Embed Size (px)
Citation preview

C
R
M
F
a
b
1
RA
I
Iobmmaa
f
oF
c
0d
ell Calcium (2008) 44, 24—35
journa l homepage: www.e lsev ier .com/ locate /ceca
EVIEW
itochondrial biogenesis and turnover
rancisca Diaza,∗, Carlos T. Moraesa,b,∗∗
Department of Neurology, University of Miami, Miller School of Medicine, 1095 NW 14th Terrace, Miami, FL 33136, USADepartment of Cell Biology and Anatomy, University of Miami, Miller School of Medicine,095 NW 14th Terrace, Miami, FL 33136, USA
eceived 6 October 2007; received in revised form 10 December 2007; accepted 12 December 2007vailable online 18 April 2008
KEYWORDSMitochondria;Biogenesis;PGC-1;Replication;
Summary Mitochondrial biogenesis is a complex process involving the coordinated expressionof mitochondrial and nuclear genes, the import of the products of the latter into the organelleand turnover. The mechanisms associated with these events have been intensively studied inthe last 20 years and our understanding of their details is much improved. Mitochondrial biogen-esis requires the participation of calcium signaling that activates a series of calcium-dependent
Transcription;Turnover
protein kinases that in turn activate transcription factors and coactivators such as PGC-1� thatregulates the expression of genes coding for mitochondrial components. In addition, mitochon-drial biogenesis involves the balance of mitochondrial fission—fusion. Mitochondrial malfunctionor defects in any of the many pathways involved in mitochondrial biogenesis can lead to degen-erative diseases and possibly play an important part in aging.
ts re
snsb
© 2008 Elsevier Ltd. All righ
ntroduction
n eukaryotes, cellular respiration, energy production andther metabolic processes take place in specialized dou-le membrane organelles, containing their own genome, the
itochondria. Mitochondria play a very important role in celletabolism, not only by energy generation but also by beingmajor site for production of reactive oxygen species (ROS)nd being a key player in apoptosis and calcium homeosta-∗ Corresponding author. Tel.: +1 305 243 4232;ax: +1 305 243 3914.∗∗ Corresponding author at: Department of Neurology, Universityf Miami, Miller School of Medicine, 1095 NW 14th Terrace, Miami,L 33136, USA. Tel.: +1 305 243 5858.
E-mail addresses: [email protected] (F. Diaz),[email protected] (C.T. Moraes).
bcihic
M
Fmm
143-4160/$ — see front matter © 2008 Elsevier Ltd. All rights reserved.oi:10.1016/j.ceca.2007.12.004
served.
is. Malfunction of mitochondria has been associated withumerous degenerative diseases and aging [1,2]. Table 1hows some of the diseases associated with mitochondrialiogenesis that are discussed in this paper.
The origin of this organelle in eukaryotes is explainedy the endo-symbiont theory, which proposes that mito-hondria originated from bacteria that were incorporatednto a host cell and maintained during evolution [3,4]. Thisypothesis is backed by substantial experimental evidence,ncluding the similarities between the bacterial and mito-hondrial translational apparatus [5].
itochondrial DNA
ig. 1A illustrates the structure and expression of the humanitochondrial DNA (mtDNA), a double stranded circularolecule of 16,569 bp encoding 2 ribosomal RNAs, 22 trans-

Mitochondrial biogenesis and turnover 25
Table 1 Diseases associated with mitochondrial biogenesis
Mutations Diseases References
mtDNA mutations Multiple respiratory chain defects Reviewed in [1,2,108]Helicase TWINKLE mutations Autosomal dominant progressive external
ophthalmoplegia (PEO). Sensory ataxicneuropathy dysarthria and ophthalmoparesis(SANDO)
[9,10]
Polymerase � mutations Catalytic subunit: PEO, SANDO, mitochondrialneurogastrointestinal encephalomyopathy(MNGIE). Pol2 subunit: PEO
Reviewed in [8]
E1a subunit of pyruvate dehydrogenase (mutationsin mitochondrial targeting sequencing)
Pyruvate dehydrogenase deficiency [46]
Alanine/glyoxylate aminotransferase(mistargeting of peroxisomal protein tomitochondria)
Alanine/glyoxylate aminotransferasedeficiency
[47]
Superoxide dismutase (defect in Import efficiency) Severe alcoholic liver disease [48]Mutation in Tim8 (affects biogenesis of inner
membrane translocase TIM23)Deafness dystonia syndrome orMohr-Tranebjaerg syndrome
[49]
Mutation on Pam18 (motor protein of TIM23complex)
Dilated cardiomyopathy with Ataxia [50]
HSP60 mutations Spastic paraplegia-13 [51]Mitofusin 2 mutations Charcot-Marie-Tooth 2A [90]
al d
(ipos(swnsmu[
tpfwHti
tcmTafa
Opa1 mutations Autosom
fer RNAs and 13 proteins that form part of the multisubunitscomplexes of the oxidative phosphorylation system (7 sub-units of complex I, 1 subunit of complex III, 3 subunits ofcomplex IV and 2 subunits of complex V).
The mammalian mtDNA contains few non-codingsequences, the largest being the D-loop or displacement-loop, which contains promoters and origins of replication(Fig. 1B). Protein-coding genes have no intronic regions[6]. Alkaline gradient centrifugation experiments allowedthe separation of the mtDNA double strands into a heavy(H-strand) and a light (L-strand) due to their differentialcontent of guanosine and cytidine. The H-strand encodesfor the 2 rRNAs, 12 of the polypeptides and 14 of thetRNAs whereas the L-strand encodes for only one of thepolypeptides (ND6) and 8 tRNAs. The genetic code ofmitochondria differs from the nuclear universal code. TheTGA codon codes for tryptophan instead of stop, the AGAand AGG code for stop instead of arginine and the ATA codesfor methionine instead of isoleucine [6].
The mtDNA is associated with several proteins packedin structures denominated nucleoids, which are also asso-ciated with the inner mitochondrial membrane. In thelast few years progress has been made on identifying thecomposition of these structures. Several studies showedthat yeast nucleoids include proteins that bind DNA andare associated with replication and transcription such asthe mitochondrial transcription factor A (TFAM), helicaseTWINKLE, polymerase � (Pol�) and the mitochondrial single-strand binding protein (mtSSB); proteins associated with
metabolism such as the E2 subunit of pyruvate dehydro-genase and �-ketoglutarate dehydrogenase, aconitase; andsome cytoskeletal proteins [7].Mutations in components of the replication machin-ery are associated with mitochondrial depletion syndromes
srta[
ominant optic atrophy [91,92]
reviewed by [8]). Mutations in the PEO1 gene encod-ng the helicase TWINKLE have been reported in someatients with autosomal dominant progressive externalphthalmoplegia (PEO) and in one patient with sen-ory ataxic neuropathy dysarthria and ophthalmoparesisSANDO) [9,10]. In addition, mutations in the catalyticubunit of polymerase � have been associated with aide range of disease such as PEO, SANDO, mitochondrialeurogastrointestinal encephalomyopathy (MNGIE), parkin-onism and Alpers syndrome (reviewed by [8]) whereasutations in the gene POL2 encoding one of the sub-
nits of Pol�, is associated with autosomal dominant PEO11].
Recently, Wang and Bogenhagen [12] found a novel pro-ein, DHX30 (DEAH-box helicase) to be present in nucleoidsurified from HeLa cells [12]. In addition, the authors alsoound ATAD3, a member of the AAA+-ATPases, associatedith nucleoids. Further studies in rat liver mitochondria bye et al. [13] found that ATAD3 binds mtDNA, preferen-ially in the D-loop region, and it appears to be involvedn nucleoid organization and segregation [13,14].
Despite these advances, understanding of the composi-ion and structure of mitochondrial nucleoids is far fromomplete. There are several proteins associated with mtDNAetabolism that are not easily detectable in nucleoids.hese include the mitochondrial transcription factors B1nd B2 and regulators of transcription from the mTERFamily [12]. The mitochondrial transcription factors B1nd B2 have been found to be required for mtDNA tran-
cription (at least one of them) and may also have aole in methylation of DNA or RNA [15]. TFAM seemso be present at relatively high levels, and by itself isble to organize the mtDNA in ‘‘nucleoid-like’’ structures16].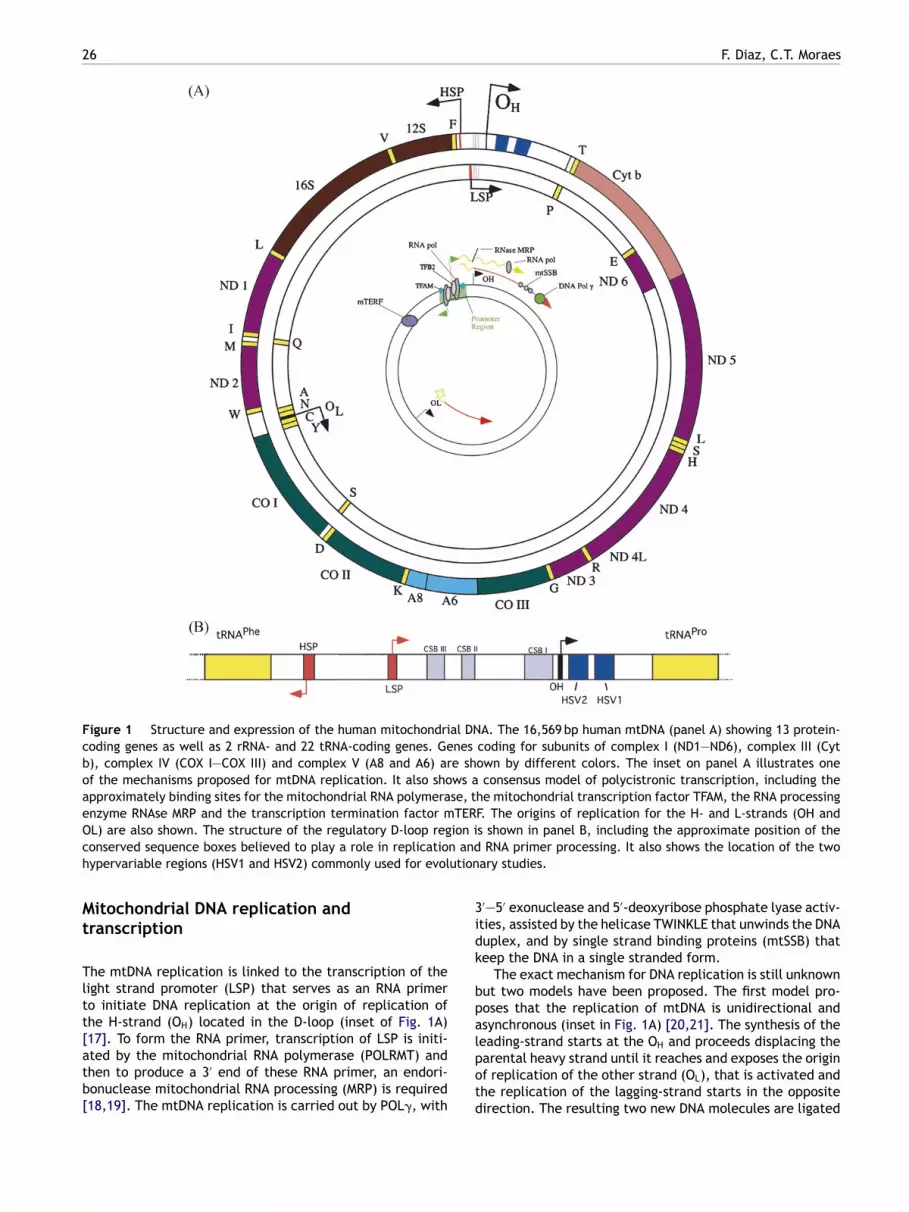
26 F. Diaz, C.T. Moraes
Figure 1 Structure and expression of the human mitochondrial DNA. The 16,569 bp human mtDNA (panel A) showing 13 protein-coding genes as well as 2 rRNA- and 22 tRNA-coding genes. Genes coding for subunits of complex I (ND1—ND6), complex III (Cytb), complex IV (COX I—COX III) and complex V (A8 and A6) are shown by different colors. The inset on panel A illustrates oneof the mechanisms proposed for mtDNA replication. It also shows a consensus model of polycistronic transcription, including theapproximately binding sites for the mitochondrial RNA polymerase, the mitochondrial transcription factor TFAM, the RNA processingenzyme RNAse MRP and the transcription termination factor mTERF. The origins of replication for the H- and L-strands (OH andO ion ic n andh ution
Mt
Tltt[atb[
3idk
bpa
L) are also shown. The structure of the regulatory D-loop regonserved sequence boxes believed to play a role in replicatioypervariable regions (HSV1 and HSV2) commonly used for evol
itochondrial DNA replication andranscription
he mtDNA replication is linked to the transcription of theight strand promoter (LSP) that serves as an RNA primero initiate DNA replication at the origin of replication ofhe H-strand (OH) located in the D-loop (inset of Fig. 1A)
17]. To form the RNA primer, transcription of LSP is initi-ted by the mitochondrial RNA polymerase (POLRMT) andhen to produce a 3′ end of these RNA primer, an endori-onuclease mitochondrial RNA processing (MRP) is required18,19]. The mtDNA replication is carried out by POL�, withlpotd
s shown in panel B, including the approximate position of theRNA primer processing. It also shows the location of the two
ary studies.
′—5′ exonuclease and 5′-deoxyribose phosphate lyase activ-ties, assisted by the helicase TWINKLE that unwinds the DNAuplex, and by single strand binding proteins (mtSSB) thateep the DNA in a single stranded form.
The exact mechanism for DNA replication is still unknownut two models have been proposed. The first model pro-oses that the replication of mtDNA is unidirectional andsynchronous (inset in Fig. 1A) [20,21]. The synthesis of the
eading-strand starts at the OH and proceeds displacing thearental heavy strand until it reaches and exposes the originf replication of the other strand (OL), that is activated andhe replication of the lagging-strand starts in the oppositeirection. The resulting two new DNA molecules are ligated
dTcctw(tpuhca(Tbipbm[
I
Tt((apmthpTtbtoTptmttbfT
I
Mtmtb
Mitochondrial biogenesis and turnover
to form the circular double strand mtDNA [20]. The secondmodel involves the coupled replication of both leading andlagging strands [22].
The mtDNA transcription starts from the promoterspresent in each strand (HSP and LSP) producing polycistronicRNAs. In the HSP there are two sites of initiation, the H1 site,that produces a short transcript containing the 12S RNA,16S RNA and the tRNAleu, and the H2 site, that producesa polycistronic transcript of the length of the full genome,whereas in the LSP there is only one initiation site [23—25].The transcription is carried out by a mitochondrial RNA poly-merase (POLRMT) assisted by a TFAM, that recruits POLRMTto the promoter site, and by mitochondrial transcription fac-tor B1 or B2 (TFB1M and TFB2M; Fig. 1A). The terminationof the H1 short transcript is dependent on the mitochondrialtranscription terminator factor (MTERF) ([26] reviewed in[27,28]). Recently, Park et al. [29] identified the function ofa new transcription terminator factor, MTERF3, as a negativeregulator of transcription [29].
Mitochondrial protein import
Mitochondrial proteomics in humans has estimated approx-imately 1500 proteins with a very small portion of theseproteins encoded by the mtDNA [30,31]. Therefore, themajority of the mitochondrial proteins are encoded by thenuclear DNA and imported into the organelle. Progress onthe knowledge of the components and the mechanisms ofprotein transport into the mitochondria have been spear-headed by studies performed in fungi (Neurospora crassa andSaccharomyces cerevisiae). In many instances, the mam-malian counterparts have not been identified yet.
Distribution of mitochondrial proteins into the appropri-ate compartment in the organelle is a very complex processand the targeting signals for sorting are not obvious. Depend-ing on the mitochondrial location of the protein (matrix,inner mitochondrial membrane, intermembrane space orouter mitochondrial membrane) there are different importpathways. We briefly describe each scenario but for specificdetails refer to recent reviews [32,33].
Matrix proteins
These proteins are synthesized in cytoplasmic ribosomesas precursors containing a targeting signal located in theamino terminal end. This signal is about 10—80 amino acidslong with no consensus in the primary structure and com-posed of hydrophobic and positively charged amino acidsthat can form amphipathic helices. Once the protein isinside the mitochondria this amino terminal sequence iscleaved by mitochondrial processing peptidases to generatethe mature form of the imported protein. There are caseswhere the targeting sequence is located internally or inthe carboxy terminal end of the protein. Protein precursorssynthesized in the cytoplasm are associated with cytoplas-mic chaperones (e.g. Hsp70 and Hsp90), which keep them
in an unfolded state and competent for translocation post-translationally into the mitochondria, while other proteinsseem to be imported cotranslationally [34].The import of the matrix proteins is mediated by twotranslocase complexes, one located in the outer mitochon-
((TTT
27
rial membrane (translocase of the outer membrane orOM) and one in the inner mitochondrial membrane (translo-ase of the inner membrane or TIM23) that function in aoncerted manner. The TOM complex is formed by seven pro-eins (Tom70, Tom40, Tom22, Tom20, Tom7, Tom6 and Tom5)ith specialized functions: (1) surface receptor function
Tom70, Tom22 and Tom20) that recognize the precursor pro-ein and interacts with cytosolic chaperones and (2) importore function (Tom40, Tom5, Tom6 and Tom7) that can formp to three pores in the holocomplex. Once the proteinas crossed the TOM complex it is recognized by the TIM23omplex that is formed by Tim50, Tim23, Tim21, Tim17nd by the motor components that drive the translocationTim44, Tim16/Pam16, Tim14/Pam18, Mge1 and mtHsp70).his translocation process is dependent on ATP and a mem-rane potential. The final step in the matrix import consistsn an active mtHSP70 function. Although it is not clear if theulling of the preprotein into the matrix by mtHSP70 is doney an active conformational change or by a Brownian move-ent ratchet [35], recent evidence has favored the latter
36].
nner membrane proteins
here are three different ways for inner membrane proteinso be translocated and inserted into the inner membrane.1) Proteins that contain several transmembrane domainswith negative charges), are synthesized with a presequencend translocated via TOM—TIM23 to the matrix where theresequence is cleaved and they are ‘‘exported’’ into theembrane by Oxa1 [37]. (2) Proteins that contain one
ransmembrane domain (usually with more charged andydrophobic residues in the carboxy terminus, than theroteins inserted via Oxa1) are translocated through theOM complex, pass to the TIM23 complex, get arrested,ransferred laterally and integrated into the inner mem-rane where the targeting sequence is cleaved. (3) Proteinshat contain hydrophobic residues and an even numberf transmembrane domains are first translocated by theOM complex and then are intercepted by the small Timroteins (small polypeptides Tim9, Tim10 and Tim12, con-aining Cx3C motifs that form hexamers) in the innerembrane space [38] and then transferred to another
ranslocase, the TIM22 complex in the inner membranehat inserts the protein into the membranes in a mem-rane potential-dependent manner. The TIM22 complex isormed by three membrane proteins Tim54, Tim22 andim18 [39].
mport of outer membrane proteins
any of these proteins contain a �-barrel structure andhey use the TOM complex to pass through the outerembrane, then are guided by the small Tim proteins
o another complex located also in the outer mem-rane, the translocase of outer membrane �-barrel proteins
TOB) or sorting and assembly machinery (SAM) complexTOB/SAM), that inserts the protein into the membrane. TheOB/SAM complex is formed by Tom55 (also called Sam50 orom50), Tob38 (Sam35 or Tom38) and by Mas37 (Sam37 orom37) [40,41].
2
I
Siarat[m
tdtamlo[dTpdc
ifz
M
RpctTcncowtt
FdtbiahTasMt
8
mport of intermembrane space proteins
ome proteins contain two signal sequences allowing thensertion into the inner membrane via TOM—TIM23 complexnd then the mature protein is formed by proteolysis andeleased into the inner membrane, whereas other proteinsre translocated by the TOM complex and then folded inhe intermembrane space by the help of Mia40 and Erv142—45]. Fig. 2 summarizes the current knowledge of theitochondrial import machinery.There are several diseases associated with errors in
he import of mitochondrial proteins such as pyruvateehydrogenase deficiency (mutations in the mitochondrialargeting sequence of subunit E1� [46]), Alanine/glyoxylateminotransferase deficiency (peroxisomal protein that isistargeted into the mitochondria [47]), severe alcoholic
iver disease (caused by defects in the import efficiencyf the genetic dimorphic manganese superoxide dismutase48]), Deafness Dystonia Syndrome or Mohr-Tranebjaerg Syn-
rome (caused by mutations in one of the small Tim proteins,im8, that result in an impaired biogenesis of the TIM23 com-lex [49]), Dilated cardiomyopathy with Ataxia (caused byefects in Pam18, one of the motor proteins of the TIM23omplex [50]) and Spastic Paraplegia-13 (caused by defectsplarr
igure 2 Protein import into mitochondria. Nuclear encoded prepending on their final destination. Matrix proteins, synthesizederminus are translocated through TOM and TIM23—Hsp70/Mge1 comy mitochondrial proteases to render the mature form. Inner membran the latter, transferred laterally into the membrane and have theire imported into the matrix by TOM and TIM23 and inserted intoydrophobic inner membrane proteins with even number of transmeIM22 via small Tim proteins and inserted into the membrane. Intermnd laterally transferred to inner membrane, where the presequencepace. Other IMS proteins are translocated through the TOM complexia40 and Erv1 chaperones. Outer membrane proteins are translocat
o the TOB/SAM complex and inserted into the membrane. See text
F. Diaz, C.T. Moraes
n the mitochondrial Hsp60 causing disruption in proteinolding in the matrix [51]). For details see review by MacKen-ie and Payne [52].
itochondrial-nuclear communication
esponses to different stimuli such as changes in tem-erature, nutritional state, energy production, hormonalhanges as well as normal homeostasis in mammals, requirehe action of both mitochondrial and nuclear genomes.hese responses involve signals from the nucleus to mito-hondria as well as from mitochondria to the nucleus. Theuclear to mitochondrial signal involves the activation ofertain transcription factors that regulate the expressionf mitochondrial genes leading to mitochondrial biogenesis,hereas the mitochondrial to nucleus signals (also referred
o as ‘‘retrograde signaling’’) sense the mitochondrial func-ional state (e.g. loss of function by decrease in membrane
otential, defects in oxidative phosphorylation, lower ATPevels, uncoupling, mtDNA mutations and hypoxia) [53]. Inddition, mitochondrial stress response to heat shock oreduced levels of certain proteins can also trigger a nuclearesponse [54]. The mechanisms involved in the retrogradeoteins are imported into mitochondria by different pathwaysas precursors containing a targeting sequence in the amino
plexes and once in the matrix the targeting sequence is cleavedne proteins are translocated by TOM and TIM23 but get arrestedr targeting sequence cleaved. Other inner membrane proteinsthe inner membrane with the help of Oxa1. In addition, verymbrane domains are translocated through TOM, then passed toembrane space (IMS) proteins are transported via TOM—TIM23is cleaved and the protein is released into the intermembraneand then folded in the intermembrane space with the help of
ed through the TOM complex, guided by the small Tim proteinsfor more details.

Apettbts
ptawid(eImfipl
csstpthpantlmdaiclco
p[c
ctbswt[
c
Mitochondrial biogenesis and turnover
signaling are not well understood, although in yeast a num-ber of transcriptional activators mediating this signalingpathway have been identified [55]. As described below,calcium participates in both antero- and retrograde mito-chondrial nuclear signaling in response to stimuli or stress(recently reviewed in [56]).
Mitochondrial biogenesis
Although the complete pathway controlling mitochondrialbiogenesis has not been elucidated, great progress in iden-tifying key players has been obtained in the last fewyears. The expression of mitochondrial proteins encodedin the nuclear genome participating in oxidative phospho-rylation, heme biosynthesis, mitochondrial protein import,and mtDNA transcription and replication, is regulated bytranscription factors and transcriptional coactivators. Themost prevalent transcription factors activating promotersof mitochondrial genes are the nuclear respiratory factor1 and 2 (NRF-1 and NRF-2) [57,58] and the estrogen-relatedreceptor (ERR�) that work in concert with transcriptionalcoactivators of the peroxisome proliferator-activated recep-tor �-coactivator-1 (PGC-1) family, PGC-1�, PGC-1� and PRC(PGC-1-related coactivator) [59]. This family of coactiva-tors regulates several metabolic pathways such as cellularrespiration, thermogenesis and hepatic glucose metabolism(reviewed in [60—62]). Although these coactivators stimu-late mitochondrial biogenesis, PGC-1� is mainly involved inthe regulation of gluconeogenesis and PGC-1� in the regu-lation of �-oxidation of fatty acids [63,64]. Srivastava et al.[65] showed that overexpression of PGC-1� and PGC-1� wasassociated with an improvement of respiration in cells withdeleterious mtDNA mutations [65].
Physiological mechanisms involved in PGC-1� regula-tion have been extensively studied in the last few years.Transgenic mice overexpressing PGC-1� showed mitochon-drial proliferation in skeletal muscle and switch in fibertype composition from the more prominent type II (gly-colytic) to type I (oxidative) [66]. In skeletal muscle,endurance exercise induces an increase in mitochondrialmass that is mediated by the increase in intracellularcalcium levels during fiber contraction [67]. This pro-cess is regulated by the orchestrated expression of bothnuclear and mitochondrial genes and the main pathwayinvolved includes the activation and action of PGC-1�.
Increased levels of intracellular calcium activatescytoplasmic protein kinases such as calcium/calmodulin-dependent protein kinase (CaMK) or protein kinase C (PKC)that in turn stimulate the expression of several nuclearand mitochondrial genes [68]. Studies in vitro in isolatedrat epitrochlearis muscle where an intracellular increaseof calcium was achieved by incubation with low concen-trations of caffeine (concentrations of 3.5 mM caffeinewere low enough to not induce muscle contraction but toincrease PGC-1� levels) and using specific kinase inhibitors
showed that the activation of CaMK occurs upstream ofthe activation of p38 mitogen-activated protein kinase(p38 MAPK) [69]. Moreover, p38 MAPK is responsible forphosphorylating, activating and inducing PGC-1� expres-sion [69,70]. In addition, Jager et al. [71] showed thataaDst
29
MP-activated protein kinase (AMPK) directly binds andhosphorylates PGC-1� [71]. The induction of PGC-1�xpression is also mediated through activating transcrip-ion factor 2 (ATF-2) that binds to the PGC-1� promoter inhe cyclic AMP-response binding protein (CREB) element-inding site [72,73]. Fig. 3 shows a general diagram ofhe calcium signaling involved in mitochondrial biogene-is.
The effect of calcium on the expression of mitochondrialroteins is not confined to muscle. Au et al. [74] had shownhe increase of COX1, ATPase6, SDH and TFAM (transcripts well as protein levels) in human granulosa cells treatedith the calcium ionophore A23187 and this effect was inhib-
ted by EGTA [74]. Likewise, Mercy et al. [75] using mtDNAepleted (rho0) cell lines, L929 (mouse fibroblast) and 143Bhuman osteosarcoma) showed that mitochondrial biogen-sis is mediated by calcium signaling through CREB [75].n addition to phosphorylation, another post-translationalodification that regulates PGC-1� activity has been identi-ed. Namely, deacetylation mediated by SIRT1 during fastingromotes the expression of mitochondrial genes involved inipid oxidation [76].
The control of mitochondrial biogenesis during the cellycle in mammalian cells remains unknown and controver-ial. Recently, Lee et al. [77] investigated mitochondria inynchronized cultures of HeLa cells at different points duringhe cell cycle and showed evidence that mitochondrial mor-hology varies during the cell cycle. During interphase, theypical mitochondrial tubular network was observed witheterogeneity in mitochondrial size and morphology, butrogression into the mitotic phase disrupted this networknd the mitochondrial population became more homoge-eous [77]. Morphological changes of mitochondria duringhe cell cycle have been reported previously [78]. The tubu-ar network seen during interphase was associated withicrotubules whereas in mitosis, the fragmented mitochon-ria no longer interacted with microtubules but rather withctin. The mitochondrial mass and membrane potentialncreased during the progression of G1 to mitosis and afterell division these parameters were reduced again. Theevels of mtDNA also increased in G1/S to G2 transition con-omitant with increase of NRF-1 levels although the levelsf TFAM and PRC were not altered [77].
Mitochondrial biogenesis, can also be stimulated by otherathways like reactive oxygen species [79], nitric oxide80,81] and hypoxia [82], which can be caused by a mito-hondrial disorder or ischemic insults.
During heat shock or in the case of a respiratory defi-iency caused by the decrease of one subunit of one ofhe respiratory complexes, an accumulation of unassem-led subunits may occur, leading mitochondria into stressignals and a cellular unfolded protein response (UPR)hereby the cell responds by increasing the amount of pro-
eins required for quality control (chaperones or proteases)54].
Mitochondrial biogenesis is commonly stimulated in mito-hondrial disorders, a group of diseases with heterogeneous
rray of clinical manifestations. These disorders are associ-ted with either mutations in the mtDNA or in the nuclearNA and often affect tissues with high energetic demandsuch as brain, heart and skeletal muscle [1,2]. In addi-ion, alterations of mtDNA (both large deletions and point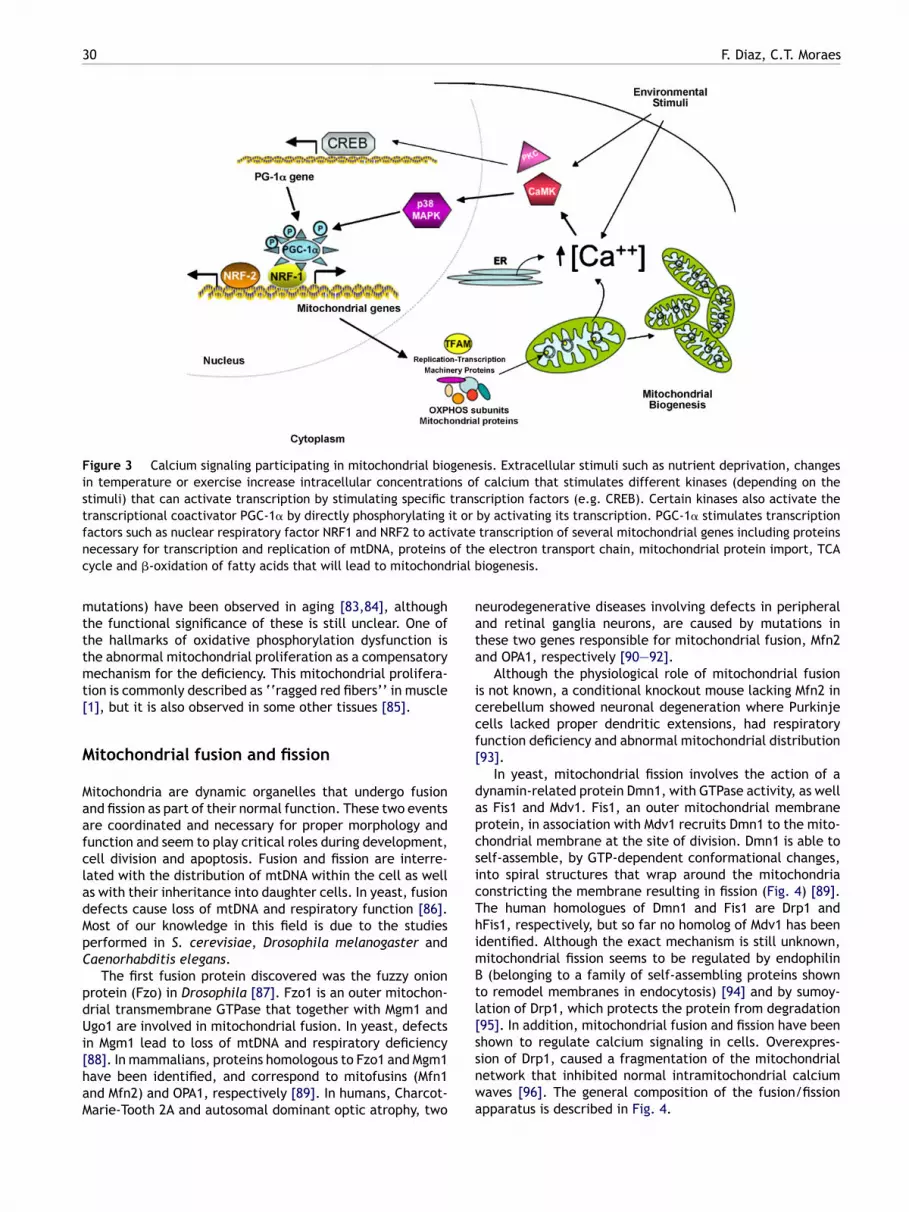
30 F. Diaz, C.T. Moraes
Figure 3 Calcium signaling participating in mitochondrial biogenesis. Extracellular stimuli such as nutrient deprivation, changesin temperature or exercise increase intracellular concentrations of calcium that stimulates different kinases (depending on thestimuli) that can activate transcription by stimulating specific transcription factors (e.g. CREB). Certain kinases also activate thetranscriptional coactivator PGC-1� by directly phosphorylating it or by activating its transcription. PGC-1� stimulates transcriptionf vaten of thc rial b
mtttmt[
M
MaafcladMpC
pdUi[haM
nata
iccf[
dapcsicThimBtl[
actors such as nuclear respiratory factor NRF1 and NRF2 to actiecessary for transcription and replication of mtDNA, proteinsycle and �-oxidation of fatty acids that will lead to mitochond
utations) have been observed in aging [83,84], althoughhe functional significance of these is still unclear. One ofhe hallmarks of oxidative phosphorylation dysfunction ishe abnormal mitochondrial proliferation as a compensatoryechanism for the deficiency. This mitochondrial prolifera-
ion is commonly described as ‘‘ragged red fibers’’ in muscle1], but it is also observed in some other tissues [85].
itochondrial fusion and fission
itochondria are dynamic organelles that undergo fusionnd fission as part of their normal function. These two eventsre coordinated and necessary for proper morphology andunction and seem to play critical roles during development,ell division and apoptosis. Fusion and fission are interre-ated with the distribution of mtDNA within the cell as wells with their inheritance into daughter cells. In yeast, fusionefects cause loss of mtDNA and respiratory function [86].ost of our knowledge in this field is due to the studieserformed in S. cerevisiae, Drosophila melanogaster andaenorhabditis elegans.
The first fusion protein discovered was the fuzzy onionrotein (Fzo) in Drosophila [87]. Fzo1 is an outer mitochon-rial transmembrane GTPase that together with Mgm1 andgo1 are involved in mitochondrial fusion. In yeast, defects
n Mgm1 lead to loss of mtDNA and respiratory deficiency88]. In mammalians, proteins homologous to Fzo1 and Mgm1ave been identified, and correspond to mitofusins (Mfn1nd Mfn2) and OPA1, respectively [89]. In humans, Charcot-arie-Tooth 2A and autosomal dominant optic atrophy, two
ssnwa
transcription of several mitochondrial genes including proteinse electron transport chain, mitochondrial protein import, TCAiogenesis.
eurodegenerative diseases involving defects in peripheralnd retinal ganglia neurons, are caused by mutations inhese two genes responsible for mitochondrial fusion, Mfn2nd OPA1, respectively [90—92].
Although the physiological role of mitochondrial fusions not known, a conditional knockout mouse lacking Mfn2 inerebellum showed neuronal degeneration where Purkinjeells lacked proper dendritic extensions, had respiratoryunction deficiency and abnormal mitochondrial distribution93].
In yeast, mitochondrial fission involves the action of aynamin-related protein Dmn1, with GTPase activity, as wells Fis1 and Mdv1. Fis1, an outer mitochondrial membranerotein, in association with Mdv1 recruits Dmn1 to the mito-hondrial membrane at the site of division. Dmn1 is able toelf-assemble, by GTP-dependent conformational changes,nto spiral structures that wrap around the mitochondriaonstricting the membrane resulting in fission (Fig. 4) [89].he human homologues of Dmn1 and Fis1 are Drp1 andFis1, respectively, but so far no homolog of Mdv1 has beendentified. Although the exact mechanism is still unknown,itochondrial fission seems to be regulated by endophilin(belonging to a family of self-assembling proteins shown
o remodel membranes in endocytosis) [94] and by sumoy-ation of Drp1, which protects the protein from degradation95]. In addition, mitochondrial fusion and fission have been
hown to regulate calcium signaling in cells. Overexpres-ion of Drp1, caused a fragmentation of the mitochondrialetwork that inhibited normal intramitochondrial calciumaves [96]. The general composition of the fusion/fissionpparatus is described in Fig. 4.
Mitochondrial biogenesis and turnover 31
Figure 4 Mitochondrial fission and fusion. In yeast, mitochondria fission involves the action of Dmn1, that can self-assemble intopolymeric spirals and is recruited into the mitochondrial membrane by Fis1 and Mdv1. Dmn1 polymers wrap around the organelleand constrict the membrane until fission occurs. In humans, Drp1 and hFis1 are the homologs of Dmn1 and Fis1 whereas no homologof Mdv1 has been found yet. Mitochondrial fusion involves the interaction of Fzo1/Ugo1 molecules located in the outer membrane oftwo mitochondria until outer membrane fuses, and then inner membrane fusion occurs through the interaction of Mgm1 molecules.In mammals, there are two homologs of Fzo1, the mitofusins 1 and 2 (Mfn1 and Mfn2) whereas the homolog of Mgm1 is OPA1. MIM,
bra
tuapacb
nab[di
C
Mcidamtttpim
mitochondrial inner membrane: MOM, mitochondrial outer memdescribed in Ref. [80].
Mitochondrial turnover
Defective mitochondria have to be eliminated to maintaincellular homeostasis. Menzies and Gold [97] estimated thatthe turnover of mitochondria in liver, heart and brain is 9.3,17.5 and 24.4 days, respectively [97]. Autophagy appears tobe the main pathway for mitochondrial turnover (mitophagy)although it has been shown that ubiquitin-mediated prote-olysis plays a major role in the elimination of the paternalmitochondria during fertilization [98,99].
Mitophagy consist in the sequestration of the defectiveorganelle into a double membrane phagosome that is laterfused with lysosomes forming autolysosomes, where thecontents are degraded and can be recycled (reviewed in[100,101]). In yeast, several genes involved in autophagy(ATG genes) have been described and their mechanism ofaction requires two novel conjugation systems that aresomehow interrelated (reviewed in [102]). In one of thesystems, Atg12, a small protein, is conjugated to a lysineon Atg5 by Atg7 and Atg10, in a way similar to the E1and E2 enzymes of the ubiquitin system, and once Atg5 istagged with Atg12 it forms a complex with Atg16 [103]. Inthe other conjugation system, Atg8, also a small protein,is conjugated to phosphatidylethanolamine (PE, membranephospholipid) by Atg7 and Atg3 [104]. In addition to the con-jugated systems, two protein kinase complexes are requiredfor autophagy. The protein kinase complexes are the Atg1kinase complex and the phosphatidylinositol 3 kinase (PI3K)complex both formed by several proteins. The four systems
described above (protein conjugation and kinase complexes)work in concert to produce the autophagosome.The mitophagy process starts by the recruitment andtransient association of the conjugated protein (Atg5—Atg12conjugated) into a precursor vesicle (PE—Atg8 conjugated)
C
N
ne. See text for more details. The cartoon is based on models
hat elongates along the perimeter of the mitochondrionntil both ends fuse forming the double membrane of theutophagosome. The PI3K complex is required for the initialrocess of autophagosome formation and its inhibition stopsutophagy [105]. The function of the Atg1 kinase is not verylear but appears to control the autophagy response inducedy nutrient deprivation [106].
The process of how an individual mitochondrion is recog-ized for degradation is not known but appears that therere some proteins such as Uth1 (mitochondrial outer mem-rane protein) involved in the selectivity of the process107]. For more details, this topic (calcium and mitochon-ria autophagy) is covered in another chapter of this specialssue of Cell Calcium.
onclusions
itochondrial biogenesis is a very complex cellular pro-ess that requires the coordination of several mechanismsnvolving nuclear-mitochondrial communication, mitochon-rial protein expression and import, mtDNA gene expression,ssembly of multisubunit enzyme complexes, regulation ofitochondrial fission and fusion as well as mitochondrial
urnover in response to various stimuli. Disruption of any ofhese processes can lead to defective mitochondrial func-ion and therefore to a disease state. Many of the pathwaysarticipating in mitochondrial biogenesis require the partic-pation of calcium as a signaling molecule, as discussed inore detail in the subsequent chapters of this book.
onflict of interest statement
one.

3
A
OtD
R
2
cknowledgments
ur work was supported by grants from the National Insti-utes of Health (NINDS, NCI and NEI), and the Muscularystrophy Association.
eferences
[1] S. DiMauro, E.A. Schon, Mitochondrial respiratory-chain dis-eases, N. Engl. J. Med. 348 (2003) 2656—2668.
[2] D. Wallace, Mitochondrial defects in neurodegenerative dis-ease, Ment. Retard. Dev. Disabil. Res. Rev. 7 (2001) 158—166.
[3] L. Margulis, Symbiotic theory of the origin of eukaryoticorganelles; criteria for proof, Symp. Soc. Exp. Biol. (1975)21—38.
[4] L. Margulis, D. Bermudes, Symbiosis as a mechanism of evo-lution: status of cell symbiosis theory, Symbiosis 1 (1985)101—124.
[5] T.E. Shutt, M.W. Gray, Bacteriophage origins of mitochon-drial replication and transcription proteins, Trends Genet. 22(2006) 90—95.
[6] S. Anderson, A.T. Bankier, B.G. Barrell, M.H. de Bruijn,A.R. Coulson, J. Drouin, I.C. Eperon, D.P. Nierlich, B.A.Roe, F. Sanger, P.H. Schreier, A.J. Smith, R. Staden, I.G.Young, Sequence and organization of the human mitochon-drial genome, Nature 290 (1981) 457—465.
[7] B.A. Kaufman, S.M. Newman, R.L. Hallberg, C.A. Slaugh-ter, P.S. Perlman, R.A. Butow, In organello formaldehydecrosslinking of proteins to mtDNA: identification of bifunc-tional proteins, Proc. Natl. Acad. Sci. U.S.A. 97 (2000)7772—7777.
[8] W.C. Copeland, Inherited mitochondrial diseases of DNA repli-cation, Annu. Rev. Med. 59 (2008).
[9] J.N. Spelbrink, F.Y. Li, V. Tiranti, K. Nikali, Q.P. Yuan, M. Tariq,S. Wanrooij, N. Garrido, G. Comi, L. Morandi, L. Santoro, A.Toscano, G.M. Fabrizi, H. Somer, R. Croxen, D. Beeson, J.Poulton, A. Suomalainen, H.T. Jacobs, M. Zeviani, C. Larsson,Human mitochondrial DNA deletions associated with muta-tions in the gene encoding Twinkle, a phage T7 gene 4-likeprotein localized in mitochondria, Nat. Genet. 28 (2001)223—231.
[10] G. Hudson, M. Deschauer, K. Busse, S. Zierz, P.F. Chinnery,Sensory ataxic neuropathy due to a novel C10Orf2 muta-tion with probable germline mosaicism, Neurology 64 (2005)371—373.
[11] M.J. Longley, S. Clark, C. Yu Wai Man, G. Hudson, S.E. Durham,R.W. Taylor, S. Nightingale, D.M. Turnbull, W.C. Copeland, P.F.Chinnery, Mutant POLG2 disrupts DNA polymerase gamma sub-units and causes progressive external ophthalmoplegia, Am.J. Hum. Genet. 78 (2006) 1026—1034.
[12] Y. Wang, D.F. Bogenhagen, Human mitochondrial DNAnucleoids are linked to protein folding machinery andmetabolic enzymes at the mitochondrial inner membrane, J.Biol. Chem. 281 (2006) 25791—25802.
[13] J. He, C.-C. Mao, A. Reyes, H. Sembongi, M. Di Re, C.Granycome, A.B. Clippingdale, I.M. Fearnley, M. Harbour, A.J.Robinson, S. Reichelt, J.N. Spelbrink, J.E. Walker, I.J. Holt,The AAA+ protein ATAD3 has displacement loop binding prop-erties and is involved in mitochondrial nucleoid organization,J. Cell Biol. 176 (2007) 141—146.
[14] I.J. Holt, J. He, C.C. Mao, J.D. Kirk-Up, P. Martinsson, H.
Sembongi, A. Reyes, J.N. Spelbrink, Mammalian mitochon-drial nucleoids: organizing an independently minded genome,Mitochondrion 7 (2007) 311—321.[15] J. Cotney, Z. Wang, G.S. Shadel, Relative abundance of thehuman mitochondrial transcription system and distinct roles
F. Diaz, C.T. Moraes
for h-mtTFB1 and h-mtTFB2 in mitochondrial biogenesis andgene expression, Nucleic Acids Res. 35 (2007) 4042—4054.
[16] B.A. Kaufman, N. Durisic, J.M. Mativetsky, S. Costantino, M.A.Hancock, P. Grutter, E.A. Shoubridge, The mitochondrial tran-scription factor TFAM coordinates the assembly of multipleDNA molecules into nucleoid-like structures, Mol. Biol. Cell18 (2007) 3225—3236.
[17] D.D. Chang, D.A. Clayton, Priming of human mitochondrialDNA replication occurs at the light-strand promoter, Proc.Natl. Acad. Sci. U.S.A. 82 (1985) 351—355.
[18] D.D. Chang, R.P. Fisher, D.A. Clayton, Roles for a pro-moter and RNA processing in the synthesis of mitochondrialdisplacement-loop strands, Biochim. Biophys. Acta 909 (1987)85—91.
[19] D.D. Chang, D.A. Clayton, A mammalian mitochondrial RNAprocessing activity contains nucleus-encoded RNA, Science235 (1987) 1178—1184.
[20] D.A. Clayton, Replication of animal mitochondrial DNA, Cell28 (1982) 693—705.
[21] T.A. Brown, C. Cecconi, A.N. Tkachuk, C. Bustamante, D.A.Clayton, Replication of mitochondrial DNA occurs by stranddisplacement with alternative light-strand origins, not via astrand-coupled mechanism, Genes Dev. 19 (2005) 2466—2476.
[22] I.J. Holt, H.E. Lorimer, H.T. Jacobs, Coupled leading- andlagging-strand synthesis of mammalian mitochondrial DNA,Cell 100 (2000) 515—524.
[23] J. Montoya, T. Christianson, D. Levens, M. Rabinowitz,G. Attardi, Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrialDNA, Proc. Natl. Acad. Sci. U.S.A. 79 (1982) 7195—7199.
[24] J. Montoya, G.L. Gaines, G. Attardi, The pattern of tran-scription of the human mitochondrial rRNA genes reveals twooverlapping transcription units, Cell 34 (1983) 151—159.
[25] G.S. Shadel, D.A. Clayton, Mitochondrial DNA maintenance invertebrates, Annu. Rev. Biochem. 66 (1997) 409—435.
[26] M. Martin, J. Cho, A.J. Cesare, J.D. Griffith, G. Attardi, Ter-mination factor-mediated DNA loop between termination andinitiation sites drives mitochondrial rRNA synthesis, Cell 123(2005) 1227—1240.
[27] M. Falkenberg, N.G. Larsson, C.M. Gustafsson, DNA replica-tion and transcription in mammalian mitochondria, Annu. Rev.Biochem. 76 (2007) 679—699.
[28] N.D. Bonawitz, D.A. Clayton, G.S. Shadel, Initiation andbeyond: multiple functions of the human mitochondrial tran-scription machinery, Mol. Cell 24 (2006) 813—825.
[29] C.B. Park, J. Asin-Cayuela, Y. Camara, Y. Shi, M. Pellegrini,M. Gaspari, R. Wibom, K. Hultenby, H. Erdjument-Bromage,P. Tempst, M. Falkenberg, C.M. Gustafsson, N.G. Larsson,MTERF3 is a negative regulator of mammalian mtDNA tran-scription, Cell 130 (2007) 273—285.
[30] M.F. Lopez, B.S. Kristal, E. Chernokalskaya, A. Lazarev, A.I.Shestopalov, A. Bogdanova, M. Robinson, High-throughputprofiling of the mitochondrial proteome using affinityfractionation and automation, Electrophoresis 21 (2000)3427—3440.
[31] S. Calvo, M. Jain, X. Xie, S.A. Sheth, B. Chang, O.A. Gold-berger, A. Spinazzola, M. Zeviani, S.A. Carr, V.K. Mootha,Systematic identification of human mitochondrial diseasegenes through integrative genomics, Nat. Genet. 38 (2006)576—582.
[32] W. Neupert, J.M. Herrmann, Translocation of proteins intomitochondria, Annu. Rev. Biochem. 76 (2007) 723—749.
[33] M. van der Laan, M. Rissler, P. Rehling, Mitochondrial prepro-tein translocases as dynamic molecular machines, FEMS YeastRes. 6 (2006) 849—861.
[34] W. Neupert, Protein import into mitochondria, Annu. Rev.Biochem. 66 (1997) 863—917.

Mitochondrial biogenesis and turnover
[35] H.C. Schneider, J. Berthold, M.F. Bauer, K. Dietmeier, B.Guiard, M. Brunner, W. Neupert, Mitochondrial Hsp70/MIM44complex facilitates protein import, Nature 371 (1994)768—774.
[36] T. Sato, M. Esaki, J.M. Fernandez, T. Endo, Comparison of theprotein-unfolding pathways between mitochondrial proteinimport and atomic-force microscopy measurements, Proc.Natl. Acad. Sci. U.S.A. 102 (2005) 17999—18004.
[37] F.U. Hartl, J. Ostermann, B. Guiard, W. Neupert, Successivetranslocation into and out of the mitochondrial matrix: tar-geting of proteins to the intermembrane space by a bipartitesignal peptide, Cell 51 (1987) 1027—1037.
[38] C.T. Webb, M.A. Gorman, M. Lazarou, M.T. Ryan, J.M. Gul-bis, Crystal structure of the mitochondrial chaperone TIM9.10reveals a six-bladed alpha-propeller, Mol. Cell 21 (2006)123—133.
[39] C. Sirrenberg, M.F. Bauer, B. Guiard, W. Neupert, M. Brun-ner, Import of carrier proteins into the mitochondrial innermembrane mediated by Tim22, Nature 384 (1996) 582—585.
[40] S.A. Paschen, T. Waizenegger, T. Stan, M. Preuss, M. Cyrklaff,K. Hell, D. Rapaport, W. Neupert, Evolutionary conservationof biogenesis of beta-barrel membrane proteins, Nature 426(2003) 862—866.
[41] N. Wiedemann, V. Kozjak, A. Chacinska, B. Schonfisch, S.Rospert, M.T. Ryan, N. Pfanner, C. Meisinger, Machinery forprotein sorting and assembly in the mitochondrial outer mem-brane, Nature 424 (2003) 565—571.
[42] M. Naoe, Y. Ohwa, D. Ishikawa, C. Ohshima, S. Nishikawa,H. Yamamoto, T. Endo, Identification of Tim40 that mediatesprotein sorting to the mitochondrial intermembrane space, J.Biol. Chem. 279 (2004) 47815—47821.
[43] N. Mesecke, N. Terziyska, C. Kozany, F. Baumann, W. Neu-pert, K. Hell, J.M. Herrmann, A disulfide relay system in theintermembrane space of mitochondria that mediates proteinimport, Cell 121 (2005) 1059—1069.
[44] N. Terziyska, T. Lutz, C. Kozany, D. Mokranjac, N. Mesecke,W. Neupert, J.M. Herrmann, K. Hell, Mia40, a novel factor forprotein import into the intermembrane space of mitochondriais able to bind metal ions, FEBS Lett. 579 (2005) 179—184.
[45] M. Rissler, N. Wiedemann, S. Pfannschmidt, K. Gabriel, B.Guiard, N. Pfanner, A. Chacinska, The essential mitochondrialprotein Erv1 cooperates with Mia40 in biogenesis of inter-membrane space proteins, J. Mol. Biol. 353 (2005) 485—492.
[46] F. Takakubo, P. Cartwright, N. Hoogenraad, D.R. Thorburn,F. Collins, T. Lithgow, H.H. Dahl, An amino acid substitutionin the pyruvate dehydrogenase E1 alpha gene, affecting mito-chondrial import of the precursor protein, Am. J. Hum. Genet.57 (1995) 772—780.
[47] C.J. Danpure, P.J. Cooper, P.J. Wise, P.R. Jennings, An enzymetrafficking defect in two patients with primary hyperoxaluriatype 1: peroxisomal alanine/glyoxylate aminotransferasererouted to mitochondria, J. Cell Biol. 108 (1989) 1345—1352.
[48] A. Sutton, H. Khoury, C. Prip-Buus, C. Cepanec, D. Pessayre,F. Degoul, The Ala16Val genetic dimorphism modulates theimport of human manganese superoxide dismutase into ratliver mitochondria, Pharmacogenetics 13 (2003) 145—157.
[49] C.M. Koehler, D. Leuenberger, S. Merchant, A. Renold, T.Junne, G. Schatz, Human deafness dystonia syndrome is amitochondrial disease, Proc. Natl. Acad. Sci. U.S.A. 96 (1999)2141—2146.
[50] K.M. Davey, J.S. Parboosingh, D.R. McLeod, A. Chan, R. Casey,P. Ferreira, F.F. Snyder, P.J. Bridge, F.P. Bernier, Mutation ofDNAJC19, a human homologue of yeast inner mitochondrial
membrane co-chaperones, causes DCMA syndrome, a novelautosomal recessive Barth syndrome-like condition, J. Med.Genet. 43 (2006) 385—393.[51] J.J. Hansen, A. Durr, I. Cournu-Rebeix, C. Georgopoulos, D.Ang, M.N. Nielsen, C.S. Davoine, A. Brice, B. Fontaine, N.
33
Gregersen, P. Bross, Hereditary spastic paraplegia SPG13 isassociated with a mutation in the gene encoding the mito-chondrial chaperonin Hsp60, Am. J. Hum. Genet. 70 (2002)1328—1332.
[52] J.A. MacKenzie, R.M. Payne, Mitochondrial protein import andhuman health and disease, Biochim. Biophys. Acta 1772 (2007)509—523.
[53] G. Amuthan, G. Biswas, H.K. Ananadatheerthavarada, C.Vijayasarathy, H.M. Shephard, N.G. Avadhani, Mitochondrialstress-induced calcium signaling, phenotypic changes andinvasive behavior in human lung carcinoma A549 cells, Onco-gene 21 (2002) 7839—7849.
[54] Q. Zhao, J. Wang, I.V. Levichkin, S. Stasinopoulos, M.T. Ryan,N.J. Hoogenraad, A mitochondrial specific stress response inmammalian cells, EMBO J. 21 (2002) 4411—4419.
[55] Z. Liu, R.A. Butow, A transcriptional switch in the expressionof yeast tricarboxylic acid cycle genes in response to a reduc-tion or loss of respiratory function, Mol. Cell. Biol. 19 (1999)6720—6728.
[56] M.T. Ryan, N.J. Hoogenraad, Mitochondrial-nuclear commu-nications, Annu. Rev. Biochem. 76 (2007) 701—722.
[57] C.A. Virbasius, J.V. Virbasius, R.C. Scarpulla, NRF-1, an acti-vator involved in nuclear-mitochondrial interactions, utilizesa new DNA-binding domain conserved in a family of develop-mental regulators, Genes Dev. 7 (1993) 2431—2445.
[58] J.V. Virbasius, C.A. Virbasius, R.C. Scarpulla, Identity of GABPwith NRF-2, a multisubunit activator of cytochrome oxidaseexpression, reveals a cellular role for an ETS domain activatorof viral promoters, Genes Dev. 7 (1993) 380—392.
[59] R.C. Scarpulla, Transcriptional activators and coactivators inthe nuclear control of mitochondrial function in mammaliancells, Gene 286 (2002) 81—89.
[60] R.C. Scarpulla, Nuclear control of respiratory gene expressionin mammalian cells, J. Cell Biochem. 97 (2006) 673—683.
[61] B. Chabi, P.J. Adhihetty, V. Ljubicic, D.A. Hood, How is mito-chondrial biogenesis affected in mitochondrial disease? Med.Sci. Sports Exerc. 37 (2005) 2102—2110.
[62] D.P. Kelly, R.C. Scarpulla, Transcriptional regulatory circuitscontrolling mitochondrial biogenesis and function, Genes Dev.18 (2004) 357—368.
[63] J. Lin, P.T. Tarr, R. Yang, J. Rhee, P. Puigserver, C.B. New-gard, B.M. Spiegelman, PGC-1beta in the regulation of hepaticglucose and energy metabolism, J. Biol. Chem. 278 (2003)30843—30848.
[64] C. Ling, P. Poulsen, E. Carlsson, M. Ridderstrale, P. Almgren,J. Wojtaszewski, H. Beck-Nielsen, L. Groop, A. Vaag, Multipleenvironmental and genetic factors influence skeletal musclePGC-1alpha and PGC-1beta gene expression in twins, J. Clin.Invest. 114 (2004) 1518—1526.
[65] S. Srivastava, J.N. Barrett, C.T. Moraes, PGC-1alpha/betaupregulation is associated with improved oxidative phospho-rylation in cells harboring nonsense mtDNA mutations, Hum.Mol. Genet. 16 (2007) 993—1005.
[66] J. Lin, H. Wu, P.T. Tarr, C.Y. Zhang, Z. Wu, O. Boss, L.F.Michael, P. Puigserver, E. Isotani, E.N. Olson, B.B. Lowell,R. Bassel-Duby, B.M. Spiegelman, Transcriptional co-activatorPGC-1 alpha drives the formation of slow-twitch musclefibres, Nature 418 (2002) 797—801.
[67] J.O. Holloszy, F.W. Booth, Biochemical adaptations toendurance exercise in muscle, Annu. Rev. Physiol. 38 (1976)273—291.
[68] D. Freyssenet, M. Di Carlo, D.A. Hood, Calcium-dependentregulation of cytochrome c gene expression in skeletal muscle
cells. Identification of a protein kinase c-dependent pathway,J. Biol. Chem. 274 (1999) 9305—9311.[69] D.C. Wright, P.C. Geiger, D.H. Han, T.E. Jones, J.O. Holloszy,Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochon-

3
4drial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation, J. Biol. Chem. 282 (2007)18793—18799.
[70] P. Puigserver, J. Rhee, J. Lin, Z. Wu, J.C. Yoon, C.Y. Zhang, S.Krauss, V.K. Mootha, B.B. Lowell, B.M. Spiegelman, Cytokinestimulation of energy expenditure through p38 MAP kinaseactivation of PPARgamma coactivator-1, Mol. Cell 8 (2001)971—982.
[71] S. Jager, C. Handschin, J. St-Pierre, B.M. Spiegelman, AMP-activated protein kinase (AMPK) action in skeletal muscle viadirect phosphorylation of PGC-1alpha, Proc. Natl. Acad. Sci.U.S.A. 104 (2007) 12017—12022.
[72] T. Akimoto, S.C. Pohnert, P. Li, M. Zhang, C. Gumbs,P.B. Rosenberg, R.S. Williams, Z. Yan, Exercise stimulatesPgc-1alpha transcription in skeletal muscle through activa-tion of the p38 MAPK pathway, J. Biol. Chem. 280 (2005)19587—19593.
[73] W. Cao, K.W. Daniel, J. Robidoux, P. Puigserver, A.V.Medvedev, X. Bai, L.M. Floering, B.M. Spiegelman, S. Collins,p38 mitogen-activated protein kinase is the central regulatorof cyclic AMP-dependent transcription of the brown fat uncou-pling protein 1 gene, Mol. Cell Biol. 24 (2004) 3057—3067.
[74] H.K. Au, T.S. Yeh, S.H. Kao, C.M. Shih, R.H. Hsieh, C.R. Tzeng,Calcium-dependent up-regulation of mitochondrial electrontransfer chain gene expressions in human luteinized granulosacells., Fertil. Steril. 84 (Suppl. 2) (2005) 1104—1108.
[75] L. Mercy, A. Pauw, L. Payen, S. Tejerina, A. Houbion, C.Demazy, M. Raes, P. Renard, T. Arnould, Mitochondrial bio-genesis in mtDNA-depleted cells involves a Ca2+-dependentpathway and a reduced mitochondrial protein import, FEBSJ. 272 (2005) 5031—5055.
[76] Z. Gerhart-Hines, J.T. Rodgers, O. Bare, C. Lerin, S.H. Kim,R. Mostoslavsky, F.W. Alt, Z. Wu, P. Puigserver, Metabolic con-trol of muscle mitochondrial function and fatty acid oxidationthrough SIRT1/PGC-1alpha, EMBO J. 26 (2007) 1913—1923.
[77] S. Lee, S. Kim, X. Sun, J.H. Lee, H. Cho, Cell cycle-dependentmitochondrial biogenesis and dynamics in mammalian cells,Biochem. Biophys. Res. Commun. 357 (2007) 111—117.
[78] N. Arakaki, T. Nishihama, H. Owaki, Y. Kuramoto, M. Suenaga,E. Miyoshi, Y. Emoto, H. Shibata, M. Shono, T. Higuti, Dynamicsof mitochondria during the cell cycle, Biol. Pharm. Bull. 29(2006) 1962—1965.
[79] K.A. Rasbach, R.G. Schnellmann, Signaling of mitochondrialbiogenesis following oxidant injury, J. Biol. Chem. 282 (2007)2355—2362.
[80] E. Nisoli, E. Clementi, C. Paolucci, V. Cozzi, C. Tonello, C.Sciorati, R. Bracale, A. Valerio, M. Francolini, S. Moncada,M.O. Carruba, Mitochondrial biogenesis in mammals: the roleof endogenous nitric oxide, Science 299 (2003) 896—899.
[81] S.C. Leary, E.A. Shoubridge, Mitochondrial biogenesis: whichpart of ‘‘NO’’ do we understand? Bioessays 25 (2003)538—541.
[82] C.J. McLeod, I. Pagel, M.N. Sack, The mitochondrial biogen-esis regulatory program in cardiac adaptation to ischemia—–aputative target for therapeutic intervention, Trends Cardio-vasc. Med. 15 (2005) 118—123.
[83] M. Corral-Debrinski, T. Horton, M.T. Lott, J.M. Shoffner, M.F.Beal, D.C. Wallace, Mitochondrial DNA deletions in humanbrain: regional variability and increase with advanced age,Nat. Genet. 2 (1992) 324—329.
[84] M. Khaidakov, R.H. Heflich, M.G. Manjanatha, M.B. Myers, A.Aidoo, Accumulation of point mutations in mitochondrial DNAof aging mice, Mutat. Res./Fundam. Mol. Mech. Mutagen. 526
(2003) 1—7.[85] F. Diaz, S. Garcia, D. Hernandez, A. Regev, A. Rebelo, J. Oca-Cossio, C.T. Moraes, Pathophysiology and fate of hepatocytesin a mouse model of mitochondrial hepatopathies, Gut 57(2008) 232—242.
F. Diaz, C.T. Moraes
[86] H. Chen, A. Chomyn, D.C. Chan, Disruption of fusion results inmitochondrial heterogeneity and dysfunction, J. Biol. Chem.280 (2005) 26185—26192.
[87] K.G. Hales, M.T. Fuller, Developmentally regulated mito-chondrial fusion mediated by a conserved, novel, predictedGTPase, Cell 90 (1997) 121—129.
[88] B.A. Jones, W.L. Fangman, Mitochondrial DNA maintenancein yeast requires a protein containing a region related tothe GTP-binding domain of dynamin, Genes Dev. 6 (1992)380—389.
[89] S. Hoppins, L. Lackner, J. Nunnari, The machines thatdivide and fuse mitochondria, Annu. Rev. Biochem. 76 (2007)751—780.
[90] S. Zuchner, I.V. Mersiyanova, M. Muglia, N. Bissar-Tadmouri,J. Rochelle, E.L. Dadali, M. Zappia, E. Nelis, A. Patitucci, J.Senderek, Y. Parman, O. Evgrafov, P.D. Jonghe, Y. Takahashi,S. Tsuji, M.A. Pericak-Vance, A. Quattrone, E. Battologlu,A.V. Polyakov, V. Timmerman, J.M. Schroder, J.M. Vance,Mutations in the mitochondrial GTPase mitofusin 2 causeCharcot-Marie-Tooth neuropathy type 2A, Nat. Genet. 36(2004) 449—451.
[91] C. Alexander, M. Votruba, U.E.A. Pesch, D.L. Thiselton, S.Mayer, A. Moore, M. Rodriguez, U. Kellner, B. Leo-Kottler, G.Auburger, S.S. Bhattacharya, B. Wissinger, OPA1, encoding adynamin-related GTPase, is mutated in autosomal dominantoptic atrophy linked to chromosome 3q28, Nat. Genet. 26(2000) 211—215.
[92] C. Delettre, G. Lenaers, J.-M. Griffoin, N. Gigarel, C. Lorenzo,P. Belenguer, L. Pelloquin, J. Grosgeorge, C. Turc-Carel, E.Perret, C. Astarie-Dequeker, L. Lasquellec, B. Arnaud, B.Ducommun, J. Kaplan, C.P. Hamel, Nuclear gene OPA1, encod-ing a mitochondrial dynamin-related protein, is mutated indominant optic atrophy, Nat. Genet. 26 (2000) 207—210.
[93] H. Chen, J.M. McCaffery, D.C. Chan, Mitochondrial fusion pro-tects against neurodegeneration in the cerebellum, Cell 130(2007) 548—562.
[94] M. Karbowski, S.Y. Jeong, R.J. Youle, Endophilin B1 is requiredfor the maintenance of mitochondrial morphology, J. CellBiol. 166 (2004) 1027—1039.
[95] Z. Harder, R. Zunino, H. McBride, Sumo1 conjugates mito-chondrial substrates and participates in mitochondrial fission,Curr. Biol. 14 (2004) 340—345.
[96] G. Szabadkai, A.M. Simoni, M. Chami, M.R. Wieckowski, R.J.Youle, R. Rizzuto, Drp-1-dependent division of the mitochon-drial network blocks intraorganellar Ca2+ waves and protectsagainst Ca2+-mediated apoptosis, Mol. Cell 16 (2004) 59—68.
[97] R.A. Menzies, P.H. Gold, The turnover of mitochondria in avariety of tissues of young adult and aged rats, J. Biol. Chem.246 (1971) 2425—2429.
[98] P. Sutovsky, R.D. Moreno, J. Ramalho-Santos, T. Dominko, C.Simerly, G. Schatten, Ubiquitin tag for sperm mitochondria,Nature 402 (1999) 371—372.
[99] P. Sutovsky, R.D. Moreno, J. Ramalho-Santos, T. Dominko,C. Simerly, G. Schatten, Ubiquitinated sperm mitochondria,selective proteolysis, and the regulation of mitochondrialinheritance in mammalian embryos, Biol. Reprod. 63 (2000)582—590.
[100] D. Mijaljica, M. Prescott, R.J. Devenish, Different fates ofmitochondria: alternative ways for degradation? Autophagy 3(2007) 4—9.
[101] I. Kim, S. Rodriguez-Enriquez, J.J. Lemasters, Selectivedegradation of mitochondria by mitophagy, Arch. Biochem.Biophys. 462 (2007) 245—253.
[102] Y. Ohsumi, N. Mizushima, Two ubiquitin-like conjugation sys-tems essential for autophagy, Semin. Cell Dev. Biol. 15 (2004)231—236.
[103] N. Mizushima, T. Noda, T. Yoshimori, Y. Tanaka, T. Ishii, M.D.George, D.J. Klionsky, M. Ohsumi, Y. Ohsumi, A protein con-

[
Biol. Chem. 279 (2004) 39068—39074.[108] D.A. Cottrell, E.L. Blakely, M.A. Johnson, G.M. Borthwick,
Mitochondrial biogenesis and turnover
jugation system essential for autophagy, Nature 395 (1998)395—398.
[104] Y. Ichimura, T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N.Ishihara, N. Mizushima, I. Tanida, E. Kominami, M. Ohsumi,T. Noda, Y. Ohsumi, A ubiquitin-like system mediates proteinlipidation, Nature 408 (2000) 488—492.
[105] A. Tassa, M.P. Roux, D. Attaix, D.M. Bechet, Class III phos-
phoinositide 3-kinase—Beclin1 complex mediates the aminoacid-dependent regulation of autophagy in C2C12 myotubes,Biochem. J. 376 (2003) 577—586.[106] A.R.J. Young, E.Y.W. Chan, X.W. Hu, R. Kochl, S.G. Crawshaw,S. High, D.W. Hailey, J. Lippincott-Schwartz, S.A. Tooze,
35
Starvation and ULK1-dependent cycling of mammalian Atg9between the TGN and endosomes, J. Cell Sci. 119 (2006)3888—3900.
107] I. Kissova, M. Deffieu, S. Manon, N. Camougrand, Uth1p isinvolved in the autophagic degradation of mitochondria, J.
P.I. Ince, D.M. Turnbull, Mitochondrial DNA mutations in dis-ease and ageing, Novartis Found Symp. 235 (2001) 234—243(discussion 243—246).





























