Embed Size (px)
Citation preview

Epilepsy Research 68S (2006) S21–S37
Review
Basic research in epilepsy and aging
Ilo E. Leppika,∗, Kevin M. Kelly b, Leyla deToledo-Morrellc, Peter R. Patrylod,Robert J. DeLorenzoe,1, Gary W. Mathernf, H. Steve Whiteg
a College of Pharmacy, University of Minnesota, 308 Harvard St. SE, Minneapolis, MN 55455, USAb Department of Neurology, Drexel University College of Medicine, Allegheny-Singer Research Institute,Allegheny General Hospital – 940 South Tower, 320 East North Avenue, Pittsburgh, PA 15212-4772, USAc Department of Neurological Sciences, Rush University Medical Center, 1653 West Congress Parkway,
Chicago, IL 60612-3833, USAd Department of Physiology, Southern Illinois University School of Medicine, Carbondale, IL 62901, USAe Departments of Neurology, Pharmacology and Toxicology, and Molecular Biophysics and Biochemistry,
Virginia Commonwealth University, P.O. Box 980599, Richmond, VA 23298, USAf Division of Neurosurgery, The Mental Retardation Research Center, and The Brain Research Institute,
David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USAg Anticonvuslant Drug Development Program, Department of Pharmacology & Toxicology,
University of Utah, 20 S. 2030 E., Rm 408, Salt Lake City, UT 84112, USA
Received 5 May 2005; received in revised form 27 July 2005; accepted 27 July 2005
ures ormals andg-term
m stroke.mbers ofepilepsy is
Available online 27 December 2005
Abstract
A PubMed search of the years 1965 to 2003 found only 30 articles that were directly related to modeling seizepilepsy in aged animals. This lack of research is disturbing but explainable because of the high cost of aged anitheir increasing infirmity. Many changes occur in the older brain: cell loss in the hippocampal formation, changes in lonpotentiation maintenance, alteration in kindling, increased susceptibility to status epilepticus, and neuronal damage froThe effect of aging on voltage-gated sodium and calcium channels has not been studied sufficiently. With increasing nuelderly persons with epilepsy needing appropriate treatment, the need to better understand the basic mechanisms ofcrucial.© 2005 Elsevier B.V. All rights reserved.
Keywords: Epilepsy; Aging; Animal models; Kindling; Stroke
∗ Corresponding author. Fax: +1 612 624 6695.E-mail addresses: [email protected] (I.E. Leppik), [email protected] (R.J. DeLorenzo).
1 Tel.: +1 804 828 8969; fax: +1 804 828 6432.
0920-1211/$ – see front matter © 2005 Elsevier B.V. All rights reserved.doi:10.1016/j.eplepsyres.2005.07.014

S22 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
Contents
1. Introduction. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S222. The state of the basic science of aging and epilepsy. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S233. Neuroanatomic and neurophysiologic changes in the aging brain. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S24
3.1. Age-related anatomical changes in mesial temporal lobe structures. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S243.2. Age-related alterations in hippocampal synaptic plasticity: long-term potentiation. . . . . . . . . . . . . . . . . . . . . . . . S253.3. Age-related alterations in hippocampal synaptic plasticity: kindling. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S25
4. The susceptibility to epileptiform activity in the aged central nervous system: in vivo and in vitro studies. . . . . . . . . S265. Poststroke epilepsy: in vivo and in vitro modeling. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S286. The aging hippocampus: surgical studies. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S317. Mechanisms of action of antiepileptic drugs: matching the drug with the older patient. . . . . . . . . . . . . . . . . . . . . . . . . . . S32
7.1. Modulation of voltage-gated ion channels. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S337.2. Enhanced inhibition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S347.3. Reduced excitation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S347.4. Implications for treating elderly patients with epilepsy. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S34References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . S35
1. Introduction
The elderly comprise the fastest growing segmentof the population in the United States; estimates sug-gest that by the first part of the 21st century 20% ofthe population will be older than 65 years (Butler,1997). Until recently, seizure disorders and epilepsyin the elderly were one of several age-related medicalproblems that generated little interest. However, sev-eral epidemiologic studies conducted within the past10–15 years have revealed that the elderly experiencethe highest incidence and prevalence of seizure dis-orders in developed countries (Luhdorf et al., 1986;Hauser et al., 1996) and this population exhibits anincreased likelihood for developing status epilepticusand status-related morbidity and mortality (DeLorenzoet al., 1992; Treiman et al., 1997).
The question of whether repeated seizures progres-sively damage or otherwise injure the brain has clinicalrelevance for elderly patients with epilepsy. Seizurecontrol is a critical aspect of patient care and if repeatedseizures cause progressive damage, those with uncon-trolled seizures would be expected to experience moreproblems than a population of similarly aged individu-als. This has proven to be a difficult issue to resolve, butrl -i ea r-
vals (Fig. 1). Patients with histories of temporal lobeepilepsy seizure exceeding 20 years showed a progres-sive loss of hippocampal neuronal densities, hippocam-pal volumes and cognitive measures that exceeded theamount expected for normal aging. It is not known,however, whether the progressive damage was sec-ondary to temporal lobe seizures, secondary general-ized events or antiepileptic drugs (AEDs). These long-term effects are important to consider in the treatmentof elderly patients with a long history of epilepsy.
Perhaps because until recently epilepsy has beenconsidered a predominantly pediatric disorder, withdecreasing incidence in adulthood, issues relating to
F tioni ssivec
ecent pathologic (Mathern et al., 2002), neuropsycho-ogic (Jokeit and Ebner, 1999), and neuroimaging studes (Tasch et al., 1999) have found that seizures can bssociated with injury to the brain over long time inte
ig. 1. Hippocampal cell density as a function of seizure duran years. There is a negative linear correlation indicting progreell loss with very long seizure histories (r =−0.193;p = 0.0025).

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S23
epilepsy in the elderly have remained relatively unex-plored. Very little basic research has attempted todevelop models of epilepsy in elderly patients; how-ever, it is hoped that the increasing recognition ofepilepsy in elderly patients will attract researchers andfunding that have previously focused on the study ofyoung animal models of epilepsy for eventual transla-tion to pediatric populations.
2. The state of the basic science of aging andepilepsy
In an effort to determine the level of basic scienceactivity that integrates the fields of aging and epilepsy,a PubMed search of the years 1965–2003, combiningthe search terms “aged” or “aging” and “epilepsy” or“seizures”, was conducted. The search identified only30 publications that were directly related to modelingseizures and/or epilepsy in aged animals (Fig. 2); allthe literature was published within the past 18 years,including four articles in both 2002 and 2003. Thesefindings corroborate the general impression that there isrelatively modest interest and limited progress in basicscience research concerning epilepsy and aging.
The question of what exactly to model whenr aryi arer ions:
F s ore lts ofa with“
(1) epilepsy and aging—the potential for changedexpression of both inherited and acquired formsof epilepsy with advancing age, or (2) aging andepilepsy—new onset epilepsy at an advanced age.The known causes of newly diagnosed epilepsy inthe elderly include stroke, degenerative disease, tumor,trauma and infection, and are therefore critical factorsto incorporate into models of aging and epilepsy, butestablished research protocols for these clinical entitiesdo not exist. In addition, epidemiologic studies revealthat up to 50% of elderly patients with seizure disordershave no identifiable antecedents; these cases are classi-fied as idiopathic or cryptogenic (Hauser, 1997). Thisobservation has lead to the hypothesis that aging of thecentral nervous system itself may affect seizure suscep-tibility ( Ng et al., 1985; Hauser et al., 1993; Hauser,1997). Additional research is also required to iden-tify risk factors associated with new onset epilepsy inthe elderly of unknown cause, and determine whetherthe aged central nervous system may be more proneto seizure generation following insults remote in time(e.g., stroke, trauma or metabolic insult).
To comprehensively model the causes of, and con-tributors to, epilepsy in the elderly, recognizing andimplementing the rich knowledge base that exists inthe neurobiology of aging literature is an importants rcha cien-t ssuesa rs tot ri-a ultso psym poth-e sult,m
ar mayi gw andh nes,m thea s ont arch.S h ask blet anet nts.
esearching epilepsy and the elderly is of primmportance. Two different conceptual approacheseasonable and raise important clinical considerat
ig. 2. Thirty articles related to animal modeling of seizurepilepsy in the elderly were published from 1965 to 2003. ResuPubMed search performed using the terms “aged” or “aging”
seizure” or “epilepsy”.
tep. A lack of familiarity with general aging reseappears to be an impediment for basic research s
ists; increased knowledge and awareness of the issociated with aging may encourage researche
ransition the focus of their work to modeling getric epilepsy. By incorporating concepts and resf neurobiological aging research, geriatric epileodeling can become more evidence based, hysis driven, mechanistically defined and, as a reore competitive for funding.Depending on the clinical issue involved in
esearch endeavor, modeling of geriatric epilepsynvolve multiple levels of investigation, includinhole animal, network (e.g., neocortex, thalamusippocampus), cellular and molecular (e.g., geRNA or protein) studies. To a significant degree,natomical level at which modeling occurs depend
he conceptual approach used to frame the resetandard models of temporal lobe epilepsy, sucindling or kainate/pilocarpine, may not be applicao many of the clinical issues that are most germo new onset symptomatic epilepsy in elderly patie

S24 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
For this reason, new and innovative models that per-tain to geriatric epilepsy should include the use ofaged brain and relevant lesions (i.e., stroke, traumaticbrain injury and neurodegeneration) that can result inepileptogenesis and the establishment of an epilepticstate in elderly patients. However, models used to studytemporal lobe epilepsy (e.g., kindling, kainate or pilo-carpine) may provide critical insight into (1) how agingof the central nervous system and peripheral systems(e.g., blood brain barrier) can affect the brain’s sus-ceptibility to a specific secondary insult and (2) howthe clinical and electroencephalographic characteris-tics of seizure disorders may change in the elderly(Tinuper, 1997). Improved methods of identifying,procuring and investigating available surgical and post-mortem brain tissue from elderly patients with epilepsyare necessary for the development of novel geriatricmodels.
When conducting animal research to study thecauses of geriatric epilepsy, choosing the method usedto produce a lesion in an aged brain is important in termsof what brain structures are compromised and to whatdegree because, as in animals of any age, lesioning mayresult in variable neuronal loss or injury. Recognizedneuronal loss in an aged animal can be due to differingdegrees of atrophy, necrosis and apoptosis; neuronali brainp es-o rts.T ateb mayr epto-g romt
its mayfi suchs . Thec aret se off onA rceD ese.A ithr nd-i withn newi
The unmistakable need for more basic scientificresearch into the mechanisms of epilepsy in the elderlywill require knowledge from the general literature onthe neurobiology of aging, the development of new andimproved models for study, increased investigation ofhuman epileptic tissue and the use of new and avail-able funding sources for pilot studies and new researchinitiatives. Invigorated and increased efforts in animalmodeling of geriatric epilepsy will herald advances inthe treatment of the disease in elderly patients.
3. Neuroanatomic and neurophysiologicchanges in the aging brain
As aging occurs, numerous changes take placein the brain, including anatomic and electrophysio-logic alterations in mesial temporal lobe structures.These brain regions are not only vulnerable to theaging process, especially age-related degenerative dis-eases, but are also involved in temporal lobe epilepsy.Because new onset temporal lobe epilepsy occurs inthe elderly, age-related studies of the mesial temporallobe have application to the study of epilepsy in theelderly.
3t
palf sial.W loss,m gicalc find-i ts ularl beend bero pop-u ucha apses(
eticr toolf y inv anda l cor-t sial
njury may be sustained as a result of decreasedlasticity or may require a protracted period for rlution when compared with younger animal cohohoughtful experimental design can help discriminetween these possible contributing factors andeveal essential pathways and components of epilenesis in the aged animal brain that may differ f
hose of younger animals.Research involving aging animals carries with
ome practical realities that many investigatorsnd onerous, thus perpetuating apprehension oftudies and the avoidance of topics related to agingost of aged animals and their increasing infirmityypical concerns associated with aging research; uunds made available through the National Instituteging, Office of Biological Resources and Resouevelopment, can mitigate concerns such as thvoiding potentially prohibitive costs associated wesearch on aged animals by utilizing existing fung sources, as well as promoting bench researchew initiatives may help to attract both veteran and
nvestigators to the field.
.1. Age-related anatomical changes in mesialemporal lobe structures
The issue of age-related cell loss in the hippocamormation in animal models is somewhat controver
hile early studies demonstrated age-related cellost recent experiments using unbiased stereolo
ell counting techniques have not replicated thesengs (Morrison and Hof, 1997). However, significanynaptic loss in both the middle and inner molecayers of the dentate gyrus of the aged rat hasemonstrated, with reductions in the mean numf synapses per neuron for the entire synapticlation and within specific synaptic categories, ss perforated and nonperforated axospinous synGeinisman et al., 1995).
Quantitative, high-resolution structural magnesonance imaging (MRI) techniques provide aor examining alterations in human brain anatomivo, allowing comparison between normal agingge-related degenerative diseases. The entorhina
ex and hippocampal formation are part of the me

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S25
Fig. 3. In vivo age- and disease-related changes in hippocampal vol-ume.
temporal lobe memory system. The entorhinal cortexconnects the neocortex with the hippocampal forma-tion via the perforant path, providing the hippocampalformation with multimodal sensory information. Theseregions of the brain have received particular investiga-tive attention in reference to the pathophysiology ofAlzheimer’s disease since memory dysfunction is oneof the earliest hallmarks of the disease.
Age-related and disease-related atrophy of the hip-pocampal formation has been extensively demon-strated by neuroimaging techniques (Fig. 3). Elderlyindividuals who complain of memory problems or whodemonstrate an isolated memory deficit (mild cog-nitive impairment) are at greater risk of developingAlzheimer’s disease. These elderly patients exhibit sig-nificant atrophy of both the entorhinal cortex and thehippocampal formation when compared to elderly con-trols (Dickerson et al., 2001; Du et al., 2001). Thevolume of the entorhinal cortex is the best predictor ofwhom among patients with mild cognitive impairmentwill progress to a diagnosis of Alzheimer’s diseasewithin three years of baseline scanning (deToledo-Morrell et al., 2004). This finding is important in lightof postmortem studies that have implicated the entorhi-nal cortex and the transentorhinal cortex as early sitesof pathology in Alzheimer’s disease and mild cognitivei al.,1 lsos phya indi-v
3.2. Age-related alterations in hippocampalsynaptic plasticity: long-term potentiation
Long-term potentiation (LTP) is defined as the long-lasting augmentation in synaptic responses observedfollowing brief trains of high frequency stimulation of amonosynaptic pathway. In the chronic preparation, LTPcan last from days to weeks. When robust stimulationparameters are used in either in vivo preparations or inhippocampal slices, there appears to be no age-relateddeficit in the induction of LTP. The lack of difference inthe extent of potentiation between young and old ani-mals has been demonstrated in the Shaffer collateral-CA1 synapse, as well as the perforant path-granule cellsynapse (Barnes, 2001). However, in marked contrastto the lack of age-related induction deficits with robuststimulation protocols, there is an age-related LTP main-tenance deficit (Barnes, 1979; deToledo-Morrell andMorrell, 1991; Geinisman et al., 1995) that is bestobserved in chronically prepared young and old ratsstudied over extended periods of time; in old, memory-impaired rats, LTP decays much faster than in youngrats. Furthermore, faster decay of LTP at the perforantpath-granule cell synapse in old rats parallels the ratesof decay of spatial memory (Barnes and McNaughton,1985).
thei ersi ratedi rma-c -t CC)d ofL iss als( if-ia in ac func-t
3s
ale lec-t ep-t ental
mpairment (Braak and Braak, 1991; Gomez-Isla et996; Kordower et al., 2001). Recent studies have ahown significant entorhinal and hippocampal atrossociated with cerebrovascular disease in elderlyiduals (Du et al., 2002).
The synaptic strength measured followingnduction of LTP with robust stimulation parametn the CA1 sector of the hippocampus can be sepanto two components that can be dissected phaologically: theN-methyl-d-aspartate (NMDA) recepor and voltage-dependent calcium channel (VDependent forms. Aging alters these two formsTP differently; NMDA receptor-dependent LTPignificantly reduced in magnitude in aged animp < 0.01) while the VDCC-dependent form is signcantly increased (p < 0.01;Shankar et al., 1998). Theggregate of these two forms of LTP may resultompound potentiation that shows no change as aion of aging.
.3. Age-related alterations in hippocampalynaptic plasticity: kindling
Kindling, a long-lasting alteration in neuronxcitability brought about by repeated, low level erical stimulation, is considered a model of epilogenesis. It can also be considered an experim

S26 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
model of the formation of an engram in the brain,albeit an engram of abnormal cellular behavior. Inde-pendent research has shown that the process of kin-dling is retarded in aged animals (deToledo-Morrellet al., 1984; Fanelli and McNamara, 1986). Researchusing perforant-path stimulation to induce hippocam-pal kindling demonstrated no change in after-dischargethreshold between young and old animals (deToledo-Morrell et al., 1984; deToledo-Morrell and Morrell,1991); however, hippocampal kindling (to a criterionof five generalized seizures) took significantly longerin old rats (95.9 trials and 59.6 trials in old andyoung rats, respectively;t26 = 3.78; p < 0.001). Addi-tionally, there was a striking relationship between rateof kindling in old animals and hippocampal-dependentspatial memory function (ρ = 0.73, p < 0.001); oldrodents with poor memory kindled slowly, whereasold rodents with good memory kindled as quickly asthe young (deToledo-Morrell et al., 1984; deToledo-Morrell and Morrell, 1991). Poor spatial mem-ory may be indicative of hippocampal neuronaldysfunction.
The development of secondary or progressiveepileptogenesis is one of the best examples of kindlingin humans. The concept of secondary epileptogene-sis suggests that an actively discharging epileptogenicr ionsw ys-m isen
en-e itsd itedtd li-h inh nd-i all.P r 25y lopswa esisi ing.T y bei typeo eredp
Table 1Factors affecting the development of an independent secondary focusin frontal lobe tumor cases
(A) Effect of duration of illness
Presence of secondary focus Duration
0–5 years 6 years and over
Positive 2 (6%) 10 (40%)Negative 30 15χ2 = 7.70,p = 0.0055
(B) Effect of age of seizure onset with a duration of illness of 5years and over
Presence of secondary focus Age
0.5–25 years 26–60 years and over
Positive 10 (56%) 0 (0%)Negative 8 12χ2 = 7.66,p = 0.0057
4. The susceptibility to epileptiform activity inthe aged central nervous system: in vivo and invitro studies
Due to the high incidence and prevalence of idio-pathic seizure disorders in the elderly, as well as theincreased likelihood of developing status epilepticus,related morbidity and mortality, recent animal studiesare beginning to focus on adult and aging rats in aneffort to apply findings to the elderly human population.Whether rodents exhibit a similar increase in seizuresusceptibility and severity during aging remains debat-able due to the contradictory nature of available data(deToledo-Morrell et al., 1984; Wozniak et al., 1991;Chiba et al., 1992; Nokubo and Kitani, 1988). However,several studies have noted an enhanced vulnerabilityto the known convulsant agents domoic acid and/orits structural analogue, kainate, in elderly humans andaged rodents (Perl et al., 1990; Teitelbaum et al., 1990;Kerr et al., 2002; Wozniak et al., 1991). Nevertheless,it remains unclear how the progression of behavioralmanifestations and electroencephalogram (EEG) activ-ity associated with status epilepticus are altered duringaging.
Kainate-induced status epilepticus in adult (7–8months) and aged (22–25 months) Fischer 344 ratsw singk eri-t nd
egion (primary focus) that has massive connectith another brain region may induce similar paroxal behavior in the cellular elements of that otherwormal network.
The incidence of human secondary epileptogsis and the important factors that contribute toevelopment were examined in three series lim
o cerebral tumors and malformations (Morrell andeToledo-Morrell, 1999). In these cases, the likeood that a similar lesion may have developedomotopic or juxtahomotopic brain regions, depe
ng on anatomical connections, was exceedingly smatients with tumors who developed seizures afteears of age were significantly less likely to deveecondary epileptogenesis (χ2 = 7.66,p = 0.0057). Thisas especially true in frontal lobe tumor cases (Table 1)nd indicated that the progression of epileptogen
n humans may be reduced as a function of agherefore, although the incidence of seizures ma
ncreased in the elderly due to various insults, thef epilepsy may be less progressive due to altlasticity.
as examined by a low-dose treatment protocol uainic acid (2.5 mg/kg per h) administered by intraponeal injection, in conjunction with behavioral a

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S27
Table 2Behavioral characteristics in adult and aged Fischer 344 rats during systemic kainic acid treatment
Seizure type Median valuesa
Adult Fischer 344 rats Aged Fischer 344 rats
Number of wet-dog shakes during the hour following the 1st injection (events/h) 25 110Latency to onset of class I/II seizures (min) 30 20Latency to onset of First Class V seizure (min) 145 60Latency to onset of sustained seizures class I/II (min) 100 35Latency to onset of sustained seizures class III (min) 170 115Latency to onset of sustained seizures class IV/V (min) 205 172.5
a Within a row, all values were different;p < 0.05.
EEG monitoring, until convulsive sustained seizureswere elicited (Darbin et al., 2004). Both adult and agedrats exhibited a progression of behavioral manifesta-tions: wet-dog shakes, class I/II seizures, sustainedseizures class I/II (nonconvulsive status epilepticusclass I/II), a decrease in motor and locomotor activ-ity, convulsive seizures (classes III, IV and V) and,finally, sustained seizures class IV/V (generalized con-vulsive status epilepticus). While similar results wereobserved in both groups of rats, the latency to onsetfor each behavioral manifestation was significantlyshorter among aged animals (p < 0.05;Table 2) and thefrequency of preseizure manifestations (i.e., wet-dogshakes) was greater for aged animals. While both adultand aged rats exhibited an increase in EEG power thatcorresponded with the time course of treatment, visualinspection and spectral analysis revealed a reduction ofthe faster frequencies (12.5–35 Hz) in the EEG of agedrats.
The differences observed between aged and adultrats during kainate-induced status epilepticus mayreflect, in part, age-related changes in limbic andextra-limbic network activity. Even though potentialcontributions from age-related changes in pharma-cokinetics and blood brain barrier permeability can-not be discounted, available data indicate that the netc te-t thina rac-t t,2 itsr ulda ses( sta-t ate-t
To further assess whether local network activityis altered during aging, a series of preliminary elec-trophysiological experiments were performed in hip-pocampal slices from aged and adult rats using whole-cell patch recordings (Patrylo et al., 2000). The dentategyrus, a region within the hippocampal formation, waschosen for examination for several reasons: (1) in vivodata revealed that the frequency of preseizure mani-festations (e.g., wet-dog shakes) was greater and thelatency to onset of behavioral symptoms associatedwith limbic seizures was shorter in aged rodents relativeto controls—both of which are characteristic of hip-pocampal involvement; (2) it has been proposed that thedentate gyrus plays a critical role in regulating the onsetand spread of seizure activity within the hippocampalformation (Lothman et al., 1992; Grecksch et al., 1995);and (3) available anatomical data suggest that the den-tate gyrus in aged subjects exhibits several anatomicalchanges, including decreases in glutamic acid decar-boxylase immunoreactive neurons (rodent data;Shettyand Turner, 1998) and increased probability for Timm’sstained fibers in the inner molecular layer (human data;Cassell and Brown, 1984), which could affect circuitactivity and subsequently the role of the dentate gyrusin regulating seizure onset and spread.
To date, data collected are consistent with theh thed ncyo wasd roma inalflAo on-t tsy-n nif-
onsequence (e.g., epileptiform activity) of kainareatment appears to depend on the activity wi
given neuronal network and the dynamic inteion between such networks (Ben-Ari and Cossar000). Subsequently, if network properties and/oregulation were modified during aging, one wonticipate that different electrophysiological responi.e., altered EEG) and altered behavioral manifeions would be elicited in aged rodents during kainreatment.
ypothesis that local circuit activity is altered inentate gyrus during aging. Specifically, the frequef spontaneous inhibitory postsynaptic potentialsecreased by approximately 50% in granule cells fged versus adult rats in normal artificial cerebrospuid using KCl-patch electrodes (Patrylo et al., 2000).dditionally, in bicuculline (30�M), the proportionf granule cells from aged rats that displayed sp
aneous prolonged large amplitude excitatory posaptic potentials (>10 mV, 100 ms duration) was sig

S28 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
icantly greater than in adult rats (Patrylo et al., 2000).In combination, these data suggest that both local exci-tatory and inhibitory circuit activity is modified duringaging.
These preliminary findings suggest that kainic acidtreatment may be a valuable model system to exam-ine the mechanisms associated with increasing thesusceptibility to excitotoxicity status epilepticus, andpotentially idiopathic seizure disorders, during aging.Although additional studies are warranted, preliminarydata suggest that changes in local circuitry activity maycontribute to the aging-related change in susceptibilityto kainate-induced status epilepticus.
5. Poststroke epilepsy: in vivo and in vitromodeling
Among the elderly, stroke is the dominant causeof epilepsy (Hauser and Hesdorffer, 1990), yet themechanisms of poststroke epileptogenesis, the processby which injured brain begins generating spontaneousrecurrent seizures, are not known (Kelly, 2002). Rela-tively few studies have been designed to examine therelationship between ischemic infarction and the poten-tial development of epilepsy in adult or aged rats. Twom teryo eenu inew ingl
esa theo rctc urya bra.I ronaln le ofi diesui hics rgesw tionso ula-t ep-t thel oni-t gh
Fig. 4. Typical lesions produced by middle cerebral artery occlu-sion (MCAO) and cortical photothrombosis. Arrows indicate areasof lesions. Note markedly increased lesion size resulting from MCAOwhen compared with photothrombosis.
this study reflected several electroclinical aspects ofthe immediate and early phases of human stroke andsuggested the potential for subsequent epileptogenic-ity in the preinfarct area, because of limited moni-toring times, no conclusion could be drawn regardingwhether animals would have become epileptic. In con-trast, Karhunen et al. (2003)performed MCAO andmonitored animals intermittently from three months toone year post-lesioning and found no evidence of eitherbehavioral or EEG seizure activity. Additional researchwith MCAO will determine whether it will be a viablemodel of poststroke epilepsy.
Cortical photothrombosis and brain infarction usingrose bengal, a photosensitive dye, is an alternative tothe MCAO model. Rose bengal is injected into an ani-mal and activated in brain vasculature by an externallight beam that is focused on and penetrates into thetranslucent skull and underlying cerebral cortex. Theactivated dye generates singlet oxygen, which causesperoxidation of endothelial cell membranes and occlu-sive platelet aggregation. Subsequent thrombus forma-tion, vascular stasis and vasogenic edema lead to focalcortical infarction and necrosis. This method is wellcharacterized, relatively noninvasive, produces corti-cal infarcts that extend to the subcortical interface,and allows for their selective placement with repro-d AO,p vol-u
odels of cerebral infarction, middle cerebral arcclusion (MCAO) and photothrombosis, have bsed in combination with EEG recording to determhether seizures and/or epilepsy can occur follow
esioning.Middle cerebral artery occlusion typically produc
large cortical and subcortical infarct ipsilateral tocclusion (Fig. 4). This is characterized by an infaore and surrounding volume of partial cellular injnd death, often referred to as the ischemic penum
t has been hypothesized that injured, aberrant neuetworks within the penumbral area may be capab
nitiating poststroke epileptogenesis. In EEG stusing adult rats (Hartings et al., 2003), MCAO resulted
n multiple generalized or ipsilateral electrograpeizures within 2 h of lesioning. These ictal dischaere unaccompanied by any behavioral manifestaf seizure activity in the unanaesthetized and amb
ory animals. In addition, periodic lateralized epiliform discharges occurred primarily ipsilateral toesion and persisted until the end of the 72-h moring period in most of the animals tested. Althou
ucible area, depth and location. Compared to MChotothrombosis results in much smaller infarctmes (∼10–20 mm3 versus∼200–300 mm3). Initial

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S29
Fig. 5. EEG recording obtained two months after photothrombo-sis. A focal 5 Hz spike-wave discharge lasting 3 s occurs over leftcentroparietal cortex adjacent to the area of infarct. Vertical cursorindicates time of mid eye blink during seizure associated with briefmotor arrest. F = frontal; P = parietal; 3 = left; 4 = right.
studies in adult rats have demonstrated that large pho-tothrombic brain infarcts can result in brief recurrentelectrical seizures arising in the perilesional cortexassociated with behavioral arrest of the animal occur-ring 1–2 months after lesioning (Fig. 5; Kelly et al.,2001; Kharmalov et al., 2003). Despite certain theo-retical limitations of photothrombosis as an alternativemodel for research focusing primarily on penumbraltissue injury following stroke, it may be a viable modelfor studies of cortical infarction and mechanisms ofsubsequent epileptogenesis.
An in vitro system was developed and characterizedby DeLorenzo’s laboratory to model the developmentof epilepsy subsequent to stroke (DeLorenzo et al.,2004; Sun et al., 2001, 2002, 2004). This model focuseson glutamate excitotoxicity injury-induced epilepto-genesis in hippocampal neuronal cultures (GET HNCmodel). Injury produced by exposure of cultured hip-pocampal neurons to glutamate resulted in a mixedpopulation of injured neurons; some neurons developedexcitotoxic neuronal death, analogous to the ischemiccore of a stroke, and others survived injury, analo-gous to the ischemic penumbra. Neurons that wereinjured, but survived the initial glutamate challenge,were substrates for the development of epilepsy. Thesurviving neurons in the GET HNC model manifesteds syn-c onsi ede atei cur-
Fig. 6. Current-clamp recording from a glutamate-injured neuronshowing spontaneous, recurrent, epileptiform discharges that per-sisted for the duration of the recording period. All five tracingsare segments of a continuous, 2.5-h current-clamp experiment. Thisneuron (−66 mV resting potential) exhibited nine independent spon-taneous, recurrent, epileptiform discharges during the recordingperiod.
rent seizures. This model has the advantage of beingaccessible for electrophysiologic, biochemical and cal-cium imaging studies to evaluate the cause of therecurrent epileptiform discharges induced by the gluta-mate injury. Whole-cell current clamp electrophysiol-ogy techniques and Fura-2 calcium imaging were usedto characterize this model and evaluate the molecu-lar mechanisms mediating epileptogenesis. A currentclamp recording from an epileptic neuron demon-strated a pattern of spontaneous, recurrent, epileptiformdischarges that occurred over a 2.5-h period (Fig. 6).These discharges terminated like seizure activity, hadidentical electrophysiologic characteristics of neuronsundergoing seizure activity, were inhibited by anticon-vulsant agents and occurred synchronously in neuronalpopulations (Sun et al., 2001). Thus, the GET HNCmodel provides a powerful tool to evaluate the molec-ular mechanisms mediating stroke-induced epileptoge-nesis.
pontaneous, recurrent, epileptiform discharges inhronized neural networks for the life of the neurn culture. Thus, the GET HNC model of acquirpilepsy provides a method to model the glutam
njury of a stroke and develop spontaneous re
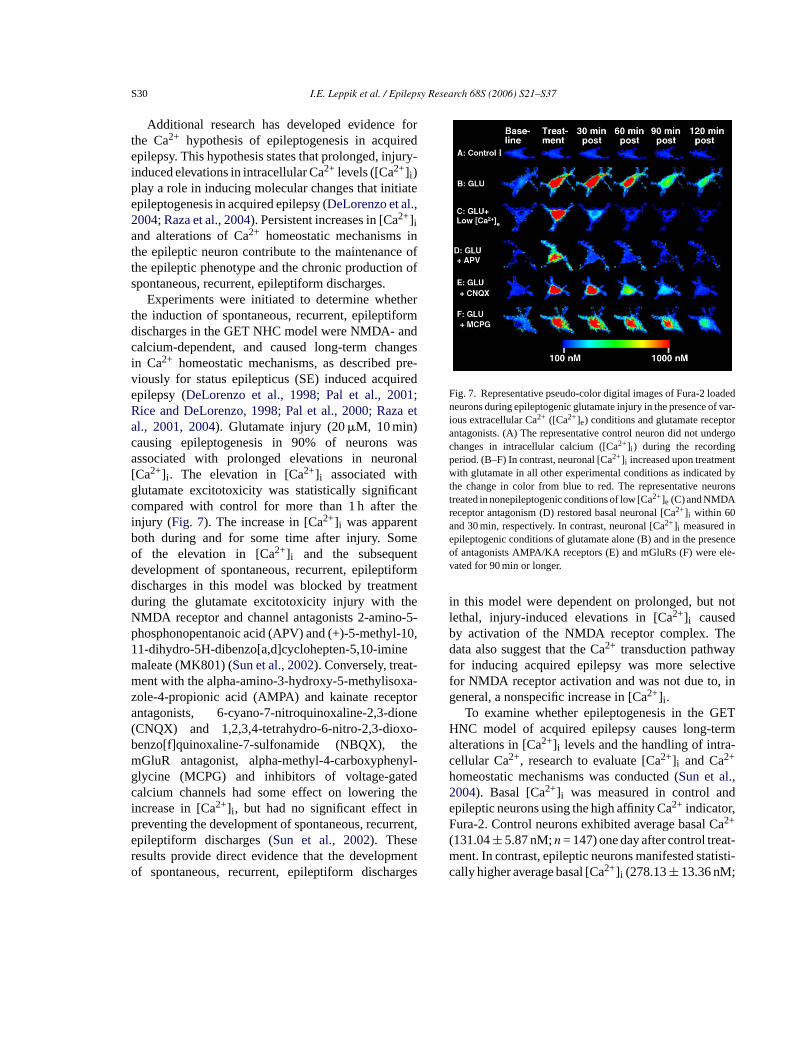
S30 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
Additional research has developed evidence forthe Ca2+ hypothesis of epileptogenesis in acquiredepilepsy. This hypothesis states that prolonged, injury-induced elevations in intracellular Ca2+ levels ([Ca2+]i )play a role in inducing molecular changes that initiateepileptogenesis in acquired epilepsy (DeLorenzo et al.,2004; Raza et al., 2004). Persistent increases in [Ca2+]iand alterations of Ca2+ homeostatic mechanisms inthe epileptic neuron contribute to the maintenance ofthe epileptic phenotype and the chronic production ofspontaneous, recurrent, epileptiform discharges.
Experiments were initiated to determine whetherthe induction of spontaneous, recurrent, epileptiformdischarges in the GET NHC model were NMDA- andcalcium-dependent, and caused long-term changesin Ca2+ homeostatic mechanisms, as described pre-viously for status epilepticus (SE) induced acquiredepilepsy (DeLorenzo et al., 1998; Pal et al., 2001;Rice and DeLorenzo, 1998; Pal et al., 2000; Raza etal., 2001, 2004). Glutamate injury (20�M, 10 min)causing epileptogenesis in 90% of neurons wasassociated with prolonged elevations in neuronal[Ca2+]i . The elevation in [Ca2+]i associated withglutamate excitotoxicity was statistically significantcompared with control for more than 1 h after theinjury (Fig. 7). The increase in [Ca2+] was apparentb meo ntd ormd entd eN o-5-p -10,1 em t-m xa-z tora one( o-b hem nyl-g edc thei inp rent,er ento rges
Fig. 7. Representative pseudo-color digital images of Fura-2 loadedneurons during epileptogenic glutamate injury in the presence of var-ious extracellular Ca2+ ([Ca2+]e) conditions and glutamate receptorantagonists. (A) The representative control neuron did not undergochanges in intracellular calcium ([Ca2+]i ) during the recordingperiod. (B–F) In contrast, neuronal [Ca2+]i increased upon treatmentwith glutamate in all other experimental conditions as indicated bythe change in color from blue to red. The representative neuronstreated in nonepileptogenic conditions of low [Ca2+]e (C) and NMDAreceptor antagonism (D) restored basal neuronal [Ca2+]i within 60and 30 min, respectively. In contrast, neuronal [Ca2+]i measured inepileptogenic conditions of glutamate alone (B) and in the presenceof antagonists AMPA/KA receptors (E) and mGluRs (F) were ele-vated for 90 min or longer.
in this model were dependent on prolonged, but notlethal, injury-induced elevations in [Ca2+]i causedby activation of the NMDA receptor complex. Thedata also suggest that the Ca2+ transduction pathwayfor inducing acquired epilepsy was more selectivefor NMDA receptor activation and was not due to, ingeneral, a nonspecific increase in [Ca2+]i .
To examine whether epileptogenesis in the GETHNC model of acquired epilepsy causes long-termalterations in [Ca2+]i levels and the handling of intra-cellular Ca2+, research to evaluate [Ca2+]i and Ca2+
homeostatic mechanisms was conducted (Sun et al.,2004). Basal [Ca2+]i was measured in control andepileptic neurons using the high affinity Ca2+ indicator,Fura-2. Control neurons exhibited average basal Ca2+
(131.04± 5.87 nM;n = 147) one day after control treat-ment. In contrast, epileptic neurons manifested statisti-cally higher average basal [Ca2+]i (278.13± 13.36 nM;
ioth during and for some time after injury. Sof the elevation in [Ca2+]i and the subsequeevelopment of spontaneous, recurrent, epileptifischarges in this model was blocked by treatmuring the glutamate excitotoxicity injury with thMDA receptor and channel antagonists 2-aminhosphonopentanoic acid (APV) and (+)-5-methyl1-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-iminaleate (MK801) (Sun et al., 2002). Conversely, treaent with the alpha-amino-3-hydroxy-5-methyliso
ole-4-propionic acid (AMPA) and kainate recepntagonists, 6-cyano-7-nitroquinoxaline-2,3-diCNQX) and 1,2,3,4-tetrahydro-6-nitro-2,3-dioxenzo[f]quinoxaline-7-sulfonamide (NBQX), tGluR antagonist, alpha-methyl-4-carboxyphelycine (MCPG) and inhibitors of voltage-gatalcium channels had some effect on loweringncrease in [Ca2+]i , but had no significant effectreventing the development of spontaneous, recurpileptiform discharges (Sun et al., 2002). Theseesults provide direct evidence that the developmf spontaneous, recurrent, epileptiform discha
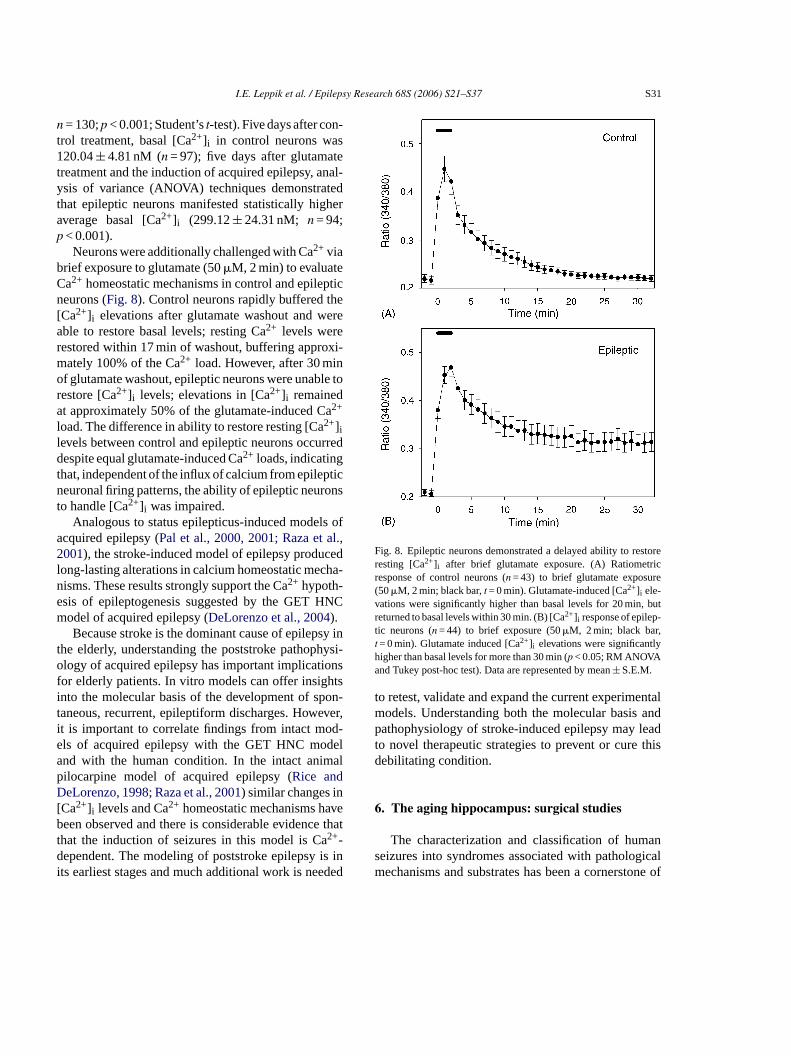
I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S31
n = 130;p < 0.001; Student’st-test). Five days after con-trol treatment, basal [Ca2+]i in control neurons was120.04± 4.81 nM (n = 97); five days after glutamatetreatment and the induction of acquired epilepsy, anal-ysis of variance (ANOVA) techniques demonstratedthat epileptic neurons manifested statistically higheraverage basal [Ca2+]i (299.12± 24.31 nM; n = 94;p < 0.001).
Neurons were additionally challenged with Ca2+ viabrief exposure to glutamate (50�M, 2 min) to evaluateCa2+ homeostatic mechanisms in control and epilepticneurons (Fig. 8). Control neurons rapidly buffered the[Ca2+]i elevations after glutamate washout and wereable to restore basal levels; resting Ca2+ levels wererestored within 17 min of washout, buffering approxi-mately 100% of the Ca2+ load. However, after 30 minof glutamate washout, epileptic neurons were unable torestore [Ca2+]i levels; elevations in [Ca2+]i remainedat approximately 50% of the glutamate-induced Ca2+
load. The difference in ability to restore resting [Ca2+]ilevels between control and epileptic neurons occurreddespite equal glutamate-induced Ca2+ loads, indicatingthat, independent of the influx of calcium from epilepticneuronal firing patterns, the ability of epileptic neuronsto handle [Ca2+]i was impaired.
Analogous to status epilepticus-induced models ofa al.,2 cedl ha-ne NCm
sy int hysi-o onsf htsi on-t ver,i d-e dela alpD n[ veb e thattd is ini ded
Fig. 8. Epileptic neurons demonstrated a delayed ability to restoreresting [Ca2+]i after brief glutamate exposure. (A) Ratiometricresponse of control neurons (n = 43) to brief glutamate exposure(50�M, 2 min; black bar,t = 0 min). Glutamate-induced [Ca2+]i ele-vations were significantly higher than basal levels for 20 min, butreturned to basal levels within 30 min. (B) [Ca2+]i response of epilep-tic neurons (n = 44) to brief exposure (50�M, 2 min; black bar,t = 0 min). Glutamate induced [Ca2+]i elevations were significantlyhigher than basal levels for more than 30 min (p < 0.05; RM ANOVAand Tukey post-hoc test). Data are represented by mean± S.E.M.
to retest, validate and expand the current experimentalmodels. Understanding both the molecular basis andpathophysiology of stroke-induced epilepsy may leadto novel therapeutic strategies to prevent or cure thisdebilitating condition.
6. The aging hippocampus: surgical studies
The characterization and classification of humanseizures into syndromes associated with pathologicalmechanisms and substrates has been a cornerstone of
cquired epilepsy (Pal et al., 2000, 2001; Raza et001), the stroke-induced model of epilepsy produ
ong-lasting alterations in calcium homeostatic mecisms. These results strongly support the Ca2+ hypoth-sis of epileptogenesis suggested by the GET Hodel of acquired epilepsy (DeLorenzo et al., 2004).Because stroke is the dominant cause of epilep
he elderly, understanding the poststroke pathoplogy of acquired epilepsy has important implicati
or elderly patients. In vitro models can offer insignto the molecular basis of the development of spaneous, recurrent, epileptiform discharges. Howet is important to correlate findings from intact mols of acquired epilepsy with the GET HNC mond with the human condition. In the intact animilocarpine model of acquired epilepsy (Rice andeLorenzo, 1998; Raza et al., 2001) similar changes i
Ca2+]i levels and Ca2+ homeostatic mechanisms haeen observed and there is considerable evidenc
hat the induction of seizures in this model is Ca2+-ependent. The modeling of poststroke epilepsy
ts earliest stages and much additional work is nee
S32 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
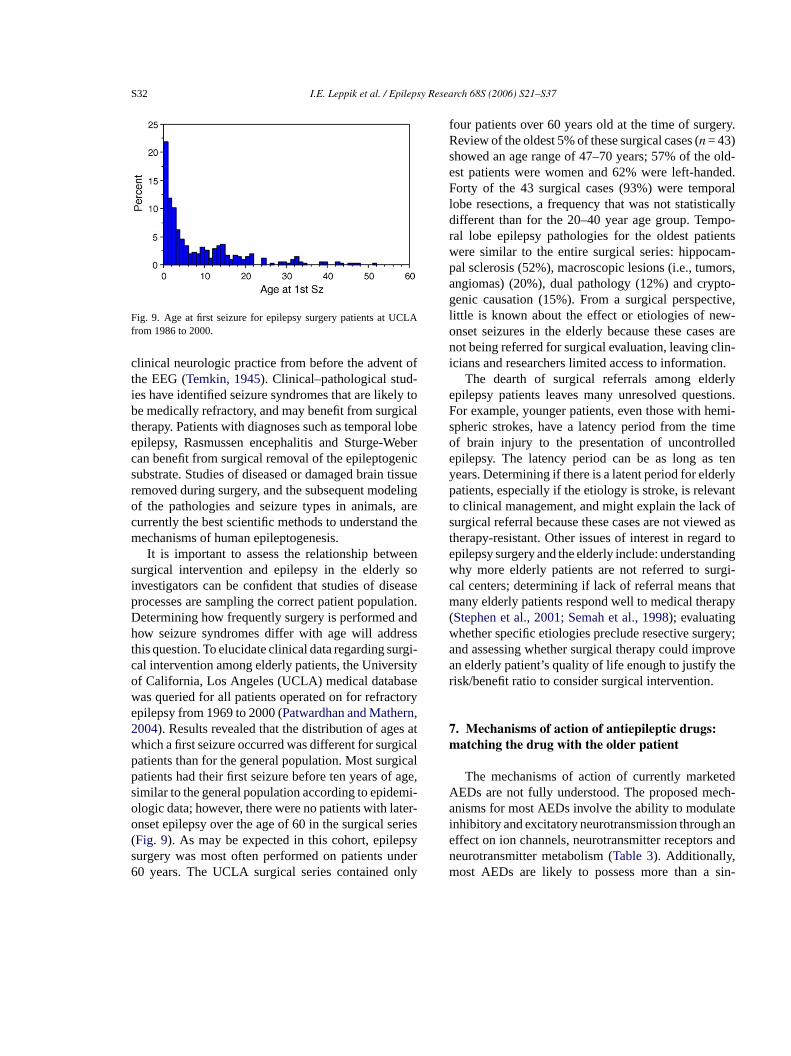
Fig. 9. Age at first seizure for epilepsy surgery patients at UCLAfrom 1986 to 2000.
clinical neurologic practice from before the advent ofthe EEG (Temkin, 1945). Clinical–pathological stud-ies have identified seizure syndromes that are likely tobe medically refractory, and may benefit from surgicaltherapy. Patients with diagnoses such as temporal lobeepilepsy, Rasmussen encephalitis and Sturge-Webercan benefit from surgical removal of the epileptogenicsubstrate. Studies of diseased or damaged brain tissueremoved during surgery, and the subsequent modelingof the pathologies and seizure types in animals, arecurrently the best scientific methods to understand themechanisms of human epileptogenesis.
It is important to assess the relationship betweensurgical intervention and epilepsy in the elderly soinvestigators can be confident that studies of diseaseprocesses are sampling the correct patient population.Determining how frequently surgery is performed andhow seizure syndromes differ with age will addressthis question. To elucidate clinical data regarding surgi-cal intervention among elderly patients, the Universityof California, Los Angeles (UCLA) medical databasewas queried for all patients operated on for refractoryepilepsy from 1969 to 2000 (Patwardhan and Mathern,2004). Results revealed that the distribution of ages atwhich a first seizure occurred was different for surgicalpatients than for the general population. Most surgicalp age,s mi-o ter-o eries( psys nder6 nly
four patients over 60 years old at the time of surgery.Review of the oldest 5% of these surgical cases (n = 43)showed an age range of 47–70 years; 57% of the old-est patients were women and 62% were left-handed.Forty of the 43 surgical cases (93%) were temporallobe resections, a frequency that was not statisticallydifferent than for the 20–40 year age group. Tempo-ral lobe epilepsy pathologies for the oldest patientswere similar to the entire surgical series: hippocam-pal sclerosis (52%), macroscopic lesions (i.e., tumors,angiomas) (20%), dual pathology (12%) and crypto-genic causation (15%). From a surgical perspective,little is known about the effect or etiologies of new-onset seizures in the elderly because these cases arenot being referred for surgical evaluation, leaving clin-icians and researchers limited access to information.
The dearth of surgical referrals among elderlyepilepsy patients leaves many unresolved questions.For example, younger patients, even those with hemi-spheric strokes, have a latency period from the timeof brain injury to the presentation of uncontrolledepilepsy. The latency period can be as long as tenyears. Determining if there is a latent period for elderlypatients, especially if the etiology is stroke, is relevantto clinical management, and might explain the lack ofsurgical referral because these cases are not viewed ast rd toe dingw rgi-c thatm apy(w ery;a rovea her
7m
tedA ch-a atei ane andnm sin-
atients had their first seizure before ten years ofimilar to the general population according to epidelogic data; however, there were no patients with lanset epilepsy over the age of 60 in the surgical sFig. 9). As may be expected in this cohort, epileurgery was most often performed on patients u0 years. The UCLA surgical series contained o
herapy-resistant. Other issues of interest in regapilepsy surgery and the elderly include: understanhy more elderly patients are not referred to sual centers; determining if lack of referral meansany elderly patients respond well to medical ther
Stephen et al., 2001; Semah et al., 1998); evaluatinghether specific etiologies preclude resective surgnd assessing whether surgical therapy could impn elderly patient’s quality of life enough to justify tisk/benefit ratio to consider surgical intervention.
. Mechanisms of action of antiepileptic drugs:atching the drug with the older patient
The mechanisms of action of currently markeEDs are not fully understood. The proposed menisms for most AEDs involve the ability to modul
nhibitory and excitatory neurotransmission throughffect on ion channels, neurotransmitter receptorseurotransmitter metabolism (Table 3). Additionally,ost AEDs are likely to possess more than a

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S33
Table 3Functional implications of proposed mechanisms of action of antiepileptic drugs
Molecular mechanism of action Antiepileptic druga Consequences of action
Na+ channel blockers CBZ, FBM, LTG, OCBZ, PHT, TPM, VPA, ZNS
1. Block action potential propagation2. Stabilize neuronal membranes3. Decrease neurotransmitter release4. Decrease focal firing5. Decrease seizure spread
Ca2+ channel blockers ESM, GBP, LEV, LTG, TPM, VPA, ZNS1. Decrease neurotransmitter release(N & P types)2. Decrease slow-depolarization(T-type)3. Decrease spike-wave discharges
GABAA receptor allosteric modulators BZD, FBM, PB, TPM, ZNS
1. Increase membranehyperpolarization2. Elevate seizure threshold3. Attenuate (BZD) spike-wavedischarges4. Aggravate (barbiturates)spike-wave discharges5. Decrease focal firing
Inhibition of GABA metabolism and re-uptake TGB, VGB, VPA
1. Increase synaptic GABA levels2. Increase membranehyperpolarization3. Decrease focal firing4. Aggravate spike-wave discharges
NMDA-receptor antagonists FBM1. Decrease slow excitatoryneurotransmission2. Decrease excitatory amino acidneurotoxicity3. Delay epileptogenesis
AMPA/kainate-receptor antagonists PB, TPM1. Decrease fast excitatoryneurotransmission2. Attenuate focal firing
a BZD = benzodiazepines; CBZ = carbamazepine; ESM = ethosuximide; FBM = felbamate; GBP = gabapentin; LEV = levetiracetam;LTG = lamotrigine; OCBZ = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; TGB = tiagabine; TPM = topiramate; VPA = valproic acid;and ZNS = zonisamide.
gle mechanism of action (Macdonald and Kelly, 1995;Meldrum, 1996; Rho and Sankar, 1999; White, 2001;Rogawski, 2002).
7.1. Modulation of voltage-gated ion channels
A seemingly large proportion of available AEDs arethought to exert an action at voltage-gated ion chan-nels: sodium channels (phenytoin, carbamazepine,valproic acid, felbamate, lamotrigine, topiramate,oxcarbazepine and zonisamide); calcium channels(valproic acid, ethosuximide, felbamate, lamotrigine,
topiramate and gabapentin) and potassium channels(topiramate). Of the drugs that modify voltage-gatedcalcium currents, valproic acid, ethosuximide and,perhaps, zonisamide are unique in that they appearto reduce voltage-dependent low threshold T-typecurrents in thalamocortical neurons. It is this actionthat is thought to contribute to the ability of thesedrugs to interrupt the thalamic oscillatory firingpatterns associated with spike-wave seizures. Actionsat both the voltage-gated sodium and calcium channelswould be predictive of a capacity to stabilize neuronalmembranes, block action potential firing and prop-

S34 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
agation, reduce neurotransmitter release and preventseizure spread. The ability of topiramate to activatepotassium currents is unique among the AEDs and thiseffect would be expected to contribute to membranehyperpolarization (Herrero et al., 2002).
7.2. Enhanced inhibition
Inhibitory neurotransmission is primarily mediatedthrough gamma-aminobutyric acid (GABA) recep-tors. Once released from the presynaptic terminal,GABA can modify activity by binding to the GABAAreceptor to increase chloride permeablity or by bind-ing to a G-protein coupled GABAB receptor toincrease potassium permeability. Several of the avail-able AEDs have demonstrated the ability to poten-tiate GABAergic neurotransmission mediated by theGABAA receptor (e.g., the benzodiazepines, barbitu-rates, felbamate, topiramate and zonisamide). SeveralAEDs have also been found to increase GABAergictone by decreasing GABA metabolism (i.e., valproateand vigabatrin), by preventing GABA reuptake (i.e.,tiagabine) or by increasing synthesis (i.e., valproate andgabapentin).
7.3. Reduced excitation
atedb con-t ta-m theser DAa
atea duceg bym -m n off d tor hapsd oci-a rast,a ep-t rya focalfi
picg eight
metabotropic receptors identified; however, none of theAEDs have been found to alter neurotransmission mod-ified by metabotropic receptors.
7.4. Implications for treating elderly patients withepilepsy
Several AEDs display a number of different phar-macologic actions that could account for anticonvul-sant properties and contribute to efficacy in vivo. Incases where multiple actions have been defined, it ishighly likely that distinct mechanisms may offer somedegree of synergy. Interestingly, several AEDs (e.g.,valproic acid, gabapentin, lamotrigine and topiramate)have been found useful in treating additional CNSdisorders, such as bipolar disorder (valproic acid andlamotrigine), migraine (valproic acid and topiramate)and neuropathic pain (gabapentin). Since AEDs targetmany of the same neuropathic mechanisms thought tobe underlying causes of other CNS disorders, this is anunderstandable outcome.
Currently, there is no sound experimental evidenceto suggest that the mechanistic basis of seizures dif-fer with the age of the patient. Unfortunately, studiesof the mechanisms of seizure activity conducted todate have not examined age as an independent vari-a pectt ec-t nts.H eptora onics oke,s eenr PAr rme-a 0;F al.,2 rgestc thee cesf lec-t iump cep-t witha pres-e tantc erlyp
Excessive excitatory neurotransmission mediy glutamte and other excitatory amino acids can
ribute to seizure activity. Within the CNS, four gluate receptor types have been identified. Three of
eceptors are coupled to ion channels: AMPA, NMnd kainate.
Of the available AEDs, felbamate and topiramre novel in that they possess a unique ability to relutamate-mediated excitatory neurotransmissionodulation of NMDA (felbamate), AMPA (topiraate) and kainate (topiramate) receptors. Actio
elbamate at the NMDA receptors would be expecteeduce slow excitatory neurotransmission and perecrease excitatory amino acid neurotoxicity assted with excessive glutamate release. In contction of topiramate at AMPA and kainate rec
ors would be predicted to modify fast excitatond neurotansmission, and perhaps attenuatering.
The fourth glutamate-binding site is a metabotrolutamate receptor. Thus far, there have been
ble. As such, there is no apriori reason to sushat any specific AED would be more or less effive for the management of seizures in elderly patieowever, emerging evidence suggests that recnd voltage-gated subunits can be modified by chreizures and the underlying pathophysiology. Strtatus epilepticus, and kindling, for example, have beported to modify the expression of GluR2 AMeceptors and potentially increase the calcium pebility of the AMPA receptor (Prince et al., 1995, 200riedman et al., 2000; Anzai et al., 2003; Liu et004). Given that cerebrovascular disease is the laontributing factor to newly diagnosed epilepsy inlderly, this finding may have practical consequen
or the treatment of older patients when subunit seive AEDs are developed that selectively target calcermeable versus calcium impermeable AMPA re
ors. In the meantime, other factors associatedging, such as renal and hepatic function, and thence of comorbidities, should be the most imporonsiderations when selecting an AED for an eldatient.

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S35
References
Anzai, T., Tsuzuki, K., Yamada, N., Hayashi, T., Iwakuma, M.,Inada, K., Kameyama, K., Hoka, S., Saji, M., 2003. Overexpres-sion of Ca2+-permeable AMPA receptor promotes delayed celldeath of hippocampal CA1 neurons following transient forebrainischemia. Neurosci. Res. 46, 41–51.
Barnes, C.A., 1979. Memory deficits associated with senescence: aneurological and behavioral study in the rat. J. Comp. Physiol.Psychol. 931, 74–104.
Barnes, C.A., 2001. Plasticity in the aging central nervous system.Int. Rev. Neurobiol. 45, 339–354.
Barnes, C.A., McNaughton, B.L., 1985. An age comparison of therates of acquisition and forgetting of spatial information in rela-tion to long-term enhancement of hippocampal synapses. Behav.Neurosci. 99, 1040–1048.
Ben-Ari, Y., Cossart, R., 2000. Kainate, a double agent that generatesseizures: two decades of progress. Trends Neurosci. 32, 580–587.
Braak, H., Braak, E., 1991. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. (Berl.) 82, 239–259.
Butler, R.N., 1997. Aging: the challenge of the twenty-first cen-tury medicine. In: Rowan, A.J., Ramsay, RE. (Eds.), Seizuresand Epilepsy in the Elderly. Butterworth-Heinemann, Boston,pp. 3–5.
Cassell, M.D., Brown, M.W., 1984. The distribution of Timm’s stainin the nonsulphide-perfused human hippocampal formation. J.Comp. Neurol. 222, 461–471.
Chiba, S., Muneoka, Y., Sato, Y., Miyagishi, T., 1992. Seizuresusceptibility in aged rats - pentylenetetrazol-induced seizuresand amygdaloid kindling seizures. No To Shinkei 44, 559–564,
D oen-uced
D ech-is of
. 105,
D ationy
hip-95,
D Sta-a 33
d s inell,am
d entpal
d .S.,RI-rom
Dickerson, B.C., Goncharova, I., Sullivan, M.P., Forchetti, C., Wil-son, R.S., Bennett, D.A., Beckett, L.A., deToledo-Morrell, L.,2001. MRI-derived entorhinal and hippocampal atrophy in incip-ient and very mild Alzheimer’s disease. Neurobiol. Aging 22,747–754.
Du, A.T., Schuff, N., Amend, D., Laakso, M.P., Hsu, Y.Y., Jagust,W.J., Yaffe, K., Kramer, J.H., Reed, B., Norman, D., Chui,H.C., Weiner, M.W., 2001. Magnetic resonance imaging of theentorhinal cortex and hippocampus in mild cognitive impairmentand Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 71,441–447.
Du, A.T., Schuff, N., Laakso, M.P., Zhu, X.P., Jagust, W.J., Yaffe,K., Kramer, J.H., Miller, B.L., Reed, B.R., Norman, D., Chui,H.C., Weiner, M.W., 2002. Effects of subcortical ischemic vas-cular dementia and AD on entorhinal cortex and hippocampus.Neurology 58, 1635–1641.
Fanelli, R.J., McNamara, J.O., 1986. Effects of age on kindlingand kindled seizure-induced increase of benzodiazepine receptorbinding. Brain Res. 362, 17–22.
Friedman, L.K., Belayev, L., Alfonso, O.F., Ginsberg, M.D., 2000.Distribution of glutamate and preproenkephalin messengerRNAs following transient focal cerebral ischemia. Neuroscience95, 841–857.
Geinisman, Y., deToledo-Morrell, L., Morrell, F., Heller, R.E.,1995. Hippocampal markers of age-related memory dysfunc-tion: behavioral, electrophysiological and morphological per-spectives. Prog. Neurobiol. 45, 223–252.
Gomez-Isla, T., Price, J.L., McKeel Jr., D.W., Morris, J.C., Growdon,J.H., Hyman, B.T., 1996. Profound loss of layer II entorhinal cor-tex neurons occurs in very mild Alzheimer’s disease. J. Neurosci.16, 4491–5000.
G PTZ-rat
H e ofand
xp.
H theandpp.
H epi-dies.
H e ofsota:
H uses,ork.
H .M.,riz-sium
J oraly. J.
Japanese.arbin, O., Naritoku, D., Patrylo, P.R., 2004. Aging alters electr
cephalographic and clinical manifestations of kainate-indstatus epilepticus. Epilepsia 45, 1219–1227.
eLorenzo, R.J., Sun, D.A., Deshpande, L.S., 2004. Cellular manisms underlying acquired epilepsy: the calcium hypothesthe induction and maintenance of epilepsy. Pharmacol. Ther229–266.
eLorenzo, R.J., Pal, S., Sombati, S., 1998. Prolonged activof theN-methyl-d-aspartate receptor-Ca2+ transduction pathwacauses spontaneous recurrent epileptiform discharges inpocampal neurons in culture. Proc. Natl. Acad. Sci. USA14482–14487.
eLorenzo, R.J., Towne, A.R., Pellock, J.M., Ko, D., 1992.tus epilepticus in children, adults, and the elderly. Epilepsi(Suppl. 4), S15–S25.
eToledo-Morrell, L., Morrell, F., 1991. Age-related alterationlong-term potentiation and susceptibility to kindling. In: MorrF. (Ed.), Kindling and Synaptic Plasticity: The Legacy of GrahGoddard. Birkhauser Press, Boston, pp. 202–216.
eToledo-Morrell, L., Morrell, F., Fleming, S., 1984. Age-dependdeficits in spatial memory are related to impaired hippocamkindling. Behav. Neurosci. 98, 902–907.
eToledo-Morrell, L., Stoub, T.R., Bulgakova, M., Wilson, RBennett, D.A., Leurgans, S., Wuu, J., Turner, D.A., 2004. Mderived entorhinal volume is a good predictor of conversion fMCI to AD. Neurobiol. Aging 25, 1197–1203.
recksch, G., Ruethrich, H., Bernstein, H.G., Becker, A., 1995.kindling after colchicines lesion in the dentate gyrus of thehippocampus. Physiol. Behav. 58, 695–698.
artings, J.A., Williams, A.J., Tortella, F.C., 2003. Occurrencnonconvulsive seizures, periodic epileptiform discharges,intermittent rhythmic delta activity in rat focal ischema. ENeurol. 179, 139–149.
auser, W.A., 1997. Epidemiology of seizures and epilepsy inelderly. In: Rowan, A.J., Ramsay, RE. (Eds.), SeizuresEpilepsy in the Elderly. Butterworth-Heinemann, Boston,7–18.
auser, W.A., Annegers, J.F., Rocca, W.A., 1996. Descriptivedemiology of epilepsy: contributions of population-based stufrom Rochester, Minnesota. Mayo. Clin. Proc. 71, 576–586
auser, W.A., Annegers, J.F., Kurland, L.T., 1993. Incidencepilepsy and unprovoked seizures in Rochester, Minne1935–1984. Epilepsia 34, 453–468.
auser, W.A., Hesdorffer, D.C., 1990. Epilepsy: Frequency, Caand Consequences. Demos Medical Publishing Inc., New Y
errero, A.I., Del Olmo, N., Gonzalez-Escalada, J.R., Solis, J2002. Two new actions of topiramate: inhibition of depolaing GABA(A)-mediated responses and activation of a potasconductance. Neuropharmacology 42, 210–220.
okeit, H., Ebner, A., 1999. Long term effects of refractory templobe epilepsy on cognitive abilities: a cross sectional studNeurol. Neurosurg. Psychiatry 67, 44–50.

S36 I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37
Karhunen, H., Pitkanen, A., Virtanen, T., Gureviciene, I., Pussinen,R., Ylinen, A., Sivenius, J., Nissinen, J., Jokkonen, J., 2003.Long-term functional consequences of transient occlusion of themiddle cerebral artery in rats: a 1-year follow-up of the develop-ment of epileptogenesis and memory impairment in relation tosensorimotor deficits. Epilepsy Res. 54, 1–10.
Kelly, K.M., 2002. Poststroke seizures and epilepsy: clinical studiesand animal models. Epilepsy Curr. 2, 173–177.
Kelly, K.M., Kharlamov, A., Hentosz, T.M., Kharlamov, E.A.,Williamson, J.M., Bertram 3rd., E.H., Kapur, J., Armstrong,D.M., 2001. Photothrombotic brain infarction results in seizureactivity in aging Fischer 344 and Sprague Dawley rats. EpilepsyRes. 47, 189–203.
Kerr, D.S., Razak, A., Crawford, N., 2002. Age-related changes intolerance to the marine algal excitotoxin domoic acid. Neurophar-macology 43, 357–366.
Kharmalov, E.A., Jukkola, P.I., Schmitt, K.L., Kelly, K.M., 2003.Electrobehavioral characteristics of epileptic rats followingphotothrombotic brain infarction. Epilepsy Res. 56, 185–203.
Kordower, J.H., Chu, Y., Stebbins, G.T., DeKosky, S.T., Cochran,E.J., Bennett, D., Mufson, E.J., 2001. Loss and atrophy of layerII entorhinal cortex neurons in elderly people with mild cognitiveimpairment. Ann. Neurol. 49, 202–213.
Liu, S., Lau, L., Wei, J., Zhu, D., Zou, S., Sun, H.S., Fu, Y., Liu,F., Lu, Y., 2004. Expression of Ca(2+)-permeable AMPA recep-tor channels primes cell death in transient forebrain ischemia.Neuron 43, 43–55.
Lothman, E.W., Stringer, J.L., Bertram, E.H., 1992. The dentategyrus as a control point for seizures in the hippocampus andbeyond. Epilepsy Res. Suppl. 7, 301–313.
L the27,
M ms
M Hip-hesis
M santuppl.
M ne-Theork,
M the
N .W.,l. 18,
N the41–
P hibi-ging.
Patwardhan, R., Mathern, G.W., 2004. Surgical treatment of therapy-resistant epilepsy. Continuum: Lifelong Learning in Neurology.Epilepsy 10 (4), 100–118.
Pal, S., Limbrick Jr., D.D., Rafiq, A., DeLorenzo, R.J., 2000. Induc-tion of spontaneous recurrent epileptiform discharges causeslong-term changes in intracellular calcium homeostatic mech-anisms. Cell Calcium 28, 181–193.
Pal, S., Sun, D., Limbrick, D., Rafiq, A., DeLorenzo, R.J., 2001.Epileptogenesis induces long-term alterations in intercellularcalcium release and sequestration mechanisms in the hippocam-pal neuronal culture model of epilepsy. Cell Calcium 30, 285–296.
Perl, T.M., Bedard, L., Kosatsky, T., Hockin, J.C., Todd, E.C., Remis,R.S., 1990. An outbreak of toxic encephalopathy caused by eatingmussels contaminated with domoic acid. N. Engl. J. Med. 332,1775–1780.
Prince, H.C., Tzingounis, A.V., Levey, A.I., Conn, P.J., 2000. Func-tional downregulation of GluR2 in piriform cortex of kindledanimals. Synapse 38, 489–498.
Prince, H.K., Conn, P.J., Blackstone, C.D., Huganir, R.L., Levey,A.I., 1995. Down-regulation of AMPA receptor subunit GluR2in amygdaloid kindling. J. Neurochem. 64, 462–465.
Raza, M., Pal, S., Rafiq, A., DeLorenzo, R.J., 2001. Long-termalteration of calcium homeostatic mechanisms in the pilocarpinemodel of temporal lobe epilepsy. Brain Res. 903, 1–12.
Raza, M., Blair, R.E., Sombati, S., Carter, D.S., Deshpande, L.S.,DeLorenzo, R.J., 2004. Evidence that injury-induced changesin hippocampal neuronal calcium dynamics during epileptoge-nesis cause acquired epilepsy. Proc. Natl. Acad. Sci. 101 (50),17522–17527.
Rho, J.M., Sankar, R., 1999. The pharmacologic basis of antiepileptic
R ringpsy.
R lep-S.,ott
S A.,ingNeu-
S iallyarea
S ress-teins394,
S se ofpilep-
S ong-n in55–
uhdorf, K., Jensen, L.K., Plesner, A.M., 1986. Epilepsy inelderly: incidence, social function, and disability. Epilepsia135–141.
acDonald, R.L., Kelly, K.M., 1995. Antiepileptic drug mechanisof action. Epilepsia 36 (Suppl. 2), S2–S12.
athern, G.W., Adelson, P.D., Cahan, L.D., Leite, J.P., 2002.pocampal neuron damage in human epilepsy: Meyer’s hypotrevisited. Prog. Brain Res. 135, 237–251.
eldrum, B., 1996. Action of established and novel anticonvuldrugs on the basic mechanisms of epilepsy. Epilepsy Res. S11, 67–77.
orrell, F., deToledo-Morrell, L., 1999. Secondary epileptogesis and brain tumors. In: Kotagal, P., Lynders, H.O. (Eds.),Epilepsies: Etiology and Prevention. Academic Press, New Ypp. 357–364.
orrison, J.H., Hof, P.R., 1997. Life and death of neurons inaging brain. Science 278, 412–419.
g, S.K., Hauser, W.A., Brust, J.C.M., Healton, E.B., Susser, M1985. Risk factors for adult-onset first seizures. Ann. Neuro153.
okubo, M., Kitani, K., 1988. Age-dependent decrease inlethal threshold of pentylenetetrazole in mice. Life Sci. 43,47.
atrylo, P.R., Lee, S., Williamson, A., 2000. The balance of intion/excitation changes in the rodent dentate gyrus during aEpilepsia 41 (Suppl. 7), 32.
drug action. Epilepsia 40, 1471–1483.ice, A.C., DeLorenzo, R.J., 1998. NMDA receptor activation du
status epilepticus is required for the development of epileBrain Res. 782, 240–247.
ogawski, M.A., 2002. General principles: principles of antiepitic drug action. In: Levy, R.H., Mattson, R.H., Meldrum, B.Perucca, E. (Eds.), Antiepileptic Drugs, fifth ed. LippincWilliams & Wilkins, Philadelphia, pp. 3–22.
emah, F., Picot, M.C., Adam, C., Broglin, D., Arzimanoglou,Bazin, B., Cavalcanti, D., Baulac, M., 1998. Is the underlycause of epilepsy a major prognostic factor for recurrence?rology 51, 1256–1262.
hankar, S., Teyler, T.J., Robbins, N., 1998. Aging differentalters forms of long-term potentiation in rat hippocampalCA1. J. Neurophysiol. 79, 334–341.
hetty, A.K., Turner, D.A., 1998. Hippocampal interneurons exping glutamic acid decarboxylase and calcium-binding prodecrease with aging in Fischer 344 rats. J. Comp. Neurol.252–269.
tephen, L.J., Kwan, P., Brodie, M.J., 2001. Does the caulocalisation-related epilepsy influence the response to antietic drug treatment? Epilepsia 42, 357–362.
un, D.A., Sombati, S., Blair, R.E., DeLorenzo, R.J., 2004. Llasting alterations in neuronal calcium homeostatis in avitro model of stroke-induced epilepsy. Cell Calcium 35, 1163.

I.E. Leppik et al. / Epilepsy Research 68S (2006) S21–S37 S37
Sun, D.A., Sombati, S., Blair, R.E., DeLorenzo, R.J., 2002. Calcium-dependent epileptogenesis in an in vitro model of stroke“induced” epilepsy. Epilepsia 43, 1296–1305.
Sun, D.A., Sombati, S., DeLorenzo, R.J., 2001. Glutamate injury-induced epileptogenesis in hippocampal neurons: an in vivomodel of stroke-induced “epilepsy”. Stroke 32, 2344–2350.
Tasch, E., Cendes, F., Li, L.M., Dubeau, F., Andermann, F., Arnold,D.L., 1999. Neuroimaging evidence of progressive neuronal lossand dysfunction in temporal lobe epilepsy. Ann. Neurol. 45,568–576.
Teitelbaum, J.S., Zatorre, R.J., Carpenter, S., Gendron, D., Evans,A.C., Gjedde, A., Cashman, N.R., 1990. Neurologic sequelae ofdomoic acid intoxication due to the ingestion of contaminatedmussels. N. Engl. J. Med. 322, 1781–1787.
Temkin, O., 1945. The Falling Sickness: A History of Epilepsy fromthe Greeks to the Beginnings of Modern Neurology. The JohnsHopkins Press, Baltimore, pp. 1–380.
Tinuper, P., 1997. The altered presentation of seizures in the elderly.In: Rowan, J.A., Ramsay, R.E. (Eds.), Seizures and Epilepsyin the Elderly. Butterworth-Heinemann, Boston, pp. 123–130.
Treiman, D.M., Meyers, P.D., Walton, N.Y., 1997. Status epilepticusin the elderly: nosology and therapy. In: Rowan, A.J., Ramsay,R.E. (Eds.), Seizures and Epilepsy in the Elderly. Butterworth-Heinemann, Boston, pp. 201–215.
White, H.S., 2001. Comparative anticonvulsant profile and proposedmechanisms of action of the established and newer antiepilepticdrugs. In: Pellock, J.M., Dodson, W.E., Bourgeois, B.F.D. (Eds.),Pediatric Epilepsy: Diagnosis and Therapy, second ed. DemosMedical Publishing Inc., New York.
Wozniak, D.F., Stewart, G.R., Miller, J.P., Olney, J.W., 1991. Age-related sensitivity to kainate neurotoxicity. Exp. Neurol. 114,250–253.





























