Embed Size (px)
Citation preview

Exploring Versatile Sulfhydryl Chemistry in the Chain End of aSynthetic PolylactideMijanur Rahaman Molla and Suhrit Ghosh*
Polymer Science Unit, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India
*S Supporting Information
ABSTRACT: Synthesis of an end-functionalized polylactideby ring-opening polymerization of lactide monomer using afunctional initiator containing pyridyl disulfide group isreported. Molecular weight of the polymer determined byGPC matched very well with that determined by end-groupanalysis using the UV/vis method, suggesting survival of theend-group functionality during polymerization. DTT-inducedreduction of the pyridyl disulfide group produced freesulfhydryl group quantitatively which was utilized for versatilechain-end modifications using various thiol-mediated high-yielding chemical transformations including thiol−ene, thiol−maleimide, and thiol−acrylate “click” reactions. This strategy was further extended to link two macromolecules by reaction ofsulfhydryl-functionalized polylactide and acrylate-terminated poly(ethylene oxide) (PEO) which produced a block copolymerwith an acid-labile β-thiopropionate linker between the two constituent blocks. This functional group could be cleaved undermild acidic condition to produce the individual parent polymers. Further as-synthesized pyridyl disulfide-terminated polylactidewas treated with thiol-functionalized sugar moiety and n-type semiconducting naphthalene diimide (NDI) chromophore whichalso generated quantitative chain-end functionalization by thiol−disulfide exchange reaction. NDI-functionalized polylactideshowed white light emission due to mixed emission from monomeric and excimer-type species. Further atomic force microscopic(AFM) studies revealed NDI-functionalized polymer formed uniform spherical aggregates upon drying of a drop-casted film onsilicon surface possibly due to solvent-evaporation-induced defined organization of the polymer chain dictated by strong π-stacking interaction among the NDI chromophores.
■ INTRODUCTION
Synthetic polymers that contain highly reactive functionalgroups1 either as pendant2 or in the chain end3 have beenstudied extensively in the recent past owing to the possibility ofgenerating structurally diverse functional polymers and variouspolymeric conjugates by high-yielding postpolymerizationchemical transformations. Postpolymerization end-group mod-ification is particularly challenging because it demands survivalof the chain-end functional group during polymerizationcondition, and at the same time the reactivity of the functionalgroup should be high to allow quantitative transformation afterpolymerization. However, this has been successfully achieved4
in various controlled chain polymerization reactions by utilizingsuitably designed functional initiators. In this context sulfhydrylgroup has been studied extensively owing to its well-establishedhigh fidelity reactions5 with number of functional groups suchas maleimide, isocyanate, alkene, alkyne, acrylate, halogenatedalkane, disulfides, and so forth. Mainly two different strategieshave been adopted for chain-end modifications using sulfhydrylchemistry. One is to generate a thiol group in the terminal of apolymer synthesized by mostly RAFT route by postpolymeriza-tion reduction of the macro-chain-transfer agent using suitablereagents6 and subsequently utilizing the free sulfhydrylfunctionality for conjugation with various small molecules andmacromolecules by different thiol-mediated reactions. The
other approach includes controlled/living chain polymerizationusing functional initiators containing a thiol-reactive function-ality such as maleimide or derivatives7 in protected form andmodification of chain end by postpolymerization reactions withdesired molecules, macromolecules, or biomacromoleculescontaining free thiol functionality. Pyridyl disulfide is aninteresting option in this context because of the following: (i)It can react with sulfhydryl groups quantitatively under verymild conditions in range of solvents starting from chloroform towater and generates a redox-sensitive disulfide bond.8 (ii) It canalso quantitatively and spontaneously generate a thiol group inthe presence of a reducing agent such as dithiothreitol (DTT).It is noteworthy that generation of thiol functionality byreducing macro-CTA in RAFT-generated polymers often leadsdisulfide bond formation during the course of the reactionitself.6 But generating thiol by cleavage of pyridyl disulfidefunctionality is not associated with this problem due to thepresence of DTT in the reaction mixture. (iii) The byproductof both these above-mentioned reactions, 2-pyridinethione, ishighly water-soluble and thus can be removed easily fromreaction medium. Further, it also exhibits a signature absorption
Received: October 11, 2012Revised: October 20, 2012Published: October 29, 2012
Article
pubs.acs.org/Macromolecules
© 2012 American Chemical Society 8561 dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−8570
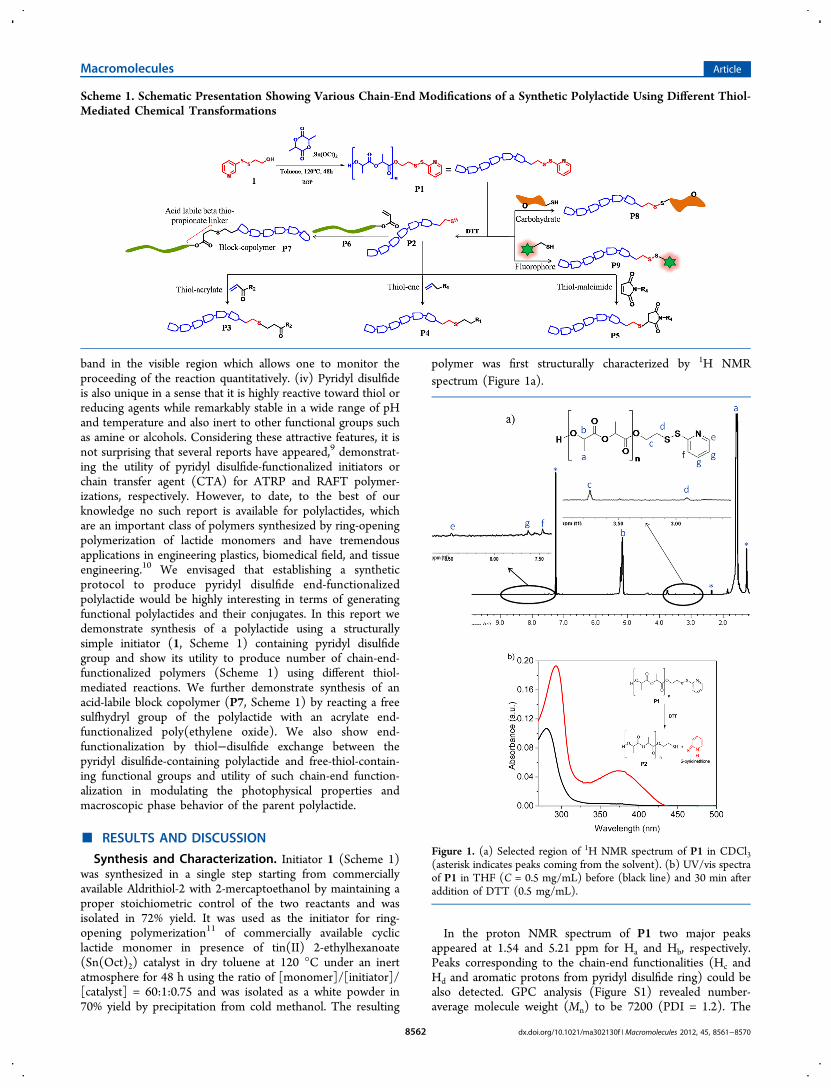
band in the visible region which allows one to monitor theproceeding of the reaction quantitatively. (iv) Pyridyl disulfideis also unique in a sense that it is highly reactive toward thiol orreducing agents while remarkably stable in a wide range of pHand temperature and also inert to other functional groups suchas amine or alcohols. Considering these attractive features, it isnot surprising that several reports have appeared,9 demonstrat-ing the utility of pyridyl disulfide-functionalized initiators orchain transfer agent (CTA) for ATRP and RAFT polymer-izations, respectively. However, to date, to the best of ourknowledge no such report is available for polylactides, whichare an important class of polymers synthesized by ring-openingpolymerization of lactide monomers and have tremendousapplications in engineering plastics, biomedical field, and tissueengineering.10 We envisaged that establishing a syntheticprotocol to produce pyridyl disulfide end-functionalizedpolylactide would be highly interesting in terms of generatingfunctional polylactides and their conjugates. In this report wedemonstrate synthesis of a polylactide using a structurallysimple initiator (1, Scheme 1) containing pyridyl disulfidegroup and show its utility to produce number of chain-end-functionalized polymers (Scheme 1) using different thiol-mediated reactions. We further demonstrate synthesis of anacid-labile block copolymer (P7, Scheme 1) by reacting a freesulfhydryl group of the polylactide with an acrylate end-functionalized poly(ethylene oxide). We also show end-functionalization by thiol−disulfide exchange between thepyridyl disulfide-containing polylactide and free-thiol-contain-ing functional groups and utility of such chain-end function-alization in modulating the photophysical properties andmacroscopic phase behavior of the parent polylactide.
■ RESULTS AND DISCUSSION
Synthesis and Characterization. Initiator 1 (Scheme 1)was synthesized in a single step starting from commerciallyavailable Aldrithiol-2 with 2-mercaptoethanol by maintaining aproper stoichiometric control of the two reactants and wasisolated in 72% yield. It was used as the initiator for ring-opening polymerization11 of commercially available cycliclactide monomer in presence of tin(II) 2-ethylhexanoate(Sn(Oct)2) catalyst in dry toluene at 120 °C under an inertatmosphere for 48 h using the ratio of [monomer]/[initiator]/[catalyst] = 60:1:0.75 and was isolated as a white powder in70% yield by precipitation from cold methanol. The resulting
polymer was first structurally characterized by 1H NMRspectrum (Figure 1a).
In the proton NMR spectrum of P1 two major peaksappeared at 1.54 and 5.21 ppm for Ha and Hb, respectively.Peaks corresponding to the chain-end functionalities (Hc andHd and aromatic protons from pyridyl disulfide ring) could bealso detected. GPC analysis (Figure S1) revealed number-average molecule weight (Mn) to be 7200 (PDI = 1.2). The
Scheme 1. Schematic Presentation Showing Various Chain-End Modifications of a Synthetic Polylactide Using Different Thiol-Mediated Chemical Transformations
Figure 1. (a) Selected region of 1H NMR spectrum of P1 in CDCl3(asterisk indicates peaks coming from the solvent). (b) UV/vis spectraof P1 in THF (C = 0.5 mg/mL) before (black line) and 30 min afteraddition of DTT (0.5 mg/mL).
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708562

polymer was also characterized by FT-IR spectra (Figure S2)wherein a characteristic sharp peak was noticed at 1752 cm−1
due to the carbonyl stretching of the backbone ester in contrastto the peak at 1765 cm−1 for the carbonyl of the cyclic estermonomer.To generate free thiol group in the chain end P1 was treated
with a reducing agent DTT, and the reaction was monitored byUV/vis spectroscopy (Figure 1b). The absorption spectrum ofP1 in THF showed a peak at 280 nm, confirming the presenceof the pyridyl disulfide functional group.9b It was then addedwith DTT and stirred for 30 min while a yellow color appeared,suggesting generation of 2-pyridinethione (Figure 1b) which isthe byproduct of the cleavage reaction. Consequently, in theUV/vis spectrum of the reaction mixture a new peak appearedwith λmax = 380 nm due to 2-pyridinethione. The extinctioncoefficient of 2-pyridinethione in THF was independentlyestimated to be 900 M−1 cm−1 at 380 nm (Figure S3), whichwas used to estimate the concentration of released 2-pyridinethione during conversion of P1 to P2 (Figure 1b).Utilizing this data and assuming DTT induced cleavagereaction to be quantitative, the molecular weight of thepolymer which was estimated to be 8900, which is in reasonablegood agreement with that found from the GPC analysis andprovides further strong support in favor of survival of the chain-end functionality (unsymmetrical pyridyl disulfide) during thepolymerization reaction. As the GPC analysis was done using aconventional calibration curve with respect to PMMA standard,it may not provide most accurate data for the molecular weight.Thus, for all studies polymer concentration was calculated usingmolecular weight determined by end-group analysis using theUV/vis method as described above.Chain-End Modification of P2 Using Various Thiol-
Reactive Chromophores. To test the possibility of variouschain-end modifications, trademark thiol-mediated reactions(Scheme 2) were attempted with polymer P2 which contains afree sulfhydryl group in the chain end.Quantification of functional groups present in the terminal of
a polymer chain with reasonably high molecular weight isalways a difficult task due to lack of appropriate experimentaltool. To overcome this problem in this study, we intentionallysynthesized various thiol reactive building blocks containing π-
conjugated chromophores so that their attachment to thepolymer chain could be traced by optical spectroscopic studieseven at relatively low concentration. First, thiol−acrylateMichael addition reaction was attempted with P2 andcompound 2 (Scheme 2) using Me2PPh as catalyst. Theresulting polymer P3 (Scheme 2) was isolated by precipitationfrom cold diethyl ether to ensure complete removal of anyunreacted/excess compound 2 or catalyst and was isolated inalmost quantitative yield.
1H NMR of P3 (Figure 2a) revealed peaks in the aromaticregion (7.2 −8.0 ppm) indicating incorporation of the naphthylgroup. The UV/vis absorption spectrum of P3 was comparedwith that of compound 2 while a sharp absorption band withλmax = 323 nm (Figure 2b) was noticed for both samples furtherconfirming attachment of the naphthyl chromophore in thepolymer chain. Photoluminescence spectrum of P3 (Figure 2b)also showed characteristic emission of naphthyl chromophore.To estimate the extent of functionalization, molecular weight ofP3 was calculated by end-group analysis using UV/vis datashown in Figure 2b. The extinction coefficient of 2 in THF wasindependently estimated to be 3536 M−1 cm−1, which indicatedconcentration of naphthyl chromophore was 0.053 mM in thepolymer solution. Assuming that each polymer chain containsone naphthyl group, it can be stated that [naphthyl] =[polymer]. It means 0.5 mg/mL = 0.053 mM, whichcorresponds to a molecular weight of 9363 for P3, whichcorroborates well with its theoretically calculated molecularweight of 8987 (molecular weight of P1 by UV/vis −pyridinethione + 2). One can expect this to happen only ifconversion of P2 to P3 is quantitative. Further thiol−ene andthiol−maleimide reactions were also tested with P2 withcompounds 3 and 4 (Scheme 2) which generated two moreend-functionalized polymers P4 and P5, respectively. In bothoccasions incorporation of the naphthyl chromophore wasconfirmed by detecting aromatic signals in their proton NMR(Figures S4 and S5) and also by UV/vis and emissionspectroscopy (Figure S6). Molecular weights of P4 and P5were estimated to be 9487 and 9074, respectively, by end-groupanalysis using the same method described above for P3. In bothcases they nearly matched with theoretically calculated values of8973 (molecular weight of P1 by UV/vis − pyridinethione + 3)
Scheme 2. Schematic Presentation Showing Various Thiol-Mediated Transformations of P2
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708563
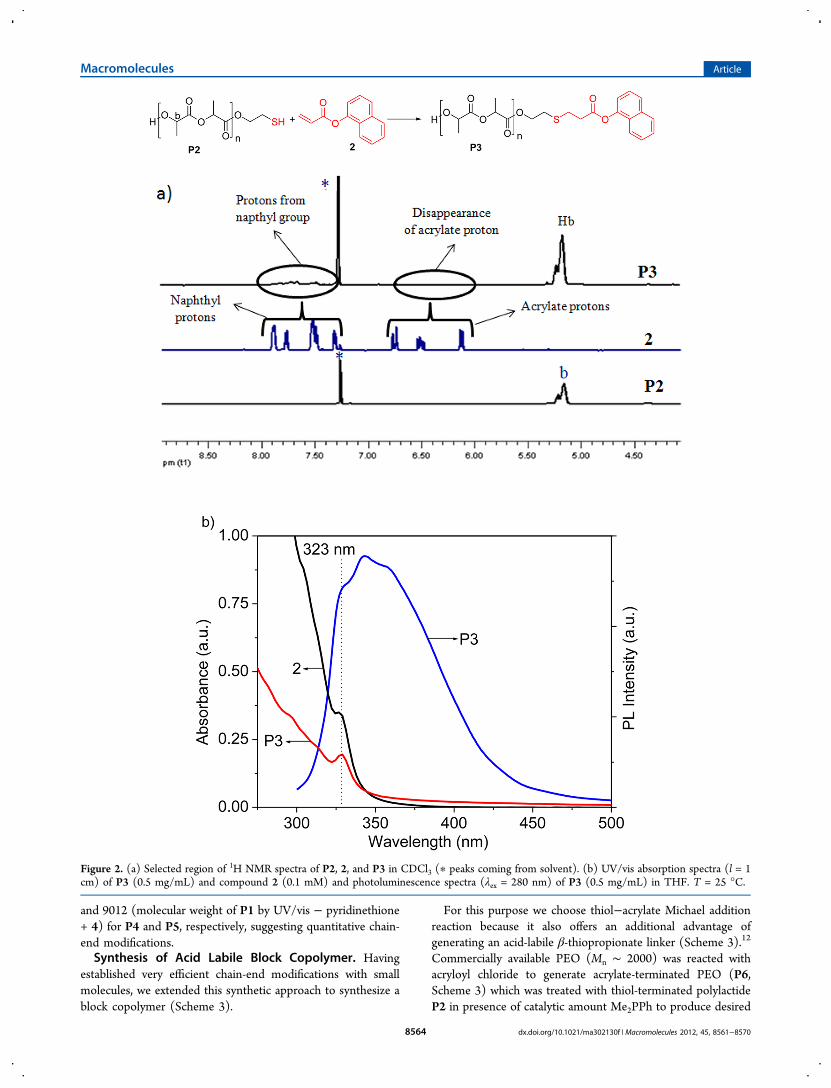
and 9012 (molecular weight of P1 by UV/vis − pyridinethione+ 4) for P4 and P5, respectively, suggesting quantitative chain-end modifications.Synthesis of Acid Labile Block Copolymer. Having
established very efficient chain-end modifications with smallmolecules, we extended this synthetic approach to synthesize ablock copolymer (Scheme 3).
For this purpose we choose thiol−acrylate Michael additionreaction because it also offers an additional advantage ofgenerating an acid-labile β-thiopropionate linker (Scheme 3).12
Commercially available PEO (Mn ∼ 2000) was reacted withacryloyl chloride to generate acrylate-terminated PEO (P6,Scheme 3) which was treated with thiol-terminated polylactideP2 in presence of catalytic amount Me2PPh to produce desired
Figure 2. (a) Selected region of 1H NMR spectra of P2, 2, and P3 in CDCl3 (∗ peaks coming from solvent). (b) UV/vis absorption spectra (l = 1cm) of P3 (0.5 mg/mL) and compound 2 (0.1 mM) and photoluminescence spectra (λex = 280 nm) of P3 (0.5 mg/mL) in THF. T = 25 °C.
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708564
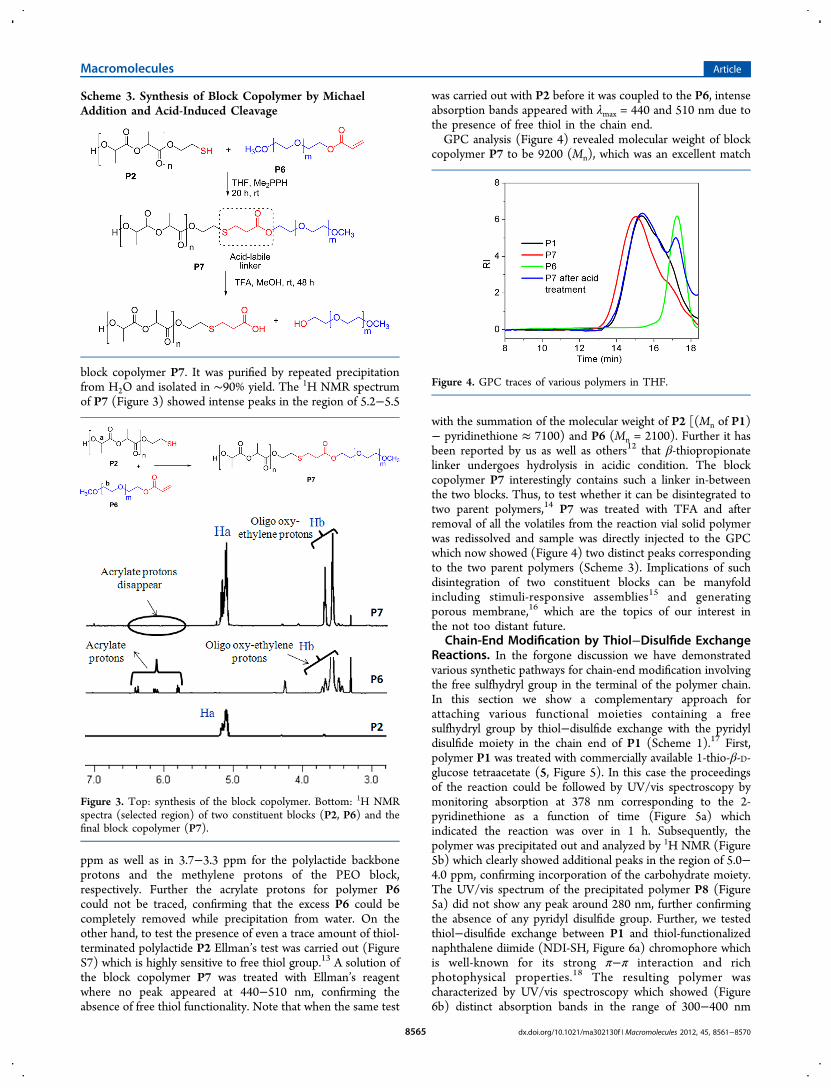
block copolymer P7. It was purified by repeated precipitationfrom H2O and isolated in ∼90% yield. The 1H NMR spectrumof P7 (Figure 3) showed intense peaks in the region of 5.2−5.5
ppm as well as in 3.7−3.3 ppm for the polylactide backboneprotons and the methylene protons of the PEO block,respectively. Further the acrylate protons for polymer P6could not be traced, confirming that the excess P6 could becompletely removed while precipitation from water. On theother hand, to test the presence of even a trace amount of thiol-terminated polylactide P2 Ellman’s test was carried out (FigureS7) which is highly sensitive to free thiol group.13 A solution ofthe block copolymer P7 was treated with Ellman’s reagentwhere no peak appeared at 440−510 nm, confirming theabsence of free thiol functionality. Note that when the same test
was carried out with P2 before it was coupled to the P6, intenseabsorption bands appeared with λmax = 440 and 510 nm due tothe presence of free thiol in the chain end.GPC analysis (Figure 4) revealed molecular weight of block
copolymer P7 to be 9200 (Mn), which was an excellent match
with the summation of the molecular weight of P2 [(Mn of P1)− pyridinethione ≈ 7100) and P6 (Mn = 2100). Further it hasbeen reported by us as well as others12 that β-thiopropionatelinker undergoes hydrolysis in acidic condition. The blockcopolymer P7 interestingly contains such a linker in-betweenthe two blocks. Thus, to test whether it can be disintegrated totwo parent polymers,14 P7 was treated with TFA and afterremoval of all the volatiles from the reaction vial solid polymerwas redissolved and sample was directly injected to the GPCwhich now showed (Figure 4) two distinct peaks correspondingto the two parent polymers (Scheme 3). Implications of suchdisintegration of two constituent blocks can be manyfoldincluding stimuli-responsive assemblies15 and generatingporous membrane,16 which are the topics of our interest inthe not too distant future.
Chain-End Modification by Thiol−Disulfide ExchangeReactions. In the forgone discussion we have demonstratedvarious synthetic pathways for chain-end modification involvingthe free sulfhydryl group in the terminal of the polymer chain.In this section we show a complementary approach forattaching various functional moieties containing a freesulfhydryl group by thiol−disulfide exchange with the pyridyldisulfide moiety in the chain end of P1 (Scheme 1).17 First,polymer P1 was treated with commercially available 1-thio-β-D-glucose tetraacetate (5, Figure 5). In this case the proceedingsof the reaction could be followed by UV/vis spectroscopy bymonitoring absorption at 378 nm corresponding to the 2-pyridinethione as a function of time (Figure 5a) whichindicated the reaction was over in 1 h. Subsequently, thepolymer was precipitated out and analyzed by 1H NMR (Figure5b) which clearly showed additional peaks in the region of 5.0−4.0 ppm, confirming incorporation of the carbohydrate moiety.The UV/vis spectrum of the precipitated polymer P8 (Figure5a) did not show any peak around 280 nm, further confirmingthe absence of any pyridyl disulfide group. Further, we testedthiol−disulfide exchange between P1 and thiol-functionalizednaphthalene diimide (NDI-SH, Figure 6a) chromophore whichis well-known for its strong π−π interaction and richphotophysical properties.18 The resulting polymer wascharacterized by UV/vis spectroscopy which showed (Figure6b) distinct absorption bands in the range of 300−400 nm
Scheme 3. Synthesis of Block Copolymer by MichaelAddition and Acid-Induced Cleavage
Figure 3. Top: synthesis of the block copolymer. Bottom: 1H NMRspectra (selected region) of two constituent blocks (P2, P6) and thefinal block copolymer (P7).
Figure 4. GPC traces of various polymers in THF.
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708565
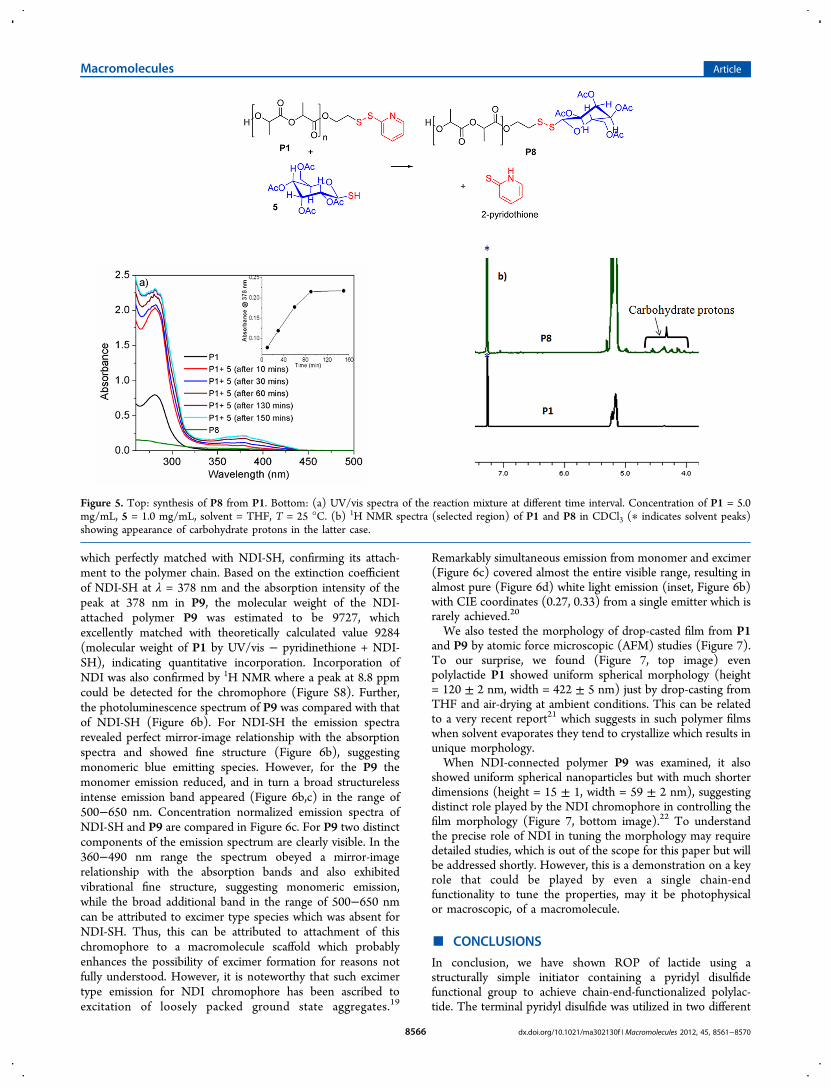
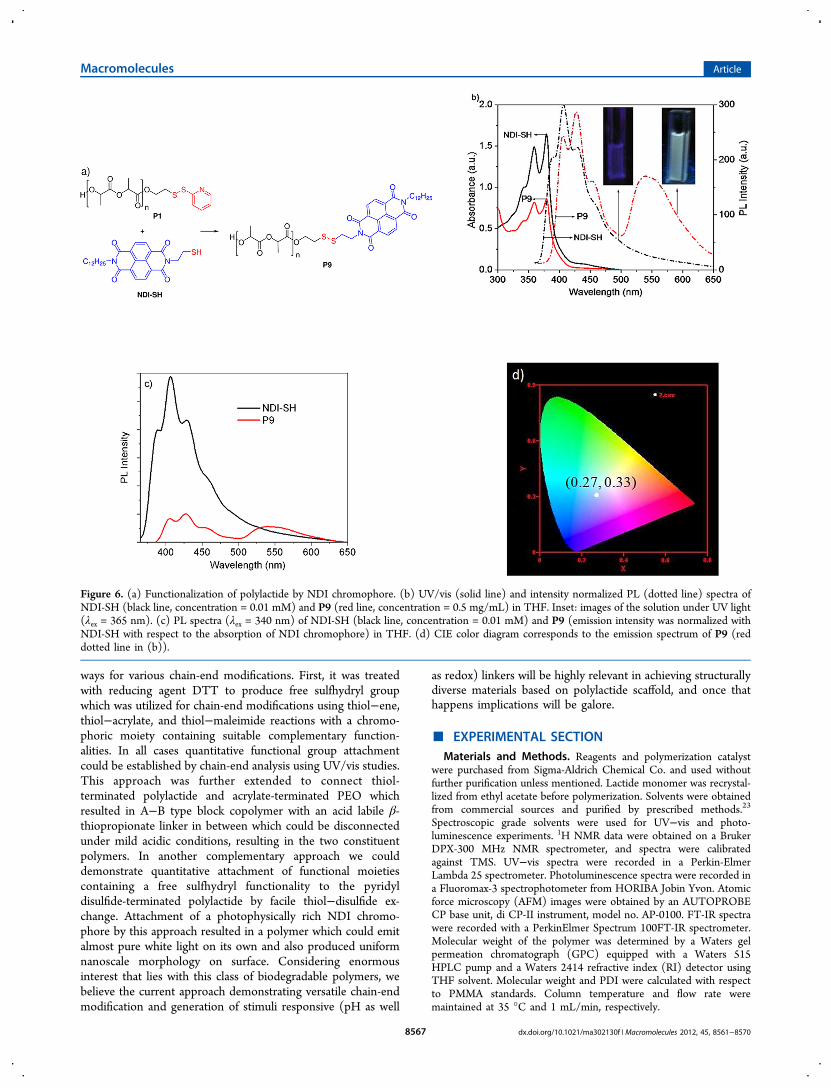
which perfectly matched with NDI-SH, confirming its attach-ment to the polymer chain. Based on the extinction coefficientof NDI-SH at λ = 378 nm and the absorption intensity of thepeak at 378 nm in P9, the molecular weight of the NDI-attached polymer P9 was estimated to be 9727, whichexcellently matched with theoretically calculated value 9284(molecular weight of P1 by UV/vis − pyridinethione + NDI-SH), indicating quantitative incorporation. Incorporation ofNDI was also confirmed by 1H NMR where a peak at 8.8 ppmcould be detected for the chromophore (Figure S8). Further,the photoluminescence spectrum of P9 was compared with thatof NDI-SH (Figure 6b). For NDI-SH the emission spectrarevealed perfect mirror-image relationship with the absorptionspectra and showed fine structure (Figure 6b), suggestingmonomeric blue emitting species. However, for the P9 themonomer emission reduced, and in turn a broad structurelessintense emission band appeared (Figure 6b,c) in the range of500−650 nm. Concentration normalized emission spectra ofNDI-SH and P9 are compared in Figure 6c. For P9 two distinctcomponents of the emission spectrum are clearly visible. In the360−490 nm range the spectrum obeyed a mirror-imagerelationship with the absorption bands and also exhibitedvibrational fine structure, suggesting monomeric emission,while the broad additional band in the range of 500−650 nmcan be attributed to excimer type species which was absent forNDI-SH. Thus, this can be attributed to attachment of thischromophore to a macromolecule scaffold which probablyenhances the possibility of excimer formation for reasons notfully understood. However, it is noteworthy that such excimertype emission for NDI chromophore has been ascribed toexcitation of loosely packed ground state aggregates.19
Remarkably simultaneous emission from monomer and excimer(Figure 6c) covered almost the entire visible range, resulting inalmost pure (Figure 6d) white light emission (inset, Figure 6b)with CIE coordinates (0.27, 0.33) from a single emitter which israrely achieved.20
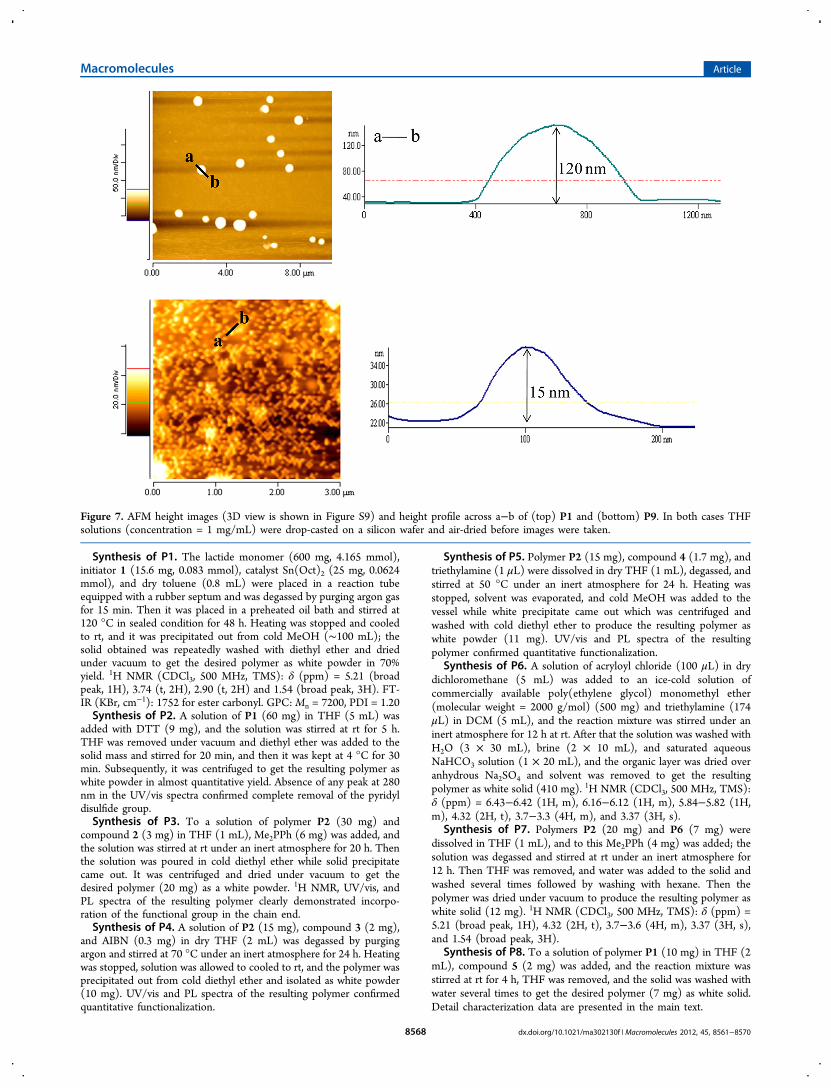
We also tested the morphology of drop-casted film from P1and P9 by atomic force microscopic (AFM) studies (Figure 7).To our surprise, we found (Figure 7, top image) evenpolylactide P1 showed uniform spherical morphology (height= 120 ± 2 nm, width = 422 ± 5 nm) just by drop-casting fromTHF and air-drying at ambient conditions. This can be relatedto a very recent report21 which suggests in such polymer filmswhen solvent evaporates they tend to crystallize which results inunique morphology.When NDI-connected polymer P9 was examined, it also
showed uniform spherical nanoparticles but with much shorterdimensions (height = 15 ± 1, width = 59 ± 2 nm), suggestingdistinct role played by the NDI chromophore in controlling thefilm morphology (Figure 7, bottom image).22 To understandthe precise role of NDI in tuning the morphology may requiredetailed studies, which is out of the scope for this paper but willbe addressed shortly. However, this is a demonstration on a keyrole that could be played by even a single chain-endfunctionality to tune the properties, may it be photophysicalor macroscopic, of a macromolecule.
■ CONCLUSIONS
In conclusion, we have shown ROP of lactide using astructurally simple initiator containing a pyridyl disulfidefunctional group to achieve chain-end-functionalized polylac-tide. The terminal pyridyl disulfide was utilized in two different
Figure 5. Top: synthesis of P8 from P1. Bottom: (a) UV/vis spectra of the reaction mixture at different time interval. Concentration of P1 = 5.0mg/mL, 5 = 1.0 mg/mL, solvent = THF, T = 25 °C. (b) 1H NMR spectra (selected region) of P1 and P8 in CDCl3 (∗ indicates solvent peaks)showing appearance of carbohydrate protons in the latter case.
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708566
ways for various chain-end modifications. First, it was treatedwith reducing agent DTT to produce free sulfhydryl groupwhich was utilized for chain-end modifications using thiol−ene,thiol−acrylate, and thiol−maleimide reactions with a chromo-phoric moiety containing suitable complementary function-alities. In all cases quantitative functional group attachmentcould be established by chain-end analysis using UV/vis studies.This approach was further extended to connect thiol-terminated polylactide and acrylate-terminated PEO whichresulted in A−B type block copolymer with an acid labile β-thiopropionate linker in between which could be disconnectedunder mild acidic conditions, resulting in the two constituentpolymers. In another complementary approach we coulddemonstrate quantitative attachment of functional moietiescontaining a free sulfhydryl functionality to the pyridyldisulfide-terminated polylactide by facile thiol−disulfide ex-change. Attachment of a photophysically rich NDI chromo-phore by this approach resulted in a polymer which could emitalmost pure white light on its own and also produced uniformnanoscale morphology on surface. Considering enormousinterest that lies with this class of biodegradable polymers, webelieve the current approach demonstrating versatile chain-endmodification and generation of stimuli responsive (pH as well
as redox) linkers will be highly relevant in achieving structurallydiverse materials based on polylactide scaffold, and once thathappens implications will be galore.
■ EXPERIMENTAL SECTIONMaterials and Methods. Reagents and polymerization catalyst
were purchased from Sigma-Aldrich Chemical Co. and used withoutfurther purification unless mentioned. Lactide monomer was recrystal-lized from ethyl acetate before polymerization. Solvents were obtainedfrom commercial sources and purified by prescribed methods.23
Spectroscopic grade solvents were used for UV−vis and photo-luminescence experiments. 1H NMR data were obtained on a BrukerDPX-300 MHz NMR spectrometer, and spectra were calibratedagainst TMS. UV−vis spectra were recorded in a Perkin-ElmerLambda 25 spectrometer. Photoluminescence spectra were recorded ina Fluoromax-3 spectrophotometer from HORIBA Jobin Yvon. Atomicforce microscopy (AFM) images were obtained by an AUTOPROBECP base unit, di CP-II instrument, model no. AP-0100. FT-IR spectrawere recorded with a PerkinElmer Spectrum 100FT-IR spectrometer.Molecular weight of the polymer was determined by a Waters gelpermeation chromatograph (GPC) equipped with a Waters 515HPLC pump and a Waters 2414 refractive index (RI) detector usingTHF solvent. Molecular weight and PDI were calculated with respectto PMMA standards. Column temperature and flow rate weremaintained at 35 °C and 1 mL/min, respectively.
Figure 6. (a) Functionalization of polylactide by NDI chromophore. (b) UV/vis (solid line) and intensity normalized PL (dotted line) spectra ofNDI-SH (black line, concentration = 0.01 mM) and P9 (red line, concentration = 0.5 mg/mL) in THF. Inset: images of the solution under UV light(λex = 365 nm). (c) PL spectra (λex = 340 nm) of NDI-SH (black line, concentration = 0.01 mM) and P9 (emission intensity was normalized withNDI-SH with respect to the absorption of NDI chromophore) in THF. (d) CIE color diagram corresponds to the emission spectrum of P9 (reddotted line in (b)).
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708567
Synthesis of P1. The lactide monomer (600 mg, 4.165 mmol),initiator 1 (15.6 mg, 0.083 mmol), catalyst Sn(Oct)2 (25 mg, 0.0624mmol), and dry toluene (0.8 mL) were placed in a reaction tubeequipped with a rubber septum and was degassed by purging argon gasfor 15 min. Then it was placed in a preheated oil bath and stirred at120 °C in sealed condition for 48 h. Heating was stopped and cooledto rt, and it was precipitated out from cold MeOH (∼100 mL); thesolid obtained was repeatedly washed with diethyl ether and driedunder vacuum to get the desired polymer as white powder in 70%yield. 1H NMR (CDCl3, 500 MHz, TMS): δ (ppm) = 5.21 (broadpeak, 1H), 3.74 (t, 2H), 2.90 (t, 2H) and 1.54 (broad peak, 3H). FT-IR (KBr, cm−1): 1752 for ester carbonyl. GPC: Mn = 7200, PDI = 1.20Synthesis of P2. A solution of P1 (60 mg) in THF (5 mL) was
added with DTT (9 mg), and the solution was stirred at rt for 5 h.THF was removed under vacuum and diethyl ether was added to thesolid mass and stirred for 20 min, and then it was kept at 4 °C for 30min. Subsequently, it was centrifuged to get the resulting polymer aswhite powder in almost quantitative yield. Absence of any peak at 280nm in the UV/vis spectra confirmed complete removal of the pyridyldisulfide group.Synthesis of P3. To a solution of polymer P2 (30 mg) and
compound 2 (3 mg) in THF (1 mL), Me2PPh (6 mg) was added, andthe solution was stirred at rt under an inert atmosphere for 20 h. Thenthe solution was poured in cold diethyl ether while solid precipitatecame out. It was centrifuged and dried under vacuum to get thedesired polymer (20 mg) as a white powder. 1H NMR, UV/vis, andPL spectra of the resulting polymer clearly demonstrated incorpo-ration of the functional group in the chain end.Synthesis of P4. A solution of P2 (15 mg), compound 3 (2 mg),
and AIBN (0.3 mg) in dry THF (2 mL) was degassed by purgingargon and stirred at 70 °C under an inert atmosphere for 24 h. Heatingwas stopped, solution was allowed to cooled to rt, and the polymer wasprecipitated out from cold diethyl ether and isolated as white powder(10 mg). UV/vis and PL spectra of the resulting polymer confirmedquantitative functionalization.
Synthesis of P5. Polymer P2 (15 mg), compound 4 (1.7 mg), andtriethylamine (1 μL) were dissolved in dry THF (1 mL), degassed, andstirred at 50 °C under an inert atmosphere for 24 h. Heating wasstopped, solvent was evaporated, and cold MeOH was added to thevessel while white precipitate came out which was centrifuged andwashed with cold diethyl ether to produce the resulting polymer aswhite powder (11 mg). UV/vis and PL spectra of the resultingpolymer confirmed quantitative functionalization.
Synthesis of P6. A solution of acryloyl chloride (100 μL) in drydichloromethane (5 mL) was added to an ice-cold solution ofcommercially available poly(ethylene glycol) monomethyl ether(molecular weight = 2000 g/mol) (500 mg) and triethylamine (174μL) in DCM (5 mL), and the reaction mixture was stirred under aninert atmosphere for 12 h at rt. After that the solution was washed withH2O (3 × 30 mL), brine (2 × 10 mL), and saturated aqueousNaHCO3 solution (1 × 20 mL), and the organic layer was dried overanhydrous Na2SO4 and solvent was removed to get the resultingpolymer as white solid (410 mg). 1H NMR (CDCl3, 500 MHz, TMS):δ (ppm) = 6.43−6.42 (1H, m), 6.16−6.12 (1H, m), 5.84−5.82 (1H,m), 4.32 (2H, t), 3.7−3.3 (4H, m), and 3.37 (3H, s).
Synthesis of P7. Polymers P2 (20 mg) and P6 (7 mg) weredissolved in THF (1 mL), and to this Me2PPh (4 mg) was added; thesolution was degassed and stirred at rt under an inert atmosphere for12 h. Then THF was removed, and water was added to the solid andwashed several times followed by washing with hexane. Then thepolymer was dried under vacuum to produce the resulting polymer aswhite solid (12 mg). 1H NMR (CDCl3, 500 MHz, TMS): δ (ppm) =5.21 (broad peak, 1H), 4.32 (2H, t), 3.7−3.6 (4H, m), 3.37 (3H, s),and 1.54 (broad peak, 3H).
Synthesis of P8. To a solution of polymer P1 (10 mg) in THF (2mL), compound 5 (2 mg) was added, and the reaction mixture wasstirred at rt for 4 h, THF was removed, and the solid was washed withwater several times to get the desired polymer (7 mg) as white solid.Detail characterization data are presented in the main text.
Figure 7. AFM height images (3D view is shown in Figure S9) and height profile across a−b of (top) P1 and (bottom) P9. In both cases THFsolutions (concentration = 1 mg/mL) were drop-casted on a silicon wafer and air-dried before images were taken.
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708568

Synthesis of P9. A solution of P1 (10 mg) in THF (1 mL) wasadded with NDI-SH (0.6 mg), and the reaction mixture was stirred atrt for 8 h and precipitated out from cold MeOH and washed with colddiethyl ether to get the desired polymer as light yellow solid (9 mg).Detail characterization data are presented in the main text.Acid-Induced Cleavage of P7. A solution of polymer P7 (5 mg)
in MeOH (1 mL) was added with TFA (20 μL), and the reactionmixture was stirred at rt for 48 h; then all the volatiles were removedunder reduced pressure, and the solid obtained was redissolved inTHF and injected in GPC.Ellman’s Test. To a solution of a polymer under investigation in
THF (0.5 mg/mL), Ellman’s reagent was added (∼5 mg) followed byaddition of Et3N (20 μL), and it was stirred for 30 min. UV/vis spectraof the solution were checked before and after Ellman’s treatment todetect presence of free thiol functionality by monitoring theabsorption band at 440 nm.Atomic Force Microscopy (AFM) Studies. In a typical AFM
experiment, 50 μL of THF solution of P1/P9 (0.5 mg/mL) was drop-casted on a silicon wafer and allowed to air-dry for 4 h before imageswere taken.
■ ASSOCIATED CONTENT*S Supporting InformationSynthesis and characterization data of various small moleculescontaining thiol containing and thiol reactive functional groupsand additional spectral data. This material is available free ofcharge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] ContributionsThe manuscript was written through contributions of allauthors. All authors have given approval to the final version ofthe manuscript.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSM.R.M. thanks IACS for a research fellowship, and S.G. thanksCSIR, New Delhi, India, for financial support (Project No. 01/2366/10/EMR11). We thank Dr. Raja Shunmugam, IISERKolkata, for GPC analysis and Mr. Rabindranath Banik forAFM measurements.
■ REFERENCES(1) For recent reviews in this area see: (a) Siegwart, D. J.; Oh, J. K.;Matyjaszewski, K. Prog. Polym. Sci. 2012, 37, 18−37. (b) Canalle, L. A.;Lowik, D. W. P. M.; van Hest, J. C. M. Chem. Soc. Rev. 2010, 39, 329−353. (c) Nicolas, J.; Mantovani, G.; Haddleton, D. M. Macromol. RapidCommun. 2010, 28, 1083−1111. (d) Iha, R. K.; Wooley, K. L.;Nystrom, A. M.; Burke, D. J.; Kade, M. J.; Hawker, C. J. Chem. Rev.2009, 109, 5620−5686. (e) Broyer, R. M.; Grover, G. N.; Maynard, H.D. Chem. Commun. 2011, 47, 2212−2226. (f) Becer, C. R.;Hoogenboom, R.; Schubert, U. S. Angew. Chem., Int. Ed. 2009, 48,4900−4908. (g) Gauthier, M. A.; Gibson, M. I.; Klok, H.-A. Angew.Chem., Int. Ed. 2009, 48, 48−58.(2) (a) Saha, A.; De, S.; Stuparu, M. C.; Khan, A. J. Am. Chem. Soc.2012, 134, 17291−17297. (b) Averick, S.; Paredes, E.; Li, W.;Matyjaszewski, K.; Das, S. R. Bioconjugate Chem. 2011, 22, 2030−2037. (c) Biedermann, F.; Rauwald, U.; Zayed, J. M.; Scherman, O. A.Chem. Sci. 2011, 2, 279−286. (d) Delaittre, G.; Pauloehrl, T.;Bastmeyer, M.; Barner-Kowollik, C. Macromolecules 2012, 45, 1792−1802. (e) Le, D.; Montembault, V.; Pascul, S.; Legoupy, S.; Fontaine,L. Macromolecules 2012, 45, 7758−7769. (f) Li, M.; De, P.; Gondi, S.R.; Sumerline, B. S. J. Polym. Sci., Part A: Polym. Chem. 2008, 46,
5093−5100. (g) Ghosh, S.; Basu, S.; Thayumanavan, S.Macromolecules2006, 39, 5595−5597. (h) Deng, J.; Zhou, Y.; Xu, B.; Mai, K.; Deng,Y.; Zhang, L.-M. Biomacromolecules 2011, 12, 642−649. (i) Onbulak,S.; Tempelaar, S.; Pounder, R. J.; Gok, O.; Sanyal, R.; Dove, A. P.;Sanyal, A. Macromolecules 2012, 45, 1715−1722. (j) Budhathoki-Uprety, J.; Novak, B. M. Macromolecules 2011, 44, 5947−5954.(j) Campos, L. M.; Killops, K. L.; Sakai, R.; Paulusse, J. M. J.; Damiron,D.; Drockenmuller, E.; Messmore, B. W.; Hawker, C. J.Macromolecules2008, 41, 7063−7070. (k) Lanj, A. S.; Neubig, A.; Sommer, M.;Thelakkat, M. Macromolecules 2010, 43, 7001−7010. (l) Yang, S. K.;Weck, M. Macromolecules 2008, 41, 346−351. (m) Sun, B.; Liu, X.;Buck, M. E.; Lynn, D. M. Chem. Commun. 2010, 46, 2016−2018.(n) Vogt, A. P.; Sumerlin, B. S. Macromolecules 2008, 41, 7368−7373.(o) De, S.; Khan, A. Chem. Commun. 2012, 48, 3130−3132.(p) Malkoch, M.; Thibault, R. J.; Drockenmuller, E.; Messerschmidt,M.; Voit, B.; Russell, T. P.; Hawker, C. J. J. Am. Chem. Soc. 2005, 127,14942−14949. (q) Wong, L.; Boyer, C.; Jia, Z.; Zareie, H. M.; Davis,T. P.; Bulmus, V. Biomacromolecules 2008, 9, 1934−1944. (r) De, S.;Stelzer, C.; Khan, A. Polym. Chem. 2012, 3, 2342−2345. (s) Brandle,A.; Khan, A. Polym. Chem. 2012, DOI: 10.1039/c2py20591b.(3) For a recent review see: Mansfeld, U.; Pietsch, C.; Hoogenboom,R.; Becer, C. R.; Schubert, U. S. Polym. Chem. 2010, 1, 1560−1598.(4) (a) Hansell, C. F.; Espeel, P.; Stamenovic, M. M.; Barker, I. A.;Dove, A. P.; Prez, F. E. D.; O’Reilly, R. K. J. Am. Chem. Soc. 2011, 133,13828−13831. (b) Potzsch, R.; Fleischmann, S.; Tock, C.; Komber,H.; Voit, B. I. Macromolecules 2011, 44, 3260−3269. (c) Tao, L.; Liu,J.; Xu, J.; Davis, T. P. Chem. Commun. 2009, 6560−6562. (d) Ryu, J.-H.; Park, S.; Kim, B.; Klaikherd, A.; Russell, T. P.; Thayumanavan, S. J.Am. Chem. Soc. 2009, 131, 9870−9871. (e) Mansfeld, U.; Hager, M.D.; Hoogenboom, R.; Ott, C.; Winter, A.; Schubert, U. S. Chem.Commun. 2009, 3386−3388. (f) Megenau, A. J. D.; Martinez-Castro,N.; Savin, D. A.; Storey, R. F. Macromolecules 2009, 42, 8044−8051.(5) For recent reviews see: (a) Hoyle, C. E.; Lowe, A. B.; Bowman,C. N. Chem. Soc. Rev. 2010, 39, 1355−1387. (b) Hoyle, C. E.;Bowman, C. N. Angew. Chem., Int. Ed. 2010, 49, 1540−1573.(c) Stamenovic, M. M.; Espeel, P.; Camp, W. V.; Prez, F. E. D.Macromolecules 2011, 44, 5619−5630. (d) Huynh, V. T.; Chen, G.;Souza, de, P.; Stenzel, M. H. Biomacromolecules 2011, 12, 1738−1751.(e) Huang, Y.; Yonghong, Z.; Yang, J.; Zhaohua, Z.; Fangming, Z.;Chen, X. Chem. Commun. 2011, 47, 7509−7511. (f) Boyer, C.;Bulmus, V.; Davis, T. P. Aust. J. Chem. 2009, 62, 830−847.(g) Dondoni, A. Angew. Chem., Int. Ed. 2008, 47, 8995−8997.(h) Hoogenboom, R. Angew. Chem., Int. Ed. 2010, 49, 3415−3417.(6) (a) Roth, P. J.; Boyer, C.; Lowe, A. B.; Davis, T. P. Macromol.Rapid Commun. 2011, 32, 1123−1143. (b) Boyer, C.; Bulmus, V.;Davis, T. P.; Ladmiral, V.; Liu, J.; Perrier, S. Chem. Rev. 2009, 109,5402−5436.(7) (a) Campos, L. M.; Killops, K. L.; Sakai, R.; Paulusse, J. M. J.;Damiron, D.; Drockenmuller, D. E.; Messmore, B. M.; Hawker, C. J.Macromolecules 2008, 41, 7063−7070. (b) Nurmi, L.; Lindqvist, J.;Randev, R.; Syrett, J.; Haddleton, D. M. Chem. Commun. 2009, 2727−2729. (c) Pounder, R. P.; Stanford, M. J.; Brooks, P.; Richards, S. P.;Dove, A. P. Chem. Commun. 2008, 5158−5160. (d) Stanford, M. J.;Dove, A. P. Macromolecules 2009, 42, 141−147.(8) Bang, E.-K.; Lista, M.; Sforazzini, G.; Sakai, N.; Matile, S. Chem.Sci. 2012, 3, 1752−1763.(9) (a) Bontempo, D.; Heredia, K. L.; Fish, B. A.; Maynard, H. D. J.Am. Chem. Soc. 2004, 126, 15372−15373. (b) Klaikherd, A.; Ghosh, S.;Thayumanavan, S. Macromolecules 2007, 40, 8518−8520. (c) Phillips,D. J.; Gibson, M. I. Biomacromolecules 2012, 13, 3200−3208.(d) Wang, F.; Klaikherd, A.; Thayumanavan, S. J. Am. Chem. Soc.2011, 133, 13496−13503. (e) Boyer, C.; Liu, J.; Bulmus, V.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M. H. Macromolecules 2008, 41,5641−5650. (f) Boyer, C.; Liu, J.; Wong, L.; Tippett, M.; Bulmus, V.;Davis, T. P. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 7207−7224.(g) Wong, L. J.; Sevimli, S.; Zareie, H. M.; Davis, T. P.; Bulmus, V.Macromolecules 2010, 43, 5365−5375.(10) (a) Poly(Lactic Acid): Synthesis, Structures, Properties, Processing,and Applications; Auras, R., Lim, L.-T., Selke, S. E. M., Tsuji, H., Eds.;
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708569

John Wiley & Sons, Inc.: Hoboken, NJ, 2010. (b) Castillo, J. A.;Borchimall, D. E.; Cheng, A. Y.; Wang, Y.; Hu, C.; García, A. J.; Weck,M. Macromolecules 2012, 45, 62−69.(11) Wolf, F. F.; Nora, F.; Frey, H. Macromolecules 2009, 42, 5622−5628.(12) (a) Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K.J. Am. Chem. Soc. 2005, 127, 1624−1625. (b) Dan, K.; Pan, R.; Ghosh,S. Langmuir 2011, 27, 612−617.(13) Velonia, K.; Rowan, A. E.; Nolte, R. J. M. J. Am. Chem. Soc.2002, 124, 4224−4225.(14) Yurt, S.; Anyanwu, U. K.; Scheintaub, J. R.; Coughlin, E. B.;Venkataraman, D. Macromolecules 2006, 39, 1670−1672.(15) (a) Elsabahy, M.; Wooley, K. L. Chem. Soc. Rev. 2012, 41,2545−2561. (b) Esser-Kahn, A. P.; Odom, S. A.; Sottos, N. R.; White,S. R.; Moore, J. S. Macromolecules 2011, 44, 5539−5553. (c) Roy, D.;Cambre, J. N.; Sumerlin, B. S. Prog. Polym. Sci. 2010, 35, 278−301.(16) (a) Mansky, P.; Liu, Y.; Huang, E.; Russell, T. P.; Hawker, C.Science 1997, 275, 1458−1460. (b) Ryu, D. Y.; Shin, K.;Drockenmuller, E.; Hawker, C. J.; Russell, T. P. Science 2005, 308,236−239. (c) Zhang, M. F.; Yang, L.; Yurt, S.; Misner, M. J.; Chen, J.T.; Coughlin, E. B.; Venkataraman, D.; Russell, T. P. Adv. Mater. 2007,19, 1571. (d) Zalusky, A. S.; Olayo-Valles, R.; Wolf, J. H.; Hillmyer, M.A. J. Am. Chem. Soc. 2002, 124, 12761−12773. (e) Lodge, T. P.; Bang,J. A.; Li, Z. B.; Hillmyer, M. A.; Talmon, Y. Faraday Discuss. 2005, 128,1−12. (f) Jirage, K. B.; Hulteen, J. C.; Martin, C. R. Science 1997, 278,655−658.(17) Lamoureux, G. V.; Whitesides, G. M. J. Org. Chem. 1993, 58,633.(18) (a) Sakai, N.; Mareda, J.; Vauthey, E.; Matile, S. Chem. Commun.2010, 46, 4237. (b) Bhosale, S. V.; Janiab, C. H.; Langford, S. J. Chem.Soc. Rev. 2008, 37, 331−342.(19) (a) Shao, H.; Nguyen, T.; Romano, N. C.; Modarelli, D. A.;Parquette, J. R. J. Am. Chem. Soc. 2009, 131, 16374−16376. (b) Bell, T.D. M.; Bhosale, S. V.; Forsyth, C. M.; Hayne, D.; Ghiggino, K. P.;Hutchison, J. A.; Jani, C. H.; Langford, S. J.; Lee, M. A.-P.; Woodward,C. P. Chem Commun. 2010, 46, 4881−4883. (c) Kumar, M.; George, S.J. Nanoscale 2011, 3, 2130−2133.(20) (a) Molla, M. R.; Ghosh, S. Chem.Eur. J. 2012, 18, 1290−1294. (b) Mazzeo, M.; Vitale, V.; Sala, F. D.; Anni, M.; Barbarella, G.;Fa-baretto, L.; Sotgiu, G.; Cingolani, R.; Gigli, G. Adv. Mater. 2005, 17,34−39.(21) Tu, S.; Wang, B-l.; Chen, Y-w.; Li, Z-m.; Luo, X-l. ACS MacroLett. 2012, 1, 933−936.(22) Hentschel, J.; Kushner, A. M.; Ziller, J.; Guan, Z. Angew. Chem.,Int. Ed. 2012, DOI: 10.1002/anie.201204840.(23) Perrin, D. D.; Armarego, W. L. F.; Perrin, D. R. Purification ofLaboratory Chemicals, 2nd ed.; Pergamon: Oxford, 1980.
Macromolecules Article
dx.doi.org/10.1021/ma302130f | Macromolecules 2012, 45, 8561−85708570





























