptamers ([PA63]7). Alternatively, [PA63]7 can form in the blood and bind ANTXR1 or ANTXR2. (4) Edema factor (EF) and lethal factor (LF) bind [PA63]7 and (5) the complexes internalize by receptor-mediated endocytosis. (6) The low pH of the endosome triggers the insertion of [PA63]7 into the membrane and translocation of EF and LF. Reprinted with permission from Collier, R. J.; Young, J. A. T. Annu. Rev. Cell Dev. Biol. 2003, 19, 45. Copyright 2003 Annual Reviews.
First, PA binds to the human cellular receptors anthrax toxin receptor 1 (ANTXR1, also
known as ATR and TEM8)13 or anthrax toxin receptor 2 (ANTXR2, also known as CMG2),14
which are widely expressed in epithelial cells.15 Cell-surface proteases cleave a 20 kDa
fragment off of PA that does not play a further role in the intoxication pathway.16,17 The
remaining 63 kDa fragment (PA63) subsequently self-assembles into toroid-shaped
prepores18 that can function as octamers,19 but are usually primarily ([PA63]7)
heptamers.20,21 Either way, the oligomerization of PA63 is necessary for LF and EF binding22
and for clustering the ANTXR on the cell surface, which then induces clathrin-mediated
endocytosis.23 Once the toxin complexes are in the acidic environment of the endosome,

3
[PA63]7 inserts seven loops into the membrane to form a 14-stranded beta-barrel channel,
allowing the enzymatic LF and EF to translocate via a charge state-dependent Brownian
ratchet mechanism.11,12,24,25
LeTx and EdTx exhibit various immunosuppressive effects, and the bacterial
expression of either toxin independently can be fatal to the host organism.26–29 EF binds
to calmodulin in the cytosol and exhibits potent adenylate cyclase activity.30,31 The
increased cAMP affects cytokine expression, leading to inhibition of neutrophil
phagocytosis of B. anthracis.32,33 LF is a zinc-dependent protease that cleaves mitogen-
activate protein kinase kinases (MAPKKs) 1-4, 6 and 7; this in turn downregulates signaling
of extracellular-signal-regulated kinases, p38 mitogen-activated protein kinases, and c-
Jun N-terminal kinases.34,35 This activity of LF affects a multitude of cell types, including T
cells, dendridic cells, macrophages, and endothelial cells, by inhibiting cytokine
expression. Inhibition of the production of Type IIA phospholipase A2 by macrophages, of
differentiation of monocytes into macrophages, of production of immunoglobulin by B
cells, of production of superoxide by neutrophils, and of neutrophil motility are also the
result of LeTx exposure.36–38 Finally, LeTx exhibits a cytotoxic effect on macrophages,
dendritic cells, and some endothelial cells, and may eventually cause morbidity due to
vascular leakage and organ failure.28,29
1.2 Influenza
1.2.1 Overview
Influenza, commonly known to the general public as simply “the flu,” is a disease caused
by a virus that infects humans and a variety of other mammals and birds. There are three
types (analogous to species) of influenza virus, known as influenza A, B, and C,39 which
are classified based on the viral proteins on the exterior of the virions and the
characterization of the RNA genomes.40–42 Phylogenetically, influenza A and B are more
closely related than C, which is an indicator that influenza C diverged from the
evolutionary tree in the more distant past.42 Another difference between the types is that
influenza B and C are essentially limited to infecting human hosts, with only a couple of

4
known exceptions.39 In contrast, the native hosts of influenza A are waterfowl.39,43 While
influenza A is normally asymptomatic in aquatic birds, it is typically the most virulent type
in humans; it should be noted that the factors which contribute to influenza pathogenicity
in avian hosts may be different from those in humans.43 The natural avian hosts sustain
the reservoir of viral genetic variation from which the virus evolves to infect other animal
species, a process known as zoonosis.42 This process often involves reassortment of viral
genomes and can lead to the sudden appearance of new, pandemic-causing influenza A
strains. Genome reassortment is restricted by the influenza type,44 and I will be focused
on influenza type A for the remainder of this document.
1.2.2 Virion morphology and infection pathway
Influenza virions are known to be pleomorphic,40,41 which means that the virions can exist
in more than one general shape. In vitro, the viral particles are typically characterized as
being spherical with a diameter of roughly 100 nm, but in vivo the virions have been
reported to take on more filamentous appearances, with diameters of ~100 nm and
lengths of up to 20 µm.41 Another three classes of virions have been described for a total
of five different classes.40 Although the functional significance of this pleomorphism is
unknown, it has been found that all forms of the virions contains just one copy of the viral
genome.45,46 The genome is packaged inside the virion as a ribonucleoprotein complex, in
which the RNA segments are coated with nucleoprotein and an RNA-dependent RNA
polymerase.44 This complex and another protein called the nuclear export protein are
enclosed in a viral capsid formed by the M1 protein.41 Enveloping the viral capsid of the
mature virion is a lipid bilayer membrane, in which three transmembrane proteins are
embedded. Two of the three proteins are hemagglutinin (HA) and neuraminidase (NA),
which form the glycoprotein “spikes” that point outward from the viral envelope, and the
third envelope protein is the matrix ion channel protein (M2). HA exists as a homotrimer
and NA exists as a homotetramer; these are immediately formed during the process of
viral protein expression. The typical center-to-center distance between the glycoprotein
spikes on the virion surface is about 11 nm on average, and a spherical virion of average
size typically displays about 350 of these spikes in a roughly 6:1 ratio of HA to NA.40

5
1.2.3 Antigenic variation
The genome of influenza consists of eight separate negative-sense, single-stranded RNA
segments.44 The ability of these segments to recombine in a host cell that has been
infected with more than one strain of influenza can create an antigenic shift, which can
sometimes result in the sudden emergence of a pandemic-causing strain. Antigenic drift
is another type of antigenic variation, but describes the more continuous changes in the
antigenic sites of the viral glycoproteins over time.47,48 Proteins which are characterized
by the ability to consistently perform their primary functions while at the same time
tolerating a wide range of antigen-altering mutations are known as having structural
plasticity. The HA and NA of influenza are some of the best examples of proteins with a
high degree of structural plasticity, and this feature enables influenza to quickly develop
mutants that escape antibody neutralization by the mammalian adaptive immune
system.
1.2.4 Infection pathway
The first step in viral infection is cell attachment. The influenza virus attaches to cells by
the binding of HA to N-acetylneuraminic (sialic) acid on the cell surface.44 Sialic acids are
monosaccharides that are common on the ends of many kinds of glycosylated proteins
and exist in the airways and lungs of mammals and birds, which explains the mechanism
of transmission. Once attached to a cell, the virion is endocytosed.44 In the endosome,
acidic pH triggers a conformational change in the hemagglutinin stem/stalk region that
exposes a cell membrane fusion peptide and draws the viral and cell membranes
together.44 Meanwhile, the M2 ion channel pumps protons from the endosome into the
viral interior in order to release the ribonucleoprotein complex from the viral matrix. After
entering the cytosol, the nuclear localization signal of the ribonucleoprotein complex
enables delivery of the viral RNA and RNA-dependent RNA polymerase into the nucleus.44
The negative-sense RNA genes are then transcribed in the nucleus into positive-sense
messenger RNA and positive-sense complementary RNA for transcribing additional copies
of the negative-sense genome.44 The messenger RNAs are translated in the endoplasmic

6
reticulum, and the viral envelope proteins are apically sorted by the golgi apparatus to
specific locations on the cell surface for virion assembly.44 The virions begin to bud off
from the host cell once a sufficient amount of viral M1 protein has accumulated along the
lipid bilayer and a full set of the segmented viral genome has been packaged.44 The NA
then cleaves any extracellular sialic acid which would otherwise prevent the virions from
disengaging from the initially-infected host cell.44
1.3 Human Immunodeficiency Virus (HIV)
1.3.1 Overview
Despite over thirty years of intense scientific effort to develop cures and vaccines, the
Joint United Nations Programme on HIV/AIDS estimates that there were 2.5 million new
HIV infections in 2011.49
1.3.2 Infection pathway
HIV targets human cells primarily via a host cell surface glycoprotein called CD4 molecule.
Interaction with CD4 tethers the HIV virion to the cell surface and allows for binding to
co-receptors C-C chemokine receptor type 5 (CCR5) or C-X-C chemokine receptor type 4
(CXCR4).50,51 The interaction with CD4 induces a conformational change in gp120 that
allows it to complex with the co-receptor, and further conformational rearrangement
causes the release of gp41 into a fusogenic hairpin structure.52–54 This glycoprotein
structure pierces the host cell membrane and brings it and the viral membrane together,
resulting in the fusion of the viral and cell membranes.
Once the virion has entered the cell, the RNA from HIV is internalized, reverse
transcribed into double-stranded DNA, and integrated into the host cell chromosome by
a combination of viral and host enzymes.50,51 The integration into the host chromosome
allows the provirus to lie dormant for some time post-infection. When the viral DNA is
transcribed, several different viral mRNAs are produced and then translated into both
regulatory and structural proteins. The self-assembly of structural proteins with the viral
genomic RNA forms a core,51 which translocates to the cell surface and buds off, acquiring

7
the viral envelope by taking from the host cell membrane. During the final stage of viral
budding, a glycosaminoglycan polyprotein precursor is cleaved by a virally-encoded
protease, which results in the morphological progression into mature virions.51
1.3.3 Current treatment strategies
Pharmaceutical companies have discovered multiple classes of small molecule drugs that
target each of the various steps in the HIV replication pathway. Currently available classes
of antiretroviral drugs include viral entry/fusion inhibitors, nucleoside and nucleotide
reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors,
integrase inhibitors, protease inhibitors, and maturation inhibitors. More experimental
methods based on RNA interference have also been shown to inhibit HIV replication,55,56
but unfortunately, each of these strategies is susceptible to the emergence of HIV strains
that are resistant.
HIV resistance is just one of several serious medical concerns with current
antiretroviral drugs. Drug resistant strains evolve due to the error-prone nature of the HIV
reverse transcriptase enzyme and the high rate of viral turnover in infected individuals.
The evolution of resistance can be greatly affected by patient compliance to the
treatment regimen, such as improper administration or missed doses. In order to combat
the emergence of drug resistance, the current treatment strategy is to prescribe a
combination of several classes of drugs (at least three) into one “cocktail,” a strategy that
is known simply as combination antiretroviral therapy (cART) or highly active
antiretroviral therapy (HAART). However, many of these drugs can produce dangerous
side effects.57 Intolerance to side effects is a serious issue that can make it difficult to find
a satisfactory combination of therapies. In addition, the need to administer multiple drugs
simultaneously can also be extremely costly, especially for individuals in the countries
where populations are at the greatest risk, because of the high rate of poverty in those
areas.
Due to the concerns associated with current HIV treatments, the best solution to
reducing the numbers of HIV infections may be to prevent the transmission of the virus
to uninfected individuals. This goal continues to drive efforts to develop an effective HIV

8
vaccine,58–60 but there have been only two unsuccessful trials conducted to date.61–63
Recent research has shown that proper adherence to a HAART regimen can significantly
help reduce the risk of spreading HIV, but this finding is still subject to the problems
mentioned above. With these factors in mind, a prophylactic microbicide that could be
applied prior to intercourse may be a very worthwhile objective. Such a prophylactic must
not only be potent, but also able to impede the evolution of viral resistance.
1.4 Multivalent therapeutics
1.4.1 Multivalency
Multivalency is one of the most notable strategies used by nature to modulate
interactions between biological machinery.64 A multivalent system is characterized by an
arrangement of multiple binding elements on one entity interacting with another (often
biological) entity. Relative to the binding strength of an individual ligand-receptor pair (a
monovalent interaction), the corresponding multivalent or polyvalent (multivalency of a
very high valency) interaction is frequently enhanced by many orders of magnitude,64,65
and there have been a multitude of reviews in the past fifteen years highlighting the
potential benefits of utilizing multivalency.64,66–73 However, multivalency can play a role
in a wide variety of biological interactions that are not merely limited to the enhancement
of binding strength. For example, multivalent interactions can affect biological
signaling64,70,74 by establishing scaled interactions, by creating combinations of biological
interactions, and by maintaining contact between two surfaces over a large area. Such
multivalent interactions may play an important role in determining cell fate.75 In addition,
potent multivalent binders can be used to inhibit undesired interactions such as viral
attachment to cells76–82 and bacterial toxin assembly.83–96 Finally, multivalency can be
used to improve the specificity of therapeutics for certain applications such as tumor cell
targeting.97,98 Due to the fact that these cells have overexpressed receptors on the cell
surface, the best specificity for a therapeutic may be one that does not use targeting
ligands that are monovalently strong binders; in this case, using a polyvalent therapeutic
in which the monovalent contacts are only very weakly interacting should provide the

9
greatest contrast between desired and off-target binding. As is evident from these
examples, many of these different types of multivalent interactions have therapeutic
implications, and there has been growing interest in implementing multivalency for
therapeutic purposes.71–73
1.4.2 Scaffolds for multivalent therapeutics
Some of the structures that have been used to support multivalent or polyvalent displays
include nanoparticles, often gold or iron oxide, and derivatives of naturally occurring
constructs, such as viral particles and liposomes. However, synthetic polymers have
advantages in that they can be used in a broad range of structures (scaffolds) to
manipulate the presentation of biomolecules, and the details of the presentation can be
sequentially modified, thus resulting in the optimization of activity through multivalency.
For all types of linear polymer therapeutics (including end-functional, side-chain-
functional linear polymers, or branched (brush) polymers), the molecular weight of the
polymer backbone is one of the primary variables in the scaffold design. Whether using
pre-synthesized polymers or controlled polymerization techniques for custom synthesis,
the molecular weight, the DP, and the polydispersity index (PDI) are measures of how
well-defined the polymer sample is. These factors affect the hydrodynamic radius and
therapeutically relevant qualities such as the enhanced permeation and retention (EPR)
effect in tumors, other pharmacokinetic and biodistribution effects, and biodegradability
or toxicity.
There are a multitude of good reviews on the synthesis of polymer conjugates,99–108
but the main strategies relevant for synthesis of multivalent polymer therapeutics are
“grafting through” and “grafting to”. The “grafting through” strategy involves the
polymerization of monomers that have been pre-conjugated with the desired ligand,
which can yield polymers that display a high density of ligands along the polymer
backbone. However, there are several disadvantages of this strategy. Lack of reaction
modularity and lack of reproducibility are major disadvantages that arise from the need
to tune the reaction conditions of each polymerization and the difficulty in handling or
accurately measuring the minuscule quantities of expensive, modified-biomolecule

10
ligands, respectively. Furthermore, the difficulties involved in characterization and
analysis of qualities such as polydispersity and ligand composition can hinder the
optimization of the product multivalent presentation. In contrast, the “grafting to”
strategy involves the attachment of ligands to the polymer scaffold post-polymerization.
By using controlled polymerization and traditional polymer characterization techniques
to reveal accurate descriptors of the pieces involved pre-conjugation, product analysis is
much easier. That information can then be used in turn to guide subsequent multivalent
designs, which can often be synthesized without complication of reaction conditions.
1.4.3 A multivalent anthrax toxin inhibitor with in vivo efficacy
One of the first examples of a polymeric multivalent conjugate that was effective in vivo
was the anthrax toxin inhibitor reported by Mourez et al.86 This polyvalent polymer
therapeutic inhibited the assembly of anthrax toxin. The researchers used phage display
to identify a peptide sequence (HTSTYWWLDGAP) that binds the [PA63]7 heptamer and
inhibits toxin assembly with a half-maximal inhibitory concentration (IC50) of 150 µM
(weakly). Multiple copies of a modified peptide containing the [PA63]7-binding sequence
and a C-terminal lysine, Ac-HTSTYWWLDGAPK-Am, were attached pendant to a
polyacrylamide polymer backbone. The polyvalent conjugation was performed by
reacting poly(N-acryloyloxysuccinimide) (pNAS), an activated ester form of polyacrylic
acid, with the amine-functionalized peptide ligand. This protocol was based on previous
reports64,78–80 on the synthesis of polyvalent displays of sialic acid along polyacrylamide
backbones. The polyvalent anthrax toxin inhibitor thus created was 7,500-fold more
potent than the monovalent peptide at inhibiting anthrax toxin complex formation in
vitro, on a per-peptide basis. Furthermore, the polyvalent inhibitor was shown to have in
vivo efficacy against a toxin challenge, preventing symptoms in a rat intoxication model.
The successful inhibition of the toxin in vivo demonstrated that the therapeutic
design strategy was sound. The study86 showed that it is possible to construct a
therapeutically effective conjugate from weakly-binding, previously-unknown ligands
without extensive modification or optimization. Ligands which have been screened from
a random library for a desired target molecule can be made significantly more efficacious

11
by multivalent display on a suitable scaffold. Our group has placed a particular emphasis
on controlling the architecture of multivalent scaffolds in order to better understand the
influence of the design parameters on the efficacy of multivalent therapeutics. Here, we
will primarily highlight our studies on polymer-based scaffolds. Specifically, we will discuss
controlling the molecular weight and pendant ligand spacing for linear polymeric
scaffolds91,109,110 We will also include some discussion on increasing the biocompatibility
and biodegradability of the conjugate through the use of known biocompatible polymers,
such as N-(2-hydroxypropyl)methacrylamide (HPMA), or synthetic polypeptide-based
scaffolds, such as poly(glutamic acid) (PGA).92 While there have been many studies on the
structure-activity relationships of potential multivalent therapeutics (see recent reviews
on multivalent therapeutics111,112 and more general reviews of polymer therapeutics113–
116), the discussion here will focus primarily on the previous work of the Kane research
group towards developing polyvalent inhibitors of anthrax toxin.
1.4.4 Multivalent anthrax toxin inhibitors with control over average ligand spacing
In the interest of investigating the role of the geometry of a multivalent display (such as
the arrangement and spacing between ligands) on the resulting therapeutic efficacy, we
pursued linear polymer modification. In the context of multivalent therapeutic design, the
molecular weight of a polymer scaffold is particularly important for determining the
potential valency and density of a conjugated ligand. Moreover, with greater precision
over DP and PDI, the degree of uncertainty in conjugate characterization can be reduced
so that structure-activity relationships and optimized therapeutic properties can be
elucidated with greater confidence. For these reasons, we91,109 and others80,117 have
investigated the effects of scaffold molecular weight on post-polymerization functional
polymers by minimizing the effects of PDI.
Two different approaches were used to create multivalent inhibitors using polymer
scaffolds with low PDI. The work by Gujraty et al.91 took advantage of the fact that
poly(tert-butyl acrylate) (pTBA) was commercially available in a range of molecular
weights (e.g., 28.4, 69, 100, and 150 kDa) with low PDIs (ranging from 1.03 to 1.2). For
each sample, the pTBA was treated to produce low-PDI pNAS (Figure 1.2). The resulting

12
polymer contained the same activated esters and polymer backbone that were used to
make the anthrax toxin inhibitor reported by Mourez et al.,86 except that in this case the
starting materials had molecular weights that were more well-defined, leading to
products that could be compared with greater confidence. An alternative approach
toward low-PDI polymer scaffolds by Yanjarappa et al.109 relied on the RAFT is a
controlled, living polymerization technique known as reversible addition-fragmentation
chain transfer (RAFT) polymerization to create an activated polymer backbone with well-
controlled molecular weight. The resulting heteropolymers of N-
methacryloyloxysuccinimide (NMS) and N-(2-hydroxypropyl)methacrylamide (HPMA)
were characterized by 1H NMR spectroscopy and size-exclusion chromatography (SEC)
confirmed the ability to control the composition and size of the polymer chains.
Figure 1.2: Synthesis of a polyvalent anthrax toxin inhibitor of controlled molecular weight.
(a) TFA/CH2Cl2. (b) N,N’-Carbonyldiimidazole, N-hydroxysuccinimide, pyridine, 110°C. (c) i. peptide, DMF. ii. NH4OH. Reprinted with permission from Gujraty, K. V; Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Biomacromolecules 2006, 7, 2082. Copyright 2006 American Chemical Society.
The controlled molecular weight of the resulting activated polymers was exploited to
compare the efficacies of a range of polyvalent inhibitors that varied in both the density
of the conjugated ligands on the backbone and in the backbone molecular weight. While
the use of commercially available pTBA allowed for the synthesis of low-PDI
homopolymers of pNAS, the use of RAFT to create activated copolymers of HPMA with
controlled molecular weight and PDI demonstrated that various polymer backbones are
generally effective for use in polyvalent therapeutics. The results (Figure 1.3) revealed
that for polyvalent inhibitors prepared from the same size backbone and various degrees
13
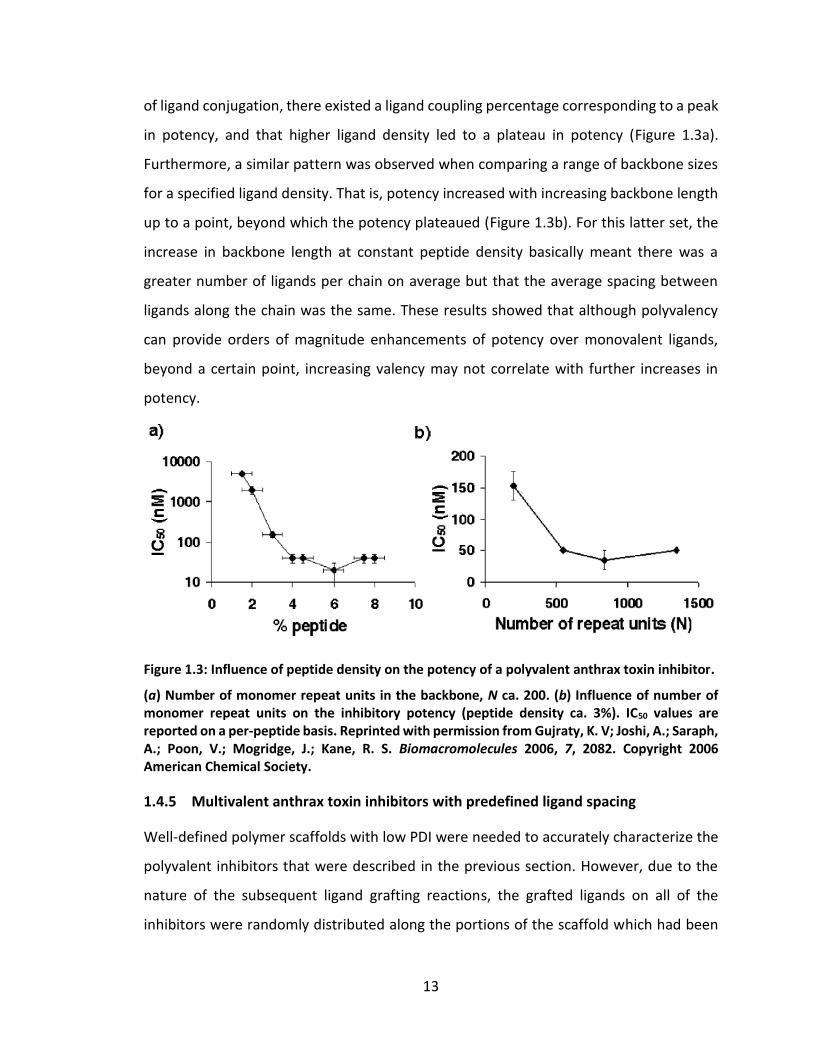
of ligand conjugation, there existed a ligand coupling percentage corresponding to a peak
in potency, and that higher ligand density led to a plateau in potency (Figure 1.3a).
Furthermore, a similar pattern was observed when comparing a range of backbone sizes
for a specified ligand density. That is, potency increased with increasing backbone length
up to a point, beyond which the potency plateaued (Figure 1.3b). For this latter set, the
increase in backbone length at constant peptide density basically meant there was a
greater number of ligands per chain on average but that the average spacing between
ligands along the chain was the same. These results showed that although polyvalency
can provide orders of magnitude enhancements of potency over monovalent ligands,
beyond a certain point, increasing valency may not correlate with further increases in
potency.
Figure 1.3: Influence of peptide density on the potency of a polyvalent anthrax toxin inhibitor.
(a) Number of monomer repeat units in the backbone, N ca. 200. (b) Influence of number of monomer repeat units on the inhibitory potency (peptide density ca. 3%). IC50 values are reported on a per-peptide basis. Reprinted with permission from Gujraty, K. V; Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Biomacromolecules 2006, 7, 2082. Copyright 2006 American Chemical Society.
1.4.5 Multivalent anthrax toxin inhibitors with predefined ligand spacing
Well-defined polymer scaffolds with low PDI were needed to accurately characterize the
polyvalent inhibitors that were described in the previous section. However, due to the
nature of the subsequent ligand grafting reactions, the grafted ligands on all of the
inhibitors were randomly distributed along the portions of the scaffold which had been
14
pre-activated. While statistical interpretations of the characterization data could lead to
deductions about the average valency and average inter-ligand spacing, it was
hypothesized that more precise control over the placement of the activated monomers
would help to gain an even more comprehensive understanding of the relationship
between structure and activity in polyvalent systems. With this goal in mind, the semi-
batch RAFT polymerization approach described previously was modified to construct
polymers with defined spacing between reactive monomers.110
Table 1.1: Polymerization of NAS at various [M]/[CTA] ratios.a
Reprinted with permission from Gujraty, K. V; Yanjarappa, M. J.; Saraph, A.; Joshi, A.; Mogridge, J.; Kane, R. S. J. Polym. Sci. A. Polym. Chem. 2008, 46, 7246. Copyright 2008 Wiley Periodicals, Inc.
The first step was to determine what distances between ligands would likely provide
a reasonable range of activity in the designed polyvalent inhibitor. To solve this problem,
a range of homopolymers of NAS with low PDIs were prepared by RAFT. The samples
differed in their DP as determined by SEC (Table 1.1). The Ac-HTSTYWWLDGAPK-Am
peptide was conjugated with each polymer sample such that the number of copies of the
ligand that were attached was controlled, and the remainder of the polymer backbone
was “quenched” by reaction with ammonium hydroxide. An average of three copies of
the peptide were conjugated to each individual polymer chain (as determined by 1H NMR
spectroscopy), but due to the differences in the backbone chain length of each sample, it
was inferred that the resulting polyvalent inhibitors would have different averages for the
number of unconjugated monomers separating each attached peptide. As illustrated in
Figure 1.4, the difference in ligand spacing could conceivably lead to a measurable

15
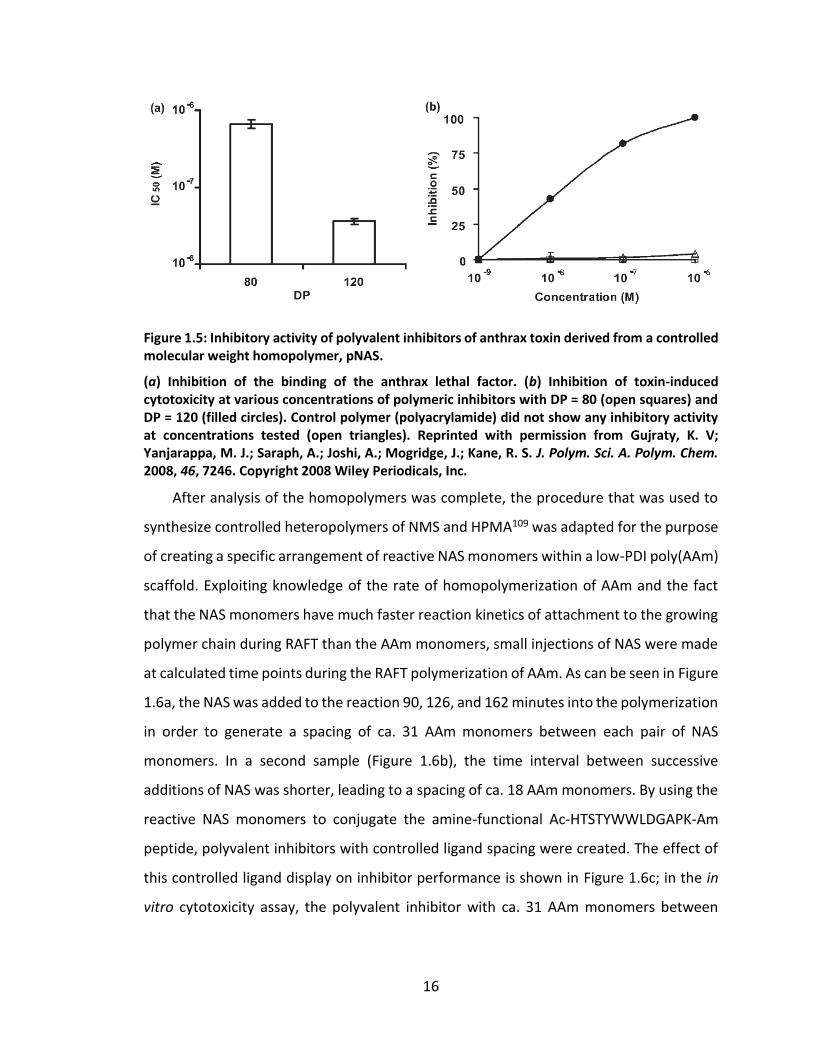
variation in sample efficacy. Indeed, for the inhibitor sample with DP = 120, the inhibition
IC50 of the binding of LF to [PA63]7 was over an order of magnitude lower than the IC50 of
the polyvalent inhibitor with DP = 80 (Figure 1.5a). Furthermore, as shown in Figure 1.5b,
the polyvalent inhibitor with DP = 120 was able to inhibit the cytotoxicity of anthrax lethal
toxin on RAW264.7 cells in vitro with an IC50 of 36 nM, yet the inhibitor with DP = 80 was
unable to inhibit cytotoxicity even at concentrations as high as 1 µM on a per-peptide
basis. With these results in hand, it was possible to design the polymers with controlled
spacing between reactive monomers so that the controlled spacing matched that of the
average spacing achieved by the homopolymer conjugation protocol. For a polyvalent
inhibitor with a number of peptides “𝑖” conjugated randomly along the polymer
backbone, the average spacing between adjacent conjugated peptides was calculated by
DP (𝑖 + 1)⁄ . Accordingly, for the polyvalent inhibitor with DP = 120 and 𝑖 = 3, the average
number of monomers of separation was estimated to be roughly 30. This spacing thus
served as the target number of non-active acrylamide (AAm) monomers to incorporate
between reactive NAS monomers in a controlled RAFT polymerization of a
heteropolymer, poly(AAm-co-NAS).
Figure 1.4: Design of polyvalent inhibitors with control over molecular weight and ligand spacing.
The linear polyvalent inhibitors displaying peptides (black ovals) are shown bound to the PA63 heptamer at the peptide-binding sites (circles). The spacing between peptides on the linear scaffold is either too short (left panel) or is sufficient (right panel) to allow a polyvalent interaction. Reprinted with permission from Gujraty, K. V; Yanjarappa, M. J.; Saraph, A.; Joshi, A.; Mogridge, J.; Kane, R. S. J. Polym. Sci. A. Polym. Chem. 2008, 46, 7246. Copyright 2008 Wiley Periodicals, Inc.
16
Figure 1.5: Inhibitory activity of polyvalent inhibitors of anthrax toxin derived from a controlled molecular weight homopolymer, pNAS.
(a) Inhibition of the binding of the anthrax lethal factor. (b) Inhibition of toxin-induced cytotoxicity at various concentrations of polymeric inhibitors with DP = 80 (open squares) and DP = 120 (filled circles). Control polymer (polyacrylamide) did not show any inhibitory activity at concentrations tested (open triangles). Reprinted with permission from Gujraty, K. V; Yanjarappa, M. J.; Saraph, A.; Joshi, A.; Mogridge, J.; Kane, R. S. J. Polym. Sci. A. Polym. Chem. 2008, 46, 7246. Copyright 2008 Wiley Periodicals, Inc.
After analysis of the homopolymers was complete, the procedure that was used to
synthesize controlled heteropolymers of NMS and HPMA109 was adapted for the purpose
of creating a specific arrangement of reactive NAS monomers within a low-PDI poly(AAm)
scaffold. Exploiting knowledge of the rate of homopolymerization of AAm and the fact
that the NAS monomers have much faster reaction kinetics of attachment to the growing
polymer chain during RAFT than the AAm monomers, small injections of NAS were made
at calculated time points during the RAFT polymerization of AAm. As can be seen in Figure
1.6a, the NAS was added to the reaction 90, 126, and 162 minutes into the polymerization
in order to generate a spacing of ca. 31 AAm monomers between each pair of NAS
monomers. In a second sample (Figure 1.6b), the time interval between successive
additions of NAS was shorter, leading to a spacing of ca. 18 AAm monomers. By using the
reactive NAS monomers to conjugate the amine-functional Ac-HTSTYWWLDGAPK-Am
peptide, polyvalent inhibitors with controlled ligand spacing were created. The effect of
this controlled ligand display on inhibitor performance is shown in Figure 1.6c; in the in
vitro cytotoxicity assay, the polyvalent inhibitor with ca. 31 AAm monomers between
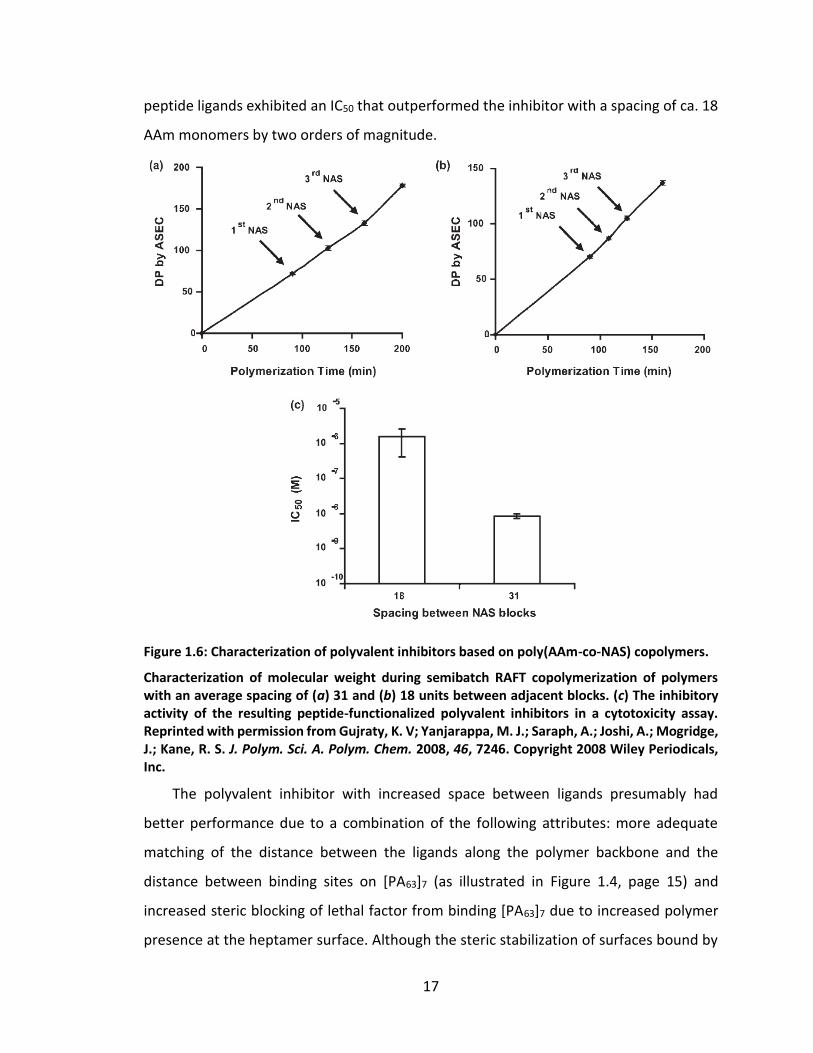
17
peptide ligands exhibited an IC50 that outperformed the inhibitor with a spacing of ca. 18
AAm monomers by two orders of magnitude.
Figure 1.6: Characterization of polyvalent inhibitors based on poly(AAm-co-NAS) copolymers.
Characterization of molecular weight during semibatch RAFT copolymerization of polymers with an average spacing of (a) 31 and (b) 18 units between adjacent blocks. (c) The inhibitory activity of the resulting peptide-functionalized polyvalent inhibitors in a cytotoxicity assay. Reprinted with permission from Gujraty, K. V; Yanjarappa, M. J.; Saraph, A.; Joshi, A.; Mogridge, J.; Kane, R. S. J. Polym. Sci. A. Polym. Chem. 2008, 46, 7246. Copyright 2008 Wiley Periodicals, Inc.
The polyvalent inhibitor with increased space between ligands presumably had
better performance due to a combination of the following attributes: more adequate
matching of the distance between the ligands along the polymer backbone and the
distance between binding sites on [PA63]7 (as illustrated in Figure 1.4, page 15) and
increased steric blocking of lethal factor from binding [PA63]7 due to increased polymer
presence at the heptamer surface. Although the steric stabilization of surfaces bound by

18
a polyvalent inhibitor is expected to play a role in inhibition of biological interactions,79
factors that enhance the binding strength of the inhibitor account for the majority of the
inhibitor’s efficacy.64 Thus, it is desirable to adjust the architecture of the polyvalent
inhibitor such that the complementarity to the target’s binding epitopes is maximized. As
an additional example, the target of the anthrax toxin inhibitors described thus far has
been [PA63]7, a heptameric protein oligomer with seven binding sites for the peptide
ligand containing the sequence HTSTYWWLDGAP. For the polyvalent inhibitors of varying
controlled molecular weights that were prepared by modifying pTBA, the sample that
displayed the greatest inhibitory activity was the one that had an average of 6 ± 2 peptide
ligands per polymer chain,91 which means the valency of the inhibitor and target were
nearly matched.
1.5 Motivation
The initial work by Mourez et al.86 showed that it is possible to construct a therapeutically
effective conjugate from weakly-binding, previously-unknown ligands without extensive
modification or optimization. While such a study was encouraging because of the
implication that the road to developing new constructs that are therapeutically active
may not be too difficult, the actual translation of such approaches into clinically relevant
treatments has yet to be realized. In addition, the previous research on multivalent
inhibitors had begun to recognize the importance of the design parameters of ligand
spacing and valency, as described in Section 1.4. Therefore I was motivated to study the
design parameters of multivalent bioconjugate synthesis with greater precision, as well
as to try to expand the approach of designing multivalent inhibitors to new applications:
inhibition of influenza virus and HIV.

19
2. Preclinical development of polyvalent inhibitors of anthrax toxin2
The Kane research group has gained a great deal of experience the field of
multivalent/polyvalent conjugate design by studying a wide range of multi/polyvalent
anthrax toxin inhibitors that show potent activity in in vitro and in vivo toxin challenges
using rodent models. However, we firmly believe that this therapeutic strategy could find
real use in the medical field. To that end, I have put considerable time and effort into
preclinical development studies of polyvalent anthrax toxin inhibitors, as described in this
chapter.
2.1 Therapeutic suitability
2.1.1 Biocompatibility
When designing a polymeric substance to be administered within the human body or
within the environment, some of the primary concerns are biocompatibility and
biodegradability. For a polymer therapeutic to be biocompatible, the polymer and all of
its metabolites should be non-toxic, and administration should not induce excessive
immunological reactions. Biodegradability describes the ability of a material to be
decomposed naturally either due to natural chemical or enzymatic processes, and is
desirable in the context of most therapeutics because the ability of a material to be
naturally cleared from the body is correlated with good biocompatibility. The ability to
use pHPMA, a proven biocompatible polymer,118,119 as an effective multivalent or
polyvalent scaffold has already been described in a previous section.109 However, HPMA
is not biodegradable. On the other hand, the polypeptide-based polymer, poly(L-glutamic
acid) (PGA), possesses several properties desirable for drug carriers, including high water
solubility, biodegradability, and low toxicity (biocompatibility).120 PGA backbones have
been used to prepare a variety of polyvalent displays.81,93,121,122 Furthermore, a paclitaxel-
PGA conjugate named XYOTAX™ has shown encouraging outcomes in clinical studies.114,123
Portions of this chapter previously appeared as: Martin, J. T.; Kane, R. S. Design of Polyvalent Polymer Therapeutics. In Functional Polymers by Post-Polymerization Modification; Theato, P.; Klok, H.-A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 267–290.

20
2.1.2 PGA inhibitor synthesis
The synthesis of anthrax toxin inhibitors based on PGA was originally described by Joshi
et al.92 This study used an ester activation strategy similar to that used for pAA to modify
the carboxyl groups on the side chains of this polypeptide with the amine-functional
[PA63]7-binding peptide ligand, Ac-HTSTYWWLDGAPK-Am, as shown in Figure 2.1.
Figure 2.1: Synthesis of a PGA-based polyvalent anthrax toxin inhibitor.
(a) EDC, H2O; (b) HOBt, DMF; (c) peptide, DMF, TEA (d) NH4OH, dialysis. The polyvalent inhibitor is a random copolymer composed of peptide conjugates (x repeat units) glutamine (y repeat units), and glutamic acid (z repeat units). Reprinted with permission from Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Bioconjug. Chem. 2006, 17, 1265. Copyright 2006 American Chemical Society.
2.1.3 PGA inhibitor efficacy in vitro
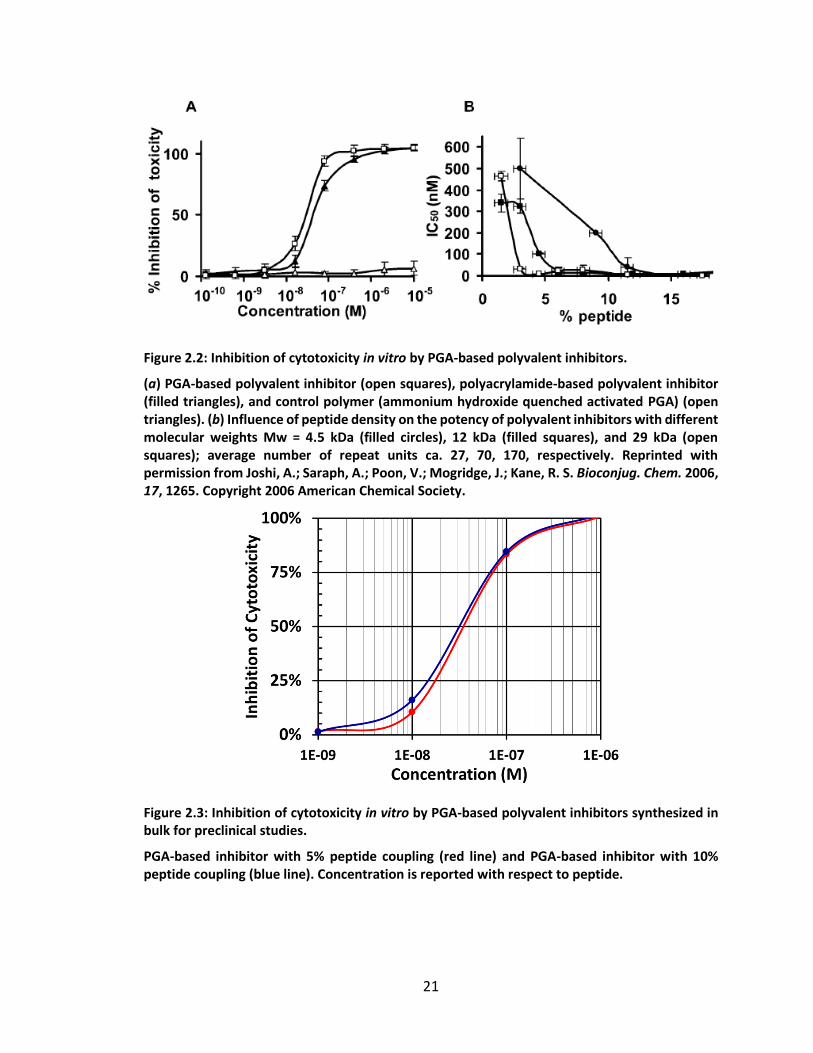
The resulting inhibitors synthesized by Joshi had similar activity to the corresponding pAA-
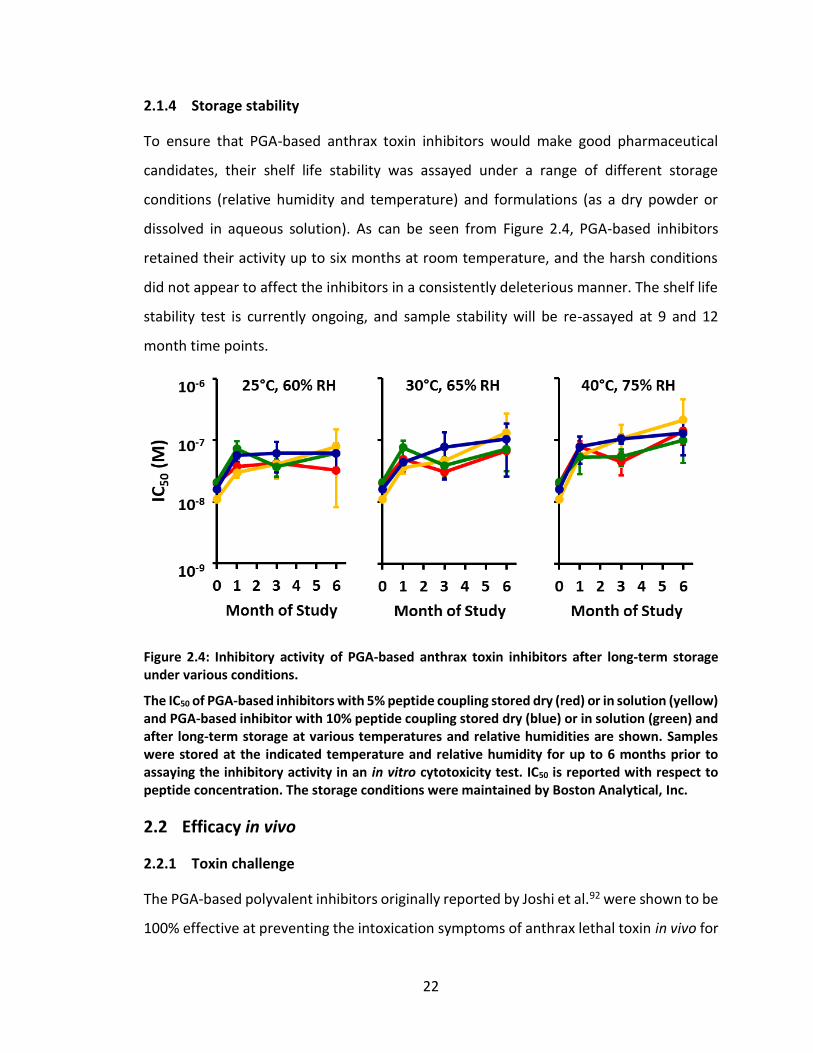
based inhibitors in an in vitro cytotoxicity assay (Figure 2.2). For the purpose of several
preclinical studies assaying the therapeutic potential of these PGA-based anthrax toxin
inhibitors, I have successfully been able to scale up the synthesis protocol to generate
hundreds of milligrams of polyvalent inhibitors per batch. We ensured that the inhibitors
I have created also show potent activity by testing in additional in vitro assays of
cytotoxicity inhibition (Figure 2.3).
21
Figure 2.2: Inhibition of cytotoxicity in vitro by PGA-based polyvalent inhibitors.
(a) PGA-based polyvalent inhibitor (open squares), polyacrylamide-based polyvalent inhibitor (filled triangles), and control polymer (ammonium hydroxide quenched activated PGA) (open triangles). (b) Influence of peptide density on the potency of polyvalent inhibitors with different molecular weights Mw = 4.5 kDa (filled circles), 12 kDa (filled squares), and 29 kDa (open squares); average number of repeat units ca. 27, 70, 170, respectively. Reprinted with permission from Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Bioconjug. Chem. 2006, 17, 1265. Copyright 2006 American Chemical Society.
Figure 2.3: Inhibition of cytotoxicity in vitro by PGA-based polyvalent inhibitors synthesized in bulk for preclinical studies.
PGA-based inhibitor with 5% peptide coupling (red line) and PGA-based inhibitor with 10% peptide coupling (blue line). Concentration is reported with respect to peptide.
22
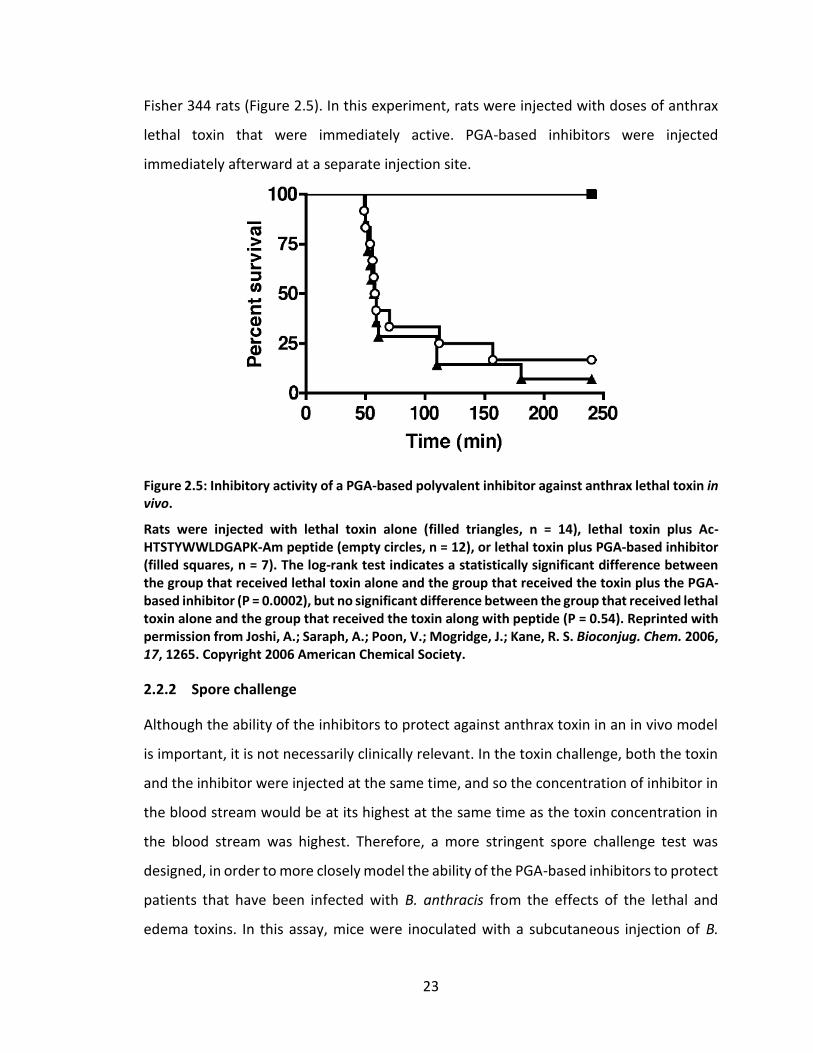
2.1.4 Storage stability
To ensure that PGA-based anthrax toxin inhibitors would make good pharmaceutical
candidates, their shelf life stability was assayed under a range of different storage
conditions (relative humidity and temperature) and formulations (as a dry powder or
dissolved in aqueous solution). As can be seen from Figure 2.4, PGA-based inhibitors
retained their activity up to six months at room temperature, and the harsh conditions
did not appear to affect the inhibitors in a consistently deleterious manner. The shelf life
stability test is currently ongoing, and sample stability will be re-assayed at 9 and 12
month time points.
Figure 2.4: Inhibitory activity of PGA-based anthrax toxin inhibitors after long-term storage under various conditions.
The IC50 of PGA-based inhibitors with 5% peptide coupling stored dry (red) or in solution (yellow) and PGA-based inhibitor with 10% peptide coupling stored dry (blue) or in solution (green) and after long-term storage at various temperatures and relative humidities are shown. Samples were stored at the indicated temperature and relative humidity for up to 6 months prior to assaying the inhibitory activity in an in vitro cytotoxicity test. IC50 is reported with respect to peptide concentration. The storage conditions were maintained by Boston Analytical, Inc.
2.2 Efficacy in vivo
2.2.1 Toxin challenge
The PGA-based polyvalent inhibitors originally reported by Joshi et al.92 were shown to be
100% effective at preventing the intoxication symptoms of anthrax lethal toxin in vivo for
23
Fisher 344 rats (Figure 2.5). In this experiment, rats were injected with doses of anthrax
lethal toxin that were immediately active. PGA-based inhibitors were injected
immediately afterward at a separate injection site.
Figure 2.5: Inhibitory activity of a PGA-based polyvalent inhibitor against anthrax lethal toxin in vivo.
Rats were injected with lethal toxin alone (filled triangles, n = 14), lethal toxin plus Ac-HTSTYWWLDGAPK-Am peptide (empty circles, n = 12), or lethal toxin plus PGA-based inhibitor (filled squares, n = 7). The log-rank test indicates a statistically significant difference between the group that received lethal toxin alone and the group that received the toxin plus the PGA-based inhibitor (P = 0.0002), but no significant difference between the group that received lethal toxin alone and the group that received the toxin along with peptide (P = 0.54). Reprinted with permission from Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Bioconjug. Chem. 2006, 17, 1265. Copyright 2006 American Chemical Society.
2.2.2 Spore challenge
Although the ability of the inhibitors to protect against anthrax toxin in an in vivo model
is important, it is not necessarily clinically relevant. In the toxin challenge, both the toxin
and the inhibitor were injected at the same time, and so the concentration of inhibitor in
the blood stream would be at its highest at the same time as the toxin concentration in
the blood stream was highest. Therefore, a more stringent spore challenge test was
designed, in order to more closely model the ability of the PGA-based inhibitors to protect
patients that have been infected with B. anthracis from the effects of the lethal and
edema toxins. In this assay, mice were inoculated with a subcutaneous injection of B.
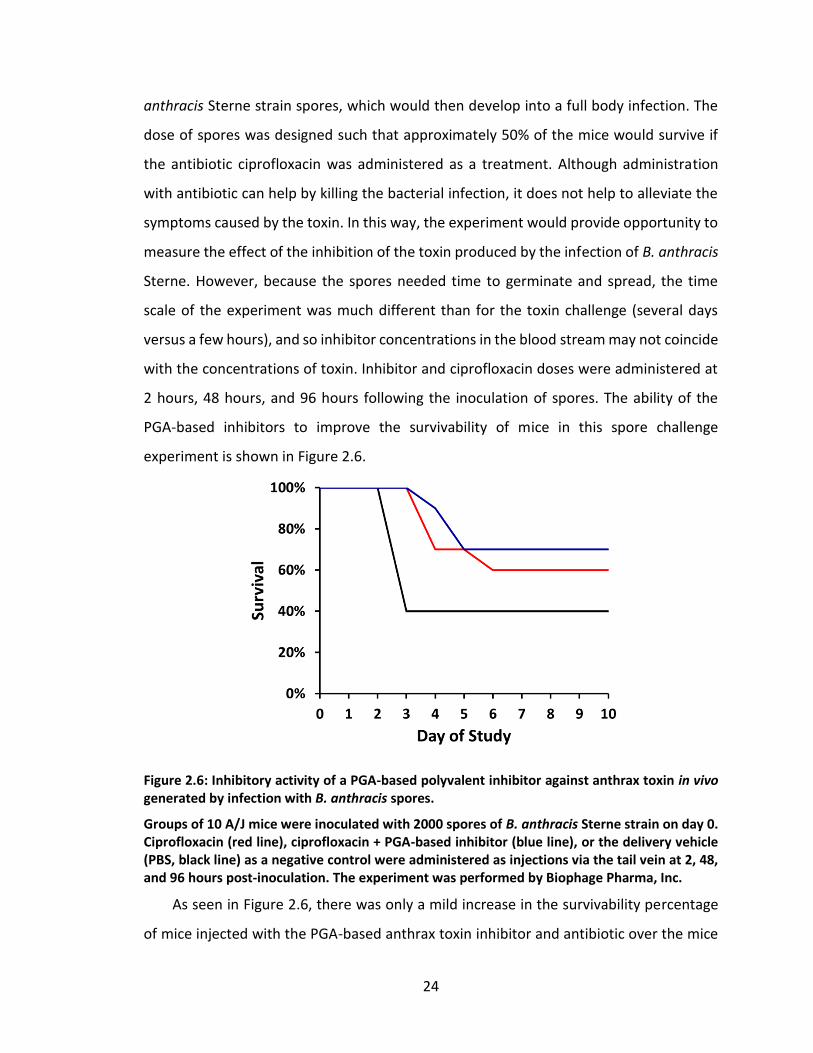
24
anthracis Sterne strain spores, which would then develop into a full body infection. The
dose of spores was designed such that approximately 50% of the mice would survive if
the antibiotic ciprofloxacin was administered as a treatment. Although administration
with antibiotic can help by killing the bacterial infection, it does not help to alleviate the
symptoms caused by the toxin. In this way, the experiment would provide opportunity to
measure the effect of the inhibition of the toxin produced by the infection of B. anthracis
Sterne. However, because the spores needed time to germinate and spread, the time
scale of the experiment was much different than for the toxin challenge (several days
versus a few hours), and so inhibitor concentrations in the blood stream may not coincide
with the concentrations of toxin. Inhibitor and ciprofloxacin doses were administered at
2 hours, 48 hours, and 96 hours following the inoculation of spores. The ability of the
PGA-based inhibitors to improve the survivability of mice in this spore challenge
experiment is shown in Figure 2.6.
Figure 2.6: Inhibitory activity of a PGA-based polyvalent inhibitor against anthrax toxin in vivo generated by infection with B. anthracis spores.
Groups of 10 A/J mice were inoculated with 2000 spores of B. anthracis Sterne strain on day 0. Ciprofloxacin (red line), ciprofloxacin + PGA-based inhibitor (blue line), or the delivery vehicle (PBS, black line) as a negative control were administered as injections via the tail vein at 2, 48, and 96 hours post-inoculation. The experiment was performed by Biophage Pharma, Inc.
As seen in Figure 2.6, there was only a mild increase in the survivability percentage
of mice injected with the PGA-based anthrax toxin inhibitor and antibiotic over the mice
25
that only received antibiotic treatment. Statistical analysis of the results (not shown)
showed that this difference was not large enough to be statistically significant. These
results showed that there may be some problems with the current design of the
polyvalent PGA-based anthrax toxin inhibitor. The next step was to try to identify what
factors might be affecting the efficacy of the PGA-based toxin inhibitor in this experiment.
2.3 Pharmacokinetics
2.3.1 Serum stability
The first hypothesis was that there may be some protease activity that is degrading the
PGA-based inhibitor in vivo on a time scale that is slow enough that allowed it to remain
efficacious in the toxin challenge (over the course of a few hours), but fast enough to
reduce inhibitor efficacy in the spore challenge (over the course of a few days). In order
to test this hypothesis, we performed a serum incubation assay. PGA-based inhibitors
were pre-incubated in serum for up to three days prior to assaying the inhibitory efficacy
in a cell culture cytotoxicity assay (Figure 2.7). From this experiment, it was evident that
the PGA-based inhibitors retain their activity at least over the course of a few days in
biological fluids.
Figure 2.7: Inhibitory activity of PGA-based inhibitors after incubation in serum.
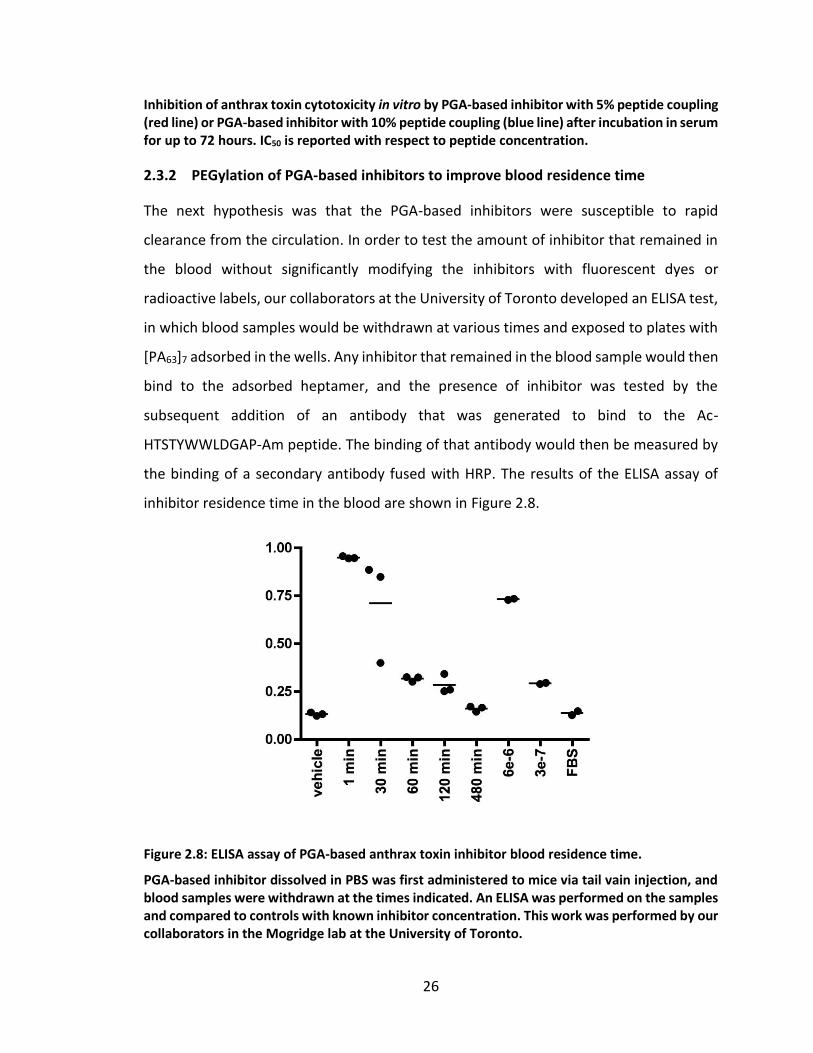
26
Inhibition of anthrax toxin cytotoxicity in vitro by PGA-based inhibitor with 5% peptide coupling (red line) or PGA-based inhibitor with 10% peptide coupling (blue line) after incubation in serum for up to 72 hours. IC50 is reported with respect to peptide concentration.
2.3.2 PEGylation of PGA-based inhibitors to improve blood residence time
The next hypothesis was that the PGA-based inhibitors were susceptible to rapid
clearance from the circulation. In order to test the amount of inhibitor that remained in
the blood without significantly modifying the inhibitors with fluorescent dyes or
radioactive labels, our collaborators at the University of Toronto developed an ELISA test,
in which blood samples would be withdrawn at various times and exposed to plates with
[PA63]7 adsorbed in the wells. Any inhibitor that remained in the blood sample would then
bind to the adsorbed heptamer, and the presence of inhibitor was tested by the
subsequent addition of an antibody that was generated to bind to the Ac-
HTSTYWWLDGAP-Am peptide. The binding of that antibody would then be measured by
the binding of a secondary antibody fused with HRP. The results of the ELISA assay of
inhibitor residence time in the blood are shown in Figure 2.8.
Figure 2.8: ELISA assay of PGA-based anthrax toxin inhibitor blood residence time.
PGA-based inhibitor dissolved in PBS was first administered to mice via tail vain injection, and blood samples were withdrawn at the times indicated. An ELISA was performed on the samples and compared to controls with known inhibitor concentration. This work was performed by our collaborators in the Mogridge lab at the University of Toronto.

27
As we hypothesized, a large percentage of the PGA-based inhibitor has been cleared
from the circulation within one hour post-injection. These results may help explain the
lack of success during the spore challenge experiment, because the low concentration of
inhibitor remaining in the blood (if any) past this time would probably not be providing
adequate inhibition of anthrax toxin being continually released by B. anthracis.
Since analyzing these results, we have decided to try to improve the
pharmacokinetics of the PGA-based inhibitor by further modification with PEG. The use
of PEG conjugation is a commonly used method for increasing the hydrodynamic volume,
and by extension the blood residence time, of a variety of therapeutic molecules. Because
our inhibitors are roughly 40 kDa in size, we used mono-functional methoxy-PEG-amines
of 20 kDa and 40 kDa. These PEGs react with the HOBt-activated esters of the glutamate
side chains of our activated PGA in the same way that the amine of the lysine side chain
of Ac-HTSTYWWLDGAPK-Am does. Due to the more difficult solubilization and
significantly larger size of these PEGs relative to the peptide, we expected a slower
conjugation rate of the methoxy-PEG-amines to the activated PGA. Thus we added the
PEG first and allowed 5-10 min reaction prior to addition of the peptide to the reaction
mixture. Successful PEGylation could be visualized by SDS-PAGE with barium iodide
staining to show the PEG attachment (see Figure 2.9).124,125 Polyvalent PGA-peptide
conjugates cannot typically by visualized by either Coomassie blue or silver staining due
to the highly negatively-charged polypeptide backbone, as shown by the silver-stained
image of the gel (left). PEG is also not stained by common gel-staining methods, but after
treatment with barium chloride followed by an iodine solution, PEG stains dark brown
against a light brown background.124,125 As is evident by the multilple bands on the gel,
there are multiple different species of PGA-PEG/peptide in the reaction mixture. The
mixture of these PEGylated PGA-peptide anthrax toxin inhibitors have been tested for
activity in the in vitro inhibition of cytotoxicity assay, and they retain their inhibitory
activity.

28
Figure 2.9: Silver-stain (left) followed by barium iodide-stain (right) of a 4-12% acrylamide SDS-PAGE of PGA-based anthrax toxin inhibitors conjugated with monofunctional PEG20k or PEG40k.
Lanes left to right: (1) Reaction targeting average attachment of three PEG20k chains per PGA35k scaffold with 10% peptide coupling, (2) Reaction targeting average attachment of three PEG40k chains per PGA35k scaffold with 10% peptide coupling, (3) Reaction targeting average attachment of three PEG20k chains per PGA120k scaffold with 10% peptide coupling, (4) Reaction targeting average attachment of one PEG40k chain per PGA120k scaffold with 10% peptide coupling, (5) Reaction targeting average attachment of three PEG40k chains per PGA120k scaffold with 10% peptide coupling, (6) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled. Both images are of the same gel, first after silver staining (left) and then after barium iodide staining of PEG (right).
2.4 Conclusions
The fact that the PEGylated polyvalent PGA-peptide anthrax toxin inhibitors retain their
inhibitory activity in vitro is promising. We are currently pursuing methods for purifying
the individual species as shown in the gel image on the right in Figure 2.9, because the
polydispersity of the PGA and the lack of control over the number of PEG chains
complicates characterization efforts. Unfortunately, purification by SEC does not provide
adequate resolution for separation of the various species of PEGylated PGA-based
anthrax toxin inhibitors. To address this difficulty, we have turned to a less commonly-
used method for purification, large-scale gel electrophoresis. The apparatus allows for
size-based and/or charge-based fractionation of material by directing a flow of liquid
along the bottom of the gel to a fractionator.126–128 In this way, large samples can be run

29
through the gel and analyzed based on the time of electrophoretic elution. Once this
purification method has been optimized, the well-fractionated, PEGylated, PGA-based
anthrax toxin inhibitors can be characterized by 1H NMR to confirm the average percent
coupling of PEG. In addition, the fractionated, PEGylated inhibitors can be compared by
in vitro cytotoxicity assay and ELISA to determine blood residence time. If the blood
residence time is significantly improved, these PEGylated anthrax toxin inhibitors may be
much more promising candidates for clinical application. In that case, another in vivo
spore challenge experiment similar to the one described in Section 2.2.2 should be
performed with the PEGylated anthrax toxin inhibitors.

30
3. Radial heptavalent inhibitors of anthrax toxin3
3.1 Abstract129
The design of multivalent molecules, consisting of multiple copies of a biospecific ligand
attached to a suitable scaffold, represents a promising approach to inhibit pathogens and
oligomeric microbial toxins. Despite the increasing interest in structure-based drug
design, few multivalent inhibitors based on this approach have shown efficacy in vivo.
Here we demonstrate the structure-based design of potent biospecific heptavalent
inhibitors of anthrax lethal toxin. Specifically, we illustrate the ability to design potent
multivalent ligands by matching the pattern of binding sites on the biological target. We
used a combination of experimental studies based on mutagenesis to identify the binding
site for an inhibitory peptide on the heptameric subunit of anthrax toxin. We developed
an approach based on copper-catalyzed azide-alkyne cycloaddition (click-chemistry) to
facilitate the attachment of seven copies of the inhibitory peptide to a β-cyclodextrin core
via a polyethylene glycol linker of an appropriate length. The resulting heptavalent
inhibitors neutralized anthrax lethal toxin both in vitro and in vivo and showed
appreciable stability in serum. Given the inherent biocompatibility of cyclodextrin and
polyethylene glycol, these potent well-defined heptavalent inhibitors show considerable
promise as anthrax anti-toxins.
3.2 Introduction
Anthrax toxin, responsible for the major symptoms and death associated with anthrax, is
composed of three proteins: lethal factor (LF) and edema factor (EF) are enzymes that act
in the mammalian cell cytosol; protective antigen (PA) binds to cell-surface receptors and
mediates toxin internalization.130 PA is proteolytically processed into a 63 kDa fragment,
PA63, which oligomerizes into heptamers ([PA63]7) that bind EF and LF. The assembled
toxin complex is internalized by receptor-mediated endocytosis and the enzymes are
This chapter previously appeared as: Joshi, A.; Kate, S.; Poon, V.; Mondal, D.; Boggara, M. B.; Saraph, A.; Martin, J. T.; McAlpine, R.; Day, R.; Garcia, A. E.; Mogridge, J.; Kane, R. S. Biomacromolecules 2011, 12, 791. Copyright 2011 American Chemical Society.

31
translocated into the cytosol when the toxin reaches an acidic compartment. The
enzymatic activities of these proteins cause a variety of cellular dysfunctions that
contribute to disease progression.5 Anthrax toxin is, therefore, an important therapeutic
target and inhibitors have been described that block different steps in this intoxication
pathway.10,13,14,86,89,90,92,94,96,131–135
A promising approach to designing potent ligands for oligomeric targets involves the
design of inhibitors that are multivalent, as multivalency or polyvalency can enhance the
affinity of interactions by several orders of magnitude.64,67,69 Some oligovalent inhibitors
have been designed without prior knowledge of the spatial relationships among the
binding sites on the target64,86 and others have been designed with architectures that
matched the geometry of binding epitopes on various bacterial toxins.83,84,88,89,131,136,137
The rational structure-based design of oligovalent inhibitors that provide structurally and
compositionally well-defined molecules may, however, be better suited for pre-clinical
and clinical development. There are but a few examples of the structure-based design of
oligovalent inhibitors that have shown efficacy in vivo.88,89
Here, we describe the rational structure-based design of an anthrax anti-toxin based
on the heptavalent display of a biospecific ligand. Our strategy to optimize the design of
the inhibitor was to attach peptides to a defined scaffold that would present the peptides
in a spatial orientation matching the peptide-binding sites of the toxin. For the core of the
scaffold, we considered using cyclodextrins, because they are cyclic oligomers of
glucopyranose138 that have a defined molecular weight and a given symmetry, and they
are commercially available in high purity. β-cyclodextrin was chosen because it has 7-fold
symmetry, as does [PA63]7 (Figure 3.1), and because cyclodextrins are widely used as
pharmaceutical agents to enhance the solubility, bioavailability, and stability of drug
molecules. Furthermore, cyclodextrin has been found to partially block the pore of
oligomeric staphylococcal alpha-hemolysin139 and small cationic β-cyclodextrin
derivatives can bind to and block the [PA63]7 pore via electrostatic interactions.89 To graft
the inhibitory peptides to the β-cyclodextrin core, we selected polyethylene glycol as a
linker. We chose an appropriate linker length using information on the location of the

32
peptide-binding sites on [PA63]7, obtained as described below. The well-defined
heptavalent inhibitor not only neutralizes anthrax lethal toxin in vitro, but also protect
animals from a toxin challenge.
Figure 3.1: Structure-based design of heptavalent anthrax toxin inhibitors.
(A) Structure of the LF-binding face of [PA63]7. Residues 184, 187, 197, and 200, which form part of the peptide-binding site are shown in purple. (B) Structure of the core, β-cyclodextrin. (C) Scheme illustrating the binding of a heptavalent inhibitor, synthesized by the attachment of seven inhibitory peptides to the β-cyclodextrin core via an appropriate polyethylene glycol linker, to [PA63]7
3.3 Materials and methods
3.3.1 Preparation of toxin components
Mutations in PA were generated using Quickchange mutagenesis according to the
manufacturer’s instructions (Stratagene). PA and LF were purified as described
previously.140
3.3.2 Cytotoxicity assay
RAW264.7 cells were seeded in 96-well plates and incubated overnight. The cells were
treated with 10-8 M PA and 10-9 M LF in the absence or presence of inhibitors. After an
incubation period of 4 h, cell viability was assessed using the MTS assay according to the
manufacturer’s instructions (Promega).

33
3.3.3 Serum stability
The heptavalent inhibitor was dissolved in PBS and incubated with mouse serum (Sigma,
St. Louis, MO, USA) (80% v/v) at 37 °C. Samples were withdrawn at different time intervals
and tested in a cytotoxicity assay as described above.
3.3.4 Rat intoxication
Animal experiments were carried out by our collaborator, Prof. Jeremy Mogridge at the
University of Toronto, and were performed under University of Toronto ethical
guidelines. A mixture of purified PA (40 µg) and LF (8 µg) mixed with either PBS,
heptavalent inhibitor (300 nmol on a per-peptide basis), or control thioglycerol-
functionalized heptavalent molecules, was used for the rat intoxication experiments.
Fisher 344 rats (Charles River Laboratories) were injected intravenously in the tail vein.
Seven rats were used per group, and the appearance of symptoms of intoxication was
monitored over 4 h. The rats were euthanized to avoid unnecessary distress when the
symptoms became pronounced.
3.4 Results and discussion
3.4.1 Structure-based design of heptavalent inhibitors
We developed a synthetic strategy for attaching the peptide to the β-cyclodextrin core
via a polyethylene glycol linker (Figure 3.2). Given the difficulties inherent in attaching
seven polymeric linkers to a single core with high yield, we developed an approach based
on “click chemistry” in which the copper-catalyzed 1,3-dipolar cycloaddition of an azide
to a terminal alkyne results in the production of a triazole with high yield.141–144 Briefly,
the secondary hydroxyl groups of the β-cyclodextrin core were first methylated and seven
terminal alkyne groups were introduced by the reaction of the primary hydroxyl groups
with propargyl bromide. Seven polyethylene glycol linkers were attached to the core by
the reaction of the alkynes with O-(2-Aminoethyl)-O′-(2-azidoethyl)decaethylene glycol
(H2N-CH2CH2-(OCH2CH2)11-N3, MW = 570.7 Da) (PEG11) – a commercially available,
monodisperse, azide-functionalized polyethylene glycol derivative. Chloroacetylation of
34
the free terminal amine, followed by reaction with the peptide HTSTYWWLDGAPC, a
cysteine-derivatized version of the inhibitory 12-mer peptide, yielded a heptavalent
inhibitor (6, Figure 3.2).
Figure 3.2: Synthesis scheme of heptavalent anthrax toxin inhibitor.
(a) TBDMSCl, pyridine, 0°C - rt. (b) NaH, MeI, THF (c) NH4F, MeOH, reflux (d) NaH, propargyl bromide, DMF, 0°C – room temperature. (e) CuSO4, sodium ascorbate, THF:H2O:BuOH (0.5:1:1), 80°C (f) Chloroacetic anhydride, triethylamine (g) Peptide, DMF, DBU, triethylamine.

35
The selection of the polyethylene glycol linker, PEG11, was based on the location of
the peptide-binding site. Mourez et al.86 used phage display to identify an inhibitory 12-
mer peptide, HTSTYWWLDGAP, that competes with LF for binding [PA63]7. A subsequent
screen identified a related peptide, HYTYWWLD, that also contains the TYWWLD
sequence, which we’ve shown is both necessary and sufficient for competing with LF.145
These experiments suggested that the peptides interact with [PA63]7 at or near the LF-
binding site. This information was used to guide the design of heptavalent inhibitors. The
distance between the peptide-binding residues (P184, L187, K197, and R200) and the
center of the lumen of [PA63]7 is ca. 30-40 Å. Recognition and inhibition of the toxin would
be facilitated by choosing a linker such that the root-mean-square distance from the
center of the cyclodextrin core to the end of the linker matched the distance from the
center of the lumen of [PA63]7 to the peptide-binding residues,83,85,146–148 i.e., by
statistically matching the heptavalent inhibitor with the heptavalent target.94 For our
inhibitor, the linker consisted of a rigid cyclodextrin core, a flexible region (consisting
primarily of ethylene glycol repeat units149), and the amino acids CPAG (since the
TYWWLD residues of the HTSTYWWLDGAPC are necessary and sufficient for binding145).
Using the method of Krishnamurthy et al.,148 we estimated that the root-mean-square
distance from the center of the cyclodextrin core to the end of the linker was ca. 30 Å for
inhibitors based on the PEG11 linker, which was consistent with the distance from the
center of the lumen to the peptide-binding residues on [PA63]7.
3.4.2 Characterization of heptavalent inhibitors
We tested the ability of this well-defined heptavalent inhibitor (6, Figure 3.2) to neutralize
anthrax lethal toxin in vitro by incubating RAW264.7 cells with a mixture of PA and LF in
the presence of several concentrations of the inhibitor. The heptavalent molecule could
inhibit cytotoxicity with a half-maximal inhibitory concentration (IC50) of ca. 10 nM on a
per-peptide basis (Figure 3.3A). Heptavalent molecules presenting only thioglycerol
showed no inhibitory activity (Figure 3.3A), and the monovalent peptide did not inhibit
cytotoxicity at concentrations as high as 2 mM. The heptavalent inhibitor therefore
provided a more than 100,000-fold enhancement in the activity of this peptide. To test
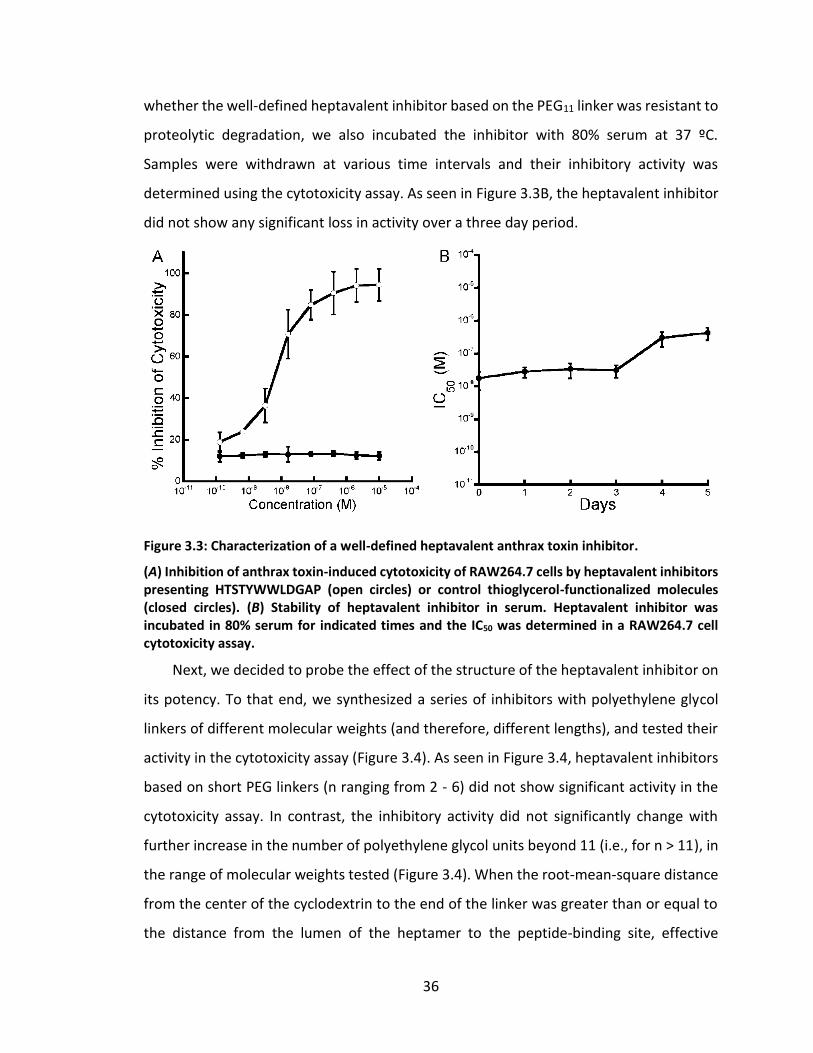
36
whether the well-defined heptavalent inhibitor based on the PEG11 linker was resistant to
proteolytic degradation, we also incubated the inhibitor with 80% serum at 37 ºC.
Samples were withdrawn at various time intervals and their inhibitory activity was
determined using the cytotoxicity assay. As seen in Figure 3.3B, the heptavalent inhibitor
did not show any significant loss in activity over a three day period.
Figure 3.3: Characterization of a well-defined heptavalent anthrax toxin inhibitor.
(A) Inhibition of anthrax toxin-induced cytotoxicity of RAW264.7 cells by heptavalent inhibitors presenting HTSTYWWLDGAP (open circles) or control thioglycerol-functionalized molecules (closed circles). (B) Stability of heptavalent inhibitor in serum. Heptavalent inhibitor was incubated in 80% serum for indicated times and the IC50 was determined in a RAW264.7 cell cytotoxicity assay.
Next, we decided to probe the effect of the structure of the heptavalent inhibitor on
its potency. To that end, we synthesized a series of inhibitors with polyethylene glycol
linkers of different molecular weights (and therefore, different lengths), and tested their
activity in the cytotoxicity assay (Figure 3.4). As seen in Figure 3.4, heptavalent inhibitors
based on short PEG linkers (n ranging from 2 - 6) did not show significant activity in the
cytotoxicity assay. In contrast, the inhibitory activity did not significantly change with
further increase in the number of polyethylene glycol units beyond 11 (i.e., for n > 11), in
the range of molecular weights tested (Figure 3.4). When the root-mean-square distance
from the center of the cyclodextrin to the end of the linker was greater than or equal to
the distance from the lumen of the heptamer to the peptide-binding site, effective
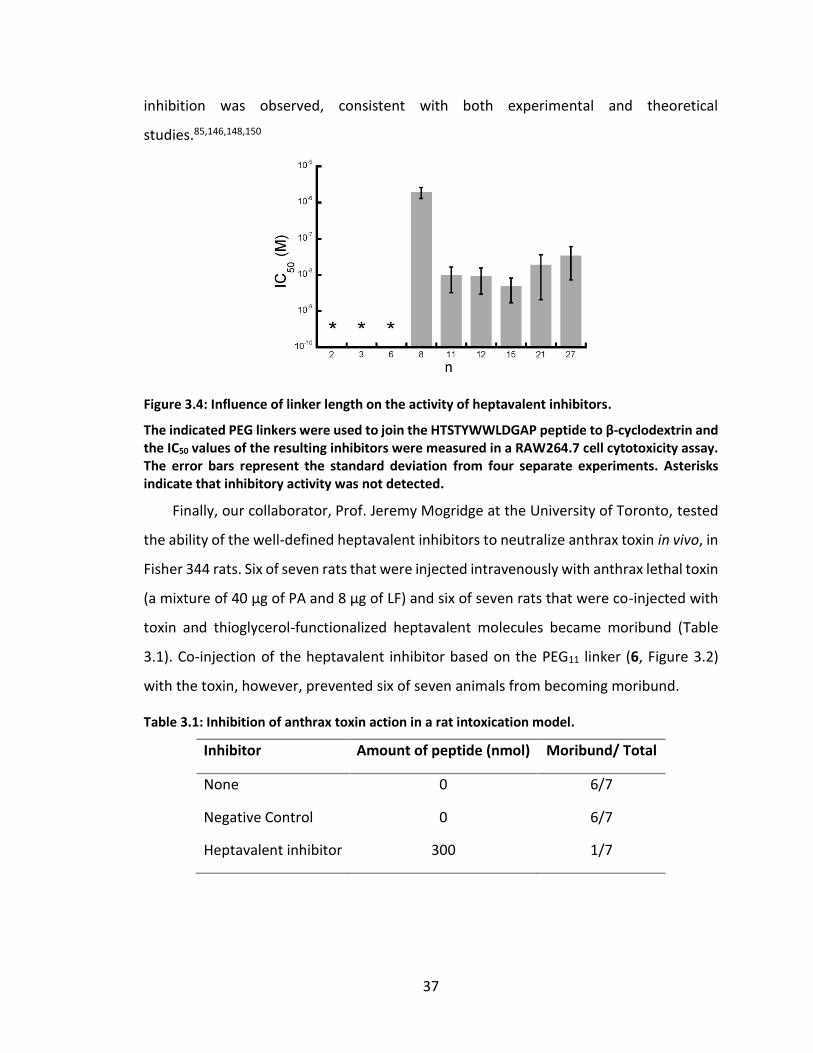
37
inhibition was observed, consistent with both experimental and theoretical
studies.85,146,148,150
Figure 3.4: Influence of linker length on the activity of heptavalent inhibitors.
The indicated PEG linkers were used to join the HTSTYWWLDGAP peptide to β-cyclodextrin and the IC50 values of the resulting inhibitors were measured in a RAW264.7 cell cytotoxicity assay. The error bars represent the standard deviation from four separate experiments. Asterisks indicate that inhibitory activity was not detected.
Finally, our collaborator, Prof. Jeremy Mogridge at the University of Toronto, tested
the ability of the well-defined heptavalent inhibitors to neutralize anthrax toxin in vivo, in
Fisher 344 rats. Six of seven rats that were injected intravenously with anthrax lethal toxin
(a mixture of 40 µg of PA and 8 µg of LF) and six of seven rats that were co-injected with
toxin and thioglycerol-functionalized heptavalent molecules became moribund (Table
3.1). Co-injection of the heptavalent inhibitor based on the PEG11 linker (6, Figure 3.2)
with the toxin, however, prevented six of seven animals from becoming moribund.
Table 3.1: Inhibition of anthrax toxin action in a rat intoxication model.
Inhibitor Amount of peptide (nmol) Moribund/ Total
None 0 6/7
Negative Control 0 6/7
Heptavalent inhibitor 300 1/7

38
3.5 Conclusions
In conclusion, we have demonstrated the structure-based design of potent biospecific
oligovalent inhibitors of anthrax lethal toxin. We used experimental studies to identify
the binding site for an inhibitory peptide on the heptameric subunit of anthrax toxin. We
developed a click-chemistry-based approach for the efficient attachment of a suitable
polymeric linker to a cyclodextrin core. Subsequent functionalization with the peptide
yielded a well-defined heptavalent inhibitor that neutralized anthrax lethal toxin both in
vitro and in vivo and showed appreciable stability in serum. Given the inherent
biocompatibility of cyclodextrin and polyethylene glycol, these potent well-defined
heptavalent anti-toxins might serve as valuable adjuncts to antibiotics for the treatment
of anthrax. The approach outlined in this work might also be broadly applicable to
designing well-defined oligovalent molecules that inhibit pathogens or other microbial
toxins in vivo.

39
4. Multivalent inhibitors of influenza virus
4.1 Rationale for the design of multivalent inhibitors of influenza virus entry
4.1.1 Targeting a conserved epitope on the hemagglutinin spike
The current standard influenza vaccines, which include both live attenuated influenza
vaccine (LAIV) and inactivated influenza vaccine (IIV) formulations, were only 62%
effective at preventing acute respiratory infections of influenza in 2013.151 Analysis has
shown that LAIVs are consistently more effective than IIVs, particularly in children,152 but
the rate of protection is still relatively low when compared to that of standard vaccines
against other diseases, such as measles (confers 95-99% immunity after one dose).153
Nonetheless, the Center for Disease Control reports that vaccination has been shown to
reduce the severity and repercussions of influenza-like illness, even when prevention
rates are only moderate.154
One of the primary causes of the inability of standard influenza vaccines to prevent
a greater proportion of confirmed influenza infections is the antigenic drift of the virus,155
as discussed in Section 1.2.3 Antigenic variation on page 5. Seasonal influenza vaccines
take time to prepare, and so the selection of influenza strains which are included in
vaccine formulations are not always accurate predictions of what end up being the
dominant circulating strains.156 By the time flu season actually hits, the circulating
influenza viruses may have undergone enough antigenic drift to escape the protective
effects of the neutralizing antibodies that were generated by the immune system in
response to the administered vaccinations.155 This disconnect has been a driving factor in
recent years behind the interest in broadly neutralizing antibodies. These antibodies are
characterized by cross-reactivity to a broad spectrum of influenza subtypes.155–159 In the
last five years, crystal structures of some of these antibodies have been published, and
the structures have shown that the antibodies bind to regions of the viral glycoproteins
that are not as structurally plastic and therefore not prone to the same rates of antigenic
drift.155,157,158 In the case of HA, these so-called “conserved” epitopes are typically found
on the stem or directly at the receptor binding site, as opposed to other parts of the
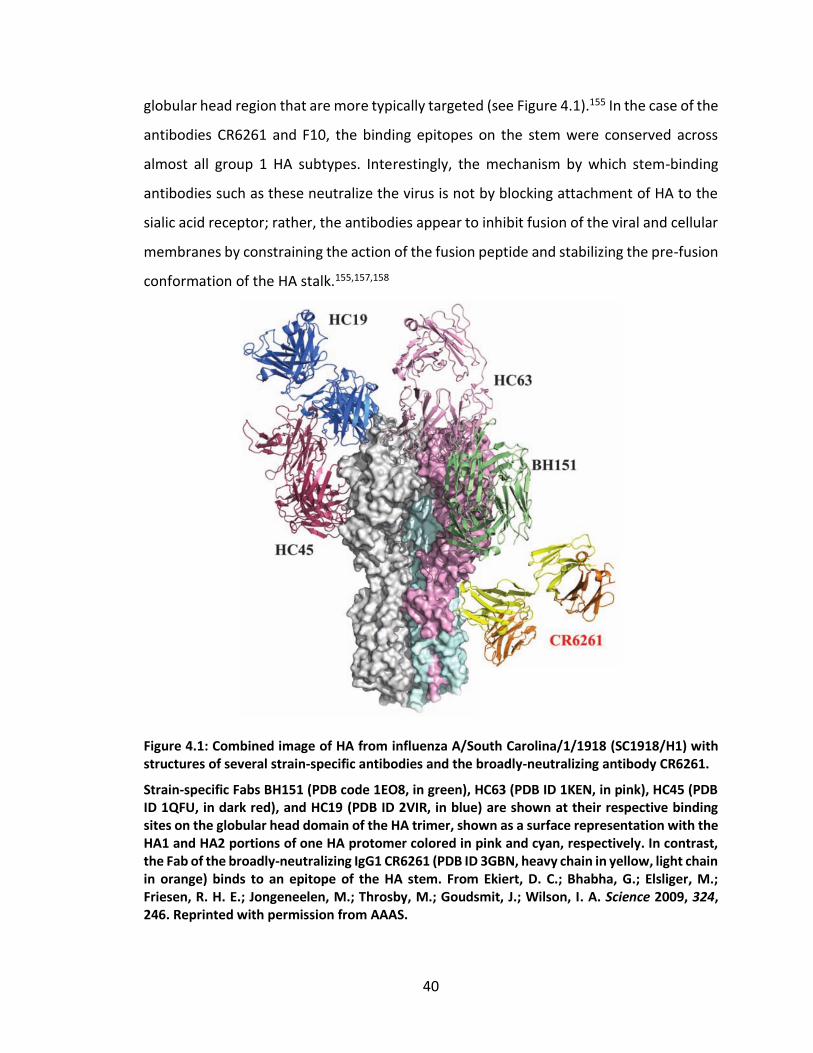
40
globular head region that are more typically targeted (see Figure 4.1).155 In the case of the
antibodies CR6261 and F10, the binding epitopes on the stem were conserved across
almost all group 1 HA subtypes. Interestingly, the mechanism by which stem-binding
antibodies such as these neutralize the virus is not by blocking attachment of HA to the
sialic acid receptor; rather, the antibodies appear to inhibit fusion of the viral and cellular
membranes by constraining the action of the fusion peptide and stabilizing the pre-fusion
conformation of the HA stalk.155,157,158
Figure 4.1: Combined image of HA from influenza A/South Carolina/1/1918 (SC1918/H1) with structures of several strain-specific antibodies and the broadly-neutralizing antibody CR6261.
Strain-specific Fabs BH151 (PDB code 1EO8, in green), HC63 (PDB ID 1KEN, in pink), HC45 (PDB ID 1QFU, in dark red), and HC19 (PDB ID 2VIR, in blue) are shown at their respective binding sites on the globular head domain of the HA trimer, shown as a surface representation with the HA1 and HA2 portions of one HA protomer colored in pink and cyan, respectively. In contrast, the Fab of the broadly-neutralizing IgG1 CR6261 (PDB ID 3GBN, heavy chain in yellow, light chain in orange) binds to an epitope of the HA stem. From Ekiert, D. C.; Bhabha, G.; Elsliger, M.; Friesen, R. H. E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I. A. Science 2009, 324, 246. Reprinted with permission from AAAS.

41
4.1.2 De novo design of protein ligands that target a conserved epitope on the hemagglutinin spike
While influenza vaccine research has been reenergized by the discovery of the conserved
epitopes of the HA stem region and the new quest to develop antigens that stimulate
antibody generation against these conserved targets, researchers in the Baker lab at the
University of Washington took a different approach and used their expertise in modeling
protein-protein interactions to computationally design new proteins to bind the
conserved HA stem epitope.160,161
By taking into consideration features such as the average solvent-exposed area on
protein surfaces that becomes buried during a typical protein-protein interaction, the
researchers began searching the protein data bank for proteins amenable to
manipulation (lacking disulfide bonds, expressed in E. coli, predicted to form monomers)
and which had structures with a high degree of shape complementarity to the
hydrophobic groove of the target epitope.161 In parallel, three key “hotspot” interactions
were identified by allowing disembodied amino acids to probe the target epitope,
optimizing potential electrostatic interactions, hydrogen binding, and Van der Waals
interactions.161 The fact that one of the three hotspots identified in this manner
resembled the interaction between the same amino acid (Tyr) observed in the crystal
structures of both the CR6261 and F10 antibodies was an early indication of the validity
of their technique.161 After the candidate protein scaffolds were identified, an iterative
process was developed for adding two or all three hotspot interactions to the scaffold. A
total of 88 new proteins based on 79 original scaffolds were designed in this way.161 Yeast
display was used to test the designed proteins for activity, which significantly narrowed
the pool down to just two candidates which reproducibly bound the HA stem. These
candidate proteins were HB36 and HB80, with HB standing for hemagglutinin-binder.
HB36 was one of the two-hotspot-binding designs, and was derived from a protein of
unknown function identified from Bacillus stearothermophilus.161 HB80 was a three-
hotspot-binding design, derived from a transcription factor from Antirrhinum majus.161
Neither of the original protein scaffolds were able to bind HA without the modifications.
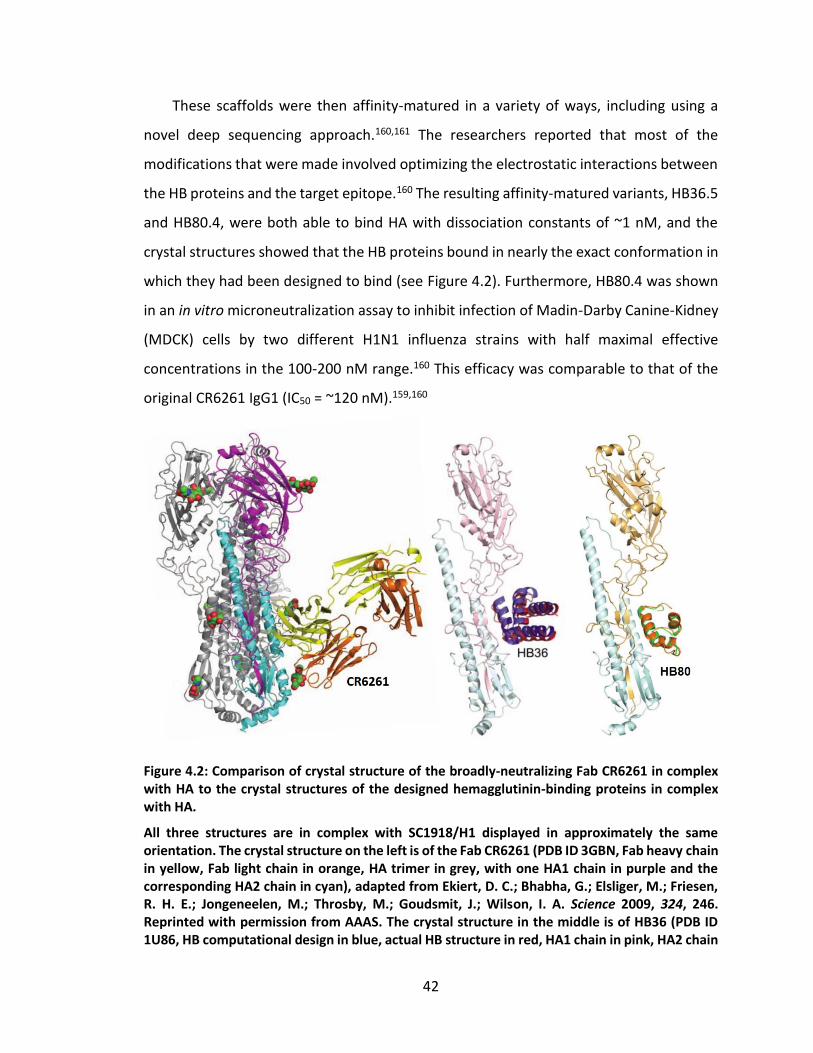
42
These scaffolds were then affinity-matured in a variety of ways, including using a
novel deep sequencing approach.160,161 The researchers reported that most of the
modifications that were made involved optimizing the electrostatic interactions between
the HB proteins and the target epitope.160 The resulting affinity-matured variants, HB36.5
and HB80.4, were both able to bind HA with dissociation constants of ~1 nM, and the
crystal structures showed that the HB proteins bound in nearly the exact conformation in
which they had been designed to bind (see Figure 4.2). Furthermore, HB80.4 was shown
in an in vitro microneutralization assay to inhibit infection of Madin-Darby Canine-Kidney
(MDCK) cells by two different H1N1 influenza strains with half maximal effective
concentrations in the 100-200 nM range.160 This efficacy was comparable to that of the
original CR6261 IgG1 (IC50 = ~120 nM).159,160
Figure 4.2: Comparison of crystal structure of the broadly-neutralizing Fab CR6261 in complex with HA to the crystal structures of the designed hemagglutinin-binding proteins in complex with HA.
All three structures are in complex with SC1918/H1 displayed in approximately the same orientation. The crystal structure on the left is of the Fab CR6261 (PDB ID 3GBN, Fab heavy chain in yellow, Fab light chain in orange, HA trimer in grey, with one HA1 chain in purple and the corresponding HA2 chain in cyan), adapted from Ekiert, D. C.; Bhabha, G.; Elsliger, M.; Friesen, R. H. E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I. A. Science 2009, 324, 246. Reprinted with permission from AAAS. The crystal structure in the middle is of HB36 (PDB ID 1U86, HB computational design in blue, actual HB structure in red, HA1 chain in pink, HA2 chain

43
in light blue), adapted from Fleishman, S. J.; Whitehead, T. A.; Ekiert, D. C.; Dreyfus, C.; Corn, J. E.; Strauch, E.-M.; Wilson, I. A.; Baker, D. Science. 2011, 332, 816. Reprinted with permission from AAAS. The crystal structure on the right is of HB80 (PDB ID 4EEF, HB computational design in green, actual HB structure in orange, HA1 in gold, HA2 in light blue). Adapted by permission from Macmillan Publishers Ltd: Nat. Biotechnol. 2012, 30, 543, copyright 2012.
4.1.3 Design of divalent inhibitors that target a conserved epitope on the hemagglutinin spike
Given our experience in designing multivalent conjugates which improve binding by
orders of magnitude, we were naturally interested in seeing if we could further enhance
the efficacy of the Baker lab’s computationally-designed monovalent HB proteins by
displaying them on appropriate multivalent scaffolds. The fact that HA is itself a trimer of
HA protomers and that these trimers are expressed polyvalently on the surface of
influenza virions made these HB proteins seem to us to be especially applicable for the
design of multivalent influenza inhibitors. While we could envision multiple different
strategies for creating multivalent conjugates that are effective at spanning multiple HB
protein binding sites, we decided to start with the most simple design first. That is, we
decided to create homodivalent conjugates by using bifunctional PEG linkers. Several
different chemistries can be used for creating homodivalent scaffolds. Commercially
available terminal functionalities include amine-reactive activated esters such as NHS,
TFP, and PFP, thiol-reactive maleimides, alkyne-reactive azides, and primary amines and
hydroxyls. Furthermore, the primary amines or terminal hydroxyl groups can be
chloroacetylated to introduce another form of thiol-reactivity, as described in Chapter 3
for the synthesis of β-cyclodextrin-based anthrax inhibitors.
In order to decide which PEG length might be appropriate for this application, I
consulted a thermodynamic analysis on the use of a flexible linker such as PEG in
divalently-binding constructs that was recently published by Kane.146 Although the
analysis specifically addresses the slightly more complex case of a heterodivalent binder,
the findings can easily be applied to homodivalent binders such as those we were
interested in designing. Consider a system consisting of a heterodivalent ligand composed
of two separate ligands, A and B, connected by a flexible linker, and interacting with a
receptor with a separate binding site specific to each ligand. The binding avidity of such a

44
heterodivalent ligand can be optimized by choosing a linker of such a length that will
maximize the probability that the two ligands are separated by an appropriate distance –
the distance, 𝑟, between the two ligand binding sites on the receptor. The derived
expression for the free energy of heterodivalent binding, ∆𝐺div0 , is given by the equation
∆𝐺div0 = ∆𝐺mono, A
0 + ∆𝐺mono, B0 − 𝑅𝑇 𝑙𝑛[𝐶eff(𝑟)], where the free energy of binding for
monovalent ligands A and B is represented by ∆𝐺mono, A0 and ∆𝐺mono, B
0 , respectively. The
term 𝐶eff(𝑟) represents an “effective concentration,” which is a function of 𝑟, and is
proportional to the probability that the two ends of the linker are a distance, 𝑟, apart.146
The behavior of a highly flexible linker in a good solvent approaches a random walk;
therefore the most probable distance separating the ends of the linker (and thus the two
ligands) is proportional to the root-mean-square (RMS) of the end-to-end distance of the
fully extended linker. Accordingly, the magnitude of 𝐶eff(𝑟) is maximized when the RMS
end-to-end distance of the linker matches the distance separating the two target binding
sites. This analysis applies for homodivalent linkers and the linear backbone between
ligands on a linear, polyvalent scaffold, and therefore the conclusions can be used to guide
the designed spacing between ligands for all types of one-dimensional multivalent
scaffolds.
To apply this analysis for designing multivalent displays of HB proteins, I first needed
to estimate the distance between binding sites. By using the PDB structure published by
the Baker group160 of HB80.4 in complex with SC1918/H1, I estimated that 6 nm was
approximately the distance that a linker would need to span in order to enable
simultaneous binding to two of the three epitopes. I calculated this distance as the arc
length between two of the binding sites, formed by a circle circumscribing the HA trimer
in the plane formed by the three binding epitopes (see Figure 4.3). Therefore, according
to the thermodynamic analysis discussed above, PEG linkers with RMS lengths in solution
that are in that range would maximize 𝐶eff and thus provide the greatest enhancement to
avidity. Discrete lengths of oligo(ethylene glycol) linkers (i.e. monodisperse PEG) are
commercially available (Quanta Biodesign) in a range of sizes from 4 to 29 ethylene glycol
repeats ((EG)n, n = 4-29), which correspond to extended lengths of 1.4 – 101.5 nm.

45
According to published correlations148,149 between the number of EG repeats (i.e. the
value of n) and the RMS length of PEG, I estimated the RMS lengths of these potential
linkers to be 1.2 – 3.1 nm. Longer homobifunctional PEGs are also available (Jenkem USA),
with MWs of 2.0, 3.5, 5.0, and 7.5 kDa, corresponding to estimated RMS lengths of
approximately 4, 5, 6, and 7.5 nm, respectively. It is noteworthy that the change in RMS
end-to-end distance of a flexible polymer like PEG is much more significant when
comparing linkers with smaller extended end-to-end distances. That is, as the distance
between target binding sites increases, the need to precisely match the RMS end-to-end
distance of the linker becomes less critical.
Figure 4.3: Estimated distance between HB protein binding sites on the HA trimer stalk.
The distance for an appropriately-sized flexible linker to span was estimated by calculating the arc length based on a circle circumscribing the HA trimer stalk in the plane of a cross-section of the trimer as shown, from the PDB ID 4EEF. Adapted by permission from Macmillan Publishers Ltd: Nat. Biotechnol. 2012, 30, 543, copyright 2012.
We decided to test three different PEG linkers differing in estimated RMS end-to-end
distance by ~2.5 nm for each size, as shown in Table 4.1. The smallest of the linkers has
an extended length that is only slightly larger than the predicted required minimum
distance, so we expected the 𝐶eff for this linker to be approximately an order of magnitude
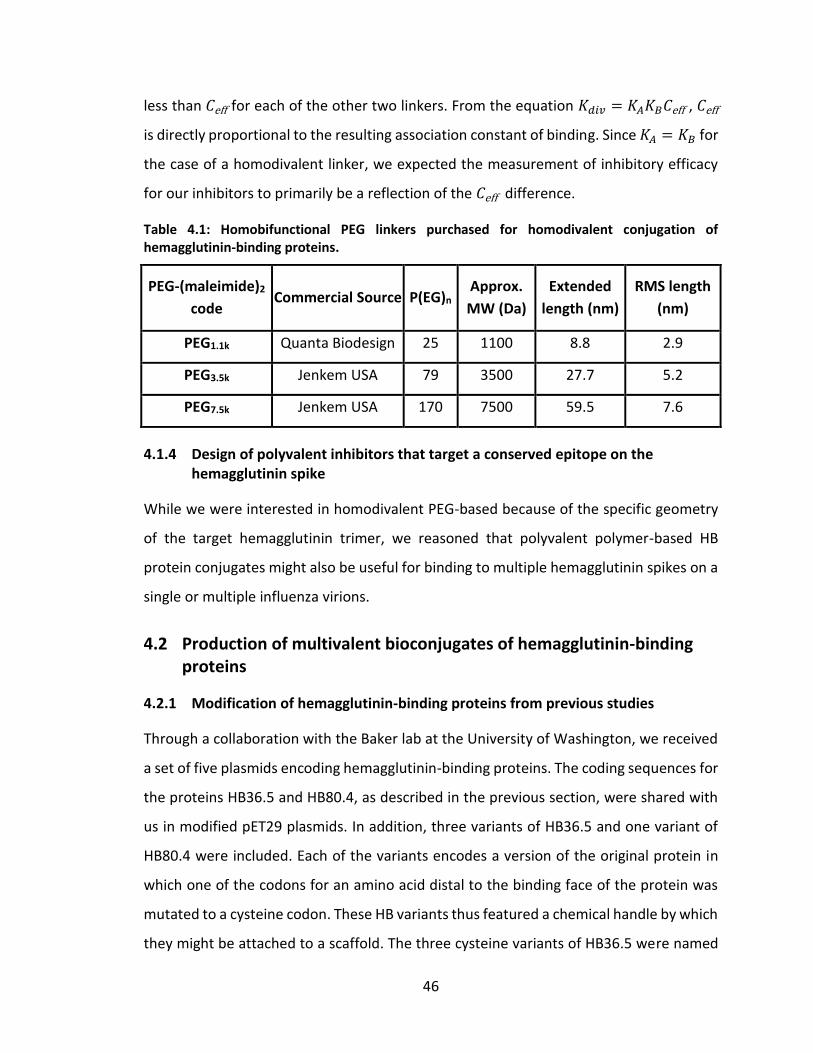
46
less than 𝐶eff for each of the other two linkers. From the equation 𝐾𝑑𝑖𝑣 = 𝐾𝐴𝐾𝐵𝐶eff , 𝐶eff
is directly proportional to the resulting association constant of binding. Since 𝐾𝐴 = 𝐾𝐵 for
the case of a homodivalent linker, we expected the measurement of inhibitory efficacy
for our inhibitors to primarily be a reflection of the 𝐶eff difference.
Table 4.1: Homobifunctional PEG linkers purchased for homodivalent conjugation of hemagglutinin-binding proteins.
PEG-(maleimide)2
code Commercial Source P(EG)n
Approx.
MW (Da)
Extended
length (nm)
RMS length
(nm)
PEG1.1k Quanta Biodesign 25 1100 8.8 2.9
PEG3.5k Jenkem USA 79 3500 27.7 5.2
PEG7.5k Jenkem USA 170 7500 59.5 7.6
4.1.4 Design of polyvalent inhibitors that target a conserved epitope on the hemagglutinin spike
While we were interested in homodivalent PEG-based because of the specific geometry
of the target hemagglutinin trimer, we reasoned that polyvalent polymer-based HB
protein conjugates might also be useful for binding to multiple hemagglutinin spikes on a
single or multiple influenza virions.
4.2 Production of multivalent bioconjugates of hemagglutinin-binding proteins
4.2.1 Modification of hemagglutinin-binding proteins from previous studies
Through a collaboration with the Baker lab at the University of Washington, we received
a set of five plasmids encoding hemagglutinin-binding proteins. The coding sequences for
the proteins HB36.5 and HB80.4, as described in the previous section, were shared with
us in modified pET29 plasmids. In addition, three variants of HB36.5 and one variant of
HB80.4 were included. Each of the variants encodes a version of the original protein in
which one of the codons for an amino acid distal to the binding face of the protein was
mutated to a cysteine codon. These HB variants thus featured a chemical handle by which
they might be attached to a scaffold. The three cysteine variants of HB36.5 were named

47
HB36.5_Q265C, HB36.5_A276C, and HB36.5_A341C, and the cysteine variant of HB80.4
was named HB80.4_K315C. The nomenclature for the mutations was based on the
residue number of the original protein scaffold that was the basis for the HB protein
design, and should not be confused with the residue number of the final HB proteins, as
the HB36.5 and HB80.4 constructs provided to us only coded for 115 and 72 amino acids,
respectively.
After some initial work using these constructs as provided, we decided that some
further modifications would be beneficial for our use. The coding sequence of the HB
protein constructs we received from the Baker lab featured the following general design:
start codon – FLAG tag – start codon – HB protein sequence (with or without cysteine) –
polyhistidine tag. The FLAG tag is an eight-residue polypeptide tag which was
trademarked by Sigma-Aldrich Co., LLC and the sequence (DYKDDDDK) was specifically
designed to be a hydrophilic, antigenic, cleavable fusion peptide for use in protein
purification and detection.162 The Baker lab reported adding the tag to their HB protein
designs because it enhanced expression during yeast display and improved protein
solubility. However, the researchers did not remove the original start codon, and I found
that the presence of this additional start codon consistently resulted in the co-production
of a contaminating amount of non-FLAG-tagged proteins, as shown in Figure 4.4. While
there is no indication that the inhibition efficacy of the non-FLAG-tagged proteins is
significantly different from the full-length FLAG-tagged proteins, these proteins could not
be removed by SEC, and interfered with characterization and purification of downstream
reaction products. Therefore, we decided to engineer the HB constructs by removing the
original start codon.
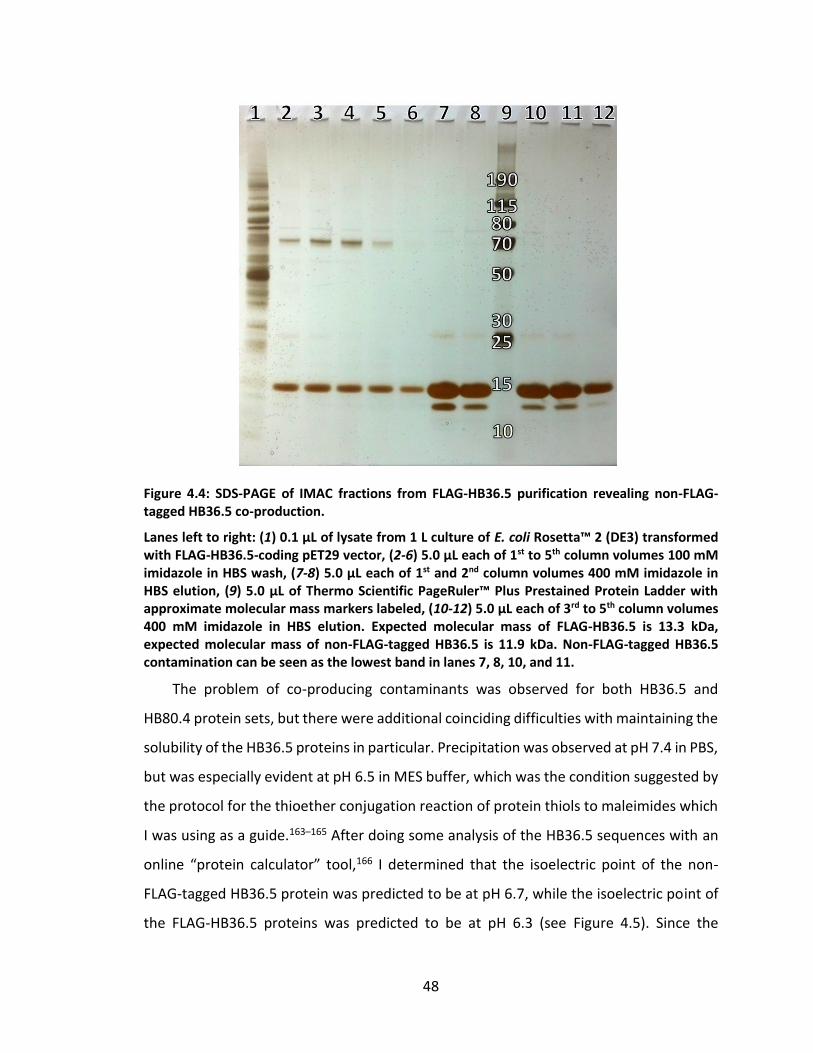
48
Figure 4.4: SDS-PAGE of IMAC fractions from FLAG-HB36.5 purification revealing non-FLAG-tagged HB36.5 co-production.
Lanes left to right: (1) 0.1 µL of lysate from 1 L culture of E. coli Rosetta™ 2 (DE3) transformed with FLAG-HB36.5-coding pET29 vector, (2-6) 5.0 µL each of 1st to 5th column volumes 100 mM imidazole in HBS wash, (7-8) 5.0 µL each of 1st and 2nd column volumes 400 mM imidazole in HBS elution, (9) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (10-12) 5.0 µL each of 3rd to 5th column volumes 400 mM imidazole in HBS elution. Expected molecular mass of FLAG-HB36.5 is 13.3 kDa, expected molecular mass of non-FLAG-tagged HB36.5 is 11.9 kDa. Non-FLAG-tagged HB36.5 contamination can be seen as the lowest band in lanes 7, 8, 10, and 11.
The problem of co-producing contaminants was observed for both HB36.5 and
HB80.4 protein sets, but there were additional coinciding difficulties with maintaining the
solubility of the HB36.5 proteins in particular. Precipitation was observed at pH 7.4 in PBS,
but was especially evident at pH 6.5 in MES buffer, which was the condition suggested by
the protocol for the thioether conjugation reaction of protein thiols to maleimides which
I was using as a guide.163–165 After doing some analysis of the HB36.5 sequences with an
online “protein calculator” tool,166 I determined that the isoelectric point of the non-
FLAG-tagged HB36.5 protein was predicted to be at pH 6.7, while the isoelectric point of
the FLAG-HB36.5 proteins was predicted to be at pH 6.3 (see Figure 4.5). Since the
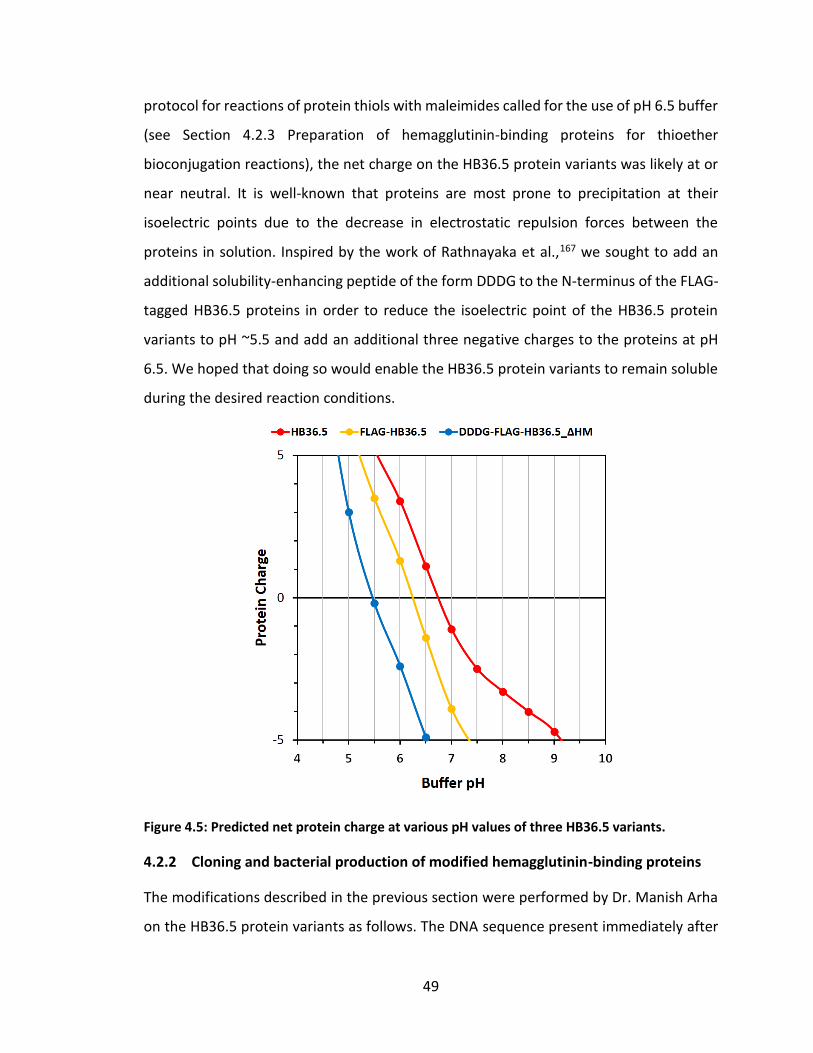
49
protocol for reactions of protein thiols with maleimides called for the use of pH 6.5 buffer
(see Section 4.2.3 Preparation of hemagglutinin-binding proteins for thioether
bioconjugation reactions), the net charge on the HB36.5 protein variants was likely at or
near neutral. It is well-known that proteins are most prone to precipitation at their
isoelectric points due to the decrease in electrostatic repulsion forces between the
proteins in solution. Inspired by the work of Rathnayaka et al.,167 we sought to add an
additional solubility-enhancing peptide of the form DDDG to the N-terminus of the FLAG-
tagged HB36.5 proteins in order to reduce the isoelectric point of the HB36.5 protein
variants to pH ~5.5 and add an additional three negative charges to the proteins at pH
6.5. We hoped that doing so would enable the HB36.5 protein variants to remain soluble
during the desired reaction conditions.
Figure 4.5: Predicted net protein charge at various pH values of three HB36.5 variants.
4.2.2 Cloning and bacterial production of modified hemagglutinin-binding proteins
The modifications described in the previous section were performed by Dr. Manish Arha
on the HB36.5 protein variants as follows. The DNA sequence present immediately after

50
the N-term FLAG tag, which coded for “His - Met” in the coding sequences of HB36.5,
HB36.5_A276C and HB36.5_Q265C, and “Ile - Met” in the coding sequence of
HB36.5_A341C, was removed by PCR amplifying the respective coding sequences from
their original plasmid. The PCR amplification also incorporated a “DDDG” tag at the 5′ end
of each of the coding sequences (after the start codon). Additionally, a new cysteine
variant of HB36.5 was created by adding a cysteine to the N-terminus of the solubility-
enhancement tag, yielding a “CDDDG” tag. For CDDDG-FLAG-HB36.5_ΔHM, the forward
primer for these modifications was (5′-CAT ATG TGT GAC GAC GAT GGA GAT TAC AAG
GAT GAC GAC GAT AAA GGA TCC TCT AAC GCA ATG GAC GGT CAA-3′) and the reverse
primer was (5′-CTC GAG TCA GTG GTG GTG GTG GTG GTG GGA TCC-3′). For DDDG-FLAG-
HB36.5_Q265C_ΔHM, DDDG-FLAG-HB36.5_A276C_ΔHM, and DDDG-FLAG-
HB36.5_A341C_ΔIM, the forward primer used was (5′-CAT ATG GAC GAC GAT GGA GAT
TAC AAG GAT GAC GAC GAT AAA GGA TCC TCT AAC GCA ATG GAC GGT CAA-3′) and the
reverse primer was (5′-CTC GAG TCA GTG GTG GTG GTG GTG GTG TTC GAG-3′). Similarly,
to remove the DNA sequence coding for the amino acids “His - Met”, which are present
immediately after the N-term FLAG tag of HB80.4 and HB80.4_K315C, the coding
sequences were PCR amplified from their respective plasmids using the forward primer
(5′-CAT ATG GAT TAC AAG GAT GAC GAC GAT AAA GGA TCC GCA TCC ACC AGA GGT TCC
GGT AGA-3′) and the reverse primer (5′-CTC GAG TCA GTG GTG GTG GTG GTG GTG TTC
GAG-3′). Each PCR-amplified sequence was then ligated into the NdeI and XhoI restriction
sites of the pET28b plasmid (modified). The pET28b vectors also contained a gene for
kanamycin resistance. I was then able to use the resulting vectors to transform the E. coli
Rosetta™2(DE3) strain (Novagen), which contains a chromosomal copy of T7 RNA
polymerase under the control of the lacUV5 promoter.
To produce the HB36.5 and HB80.4 proteins, the transformants were inoculated from
starter cultures at 1:100 dilution into multiple baffled Fernbach flasks each containing 1 L
of auto-induction media supplemented with 100 µg/mL kanamycin, as described by
Studier.168 After 6 hours of incubation at 37°C followed by 24 hours at 18°C and a constant
215 RPM, the cells were pelleted by centrifugation at 4°C for 30 minutes, and then

51
resuspended with a homogenizer into lysis buffer (pH 8.5, 100 mM HEPES, 100 mM NaCl,
25 mM imidazole, 1 mM TCEP, 1 mM PMSF, 2 mg/mL lysozyme, 0.2 mg/mL DNase) and
lysed by passing through a microfluidizer twice. The resulting lysate was clarified of
insoluble material by centrifugation at 4°C and 25,000 g for 30 minutes, and finally filtered
through a 0.45 µm filter. A disposable plastic column was packed with ~2.5 mL Ni-NTA
agarose resin for each liter of bacterial culture, and the clarified, filtered lysate was passed
through by gravity flow. The His-tagged HB protein variants exhibited strong affinity for
the immobilized metal resin, and were cleared of most contaminants by washes with a
step gradient of imidazole (25 mM, 50 mM, 100 mM for 5 column volumes each) in pH
8.5 HBS. The HB variants were then eluted from the columns with up to 10 column
volumes of 400 mM imidazole in pH 8.5 HBS. Reducing SDS-PAGE with silver staining was
performed on the IMAC fractions to confirm the successful expression and elimination of
the majority of contaminating native bacterial proteins, as shown in Figure 4.6. The results
of the gels were used to guide the choice of fractions to be pooled and concentrated by
3 kDa MWCO centrifugal filtration. These were subsequently purified by SEC on a
prepacked GE Tricorn 10/300 GL Superdex75 column in pH 7.5 MBS with TCEP and EDTA
for preventing the formation of disulfide bonds between proteins. The chromatogram of
UV absorbance at 280 nm featured two major peaks, even for non-cysteine-containing
variants of HB36.5, as shown in Figure 4.7. As reported by the Baker research group, the
HB36.5 variants exhibited a tendency to associate non-covalently as dimers during SEC,161
and this was confirmed by running the apparent dimer peak on non-reducing SDS-PAGE.
The fractions corresponding to the pure HB protein, as revealed by non-reducing SDS-
PAGE, were pooled and sterilized by vacuum filtration through 0.2 µm filters. Finally, the
protein solutions were concentrated by 3 kDa MWCO centrifugal filtration and stored at
4°C until further use in various assays and conjugation reactions.
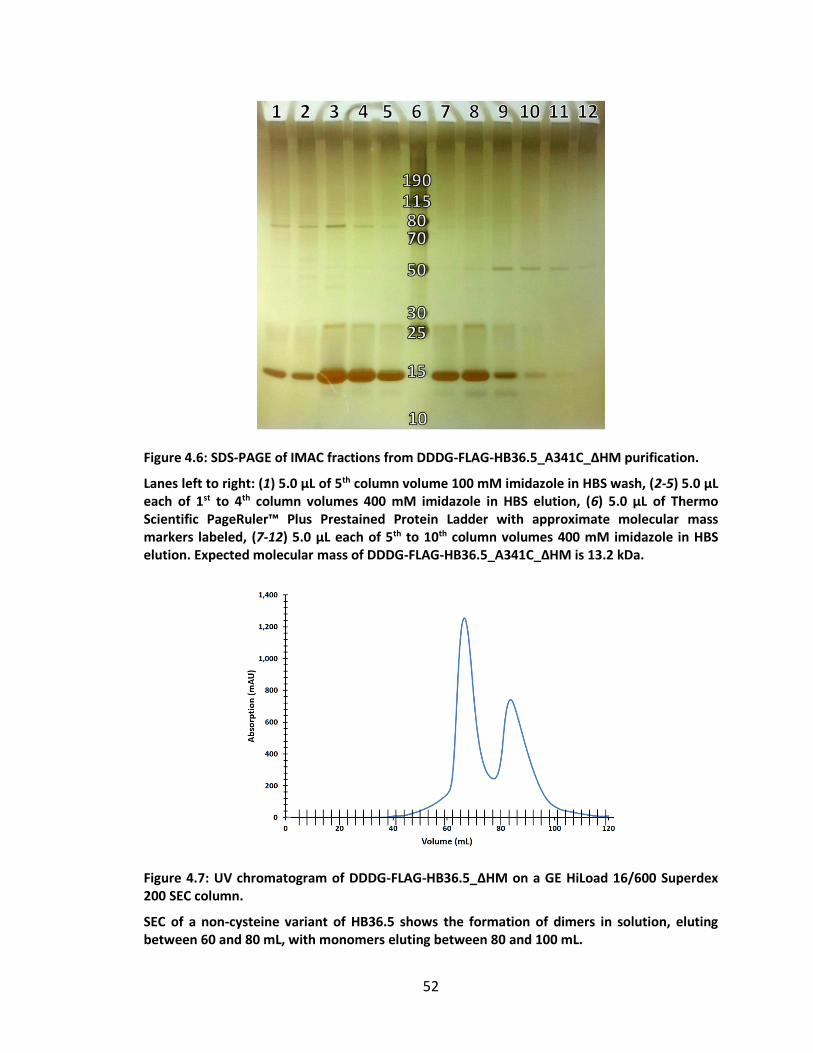
52
Figure 4.6: SDS-PAGE of IMAC fractions from DDDG-FLAG-HB36.5_A341C_ΔHM purification.
Lanes left to right: (1) 5.0 µL of 5th column volume 100 mM imidazole in HBS wash, (2-5) 5.0 µL each of 1st to 4th column volumes 400 mM imidazole in HBS elution, (6) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (7-12) 5.0 µL each of 5th to 10th column volumes 400 mM imidazole in HBS elution. Expected molecular mass of DDDG-FLAG-HB36.5_A341C_ΔHM is 13.2 kDa.
Figure 4.7: UV chromatogram of DDDG-FLAG-HB36.5_ΔHM on a GE HiLoad 16/600 Superdex 200 SEC column.
SEC of a non-cysteine variant of HB36.5 shows the formation of dimers in solution, eluting between 60 and 80 mL, with monomers eluting between 80 and 100 mL.

53
4.2.3 Preparation of hemagglutinin-binding proteins for thioether bioconjugation reactions
I have optimized a protocol for synthesizing multivalent conjugates of any biomolecule
containing a single reactive thiol functionality with a scaffold presenting maleimide
functionalities. The important considerations for this type of conjugation reaction are
keeping the thiols from forming disulfide side products and preventing the hydrolysis of
the reactive maleimide during the conjugation reaction. First, the biomolecules of interest
are prepared for the reaction by reduction of disulfide-bonded biomolecules in the stock
solution to free thiols. This can be accomplished by incubating the biomolecules with a
high concentration of a reducing agent for at least 30 minutes. The use of dithiothreitol
(DTT), β-mercaptoethanol (ME), and other thiol-containing reducing agents is to be
avoided because these small-molecule thiols will directly compete with the biomolecule-
thiol during the subsequent conjugation reaction. Tris(2-carboxyethyl)phosphine (TCEP)
and tris(hydroxypropyl)phosphine (THP) are trialkylphosphine reducing agents that do
not contain thiols and that catalyze the conversion of disulfides into thiols by nucleophilic
attack of the phosphine towards the disulfide.169–172 Trialkylphosphines are not stable in
phosphate buffer, so MBS should be used.169
Once all disulfide bonds have been reduced to free thiols, it is important to prevent
the oxidation reaction that converts the thiols back into disulfides. The degassing of
dissolved oxygen from the buffer is an easily-overlooked consideration, but it is an
important step for that purpose.171 The easiest method for degassing oxygen is simply to
bubble high purity nitrogen or argon gas through the buffer for at least 30 minutes
immediately prior to its use.171 The pH of the buffer should be slightly acidic (~pH 6.5) to
keep the thiols protonated, which also helps prevent the reformation of disulfides.171
Trace metal ions may be another catalytic source of thiol oxidation,171,173 therefore the
MBS should also contain 5-10 mM EDTA in order to chelate any such metal ions. However,
the presence of chelating agents has also been shown to catalyze the oxidation of
trialkylphosphine reducing agents,170,171 so the combination of TCEP or THP with EDTA
should be avoided.

54
Although TCEP is frequently reported as being compatible with conjugation reactions
of thiols,170 I have found in multiple situations that concentrations of TCEP greater than 1
mM actually inhibit reactions with thiols. Therefore I remove the excess TCEP prior to the
biomolecule conjugation reaction by passing the reduced biomolecule stock through a
desalting column which has been equilibrated with buffer that contains 10 mM EDTA.
I have found that biomolecules prepared in this manner will react to the fullest extent
possible in a matter of minutes when combined at sufficient concentrations with the
chosen maleimide-activated scaffold in degassed pH 6.5 MBS with EDTA. If the
reformation of disulfide-bonded side products appears to be a significant limitation on
the reaction success, as determined by non-reducing SDS-PAGE, I recommend spiking the
reaction with a small amount (50 – 500 µM) of trialkylphosphine reducing agent after ~1
hour.
4.2.4 Synthesis of divalent hemagglutinin-binding protein bioconjugates
The synthesis scheme for homodivalent PEG-HB protein conjugates is shown in Figure 4.8.
PEG linkers used for homodivalent inhibitor synthesis were named according to their
molecular masses: PEG1.1k, PEG3.5k, and PEG7.5k (Table 4.1). Purified HB36.5 variants
CDDDG-FLAG-HB36.5_ΔHM and DDDG-FLAG-HB36.5_A341C_ΔHM, which for simplicity
will be referred to as HBA and HBB, respectively, from this point forward, were prepared
as described above for reactions of biomolecule-thiols with scaffold-maleimides. The
reduced HB proteins were then added to the scaffold at a 10% molar excess relative to
the linker maleimides (i.e. 220 mol% relative to the linker), in order to promote
attachment at both ends of the PEG. The reaction buffer was degassed pH 6.5 MBS with
EDTA, and the concentration of HB proteins was maintained as high as possible, typically
in the range of 300-500 µM. The reaction was left at ambient temperature for a few hours
before checking the conjugation efficiency by non-reducing SDS-PAGE. If a significant
amount of disulfide-bonded dimer appeared in the PEG reaction, the reaction was spiked
with 500 µM TCEP (pH 6.5) and left for an additional few hours. Under these conditions,
the homodivalent products were produced in significant quantity as characterized by non-
reducing SDS-PAGE (Figure 4.11 lanes 1-3 and 16-18) and subsequent SEC purification.

55
Figure 4.8: Synthesis scheme for homodivalent PEG-protein bioconjugation.
4.2.5 Synthesis of polyvalent hemagglutinin-binding protein bioconjugates
4.2.5.1 Activation of biocompatible polymer scaffolds by reaction with aminoethyl-maleimide
The synthesis of polyvalent protein conjugates is more complicated than the PGA-peptide
conjugation methods described in Chapters 1 and 2 for multiple reasons. First, the use of
organic solvents is not conducive to the attachment of proteins due to the denaturing
effects they have on protein folding. While some proteins may exhibit accurate refolding
behavior when returned to an aqueous buffer, this is not a given, and the refolding
process could be further complicated by the proximity of the scaffold and other nearby
unfolded proteins post-conjugation. Furthermore, proteins feature many free amines and
carboxylates on their surfaces and at their termini, which complicates the strategy of
using activated ester chemistry. Successful polyvalent conjugation requires the
attachment of multiple large biomolecules (each on the order of tens to hundreds of
kiloDaltons) to a single scaffold, which can be inherently difficult from a steric viewpoint.
Prima facie, using the highest possible concentrations of each of the components is the
most logical way to address this issue, but that approach may be difficult in and of itself
due to issues with high viscosity or precipitation.
In order to activate the biocompatible polymeric PGA and HyA scaffolds, I combined
and modified previously-published protocols for the polyvalent attachment of amido-PEG
to PGA174 or sonic hedgehog protein to HyA via thio-ether coupling.163 The synthesis
scheme is shown in Figure 4.9. Using degassed pH 6.5 MBS with 10 mM EDTA as a buffer,
I prepared stocks of various MW PGA at concentrations of 13.2 mg/mL (100 mM
glutamate monomer) or 3 mg/mL HyA (8 mM disaccharide monomer). I then added 4-
(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) up to 150

56
mol% relative to glutamate monomer or up to 1000 mol% relative to disaccharide
monomer, followed by up to 33% mol% AEM relative to glutamate monomer or up to 500
mol% relative to disaccharide monomer. DMTMM is a water-soluble amide-bond
coupling reagent that creates activated esters in situ,175,176 similar to the more commonly-
used EDC-NHS coupling chemistry. While a more basic pH might enhance the rate of
nucleophilic attack of the AEM amine on the activated esters, maleimides are known to
be prone to high rates of hydrolysis at high pH, so the reaction was performed at pH
6.5.177–180 The activated polymer scaffolds were either dialyzed extensively against water
for subsequent lyophilization and characterization by NMR (Figure 4.10), or dialyzed
extensively against pH 6.5 MBS with 10 mM EDTA. In the latter case, the concentration of
AEM in the sample was measured by UV absorbance at 290 nm in order to estimate the
percent maleimide coupling to the scaffold.
Figure 4.9: Synthesis schemes for polyvalent polymer-maleimide-protein bioconjugation.
(a) DMTMM, AEM, H2O; (b) dialysis; (c) pH 6.5 MBS, EDTA, thiol-biomacromolecules; (d) 100 mM MoO4 1-2 h. The polyvalent conjugate is a random copolymer composed of unconjugated monomers (x repeat units) and protein bioconjugates (y repeat units) (omitted from HyA scheme for simplicity).

57
Figure 4.10: NMR spectra of three different PGA samples functionalized with increasing percentages of maleimide.
Peaks with integration highlighted in green correspond to the two aromatic protons of the maleimide. Analysis shows the red spectrum corresponds to 2.3% coupling, the black spectrum corresponds to 15.0% coupling, and the blue spectrum corresponds to 37.5% coupling.
4.2.5.2 Thioether bioconjugation with scaffolds presenting reactive maleimides
For proof of concept, I first synthesized polyvalent PGA-based anthrax toxin inhibitors by
using PGA activated with maleimide and the Ac-HTSTYWWLDGAPC-Am [PA63]7-binding
peptide used in the cyclodextrin-based anthrax toxin inhibitors. Prior to peptide
conjugation, the percentage of PGA monomers featuring reactive maleimide
functionalities on their side chains was 10%, as analyzed by 1H NMR in D2O. Peptide
conjugation was performed in 50% DMF to dissolve and maintain the solubility of the
peptide. The reaction was left overnight, and the following morning, both unconjugated
and peptide-conjugated maleimide rings were hydrolyzed by dialyzing in the presence of

58
100 mM molybdate oxyanion.180 Hydrolysis of the maleimide ring prevents further
reaction of both unconjugated and biomolecule-conjugated maleimides with small-
molecule thiols such as free cysteine or glutathione.178,179,181,182 Analysis of the 1H NMR
spectra of polyvalent peptide-based anthrax toxin inhibitors synthesized by this method
showed attachment in the expected range (5% or 10% of PGA monomers featured
peptide conjugation), and these conjugates were as effective at inhibiting cytotoxicity in
an in vitro lethal toxin assay as inhibitors synthesized using the previously-established
approach with the same peptide coupling percentages (IC50 ~50 nM).
The synthesis of polyvalent conjugates of HBA and HBB with PGA60k, PGA120k, and HyA
scaffolds was performed under similar conditions to the syntheses of homodivalent PEG-
HB protein conjugates in Section 4.2.4. A bioengineered protein polymer, named
[(SE)10K]10W3H10 to convey the details of its repetitive amino acid sequence, was used as
an additional scaffold as well. This bioengineered polypeptide is described in further
detail in Section 7.3.2 Bioengineering scaffolds. It features 11 reactive amines (10 lysines
and the N-terminus) which were first activated by reaction with a 500 mol% excess (with
respect to amine) amount of the bifunctional crosslinking agent SMCC (Thermo Scientific
Pierce). This crosslinker is a short (8.3 Å) molecule with an amine-reactive sulfo-NHS
activated ester at one end and a thiol-reactive maleimide at the other end. A silver-
stained non-reducing SDS-PAGE of the reaction mixtures after overnight incubation is
shown in Figure 4.11. The observed formation of discrete bands in the gel in the size range
of 30 – 200 kDa (by comparison to the protein standard ladder) is typical for all kinds of
polyvalent conjugation reactions involving the attachment of large molecules to PGA. For
further proof of this phenomenon, compare the multivalent conjugation of large,
monofunctional PEGs to PGA in Chapter 2 (Figure 2.9 on page 28), the similar polyvalent
conjugation of proteins to PGA in Chapter 5 (Figure 5.3 on page 73 and Figure 5.4 on page
75), and the polyvalent conjugation of ssDNA aptamers to PGA in Chapter 6 (Figure 6.1
on page 83 and Figure 6.2 on page 84). In my experience, such results are typically
observed by SDS-PAGE even when the samples are loaded on the gel only 1 hour after the
initial reaction setup. It should be noted that although lane 5 appears at first glance to

59
have been an unsuccessful polyvalent conjugation of HBA, in fact, the HyA-HBA conjugate
is too large to enter the gel (assuming the target of 13% percent of HyA disaccharide
monomers were conjugated with HBA, the resulting polyvalent conjugate MW would be
~9,100 kDa). Upon close inspection, there is a bit of staining at the very top of lane 5,
which might actually be due to some HyA-HBA conjugate being stuck to the bottom of the
well.
Figure 4.11: Silver-stained non-reducing SDS-PAGE of conjugation reactions of HBA (lanes 1-12) and HBB (lanes 13-25) before purification, 5.0 µg total HB protein per lane.
Lanes left to right: (1-3) Reactions of HBA with PEG1.1k, PEG3.5k, or PEG7.5k (expected homodivalent conjugate MWs = 28.2 kDa, 37.8 kDa, or 41.6 kDa), (4) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (5) Reaction targeting 13% coupling of HyA monomers with HBA, (6) Reaction targeting coupling 11 copies of HBA to a bioengineered scaffold named [(SE)10K]10W3H10, (7-9) Reactions targeting 2%, 5%, or 13% coupling of PGA60k monomers with HBA, (10-12) Reactions targeting 2%, 5%, or 13% coupling of PGA120k monomers with HBA, (13) HBB-AEM (HBB with the reactive thiol quenched by reaction with excess AEM to prevent disulfide bond formation – expected MW = 13.4 kDa), (14) HBB in the presence of 5 mM DHA (attempt to force disulfide bond formation – expected MW = 26.5 kDa), (15) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (17-19) Reactions of HBB with PEG1.1k, PEG3.5k, or PEG7.5k (expected homodivalent conjugate MWs = 27.6 kDa, 37.1 kDa, or 41.0 kDa), (19-21) Reactions targeting 2%, 5%, or 13% coupling of PGA60k monomers with HBB, (22-24) Reactions targeting 2%, 5%, or 13% coupling of PGA120k monomers with HBB, (25) Reaction targeting coupling of 11 copies of HBB to a bioengineered scaffold named [(SE)10K]10W3H10. Molecular mass of unconjugated HBA is 13.6 kDa and unconjugated HBB is 13.2 kDa.
4.2.6 Purification of multivalent hemagglutinin-binding protein bioconjugates
After the conjugation has been deemed complete, any remaining unreacted maleimide
functionality can be easily quenched by reaction with excess ME or thioglycerol. However,

60
due to the reported susceptibility of maleimide conjugates to exchange with small-
molecule thiols,178,179,182 I prefer to hydrolyze the maleimide rings with molybdate.
Therefore, after overnight incubation at ambient temperature, all HB-protein thioether
conjugation reaction products were treated with 100 mM molybdate and 20 mM TCEP in
MBS for 1-2 hours to hydrolyze the maleimide ring and reduce any disulfide-bonded HB
proteins that formed during the reaction. By hydrolyzing the maleimide, further
conjugation was quenched.
As can be seen by SDS-PAGE of the reaction product, there is often a significant
amount of unreacted biomolecule leftover in the sample (Figure 4.11, lanes 1-12 and 16-
25). In order to most accurately determine the effect of polyvalent attachment of the
biomolecule of interest, the remaining unconjugated biomolecule must be removed. If
the MW of the scaffold is sufficiently larger than the MW of the conjugated biomolecule
(e.g. for HyA conjugates – the HyA backbone MW is ~1,500 kDa), the easiest purification
strategy is definitely centrifugal filtration. To facilitate removal of the unconjugated
biomolecule, the largest possible MWCO pore size filter should be used that will not
permit loss of the conjugation product to the filtrate. Therefore, the HyA-based
polyvalent HB protein conjugates were purified by centrifugal filtration on 100 kDa
MWCO filters (Millipore) by using ~50-100 times the original reaction volume of pH 7.4
PBS for three times each.
SEC is another size-based method for separating the conjugation products from
unconjugated reagent biomolecule. Less volume of eluent is required to elute larger
molecules from the SEC column due to the fact that their large size “excludes” them from
accessing much of the column volume. Smaller molecules are able to diffuse into the
pores of the SEC resin and access a larger percentage of the column volume, and therefore
they require a larger volume of eluent to be eluted from the column. Prior to SEC
purification, the bioconjugation reactions were spiked with a high concentration of TCEP
to again ensure the elimination of disulfide-bonded dimers. PGA-based polyvalent
conjugates and PEG-based homodivalent conjugates were then purified by SEC on a 24
mL Superdex75 column with pH 7.4 PBS as the eluent. After the appropriate SEC fractions

61
were pooled, the samples were concentrated so that an accurate HB concentration could
be detected with a NanoDrop UV-vis spectrophotometer. Each sample was then stored
at 4°C until further testing.
4.3 Characterization of multivalent bioconjugates of hemagglutinin-binding proteins
4.3.1 In vitro microneutralization assay of influenza virus infection inhibition
In order to assess the ability of the modified HB proteins and conjugates to inhibit
influenza infection, an in vitro assay was adapted from established protocols.183,184 First,
serial dilutions of HB protein-containing samples were prepared over a range of
concentrations of several orders of magnitude; in a typical experiment, half-log dilutions
of the samples were prepared in duplicate in 50 µL PBS per well in 96-well microtiter
plates. These dilutions were then incubated at 37°C for 1 h with a fixed amount of virus,
typically 25 times the TCID50 (50% tissue culture infective dose) in 50 µL culture medium
(DMEM + 10% FBS), so that the final concentration of HB protein spanned the range from
10 µM down to 100 pM. The virus strain tested was H1N1 A/CA/4/2009 (Virapur, CA), a
pandemic strain isolated from a child patient in San Diego, CA during the H1N1 outbreak
of 2009. After the incubation with virus particles, approximately 15,000 MDCK cells in 100
µL culture medium were added to each well and further incubated for another 18-20
hours (overnight) at 37°C under an atmosphere of 5% CO2. During that time the cells
adhered to the plate bottom, and if infected, began the expression of viral proteins. After
the overnight incubation, the cells were fixed with a mixture of 4:1 acetone:PBS for 10-
15 minutes, the fixing solution was removed, and the plates were left to dry. Finally, an
ELISA was performed to detect the relative concentration of influenza nucleoprotein in
each well. An anti-nucleoprotein murine antibody (MAB8257, Millipore) and an anti-
mouse goat antibody-HRP conjugate (115-035-146, Jackson Immunoresearch, PA) were
used in the ELISA, and the absorbance of each well relative to the negative control
(uninfected cells) and positive control (cells + virus without inhibitor) was used to
calculate percent viral inhibition.
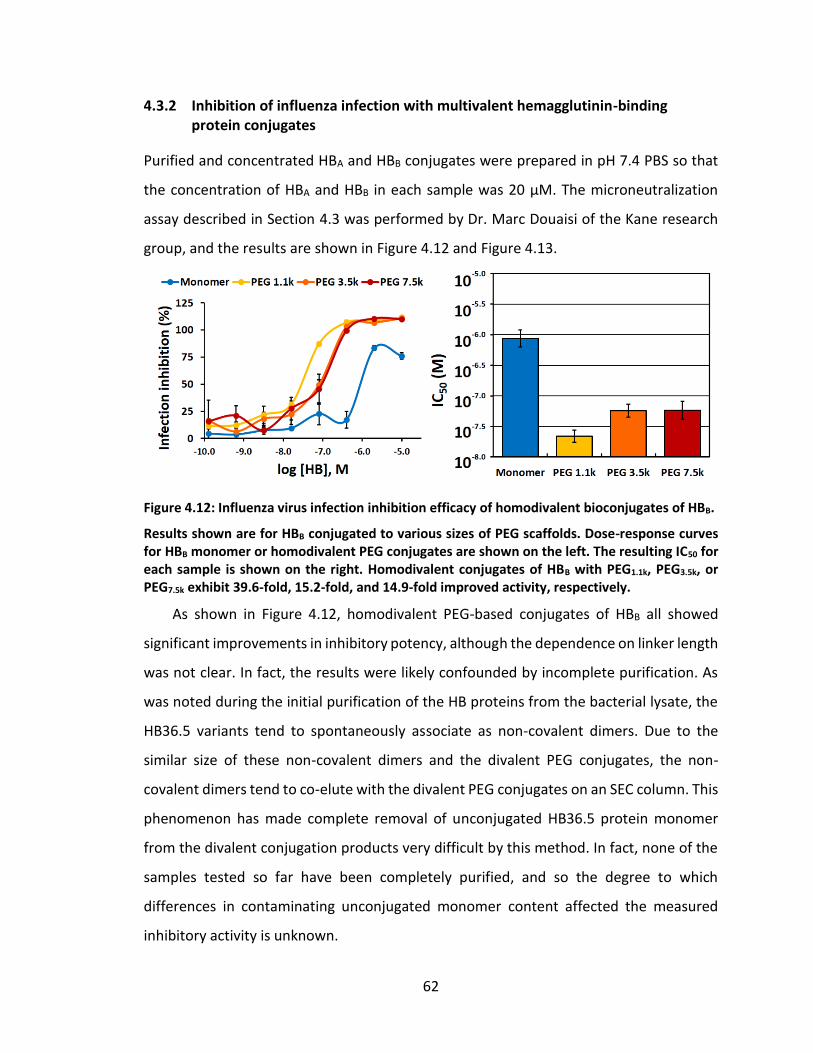
62
4.3.2 Inhibition of influenza infection with multivalent hemagglutinin-binding protein conjugates
Purified and concentrated HBA and HBB conjugates were prepared in pH 7.4 PBS so that
the concentration of HBA and HBB in each sample was 20 µM. The microneutralization
assay described in Section 4.3 was performed by Dr. Marc Douaisi of the Kane research
group, and the results are shown in Figure 4.12 and Figure 4.13.
Figure 4.12: Influenza virus infection inhibition efficacy of homodivalent bioconjugates of HBB.
Results shown are for HBB conjugated to various sizes of PEG scaffolds. Dose-response curves for HBB monomer or homodivalent PEG conjugates are shown on the left. The resulting IC50 for each sample is shown on the right. Homodivalent conjugates of HBB with PEG1.1k, PEG3.5k, or PEG7.5k exhibit 39.6-fold, 15.2-fold, and 14.9-fold improved activity, respectively.
As shown in Figure 4.12, homodivalent PEG-based conjugates of HBB all showed
significant improvements in inhibitory potency, although the dependence on linker length
was not clear. In fact, the results were likely confounded by incomplete purification. As
was noted during the initial purification of the HB proteins from the bacterial lysate, the
HB36.5 variants tend to spontaneously associate as non-covalent dimers. Due to the
similar size of these non-covalent dimers and the divalent PEG conjugates, the non-
covalent dimers tend to co-elute with the divalent PEG conjugates on an SEC column. This
phenomenon has made complete removal of unconjugated HB36.5 protein monomer
from the divalent conjugation products very difficult by this method. In fact, none of the
samples tested so far have been completely purified, and so the degree to which
differences in contaminating unconjugated monomer content affected the measured
inhibitory activity is unknown.
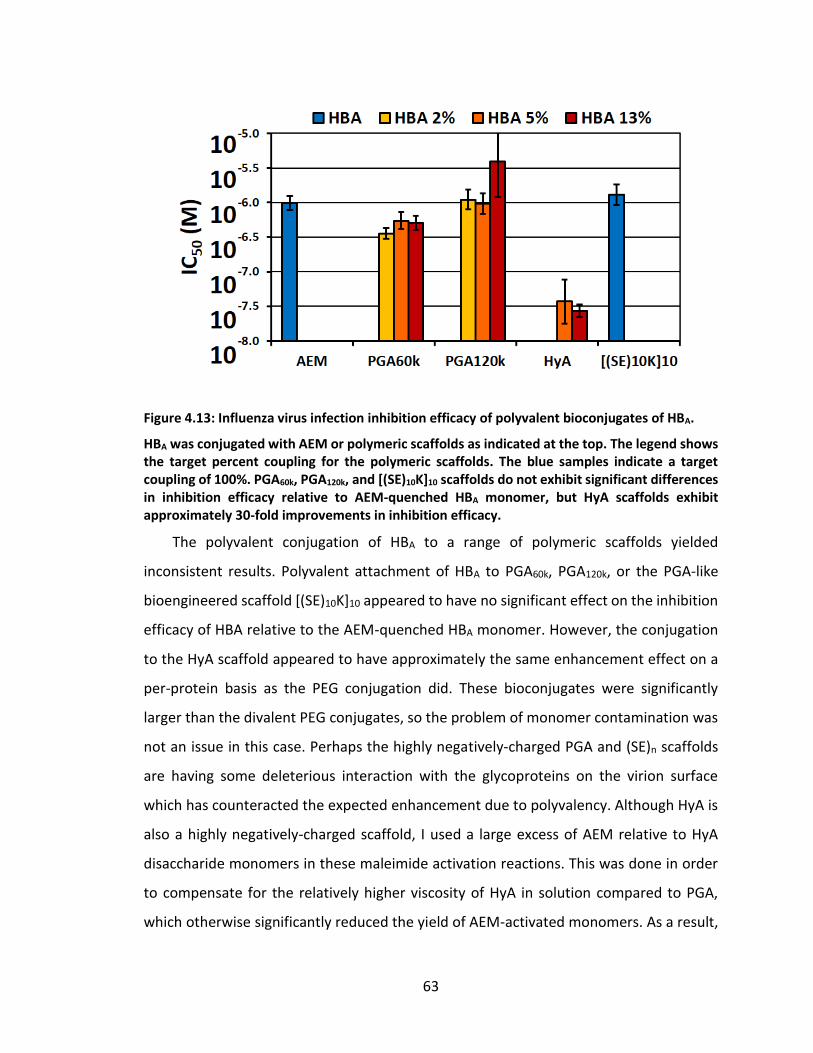
63
Figure 4.13: Influenza virus infection inhibition efficacy of polyvalent bioconjugates of HBA.
HBA was conjugated with AEM or polymeric scaffolds as indicated at the top. The legend shows the target percent coupling for the polymeric scaffolds. The blue samples indicate a target coupling of 100%. PGA60k, PGA120k, and [(SE)10K]10 scaffolds do not exhibit significant differences in inhibition efficacy relative to AEM-quenched HBA monomer, but HyA scaffolds exhibit approximately 30-fold improvements in inhibition efficacy.
The polyvalent conjugation of HBA to a range of polymeric scaffolds yielded
inconsistent results. Polyvalent attachment of HBA to PGA60k, PGA120k, or the PGA-like
bioengineered scaffold [(SE)10K]10 appeared to have no significant effect on the inhibition
efficacy of HBA relative to the AEM-quenched HBA monomer. However, the conjugation
to the HyA scaffold appeared to have approximately the same enhancement effect on a
per-protein basis as the PEG conjugation did. These bioconjugates were significantly
larger than the divalent PEG conjugates, so the problem of monomer contamination was
not an issue in this case. Perhaps the highly negatively-charged PGA and (SE)n scaffolds
are having some deleterious interaction with the glycoproteins on the virion surface
which has counteracted the expected enhancement due to polyvalency. Although HyA is
also a highly negatively-charged scaffold, I used a large excess of AEM relative to HyA
disaccharide monomers in these maleimide activation reactions. This was done in order
to compensate for the relatively higher viscosity of HyA in solution compared to PGA,
which otherwise significantly reduced the yield of AEM-activated monomers. As a result,

64
it is likely that nearly all of the carboxylates on the HyA scaffold were replaced by
maleimide functionalities, which would have neutralized the polyanionic character of the
scaffold.
4.4 Conclusions
As we hoped, the principles of multivalent inhibition were successfully adapted to a new
target, and over an order of magnitude enhancement in inhibitory efficacy was observed
with two different types of scaffolds, PEG and HyA. However, there were limitations that
hampered the ability to accurately assess the effects of the scaffold on the inhibition
efficacy. The activity of the divalent PEG-HB bioconjugates may have been strongly
affected by differences in amounts of contaminating unconjugated monomer, which co-
elutes when purified by SEC. To address this issue, other methods of purification are
currently being considered, such as hydrophobic interaction column chromatography. In
addition, production of the other HB protein inhibitor, HB80.4, is underway.
Unconjugated HB80.4 has already similar shown inhibitory activity to HB36.5 by our
microneutralization assay, and this protein is not known to form dimers in solution.
Therefore, divalent PEG bioconjugates of HB80.4 may be much easier to purify, and thus
to determine the effect of the size of the PEG linker on inhibition efficacy.
Continued work on the synthesis of polyvalent HyA-bioconjugates of HB-proteins is
also ongoing. To help to elucidate the effect of HB protein spacing, a commercially-
available, enzymatically-synthesized, monodisperse HyA scaffold will be used, and a series
of bioconjugates with a range of HB-protein coupling percentages will be synthesized. A
worthwhile extension of this project in future work may also be to bioengineer a single
protein polymer with several tandem repeats of the HB protein included in-line (similar
to the recent work with bioengineered anthrax toxin inhibitors,185 see Appendix B).
Furthermore, it would be exciting to see if the inhibitors could work in vivo, in an animal
model of influenza infection.

65
5. Multivalent HIV entry inhibitors
5.1 Rationale for the design of multivalent HIV entry inhibitors
5.1.1 Targeting the cellular receptor CCR5
HIV infections are particularly difficult to treat and prevent, due in large part to the
propensity of the virus for mutation. As mentioned in Section 1.3.3 on page 7, current
treatment and prevention strategies often involve the administration of small-molecule
antiretroviral drugs that target viral proteins such as reverse transcriptase, integrase, and
protease. Due to the selection pressure of these treatments and the high mutation rate
of retroviruses, HIV can develop resistance to these drugs over a short time of exposure.
On a related note, the explosion of the problem of antibiotic resistance has demonstrated
that the evolution of resistant microorganisms is virtually inevitable when therapeutic
strategies are used that directly target the proteins and other structural features of
pathogens with short life cycles. While the simultaneous administration of multiple
different antiretroviral drugs (a drug “cocktail”) can decrease the rate of evolution to viral
drug resistance, the strong selection pressure and propensity for mutation remains, so
the emergence of multi-drug-resistant viral strains is still possible.
The strategy of targeting host receptors may help overcome these problems. Host
cellular receptors are conserved targets, not prone to the same high mutation rate or
variability between different serotypes that may be the case for bacterial and viral
proteins. Moreover, targeting host receptors may be more conducive to preventative
medical strategies. These factors have led me to pursue the design of HIV entry inhibitors
that target the host cell receptors involved in the infection pathway, specifically CCR5 (See
Section 1.3.2 Infection pathway on page 6).
HIV infection requires that the viral envelope glycoprotein gp120 first binds to the
human glycoprotein CD4 and then to a co-receptor.52–54 The chemokine receptors CCR5
and CXCR4 are the primary co-receptors used by HIV; viral strains that use the CCR5 co-
receptor are referred to as R5 strains, whereas those that use the CXCR4 co-receptor are
referred to as X4 strains. Typically, CCR5-tropic viruses are the dominant strains during

66
the initial stages of infection, while CXCR4 (X4) tropism is a hallmark of viral evolution
during prolonged infection. A noteworthy corollary is that individuals who harbor a
genetic mutation in the gene for CCR5, known as CCR5-∆32, do not express CCR5 and are
highly resistant to HIV infection,186 but are otherwise healthy. Furthermore, the known
natural chemokine ligands of CCR5 all show the ability to inhibit HIV infection, and the
data suggest that the mechanism of inhibition may be primarily due to inducing receptor
internalization rather than by steric inhibition.187 Based on the health of individuals with
the CCR5-∆32 phenotype, the role of CCR5 in normal immune system operation appears
to be non-essential, and some studies actually suggest that the natural functions of CCR5
actually facilitates HIV infection by inducing immune system activation in ways that
attract additional HIV-permissive cells to the site of infection.188 As CCR5 is a host cell
receptor rather than a viral protein, CCR5 is a static target and is not prone to the
variations that occur naturally or that may evolve between different viral strains and
mutants. Furthermore, inhibitors that target CCR5 may also be effective when
administered prior to HIV exposure, and would therefore be a particularly useful means
for inhibiting viral transmission.
CCR5 is a G protein-coupled receptor that forms a 7-transmembrane-domain
structure, making it inherently difficult to analyze via X-ray crystallography or NMR
spectroscopy. Consequently, there has been a lack of high resolution structural
information that has prevented the rational design of CCR5-targeting HIV entry inhibitors.
However, there are a few known natural chemokine ligands of CCR5, and each has been
referred to in the literature by multiple different naming conventions. The most well-
known CCR5 ligand was called RANTES, an acronym standing for “regulated on activation,
normal T-cell expressed and secreted,” and is now officially named C-C chemokine ligand
5 (CCL5). Macrophage inflammatory protein 1α and 1β (MIP-1α and MIP-1β, respectively)
are also natural ligands, officially named CCL3 and CCL4, respectively. Although these
ligands of CCR5 are naturally able to inhibit HIV infection, their primary roles are to
activate various functions of the immune system, which may prohibit their direct use as
therapeutics.

67
5.1.2 Leukotoxin E is a protein ligand of CCR5
Many bacterial species of the genus Staphylococcus produce pore-forming toxins,
including γ-hemolysin and various enterotoxins and leukotoxins (also called leukocidins).
The leukotoxins help the bacteria evade the host immune system response during an
infection by causing osmotic imbalances that lead to cell death and lysis of leukocytes,
especially the neutrophils that engulf invading bacteria.189 Several types of leukotoxins
have been described in the literature, all comprised of two separately-secreted water-
soluble proteins of slightly different molecular mass, which self-assemble into octameric
toroids with alternating subunits.189–194 The naming convention for these bi-component
leukotoxins has been to refer to the two components as one unit, with each component
represented by a single letter so that the subunit with the lower molecular mass is
reported first. Leukotoxin AB is one example, where subunit A is the slower-eluting
component on a gel filtration column (“S-component”) and subunit B is the faster-eluting
component (“F-component”), therefore leukotoxin subunit A is of lower molecular mass
than subunit B.
In general, the fact that leukotoxins kill leukocytes has been known for decades, but
it has only been very recently that some light has been shed on which host factors
function as the toxin receptors. For example, Leukotoxin ED (LukED) was originally
described in 1998,195 but it wasn’t until 2013 that it was reported by the Torres research
group that host cell expression of CCR5, CXC chemokine receptor 1 (CXCR1, formerly
known as Interleukin 8 receptor alpha, or IL8RA), or CXC chemokine receptor 2 (CXCR2,
formerly known as Interleukin 8 receptor beta, or IL8RB) are required for LukED
cytotoxicity.196,197 While both toxin subunits were required for the cytotoxic effect, it was
shown by the use of green fluorescent protein (GFP) fusions that only subunit E (LukE)
mediates binding to CCR5, CXCR1, and CXCR2.196,197 In addition, the researchers were able
to use the wide array of known CCR5-binding antibodies to begin to probe which region
of CCR5 interacts with LukE. Of particular interest to my research was the fact that LukE
binding to CCR5 was inhibited by the antibody 45531, which has also been shown to
inhibit HIV infection in vitro.196,198 Furthermore, the researchers showed that LukE did not

68
activate CCR5-mediated cell signaling and even slightly suppressed signaling by CCL5.196
This result is consistent with the leukotoxin function enabling immune system evasion,
but it is also evidence that LukE might serve as a useful ligand for inhibition of HIV cell
entry.
5.1.3 Design of multivalent inhibitors that target CCR5
Although synthetic inhibitors of HIV entry via the CCR5 co-receptor have already been
identified, these have primarily been small molecule inhibitors that are prone to
complications associated with inhibiting protein-protein interactions. This is because the
large contact areas that are typical of protein-protein interactions may allow for partial
retention of the binding interface even when a competing small molecule drug or peptide
ligand is present. The retention of a partial binding interface may even enable the
acquisition of mutations that allow the virus to recognize the receptor-inhibitor complex.
Moreover, if the two interacting proteins are already bound, it may be unlikely that a
small ligand will gain access to the target site, even if the drug has a lower dissociation
constant. In the case of HIV, these types of concerns may be more problematic because
the virion is coated with a polyvalent display of the gp120 glycoprotein. In addition,
studies have shown that viral resistance to such small-molecule CCR5-targeting entry
inhibitors occurs through the acquisition of mutations that increase gp120’s affinity for
CCR5 and/or that bind to the CCR5-drug complex.199
Several factors suggest that multivalent inhibitors might be well suited to
counteracting the challenges of HIV antigenic drift and resistance to other methods of
entry inhibition. First, the inherently larger structure of multivalent binders should be
more effective than a monovalent ligand at creating steric hindrance to block access of
the HIV glycoprotein to CCR5. This larger size, along with the fact that CCR5 can diffuse
laterally on the cell surface, should allow for a multivalent inhibitor to bind to multiple
CCR5 molecules simultaneously and with high avidity. The ability to tightly bind multiple
CCR5 receptors may play an important role in thermodynamically outcompeting HIV for
access to the receptors, thus preventing the emergence of resistant strains and reducing
the consequences of viral mutations. Additionally, the ability to bind multiple CCR5

69
receptors and draw them together may induce endocytosis, effectively downregulating
the expression of CCR5 on the cell surface and producing a virtual CCR5-∆32 phenotype.
The Kane research group92 and others75,81,93,121,164 have demonstrated the potential
value of polyvalent ligands based on PGA81,93,121 or hyaluronic acid75,164 (HyA) scaffolds.
We reasoned that these biocompatible scaffolds should also support the synthesis of
polyvalent conjugates of LukE or GFP-LukE. Based on our previous research experience in
designing polyvalent inhibitors, we expected polyvalent conjugates of LukE or GFP-LukE
to be more efficacious on a per-ligand basis than the monovalent LukE or GFP-LukE at
binding CCR5. Furthermore, we hypothesized that these polyvalent conjugates would be
able to compete with other known CCR5-binders and potentially inhibit HIV attachment
and entry into CCR5+ cells. Therefore I have synthesized polyvalent bioconjugates using
PGA and HyA as the scaffolds for polyvalent display of LukE. The polyvalent conjugates
were synthesized by using the free thiol of LukE engineered with a C-terminal cysteine for
attachment to PGA or HyA scaffolds pre-activated by the polyvalent attachment of
aminoethylmaleimide (AEM). The resulting conjugates were characterized by SDS-PAGE
and SEC to determine the quality of the polyvalent attachment, and flow cytometry to
determine the ability to bind and compete for CCR5 on HEK-293T cells.
5.2 Production of polyvalent bioconjugates of leukotoxin E
5.2.1 Cloning and bacterial production of leukotoxin E variants and fusion proteins
The bacterial genome of the S. aureus strain Newman was purchased from ATCC, and the
gene encoding LukE was PCR amplified using the forward primer (5’-TCT AGA AAT AAT
TTT GTT TAA CTT TAA GAA GGA GAT ATA CAT ATG AAT ACT AAT ATT GAA AAT ATT GGT
GAT GGT G-3’) and either the reverse primer (5’-CTC GAG TTA GTG ATG ATG ATG ATG
ATG ATT ATG TCC TTT CAC TTT AAT TTC GT-3’) to add a C-terminal hexahistadine tag (His-
tag), or (5’-CTC GAG TTA ACA GTG ATG ATG ATG ATG ATG ATT ATG TCC TTT CAC TTT AAT
TTC GT-3') to add an additional cysteine to the end of the His-tag. The addition of the His-
tag facilitates purification by an immobilized metal affinity column (IMAC). It should be
noted that the wild type LukE sequence does not contain any cysteines, so the addition

70
of a cysteine at the C-terminus provides the LukE variant with a single thiol as a chemical
handle for conjugation reactions. The two variants were each cloned into pET28b vectors
between the XbaI and XhoI restriction sites. The pET28b vectors also contained a gene for
kanamycin resistance. The resulting constructs were then used to transform the E. coli
Rosetta™2(DE3) strain (Novagen), which contains a chromosomal copy of T7 RNA
polymerase under the control of the lacUV5 promoter.
In order to reproduce the GFP-LukE fusion proteins used by Alonzo III et al., a GFP
coding sequence from the plasmid pCIBN(deltaNLS)-pmGFP (Addgene Plasmid #26867)
was amplified by PCR. During amplification, the forward primers (5’-CAT ATG CAT CAT
CAT CAT CAT CAC GTG AGC AAG GGC GAG GAG C-3’) or (5’-CAT ATG TGT CAT CAT CAT
CAT CAT CAC GTG AGC AAG GGC GAG GAG C-3’) and the reverse primer (5’-CTC GAG TTG
TTG TTG TTG GGA TCC CTT ATA CAG CTC GTC CAT GCC GAG AGT GAT CC-3’) were used to
introduce a BamHI restriction site at the 3’ end and either a hexahistidine or a Cys-His6
tag at the N-terminus. These were ligated into the NdeI and XhoI restriction sites of a
modified pET28b plasmid, in which the N-terminal hexahistidine coding sequence and
NcoI restriction site had been previously removed. The LukE coding sequence was again
PCR amplified from the S. aureus genomic DNA, this time with the forward primer (5’-
GGA TCC AAT ACT AAT ATT GAA AAT ATT GGT GAT GGT G-3’) and the reverse primer (5’-
CTC GAG TTA ATT ATG TCC TTT CAC TTT AAT TTC GT-3’), and ligated downstream of GFP
into the BamHI and XhoI restriction sites of the modified pET28b plasmid, which also
conferred kanamycin resistance. The LukE and GFP-LukE cloning work described in this
section was performed by Dr. Manish Arha, and I used the resulting constructs to
transform E. coli Rosetta™2(DE3) for bacterial production of the proteins.
The transformants were inoculated from starter cultures at 1:100 dilution into
multiple baffled Fernbach flasks, each containing 1 L of auto-induction media supple-
mented with 100 µg/mL kanamycin, as described by Studier.168 After 6 hours of incubation
at 37°C followed by 24 hours at 18°C and a constant 215 RPM, the cells were pelleted by
centrifugation at 4°C for 30 minutes, then resuspended with a homogenizer into lysis
buffer (pH 8.5, 100 mM HEPES, 100 mM NaCl, 25 mM imidazole, 1 mM TCEP, 1 mM PMSF,

71
2 mg/mL lysozyme, 0.2 mg/mL DNase) and lysed by passing through a microfluidizer
twice. The resulting lysate was clarified of insoluble material by centrifugation at 4°C and
25,000 g for 30 minutes, and finally filtered through a 0.45 µm filter. A disposable plastic
column was packed with ~2.5 mL Ni-NTA agarose resin for each liter of bacterial culture,
and the clarified, filtered lysate was passed through by gravity flow. The His-tagged LukE
variants exhibited strong affinity for the immobilized metal resin, and were cleared of
most contaminants by washes with a step gradient of imidazole (25 mM, 50 mM, 100 mM
for 5 column volumes each) in HEPES buffered saline (HBS). LukE variants were eluted
from the columns with 5 column volumes of 400 mM imidazole in HBS. Reducing SDS-
PAGE with silver staining was performed to confirm the successful expression and
elimination of the majority of contaminating native bacterial proteins, as shown in Figure
5.1 for LukE-H6-C and Figure 5.2 for C-H6-GFP-LukE. The results of the gels were used to
guide the choice of fractions to be pooled and concentrated by 10 kDa molecular weight
cut-off (MWCO) centrifugal filtration and subsequent size-based purification on a
prepacked GE HiLoad 16/600 Superdex200 prep grade SEC column. The cysteine-
containing protein variants were purified in pH 7.5 MES buffered saline (MBS) with TCEP
and EDTA for preventing the formation of disulfide bonds between proteins, and the non-
reactive protein variants were purified in pH 7.4 PBS. The chromatogram of UV
absorbance at 280 nm produced a single peak in the expected elution volume for each
protein. The purity of the fractions corresponding to the LukE or GFP-LukE variant peak
was confirmed by non-reducing SDS-PAGE, and the corresponding fractions were pooled
and sterilized by vacuum filtration through 0.2 µm filters. Finally, the protein solutions
were concentrated by 10 kDa MWCO centrifugal filtration and stored at 4°C until further
use in various assays and conjugation reactions.
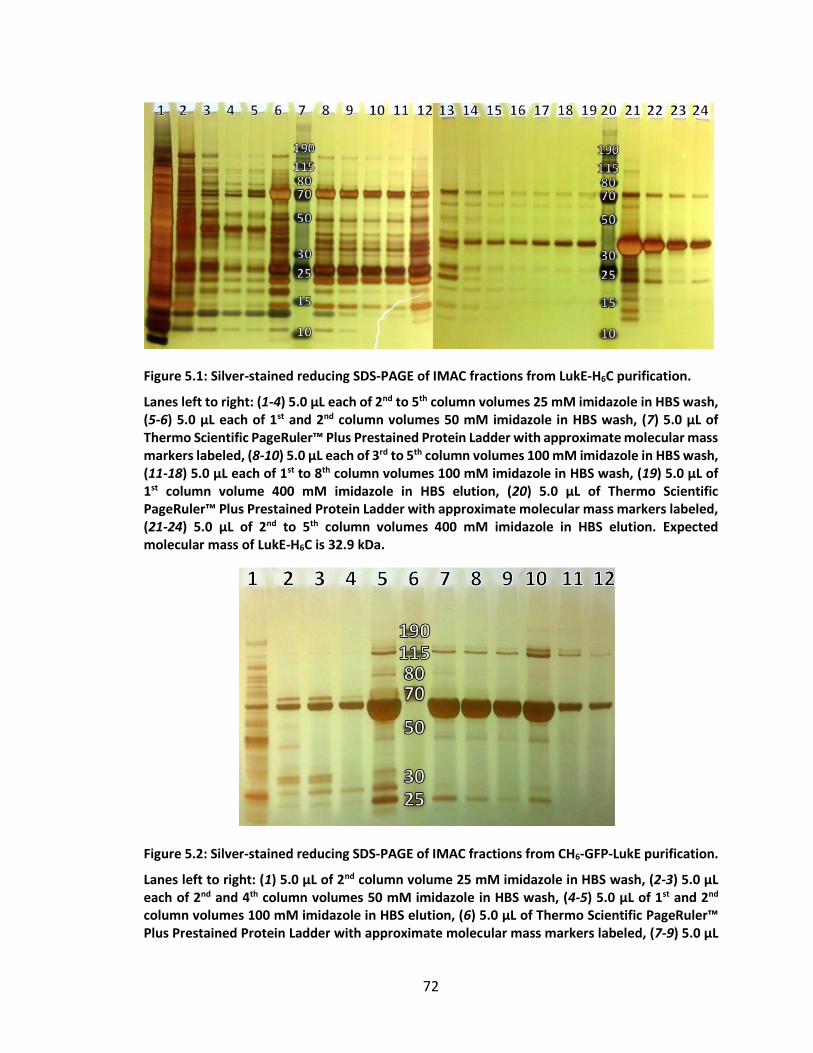
72
Figure 5.1: Silver-stained reducing SDS-PAGE of IMAC fractions from LukE-H6C purification.
Lanes left to right: (1-4) 5.0 µL each of 2nd to 5th column volumes 25 mM imidazole in HBS wash, (5-6) 5.0 µL each of 1st and 2nd column volumes 50 mM imidazole in HBS wash, (7) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (8-10) 5.0 µL each of 3rd to 5th column volumes 100 mM imidazole in HBS wash, (11-18) 5.0 µL each of 1st to 8th column volumes 100 mM imidazole in HBS wash, (19) 5.0 µL of 1st column volume 400 mM imidazole in HBS elution, (20) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (21-24) 5.0 µL of 2nd to 5th column volumes 400 mM imidazole in HBS elution. Expected molecular mass of LukE-H6C is 32.9 kDa.
Figure 5.2: Silver-stained reducing SDS-PAGE of IMAC fractions from CH6-GFP-LukE purification.
Lanes left to right: (1) 5.0 µL of 2nd column volume 25 mM imidazole in HBS wash, (2-3) 5.0 µL each of 2nd and 4th column volumes 50 mM imidazole in HBS wash, (4-5) 5.0 µL of 1st and 2nd column volumes 100 mM imidazole in HBS elution, (6) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (7-9) 5.0 µL

73
each of 3rd to 5th column volumes 100 mM imidazole in HBS elution, (10-12) 5.0 µL each of 2nd, 3rd, and 5th column volumes 400 mM imidazole in HBS elution. Expected molecular mass of CH6-GFP-LukE is 59.9 kDa.
5.2.2 Synthesis of polyvalent leukotoxin E bioconjugates
Polyvalent bioconjugates of LukE-H6C were synthesized by the same polyvalent
bioconjugation protocols described previously in Section 4.2.5 Synthesis of polyvalent
hemagglutinin-binding protein bioconjugates. The silver-stained non-reducing SDS-PAGE
of a typical conjugation set of LukE-H6C with two different MW PGA scaffolds is shown in
Figure 5.3. Each lane contains the same quantity of LukE (5.0 µg, or 152 pmol). Lanes 2
and 3 are useful guides to show the positions of LukE monomer and disulfide-bonded
dimer, respectively. The high molecular mass bands in lanes 4 through 8 are indicative of
the formation of large polyvalent conjugates. Before purifying out the unreacted LukE
from these bioconjugate reaction products, the maleimide rings were hydrolyzed by
treatment with 100-200 mM molybdate oxyanion for 1-2 hours.
Figure 5.3: Silver-stained non-reducing SDS-PAGE of LukE-H6C conjugation reaction products before purification, 5.0 µg total LukE protein per lane.

74
Lanes left to right: (1) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (2) LukE-H6C-AEM (LukE with the reactive thiol quenched by reaction with excess AEM to prevent disulfide bond formation – expected MW = 33.1 kDa), (3) LukE-H6C in the presence of 5 mM DHA (attempt to force disulfide bond formation – expected MW = 65.8 kDa), (4-5) Reactions targeting 5% or 13% coupling of PGA60k monomers with LukE-H6C, (6-7) Reactions targeting 5% or 13% coupling of PGA120k monomers with LukE-H6C. Molecular mass of unconjugated LukE-H6C is 32.9 kDa.
5.2.3 Purification of polyvalent leukotoxin E bioconjugates
Each of the PGA-LukE conjugation reaction samples from Figure 5.3 were treated with 20
mM TCEP in MBS with 100 mM molybdate for 1-2 hours and then run on a 24 mL
Superdex200 column with pH 7.4 PBS as the eluent. As is evident from the
chromatograms, there was a significant amount of unreacted LukE in each sample,
corresponding to a large peak relative to the low shallow hump from the polyvalent
conjugate. However, by pooling all elution fractions collected up until the unreacted
monomer peak and running SDS-PAGE (Figure 5.4), it was clear that the unreacted LukE
was successfully removed (compare the position of the AEM-quenched LukE monomer in
lane 10 to lanes 1-4 and 6-9). After confirming the successful removal of unconjugated
LukE from the desired conjugation products, I next concentrated the samples by
centrifugal filtration with a 3 kDa MWCO filter, measured the concentration of LukE in
each sample by recording the UV absorbance at 280 nm on a NanoDrop
spectrophotometer, and adjusted each sample to a set concentration in preparation for
the flow cytometry assay of CCR5-binding inhibition.

75
Figure 5.4: Silver-stained non-reducing SDS-PAGE of LukE-H6C conjugation reaction products after SEC purification on a Superdex200 column.
Lanes left to right: (1) Highest MW SEC fractions of LukE conjugated to PGA60k (target 5% coupling), (2) Second highest MW SEC fractions of LukE conjugated to PGA60k (target 5% coupling), (3) Highest MW SEC fractions of LukE conjugated to PGA60k (target 13% coupling), (4) Second highest MW SEC fractions of LukE conjugated to PGA60k (target 13% coupling), (5) 5.0 µL of Thermo Scientific PageRuler™ Plus Prestained Protein Ladder with approximate molecular mass markers labeled, (6) Highest MW SEC fractions of LukE conjugated to PGA120k (target 5% coupling), (7) Second highest MW SEC fractions of LukE conjugated to PGA120k (target 5% coupling), (8) Highest MW SEC fractions of LukE conjugated to PGA120k (target 13% coupling), (9) Second highest MW SEC fractions of LukE conjugated to PGA120k (target 13% coupling), (10) Monomer SEC fractions of LukE-H6C-AEM (LukE with the reactive thiol quenched by reaction with excess AEM, to prevent disulfide bond formation – expected MW = 33.1 kDa).
5.3 Characterization of polyvalent conjugates of leukotoxin E
5.3.1 Flow cytometry assay of CCR5-binding inhibition
The LukE variants were tested by flow cytometry for their ability to bind to CCR5-
expressing HEK-293T cells (Human embryonic kidney cells containing the SV40 large T
antigen, which enables the cell to replicate transfected plasmids that contain the SV40
origin of replication during cell division, thus allowing for the inherited expression of a

76
non-native gene such as CCR5). Approximately 100,000 cells were incubated with 300 nM
LukE or GFP-LukE at 4°C for 30 minutes, followed by the addition of one of a variety of
competing CCR5-specific binders or a control antibody (HLA-ABC, APC-conjugate, 562006,
Becton Dickinson, CA) that binds to MHC class I molecules (which are also expressed by
the cells). The phycoerythrin-conjugated monoclonal antibodies 2D7 (555993, Becton
Dickinson, CA) or 45531 (FAB182P, R&D Systems, MN), which also bind to the ECL2 of
CCR5, or 3A9 (560635, Becton Dickinson, CA), which binds to the N-terminus of CCR5,
were added at a concentration of 1 nM, 4.2 nM, or 8.3 nM, respectively. Alternatively,
FLSC-Ig, a fusion protein mimic of the HIV gp120 protein in complex with CD4 (i.e. HIV
gp120 in its CCR5-binding conformation) was added at a concentration of 23 nM. FLSC-Ig
stands for full length single chain immunoglobulin, and consists of the HIV gp120 from the
strain Ba-L, the D1 and D2 domains of the human glycoprotein CD4, and a human
immunoglobulin Fc fragment IgG1. Therefore, FLSC-Ig might represent the binding
capability of HIV for CCR5 more accurately. In the presence of each competing binder (or
control antibody), the LukE or GFP-LukE remained at an excess of 13-300 fold, and the
combined solution was incubated with the cells at 4°C for an additional 30 minutes. After
two washes with 250 µL of PBS + 1% FBS, FLSC-Ig was detected with 2 µg/mL
AlexaFluor647-conjugated anti-human IgG goat antibody (A21445, Invitrogen, CA) at 4°C
for another 20 minutes. Finally the cells were washed twice in 250 µL PBS + 1% FBS and
then resuspended and fixed in 1% paraformaldehyde (in PBS) prior to acquisition with a
LSRII flow cytometer (Becton Dickinson, CA).
5.3.2 Inhibition of CCR5-binding with polyvalent bioconjugates of leukotoxin E
Purified LukE and GFP-LukE conjugates with PGA60k, PGA120k, and HyA were prepared in
pH 7.4 PBS so that the concentration of LukE or GFP-LukE in each sample was 2 µM. AEM-
quenched LukE and GFP-LukE were also prepared at the same concentration. The assay
described in Section 5.3 was performed by Dr. Marc Douaisi of the Kane research group,
and the results are shown in Figure 5.5 and Figure 5.6. In both figures, the amount of
CCR5-binding antibody detected when in competition with the LukE sample is reported
as a percentage of the amount of antibody detected when no LukE was present. In
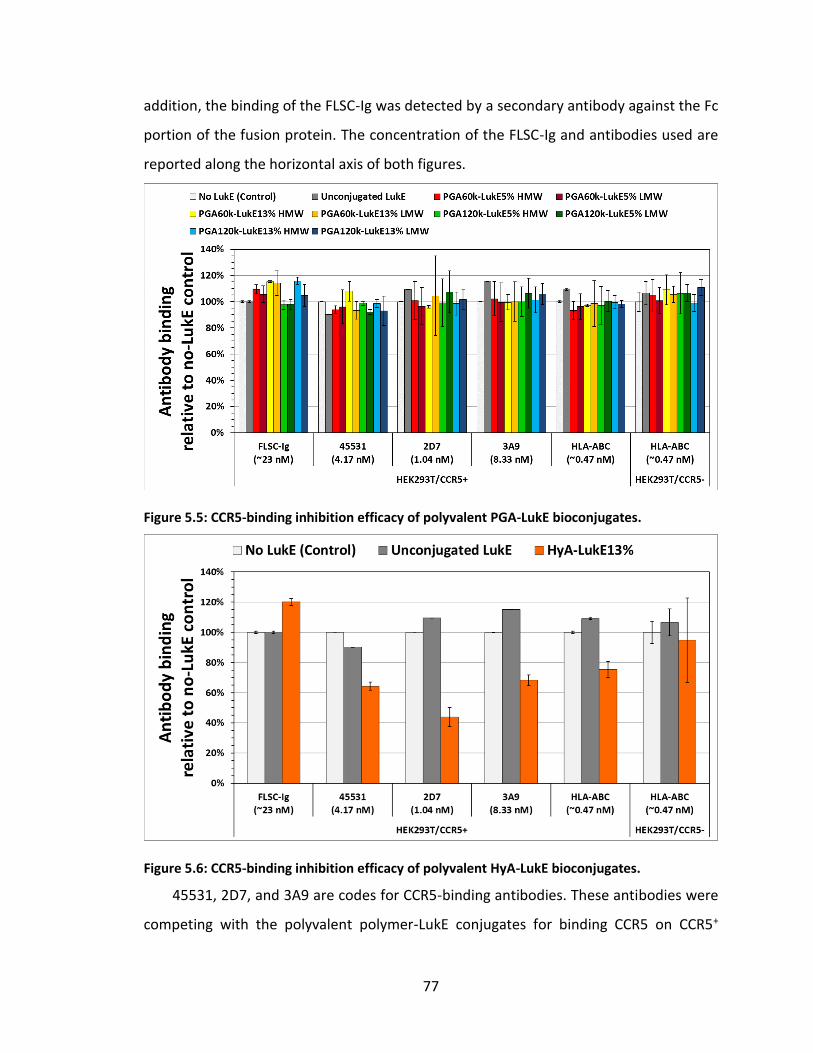
77
addition, the binding of the FLSC-Ig was detected by a secondary antibody against the Fc
portion of the fusion protein. The concentration of the FLSC-Ig and antibodies used are
reported along the horizontal axis of both figures.
Figure 5.5: CCR5-binding inhibition efficacy of polyvalent PGA-LukE bioconjugates.
Figure 5.6: CCR5-binding inhibition efficacy of polyvalent HyA-LukE bioconjugates.
45531, 2D7, and 3A9 are codes for CCR5-binding antibodies. These antibodies were
competing with the polyvalent polymer-LukE conjugates for binding CCR5 on CCR5+

78
HEK293T cells. 45531 and 2D7 bind specifically to separate epitopes on the second extra-
cellular loop of CCR5 (ECL2), and 3A9 binds specifically to the N-terminus of CCR5.198 LukE
was originally reported to bind to ECL2 based on the fact that antibody 45531 could
directly outcompete LukE for binding CCR5.196 In contrast, neither 3A9 nor 2D7 were
shown to inhibit LukE binding, despite the fact that 2D7 binds to the ECL2 of CCR5 near
the binding epitope of 45531.196 HLA-ABC is the generic name for a control antibody that
binds to the unrelated MHC class I proteins that are also expressed by the HEK293T cells.
The binding of HLA-ABC thus serves as a control. In Figure 5.5, none of the PGA-LukE
conjugates appear to have any effect on the ability of any of the antibodies or the FLSC-
Ig to bind to CCR5. Unconjugated LukE does not seem to have much effect either, other
than perhaps a ~10% inhibition of antibody 45531, the antibody reported to compete
most directly with LukE for binding CCR5. In contrast, HyA-LukE13% (13% of disaccharide
monomers were targeted for LukE coupling) appears to be able to inhibit all three CCR5-
binding antibodies in the range of 30-60% (Figure 5.6). Surprisingly, the binding of HLA-
ABC to CCR5+ cells was also inhibited, although to a slightly lesser extent. However, the
binding of this antibody was not inhibited on CCR5- cells, indicating that the inhibition of
HLA-ABC is not due to non-specific binding of the HyA-LukE conjugate to the cells. I
hypothesize that the inhibition of HLA-ABC on CCR5+ cells might be due to steric
hindrance by the large HyA backbone. On the other hand, the binding of FLSC-Ig was not
inhibited by HyA-LukE either.
5.4 Conclusions
The ability of HyA-LukE to block three different CCR5-binding antibodies is a very
promising result, especially because each of these antibodies is itself able to inhibit HIV
infection. As with the HyA-HB bioconjugates described in Chapter 4, additional work
synthesizing HyA-LukE bioconjugates with more well-defined HyA scaffolds and coupling
percentages is underway. This work should help to elucidate the optimal spacing between
individual, polyvalently-conjugated LukE proteins on a linear scaffold. In addition, GFP-
LukE fusion proteins have been successfully produced, and polyvalent HyA bioconjugates

79
are in the process of being synthesized. The CCR5-binding capacity of these HyA-GFP-LukE
bioconjugates should be detectable by flow cytometry directly via the GFP fluorescent
signal. Once the binding inhibition of these HyA-LukE and HyA-GFP-LukE bioconjugates
has been optimized, testing them directly in HIV infection inhibition assays is the next
logical step.

80
6. Multivalent oligonucleotide aptamer bioconjugates
6.1 Rationale for the use of oligonucleotide aptamers as ligands
A potential downside of the bioconjugates described up to this point is the use of peptides
or proteins as ligands. These types of ligands might suffer from problems in therapeutic
applications due to the tendency of non-native peptides and proteins to provoke an
immunogenic response. Such undesired side-effects can be a concern especially if the
therapeutics are to be administered as prophylactics. In addition, peptide ligands in
particular often exhibit only weak affinity for their targets, partly due to their small size.
Although this can be overcome by multivalent presentation, peptides may not be the best
choice of ligand in applications where very strong avidity with the target is desired.
Furthermore, peptide ligands may share some of the same difficulties associated with
inhibiting protein-protein interactions as small-molecule drugs. Finally, manufacture of
synthetic peptides is typically limited to short sequences, partly due to the high cost of
reagents and partly due to the increased likelihood of errors during the synthesis of longer
peptide sequences. Recombinant production of protein ligands can be even more
complicated and costly. Proteins which require post-translational modifications such as
glycosylation or the formation of internal disulfide bonds can be especially difficult, and
proteins often require special storage conditions to prevent their misfolding, aggregation,
or precipitation.
Oligonucleotide aptamers have a number of advantages over peptide and protein
antagonists, including their low cost of production and predisposition for synthesis by
“good manufacturing practices”. Oligonucleotide aptamer-based therapeutics may also
have a better outlook for approval by the US FDA, because nucleic acids are typically non-
immunogenic. A case in point is the FDA-approved RNA aptamer, Macugen, which is
prescribed for the treatment of neovascular age-related macular degeneration. In
addition, even short oligonucleotides are typically much larger in size and molecular mass
than peptides, and this increased size may enhance their ability to inhibit protein-protein
interactions. The increased contact area between oligonucleotide aptamers and their
targets also allows for increased affinity and specificity relative to peptide ligands. For

81
example, some oligonucleotide aptamers have been characterized as binding their targets
so strongly that even the dissociation constants of monovalent complexes are in the
picomolar range. Taken together, these characteristic features of oligonucleotide
aptamers make them ideal ligands for the design of multivalent inhibitors.
6.2 Production of polyvalent bioconjugates of the ssDNA aptamer sgc8c
Our previous work and the literature on polyvalent therapeutics suggested that we would
be able to significantly enhance the efficacy of the monovalent aptamers using
polyvalency.
To test this hypothesis, I synthesized polyvalent conjugates of a previously-
published200–202 ssDNA aptamer called sgc8c with PGA scaffolds for polyvalent ligand
display (see Figure 6.1, Figure 6.2, Figure 6.3, and Figure 6.4). The sgc8c aptamer is 42
bases long and was first identified by whole cell-SELEX. It binds to a cellular receptor on
lymphocytes,203 so it is a great model for the kind of targeting ligand we are interested in
moving to in the future. Although we (and others) have already reported the successful
use of polyvalent inhibitors based on PGA scaffolds,81,93,121,122 these previously
synthesized inhibitors have not made use of oligonucleotide aptamer ligands.
6.2.1 Synthesis of polyvalent bioconjugates of the ssDNA aptamer sgc8c
One of the challenges of polyvalent conjugation of oligonucleotide aptamers to PGA is the
fact that each of the biomolecules in question is polyanionic. The behavior of
polyelectrolytes in aqueous solution is extremely complex204 (especially for polyanions in
solution with a mixture of monovalent and divalent cations, e.g. Na+, Ca2+, and Mg2+), and
charge repulsion between the scaffold and oligonucleotide ligands may restrain the ability
to use some conjugation strategies.
Anticipating these challenges, I first optimized the bioconjugation conditions using
much less expensive single-stranded DNA (ssDNA) oligonucleotides, 25 bases long, as
model nucleic acid aptamers, and then synthesized polyvalent PGA-based conjugates of
the ssDNA aptamer sgc8c. Both model ssDNA oligonucleotides were custom-synthesized
by a commercial supplier (Integrated DNA Technologies, Inc.) with a 5’-thiol functionality,

82
which was provided protected by a disulfide-linked hexane cap. In order to expose the
free thiol, the ssDNA stock was incubated with 10 mM trialkylphosphine reducing agent
for 1 hour at pH 6.5, similar to the preparation of protein-thiols for maleimide conjugation
described in Section 4.2.3. The cleaved hexane-thiol cap is then removed from the ssDNA-
thiol solution by passing through a size-exclusion spin column.
After preparing ssDNA oligonucleotides by the deprotection/purification protocol,
the ssDNA and functionalized PGA scaffold are combined in a reaction buffer the same as
that described in Section 4.2.5.2 for polyvalent HB conjugation to PGA. The final
concentration of oligonucleotide was adjusted to 200 µM in order to be consistent with
the concentration we expect to be able to achieve with our 5’ modified, transcribed RNA
aptamers. As shown in Figure 6.1 for the ssDNA model oligonucleotides and in Figure 6.2
for the sgc8c aptamer, under these conditions, my reactions result in high conjugation
efficiency. Keeping in mind that the molecular weight of the polymer backbone may
influence inhibitory potency, I have functionalized PGA scaffolds from two different
commercially available molecular weights (Mw 35 kDa and 120 kDa).92 After the
conjugation was completed, the remaining maleimide functionality were hydrolyzed by
incubation with 100 mM MoO4 for 1-2 hours.
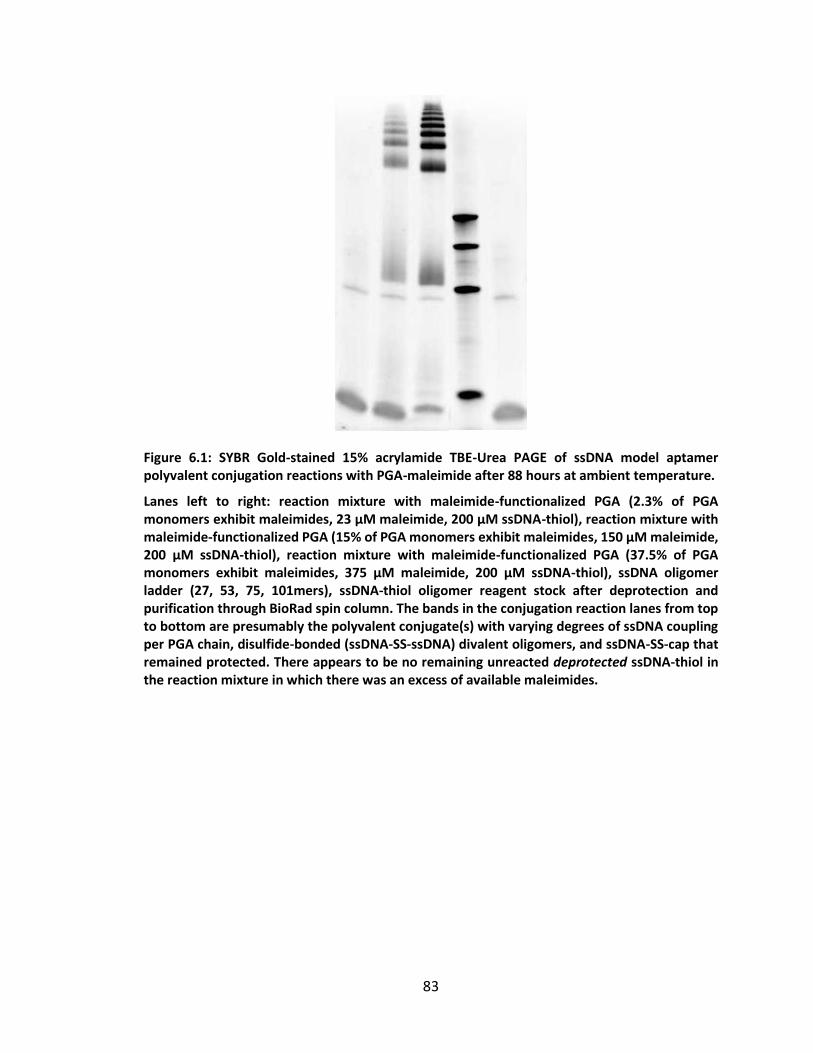
83
Figure 6.1: SYBR Gold-stained 15% acrylamide TBE-Urea PAGE of ssDNA model aptamer polyvalent conjugation reactions with PGA-maleimide after 88 hours at ambient temperature.
Lanes left to right: reaction mixture with maleimide-functionalized PGA (2.3% of PGA monomers exhibit maleimides, 23 µM maleimide, 200 µM ssDNA-thiol), reaction mixture with maleimide-functionalized PGA (15% of PGA monomers exhibit maleimides, 150 µM maleimide, 200 µM ssDNA-thiol), reaction mixture with maleimide-functionalized PGA (37.5% of PGA monomers exhibit maleimides, 375 µM maleimide, 200 µM ssDNA-thiol), ssDNA oligomer ladder (27, 53, 75, 101mers), ssDNA-thiol oligomer reagent stock after deprotection and purification through BioRad spin column. The bands in the conjugation reaction lanes from top to bottom are presumably the polyvalent conjugate(s) with varying degrees of ssDNA coupling per PGA chain, disulfide-bonded (ssDNA-SS-ssDNA) divalent oligomers, and ssDNA-SS-cap that remained protected. There appears to be no remaining unreacted deprotected ssDNA-thiol in the reaction mixture in which there was an excess of available maleimides.
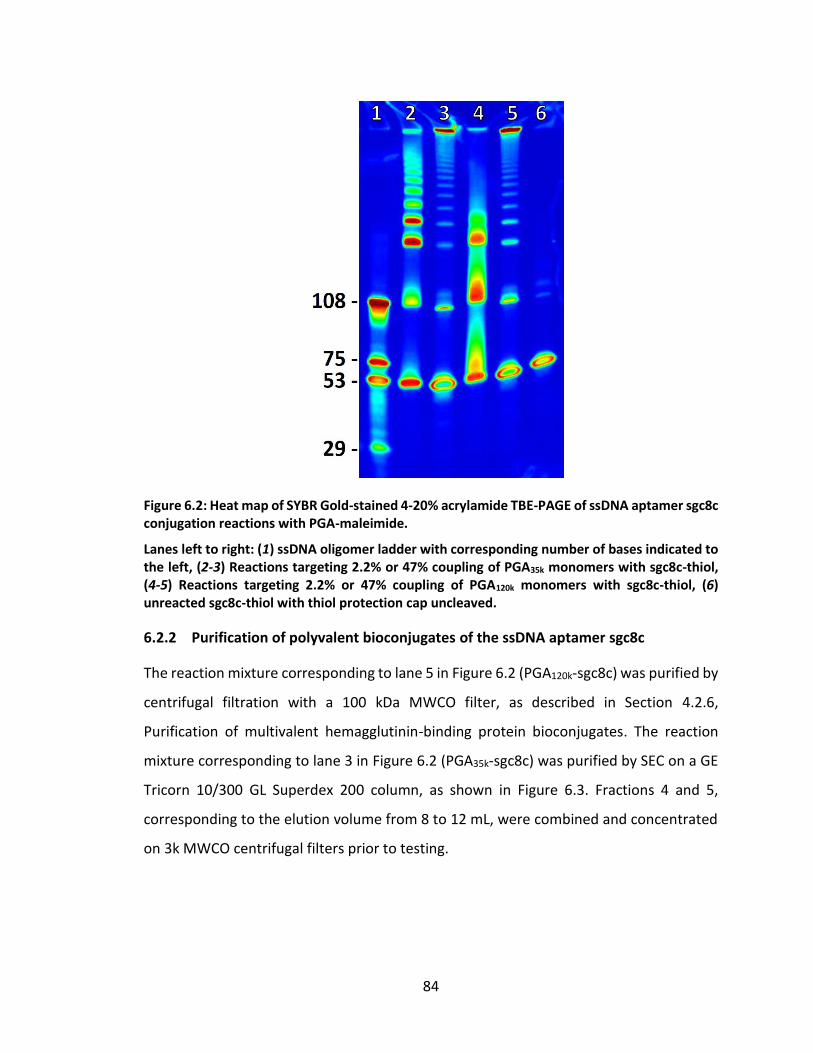
84
Figure 6.2: Heat map of SYBR Gold-stained 4-20% acrylamide TBE-PAGE of ssDNA aptamer sgc8c conjugation reactions with PGA-maleimide.
Lanes left to right: (1) ssDNA oligomer ladder with corresponding number of bases indicated to the left, (2-3) Reactions targeting 2.2% or 47% coupling of PGA35k monomers with sgc8c-thiol, (4-5) Reactions targeting 2.2% or 47% coupling of PGA120k monomers with sgc8c-thiol, (6) unreacted sgc8c-thiol with thiol protection cap uncleaved.
6.2.2 Purification of polyvalent bioconjugates of the ssDNA aptamer sgc8c
The reaction mixture corresponding to lane 5 in Figure 6.2 (PGA120k-sgc8c) was purified by
centrifugal filtration with a 100 kDa MWCO filter, as described in Section 4.2.6,
Purification of multivalent hemagglutinin-binding protein bioconjugates. The reaction
mixture corresponding to lane 3 in Figure 6.2 (PGA35k-sgc8c) was purified by SEC on a GE
Tricorn 10/300 GL Superdex 200 column, as shown in Figure 6.3. Fractions 4 and 5,
corresponding to the elution volume from 8 to 12 mL, were combined and concentrated
on 3k MWCO centrifugal filters prior to testing.
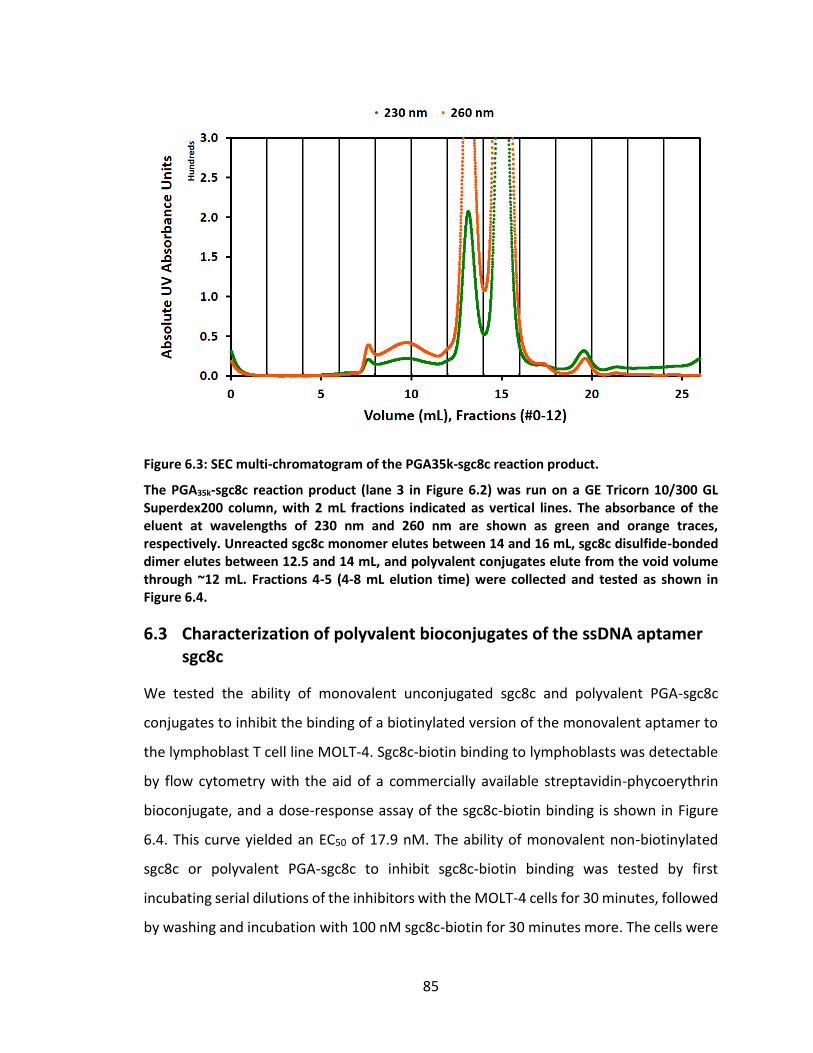
85
Figure 6.3: SEC multi-chromatogram of the PGA35k-sgc8c reaction product.
The PGA35k-sgc8c reaction product (lane 3 in Figure 6.2) was run on a GE Tricorn 10/300 GL Superdex200 column, with 2 mL fractions indicated as vertical lines. The absorbance of the eluent at wavelengths of 230 nm and 260 nm are shown as green and orange traces, respectively. Unreacted sgc8c monomer elutes between 14 and 16 mL, sgc8c disulfide-bonded dimer elutes between 12.5 and 14 mL, and polyvalent conjugates elute from the void volume through ~12 mL. Fractions 4-5 (4-8 mL elution time) were collected and tested as shown in Figure 6.4.
6.3 Characterization of polyvalent bioconjugates of the ssDNA aptamer sgc8c
We tested the ability of monovalent unconjugated sgc8c and polyvalent PGA-sgc8c
conjugates to inhibit the binding of a biotinylated version of the monovalent aptamer to
the lymphoblast T cell line MOLT-4. Sgc8c-biotin binding to lymphoblasts was detectable
by flow cytometry with the aid of a commercially available streptavidin-phycoerythrin
bioconjugate, and a dose-response assay of the sgc8c-biotin binding is shown in Figure
6.4. This curve yielded an EC50 of 17.9 nM. The ability of monovalent non-biotinylated
sgc8c or polyvalent PGA-sgc8c to inhibit sgc8c-biotin binding was tested by first
incubating serial dilutions of the inhibitors with the MOLT-4 cells for 30 minutes, followed
by washing and incubation with 100 nM sgc8c-biotin for 30 minutes more. The cells were
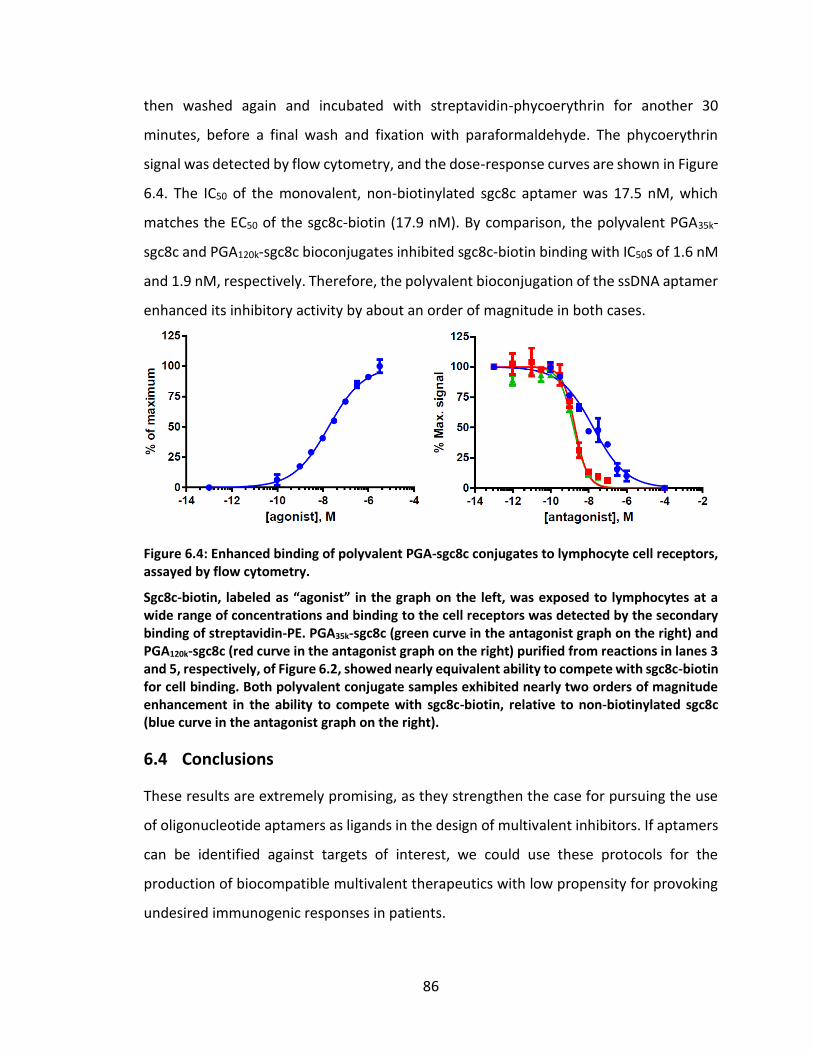
86
then washed again and incubated with streptavidin-phycoerythrin for another 30
minutes, before a final wash and fixation with paraformaldehyde. The phycoerythrin
signal was detected by flow cytometry, and the dose-response curves are shown in Figure
6.4. The IC50 of the monovalent, non-biotinylated sgc8c aptamer was 17.5 nM, which
matches the EC50 of the sgc8c-biotin (17.9 nM). By comparison, the polyvalent PGA35k-
sgc8c and PGA120k-sgc8c bioconjugates inhibited sgc8c-biotin binding with IC50s of 1.6 nM
and 1.9 nM, respectively. Therefore, the polyvalent bioconjugation of the ssDNA aptamer
enhanced its inhibitory activity by about an order of magnitude in both cases.
Figure 6.4: Enhanced binding of polyvalent PGA-sgc8c conjugates to lymphocyte cell receptors, assayed by flow cytometry.
Sgc8c-biotin, labeled as “agonist” in the graph on the left, was exposed to lymphocytes at a wide range of concentrations and binding to the cell receptors was detected by the secondary binding of streptavidin-PE. PGA35k-sgc8c (green curve in the antagonist graph on the right) and PGA120k-sgc8c (red curve in the antagonist graph on the right) purified from reactions in lanes 3 and 5, respectively, of Figure 6.2, showed nearly equivalent ability to compete with sgc8c-biotin for cell binding. Both polyvalent conjugate samples exhibited nearly two orders of magnitude enhancement in the ability to compete with sgc8c-biotin, relative to non-biotinylated sgc8c (blue curve in the antagonist graph on the right).
6.4 Conclusions
These results are extremely promising, as they strengthen the case for pursuing the use
of oligonucleotide aptamers as ligands in the design of multivalent inhibitors. If aptamers
can be identified against targets of interest, we could use these protocols for the
production of biocompatible multivalent therapeutics with low propensity for provoking
undesired immunogenic responses in patients.

87
7. Suggestions for future work4
7.1 Identification of oligonucleotide aptamers that target cell receptors
7.1.1 Rationale for the search for oligonucleotide aptamers that target cell receptors
We have already shown that targeting [PA63]7 can be a successful therapeutic strategy for
inhibiting the pathogenicity of B. anthracis.205,206 Additionally, I have more recently shown
that we can successfully inhibit influenza infection in vitro by targeting the viral HA
protein. However, there are some potential downsides for these strategies due to their
reliance on peptide and protein targeting ligands.
As was already discussed in Section 6.1 Rationale for the use of oligonucleotide
aptamers as ligands, oligonucleotide aptamer ligands have many advantages over peptide
and protein ligands. Furthermore, as was discussed in Section 5.1.1 Targeting the cellular
receptor CCR5, the host receptors that are exploited by a pathogen may be the most
logical targets when designing inhibitors of that pathogen. These factors have led me to
pursue biopanning for new anthrax toxin inhibitors that target the host cell receptors
involved in the intoxication pathway, specifically ANTXR2 (See Section 1.1.2 Intoxication
pathway, page 1).
7.1.2 Screening libraries of oligonucleotide aptamers
7.1.2.1 Systematic Evolution of Ligands by EXponential enrichment (SELEX)
Protocols for the identification of oligonucleotide aptamers were first reported in 1990
by two research groups independently.207,208 Systematic Evolution of Ligands by
EXponential enrichment (SELEX) is a type of iterative biopanning procedure for identifying
aptamers with desired binding properties. Many different adaptations of this protocol
have been reported in the past ~20 years,209–211 but the general procedure (depicted in
Figure 7.1) of almost every variant involves a progressive cycling of the following steps:
Portions of this chapter previously appeared as: Martin, J. T.; Kane, R. S. Design of Polyvalent Polymer Therapeutics. In Functional Polymers by Post-Polymerization Modification; Theato, P.; Klok, H.-A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 267–290.

88
exposure of a large, randomized, undefined “library” of unique sequences of potential
oligonucleotide ligands to a target molecule, isolation of binders, and amplification of the
binder sequences to regenerate the oligonucleotide pool. This amplification step (and
additional translation steps in the case of RNA aptamer panning) requires the inclusion of
predefined primer sequences in the oligonucleotide sequence (see Figure 7.2). The
primers enable the enzymatic amplification of the oligonucleotide pool by polymerase
chain reaction (PCR), as well as forward and reverse in vitro transcriptions of the pool
between RNA and DNA forms, for the cases in which RNA aptamers are the desired
product. As a result, the standard library design involves a string of ~30-40 randomized
nucleotides, flanked on either end by fixed primer sequences (Figure 7.2).
Figure 7.1: General SELEX procedure for screening RNA aptamers.
Reprinted from Int. J. Parasitol., 33, Göringer, H. U.; Homann, M.; Lorger, M., In vitro selection of high-affinity nucleic acid ligands to parasite target molecules, 1309-1317, 2003, with permission from Elsevier.

89
Figure 7.2: The sequence of the initial DNA library and a representative mfold prediction.
The sequence of the initial DNA library for a typical SELEX screen with 40 random nucleotides flanked by primer-derived sequences is above an mfold-predicted secondary structure of a model aptamer, showing the interference from the flanking regions.
7.1.2.2 A modified SELEX protocol for identifying short oligonucleotide aptamers
Aptamers which have been identified by screening standard SELEX libraries as described
above may not be feasible for use in therapeutics due to their excessive length. The fixed
primer sequences can contribute to the RNA aptamer secondary structure (Figure 7.2)
and consequently influence target binding; thus, the random portion of the sequence
probably cannot be isolated and used independently. As a result, it may be necessary to
continue using the entire sequence, primers included, that was present for the screen
(typically ~75 bases or more). In order to reduce the high synthesis costs of such long
aptamers that may be prohibitive for therapeutic applications, it might be possible to
perform 5’ and 3’ truncations, but these are essentially time-consuming trial-and-error
studies, and are unlikely to yield significantly shorter aptamers that retain their binding
character. These limitations have motivated our lab group to develop a modified SELEX-
based approach to overcome the limitations of oligonucleotide aptamers panned from a
standard SELEX library.
The goal of developing a modified SELEX protocol was to enable the identification of
short oligonucleotide aptamers. A published technique for analyzing RNA structure212,213

90
provided some inspiration. This technique takes advantage of the ability to design RNA
sequences that self-hybridize into stable hairpins. Single-stranded DNA libraries are
commercially synthesized such the primer sequences are contained within 5’ and 3’
cassettes (Figure 7.3) that flank the random portion of the oligonucleotide sequence. The
hairpins in the cassettes are individually stable secondary structures and thus significantly
diminish the probability of interfering with the secondary structure of the random region.
We opted for random sequences that are 25 or 30 nucleotides long, depending on the
library; this should enable the identification of RNA aptamers no longer than 30
nucleotides that can be isolated from the flanking cassettes and that are well-suited for
synthesizing polyvalent bioconjugates (Figure 7.3). I applied this method in order to
screen large libraries of nuclease-resistant RNA for short aptamer sequences that bind to
ANTXR2, and my colleagues in the Kane group performed screens against CCR5.
Figure 7.3: Schematic representation of the new library design for screening of short oligonucleotide aptamers.
(a) Random region is flanked by 5’ and 3’ cassettes, which have a stem-loop structure where the tetraloop autonomously folds onto itself and eliminates any alternative folding.
Inhibitors composed of aptamer ligands will need to function in vivo, therefore the
conditions used in the binding screens were chosen to match the conditions in the blood

91
serum. A temperature of 37°C and a solution of 20 mM HEPES-NaOH buffer, pH 7.4,
containing 150 mM NaCl, 1.5 mM CaCl2, and 0.5 mM MgCl2 were maintained for each of
the steps in which the binding capacity of the pool was important. In order to protect the
RNA against nuclease degradation, 2’-fluoropyrimidines were used in the in vitro
transcription reactions. Yeast t-RNA was also added to the buffered RNA aptamer pool in
order to reduce the effects of potential non-specific (e.g. electrostatic) RNA binding
interactions during screening.
The folded RNA pool was exposed to one positive and one negative selection during
each round of the screen. The positive screen was against ANTXR2, which was incubated
overnight in protein-adsorbing tubes. Potentially uncoated surfaces were “blocked” by
exposure to a high concentration of bovine serum albumin (BSA) before exposure to the
RNA pool. The presence of BSA necessitated a negative screen against tubes that had only
BSA adsorbed. The negative screen was carried out first, in order to deplete the pool of
aptamers that bound BSA (or that bound nonspecifically).
Each round of screening consisted of in vitro RNA transcription, negative and positive
selections, reverse transcription, and amplification. The two single-stranded DNA libraries
were combined and amplified by polymerase chain reaction (PCR), then transcribed in
vitro (using 2’-fluoropyrimidines) to generate the initial RNA library. The starting RNA pool
was unfolded at 96°C and then allowed to refold at 37°C in the presence of the buffer
described above. The refolded RNA pool was allowed to incubate in the BSA-coated tubes
for 10 minutes, and the supernatant, containing aptamers that did not bind, was then
transferred to the tubes coated with ANTXR2. After a second incubation period, the
ANTR2-coated tubes were extensively washed with buffer to remove unbound or weakly
bound RNA. A 20 mM EDTA dissociation solution was then used to remove the divalent
cations that help give the RNA its secondary structure, thereby eluting the remaining
binders. Phenol-chloroform extraction was used to separate the recovered RNA from any
protein in the solution. Finally, the RNA pool was regenerated by reverse transcription,
PCR amplification and in vitro transcription. The process was repeated for five rounds.

92
7.1.3 Characterization of screened oligonucleotide aptamers
The final pool of aptamer sequences should be enriched with sequences that bind
specifically to the target, in this case ANTXR2. In order to determine which sequences
have been enriched, we have submitted the round 5 aptamer pool and the initial library
for Illumina deep sequencing. We are currently analyzing the sequencing results to
identify sequences which are promising candidates for further testing. Ideally, up to 10
unique aptamer sequences should be ordered and tested for their ability to bind
specifically to ANTXR2 and for their ability to inhibit the binding of PA. The aptamers that
show the best monovalent activity could then be used further for the synthesis of
polyvalent inhibitor constructs.
Modification of the oligonucleotide with an attachment chemistry functionality at
either the 5’ or 3’ end can be accomplished synthetically, by ordering commercially
available modified oligonucleotides. However, in the case of RNA aptamers, which often
include 2’-fluoropyrimidines for nuclease-resistance, even short aptamer sequences can
become quite costly. For this reason, RNA aptamers of interest could be functionalized at
the 5’-end with a modified GMP, using established procedures.214–216 Essentially, RNA
aptamers can be generated by in vitro transcription with T7 RNA polymerase in the
presence of a large molar excess of a modified GMP relative to GTP, such that the GMP is
preferentially incorporated at the 5’-end of the transcript. GMP-S and GMP-NH2 are
commercially available, and I have synthesized a modified GMP-alkyne from GMP using a
published synthesis strategy.217 The availability of these various options should allow for
attachment to a scaffold using whichever chemistry proves to be most convenient for the
exact application at hand.
7.2 Alternative designs for multivalent HIV entry inhibitors
7.2.1 Peptide ligand derivatives of CCL5
The natural ability of the C-C chemokine ligands to inhibit HIV infection has fueled a
number of studies with the aim of adapting CCL5 to improve its therapeutic potential. An
important thrust of much of this research was the attempt to uncouple the

93
agonistic/inflammatory activity of CCL5 from the binding affinity/inhibitory efficacy.218 Of
particular interest to us were the studies aimed at identifying the structural determinants
of CCR5 recognition and antiviral activity.218–220 Although three different research groups
performed similar peptide scanning studies using sequential CCL5-derived peptide
fragments, they produced conflicting reports as to which domains of CCL5 showed
binding activity and HIV inhibitory activity. However, it now seems that the field supports
the work of Lusso and colleagues,218,221–225 who initially identified the N-loop and β1-
strand (residues 11-29, CFAYIARPLPRAHIKEYFY) as the structural determinants of CCL5
that are most important for CCR5 binding and HIV inhibition (Figure 7.4).218,224
Interestingly, they also reported that an analogous region of CCL4 (residues 12-30,
CFSYTARKLPRNFVVDYYE) showed similar binding and inhibitory activity.221 A later study
on the binding surfaces of full-length CCL5 also found a second region they called the 40’
loop (residues 43-48, TRKNRQ) that was close to the key hydrophobic regions of CCL5 and
that was important for binding to the N-terminus of CCR5 (Figure 7.4).225
Figure 7.4: Structure of CCL5 monomer (PDB ID 1HRJ), with regions important for binding CCR5 highlighted.
The sequence of the N-loop / β1-strand, CFAYIARPLPRAHIKEYFY (residues 11-29), is highlighted in yellow and green. Yellow indicates the hydrophobic regions that appear crucial for CCR5 binding. The 40’ loop region, identified as an additional part of the binding surface by Duma et al.225 is highlighted in blue. Reprinted from Chem. Biol. 19, Secchi, M.; Longhi, R.; Vassena, L.; Sironi, F.; Grzesiek, S.; Lusso, P.; Vangelista, L., 1579-1588, 2012, with permission from Elsevier.

94
Since their initial report,218 the researchers have worked extensively to develop their
lead peptide candidate by sequential residue mutations and truncations.221–223 Additional
work on the structural interactions between CCL5 and CCR5 showed that the natural
chemokine ligands and the CCL5-derived peptides bind to the solvent-exposed N-terminal
region of CCR5. Furthermore, when the cysteine at the N-terminus of the peptide ligand
was used to create peptide dimers (via the formation of a disulfide bond), the key binding
surface area was increased. Accordingly, the activity of peptide dimers was enhanced 10-
fold relative to the corresponding monomers.221 However, NMR showed no interactions
between the monomers,222 which suggests that the efficacy enhancement by
dimerization may be purely a function of multivalency. Although Lusso and colleagues
stress the importance of peptide dimer formation for their reported inhibitory activities,
the researchers make no references to investigating the addition of a linker for increased
binding range or incorporating these peptide monomers into higher-order multivalent
ligand conjugates.221
In prior unpublished work, our group (in collaboration with the Gray-Owen group at
the University of Toronto) found that liposomal polyvalent conjugates of one of the initial
lead peptides (Ac-CFAYIARPLPRA-Am) reported by the Lusso group218 showed some
ability to inhibit HIV infection, but the activity was not very impressive, especially for a
polyvalent inhibitor (Figure 7.5). Recently, however, an improved version of the peptide
ligand with the sequence Ac-CFPYITRPGTYHDWWYTRKNRQ was reported to have three
to four orders of magnitude better activity.221 This peptide was able to inhibit HIV
envelope protein-mediated cell fusion with an IC50 of 20 nM and was able to inhibit HIV
infection of peripheral blood mononuclear cells (PBMCs) with an IC50 of 167 nM. Also
noteworthy is the fact that the authors found that removing the C-terminal amidation
improved solubility without affecting activity.221 Since lack of peptide water-solubility is a
factor that we have noticed in our preliminary work with Ac-CFAYIARPLPRA-Am, using
peptides without C-terminal amidation may be especially important for the performance
of our polyvalent inhibitors.
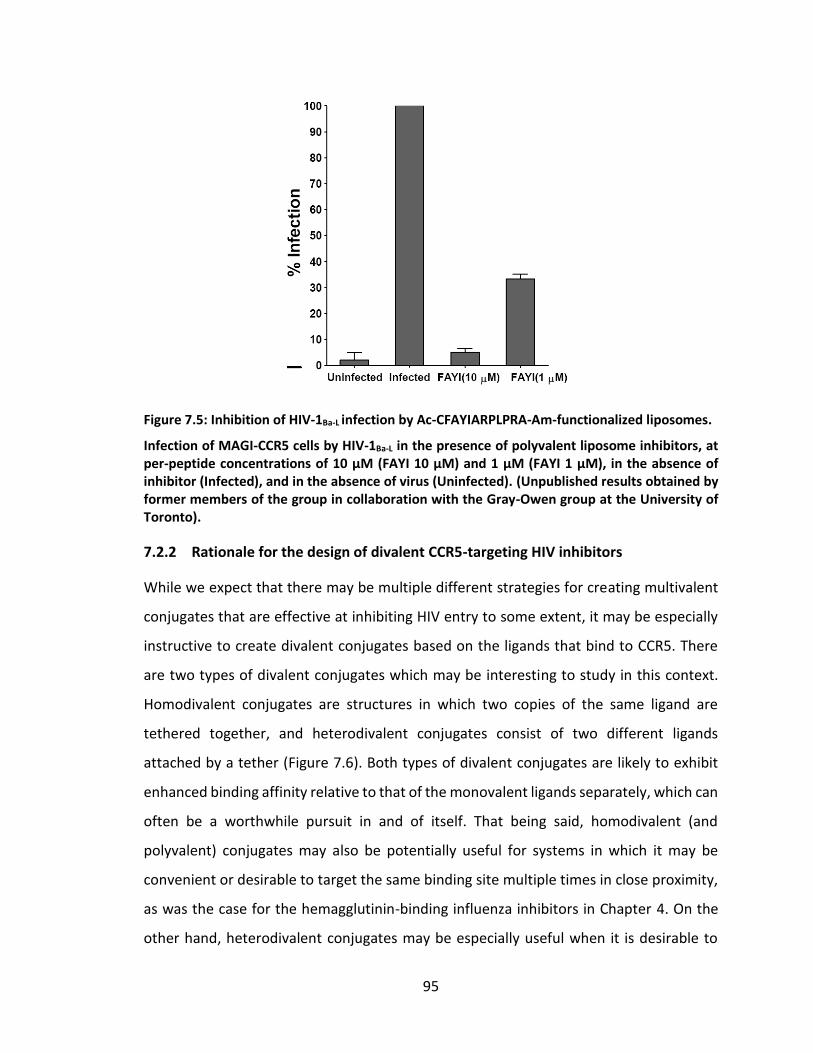
95
Figure 7.5: Inhibition of HIV-1Ba-L infection by Ac-CFAYIARPLPRA-Am-functionalized liposomes.
Infection of MAGI-CCR5 cells by HIV-1Ba-L in the presence of polyvalent liposome inhibitors, at per-peptide concentrations of 10 µM (FAYI 10 µM) and 1 µM (FAYI 1 µM), in the absence of inhibitor (Infected), and in the absence of virus (Uninfected). (Unpublished results obtained by former members of the group in collaboration with the Gray-Owen group at the University of Toronto).
7.2.2 Rationale for the design of divalent CCR5-targeting HIV inhibitors
While we expect that there may be multiple different strategies for creating multivalent
conjugates that are effective at inhibiting HIV entry to some extent, it may be especially
instructive to create divalent conjugates based on the ligands that bind to CCR5. There
are two types of divalent conjugates which may be interesting to study in this context.
Homodivalent conjugates are structures in which two copies of the same ligand are
tethered together, and heterodivalent conjugates consist of two different ligands
attached by a tether (Figure 7.6). Both types of divalent conjugates are likely to exhibit
enhanced binding affinity relative to that of the monovalent ligands separately, which can
often be a worthwhile pursuit in and of itself. That being said, homodivalent (and
polyvalent) conjugates may also be potentially useful for systems in which it may be
convenient or desirable to target the same binding site multiple times in close proximity,
as was the case for the hemagglutinin-binding influenza inhibitors in Chapter 4. On the
other hand, heterodivalent conjugates may be especially useful when it is desirable to
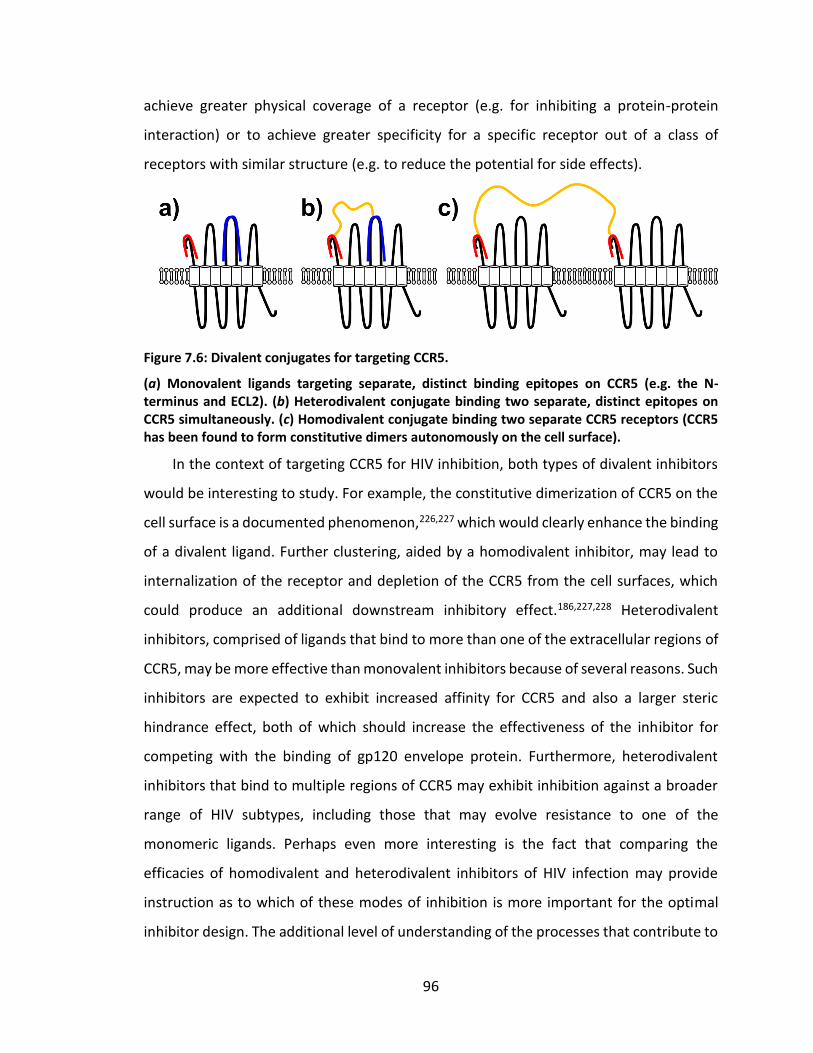
96
achieve greater physical coverage of a receptor (e.g. for inhibiting a protein-protein
interaction) or to achieve greater specificity for a specific receptor out of a class of
receptors with similar structure (e.g. to reduce the potential for side effects).
Figure 7.6: Divalent conjugates for targeting CCR5.
(a) Monovalent ligands targeting separate, distinct binding epitopes on CCR5 (e.g. the N-terminus and ECL2). (b) Heterodivalent conjugate binding two separate, distinct epitopes on CCR5 simultaneously. (c) Homodivalent conjugate binding two separate CCR5 receptors (CCR5 has been found to form constitutive dimers autonomously on the cell surface).
In the context of targeting CCR5 for HIV inhibition, both types of divalent inhibitors
would be interesting to study. For example, the constitutive dimerization of CCR5 on the
cell surface is a documented phenomenon,226,227 which would clearly enhance the binding
of a divalent ligand. Further clustering, aided by a homodivalent inhibitor, may lead to
internalization of the receptor and depletion of the CCR5 from the cell surfaces, which
could produce an additional downstream inhibitory effect.186,227,228 Heterodivalent
inhibitors, comprised of ligands that bind to more than one of the extracellular regions of
CCR5, may be more effective than monovalent inhibitors because of several reasons. Such
inhibitors are expected to exhibit increased affinity for CCR5 and also a larger steric
hindrance effect, both of which should increase the effectiveness of the inhibitor for
competing with the binding of gp120 envelope protein. Furthermore, heterodivalent
inhibitors that bind to multiple regions of CCR5 may exhibit inhibition against a broader
range of HIV subtypes, including those that may evolve resistance to one of the
monomeric ligands. Perhaps even more interesting is the fact that comparing the
efficacies of homodivalent and heterodivalent inhibitors of HIV infection may provide
instruction as to which of these modes of inhibition is more important for the optimal
inhibitor design. The additional level of understanding of the processes that contribute to

97
HIV entry inhibition that can be gained by comparing and contrasting these two types of
divalent inhibitors will undoubtedly be important for the intelligent design of conjugates
with higher levels of multivalency or complexity. For these reasons, I have completed
preliminary work on the synthesis and characterization both of homodivalent and
heterodivalent conjugates (see Figure 7.6), as well as polyvalent conjugates of the various
ligands identified in Section 7.2.1 above.
7.2.3 Synthesis of homodivalent bioconjugates of aptamers that bind CCR5
In preliminary work using the model 5’-thiol-functionalized ssDNA sequences first
described in Section 6.1, I have confirmed that the protocol for conjugation to a
homobifunctional PEG linker can be applied for thiol-functionalized oligonucleotide
aptamers. The typical conjugation strategy is depicted in Figure 7.7 below.
Figure 7.7: Synthesis scheme for homodivalent PEG-aptamer bioconjugates.
The PEG linker used for the conjugation optimization experiment was a maleimide-(EG)11-
maleimide linker. The purified, de-protected ssDNA was then added to the reaction buffer
at a 2.2:1 molar ratio relative to the linker. The reaction buffer is the same as for the
synthesis of homodivalent PEG-protein conjugates described in Section 4.2.4, degassed
pH 6.5 MBS with 10 mM EDTA at ambient temperature. The concentration of the ssDNA
in the reaction buffer was adjusted to 200 µM. As shown in Figure 7.8, under these
conditions, there is evidence for nearly complete divalent conjugation within 3.5 days.

98
Figure 7.8: SYBR Gold-stained 15% acrylamide TBE-Urea PAGE of ssDNA model aptamer homodivalent PEG conjugation reactions after 88 hours at ambient temperature.
Lanes left to right: ssDNA ladder (27, 53, 75, 101mers), ssDNA-thiol oligomer reagent stock after deprotection and purification through BioRad spin column, conjugation reaction mixture after 88 hours at ambient temperature. The bands in the conjugation reaction lane from top to bottom are presumably the homodivalent conjugate, disulfide-bonded (ssDNA-SS-ssDNA) divalent oligomers, monovalent conjugate, and ssDNA-SS-cap that remained protected. There appears to be no remaining unreacted deprotected ssDNA. Stained with SYBR Gold.
7.2.4 Synthesis of heterodivalent inhibitors that bind CCR5
CCR5-targeting heterodivalent inhibitors can be created by the combination of two site-
specific ligands (either peptides or oligonucleotides) connected by an appropriately-sized
PEG linker (Figure 7.6 and Figure 7.9). For optimizing reaction conditions for
heterodivalent inhibitors, I chose to experiment with the heterobifunctional linker
sulfosuccinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (sulfo-SMCC) as
proof of concept. Sulfo-SMCC is a commonly used, water-soluble, short (8.3 Å) spacer
with amine and thiol reactivity (sulfo-NHS and maleimide functionalities). The same
deprotection protocol and buffer conditions as used for homodivalent conjugation were
used here, and both 5’-thiol and 5’-amine functional ssDNA oligonucleotides were added
into the reaction buffer with sulfo-SMCC, in a 1:1:1 molar ratio. The formation of the
heterodivalent conjugate was confirmed by mass spectrometry. These kinds of divalent

99
conjugates will be interesting in future studies if multiple aptamers against the same
screening target are identified, especially if it can be determined that the aptamers bind
to different regions on the same target molecule.
Figure 7.9: Synthesis scheme for heterodivalent PEG-aptamer bioconjugates.
(a) aptamer 1 (5’-amine), aptamer 2 (5’-thiol), TCEP, NaCl, pH 6.5 MBS.
7.3 Bioengineered protein polymer scaffolds
7.3.1 Using proteins as monodisperse polymer scaffolds
Monodisperse, functional polymer scaffolds are readily available from nature in the form
of proteins. Amine (lysine), carboxylic acid (aspartic acid and glutamic acid), thiol
(cysteine), and hydroxyl (serine and threonine) are naturally occurring functional handles
on the side chains of peptides. The research of Yang et al.229 demonstrated as a proof of
concept the ability to acylate lysine side chains with acetic anhydride in three different
proteins: ubiquitin (8.6 kDa, 7 lysines, 1 N-terminal amine), lysozyme (14.3 kDa, 6 lysines,
1 N-terminal amine), and bovine carbonic anhydrase II (BCA) (29 kDa, 18 lysines, post-
translationally acetylated N- terminal amine). The ability to acylate the lysine side chains
depended on the size of the protein; denaturation with SDS was required for complete
acylation of the lysine residues in BCA, but there was no reported difference between the
native or denatured reactivity of the lysine residues in ubiquitin. Furthermore, the ability
to react ubiquitin with hydrophilic triethylene glycol carboxylate, hydrophobic benzoate,
anionic glutarate, and chemically reactive iodoacetate demonstrated both the generality
of the procedure and the range of chemical properties that might be introduced into
protein scaffolds. The approach is interesting, but the propensity for the proteins to fold
into tertiary structures should still be considered.
O
n
Na
O
O
O
O
N
O
O
O
n
N
O
O
HN
O
aptamer S
aptamer

100
7.3.2 Bioengineering scaffolds for precise control over multivalent architecture
Recombinant protein engineering methods can be used to take advantage of nature’s
polymer synthesis machinery in order to create scaffolds that are both monodisperse and
designed to have functional handles at predetermined locations.230 The Kiick research
group has employed these methods95,231,232 to make well-defined scaffolds into
polyvalent inhibitors of the cholera toxin B pentamer subunit. The researchers
demonstrated that by altering the composition of the polypeptide, they could control the
inter-ligand spacing, the valency, and even the conformation (α-helical versus random-
coil) of the inhibitors. In addition to observing improvements in inhibitors with inter-
ligand spacing and valency that matched the target toxin, the results revealed more
potent inhibition using α-helical scaffolds than random-coil scaffolds. The authors
hypothesized that the α-helical conformation may have increased the accessibility of the
pendant ligands and/or reduced the loss of conformational entropy upon binding, thus
leading to the measured improvement in potency. Using oligovalent proteins as
supramolecular templates for pre-arranging the displays of heterobifunctional ligands on
a polyvalent polymer scaffold have also been shown to yield vast improvements in
potency.233,234
As a demonstration of the power of using bioengineering techniques to design
multivalent binders, we have designed and tested monodisperse anthrax toxin inhibitors
based on polypeptide scaffolds. A manuscript describing this research185 was very
recently accepted for publication in Angewandte Chemie (see Appendix). Specifically, my
colleagues in the Kane group created polypeptides with the sequence
H10(SE)[HTSTYWWLDGAP(SE5)]n(SE)4, in which an (SE)5 linker separates adjacent copies of
the peptide HTSTYWWLDGAP that binds to the heptameric receptor-binding subunit of
anthrax toxin. These multivalent inhibitors were expressed in E. coli and purified using
immobilized metal affinity chromatography. The activity of the best polypeptide inhibitor
(n=8) had an IC50 of ~2 nM on a per-peptide basis.
We have also designed and begun expression and purification of well-defined
polypeptide scaffolds with the sequence [(GE)mK]n(GE)mH10 and [(SE)mK]n(SE)mH10,

101
where the K (lysine) residues will serve as reactive sites for ligand conjugation (Fig 5).
(GE)m or (SE)m serve as linkers whose length can be tuned by varying the value of “m”, “n”
represents the number of reactive sites in the scaffold (and provides control over the
valency of the multivalent ligand), and the terminal decahistidine sequence will help with
purification. Our prior results and a recent thermodynamic model emphasize the
importance of the root-mean-square (RMS) end-to-end distance of the linker as a design
parameter. The RMS end-to-end distance for the linker ((GE)m or (SE)m), however, will be
significantly larger in multivalent conjugates with bulky protein ligands (e.g., HB36.5 or
LukE) than in “free” unconjugated polypeptide scaffolds due to steric effects, particularly
for short linkers with an extended length comparable to the diameter of the protein
ligand. We have therefore initially designed scaffolds with 3 values of m – 10, 20, and 30
– corresponding to linkers composed of 20, 40, and 60 amino acids respectively. The
smallest value of m corresponds to a linker with an extended length between 7 and 8 nm.
Our simulations indicate that the intermediate linkers (m=20) would have RMS end-to-
end distances of ~6 nm, whereas the longest linkers (m=30) would have RMS end-to-end
distances of ~8.5 nm and an extended length of more than 21 nm. Three different values
for the valency, n, were initially chosen: 10, 20, and 30. These and other bioengineered
scaffolds should prove extremely useful for elucidating the most efficacious designs of
future polyvalent bioconjugates.

102
8. Conclusions
As we have shown in extensive work on the inhibition of a variety of different pathogenic
processes, multivalency can be a powerful tool for enhancing the efficacy of bioactive
molecules. This document specifically described the design principles that guided the
synthesis of multivalent biomolecular conjugates with enhanced activity in three different
applications with broad public interest: the inhibition of anthrax intoxication, the
inhibition of influenza infection, and the inhibition of HIV infection. Each of these
inhibitors has been designed to target a stage in the pathogenesis that is conserved or
that will not engender strong selection pressure. The protocols optimized during the
course of these studies could be used to synthesize the next-generation of multivalent
therapeutics, which may use a wide variety of new binding ligands, including
oligonucleotide aptamers. The successful use of protein engineering to bioengineer
scaffolds that have built-in chemical functionality, allowing for the attachment of
bioactive molecules at precisely defined locations and spacing, will be a crucial tool for
synthesizing bioconjugates that can clarify the complex mechanisms behind the actions
of various biological systems. By using biocompatible materials and methods for
bioconjugate synthesis such as those described here, there will be great potential for
clinical use of multivalent therapeutics in the near future.

103
REFERENCES
(1) Martin, J. T.; Kane, R. S. Design of Polyvalent Polymer Therapeutics. In Functional Polymers by Post-Polymerization Modification; Theato, P.; Klok, H.-A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 267–290.
(2) Coleman, M. E.; Thran, B.; Morse, S. S.; Hugh-Jones, M.; Massulik, S. Biosecur. Bioterror. 2008, 6, 147.
(3) Jernigan, D. B.; Raghunathan, P. L.; Bell, B. P.; Brechner, R.; Bresnitz, E. A.; Butler, J. C.; Cetron, M.; Cohen, M.; Doyle, T.; Fischer, M.; Greene, C.; Griffith, K. S.; Guarner, J.; Hadler, J. L.; Hayslett, J. A.; Meyer, R.; Petersen, L. R.; Phillips, M.; Pinner, R.; Popovic, T.; Quinn, C. P.; Reefhuis, J.; Reissman, D.; Rosenstein, N.; Schuchat, A.; Shieh, W.-J.; Siegal, L.; Swerdlow, D. L.; Tenover, F. C.; Traeger, M.; Ward, J. W.; Weisfuse, I.; Wiersma, S.; Yeskey, K.; Zaki, S.; Ashford, D. A.; Perkins, B. A.; Ostroff, S.; Hughes, J.; Fleming, D.; Koplan, J. P.; Gerberding, J. L. Emerg. Infect. Dis. 2002, 8, 1019.
(4) Makino, S.; Uchida, I.; Terakado, N.; Sasakawa, C.; Yoshikawa, M. J. Bacteriol. 1989, 171, 722.
(5) Collier, R. J.; Young, J. A. T. Annu. Rev. Cell Dev. Biol. 2003, 19, 45.
(6) Rainey, G. J. A.; Young, J. A. T. Nat. Rev. Microbiol. 2004, 2, 721.
(7) Meyerhoff, A.; Albrecht, R.; Meyer, J. M.; Dionne, P.; Higgins, K.; Murphy, D. Clin. Infect. Dis. 2004, 39, 303.
(8) Dixon, T. C.; Meselson, M.; Guillemin, J.; Hanna, P. C. N. Engl. J. Med. 1999, 341, 815.
(9) Abrami, L.; Lindsay, M.; Parton, R. G.; Leppla, S. H.; van der Goot, F. G. J. Cell Biol. 2004, 166, 645.
(10) Rainey, G. J. A.; Wigelsworth, D. J.; Ryan, P. L.; Scobie, H. M.; Collier, R. J.; Young, J. A. T. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 13278.
(11) Krantz, B. A.; Melnyk, R. A.; Zhang, S.; Juris, S. J.; Lacy, D. B.; Wu, Z.; Finkelstein, A.; Collier, R. J. Science 2005, 309, 777.
(12) Pentelute, B. L.; Sharma, O.; Collier, R. J. Angew. Chem. Int. Ed. Engl. 2011, 50, 2294.

104
(13) Bradley, K. A.; Mogridge, J.; Mourez, M.; Collier, R. J.; Young, J. A. Nature 2001, 414, 225.
(14) Scobie, H. M.; Rainey, G. J. A.; Bradley, K. A.; Young, J. A. T. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5170.
(15) Bonuccelli, G.; Sotgia, F.; Frank, P. G.; Williams, T. M.; de Almeida, C. J.; Tanowitz, H. B.; Scherer, P. E.; Hotchkiss, K. A.; Terman, B. I.; Rollman, B.; Alileche, A.; Brojatsch, J.; Lisanti, M. P. Am. J. Physiol. Cell Physiol. 2005, 288, C1402.
(16) Molloy, S. S.; Bresnahan, P. A.; Leppla, S. H.; Klimpel, K. R.; Thomas, G. J. Biol. Chem. 1992, 267, 16396.
(17) Gordon, V. M.; Klimpel, K. R.; Arora, N.; Henderson, M. A.; Leppla, S. H. Infect. Immun. 1995, 63, 82.
(18) Petosa, C.; Collier, R. J.; Klimpel, K. R.; Leppla, S. H.; Liddington, R. C. Nature 1997, 385, 833.
(19) Kintzer, A. F.; Thoren, K. L.; Sterling, H. J.; Dong, K. C.; Feld, G. K.; Tang, I. I.; Zhang, T. T.; Williams, E. R.; Berger, J. M.; Krantz, B. A. J. Mol. Biol. 2009, 392, 614.
(20) Milne, J. C.; Furlong, D.; Hanna, P. C.; Wall, J. S.; Collier, R. J. J. Biol. Chem. 1994, 269, 20607.
(21) Ezzell, J. W.; Abshire, T. G. J. Gen. Microbiol. 1992, 138, 543.
(22) Mogridge, J.; Cunningham, K.; Lacy, D. B.; Mourez, M.; Collier, R. J. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7045.
(23) Abrami, L.; Liu, S.; Cosson, P.; Leppla, S. H.; van der Goot, F. G. J. Cell Biol. 2003, 160, 321.
(24) Krantz, B. A.; Finkelstein, A.; Collier, R. J. J. Mol. Biol. 2006, 355, 968.
(25) Basilio, D.; Juris, S. J.; Collier, R. J.; Finkelstein, A. J. Gen. Physiol. 2009, 133, 307.
(26) Firoved, A. M.; Miller, G. F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J. F.; McNally, E. M.; Tang, W.-J.; Leppla, S. H. Am. J. Pathol. 2005, 167, 1309.
(27) Lovchik, J. A.; Drysdale, M.; Koehler, T. M.; Hutt, J. A.; Lyons, C. R. Infect. Immun. 2012, 80, 2414.

105
(28) Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S. M.; Leppla, S. H.; Eichacker, P. Q. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699.
(29) Moayeri, M.; Haines, D.; Young, H. A.; Leppla, S. H. J. Clin. Invest. 2003, 112, 670.
(30) Guo, Q.; Shen, Y.; Zhukovskaya, N. L.; Florián, J.; Tang, W.-J. J. Biol. Chem. 2004, 279, 29427.
(31) Leppla, S. H. Proc. Natl. Acad. Sci. 1982, 79, 3162.
(32) O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Infect. Immun. 1985, 47, 306.
(33) Hoover, D. L.; Friedlander, A. M.; Rogers, L. C.; Yoon, I. K.; Warren, R. L.; Cross, A. S. Infect. Immun. 1994, 62, 4432.
(34) Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Biochem. J. 2000, 352, 739.
(35) Duesbery, N. S.; Webb, C. P.; Leppla, S. H.; Gordon, V. M.; Klimpel, K. R.; Copeland, T. D.; Ahn, N. G.; Oskarsson, M. K.; Fukasawa, K.; Paull, K. D.; Vande Woude, G. F. Science 1998, 280, 734.
(36) Banks, D. J.; Ward, S. C.; Bradley, K. A. Expert Rev. Mol. Med. 2006, 8, 1.
(37) Turk, B. E. Biochem. J. 2007, 402, 405.
(38) Baldari, C. T.; Tonello, F.; Paccani, S. R.; Montecucco, C. Trends Immunol. 2006, 27, 434.
(39) Suzuki, Y. Biol. Pharm. Bull. 2005, 28, 399.
(40) Harris, A.; Cardone, G.; Winkler, D. C.; Heymann, J. B.; Brecher, M.; White, J. M.; Steven, A. C. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19123.
(41) Rossman, J. S.; Lamb, R. A. Virology 2011, 411, 229.
(42) Webster, R. G.; Bean, W. J.; Gorman, O. T.; Chambers, T. M.; Kawaoka, Y. Microbiol. Rev. 1992, 56, 152.
(43) Zambon, M. C. Rev. Med. Virol. 2001, 11, 227.
(44) Bouvier, N. M.; Palese, P. Vaccine 2008, 26, D49.

106
(45) Calder, L. J.; Wasilewski, S.; Berriman, J. A.; Rosenthal, P. B. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 10685.
(46) Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R. H.; Kawaoka, Y. Nature 2006, 439, 490.
(47) Smith, D. J.; Lapedes, A. S.; de Jong, J. C.; Bestebroer, T. M.; Rimmelzwaan, G. F.; Osterhaus, A. D. M. E.; Fouchier, R. A. M. Science 2004, 305, 371.
(48) Garten, R. J.; Davis, C. T.; Russell, C. A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W. M.; Xu, X.; Skepner, E.; Deyde, V.; Okomo-Adhiambo, M.; Gubareva, L.; Barnes, J.; Smith, C. B.; Emery, S. L.; Hillman, M. J.; Rivailler, P.; Smagala, J.; de Graaf, M.; Burke, D. F.; Fouchier, R. A. M.; Pappas, C.; Alpuche-Aranda, C. M.; López-Gatell, H.; Olivera, H.; López, I.; Myers, C. A.; Faix, D.; Blair, P. J.; Yu, C.; Keene, K. M.; Dotson, P. D.; Boxrud, D.; Sambol, A. R.; Abid, S. H.; St. George, K.; Bannerman, T.; Moore, A. L.; Stringer, D. J.; Blevins, P.; Demmler-Harrison, G. J.; Ginsberg, M.; Kriner, P.; Waterman, S.; Smole, S.; Guevara, H. F.; Belongia, E. A.; Clark, P. A.; Beatrice, S. T.; Donis, R.; Katz, J.; Finelli, L.; Bridges, C. B.; Shaw, M.; Jernigan, D. B.; Uyeki, T. M.; Smith, D. J.; Klimov, A. I.; Cox, N. J. Science 2009, 325, 197.
(49) Global report: UNAIDS report on the global AIDS epidemic 2012; Joint United Nations Programme on HIV/AIDS (UNAIDS): Geneva, Switzerland, 2012.
(50) Eckert, D. M.; Kim, P. S. Annu. Rev. Biochem. 2001, 70, 777.
(51) Garg, R.; Gupta, S. P.; Gao, H.; Babu, M. S.; Debnath, A. K.; Hansch, C. Chem. Rev. 1999, 99, 3525.
(52) Berger, E. A.; Murphy, P. M.; Farber, J. M. Annu. Rev. Immunol. 1999, 17, 657.
(53) Doms, R. W.; Peiper, S. C. Virology 1997, 235, 179.
(54) Siciliano, S. J.; Kuhmann, S. E.; Weng, Y.; Madani, N.; Springer, M. S.; Lineberger, J. E.; Danzeisen, R.; Miller, M. D.; Kavanaugh, M. P.; DeMartino, J. A.; Kabat, D. J. Biol. Chem. 1999, 274, 1905.
(55) Jacque, J.-M.; Triques, K.; Stevenson, M. Nature 2002, 418, 435.
(56) Pomerantz, R. J. Nat. Med. 2002, 8, 659.
(57) Joshi, D.; O’Grady, J.; Dieterich, D.; Gazzard, B.; Agarwal, K. Lancet 2011, 377, 1198.
(58) Letvin, N. L. Science 2009, 326, 1196.

107
(59) Baltimore, D. Science 2002, 296, 2297.
(60) Richman, D. D.; Little, S. J.; Smith, D. M.; Wrin, T.; Petropoulos, C.; Wong, J. K. Trans. Am. Clin. Climatol. Assoc. 2004, 115, 289.
(61) Munier, C. M. L.; Andersen, C. R.; Kelleher, A. D. Drugs 2011, 71, 387.
(62) Uberla, K. PLoS Pathog. 2008, 4, e1000114.
(63) Vaccari, M.; Poonam, P.; Franchini, G. Expert Rev. Vaccines 2010, 9, 997.
(64) Mammen, M.; Choi, S.-K.; Whitesides, G. M. Angew. Chemie Int. Ed. 1998, 37, 2754.
(65) Rao, J.; Lahiri, J.; Isaacs, L.; Weis, R. M.; Whitesides, G. M. Science 1998, 280, 708.
(66) Kiessling, L. L.; Pohl, N. L. Chem. Biol. 1996, 3, 71.
(67) Kiessling, L. L.; Strong, L. E.; Gestwicki, J. E. Annu. Rep. Med. Chem. 2000, 35, 321.
(68) Mulder, A.; Huskens, J.; Reinhoudt, D. N. Org. Biomol. Chem. 2004, 2, 3409.
(69) Badjić, J. D.; Nelson, A.; Cantrill, S. J.; Turnbull, W. B.; Stoddart, J. F. Acc. Chem. Res. 2005, 38, 723.
(70) Kiessling, L. L.; Gestwicki, J. E.; Strong, L. E. Angew. Chem. Int. Ed. Engl. 2006, 45, 2348.
(71) Joshi, A.; Vance, D.; Rai, P.; Thiyagarajan, A.; Kane, R. S. Chemistry 2008, 14, 7738.
(72) Vance, D.; Shah, M.; Joshi, A.; Kane, R. S. Biotechnol. Bioeng. 2008, 101, 429.
(73) Vance, D.; Martin, J.; Patke, S.; Kane, R. S. Adv. Drug Deliv. Rev. 2009, 61, 931.
(74) Maheshwari, G.; Brown, G.; Lauffenburger, D. A.; Wells, A.; Griffith, L. G. J. Cell Sci. 2000, 113, 1677.
(75) Wall, S. T.; Saha, K.; Ashton, R. S.; Kam, K. R.; Schaffer, D. V; Healy, K. E. Bioconjug. Chem. 2008, 19, 806.
(76) Matrosovich, M. N.; Mochalova, L. V; Marinina, V. P.; Byramova, N. E.; Bovin, N. V. FEBS Lett. 1990, 272, 209.
(77) Spevak, W.; Nagy, J. O.; Charych, D. H.; Schaefer, M. E.; Gilbert, J. H.; Bednarski, M. D. J. Am. Chem. Soc. 1993, 115, 1146.

108
(78) Mammen, M.; Dahmann, G.; Whitesides, G. M. J. Med. Chem. 1995, 38, 4179.
(79) Choi, S. K.; Mammen, M.; Whitesides, G. M. Chem. Biol. 1996, 3, 97.
(80) Sigal, G. B.; Mammen, M.; Dahmann, G.; Whitesides, G. M. J. Am. Chem. Soc. 1996, 118, 3789.
(81) Kamitakahara, H.; Suzuki, T.; Nishigori, N.; Suzuki, Y.; Kanie, O.; Wong, C.-H. Angew. Chemie Int. Ed. 1998, 37, 1524.
(82) Honda, T.; Yoshida, S.; Arai, M.; Masuda, T.; Yamashita, M. Bioorg. Med. Chem. Lett. 2002, 12, 1929.
(83) Fan, E.; Zhang, Z.; Minke, W. E.; Hou, Z.; Verlinde, C. L. M. J.; Hol, W. G. J. J. Am. Chem. Soc. 2000, 122, 2663.
(84) Kitov, P. I.; Sadowska, J. M.; Mulvey, G.; Armstrong, G. D.; Ling, H.; Pannu, N. S.; Read, R. J.; Bundle, D. R. Nature 2000, 403, 669.
(85) Gargano, J. M.; Ngo, T.; Kim, J. Y.; Acheson, D. W.; Lees, W. J. J. Am. Chem. Soc. 2001, 123, 12909.
(86) Mourez, M.; Kane, R. S.; Mogridge, J.; Metallo, S.; Deschatelets, P.; Sellman, B. R.; Whitesides, G. M.; Collier, R. J. Nat. Biotechnol. 2001, 19, 958.
(87) Nishikawa, K.; Matsuoka, K.; Kita, E.; Okabe, N.; Mizuguchi, M.; Hino, K.; Miyazawa, S.; Yamasaki, C.; Aoki, J.; Takashima, S.; Yamakawa, Y.; Nishijima, M.; Terunuma, D.; Kuzuhara, H.; Natori, Y. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7669.
(88) Mulvey, G. L.; Marcato, P.; Kitov, P. I.; Sadowska, J.; Bundle, D. R.; Armstrong, G. D. J. Infect. Dis. 2003, 187, 640.
(89) Karginov, V. A.; Nestorovich, E. M.; Moayeri, M.; Leppla, S. H.; Bezrukov, S. M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15075.
(90) Basha, S.; Rai, P.; Poon, V.; Saraph, A.; Gujraty, K.; Go, M. Y.; Sadacharan, S.; Frost, M.; Mogridge, J.; Kane, R. S. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13509.
(91) Gujraty, K. V; Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Biomacromolecules 2006, 7, 2082.
(92) Joshi, A.; Saraph, A.; Poon, V.; Mogridge, J.; Kane, R. S. Bioconjug. Chem. 2006, 17, 1265.

109
(93) Polizzotti, B. D.; Kiick, K. L. Biomacromolecules 2006, 7, 483.
(94) Rai, P.; Padala, C.; Poon, V.; Saraph, A.; Basha, S.; Kate, S.; Tao, K.; Mogridge, J.; Kane, R. S. Nat. Biotechnol. 2006, 24, 582.
(95) Polizzotti, B. D.; Maheshwari, R.; Vinkenborg, J.; Kiick, K. L. Macromolecules 2007, 40, 7103.
(96) Rai, P. R.; Saraph, A.; Ashton, R.; Poon, V.; Mogridge, J.; Kane, R. S. Angew. Chem. Int. Ed. Engl. 2007, 46, 2207.
(97) Carlson, C. B.; Mowery, P.; Owen, R. M.; Dykhuizen, E. C.; Kiessling, L. L. ACS Chem. Biol. 2007, 2, 119.
(98) Ohlson, S. Drug Discov. Today 2008, 13, 433.
(99) Lutz, J.-F.; Börner, H. G. Prog. Polym. Sci. 2008, 33, 1.
(100) Hartmann, L.; Börner, H. G. Adv. Mater. 2009, 21, 3425.
(101) Hawker, C. J.; Wooley, K. L. Science 2005, 309, 1200.
(102) Börner, H. G. Macromol. Rapid Commun. 2011, 32, 115.
(103) Dehn, S.; Chapman, R.; Jolliffe, K.; Perrier, S. Polym. Rev. 2011, 51, 214.
(104) Canalle, L. A.; Löwik, D. W. P. M.; van Hest, J. C. M. Chem. Soc. Rev. 2010, 39, 329.
(105) Gauthier, M. A.; Klok, H.-A. Chem. Commun. (Camb). 2008, 2591.
(106) Klok, H.-A. Macromolecules 2009, 42, 7990.
(107) Le Droumaguet, B.; Nicolas, J. Polym. Chem. 2010, 1, 563.
(108) Gauthier, M. A.; Gibson, M. I.; Klok, H.-A. Angew. Chem. Int. Ed. Engl. 2009, 48, 48.
(109) Yanjarappa, M. J.; Gujraty, K. V; Joshi, A.; Saraph, A.; Kane, R. S. Biomacromolecules 2006, 7, 1665.
(110) Gujraty, K. V; Yanjarappa, M. J.; Saraph, A.; Joshi, A.; Mogridge, J.; Kane, R. S. J. Polym. Sci. A. Polym. Chem. 2008, 46, 7246.
(111) Pieters, R. J. Org. Biomol. Chem. 2009, 7, 2013.

110
(112) Top, A.; Kiick, K. L. Adv. Drug Deliv. Rev. 2010, 62, 1530.
(113) Duncan, R. Nat. Rev. Drug Discov. 2003, 2, 347.
(114) Twaites, B.; de las Heras Alarcón, C.; Alexander, C. J. Mater. Chem. 2005, 15, 441.
(115) Kiick, K. L. Science 2007, 317, 1182.
(116) Liu, S.; Maheshwari, R.; Kiick, K. L. Macromolecules 2009, 42, 3.
(117) Gordon, E. J.; Gestwicki, J. E.; Strong, L. E.; Kiessling, L. L. Chem. Biol. 2000, 7, 9.
(118) Kopecek, J.; Kopecková, P.; Minko, T.; Lu, Z. Eur. J. Pharm. Biopharm. 2000, 50, 61.
(119) Kopecek, J.; Kopecková, P. Adv. Drug Deliv. Rev. 2010, 62, 122.
(120) Li, C. Adv. Drug Deliv. Rev. 2002, 54, 695.
(121) Totani, K.; Kubota, T.; Kuroda, T.; Murata, T.; Hidari, K. I.-P. J.; Suzuki, T.; Suzuki, Y.; Kobayashi, K.; Ashida, H.; Yamamoto, K.; Usui, T. Glycobiology 2003, 13, 315.
(122) Zeng, X.; Murata, T.; Kawagishi, H.; Usui, T.; Kobayashi, K. Biosci. Biotechnol. Biochem. 1998, 62, 1171.
(123) Sunderland, C. J.; Steiert, M.; Talmadge, J. E.; Derfus, A. M.; Ã, S. E. B. Drug Dev. Res. 2006, 67, 70.
(124) Natarajan, A.; Xiong, C.-Y.; Albrecht, H.; DeNardo, G. L.; DeNardo, S. J. Bioconjug. Chem. 2005, 16, 113.
(125) Kurfürst, M. M. Anal. Biochem. 1992, 200, 244.
(126) Kastenholz, B. Protein Pept. Lett. 2006, 13, 503.
(127) Ly, M.; Wang, Z.; Laremore, T. N.; Zhang, F.; Zhong, W.; Pu, D.; Zagorevski, D. V; Dordick, J. S.; Linhardt, R. J. Anal. Bioanal. Chem. 2011, 399, 737.
(128) Seelert, H.; Krause, F. Electrophoresis 2008, 29, 2617.
(129) Joshi, A.; Kate, S.; Poon, V.; Mondal, D.; Boggara, M. B.; Saraph, A.; Martin, J. T.; McAlpine, R.; Day, R.; Garcia, A. E.; Mogridge, J.; Kane, R. S. Biomacromolecules 2011, 12, 791.
(130) Young, J. A. T.; Collier, R. J. Annu. Rev. Biochem. 2007, 76, 243.

111
(131) Moayeri, M.; Robinson, T. M.; Leppla, S. H.; Karginov, V. A. Antimicrob. Agents Chemother. 2008, 52, 2239.
(132) Maynard, J. A.; Maassen, C. B. M.; Leppla, S. H.; Brasky, K.; Patterson, J. L.; Iverson, B. L.; Georgiou, G. Nat. Biotechnol. 2002, 20, 597.
(133) Numa, M. M. D.; Lee, L. V; Hsu, C.-C.; Bower, K. E.; Wong, C.-H. Chembiochem 2005, 6, 1002.
(134) Panchal, R. G.; Hermone, A. R.; Nguyen, T. L.; Wong, T. Y.; Schwarzenbacher, R.; Schmidt, J.; Lane, D.; McGrath, C.; Turk, B. E.; Burnett, J.; Aman, M. J.; Little, S.; Sausville, E. A.; Zaharevitz, D. W.; Cantley, L. C.; Liddington, R. C.; Gussio, R.; Bavari, S. Nat. Struct. Mol. Biol. 2004, 11, 67.
(135) Sellman, B. R.; Mourez, M.; Collier, R. J. Science 2001, 292, 695.
(136) Ragle, B. E.; Karginov, V. A.; Bubeck Wardenburg, J. Antimicrob. Agents Chemother. 2010, 54, 298.
(137) Fan, E.; Merritt, E. A.; Verlinde, C. L.; Hol, W. G. Curr. Opin. Struct. Biol. 2000, 10, 680.
(138) Szejtli, J. Chem. Rev. 1998, 98, 1743.
(139) Gu, L. Q.; Braha, O.; Conlan, S.; Cheley, S.; Bayley, H. Nature 1999, 398, 686.
(140) Liao, K.-C.; Mogridge, J. Infect. Immun. 2009, 77, 4455.
(141) Kolb, H. C.; Finn, M.; Sharpless, K. B. Angew. Chemie Int. Ed. 2001, 40, 2004.
(142) Rostovtsev, V. V; Green, L. G.; Fokin, V. V; Sharpless, K. B. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596.
(143) Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057.
(144) Lutz, J.-F. Angew. Chem. Int. Ed. Engl. 2007, 46, 1018.
(145) Gujraty, K.; Sadacharan, S.; Frost, M.; Poon, V.; Kane, R. S.; Mogridge, J. Mol. Pharm. 2005, 2, 367.
(146) Kane, R. S. Langmuir 2010, 26, 8636.
(147) Kramer, R. H.; Karpen, J. W. Nature 1998, 395, 710.

112
(148) Krishnamurthy, V. M.; Semetey, V.; Bracher, P. J.; Shen, N.; Whitesides, G. M. J. Am. Chem. Soc. 2007, 129, 1312.
(149) Knoll, D.; Hermans, J. J. Biol. Chem. 1983, 258, 5710.
(150) Diestler, D.; Knapp, E. Phys. Rev. Lett. 2008, 100, 1.
(151) Centers for Disease Control and Prevention. Early estimates of seasonal influenza vaccine effectiveness--United States, January 2013; MMWR 2013; Vol. 62; U.S. Department of Health and Human Services: Atlanta, GA, 2013.
(152) Osterholm, M. T.; Kelley, N. S.; Sommer, A.; Belongia, E. A. Lancet Infect. Dis. 2012, 12, 36.
(153) Cohen, C.; White, J. M.; Savage, E. J.; Glynn, J. R.; Choi, Y.; Andrews, N.; Brown, D.; Ramsay, M. E. Emerg. Infect. Dis. 2007, 13, 12.
(154) Centers for Disease Control and Prevention. Prevention and control of seasonal influenza with vaccines. Recommendations of the Advisory Committee on Immunization Practices--United States, 2013-2014; MMWR 2013; Vol. 62; U.S. Department of Health and Human Services: Atlanta, GA, 2013.
(155) Laursen, N. S.; Wilson, I. A. Antiviral Res. 2013, 98, 476.
(156) Lee, P. S.; Yoshida, R.; Ekiert, D. C.; Sakai, N.; Suzuki, Y.; Takada, A.; Wilson, I. A. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17040.
(157) Ekiert, D. C.; Bhabha, G.; Elsliger, M.; Friesen, R. H. E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I. A. Science 2009, 324, 246.
(158) Sui, J.; Hwang, W. C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; Wan, H.; Murakami, A.; Yammanuru, A.; Han, T.; Cox, N. J.; Bankston, L. A.; Donis, R. O.; Liddington, R. C.; Marasco, W. A. Nat. Struct. Mol. Biol. 2009, 16, 265.
(159) Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L. L. M.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; Cinatl, J.; ter Meulen, J.; Lasters, I.; Carsetti, R.; Peiris, M.; de Kruif, J.; Goudsmit, J. PLoS One 2008, 3, e3942.
(160) Whitehead, T. A.; Chevalier, A.; Song, Y.; Dreyfus, C.; Fleishman, S. J.; De Mattos, C.; Myers, C. A.; Kamisetty, H.; Blair, P.; Wilson, I. A.; Baker, D. Nat. Biotechnol. 2012, 30, 543.

113
(161) Fleishman, S. J.; Whitehead, T. A.; Ekiert, D. C.; Dreyfus, C.; Corn, J. E.; Strauch, E.-M.; Wilson, I. A.; Baker, D. Science 2011, 332, 816.
(162) Einhauer, A.; Jungbauer, A. J. Biochem. Biophys. Methods 2001, 49, 455.
(163) Wall, S. T.; Saha, K.; Ashton, R. S.; Kam, K. R.; Schaffer, D. V; Healy, K. E. Bioconjug. Chem. 2008, 19, 806.
(164) Conway, A.; Vazin, T.; Spelke, D. P.; Rode, N. A.; Healy, K. E.; Kane, R. S.; Schaffer, D. V. Nat. Nanotechnol. 2013.
(165) Hermanson, G. Bioconjugate Techniques; 2nd ed.; Academic Press: Boston, MA, 2008.
(166) Protein Calculator v3.4. http://protcalc.sourceforge.net/ (accessed Mar 20, 2014).
(167) Rathnayaka, T.; Tawa, M.; Nakamura, T.; Sohya, S.; Kuwajima, K.; Yohda, M.; Kuroda, Y. Biochim. Biophys. Acta 2011, 1814, 1775.
(168) Studier, F. W. Protein Expr. Purif. 2005, 41, 207.
(169) Han, J. C.; Han, G. Y. Anal. Biochem. 1994, 220, 5.
(170) Getz, E. B.; Xiao, M.; Chakrabarty, T.; Cooke, R.; Selvin, P. R. Anal. Biochem. 1999, 273, 73.
(171) Hansen, R. E.; Winther, J. R. Anal. Biochem. 2009, 394, 147.
(172) Burns, J. A.; Butler, J. C.; Moran, J.; Whitesides, G. M. J. Org. Chem. 1991, 56, 2648.
(173) Bagiyan, G.; Koroleva, I. Russ. Chem. Bull. 2003, 52, 1135.
(174) Ochs, C. J.; Such, G. K.; Stadler, B.; Caruso, F. Biomacromolecules 2008, 9, 3389.
(175) Jastrzabek, K. G.; Subiros-Funosas, R.; Albericio, F.; Kolesinska, B.; Kaminski, Z. J. J. Org. Chem. 2011, 76, 4506.
(176) Kamiński, Z. J.; Kolesińska, B.; Kolesińska, J.; Sabatino, G.; Chelli, M.; Rovero, P.; Błaszczyk, M.; Główka, M. L.; Papini, A. M. J. Am. Chem. Soc. 2005, 127, 16912.
(177) Khan, M. N. J. Pharm. Sci. 1984, 73, 1767.
(178) Baldwin, A. D.; Kiick, K. L. Bioconjug. Chem. 2011, 22, 1946.

114
(179) Baldwin, A. D.; Kiick, K. L. Polym. Chem. 2013, 4, 133.
(180) Kalia, J.; Raines, R. T. Bioorg. Med. Chem. Lett. 2007, 17, 6286.
(181) Lin, D.; Saleh, S.; Liebler, D. C. Chem. Res. Toxicol. 2008, 21, 2361.
(182) Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K. L.; Tien, J.; Yu, S.-F.; Mai, E.; Li, D.; Tibbitts, J.; Baudys, J.; Saad, O. M.; Scales, S. J.; McDonald, P. J.; Hass, P. E.; Eigenbrot, C.; Nguyen, T.; Solis, W. A.; Fuji, R. N.; Flagella, K. M.; Patel, D.; Spencer, S. D.; Khawli, L. A.; Ebens, A.; Wong, W. L.; Vandlen, R.; Kaur, S.; Sliwkowski, M. X.; Scheller, R. H.; Polakis, P.; Junutula, J. R. Nat. Biotechnol. 2012, 30, 184.
(183) Manual for the laboratory diagnosis and virological surveillance of influenza; WHO Press: Geneva, Switzerland, 2011.
(184) Klimov, A.; Balish, A.; Veguilla, V.; Sun, H.; Schiffer, J.; Lu, X.; Katz, J. M.; Hancock, K. Influenza Virus Titration, Antigenic Characterization, and Serological Methods for Antibody Detection; Kawaoka, Y.; Neumann, G., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2012; Vol. 865, pp. 25–51.
(185) Patke, S.; Boggara, M.; Maheshwari, R.; Srivastava, S. K.; Arha, M.; Douaisi, M.; Martin, J. T.; Harvey, I. B.; Brier, M.; Rosen, T.; Mogridge, J.; Kane, R. S. Angew. Chemie [Online Early Access]. DOI: 10.1002/ange.201400870. Published Online: Apr 6, 2014. http://onlinelibrary.wiley.com.libproxy.rpi.edu/doi/10.1002/ange.201400870/full (accessed May 7, 2014).
(186) Agrawal, L.; Lu, X.; Qingwen, J.; VanHorn-Ali, Z.; Nicolescu, I. V.; McDermott, D. H.; Murphy, P. M.; Alkhatib, G. J. Virol. 2004, 78, 2277.
(187) Signoret, N.; Pelchen-Matthews, A.; Mack, M.; Proudfoot, A. E.; Marsh, M. J. Cell Biol. 2000, 151, 1281.
(188) Muniz-Medina, V. M.; Jones, S.; Maglich, J. M.; Galardi, C.; Hollingsworth, R. E.; Kazmierski, W. M.; Ferris, R. G.; Edelstein, M. P.; Chiswell, K. E.; Kenakin, T. P. Mol. Pharmacol. 2009, 75, 490.
(189) Foster, T. J. Nat. Rev. Microbiol. 2005, 3, 948.
(190) Galy, R.; Bergeret, F.; Keller, D.; Mourey, L.; Prévost, G.; Maveyraud, L. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2012, 68, 663.

115
(191) Joubert, O.; Viero, G.; Keller, D.; Martinez, E.; Colin, D. A.; Monteil, H.; Mourey, L.; Dalla Serra, M.; Prévost, G. Biochem. J. 2006, 396, 381.
(192) Viero, G.; Cunaccia, R.; Prévost, G.; Werner, S.; Monteil, H.; Keller, D.; Joubert, O.; Menestrina, G.; Dalla Serra, M. Biochem. J. 2006, 394, 217.
(193) Jayasinghe, L.; Bayley, H. Protein Sci. 2005, 14, 2550.
(194) Yamashita, K.; Kawai, Y.; Tanaka, Y.; Hirano, N.; Kaneko, J.; Tomita, N.; Ohta, M.; Kamio, Y.; Yao, M.; Tanaka, I. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 17314.
(195) Gravet, A.; Colin, D. A.; Keller, D.; Girardot, R.; Monteil, H.; Prévost, G.; Giradot, R. FEBS Lett. 1998, 436, 202.
(196) Alonzo, F.; Kozhaya, L.; Rawlings, S. A.; Reyes-Robles, T.; DuMont, A. L.; Myszka, D. G.; Landau, N. R.; Unutmaz, D.; Torres, V. J. Nature 2013, 493, 51.
(197) Reyes-Robles, T.; Alonzo, F.; Kozhaya, L.; Lacy, D. B.; Unutmaz, D.; Torres, V. J. Cell Host Microbe 2013, 14, 453.
(198) Lee, B.; Sharron, M.; Blanpain, C.; Doranz, B. J.; Vakili, J.; Setoh, P.; Berg, E.; Liu, G.; Guy, H. R.; Durell, S. R.; Parmentier, M.; Chang, C. N.; Price, K.; Tsang, M.; Doms, R. W. J. Biol. Chem. 1999, 274, 9617.
(199) Westby, M.; Smith-Burchnell, C.; Mori, J.; Lewis, M.; Mosley, M.; Stockdale, M.; Dorr, P.; Ciaramella, G.; Perros, M. J. Virol. 2007, 81, 2359.
(200) Xiao, Z.; Shangguan, D.; Cao, Z.; Fang, X.; Tan, W. Chemistry 2008, 14, 1769.
(201) Zhang, Y.; Chen, Y.; Han, D.; Ocsoy, I.; Tan, W. Bioanalysis 2010, 2, 907.
(202) Hicke, B. J.; Marion, C.; Chang, Y. F.; Gould, T.; Lynott, C. K.; Parma, D.; Schmidt, P. G.; Warren, S. J. Biol. Chem. 2001, 276, 48644.
(203) Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z. C.; Chen, H. W.; Mallikaratchy, P.; Sefah, K.; Yang, C. J.; Tan, W. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11838.
(204) Yethiraj, A. J. Phys. Chem. B 2009, 113, 1539.
(205) Kolesnikov, A. V.; Kozyr’, A. V.; Shemyakin, I. G. Mol. Genet. Microbiol. Virol. 2012, 27, 49.
(206) Lauridsen, L. H.; Veedu, R. N. Nucleic Acid Ther. 2012, 22, 371.

116
(207) Ellington, A. D.; Szostak, J. W. Nature 1990, 346, 818.
(208) Tuerk, C.; Gold, L. Science 1990, 249, 505.
(209) Mendonsa, S. D.; Bowser, M. T. J. Am. Chem. Soc. 2004, 126, 20.
(210) Cerchia, L.; Ducongé, F.; Pestourie, C.; Boulay, J.; Aissouni, Y.; Gombert, K.; Tavitian, B.; de Franciscis, V.; Libri, D. PLoS Biol. 2005, 3, e123.
(211) Lou, X.; Qian, J.; Xiao, Y.; Viel, L.; Gerdon, A. E.; Lagally, E. T.; Atzberger, P.; Tarasow, T. M.; Heeger, A. J.; Soh, H. T. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2989.
(212) Merino, E. J.; Wilkinson, K. A.; Coughlan, J. L.; Weeks, K. M. J. Am. Chem. Soc. 2005, 127, 4223.
(213) Wilkinson, K. A.; Merino, E. J.; Weeks, K. M. Nat. Protoc. 2006, 1, 1610.
(214) Hendrix, M.; Priestley, E. S.; Joyce, G. F.; Wong, C. H. J. Am. Chem. Soc. 1997, 119, 3641.
(215) Wu, C.; Eder, P. S.; Gopalan, V.; Behrman, E. J. Bioconjug. Chem. 2001, 12, 842.
(216) Wecker, M.; Smith, D.; Gold, L. RNA 1996, 2, 982.
(217) Seelig, B.; Jäschke, A. Bioconjug. Chem. 1999, 10, 371.
(218) Nardese, V.; Longhi, R.; Polo, S.; Sironi, F.; Arcelloni, C.; Paroni, R.; DeSantis, C.; Sarmientos, P.; Rizzi, M.; Bolognesi, M.; Pavone, V.; Lusso, P. Nat. Struct. Biol. 2001, 8, 611.
(219) Nishiyama, Y.; Murakami, T.; Shikama, S.; Kurita, K.; Yamamoto, N. Bioorg. Med. Chem. 2002, 10, 4113.
(220) Ramnarine, E.; DeVico, A. L.; Vigil-Cruz, S. C. Lett. Pept. Sci. 2003, 10, 637.
(221) Secchi, M.; Longhi, R.; Vassena, L.; Sironi, F.; Grzesiek, S.; Lusso, P.; Vangelista, L. Chem. Biol. 2012, 19, 1579.
(222) Lusso, P.; Vangelista, L.; Cimbro, R.; Secchi, M.; Sironi, F.; Longhi, R.; Faiella, M.; Maglio, O.; Pavone, V. FASEB J. 2011, 25, 1230.
(223) Vangelista, L.; Secchi, M.; Lusso, P. Vaccine 2008, 26, 3008.

117
(224) Vangelista, L.; Longhi, R.; Sironi, F.; Pavone, V.; Lusso, P. Biochem. Biophys. Res. Commun. 2006, 351, 664.
(225) Duma, L.; Häussinger, D.; Rogowski, M.; Lusso, P.; Grzesiek, S. J. Mol. Biol. 2007, 365, 1063.
(226) Issafras, H.; Angers, S.; Bulenger, S.; Blanpain, C.; Parmentier, M.; Labbé-Jullié, C.; Bouvier, M.; Marullo, S. J. Biol. Chem. 2002, 277, 34666.
(227) Bennett, L. D.; Fox, J. M.; Signoret, N. Immunology 2011, 134, 246.
(228) Lin, Y.-L.; Mettling, C.; Portales, P.; Reynes, J.; Clot, J.; Corbeau, P. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15590.
(229) Yang, J.; Gitlin, I.; Krishnamurthy, V. M.; Vazquez, J. A.; Costello, C. E.; Whitesides, G. M. J. Am. Chem. Soc. 2003, 125, 12392.
(230) Davis, N. E.; Karfeld-Sulzer, L. S.; Ding, S.; Barron, A. E. Biomacromolecules 2009, 10, 1125.
(231) Wang, Y.; Kiick, K. L. J. Am. Chem. Soc. 2005, 127, 16392.
(232) Liu, S.; Kiick, K. L. Macromolecules 2008, 41, 764.
(233) Kitov, P. I.; Mulvey, G. L.; Griener, T. P.; Lipinski, T.; Solomon, D.; Paszkiewicz, E.; Jacobson, J. M.; Sadowska, J. M.; Suzuki, M.; Yamamura, K.-I.; Armstrong, G. D.; Bundle, D. R. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16837.
(234) Cui, L.; Kitov, P. I.; Completo, G. C.; Paulson, J. C.; Bundle, D. R. Bioconjug. Chem. 2011, 22, 546.

118
APPENDIX A – List of abbreviations
AEM Aminoethyl-maleimide
ANTXR1 Anthrax toxin receptor 1, also referred to by the acronyms ATR, for
“anthrax toxin receptor,” and TEM8, for “tumor endothelial marker 8”
ANTXR2 Anthrax toxin receptor 2, also referred to by the acronym CMG2, for
human capillary morphogenesis protein 2
CCL5 Chemokine (C-C motif) ligand 5, also referred to by the acronym RANTES,
for “regulated on activation, normal T cell expressed and secreted”
CCR5 C-C chemokine receptor type 5
CDC Center for Disease Control
DHA dehydroascorbic acid, an oxidizing agent
DMEM Dulbecco’s modified Eagle’s medium
DMTMM 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride
DTT dithiothreitol, a reducing agent
EC50 Half maximal effective concentration
EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, a water-soluble reagent
for activating carboxylates for attack by amines to create amide bond
linkages
EDTA Ethylenediaminetetraacetic acid
EdTx Anthrax edema toxin (edema factor + protective antigen)
EF Anthrax edema factor
Fab Fragment, antigen binding. The antigen-binding antibody fragment
composed of one constant and one variable domain of each of
the heavy and the light chain
FBS Fetal bovine serum
HA Hemagglutinin
HBS HEPES-buffered saline
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
HIV Human immunodeficiency virus

119
HOBt 1-hydroxybenzotriazole, a peptide-coupling reagent used to create
activated acids, similar to NHS, but more hydrophobic
HyA Hyaluronic acid, a polyanionic, unsulfated glycosaminoglycan
IC50 Half maximal inhibitory concentration
LAIV Live attenuated influenza vaccine
LeTx Anthrax lethal toxin (lethal factor + protective antigen)
LF Anthrax lethal factor
MBS MES-buffered saline
MDCK Madin-Darby Canine-Kidney cells
ME β-mercaptoethanol, a reducing agent
MES 2-(N-morpholino)ethanesulfonic acid
MWCO Molecular weight cut-off
NA Neuraminidase
NHS N-Hydroxysuccinimide, a reagent often used with EDC for creating an
activated acid (an ester with a good leaving group) to create amide bonds
PA Anthrax protective antigen
PA63 63 kDa fragment of anthrax protective antigen
[PA63]7 Toroid-shaped, self-assembled heptamer of PA63
PCR Polymerase chain reaction
PDB Protein data bank
PEG Poly(ethylene glycol)
PGA Poly(L-glutamic acid)
RMS Root-mean square
RNP Ribonucleoprotein complex of influenza virus, composed of viral RNA
coated with nucleoprotein and the RNA-dependent RNA polymerase
TCEP tris(2-carboxyethyl)phosphine, a reducing agent
TCID50 50% tissue culture infective dose (the dose that will infect 50% of
inoculated tissue cultures)
THP tris(hydroxypropyl)phosphine, a reducing agent





























