Embed Size (px)
Citation preview

1
Aix-Marseille Université
Faculté de médecine de Marseille
École Doctorale Sciences de la Vie et de la Santé
Thèse de doctoratSpécialité Maladies infectieuses
Contribution à l’analyse des facteursdéterminant les fibroses graves du foie
(bilharziose) et de la peau (tissus chéloïdes)
Présentée et soutenue par Mr Nicolas Duflot
Membres du jury de Thèse:
Pr. Dulce Papy-Garcia Rapporteur et présidente du jury
Pr. Raymond Pierce Rapporteur
Dr. Audrey Romano Examinatrice
Dr. Antoine Petit Examinateur
Pr. Laurent Abel Examinateur
Pr. Alain Dessein Directeur de thèse
Unité INSERM 906 & Laboratoire de Recherche Bilhi Genetics

2
RésuméLes fibroses anormales sont responsables de plus de 40% des décès pour raison médicale ;
elles se développent suite à une inflammation chronique et causent souvent une perte de fonction del’organe affecté. Alain Dessein et son équipe à l’INSERM ont montré que les fibroses hépatiquescausées par les schistosomes et par le virus HCV sont en grande partie déterminées par la génétiquedu malade. Cette thèse a consisté à poursuivre le travail de caractérisation du déterminisme génétiquedes fibroses hépatiques et cutanées afin de contribuer à la mise au point de tests prédictifs desfibroses sévères.
La première partie de cette thèse, correspond à l’étude informatique et statistique des donnéesde génotypage de 600 Brésiliens qui présentent une fibrose hépatique bilharzienne grave. Cesindividus et un nombre équivalent de contrôles ont été génotypés (2.5 millions de SNPs parmicroarray ILLUMINA). Après purification des données de génotypage, nous avons ajusté celles-cisur les “ composantes principales ” qui contribuent à l’hétérogénéité des données, en particulierl’origine ethnique des patients. Sur ces données épurées et ajustées, nous avons alors sélectionné 180SNPs qui montraient une association significative (p < 5.10-7) ou suggestive (p < 10-6) avec lesfibroses graves. Ces SNPs ont été ensuite testés sur une cohorte de 460 pêcheurs ougandais exposés àSchistoma mansoni dont la moitié présentait des fibroses hépatiques graves. Cette analyse a confirmél’association de 4 SNPs dans 4 gènes différents avec la fibrose. Une seconde cohorte brésilienne aété recrutée pour compléter cette étude de confirmation. Parmi les SNPs présentant une associationsuggestive, certains affectent les gènes des voies Wnt. Un retour sur les données GWAS indiquentque des SNPs dans plusieurs gènes de la voie Wnt pourraient être associées aux fibroses graves.
Dans la deuxième partie de la thèse, est exposé l’analyse génomique (transcriptome etgénétique) des mécanismes responsables des fibroses anormales de la peau de sujets affectés par desfibroses chéloïdes. Nous avons effectué une analyse des gènes (par RNASeq) exprimés de manièredifférente entre tissus chéloïdes (n= 20) et tissus sains (n= 7) ou tissus affectés par des cicatriceshypertrophiques (n= 7). Cette analyse montre que le développement des chéloïdes est la conséquenced’une stimulation anormale des voies de cicatrisation, qui sont également stimulées à un moindredegré dans les cicatrices hypertrophiques lesquelles régressent spontanément. Les comparaisons dutranscriptome des tissus chéloïdes et des cicatrices hypertrophiques ainsi que la comparaison deschéloïdes sévères avec des chéloïdes moins agressives, suggèrent que cette forte stimulation est enpartie due à l’activation de la voie Wnt βcatenin et Wnt PCP. Afin de conforter cette hypothèse nousavons effectué une analyse génétique de la voie Wnt dans deux cohortes de 270 sujets chéloïdes et560 contrôles en testant les SNPs dans les gènes des voies Wnt sélectionnés à partir de l’analyseGWAS. L’analyse statistique des résultats montre que des polymorphismes dans 5 gènes de la voieWnt / βcatenin contribuent au développement des fibroses chéloïdes. Nous démontrons ainsi que ledéveloppement de la fibrose chéloïdes est, au moins en partie, la conséquence de mutations quialtèrent le fonctionnement de la voie Wnt / βcatenin. L’ensemble de ces résultats est utilisé pouraméliorer les tests génétiques développés par notre groupe Bilhi Genetics dans le but de prédire lescicatrisations cutanées anormales et les fibroses sévères du foie.

3
Table des matièresIntroduction.
A) La cicatrisation normale: ses mécanismes
a) Aspects généraux, fibrose
b) Le foie
c) La peau
B) La cicatrisation pathologique et la fibrose
a) Aspects généraux
b) Le foie
c) La peau
C) Voies de signalisation de la fibrose
a) Voie Wnt/β-caténineb) Voie du TGFB & BMP7
D) Quelles approches utiliser pour l’étude des mécanismes de cicatrisation ?
a) Analyse génomique, séquençage et génotypage ?
b) Analyse du transcriptome
c) Analyse de l’épi-génome
d) Analyse bio-informatique
Matériels et Méthodes.
A) Cohortes et recrutement des patients
a) Cohortes fibrose de le peau
b) Cohortes fibrose du foie
B) Biologie moléculaire
a) Préparation ADN et ARN
b) Nouvelles technologies de séquençage haut débit (NGS)
c) Génotypages TaqMan
C) Analyse des données
a) Analyse d'enrichissement et recherche de voies métaboliques associées
b) Classification hiérarchique.
c) Extraction des sous jeux de données provenant du GWAS pour analyse génétique ciblée
d) Analyse bio-informatique des séquences.

4
Résultats
A) Déterminisme génétique des fibroses hépatiques causées par les schistosomes : Recherched’association sur le génome entier (GWAS) de sujets brésiliens
1) Description de la cohorte de découverte et du microarray utilisé dans l’analyse GWAS
2) Contrôle des données
a) Contrôle par individu: Exclusion des patients définis comme apparentés, confirmationdu sexe civil par inférence génétique, taux de génotypage par patient, taux d'hétérozygotie
b) Contrôle par variant: Suppression des SNP dupliqués et des chromosomes sexuels, tauxde génotypage par SNP, test de Hardy Weinberg, Filtrage sur la fréquence allélique
3) Étude de l’hétérogénéité de la cohorte à l’échelle mondiale et structure à l'échelle locale
4) Analyses statistiques des données du génotypage sur l’ensemble des sujets de la cohorte dedécouverte
a) Stratégies d’analyse possibles
b) Régression logistique binaire et modèle statistique linéaire mixte
c) Résultats: Q-Q plots et Manhattan plots
5) Évaluation des associations dans les deux cohortes de confirmation recrutées au Brésil et enOuganda
B) Cicatrisation anormale de la peau: analyse du transcriptome et analyse génétique descicatrices hypertrophiques et des tissus chéloïdes.
1) Voies de signalisation dans les cicatrisations anormales de la peau.
a) Classification / agrégation hiérarchique des patients basée sur l’analyse du transcriptomedes tissus. Homogénéité des groupes cliniques.
b) Analyse des voies métaboliques exprimées de manière différente entre les tissuschéloïdes et les tissus sains.
c) Analyse des voies métaboliques exprimées de manière différente entre les tissushypertrophiques et les tissus sains
d) Gènes et voies différemment exprimés entre tissus hypertrophiques et tissus chéloïdes.
e) Comparaison du transcriptome des tissus chéloïdes provenant du thorax ou des oreilles.
2) Analyse génétique de la voie Wnt/βcatenin dans le développement des tissus chéloïdes dansune cohorte africaine
Discussion et Conclusion
Bibliographie

5
Introduction.
Chez certaines personnes lors de la cicatrisation, une accumulation anormale de tissus cicatricielsfibreux peut entrainer une cicatrisation pathologique. Le premier axe de notre travail porte sur lesdeux formes les plus connues des pathologies de la cicatrisation liées à la fibrose : les cicatriceschéloïdes et hypertrophiques (Ref. 1, Figures 1 et 2). La cicatrice chéloïde peut être initiée parn’importe quelle petite altération de la peau : une blessure, coupure ou même brûlure. Pour desraisons encore mal comprises, les chéloïdes semblent se développer préférentiellement sur certainesparties du corps plutôt que sur d’autres : Le cou, les épaules, le thorax ou encore les oreilles (2). Ellesrésultent de l'accumulation anormale de tissus fibreux cicatriciels. A l'image d'un cancer bénin, leurcroissance verticale et horizontale est continue et tend à perdurer après traitement. Chez lespersonnes les plus prédisposées, elles peuvent se développer sur tout le corps entraînant ainsi desproblèmes esthétiques. Les cicatrices chéloïdes sont généralement actives et peuvent causer desdémangeaisons (prurit) et douleurs qui peuvent être extrêmement ennuyeuses et affecter la qualité devie du patient (3). A l'inverse, la cicatrice hypertrophique est une pathologie moins grave, elle auratendance à régresser au bout de quelques années (2 ans) et n’envahira pas les tissus.
A ce jour, il n’existe pas encore de traitement efficace contre ces fibroses de la peau. Chirurgie,cryothérapie ou encore traitement au laser ne permettent pas d'enrayer le développement de cescicatrices efficacement et qui finissent toujours pas réapparaître avec le temps (4 , 5).
Les mécanismes moléculaires sous-susjacents au développement des cicatrices chéloïdes tout commehypertrophiques sont encore mal caractérisés. Pour se former, le tissu chéloïdien ferait intervenir lesprocessus de fibrose, d’angiogenèse et les mécanismes du développement des tumeurs (6, 7, 8, 9). Ilest donc apparu nécessaire d’étudier ces mécanismes afin de mieux caractériser ces pathologies. Plusencore, face aux effets secondaires récurrents que posent les traitements proposés pour ces cicatrices,la nécessité de prévenir leur apparition en amont s’est imposée. La fibrose, dont celle de la peau,présente un fort prédéterminisme génétique. Elle se transmet de générations en générations au seinde la famille (10) et leur prévalence est plus importante chez certaines populations, comme chez lesasiatiques ou les peaux fortement pigmentées (noires ou métissées) (11, 12). Les recherches engénétique et plus particulièrement les études d’associations ont mise en évidence de nombreuxpolymorphismes associés aux cicatrices chéloïdes (13, 14, 15, 16) ainsi que des voies designalisations comme la voie de l’IL6 (17) ou encore la voie Wnt (18) et celle du TGF-β (19).
Le second axe de notre travail porte sur la fibrose du foie bilharzienne. La bilharziose est uneparasitose chronique causée par un vers plathelminthe dont cinq espèces sont connues pour êtrepathogènes chez l’homme. Cette pathologie est souvent négligée du fait qu’elle constitue unemaladie tropicale ou encore du fait qu’elle touche des populations qui ne sont pas la ciblecommerciale privilégiée des laboratoires pharmaceutiques. Cependant, l’exposition au parasite dansles pays endémiques est permanente avec en particulier les pays d’Afrique et d’Amérique du sudcomme le Brésil. Elle représente ainsi une des pathologies parasitaires les plus répandue dans lemonde, particulièrement dans les régions tropicales et subtropicales. Le fait que cette pathologie soitrépandue dans des zones du monde reculées, délaissées par la médecine moderne, explique sa forteprévalence chez les populations touchées. Elle représente non seulement un problème grave dans lespays touchés, mais constituera également à terme un problème de santé publique à l’échellemondiale si les zones endémiques s’élargissent à d’autres parties du globe, par exemple sous l’effetdu réchauffement climatique.
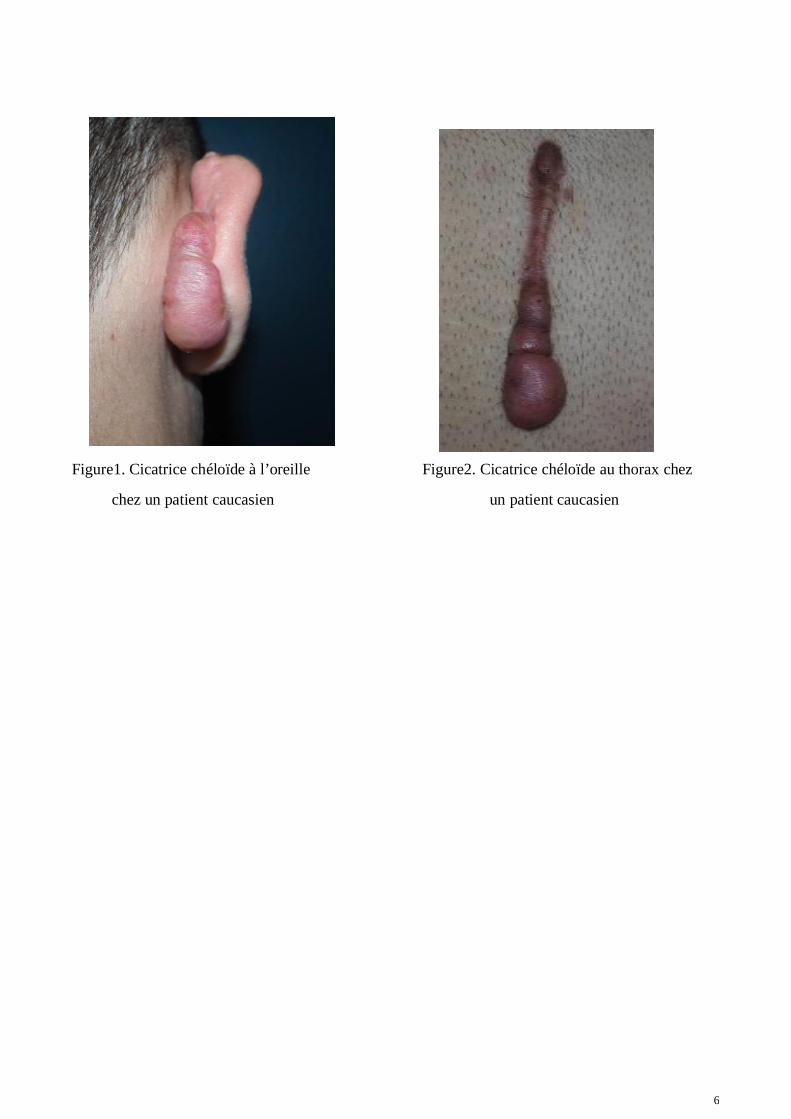
6
Figure1. Cicatrice chéloïde à l’oreille Figure2. Cicatrice chéloïde au thorax chez
chez un patient caucasien un patient caucasien

7
Notre étude porte sur l’espèce Schistosoma mansoni, causant la bilharziose intestinale et hépatique.En Afrique, on le retrouve en Libye, au Soudan, mais aussi en Ouganda. Ce dernier pays constituenotre premier terrain d’étude. On le retrouve également en Amérique du sud comme au Brésil, notresecond terrain d’étude. Le cycle parasitaire (Figure 3) se décompose en deux sous-ensemblescaractérisés par une phase de reproduction sexuée chez l’hôte définitif et une phase asexuée chezl’hôte intermédiaire. L’hôte intermédiaire et définitif sont nécessaires à l’établissement du cycle,l’homme étant l’hôte définitif du parasite. L’hôte intermédiaire est un mollusque escargot d’eaudouce. Chez l’hôte définitif, le parasite rejette ses œufs dans les excréments qui sont ainsi libérésdans le milieu aquatique local. Quand ceux-ci éclosent, ils libèrent une larve appelée miracidium quiva infecter l’hôte intermédiaire, le mollusque. Appelé alors sporocyste, il est capable de se reproduireet de produire des centaines de larves dites cercaires capables d’infecter à leur tour l’hôte définitif,l’homme. L’homme s’infecte durant ses activités quotidiennes quand il rentre en contact cutané avecl’eau douce du milieu contaminé par des cercaires. Après contact cutané, les cercaires quittentl’épiderme et se dirige vers le foie en utilisant le système sanguin. Le parasite subira un ensemble demodifications morphologiques durant sa transition vers le foie, modifications aboutissant à sa formefinale de schistosome infectant l’hôte humain pendant des dizaines d’années. Les mâles et lesfemelles vont alors se loger dans le plexus mésentérique veineux. La reproduction du parasite dansl’hôte définitif s’effectue de manière sexuée et les femelles pondent leurs œufs dans la circulationsanguine afin qu’ils soient libérés dans les selles de l’hôte. Du fait que la veine mésentériqueinférieure draine le sang de l’intestin vers la veine splénique et ensuite le foie, certains œufs sontentrainés vers le foie et se logent alors dans les capillaires hépatiques où ils sont incapables decirculer du fait de leur trop gros diamètre. Le foie forme alors des granulomes autour des œufs afinde protéger le tissu hépatique du corps étranger. Ces granulomes vont perdurer dans le temps dans lefoie de l’hôte et affectent le bon fonctionnement de l’organe sur plusieurs années. Il faut ainsi desannées avant que l’accumulation des œufs devienne pathologique, on dit alors que le sujet passe enphase chronique de la pathologie. Cette forme chronique est caractérisée par une forte réponseinflammatoire autour des granulomes pouvant entrainer des processus symptomatiques comme lafibrose. C’est sur la fibrose causée par ces œufs de schistosomes, qui est péri-portale et hépatique,que nous axons ce travail.
C’est la fibrose qui entraine l’apparition de symptômes chez le patient tels que l’hypertension portaleou encore les varices œsophagiennes causant des vomissements de sang chez ces mêmes patients. Legroupe de recherche du professeur Dessein a également démontré la présence de nombreux facteursgénétiques liés à la fibrose chez différentes populations et pathologies fibrosantes comme la fibrosecausée par le schistosome (20, 21) et le virus de l’hépatite C (22). Nous avons donc émis l’hypothèsequ’il était possible de prédire la susceptibilité des personnes à développer ces cicatrices. Enprévenant ces cicatrices, on informe le patient des risques et on permet aux praticiens hospitaliersd'adapter leurs soins. C'est un premier pas vers la médecine personnalisée, la médecine de demain.
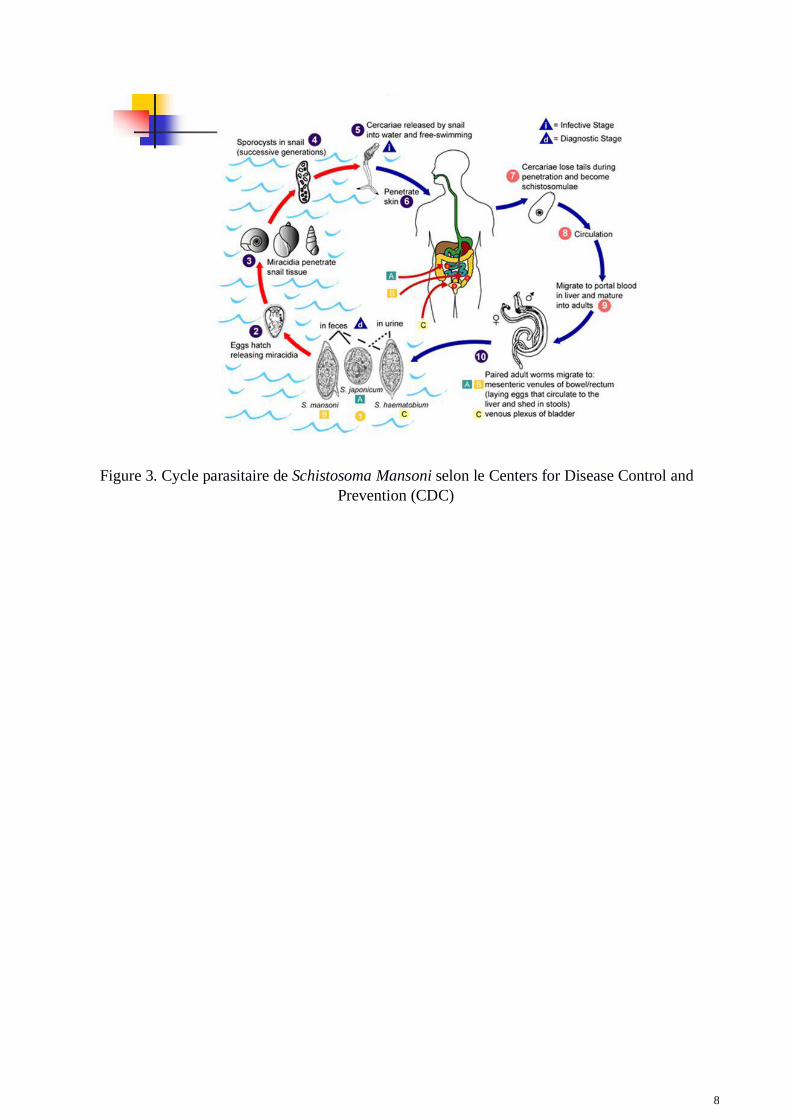
8
Figure 3. Cycle parasitaire de Schistosoma Mansoni selon le Centers for Disease Control andPrevention (CDC)

9
A) La cicatrisation normale, ses mécanismesa) Aspects généraux, fibrose
Pour comprendre les mécanismes impliqués dans la cicatrisation anormale tels que la fibrose, ilconvient dans un premier temps de comprendre le processus de cicatrisation normal. La cicatrisationnormale, qui intervient quand une plaie aiguë se forme sur un organe en contact avec le milieuextérieur (peau) ou interne (foie) se décompose en quatre phases précises et ordonnées (23) :l'hémostase, l’inflammation, la prolifération et enfin le remodelage (Figure 4).
Le terme de plaie est défini comme une perturbation de la structure et de la fonction anatomiquenormale d’un organe. Par conséquent la cicatrisation, c'est-à-dire le processus complexe etdynamique qui aboutit à la guérison de la plaie, permet la restauration du tissu et de sa fonctionanatomique à l'identique du tissu qui était présent avant la blessure (24).
Au moment où le tissu est blessé, les composants du sang se répandent au niveau de la lésion. Lesthrombocytes ou plaquettes sanguines sont les premières cellules à arriver sur site. Les plaquettessanguines entrent en contact avec les éléments de la matrice extracellulaire : c’est l’hémostase. Cecontact entraîne la libération de facteurs de coagulation, la formation d'agrégats de fibrine et unevasoconstriction des vaisseaux sanguins afin de stopper le saignement et limiter la perte de sang. Lecaillot de sang nouvellement créé joue le rôle de matrice de cicatrisation provisoire et lavasodilatation qui s'en suit permet la migration des cellules nécessaires à la suite du processus. Lerecrutement des plaquettes est essentiel à ce stade, il fournit le point de départ des cascades designalisations cellulaires et facteurs de croissances nécessaires à l'ensemble de la suite du processus(25). Elles activent l'ensemble des voies de signalisation nécessaires à l'initialisation du processus decicatrisation et de l’inflammation qui s'en suit en libérant des facteurs de croissance aussi appeléscytokines, comme par exemple les molécules PDGF et TGF-β (26, 27, 28).
Ensuite, au cours de l’inflammation, le système immunitaire inné entre en action pour éliminer lesdébris cellulaires et prévenir l'infection locale. Neutrophiles et monocytes entrent alors en jeu auniveau de la plaie afin de phagocyter les corps étrangers et bactéries qui auraient pu pénétrer dumilieu extérieur dans le cas de la peau ou provenant de l’intestin dans le cas de la fibrose du foie. Lesystème immunitaire élimine également les cellules qui auraient pu être endommagées suite à lalésion (29, 30).
L’objectif de la phase de prolifération est de reconstituer le tissu endommagé le plus rapidementpossible. Le fibroblaste est connu depuis plus de 50 ans comme étant la principale celluleresponsable du dépôt de collagène qui est nécessaire pour réparer la lésion tissulaire (31). Lesfibroblastes migrent alors sur le site de la plaie pour former et y déposer une nouvelle matriceextracellulaire.
La prolifération du collagène, principal constituant de la matrice extracellulaire, est nécessaire pourrestaurer la structure anatomique et la fonction de la peau blessée. Cette étape est particulièrementgourmande en énergie et en consommation d'oxygène. La formation de nouveaux vaisseaux sanguinsest donc essentielle. L’angiogenèse est principalement déclenchée par les molécules VEGF, FGF etTGF-β (32, 33).
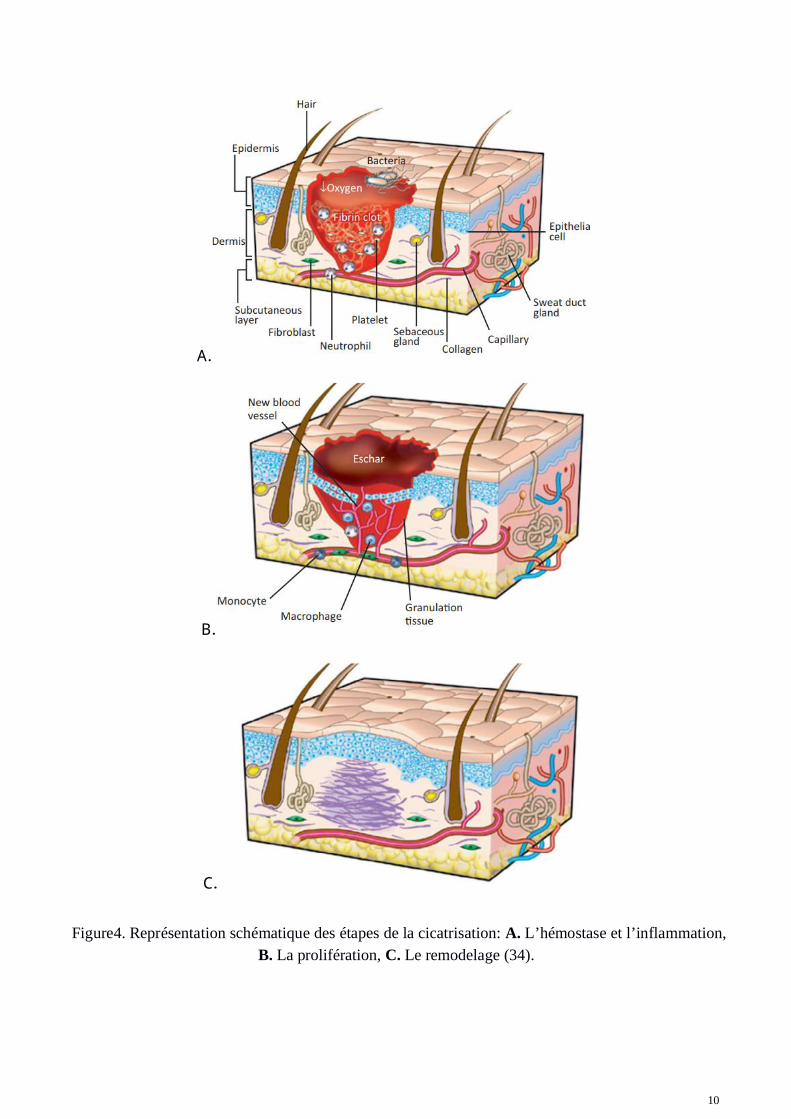
10
A.
B.
C.
Figure4. Représentation schématique des étapes de la cicatrisation: A. L’hémostase et l’inflammation,B. La prolifération, C. Le remodelage (34).

11
La matrice extra-cellulaire est un réseau complexe, dense et organisé de fibres de collagène,élastiques et réticulaires. Elle assure l'intégrité cellulaire des organes ainsi que leur résistanceélastique. Le collagène est la protéine la plus abondante dans le règne animal, représentant 30% desprotéines totales dans le corps humain (35). Il existe différents types de fibres de collagène mais lamatrice extra-cellulaire en rassemble 3 types spécifiques. Dans la peau les fibres de collagène de type1, 3 et 5 sont les plus présentes. La synthèse des fibres de collagène commence par la formation dupro-collagène. Soluble, le pro-collagène est assemblé par les fibroblastes et prend ainsi sa formationen triple hélices d'acides aminés (36). Il existe une trentaine de chaînes d'acides aminés pouvantconstituer une vingtaine de collagènes différents nommés avec des chiffres précédés de «COL». Parla suite, c'est l’excision des peptides C et N terminaux du pro-collagène qui le transforme encollagène. Insoluble, il s’auto-assemble alors au sein du maillage de collagène environnant formantun réseau qui s'étend sur 3 dimensions dans des zones bien définies de l’organe occupant ainsi toutl’espace du tissu dans lequel il se déploie.
La cicatrisation normale de la peau nécessite un équilibre entre les phases de prolifération quiforment la cicatrice et de remodelage. Cet équilibre permet la transformation de la cicatrice en peaunormale. Durant cette phase, la prolifération stoppe progressivement tandis que la cicatrice serésorbe pour rendre à la peau son aspect et sa fonction initiale. Pour cela, les fibrilles de collagèneprésentes pour la cicatrisation sont remodelées. La peau normale se reconstitue et les cellulesdevenues inutiles sont éliminées. La nouvelle matrice de collagène devient alors réticulée etorganisée.
Pour que ce processus de réparation efficace et hautement contrôlé ait lieu, de nombreux événementsde signalisation et migration cellulaire sont nécessaires. Tout dérèglement entre ces deux phases (ieprolifération versus remodelage) peut être interprété comme une réaction pathologique à lacicatrisation. En cas de cicatrisation excessive, le dépôt massif de tissu conjonctif se traduit par unestructure altérée et par conséquent par une perte de l'aspect initial de la peau (37). La fibrose descicatrices chéloïdes et hypertrophiques en sont une conséquence (38, 39). Dans la formation descicatrices chéloïdes autant que hypertrophiques, la phase de prolifération ne s’arrête pas et peut durerparfois des années. Cela va donc entraîner une cicatrisation anormale, abondante, visuellement nonfinie et parfois esthétiquement gênante.
Dans la pathologie de la fibrose du foie, les processus amenant à la fibrose sont similaires. Commedans la fibrose de la peau, la fibrose hépatique résulte de la capacité du foie à cicatriser après unelésion. A l’inverse de la peau où la fibrose est causée par une lésion souvent unique, la fibrose dufoie est causée par des lésions multiples et chroniques. Ces agressions chroniques peuvent avoirdiverses origines comme la présence de certains virus (hépatite C), de corps étrangers comme desparasites (œufs de schistosomes) ou encore après une intoxication chimique (alcoolisme,médicaments hors dose). Difficile à diagnostiquer, la fibrose hépatique naissante ne présente aucunsymptôme chez le patient. A un stade précoce, traiter correctement la pathologie sous-jacente auxlésions du foie peut permettre à la fibrose de régresser. Mais en absence de symptômes visibles, elleest souvent détectée plusieurs années voire dizaines d’années après son apparition.

12
La fibrose devient alors chronique et le patient présente le plus souvent les symptômes despathologies dont elle est la cause, comme la cirrhose ou encore le cancer hépatocellulaire. A ce stade,il est trop tard pour enrayer la fibrose (40). Le foie normal est composé de 3% de matrice extracellulaire formée de fibres de collagène de type 1, 3, 4 et 5 (41). La matrice extra cellulaire du foiene présente normalement pas d’altération dans l’organe adulte en bonne santé. Cependant celui-ci estcapable de cicatrisation cellulaire, la prolifération et le remodelage de la matrice extra-cellulaire sontdonc importants. Les éléments effecteurs de la fibrose du foie sont similaires à ceux de la fibrose dela peau. Les molécules métalloprotéinases de la matrice et leurs inhibiteurs tissue inhibitors ofmetalloproteinases (TIMPs), capables de dégrader et reformer la matrice extra cellulaire, sontimpliqués dans la fibrose du foie également (41).
b) Le foie
Une des principales caractéristiques qui différencie le foie des autres organes est sa capacité à serégénérer et même à reprendre sa forme initiale en cas de grave lésion. Le processus de cicatrisationdu foie tout comme celui de la peau, nécessite la formation de tissu cicatriciel, le recrutement defibroblastes et le dépôt de matrice extra-cellulaire.
Comme dans tous les processus de cicatrisation, ce sont les molécules produites par les celluleslésées qui activent le système immunitaire en premier et les cascades de signalisation nécessaires auprocessus d’inflammation. Dans le foie, le principal type cellulaire mis en jeu, recruté et impliquélors de inflammation sont les cellules hépatiques étoilées (Hepatic Stellate Cell HSC ou encorecellules stellaires). Situées au niveau de l’espace de Disse, les HSC ont initialement été découvertescomme réservoir de la vitamine A dans le foie et dans le pancréas. Elles représentent 5 à 8 % descellules du foie (42). Ce n’est que plus tard que leurs implications dans la régénération des tissus aété démontrées. Les HSC sont situées au niveau de l’endothélium vasculaire des vaisseaux sanguinshépatiques et ont une origine mésenchymateuse (figure 5). Les interactions entre les cellulesépithéliales et le mésenchyme jouent un important rôle dans établissement des fonctions des organeslors du développement de ceux-ci. La lame basale des cellules épithéliales et la matriceextra-cellulaire de l’organe assurent le maintien des caractéristiques des vaisseaux sanguins. Dans lefoie, la matrice extra-cellulaire située dans l’espace de Disse contrôle l’homéostasie. Toutes lescellules situées dans l’espace de Disse produisent du collagène de type 1. Les cellules endothélialesproduisent du collagène de type 4. Enfin les hépatocytes produisent de la fibronèctine et les cellulesstellaires, du collagène de type 3 et 4 (43). Cette production de matrice extra-cellulaire s’effectue enparallèle de sa dégradation. Cette dégradation est assurée par les métallo protéines de la matrice(MMP), elles-même régulées par les inhibiteurs des MMP appelées TIMP (44). Quand lacicatrisation est nécessaire, cet équilibre entre dégradation et production de matrice est rompu.Certains facteurs comme les molécules “A disintegrin and metalloproteinase” (ADAMs) (45)participent alors à ce changement et à l’activation des HSC. L’élément pivot de l’activation des HSCest leur capacité à se différencier en myo-fibroblastes.

13
Figure 5. Illustration des types cellulaires hépatiques et de leur agencement au niveau des sinusoïdeshépatiques (46)

14
Cette différenciation peut se faire sous effet de signaux activateurs comme la présence des facteursIL-22, IL-9 et IL-17A sécrétés par les cellules du système immunitaire tel que les cellules T CD4+.La présence de cellules B sécrétant les facteurs TNF Alpha ou encore les cellules NKT (NaturalKiller) sécrétant les molécules IL-4 et IL-13 permettent aussi cette différenciation. Elles pourront sedifférencier avec la présence de macrophages (cellules de Kupffer) capables de sécréter les facteursde croissance PDGF, TGFβ, TNF Alpha, et IL-1β. Enfin, cette différenciation est égalementauto-entretenue par les myo-fibroblastes eux-mêmes. Parmi tous ces facteurs et voies de signalisationimpliqués, la voie du TGF-β / BMP7 constitue un des mécanismes d’activation, celle-ci est décriteplus en détail, plus loin dans le manuscrit. Elle est aussi appelée voie des SMADs.
L’origine embryonnaire des HSC reste pour l’instant peu connue. L’étude de Keiko et son équipe (47)a tenté de déterminer la lignée cellulaire embryonnaire à partir de laquelle les cellules stellaires seforment en isolant des cellules du mésenchyme du foie fœtal chez la souris. Ceux-ci ont pu observerque leur origine provient probablement des cellules sous-mésothéliales et du mésoderme au stadeembryonnaire. D’autres études postulent une origine endodermique (48) ou encore neuroectodermique (49). Pour agir, les HSC différenciées en myofibroblastes migrent au sein du tissuhépatique. Les myo-fibroblastes sont absents dans les tissus sains. La migration cellulaire est unmécanisme et évènement essentiel chez les organismes multi-cellulaires (50). Il a été montré que lesmyo-fibroblastes présents dans le foie dérivent en premier de la différenciation des HSC puis, ensecond, des fibroblastes de la veine porte et des cellules mésothéliales (51). Dans le foie, ces cellulessont ainsi capables de synthétiser des fibres de collagène et participent alors, lors de la réparationnormale du tissu hépatique, au dépôt de la nouvelle matrice extra-cellulaire et à la formation desfibres de collagène. Ces fibres de collagène, dont le rôle est d’améliorer la cohésion cellulaire aprèsune lésion, disparaissent ensuite. Les HSC activées sont également éliminées par apoptose oueffectuent un retour à leur stade initial indifférenciée (52). Il existe donc des mécanismes de rétroaction capable de stopper l’activité de ces cellules. Le parallèle est à faire avec la cicatrisation de lapeau, qui, quand elle se produit normalement, doit maintenir un équilibre entre prolifération etremodelage pour que son processus soit optimal. Non seulement impliquées dans la production decette matrice extra cellulaire, les cellules étoilées jouent également un rôle dans son remodelage.
Les cellules étoilées sont capables de sécréter des molécules de la famille des métallo protéinasescomme MMP2, 9 et 13 dont le rôle est de dégrader les composants de cette matrice. A l’inverse,TIMP1 et 2 dont le rôle est d’inhiber l’activité de dégradation des MMPs, peuvent également êtresécrétées par ces même cellules. Afin de mieux comprendre la différenciation des HSC, il convientde définir la transition épithélio-mésenchymateuse. Ce mécanisme, qui ne se limite pas à l’étude descellules stellaires épithéliales, concerne la transformation des cellules épithéliales enmyo-fibroblastes. L’endothélium vasculaire est un des tissus cellulaires qui compose les vaisseauxsanguins. Il est situé directement au contact du sang, interne au vaisseau sanguin. Le mésenchyme,parfois appelé parenchyme à tort, est un tissu embryonnaire, précurseur de plusieurs autres tissusplus spécifiques comme les muscles, le cartilage ou les vaisseaux sanguins. Tous les deux dériventdu mésoderme lors des stades embryonnaires plus précoces. La transitionépithélio-mésenchymateuse concerne avant tout un changement de propriété et d’organisation destissus.

15
L’endothélium vasculaire, de par sa position dans le vaisseau sanguin et son contact avec le sang,joue plusieurs rôles bien précis. Tel une barrière naturelle, il est responsable de l’étanchéité desvaisseaux sanguins mais aussi de sa perméabilité à certains types cellulaires précis pour lesquels leurmigration dans les organes est nécessaire agissant ainsi comme un filtre. Pour remplir leurs fonctions,les cellules de l’endothélium sont polarisées et leur cohésion est assurée par leur adhérence à la lamebasale de collagène. Les desmosomes assurent également leurs cohésions.
La transition épithélio-mésenchymateuse, qui est un processus réversible, change radicalement lapropriété de ces cellules. La dégradation de la E-cadhérine au profit de la N-cadhérine entraine laperte de la cohésion et polarisation cellulaire leur permettant ainsi de migrer dans les tissusenvironnants. La β-caténine qui n’intéragit alors plus avec la E-cadhérine est libérée dans lecytoplasme cellulaire. Si la voie Wnt est activée, celle-ci joue un rôle de médiateur du signal de lavoie via sa translocation vers le noyau. Dans le cas contraire, elle sera dégradée. La voie Wnt amême été associée avec le TGF-β à la transition épithélio-mésenchymateuse dans le cadre de lafibrose pulmonaire (53).
Plus généralement, après sa transition, la cellule acquiert la propriété de pouvoir réagir aux différentssignaux extérieurs comme Wnt mais aussi d’autres molécules pro-inflammatoires, ce qui lui confèrela possibilité de sécréter les différents facteurs à l’origine de la prolifération cellulaire et de lacicatrisation / remodelage des tissus que nous avons vus précédemment.
c) La peau
La peau est l'organe le plus étendu du corps. Elle est composée de 3 sous-ensembles ou couchesappelées épiderme, derme et hypoderme. Entre le derme et l’épiderme se situe une lame basalecellulaire. En contact permanent avec l'environnement extérieur, la peau possède des caractéristiquesspécifiques pour protéger le corps de cet environnement. Elle est imperméable à l'eau, constitue unebarrière contre les pathogènes, protège des rayons ultraviolets et est capable de réparer les dommagesque ceux-ci pourraient causer à l'ADN. Elle participe également à la régulation de la température ducorps. C'est elle qui possède la pigmentation qui détermine notre couleur de peau. La figure 7présente la localisation des différents type cellulaires dont nous allons parler ci-après.
L'épiderme, la couche la plus externe, est directement en contact avec l’extérieur. Il protège doncl’organisme contre les éléments du milieu extérieur. Couche principalement visible de l’extérieur,c’est elle qui détermine la couleur de notre peau. Elle comporte des caractéristiques spécifiquescomme des pores qui permettent l’épanchement de sécrétions comme la transpiration, mais aussi desorifices pilaires pour la sortie des poils vers l’extérieur. Elle est parsemée de villosités naturelles quidonnent naissance aux empreintes digitales sur les doigts par exemple. Son aspect se modifie avecl’âge comme lors de l’apparition des rides en vieillissant.
C'est un épithélium stratifié avec, à sa base, une lame basale où s’effectue la mitose qui alimentecette couche en nouvelles cellules, qui remontent ensuite vers la partie externe au rythme desmitoses.

16
Au cours de leur migration du bas vers le haut, elles subissent des transformations morphologiques et,à l’extrémité externe de la peau (couche cornée), les plus vieilles cellules desquament au quotidiendans le milieu extérieur. Ces transformations permettent de définir des sous couches dans l’épiderme.Ces sous couches sont composées de kératinocytes uniquement qui présentent des caractéristiquesanatomiques distinctes. Les kératinocytes sont alimentés en oxygène par diffusion via le derme. Onretrouve, du bas vers le haut de la couche: la lame basale, le corps muqueux, la couche granuleuse etenfin la couche cornée qui est la plus externe. La lame basale est composée d’une seule couche dekératinocytes, les seuls pouvant effectuer une mitose intense et ainsi alimenter en nouvelles cellulesles couches supérieures de l’épiderme. Afin de pouvoir conserver la conformation de couchecellulaire unie, les kératinocytes sont liés à une membrane basale via des émi-desmosomes maiségalement entres elles par les desmosomes. Le corps muqueux est composé de kératinocytesnouvellement créés dont la forme est étirée et tend à s’aplatir à son sommet. Ils assurent en partiel’élasticité de la peau de par les liaisons avec les desmosomes et aussi par le fait que la kératine, quiles constitue, est jeune et malléable. Les cellules commencent à constituer leur réserve de mélanineau sein du cytoplasme. Ensuite on retrouve la couche granuleuse composée de kératinocytes aplatis.Leur cytoplasme perd progressivement ses organites pour ne conserver que des tonofilamentscomposés de kératine. En dernier, la couche cornée est composée de couches cellulaires totalementaplaties et kératinisées. Les grains de kératohyaline contenus dans le cytoplasme se réunissent avecles tonofilaments pour former une structure rigide, imperméable et extrêmement résistante. Ce sontles cellules de cette couche qui desquament. L'épiderme est également constitué d’autres typescellulaires, principalement des cellules de Merkel, mélanocytes et cellules de Langerhans. Lesmélanocytes, localisés au niveau de la couche basale, ont pour principale fonction de produire lamélanine qui sera ensuite stockée dans le cytoplasme des kératinocytes. Les cellules de Langerhans,rarement rencontrées dans la lame basale et absentes de la couche cornée, se localisent dans lescouches intermédiaires de la peau. Ces cellules dendrytiques font parties du système immunitaire,jouant un rôle phagocytaire et de reconnaissance d’antigène. Les cellules de Merkel participent ausystème nerveux, elles permettent les sensations du toucher en tant que récepteur sensitif. Situées surtout le corps, elles sont réparties de manière irrégulière. Elles sont plus présentes au niveau des zonesles plus sensibles de la peau, comme les lèvres ou les doigts. Elles sont situées au niveau de la lamebasale.
Le derme, juste en dessous, assure en priorité la fonction élastique de la peau et sa réponse au stresslors des déformations. D'une épaisseur de 1 à 2 mm, c'est un tissu conjonctif élastique qui contientles terminaisons nerveuses nécessaires au sens du toucher et de la chaleur. Il contient en autre lesfollicules pileux, les glandes sudoripares, sébacées et autres glandes apocrines, les vaisseauxlymphatiques et sanguins de l'organe. Il a un rôle de soutien de la peau, métabolique et fonctionnel.C’est la strate qui est mise en jeu en cas de cicatrisation et qui contient les cellules immunitaires de lapeau essentielles à la formation de la cicatrice. Le derme constitue la principale barrière du corpscontre les pathogènes extérieurs grâce aux cellules dendritiques. La principale population cellulairedu système immunitaire représenté ici est celle des macrophages. Produits dans la moelle osseuse,différenciés en monocytes dans le sang puis en macrophages dans le derme, ils sont capablesd'éliminer les débris cellulaires ou les pathogènes par phagocytose.

17
Ils peuvent aussi déclencher une réponse immunitaire spécifique si nécessaire.
Le derme se subdivise en deux sous parties, la région papillaire et réticulaire. La région papillaire estla plus externe, elle est connectée à l'épiderme par sa membrane basale. Composée en grande partiede matrice extra-cellulaire, c'est dans cette région que les fibroblastes, fibrocytes et myo-fibroblastessont les plus actifs.
D'une forme fusiforme ou étoilée avec de longs prolongements cellulaires, ceux-ci ont pour tâched'organiser la matrice extra-cellulaire. Grâce à leur capacité à produire ou dégrader la matriceextra-cellulaire, ils déterminent la structure du derme et lui confère sa résistance. Pour cela, ils sontcapables de sécréter différents types de collagènes, élastines, protéoglycanes et glycoprotéines : lescomposants essentiels à l’établissement de la structure de la matrice extra-cellulaire. Ce tissuconjonctif élastique est organisé par les fibres de collagène de type 1 et 3 orientéesperpendiculairement à la jonction derme/épiderme. Le collagène représente la majorité des protéinesde la peau. Les élastines confèrent également une résistance physique à la peau mais aussi chimique.Les protéoglycanes et glycoprotéines assurent, avec l’eau et les sels minéraux, l’ancrage des cellulesdans le maillage fibreux. Située en dessous, la région réticulaire composée d'un tissu conjonctif plusdense contient beaucoup moins de fibres de collagènes de type 3 que la strate précédente. A sa base,la transition entre un tissu fibreux et un tissu adipeux marque la frontière entre le derme etl’hypoderme. Enfin, le derme assure la régulation physiologique de la peau. Il assure la sécrétion dediverses molécules comme des cytokines, facteurs de croissance qui peuvent activer ou réprimertoute une machinerie cellulaire complexe de voies de signalisation. Il contient aussi différentesglandes comme les glandes sudorales produisant la sueur dont le principal objectif est de réguler latempérature du corps ou encore les glandes sébacées.
L’hypoderme, la couche la plus interne de la peau aussi appelée tissu adipeux, est maintenue enplace au niveau de la partie la plus profonde de la peau par les fibres de collagène. L’hypoderme «attache » la peau aux os et muscles sous-jacents. Il possède différents rôles. Un rôle métabolique : ilassure la régulation des lipides, principale source d'énergie de l'organisme. Il est capable se stockerou de libérer ces lipides sur demande. Il assure aussi un rôle mécanique pour la protection desorganes situés directement au-dessous le lui. C’est la graisse hypodermique qui détermine notresilhouette extérieure, qui peut varier avec le temps, notamment lors du vieillissement par exemple. Ilassure également l'homéostasie de l'organisme comme la thermorégulation. Cette dernière couche estvascularisée, alimentée par les artères sous cutanées qui se répartissent sous la peau.
La cicatrisation de la peau se produit naturellement quand une lésion des couches décrites ci-dessussurvient. Son objectif est de refermer la plaie au plus vite par la formation d’une suture cutanée. Pourcela, les mécanismes de formation de la cicatrice (phase de prolifération) mais aussi d’épidermisation(phase de remodelage) sont mis en jeu. La cicatrisation normale requiert un enchaînement précisentre les différentes phases de la cicatrisation évoquées précédemment : l’hémostase, l’inflammation,la prolifération et le remodelage. Ces phases mettent en jeu diverses molécules dont la production oul’inhibition est finement régulée par l’organisme. Durant la phase de l’hémostase, ce sont les facteursPDGF, les FGF et les VEGF qui permettent le recrutement des plaquettes sanguines au niveau de laplaie (54).
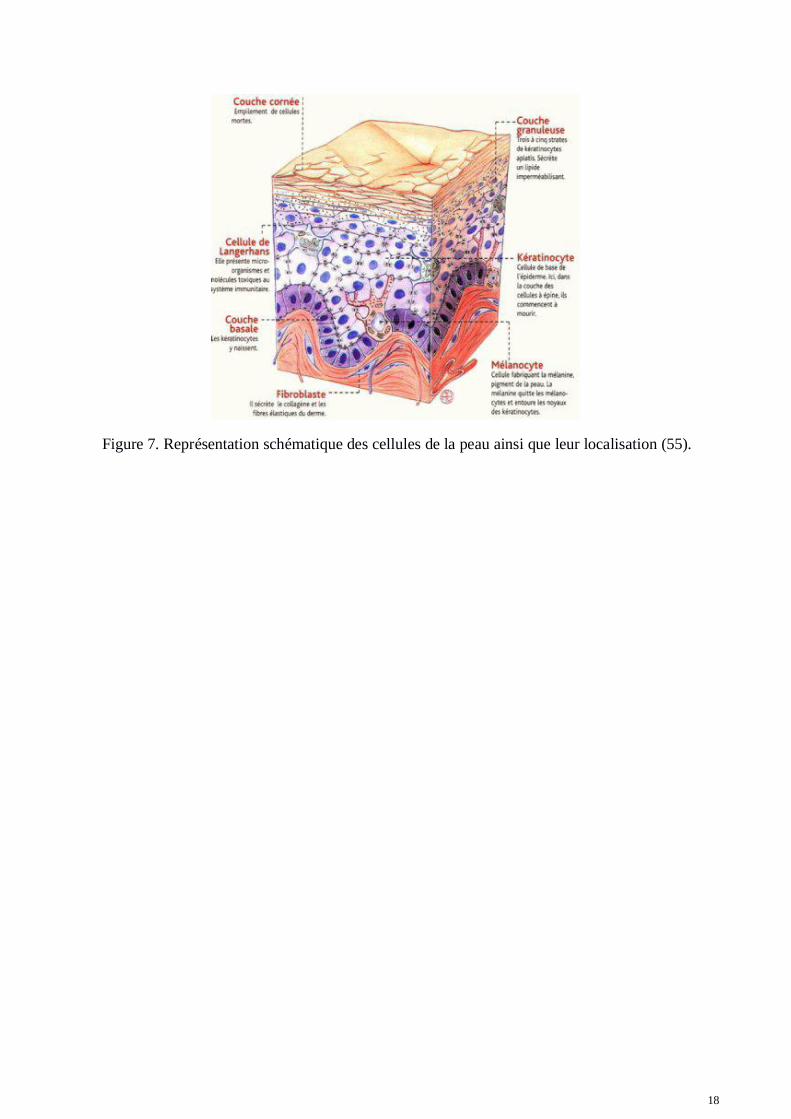
18
Figure 7. Représentation schématique des cellules de la peau ainsi que leur localisation (55).

19
L’inflammation de la peau peut durer jusqu’à deux semaines après la lésion. Physiquement elle seressent par des douleurs et rougeurs autour et sur la plaie. Les cellules du système immunitairecomme les macrophages, les neutrophiles et les lymphocytes T sont recrutés au niveau de la plaie(56). Des molécules d’adhésion cellulaire (CAM, Cell Adhésion Molecules) permettent lerecrutement de ces types cellulaires. Ces CAM sont notamment portées par les fibroblastes recrutéspar la présence des molécules TNF-α et IL-1 sécrétés par les neutrophiles ainsi que l’IL-2 sécrété parles lymphocytes. Les macrophages produisent de nombreux facteurs de croissance comme le PDGF,les TGF-β, des FGF, IL-1 et IL-6. Ces derniers facteurs permettent à la plaie d’entrer en phase deprolifération. Durant la prolifération, c’est majoritairement le PDGF qui stimule la prolifération desfibroblastes, la production des collagènes et la sécrétion de métalloprotéinases (MMPs). Ainsi lesfibres de collagène, molécules structurales de la peau, sont assemblés et déposées dans le tissu. Dansle même temps, les MMPs dont l’activité principale est la dégradation des composants de la matriceextra cellulaire, permettent de maintenir l’action des fibroblastes au front de la plaie. Durant la phasede remodelage, les fibroblastes situés dans les régions où la plaie est refermée sont ainsi dégradés etles types cellulaires devenus inutiles au fonctionnellement de la peau normale sont également lysés.Le TGF-β et le CTGF sont impliqués dans cette transition entre peau cicatricielle et peau normale.Le facteur VEGF contribue à l’angiogenèse, permettant la reconstruction des vaisseaux sanguins dela peau. L’épidermisation se termine par la migration des cellules épithéliales dans la matricenouvellement formée permettant ainsi à la peau de retrouver ses fonctions normales. Cette migrationest permise par les facteurs EGF et les keratinocyte growth factors (KGFs). La peau normale, aprèsreconstruction, doit atteindre 80% de l’élasticité attendue sur une peau n’ayant jamais cicatrisée pourvalider sa bonne reconstruction (54)

20
B) La cicatrisation pathologique et la fibrosea) Aspects généraux
La fibrose peut concerner n'importe quel tissu de n'importe quel organe dans le corps avec desmécanismes de mise en place différents. Quel que soit le tissu, la fibrose se caractérise par uneanomalie quantitative des cellules de la matrice extra-cellulaire entraînant des structures fibreuses quis'accumulent dans les tissus et organes atteints. Cette anomalie quantitative est caractérisée par deuxdérèglements distincts, celui entraînant une synthèse trop importante de matrice extra-cellulaire maisaussi à une capacité, quasiment absente, à dégrader cette matrice. C'est tout le processus deremodelage du tissu qui est atteint.
L'accumulation si spécifique à la fibrose provient en premier lieu d'un dérèglement des fibroblastesqui possèdent ainsi une activité de prolifération supérieure à la normale. Ces fibroblastes sontcaractérisés par un réticulum endoplasmique rugueux prononcé et des fibres stressées avec un largenoyau (57)
Ce dérèglement provient de facteurs dit « pro-fibrosant ». Ce terme englobe l'ensemble des facteursde croissance qui peuvent être sécrétés, de manière autocrine ou paracrine, par l'ensemble des typescellulaires impliqués lors de l’inflammation. Initialement appelés facteurs de croissance car ils ontété associés à la croissance cellulaire dès leurs découvertes, ces molécules sont aujourd'hui bienconnues pour jouer des rôles physiologiques plus vastes comme dans la fibrose, les cancers et laprolifération cellulaire (58, 59).
De nombreux facteurs de croissance ont été associés à la fibrose. Le Transcription Growth Factorbeta (TGF-β), par exemple, est impliqué dans la cicatrisation anormale et le Platelet Derived GrowthFactor (PDGF), composé de ses chaînes A et B, joue un rôle dans la prolifération, migration et lechimiotactisme cellulaire. Il est ainsi capable d'initier le recrutement des cellules des muscles lisseset des fibroblastes, acteurs principaux de la fibrose. Les Fibroblast Growth Factors (FGF) constituentun groupe de 23 molécules différentes nommées de FGF 1 à 23 qui agissent via les récepteurs FGFR1 à 5. Ils possèdent un effet fortement fibrosant et sont impliqués dans la prolifération cellulaire etl’angiogenèse. Certaines Interleukines (IL) et des molécules ayant un effet vaso-constricteur commel'angiotensine ou l'endothéline contribueraient à la fibrose.
L’identification des fibroblastes produisant anormalement du collagène sous l'effet de ces moléculesse fait par l’utilisation d’anticorps spécifiques des fibres Alpha des muscles lisses, c'est pourquoil’appellation « fibroblastes » et « myo-fibroblastes » est souvent équivalente dans la littérature. Bienque de nombreux types de fibroblastes existent, seuls ceux exprimant la Fibroblast specific protein 1(FSP1) ont été clairement identifiés comme étant directement impliqués dans la fibrose de diversorganes. FSP1 pourrait même servir de marqueur pour détecter la fibrose (60). De plus, différentstypes cellulaires peuvent se différentier en fibroblastes donnant ce qui peut jouer sur leur capacité deprolifération.
Enfin, de nombreuses enzymes cataboliques de la matrice extra-cellulaire sont également impliquées,comme les Matrix Metallo protéases (MMP) (61, 62, 63, 64).

21
Le nombre important des molécules et types cellulaires impliqués dans la fibrose nous amène àpenser que le processus de fibrose est vaste et reste encore largement incompris par la communautéscientifique. Un tissu présentant une fibrose anormale arbore un aspect macroscopique pâle, uneraréfaction microvasculaire et n’a pas la capacité à se régénérer correctement ni même à former denouveaux vaisseaux sanguins. Cela entraîne une hypoxie chronique du tissu. Le facteur hypoxiqueest souvent retrouvé dans les fibroses de type chéloïde. En cas de faible quantité d'oxygène dans letissu, le facteur Hypoxia Inducible Factor (HIF) est induit par les cellules endothéliales vasculaires(65). Ce facteur est capable de réguler l'expression du Vascular Endothelial Growth Factor (VEGF)et de stimuler ainsi l'angiogénèse. Différentes études sur les conséquences de la fibrose du foie et dela peau attestent que l’inflammation et le manque d’oxygène présentés par les tissus fibrotiques sontdes facteurs stimulant la formation de nouveaux vaisseaux sanguins (66, 67).
Le tissu fibreux présente souvent une inflammation chronique dite stérile, la plupart du temps causéepar la présence de cellules en apoptose au sein du tissu qui se dégrade. Mais cela n'est pas toujours lecas, par exemple quand la fibrose est causée par une infection comme dans le foie. Le tissu fibreuxpossède un aspect plus consistant, dur et rigide pouvant amener à la déformation de l'organe quiaugmente parfois en volume. La fibrose n'est pas un processus physiologique réversible, les tissusfibreux régressent rarement et ce, même si le facteur déclenchant peut être traité. Pire encore, lafibrose peut même avoir la capacité de s'auto-entretenir au point que certaines fibroses peuvent durerdes années (68).
On peut distinguer 2 types de fibroses selon leurs âges :
● Les fibroses jeunes ou récentes, avec des tissus richement vascularisés, une matrice encore peudense et une forte inflammation.
● Les fibroses plus anciennes dites chroniques, avec toutes les caractéristiques évoquées ci-dessuscomme une faible vascularisation, une inflammation décroissante et des fibres de collagènesorganisées en faisceaux, certaines fois même calcifiées.
La dangerosité en termes de santé publique et de mortalité des patients atteints de fibroses graves,n'est pas due à la fibrose elle-même, mais bien à ses conséquences. C'est la perte de fonctionprogressive de l'organe atteint qui cause un réel problème chez les patients, le tissu fibreux invasifempêchant ainsi l'accomplissement des fonctions principales de l'organe touché.
La gravité de la fibrose en termes de pronostic vital dépend donc uniquement de la localisation decelle-ci. Les chéloïdes qui sont des fibroses bénignes de la peau survenant au cours du processus decicatrisation n’engagent généralement jamais de pronostic vital. A L'inverse, une fibrose du foie peutcauser la mort du patient par insuffisance hépatique.

22
b) Le foie
Comme nous l’avons vu précédemment, le foie est un organe avec une capacité de cicatrisation horsdu commun. Mais cette capacité peut entraîner des problèmes dès lors qu’elle est trop souventsollicitée, par exemple, en cas de lésion régulière ou inflammation chronique de l’organe suite auvirus de l’hépatite C ou B, un problème métabolique, la stéatose non alcoolique (NASH) ou encorel’alcool. Ainsi, si les lésions sont régulières et trop importantes, ce processus de cicatrisation peutremplacer les cellules hépatiques par une accumulation de tissus cicatriciels. Ces tissus cicatriciels necontribuant pas aux fonctions hépatiques, l’organe perd progressivement ses capacités hépatiques. Lecancer hépato cellulaire est le 5 ième cancer le plus retrouvé dans le monde et il représente la 3 ièmecause de mortalité des cancers dans le monde (69). Il survient le plus souvent chez les patientspossédant une fibrose du foie avancée et persistante que l’on appelle cirrhose (70). Cependant,certains patients peuvent avoir une fibrose avancée du foie sans cancer et d’autres développeront uncancer rapidement malgré un faible niveau de fibrose. Dans cette étude, nous faisons l’hypothèsequ’une large composante génétique pourrait expliquer cette prédisposition chez certains sujets plutôtque d’autres.
Ces fibroses donnent un aspect rigide au foie, entraînent la constriction des vaisseaux sanguinshépatiques dont la circulation est altérée donnant parfois lieu à des hémorragies dans le systèmegastrique. Dans le foie, c’est l’activation des HSC et la transition épithélio-mésenchymateuse quipermet la cicatrisation. Cette transition, normale à l’état embryonnaire, est le plus souvent associée àla formation de métastases en cas de cancer chez l’adulte. Son implication dans les processus defibrose est aujourd’hui bien connue comme dans le cadre de la fibrose cardiaque (71). A l’inverse, ila été démontré que BMP7 était capable de contrer l’effet du TGF-β sur la transitionépithélio-mésenchymateuse en induisant à nouveau la production de E-cadherin nécessaire à laréparation de la cohésion cellulaire spécifique à l’épithélium vasculaire (72).
Les cellules stellaires, qui font partie de l’endothélium, sont normalement caractérisées par un étatquiescent contenant des gouttelettes lipidiques cytoplasmiques. Elles se différencient enmyo-fibroblastes uniquement lors de l’apparition d’une lésion. Une fois la cicatrisation terminée,elles perdent leur activité fibrosante, soit en retournant dans un état inactivé, soit en entrant enapoptose. Lorsque des dérèglements sur ces processus surviennent, les myo-fibroblastes nes’inactivent pas et continuent de jouer leur rôle de manière incontrôlée. Leurs caractéristiqueschangent, elles prolifèrent, deviennent contractiles et présentent une sécrétion importante d’élémentsde la matrice extra-cellulaire qui s’accumulent anormalement.
S’en suit alors une accumulation de fibres de collagène de type 1 et 3 formant des fibres stressées etrigides au sein de l’organe entraînant la fibrose hépatique (73). Dans certains cas, une fibrose de laveine porte dite fibrose péri-portale apparait. Il a été récemment proposé que le type de microbioteintestinal ou encore le stress oxydatif (notamment dans les cirrhoses non alcooliques de type NASH)joueraient également un rôle dans la cicatrisation excessive contribuant au maintien del’environnement pro-fibrotique.

23
c) La peau
Les principales fibroses au niveau de la peau se forment lors de cicatrisations anormales qui sedéclinent en deux pathologies : les cicatrices chéloïdes et les cicatrices hypertrophiques. Ce sont destumeurs cutanées bénignes qui se forment pendant le processus de cicatrisation de la plaie en réponseà une lésion cutanée ou après grattage, opération chirurgicale, piercing de l'oreille ou brûlures. Ellesconcernent toutes les deux un dérèglement aboutissant à un processus de fibrose. À ce jour, aucunesolution thérapeutique satisfaisante n'a encore été découverte ou proposée pour enrayer cesproblèmes de cicatrisation (74, 75, 76, 77). Les fibres de collagène sont l'entité la plus abondantedans la peau. Les fibres de collagène de type 1 représentent jusqu’à 80 % des collagènes retrouvésdans la peau, suivi par le type 3 à environ 20 % et enfin le type 5 à mois de 2 %. Il a été démontréque les fibroblastes isolés à partir d'un tissu chéloïdien produisent 2 à 3 fois plus de collagène que lesfibroblastes qui composent la peau normale des mêmes patients (78). Dans le derme normal, lesfibrilles de collagène situées dans les espaces interstitiels du tissu dermique sont organisées engrappes de tailles variables. Par contre, en cas de cicatrisation anormales chéloïdes ouhypertrophiques, les fibrilles de collagène présentent un alignement unidirectionnel, stressées parmanque de remodelage et par la croissance au-delà de la cicatrice originale. Pour les cas les moinsprononcés, il est quasiment impossible de différentier visuellement une cicatrice chéloïde d'unecicatrice hypertrophique. Il existe cependant des différences fondamentales entres les deuxpathologies. Les cicatrices hypertrophiques régressent généralement quelques années après leursapparitions (1 à 2 ans) tandis que les cicatrices chéloïdes ne régressent généralement passpontanément (79). Les cicatrices chéloïdes se développement au-delà de la cicatrice initiale pouvantdevenir assez proéminentes pour certaines, lui confèrent un aspect invasif (80). Au niveauanatomique, les cicatrices chéloïdes présentent une absence de myofibroblastes alors que ceux-cisont abondants dans les cicatrices hypertrophiques (80). La cicatrice chéloïde ne régresse passpontanément et continue à croître de façon aberrante au-delà de la frontière originale de la coupure.
Les cicatrices chéloïdes sont caractérisées par un taux élevé de proliférations fibroblastiques (81, 82,83), par l'accumulation de matrice extra-cellulaire et par le dépôt excessif de collagène, en particulierle collagène de type I et III (84, 85, 86, 87). Pour notre étude, nous avons travaillé à partir debiopsies de peau humaine provenant de tissus cicatriciels de cicatrices chéloïdes, de tissus cicatricielsprovenant de cicatrices hypertrophiques, de cicatrices normales et de peaux normales (Table 1). Lacicatrisation exubérante peut apparaître n'importe où sur la peau. De nombreux facteurs tels quel'hypertension cutanée (88, 89), l'infection de la plaie (90), la pilosité, les hormones (91) ont étéavancées pour expliquer l’appartition de ces lésions fibro-prolifératives dermiques. Maisl'appartenance à une population et la prédisposition génétique sont de loin l'hypothèse la plusplausible pour expliquer ces cicatrices. En effet les personnes à peaux noires auraient entre 5 à 10fois plus de prévalence à développer des cicatrices chéloïdes que les peaux blanches (92). Il estaujourd'hui bien établi que la prédisposition à la formation de chéloïdes est retrouvée principalementchez les populations africaines et asiatiques. La prévalence dans les populations africaines etchinoises est d'une personne atteinte sur 30 contre seulement 1 sur 625 aux États-Unis (93).L'héritabilité de la pathologie au sein d'une même famille est également très forte (94). Nouspouvons fortement supposer qu'il existerait donc des prédispositions héréditaires à cette pathologie.
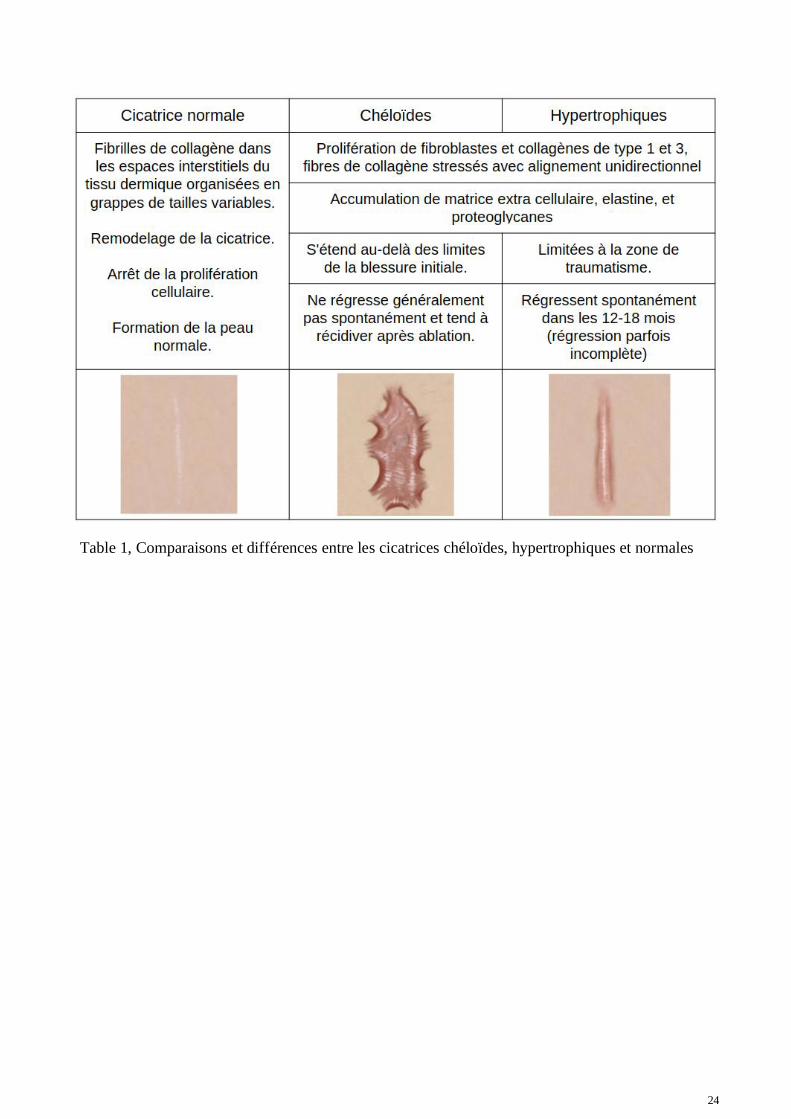
24
Table 1, Comparaisons et différences entre les cicatrices chéloïdes, hypertrophiques et normales

25
Cela suppose naturellement un fort déterminisme génétique pour ces pathologies. Cependant, aucungène spécifique ou mécanisme moléculaire expliquant la formation des chéloïdes n'a encore étédécouvert. La cicatrisation de type chéloide semble correspondre à une maladie multigénique et doncprésentant une pathogénie complexe. Cela rend plus difficile la mise en œuvre d'un traitementsatisfaisant ainsi qu'une prise en charge clinique efficace.
Les traitements disponibles pour enrayer la cicatrisation pathologique de type chéloïdes restent peuefficaces et limités (95, 96, 97, 98, 99). Une approche génétique serait donc indiquée pour améliorerla prise en charge des patients face aux risques de développer ces cicatrices aberrantes. Lesmécanismes de résistance aux thérapies actuelles et leur inefficacité due à un taux de récidive élevémême après traitement restent inconnus. Malgré le fait que les mécanismes de ces type decicatrisation soient encore très mal connus, de nombreuses molécules, voies de signalisationsgénomiques et mutations dans ces voies (mutations uniques dit SNP pour Single NucléotidePolymorphism) ont été identifiés. Ces cibles peuvent jouer un rôle important dans la formation descicatrices chéloïdes, la prolifération des fibroblastes et l'accumulation de la matrice extra-cellulaire.De nombreuses études sur les cicatrices chéloïdes et hypertrophiques ont été réalisées à ce jour.Parmi elles, certaines études ont mis en évidence des régions génomiques au sein desquelles desmutations pouvaient constituer de bons marqueurs de la pathologie. Les études d’expression par« Microarray » sont utilisées pour mesurer les niveaux d'expression d’un panel de gènes présents surla puce. L’analyse consiste à comparer les profils d'expression de biopsies provenant de sujetsregroupés en deux catégories : les sujets présentant une cicatrice chéloïde et les témoins. Les biopsiesprélevèes sur le premier groupe de patients, sont effectués sur les tissus chéloïdiens. En ce quiconcerne les biopsies témoins, elles correspondent à des prélèvements sur une peau normale de sujetsne présentant pas de cicatrices chéloïdes ou encore une peau normale prélevée à côté de la cicatricechéloïde sur le même patient. Les résultats obtenus définissent une liste de gènes dérégulés, pourcertains sur-exprimés et pour d’autres sous-exprimés dans le tissu de la cicatrice chéloïde. La table 2présente un ensemble de gènes ayant été observés dans la littérature comme impliqués dans lapathologie des chéloïdes. Dans la table 3 sont énoncés des variants génétiques SNPs présentant uneassociation avec la pathologie des chéloïdes (liste non exhaustive).
Dans l'article de Smith et de son équipe (100), la comparaison du profil transcriptomique defibroblastes chéloïdiens en culture avec et sans hydrocortisone (réduit l’inflammation) a mis enévidence 175 gènes sur-exprimés et 559 gènes sous-exprimés. Ils mettent ainsi en évidencel’implication des gènes HOX des complexes A, B, C et D situés dans les régions 7p14 et 12q13.3.Une analyse d’expression similaire entre un tissu chéloïde traité avec des radiations et un tissunormal révèle l'implication de la voie de l’interleukine 6 (IL-6) et a mis en évidence 28 gènessur-exprimés et 68 sous-exprimés montrant ainsi la répression de nombreux gènes sous l’effet desradiations, traitement potentiel contre les cicatrices chéloïdes (101).
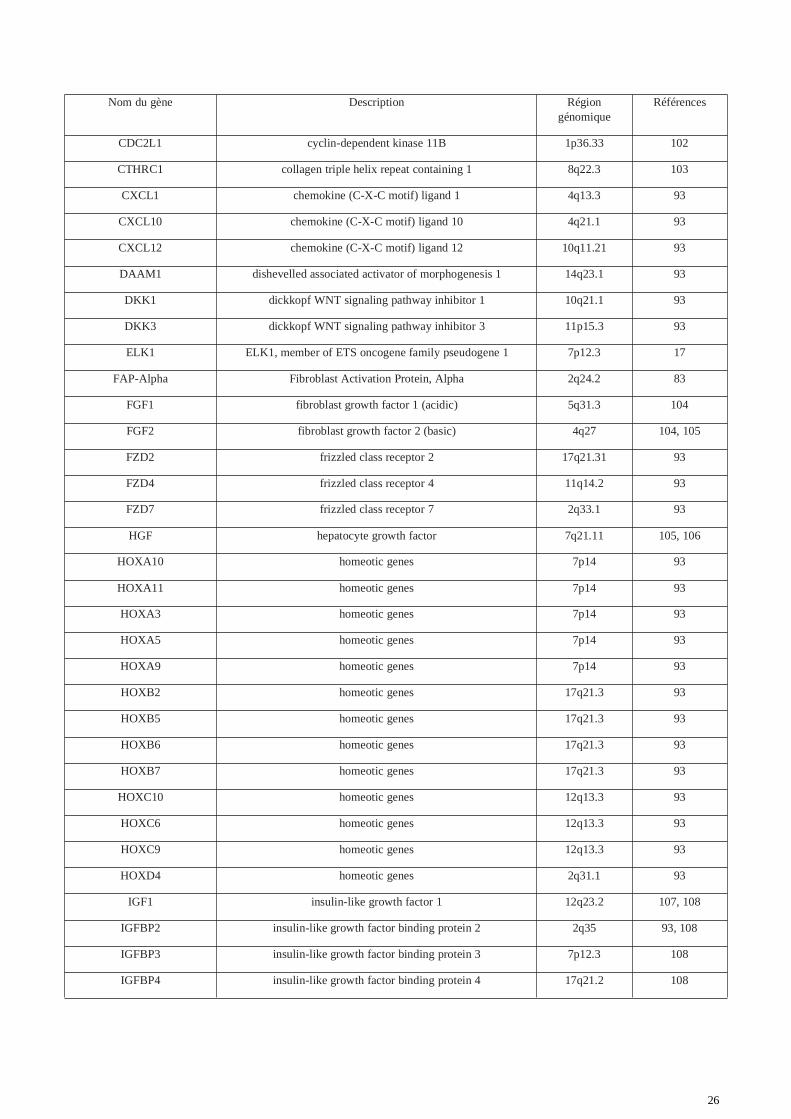
26
Nom du gène Description Régiongénomique
Références
CDC2L1 cyclin-dependent kinase 11B 1p36.33 102
CTHRC1 collagen triple helix repeat containing 1 8q22.3 103
CXCL1 chemokine (C-X-C motif) ligand 1 4q13.3 93
CXCL10 chemokine (C-X-C motif) ligand 10 4q21.1 93
CXCL12 chemokine (C-X-C motif) ligand 12 10q11.21 93
DAAM1 dishevelled associated activator of morphogenesis 1 14q23.1 93
DKK1 dickkopf WNT signaling pathway inhibitor 1 10q21.1 93
DKK3 dickkopf WNT signaling pathway inhibitor 3 11p15.3 93
ELK1 ELK1, member of ETS oncogene family pseudogene 1 7p12.3 17
FAP-Alpha Fibroblast Activation Protein, Alpha 2q24.2 83
FGF1 fibroblast growth factor 1 (acidic) 5q31.3 104
FGF2 fibroblast growth factor 2 (basic) 4q27 104, 105
FZD2 frizzled class receptor 2 17q21.31 93
FZD4 frizzled class receptor 4 11q14.2 93
FZD7 frizzled class receptor 7 2q33.1 93
HGF hepatocyte growth factor 7q21.11 105, 106
HOXA10 homeotic genes 7p14 93
HOXA11 homeotic genes 7p14 93
HOXA3 homeotic genes 7p14 93
HOXA5 homeotic genes 7p14 93
HOXA9 homeotic genes 7p14 93
HOXB2 homeotic genes 17q21.3 93
HOXB5 homeotic genes 17q21.3 93
HOXB6 homeotic genes 17q21.3 93
HOXB7 homeotic genes 17q21.3 93
HOXC10 homeotic genes 12q13.3 93
HOXC6 homeotic genes 12q13.3 93
HOXC9 homeotic genes 12q13.3 93
HOXD4 homeotic genes 2q31.1 93
IGF1 insulin-like growth factor 1 12q23.2 107, 108
IGFBP2 insulin-like growth factor binding protein 2 2q35 93, 108
IGFBP3 insulin-like growth factor binding protein 3 7p12.3 108
IGFBP4 insulin-like growth factor binding protein 4 17q21.2 108
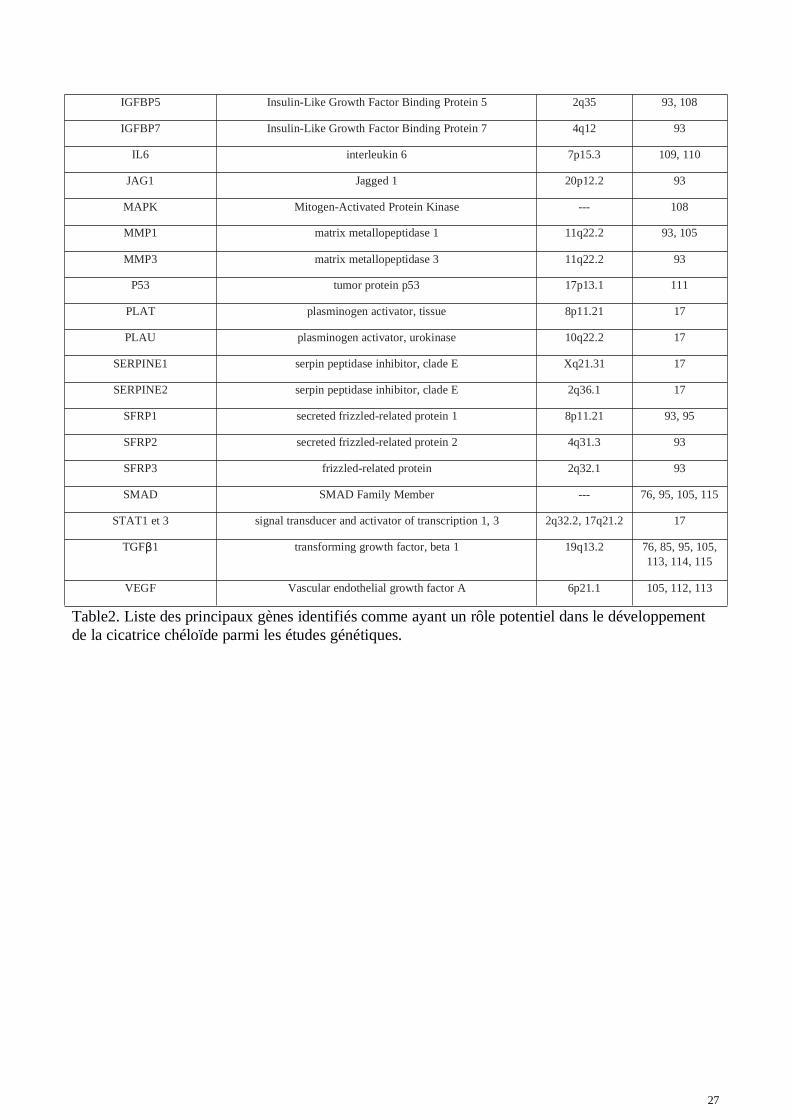
27
IGFBP5 Insulin-Like Growth Factor Binding Protein 5 2q35 93, 108
IGFBP7 Insulin-Like Growth Factor Binding Protein 7 4q12 93
IL6 interleukin 6 7p15.3 109, 110
JAG1 Jagged 1 20p12.2 93
MAPK Mitogen-Activated Protein Kinase --- 108
MMP1 matrix metallopeptidase 1 11q22.2 93, 105
MMP3 matrix metallopeptidase 3 11q22.2 93
P53 tumor protein p53 17p13.1 111
PLAT plasminogen activator, tissue 8p11.21 17
PLAU plasminogen activator, urokinase 10q22.2 17
SERPINE1 serpin peptidase inhibitor, clade E Xq21.31 17
SERPINE2 serpin peptidase inhibitor, clade E 2q36.1 17
SFRP1 secreted frizzled-related protein 1 8p11.21 93, 95
SFRP2 secreted frizzled-related protein 2 4q31.3 93
SFRP3 frizzled-related protein 2q32.1 93
SMAD SMAD Family Member --- 76, 95, 105, 115
STAT1 et 3 signal transducer and activator of transcription 1, 3 2q32.2, 17q21.2 17
TGFβ1 transforming growth factor, beta 1 19q13.2 76, 85, 95, 105,113, 114, 115
VEGF Vascular endothelial growth factor A 6p21.1 105, 112, 113
Table2. Liste des principaux gènes identifiés comme ayant un rôle potentiel dans le développementde la cicatrice chéloïde parmi les études génétiques.
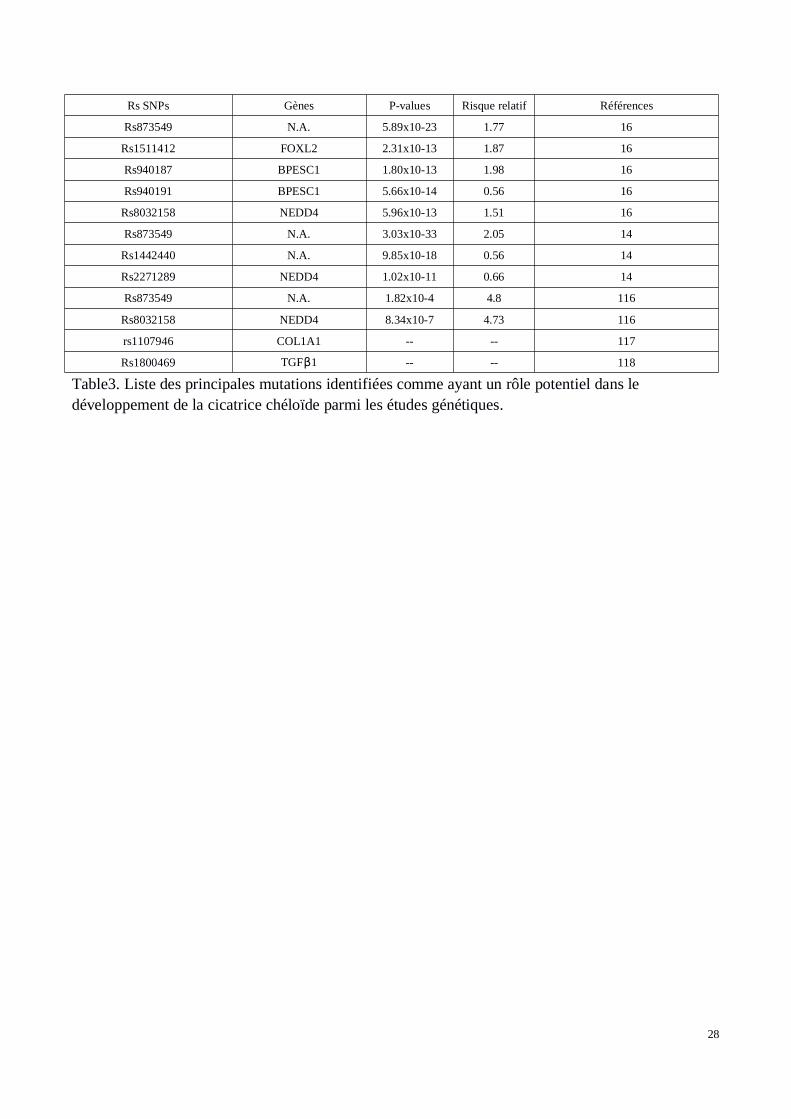
28
Rs SNPs Gènes P-values Risque relatif Références
Rs873549 N.A. 5.89x10-23 1.77 16
Rs1511412 FOXL2 2.31x10-13 1.87 16
Rs940187 BPESC1 1.80x10-13 1.98 16
Rs940191 BPESC1 5.66x10-14 0.56 16
Rs8032158 NEDD4 5.96x10-13 1.51 16
Rs873549 N.A. 3.03x10-33 2.05 14
Rs1442440 N.A. 9.85x10-18 0.56 14
Rs2271289 NEDD4 1.02x10-11 0.66 14
Rs873549 N.A. 1.82x10-4 4.8 116
Rs8032158 NEDD4 8.34x10-7 4.73 116
rs1107946 COL1A1 -- -- 117
Rs1800469 TGFβ1 -- -- 118
Table3. Liste des principales mutations identifiées comme ayant un rôle potentiel dans ledéveloppement de la cicatrice chéloïde parmi les études génétiques.

29
C) Voies de signalisation de la fibrose
a) Voie Wnt/β-caténine
La voie Wnt joue un rôle important dans la cicatrisation et les cancers. Découverte il y a maintenantplus de 40 ans, la première mise en évidence du processus Wnt fut chez Drosophila melanogaster.Ces découvertes ont mis en avant son action sur le développement (119). Ça n’est que des annéesplus tard et à l’aide du modèle murin que l’implication de Wnt fut observée dans les processustumoraux et les développements cellulaires anormaux (120).
Le terme Wnt regroupe 19 gènes chez l’homme. Chacun d’eux sont capables d’influencer, stimulerles différentes voies de signalisation propres au processus Wnt. Il existe deux principales cascades designalisations cellulaires pouvant être stimulées par les molécules Wnt : la voie canonique et lesvoies non canoniques. Parmi les voies non canoniques on retrouve la voie Planar Cell Polarity (PCP)et du calcium (Ca2+) (121). Les molécules Wnt se fixent sur leurs récepteurs membranaires composésde LRP5, LRP6 et Frizzled (Fzd). Fzd est capable de lier toutes les molécules Wnt et d’activer toutesles cascades de signalisation Wnt en aval (122). Cependant, chacune des différentes molécules Wntagira préférentiellement sur une voie plutôt qu’une autre. Cela est principalement dû au fait qu’ilexiste plus de 15 récepteurs des molécules Wnt différents, chacun ayant plus ou moins d’affinitéavec les Wnts (122). C’est la combinaison des récepteurs Wnt entre eux et la fixation du Wnt àceux-ci qui déterminent l’activation en aval d’une cascade de signalisation plutôt qu’une autre.
La voie canonique Wnt (Figure 7) est associée à la molécule β-caténine qui y joue un rôleessentiel. C’est historiquement la voie la mieux décrite et la plus connue. En absence de fixation dela molécule Wnt sur son récepteur trans-membranaire, la β-caténine intra-cellulaire fait partie d’uncomplexe constitué des molécules APC, GSK-3B, PP2A, AXIN-1 et CK-1 qui entraînent sadégradation par le protéasome. CK1 a pour rôle de phosphoryler la β-catenine en Ser45 et GSK-3Ben Ser33 et Ser37. Cette phosphorylation permet la fixation de β-TRCP amenant à l’ubiquitinisationet dégradation de la β-catenine (123).
La fixation de Wnt1, Wnt3A ou Wnt8 à LRP5, 6 et Fzd, active la cascade de signalisation Wnt grâceau recrutement de DVL (Dishevelled). LRP5 et 6 sont alors phosphorylés et permettent la fixation deAXIN1 à ce complexe trans-membranaire. Ainsi la dégradation de la β-caténine est inhibée. D’unpoint de vue biologique, la formation du complexe fixé aux molécules trans-membranaires n’est passuffisante pour expliquer l’absence de formation du complexe de dégradation de la β-caténine.GSK3B, recruté au niveau du récepteur pour phosphoryler LRP5 et 6, reste toujours capable deformer le complexe qui amènera à la dégradation de la β-catenine. L’hypothèse la plus probable estque GSK3B soit inhibé par DVL afin que la β-caténine reste intacte. Ce mécanisme est pour l’instantencore mal connu.
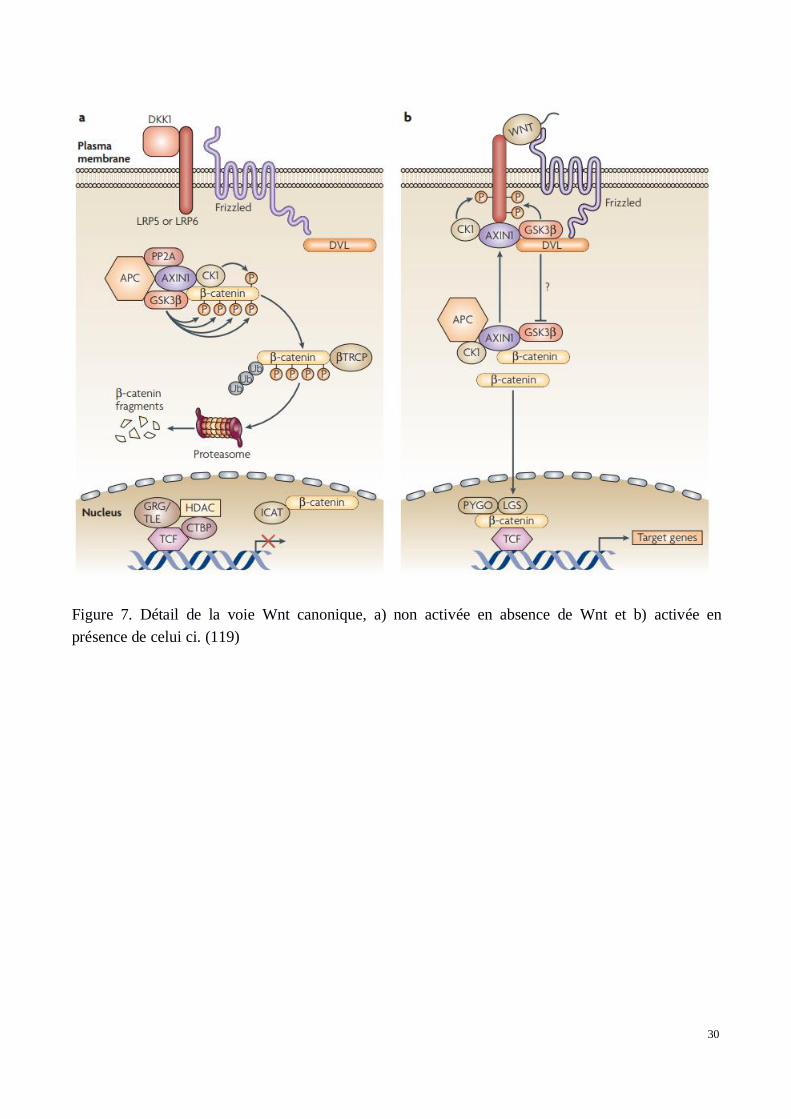
30
Figure 7. Détail de la voie Wnt canonique, a) non activée en absence de Wnt et b) activée enprésence de celui ci. (119)

31
Malgré la multiplicité des protéines mises en jeu dans la voie Wnt, seule la β-caténine est capable detransmettre le signal de transcription vers le noyau. Son accumulation dans le cytoplasme cellulaireest associée à sa translocation vers le noyau. Elle agit alors sur les facteurs de transcription TCF/LEFqui se déclinent en une famille de quatre membres chez les vertébrés. Ces facteurs possèdent unmotif high-mobility group (HMG) ainsi que ce qui est appelé la “basic tail”, petit peptide de résidusprotéiques associés qui assurent le rôle d’ancrage à l’ADN. En effet, TCF/LEF peuvent jouer un rôleactivateur ou inhibiteur selon la présence ou l’absence de la β-caténine. Il possède aussi la capacitéde plier la double hélice d’ADN. Les motifs protéiques de la “basic tail” assurent une haute affinitépour la fixation à l’ADN et permettent la conservation du facteur de transcription au sein du noyau.TCF4 serait, parmi tous les TCF, celui qui aurait conservé lors de l’évolution la plus forte capacité defixation à la β-caténine et donc la plus forte réponse à celle-ci. Le domaine de fixation de laβ-caténine sur les molécules TCF est nommé “C clamp”. Son mode de fonctionnement est encoremal connu. En absence de β-caténine, l’activité de TCF est réprimée par divers co-répresseurscomme MTGR1, Coop ou encore les molécules de la famille Grucho TLE. Ces mécanismes sontbien connus chez la drosophile mais restent encore à clarifier chez les vertébrés.
Parmi les processus impliqués par la stimulation de ces gènes, on retrouve l’adhésion et proliférationcellulaire ainsi que divers effets anti-apoptotiques. Enfin, cette voie de signalisation Wnt canoniqueest soumise à une régulation négative qui, même en présence de la molécule Wnt, empêche etréprime son activation. Les molécules DKK1, TCF, LEF sont capables d’altérer la signalisation de lavoie Wnt. DKK1 se fixe sur LRP5 et 6 empêchant ainsi leur fonctionnement avec Wnt. LEF et TCFse lient à la β-caténine et empêchent son action au niveau du noyau.
Les voies Wnt non canoniques contrastent avec la voie canonique car elles n’impliquent pas laβ-caténine dans la transduction du signal, ni même LRP5 et 6 comme récepteur. Plusieurs voies noncanoniques existent comme la voie Ca2+ (Figure 8) ou la voie PCP (Figure 9). Ces voies présententdes rôles bien plus importants que la voie canonique, notamment pour la voie PCP chez les vertébrés(124). La voie PCP est stimulée par Wnt5A et Wnt11 via Fzd, ROR et PTK7 comme co-récepteurs.Elle n’implique pas LRP et ne nécessite pas la présence d’une protéine G contrairement à la voieCa2+. Elle implique les GTPases RAC1, RHOA et JUN-N-Terminal kinase (JNK). Elle estprincipalement impliquée dans la réorganisation du cytosquelette et donc la forme de la cellule, maisaussi la migration cellulaire en réponse à divers stress. Elle a été décrite comme jouant un rôleimportant au niveau de la formation des métastases, migrations cellulaires des cellules cancéreusesde la tumeur initiale. La seconde voie non canonique dite Ca+ est une des plus récente à avoir étédécouverte. Principalement stimulée par Wnt5A et Frizzled2 (Fzd2) qui sont capables de stimuler laphospholipase C à l’aide d’une protéine G qui active à son tour la protéine kinase C capable de cliverle phosphatidylinositol. Celle-ci une fois clivée, Ca+ peut se fixer à son récepteur empêchant sadégradation et donc entraînant son accumulation intra-cellulaire et l’activation de kinases calciumdépendantes. Ces kinases stimulent alors la transcription au niveau du noyau via des facteurs commele Nuclear Factor of Activated T Cells (NFAT).
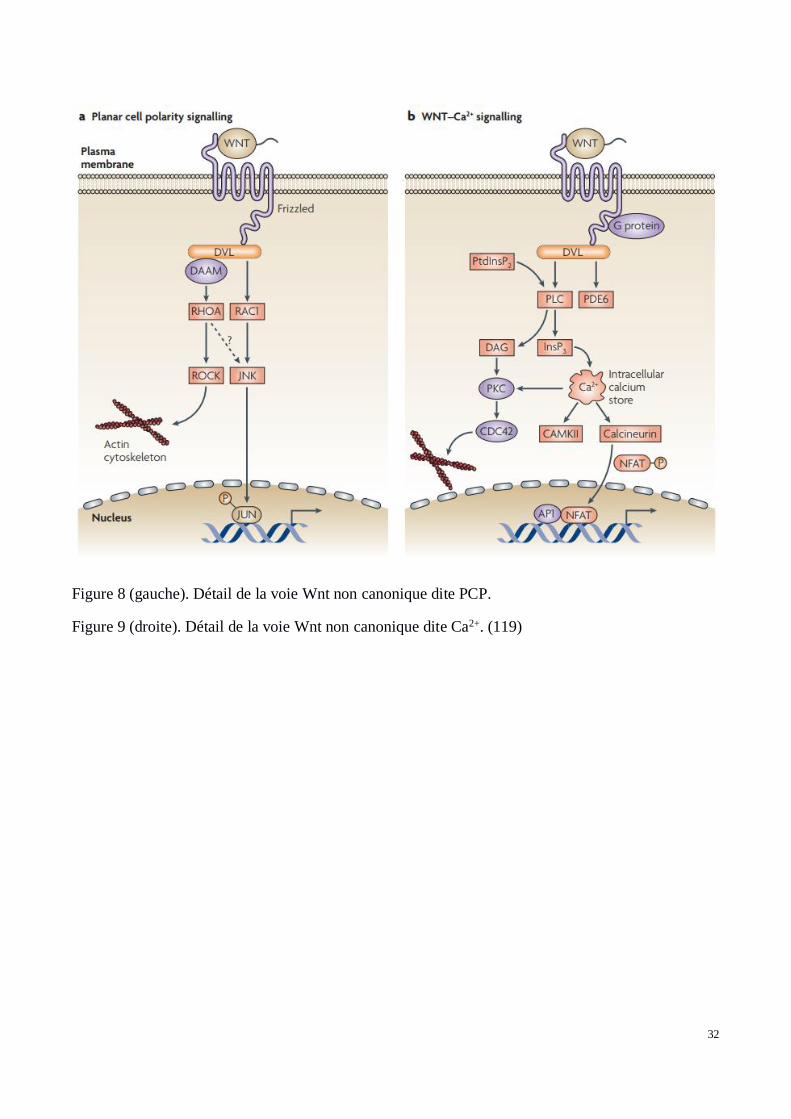
32
Figure 8 (gauche). Détail de la voie Wnt non canonique dite PCP.
Figure 9 (droite). Détail de la voie Wnt non canonique dite Ca2+. (119)

33
La stimulation des voies non canoniques a pour conséquence d’inhiber la production de β-Caténine.Cette inhibition s’effectue par compétitivité au niveau des récepteurs Fzd, la fixation de Wnt5Acapable d’activer la cascade PCP empêche ainsi la fixation de Wnt3A au même récepteur réprimantainsi l’activation de la voie canonique (125). La voie PCP fait intervenir la polarisation des tissus etdes cellules de ces tissus. Cette polarisation est également essentielle à la formation des épithéliumscomme la peau (126). Bien connu dans tous les processus liés au développement embryonnaire chezla souris et la drosophile, le mécanisme PCP utilise l’action commune de gradients moléculaires deconcentration et du regroupement spécifique de récepteurs trans-membranaires.
La polarisation de la cellule est assurée par un regroupement de récepteurs trans-membranaires surune zone spécifique de la membrane (Frizzled (Fzd) et Flamingo (Fmi)). Le complexe moléculaireDishevelled (Dsh) et Diego (Dgo) permet ce regroupement. Les récepteurs Fzd et Fmi ainsiregroupés vont se lier à leurs récepteurs complémentaires situés sur la cellule voisine (Fmi et VanGogh (Vang)). L’accumulation du complexe Vang-Fmi sur une zone ciblée de la membranecellulaire entraîne l’accumulation sur la même cellule du complexe Fzd-Fmi au pôle opposé.L’accumulation d’un complexe plutôt qu’un autre à l’un des pôles ne suffit pas à expliquerl’exclusion mutuelle du second complexe. Vang serait capable de recruter Prickle (Pk) qui, on lesuppose, empêche l’association de Dsh avec Fmi-Vang. Dsh et Dgo étant responsable del’accumulation du complexe Fzd-Fmi, son accumulation devient alors impossible quand Fmi-Vangest présent. La polarisation des cellules ne permet pas uniquement une orientation, elle entraîneégalement des migrations de molécules cytoplasmiques, l’apparition de gradients moléculaires et deforces mécaniques capables de ré-arranger les cellules entres elles. Elle permet des mouvementscellulaires ainsi que la stratification des tissus. La peau est un épithélium stratifié en derme,hypoderme et épiderme et les mouvements et migrations cellulaires sont des mécanismes essentielsdu processus de cicatrisation. Rab11 et Ankrd6 apparaissent comme étant polarisés de par l’actionPCP, jouant ainsi un rôle dans la cicatrisation, notamment au niveau du remodelage des tissus et pourle maintien des limites de la cicatrice (127). Ces constatations amènent à penser que la stimulation dela voie PCP pourrait jouer un rôle dans le développement de la cicatrice chéloïde et dans sesdifférences de comportement avec le caractère hypertrophique.

34
b) Voie du TGF-β & BMP7
Le TGF-β est représenté par une famille de trois facteurs de croissance numérotés de 1 à 3. Cesrécepteurs ont la capacité de se lier aux récepteurs membranaires TGFβ-R1, R2 et R3. La liaisond’un des TGF-β aux récepteurs 1 et 2 permet la formation d’un complexe avec le troisième récepteuret TGFβ-R1 est phosphorylé. La cascade de signalisation (Figure 10) est alors activée grâce à laphosphorylation de SMADs 2 et -3 qui forment, avec SMAD4, un complexe hétéromérique. Ceshétéromères situés dans le cytoplasme sont transloqués dans le noyau. SMAD7, un compétiteur deSMAD4, est capable de le remplacer dans le complexe diminuant ainsi la translocation du complexeSMAD vers le noyau. Il existe une seconde régulation négative : BAMBI et FKBP1A entrent encompétition avec le récepteur TGF-β de type 1 l’obligeant à conserver une conformation inactive etempêchant ainsi la fixation du TGF-β et donc la diffusion du signal au niveau intra-cellulaire. Lavoie BMP7 a également été décrite comme ayant un effet similaire à celle du TGF-β. BMP7 possèdesa propre voie de signalisation qui nécessite également l’intervention de certains SMADs. MaisBMP7, capable de se lier aussi aux récepteurs du TGF-β, peut également stimuler la voie du TGF-β.Dans la voie BMP7, celui-ci se lie à ses récepteurs privilégiés : BMPR-1A, BMPR-1B et BMP-R2.BMP7 est lui-même en compétition avec BMP2 et 4, qui constituent des ligands de moindre affinité.Il a donc lui aussi la capacité d’activer la voie du TGF-β via la phosphorylation des SMADs 1, 5 et 8.Les SMADs 2, 3, 1, 5 et 8 sont appelés des R-SMADs (Récepteurs SMADs) tandis que les SMADs 6et 7 sont appelés des I-SMADs (Inhibiteurs SMADs). Avec l’aide de SMAD4 ils peuvent égalementtransmettre le signal de transcription au noyau. De manière homologue à SMAD7, SMAD6 entrenten compétition avec SMAD4 et réduisent la translocation du complexe vers le noyau.
Il existe plusieurs mécanismes de translocation des SMADs vers le noyau de la cellule (128). Lespremières études ont montré que SMAD 4 contient une séquence protéique de localisation nucléaire(NLS, nuclear localisation sequence) qui constitue un site de fixation pour l’importinβ capabled’effectuer la translocation vers le noyau. Cette séquence protéique de localisation nucléaire sembleconservée chez tous les R-SMADS. Les R-SMADS possèdent également des séquences d’exportnucléaire dites “CRM1 dépendantes” (Chromosomal Maintenance 1) dont le rôle assure la sortie ducomplexe à extérieur du noyau. Ainsi, CRM1 étant toujours actif, il empêche en permanencel’accumulation nucléaire des SMADs. Ce n’est qu’une fois la voie stimulée et que les R-SMADs etSMAD4 sont phosphorylés qu’un changement de conformation intervient permettant lafonctionnalité du domaine NLS et donc une accumulation nucléaire. Ce mécanisme de translocationfait également intervenir d’autres interactions cytoplasmiques.
L’étude de Dong et son équipe (129) montre que les microtubules situés dans le cytoplasmeparticiperaient à la rétention des SMAD2, -3 et -7. Le TGF-β entraînerait alors la libération desSMADs pour favoriser leur migration dans le noyau.
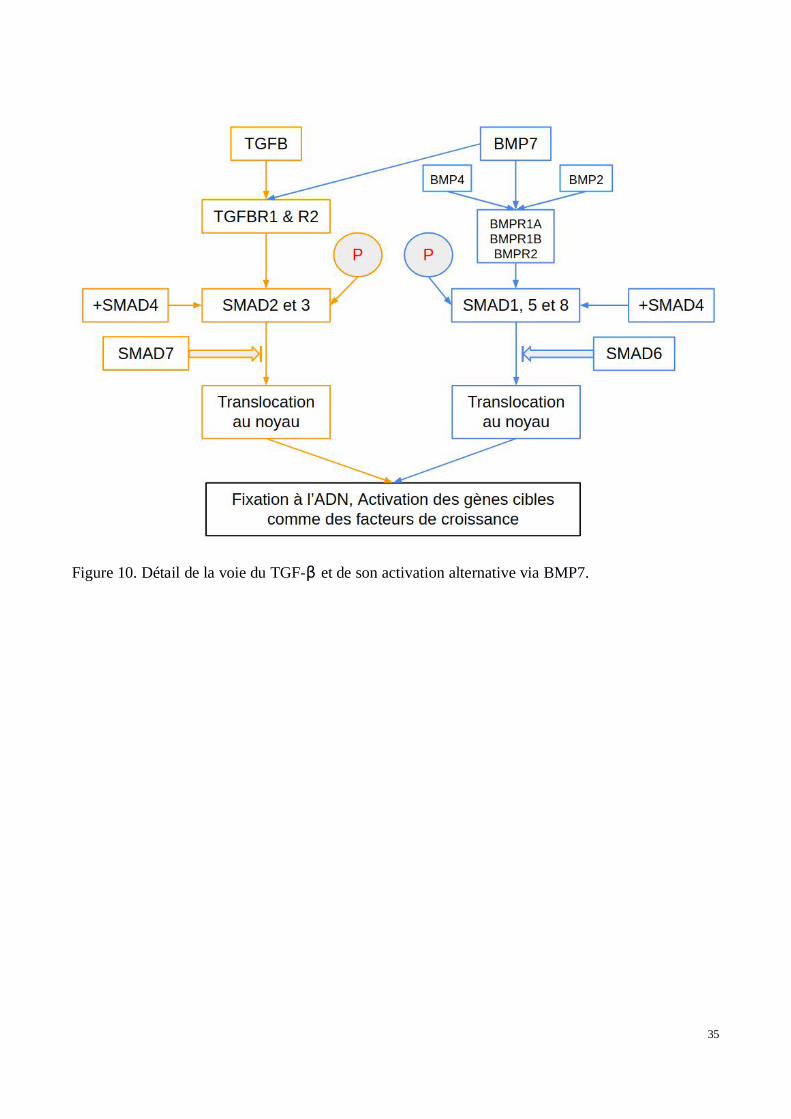
35
Figure 10. Détail de la voie du TGF-β et de son activation alternative via BMP7.

36
De plus, une déstabilisation du réseau de microtubules constituerait un facteur aggravant dans le casoù celui-ci serait cumulé avec une sur-stimulation de la voie du TGF-β. Une fois dans le noyau, lesSMADs se fixent à l’ADN pour activer la transcription de gènes cibles. Les SMAD3 et SMAD4possèdent un domaine MH1 (Mad Homology 1) capable de reconnaitre un motif nucléotidiquespécifique (GTCTAGAC) et donc d’assurer sa fixation sur l’ADN (130). SMAD2 n’est pas capablede se fixer à l’ADN tandis que SMAD 1 et -3 sont capables de reconnaitre le motif même si celui-cin’est présent qu’à moitié, donc soit GTCT, soit AGAC. En théorie, dès lors que cette séquence estprésente sur les génomes, les SMADs sont capables de déclencher la transcription des gènes dont lespromoteurs sont dotés d’éléments de réponse correspondants. Cependant, ces mécanismes sontencore mal connus et il est pour l’instant impossible de lister clairement tous les sites de fixationSMAD sur l’ADN. La liste exacte des gènes régulés par le TGF-β n’est pas connue de manièreexhaustive et de nombreux gènes ont été décrits comme tels dans la littérature sans pour autantprouver précisément de manière moléculaire la relation de cause à effet.
De plus, les sites de fixation SMADs sur l’ADN sont nombreux et la capacité de fixation aux unsplutôt qu’aux autres varie selon le tissu dans lequel la voie est activée mais également en fonctiondes autres voies activées simultanément dans ces mêmes tissus entrainant des réponses cellulairesparfois paradoxales. En ce sens, la dualité du rôle de la voie TGF-β / BMP7 à la fois pro- etanti-cancéreuse peut se comprendre aisément. L’étude de Ranganathan et collaborateurs (131) meten évidence une liste de 917 gènes sur-exprimés et 83 gènes sous-exprimés en comparant descellules traitées avec ou sans TGF-β1. L’étude met en évidence la régulation par le TGF-β del’Integrin αV, la thrombospondin 1 et la α2-macroglobulin responsable en partie de la survie destumeurs et des métastases. Ce même TGF-β régulerait également WT1 (Wilms tumor protein 1)agissant comme un suppresseur de tumeur et soulignant la dualité de la signalisation du TGF-β /BMP7.

37
D) Quelles approches utiliser pour l’étude des mécanismes de
cicatrisation ?
a) Analyse génomique, séquençage ou génotypage ?
En génétique prédictive, l’activité principale consiste à rechercher des SNP capables de prédire lapathologie d’étude. Le but n’est pas de découvrir de nouveaux SNP, mais de rechercher plutôt ceuxqui possèdent un effet marginal permettant de discriminer les cas des contrôles parmi les patients dela cohorte d’étude. Les SNPs correspondants à cette définition sont appelés des marqueursgénétiques. L’analyse statistique qui permet la découverte de ces marqueurs est appelée analysed’association. On dit alors que le marqueur est “associé” à la pathologie d’étude.
Le terme GWAS désigne en anglais : Étude d’association sur le génome entier (Genome WideAssociation Study). Il consiste à obtenir les génotypes de millions de SNPs via une puce uniqueappelée Microarray. Le nombre de SNPs testés sur une puce varie selon le modèle de la puce choisi.Bien que l’ensemble du génome humain compte 84,4 millions de SNP (Projet 1000 Génomes,Phase3) et que ceux-ci ne soient pas tous couverts par la puce GWAS, les SNPs génotypés sur lalame GWAS sont dit représentatifs de cet ensemble. Ils couvrent au maximum, par déséquilibre deliaison, l’ensemble du génome. Le déséquilibre de liaison vient de la caractéristique que deux ouplusieurs allèles peuvent être toujours transmis ensemble à la descendance. Généralement ces SNPssont situés proches les uns des autres sur le génome et regroupés sur une région génomique restreinte,c’est ce qui leur confère le pouvoir d’être toujours transmis ensemble. Ainsi, on dit que deux ouplusieurs SNP sont en déséquilibre de liaison quand la présence d’un allèle sur un des SNPs permetde prédire, par exemple avec 80% de certitude, l’allèle des autres. Obtenir le génotype d’un SNP dugroupe en déséquilibre de liaison revient alors à obtenir, avec une probabilité estimant un tauxd’erreur, les génotypes des autres SNPs. L’objectif principal de cette technique n’est pas tant deconnaitre les génotypes des SNPs de tout le groupe d’association, mais bien de pouvoirstatistiquement, en testant un seul SNP, savoir si parmi ceux-ci sont présents un ou plusieurs SNPscausaux de la pathologie. Donc si un SNP Tag est associé à la présence d’une pathologie, il n’en estpas forcément causal mais nous renseigne sur le fait que un des SNP du groupe l’est sûrement.
En génétique prédictive, nous nous concentrons sur la recherche de marqueurs, SNPs prédictifs de lapathologie. Comme nous ne détectons qu’une partie des SNPs du génome, un marqueur n’est pasforcément causal de la pathologie, mais il sera sûrement en déséquilibre de liaison avec d’autresvariants qui influent sur le développement de la pathologie. La recherche de marqueurs identifieprincipalement ceux qui ont un effet marginal, c’est à dire un effet fort. Le nombre important de testsrépétés qui consistent à tester la même hypothèse pour chacun des variants nécessite d’appliquer unecorrection de Bonferroni dite des “tests multiples”.

38
L’idée étant que au plus le nombre de test réalisé est grand, au plus la probabilité de trouver uneassociation significative par erreur augmente. Afin de corriger cela, le seuil de significativité pour laP-value est rabaissé à 5*10-7, empêchant du même coup la détection de variants à effet plus faibles. Ilest possible de détecter des variants à faible effet en augmentant la taille de la cohorte d’étude ouencore en ayant une sélection de patients ethniquement homogènes entre cas et contrôles. Notreobjectif n’est pas tant de découvrir le ou les SNPs causaux de la pathologie, mais la découverte d’unou plusieurs SNPs marqueurs prédictifs de la pathologie. Ces marqueurs prédictifs sont le plussouvent en déséquilibres de liaison avec un des SNPs causal de la pathologie. Cependant la recherchede SNP causaux de la pathologie est quant à elle plus difficile à faire à partir de données provenantde GWAS et nécessite la réalisation d’études fonctionnelles. Ces SNPs doivent être aussi capablesd’intégrer un modèle prédictif plus large composé d’autres tags SNP.
En dehors de la correction pour les tests multiples, l’analyse GWAS pose également des limites. Unemultitude de variants rares peuvent être impliqués dans la pathologie d’étude et cela sous différentsmodèles (Récessif, dominant etc.…). L’environnement dans lequel évoluent les patients de l’étudepeut également influer sur le développement de la pathologie induisant un biais dans l’étude. Ce biaisaura tendance à affaiblir notre puissance de détection de variant, leur P-value sera moins élevée etceux-ci risquent de ne pas être significatifs. (132). De plus, tester uniquement des SNPs communs(fréquents) et aléatoirement répartis sur le génome peut ne pas être suffisant pour caractériser lesmarqueurs impliqués dans la pathologie. Enfin, il est probable que la pathologie étudiée possède unecomposante explicative autre que génétique. Même faible, cette composante qui pourrait êtreenvironnementale ou même sociale, rendrait plus difficile l’identification des polymorphismes ou desrégions de susceptibilité. Cette recherche devrait alors prendre en compte ces facteurs dans l’analyse,mais ceux-ci ne sont malheureusement pas toujours connus ou quantifiables. L’analyse GWAS peutcependant prendre en compte l’effet de co-variables, cryptiques ou non, et les intégrer à l’analysecomme nous le verrons plus tard.

39
b) Analyse du transcriptome
La technique choisie pour l’étude du transcriptome est le RNA-Seq. Bien qu'il existe d'autrestechniques permettant d'étudier l'expression des gènes comme les Microarray, nous avons choisi leRNA-Seq pour différentes raisons :
● Le séquençage n'est pas spécifique. Le RNA-Seq permet le séquençage de l'ensemble des
ARN transcrits matures présents au sein du tissu au moment de son exérèse. Il dissocie ladétection des mesures d’expression de la technique d’obtention de ces mesures. En Microarray,la technique vise à mesurer l’expression des gènes à partir d'une liste de sondes de détectionconnue, placées sur une lame de verre. Il peut y avoir plusieurs sondes pour détecter un gènemais un gène qui ne possède pas de sonde ne pourra pas être détecté. En séquençage, la détectionde l'expression des gènes est indépendante de l'obtention des séquences permettant ainsid'obtenir toutes les séquences de tous les ARN présents et non pas uniquement de ceuxdisponibles à la détection. Cette technique permet la réalisation d’une étude moins biaisée.
● Le séquençage permet une finesse de détection. La quantité de séquences obtenues pour un
gène est proportionnelle au taux d'expression de ce gène présent dans le tissu car le séquençagene limite pas le nombre de séquences pouvant être obtenu pour un seul et même gène. La finessede détection est telle, que si un seul ARN est présent pour un gène, celui-ci peut être séquencé etdonc être comptabilisé dans la mesure du gène qu'il concerne. Il n'y a pas de seuil de détection,aucune limite définie par un capteur ou autre instrument de mesure.
● Le séquençage permet l’obtention de données autres que l'expression. En RNA-Seq, le taux
d'expression d'un gène correspond au nombre de séquences retrouvées pour ce gène. Cependantces séquences peuvent nous apporter d'autres informations. Comparées au génome humain deréférence, les mutations et variants peuvent être recherchés à partir de ces séquences. Il n'estcependant pas possible d'observer les mutations pour des gènes dont l'expression est faible ouabsente, par manque de séquences ou encore si celles-ci sont situées au niveau des régionsinter-géniques et introniques.
Il existe certaines limitations aux études par séquençage. Ces limitations constituent principalementdes biais de technique. Le séquençage génère en effet un ensemble de séquences aléatoirementobtenues sur l’ensemble du génome. Plusieurs étapes bio-informatiques sont donc nécessaires pouridentifier l’appartenance de ces séquences aux gènes connus du génome. Cette identification, diteétape d’alignement sur le génome de référence, est une étape cruciale de l’analyse. C’est elle quidéterminera le nombre de séquences attribué à chaque gène et donc leur taux d’expression. Il peutarriver cependant que des erreurs d’alignement se produisent. Du fait que la technique de séquençageIllumina produise des bases de moindre qualité en fin des séquences ou que certaines séquencespeuvent être trop courtes pour pouvoir avoir un alignement sans ambiguïté sur le génome, certainesattributions séquences / gènes peuvent présenter des erreurs.

40
c) Analyse de l’épi-génome
L’épi-génétique provient du constat initial que nos cellules sont capables de jouer des rôles différents,d’exprimer des cocktails de gènes précis selon les besoins. Bien que les cellules possèdent le mêmegénome, la méthylation de l’ADN, qui consiste en l’addition de groupes méthyle (CH3) aux cytosinesde l’ADN, est capable d’altérer l’expression des gènes sans altérer la séquence d’ADN.L’hyper-méthylation répartie en ilots CpG (cytosine-phosphodiester bondguanine) permetl’inactivation de gènes. A l’inverse, l’hypo-méthylation entraîne l’activation de certains gènes. Cesprocessus rajoutent une couche de complexité à l’explication des pathologies par la génétique seule.Dans les pathologies cancéreuses, la méthylation aberrante de gènes précis est généralementobservée tandis qu’une faible méthylation sur tout le reste du génome est présente (133).
Des études à large échelle sur la méthylation du génome de cellules chéloïdes commencent àémerger. Il a été montré que la prédominance de l’hypo-méthylation suggérait une activationd’oncogènes plus que l’activation de gènes suppresseurs de tumeurs (134). Cette méthylationfavoriserait les régions non promotrices des gènes. D’autres études soulignent le fait que les étudesgénétiques d’association à larges échelles (GWAS) sont limitées. Elles permettent en effet dedécouvrir des régions génomiques cibles jouant un rôle dans la pathologie mais ne sont pas capablesde justifier les actions et mécanismes impliqués dans les gènes identifiés. La génétique ne peut pasnon plus expliquer les actions des molécules mises en jeu par ces régions. L’épigénétique a aussi lacapacité de prendre en compte des facteurs difficilement quantifiables.
Modulée par l’environnement de vie du sujet, elle reflète l’action de celui-ci sur notre génome, ce quine peut pas être observé en génétique. Bien que nous n’ayons pas étudié cet aspect génomique dansnotre travail, ces études permettent un regard nouveau sur la pathologie et étoffent notre réflexionquant à la recherche de marqueurs génétiques pouvant expliquer la pathologie.
d) Analyse bio-informatique
L’avènement des nouvelles techniques et méthodes de recherche en biologie nécessite l’utilisation deprocessus informatiques complexes pour le traitement des données générées. Au cours des 20dernières années, les technologies développées à la paillasse ont décuplé la quantité de donnéesobtenues au point où il n’est désormais plus possible de les traiter manuellement. Les nouvellestechnologies de séquençages haut débits (NGS) ou même les études d’association à partir de donnéesde génotypage haut débit nécessitent une automatisation du traitement. C’est dans ce cadre, et grâceà mes compétences de développeur Python, que j’ai pu mettre en place un certain nombres d’outilsfacilitant l’automatisation de ces traitements.

41
Matériel et Méthodes.
B) Cohorte et recrutement des patients
a) Cohortes fibrose de le peau
Pour notre étude sur la fibrose de la peau, notre groupe a constitué 2 cohortes de patients prélevés sur3 localités entre 2012 et 2016. Une première cohorte a été recrutée en France, à Marseille (APHM -Professeur Casanova et Pr Magalon) et Paris, recrutement auquel j’ai participé avec le Dr. Petit(APHP – Dr Petit). Une seconde cohorte a été recrutée en Ouganda (Pr Alain Dessein et son équipe)au sein de la population Allur. Au niveau clinique, les critères choisis pour inclure un patient àl'étude cicatrisant anormalement en France sont les suivants : un cas possédant des chéloïdesprésente une ou plusieurs cicatrices chéloïdes constatée(s), évoluant sans régresser depuis plus de 2ans, se développant au-delà des bords originels de la cicatrice. Un patient possédant deshypertrophiques présente une ou plusieurs cicatrices épaisses qui, en largeur, ne dépassent pas leslimites de la cicatrice d'origine, évoluant depuis moins de 2 ans avec une tendance à régresser.Généralement, les cicatrices hypertrophiques sont moins symptomatiques (douleurs,prurits/démangeaisons) que les chéloïdes.
Les cicatrices évoluant depuis moins de 2 ans et qui dépassent les limites de la cicatrice originellen'ont pas été prélevées. Une fois la biopsie réalisée sur le patient, une partie du tissu estimmédiatement conservée à l’aide du RNA-later et une seconde partie dans du formol. Le tissu enRNA-later est ensuite conservé entre 1 à 5 jours à 4°C pour que la biopsie s'imprègne du liquideavant d’être placée à -20°C. Le tissu sous formol est envoyé à un anatomo-pathologiste qui confirmele diagnostic chéloïde ou hypertrophique. Histologiquement, le tissu chéloïdien présente uneprolifération aléatoire de fibres de collagène épaisses sous forme d’amas, caractéristiques d’unefibrose importante. La cicatrice hypertrophique présente également cette caractéristique mais lesfibres de collagène sont alignées, allongées et mieux structurées. La cicatrice hypertrophique est peuvascularisée et va parfois présenter des calcifications pour les cas les plus prononcés. Contrairementaux hypertrophiques, les chéloïdes présentent une accumulation faible de fibres lisses musculairesAlpha. La table 4 détaille les prélèvements que nous avons eus à disposition une fois les extractionsd'ADN ou d'ARN réalisées. L’ADN est extrait à partir de prélèvements sanguins et des biopsies,tandis que l’ARN provient des biopsies uniquement. La cohorte des chéloïdes français est composéede patients provenant de nombreux pays. Sur les 219 prélèvements effectués, 54 sont européens, ilsreprésentent 25 % de la cohorte. Les patients de pays Africains représentent 53% (n=117) desprélèvements, 13% (n=29) pour l’Amérique du sud et 4,5% (n=10) pour l’Asie et Indonésie. Pour lescontrôles de la cohorte française, les ADN proviennent à la fois de biopsies de peaux mais aussi dedons de sang aux établissements Français du sang (EFS). Pour ces derniers, l’absence totale dechéloïdes n’a pas pu être vérifiée, ces dons étant anonymes.

42
Table 4, Nombres et origines géographiques (continents) des patients présentant des cicatriceschéloïdes, hypertrophiques et contrôles recrutés et utilisés dans notre travail.

43
b) Cohortes fibrose du foie
Pour notre étude sur la fibrose bilharzienne du foie, notre groupe a constitué 3 cohortes de patientsprélevés sur 2 pays différents : le Brésil (2 campagnes) et l’Ouganda.
L’échantillon 1 (Brésil 1) et 2 (Brésil 2) constituent respectivement la cohorte de découverte et deconfirmation recrutées au Brésil dans divers villages de l’état de Pernambucco par les Pr A.Coutinho(AC) et Pr A. Dessein (AD). L’échantillon de découverte est constitué de 525 sujets affectés d’unefibrose hépatique sévère (SHF) et 576 individus présentant une fibrose hépatique légère (LHF)malgré une exposition prolongée (>20 ans) au parasite Schistosoma mansoni. L’échantillon deconfirmation au Brésil est actuellement en préparation pour le génotypage et n’est donc pas inclusdans cette thèse. AD a assuré l’homogénéité du recrutement entre les échantillons Brésiliens etOugandais qui constituent la cohorte d’extension pour l’étude. AC est une experte reconnue del’évaluation des fibroses causées par les schistosomes. AD a évalué la fibrose hépatique en utilisantl’échographie (>15000 patients). Les critères d’exclusion ont été : alcoolisme avéré, infections par leVHB ou le VHC, stéatose hépatique et NASH ainsi que toutes autres causes de cirrhoses hépatiques(hépatite auto-immune, cirrhose biliare etc..).
Les SHFs ont été recrutés parmi les patients suivis à la clinique d’AC en utilisant l’échographie. Lesdegrés de fibrose sont établis suivant l’échelle proposée par l’OMS (en particulier la plus récenteédition). Les SHFs sont porteurs d’une fibrose de degré D, E or F. Parmi les cas vus à l’hôpital parAC, 22.6, 47.6 et 26.2% sont D, E ou F. Tous les SHFs présentent des signes évidents d’hypertensionporte avant traitement (diamètres anormaux de la veine porte et de la veine splénique, présence devarices). Environ 1/3 des patients présentaient une fibrose décompensée et étaient en phased’évolution vers une cirrhose. Des varices œsophagiennes ont été observées chez 80% des SHFsavec des saignements chez la moitié d’entre eux. Vingt-huit pour cent de ces patients ont étésplénectomisés. Les ascites étaient peu fréquentes sauf chez les sujets décompensés.
Les LHFs ont été recrutés dans les mêmes villages ou proches de ceux où vivaient les cas en utilisantdes ultrasons portables. L’évaluation échographique a été effectuée deux fois à deux intervalles detemps différents parmi les patients âgés de 40 à 65 ans et ayant au moins plus de 20 annéesd’exposition à S mansoni. Les LHFs ont été recrutés parmi les individus qui présentaient, soit aucunefibrose hépatique, soit des fibroses de type B ou C. Ils ne devaient pas avoir été traités plus d’unefois avec le Praziquantel dans les dix dernières années. La sélection des LHFs a requis l’évaluationde 3000 sujets vivants dans les villages endémiques de l’état de Pernambucco (particulièrement dansla région de Vicença)
L’échantillon Ougandais (120 SHFs, / 449 LHFs) a été recruté suivant les critères décritsprécédemment pour le recrutement des échantillons Brésiliens, parmi la population de pêcheurs dulac Albert. Cette population est affectée par une bilharziose très sévère due aux conditions extrêmesde transmission de S mansoni dans cette région. Cette population a été choisie du fait de sa forteexposition à S mansoni et de sa grande homogénéité ethnique car toutes les familles vivant sur lesrives de ce lac (dans un rayon de 50 km de Pakwash) sont des Allurs. Cette homogénéité ethnique estrequise pour éviter les biais de sélection qui pourraient affecter l’analyse génétique.

44
Du fait de la plus grande résistance de cette population au développement des fibroses hépatiques(les africains sont exposés depuis de nombreuses générations au schistosome) les seuils d’expositionpour choisir les contrôles ont été relevés à 25 ans pour les hommes et 40 ans pour les femmes aprèsanalyse des données cliniques (échographie) des 5000 individus riverains du lac. L’échantilloncomprenait 1/3 de femmes et 2/3 d’hommes. L’échographie de ces patients a été effectuée par deuxéchographistes professionnels habitués à l’évaluation de la fibrose bilharzienne. Chaque patient a étéévalué au moins 4 fois à deux temps différents par les deux échographistes. En cas de désaccordentre les résultats, l’analyse du patient était effectuée à nouveau en présence d’AD. Quand le doutepersistait le patient n’était pas inclus dans l’étude.

45
B) Biologie moléculaire
a) Préparation ADN et ARN
Les extractions d'ARN ont été réalisées à partir des biopsies de peaux de cicatrices chéloïdes,hypertrophiques et peaux saines à l'aide du kit Qiagen RNeasy mini kit.
Les concentrations et ratios A260 / A280 & A260 / A230 ont été mesurés à l'aide du NanoVue etl'ensemble des ratios étaient dans les spécifications de qualités attendues.
Les RINs (Rna Integrity Number) ont été réalisés avec le Bioanalyser (Agilent). Tous leséchantillons utilisés pour les expérimentations de cette étude possèdent un RIN supérieur à 7.5.
Les prélèvements d'ADN ont été réalisés à l'aide de tubes EDTA. L'ADN des prélèvements sanguinsréalisés lors des campagnes (Ouganda et Brésil) on été extraits directement sur place en laboratoirepar des techniciens envoyés en mission. Les prélèvements effectués en France (Paris et Marseille)ont été transportés au laboratoire du professeur Dessein où l'ADN a également été extrait.
Deux types de kit Qiagen ont été utilisés que ce soit à l’étranger ou en France :
● Le QIAmp DNA Blood Midi Kit, extraction à partir de 1 à 2 ml de sang.
● Le QIAmp DNA Blood Mini Kit, extraction à partir de 200 à 400 µl de sang.
Les dosages des concentrations en matériel génétique obtenues après extraction ont été réalisés àl'aide de deux techniques pour chaque patient : par spectrophotométrie au Nanovue (GE Healthcare)et par fluorescence avec le kit Qubit (Thermo Fischer), spécifique de l'ADN double brin. La lecturede fluorescence a été réalisée à l'aide du 7900HT Fast Real Time PCR system (Applied Biosystem)sur plaques 96 puits de 0.2 ml. La conversion des données de fluorescence en concentrations réellesest réalisée à l'aide d'un script R par régression linéaire avec comme référence les deux mesuresstandards, une correspondant à 0 ng/µl (Standard S1) et une seconde à 100 ng/µl (Standard S2).

46
b) Nouvelles technologies de séquençage haut débit (NGS)
Les séquençages du RNA total ainsi que la préparation des librairies de nos individus ont été réaliséspar le prestataire externe Genewiz.
Les librairies ont été préparées à l'aide du kit TruSeq® Stranded mRNA Sample Preparation afind'obtenir un ensemble d'ADNc purifié, enrichi et représentatif du transcriptome de l'échantillonassocié. La taille moyenne des séquences de la librairie est de 260 paires de bases.
Les différentes étapes des préparatifs des librairies sont décrites figure 11. La purification de l'ARNtotal sur billes magnétiques possédant les sites de fixation poly T permet la fixation des ARNpossédant une queue poly A spécifique des ARNs messagers. Seuls les transcrits matures sont doncconservés. Ces librairies sont dites « stranded », c'est à dire préparées à partir du brin d'ADN lutoujours dans le même sens. Nous savons que dans le génome humain, une portion génomique peutcoder pour un ensemble de gènes quand elle est lue dans le sens 5' ver 3' et pour un autre ensemblede gènes quand elle est transcrite dans l'autre sens. Ceci est d'une importance capitale pourl'alignement des séquences.
Il n'est pas encore possible de séquencer l'ARN directement, il est donc nécessaire de réaliser uneRT-PCR à l'aide d'une RNase H. L'ajout ensuite d'adaptateurs spécifiques à la technologie Illuminapermet à la séquence de pouvoir s'hybrider sur la lame illumina où va être effectué le séquençage.
Le séquençage a été réalisé sur la plateforme Illumina HiSeq par la méthode dite « par synthèse »avec la technologie two-channel SBS. A chaque incorporation d'une nouvelle base, une image de lafluorescence est réalisée et les signaux lus servent à obtenir la séquence aussi appelée lecture («Reads »). Cette image est basée sur la détection de deux fluorochromes. Un dans le rouge pour labase C, un dans le vert pour la base T, un orange (rouge+vert) pour la base A et enfin une absence defluorescence pour le G.
Le séquençage est réalisé en “Paired End”, c'est à dire que chaque séquence sera « lue » par leséquenceur par chaque extrémité en remontant vers le centre de la séquence. Le brin séquencé étantplus long que la capacité de lecture du séquenceur, les deux lectures ne se rejoignent pas pour sechevaucher. Cependant l'information comme quoi ces deux lectures proviennent du même brin lu en« paired end » est conservée dans les fichiers fastaq. Nous obtenons des lectures de 150 paires debases pour environ 35 millions de paires de lecture par individus. Le prestataire fournit les fichiersFastaq des séquences pour analyse bio-informatique.
47
Figure 11. Présentation des différentes étapes nécessaires à la préparation des librairies pourséquençage Illumina à l’aide du kit TruSeq® Stranded.

48
c) Génotypages TaqMan
Le génotypage correspond à l'action d'obtenir l’état du génotype pour un patient pour un variantunique de l'ADN.
Un SNP (Single Nucléotide Polymorphism) correspond à une variation d'un seul nucléotide dans laséquence d'ADN à une position donnée sur le génome appelée locus. Cette variation peut apparaîtredans des régions codantes ou non codantes. Les modalités de cette variation sont appelées allèles.Ainsi pour un locus particulier, une base peut prendre les allèles A, T, G ou C. Généralement, lavariation allélique prendra le plus souvent 2 modalités (A ou G par exemple). Cependant, 3 voir 4bases peuvent aussi être retrouvées dans une même variation (SNP mosaïques). Donc génotyper unindividu équivaut à obtenir la valeur de son génotype à un locus donné. Cette variation peut êtreprésente au sein d'une population avec différentes fréquences alléliques. Pour un locus donné, onexprime la MAF (Minimum Allélic Fréquency), qui correspond à la fréquence de l'allèle qui est lemoins fréquent dans la population. Pour un même SNP, ces fréquences alléliques peuvent varier ausein des populations réparties autour du globe du fait de leurs origines évolutives ancestralesdifférentes. L'évolution génétique des populations, l'adaptation à un environnement différent,l'histoire des populations elles-mêmes (migrations, contraintes…) entraînent des différences dansnotre génome et donc au niveau des SNP. Ainsi, un SNP présent dans une population africainepourra ne pas être observé chez les européens et inversement. De même un SNP présent dans 2populations différentes pourra avoir des MAF différentes, voire même inversées par rapport auxallèles rares. (Ex : 95% de A et 5% de T pour une population pour un SNP et pour le même SNPdans une population différente : 95% de T et 5% de A).
Nous avons utilisé ici la technique de discrimination allélique par méthode TaqMan pour obtenir lesgénotypes de nos patients. Cette technique permet de tester une variation à seulement 2 modalitésappelées les allèles à une position génomique unique. Par exemple un SNP avec les bases A ou T.Durant la détection, l'ADN génomique spécifique de la région où est situé le variant testé estamplifiée par PCR à l'aide d'amorces spécifiques. La détection des allèles est assurée par la présencede deux sondes spécifiques capables de se fixer au niveau du variant testé par complémentarité àl'ADN rendu simple brin au cours des étapes de la PCR. On peut considérer qu'il est assez rare que 2polymorphismes soient situés proches l'un de l'autre. Sur une portion génomique restreinte à la zonedu variant et présentant un seul polymorphisme génétique, il est alors possible de créer 2 sondesidentiques en tout point sauf au niveau de la base d'ADN qui porte le polymorphisme.
Quand l'ADN génomique d'un patient est testé, celui-ci est mis en présence des deux sondes qui vontpouvoir se fixer ou pas sur la portion d'ADN reconnue selon l'allèle porté. Pour sa détection, chaque

49
sonde porte un fluorochrome différent, c'est le couple VIC (Absorption : 583 nm, Émission: 554nm)et FAM (Absorption: 494 nm, Émission: 518 nm) qui est utilisé.
La sonde comporte également un Quencheur, capable d’empêcher l'émission de fluorescence desfluorophores tant que les deux sont proches l'un de l'autre. Une fois la sonde fixée, celle-ci seretrouve sur le passage de la polymérase qui amplifie toute la région du variant à chaque cycle PCR.La sonde est alors détruite de par l'activité exo-nuclasique de cette polymérase, libérant lefluorophore du quencheur et nous permettant de détecter la fluorescence correspondante à l'allèleprésent.
En fin de PCR, une lecture en point final est réalisée et les valeurs de fluorescence pour VIC et FAMde chaque patient testé sont reportées sur un graphique, un fluorophore par axe. Trois nuages depoint (figure 12) vont alors apparaître, parfois avec des délimitations plus ou moins précises quel'utilisateur doit déterminer à la main. Ils correspondent aux trois génotypes présents possibles, parexemple, avec les allèles A ou T, nous aurons AA, AT et TT. C'est en fonction de la position desnuages les uns par rapport aux autres et des valeurs des fluorescences sur chaque axe, que l'ondétermine ensuite à quel nuage appartient quel génotype.
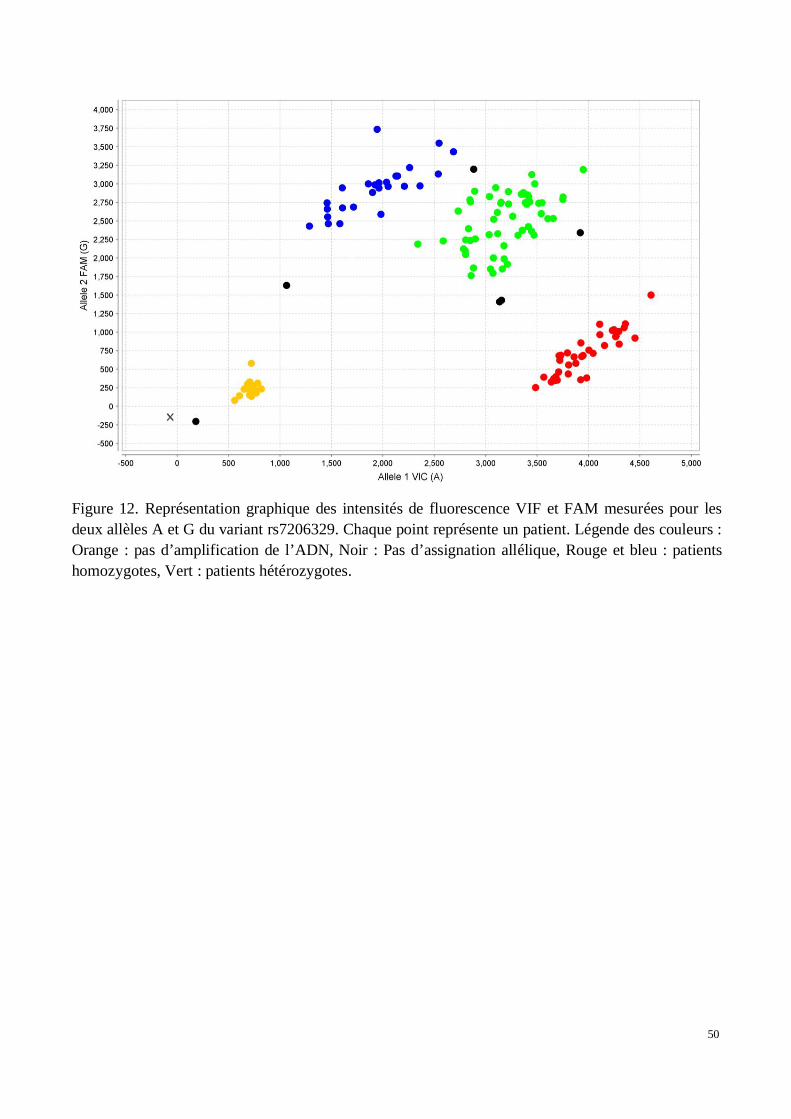
50
Figure 12. Représentation graphique des intensités de fluorescence VIF et FAM mesurées pour lesdeux allèles A et G du variant rs7206329. Chaque point représente un patient. Légende des couleurs :Orange : pas d’amplification de l’ADN, Noir : Pas d’assignation allélique, Rouge et bleu : patientshomozygotes, Vert : patients hétérozygotes.

51
C) Analyse des données
a) Analyse d'enrichissement et recherche de voies métaboliques associées
L'enrichissement est un terme large, qui désigne l'ajout de données à une base de donnéespré-existante. L’enrichissement génomique d'une liste de gènes, de variants ou toutes autres entitésgénomiques, désigne la recherche intelligente et automatisée (par algorithme d'enrichissement) dessous-ensembles de données sur-représentées dans une liste de départ.
Ainsi, si dans une liste de gènes une proportion de ces gènes correspondant, par exemple, aux voiesmétaboliques (au sens large) « cycle cellulaire » est similaire à la proportion qui aurait pu être tiréealéatoirement au sein du génome humain complet (base de données publique), la liste de départ n'estpas considérée comme enrichie en gènes du “cycle cellulaire” même si certains des gènes de la listesont associés (statistiquement) au phénotype étudié.
Sur ce principe, une P-value est calculée à l'aide du test exact de Fischer en fonction des différentsgroupes observables et connus : voies de signalisation, maladies etc.
Par exemple, la documentation IPA explique :
● Liste de gènes 1 : 5 gènes sont reliés à l’hématopoïèse alors que 50 gènes sont reliés à
l’hématopoïèse dans la référence (base de données publiques).
● Liste de gènes 2 : 5 gènes sont reliés à l’hématopoïèse alors que 10 gènes sont reliés à
l’hématopoïèse dans la référence (base de données publiques).
Dans ce cas, la P-value du processus biologique de l’hématopoïèse dans la liste 2 sera plussignificative que la P-value dans la liste 1 du fait de la plus grande représentation des gènes liés auprocessus en 1.
b) Classification hiérarchique.
Le clustering hiérarchique est une méthode de classification ou stratification qui groupe les objets(échantillons) par similitude selon un caractère défini et sans connaissance préalable de touséléments pouvant influencer la classification.
Par exemple, les différences d'expression (transcriptome) entre individus pris deux à deux sontcomptabilisées en fonction de l'ensemble de l'expression des gènes de manière itérative. Ainsi, àl'itération 1, la paire d'individus la plus proche est considérée comme formant un groupe. A l'itération2, l’algorithme recalcule les différences entre les individus sauf pour les 2 regroupés à l'itération 1qui sont considérés comme une seule entité dont l'expression des gènes est égale à la moyenne de

52
l'expression des gènes des individus du groupe. Deux nouveaux individus peuvent être ainsiregroupés à l'itération 2, un individu peut aussi être groupé avec le groupe formé à l'itération 1. Ainsi,à l'itération N, on recalcule les distances entre les individus et groupes formés à l'itération N-1jusqu’à ce qu'il n'y ait plus d'individus encore non inclus dans les groupes. Ces données numériquespermettent alors de positionner les individus étudiés sur un arbre appelé arbre des distances dont lalongueur des bras est d’autant plus importante que ces individus sont plus éloignés dans l’analyse. Lelogiciel R a été utilisé pour le calcul des distances et la représentation graphique de l'arbre. L'arbreest réalisé à partir des données d'expression normalisées par le package R « samr ». Les distancessont exprimées en comptage des lectures différentes entre deux patients.
c) Extraction des sous jeux de données provenant du GWAS pour analyse génétique ciblée.
Afin de réaliser des analyses d’associations génétique plus ciblées à partir des données du GWAS,une méthodologie d’extraction des données de génotypage à partir des résultats du GWAS a été miseen place. Pour une liste de gènes cibles, nous listons l’ensemble des SNP existants et disponiblessitués dans leur région génomique en utilisant la base de données 1000 Génomes Phase 3 sur lespopulations africaines disponibles. Nous définissons la “région génomique” d’un gène comme situéeentre les positions chromosomiques de début et fin du gène majorées de 10 000 paires de bases enamont et en aval. Cette méthode nous permet d’englober également les régions régulatrices du gène,et de rechercher également ses mutations. A partir de cette liste de variants génétiques, les groupesde déséquilibre de liaison sont calculés. Pour définir que deux variants sont en déséquilibre de liaison,le seuil du coefficient de corrélation choisi (R²) est fixé à 0,8. Au-dessous de ce seuil, les deuxmarqueurs sont considérés comme indépendants et donc n’apparentant pas au même groupe. Le seuilde la fréquence allélique minimum (MAF) est fixé à 5%, les variants présents à moins de 5% dansles bases de données de 1000 génomes ont été exclus. A partir de ces résultats et pour chaque groupede déséquilibre de liaison, un variant représentatif du groupe est choisi aléatoirement. Ce variant estensuite recherché dans les données du GWAS réalisé par le laboratoire. Si le variant n’est pas présentdans les données du GWAS, la méthode autorise le tirage aléatoire d’autres SNPs du même groupede déséquilibre de liaison en remplacement de celui-ci. Le tirage s’effectue jusqu’à épuisement desSNPs disponibles dans le groupe si le SNP tiré n’est toujours pas retrouvé dans les données duGWAS. Si aucun SNPs du groupe n’est présent dans le GWAS, celui-ci ne peut pas être étudié. Si leSNP est présent dans le GWAS, alors les génotypes obtenus pour tous les patients pour ce SNP sontreportés dans un fichier tableau pour une analyse d’association indépendante. Cette étuded’association indépendante permet de sélectionner un sous ensemble de SNPs présentant uneassociation ou une tendance à l’association avec lesquels nous souhaitons continuer en génotypageafin de tester ce sous ensemble sur d’autres patients et d’autres populations.

53
d) Analyse bio-informatique des séquences
Aprés réception des séquences au format fastaq provenant de notre prestataire de séquençage, uncontrôle qualité des séquences par individu est d'abord réalisé sur les fastaq directement en sortie deséquençage avec le logiciel FastQC (Babraham Bioinformatics). Cet outil est capable de générer unrapport qualité selon des critères précis pour chaque individu. Du fait du grand nombre de patientsdans cette étude, un second outil nommé MultiQC est utilisé. Il permet de générer un rapport qualitéunique à partir des rapports individuels de FastQC pour tous les patients de l'étude et donc visualiserl'ensemble sur un seul tableau ou graphique pour chaque critère. C'est un contrôle qualité passif,permettant uniquement de valider les différents points de qualité mais n'agissant en aucun cas sur lesfichiers fastaq contenant les séquences.
Les différents critères contrôlés avec attention sont les suivants :
● La technologie Illumina a plutôt tendance à produire des bases de qualité plus faible en fin de
séquence, il est donc nécessaire de contrôler la qualité des bases le long des séquences (Figure13). Le logiciel AdapterRemoval utilisé ensuite est capable de corriger ce problème sinécessaire.
● La qualité d’une séquence correspond à la moyenne de toutes les valeurs de qualité des bases qui
la compose. Il est ainsi possible de réaliser la distribution des qualités de toutes les séquences duprojet (Figure 14). Les données présentent un pic à un score phred entre 39 et 40, soit uneprobabilité d’avoir identifié une base par erreur située entre 0,00013% et 0,00010%.
● Longueur et nombre de séquences obtenues. Dans nos fichiers, le nombre et la taille des
séquences sont deux des paramètres les plus importants et cela pour plusieurs raisons, toutd'abord pour l'intégrité de nos données. Du fait que nous travaillons en paired end, il estnécessaire que dans les fichiers fastaq des lectures sens et anti-sens, le nombre de lectures soitidentique et organisé par paires concordantes. Sans cela, il est impossible de travailler avec lesfichiers, les résultats sont dit corrompus. Enfin, pour la qualité de l’analyse qui s'en suit, il estnécessaire que les lectures respectent les spécifications demandées au prestataire pour le projet,c'est à dire environ 35 millions de séquences par individus avec en moyenne 150 paires de basespour chaque lecture (Figure 15).
54
Figure 13. Distribution du score Phred moyen sur toutes les lectures pour chacune des bases obtenuesen fonction de leur position le long de la lecture
Figure 14. Distribution du nombre de lectures en fonction de leur scores Phred moyen calculé surtoutes les bases de la séquence.
Figure 15. Distribution de la longueur des séquences pour l’ensemble des fichiers FastaQ obtenuslors des séquençages RNA-Seq

55
L’outil AdapterRemoval permet un contrôle qualité actif. AdapterRemoval est capable d'éliminer lesbases de faible qualité en fin de séquence, problème chronique de la technologie Illumina. Si desbases ambiguës (N) ou de qualité supérieure à 5% d’erreur sont retrouvées en 5 prime ou 3 prime,alors celles-ci sont retirées. Les séquences trop courtes (20 bases) sont également retirées du fichier,celles-ci pouvant provenir d'un problème lors du séquençage.
Le contrôle qualité des données s’effectue donc en trois étapes. Dans un premier temps, nousgénérons un rapport qualité sur les données brutes reçues, puis nous « épurons » le jeu de donnéesavec AdapterRemoval et enfin nous générons à nouveau un rapport de qualité sur les nouveauxfichiers.
Le traitement des données a ensuite été réalisé par nos soins. Bien que les algorithmes développéspour le traitement des données de séquençage soient de plus en plus perfectionnés et que lesmachines à notre disposition soient de plus en plus puissantes, le traitement des données restetoujours l'étape la plus longue et celle qui nécessite le plus de ressources informatiques en termes depuissance de calcul, mais également de stockage et gestion des grandes quantités de donnéesgénérées. Savoir traiter ces données n'est donc pas suffisant, il faut aussi savoir les compresser aubon moment pour optimiser l'espace disque, savoir identifier quels sont les fichiers essentiels àconserver et quels sont les fichiers intermédiaires qu'il n'est pas nécessaire de conserver pour la suite.Sans cette gestion, un disque dur de 1To (1024Go) sera rapidement en manque d'espace disque et cepour une simple étude comprenant 20 patients.
Pour notre étude nous avons utilisé un serveur dédié en location chez OVH. Ce serveur a été loué surle sol Français pour conserver nos données sous la juridiction des lois françaises bien plusprotectrices pour nos données que celles des autres pays, États Unis par exemple. Il possède deuxprocesseurs Intel(R) Xeon(R) CPU E5-2670, 256 Go de RAM et 2To de stockage redondé sur 2disques en soft RAID 1. Ce serveur a été installé sous Ubuntu serveur 16.04 LTS (Long Timesupport) pour plus de stabilité. Il a également été mis en place par mes soins l'ensemble des outilsgénomiques nécessaires à l'analyse ainsi que des outils de sécurité anti intrusion & piratage.Préalablement à l’alignement de nos séquences sur le génome de référence, celui-ci doit être préparéet mis en forme pour pouvoir être utilisé. L'ensemble des séquences chromosomiques sont réunies enun seul fichier fasta qui est ensuite indexé pour pouvoir servir de génome de référence. Le logicielBowtie2, une référence dans le monde de la génomique, est utilisé pour confectionner notre génomede référence. Le génome de référence correspond à l'ensemble des séquences chromosomiquesobtenues pour une espèce donnée. Celui-ci est obtenu à partir de séquençages de plusieursvolontaires anonymes (13 provenant de New York pour la dernière version du génome humain).Leur génome est séquencé puis assemblé de novo sans présupposé de référence quelconque. Ilconstitue alors le génome de référence de l'espèce utilisé par toute la communauté scientifique,faisant de la dite espèce, un organisme « modèle ».

56
Les deux dernières versions de référence du génome humain sont HG19, sorties en février 2009 etHG38 en décembre 2013. Celles-ci sont disponibles en libre-service sur les serveurs de l'UCSC(University of California Santa Cruz) par exemple.
Les outils d'assemblages devenant toujours plus performants avec le temps, par rapport à HG19,HG38 a été assemblé et réalisé avec des outils plus modernes. Le génome est donc mieux “fini” etplus complet ce qui permettra une meilleure identification des séquences. Cependant, du fait qu'ilsoit plus récent que HG19, celui-ci est moins bien annoté que son homologue HG19 encorebeaucoup utilisé par la communauté scientifique. Pour cette étude, nous avons choisi d'utiliser laversion HG38 du génome humain comme référence.
Le logiciel utilisé pour l'alignement est TopHat2, qui utilise en interne l’algorithme d'alignement deBowtie2, est conçu pour aligner des séquences de génome entières comme l'on peut retrouver enséquençage d'exomes ou de génome total. Dans le cas du RNA-Seq, il est nécessaire de prendre encompte l'épissage alternatif. Si sur la séquence que nous essayons d'aligner se trouve une jonctionExon-Exon, alors cette séquence ne s'alignera jamais parfaitement sur une région génomique carnotre génome de référence contient aussi les introns. Bowie2 n'est pas capable d'identifier les sites decoupure dans nos séquences d'ARN afin qu’elles puissent être éventuellement coupées et répartiescorrectement entre exons d'un même gène. TopHat2 ajoute une sur-couche de fonctionnalité àBowtie2 et le rend capable de prendre en compte l'épissage alternatif des lectures.
Par individu, les 2 fichiers fastaq R1 et R2 (lectures sens et anti sens) sont spécifiés au logiciel.Comme nous l'avons vu à l'étape de préparation des librairies, nous ne travaillons pas avec des pairesde séquence concordante entre les deux fichiers, il faut donc le spécifier au logiciel.
En termes de spécificité, la probabilité qu'une séquence de 150 paires de bases s'aligne sur plusieursrégions génomiques est non négligeable. C'est ce que l'on appelle un alignement multiple. Cetteprobabilité est fortement réduite avec le système “paired end” car il impose que les 2 lectures R1 etR2 soient alignées l’une par rapport à l’autre. Il va cependant rester un faible pourcentaged'alignement multiple sur nos patients. Nous spécifions au logiciel d’alignement que nos séquencessont dites « stranded » ou “sens spécifiques” sur le brin pour l'alignement. Ainsi, il alignera lesséquences toujours dans le même sens sur le brin et n’essaiera pas de les « retourner » et de lesaligner par symétrie sur le même brin d'ADN. Ainsi, notre alignement est précis en termesd'assignation de gènes, sans cette option, il est également impossible d'identifier les exons plus oumoins exprimés en fonction de l'épissage alternatif. A ce stade de l'analyse, seul l’alignement sur legénome de référence est réalisé. L'information sur l'annotation, c'est à dire le fait de savoir quelleséquence a été alignée sur quel gène, n'est pas connue. Cette information n'est pas contenue dans lefichier de sortie final de l'alignement, le fichier BAM (Binary Alignment Map).

57
A partir des fichiers BAM, le nombre de séquences obtenues pour un gène est assigné par comptageà l'aide de l'outil Feature Count et directement à partir du fichier BAM. L’annotation du génome deréférence HG38 est alors fournie à Feature Count qui, à partir de ces positions génomiques estcapable de compter les séquences d’ARN s’alignant sur la portion de génome considérée. La sortiede ce logiciel consiste en un tableau double entrée au format texte récapitulatif. Indiquant le nombrede séquences obtenues par gène et par individu, il constitue l’information de base utilisée pourl’analyse statistique qui s’en suit.
L’obtention de la liste des gènes différentiellement exprimés se fait à l'aide de la méthode statistiqueSam-Seq implémentée par le paquet R samr. Les données sont normalisées puis le test SAM estexécuté avec un taux de faux positif (Q-value) de 5 %.
Le test SAM (Significant Analysis of Microarray) a été établi en 2001 par Virginia Tusher, RobertTibshirani et Gilbert Chuest et constitue un des tests le plus utilisé pour l'analyse d'expression enMicroarray. Pour un gène donné, il utilise le test-T pour calculer une statistique qui mesure la forcede la relation entre l'expression et la variable biologique associée, qui définit les conditionsexpérimentales. C'est la statistique T observée. Des permutations aléatoires du jeu de données sontensuite réalisées et le test-T est exécuté pour chaque permutation permettant de calculer unestatistique T théorique (attendue) sous l'hypothèse nulle, qui suppose que la variable biologiqueassociée testée n'est pas la seule à faire ressortir des différences d'expression. Nous utilisons 1500permutations. Les deux valeurs, T observé et T attendue, sont ensuite comparés à 5 % d’erreur etdéterminent quels sont les gènes dont le T observé est suffisamment éloigné du T attendu pourestimer que la variable biologique testée explique les différences d'expression entre les gènes.

58
Résultats
A) Déterminisme génétique des fibroses hépatiques causées par les schistosomes : Recherched’association sur le génome entier (GWAS) de sujets brésiliens
Les premières étapes de l’analyse des données d’un GWAS consistent à contrôler la qualité desdonnées. La figure 16 représente les différentes étapes qui vont constituer le contrôle qualité réaliséainsi que les SNPs et sujets d’étude qui n’ont pas passé ces contrôles.
1) Description de la cohorte et du microarray utilisé dans l’analyse GWAS
Dans les zones endémiques où le schistosome se développe, l'exposition au parasite pour lespopulations locales est élevée. La sélections des cas et des contrôles est basée sur la détermination duniveau de fibrose du foie. Pour cela, une échographie du foie est réalisée sur les patients infectés parle schistosome et le niveau de fibrose est déterminé à l'aide de la classification de Niamey définie en1996 comme étant la première classification de la fibrose par imagerie standardisée (135) et permetde classifier les différents niveaux de fibroses.
Les différents niveaux vont de A, absence de fibrose, à F fibroses très grave, avec des variantes plusou moins graves pour chaque classe. Ainsi un D+ est plus proche d'un E qu'un D- qui sera plusproche d’un C. Les individus possédant les niveaux de fibroses A, B et C sont décrits commepossédant une fibrose faible voire inexistante malgré l'exposition et l’infection prolongée auschistosome. Elles sont considérées comme des fibroses non graves qui n’évoluent pas vers desformes graves.
Les foies présentant des fibroses de type E et F sont les plus gravement atteints. La classification Ecorrespond à un foie présentant une fibrose dans sa région centrale qui s’étend au sein duparenchyme. La classification F, la plus grave, correspond à une fibrose s’étendant au-delà de lapériphérie du foie et entrainant une rétractation de l’organe. Les formes D ont toujours été difficiles àclasser. Elles ne représentent pas toujours un stade de fibroses intermédiaires entre les fibrosesgraves et bénignes. Les patients peuvent, soit rester sous forme D malgré l'exposition au parasite, soitévoluer plus ou moins rapidement vers des fibroses plus graves. Il est donc difficile de les classifierau moment même de la campagne de prélèvement. Il faudrait pour cela suivre leur évolution dans letemps (mois, voire années) ce qui est parfois difficile à réaliser. Ces formes D ont été exclues denotre étude.
Afin de réaliser cette étude, la puce GWAS illumina de type omni25 a été utilisée. Le génotypage aété réalisé au Centre National de Génotypage (CNG). Les génotypes pour 2 329 163variant/mutations uniques de l'ADN (SNP) pour 1157 patients. L’analyse des données a été réaliséeen collaboration avec l'institut des maladies génétiques IMAGINE à Paris, avec l'équipebio-informatique de Laurent Abel.

59
Figure 16. Étapes du contrôle qualité réalisées sur les données brutes.

60
2) Contrôle des données
a) Contrôle par individu : exclusion des patients définis comme apparentés, confirmation dusexe civil par inférence génétique, taux de génotypage par patients, taux d'hétérozygotie
La cohorte d’étude doit répondre au critère d'indépendance génétique des individus. La premièreétape consiste à estimer les liens familiaux entre individus en utilisant le coefficient de Kinship quiestime le paramètre d'Identity by descent (IBD). Le coefficient de Kinship sera élevé entre deuxindividus s’ils partagent des allèles similaires tout au long de leur génome. Cette situation suggèrealors un ancêtre commun entre ces deux patients et donc un lien de parenté. Les paires d'individusqui possèdent un coefficient supérieur à 0,49 correspondent à des jumeaux ou à un seul et mêmeindividu inclus 2 fois dans le génotypage. Vingt-quatre individus ne passent pas ce filtre et ont étéexclus.
Le sexe peut être estimé génétiquement à partir de la présence ou absence du chromosome Y. Lesexe ainsi inféré peut être comparé au sexe enregistré lors du recrutement des patients. Cette étapepeut détecter un manque de fiabilité dans nos données ou encore des erreurs de manipulation deséchantillons. Toutes discordances entre le sexe obtenu par interview et le sexe déduit du génotypagesuggère une erreur qui conduit à éliminer l’ADN correspondant. Le taux d'hétérozygotie duchromosome X présent en une seule copie chez les hommes (XY) doit être faible, contrairement chezles femmes qui présentent des taux plus élevés du fait du double X (XX). Le décompte du nombre demarqueurs présentant des génotypes sur le chromosome Y (nommé ici décompte Y) permetd’estimer le sexe. Les hommes présentent un décompte plus élevé. La figure 17 montre les valeurs dedécompte Y en ordonnée et le taux d'hétérozygotie en abscisse. Les points noirs représentent leshommes et les points rouges correspondent aux femmes. La cohorte GWAS est composée de 574hommes et 583 femmes avant le contrôle qualité. Cinquante patients présentent une discordanceentre leur sexe génétique et leur sexe enregistré lors du recrutement. Un patient (en vert) parmi ces50 peut être considéré comme homme et femme à la fois (XXY), c'est à dire qu'il possède un nombreélevé de SNPs génotypé sur le chromosome Y tout en possédant en même temps un tauxd’hétérozygotie important sur les deux chromosomes X. Cela pourrait indiquer un mélange de deuxéchantillons lors des manipulations. Ce patient a été exclu. Pour les 49 autres, nous conservons lesexe inféré génétiquement.
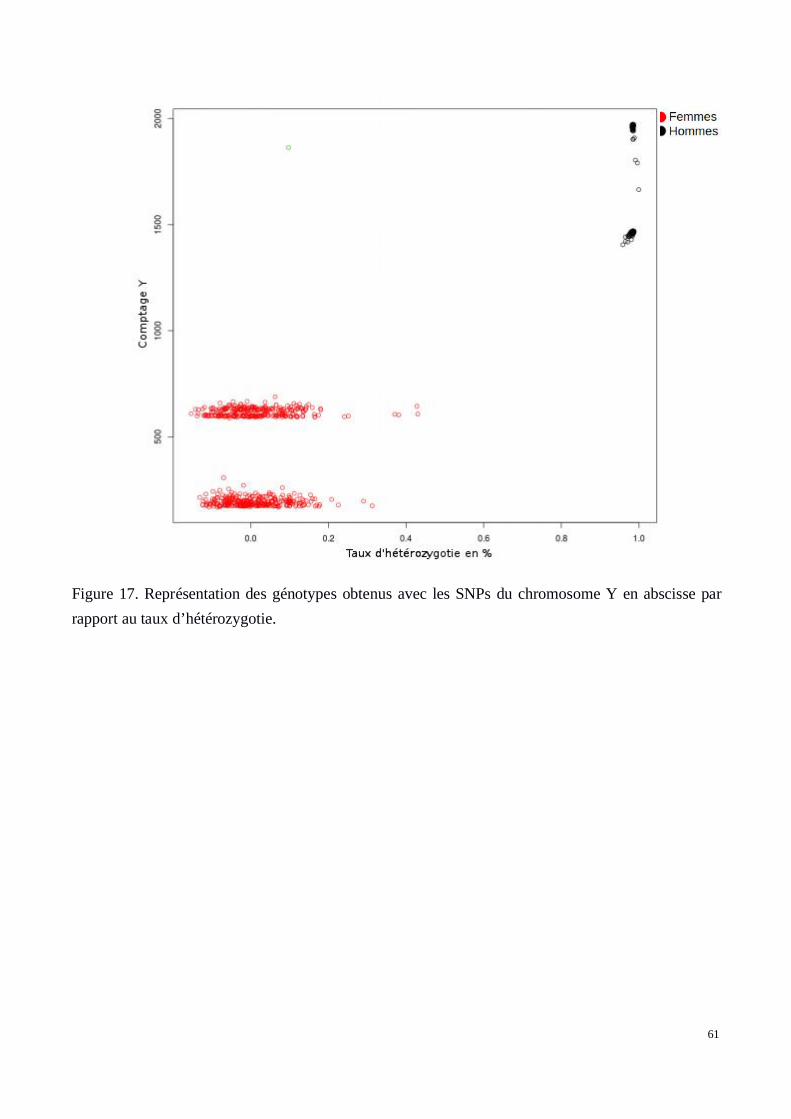
61
Figure 17. Représentation des génotypes obtenus avec les SNPs du chromosome Y en abscisse parrapport au taux d’hétérozygotie.

62
Le taux moyen de génotypage pour les 1157 patients est de 98.93%. Pour six patients, 5 % desgénotypes sont manquants. Un taux de génotypage inférieur à 95 % pourrait indiquer un manque defiabilité dans les résultats obtenus pour cet ADN. Les ADN ayant un taux de génotypage inférieur à95% sont donc exclus.
Le taux d'hétérozygotie permet d'estimer la variabilité et la diversité génétique dans l’échantillon. Letaux d’hétérozygotie observé correspond à la probabilité que deux gènes tirés aléatoirement aient desallèles similaires ou différents sous l’hypothèse d’Hardy-Weinberg. Les taux d'hétérozygotieobservés et estimés sur notre échantillon sont ainsi calculés et comparés. Si une trop grandedifférence est retrouvée entre ces taux, alors cela indique un problème pour l’échantillon qui doit êtreexclu. Un taux élevé peut indiquer que les ADN proviennent de plusieurs patients et auraient pu êtremélangés tandis qu'un taux bas peut indiquer un problème dans le génotypage. Classiquement, leseuil défini par la littérature, qui suggère qu’un ADN a un profil anormal, est 3 fois l'écart typeau-dessus et au-dessous du taux moyen d'hétérozygotie de la cohorte. Quinze individus ont été ainsiexclus.
b) Contrôle par variant : Suppression des SNP dupliqués et des chromosomes sexuels, taux degénotypage par SNP, test de HardyWeinberg, Filtrage sur la fréquence allélique
Certains SNPs apparaissent sous plusieurs nomenclatures différentes et peuvent donc êtremultiplexés sur la puce GWAS illumina. Ces SNPs “doublons” doivent être éliminés. Neuf milletrois cents soixante-huit (9 368) variants redondants sont exclus. Ne sont conservés uniquement lesSNPs dont le nombre de génotypes obtenu est le plus élevé.
Les SNPs situés sur les chromosomes sexuels sont également exclus. Les chromosomes sexuelspartagent des régions génomiques similaires avec d'autres régions situées sur les autosomes. Cesrégions, situées aux extrémités des chromosomes X et Y, sont également homologues entre X et Y.Les variants qui les constituent présentent une homologie forte entre les hommes et les femmes,contrastant avec l’héritabilité spécifique des variants du chromosome Y et X. Ces régionspseudo-autosomales, qui ne figurent pas sur les cartes génomiques, peuvent alors causer desproblèmes lors de l'analyse statistique. Les SNPs présents sur les chromosomes sexuels ne peuventpas être analysés avec les tests statistiques utilisés pour l’analyse des autosomes. Ils sont donc retirésde l’analyse : 53 435 SNPs sont exclus.
Le filtre du taux de génotypage par SNPs consiste à retirer les SNPs dont le taux de génotypage estinférieur à 95 %. Cinquante-deux mille cinq cent soixante-quatre SNPs sont inférieurs à ce seuil et nesont pas considérés comme fiables. Le même taux de génotypage est ensuite calculé chez les cas etles témoins de manière indépendante afin d'analyser, non pas le taux de génotypage global, maiscelui des cas et celui des témoins indépendamment.

63
Si, pour un SNP donné, un de ces deux taux est inférieur à 95 %, alors le SNP ne passe pas le filtre.Deux milles quatre cent cinq SNPs ont été ainsi exclus.
Le modèle de Hardy-Weinberg postule que nous sommes en présence d'une population assez grandepour être considérée comme idéale. La notion de population théorique idéale en génétique faitréférence à des caractéristiques précises. Dans une population idéale, il n’y aurait pas d’apport denouveaux patrimoines génétiques qui pourraient provenir de la migration d’individus ou encore del’apparition de nouvelles mutations de novo. La sélection naturelle n’interviendrait pas : tous lesindividus auraient la même chance de se reproduire et de transmettre leur patrimoine génétique à lagénération suivante. Enfin la population doit être considérée infinie afin que tous les allèles soientreprésentés, même les plus rares. Dans ce cadre, l’équilibre de Hardy Weinberg est défini selon laformule suivante :
p² + q² + 2pq = 1
Pour un SNP donné, p correspond à la fréquence d’apparition dans la population d’un allèle et q lafréquence de l’autre. p² et q² correspondent alors à la fréquence des deux génotypes homozygotes et2pq la fréquence des génotypes hétérozygotes. Si la somme de ces fréquences est égale à 1 alors lesfréquences alléliques sont conservées d'une génération à l'autre. Un écart important à cet équilibre estsouvent l’indication d’un biais dans le génotypage. La technique de détection allélique du GWAS parInfinitum Assay est basée sur l'intensité de deux fluorochromes, un par allèle, détectée en mêmetemps lors de la lecture du signal pour un SNP donné. La discrimination allélique se fait ensuite parla dissociation d'intensité des deux signaux grâce à un algorithme automatisé. Certains allèlesdifficiles à détecter peuvent présenter des signaux difficiles à discriminer. Ce problèmed'identification entraîne parfois la fausse attribution d'un groupe d'allèle pour un autre. Cela entraîneun écart à l’équilibre d'Hardy Weinberg pour le marqueur concerné. Les résultats pour ces SNPs làne sont donc pas fiables. Le test de HardyWeinberg est appliqué sur les données.
Les SNPs présentant une P-value inférieure à 10-5 pour notre test de Hardy Weinberg sont retirés.Deux cents SNPs sont concernés.
Lors de l’assignation allélique, l’absence de représentation d’un allèle peut signifier un problème dedétection de signal lors du génotypage. En revanche, si ce n’est pas un biais de la technique, celapeut indiquer un allèle réellement peu représenté dans la population pour le SNP en question (Allèlepeu fréquent <5% ou rare <1%). L’analyse GWAS n’est pas adaptée (puissance statistiqueinsuffisante) pour détecter des SNPs rares dont la fréquence est inférieure à 1%. Nous recherchonsdonc des marqueurs communs entre tous les patients (>1%). Nous avons donc fixé un seuil de MAFà 0,5 % pour définir et exclure les allèles rares, 421 124 SNPs sont exclus.

64
Au terme de ces contrôles qualité, ont été exclus 46 individus pour n’en conserver que 1111, 555femmes et 556 hommes, 493 cas et 567 témoins. Quarante-neuf pour cent (n= 241) des cas sont deshommes tandis que 51% (n= 290) des témoins le sont également. Par ailleurs, 539 096 marqueurs ontété exclus et 1 816 152 SNP ont été conservés. Ont été exclus 150 patients supplémentaires de nosdonnées, majoritairement des cas, pour arriver à 961 patients au total. Pour ces patients, le phénotypeà été confirmé et validé. La nouvelle cohorte d’étude est composée de 477 hommes et 484 femmes,343 cas et 567 contrôles.
3) Étude de l’hétérogénéité de la cohorte à l’échelle mondiale et structure à l’échelle locale
Une Analyse en composantes principales (ACP) a été réalisée à partir des 961 patients ayant passéles filtres du contrôle qualité. L’ACP permet un changement de repère. Ces nouveaux repères dansl’espace, appelés composantes, déterminent des axes dont l’objectif est de mettre en évidence au seindes données et sans a priori, les variables capables de différentier au mieux les résultats. L’ACP nerenseigne pas sur la nature de ces variables obtenues ni sur ce qu’elles représentent réellement. Cesvariables sont hiérarchisées, la première étant celle qui représente la plus grande variabilité observéedans les données et la dernière la moins grande. Ainsi les premières composantes sont ditesprincipales. L’ACP de notre étude intègre les données disponibles pour 2504 individus provenant dela base de données publiques 1000 génomes phase 3 représentant 26 populations distinctesregroupées en 5 continents. Elle contient 84.4 millions de SNPs. Seuls les SNPs communs à nosdonnées et à celles de 1000 génomes ont été conservés, c'est à dire 1 433 287.
Généralement les 2 premières composantes principales trouvées expriment la variation génétique dueà la géographie, c'est à dire aux différences génétiques retrouvées entre les populations de référence.Les deux premières composantes principales de cette ACP sont représentées en figure 18. Lesdonnées du GWAS, ici en bleu ciel, montrent une hétérogénéité allant de la population Africaine àEuropéenne. Le profil génétique des individus de l’étude est donc intermédiaire entre ces deuxpopulations, certains étant uniquement africains et d'autres uniquement européens. La populationd’étude présente une forte hétérogénéité qui devra être prise en compte lors de l'analyse statistiquedes données.
Sur la représentation graphique des deux premières composantes principales, les sexes des individussont répartis de manière homogène. Ils ne produisent donc pas une stratification supplémentaire denotre population (données non présentées).
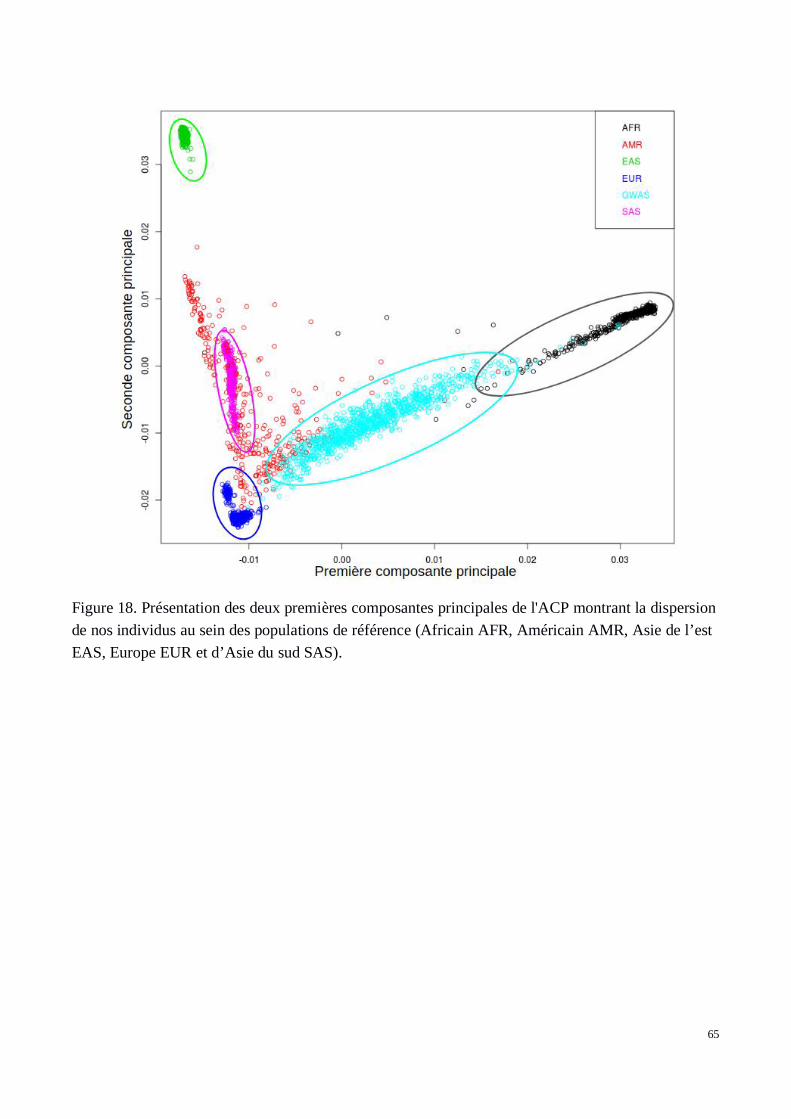
65
Figure 18. Présentation des deux premières composantes principales de l'ACP montrant la dispersionde nos individus au sein des populations de référence (Africain AFR, Américain AMR, Asie de l’estEAS, Europe EUR et d’Asie du sud SAS).

66
A l'échelle locale, un biais semble persister : les liens de parentés entre les individus. Il existe deuxtypes de liens, ceux qui peuvent être calculés via l'Identity By Descent comme vu précédemmentavec King, et ceux dit « cryptiques » qui ne peuvent être modélisés qu'à l'aide d'une ACP.
Précédemment, nous avions retiré les individus quasiment identiques de type « jumeaux » à l'aide ducoefficient de Kingship. C'est à l'aide de ce même coefficient que sont éliminés les individus demanière plus large en étendant le lien de parenté à la famille. Enfin, l'homogénéité de notrepopulation a été contrôlée à l'aide d'une ACP, cette fois_ci avec seulement nos échantillons.
Ont été représentées sur la figure 19 les 3 premières composantes principales résultantes de cetteACP. Ces représentations permettent de visualiser les biais de sélection des patients qui n'auraientpas été corrigés et qui diviseraient notre population en groupes génétiques distincts qui pourraientfausser l’analyse. La cohorte finale présente un aspect homogène excepté pour certains individus dits« outliers ». Aux vues des graphes, un seuil arbitraire est défini, celui-ci permettra d'éliminer lespatients s'écartant trop de notre groupe homogène. Cent quatre-vingt-cinq individus partagent un liende parenté à divers degré plus ou moins éloigné.
4) Analyses statistiques des données du génotypage sur l’ensemble des sujets de la cohorte dedécouverte
a) Stratégies d’analyses possibles
La figure 20 représente le cheminement défini pour analyser les données (liste des possibilités nonexhaustives). Les couleurs correspondent au cheminement envisagé, en vert ce qui a été choisi pourl’analyse, en rouge ce qui a été exclu. Les notations i, ii, iii et iiii sont utilisées par la suite dans letexte pour situer sur la figure l'étape qui est décrite.
Un test allélique (ie D versus d, avec D l'allèle ancestral et d l'allèle mutant) devra prendre en compte2 allèles indépendants par génotype. Cela peut engendrer des biais statistiques, il est donc préférablede faire une analyse avec les génotypes (i). Nous utilisons alors un test de tendance (Cochran –Armitage) (ii). Le test de tendance permet d’établir une relation linéaire entre le phénotype et lesgénotypes, le phénotype (malades / témoins) étant la variable que nous cherchons à expliquer à l’aidedes génotypes, les variables explicatives. La relation linéaire permet ensuite de déterminer la part duphénotype expliqué par d’autres variables.
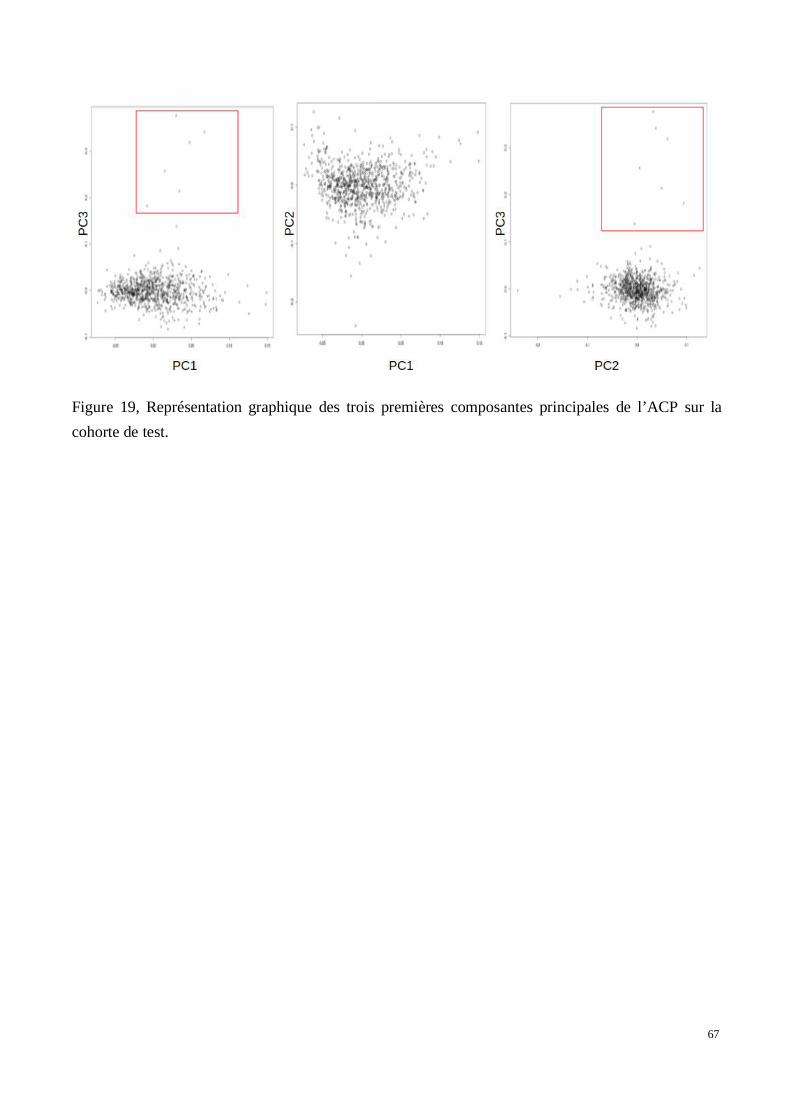
67
Figure 19, Représentation graphique des trois premières composantes principales de l’ACP sur lacohorte de test.
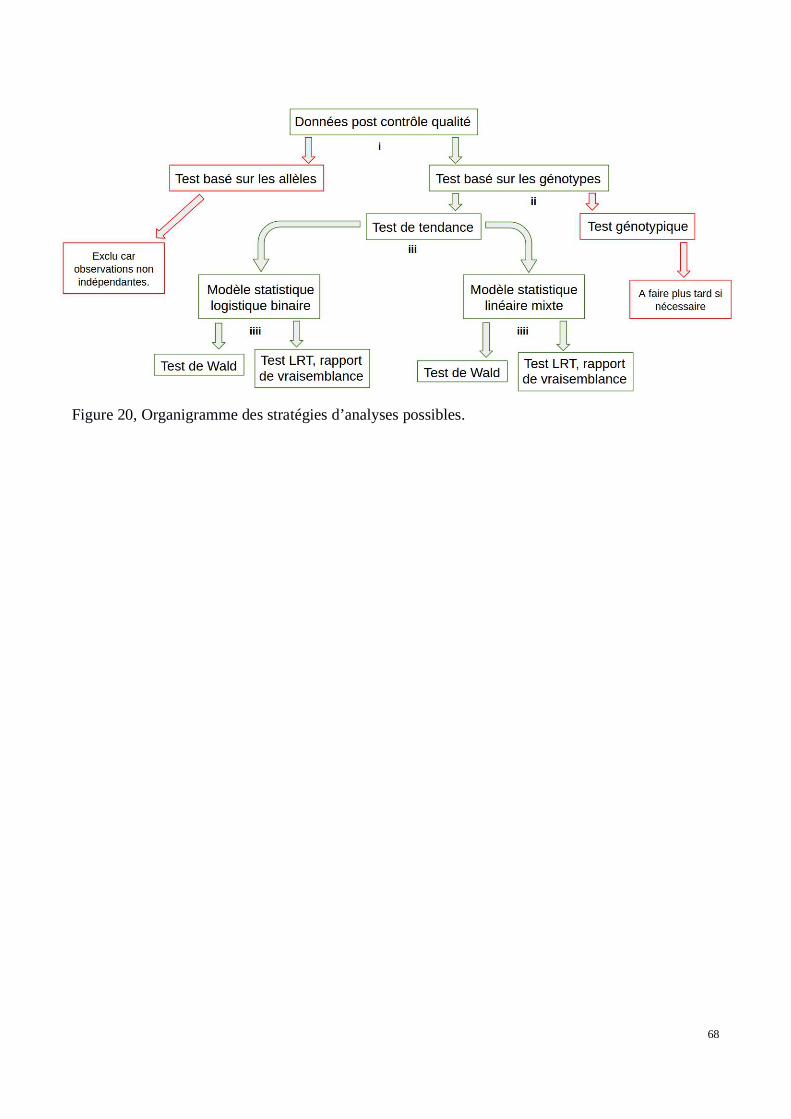
68
Figure 20, Organigramme des stratégies d’analyses possibles.

69
Le test de tendance prend en compte différents paramètres :
● Le modèle du test : Les génotypes sont encodés sous forme binaire. Il existe 3 génotypespossibles avec 2 homozygotes (DD et dd) et 1 hétérozygote (Dd). Nous pouvons donc réaliser letest sous 3 modèles différents :
Dominant : (DD, Dd) versus dd (codage 1,2)
Récessif : DD versus (Dd, dd) (codage 1,2)
Additif : DD versus Dd versus dd (codage 0,1,2)
● L'ajustement sur les co-variables : Il s'agit ici de prendre en compte un ensemble de variablesliées aux individus et pouvant affecter les résultats des tests réalisés. Nous pouvons ajuster sur lesexe, l'âge ou encore la présence ou absence d'autres pathologies. Ces facteurs peuventengendrer des biais dans l’étude et dans certains cas faire ressortir des SNPs de manièresignificative. Il est donc nécessaire de les prendre en compte pour limiter les faux positifs.
● L'ajustement sur les composantes principales : il s’agit ici de prendre en considération lesderniers biais qui n’auraient pas été identifiés dans la population d’étude. Une population d'étudeest rarement idéale. Celle-ci peut présenter une stratification génétique comme nous le montrel’analyse en composante principale. Certains liens de parentés, même lointains, peuventégalement persister entre les individus. Lors des tests, un ajustement est également réalisé àl'aide des trois premières composantes principales obtenues par ACP sur les données en fin decontrôle qualité.
La question se pose toujours de savoir combien de composantes principales doivent être utiliséesdans le test et cette question est souvent soumise à débat. C'est pour cela que nous testons plusieurspossibilités d'ajustement et ferons un consensus des résultats obtenus.
Dans un premier temps nous avons donc réalisé pour les 3 modèles :
● Une régression avec les phénotypes seuls.
● Une régression avec les phénotypes + un ajustement sur le sexe ainsi que les 3 premières
composantes principales.
Nous avons vu les différents paramètres à inclure dans un test de tendance, nous allons voirmaintenant qu'il existe plusieurs tests possibles. Nous avons deux possibilités pour le test detendance (iii) : (1) Modèle statistique logistique binaire et (2) Modèle statistique linéaire mixte.

70
b) Régression logistique binaire et modèle statistique linéaire mixte
Dans le cas d'un phénotype binaire (codé 0 et 1) on peut modéliser la relation phénotype / génotypepar un modèle de régression logistique et appliquer le test de tendance en testant le paramètre associéau génotype. La modélisation se fait à partir d'une combinaison linéaire de paramètres explicatifs dela pathologie qui sont nos SNPs.
Logit (P(Y=1)) = A + B X génotype + C X covar1 +….
L’expression ci-dessus montre le modèle du test de tendance avec A une constante, B la valeurcorrespondant au « poids » du génotype dans le modèle et ensuite les différentes co-variablesincluses. Nous nous intéressons plus particulièrement au B via un test de nullité. L'hypothèse testéeest B = 0, c'est à dire que le génotype n'a pas d’effet dans notre modèle. S’il est différent de 0 alorsc'est qu'il y a un effet du génotype, c'est ce qui est recherché. C'est la p-value de ce test de nullité quinous dira si l’effet du variant est significatif ou pas pour notre pathologie et donc si on le considèrecomme associé.
Ce test de nullité peut dans ce cadre se faire en utilisant différentes statistiques (iiii) :
● Le test de Wald : il est plutôt adapté à un échantillon tendant vers l'infini, ce n'est pas le cas denotre cohorte dont le nombre d'individus conservé au final est autour de 900.
● Le rapport de vraisemblance (LRT) : il compare l’adéquation de deux modèles pour réaliser lastatistique de test. Un des modèles recherche l’effet des variables SNPs sous l’hypothèse nulletandis que le second prend en compte des paramètres libres. Le test estime ensuite si ladifférence entre les deux modèles (absence ou présence de l’effet des variants génétiques) peutexpliquer le phénotype.
Par défaut le logiciel Plink utilise le test de Wald mais ne permet pas de faire le rapport devraisemblance, ce second est donc implémenté en R sous un environnement Plink. Les scripts R ontété réalisés par les bio-statisticiens de l'équipe du Professeur Abel. Ici, nous privilégions le rapport devraisemblance pour l'estimation de notre P-value, le test de Wald étant beaucoup moins adapté etmoins robuste.
Nous avons vu lors du calcul des liens de parenté (Identity be Descent IBD) que 24 individus étaientconsidérés comme des jumeaux et 185 partageaient un lien génétique à divers degrés de parenté plusou moins éloignés. Ceux-ci ont donc été exclus de l'analyse avec le modèle de statistique logistique.En effet, bien que ce modèle puisse intégrer des co-variables, les liens familiaux cryptiques nepeuvent être estimés via celles-ci. Nous perdons de ce fait de la puissance statistique. Ces lienscryptiques peuvent s’apparenter à des données en parties répétées. Le modèle mixte est généralementutilisé pour l'analyse de plusieurs mesures pour un même individu à différents temps par exemple.Ce n'est bien sûr pas notre cas, car nous travaillons avec un seul génotype par SNP et par patient.

71
Cependant des liens familiaux forts qui rendraient proches génétiquement 2 individus peuvents’apparenter à des mesures différentes sur une même famille et donc à des données répétées. Nousallons pour cela utiliser le modèle statistique linéaire mixte.
Nous allons donc travailler sur ce modèle mixte avec l'ensemble des individus, quel que soit leurrelation de parenté. Les autres étapes du contrôle qualité sont conservées. Les 24 “duplicata”génétiques seront exclus (valeurs de King supérieures à 0,49).
Cette modélisation est réalisée avec le logiciel GEMMA. GEMMA permet d'estimer la p-value avecle test de Wald ainsi qu'avec le rapport de vraisemblance à la différence de Plink qui nécessite Rpour le rapport de vraisemblance.
c) Résultats : Q-Q plots et Manhattan plots
Compte tenu du grand nombre de P-value obtenu à la suite d'une telle analyse, il est impossible deles représenter sous forme de tableau. Le Manhattan plot est utilisé pour représenter graphiquementle log négatif des P-values le long des 22 chromosomes humains (figure 21). Ce type de graphiquepermet de visualiser le profil de l'analyse réalisée, et d'identifier rapidement quels sont les variantsprésentant les associations les plus fortes.
Afin de d'estimer l’erreur de type 1, c'est à dire celle qui pourrait faire apparaître des faux positifsdans nos résultats, un Quantile-Quantile (Q-Q) plot est également réalisé. Chaque point sur ce Q-Qplot représente un SNP pour lequel est représenté sa valeur statistique observée par rapport à savaleur estimée sous l'hypothèse nulle. Dans le profil classique de cette représentation, les points sontclassés par P-values estimées croissantes. Les points représentants les SNP non significatifs doiventsuivre la droite Y=X tandis que ceux qui sont significatifs, avec les P-values les plus hautes, dévientde la droite Y=X (figure 22).
Si la majorité des points dévient de la droite Y=X, alors un biais est présent dans notre analyse.Généralement, quand ce type de situation se présente, l'ajout des co-variables pour ajustement lors dutest améliore les résultats. Un consensus entre toutes les analyses effectuées permet de mettre enévidence 215 SNPs qui présentent une association (p<10-7, n= 17) ou suggestion d’association(p<10-6, n=198) avec la fibrose hépatique grave.
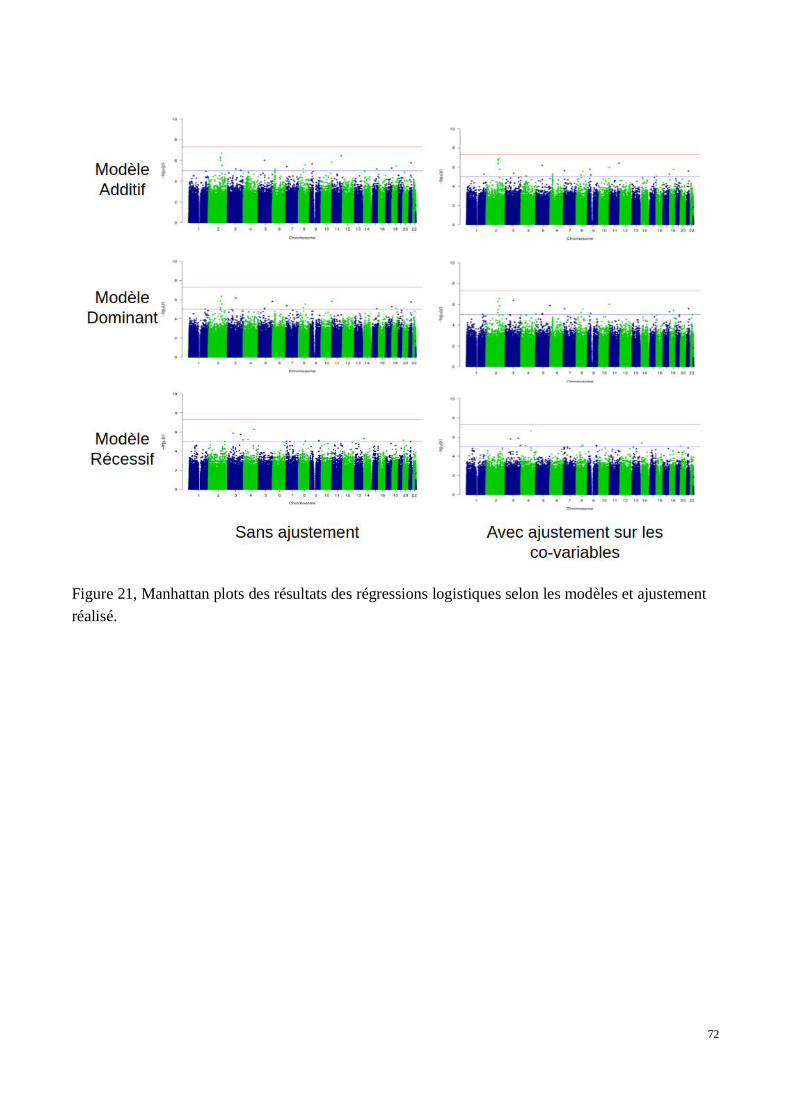
72
Figure 21, Manhattan plots des résultats des régressions logistiques selon les modèles et ajustementréalisé.
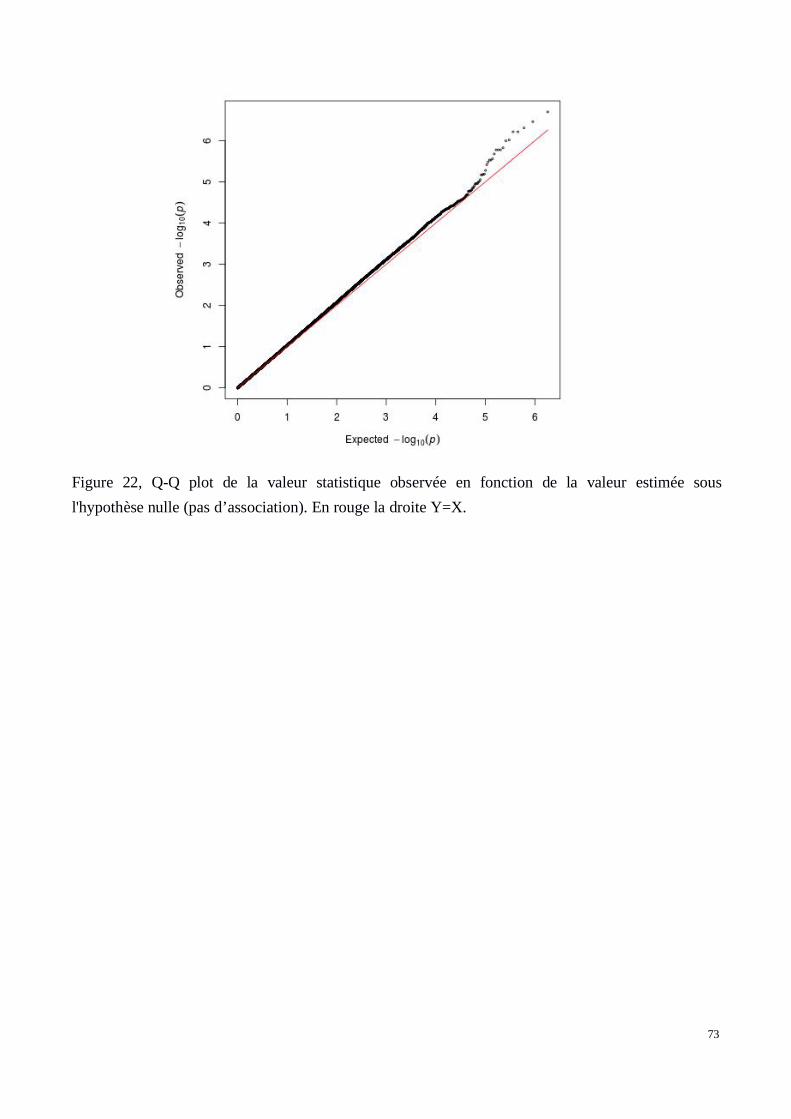
73
Figure 22, Q-Q plot de la valeur statistique observée en fonction de la valeur estimée sousl'hypothèse nulle (pas d’association). En rouge la droite Y=X.

74
5) Évaluation des associations dans une cohorte de confirmation recrutée en Ouganda
Parmi les associations observées dans l’analyse GWAS, nous en avons sélectionnés 180 pourvalidation dans une cohorte indépendante. Celle-ci a été recrutée auprès de la population Allur vivantsur les bords du lac Albert en Ouganda d’origine ethnique sud soudan. Les principalescaractéristiques de cette cohorte sont qu’elle est composée d’individus génétiquement unis, cas etcontrôles appartenant à la population proche. Elle est composée de pêcheurs exposés à S mansonidont la moitié présentait des fibroses hépatiques graves.
Le test d’association (Tableau 5) uni-varié réalisé sur cette cohorte montre l’association de 4 SNPsdans les gènes DAB1, DLX2AS1, HPSE2 et LSAMP. Un second test d’association a été réalisé(Tableau 6) en analyse multi-variée. L’analyse multi-variée permet de calculer la P-value de chacundes SNPs en calculant l’effet des uns par rapport aux autres. Le logiciel IBM SPSS incorpore aumodèle l’effet de chacun des SNPs selon une méthode d’ascendance (SNPs incorporés les un aprèsles autres) ou descendante (tous sont incorporés dès le départ et retirés les un après les autres). LesP-values sont recalculées à chacun des ajouts ou retraits des SNPs jusqu’à ne conserver que les SNPsprésentant, ensemble, une bonne significativité dans le modèle. Les marqueurs des gènes DLX2AS1,HPSE2, LSAMP et DAB1 sont conservés dans le modèle et apportent un pouvoir prédictif de lapathologie. Tout comme dans l’analyse GWAS, l’âge et le sexe des patients ont été inclus en tant queco-variables pour la prédiction.
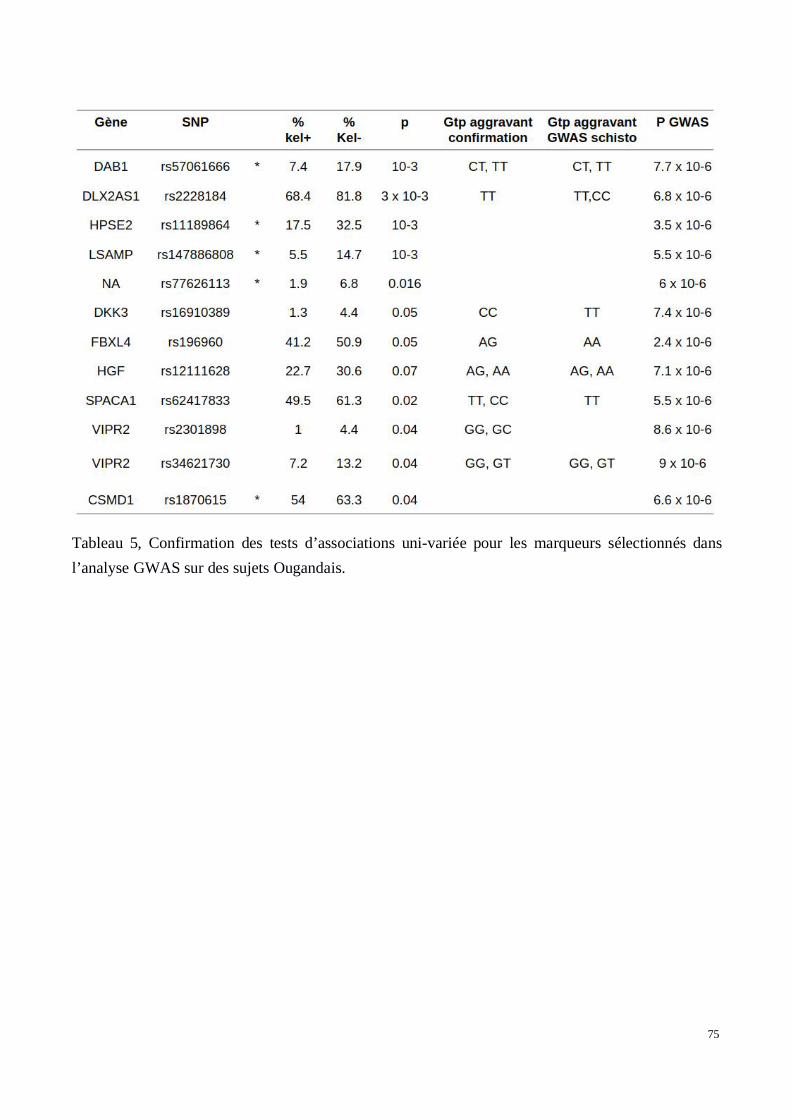
75
Tableau 5, Confirmation des tests d’associations uni-variée pour les marqueurs sélectionnés dansl’analyse GWAS sur des sujets Ougandais.
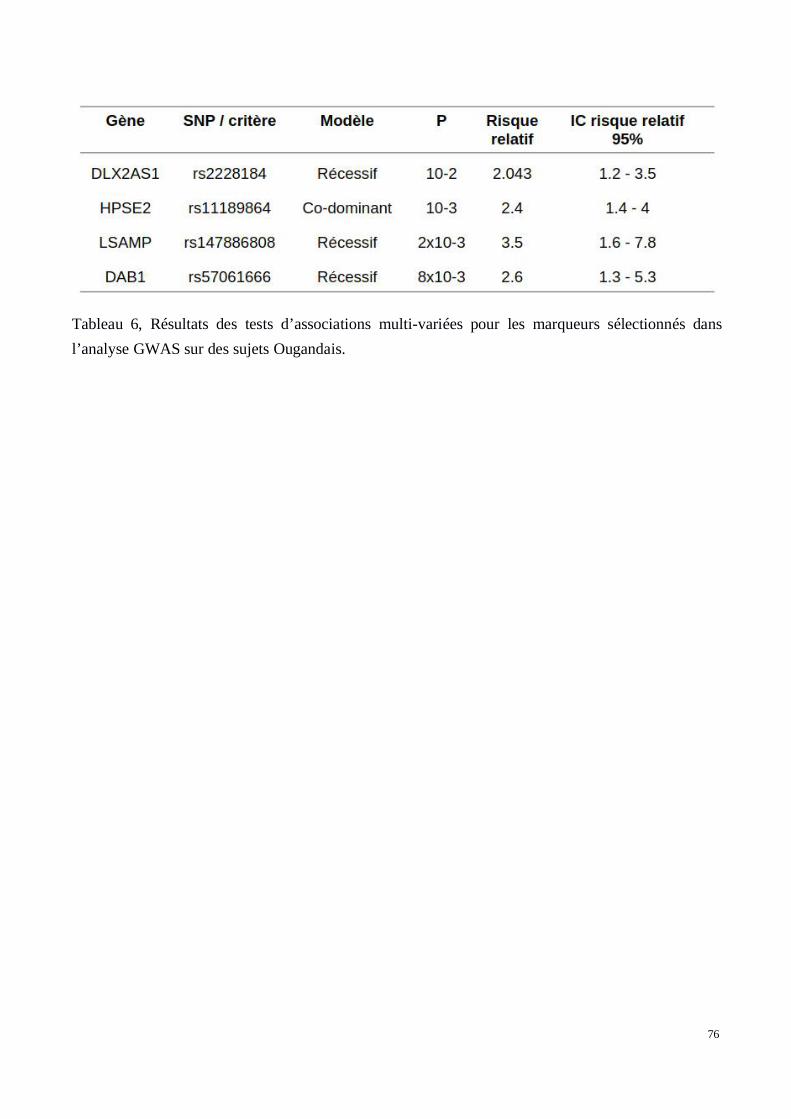
76
Tableau 6, Résultats des tests d’associations multi-variées pour les marqueurs sélectionnés dansl’analyse GWAS sur des sujets Ougandais.

77
B) Cicatrisation anormale de la peau : analyse du transcriptome et analyse génétique descicatrices hypertrophiques et des tissus chéloïdes.
1) Voies de signalisation dans les cicatrisations anormales de la peau.
a) Classification / agrégation hiérarchique des patients basée sur l’analyse du transcriptomedes tissus. Homogénéité des groupes cliniques.
Dans la seconde partie de la thèse, nous avons voulu clarifier les mécanismes biologiques mis en jeudans les fibroses cutanées causées par la cicatrisation pathologique de type cicatrice chéloïde ethypertrophique. Pour cela, avant de rechercher les mutations qui pourraient expliquer ces problèmesde cicatrisation, nous avons souhaité analyser l’expression des gènes dans ces tissus. L’expressiondes gènes dans 20 tissus cicatriciels chéloïdes (Kx, avec x le numéro du patient) et 7 tissushypertrophiques (HYPx) a été comparée à l’expression des gènes retrouvés dans 7 tissus de peaunormale (NSx) provenant de patients sans problème cicatriciel connu. Les sujets de l’étude ont étéclassés en fonction des similitudes observées dans l’expression de leur génome (Figure 23). Lespatients présentant le plus grand degré de similitude étant les plus proches sur l’arbre et inversement,les patients qui présentent les plus grandes différences dans leur transcriptome sont les plus éloignés.Dans cette classification hiérarchique, les sujets affectés de cicatrices chéloïdes (KEL) sontclairement éloignés des sujets HYP et des sujets sans cicatrices NS. Au sein du groupe KEL lesindividus sont proches au regard de la distance qui sépare KEL avec le groupe HYP et NS. Cecijustifie pleinement de considérer ce groupe KEL comme un groupe distinct des HYP et des NS; lesindividus KEL peuvent donc être analysés ensemble. On notera néanmoins une certaine tendance desKEL localisées aux oreilles et des KEL localisées au thorax à ségréguer comme des groupes séparés.Le groupe formé par K051, K063, K076, K043, K055, K058 et K059 est composé de KEL localiséesaux oreilles à l’exception du K055 qui est une KEL localisée au thorax. Le groupe K086, K021,K057, K052 et K089 est composé de KEL localisées au thorax à l’exception du K052 qui est uneKEL à l'oreille. Ceci nous conduira ultérieurement à tenter une analyse comparative du transcriptomede ces deux sous-groupes de KEL. Les HYP apparaissent proches des NS, certains comme HYP 11,14, 15, ne pouvant pas être distingués des sujets contrôles. Cela confirme que ces lésionshypertrophiques sont moins graves que les cicatrices chéloïdes. Elles sont en effet non invasives etrégressent assez rapidement alors que les KEL sont extrêmement invasives et tendent à récidiveraprès ablation.
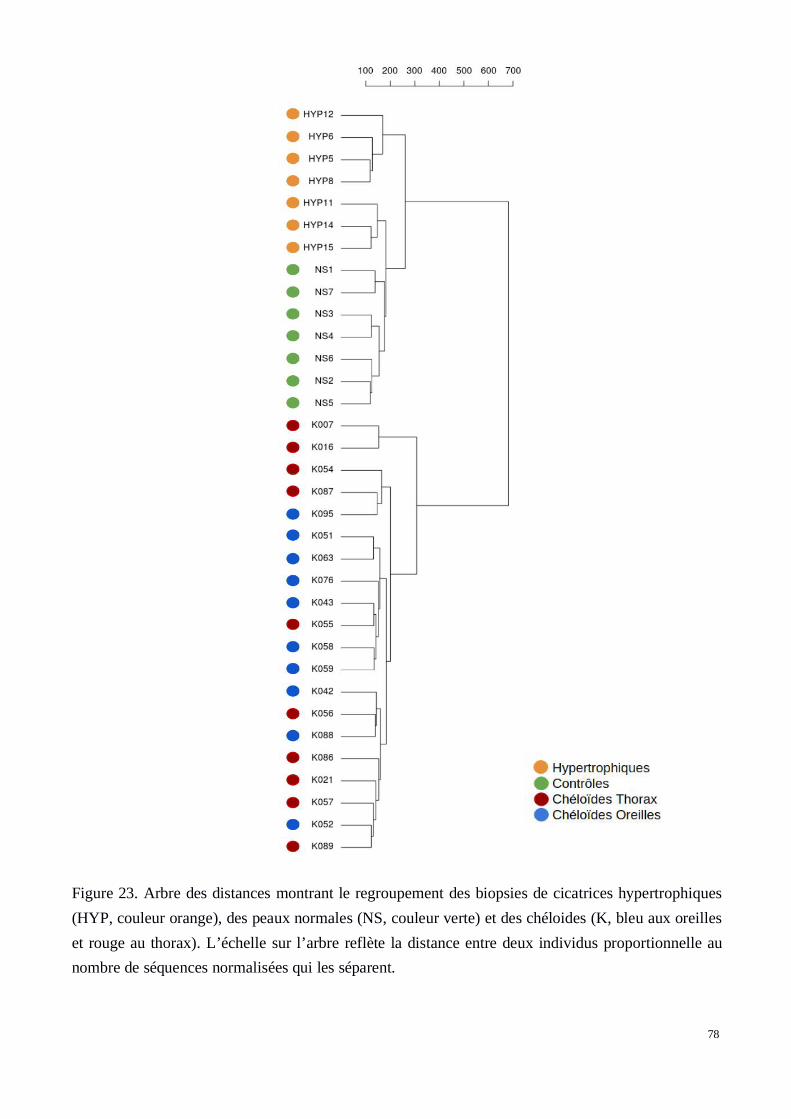
78
Figure 23. Arbre des distances montrant le regroupement des biopsies de cicatrices hypertrophiques(HYP, couleur orange), des peaux normales (NS, couleur verte) et des chéloides (K, bleu aux oreilleset rouge au thorax). L’échelle sur l’arbre reflète la distance entre deux individus proportionnelle aunombre de séquences normalisées qui les séparent.

79
b) Analyse des voies métaboliques exprimées de manière différente entre les tissus chéloïdes etles tissus sains.
La comparaison du transcriptome des peaux saines et des tissus chéloides (oreilles et thorax) révèleque 6892 gènes sont exprimés différemment entre ces deux groupes en acceptant un taux de fauxpositif (Q-value) de 5 % et en prenant en considération des variations d'expression égales ousupérieures à deux. L’analyse avec le logiciel I.P.A qui recherche les voies dans lesquels ladistribution des gènes ne semble pas être aléatoire, identifie 52 voies de signalisation dontl’activation ou l’inhibition sont associées aux KEL (p > 10-2). Comme plusieurs voies partagentcertains gènes, l’information IPA est en partie redondante faisant référence à des processus communs.Nous avons donc regroupé les voies qui correspondent à des fonctions biologiques similaires ou quiparticipent à une même fonction plus large. Nous avons ainsi pu regrouper les voies identifiées parIPA en 10 groupes (Table 7). Le groupe fibrose, matrice et jonction cellulaire fait directementréférence aux processus de fibrose avec la voie de référence de la fibrose hépatique. La présence dela voie de l’inhibition des métalloprotéinase matricielle confirme son rôle important dans leprocessus de cicatrisation.
La réponse inflammatoire constitue un large groupe avec 11 voies de signalisation. La signalisationWnt, présentant des molécules sur- et sous-exprimées, est observée uniquement sous sa formecanonique (β-caténine dépendante). Les voies des cancers sont également bien représentées.Parmi les autres groupes on retrouve aussi le groupe des dermatan / chondroitin sulfate,développement neuronal, musculaire et squelettique, arthrite, réponse à l’hypoxie et métabolisme duglucose. La réponse à l'hypoxie et le métabolisme du glucose sont connus comme impliqués dans lesprocessus tumoraux. La croissance tumorale nécessite une forte alimentation en éléments nutritifs,glucose et oxygène pour les cellules. La tumeur grossissant rapidement, la ressource en élémentsnécessaires aux cellules tumorales n’est pas toujours assurée correctement ce qui entraine parfois unehypoxie. Afin de pallier à cela, la tumeur est capable de former de nouveaux vaisseaux sanguinsgarantissant ainsi son apport en ressource nutritive.
La diapédèse et la migration des granulocytes et agranulocytes joue un rôle dans l'initiation duprocessus inflammatoire. Elle permet le recrutement sur le site de l’infection des groupes cellulairesnécessaires à limiter l’infection de la plaie. L’IL-1 est également un facteur important permettantcette migration. La putréscine, produit de la dégradation des éléments vivants suite à l’infection, doitêtre dégradée car elle est toxique et dangereuse à forte dose pour les tissus, d’où la présence de savoie de dégradation. L’ensemble de ces voies montre une relation étroite avec les processusimpliqués dans la cicatrisation et la fibrose. Le fait que ces voies soient différentiellement réguléesentre les conditions de notre étude valide notre approche, confortant le fait que nos résultats sont enrapport avec la pathologie étudiée.
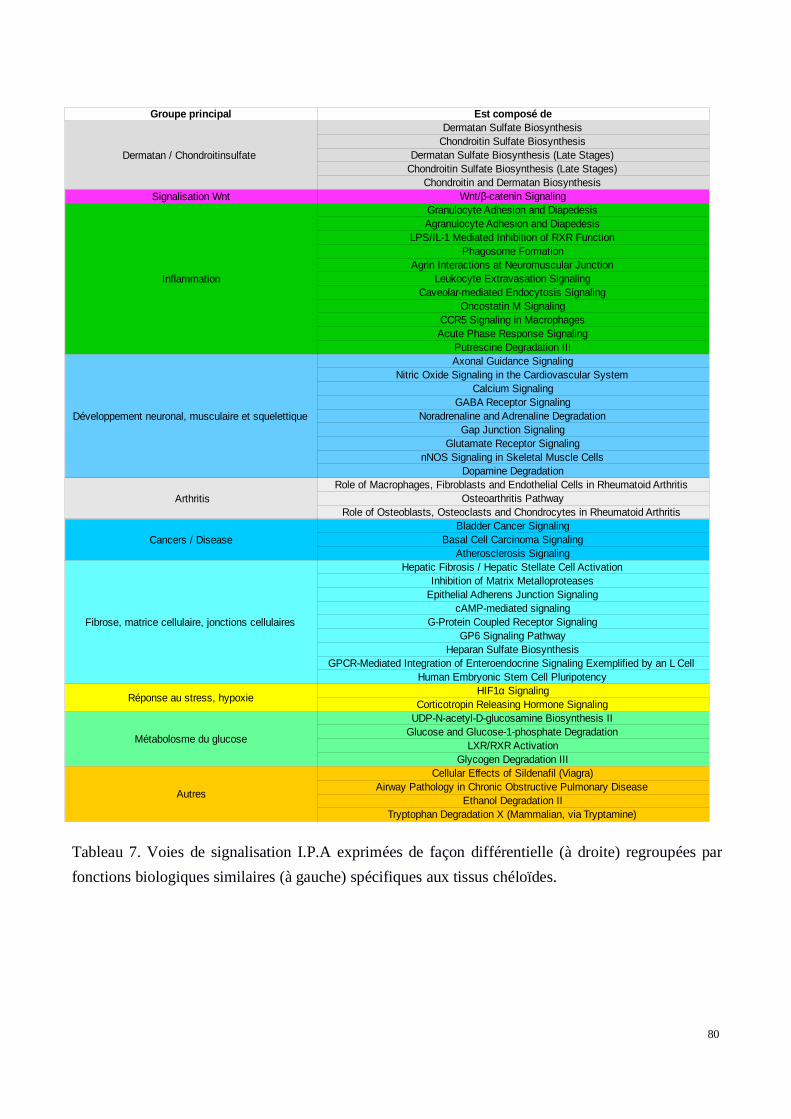
80
Tableau 7. Voies de signalisation I.P.A exprimées de façon différentielle (à droite) regroupées parfonctions biologiques similaires (à gauche) spécifiques aux tissus chéloïdes.
Groupe principal Est composé de
Dermatan / Chondroitinsulfate
Dermatan Sulfate BiosynthesisChondroitin Sulfate Biosynthesis
Dermatan Sulfate Biosynthesis (Late Stages)Chondroitin Sulfate Biosynthesis (Late Stages)
Chondroitin and Dermatan BiosynthesisSignalisation Wnt Wnt/β-catenin Signaling
Inflammation
Granulocyte Adhesion and DiapedesisAgranulocyte Adhesion and Diapedesis
LPS/IL-1 Mediated Inhibition of RXR FunctionPhagosome Formation
Agrin Interactions at Neuromuscular JunctionLeukocyte Extravasation Signaling
Caveolar-mediated Endocytosis SignalingOncostatin M Signaling
CCR5 Signaling in MacrophagesAcute Phase Response Signaling
Putrescine Degradation III
Développement neuronal, musculaire et squelettique
Axonal Guidance SignalingNitric Oxide Signaling in the Cardiovascular System
Calcium SignalingGABA Receptor Signaling
Noradrenaline and Adrenaline DegradationGap Junction Signaling
Glutamate Receptor SignalingnNOS Signaling in Skeletal Muscle Cells
Dopamine Degradation
ArthritisRole of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis
Osteoarthritis PathwayRole of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis
Cancers / DiseaseBladder Cancer Signaling
Basal Cell Carcinoma SignalingAtherosclerosis Signaling
Fibrose, matrice cellulaire, jonctions cellulaires
Hepatic Fibrosis / Hepatic Stellate Cell ActivationInhibition of Matrix Metalloproteases
Epithelial Adherens Junction SignalingcAMP-mediated signaling
G-Protein Coupled Receptor SignalingGP6 Signaling Pathway
Heparan Sulfate BiosynthesisGPCR-Mediated Integration of Enteroendocrine Signaling Exemplified by an L Cell
Human Embryonic Stem Cell Pluripotency
Réponse au stress, hypoxieHIF1α Signaling
Corticotropin Releasing Hormone Signaling
Métabolosme du glucose
UDP-N-acetyl-D-glucosamine Biosynthesis IIGlucose and Glucose-1-phosphate Degradation
LXR/RXR ActivationGlycogen Degradation III
Autres
Cellular Effects of Sildenafil (Viagra)Airway Pathology in Chronic Obstructive Pulmonary Disease
Ethanol Degradation IITryptophan Degradation X (Mammalian, via Tryptamine)

81
c) Analyse des voies métaboliques exprimées de manière différentes entre les tissushypertrophiques et les tissus sains
La comparaison des transcriptomes des tissus de peaux saines et des tissus des cicatriceshypertrophiques révèle 1546 gènes exprimés différemment en prenant en considération desvariations d'expression égales ou supérieures à deux avec un taux de faux positif (Q-value) de 5 %.
Vingt-trois voies de signalisation significatives (p < 10-2) sont mises en évidence dans l’analysed’enrichissement IPA (Table 8). Les voies de signalisation ont été regroupées par similarité commeprécédemment. Ces résultats ne montrent pas de grandes différences avec les voies observées dans leparagraphe précédent. Cette nouvelle comparaison semble moins présenter de voies que pour lesanalyses précédentes, principalement du fait que le nombre de gènes retrouvé est moins important. Ilsemble donc que les processus impliqués dans la cicatrisation hypertrophique soient similaires maismoins prononcés que lors de la cicatrisation des chéloïdes.
Les deux analyses précédentes montrent que la totalité des voies de signalisation activées dans lestissus hypertrophiques sont également activées dans les tissus chéloïdes. Il semble que peu ou pas devoies de signalisation soient sélectivement activées ou inhibées dans les tissus chéloïdes.

82
Tableau 8. Voies de signalisation I.P.A exprimées de façon différentielle (à droite) regroupées parfonctions biologiques similaires (à gauche) spécifiques aux tissus hypertrophiques.
Groupe principalSignalisation Wnt Wnt/β-catenin Signaling
InflammationGranulocyte Adhesion and DiapedesisAgranulocyte Adhesion and Diapedesis
Putrescine Degradation III
Développement neuronal, musculaire et squelettique
Axonal Guidance SignalingCardiac β-adrenergic Signaling
Nitric Oxide Signaling in the Cardiovascular SystemDopamine Degradation
Arthritis Osteoarthritis PathwayRole of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis
Cancers / DiseaseBladder Cancer Signaling
Inhibition of Angiogenesis by TSP1
Fibrose, matrice cellulaire, jonctions cellulaires
Hepatic Fibrosis / Hepatic Stellate Cell ActivationInhibition of Matrix Metalloproteases
Relaxin SignalingcAMP-mediated signaling
G-Protein Coupled Receptor SignalingGP6 Signaling Pathway
GPCR-Mediated Integration of Enteroendocrine Signaling Exemplified by an L CellHuman Embryonic Stem Cell Pluripotency
Réponse au stress, hypoxie HIF1α Signaling
AutresCellular Effects of Sildenafil (Viagra)
Tryptophan Degradation X (Mammalian, via Tryptamine)

83
d) Gènes et voies différemment exprimées entre tissus hypertrophiques et tissus chéloides.
Un des objectifs de ce travail est d’identifier les voies métaboliques qui sont anormalement activéesou réprimées dans les tissus de cicatrices chéloides, la comparaison des tissus chéloïdes avec descicatrices normales pourraient donc aussi apporter des informations très utiles. Toutefois il estdifficile d’obtenir des biopsies pour ces cicatrices car l’état de ces dernières ne nécessite pas uneexcision chirurgicale. De plus, les cicatrices normales (n=4) que nous avons pu étudier étaientbiologiquement très peu actives et ne donnaient probablement qu’une image inexacte des voiesactivées lors du processus de cicatrisation normale. Dans ce contexte, l’analyse comparative destranscriptomes des tissus chéloïdes et des tissus hypertrophiques a un grand intérêt car elle révèle lesvoies dont l’activation / inhibition est associée aux cicatrices chéloides.
Cette analyse montre que 3822 gènes sont exprimés différemment entre cicatrices chéloïdes ethypertrophiques. Les gènes sur-exprimés dans les tissus chéloïdes par rapport aux hypertrophiquessont plus nombreux que les gènes sous exprimés. Ces gènes ne semblent pas indiquer des voies designalisation nouvelles par rapport aux résultats obtenus avec les biopsies chéloïdes, mais plutôtrefléter une activation plus grande de voies déjà activées dans les tissus HYP, dans les tissuschéloïdes. Cette observation est cohérente avec les observations faites précédemment.
Les cicatrices chéloïdes sont toutefois associées avec l’activation des voies Wnt canonique et noncanoniques (Table 9). Alors que les comparaisons des tissus chéloïdes avec les peaux normales ontfait ressortir la voie Wnt canonique β-caténine uniquement, les voies non canonicales Wnt: « PCPpathway » (-log(P) = 4,05) et “Wnt/Ca+ pathway” (-log(P) = 2,03) soulignent un dérèglementimportant de la signalisation Wnt dans les cicatrices chéloïdes. Ces voies non canonicales pourraientdonc contribuer à l’aggravation du processus de cicatrisation.
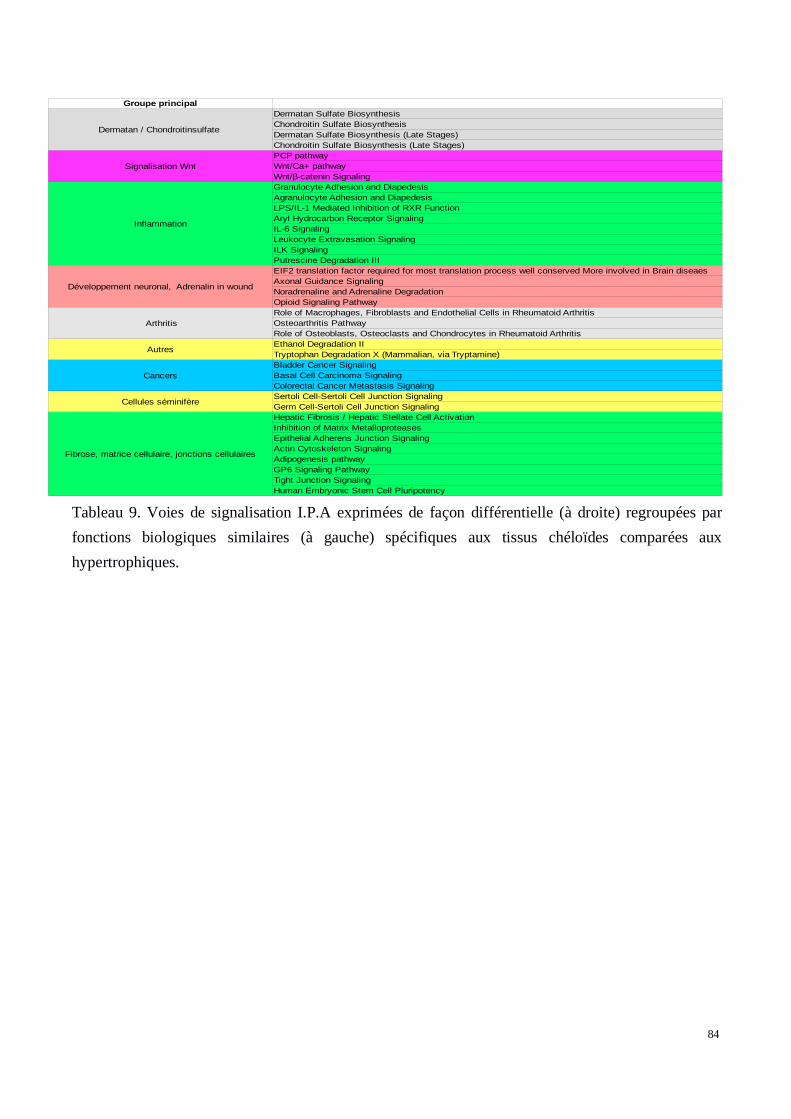
84
Tableau 9. Voies de signalisation I.P.A exprimées de façon différentielle (à droite) regroupées parfonctions biologiques similaires (à gauche) spécifiques aux tissus chéloïdes comparées auxhypertrophiques.
Groupe principal
Dermatan / Chondroitinsulfate
Dermatan Sulfate BiosynthesisChondroitin Sulfate BiosynthesisDermatan Sulfate Biosynthesis (Late Stages)Chondroitin Sulfate Biosynthesis (Late Stages)
Signalisation WntPCP pathwayWnt/Ca+ pathwayWnt/β-catenin Signaling
Inflammation
Granulocyte Adhesion and DiapedesisAgranulocyte Adhesion and DiapedesisLPS/IL-1 Mediated Inhibition of RXR FunctionAryl Hydrocarbon Receptor SignalingIL-6 SignalingLeukocyte Extravasation SignalingILK SignalingPutrescine Degradation III
Développement neuronal, Adrenalin in wound
EIF2 translation factor required for most translation process well conserved More involved in Brain diseaesAxonal Guidance SignalingNoradrenaline and Adrenaline DegradationOpioid Signaling Pathway
ArthritisRole of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid ArthritisOsteoarthritis PathwayRole of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis
AutresEthanol Degradation IITryptophan Degradation X (Mammalian, via Tryptamine)
CancersBladder Cancer SignalingBasal Cell Carcinoma SignalingColorectal Cancer Metastasis Signaling
Cellules séminifèreSertoli Cell-Sertoli Cell Junction SignalingGerm Cell-Sertoli Cell Junction Signaling
Fibrose, matrice cellulaire, jonctions cellulaires
Hepatic Fibrosis / Hepatic Stellate Cell ActivationInhibition of Matrix MetalloproteasesEpithelial Adherens Junction SignalingActin Cytoskeleton SignalingAdipogenesis pathwayGP6 Signaling PathwayTight Junction SignalingHuman Embryonic Stem Cell Pluripotency

85
e) Comparaison des transcriptomes de tissus chéloides provenant du thorax ou des oreilles
L’agrégation hiérarchique des patients chéloïdes entre eux sur l’arbre des distances suggère qu’ilexiste bien une grande similarité de profil d’expression entre toutes les cicatrices chéloides.Cependant, au sein de ces chéloïdes deux sous-groupes semblent se différencier: le groupe descicatrices chéloïdes localisées au thorax (KelT) et le groupe des cicatrices cheloïdes loalisées auxoreilles (KelO).
Cette observation est cohérente avec le fait que les chéloïdes qui se forment aux oreilles se présententsous forme de nodules qui sont localisés au lobe de l'oreille et sont peu douloureuses. Les chéloïdesqui se forment sur le thorax se présentent souvent sous forme de gros cordons fibreux, qui peuvents’étendre à tout le thorax et être très douloureuses.
L'analyse d’enrichissement génomique des voies de signalisation obtenues montre que les voiesassociées aux KelT et KetO par comparaison aux peaux saines sont similaires. Les 5 voies qui sontles plus significativement associées aux listes de gènes sont les mêmes.
Toutefois, les voies Wnt/βcatenin, VEGF et IL8 ressortent dans l’analyse des chéloïdes au thoraxalors qu’elles ne ressortent pas dans l’analyse comparant les KelO avec les peaux saines. Ces mêmesKelO semblent plus fortement associées à des voies de signalisation du système nerveux comme lesvoies “Agrin interactions at neuromuscular junction”, “Calcium signaling” et “GABA receptorSignaling”.
Considérons maintenant les comparaisons KelT et KelO avec les cicatrices hypertrophiques : lesvoies de signalisation impliquées dans le développement des cicatrices KelO et KelT sontsensiblement différentes. En particulier, les KelT sont associées aux voies de signalisation PCPpathway (Wnt Ca+ non canonique), TGF-β, IL-6, BMP signaling, WNT/Ca2+ pathway (Voie Wntnon canonique). Ces voies ne sont donc pas associées avec les KelO. A l’inverse, la voie LPS/IL-1,la voie des jonctions des cellules se Sertoli ou encore la voie de sugnalisation des opioïdes, semblentêtre retrouvées uniquement chez les KelO. Ainsi, les chéloïdes aux oreilles et au thorax semblentmettre en jeu nombre de voies communes mais les KelT montrent une activation spécifique des voiesde signalisation Wnt canonique et non canoniques. La voie Wnt/βcatenine est également activéedans les cicatrices hypertrophiques mais les voies Wnt non canoniques ne le sont pas. Les voiesBMP et TGF-β participent probablement à l’activation des voies Wnt. Les voies VEGF et IL8 quidifférencient les profils de transcription KelT / Peaux saines et KelO / Peaux saines n’apparaissentpas quand l’analyse est faite par rapport aux cicatrices hypertrophiques. Elles ne sont doncprobablement pas sélectivement associées aux KelT. Cette analyse souligne donc une associationsélective de deux voies Wnt non canoniques (que nous regroupons avec les voies de signalisationTGF-β et BMP) et de la voie de l’IL-6 avec les tissus chéloïdiens du thorax représentant les formesles plus graves de cicatrisation étudiées dans notre étude.

86
Nous avons ensuite comparé les listes de gènes KelO et KelT avec les cicatrices HYP. Nous faisonsainsi l'hypothèse qu’en réalisant l’intersection entre ces deux listes de gènes, nous faisons ressortiruniquement les gènes qui sont spécifiques aux processus communs entre KelO et KelT.
Une liste de 2208 gènes en commun est obtenue et une analyse d'enrichissement sur les pathologiespouvant être reliées à ces gènes a été réalisée. Les 50 premières pathologies significatives ont étéclassées en 4 grandes catégories : cancers (25 pathologies), connexions cellulaires (12 pathologies),mouvement cellulaire (6 pathologies), troubles squelettiques et musculaires (5 pathologies) (table 10).Deux pathologies n'ont pas pu être classées dans ces groupes, elles sont trop généralistes. Unesous-catégorie dans les cancers se démarque, ce sont les cancers gastro intestinaux, “G.I. Tracts”. Ilsreprésentent 17 des 25 pathologies de cancers retrouvés et sont indiqués à part sur la table 7.
Le sous-groupe des G.I. tracts présente le plus d’homogénéité avec 519 gènes commun aux 17pathologies de cancer intestinaux reconnus par le logiciel I.P.A. Les catégories des pathologies dutrouble squelettique et musculaire et des connexions tissulaires sont celles qui partagent le moins degènes en commun avec respectivement 10 et aucun gène en commun pour les connexions cellulaires.
Les gènes communs aux mécanismes des KelO autant que KelT montrent clairement l'associationaux cancers. Bien que les chéloïdes ne soient pas toujours considérées comme des tumeurs du faitqu'elles soient bénignes, elles partagent les mêmes mécanismes cancéreux que ceux retrouvés pourdes tumeurs plus graves. L'association au mouvement cellulaire et à la connexion tissulaire faitréférence au processus de cicatrisation qui nécessite le recrutement de nombreux types cellulairescomme des cellules du système immunitaire pour limiter l’infection et les fibroblastes pour lareconstruction de la peau. Enfin, l'association aux troubles squelettiques et musculaires présentemoins de relation avec la cicatrisation ou la fibrose. Cependant, les voies de signalisation montrentun fort intérêt pour le développement neuronal, système nerveux et musculaire. Cela pourraitsignifier une nouvelle approche pour l'analyse des chéloïdes.
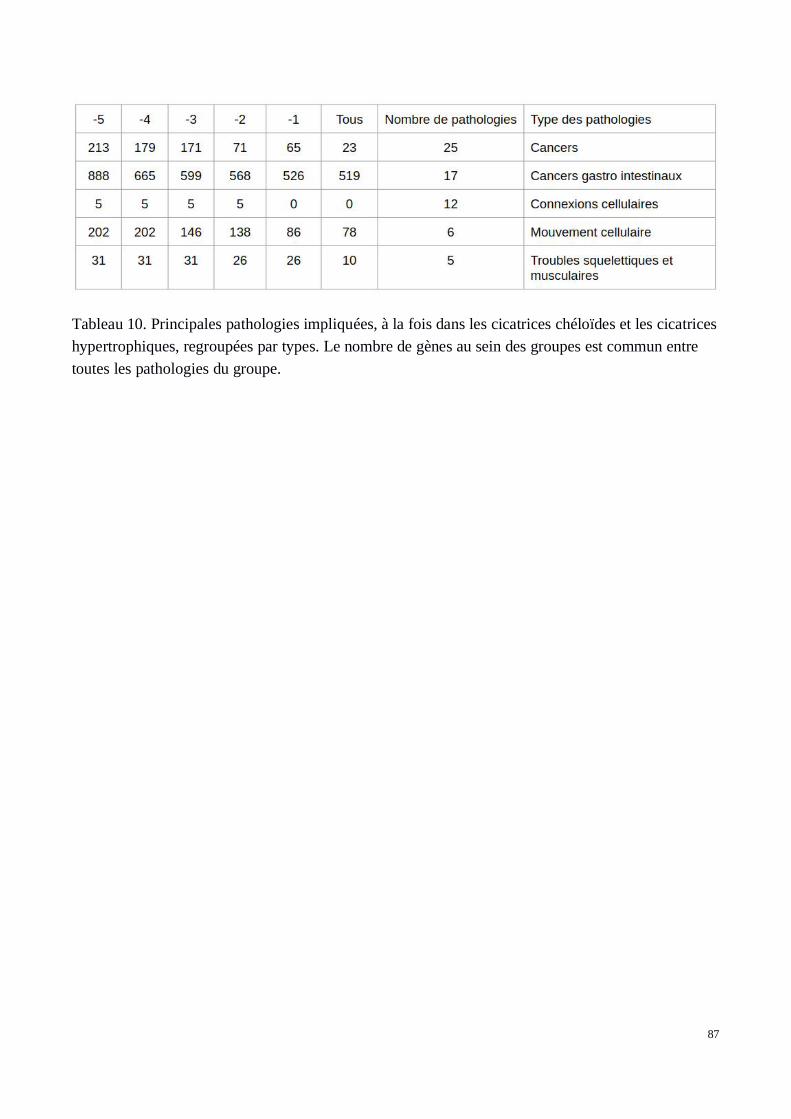
87
Tableau 10. Principales pathologies impliquées, à la fois dans les cicatrices chéloïdes et les cicatriceshypertrophiques, regroupées par types. Le nombre de gènes au sein des groupes est commun entretoutes les pathologies du groupe.

88
2) Analyse d’expression et d’association génétique de la voie Wnt/βCatenine dans la fibrose
chéloïde dans une cohorte africaine
Afin de confirmer l’hypothèse de l’implication de la voie Wnt canonique retrouvée dans les résultats
d’expression, une analyse génétique de la voie de la Wnt/β-caténine a été réalisée dans des
cohortes africaines d’Ouganda. Bien que nous ayons les ADNs des individus de la cohorte chéloïde
française, nous faisons face à un manque d’homogénéité entre les ethnies des patients pour cette
cohorte. En effet, les témoins recrutés pour cette cohorte sont des donneurs de sang anonymes
provenant des établissements français du sang. Pour ceux-ci, nous n’avons aucune informations
permettant de connaitre leur origine ethnique, ni même confirmant l’absence totale de chéloïdes. De
plus, les patients français sujets aux chéloïdes sont pour la plupart originaires de pays d’Afrique,
d’Amérique du Sud ou d’Asie. Nous parlons de la cohorte des cicatrices chéloïdes françaises mais
nous ne pouvons pas affirmer que les patients soient d’origine française unie. Nous avons donc
souhaité effectuer nos analyses génétiques sur une population Ougandaise génétiquement homogène
par méthode TaqMan. Cette cohorte a été recrutée en Ouganda dans la population Allur. La
prévalence des cicatrices chéloïdes dans les populations africaines étant plus élevée qu’en Europe.
L’ensemble des SNPs de 39 des gènes régulateurs (Table 11), effecteurs, stimulateurs et
transducteurs de signal des voies Wnt ont ainsi été listés, le calcul des déséquilibres de liaison a été
réalisé et les SNPs sélectionnés en conséquence. Les génotypes pour ces SNPs ont été extraits des
données du GWAS pour une analyse ciblée. La table 11 montre dans la colonne de droite le nombre
de marqueurs pour chacun des gènes qui sont associés à une fibrose hépatique sévère sur la
population brésilienne du GWAS avec une P-value inférieure à 10-2. Sur ces SNPs Un premier test
d'association uni-varié a d'abord été réalisé sur une cohorte Ougandaise en utilisant une régression
logistique ; il a montré que les tags SNPs de 5 gènes différents présentent une association avec le
phénotype chéloïdes LEF1 (p=0.004), SFRP1 (p=0,03), SFRP4 (p=0,02), TGFBR1 (p=0,002) et
LRP5 (p=0,03). Un SNP sur le gène AXIN1 présentait également une suggestion d’association :
(p=0,18) (Table 12).
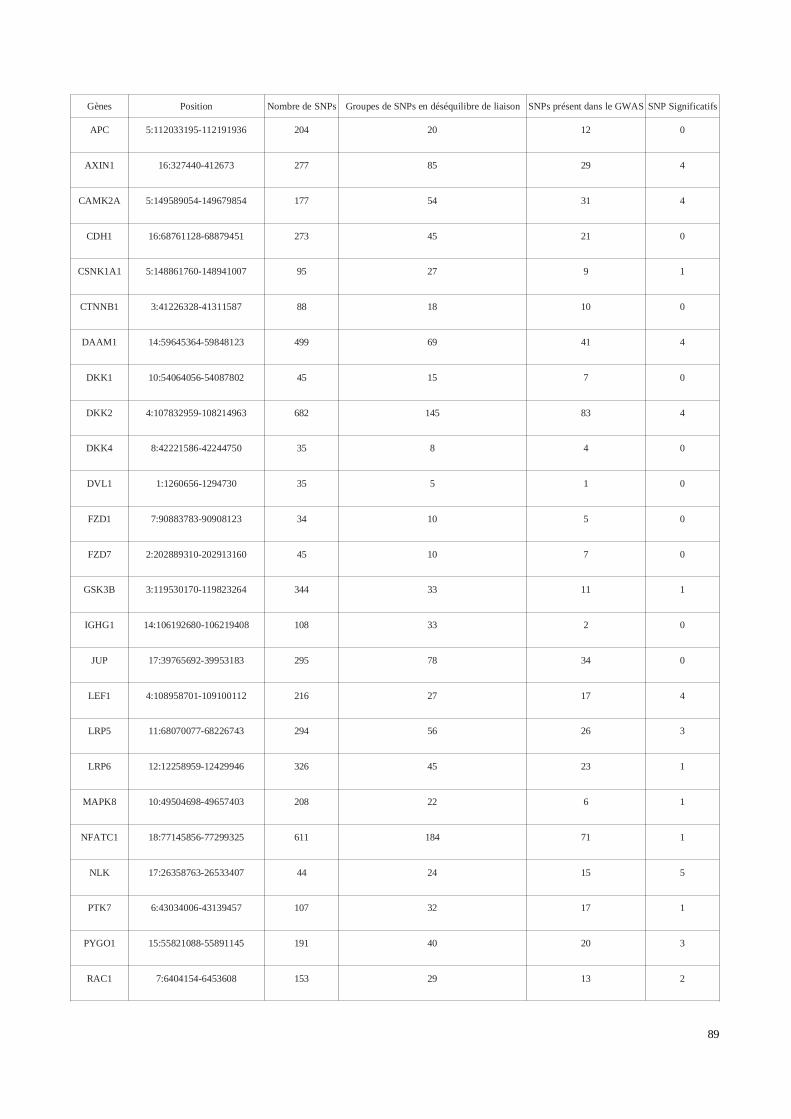
89
Gènes Position Nombre de SNPs Groupes de SNPs en déséquilibre de liaison SNPs présent dans le GWAS SNP Significatifs
APC 5:112033195-112191936 204 20 12 0
AXIN1 16:327440-412673 277 85 29 4
CAMK2A 5:149589054-149679854 177 54 31 4
CDH1 16:68761128-68879451 273 45 21 0
CSNK1A1 5:148861760-148941007 95 27 9 1
CTNNB1 3:41226328-41311587 88 18 10 0
DAAM1 14:59645364-59848123 499 69 41 4
DKK1 10:54064056-54087802 45 15 7 0
DKK2 4:107832959-108214963 682 145 83 4
DKK4 8:42221586-42244750 35 8 4 0
DVL1 1:1260656-1294730 35 5 1 0
FZD1 7:90883783-90908123 34 10 5 0
FZD7 2:202889310-202913160 45 10 7 0
GSK3B 3:119530170-119823264 344 33 11 1
IGHG1 14:106192680-106219408 108 33 2 0
JUP 17:39765692-39953183 295 78 34 0
LEF1 4:108958701-109100112 216 27 17 4
LRP5 11:68070077-68226743 294 56 26 3
LRP6 12:12258959-12429946 326 45 23 1
MAPK8 10:49504698-49657403 208 22 6 1
NFATC1 18:77145856-77299325 611 184 71 1
NLK 17:26358763-26533407 44 24 15 5
PTK7 6:43034006-43139457 107 32 17 1
PYGO1 15:55821088-55891145 191 40 20 3
RAC1 7:6404154-6453608 153 29 13 2
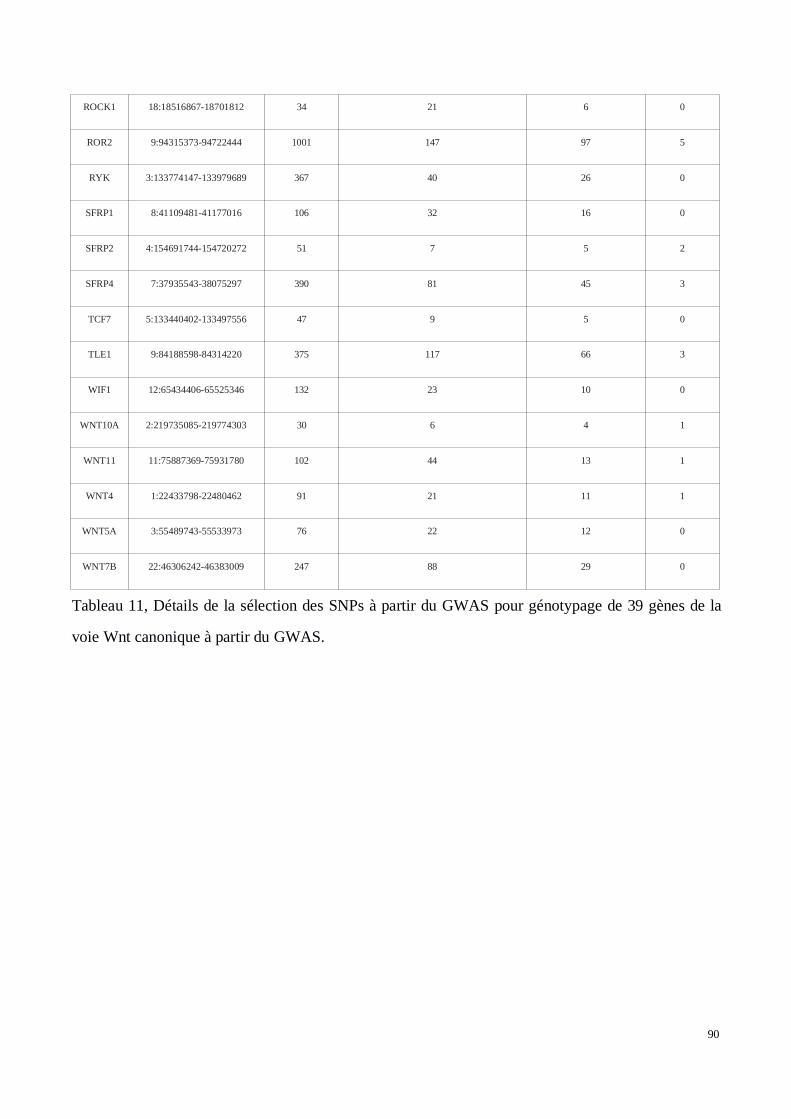
90
ROCK1 18:18516867-18701812 34 21 6 0
ROR2 9:94315373-94722444 1001 147 97 5
RYK 3:133774147-133979689 367 40 26 0
SFRP1 8:41109481-41177016 106 32 16 0
SFRP2 4:154691744-154720272 51 7 5 2
SFRP4 7:37935543-38075297 390 81 45 3
TCF7 5:133440402-133497556 47 9 5 0
TLE1 9:84188598-84314220 375 117 66 3
WIF1 12:65434406-65525346 132 23 10 0
WNT10A 2:219735085-219774303 30 6 4 1
WNT11 11:75887369-75931780 102 44 13 1
WNT4 1:22433798-22480462 91 21 11 1
WNT5A 3:55489743-55533973 76 22 12 0
WNT7B 22:46306242-46383009 247 88 29 0
Tableau 11, Détails de la sélection des SNPs à partir du GWAS pour génotypage de 39 gènes de la
voie Wnt canonique à partir du GWAS.

91
Un modèle génétique a été constitué à partir de ces marqueurs à l’aide d’un test d’association
multi-varié et les risques relatifs ont été calculés pour chacun des marqueurs (Table 13). Les
nouvelles Pp-values obtenues montrent que le gène de l’AXIN1 présente désormais une association
plus que suggestive. C’est le variant du gène SFRP1 qui présente le risque relatif le plus fort avec un
intervalle de confiance à ce risque relatif assez large ,ce qui indique que le risque relatif calculé pour
ce variant est peu précis. Les génotypes aggravants retrouvés sont cohérents avec ceux obtenus dans
l’analyse multi-variée. Il est intéressant de noter que l'analyse du transcriptome dans le chapitre
précédent, réalisée indépendamment de l'analyse génétique, a montré que LEF1 et TGFβR1 étaient
régulés dans les cicatrices chéloïdes par rapport aux peaux normales.
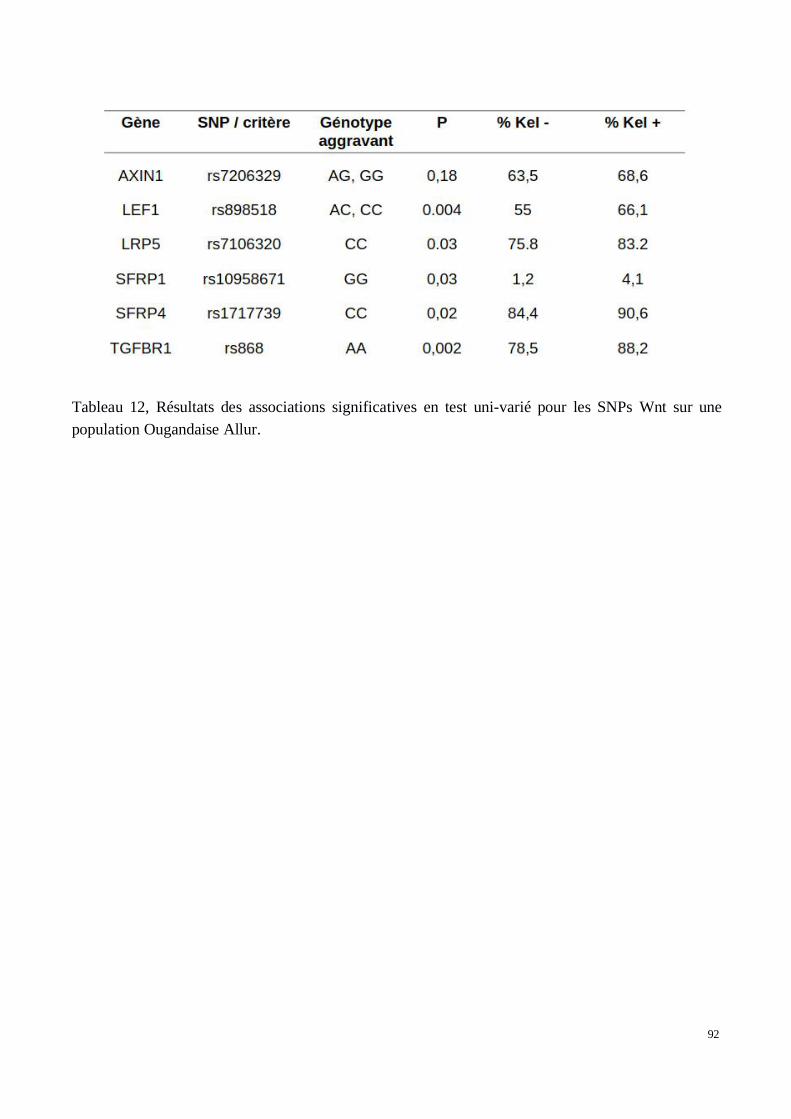
92
Tableau 12, Résultats des associations significatives en test uni-varié pour les SNPs Wnt sur unepopulation Ougandaise Allur.
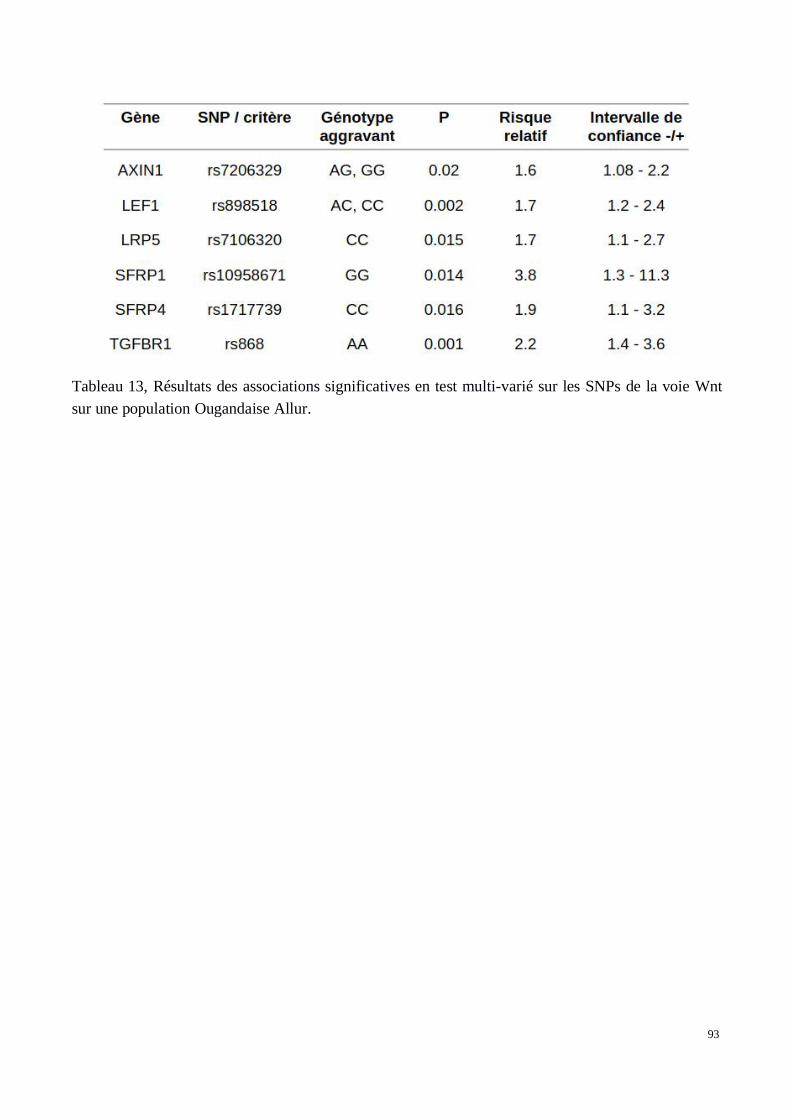
93
Tableau 13, Résultats des associations significatives en test multi-varié sur les SNPs de la voie Wntsur une population Ougandaise Allur.

94
Discussion sur l’analyse génétique des fibroses du foie bilharzienne par méthode GWAS.
Avant d’analyser les données du GWAS, la puissance statistique et le risque relatif associé auxvariants obtenus ont été estimés. Les principaux facteurs qui peuvent impacter les résultats desanalyses d’association sont : la prévalence du trait, la fréquence allélique (MAF) et le risque associéà chaque allèle. La taille et la composition de la cohorte sont déterminées en fonction de cesparamètres pour optimiser la puissance statistique de l’analyse. Cette puissance se mesure par lecoefficient β qui, dans les conditions optimales, doit être supérieur ou égal à 80%. Quand β vaut 0,8cela signifie que 8 cohortes sur 10 répliquent les associations observées. Ainsi, si une cohorte nepermet pas une puissance de 0,8 et que les associations observées ne sont pas répliquées, cela nesignifie pas pour autant que cette association n’existe pas. Comme la taille de la cohorte est souventlimitée par les conditions du recrutement, il est parfois nécessaire de poser des limites à la fréquenceallélique et au risque associé. Dans notre cas, la MAF est fixée supérieure ou égale à 5% et le risquerelatif supérieur à 1,8. La figure 24 montre l’évolution du nombre de cas nécessaire en fonction de lapuissance statistique de l’étude. La MAF, que nous avons fait varier de 1% à 20 %, ainsi que lerisque relatif, que nous avons fait varier de 1,5 à 2, sont également deux facteurs qui jouent sur lapuissance de l’étude. Pour la taille de notre cohorte, un risque relatif de 1,8 et une MAF de 5%, βvaudra 57%. β atteindrait les 80% pour un nombre de cas égal à 900. La figure 25 montre que, avecle même nombre de cas et 4 fois plus de contrôles, la puissance statistique obtenue est de 80%.L’article de Eun Pyo Hong et collaborateurs, (136) estiment que pour un risque relatif détecté à 2 etune MAF à 5%, détecter des associations significatives en testant 500 000 SNP nécessite au moins1206 cas et autant de contrôles tandis que pour 1 million de SNP, 1255 cas et autant de contrôles sontnécessaires. Ceci montre que le modèle allélique dominant requiert une taille de cohorte plus faibleque le modèle récessif pour détecter des associations à puissance statistique égale. Il est possibleégalement de sélectionner une MAF supérieure à 5% quand on travaille sur un trait fréquent commecelui de cette étude. Enfin ces auteurs montrent que l’augmentation du rapport cas / contrôlesau-dessus de 4 n’a pas d’effet net sur la puissance statistique du test.
La discussion ci-dessus s’applique à un test sur quelques SNPs avec une valeur seuil de α = 0,05. Siles tests sont nombreux, la possibilité de détecter de fausses associations augmente. Pour éviter ceserreurs dites de type 2, il faut réduire le seuil de significativité et différentes méthodes ont étéproposées. La plus simple étant celle de Bonférroni qui consiste à diviser Alpha par le nombre de testindépendant réalisé. Dans notre étude, nous avons testé 1 816 152 variants. Si ces variants étaienttous indépendants, alors la P-value corrigée serait située autour de 10-8. Cependant, les tests effectuésne sont pas indépendants car certains SNPs sont en déséquilibre de liaison, ce qui permet de minorerla correction de Bonferroni. Deux SNPs sont indépendants s’ils n’ont pas de déséquilibre de liaisonsupérieur à 0,6. Les résultats obtenus à partir de la cohorte de découverte n’ont cependant pas permisde détecter des associations présentant des P-values inférieures à 10-7.
95
Figure 24, Puissance statistique en fonction de la taille de la cohorte et du rapport entre le nombre decas et de contrôles.
Figure 25, Taille de la cohorte en fonction du risque relatif attendu en statistique selon différentsrapports entre le nombre de cas et de contrôles.

96
Randall C Johnson et collaborateurs (137) indiquent que la correction de Bonférroni n’est pastoujours le bon choix et qu’il existe d’autres méthodes de correction. Par exemple à l’aide d’uneACP ou encore de permutations.
La régression logistique binaire utilise le principe de la régression linéaire réalisée dans ce cas surdes données binaires. Meurer et son équipe (138) expliquent que cette technique permet de prédire lerisque de maladie en se basant sur des variables prédictives qui leur sont propres. En génétique, cesont les génotypes. La régression modélise la relation entre les variables prédictives et le devenir dupatient face au développement d’une pathologie future. Le modèle attribue un risque relatif quipondère la présence ou l’absence des variables explicatives, d’où le caractère binaire. Le modèle peutintégrer des variables binaires et des variables continues comme l’âge des sujets.
Les coefficients du modèle de régression permettent le calcul de l’odd ratio (OR) qui permet uneapproximation du risque relatif (RR), c’est-à-dire du risque associé à la variable en prenant encompte les effets des autres variables.
R1 ( 1 - R1 ) 1 - R0
OR = ------------------ RR = -------------
R0 ( 1 - R0 ) 1 - R1
R1 correspond à la fréquence du trait chez les porteurs de l’allèle à risque testé et R0 pour l’autreallèle. Si la pathologie est rare, alors R0 et R1 sont faibles et on peut dire que OR = RR. L’OR estcalculable quand la prévalence de la maladie n’est pas respectée, c’est à dire dans les enquêtes castémoins où le quota de malade / non malade est connu d’avance, contrairement aux cohortesconstituées d’un échantillon représentatif de la population générale.
Il existe au moins trois méthodes pour construire le modèle : la méthode “saturée” ou au contraire “àvide”. Dans la méthode saturée, toutes les variables sont intégrées au modèle dès le départ et sontensuite retirées une par une pour arriver à un modèle optimal. A l’inverse, dans la méthode “à vide”,les variables sont ajoutées les unes après les autres et celles qui perdent leur significativité au coursdes ajouts sont retirées. Ces méthodes présentent chacune des désavantages. La méthode “à vide”présuppose un ordre dans lequel les variables sont ajoutées au modèle. La dépendance de cet ordred’entrée induit un biais étant donné que des variables qui auraient été pertinentes à conserver dans lemodèle final peuvent être éliminées dès le départ par manque de vue d’ensemble. La méthode saturéeinclut trop de variables au modèle dès le départ. La puissance statistique à détecter les associationsfortes est donc réduite. La principale difficulté rencontrée par les modèles de régression logistiquequand beaucoup de variables sont testées est le risque de sur-apprentissage. Une variable peutapparaitre explicative sans avoir une réelle relation d’effet avec la pathologie. Ces variables ne sontpas confirmées dans des tests ultérieurs sur d’autres cohortes. Le sur-apprentissage peut être évité entravaillant avec des cohortes dont la taille est 10 fois le nombre de variables testés (139). Quand celan’est pas le cas, il est mieux d’effectuer une pré-sélection des variables afin de n‘intégrer au modèleque les variables utiles au modèle prédictif.

97
Ces méthodes s’apparentent aux nouvelles méthodes de deep learning ou encore machine learning.Ces analyses deviennent puissantes dès lors qu’elles sont réalisées à grande échelle. Wenfa Li et sonéquipe (140) ont développé une solution mathématique disponible sur de grandes puissances decalculs réparties sur plusieurs machines (Clusters de calculs) associées à un protocole de sécuritécryptographique développé sur mesure garantissant la protection des données génétiques traitées.Cette solution permet d’appliquer des régressions logistiques sur de larges cohortes interconnectantles bases de données des différents grands consortiums. Ce travail confirme l’intérêt croissant de cesanalyses pour la communauté scientifique et préfigure ce qui doit être la recherche d’aujourd’hui.
D’autres modèles existent comme le modèle linéaire mixte qui a également été utilisé sur les données.Le modèle mixte dérive des modèles linéaires plus simples et reste dédié à l’analyse des donnéesbinaires. Tout comme la régression logistique, il est capable de prendre en compte des variables àeffet fixe comme les SNPs. Il prend en compte également la notion de facteurs aléatoires, notion plusrécente en statistique. Il permet l’analyse de mesures répétées, acquisitions non indépendantes, sansinduire de biais. Pour cela, une matrice de co-variables prenant en compte ces facteurs aléatoires estcalculée et prise en compte dans l’analyse.
Carl A. Anderson et collaborateurs (141) expliquent que le contrôle qualité des données degénotypage est nécessaire pour limiter les faux positifs lors de l’analyse. Les méthodestraditionnelles de génotypage bas débit permettent encore un contrôle manuel des données dues aufaible volume des génotypes obtenus. Les nouvelles méthodes de génotypage haut débit, telles quecelles utilisant les puces de GWAS Illumina, permettent de génotyper plus de 2 millions de SNPs.Ces techniques ne permettent plus un contrôle manuel des données. Des biais dont les effets peuventsembler restreints sur des analyses réalisées sur quelques SNPs, doivent être bien mieux maitriséslors de l’analyse des données en haut débit. Dès lors que l’on considère 2 millions de tests, il estnécessaire de minimiser au mieux le taux de faux positif et ainsi maximiser la puissance de l’analyse.Des logiciels permettent de réaliser les étapes du contrôle qualité de manière automatisée.
Cependant, un contrôle automatisé trop restrictif aura pour effet de retirer de l’étude trop de SNPs oude patients, ce qui fera perdre de la puissance statistique à l'analyse. A l'inverse, un contrôle trop peurestrictif, conservera dans les données des SNPs ou patients dont la qualité des résultats n'est pasconvenable. Ces résultats réduiront alors l’information apportée par les SNPs associés à la pathologie,réduisant la puissance de l'analyse. Il est donc nécessaire de conduire un contrôle de qualité juste etprécis. Carl A. Anderson et son équipe suggèrent même qu'il est préférable de retirer un patientplutôt qu'un SNPs, car ce dernier pourrait être prédictif de la pathologie. Il est donc conseillé decommencer en premier le contrôle qualité des patients, ensuite celui des SNPs. Réaliser un premiernettoyage des données permet d’améliorer le contrôle des SNPs, et donc de minimiser le nombred'exclusion.
Le contrôle qualité par patient est composé de 4 filtres principaux : filtrage des individus présentantdes discordances sur l’établissement de leur sexe, filtre sur les individus possédant des génotypesmanquants ou un taux d’hétérozygotie anormal, filtre sur les individus dupliqués ou présentant desliens de parentés.

98
Du fait que les hommes possèdent un chromosome X et Y, ils ne doivent pas présenter de résultatshétérozygotes pour les marqueurs situés dans la zone pseudo autosomale du chromosome X. Il estdonc attendu que les hommes aient un taux d'homozygotie de 1, tandis que ce même taux doit êtreinférieur à 0,2 chez les femmes. Une discordance entre le sexe donné par la génétique et le sexe civildu patient peut indiquer une erreur dans l’échantillon. Retirer les échantillons pour lesquels un douteest présent permet d'augmenter la puissance statistique de l'analyse. Le taux de génotype obtenu ouencore le taux d'hétérozygotie renseignent sur la qualité de l'ADN utilisé pour l'étude. Il est conseilléde retirer les patients dont le taux de génotype manquant dépasse les 3 à 7% en fonction du jeu dedonnées. Un manque de résultat pour un patient indique un ADN de mauvaise qualité et donc desrésultats peu fiables. Le taux d'hétérozygotie, quant à lui, permet de détecter une contaminationpotentielle d'un ADN par un autre ADN ou encore un patient avec une forte consanguinité.
Une des principales nécessités pour l'analyse statistique qui fait suite au contrôle qualité estl'indépendance des individus. Si des individus sont dupliqués ou possèdent des liens de parentés, celaintroduira un biais statistique. Du fait que les génomes sont liés au sein d'une famille, tropd'individus apparentés affectent les fréquences alléliques du jeu de données qui ne constituera alorsplus un échantillonnage représentatif de la population. Pour cela, l'identité par état (IBS, Identity byState) est utilisée. Pour son calcul, elle nécessite un jeu de données dans lequel les SNPs sontindépendants. Le jeu de données sera dit “taillé”. Pour une portion de génome donné, tous les SNPsen déséquilibre de liaison (R² > 0,2) sont retirés pour ne conserver que des SNPs indépendants entreeux. Ce travail est effectué de proche en proche sur tout le génome.
Un des biais confondants qui peut encore exister est une stratification différente des cas et destémoins. La population est stratifiée quand des différences fortuites entre les individus sont présentes,autres que celles définissant les cas et les témoins de l'étude. Les cohortes sélectionnées pour cesétudes doivent être de population homogène. Il subsiste cependant des sous structures cryptiques quiposent problème dès lors qu'elles ne sont pas équi-réparties selon les groupes de l’étude. Un SNPretrouvé associé au trait étudié pourrait alors refléter la présence de ces sous structures au lieu d’uneréelle association avec le trait. Pour estimer l’homogénéité de la population, sa stratification peut êtrereprésentée graphiquement à l’aide des valeurs obtenues par une analyse en composante principale(ACP). La représentation graphique des 2 premières composantes principales, l'une par rapport àl'autre, permet de visualiser la provenance géographique des individus selon les populations deréférence : africains, européens, asiatique etc... La présence d'individus rompant avec l'homogénéitéde la cohorte est ainsi identifiable et ces patients peuvent être retirés pour éliminer ces biaisconfondants.
Dans un second temps, le contrôle qualité des SNPs a été réalisé. Cette étape permet d'éliminer lesSNPs qui présentent des génotypes peu robustes et ainsi éviter la présence de fausses associationslors de l'analyse. Les paramètres des filtres qui sont appliqués aux variants sont : la MAF, le nombrede génotypes manquant globalement et séparément pour les cas et témoins, et enfin l'équilibre deHW.

99
Dans l’analyse, les variants présents sur les chromosomes sexuels X et Y ont été excluspréalablement à ces contrôles. Pour autant, la microarray de génotypage utilisée pour l’analyse inclutdes génotypes sur ces deux chromosomes. Bien que la taille du chromosome X (155 Mb, 1669 gènesconnus) soit similaire à celle du chromosome 7, les variants analysés sur X sont principalementinter-géniques. Par conséquent, on observera moins de mutations non-sens ou faux-sens, et unehérédité non soumise aux mêmes pressions de sélection que pour les autosomes. De plus, lesmicroarrays de génotypage GWAS présentent parfois une qualité de génotypage moindre du fait queces variants sont moins connus, avec des caractéristiques plus irrégulières et des déséquilibres deliaison moins bien caractérisés. Les analyses d’association impliquant des variants sur lechromosome X présentent le plus souvent des P-values élevées, ce qui traduit la présence des biaisprécédents évoqués. Avant même toute analyse, les chromosomes sexuels sont donc exclus dès lespremières étapes du contrôle qualité. Chez la femme (X,X), le phénomène d’inactivation aléatoirecause de manière non prédictible l’inactivation transcriptionnelle de zones génomiques entières surl’un des deux chromosomes X. Ce phénomène rend plus difficile l’interprétation des résultats etquestionne sur la vraie signification des P values observées avec les variants localisés sur ceschromosomes (142). De nouvelles méthodes ont été mises en place pour l’analyse sur ceschromosomes et pourraient être utilisées sur nos données (142).
Au cours du contrôle qualité, les variants ne respectant pas l’équilibre de Hardy Weinberg (HW) sontexclus. Nous pouvons nous demander s’il est vraiment pertinent de filtrer les variants sur ce critère.Quand l’équilibre de HW est vérifié, cela suppose que notre population est dite “idéale”. Elle réponddonc à des critères précis comme la panmixie, une sélection naturelle absente ou encore absence demigration d’individus. Le premier frein à la panmixie serait l’étude d’une pathologie entrainant ledécès d’un nombre de patient non négligeable avant l’âge de la reproduction (19/20 ans). Un biaisimportant serait alors apporté à l’étude qui ne pourrait être réalisée uniquement sur les patients lesplus résistants, ceux dont la survie serait possible au delà de l’age de procréation. Mais cela n’est pasle cas pour la bilharziose. Même chez les patients infectés étant jeunes, la fibrose du foie met desannées à se développer. Bien que les sujets infectés étant enfant meurent souvent au début de l’ageadulte, la bilharziose n’empêche pas la procréation. D’un point de vue fonctionnel et biologique, rienne peut limiter la panmixie au niveau reproduction. Mais pour cela il faudrait aussi montrer que lesmariages dans les communautés s’effectuent au hasard, ce que nous ne pouvons pas prouver. Celarelève des mœurs spécifiques aux ethnies que nous n’avons pas étudiées. Au niveau génétique, uneélimination de gènes liés à une autre pathologie que celle de l’étude pourrait également mettre encause l’équilibre de HW si les gènes touchés sont communs avec notre pathologie. Cela pourraitainsi créer un biais de sélection anormal dans la population influençant les résultats génétiques dansnotre population d’étude.
Ces biais seraient alors spécifiques à notre population d’étude et réduiraient le caractère universel denos analyses. Nous ne pouvons pas répondre à ces interrogations. Il est donc nécessaire de retirer lesvariants pour lesquels l’équilibre de HW n’est pas vérifié.

100
Un écart à l'équilibre de Hardy Weinberg peut signifier soit un problème dans le génotypage : lesgénotypes obtenus sont faux et ne reflètent pas le génome du patient; soit que des pressions externesse sont exercées sur les individus, comme une pression de sélection. Il est évident que les SNPsassociés à la pathologie peuvent présenter un écart à cet équilibre du fait que leur présence/absenceagit directement sur l'état de santé du sujet. L'équilibre de Hardy Weinberg est donc testé uniquementsur les contrôles.
Retirer les SNPs dont la MAF est inférieure à un seuil défini pour l'analyse permet d'augmenter lapuissance de l'analyse comme décrit précédemment.
L’objectif de ce travail était de rechercher de nouveaux marqueurs génétiques des fibroses du foieinduites par la schistosomiase par méthode GWAS. Les résultats ont mis en évidence 215 SNPs quiprésentent une association (p<10-7, n= 17) ou suggestion d’association (p<10-6, n=198) avec lafibrose hépatique grave induite par le schistosome. Aucun variant n’atteignait un seuil designificativité P= 10-8 très probablement dû à un manque de puissance statistique. Nous avonscependant sélectionné des variants en utilisant un seuil P > 10-6. Parmi ces gènes, IL10RA, SMAD4,CERS6 et JADE1 possèdent des propriétés intéressantes au regard de la fibrose.
L’IL10RA est un gène de 15 Kilos bases localisé sur le chromosome 11. Il code pour la protéinealpha du récepteur principal de l’IL-10. Il agit en tant que médiateur dans la synthèse des cytokinespro-inflammatoires qui affectent la réponse des lymphocytes Th1 et Th2. Un manque d’IL-10 induitl’inflammation des tissus et favorise la croissance tumorale (119). La fixation de l’IL-10 à sonrécepteur entraine la phosphorylation de JAK1 (Janus Kinase 1) et TYK2 (Tyrosine Kinase 2),entrainant alors la phosphorylation de STAT3 (Signal Transducer and Activator of Transcription-3).STAT3 agit sur l’ADN au niveau du noyau avec la transcription de facteurs anti-apoptotique etrégulant le cycle cellulaire. Des mutations dans les récepteurs de l’IL-10 pourraient donc aggraverl’inflammation hépatique et la fibrose. L’IL-10 présente 25 % de similarité avec l’IL-22 dans sachaine protéique. L’IL-22 peut aussi se lier au second récepteur de l’IL-10, l’IL-10RB (144, 145). Legroupe du Pr. Dessein a également mis en évidence l’IL-22 comme impliqué dans le devenir de lafibrose et de la cirrhose hépatique en cas d’infection par l’hépatite C et plusieurs espèces de parasiteSchistosoma (146). Nakajima S et son équipe (147) identifient la mutation rs2228055 dans l’IL10RAcomme un marqueur candidat au développement des fibroses hépatiques dans la stéatohépatitenon-alcoolique (NASH). Ils ne concluent cependant pas que cette variation puisse avoir un effetfonctionnel. En revanche, ce SNP a été décrit dans d’autres pathologies comme le troubleneuro-rétinien en cas d’infection par l’hépatite C (148) suggérant une altération de la voie de l’IL-10.
SMAD4 est un gène de 117 Kilos bases situé sur le chromosome 18. Il joue un rôle dans latransduction du signal au niveau cytoplasmique dans la voie du TGF-β / BMP7. Quand le TGF-β estlié à ses récepteurs, le signal est transmis au niveau cellulaire vers le noyau par le complexe SMAD2,3 et 4. SMAD4 est nécessaire à la translocation de ce complexe vers le noyau car il contient uneséquence protéique spécifique à cette fonction.

101
Des mutations dans SMAD4 sont impliquées dans les cancers du colon (149), de la thyroïde (150), dufoie (151), et sont associées à un faible taux de survie pour ces même cancers (152).
CERS6, aussi appelée céramide, est un gène de 319 Kilos bases situé sur le chromosome 2. Il estresponsable de la sécrétion de plusieurs acides gras dont certains ont des implications dans lescancers (153). Il existe 6 molécules CERS numérotées de 1 à 6. La principale fonction de CERS6 estde synthétiser les céramides C14 et C16. La structure de base des céramides chez les mammifères estconstituée d’un acide gras lié par liaison amide à des sous types de sphingoïde comme : ledihydrosphingosine, la sphingosine, le 6-hydroxy sphingosine et le phytosphingosine. CERS6 estcapable de stimuler la production du TNF, impliqué dans l’inflammation et la nécrose tumorale. Laproduction de P38, des Mitogen-activated protein kinases (MAPK), est également stimulée parCERS6, impliqué dans la différenciation cellulaire et l’apoptose (154). Yih-Huei Uen et son équipeont montré que CERS6 est impliqué dans les cancers gastriques. Sa sur-expression est associée avecdes paramètres cliniques indiquant un faible taux de survie du patient (155). Il agirait sur la voie designalisation STAT3 qui peut également être stimulée par certaines interleukines comme l’IL-6. Lavoie STAT3 est impliquée dans de nombreux cancers et régule la prolifération cellulaire (156). Dansle cancer gastrique, l’expression de CERS6 est également corrélée à celle de MMP2 et MMP9,impliqués dans la dégradation de la matrice extracellulaire et les métastases.
JADE1, gène de 65 mégas bases situé sur le chromosome 4, fait partie d’une famille de 3molécules à doigts de zinc nommées de 1 à 3. JADE1 est impliqué dans la régulation du cyclecellulaire des épithéliums (157). La fonction principale de JADE1 est l’acétylation des histones H4.Corrélé à la gestion de la chromatine, son rôle dans la prolifération cellulaire est plus que suggéré(158). JADE1 se lie à HBO1 faisant partie du complexe multi protéique de l’acetyl transférase,responsable de l’acétylation (159). Cependant, le mécanisme par lequel cette liaison contrôle leshistones est encore mal connu. JADE1, tel un catalyseur, augmenterait l’affinité de HBO1 avec sonsubstrat sur les histones H3 et H4. Sa capacité à acétyler les histones est donc augmentée (160). Sonniveau d’expression est faible dans les cellules cancéreuses rénales tandis qu’il est élevé dans lescellules rénales normales (161). Il supprime la croissance cellulaire des cellules cancéreuses enculture en permettant l’apoptose. Il affecterait les caractéristiques des cellules en croissance, altérantl’adhésion cellulaire au collagène de la matrice extra-cellulaire, altérant de ce fait l’organisation destissus. JADE1 régule aussi la voie Wnt en altérant la formation de la β-caténine, indispensable à latransduction du signal Wnt du cytoplasme vers le noyau (162). La transduction du signal Wnt est liéeà la prolifération cellulaire et aux processus de formation des tumeurs. JADE1 favorise ladégradation de la β-caténine par le protéasome et interagit préférentiellement avec la β-caténineen absence de stimulation, celle-ci étant uniquement cytoplasmique quand la voie Wnt n’est passtimulée. JADE1 est donc un inhibiteur de la voie Wnt. La voie Wnt, fortement stimulée parl’inflammation induite par les œufs du schistosome dans le foie, ne pourra pas être réguléenégativement par JADE1.

102
Ces variants ont ensuite été testés dans une nouvelle cohorte. Celle validation est effectuée par laméthode de génotypage TaqMan. Cette méthode présente plusieurs contraintes. Tout d’abord, elle nepermet de tester que deux modalités (allèles) pour un variant. De plus, il n’est pas toujours possiblede synthétiser des sondes pour détecter certains variants. Parmi les 215 variants obtenus à partir desdonnées du GWAS, la technique TaqMan a permis d’en tester seulement 180 pour validation. Eneffet, les contraintes de la technologie TaqMan ne sont pas les mêmes que celles de la puce Illuminadu GWAS. La validation du design des sondes par Thermo Fischer n’a pas toujours été un succès etcertains variants n’ont pas été compatibles avec notre technique de validation. Parmi ceux-ci,certains variants analysés ne correspondent ainsi pas à ceux qui ont été testés dans le GWAS mais ilsont été sélectionnés comme étant en déséquilibre de liaison avec ces premiers. Sur ces 180 SNPs,122 sont polymorphiques, ont un taux de génotypage supérieur à 95 % et sont en équilibre de HW.Le test d’association analysant chaque SNP séparément montre une association significative pour 15variants.
CSMD1 (CUB and sushi multiple domains-1) est un gène de 2 Méga bases contenant 71 exonssur le chromosome 8. Il code pour un ensemble protéique encore peu connu. Il jouerait cependant unrôle majeur dans les cancers, notamment associés aux cancers du sein, de la prostate et du colon. Untrop faible niveau d’expression serait associé à une stimulation accrue de la prolifération, l’invasionet migration cellulaire amenant à une augmentation de la mortalité du cancer du sein (163). La régiondu chromosome 8, dans laquelle est situé le gène, serait souvent inactivée lors des cancers (164). Ensa présence, il agirait comme un suppresseur de tumeur (165). La migration cellulaire est unprocessus qui nécessite une étape de polarisation cellulaire, formation de laméllipodium (via l’actin)et adhésion focale. CSMD1 entrainerait l’absence de la formation de ces laméllipodium, suggérantune interaction avec l’actine qui les compose. A l’inverse, en présence de CSMD1, l’adhésion focaleserait stimulée, permettant cette migration. CSMD1 interagirait avec SMAD1, 2 et 3 en les activantet stimulerait la production de SMAD4 (166). Celui-ci pourrait donc stimuler la voie du TGF-β quicomprend SMAD2, 3 et 4. Celle-ci est également connue pour être impliquée dans la proliférationcellulaire, notamment dans la cicatrisation pathologique chéloïde que nous avons étudié dans cettethèse.
HPSE2 (héparanase 2) est un gène de 780 méga bases situé sur le chromosome 10. Il code pourune endoglycosidase capable de dégrader les protéoglycanes de la matrice extracellulaire. Elle estconnue pour être impliquée dans les mécanismes de remodelage de la matrice extracellulaire (167)ainsi que l’angiogenèse et la progression des tumeurs (168). L’article de Cintia Pereira de Oliveira etcollaborateurs, (167) explique que HPSE1 est capable de cliver les chaines glycosaminoglycaned’héparanase sulfate dont le rôle est l’organisation de la matrice extracellulaire, l’invasion cellulaireet l’inflammation. Le rôle de HPSE2 n’est pas encore connu. Junjie Fu et collaborateurs proposentun mécanisme d’action pour HPSE2 (168).

103
HPSE2, en compétition avec HPSE1, est aussi capable de se lier à l’héparanase sulfate empêchantHPSE1 de dégrader la matrice extracellulaire. Dans la pathologie du cancer du sein MMP11, àl’inverse, dégrade cette même matrice (Figure 26). Des perturbations dans l’expression des gènesMMP11, sur exprimé, et HPSE2, sous exprimé, entrainent une dynamique anormale de la matriceextra-cellulaire et une absence de sa dégradation.
LSAMP est un gène de 2,1 Mégas bases situé sur le chromosome 3 sur le brin anti sens. Ilintervient dans le système limbique du cerveau. Cette partie de l’encéphale contrôle le comportementet les émotions comme la peur, l’agressivité ou le plaisir ainsi que la formation de la mémoire. Celaconfirme le rapprochement avec les voies de signalisation du développement neuronal retrouvé dansles analyses IPA bien que le rapport avec la cicatrisation soit encore inconnu. LSAMP-AS3 etLSAMP-AS4 semblent jouer un rôle en tant que répresseurs des tumeurs, ayant un effet sur larapidité de la progression du cancer (169, 170, 171). Tale Barøy et son équipe (170) montrent queces deux molécules sont exprimées faiblement, que ce soit dans les cellules d’ostéosarcomes ou dansles cellules saines témoins, ce qui correspondrait à son niveau d’expression normal. La variationgénétique mise en évidence par notre étude est située au sein du gène LSAMP et non en amont ou enaval où nous pourrions penser trouver les régions promotrices du gène. L’hypothèse serait quel’expression de LSAMP serait inhibée de manière épigénétique.
DAB1 (DIABLO) est un gène de 1,5 mégas bases situé sur le chromosome 1. La moléculeDAB1 intervient dans la transduction du signal de la reelin. DAB1 se lie aux queues intracellulairesdes récepteurs transmembranaires de la reelin : VLDLR (very low-density lipoprotein receptor) etApoER2 (apolipoprotein receptor 2). VLDLR et ApoER2 sont des lipoprotéines homologuespartageant environ 50% d’homologie (172). Elles possèdent des domaines en commun comme undomaine extra-cellulaire N terminal riche en cystéine, un domaine capable de reconnaitre lesmolécules Epidermal Growth Factor (EGF), une unique séquence traversant la membrane cellulaireet un domaine cytoplasmique composé de motifs NPxY (172). Ensemble, ils constituent lesrécepteurs exclusifs de la reelin tandis que leur liaison avec DAB1 constitue la seule voie possiblepour la transmission du signal extracellulaire de la reelin. La reelin est une glycoprotéineextra-cellulaire de 420 kDa qui joue un rôle durant le développement embryonnaire et à l’âge adulte.Durant le développement embryonnaire, elle permet la mise en place du système nerveux et ledéveloppement du cerveau. Elle assure la migration de neurones vers leur emplacement définitifrégulant ainsi le positionnement neuronal chez l’embryon (173). Les gradients de concentration de lareelin permettent la différenciation des cellules gliales radiales, progénitrices des neurones, selon uneorientation particulière définissant ainsi des connexions neuronales précises en favorisant lamaturation des dendrites. Par le contrôle des interactions cellulaires, elle assure ainsi la migration etle positionnement neuronal.
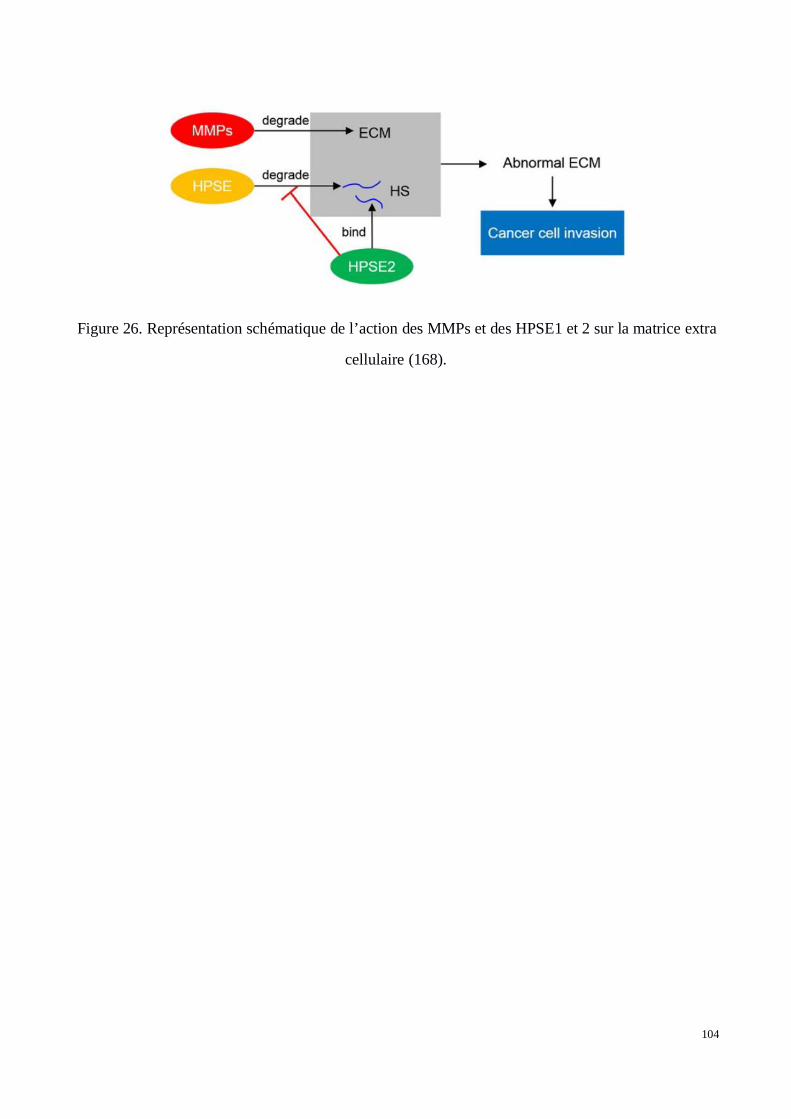
104
Figure 26. Représentation schématique de l’action des MMPs et des HPSE1 et 2 sur la matrice extra
cellulaire (168).

105
A ce stade embryonnaire, elle est aussi observée dans beaucoup d’autres organes qui n’ont pas derapport avec le système nerveux, mais son rôle dans ceux-ci reste encore obscur. Une fois le stade dudéveloppement neuronal terminé, la reelin n’est plus exprimée. A l’âge adulte, la reelin est aussiprésente dans le foie, la glande thyroïde, la glande surrénale et le sein. Présente en faibleconcentration, elle est un indicateur de la bonne santé de l’organe. Dans le système nerveux adulte,elle continue à jouer son rôle dans la neurogenèse, dans la zone sous-ventriculaire et le gyrus denté.L’étude de Khialeeva (174) montre qu’une déficience en reelin dans le cancer du sein est associéeavec un faible taux de survie. Dans le foie également, un taux réduit de reelin est associé à un plusfort risque de cancer hépato cellulaire, lesquels se développent préférentiellement chez les patientscirrhotiques (175). Ces études suggèrent que l’expression de la reelin serait régulée négativementdans les cancers ce qui permettrait aux cellules d’augmenter leur capacité à métastaser.
La reelin joue également un rôle dans l’athérosclérose, une pathologie entrainant le durcissement desparois des artères accompagnée du recrutement de leucocytes et monocytes, acteurs del’inflammation tissulaire. La reelin favorise l'athérosclérose via l’adhésion et le recrutement desleucocytes à l’endothélium vasculaire (176). Les résultats suggèrent que la reelin pourrait intervenirdans l’inflammation chronique hépatique et dans les processus de cicatrisation pathologique. Cettehypothèse est également en accord avec de récentes études montrant un taux élevé de reelin lorsd’événements cellulaires répondant à une blessure, comme l’inflammation et le remodelage cellulaire(177). Dominik Kobold et son équipe (178) décrivent la reelin comme dérégulée dans la matriceextra-cellulaire du foie blessé fibrosé. Leur étude montre une forte concentration en reelin lors del’apparition de lésions hépatiques chez la souris. De plus, les souris déficientes en reelin présententune qualité de cicatrisation hépatique accrue et sans fibrose, suggérant le rôle de la reelin dans lacicatrisation (178, 179). La reelin a aussi été identifiée chez le rat, en particulier dans les HSCs, alorsqu’elle est absente dans les myo-fibroblastes (178). En 2017, une corrélation significative étéobservée entre l'expression de la reelin et le stade de la fibrose hépatique (180). Son expression a étéretrouvée dans les cellules étoilées du foie de patients atteints par le virus de l’hépatite C. Les auteursconcluent cependant que la reelin serait plus corrélée avec les cellules CRBP-1 plutôt que les fibresmusculaires lisses d’actines et donc ne serait pas à considérer comme un marqueur de la fibrose. Lefait que la reelin et dab1 soient le plus souvent associés à la fibrose du foie, aux HSCs, montre quecelle-ci pourrait intervenir dans la régulation de la fibrose. Le fait que la reelin joue un rôle dans lamigration et l’organisation des cellules neuronales à l’état embryonnaire pourrait nous orienter versl’hypothèse que celle-ci participerait de manière similaire à la migration des HSC vers les tissushépatiques lésés où elles se différencient en myo-fibroblastes.

106
Discussion sur l’analyse d’expression des voies de signalisation impliquées dans la cicatrisationpathologique.
Au cours du processus de cicatrisation cutanée, les cellules épidermiques produisent une plus grandequantité de facteurs de croissance, de cytokines et de protéines de la matrice extra-cellulaire. Cesdernières organisent et régulent la ré-épithélialisation de la plaie par les kératinocytes ainsi que laréparation tissulaire en quelques jours ou semaines. Dans de rares cas, ces processus biologiques nese résorbent pas rapidement et entraînent une accumulation anormale de tissu cicatriciel. Lesprincipaux composants des cicatrices anormales sont les fibroblastes activés qui produisent desquantités importantes de protéines fibrillaires de la matrice extra-cellulaire. On appellehypertrophiques les cicatrices qui ne recouvrent pas la plaie et disparaissent un ou deux ans aprèsleurs apparitions. Les cicatrices dites chéloïdes s'étendent au-delà de la limite de la blessure etpersistent sur plusieurs années. Les cicatrices chéloïdes peuvent parfois apparaître spontanément enl'absence de blessures identifiées. Elles sont souvent associées à un prurit et parfois même à desdouleurs. Elles peuvent, comme les tumeurs bénignes, se propager sur de très grandes parties ducorps, principalement sur le cou, le thorax et l'abdomen. Elles n’engagent pas le pronostic vital maispeuvent rapidement constituer un handicap permanent chez les patients les plus susceptibles et avoirdes répercutions socio-psychologiques importantes.
Il n’existe pas à ce jour de bon modèle animal permettant l’étude des cicatrices anormales (chéloïdesou hypertrophiques). De ce fait les études in vivo sont limitées et les mécanismes responsables dudéveloppement des cicatrices chéloïdes sont actuellement mal connus. Le second objectif de cetravail était de préciser les mécanismes biologiques impliqués dans le développement des tissuschéloïdes. Nous avons mené une étude comparative du transcriptome par méthode de séquençageRNA-Seq impliquant des tissus de peau normale, de cicatrices chéloïdes et hypertophiques etidentifié au moins 54 voies de signalisation différentiellement activées.
Le barème d’Ogawa R (181) permet d’établir un score en fonction de l’âge d’apparition de lapremière cicatrice chéloïde, la région du corps sur laquelle elle(s) se développe(nt) ou encorel’origine ethnique du patient. Cette classification, basée sur des observations cliniques et non sur lacaractéristique physiologique des cicatrices, constitue une aide à la caractérisation des patientsprésentant une ou plusieurs cicatrices chéloïdes. Un patient sera inclus dans le groupe des patientsavec cicatrices chéloïdes s’il présente un score supérieur à 15 et si la cicatrice est en croissancedepuis plus de deux ans. En effet, les cicatrices ayant moins de deux ans peuvent être confonduesavec des cicatrices hypertrophiques et ainsi induire une erreur de diagnostique. Histologiquement, lesdifférences entre cicatrices hypertrophiques et chéloïdes portent principalement sur la disposition desfibres de collagène. Dans les cicatrices hypertrophiques, ces amas sont moins proéminents, plusallongés et mieux alignés entre eux. Une cohérence qui rappelle une structure bien plus proche de lapeau normale. Les hypertrophiques présentent une accumulation plus importante de fibres lissesmusculaires α faisant le lien avec leurs capacités contractiles permettant de refermer la plaie,contrairement à la forme chéloïde où la cicatrice s’étend au-delà de la plaie originelle. Cependant, ilest parfois difficile d’observer une claire différence histologique entre chéloïde et hypertrophique.Parfois les tissus peuvent présenter des traits propres aux deux types de cicatrices. De plus, pour desraisons techniques et pratiques, il n’a pas toujours été possible d’envoyer une parti du tissus prélevé àl’anapathologiste. L’information est donc manquante pour certains des tissus de l’étude.

107
Un des biais principaux présents dans le recrutement de cette cohorte est l’origine ethnique et le sexedes patients. La forte prévalence des cicatrices chéloïdes chez les populations à peau noire entraine lerecrutement de sujets le plus souvent Africains et Sud-Américains. A l’inverse, nos témoins sont despatientes ayant réalisé un acte de chirurgie esthétique dans le cadre de réduction ou pose de prothèsesmammaires. Ce sont donc toutes des femmes issues d’un milieu aisé, la plupart ayant la peau blancheet donc une faible prévalence à la cicatrisation pathologique. Les biopsies témoins correspondent àdes tissus sains. Or une peau normale constitue un tissu peu actif où la division cellulaire est faible,contrairement aux cicatrices anormales. Ainsi, la comparaison entre une cicatrice anormale et untissu sain apporte de nombreuses informations qui ne sont pas spécifiques de la forme chéloïde, maisplus probablement du fait des processus de cicatrisation. L’utilisation de biopsies de peau saine n’estpas idéale. Les tissus cicatriciels normaux auraient constitué de meilleurs contrôles. Il est cependantdifficile, pour des raisons éthiques, de réaliser une biopsie de peau chez une personne cicatrisantnormalement. C’est pour cette raison que nous avons croisé les données issues des comparaisonsentre les tissus hypertrophiques et chéloïdes avec les comparaisons entre les peaux saines etcicatrices chéloïdes.
Le contrôle qualité des données est une étape cruciale et répétée à différents moments du traitementdes données. Les données brutes, l’alignement sur le génome de référence et enfin le calcul del’expression des gènes sont soumis au contrôle qualité. Un contrôle qualité partiel, réaliséuniquement sur les données brutes, ne garantit pas que l’alignement sur le génome de référence serade qualité (182). Lors du séquençage de l’ADN, la couverture du génome, c’est-à-dire le nombre defois que chaque base a été séquencée, permet de définir la qualité après l’alignement. En RNA-Seq lacouverture dépend du nombre de séquences obtenues pour chaque gène et donc de son expression.La qualité de la détection des niveaux d’expression des gènes dépendra du nombre de séquencesobtenues lors du séquençage. Pour un séquençage de l’ARN codant total, il est conseillé d’obtenirentre 30 et 40 millions de séquences par échantillon (182). Ce nombre dépend de la qualité de l’ARNutilisé lors de la préparation des librairies et des caractéristiques du séquenceur. Il est égalementpossible de séquencer plusieurs individus lors d’un même séquençage, on appelle alors cela lemultiplexage. Cela impose de dé-multiplexer les séquences obtenues, attribuer chaque séquence àl’individu auquel il appartient. Le contrôle qualité doit s’assurer que cette étape est correctementréalisée. La qualité des bases doit également être contrôlée. Ce contrôle détermine la probabilitéd’avoir identifié correctement chacune des bases séquencées. Les séquences dont les bases sont, enmoyenne, de mauvaises qualités doivent être retirées. La baisse de qualité des bases peut s’expliquerpar des soucis de détections lors du séquençage ou encore dû à une baisse de la qualité de la réactionchimique en fin de séquences. La longueur des séquences est également un paramètre à contrôler.Représentée sous la forme d’un diagramme de distribution, tous les individus doivent présenter lemême profil cohérent avec les caractéristiques du séquenceur. Le contrôle qualité après alignementsur le génome de référence permet d’identifier d’éventuelles contaminations des échantillons durantla manipulation. Le pourcentage de séquences alignées sur le génome ainsi que le pourcentaged’alignement multiple sont également contrôlés. Certaines séquences peuvent s’aligner à demultiples endroits sur le génome. Il est donc nécessaire de conserver cette information pour ne pasbiaiser le calcul des niveaux d’expression par la suite. L’effet « batch », caractérisé par un meilleuralignement pour certains individus par rapport à d’autres, est également estimé à cette étape. Malgréces différents contrôles, d’autres biais peuvent néanmoins subsister.

108
Une fois le niveau d’expression de chaque gène quantifié, il est nécessaire de déterminer si lesgroupes d’étude définis par les phénotypes cliniques sont homogènes selon leur expression, nousavons donc réalisé une agréation hiérarchique basé sur l’ensemble du transcriptome.
Un échantillon peut alors être situé hors du groupe formé par les individus ayant le même phénotype.Dans ce cas, cela peut signifier que le phénotype de ce patient a été mal évalué, que l’expressiondétectée pour ce patient présente un profil particulier qui n’a pas de rapport avec son phénotype, unecontamination, ou encore une erreur dans l’identification de l’échantillon séquencé. C’est en évaluantces possibilités qu’il est décidé si l’échantillon doit être retiré ou conservé dans l’analyse. Aucunpatient n’a été retiré pour ce motif.
Par l’analyse d’expression, nous avons pu identifier plusieurs centaines de gènes différentiellementexprimés de manière significative ce qui rend l’étude des gènes individuelle impossible. Le logicield’enrichissement génomique Ingenuity Pathway Analysis (I.P.A, Qiagen) a donc été choisi commeaide à l’interprétation des listes de gènes obtenues. Toutefois ce logiciel présente un biais majeurdans l’interprétation des données. En effet, il est dépendant de la base de données de connaissancequ’utilise le logiciel. Ces bases de données sont complétées manuellement par des équipes si bienque deux équipes n’ayant pas les mêmes méthodes de travail et présentant deux logiciels distinctspeuvent interpréter les données de manières différentes. L’impact de certains processus pourrait êtremis en avant de manière abusive ou sous-estimé par le logiciel. De plus, ces bases de données secontentent de refléter l’état actuel des connaissances scientifiques dans le domaine d’étude. Elles nepermettent pas la découverte de nouveaux processus innovants. Bien que utiles, les logicielsd’enrichissement génomique ne permettent pas de conclusion objective.
Les comparaisons du transcriptome du groupe témoin avec celui des cicatrices chéloïdes ou letranscriptome des cicatrices hypertrophiques ont mis en évidence des voies de signalisationsimilaires. Parmi ces voies de signalisation, on retrouve les processus de fibrose, des jonctionscellulaires et de l’inflammation. On retrouve également la diapédèse et la migration des leucocytessur le site de l’infection. Seule la voie Wnt canonique est présente. Lorsque l’on compare lestranscriptomes des biopsies témoins avec les biopsies des cicatrices chéloides, la voie des processusde la fibrose est la plus significative. Cette observation est inattendue dans un mécanisme decicatrisation car bien d’autres voies qui font, par exemple, entrer les facteurs de croissance tels queles EGF et les mécanismes de réépithélialisation devraient apparaître au premier plan dans l’analyse.EGF participe à la prolifération et à la mobilité cellulaire au niveau des fibroblastes des chéloïdes(183). Dans une cicatrice de type chéloïde, les fibroblastes représentent le type cellulaire le plusabondant et leur capacité de réponse aux modulateurs de la production de collagène est altérée. Lacicatrice chéloïde n’a pas la capacité de remodelage que l’on pourrait retrouver dans un tissu normal.
La cicatrisation est permanente avec absence de nouvelle formation de peau normale. La peau est unépithélium stratifié possédant une organisation précise. Sa formation nécessite donc une réorganisation du tissu cicatriciel. De plus, le rétablissement de la peau normale ne nécessite pas laprésence de tous les éléments cellulaires nécessaires à la cicatrisation. Les cellules du systèmeimmunitaire recrutées lors des premiers stades (inflammation) de cicatrisation ne sont probablementplus nécessaires. Tout comme les fibroblastes, recrutés en grande quantité lors de la proliférationcicatricielle, seule la quantité nécessaire au maintien de la peau doit subsister après remodelage.

109
Il se pourrait donc que la polarisation du tissu perturbe les migrations cellulaires au niveau de lacicatrice entrainant une sur-réaction du système immunitaire et l’accumulation des fibroblastes (184,185).
Lors des prélèvements, les observations cliniques montrent le plus souvent des cicatricessymptomatiques avec démangeaisons, prurits voir douleurs, potentiellement dues à une inflammationanormale du tissu. L’impossibilité de se “gratter” pour limiter les démangeaisons sans risquerd’aggraver la cicatrice rendent ces démangeaisons à la limite du supportable pour le quotidien decertains patients. La mobilisation anormale des cellules du système immunitaire pourrait expliquercette inflammation permanente, même au niveau des tissus sur lesquels la chéloïde s’est répandueaprès lésion (tissus sans cicatrisation initiale).
Les cicatrices chéloïdes se forment par prolifération de tissus conjonctifs au niveau du derme. A ladifférence de la cicatrice hypertrophique qui est capable de régresser, même de manière incomplète,la chéloïde se comporte plus comme une tumeur. Les chéloïdes sont plus actives que leshypertrophiques : le nombre de gènes impliqués comparés à de la peau normale, par définition àactivité réduite, ressort bien cette information. Il n’est pas surprenant que les voies impliquées dansles comparaisons avec les peaux saines soient similaires. La présence d’un plus grand nombre degènes dérégulés dans les tumeurs chéloïdes par rapport aux cicatrices hypertrophiques montre lesdifférences d’activités de chacun des tissus. En termes de comportement, les chéloïdes s’apparententplus à des tumeurs invasives (même si elles n’en ont pas les caractéristiques physiologiques), tandisque les hypertrophiques s’apparentent plus à des petites cicatrices qui perdurent anormalement dansle temps. Cela appuie l’hypothèse comme quoi la chéloïde n’est pas un tissu en cicatrisation. Laprincipale observation qui permet de différentier la cicatrice chéloïde des autres pathologies decicatrisation aberrante est leur capacité à pouvoir se développer au-delà des limites de leur cicatriceinitiale. Cela implique que le tissu cicatriciel ne présente plus de limites physiques à sa croissance.Suite à cette levée de frontière, la migration cellulaire intervient probablement pour envahir la peaunormale environnante.
Une des caractéristiques des chéloïdes du thorax est leur capacité à coloniser le tissu qui les entouretel une tumeur bénigne. Cet aspect très invasif est également supporté par les voies de cancérisation,qui sont activées dans les chéloïdes et qui ressortent tout particulièrement dans la comparaisonchéloïdes thorax (très invasives) versus chéloïdes oreilles (peu invasives). La recherche despathologies partageant le plus de voies communes avec les chéloïdes montre que parmi les cinqgroupes de pathologies cités, les cancers et tumeurs sont en très bonne place. Il est sans doute peuapproprié de parler de cicatrices chéloïdes mais plutôt d’utiliser les termes de tissu chéloïdes oumême de fibrose chéloidienne.
L’agrégation hiérarchique des patients présentant des chéloïdes oreilles et thorax suggère l’hypothèseque la localisation de ces cicatrices forme des groupes distincts. Les chéloïdes qui se forment sur lethorax présentent le plus souvent les cas de chéloïdes les plus graves. La capacité de ces cicatrices àproliférer et se répandre sur l’ensemble du thorax diffère du comportement des chéloïdes aux oreilles,le plus souvent présenté sous forme de petits nodules. La cicatrisation chéloïde pourrait êtresubdivisée en deux types selon leur localisation.

110
Il est inattendu que jamais les kératinocytes et leur rôle central dans la cicatrisation n’apparaissentdans les voies qui regroupent les gènes exprimés différemment entre tissus sains et chéloides. Celasouligne toute l’importance de la comparaison avec les tissus sains car si le transcriptome des tissuschéloides avait été seulement comparé à celui des cicatrices normales, on pourrait penser que cesmécanismes sont communs et ils auraient été soustraits dans cette comparaison.
Ces deux observations nous amènent à une conclusion qui est importante pour la compréhension dece que sont les tissus chéloides : les chéloides ne sont pas des tissus en voie de cicatrisation, ni mêmeen voie de cicatrisation très retardée. Ce sont d’abord un phénomène de fibrose massive des tissuscutanés.
Cette conclusion est en accord avec les études effectuées par notre groupe dans les populationsafricaines : les chéloïdes sont parfois initiées par une blessure rituelle ou causée par un guérisseur,mais une fraction importante (environ la moitié) des chéloïdes se développent en l’absence de touteslésions, souvent à partir d’une zone de prurit qui conduit le patient à se gratter. Il n’y a pas, en ce cas,de blessure mais simplement une zone d’irritation due au grattage. Si les chéloïdes peuvent êtreinitiées par un processus de cicatrisation des blessures, ce n’est pas toujours le cas. L’analyse destissus chéloides supporte que ceux-ci ne mettent pas (ou ne mettent plus) en jeu les mécanismes lesplus centraux de la cicatrisation. On peut argumenter que les processus de migration cellulaire desgranulocytes et agranulocytes, de diapédèse de ces mêmes cellules, les voies impliquant les jonctionscellulaires, se rapportent à la cicatrisation. Cependant ce lien est peu spécifique car ces voies sontactivées dans de nombreux autres processus comme par exemple dans l’inflammation ou dans leprocessus d‘invasion des tissus par la cicatrice chéloïde.
Ceci nous amène à soulever deux questions : Comment expliquer que le processus de cicatrisationnormal ait été stoppé dans le cas des chéloïdes ayant pour point départ une blessure ? et quelsmécanismes expliquent cette fibrose massive et cette capacité invasive si marquée des chéloïdes ?Nous proposons une explication à cette cicatrisation anormale basée sur un dérèglement génétique dela voie Wnt.
Les résultats obtenus montrent une activation des voies Wnt : la voie Wnt canonique et les noncanoniques : la voie planar cell polarity (PCP) et la voie Wnt Ca2+. Les voies Wnt jouent un rôledéterminant dans de nombreux cancers comme le cancer du foie (185), ou encore le cancer des os(186) et des processus neuronaux (187).
Le fait que la cicatrice chéloïde n’ait pas la capacité de remodelage permettant la formation d’untissu normal permet le rapprochement avec la voie Wnt PCP responsable du phénomène de polaritécellulaire, du mouvement et de l’orientation de la division cellulaire (188). Les molécules Wnt sontnécessaires à l’établissement de la polarité. C’est l’effet conjoint des gradients des moléculesintracellulaires et des ligands extracellulaires qui permet l’ensemble du mécanisme. Chez le xénope,les molécules Wnt5A et Wnt11 sont en effet capables d’orienter l’accumulation des molécules Pk etVang dans la cellule en les repoussant à l’opposé de la source d’expression de leur ligand durant lestade neurula, entrainant l’orientation de l’axe antéro-postérieur (189). Chez l’homme, en se liant aurécepteur Fzd, ces mêmes molécules activent la voie Wnt PCP.
Si l’on considère les différences d’expression entre les tissus hypertrophiques, qui sont de meilleurscontrôles que les peaux saines, et les chéloïdes, seule la voie Wnt canonique est activée. Nous

111
regroupons la voie de signalisation Wnt avec celle du TGF-β et BMP. Il a été montré que les voiesdu TGF-β et Wnt présentent une interaction spécifique (190, 191). Une surexpression de TGF-β estsouvent associée à la fibrose et régulièrement impliquée dans la cicatrisation anormale. Fonctionnantavec SMAD 2 et 3 comme transducteurs de signal intra-cellulaire, ceux-ci sont phosphorylés en casde stimulation par TGFβ. TGF-β pourrait être considéré comme un stimulateur positif de la voie Wntcanonique (192).
Alors que l’expression de SMAD 2 et 3 est similaire dans les tissus chéloïdes et peaux normales,TGFβ1 présente une expression plus forte au sein des tissus chéloïdes. De plus, les auteurs notentque cette différence d’expression n’est pas retrouvée dès lors que l’on compare les tissus chéloïdesavec des cicatrices hypertrophiques. TGFβ1 semble donc avoir le même comportement dans les deuxtypes de cicatrices (193). Les auteurs montrent également que, en présence de TGF-β1, laconcentration en β-caténine est retrouvée significativement plus élevée dans les cicatriceschéloïdes et hypertrophiques par rapport aux peaux normales. En revanche, quand la stimulation enTGF-β1 augmente encore, l’effet inverse est observé sur la β-caténine dont la concentration se met àdiminuer, comme sous l’effet d’une rétro-action. La variation observée est d’autant plus prononcéechez les peaux contrôles que chez les cicatrices anormales, où les taux de β-caténine ont tendance àrester élevés.
Une trop grande accumulation de ces deux molécules pro-fibrosantes présente un danger pourl’intégrité des tissus et ce mécanisme de rétro-action permet d’éviter cette situation. Le fait que lescicatrices anormales soient capables d’accumuler à la fois des concentrations élevées de β-caténine etde TGF-β, montre clairement un dérèglement de ces mécanismes. Notre étude n’a pas pour seulobjectif d’identifier les gènes et voies de signalisation impliqués dans les pathologies de lacicatrisation anormale. Nous avons également recherché quelles pourraient être les causes de cesdérèglements d’expression.
Le développement de la cicatrisation anormale des cicatrices chéloïdes montre une plus forteprévalence chez les personnes à peau noire ou les asiatiques. Certaines populations sont donc plussujettes à cette pathologie. De plus, l’équipe du Pr. Dessein a montré que de forts facteurs génétiquesétaient responsables des fibroses graves comme l’IL22 dont certaines mutations présentes seraientcapables de réguler la progression de la fibrose et cirrhose en cas d’infection par l’hépatite C.Certains variants du gène CTGF sont également impliqués dans la fibrose hépatique causée par leschistosome (20, 194). Dans cette thèse nous avons donc recherché si de tels facteurs génétiquespouvaient également expliquer la présence des cicatrices chéloïdes. Nous pouvons donc supposerque de tels mécanismes aussi critiques pour le fonctionnement de l’organisme aient été hautementconservés au cours de l’évolution. Toute mutation non-synonyme ou non-sens entrainant une pertede fonction des mécanismes Wnt ne serait donc pas transmise à la génération suivante (descendance).La redondance et l’antagonisme des différentes signalisations Wnt appuient cette hypothèse, dans lesens ou un un tel système ne pourrait se permettre de ne pas être redondé en cas de défaillance.Nombreux sont les facteurs qui pourraient affecter le bon fonctionnement de la voie Wnt.
L’hypothèse principale serait la présence de mutations génétiques au sein des éléments de la voie.Une mutation qui interviendrait au sein d’un des éléments effecteur, récepteur, ligand stimulateur ourépresseur, peut perturber toute la signalisation et entrainer l’expression de molécules non souhaitéesou dans des quantités anormales pour un fonctionnement cellulaire optimal (195).

112
Une des premières mutations affectant les voies Wnt a été observée en 1991 sur le gène adenomatouspolyposis coli (APC), mettant en évidence les causes de l’hérédité du cancer du colon et des polypes.La pathologie de la leucémie myéloïde aiguë, leucémie la plus répandue chez les adultes, ou encorele développement des mélanomes est également une conséquence de cette voie. Une signalisationWnt activée a également été reportée comme présente dans plus de 50% des cancers du sein. Denombreuses études sur les cicatrices chéloïdes visant à étudier l’effet de quelques mutations ont étéréalisées (81, 105, 113).
Cependant, compte tenu du nombre important de SNPs présents dans le génome humain, cesapproches restent limitées. Des études plus larges, GWAS, ont été réalisées pour tenter de percer lemystère des cicatrices chéloïdes (14). Il a ainsi été identifié, chez les populations chinoises, les loci1q41, 3q22.3 et 15q21.3 comme étant des régions génomiques cruciales dans le développement descicatrices chéloïdes. D'autres régions génomiques comme les région 2q23 (chez les asiatiques) et7p11 chez les européens ont été mises en évidence par criblage du génome (143).
L’étude génétique présentée dans notre thèse, montre que des polymorphismes dans 5 gènes (dont 3sont sur-exprimés dans l’analyse transcriptome) sont associés au développmeent de cicatriceschéloïdes. Les figures 27 et 28 montrent les gènes différentiellement exprimés dans les tissuschéloïdes et tissus hypertrophiques. Une forte stimulation des molécules agissant au niveau de lamembrane cellulaire est observée. Tous les éléments extra-cellulaires pour une initiation de lacascade cytoplasmique Wnt sont réunis.
Lors de l’étude trascriptomique, Wnt5A est retrouvé (variation de l’expression fold change (FC)observé entre les tissus chéloïdes et les tissus sain = 29,64) et son récepteur Frizzled 2 (FC= 23,7) ouencore le TGF-β3 (FC= 21,1) ainsi que son récepteur TGFBR qui constituent deux signalisationsdifférentes pour l’activation de la transcription des gènes cibles. Au sein de la cellule, les moléculesformant ou participale à la formation du complexe qui permet la dégradation de la β-caténine sontobservées. PP2A est sous exprimé dans les tissus chéloïdes et hypertrophiques. APC ainsi que CK1sont retrouvés sur exprimés uniquement pour les chéloïdes. Ces observations indiquent que lesmolécules nécessaires à la formation du complexe de dégradation de la β-caténine sont bienimpliquées dans la pathologie des chéloïdes. La transduction du signal Wnt du cytoplasme vers lenoyau peut donc être altérée.
La présence des facteurs de transcription au sein du noyau nous montre cependant que la cascade designalisation de la voie est bien activée. Nous retrouvons le facteur LEF sur-exprimé ainsi que SOX,un inhibiteur du complexe de transcription qui est sous-exprimé. Certaines molécules transcrites parl’activation de cette voie, comme TCF4 ou BMP7, qui jouent un rôle dans l’inflammation et ladifférenciation des myo-fibroblastes sont observées. D’autres arguments sont en faveur del’activation de la voie Wnt. On remarque une levée de l’inhibition de la voie : WIF 1 (FC= -250) etSFRP5 (FC= -9,4) sont retrouvés sous exprimés. Ces deux molécules sont des compétiteurs de Wntcar ils peuvent eux aussi se fixer au récepteur Frizzled. Le gène DKK2 (FC= 3), également uninhibiteur de la voie de la β-caténine, est retrouvé quant à lui sur-exprimé comme dans la plupartdes cancers. Pour agir, il doit se fixer à LRP5 et 6, eux aussi trans-membranaires. Ils jouent un rôlestratégique et sont tous les deux sujets à des variations d’expression.
Dans un second temps, nous avons souhaité vérifier si des mutations dans les gènes de la voie Wntpouvaient expliquer ces dérèglements. Parmi les 60 SNPs que nous avons testés sur les gènes de

113
cette voie, nous avons mis en évidence six mutations dans six gènes différents. Ces mutations sontretrouvées dans des gènes différentiellement exprimés autant que dans des gènes qui ne sont pas misen évidence en étude d’expression. Elles sont présentes sur des gènes clés de la voie, tant au niveaurécepteur que sur la signalisation interne.
Deux mutations dans les gènes SFRP, les SFRP1 et 4, sont retrouvées associées à la pathologie descicatrices chéloïdes, confirmant le rôle de l’inhibiteur de Wnt qui est sous exprimé. Nous pourrionspenser que ce sont ces mutations qui peuvent être responsables de la variation d’expression dans cesgènes SFRP. LRP5 et TGFβR1 présentent également une mutation.
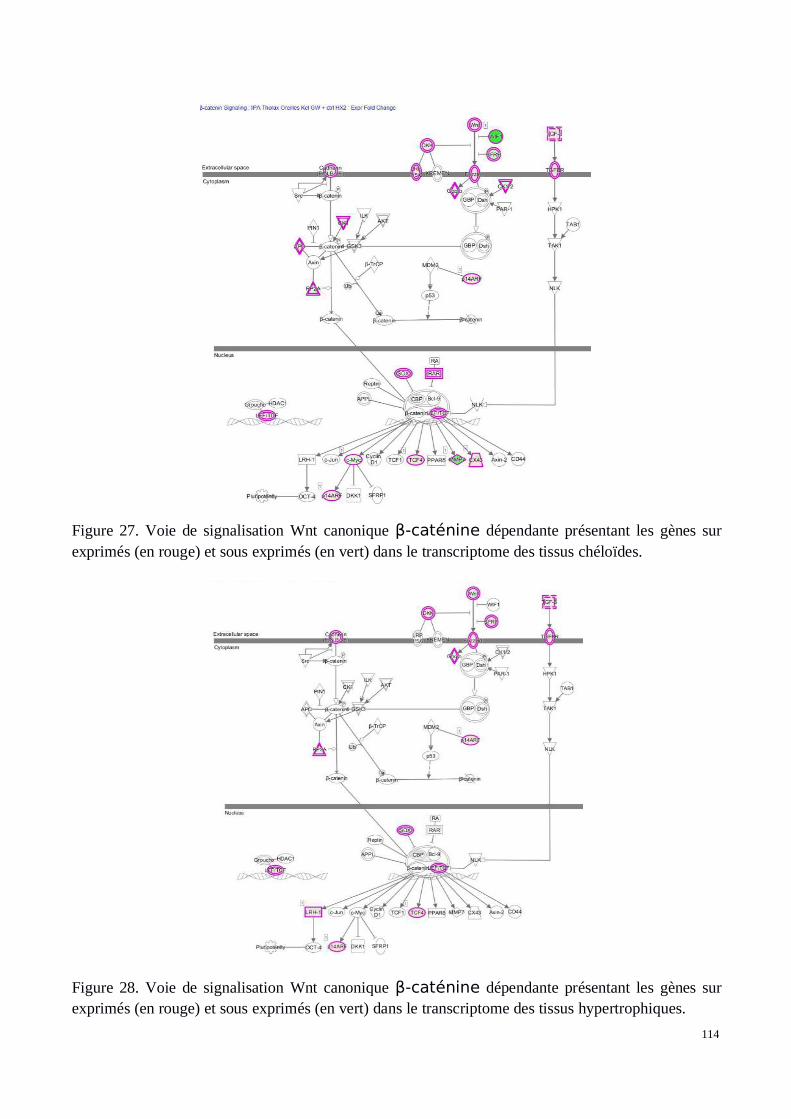
114
Figure 27. Voie de signalisation Wnt canonique β-caténine dépendante présentant les gènes surexprimés (en rouge) et sous exprimés (en vert) dans le transcriptome des tissus chéloïdes.
Figure 28. Voie de signalisation Wnt canonique β-caténine dépendante présentant les gènes surexprimés (en rouge) et sous exprimés (en vert) dans le transcriptome des tissus hypertrophiques.

115
Ceux-ci font partie des récepteurs membranaires capables de stimuler ou réprimer la voie. Desmutations dans ces récepteurs peuvent impacter l’activation de la signalisation intra-cellulaire quidécoule et activer anormalement la transcription. Une mutation sur le gène de l’AXIN1 estégalement observée. Cette molécule joue un rôle pivot dans la formation du complexe de dégradationcytoplasmique de la β-caténine. Quand LRP5 et 6 sont phosphorylés, ils donnent la capacité àl’AXIN1 de bloquer la formation du complexe de dégradation de la β-caténine qui va alorss’accumuler dans le cytoplasme pour ensuite être transloqué dans le noyau.
Enfin, une mutation est observée sur le gène LEF1. La molécule produite par ce gène correspond aufacteur sollicité par la β-caténine pour activer la transcription des gènes cibles de la voie Wnt surl’ADN. Parmi les gènes cibles activés, notre transcriptome indique une augmentation de l’expressiondes gènes CX43, TCF4 (FC= 3) et C-Myc et une diminution de l’expression de MMP7 (FC= -26,3).Seul TCF4 est retrouvé sur exprimé dans les tissus hypertrophiques.
L’étude génétique a permis de montrer que des mutations étaient présentes dans les gènes clés de lavoie Wnt. Ces mutations sont retrouvées dans des gènes codant pour les récepteurstrans-membranaires comme LRP5 autant que dans ceux qui codent pout pour les inhibiteurs de Wntcomme SFRP1.
L’axine 1 est un gène de 65 kilo bases situé sur le chromosome 16. Il code pour une moléculecapable d’inhiber la formation du complexe de dégradation de la β-caténine. La β-caténine setransloquerait ainsi dans le noyau. Ce que nous confirmons dans notre étude par le facteur detranscription LEF qui est retrouvé différentiellement exprimé et pour lequel nous retrouvonségalement une mutation. D’autres mécanismes de stimulation de la voie Wnt sont égalementpossibles comme une activation via le TGFβ, molécule qui est retrouvée sur exprimée dans notreétude du transcriptome. Une mutation dans son récepteur, le TGFβR1 est observée, pouvant soitsignifier un blocage de l’activité du récepteur soit une sur stimulation de celui-ci.
Les effecteurs de la voie Wnt semblent être modulés à différents niveaux de sa signalisation. Leprincipal effecteur intra-cellulaire de la voie, la β-caténine, n’est cependant pas altéré. Nos résultatsmontrent une modification au niveau génétique, par la découverte de nouvelles mutations associées àla cicatrisation pathologique, mais aussi au niveau transcriptionnel, par la mise en évidence de gènesdont l’expression est modulée par cette cicatrisation. Il est intéressant de noter que les mutationsobservées sont situées dans des gènes dont l’expression varie dans la pathologie. Cependant l’étudene nous renseigne pas sur l’effet de ces mutations sur les gènes ni même si elles sont responsables deleur différences d’expression.

116
Conclusion
Lors de ce travail de thèse, les fibroses du foie bilharzienne causées par la parasite S.Mansoni et lesfibroses de la peau liées à la cicatrisation anormale de type chéloïde et hypertrophique ont étéétudiées selon deux hypothèses fondamentales :
● Toutes les fibroses, quelques soit l’organe quelles concernent, présentent des mécanismes de
développement commun.
● Certaines personnes sont susceptibles de développer des fibroses plus rapidement et plus graves
que d’autres ; Cette suscèptibilité présente un fort déterminisme génétique.
Pour étudier cette prédisposition à la fibrose du foie bilharzienne, une étude par méthode GWAS aété réalisée sur des sujets brésiliens. Cette première étude a mis en évidence des marqueursgénétiques associés à la pathologie et 12 d’entre eux ont été confirmés dans une cohorteindépendante de sujets ougandais atteints de fibroses bilharzienne plus ou moins graves. Cette étudea également permise d’analyser les variants génétiques présents dans les gènes des voies Wnt, connupour participer à la fibrose. Certains des variants génétiques des voies Wnt sont associés aux fibroseshépatiques graves dans nos analyses.
L’étude transcriptomique des cicatrices chéloïde et hypertrophique a, quant à elle, permise de mettreen évidence des voies de signalisation génomique dont les mécanismes pourrait être altérés, causantainsi ces fibroses cutanées. Ces mêmes voies Wnt sont retrouvées parmi les voies de signalisationmises en jeu dans la pathologie des chéloïdes. Selon l’hypothèse que les mécanismes d’établissementdes fibroses sont similaires entre le foie et la peau, nous avons testé si les variants génétiquesidentifiés dans les voies Wnt lors de l’étude sur la fibrose bilharzienne pouvaient également êtreassociés à la formation des fibroses cutanées.
Les résultats d’association obtenu pour ces variants sur une cohorte africaine de sujets affectes parles chéloïdes montrent 6 variants pouvant êtres utilisés ensemble pour l’élaboration d’un testdéterminant la prédisposition à la pathologie des chéloïdes.
Notre travail sur la cicatrisation chéloïde, les études menées et les rencontres avec les patientsfrançais et les sujets ougandais atteints par les chéloïdes ont soulevés deux questions: Commentexpliquer que le processus de cicatrisation normal ait été stoppé dans le cas des cicatrices chéloïdesayant pour point départ une blessure ? ; Quels mécanismes expliquent cette fibrose massive et cettecapacité invasive si marquée des cicatrices chéloïdes ?
A la première question notre travail n’apporte pas de réponse satisfaisante et il faudra probablementapprofondir l’analyse génétique pour y répondre.
La littérature fournit toutefois un début de réponse à cette question : il se pourrait que lesmécanismes de cicatrisation soient simplement stoppés, « étouffés », par la fibrose massive des tissus(196).

117
Les travaux conduit sur la régénération des voies nerveuses montrent en effet que cette régénérationest liée à l’accumulation du collagène et des fibroblastes (197). Peut-être en est-il de même pourd’autres mécanismes de réparation en particulier pour la recolonisation des tissus lésés par leskératinocytes.
Les études effectuées sur les fibroses hépatiques massives supportent aussi l’idée qu’une fibrosemassive bloque la cicatrisation. Un développement massif de fibrose favorise le développement detumeurs (198). Le caractère invasif de la cicatrice chéloïde est sans doute favorisé par la fibrosemassive du tissu cutané. La deuxième question traite des causes de la fibrose cutanée observée dansles cicatrices cheloïdes. Notre travail apporte une réponse au travers de la voie Wnt, qui n’estprobablement pas complète mais qui est convaincante du fait des résultats de l’analyse génétique.
La cohorte ougandaise de réplication n’est pas toujours représentative de la population du pays. LesOugandais Allur de notre étude ont été sélectionnés car ils vivent et pêchent proche du lac Albert,source d’exposition aux schistosomes. Le fait que cette population soit génétiquement homogèneréduit le risque de biais entre cas et contrôles. Cela pose cependant le problème de l’universalité desrésultats génétiques obtenus. La cohorte Ougandaise a été utilisée pour valider les suggestionsd’association obtenues à partir du GWAS réalisé sur des sujets Brésiliens infectés par lesschistosomes mais aussi pour valider les associations observées sur les tissus chéloïdes de la cohortefrançaises. Elle montre que ces associations sont présentes dans des populations génétiquement trèsdifférentes. Toutefois certains variants dans la population brésilienne ne sont pas polymorphiques enAfrique. C’est le cas pour 16 des 180 polymorphismes obtenus en GWAS que nous avons testés enOuganda. Notre étude présente des biais et peut être améliorée. L’étude du transcriptome à partir desdonnées de séquençage est une discipline nouvelle. Les bases de données transcriptomiquesconstituées par les consortiums comme le National Center of Biotechnology Information (NCBI, 199)ou encore la base Ensembl et le 1000 génomes project (200) cataloguent l’ensemble des transcritsARN annotés des organisme modèles. Ces bases de données sont parfois incomplètes.
C’est à l’aide d’Ensembl que l’analyse des données a été conduite dans l’étude. De plus, l’analyse del’ARN ne peut se limiter aux ARN messagers uniquement. D’autres ARN existent comme les ARNslongs non codants ou encore les petits ARNs. L’étude de l’ARN renseigne aussi sur la structure desgènes, type Intron-Exon, ainsi que les différents transcrits issus de la combinaison de ces exons.
L’avantage de l’analyse du transcriptome par séquençage est que sa capacité de détection ne se limitepas uniquement à une liste de transcrits définie. La détection de l’expression ne se limite pas àl’échelle du gène mais peut être plus précise en étant mesurée au niveau des transcrits de chaquegène. Le RNA-Seq permet donc une exploitation plus précise des données d’expression.
Nous pourrions donc approfondir encore les résultats en ciblant les gènes d’intérêts de l’étude afin,soit de rechercher de nouvelles mutations, soit d’étudier la structure des transcrits issus de l’épissagealternatif. Il est également possible de conduire des études fonctionnelles sur les protéines de cesgènes, leurs produits, leurs interactions et leur implications dans les processus de fibrose.

118
Remerciements
Je souhaiterai tout d’abord remercier le professeur Alain Dessein pour avoir accepté de diriger et dem’encadrer tout au long de mes 5 années de doctorat. Son expérience dans la rechercher et saméthodologie de travail m’ont permis d’acquérir de nouvelles compétences en gestion de projet, enbiologie humaine et bien sur en génétique.
Je remercie également tous les membres du laboratoire UMR906 pour m’avoir accueilli pendant cesannées et pour m’avoir assisté au cour de ma formation. Je pense en particulier à Mr Laurent Argiroet Mme Sandrine Cabantous pour m’avoir encadré et formé aux manipulations de laboratoire enbiologie molléculaire, un domaine qui m’était inconnu jusque là.
Je remercie tous les membres de l’entreprise Genepred Biotechnologies, renommée Bilhi Geneticsdurant ma thèse. Sans eux cette thèse n’aurait pas été possible. Je remercie tout particulièrement lesmembres de la direction de Bilhi Genetics qui ont su me faire confiance en finançant mon travail dethèse durant ces 5 années et qui ont su apprécier mes compétences en informatique à leur justevaleur.
Je remercie ma famille pour m’avoir soutenu tout au long de mes études. Je remercieparticulièrement ma grand mère Marcelle Duflot, chercheuse renommée et reconnue en astronomie,pour m’avoir présenté auprès du professeur Dessein. Sans elle, cette thèse n’aurait pas été possiblenon plus.
Enfin, je ne peux pas conclure sans avoir une pensée pour mon ami et co-locataire Arnaud et macopine et future femme Colombe qui ont joués un rôle majeur dans les derniers mois de mon doctoraten me soutenant et en étant à mes coté dans les moments les plus difficiles.

119
Bibliographie
1 Datubo-Brown, D. D. Keloids: a review of the literature. Br J Plast Surg. 1990 Jan;43(1):70-7.
2. Butzelaar L, Niessen FB, Talhout W, Schooneman DPM, Ulrich MM, Beelen RHJ, Mink van der
Molen AB. Different properties of skin of different body sites: The root of keloid formation?.Wound Repair Regen. 2017 Sep;25(5):758-766.
3. Bock O, Schmid-Ott G, Malewski P, Mrowietz U. Quality of life of patients with keloid and
hypertrophic scarring. Arch Dermatol Res. 2006 Apr;297(10):433-8. Epub 2006 Mar 10.
4. Al-Attar A, Mess S, Thomassen JM, Kauffman CL, Davison SP. Keloid Pathogenesis and
Treatment. Plast Reconstr Surg. 2006 Jan;117(1):286-300.
5. Coentro JQ, Pugliese E, Hanley G, Raghunath M, Zeugolis DI. Current and upcoming
therapies to modulate skin scarring and fibrosis. Adv Drug Deliv Rev. 2018 Aug 30. pii:S0169-409X(18)30207-2.
6. Gál P, Varinská L, Fáber L, Novák Š, Szabo P, Mitrengová P, Mirossay A, Mučaji P, Smetana K.
How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair.Molecules. 2017 Oct 26;22(11). pii: E1818.
7. Zhang Z, Nie F, Chen X, Qin Z, Kang C, Chen B, Ma J, Pan B, Ma Y. Upregulated periostin
promotes angiogenesis in keloids through activation of the ERK 1/2 and focal adhesion kinasepathways, as well as the upregulated expression of VEGF and angiopoietin‑1. Mol Med Rep.2015 Feb;11(2):857-64.
8. Sakaguchi M, Fukumoto T, Fujishima F, Fukuda K, Kozaru T, Ban M, Oka M. Bilateral breast
keloids in an elderly woman associated with bilateral breast cancers and high concentration ofserum tumor growth factor-β. J Dermatol. 2017 Nov;44(11):1303-1308.
9. Kiritsi D, Nyström A. The role of TGF-β in wound healing pathologies. Mech Ageing Dev.
2018 Jun;172:51-58.
10. Marneros AG, Norris JE, Olsen BR, Reichenberger E. Clinical genetics of familial keloids.
Arch Dermatol. 2001 Nov;137(11):1429-34.
11. Sun LM, Wang KH, Lee YC. Keloid incidence in Asian people and its comorbidity with
other fibrosis-related diseases: a nationwide population-based study. Arch Dermatol Res. 2014Nov;306(9):803-8.
12. Velez Edwards DR, Tsosie KS, Williams SM, Edwards TL, Russell SB. Admixture mapping

120
identifies a locus at 15q21.2-22.3 associated with keloid formation in African Americans. HumGenet. 2014 Dec;133(12):1513-23.
13. Halfon P, Bourliere M, Ouzan D, Maor Y, Renou C, Wartelle C, Pénaranda G, Tran A, Botta D,
Oules V, Castellani P, Portal I, Argiro L, Dessein A. A single IL28B genotype SNP rs12979860determination predicts treatment response in patients with chronic hepatitis C Genotype 1virus. Eur J Gastroenterol Hepatol. 2011 Oct;23(10):931-5.
14. Zhu F, Wu B, Li P, Wang J, Tang H, Liu Y, Zuo X, Cheng H, Ding Y, Wang W, Zhai Y, Qian
F, Wang W, Yuan X, Wang J, Ha W, Hou J, Zhou F, Wang Y, Gao J, Sheng Y, Sun L, Liu J, Yang S,Zhang X. Association study confirmed susceptibility loci with keloid in the Chinese Hanpopulation. PLoS One. 2013 May 7;8(5):e62377.
15. Shih B, McGrouther DA, Bayat A. Identification of novel keloid biomarkers through
Profiling of Tissue Biopsies versus Cell Cultures in Keloid Margin specimens Compared toadjacent Normal Skin. Eplasty. 2010 Apr 7;10:e24.
16. Nakashima M, Chung S, Takahashi A, Kamatani N, Kawaguchi T, Tsunoda T, Hosono N, Kubo
M, Nakamura Y, Zembutsu H. A genome-wide association study identifies four susceptibility locifor keloid in the Japanese population. Nat Genet. 2010 Sep;42(9):768-71.
17. Tosa M1, Ghazizadeh M, Shimizu H, Hirai T, Hyakusoku H, Kawanami O. Global Gene
Expression Analysis of Keloid Fibroblasts in Response to Electron Beam Irradiation Revealsthe Involvement of Interleukin-6 Pathway. J Invest Dermatol. 2005 Apr;124(4):704-13
18. Igota S1, Tosa M, Murakami M, Egawa S, Shimizu H, Hyakusoku H, Ghazizadeh M.
Identification and characterization of Wnt signaling pathway in keloid pathogenesis. Int J MedSci. 2013;10(4):344-54.
19. Jagadeesan J, Bayat A. Transforming growth factor beta (TGFbeta) and keloid disease. Int
J Surg. 2007 Aug;5(4):278-85.
20. Dessein A, Arnaud V, He H, Li J, Dessein H, Hou X, Luo X, Li Y. Genetic analysis of human
predisposition to hepatosplenic disease caused by schistosomes reveals the crucial role ofconnective tissue growth factor in rapid progression to severe hepatic fibrosis. Pathol Biol(Paris). 2013 Jan;61(1):3-10.
21. Dessein AJ, Hillaire D, Elwali NE, Marquet S, Mohamed-Ali Q, Mirghani A, Henri S,
Abdelhameed AA, Saeed OK, Magzoub MM, Abel L. Severe hepatic fibrosis in Schistosomamansoni infection is controlled by a major locus that is closely linked to the interferon-gammareceptor gene. Am J Hum Genet. 1999 Sep;65(3):709-21.

121
22. Sertorio M1, Hou X, Carmo RF, Dessein H, Cabantous S, Abdelwahed M, Romano A,
Albuquerque F, Vasconcelos L, Carmo T, Li J, Varoquaux A, Arnaud V, Oliveira P, Hamdoun A, HeH, Adbelmaboud S, Mergani A, Zhou J, Monis A, Pereira LB, Halfon P, Bourlière M, Parana R, DosReis M, Gonnelli D, Moura P, Elwali NE, Argiro L, Li Y, Dessein A. IL-22 and IL-22 bindingprotein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosomeinfections. Hepatology. 2015 Apr;61(4):1321-31.
23. Landén NX, Li D, Ståhle M. Transition from inflammation to proliferation: a critical step
during wound healing. Cell Mol Life Sci. 2016 Oct;73(20):3861-85. doi:10.1007/s00018-016-2268-0. Epub 2016 May 14.
24. Lazarus GS, Cooper DM, Knighton DR, Margolis DJ, Percoraro RE, Rodeheaver G, et al.
Definitions and guidelines for assessment of wounds and evaluation of healing. Wound RepairRegen. Arch Dermatol. 1994 Apr;130(4):489-93.
25. Clark RA f. Fibrin and Wound Healing. Ann N Y Acad Sci. 2001 Jun 1;936(1):355–67.
26. Lawrence WT, Diegelmann RF. Growth factors in wound healing. Clin Dermatol. 1994 Jan
1;12(1):157–69.
27. Kim WJ, Gittes GK, Longaker MT. Signal transduction in wound pharmacology. Arch
Pharm Res. 1998 Oct;21(5):487–95.
28. Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001 Apr
1;69(4):513–21.
29. Hart J. Inflammation 1: its role in the healing of acute wounds. J Wound Care. 2002 Jun
1;11(6):205–9.
30. Sylvia C j. The role of neutrophil apoptosis in influencing tissue repair. J Wound Care. 2003
Jan 1;12(1):13–6.
31. Ross R.Wound healing. Sci Am. 1969 Jun;220(6):40–50.
32. Tonnesen MG, Feng X, Clark RAF. Angiogenesis in Wound Healing. J Investig Dermatol
Symp Proc. 2000 Dec;5(1):40–6.
33. Battegay EJ. Angiogenesis: mechanistic insights, neovascular diseases, and therapeutic
prospects. J Mol Med Berl Ger. 1995 Jul;73(7):333–46.
34. Stein C, Küchler S. Targeting inflammation and wound healing by opioids. Trends

122
Pharmacol Sci. 2013 Jun;34(6):303-12.
35. Prockop DJ, Kivirikko KI. Collagens: Molecular Biology, Diseases, and Potentials for
Therapy. Annu Rev Biochem. 1995;64(1):403–34.
36. Blumenkrantz N, Assad R, Peterkofsky B. Characterization of collagen hydroxylysyl
glycosyltransferases as mainly intramembranous microsomal enzymes. J Biol Chem. 1984 Jan25;259(2):854–9.
37. nan TP. Keloid Scarring: Understanding the Genetic Basis, Advances, and Prospects. Arch
Plast Surg. 2012 May;39(3):184–9.
38. Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: The paradigm of skin
fibrosis — Pathomechanisms and treatment. Matrix Biol. 2016 Apr;51:37–46.
39. Rahban SR, Garner WL. Fibroproliferative scars. Clin Plast Surg. 2003 Jan;30(1):77–89.
40. Jesse M. Civan, MD, Thomas Jefferson. Fibrose hépatique. Le manuel MSD, 2018.
41. Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and
fibrosis.Matrix Biol. 2015 May-Jul;44-46:147-56
42. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development,
regeneration, and cancer. J Clin Invest. 2013 May;123(5):1902-10.
43. Moreira RK. Hepatic Stellate Cells and Liver Fibrosis. Arch Pathol Lab Med. 2007
Nov;131(11):1728-34.
44. Knittel T1, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of
matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells ofrat liver: Regulation by TNF-alpha and TGF-beta1. J Hepatol. 1999 Jan;30(1):48-60.
45. Oikawa H, Maesawa C, Tatemichi Y, Nishinari Y, Nishiya M, Mizugai H, Ikeda A, Oikawa K,
Takikawa Y, Masuda T. A disintegrin and metalloproteinase 17 (ADAM17) mediates epidermalgrowth factor receptor transactivation by angiotensin II on hepatic stellate cells. Life Sci. 2014Mar 3;97(2):137-44.
46. Scott L. Friedman. Macrophages– the double-edged sword of hepatic fibrosis. The Journal
of Clinical Investigation, 2005.
47. Keiko Iwaisako, Chunyan Jiang, Mingjun Zhang, Min Cong, Thomas Joseph Moore-Morris,
Tae Jun Park, Xiao Liu, Jun Xu, Ping Wang, Yong-Han Paik, Fanli Meng, Masataka Asagiri, Lynne

123
A. Murray, Alan F. Hofmann, Takashi Iida,j Christopher K. Glass,k David A. Brenner, and TatianaKisseleva. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014Aug 12; 111(32): E3297–E3305.
48. Kiassov AP, Van Eyken P, van Pelt JF, Depla E, Fevery J, Desmet VJ, Yap SH. Desmin
expressing nonhematopoietic liver cells during rat liver development: an immunohistochemicaland morphometric study. Differentiation. 1995 Nov;59(4):253-8.
49. Eng FJ, Friedman SL. Fibrogenesis I. New insights into hepatic stellate cell activation: the
simple becomes complex. Am J Physiol Gastrointest Liver Physiol. 2000 Jul;279(1):G7-G11.
50. Franz CM, Jones GE, Ridley AJ. Cell migration in developpement and disease. Dev Cell.
2002 Feb;2(2):153-8.
51. Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017
Mar;65(3):1039-1043.
52. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M,
Gabbiani G. Recent développements in myofibroblast biology: paradigms for connective tissueremodeling. Am J Pathol. 2012 Apr;180(4):1340-55.
53. Baarsma HA1, Spanjer AI, Haitsma G, Engelbertink LH, Meurs H, Jonker MR, Timens W,
Postma DS, Kerstjens HA, Gosens R. Activation of Wnt / Beta caténin signaling in pulmonaryfibroblast by TGF beta is increasing chronic obstructive pulmonary disease. PLoS One.2011;6(9):e25450.
54. Jumaat Mohd. Yussof Shah, Effat Omar, Dinker R. Pai, and Suneet Sood. Cellular events and
biomarkers of wound healing, Indian J Plast Surg. 2012 May-Aug; 45(2): 220–228.
55. Extrait de http://planete.gaia.free.fr/animal/homme/corps/epiderme.peau.html#89
56. Chin GC, Diegelmann RF, Schultz GS. Cellular and molecular regulation of wound healing.
Taylor & Francis Group; 2005. pp. 17–37.
57. Michael Zeisberg and Raghu Kalluri, Cellular Mechanisms of Tissue Fibrosis. 1. Common and
organ-specific mechanisms associated with tissue fibrosis, Am J Physiol Cell Physiol. 2013 Feb 1;304(3): C216–C225.
58. Wozney JL, Antonarakis ES. Growth factor and signaling pathways and their relevance to
prostate cancer therapeutics. Cancer Metastasis Rev. 2014 Sep;33(0):581.

124
59. Witsch E, Sela M, Yarden Y. Roles for Growth Factors in Cancer Progression. Physiol
Bethesda Md. 2010 Apr;25(2):85.
60. William E. Lawson , Vasiliy V. Polosukhin , Ornella Zoia , Georgios T. Stathopoulos , Wei
Han , David Plieth , James E. Loyd , Eric G. Neilson , and Timothy S. Blackwell, Characterizationof Fibroblast-specific Protein 1 in Pulmonary Fibrosis, American Journal of Respiratory andCritical Care Medicine, 2005
61. Pilcher BK, Wang M, Qin X-J, Parks WC, Senior RM, Welgus HG. Role of Matrix
Metalloproteinases and Their Inhibition in Cutaneous Wound Healing and Allergic ContactHypersensitivity. Ann N Y Acad Sci. 1999 Jun 1;878(1):12–24.
62. Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis.
Dis Model Mech. 2014 Feb;7(2):193.
63. Gill SE, Parks WC.Metalloproteinases and Their Inhibitors: Regulators of Wound Healing.
Int J Biochem Cell Biol. 2008;40(6-7):1334.
64. Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol.
2015 May;44–46:113–21.
65. Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two
vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated byhypoxia. J Biol Chem. 1997 Sep 19;272(38):23659–67.
66. ZHE ZHANG, FANGFEI NIE, XINLEI CHEN, ZELIAN QIN, CHUNFU KANG, BIN CHEN,
JIANXUN MA, BOLIN PAN, and YONGGUANG MA, Upregulated periostin promotesangiogenesis in keloids through activation of the ERK 1/2 and focal adhesion kinase pathways,as well as the upregulated expression of VEGF and angiopoietin-1, Mol Med Rep. 2015 Feb;11(2): 857–864.
67. Gülsüm Özlem Elpek, Angiogenesis and liver fibrosis, World J Hepatol. 2015 Mar 27; 7(3):
377–391
68. Gulcen Yeldag, Alistair Rice and Armando del Río Hernández, Chemoresistance and the
Self-Maintaining Tumor Microenvironment, Cancers 2018
69. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA
Cancer J Clin. 2011 Mar-Apr;61(2):69-90.

125
70. Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis.
Hepatology. 2012 Aug;56(2):769-7.
71. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker
A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R.Endothelial-to-mesenchymal transition contributes to cardicac fibrosis. Nat Med. 2007Aug;13(8):952-61.
72. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7
counteracts TGFBeta1 induced epithelial-to-mesenchymal transition and reverses chronicrenal enjury. Nat Med. 2003 Jul;9(7):964-8.
73. Birukawa NK, Murase K, Sato Y, Kosaka A, Yoneda A, Nishita H, Fujita R, Nishimura M,
Ninomiya T, Kajiwara K, Miyazaki M, Nakashima Y, Ota S, Murakami Y, Tanaka Y, Minomi K,Tamura Y, Niitsu Y. Activated hepatic stellate cells are dependent on self-collagen, cleaved bymembrane type 1 matrix metalloproteinase for their growth. J Biol Chem. 2014 Jul18;289(29):20209-21.
74. Shaffer JJ, Taylor SC, Cook-Bolden F. Keloidal scars: a review with a critical look at
therapeutic options. J Am Acad Dermatol. 2002 Feb;46(2 Suppl Understanding):S63–97.
75. Tiede S, Ernst N, Bayat A, Paus R, Tronnier V, Zechel C. Basic fibroblast growth factor: A
potential new therapeutic tool for the treatment of hypertrophic and keloid scars. Ann Anat -Anat Anz. 2009 Jan;191(1):33–44.
76. Hsu Y-C, Chen M-J, Yu Y-M, Ko S-Y, Chang C-C. Suppression of TGF-β1/SMAD pathway
and extracellular matrix production in primary keloid fibroblasts by curcuminoids: itspotential therapeutic use in the chemoprevention of keloid. Arch Dermatol Res. 2010 Dec1;302(10):717–24.
77. Wolfram D, Tzankov A, Pülzl P, Piza-Katzer H. Hypertrophic Scars and Keloids—A Review of
Their Pathophysiology, Risk Factors, and Therapeutic Management. Dermatol Surg. 2009 Feb1;35(2):171–81.
78. Diegelmann RF, Cohen IK, McCoy BJ. Growth kinetics and collagen synthesis of normal
skin, normal scar and keloid fibroblasts in vitro. J Cell Physiol. 1979 Feb;98(2):341–6.
79. Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic Scarring and
Keloids: Pathomechanisms and Current and Emerging Treatment Strategies. Mol Med. 2011Jan-Feb;17(1-2):113-25.

126
80. Ehrlich HP, Desmoulière A, Diegelmann RF, Cohen IK, Compton CC, Garner WL, et al.
Morphological and immunochemical differences between keloid and hypertrophic scar. Am JPathol. 1994 Jul;145(1):105.
81. Wu W-S, Wang F-S, Yang KD, Huang C-C, Kuo Y-R. Dexamethasone Induction of Keloid
Regression through Effective Suppression of VEGF Expression and Keloid FibroblastProliferation. J Invest Dermatol. 2006 Mar 30;126(6):1264–71.
82. Szulgit G, Rudolph R, Wandel A, Tenenhaus M, Panos R, Gardner H. Alterations in
Fibroblast α1β1 Integrin Collagen Receptor Expression in Keloids and Hypertrophic Scars. JInvest Dermatol. 2002 Mar;118(3):409–15.
83. Dienus K, Bayat A, Gilmore BF, Seifert O. Increased expression of fibroblast activation
protein-alpha in keloid fibroblasts: implications for development of a novel treatment option.Arch Dermatol Res. 2010 Dec 1;302(10):725–31.
84. Ala-Kokko L, Rintala A, Savolainen E-R. Collagen Gene Expression in Keloids: Analysis of
Collagen Metabolism and Type I. III, IV, and V Procollagen mRNAs in Keloid Tissue andKeloid Fibroblast Cultures. J Invest Dermatol. 1987 Sep;89(3):238–44.
85. Li J, Cao J, Li M, Yu Y, Yang Y, Xiao X, et al. Collagen triple helix repeat containing-1
inhibits transforming growth factor-β1-induced collagen type I expression in keloid. Br JDermatol. 2011 May 1;164(5):1030–6.
86. Russell SB, Trupin JS, Myers JC, Broquist AH, Smith JC, Myles ME, et al. Differential
glucocorticoid regulation of collagen mRNAs in human dermal fibroblasts. Keloid-derived andfetal fibroblasts are refractory to down-regulation. J Biol Chem. 1989 Aug 15;264(23):13730–5.
87. Chen J, Zhao S, Liu Y, Cen Y, Nicolas C. Effect of captopril on collagen metabolisms in
keloid fibroblast cells. ANZ J Surg. 2014 Jun 1;n/a – n/a.
88. Huang C, Ogawa R. The link between hypertension and pathological scarring: Does
hypertension cause or promote keloid and hypertrophic scar pathogenesis? Wound RepairRegen. 2014 Jul 1;22(4):462–6.
89. Ogawa R, Arima J, Ono S, Hyakusoku H. CASE REPORT Total Management of a Severe
Case of Systemic Keloids Associated With High Blood Pressure (Hypertension): ClinicalSymptoms of Keloids May Be Aggravated by Hypertension. Eplasty [Internet]. 2013 Jun 3 [cited2014 Jul 24];13

127
90. Alonso PE, Rioja LF, Pera C. Keloids: A viral hypothesis. Med Hypotheses.
2008;70(1):156–66.
91. Seifert O, Mrowietz U. Keloid scarring: bench and bedside. Arch Dermatol Res. 2009
Apr;301(4):259–72.
92. LeFlore IC. Misconceptions regarding elective plastic surgery in the black patient. J Natl
Med Assoc. 1980 Oct;72(10):947–8.
93. Smith JC, Boone BE, Opalenik SR, Williams SM, Russell SB. Gene profiling of keloid
fibroblasts shows altered expression in multiple fibrosis-associated pathways. J Invest Dermatol.2008 May;128(5):1298–310.
94. Marneros AG, Norris JEC, Olsen BR, Reichenberger E. Clinical Genetics of Familial Keloids.
Arch Dermatol. 2001 Nov 1;137(11):1429–34.
95. Lam AP, Gottardi CJ. Beta-catenin signaling: a novel mediator of fibrosis and potential
therapeutic target. Curr Opin Rheumatol. 2011 Nov;23(6):562–7.
96. Eke U, Diaz C, Abdullah A. Keloid scars in type VI skin successfully treated with combined
surgery and pulsed dye laser therapy. Br J Dermatol. 2013 Jun 1;168(6):1360–2.
97. Patel PA, Bailey JK, Yakuboff KP. Treatment outcomes for keloid scar management in the
pediatric burn population. Burns J Int Soc Burn Inj. 2012 Aug;38(5):767–71.
98. Van den Broek LJ, Limandjaja GC, Niessen FB, Gibbs S. Human hypertrophic and keloid
scar models: principles, limitations and future challenges from a tissue engineering perspective.Exp Dermatol. 2014 Jun 1;23(6):382–6.
99. Ogawa R. The Most Current Algorithms for the Treatment and Prevention of
Hypertrophic Scars and Keloids. Plast Reconstr Surg. 2010 Feb;125(2):557–68.
100. Smith, Joan C, Braden E Boone, Susan R Opalenik, Scott M Williams, and Shirley B Russell.
Gene Profiling of Keloid Fibroblasts Shows Altered Expression in Multiple Fibrosis-AssociatedPathways. Journal of Investigative Dermatology 128, no. 5 (May 2008): 1298–1310.
101. Tosa, Mamiko, Mohammad Ghazizadeh, Hajime Shimizu, Takashi Hirai, Hiko Hyakusoku,
and Oichi Kawanami. Global Gene Expression Analysis of Keloid Fibroblasts in Response toElectron Beam Irradiation Reveals the Involvement of Interleukin-6 Pathway. Journal ofInvestigative Dermatology 124, no. 4 (April 2005): 704–13.

128
102. Zhang, G., J. Jiang, S. Luo, S. Tang, J. Liang, and P. Yao. “Analyses of CDC2L1 Gene
Mutations in Keloid Tissue.” Clinical and Experimental Dermatology 37, no. 3 (April 1, 2012):277–83.
103. Li, J., J. Cao, M. Li, Y. Yu, Y. Yang, X. Xiao, Z. Wu, L. Wang, Y. Tu, and H. Chen.
“Collagen Triple Helix Repeat Containing-1 Inhibits Transforming Growth Factor-β1-InducedCollagen Type I Expression in Keloid.” British Journal of Dermatology 164, no. 5 (May 1, 2011):1030–36.
104. Tan, E. M., S. Rouda, S. S. Greenbaum, J. H. Moore, Jr, J. W. Fox, 4th, and S. Sollberg.
“Acidic and Basic Fibroblast Growth Factors down-Regulate Collagen Gene Expression inKeloid Fibroblasts.” The American Journal of Pathology 142, no. 2 (February 1993): 463
105. Naim, R., A. Naumann, J. Barnes, A. Sauter, K. Hormann, D. Merkel, W. Aust, T. Braun, and
M. Bloching. Transforming Growth Factor-β1-Antisense Modulates the Expression ofHepatocyte Growth Factor/Scatter Factor in Keloid Fibroblast Cell Culture. Aesthetic PlasticSurgery 32, no. 2 (March 1, 2008): 346–52.
106. Eto, Hitomi, Hirotaka Suga, Noriyuki Aoi, Harunosuke Kato, Kentaro Doi, Shinichiro Kuno,
Yasuhiko Tabata, and Kotaro Yoshimura. “Therapeutic Potential of Fibroblast Growth Factor-2for Hypertrophic Scars: Upregulation of MMP-1 and HGF Expression.” LaboratoryInvestigation 92, no. 2 (February 2012): 214–23.
107. Kashiyama, Kazuya, Norisato Mitsutake, Michiko Matsuse, Tomoo Ogi, Vladimir A. Saenko,
Kenta Ujifuku, Atsushi Utani, Akiyoshi Hirano, and Shunichi Yamashita. “miR-196aDownregulation Increases the Expression of Type I and III Collagens in Keloid Fibroblasts.”Journal of Investigative Dermatology 132, no. 6 (June 2012): 1597–1604.
108. Phan, Toan-Thang, Ivor Jiun Lim, Boon Huat Bay, Robert Qi, Michael Thornton Longaker,
Seng-Teik Lee, and Hung Huynh. “Role of IGF System of Mitogens in the Induction ofFibroblast Proliferation by Keloid-Derived Keratinocytes in Vitro.” American Journal ofPhysiology - Cell Physiology 284, no. 4 (April 1, 2003): C860–C869.
109. Xue, Hui, Robert L. McCauley, and Wenru Zhang. “Elevated Interleukin-6 Expression in
Keloid Fibroblasts.” Journal of Surgical Research 89, no. 1 (March 2000): 74–77.
110. Ghazizadeh, Mohammad, Mamiko Tosa, Hajime Shimizu, Hiko Hyakusoku, and Oichi
Kawanami. “Functional Implications of the IL-6 Signaling Pathway in Keloid Pathogenesis.”Journal of Investigative Dermatology 127, no. 1 (October 5, 2006): 98–105.

129
111. Heitzer, Ellen, Hannes Seidl, Isabella Bambach, Ulrike Schmidbauer, Lorenzo Cerroni, and
Peter Wolf. “Infrequent p53 Gene Mutation but UV Gradient-like p53 Protein Positivity inKeloids.” Experimental Dermatology 21, no. 4 (April 1, 2012): 277–80.
112. Wu, Wen-Sheng, Feng-Sheng Wang, Kuender D. Yang, Chao-Cheng Huang, and Yur-Ren
Kuo. “Dexamethasone Induction of Keloid Regression through Effective Suppression of VEGFExpression and Keloid Fibroblast Proliferation.” Journal of Investigative Dermatology 126, no. 6(March 30, 2006): 1264–71.
113. Fujiwara, Masao, Yasuteru Muragaki, and Akira Ooshima. Upregulation of Transforming
Growth Factor-β1 and Vascular Endothelial Growth Factor in Cultured Keloid Fibroblasts:Relevance to Angiogenic Activity. Archives of Dermatological Research 297, no. 4 (October 1,2005): 161–69.
114. Rognoni, Emanuel, Moritz Widmaier, Madis Jakobson, Raphael Ruppert, Siegfried Ussar,
Despoina Katsougkri, Ralph T. Böttcher, et al. “Kindlin-1 Controls Wnt and TGF-Β Availabilityto Regulate Cutaneous Stem Cell Proliferation.” Nature Medicine 20, no. 4 (April 2014): 350–59.doi:10.1038/nm.3490.
115. Wang, Zimin, Zhongyu Gao, Yi Shi, Yi Sun, Zihao Lin, Hua Jiang, Tiesheng Hou, et al.
“Inhibition of Smad3 Expression Decreases Collagen Synthesis in Keloid Disease Fibroblasts.”Journal of Plastic, Reconstructive & Aesthetic Surgery 60, no. 11 (November 2007): 1193–99.
116. Ogawa, Rei, Atsushi Watanabe, Banyar Than Naing, Motoko Sasaki, Atsushi Fujita, Satoshi
Akaishi, Hiko Hyakusoku, and Takashi Shimada. “Associations between Keloid Severity andSingle-Nucleotide Polymorphisms: Importance of rs8032158 as a Biomarker of KeloidSeverity.” Journal of Investigative Dermatology 134, no. 7 (July 2014): 2041–43.
117. A Linjawi S, E Tork S, M Shaibah R. Genetic association of the COL1A1 gene promoter
-1997 G/T (rs1107946) andSp1 +1245 G/T (rs1800012) polymorphisms and keloid scars in aJeddah population. Turk J Med Sci. 2016 Feb 17;46(2):414-23.
118. Kulawczuk P, Czapla N, Bińczak-Kuleta A, Safranow K, Jaworska-Kulawczuk A, Gajewska
D, Agata K, Brykczyński M, Bargiel P. Genetic basis of keloid formation in wounds after cardiacsurgery. Kardiochir Torakochirurgia Pol. 2014 Sep;11(3):273-7. doi: 10.5114/kitp.2014.45676.
119. Staal FJ, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is
spreading its wings. Nat Rev Immunol. 2008 Aug;8(8):581-93.

130
120. T Zhan, N Rindtorff and M Boutros. Wnt signaling in cancer. Oncogene. 2017 Mar; 36(11):
1461–1473.
121. G Corda and A Sala. Non-canonical WNT/PCP signalling in cancer: Fzd6 takes centre
stage. Oncogenesis. 2017 Jul; 6(7): e364.
122. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012
Dec;13(12):767-79.
123. Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, Zhang F, Mohammed MK, Haydon RC,
Luu HH, Bi Y, He TC. The evolving roles of canonical WNT signaling in stem cells andtumorigenesis: Implications in targeted cancer therapies. Lab Invest. 2016 Feb;96(2):116-36.
124. Sergei Y. Sokol. Spatial and temporal aspects of Wnt signaling and planar cell polarity
during vertebrate embryonic development. Semin Cell Dev Biol. Author manuscript; available inPMC 2016 Jun 1.
125. Akira Sato, Hideki Yamamoto, Hiroshi Sakane, Hirofumi Koyama, and Akira Kikuchi.
Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 2010 Jan 6;29(1): 41–54.
126. Mitchell T. Butler and John B. Wallingford1. Planar cell polarity in development and
disease. Nat Rev Mol Cell Biol. Author manuscript; available in PMC 2018 Feb 26.
127. Olga Ossipova, Kyeongmi Kim, Blue B. Lake, Keiji Itoh, Andriani Ioannou, and Sergei Y.
Sokol. Role of Rab11 in planar cell polarity and apical constriction during vertebrate neuraltube closure. Nat Commun. Author manuscript; available in PMC 2014 Nov 13.
128. Reguly T1, Wrana JL. In or out ? The dynamics of Smad nucleocytoplasmic shuttling.
Trends Cell Biol. 2003 May;13(5):216-20.
129. Dong C, Li Z, Alvarez R Jr, Feng XH, Goldschmidt-Clermont PJ. Microtubule binding to
Smads may regulate TGF beta activity.Mol Cell. 2000 Jan;5(1):27-34.
130. Morikawa M, Koinuma D, Miyazono K, Heldin CH. Genome-wide mechanisms of Smad
binding. Oncogene. 2013 Mar 28;32(13):1609-15.
131. Ranganathan P, Agrawal A, Bhushan R, Chavalmane AK, Kalathur RK, Takahashi T,
Kondaiah P. Expression profiling of genes regulated by TGF-beta: Differential regulation innormal and tumour cells. BMC Genomics. 2007 Apr 11;8:98.

131
132. Eun Pyo Hong and Ji Wan Park. Sample Size and Statistical Power Calculation in Genetic
Association Studies. Genomics Inf. 2012 Jun; 10(2): 117–122.
133. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007 Feb 23;128(4):683-92.
134. Jones LR, Young W, Divine G, Datta I, Chen KM, Ozog D, Worsham MJ. Genome-Wide
Scan for Methylation Profiles in Keloids. Dis Markers. 2015;2015:943176.
135. Germana Titonelli Santos; Danilo Moulin Sales; Alberto Ribeiro de Souza Leão; José Eduardo
Mourão Santos; Luciane Aparecida Kopke de Aguiar; Paulo Eugênio Brant; David Carlos Shigueoka;Ramiro Colleoni Neto; Giuseppe D'Ippolito. Reproducibility of ultrasonography in theassessment of periportal fibrosis according to Niamey criteria in patients with schistosomiasismansoni, Radiologia Brasileira 2007.
136. Hong EP, Park JW. Sample Size and Statistical Power Calculation in Genetic Association
Studies. Genomics Inform. 2012 Jun;10(2):117-22.
137. Johnson RC, Nelson GW, Troyer JL, Lautenberger JA, Kessing BD, Winkler CA, O'Brien SJ.
Accounting for multiple comparisons in a genome-wide association study (GWAS). BMCGenomics. 2010 Dec 22;11:724.
138. Meurer WJ, Tolles J. Understanding How Well a Model Predicts Outcomes. JAMA. 2017
Mar 14;317(10):1068-1069.
139. Sandro Sperandei. Understanding logistic regression analysis. Biochem Med (Zagreb). 2014
Feb; 24(1): 12–18.
140. Wenfa Li, Hongzhe Liu, Peng Yang, and Wei Xie. Supporting Regularized Logistic
Regression Privately and Efficiently. PLoS One. 2016; 11(6): e0156479.
141. Carl A. Anderson, Fredrik H Pettersson, Geraldine M Clarke, Lon R Cardon, Andrew P.
Morris, and Krina T. Zondervan. Data quality control in genetic case-control association studies.Nat Protoc. Author manuscript; available in PMC 2011 Mar 1.
142. Anastasia L. Wise, Lin Gyi, and Teri A. Manolio. eXclusion: Toward Integrating the X
Chromosome in Genome-wide Association Analyses. Am J Hum Genet. 2013 May 2; 92(5):643–647.
143. Zheng G, Joo J, Zhang C, Geller NL. Testing association for markers on the X
chromosome. Genet. Genet Epidemiol. 2007 Dec;31(8):834-43.

132
144. Hennig BJ, Frodsham AJ, Hellier S, Knapp S, Yee LJ, Wright M, Zhang L, Thomas HC,
Thursz M, Hill AV. Infuence of IL-10RA and IL-22 polymorphisms on outcome of hepatitis Cvirus infection. Liver Int. 2007 Oct;27(8):1134-43.
145. Acuner-Ozbabacan ES, Engin BH, Guven-Maiorov E, Kuzu G, Muratcioglu S, Baspinar A,
Chen Z, Van Waes C, Gursoy A, Keskin O, Nussinov R. The structural network of Interleukin-10and its implications in inflammation and cancer. BMC Genomics. 2014;15 Suppl 4:S2.
146. Sertorio M, Hou X, Carmo RF, Dessein H, Cabantous S, Abdelwahed M, Romano A,
Albuquerque F, Vasconcelos L, Carmo T, Li J, Varoquaux A, Arnaud V, Oliveira P, Hamdoun A, HeH, Adbelmaboud S, Mergani A, Zhou J, Monis A, Pereira LB, Halfon P, Bourlière M, Parana R, DosReis M, Gonnelli D, Moura P, Elwali NE, Argiro L, Li Y, Dessein A. IL-22 and IL-22 bindingprotein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosomeinfections. Hepatology. 2015 Apr;61(4):1321-31
147. Nakajima S, Tanaka H, Sawada K, Hayashi H, Hasebe T, Abe M, Hasebe C, Fujiya M,
Okumura T. Polymorphism of receptor-type tyrosine-protein phosphatase delta gene in thedevelopment of non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2018 Jan;33(1):283-290.
148. Branch AD, Drye LT, Van Natta ML, Sezgin E, Fishman SL, Dieterich DT, Meinert CL, Jabs
DA. Evaluation of hepatitis C virus as a risk factor for HIV-associated neuroretinal disorder.Clin Infect Dis. 2013 Dec;57(11):1618-25.
149. Fleming NI, Jorissen RN, Mouradov D, Christie M, Sakthianandeswaren A, Palmieri M, Day
F, Li S, Tsui C, Lipton L, Desai J, Jones IT, McLaughlin S, Ward RL, Hawkins NJ, Ruszkiewicz AR,Moore J, Zhu HJ, Mariadason JM, Burgess AW, Busam D, Zhao Q, Strausberg RL, Gibbs P, SieberOM. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013 Jan15;73(2):725-35.
150. Nikolic A, Ristanovic M, Zivaljevic V, Rankov AD, Radojkovic D, Paunovic I. SMAD4 gene
promoter mutations in patients with thyroid tumors. Exp Mol Pathol. 2015 Aug;99(1):100-3. doi:10.1016/j.yexmp.2015.06.005.
151. Moussa MM, Helal NS, Youssef MM. Significance of pSmad2/3 and Smad4 in hepatitis C
virus-related liver fibrosis and hepatocellular carcinoma. APMIS. 2018 Jun;126(6):477-485. doi:10.1111/apm.12844.
152. Yan P, Klingbiel D, Saridaki Z, Ceppa P, Curto M, McKee TA, Roth A, Tejpar S, Delorenzi
M, Bosman FT, Fiocca R. Reduced Expression of SMAD4 Is Associated with Poor Survival inColon Cancer. Clin Cancer Res. 2016 Jun 15;22(12):3037-47.

133
153. Saddoughi SA, Ogretmen B. Diverse Functions of Ceramide in Cancer Cell Death and
Proliferation. Adv Cancer Res. 2013;117:37-58.
154. Kim MH, Ahn HK, Lee EJ, Kim SJ, Kim YR, Park JW, Park WJ. Hepatic inflammatory
cytokine production can be regulated by modulating sphingomyelinase and ceramide synthase6. Int J Mol Med. 2017 Feb;39(2):453-462
155. Uen YH, Fang CL, Lin CC, Hseu YC, Hung ST, Sun DP, Lin KY. Ceramide synthase 6
predicts the prognosis of human gastric cancer: it functions as an oncoprotein by dysregulatingthe SOCS2/JAK2/STAT3 pathway. Mol Carcinog. 2018 Dec;57(12):1675-1689.
156. Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review.
Int J Cancer. 2016 Jun 1;138(11):2570-8. doi: 10.1002/ijc.29923
157. Siriwardana NS1, Meyer RD, Panchenko MV. The novel function of JADE1S in cytokinesis
of epithelial cells. Cell Cycle. 2015;14(17):2821-34.
158. Panchenko MV. Structure, function and regulation of Jade Family PHD Finger 1
(JADE1). Gene. 2016 Sep 1;589(1):1-11.
159. Han J, Lachance C, Ricketts MD, McCullough CE, Gerace M, Black BE, Côté J, Marmorstein
R. The scaffolding protein JADE1 physically links the acetyltransferase subunit HBO1 with itshistone H3/H4 substrate. J Biol Chem. 2018 Mar 23;293(12):4498-4509.
160. Han J, Lachance C, Ricketts MD, McCullough CE, Gerace M, Black BE, Côté J, Marmorstein
R. The scaffolding protein JADE1 physically links the acetyltransferase subunit HBO1 with itshistone H3/H4 substrate. J Biol Chem. 2018 Mar 23;293(12):4498-4509.
161. Zhou MI, Foy RL, Chitalia VC, Zhao J, Panchenko MV, Wang H, Cohen HT. Jade-1, a
candidate renal tumor suppressor that promotes apoptosis. Proc Natl Acad Sci U S A. 2005 Aug2;102(31):11035-40
162. Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, Bharti A, Seldin
DC, Lecker SH, Dominguez I, Cohen HT. Jade-1 inhibits Wnt signaling by ubiquitinatingβ-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008Oct;10(10):1208-16.
163. Escudero-Esparza A, Bartoschek M, Gialeli C, Okroj M, Owen S, Jirström K, Orimo A, Jiang
WG, Pietras K, Blom AM. Complement inhibitor CSMD1 acts as tumor suppressor in humanbreast cancer. Oncotarget. 2016 Nov 22;7(47):76920-76933.

134
164. Sun PC1, Uppaluri R, Schmidt AP, Pashia ME, Quant EC, Sunwoo JB, Gollin SM, Scholnick
SB. Transcript map of the 8p23 putative tumor suppressor region. Genomics. 2001Jul;75(1-3):17-25.
165. Kamal M, Holliday DL2, Morrison EE1, Speirs V2, Toomes C1, Bell SM. Loss of CSMD1
expression disrupts mammary duct formation while enhancing proliferation, migration andinvasion. Oncol Rep. 2017 Jul;38(1):283-292.
166. Tang MR1, Wang YX, Guo S, Han SY, Wang D. CSMD1 exhibits antitumor activity in
A375 melanoma cells through activation of the Smad pathway. Apoptosis. 2012Sep;17(9):927-37.
167. Cintia Pereira de Oliveira, Luciano Miller Reis Rodrigues, Maria Vitória Ventura Dias Fregni,
Alberto Gotfryd, Ana Maria Made, and Maria Aparecida da Silva Pinhal. Extracellular matrixremodeling in experimental intervertebral disc degeneration. Acta Ortop Bras. 2013 May-Jun;21(3): 144–149.
168. Fu J, Khaybullin R, Zhang Y, Xia A, Qi X. Gene expression profiling leads to discovery of
correlation of matrix metalloproteinase 11 and heparanase 2 in breast cancer progression.BMC Cancer. 2015 Jun 18;15:473. doi: 10.1186/s12885-015-1410-y.
169. Petrovics G, Li H, Stümpel T, Tan SH, Young D, Katta S, Li Q, Ying K, Klocke B,
Ravindranath L, Kohaar I, Chen Y, Ribli D, Grote K, Zou H, Cheng J, Dalgard CL, Zhang S, CsabaiI, Kagan J, Takeda D, Loda M, Srivastava S, Scherf M, Seifert M, Gaiser T, McLeod DG, Szallasi Z,Ebner R, Werner T, Sesterhenn IA, Freedman M, Dobi A, Srivastava S. A novel genomic alterationof LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine.2015 Oct 31;2(12):1957-64.
170. Barøy T, Kresse SH, Skårn M, Stabell M, Castro R, Lauvrak S, Llombart-Bosch A, Myklebost
O, Meza-Zepeda LA. Reexpression of LSAMP inhibits tumor growth in a preclinicalosteosarcoma model. Mol Cancer. 2014 Apr 28;13:93.
171. Coccaro N, Zagaria A, Tota G, Anelli L, Orsini P, Casieri P, Cellamare A, Minervini A,
Impera L, Minervini CF, Brunetti C, Mestice A, Carluccio P, Cumbo C, Specchia G, Albano F.Overexpression of the LSAMP and TUSC7 genes in acute myeloid leukemia followingmicrodeletion/duplication of chromosome 3. Cancer Genet. 2015 Oct;208(10):517-22.
172. Reddy SS, Connor TE, Weeber EJ, Rebeck W. Similarities and differences in structure,
expression, and functions of VLDLR and ApoER2. Mol Neurodegener. 2011 May 9;6:30.

135
173. Rice DS, Curran T. Role of the Reelin signaling pathway in central nervous system
development. Annu Rev Neurosci. 2001;24:1005-39.
174. Khialeeva E, Chou JW, Allen DE, Chiu AM, Bensinger SJ, Carpenter EM. Reelin Deficiency
Delays Mammary Tumor Growth and Metastatic Progression. J Mammary Gland Biol Neoplasia.2017 Mar;22(1):59-69
175. Okamura Y, Nomoto S, Kanda M, Hayashi M, Nishikawa Y, Fujii T, Sugimoto H, Takeda S,
Nakao A. Reduced Expression of Reelin (RELN) Gene Is Associated With High RecurrenceRate of Hepatocellular Carcinoma. Ann Surg Oncol. 2011 Feb;18(2):572-9.
176. Ding Y, Huang L, Xian X, Yuhanna IS, Wasser CR, Frotscher M, Mineo C, Shaul PW, Herz J.
Loss of Reelin protects against atherosclerosis by reducing leukocyte–endothelial cell adhesionand lesion macrophage accumulation. Sci Signal. 2016 Mar 15;9(419):ra29.
177. Micera A, Balzamino BO, Biamonte F, Esposito G, Marino R, Fanelli F, Keller F. Current
progress of Reelin in development, inflammation and tissue remodeling: from nervous to visualsystems. Curr Mol Med. 2016 Aug 5.
178. Kobold D, Grundmann A, Piscaglia F, Eisenbach C, Neubauer K, Steffgen J, Ramadori G,
Knittel T. Expression of reelin in hepatic stellate cells and during hepatic tissue repair: a novelmarker for the differentiation of HSC from other liver myofibroblast. J Hepatol. 2002May;36(5):607-13.
179. Botella-Lopez A, de Madaria E, Jover R, Bataller R, Sancho-Bru P, Candela A, Compañ A,
Pérez-Mateo M, Martinez S, Sáez-Valero J. Reelin is overexpressed in the liver and plasma of bileduct ligated rats and its levels and glycosylation are altered in plasma of humans with cirrhosis.Int J Biochem Cell Biol. 2008;40(4):766-75.
180. Carotti S, Perrone G, Amato M, Vespasiani Gentilucci U, Righi D, Francesconi M, Pellegrini
C, Zalfa F, Zingariello M, Picardi A, Onetti Muda A, Morini S. Reelin expression in human liverof patients with chronic hepatitis C infection. Eur J Histochem. 2017 Mar 17;61(1):2745. doi:10.4081/ejh.2017.2745.
181. Ogawa R, Akaishi S. Endothelial dysfunction may play a key role in keloid and
hypertrophic scar pathogenesis - Keloids and hypertrophic scars may be vascular disorders.Med Hypotheses. 2016 Nov;96:51-60
182. Sheng Q, Vickers K, Zhao S, Wang J, Samuels DC, Koues O, Shyr Y, Guo Y.
Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief Funct

136
Genomics. 2017 Jul 1;16(4):194-204.
183. Satish L, Babu M, Tran KT, Hebda PA, Wells A. Keloid fibroblast responsiveness to
epidermal growth factor and activation of downstream intracellular signaling pathways.Wound Repair Regen. 2004 Mar-Apr;12(2):183-92.
184. Abad C, Tan YV. Immunomodulatory Roles of PACAP and VIP: Lessons from Knockout
Mice. J Mol Neurosci. 2018 Sep;66(1):102-113.
185. Liu LJ, Xie SX, Chen YT, Xue JL, Zhang CJ, Zhu F. Aberrant regulation of Wnt signaling
in hepatocellular carcinoma. World J Gastroenterol. 2016 Sep 7;22(33):7486-99. Front Biosci(Landmark Ed). 2014 Jan 1;19:379-407.
186. Wang Y, Li YP, Paulson C, Shao JZ, Zhang X, Wu M, Chen W. Wnt and the Wnt signaling
pathway in bone development and disease. Front Biosci (Landmark Ed). 2014 Jan 1;19:379-407.
187. Chavali M, Klingener M, Kokkosis AG, Garkun Y, Felong S, Maffei A, Aguirre A.
Non-canonical Wnt signaling regulates neural stem cell quiescence during homeostasis andafter demyelination. Nat Commun. 2018 Jan 2;9(1):36
188. Babayeva S, Rocque B, Aoudjit L, Zilber Y, Li J, Baldwin C, Kawachi H, Takano T, Torban
E. Planar Cell Polarity Pathway Regulates Nephrin Endocytosis in Developing Podocytes. JBiol Chem. 2013 Aug 16;288(33):24035-48.
189. Ossipova O, Kim K, Lake BB, Itoh K, Ioannou A, Sokol SY. Role of Rab11 in planar cell
polarity and apical constriction during vertebrate neural tube closure. Nat Commun. 2014 May13;5:3734
190. Beljaars L, Daliri S, Dijkhuizen C, Poelstra K, Gosens R. WNT-5A regulates TGF-β-related
activities in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2017 Mar 1;312(3):G219-G227.
191. Vallée A, Lecarpentier Y, Guillevin R, Vallée JN. Interactions between TGF-β1, canonical
WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget. 2017 Sep23;8(52):90579-90604.
192. Sun Q, Guo S, Wang CC, Sun X, Wang D, Xu N, Jin SF, Li KZ. Cross-talk between
TGF-β/Smad pathway and Wnt/β-catenin pathway in pathological scar formation. Int J ClinExp Pathol. 2015 Jun 1;8(6):7631-9. eCollection 2015.
193. Sato M1. Upregulation of the Wnt/beta-catenin pathway induced by transforming
growth factor-beta in hypertrophic scars and keloids. Acta Derm Venereol. 2006;86(4):300-7.

137
194. Dessein A, Chevillard C, Arnaud V, Hou X, Hamdoun AA, Dessein H, He H, Abdelmaboud
SA, Luo X, Li J, Varoquaux A, Mergani A, Abdelwahed M, Zhou J, Monis A, Pitta MG,Gasmelseed N, Cabantous S, Zhao Y, Prata A, Brandt C, Elwali NE, Argiro L, Li Y. Variants ofCTGF are associated with hepatic fibrosis in Chinese, Sudanese, and Brazilians infected withSchistosomes. J Exp Med. 2009 Oct 26;206(11):2321-8.
195. Katoh M, Katoh M. Molecular genetics and targeted therapy of WNT-related human
diseases (Review). Int J Mol Med. 2017 Sep;40(3):587-606.
196. Atala A, Irvine DJ, Moses M, Shaunak S.Wound Healing Versus Regeneration: Role of the
Tissue Environment in Regenerative Medicine. MRS Bull. 2010 Aug 1;35(8).
197. Lv D, Zhou L, Zheng X, Hu Y. Sustained release of collagen VI potentiates sciatic nerve
regeneration by modulating macrophage phenotype. Eur J Neurosci. 2017May;45(10):1258-1267
198. Wen-Ce Zhou, Quan-Bao Zhang, and Liang Qiao. Pathogenesis of liver cirrhosis. World J
Gastroenterol. 2014 Jun 21; 20(23): 7312–7324.
199. https://www.ncbi.nlm.nih.gov/
200. http://grch37.ensembl.org/index.html
201. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat
Rev Genet. 2009 Jan;10(1):57-63.

138
Résumé Français
Les fibroses anormales sont responsables de plus de 40% des décès pour raison médicale ; elles se développent
suite à une inflammation chronique et causent souvent une perte de fonction de l’organe affecté. Les fibroseshépatiques causées par les schistosomes et par le virus HCV sont en grande partie déterminées par la génétique du
malade. Notre thèse a consisté à poursuivre le travail de caractérisation du déterminisme génétique des fibroseshépatiques et cutanées afin de contribuer à la mise au point de tests prédictifs des fibroses sévères. La premièrepartie de notre thèse, est l’étude informatique et statistique des données de génotypage GWAS de Brésiliens qui
présentent une fibrose hépatique bilharzienne grave sur plus de 2,5 millions de SNPs. 180 SNPs qui montraient uneassociation suggestive avec les fibroses graves ont été sélectionnés, dont certains affectent les gènes des voies Wnt.
Ces SNPs ont été testés sur une cohorte de 460 pêcheurs ougandais exposés à S.mansoni et nous avons confirmél’association avec la fibrose de 4 SNPs. La deuxième partie de notre thèse est l’analyse génomique (transcriptome
et génétique) des mécanismes responsables des fibroses anormales de la peau de sujets affectés par des fibroseschéloïdes. Nous avons effectué une analyse différentielle d’expression par séquençage RNASeq entre 20 tissuschéloïdes, 7 tissus sains et 7 tissus affectés par des cicatrices hypertrophiques. Cette analyse montre que le
développement des chéloïdes est la conséquence d’une stimulation anormale des voies de la cicatrisation. Notreétude suggèrent que cette forte stimulation cicatricielle est en partie due à l’activation de la voie Wnt βcatenin et
Wnt PCP. Pour conforter cette proposition, nous avons effectué une analyse génétique de la voie Wnt dans deuxcohortes indépendantes. L’analyse statistique montre que des SNPs dans 6 gènes de la voie Wnt βcatenincontribuent au développement des fibroses chéloïdes. L’ensemble de ces résultats est utilisé pour améliorer les tests
génétiques développés par notre groupe Bilhi Genetics dans le but de prédire les cicatrisations cutanées anormaleset les fibroses sévères du foie.
Résumé Anglais
Abnormal fibrosis is responsible for more than 40% of medical deaths. They develop as a result of chronicinflammation and often cause a loss of function of the affected organ. Hepatic fibroses caused by schistosomes and
HCV virus are largely determined by the genetic background of the patient. Our thesis consisted of continuing thework of characterizing the genetic determinism of liver and skin fibrosis in order to contribute to the developmentof predictive tests for severe fibrosis. The first part of our thesis is the computer and statistical study of GWAS
genotyping data of Brazilians who have severe bilharzeal liver fibrosis on more than 2.5 million SNPs. 180 SNPsthat showed suggestive association with severe fibrosis were selected, some of which affect the Wnt pathway.
These SNPs were then tested on a cohort of 460 Ugandan fishers exposed to S.mansoni and the results confirmedthe association of 4 SNPs with fibrosis. The second part of our thesis is the genomic analysis (transcriptome and
genetics) of the mechanisms responsible for the abnormal skin fibrosis with subjects affected by keloid scars. Weperformed an analysis of genes (RNASeq) expressed differently between 20 keloids, 7 healthy tissues and 7 tissuesaffected by hypertrophic scars. This analysis shows that the development of keloids is the consequence of an
abnormal stimulation of cicatrization pathways with strong activation of the Wnt βcatenin and Wnt PCP pathway.To support this proposal, we performed a genetic analysis of the Wnt pathway in two independant cohorts. The
statistical analysis of the results shows that polymorphisms in 6 genes of the Wnt βcatenin pathway contribute tothe development of keloid fibrosis. All of these results are used to improve the genetic tests developed by our groupBilhi Genetics in order to predict abnormal skin scarring and severe fibrosis of the liver.





























