Embed Size (px)
Citation preview

of December 18, 2013.This information is current as
Enhancer Activity′ 3κTransactivation and IgProtein Acetylation Regulates Both PU.1
Michael L. AtchisonYuchen Bai, Lakshmi Srinivasan, Leslie Perkins and
http://www.jimmunol.org/content/175/8/51602005; 175:5160-5169; ;J Immunol
Referenceshttp://www.jimmunol.org/content/175/8/5160.full#ref-list-1
, 33 of which you can access for free at: cites 56 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 18, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from

Protein Acetylation Regulates Both PU.1 Transactivation andIg� 3� Enhancer Activity1
Yuchen Bai,2 Lakshmi Srinivasan, Leslie Perkins, and Michael L. Atchison3
Ig� gene expression and chromatin structure change during B cell development. At the pre-B cell stage, the locus is relativelyhypoacetylated on histone H3, whereas it is hyperacetylated at the plasma cell stage. We find in this study that the histonedeacetylase inhibitor, trichostatin A (TSA) stimulated 3� enhancer activity through the PU.1 binding site. TSA also stimulated PU.1transactivation potential. PU.1 activity was increased by the coactivator acetyltransferase protein, p300, and p300 physicallyinteracted with PU.1 residues 7–30. PU.1 served as a substrate for p300 and was acetylated on lysine residues 170, 171, 206, and208. Mutation of PU.1 lysines 170 and 171 did not affect PU.1 DNA binding, but did lower the ability of PU.1 to activatetranscription in association with p300. Lysine 170 was acetylated in pre-B cells and plasmacytoma cells, but TSA treatment didnot stimulate PU.1 acetylation at this residue arguing that a second mechanism can stimulate 3� enhancer activity. Using chromatinimmunoprecipitation assays we found that TSA caused preferential acetylation of histone H3 at the 3� enhancer. The relevanceof these studies for PU.1 function in transcription and hemopoietic development is discussed. The Journal of Immunology, 2005,175: 5160–5169.
I mmunoglobulin gene rearrangement is an ordered process inwhich H chain gene rearrangement precedes L chain generearrangement (1). H chain DJ rearrangement occurs in pro-B
cells, followed by VDJ rearrangement, whereas L chain genes un-dergo VJ rearrangement at the pre-B cell stage (1). B cells aredefined as having both H and L chain genes rearranged. The sameenzymatic machinery and DNA recognition sequences are used forrearrangement of H and L chain genes, and therefore, the mecha-nism of temporal specificity between the loci remains unclear.Transcriptional activation or chromatin accessibility models havebeen proposed to account for these differences (2).
The Ig� locus is controlled by two stage-specific enhancers, theintron enhancer, and the 3� enhancer (3). Both enhancers are in-active at the pro-B and pre-B cell stages, but are functional at theB cell and plasma cell stages (4–6). Both enhancers also contrib-ute to the ability to rearrange the � locus in vivo (3, 7). Coincidentwith the development of enhancer activity and subsequent tran-scription, DNA at the locus changes from a heavily methylated,DNase I-resistant structure, to a hypomethylated, DNase I-sensi-tive structure (8–13). The factors directly responsible for thesechanges are unknown, but presumably include the transcriptionfactors that bind to the Ig� enhancers.
A number of studies explored chromatin accessibility at variousdevelopmental stages (3, 7, 10, 14). These studies revealed somesite-specific changes in protein occupancy during development.For instance, hypersensitivity at certain sites in the 3� enhancer is
increased in pre-B cells compared with pro-B cells (14), and thesechanges may contribute to control of � locus expression. However,simple protein accessibility to the � locus is not a sufficient mech-anism for controlling transcription or recombination in pro-B cells,because in vivo footprinting studies showed proteins bound at the� enhancers before the onset of transcriptional or recombinationalactivity (14). Instead, increased activity of the 3� enhancer at laterB cell stages must be controlled by the composition of proteinsbound to the regulatory elements or to post-translational modifi-cations of histones or regulatory proteins.
An important motif in the 3� enhancer is the PU.1/Pip (IFNregulatory factor-4) composite element. This motif is dependentupon transcription factor PU.1, which is necessary for the recruit-ment of Pip to DNA (15–18). PU.1 is an erythroblast transforma-tion-specific (ETS)4 domain transcription factor necessary for de-velopment of multiple hemopoietic lineages (19–28). PU.1homozygous mutants fail to develop lymphoid, myeloid, and gran-ulocyte lineages and die in late gestation (27). Levels of PU.1 areimplicated in regulating the lymphoid and myeloid cell fates (23),and PU.1 is down-regulated during erythrogenesis (26, 29, 30).Reduced levels of PU.1 also contribute to acute myeloid leukemia(31). PU.1 can physically interact with CREB binding protein(CBP) and elevated PU.1 expression can lead to changes in globalhistone acetylation levels in vivo (32, 33). PU.1 is also implicatedin controlling chromatin structure at the Ig H chain enhancer (34).Therefore, PU.1 may influence both global histone acetylation pat-terns as well as local chromatin structure.
The factors that control � locus chromatin structure are pres-ently unknown, although Pax5 has been suggested to play an im-portant role (35). Previously we showed that H3 acetylation at the3� enhancer is increased greatly in pre-B cells by treatment withthe histone deacetylase inhibitor, trichostatin A (36). We find inthis study that trichostatin A (TSA) treatment increased 3� en-hancer activity, and this increase was largely through the PU.1/Pipcomposite element. The PU.1 transactivation potential was also
Department of Animal Biology, University of Pennsylvania School of VeterinaryMedicine, Philadelphia, PA 19104
Received for publication January 19, 2005. Accepted for publication August 11, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grant RO1GM42415 (toM.L.A.).2 Current address: 145 King of Prussia Road, Wyeth-Ayrest Research Laboratories,Radnor, PA 19087.3 Address correspondence and reprint requests to Dr. Michael Atchison, University ofPennsylvania School of Veterinary Medicine, 3800 Spruce Street, Philadelphia, PA19104. E-mail address: [email protected]
4 Abbreviations used in this paper: ETS, erythroblast transformation specific; CBP,CREB binding protein; TSA, trichostatin A; HAT, histone acetyltransferase; ChIP,chromatin immunoprecipitation; P/CAF, p300/CBP-associated factor.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

increased in the presence of TSA or when cotransfected with thehistone acetyltransferase (HAT) protein, p300. We find that PU.1physically interacts with p300 and is a substrate for p300 acety-lation with acetylation sites at lysines 170, 171, 206, and 208. Wefound that PU.1 is acetylated in vivo, and this acetylation may beimportant for PU.1 transactivation with p300. However, TSA treat-ment did not increase PU.1 lysine 170 acetylation, arguing thateither other PU.1 lysines are targeted, or a second mechanism isneeded for TSA induction of enhancer activity. Using chromatinimmunoprecipitation methods we found that TSA causes prefer-ential acetylation of histone H3 at the 3� enhancer. Our resultsprovide evidence of two mechanisms for controlling 3� enhanceractivity.
Materials and MethodsChromatin immunoprecipitation (ChIP) assays
S194 cells (3.4 � 108) and 11 � 108 3-1 cells were fixed with 1.1% (v/v)formaldehyde, 100 mM NaCl, 0.5 mM EGTA, and 50 mM Tris-HCl (pH8.0) in growth medium at 37°C for 10 min, then at 4°C for 50 min. Form-aldehyde was quenched by adding 0.05 vol 2.5 M glycine. Fixed cells werewashed with PBS, incubated for 15 min in 15 ml of 10 mM Tris-HCl (pH8.0), 10 mM EDTA, 0.5 mM EGTA, and 0.25% (v/v) Triton X-100, fol-lowed by 15 min in 15 ml of 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.5mM EGTA, and 200 mM NaCl, and finally sonicated in 1 ml of 10 mMTris-HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 1% (w/v) SDS plus 1mM PMSF and a protease inhibitor mixture (1/100; P8340; Sigma-Aldrich). After sonication, cell debris was removed by centrifugation.Chromatin extracts were diluted to 6 OD 260 U/ml in IP buffer (140 mMNaCl, 1% (w/v) Triton X-100, 0.1% (w/v) sodium deoxycholate, 1 mMPMSF, 100 �g/ml yeast tRNA, and 100 �g/ml BSA); preincubated for 1 hat 4°C with 10 �l/ml 50% (v/v) protein A-agarose (Invitrogen Life Tech-nologies); reconstituted in PBS, and washed several times in IP buffer.Aliquots (600 �l) were incubated with 24 �g of preimmune, anti-acetylH3,
anti-acetyl H4 (Upstate Biotechnology), or p300 (Santa Cruz Biotechnol-ogy) Abs and incubated overnight at 4°C. Complexes were separated intobound and unbound complexes with protein A-agarose and cross-linkswere reversed by treatment at 65°C overnight. After treatment with RNaseA and proteinase K, samples were extracted first with phenol/chloroform,then with chloroform, and precipitated with 2 vol of ethanol and 10 �g ofglycogen (Roche). PCR was performed on 5-, 10-, and 20-ng DNA aliquotsat 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s usingprimers specific for the Ig� 3� enhancer, the intron enhancer, or the C�constant region (see Table I). Samples were dot-blotted to Hybond-N�
(Amersham Biosciences) and hybridized to a 1.1-kb EcoRI-SacI DNAfragment containing the 3� enhancer, a 473-bp AluI fragment containing theintron enhancer, or a 1.7-kb HindIII-BglII fragment containing the constantregion.
Plasmids and recombinant proteins
Various full-length or mutant PU.1 plasmids were originally generatedfrom a pBluescript KS� PU.1 plasmid. PU.1 site-directed mutations weregenerated by overlap extension PCR (37) with primers containing the mu-tated sequences (Table I), followed by digestion with appropriate restric-tion enzymes. PCRs contained 30 ng of template DNA; 300 ng of eachprimer, 50 mM each of dATP, dCTP, dGTP, and dTTP; 10 mM Tris-HCl(pH 8.3); 50 mM KCl; 1.5 mM MgCl2; 0.001% (w/v) gelatin (PerkinElmer/Cetus); 2.5 U of AmpliTaq polymerase; and 2.5 U of Taq extender (Strat-agene). The amplification cycle consisted of 35 cycles of 1 min at 95°C, 1min at 50°C, and 2 min at 72°C, followed by one cycle of extension for 4min at 72°C.
PCR products were either subcloned into KS� pBluescript or directlycloned into pGEX-2T bacterial expression vector in-frame with the GST cod-ing region. The pGEX plasmid PU.1 �201–272 was generated from pGEX-PU.1 by digestion with KpnI, followed by religation. The pGEX plasmidsPU.1 �223–272 and �243–272 were generated by PCR with 3� primers p222and p242, respectively (Table I). Plasmids PU.1 �7–30, �30–100, �119–160,and �245–272 were described by Pongubala et al. (15); �201–272 and �255–272 were described by Perkel and Atchison (38).
Table I. Primers used for construction of mutant plasmids or for ChIP PCR
Primers Sequences
5� PU.1 K169R GGGGAGACAGGCAGCCGCAAAAAGATTCGCCTG3� PU.1 K169R CAGGCGAATCTTTTTGCGGCTGCCTGTCTCCCC5� PU.1 K170R GAGACAGGCAGCAAGCGCAAGATTCGCCTGTAC3� PU.1 K170R GTACAGGCGAATCTTGCGCTTGCTGCCTGTCTC5� PU.1 K171R ACAGGCAGCAAGAAACGCATTCGCCTGTACCAG3� PU.1 K171R CTGGTACAGGCGAATGCGTTTCTTGCTGCCTGT5� PU.1 K169,170R GGGGAGACAGGCAGCCGCCGCAAGATTCGCCTGTACCAG3� PU.1 K169,170R CTGGTACAGGCGAATCTTGCGGCGGCTGCCTGTCTCCCC5� PU.1 K169,171R GGGGAGACAGGCAGCCGCAAACGCATTCGCCTGTACCAG3� PU.1 K169,171R CTGGTACAGGCGAATGCGTTTGCGGCTGCCTGTCTCCCC5� PU.1 K170,171R GGGGAGACAGGCAGCAAGCGCCGCATTCGCCTGTACCAG3� PU.1 K170,171R CTGGTACAGGCGAATGCGGCGCTTGCTGCCTGTCTCCCC5� PU.1 K169,170,171R GGGGAGACAGGCAGCCGCCGCCGCATTCGCCTGTACCAG3� PU.1 K169,170,171R CTGGTACAGGCGAATGCGGCGGCGGCTGCCTGTCTCCCC5� PU.1 K196R TGGTGGGTGGACCGCGACAAAGGTACCTTCCAG3� PU.1 K196R CTGGAAGGTACCTTTGTCGCGGTCCACCCACCA5� PU.1 K198R TGGTGGGTGGACAAGGACCGCGGTACCTTCCAG3� PU.1 K198R CTGGAAGGTACCGCGGTCCTTGTCCACCCACCA5� PU.1 K196,198R TGGTGGGTGGACCGCGACCGCGGTACCTTCCAG3� PU.1 K196,198R CTGGAAGGTACCGCGGTCGCGGTCCACCCACCA5� PU.1 K206R TTCCAGTTCTCGTCCCGCCACAAGGAGGCGCTG3� PU.1 K206R CAGCGCCTCCTTGTGGCGGGACGAGAACTGGAA5� PU.1 K208R TTCTCGTCCAAGCACCGCGAGGCGCTGGCGCAC3� PU.1 K208R GTGCGCCAGCGCCTCGCGGTGCTTGGACGAGAA5� PU.1 K206,208R CAGTTCTCGTCCCGCCACCGCGAGGCGCTGGCG3� PU.1 K206,208R CGCCAGCGCCTCGCGGTGGCGGGACGAGAACTG3� PU.1 p222 CTAGATATCTTAGCGGTTGCCCTTCTGGATGCCCCA3� PU.1 p242 CTAGATATCTTACACCTCGCCTGTCTTGCCGTAGTT3� Enhancer F GCACAGAGTACCCACCCATATCTC;3� Enhancer R CTTGAAAGGGTGTGGAGTGCACCAIntron enhancer F CCTCTGTCACCCAAGAGTTGGCATIntron enhancer R AGCCAGGGTCTGTATTTGGGTGTC� constant F CCCACCATCCAGTGAGCAGTTAAC� constant R GAGCTGGTGGTGGCGTCTCAGGAC
5161The Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

Plasmid CMV-p300 was a gift from Dr. P. Liberman (Wistar Institute,Philadelphia, PA). CMV-PU.1 was described by Pongubala et al. (16).(Oligo2)4TKCAT, (Oligo5)4TKCAT, (Oligo7)4TKCAT, and GALTKCATwere described by Pongubala and Atchison (4).
In vitro acetylation assays
The HAT domain of p300 or p300/CBP-associated factor (P/CAF) fused toGST were expressed in Escherichia coli (BL21), bound to glutathione-Sepharose beads, and eluted with buffer containing 25 mM glutathione.Eluted proteins were further purified on PD-10 columns (SephadexG-25M; Amersham Biosciences), and eluted with buffer containing 20 mMTris-HCl (pH 8.0), 0.5 mM EDTA, 100 mM KCl, 20% glycerol, 0.5 mMDTT, and 1 mM PMSF. Before storage at �80°C proteins were concen-trated by Centricon (YW30; Amicon). Substrate proteins (GST fusions orchicken erythrocyte histones; Sigma-Aldrich) were incubated with 0.25�Ci of [3H]acetyl-coenzyme A (Amersham Biosciences) and 0.2 �g ofpurified enzyme in 30 �l of acetylation buffer containing 50 mM sodiumbutyrate. Reaction mixtures were incubated at 30°C for 30 min, stopped byaddition of Laemmli buffer, and resolved by electrophoresis on 10% SDS-polyacrylamide gels. Gels were stained with Coomassie Blue to verifyprotein quantity, then subjected to autoradiography to evaluate acetylationactivity.
EMSAs
Nuclear extracts were prepared from S194 plasmacytoma cells essentiallyas previously described (39), and binding reactions were performed aspreviously described (15). Briefly, EMSA was performed with 0.1 ng oflabeled DNA probe (10,000 cpm) in a 20-�l reaction mixture containing 2�g poly(dI-dC), 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1mM DTT, 5% glycerol, and 8 �g of nuclear extract or protein made by invitro translation. Proteins made by in vitro transcription and translationwere prepared using a coupled transcription and translation kit (Promega)with T3 or T7 RNA polymerase. The PU.1-Pip-binding site from the �3�enhancer used as probe is CTTTGAGGAACTGAAAACAGAACCT(Oligo 5). Samples were electrophoresed on 4% polyacrylamide gels in 6.7mM Tris-HCl (pH 7.5), 3.3 mM sodium acetate, and 1 mM EDTA.
Cell culture and transfections
NIH-3T3 cells were grown in DMEM supplemented with 10% FCS andtransfected by the calcium phosphate coprecipitation method (40). Cellswere harvested 48 h after transfection. Each transfection contained 1 �g ofa �-galactosidase-expressing plasmid to normalize transfection efficien-cies, 3–5 �g of reporter plasmid, and 3–5 �g of either empty expressionvector (CMV) or CMV effectors. Transfection efficiencies were normalizedusing �-galactosidase activity, and CAT assays, followed by TLC, wereperformed as described by Gorman et al. (41). The percent CAT activitywas calculated by scintillation counting of the acetylated product and sub-strate spots. In each case, transfections were performed three to five times.S194 and 3-1 cells were grown and transfected as previously described (4).TSA treatment was performed for 24 h at a concentration of 33 nM.
Preparation of GST fusion proteins
GST fusion proteins were prepared essentially as previously described(42). Ten-milliliter cultures of E. coli BL21 cells containing the appropriateplasmid were inoculated overnight. The following morning, cells were di-luted 20 times and incubated for another 3 h. Cultures were then inducedwith 0.25 mM isopropyl �-D-thiogalactoside for 2–3 h at 30°C. Cells werespun down and subjected to a freeze-thaw cycle, followed by resuspensionin 20 mM Tris-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, 0.5% NonidetP-40 (NETN) containing lysozyme, PMSF, leupeptin, and aprotinin. Afterincubation for 20 min on ice, cells received five 15-s sonication bursts. Thesuspension was centrifuged at 10,000 rpm in a Sorvall RTH-750 rotor for10 min, and the remaining supernatant was incubated at 4°C with 0.5 ml ofglutathione beads (50% slurry) for 2 h. The beads were spun down andwashed three times with 5 ml of NETN before storage at 4°C.
GST chromatography
Reactions consisted of GST fusion protein or an equivalent amount of GSTprotein alone incubated with 5–15 �l of 35S-labeled protein prepared by invitro transcription and translation in a 100-�l reaction containing NETN.Samples were rocked for 2 h at 4°C and washed at least five times with 450�l of NETN. Samples were electrophoresed on 10% SDS-polyacrylamidegels for 1 h at 160 V, dried, and subjected to autoradiography.
Coimmunoprecipitation assays
NIH-3T3 cells were transfected with the appropriate plasmids and lysed inbuffer containing 20 mM Tris (pH 7.5), 100 mM NaCl, 0.5% Nonidet P-40,0.5 mM EDTA, and protease inhibitors. After removal of cell debris bycentrifugation, lysates were immunoprecipitated with anti-FLAG M2 af-finity agarose (Sigma-Aldrich) for 3 h at 4°C. Proteins bound to the affinitybeads were washed extensively with lysis buffer, separated by SDS-PAGE,and then subjected to a Western blot procedure with anti-PU.1 and anti-p300 (Santa Cruz Biotechnology) Abs.
Generation of PU.1 acetyl lysine 170-specific antiserum
Rabbit antisera was prepared by Research Genetics against keyhole limpethemocyanin-conjugated PU.1 peptide CGFTGSK-acetylK-KIRLY. The re-sulting antiserum was affinity purified, then cross-absorbed against the non-acetylated peptide. The specificity of the serum for acetylated PU.1 wastested by Western blot analysis with acetylated and unacetylated PU.1 (seeFig. 5C).
Metabolic labeling experiments
S194 cells were labeled for 90 min with [3H]acetate (16 Ci/mmol; ICNBiomedical) in the presence of 33 nM TSA. Cells were lysed in 2 ml of 350mM NaCl, 50 mM Tris-HCl (pH 7.5), 0.5% IGEPAL (Rhone-Poulenc), 1mM EDTA, 0.5 mM DTT, 10 mM sodium butyrate, 0.2 mM PMSF, and 1�g/ml each of aprotinin, pepstatin, and leupeptin. The sample was centri-fuged at 10,000 � g for 5 min and diluted to 150 mM NaCl with 1 mMEDTA, 0.5 mM DTT, 10 mM sodium butyrate, and protease inhibitors.Equal quantities of cell lysate were precipitated with either preimmune oranti-PU.1 Abs coupled to protein A-agarose. Precipitates were washed fivetimes with 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.5mM DTT, 10 mM sodium butyrate, and protease inhibitors. Samples werefractionated by SDS-PAGE, and the gel was treated with Autofluor (Am-ersham Biosciences), dried, and exposed to x-ray film.
ResultsTSA induces 3� enhancer activity and PU.1 transactivation
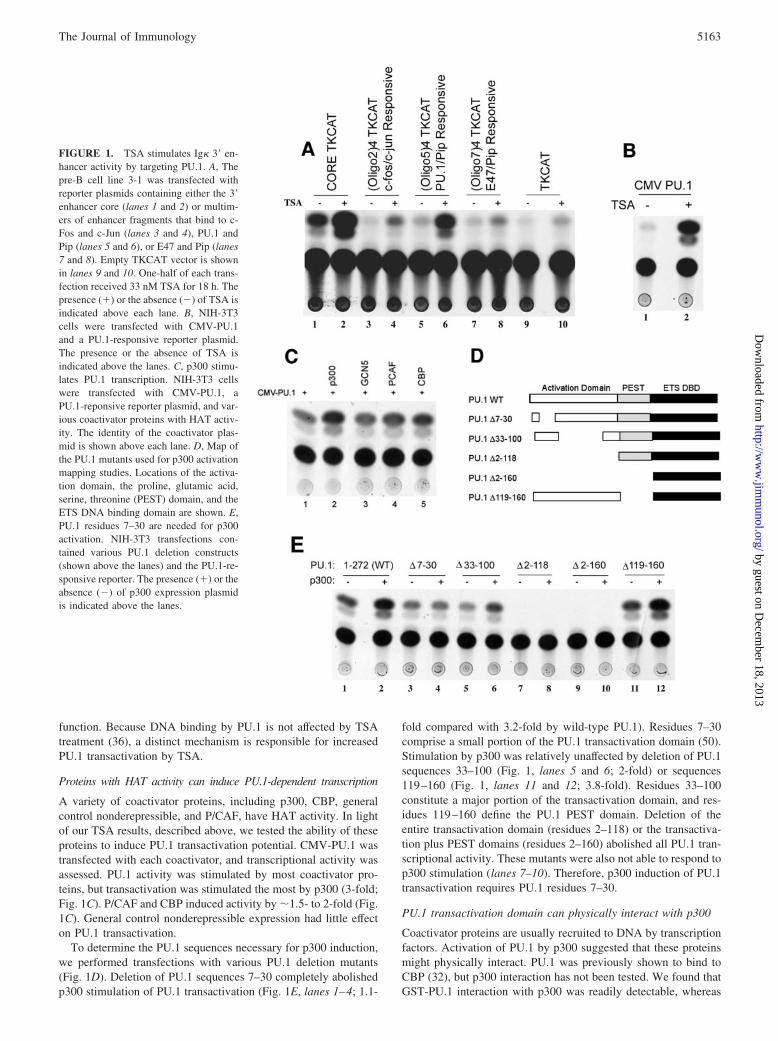
Previously, we showed that TSA could stimulate histone H3 acet-ylation at the Ig� 3� enhancer (36). The 3� enhancer is implicatedin numerous important functions, including transcription, somaticrearrangement, and somatic mutation (6, 43–47). To explorewhether TSA treatment could augment 3� enhancer activity, wetransfected the pre-B cell line 3-1 with a reporter plasmid contain-ing the 3� enhancer 132-bp core segment driving the CAT gene(CORETKCAT) (4). This reporter construct shows weak activityin pre-B cells (4). After transfection, cells were treated with either33 nM TSA or vehicle control. Interestingly, TSA treatmentcaused a dramatic 7-fold increase in enhancer activity (Fig. 1A,lanes 1 and 2). The 3� enhancer contains several segments thatshow enhancer activity when multimerized (4, 48). These seg-ments bind to c-Fos and c-Jun, PU.1 and Pip, or E47 and Pip (4,48, 49). To determine the TSA-responsive region of the 3� en-hancer, we performed transfections with reporter plasmids con-taining multimers of these enhancer elements in the presence or theabsence of TSA. Weak TSA induction (2- to 3-fold) was observedwith the c-Fos/c-Jun and the E47/Pip reporters (Fig. 1, lanes 3, 4,7, and 8). This induction level was similar to that of the emptyTKCAT reporter (2-fold; lanes 9 and 10). However, the activity ofthe PU.1/Pip reporter was very dramatically increased by TSAtreatment (10-fold; Fig. 1, lanes 5 and 6). Because the PU.1/Pipreporter, but not the E47/Pip reporter, was induced by TSA, wereasoned that PU.1 was the most likely target of TSA induction. Toconfirm this, we performed transfections in NIH-3T3 cells (whichlack PU.1) with the PU.1/Pip reporter plasmid and a plasmid ex-pressing PU.1 (CMV-PU.1). TSA treatment resulted in a very dra-matic increase in PU.1 transcriptional activity (10- to 20-fold; Fig.1B). Therefore, PU.1 transcriptional activity can be greatly stim-ulated by an agent that inhibits histone deacetylase activity. Insummary, our results show that histone deacetylase inhibitionstimulated 3� enhancer activity and increased PU.1 transactivation
5162 PU.1 AND H3 ACETYLATION
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
function. Because DNA binding by PU.1 is not affected by TSAtreatment (36), a distinct mechanism is responsible for increasedPU.1 transactivation by TSA.
Proteins with HAT activity can induce PU.1-dependent transcription
A variety of coactivator proteins, including p300, CBP, generalcontrol nonderepressible, and P/CAF, have HAT activity. In lightof our TSA results, described above, we tested the ability of theseproteins to induce PU.1 transactivation potential. CMV-PU.1 wastransfected with each coactivator, and transcriptional activity wasassessed. PU.1 activity was stimulated by most coactivator pro-teins, but transactivation was stimulated the most by p300 (3-fold;Fig. 1C). P/CAF and CBP induced activity by �1.5- to 2-fold (Fig.1C). General control nonderepressible expression had little effecton PU.1 transactivation.
To determine the PU.1 sequences necessary for p300 induction,we performed transfections with various PU.1 deletion mutants(Fig. 1D). Deletion of PU.1 sequences 7–30 completely abolishedp300 stimulation of PU.1 transactivation (Fig. 1E, lanes 1–4; 1.1-
fold compared with 3.2-fold by wild-type PU.1). Residues 7–30comprise a small portion of the PU.1 transactivation domain (50).Stimulation by p300 was relatively unaffected by deletion of PU.1sequences 33–100 (Fig. 1, lanes 5 and 6; 2-fold) or sequences119–160 (Fig. 1, lanes 11 and 12; 3.8-fold). Residues 33–100constitute a major portion of the transactivation domain, and res-idues 119–160 define the PU.1 PEST domain. Deletion of theentire transactivation domain (residues 2–118) or the transactiva-tion plus PEST domains (residues 2–160) abolished all PU.1 tran-scriptional activity. These mutants were also not able to respond top300 stimulation (lanes 7–10). Therefore, p300 induction of PU.1transactivation requires PU.1 residues 7–30.
PU.1 transactivation domain can physically interact with p300
Coactivator proteins are usually recruited to DNA by transcriptionfactors. Activation of PU.1 by p300 suggested that these proteinsmight physically interact. PU.1 was previously shown to bind toCBP (32), but p300 interaction has not been tested. We found thatGST-PU.1 interaction with p300 was readily detectable, whereas
FIGURE 1. TSA stimulates Ig� 3� en-hancer activity by targeting PU.1. A, Thepre-B cell line 3-1 was transfected withreporter plasmids containing either the 3�enhancer core (lanes 1 and 2) or multim-ers of enhancer fragments that bind to c-Fos and c-Jun (lanes 3 and 4), PU.1 andPip (lanes 5 and 6), or E47 and Pip (lanes7 and 8). Empty TKCAT vector is shownin lanes 9 and 10. One-half of each trans-fection received 33 nM TSA for 18 h. Thepresence (�) or the absence (�) of TSA isindicated above each lane. B, NIH-3T3cells were transfected with CMV-PU.1and a PU.1-responsive reporter plasmid.The presence or the absence of TSA isindicated above the lanes. C, p300 stimu-lates PU.1 transcription. NIH-3T3 cellswere transfected with CMV-PU.1, aPU.1-reponsive reporter plasmid, and var-ious coactivator proteins with HAT activ-ity. The identity of the coactivator plas-mid is shown above each lane. D, Map ofthe PU.1 mutants used for p300 activationmapping studies. Locations of the activa-tion domain, the proline, glutamic acid,serine, threonine (PEST) domain, and theETS DNA binding domain are shown. E,PU.1 residues 7–30 are needed for p300activation. NIH-3T3 transfections con-tained various PU.1 deletion constructs(shown above the lanes) and the PU.1-re-sponsive reporter. The presence (�) or theabsence (�) of p300 expression plasmidis indicated above the lanes.
5163The Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
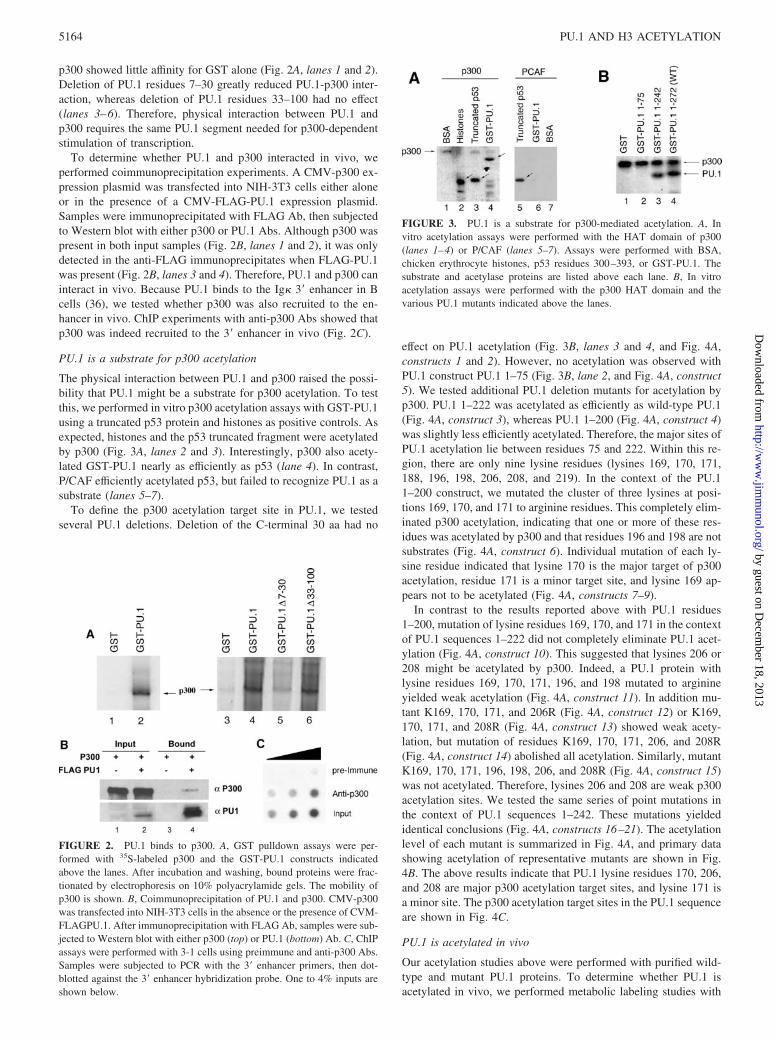
p300 showed little affinity for GST alone (Fig. 2A, lanes 1 and 2).Deletion of PU.1 residues 7–30 greatly reduced PU.1-p300 inter-action, whereas deletion of PU.1 residues 33–100 had no effect(lanes 3–6). Therefore, physical interaction between PU.1 andp300 requires the same PU.1 segment needed for p300-dependentstimulation of transcription.
To determine whether PU.1 and p300 interacted in vivo, weperformed coimmunoprecipitation experiments. A CMV-p300 ex-pression plasmid was transfected into NIH-3T3 cells either aloneor in the presence of a CMV-FLAG-PU.1 expression plasmid.Samples were immunoprecipitated with FLAG Ab, then subjectedto Western blot with either p300 or PU.1 Abs. Although p300 waspresent in both input samples (Fig. 2B, lanes 1 and 2), it was onlydetected in the anti-FLAG immunoprecipitates when FLAG-PU.1was present (Fig. 2B, lanes 3 and 4). Therefore, PU.1 and p300 caninteract in vivo. Because PU.1 binds to the Ig� 3� enhancer in Bcells (36), we tested whether p300 was also recruited to the en-hancer in vivo. ChIP experiments with anti-p300 Abs showed thatp300 was indeed recruited to the 3� enhancer in vivo (Fig. 2C).
PU.1 is a substrate for p300 acetylation
The physical interaction between PU.1 and p300 raised the possi-bility that PU.1 might be a substrate for p300 acetylation. To testthis, we performed in vitro p300 acetylation assays with GST-PU.1using a truncated p53 protein and histones as positive controls. Asexpected, histones and the p53 truncated fragment were acetylatedby p300 (Fig. 3A, lanes 2 and 3). Interestingly, p300 also acety-lated GST-PU.1 nearly as efficiently as p53 (lane 4). In contrast,P/CAF efficiently acetylated p53, but failed to recognize PU.1 as asubstrate (lanes 5–7).
To define the p300 acetylation target site in PU.1, we testedseveral PU.1 deletions. Deletion of the C-terminal 30 aa had no
effect on PU.1 acetylation (Fig. 3B, lanes 3 and 4, and Fig. 4A,constructs 1 and 2). However, no acetylation was observed withPU.1 construct PU.1 1–75 (Fig. 3B, lane 2, and Fig. 4A, construct5). We tested additional PU.1 deletion mutants for acetylation byp300. PU.1 1–222 was acetylated as efficiently as wild-type PU.1(Fig. 4A, construct 3), whereas PU.1 1–200 (Fig. 4A, construct 4)was slightly less efficiently acetylated. Therefore, the major sites ofPU.1 acetylation lie between residues 75 and 222. Within this re-gion, there are only nine lysine residues (lysines 169, 170, 171,188, 196, 198, 206, 208, and 219). In the context of the PU.11–200 construct, we mutated the cluster of three lysines at posi-tions 169, 170, and 171 to arginine residues. This completely elim-inated p300 acetylation, indicating that one or more of these res-idues was acetylated by p300 and that residues 196 and 198 are notsubstrates (Fig. 4A, construct 6). Individual mutation of each ly-sine residue indicated that lysine 170 is the major target of p300acetylation, residue 171 is a minor target site, and lysine 169 ap-pears not to be acetylated (Fig. 4A, constructs 7–9).
In contrast to the results reported above with PU.1 residues1–200, mutation of lysine residues 169, 170, and 171 in the contextof PU.1 sequences 1–222 did not completely eliminate PU.1 acet-ylation (Fig. 4A, construct 10). This suggested that lysines 206 or208 might be acetylated by p300. Indeed, a PU.1 protein withlysine residues 169, 170, 171, 196, and 198 mutated to arginineyielded weak acetylation (Fig. 4A, construct 11). In addition mu-tant K169, 170, 171, and 206R (Fig. 4A, construct 12) or K169,170, 171, and 208R (Fig. 4A, construct 13) showed weak acety-lation, but mutation of residues K169, 170, 171, 206, and 208R(Fig. 4A, construct 14) abolished all acetylation. Similarly, mutantK169, 170, 171, 196, 198, 206, and 208R (Fig. 4A, construct 15)was not acetylated. Therefore, lysines 206 and 208 are weak p300acetylation sites. We tested the same series of point mutations inthe context of PU.1 sequences 1–242. These mutations yieldedidentical conclusions (Fig. 4A, constructs 16–21). The acetylationlevel of each mutant is summarized in Fig. 4A, and primary datashowing acetylation of representative mutants are shown in Fig.4B. The above results indicate that PU.1 lysine residues 170, 206,and 208 are major p300 acetylation target sites, and lysine 171 isa minor site. The p300 acetylation target sites in the PU.1 sequenceare shown in Fig. 4C.
PU.1 is acetylated in vivo
Our acetylation studies above were performed with purified wild-type and mutant PU.1 proteins. To determine whether PU.1 isacetylated in vivo, we performed metabolic labeling studies with
FIGURE 2. PU.1 binds to p300. A, GST pulldown assays were per-formed with 35S-labeled p300 and the GST-PU.1 constructs indicatedabove the lanes. After incubation and washing, bound proteins were frac-tionated by electrophoresis on 10% polyacrylamide gels. The mobility ofp300 is shown. B, Coimmunoprecipitation of PU.1 and p300. CMV-p300was transfected into NIH-3T3 cells in the absence or the presence of CVM-FLAGPU.1. After immunoprecipitation with FLAG Ab, samples were sub-jected to Western blot with either p300 (top) or PU.1 (bottom) Ab. C, ChIPassays were performed with 3-1 cells using preimmune and anti-p300 Abs.Samples were subjected to PCR with the 3� enhancer primers, then dot-blotted against the 3� enhancer hybridization probe. One to 4% inputs areshown below.
FIGURE 3. PU.1 is a substrate for p300-mediated acetylation. A, Invitro acetylation assays were performed with the HAT domain of p300(lanes 1–4) or P/CAF (lanes 5–7). Assays were performed with BSA,chicken erythrocyte histones, p53 residues 300–393, or GST-PU.1. Thesubstrate and acetylase proteins are listed above each lane. B, In vitroacetylation assays were performed with the p300 HAT domain and thevarious PU.1 mutants indicated above the lanes.
5164 PU.1 AND H3 ACETYLATION
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

tritiated acetate in S194 cells. Cell lysates were prepared, and sam-ples were incubated with either preimmune or anti-PU.1 Abs. Theanti-PU.1 Ab precipitated a labeled protein with the electro-phoretic mobility expected for PU.1 (�43 kDa; Fig. 5A), indicat-ing that PU.1 can be acetylated in vivo.
Because PU.1 lysine 170 is a major acetylation site in vitro, weprepared antiserum specific for PU.1 acetylated on lysine 170 andused this antiserum to determine the acetylation status of PU.1lysine 170 at the pre-B and plasma cell stages. Western blots ofwhole cell extracts from 3-1 and S194 cells were probed in parallelwith PU.1 antisera and acetyl lysine 170-specific antisera. Inter-estingly, PU.1 was acetylated on lysine 170 at both developmentalstages (Fig. 5B). The specificity of the Ab for acetylated PU.1 wasshown by observing greater reactivity against p300 acetylatedPU.1 than with unacetylated PU.1 protein (Fig. 5C).
Functional consequences of PU.1 acetylation
The major PU.1 acetylation sites that we identified in this study residewithin the PU.1 DNA binding domain. Therefore, acetylation couldpotentially alter the ability of PU.1 to bind to DNA. We tested theDNA-binding ability of acetylated and unacetylated PU.1 by EMSAand found no discernable difference (although the fraction of acety-lated PU.1 molecules was uncertain; data not shown). We also testedwhether mutation of the PU.1 acetylation sites would alter DNA bind-ing. PU.1 mutant proteins K170,171R and K206,208R were tested byEMSA for binding to the PU.1 site in the Ig� 3� enhancer and for theirability to recruit Pip to its adjacent DNA binding site. MutantK170,171R bound to DNA and recruited Pip as efficiently as wild-type PU.1 (Fig. 6A, lanes 1–4). In contrast, mutant K206,208R boundto DNA poorly and was inefficient at recruiting Pip to DNA (lanes 5and 6).
FIGURE 4. Identification of p300acetylation sites in PU.1. A, PU.1 con-structs used for p300 acetylation assaysare diagrammed. The diagram showsPU.1 amino acid residues present in eachconstruct, and Xs mark the positions oflysine residues mutated to arginine. Theright panel indicates the relative level ofp300 acetylation of each mutant con-struct as well as the specific lysine resi-dues that are mutated to arginine. Rela-tive acetylation was determined bycomparison of multiple film exposures ofHAT assay gels. Each � represents�25% of the level observed with wild-type PU.1, which was defined as 100%.B, Representative HAT results showingrelative acetylation levels. C, PU.1 se-quences 161–220. �, Major p300 acety-lation sites are marked by an asterisk (ly-sines 170, 206, and 208); E, the minorsite at lysine 171.
5165The Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
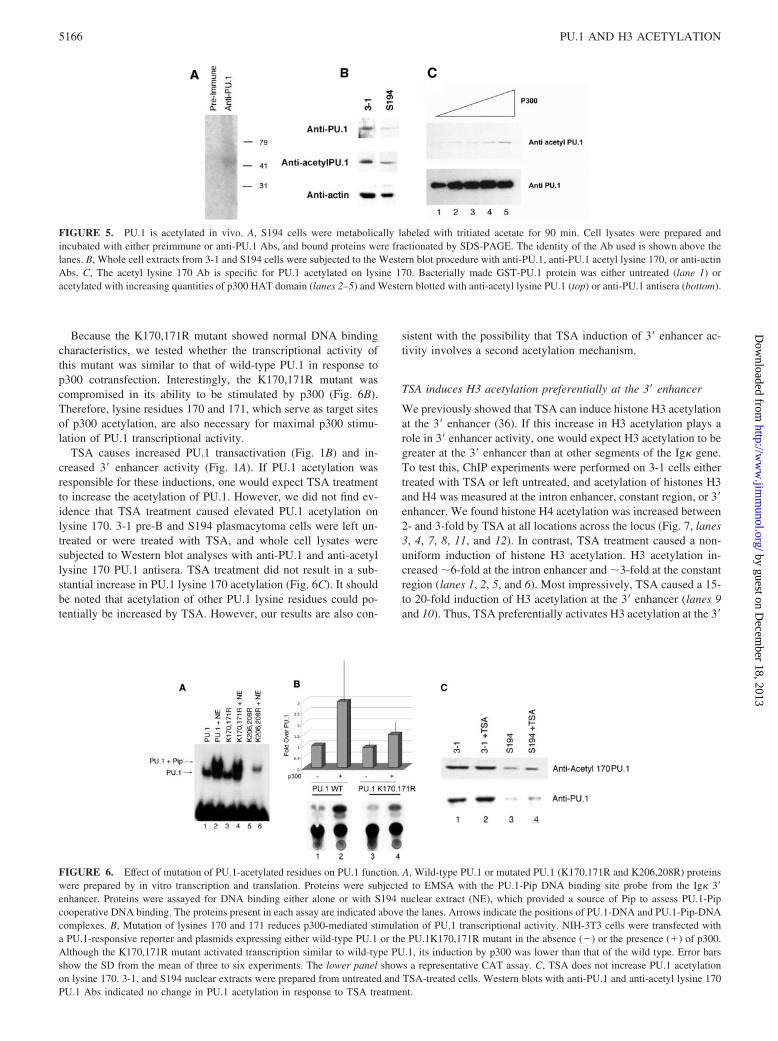
Because the K170,171R mutant showed normal DNA bindingcharacteristics, we tested whether the transcriptional activity ofthis mutant was similar to that of wild-type PU.1 in response top300 cotransfection. Interestingly, the K170,171R mutant wascompromised in its ability to be stimulated by p300 (Fig. 6B).Therefore, lysine residues 170 and 171, which serve as target sitesof p300 acetylation, are also necessary for maximal p300 stimu-lation of PU.1 transcriptional activity.
TSA causes increased PU.1 transactivation (Fig. 1B) and in-creased 3� enhancer activity (Fig. 1A). If PU.1 acetylation wasresponsible for these inductions, one would expect TSA treatmentto increase the acetylation of PU.1. However, we did not find ev-idence that TSA treatment caused elevated PU.1 acetylation onlysine 170. 3-1 pre-B and S194 plasmacytoma cells were left un-treated or were treated with TSA, and whole cell lysates weresubjected to Western blot analyses with anti-PU.1 and anti-acetyllysine 170 PU.1 antisera. TSA treatment did not result in a sub-stantial increase in PU.1 lysine 170 acetylation (Fig. 6C). It shouldbe noted that acetylation of other PU.1 lysine residues could po-tentially be increased by TSA. However, our results are also con-
sistent with the possibility that TSA induction of 3� enhancer ac-tivity involves a second acetylation mechanism.
TSA induces H3 acetylation preferentially at the 3� enhancer
We previously showed that TSA can induce histone H3 acetylationat the 3� enhancer (36). If this increase in H3 acetylation plays arole in 3� enhancer activity, one would expect H3 acetylation to begreater at the 3� enhancer than at other segments of the Ig� gene.To test this, ChIP experiments were performed on 3-1 cells eithertreated with TSA or left untreated, and acetylation of histones H3and H4 was measured at the intron enhancer, constant region, or 3�enhancer. We found histone H4 acetylation was increased between2- and 3-fold by TSA at all locations across the locus (Fig. 7, lanes3, 4, 7, 8, 11, and 12). In contrast, TSA treatment caused a non-uniform induction of histone H3 acetylation. H3 acetylation in-creased �6-fold at the intron enhancer and �3-fold at the constantregion (lanes 1, 2, 5, and 6). Most impressively, TSA caused a 15-to 20-fold induction of H3 acetylation at the 3� enhancer (lanes 9and 10). Thus, TSA preferentially activates H3 acetylation at the 3�
FIGURE 6. Effect of mutation of PU.1-acetylated residues on PU.1 function. A, Wild-type PU.1 or mutated PU.1 (K170,171R and K206,208R) proteinswere prepared by in vitro transcription and translation. Proteins were subjected to EMSA with the PU.1-Pip DNA binding site probe from the Ig� 3�enhancer. Proteins were assayed for DNA binding either alone or with S194 nuclear extract (NE), which provided a source of Pip to assess PU.1-Pipcooperative DNA binding. The proteins present in each assay are indicated above the lanes. Arrows indicate the positions of PU.1-DNA and PU.1-Pip-DNAcomplexes. B, Mutation of lysines 170 and 171 reduces p300-mediated stimulation of PU.1 transcriptional activity. NIH-3T3 cells were transfected witha PU.1-responsive reporter and plasmids expressing either wild-type PU.1 or the PU.1K170,171R mutant in the absence (�) or the presence (�) of p300.Although the K170,171R mutant activated transcription similar to wild-type PU.1, its induction by p300 was lower than that of the wild type. Error barsshow the SD from the mean of three to six experiments. The lower panel shows a representative CAT assay. C, TSA does not increase PU.1 acetylationon lysine 170. 3-1, and S194 nuclear extracts were prepared from untreated and TSA-treated cells. Western blots with anti-PU.1 and anti-acetyl lysine 170PU.1 Abs indicated no change in PU.1 acetylation in response to TSA treatment.
FIGURE 5. PU.1 is acetylated in vivo. A, S194 cells were metabolically labeled with tritiated acetate for 90 min. Cell lysates were prepared andincubated with either preimmune or anti-PU.1 Abs, and bound proteins were fractionated by SDS-PAGE. The identity of the Ab used is shown above thelanes. B, Whole cell extracts from 3-1 and S194 cells were subjected to the Western blot procedure with anti-PU.1, anti-PU.1 acetyl lysine 170, or anti-actinAbs. C, The acetyl lysine 170 Ab is specific for PU.1 acetylated on lysine 170. Bacterially made GST-PU.1 protein was either untreated (lane 1) oracetylated with increasing quantities of p300 HAT domain (lanes 2–5) and Western blotted with anti-acetyl lysine PU.1 (top) or anti-PU.1 antisera (bottom).
5166 PU.1 AND H3 ACETYLATION
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
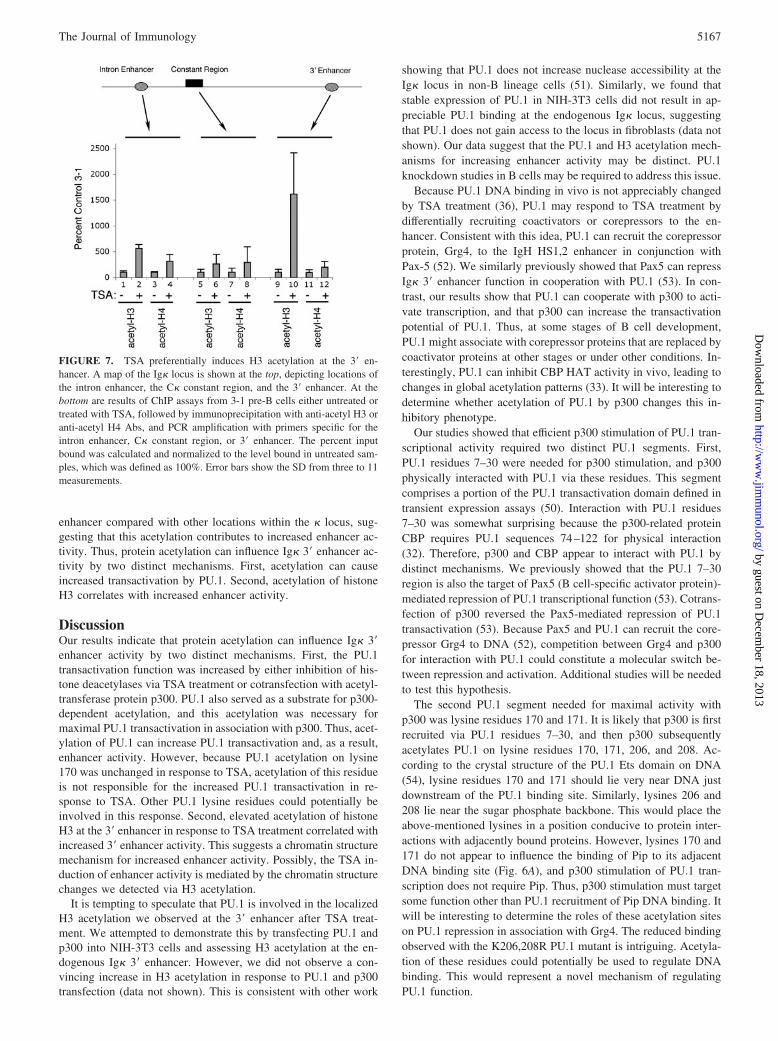
enhancer compared with other locations within the � locus, sug-gesting that this acetylation contributes to increased enhancer ac-tivity. Thus, protein acetylation can influence Ig� 3� enhancer ac-tivity by two distinct mechanisms. First, acetylation can causeincreased transactivation by PU.1. Second, acetylation of histoneH3 correlates with increased enhancer activity.
DiscussionOur results indicate that protein acetylation can influence Ig� 3�enhancer activity by two distinct mechanisms. First, the PU.1transactivation function was increased by either inhibition of his-tone deacetylases via TSA treatment or cotransfection with acetyl-transferase protein p300. PU.1 also served as a substrate for p300-dependent acetylation, and this acetylation was necessary formaximal PU.1 transactivation in association with p300. Thus, acet-ylation of PU.1 can increase PU.1 transactivation and, as a result,enhancer activity. However, because PU.1 acetylation on lysine170 was unchanged in response to TSA, acetylation of this residueis not responsible for the increased PU.1 transactivation in re-sponse to TSA. Other PU.1 lysine residues could potentially beinvolved in this response. Second, elevated acetylation of histoneH3 at the 3� enhancer in response to TSA treatment correlated withincreased 3� enhancer activity. This suggests a chromatin structuremechanism for increased enhancer activity. Possibly, the TSA in-duction of enhancer activity is mediated by the chromatin structurechanges we detected via H3 acetylation.
It is tempting to speculate that PU.1 is involved in the localizedH3 acetylation we observed at the 3� enhancer after TSA treat-ment. We attempted to demonstrate this by transfecting PU.1 andp300 into NIH-3T3 cells and assessing H3 acetylation at the en-dogenous Ig� 3� enhancer. However, we did not observe a con-vincing increase in H3 acetylation in response to PU.1 and p300transfection (data not shown). This is consistent with other work
showing that PU.1 does not increase nuclease accessibility at theIg� locus in non-B lineage cells (51). Similarly, we found thatstable expression of PU.1 in NIH-3T3 cells did not result in ap-preciable PU.1 binding at the endogenous Ig� locus, suggestingthat PU.1 does not gain access to the locus in fibroblasts (data notshown). Our data suggest that the PU.1 and H3 acetylation mech-anisms for increasing enhancer activity may be distinct. PU.1knockdown studies in B cells may be required to address this issue.
Because PU.1 DNA binding in vivo is not appreciably changedby TSA treatment (36), PU.1 may respond to TSA treatment bydifferentially recruiting coactivators or corepressors to the en-hancer. Consistent with this idea, PU.1 can recruit the corepressorprotein, Grg4, to the IgH HS1,2 enhancer in conjunction withPax-5 (52). We similarly previously showed that Pax5 can repressIg� 3� enhancer function in cooperation with PU.1 (53). In con-trast, our results show that PU.1 can cooperate with p300 to acti-vate transcription, and that p300 can increase the transactivationpotential of PU.1. Thus, at some stages of B cell development,PU.1 might associate with corepressor proteins that are replaced bycoactivator proteins at other stages or under other conditions. In-terestingly, PU.1 can inhibit CBP HAT activity in vivo, leading tochanges in global acetylation patterns (33). It will be interesting todetermine whether acetylation of PU.1 by p300 changes this in-hibitory phenotype.
Our studies showed that efficient p300 stimulation of PU.1 tran-scriptional activity required two distinct PU.1 segments. First,PU.1 residues 7–30 were needed for p300 stimulation, and p300physically interacted with PU.1 via these residues. This segmentcomprises a portion of the PU.1 transactivation domain defined intransient expression assays (50). Interaction with PU.1 residues7–30 was somewhat surprising because the p300-related proteinCBP requires PU.1 sequences 74–122 for physical interaction(32). Therefore, p300 and CBP appear to interact with PU.1 bydistinct mechanisms. We previously showed that the PU.1 7–30region is also the target of Pax5 (B cell-specific activator protein)-mediated repression of PU.1 transcriptional function (53). Cotrans-fection of p300 reversed the Pax5-mediated repression of PU.1transactivation (53). Because Pax5 and PU.1 can recruit the core-pressor Grg4 to DNA (52), competition between Grg4 and p300for interaction with PU.1 could constitute a molecular switch be-tween repression and activation. Additional studies will be neededto test this hypothesis.
The second PU.1 segment needed for maximal activity withp300 was lysine residues 170 and 171. It is likely that p300 is firstrecruited via PU.1 residues 7–30, and then p300 subsequentlyacetylates PU.1 on lysine residues 170, 171, 206, and 208. Ac-cording to the crystal structure of the PU.1 Ets domain on DNA(54), lysine residues 170 and 171 should lie very near DNA justdownstream of the PU.1 binding site. Similarly, lysines 206 and208 lie near the sugar phosphate backbone. This would place theabove-mentioned lysines in a position conducive to protein inter-actions with adjacently bound proteins. However, lysines 170 and171 do not appear to influence the binding of Pip to its adjacentDNA binding site (Fig. 6A), and p300 stimulation of PU.1 tran-scription does not require Pip. Thus, p300 stimulation must targetsome function other than PU.1 recruitment of Pip DNA binding. Itwill be interesting to determine the roles of these acetylation siteson PU.1 repression in association with Grg4. The reduced bindingobserved with the K206,208R PU.1 mutant is intriguing. Acetyla-tion of these residues could potentially be used to regulate DNAbinding. This would represent a novel mechanism of regulatingPU.1 function.
FIGURE 7. TSA preferentially induces H3 acetylation at the 3� en-hancer. A map of the Ig� locus is shown at the top, depicting locations ofthe intron enhancer, the C� constant region, and the 3� enhancer. At thebottom are results of ChIP assays from 3-1 pre-B cells either untreated ortreated with TSA, followed by immunoprecipitation with anti-acetyl H3 oranti-acetyl H4 Abs, and PCR amplification with primers specific for theintron enhancer, C� constant region, or 3� enhancer. The percent inputbound was calculated and normalized to the level bound in untreated sam-ples, which was defined as 100%. Error bars show the SD from three to 11measurements.
5167The Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

PU.1 is implicated in numerous developmental processes, in-cluding differentiation of B cells, T cells, macrophages, and ery-throid cells (19–23, 25–30, 55). The interplay between PU.1 andeither GATA-1 or Pax5 (B cell-specific activator protein) has beenproposed to regulate the development of either the erythroid ormacrophage lineage, respectively (23, 29, 30, 53, 56). More re-cently, the effects of modified PU.1 expression levels have beendescribed (23, 31). Low PU.1 expression levels in progenitor cellsgenerally give rise to B cell development, whereas higher levelsyield macrophage development (23). In addition, reducing PU.1expression levels in vivo to 20% of wild-type levels leads to a highincidence of acute myeloid leukemia (31). Thus, changing PU.1activity by either expression level or, perhaps, its function via pro-tein interactions or post-translational modifications could have avery dramatic impact on biological processes. Exploring how thep300-dependent acetylation sites identified in this study relate tovarious PU.1 functions will be an important goal.
AcknowledgmentsWe are very grateful for the advice and reagents provided by Shelly Bergerand Lin Liu for p300 in vitro acetylation assays. We thank Paul Liebermanfor coactivator expression plasmids. We also thank Barbara Nikolajczykfor helpful comments on the manuscript.
DisclosuresThe authors have no financial conflict of interest.
References1. Hardy, R. R., and K. Hayakawa. 2001. B cell developmental pathways. Annu.
Rev. Immunol. 19: 595–621.2. Krangel, M. S. 2003. Gene segment selection in V(D)J recombination: accessi-
bility and beyond. Nat. Immunol. 4: 624–630.3. Liu, X., A. Prabhu, and B. Van Ness. 1999. Developmental regulation of the �
locus involves both positive and negative sequence elements in the 3� enhancerthat affect synergy with the intron enhancer. J. Biol. Chem. 274: 3285–3293.
4. Pongubala, J. M. R., and M. L. Atchison. 1991. Functional characterization of thedevelopmentally controlled immunoglobulin � 3� enhancer: regulation by Id, arepressor of helix-loop-helix transcription factors. Mol. Cell. Biol. 11:1040–1047.
5. Klug, C. A., S. J. Gerety, P. C. Shah, Y. Y. Chen, N. R. Rice, N. Rosenberg, andH. Singh. 1994. The v-abl tyrosine kinase negatively regulates NF-�B/Rel factorsand blocks � gene transcription in pre-B lymphocytes. Genes Dev. 8: 678–687.
6. Meyer, K. B., Y. M. Teh, and M. S. Neuberger. 1996. The Ig� 3�-enhancertriggers gene expression in early B lymphocytes but its activity is enhanced on Bcell activation. Int. Immunol. 8: 1561–1568.
7. Inlay, M., F. W. Alt, D. Baltimore, and Y. Xu. 2002. Essential roles of the � lightchain intronic enhancer and 3� enhancer in � rearrangement and methylation. Nat.Immunol. 3: 463–468.
8. Mather, E. L., and R. P. Perry. 1983. Methylation status and DNase I sensitivityof immunoglobulin genes: changes associated with rearrangement. Proc. Natl.Acad. Sci. USA 80: 4689–4693.
9. Parslow, T. G., and D. K. Granner. 1982. Chromatin changes accompany immu-noglobulin � gene activation: a potential control region within the gene. Nature299: 449–451.
10. Roque, M. C., P. A. Smith, and V. C. Blazquez. 1996. A developmentally mod-ulated chromatin structure at the mouse immunoglobulin � 3� enhancer. Mol.Cell. Biol. 16: 3138–3155.
11. Rose, S. M., and W. T. Garrard. 1984. Differentiation-dependent chromatin al-terations precede and accompany transcription of immunoglobulin light chaingenes. J. Biol. Chem. 259: 8534–8544.
12. Storb, U., and B. Arp. 1983. Methylation patterns of immunoglobulin genes inlymphoid cells: correlation of expression and differentiation with methylation.Proc. Natl. Acad. Sci. USA 80: 6642–6646.
13. Xu, M., M. B. Barnard, S. M. Rose, P. N. Cockerill, S. Huang, and W. T. Garrard.1986. Transcription termination and chromatin structure of active immunoglob-ulin � gene locus. J. Biol. Chem. 261: 3838–3845.
14. Shaffer, A. L., A. Peng, and M. S. Schlissel. 1997. In vivo occupancy of the �light chain enhancers in primary pro- and pre-B cells: a model for � locus acti-vation. Immunity 6: 131–143.
15. Pongubala, J. M. R., S. Nagulapalli, M. J. Klemsz, S. R. McKercher, R. A. Maki,and M. L. Atchison. 1992. PU.1 recruits a second nuclear factor to a site impor-tant for immunoglobulin � 3� enhancer activity. Mol. Cell. Biol. 12: 368–378.
16. Pongubala, J. M. R., C. Van Beveren, S. Nagulapalli, M. J. Klemsz,S. R. McKercher, R. A. Maki, and M. L. Atchison. 1993. Effect of PU.1 phos-phorylation on interaction with NF-EM5 and transcriptional activation. Science259: 1622–1625.
17. Eisenbeis, C. F., H. Singh, and U. Storb. 1993. PU.1 is a component of a mul-tiprotein complex which binds an essential site in the murine immunoglobulin�2–4 enhancer. Mol. Cell. Biol. 13: 6452–6461.
18. Eisenbeis, C. F., H. Singh, and U. Storb. 1995. Pip, a novel IRF family member,is a lymphoid-specific PU.1-dependent transcriptional activator. Genes Dev. 9:1377–1387.
19. Voso, M. T., T. C. Burn, G. Wulf, B. Lim, G. Leone, and D. G. Tenen. 1994.Inhibition of hematopoiesis by competitive binding of transcription factor PU.1.Proc. Natl. Acad. Sci. USA 91: 7932–7936.
20. Scott, E. W., R. C. Fisher, M. C. Olson, E. W. Kehrli, M. C. Simon, and H. Singh.1997. PU.1 functions in a cel-autonomous manner to control the differentiation ofmultipotential lymphoid-myeloid progenitors. Immunity 6: 437–447.
21. Olson, M. C., E. W. Scott, A. A. Hack, G. H. Su, Tenen, D. G. H. Singh, andM. C. Simon. 1995. PU.1 is not essential for early myeloid gene expression butis required for terminal myeloid differentiation. Immunity 3: 703–714.
22. Celada, A., F. E. Borras, C. Soler, J. Lloberas, M. Klemsz, C. van Beveren,S. McKercher, and R. A. Maki. 1996. The transcription factor PU.1 is involvedin macrophage proliferation. J. Exp. Med. 184: 61–69.
23. DeKoter, R. P., and H. Singh. 2000. Regulation of B lymphocyte and macrophagedevelopment by graded expression of PU.1. Science 288: 1439–1441.
24. DeKoter, R. P., J. C. Walsh, and H. Singh. 1998. PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progeni-tors. EMBO J. 17: 4456–4468.
25. Nerlov, C., and T. Graf. 1998. PU.1 induces myeloid lineage commitment inmultipotent hematopoietic progenitors. Genes Dev. 12: 2403–2412.
26. Schuetze, S., R. Paul, B. C. Gliniak, and D. Kabat. 1992. Role of the PU.1transcription factor in controlling differentiation of Friend erythroleukemia cells.Mol. Cell. Biol. 12: 2967–2975.
27. Scott, E. W., M. C. Simon, J. Anastasi, and H. Singh. 1994. Requirement oftranscription factor PU.1 in the development of multiple hematopoietic lineages.Science 265: 1573–1577.
28. McKercher, S. R., B. E. Torbett, K. L. Anderson, G. W. Henkel, D. J. Vestal,H. Baribault, M. Klemsz, A. J. Feeney, G. E. Wu, C. J. Paige, et al. 1996.Targeted disruption of the PU.1 gene results in multiple hematopoietic abnor-malities. EMBO J. 15: 5647–5658.
29. Rekhtman, N., F. Radparvar, T. Evans, and A. I. Skoultchi. 1999. Direct inter-action of hematopoietic transcription factors PU.1 and GATA-1: functional an-tagonism in erythroid cells. Genes Dev. 13: 1398–1411.
30. Zhang, P., G. Behre, J. Pan, A. Iwama, N. Wara-Aswapati, H. S. Radomska,P. E. Auron, D. G. Tenen, and Z. Sun. 1999. Negative cross-talk between he-matopoietic regulators: GATA proteins repress PU.1. Proc. Natl. Acad. Sci. USA96: 8705–8710.
31. Rosenbauer, F., K. Wagner, J. Kutok, H. Iwasaki, M. M. Le Beau, Y. Okuno,K. Asashi, S. Fiering, and D. G. Tenen. 2004. Acute myeloid leukemia inducedby graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet.36: 624–630.
32. Yamamoto, H., F. Kihara-Negishi, T. Yamada, Y. Hashimoto, and T. Oikawa.1999. Physical and functional interactions between the transcription factor PU.1and the coactivator CBP. Oncogene 18: 1495–1501.
33. Hong, W., A. Y. Kim, S. Ky, C. Rakowski, S. B. Seo, D. Chakravarti,M. Atchison, and G. A. Blobel. 2002. Inhibition of CBP-mediated protein acet-ylation by the Ets family oncoprotein PU.1. Mol. Cell. Biol. 22: 3729–3743.
34. Nikolajczyk, B. S., A. Sanchez, and R. Sen. 1999. ETS protein-dependent ac-cessibility changes at the immunoglobulin � heavy chain enhancer. Immunity 11:11–20.
35. Sato, H., F. Saito-Ohara, J. Inazawa, and A. Kudo. 2004. Pax-5 is essential for �sterile transcription during Ig� chain gene rearrangement. J. Immunol. 172:4858–4865.
36. McDevit, D. C., L. Perkins, M. L. Atchison, and B. S. Nikolajczyk. 2005. TheIg�3� enhancer Is activated by gradients of chromatin accessibility and proteinassociation. J. Immunol. 174: 2834–2842.
37. Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Sitedirected mutagenesis by overlap extension using the polymerase chain reaction.Gene 77: 51–59.
38. Perkel, J. M., and M. L. Atchison. 1998. A two-step mechanism for recruitmentof Pip by PU.1. J. Immunol. 160: 241–252.
39. Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcriptioninitiation by RNA polymerase II in a soluble extract from isolated mammaliannuclei. Nucleic Acids Res. 11: 1475–1489.
40. Graham, F. L., and A. J. Van der Eb. 1973. A new technique for the assay ofinfectivity of human adenovirus 5 DNA. Virology 52: 456–467.
41. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant genomeswhich express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell.Biol. 2: 1044–1051.
42. Kaelin, W. G. J., D. C. Pallas, J. A. DeCaprio, F. J. Kaye, and D. M. Livingston.1991. Indentification of cellular proteins that can interact specifically with theT/E1A-binding region of the retinoblastoma gene product. Cell 64: 521–532.
43. Takeda, S., Y. R. Zou, H. Bluethmann, D. Kitamura, U. Muller, and K. Rajewsky.1993. Deletion of the immunoglobulin � chain intron enhancer abolishes � chaingene rearrangement in cis but not � chain gene rearrangement in trans. EMBO J.12: 2329–2336.
44. Gorman, J. R., N. van der Stoep, R. Monroe, M. Cogne, L. Davidson, andF. W. Alt. 1996. The Ig� 3� enhancer influences the ratio of Ig� versus Ig� Blymphocytes. Immunity 5: 241–252.
45. Xu, Y., L. Davidson, F. W. Alt, and D. Baltimore. 1996. Deletion of the Ig� lightchain intronic enhancer/matrix attachment region impairs but does not abolishV�J� rearrangement. Immunity 4: 377–385.
5168 PU.1 AND H3 ACETYLATION
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

46. Hiramatsu, R., K. Akagi, M. Matsuoka, K. Sakumi, H. Nakamura, L. Kingsbury,C. David, R. R. Hardy, K.-i. Yamamura, and H. Sakano. 1995. The 3� enhancerregion determines the B/T specificity and pro-B/pre-B specificity of immuno-globulin V�-J� joining. Cell 83: 1113–1123.
47. Betz, A. G., C. Milstein, A. Gonzalel-Fernandez, R. Pannell, T. Larson, andM. S. Neuberger. 1994. Elements regulating somatic hypermutation of an Ig�gene: critical role of the intron enhancer/matrix attachment region. Cell 77:239–248.
48. Pongubala, J. M. R., and M. L. Atchison. 1995. Activating transcription factor 1and cyclic AMP response element modulator can modulate the activity of theimmunoglobulin � 3� enhancer. J. Biol. Chem. 270: 10304–10413.
49. Nagulapalli, S., and M. L. Atchison. 1998. Transcription factor Pip can enhanceDNA binding by E47, leading to transcriptional synergy involving multiple pro-tein domains. Mol. Cell. Biol. 18: 4639–4650.
50. Klemsz, M. J., and R. A. Maki. 1996. Activation of transcription by PU.1 requiresboth acidic and glutamine domains. Mol. Cell. Biol. 16: 390–397.
51. Marecki, S., K. McCarthy, and B. S. Nikolajczyk. 2004. PU.1 as a chromatinaccessibility factor for immunoglobulin genes. Mol. Immunol. 40: 723–731.
52. Linderson, Y., D. Eberhard, S. Malin, A. Johansson, M. Busslinger, andS. Pettersson. 2004. Corecruitment of the Grg4 repressor by PU.1 is critical forPax5-mediated repression of B-cell-specific genes. EMBO Rep. 5: 291–296.
53. Maitra, S., and M. Atchison. 2000. BSAP can repress enhancer activity by tar-geting PU.1 function. Mol. Cell. Biol. 20: 1911–1922.
54. Kodandapani, R., F. Pio, C.-Z. Ni, G. Piccialli, M. Klemsz, S. McKercher,R. A. Maki, and K. R. Ely. 1996. A new pattern for helix-turn-helix recognitionrevealed by the PU.1 ETS-domain-DNA complex. Nature 380: 456–460.
55. Henkel, G. W., S. R. McKercher, P. J. Leenen, and R. A. Maki. 1999. Commit-ment to the monocytic lineage occurs in the absence of the transcription factorPU.1. Blood 93: 2849–2858.
56. Chiang, M. Y., and J. G. Monroe. 1999. BSAP/Pax5A expression blocks survivaland expansion of early myeloid cells implicating its involvement in maintainingcommitment to the B-lymphocyte lineage. Blood 94: 3621–3632.
5169The Journal of Immunology
by guest on Decem
ber 18, 2013http://w
ww
.jimm
unol.org/D
ownloaded from





























