Embed Size (px)
Citation preview

Integrative Bioinformatics Analysis Reveals New PrognosticBiomarkers of Clear Cell Renal Cell Carcinoma
Henriett Butz,1,2 Peter M. Szabo,3 Roy Nofech-Mozes,1 Fabio Rotondo,1 Kalman Kovacs,1 Lorna Mirham,1
Hala Girgis,1 Dina Boles,1 Attila Patocs,4 and George M. Yousef1,2*
BACKGROUND: The outcome of clear cell renal cell car-cinoma (ccRCC) is still unpredictable. Even with newtargeted therapies, the average progression-free sur-vival is dismal. Markers for early detection and pro-gression could improve disease outcome.
METHODS: To identify efficient and hitherto unrecog-nized pathogenic factors of the disease, we performed auniquely comprehensive pathway analysis and built agene interaction network based on large publicly avail-able data sets assembled from 28 publications, com-prising a 3-prong approach with high-throughputmRNA, microRNA, and protein expression profiles of593 ccRCC and 389 normal kidney samples. We vali-dated our results on 2 different data sets of 882 ccRCCand 152 normal tissues. Functional analyses were doneby proliferation, migration, and invasion assays follow-ing siRNA (small interfering RNA) knockdown.
RESULTS: After integration of multilevel data, we iden-tified aryl-hydrocarbon receptor (AHR), grainyhead-like-2 (GRHL2), and KIAA0101 as new pathogenic fac-tors. GRHL2 expression was associated with higherchances for disease relapse and retained prognosticutility after controlling for grade and stage [hazard ra-tio (HR), 3.47, P � 0.012]. Patients with KIAA0101-positive expression suffered worse disease-free survival(HR, 3.64, P � 0.001), and in multivariate analysisKIAA0101 retained its independent prognostic signifi-cance. Survival analysis showed that GRHL2- andKIAA0101-positive patients had significantly lowerdisease-free survival (P � 0.002 and P � 0.001). Wealso found that KIAA0101 silencing decreased kidneycancer cell migration and invasion in vitro.
CONCLUSIONS: Using an integrative system biology ap-proach, we identified 3 novel factors as potential bio-markers (AHR, GRHL2 and KIAA0101) involved inccRCC pathogenesis and not linked to kidney cancerbefore.© 2014 American Association for Clinical Chemistry
Kidney cancer is among the 10 most frequently occur-ring cancers in Western communities and its incidencehas been steadily increasing by 2%– 4% each year(1 ). About 90% of adult kidney cancers are renal cellcarcinomas (RCC),5 and the majority of these (70%–85%) are of the clear cell subtype (ccRCC) (1 ). In theabsence of symptoms, about 30% of patients are diag-nosed with disease that is already in the metastatic stage(1 ). Localized kidney cancer can be cured surgically,whereas patient survival drops sharply if the diseasebecomes metastatic. Recently, targeted therapies havecome to be available [multikinase inhibitors, anti-VEGF (anti–vascular endothelial growth factor) anti-bodies, and mTOR (mammalian target of rapamycin)inhibitors]. Although these new therapies have im-proved patient survival, and the median progression-free and overall survival are approaching 2 years (2 ),most patients eventually develop resistance and sur-render to the disease. A more thorough understand-ing of ccRCC pathogenesis is important for the dis-covery of novel, more effective biomarkers and/ortherapeutic targets.
Over 90% of sporadic ccRCC cases show typicalcytogenetic changes in the short arm of chromosome3 (complete loss, translocation, deletion) typicallyleading to von Hippel-Lindau tumor suppressor, E3
1 Department of Laboratory Medicine and the Keenan Research Centre forBiomedical Science of St. Michael’s Hospital, Toronto, Canada; 2 Department ofLaboratory Medicine and Pathobiology, University of Toronto, Toronto, Canada;3 Biometric Research Branch, Division of Cancer Treatment and Diagnosis,National Cancer Institute, National Institutes of Health, Bethesda, MD; 4 HAS-SE“Lendulet” Hereditary Endocrine Tumors Research Group, Hungarian Academyof Sciences, Budapest, Hungary.
* Address correspondence to this author at: Department of Laboratory Medicine,St. Michael’s Hospital, 30 Bond St., Toronto, ON, M5B 1W8, Canada. Fax416-864-5648; e-mail [email protected].
Received April 8, 2014; accepted July 17, 2014.Previously published online at DOI: 10.1373/clinchem.2014.225854© 2014 American Association for Clinical Chemistry5 Nonstandard abbreviations: RCC, renal cell carcinomas; ccRCC, clear cell RCC;
miRNA, microRNA; siRNA, small interfering RNA; TMA, tissue microarray; AHR,aryl-hydrocarbon receptor; TCGA, the Cancer Genome Atlas; AUC, area underthe ROC curve; GRHL2, grainyhead-like-2; HR, hazard ratio; IPA, ingenuitypathway analysis; EMT, epithelial mesenchymal transition; TGF-�, transforminggrowth factor �.
Clinical Chemistry 60:101314–1326 (2014)
Molecular Diagnostics and Genetics
1314

ubiquitin protein ligase (VHL)6 inactivation (3 ). Anumber of additional molecular changes have been re-cently reported in ccRCC at the mRNA, protein, andmicroRNA (miRNA) levels; although these studiesrepresent a step forward, they focus on only one type ofmolecule, whereas cancer development necessitates aninteraction between different levels of molecularchanges. More recently, “integrated genomics” holdsthe promise of a much better understanding of thepathogenesis of ccRCC through analyzing the interac-tion between the different levels of molecular changes.Furthermore, integrated analyses could help in over-coming the problem of tumor and patient heterogene-ity, a recently recognized challenge in cancer research(4 ). A number of recent studies have shown the greatadvantages of the integrating approach in RCC (5 )(6, 7 ).
The goal of this study was to obtain greater knowl-edge of the complex biological background of kidneycancer tumor development by using a systems biologyapproach to integrate molecular changes of ccRCC atthe mRNA, miRNA, and protein expression levels toidentify novel pathways and new pathogenetic factorsthat can be of clinical utility as prognostic biomarkersor potential therapeutic targets.
Materials and Methods
PATIENT COHORTS
The discovery set included high-throughput gene,miRNA, and protein expression data from 593 ccRCCand 389 normal kidney samples. The first validation setcomprised the Cancer Genome Atlas data of 470ccRCC and 69 normal kidneys (mRNA sequencingdata) and 499 ccRCC and 66 normal samples (miRNAsequencing data). For the second validation set, weconstructed a tissue microarray of 383 primary ccRCC,and 85 matched normal tissues (Fig. 1A).
Details of the applied data sets, RNA isolation,quantitative PCR, and bioinformatics analysis are pre-sented in Fig. 1, B and C, and the Supplementary Ma-terials and Methods file and Supplementary Table 1 inthe Data Supplement that accompanies the online versionof this report at http://www.clinchem.org/content/vol60/issue10.
CELL PROLIFERATION, MIGRATION, AND INVASION ASSAYS
FOLLOWING siRNA TRANSFECTION
The primary 786-O and metastatic CAHN and Caki-2kidney cancer cell lines were purchased from AmericanType Culture Collection. Cells were transfected with 1of 2 different Locked Nucleic Acid® small interferingRNAs (siRNAs), an siRNA against KIAA0101 (SilencerSelect s18861, s18863) (30 nmol/L) (Life Technolo-gies), or a negative-control siRNA (30 nmol/L) usingLipofectamine RNAiMAX (Life Technologies). Trans-fections were optimized by BLOCK-it fluorescent oligo(Life Technologies). Gene knockdown was verified byquantitative PCR using the previously published prim-ers 5�-AGGTTGTCCCCTAAAGATTCTG-3� and 5�-CAGGTTGCAAAGGACATGC-3� (8 ) after RNA iso-lation (miRNAs kit, Qiagen) and by Western blot usinga published mouse monoclonal antibody againstKIAA0101 (H00009768-M01; Abnova) (9 ). Cell pro-liferation/viability was controlled by WST-1 cell prolif-eration reagent (Roche Applied Science) according tothe manufacturer’s protocol, and cell number wascounted after trypsinization. Cell behavior was investi-gated by migration and invasion assays (BD-Bio coat),as previously described, on 8.0-�m chambers (10 ).
6 Human genes: VHL, Von Hippel-Lindau tumor suppressor, E3 ubiquitin proteinligase (von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase, isinvolved in the ubiquitination and degradation of hypoxia-inducible factor (HIF).The germline mutation on VHL is a cause of von Hippel-Lindau syndrome);CCND1, cyclin D1 (cyclin D1 controls cell cycle by regulating CDK4 and CDK6activity which is required for G1/S transition); CDKN2A, cyclin dependent kinaseinhibitor 2A (cyclin dependent kinase inhibitor 2A regulates cell cycle G1progression by inhibiting CDK4 kinase. It also stabilizes p53); ETS1, v-ets avianerythroblastosis virus E26 oncogene homolog 1 (v-ets avian erythroblastosisvirus E26 oncogene homolog 1 is a transcription factor that controls theexpression of numerous genes involved in stem cell development, cell senes-cence and death, and tumorigenesis); ISG15, ubiquitin-like modifier (ISG15ubiquitin-like modifier, the product of this gene is an ubiquitin-like protein thatis conjugated to intracellular target proteins upon activation by interferon-alphaand beta); KIAA0101, KIAA0101 (also called PAF (PCNA-associated factor,regulates DNA repair during DNA replication); FBXO21, F-box protein 21 (F-boxprotein 21 is a member of the F-box protein family and constitutes a subunit ofubiquitin protein ligase complex, which functions in phosphorylation-dependentubiquitination); GRHL2, grainyhead like-2 (grainyhead like-2, a transcriptionfactor, plays a role in development, and its defect leads to sensorineuraldeafness); TCF4, transcription factor 4 (transcription factor 4, a basic helix-loop-helix transcription factor that is broadly expressed, plays an important role innervous system development); ZEB1, zinc finger E-box binding homeobox 1(zinc finger E-box binding homeobox 1 is a transcription factor that amongother functions represses E-cadherin promoter and induces an epithelial-mesenchymal transition. It promotes tumorigenicity by repressing stemness-inhibiting miRNAs); CCND2, cyclin D2 (cyclin D2 forms a complex with andregulates CDK4 and CDK6 in cell cycle G1/S transition. In a complex with CDK4it phosphorylates and inhibits retinoblastoma 1 (RB1) protein to control therestriction point of the cell cycle); AHR, aryl hydrocarbon receptor (aryl hydro-carbon receptor is an intracellular ligand activated receptor and transcriptionfactor, and regulates xenobiotic-metabolizing enzymes expression); SMAD2,SMAD family member 2; SMAD3, SMAD family member 3 SMAD family mem-bers are intracellular signal transducers and mediate the signal of TGF-�.SMAD2 and -3 are receptor-regulated SMADs activated by TGF-� and activintype 1; then, in a complex with other molecules, they bind the SBE (SMAD-binding elements) of DNA sequences; CDK6, cyclin-dependent kinase 6; SOX9,SRY (sex determining region Y)-box 9; CXCR4, chemokine (C-X-C motif) receptor4 (the receptor for the C-X-C chemokine CXCL12/SDF-1, plays roles in manydiverse cellular functions, including embryogenesis, immune surveillance, in-flammation response, tissue homeostasis, and tumor growth and metastasis);LAMC1, laminin, gamma 1; ITGB1, integrin, beta 1 (fibronectin receptor, betapolypeptide, antigen CD29 includes MDF2, MSK12); FOS, FBJ murine osteosar-coma viral oncogene homolog (a transcription factor and can dimerize withproteins of the JUN family, thereby forming the transcription factor complexAP-1. It has been implicated as a regulator of cell proliferation, differentiation,and transformation).
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1315
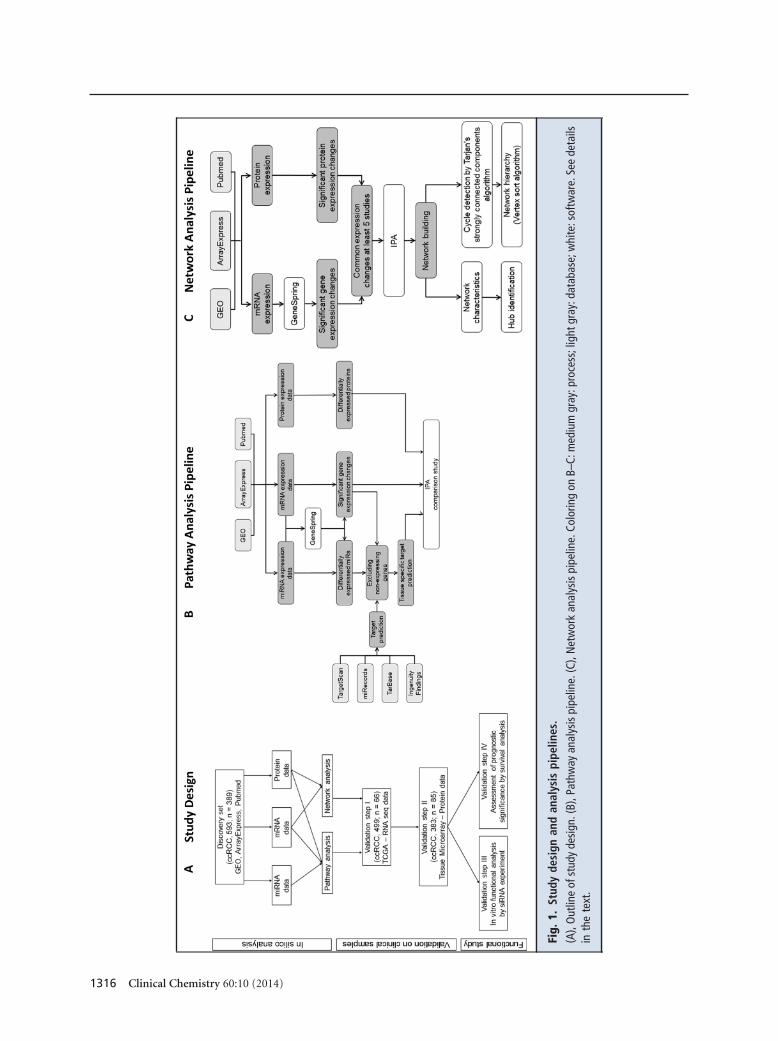
Fig.
1.St
udy
desi
gnan
dan
alys
ispi
pelin
es.
(A),
Out
line
ofst
udy
desi
gn.(
B),P
athw
ayan
alys
ispi
pelin
e.(C
),N
etw
ork
anal
ysis
pipe
line.
Colo
ring
onB–
C:m
ediu
mgr
ay:p
roce
ss;l
ight
gray
:dat
abas
e;w
hite
:sof
twar
e.Se
ede
tails
inth
ete
xt.
1316 Clinical Chemistry 60:10 (2014)

TISSUE MICROARRAY
Tissue microarrays (TMAs) were constructed from 383primary cSrc and 85 matched normal tissues. Sampleswere collected from St. Michael’s Hospital (Toronto,Canada) after we obtained Research Ethics Board ap-proval. For TMA construction, two 1.0-mm cores wereobtained from each tumor. Immunohistochemistrywas performed using primary antibodies against aryl-hydrocarbon receptor (AHR) (Novus Biologicals),grainyhead-like-2 (GRHL2) (Novus Biologicals), andKIAA0101 (Abnova). The detailed description of TMAconstruction, immunostaining, and scoring is docu-mented in the online Supplementary Materials andMethods.
Results
PATHWAY ANALYSIS
First, we identified mRNAs, proteins, and miRNAs thatare commonly differentially expressed between ccRCCand normal kidney tissues (see online SupplementaryTables 2 and 3). After performing pathway analysis, wefound that the most commonly altered signaling weremetabolic pathways related to glucose metabolism(“glycolysis”), fatty acid-retinoic acid (“LXR/RXR ac-tivation”), and amino-acid metabolism (“valine degra-dation”) (see online Supplementary Table 4). We alsoidentified “renal cell cancer,” “hypoxia signaling,” and“angiopoietin signaling” among the significant path-ways, and “cellular movement” and “cell death andsurvival” on the basis of the gene expression profile,with the functional terms “renal cancer,” “kidney de-velopment,“ “angiogenesis,” “migration and prolifera-tion of vascular endothelial cells,” and “vasculariza-tion” among BioFunction categories.
“AHR signaling” was also identified as significant(Fig. 2; also see online Supplementary Fig. 1 for a ver-sion of this image that can be enlarged). This pathwayis involved in xenometabolism, cell cycle, differentia-tion, and apoptosis, but has not yet been linked toccRCC pathogenesis; thereby we selected it for furthervalidation.
NETWORK ANALYSIS
We built an interaction network using the most sig-nificant molecules. This network is based on an un-biased approach that relies only on molecular inter-actions and thus differs from pathway analysis thatperforms gene set enrichment on a predefined geneset. The network structure is formed of basic ele-ments (mRNAs or proteins), designated nodes, andthe physical interactions or relationships betweenthese nodes (edges) (11 ). Interactions are direction-ally based on their type (such as activation and inhibi-tion). The relationships between different members are
predicted by computational algorithms and/or literature-documented experimentally validated interactions(e.g., protein–protein, protein–DNA, RNA–miRNA).The highest-degree nodes based on the number of inter-actions are generally defined as hubs. In our ccRCC net-work, 5 nodes were determined as hubs (cyclin D1[CCND1], cyclin dependent kinase inhibitor 2A[CDKN2A]), v-ets avian erythroblastosis virus E26 onco-gene homolog 1 [ETS1], ubiquitin-like modifier [ISG15],and KIAA0101 [KIAA0101]). In the functional annota-tion of the network members, we found “proliferation ofkidney cells” and “angiogenesis” categories to be signifi-cant (data are not shown).
Based on directional interactions, a network hier-archy contains 3 (or more) layers of nodes (see onlineSupplementary Fig. 2A). Genes in the “top” layer(s) areconsidered to be the master regulators of the networkbecause they are not affected by other nodes, and thesegenes act as regulators of all others, and as such, theyshould influence the whole network through theirdownstream targets. The second, “core” layer(s) usu-ally contain the majority of the hubs, which determinethe basic structure of the network. This layer plays acentral role in the regulation of signal propagation andmodulation (the signal can be enhanced, attenuated, orbuffered). Genes in the third, “bottom” layer(s) are di-rectly regulated effector molecules (12 ). The top-layergenes are potential drug targets, being “directors” ofthe network and having only a few further connections;therefore, targeting of these molecules should be asso-ciated with fewer side effects (13 ). Regarding our net-work among top members, the only individual geneswere F-box protein (FBXO21) and grainyhead like-2(GRHL2), whereas the other members were moleculargroups/families (Fig. 3), and the core layer was themost abundant layer, containing all of the hubs.
miRNA CONTRIBUTION TO ccRCC PATHOGENESIS
Integrating another level of regulation, we subse-quently identified tissue-specific miRNAs that are pre-dicted to target network members (see online Supple-mentary Table 5). The miRNAs miR-124, miR-139-5p,and miR-204-5p had the most important effect on theexpression of network members, with each of them tar-geting 4 different transcripts (see online Supplemen-tary Fig. 2B). Transcription factor 4 (TCF4), zinc fingerE-box binding homeobox 1 (ZEB1), and cyclin D2(CCND2) were the genes found to be most influencedby these miRNAs.
We also identified 11 miRNAs that are predictedto target the AHR signaling pathway, among these,miR-203–3p and miR-124 –3p can target the aryl hy-drocarbon receptor (AHR) gene itself (Fig. 2).
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1317

Fig.
2.A
HR
Sign
alin
gin
ccRC
C.
Cont
inue
don
page
1319
1318 Clinical Chemistry 60:10 (2014)

VALIDATION OF GENE DEREGULATION IN ccRCC
For validation of our results, we selected 3 criticalgenes, AHR, which is a major component of AHR sig-naling that was significantly affected in ccRCC in ourpathway analysis, GRHL2, which is located in the toplevel of the network, and KIAA0101, which is amongthe highest-degree hubs in our network.
In the first step, we used the Cancer Genome Atlas(TCGA) data set to assess gene expression at the mRNAlevels between normal and cancerous tissues. AHR andKIAA0101 expressions were significantly higher inccRCC than in normal kidney [fold change, 1.72 (P �0.0001) and fold change, 5.2 (P � 0.0001)] (Fig. 4, Aand K); and GRHL2 downregulation was confirmed inccRCC with a 16.1-fold change (P � 0.0001) (Fig. 4F).We also validated the downregulation of miR-124,miR-204 –5p, miR-139 –5p, miR-141, and miR-199aand the upregulation of miR-23a in kidney cancercompared to normal tissue (see online SupplementaryFig. 2C).
We next investigated the differential expression ofthese genes at the protein level using immunohisto-chemical analysis on our individual sample set using aTMA format. As shown in Fig. 4, D–E, and online Sup-plementary Table 6, expression of AHR was upregu-lated in 75% of patients in comparison to normaltissue. Expression of GRHL2 was found to be signif-icantly downregulated in 90% of cancerous samplescompared to normal tissue (Fig. 4I). KIAA0101 ex-pression was higher in ccRCC than in normal counter-parts in 56% of cases (P � 0.003) (Fig. 4N; also seeonline Supplementary Table 6). We also confirmed
KIAA0101 upregulation in ccRCC using Western blotanalysis (Fig. 4M).
DIFFERENTIAL EXPRESSION OF AHR, GRHL2, AND KIAA0101 IN
ccRCC COMPARED TO NORMAL KIDNEY TISSUE
ROC analysis showed the potential utility of AHR todistinguish cancer from normal kidney tissues on thebasis of mRNA expression levels [area under the ROCcurve (AUC) 0.825 with 80% sensitivity and 80% spec-ificity at a cutoff of expression of 1413.4 (read num-ber)] (Fig. 4B). Also, the lower mRNA expression ofGRHL2 was highly specific for the tumors [AUC, 0.963(P � 0.0001); cutoff of expression, 130.2 (read num-ber)] (Fig. 4G). KIAA0101 was also found to be a po-tential diagnostic biomarker (AUC, 0.956, P � 0.001)[at cutoff of expression 30.9 (read number), 94.4% sen-sitivity and 91.3% specificity] (Fig. 4L).
THE IDENTIFICATION OF POTENTIAL PROGNOSTIC
MARKERS FOR ccRCC
We assessed the prognostic significance of AHR,KIAA0101, and GRHL2 in ccRCC at both mRNA andprotein levels.
GRHL2 AS A PROGNOSTIC MARKER IN ccRCC
Lower mRNA expression levels for GRHL2 were asso-ciated with significantly better survival (Fig. 4H).GRHL2 protein expression in cancer tissues is posi-tively associated with tumor grade (P � 0.001) (seeonline Supplementary Table 7) and with tumor size(GRHL2-positive staining in 19% of tumors �4 cm vs48% in those �4 cm, P � 0.001) (Supplementary Table
Fig. 2. Continued.
Significant numbers of members of this pathway were frequently reported as significantly dysregulated in ccRCC, as shown inthe color legend. The color intensity corresponds to the number of studies showing the dysregulation. After ligand binding, AHRtranslocates to the nucleus, where it transactivates/represses several genes. This figure can be also found as onlineSupplementary Fig. 1, where it can be enlarged. NF-�B, nuclear factor �B; IL-1, interleukin 1; TNF, tumor necrosis factor; HSP90,heat shock protein 90; XAP2, X-associated protein 2; TEBP, telomere end binding protein; Src, Src protein; ESR1, estrogenreceptor 1; Rb, retinoblastoma protein; ATM, ataxia telangiectasia mutated protein; ATR, ataxia telangiectasia and Rad3-relatedprotein; CDK, cyclin dependent kinase; AHRR, aryl hydrocarbon receptor repressor; ARNT, AhR nuclear translocator; MDM2,mouse double minute 2 homolog; ChK2, checkpoint kinase 2; RelA, v-rel avian reticuloendotheliosis viral oncogene homologA; ERK, extracellular signal-regulated kinase; JNK, c-Jun amino-terminal kinase; NR2F1, nuclear receptor subfamily 2, group F,member 1; NR0B2, orphan nuclear receptor DAX-1; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor;NCOA7, nuclear receptor coactivator 7; NEDD8, neural precursor cell expressed, developmentally down-regulated 8; RIP140,receptor-interacting protein 140; DRE, DNA replication-related element; XRE, xenobiotic response element; E2F1, E2F tran-scription factor 1; RAR, retinoic acid receptor; RXR, retinoid X receptor; RARE, RAR binding element; RXRE, RXR binding element;TGM2, transglutaminase 2; Arf, ADP-ribosylation factor; Apaf1, apoptotic peptidase activating factor 1; c-Myc, c-v-myc avianmyelocytomatosis viral oncogene homolog; p27Kip1, cyclin-dependent kinase inhibitor 1B (p27, Kip1); p21Cip1, cyclin-dependent kinase inhibitor 1A (p21, Cip1); BAX, BCL2-associated X protein; Fas, fatty acid synthase; CYP1B1, cytochrome P450,family 1, subfamily B, polypeptide 1; ALDH, aldehyde dehydrogenase; NQO, NAD(P)H:quinone acceptor oxidoreductase; GST,glutathionine S-transferase; NRF2, nuclear factor erythroid 2-related factor; ARE, antioxidant response element.
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1319

Fig.
3.H
iera
rchi
calm
olec
ular
netw
ork
ofcc
RCC.
Cont
inue
don
page
1321
1320 Clinical Chemistry 60:10 (2014)

7B). Higher protein expression levels were associatedwith higher tumor stages with 26% positivity in stage 1tumors compared to 51% in higher stages (P � 0.008)(see online Supplementary Table 8).
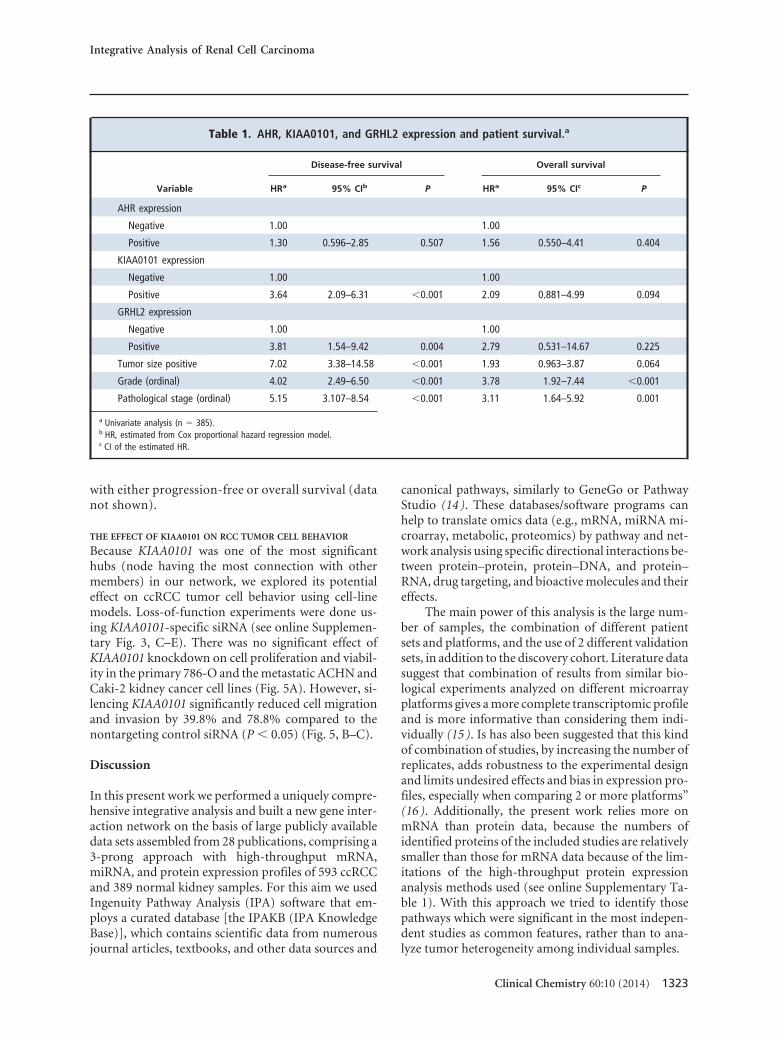
In univariate analysis, GRHL2 protein expressionwas associated with higher chances for disease relapse[hazard ratio (HR), 3.81, 95% CI, 1.54 –9.42, P �0.004] (Table 1). The GRHL2 protein expression asso-ciation with shorter overall survival, although positive,did not reach statistical significance (HR, 2.79; 95% CI,0.531–14.67; P � 0.225). In multivariate analysis,GRHL2 retained its prognostic utility after controllingfor grade and stage (HR, 3.47; 95% CI, 1.32–9.15; P �0.012). Kaplan–Meier survival analysis showed thatGRHL2-positive patients had significantly lowerdisease-free survival than those who were GRHL2 neg-ative (P � 0.002) (Fig. 4J). No association was foundbetween GRHL2 expression and overall survival (datanot shown).
KIAA0101 AS A PROGNOSTIC MARKER IN ccRCC
KIAA0101 had significant influence on prognosis at themRNA level: higher expression was associated withworse overall survival (P � 0.0003) (see online Supple-mentary Fig. 3A). On the protein level, no statistically
significant association was found between KIAA0101expression and tumor grade (see online Supplemen-tary Table 7). Univariate analysis showed thatKIAA0101-positive patients had worse disease-freesurvival (HR, 3.64; 95% CI, 2.09 – 6.31, P � 0.001) (Ta-ble 1). Although there was an association betweenKIAA0101 and worse overall survival, this was not sta-tistically significant (P � 0.094) (Table 1). In the mul-tivariate analysis, KIAA0101 retained its independentprognostic significance after controlling for tumor sizeand grade (HR, 2.76, P � 0.001) or grade and stage(HR, 2.73, P � 0.002). Kaplan–Meier survival analysisindicated that KIAA0101-positive patients had re-duced disease-free survival (P � 0.001) (Fig. 4O).Those patients also had lower overall survival, althoughthis was not statistically significant (P � 0.087) (seeonline Supplementary Fig. 3B).
AHR AS A PROGNOSTIC MARKER IN ccRCC
At the mRNA level, higher expression of AHR wasassociated with better overall survival (Fig. 4C).AHR protein expression in cancer tissues was nega-tively associated with tumor grade (P � 0.047) (seeonline Supplementary Table 7). Univariate and mul-tivariate analyses showed no significant association
Fig. 3. Continued.
Hubs (nodes with the most significant number of interactions) are bolded. After the hierarchy of the nodes was determined onthe basis of their directed relationships, nodes were categorized into top, core, and bottom levels. See text for more discussion.acad, acyl-CoA dehydrogenase; SLC16A1, solute carrier family 16 (monocarboxylate transporter), member 1; SHMT2, serinehydroxymethyltransferase 2 (mitochondrial); SUCLG2, succinate-CoA ligase, GDP-forming, beta subunit; APOM, apolipoproteinM; SLIT2, slit homolog 2 (Drosophila); CTSH, cathepsin H;HSPA6, heat shock 70 kDa protein 6; RNF213, ring finger protein 213;DEGS1, delta(4)-desaturase, sphingolipid 1; FAM171A1, family with sequence similarity 171, member A1; FHIT, fragile histidinetriad; RALYL, RALY RNA binding protein-like; LMO3, LIM domain only 3 (rhombotin-like 2); RUNX3, runt-related transcriptionfactor 3; CSTA, cystatin A (stefin A); GLT25D1, glycosyltransferase 25 domain containing 1; CEP170, centrosomal protein170kDa; GATA3, GATA binding protein 3; INSIG2, insulin-induced gene 2 protein; CDKN1C, cyclin-dependent kinase inhibitor1C; DSP, desmoplakin; IFI44, interferon-induced protein 44; DDX41, DEAD (Asp-Glu-Ala-Asp) box polypeptide 41; ZC3HAV1L,zinc finger CCCH-type, antiviral 1-like; PFKP, phosphofructokinase, platelet; ALDOA, aldolase A, fructose-bisphosphate; KHK,ketohexokinase (fructokinase); LYZ, lysozyme; IGF2BP2, insulin-like growth factor 2 mRNA binding protein 2; C21orf33,chromosome 21 open reading frame 33; LDHB, lactate dehydrogenase B, PKM, pyruvate kinase; AZGP1, alpha-2-glycoprotein1, zinc-binding; WFDC2, WAP four-disulfide core domain protein 2; PHGDH, phosphoglycerate dehydrogenase; PLOD3,procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3; LTF, lactotransferrin; IKZF1, IKAROS family zinc finger 1 protein; Ctbp,C-terminal binding protein; ACLY, ATP citrate lyase; MYOF, myoferlin; EGLN3, egl-9 family hypoxia-inducible factor 3; ELF4,E74-like factor 4 (ets domain transcription factor); ANXA3, annexin A3; IKBIP, Inhibitor of nuclear factor kappa-B kinase-interacting protein, partial; CP, ceruloplasmin (ferroxidase); MPPED2, metallophosphoesterase domain containing 2; PRAP1,proline-rich acidic protein 1; STRA6, stimulated by retinoic acid 6; LRP2, low density lipoprotein receptor-related protein 2;MEST, mesoderm specific transcript; SLC39A4, solute carrier family 39 (zinc transporter), member 4;PMEPA1, prostatetransmembrane protein, androgen induced 1; ABCC1, ATP-binding cassette, sub-family C (CFTR/MRP), member 1; HPGD,hydroxyprostaglandin dehydrogenase 15-(NAD); RHOBTB3, rho-related BTB domain-containing protein 3; RBP4, retinol bindingprotein 4, plasma; PLAT, plasminogen activator, tissue; ANPEP, alanyl (membrane) aminopeptidase; MPZl2, Myelin proteinzero-like protein 2; MELK, maternal embryonic leucine zipper kinase; TBXAS1, thromboxane-A synthase; ARL4C, ADP-ribosylation factor-like 4C; CMTM3, CKLF-like MARVEL transmembrane domain-containing protein 3; SERPINE1, serpinpeptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1.
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1321

Fig.
4.A
HR,
GRH
L2,a
ndKI
AA
0101
expr
essi
on.
(A,F
,K),
AHR,
GHR
L2,a
ndKI
AA01
01ex
pres
sion
incc
RCC
vsno
rmal
tissu
e.(B
,G,L
),RO
Can
alys
isfo
rAHR
,GRH
L2,a
ndKI
AA01
01.(
C,H)
,Sur
viva
lana
lysis
ofAH
Ran
dG
RHL2
.(D,
I,N
),AH
R,G
RHL2
,and
KIAA
0101
imm
unos
tain
ing.
(E),
AHR
prot
ein
expr
essio
nw
ashi
gher
incc
RCC
(bla
ckbo
xes)
com
pare
dto
norm
altis
sue
(ope
nbo
xes)
.(J,
O)G
RHL2
and
KIAA
0101
stai
ning
isas
socia
ted
with
wor
sepr
ogno
sis.(
M),
KIAA
0101
Wes
tern
blot
ofcc
RCC
vsno
rmal
tissu
e.*,
Stat
istica
lsig
nific
ance
;N,n
orm
al;C
,can
cer;
DFS,
dise
ase-
free
surv
ival
;OS,
over
alls
urvi
val.
1322 Clinical Chemistry 60:10 (2014)
with either progression-free or overall survival (datanot shown).
THE EFFECT OF KIAA0101 ON RCC TUMOR CELL BEHAVIOR
Because KIAA0101 was one of the most significanthubs (node having the most connection with othermembers) in our network, we explored its potentialeffect on ccRCC tumor cell behavior using cell-linemodels. Loss-of-function experiments were done us-ing KIAA0101-specific siRNA (see online Supplemen-tary Fig. 3, C–E). There was no significant effect ofKIAA0101 knockdown on cell proliferation and viabil-ity in the primary 786-O and the metastatic ACHN andCaki-2 kidney cancer cell lines (Fig. 5A). However, si-lencing KIAA0101 significantly reduced cell migrationand invasion by 39.8% and 78.8% compared to thenontargeting control siRNA (P � 0.05) (Fig. 5, B–C).
Discussion
In this present work we performed a uniquely compre-hensive integrative analysis and built a new gene inter-action network on the basis of large publicly availabledata sets assembled from 28 publications, comprising a3-prong approach with high-throughput mRNA,miRNA, and protein expression profiles of 593 ccRCCand 389 normal kidney samples. For this aim we usedIngenuity Pathway Analysis (IPA) software that em-ploys a curated database [the IPAKB (IPA KnowledgeBase)], which contains scientific data from numerousjournal articles, textbooks, and other data sources and
canonical pathways, similarly to GeneGo or PathwayStudio (14 ). These databases/software programs canhelp to translate omics data (e.g., mRNA, miRNA mi-croarray, metabolic, proteomics) by pathway and net-work analysis using specific directional interactions be-tween protein–protein, protein–DNA, and protein–RNA, drug targeting, and bioactive molecules and theireffects.
The main power of this analysis is the large num-ber of samples, the combination of different patientsets and platforms, and the use of 2 different validationsets, in addition to the discovery cohort. Literature datasuggest that combination of results from similar bio-logical experiments analyzed on different microarrayplatforms gives a more complete transcriptomic profileand is more informative than considering them indi-vidually (15 ). Is has also been suggested that this kindof combination of studies, by increasing the number ofreplicates, adds robustness to the experimental designand limits undesired effects and bias in expression pro-files, especially when comparing 2 or more platforms”(16 ). Additionally, the present work relies more onmRNA than protein data, because the numbers ofidentified proteins of the included studies are relativelysmaller than those for mRNA data because of the lim-itations of the high-throughput protein expressionanalysis methods used (see online Supplementary Ta-ble 1). With this approach we tried to identify thosepathways which were significant in the most indepen-dent studies as common features, rather than to ana-lyze tumor heterogeneity among individual samples.
Table 1. AHR, KIAA0101, and GRHL2 expression and patient survival.a
Variable
Disease-free survival Overall survival
HRa 95% CIb P HRa 95% CIc P
AHR expression
Negative 1.00 1.00
Positive 1.30 0.596–2.85 0.507 1.56 0.550–4.41 0.404
KIAA0101 expression
Negative 1.00 1.00
Positive 3.64 2.09–6.31 �0.001 2.09 0.881–4.99 0.094
GRHL2 expression
Negative 1.00 1.00
Positive 3.81 1.54–9.42 0.004 2.79 0.531–14.67 0.225
Tumor size positive 7.02 3.38–14.58 �0.001 1.93 0.963–3.87 0.064
Grade (ordinal) 4.02 2.49–6.50 �0.001 3.78 1.92–7.44 �0.001
Pathological stage (ordinal) 5.15 3.107–8.54 �0.001 3.11 1.64–5.92 0.001
a Univariate analysis (n � 385).b HR, estimated from Cox proportional hazard regression model.c CI of the estimated HR.
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1323

Our findings are in keeping with previous data in-dicating that metabolic pathways are critical in ccRCCpathogenesis (17 ), and we showed that AHR signalingis altered in ccRCC. We identified and validated AHRas an overexpressed gene that is regulated by the under-expressed miR-124 in ccRCC, an interaction that al-ready has been experimentally validated in other tu-mors (18 ). AHR can mediate either tumor-promotingor -inhibiting effects. In mammary tumors, high levelsAHR were detected (19 ). In addition, overexpressionof AHR resulted in increased hepatic and stomach tu-mor development, and its knockdown reduced tumorgrowth and metastasis. In contrast, AHR null micehave hyperplastic phenomena (hyperkeratosis of theskin, hyperproliferation of hair follicles, hyperplasia inthe gastric pylorus, hyperproliferation of portal bloodvessels) and develop liver and lung adenocarcinoma(20 ). Active AHR expression led to growth inhibition
and apoptosis, and caused cell-cycle arrest (21, 22 ). Itwas also reported that AHR activation has a promigra-tory effect in epithelial cells and enhances cancer inva-sion in urothelial cancer (23 ). AHR can also represent atherapeutic target. Callero et al. reported that activat-ing AHR with an agonist inhibited cell growth in adose-dependent manner in RCC cell lines (24 ). It wasalso found that sunitinib, a tyrosine kinase inhibitorused for metastatic ccRCC, induced CYP1A1 expres-sion through AHR in breast cancer (25 ). Aminofla-vone, which has an antiproliferative effect on humanbreast cancer cells mediated by AHR, is also applied inclinical trials as a new anticancer drug. It was found toinhibit cell growth and to induce apoptosis in severalbut not all renal cancer cell lines as well (24 ).
By characterizing the ccRCC disease network, weidentified the most significant molecules in the patho-genesis as hubs (KIAA0101, CCND1, ETS1, CDKN2A).
Fig. 5. In vitro functional validation of the role of KIAA0101 in RCC cells.
(A), KIAA0101 knockdown has no effect on proliferation and viability. (B, C), Inhibition of KIAA0101 function by siRNAsignificantly decreased the migration and invasion ability of RCC cells (P � 0.05). siKIAA0101, siRNA against KIAA0101.
1324 Clinical Chemistry 60:10 (2014)

Among these, CCND1 and ETS1 were already linked tokidney cancer. ETS1 was found to correlate with mi-crovascular density in ccRCC (26 ). We found thatGRHL2 was underexpressed in tumors compared tonormal kidney both for mRNA and protein levels andcan be a potential prognostic marker based on immu-nostaining. GRHL2 is a transcription factor playingroles in development, regulation of epithelial mesen-chymal transition (EMT), and restoring the sensitivity toanoikis (27). GRHL2 overexpression was detected in sev-eral cancers; however, in certain subclasses of breast can-cer, GRHL2 was downregulated (27, 28 ). This dual,context-dependent effect of GRHL2 might be ex-plained by operation through transforming growthfactor � (TGF-�) signaling, which also has both tumor-promoting and -suppressing effects in a context-dependent manner. It was shown that GRHL2 inter-feres with TGF-� signaling by repressing the ZEB1promoter and by interfering with SMAD family mem-ber 2 (SMAD2)- and SMAD family member 3 (SMAD3)-mediated transcriptional activation (27 ). Its over-expression suppressed primary tumor growth in xeno-graft assay and tumor cells sensitized to chemotherapy-induced cytotoxicity (27 ). In breast cancer, GRHL2was found to be downregulated in the context of TGF-�/EMT-driven tumor types and its loss was associatedwith a mesenchymal phenotype (27 ); thereby, GRHL2was considered as an “oncogenic restriction point”(27 ). The role of GRHL2 downregulation in ccRCC hasnot been investigated, but its target, ZEB1, which is adirect suppressor of GRHL2, is overexpressed in kidneycancer. The GRHL2 targets miR-200a and-b are under-expressed in ccRCC samples (29 ), which supports therole of GRHL2 in ccRCC. These, and GRHL2 being atop-layer gene in the network, suggest that restoring ofthe function of GRHL2 (including its downstream tar-gets) may be a new, potentially therapeutic direction inaddition to being a potential diagnostic marker.KIAA0101 encodes a PCNA (proliferating cell nuclearantigen)-associated factor, also referred to as PAF15(proliferating cell unclear antigen-associated factor).The KIAA0101 protein plays a role in controlling cell-cycle progression through affecting DNA replication.Its role in the cell cycle and DNA damage response wasalso described (30 ). The APC/C (cell cycle anaphase-promoting complex/cyclosome) controls degradationof substrate proteins, such as KIAA0101, at mitotic exitand throughout the G1 phase (30 ). This may explainthe relationship between high KIAA0101 expressionand cancer. KIAA0101 was found to be overexpressedin several malignancies, such as breast, gastric, hepato-cellular, adrenal, lung, and pancreatic carcinoma. Wefound that KIAA101 was significantly overexpressed atboth the mRNA and protein levels in ccRCC, similarlyto other cancers. In line with previous reports, we
found that knockdown of KIAA0101 led to reducedmigration and invasion ability of kidney cells but didnot affect the cell proliferation similarly to adrenal can-cer (31 ). In several cell types, proliferation and migra-tion/invasion are distinct, supporting the “go or grow”hypothesis (32 ). Our results suggest that KIAA0101 isinvolved in malignant behavior and development ofmetastasis of ccRCC cells rather than tumorigenesis.We also showed that the increased expression ofKIAA0101 protein was positively associated with poorprognosis, in agreement with recent reports in othertumors (33 ). Interestingly, circulating KIAA0101mRNA has also been shown to be a predictive markerfor hepatic cancer (33 ).
MiR-124, miR-139, and miR-204 were downregu-lated and we considered them to be the most critical miR-NAs influencing our ccRCC network having 4 targetseach. MiR-124 was characterized as tumor suppressor inseveral tumor types. It has an essential role in differentia-tion of neural stems cells and neural progenitor cells (34),and its downregulation promotes growth, invasiveness,and metastasis (35) by targeting laminin, gamma 1(LAMC1), integrin, beta 1 (fibronectin receptor, betapolypeptide, antigen CD29 includes MDF2, MSK12)(ITGB1), cyclin-dependent kinase 6 (CDK6), and SRY(sex determining region Y)-box 9 (SOX9) (36). Similarly,miR-204 loss enhanced migration and stem cell pheno-type in vivo (37). MiR-139 expression was shown to differbetween different kidney cancer subtypes (38) and wasalso linked to metastasis and prognosis in ccRCC (39). Inour network, miR-139 targets ETS1, TCF4, ZEB1, andCCND2. This miRNA was found to be downregulated inother cancers and target chemokine (C-X-C motif) recep-tor 4 (CXCR4) and FBJ murine osteosarcoma viral onco-gene homolog (FOS) (40, 41). All 3 miRNAs (miR-124,miR-204, and miR-139) target TCF4 and CCND2. Theoverexpressed ETS1 and ZEB1 are both targeted by 2of these three miRNAs. On the basis of their role inthe RCC disease network, we propose that these 3 miR-NAs have the most important role in ccRCCpathogenesis.
In conclusion, using an integrative systems biol-ogy approach, we determined that AHR signaling, anda number of critical molecules, including GRHL2 andKIAA0101, are involved in ccRCC pathogenesis. To thebest of our knowledge, these pathogenic factors havenot previously been linked to kidney cancer.
Author Contributions: All authors confirmed they have contributed tothe intellectual content of this paper and have met the following 3 re-quirements: (a) significant contributions to the conception and design,acquisition of data, or analysis and interpretation of data; (b) draftingor revising the article for intellectual content; and (c) final approval ofthe published article.
Integrative Analysis of Renal Cell Carcinoma
Clinical Chemistry 60:10 (2014) 1325

Authors’ Disclosures or Potential Conflicts of Interest: Upon man-uscript submission, all authors completed the author disclosure form.Disclosures and/or potential conflicts of interest:
Employment or Leadership: G.M. Yousef, St. Michael’s Hospital.Consultant or Advisory Role: None declared.Stock Ownership: None declared.Honoraria: None declared.Research Funding: G.M. Yousef, grants from Canadian Instituteof Health Research (MOP 119606), Kidney Foundation of Canada
(KFOC130030), the Kidney Cancer Research Network of Canada,and Prostate Cancer Canada Movember Discovery Grants(D2013-39).Expert Testimony: None declared.Patents: None declared.
Role of Sponsor: The funding organizations played no role in thedesign of study, choice of enrolled patients, review and interpretationof data, or preparation or approval of manuscript.
References
1. Cairns P. Renal cell carcinoma. Cancer Biomark2010;9:461–73.
2. Vera-Badillo FE, Templeton AJ, Duran I, Ocana A, deGouveia P, Aneja P, et al. Systemic therapy for non-clear cell renal cell carcinomas: a systematic reviewand meta-analysis. Eur Urol [Epub ahead of print 2014May 29].
3. Lopez JI. Renal tumors with clear cells. A review.Pathol Res Pract 2013;209:137–46.
4. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endes-felder D, Gronroos E, et al. Intratumor heterogeneityand branched evolution revealed by multiregionsequencing. N Engl J Med 2012;366:883–92.
5. Chen J, Zhang D, Zhang W, Tang Y, Yan W, GuoL, Shen B. Clear cell renal cell carcinoma associ-ated microRNA expression signatures identifiedby an integrated bioinformatics analysis. J TranslMed 2013;11:169.
6. Dondeti VR, Wubbenhorst B, Lal P, Gordan JD,D’Andrea K, Attiyeh EF, et al. Integrative genomicanalyses of sporadic clear cell renal cell carci-noma define disease subtypes and potential newtherapeutic targets. Cancer Res 2012;72:112–21.
7. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S,Okuno Y, Kamura T, et al. Integrated molecularanalysis of clear-cell renal cell carcinoma. NatGenet 2013;45:860–7.
8. Mohammed N, Rodriguez M, Garcia V, Garcia JM,Dominguez G, Pena C, et al. EPAS1 mRNA inplasma from colorectal cancer patients is associ-ated with poor outcome in advanced stages. On-col Lett 2011;2:719–24.
9. Chang CN, Feng MJ, Chen YL, Yuan RH, Jeng YM.p15(PAF) is an Rb/E2F-regulated S-phase proteinessential for DNA synthesis and cell cycle pro-gression. PLoS One 2013;8:e61196.
10. Khella HW, Bakhet M, Allo G, Jewett MA, GirgisAH, Latif A, et al. miR-192, miR-194 and miR-215: a convergent microRNA network suppress-ing tumor progression in renal cell carcinoma.Carcinogenesis 2013;34:2231–9.
11. Vidal M, Cusick ME, Barabasi AL. Interactome net-works and human disease. Cell 2011;144:986–98.
12. Jothi R, Balaji S, Wuster A, Grochow JA, Gsponer J,Przytycka TM, et al. Genomic analysis reveals a tightlink between transcription factor dynamics and regu-latory network architecture. Mol Syst Biol 2009;5:294.
13. Liu YY, Slotine JJ, Barabasi AL. Control centralityand hierarchical structure in complex networks.PLoS One 2012;7:e44459.
14. Henderson-Maclennan NK, Papp JC, Talbot CC Jr,McCabe ER, Presson AP. Pathway analysissoftware: annotation errors and solutions. MolGenet Metab 2010;101:134–40.
15. Hockley SL, Mathijs K, Staal YC, Brewer D, Gid-dings I, van Delft JH, Phillips DH. Interlaboratory
and interplatform comparison of microarray geneexpression analysis of HepG2 cells exposed tobenzo (a) pyrene. OMICS 2009;13:115–25.
16. Severgnini M, Bicciato S, Mangano E, Scarlatti F,Mezzelani A, Mattioli M, et al. Strategies for com-paring gene expression profiles from different mi-croarray platforms: application to a case-controlexperiment. Anal Biochem 2006;353:43–56.
17. Yang OC, Maxwell PH, Pollard PJ. Renal cellcarcinoma: translational aspects of metabolismand therapeutic consequences. Kidney Int 2013;84:667–81.
18. Huang TC, Chang HY, Chen CY, Wu PY, Lee H,Liao YF, et al. Silencing of miR-124 induces neu-roblastoma SK-N-SH cell differentiation, cell cyclearrest and apoptosis through promoting AHR.FEBS Lett 2011;585:3582–6.
19. Schlezinger JJ, Liu D, Farago M, Seldin DC, Bel-guise K, Sonenshein GE, Sherr DH. A role for thearyl hydrocarbon receptor in mammary gland tu-morigenesis. Biol Chem 2006;387:1175–87.
20. Gonzalez FJ, Fernandez-Salguero P. The aryl hy-drocarbon receptor: studies using the AHR-nullmice. Drug Metab Dispos 1998;26:1194–8.
21. Abel J, Haarmann-Stemmann T. An introductionto the molecular basics of aryl hydrocarbon re-ceptor biology. Biol Chem 2010;391:1235–48.
22. Marlowe JL, Fan Y, Chang X, Peng L, Knudsen ES,Xia Y, Puga A. The aryl hydrocarbon receptorbinds to E2F1 and inhibits E2F1-induced apopto-sis. Mol Biol Cell 2008;19:3263–71.
23. Ishida M, Mikami S, Kikuchi E, Kosaka T, MiyajimaA, Nakagawa K, et al. Activation of the aryl hydro-carbon receptor pathway enhances cancer cell in-vasion by upregulating the MMP expression and isassociated with poor prognosis in upper urinary tracturothelial cancer. Carcinogenesis 2010;31:287–95.
24. Callero MA, Suarez GV, Luzzani G, Itkin B,Nguyen B, Loaiza-Perez AI. Aryl hydrocarbon re-ceptor activation by aminoflavone: new molecu-lar target for renal cancer treatment. Int J Oncol2012;41:125–34.
25. Maayah ZH, Ansari MA, El Gendy MA, Al-Arifi MN,Korashy HM. Development of cardiac hypertrophyby sunitinib in vivo and in vitro rat cardiomyocytesis influenced by the aryl hydrocarbon receptor sig-naling pathway. Arch Toxicol 2013;88:725–38.
26. Mikami S, Oya M, Mizuno R, Murai M, Mukai M,Okada Y. Expression of Ets-1 in human clear cellrenal cell carcinomas: implications for angiogen-esis. Cancer Sci 2006;97:875–82.
27. Cieply B, Farris J, Denvir J, Ford HL, Frisch SM.Epithelial-mesenchymal transition and tumor sup-pression are controlled by a reciprocal feedbackloop between ZEB1 and Grainyhead-like-2. Can-cer Res 2013;73:6299–309.
28. Chen W, Xiao LZ, Oh JE, Shin KH, Kim RH, Jiang M,et al. Grainyhead-like 2 (GRHL2) inhibits keratino-cyte differentiation through epigenetic mecha-nism. Cell Death Dis 2012;3:e450.
29. Xiang X, Deng Z, Zhuang X, Ju S, Mu J, Jiang H,et al. Grhl2 determines the epithelial phenotypeof breast cancers and promotes tumor progres-sion. PLoS One 2012;7:e50781.
30. Emanuele MJ, Ciccia A, Elia AE, Elledge SJ. Prolif-erating cell nuclear antigen (PCNA)-associatedKIAA0101/PAF15 protein is a cell cycle-regulatedanaphase-promoting complex/cyclosome substrate.Proc Natl Acad Sci U S A 2011;108:9845–50.
31. Jain M, Zhang L, Patterson EE, Kebebew E. KIAA0101is overexpressed, and promotes growth and invasionin adrenal cancer. PLoS One 2011;6:e26866.
32. Hoek KS, Eichhoff OM, Schlegel NC, Dobbeling U,Kobert N, Schaerer L, et al. In vivo switching ofhuman melanoma cells between proliferative andinvasive states. Cancer Res 2008;68:650–6.
33. Su X, Zhang T, Cheng P, Zhu Y, Li H, Li D, et al.KIAA0101 mRNA overexpression in peripheralblood mononuclear cells acts as predictive markerfor hepatic cancer. Tumour Biol 2013;35:2681–6.
34. Papagiannakopoulos T, Kosik KS. MicroRNAs:regulators of oncogenesis and stemness. BMCMed 2008;6:15.
35. Zhao WH, Wu SQ, Zhang YD. Downregulation ofmiR-124 promotes the growth and invasivenessof glioblastoma cells involving upregulation ofPPP1R13L. Int J Mol Med 2013;32:101–7.
36. Lee MR, Kim JS, Kim KS. miR-124a is important formigratory cell fate transition during gastrulation ofhuman embryonic stem cells. Stem Cells 2010;28:1550–9.
37. Ying Z, Li Y, Wu J, Zhu X, Yang Y, Tian H, et al.Loss of miR-204 expression enhances glioma mi-gration and stem cell-like phenotype. Cancer Res2013;73:990–9.
38. Fridman E, Dotan Z, Barshack I, David MB, Dov A,Tabak S, et al. Accurate molecular classificationof renal tumors using microRNA expression. J MolDiagn 2010;12:687–96.
39. Wu X, Weng L, Li X, Guo C, Pal SK, Jin JM, et al.Identification of a 4-microRNA signature for clearcell renal cell carcinoma metastasis and progno-sis. PLoS One 2012;7:e35661.
40. Bao W, Fu HJ, Xie QS, Wang L, Zhang R, Guo ZY,et al. HER2 interacts with CD44 to up-regulate CXCR4via epigenetic silencing of microRNA-139 in gastriccancer cells. Gastroenterology 2011;141:2076–87.
41. Fan Q, He M, Deng X, Wu WK, Zhao L, Tang J,et al. Derepression of c-Fos caused by microRNA-139 down-regulation contributes to the metasta-sis of human hepatocellular carcinoma. CellBiochem Funct 2013;31:319–24.
1326 Clinical Chemistry 60:10 (2014)





























