Embed Size (px)
Citation preview

Adenovirus lacking the 19-kDa and 55-kDa E1B genesexerts a marked cytotoxic effect in human malignant cellsPilar Martın Duque,1 Covadonga Alonso,2 Ricardo Sanchez-Prieto,1 Matilde Lleonart,1 CarlosMartınez,3 Gonzalo Gonzalez de Buitrago,3 Amparo Cano,4 Miguel Quintanilla,4
and Santiago Ramon y Cajal1
1Department of Pathology, Clınica Puerta de Hierro, Madrid, Spain; 2Centro de Investigacion en SanidadAnimal; 3Centro Nacional de Biotecnologıa; and 4Instituto de Investigaciones Biomedicas, Consejo Superiorde Investigaciones Cientificas, Madrid, Spain.
The adenovirus (Ad) E1A gene exerts an antitumor effect and can induce sensitivity to treatment with DNA-damaging agents. Incontrast, the Ad 19-kDa E1B protein inhibits E1A-mediated apoptosis and the 55-kDa E1B inactivates the p53 protein. In this paper,we study the in vitro and in vivo effects of a 19-kDa and 55-kDa E1B-defective Ad in several malignant human tumor cell lines.
Materials and Methods. Nontumorigenic human fibroblasts (CCD-45SK and Hs67), peripheral blood lymphocytes, and severalhuman tumor cell lines derived from cervix, colon, and breast carcinomas, epidermoid carcinoma, and osteosarcoma (HeLa, HT29,MCF7, Saos-2, and A431 cell lines) were studied. Wild-type (wt) Ad type 5 and H5 dL118 Ad, a mutant with the deleted E1B region,were employed. The cells were infected at 20 plaque-forming units, and cell viability was evaluated by the crystal violet method.In the in vivo experiments, 2 3 106 cells from the carcinoma cell lines HeLa, A431. and HT29 were injected into nude mice. Thetumorigenicity of previously infected cells and after an intratumoral injection of Ad was analyzed. The mice received whole-bodyg-irradiation.
Results. The H5 dL118 mutant produced a marked cytopathic effect in all of the malignant cells, surpassing that of the wt Ad;viability at 72 hours ranged from 11% to 20% for H5 dL118 Ad and from 70% to 93% for the wt Ad with respect to uninfectedcontrols. In the in vivo experiments, a total inhibition of tumorigenicity was detected when cells were infected prior to injection anda partial and transitory decrease in tumorigenicity was detected when the mutant H5 dL118 was injected intratumorally.g-irradiation enhanced the in vivo antitumor effects.
Conclusions. These results indicate that infection with completely E1B-deficient Ads induced a marked cytopathic effect onmalignant cells that was higher than that seen for wt Ads; in addition, infection with such Ads exerts a tumor suppressor effect invivo.
Key words: E1B-defective adenovirus; gene therapy; tumorigenicity; adenovirus E1A gene; adenovirus E1B.
Malignant tumors are characterized by the greatheterogeneity of cell types, pathological features,
and clinical behaviors.1–3 Malignant cells carry multiplegenetic alterations that accumulate during the process ofcarcinogenesis, leading to more “malignant” or “aggres-sive” phenotypes.4–7 Many oncogenic alterations are stillunknown, and it is currently accepted that, except forsome lymphoproliferative and leukemic diseases andspecific childhood round-cell tumors, there are no spe-cific oncogenic alterations.8–10
Based on the knowledge of the molecular biology ofmalignant cells, numerous approaches are being under-
taken in the development of new therapeutic proto-cols.10–14 Most of these approaches have focused on thereplacement of genes, such as p53, that have undergonemutation or deletion.14–18 Nevertheless, because of theamazing variety of genes involved in cell transformation,other studies have focused on the use of genes thatinduce cell death or increase cell susceptibility to apo-ptosis after exposure to DNA-damaging agents, regard-less of the multiple genetic alterations activated in thecells.4,19–21 One of these genes is the adenovirus (Ad)E1A gene. Early region 1A of human Ad type 5 (Ad5)encodes two major proteins of 289R and 243R, synthe-sized after infection, that have myriad cellular effects.These proteins provide an optimal environment for viralreplication and can activate and repress the transcriptionof multiple genes (reviewed in Refs. 22, 23, and 24).Most of their effects are modulated by binding todifferent cellular proteins, including the cyclic adenosinemonophosphate-response-element-binding protein and
Received August 21, 1998; accepted December 12, 1998.Address correspondence and reprint requests to Dr. Santiago Ramon y
Cajal, Departamento de Patologıa, Clınica Puerta de Hierro, c/SanMartın de Porres 4, 28035 Madrid, Spain. E-mail address: [email protected]
© 1999 Stockton Press 0929-1903/99/$12.00/10
Cancer Gene Therapy, Vol 6, No 6, 1999: pp 554–563554

a related protein p300, pRb and related proteins p130and p107, p60/cyclin A, p33cdk2, BS69, and CtBB. Thus,Ad5 E1A can repress HER-2/neu, p27/kip1, p21/waf1/cip1, p15/INK41, and other genes.25–29 Interestingly, theE1A gene appears to have a dual and paradoxical effecton cells. E1A gene can activate DNA synthesis and cellproliferation when cells are growing in optimal condi-tions or can drive cells rapidly to apoptosis in sera-freemedium or after DNA damage. These opposing pro-cesses involved in cell proliferation and death can begeneralized to virtually all other promoters of cell pro-liferation such as c-myc, the G1 progression transcrip-tion factor E2F1, and the Cdc25a phosphatase.30–32
The antitumor effect of Ad E1A was initially de-scribed by Frisch,33 who observed it in malignant cellstransfected by E1A-12S. Recently, this effect has alsobeen obtained after the injection of retrovirus E1Aproducer cells into the tumors.34 Some authors haveassociated the antitumor effect of Ad E1A with thedown-regulation of the transcription of HER-2/neu35,36
or the conversion of tumor cells into an epithelialphenotype.37 In addition, in many cases, E1A expressionprevents malignant cells from metastasizing.38
We reported previously that the Ad E1A gene inducesan increased sensitivity to DNA-damaging agents inmurine keratinocytes and multiple carcinoma cell lines,regardless of p53 status and other genetic alterations ofthe malignant cells.21,39–41 Moreover, we affirmed that inseveral E1A mutant-expressing cells, sensitivity to DNA-damaging agents requires binding to p60 pRb and p300cellular proteins independent of the level of expressionof the p53 protein. We postulated that other cellularmechanisms in addition to p53 must be involved in thesensitization of cells to the DNA-damaging agents in-duced by the wild-type (wt) E1A gene.40 Recently, wehave shown that the Ad E1A gene decreases the tumor-igenicity of carcinoma cells in cell lines with a constitu-tive E1A expression and after intratumoral (i.t.) injec-tion of retrovirus-E1A producer cells.34 Other studieswith recombinant Ads have shown that E1B-defectiveAd5 also induces p53-dependent and -independent ap-optosis, and that the expression of other Ad early geneproducts, such as an E4 product, may be essential forE1A-induced, p53-independent apoptosis.42
Interestingly, 55-kDa E1B-defective Ad5 was reportedto replicate selectively in p53-deficient human tumorcells43 and, more recently, the same vector ONYX-015was observed to be capable of destroying numerouscarcinoma cell lines with either a mutated or normal p53gene.44 These promising approaches to cancer genetherapy are already undergoing clinical trials.
Recently, we reported that infection with Ads lackingboth E1B genes (19-kDa E1B and 55-kDa E1B) could bea more efficient method of inducing lethality acting onmalignant cells.45 Both E1B proteins encoded by theE1B region seem to be involved in controlling most ofthe effects mediated by the E1A gene. In fact, the19-kDa E1B gene may prevent the premature death ofAd-infected cells and inhibit the apoptotic pathwaysinduced by expression of the Ad E1A protein, whereas
the 55-kDa E1B protein leads to the stabilization of p53and may inhibit the transcriptional activation potentialof p53.46 We hypothesized that Ads lacking expressionof the 19-kDa and 55-kDa E1B proteins and havinginherent E1A function could combine the antitumoreffects of Ad E1A protein and the selective replication ofE1B-defective Ads. We show here that the mutant AdH5 dL118 exerts a marked cytopathic effect on severalmalignant human cell lines and a partial reduction oftumorigenicity in vivo. Moreover, we show that thismutant Ad can enhance chemosensitivity to DNA-dam-aging agents in vitro and in vivo.
MATERIALS AND METHODS
Cell lines
Several cell lines derived from the most common humantumors were studied, including HeLa cells (from cervicalcarcinoma carrying human papilloma virus, with wt but inac-tivated p53), A431 cells (epidermoid carcinoma; mutated p53),HT29 cells (colon adenocarcinoma; mutated p53), MCF7 cells(breast adenocarcinoma; wt p53), Saos-2 cells (osteosarcoma;absence of p53), and three uveal melanoma cell lines: MKTBr(mutated p53), OCM1 (mutated p53), and Sp6 (wt p53). Thecells were grown in Dulbecco’s modified Eagle’s medium (LifeTechnologies, Grand Island, NY) supplemented with 10%fetal calf sera and antibiotics, with the exception of themelanoma cell lines, which grew in RPMI 1640 medium.
As nontumorigenic cells, the fibroblastic cell lines Hs67 andCCD-45SK, uveal melanocytes UW3, lymphocytes (from adonor), and the Vero cell line were used. Moreover, a sponta-neously transformed set of Hs67 fibroblasts was analyzed (wt 53).
Viruses
wt Ad5 (wt 300) and a recombinant, replication-deficient AdH5 dL118 (provided by Dr. Ginsberg, National Institutes ofHealth, Rockville, MD) were employed. H5 dL118 contains adeletion in the E1B region and does not synthesize the 19-kDaor 55-kDa E1B proteins.47 Both wt 300 and H5 dL118 carry asmall deletion in the E3 region. Ads were grown on the humanembryonic kidney cell line 293.48,49 Viruses were purified,titrated by plaque assay, and stored in 10% glycerol/phosphate-buffered saline (PBS) at 270°C as described previously.35,48
The 293 cells were grown in Dulbecco’s modified Eagle’smedium supplemented with 0.01 M L-glutamine, antibiotics,and 10% fetal calf sera. During viral infection, 5% horse serawas added.
Replication assays
Two different methods were used to analyze the replicationefficiency of H5 dL118 and wt Ads in the A431 and HeLa cells.First, virus supernatants were tested at serial dilutions to infectthe 293 cells (plaque assays), as described previously.49 Sec-ond, we compared the replication of the viruses in both 293cells and the carcinoma cell lines A431 and MCF7 (that mostsensitive and that most resistant to infection with these Ads,respectively). We infected them at a multiplicity of infection(MOI) of 20 per cell and obtained lysates at 1, 2, 3, and 5 dayspostinfection by three cycles of freezing and thawing. Wesubsequently infected 293 cells at 80% confluency with serialdilutions of the lysates obtained from 293, A431, and MCF7cell lines. At 48 hours postinfection, the wells were fixed inglutaraldehyde and stained by the crystal violet method.
MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS 555
Cancer Gene Therapy, Vol 6, No 6, 1999

Sensitivity to cisplatin and radiotherapy
Cisplatin (CDDP, Bristol-Myers, Syracuse, NY) was used atdoses ranging from 0.01 to 10 mg/mL. All dilutions wereperformed in PBS. In irradiation assays, cells were g-irradiatedwith a 60Co source at a skin surface distance of 57 cm and adose rate of 238 cGy/min.
A total of 4 3 104 cells/well were plated in 24-multiwellculture plates; drugs were added 48 hours after plating. Cellviability was measured after different times of exposure toCDDP or doxorubicin. Cell density was evaluated by thecrystal violet method.50 Briefly, cells were fixed with 1%glutaraldehyde for 10 minutes, washed twice in PBS, andstained with 1.5 mL of 0.1% crystal violet solution for 30minutes. Wells were rinsed in a beaker with a slow stream ofdistilled water until the dye was washed off and left to dryovernight. Absorbance was read at 590 nm by dye uptake in10% acetic acid. For 7-day experiments, 5 3 103 cells/well wereplated 24 hours prior to being irradiated at doses ranging from1 to 8 Gy.
Immunohistochemistry for Ad protein expression
For immunohistochemistry, the different cell lines were placedon glass slides, as described previously. The antibody (Ab) M73(anti-E1A, Oncogene Science, Uniondale, NY) and monoclo-nal Ab 805 (anti-Ad capsid, Chemicon International, Te-mecula, Calif) were applied at dilutions of 1/50 and 1/100,respectively. The cells were incubated for 1 hour with theprimary Ab, followed by a biotinylated goat anti-mouse sec-ondary Ab and the streptavidin-horseradish peroxidase conju-gate. Diaminobenzidine was used as the chromogen, and slideswere counterstained with hematoxylin.
Western blot analysis of Ad E1A protein expression
Cells were collected in a buffer (50 mM tris(hydroxymethyl)aminomethane/HCl, 150 mM NaCl, 0.1% sodium deoxy-cholate, and 1% Triton X-100) with protease inhibitors (10mg/mL aprotinin and 100 mM phenylmethylsulfonyl fluoride).The protein content was determined by the Bradford assay(Bio-Rad, Richmond, Calif). A total of 50 mg of protein wasdissolved in the sample buffer, heated at 90°C for 5 minutes,and analyzed by 10% sodium dodecyl sulfate-polyacrylamidegel electrophoresis. For immunoblotting, we used the M73 Ab(Oncogene Science) to detect E1A gene products. Blots wererevealed by the enhanced chemiluminescence method (Amer-sham, Arlington Heights, Ill). Blots were analyzed using Na-tional Institutes of Health Image 1.5 software.
Analysis of caspasesPreparation of cytosols. After infection with 20 plaque-formingunits (PFU) of wt and mutant H5 dL118 Ads, cells wereallowed to sediment at 250 3 g for 10 minutes, washed twice inPBS, and resuspended in extraction buffer (50 mM tris(hy-droxymethyl)aminomethane/HCl (pH 7.6), 150 mM NaCl, 0.5mM ethylenediaminetetraacetic acid, 10 mM NaH2PO4, 10mM Na2HPO4, and 1% Nonidet P-40, supplemented immedi-ately before use with 0.4 mM Na3VO4, 1 mM phenylmethyl-sulfonyl fluoride, 10 g mL-1 aprotinin, and 10 g mL-1 leupep-tin). After a 30-minute incubation on ice, the cell lysate wascentrifuged at 20,000 3 g for 30 minutes, and the supernatantwas used as cytosolic extract.
Fluorogenic assays. The cytosolic extract (aliquots containing 1mg mL-1 of cytosolic protein estimated by the bicinchoninicacid method51) was diluted five times with the assay buffer (25
mM N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid (pH7.5), 0.1% 3-([3-cholamidopropyl]dimethylammonio)-1-pro-panesulfonate (w/v), 10% sucrose, 10 mM dithiothreitol, and0.1 mg mL-1 ovalbumin) and incubated at 37°C for 2 hourswith a 10-mM concentration of the fluorescent substrate (AC-YVAD-7-amino-4-methylcoumarin (AMC), AC-DEVD-AMC, and AC-VEID-AMC for interleukin-1b-converting en-zyme (ICE)-, CPP32-, and Mch2-like activity, respectively).The reaction was stopped by the addition of high performanceliquid chromatography buffer (water/acetonitrile, 75/25; 0.1%trifluoroacetic acid). Cleaved substrate fluorescence was deter-mined by C18 reverse-phase high performance liquid chroma-tography using fluorescence detection (338 nm excitation and455 nm emission). Control experiments (data not shown)confirmed that the release of substrate was linear with timeand with protein concentration under the conditions specified.
Tumorigenicity assays
HeLa and A431 cells were infected with H5 dL118 and wt 300at an MOI of 20. An equal amount of cells was treated withmedium as a control. After 30 minutes of infection, the treatedcells were harvested and rinsed with PBS. For each injection,1 3 106 cells in a volume of 100 mL were injected subcutane-ously into nude (nu/nu) mice aged 4–5 weeks. Five mice wereused for each treatment; they were examined every other daywith calipers to determine two perpendicular diameters. Inother experiments, 1 3 106 HeLa and A431 cells were mixed invitro with 1 3 106 293 producer cells that had been infectedpreviously with both Ads.
Inhibition of tumor growth in vivo
When the tumors reached a diameter of 2–3 mm, 100 mL of H5dL118 (2 3 1011 PFU) or of PBS alone were injected i.t. andevery other day for a total of three times. Mice were irradiatedon the following days to the injection of Ads. The tumors weremeasured every other day with calipers.
RESULTS
Immunodetection of viral proteins
Immunohistochemistry with anti-E1A (M73) and an-ti-Ad capsid (monoclonal Ab 805) Abs revealed viralprotein expression by 24 hours postinfection. The cellsinfected with the wt Ad at an MOI of 20 were positivefor both Abs, with an intensity similar to those infectedwith mutant H5 dL118.
To correlate the cellular effects with E1A proteinexpression, we performed Western blot analysis of sev-eral lysates in the days immediately following infection.As shown in Figures 1 and 2, marked E1A proteinexpression was detected at 48 hours after infection withthe recombinant H5 dL118 Ad. Nevertheless, the E1Aprotein could be detected for a period of 7 days (datanot shown). Because the effects of these Ads on malig-nant cells could be due both to viral replication andcytolytic effects and to E1A protein expression, weanalyzed the level of E1A protein in nontumorigenichuman cells (Fig 2). As can be seen, the Ad infected thecells properly, with a high content of E1A protein at24–48 hours postinfection.
556 MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS
Cancer Gene Therapy, Vol 6, No 6, 1999

Cell viability after Ad infection
Primary, nonimmortalized human fibroblasts and lym-phocytes and the monkey Vero cell line were resistant tocytolysis with the Ad H5 dL118 after 3 days of exposureat an MOI of 20. Conversely, most of the malignant cellsshowed a marked decrease in cell viability at 48 hourspostinfection. As shown in Figures 3 and 4, only thebreast carcinoma MCF7 and the uveal melanoma celllines were more resistant, although less so than thecontrol cells. Interestingly, the spontaneously trans-formed fibroblastic cell line Hs67 (wt p53) showed nodifferences in cell viability for the first 5 days, butpresented a marked decrease thereafter.
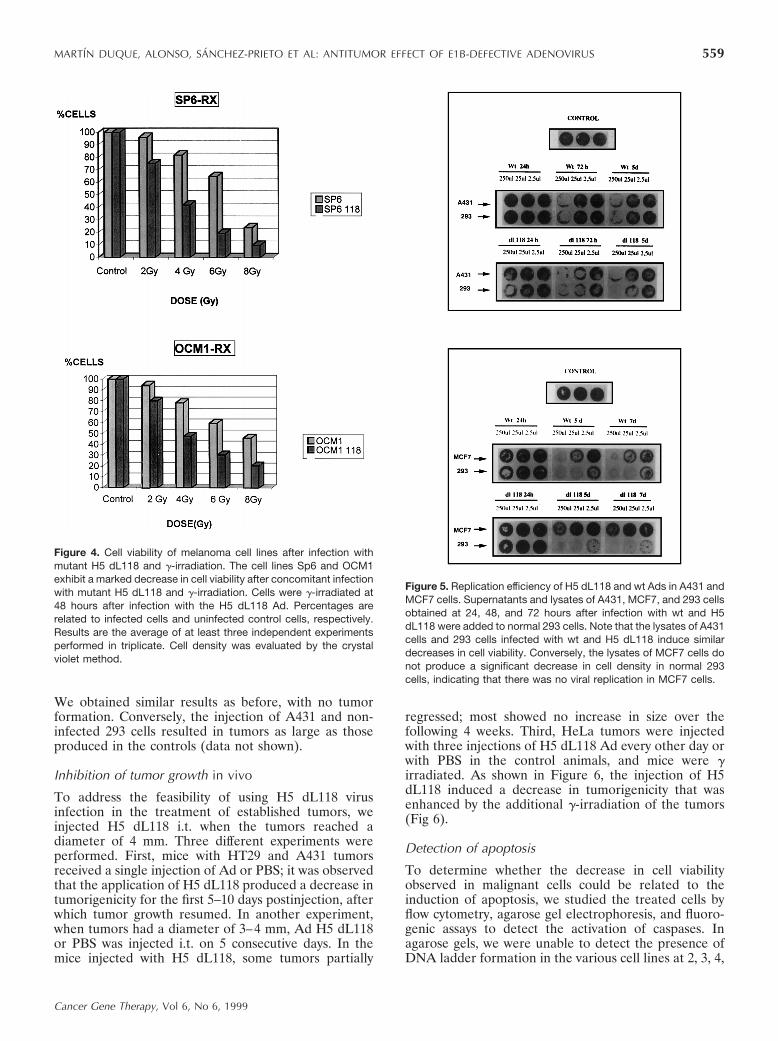
Cell viability differed very significantly in the malig-nant cells depending upon whether they had been ex-posed to wt Ad or H5 dL118 Ad infection. At similarMOIs, the cellular effects and the decrease in cellviability were significantly more marked in the cellsinfected with mutant H5 dL118 than in the cells infectedwith the wt Ad over the first days after infection. Asshown in Figure 3, after infection with the Ad H5 dL118,the percentage of cell density was 2–10 times less than inthe same cells infected with wt Ads. This cytotoxic effectof the recombinant H5 dL118 Ad was also detected inhuman malignant cells with wt p53 (exons 5–9), espe-cially in HeLa cells and uveal melanoma Sp6 cells.Moreover, the melanocytic cell lines Sp6 and OCM1infected with H5 dL118 showed an increased sensitivityto g-irradiation (Fig 4).
Viral replication
To determine whether the cellular effects were due toviral replication, we studied the viral replication by anindirect method,35 taking diluted concentrations of themedium and cell lysates of the infected human malig-nant and 293 cells several days after infection; next, weadded the medium to noninfected 293 cells. As shown inFigure 5, the cytopathic effect and the decrease in cellviability were rapidly detected at 3 days after infectionwith the supernatant of the previously infected A431(very sensitive to H5 dL118) and MCF7 (more resistantto H5 dL118) cell lines. These results confirm the factthat H5 dL118 replicates very actively in the carcinomacell line A431 and ;100 times less efficiently in MCF7cells. To study whether the titer of the H5 dL118 Adincreases in infected human cells, we took supernatantsat 24 hours, 48 hours, and 5 days and compared themwith a known virus titer of the H5 dL118. In theseexperiments, we detected a significant increase of thetiter of H5 dL118 in A431-infected cells, with titersranging from 1.5 3 105 at 24 hours postinfection to 1.5 3108 after 5 days postinfection. Conversely, we did notobserve an increase of the titer in the supernatants takenfrom MCF7 infected cells. In addition, infection with H5dL118 Ads at MOIs of 0.1, 1, and 10 showed thatmalignant cells were killed much more efficiently withlower MOIs per cell than nontumorigenic cells (data notshown).
Inhibition of tumorigenicity
To examine whether the H5 dL118 virus could inhibitthe tumorigenicity of the human malignant cell lines,nude mice were injected subcutaneously with A431,HeLa, and HT29 cells in two different experiments toinitiate tumor formation. First, we injected the malig-nant cells (2 3 106 cells) that had been infected with wtor H5 dL118 Ads at an MOI of 20 for 45 minutes. Cellstreated with medium alone were used as noninfectedcontrols. Interestingly, none of the cell lines infectedwith both the wt and mutant H5 dL118 developedtumors. Moreover, we injected A431 concomitantly withthe 293 producer cells infected previously with the Ads.
Figure 1. Expression of E1A protein in malignant cells infected withH5 dL118. Western blot analysis demonstrated E1A protein expres-sion at 24 hours postinfection in all cell lines infected with themutant H5 dL118. The highest level was detected at 48 hours. A:The melanoma cell line MKTBr. B: The epidermoid carcinoma cellline A431. Control indicates cells transfected with the E1A gene.Hours postinfection are shown in brackets. MKTBr 312 are extractsof the melanocytic cell line infected with the mutant Ad H5 dL312,which do not express E1A proteins.
MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS 557
Cancer Gene Therapy, Vol 6, No 6, 1999
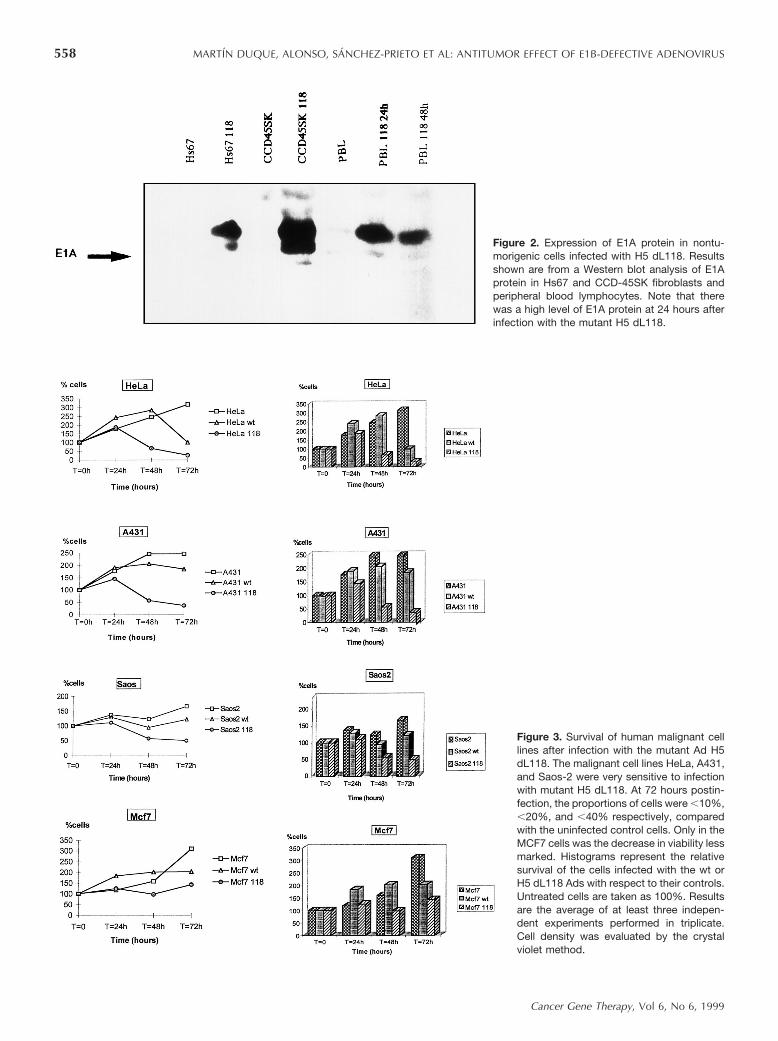
Figure 2. Expression of E1A protein in nontu-morigenic cells infected with H5 dL118. Resultsshown are from a Western blot analysis of E1Aprotein in Hs67 and CCD-45SK fibroblasts andperipheral blood lymphocytes. Note that therewas a high level of E1A protein at 24 hours afterinfection with the mutant H5 dL118.
Figure 3. Survival of human malignant celllines after infection with the mutant Ad H5dL118. The malignant cell lines HeLa, A431,and Saos-2 were very sensitive to infectionwith mutant H5 dL118. At 72 hours postin-fection, the proportions of cells were ,10%,,20%, and ,40% respectively, comparedwith the uninfected control cells. Only in theMCF7 cells was the decrease in viability lessmarked. Histograms represent the relativesurvival of the cells infected with the wt orH5 dL118 Ads with respect to their controls.Untreated cells are taken as 100%. Resultsare the average of at least three indepen-dent experiments performed in triplicate.Cell density was evaluated by the crystalviolet method.
558 MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS
Cancer Gene Therapy, Vol 6, No 6, 1999
We obtained similar results as before, with no tumorformation. Conversely, the injection of A431 and non-infected 293 cells resulted in tumors as large as thoseproduced in the controls (data not shown).
Inhibition of tumor growth in vivo
To address the feasibility of using H5 dL118 virusinfection in the treatment of established tumors, weinjected H5 dL118 i.t. when the tumors reached adiameter of 4 mm. Three different experiments wereperformed. First, mice with HT29 and A431 tumorsreceived a single injection of Ad or PBS; it was observedthat the application of H5 dL118 produced a decrease intumorigenicity for the first 5–10 days postinjection, afterwhich tumor growth resumed. In another experiment,when tumors had a diameter of 3–4 mm, Ad H5 dL118or PBS was injected i.t. on 5 consecutive days. In themice injected with H5 dL118, some tumors partially
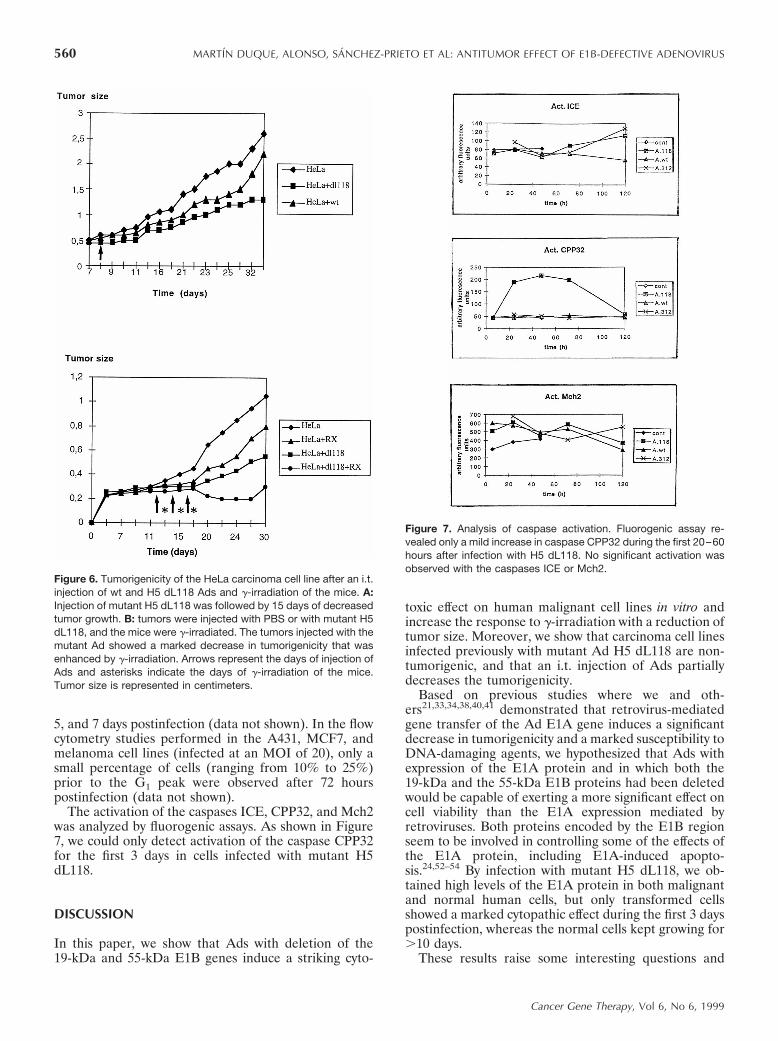
regressed; most showed no increase in size over thefollowing 4 weeks. Third, HeLa tumors were injectedwith three injections of H5 dL118 Ad every other day orwith PBS in the control animals, and mice were girradiated. As shown in Figure 6, the injection of H5dL118 induced a decrease in tumorigenicity that wasenhanced by the additional g-irradiation of the tumors(Fig 6).
Detection of apoptosis
To determine whether the decrease in cell viabilityobserved in malignant cells could be related to theinduction of apoptosis, we studied the treated cells byflow cytometry, agarose gel electrophoresis, and fluoro-genic assays to detect the activation of caspases. Inagarose gels, we were unable to detect the presence ofDNA ladder formation in the various cell lines at 2, 3, 4,
Figure 4. Cell viability of melanoma cell lines after infection withmutant H5 dL118 and g-irradiation. The cell lines Sp6 and OCM1exhibit a marked decrease in cell viability after concomitant infectionwith mutant H5 dL118 and g-irradiation. Cells were g-irradiated at48 hours after infection with the H5 dL118 Ad. Percentages arerelated to infected cells and uninfected control cells, respectively.Results are the average of at least three independent experimentsperformed in triplicate. Cell density was evaluated by the crystalviolet method.
Figure 5. Replication efficiency of H5 dL118 and wt Ads in A431 andMCF7 cells. Supernatants and lysates of A431, MCF7, and 293 cellsobtained at 24, 48, and 72 hours after infection with wt and H5dL118 were added to normal 293 cells. Note that the lysates of A431cells and 293 cells infected with wt and H5 dL118 induce similardecreases in cell viability. Conversely, the lysates of MCF7 cells donot produce a significant decrease in cell density in normal 293cells, indicating that there was no viral replication in MCF7 cells.
MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS 559
Cancer Gene Therapy, Vol 6, No 6, 1999
5, and 7 days postinfection (data not shown). In the flowcytometry studies performed in the A431, MCF7, andmelanoma cell lines (infected at an MOI of 20), only asmall percentage of cells (ranging from 10% to 25%)prior to the G1 peak were observed after 72 hourspostinfection (data not shown).
The activation of the caspases ICE, CPP32, and Mch2was analyzed by fluorogenic assays. As shown in Figure7, we could only detect activation of the caspase CPP32for the first 3 days in cells infected with mutant H5dL118.
DISCUSSION
In this paper, we show that Ads with deletion of the19-kDa and 55-kDa E1B genes induce a striking cyto-
toxic effect on human malignant cell lines in vitro andincrease the response to g-irradiation with a reduction oftumor size. Moreover, we show that carcinoma cell linesinfected previously with mutant Ad H5 dL118 are non-tumorigenic, and that an i.t. injection of Ads partiallydecreases the tumorigenicity.
Based on previous studies where we and oth-ers21,33,34,38,40,41 demonstrated that retrovirus-mediatedgene transfer of the Ad E1A gene induces a significantdecrease in tumorigenicity and a marked susceptibility toDNA-damaging agents, we hypothesized that Ads withexpression of the E1A protein and in which both the19-kDa and the 55-kDa E1B proteins had been deletedwould be capable of exerting a more significant effect oncell viability than the E1A expression mediated byretroviruses. Both proteins encoded by the E1B regionseem to be involved in controlling some of the effects ofthe E1A protein, including E1A-induced apopto-sis.24,52–54 By infection with mutant H5 dL118, we ob-tained high levels of the E1A protein in both malignantand normal human cells, but only transformed cellsshowed a marked cytopathic effect during the first 3 dayspostinfection, whereas the normal cells kept growing for.10 days.
These results raise some interesting questions and
Figure 6. Tumorigenicity of the HeLa carcinoma cell line after an i.t.injection of wt and H5 dL118 Ads and g-irradiation of the mice. A:Injection of mutant H5 dL118 was followed by 15 days of decreasedtumor growth. B: tumors were injected with PBS or with mutant H5dL118, and the mice were g-irradiated. The tumors injected with themutant Ad showed a marked decrease in tumorigenicity that wasenhanced by g-irradiation. Arrows represent the days of injection ofAds and asterisks indicate the days of g-irradiation of the mice.Tumor size is represented in centimeters.
Figure 7. Analysis of caspase activation. Fluorogenic assay re-vealed only a mild increase in caspase CPP32 during the first 20–60hours after infection with H5 dL118. No significant activation wasobserved with the caspases ICE or Mch2.
560 MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS
Cancer Gene Therapy, Vol 6, No 6, 1999

observations. First, the application of the E1B-defectiveAd, with an absence of both E1B proteins, also producesa marked and selective antitumor effect similar to thatreported previously with the 55-kDa E1B-defective AdONYX-015.44 The cytotoxic effects are also observedpredominantly in malignant cells with a mutated or wtp53 gene.44 Interestingly, the cytopathic effect of thevector ONYX-015 was similar to the wt Ads, whereasthe mutant H5 dL118 kills the malignant cells muchfaster than the wt Ad. This “faster” and selective effectcould be related to a more efficient Ad replication, to theabsence of the 19-kDa E1B protein, which is a verypotent inhibitor of apoptosis,54,55 and/or to E1A proteinexpression in the cells.
Second, infection with H5 dL118 increases the in vitroand in vivo cellular response to g-irradiation in some celllines. This effect may be related to E1A expression, asreported previously.21,39,41
Third, we detected a scarce apoptotic cell populationand moderate activation of the caspase CPP32 in H5dL118-infected cells by flow cytometry analyses andfluorogenic assays. These results would be similar tothose pointed out previously by Zhang et al.35 It isknown that Ad infection can induce both apoptotic andnecrotic types of cell death, depending upon the celltype.56 Moreover, Ad-induced cell death can occurindependently of p53 expression,42 and E1A productscan induce p53-independent and p53-dependent apo-ptosis.21,40,52,55 Previous reports indicated that p53-inde-pendent apoptotic cell death was induced only by the289R E1A protein, and that an E4 product was essentialfor E1A-induced p53-independent apoptosis.42 There-fore, after infection with Ad H5 dL118, we cannotconclude that cell death is mediated predominantly byapoptotic or necrotic mechanisms. It seems that cellsmay die by either or both pathways, because we coulddetect a small population of apoptotic cells by flowcytometry and electron microscopy as well as a moderateactivation of caspase CPP32.
The 19-kDa E1B protein may block both types of celldeath and, in fact, may facilitate virus multiplicity bypreventing premature cell death. Interestingly, it hasbeen shown that Ad type 2 19-kDa deletion mutants caninduce a necrotic type of cell death in permissive humancells and an apoptotic type in nonpermissive cells with amuch lower production of Ads.56 In fact, we detectedonly a small population of apoptotic cells after infectionwith 10–20 PFU of the mutant H5 dL118 Ad and noapoptosis whatsoever at high Ad concentrations. Impor-tantly, previous reports56 indicated that infection withthe 19-kDa-deleted Ad provoked a marked decrease incell viability in vitro that was almost as prominent as thatobserved with the 19-kDa/55-kDa mutant Ad H5dL118.56 These results indicate that the cellular effect ofthe double-mutant H5 dL118 is not only related to theselective replication of this vector in p53-deficient cells,as proposed for the 55-kDa mutant Ad ONYX-015. Infact, a comparison of the cell viability and the viralreplication of the 19-kDa/55-kDa mutant H5 dL118 withthose of the 19-kDa mutant Ad and of the 55-kDa
mutant Ad ONYX-015 seems to suggest that absence ofthe 19-kDa E1B protein may be related to the strikinglydecreased cell viability of malignant cells and to therandom DNA fragmentation in human cell lines. Nev-ertheless, the higher cytotoxicity of H5 dL118 whencompared with that reported for the 19-kDa mutant Admay be due to the concomitant absence of the 55-kDaE1B protein. It has been proposed recently that thepathways driving cells to apoptosis and necrosis are notas independent as previously supposed.57 The availabil-ity of apoptogenic proteases, activated following mito-chondrial permeability transition, could dictate themode of cell death.58 In fact, after infection with thecompletely E1B-defective Ad H5 dL118, we could onlydetect activation of the CPP32 caspase and mixed cellpopulations of necrotic and apoptotic cells. The fact thatwe infected proliferating cells could explain the absenceof E1A-mediated apoptosis.59
The fact that infection with defective Ads sensitizesmalignant cells to g-irradiation is the most relevantresult obtained in vivo. This result may be due to theexpression of the E1A protein and the absence of the19-kDa E1B protein, as reported previously in retrovi-ral-mediated systems. The enhanced response to radio-therapy could be the basis for future clinical trials,combining injection of these completely E1B-defectiveAds and g-irradiation. Importantly, no significant sideeffects could be detected in mice after repeated injec-tions of mutant H5 dL118.
The prospects for gene therapy with defective Ads arecontroversial. Most of the clinical trials in progress focuson the application of E1-Ad carrying the wt p53 gene,the p16 gene, or suicide genes.10,12,14 An overexpressionof p53 may induce apoptosis in carcinomas with mutatedor wt p53 protein, and preliminary clinical data point toan in vivo antitumor effect of these Ads.14,18
With the completely E1B-defective Ads, which com-bine the selective viral replication of the 55-kDa E1BAd, the potent cytopathic effect of the 19-kDa mutantAd, and high-level E1A protein expression, a significantantitumor response may be expected in human tumors.The concomitant g-irradiation of those tumors may be anovel approach in the treatment of human cancers.
ACKNOWLEDGMENTS
We thank H.S. Ginsberg for generously providing the H5dL118 and H5 dL312 adenoviruses, Maria Antonia Saornil forproviding the melanoma cell lines MKTBr, Sp6, and OCM1,Jesus Romero for his help in the g-irradiation experiments,Eloy Blanco for his assistance with the photographs, andMartha Messman for preparing the manuscript. This work wassupported by grants from the Fondo de Investigaciones Sani-tarias del Ministerio de Sanidad de Espana (FIS 94/395 and96/2174) and by Grants SAF95-1547 and SAF95-0818 from theComision Interministerial de Ciencia y Tecnologia. C.A. wassupported by grants PB97-0552 and CS96-066. R.S.-P. was apostdoctoral fellow under the RICH foundation. P.M.D.was a predoctoral fellow under the Fondo de InvestigacionesSanitarias.
MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS 561
Cancer Gene Therapy, Vol 6, No 6, 1999

REFERENCES
1. Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–248.
2. Ramon y Cajal S, Suster S, Halaban R, Filvaroff E, DottoGP. Induction of different morphological patterns of ma-lignant melanoma in mice after transformation with bFGF,H-ras, myc, neu, and E1a oncogenes. Am J Pathol. 1991;138:349–358.
3. Woodruff MFA. Cellular heterogeneity in tumors. Br JCancer. 1983;47:589–594.
4. Freeman SM, Whartenby KA, Freeman JL, et al. In situuse of suicide genes for cancer therapy. Semin Oncol.1996;23:31–45.
5. Hinds PW, Weinberg RA. Tumor suppressor genes. CurrOpin Genet Dev. 1994;4:135–141.
6. Hunter T. Oncoprotein networks. Cell. 1997;88:333–346.7. Kinzler KW, Vogelstein V. Gatekeepers and caretakers.
Nature. 1997;386:761–763.8. Desmaze C, Brizard F, Turc-Carel C, et al. Multiple
chromosomal mechanisms generate an EWS/FLI1 or anEWS/ERG fusion gene in Ewing tumors. Cancer GenetCytogenet. 1997;97:12–19.
9. Rabbits TH. The clinical significance of fusion oncogenesin cancer. N Engl J Med. 1998;338:192–194.
10. Anderson WF. Human gene therapy. Science. 1992;256:808–813.
11. Barinaga M. Designing therapies that target tumor bloodvessels. Science. 1997;275:482–484.
12. Blau HM, Springer ML. Gene therapy: a novel form ofdrug delivery. N Engl J Med. 1995;333:1204–1207.
13. Cournoyer D, Caskey CT. Gene therapy of the immunesystem. Annu Rev Immunol. 1993;11:297–329.
14. Carbone DP, Minna JD. In vivo gene therapy of humanlung cancer using wild-type p53 delivered by retrovirus.J Natl Cancer Inst. 1994;86:143–144.
15. Fujiwara T, Grimm EA, Mukhopadhyay T, et al. Inductionof chemosensitivity in human lung cancer cells in vivo byadenovirus-mediated transfer of the wild-type p53 gene.Cancer Res. 1994;54:2287–2291.
16. Huang HJS, Yee JK, Shew JY, et al. Suppression of theneoplastic phenotype by replacement of the RB gene inhuman cancer cells. Science. 1988;242:1563–1566.
17. Levine AJ. p53, the cellular gatekeeper for growth anddivision. Cell. 1997;88:323–331.
18. Liu TJ, Zhang WW, Taylor DL, et al. Growth suppressionof human head and neck cells by introduction of awild-type p53 gene via a recombinant adenovirus. CancerRes. 1994;54:3662–3667.
19. Molten FL. Drug sensitivity (suicide) genes for selectivecancer chemotherapy. Cancer Gene Ther. 1994;1:279–287.
21. Sanchez-Prieto R, Quintanilla M, Cano A, et al. Carci-noma cell lines become sensitive to DNA-damaging agentsby the expression of the adenovirus E1A gene. Oncogene.1996;13:1083–1092.
22. Bayley ST, Mymryk JS. Adenovirus E1A proteins andtransformation. Int. J Oncol. 1994;5:425–444.
23. Dyson N, Harlow E. Adenovirus E1A targets key regula-tors of cell proliferation. Cancer Surv. 1992;12:161–195.
24. Mymryk JS. Tumor suppressive properties of the adenovi-rus 5 E1A oncogene. Oncogene. 1996;13:1581–1589.
25. Datto MB, Hu PP, Kowalik TF, Yingling J, Wang XF. Theviral oncoprotein E1A blocks transforming growth factor
b-mediated induction of p21/WAF1/Cip1 and p15/INK4B.Mol Cell Biol. 1997;17:2030–2037.
26. Frisch SM, Francis H. Disruption of epithelial cell matrixinteractions induces apoptosis. J Cell Biol. 1994;124:619–626.
27. Liu F, Green MR. Promoter targeting by adenovirus E1Athrough interaction with different cellular DNA-bindingdomains. Nature. 1994;368:520–525.
28. Mal A, Poon RY, Howe PH, Toyoshima H, Hunter T,Harter ML. Inactivation of p27Kip1 by the viral E1Aoncoprotein in TGFb-treated cells. Nature. 1996;380:262–265.
29. Somasundaram K, El-Deiry WS. Inhibition of p53-medi-ated transactivation and cell cycle arrest by E1A throughits p300/CBP-interacting region. Oncogene. 1997;14:1047–1057.
30. Evan G. Cancer, a matter of life and cell death. Int JCancer. 1997;71:709–711.
31. Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phos-phatase as a target of c-myc. Nature. 1996;382:511–517.
32. Qin XQ, Livingston DM, Kaelin WG, Adams PD. Dereg-ulated transcription factor E2F-1 expression leads S-phaseentry and p53-mediated apoptosis. Proc Natl Acad SciUSA. 1994;91:10918–10922.
33. Frisch SM. Antioncogenic effect of adenovirus E1A inhuman tumor cells. Proc Natl Acad Sci USA. 1991;88:9077–9081.
34. Sanchez-Prieto R, Quintanilla M, Martın P, et al. In vivotumor suppressor effect of retrovirus gene transfer of theadenovirus E1a gene. Cancer Gene Ther. 1998;5:215–224.
35. Zhang Y, Yu D, Xia W, Hung MC. HER-2/neu-targetingcancer therapy via adenovirus-mediated E1A delivery inan animal model. Oncogene. 1995;10:1947–1954.
36. Chang JY, Xia W, Shao R, et al. The tumor suppressionactivity of E1A in HER-2/neu-overexpressing breast can-cer. Oncogene. 1997;14:561–568.
37. Frisch SM. The epithelial cell default-phenotype hypoth-esis and its implications for cancer. Bioassays. 1997;19:705–709.
38. Frisch SM, Reich R, Collier IE, Genrich T, Martin G,Goldberg GI. Adenovirus E1A represses protease geneexpression and inhibits metastasis of human cells. Onco-gene. 1997;5:75–83.
39. Marcehetti E, Romero J, Sanchez R, Vargas JA, Lacal JC,Ramon y Cajal S. Oncogenes and cellular sensitivity toradiotherapy: a study on murine keratinocytes transformedby v-H-ras, v-myc, neu, adenovirus E1a, and p53 mutant.Int J Oncol. 1994;5:611–618.
40. Sanchez R, Lleonart M, Ramon y Cajal S. Lack ofcorrelation between p53 protein level and sensitivity toDNA-damaging agents in keratinocytes carrying adenovi-rus E1A mutants. Oncogene. 1995;11:675–682.
41. Sanchez-Prieto R, Vargas JA, Carnero A, et al. Modula-tion of cellular chemoresistance in keratinocytes by acti-vation of different oncogenes. Int J Cancer. 1995;60:235–243.
42. Marcellus RC, Teodoro JG, Wu T, et al. Adenovirus type5 early region 4 is responsible for E1A-induced p53-independent apoptosis. J Virol. 1996;70:6207–6215.
43. Bischoff JR, Kirn DH, Williams A, et al. An adenovirusmutant that replicates selectively in p53-deficient humantumor cells. Science. 1996;274:373–376.
44. Heise C, Sampson-Johannes A, Williams A, et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-
562 MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS
Cancer Gene Therapy, Vol 6, No 6, 1999

specific cytolysis and antitumoral efficacy that can beaugmented by standard chemotherapeutic agents. NatMed. 1997;3:639–645.
45. Martin P, Alonso C, Sanchez-Prieto R, Quintanilla M,Ramon y Cajal S. Antitumoral effect of E1B-defectiveadenoviruses in human malignant cells. Gene Ther. 1998;5:286–287.
46. Yew PR, Berk AJ. Inhibition of p53 transactivation re-quired for transformation by adenovirus early 1B protein.Nature. 1992;357:82–85.
47. Babiss LE, Fisher PB, Ginsberg HS. Effect on transforma-tion of mutations in the early region 1b-encoded 21 and55-kilodalton proteins of adenovirus 5. J Virol. 1984;52:389–395.
48. Graham FL, Smiley J, Russell WC, Nairn R. Characteris-tics of a human cell line transformed by DNA from humanadenovirus type 5. J Gen Virol. 1977;36:59–72.
49. Graham FL, Prevec L. Manipulation of adenovirus vec-tors. In: Murray EJ, ed. Methods in Molecular Biology:Gene transfer and Expression Protocols. Clifton, New Jer-sey: The Human Press; 1991:109–128.
50. Gillies RJ, Didier N, Denton M. Determination of cellnumber in monolayer cultures. Anal Biochem. 1986;159:109–113.
51. Smith PK, Krohn RI, Hermanson GT, et al. Measurementof protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85.
52. Teodoro JG, Shore GC, Branton PE. Adenovirus E1Aproteins induce apoptosis by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:467–474.
53. Teodoro JG, Branton PE. Regulation of p53-dependentapoptosis, transcriptional repression, and cell transforma-tion by phosphorylation of the 55-kilodalton E1B proteinof human adenovirus type 5. J Virol. 1997;71:3620–3627.
54. White E, Cipriani R, Sabattini P, Danton A. AdenovirusE1B 19-kilodalton protein overcomes the cytotoxicity ofE1A proteins. J Virol. 1991;65:2968–2978.
55. Chiou S, White E. p300 binding by E1A cosegregates withp53 induction but is dispensable for apoptosis. J Virol.1997;71:3515–3525.
56. Subramanian T, Tarodi B, Chinnadurai G. p53-indepen-dent apoptotic and necrotic cell deaths induced by adeno-virus infection: suppression by E1B 19K and Bcl-2 pro-teins. Cell Growth Differ. 1995;6:131–137.
57. Schwartz LM, Smith SW, Jones MEE, Osborne BA. Do allprogrammed cell deaths occur via apoptosis? Proc NatlAcad Sci USA. 1993;90:980–984.
58. Hirsch T, Marchetti P, Susin Santos A, et al. The apopto-sis-necrosis paradox: apoptogenic proteases activated aftermitochondrial permeability transition determine the modeof cell death. Oncogene. 1997;15:1573–1581.
59. Mymryk JS, Shire K, Bayley ST. Induction of apoptosis byadenovirus type 5 E1A in rat cells requires a proliferationblock. Oncogene. 1994;9:1187–1193.
MARTIN DUQUE, ALONSO, SANCHEZ-PRIETO ET AL: ANTITUMOR EFFECT OF E1B-DEFECTIVE ADENOVIRUS 563
Cancer Gene Therapy, Vol 6, No 6, 1999





























