Embed Size (px)
Citation preview

Mat. Res. Bull. Vol. 2, pp. 165-184, 1967. Pergamon Pres s , Inc. Printed in the United States.
CHARACTERIZATION OF d ELECTRONS IN SOLIDS BY STRUCTURE II. Spontaneous Crystallographic Distortions
John B. Goodenough Lincoln Laboratory,* Massachusetts Institute of Technology
Lexington, Massachusetts
ABSTRACT
(Received December 5, 1966; Communicated by author)
Spontaneous crystallographic distortions to a lower symmetry phase at lower temperatures may be induced either by localized _d electrons or by narrow-band electrons. Band electrons induce changes in trans- lational symmetry that introduce planes of energy dis- continuity at the Fermi surface; localized electrons induce changes in local symmetry that leave the transition-metal cations in the centers of symmetry of their anionic interstices. Ferroelectric and semi- conducting = metallic transitions illustrate distor- tions induced by collective electrons. It is also shown how the shear structures found in nonstoichio- metric, homologous series of transition-metal oxides may be induced by ferroelectric-type distortions. Non. stoichiometric compounds having localized d electrons exhibit ordering of point charges and point defects. Ferroelectric distortions induced by heavy atoms in a lower valence state are also discussed.
Theory
Many transition-metal oxides exhibit spontaneous crystallo-
graphic distortions to a lower symmetry phase at lower tempera-
tures (or have a low symmetry that can be related to a higher-
symmetry phase via a diffusionless distortion). Others exhibit
*Operated with support from the U. S. Air Force.
This paper was presented at the International Conference on the Characterization of Materials, State College. Pa . , U.S.A., November 16- 18, 1966.
165

166 d ELECTRONS IN SOLIDS Vol. 2, No. 2
order-disorder transitions that may involve defect diffusion as
well as electron ordering. Since the character of these
structural changes depends upon the presence or absence of a
Fermi surface, the syn~etry of the low-temperature phase provides
a sensitive indicator of the nature of the d electrons, localized
vs collective, in the higher-symmetry phase.
A. Localized Electrons
Crystallographic distortions due to localized electrons are
characteristically local deformations, but if there is a large
enough density of these local deformations to couple elastically
with one another via VA of Eq. (3), Part I, these deformations
give a cooperative distortion of the crystal as a whole.
There are two types of local deformations: (a) ordering of
electrons among localized orbitals that produces a less symmetric
charge distribution, thus deforming the site (point symmetry) to
stabilize the order, and (b) trapping of charge carriers at
specific cationic sites via atomic deformations that create a
unique potential at those sites. The first type is illustrated
by Jahn-Teller distortions and by magnetostrictive distortions
due to spin-orbit coupling. The second is illustrated by the
formation of small polarons (Landau trapping) in compounds having
the same atom in two different valence states. For the first
type, cooperative distortions are achieved by long-range ordering
of the unique axes of the asymmetric charge distributions at
individual cations so as to minimize the elastic energy. VI of
Eq. (3), Part I, represents the elastic coupling energy between
individually distorted sites. For the second type, cooperative
distortions are achieved by ordering the small polarons - and any

Vol. 2, No. 9. d ELECTRONS IN SOLIDS 167
associated defects - so as to optimize the sum of the elastic
and electrostatic energies.
Although these local distortions may be cooperative~ thus
inducing a macroscopic distortion of the entire crystal, never-
theless the fact that localized orbit~Is are always centro-
syn~netric with respect to their cationic nucleus wherever the
higher-symmetry phase has a centrosymmetric arrangement of near-
neighbor ions imposes a necessary (but not sufficient I condition
on the symmetry of the low-temperature phase: The cations must
remain in the centers of symmetr~ of their anionic interstices (I)
Further~ these distortipns do not require an increase in the
number of molecules per primitive unit cell.
Exchange striction below a magnetic-ordering temperature is a
many-body effect that might be expected to provide an exception to
this general conclusion. However, the most general spin con-
figuration is a spiral, which gives centrosymmetric exchange
forces if the cation is centrosymmetric to the near-neighbor
cations with which it interacts. Therefore any distortions due to
interatomic exchange forces also leave the cations in the centers
of symmetry of their anionic interstices.
B. Collective Electrons
The energy of a free electron of momentum ~ ~ that is moving
in a periodic potential is
E k = E o + (~2k2/2m*)
where m* is the effective mass of the electron.
is characteristic of wave propagation in periodic structures.
(1)
Bragg reflection
The
vectors that satisfy the Bragg reflection conditions define

168 d ELECTRONS IN SOLrDS Vol. 2, No. 2
energy surfaces in momentum space across which there is an energy
discontinuity Eg z in Ek vs ~. The magnitude of E z depends upon
the periodic potential. This potential at any point r in crystal
space may be expanded as a Fourier series
U(r) = 7~ k U k exp(i k.r) (2)
For an energy discontinuity that is small relative to the
bandwidth, Brillouin has shown that
s
Eg z = -2elUkl = -2elX Aktexp(i k-nt) l (3) t=l
where ~t gives the positions in real space of the ~ atoms of the
unit cell. If all the atoms are identical, then
s
U k = A k ? exp(i k.n ) = AkS k
where S k is the usual structure factor of x-ray intensities.
(4)
i. Semiconducting = metallic transitions
The resultant E k vs k curve for a linear chain of N atoms
e v e n l y s p a c e d a d i s t a n c e ~ i s shown i n F i g . 1. I f t h e c o l l e c t i v e -
¢ -_
~, a _1 r I
B A N D W I D T H
(a)
Ek
/ /
T ~ 0 ~ T - o" - 2-~ 2"~ o"
0
(b)
FIG. 1
Energy vs wave vector
for a linear chain of
atoms having one over-
lapping orbital per atom.
Fermi energy Ef corres-
ponds to a half-filled
k band, or one electron
per atom.

Vol. 2, No. 2 d E L E C T R O N S IN SOLIDS 169
electron orbitals are formed from one-each atomic orbitals and if
there is one electron per atom~ the Fermi energy is at k = ±~/2~
and the band is half-filled. Clearly any change in the trans-
lational symmetry that introduces a new energy discontinuity at
the Fermi surface will stabilize occupied states at the expense
of unoccupied states~ thereby reducing the energy Zk(E_k - Eo ) .
The l i n e a r c h a i n o f F i g . 1 may b e r e p r e s e n t e d b y two~
interpenetrating subchains each consisting of alternate ions. A
displacement of each subchain toward the other a distance 6
results in atomic pairs spaced a-26 within pairs and a+26 between
pairs. Such a displacement creates an energy discontinuity at
the Fermi surface (~ = ±~/2~) having a structure factor
S k = exp(i~6/a) + exp(i~)exp(-in6/a) = 2i sin(~6/a)
which means that
(5)
E = A6+... (6) gz
Since the elastic restoring forces are proportional to ~2 a
spontaneous static distortion requires a stabilization of the
occupied states that is linear in the displacements. Initially~
where Eg z is small compared to the bandwidth ~, the number of
states n k that are stabilized is proportional to Egz, so that the
total stabilization energy is
= -A' 62+ .... (7) = -½f Eg z
and no spontaneous distortion is anticipated. This is why semi-
conducting = metallic transitions are not a general phenomenon in
broad-band metals. However~ if the bands are so narrow that
Eqz > ~¢b for relatively small ~ a 6c~ then A' is large and n k -
saturates for 6 > 6 . In this case -- c
~E =-A'62 - -B6 for 6 > 6 c (8)

170 d ELECTRONS IN SOLIDS Vol. 2, No. 2
and the conditions for a finite, spontaneous distortion are met.
Further, note that for Eg z > ~¢b' the widths of the two half
bands have been reduced to nearly zero: This corresponds to
trapping the conducting electrons into homopolar, metal-metal
bonds in the linkages of length a-26 along the chain.
This type of distortion represents a semiconducting
metallic transition, since the low-symmetry phase contains a
filled band (or level) separated by a finite energy gap from an
empty band (or level) whereas the high-symmetry phase has a
partially filled band. It is also possible to show that such a
transition is first-order. It has been found in transition-metal
compounds having partially filled, cation-sublattice d bands
(Class (1) metallic oxides). The characteristic feature of these
distortions is a displacement of the cations from the centers of
s3nmnetry of their anionic interstices toward one or more near-
neighbor cations. This is accompanied by an increase in the
number of molecules per primitive unit cell. The displacements
commonly result in the formation of cationic clusters - pairs,
triangles, squares, ... - the particular cluster formation de-
pending upon the higher-symmetry structure and the number of
electrons per cation in the partially filled bands.
In the case of partially filled, crystalline d bands (Class
(2) metallic oxides), such a semiconducting = metallic transition
could be manifested as a disproportionation into two valence
states for the ions. On the other hand, there may simply be a
change in the number of molecules per primitive unit cell. In
either case the cations are not displaced from the center of
symmetry of their anionic interstice, but a change in the number
Vol. 2, No. 2 d ELECTRONS IN SOLE)S 171
of molecules per primitive unit_cell is required.
Thus distortions due to semiconducting = metallic transitions
can almost always be distinguished from those due to localized-
electron ordering~ and they represent a Fermi-surface-dependent
property that is most prominant where the bands are narrowest. A
similar statement holds for the ferroelectric-type distortions.
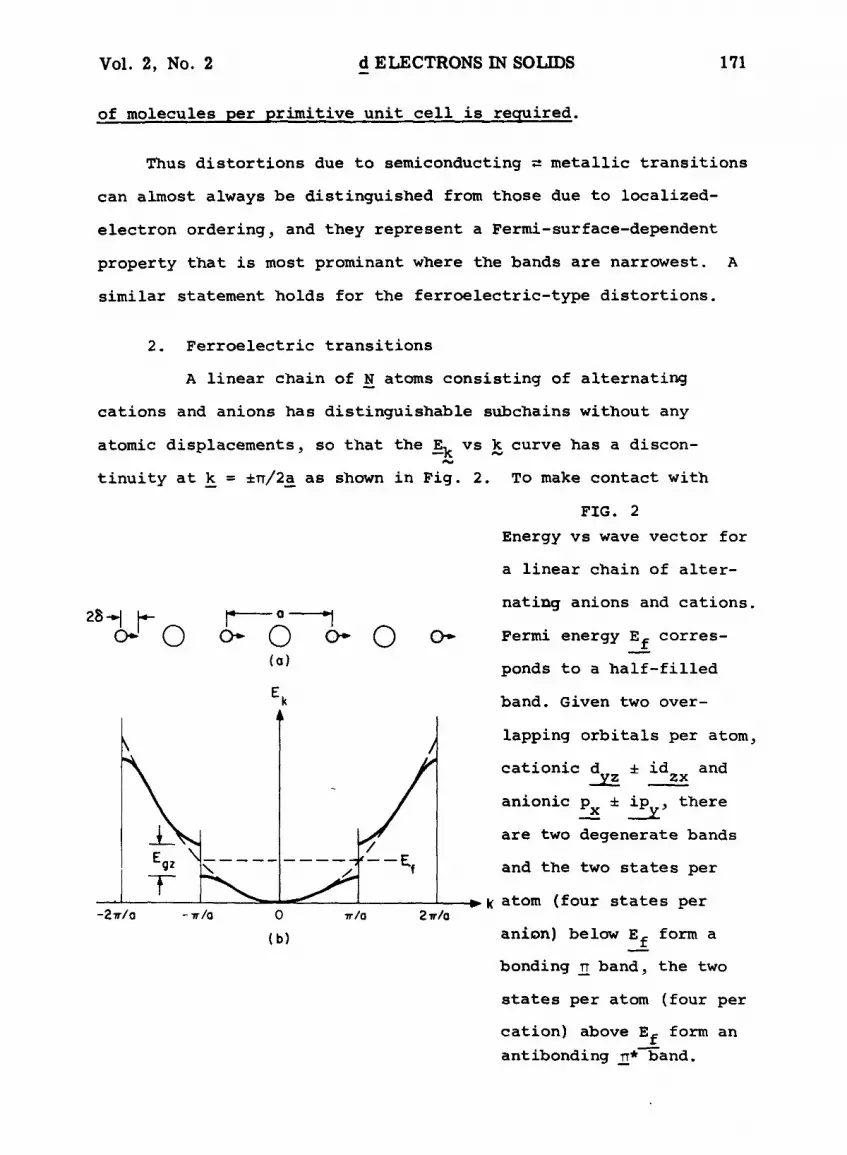
2. Ferroelectric transitions
A linear chain of N atoms consisting of alternating
cations and anions has distinguishable subchains without any
atomic displacements~ so that the ~k vs ~ curve has a discon-
tinuity at ~ = e~/2~ as shown in Fig. 2. To make contact with
FIG. 2
Energy vs wave vector for
a linear chain of alter-
o- - '0 o- . -0 o- . -0 o-- ( a )
E k
• ~ , / Y
-27r/0 - -,r/o 0 ~/o 2 ~/a
(b)
nating anions and cations.
Fermi energy Ef corres-
ponds to a half-filled
band. Given two over-
lapping orbitals per atom~
cationic d ~ id and _y~ zx
anionic P xx ~ ipy~ there
are two degenerate bands
and the two states per
k atom (four states per
anion) below Ef form a
bonding ~ band~ the two
states per atom (four per
cation) above Ef form an
antibonding n* band.

1"t2 d ELECTRONS IN SOL.'~S Vol. 2, No. 2
the perovskite BaTiO3, consider only the two anion Pn orbitals Px
and py (z-axis along chain) and the two cationic d orbitals dz__~ x
and dy z. Because of the difference in potential at the cations
and anions, the ~ bands represented by Fig. 2 consist of a bonding
band for -n/2~ < k < ~/2~ and an antibonding Z* band for
[~/2~l<k<In/~ [ . The bonding orbitals are primarily anionic in
character, the antibonding orbitals are crystalline d orbitals.
With two electrons per atom (four electrons per cation-anion
molecule), the ~ band is filled and the ~* band is empty. From
Eq. (3), the energy gap between ~ and ~* bands is
= -2elAM-AoJ (9) Egz
where A M and A 0 are the A k for the cation and anion, respectively.
If the two sublattices are displaced toward one another a distance
~, then this energy increases:
Eg z = -2e{ IAM-AoI + (AM+Ao)q6+...] (10)
Therefore if the two bands are so narrow that the number of states
that are stabilized by the distortion is n k - 2N for small ~,
then a finite, spontaneous distortion occurs at lower T. Note
also that if ~ additional electrons are introduced, the
stabilization energy is reduced because ~ occupied ~* states are
destabilized by the transition. Therefore additional electrons
either reduce the transition temperature or suppress the
transition entirely.
This type of distortion represents a ferroelectric or anti-
ferroelectric transition, since it is characterized by a dis-
placement of the cations from the centers of symmetry of their
anionic interstices toward one or more near-neighb_or anions. This
need not be accompanied by an increase in the number of molecules

Vol. 2, No. 2 d ELECTRONS IN SOT.TDS 173
per primitive unit cell.
3. Shear structures
Several nonstoichiometric oxide systems have been shown
to consist of a homologous series of discrete phases rather than
a broad range of solid solubility with randomly distributed de-
fects. Magn~li and his coworkers have identified, for example,
the series TinO2n_l (2), (W,M)nO3n_2 with M = Nb or Ta (3), and
(W,MO)nO3n_l (4). These homologous series are to be distinquished
from other ordered-defect phases, such as are found in PRO2_ x or
FeOl+x, by the characteristic occurrence of crystal shear at
regularly spaced intervals. The other compounds, which contain
localized ~ or d electrons, are characterized by ordered point
defects. The systems exhibiting shear structures, on the other
hand, appear to contain collective d ombitals, which suggests
that the surprising regularity of the spacings between shear
planes is due to the existence of a Fermi surface and the energy
stabilization to be achieved by either creating, or increasing the
energy discontinuity across, a Brillouin-zone surface at the
Fermi surface.
Magn~li (5) has already pointed out how condensation of the
defects into common planes minimizes the elastic energy of the
crystal, but this does not explain why these planes are
regularly spaced. However, it does show that any stabilization
due to the creation of a new periodicity via regular spacing only
has to be greater than the entropic energy at temperatures high
enough for diffusion to occur. This stabilization is greater the
narrower the bands, just as for the semiconducting = metallic
transitions and the ferroelectric transitions.

174 d ELECTRONS IN SOLIDS Vol. 2, No. 2
To illustrate this concept, consider TinO2n_l ~ which has
rutile slabs ~ titanium atoms thick that are connected by shear
planes across which the titanium octahedra share common faces~ as
shown in Fig. 3. These shear planes are so spaced that if the
titanium ions are given formal charges +3 and +4, all of the Ti
SHEAR PLANE (lOT)
3+
X OCTAH EDRAL- SITE VACANCl ES
• TITANIUM OF UPPER SLAB
"T • T,TAN,u. OF LOWER SLAB
O OXYGEN SLAB
n+½)a TH CK
c, a, (lOT) REFER TO AXES OF RUTILE SLABS
FIG. 3
Shear plane in TinO2n_l structure.
ions could face one another across the shared octahedral-site
faces. Howeverj with the exception of low-temperature Ti305, the
Ti-Ti spacing across these faces is relatively large, indicating
that Ti3+-Ti 3+ homopolar bonding across the shear planes does not

Vol. 2, No. 2 d ELECTRONS IN SOLIDS 175
take place at room temperature. This means that at high tempera-
tures the semiconductor = metallic transition is not competitive
with a ferroelectric-type transition, which displaces the cations
away from each other across the shear planes. Displacements of
the cations towards one another would reduce the electrostatic
repulsion across a shared face by concentrating electronic charge
in homopolar Ti3+-Ti 3+ bonds. Displacements away from one
another reduces the electrostatic repulsion by increasing both
the cation-cation separation and the anionic shielding. In
either mode, the effect is enhanced by a periodic spacing~ since
this concentrates cooperatively the influence of the distortions
to a definite group of collective-electron orbitals, stabilizing
some at the expense of others. (Localized orbitals would have
only a short-range influence.) In the "ferroelectric mode," a few
unoccupied orbitals are destabilized a great deal whereas a larger
number of occupied orbitals are each stabilized a smaller amount.
In the "semiconductor = metallic mode", a smaller number of
occupied orbitals (2/n per TiO 2 molecule) are stabilized a greater
amount. Since the energy gap between those occupied and un-
occupied orbitals influenced by the transition is larger for the
ferroelectric mode~ this mode dominates at high temperatures
where the periodic structures are initially formed. In a case
like W35Ta4Ol15, the crystalline d bands are all empty~ so that
stabilization via the ferroelectric mode is alone possible. How-
ever, in the system TinO2n_l it is necessary to entertain the
possibility of a semiconductor = metallic transition at lower
temperatures. This would be accompanied by a reversal in the
sign of the Ti-atom displacements at the shear planes.

176 d ELECTRONS IN SOI/DS Vol. 2, No. 2
4. Heavy-atom ferroelectric distortions
In a free atom or ion, splitting between ~ and ~ orbitals
of the same pricipal quantum number is due to different screening
by the core electrons. Therefore the splitting is small for
light atoms, but increases with atomic number until, in the
heavier atoms, it may become comparable to or greater than the
width of the collective-electron ~ and ~ bands. This is why the
heavier atoms may occur in a low formal-valence state. The ions
pb 2+ and Bi 3+, for example, have core electrons (6~ 2 in the free
ions) of large radial extension and of energy intermediate between
bonding and antibonding ~ and ~ orbitals. Because the core
electrons have the same principal quantum number as the cationic
orbitals primarily responsible for binding, they present a core-
repulsion potential of such relatively large radial extension
that the overlap integrals of the bonding and antibonding orbitals
are relatively small. This, coupled with a large electro-
negativity difference between ions as in oxides, can mean narrow
and ~ bands, which provides the possibility of a ferroelectric
transition in which cooperative cationic displacements relative
to the anionic subarray increase the energy gap between bonding
and antibonding states. This possibility is further enhanced by
the large polarizability of the outer core electrons, which
strongly reduces the elastic restoring forces. This
polarizability is due to the relatively small separation between
6~ and 6~ orbitals, which allows for the formation of hybridized
2 6(~sin2~ + ~ cos ~) core orbitals that concentrate the core
electrons on the side of the cation opposite to a nearest-neighbor
anion. This is the reason that spontaneous ferroelectric dis-
tortions may be induced by the heavy atoms Pb 2+ and Bi 3+ even

Vol. 2, No. 2 d ELECTRONS IN SOLIDS 177
though the more obvious source of narrow-band orbitals~ viz.
orbitals~ is absent. Although we are rarely in doubt about the
collective-electron character of outer ~ and ~ electrons~ it is
impDi-tant to appreciate the generality of our concept of the
origin of the ferroelectric-type distortion.
C. Magnetic Order
Although this paper is not concerned with characterization or
the d electrons from magnetic properties~ it is relevant to point
out that the periodicity seen by an electron includes the magnetic
structure. Therefore it is the magnetic space group given by
neutrons~ not the crystallographic space group given by x-rays~
that determines the scattering factors S k and hence the surfaces
of energy discontinuity. In the linear-chain situation of Fig. i~
for example~ an energy discontinuity can be introduced at k =
±~/2~ by the introduction of an antiferromagnetic spin-density
wave (6) ~ since this doubles the number of atoms per unit cell.
The atomic moment created by a spin-density wave is usually small
(<0.5UB) compared to localized-electron moments. Because screen-
ing effects damp out spin-density waves (7)~ this effect has been
unambiguously identified only in a few materials with specialized
conditions. However~ onset of a semiconductor = metallic transi-
tion may stabilize a spin-density wave in the low-temperature
phase~ the magnetic periodicity simply enhancing the energy dis-
continuity introduced by the new lattice periodicity (8). The
significant point is that the appearance of long-range magnetic
order below a semiconducting = metallic transition does not
distinguish a magnetic vs crystallographic origin for the
transition. Such transitions may have a purely magnetic origin~

178 d ELECTRONS Eq SOT,TnS Vol. 2, No. 2
a purely crystallographic origin, or may be due to both mechanisms
operating cooperatively. For our purposes, the point to be
emphasized is that such transitions signal the presence of
collective electrons. In particular~ although the electrons may
be trapped in molecular-cluster orbitals at low temperatures~
these transitions do not reflect localized-electron = collective-
electron transitions in the sense of crystal-field-orbital =
molecular-orbital transitions.
Application
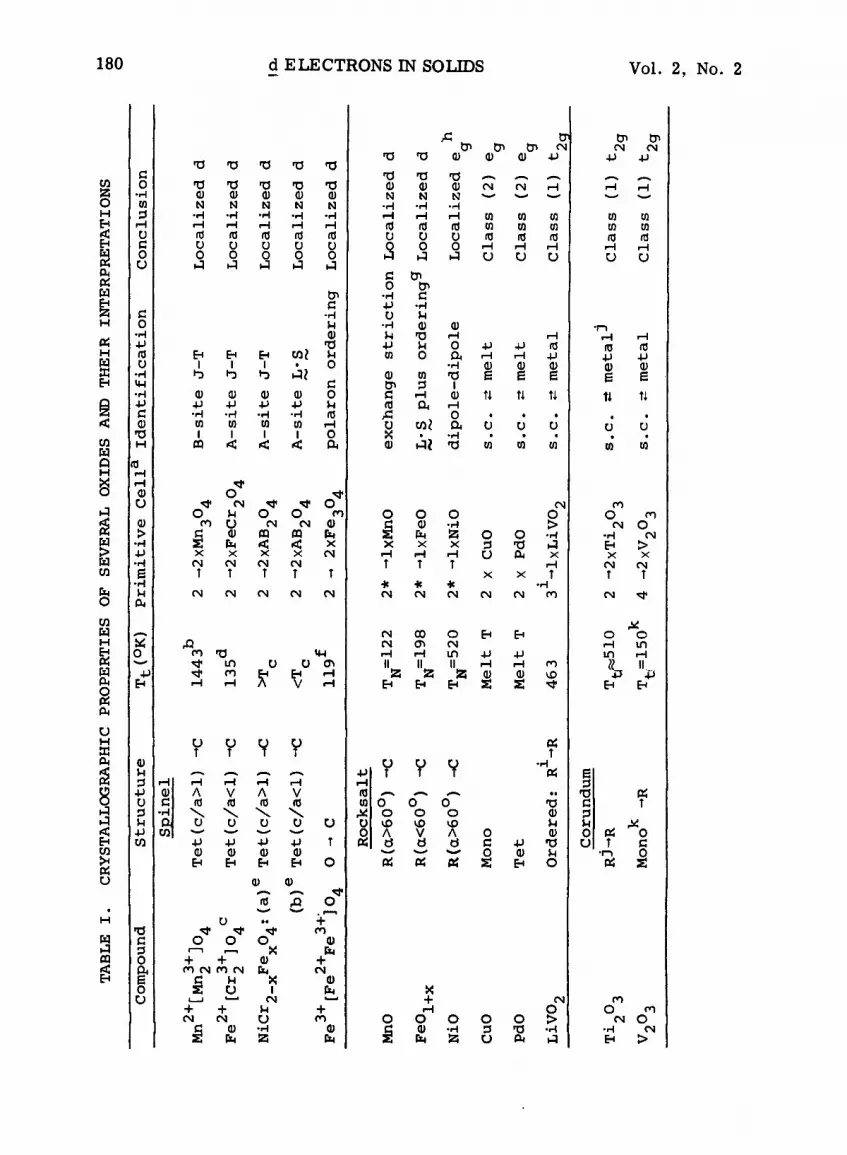
Table I lists some relevant crystallographic properties of
several transition-metal oxides. These have been chosen to
illustrate the several kinds of spontaneous crystallographic dis-
tortions that can be encountered and the type of information about
the d electrons that they provide.
Distortions induced by localized d electrons are illustrated
by several types of Jahn-Teller distortions (9),spin-orbit dis-
tortions (i0), Jahn-Teller vs spin-orbit distortions (ii), small-
polaron ordering, exchange striction, dipole-dipole interactionsj
and high-spin = low-spin transitions (12). The last three items,
however, are not exclusive to localized electrons~ so that the
conclusions that MnO~ CrO 2 and NiO contain localized d electrons
follow from supplementary magnetic data. The compound LaNiO3, on
the other hand, is included because octahedral-site, low-spin Ni 3+
ions are Jahn-Teller ions if the single e electron per ion is q
localized. It was the absence of a Jahn-Teller distortion that led
to the prediction and subsequent discovery (13) that LaNiO 3 is a
metallic oxide.

Vol. 2, No. 2 d ELECTRONS IN SOLIDS 179
Distortions induced by collective d electrons are illustrated
by semiconductor = metallic transitions~ with and without coopera-
tive spin-density-wave formation~ and various ferroelectric-type
distortions. VO 2 represents cooperative semiconductor = metallic
and ferroelectric-type transitions.
BiFeO 3 is included because it represents a ferroelectric
compound having localized d electrons~ the ferroelectric dis-
tortion being due to the existence of a heavy ion Bi 3+ in a low-
valence state. The compound LaCoO 3 illustrates a first-order
semiconductor = metallic transition~ and the key to this
identification was a structural study (12). The metallic compound
PbRu03~ which has a defect pyrochlore structure~ illustrates trap-
mediated Pb-Pb bonding~ which has again been identified by mainly
structural considerations (14).
Although this summary of illustrations is brief~ it does
reveal the power of structural analysis as an aid in the
characterization of the thermodynamic state of the outer
electrons in transition-metal compounds.
TABLE I. CRYSTALLOGRAPHIC PROPERTIES OF SEVERAL OXIDES AND THEIR INTERPRETATIONS
Nomenclature: Mono~ 0 or 0'~ R~ Tet~ C = monoclinc~ orthorhombic
(~<c_//2<b or c_//2<a<b) ~ rhombohedral~ tetragonal~ and cubic. Tt~
TN~ T c = transition~ N~el~ and Curie temperature. J-T = Jahn-
Teller distortion~ L'S = cooperative spin-orbit-coupling ordering
via magnetic ordering with a collinear-spin configuration; B-site
and A-site = octahedral and tetrahedral cations; polarons = small
polarons; s.c. m metal = semiconductor m metallic transition; Co 3+
vs Co III= high-spin vs low-spin trivalent cobalt; ferroel. =
ferroelectric-type transition;ior~cation or anion vacancies; SDW
= spin-density wave. Class (i) = cation-sublattice d bands; Class
(2) = crystalline d bands.
180 d ELECTRONS IN SOI3"DS Vol. 2, No. 2
O IM
I-I
I-I r~
<
r~
I-4
D~
r~ O
I-I
0
8 <
C O
-,-I a}
,M
o C o O
C O
-,-I
r0 o
-,-I tW .,~
'O I-..I
,.-I ,..4
O
.,-I
.M
.,-I
o~
4a
O
a~ O
C
0
R 0
'O 'O 'O 'O 'O • • • • • N N N N N
• M -,-~ .M .M -M
O O O D O O O O O O
C -M
o
I I 0
C • • • • O
4a o 4J .~ • ~ .,"l .,-I -,.-I t~
I I I I 0
O
O ~ O O on m O ~ ~ •
X X X X
T T T T T
~1' m ~ ~ ,-4 ,--4 A V
v ^ v
-IJ ~ 4-; ~ T
~ ~ ~ 0
" " " " 0"*
O O O
+ + • +
+ + ~ +
t r
,10 , o • • • a~
' o ' o ' o - - . .--. ---. • • • e~ ~ ,--4 N N N v ..... • ,-I -M -M
O O O • r0 tO o o ,...4 ,~ ,~
O O~ -,-I izl
.,-4
• ,~ • •
4.J 1,4 0 4-s 4.J ~1
-,4 • • •
O~ ~ I
O o m~o~ d 6 d x - .~ • • •
o o o o ~
o o x x x ~ ~
,-I ,-4 ,-I O 11~ x T T T
x x T • )¢ .N 'I~ -,--I
,-t ,-4 u~ 4.a 4a I I II Z I I • •
O~
T
0 0 o
~ ~ ~ ~ 0
X +
o 0 0 0 0 0 ~>
C,,l J~
A A
,M ,-4
U} tn tO
,-I ,-I O O
.%.-% ,-.-I , - I n~ to
t~ t~
i
d o
t '~ O m
r~ O • M {~I
X X {",I
T T
o o ,.-4 U~ u~ ,-4 ~b I I
.r'~ O
t ~ O m
e~ O

Vol. 2, No. 2 _d ELECTRONS IN SOLIDS 181
C O
.,-I
O c o O
c o .,~ . o
o -,-I
.H 4~
o ~O
tu r-4
o o
o >
-,-I
-,-I
~h
O
O
O
O
C
o
O
C -H 0 -H 0
4~ 4~ 4J
m m m tu a~ t~ O
,-.I ,--I ,--I O r.9 ro ro .-3
O
-W O
4-1 m
• • /
• 1'1 • c O O a~
o $4 o • • :~
tl-I
04 O . ~ c~ O
O > O
x x X
O4 ~
(~ c~
o m
o
0J
@ @
,-4 I
txl O
C e~ O -,~ O
> o
O~ ~n O~ c~ e~ O~ (]; 013
c~ • 0) • C • • c~
.H -H .H ,Q .H -H
m tU ~ ~ 0 ~ • ,-4 m m o o o I o o O ,-4 0 0 0 -.4 0 0
E-I I I-~
i-~ • ,, .-~
@ 0 ,'0 1.-i i.-i .iJ O H O C
• ~ rO m • o • ~ II , 0 0 I-I 0 • "~
• - - I 0 • fl/ 0 I 0 0 I
0'3 t ~ ¢ q 0 3 t 'q t n O O O O O
O C • • O O l> ~ t n r~ [~ o o
o • a~ o -,~ . ~ • a~
E~ x x < x x x x ~ ~-4 ~ ¢xl txl O4 T I' x T T T T
X
O3 I ~ t ~ 0 3
ro cq ,-4 O ,-~ ,--4 k0 O
O II o II II O ,--4 o ~ o O ~ u'~
A ,--4
0 V o T O ~n t
C@ O
O ,--4 ~ v • I~
ml 0 ~ .- t,.
I -~ ,I~ o
~ , .a O
~o O
+
+
O m O ~ O O O • ,~ O c ~ • O .,-~
UI IN
C
t~ C
-,-t
o 0
D
O
X
t~ o
u~
o @ -,-I
o 0 -,-4
o @ 0
-,-I
o
0
ix]
O C
o

182 d ELECTRONS IN SOLIDS Vol. 2, No. 2
TABLE I (CONT'D)
a. Number of molecules per primitive unit cell. Asterisk refers
to magnetic cell, where different from crystalline cell.
b. T = 43°K. c
c. The system Fe3_xCrxO 4 exhibits Tet(c/a<l)~ orthorhombic, and
Tet(c/a>l) symmetries for different values of ~, and these all
correspond to different Jahn-Teller modes for A-site Fe 2+
ions (15).
d. T = 90°K. _c3+..2+ ~+ 3+
e. re x ~ll_ x [Ni Cr2_x]O 4. (a) x < 0.2, (b) x > 0.25.
f. T = 858OK c
g. Two ordered FeOl+ x phases have been identified (16). D and
Fe 3+ ions order so as to minimize the Madelung energy.
h. Although structure alone cannot distinguish antiferromagnetic~
localized e electrons from collective e electrons with a g g
spin-density wave~ the contrast with the PdO structure plus
the nearly spin-only atomic moment indicate these electrons
are localized. Nevertheless~ the high N~el temperature indi-
cates Aca c ~ Ac~ which may account for the ambiguity of the
> A c has been confirmed; transport data. In La2Ni04~ a Aca c
it is Pauli paramagnetic and metallic (17). The Ni-O-Ni
distances in Ni0 and La2NiO 4 are 4.168~ and 3.865A,
respectively.
i. The assumption that triangular clusters are formed in (iii)
planes of V 3+ ions has not yet been established by x-rays.
j. A decrease in c/a ratio with decreasing temperature indicates
stabilization of t o orbitals relative to te orbitals. In this
compound a s.c. = metal transition does not require a change in
symmetry since there is no inversion symmetry about cations
(these are in pairs along c-axis), and it can therefore be
second-order. There is nc antiferromagnetic order below
T t (18).
k. Here s.c. = metal transition causes formation of cation-cation
pairs in basal planes, so that each V 3+ ion has two nearest-
neighbor V 3+ ions and two at a larger separation. T t = 180°K
for rising temperature.
i. At low temperatures a s.c. = metal transition might compete
with the ferroelectric distortion~ the conducting d electrons

Vol. 2, No. 2 d ELECTRONS IN SOLIDS 183 m
becoming trapped in Ti-Ti bonds across the shear planes.
m. At low temperatures, c-axis V-V chains break up into 2.65~ V-V
bonds separated by 3.12~ V-V distances. Hence, there is a
s.c. = metal transition like that of Fig. 1 for the t o orbitals
directed along the c-axis. There is also a tilting of the
pairs to make one shortest Ti-O bond, which indicates a
simultaneous ferroelectric-type transition for the t± orbitals,
the two remaining t2g orbitals that overlap with a free,
anionic p~ orbital.
n. Below the ferromagnetic Curie temperature, there is an
anomalous increase in the c-axis with decreasing temperature.
This indicates half-filled t O orbitals, which would be
stabilized by antiferromagnetic coupling, are forced to have
parallel spins and hence to be localized. The metallic con-
ductivity in this compound occurs because of Class (2) t±
orbitals. (One-electron energies for CrO 2 are given in Ref.
19,
o. One-electron energies for the perovskite structure may be
found in Ref. 20.
p T N 100°K. Although • = Aca c ~ A c and the low-temperature phase
has an enlarged primitive unit cell, the magnetic order and
the distortions are both well interpreted on a crystal-field
model with Jahn-Teller distortions to an orthorhombic (rather
than tetragonal) local-site symmetry [21).
REFERENCES
i. This is clearly not applicable where there are displacements from the centers of symmetry of the anionic interstices in the high-symmetry phase, as in A1203, as a result of a non-centro- symmetric ionic arrangement about the cations.
2. S. Andersson and A• Magn~li, Naturwis 43, 495 (1956); Acta Chem. Scand. i i~ 164 (1950).
3. P. Gado~ B. Holmberg, and A. Magn~li~ Acta Chem. Scand 19~ 2010 (1965).
4. A. Magn~li~ Acta Cryst. ~, 495 (1953)•
5. A. Magn~li~ Nature I15~ 356 (1950); Arkiv Kemi ~, 513 (1950).
6. A. W. Overhauser, Phys. Rev. 128, 1437 (1962)•
7. P. A. Fedders and P. C. Martin, Phys. Rev. 143, 245 (1966).

184 d ELECTRONS ~ SOLIDS Vol. 2 No. 2
8. Alternately~ the magnetic coupling may be treated as strong coupling within cationic clusters and weak coupling between clusters. In the linear-chain case of Fig. 3~ the electrons form ideal homopolar (singlet state) bonds only if there is no interaction between pairs. Interaction between pairs introduces a finite atomic spin density and long-range mag- netic order. The magnitude of the atomic moments depends upon the ratio of the coupling within a pair to that between pairs.
9. J. B. Goodenough~ J. Phys. Chem. Solids 25~ 151 (1964).
i0. J. Kanamori~ Progr. Theoret. Phys. (Kyoto) 17~ 177~ 197 (1957)
ii. J. B. Goodenough~ J. Phys. Soc. Japan 17~ Suppl. B-I~ 185 (1962).
12. P. M. Raccah and J. B. Goodenough~ Phys. Rev. (to be published).
13. J. B. Goodenough and P. M. Raccah~ J. Appl. Phys. Suppl. 36~ 1031 (1965).
14. J. M. Longo~ P. M. Raccah~ and J. B. Goodenough~ J. Chem. Phys. (to be published).
15. M. H. Francombe~ J. Phys. Chem. Solids ~ 37 (1957).
16. P. Vallet and P. M. Raccah~ C.R. Acad. Sci. Paris 258~ 3679 (1964); C. Carel~ D. Weigel and P. Vallet~ C.R. Acad. Sci. Paris 260~ 4325 (1965).
17. J. M. Longo and P. M. Raccah (unpublished research).
18. A. Arrott (personal communication) has shown that the report of antiferromagnetic order by S. C. Abrahams~ Phys. Rev. 130, 2230 (1963)~ is incorrect. J. M. Honig and L. VanZandt also report (unpublished research) that the transport data is incompatible with antiferromagnetic order.
19. J. B. Goodenough~ Bull. Soc. Chim. France~ 1200 (1965).
20. J. B. Goodenough~ J. Appl. Phys. 37~ 1415 (1966).
21. J. B. Goodenoughj Phys. Rev. i00~ 564 (1965); Magnetism and the Chemical Bond~ (Interscience, John Wiley and Sons~ New York-London~ 1963).





























