Embed Size (px)
Citation preview

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/262053404
Excitation/inhibitionbalanceandlearningaremodifiedbyDyrk1agenedosage
ARTICLEinNEUROBIOLOGYOFDISEASE·SEPTEMBER2014
ImpactFactor:5.08·DOI:10.1016/j.nbd.2014.04.016
CITATIONS
13
READS
122
15AUTHORS,INCLUDING:
FaycalGuedj
TuftsMedicalCenter
18PUBLICATIONS210CITATIONS
SEEPROFILE
YannHerault
InstitutdeGénétiqueetdeBiologieMolécu…
119PUBLICATIONS2,076CITATIONS
SEEPROFILE
NathalieJanel
ParisDiderotUniversity
77PUBLICATIONS1,179CITATIONS
SEEPROFILE
JeanMDelabar
FrenchNationalCentreforScientificResea…
155PUBLICATIONS3,634CITATIONS
SEEPROFILE
Allin-textreferencesunderlinedinbluearelinkedtopublicationsonResearchGate,
lettingyouaccessandreadthemimmediately.
Availablefrom:IgnasiSahúnAbizanda
Retrievedon:04February2016

Neurobiology of Disease 69 (2014) 65–75
Contents lists available at ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r .com/ locate /ynbd i
Excitation/inhibition balance and learning are modified by Dyrk1agene dosage
Benoit Souchet a,1, Fayçal Guedj a,1, Ignasi Sahún b,c, Arnaud Duchon d, Fabrice Daubigney a, Anne Badel e,Yuchio Yanagawa f, Maria Jose Barallobre g, Mara Dierssen b,c, Eugene Yu h, Yann Herault d, Mariona Arbones c,g,Nathalie Janel a, Nicole Créau a,⁎, Jean Maurice Delabar a,⁎a Univ Paris Diderot, Sorbonne Paris Cité, Adaptive Functional Biology, UMR CNRS 8251, 75205 Paris, Franceb Genomic Regulation Center, Barcelona, Spainc Centro de Investigación Biomédica en Red de Enfermedades Raras, Barcelona, Spaind IGBMC, CNRS, INSERM, UMR7104, UMR964, Illkirch, Francee MTI, Univ Paris Diderot, Sorbonne Paris Cité, Francef Department of Genetic and Behavioral Neuroscience, Gunma University Graduate School of Medicine and JST, CREST, Japang Plataforma de Recerca Aplicada en Animal de Laboratori (PRAAL), Parc Científic de Barcelona (PCB), Spainh Children's Guild Foundation Down Syndrome Research Program, Department of Cancer Genetics, Roswell Park Cancer Institute, Elm and Carlton Streets, Buffalo, NY 14263, USA
⁎ Corresponding authors.E-mail addresses: [email protected] (N. Créa
[email protected] (J.M. Delabar).Available online on ScienceDirect (www.sciencedir
1 These authors contributed equally to this work.
http://dx.doi.org/10.1016/j.nbd.2014.04.0160969-9961/© 2014 Elsevier Inc. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 9 March 2014Revised 17 April 2014Accepted 24 April 2014Available online 4 May 2014
Keywords:Down syndromeDYRK1AExcitationInhibition
Cognitive deficits in Down syndrome (DS) have been linked to increased synaptic inhibition, leading to an imbal-ance of excitation/inhibition (E/I). Various mouse models and studies from human brains have implicated anHSA21 gene, the serine/threonine kinase DYRK1A, as a candidate for inducing cognitive dysfunction. Here, con-sequences of alterations inDyrk1a dosagewere assessed inmousemodelswith varying copy numbers of Dyrk1a:mBACtgDyrk1a, Ts65Dn and Dp(16)1Yey (with 3 gene copies) and Dyrk1a(+/−) (one functional copy).Molecular (i.e. immunoblotting/immunohistochemistry) and behavioral analyses (e.g., rotarod, Morris watermaze, Y-maze) were performed in mBACtgDyrk1a mice. Increased expression of DYRK1A in mBACtgDyrk1a in-duced molecular alterations in synaptic plasticity pathways, particularly expression changes in GABAergic andglutaminergic related proteins. Similar alterations were observed in models with partial trisomy of MMU16,Ts65Dn andDp(16)1Yey, andwere reversed in the Dyrk1a(+/−)model. Dyrk1a overexpression produced an in-creased number and signal intensity of GAD67 positive neurons, indicating enhanced inhibition pathways inthree different models: mBACtgDyrk1a, hYACtgDyrk1a and Dp(16)1Yey. Functionally, Dyrk1a overexpressionprotected mice from PTZ-induced seizures related to GABAergic neuron plasticity. Our study showsthat DYRK1A overexpression affects pathways involved in synaptogenesis and synaptic plasticity and influencesE/I balance toward inhibition. Inhibition of DYRK1A activity offers a therapeutic target for DS, but its inhibition/activation may also be relevant for other psychiatric diseases with E/I balance alterations.
© 2014 Elsevier Inc. All rights reserved.
Introduction
An imbalance of excitation and inhibition (E/I) is thought to underlieseveral neurological diseases, including autism (Rubenstein andMerzenich, 2003), Tourette's syndrome (Singer and Minzer, 2003),and schizophrenia (Wassef et al., 2003). Cognitive deficits in Downsyndrome (DS), or trisomy 21, have been proposed to result from anexcess of inhibition. However, chromosome 21 genes responsible forsuch defects have not been definitively identified.
u),
ect.com).
Excessive inhibition has also been observed in the Ts65Dn mouse,the most widely studied model of DS. Ts65Dn has partial trisomy ofMMU16 in the region syntenic to a 13.9-Mb HSA21 region containingDyrk1a and 98 other genes (Davisson et al., 1993; Reinholdt et al.,2011). These mice recapitulate the hallmarks of the DS phenotype,including a serious cognitive impairment (Escorihuela et al., 1995;Reeves et al., 1995), and their learning defects appear to be relatedto excessive levels of inhibition in temporal lobe and hippocampalcircuitry (Kleschevnikov et al., 2004; Siarey et al., 1997). Indeed,markedmorphological changes are evident in the hippocampus and cerebralcortex, with selective enlargement of the active zones of symmetric syn-apses and increased immunoreactivity for synaptic proteins that markinhibitory synapses (P.V. Belichenko et al., 2009; Perez-Cremadeset al., 2010). Additionally, increased efficiency of GABA-A and GABA-Breceptor-mediated neurotransmission has been reported for Ts65Dn

66 B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
(Kleschevnikov et al., 2012b) and the GABA(B)/GABA(A) ratios evokedby stimulation within the stratum lacunosum moleculare of Ts65Dnhippocampus were found to be significantly altered (Best et al., 2012).A more recent DS model including the genes of MMU16 present in 3copies in Ts65Dn, Dp(16)1Yey (Li et al., 2007), also shows altered hip-pocampal long-term potentiation (LTP) and deficits in visuo-spatiallearning memory (Yu et al., 2010) suggesting common molecularpathways.
The molecular links between increased copy number of specificHSA21 genes and these neural defects remain unknown. In human,altered copy number for segments of chromosome 21 that resultsin either deletion or duplication of the gene DYRK1A can inducemorphological defects and cognitive impairments (Delabar et al.,1993; Oegema et al., 2010; Rahmani et al., 1989; Ronan et al., 2007;van Bon et al., 2011). Mouse models have again been invaluable ingaining insight to these phenotypes. A phenotype rescue experimentbreeding Ts65Dn mice, which have three copies of Dyrk1a, with micemonosomic for a chromosomal segment containing 33 genes includingDyrk1a (Ms1Rhr) produced progeny with 2 copies of the 33-gene seg-ment and a normal learning phenotype, indicating that duplication ofthis small region is necessary to produce the cognitive deficit (Olsonet al., 2007). Further, trisomy of this region (Ts1Rhr mouse) is sufficientto produce significant alterations in results of an open field test, a novelobject recognition test and a T-maze task. As in Ts65Dn and Ts1Cjemice (another model of segmental trisomy 21), LTP in fascia dentata(FD) of Ts1Rhr can be induced only after blocking GABA(A)-dependentinhibitory neurotransmission. In addition, widespread enlargement ofdendritic spines and decreased density of spines in FD are preservedin Ts1Rhr (N.P. Belichenko et al., 2009).
Among the genes from this 33-gene region, Dyrk1a is an attractivecandidate for inducing cognitive impairment phenotypes. DYRK1A(dual-specificity tyrosine phosphorylated and regulated kinase 1A), themammalian ortholog of Drosophila minibrain kinase (mnb) (Tejedoret al., 1995), encodes a proline/arginine-directed serine/threoninekinase. Both in trisomic mice and in individuals with DS, brain levels ofDYRK1A are increased approximately 1.5-fold, indicating that this pro-tein is overexpressed in a gene dosage-dependent manner (Dowjatet al., 2007).
We have already reported similarities and differences between brainmorphological alterations found in our BAC-transgenic Dyrk1a overex-pression model (mBACtgDyrk1a) and Ts65Dn models (Guedj et al.,2012). Additionally, hYACtgDyrk1a transgenic mice with three func-tional copies as well as Dyrk1a(+/−) mice with one functional copyof Dyrk1a have been shown to presentmorphological alterations, learn-ing and memory impairments (Arque et al., 2008; Fotaki et al., 2002;Guedj et al., 2009; Smith et al., 1997) suggesting that an altered levelof DYRK1A is associated with abnormal brain function. To explore po-tential molecular links between DYRK1A level and these alterations,we performed a set of assessments of learning and memory in ournewmBACtgDyrk1a, which contains three copies of the full-lengthmu-rine gene encoding DYRK1A (Guedj et al., 2012). Then we exhaustivelyinvestigated molecular alterations of biochemical components of learn-ing pathways present in this model, and in two models with segmentaltrisomy of MMU16, Ts65Dn and Dp(16)1Yey (Li et al., 2007) as well asin the loss-of-function model Dyrk1a (+/−).
Materials and methods
Animals: handling and genotyping
Mice were housed in standard cages with access to food and waterad libitum, under a controlled environment (temperature = 20 ±1 °C; humidity = 60%), and with a light/dark cycle of 12 h.
All experimentswere conducted in accordancewith the ethical stan-dards of French and European regulations (European CommunitiesCouncil Directive, 86/609/EEC). Official authorization from the French
Ministry of Agriculture was granted to carry out research and experi-ments on animals (authorization number 75–369), and the study wasapproved by the local ethical committee (Univ Paris-Diderot). Ts65Dnmice (Davisson et al., 1993) were maintained on a B6/C3H backgroundand genotyped as described previously (Reinholdt et al., 2011). Micecarrying the murine BAC containing one copy of Dyrk1A were main-tained on a C57Bl/6J background and genotyped as described (Guedjet al., 2012). Dp(16)1Yey mice were maintained on a C57Bl/6J back-ground and genotyped as described (Li et al., 2007). The GAD67-GFPknock-in delta-neomouse wasmaintained on a CD1 background, geno-typed as previously described (Tamamaki et al., 2003), and femaleswere crossed with males of the hYACtgDyrk1a (Guedj et al., 2009;Smith et al., 1997) model (on C57Bl/6J background) or of the Dp(16)1Yey (C57Bl/6J) to obtain F1 generation mice that were genotyped forthe three genotypes as previously described (Guedj et al., 2009; Liet al., 2007; Tamamaki et al., 2003). (Primer information for genotypingis included in Supplementary Table 1.) Dyrk1a(+/−) mice were main-tained on a CD1 background and genotyped as previously described(Fotaki et al., 2002). All analyses were performed comparing thetransgenic or trisomic mice with their wildtype (WT) littermates.
Behavioral analyses
Parameters of motor coordination (Rotarod), spontaneous alterna-tion behavior (Y-maze) and visuo-spatial learning andmemory (Morriswater maze) were recorded for the two groups of male WT andtransgenic animals as described in the Supplemental Methods section.
Tissue collection
Male mice [3–4 months old for mBACtgDyrk1a, Dyrk1a(+/−),Dp(16)1Yey, F1 (GAD67-GFPxhYACDyrk1a) or F1(GAD67-GFPxDp(16)1Yey); 5–6 months old for Ts65Dn] were euthanized by decapitation,and brain tissue was rapidly removed. For immunoblotting, tissue wascooled on an ice block, dissected in less than 3 min, and snap-frozen inliquid nitrogen. For immunohistochemistry, tissue was post-fixed in 4%paraformaldehyde in PBS for 48 h, cryoprotected in 30% sucrose in PBSand slowly frozen onto liquid nitrogen in Tissuetek before storage at−80 °C.
Immunohistochemistry and stereology
Serial sagittal brain cryosections (50 μm) were cut on a cryostat andimmunohistochemistry was performed as described in the Supplemen-tal Methods section.
NeuN-positive and GAD67-GFP-positive neuron densities wereassessed with StereoInvestigator (MBF) in hippocampal CA1 stratumradiatum and prelimbic cortex in parasagittal slices (+2.0 mm and+0.3 mm respectively) (see Supplementary Fig. 1). Preliminary cellcount was performed to determine a suitable number of countingframes to produce a coefficient of error of Gundersen (m = 1) lessthan 0.05. Once parameters were defined for each brain region, cellcounting was performed in a random, systematic fashion using the op-tical fractionator with a dissector height of 40 μm and a guard zone of5 μm. The percentage of GAD67-GFP positive neurons in NeuN positiveneuronswere calculated for each genotype. The results are presented asthe ratio to mean GAD67-GFP (+/−) littermates level.
Immunoblotting
Immunoblotting was performed following standard protocols usingslot blot (Guedj et al., 2012) after testing the specificity of antibodies byWestern blotting. Antibodies are listed in Supplementary Table 2. Digi-tized images of immunoblots were obtained using an LAS-3000 imagingsystem (Fuji Photo Film Co. Ltd.), and densitometry measurementswere collected with an image analyzer (UnScan It software, Silk

67B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
Scientific Inc.). Normalization was performed relative to total proteinslevel using Ponceau staining.
Seizure assessment
Seizures were induced through intraperitoneal injection of PTZ(50 mg/kg), and convulsion activity was recorded for 15 min followinginjection (Braudeau et al., 2011). Genotype effect was assessed in males(WT, n = 8, TG, n = 7) and females (WT, n = 5, TG n = 5) (3–5months) mBACtgDyrk1a, and on males (WT, n = 4, Het, n = 4)Dyrk1a(+/−).
Statistical analysis
All data passed the normality test of Kolmogorov–Smirnov. Forcomparisons between two groups, unpaired t-tests were performedand p values were considered significant for p b 0.05. The results areexpressed as means ± SEM. Graphs were plotted as box and whiskerswith minimum to maximum. All statistical analyses were performedusing GraphPad6 software package.
Results
Motor function, learning, and memory in mBACtgDyrk1a
Transgenesis of a murine BAC clone containing full-length Dyrk1aproduces an ~50% increase in Dyrk1a mRNA and protein expression(Guedj et al., 2012). Primary screening of mBACtgDyrk1a and WTmice using the SHIRPA protocol (Rogers et al., 2001) indicated nodifferences for autonomic nervous system, pain sensitivity, vision,habituation, activity in openfield, or day/night activity. However a slightincrease in response to auditory stimuli and a slight decrease in gripstrengthwas observed formBACtgDyrk1amice (data not shown). To as-sess motor coordination and learning of a motor task we used a rotarodparadigm. At fixed rotation speeds mBACtgDyrk1a mice exhibited adeficit that was more marked at higher rotating speeds (Fig. 1A). In anaccelerating rod paradigm, during the first session, mBACtgDyrk1aperformed similarly to WT littermates; however, in the second sessiona significant impairment was observed in mBACtgDyrk1a animals(Supplementary Fig. 2).
To assess short-termmemory and spatial learning, we used a Y-mazeparadigm. Spontaneous alternation was significantly decreased inmBACtgDyrk1a mice (Fig. 1B). Long-term memory and spatial learningwere assessed using the Morris water maze task. Escape latency wasreduced over the 7 sessions of the acquisition phase for the WT mice(Fig. 1C) indicating efficient learning of the platformposition; in contrast,escape latency did not improve for mBACtgDyrk1amice, and percentageof time spent in the platform quadrant (NE) did not increase along thesessions (Fig. 1D). mBACtgDyrk1a mice did not differ from WT mice inperformance during the cued session, thus discarding motor or motiva-tional factors as underlying the phenotypes. Following platform removal,the percentage of time spent in center periphery and critical zones(target quadrant) was significantly different for WT and mBACtgDyrk1aanimals (Fig. 1E). mBACtgDyrk1a mice were more thigmotaxic andspent less time in the trained quadrant, indicating poor spatial memory.In the reversal (rev) test, which evaluates cognitive flexibility, we ob-served a significant difference between the two groups in time spent inthe NE quadrant at day 9 (rev 1) and in NW quadrant at day 10 (rev 2)(Fig. 1F).
Alteration in synaptic plasticity pathways in mBACtgDyrk1a
Expression of proteins of signaling pathways involved in synapticplasticity was measured in different brain regions of mBACtgDyrk1a(cortex, hippocampus and cerebellum): five different sets ofexperiments were performed on adult males from this transgenic line.
Immunoblotting was used to measure relative expression level ofeach marker and was assessed in at least 3 different groups of WTand transgenic mice (Table 1). Unsurprisingly, quantitative slotblot analysis showed increased relative DYRK1A expression inmBACtgDyrk1a; this overexpression was stronger in hippocampusthan in cortex (p = 0.0025).
In the GABAergic pathway, which functions in inhibition, relativeprotein expression was generally increased. GAD67 and GAD65, key el-ements of GABA production, and VGAT1, the major vesicular GABAtransporter, had significantly increased expression in cortex (GAD67/GAD65/VGAT1: +34.1%/+39.5%/+23.2%), hippocampus (+41.4%/+25.8%/+25.3%), and cerebellum (+60.4%/+45.8%/+21.9%), (Table 1,Fig. 2 anddata not shown). In contrast, someproteins of the glutaminergicpathway, which functions in excitation, exhibited reduced relativeexpression. Levels of two AMPA-type glutamate receptors, GLUR1 andGLUR2, their phosphorylated forms, and the vesicular glutamate trans-porter VGLUT1 varied by region according to genotype. While GLUR1levels were no different in the cortex and hippocampus between WTand Tg mice, relative GLUR2 levels were lower in hippocampus (−17%)and cerebellum (−19.3%) of Tg mice compared with WT. In contrast,the phosphorylated forms of both GLUR1 and GLUR2 were detected athigher levels in hippocampus (+12.8% and +24%, respectively) of Tgmice. VGLUT1 level was significantly lower in cortex (−15.8%) and cere-bellum (−14.7%) but was not different in hippocampus of Tg relative toWT. To characterize a shift in E/I balance we assessed the VGAT1/VGLUT1 ratios. Themean individual ratios of VGAT1/VGLUT1were higherin cortex (+55.7%), cerebellum (+40.1%), and hippocampus (+22.5%)of Tg mice. We also assessed expression of two components of long-term potentiation (LTP): NMDA receptor with two of its subunits, NR1and NR2a, and CAMKII. NR1was significantly lower only in hippocampus(−11.6%) of Tgmice relative toWT. NR2awas lower in cortex (−12.4%),hippocampus (−19.4%) and cerebellum (−18%). The ratio of pCAMKII/CAMKII was also lower in cortex (−12.8%; p = 0.05) and significantlylower in hippocampus (−20.7%; p b 0.0001) of mBACtgDyrk1a relativeto WT.
Alterations in synaptic plasticity pathways in Ts65Dn, Dp(16)1Yey andDyrk1a(+/−)
Dyrk1a triplicationTo study the effect of Dyrk1a overexpression in the context of triso-
my, we assessed protein levels in mice with segmental trisomy ofMMU16, Ts65Dn and Dp(16)1Yey, and compared these results tomBACtgDyrk1a. Unsurprisingly, quantitative slot blot analysis showedhigher relative DYRK1A expression in cortex, hippocampus, and cere-bellum of Ts65Dn and Dp(16)1Yey compared to WT, as expected fromthe gene dosage (Table 2). GAD67, which was higher in mBACtgDyrk1abrain, was also higher in cortex (+41% and +43.7%, respectively forTs65Dn and Dp(16)1Yey), hippocampus (tendency for Ts65Dn and+33%, for Dp(16)1Yey), and cerebellum (+52.2% and +45%, respec-tively) of trisomic mice relative to WT littermates. Though the effectswere similar in the two trisomic DS models, they were statistically sig-nificant in all three brain regions in Dp(16)1Yey mice, which showedless variability overall. VGAT1was significantly higher in the cerebellumof Ts65Dn (+44.3%) and in the cortex and hippocampus of Dp(16)1Yey(+23.3% and+16%, respectively) relative toWT. Other regions (cortexand hippocampus of Ts65Dnmice; cerebellum of Dp(16)1Yey) showedonly a trend toward higher levels, resulting from either variability or alower effect. Proteins in the glutaminergic pathway were similarly af-fected in the trisomic models as in the Tg model. GLUR1 and GLUR2were significantly lower in cortex of Ts65Dn (−15.8% and −22.9%, re-spectively) and in hippocampus of Dp(16)1Yey (−10.2% and −12.2%,respectively) relative to WT. VGLUT1 was lower in cortex (−8.8%)and hippocampus (−17.2%) of Ts65Dn mice relative to WT. Finally,the ratio of VGAT1/VGLUT1 was significantly higher in hippocampusof both Ts65Dn (+42.5%) and Dp(16)1Yey (+24.7%) relative to WT.
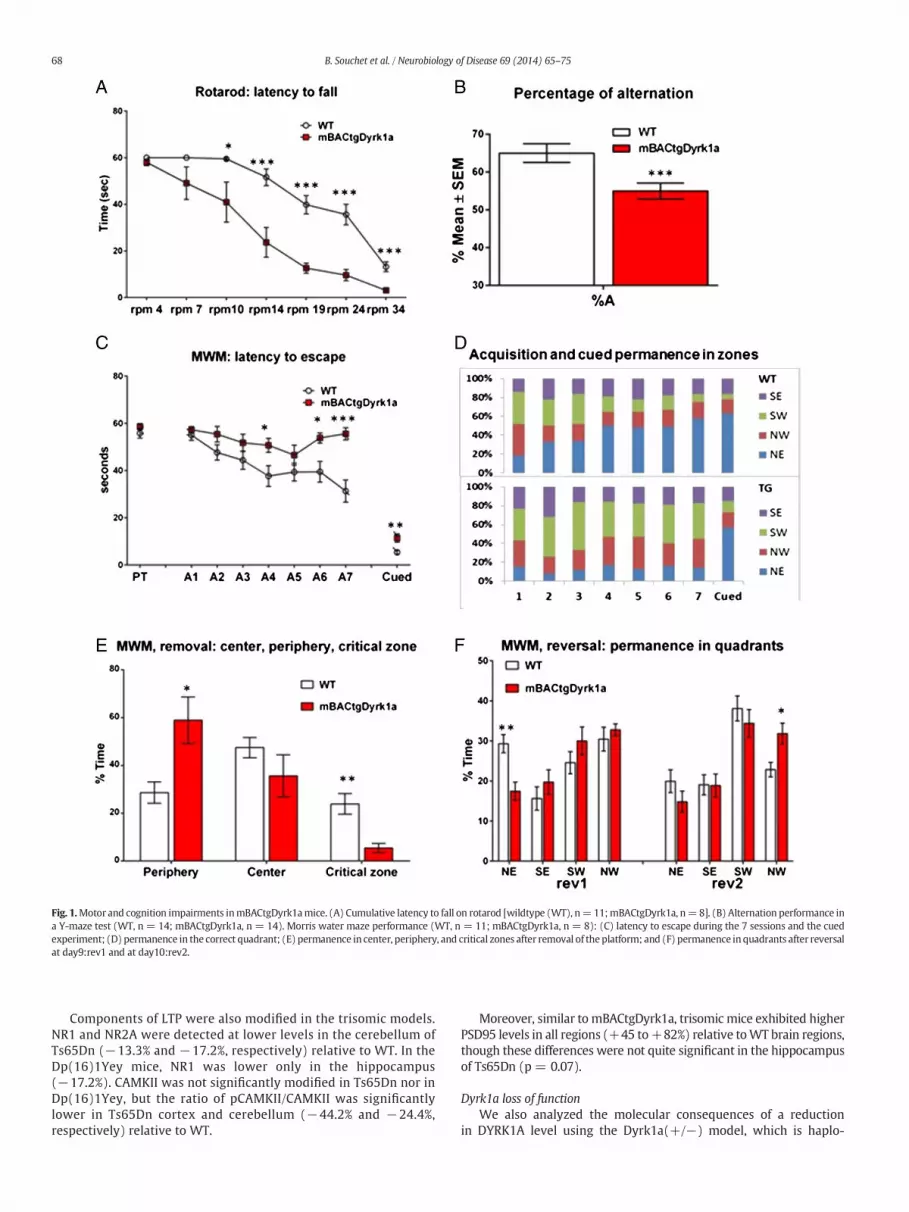
Fig. 1.Motor and cognition impairments inmBACtgDyrk1amice. (A) Cumulative latency to fall on rotarod [wildtype (WT), n=11;mBACtgDyrk1a, n=8]. (B) Alternation performance ina Y-maze test (WT, n = 14; mBACtgDyrk1a, n = 14). Morris water maze performance (WT, n = 11; mBACtgDyrk1a, n = 8): (C) latency to escape during the 7 sessions and the cuedexperiment; (D) permanence in the correct quadrant; (E) permanence in center, periphery, and critical zones after removal of the platform; and (F) permanence inquadrants after reversalat day9:rev1 and at day10:rev2.
68 B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
Components of LTP were also modified in the trisomic models.NR1 and NR2A were detected at lower levels in the cerebellum ofTs65Dn (−13.3% and −17.2%, respectively) relative to WT. In theDp(16)1Yey mice, NR1 was lower only in the hippocampus(−17.2%). CAMKII was not significantly modified in Ts65Dn nor inDp(16)1Yey, but the ratio of pCAMKII/CAMKII was significantlylower in Ts65Dn cortex and cerebellum (−44.2% and −24.4%,respectively) relative to WT.
Moreover, similar to mBACtgDyrk1a, trisomic mice exhibited higherPSD95 levels in all regions (+45 to+82%) relative toWT brain regions,though these differences were not quite significant in the hippocampusof Ts65Dn (p = 0.07).
Dyrk1a loss of functionWe also analyzed the molecular consequences of a reduction
in DYRK1A level using the Dyrk1a(+/−) model, which is haplo-

Table 1Protein levels ofmarkers of inhibition and excitation pathways forWT andmBACtgDyrk1a(Tg) in cortex, hippocampus and cerebellum.Protein levels relative to the levels detected in wildtype animals (WT: 100). Meanexpression relative to WT level ± standard error of the mean (SEM), number of mice(N). p Values below 0.05 are in bold characters. CTX: cortex; HPC: hippocampus; CRB: cer-ebellum. GAD65 levels were recorded in a smaller number of mice for cortex (+39.5%,p = 0.003; WT, n = 10; Tg, n = 9), hippocampus (+25%, p = 0.0001; WT, n = 10;Tg, n = 9) and cerebellum (+45.8%, p = 0.001; WT, n = 9; Tg, n = 9).
CTX WT TG
DYRK1A 100.0 ± 2.9. n = 49 163.9 ± 4.4. n = 41 b0.0001GAD67 100.0 ± 3.8. n = 38 134.1 ± 5.6. n = 32 b0.0001VGAT1 100.0 ± 1.9. n = 49 123.2 ± 3.2. n = 39 b0.0001PSD95 100.0 ± 5.0. n = 38 141.1 ± 7.6. n = 31 b0.0001GLUR1 100.0 ± 3.3. n = 30 97.64 ± 4.6. n = 25 0.67pGLUR1/GLUR1 100.0 ± 3.4. n = 30 96.63 ± 6.1. n = 23 0.5GLUR2 100.0 ± 2.6. n = 29 104.8 ± 4.5. n = 24 0.26pGLUR2/GLUR2 100.0 ± 2.5. n = 28 94.17 ± 2.2. n = 24 0.14VGLUT1 100.0 ± 2.5. n = 39 84.21 ± 1.8. n = 32 b0.0001NR1 100.0 ± 3.1. n = 29 91.52 ± 3.7. n = 22 0.05NR2A 100.0 ± 2.5. n = 37 87.62 ± 2.8. n = 31 0.0015pCAMKII/CAMKII 100.0 ± 4.0. n = 37 87.87 ± 4.0. n = 31 0.05VGAT1/VGLUT 100.0 ± 3.3. n = 39 155.7 ± 8.4. n = 31 b0.0001
HPC WT TG p Value
DYRK1A 100.0 ± 4.1. n = 40 192.7 ± 11.9. n = 33 b0.0001GAD67 100.0 ± 3.3. n = 36 141.4 ± 6.5. n = 27 b0.0001VGAT1 100.0 ± 2.2. n = 38 125.3 ± 3.9. n = 33 b0.0001PSD95 100.0 ± 4.8. n = 39 159.7 ± 9.017. n = 32 b0.0001GLUR1 100.0 ± 2.6. n = 28 100.1 ± 2.8. n = 24 0.93pGLUR1/GLUR1 100.0 ± 2.2. n = 28 112.8 ± 3.7. n = 24 0.0039GLUR2 100.0 ± 2.6. n = 27 83.06 ± 2.4. n = 22 b0.0001pGLUR2/GLUR2 100.0 ± 4.8. n = 27 124.0 ± 5.9. n = 22 0.0065VGLUT1 100.0 ± 1.5. n = 39 106.4 ± 3.1. n = 30 0.075NR1 100.0 ± 3.3. n = 47 88.42 ± 3.4. n = 36 0.0238NR2A 100.0 ± 4.5. n = 37 80.63 ± 2.8. n = 30 0.0011pCAMKII/CAMKII 100.0 ± 2.8. n = 36 79.33 ± 3.3. n = 26 b0.0001VGAT1/VGLUT 100.0 ± 2.5. n = 38 122.5 ± 5.3. n = 30 0.0002
CRB WT TG p Value
DYRK1A 100.0 ± 4.5. n = 19 172.2 ± 12.8. n = 18 b0.0001GAD67 100.0 ± 7.2. n = 20 160.4 ± 6.6. n = 18 b0.0001VGAT1 100.0 ± 1.8. n = 20 121.9 ± 3.5. n = 17 b0.0001PSD95 100.0 ± 9.6. n = 19 168.6 ± 8.3. n = 16 b0.0001GLUR1 100.0 ± 3.5. n = 19 80.25 ± 4.8. n = 18 0.0022pGLUR1/GLUR1 100.0 ± 3.3. n = 19 126.5 ± 9.4. n = 18 0.0111GLUR2 100.0 ± 3.2. n = 19 80.67 ± 5.2. n = 17 0.0029pGLUR2/GLUR2 100.0 ± 4.4. n = 18 98.47 ± 5.2. n = 18 0.82VGLUT1 100.0 ± 2.9. n = 20 85.29 ± 2.9. n = 17 0.0011NR1 100.0 ± 4.0. n = 20 98.43 ± 2.2. n = 17 0.74NR2A 100.0 ± 4.8. n = 19 81.97 ± 4.0. n = 18 0.0078pCAMKII/CAMKII 100.0 ± 4.1. n = 20 89.30 ± 6.1. n = 18 0.15VGAT1/VGLUT 100.0 ± 3.6. n = 20 140.1 ± 6.4. n = 18 b0.0001
69B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
insufficient (Table 2). In contrast to trisomic mice, mice with one func-tional copy of Dyrk1a exhibited a lower level of GAD67, with the largestdifference in hippocampus (−43.4%) and cerebellum (−43%), relativeto WT. We observed higher VGAT1 expression in cortex (+28.5%) andno change in hippocampus and cerebellum. Further, higher levels ofproteins of the glutaminergic pathway were detected: GLUR1 was sig-nificantly higher in cortex (+21.7%) and hippocampus (+26%), andGLUR2was significantly higher in hippocampus (+32.4%) and cerebel-lum (+25.9%) of Dyrk1a(+/−) relative to WT. VGLUT1, however, wasnot different. NR1 and NR2A were both higher in Dyrk1a(+/−) hippo-campus (+47.1% and+49.2%, respectively), but in the cerebellum onlyNR2A was higher in this single-copy model (+27.3%) relative to WT.The pCaMKII/CaMKII ratio was higher only in cortex of Dyrk1a(+/−)mice (+45.9%). Lower PSD95 was detected in all regions (−32%to −48.8%), though the change was not quite significant in the cortex(p = 0.06).
GAD67-GFP signal and number/density of GAD67 neurons inmBACtgDyrk1a and in double transgenic F1(hYACtgDyrk1a × GAD67-GFP),F1(Dp(16)1Yey × GAD67-GFP)
As proteins of the GABAergic pathway appear to be regulated inrelation to Dyrk1a dosage, we analyzed the number of GAD67-positive neurons in hippocampus of mBACtgDyrk1a mice. Usingstereology, the density of GAD67-positive neurons was assessed byimmunohistochemistry on sagittal sections of hippocampal CA1stratum radiatum in mBACtgDyrk1a and WT mice. The density ofGAD67-positive neurons was 38% higher in Tg mice (Fig. 3A). Tofurther assess the alterations in the GABAergic pathway induced byDyrk1a triplication in a more complex genetic condition, we crossedhYACtgDyrk1a (human DYRK1A and 4 other genes) or Dp(16)1Yey(mouse Dyrk1a and 114 other genes) mice with GAD67-GFP mice.GAD67-GFP mice express GFP under the control of the GAD67 pro-moter, inducing fluorescence in any neurons expressing GAD67.We first visually assessed signal intensity in sagittal sections ofWT-GAD67-GFP versus hYACtgDyrk1a-GAD67-GFP; higher signalintensity was detected in hYACtgDyrk1a-GAD67-GFP vs the WT-GAD67-GFP littermates. Then, the density of GAD67-GFP-positiveneurons was measured; the density of GAD67-GFP-positive neuronswas significantly higher in both CA1 stratum radiatum (Fig. 3B) andprelimbic cortex (Supplementary Fig. 3) of hYACtgDyrk1a-GAD67-GFP mice. In Dp(16)1Yey-GAD67-GFP mice, the density of GAD67-GFP-positive neurons was assessed on sagittal sections in CA1stratum radiatum and was higher (Fig. 3C–E) in trisomic mice thanin WT littermates.
PTZ-induced seizures in mice with altered Dyrk1a copy number
To further evaluate the consequences of DYRK1A overexpression inmodifying the level of proteins involved in GABA production at synap-ses, we tested if these alterations may induce change in sensitivity toinduced seizures,which are regulated by functional GABA.We thereforechallengedWT, mBACtgDyrk1a, and Dyrk1a(+/−) mice to the seizure-provoking agent PTZ to determine whether changes in Dyrk1a copynumber modified susceptibility to seizure through alterations insynaptic plasticity. PTZ decreases the potency of GABA-mediated inhibi-tion in the brain and, depending on dosage, can produce myoclonicjerks and tonic-clonic convulsions with or without tonic seizures(Braudeau et al., 2011). We administered 50 mg/kg PTZ and scoredseizure activity in 3 stages: stage I (tail upward), stage II (tail curved to-ward the head), and stage III (falling on side and convulsing). MostmBACtgDyrk1a did not reach stage III (male, 29%; female, 0%) comparedto their WT littermates (male, 63%; female, 80%) (Fig. 4A). ForDyrk1a(+/−) mice, we scored the latency to reach stage III since allDyrk1a(+/−) reached this stage eventually. Latency to reach stage IIIwas significantly decreased in PTZ-injected Dyrk1a(+/−) mice(Fig. 4B).
Discussion
Here, we report the consequences of overexpression of murineDyrk1a on behavioral and molecular alterations involving, particu-larly, synaptic plasticity and E/I balance, in the mBACtgDyrk1atransgenic.
Increased copy number of murine Dyrk1a alters learning, memory, andmotor function
Presence of the murine BAC transgene results in overexpressionof Dyrk1a similar to that found in Ts65Dn and Dp(16)1Yey brains.Dyrk1a overexpression is sufficient to induce strong alterations inthe nervous system: motor learning and motor function (rotarod),short-term memory (Y-maze), and long-term memory and spatial

Fig. 2. Level of synaptic markers altered in both cortex and hippocampus of mBACtgDyrk1a. Immunoblottingwas used to determine relative protein levels in specified brain regions. Box-plots of expression relative to WT level for GAD67, VGAT1, NR2A, and VGAT1/VGLUT1; **p b 0.01, ***p b 0.001.
70 B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
learning (Morris water maze) are all reduced in mBACtgDyrk1amice. It was previously shown that DYRK1A overexpression inducesfunctional alterations in prefrontal cortex of mBACtgDyrk1a micewith dendritic alterations and anomalous NMDAR-mediated long-term potentiation (Thomazeau et al., 2014). The first model overex-pressing Dyrk1a was constructed using a rat cDNA under the controlof a metallothionein promoter (Altafaj et al., 2001); that model alsoexhibits motor alterations in open field activity but the phenotypesobserved using the Morris paradigm were less significant inTgDyrk1a than those highlighted in our murine BAC model. A secondmodel, constructed using a human BAC (Ahn et al., 2006), does notshow any deficit in rotarod testing, but exhibits impairment in theMorris paradigm, with almost no learning after 8 sessions. Thus,our observations are consistent with these previous models, butthe motor and learning phenotypes seem to be more pronouncedin our model.
Comparing the mBACtgDyrk1a behavioral deficits with other DSmodels suggest a role of increased level of DYRK1A in the phenotypes.Such deficits were previously observed in Ts65Dn and in Dp(16)Yey(% of alternation, latency in Morris water maze), and in Ts1Rhr (openfield, novel object recognition and T-maze tasks), each of which con-tains three copies of Dyrk1a and a varied number of other MMU16genes. These results emphasize the involvement of DYRK1A in mecha-nisms controlling cognition and synaptic plasticity. However alterationsof these mechanisms observed in Ts65Dnmice are the consequences ofa large segmental trisomy: the segment encodes proteins such as SYNJ1(Voronov et al., 2008), or OLIG1 and OLIG2 (Chakrabarti et al., 2010),normalization of which rescue also some phenotypic defects found inTs65Dn. Though the conclusion from studies on partial trisomies isthat the effects of individual genes are not strictly additive, phenotypesobserved in Ts65Dn and Dp(16)Yey are the consequences of the dosageof genes in 3 copies, including Dyrk1a.
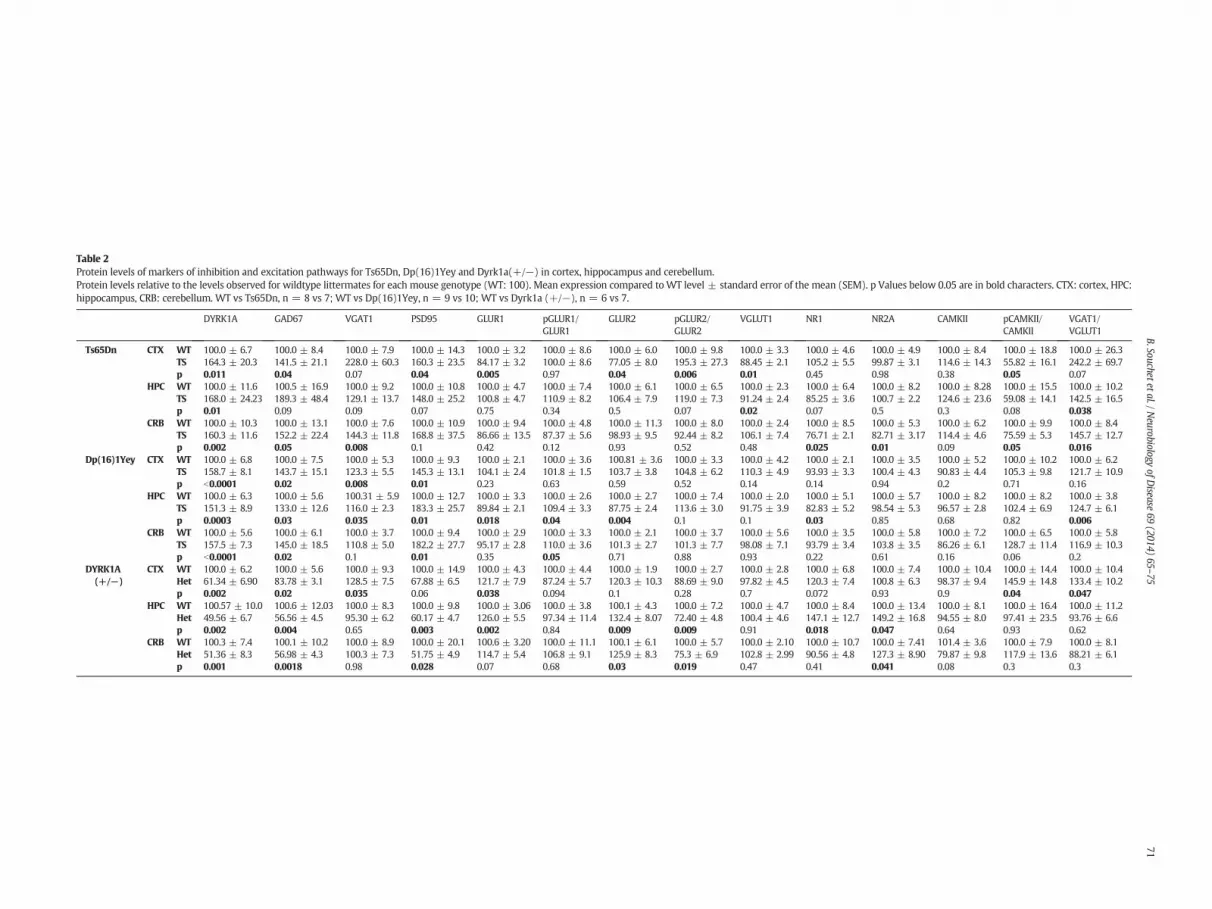
Table 2Protein levels of markers of inhibition and excitation pathways for Ts65Dn, Dp(16)1Yey and Dyrk1a(+/−) in cortex, hippocampus and cerebellum.Protein levels relative to the levels observed for wildtype littermates for each mouse genotype (WT: 100). Mean expression compared toWT level ± standard error of the mean (SEM). p Values below 0.05 are in bold characters. CTX: cortex, HPC:hippocampus, CRB: cerebellum. WT vs Ts65Dn, n = 8 vs 7; WT vs Dp(16)1Yey, n = 9 vs 10; WT vs Dyrk1a (+/−), n = 6 vs 7.
DYRK1A GAD67 VGAT1 PSD95 GLUR1 pGLUR1/GLUR1
GLUR2 pGLUR2/GLUR2
VGLUT1 NR1 NR2A CAMKII pCAMKII/CAMKII
VGAT1/VGLUT1
Ts65Dn CTX WT 100.0 ± 6.7 100.0 ± 8.4 100.0 ± 7.9 100.0 ± 14.3 100.0 ± 3.2 100.0 ± 8.6 100.0 ± 6.0 100.0 ± 9.8 100.0 ± 3.3 100.0 ± 4.6 100.0 ± 4.9 100.0 ± 8.4 100.0 ± 18.8 100.0 ± 26.3TS 164.3 ± 20.3 141.5 ± 21.1 228.0 ± 60.3 160.3 ± 23.5 84.17 ± 3.2 100.0 ± 8.6 77.05 ± 8.0 195.3 ± 27.3 88.45 ± 2.1 105.2 ± 5.5 99.87 ± 3.1 114.6 ± 14.3 55.82 ± 16.1 242.2 ± 69.7p 0.011 0.04 0.07 0.04 0.005 0.97 0.04 0.006 0.01 0.45 0.98 0.38 0.05 0.07
HPC WT 100.0 ± 11.6 100.5 ± 16.9 100.0 ± 9.2 100.0 ± 10.8 100.0 ± 4.7 100.0 ± 7.4 100.0 ± 6.1 100.0 ± 6.5 100.0 ± 2.3 100.0 ± 6.4 100.0 ± 8.2 100.0 ± 8.28 100.0 ± 15.5 100.0 ± 10.2TS 168.0 ± 24.23 189.3 ± 48.4 129.1 ± 13.7 148.0 ± 25.2 100.8 ± 4.7 110.9 ± 8.2 106.4 ± 7.9 119.0 ± 7.3 91.24 ± 2.4 85.25 ± 3.6 100.7 ± 2.2 124.6 ± 23.6 59.08 ± 14.1 142.5 ± 16.5p 0.01 0.09 0.09 0.07 0.75 0.34 0.5 0.07 0.02 0.07 0.5 0.3 0.08 0.038
CRB WT 100.0 ± 10.3 100.0 ± 13.1 100.0 ± 7.6 100.0 ± 10.9 100.0 ± 9.4 100.0 ± 4.8 100.0 ± 11.3 100.0 ± 8.0 100.0 ± 2.4 100.0 ± 8.5 100.0 ± 5.3 100.0 ± 6.2 100.0 ± 9.9 100.0 ± 8.4TS 160.3 ± 11.6 152.2 ± 22.4 144.3 ± 11.8 168.8 ± 37.5 86.66 ± 13.5 87.37 ± 5.6 98.93 ± 9.5 92.44 ± 8.2 106.1 ± 7.4 76.71 ± 2.1 82.71 ± 3.17 114.4 ± 4.6 75.59 ± 5.3 145.7 ± 12.7p 0.002 0.05 0.008 0.1 0.42 0.12 0.93 0.52 0.48 0.025 0.01 0.09 0.05 0.016
Dp(16)1Yey CTX WT 100.0 ± 6.8 100.0 ± 7.5 100.0 ± 5.3 100.0 ± 9.3 100.0 ± 2.1 100.0 ± 3.6 100.81 ± 3.6 100.0 ± 3.3 100.0 ± 4.2 100.0 ± 2.1 100.0 ± 3.5 100.0 ± 5.2 100.0 ± 10.2 100.0 ± 6.2TS 158.7 ± 8.1 143.7 ± 15.1 123.3 ± 5.5 145.3 ± 13.1 104.1 ± 2.4 101.8 ± 1.5 103.7 ± 3.8 104.8 ± 6.2 110.3 ± 4.9 93.93 ± 3.3 100.4 ± 4.3 90.83 ± 4.4 105.3 ± 9.8 121.7 ± 10.9p b0.0001 0.02 0.008 0.01 0.23 0.63 0.59 0.52 0.14 0.14 0.94 0.2 0.71 0.16
HPC WT 100.0 ± 6.3 100.0 ± 5.6 100.31 ± 5.9 100.0 ± 12.7 100.0 ± 3.3 100.0 ± 2.6 100.0 ± 2.7 100.0 ± 7.4 100.0 ± 2.0 100.0 ± 5.1 100.0 ± 5.7 100.0 ± 8.2 100.0 ± 8.2 100.0 ± 3.8TS 151.3 ± 8.9 133.0 ± 12.6 116.0 ± 2.3 183.3 ± 25.7 89.84 ± 2.1 109.4 ± 3.3 87.75 ± 2.4 113.6 ± 3.0 91.75 ± 3.9 82.83 ± 5.2 98.54 ± 5.3 96.57 ± 2.8 102.4 ± 6.9 124.7 ± 6.1p 0.0003 0.03 0.035 0.01 0.018 0.04 0.004 0.1 0.1 0.03 0.85 0.68 0.82 0.006
CRB WT 100.0 ± 5.6 100.0 ± 6.1 100.0 ± 3.7 100.0 ± 9.4 100.0 ± 2.9 100.0 ± 3.3 100.0 ± 2.1 100.0 ± 3.7 100.0 ± 5.6 100.0 ± 3.5 100.0 ± 5.8 100.0 ± 7.2 100.0 ± 6.5 100.0 ± 5.8TS 157.5 ± 7.3 145.0 ± 18.5 110.8 ± 5.0 182.2 ± 27.7 95.17 ± 2.8 110.0 ± 3.6 101.3 ± 2.7 101.3 ± 7.7 98.08 ± 7.1 93.79 ± 3.4 103.8 ± 3.5 86.26 ± 6.1 128.7 ± 11.4 116.9 ± 10.3p b0.0001 0.02 0.1 0.01 0.35 0.05 0.71 0.88 0.93 0.22 0.61 0.16 0.06 0.2
DYRK1A(+/−)
CTX WT 100.0 ± 6.2 100.0 ± 5.6 100.0 ± 9.3 100.0 ± 14.9 100.0 ± 4.3 100.0 ± 4.4 100.0 ± 1.9 100.0 ± 2.7 100.0 ± 2.8 100.0 ± 6.8 100.0 ± 7.4 100.0 ± 10.4 100.0 ± 14.4 100.0 ± 10.4Het 61.34 ± 6.90 83.78 ± 3.1 128.5 ± 7.5 67.88 ± 6.5 121.7 ± 7.9 87.24 ± 5.7 120.3 ± 10.3 88.69 ± 9.0 97.82 ± 4.5 120.3 ± 7.4 100.8 ± 6.3 98.37 ± 9.4 145.9 ± 14.8 133.4 ± 10.2p 0.002 0.02 0.035 0.06 0.038 0.094 0.1 0.28 0.7 0.072 0.93 0.9 0.04 0.047
HPC WT 100.57 ± 10.0 100.6 ± 12.03 100.0 ± 8.3 100.0 ± 9.8 100.0 ± 3.06 100.0 ± 3.8 100.1 ± 4.3 100.0 ± 7.2 100.0 ± 4.7 100.0 ± 8.4 100.0 ± 13.4 100.0 ± 8.1 100.0 ± 16.4 100.0 ± 11.2Het 49.56 ± 6.7 56.56 ± 4.5 95.30 ± 6.2 60.17 ± 4.7 126.0 ± 5.5 97.34 ± 11.4 132.4 ± 8.07 72.40 ± 4.8 100.4 ± 4.6 147.1 ± 12.7 149.2 ± 16.8 94.55 ± 8.0 97.41 ± 23.5 93.76 ± 6.6p 0.002 0.004 0.65 0.003 0.002 0.84 0.009 0.009 0.91 0.018 0.047 0.64 0.93 0.62
CRB WT 100.3 ± 7.4 100.1 ± 10.2 100.0 ± 8.9 100.0 ± 20.1 100.6 ± 3.20 100.0 ± 11.1 100.1 ± 6.1 100.0 ± 5.7 100.0 ± 2.10 100.0 ± 10.7 100.0 ± 7.41 101.4 ± 3.6 100.0 ± 7.9 100.0 ± 8.1Het 51.36 ± 8.3 56.98 ± 4.3 100.3 ± 7.3 51.75 ± 4.9 114.7 ± 5.4 106.8 ± 9.1 125.9 ± 8.3 75.3 ± 6.9 102.8 ± 2.99 90.56 ± 4.8 127.3 ± 8.90 79.87 ± 9.8 117.9 ± 13.6 88.21 ± 6.1p 0.001 0.0018 0.98 0.028 0.07 0.68 0.03 0.019 0.47 0.41 0.041 0.08 0.3 0.3
71B.Souchetetal./N
eurobiologyofD
isease69
(2014)65
–75

Fig. 3.GAD67-positive neuron density in the stratum radiatum of mouse models with 3 copies of Dyrk1a. (A–C) Density of GAD67-immunolabeled neurons (neurons/mm3) (ver-sus NeuN + neurons) in the hippocampal CA1 stratum radiatum (SR). Box-plots of levels in (A) mBACtgDyrk1a; (B) F1(hYACtgDyrk1a × GAD67-GFP); (C) F1(Dp(16)1Yey ×GAD67-GFP) relative to those of WT littermates. (d and e) Images of GFP + neurons in the stratum radiatum of GAD67-GFP-WT (D) and GAD67-GFP-Dp(16)1Yey (E), illustrating theincrease in density of GAD67-positive neurons in the SR of Dp(16)1Yey. In this model, total co-labeling of GFP with GAD67 was checked in the two genotypes by confocal microscopy(data not shown). *p b 0.05, **p b 0.01, ***p b 0.001.
Fig. 4. Sensitivity to PTZ-induced seizures relative to Dyrk1a gene copy number in mBACtgDyrk1a and Dyrk1a(+/−). (A) Distribution of the percentages of PTZ-injected mice in the 3stages of convulsion (I, II, III) in female (left) and male (right) mBACtgDyrk1a (n = 22, red) and their WT littermates (n = 22). (B) Latency (in sec) to reach stage III in (green)Dyrk1a(+/−) and their WT littermates (n = 10 per genotype). **p b 0.01.
72 B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75

73B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
DYRK1A is involved in the maintenance of E/I balance
We found that DYRK1A protein level is associated with expressionlevels of proteins involved in synaptic plasticity. Specifically, enzymesinvolved in decarboxylation of glutamate to produce GABA and in vesic-ular transport of GABA are higher inmicewith three copies ofDyrk1a. InDyrk1a single-copy mice, only GABA-producing enzymes are detectedat lower levels than in WT. The increase of VGAT1 in cortex may bedue to a compensation mechanism that has been already observed inculture of neurons lacking GAD67 (Lau and Murthy, 2012),and sug-gested to happen in face of a too low level of cytosolic GABA. In contrast,in the cerebellumGAD67 and VGAT1 levels are changed in the same di-rection. Thus, molecular data suggest that increasing Dyrk1a dosage in-duces activation of the GABA pathway with an increased productionand transport of GABA; decreasing the level of Dyrk1a induces a de-crease in both GADs. These molecular changes offer support for behav-ioral phenotypes observed in mouse models. Dyrk1a(+/−) mice showimpairment in the development of spatial learning strategies in ahippocampal-dependent memory task (Arque et al., 2008). In Ts65Dn,excessive GABAergic neurotransmission results in local over-inhibitionof hippocampal circuits, which dampens hippocampal synaptic plastic-ity and contributes to cognitive impairments; treatment with severalGABA(A) receptor antagonists results in increased plasticity andimprovedmemorydeficits in Ts65Dnmice (Fernandez et al., 2007). Def-icits in cognition and synaptic plasticity in Ts65Dn are also amelioratedby a selective inverse agonist of GABA-A receptorα5 subtype (Braudeauet al., 2011) or by GABA-B receptor antagonists (Kleschevnikov et al.,2012a).
Modification of GABAergic pathways in mBACtgDyrk1a mice sug-gested that PTZ, an inhibitor of GABA-A receptor (Kalueff, 2007),might produce different responses in WT and mBACtgDyrk1a mice.Indeed, mice with increased dosage of Dyrk1a are protected againstPTZ-induced seizures, reminiscent of findings from a similar approachin Ts65Dn (Braudeau et al., 2011) with a difference between male andfemale as previously reported (Medina et al., 2001). Mice with onecopy of Dyrk1a, however, display more convulsions following PTZ thanWT mice of the same background, confirming the protective effect ofDyrk1a, which likely functions by elevating GABAergic inhibition. Inter-estingly prominent epilepsy in addition to developmental delay and IDare associated to deletion or truncation of the DYRK1A gene in human(Courcet et al., 2012;Moller et al., 2008; van Bon et al., 2011). Moreovertruncating mutations in the gene have been found in cases with autism(O'Roak et al., 2012). GABAergic pathway alterations are also evidentthrough changes in GAD67 expression when DYRK1A is overexpressed.GAD67-GFP mice with 3 copies of Dyrk1a had more GAD67-positiveneurons than GAD67-GFP mice with 2 copies of Dyrk1a. Indeed,GAD67-positive neuronal density is increased over NeuN-positive neu-ronal density in Dyrk1a mBACtgDyrk1a mice, demonstrating thatDYRK1A has an effect very early during neuronal differentiation andcan shift the 1/5 balance (approximately 1/5 neurons is GABAergic inmany neocortical areas) (Sahara et al., 2012) toward a more GABAergicfate. DYRK1A overexpression may also affect the amount of Gad67mRNA produced in each GABAergic neuron. Interestingly an increasein GAD67 immunoreactivity has been previously reported in thesomatosensory cortex (Perez-Cremades et al., 2010) and in the dentateinner molecular layer of hippocampus (Martinez-Cue et al., 2013) ofTs65Dn mice. Additionally an increase in Parvalbumin-positive inter-neurons in the CA1 pyramidal layer of Ts65Dn hippocampus has beendescribed (Chakrabarti et al., 2010).
In general, expression of proteins involved in glutamate transportis decreased when DYRK1A is increased and increasedwhen DYRK1Ais decreased. Ts65Dn mice exhibit significantly reduced vesicularglutamate transporter-1 (VGLUT1) labeling compared to euploidcontrol mice (Rueda et al., 2010). In parallel with these results, weobserved an effect of Dyrk1a dosage on NR1, NR2A, GLUR1, GLUR2,and the activated form of CAMKII, components of the synaptic
machinery that are generally decreased in mBACtgDyrk1a and in-creased in Dyrk1a(+/−). We observed that other synaptic proteins,like PSD95, a scaffold protein involved in synaptic scaling that hasbeen proposed to block scaling down of the synapses (Sun andTurrigiano, 2011), are strongly altered by Dyrk1a dosage. These var-iations are also seen in brain samples from the two trisomic DSmodels Ts65Dn and Dp(16)1Yey mice. Moreover, GAD67 expressionis increased in both models in cortex, hippocampus and cerebellum.Though a trend toward increase in other proteins involved in theinhibition pathway and toward decrease in proteins involved in theexcitation pathway was observed, some differences could be dueeither to the number of genes in 3 copies (99 in the Ts65Dn versus115 genes in the Dp(16)1Yey), or to the 5.8 Mb additional trisomyfor MMU17 genes not orthologous to HSA21 genes, and transmittedin the Ts65Dn. We also cannot exclude that the difference of geneticbackground between these models may play a role, thoughmBACtgDyrk1a and Dp(16)1Yey were both developed on C57Bl/6J.Globally, these variations might correspond to an increase inGABAergic neurotransmission and a decrease in glutaminergictransmission. Moreover, preliminary data (not shown) from cortexof mice resulting from breeding Ts65Dn with Dyrk1a(+/−) wereobtained for the three genotypes [Ts65Dn, Dyrk1a(+/−) andTs65Dn-Dyrk1a(+/−)]: they showed significant Spearman correla-tions, positive for DYRK1A/GAD67 (r = 0.61, p = 0.02), and nega-tive for DYRK1A/GLUR2 (r = −0.56, p = 0.03). Our results providea link between an alteration in E/I balance and the molecular and be-havioral defects observed inmBACtgDyrk1a, Ts65Dn and Dp(16)Yey.Further, these findings potentially explain comprehensive deficits indeclarative learning and memory, which are believed to relate to ex-cessive inhibition, observed in individuals with DS.
The relationship between cognitive alterations and increased in-hibition has been established by many studies (Braudeau et al.,2011; Faizi et al., 2011; Fernandez et al., 2007; Kleschevnikov et al.,2004; Siarey et al., 1997) (for a review see Creau (2012); Mohler(2012); Potier et al. (2013)). Inhibiting GABA receptors induces im-provement in the cognitive functions of Ts65Dn (Braudeau et al.,2011; Fernandez et al., 2007). A direct measure of GABA releaseshows an increase in hippocampal and visual cortex synaptosomesof Ts65Dn mice compared to WT animals (Begenisic et al., 2011).However, a direct molecular link between the presence of three cop-ies of 99 genes in Ts65Dn and increased inhibition has not been iden-tified. It was hypothesized that KIR3.2 subunits (KCNJ6, GIRK2),which are triplicated in Ts65Dn, might result in increased inhibition(Cramer et al., 2010; Kleschevnikov et al., 2012a, 2012b); however, treat-ment targeting GABA-B receptors does not correct spontaneous locomo-tor activity nor T-maze performance (Kleschevnikov et al., 2012a).Similarly, a drug targeting Kir3.2 channels improves neither sensorimo-tor abilities nor Morris water maze performance (Vidal et al., 2012).The relationship between increased inhibition and cognition defects isfurther complicated by the observation that treatment with fluoxetine,a selective serotonin reuptake inhibitor known to lower GABAergic inhi-bition in adult rodent brains, has adverse effects on Ts65Dn adult mice(Heinen et al., 2012). Here, we show that DYRK1A is a major player inregulating the E/I balance through different synaptic pathways.
Recently, Ts65Dnmice were shown to be hypersensitive to the lo-comotor stimulatory effects of the high-affinity N-methyl-D-aspartate (NMDA) receptor (NMDAR) channel blocker, MK-801,when compared with euploid control mice (Costa et al., 2008): Inter-estingly, the molecular study of the effects of MK-801 treatmentshowed that treated mice present a strong decrease in the amountof DYRK1A in cortex and hippocampus (Siddiqui et al., 2008) sug-gesting that part of the action of MK-801might be through downreg-ulating DYRK1a. MK-801 treatment has already been shown to resultin decreased Gad67 expression in cortical areas through an unknownmechanism (Turner et al., 2010). This observation raises the possibil-ity that some beneficial effects of memantine, another uncompetitive

74 B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
N-methyl-D-aspartate (NMDA) receptor antagonists, on TS65Dncognitive alterations (Lockrow et al., 2011) might be linked to adecreased level of DYRK1A.
Recently, we have shown that the green tea flavonolepigallocatechin-gallate (EGCG), a DYRK1A inhibitor, rescues thecognitive deficits, observed using a MWM paradigm, of both segmentaltrisomy 16 (Ts65Dn) and transgenic mice overexpressing Dyrk1A in atrisomic or disomic genetic background, respectively (De la Torreet al., 2014). The panel of markers that have been characterized in thepresent study will be used to assess the molecular corrections thatcould be associated with this cognitive rescue.
In conclusion, our results represent the first experimental evidenceestablishing DYRK1A as one of the major targets for controlling E/Ibalance in diseases with altered interneuron functions and increasedinhibition, such as Down syndrome; it may be also an important playerin diseases with increased excitability (schizophrenia) or decreasedinhibition (autism) (Krumm et al., 2014).
Acknowledgments
We thank CNRS, INSERM, and the European commission (AnEUploidyproject: LSHG-CT-2006-037627) for support. Thisworkwas also support-ed by grants ANR-2009-DSTHER; Lejeune foundation-2010-2012;2009SGR1313; SAF2010-16427.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.04.016.
References
Ahn, K.J., et al., 2006. DYRK1A BAC transgenic mice show altered synaptic plasticity withlearning and memory defects. Neurobiol. Dis. 22, 463–472.
Altafaj, X., et al., 2001. Neurodevelopmental delay,motor abnormalities and cognitive def-icits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model ofDown's syndrome. Hum. Mol. Genet. 10, 1915–1923.
Arque, G., et al., 2008. Impaired spatial learning strategies and novel object recognition inmice haploinsufficient for the dual specificity tyrosine-regulated kinase-1A (Dyrk1A).PLoS One 3, e2575.
Begenisic, T., et al., 2011. Environmental enrichment decreases GABAergic inhibition andimproves cognitive abilities, synaptic plasticity, and visual functions in a mousemodel of Down syndrome. Front. Cell. Neurosci. 5, 29.
Belichenko, N.P., et al., 2009a. The “Down syndrome critical region” is sufficient in themouse model to confer behavioral, neurophysiological, and synaptic phenotypescharacteristic of Down syndrome. J. Neurosci. 29, 5938–5948.
Belichenko, P.V., et al., 2009b. Excitatory-inhibitory relationship in the fascia dentata inthe Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 512, 453–466.
Best, T.K., et al., 2012. Dysfunctional hippocampal inhibition in the Ts65Dn mouse modelof Down syndrome. Exp. Neurol. 233, 749–757.
Braudeau, J., et al., 2011. Specific targeting of the GABA-A receptor alpha5 subtype by aselective inverse agonist restores cognitive deficits in Down syndrome mice. J.Psychopharmacol. 25, 1030–1042.
Chakrabarti, L., et al., 2010. Olig1 and Olig2 triplication causes developmental brain de-fects in Down syndrome. Nat. Neurosci. 13, 927–934.
Costa, A.C., et al., 2008. Acute injections of the NMDA receptor antagonist memantine res-cue performance deficits of the Ts65Dn mouse model of Down syndrome on a fearconditioning test. Neuropsychopharmacology 33, 1624–1632.
Courcet, J.B., et al., 2012. The DYRK1A gene is a cause of syndromic intellectual disabilitywith severe microcephaly and epilepsy. J. Med. Genet. 49, 731–736.
Cramer, N.P., et al., 2010. GABAB-GIRK2-mediated signaling in Down syndrome. Adv.Pharmacol. 58, 397–426.
Créau, N., 2012. Molecular and cellular alterations in Down syndrome: toward the identi-fication of targets for therapeutics. Neural Plast. 2012, 171639.
Davisson, M.T., et al., 1993. Segmental trisomy as a mouse model for Down syndrome.Prog. Clin. Biol. Res. 384, 117–133.
De la Torre, R., et al., 2014. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cogni-tive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res.58, 278–288.
Delabar, J.M., et al., 1993. Molecular mapping of twenty-four features of Down syndromeon chromosome 21. Eur. J. Hum. Genet. 1, 114–124.
Dowjat, W.K., et al., 2007. Trisomy-driven overexpression of DYRK1A kinase in the brainof subjects with Down syndrome. Neurosci. Lett. 413, 77–81.
Escorihuela, R.M., et al., 1995. A behavioral assessment of Ts65Dn mice: a putative Downsyndrome model. Neurosci. Lett. 199, 143–146.
Faizi, M., et al., 2011. Comprehensive behavioral phenotyping of Ts65Dn mouse model ofDown syndrome: activation of beta1-adrenergic receptor by xamoterol as a potentialcognitive enhancer. Neurobiol. Dis. 43, 397–413.
Fernandez, F., et al., 2007. Pharmacotherapy for cognitive impairment in a mouse modelof Down syndrome. Nat. Neurosci. 10, 411–413.
Fotaki, V., et al., 2002. Dyrk1A haploinsufficiency affects viability and causes develop-mental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 22,6636–6647.
Guedj, F., et al., 2009. Green tea polyphenols rescue of brain defects induced by overex-pression of DYRK1A. PLoS One 4, e4606.
Guedj, F., et al., 2012. DYRK1A: a master regulatory protein controlling brain growth.Neurobiol. Dis. 46, 190–203.
Heinen, M., et al., 2012. Adult-onset fluoxetine treatment does not improve behavioralimpairments and may have adverse effects on the Ts65Dn mouse model of Downsyndrome. Neural Plast. 2012, 467251.
Kalueff, A.V., 2007. Mapping convulsants' binding to the GABA-A receptor chloride iono-phore: a proposed model for channel binding sites. Neurochem. Int. 50, 61–68.
Kleschevnikov, A.M., et al., 2004. Hippocampal long-term potentiation suppressed by in-creased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J.Neurosci. 24, 8153–8160.
Kleschevnikov, A.M., et al., 2012a. Deficits in cognition and synaptic plasticity in a mousemodel of Down syndrome ameliorated by GABAB receptor antagonists. J. Neurosci.32, 9217–9227.
Kleschevnikov, A.M., et al., 2012b. Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome.Neurobiol. Dis. 45, 683–691.
Krumm, N., et al., 2014. A de novo convergence of autism genetics and molecular neuro-science. Trends Neurosci. 37, 95–105.
Lau, C.G., Murthy, V.N., 2012. Activity-dependent regulation of inhibition via GAD67. J.Neurosci. 32, 8521–8531.
Li, Z., et al., 2007. Duplication of the entire 22.9 Mb human chromosome 21 syntenic re-gion onmouse chromosome 16 causes cardiovascular and gastrointestinal abnormal-ities. Hum. Mol. Genet. 16, 1359–1366.
Lockrow, J., et al., 2011. Effects of long-term memantine on memory and neuropa-thology in Ts65Dn mice, a model for Down syndrome. Behav. Brain Res. 221,610–622.
Martinez-Cue, C., et al., 2013. Reducing GABAA alpha5 receptor-mediated inhibitionrescues functional and neuromorphological deficits in a mouse model of Down syn-drome. J. Neurosci. 33, 3953–3966.
Medina, A.E., et al., 2001. Sex differences in sensitivity to seizures elicited by pentylenetet-razol in mice. Pharmacol. Biochem. Behav. 68, 591–596.
Möhler, H., 2012. Cognitive enhancement by pharmacological and behavioralinterventions: the murine Down syndrome model. Biochem. Pharmacol. 84,994–999.
Moller, R.S., et al., 2008. Truncation of the Down syndrome candidate gene DYRK1A intwo unrelated patients with microcephaly. Am. J. Hum. Genet. 82, 1165–1170.
Oegema, R., et al., 2010. Distinctive phenotypic abnormalities associated with submicro-scopic 21q22 deletion including DYRK1A. Mol. Syndromol. 1, 113–120.
Olson, L.E., et al., 2007. Trisomy for the Down syndrome 'critical region' is necessary butnot sufficient for brain phenotypes of trisomic mice. Hum. Mol. Genet. 16, 774–782.
O'Roak, B.J., et al., 2012. Multiplex targeted sequencing identifies recurrently mutatedgenes in autism spectrum disorders. Science 338, 1619–1622.
Perez-Cremades, D., et al., 2010. Alteration of inhibitory circuits in the somatosensorycortex of Ts65Dn mice, a model for Down's syndrome. J. Neural Transm. 117,445–455.
Potier, M.C., et al., 2014 Feb. Reducing GABAergic inhibition restores cognitive functions ina mouse model of Down syndrome. CNS Neurol. Disord. Drug Targets 13 (1), 8–15.
Rahmani, Z., et al., 1989. Critical role of the D21S55 region on chromosome 21 in the path-ogenesis of Down syndrome. Proc. Natl. Acad. Sci. U. S. A. 86, 5958–5962.
Reeves, R.H., et al., 1995. A mouse model for Down syndrome exhibits learning and be-haviour deficits. Nat. Genet. 11, 177–184.
Reinholdt, L.G., et al., 2011. Molecular characterization of the translocation breakpoints inthe Down syndrome mouse model Ts65Dn. Mamm. Genome 22, 685–691.
Rogers, D.C., et al., 2001. SHIRPA, a protocol for behavioral assessment: validationfor longitudinal study of neurological dysfunction in mice. Neurosci. Lett. 306,89–92.
Ronan, A., et al., 2007. Familial 4.3 Mb duplication of 21q22 sheds new light on the Downsyndrome critical region. J. Med. Genet. 44, 448–451.
Rubenstein, J.L., Merzenich, M.M., 2003. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Rueda, N., et al., 2010. Memantine normalizes several phenotypic features in the Ts65Dnmouse model of Down syndrome. J. Alzheimers Dis. 21, 277–290.
Sahara, S., et al., 2012. The fraction of cortical GABAergic neurons is constant from nearthe start of cortical neurogenesis to adulthood. J. Neurosci. 32, 4755–4761.
Siarey, R.J., et al., 1997. Altered long-term potentiation in the young and old Ts65Dnmouse, a model for Down Syndrome. Neuropharmacology 36, 1549–1554.
Siddiqui, A., et al., 2008. Molecular responses of the Ts65Dn and Ts1Cje mouse models ofDown syndrome to MK-801. Genes Brain Behav. 7, 810–820.
Singer, H.S., Minzer, K., 2003. Neurobiology of Tourette's syndrome: concepts of neuroan-atomic localization and neurochemical abnormalities. Brain Dev. 25 (Suppl. 1),S70–S84.
Smith, D.J., et al., 1997. Functional screening of 2 Mb of human chromosome 21q22.2 intransgenic mice implicates minibrain in learning defects associated with Downsyndrome. Nat. Genet. 16, 28–36.
Sun, Q., Turrigiano, G.G., 2011. PSD-95 and PSD-93 play critical but distinct roles in synap-tic scaling up and down. J. Neurosci. 31, 6800–6808.

75B. Souchet et al. / Neurobiology of Disease 69 (2014) 65–75
Tamamaki, N., et al., 2003. Green fluorescent protein expression and colocalization withcalretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J.Comp. Neurol. 467, 60–79.
Tejedor, F., et al., 1995. minibrain: a new protein kinase family involved in postembryonicneurogenesis in Drosophila. Neuron 14, 287–301.
Thomazeau, A., et al., 2014. Prefrontal deficits in a murine model overexpressing thedown syndrome candidate gene dyrk1a. J. Neurosci. 34, 1138–1147.
Turner, C.P., et al., 2010. Postnatal exposure to MK801 induces selective changes inGAD67 or parvalbumin. Exp. Brain Res. 201, 479–488.
van Bon, B.W., et al., 2011. Intragenic deletion in DYRK1A leads to mental retardation andprimary microcephaly. Clin. Genet. 79, 296–299.
Vidal, V., et al., 2012. Lack of behavioral and cognitive effects of chronic ethosuximide andgabapentin treatment in the Ts65Dnmouse model of Down syndrome. Neuroscience220, 158–168.
Voronov, S.V., et al., 2008. Synaptojanin 1-linked phosphoinositide dyshomeostasis and cog-nitive deficits in mouse models of Down's syndrome. Proc. Natl. Acad. Sci. U. S. A. 105,9415–9420.
Wassef, A., et al., 2003. GABA and schizophrenia: a review of basic science and clinicalstudies. J. Clin. Psychopharmacol. 23, 601–640.
Yu, T., et al., 2010. Effects of individual segmental trisomies of human chromosome 21syntenic regions on hippocampal long-term potentiation and cognitive behaviors inmice. Brain Res. 1366, 162–171.





























