Embed Size (px)
Citation preview

Matrix metalloproteinase inhibitors*/an emphasis on gastrointestinalmalignancies
Ian Chau, Anne Rigg, David Cunningham *
Gastrointestinal Unit, Department of Medicine, Royal Marsden Hospital, Downs Road, Sutton, London, Surrey SM2 5PT, UK
Accepted 9 February 2002
* Corresponding author. Tel.: �/44-208-661-3156; fax: �/44-208-643-9414.
E-mail address: [email protected] (D. Cunningham).
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
2. The importance of proteases for tumour growth and metastasis . . . . . . . . . . . . . . . . 152
2.1. The matrix metalloproteinases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154
2.1.1. Structure of matrix metalloproteinases . . . . . . . . . . . . . . . . . . . . . . . . 154
2.1.2. Genomic and post transcriptional control of MMPs . . . . . . . . . . . . . . . . 155
2.2. Tissue inhibitors of metalloproteinases . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
2.2.1. Structure of TIMPs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
2.2.2. Interaction between MMPs and TIMPs . . . . . . . . . . . . . . . . . . . . . . . 159
2.2.3. Other functions of TIMPs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
3. Therapeutic approaches to alter the MMP/TIMP balance in cancer . . . . . . . . . . . . . . . 161
3.1. Synthetic MMP inhibitors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.2. Gene therapy using TIMPs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
4. Problems encountered in designing MMPI clinical trials . . . . . . . . . . . . . . . . . . . . . 164
4.1. Tools to assess efficacy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4.1.1. Biomarker assessment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4.1.2. Histological assessment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
4.1.3. Non invasive imaging . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
4.2. Clinical trial design . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
5. Matrix metalloprotenase inhibitors in clinical development . . . . . . . . . . . . . . . . . . . . 167
5.1. Batimastat (BB-94) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
5.2. Marimastat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
5.2.1. Phase II studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
5.2.2. Phase III studies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
5.2.3. Combination with other cytotoxic and angiogenesis therapy . . . . . . . . . . . . 169
5.2.4. Adjuvant therapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
5.2.5. Safety profiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
5.3. BAYER 12-9566 (Tanomastat) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
5.4. MMI270 (CGS27023A) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
5.5. AG3340 (Prinomastat) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
5.6. COL-3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
5.7. BMS-275291 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
6. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
Reviewers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
Biography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
Critical Reviews in Oncology/Hematology 45 (2003) 151�/176
www.elsevier.com/locate/critrevonc
1040-8428/02/$ - see front matter # 2002 Elsevier Science Ireland Ltd. All rights reserved.
PII: S 1 0 4 0 - 8 4 2 8 ( 0 2 ) 0 0 0 1 5 - X

Abstract
Gastrointestinal malignancies are the commonest sites of human cancer collectively. Improved understanding of tumour biology
in the last few decades has allowed the identification of cellular pathways responsible for the autonomous growth and replication in
cancer cells. There is considerable preclinical evidence implicating matrix metalloproteinases (MMPs) in cancer dissemination and
tumour angiogenesis. Effective MMP inhibitors (MMPIs) may, therefore, hold an important key in the treatment of gastrointestinal
cancers. MMPIs are cytostatic agents and traditional values of tumour regression may not be the best measures of treatment
efficacy. Biological correlation studies are increasingly being incorporated into the early development of these agents, but many of
these studies lack preclinical validation and are often chosen on availability rather than biological plausibility. Disappointing results
with many MMPIs that have entered phase III testing so far would prompt for identification of reliable surrogate biomarkers and
incorporation of functional imaging in the clinical development of matrix metalloproteinase inhibitors in gastrointestinal
malignancies. In this review, the integral part in which MMPs are involved in cancer growth and metastases will be presented.
This is then followed by a discussion of the challenges that clinicians are facing in assessing the efficacy of MMPIs and finally a
review of the clinical studies of the synthetic MMPIs in development.
# 2002 Elsevier Science Ireland Ltd. All rights reserved.
Keywords: Matrix metalloproteinases; Matrix metalloproteinase inhibitors; Metastasis; Angiogenesis; Cytostatic agents
1. Introduction
Gastrointestinal malignancies are the commonest sites
of human cancer collectively. Colorectal, oesophageal,
gastric and pancreatic cancers are among the top ten
cancer killers in the world accounting for over 2.5
million cases in 2000 [1]. Five year survivals are poor
in these diseases ranging from 40 to 60% in colorectal
cancers to less than 5% in pancreatic cancers [2]. Newer
cytotoxic drugs such as irinotecan, oxaliplatin, gemci-
tabine, taxanes and oral fluopyrimidines have at best
made a very modest (if any) impact on the survival of
these patients. There is, therefore, an urgent need to
identify novel agents to complement these tumoricidal
drugs. Improved understanding of tumour biology in
the last few decades has allowed the identification of
cellular pathways responsible for the autonomous
growth and replication in cancer cells. Several strategies
have emerged to target these abnormal processes
therapeutically. These targets include tumour angiogen-
esis, genetic mutation encoding signal transduction
pathway, overexpression of membrane receptors (e.g.
epidermal growth factor receptor family) and molecules
involved in tumour growth and metastasis. There is
considerable preclinical evidence implicating matrix
metalloproteinases (MMPs) in cancer dissemination
and tumour angiogenesis. Effective MMP inhibitors
(MMPIs) may, therefore, hold an important key in the
treatment of gastrointestinal cancers. In this review, the
integral part in which MMPs are involved in cancer
growth and metastases will be presented. This is then
followed by a discussion of the challenges that clinicians
are facing in assessing the efficacy of MMPIs and finally
a review of the clinical studies of the synthetic MMPIs in
development.
2. The importance of proteases for tumour growth and
metastasis
An integral feature of cancer is local invasion
accompanied by spread to distant sites: the process of
metastasis. Much work has been done to investigate the
mechanisms by which the metastatic process occurs.
Steps in this process are the escape of malignant cells
from the primary tumour, entry of the cells into the
vascular or lymphatic circulation (intravasation), survi-
val and transport in the circulation, escape of the cells
from the circulation (extravasation) and the growth of
cells at the new site to form a secondary tumour.
Mechanisms of avoiding immune destruction are re-
quired at all stages.
Proteolysis in the tumour environment degrades the
components of the extracellular matrix (ECM) and
basement membranes, allowing tumour cells to invade.
The key proteases of the ECM are the serine proteases
(plasmin) and the MMPs as these enzymes function at
neutral pH. There is considerable evidence that in-
creased levels of plasmin and MMPs occur in tumours
and that the level directly correlates with the tumour
stage [3�/8]. Rat embryo fibroblasts over-expressing
MMP-9 caused more metastases in nude mice than the
unmodified cells [9]. Likewise, rat bladder carcinoma
cells over-expressing MMP-2 produced an increased
area of lung metastases when injected into nude mice
[10].
As it became obvious that proteases were crucial for
tumour metastasis it was of interest to identify the
specific cellular components that were producing these
enzymes. Initially, it was expected that the tumour cells
alone would produce the proteases. However, a different
picture emerged when human tumour specimens were
studied. For human colon cancer, urokinase plasmino-
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176152

gen activator (uPA) was expressed by stromal fibro-
blasts around the tumour whereas uPA receptor (uPAR)
was expressed by cancer cells adjacent to the stroma and
tumour-infiltrating macrophages [11,12]. Plasminogen
activator inhibitor 1 (PAI-1) was shown to be present in
endothelial cells in tumour stroma, but not endothelial
cells of normal tissue [13]. It has been proposed that this
PAI-1 distribution may protect the cancer tissue from
uPA-mediated matrix degradation. This fits with the
observation from clinical studies that high serum PAI-1
levels correlate with a poor prognosis [5,14].
A very similar pattern is seen with MMP expression in
colorectal tumours. MMP-2 is restricted to fibroblast-
like stromal cells, as are MMP-3 and membrane-type 1-
matrix metalloproteinase (MT1-MMP). MMP-9 is pre-
sent in macrophages at the invasive edges of the tumour
and MMP-7 is found in the cancer cells themselves [15�/
17]. There is also evidence that tissue inhibitors of
matrix metalloproteinases (TIMPs) are overexpressed
by some tumours and that, like PAI-1, they may be
protecting the cancer from excessive protease activity.
Pancreatic carcinomas have been shown to over-express
several MMPs. MMP-2 and MMP-9 were expressed in
75% of pancreatic tumour samples [18] and the level of
MMP-2 has been directly correlated with the degree of
disruption of the basement membrane [19]. MMP-2 was
found to be present in both tumour epithelial cells and
the cellular elements of the stroma. MT1-MMP, MMP-
7, TIMP-1 and TIMP-2 expression was confined to the
tumour epithelium [20]. Ultimately, it is the net balance
between the proteases and their inhibitors that is critical
to their effect on the environment.
It would appear that cancer cells recruit stromal cellsvia growth factors and control the specific proteases that
they produce. Therefore, the ability to invade is a result
of the properties of the stromal cells as well as the
cancerous cells themselves.
There is additional work with knock-out mouse
models. Lewis lung carcinoma and B16F10 melanoma
cells injected into MMP-2-deficient mice showed re-
duced lung metastasis, although, the tumour cellsthemselves did produce some MMP-2 [21].
The overall conclusion from these studies is that the
majority of cancers over-express a variety of proteases
and inhibitors. It is the stromal cells within a tumour
that are integral to the production of proteases under
the control of the tumour cells. The involvement of the
stromal cells may explain the pattern of organ metas-
tasis for a particular tumour and why there is often alatent period between the appearance of a primary
tumour and the development of metastases. Fig. 1
shows a schematic representation of the metastatic
process and the points at which MMPs are involved.
2.1. The matrix metalloproteinases
The MMPs are a family of zinc-dependent endopep-
tidases that degrade various components of the ECM.
Fig. 1. A schematic representation of the metastatic process and the points at which MMPs are involved. Adapted from Chambers and Matrisian
[147].
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 153

Twenty MMPs have been identified to date. Theseenzymes function at physiological pH and are either
secreted or membrane-bound. There are three main
groups of MMPs: collagenases that degrade fibrillar
collagens at a specific locus of the helix, stromelysins
that degrade proteoglycans and glycoproteins, and
gelatinases that degrade non-fibrillar and denatured
collagens (gelatins). The family members can be cate-
gorised depending on substrate specificity, cellularsources and inducibility (Table 1).
2.1.1. Structure of matrix metalloproteinases
All the MMPs share a basic structure. This consists of
a signal peptide, a propeptide domain that has to becleaved to ensure enzyme activity, a catalytic domain
with a zinc-binding site and a C-terminal domain. The
MMP family members differ in the presence or absence
of additional domains that are involved in activities such
as membrane binding, inhibitor binding and substrate
specificity (Fig. 2).
The signal peptide sequence directs the translated
protein to the endoplasmic reticulum. The N-terminalpropeptide domain is 77�/87 amino acids in length and
contains a conserved amino acid sequence PRCGXPDV
(proline/arginine/cysteine/glycine/ (valine or aspara-
gine)/proline/aspartic acid/valine). The cysteine residue
is unpaired and able to interact with the zinc atom at the
catalytic site of the enzyme. This cysteine�/zinc interac-
tion maintains the proenzyme in an inactive state.
MMPs share a common catalytic domain containing
the HEYGH motif (histadine/glutamic acid/phenylala-
nine, leucine, or isoleucine/glycine/histadine) responsible
for binding zinc that is essential for functional enzymatic
activity.
Specific MMPs have additions or deletions to this
basic structure as illustrated in Fig. 2. For example both
gelatinases MMP-2 and MMP-9 have a fibronectin-like
region inserted into the catalyic domain and MMP-9 has
an additional collagen-like region situated at the C-
terminal end of the catalytic domain. These additional
domains are thought to be involved in binding of
gelatins as they resemble the gelatin-binding domains
of fibronectin.
The C-terminal domain appears to mediate binding of
the enzyme to its substrate although it does not actively
cleave in the absence of the catalytic domain. Also the
C-terminal domain appears to be involved in binding of
the TIMPs as loss of this domain prevents proMMP-2
binding to TIMP-2 and MMP-2 binding to TIMP-1 or
TIMP-2 [22,23]. The functions of TIMPs will be
Table 1
MMPs and their cellular sources and major substrates
MMP Other names Cell sources Major substrates
1 Collagenase-1 Connective tissue cells Collagen I, II, III, VII, VIII, X, gelatins, aggrecan
2 Gelatinase A Most cell types and tumour cells Gelatin I, II, III collagen IV, V, VII, X, fibronectin, elastin, plasminogen
3 Stromelysin-1 Connective tissue cells, macrophages Proteoglycan, fibronectin, laminin, collagen III, IV, V, IX, gelatin I, III,
IV, V, activates procollagenase
7 Matrilysin, PUMP1 Immature monocytes, measngial Gelatin I, III, IV, V, proteoglycan, fibronectin
8 Collagenase-2 Neutrophils, chondrocytes, synovial
fibroblasts, endothelial cells
Collagen I, II, III, aggrecan core protein
9 Gelatinase B Monocytes, connective tissue cells Gelatin I, V, collagen IV, V
10 Stromelysin-2 Fibroblasts, macrophages Gelatin I, III, IV, V, collagen III, IV, V, activates procollagenase
11 Stromelysin-3 Stromal cells of tumours Serine protease inhibitors such as a1-proteinase inhibitor and a1-
antitrypsin
12 Metalloelastase Macrophages Elastin, fibronectin
13 Collagenase-3 Connective tissue cells, tumour cells Collagen I, II, III, IV, X, XI, gelatin, laminin, tenascin, aggrecan,
fibronectin
14 MT1-MMP Malignant and stromal cells within a
tumour
Pro-MMP2, proteoglycan, fibrillar collagen, gelatin, fibronectin,
laminin, vitronectin, aggrecan
15 MT2-MMP Collagen I, II, III, gelatin, fibronectin, laminin, vitronectin, aggrecan,
ProMMP2
16 MT3-MMP Pro-MMP2, fibronectin. Collagen III
17 MT4-MMP ND
18 Collagenase 4 ND
19 Placenta, lung, panreas, ovary, spleen,
intestine
ND
20 Enamelysin Odontoblastic cells Dental amelogenin
21 XMMP (xenopus) ND
22 CMMP (chicken) ND
23 MT5-MMP Brain, kidney, pancreas, lung ProMMP2
Note the MMPs initially described as 4, 5 and 6 were found to be identical to other family members, hence 4, 5 and 6 are redundant nomenclature.
ND, not determined.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176154

discussed in Section 2.2. Four membrane-bound MMPs
have been described. They differ from the secreted
MMPs in that they have a transmembrane domain atthe carboxy terminus and a recognition site for the
furin-like enzymes at the end of the propeptide domain.
Therefore, membrane-bound MMPs can be activated in
the intracellular compartment by Golgi-associated
furin-like proteases [24]. Of the four, MT1-MMP has
been most extensively studied [25]. MT1-MMP is over-
expressed in a variety of human tumours and its level
correlates with the activation rate of MMP-2 in thetissues.
2.1.2. Genomic and post transcriptional control of MMPs
The genes encoding MMPs are not usually continu-
ously expressed, but become transcriptionally activewhen tissue remodelling is required for physiological
or pathological reasons. MMP gene transcription can be
induced by a variety of factors including cytokines,
growth factors, and exposure to components of the
ECM (Table 2). Intracellular signal transduction trig-
gered by the cell surface interactions with growth
factors/cytokines, other cells or the ECM leads to the
phosphorylation of transcription factors via the mito-
gen-activated protein kinase (MAPK) and serine/threo-
nine kinase pathways.
One of the most stringent levels of control over MMP
enzymatic function is the activation of the proenzyme
that takes place in the extracellular space. In all cases
this involves the ‘cysteine-switch’ hypothesis of cleavage
of a proportion of the propeptide domain so that the
cysteine�/zinc interaction is disrupted and a H2O mole-
cule can interact with the zinc atom. This then allows
auto-catalytic cleavage of the remainder of the propep-
tide domain. The majority of proMMPs are activated by
plasmin. Pro-urokinase plasminogen activator (pro-
uPA) and pro-tissue plasminogen activator (pro-tPA)
are secreted by cells in a latent form and bind to cell
Fig. 2. Domain structure of the members of the matrix metalloproteinase family. The conserved motif of the propeptide domain, PRCGXPD, is
involved with maintaining latency of the pro-enzyme. The zinc-binding motif of the catalytic domain is HEYGH. MMPs 2 and 9 have an additional
fibronectin-like domain in the catalytic domain, and MMP9 has another extra domain called the collagen-like region.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 155
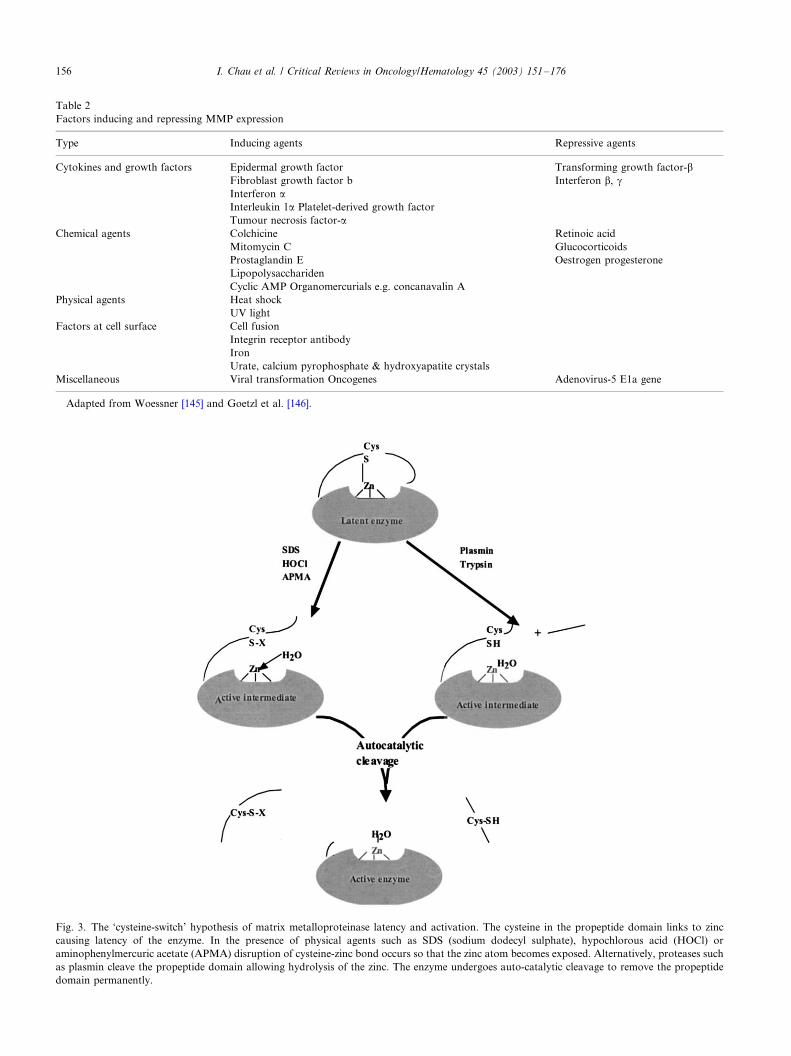
Fig. 3. The ‘cysteine-switch’ hypothesis of matrix metalloproteinase latency and activation. The cysteine in the propeptide domain links to zinc
causing latency of the enzyme. In the presence of physical agents such as SDS (sodium dodecyl sulphate), hypochlorous acid (HOCl) or
aminophenylmercuric acetate (APMA) disruption of cysteine-zinc bond occurs so that the zinc atom becomes exposed. Alternatively, proteases such
as plasmin cleave the propeptide domain allowing hydrolysis of the zinc. The enzyme undergoes auto-catalytic cleavage to remove the propeptide
domain permanently.
Table 2
Factors inducing and repressing MMP expression
Type Inducing agents Repressive agents
Cytokines and growth factors Epidermal growth factor
Fibroblast growth factor b
Interferon aInterleukin 1a Platelet-derived growth factor
Tumour necrosis factor-a
Transforming growth factor-bInterferon b, g
Chemical agents Colchicine
Mitomycin C
Prostaglandin E
Lipopolysacchariden
Cyclic AMP Organomercurials e.g. concanavalin A
Retinoic acid
Glucocorticoids
Oestrogen progesterone
Physical agents Heat shock
UV light
Factors at cell surface Cell fusion
Integrin receptor antibody
Iron
Urate, calcium pyrophosphate & hydroxyapatite crystals
Miscellaneous Viral transformation Oncogenes Adenovirus-5 E1a gene
Adapted from Woessner [145] and Goetzl et al. [146].
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176156

surface receptors. Once bound to the receptors they are
activated and able to convert extracellular plasminogen
into plasmin [26,27]. Plasmin is then able to cleave a
portion of the propeptide domain of proMMPs as
demonstrated in Fig. 3.
Unlike the other MMPs, MMP-2 appears to be
refractory to ‘classical’ activators such as plasmin and
is activated by a unique cell-mediated mechanism. A
series of experiments using recombinant MT1-MMP
immobilised on agarose beads to mimic the cell surface
showed that MT1-MMP binds TIMP-2 and in turn the
MT1-MMP/TIMP-2 complex acts as a receptor for
proMMP-2 [28]. Then an adjacent free MT1-MMP
molecule cleaves the propeptide domain from the
proMMP2/TIMP2/MT1-MMP complex. Thus when
levels of TIMP-2 are low some MT1-MMP molecules
form the complex with TIMP-2 and proMMP-2 whileother MT1-MMP molecules are free and able to activate
proMMP2. However, when TIMP-2 levels are high all
the MT1-MMP molecules bind TIMP-2 (and
proMMP2) so there are no adjacent free molecules to
activate the proMMP2. Therefore, activation of
proMMP-2 requires at least two MT1-MMP molecules,
one that functions as the receptor and another as an
activator (Fig. 4).There is evidence that MMP-13 is activated by MT1-
MMP and MMP-2. Kinetic studies show that MMP-13
does not bind to the C-terminal domain of TIMP-2
suggesting that the mechanism is different to that for
proMMP2 [29]. MT-MMPs are known to be activated
in the intracellular space by furin-like proteases.
2.2. Tissue inhibitors of metalloproteinases
To date, four TIMPs have been identified (Table 3).
TIMP-1 was first described in 1985 and its sequence
found to be identical to the growth factor erythroid-
potentiating activity [30]. Four years later TIMP-2 was
identified [31]. TIMP-3 was initially cloned from Rous
sarcoma virus transformed chick embryo fibroblasts [32]
and subsequently the human homologue has been
identified [33]. TIMP-4 has now been cloned fromhuman and murine genomes [34,35]. TIMP-1 and
TIMP-2 are secreted proteins located in the vicinity of
the cell surface and the ECM. TIMP-3 is unusual in that
it appears to be bound to components of the ECM.
These molecules are specifically able to inhibit the
activity of the MMPs. All four TIMPs can inhibit all
forms of active MMPs. In addition, TIMP-1 can bind to
Fig. 4. Activation of pro-MMP2. Pro-MMP2 forms a triple complex
with TIMP2 and MTI-MMP at the cell surface to aallow an adjacent
free MTI-MMP molecule to cleave toe propeptide domain of pro-
MMP2. If excessive TIMP2 is present pro-MMP2 cannot be activated
as all the MT1-MMP molecules have TIMP2 bound. Zn, MMP
catalytic domain; Hx, MMP hemopexin-like domain; N, TIMP2 N-
terminal domain; C, TIMP2 C-terminal domain.
Table 3
General characteristics of the tissue inhibitors of metalloproteinases
TIMP1 TIMP2 TIMP3 TIMP4
Messenger RNA
(kb)
0.9 1.1, 3.5 2.2, 2.5, 4.4 0.9, 1.4, 2.1,
4.1
Amino acids 184 194 188 195
Molecular
weight (kDa)
28 glycosylated 21 24 glycosylated 22
Complex forma-
tion
ProMMP9 ProMMP2 ProMMP2 �/ ECM ProMMP2
MMPs inhibited All All All All
Localisation Soluble Soluble ECM Soluble
Gene Xp11.23-11.4 17q2.3-2.5 22q12.1-13.2 3p25
Regulation Induced by TPA TGFb IL-1 & Il-6, serum
retinoic acid progesterone oncostatin M bFGF
EGF PDGF
Constitutive upregulated by
cAMP and retinoic acid
Induced by TPA dexamethasone
TGFb serum oncostatin M
Maximum mur-
ine
Ovary Placenta Kidney Heart
Tissue expres-
sion
Bone Brain
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 157
and stabilise the pro-enzyme form of MMP-9
(proMMP-9) and likewise TIMP-2 for proMMP-2.
2.2.1. Structure of TIMPs
Although these proteins have been described as
inhibitors for MMPs their role extends beyond this
with effects on cell morphology, enhanced growth of
certain cell types while being pro-apoptotic and anti-angiogenic for others. TIMP-2 is also implicated in the
activation of proMMP2.
TIMP proteins consist of 184�/195 amino acids.
TIMPs contain 12 conserved cysteine residues that
divide the protein into two distinct domains, each with
three internal disulphide-bonded loops. This six loop
structure confers stability to pH and temperature [36].
TIMPs consist of two domains: the N-terminal domain
that can bind to the active site of a MMP molecule and
the C-terminal domain that has the ability to bind to aregion of the C-terminal domain of certain MMPs [37].
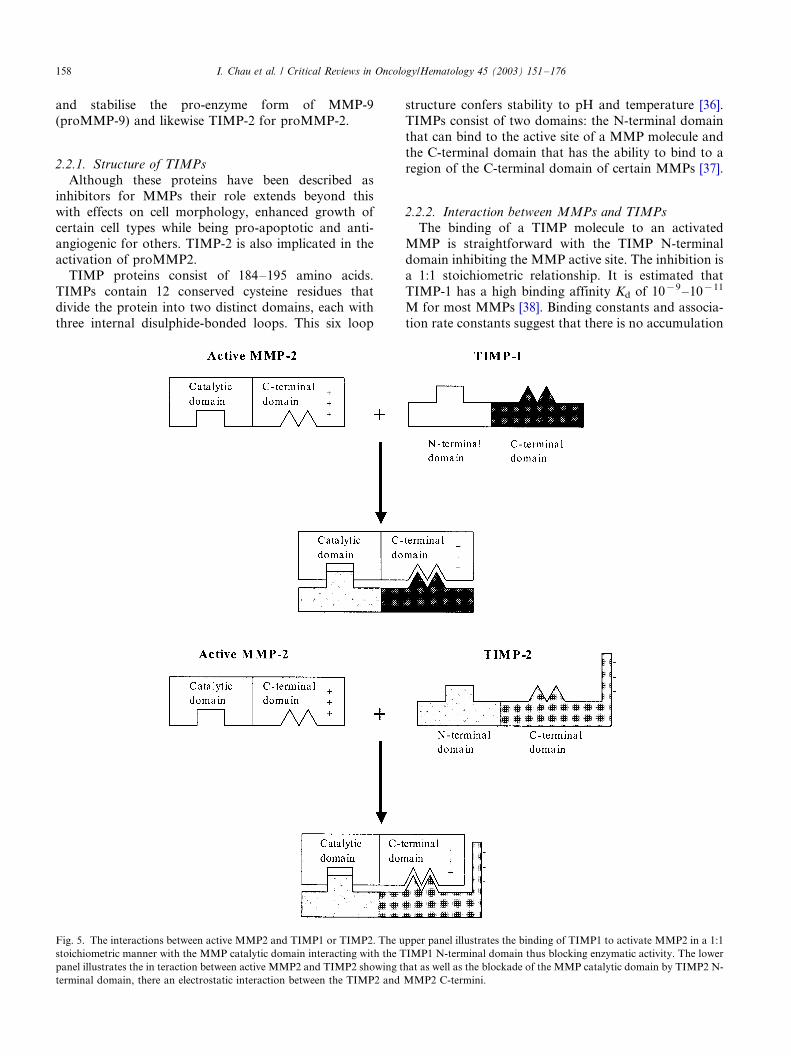
2.2.2. Interaction between MMPs and TIMPs
The binding of a TIMP molecule to an activatedMMP is straightforward with the TIMP N-terminal
domain inhibiting the MMP active site. The inhibition is
a 1:1 stoichiometric relationship. It is estimated that
TIMP-1 has a high binding affinity Kd of 10�9�/10�11
M for most MMPs [38]. Binding constants and associa-
tion rate constants suggest that there is no accumulation
Fig. 5. The interactions between active MMP2 and TIMP1 or TIMP2. The upper panel illustrates the binding of TIMP1 to activate MMP2 in a 1:1
stoichiometric manner with the MMP catalytic domain interacting with the TIMP1 N-terminal domain thus blocking enzymatic activity. The lower
panel illustrates the in teraction between active MMP2 and TIMP2 showing that as well as the blockade of the MMP catalytic domain by TIMP2 N-
terminal domain, there an electrostatic interaction between the TIMP2 and MMP2 C-termini.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176158

of intermediate products and that the interaction is a
simple bimolecular collision. All the MMPs were
inhibited by the N-terminal domain of either TIMP-1
or TIMP-2 [37]. Mutants of TIMP-1 showed that therewas no one site within the TIMP-1 N-terminal domain
that is critical to the interaction, and suggested that
there are numerous sites of contact between MMP and
TIMP-1 [39]. Of interest is the observation that TIMP-2
inhibits MMP-2 more rapidly than TIMP-1. However, if
the C-terminus of the TIMP-2 molecule is deleted then
its ability to inhibit MMP-2 is reduced 4-fold. Also if
full-length TIMP-2 and MMP-2 interact in different saltconcentration, increasing ionic strength leads to decreas-
ing TIMP-2 inhibition, whereas salt concentrations have
no effect on TIMP-1, nor TIMP-2 lacking the C-
terminus [40]. This suggests that there is a charged
region at the C-terminal end of the TIMP-2 molecule
that enables docking of the TIMP-2 to MMP-2 in the
correct alignment (Fig. 5). There may also be electro-
static interactions between TIMP-2 and MMP-2 N-terminal domains but this phenomenon has not been
fully described. In addition to the charged C-terminus of
the TIMP-2 C-terminal domain, there have been ob-
servations that if the whole C-terminal domain is
removed from either TIMP-1 or TIMP-2 then the
inhibitory ability of these molecules for MMP-2 is
reduced. It is thought that the interaction between the
TIMP C-terminal domain and the MMP-2 C-terminaldomain is distinct from the TIMP-2-specific binding site
in the C-terminal domain of proMMP2 [41].
Recently, the crystal structure of the complex formed
between human MMP-3 and TIMP-1 has been discov-
ered [42]. The catalytic domain of an MMP is known to
have an active site cleft that is relatively flat on the left-
hand side, contains the catalytic zinc at the centre and
has a deep pocket on the right-hand side. When asubstrate approaches the catalytic domain of an MMP it
becomes fixed to the MMP by inter-main chain hydro-
gen bonds. Its scissile peptide bond carbonyl group is
then directed towards the catalytic zinc to be polarised.
In this protease-substrate encounter the water molecule
localised to the catalytic zinc is squeezed between the
two and attacks the scissile peptide of the substrate. As a
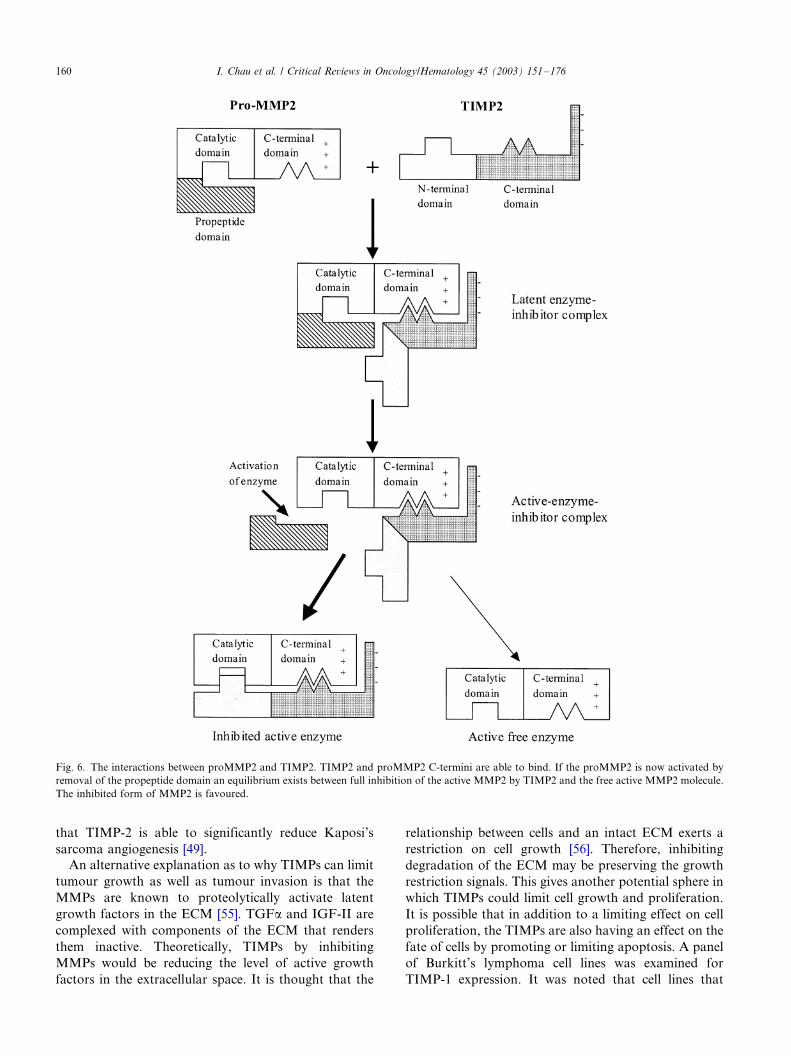
result the substrate decomposes into two fragments.The relationship between TIMP-2 and proMMP2/
MMP-2 has been the most extensively investigated as it
is known that TIMP-2 can bind to either the catalytic
domain active site or the C-terminal domain. Mutant
proMMP2 lacking a C-terminal domain cannot bind
TIMP-2 [41]. Therefore, when TIMP-2 binds to
proMMP2 it must be via the C-terminal domain as the
catalytic site is blocked by the propeptide domain.Recent mutagenesis studies demonstrate that TIMP-2
binds to the upper surface of the proMMP2 C-terminal
domain at the junction of hemopexin modules III and
IV [43]. ProMMP2 complexed to TIMP-2 via C-
terminal domain interactions can be activated by
organomercurials such as concavalin A. The active
MMP-2 still complexed to TIMP-2 only has 5�/10% of
the gelatinolytic activity of free activated MMP2 [41].The active MMP-2/TIMP-2 complex is in equilibrium
between the inhibited and non-inhibited forms of the
complex with a tendency toward inhibition [40]. There is
evidence that when the active site becomes available
after activation that the TIMP-2 is now in a position to
bind there resulting in a completely and irreversibly
inactivated complex. It is not yet clear whether both
TIMP-2 domains may interact with the two binding siteson MMP-2 simultaneously (Fig. 6). It is thought that the
interaction between TIMP-1 and proMMP-9 is of a
similar nature [44].
2.2.3. Other functions of TIMPs
There appears to be increasing evidence that TIMPs
have additional activities to MMP inhibition. TIMP-1
was originally identified as the growth factor witherythroid-potentiating activity that stimulates the
growth of erythroid precursor cells [30,45]. This function
of TIMP-1 is thought to be distinct from its activity as
an MMP inhibitor. TIMP-1 and TIMP-2 have been
shown to potentiate growth for a number of human cell
types in vitro, including aortic smooth muscle, skin
epithelia, gingival fibroblasts, Raji Burkitt’s lymphoma
and MCF-7 breast adenocarcinoma [46]. Overall thereported growth effects have been modest with only
1.5�/2.5-fold increase in proliferation as assessed by 3H
thymidine incorporation or DNA content. Overall the
reported growth effects have been modest with only
1.5�/2.5 fold increase in proliferation as assessed by 3H
thymidine incorporation of DNA content. A recent
study has demonstrated that a chimeric protein of
TIMP-1 and green fluorescent protein (GFP) binds tothe cell surface of MCF-7 breast adenocarcinoma cells,
but not HBL-100 breast epithelial cells. After 72 h of co-
culture the TIMP-GFP had localised to the nuclei of the
MCF-7 cells [47]. There has also been a suggestion that
TIMP-1 acts as an autocrine growth factor in the
fibrosis of systemic sclerosis [48].
TIMPs and synthetic protease inhibitors are able to
inhibit angiogenesis. In vitro there is a reduction ofendothelial cell proliferation in the presence of TIMP-1
and TIMP-2. In vivo, angiogensis is abrogated by
TIMP-1, TIMP-2 and TIMP-3. Studies include work
with normal endothelial cells in the chick yolk sac
membrane and endothelial cells in the context of a
tumour [49�/54]. TIMP-2 was shown to inhibit the
proliferation of cultured endothelial cells while the
protease inhibitor BB94 did not, suggesting thatTIMP-2 may be having effects beyond the role of
protease inhibition [52]. Kaposi’s sarcoma is a highly
vascular tumour and in vivo experiments demonstrated
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 159
that TIMP-2 is able to significantly reduce Kaposi’s
sarcoma angiogenesis [49].
An alternative explanation as to why TIMPs can limit
tumour growth as well as tumour invasion is that the
MMPs are known to proteolytically activate latent
growth factors in the ECM [55]. TGFa and IGF-II are
complexed with components of the ECM that renders
them inactive. Theoretically, TIMPs by inhibiting
MMPs would be reducing the level of active growth
factors in the extracellular space. It is thought that the
relationship between cells and an intact ECM exerts a
restriction on cell growth [56]. Therefore, inhibiting
degradation of the ECM may be preserving the growth
restriction signals. This gives another potential sphere in
which TIMPs could limit cell growth and proliferation.
It is possible that in addition to a limiting effect on cell
proliferation, the TIMPs are also having an effect on the
fate of cells by promoting or limiting apoptosis. A panel
of Burkitt’s lymphoma cell lines was examined for
TIMP-1 expression. It was noted that cell lines that
Fig. 6. The interactions between proMMP2 and TIMP2. TIMP2 and proMMP2 C-termini are able to bind. If the proMMP2 is now activated by
removal of the propeptide domain an equilibrium exists between full inhibition of the active MMP2 by TIMP2 and the free active MMP2 molecule.
The inhibited form of MMP2 is favoured.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176160

were TIMP1 positive were resistant to cold shock-
induced apoptosis. The resistance to apoptosis was
reversed with anti-TIMP-1 antibodies. TIMP-1 up-regulation induced expression of Bcl-XL but not Bcl-2
[57]. There have been several experiments with transfec-
tion of tumour cells with TIMP-1, TIMP-2 or TIMP-3
using a variety of delivery vectors. TIMP-3 has been
shown to be pro-apoptotic in melanoma, cervical
carcinoma and vascular smooth muscle cells [58,59].
TIMP-3-mediated cell death is independent of MMP
inhibitory activity as the synthetic inhibitor BB-94 doesnot have the same effect.
3. Therapeutic approaches to alter the MMP/TIMP
balance in cancer
The functions of the MMP enzymes and their
inhibitors TIMPs have been described. Given that
disturbances of the levels of the MMPs and TIMPsare implicated in tumour growth and metastasis it is
logical to attempt to correct the balance by reducing the
level of MMP and/or increasing the level of TIMP.
There are several potential points of intervention as
illustrated in Fig. 7. The majority of attention has been
focused on the inhibition of the MMP enzyme by
synthetic inhibitors or TIMP gene therapy and these
will be discussed in detail. There is some experimentalevidence of intervention in the earlier steps of the MMP
production pathway. For example, squamous cancer
cells transfected with an antisense expression construct
for the transcription factor E1AF produce less MMP-1,
MMP-3 and MMP-9 and this correlates with reduced
invasion by the tumour cells in vitro and in vivo [60].Another point of intervention is the use of synthetic
furin inhibitors to prevent the intracellular activation of
the MT-MMPs by furin-like enzymes [61].
3.1. Synthetic MMP inhibitors
One approach to limit the activity of the MMPs in
cancer has been the development of synthetic inhibitors.
The first generation of MMPIs was designed to mimicthe part of the collagen peptide sequence that is initially
cleaved by MMP-1. Therefore, the inhibitor fits tightly
into the active site of the MMP in a stereo-specific
manner. The inhibitor includes a zinc-binding group
such as hydroxamate or carboxylate. This then binds to
the zinc at the MMP active site. The compound
marimastat (British Biotech) is an example of a pep-
tide-mimetic MMP inhibitor with a hydroxamate zinc-binding group. Marimastat is a broad-spectrum inhibi-
tor for the MMP family with low nanomolar IC50s
against all the MMPs except MMP-3.
Most of the peptide-mimetic MMPIs have poor oral
bioavailability so research focused on the development
of a non-peptidic inhibitor based on the newly available
X-ray crystallographic information about the MMP
active site. CGS 27023A (Novartis), AG3340 (Agouron)and BAY 12-9566 (Bayer) are examples for these non-
peptidic inhibitors. As yet it is not possible to design an
MMP inhibitor specific for just one MMP, but com-
Fig. 7. Potential levels of intervention to alter the MMP/TIMP balance with therapeutic intent.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 161

pounds can be made that favour the inhibition of MMPs
with a ‘deep pocket’ at the active site (MMP-2, MMP-9,
MMP-3) at the expense of MMPs with ‘shallow pockets’
(MMP-1 and MMP-7). AG3340 is a compound thatappears to preferentially inhibit the gelatinases, while
Ro 32-3555 (Roche) inhibits MMP-1 fifty times more
potently than MMP-2.
Many of the synthetic MMPIs have been widely
tested for a variety of cancer in vivo models. There is
much evidence that they can inhibit the growth of both
primary tumours and metastatic deposits. Animals were
established with a primary tumour adjacent to themammary fat pad. The animals were then commenced
on batimastat (British Biotech) for short (7 days) or long
(58 days) courses just before the primary tumour was
resected. Animals treated with the short course of
batimastat had a reduction of pulmonary metastases
compared with untreated controls, but still developed
lymphatic metatases. However, the animals that re-
ceived the long course of batimastat did not developlymphatic metastases as any tumour cells remained as
silent micrometastases [62].
The effect of synthetic MMPIs on human cancers
established as xenografts in nude mice has also been
investigated. In vivo models of intra-peritoneal human
colorectal and ovarian carcinomas in mice have shown
significant reductions in tumour burden and ascites, and
increased survival when batimastat was administered asan intra-peritoneal agent [63�/65]. The ‘selective’ gelati-
nase inhibitor AG3340 has shown tumour inhibition for
human gliomas, human colon carcinomas and Lewis
lung carcinomas in vivo [66,67]. Both broad-spectrum
MMPIs like batimastat and ‘selective’ MMPIs like BAY
12-9566 have shown anti-angiogenic activity when tested
in a matrigel implant model [68�/70]. Another interesting
approach has been to combine a synthetic MMPinhibitor with a cytotoxic drug producing additive
benefits without a significant increase in toxicity. This
has been observed for batimastat in combination with
cisplatin in a human ovarian cancer model, AG3340 in
combination with carboplatin for a pulmonary metas-
tasis model, and for CT1476 (Celltech) with cisplatin or
cyclophosphamide in Lewis lung carcinoma models
[3,71,72].The first MMP inhibitor to be tested in a cancer
clinical trial was batimastat for patients with ascites
secondary to ovarian cancer. Batimastat had to be
administered parenterally as it has poor oral bioavail-
ability. Newer MMPIs such as marimastat, AG3340
(prinomastat), BAY 12-9566 (tanomastat), Ro 32-3555,
Col 3 and CGS27023A demonstrate high plasma con-
centrations after oral administration. There has beeninterest in developing peptide inhibitors to the MMPs.
Synthetic peptides mimicking the conserved
PRCGXPDV motif of the propeptide inhibited proteo-
lytic activity and inhibited invasion of tumour cells. A
recent report has demonstrated that by screening a
library of peptides a candidate that specifically inhibits
gelatinases MMP-2 and MMP-9 could be identified. The
candidate peptide is cyclic and contains the motifHWGF. In the presence of this peptide tumour growth
and invasion are reduced in vitro and in vivo [73].
Synthetic MMPIs have shown promising pre-clinical
evidence of anti-tumour effects in a variety of solid
tumour models. The availability of oral formulation
allows chronic administration in human subjects. These
agents have been brought forward for large scale clinical
testing (Section 4).
3.2. Gene therapy using TIMPs
As an alternative to synthetic MMPIs several groups
have opted to utilise the TIMPs themselves as anti-
cancer agents. Initially recombinant TIMP proteins
were used, but the approach has evolved into the gene
transfer of cDNA encoding the required TIMP gene into
the target cells. A variety of gene transfer vectors havebeen used to achieve this. The advantage of TIMP gene
therapy is definitive MMP inhibition by the naturally
occurring substance that can be produced in excess by
cells at the desired location. Genetic manipulation of
TIMP levels locally may also avoid the toxicity asso-
ciated with synthetic MMPIs. With the advent of tissue/
tumour specific promoters and targeted vectors gene
delivery is becoming increasingly more accurate.The first reports of the use of TIMPs as anti-cancer
agents centred on recombinant TIMP-1. It was demon-
strated that the recombinant protein interfered with the
invasion of B16-F10 melanoma cells through an amnio-
tic membrane. Interestingly, the tumour cells were able
to adhere to the membrane but appeared to be less able
to invade through it in the presence of TIMP-1. The
same melanoma cells were then injected into the tail veinor subcutaneous tissue of mice and intra-peritoneal
recombinant TIMP-1 was administered. TIMP-1 sig-
nificantly reduced the number of lung metastases
following tail vein injection of melanoma cells, but
those tumours that did develop were of the same size as
tumours in the control mice. This suggested that TIMP-
1 was inhibiting the invasion step rather than tumour
growth and this was corroborated by the fact that thesubcutaneous tumours were not significantly affected by
recombinant TIMP-1 [74]. A highly metastatic rat
embryo cell line transformed with H-ras (4R cells) had
reduced ability to colonise the lungs after tail vein
injection into nude mice if recombinant TIMP-1 was
administered into the peritoneal cavity. Again, if lung
metastases did develop they were the same size as those
in control mice [75]. Detailed in vitro studies of theeffects of recombinant TIMP-2 on tumour cell lines
indicated that there was an inhibition of gelatinolytic
activity that was independent of cell growth rates.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176162

TIMP-2 did not appear to interfere with cell attachment
nor mobility but did prevent the invasion of tumour
cells through a porous membrane [76,77]. Recombinant
TIMP-1 and TIMP-2 were shown to reduce angiogen-esis in chick yolk sac membranes that had been induced
with spermine [54].
DeClerck et al. transfected 4R cells with a plasmid
expressing TIMP-2 from a constitutive promoter. When
given as an intravenous injection to nude mice only one
4R TIMP2-expressing clone demonstrated a reduction
in the number of lung metastases formed, although this
clone was the highest TIMP-2 expresser. Another cohortof nude mice received 4R-TIMP2 cells in one flank and
parental 4R cells in the other flank as subcutaneous
injections. The TIMP2-expressing cells formed signifi-
cantly smaller tumours than the control cells. Control
tumours had invaded through the abdominal muscle
into the peritoneal cavity. However, the TIMP-2 tu-
mours were confined to the subcutaneous tissue and
surrounded by a capsule of dense connective tissue.When parental cells were implanted and recombinant
TIMP-2 was injected around the tumour site a similar
peri-tumoral connective tissue capsule was seen con-
firming that local TIMP-2 production is causing this
response [78]. This reduction in tumour growth as well
as reduction of invasion contrasts with the findings for
recombinant TIMP proteins. Similarly in mice, TIMP1-
expressing tumour cells injected intravenously producedfewer and smaller lung tumours [79]. Another group
used videomicroscopy to observe the movement of B16-
F10 melanoma cells across the chick embryo chorioal-
lantoic membrane. They found B16-F10 cells over-
expressing TIMP-1 extravasated as quickly as parental
cells across the membrane, but led to smaller and fewer
established metastases at 7 days [80]. Again there was
the suggestion that TIMPs were exerting their effect bylimiting tumour growth post-extravasation.
A highly metastatic human melanoma cell line
M24net was transfected by electroporation with a
eukaryotic expression vector containing the TIMP-2
cDNA under the control of the CMV promoter. High
TIMP2-expressing clones were identified by reverse
zymography. In vitro the high-expressing clones had a
reduced growth rate when embedded in a three-dimen-sional collagen matrix compared with the wild-type
cells. However, on collagen-coated plates there was no
reduction in growth rate. If the collagen concentration
on the plate was increased the growth-inhibitory effect
was seen again. This suggested that type I collagen
might suppress the early growth of M24net cells and
that elevated TIMP-2 levels perpetuate the growth
inhibition by preventing localised collagen degradation.It was also noted that the TIMP2-expressing clones
maintained morphology with multiple dendrites when in
the collagen matrix. In a murine model, TIMP2-
transfected M24net tumours grew significantly more
slowly than wild-type M24net tumours. There was no
evidence of tumour-associated capsules around the
TIMP2-expressing tumours in contrast to the findings
of other experiments. All tumours metastasised to thelungs and lymph nodes at equal rates although by
reverse zymography it could still be shown that TIMP-2
was being over-expressed [81].
The same group went on to develop retroviral vector
producing cells (VPCs) that release retroviruses encod-
ing the TIMP-2 cDNA under the control of the retro-
viral LTR. H-ras transformed rat embryo fibroblasts
were mixed with irradiated VPCs at ratios of 1:5 and1:10 and injected into the flanks of nude mice. There was
a significant reduction in the growth of tumours injected
with VPCs releasing TIMP-2 compared with VPCs
secreting galactosidase. Transduction efficiency of tu-
mour cells was 13%. In addition, the TIMP2-exposed
tumour cells were confined to the subcutaneous layer
and had apparently been unable to invade through
muscle while the control tumours had invaded throughthe muscle wall to the peritoneal cavity [82]. Recent data
shows that TIMP-1 or TIMP-2 can be successfully
delivered to the peritoneum of nude mice by an
adenoviral vector. Nude mice harbouring human pan-
creatic carcinomas had significantly improved survival if
treated with the adenoviral-TIMP-1 or TIMP-2 vector
than untreated controls [83].
There have been recent reports of TIMP-3 being usedfor gene therapy and also inhibiting invasion in vitro
[58,59] when delivered by an adenoviral vector. As yet
there are no in vivo data for TIMP-3. Another area of
interest has been that TIMPs may not be inducing an
anti-tumour effect by pure MMP inhibition. The effects
of TIMPs on apoptosis and angiogenesis have already
been discussed and are relevant to the gene transfer of
TIMP cDNA to target cells.Gene therapy targeting TIMPs may avoid toxicity
associated with synthetic MMPIs. However, there are
limitations to gene therapy, which include low efficiency
of gene transfer, poor specificity of response and
methods to accurately evaluate responses, and lack of
truly tumour-specific targets at which to aim. As with all
new therapies, we are climbing a steep learning curve in
terms of encountering treatment-related toxicities, aswell as profound ethical and regulatory issues. Preclini-
cal data are promising, but they will require confirma-
tion by large scale clinical testing.
4. Problems encountered in designing MMPI clinical
trials
One of the major obstacles for clinical trials ofsynthetic MMPIs in cancer patients is that the agents
are tumoristatic and so traditional values of tumour
regression may not be the best measures of treatment
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 163

efficacy. The trials conducted to date have focused on
tumour marker levels, MMP concentrations, growth
factors such as VEGF and urinary levels of collagen
breakdown products-all of which has led to debateregarding the value of these surrogate markers [84]. The
most objective endpoint would be survival, but as in
accordance with clinical trial ethics, synthetic MMPIs
have mainly been tested in patients with advanced
cancer for whom any treatment is unlikely to have a
great impact.
4.1. Tools to assess efficacy
Although survival would remain as the most objective
endpoint, two other approaches have been adopted to
assess the efficacy of MMPIs. Use of serum tumour
markers and serial histological assessment have been
used as surrogate markers of the biological activity of
the underlying tumour. Serum tumour markers have
been used for measurement of tumour activity anddetection of recurrence for a number of years. Carbohy-
drate antigen (CA) 19-9 and carcinoembryonic antigen
(CEA) are the two most commonly used markers in
gastrointestinal cancer. A change of CA19-9 has been
shown to predict response and survival in patients with
inoperable carcinoma of pancreas. [85�/87] Most of
these studies, however, involved only small number of
patients. In addition, interpretation of CA19-9 levelchanges is particularly problematic in patients with
obstructive jaundice [88] and a proportion of patients
with advanced pancreatic cancer may develop a degree
of biliary obstruction over the course of their treatment.
Serial measurement of CEA levels can add to the clinical
decision-making process in patients with metastatic
colorectal cancer. However, the presence of tumour
clone that do not express CEA marker would result in alack of rising CEA levels in some patients with
progressive disease. Therefore although tumour marker
measurement can reflect biological effects (BE) of the
antitumour agents, these markers are still suboptimal in
their sensitivity and specificity.
The second approach to assess biological activity of
MMPIs would involve serial macroscopic and micro-
scopic assessment of tumour, but this requires sites ofdisease which are readily accessible for repeated biop-
sies. Upper gastrointestinal endoscopy has relatively few
associated morbidity and mortality and the primary
tumour is amenable to biopsy during the procedure.
This strategy has therefore been adopted in MMPI trials
in gastric cancer. This approach would be impractical in
pancreatic cancer patients as the primary tumours are
often difficult to biopsy. In colorectal cancers, serialcolonscopies or tumour biopsies from colon or liver
would not be acceptable in large scale clinical trials in
view of their associated morbidity.
4.1.1. Biomarker assessment
In general, the implementation of correlative biologi-
cal studies has been a rather disorganised process in
which assays, that are often chosen on the basis ofavailability rather than relevance, are utilised in clinical
studies without a strong rationale and with minimal, if
any, preclinical validation [89]. In the clinical studies of
MMPIs, plasma concentration of angiogenic growth
factors such as vascular endothelial growth factor
(VEGF) and basic fibroblast growth factor (bFGF),
urinary secretion of collagen degradation products such
as pyridinoline and deoxypyridinoline as well as plasmaMMP activity have been measured as surrogate markers
for the BEs of MMPIs. However, results from these
biological correlative studies have often been inconclu-
sive (Table 4). This may be due to the lack of preclinical
evidence supporting that the MMPI being studied
impacts on these parameters or that changes in plasma
concentration of a given marker correlates with changes
in tumour tissue. Most investigators determined MMPexpression and secretion at the protein level using
ELISA. Duivenvoorden et al. tried to overcome the
limitations of these assays through the measurement of
the true activity of MMPs found in human plasma
sample using gelatin enzymography and fluorimetric
degradation assays [90]. Fluorimetric degradation assay
is truly quantitative in contrast to the method of gelatin
enzymography, but this assay determines total gelati-nase activity and is thus unable to distinguish between
the activities of a single MMP. By using gelatin
enzymography in combination with densitometry, the
authors could make a distinction between individual
MMP activity and at least semi-quantitative results
could be obtained using this approach.
4.1.2. Histological assessment
The development of MMPI would be accelerated bythe availability of reliable and reproducible surrogate
end-points to determine efficacy of treatment. As
discussed before, there is now growing evidence that
MMPs and TIMPs are involved in angiogenesis [91].
Indeed MMP activity is an early event in the angiogenic
response and much effort has been put in to identify
specific MMPs that mediate the angiogenic response in
order to target MMPIs to disrupt tumour neovascular-isation and subsequent dissemination.
The most widely used method to assess neovascular-
isation in human neoplasm is the quantitative analysis
of intratumoral microvessel density (IMD) using im-
munohistochemical methods and specific markers for
endothelial cells [92].
Assessment of IMD can be performed using three
different methods: (a) IMD in vascular hot spotsaccording to Weidner’s method [93]; (b) Chalkly count-
ing [94]; (c) multiparametric computerised image analy-
sis system [95�/98]. These methods all identify the
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176164
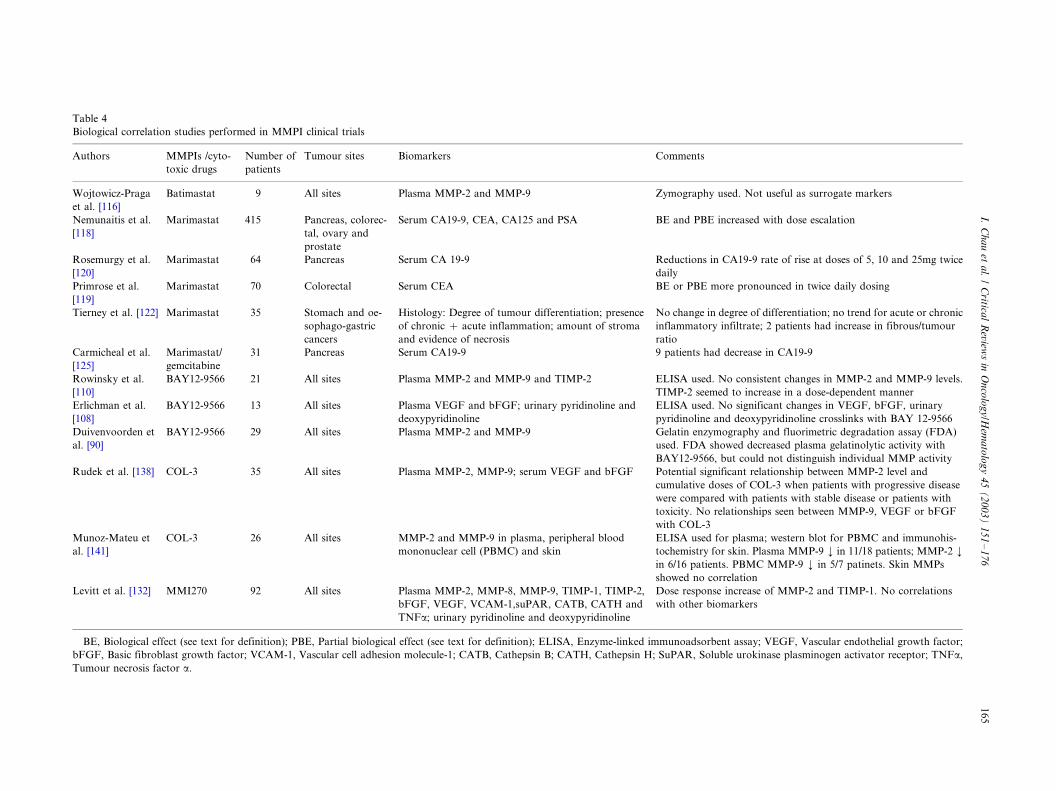
Table 4
Biological correlation studies performed in MMPI clinical trials
Authors MMPIs /cyto-
toxic drugs
Number of
patients
Tumour sites Biomarkers Comments
Wojtowicz-Praga
et al. [116]
Batimastat 9 All sites Plasma MMP-2 and MMP-9 Zymography used. Not useful as surrogate markers
Nemunaitis et al.
[118]
Marimastat 415 Pancreas, colorec-
tal, ovary and
prostate
Serum CA19-9, CEA, CA125 and PSA BE and PBE increased with dose escalation
Rosemurgy et al.
[120]
Marimastat 64 Pancreas Serum CA 19-9 Reductions in CA19-9 rate of rise at doses of 5, 10 and 25mg twice
daily
Primrose et al.
[119]
Marimastat 70 Colorectal Serum CEA BE or PBE more pronounced in twice daily dosing
Tierney et al. [122] Marimastat 35 Stomach and oe-
sophago-gastric
cancers
Histology: Degree of tumour differentiation; presence
of chronic �/ acute inflammation; amount of stroma
and evidence of necrosis
No change in degree of differentiation; no trend for acute or chronic
inflammatory infiltrate; 2 patients had increase in fibrous/tumour
ratio
Carmicheal et al.
[125]
Marimastat/
gemcitabine
31 Pancreas Serum CA19-9 9 patients had decrease in CA19-9
Rowinsky et al.
[110]
BAY12-9566 21 All sites Plasma MMP-2 and MMP-9 and TIMP-2 ELISA used. No consistent changes in MMP-2 and MMP-9 levels.
TIMP-2 seemed to increase in a dose-dependent manner
Erlichman et al.
[108]
BAY12-9566 13 All sites Plasma VEGF and bFGF; urinary pyridinoline and
deoxypyridinoline
ELISA used. No significant changes in VEGF, bFGF, urinary
pyridinoline and deoxypyridinoline crosslinks with BAY 12-9566
Duivenvoorden et
al. [90]
BAY12-9566 29 All sites Plasma MMP-2 and MMP-9 Gelatin enzymography and fluorimetric degradation assay (FDA)
used. FDA showed decreased plasma gelatinolytic activity with
BAY12-9566, but could not distinguish individual MMP activity
Rudek et al. [138] COL-3 35 All sites Plasma MMP-2, MMP-9; serum VEGF and bFGF Potential significant relationship between MMP-2 level and
cumulative doses of COL-3 when patients with progressive disease
were compared with patients with stable disease or patients with
toxicity. No relationships seen between MMP-9, VEGF or bFGF
with COL-3
Munoz-Mateu et
al. [141]
COL-3 26 All sites MMP-2 and MMP-9 in plasma, peripheral blood
mononuclear cell (PBMC) and skin
ELISA used for plasma; western blot for PBMC and immunohis-
tochemistry for skin. Plasma MMP-9 ¡/ in 11/18 patients; MMP-2 ¡/
in 6/16 patients. PBMC MMP-9 ¡/ in 5/7 patinets. Skin MMPs
showed no correlation
Levitt et al. [132] MMI270 92 All sites Plasma MMP-2, MMP-8, MMP-9, TIMP-1, TIMP-2,
bFGF, VEGF, VCAM-1,suPAR, CATB, CATH and
TNFa; urinary pyridinoline and deoxypyridinoline
Dose response increase of MMP-2 and TIMP-1. No correlations
with other biomarkers
BE, Biological effect (see text for definition); PBE, Partial biological effect (see text for definition); ELISA, Enzyme-linked immunoadsorbent assay; VEGF, Vascular endothelial growth factor;
bFGF, Basic fibroblast growth factor; VCAM-1, Vascular cell adhesion molecule-1; CATB, Cathepsin B; CATH, Cathepsin H; SuPAR, Soluble urokinase plasminogen activator receptor; TNFa,
Tumour necrosis factor a.
I.C
ha
uet
al.
/C
ritical
Review
sin
On
colo
gy
/Hem
ato
log
y4
5(
20
03
)1
51�
/17
61
65

vascular hot spot defined as the area of highest
neovascularisation within a tumour section. Such highly
neovascular areas may occur anywhere within the
tumour but are generally located near its edges.The Weidner method and Chalkley point counting are
reproducible and require little time for assessment of
IMD with low associated costs. However, both methods
require manual counting which may give rise to inter-
observer variability particularly for the selection of the
vascular hot spot. In an attempt to automate the
counting procedure, improve the reproducibility and
reduce the variability associated with manual counting,computerised image analysis system has been developed.
However, the systems still require a high degree of
operator interaction and are more expensive than
manual counting.
Although histological microvessel density technique is
the current gold standard to characterise tumour
angiogenesis, it may not be ideal for clinical purposes,
because, it requires histological material and does notassess dynamic angiogenic activity. In vivo functional
imaging would, therefore, be a much more attractive
tool in clinical studies.
4.1.3. Non invasive imaging
Non invasive imaging methods, capable of providing
information on the physiological changes occurring in atumour during therapy, are being developed. Since
many cytostatic agents such as MMPIs are being tested
in clinical trials, there is an increasing need to utilise
functional measures of response rather than changes in
lesion dimension [99]. These functional imaging techni-
ques can be used to measure tumour blood flow, tumour
blood volume, changes in tumour metabolism and
changes in tumour vascular density. Among thesetechniques, positron emission tomography (PET) and
magnetic resonance imaging (MRI) appear to hold early
promises.
PET has been used in a variety of clinical settings to
measure both blood flow and blood volume [100�/102],
as well as rates of glucose metabolism [103,104]. Several
studies have demonstrated malignant lesions undergo
elevated glycolysis when compared with normal tissues.
Increased uptake of [18F] fluorodeoxyglucose (18FDG)
in cancer tissues has been demonstrated in several
studies, therefore, allowing PET evaluation. Why tu-
mours accumulate glucose is only partly understood.
The energy requirement for rapidly dividing cells
obviously plays a role. In addition, if the blood flow
to a tumour is decreased, the supply of oxygen and
nutrients should be decreased as well. The decrease in
oxygen should result in a shift from aerobic to anaerobic
metabolism, which may also have an impact on glucose
metabolism of the tumour cells. For these reasons, it
may be important to measure both tumour blood flow
and metabolism. The ability of PET scanning to per-
form such functional imaging (i.e. assessing both
tumour physiology and biochemical function) may
hold future promise as an important adjunct to conven-
tional imaging methods such as CT, which portray
primarily anatomy, in the assessment of new MMPIs.
Dynamic enhanced MRI involves the rapid adminis-
tration of a gadolinium-based contrast agent followed
by rapid analysis of signal intensity. Patterns of MRI
contrast uptake within tumour correlate with micro-
vessel density. Although this correlation may have some
variability, it allows investigators to monitor relative
changes in microvessel density, although absolute den-
sities cannot be determined. Using image-processing
algorithms established for the detection of malignancy
by MRI, it might be possible to assess changes in
perfusion and microvessel density over time in lesions
being treated with angiosuppressive agents.
Functional MRI is based on imaging differences in
oxygenated and deoxygenated haemoglobin. Carbogen
is a gas mixture of 95% O2 and 5% CO2 and allows
measurement of tissue oxygenation and perfusion with
MRI after administration to patients. Carbogen may
have diagnostic value, because, tissue oxygenation may
Table 5
Challenges in developing MMPI
Phase of clinical
study
Potential problems in trial design
Phase I Dose finding: Biological dose vs. maximum toler-
ated dose based on toxicity
Phase II Efficacy: Tumour regression vs. progression-free.
Clinical benefit vs. valid biological end points
Phase III Disease setting: Metastatic disease vs. minimal
volume disease (i.e. adjuvant or maximal response
after chemotherapy). Treatment strategies: MMPI
vs. placebo vs. chemotherapy alone vs. combination
of chemotherapy and MMPI
Table 6
Matrix metalloproteinase inhibitors (MMPI) tested in gastrointestinal
malignancies
MMPI MMP Inhibition Recommended doses for
phase II and III testing
Batimastat MMP-1,-2, -3, -7 and -9 No further testing
Marimastat MMP-1,-2, -3, -7 and -9 5, 10 and 25 mg b.i.d.
Prinomastat
(AG3340)
MMP-2, -3, -9 and -13 5, 10 and 15 mg b.i.d.
BAY 12-9566 MMP-2, -3 and -9 800 mg b.i.d.
CGS27023A Broad spectrum 300 mg b.i.d.
COL-3 MMP-2 and -9 36 mg/m2 per day
BMS275291 Broad spectrum including
MMP-2 and -9; shed-
dases sparing
1200 mg/day
b.i.d., twice daily.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176166

reflect on the angiogenesis stimulus and response to
therapy [105,106].
To assess efficacy of MMPIs, it would require parallel
effort to develop valid assessment tools that arepractical in clinical trials. At present, the search for
sensitive and specific biomarkers for assessing the
efficacy of MMPI continues. Histological assessment is
still the gold standard for angiogenesis assessment, but
non invasive functional imaging techniques holds early
promise and may represent a significant advance.
4.2. Clinical trial design
Novel clinical trial designs are needed to properly
assess the potential beneficial effects of MMPIs (Tables
5 and 6). Unlike the cytotoxic agents, many MMPIs in
development do not cause unacceptable toxicity at doses
that achieve concentrations with desirable BEs. In
designing phase I studies for MMPI, the traditional
goal for achieving maximum tolerated dose based on
toxicity is no longer appropriate, rather the optimumbiological dose required to obtain the hypothesised
cellular effects needs to be determined. The use of these
biological end points is based on preclinical data, but as
discussed before, the measurement of these end points in
clinical trials is often based on availability rather than
plausibility. It may be difficult to define and validate an
appropriate end point to measure and agents may have
additional antitumour mechanisms than those initiallyhypothesised [107]. Furthermore, the measurement of
plasma concentrations of MMPs in clinical studies is
often carried out, but this does not necessarily reflect the
expression of MMPs at the tumour site. Obtaining
adequate tumour tissues for testing MMP expression is
difficult to achieve in large-scale clinical trials. As a
result, the actual BEs of MMPI at the tumour site may
not be able to be determined.Although one assumes that higher doses of agents, if
tolerable, will provide at least the same effect, if not
more, there is no rationale to escalate the dose any
further if pharmacokinetic analysis implies saturable
absorption beyond a certain dose. The phase II�/III
doses of BAY 12-9566 and prinomastat were both
determined from this rationale using pharmacokinetic
data [108,109]. As there was no valid surrogate markersof BE or any knowledge about the effect of prolonged
inhibition of MMPs on MMP regulation and cross
reactivity, the investigators related plasma steady state
trough levels of BAY12-9566 to the inhibiting concen-
trations for the target MMPs i.e. MMP-2, MMP-3 and
MMP-9 [108�/110]. These trough levels were of multiple
orders of magnitude higher than the inhibiting concen-
trations for the target MMPs. As BAY12-9566 was99.9% protein bound, these plasma concentrations
included both free and bound drug. It was unclear
what levels of free drug were required to achieve optimal
concentrations of drug in tumour tissue and inhibition
of MMPs in the tumour milieu.
Phase II trial designs are also problematic for MMPI
although many agents appear to move directly fromphase I to phase III. Rather than assessing tumour
shrinkage, a different clinical end point (e.g. progression
free survival) may be more meaningful. One could still
use a standard two stage design that targets progression
free survival or time to treatment failure. However, one
potential problem in a two-stage design with an out-
come that takes 6 months or 1 year to evaluate is that
the accrual of the first stage can be completed for sometime before it is known whether there are sufficient
clinical efficacy to continue on to the second stage of the
trial. One of the solutions would be allowance for a
slight over-accrual to the first stage [111] or incorporat-
ing tumour response as an additional criterion to
proceed to second stage such as the Zee’s design [112].
Many MMPIs moved directly to large definitive phase
III testing where survival is the primary endpoint. Mosttrials reported so far failed to make any impact on
survival either compared with chemotherapy or in
combination with chemotherapy. These trials are re-
source intensive in terms of patients, funding and
planning time. As such the pharmaceutical companies
and the clinicians have learnt a very dear lesson. Before
moving onto large scale phase III trials, smaller
randomised screening phase II�/III trials could beplanned to determine optimum situations in which these
MMPIs can be tested.
5. Matrix metalloprotenase inhibitors in clinical
development
5.1. Batimastat (BB-94)
The development of metalloproteinase inhibitors has
been hindered originally by the lack of non-invasive
route of administration. Batimastat (BB-94), one of the
first MMPI developed, was administered directly into
peritoneum or pleural space of patients with malignant
effusion [113�/116]. As batimastat can only be adminis-
tered invasively, widespread use is impractical and its
development has been halted.
5.2. Marimastat
It is a broad spectrum MMPI, which inhibits all the
major MMPs (collagenase, stromelysin, gelatinase A,
gelatinase B and matrilysin). It is the most extensively
tested MMPI among all the current available MMPIs. It
has a good oral bioavailability in contrast to batimastat.In healthy volunteers marimastat was well tolerated at
doses of up to 200 mg twice daily for a week, with a
small rise in transaminases being the only abnormality
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 167

noted in a minority of the patients [117]. Cancer patients
registered higher plasma concentrations of marimastat
than healthy volunteers for the same dosage possibly
due to increased plasma protein binding or alteredhepatic/renal function. Some cancer patients who re-
ceived marimastat for 1�/2 months were noted to
experience arthralgia, tendonitis or myalgia. These side
effects were reversible [118]. About 60% of the drug was
metabolised by the liver and 40% excreted unchanged
via the kidney.
5.2.1. Phase II studies
Six studies have been conducted to assess effects of
marimastat in advanced cancers using serum tumour
markers. A combined analysis of all six studies have
been reported involving 415 patients [118]. Patients with
advanced, serologically progressive pancreatic, color-
ectal, ovarian and prostatic cancers were recruited into
six nonrandomised, dose ranging, multicenter clinical
trials in North America and Europe. Two studies wereconducted in advanced colorectal cancer, one in pan-
creatic cancer, two in ovarian and one in prostatic
cancers. CEA and CA19.9 were used as tumour markers
for colorectal and pancreatic cancer studies. Histologi-
cally proven cancers were required for patients entering
the colorectal study whereas that was not a prerequisite
for pancreatic cancer study. Patients were selected for
these studies on the basis of serum tumour markersabove prespecified levels (�/5 ng/ml CEA and �/37 Iu/
ml CA19-9) and values rising by 25% or more over 4-
week period before entry into the study. An exception
was the North American colorectal cancer study, in
which the required rate of rise of CEA was 25% over 12
weeks. All patients had failed conventional first-line
treatment where offered.
Patients were recruited in sequential dose groups ofeight to ten patients starting at 25 mg twice a day with a
view to dose escalate to 100 mg twice daily. It became
apparent that doses beyond 50 mg twice daily were
poorly tolerated. Thereafter, doses were titrated down-
ward and patients were recruited at 10 and 5 mg twice
daily and 5�/25 mg once daily. Patients received
marimastat for 4 weeks, except in the North American
colorectal cancer study, in which treatment was for 12weeks. Patients who benefited from marimastat treat-
ment were allowed into a long term continuation arm in
which they could continue for as long as clinical benefits
continued.
A BE was defined as a rate of rise of tumour marker
at the end of treatment period of B/0% (i.e. tumour
marker level no greater than that on day 0). A partial
biological effect (PBE) was defined as a rate of rise oftumour marker of between 0 and 25%. Any patients
with rise of tumour markers �/25% were classified as
non-responders.
However, practical difficulties during these studies
meant that blood samples were not always taken at
scheduled times or according to the protocol. A pre-
analysis algorithm was devised that selected those
tumour marker measurements that adhered most closely
to the protocol for analysis. Both per-algorithm and
intention-to-treat data were presented in the paper.
About 54% were eligible for tumour marker response
analysis per algorithm. Only 75% of patients were
included in the intention-to-treat analysis with a total
of 103 patients excluded from any analysis. These
studies represented an unconventional way of presenting
clinical data and interpretation was, therefore, proble-
matic.
A total of 131 patients and 64 patients were enrolled
into colorectal and pancreatic cancer studies, respec-
tively. Although one of the colorectal studies and the
pancreatic cancer study was published separated
[119,120], results in this paper were presented as
combined analysis for all solid tumour groups. Propor-
tion of patients showing a BE and PBE increased as
dose increased. No correlation was found between
trough levels of marimastat and BE or PBE. Survival
was significantly different in favour of those patients
showing a BE or PBE, but patients suffering early
deaths are excluded from the nonresponders to avoid an
exaggeration of the poorer survival curve. Based on the
combined analysis of biological activity described,
pharmacokinetic data and the observation of dose
related musculoskeletal pain, dose range of 5�/25 mg
twice daily was identified for future studies.
The analysis in pancreatic cancer has been published
separately in full [120]. The most pronounced reduction
in CA19-9 rate of rise was observed at a dose of 10 mg
twice daily. As expected, no objective tumour response
was observed on CT scan. Overall median survival was
160 days and 1 year survival was 21%. The group of
patients was heterogeneous with regard to exposure to
cytotoxic agents and therefore, it would be difficult to
make direct comparison with published studies of
cytotoxic chemotherapy. No correlation between anti-
gen response and survival was found.
Another multicentre phase II study of marimastat in
advanced pancreatic cancer evaluated 113 patients in
three different doses. The majority of patients received
25 mg once daily. Survival was better in patients with
stable or falling CA19-9 level than those with rising
CA19-9 (245 vs. 128 days, respectively). Half of the
patients who completed the study had stabilisation or
reduction in pain, mobility and analgesia scores [121].
The European colorectal study has also been pub-
lished in full [119]. A total of 70 patients were recruited.
Highest percentages of patients achieving BE and PBE
were found in 10 and 50 mg twice daily dose groups.
Changes in CEA were more substantial in the higher
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176168

dose groups and more evident in twice daily dosing of
marimastat.
Tierney et al. reported a study of marimastat in
gastric cancer [122]. Patients with inoperable, histologi-cally proven primary or recurrent gastric or gastro-
oesophageal carcinoma were enrolled. Evidence of
biological activity for marimastat was assessed by
changes in the endoscopic and histological appearance
of the tumour. Study medication dose was originally
intended to be 50 mg twice daily for 28 days, but was
subsequently reduced to 25 mg once daily after realisa-
tion of higher trough level in these groups of patientscompared with healthy volunteers and the development
of musculoskeletal events (MSE) at higher doses. All
patients underwent endoscopic examination at days 0
and 28. Thirty-five patients were enrolled in this study.
No macroscopic changes in tumour size were noted. Ten
patients had a definite increased fibrous cover. Micro-
scopically, no changes in degree of differentiation of
tumour, necrosis, inflammatory infiltrate of fibroustissue were noted. Although authors remarked that the
apparent increase in fibrotic tissue in the gastric tumours
of some patients was one of the most striking evidence
of marimastat’s biological activity, no direct indication
of tumour apoptosis or arrest of angiogenic process was
shown. This again emphasises the importance of better
histological and molecular markers in assessing this
novel class of cytostatic drugs.
5.2.2. Phase III studies
Bramhall et al. reported a randomised study compar-
ing marimastat to gemcitabine as first line therapy in
patients with non resectable pancreatic cancer [123]. As
discussed before, phase II studies have shown the
feasibility of a dose range of 5�/25 mg twice daily for
marimastat, but no single dose has emerged as the most
efficacious. This study, therefore, randomised patientsinto three different doses of marimastat (5, 10 and 25 mg
twice daily) or gemcitabine. Comparison between mar-
imastat and gemcitabine was open-labelled, whereas
comparison between the marimastat dose levels were
double-blind. A total of 414 patients were enrolled.
There was no difference in overall survival between the
marimastat 25 mg group and the gemcitabine group.
Overall survivals of marimastat 5 and 10 mg groupswere poorer than gemcitabine. The multivariate analysis
using Cox proportional hazard model confirmed this. In
addition, a statistically worse progression free survival
was found with each of the three marimastat treatment
groups compared with gemcitabine. Further exploratory
analyses revealed that patients who received marimastat
enjoyed better survival if they had non-metastatic
disease (stages I, II and III) compared with metastaticdisease (stage IV). Tolerability was similar except more
MSE were reported with marimastat and more haema-
tological toxicity with gemcitabine.
Limited data are available on a further study of
gemcitabine with or without marimastat (10 mg twice
daily) in advanced pancreatic cancer. About 239
patients have been recruited and the combination ofmarimastat and gemcitabine failed to show a significant
improvement in survival. Moreover analyses of second-
ary end-points did not reveal significant benefit attribu-
table to marimastat. No quality of life advantages were
seen, but there was a trend indicating that patients with
less extensive disease responded better to marimastat.
Marimastat has also been tested in a randomised trial
in gastric cancer [124]. Three hundred and sixty-sevenpatients were recruited in 37 centres. Patients with
inoperable gastric carcinoma were randomised to mar-
imastat 10 mg twice daily or placebo. The primary
endpoint was overall survival and the secondary end-
points were progression-free survival, overall survival
for patients who received chemotherapy, quality of life
and safety. There was a trend towards survival advan-
tage in the marimastat group at censored date and withfurther follow up a significant survival advantage
emerged in the marimastat group. In subset analysis,
patients who received prior chemotherapy had better
survival in the marimastat group, especially in those
who had no evidence of distant metastasis. Progression
free survival showed similar trend. About 1 year survival
was 20% for marimastat and 14% for placebo. This is
the first randomised trial that has shown a favourablesurvival outcome for patients treated with MMPI in
gastrointestinal cancers.
5.2.3. Combination with other cytotoxic and angiogenesis
therapy
No combination therapy trials have been published in
full including the aforementioned phase III study with
gemcitabine in advanced pancreatic cancer. Phase I dose
escalation study of marimastat with gemcitabine inunresectable pancreatic cancer was reported by Carmi-
chael et al. [125]. Sequential dose levels of marimastat of
5, 10, 15 and 20 mg twice daily with gemcitabine at 1000
mg/m2 weekly for 3 out of 4 weeks were studied. Thirty-
one patients were enrolled and grade 3 and 4 toxicities
included deranged liver function, myelosuppression and
back pain. About two responses out of 11 were noted
and six had stable disease.Another study combining 5-FU with marimastat was
reported [126]. Thirteen patients with solid tumour
malignancies were enrolled. 5-FU was administered
either by continuous infusion (300 mg/m2 per day) or
by bolus Mayo schedule (425 mg/m2 per day with
leucovorin 20 mg/m2 per day for 5 days every 4 weeks).
Marimastat was started 1 week prior to 5-FU. The
starting dose was 5 mg twice daily escalating to 10 mgtwice daily. Only one patient with grade 3 neck pain was
noted who had a past history of cervical spondylosis and
toxicity resolved with discontinuation of marimastat
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 169

and subsequent retreatment with 50% dose reduction
was well tolerated.
Marimastat has also been combined with other
putative antiangiogenesis therapy. Jones et al. reportedon the use of marimastat 10 mg twice daily, dalteparin
and captopril in 17 patients with colorectal and other
solid tumours [127]. Plasma VEGF, thrombin�/antith-
rombin complexes (TAT) and tissue factor (TF) were
measured at baseline and after 1 month of treatment as
surrogate markers of angiogenesis. This combination
was well tolerated with the most common toxicity being
musculoskeletal pain. VEGF fell in five patients. TATand TF increased in five patients. These data are
preliminary, but support the increasing recognition of
the need to employ alternative biochemical and mole-
cular outcome measures in assessing these novel drugs.
5.2.4. Adjuvant therapy
Preclinical and clinical studies suggested that marima-
stat works best in patients with less extensive disease and
smaller tumour volume. MMPI would, therefore, beideal in adjuvant setting. Its favourable tolerability as
oral medication would also be an distinct advantage for
long term use. No published data have been identified in
gastrointestinal tumours. However, Miller et al. re-
ported a randomised phase II trial of adjuvant marima-
stat in patients with early breast cancer [128]. Patients
with high risk node negative or node positive breast
cancer were randomised to receive either 5 or 10 mgmarimastat twice daily for 12 months. Marimastat was
given either as single agent following completion of
adjuvant chemotherapy with doxorubicin and cyclopho-
sphamide or concurrently with tamoxifen. Sixty-three
patients were recruited so far. Main toxicity encountered
was musculoskeletal with 17% of patient required dose
interruptions or reductions. Recurrence and overall
survival have not been reported, but the data would betoo premature at this stage.
5.2.5. Safety profiles
The most commonly reported adverse events of
marimastat were MSE, particularly arthralgia, myalgia
and back pain. These events appeared to be dose-
related. The symptoms very often started in the small
joints in the hand, as well as the shoulder girdle, oftenon the dominant side. If dosing continued unchanged,
the symptoms would spread to involve other joints as
well. These symptoms responded poorly to non-steroi-
dal anti-inflammatory drugs. These side effects also
became more frequent beyond 28 days of treatment,
particularly at doses of 25 mg twice daily and higher.
MMP activation is known to be important in wound
healing and bone resorption. There is increasing evi-dence that MMPs are important in the pathogenesis of
rheumatoid arthritis. Not surprisingly marimastat,
being a broad spectrum MMPI, would interfere with
the normal bone remodelling process. However, these
MSE often subside following a brief period of absti-
nence and lower restarting dose of marimastat.
Safety review of marimastat has been presented withover 1109 patients enrolled in four ongoing placebo
controlled studies [129]. About 10 mg twice daily dose
has been utilised in these ongoing studies. At least 70
patients received more than 6 months of therapy. MSE
emerged as the only observable toxicity. About 5% of
patients required discontinuation of therapy due to
adverse events. 66.3% of patients receiving marimastat
developed any MSE with 3.1% classified as seriousadverse events. There was a high level of background
symptoms in these patients with advanced cancers as
evidenced by the high percentage of MSE in the placebo
group. Other toxicities noted in phase II studies such as
liver dysfunction have not been confirmed.
5.3. BAYER 12-9566 (Tanomastat)
This is a novel nonpeptidic biphenyl compound whichinhibits MMP-2 and MMP-9. Four phase I studies have
now published [108�/110,130]. Doses up to 1600 mg per
day were well tolerated with biologically relevant plasma
steady state concentrations. The predominant toxicities
were asymtomatic thrombocytopenia, anaemia and
hepatic transaminase elevation. There is a lack of
MSE with BAY 12-9566. A dose of 800 mg twice a
day was recommended in further studies.Biological correlative studies were undertaken during
these phase I studies. There was no consistent effects of
BAY 12-9566 on the plasma concentrations of MMP-2
and MMP-9 over the continuous dosing period at any
dose schedule level using ELISA assay. However,
plasma levels of TIMP-2 seemed to increase in a dose-
dependent manner [109]. In a separate study, changes in
plasma VEGF, bFGF and urinary pyridinoline anddeoxypyridinoline crosslinks were measured and no
significant association was found with BAY12-9566
administration [108]. Duivenvoorden et al. showed
that BAY12-9566 was effective in lowering the plasma
MMP-2 and MMP-9 activity using fluorimetric gelati-
nase assay [90].
In a phase III study in chemonaıve patients with
metastatic pancreatic carcinoma, a dose of 800 mg twicea day was used [131]. Patients were randomised between
gemcitabine (G) and BAY 12-9566 (BAY). Patients who
progressed on BAY 12-9566 were permitted to cross
over to gemcitabine but not vice versa. Study was
terminated prematurely after the second interim analysis
when 277 patients had been recruited. Two hundred and
thirty-nine deaths had occurred at final analysis. Base-
line patient characteristics were balanced. Haematolo-gical toxicity was significantly more frequent in
gemcitabine arm. There were no treatment related
deaths. However, median progression free survival and
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176170

overall survival were both superior in gemcitabine
compared with BAY 12-9566 [Median progression free
survival: 3.4 (G) vs. 1.68 months (BAY) P�/0.0001
Median overall survival: 6.67 (G) vs. 3.74 months(BAY) P�/0.0001]. One third of the patients who
progressed on BAY 12-9566 received gemcitabine sal-
vage. Multivariate analysis identified treatment with
BAY 12-9566, pain, metastatic disease, elevated aspar-
tate transaminase and alkaline phosphatase to be
adverse prognostic markers in this study. In view of
these highly significant adverse results in this study and
excessive deaths occurring in the BAY 12-9566 arm inanother small cell lung cancer trial, all clinical studies
with BAY 12-9566 had been suspended temporarily.
5.4. MMI270 (CGS27023A)
This is another orally administered, broad spectrum
MMPI with antitumour activity in preclinical models. It
inhibits MMP-1, 2, 3, 9 and 13. In a phase I study, 92
patients were enrolled (26 with colorectal and five withgastric cancers) [132]. No myelosuppression was seen.
Two main non-haematological toxicities were maculo-
papular rash and MSE. The rash was symmetrical
affecting trunk and arms generally. The maximum
tolerated dose was 300 mg twice daily. At all dose levels
higher than this, there was a marked increase in both the
incidence and severity of rash. Biological correlative
studies were undertaken. Although there was a dose-response increase of MMP-2 and TIMP-1 with MMI-
270, no correlation was seen with other biomarkers
being investigated (see Table 4). Clear interpretation of
the data was problematic reinforcing the need for better
surrogate marker for MMPIs.
Another phase I study in combination with 5-FU and
folinic acid (according to de Gramont’s regimen) has
been reported in which 18 patients with colorectalcancer were enrolled. Eleven had received previous
fluropyrimidine chemotherapy. Grade 3 toxicity oc-
curred in MSE. Two partial response and ten stable
diseases were seen. It was feasible to administer
MMI270 at 300 mg twice daily in combination with 5-
FU and leucovorin [133].
5.5. AG3340 (Prinomastat)
It is a potent selective inhibitor for MMP-2 (gelati-
nase A), MMP-3, MMP-9 and MMP-13. As MMP-1 is
believed to be associated with joint related toxicities,
prinomastat has been designed selectivity to inhibit
MMPs that are most commonly associated with growing
and invasive tumours while sparing the musculoskeletal
system. Phase I studies showed that doses below 25 mgtwice daily were well tolerated. Prinomastat has also
been tested in combination with a number of chemother-
apy drugs in solid tumour and toxicities attributable to
prinomastat was minimal [134,135]. Although no phase
III trials have been reported in GI tumours, two phase
III studies were presented recently in advanced non-
small cell lung cancer (NSCLC) [136] and metastatichormone refractory prostate cancer (HRPC) [137]. Both
studies involved concomitant administration of prino-
mastat with chemotherapy compared with chemother-
apy alone. When prinomastat at different doses was
combined with paclitaxel and carboplatin in chemonaıve
patients with advanced NSCLC, no differences were
observed among treatment groups in overall or 1-year
survival, progression-free survival, symptomatic pro-gression free survival or response rate. Adding prinoma-
stat to cytotoxic drug combination did not, therefore,
enhance efficacy. Similar results were found when
different doses of prinomastat were added to mitoxan-
trone and prednisolone in chemonaıve patients with
metastatic HRPC.
5.6. COL-3
Tetracycline and its derivatives inhibit collagenase
activity in a range of cell types. Probable mechanisms
for inhibiting MMP include divalent cation chelation of
zinc at the active site of the MMPs, down regulation of
the production of the proenzyme, inhibition of the
oxidative activation of the proenzyme and an increase
in the degradation of the proenzyme. COL-3 wasproduced by modifying the basic tetracycline structure.
The anitmicrobial properties of the molecule were
eliminated whereas the MMP inhibitory properties
were retained. COL-3 is a competitive inhibitor of
MMP-2 and MMP-9 [138].
In a phase I study, 35 patients with advanced
refractory metastatic cancer were recruited [138]. Cuta-
neous phototoxicity was dose-limiting at 98mg/m2 perday. As grade 2 phototoxicity was observed at the first
dose level, mandatory routine use of sunblock with a
sun protection factor of 28 was implemented with dose
level 2. The sunblock used protected against ultraviolet
(UV)A and UVB. In addition, sun avoidance and
covering sun-exposed areas with clothing were advised.
The phototoxicity resolved with time at lower doses
despite continuation of treatment. Two other unusualside effects were noted with COL-3. Three patients
developed drug induced lupus [139]. These patients
developed sunburnlike eruptions accompanied by fever
and a positive antinuclear antibody titer. Two out of
three had positive antihistone antibody levels and
arthralgia. One patient had marked systemic manifesta-
tions including pulmonary infiltrates. Another unusual
side effect was the development of sideroblastic anaemiain three patients confirmed on bone marrow examina-
tion [140]. Efficacy appeared to be more marked in
patients with tumours of nonepithelial origin.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 171

Plasma MMP-2 and MMP-9 and serum VEGF and
bFGF levels were determined using ELISA kit in this
study. There was a possible significant relationship
between changes in plasma MMP-2 levels and cumula-tive doses of drug when progressive disease patients
were compared with those with stable disease.
In another phase 1 study with 26 patients, principal
toxicities were photosensitivity skin reactions and asth-
enia [141]. Correlative biological studies were performed
assessing MMP-2, MMP-9 expression in peripheral
blood mononuclear cells and skin biopsies using ELISA,
Western Blot and immunohistochemical techniques.Plasma concentrations of MMP-2 and MMP-9 were
decreased in a proportion of patients as well as the
expression of MMP-9 in peripheral blood mononuclear
cells. No changes were observed in MMPs expression in
serial skin biopsies.
5.7. BMS-275291
This is a novel, non-hydroxamate MMPI designed toinhibit a broad spectrum of MMPs (including MMP2
and MMP9) and spare sheddases. Sheddase inhibition is
hypothesised to induce the dose limiting arthritis
observed with earlier hydroxamate based MMPIs. In
healthy subjects, BMS-275291 was safe and very well
tolerated across all dose levels [142]. In cancer patients,
maximum tolerated dose was not reached in two phase I
studies [143,144]. One thousand and two hundred mgper day was chosen for phase II studies as the trough
concentration of BMS-275291 exceed IC90s (concentra-
tion that causes 90% of enzyme inhibition) of multiple
MMPs.
6. Conclusion
MMPs are novel targets for gastrointestinal malig-
nancies. Matrix metalloproteinase inhibitors remain an
important and potentially useful class of drugs for
cancer therapy, but they have not clearly demonstrated
clinically significant activity when used as single agents
in patients with advanced stage gastrointestinal cancers.
They have not been adequately tested in early stage
(adjuvant) settings or in combination with chemother-apy or radiation, where their impact may be the greatest.
The recent lack of success from large phase III MMPI
studies in solid tumours would obviously dampen the
enthusiasm of both the clinicians and the pharmaceu-
tical industries in investing further MMPI studies.
However, the superior results of marimastat in advanced
gastric cancer patients who have received chemotherapy
are encouraging. The challenges in future studies wouldlie at targeting the correct matrix metalloproteinase at
the correct time point in tumour growth and metastasis,
and the correct sequence of using MMPIs with other
cytotoxic agents. Identification of reliable surrogate
biomarkers and incorporation of functional imaging
will be of high priority in the clinical development of
matrix metalloproteinase inhibitors in gastrointestinalmalignancies.
Reviewers
Nicholas R. Lemoine, Professor of Molecular Pathol-
ogy, Head of Molecular Oncology, Imperial Collage
School of Medicine, Department of Cancer Medicine,Hammersmith Campus, Du Cane Road, London W12
0NN, UK.
Dr Arnaud Roth, Department de chirurgie, Onco-
chirurgie, Hopitaux Universitaires de Geneve 24, rue
Micheli-du-Crest, CH-1211 Geneve 4, Switzerland.
Professor K. Kesteloot, K. U. Leuven. Centre for
Health Sciences & Nursing Research, Kapucynenvoer
35, B-3000 Leuven, Belgium.
Acknowledgements
The Organizing Committee of the ICACT Conference
on ‘New Drugs in GI Malignancies’ held from 30
January to 1 February 2000 in Paris.
References
[1] Ferlay J, Bray F, Pisani P, Parkin DM. GLOBOCAN 2000-Cancer
Incidence, Mortality and Prevalence Worldwide, Version 1.0.
2001.
[2] Office for National Statistics. Cancer Survival Trends in England
and Wales 1971�/1995. 1999.
[3] Anderson IC, Shipp MA, Docherty AJP, Teicher BA. Combina-
tion therapy including a gelatinase inhibitor and cytotoxic agent
reduces local invasion and metastasis of murine Lewis lung
carcinoma. Cancer Res 1996;56:715�/8.
[4] Foekens JA, Portengen H, Look MP, et al. Relationship of PS2
with response to tamoxifen therapy in patients with recurrent
breast cancer. Br J Cancer 1994;70:1217�/23.
[5] Grondahl-Hansen J, Christensen IJ, Rosenquist C, et al. High
levels of urokinase-type plasminogen activator and its inhibitor
PAI-1 in cytosolic extracts of breast carcinomas are associated
with poor prognosis. Cancer Res 1993;53:2513�/21.
[6] Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie
S. Metastatic potential correlates with enzymatic degradation of
basement membrane collagen. Nature 1980;284:67�/8.
[7] Stetler-Stevenson WG. Type IV collagenases in tumor invasion
and metastasis. Cancer Metastasis Rev 1990;9:289�/303.
[8] van der Stappen JW, Hendriks T, Wobbes T. Correlation
between collagenolytic activity and grade of histological differ-
entiation in colorectal tumors. Int J Cancer 1990;45:1071�/8.
[9] Bernhard EJ, Gruber SB, Muschel RJ. Direct evidence linking
expression of matrix metalloproteinase 9 (92-kDa gelatinase/
collagenase) to the metastatic phenotype in transformed rat
embryo cells. Proc Natl Acad Sci USA 1994;91:4293�/7.
[10] Kawamata H, Kameyama S, Kawai K, et al. Marked accelera-
tion of the metastatic phenotype of a rat bladder carcinoma cell
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176172

line by the expression of human gelatinase A. Int J Cancer
1995;63:568�/75.
[11] Grondahl-Hansen J, Ralfkiaer E, Kirkeby LT, Kristensen P,
Lund LR, Dano K. Localization of urokinase-type plasminogen
activator in stromal cells in adenocarcinomas of the colon in
humans. Am J Pathol 1991;138:111�/7.
[12] Pyke C, Kristensen P, Ralfkiaer E, et al. Urokinase-type
plasminogen activator is expressed in stromal cells and its
receptor in cancer cells at invasive foci in human colon
adenocarcinomas. Am J Pathol 1991;138:1059�/67.
[13] Pyke C, Kristensen P, Ralfkiaer E, Eriksen J, Dano K. The
plasminogen activation system in human colon cancer: messen-
ger RNA for the inhibitor PAI-1 is located in endothelial cells in
the tumor stroma. Cancer Res 1991;51:4067�/71.
[14] Pedersen H, Grondahl-Hansen J, Francis D, et al. Urokinase
and plasminogen activator inhibitor type 1 in pulmonary
adenocarcinoma. Cancer Res 1994;54:120�/3.
[15] McDonnell S, Navre M, Coffey RJ, Jr, Matrisian LM. Expres-
sion and localization of the matrix metalloproteinase pump-1
(MMP-7) in human gastric and colon carcinomas. Mol Carcinog
1991;4:527�/33.
[16] Okada A, Bellocq JP, Rouyer N, et al. Membrane-type matrix
metalloproteinase (MT-MMP) gene is expressed in stromal cells
of human colon, breast, and head and neck carcinomas. Proc
Natl Acad Sci USA 1995;92:2730�/4.
[17] Pyke C, Ralfkiaer E, Tryggvason K, Dano K. Messenger RNA
for two type IV collagenases is located in stromal cells in human
colon cancer. Am J Pathol 1993;142:359�/65.
[18] Gress TM, Muller-Pillasch F, Lerch MM, Friess H, Buchler M,
Adler G. Expression and in-situ localization of genes coding for
extracellular matrix proteins and extracellular matrix degrading
proteases in pancreatic cancer. Int J Cancer 1995;62:407�/13.
[19] Satoh K, Ohtani H, Shimosegawa T, Koizumi M, Sawai T,
Toyota T. Infrequent stromal expression of gelatinase A and
intact basement membrane in intraductal neoplasms of the
pancreas. Gastroenterology 1994;107:1488�/95.
[20] Bramhall SR, Neoptolemos JP, Stamp GW, Lemoine NR.
Imbalance of expression of matrix metalloproteinases (MMPs)
and tissue inhibitors of the matrix metalloproteinases (TIMPs) in
human pancreatic carcinoma. J Pathol 1997;182:347�/55.
[21] Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H,
Itohara S. Reduced angiogenesis and tumor progression in
gelatinase A-deficient mice. Cancer Res 1998;58:1048�/51.
[22] Fridman R, Fuerst TR, Bird RE, et al. Domain structure of
human 72-kDa gelatinase/type IV collagenase. Characterization
of proteolytic activity and identification of the tissue inhibitor of
metalloproteinase-2 (TIMP-2) binding regions. J Biol Chem
1992;267:15398�/405.
[23] Murphy G, Allan JA, Willenbrock F, Cockett MI, O’Connell JP,
Docherty AJ. The role of the C-terminal domain in collagenase
and stromelysin specificity. J Biol Chem 1992;267:9612�/8.
[24] Sato H, Kinoshita T, Takino T, Nakayama K, Seiki M.
Activation of a recombinant membrane type 1-matrix metallo-
proteinase (MT1-MMP) by furin and its interaction with tissue
inhibitor of metalloproteinases (TIMP)-2. FEBS Lett
1996;393:101�/4.
[25] Sato H, Takino T, Okada Y, et al. A matrix metalloproteinase
expressed on the surface of invasive tumour cells. Nature
1994;370:61�/5.
[26] Mignatti P, Rifkin DB. Biology and biochemistry of proteinases
in tumor invasion. Physiol Rev 1993;73:161�/95.
[27] Murphy G, Atkinson S, Ward R, Gavrilovic J, Reynolds JJ. The
role of plasminogen activators in the regulation of connective
tissue metalloproteinases. Ann New York Acad Sci 1992;667:1�/
12.
[28] Kinoshita T, Sato H, Okada A, et al. TIMP-2 promotes
activation of progelatinase A by membrane-type 1 matrix
metalloproteinase immobilized on agarose beads. J Biol Chem
1998;273:16098�/103.
[29] Knauper V, Cowell S, Smith B, et al. The role of the C-terminal
domain of human collagenase-3 (MMP-13) in the activation of
procollagenase-3, substrate specificity, and tissue inhibitor of
metalloproteinase interaction. J Biol Chem 1997;272:7608�/16.
[30] Docherty AJ, Lyons A, Smith BJ, et al. Sequence of human
tissue inhibitor of metalloproteinases and its identity to ery-
throid-potentiating activity. Nature 1985;318:66�/9.
[31] Stetler-Stevenson WG, Krutzsch HC, Liotta LA. Tissue inhi-
bitor of metalloproteinase (TIMP-2). A new member of the
metalloproteinase inhibitor family. J Biol Chem
1989;264:17374�/8.
[32] Pavloff N, Staskus PW, Kishnani NS, Hawkes SP. A new
inhibitor of metalloproteinases from chicken: ChIMP-3. A third
member of the TIMP family. J Biol Chem 1992;267:17321�/6.
[33] Uria JA, Ferrando AA, Velasco G, Freije JM, Lopez-Otin C.
Structure and expression in breast tumors of human TIMP-3, a
new member of the metalloproteinase inhibitor family. Cancer
Res 1994;54:2091�/4.
[34] Greene J, Wang M, Liu YE, Raymond LA, Rosen C, Shi YE.
Molecular cloning and characterization of human tissue inhi-
bitor of metalloproteinase. J Biol Chem 1996;271:30375�/80.
[35] Leco KJ, Apte SS, Taniguchi GT, et al. Murine tissue inhibitor
of metalloproteinases-4 (TIMP-4): cDNA isolation and expres-
sion in adult mouse tissues. FEBS Lett 1997;401:213�/7.
[36] Williamson RA, Marston FA, Angal S, et al. Disulphide bond
assignment in human tissue inhibitor of metalloproteinases
(TIMP). Biochem J 1990;268:267�/74.
[37] Murphy G, Houbrechts A, Cockett MI, Williamson RA, O’Shea
M, Docherty AJ. The N-terminal domain of tissue inhibitor of
metalloproteinases retains metalloproteinase inhibitory activity.
Biochemistry 1991;30:8097�/102.
[38] Murphy GJ, Murphy G, Reynolds JJ. The origin of matrix
metalloproteinases and their familial relationships. FEBS Lett
1991;289:4�/7.
[39] O’Shea M, Willenbrock F, Williamson RA, et al. Site-directed
mutations that alter the inhibitory activity of the tissue inhibitor
of metalloproteinases-1: importance of the N-terminal region
between cysteine 3 and cysteine 13. Biochemistry
1992;31:10146�/52.
[40] Willenbrock F, Crabbe T, Slocombe PM, et al. The activity of
the tissue inhibitors of metalloproteinases is regulated by C-
terminal domain interactions: a kinetic analysis of the inhibition
of gelatinase A. Biochemistry 1993;32:4330�/7.
[41] Murphy G, Willenbrock F, Ward RV, Cockett MI, Eaton D,
Docherty AJ. The C-terminal domain of 72 kDa gelatinase A is
not required for catalysis, but is essential for membrane
activation and modulates interactions with tissue inhibitors of
metalloproteinases. Biochem J 1992;283(Pt 3):637�/41.
[42] Gomis-Ruth FX, Maskos K, Betz M, et al. Mechanism of
inhibition of the human matrix metalloproteinase stromelysin-1
by TIMP-1. Nature 1997;389:77�/81.
[43] Overall CM, King AE, Sam DK, et al. Identification of the tissue
inhibitor of metalloproteinases-2 (TIMP-2) binding site on the
hemopexin carboxyl domain of human gelatinase A by site-
directed mutagenesis. The hierarchical role in binding TIMP-2 of
the unique cationic clusters of hemopexin modules III and IV. J
Biol Chem 1999;274:4421�/9.
[44] Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA,
Goldberg GI. SV40-transformed human lung fibroblasts secrete
a 92-kDa type IV collagenase which is identical to that secreted
by normal human macrophages. J Biol Chem 1989;264:17213�/
21.
[45] Gasson JC, Golde DW, Kaufman SE, et al. Molecular char-
acterization and expression of the gene encoding human
erythroid-potentiating activity. Nature 1985;315:768�/71.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 173

[46] Hayakawa T, Yamashita K, Tanzawa K, Uchijima E, Iwata K.
Growth-promoting activity of tissue inhibitor of metalloprotei-
nases-1 (TIMP-1) for a wide range of cells. A possible new
growth factor in serum. FEBS Lett 1992;298:29�/32.
[47] Ritter LM, Garfield SH, Thorgeirsson UP. Tissue inhibitor of
metalloproteinases-1 (TIMP-1) binds to the cell surface and
translocates to the nucleus of human MCF-7 breast carcinoma
cells. Biochem Biophys Res Commun 1999;257:494�/9.
[48] Kikuchi K, Kadono T, Furue M, Tamaki K. Tissue inhibitor of
metalloproteinase 1 (TIMP-1) may be an autocrine growth
factor in scleroderma fibroblasts. J Invest Dermatol
1997;108:281�/4.
[49] Albini A, Fontanini G, Masiello L, et al. Angiogenic potential in
vivo by Kaposi’s sarcoma cell-free supernatants and HIV-1 tat
product: inhibition of KS-like lesions by tissue inhibitor of
metalloproteinase-2. AIDS 1994;8:1237�/44.
[50] Anand-Apte B, Pepper MS, Voest E, et al. Inhibition of
angiogenesis by tissue inhibitor of metalloproteinase-3. Invest
Ophthalmol Vis Sci 1997;38:817�/23.
[51] Johnson MD, Kim HR, Chesler L, Tsao-Wu G, Bouck N,
Polverini PJ. Inhibition of angiogenesis by tissue inhibitor of
metalloproteinase. J Cell Physiol 1994;160:194�/202.
[52] Murphy AN, Unsworth EJ, Stetler-Stevenson WG. Tissue
inhibitor of metalloproteinases-2 inhibits bFGF-induced human
microvascular endothelial cell proliferation. J Cell Physiol
1993;157:351�/8.
[53] Schnaper HW, Grant DS, Stetler-Stevenson WG, et al. Type IV
collagenase(s) and TIMPs modulate endothelial cell morphogen-
esis in vitro. J Cell Physiol 1993;156:235�/46.
[54] Takigawa M, Nishida Y, Suzuki F, Kishi J, Yamashita K,
Hayakawa T. Induction of angiogenesis in chick yolk-sac
membrane by polyamines and its inhibition by tissue inhibitors
of metalloproteinases (TIMP and TIMP-2). Biochem Biophys
Res Commun 1990;171:1264�/71.
[55] Fowlkes JL, Enghild JJ, Suzuki K, Nagase H. Matrix metallo-
proteinases degrade insulin-like growth factor-binding protein-3
in dermal fibroblast cultures. J Biol Chem 1994;269:25742�/6.
[56] Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R.
Fibrillar collagen inhibits arterial smooth muscle proliferation
through regulation of Cdk2 inhibitors. Cell 1996;87:1069�/78.
[57] Guedez L, Stetler-Stevenson WG, Wolff L, et al. In vitro
suppression of programmed cell death of B cells by tissue
inhibitor of metalloproteinases-1. J Clin Invest 1998;102:2002�/
10.
[58] Ahonen M, Baker AH, Kahari VM. Adenovirus-mediated gene
delivery of tissue inhibitor of metalloproteinases-3 inhibits
invasion and induces apoptosis in melanoma cells. Cancer Res
1998;58:2310�/5.
[59] Baker AH, George SJ, Zaltsman AB, Murphy G, Newby AC.
Inhibition of invasion and induction of apoptotic cell death of
cancer cell lines by overexpression of TIMP-3. Br J Cancer
1999;79:1347�/55.
[60] Hida K, Shindoh M, Yasuda M, et al. Antisense E1AF
transfection restrains oral cancer invasion by reducing matrix
metalloproteinase activities. Am J Pathol 1997;150:2125�/32.
[61] Maquoi E, Noel A, Frankenne F, Angliker H, Murphy G,
Foidart JM. Inhibition of matrix metalloproteinase 2 maturation
and HT1080 invasiveness by a synthetic furin inhibitor. FEBS
Lett 1998;424:262�/6.
[62] Eccles SA, Box GM, Court WJ, Bone EA, Thomas W, Brown
PD. Control of lymphatic and hematogenous metastasis of a rat
mammary carcinoma by the matrix metalloproteinase inhibitor
batimastat (BB-94). Cancer Res 1996;56:2815�/22.
[63] Davies B, Brown PD, East N, Crimmin MJ, Balkwill FR. A
synthetic matrix metalloproteinase inhibitor decreases tumor
burden and prolongs survival of mice bearing human ovarian
carcinoma xenografts. Cancer Res 1993;53:2087�/91.
[64] Watson SA, Morris TM, Robinson G, Crimmin MJ, Brown PD,
Hardcastle JD. Inhibition of organ invasion by the matrix
metalloproteinase inhibitor batimastat (BB-94) in two human
colon carcinoma metastasis models. Cancer Res 1995;55:3629�/
33.
[65] Watson SA, Morris TM, Parsons SL, Steele RJ, Brown PD.
Therapeutic effect of the matrix metalloproteinase inhibitor,
batimastat, in a human colorectal cancer ascites model. Br J
Cancer 1996;74:1354�/8.
[66] Price A, Shi QA, Morris D, et al. Marked inhibition of tumor
growth in a malignant glioma tumor model by a novel synthetic
matrix metalloproteinase inhibitor AG3340. Clin Cancer Res
1999;5:845�/54.
[67] Santos O, McDermott CD, Daniels RG, Appelt K. Rodent
pharmacokinetic and anti-tumor efficacy studies with a series of
synthetic inhibitors of matrix metalloproteinases. Clin Exp
Metastasis 1997;15:499�/508.
[68] Gatto C, Rieppi M, Borsotti P, et al. BAY 12-9566, a novel
inhibitor of matrix metalloproteinases with antiangiogenic
activity. Clin Cancer Res 1999;5:3603�/7.
[69] Hibner B, Bull C, Flynn C, et al. Activity of the matrix
metalloproteinase inhibitor BAY 12-9566 against murine sub-
cutaneous and metastatic in vivo models. Ann Oncol 1998;9:283.
[70] Taraboletti G, Garofalo A, Belotti D, et al. Inhibition of
angiogenesis and murine hemangioma growth by batimastat, a
synthetic inhibitor of matrix metalloproteinases. J Natl Cancer
Inst 1995;87:293�/8.
[71] Giavazzi R, Garofalo A, Ferri C, et al. Batimastat, a synthetic
inhibitor of matrix metalloproteinases, potentiates the antitumor
activity of cisplatin in ovarian carcinoma xenografts. Clin
Cancer Res 1998;4:985�/92.
[72] Neri A, Goggin B, Kolis S, Brekken J, Khelemskaya N, Gabriel
L. Pharmacokinetics and efficacy of a novel matrix metallopro-
teinase inhibitor AG3340, in single agent and combination
therapy against B16-F10 melanoma tumors developing in the
lung after IV tail vein implantation in C57BL/6 mice. Proc Am
Assoc Cancer Res 1998;39:302.
[73] Koivunen E, Arap W, Valtanen H, et al. Tumor targeting with a
selective gelatinase inhibitor. Nat Biotechnol 1999;17:768�/74.
[74] Schultz RM, Silberman S, Persky B, Bajkowski AS, Carmichael
DF. Inhibition by human recombinant tissue inhibitor of
metalloproteinases of human amnion invasion and lung coloni-
zation by murine B16-F10 melanoma cells. Cancer Res
1988;48:5539�/45.
[75] Alvarez OA, Carmichael DF, DeClerck YA. Inhibition of
collagenolytic activity and metastasis of tumor cells by a
recombinant human tissue inhibitor of metalloproteinases. J
Natl Cancer Inst 1990;82:589�/95.
[76] Albini A, Melchiori A, Santi L, Liotta LA, Brown PD, Stetler-
Stevenson WG. Tumor cell invasion inhibited by TIMP-2. J Natl
Cancer Inst 1991;83:775�/9.
[77] DeClerck YA, Yean TD, Chan D, Shimada H, Langley KE.
Inhibition of tumor invasion of smooth muscle cell layers by
recombinant human metalloproteinase inhibitor. Cancer Res
1991;51:2151�/7.
[78] DeClerck YA, Perez N, Shimada H, Boone TC, Langley KE,
Taylor SM. Inhibition of invasion and metastasis in cells
transfected with an inhibitor of metalloproteinases. Cancer Res
1992;52:701�/8.
[79] Khokha R. Suppression of the tumorigenic and metastatic
abilities of murine B16-F10 melanoma cells in vivo by the
overexpression of the tissue inhibitor of the metalloproteinases-
1. J Natl Cancer Inst 1994;86:299�/304.
[80] Koop S, Khokha R, Schmidt EE, et al. Overexpression of
metalloproteinase inhibitor in B16F10 cells does not affect
extravasation but reduces tumor growth. Cancer Res
1994;54:4791�/7.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176174

[81] Montgomery AM, Mueller BM, Reisfeld RA, Taylor SM,
DeClerck YA. Effect of tissue inhibitor of the matrix metallo-
proteinases-2 expression on the growth and spontaneous metas-
tasis of a human melanoma cell line. Cancer Res 1994;54:5467�/
73.
[82] Imren S, Kohn DB, Shimada H, Blavier L, DeClerck YA.
Overexpression of tissue inhibitor of metalloproteinases-2 retro-
viral-mediated gene transfer in vivo inhibits tumor growth and
invasion. Cancer Res 1996;56:2891�/5.
[83] Rigg A, Lemoine NR. Adenoviral delivery of TIMP 1 or TIMP 2
can modify the invasive behaviour of pancreatic cancer and
prolong the survival of mice harboring a human pancreatic
tumor, Cancer Gene Ther 2001;11:869�/78.
[84] Gore M, A’Hern R, Stankiewicz M, Slevin M. Tumour marker
levels during marimastat therapy. Lancet 1996;348:263�/4.
[85] Gogas H, Lofts FJ, Evans TR, Daryanani S, Mansi JL. Are
serial measurements of CA19-9 useful in predicting response to
chemotherapy in patients with inoperable adenocarcinoma of the
pancreas. Br J Cancer 1998;77:325�/8.
[86] Halm U, Schumann T, Schiefke I, Witzigmann H, Mossner J,
Keim V. Decrease of CA 19-9 during chemotherapy with
gemcitabine predicts survival time in patients with advanced
pancreatic cancer. Br J Cancer 2000;82:1013�/6.
[87] Ishii H, Okada S, Sato T, et al. CA 19-9 in evaluating the
response to chemotherapy in advanced pancreatic cancer.
Hepatogastroenterology 1997;44:279�/83.
[88] Mann DV, Edwards R, Ho S, Lau WY, Glazer G. Elevated
tumour marker CA19-9: clinical interpretation and influence of
obstructive jaundice. Eur J Surg Oncol 2000;26:474�/9.
[89] Hidalgo M, Eckhardt SG. Matrix metalloproteinase inhibitors:
How can we optimize their development. Ann Oncol
2001;12:285�/7.
[90] Duivenvoorden WCM, Hirte HW, Singh G. Quantification of
matrix metalloproteinase activity in plasma of patients enrolled
in a BAY 12-9566 phase I study. Int J Cancer 2001;91:857�/62.
[91] Stetler-Stevenson WG. Matrix metalloproteinases in angiogen-
esis: a moving target for therapeutic intervention. J Clin Invest
1999;103:1237�/41.
[92] Fanelli M, Locopo N, Gattuso D, Gasparini G. Assessment of
tumor vascularization: immunohistochemical and non- invasive
methods. Int J Biol Markers 1999;14:218�/31.
[93] Weidner N, Semple JP, Welch WR, Folkman J. Tumor
angiogenesis and metastasis*/correlation in invasive breast
carcinoma. New Engl J Med 1991;324:1�/8.
[94] Fox SB, Leek RD, Weekes MP, Whitehouse RM, Gatter KC,
Harris AL. Quantitation and prognostic value of breast cancer
angiogenesis: comparison of microvessel density, Chalkley
count, and computer image analysis. J Pathol 1995;177:275�/83.
[95] Axelsson K, Ljung BM, Moore DH, et al. Tumor angiogenesis
as a prognostic assay for invasive ductal breast carcinoma. J Natl
Cancer Inst 1995;87:997�/1008.
[96] Barbareschi M, Gasparini G, Morelli L, Forti S, Dalla PP.
Novel methods for the determination of the angiogenic activity
of human tumors. Breast Cancer Res Treat 1995;36:181�/92.
[97] Visscher DW, Smilanetz S, Drozdowicz S, Wykes SM. Prog-
nostic significance of image morphometric microvessel enumera-
tion in breast carcinoma. Anal Quant Cytol Histol 1993;15:88�/
92.
[98] Wakui S, Furusato M, Itoh T, et al. Tumour angiogenesis in
prostatic carcinoma with and without bone marrow metastasis: a
morphometric study. J Pathol 1992;168:257�/62.
[99] Libutti SK, Choyke P, Carrasquillo JA, Bacharach S, Neumann
RD. Monitoring responses to antiangiogenic agents using
noninvasive imaging tests. Cancer J Sci Am 1999;5:252�/6.
[100] Leenders KL. PET: blood flow and oxygen consumption in brain
tumors. J Neurooncol 1994;22:269�/73.
[101] Mineura K, Sasajima T, Itoh Y, et al. Blood flow and
metabolism of central neurocytoma: a positron emission tomo-
graphy study. Cancer 1995;76:1224�/32.
[102] Schelbert HR. Blood flow and metabolism by PET. Cardiol Clin
1994;12:303�/15.
[103] Ito K, Kato T, Tadokoro M, et al. Recurrent rectal cancer and
scar: differentiation with PET and MR imaging. Radiology
1992;182:549�/52.
[104] Valk PE, Abella-Columna E, Haseman MK, et al. Whole-body
PET imaging with [18F]fluorodeoxyglucose in management of
recurrent colorectal cancer. Arch Surg 1999;134:503�/11.
[105] Robinson SP, Howe FA, Griffiths JR. Noninvasive monitoring
of carbogen-induced changes in tumor blood flow and oxygena-
tion by functional magnetic resonance imaging. Int J Radiat
Oncol Biol Phys 1995;33:855�/9.
[106] Robinson SP, Howe FA, Rodrigues LM, Stubbs M, Griffiths
JR. Magnetic resonance imaging techniques for monitoring
changes in tumor oxygenation and blood flow. Semin Radiat
Oncol 1998;8:197�/207.
[107] Korn EL, Arbuck SG, Pluda JM, Simon R, Kaplan RS,
Christian MC. Clinical trial designs for cytostatic agents: are
new approaches needed. J Clin Oncol 2001;19:265�/72.
[108] Erlichman C, Adjei AA, Alberts SR, et al. Phase I study of the
matrix metalloproteinase inhibitor, BAY 12-9566. Ann Oncol
2001;12:389�/95.
[109] Hirte H, Goel R, Major P, et al. A phase I dose escalation study
of the matrix metalloproteinase inhibitor BAY 12-9566 adminis-
tered orally in patients with advanced solid tumours. Ann Oncol
2000;11:1579�/84.
[110] Rowinsky EK, Humphrey R, Hammond LA, et al. Phase I and
pharmacologic study of the specific matrix metalloproteinase
inhibitor BAY 12-9566 on a protracted oral daily dosing
schedule in patients with solid malignancies. J Clin Oncol
2000;18:178�/86.
[111] Herndon JE. A design alternative for two-stage, phase II,
multicenter cancer clinical trials. Control Clin Trials
1998;19:440�/50.
[112] Zee B, Melnychuk D, Dancey J, Eisenhauer E. Multinomial
phase II cancer trials incorporating response and early progres-
sion. J Biopharm Stat 1999;9:351�/63.
[113] Beattie GJ, Smyth JF. Phase I study of intraperitoneal metallo-
proteinase inhibitor BB94 in patients with malignant ascites.
Clin Cancer Res 1998;4:1899�/902.
[114] Macaulay VM, O’Byrne KJ, Saunders MP, et al. Phase I study
of intrapleural batimastat (BB-94), a matrix metalloproteinase
inhibitor, in the treatment of malignant pleural effusions. Clin
Cancer Res 1999;5:513�/20.
[115] Parsons SL, Watson SA, Steele RJ. Phase I/II trial of batimastat,
a matrix metalloproteinase inhibitor, in patients with malignant
ascites. Eur J Surg Oncol 1997;23:526�/31.
[116] Wojtowicz-Praga S, Low J, Marshall J, et al. Phase I trial of a
novel matrix metalloproteinase inhibitor batimastat (BB-94) in
patients with advanced cancer. Invest New Drugs 1996;14:193�/
202.
[117] Millar AW, Brown PD, Moore J, et al. Results of single and
repeat dose studies of the oral matrix metalloproteinase inhibitor
marimastat in healthy male volunteers. Br J Clin Pharmacol
1998;45:21�/6.
[118] Nemunaitis J, Poole C, Primrose J, et al. Combined analysis of
studies of the effects of the matrix metalloproteinase inhibitor
marimastat on serum tumor markers in advanced cancer:
selection of a biologically active and tolerable dose for longer-
term studies. Clin Cancer Res 1998;4:1101�/9.
[119] Primrose JN, Bleiberg H, Daniel F, et al. Marimastat in
recurrent colorectal cancer: exploratory evaluation of biological
activity by measurement of carcinoembryonic antigen. Br J
Cancer 1999;79:509�/14.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176 175

[120] Rosemurgy A, Harris J, Langleben A, Casper E, Goode S,
Rasmussen H. Marimastat in patients with advanced pancreatic
cancer: a dose-finding study. Am J Clin Oncol 1999;22:247�/52.
[121] Evans JD, Stark A, Johnson CD, et al. A phase II trial of
marimastat in advanced pancreatic cancer. Br J Cancer
2001;85:1865�/70.
[122] Tierney GM, Griffin NR, Stuart RC, et al. A pilot study of the
safety and effects of the matrix metalloproteinase inhibitor
marimastat in gastric cancer. Eur J Cancer 1999;35:563�/8.
[123] Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA.
Marimastat as first-line therapy for patients with unresectable
pancreatic cancer: a randomized trial. J Clin Oncol
2001;19:3447�/55.
[124] Fielding J, Scholefield J, Stuart R, et al. A ramdomized double-
blind placebo-controlled study of marimastat in patients with
inoperable gastric adenocarcinoma. Proc Am Soc Clin Oncol
2000;19:240a.
[125] Carmichael J, Ledermann J, Woll P, Gulliford T, Russell R.
Phase Ib study of concurrent administration of marimastat and
gemcitabine in non-resectable pancreatic cancer. Proc Am Soc
Clin Oncol 1998;17:232a.
[126] O’Reilly S, Mani S, Ratain M, et al. Schedules of 5FU and the
matrix metalloproteinase inhibitor marimastat: a phase I study.
Proc Am Soc Clin Oncol 1998;17:217a.
[127] Jones P, Elliot M, Dobbs N, et al. Phase I/II study of
combination antiangiogenesis therapy with Marimastat, Capto-
pril and Fragmin. Proc Am Soc Clin Oncol 1999;18:447a.
[128] Miller K, Gradishar W, Schuchter S, et al. A randomized phase
II pilot trial of adjuvant marimastat in patients with early breast
cancer. Proc Am Soc Clin Oncol 2000;19:96a.
[129] Rasmussen H, Rugg T, Brookes C, Baillet M, Harris J. First
placebo controlled safety review of marimastat: a potential new
therapeutic class. Proc Am Soc Clin Oncol 1999;18:193a.
[130] Heath EI, O’Reilly S, Humphrey R, et al. Phase I trial of the
matrix metalloproteinase inhibitor BAY12-9566 in patients with
advanced solid tumors. Cancer Chemother Pharmacol
2001;48:269�/74.
[131] Moore MJ, Hamm J, Eisenberg PD, et al. A comparison
between gemcitabine and the matrix metalloproteinase inhibitor
BAY12-9566 in patients with advanced pancreatic cancer. Proc
Am Soc Clin Oncol 2000;19:240a.
[132] Levitt NC, Eskens FA, O’Byrne KJ, et al. Phase I and
pharmacological study of the oral matrix metalloproteinase
inhibitor, MMI270 (CGS27023A), in patients with advanced
solid cancer. Clin Cancer Res 2001;7:1912�/22.
[133] Eatock M, Cassidy J, Johnson J, et al. A Phase 1 Study of the
Matrix Metalloproteinase Inhibitor MMI270 (Previously
Termed CGS27023A) with 5FU and Folinic Acid. Proc Am
Soc Clin Oncol 1999;18:209a.
[134] Collier M, Shepherd FA, Ahmann F, et al. A novel approach to
studying the efficacy of AG3340, a selective inhibitor of matrix
metalloproteases (MMPs). Proc Am Soc Clin Oncol
1999;18:482a.
[135] D’Olimpio J, Hande K, Collier M, Michelson G, Paradiso L,
Clendeninn N. Phase I study of the matrix metalloprotease
inhibitor AG3340 in combination with paclitaxel and carbopla-
tin for the treatment of patients with advanced solid tumors.
Proc Am Soc Clin Oncol 1999;18:160a.
[136] Smylie M, Mercier R, Aboulafia D, et al. Phase III study of the
matrix metalloprotease (MMP) inhibitor prinomastat in patients
having advanced non-small cell lung cancer (NSCLC). Proc Am
Soc Clin Oncol 2001;20:307a.
[137] Ahmann F, Saad F, Mercier R, et al. Interim results of a phase
III study of the matrix metalloprotease inhibitor prinomastat in
patients having metastatic, hormone refractory prostate cancer
(HRPC). Proc Am Soc Clin Oncol 2001;20:174a.
[138] Rudek MA, Figg WD, Dyer V, et al. Phase I clinical trial of oral
COL-3, a matrix metalloproteinase inhibitor, in patients with
refractory metastatic cancer. J Clin Oncol 2001;19:584�/92.
[139] Ghate JV, Turner ML, Rudek MA, et al. Drug-induced lupus
associated with COL-3-report of 3 cases. Arch Dermatol
2001;137:471�/4.
[140] Rudek MA, Horne M, Figg WD, et al. Reversible sideroblastic
anemia associated with the tetracycline analogue COL-3. Am J
Hematol 2001;67:51�/3.
[141] Munoz-Mateu M, de’Grafenried L, Eckhardt G, et al. Pharma-
codynamic Studies of Col-3, a Novel Matrix Metalloproteinase
Inhibitor, in Patients with Advanced Cancer. Proc Am Soc Clin
Oncol 2001;20:76a.
[142] Daniels R, Gupta E, Kollia G, et al. Safety and pharmacoki-
netics of BMS-275291, a novel matrix metalloproteinase inhi-
bitor in healthy subjects. Proc Am Soc Clin Oncol 2001;20:100a.
[143] Hurwitz H, Humphrey J, Williams K, et al. A Phase I Trial of
BMS-275291: a novel, non-hydroxamate, sheddase-sparing ma-
trix metalloproteinase inhibitor (MMPI) with no dose-limiting
arthritis. Proc Am Soc Clin Oncol 2001;20:98a.
[144] Gupta E, Huang M, Mao Y, et al. Pharmacokinetic (PK)
evaluation of BMS-275291, a matrix metalloproteinase (MMP)
inhibitor, in cancer patients. Proc Am Soc Clin Oncol
2001;20:76a.
[145] Woessner JF, Jr. Matrix metalloproteinases and their inhibitors
in connective tissue remodeling. FASEB J 1991;5:2145�/54.
[146] Goetzl EJ, Banda MJ, Leppert D. Matrix metalloproteinases in
immunity. J Immunol 1996;156:1�/4.
[147] Chambers AF, Matrisian LM. Changing views of the role of
matrix metalloproteinases in metastasis. J Natl Cancer Inst
1997;89:1260�/70.
Biography
Dr D. Cunningham is a Counsultant in Medical
Oncology at the Royal Mardsen Hospital in London
and Surrey, and he is Head of the GI and Lymphoma
Units. He qualified from Glasgow University and was a
Cancer Research Campaign Fellow there before moving
to London where he completed his training. Before
talking up his current post, he was Senior Lecturer in the
Institute of Cancer Research. His main research inter-
ests are clinical trials in GI cancer and lymphoma, along
with molecular therapies.
I. Chau et al. / Critical Reviews in Oncology/Hematology 45 (2003) 151�/176176





























