Embed Size (px)
Citation preview

MASARYK UNIVERSITY
FACULTY OF SCIENCE
DISSERTATION THESIS
MARTIN SOJKA
BRNO 2019

MASARYK UNIVERSITY FACULTY OF SCIENCE
NOVEL PT AND RU COMPLEXES FROM THE PERSPECTIVES OF THEORY
AND MEDICINAL APPLICATIONS
DISSERTATION THESIS
Martin Sojka
Supervisor:
Dr Marek Nečas
DEPARTMENT OF CHEMISTRY
BRNO 2019

3
BIBLIOGRAFICKÝ ZÁZNAM AUTOR Mgr. Martin Sojka
NÁZEV PRÁCE Novel Pt and Ru complexes from the perspectives of theory and medicinal applications
STUDIJNÍ PROGRAM Chemie
SPECIALIZACE Anorganická chemie
ŠKOLITEL doc. Mgr. Marek Nečas, Ph.D.
AKADEMICKÝ ROK 2019/2020
POČET STRAN 227
KLÍČOVÁ SLOVA platina, ruthenium, krystalografie, rakovina, metaloléčiva, NMR, paramagnetismus, cyklometalace

4
BIBLIOGRAPHIC ENTRY AUTHOR Martin Sojka, M.Sc.
TITLE OF THESIS Novel Pt and Ru complexes from the perspectives of theory and medicinal applications
DEGREE PROGRAMME Chemistry
SPECIALISATION Inorganic chemistry
SUPERVISOR Marek Nečas, PhD.
AKADEMIC YEAR 2019/2020
NUMBER OF PAGES 227
KEYWORDS platinum, ruthenium, crystallography, cancer, metallodrugs, NMR, paramagnetism, cyclometalation

5
ABSTRAKT
Tato disertační práce popisuje strukturní a fyzikálně-chemické vlastnosti a biologickou aktivitu koordinačních sloučenin Pt a Ru s P-a N-donorovými ligandy. Komplexy byly připraveny a studovány běžnými analytickými metodami, zejména pomocí vícejaderné (1H, 13C, 31P, 195Pt) NMR spektroskopie a rentgenové difrakce. Experimentálně získaná data byla v řadě případů doplněna výpočetními metodami jako jsou DFT a QTAIM.
Úvodní část práce zahrnuje témata z koordinační chemie Pt a Ru a jejich komplexů používaných jako protinádorová léčiva. Dále jsou diskutovány fosfanové ligandy a cyklometalační reakce. Hlavní část pak shrnuje výsledky práce, které byly publikovány nebo jsou připraveny k odeslání.
První soubor prací se zabývá syntézou komplexů Pt(II) s fosfanovými ligandy. Vliv intramolekulární vodíkové vazby na strukturu komplexů byl zkoumán s využitím množství strukturních dat a výpočtů QTAIM. Kromě toho byl zkoumán a popsán vliv vodíkové vazby na reaktivitu a cyklometalaci příslušných komplexů. Druhý soubor prací je zaměřen na syntézu komplexů Ru(III) a Ru(II) jako potenciálních protinádorových látek. Byly připraveny a charakterizovány komplexy Ru(III) s pyridinovými ligandy, přičemž vzniklá řada byla použita pro systematické zkoumání vztahu mezi elektronovou strukturou a chemickými posuny NMR těchto paramagnetických systémů. Organokovové supramolekulární komplexy Ru(II) na bázi kukurbiturilu byly studovány jako látky s antimetastatickou aktivitou. Čtyři nové supramolekulární sloučeniny Ru(II) byly charakterizovány a potvrzeny jako účinné látky proti rakovinným buněčným liniím MCF-7 a MDA-MB-231.

6
ABSTRACT
The present work describes the synthesis and investigation of structural and physicochemical properties and biological activity of Pt and Ru coordination compounds with P- and N-donor ligands. The complexes were prepared and studied by analytical methods, mainly utilising multinuclear (1H, 13C, 31P, 195Pt) solution NMR spectroscopy and X-ray crystallography. The experimental findings were supported extensively by computational methods, for instance, by DFT and QTAIM calculations.
The introductory part covers topics on the coordination chemistry of Pt and Ru and their complexes used as anticancer agents as well as general information on phosphine ligands and cyclometalation reactions. The central part summarises the published and submitted results and comments on them. The platinum part deals with the synthesis of Pt(II) complexes with various phosphine ligands. The impact of intramolecular hydrogen bonding on the structure of the complexes was investigated using a substantial amount of structural data and the results of the QTAIM calculations. Also, the influence of the hydrogen bonding on the reactivity and cyclometalation of the respective complexes was investigated and described. The second part is focused on the synthesis of Ru(III) and Ru(II) complexes as potential anticancer agents. Various Ru(III) complexes with pyridine ligands were prepared and characterised, and the resulting series was used for systematic investigation of the link between the electronic structure and the NMR chemical shifts in paramagnetic systems. Further, the organometallic Ru(II) complexes built on a cucurbituril-based supramolecular core were evaluated as antimetastatic agents. The four novel supramolecular Ru(II) compounds were characterised and confirmed as potent agents against MCF-7 and MDA-MB-231 cancerous cell lines.

7
ACKNOWLEDGEMENTS
In the following lines, I would like to express my sincere gratitude to my family and co-workers. Thanks to those, I have been able to follow my research ideas freely with passion.
Here, I want to thank all teachers and students of inorganic chemistry at our faculty, namely to Assoc. Prof. J. Toužín, who introduced me to inorganic chemistry, to Assoc. Prof. J. Novosad for his support, advice and comments, to Prof. J. Pinkas for his counselling regarding general chemistry and NMR spectroscopy, to Dr M. Černík for his inspiring precision, countless discussions and immense knowledge, to Assoc. Prof. M. Munzarová for she was able to introduce me painlessly to the basics of quantum chemistry and to Prof. J. Příhoda for his inspiring spirit and enthusiasm.
I am grateful to Prof. W. Plass from Jena for enlightening me the first glance of studying abroad and for valuable advice regarding my scientific writings. My thanks go to all good friends from his research group, for I spent such an excellent time over there. Also, I am happy to say, I was given a chance to cooperate with Prof. R. Marek on many scientific projects during my studies. His admirable leadership and high level of research standards provided me with powerful motivation for future work.
I was fortunate having a chance to enjoy such a great time with my colleagues from the inorganic chemistry department, Z. Moravec, I. Doroshenko, M. Babiak, A. Stýskalík, D. Škoda, J. Podhorský, V. Vykoukal, V. Matuška, and others. Your professionality, friendship, and constant optimistic attitude were very helpful. I am forever indebted to Dr L. Jeremias for his friendship, loyalty, calm and positive attitude, and countless short and long (not always scientific) discussions.
Last but not least, I would like to thank my family for supporting me spiritually, for their patience and understanding. Mainly, I thank my mom for she was willing and able to motivate me and bring me to my senses in the times of struggle. I also thank my wife Tereza for her patience, support, and for that, she saw and accepted the reasons for my doings. Foremost, I would like to thank my supervisor,

8
Assoc. Prof. M. Nečas for his continuous support of my research, for his patience, inspiration, motivation, friendship and rationality. His guidance helped me in the time of my early studies, later in writings and research activities, and I could hardly imagine having a better advisor and mentor. I am more than grateful for all the experience I could gain from him, and for all the fun we have had in the past years.

9
solve et coagula

10
Copyright ©
Martin Sojka

11
TABLE OF CONTENTS
PREFACE 13
CHAPTER 1: COORDINATION CHEMISTRY OF PT AND RU 14
1. INTRODUCTION 15 2. COORDINATION CHEMISTRY OF PLATINUM 16 3. COORDINATION CHEMISTRY OF RUTHENIUM 20 4. BIOACTIVE COMPLEXES OF PLATINUM AND RUTHENIUM 23 REFERENCES 32
CHAPTER 2: PHOSPHINE LIGANDS 37
1. INTRODUCTION 38 2. PHOSPHINOAMINE LIGANDS 40 3. PHOSPHINOAMINES IN COORDINATION CHEMISTRY 50 REFERENCES 55
CHAPTER 3: CYCLOMETALATION REACTIONS 60
1. CYCLOMETALATION REACTIONS 61 2. CYCLOMETALATED COMPLEXES OF PGMS 66 REFERENCES 69
CHAPTER 4: DISCUSSION OF PUBLISHED RESULTS 72
1. GENERAL REMARKS 73 2. SUMMARY OF THE RESULTS 73 3. CONCLUDING REMARKS AND FUTURE OUTLOOK 79 4. WORK ON PLATINUM COMPLEXES 81 5. WORK ON RUTHENIUM COMPLEXES 82

12
LIST OF ABBREVIATIONS
BINAP 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl Bp boiling point bpy bipyridine Bu butyl cbdca 1,1-cyclobutanedicarboxylate COD 1,5‐cyclooctadiene Cy cyclopentadienyl
DIOP 2,3‐O‐isopropylidene‐2,3‐dihydroxy‐1,3‐bis(diphenylphosphino)butane
DMAP N,N-dimethyl-4-aminopyridine en ethylenediamine Et ethyl Et2O diethyl ether Et3N triethylamine EtAc ethyl acetate FDA The Food and Drug Administration US federal agency HMDS hexamethyldisilazane / bis(trimethylsilyl)amine IR infrared spectroscopy J coupling constant L-dach trans-L-diaminocyclohexane Me methyl MeOH methanol MLCT metal-to-ligand charge transfer MO molecular orbital Mp, MP melting point MRI Magnetic resonance imaging NMR nuclear magnetic resonance PET positron emission tomography PGM, PGMs platinum group metal(s) Ph phenyl py pyridine R any hydrocarbon SPECT single-photon emission computed tomography tBu tert-butyl tBuONa sodium tert-butoxide THF tetrahydrofuran vs, s, m, w IR bands intensity XRD X-ray diffraction Y yield δ chemical shift η hapto

13
PREFACE
Whether we want it or not, chemistry interferes with our lives from all sides. Many chemistry disciplines are not commonly encountered, but it is good to be aware of the overlap of basic and applied research with society. Coordination chemistry usually dates back to the times of Alfred Werner, humans, however, encountered coordination compounds since time immemorial, without any idea of it, and the extensive development of the discipline over the past half-century has shown its potential.
In bachelor and diploma work, I focused on the synthesis of multifunctional phosphine ligands and related complexes with various transition metals. Naturally, this resulted in the desire for a more in-depth examination of the work in progress, which brought several exciting insights into the structure and reactivity of platinum complexes. In the following years, I was allowed to participate in two grant projects as a coordination chemist. The projects were focused on the development of platinum and ruthenium anticancer agents because both the platinum and ruthenium represent the most prominent groups of anticancer agents being developed.
The thesis aims to give the reader a literary overview in the form of three summarising chapters concerning the published works. A brief description of the coordination chemistry of platinum and ruthenium, as well as the corresponding anticancer metallodrugs, is provided concerning different oxidation states and biological activity of both metals. The next chapter is focused on phosphine ligands, their properties, synthesis and utilisation in coordination chemistry. The third chapter summarises the findings on cyclometalation reactions of transition metal complexes.

14
CHAPTER 1
COORDINATION CHEMISTRY OF PT AND RU
TABLE OF CONTENT
1. Introduction 15 2. Coordination chemistry of platinum 16 2.1. General considerations 16 2.2. Platinum coordination compounds 17 3. Coordination chemistry of ruthenium 20 3.1. General considerations 20 3.2. Ruthenium coordination compounds 20 4. Bioactive complexes of platinum and ruthenium 23 4.1. Cancer 24 4.2. Anticancer complexes of platinum 25 4.3. Anticancer complexes of ruthenium 28 References 31

15
1. INTRODUCTION
Metal ions play an essential role in both synthetic1–4 and medicinal chemistry,5–9 with applications ranging from industrial transformations to therapy and diagnosis. The platinum group metals (PGMs) represent, without a doubt, one of the essential classes of heavy elements with significant impact on society on a large scale. Ruthenium and platinum stand out because of their prominent and well-established applications in electrochemistry, photovoltaics, catalyses, and chemotherapy. Stability, accessible oxidation states, geometrical preferences, and variability in the choice of ligands result in an enormous number of references on both metals. Following sections describe their properties, coordination chemistry, and applications.
Platinum has been found in ancient Egyptian and pre-Columbian artefacts, while the first European description of the element comes from the 18th century.10 Russia, Columbia, Borneo, Burma, and Canada are the main producers, while the overall demand between 2016 – 2018 raised by 7 t from 51 to 58 per year.11,12 Procedures for industrial extraction are usually a subject of a trade secret, however, recovering Pt from laboratory wastes have been described.13 Platinum has six isotopes, of which the 194Pt (32.9 %) and 195Pt (33.8%) are the most abundant. Also, the 195Pt has a nuclear spin ½ and is NMR active, which can be exploited for investigation of its compounds by measurements of platinum spectra or by observing couplings with other nuclei. Platinum is known for being inert in the presence of mineral acids, but it is attacked by aqua regia, fused alkalis, or chlorine and fluorine when in red heat.12 The usual oxidation states of platinum in compounds are Pt(II) and Pt(IV). The Pt(I) and Pt(III) are less common and are often stabilised by metal-metal bonding in bi- or polymetallic species. Elemental Pt is successfully employed in petroleum reforming, hydrogenation and hydrosilylation of unsaturated hydrocarbons, production of HNO3, and removing pollutants from power generators.14–16 Pt is usually applied in a colloidal form on aluminium oxide, graphite, or other high surface area support improving its catalytic activity. Also, in addition to numerous biomedical devices, Pt compounds are used clinically in important antitumor agents such as cisplatin cis-[PtCl2(NH3)2],

16
carboplatin cis-[Pt(cbdca)(NH3)2], and oxaliplatin [Pt(ox)(L-dach)] (see Fig. A1). The PtCl2, K2[PtCl4], PtCl2(cod), K[PtCl3(en)], and H2PtCl6 represent most common starting compounds used for the synthesis of various platinum complexes.17
Figure A1. Representative examples of Pt and Ru complexes; for Pt the FDA approved anticancer drug oxaliplatin (left), and for Ru the Grubbs catalyst (right).
Ruthenium was recognised as an element in the 19th century by K. E. Clauss, who named it after the country of his origin, Russia (lat. Ruthenia). Ruthenium is (after Rh) the second least abundant PGM (10−3 to 10−4 ppm), and yet the demand for Ru is on the rise because of the increased interest from the electronics industry. Because of high demand and low incidence, Ru can become a strategic raw material, with significant deposits located in Russia, South America and South Africa. Ruthenium has seven stable isotopes, with the 102Ru (31.6 %) being the most abundant of all. Among the PGMs, Ru shows the broadest range of oxidation states, from Ru(−II) to Ru(VIII), the essential being Ru(II) and Ru(III).18,19 The very rich coordination and organometallic chemistry of Ru have tremendous application potential in electro- and photochemistry, catalyses, and biology.20,21 For instance, R. Noyori obtained the Nobel Prize in Chemistry in 2001 for hydrogenations chirally catalysed by Ru((S)-BINAP)(OAc)2, and the Grubbs catalyst (RuCl2(PhCH)(PCy3)2) yielded a Nobel Prize for catalysis of olefin metathesis in 2005.
2. COORDINATION CHEMISTRY OF PLATINUM
2.1. GENERAL CONSIDERATIONS
The application potential of produced compounds stimulates the development of Pt coordination chemistry. Also, many essential concepts in coordination chemistry, such as square-planar coordination or the trans effect, were first discussed on Pt complexes. The coordination number six

17
is rarely exceeded and is usually found in either complicated heterometallic structures or with carbonyl and carborane ligands (e.g. BABYEI, KOMJEB, from CSD22). In general, the coordination number six is typical for Pt(IV), but examples with Pt(II) are also available.23,24 For the Pt(II) complexes, however, the coordination number four with 16-electron square-planar geometry is usually found.
Figure A2. Subsequent dissociation of PPh3 molecules from the tetrahedral [Pt(PPh3)4] complex in solution and the formation of complexes with different geometries.
An exciting group of complexes features the complexes of Pt(0), which can be tetrahedral, planar three-coordinate, or linear two-coordinate. Soft and π-acceptor ligands such as acetylenes, olefins, and especially phosphines are necessary to stabilise the zerovalent Pt. A wide range of Pt(0) complexes is known as derivatives of the tetrahedral [Pt(PPh3)4], which is formed by the reduction of [PtCl2(PPh3)2] by hydrazine or KOH in alcohol. The [Pt(PPh3)4] dissociates readily in solution by sequential loss of PPh3 (see Fig. A2). The tris(phosphine) Pt complex is planar, while the bis(phosphine) (analogous to isolable [Pt(PCy3)2] species25) represents one of the few linear Pt complexes.26–28 The CSD22 offers many examples of seemingly unusual oxidation states of platinum (I, III) with Pt–Pt bonds. Such compounds tend to preserve the square-planar and octahedral geometries, although these are usually deformed. Examples of these complexes are shown in Fig. A3. Pt(III) complexes are often mixed Pt(II)/Pt(IV) complexes or chain structures.29–31
2.2. PLATINUM COORDINATION COMPOUNDS
Pt(II) represents the most stable oxidation state of platinum. The complexes of Pt(II) adopt almost always square planar geometry as expected for a d8 metal, but rare five-coordinate species can also be formed (see Fig. A3).32,33 Pt(II) forms a wide range of Pt–E bonds where E represents almost any element (metals included). Also, many square-planar Pt complexes can stack via Pt–Pt interaction in the solid-state, resulting in compounds with semiconducting and photoconducting

18
properties,34,35 luminescent thermochromism,36 and vapoluminiscence.37–
40 Some of these compounds are also employed as hydrogenation catalyst for alkynes.41,42
Figure A3. Examples of Pt complexes with rather unusual oxidation states and geometries of Pt coordination polyhedra; hydrogens are omitted for clarity. Both the binuclear square-planar43 Pt(I) and octahedral44 Pt(III) complexes are neutral; the tetragonal-pyramidal33 and the trigonal bipyramidal45 Pt(II) fragments are complex cations (the anions are omitted from the picture).
Substitution reactions in Pt(II) complexes usually take an associative pathway via an 18-electron five-coordinate intermediate.11 Some ligands such as thiourea, CO or CN− favour substitutions trans to themselves (see Fig. A4). The situation described relates to a more general phenomenon driven by electronic effects of the ligands, i.e. the trans effect,46,47 described on cis and trans isomers of [PtCl2(NH3)2].48
The amine complexes form the most prominent group of Pt(II) complexes. Not only they were important in A. Werner’s studies,49

19
but the cis and trans complexes [PtCl2(NH3)2] have taken on particular importance as anticancer agents that have led to sharp falls in mortality from a variety of cancers.50 Apart from the abovementioned isomers Pt(II) form halo-bridged complexes of the type [(amine)XPt(µ-X)2PtX(amine)], complexes with chelating ligands like bipy, and porphyrin, or soluble nitrile complexes [PtCl2L2] (L = RCN), which are useful as precursors in synthetic chemistry.51
Figure A4. The trans effect demonstrated on a ligand substitution reaction using a general formula of square-planar Pt(II) complex with trans-directing ligand T. Note, that the intensity of the trans effect follows the exemplary sequence: F−, H2O, OH− < NH3 < py < Cl− < Br− < I−, SCN−, NO2−, SC(NH2)2, Ph− < SO3
2− < PR3, AsR3, SR2, CH3−
< H−, NO, CO, CN−, C2H4.
The two most important classes of Pt(II) complexes represent mononuclear [PtX2L2], and dinuclear [LXPt(µ-X)2PtXL], where X = halide and L = phosphines, arsines, and stibines. For the mononuclear complexes, both the cis and trans isomers can usually be prepared and isolated. The dinuclear species readily undergo bridge-splitting with a variety of ligands to give related mononuclear products. Complexes are usually air-stable and soluble in organic solvents, the solubility, however, can be adjusted by appropriate choice of ligands.10,52,53 Introducing phosphine ligands enables the possibility for investigation of complexes utilising 31P NMR spectroscopy; 31P – 195Pt nuclei interactions show 1JPtP coupling constants in the range from 3000 to 6000 Hz. Notably, the cis and trans isomers can be distinguished by different ranges of the 1JPtP coupling constants.54,55
While Pd only reluctantly forms MIV complexes, the tetravalent state is typical for platinum. It is likely because of lower ionisation energy and stronger M–L bonds.19 The d6 Pt(IV) complexes adopt octahedral geometry formed by a dissociative pathway via a 16-electron five-coordinate intermediates.56,57 Although the coordination chemistry of Pt(IV) is not so rich as of Pt(II), the late interest in Pt(IV) lies mainly in

20
the potent biological activity of the Pt(IV) complexes.58,59 Important aspect of the Pt(IV) complexes is their susceptibility to reduction to Pt(II) while both the axial ligands leave the structure (see Fig. A5). This principle becomes one of the significant sales pitches of the Pt(IV)-based anticancer agents (see part 4.2. Anticancer complexes of platinum).60
3. COORDINATION CHEMISTRY OF RUTHENIUM
3.1. GENERAL CONSIDERATIONS
Coordination chemistry of ruthenium mainly revolves around two oxidation states, Ru(II) and Ru(III).61,62 It comprises a plethora of coordination and organometallic compounds with application ranging from photochemistry to catalyses. However, most of them usually starting with ruthenium trichloride, RuCl3.nH2O, as a universal precursor in Ru coordination chemistry (see Fig. A5).19 Although ruthenium adopts every oxidation state, the most common is the Ru(II), Ru(III), and Ru(IV) states with octahedral geometry, while the unusual oxidation states (e.g., –II, 0, I) are accompanied by tetrahedral or trigonal-bipyramidal geometry, for instance, in tetrahedral d10 [Ru(CO)4]2– and d1 RuO4–, or trigonal-bipyramidal d8 Ru(CO)5 and d7 Ru(CO)2(Pt-Bu2Me)2. The lower oxidation states of ruthenium mainly involve π-acceptor ligands (PR3, CO)63 and nonclassical bonds with various olefins like cyclooctadiene (see Dewar–Chatt–Duncanson MO description64,65). It should be noted that almost all the low-valent Ru complexes are sensitive to oxygen and moisture.
3.2. RUTHENIUM COORDINATION COMPOUNDS
The Ru(0) complexes are coordinatively unsaturated and prone to a formation of clusters with M–M bonds. Although the Ru(I) complexes might be of interest because they are paramagnetic, they are inherently unstable and decompose rapidly. Interestingly, many compounds can undergo reversible conversions between them while retaining their geometry making ruthenium complexes attractive for reversible redox chemistry.66 On the other hand, the chemistry of the higher oxidation states is dominated by metal-ligand multiple bonds, with the oxo ligand being the ligand of choice.

21
Figure A5. The RuCl3.nH2O as a versatile starting material in ruthenium coordination
chemistry. Examples of various methods of preparation of Ru(II) complexes.
The d6 Ru(II) complexes are diamagnetic, reasonably labile, and tend to preserve the configuration during substitution reactions suggesting an associative mechanism. Ru(II) is prepared usually from aqueous and alcoholic media via a blue solution (the nature of which is yet undetermined),67 which is usually used as starting material for the syntheses of Ru(II) complexes. Numerous complexes were described containing π-acid ligands, halides, ammines, and interestingly also molecular nitrogen, which only illustrate excellent π-bonding characteristics of Ru. The ligand exchange rates for small inorganic and biomolecular ligands were investigated employing various spectroscopic methods.68–73
The established synthetic protocols with N- and P-ligands along with an extensive application of analytical and biological methods stimulate the research of various catalysts and metallodrugs (see part 4.3 Anticancer complexes of ruthenium). The Ru complexes are considered less active than those of Ir or Rh, but this is an advantage in chemoselective hydrogenations (e.g. synthesis of Naproxen75),

22
many Ru(II) compounds have been successfully applied to catalytic transformations,76–78 such as Roper’s79 [Ru(PPh3)3(CO)2] and Grubbs catalysts. With sterically less-hindering ligands the Ru(II) can be oxidised by air to give the Ru(III). The latter is usually not feasible with organometallic Ru(II) complexes bearing arene-type ligands (e.g., η5-C5R5, η6-C6R6, R = H, alkyl or aryl), which constitute a considerable group of Ru(II) coordination compounds. The organometallic complexes are considered more stable than the traditional coordination compounds (although they are generally labile enough to be substituted by moderately nucleophilic ligands). The properties of a bulky arene system in conjunction with the unique properties of ruthenium have also been incorporated in the concept of electron-fuelled molecular rotary motors.66,80 Significant part of Ru(II) chemistry is concerned with the luminescence of various bipyridine and polypyridine complexes, which are tested as photocatalysts,81 light antennas,82 or luminescent probes.83–86
The Ru(III) has equally great coordination chemistry with both π-acceptors and -donor ligands, in which it predominantly adopts an octahedral geometry. Significant difference plays the d5 low spin configuration resulting in paramagnetic behaviour of Ru(III) compounds and thus employment of additional analytical techniques (e.g. EPR, SQUID). Apart from traditional methods, several groups are also interested in the development of paramagnetic NMR87–93 for investigation of systems with unpaired electrons such as Ru(III). An excellent review came out recently in this regard.94 For the preparation of Ru(III) complexes, the RuCl3⋅nH2O is also a starting material. As a harder acceptor, Ru(III) forms various oxo-bridged polynuclear complexes (e.g. [Ru3(3-O)(2-OAc)6ROH]+),95,96 and similarly to Ru(II), Ru(III) forms a large variety of complexes with N-donor ligands (e.g. NO, en, py, bpy) with exciting properties. For instance, the complexes bearing NO were studied as light-triggered NO-releasing therapeutic agents for blood pressure regulation,97,98 while many pyridines (and other N-heterocyclic) derivatives were investigated for antimetastatic properties.99–104 Many complexes with a variety of S- and O-donor ligands (e.g. oxalates, acetylacetonates, thiols, sulfoxides) are also investigated.105

23
4. BIOACTIVE COMPLEXES OF PLATINUM AND RUTHENIUM
Among all different branches of chemistry, the medicinal inorganic (sometimes bioinorganic) chemistry can be considered as a rather young discipline.9 It is, however, contrary to the historical use of metals in pharmaceutical potions by the ancient civilisations of Mesopotamia, Egypt, India, and China.8,106 In modern era, one of the first metallodrugs was salvarsan, developed by P. Ehrlich in 1912 providing an effective treatment against syphilis.107,108 Bioinorganic chemistry deals with the development of bioactive metal complexes (metallodrugs) and their introduction into biological systems, investigates their speciation and the impact on treatment of various diseases.109,110 Thus the introduction of metallodrugs into biological systems is useful for both diagnostic and therapeutic purposes. Some of the FDA approved metallodrugs are shown in Figure A6. The overall impact of metallodrugs on human society can be demonstrated on the contrast agents, which have contributed to an increased understanding and early detection of cancer or cardiologic diseases by SPECT, PET, and MRI techniques.
Figure A6. Examples of FDA approved metallodrugs. The 99Tc-based Cardiolite® for diagnosis of a heart condition and stress test,116 Gd(III)-based Magnevist117 as an MRI contrast agent. Structural data were obtained from the CSD (AMUCAM, HEQBUA01,).22
Although the creation of salvarsan is widely recognised as the birth of modern chemotherapy by metallodrugs, the cisplatin (discovered

24
unexpectedly by B. Rosenberg and L. VanCamp in 1965) remains the prodigy in the field.111,112 Studies on cisplatin revealed its antitumor activity, which was patented and approved by the FDA for cancer treatment in 1978.113 From that time on, many patients with oncological diseases are treated using combined therapies which usually constitute combination of various organic (e.g. paclitaxel, gemcitabine) and Pt-based (e.g. cisplatin, carboplatin, oxaliplatin)50,114,115 anticancer drugs.
The used platinum chemotherapeutics, however, introduce severe side effects (e.g. nephrotoxicity118 and ototoxicity, neurotoxicity, or myelosuppression)119 originating from both cumulative systemic toxicity and non-specific mechanism of action. It is why the development of a new generation of platinum and non-platinum based anticancer metallodrugs50,120 is a very substantial part of current research in both coordination and medicinal chemistry. Although the Pt core in cisplatin proved useful, in general, all the PGMs offer unique possibilities because of their wide range of geometries, coordination numbers, and oxidation states which can be exploited in tailored design towards biologically active compounds.
4.1. CANCER
Cancer does not represent a single disease but a broad group characterised by malignant cells that distinguish themselves from healthy cells by uncontrolled growth.121 The process of uncontrolled growth is triggered by abnormalities in the sequence and expression of genes, notably oncogenes and tumour suppressor genes resulting in deregulation of critical biochemical pathways that control proliferation, the cell cycle, apoptosis, angiogenesis, invasion and metastasis.122 Malignant growth of cells results in tumour development, which can be accompanied by additional processes (e.g. detachment of cells from the primary tumour, entrance/exit from the circulatory system) leading to the infiltration of distant tissues, and thus metastasis development.123
Although cancer accompanied humans throughout their history, its various types remain a public health issue and a significant challenge for the scientific community to overcome.122,124 Even in the 21st century, cancer holds the 2nd place among the top ten causes of death worldwide (e.g. HIV, tuberculosis, or malaria), being more pronounced in upper-,

25
middle-, and high-income countries125 with lung (2.09 mil), breast (2.09 mil), and colorectal (1.80 mils) cancer is the leading variant of the disease. Despite the number of treatment strategies developed (phototherapy,126 use of stem cells,127 antibody targeting, magnetic hyperthermia,128,129 deep learning drug design130), the WHO expects future patient growth.
4.2. ANTICANCER COMPLEXES OF PLATINUM
The key Pt-based anti-cancer drug, cisplatin, is still prepared by very efficient multi-step procedure reported by Dhara in 1970 (see Fig. A7).131 The aqueous K2[PtCl4] is treated with an excess of KI to form K2[PtI4]. The aqueous NH3 is then added to the dark brown solution of iodide inducing precipitation of yellow cis-[Pt(NH3)2I2], which is collected and dried (that second step is governed by trans effect of iodide). The two iodide ligands are removed from Pt by addition of two equivalents of aqueous AgNO3 resulting in a water-soluble [Pt(NH3)2(H2O)2](NO3)2 while the AgI precipitates from the solution. The filtrate with nitrate Pt complex is treated with an excess of KCl to get pure yellow cisplatin, cis-[Pt(NH3)2Cl2].
Figure A7. The Dhara’s multi-step synthetic procedure of preparation of pure cisplatin from the K2[PtCl4]. The resulting cisplatin can be further purified by recrystallisation from hot 0.10 M HCl or 0.15 M NaCl.
The increasing number of cancerous patients, severe side-effects of the treatment with cisplatin (e.g. severe kidney damage, anaphylactic reactions, decreased immunity, gastrointestinal disorders, haemorrhage, hearing loss), and the resistance towards the treatment, all those factors stimulated the pursuit of a better Pt metallodrug.113 Along with the preclinical and clinical development of cisplatin, several thousand analogues have been prepared and tested. However, only 12 of these analogues have been evaluated in clinical trials, and only a few

26
(e.g. oxaliplatin, carboplatin, satraplatin, see Fig. A8) offer an advantage over the cisplatin.
Figure A8. Two families of Pt(II)- and Pt(IV)-based metallodrugs including world-wide used cisplatin, oxaliplatin, and carboplatin and other examples of promising candidates.
Cisplatin is used for the treatment of numerous human cancers, including bladder, head and neck, lung, ovarian, and testicular, germ cell tumours, lymphomas, and sarcomas. Therefore, designing a new anticancer metal-based drug or strategy for improving its chemotherapeutic effectiveness relies on understanding the mechanisms of its biological effect. The biological effect, however, is a very complex process with many variables (e.g. biodistribution, cellular accumulation, toxicity, resistance) involving different biological targets. The determination of the mechanism of action for Pt-based metallodrugs (including cisplatin) is in the scope of many research groups; however, to some extent, it remains surrounded by a few secrets.132 For cisplatin in general, the mechanism of action has been linked with its ability to crosslink the purine bases of the DNA, interfering with the DNA repair

27
mechanisms resulting in DNA damage, and subsequently induction of apoptosis in cancer cells. A more detailed description of the mechanism of action of cisplatin and other Pt-based metallodrugs is covered by several articles and reviews.50,60,113,132
The Pt(IV) complexes were introduced as an alternative to the traditional Pt(II) as a concept of less active pro-drug which needs to be reduced before releasing the desired active compound. The advantage of the Pt(IV) complexes is their slow ligand exchange kinetics and interaction with biomolecules in the bloodstream and thus lower overall toxicity. Pt(IV) complexes are usually built on the previously successful Pt(II) candidates (oxaliplatin, carboplatin) which are oxidised by H2O2 resulting in the octahedral Pt(IV) complex with labile OH groups in axial positions. The hydroxyl groups can be subsequently substituted by the same or by different ligands, such as biological vectors or modulators of solubility (see Fig. A9).
Figure A9. Example of Pt(IV)-based metallodrug satraplatin showing a general synthetic approach via oxidation with H2O2 and subsequent replacement of resulting axial OH groups with acetate moieties.
The pursuit of better metallodrugs has led to a wide range of coordination compounds based on Pt(II), Pt(IV),133,134 and other135,136 metals (e.g., Ru, Au). Various strategies137–142 for improving the drug delivery, including encapsulating Pt complexes in macrocyclic carriers (e.g. cucurbiturils,143 cyclodextrins144) have been investigated.
Two examples of encapsulated platinum host-guest complexes are depicted in Fig. A10. The dinuclear platinum complex with dipyrazolylmethane ligand forming an inclusion complex with CB[7] belongs to a group of multinuclear platinum complexes designed for encapsulation within the cavities of both CB[7] and CB[8]. Resulting
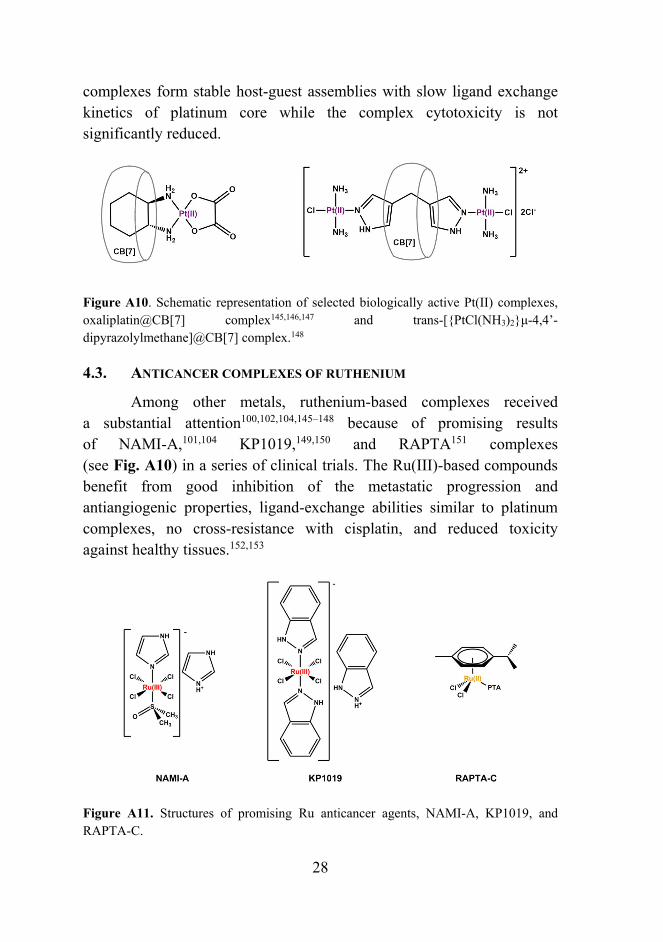
28
complexes form stable host-guest assemblies with slow ligand exchange kinetics of platinum core while the complex cytotoxicity is not significantly reduced.
Figure A10. Schematic representation of selected biologically active Pt(II) complexes, oxaliplatin@CB[7] complex145,146,147 and trans-[{PtCl(NH3)2}μ-4,4’-dipyrazolylmethane]@CB[7] complex.148
4.3. ANTICANCER COMPLEXES OF RUTHENIUM
Among other metals, ruthenium-based complexes received a substantial attention100,102,104,145–148 because of promising results of NAMI-A,101,104 KP1019,149,150 and RAPTA151 complexes (see Fig. A10) in a series of clinical trials. The Ru(III)-based compounds benefit from good inhibition of the metastatic progression and antiangiogenic properties, ligand-exchange abilities similar to platinum complexes, no cross-resistance with cisplatin, and reduced toxicity against healthy tissues.152,153
Figure A11. Structures of promising Ru anticancer agents, NAMI-A, KP1019, and RAPTA-C.

29
Although NAMI-A possesses a very low general toxicity,102,154,155 and reacts against metastases,156,157 it does not affect primary tumour growth136,155,157 and does not exhibit cytotoxicity against tumour cells in vitro.158 On the other hand, the KP1019 was found to exhibit antiproliferative activity in vitro in human colon carcinoma cell lines.159 The exact molecular mechanism of abovementioned Ru prodrugs, however, remains enigmatic101,160,161 because of the complexity of the processes involving many cellular/non-cellular components, micro-environmental elements, and their interplay.
On the other hand, a controlled application of Ru(III) coordination compounds is somewhat hampered by their biochemical instability and complicated ligand exchange chemistry.162 The RuPy complexes with various pyridine derivatives were extensively investigated by Walsby and co-workers, who intend to enhance the bioavailability of the metallodrug via ligand hydrophobic interactions with transportation proteins.99
Figure A12. Structures of organometallic Ru(II) anticancer agents of RAPTA- and RAED-type.
Organoruthenium Ru(II) “piano stool” complexes have been introduced as potent in vitro and in vivo drug candidates.100,151,163,164 The huge structural variability of Ru organometallic complexes resulted in a plethora of half-sandwich complexes, pioneered by Sadler and Dyson with reports on RAED- and RAPTA-type complexes. The RAED complexes contain a geminal diamine ligand (e.g. ethylenediamine, 1,2-diaminocyclohexane), η5- and η6-arene rings, and additional (usually anionic) ligand (see Fig. A12). As a result, the RAED complexes
30
are charged. Notably, the RAED complexes are usually highly cytotoxic, some of which achieving the concentration levels of cisplatin (4 – 20 M). The RAPTA-type complexes have two distinct components, the η6-arene rings (e.g. benzene, cymene), which represent the hydrophobic part facilitating the entry of Ru(II) complexes into cells, and the PTA ligand ensuring solubility of the complex in water. RAPTA derivatives possess low toxicity (IC50 < 300 M) and inhibit lung metastasis growth in CBA mice with MCa mammary carcinoma, while mildly affecting the primary tumour.165 Also, the RAPTA-C was reported to reduce the growth of primary tumours in preclinical models for ovarian and colorectal carcinomas.166,167
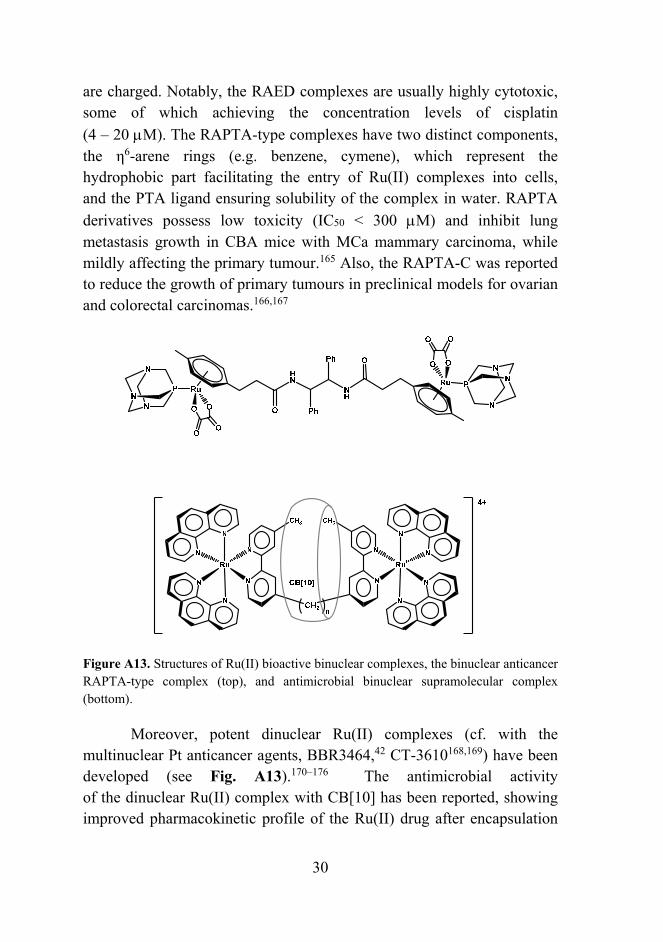
Figure A13. Structures of Ru(II) bioactive binuclear complexes, the binuclear anticancer RAPTA-type complex (top), and antimicrobial binuclear supramolecular complex (bottom).
Moreover, potent dinuclear Ru(II) complexes (cf. with the multinuclear Pt anticancer agents, BBR3464,42 CT-3610168,169) have been developed (see Fig. A13).170–176 The antimicrobial activity of the dinuclear Ru(II) complex with CB[10] has been reported, showing improved pharmacokinetic profile of the Ru(II) drug after encapsulation

31
by the CB macrocycle.177 Besides, biological properties of similar supramolecular Ru assemblies with CB[n] carriers have been investigated only recently.178,179 Similarly to platinum drugs, various delivery strategies and mechanisms for ruthenium-based systems have been suggested and explored in recent years.180–183

32
REFERENCES
1. Lee, J.; Farha, O. K.; Roberts, J.; Scheidt, K. A.; Nguyen, S. T.; Hupp, J. T. Chem. Soc. Rev. 2009, 38, 1450.
2. Heitbaum, M.; Glorius, F.; Escher, I. Angew. Chem. Int. Ed. 2006, 45, 4732. 3. Albrecht, M.; Koten, G. van Angew. Chem. Int. Ed. 2001, 40, 3750. 4. González-Sebastián, L.; Morales-Morales, D. J. Organomet. Chem. 2019, 893, 39. 5. Nandanwar, S. K.; Kim, H. J. ChemistrySelect 2019, 4, 1706. 6. Arjmand, F.; Afsan, Z.; Sharma, S.; Parveen, S.; Yousuf, I.; Sartaj, S.; Siddique, H. R.; Tabassum, S.
Coord. Chem. Rev. 2019, 387, 47. 7. Medici, S.; Peana, M.; Nurchi, V. M.; Lachowicz, J. I.; Crisponi, G.; Zoroddu, M. A. Coord. Chem.
Rev. 2015, 284, 329. 8. Mjos, K. D.; Orvig, C. Chem. Rev. 2014, 114, 4540. 9. Guo, Z.; Sadler, P. J. Angew. Chem. Int. Ed. 1999, 38, 1512. 10. McDonald, D.; Hunt, L. B. A history of platinum and its allied metals; Johnson Matthey : Distributed
by Europa Publications: London, 1982. 11. Comprehensive coordination chemistry II: from biology to nanotechnology; McCleverty, J. A.;
Meyer, T. J., Eds.; 1st ed.; Elsevier Pergamon: Amsterdam ; Boston, 2004. 12. Greenwood, N. N.; Earnshaw, A. Chemistry of the elements; 2nd ed.; Butterworth-Heinemann:
Oxford ; Boston, 1997. 13. Brauer, G. Handbook of preparative inorganic chemistry. Vol. 2 Vol. 2; Academic Press: Place of
publication not identified, 1965. 14. Troegel, D.; Stohrer, J. Coord. Chem. Rev. 2011, 255, 1440. 15. Rao, C. R. K.; Trivedi, D. C. Coord. Chem. Rev. 2005, 249, 613. 16. Obuchi, A.; Ohi, A.; Nakamura, M.; Ogata, A.; Mizuno, K.; Ohuchi, H. Appl. Catal. B Environ. 1993,
2, 71. 17. Inorganic Syntheses; Darensbourg, Marcetta. Y., Ed.; Inorganic Syntheses; John Wiley & Sons, Inc.:
Hoboken, NJ, USA, 1998; Vol. 32. 18. Cotton, S. A. Chemistry of precious metals; 1st ed.; Blackie Academic & Professional: London ; New
York, 1997. 19. Housecroft, C. E. Inorganic chemistry; Fifth edition.; Pearson: Harlow, England, 2018. 20. Chakrabortty, S.; Agrawalla, B. K.; Stumper, A.; Vegi, N. M.; Fischer, S.; Reichardt, C.; Kögler, M.;
Dietzek, B.; Feuring-Buske, M.; Buske, C.; Rau, S.; Weil, T. J. Am. Chem. Soc. 2017, 139, 2512. 21. Grubbs, R. H.; Trnka, T. M. In Ruthenium in Organic Synthesis; John Wiley & Sons, Ltd, 2005; pp
153. 22. Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. Acta Crystallogr. Sect. B Struct. Sci. Cryst.
Eng. Mater. 2016, 72, 171. 23. Puddephatt, R. J.; Rashidi, M.; Vittal, J. J. J. Chem. Soc. Dalton Trans. 1991, 2835. 24. Zhao, S.-B.; Wang, R.-Y.; Nguyen, H.; Becker, J. J.; Gagné, M. R. Chem. Commun. 2011, 48, 443. 25. Immirzi, A.; Musco, A.; Zambelli, P.; Carturan, G. Inorganica Chim. Acta 1975, 13, L13. 26. Otsuka, S.; Yoshida, T.; Matsumoto, M.; Nakatsu, K. J. Am. Chem. Soc. 1976, 98, 5850. 27. Braunschweig, H.; Brenner, P.; Dewhurst, R. D.; Guethlein, F.; Jimenez‐Halla, J. O. C.; Radacki, K.;
Wolf, J.; Zöllner, L. Chem. – Eur. J. 2012, 18, 8605. 28. Phelps, J.; Butikofer, J. L.; Thapaliya, B.; Gale, E.; Vowell, C. L.; Debnath, S.; Arulsamy, N.;
Roddick, D. M. Polyhedron 2016, 116, 197. 29. Uson, Rafael.; Fornies, Juan.; Tomas, Milagros.; Menjon, Babil. Organometallics 1985, 4, 1912. 30. Blake, A. J.; Gould, R. O.; Holder, A. J.; Hyde, T. I.; Lavery, A. J.; Odulate, M. O.; Schröder, M. J.
Chem. Soc. Chem. Commun. 1987, 118. 31. Wilson, J. J.; Lippard, S. J. Inorg. Chem. 2012, 51, 9852. 32. Mair, G. A.; Powell, H. M.; Venanzi, L. M. Proc. Chem. Soc. 1961, 153. 33. Ainscough, E. W.; Brodie, A. M.; Burrell, A. K.; Derwahl, A.; Jameson, G. B.; Taylor, S. K.
Polyhedron 2004, 23, 1159. 34. Atkinson, L.; Day, P.; Williams, R. J. P. Nature 1968, 218, 668. 35. Kim, E.-G.; Schmidt, K.; Caseri, W. R.; Kreouzis, T.; Stingelin‐Stutzmann, N.; Brédas, J.-L. Adv.
Mater. 2006, 18, 2039. 36. Kato, M.; Kosuge, C.; Morii, K.; Ahn, J. S.; Kitagawa, H.; Mitani, T.; Matsushita, M.; Kato, T.; Yano,
S.; Kimura, M. Inorg. Chem. 1999, 38, 1638. 37. Shigeta, Y.; Kobayashi, A.; Yoshida, M.; Kato, M. Inorg. Chem. 2019, 58, 7385. 38. Yoshida, M.; Kato, M. Coord. Chem. Rev. 2018, 355, 101. 39. Chen, T.; Li, M.; Liu, J. Cryst. Growth Des. 2018, 18, 2765.

33
40. Yam, V. W.-W.; Wong, K. M.-C.; Zhu, N. J. Am. Chem. Soc. 2002, 124, 6506. 41. Buchwalter, P.; Rosé, J.; Braunstein, P. Chem. Rev. 2015, 115, 28. 42. Adams, R. D.; Li, Z.; Swepston, P.; Wu, W.; Yamamoto, J. J. Am. Chem. Soc. 1992, 114, 10657. 43. Leopold, H.; Tenne, M.; Tronnier, A.; Metz, S.; Münster, I.; Wagenblast, G.; Strassner, T. Angew.
Chem. 2016, 128, 16011. 44. Arnal, L.; Fuertes, S.; Martín, A.; Baya, M.; Sicilia, V. Chem. – Eur. J. 2018, 24, 18743. 45. Fernández-Anca, D.; García-Seijo, M. I.; García-Fernández, M. E. Dalton Trans. 2013, 42, 10221. 46. Coe, B. J.; Glenwright, S. J. Coord. Chem. Rev. 2000, 203, 5. 47. Kauffman, G. B. J. Chem. Educ. 1977, 54, 86. 48. Kauffman, G. B.; Cowan, D. O.; Slusarczuk, G.; Kirschner, S. In Inorganic Syntheses; John Wiley &
Sons, Ltd, 2007; pp 239. 49. Kauffman, G. B. In Encyclopedia of Inorganic Chemistry; American Cancer Society, 2006. 50. Brabec, V.; Hrabina, O.; Kasparkova, J. Coord. Chem. Rev. 2017, 351, 2. 51. Crabtree, R. H.; Torrens, H. In Encyclopedia of Inorganic and Bioinorganic Chemistry; American
Cancer Society, 2011. 52. Liu, P.; Lu, Y.; Gao, X.; Liu, R.; Zhang-Negrerie, D.; Shi, Y.; Wang, Y.; Wang, S.; Gao, Q. Chem.
Commun. 2013, 49, 2421. 53. Moradell, S.; Lorenzo, J.; Rovira, A.; van Zutphen, S.; Avilés, F. X.; Moreno, V.; de Llorens, R.;
Martinez, M. A.; Reedijk, J.; Llobet, A. J. Inorg. Biochem. 2004, 98, 1933. 54. Hill, W. E.; Minahan, D. M. A.; Taylor, J. G.; McAuliffe, C. A. J. Am. Chem. Soc. 1982, 104, 6001. 55. Power, W. P.; Wasylishen, R. E. Inorg. Chem. 1992, 31, 2176. 56. Bernhardt, P. V.; Gallego, C.; Martinez, M.; Parella, T. Inorg. Chem. 2002, 41, 1747. 57. Julliard, M.; Chanon, M. Chem. Rev. 1983, 83, 425. 58. Hall, M. D.; Hambley, T. W. Coord. Chem. Rev. 2002, 232, 49. 59. Wexselblatt, E.; Gibson, D. J. Inorg. Biochem. 2012, 117, 220. 60. Deo, K. M.; Ang, D. L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.;
Bjelosevic, A.; Pages, B.; Gordon, C.; Aldrich-Wright, J. R. Coord. Chem. Rev. 2018, 375, 148. 61. Sabo-Etienne, S.; Grellier, M. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R. A.,
Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2011; p eibc0192. 62. Lawrence, M. A. W.; Bullock, J. L.; Holder, A. A. In Ruthenium Complexes; John Wiley & Sons, Ltd,
2017; pp 25. 63. Bruce, M. I.; Hambley, T. W.; Snow, M. R.; Swincer, A. G. Organometallics 1985, 4, 501. 64. Chatt, J.; Duncanson, L. A. J. Chem. Soc. Resumed 1953, 2939. 65. Jean, Y. Molecular orbitals of transition metal complexes; Oxford University Press: Oxford ; New
York, 2005. 66. Holder, A. A.; Lilge, L. D.; Browne, W. R.; Lawrence, M. A. W.; Bullock, J. L. Ruthenium
complexes; 2018. 67. Advanced inorganic chemistry; Cotton, F. A.; Wilkinson, G.; Murilo, C. A.; Bochman, M., Eds.; 6th
ed.; Wiley: New York, 1999. 68. Rapaport, I.; Helm, L.; Merbach, A. E.; Bernhard, P.; Ludi, A. Inorg. Chem. 1988, 27, 873. 69. Helm, L.; Merbach, A. E. Chem. Rev. 2005, 105, 1923. 70. Cannizzo, A.; Mourik, F. van; Gawelda, W.; Zgrablic, G.; Bressler, C.; Chergui, M. Angew. Chem.
Int. Ed. 2006, 45, 3174. 71. Jain, A.; Wyland, K. R.; Davis, D. H. J. Coord. Chem. 2018, 71, 231. 72. Baranoff, E.; Collin, J.-P.; Furusho, J.; Furusho, Y.; Laemmel, A.-C.; Sauvage, J.-P. Inorg. Chem.
2002, 41, 1215. 73. Matsuo, H.; Toganoh, M.; Ishida, M.; Mori, S.; Furuta, H. Inorg. Chem. 2017, 56, 13842. 74. Traeger, J. C. Int. J. Mass Spectrom. 2000, 200, 387. 75. Ohta, T.; Takaya, H.; Kitamura, M.; Nagai, K.; Noyori, R. J. Org. Chem. 1987, 52, 3174. 76. Crochet, P.; Cadierno, V. Dalton Trans. 2014, 43, 12447. 77. Francos, J.; Menéndez-Rodríguez, L.; Tomás-Mendivil, E.; Crochet, P.; Cadierno, V. RSC Adv. 2016,
6, 39044. 78. Battistin, F.; Balducci, G.; Milani, B.; Alessio, E. Inorg. Chem. 2018, 57, 6991. 79. Cavit, B. E.; Grundy, K. R.; Roper, W. R. J. Chem. Soc. Chem. Commun. 1972, 60b. 80. Vives, G.; Carella, A.; Launay, J.-P.; Rapenne, G. Coord. Chem. Rev. 2008, 252, 1451. 81. Teegardin, K.; Day, J. I.; Chan, J.; Weaver, J. Org. Process Res. Dev. 2016, 20, 1156. 82. Cao, K.; Lu, J.; Cui, J.; Shen, Y.; Chen, W.; Alemu, G.; Wang, Z.; Yuan, H.; Xu, J.; Wang, M.;
Cheng, Y. J. Mater. Chem. A 2014, 2, 4945. 83. Boynton, A. N.; Marcélis, L.; McConnell, A. J.; Barton, J. K. Inorg. Chem. 2017, 56, 8381. 84. Gill, M. R.; Thomas, J. A. Chem. Soc. Rev. 2012, 41, 3179. 85. Lo, K. K.-W.; Lee, T. K.-M.; Lau, J. S.-Y.; Poon, W.-L.; Cheng, S.-H. Inorg. Chem. 2008, 47, 200. 86. Bhat, S. S.; Kumbhar, A. S.; Lönnecke, P.; Hey-Hawkins, E. Inorg. Chem. 2010, 49, 4843.

34
87. Jeremias, L.; Novotný, J.; Repisky, M.; Komorovsky, S.; Marek, R. Inorg. Chem. 2018, 57, 8748. 88. Gendron, F.; Autschbach, J. J. Chem. Theory Comput. 2016, 12, 5309. 89. Ravera, E.; Parigi, G.; Luchinat, C. J. Magn. Reson. 2017, 282, 154. 90. Rouf, S. A.; Mareš, J.; Vaara, J. J. Chem. Theory Comput. 2017, 13, 3731. 91. Benda, L.; Mareš, J.; Ravera, E.; Parigi, G.; Luchinat, C.; Kaupp, M.; Vaara, J. Angew. Chem. Int. Ed.
2016, 55, 14713. 92. Novotný, J.; Přichystal, D.; Sojka, M.; Komorovsky, S.; Nečas, M.; Marek, R. Inorg. Chem. 2018, 57,
641. 93. Novotný, J.; Sojka, M.; Komorovsky, S.; Nečas, M.; Marek, R. J. Am. Chem. Soc. 2016, 138, 8432. 94. Pell, A. J.; Pintacuda, G.; Grey, C. P. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1. 95. Tauchman, J.; Paul, L. E. H.; Furrer, J.; Therrien, B.; Süss-Fink, G. Inorganica Chim. Acta 2014, 423,
16. 96. Almog, O.; Bino, A.; Garfinkel-Shweky, D. Inorganica Chim. Acta 1993, 213, 99. 97. Rose, M. J.; Olmstead, M. M.; Mascharak, P. K. J. Am. Chem. Soc. 2007, 129, 5342. 98. Rose, M. J.; Fry, N. L.; Marlow, R.; Hinck, L.; Mascharak, P. K. J. Am. Chem. Soc. 2008, 130, 8834. 99. Webb, M. I.; Wu, B.; Jang, T.; Chard, R. A.; Wong, E. W. Y.; Wong, M. Q.; Yapp, D. T. T.; Walsby,
C. J. Chem. - Eur. J. 2013, 19, 17031. 100. Coverdale, J. P. C.; Laroiya-McCarron, T.; Romero-Canelón, I. Inorganics 2019, 7, 31. 101. Brabec, V.; Kasparkova, J. Coord. Chem. Rev. 2018, 376, 75. 102. Alessio, E.; Messori, L. In Metallo-Drugs: Development and Action of Anticancer Agents; De
Gruyter: Berlin, Boston, 2018; Vol. 18. 103. Leijen, S.; Burgers, S. A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.;
Beijnen, J. H.; Schellens, J. H. M. Invest. New Drugs 2015, 33, 201. 104. Alessio, E. Eur. J. Inorg. Chem. 2017, 2017, 1549. 105. Vahrenkamp, H. Angew. Chem. Int. Ed. Engl. 1975, 14, 322. 106. Magner, L. N. A history of medicine; 2nd ed.; Taylor & Francis: Boca Raton, 2005. 107. Lloyd, N. C.; Morgan, H. W.; Nicholson, B. K.; Ronimus, R. S. Angew. Chem. Int. Ed. 2005, 44, 941. 108. Ehrlich, P.; Bertheim, A. Berichte Dtsch. Chem. Ges. 1912, 45, 756. 109. Thompson, K. H.; Orvig, C. Science 2003, 300, 936. 110. Finney, L. A.; O’Halloran, T. V. Science 2003, 300, 931. 111. Rosenberg, B.; Camp, L. V.; Krigas, T. Nature 1965, 205, 698. 112. Rosenberg, B.; Vancamp, L.; Trosko, J. E.; Mansour, V. H. Nature 1969, 222, 385. 113. Ghosh, S. Bioorganic Chem. 2019, 88, 102925. 114. Ho, G. Y.; Woodward, N.; Coward, J. I. G. Crit. Rev. Oncol. Hematol. 2016, 102, 37. 115. Stein, A.; Arnold, D. Expert Opin. Pharmacother. 2012, 13, 125. 116. Norman, J. G. jr. Method and kit for locating hyperactive parathyroid tissue or adenomatious tissue in
a patient and for removal of such tissue 2001. 117. Gries, H.; Rosenberg, D.; Weinmann, H.-J.; Speck, U.; Mutzel, W.; Hoyer, G.-A.; deceased, H. P.
Method of enhancing NMR imaging using chelated paramagnetic ions bound to biomolecules 1991. 118. Karasawa, T.; Steyger, P. S. Toxicol. Lett. 2015, 237, 219. 119. Oun, R.; Moussa, Y. E.; Wheate, N. J. Dalton Trans. 2018, 47, 6645. 120. Han Ang, W.; Dyson, P. J. Eur. J. Inorg. Chem. 2006, 2006, 4003. 121. Workman, P. New Drug Targets for Genomic Cancer Therapy Successes, Limitations, Opportunities
and Future Challenges. Curr. Cancer Drug Targets. http://www.eurekaselect.com/65208/article (accessed June 27, 2019).
122. Weinberg, R. A. The biology of cancer; Second edition.; Garland Science, Taylor & Francis Group: New York, 2014.
123. Introduction to cancer metastasis; Ahmad, A., Ed.; Cancer metastasis; Academic Press : Elsevier: Amsterdam, 2017.
124. Biotechnology and Production of Anti-Cancer Compounds; Malik, S., Ed.; Springer International Publishing: Cham, 2017.
125. The top 10 causes of death. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed June 27, 2019).
126. Liu, Y.; Bhattarai, P.; Dai, Z.; Chen, X. Chem. Soc. Rev. 2019, 48, 2053. 127. Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J. D. Cell Stem Cell 2019, 24, 25. 128. Dulińska-Litewka, J.; Łazarczyk, A.; Hałubiec, P.; Szafrański, O.; Karnas, K.; Karewicz, A. Materials
2019, 12, 617. 129. Vines, J. B.; Yoon, J.-H.; Ryu, N.-E.; Lim, D.-J.; Park, H. Front. Chem. 2019, 7, 167. 130. Topol, E. J. Nat. Med. 2019, 25, 44. 131. Dhara, S. C. Indian J Chem 1970, 8, 193. 132. Dasari, S.; Bernard Tchounwou, P. Eur. J. Pharmacol. 2014, 740, 364. 133. Dilruba, S.; Kalayda, G. V. Cancer Chemother. Pharmacol. 2016, 77, 1103.

35
134. Hu, X.; Li, F.; Noor, N.; Ling, D. Sci. Bull. 2017, 62, 589. 135. Muhammad, N.; Guo, Z. Curr. Opin. Chem. Biol. 2014, 19, 144. 136. Bergamo, A.; Sava, G. Chem. Soc. Rev. 2015, 44, 8818. 137. Lu, C.; Perez-Soler, R.; Piperdi, B.; Walsh, G. L.; Swisher, S. G.; Smythe, W. R.; Shin, H. J.; Ro, J.
Y.; Feng, L.; Truong, M.; Yalamanchili, A.; Lopez-Berestein, G.; Hong, W. K.; Khokhar, A. R.; Shin, D. M. J. Clin. Oncol. 2005, 23, 3495.
138. Dragovich, T.; Mendelson, D.; Kurtin, S.; Richardson, K.; Hoff, D. V.; Hoos, A. Cancer Chemother. Pharmacol. 2006, 58, 759.
139. Rice, J. R.; Gerberich, J. L.; Nowotnik, D. P.; Howell, S. B. Clin. Cancer Res. 2006, 12, 2248. 140. Domínguez, C. S. H.; Hernández, P. Am. J. Anal. Chem. 2013, 04, 314. 141. Jun, Y. J.; Kim, J. I.; Jun, M. J.; Sohn, Y. S. J. Inorg. Biochem. 2005, 99, 1593. 142. Gao, C.; Fei, F.; Wang, T.; Yang, B.; Gou, S.; Yang, J.; Liao, L. J. Coord. Chem. 2013, 66, 1068. 143. Wheate, N. J. J. Inorg. Biochem. 2008, 102, 2060. 144. Komeda, S.; Uemura, M.; Yoneyama, H.; Harusawa, S.; Hiramoto, K. Inorganics 2019, 7, 5. 145. Keppler, B. K.; Henn, M.; Juhl, U. M.; Berger, M. R.; Niebl, R.; Wagner, F. E. In Ruthenium and
Other Non-Platinum Metal Complexes in Cancer Chemotherapy; Baulieu, E.; Forman, D. T.; Ingelman-Sundberg, M.; Jaenicke, L.; Kellen, J. A.; Nagai, Y.; Springer, G. F.; Träger, L.; Will-Shahab, L.; Wittliff, J. L., Eds.; Progress in Clinical Biochemistry and Medicine; Springer Berlin Heidelberg, 1989; pp 41.
146. Dyson, P. J.; Sava, G. Dalton Trans. 2006, 0, 1929. 147. Jakupec, M. A.; Galanski, M.; Arion, V. B.; Hartinger, C. G.; Keppler, B. K. Dalton Trans. 2007, 0,
183. 148. Allardyce, C. S.; Dyson, P. J. Ruthenium in Medicine: Current Clinical Uses and Future Prospects.
https://www.ingentaconnect.com/content/matthey/pmr/2001/00000045/00000002/art00005 (accessed July 4, 2019).
149. Hartinger, C. G.; Jakupec, M. A.; Zorbas‐Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P. J.; Keppler, B. K. Chem. Biodivers. 2008, 5, 2140.
150. Hartinger, C. G.; Zorbas-Seifried, S.; Jakupec, M. A.; Kynast, B.; Zorbas, H.; Keppler, B. K. J. Inorg. Biochem. 2006, 100, 891.
151. Murray, B. S.; Babak, M. V.; Hartinger, C. G.; Dyson, P. J. Coord. Chem. Rev. 2016, 306, 86. 152. Dyson, P. J. Ruthenium; A Non-essential Element that May Become Essential in Treating
Chemoresistant Cancers. https://www.ingentaconnect.com/content/scs/chimia/2019/00000073/00000004/art00022 (accessed July 4, 2019).
153. Webb, M. I.; Walsby, C. J. Dalton Trans. 2011, 40, 1322. 154. Gagliardi, R.; Sava, G.; Pacor, S.; Mestroni, G.; Alessio, E. Clin. Exp. Metastasis 1994, 12, 93. 155. Sava, G.; Capozzi, I.; Clerici, K.; Gagliardi, G.; Alessio, E.; Mestroni, G. Clin. Exp. Metastasis 1998,
16, 371. 156. Cocchietto, M.; Zorzet, S.; Sorc, A.; Sava, G. Invest. New Drugs 2003, 21, 55. 157. Sava, G.; Bergamo, A.; Zorzet, S.; Gava, B.; Casarsa, C.; Cocchietto, M.; Furlani, A.; Scarcia, V.;
Serli, B.; Iengo, E.; Alessio, E.; Mestroni, G. Eur. J. Cancer 2002, 38, 427. 158. Sava, G.; Gagliardi, R.; Cocchietto, M.; CLERICI, K.; Capozzi, I.; Marrella, M.; Alessio, E.;
Mestroni, G.; Milanino, R. Pathol. Oncol. Res. 1998, 4, 30. 159. Alessio, E.; Messori, L. Molecules 2019, 24, 1995. 160. Pacor, S.; Zorzet, S.; Cocchietto, M.; Bacac, M.; Vadori, M.; Turrin, C.; Gava, B.; Castellarin, A.;
Sava, G. J. Pharmacol. Exp. Ther. 2004, 310, 737. 161. Bacac, M.; Hotze, A. C. G.; Schilden, K. van der; Haasnoot, J. G.; Pacor, S.; Alessio, E.; Sava, G.;
Reedijk, J. J. Inorg. Biochem. 2004, 98, 402. 162. Abid, M.; Shamsi, F.; Azam, A. Mini-Rev. Med. Chem. 2016, 16, 772. 163. Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T. J.;
Sava, G.; Dyson, P. J. J. Med. Chem. 2005, 48, 4161. 164. Thota, S.; Rodrigues, D. A.; Crans, D. C.; Barreiro, E. J. J. Med. Chem. 2018, 61, 5805. 165. Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T. J.;
Sava, G.; Dyson, P. J. J. Med. Chem. 2005, 48, 4161. 166. Morris, R. E.; Aird, R. E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N. D.; Parsons,
S.; Parkin, A.; Boyd, G.; Jodrell, D. I.; Sadler, P. J. J. Med. Chem. 2001, 44, 3616. 167. Süss-Fink, G. Dalton Trans. 2010, 39, 1673. 168. Gatti, L.; Perego, P.; Leone, R.; Apostoli, P.; Carenini, N.; Corna, E.; Allievi, C.; Bastrup, U.; De
Munari, S.; Di Giovine, S.; Nicoli, P.; Grugni, M.; Natangelo, M.; Pardi, G.; Pezzoni, G.; Singer, J. W.; Zunino, F. Mol. Pharm. 2010, 7, 207.
169. Manzotti, C.; Pratesi, G.; Menta, E.; Domenico, R. D.; Cavalletti, E.; Fiebig, H. H.; Kelland, L. R.; Farrell, N.; Polizzi, D.; Supino, R.; Pezzoni, G.; Zunino, F. Clin. Cancer Res. 2000, 6, 2626.

36
170. Davey, G. E.; Adhireksan, Z.; Ma, Z.; Riedel, T.; Sharma, D.; Padavattan, S.; Rhodes, D.; Ludwig, A.; Sandin, S.; Murray, B. S.; Dyson, P. J.; Davey, C. A. Nat. Commun. 2017, 8.
171. Eichhorn, T.; Hey-Hawkins, E.; Maksimović-Ivanić, D.; Mojić, M.; Schmidt, J.; Mijatović, S.; Schmidt, H.; Kaluđerović, G. N. Appl. Organomet. Chem. 2015, 29, 20.
172. Iengo, E.; Mestroni, G.; Geremia, S.; Calligaris, M.; Alessio, E. J. Chem. Soc. Dalton Trans. 1999, 0, 3361.
173. Billecke, C.; Finniss, S.; Tahash, L.; Miller, C.; Mikkelsen, T.; Farrell, N. P.; Bögler, O. Neuro-Oncol. 2006, 8, 215.
174. Harris, A. L.; Yang, X.; Hegmans, A.; Povirk, L.; Ryan, J. J.; Kelland, L.; Farrell, N. P. Inorg. Chem. 2005, 44, 9598.
175. Hegmans, A.; Kasparkova, J.; Vrana, O.; Kelland, L. R.; Brabec, V.; Farrell, N. P. J. Med. Chem. 2008, 51, 2254.
176. Li, X.; Gorle, A. K.; Sundaraneedi, M. K.; Keene, F. R.; Collins, J. G. Coord. Chem. Rev. 2018, 375, 134.
177. Li, F.; Gorle, A. K.; Ranson, M.; Vine, K. L.; Kinobe, R.; Feterl, M.; Warner, J. M.; Keene, F. R.; Collins, J. G.; Day, A. I. Org. Biomol. Chem. 2017, 15, 4172.
178. Sun, B.; Musgrave, I. F.; Day, A. I.; Heimann, K.; Keene, F. R.; Collins, J. G. Front. Chem. 2018, 6. 179. Shinde, M. N.; Rao, S. S.; Gejji, S. P.; Kumbhar, A. A. Dalton Trans. 2018, 47, 3857. 180. Blunden, B. M.; Stenzel, M. H. J. Chem. Technol. Biotechnol. 2015, 90, 1177. 181. Moreira, T.; Francisco, R.; Comsa, E.; Duban-Deweer, S.; Labas, V.; Teixeira-Gomes, A.-P.;
Combes-Soia, L.; Marques, F.; Matos, A.; Favrelle, A.; Rousseau, C.; Zinck, P.; Falson, P.; Garcia, M. H.; Preto, A.; Valente, A. Eur. J. Med. Chem. 2019, 168, 373.
182. Lu, M.; Chen, F.; Noy, J.-M.; Lu, H.; Stenzel, M. H. Macromol. Biosci. 2017, 17, 1600513. 183. Lu, M.; Henry, C. E.; Lai, H.; Khine, Y. Y.; Ford, C. E.; Stenzel, M. H. Biomater. Sci. 2019, 7, 1652.

37
CHAPTER 2
PHOSPHINE LIGANDS TABLE OF CONTENTS
1. Introduction 38 2. Phosphinoamine ligands 40 2.1. Characterisation 42 2.1.1. NMR spectra 42 2.1.2. X‐ray structure analyses 44 2.1.3. IR spectra and other analytical methods 44 2.2. Synthesis 45 2.2.2. Scrambling reaction and transamination 49 3. Phosphinoamines in coordination chemistry 50 References 55
38
1. INTRODUCTION
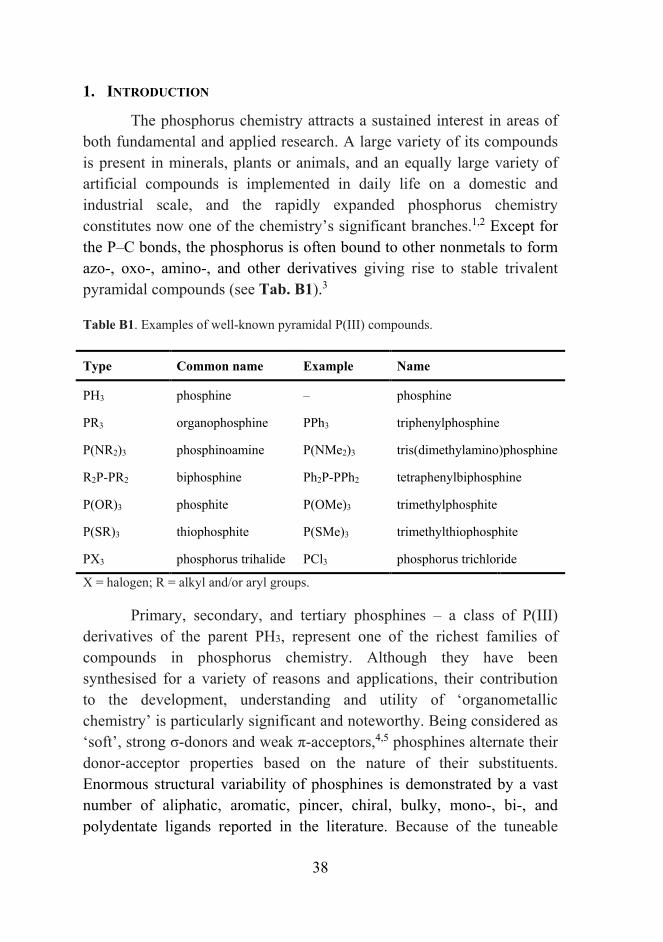
The phosphorus chemistry attracts a sustained interest in areas of both fundamental and applied research. A large variety of its compounds is present in minerals, plants or animals, and an equally large variety of artificial compounds is implemented in daily life on a domestic and industrial scale, and the rapidly expanded phosphorus chemistry constitutes now one of the chemistry’s significant branches.1,2 Except for the P–C bonds, the phosphorus is often bound to other nonmetals to form azo-, oxo-, amino-, and other derivatives giving rise to stable trivalent pyramidal compounds (see Tab. B1).3
Table B1. Examples of well-known pyramidal P(III) compounds.
Type Common name Example Name
PH3 phosphine – phosphine
PR3 organophosphine PPh3 triphenylphosphine
P(NR2)3 phosphinoamine P(NMe2)3 tris(dimethylamino)phosphine
R2P-PR2 biphosphine Ph2P‐PPh2 tetraphenylbiphosphine
P(OR)3 phosphite P(OMe)3 trimethylphosphite
P(SR)3 thiophosphite P(SMe)3 trimethylthiophosphite
PX3 phosphorus trihalide PCl3 phosphorus trichloride
X = halogen; R = alkyl and/or aryl groups.
Primary, secondary, and tertiary phosphines – a class of P(III) derivatives of the parent PH3, represent one of the richest families of compounds in phosphorus chemistry. Although they have been synthesised for a variety of reasons and applications, their contribution to the development, understanding and utility of ‘organometallic chemistry’ is particularly significant and noteworthy. Being considered as ‘soft’, strong σ‐donors and weak π‐acceptors,4,5 phosphines alternate their donor-acceptor properties based on the nature of their substituents. Enormous structural variability of phosphines is demonstrated by a vast number of aliphatic, aromatic, pincer, chiral, bulky, mono-, bi-, and polydentate ligands reported in the literature. Because of the tuneable
39
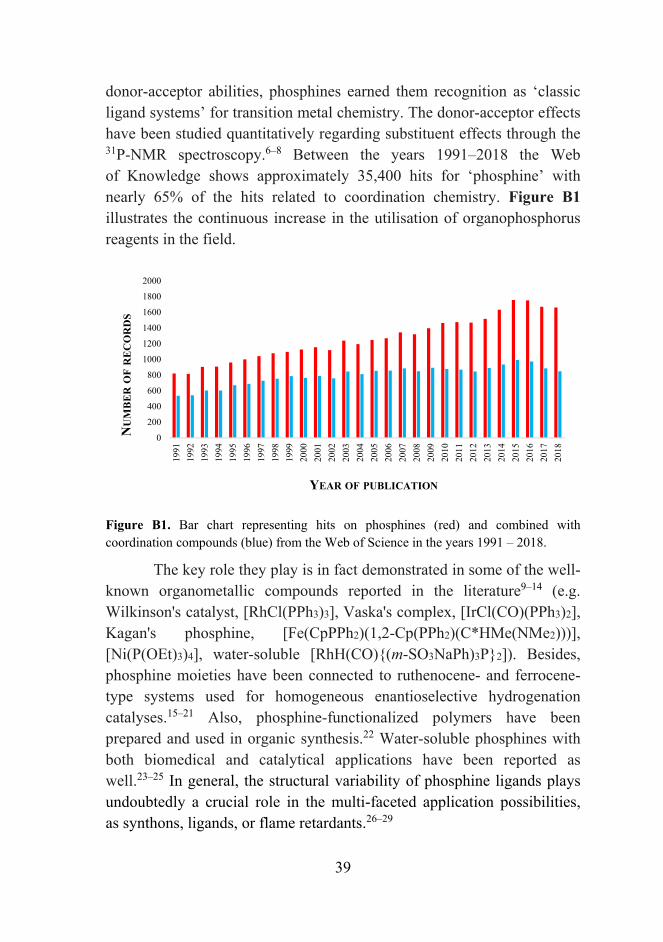
donor‐acceptor abilities, phosphines earned them recognition as ‘classic ligand systems’ for transition metal chemistry. The donor‐acceptor effects have been studied quantitatively regarding substituent effects through the 31P‐NMR spectroscopy.6–8 Between the years 1991–2018 the Web of Knowledge shows approximately 35,400 hits for ‘phosphine’ with nearly 65% of the hits related to coordination chemistry. Figure B1 illustrates the continuous increase in the utilisation of organophosphorus reagents in the field.
Figure B1. Bar chart representing hits on phosphines (red) and combined with coordination compounds (blue) from the Web of Science in the years 1991 – 2018.
The key role they play is in fact demonstrated in some of the well‐known organometallic compounds reported in the literature9–14 (e.g. Wilkinson's catalyst, [RhCl(PPh3)3], Vaska's complex, [IrCl(CO)(PPh3)2], Kagan's phosphine, [Fe(CpPPh2)(1,2‐Cp(PPh2)(C*HMe(NMe2)))], [Ni(P(OEt)3)4], water‐soluble [RhH(CO){(m‐SO3NaPh)3P}2]). Besides, phosphine moieties have been connected to ruthenocene- and ferrocene-type systems used for homogeneous enantioselective hydrogenation catalyses.15–21 Also, phosphine‐functionalized polymers have been prepared and used in organic synthesis.22 Water‐soluble phosphines with both biomedical and catalytical applications have been reported as well.23–25 In general, the structural variability of phosphine ligands plays undoubtedly a crucial role in the multi-faceted application possibilities, as synthons, ligands, or flame retardants.26–29
0
200
400
600
800
1000
1200
1400
1600
1800
2000
1991
1992
1993
1994
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
2010
2011
2012
2013
2014
2015
2016
2017
2018
NU
MB
ER
OF
RE
CO
RD
S
YEAR OF PUBLICATION

40
The on-going research in phosphine chemistry is mainly focused on a tailored design of phosphine ligands for the synthesis of metal complexes with improved catalytic and biological activity.30–34 The following text is focused on structure, reactivity and applications of azo- subgroup of phosphines, the phosphinoamines (phosphinous amide), with comments on similarities and differences with traditional P–C phosphines and P–O phosphinites, their structure, reactivity, and application.
2. PHOSPHINOAMINE LIGANDS
Although both azo- and oxo- derivatives represent tertiary phosphines (see Fig. B2), the bridging atoms change their reactivity and properties. For instance, with P–N bonds the phosphinoamines are both air- and moisture-sensitive compounds, while P–O phosphinites represent more robust, mostly only air-sensitive compounds. Also, in phosphinoamines the nitrogen centres may act as additional donor towards metal ions, which can be demonstrated by over 160 examples of structures with both P and N donor atoms; analogous complexes with P, O donor atoms were not observed with phosphinites.35 Also, the phosphorus-nitrogen bonding offers structural motive (P=N) not feasible in oxo-derivatives. The corresponding group of P=N derivatives (e.g. phosphazenes / imidophosphoranes)36–39 is, however, beyond the scope of this introduction. In addition, many P, N ligands without direct P–N bond represent a particularly important group of heterofunctional polydentate ligands in coordination chemistry and catalysis.40,41
One of the first examples of phosphinoamines was discovered at the beginning of the 20th century.42 Their extensive involvement in coordination chemistry and catalysis, in the chemistry of heterocycles or peptides, the ability to form chalcogenides, or bioactivity improvement of anticancer agents – thanks to all the research in phosphinoamine chemistry gains a momentum day‐by‐day.41,43–49 The phosphinoamines are usually either colourless liquids or solids, which are highly soluble in common organic solvents. Most of them are thermally stable compounds, particularly sensitive to air oxidation and attack of moisture. Thus handling and storage of phosphinoamines demands dry and inert atmosphere and employment of standard Schlenk techniques

41
(inert atmosphere manipulations, vacuum line techniques).26,48,50,51 Perfectly dried, and purified solvents and reagents should be used.52 The fact that the basicity of phosphine is affected by the presence of adjacent nitrogen centres is an interesting issue. Although the basicity of P–C tertiary phosphines were determined using potentiometric titrations without many complications,53–56 the determination of pKa values for phosphinoamines poses problems because of the competition of nitrogen for protonation. However, the reports describe phosphinoamines as powerful bases like tertiary alkyl amines and tertiary alkyl phosphines. Kinetic studies on reactions of phosphinoamines with CH3I indicate the nucleophilicity of phosphinoamines is mainly governed by the free electron pair available on phosphorus rather than the π‐electron transfer from neighbouring nitrogen atoms.57
Figure B2. Classes of ligands with P(III) donor atoms illustrated by representative examples.
Phosphinoamines, like all other P(III) phosphines, are prone to a formation (i.e. oxidation) of various chalcogenides (R2N)3PE (E = O, S, Se, Te), which are usually (c.f. ref. 58) more stable compared to the corresponding phosphinoamines. The oxidation, however, changes the phosphinoamine properties, such as solubility, thermal stability.59 Most importantly, the oxidation by oxygen affects the ability

42
to coordinate soft acceptors such as Au(I) or Pt(II). The resulting oxides can be used for coordination of metal ions such as Zr60 or lanthanides,61 which are more typical acceptors for traditional phosphine oxides.
2.1. CHARACTERISATION
Phosphinoamines represent no particular exception in terms of characterisation, although some techniques can be exploited as particularly helpful (e.g. 31P NMR). A variety of techniques like infrared spectroscopy (IR), electron spin resonance (ESR), mass spectrometry (MS), and photoelectron spectroscopy (PES) techniques have been employed for their characterisation.
2.1.1. NMR SPECTRA
The nuclear magnetic resonance represents a powerful analytical tool, which can be exploited in both biology and chemistry for investigation of intra- and intermolecular interactions and elucidation of molecular structure. The phosphinoamines offer excellent scope for multinuclear NMR spectroscopy, where usually all the 1H, 13C, 15N, and 31P can be exploited for inspection of the structure of for example the (Me2N)3P.
Table B2. 31P NMR values of the chemical shift δP for various types of P–N systems (R = alkyl or aryl group)
Type of phosphine δP (ppm)
(R2N)3P 110 – 130
(R2N)2PCl 130 – 150
(R2N)PCl2 150 – 170
Ph(R2N)PCl 130 – 140
Ph(R2N)2P 50 – 100
Ph2(R2N)P 40 – 70
(OR)2(R′2N)P 140 – 160
(OR)(R′2N)2P 120 – 140
The 1H, 13C and 15N NMR of phosphinoamines provide
information on their structure and dynamic behaviour in solution.62–65

43
It also allows considerations about their properties as follows: (i) the influence of the solvent on the NH proton resonance; (ii) the determination of rotational barriers in terms of steric effects;66–69 (iii) temperature-dependent investigations of chirality.63,64
The 31P NMR reported by Knight70 in 1949 was used for probing and identifying organophosphorus compounds.66 Since then, a large number of the 31P NMR chemical shift values (δ) were compiled in the literature (e.g. Tab. B2) 71,72 for a large number of phosphinoamines apart from other phosphines.
Table B3. Effect of various substituents on phosphine on the values of δP.74
Phosphine δP (ppm)
PCl3 223.0
PhPCl2 159.8
Ph[(C6H11)2N]PCl 131.7
Ph[(C6H11)2N](Me2N)P 78.4
Ph[(C6H11)2N](MeNH)P 67.9
(i‐Pr2N)PCl2 168.4
(i‐Pr2N)(n‐Bu2N)PCl 144.0
(i‐Pr2N)(n‐Bu2N)(Et2N)P 104.3
(i‐Pr2N)(n‐Bu2N)(C5H10N)P 103.9
(i‐Pr2N)(n‐Bu2N)(OC4H8N)P 103.8
(n‐Bu2N)2PCl 150.0
(n‐Bu2N)[(C6H11)2N]PCl 142.1
(CH2)3(N‐t‐Bu)2PCl 161.1
(o‐C6H4O2)PCl 172.3
(o‐C6H4O2)(i‐Pr2N)P 154.2
Structural analyses of phosphines using 31P NMR proved to be a useful tool given the sensitivity of the monoisotopic 31P nucleus with the spin ½. The temperature-dependent 31P NMR was used to investigate restricted rotations around the P–N bond of phosphinoamines. Also, the stereochemical preferences of lone pairs of nitrogen and phosphorus resulting in conformational subtleties of bis(phosphino)amines

44
(i‐Pr)N[P(o‐C6H4O2)]2 and (i‐Pr)N[PPh2][P(o‐C6H4O2)] were studied using dynamic 31P NMR.73
Table B3 shows a set of δP values for phosphinoamines that illustrates the effect of varying groups on the phosphorus centre. Subsequent replacement of halogen groups causes shielding effect around the phosphorus bringing the δP values in the upfield region. Also, the substituent steric effects play an essential role in the influence of the δP values suggesting that the increase in substituent steric bulk exerts a more significant shielding effect.
2.1.2. X‐RAY STRUCTURE ANALYSES
Although the X‐ray diffraction analyses of phosphinoamines are mostly exploited in their related complexes of transition and late-transition metals, a plethora of phosphinoamines have been characterised structurally. For example, the X-ray diffraction was particularly successfully utilised in solving problems regarding the tris(pyrrolyl)phosphine, (C4H4N)3P.75 In this case, the nitrogen was found to be bonded to a phosphorus centre, while the earlier studies offered different suggestions.76 Various representatives were investigated including both the linear and cyclic structures with aliphatic and aromatic sidechains.37,62,77–82 As with other amines, the knowledge of the phosphinoamine molecule conformation brings more detailed information on the relationship between the nature of bonds and its reactivity. In this regard, the phosphinoamines were thoroughly early examined using a combination of theoretical and experimental approach by Cowly83–85 and later on by Belyakov.86,87
2.1.3. IR SPECTRA AND OTHER ANALYTICAL METHODS
Infrared spectra represent one of the fundamental and early-on methods of phosphinoamine characterisation, with several reports on vibrational assignments.26,89–94 Very sharp and strong bands are observed for υPN of P–N in the range 780 – 1100 cm−1. The relatively wide span of vibrations is caused by the influence of substituents on both P and N. The υNH band of N–H bonds in phosphinoamines is located around 3200 cm-1. IR studies were carried out for a series of phosphinoamines of the type X2P–N(H)R to investigate the effect of

45
substituent X on the s character of the N–H nitrogen bond orbital, based on mechanical anharmonicity and hydrogen bond donor ability.95
Several other methods were employed in the investigation of phosphinoamines, including ESR spectral studies for half-life determination of different radicals,96–98 mass spectrometry addressing relative stabilities and fragmentation,99 or photoelectron and Raman spectroscopy for prediction of molecular conformations in the gas phase. However, the synthetic chemists usually go for more straightforward methods like the NMR or X-ray crystallography.
2.2. SYNTHESIS
In general, the synthesis of phosphinoamines depends on the requirement to form one, two or three P–N bonds around the phosphorus centre.
(R2N)PCl2
(R2N)3P
(R2N)2PCl
OXIDATION
HYDROLYSIS
ALCOHOLYSIS
METATHESIS(R2N)PF2
(R2N)P(O)Cl2
(R2N)HP(O)OH
(R2N)P(OR')2
O3
ZnF2R'OH
H2O
CONDENSATION
R2NH
REDUCTIVE COUPLING
(R2N)2P-P(NR2)2
(R2N)2PHLiBH4
ALKYLATION(R2N)2PR'
Na R'Li
CONDENSATIONR2NH
TRANSAMINATION
COORDINATIONDEAMINATION
(R'2N)3P
HX MX2
SALT FORMATION[(R2N)3PR']X
R'X R'2NH
(R2N)2PX X2M[(R2N)3P]n
REDUCTION
Figure B4. Scheme illustrating the reactivity and formation of different products from primary, secondary, and tertiary phosphinoamines regarding the P–Cl reactive moiety. Note that oxidation is valid for all the derivatives, likewise for alcoholysis, hydrolysis, or reduction, for both the (R2N)PCl2 and the (R2N)2PCl derivatives.
Although other synthetic methods like scrambling or transamination reactions were reported, the condensation route remains the most used and likely one of the best for preparation of a variety of P–N compounds. The reactivity of phosphinoamines represents one of their most appreciated features. However, some structural aspects

46
are of utmost importance regarding the reactivity of phosphinoamines, mainly the presence/absence of P–X (X = Cl, Br) bonds. It is worth to mention that the reactivity of fluoro-derivatives is somewhat reduced compared to other derivatives. It is most likely because of the significant difference of the bond dissociation energies for the P–F (489 kJ.mol−1) and for example, the P–Cl (318 kJ.mol−1). Figure B4 illustrates different types of reactions reported for dichlorophosphinoamines, (R2N)PCl2, chlorophosphinodiamines, (R2N)2PCl, and phosphinotriamines, (R2N)3P. Information on the reactivity of the former two is relatively scarce compared to the latter, because of considerable application potential of the (R2N)3P and high reactivity of the former chloro-derivatives.
2.2.1. CONDENSATION REACTION
The condensation reaction has been exploited to a very large extent.26,37,46,78,100–103 It is usually carried out by a reaction of phosphorus halide and a primary or a secondary amine under a variety of reaction conditions. The reaction arrangement usually features cheap and readily available starting materials with high reactivity, straightforward separation of the products and by‐products, and wide possibilities of control over the reaction conditions. The P–N bonds are usually formed from halogenophosphine (e.g. Ph2PCl) and amine (RNH2, R2NH, R’RNH) in the presence of a base, which acts as a scavenger of HCl resulting from the condensation of the phosphine and amine. Notably, amines can be used as both substrates and reagents (base) adjusting the reaction conditions. Figure B5 summarises the synthetic routes towards monodentate phosphinoamines, for additional details on syntheses of polydentate phosphinoamines, please see ref. 26,37,61.
Many reaction parameters (e.g., reaction temperature and time, the molar ratio of reagents and mode of their addition, presence/absence and the choice of a hydrogen chloride scavenger, choice of solvent) can affect both the formation and the yield of the desired products. The reaction setup depends on the system of interest; however, the work of Scopelliti and co-workers exemplifies the effects described.37 Typically, non‐polar solvents (e.g. diethyl ether, toluene) are used for the reaction, mainly for effective separation of poorly soluble by‐products (ammonium halides).

47
However, many examples of the use of less (benzene, hexane) or more (tetrahydrofuran, chloroform) polar solvents can be found in the literature, which only confirms the overall variability and applicability of the condensation method. The yields are found to be generally high, in the range 50 – 80%, depending upon the number of subsequent reaction steps carried out.
Figure B5. Schematic description of the condensation reaction between phosphorus trichloride and various secondary amines. Note that the R–P bonds are formed typically by Grignard reagents; however, the Friedel-Crafts reactions104–106 with AlCl3 and Michaelis-Arbuzov reactions107–109 can also be employed. Both R and R’ stand for aliphatic or aromatic hydrocarbons.
Figure B6. Example of reaction between phosphine chloride (e.g. Ph2PCl) and trimethylsilylamine (e.g. PhNHSiMe3) resulting in the formation of phosphinoamine and volatile trimethylsilylchloride.
Traces of ammonium chlorides represent persistent impurities in the products, and repeated recrystallisation of phosphines needs to be employed, usually carried out in hexane or benzene. This problem can be overcome by the use of organometallic reagents like n‐BuM (M = Li, Na, K) instead of amine-based HCl scavenger. However, handling of the organometallic reagents is slightly tricky because of their susceptibility to aerial and hydrolytic attacks, the formation of reactive intermediates in the reaction, besides being more expensive. Also, the use

48
of n-BuM reagents (n-BuLi in particular) is accompanied by the formation of fine alkaline halide suspensions, which are usually difficult to separate.
Introduction of silylamines brings the advantage of N–Si bond lability. It can be exploited for the synthesis of various bisphosphinoamines under mild conditions,26,77 in particular, various R’NHSiMe3 derivatives with phosphorus chlorides can be employed in this regard,110 as shown in Figure B6.
Figure B7. The variability of condensation reaction demonstrated on four different outcoming product resulting from different reaction conditions and reaction setups.
The resulting by‐product (Me3SiCl) is volatile and can be easily separated by distillation, which leads to a nearly quantitative yield of a very pure product. The starting silylamine must be usually synthesised, which imposes some limitations to this approach.
The complexity of the condensation method for the formation of phosphinoamines can be demonstrated on the reaction of PCl3 with primary amine (Fig. B7). Generally, the reaction can result in many products because of the inter‐ and intramolecular reactions, mainly because of the three reactive P–Cl bonds. Additional reaction steps can result in a formation of cyclic, cage-like, and polymeric products, namely chlorocyclophosphazanes (RNPCl)n [n = 2, 3, 4,…] apart from other

49
molecular species. A controlled condensation reaction between RNH2 and PCl3 in the presence of a base can afford acyclic diphosphazanes, RN(PCl2)2, however, when the group R is bulky (i‐Pr, t‐Bu, neo-pentyl), then only cyclodiphosphazanes, (RNPCl)2 are obtained.36,38,39
2.2.2. SCRAMBLING REACTION AND TRANSAMINATION
The two other methods, scrambling reaction and transamination, represent reaction procedures resulting in re-arrangement of substituents on reaction partners and thus, although both methods involve different phosphines, are in principle similar.
An example of scrambling reaction can be demonstrated on the reaction between a phosphinotriamine and phosphorus trichloride, which leads to a formation of P–N bonded phosphines or (chloro)phosphinoamines (R2N)xPCl3−x (x = 1, 2) in particular (Fig. B8).111 The reaction is carried out usually at higher temperatures and the products are then separated based on their different boiling points. Therefore, this method has its limitations because of the separation process, especially in the case of newly synthesised compounds with unknown properties.
Figure B8. Example of transamination between phosphorus trichloride and phosphinotriamine resulting in chlorophosphinoamines.
The transamination represents another form of scrambling reaction where the tris(amino)phosphine is treated with a primary or secondary amine at a higher temperature, which results in amine exchanged (Fig. B9). The reaction proceeds favourably only when the product amine is a low-weight and volatile compound.
Figure B9. Transamination reaction of phosphinotriamine and secondary amine resulting in exchange of amino groups on phosphorus centre and hence a form of corresponding triamine derivative.

50
Although the transamination has a somewhat limited synthetic scope (because of the limited control throughout the reaction), it offers advantages in production of less complicated reaction mixtures without any issues with ammonium chloride impurities. The transamination method was successfully exploited for reactions of (Et2N)3P with primary aromatic di‐ and polyamines, which led to a formation of a structurally interesting product. In addition, structurally complicated amines were used in transaminations,45,112 leading to important classes of compounds exhibiting superbase behaviour (e.g. Verkade's bases113–115).
3. PHOSPHINOAMINES IN COORDINATION CHEMISTRY
Without a doubt, phosphines represent one of the most prominent group compounds well established in coordination chemistry. Although the phosphinoamines are not as established as traditional phosphines, they are employed successfully in many subareas of coordination chemistry. Needless to say, whenever phosphinoamines are reported, the report is likely focused on the preparation of related coordination compounds with either catalytical or biological activity.116–122 The donor properties of both phosphorus and nitrogen can be varied with the choice of substituents, which is further manifested in the properties of resulting coordination compounds.123–126 The influence of steric crowding (resulting from the arrangement of substituents on the phosphorus centre) on the coordination of phosphine ligands were reported based on ligand exchange studies, molecular mechanics methods, and NMR spectral data. The Tolman cone angle concept is helpful for monodentate phosphines, whereas for multidentate phosphines, the bite angle concept is of importance.4,7,127–133 The term hybrid electron‐rich phosphinoamine can be encountered in the coordination chemistry of phosphinoamines for those containing two amino groups and one alkyl or aryl substituent.26,77–
79,134 The crystallisation of prepared molecular complexes is usually straightforward since their solubility in either protic or aprotic solvents is good. Therefore, the X-ray crystallography represents an integral part of analyses of phosphinoamine complexes. In the case of NMR-active transition metal nuclei, the values of coupling constants are beneficial for elucidating the structures, for Pt complexes in particular.77,135,136

51
Figure B10. Two examples of mono- and dinuclear molybdenum complex with carbonyls and phosphinoamines acting as bridging (left) and chelating (right) ligands.
Phosphinoamines bind easily to metals under relatively mild conditions and in almost all cases, the ligands bind only through the phosphorus atom.79,117,118,120,137–142 As suggested, the reason might be that the nitrogen lone pair is involved in negative hyperconjugation with orbitals of phosphorus. Phosphinoamines usually coordinate soft metal centres with lower oxidation states such as CuI, Mo0, RuII, RhI, PdII, AgI, AuI, and PtII. Depending on the denticity, the phosphinoamines coordinate in mono-, bidentate, or polymeric fashion towards the respective metal centre creating a vast variety of structural motives (e.g. Fig. B10).143–146 Similar carbonyl complexes have been useful for investigation of σ‐donor/π‐acceptor properties of phosphinoamines using infrared spectroscopy. Measurement of the υco values led to the determination of the σ‐donor strength of the ligand.147 Examples of metal complexes MXnL dimerising to [MXnL]2 are also common, usually because of coordinative unsaturation.110,143,148,149 Also, an increased steric crowding on phosphorus centre (e.g. by mesityl groups) usually leads to a formation of di‐ and tri‐nuclear complexes.150
As mentioned before, changes of substituents on phosphorus can remarkably change the properties of the resulting coordination compounds. For instance, the change of electronic properties by introducing a variety of phosphine ligands was studied on a series of RuII complexes with supplementary N-heterocyclic ligands (terpyridine, bipyridine).151–155 The electronic properties were studied by cyclic

52
voltammetry, absorption and luminescence spectroscopy, which showed appreciable differences originating from MLCT states.
Figure B11. The general structure of flexible bisphosphine (left) and rigid pincer-type (right) ligands which are referenced to in the text.
Apart from the monodentate phosphinoamines, two groups of bidentate ligands are of interest – the bisphosphinoamines and pincer-type ligands (see Fig. B11). The bisphosphines feature a higher degree of flexibility, which helps to overcome strains that may otherwise prevent coordination. The flexibility provides two possibilities – either the formation of a chelate or bridging of two or more centres (cf. Fig. B10). The bisphosphine ligands proved to be useful in the design of catalysts, mainly because they offer multidentate coordination. Indeed, some of the analogues of Wilkinson’s catalyst exhibit high stereoselectivity in hydrogenation applications.156–159 Also, some of the bisphoshinoamines can be oxidised in symmetrical or unsymmetrical fashion.26,160 This feature was thoroughly exploited for the preparation of a structurally rich group of transition metal complexes.161–163
Figure B12. Formation of the palladacycle from pincer phosphinoamine ligand refluxed
in toluene with PdCl2(cod). Formation of the respective Pd–C bond is highlighted in red.
The pincer-type ligands possess a rigid pincer-like structure, which leads to the formation of the chelate complex. Although many of the very famous phosphine ligands (e.g. BINAP) adapt similarly rigid

53
orientation of the phosphine moieties, the pincer phosphines traditionally offer a formation of a direct carbon-metal bond, i.e. cyclometallation (see Fig. B12). Once the respective pincer complex (metallacycle) is formed, it exhibits high hydrolytic and thermal stability despite the (otherwise) labile P−N bond. Pincer complexes proved to be useful particularly in the preparation of efficient catalysts based on Ni, Pd or Ir.164–168 For instance, the nickel complexes have been utilised for catalysing polymerisation reactions of ethylene affording only linear polyethylenes.169,170
Figure B13. Example of the reaction catalysed by platinum complex PtCl2(PPh3) with two tertiary phosphines resulting in a formation of carboxylic acid ester.176
Although the coordination chemistry of phosphines is vibrant, as soft donors, they are typical ligands for platinum group metals. Thus they have been studied since 1857 when von Hofmann prepared the first Au and Pt complexes with phosphines, arsanes and stibanes.171 The platinum complexes with phosphines were employed as catalysts (e.g. Fig. B13) for various reactions,172,173 or as anticancer agents.174,175 Besides, the coordination of phosphines to Pt salts changes their properties, for example, it increases their solubility in organic solvents for homogeneous catalysis.
Despite sensitive P–N bonds, phosphinoamines are vital for everyday life.177–180 As ligands, they play a substantial role in organic conversions such as asymmetric allylations, conjugate additions, enantioselective allylic substitutions, hydroaminations, hydroborations, hydroformylations, hydrogenations, hydrosilylations, Suzuki–Miyaura, cross‐coupling, and Sonogashira reactions.100,181–187 Besides, chiral phosphinoamines with N‐stereogenic centres were utilised for homogeneous and regioselective catalysis.188–191 The phosphinoamine P(NMe2)3 catalyses the reaction of thiodiglycol (S(CH2CH2OH)2) and triethylene glycol resulting in polyethers with shorter reaction times and produces higher yields of products. The P(NEt2)3, was found to significantly reduce the polymer decomposition rate. Also, phosphine‐ and phosphinoamine‐containing metal complexes have shown their

54
potential in electro‐ and photoluminescent applications (e.g. complexes of EuIII and TbIII with HN(P(O)Ph2)2 in photonic devices and sensors),192–194 displays, detectors, photovoltaic cells, solid‐state lighting and other electronic devices.195–199 The phosphinoamines were utilised as useful reagents for synthesising peptides,200 as antimicrobial agents, fire retardants,201 chemotherapeutic agents202,203 and insecticides.2 However, some phosphinoamines were found to induce cancer and hence they should be handled with caution.204

55
REFERENCES
1. Phosphorus: Chemistry, Biochemistry and Technology, Sixth Edition. CRC Press. https://www.crcpress.com/Phosphorus-Chemistry-Biochemistry-and-Technology-Sixth-Edition/Corbridge/p/book/9781439840887 (accessed June 1, 2019).
2. Toy, A. D. F. Phosphorus Chem. Everyday Living 1976. 3. Phosphorus(III) ligands in homogeneous catalysis: design and synthesis; Kamer, P. C. J., Ed.; Wiley:
Chichester, 2012. 4. Mitoraj, M. P.; Michalak, A. Inorg. Chem. 2010, 49, 578. 5. Pacchioni, G.; Bagus, P. S. Inorg. Chem. 1992, 31, 4391. 6. Andersen, N. G.; Keay, B. A. Chem. Rev. 2001, 101, 997. 7. Rahman, M. Matiur.; Liu, H. Ye.; Eriks, Klaas.; Prock, Alfred.; Giering, W. P. Organometallics 1989,
8, 1. 8. Derencsenyi, T. T. Inorg. Chem. 1981, 20, 665. 9. Vaska, L. J. Am. Chem. Soc. 1966, 88, 5325. 10. Kagan, H. B.; Langlois, N.; Phat Dang, T. J. Organomet. Chem. 1975, 90, 353. 11. Rix, C. J.; Verkade, J. G. Coord. Chem. Rev. 1975, 16, 129. 12. Schmid, R.; Cereghetti, M.; Heiser, B.; Schönholzer, P.; Hansen, H.-J. Helv. Chim. Acta 1988, 71,
897. 13. Herrmann, W. A.; Kohlpaintner, C. W. Angew. Chem. Int. Ed. Engl. 1993, 32, 1524. 14. Osborn, J. A.; Wilkinson, G.; Mrowca, J. J. In Inorganic Syntheses; John Wiley & Sons, Ltd, 2007; pp
67. 15. Schulz, J.; Leitner, Z.; Císařová, I.; Štěpnička, P. Molecules 2018, 23, 2054. 16. Kenaree, A. R.; Cuthbert, T. J.; Barbon, S. M.; Boyle, P. D.; Gillies, E. R.; Ragogna, P. J.; Gilroy, J.
B. Organometallics 2015, 34, 4272. 17. Kuklin, S. A.; Sheloumov, A. M.; Dolgushin, F. M.; Ezernitskaya, M. G.; Peregudov, A. S.;
Petrovskii, P. V.; Koridze, A. A. Organometallics 2006, 25, 5466. 18. Arrayás, R. G.; Adrio, J.; Carretero, J. C. Angew. Chem. Int. Ed. 2006, 45, 7674. 19. Atkinson, R. C. J.; Gibson, V. C.; Long, N. J. Chem. Soc. Rev. 2004, 33, 313. 20. Štěpnička, P. Coord. Chem. Rev. 2017, 353, 223. 21. Schulz, J.; Horký, F.; Císařová, I.; Štěpnička, P. Catalysts 2017, 7, 167. 22. Guinó, M.; Hii, K. K. (Mimi) Chem. Soc. Rev. 2007, 36, 608. 23. Basallote, M. G.; Fernández-Trujillo, M. J.; Pino-Chamorro, J. Á.; Beltrán, T. F.; Corao, C.; Llusar,
R.; Sokolov, M.; Vicent, C. Inorg. Chem. 2012, 51, 6794. 24. Grabulosa, A.; Muller, G.; Ordinas, J. I.; Mezzetti, A.; Maestro, M. Á.; Font-Bardia, M.; Solans, X.
Organometallics 2005, 24, 4961. 25. Katti, K. V.; Gali, H.; Smith, C. J.; Berning, D. E. Acc. Chem. Res. 1999, 32, 9. 26. Appleby, T.; Derek Woollins, J. Coord. Chem. Rev. 2002, 235, 121. 27. Boaz, N. W. Preparation of aminophosphines 2005. 28. Schartel, B. Materials 2010, 3, 4710. 29. Ren, H.; Sun, J.; Wu, B.; Zhou, Q. Polym. Degrad. Stab. 2007, 92, 956. 30. Meyer, A.; Bagowski, C. P.; Kokoschka, M.; Stefanopoulou, M.; Alborzinia, H.; Can, S.; Vlecken, D.
H.; Sheldrick, W. S.; Wölfl, S.; Ott, I. Angew. Chem. Int. Ed. 2012, 51, 8895. 31. Vergara, E.; Casini, A.; Sorrentino, F.; Zava, O.; Cerrada, E.; Rigobello, M. P.; Bindoli, A.; Laguna,
M.; Dyson, P. J. ChemMedChem 2010, 5, 96. 32. Scheffler, H.; You, Y.; Ott, I. Polyhedron 2010, 29, 66. 33. Mirzadeh, N.; Reddy, T. S.; Bhargava, S. K. Coord. Chem. Rev. 2019, 388, 343. 34. Frik, M.; Martínez, A.; Elie, B. T.; Gonzalo, O.; Ramírez de Mingo, D.; Sanaú, M.; Sánchez-Delgado,
R.; Sadhukha, T.; Prabha, S.; Ramos, J. W.; Marzo, I.; Contel, M. J. Med. Chem. 2014, 57, 9995. 35. Groom, C. R.; Bruno, I. J.; Lightfoot, M. P.; Ward, S. C. Acta Crystallogr. Sect. B Struct. Sci. Cryst.
Eng. Mater. 2016, 72, 171. 36. Ullah, R. S.; Wang, L.; Yu, H.; Abbasi, N. M.; Akram, M.; Zain -ul-Abdin; Saleem, M.; Haroon, M.;
Khan, R. U. RSC Adv. 2017, 7, 23363. 37. Fei, Z.; Scopelliti, R.; Dyson, P. J. Dalton Trans. 2003, 0, 2772. 38. Chandrasekhar, V.; Thilagar, P.; Murugesa Pandian, B. Coord. Chem. Rev. 2007, 251, 1045. 39. Rothemund, S.; Teasdale, I. Chem. Soc. Rev. 2016, 45, 5200. 40. Bálint, E.; Tajti, Á.; Tripolszky, A.; Keglevich, G. Dalton Trans. 2018, 47, 4755. 41. Carroll, M. P.; Guiry, P. J. Chem. Soc. Rev. 2014, 43, 819. 42. Michaelis, A. Justus Liebigs Ann. Chem. 1903, 326, 129. 43. Pandey, M. K.; Mague, J. T.; Balakrishna, M. S. Inorg. Chem. 2018, 57, 7468.

56
44. Nifant’ev, I. E.; Vinogradov, A. A.; Vinogradov, A. A.; Roznyatovsky, V. A.; Grishin, Y. K.; Ivanyuk, A. V.; Sedov, I. V.; Churakov, A. V.; Ivchenko, P. V. Organometallics 2018, 37, 2660.
45. Tessier, M. D.; De Nolf, K.; Dupont, D.; Sinnaeve, D.; De Roo, J.; Hens, Z. J. Am. Chem. Soc. 2016, 138, 5923.
46. Fliedel, C.; Ghisolfi, A.; Braunstein, P. Chem. Rev. 2016, 116, 9237. 47. Li, W.; Zhang, J. Chem. Soc. Rev. 2016, 45, 1657. 48. Gopalakrishnan, J. Appl. Organomet. Chem. 2009, 23, 291. 49. Scolaro, C.; Chaplin, A. B.; Hartinger, C. G.; Bergamo, A.; Cocchietto, M.; Keppler, B. K.; Sava, G.;
Dyson, P. J. Dalton Trans. 2007, 0, 5065. 50. Shriver, D. F.; Drezdzon, M. A. The manipulation of air-sensitive compounds; 2nd ed.; Wiley: New
York, 1986. 51. Errington, R. J. Advanced practical inorganic and metalorganic chemistry; 1. ed.; Blackie Acad. &
Professional: London, 1997. 52. Armarego, W. L. F. Purification of laboratory chemicals; Eighth edition.; Butterworth-Heinemann is
an imprint of Elsevier: Amsterdam, 2017. 53. Streuli, C. A. Anal. Chem. 1960, 32, 985. 54. Henderson, Wm. A.; Streuli, C. A. J. Am. Chem. Soc. 1960, 82, 5791. 55. Li, J.-N.; Fu, Y.; Liu, L.; Guo, Q.-X. Tetrahedron 2006, 62, 11801. 56. O’Connor, A. R.; Nataro, C. Organometallics 2004, 23, 615. 57. Thorstenson, T.; Songstad, J.; Mäki, A.; Pohjonen, M.-L.; Koskikallio, J. Acta Chem. Scand. 1976,
30a, 781. 58. Jeremias, L.; Babiak, M.; Kubát, V.; Calhorda, M. J.; Trávníček, Z.; Novosad, J. Inorganica Chim.
Acta 2016, 443, 230. 59. Fliedel, C.; Poli, R. Coord. Chem. Rev. 2018, 355, 1. 60. Zi, G.; Zhang, F.; Xiang, L.; Chen, Y.; Fang, W.; Song, H. Dalton Trans. 2010, 39, 4048. 61. Magennis, S. W.; Parsons, S.; Pikramenou, Z. Chem. – Eur. J. 2002, 8, 5761. 62. Gopalakrishnan, J.; Rao, M. N. S. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 754. 63. Cowley, A. H.; Dewar, M. J. S.; Jackson, W. R. J. Am. Chem. Soc. 1968, 90, 4185. 64. Cowley, A. H.; Dewar, M. J. S.; Jackson, W. Roy.; Jennings, W. Brian. J. Am. Chem. Soc. 1970, 92,
5206. 65. Neilson, R. H.; Lee, R. C.-Y.; Cowley, A. H. J. Am. Chem. Soc. 1975, 97, 5302. 66. Gorenstein, D. G. Phosphorous-31 NMR: Principles and Applications.; Elsevier Science: St. Louis,
2014. 67. Dewar, M. J. S.; Jennings, B. J. Am. Chem. Soc. 1969, 91, 3655. 68. Neilson, R. H.; Lee, R. C.-Yi.; Cowley, A. H. Inorg. Chem. 1977, 16, 1455. 69. Samuel, R. C.; Kashyap, R. P.; Krawiec, M.; Watson, W. H.; Neilson, R. H. Inorg. Chem. 2002, 41,
7113. 70. Knight, W. D. Phys. Rev. 1949, 76, 1259. 71. Tebby, J. C. CRC handbook of phosphorus-31 nuclear magnetic resonance data; 2018. 72. Quin, L. D. A guide to organophosphorus chemistry; Wiley: New York, 2000. 73. Babu, R. P. K.; Krishnamurthy, S. S.; Nethaji, M. Heteroat. Chem. 1991, 2, 477. 74. Gopalakrishnan, J. Appl. Organomet. Chem. 2009, 23, 291. 75. Atwood, J. L.; Cowley, A. H.; Hunter, W. E.; Mehrotra, S. K. Inorg. Chem. 1982, 21, 1354. 76. Issleib, K.; Brack, A. Z. Für Anorg. Allg. Chem. 1957, 292, 245. 77. Aucott, S. M.; Clarke, M. L.; Slawin, A. M. Z.; Woollins, J. D. J. Chem. Soc. Dalton Trans. 2001,
972. 78. Slawin, A. M. Z.; Woollins, J. D.; Zhang, Q. J. Chem. Soc. Dalton Trans. 2001, 0, 621. 79. Gaw, K. G.; Smith, M. B.; Wright, J. B.; Slawin, A. M. Z.; Coles, S. J.; Hursthouse, M. B.; Tizzard,
G. J. J. Organomet. Chem. 2012, 699, 39. 80. Broomfield, L. M.; Wu, Y.; Martin, E.; Shafir, A. Adv. Synth. Catal. 2015, 357, 3538. 81. Wrackmeyer, B.; Köhler, C.; Milius, W.; Grevy, J. M.; García-Hernández, Z.; Contreras, R. Heteroat.
Chem. 2002, 13, 667. 82. Milton, H. L.; Wheatley, M. V.; Slawin, A. M. Z.; Woollins, J. D. Polyhedron 2004, 23, 2575. 83. Cowley, A. H.; Dewar, M. J. S.; Goodman, D. W.; Schweiger, J. R. J. Am. Chem. Soc. 1973, 95, 6506. 84. Rømming, C.; Songstad, J.; Kjekshus, A.; Sekutowski, J. C.; Bastiansen, O.; Fernholt, L.; Gundersen,
G.; Nielsen, C. J.; Cyvin, B. N.; Cyvin, S. J. Acta Chem. Scand. 1978, 32a, 689. 85. Forti, P.; Damiani, D.; Favero, P. G. J. Am. Chem. Soc. 1973, 95, 756. 86. Belyakov, A. V.; Haaland, A.; Shorokhov, D. J.; Sokolov, V. I.; Swang, O. J. Mol. Struct. 1998, 445,
303. 87. Durig, J. R.; Panikar, S.; Zhou, X.; El Defrawy, A. M. Spectrochim. Acta. A. Mol. Biomol. Spectrosc.
2008, 69, 715. 88. Bulloch, G.; Keat, R.; Rycroft, D. S. J Chem Soc Dalton Trans 1978, 764.

57
89. Stepanova, V. A.; Smoliakova, I. P. Curr. Org. Chem. 2012, 16, 2893. 90. Fei, Z.; Păunescu, E.; Ang, W. H.; Scopelliti, R.; Dyson, P. J. Eur. J. Inorg. Chem. 2014, 2014, 1745. 91. Gongoll, M.; Müller, B. H.; Peitz, S.; Aluri, B. R.; Peulecke, N.; Spannenberg, A.; Rosenthal, U.; Al-
Hazmi, M. H.; Wöhl, A.; Müller, W. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1845. 92. Lane, A. P.; Morton-Blake, D. A.; Payne, D. S. J Chem Soc A 1967, 0, 1492. 93. Biricik, N.; Durap, F.; Kayan, C.; Gümgüm, B. Heteroat. Chem. 2007, 18, 613. 94. Biricik, N.; Kayan, C.; Gümgüm, B.; Fei, Z.; Scopelliti, R.; Dyson, P. J.; Gürbüz, N.; Özdemir, İ.
Inorganica Chim. Acta 2010, 363, 1039. 95. Mathis, R.; Mathis, F.; Ayed, N.; Borgi, A. E.; Baccar, B.-G. J Chem Soc Chem Commun 1977, 614. 96. Roberts, B. P.; Singh, K. J Chem Soc Perkin Trans 2 1981, 866. 97. Power, P. P. Chem. Rev. 2003, 103, 789. 98. Griller, D.; Roberts, B. P.; Davies, A. G.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 554. 99. Malavaud, C.; Boisdon, M. T.; Charbonnel, Y.; Barrans, J. Tetrahedron Lett. 1979, 20, 447. 100. Wang, P.; Liu, H.; Li, Y.-Q.; Zhao, X.-L.; Lu, Y.; Liu, Y. Catal. Sci. Technol. 2016, 6, 3854. 101. Mohan, T.; Sudheendra Rao, M. N.; Aravamudan, G. Tetrahedron Lett. 1989, 30, 4871. 102. Ewart, G.; Lane, A. P.; McKechnie, J.; Payne, D. S. J. Chem. Soc. Resumed 1964, 1543. 103. Biricik, N.; Durap, F.; Kayan, C.; Gümgüm, B. Heteroat. Chem. 2007, 18, 613. 104. Fukazawa, A.; Usuba, J.; Adler, R. A.; Yamaguchi, S. Chem. Commun. 2017, 53, 8565. 105. Sollott, G. P.; Mertwoy, H. E.; Portnoy, S.; Snead, J. L. J. Org. Chem. 1963, 28, 1090. 106. Xiao, C. B. and J. Catalytic Synthesis of Phosphines and Related Compounds. Curr. Org. Chem.
http://www.eurekaselect.com/63152/article (accessed June 1, 2019). 107. Dhokale, R. A.; Mhaske, S. B. Org. Lett. 2013, 15, 2218. 108. Montchamp, J.-L.; Tian, F.; Frost, J. W. J. Org. Chem. 1995, 60, 6076. 109. Juge, S.; Genet, J. P. Tetrahedron Lett. 1989, 30, 2783. 110. Contreras, R.; Grevy, J. M.; García‐Hernández, Z.; Göizado‐Rodriguez, M.; Wrackmeyer, B.
Heteroat. Chem. 2001, 12, 542. 111. VanWazer, J. R.; Maier, Ludwig. J. Am. Chem. Soc. 1964, 86, 811. 112. Liu, Z.; Li, X.; Zhang, J. Phosphorous Sulfur Relat. Elem. 1988, 40, 215. 113. Nifantiev, E. E.; Grachev, M. K.; Burmistrov, S. Yu. Chem. Rev. 2000, 100, 3755. 114. Milbrath, D. S.; Verkade, J. G. J. Am. Chem. Soc. 1977, 99, 6607. 115. Ullah, S. S.; Rohman, S. S.; Kashyap, C.; Guha, A. K. New J. Chem. 2019, 43, 2575. 116. Yang, Y.; Guo, L.; Tian, Z.; Ge, X.; Gong, Y.; Zheng, H.; Shi, S.; Liu, Z. Organometallics 2019, 38,
1761. 117. Broomfield, L. M.; Alonso-Moreno, C.; Martin, E.; Shafir, A.; Posadas, I.; Ceña, V.; Castro-Osma, J.
A. Dalton Trans. 2017, 46, 16113. 118. Du, Q.; Yang, Y.; Guo, L.; Tian, M.; Ge, X.; Tian, Z.; Zhao, L.; Xu, Z.; Li, J.; Liu, Z. Dyes Pigments
2019, 162, 821. 119. Benito-Garagorri, D.; Bocokić, V.; Mereiter, K.; Kirchner, K. Organometallics 2006, 25, 3817. 120. Dutta, D. K.; Woollins, J. D.; Slawin, A. M. Z.; Konwar, D.; Sharma, M.; Bhattacharyya, P.; Aucott,
S. M. J. Organomet. Chem. 2006, 691, 1229. 121. Biricik, N.; Durap, F.; Kayan, C.; Gümgüm, B.; Gürbüz, N.; Özdemir, İ.; Ang, W. H.; Fei, Z.;
Scopelliti, R. J. Organomet. Chem. 2008, 693, 2693. 122. Ioannou, P.-C.; Arbez-Gindre, C.; Zoumpanioti, M.; Raptopoulou, C. P.; Psycharis, V.; Kostas, I. D.;
Kyritsis, P. J. Organomet. Chem. 2019, 879, 40. 123. Tolman, C. A. J. Am. Chem. Soc. 1970, 92, 2953. 124. Yazdani, S.; Silva, B. E.; Cao, T. C.; Rheingold, A. L.; Grotjahn, D. B. Polyhedron 2019, 161, 63. 125. Scherpf, T.; Schwarz, C.; Scharf, L. T.; Zur, J.-A.; Helbig, A.; Gessner, V. H. Angew. Chem. Int. Ed.
2018, 57, 12859. 126. Niemeyer, Z. L.; Milo, A.; Hickey, D. P.; Sigman, M. S. Nat. Chem. 2016, 8, 610. 127. Ardizzoia, G. A.; Brenna, S. Phys. Chem. Chem. Phys. 2017, 19, 5971. 128. Donahue, C. M.; McCollom, S. P.; Forrest, C. M.; Blake, A. V.; Bellott, B. J.; Keith, J. M.; Daly, S.
R. Inorg. Chem. 2015, 54, 5646. 129. Snelders, D. J. M.; van Koten, G.; Klein Gebbink, R. J. M. Chem. - Eur. J. 2011, 17, 42. 130. Morris, A. L.; York, J. T. J. Chem. Educ. 2009, 86, 1408. 131. Trogler, W. C.; Marzilli, L. G. Inorg. Chem. 1975, 14, 2942. 132. DeSanto, J. T.; Mosbo, J. A.; Storhoff, B. N.; Bock, P. L.; Bloss, R. E. Inorg. Chem. 1980, 19, 3086. 133. Brown, T. L.; Lee, K. J. Coord. Chem. Rev. 1993, 128, 89. 134. Magee, M. P.; Luo, W.; Hersh, W. H. Organometallics 2002, 21, 362. 135. Hill, W. E.; Minahan, D. M. A.; Taylor, J. G.; McAuliffe, C. A. J. Am. Chem. Soc. 1982, 104, 6001. 136. Power, W. P.; Wasylishen, R. E. Inorg. Chem. 1992, 31, 2176. 137. Brunel, J. M.; Heumann, A.; Buono, G. Angew. Chem. Int. Ed. 2000, 39, 1946.

58
138. Clarke, M. L.; Holliday, G. L.; Slawin, A. M. Z.; Woollins, J. D. J. Chem. Soc. Dalton Trans. 2002, 1093.
139. Wilkins, L. C.; Melen, R. L.; Platts, J. A.; Newman, P. D. Dalton Trans 2017, 46, 14234. 140. Slawin, A. M. Z.; Smith, M. B.; Woollins, J. D. J. Chem. Soc. Dalton Trans. 1997, 3397. 141. Slawin, A. M. Z.; Milton, H. L.; Wheatley, J.; Woollins, J. D. Polyhedron 2004, 23, 3125. 142. Gaw, K. G.; Slawin, A. M. Z.; Smith, M. B. Organometallics 1999, 18, 3255. 143. Balakrishna, M. S.; Suresh, D.; George, P. P.; Mague, J. T. Polyhedron 2006, 25, 3215. 144. Aydemir, M.; Baysal, A.; Sahin, E.; Gumgum, B.; Ozkar, S. Inorganica Chim. Acta 2011, 378, 10. 145. Balakrishna, M. S.; Naik, S.; Mobin, S. M. Inorganica Chim. Acta 2010, 363, 3010. 146. Pandey, M. K.; Mague, J. T.; Balakrishna, M. S. Inorg. Chem. 2018, 57, 7468. 147. Clarke, M. L.; Cole-Hamilton, D. J.; Slawin, A. M. Z.; Woollins, J. D. Chem. Commun. 2000, 2065. 148. Li, Y.; Li, Z.; Hou, Y.; Fan, Y.-N.; Su, C.-Y. Inorg. Chem. 2018, 57, 13235. 149. Angurell, I.; Martínez-Ruiz, I.; Rossell, O.; Seco, M.; Gómez-Sal, P.; Martín, A. Chem Commun
2004, 1712. 150. Majoumo‐Mbe, F.; Lönnecke, P.; Hey‐Hawkins, E. Z. Für Anorg. Allg. Chem. 2008, 634, 2385. 151. Dixon, I. M.; Lebon, E.; Loustau, G.; Sutra, P.; Vendier, L.; Igau, A.; Juris, A. Dalton Trans. 2008,
5627. 152. Dixon, I. M.; Lebon, E.; Sutra, P.; Igau, A. Chem. Soc. Rev. 2009, 38, 1621. 153. Lebon, E.; Bastin, S.; Sutra, P.; Vendier, L.; Piau, R. E.; Dixon, I. M.; Boggio-Pasqua, M.; Alary, F.;
Heully, J.-L.; Igau, A.; Juris, A. Chem. Commun. 2011, 48, 741. 154. Kosgei, G. K.; Breen, D. J.; Lamb, R. W.; Livshits, M. Y.; Crandall, L. A.; Ziegler, C. J.; Webster, C.
E.; Rack, J. J. J. Am. Chem. Soc. 2018, 140, 9819. 155. Lebon, E.; Sylvain, R.; Piau, R. E.; Lanthony, C.; Pilmé, J.; Sutra, P.; Boggio-Pasqua, M.; Heully, J.-
L.; Alary, F.; Juris, A.; Igau, A. Inorg. Chem. 2014, 53, 1946. 156. Miyano, S.; Nawa, M.; Mori, A.; Hashimoto, H. Bull. Chem. Soc. Jpn. 1984, 57, 2171. 157. Vineyard, B. D.; Knowles, W. S.; Sabacky, M. J.; Bachman, G. L.; Weinkauff, D. J. J. Am. Chem.
Soc. 1977, 99, 5946. 158. Czauderna, C. F.; Slawin, A. M. Z.; Cordes, D. B.; van der Vlugt, J. I.; Kamer, P. C. J. Tetrahedron
2019, 75, 47. 159. Jia, J.; Fan, D.; Zhang, J.; Zhang, Z.; Zhang, W. Adv. Synth. Catal. 2018, 360, 3793. 160. Bhattacharyya, P.; Woollins, J. D. Polyhedron 1995, 14, 3367. 161. Necas, M.; St J. Foreman, M. R.; Marek, J.; Derek Woollins, J.; Novosad, J. New J. Chem. 2001, 25,
1256. 162. Zubiri, M. R. I.; Milton, H. L.; Cole-Hamilton, D. J.; Slawin, A. M. Z.; Derek Woollins, J. Polyhedron
2004, 23, 693. 163. Slawin, A. M. Z.; Wainwright, M.; Woollins, J. D. J. Chem. Soc. Dalton Trans. 2002, 513. 164. Benito-Garagorri, D.; Bocokić, V.; Mereiter, K.; Kirchner, K. Organometallics 2006, 25, 3817. 165. Leveson-Gower, R. B.; Webb, P. B.; Cordes, D. B.; Slawin, A. M. Z.; Smith, D. M.; Tooze, R. P.;
Liu, J. Organometallics 2018, 37, 30. 166. Murugesan, S.; Stöger, B.; Carvalho, M. D.; Ferreira, L. P.; Pittenauer, E.; Allmaier, G.; Veiros, L. F.;
Kirchner, K. Organometallics 2014, 33, 6132. 167. van der Boom, M. E.; Milstein, D. Chem. Rev. 2003, 103, 1759. 168. Beletskaya, I. P.; Cheprakov, A. V. J. Organomet. Chem. 2004, 689, 4055. 169. Dolinsky, M. C. B.; Lin, W. O.; Dias, M. L. J. Mol. Catal. Chem. 2006, 258, 267. 170. Gwynne, E. A.; Stephan, D. W. Organometallics 2011, 30, 4128. 171. Hofmann, A. W. Justus Liebigs Ann. Chem. 1857, 103, 357. 172. Kondo, T.; Sone, Y.; Tsuji, Y.; Watanabe, Y. J. Organomet. Chem. 1994, 473, 163. 173. Takeuchi, R.; Tsuji, Y.; Fujita, M.; Kondo, T.; Watanabe, Y. J. Org. Chem. 1989, 54, 1831. 174. Johnstone, T. C.; Suntharalingam, K.; Lippard, S. J. Chem. Rev. 2016, 116, 3436. 175. Ghosh, A. K.; Matsuda, H. Org. Lett. 1999, 1, 2157. 176. Kondo, T.; Tsuji, Y.; Watanabe, Y. Tetrahedron Lett. 1988, 29, 3833. 177. Miyamoto, T.; Kasagami, T.; Asai, M.; Yamamoto, I. Pestic. Biochem. Physiol. 1999, 63, 151. 178. Bandyopadhyay, P.; Sathe, M.; Tikar, S. N.; Yadav, R.; Sharma, P.; Kumar, A.; Kaushik, M. P.
Bioorg. Med. Chem. Lett. 2014, 24, 2934. 179. John, H.; Thiermann, H. J. Chromatogr. B 2010, 878, 1205. 180. Coover, J. H. W. Three-component aluminum-titanium tetrahalide catalyst for olefin polymerization
therewith 1960. 181. Cheng, J.; Sun, Y.; Wang, F.; Guo, M.; Xu, J.-H.; Pan, Y.; Zhang, Z. J. Org. Chem. 2004, 69, 5428. 182. Durap, F.; Biricik, N.; Gümgüm, B.; Özkar, S.; Ang, W. H.; Fei, Z.; Scopelliti, R. Polyhedron 2008,
27, 196. 183. Tye, H.; Smyth, D.; Eldred, C.; Wills, M. Chem. Commun. 1997, 1053. 184. Wu, H.; Xie, F.; Wang, Y.; Zhao, X.; Liu, D.; Zhang, W. Org. Biomol. Chem. 2015, 13, 4248.

59
185. Priya, S.; Balakrishna, M. S.; Mobin, S. M.; McDonald, R. J. Organomet. Chem. 2003, 688, 227. 186. Peng, Z.-Y.; Wang, J.-P.; Cheng, J.; Xie, X.; Zhang, Z. Tetrahedron 2010, 66, 8238. 187. Fei, Z.; Dyson, P. J. Coord. Chem. Rev. 2005, 249, 2056. 188. Zijp, E. J.; van der Vlugt, J. I.; Tooke, D. M.; Spek, A. L.; Vogt, D. Dalton Trans. 2005, 512. 189. Chikkali, S. H.; Bellini, R.; de Bruin, B.; van der Vlugt, J. I.; Reek, J. N. H. J. Am. Chem. Soc. 2012,
134, 6607. 190. Ionescu, G.; van der Vlugt, J. I.; Abbenhuis, H. C. L.; Vogt, D. Tetrahedron Asymmetry 2005, 16,
3970. 191. Clavero, P.; Grabulosa, A.; Rocamora, M.; Muller, G.; Font-Bardia, M. Dalton Trans. 2016, 45, 8513. 192. Magennis, S. W.; Parsons, S.; Pikramenou, Z.; Corval, A.; Woollins, J. D. Chem. Commun. 1999, 61. 193. Pietraszkiewicz, M.; Mech-Piskorz, J.; Pietraszkiewicz, O. Opt. Mater. 2017, 74, 183. 194. Katkova, M. A.; Burin, M. E.; Logunov, A. A.; Ilichev, V. A.; Konev, A. N.; Fukin, G. K.;
Bochkarev, M. N. Synth. Met. 2009, 159, 1398. 195. Lecloux, D. D.; Radu, N. S.; Wang, Y. Compounds comprising phosphorus-containing metal
complexes 2005. 196. Aiello, I.; Ghedini, M.; La Deda, M. J. Lumin. 2002, 96, 249. 197. Bischoff, L.; Baudequin, C.; Hoarau, C.; Urriolabeitia, E. P. In Advances in Organometallic
Chemistry; Pérez, P. J., Ed.; Academic Press, 2018; Vol. 69, pp 73. 198. Roundhill, D. M.; Gray, H. B.; Che, C. M. Acc. Chem. Res. 1989, 22, 55. 199. van der Veen, R. M.; Milne, C. J.; El Nahhas, A.; Lima, F. A.; Pham, V.-T.; Best, J.; Weinstein, J. A.;
Borca, C. N.; Abela, R.; Bressler, C.; Chergui, M. Angew. Chem. Int. Ed. 2009, 48, 2711. 200. Anderson, G. W.; Blodinger, J.; Young, R. W.; Welcher, A. D. J. Am. Chem. Soc. 1952, 74, 5304. 201. Asrar, J. Flame retardant recycled polyester copolymers 1995. 202. Wang, D.; Wang, H. Acta Pharm. Sin. B 2012, 2, 107. 203. Vredenburg, G.; den Braver-Sewradj, S.; van Vugt-Lussenburg, B. M. A.; Vermeulen, N. P. E.;
Commandeur, J. N. M.; Vos, J. C. Toxicol. Lett. 2015, 232, 182. 204. MIHAL, C. P. Am. Ind. Hyg. Assoc. J. 1987, 48, 997.

60
Chapter 3
CYCLOMETALATION REACTIONS
TABLE OF CONTENTS
1. Cyclometalation reactions 61 1.1. Mechanisms of cyclometalation 63 1.2. Investigation of cyclometalation mechanism 64 2. Cyclometalated complexes of PGMs 66 References 69

61
1. CYCLOMETALATION REACTIONS
The cyclometalation is described as transition metal-mediated activation of a C−H or C−R bond resulting in a formation of a metallacycle with a metal-carbon σ bond.1 First, the transition metal coordinates to the donor group of the ligand (E), and then the intramolecular activation of the C−H (C−R) bond occurs resulting in a formation of a metallacycle (Fig. C1). Despite the labile C−M σ bond, the cyclometalated complexes are stable because of the chelate effect, which is even more pronounced when two cycles are formed.2,3 While the five-membered metallacycles are considered most stable, the four- or more than six-membered rings are somewhat unstable and uncommon.4 Therefore five-member rings are prepared with most transition metals as highly active catalysts for organic synthesis. Despite the remarkable stability of metallacycles, investigation of C−H (sp2) activation mechanisms revealed that formation of some four-membered metallacycles is reversible.5–7
Figure C1. General chart of cyclometalation reaction showing ligand coordinating via the donating group E and subsequent activation of the C−H bond and formation of metallacycle.
Cyclometalation in which the relatively stable C−H (or C−R) bond is activated and cleaved, is of interest for half of the century. It was successfully employed for the preparation of complexes with platinum group metals (Ru, Rh, Pd, Os, Ir, Pt).1 The positioning of both the donor atom E and the C−H (C−R) moiety on the ligand influence the cyclometalation reaction, thus the bond activation is a heteroatom-assisted

62
process, involving donors E such as N, O, P, S, and Se. Consequently, the metallacycle often consists of heteroatoms other than the metal.
The selection of a metal precursor also influences the cyclometalation process. The precursor usually contains weakly bonded ligands (e.g. C2H4, cod) that are replaced by the desired ligand (note, that chlorides are replaced in K2[PtCl4]). Cyclometalation is also promoted by metal precursors bearing ligands suitable for the abstraction of the C−H activated proton (e.g. OAc, Cl). PGMs offer a wide range of precursors, either molecular (Pd(OAc)2, [PtCl2(SEt2)2], [Ir(PPh3)2H5]) or oligomeric ([RhCl(cod)]2, [RuCl2(CO)2]∞).8 The electronic configuration at the metal centre together with the nature of the C−H bond, particularly the hybridization at carbon, determine which reaction mechanism takes place. Also, subtle changes in the ligand structure (e.g. in the donor site E) may influence the stability of particular conformations along the reaction coordinate and may transform a potential intermediate into a transition state and vice versa. The exact mechanism of cyclometalation is not well understood, and experimental data often provide more diverging rather than converging picture.
Usually, three mechanisms of cyclometalation are distinguished: (i) electrophilic C−H bond activation; (ii) oxidative addition; (iii) σ-bond metathesis.1,8–13 Apart from cyclometalation, several methods were developed for the preparation of metallacycles, in particular, the C−X bond activation14 (X = F, Cl, Br, I, OTf), and transmetalation.15,16 In addition, metallacycles can be generated by elimination reactions,17 cycloadditions,18 and by hydrometalations.19
Cyclometalation represents a highly versatile pathway for the generation of metallacycles as very efficient catalysts, materials for energy conversion,20,21 and biomedical applications.22–24 Besides, cyclometalation is useful for the ortho-functionalization of substituted arenes or heterocycles. Also, in a context of phosphine-containing complexes, the PGM cyclometalated compounds exhibit very intriguing spectroscopic properties,25,26 which can be used as luminescent probes for the investigation of bio-related environments. Considering the vast application potential of metallacycles for C−H bond activation in organic synthesis27–31, as well as its limitations, the further progress

63
in cyclometalation will provide novel and unprecedented synthetic transformations.
1.1. MECHANISMS OF CYCLOMETALATION
The two main mechanisms of cyclometalation are the electrophilic bond activation and oxidative addition. The former is typical for complexes containing 10th − 12th group d-metals (Pd, Ag, Pt).1 The latter is mainly subject to complexes with electron-rich (i.e. heavy) metals (Os, Ru, Re, Ir, Pt).32–36 Notably, the IrI and RhI complexes are most frequently cyclometalated by this mechanism.37 The electrophilic bond activation mechanism is accelerated by the presence of electron-donating substituents. Thus this type of cyclometalation can be seen as an organic electrophilic substitution.38,39
Figure C2. Proposed mechanism of cyclometalation by electrophilic bond activation. Formation of both π- and σ-complex results in a formation of five-membered metallacycle. The substituent R represents both the strong and weak electron-donating group, while E represents donor atom (N, P, As, see Fig. C1).
According to electrophilic aromatic substitution, the π-complex (i. e. interaction of π-electrons of the aromatic system with the metal) is formed, which then changes to the more favourable σ-complex accompanied by proton cleavage. Later, however, was found that the cyclometalation of some complexes (e.g. metal centres bearing carboxylates or carbonates) does not involve the formation of the π-complex, but the σ-complex is formed directly (see Fig. C2).38,40 Potent electron-donating substituents promote the cyclometalation via

64
arenium intermediate (π-complex) because of the stabilization of the delocalized positive charge, on the other hand, weaker donors promote direct formation of the σ -complex. The determination of the cyclometalation proceedings is difficult.
In the oxidative addition, the metal centre oxidises and releases (usually two) electrons into the anti-bonding σ-orbital of the C−H bond, which is then activated. The cleaved C−H hydrogen can either remain on the metal or leave with the anion released from the metal (Fig. C3) with subsequent reductive elimination.35,2 The process of reductive elimination is determined by many factors, such as the stability of the oxidation state of the metal, temperature, and basicity of the leaving ligand.
Figure C3. Mechanism of cyclometalation by oxidative addition. Both the oxidation of metal centre M → MII and the transfer of the C−H activated hydrogen to the metal centre take place in the first step, which is followed by subsequent reductive elimination MII → M and release of HX molecule.
Cyclometalations proceeding on the ligand aryl moieties are generally referred to as ortho-metalations and those usually take place via oxidative addition mechanism. The C−H bond activation on the aromatic ring during oxidative addition is enhanced by electron acceptor groups, and thus, the mesomeric and induction effects on the aromatic ring strongly influence the particular mechanism of cyclometalation.
1.2. INVESTIGATION OF CYCLOMETALATION MECHANISM
The development of computational techniques in the past twenty years enabled the chemists to pursue the search for understanding the mechanisms of various reactions. The contribution of some groups to understanding the experimental observations explained by chemical calculations is indeed essential. Mostly the work of B. Milstein,41–43 D. L. Davies,44 S.A. MacGregor,45 and E. Laga46 provide an exciting insight into the understanding of the reaction mechanism. Recent review44 discusses the issue of C-H bond activation from a perspective

65
of carboxylate-assisted C–H bond activation in PGM complexes. Notably, the PGM complexes with acetate ligands represent a usual subject of investigation of cyclometalation mechanism47,48 and can be used as an example for a demonstration of the complexity of the reaction process.
Figure C4. The energetic pathway and depiction of calculated structures of cyclometalation. The figure shows the acetate-assisted C–H bond activation and proton transfer in Pd(OAc)2(DMBA) described by three separate transition states (TS) and energy values in kcal/mol.
The DFT calculations were employed to probe the role of acetate in the Pd complex with two acetates and dimethylbenzylamine (DMBA).40 The reaction proceeds via an acetate-assisted H-transfer process involving a transition state with an agostic49,50 C…H intermediate (Fig. C4). The proton transfer occurs via a six-membered transition state which is initiated by a displacement of one arm of the η2-acetate by C−H bond in TS1. The change in conformation subsequently leads to the formation of an agostic intermediate TS2. In the TS2, the hydrogen bonding interaction between the DMBA ortho-H and the displaced acetate (C−H···OC = 2.04 Å) results in an ideal set up for proton transfer with low activation barrier (TS3). In the cyclometalated product, the acetate (proton acceptor) twists away from the Pd−C bond and shares the accepted hydrogen with the second acetate ligand. The formation of the cyclometalated product is both thermodynamically favoured and kinetically accessible.

66
Later, the acetate-assisted cyclometalation was inspected in a series of [MCl2Cp*]2 dimers (M = Ir, Rh) with meta- and para-substituted 1-phenylpyrazoles.45 The reactions of the dimers and substituted phenylpyrazoles in the presence of NaOAc resulted in a formation of cyclometalated Cp*M(phpyz)Cl. The reaction proceedings were studied both experimentally and using DFT calculations. Interestingly, the time-course and proton/deuterium exchange experiments indicate that the formation of the cyclometalated product can be reversible or irreversible depending on the metal, the substituents, and the reaction conditions. The experiments with ligands further suggest that the kinetic selectivity favours electron-donating substituents, but surprisingly the thermodynamic selectivity is opposite, with substrates with electron-withdrawing groups being favoured. Also, the impact of electronic and steric effects on the course of the cyclometalation was evaluated for various platinum complexes recently.51 The trends were reproduced with DFT calculations showing the C–H activation proceeding via ambiphilic metal-ligand activation (AMLA)52,53 mechanism. The mechanistic study indicates that the use of substituent effects to assign the mechanism of C–H activation may be misleading unless both the energetics of the C–H cleavage and the subsequent reactions are considered.
2. CYCLOMETALATED COMPLEXES OF PGMS
Platinum group metals (Ru, Os, Rh, Ir, Pd, and Pt) represent one of the most favoured groups of metals studied for cyclometalations. Their rich coordination chemistry leads to a formation of many metallacycles prepared by heteroatom-assisted C−H bond activation. The result of PGMs diversity is a vast range of references to each element, yet cyclopalladation and cycloruthenation are by far the most extensively studied. The following section aims at general aspects of PGMs cyclometalation and some of the new aspects of this chemistry.
Cyclometalated Ru complexes are proving their enormous application potential (e.g. Fig. C5) in photophysical and electrochemical applications such as solar cells54,55 or electron-transfer systems.56–59 Also, cyclometalated Ru complexes are among the most active catalysts60–63 and bioactive compounds64,65 known to date. A review66 compiling

67
a range of settings that can be employed for cycloruthenation leads to a conclusion that cycloruthenation is exceptionally versatile and of comprehensive scope.
Figure C5. Two examples of cyclometalated ruthenium complexes suitable as a sensitiser of solar cells (left) and active as antitumoral metallodrugs (right).
The numbers of reports on cyclometalations of Rh, Ir, and Os are somehow unbalanced, with the latter receiving far less attention than others.67 For instance, the C−H bond activation in Rh complexes is known for half a century,68–70 resulting in many exciting compounds, for example Rh(CH3)(PPh3)3,71 an analogue of Wilkinson’s catalyst. Investigation of cycloiridation is stimulated by the exceptional performance of iridacycles in catalysis and for photophysical applications.26,72–74 Several IrIII complexes have been recognized as powerful organic light-emitting diodes, OLEDs, and as dopants for tuning the excitation energy and emission wavelength through modifications in the cyclometalated ligand.75–77
Cyclopalladation is extensively reported because of its broad application potential.8,13,78 As enormously versatile compounds, palladacycles have been employed mostly as catalysts79,80 with a variety of ligands, including amines, phosphines, or sulfides.78,81 Also platinum readily form cyclometalated complexes with many (typically pincer) ligands bearing soft donor groups (N, P, S).8,78 The high electron density at Pt core stabilises higher oxidation states and hence the cyclometalated complexes containing PtIV are relatively stable and can be isolated.82

68
Cyclometalation in Pt(II) proceeds via the oxidative addition mechanism because of higher electron density (i. e. the lower electrophilicity of Pt(II) core reduces the scope of electrophilic mechanisms for C−H bond activation)83 especially when electron acceptor groups are present on the aromatic backbone of the ligand. The generally lower cyclometalation activity of PtII is, however, overpassed employing pincer ligands84,85 promoting the C-H bond activation and formation of double metallacycle.86

69
REFERENCES
1. Albrecht, M. Chem. Rev. 2010, 110, 576. 2. Beck, R.; Camadanli, S.; Klein, H.-F. Eur. J. Inorg. Chem. 2018, 2018, 608. 3. McGraw-Hill encyclopedia of science & technology: an international reference work in twenty
volumes including an index; 10. ed. ff.; McGraw-Hill: New York, NY, 2007. 4. Martín, R.; Crespo, M.; Font-Bardia, M.; Calvet, T. Organometallics 2009, 28, 587. 5. Albrecht, M.; Dani, P.; Lutz, M.; Spek, A. L.; van Koten, G. J. Am. Chem. Soc. 2000, 122, 11822. 6. Bessel, C. A.; Aggarwal, P.; Marschilok, A. C.; Takeuchi, K. J. Chem. Rev. 2001, 101, 1031. 7. Baltensperger, U.; Guenter, J. R.; Kaegi, S.; Kahr, G.; Marty, W. Organometallics 1983, 2, 571. 8. Omae, I. Cyclometalation Reactions Five-Membered Ring Products as Universal Reagents; 2014. 9. Vicente, J.; Saura-Llamas, I. Comments Inorg. Chem. 2007, 28, 39. 10. Ryabov, A. D. Chem. Rev. 1990, 90, 403. 11. Canty, A. J.; van Koten, G. Acc. Chem. Res. 1995, 28, 406. 12. Parshall, G. W. Acc. Chem. Res. 1970, 3, 139. 13. Omae, I. J. Organomet. Chem. 2018, 869, 88. 14. Grove, D. M.; Van Koten, G.; Ubbels, H. J. C.; Zoet, R.; Spek, A. L. Organometallics 1984, 3, 1003. 15. Longoni, G.; Fantucci, P.; Chini, P.; Canziani, F. J. Organomet. Chem. 1972, 39, 413. 16. Kumar, S.; Jana, O.; Subramaniyan, V.; Mani, G. Inorganica Chim. Acta 2018, 480, 113. 17. Poznjak, A. L.; Pawlowski, V. I.; Schkolnikowa, L. M.; Djatlowa, N. M.; Iljuchin, A. B. J.
Organomet. Chem. 1986, 314, C59. 18. Wenzel, A. G.; Grubbs, R. H. J. Am. Chem. Soc. 2006, 128, 16048. 19. Bijpost, E. A.; Zuideveld, M. A.; Meetsma, A.; Teuben, J. H. J. Organomet. Chem. 1998, 551, 159. 20. Wu, Q.; Cheng, Y.; Xue, Z.; Gao, X.; Wang, M.; Yuan, W.; Huettner, S.; Wan, S.; Cao, X.; Tao, Y.;
Huang, W. Chem. Commun. 2019, 55, 2640. 21. Giménez, N.; Lalinde, E.; Lara, R.; Moreno, M. T. Chem. – Eur. J. 2019, 25, 5514. 22. Millán, G.; Giménez, N.; Lara, R.; Berenguer, J. R.; Moreno, M. T.; Lalinde, E.; Alfaro-Arnedo, E.;
López, I. P.; Piñeiro-Hermida, S.; Pichel, J. G. Inorg. Chem. 2019, 58, 1657. 23. Martínez‐Alonso, M.; Busto, N.; Aguirre, L. D.; Berlanga, L.; Carrión, M. C.; Cuevas, J. V.;
Rodríguez, A. M.; Carbayo, A.; Manzano, B. R.; Ortí, E.; Jalón, F. A.; García, B.; Espino, G. Chem. – Eur. J. 2018, 24, 17523.
24. Clemente, M.; Polat, I. H.; Albert, J.; Bosque, R.; Crespo, M.; Granell, J.; López, C.; Martínez, M.; Quirante, J.; Messeguer, R.; Calvis, C.; Badía, J.; Baldomà, L.; Font-Bardia, M.; Cascante, M. Organometallics 2018, 37, 3502.
25. Ionescu, A.; Godbert, N.; Ricciardi, L.; La Deda, M.; Aiello, I.; Ghedini, M.; Rimoldi, I.; Cesarotti, E.; Facchetti, G.; Mazzeo, G.; Longhi, G.; Abbate, S.; Fusè, M. Inorganica Chim. Acta 2017, 461, 267.
26. Lai, P.-N.; Teets, T. S. Chem. – Eur. J. 2019, 25, 6026. 27. Periana, R. A.; Mironov, O.; Taube, D.; Bhalla, G.; Jones, C. J. Science 2003, 301, 814. 28. Meier, S. K.; Young, K. J. H.; Ess, D. H.; Tenn, W. J.; Oxgaard, J.; Goddard, W. A.; Periana, R. A.
Organometallics 2009, 28, 5293. 29. Young, K. J. H.; Meier, S. K.; Gonzales, J. M.; Oxgaard, J.; Goddard, W. A.; Periana, R. A.
Organometallics 2006, 25, 4734. 30. Young, K. J. H.; Mironov, O. A.; Periana, R. A. Organometallics 2007, 26, 2137. 31. Aguilar, D.; Aragüés, M. A.; Bielsa, R.; Serrano, E.; Navarro, R.; Urriolabeitia, E. P. Organometallics
2007, 26, 3541. 32. Higashi, T.; Ando, H.; Kusumoto, S.; Nozaki, K. J. Am. Chem. Soc. 2019, 141, 2247. 33. Chapp, S. M.; Schley, N. D. Organometallics 2017, 36, 4355. 34. Garner, M. E.; Parker, B. F.; Hohloch, S.; Bergman, R. G.; Arnold, J. J. Am. Chem. Soc. 2017, 139,
12935. 35. Camadanli, S. Cyclometalation and Bicyclometalation Reactions of Trimethylphosphine Supported
Iron, Cobalt and Nickel Compounds via C-H Activation with Imine and Carbonyl Anchoring Groups; Technischen Universität Darmstadt: Darmstadt; Vol. 2005.
36. Janowicz, A. H.; Bergman, R. G. J. Am. Chem. Soc. 1982, 104, 352. 37. Harper, T. G. P.; Desrosiers, P. J.; Flood, T. C. Organometallics 1990, 9, 2523. 38. Albrecht, M.; Spek, A. L.; van Koten, G. J. Am. Chem. Soc. 2001, 123, 7233. 39. Polukeev, A. V.; Marcos, R.; Ahlquist, M. S. G.; Wendt, O. F. Chem. Sci. 2015, 6, 2060. 40. Davies, D. L.; Donald, S. M. A.; Macgregor, S. A. J. Am. Chem. Soc. 2005, 127, 13754. 41. Barrios-Francisco, R.; Balaraman, E.; Diskin-Posner, Y.; Leitus, G.; Shimon, L. J. W.; Milstein, D.
Organometallics 2013, 32, 2973.

70
42. Frech, C. M.; Leitus, G.; Milstein, D. Organometallics 2008, 27, 894. 43. van der Boom, M. E.; Milstein, D. Chem. Rev. 2003, 103, 1759. 44. Davies, D. L.; Macgregor, S. A.; McMullin, C. L. Chem. Rev. 2017, 117, 8649. 45. Alharis, R. A.; McMullin, C. L.; Davies, D. L.; Singh, K.; Macgregor, S. A. J. Am. Chem. Soc. 2019,
141, 8896. 46. Laga, E.; García-Montero, A.; Sayago, F. J.; Soler, T.; Moncho, S.; Cativiela, C.; Martínez, M.;
Urriolabeitia, E. P. Chem. - Eur. J. 2013, 19, 17398. 47. Sajjad, M. A.; Harrison, J. A.; Nielson, A. J.; Schwerdtfeger, P. Organometallics 2018, 37, 3659. 48. Cocco, F.; Zucca, A.; Stoccoro, S.; Serratrice, M.; Guerri, A.; Cinellu, M. A. Organometallics 2014,
33, 3414. 49. Sajjad, M. A.; Harrison, J. A.; Nielson, A. J.; Schwerdtfeger, P. Organometallics 2017, 36, 4231. 50. Crosby, S. H.; Clarkson, G. J.; Rourke, J. P. Organometallics 2011, 30, 3603. 51. Maidich, L.; Dettori, G.; Stoccoro, S.; Cinellu, M. A.; Rourke, J. P.; Zucca, A. Organometallics 2015,
34, 817. 52. Boutadla, Y.; Davies, D. L.; Macgregor, S. A.; Poblador-Bahamonde, A. I. Dalton Trans. 2009, 5820. 53. Boutadla, Y.; Al-Duaij, O.; Davies, D. L.; Griffith, G. A.; Singh, K. Organometallics 2009, 28, 433. 54. Soellner, J.; Císařová, I.; Strassner, T. Organometallics 2018, 37, 4619. 55. Bessho, T.; Yoneda, E.; Yum, J.-H.; Guglielmi, M.; Tavernelli, I.; Imai, H.; Rothlisberger, U.;
Nazeeruddin, M. K.; Grätzel, M. J. Am. Chem. Soc. 2009, 131, 5930. 56. Yang, Y.-Y.; Zhu, X.-Q.; Hu, S.-M.; Su, S.-D.; Zhang, L.-T.; Wen, Y.-H.; Wu, X.-T.; Sheng, T.-L.
Angew. Chem. Int. Ed. 2018, 57, 14046. 57. Beley, M.; Collin, J. P.; Louis, R.; Metz, B.; Sauvage, J. P. J. Am. Chem. Soc. 1991, 113, 8521. 58. Launay, J.-P. Chem. Soc. Rev. 2001, 30, 386. 59. Shao, J.-Y.; Gong, Z.-L.; Zhong, Y.-W. Dalton Trans. 2017, 47, 23. 60. Simonetti, M.; Cannas, D. M.; Just-Baringo, X.; Vitorica-Yrezabal, I. J.; Larrosa, I. Nat. Chem. 2018,
10, 724. 61. Sun, R.; Chu, X.; Zhang, S.; Li, T.; Wang, Z.; Zhu, B. Eur. J. Inorg. Chem. 2017, 2017, 3174. 62. Dani, P.; Karlen, T.; Gossage, R. A.; Gladiali, S.; Koten, G. van Angew. Chem. Int. Ed. 2000, 39, 743. 63. Baratta, W.; Ros, P. D.; Zotto, A. D.; Sechi, A.; Zangrando, E.; Rigo, P. Angew. Chem. Int. Ed. 2004,
43, 3584. 64. Erxleben, A. Curr. Med. Chem. 2019, 26, 694. 65. Elumalai, P.; Jeong, Y. J.; Park, D. W.; Kim, D. H.; Kim, H.; Kang, S. C.; Chi, K.-W. Dalton Trans.
2016, 45, 6667. 66. Djukic, J.-P.; Sortais, J.-B.; Barloy, L.; Pfeffer, M. Eur. J. Inorg. Chem. 2009, 2009, 817. 67. Chi, Y.; Chou, P.-T. Chem. Soc. Rev. 2007, 36, 1421. 68. Huang, X.; Meggers, E. Acc. Chem. Res. 2019, 52, 833. 69. Han, Y.-F.; Jin, G.-X. Acc. Chem. Res. 2014, 47, 3571. 70. Omae, I. J. Organomet. Chem. 2017, 841, 12. 71. Keim, W. J. Organomet. Chem. 1968, 14, 179. 72. Feng, Z.; Yu, Y.; Yang, X.; Zhong, D.; Song, D.; Yang, H.; Chen, X.; Zhou, G.; Wu, Z. Inorg. Chem.
2019, 58, 7393. 73. Yang, J.; Brookhart, M. J. Am. Chem. Soc. 2007, 129, 12656. 74. Jensen, C. M. Chem. Commun. 1999, 2443. 75. Okamura, N.; Ishiguro, K.; Maeda, T.; Yagi, S. Chem. Lett. 2017, 46, 1086. 76. Chou, P.-T.; Chi, Y. Chem. – Eur. J. 2007, 13, 380. 77. Salinas, S.; Soto-Arriaza, M. A.; Loeb, B. Polyhedron 2011, 30, 2863. 78. Palladacycles: synthesis, characterization and applications; Dupont, J., Ed.; Wiley-VCH: Weinheim,
2008. 79. Ng, J. K.-P.; Tan, G.-K.; Vittal, J. J.; Leung, P.-H. Inorg. Chem. 2003, 42, 7674. 80. Yao, Q.; Kinney, E. P.; Zheng, C. Org. Lett. 2004, 6, 2997. 81. Sonia Pérez, †; Concepción López, *; Amparo Caubet, †; Ramon Bosque, †; Xavier Solans, ‡; Mercè
Font Bardía, ‡; Anna Roig, § and; Molins§, E. Novel Five-Membered Pallada- and Platinacycles Containing a [C(sp2, ferrocene), N, S]- Terdentate Ligand. Theoretical Interpretation of Their Electrochemical and Electronic Properties Based on Density Functional Calculations. https://pubs.acs.org/doi/abs/10.1021/om030520d (accessed June 21, 2019).
82. Puddephatt, R. J. Coord. Chem. Rev. 2001, 219–221, 157. 83. Principles and applications of organotransition metal chemistry; Collman, J. P., Ed.; University
Science Books: Mill Valley, Calif, 1987. 84. Moulton, C. J.; Shaw, B. L. J. Chem. Soc. Dalton Trans. 1976, 1020. 85. Poverenov, E.; Efremenko, I.; Frenkel, A. I.; Ben-David, Y.; Shimon, L. J. W.; Leitus, G.;
Konstantinovski, L.; Martin, J. M. L.; Milstein, D. Nature 2008, 455, 1093.

71
86. Soro, B.; Stoccoro, S.; Minghetti, G.; Zucca, A.; Cinellu, M. A.; Manassero, M.; Gladiali, S. Inorganica Chim. Acta 2006, 359, 1879.
87. Brooks, J.; Babayan, Y.; Lamansky, S.; Djurovich, P. I.; Tsyba, I.; Bau, R.; Thompson, M. E. Inorg. Chem. 2002, 41, 3055.
88. Hirani, B.; Li, J.; Djurovich, P. I.; Yousufuddin, M.; Oxgaard, J.; Persson, P.; Wilson, S. R.; Bau, R.; Goddard, W. A.; Thompson, M. E. Inorg. Chem. 2007, 46, 3865.
89. Baar, C. R.; Jenkins, H. A.; Vittal, J. J.; Yap, G. P. A.; Puddephatt, R. J. Organometallics 1998, 17, 2805.

72
CHAPTER 4
DISCUSSION OF PUBLISHED RESULTS
TABLE OF CONTENTS
1. General remarks 73 2. Summary of the results 73 2.1 Pt coordination compounds 76 2.2 Ru coordination compounds 79 3. Concluding remarks and future outlook 79 4. Work on platinum complexes 81 4.1. Bifurcated hydrogen bonds in platinum(II) complexes Chyba! Záložka není definována. 4.2. Quantum chemical calculations of 31P NMR chemical shifts Chyba! Záložka není definována. 4.3. Thermally triggered cycloplatination in platinum(II) complexes Chyba! Záložka není definována. 5. Work on ruthenium complexes 82 5.1. Interpreting the pNMR spectra of potential Ru(III) metallodrugs Chyba! Záložka není definována. 5.2. Hyperfine effects in ligand NMR in paramagnetic Ru(III) complexes Chyba! Záložka není definována. 5.3. Antimetastatic Ru(II)-capped cucurbit[n]uril-based rotaxanes Chyba! Záložka není definována.

73
1. GENERAL REMARKS
The presented work is split into two groups, with regard to the transition metal (i.e. Pt or Ru) involved. Discussed are the chapters of work which were published, accepted, or submitted to impacted chemistry journals. In general, the main contribution of the author of the thesis was the synthesis of molecular and supramolecular systems described in the manuscripts and their characterisation by various analytical methods as well as the elaboration of provided data and preparation of the manuscripts for publication.
2. SUMMARY OF THE RESULTS
2.1. PT COORDINATION COMPOUNDS
The first part deals with the synthesis and characterisation of platinum(II) complexes with phosphine ligands. The three somewhat separate manuscripts give a comprehensive overview of spectroscopic properties, structural features, and reactivity of the prepared Pt systems, mainly based on recorded 31P NMR spectra, data from X-ray crystallography, and theoretical calculations. The molecular structures were investigated for noncovalent interactions, while the reactivity, thermal behaviour and cyclometalation of the complexes were investigated using 31P NMR.
Figure D1. General chart of the Pt complexes which are referred to in the discussion and the attached manuscripts. The cis (left) and trans (right) complexes were prepared by different synthetic procedures. Note that the aromatic derivatives are connected via different groups X (X = NH, O).

74
In general, phosphinites and phosphinoamines were used as ligands bearing additional functional groups with different electronic effects (e.g. COOCH3, F, CN). The resulting group of PtII complexes consisted of both cis/trans isomers of PtCl2L2 (L = phosphine) with both ortho/para isomers of ligands attached to the Pt core (see Fig. D1).
In the crystal structures of the reported Pt/phosphinoamine complexes, the hydrogen bonds (HBs) were observed between NH groups and Cl and A1 acceptors (A1 = COOCH3, C(O)CH3, F, CN). In the subchapter 4.1, we report on the competition between the two acceptors in bifurcated intramolecular HB arrangements and its influence on conformations of the complexes. Relatively straightforward methods were employed for quantification of the energy contributions of the NH···ClPt interactions. The calculations confirmed that the presence of additional ortho substituent on the ligand moiety decreases significantly the stability of the NH···ClPt interactions, in particular when the C=O group is present. In other words, although the carbonyl-mediated HB stabilises the structures of the free ligands, the bifurcated HB arrangement in the described complexes is destabilised in its presence. The structural data from X-ray diffraction were linked to calculated conformational energies from DFT and topological analysis obtained from QTAIM calculations. The most prominent NH···ClPt interactions were found in the cis complexes, which show delocalisation index (DI) of about 0.09; the presence of the ligand ortho substituent decreases the DI value down to zero for C(O)OCH3 and C(O)CH3 moieties, respectively. The somewhat disturbed intramolecular HB may have a further impact on the stability or reactivity of such systems. We used the findings to support our subsequent investigation of cyclometalation within the closely related Pt complexes (see the subchapter 4.3). We further suggest that the intramolecular bifurcated HBs may enable particular conformations, which may be exploited in crystal engineering.
The aim of the subchapter 4.2 was to find the most suitable method for the calculation of 31P NMR chemical shifts of reported platinum complexes. In general, the influence of a basis set, DFT functional, solvent effects, and SO interactions on the calculated chemical shifts were evaluated in this work. Variety of combinations of DFT functionals (e.g. BP86, B3LYP, PBE0) with all-electron basis sets

75
(e.g. 6-31G, 6-31G(d)) and ECP basis sets were compared. The obtained data were compared to those of real 31P NMR measurements. The comparison of all the electron basis sets shows the importance of the inclusion of the d polarisation functions in the geometry optimisation. Acceptable results were obtained even with the 6-31G(d) basis, while the best results were obtained with the 6-311G(d,p) basis set. The best performance was observed for PBE0, TPSSh and CAM-B3LYP functionals. As might be expected, the inclusion of solvent effects also improved the results. Chemical shielding constants were calculated using BP86, PBE0 and B3LYP functionals in combination with the TZ2P basis and the magnitude of spin-orbit interactions was also evaluated. The comparison of DFT functionals used in the chemical shielding constants calculations revealed good performance of pure DFT functional BP86 that was slightly better than the performance of hybrid functionals PBE0 and B3LYP. The magnitude of SO interactions was in the range of 6 – 40 ppm and showed the necessity of the inclusion of SO interactions in the chemical shielding constants calculations. The work provides valuable findings for both synthetic and theoretical chemists and can also be further used for the investigation of similar systems by our group.
The last subchapter, 4.3, covers an investigation of reactivity of the prepared platinum complexes, mainly their tendency to cyclometalate. The study reports on a different susceptibility of platinum cis complexes to an intramolecular C–H bond activation and formation of five-membered metallacycles. In the study, we used several Pt cis complexes with phosphinoamine and phosphinite ligands (Fig. D1) and boiled them in CHCl3 and toluene for cyclometalation. We monitored the reactions employing 31P NMR spectroscopy, which revealed the best (nearly quantitative) proceeding towards cyclometalation with complexes bearing ligands with X = NH and A1 = 2-COOCH3, while with X = NH and A2 = 4-COOCH3 or H the reaction was almost prohibited. The deployment of the DFT methods for investigation of reaction pathways revealed that the formation of five-membered metallacycles proceeds through the AMLA(4) pathway featuring a 4-membered transition state with approximately pyramidal Pt geometry. Also, as shown by DFT calculations, the differences in reaction and activation

76
energies were not enough to explain the different reactivity. We postulate, however, that intramolecular hydrogen NH···ClPt interactions in particular, complexes are responsible for the low incidence of starting conformations that are required for a successful cyclometalation (cf. 4.1).
2.2. RU COORDINATION COMPOUNDS
The second part deals with the synthesis and characterisation of ruthenium complexes of medicinal interest, mainly the NAMI- and RAPTA-type complexes (see Fig. D2).
Figure D2. Representative examples of biologically active complexes with Ru(III) (NAMI-A, RuPy) and Ru(II) (RAPTA-C) centres.
The first two subchapters 5.1 and 5.2 are dedicated to the preparation of octahedral NAMI-type Ru(III) complexes with various pyridine ligands, which were used for the development of methodology of paramagnetic NMR. The third subchapter 5.3 is focused on the synthesis of supramolecular organometallic RAPTA-type complexes and the evaluation of their biological activity.
The first two subchapters are focused on paramagnetic analogues of the NAMI-A with a general structure [R-pyH][RuCl4(DMSO)(R-py)], (R-py = 4- or 3-substituted pyridine). The complexes were synthesised and characterised by traditional analytical methods (e.g. EA, IR, 1H NMR) for confirmation of their identity, including X-ray structural analyses of several of the complexes. The paramagnetic systems are generally considered difficult to characterise in detail by NMR spectroscopy, and thus a systematic structural and methodological NMR study was performed. The effect of the unpaired electron on the 1H and

77
13C NMR chemical shifts was investigated in the pilot study 5.1 using temperature-dependent NMR experiments and DFT calculations. A relativistic two-component DFT approach was used to understand the electronic factors influencing the orbital (δorb, temperature-independent) and paramagnetic (δpara, temperature-dependent) contributions to the total NMR chemical shifts. The paramagnetic contributions to the 13C NMR chemical shifts were correlated with the distribution of spin density in the ligand moiety and the 13C isotropic hyperfine coupling constants, Aiso(13C), for the carbon atoms. The experimental NMR chemical shifts and their orbital (temperature-independent) and paramagnetic (temperature-dependent) contributions were estimated from 1D NMR spectra measured in the temperature range 233–323 K and analysed using Curie plots. The orbital contributions to δ for the individual atoms in the Ru complexes were compared with the total NMR chemical shifts in diamagnetic Rh(III) analogues, which were also prepared and characterised. This methodological study contributes to our understanding of the factors influencing the distribution of spin density in transition metal complexes that is generally responsible for the paramagnetic contribution to the NMR shifts, and to stimulate further combined experimental-theoretical investigations of open-shell systems including metallodrugs. The structural and experimental data and the elaborated calculation procedures were already exploited in several other publications.
The following study shown in the subchapter 5.2 focuses on the further development of a tool for paramagnetic NMR using analogues of NAMI bearing 3-substituted pyridines instead of imidazole. Another systematic study of the 1H and 13C NMR chemical shifts was performed on a series of new Ru(III) complexes. Apart from other analytical techniques, the molecular structure of 3 compounds was determined by X-ray crystallography. Several relativistic DFT approaches to calculate the electronic g-tensors, hyperfine coupling tensors, and total NMR chemical shifts were evaluated. The four-component DKS approach was used to calculate and analyse the physical contributions to hyperfine coupling tensors and, subsequently, δHF for the individual ligand atoms. In order to understand the experimental hyperfine 1H and 13C NMR shifts, the relativistic spin-orbit and solvent effects on the distribution of spin

78
density and the hyperfine contributions to the NMR shifts were also demonstrated. The reported work was further exploited in an investigation of long-range substituent effects and spin-orbit relativistic contributions to the hyperfine NMR shifts.
Figure D3. Schematic representation of the synthesis of reported Ru(II)-capped rotaxane complexes with pseudorotaxane ligands 1@CB[6] and 1@CB[7].
The last subchapter 5.3 reports the first coupling of Ru(II) organometallic units with cucurbit[6/7]uril-based pseudorotaxane ligands, which are meant for biological application (Fig. D3). The resulting ruthenium-capped rotaxanes were characterised by 1H, 13C NMR, ESI-MS, IR, and the structure of one of the supramolecular systems was determined by X-ray diffraction. The ligand-exchange processes were investigated using the effect of salt concentration on the hydrolysis of chlorides through 1H NMR spectroscopy and UV-vis. As possible antimetastatic metallodrugs, the complexes were evaluated for biological activity performed on three selected mammalian breast cell lines, HBL-100, MCF-7, and MDA-MB-231. The antimetastatic activity of the assembled cationic Ru(II)-rotaxanes evaluated in migration inhibition assays against MCF-7 and MDA-MB-231 cell lines was notably enhanced compared to that of the RAPTA-C, which was used as a reference. The indicated synergistic effect of combining Ru(II) organometallic units with supramolecular pseudorotaxane opens a new direction in searching for anticancer metallodrugs.

79
3. CONCLUDING REMARKS AND FUTURE OUTLOOK
The work on Pt and Ru complexes represents just a part of a whole which is yet to be completed in the future. Apart from the reported publications, the results of the work were presented at several domestic and international conferences and meetings. Although many new platinum complexes were synthesised, characterised, and already published, many others remain a part of other on-going projects in our laboratory. The presented results highlight the tight relationship between structure and reactivity, and the discussion of the topic is approached through the perspective of structural and theoretical chemistry. The structural data and the studied non-covalent interactions might provide a base for future investigation by crystal engineering. The obtained results already stimulated a follow-up project dealing with the synthesis of catalytically active cyclometalated and pincer-like Pd complexes.12 Besides the synthesis of new compounds, the resulting theses provided an initial evaluation of the catalytic activity of the prepared complexes.
The development of paramagnetic ruthenium complexes started as a part of the Czech science foundation project but continued after its end with the development of organometallic Ru antimetastatic supramolecular complexes. The development of paramagnetic NMR represents a contribution to a very current topic of theoretical and coordination chemistry (as suggested by a very recent review3 by Pell). The results were further exploited in our group resulting in two manuscripts in high-impact journals.4,5 On the other hand, the prepared paramagnetic complexes were not evaluated for their biological activity up to date. It might also provide a lead for the following project, in particular if the N-heterocyclic cations in complexes will be replaced by sodium or potassium cations. The introduction of supramolecular systems in the chemistry of ruthenium-based metallodrugs resulted in an up-and-coming platform suitable for further development. The considerable variability
1 Štěpánek, Z. Bachelor’s thesis, Masaryk University, Brno, 2016. 2 Štěpánek, Z. Master’s thesis, Masaryk University, Brno, 2018. 3 Pell, A. J.; Pintacuda, G.; Grey, C. P. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1. 4 Chyba, J.; Novák, M.; Munzarová, P.; Novotný, J.; Marek, R. Inorg. Chem. 2018, 57, 8735. 5 Jeremias, L.; Novotný, J.; Repisky, M.; Komorovsky, S.; Marek, R. Inorg. Chem. 2018, 57, 8748.

80
of the piano-stool organometallic Ru moiety provides many possibilities for synthetic modifications. Given the results of the antimetastatic activity, the binuclear supramolecular complexes call for further development aimed at the tuning of the selectivity towards various cancer cell lines or improvement of the metallodrug efficacy.

81
4. WORK ON PLATINUM COMPLEXES
4.1. Published paper
Martin Sojka; Jaromir Tousek; Zahra Badri; Cina Foroutan-Nejad; Marek Necas Polyhedron 2019, 170, 593-601; DOI: 10.1016/j.poly.2019.06.014 (IF = 2.28, times cited: 0)
Author’s contribution: Synthesis, crystallisation, and characterisation of the Pt complexes, discussion of crystal structures, Hirshfeld surface analysis, manuscript preparation and finalisation, elaboration of the revised manuscript.
4.2. Manuscript under consideration, revision requested
Martin Sojka; Marek Necas, Jaromir Tousek J. Mol. Mod. 2019; revision requested
Author’s contribution: Synthesis and characterisation of the Pt complexes, crystallisation, NMR measurements, preparation of related parts of the manuscript.
4.3. Manuscript in preparation
Martin Sojka; Marek Necas; Cyclometalation in Pt(II) complexes with phosphinoamine ligands; manuscript in preparation
Author’s contribution: Synthesis and characterisation of compounds, NMR experiments, crystallisation and determination of crystal structures, preparation of the manuscript and its graphical layout

82
5. WORK ON RUTHENIUM COMPLEXES
5.1. Published paper
Jan Novotny; Martin Sojka; Stanislav Komorovsky; Marek Necas; Radek Marek J. Am. Chem. Soc. 2016, 138, 8432–8445; DOI: 10.1021/jacs.6b02749 (IF = 14.69, times cited: 18)
Author’s contribution: Synthesis and characterisation of the series of Ru and Rh complexes, crystallisation and determination of crystal structures, preparation of related parts of the manuscript (synthesis, figures, discussion of crystal structures).
5.2. Published paper
Jan Novotny; David Prichystal; Martin Sojka; Stanislav Komorovsky; Marek Necas; Radek Marek Inorg. Chem. 2018, 57, 641–652; DOI: 10.1021/acs.inorgchem.7b02440 (IF = 4.85, times cited: 6)
Author’s contribution: Synthesis and characterisation of Ru paramagnetic complexes, crystallisation of compounds, preparation of related parts of the manuscript (synthesis, discussion of crystal structures).
5.3. Accepted paper
Martin Sojka; Michaela Fojtu; Jindriska Fialova, Michal Masarik, Marek Necas; Radek Marek Inorg. Chem. 2019; DOI: 10.1021/acs.inorgchem.9b01203. (IF = 4.85, times cited: 0)
Author’s contribution: Design, synthesis, and characterisation of supramolecular Ru metallodrugs, UV-Vis measurements, NMR experiments, preparation of the manuscript and its graphical layout, finalising the text after comments of the co-authors and revision requests of the referees.





























