Embed Size (px)
Citation preview

Current Paediatrics (1996) 6, 13~144 ©1996 Pearson Professional Ltd
Mini-symposium; Cardiology
Cardiomyopathies in infants and children
R. W. M. Yates, J. E N. Taylor
Introduction
Structural and functional disease of the myocardium has long been recognized as being distinct from myocardial disease associated with valvular or con- genital heart disease, pulmonary vascular disease or coronary artery disease. Subsequently, this broad definition of cardiomyopathy has been subdivided into primary or idiopathic disease within the heart and diseases of other organs which cause secondary disease of the heart muscle.
This review will concentrate on the primary car- diomyopathies as they affect infants and children. The main (but not exclusive) causes of secondary cardiomyopathies are listed in Table 1.1
Both primary and secondary cardiomyopathies may have similar pathophysiological consequences for the patient and achieving symptomatic relief will require common therapeutic strategies. Goodwin's 2 original classification of primary cardiomyopathies was based on morphology and function and was primarily descriptive. He listed dilated, hypertrophic and restrictive forms of primary cardiomyopathy and the major features of each of these are pre- sented in Table 2.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is characterized by biventricular dilatation, associated with decreased myocardial pump function and a decreased cardiac output.
R.W.M. Yates MRCP, Registrar in Paediatric Cardiology, J.F.N. Taylor MA, MD, FRCP, Consultant in Paediatfic Cardiology, Cardiothoracic Unit, Cardiac Wing, Great Ormond Street Hospital, London WCIN 3JH.
Correspondence and requests for offprints to JFNT.
Aetiology
As with other idiopathic diseases, multifactorial causation has been proposed for the pathogenesis of DCM. The high familial incidence of DCM, and the fact that certain populations are predisposed to cardiomyopathies, suggests a genetic influence although nutritional and environmental factors may also play a role. Familial DCM is a heterogeneous group of conditions with reports of genomic abnor- malities of mitochondria documented in some cases and abnormalities of the contractile proteins described in others. 3
The presence of a significant mononuclear cell infiltrate within the myocardium in about one third of cases presenting with DCM suggests a possible rela- tionship between myocarditis and DCM. Increasingly sensitive molecular genetic techniques have enabled the detection of viral particles in a significant propor- tion of cases presenting with DCM. 4 A viral infection is thought to trigger a chronic inflammatory myocar- dial response or in some cases precipitate an organ specific autoimmune process directed against the myocardium in patients with genetic susceptibility.
Both persistent and intermittent chronic tachy- arrhythmias are now known to cause DCM. Most commonly these are of supraventricular origin and once the arrhythmia is controlled, the DCM is likely to resolve.
Presentation
Signs and symptoms of congestive cardiac failure secondary to myocardial dysfunction precipitate the need for medical intervention. A history of poor weight gain with respiratory distress during feeding is common in infants whereas the older child presents with genera lmala i se and lethargy. Examination usually reveals an unwell child with skin pallor and low volume peripheral pulses. There
137

138 Current Paediatrics
Table 1 Causes of secondary cardiomyopathies
Infections Viral Coxsackie
Echo Rubella Varicella HIV
Bacterial Meningococcal Pneumococcal Gonnococcal Diphtheria
Fungal Candida Aspergillus
Parasitic Chagas disease Toxoplasmosis
Metabolic Endocrine
Storage d~eases
Musculosketetal diseases
Systemic diseases
Haematological Toxicity sensitivity
Arrhythmias
Hyper/Hypothyroidism Diabetes Phaeochromocytoma Neuroblastoma Nesidioblastosis Glycogen storage diseases (type II, III, IV, V, VI) Mucopolysaccharidoses (all forms) Sphingolipidoses (Gaucher's, Neimann Pick, Tay-Sach's, Refsum's, Fabry's etc.) Nutritional deficiency (kwashiorkor, beriberi, carnitine deficiency, iron deficiency with chronic anaemia) Muscular dystrophies (Duchenne's, Erb's, Steinert) Myopathies (spinal muscular atrophy, nemaline myopathy) Neuromuscular disorders (Friedrich's ataxia, multiple lentiginosis) Connective tissue diseases (SLE, juvenile rheumatoid arthritis, PAN, pseudoxanthoma elasticum) Vasculitides (Kawasaki's) Others (mitochondrial cytopathy, Reye's syndrome, Noonan's, osteogenesis imperfecta, haemolytic uraemic syndrome) Haemoglobinopathies (thalassaemia, sickle cell disease) Antibiotics (sulphonamides, penicillin, chloramphenicol) Chemotherapy (anthracyclines) Iron (haemochromotosis) Atrial flutter SVT Ventricular tachycardia
Adapted from reference 1.
is tachypnoea, though crepitations are uncommon even in the presence of pulmonary oedema. The heart sounds are quiet with additional heart sounds producing a gallop rhythm often detectable by care- ful auscultation. Cardiac murmurs are unusual at presentation but a soft pansystolic murmur due to mitral regurgitation may appear after a few days of medical treatment. The liver is often tender and palpable well below the costal margin due to hepatic congestion. Occasionally, this may occur rapidly causing abdominal pain or be so severe as to produce frank hepatic failure as the predominant presenting condition.
Characteristically, the electrocardiogram demon- strates a sinus tachycardia with a normal QRS axis. There is frequently left ventricular hypertrophy with ST segment and T wave abnormalities.
The chest X-ray shows marked cardiomegaly (Fig. 1) with pulmonary venous congestion. Left atrial enlargement may produce widening of the carina and left-sided basal collapse due to compression of the left main bronchus.
An echocardiogram confirms a structurally normal heart with marked dilatation of both the left atrium and ventricle. Doppler studies may show evidence of mitral regurgitation due to enlargement of the mitral valve
Table 2 Characteristics of the different types of cardiomyopathy
Characteristic Dilated Hypertrophic Restrictive
Heart size Increased Increased Cavity dimensions Increased (LV/RV) Normal/decreased Muscle mass Increased Increased (++ in LV) Contractility Decreased Increased/normal Outflow obstruction Not present In LV (and RV in neonates) AV valve regurgitation Yes, MV>TV Yes, MV Thrombi In LV (+ occ. in RV) Not reported
Increased Decreased Normal/increased Normal Not present Yes both MV and TV In LV and RV
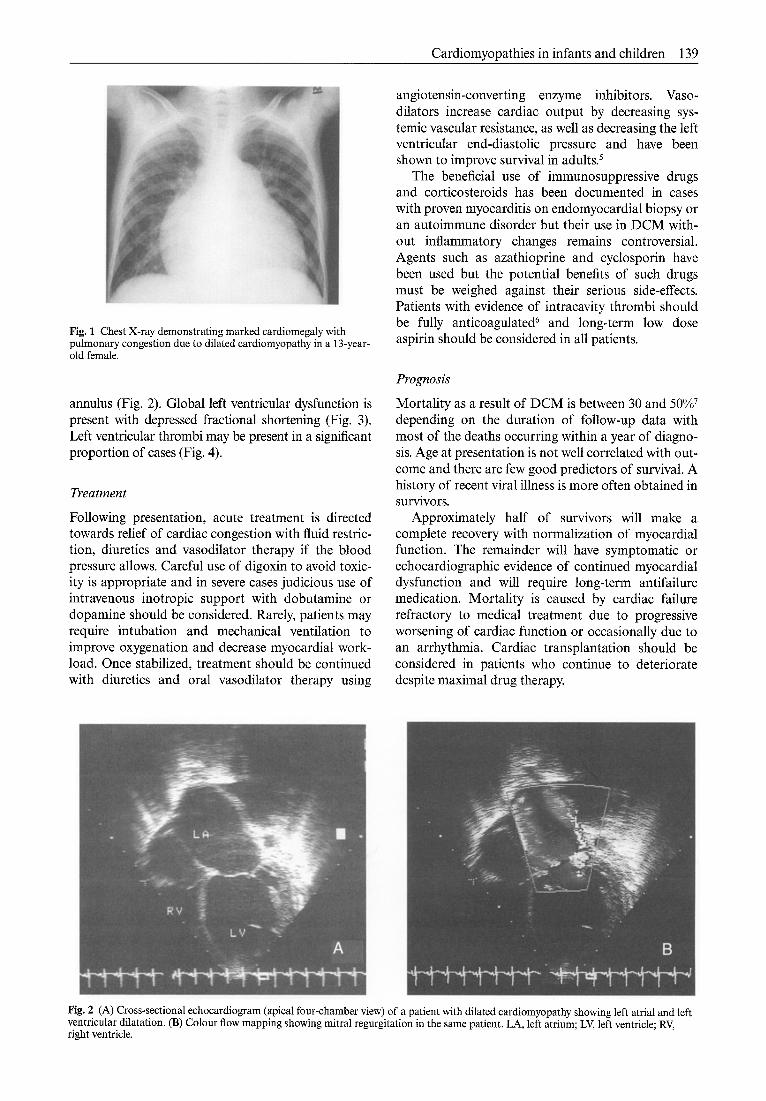
Cardiomyopathies in infants and children 139
Fig. 1 Chest X-ray demonstrating marked cardiomegaly with pulmonary congestion due to dilated cardiomyopathy in a 13-year- old female.
annulus (Fig. 2). Global left ventricular dysfunction is present with depressed fractional shortening (Fig. 3). Left ventricular thrombi may be present in a significant proportion of cases (Fig. 4).
Treatment
Following presentation, acute treatment is directed towards relief of cardiac congestion with fluid restric- tion, diuretics and vasodilator therapy if the blood pressure allows. Careful use of digoxin to avoid toxic- ity is appropriate and in severe cases judicious use of intravenous inotropic support with dobutamine or dopamine should be considered. Rarely, patients may require intubation and mechanical ventilation to improve oxygenation and decrease myocardial work- load. Once stabilized, treatment should be continued with diuretics and oral vasodilator therapy using
angiotensin-converting enzyme inhibitors. Vaso- dilators increase cardiac output by decreasing sys- temic vascular resistance, as well as decreasing the left ventricular end-diastolic pressure and have been shown to improve survival in adults?
The beneficial use of immunosuppressive drugs and corticosteroids has been documented in cases with proven myocarditis on endomyocardial biopsy or an autoimmune disorder but their use in DCM with- out inflammatory changes remains controversial. Agents such as azathioprine and cyclosporin have been used but the potential benefits of such drugs must be weighed against their serious side-effects. Patients with evidence of intracavity thrombi should be fully anticoagulated 6 and long-term low dose aspirin should be considered in all patients.
Prognosis
Mortality as a result of DCM is between 30 and 50% 7 depending on the duration of follow-up data with most of the deaths occurring within a year of diagno- sis. Age at presentation is not well correlated with out- come and there are few good predictors of survival. A history of recent viral illness is more often obtained in survivors.
Approximately half of survivors will make a complete recovery with normalization of myocardial function. The remainder will have symptomatic or echocardiographic evidence of continued myocardial dysfunction and will require long-term antifailure medication. Mortality is caused by cardiac failure refractory to medical treatment due to progressive worsening of cardiac function or occasionally due to an arrhythmia. Cardiac transplantation should be considered in patients who continue to deteriorate despite maximal drug therapy.
Fig. 2 (A) Cross-sectional echocardiogram (apical four-chamber view) of a patient with dilated cardiomyopathy showing left atrial and left ventricular dilatation. (B) Colour flow mapping showing mitral regurgitation in the same patient. LA, left atrium; LV, left ventricle; RV, right ventricle.
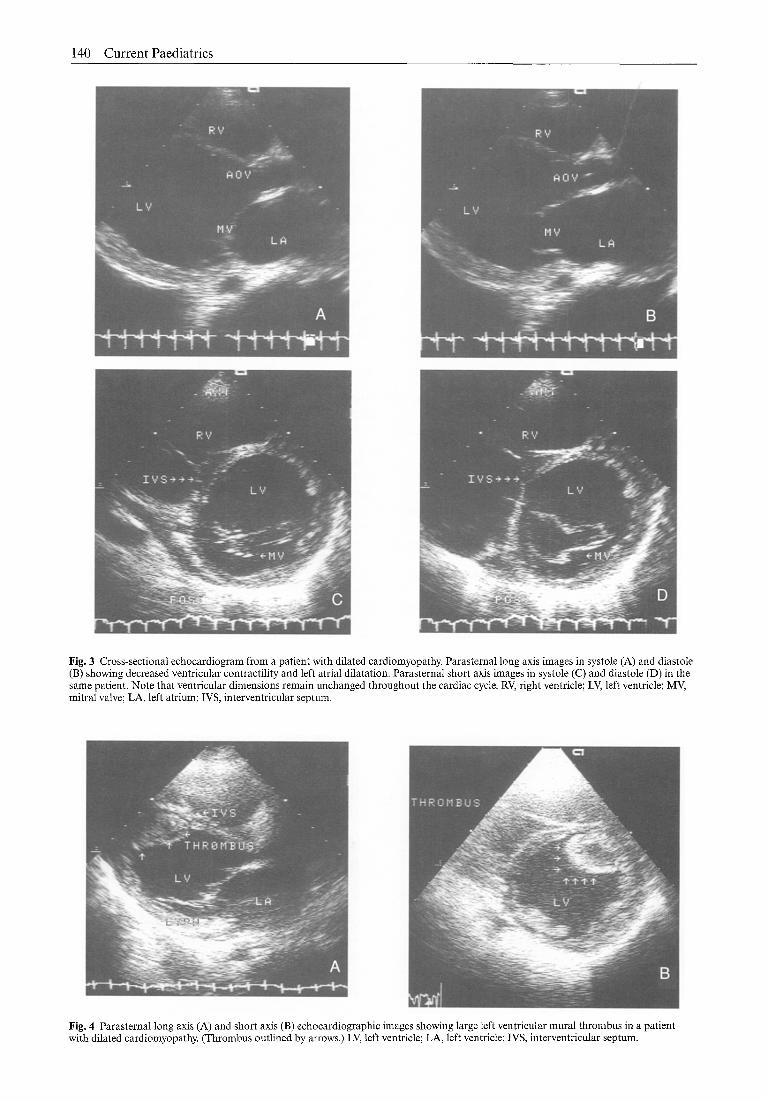
140 C u r r e n t Paedia t r ics
Fig. 3 Cross-sectional echocardiogram from a patient with dilated cardiomyopathy. Parasternal long axis images in systole (A) and diastole (B) showing decreased ventricular contractility and left atrial dilatation. Parasternal short axis images in systole (C) and diastole (D) in the same patient. Note that ventricular dimensions remain unchanged throughout the cardiac cycle. RV, right ventricle; LV, left ventricle; MV, mitral valve; LA, left atrium; IVS, interventricular septum.
Fig, 4 Parasternal long axis (A) and short axis (B) echocardiographic images showing large left ventricular mural thrombus in a patient with dilated cardiomyopathy. (Thrombus outlined by arrows.) LV, left ventricle; LA, left ventricle; IVS, interventricular septum.

Cardiomyopathies in infants and children 141
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is character- ized by asymmetrical cardiac hypertrophy involving the left ventricle and is associated with a variable degree of left ventricular outflow tract obstruction. There is frequently disproportionate hypertrophy of the interventricular septum.
Aetiology
A variety of conditions fall into the above classification (Table 3) but this discussion will concentrate on the genetic form of this disease. This is now known to be a familial disease which is caused by any one of at least four genes located on four different chromosomes? Inheritance is autosomal dominant with variable pene- trance. Genetic abnomalities coding for the myofibril- lary proteins results in abnormal spatial arrangement of the myofibrils. The myofibrillary disorganization interferes with the normal cellular relationships giving the myocytes a bizarre misshapen appearance. Hyper- trophy of the myocardium is thought to occur in response to altered wall stress caused by the myofibril disarray. At a cellular level, this may cause localized hypertrophy and enhance wall stress inequalities thus setting up a vicious circle. The genetic heterogeneity of this condition accounts only in part for the wide range of clinical variability encountered. It is known that an identical genetic defect may produce a range of effects in different family members 9 but the environmental and haemodynamic parameters which might cause such phenotypic variability are not clearly defined.
Presentation
Symptoms are usually dependent on age at presenta- tion. Neonates and infants develop signs of congestive cardiac failure and present with tachypnoea and poor feeding. The majority will have left ventricular out- flow tract obstruction and their prognosis is poor with death often occurring during the first year of life. Children over a year and adolescents are more com- monly asymptomatic. Presenting complaints include chest pain, syncope and palpitations and in a small proportion sudden death will occur following exercise often without preceding symptoms.
Typical physical signs include normal heart sounds unless there is severe left ventricular outflow tract obstruction which results in paradoxical splitting of the second heart sound. Third and fourth heart sounds are frequently heard and a systolic ejection murmur is audible in about 50% of patients. The mur- mur will be accentuated after exercise or following administration of agents that increase contractility. It is also accentuated by assuming a squatting position. This forms the basis of a useful clinical feature in diagnosis.
The chest X-ray almost invariably demonstrates cardiomegaly, but pulmonary congestion is absent. The electrocardiogram is universally abnormal in
Table 3 Conditions which predispose to disproportionate left ventricular hypertrophy
• Familial hypertrophic cardiomyopathy • Noonan's syndrome e Friedrichs ataxia • Multiple lentiginosis • Glycogen storage diseases • Infants of diabetic mothers • Phaeochromocytoma
infants, and almost always in children, but is not pre- dictive of clinical status. Infants show biventricular hypertrophy in the majority of cases whereas children are more likely to demonstrate left ventricular hyper- trophy with abnormal Q waves. Both groups have non-specific ST segment and T wave changes. Holter monitoring and exercise testing may reveal arrhyth- mias including supraventricular tachycardia, atrial flutter and heart block.
Cross-sectional echocardiography is used to demonstrate the hypertrophy (Fig. 5). Systolic func- tion is normal or decreased despite an increased fractional shortening but abnormalities of diastolic function are frequent and probably contribute to clin- ical presentation. Doppler studies will demonstrate an abnormal ejection pattern with the majority of the stroke volume occurring in early systole. It can also be used to define the extent of left ventricular outflow tract obstruction and in neonates commonly confirms the presence of right ventricular outflow tract obstruction.
M-mode measurements are used to define the extent of the hypertrophy and to demonstrate the characteristic systolic anterior motion of the mitral valve (Fig. 6) which contributes to the outflow obstruction and the typical midsystolic closure of the aortic valve.
Treatment
The use of inotropic agents may cause significant clinical deterioration rather than improvement. Symptomatic children should be started on beta blocking drugs provided there is no contraindication to their use. In patients who cannot be given beta blockers, calcium channel blocking agents have been used; particularly nifedipine and verapamil. Both classes of drugs will cause a decrease in contractility and improved diastolic function and have been associ- ated with an improved exercise tolerance. There is no evidence to suggest that either form of medication will have an effect on the myocardial hypertrophy. Patients with symptomatic arrhythmias require treatment either with propranolol or with long-term amio- darone. Children with significant bradycardias may need pacemaker implantation.
Surgical treatment has been used in cases in which medical treatment has failed to produce relief of symptoms. Left ventricular myomectomy has been
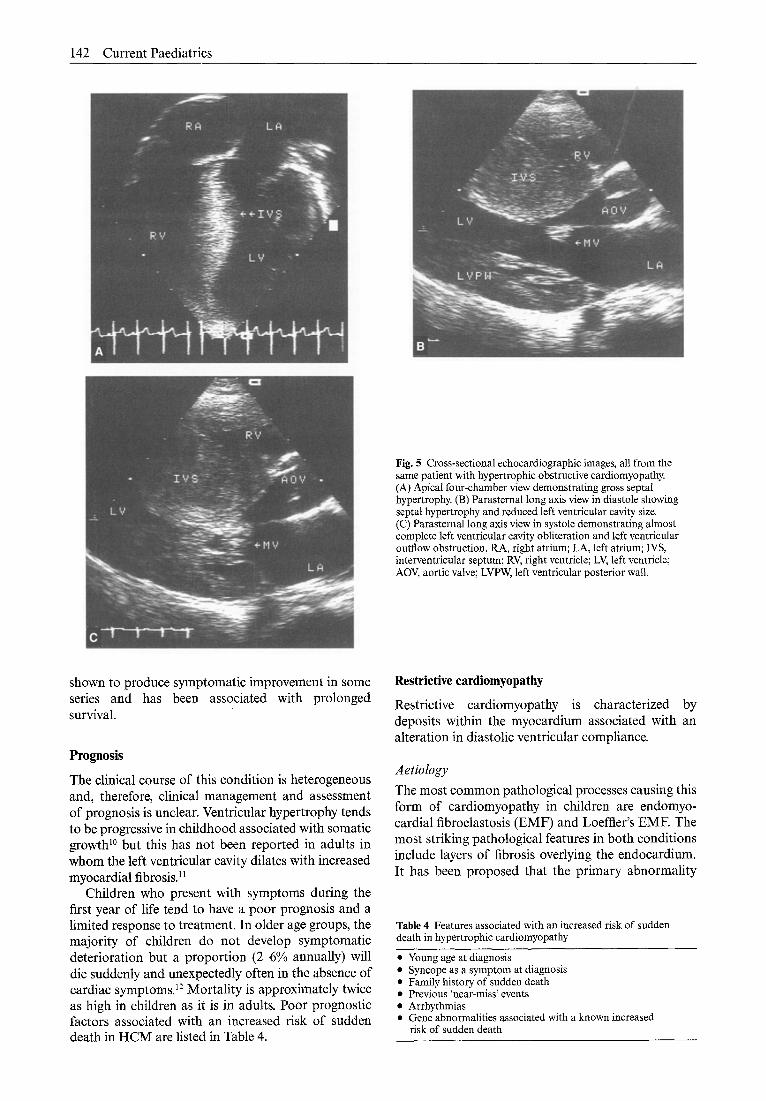
142 Current Paediatrics
Fig. 5 Cross-sectional echocardiographic images, all from the same patient with hypertrophic obstructive cardiomyopathy. (A) Apical four-chamber view demonstrating gross septal hypertrophy. (B) Parasternal long axis view in diastole showing septal hypertrophy and reduced left ventricular cavity size. (C) Parasternal long axis view in systole demonstrating almost complete left ventricular cavity obliteration and left ventricular outflow obstruction. RA, right atrium; LA, left atrium; IVS, interventricular septum; RV, right ventricle; LV, left ventricle; AOV, aortic valve; LVPW, left ventricular posterior wall.
shown to produce symptomatic improvement in some series and has been associated with prolonged survival.
Prognosis
The clinical course of this condition is heterogeneous and, therefore, clinical management and assessment of prognosis is unclear. Ventricular hypertrophy tends to be progressive in childhood associated with somatic growth 1° but this has not been reported in adults in whom the left ventricular cavity dilates with increased myocardial fibrosis.II
Children who present with symptoms during the first year of life tend to have a poor prognosis and a limited response to treatment. In older age groups, the majority of children do not develop symptomatic deterioration but a proportion (2-6% annually) will die suddenly and unexpectedly often in the absence of cardiac symptoms? 2 Mortality is approximately twice as high in children as it is in adults. Poor prognostic factors associated with an increased risk of sudden death in HCM are listed in Table 4.
Restrictive eardiomyopathy
Restrictive cardiomyopathy is characterized by deposits within the myocardium associated with an alteration in diastolic ventricular compliance.
Aetiology The most common pathological processes causing this form of cardiomyopathy in children are endomyo- cardial fibroelastosis (EMF) and Loeffler's EME The most striking pathological features in both conditions include layers of fibrosis overlying the endocardium. It has been proposed that the primary abnormality
Table 4 Features associated with an increased risk of sudden death in hypertrophic cardiomyopathy
• Young age at diagnosis • Syncope as a symptom at diagnosis • Family history of sudden death • Previous 'near-miss' events • Arrhythmias • Gene abnormalities associated with a known increased
risk of sudden death

Cardiomyopathies in infants and children 143
Fig. 6 M-rnode echocardiograrn from the same patient with hypertrophic cardiornyopathy which demonstrates the systolic anterior motion (SAM) indicated by arrows, of the anterior leaflet of the mitral valve associated with left ventricular outflow tract obstruction in this condition. IVS, interventricular septurn; ALMV, anterior leaflet of mitral valve; LVPW, left ventrieular posterior wall.
may be vasculitic narrowing of the subendocardial coronary blood supply causing fibrotic replacement of the ischaemic endocardium. 13 In Loeflter's EMF the cardiac involvement is associated with an eosinophilia which has been implicated in the pathogenesis of the condition through immunological mediators. 14 As the pathological features are similar, it is possible that these are different stages of the same condition. The cause of the eosinophilia and the possible abnormali- ties in coronary blood supply remain unclear.
Presentation
The eosinophilic form of the disease tends to affect younger children and is more common in tropical areas. EMF without eosinophilia is usually a disease of adolescents and young adults but has been reported in fetal life.15 Symptoms and signs depend on whether there is right, left or biventricular involve- ment. Right-sided involvement produces oedema, ascites with raised venous pressures and hepato- megaly with tricuspid regurgitation. Left ventricular EMF most commonly produces features of significant mitral insufficiency. Biventricular disease produces a combination of these features. The fibrosis may be unevenly distributed in severity with apical cavity obliteration or obstruction to ventricular outflow occurring particularly in the right ventricle.
The chest X-ray demonstrates cardiomegaly largely due to atrial enlargement and occasionally due to the presence of a pericardial effusion. There may be evidence of pulmonary congestion.
The electrocardiogram suggests right and or left atrial enlargement which may occur without associated
ventricular hypertrophy. Non-specific ST segment and T wave changes are present. Conduction disturbances are common and include atrial fibrillation and AV block.
Echocardiographic features include normal or reduced right and left ventricular cavity dimensions depending on site of involvement with atrial dilatation. Shortening fraction is usually within normal limits. The endocardium and papillary muscles may appear echogenic and doppler demonstrates atrioventricular valve regurgitation. A significant proportion of cases will have intracavity thrombi and irregular cavity obliteration may be evident.
Prognosis
Lack of available data means that the natural history of this condition is not known. Therapy commonly used for heart failure is unlikely to be successful since contractility is usually normal and diuretics will pro- duce hypotension associated with decreased preload. Surgical removal of fibrosis has been used and the use of beta blockers and calcium channel blockers has been proposed.
REFERENCES
1. Goodwin J F, Oakley C M. The cardiomyopathies. Br Heart J 1972; 34: 545-551.
2. Pacquet M, Hanna B D. Cardiomyopathy. In: Garson A Jr, Bricker J T, McNamara D G, eds. The Science and Practice of Pediatric Cardiology, vol. III. Philadelphia: Lea and Feibiger, 1990: 1617-1645.
3. Mestroni L, Krajinovic M, Severini G M, Giacca M, Falaschi A. Familial dilated cardiomyopthy. Br Heart J 1994; 72: $35-41.
4. Keeling P J, Tracy S. Link between enteroviruses and dilated cardiornyopathy: serological and molecular data. Br Heart J 1994; 72: $25-29.
5. The SOLVD Investigators. The effect of Enalapril on survival in patients with reduced left ventricular ejection fraction and congestive cardiac failure. N Engl J Med 1991; 325: 293-302.
6. Falk R H, Foster E, Coats M H. Ventricular thrombi and thrornboembolism in dilated cardiomyopathy; a prospective follow up study. Am Heart J 1992; 123: 136-142.
7. Talierco C P, Seward J B, Driseoll D J, Fisher L D, Gersh B J, Tajik A J. Idiopathic dilated cardiomyopathy in the young: clinical profile and natural history. J Am Coll Cardiol 1985; 6: 1126-1131.
8. Watkins H. Multiple disease genes cause hypertrophic cardiomyopathy. Br Heart J 1994; 72: $4-9.
9. Ciro E, Nichols P F, Maron B J. Heterogeneous morphological expression of genetically transmitted hypertrophic eardiornyopathy: two-dimensional echocardiographic analysis. Circulation 1983; 67: 122%1233.
10. Spirito P, Bellone E Natural history of hypertrophic cardiomyopathy. Br Heart J 1994; 72: S10-12.
11. Spirito P, Maron B J, Bonow R O, Epstein R E. Occurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in patients with hypertrophic cardiomyopathy. Am J Cardiol 1987; 60: 123-129.
12. Maron B J, Cecchi E McKenna W J. Risk factors and stratification for sudden cardiac death in patients with hypertrophic cardiomyopathy. Br Heart J 1994; 72: S13-18.
13. Harris L C, Ngheirn Q X. Cardiornyopathies in infants and children. Prog Cardiovasc Dis 1972; 15: 255-259.

144 Current Paediatrics
14. Bertrand E, Metras D. Paediatric cardiology in the tropics. In: Anderson R A, Macartney F J, Shinebourne E A, Tyrian M J, eds. Paediatric Cardiology. London: Churchill Livingstone, 1987: 1321-1341.
15. Ben-Ami M, Shalev E, Romano S, Zukerman S. Midtrimester diagnosis of endocardial fibroelastosis and atrial septal defect: a case report. Am J Obstet Gynecol 1986; 155: 662-663.





























