Embed Size (px)
Citation preview

Phototransduction: crystal clearKevin D. Ridge1, Najmoutin G. Abdulaev1, Marcelo Sousa2 and Krzysztof Palczewski3
1Center for Advanced Research in Biotechnology, National Institute of Standards and Technology and the University of Maryland
Biotechnology Institute, Rockville, MD 20850, USA2Department of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309, USA3Departments of Ophthalmology, Chemistry, and Pharmacology, University of Washington, Seattle, WA 98195, USA
Vertebrate visual phototransduction represents one
of the best-characterized G-protein-coupled receptor-
mediated signaling pathways. Structural analyses of
rhodopsin, G protein, arrestin and several other photo-
transduction components have revealed common folds
and motifs that are important for function. Static and
dynamic information has been acquired through the
application of X-ray diffraction, solution and solid-state
nuclear magnetic resonance spectroscopy’s, electron
and atomic force microscopy’s, and a host of indirect
structural methods. A comprehensive understanding of
phototransduction requires further structural work on
individual components and their relevant complexes in
solution and the native disk membrane. Given the
accelerated pace of structure determination, it is antici-
pated that this will be the first G-protein-coupled path-
way for which a complete molecular description is
ultimately available.
Vertebrate visual phototransduction begins with lightabsorption by rhodopsin in the disk membrane (DM) andculminates in plasma-membrane hyperpolarization.These two events are linked by a highly sophisticatedand elegantly organized biochemical signaling machinery[1]. For the past several decades, multidisciplinaryapproaches have been employed to unravel both theprotein constituents of the rod outer segment (ROS) andthe key players involved in signaling (Box 1). These effortshave greatly contributed to our understanding of photo-transduction. This sustained progress has been recentlyhighlighted by the successful crystallization and structuredetermination of bovine rhodopsin [2], leading to arenewed emphasis on structural studies of phototransduc-tion components. To date, more than ten distinct proteinsof the phototransduction pathway have been analyzed inmolecular detail (Table 1). These include representativecomponents of signal propagation (see Fig. Ia in Box 1),light-stimulated rhodopsin inactivation (see Fig. Ib inBox 1) and the retinal G protein transducin (Gt) a-subunitinactivation (see Fig. Ic in Box 1). Most of the reportedstructures are crystal structures of individual proteins,domains of proteins, or complexes of constituent subunits.Some physiologically relevant protein–protein complexes,such as the phosducin–Gtbg complex (see Fig. Ic in Box 1and Table 1), have also been analyzed in structural detail[3,4]. For Gt, a structural description of GTP–GDP
exchange and the GTPase cycle is almost complete [5–8](see Fig. Ia in Box 1 and Table 1). Among those proteinswithout high-resolution structures, rhodopsin kinase (RK,also known as GRK1) and cGMP phosphodiesterase(PDE6) are perhaps the most eagerly anticipated.Although insights into the modular nature of RK havebeen provided from the crystal structure of GRK2 [9], themolecular organizations of PDE6 abd- and abg2-subunitcomplexes have been revealed by electron microscopy (EM)and image processing [10,11]. These latter methods havealso been employed to study the molecular architecture ofthe cGMP-gated channel [12]. In the phototransductionrecovery phase (see Fig. Ic in Box 1), crystal structures ofthe regulator of G-protein signaling (RGS)-9–1 RGSdomain alone and in complex with Gat/i1 [a chimeric Gprotein of Gta and the a subunit of the inhibitory G protein(Gia)] and a fragment of PDE6g have been determined [13].The structure of non-myristoylated guanylate cyclase-activating protein-2 (GCAP-2) with three bound Ca2þ ionshas also been determined by nuclear magnetic resonance(NMR) [14]. Although we have categorized these proteinsin terms of amplification, desensitization and recovery (seeFig. Ia in Box 1), the intricate blending and interplaybetween each of these components is essential for efficientsignaling. It should also be noted that two accessorypathways are required for proper functioning of thephototransduction machinery: the guanine nucleotideand retinoid cycles. A partial structural description ofGTP and GDP metabolism has been provided through thecrystallization of two isoforms of retinal nucleoside dipho-sphate kinase (NDPK) [15] (See Fig. Ia in Box 1).Structural aspects of the retinoid cycle [16], whose majorcomponents have not yet been fully identified, have alsobegun to be realized [17].
Major methods contributing to a structural
understanding of phototransduction
One of the strengths of phototransduction research hasbeen the numerous methodologies and approachesemployed to acquire static and dynamic structuralinformation about key components. The most directmethods of structural analysis are clearly X-ray diffrac-tion, solution and solid-state NMR, EM and, more recently,atomic force microscopy (AFM). X-ray crystallography hasbeen used to solve the structure of the Gat/i1–RGS9–1 RGSdomain–PDE6g heterotrimeric complex [13], providingour first glimpse into the molecular mechanism of Ga
inactivation (Fig. 1a). Solid-state magic-angle spinningCorresponding author: Kevin D. Ridge ([email protected]).
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003 479
http://tibs.trends.com 0968-0004/$ - see front matter q 2003 Elsevier Ltd. All rights reserved. doi:10.1016/S0968-0004(03)00172-5

Box 1. Vertebrate visual phototransduction
The phototransduction pathway with the various components categor-
ized into signal propagation, inactivation of Rp and inactivation of G
protein transducin (Gt) a subunit (Gta) for clarity is shown in Fig. I.
Protein structures determined by X-ray crystallography or nuclear
magnetic resonance (NMR) are shown in different shades of blue,
including rhodopsin (R), the Gt abg heterotrimer and constituent
subunits, Ca2þ-bound guanylate cyclase-activating protein (GCAP)-2,
Ca2þ-free and Ca2þ-bound myristoylated recoverin (Rec), arrestin (Arr),
the regulator of G-protein signaling (RGS)-9–1 RGS domain, the Gat/i1–
RGS9–1 RGS domain–cGMP phosphodiesterase (PDE6) g subunit
fragment complex, and a phosducin (Ph) -Gt bg-subunit complex.
Outstanding protein structures or those available only at low-resolution
are shown in different shades of gray, including the rhodopsin
photointermediates (such as Rp and others that are not shown [38]),
the cGMP-gated channel holoenzyme and constituent a and b subunits,
rhodopsin kinase (RK), PDE6 holoenzyme and constituent a, b, g and d
subunits, guanylate cyclase (GC), Gb5L and R9AP. Note that R, Rp and
opsin (O) are depicted as distinctly shaded dimers to emphasize their
proposed higher-order organization. Although direct atomic force
microscopy (AFM) evidence exists for the dimeric state of murine
rhodopsin (R1R2) and opsin (O1O2) in the disk membrane (DM) [20,21],
these data are at variance with earlier translational and rotational
diffusion measurements on bovine rhodopsin [70,71]. The existence of
Rpdimers interacting with Gt and Arr is based on conceptual models that
take into account the accessible binding surface areas [21]. The
existence of Rp dimers interacting with RK is hypothetical.
Signal propagationRp in the DM catalytically promotes the exchange of GDP for GTP in
hundreds of Gt molecules to produce an active, GTP-bound form of Gta
(Fig. Ia). Activated Gta interacts with theg subunits of PDE6, removing an
inhibitory constraint from the catalytic a or b subunits and resulting in
the hydrolysis of cGMP. Fast depletion of cGMP in the rod outer
segment (ROS) results in the closure of cGMP-gated channels in the
plasma membrane (PM). A drastic reduction in the circulating ‘dark’
current – due to a blockage of Naþ and Ca2þ entry into the rod cell and
Ca2þ depletion of the ROS as a result of continuous function of the PM
Naþ–Ca2þ, Kþ exchanger – activates the Ca2þ-binding protein GCAP.
Low intracellular Ca2þ promotes activation of GC by Ca2þ-free GCAP,
catalyzing accelerated synthesis of cGMP from GTP supplied by the
guanine nucleotide cycle, which comprises guanylate kinase (GK) and
nucleoside diphosphate kinase (NDPK). Release of Ca2þ from calmo-
dulin (CaM) leads to its dissociation from the cGMP-gated channel
conferring a lower affinity for cGMP. Therefore, the photoactivation of
rhodopsin leads to changes in the net production of cGMP by perturbing
two opposing catalytic activities: degradation by PDE6 and synthesis by
GC. Because cGMP levels govern how many cation channels are open,
the light signal is transformed from a physical stimulus to a biochemical
chain of reactions that culminates in the hyperpolarization of the PM.
Inactivation of Rp
In the dark, when the intracellular Ca2þ concentration is high,
myristoylated Rec is in the Ca2þ-bound state and forms a complex
with RK at the DM, preventing phosphorylation of Rp (Fig. Ib). When the
Ca2þ concentration in the ROS drops as a result of the light response,
Rec releases its bound Ca2þ and dissociates from RK (note that this
mechanism is still controversial [1]). The interaction between Rp and RK
leads to rapid phosphorylation of Rp. Subsequent binding of Arr
completely blocks Rp signaling via Gt. Release of all-trans-retinal,
accompanied by enzymatic dephosphorylation by protein phosphatase
2A (PP2A), prepares opsin for 11-cis-retinal binding and restoration of its
inactive ground state (R).
Fig. I. Vertebrate visual phototransduction. Protein structures determined by X-ray crystallography or nuclear magnetic resonance (NMR) are shown in different shades
of blue. Outstanding protein structures or those available only at low-resolution are shown in different shades of purple. (a) Signal propagation. (b) Inactivation of Rp.
(c) Inactivation of Gta–GTP.
(a)
α
cGMP-gatedchannel closed
cGMP-gatedchannel open
Exchanger4 Na+
Ca2+
Ca2+
Ca2+
Ca2+
Ca2+, K+
Na+, Ca2+
CaM
α α
cGMP GMP
hν
R1R2 R*
PDE
α β
ATP
ATP
NDPK
GK
PM
DM
DM DM
δ GC
γ γα β
αα
β
α
αα
β
δ
γ γ
β αγ
β
GTP GTP cGMPGTP GDP
GDP
GDPGTP GTP
R1
PDE
R* O1O2
Ti BS
RK
ATP
RGS9-1
Gβ5L
(b) (c)
Arr
Arr
Rec
Rec Rec
all-trans-retinal
Ca2+
Ca2+ Ca2+
R9APPP
11-cis-retinal
PP2A
GDP
R* R*
βPHγ
α ααα βγ γGTP GTPPDE
α αα β
GDP GDPPDE
α αγ
δGβ5L
R9AP
γ γ
GCAPGCAP
RK
CaM
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003480
http://tibs.trends.com

NMR (Fig. 1b) has been useful for probing non-bondinginteractions between the protons of the methyl groups ofthe 11-cis-retinylidene b-ionone ring and the opsin protein[18]. Solution NMR methods have been applied to examinethe structures of synthetic peptides corresponding todifferent regions of opsin, providing insights into theconformations adopted by individual components [19]. Thestructural organization of rhodopsin and opsin inthe native DM has been revealed by AFM [20,21]. Theparacrystalline arrangement of rhodopsin dimers hasprofound implications for understanding signal transfervia Gt and signal quenching by arrestin. EM has beenapplied to several phototransduction components thateither reside in the membrane and/or form multi-subunitcomplexes. A low-resolution model for PDE6 abd obtainedby EM and image processing [10] affords remarkableinsights into the modular nature of this effector enzyme
(Fig. 2c). Although these direct methods of structuralanalysis are preferable, in most instances the applicationof more indirect methods has also yielded useful infor-mation. For example, UV-visible spectroscopy has beenused to examine rhodopsin assembly from complementary,non-overlapping expressed fragments [22]. More sophisti-cated spectroscopic techniques, such as Fourier transforminfrared, have been used to probe differences in theenvironment of specific amino-acid residues involved inchromophore–protein interactions between the dark andlight-activated states of rhodopsin [23,24]. The majoradvantage of this technique is a global view of proteinconformational changes at acceptable kinetic resolution.One of the more informative spectroscopic methods used tostudy conformational changes accompanying rhodopsinactivation is the combined use of cysteine scanningmutagenesis, chemical modification with thiol-reactive
Inactivation of Gta–GTPThe intrinsic GTPase activity of Gta–GTP is dramatically enhanced
upon interaction with the g-subunit of PDE (Fig. Ic). Removal of the
terminal phosphate is further increased upon interaction with the
GTPase accelerator protein (RGS9–1–Gb5L) and its DM anchor
R9AP. The resulting complex consisting of at least five distinct
components (Gta-GTP–PDEg–RGS9–1–Gb5L–R9AP) supports a
physiologically relevant rate of GTP hydrolysis and Gta inactivation.
Dissociation of Gta–GDP from PDE simultaneously inactivates both
proteins.
Table 1. Currently available structures of phototransduction componentsa
Protein Method PDB code Comments Refs
Rhodopsin X-ray 1F88 2.8 A resolution; two monomers (A and B) per asymmetric unit; key regions of the second
and third cytoplasmic loops and C terminus not resolved in A and B
[2]
Rhodopsin X-ray 1HZX Refined model of 1F88; additional amino-acid residues and palmitoyl chains added; no
major differences from 1F88
[40]
Rhodopsin X-ray 1L9H 2.6 A resolution; as in 1F88, but positions of water molecules were corrected; the
differences between the three rhodopsin models were recently discussed in Ref. [45]
[41]
G at/i1/Gt bg subunits X-ray 1GOT Chimeric Ga complexed with GDP and Mg2þ; first 5 and last seven residues of Ga not
resolved; three residues and farnesyl group removed from C terminus of Gtg
[5]
Gt a-subunit X-ray 1TAG 1.8 A resolution; 25 residues removed from N terminus; last ten residues not resolved;
complexed with GDP and Mg2þ
[6]
Gt a-subunit X-ray 1TND 2.2 A resolution; 25 residues removed from N terminus; last eight residues not resolved;
complexed with GTPgS and Mg2þ
[7]
Gt a-subunit X-ray 1TAD 1.7 A resolution; as in 1TND but complexed with GDP, AlF42, and Mg2þ [8]
Gt bg-subunits X-ray 1TBG 2.1 A resolution; Gtb seven-bladed b-propeller fold; three residues and farnesyl group
removed from C terminus of Gtg
[67]
Arrestin X-ray 1AYR 3.3 A resolution; four monomers (A,B, C and D) per asymmetric unit; last 38 residues of A
and C and 41 residues of B and D not resolved
[50]
Arrestin X-ray 1CF1 2.8 A resolution; four monomers (A,B, C and D) per asymmetric unit; last 42 residues of A
and 32 residues of B, C and D not resolved; different assignment for the C-terminal portion
[51]
Recoverin X-ray 1REC 1.9 A resolution; first seven and last four residues not resolved; one bound Ca2þ; non-
myristolyated
[53]
Recoverin NMR 1IKU RMSD of 0.44 ^ 0.04 A for backbone atoms (22 structures) of the Ca2þ-free form; last 13
residues not resolved; sequestered myristoyl group
[64]
Recoverin NMR 1JSA RMSD of 0.80 ^ 0.10 A for backbone atoms (24 structures) of the Ca2þ-bound form
(2 Ca2þ); last 13 residues not resolved; exposed myristoyl group
[65]
GCAP-2 NMR 1JBA RMSD of 0.88 ^ 0.10 A for backbone atoms (22 structures) of the Ca2þ-bound form
(3 Ca2þ); last 14 residues not resolved; non-myristoylated
[14]
Phosducin/Gt bg X-ray 2TRC 2.4 A resolution; residues 36–66 of phosducin not resolved; three residues and farnesyl
group removed from C terminus of Gtg; conserved thioredoxin fold in phosducin
[3]
Phosducin/Gt bg X-ray 1AOR 2.8 A resolution; residues 38–67 of phosducin not resolved; farnesylated Gtg; structural
changes in Gtbg upon phosducin binding
[4]
RGS9–1 X-ray 1FQI 1.94 A resolution; residues 276–422 (RGS domain) [13]
Gat/i1/RGS9–1 X-ray 1FQK 2.3 A resolution; 25 residues removed from N terminus of chimeric Ga complexed with
GDP, AlF42, and Mg2þ; RGS domain
[13]
Gat/i1/RGS9–1/PDE6g X-ray 1FQJ 2.02 A resolution; as in 1FQK but in complex with a fragment comprising residues 46–87
from PDE6g
[13]
NDPK X-ray 1BHN 2.4 A resolution; hexamer of isoform A complexed with GDP [15]
NDPK X-ray 1BE4 2.4 A resolution; hexamer of isoform B complexed with cGMP [15]
aAbbreviations: AlF42, aluminium fluoride; Gt, G protein transducin; GCAP-2, guanylate cyclase-activating protein-2; NDPK, nucleoside diphosphate kinase; NMR, nuclear
magnetic resonance; RGS, regulator of G-protein signaling; RMSD, root-mean-square deviation.
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003 481
http://tibs.trends.com

spin labels [known as site-directed spin labeling (SDSL)]and electron paramagnetic resonance (EPR) spectroscopy[25] (Fig. 1d). This approach has also been successfullyextended by others [26] using site-specific labeling ofcysteine residues with fluorescent labels to monitor light-dependent structural changes in rhodopsin. Finally,circular dichroism (CD) spectroscopy has been used toexamine the secondary structure of a synthetic peptidecorresponding to helix eight (H8) of rhodopsin. Suchstudies have indicated that H8 acts as a membrane-dependent conformational switch [27], a finding that isconsistent with subsequent mutagenesis work [28].
Chronology of a structure for rhodopsin
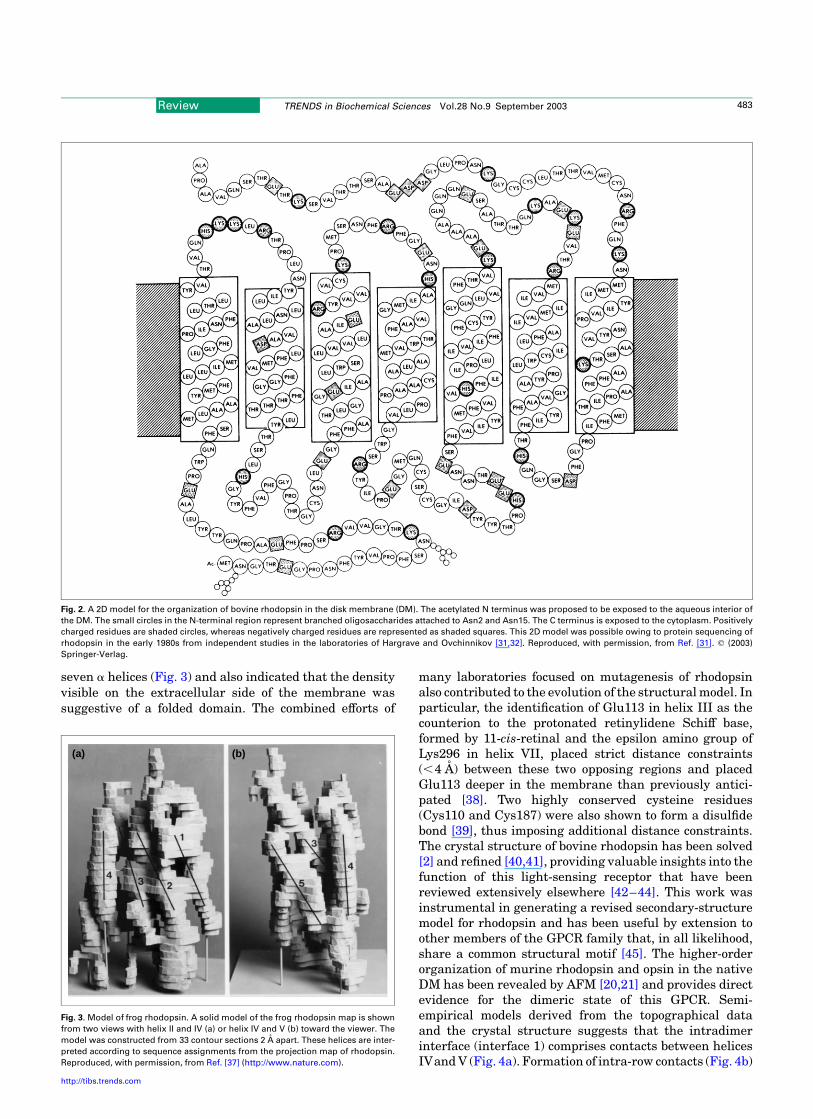
An understanding of the visual process dates back to the19th century. Extraction of the visual pigment rhodopsinfrom bovine retina with bile salts by Kuhne [29] deepenedinterest in understanding the key light receptor involvedin this process. Wald provided the first evidence that thevisual pigment is composed of a protein conjugated with11-cis-retinal [30]. More detailed information on bovinerhodopsin was obtained when the protein was sequencedand painstakingly assembled from different overlappingpeptide-fragment sequences [31,32]. The availability of theprimary structure provided the first clues into the overallorganization of rhodopsin. This initial model (Fig. 2)
verified the orientation of rhodopsin in the lipid bilayerpreviously inferred from biochemical studies. Theoreticalconsiderations predicted that rhodopsin spans the mem-brane seven times, exposing the C and N termini to thecytoplasm and extracellular (intradiscal) space, respect-ively, and that the transmembrane portion is formed by theputative a-helical domain [33]. An accurate projection mapof rhodopsin showing the positions of the helices was notdetermined until the work of Schertler and colleagues [34].This map revealed that the helices are organized like ‘aboat with a sail’. Further reconstruction models based oncryo-EM data, homology and conservation of residues inG-protein-coupled receptors (GPCRs) revealed a three-dimensional organization for rhodopsin [35]. Indepen-dently, a theoretical model of exceptional accuracy wascalculated by others [36] using an iterative distancegeometry refinement with an evolving system of hydrogenbonds, formed by intra-membrane polar side chains invarious rhodopsin-like proteins of the GPCR family andcollectively applied as distance constraints. This modeldiffered from the crystal-structure model in the trans-membrane region by an root-mean-square deviation(RMSD) of 1.63 A (PDB code: 1F88 versus 1BOK). Furtherprogress was made using electron micrographs obtainedfrom frozen-hydrated two-dimensional frog rhodopsincrystals [37]. These data unveiled tilting angles for the
Fig. 1. Methodologies used to gain structural information about phototransduction. (a) X-ray crystal structure of the Gat/i1 [a chimeric G protein of Gta and the a subunit of
the inhibitory G protein (Gia)]–RGS9–1 (regulator of G-protein signaling)–phosphodiesterase (PDE)-6 g subunit heterotrimeric complex. Ribbon drawing of Gat/i1 (green)
with bound GDP–aluminium fluoride (AlF42; ball-and-stick models), a fragment of PDEg (red), and the RGS9–1 RGS domain (blue). The structure provides the first molecular
description of the process of Ga inactivation. Image courtesy of Kevin Slep and Ted Wensel; based on work from Ref. [13]. (b) Solid-state magic-angle-spinning nuclear
magnetic resonance (NMR) of chromophore-protein interactions in rhodopsin. The color-encoded NMR ligation shifts arising from high-resolution distance constraints
emphasize the local chromophore-protein interactions in rhodopsin. The structure provides the first high-resolution view of the chromophore. Image courtesy of Huub de
Groot and Willem DeGrip; based on work from Ref. [18]. (c) Surface representation of the 3D model of PDE6 based on electron microscopy and image analysis. The dimeric
structure and the domain organization of PDE6 are shown at 2.8 nm resolution in three models rotated 458 around the vertical axis. The GAFa and GAFb domains and the
catalytic-domain fold into independent modules, consistent with X-ray studies of recombinant PDE2 GAFa or GAFb [68] and PDE4 catalytic fragments [69]. Interaction of
the inhibitory g subunit or the 17-kDa prenyl-binding protein (d subunit) with the catalytic dimer cannot be discriminated. The electron microscopy (EM) structure affords a
remarkable view into the molecular organization of PDE6. Image courtesy of Rick Cote and Patrick Schultz; based on work from Ref. [10]. (d) Electron paramagnetic reson-
ance (EPR) spectra of rhodopsin cysteine mutants modified with a sulfhydryl reactive spin label. (i) The EPR spectra show light-dependent changes in the environment of
attached nitroxide spin labels introduced at different positions (227, 250 and 252) in the cytoplasmic ends of helices V (TM5) and VI (TM6) of rhodopsin. (ii) A ribbon diagram
of rhodopsin (viewed from the cytoplasmic surface) showing the location of the nitroxide side chains in the dark state. The outward direction of the arrow illustrates the
light-dependent rigid-body tilt of TM6 away from the center of rhodopsin. EPR provides information related to local changes in the environment of the spin label as a result
of conformational changes in the protein. Image courtesy of Cheryl Hubbell, Wayne Hubbell and Christian Altenbach; based on work reviewed in Ref. [25].
(a)
(c)
(i) (ii)
PDEγ
RGS9–1
Gαt/i1
(b)
(d)
ppm (13C)
ppm (1H)–1.5
–5.5
+1.5
+5.5
Catalyticdomain
GAF bdomain
GAF adomain
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003482
http://tibs.trends.com
seven a helices (Fig. 3) and also indicated that the densityvisible on the extracellular side of the membrane wassuggestive of a folded domain. The combined efforts of
many laboratories focused on mutagenesis of rhodopsinalso contributed to the evolution of the structural model. Inparticular, the identification of Glu113 in helix III as thecounterion to the protonated retinylidene Schiff base,formed by 11-cis-retinal and the epsilon amino group ofLys296 in helix VII, placed strict distance constraints(,4 A) between these two opposing regions and placedGlu113 deeper in the membrane than previously antici-pated [38]. Two highly conserved cysteine residues(Cys110 and Cys187) were also shown to form a disulfidebond [39], thus imposing additional distance constraints.The crystal structure of bovine rhodopsin has been solved[2] and refined [40,41], providing valuable insights into thefunction of this light-sensing receptor that have beenreviewed extensively elsewhere [42–44]. This work wasinstrumental in generating a revised secondary-structuremodel for rhodopsin and has been useful by extension toother members of the GPCR family that, in all likelihood,share a common structural motif [45]. The higher-orderorganization of murine rhodopsin and opsin in the nativeDM has been revealed by AFM [20,21] and provides directevidence for the dimeric state of this GPCR. Semi-empirical models derived from the topographical dataand the crystal structure suggests that the intradimerinterface (interface 1) comprises contacts between helicesIVand V (Fig. 4a). Formation of intra-row contacts (Fig. 4b)
Fig. 2. A 2D model for the organization of bovine rhodopsin in the disk membrane (DM). The acetylated N terminus was proposed to be exposed to the aqueous interior of
the DM. The small circles in the N-terminal region represent branched oligosaccharides attached to Asn2 and Asn15. The C terminus is exposed to the cytoplasm. Positively
charged residues are shaded circles, whereas negatively charged residues are represented as shaded squares. This 2D model was possible owing to protein sequencing of
rhodopsin in the early 1980s from independent studies in the laboratories of Hargrave and Ovchinnikov [31,32]. Reproduced, with permission, from Ref. [31]. q (2003)
Springer-Verlag.
Fig. 3. Model of frog rhodopsin. A solid model of the frog rhodopsin map is shown
from two views with helix II and IV (a) or helix IV and V (b) toward the viewer. The
model was constructed from 33 contour sections 2 A apart. These helices are inter-
preted according to sequence assignments from the projection map of rhodopsin.
Reproduced, with permission, from Ref. [37] (http://www.nature.com).
(a) (b)
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003 483
http://tibs.trends.com

are largely facilitated by interactions between helices Iand II with an adjacent dimer (interface 2), whereascontacts between rows of dimers (Fig. 4b) are maintainedthrough helix I (interface 3).
Conservation of common structural folds in
phototransduction components
The crystal structure of the G-protein heterotrimer revealstwo non-overlapping regions of contact between the a andb subunits, an extended interface between the b and g
subunits, but only minimal contact between the a and g
subunits. The a subunit of Gt – like that found in all Gproteins – has a two-domain structure composed of ana-helical bundle and the GTP-binding a–b sandwich,referred to as a ‘Rossmann-fold’ domain [46]. The latter is asuper-secondary structural element present in manymononucleotide- and dinucleotide-binding enzymes that
is conformationally well conserved. Superposition ofthe structures for Gta, Gi1a and the a subunit of thestimulatory G protein (Gsa) show little conformationalvariance in the core region of the protein (Fig. 5a). Afurther comparison of Gta with ADP-ribosylation-factor-1 – a small GTP-binding protein – shows that,in spite of the low sequence similarity between thesetwo proteins (,17.9%), the structures of the Ross-mann-fold domain superimpose with an RMSD of 2.5 A(Fig. 5b). This remarkable structural similarity,accompanied by low sequence identity (in some casesas low as 2–3%), has prompted speculation thatRossmann folds are an example of convergent evol-ution. However, the fact that the nucleotides arealways bound in a similar manner suggests divergencefrom a common ancestor. Therefore, the evolutionaryorigin of the fold remains controversial.
In Gtb, the most apparent structural feature is theseven-bladed b-propeller fold. b propellers are modularstructures constructed by repetition of four to eight ‘blades’organized around a central symmetry axis, with eachblade comprising a four-stranded b-sheet. b-propellerproteins support vast arrays of functions. They rangefrom enzymes catalyzing various chemistries to proteinsmediating signaling, scaffolding and ligand binding [47].The sequence conservation among the blades of a b
propeller varies widely. The blade sequences can be nearlyidentical as in tachylectin-2 [48], or weakly conserved, asin Gtb, which contains WD (Trp-Asp) repeats. In thisfamily of WD-repeat proteins, the conservation isrestricted to a GH (Gly-His) pair and a WD pair separatedby hydrophobic residues at specific locations [49].
Visual arrestin (Arr) binds to phosphorylated, light-activated rhodopsin (Rp, also known as metarhodopsin IIor Meta II) thereby shutting down signaling via Gt
(see Fig. Ib in Box 1). Crystal structures of Arr [50,51]show that it has a similar fold to the non-visual arrestinsarrestin-2 and b-arrestin (RMSD of 1.2 A and 1.8 A,respectively). They consist of two immunoglobulin-likefolds packed head-to-head, which might bind to onephosphorylated rhodopsin (or GPCR) dimer [21]. Theimmunoglobulin-like fold is a b sandwich made up of twoanti-parallel b-sheets packing against each other, and ispresent in many proteins. An example of the structuralconservation of this fold can be found in FimC (Fig. 5c), abacterial chaperone protein whose structure superimposeswell with one of the immunoglobulin-like domains of Arr(sequence identity 13.5%, RMSD of 3.2 A).
Recoverin (Rec) appears to serve as a Ca2þ sensor in rodcells. The Ca2þ-bound form of the protein is thought toprolong the lifetime of Rp by inhibiting RK [1,52] (see Fig. Ibin Box 1). Rec belongs to the EF-hand superfamily ofproteins, of which calmodulin (CaM) is the best-knownmember. These proteins have four EF hands arranged inpairs that give rise to N- and C-terminal domains. Thecrystal structure of Rec [53] shows that the relativepositions of these two domains are very different fromthose found in CaM. In CaM, the two domains are linkedby a long helix generating a dumbbell-like shapedmolecule, whereas Rec adopts a more compact confor-mation due to the short linker between the two domains.
Fig. 4. Models for the packing arrangement of rhodopsin in the native disk mem-
brane (DM). (a) Rhodopsin assembles into dimers through contacts provided by
helices IV and V (interface 1). (b) View from the cytoplasmic surface showing rows
of dimers that form as a result of contacts between helices I and II and an adjacent
dimer (interface 2). Rows assemble through extracellular contacts formed by helix
I (interface 3). The images were generated using PDB code 1N3M and are based on
atomic force microscopy data on murine rhodopsin in the DM and the crystal
structure of bovine rhodopsin [2,21].
2
1
1
3
(a)
(b)
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003484
http://tibs.trends.com

This compact conformation (Fig. 5d) is shared with otherCa2þ-binding proteins such as neurocalcin, frequenin andthe GCAPs [14]. The Ca2þ-binding EF-hand motifs adoptstrikingly different conformations in the Ca2þ-bound andCa2þ-free forms, and these local conformational changesare propagated throughout the molecule.
Structural changes in phototransduction components
Signal propagation during phototransduction dependson changes in the concentration of cGMP and confor-mational changes in proteins that induce the formationof new protein–protein interactions or the breakingof existing complexes. Because fluctuations in cGMP,Ca2þ and cation levels are one way of communicatingand controlling the signal (see Fig. I in Box 1), the key
question as to how the G-protein-mediated signal ispropagated during phototransduction requires knowl-edge of the protein conformational changes involved inthis process.
Extensive work from the laboratories of Hubbell andKhorana using SDSL and EPR spectroscopy and disulfidecrosslinking has contributed significantly to our currentunderstanding of rhodopsin activation [25]. One promi-nent conclusion from this work is the outward movementof the cytoplasmic ends of helices III and VI together with aclockwise rotation of helix VI around its helical axis(Fig. 1d). Evidence for movement of the cytoplasmicend of helix VII upon rhodopsin activation has alsobeen provided by antibody binding [54] and muta-genesis [28] studies. All of these changes are intimatelylinked to the light-induced change in retinal geometryand are thought to remove inhibitory constraints in theseven-helical bundle that open up cytoplasmic sites forinteraction with Gt [25,42–44].
Although details of the GTP-dependent conformationalrearrangements in Gt have been largely revealed throughcrystallographic studies [5–8], our current understandingof how structural changes in Gt are coupled tosignaling by Rp have come from NMR experiments.Peptide competition studies have shown that the lasteleven amino acids of Gta (Ile340 to Phe350) and Gtg
(Asp60 to farnesylated Cys71), which are encompassedin unresolved regions of the reported crystal structures(Table 1), are major rhodopsin-interaction sites [55,56].Application of transferred nuclear Overhauser enhance-ment spectroscopy (TrNOESY) methods to examine theinteraction between a C-terminal Gta(340–350) peptideanalog and Rp have revealed structural transitions at boththe N and C termini of the bound peptide [57]. SimilarTrNOESY studies using a C-terminal Gta(340–350)peptide corresponding to the native Gta sequence oranother peptide analog have indicated the formation of areverse aL-type C-cap motif in the Rp-bound state of thesepeptides [58,59]. Remarkably, this same structural tran-sition has also been observed upon Gta(340–350)-peptidebinding to a soluble mimic of Rp [60]. More recently,TrNOESY methods have also been applied to studystructural transitions in Rp bound Gtg C-terminal peptides[61]. Models to explain the molecular mechanism govern-ing the release of GDP from Gta – the rate-limiting stepin transmission of the signal from Rp to Gt – are beyondthe scope of this review and have been describedelsewhere [62,63].
In addition to the conserved Ca2þ-binding motifs, Recalso contains an N-terminal myristoyl group. The bindingof two Ca2þ ions to myristoylated Rec leads to itstranslocation from the cytosol to the DM (see Fig. Ib inBox 1). Comparisons between the Ca2þ-free and the Ca2þ-bound forms of myristoylated Rec [64,65] show that, in theCa2þ-free form of the protein, the myristoyl group issequestered in a hydrophobic cavity where it is in contactwith several aromatic amino-acid residues contributed bythree of the four EF hands. The binding of Ca2þ inducesthe release and extrusion of the myristoyl group, enablinginteraction with the DM. This rather remarkable Ca2þ-induced transition is also accompanied by a 458 rotation of
Fig. 5. Similarity of structural motifs used by phototransduction proteins. (a)
Superposition of G-protein a-subunit structures. The structures of G protein trans-
ducin a subunit (Gta; PDB code: 1TND), the a subunit of the inhibitory G protein
(Gia1; PDB code: 1GIA), and the a subunit of the stimulatory G protein (Gsa; PDB
code: 1CUL) are superimposed. The core of the structures showing little confor-
mational variance is colored in yellow. The parts displaying different confor-
mations are highlighted for Gta (blue), Gia1 (green), and Gsa (magenta). The
activating ligand, GTPgS, is shown as a CPK model. The RMSDs for Gia1 and Gsa
compared with Gta are 1.1 A and 1.5 A, respectively, whereas the sequence simi-
larities are 69.4% and 43.1%, respectively. (b) Use of the Rossmann fold by
GTP-binding proteins. Ribbon drawings of a small GTP-binding protein [ADP ribo-
sylation factor 1 (magenta); PDB code: 1RRG] superimposed on Gta (PDB code:
1TND; yellow) to highlight the conservation of the Rossmann fold. This motif is
conserved among many mononucleotide- and dinucleotide-binding proteins. (c)
Superposition of arrestin and the FimC chaperone from Escherichia coli. The two
immunoglobulin-like b-sandwich domains in Arr (green and yellow) are superim-
posed with the FimC chaperone (magenta). The RMSD for FimC (PDB code: 1QUN)
compared with Arr (PDB code: 1AYR) is 3.2 A and the sequence identity is 13.5%.
(d) The structure of Ca2þ-binding proteins. Superposition of the Ca2þ-free
forms of Rec (red; PDB code: 1REC), neurocalcin (yellow; PDB code: 1BJF),
frequenin (blue; PDB code: 1G8I) and GCAP-2 (green; PDB code: 1JBA). All of these
Ca2þ-binding proteins share a compact conformation that is distinct from that
found in calmodulin.
(a) (b)
(c) (d)
Helicaldomain
Rossmann fold
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003 485
http://tibs.trends.com

the N-terminal domain relative to the C-terminal domain,resulting in the exposure of many hydrophobic residues.As the ‘calcium-myristoyl switch’ is an important deter-minant for DM localization by Rec, it might also representa mechanism for controlling the localization of otherstructurally similar Ca2þ-binding proteins.
Concluding remarks
Our understanding of vertebrate visual phototransductionis very mature, and represents by far the best-understoodGPCR-mediated signal transduction pathway. Most of themajor components have now been identified throughbiochemical and molecular biological approaches, andclear pathways defined with the aid of biochemicalreconstitution assays, mouse genetics and electrophysi-ology. What remains unanswered is a comprehensivestructural understanding that can fully explain howphoton absorption leads to changes in neuronal trans-mission. The importance of structural work results fromthe simple fact that ‘seeing is believing.’ The availability ofa crystal structure for rhodopsin has had a stunning effecton the formulation of new ideas regarding receptoractivation [25,28,42–44], providing clues into the speci-ficity of the ligand (chromophore)-binding site [45], and, incombination with AFM, how rhodopsin is self-organizingin the DM [20,21]. As a structural understanding ofphototransduction emerges, we also gain new insights intothe mechanisms leading to various human retinopathies.It is predicted that the sustained progress on thestructural analysis of individual phototransduction com-ponents and their relevant complexes will continue.However, it is also evident that the combined use ofmany techniques will be a prerequisite for understandingthe structural basis of function. Ultimately, a crystal-clearpicture of phototransduction will require further dramaticprogress to assemble these proteins into larger macromol-ecular complexes. A new spectacular accomplishmentusing cryo-electron tomography [66], which provides a‘birds-eye view’ of cellular structure, offers promisingevidence that such possibilities are within our grasp.
Acknowledgements
We thank many of our colleagues who helped us with incorporating theirwork into this review. We also thank John Spudich for insightfulcomments, and Yunie Kim and Jaya Jagadeesh for assistance with articlepreparation. This work was supported in part by U.S. Public HealthService grants EY0861 and EY13286 and from the National Eye Institute,an unrestricted Grant from Research to Prevent Blindness to theDepartment of Ophthalmology at the University of Washington, and agrant from the E.K. Bishop Foundation.
References
1 Polans, A. et al. (1996) Turned on by Ca2þ! The physiology andpathology of Ca2þ-binding proteins in the retina. Trends Neurosci. 19,547–554
2 Palczewski, K. et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745
3 Gaudet, R. et al. (1996) Crystal structure at 2.4 A resolution of thecomplex of transducin bg and its regulator, phosducin. Cell 87,577–588
4 Loew, A. et al. (1998) Phosducin induces a structural change intransducin bg. Structure 6, 1007–1019
5 Lambright, D.G. et al. (1996) The 2.0 A crystal structure of aheterotrimeric G protein. Nature 379, 311–319
6 Lambright, D.G. et al. (1994) Structural determinants for activation ofthe a-subunit of a heterotrimeric G protein. Nature 369, 621–628
7 Noel, J.P. et al. (1993) The 2.2 A crystal structure of transducin-acomplexed with GTPgS. Nature 366, 654–663
8 Sondek, J. et al. (1994) GTPase mechanism of G proteins from the 1.7-Acrystal structure of transducin a-GDP–AlF4. Nature 372, 276–279
9 Lodowski, D.T. et al. (2003) Keeping G proteins at bay: a complexbetween G protein-coupled receptor kinase 2 and Gbg. Science 300,1256–1262
10 Kameni Tcheudji, J.F. et al. (2001) Molecular organization of bovinerod cGMP-phosphodiesterase 6. J. Mol. Biol. 310, 781–791
11 Kajimura, N. et al. (2002) Three-dimensional structure of non-activated cGMP phosphodiesterase 6 and comparison of its imagewith those of activated forms. J. Struct. Biol. 139, 27–38
12 Higgins, M.K. et al. (2002) Molecular architecture of a retinal cGMP-gated channel: the arrangement of the cytoplasmic domains. EMBO J.21, 2087–2094
13 Slep, K.C. et al. (2001) Structural determinants for regulation ofphosphodiesterase by a G protein at 2.0 A. Nature 409, 1071–1077
14 Ames, J.B. et al. (1999) Three-dimensional structure of guanylylcyclase activating protein-2, a calcium-sensitive modulator of photo-receptor guanylyl cyclases. J. Biol. Chem. 274, 19329–19337
15 Abdulaev, N.G. et al. (1998) Nucleoside diphosphate kinase frombovine retina: purification, subcellular localization, molecular cloning,and three-dimensional structure. Biochemistry 37, 13958–13967
16 McBee, J.K. et al. (2001) Confronting complexity: the interlink ofphototransduction and retinoid metabolism in the vertebrate retina.Prog. Retin. Eye Res. 20, 469–529
17 Loew, A. and Gonzalez-Fernandez, F. (2002) Crystal structure of thefunctional unit of interphotoreceptor retinoid binding protein.Structure 10, 43–49
18 Creemers, A.F. et al. (2002) 1H and 13C MAS NMR evidence forpronounced ligand-protein interactions involving the ionone ring ofthe retinylidene chromophore in rhodopsin. Proc. Natl. Acad. Sci.U. S. A. 99, 9101–9106
19 Yeagle, P.L. et al. (1997) The first and second cytoplasmic loops of theG-protein receptor, rhodopsin, independently form b-turns. Biochem-istry 36, 3864–3869
20 Fotiadis, D. et al. (2003) Atomic-force microscopy: rhodopsin dimers innative disc membranes. Nature 421, 127–128
21 Liang, Y. et al. (2003) Organization of the G protein-coupled receptorrhodopsin and opsin in native membranes. J. Biol. Chem. 278,21655–26162
22 Ridge, K.D. et al. (1996) Examining rhodopsin folding and assemblythrough expression of polypeptide fragments. J. Biol. Chem. 271,7860–7867
23 Kuksa, V. et al. (2002) Biochemical and physiological properties ofrhodopsin regenerated with 11-cis-6-ring- and 7-ring-retinals. J. Biol.Chem. 277, 42315–42324
24 Fan, G. et al. (2002) Rhodopsin with 11-cis-locked chromophore iscapable of forming an active state photoproduct. J. Biol. Chem. 277,40229–40234
25 Hubbell, W.L. et al. (2003) Rhodopsin structure, dynamics, andactivation: a perspective from crystallography, site-directed spinlabeling, sulfhydryl reactivity, and disulfide cross-linking. Adv.Protein Chem. 63, 243–290
26 Dunham, T.D. and Farrens, D.L. (1999) Conformational changes inrhodopsin. Movement of helix F detected by site-specific chemicallabeling and fluorescence spectroscopy. J. Biol. Chem. 274, 1683–1690
27 Krishna, A.G. et al. (2001) Evidence that helix 8 of rhodopsin acts as amembrane-dependent conformational switch. Biochemistry 41,8298–8309
28 Fritze, O. et al. (2003) Role of the conserved NPxxY(x)5,6F motif in therhodopsin ground state and during activation. Proc. Natl. Acad. Sci.U. S. A. 100, 2290–2295
29 Kuhne, W. (1878) Ueber den Sehpurpur, Untersuchungen aus demPhysiologische Institut der Universitaet Heidelberg, Heidelberg,pp. 15
30 Hubbard, R. and Wald, E. (1999) George Wald memorial talk. NovartisFound. Symp. 224, 5–20
31 Hargrave, P.A. et al. (1983) The structure of bovine rhodopsin.Biophys. Struct. Mech. 9, 235–244
32 Ovchinnikov, Yu.A. et al. (1983) Bioorg. Khim. 9, 1331–1340
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003486
http://tibs.trends.com

33 Argos, P. et al. (1982) Structural prediction of membrane-boundproteins. Eur. J. Biochem. 128, 565–575
34 Schertler, G.F.X. et al. (1993) Projection structure of rhodopsin. Nature362, 770–772
35 Baldwin, J.M. et al. (1997) An a-carbon template for the transmem-brane helices in the rhodopsin family of G-protein-coupled receptors.J. Mol. Biol. 272, 144–164
36 Pogozheva, I.D. et al. (1997) The transmembrane 7-a-bundle ofrhodopsin: distance geometry calculations with hydrogen bondingconstraints. Biophys. J. 72, 1963–1985
37 Unger, V.M. et al. (1997) Arrangement of rhodopsin transmembrane a-helices. Nature 389, 203–206
38 Shichida, Y. and Imai, H. (1998) Visual pigment: G-protein-coupledreceptor for light signals. Cell. Mol. Life Sci. 54, 1299–1315
39 Karnik, S.S. and Khorana, H.G. (1990) Assembly of functionalrhodopsin requires a disulfide bond between cysteine residues 110and 187. J. Biol. Chem. 265, 17520–17524
40 Teller, D.C. et al. (2001) Advances in determination of a high-resolutionthree-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry 40, 7761–7772
41 Okada, T. et al. (2002) Functional role of internal water molecules inrhodopsin revealed by X-ray crystallography. Proc. Natl. Acad. Sci.U. S. A. 99, 5982–5987
42 Okada, T. et al. (2001) Activation of rhodopsin: new insights fromstructural and biochemical studies. Trends Biochem. Sci. 26, 318–324
43 Meng, E.C. and Bourne, H.R. (2001) Receptor activation: what does therhodopsin structure tell us? Trends Pharmacol. Sci. 22, 587–593
44 Filipek, S. et al. (2003) G-protein-coupled receptor rhodopsin: aprospectus. Annu. Rev. Physiol. 65, 851–879
45 Mirzadegan, T. et al. (2003) Sequence analyses of G-protein-coupledreceptors: similarities to rhodopsin. Biochemistry 42, 2759–2767
46 Rao, S.T. and Rossmann, M.G. (1973) Comparison of super-secondarystructures in proteins. J. Mol. Biol. 76, 241–256
47 Jawad, Z. and Paoli, M. (2002) Novel sequences propel familiar folds.Structure 10, 447–454
48 Beisel, H.G. et al. (1999) Tachylectin-2: crystal structure of a specificGlcNAc/GalNAc-binding lectin involved in the innate immunity hostdefense of the Japanese horseshoe crab Tachypleus tridentatus. EMBOJ. 18, 2313–2322
49 Smith, T.F. et al. (1999) The WD repeat: a common architecture fordiverse functions. Trends Biochem. Sci. 24, 181–185
50 Granzin, J. et al. (1998) X-ray crystal structure of arrestin from bovinerod outer segments. Nature 391, 918–921
51 Hirsch, J.A. et al. (1999) The 2.8 A crystal structure of visual arrestin:a model for arrestin’s regulation. Cell 97, 257–269
52 Kawamura, S. (1993) Rhodopsin phosphorylation as a mechanism ofcyclic GMP phosphodiesterase regulation by S-modulin. Nature 362,855–857
53 Flaherty, K.M. et al. (1993) Three-dimensional structure of recoverin, acalcium sensor in vision. Cell 75, 709–716
54 Abdulaev, N.G. and Ridge, K.D. (1999) Light-induced exposure of thecytoplasmic end of transmembrane helix seven in rhodopsin. Proc.Natl. Acad. Sci. U. S. A. 95, 12854–12859
55 Hamm, H.E. et al. (1988) Site of G protein binding to rhodopsinmapped with synthetic peptides from the a subunit. Science 241,832–835
56 Kisselev, O.G. et al. (1994) A farnesylated domain in the G protein g
subunit is a specific determinant of receptor coupling. J. Biol. Chem.269, 21399–21402
57 Dratz, E.A. et al. (1993) NMR structure of a receptor-bound G-proteinpeptide. Nature 363, 276–281.Erratum in (1997) Nature 390, 424
58 Kisselev, O.G. et al. (1998) Light-activated rhodopsin inducesstructural binding motif in G protein a subunit. Proc. Natl. Acad.Sci. U. S. A. 95, 4270–4275
59 Koenig, B.W. et al. (2002) Structure and orientation of a G proteinfragment in the receptor bound state from residual dipolar couplings.J. Mol. Biol. 322, 441–461
60 Brabazon, D.M. et al. (2003) Evidence for structural changes incarboxyl-terminal peptides of transducin a-subunit upon binding asoluble mimic of light-activated rhodopsin. Biochemistry 42, 302–311
61 Kisselev, O.G. and Downs, M.A. (2003) Rhodopsin controls aconformational switch on the transducin g subunit. Structure 11,367–373
62 Iiri, T. et al. (1998) G-protein diseases furnish a model for the turn-onswitch. Nature 394, 35–38
63 Cherfils, J. and Chabre, M. (2003) Activation of G-protein Ga subunitsby receptors through Ga–Gb and Ga–Gg interactions. TrendsBiochem. Sci. 28, 13–17
64 Tanaka, T. et al. (1995) Sequestration of the membrane-targetingmyristoyl group of recoverin in the calcium-free state. Nature 376,444–447
65 Ames, J.B. et al. (1997) Molecular mechanics of calcium-myristoylswitches. Nature 389, 198–202
66 Medalia, O. et al. (2002) Macromolecular architecture in eukaryoticcells visualized by cryoelectron tomography. Science 298, 1209–1213
67 Sondek, J. et al. (1996) Crystal structure of a G-protein bg dimer at2.1 A resolution. Nature 379, 369–374
68 Martinez, S.E. et al. (2002) The two GAF domains in phosphodiester-ase 2A have distinct roles in dimerization and in cGMP binding. Proc.Natl. Acad. Sci. U. S. A. 99, 13260–13265
69 Xu, R.X. et al. (2000) Atomic structure of PDE4: insights intophosphodiesterase mechanism and specificity. Science 288, 1822–1825
70 Liebman, P.A. et al. (1982) Lateral diffusion of visual pigment in roddisk membranes. Methods Enzymol. 81, 660–668
71 Cone, R.A. (1972) Rotational diffusion of rhodopsin in the visualreceptor membrane. Nat. New Biol. 236, 39–43
Do you want to reproduce material from a Trends journal?This publication and the individual contributions within it are protected by the copyright of Elsevier. Except as outlined in the termsand conditions (see p. ii), no part of any Trends journal can be reproduced, either in print or electronic form, without writtenpermission from Elsevier. Please address any permission requests to:
Rights and Permissions,Elsevier Ltd,
PO Box 800, Oxford, UK OX5 1DX.
Review TRENDS in Biochemical Sciences Vol.28 No.9 September 2003 487
http://tibs.trends.com





























