Embed Size (px)
Citation preview

1220
I ncreased plasma lipoprotein(a) [Lp(a)] concentrations correlate with atherothrombotic diseases and evidence is
accumulating that this association might be causal.1–5 Plasma Lp(a) levels are highly variable among individuals ranging from 1 mg/dL to more than 200 mg/dL and the consensus report of the European Atherosclerosis Society recommends a cut- off for aggressive treatment at Lp(a) levels of 50 mg/dL.6 Unfortunately, only very few drugs affect plasma Lp(a) levels, except for nicotinic acid, that maximally reduces Lp(a) by 30% with an unknown mode of action.7
see accompanying article on page 1060
Lp(a) is produced only in liver of primates.8 It is com-posed of a LDL core and the glycoprotein apolipoprotein(a) [APOA], that is bound to LDL via a disulfide bridge.9 Turnover studies in man clearly established that Lp(a) plasma concentrations are mainly controlled by the rate of APOA de novo biosynthesis, whereas Lp(a) catabolism might have only small effects.10–11 It is therefore of impor-tance to uncover mechanisms controlling Lp(a) biosynthesis and APOA expression in detail to develop strategies to lower elevated plasma Lp(a).
Recently we reported that patients suffering from obstructive cholestasis had strongly reduced plasma Lp(a) concentrations.12 This was also verified in transgenic APOA (tg-APOA) mice expressing the human APOA gene controlled by its native promoter, undergoing bile duct ligation as well as in tg- APOA mice fed a chow containing 0.2% cholic acid where the transcription of APOA in the liver and the con-centration of plasma APOA were dramatically reduced. In a series of experiments we finally provided evidence that the DR-1 at 2826/2814bp of the APOA promoter functions as a negative FXR response element. However, the downregula-tion of APOA transcription by this DR-1 element did not fully account for the almost complete disappearance of plasma APOA in tg- APOA mice fed a CA containing chow. Therefore we searched for additional negative regulators mediated by FXR signaling.
FGF15/19 plays an important regulatory role in hepatic bile acid metabolism. The key enzyme in bile acid biosynthesis CYP7A1 was shown to be downregulated indirectly by FXR after intestinal induction of FGF19 expression in humans13 and its ortholog FGF15 in mice.14 FGF19 binds to FGFR4
FGF19 signaling Cascade suppresses APOA Gene expression
Indumathi Chennamsetty, Thierry Claudel, Karam M. Kostner, Michael Trauner, Gert M. Kostner
Objective—Lipoprotein(a) is a highly atherogenic lipoprotein, whose metabolism is poorly understood. Currently no safe drugs exists that lower elevated plasma lipoprotein(a) concentrations. We therefore focused on molecular mechanisms that influence apolipoprotein(a) (APOA) biosynthesis.
Methods and Results—Transgenic human APOA mice ( tg- APO mice) were injected with 1 mg/kg of recombinant human fibroblast growth factor 19 (FGF19). This led to a significant reduction of plasma APOA and hepatic expression of APOA. Incubation of primary hepatocytes of tg- APOA mice with FGF19 induced ERK1/2 phosphorylation and, in turn, downregulated APOA expression. Repression of APOA by FGF19 was abrogated by specific ERK1/2 phosphorylation inhibitors. The FGF19 effect on APOA was attenuated by transfection of primary hepatocytes with siRNA against the FGF19 receptor 4 (FGFR4). Using promoter reporter assays, mutation analysis, gel shift, and chromatin immune- precipitation assays, an Ets-1 binding element was identified at 21630/21615bp region in the human APOA promoter. This element functions as an Elk-1 binding site that mediates repression of APOA transcription by FGF19.
Conclusion—These findings provide mechanistic insights into the transcriptional regulation of human APOA by FGF19. Further studies in the human system are required to substantiate our findings and to design therapeutics for hyper lipoprotein(a). (Arterioscler Thromb Vasc Biol . 2012;32:1220-1227 .)
Key Words: apolipoproteins fibrinolysis gene expression lipoproteins molecular biology
Received on: August 2, 2011; final version accepted on: January 6, 2012.From the Institute of Molecular Biology and Biochemistry (I.C., G.M.K.), Center of Molecular Medicine, and Laboratory of Experimental and Molecular
Hepatology (T.C., M.T.), Division of Gastroenterology and Hepatology, Department of Internal Medicine, Medical University of Graz, Austria; Division of Gastroenterology and Hepatology (T.C., M.T.), Department of Internal Medicine III, Medical University of Vienna, Austria; Department of Cardiology (K.M.K.), University of Queensland, Mater Adult Hospital, Brisbane, Australia.
the online- only data supplement is available with this article at http://atvb .ahajournals .org/lookup/suppl/doi:10 .1161/atVBaHa .111 .243055/-/dC1.Correspondence to Gert M. Kostner, Institute of Molecular Biology and Biochemistry, Center of Molecular Medicine, Medical University of Graz, 8010
Graz, Harrachgasse 21, Austria. E-mail [email protected]© 2012 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb .ahajournals .org dOi: 10 .1161/atVBaHa .111 .243055
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

Chennamsetty et al FGF19 inhibits aPOa expression 1221
on liver cells15–16 and suppresses the expression CYP7A1 in human hepatocytes in a signaling cascade involving the MAPK/ERK1/2 pathway.17 These findings prompted us to test whether this pathway might be also operative in APOA suppression.
In the present study we show that APOA transcription is indeed suppressed via the FXR-FGF15/19-FGFR4 axis. FGF15/19 binding to its receptor FGFR4 on liver cells acti-vates MAPK/ERK1/2 that in turn displaces phosphorylated Elk-1 to the nucleus. Elk-1 binds to a negative control element located at 21630/21615bp of the human APOA promoter thereby suppressing APOA transcription and plasma APOA concentrations.
Materials and MethodsChemicalsRecombinant FGF19 was purchased from R&D systems (Vienna, Austria), and cholic acid was from Sigma (Vienna, Austria). The reagents PD98059, SB203580, and SP600125 were from CalBiochem (Germany); U0126 was from Upstate Biotech (Lake Placid, NY). Collagenase was from Worthington Corporation (NJ).
animal experimentsAll animal experiments were performed following approval of the protocol by the Austrian Federal Ministry of Science and Research (Vienna, Austria). tg- APOA mice carrying human APOA gene con-trolled by its native flanking region in YAC18 were hosted under stan-dard 12 hour light/12 hour dark cycle and fed standard rodent chow diet and water ad libitum. Ten- to 12-week-old female tg- APOA mice were injected intraperitoneally recombinant FGF19 (1 mg/kg body weight) (n55 mice) or vehicle (PBS) (n56 mice), followed by plasma and tissue harvesting after 16 hours. Plasma concentrations of APOA were measured enzymatically by an in- house DELFIA method.12
PlasmidsExpression plasmids encoding human Elk-1 (pcDNA3-Elk-1) was generously provided by Dr Robert Hipskind (IGM Montpellier, France). The human APOA promoter constructs were obtained by PCR amplification as a template as described previously.12
transient transfection and reporter Gene assaysReporter gene assays were performed in HepG2 cells. Cells at 60% to 70% confluence were transiently transfected with the indicated reporter or expression plasmids using FuGENE 6 reagent as described previously.12
site- directed MutagenesisMutagenesis was performed using the Quik Change site- directed mutagenesis system (Stratagene, La Jolla, CA), according to the manufacturer’s protocol. The oligonucleotide Mut (5- GCGGTAGGTTTTCACCATTATCATTATGTTTGCCTTGCTC-3) was used to introduce mutations into the full length hAPOA 21952/152. Mutated bases are in bold.
electrophoretic Mobility shift assaysHuman Elk-1 protein was synthesized in vitro using the TNT® T7 Quick Coupled Transcription/Translation System (Promega, Madison, WI). The sense and antisense oligonucleotide probes of Elk-1 Wt (5-ACCATTATCAGGatGTTTGCCTTG-3), Elk-1 Mut (5- ACCATTATCAttatGTTTGCCTTG-3), and an Elk-1 consensus element- containing oligonucleotide (5-GGTCCTAAGCGGACCGGaaGTTCGTCAAGTTTCA-3) were annealed and radioactively labeled. In vitro translated human Elk-1 (2.0 L) was incubated for
20 minutes at room temperature in a total volume of 10 L with bind-ing buffer (Gel shift assay system, Promega, Madison, WI) before the labeled probe was added. Binding reactions were further incubated for 30 minutes and resolved by 6% nondenaturing polyacrylamide gel electrophoresis in 0.25X Tris- Borate- EDTA buffer at room tempera-ture and 120 V for 4.0 hours. The gel was dried and exposed to an X- ray film. In supershift assays, anti-- Elk-1 antibody (sc-355x, Santa Cruz Biotechnology)19 was added for 1 hour on ice prior to the addition of probes. For competition experiments, unlabeled probes were included in the binding reaction at the indicated excess concentrations.
Chromatin immunoprecipitationChromatin immunoprecipitation (ChIP) assay was performed with primary mouse hepatocytes treated with vehicle or FGF19 for 6 hours using ChIP- IT Express kit (Active Motif, Rixensart, Belgium) according to the manufacturer’s instructions. Chromatin was immunoprecipitated using 2 g anti-- Elk-1 and 1 g anti- IgG antibody. DNA extractions were PCR amplified using the fol-lowing flanking primers covering Ets-1 element and the PCR products (166bp) were analyzed by agarose gel electrophoresis. (ChIP FWD 5 CTCTATGTCGGCCACTGGAT 3; ChIP REV 5 AGGGTGCATCACCTGGACTA 3).
statisticsStatistical analyses of the experiments were performed with GraphPad Prism 5.0. Two- tailed, unpaired Student t- test was applied to deter-mine statistical significance (***P0.001; **P0.01; *P0.05).
Cell isolation, culture, treatments, real- time PCR, immunoblotting, and siRNA transfections are described in detail in the online- only Data Supplement.
resultsFGF19 inhibits aPOa expression in Vitro and in Vivo in transgenic aPOa MiceWe showed previously that cholic acid feeding dramatically suppressed APOA expression in liver and APOA abundance in plasma of tg- APOA mice.12 The in vitro effect observed when primary mouse hepatocytes were incubated with FXR ligands, however, was less pronounced. We therefore considered the possibility, that FGF15/19 might have an additive effect.
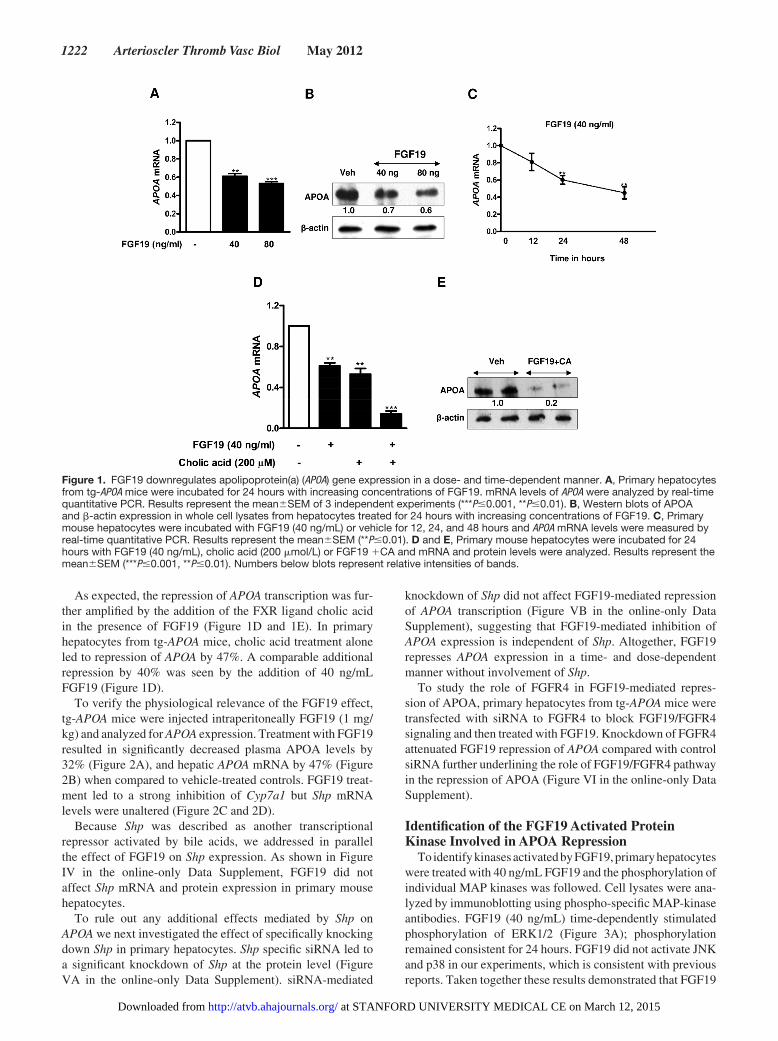
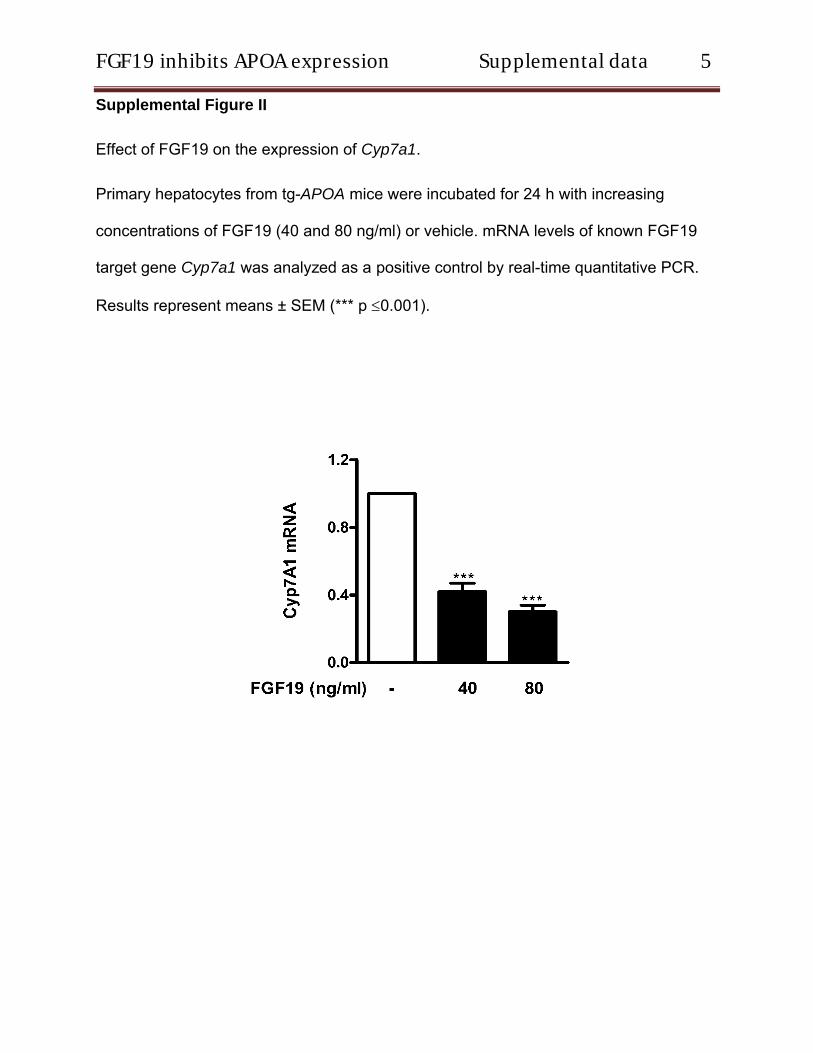
To verify the effect of FGF19 on human APOA expression, primary hepatocytes isolated from tg- APOA mice were incu-bated with different concentrations of recombinant FGF19. Analysis of mRNA levels by real- time quantitative PCR revealed a significant dose- and time- dependent decrease in APOA mRNA levels suggesting a transcriptional effect (Figure 1A and 1C). Western blot analysis confirmed that this FGF19-mediated repression also occurs at the protein level in cell lysates (Figure 1B). In addition, APOA secre-tion into the medium was reduced by 48% (Figure I in the online- only Data Supplement). Cell viability assessed by trypan blue exclusion test revealed that all concentrations of FGF19 were well tolerated (data not shown). As a posi-tive control, we measured the expression levels of the known FGF19 target gene Cyp7a1. As expected FGF19 treatment dose- dependently downregulated Cyp7a1 mRNA levels (Figure II in the online- only Data Supplement). Furthermore, FGF19 treatment also suppressed the APOA expression in primary hepatocytes isolated from tg-APOA/fxr2/2 mice con-firming a direct role of FGF19 on APOA repression (Figure III in the online- only Data Supplement).
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from
1222 Arterioscler Thromb Vasc Biol May 2012
As expected, the repression of APOA transcription was fur-ther amplified by the addition of the FXR ligand cholic acid in the presence of FGF19 (Figure 1D and 1E). In primary hepatocytes from tg- APOA mice, cholic acid treatment alone led to repression of APOA by 47%. A comparable additional repression by 40% was seen by the addition of 40 ng/mL FGF19 (Figure 1D).
To verify the physiological relevance of the FGF19 effect, tg- APOA mice were injected intraperitoneally FGF19 (1 mg/kg) and analyzed for APOA expression. Treatment with FGF19 resulted in significantly decreased plasma APOA levels by 32% (Figure 2A), and hepatic APOA mRNA by 47% (Figure 2B) when compared to vehicle- treated controls. FGF19 treat-ment led to a strong inhibition of Cyp7a1 but Shp mRNA levels were unaltered (Figure 2C and 2D).
Because Shp was described as another transcriptional repressor activated by bile acids, we addressed in parallel the effect of FGF19 on Shp expression. As shown in Figure IV in the online- only Data Supplement, FGF19 did not affect Shp mRNA and protein expression in primary mouse hepatocytes.
To rule out any additional effects mediated by Shp on APOA we next investigated the effect of specifically knocking down Shp in primary hepatocytes. Shp specific siRNA led to a significant knockdown of Shp at the protein level (Figure VA in the online- only Data Supplement). siRNA-mediated
knockdown of Shp did not affect FGF19-mediated repression of APOA transcription (Figure VB in the online- only Data Supplement), suggesting that FGF19-mediated inhibition of APOA expression is independent of Shp. Altogether, FGF19 represses APOA expression in a time- and dose- dependent manner without involvement of Shp.
To study the role of FGFR4 in FGF19-mediated repres-sion of APOA, primary hepatocytes from tg- APOA mice were transfected with siRNA to FGFR4 to block FGF19/FGFR4 signaling and then treated with FGF19. Knockdown of FGFR4 attenuated FGF19 repression of APOA compared with control siRNA further underlining the role of FGF19/FGFR4 pathway in the repression of APOA (Figure VI in the online- only Data Supplement).
identification of the FGF19 activated Protein Kinase involved in aPOa repression
To identify kinases activated by FGF19, primary hepatocytes were treated with 40 ng/mL FGF19 and the phosphorylation of individual MAP kinases was followed. Cell lysates were ana-lyzed by immunoblotting using phospho- specific MAP- kinase antibodies. FGF19 (40 ng/mL) time- dependently stimulated phosphorylation of ERK1/2 (Figure 3A); phosphorylation remained consistent for 24 hours. FGF19 did not activate JNK and p38 in our experiments, which is consistent with previous reports. Taken together these results demonstrated that FGF19
Figure 1. FGF19 downregulates apolipoprotein(a) (APOA) gene expression in a dose- and time- dependent manner. A, Primary hepatocytes from tg- APOA mice were incubated for 24 hours with increasing concentrations of FGF19. mRNA levels of APOA were analyzed by real- time quantitative PCR. Results represent the meanSEM of 3 independent experiments (***P0.001, **P0.01). B, Western blots of APOA and -actin expression in whole cell lysates from hepatocytes treated for 24 hours with increasing concentrations of FGF19. C, Primary mouse hepatocytes were incubated with FGF19 (40 ng/mL) or vehicle for 12, 24, and 48 hours and APOA mRNA levels were measured by real- time quantitative PCR. Results represent the meanSEM (**P0.01). D and E, Primary mouse hepatocytes were incubated for 24 hours with FGF19 (40 ng/mL), cholic acid (200 mol/L) or FGF19 1CA and mRNA and protein levels were analyzed. Results represent the meanSEM (***P0.001, **P0.01). Numbers below blots represent relative intensities of bands.
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

Chennamsetty et al FGF19 inhibits aPOa expression 1223
specifically activated the MEK/ERK1/2 pathway in primary hepatocytes from tg- APOA mice.
erK1/2 inhibitors attenuate FGF19-Mediated inhibition of aPOa expression in Mouse Primary HepatocytesTo further investigate the signaling pathways involved in FGF19-mediated inhibition of APOA gene transcription, mouse primary hepatocytes were treated with several spe-cific inhibitors of MAP kinases. U0126 and PD98059 alone, inhibitors of the MEK/ERK1/2 pathway, strongly stimulated APOA mRNA expression. Pretreatment with U0126 and PD98059 attenuated the inhibitory effect of FGF19 on APOA expression in mouse primary hepatocytes. SP600125, a JNK inhibitor, and SB203580, a p38 kinase inhibitor, had no effect on FGF19-mediated repression of APOA (Figure 3B and 3C). Taken together these results demonstrate that MEK/ERK1/2 inhibitors, but not JNK and p38MAPK inhibitors, blocked FGF19-mediated inhibition of APOA expression.
Mapping of the Promoter region responsive to FGF19 signalingTo identify promoter elements responsible for the observed FGF19 effects, a 2-kb fragment of human APOA promoter (hAPOA 21952/152) was cloned into a pGL3-luciferase reporter plasmid. In addition a series of 5 deletion con-structs were generated as shown in Figure 4A. HepG2 cells were then transiently transfected with these constructs of the hAPOA promoter in the absence or presence of FGF19 (40
Figure 2. FGF19 decreases plasma levels and hepatic expres-sion of apolipoprotein(a) (APOA). Transgenic human APOA (tg-APOA) mice expressing human APOA were treated with 1 mg/kg recombinant FGF19 (n55 mice) or PBS control (n56 mice). A, Plasma levels of APOA were measured by DELFIA and expressed as meanSD **P0.01 when compared to PBS control mice. Hepatic APOA (B) and Cyp7a1 (C) and Shp (D) mRNA levels were analyzed by real- time quantitative PCR, normalized to cyclo-philin and expressed relative to control mice. Results represent meansSEM (***P0.001).
Figure 3. Effect of FGF19 on mitogen- activated protein kinase (MAPK) phosphorylation in primary hepatocytes. A, Primary hepatocytes from transgenic human apolipoprotein(a) (tg-APOA) mice were treated with FGF19 (40 ng/mL) for the indicated time periods, and whole cell extracts (50 g/lane) were analyzed by Western blotting with indicated antibodies. Numbers below blots represent relative intensities of the bands. B, Effects of protein kinase inhibitors on APOA expression. Primary hepatocytes from tg- APOA mice were pretreated with the specific mitogen- activated protein kinase (MAPK) inhibitors for 1 hour, followed by the treatment with vehicle or FGF19 (40 ng/mL) for 24 hours. Total RNA was extracted and APOA mRNA levels were measured by real- time quantitative PCR. Results represent the meanSEM of 3 independent experiments (**P0.01, *P0.05, treated vs vehicle control). C, Primary mouse hepatocytes were pretreated with the specific MAPK inhibitors for 1 hour, followed by the treat-ment with vehicle or FGF19 (40 ng/mL) for 24 hours. Whole cell extracts were then analyzed by immunoblotting for APOA protein expression. Protein kinase inhibitors used were U0126 (U, 10 mol/L), PD98059 (PD, 25 mol/L) for MEK/ERK1/2, SP600125 (SP, 25 mol/L) for JNK, SB203580 (SB, 25 mol/L) for p38 kinase.
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

1224 Arterioscler Thromb Vasc Biol May 2012
ng/mL). Incubation with FGF19 strongly lowered the activ-ity of the hAPOA 21952/152 promoter by 48% (Figure 4B). However, the repression was relieved for 21446, 2757, 2657, 2477, and 2148 promoter constructs indicating that the region between 21952 to 21446bp of the human APOA promoter contains a response element mediating the suppres-sion of APOA transcriptional activity by FGF19.
Next, to study the effect of kinase inhibitors on human APOA promoter activity, HepG2 cells were transfected with the full length hAPOA 21952/152 promoter reporter plasmid pretreated for 1 hour with specific MAP kinase inhibitors, fol-lowed by treatment with vehicle or FGF19. Pretreatment with MEK/ERK1/2 inhibitors U0126 and PD98059, but not the other inhibitors, reversed the inhibitory effects of FGF19 on human APOA promoter activity (Figure 4C).
Notably, in silico Matinspector promoter analysis20 sug-gested the presence of an Ets-1 binding motif located between nucleotides 21630 and 21615 of the APOA gene promoter. To test whether this Ets-1 site could mediate FGF19 depen-dent repression of human APOA promoter, we introduced mutations in the full- length human APOA 21952/152
promoter (21952/152 Wt) and generated the mutant con-struct (21952/152 Mut) as shown in Figure 5A. Mutation of this site completely abolished the FGF19-mediated repres-sion of APOA promoter activity (Figure 5B). Taken together, these results demonstrate that the Ets-1 response element located between nucleotides 21630 and 21615 of human APOA promoter is a negative response element responsible for FGF19-mediated repression of human APOA promoter activ-ity involving MAPK/ERK1/2.
elk-1 Binds to an ets-1 Motif in the Human aPOa PromoterElk-1 has been shown to be a well- characterized common nuclear substrate for activated ERK1/221–22 that belongs to the family of Ets domain containing transcription factors. By Western blot analysis we ascertained that Elk-1 indeed is phosphorylated by FGF19 in primary mouse hepatocytes (Figure 6A). To provide additional evidence that Elk-1 indeed binds to the Ets-1 binding motif at the 21630 and 21615bp region of the human APOA promoter, gel shift assays with in
Figure 4. FGF19 downregulates human apolipoprotein(a) (APOA) promoter activity in HepG2 cells. A, Scheme of the deletion constructs of the human APOA (hAPOA) promoter used in the luciferase reporter assay. B, HepG2 cells were transfected with the indicated hAPOA promoter reporter plasmids (150 ng). Cells were subsequently treated for 36 hours with vehicle or with FGF19 (40 ng/mL) in serum- free DMEM. Transfections were performed in triplicates, and each experiment was repeated at least 3 times. Values are normalized to internal control -galactosidase activity and expressed in percentage. Data are presented as meanSD (**P0.01). RLU indicates relative light units. C, Effect of kinase inhibitors on human APOA promoter activity. HepG2 cells were transfected with full length hAPOA 21952/152 promoter reporter plasmid (150 ng) pretreated with the specific mitogen- activated protein kinase (MAPK) inhibitors for 1 hour, followed by the treatment with vehicle or FGF19 (40 ng/mL) in serum free DMEM for 36 hours. Values are normalized to internal control -galac-tosidase activity and expressed in percentage. Protein kinase inhibitors used were U0126 (U,10 mol/L), PD98059 (PD, 25 mol/L) for MEK/ERK1/2, SP600125 (SP, 25 mol/L) for JNK, SB203580 (SB, 25 mol/L) for p38 kinase. Data are presented as meanSD (**P0.01, *P0.05, treated vs. vehicle control).
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

Chennamsetty et al FGF19 inhibits aPOa expression 1225
vitro translated human recombinant Elk-1 were performed using probes that cover the Ets-1 element. Consensus Elk-1 probe was used as a positive control. Elk-1 bound the labeled consensus probe (Figure 6B, lane 2) and to the Elk-1 wild type (Wt) probe (Figure 6B, lane 5), but not to the probe carrying the mutated Elk-1 element (Mut) (Figure 6B, lane 6). The for-mation of Elk-1–DNA complex was specifically competed by unlabeled cold Wt- probe (Figure 6C, lanes 3 and 4), whereas the Mut2 probe did not compete for binding (Figure 6C, lanes 5 and 6). The intensity of Elk-1–DNA complex formation was decreased by the addition of a specific anti– Elk-1 antibody.
To further confirm the interaction of the transcription fac-tors Elk-1 with the Ets-1 element in the APOA promoter, we performed a ChIP experiment with primary mouse hepatocytes treated with FGF19. FGF19 treatment led to occupancy of the response element by Elk-1 (Figure 6D). As a negative control, equivalent amount of chromatin precipitated with IgG anti-body resulted in no signal. These results further confirmed that Elk-1 binds to the response element at the 21630/21615bp region of human APOA promoter.
Collectively, these data provide evidence that APOA is strongly repressed by the FGF19/MAPK/ERK1/2-Elk-1 sig-naling cascade.
discussionLp(a) has been recognized as an important risk factor for cardiovascular diseases by interfering with several steps of hemostasis and fibrinolysis,23 by accumulating in the arte-rial intima because of its high affinity to proteoglycans and in addition by structural alterations under high oxidative stress thereby stimulating numerous inflammatory and immuno-logic pathways.24 Unfortunately, a final proof of concept by prospective intervention studies with Lp(a) lowering drugs
are missing because there is currently no safe and effective medication available. Detailed knowledge of the Lp(a) and APOA metabolism might circumvent this problem and help to design more efficacious drugs for patients at increased risk for athero- thrombotic diseases.
As plasma Lp(a) levels are mainly controlled by its rate of biosynthesis,10,11 we focused our research to the transcriptional regulation of APOA. From previous work we knew that FXR ligands have a profound influence on APOA transcription and we identified a negative FXR response element at 2830 to 2815bp region of the APOA promoter that reduced APOA expression up to 60%.12 Promoter studies in combination
Figure 6. FGF19 promotes Elk-1 phosphorylation and Elk-1 binds to an Ets-1 motif of human apolipoprotein(a) (hAPOA) promoter in vitro. A, Primary mouse hepatocytes were treated with FGF19 (40 ng/mL) for the indicated time periods, and whole cell extracts were analyzed by Western blotting. Numbers below blots represent relative intensities of the bands. B, Electropho-retic mobility shift assays (EMSA) were performed with radio-labeled Elk-1 consensus probe (Cons) (lanes 1–4), Elk-1 wild type (Wt) (lane 5) and Elk-1 mutant (Mut) (lane 6) probes using in vitro transcribed/translated Elk-1 (lanes 2–7) or unprogrammed reticulocyte lysate (lane 1). Competition EMSAs on radiolabeled consensus probe was performed by adding 50-fold, 100-fold molar excess of the indicated cold consensus probe (lanes 3, 4). Supershift assay, performed with anti- Elk-1 antibody, reduced the intensity of shift band (lane 7). C, Competition EMSAs on radiolabeled Wt- probe was performed by adding 50-fold, 100-fold molar excess of the indicated cold Wt (lanes 3,4) and cold Mut probe (lanes 5, 6). Supershift assay with specific Elk-1 anti-body reduced the intensity of shift band (lane 7). D, ChIP assay of Elk-1 binding to the APOA promoter in primary mouse hepato-cytes treated with FGF19.
Figure 5. Mutation at 21630/21615bp abolishes FGF19-medi-ated repression of human apolipoprotein(a) (hAPOA) promoter. A, Scheme showing wild type (Wt) and mutant (Mut) sequences. B, HepG2 cells were transfected with the wild type ( 21952/152) and mutant (21952/152) hAPOA promoter reporter plasmids (150 ng). Cells were then treated with FGF19 (40 ng/mL) or vehicle for 36 hours. Values are normalized to internal control - galactosidase activity and expressed in percentage. Data are presented as meanSD (**P0.01).
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

1226 Arterioscler Thromb Vasc Biol May 2012
with in vitro findings however suggested that this negative FXR response element accounted only for part of the bile acid mediated transcriptional repression. FXR- signaling has been intensively studied with regard to bile acids metabo-lism. Among other pathways that are still under debate, ligand- activated FXR binds to promoter elements of small heterodimer partner and drives its transcription; small het-erodimer partner in turn represses the activity of key genes such as CYP7A1. Independently of this pathway, FXR was found to transactivate mouse Fgf15, a gene that is highly expressed in the terminal ileum, and its human ortholog, FGF19, expressed in small intestine and liver. In our previous report we demonstrated that the overexpression of Shp did not affect APOA promoter activity. In the current study, silencing of Shp in primary mouse hepatocytes did not influence APOA expression (Figure V in the online- only Data Supplement). We therefore focused in the present work on a possible regula-tion by FGF15/19.
FGF15/19 belongs to the hormone- like endocrine sub-family of fibroblast growth factors.25 FGF19 and the mouse ortholog FGF15 are highly expressed in the ileum and cir-culate to the liver. Activated liver cells of human but not of mouse origin have also been found to express FGF19.13,14
FGF15/19 binds to its cognate receptor FGFR4 on liver cells repressing the transcription of several proteins involved in bile acid metabolism.15,16 In addition FGF15/19 plays important roles in hepatic lipid, protein and glycogen metabolism and is most relevant for the pathophysiology of type-2 diabetes mel-litus and metabolic syndrome26–27 FGF19 on the other hand has been related to tumorigenesis in humans and thus any medication influencing FG19 expression needs to be carefully monitored for possible adverse effects.28
FGF15/19 expression in the intestine is under the con-trol of FXR.14 Binding of bile acids, the natural ligands of FXR strongly induces FGF15/19 biosynthesis that bind to the tyrosine kinase receptor FGFR4 on liver cells. FGFR4 phosphorylation activates 2 distinct intracellular substrates: phospholipase C g1 also named FGF receptor substrate FRS1 and FGFR substrate 2, named FSR2. In the FSR2 signaling cascade Ras- mitogen- activated protein kinase 1 and 2 (MEK) is phosphorylated which in turn activates further downstream substrates.25 Of particular interest for our work is the signal-ing cascade FGF19/FGFR4/ERK1/2. ERK1/2 phosphorylated by MEK binds and phosphorylates the ETS domain contain-ing transcription factor Elk-1. Phosphorylated Elk-1 in turn binds to the ETS site in the promoter thereby regulating the transcript ional activity of target genes. Activation of Elk-1 was found to suppress promoter activity by recruiting core-pressors and histone deacetylases, yet other pathways are not excluded.29–30 Conceivably, a similar mechanism could be responsible for the inhibition of APOA expression.
Because recombinant mouse FGF15 is unstable, we used recombinant human FGF19 for our studies. As shown in Figure 1, recombinant human FGF19 at a concentration of 40 ng/mL reduced APOA mRNA abundance by 40% in primary hepatocytes isolated from tg- APOA mice. This downregula-tion was time- dependent and reached maximal levels at 48 hours. The addition of the FXR ligand CA at a concentration of 200 mol/L downregulated APOA mRNA abundance by
additional 47% indicating that 2 FXR- mediated mechanisms act in parallel. FGF19 on the other hand did not affect the Shp mRNA expression in our experiments indicating that the FGF19 effect on APOA was small heterodimer partner inde-pendent. This is further corroborated by our observation that downregulation of baseline levels of Shp by specific siRNA did not influence APOA mRNA abundance (Figure V in the online- only Data Supplement).
FGF15/19-FGFR4 signaling is suggested to involve either one of the ERK1/2-JNK2 or P38 MAPK path-ways.25 To find out which of these MAP- kinases might be involved in FGF19-mediated APOA repression, primary hepatocytes from tg- APOA mice were preincubated for 1 hour with specific MAPK inhibitors followed by FGF19 incubation for 24 hours. The results displayed in Figure 3 proved that the effects of FGF19 on APOA repression was ERK1/2-mediated. FGF19-activated MEK/ERK1/2 in turn phosphorylated the downstream transcription factor Elk-1 (Figure 6A). Phosphorylated Elk-1 then suppressed APOA expression by binding to an Ets-1 motif in the promoter. Luciferase reporter assays using full-length (21952/152) and several 5 truncations of the APOA promoter revealed that the regulatory sequence responding to FGF19-mediated signaling resides in the region 21630/21615bp with the Ets-1 motif GGAT. Electrophoretic mobility shift assays and ChIP assays provide a final proof that the transcription factor Elk-1 binds the Ets-1 element at 21630/21615bp region of human APOA promoter and is responsible for the FGF19-mediated repression of APOA transcription. There is however 1 caveat that needs to be considered: The results described here were obtained in a heterolo-gous system, transgenic mice expressing human gene, and confirmation by studies in the human system will be neces-sary. Although we are working on this we were not able so far to get satisfactory results in primary human liver cell cultures because they quickly lose the expressional activity of APOA.
Taken together we propose a dual regulatory mechanism of transcriptional APOA suppression by FXR signaling. One pathway operates via competition of activated FXR binding with HNF4a binding to the DR-1 element at 2826/2814bp.12
The second pathway involves FGF15/19 binding to the FGFR4 on liver cells, the ERK1/2 phosphorylation cascade, and Elk-1 binding to its motif at 21630/21615bp. We believe that these findings may serve as a basis for strategies in developing Lp(a) lowering drugs.
acknowledgmentsThe technical assistance of Anton Ibovnik is appreciated.
sources of FundingThis work was supported by the Medical University of Graz, PhD pro gram “Molecular Medicine,” the Austrian Science Fund FWF (SFB-LIPOTOX F3008 and DK-MCD W 1226), and the Investigator initiated Study Grant from MSD # 37997.
disclosuresNone.
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

Chennamsetty et al FGF19 inhibits aPOa expression 1227
references 1. Kostner GM, Avogaro P, Cazzolato G, Marth E, Bittolo- Bon G, Qunici GB.
Lipoprotein Lp(a) and the risk for myocardial infarction. Atherosclerosis. 1981;38:51–61.
2. Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel- Nitschke P, Perret C, DeSuremain M, Meitinger T, Wright BJ, Preuss M, Balmforth AJ, Ball SG, Meisinger C, Germain C, Evans A, Arveiler D, Luc G, Ruidavets JB, Morrison C, van der Harst P, Schreiber S, Neureuther K, Schafer A, Bugert P, El Mokhtari NE, Schrezenmeir J, Stark K, Rubin D, Wichmann HE, Hengstenberg C, Ouwehand W, Ziegler A, Tiret L, Thompson JR, Cambien F, Schunkert H, Samani NJ. Genome- wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285.
3. Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M. Genetic variants associ-ated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528.
4. Kamstrup PR, Tybjaerg- Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339.
5. Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, Danesh J. Apolipoprotein(a) isoforms and the risk of vascu-lar disease: systematic review of 40 studies involving 58 000 participants. J Am Coll Cardiol. 2010;55:2160–2167.
6. Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg- Hansen A. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853.
7. Kostner KM, Kostner GM. Therapy of hyper-Lp(a). Handb Exp Pharmacol. 2005:519–536.
8. Hobbs HH, White AL. Lipoprotein(a): intrigues and insights. Curr Opin Lipidol. 1999;10:225–236.
9. Gaubatz JW, Heideman C, Gotto AM. Morrisett JD, Dahlen GH. Human plasma lipoprotein [a]. Structural properties. J Biol Chem. 1983;258:4582–4589.
10. Krempler F, Kostner GM, Bolzano K, Sandhofer F. Turnover of lipoprotein(a) in man. J Clin Invest. 1980;65:1483–1490.
11. Rader DJ, Cain W, Ikewaki K, Talley G, Zech LA, Usher D, Brewer HB, Brewer HB. The inverse association of plasma lipoprotein(a) concen-trations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest. 1994;93:2758–2763.
12. Chennamsetty I, Claudel T, Kostner KM, Baghdasaryan A, Kratky D, Levak- Frank S, Frank S, Gonzalez FJ, Trauner M, Kostner GM. Farnesoid X receptor represses hepatic human APOA gene expression. J Clin Invest. 2011;121:3724–3734.
13. Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA. Definition of a novel growth factor- dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591.
14. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225.
15. Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, Foster J, Liang J, Brush J, Gu Q, Hillan K, Goddard A, Gurney AL. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine. 1999;11:729–735.
16. Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–2672.
17. Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7 alpha- hydroxylase gene expression. Hepatology. 2009;49: 297–305.
18. Frazer KA, Narla G, Zhang JL, Rubin EM. The apolipoprotein(a) gene is regulated by sex hormones and acute- phase inducers in YAC transgenic mice. Nat Genet. 1995;9:424–431.
19. Fernandez- Alvarez A, Soledad Alvarez M, Cucarella C, Casado M. Characterization of the human insulin- induced gene 2 (INSIG2) promoter: the role of Ets- binding motifs. J Biol Chem. 2010;285:11765–11774.
20. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: pro-moter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942.
21. Liao J, Hodge C, Meyer D, Ho PS, Rosenspire K, Schwartz J. Growth hormone regulates ternary complex factors and serum response fac-tor associated with the c- fos serum response element. J Biol Chem. 1997;272:25951–25958.
22. Yang SH, Whitmarsh AJ, Davis RJ, Sharrocks AD. Differential targeting of MAP kinases to the ETS- domain transcription factor Elk-1. The EMBO journal. 1998;17:1740–1749.
23. Hancock MA, Boffa MB, Marcovina SM, Nesheim ME, Koschinsky ML. Inhibition of plasminogen activation by lipoprotein(a): critical domains in apolipoprotein(a) and mechanism of inhibition on fibrin and degraded fibrin surfaces. J Biol Chem. 2003;278:23260–23269.
24. Tsimikas S, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, Sandhu MS, Miller ER, Benessiano J, Tedgui A, Witztum JL, Khaw KT, Boekholdt SM. Oxidation- specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56: 946–955.
25. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253.
26. Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell- Braxton L, French D, Stewart TA. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143: 1741–1747.
27. Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino- Powell K, Xu HE, Shulman GI, Kliewer SA, Mangelsdorf DJ. FGF19 as a postprandial, insulin- independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624.
28. Wang H, Venkatesh M, Li H, Goetz R, Mukherjee S, Biswas A, Zhu L, Kaubisch A, Wang L, Pullman J, Whitney K, Kuro- o M, Roig AI, Shay JW, Mohammadi M, Mani S. Pregnane X receptor activation induces FGF19-dependent tumor aggressiveness in humans and mice. The J Clin Invest. 2011;121:3220–3232.
29. Buchwalter G, Gross C, Wasylyk B. Ets ternary complex transcription factors. Gene. 2004;324:1–14.
30. Yang SH, Vickers E, Brehm A, Kouzarides T, Sharrocks AD. Temporal recruitment of the mSin3 A- histone deacetylase corepressor com-plex to the ETS domain transcription factor Elk-1. Mol Cell Biol. 2001;21:2802–2814.
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from

FGF19 inhibits APOA expression Supplemental data 1
Supplemental Material:
Cell cultures.
Mouse primary hepatocytes were prepared from 7-9-week old tg-APOA mice. The
mouse liver was perfused with collagenase solution and liver cells were collected. After
filtration and centrifugation, the isolated hepatocytes were resuspended in DMEM
(Gibco, Invitrogen, Lofer, Austria) supplemented with 20% (v/v) FCS (Sigma-Aldrich
Chemie GmbH, Vienna, Austria), 100 units/ml penicillin, and 100 units/ml streptomycin,
and plated in 6-well collagen-coated dishes (BD Biosciences, Erembodegem, Belgium)
at a density of 1 x 105 cells/well at 37°C in an atmosphere of 5% CO2 for 4 h.
Thereafter, cells were cultured in DMEM supplemented with 10% FCS, 100 units/ml
penicillin/streptomycin for 16 h. Hepatocyte viability was monitored before plating by
trypan blue exclusion, more than 85% of cells were consistently viable. Experiments
were performed in serum-free DMEM supplemented with various concentrations of
FGF19 (R&D Systems) for 24 h and harvested for RNA analysis. In some experiments
cells were pre-treated with various kinase inhibitors for 1 h before the incubation with
FGF19. Primary hepatocytes isolated from tg-APOA/Fxr-/- mice were treated with
FGF19 for 24 h and harvested for RNA analysis.
HepG2 cells were obtained from the American Type Culture Collection (Rockville,
Maryland). The cells were maintained in DMEM containing 10% FCS, 100 units/ml
penicillin/streptomycin.
RNA extraction, reverse transcription and real-time PCR.
Total RNA from cells and mouse tissues were isolated using Trizol (Invitrogen, Lofer,
Austria) according to the manufacturer’s protocol. Quantitative real-time PCR was

FGF19 inhibits APOA expression Supplemental data 2
performed on a Light Cycler 480 instrument (Roche Diagnostics, Mannheim, Germany)
using the QuantiFastTM SYBR® Green PCR Kit (Qiagen, Hilden, Germany). Primer
sequences were identical to those published previously 1. The gene expression values
were normalized to cyclophilin-A as a housekeeping gene. The data were analyzed by
the public domain program Relative Expression Software Tool – REST 2. Values are
presented as mean ± SEM.
Immunoblotting.
Equivalent amounts of protein homogenates were resolved by SDS-PAGE, transferred
to a nitrocellulose membrane, and probed with rabbit polyclonal antibodies to human
APOA (1:1250). Antibodies against SHP, Elk-1 and β-actin were from Santa Cruz
Biotechnology (Santa Cruz, CA). Antibodies against ERK1/2, phospho-ERK1/2, JNK,
phospho-JNK, p38, phospho-p38, phospho-Elk-1 were from Cell Signaling Technology
(Beverly, MA). The immunoblots were visualized by PierceR ECL chemiluminescence
detection system (Thermo Scientific, Rockford, IL, USA). Densitometric analysis of the
gels was carried out using ImageJ software.
siRNA transfections.
In siRNA experiments, mouse primary hepatocytes were transfected with 100 nM
human synthetic predesigned short interfering RNA (siRNA) targeting FGFR4, SHP or
non silencing siRNA (control) (Qiagen, Maryland, USA) using Hi-PerFect transfection
reagent (Qiagen, Maryland, USA) following the manufacturer’s recommended protocol.
Cells were then treated with FGF19 for 24 h and total RNA was prepared for real-time
quantitative PCR analysis. SHP protein levels were analyzed by western blotting.

FGF19 inhibits APOA expression Supplemental data 3
References:
1. Chennamsetty I, Claudel T, Kostner KM, Baghdasaryan A, Kratky D, Levak-
Frank S, Frank S, Gonzalez FJ, Trauner M, Kostner GM. Farnesoid X receptor
represses hepatic human APOA gene expression. J Clin Invest. 2011;121:3724-3734.
2. Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for
group-wise comparison and statistical analysis of relative expression results in real-time
PCR. Nucleic Acids Res. 2002;30:e36.

FGF19 inhibits APOA expression Supplemental data 4
Supplemental Figure I
APOA secretion from mouse primary hepatocytes:
Primary hepatocytes from tg-APOA mice were incubated for 24 h with 40 ng/ml of
FGF19 or with vehicle. APOA levels in the medium were analyzed by DELFIA and
expressed as mean ± SD from three independent experiments. ***p 0.001 when
compared to vehicle treated control group.
FGF19 inhibits APOA expression Supplemental data 5
Supplemental Figure II
Effect of FGF19 on the expression of Cyp7a1.
Primary hepatocytes from tg-APOA mice were incubated for 24 h with increasing
concentrations of FGF19 (40 and 80 ng/ml) or vehicle. mRNA levels of known FGF19
target gene Cyp7a1 was analyzed as a positive control by real-time quantitative PCR.
Results represent means ± SEM (*** p 0.001).

FGF19 inhibits APOA expression Supplemental data 6
Supplemental Figure III
Effect of FGF19 on the expression of APOA in primary hepatocytes from tg-APOA/fxr-/-
mice.
Primary hepatocytes from tg-APOA/fxr-/- mice were incubated with FGF19 (40 ng/ml) or
vehicle for 24 h. APOA mRNA levels were analyzed by real-time quantitative PCR.
Results represent means ± SEM (** p 0.01).

FGF19 inhibits APOA expression Supplemental data 7
Supplemental Figure IV
Effect of FGF19 on Shp expression.
Primary hepatocytes from tg-APOA mice were incubated for 24 h with increasing
concentrations of FGF19 (40 and 80 ng/ml) or vehicle. (A) mRNA levels of Shp were
analyzed by real-time quantitative PCR. Results represent means ± SEM of three
independent experiments (*** p 0.001). (B) Western blot analysis of Shp protein
expression in whole cell lysates from hepatocytes treated for 24 h with increasing
concentrations of FGF19. β-actin expression was used as loading control.

FGF19 inhibits APOA expression Supplemental data 8
Supplemental Figure V
Silencing of SHP does not affect APOA expression.
Primary mouse hepatocytes were transfected with 100 nM siRNA targeting SHP and
control siRNA using hi-perfect transfection reagent and subsequently treated with or
without FGF19 (40 ng/ml) for 24 h. (A) SHP protein expression in whole cell lysates was
analyzed by Western blotting. (B) mRNA levels of APOA in primary hepatocytes treated
with siRNA against SHP relative to control siRNA treatment. Data are expressed as
means ± SEM (** p 0.01).

FGF19 inhibits APOA expression Supplemental data 9
Supplemental Figure VI
Knockdown of FGFR4 abolished FGF19 effect on APOA expression.
Primary mouse hepatocytes were transfected with 100 nM siRNA targeting FGFR4 and
control siRNA using hi-perfect transfection reagent and subsequently treated with or
without FGF19 (40 ng/ml) for 24 h. mRNA levels of APOA were analyzed relative to
vehicle treated control siRNA treatment. Data are expressed as mean ± SEM (** p
0.01).

KostnerIndumathi Chennamsetty, Thierry Claudel, Karam M. Kostner, Michael Trauner and Gert M.
Gene ExpressionAPOAFGF19 Signaling Cascade Suppresses
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2012 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.111.2430552012;
2012;32:1220-1227; originally published online January 19,Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/32/5/1220World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2012/01/19/ATVBAHA.111.243055.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at STANFORD UNIVERSITY MEDICAL CE on March 12, 2015http://atvb.ahajournals.org/Downloaded from





























