Embed Size (px)
Citation preview

Matters Arising
IGFBP7 Is Not Required forB-RAF-Induced Melanocyte SenescenceLyndee L. Scurr,1,* Gulietta M. Pupo,1 Therese M. Becker,1 Ken Lai,2,4 David Schrama,3 Sebastian Haferkamp,1
Mal Irvine,1 Richard A. Scolyer,2,4,5 Graham J. Mann,1 Jurgen C. Becker,3 Richard F. Kefford,1,2 and Helen Rizos1,*1Westmead Institute for Cancer Research, University of Sydney at Westmead Millennium Institute, Westmead Hospital, Westmead, New
South Wales 2145, Australia2Melanoma Institute Australia (incorporating the Sydney Melanoma Unit), Sydney, New South Wales 2050, Australia3Department of Dermatology, University Hospital Wurzburg, Wurzburg D-97080, Germany4Department of Anatomical Pathology, Royal Prince Alfred Hospital, Camperdown, Sydney, New South Wales 2050, Australia5Discipline of Pathology, Faculty of Medicine, University of Sydney, Sydney, New South Wales 2006, Australia
*Correspondence: [email protected] (L.L.S.), [email protected] (H.R.)DOI 10.1016/j.cell.2010.04.021
SUMMARY
Induction of senescence permanently restrictscellular proliferation after oncogenic stimulationthereby acting as a potent barrier to tumor develop-ment. The relevant effector proteins may therefore befundamental to cancer development. A recent studyidentified IGFBP7 as a secreted factor mediatingmelanocyte senescence induced by oncogenicB-RAF, which is found commonly in cutaneousnevi. In contrast to the previous report, we demon-strate that B-RAF signaling does not induce IGFBP7expression, nor the expression of the IGFBP7targets, BNIP3L, SMARCB1, or PEA15, in humanmelanocytes or fibroblasts. We also found no corre-lation between B-RAF mutational status and IGFBP7protein expression levels in 22 melanoma cell lines,90 melanomas, and 46 benign nevi. Furthermore,using a lentiviral silencing strategy we show thatB-RAF induces senescence in melanocytes andfibroblasts, irrespective of the presence of IGFBP7.Therefore, we conclude that the secreted proteinIGFBP7 is dispensable for B-RAFV600E-inducedsenescence in human melanocytes.
INTRODUCTION
Cutaneous melanoma is an aggressive form of cancer that is
highly resistant to conventional therapies with fewer than 10%
of patients with visceral metastases surviving 2 years (reviewed
in Thompson et al., 2005; Herlyn, 2007). It may arise de novo
from melanocytes or their precursors in the skin (Kelly et al.,
1997) or from common benign clonal expansions of cutaneous
melanocytes, known as nevi (Chin et al., 2006; reviewed in
Bennett, 2008). Although the presence of high numbers of nevi
is strongly associated with melanoma risk, the majority of benign
nevi lack proliferative activity and never progress into malig-
nancy despite harboring oncogenic alterations. Indeed, consti-
tutively activating mutations affecting the N-RAS or B-RAF
kinase components of the mitogen-activated protein kinase
(MAPK) pathway are found in over 80% of benign nevi (Bauer
et al., 2007; Papp et al., 1999; Pollock et al., 2003). The prevailing
view is that nevi have initiated a permanent form of proliferative
arrest known as oncogene-induced senescence (reviewed in
Prieur and Peeper, 2008). Certainly, benign nevi display several
markers of senescence growth arrest, including increased
senescence-associated b-galactosidase (SA-b-Gal) activity
and p16INK4a expression (Gray-Schopfer et al., 2006;
Michaloglou et al., 2005). Although the presence of senescent
cells in human melanocytic nevi remains controversial (Cotter
et al., 2007), accumulating evidence suggests that senescence
occurs in vivo and acts as an effective barrier to tumor formation
(reviewed in Prieur and Peeper, 2008). In this regard, identifica-
tion of the molecules that enforce oncogene-induced arrest in
human melanocytes is of prime importance as their loss may
be a critical step in the escape of malignant clones and the
consequent development of melanoma. These molecules are
also potential therapeutic targets, as reinstating their functions
may stimulate entry into senescence.
In an attempt to identify these molecules, a genome-wide
loss-of-function screen was used to define essential regulators
of B-RAFV600E-induced senescence (Wajapeyee et al., 2008).
Notably, this oncogenic form of B-RAF occurs in 40%–50% of
melanomas (reviewed in Platz et al., 2008) and its expression is
sufficient to induce senescence and apoptosis in human diploid
fibroblasts and melanocytes (Gray-Schopfer et al., 2006;
Michaloglou et al., 2005; Wajapeyee et al., 2008). Seventeen
genes were identified and the suppression of each permitted
diploid human fibroblasts to proliferate and survive in the pres-
ence of oncogenic B-RAFV600E. Among these were p53 and
the insulin-like growth factor (IGF) binding protein 7 (IGFBP7).
IGFBP7 is a secreted protein that reduces the bioavailability of
insulin/IGF in peripheral blood and has been implicated in tumor
suppression of breast, colon, lung, and pancreatic cancers
(Burger et al., 1998; Chen et al., 2007; Landberg et al., 2001;
Ruan et al., 2007; Swisshelm et al., 1995; Ye et al., 2007).
B-RAFV600E was also shown to upregulate IGFBP7 expression
via the B-RAF-MEK-ERK signaling cascade. IGFBP7 inhibited
Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc. 717
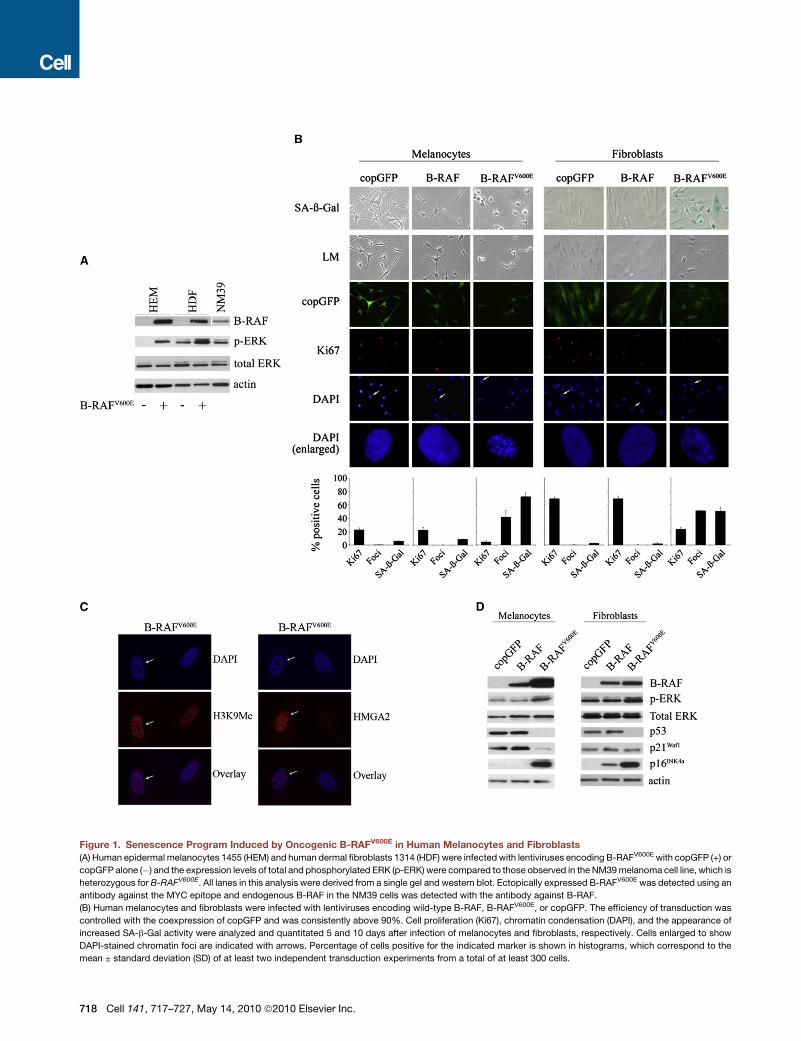
Figure 1. Senescence Program Induced by Oncogenic B-RAFV600E in Human Melanocytes and Fibroblasts
(A) Human epidermal melanocytes 1455 (HEM) and human dermal fibroblasts 1314 (HDF) were infected with lentiviruses encoding B-RAFV600E with copGFP (+) or
copGFP alone (�) and the expression levels of total and phosphorylated ERK (p-ERK) were compared to those observed in the NM39 melanoma cell line, which is
heterozygous for B-RAFV600E. All lanes in this analysis were derived from a single gel and western blot. Ectopically expressed B-RAFV600E was detected using an
antibody against the MYC epitope and endogenous B-RAF in the NM39 cells was detected with the antibody against B-RAF.
(B) Human melanocytes and fibroblasts were infected with lentiviruses encoding wild-type B-RAF, B-RAFV600E, or copGFP. The efficiency of transduction was
controlled with the coexpression of copGFP and was consistently above 90%. Cell proliferation (Ki67), chromatin condensation (DAPI), and the appearance of
increased SA-b-Gal activity were analyzed and quantitated 5 and 10 days after infection of melanocytes and fibroblasts, respectively. Cells enlarged to show
DAPI-stained chromatin foci are indicated with arrows. Percentage of cells positive for the indicated marker is shown in histograms, which correspond to the
mean ± standard deviation (SD) of at least two independent transduction experiments from a total of at least 300 cells.
718 Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc.

phosphorylation of MEK by B-RAF, initiating a negative-feed-
back loop. Consistent with the hypothesis that IGFBP7
suppresses early B-RAF-driven tumor formations in vivo,
expression of IGFBP7 was highly expressed in B-RAFV600E-posi-
tive nevi and in primary melanomas and melanoma cell lines
lacking activated B-RAF but was lost in B-RAFV600E-positive
melanomas and melanoma cell lines. Furthermore, purified re-
combinant IGFBP7 potently suppressed the growth of
B-RAFV600E-positive human melanomas but had no effect on
tumors with wild-type B-RAF (Wajapeyee et al., 2008). Taken
together these observations suggest that IGFBP7 is essential
in constraining B-RAFV600E-stimulated cell proliferation.
In this work we readdressed the role of IGFBP7 in B-RAFV600E-
induced senescence in diploid human melanocytes. This was
particularly important as two recent reports did not detect
a correlation between B-RAF mutation status and IGFBP7
expression in a panel of primary melanomas and metastases
(Schrama et al., 2009; Wajapeyee et al., 2009). We also found
no correlation between B-RAF mutational status and IGFBP7
expression levels in a series of melanoma cell lines, melanomas,
and benign nevi. Furthermore, in contrast to the previous report
(Wajapeyee et al., 2008), we demonstrate that B-RAF signaling
does not induce IGFBP7 expression in either human melano-
cytes or fibroblasts, and using a lentiviral silencing strategy we
show that B-RAF potently induces senescence in the presence
and absence of IGFBP7. Therefore, we conclude that the
secreted protein IGFBP7 is dispensable for B-RAFV600E-induced
senescence in human melanocytes.
RESULTS
Expression of Oncogenic B-RAF Induces CellularSenescence that Is Associated with IGFBP7 RepressionTo evaluate the response of human cells to oncogenic B-RAF,
the melanoma-associated B-RAFV600E mutant was stably trans-
duced into primary diploid HEM1455 melanocytes and HDF1314
epidermal fibroblasts using lentiviral vectors coexpressing
Copepod GFP (copGFP). Viral titers were selected to provide
an efficiency of infection above 90% and activation of the ERK
pathway that was comparable to human melanoma cells ex-
pressing endogenous B-RAFV600E (Figure 1A).
As expected from previous studies, expression of oncogenic
B-RAF efficiently blocked cellular proliferation and induced SA-
b-Gal activity as early as 5 days after infection (Figure 1B). The
majority of B-RAFV600E-transduced melanocytes and fibroblasts
displayed several markers of oncogene-driven senescence,
increased SA-b-Gal expression, senescence-associated hetero-
chromatin foci (SAHF), and significantly decreased Ki67 expres-
sion (Figure 1B). Each focus in a senescent cell results from
condensation of an individual chromosome and is enriched for
common markers of heterochromatin, including histone H3
methylated at lysine 9 (H3K9Me) and the nonhistone chromatin
(C) Representative examples of chromatin condensation (DAPI) in human epider
HMGA2. Cells showing DAPI-stained chromatin foci are indicated with arrows.
(D) Expression of the indicated proteins was determined by western blot analys
copGFP, wild-type B-RAF, or B-RAFV600E.
protein HMGA2 (Figure 1C) (reviewed in Adams, 2007). The
accumulation of oncogenic B-RAFV600E in both melanocytes
and fibroblasts was associated with increased levels of phos-
phorylated ERK and induction of the CDK inhibitor p16INK4a. In
addition, the levels of p53 were significantly repressed and
expression of the p53-transcription target p21Waf1 remained
low or was reduced by the accumulation of B-RAFV600E
(Figure 1D).
It has been suggested that senescence triggered by onco-
genic B-RAF is associated with the transcriptional upregulation
of IGFBP7 (Wajapeyee et al., 2008). We therefore reanalyzed
the expression of IGFBP7 and conclusively show that oncogenic
B-RAF represses, rather than induces, IGFBP7 expression in
both human melanocytes and fibroblasts (Figure 2A and
Figure S1 available online). Indeed, B-RAFV600E-transduced
melanocytes and fibroblasts displayed diminished levels of
cellular and secreted IGFBP7 compared to the control
copGFP-transduced cells (Figure 2A and Figure S1). The deple-
tion of IGFBP7 was routinely observed as early as 2 days post-B-
RAFV600E transduction and was maintained for the duration of
the transduction experiments (5 days and 15 days post-trans-
duction of melanocytes and fibroblasts, respectively). The slight
increase in secreted IGFBP7 in melanocytes, 2 days post-trans-
duction with oncogenic B-RAFV600E, was not consistent over
replicate experiments and did not correspond with an increase
in cellular IGFBP7 at this time point (Figure 2A). The depletion
of IGFBP7 occurred earlier than the onset of BRAFV600E-induced
senescence (see Figure 1B). Furthermore, the specific silencing
of oncogenic B-RAF or the addition of a MEK inhibitor led to the
accumulation of IGFBP7 in melanoma cell lines expressing the
B-RAFV600E variant (Figure 2B).
B-RAF Mutation Status Does Not Correlate with IGFBP7ExpressionTo further scrutinize the correlation of IGFBP7 expression with
B-RAF mutation status, we analyzed a series of 22 melanoma
cell lines for IGFBP7 expression. In this series, we did not find
a correlation between IGFBP7 expression and B-RAF mutation
status, as IGFBP7 was detected in 10/14 B-RAF mutant and
3/8 B-RAF wild-type tumor lines (Figure 2C). Further, we found
no correlation between B-RAF mutation status and IGFBP7 tran-
script levels in gene expression microarray analyses of 99 meta-
static melanoma tumors (Figure 3A, Table 1). This was confirmed
at the protein level by tissue microarray analyses of 90 of these
tumors (Figure 3B, Table 2). Consistent with these data, we did
not observe increased expression of several IGFBP7-regulated
gene products, including SMARCB1, BNIP3L, and PEA15
(Wajapeyee et al., 2008), in B-RAFV600E-transduced fibroblasts
or melanocytes (Figure 3C), nor did the expression of these
IGFBP7-regulated transcripts correlate with B-RAF mutation
status in our microarray analyses of 99 metastatic melanomas
(Figure 3C, Table 1). Finally, IGFBP7 protein expression did not
mal melanocytes expressing B-RAFV600E (day 5) and staining for H3K9Me and
is after infection of melanocytes and fibroblasts with lentiviruses expressing
Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc. 719

Figure 2. Oncogenic B-RAFV600E Expres-
sion Does Not Correlate with IGFBP7 Accu-
mulation
(A) Immunoblot analysis monitoring the accumula-
tion of IGFBP7 in human melanocytes and
fibroblasts infected with copGFP control lentivirus
(�) or lentivirus expressing B-RAFV600E (+). Protein
expression was analyzed at 2, 3, 5, 10, and 15
days post-transduction (PT), as shown (see also
Figure S1).
(B) Immunoblot analysis monitoring cellular
IGFBP7 expression in melanoma cell lines 5 days
post-transduction with control (�) or BRAFV600E-
specific (+) shRNA molecules, as indicated. This
figure is compiled from duplicate immunoblots.
Cells were also treated with the MEK inhibitor
PD98059 for 72 hr, as shown. The WMM1175
melanoma cells carry wild-type B-RAF, and the
ME1042 and NM39 melanoma cells are heterozy-
gous for B-RAFV600E.
(C) Immunoblot analysis monitoring cellular
IGFBP7 levels in a panel of human melanoma cell
lines, primary human melanocytes (HEM1455),
three fibroblasts (HDF1314, BJ, and WS1), and
three other cancer cell lines. The mutation status
of B-RAF and N-RAS is indicated.
correlate with B-RAF mutation status in a series of human benign
nevi (Figure 3D, Table 2). Collectively these data indicate that
B-RAFV600E does not upregulate IGFBP7 expression in fibro-
blasts or melanocytes nor is the loss of IGFBP7 required for
the development of B-RAFV600E-positive melanomas.
IGFBP7 Is Not Required for B-RAFV600E-InducedSenescenceTo specifically examine the contribution of IGFBP7 to B-RAF-
mediated senescence, we applied lentiviral shRNA vectors that
specifically suppress the expression of IGFBP7. To minimize
confounding effects of shRNA off-target silencing two indepen-
dent silencing molecules targeting IGFBP7 were generated
(Wajapeyee et al., 2008). These silencing constructs were highly
effective in silencing the expression of IGFBP7. The role of
720 Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc.
IGFBP7 in oncogene-induced senes-
cence was evaluated in human melano-
cytes (HEM1455, nHEM) and fibroblasts
(HDF1314, WS1, and BJ), and the results
for HDF1314 and HEM1455 are depicted
in detail. Cells were transduced with an
IGFBP7 shRNA molecule coexpressing
copGFP and 3 days post-infection were
retransduced with lentiviral vectors
expressing B-RAFV600E with copGFP
or copGFP alone. All experiments
included a negative control shRNA mole-
cule without homology to any human
gene. Just as in our previous single trans-
duction experiments, transduction
efficiencies reached at least 90% in all
cells (Figure S2A) and there was no
need for antibiotic selection. This strategy proved effective as
the depletion of pRb along with p53 using highly effective
short-hairpin RNAs (Haferkamp et al., 2009a) overcame
B-RAFV600E-induced melanocyte cell-cycle arrest (Figure S2B).
As expected, all melanocytes and fibroblasts transduced with
the combination of control shRNA and B-RAFV600E expressed
markers associated with the onset of senescence. These
included significantly reduced levels of the proliferation marker
Ki67, accumulation of SAHF, and increased SA-b-Gal activity
(Figures 4A and 4B). Importantly, inhibition of IGFBP7 expression
did not alter the growth arrest induced by oncogenic
B-RAFV600E, and cells were arrested after infection, stained posi-
tive for SA-b-Gal, and accumulated DAPI-stained foci regardless
of the expression level of IGFBP7 (Figures 4A and 4B). In addi-
tion, the induction of p16INK4a associated with oncogenic
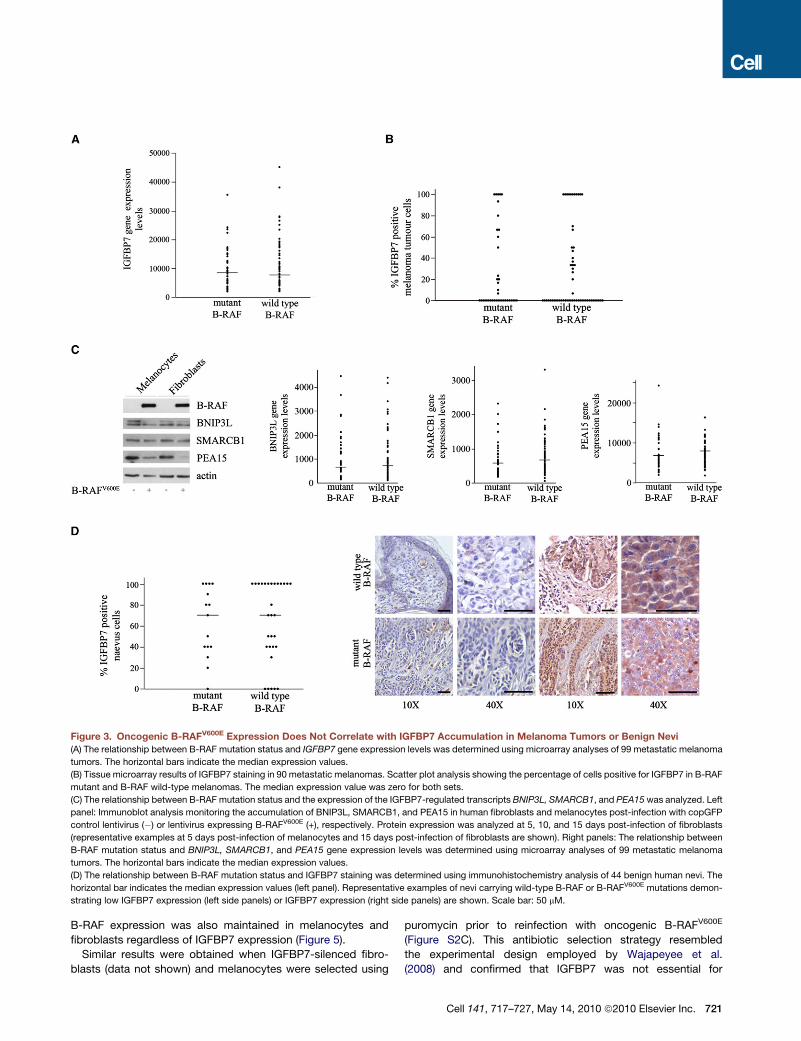
Figure 3. Oncogenic B-RAFV600E Expression Does Not Correlate with IGFBP7 Accumulation in Melanoma Tumors or Benign Nevi
(A) The relationship between B-RAF mutation status and IGFBP7 gene expression levels was determined using microarray analyses of 99 metastatic melanoma
tumors. The horizontal bars indicate the median expression values.
(B) Tissue microarray results of IGFBP7 staining in 90 metastatic melanomas. Scatter plot analysis showing the percentage of cells positive for IGFBP7 in B-RAF
mutant and B-RAF wild-type melanomas. The median expression value was zero for both sets.
(C) The relationship between B-RAF mutation status and the expression of the IGFBP7-regulated transcripts BNIP3L, SMARCB1, and PEA15 was analyzed. Left
panel: Immunoblot analysis monitoring the accumulation of BNIP3L, SMARCB1, and PEA15 in human fibroblasts and melanocytes post-infection with copGFP
control lentivirus (�) or lentivirus expressing B-RAFV600E (+), respectively. Protein expression was analyzed at 5, 10, and 15 days post-infection of fibroblasts
(representative examples at 5 days post-infection of melanocytes and 15 days post-infection of fibroblasts are shown). Right panels: The relationship between
B-RAF mutation status and BNIP3L, SMARCB1, and PEA15 gene expression levels was determined using microarray analyses of 99 metastatic melanoma
tumors. The horizontal bars indicate the median expression values.
(D) The relationship between B-RAF mutation status and IGFBP7 staining was determined using immunohistochemistry analysis of 44 benign human nevi. The
horizontal bar indicates the median expression values (left panel). Representative examples of nevi carrying wild-type B-RAF or B-RAFV600E mutations demon-
strating low IGFBP7 expression (left side panels) or IGFBP7 expression (right side panels) are shown. Scale bar: 50 mM.
B-RAF expression was also maintained in melanocytes and
fibroblasts regardless of IGFBP7 expression (Figure 5).
Similar results were obtained when IGFBP7-silenced fibro-
blasts (data not shown) and melanocytes were selected using
puromycin prior to reinfection with oncogenic B-RAFV600E
(Figure S2C). This antibiotic selection strategy resembled
the experimental design employed by Wajapeyee et al.
(2008) and confirmed that IGFBP7 was not essential for
Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc. 721

Table 1. Median and Interquartile Ranges of Transcript Expression by B-RAF Status in Metastatic Melanomas Together with
Associated p Values (Mann-Whitney Test)
B-RAF Status
Wild-Type Mutant
Transcripts Median (LQ, UQ) Median (LQ, UQ) p Value
BNIP3L 818 402, 1556 768 262, 1447 0.591
IGFBP7 7461 5057, 14312 8290 4978, 12647 0.886
PEA15 7423 5369, 9105 6125 4057, 9973 0.129
SMARCB1 1070 667, 2032 772 562, 1528 0.109
TP53 301 181, 586 219 135, 520 0.124
UQ, upper quartile; LQ, lower quartile.
B-RAFV600E-induced senescence as its depletion did not prevent
cell-cycle arrest, formation of DAPI-stained foci, or the induction
of SA-b-Gal activity (Figure S2C).
DISCUSSION
The detection of senescent cells in vivo suggests that senes-
cence is a genuine, physiological tumor suppressor mechanism,
and this notion has intensified the search for the molecules that
initiate and maintain this proliferative arrest.
In a landmark study, a genome-wide shRNA screen identified
17 genes that were essential for B-RAFV600E-induced senes-
cence in human fibroblasts and melanocytes. Two of these
essential candidates were p53 and the secreted protein IGFBP7
(Wajapeyee et al., 2008).
We and others have shown that p53 levels are not induced by
oncogenic B-RAF in human melanocytes (Gray-Schopfer et al.,
2006; Zhuang et al., 2008) or in senescent normal melanocytes
(Sviderskaya et al., 2003). Furthermore, several reports have
confirmed that inactivation of p53 alone does not prevent B-
RAF-induced senescence in melanocytes (Denoyelle et al.,
2006; Zhuang et al., 2008). In keeping with these findings,
immortal melanocytes accumulating oncogenic B-RAF and
silenced for p53 expression did arise in one investigation.
However, these immortal cells had acquired a substantial dele-
tion in chromosome 13, which encodes the pRb1 tumor
suppressor gene (Yu et al., 2009). Indeed, loss of both pRb
and p53 is sufficient to overcome both N-RASQ61K- (Haferkamp
et al., 2009a) and B-RAFV600E-induced (Figure S2B) senescence
in melanocytes. Collectively these data strongly indicate that p53
is not required for B-RAF-induced senescence in melanocytes
in vitro and in vivo.
Table 2. Median and Interquartile Ranges of IGFBP7 Protein Expres
Together with Associated p Values (Mann-Whitney Test)
B-RAF status
Wild type
IGFBP7 Protein Median (LQ, UQ)
Melanoma 0 0, 38
Nevi 70 40, 100
UQ, upper quartile; LQ, lower quartile.
722 Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc.
Because IGFBP7 might also have clinical potential, we used
four independent experimental strategies to reanalyze the
reported contribution of IGFBP7 to B-RAF-induced senescence.
First, we examined the regulation of IGFBP7 by oncogenic
B-RAF in several melanocyte and fibroblast cells. We did not
detect any significant upregulation of IGFBP7 in response
to oncogenic B-RAF. In fact we saw significant reduction in
IGFBP7 in all primary cells tested, and a slight IGFBP7 upregula-
tion was only seen in response to silencing of B-RAFV600E
or inhibiting the MAPK pathway using a MEK inhibitor in mela-
noma cell lines harboring oncogenic B-RAFV600E. IGFBP7 is tran-
scriptionally regulated by p53 (Suzuki et al., 2009), and the
depletion of IGFBP7 in response to oncogenic B-RAF may be
due to the transcriptional repression of p53. Second, two inde-
pendent and highly effective IGFBP7 shRNA molecules were
employed to examine the role of IGFBP7 in B-RAF-induced
senescence. We found that B-RAF effectively induced senes-
cence regardless of IGFBP7 expression in the two melanocyte
and three fibroblast strains tested. Third, there was no correla-
tion between IGFBP7 expression and B-RAF mutation status in
a panel of 22 melanoma cell lines. Fourth, immunohistochemical
analysis of human nevi and melanoma samples with known
B-RAF mutation status confirmed that IGFBP7 expression was
not related to B-RAF activation in vivo. The interpretation of the
melanoma tumor data is not straightforward, however, as it
is not possible to rule out the influence of environmental
and genetic risk factors present in the Australian melanoma
patients. The Australian population is generally exposed to
high levels of ultraviolet radiation and develops melanocytic
nevi early in life and in large numbers (Harrison et al., 2008).
There is evidence that interactions among phenotypic, genetic,
and environmental risk factors may influence the mechanisms
sion by B-RAF Status in Metastatic Melanomas or Benign Nevi
Mutant
Median (LQ, UQ) P Value
0 0, 50 0.894
70 40, 100 0.655
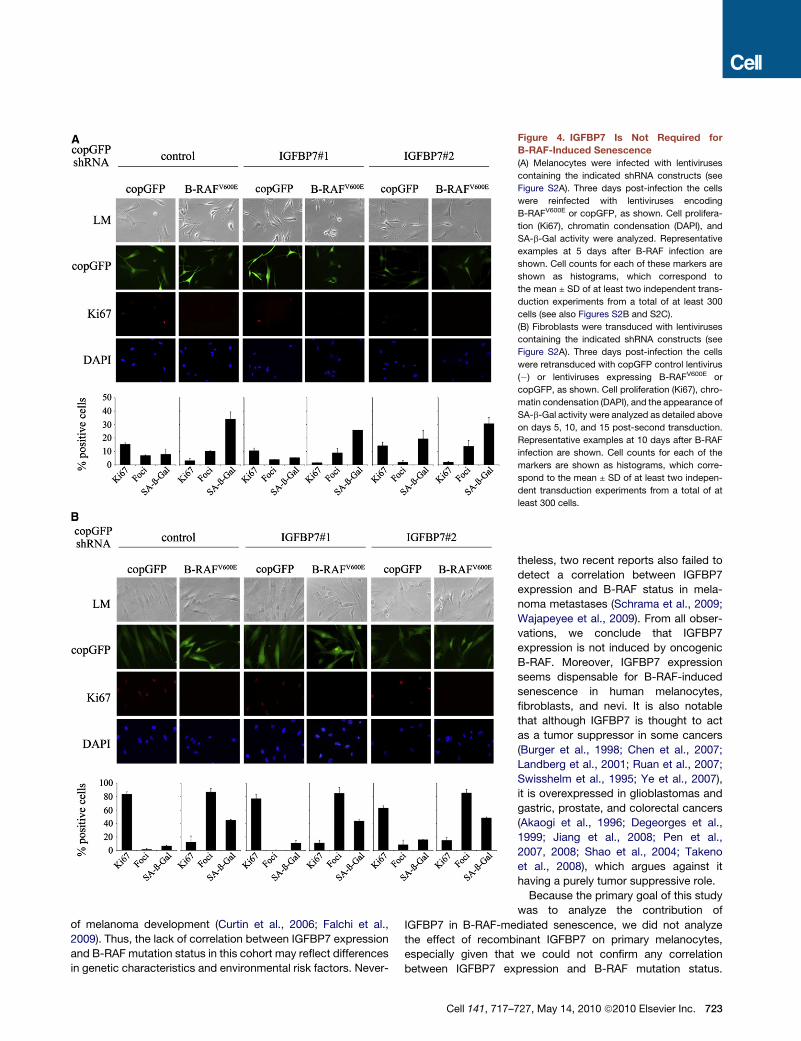
Figure 4. IGFBP7 Is Not Required for
B-RAF-Induced Senescence
(A) Melanocytes were infected with lentiviruses
containing the indicated shRNA constructs (see
Figure S2A). Three days post-infection the cells
were reinfected with lentiviruses encoding
B-RAFV600E or copGFP, as shown. Cell prolifera-
tion (Ki67), chromatin condensation (DAPI), and
SA-b-Gal activity were analyzed. Representative
examples at 5 days after B-RAF infection are
shown. Cell counts for each of these markers are
shown as histograms, which correspond to
the mean ± SD of at least two independent trans-
duction experiments from a total of at least 300
cells (see also Figures S2B and S2C).
(B) Fibroblasts were transduced with lentiviruses
containing the indicated shRNA constructs (see
Figure S2A). Three days post-infection the cells
were retransduced with copGFP control lentivirus
(�) or lentiviruses expressing B-RAFV600E or
copGFP, as shown. Cell proliferation (Ki67), chro-
matin condensation (DAPI), and the appearance of
SA-b-Gal activity were analyzed as detailed above
on days 5, 10, and 15 post-second transduction.
Representative examples at 10 days after B-RAF
infection are shown. Cell counts for each of the
markers are shown as histograms, which corre-
spond to the mean ± SD of at least two indepen-
dent transduction experiments from a total of at
least 300 cells.
of melanoma development (Curtin et al., 2006; Falchi et al.,
2009). Thus, the lack of correlation between IGFBP7 expression
and B-RAF mutation status in this cohort may reflect differences
in genetic characteristics and environmental risk factors. Never-
Cell 141, 717–
theless, two recent reports also failed to
detect a correlation between IGFBP7
expression and B-RAF status in mela-
noma metastases (Schrama et al., 2009;
Wajapeyee et al., 2009). From all obser-
vations, we conclude that IGFBP7
expression is not induced by oncogenic
B-RAF. Moreover, IGFBP7 expression
seems dispensable for B-RAF-induced
senescence in human melanocytes,
fibroblasts, and nevi. It is also notable
that although IGFBP7 is thought to act
as a tumor suppressor in some cancers
(Burger et al., 1998; Chen et al., 2007;
Landberg et al., 2001; Ruan et al., 2007;
Swisshelm et al., 1995; Ye et al., 2007),
it is overexpressed in glioblastomas and
gastric, prostate, and colorectal cancers
(Akaogi et al., 1996; Degeorges et al.,
1999; Jiang et al., 2008; Pen et al.,
2007, 2008; Shao et al., 2004; Takeno
et al., 2008), which argues against it
having a purely tumor suppressive role.
Because the primary goal of this study
was to analyze the contribution of
IGFBP7 in B-RAF-mediated senescence, we did not analyze
the effect of recombinant IGFBP7 on primary melanocytes,
especially given that we could not confirm any correlation
between IGFBP7 expression and B-RAF mutation status.
727, May 14, 2010 ª2010 Elsevier Inc. 723

Figure 5. Oncogenic B-RAF Signaling Induces
ERK Activation and p16INK4a Expression and
Downregulates p53 in the Presence and Absence
of IGFBP7 Expression
Expression of the indicated proteins was determined by
western blot analysis after infection of human epidermal
melanocytes and fibroblasts with the indicated shRNA
constructs and either lentiviruses encoding B-RAFV600E
or the copGFP (�) control. Representative examples at
5 and 10 days after B-RAF infection of melanocytes and
fibroblasts, respectively, are shown.
However, although recombinant IGFBP7 induced apoptosis in
BRAF mutation-positive cancer cell lines (Wajapeyee et al.,
2008), it also promoted cell death in a subset of human cancer
cells with wild-type B-RAF that did not respond to the MEK inhib-
itor U0126 (Wajapeyee et al., 2009). These findings suggest that
the impact of recombinant IGFBP7 may be independent of the
MAPK pathway.
The most likely explanation for the disparate results reported
in the previous study by Wajapeyee et al. (2008) is that the tissue
culture conditions required for the loss-of-function screen re-
sulted in the selective outgrowth of antibiotic-resistant cell
clones that had escaped oncogene-induced senescence. Active
oncogenes promote DNA hyper-replication and damage
(Bartkova et al., 2006; Di Micco et al., 2006), and immortal
melanocytic clones harboring oncogenes have been shown to
accumulate additional secondary genetic changes (Sviderskaya
et al., 2003; Yu et al., 2009). Consequently, we have limited the
contribution of additional genetic alterations in our study by
(1) excluding long-term antibiotic selection steps and reducing
the duration of our experiments, (2) analyzing pooled cell
populations rather than cell clones, and (3) repeating our short-
term experiments in several different primary human
cell cultures. In support of this approach, p53-deficient melano-
cytes behaved as expected and responded to oncogenic B-RAF
by inducing p16INK4a (Zhuang et al., 2008), whereas the equiva-
lent antibiotic-selected cell clones generated by Wajapeyee
et al. (2008) showed no induction of p16INK4a. As there is no
evidence that p53 loss interrupts the regulation of p16INK4a
by the MAPK pathway, we are led to speculate that
the p16INK4a gene had been altered in those clones, especially
given that both wild-type and p53 null melanocytes should
respond to oncogenic N-RAS by induction of p16INK4a expres-
sion (Haferkamp et al., 2009a).
In conclusion, despite the use of several independent experi-
mental strategies, we were unable to find evidence to support
a critical role of IGFBP7 in mediating B-RAF-induced senes-
cence in melanocytes, in contrast to the recent report in this
journal. There is little doubt, however, that secreted factors
play a significant role in senescence. Key components of the
Wnt1, transforming growth factor b, plasmin, and interleukin
signaling cascades reinforce senescence and potentially sensi-
tize neighboring cells to proliferative arrest (reviewed in Acosta
724 Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc.
et al., 2008; Kuilman and Peeper, 2009). Paradoxically, some
of these secreted factors can also exert protumorigenic effects
depending on the cell type and tumor stage. This has important
implications when considering the therapeutic applicability of
agonists that promote senescence, and it is critical that the
contribution and in vivo interactions of these secreted effectors
are defined.
EXPERIMENTAL PROCEDURES
Specimen Collection
Melanoma patients with available fresh-frozen metastatic lymph node disease
were identified from the Sydney Melanoma Unit Biospecimen Bank/Depart-
ment of Anatomical Pathology, Royal Prince Alfred Hospital. Specimen
collection is from routine surgical resections. All tissue specimens are snap
frozen in liquid nitrogen and stored at �80�C until required for use. Melanoma
tumors were obtained between 1996–2006 under a sample procurement
protocol approved by the Sydney South West Area Health Service institutional
ethics review committee (RPAH Zone) Protocol No. X08-0155 and HREC
Ref. 08/RPAH/262—‘‘Histological and Immunohistological Analysis of Mela-
nocytic Tumours.’’ The histopathologic features of each sample were reviewed
by a pathologist (R.A.S.) to confirm diagnosis and tumor content.
Paraffin0embedded samples from 19 junctional, 10 compound, and 17
congenital nevi were obtained by surgical excision. All nevi have undergone
routine histology for diagnosis.
RNA Extraction and Microarray Gene Expression Analysis
Total RNA was extracted from 10–20 mg of tumor. Tissue samples were
homogenized using a high-speed agitation Polytron blender (Kinematica,
Luzern, Switzerland) in the presence of Trizol (Invitrogen Life Technologies,
Carlsbad, CA, USA). Following homogenization, chloroform was added and
the sample centrifuged. The upper phase was removed and mixed with 70%
ethanol. The RNA was then isolated and purified with an RNeasy purification
kit with DNase I digestion on the column (QIAGEN, Valencia, CA, USA). The
quality of the RNA preparations was assessed using a Agilent 2100 Bioana-
lyser (Agilent Technologies, Palo Alto, CA, USA). The extracted total RNA
was of high quality (RNA integrity number: 8–10). cRNA amplification and
labeling with biotin were performed using the Illumina TotalPrep RNA amplifi-
cation kit (Ambion, Austin, TX, USA) according to the manufacturer’s direc-
tions starting with 250 ng total RNA. Gene expression analysis was performed
using the Sentrix HumanRef-6 v.3.0 Expression BeadChip (Illumina,
San Diego, CA, USA) and BeadStation system from Illumina according to
manufacturer’s instructions.
DNA Extraction and Genotyping
DNA was extracted from a 10–20 mg tumor tissue sample using the QIAGEN
QIAamp DNA mini kit (QIAGEN). Briefly tissue was pulverized using liquid

nitrogen, then incubated with ALT buffer (QIAGEN) and proteinase K for 96 hr
at 56�C for complete digestion. The following B-RAF and N-RAS single-nucle-
otide polymorphisms (SNPs) were genotyped in tumors and cell lines as part of
the Sequenom OncoCarta Assay Panel v1.0: B-RAF SNPs: G464R, G464V/E,
G466R, F468C, G469S, G469E, G469A, G469V, G469R, D594V/G, F595L,
G596R, L597S, L597R, L597Q, L597V, T599I, V600E, V600K, V600R, V600L,
K601N, K601E. N-RAS SNPs: G12V/A/D, G12C/R/S, G13V/A/D, G13C/R/S,
A18T, Q61L/R/P, Q61H, Q61E/K.
DNA of nevi from three consecutive sections from the paraffin blocks was
isolated using a DNA isolation kit (QIAGEN) according to the manufacturer’s
recommendations. Subsequently, the B-RAFV600E mutation was detected by
real-time PCR as previously described (Benlloch et al., 2006).
Cell Culture
Human neonatal dermal fibroblast (HDF1314) and human neonatal epidermal
melanocytes (HEM1455) were obtained from Cell Applications (San Diego, CA,
USA). Melanocytes, including nHEM cells (Invitrogen), were grown in HAM’s
F10 media, supplemented with ITS premix (Becton Dickinson, Franklin Lakes,
NJ, USA), 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma-Aldrich, St.
Louis, MO, USA), IBMX (Becton Dickinson), cholera toxin (List Biological Labo-
ratories, Campbell, CA, USA), 20% fetal bovine serum, and glutamine (GIBCO
BRL, Carlsbad, CA, USA) (modified from Halaban et al., 1986). HEK293T, fibro-
blasts, and all melanoma cell lines were grown in Dulbecco’s modified Eagle’s
medium (GIBCO BRL) supplemented with 10% fetal bovine serum, HEPES,
and glutamine. All cells were cultured in a 37�C incubator with 5% CO2. Cells
were treated with media containing 40 mM PD98059 for 72 hr and the media
with MEK inhibitor were replenished every 24 hr.
Lentiviral Transductions
Lentiviruses were produced in HEK293T cells as described previously
(Haferkamp et al., 2009b). Cells were infected using a multiplicity of infection
(moi) between 5 and 10 to provide an efficiency of infection above 90%.
Constructs
The wild-type and mutant B-RAF cDNAs were kindly provided by Professor
R. Marais (The Institute of Cancer Research, UK). An additional missense
mutation in the wild-type B-RAF clone (C > G at nucleotide 634;
NM_004333) was repaired using PCR overlap mutagenesis. The MYC-tagged
wild-type and mutant B-RAF cDNAs were each cloned into the pCDH-CMV-
MCS-EF1-copGFP lentiviral vector, which coexpresses copGFP (System
Biosciences, Mountain View, CA, USA). The IGFBP7 shRNA sequences corre-
spond to nucleotides 963–983 and 865–885 (NM_001553; Wajapeyee et al.,
2008). The V600E-specific B-RAF shRNA sequence corresponds to nucleo-
tides 1853–1871 (NM_004333). The pRb- and p53-specific shRNAs have
been described previously (Haferkamp et al., 2009a). All shRNAs were cloned
into the pSIH-H1-EF1-copGFP and pSIH-H1-Puro lentiviral vectors, which
express copGFP or puromycin resistance, respectively (System Biosciences).
The nonsilencing negative control shRNA did not show complete homology to
any known human transcript and had the following sequence: 50-TTAGAGGC-
GAGCAAGACTA-30.
Western Blotting
Total cellular proteins were extracted at 4�C using RIPA lysis buffer containing
protease inhibitors (Roche, Basel, Switzerland). Proteins (30–50 mg) were
resolved on 12% SDS-polyacrylamide gels and transferred to Immobilon-P
membranes (Millipore, Bedford, MA, USA). Secreted proteins were collected
from the culture media, which were initially cleared by centrifugation. Four
milliliters of the cleared media were centrifuged through 15 ml centricon
centrifugal filter units (50 kDa cut off) (Millipore) at 4000 3 g for 30 min and
the resulting supernatants were loaded onto 10 kDa centricon units and recen-
trifuged. The concentrated sample (100–200 ml) containing soluble IGFBP7
(�37 kDa) was collected and proteins (40 mg) were resolved on 12% SDS-poly-
acrylamide gels and transferred to Immobilon P-membranes (Millipore).
Western blots were probed with antibodies against p16INK4a (N20; Santa
Cruz, Santa Cruz, CA, USA), IGFBP7 (W18R; Santa Cruz), BNIP3L (aa 77–92
from human; ProSci Incorporated, Poway, CA, USA), SMARCB1 (3E10; Ab-
nova, Taipei, Taiwan), PEA15 (H80; Santa Cruz), p21Waf1 (C-19; Santa Cruz),
p53 (DO-1; Santa Cruz), c-MYC (A14; Santa Cruz), phosphorylated ERK (E4;
Santa Cruz), total ERK (137F5; Cell Signaling, Danvers, MA, USA), B-RAF
(L12G7, Cell Signaling), pRb (G3-245; Becton Dickinson), and b-actin
(AC-74; Sigma-Aldrich).
Indirect Immunofluorescence
Cells were seeded on coverslips in 12-well plates at 3 3 104 cells per well at
each time point and incubated overnight. Cells were washed in phosphate-
buffered saline (PBS) and fixed with 4% formaldehyde/PBS for 15 min at
room temperature. Cells were rinsed three times with PBS, then permeabilized
with 0.2% Triton X-100/PBS for 10 min, then rinsed and blocked in 10%
FCS/PBS for 1 hr. Cells were incubated with primary antibodies for 50 min,
washed then incubated with Alexa Fluor 594-conjugated secondary IgG
(Molecular Probes, Carlsbad, CA, USA) and 1 mg/ml of the nuclear DNA stain
40,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 50 min. Primary anti-
bodies used were Ki67 (MIB-1; DAKO, Glostrup, Denmark), trimethyl-histone
H3 (Lys 9) (07-442; Millipore), and HMGA2 (HMG1-C, FL109; Santa Cruz).
SA-b-Gal activity was detected as described previously (Dimri et al., 1995).
Immunohistochemistry
Tissue arrays of nevi or melanomas were dewaxed before they were rehy-
drated by washing twice with absolute ethanol, two times with 70% ethanol,
and finally by one rinse with bi-distilled water. Samples were antigen retrieved
by heating at 90�C for 20 min in antigen retrieval solution (pH 6) (S1699; DAKO)
or by microwaving for 10 min in 10 mM sodium citrate (pH 6.0) and then cooled
for 20 min. After blocking endogenous peroxidase the slides were incubated
with goat anti-IGFBP7 antibody (C16; Santa Cruz) diluted 1:100 for 25 min to
2 hr, then washed with PBS. Subsequently, the slides were incubated with bio-
tinylated anti-goat antibody (BA-5000; Vector Laboratories, Burlingame, USA
or P0449; DAKO) for 25 min, slides were washed with PBS, incubated in strep-
tavidin-HRP (K0690; DAKO) or streptavidin/biotin-HRP (Invitrogen) for 25 min,
and placed in Vector NovaRed or 3,30-diaminobenzidine substrate (DAB) for
10–15 min (Vector Laboratories). Sections were counterstained with hematox-
ylin and mounted. A pathologist performed the evaluation of the staining in
a blinded manner.
Statistical Analysis
Scatter plots were used to illustrate the distribution of gene expression by
B-RAF mutation status. Medians and interquartile ranges were applied
to summarize the distributions, and the Mann-Whitney test was used to deter-
mine the differences between the B-RAF wild-type and B-RAF mutant
populations.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures and
two figures and can be found with this article online at doi:10.1016/j.cell.
2010.04.021.
ACKNOWLEDGMENTS
This work is supported by Program Grant 402761, project grants of the
National Health and Medical Research Council of Australia (NHMRC), and an
infrastructure grant to Westmead Millennium Institute by the Health Depart-
ment of NSW through Sydney West Area Health Service. Westmead Institute
for Cancer Research is the recipient of capital grant funding from the Australian
Cancer Research Foundation. L.L.S. is a Cameron Melanoma Research
Fellow, supported by the Melanoma and Skin Cancer Research Institute,
University of Sydney. H.R. is a Cancer Institute New South Wales Research
Fellow. R.A.S. is a Cancer Institute New South Wales Clinical Research Fellow.
We thank Eva-Maria Stuhl, Claudia Siedel, James Wilmott, Suzanah Philipsz,
Branka Mijatov, Svetlana Pianova, and Jason Todd for excellent technical
assistance and Dr. Hermann Kneitz for staining evaluation. The support of
the Melanoma Foundation of the University of Sydney and colleagues from
the Melanoma Institute Australia (incorporating the Sydney Melanoma Unit)
Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc. 725

and the Department of Anatomical Pathology at the Royal Prince Alfred
Hospital are also gratefully acknowledged.
Received: July 25, 2009
Revised: November 21, 2009
Accepted: April 15, 2010
Published: May 13, 2010
REFERENCES
Acosta, J.C., O’Loghlen, A., Banito, A., Raguz, S., and Gil, J. (2008). Control of
senescence by CXCR2 and its ligands. Cell Cycle 7, 2956–2959.
Adams, P.D. (2007). Remodeling of chromatin structure in senescent cells and
its potential impact on tumor suppression and aging. Gene 397, 84–93.
Akaogi, K., Sato, J., Okabe, Y., Sakamoto, Y., Yasumitsu, H., and Miyazaki, K.
(1996). Synergistic growth stimulation of mouse fibroblasts by tumor-derived
adhesion factor with insulin-like growth factors and insulin. Cell Growth Differ
7, 1671–1677.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N.,
Vassiliou, L.V., Kolettas, E., Niforou, K., Zoumpourlis, V.C., et al. (2006). Onco-
gene-induced senescence is part of the tumorigenesis barrier imposed by
DNA damage checkpoints. Nature 444, 633–637.
Bauer, J., Curtin, J.A., Pinkel, D., and Bastian, B.C. (2007). Congenital melano-
cytic nevi frequently harbor NRAS mutations but no BRAF mutations. J. Invest.
Dermatol. 127, 179–182.
Benlloch, S., Paya, A., Alenda, C., Bessa, X., Andreu, M., Jover, R., Castells,
A., Llor, X., Aranda, F.I., and Massuti, B. (2006). Detection of BRAF V600E
mutation in colorectal cancer: comparison of automatic sequencing and
real-time chemistry methodology. J. Mol. Diagn. 8, 540–543.
Bennett, D.C. (2008). How to make a melanoma: what do we know of the
primary clonal events? Pigment Cell Melanoma Res. 21, 27–38.
Burger, A.M., Zhang, X., Li, H., Ostrowski, J.L., Beatty, B., Venanzoni, M.,
Papas, T., and Seth, A. (1998). Down-regulation of T1A12/mac25, a novel
insulin-like growth factor binding protein related gene, is associated with
disease progression in breast carcinomas. Oncogene 16, 2459–2467.
Chen, Y., Pacyna-Gengelbach, M., Ye, F., Knosel, T., Lund, P., Deutschmann,
N., Schluns, K., Kotb, W.F., Sers, C., Yasumoto, H., et al. (2007). Insulin-like
growth factor binding protein-related protein 1 (IGFBP-rP1) has potential
tumour-suppressive activity in human lung cancer. J. Pathol. 211, 431–438.
Chin, L., Garraway, L.A., and Fisher, D.E. (2006). Malignant melanoma:
genetics and therapeutics in the genomic era. Genes Dev. 20, 2149–2182.
Cotter, M.A., Florell, S.R., Leachman, S.A., and Grossman, D. (2007). Absence
of senescence-associated beta-galactosidase activity in human melanocytic
nevi in vivo. J. Invest. Dermatol. 127, 2469–2471.
Curtin, J.A., Busam, K., Pinkel, D., and Bastian, B.C. (2006). Somatic activation
of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 24, 4340–4346.
Degeorges, A., Wang, F., Frierson, H.F., Jr., Seth, A., Chung, L.W., and Sikes,
R.A. (1999). Human prostate cancer expresses the low affinity insulin-like
growth factor binding protein IGFBP-rP1. Cancer Res. 59, 2787–2790.
Denoyelle, C., Abou-Rjaily, G., Bezrookove, V., Verhaegen, M., Johnson, T.M.,
Fullen, D.R., Pointer, J.N., Gruber, S.B., Su, L.D., Nikiforov, M.A., et al. (2006).
Anti-oncogenic role of the endoplasmic reticulum differentially activated by
mutations in the MAPK pathway. Nat. Cell Biol. 8, 1053–1063.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C.,
Schurra, C., Garre, M., Nuciforo, P.G., Bensimon, A., et al. (2006). Oncogene-
induced senescence is a DNA damage response triggered by DNA hyper-
replication. Nature 444, 638–642.
Dimri, G.P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano,
E.E., Linskens, M., Rubelj, I., Pereira-Smith, O., et al. (1995). A biomarker that
identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl.
Acad. Sci. USA 92, 9363–9367.
Falchi, M., Bataille, V., Hayward, N.K., Duffy, D.L., Bishop, J.A., Pastinen, T.,
Cervino, A., Zhao, Z.Z., Deloukas, P., Soranzo, N., et al. (2009). Genome-
726 Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc.
wide association study identifies variants at 9p21 and 22q13 associated
with development of cutaneous nevi. Nat. Genet. 41, 915–919.
Gray-Schopfer, V.C., Cheong, S.C., Chong, H., Chow, J., Moss, T.,
Abdel-Malek, Z.A., Marais, R., Wynford-Thomas, D., and Bennett, D.C.
(2006). Cellular senescence in naevi and immortalisation in melanoma: a role
for p16? Br. J. Cancer 95, 496–505.
Haferkamp, S., Tran, S., Becker, T.M., Scurr, L.L., Kefford, R.F., and Rizos, H.
(2009a). The relative contributions of the p53 and pRb pathways in oncogene-
induced melanocyte senescence. Aging 1, 542–556.
Haferkamp, S., Scurr, L.L., Becker, T.M., Frausto, M., Kefford, R.F., and Rizos,
H. (2009b). Oncogene-induced senescence does not require the p16(INK4a) or
p14ARF melanoma tumor suppressors. J. Invest. Dermatol. 128, 1983–1991.
Halaban, R., Ghosh, S., Duray, P., Kirkwood, J.M., and Lerner, A.B. (1986).
Human melanocytes cultured from nevi and melanomas. J. Invest. Dermatol.
87, 95–101.
Harrison, S.L., MacLennan, R., and Buettner, P.G. (2008). Sun exposure and
the incidence of melanocytic nevi in young Australian children. Cancer Epide-
miol. Biomarkers Prev. 17, 2318–2324.
Herlyn, M. (2007). Farming cells to rebuild skin and melanoma. Cancer Biol.
Ther. 6, 467–471.
Jiang, W., Xiang, C., Cazacu, S., Brodie, C., and Mikkelsen, T. (2008). Insulin-
like growth factor binding protein 7 mediates glioma cell growth and migration.
Neoplasia 10, 1335–1342.
Kelly, J.W., Yeatman, J.M., Regalia, C., Mason, G., and Henham, A.P. (1997).
A high incidence of melanoma found in patients with multiple dysplastic naevi
by photographic surveillance. Med. J. Aust. 167, 191–194.
Kuilman, T., and Peeper, D.S. (2009). Senescence-messaging secretome:
SMS-ing cellular stress. Nat. Rev. Cancer 9, 81–94.
Landberg, G., Ostlund, H., Nielsen, N.H., Roos, G., Emdin, S., Burger, A.M.,
and Seth, A. (2001). Downregulation of the potential suppressor gene
IGFBP-rP1 in human breast cancer is associated with inactivation of the reti-
noblastoma protein, cyclin E overexpression and increased proliferation in
estrogen receptor negative tumors. Oncogene 20, 3497–3505.
Michaloglou, C., Vredeveld, L.C., Soengas, M.S., Denoyelle, C., Kuilman, T.,
van der Horst, C.M., Majoor, D.M., Shay, J.W., Mooi, W.J., and Peeper, D.S.
(2005). BRAFE600-associated senescence-like cell cycle arrest of human
naevi. Nature 436, 720–724.
Papp, T., Pemsel, H., Zimmermann, R., Bastrop, R., Weiss, D.G., and Schiff-
mann, D. (1999). Mutational analysis of the N-ras, p53, p16INK4a, CDK4,
and MC1R genes in human congenital melanocytic naevi. J. Med. Genet.
36, 610–614.
Pen, A., Moreno, M.J., Martin, J., and Stanimirovic, D.B. (2007). Molecular
markers of extracellular matrix remodeling in glioblastoma vessels: microarray
study of laser-captured glioblastoma vessels. Glia 55, 559–572.
Pen, A., Moreno, M.J., Durocher, Y., Deb-Rinker, P., and Stanimirovic, D.B.
(2008). Glioblastoma-secreted factors induce IGFBP7 and angiogenesis by
modulating Smad-2-dependent TGF-beta signaling. Oncogene 27,
6834–6844.
Platz, A., Egyhazi, S., Ringborg, U., and Hansson, J. (2008). Human cutaneous
melanoma; a review of NRAS and BRAF mutation frequencies in relation to
histogenetic subclass and body site. Mol. Oncol. 1, 395–405.
Pollock, P.M., Harper, U.L., Hansen, K.S., Yudt, L.M., Stark, M., Robbins,
C.M., Moses, T.Y., Hostetter, G., Wagner, U., Kakareka, J., et al. (2003).
High frequency of BRAF mutations in nevi. Nat. Genet. 33, 19–20.
Prieur, A., and Peeper, D.S. (2008). Cellular senescence in vivo: a barrier to
tumorigenesis. Curr. Opin. Cell Biol. 20, 150–155.
Ruan, W., Xu, E., Xu, F., Ma, Y., Deng, H., Huang, Q., Lv, B., Hu, H., Lin, J.,
Cui, J., et al. (2007). IGFBP7 plays a potential tumor suppressor role in colo-
rectal carcinogenesis. Cancer Biol. Ther. 6, 354–359.
Schrama, D., Kneitz, H., Willmes, C., Adam, C., Houben, R., and Becker, J.C.
(2009). Lack of correlation between IGFBP7 expression and BRAF mutational
status in melanoma. J. Invest. Dermatol. 130, 897–898.

Shao, L., Huang, Q., Luo, M., and Lai, M. (2004). Detection of the differentially
expressed gene IGF-binding protein-related protein-1 and analysis of its rela-
tionship to fasting glucose in Chinese colorectal cancer patients. Endocr.
Relat. Cancer 11, 141–148.
Suzuki, H., Igarashi, S., Nojima, M., Maruyama, R., Yamamoto, E., Kai, M.,
Akashi, H., Watanabe, Y., Yamamoto, H., Sasaki, Y., et al. (2009). IGFBP7 is
a p53 responsive gene specifically silenced in colorectal cancer with CpG
island methylator phenotype. Carcinogenesis 31, 342–349.
Sviderskaya, E.V., Gray-Schopfer, V.C., Hill, S.P., Smit, N.P., Evans-Whipp,
T.J., Bond, J., Hill, L., Bataille, V., Peters, G., Kipling, D., et al. (2003). p16/Cy-
clin-dependent kinase inhibitor 2A deficiency in human melanocyte senes-
cence, apoptosis, and immortalization: possible implications for melanoma
progression. J. Natl. Cancer Inst. 95, 723–732.
Swisshelm, K., Ryan, K., Tsuchiya, K., and Sager, R. (1995). Enhanced expres-
sion of an insulin growth factor-like binding protein (mac25) in senescent
human mammary epithelial cells and induced expression with retinoic acid.
Proc. Natl. Acad. Sci. USA 92, 4472–4476.
Takeno, A., Takemasa, I., Doki, Y., Yamasaki, M., Miyata, H., Takiguchi, S.,
Fujiwara, Y., Matsubara, K., and Monden, M. (2008). Integrative approach
for differentially overexpressed genes in gastric cancer by combining large-
scale gene expression profiling and network analysis. Br. J. Cancer 99,
1307–1315.
Thompson, J.F., Scolyer, R.A., and Kefford, R.F. (2005). Cutaneous mela-
noma. Lancet 365, 687–701.
Wajapeyee, N., Serra, R.W., Zhu, X., Mahalingam, M., and Green, M.R. (2008).
Oncogenic BRAF induces senescence and apoptosis through pathways medi-
ated by the secreted protein IGFBP7. Cell 132, 363–374.
Wajapeyee, N., Kapoor, V., Mahalingam, M., and Green, M.R. (2009). Efficacy
of IGFBP7 for treatment of metastatic melanoma and other cancers in mouse
models and human cell lines. Mol. Cancer Ther. 8, 3009–3014.
Ye, F., Chen, Y., Knosel, T., Schluns, K., Pacyna-Gengelbach, M., Deutsch-
mann, N., Lai, M., and Petersen, I. (2007). Decreased expression of insulin-
like growth factor binding protein 7 in human colorectal carcinoma is related
to DNA methylation. J. Cancer Res. Clin. Oncol. 133, 305–314.
Yu, H., McDaid, R., Lee, J., Possik, P., Li, L., Kumar, S.M., Elder, D.E., Van
Belle, P., Gimotty, P., Guerra, M., et al. (2009). The role of BRAF mutation
and p53 inactivation during transformation of a subpopulation of primary
human melanocytes. Am. J. Pathol. 74, 2367–2377.
Zhuang, D., Mannava, S., Grachtchouk, V., Tang, W.H., Patil, S., Wawrzyniak,
J.A., Berman, A.E., Giordano, T.J., Prochownik, E.V., Soengas, M.S., et al.
(2008). C-MYC overexpression is required for continuous suppression of
oncogene-induced senescence in melanoma cells. Oncogene 27, 6623–6634.
Cell 141, 717–727, May 14, 2010 ª2010 Elsevier Inc. 727





























