Embed Size (px)
Citation preview

Université Pierre et Marie Curie
École Doctorale Chimie physique et chimie analytique de Paris Centre
T H È S Epour obtenir le titre de
Docteur en Sciences de l’Université Pierre et Marie Curie
Spécialité : Biophysique
Par
Lucile SENICOURT
Études des protéines membranairesTSPO
Thèse dirigée par Jean-Jacques Lacapèresoutenance prévue le 27 septembre 2016
Jury :
Rapporteurs : Laura Baciou - Directrice de Recherche CNRSChristina Sizun - Chargée de Recherche CNRS
Examinateurs : Mariano Anibal Ostuni - Professeur Paris DiderotGermain Trugnan - Professeur UPMC
Directeur de thèse : Jean-Jacques Lacapère - Directeur de Recherche CNRS


Remerciements


Table des matières
Abréviations et acronymes 1
1 Les protéines TSPO 71.1 La famille TSPO : des protéines membranaires anciennes et conservées à travers
l’évolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.2 La TSPO animale : TranSlocator PrOtein 18 kDa . . . . . . . . . . . . . . . . . 12
1.2.1 Historique et découverte . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121.2.2 Localisation et distribution . . . . . . . . . . . . . . . . . . . . . . . . . . 131.2.3 Information génétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141.2.4 Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 151.2.5 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 241.2.6 Adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 421.2.7 Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
1.3 La TSPO2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 521.3.1 Localisation cellulaire et subcellulaire . . . . . . . . . . . . . . . . . . . . 541.3.2 Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 541.3.3 Fonction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 541.3.4 Structure et adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
1.4 La TSPO végétale : l’AtTSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571.4.1 Structure génétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571.4.2 Expression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571.4.3 Biodégradation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 601.4.4 Localisation de l’AtTSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . 621.4.5 Adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 631.4.6 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 651.4.7 La TSPO chez la mousse Physcomitrella patens . . . . . . . . . . . . . . . 68
1.5 La TSPO bactérienne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 691.5.1 Information génétique et localisation cellulaire . . . . . . . . . . . . . . . 691.5.2 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 691.5.3 Structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
1.6 Objectifs de la thèse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
2 Matériels et Méthodes 852.1 Expression et Production des TSPO . . . . . . . . . . . . . . . . . . . . . . . . . 86
2.1.1 Les souches bacteriennes d’Escherichia coli . . . . . . . . . . . . . . . . . 862.1.2 Les vecteurs d’expression . . . . . . . . . . . . . . . . . . . . . . . . . . . 87

iv Table des matières
2.1.3 Les milieux de cultures . . . . . . . . . . . . . . . . . . . . . . . . . . . . 892.1.4 Transformation des bactéries en bactéries compétentes . . . . . . . . . . . 912.1.5 Transgénèse des bactéries . . . . . . . . . . . . . . . . . . . . . . . . . . . 912.1.6 Amplification et purification des plasmides . . . . . . . . . . . . . . . . . 922.1.7 Culture des bactéries . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 952.1.8 Production acellualire ou cell free . . . . . . . . . . . . . . . . . . . . . . . 97
2.2 Purification des TSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1012.2.1 Les détergents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1012.2.2 Extraction des protéines . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1032.2.3 Purification sur Résine Ni-NTA . . . . . . . . . . . . . . . . . . . . . . . . 1042.2.4 Purification du peptide Nter d’AtTSPO . . . . . . . . . . . . . . . . . . . 106
2.3 Caractérisation des TSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1082.3.1 Gel SDS-PAGE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1082.3.2 Western Blot . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1112.3.3 Spectrométrie de masse . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1152.3.4 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1172.3.5 Fluorescence intrinsèque . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1182.3.6 Liaisons des Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1192.3.7 Mesure de la liaison des ligands par séparation des ligands libres et liés
sur Résine Ni-NTA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1192.3.8 Liaison de ligands radioactifs . . . . . . . . . . . . . . . . . . . . . . . . . 1202.3.9 Les lipides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1202.3.10 Reconstitution de la mTSPO1 dans des protéoliposomes . . . . . . . . . . 122
2.4 La RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1252.4.1 Principe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1252.4.2 La relaxation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1262.4.3 Le signal RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1272.4.4 La RMN du liquide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1292.4.5 La RMN du solide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134
Résultats et discussions 149
3 La mTSPO1 1493.1 Production de la mTSPO1 recombinante . . . . . . . . . . . . . . . . . . . . . . . 149
3.1.1 Expression dans E.coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1503.1.2 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
3.2 Caractérisation de la mTSPO1 recombinante en détergent . . . . . . . . . . . . . 1573.2.1 Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1573.2.2 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158

Table des matières v
3.2.3 Repliement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1593.2.4 RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.3 Effets des ligands sur la mTSPO1 recombinante . . . . . . . . . . . . . . . . . . . 1623.3.1 Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1633.3.2 Séparation des ligands libres et liés . . . . . . . . . . . . . . . . . . . . . . 1643.3.3 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1653.3.4 RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1663.3.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
3.4 La mTSPO1 en RMN du Solide . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1723.4.1 Reconstitution de la mTSPO1 dans des liposomes . . . . . . . . . . . . . 1723.4.2 Remplissage du rotor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1773.4.3 Expériences CP-MAS 1D 13C . . . . . . . . . . . . . . . . . . . . . . . . . 1823.4.4 Expériences 13C-13C PDSD . . . . . . . . . . . . . . . . . . . . . . . . . . 1893.4.5 Expériences 2D 15N-13C NCO et NCA . . . . . . . . . . . . . . . . . . . . 1903.4.6 Conclusions, discussions et perspectives . . . . . . . . . . . . . . . . . . . 191
4 La hTSPO2 1954.1 Expression et Production de la hTSPO2 dans E. coli . . . . . . . . . . . . . . . . 195
4.1.1 Intégrité et qualité du plasmide pET15b-hTSPO2 . . . . . . . . . . . . . 1954.1.2 Expression dans les bactéries BL21 (DE3) pLys S . . . . . . . . . . . . . . 1974.1.3 Expression dans les bactéries BL21 (DE3) classiques . . . . . . . . . . . . 2064.1.4 Optimisations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2094.1.5 Conclusions, discussions et perspectives . . . . . . . . . . . . . . . . . . . 209
4.2 Production de la protéine hTSPO2 en système acellulaire ou Cell Free . . . . . . 2134.2.1 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2134.2.2 Caractérisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
4.3 Conclusions et Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 218
5 Le peptide Nter d’AtTSPO 2195.1 Un nouveau rôle pour l’ATtspo ? . . . . . . . . . . . . . . . . . . . . . . . . . . . 2195.2 Production du Nter marqué 15N,13C . . . . . . . . . . . . . . . . . . . . . . . . . 223
5.2.1 Expression dans E. coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2235.2.2 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2235.2.3 Confirmation de la présence du peptide Nter . . . . . . . . . . . . . . . . 228
5.3 Caractérisation du peptide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2295.3.1 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2305.3.2 Résolution de la structure du peptide par RMN . . . . . . . . . . . . . . . 2305.3.3 Interaction du peptide avec le calcium . . . . . . . . . . . . . . . . . . . . 233
5.4 Interaction du peptide avec les lipides . . . . . . . . . . . . . . . . . . . . . . . . 2355.4.1 Bicelles de DMPG/DHPC . . . . . . . . . . . . . . . . . . . . . . . . . . . 235

vi Table des matières
5.4.2 Micelles de PI(4,5)P2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2395.5 Conclusions, perspectives et discussions . . . . . . . . . . . . . . . . . . . . . . . 244
6 Conclusions et Perspectives 253
Annexe 261
A Pourcentages d’identité entre les séquences de différents homologues TSPO261
B Knock-out global, conditionnel et Knock-down 263B.1 Le Knock-out global . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263B.2 Le Knock-out conditionnel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 265B.3 Le Knock-down . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 265
C Les végétaux face au stress abiotiques 267
D L’échange chimique en RMN 271
E Articles 273
Bibliographie 319

Abréviations et acronymes
a.a acide aminéABA Acide AbscissiqueACBD3 Acyl-Coenzyme A Binding Domain containing 3 proteinAC Anti-corpsACN ACétoNitrileADNc ADN complémentaires des ARN messagersADP Adénosine DiPhosphateAG Appareil de GolgiALA Acide δ-aminolévuliniqueAMPc Adénosine MonoPhosphate cycliqueANT Adénine Nucléotide TranslocaseATAD3A ATPase family AAA Domain-containing protein 3ATP Adénosine TriPhosphateBB BioBeadsBchl BactériochlorophylleBCIP BromoChlorylIndoloPhosphateBZ BenzodiazépineCAM ChlorAMphénicolcAMP cyclic Adenosine MonoPhosphateCBR Central Benzodiazepin Receptor - Récepteur Central aux BenzodiazépinesCI Corps d’Inclusioncmc Concentration Micellaire CritiqueCP Cross Polarisation - Polarisation croiséeCRAC Cholesterol Recognition Amino Acid ConsensusCSA Chemical Shift AnisotropyCSD Chemical Shift DeviationCSP Chemical Shift PerturbationCYP11A1 cytochrome P450 side chain cleavage enzymeCyt CytosolDBI Diazepam Binding InhibitorDC Dichroïsme CirculaireDDM n-DoDécyl–D-MaltosideDHPC 1,2-DiHexanoyl-sn-glycéro-3-PhosphoCholineDLS Diffusion Light Scattering - Diffusion de lumièreDMPC 1,2-DiMyristoyl-sn-glycéro-3-PhosphoCholineDMPE 1,2-Dimyristoyl-sn-glycero-3-phosphoethanolamineDMPG 1,2-DiMyristoyl-sn-glycéro-3-[Phospho-rac-(1-Glycérol)]DO Densité OptiqueDPC Dodécyl PhosphoCholineDSS 2,2-DimethylSilapentane-5-Sulfonic acid

2 Abréviations et acronymes
DTT DiThioThréitolECL Enhanced ChemiluminescenceFAO Fatty Acid OxydationFT Facteur de TranscriptionGdmCl chlorure de guanidiniumGH GHost - fantôme de globule rougeGR Globule RougeGST Glutathion-S-TransferaseGTP Guanosine TriphosPhateHDL High Density LipoproteinHMG-CoA Hydroxyméthylglutaryl-CoAHRP HorseRadish Peroxidase - peroxydase de raifortHSQC Heteronuclear Single Quantum CoherenceIAA IodoAcétAmideIgG Immunoglobuline GIMAC Immobilized Metal Affinity ChromatographyIMS InterMembrane Space - l’espace intermembranaire mitochondrialIPTG IsoPropyl–D-Thio-GalactosideKO Knock-OutLCP Lipidic Cubic PhaseLDL Low Density LipoproteinLMPE 1-Myristoyl-2-hydroxy-sn-glycéro-3-PhosphoEthanolamineLPR Rapport Lipide/ProtéineMAS Magic Angle Spinning - Rotation à l’angle magiqueMb MembraneME Microscopie ÉlectroniqueM&M Matériels et MéthodesMME Membrane Mitochondriale ExterneMMI Membrane Mitochondriale InterneMP Membrane PlasmiqueNAD Nicotinamide Adénine DinucléotideNADP Nicotinamide Adénine Dinucléotide PhosphoryléNADPH forme réduite de la NADPNBT Nitro-Blue TetrazoniumNCC Non fluorescent Chlorophyll CataboliteNi-NTA Nickel- NiTriloacetic Acid (Acide NitriloTriAcétique)NOX2 NADPH oxydase 2NTP Nucleoside TriPhosphateODN OctaDécaNeuropeptideP450scc cytochrome P450 side chain cleavage enzymepb paire de basesPBR Peripheral Benzodiazepine Receptor - récepteur périphérique aux benzodiazépinesPC PhosphatidylCholinePE PhosphatidylEthanolamine

Abréviations et acronymes 3
PEG PolyEthylène GlycolPEP PhosphoEnolPyruvatePI PhosphatidylinoditolPIP Phosphatidylinositol PhosphatePITP PhosphatIdylinositol Transfer ProteinPKA-RI PKA-RIα : cAMP-dependent protein kinase regulatory subunit IαPK11195 1-(2-chlorophényl)-N-méthyl-N-(1-méthyl-propyl)-3-isoquinoléinecarboxamidePM Poids MoléculairePMb Protéine MembranairePMPT Pore Mitochondrial de Perméabilité TransitoirePPIX ProtoPorphyrine IXPVC PreVacuolarPVDB PolyVinyliDene FluorideRE Réticulum EndoplasmiqueRMN Résonance Magnétique NucléaireRNC Ribosome Nascent ChainRo5-4864 4-chlorodiazépam ou
7-chloro-5-(4-chlorophényl)-1,3-dihydro-1-méthyl-2H-1,4-benzodiazépin-2-oneROS Reactive Oxygen SpeciesSCM Site de Contact MembranaireSDS Sodium Dodécyl Sulfate ou laurylsulfate de sodiumSDS-PAGE SDS PolyAcrylamide Gel ElectrophoresisSE Souches EmbryonnairesSNC Système Nerveux CentralSP Système PhotosynthétiqueSRP Signal Recognition ParticuleSSB Spinning Side Bands - bandes de rotation latéralesssNMR solid state NMR - RMN à l’état solideSSP Secondary Structure PropensityStAR Steroidogenic Acute Regulatory proteinSU SurnageantTCA TriChloroacEtic AcideTEP Tomographie par Emission de PositronsTEV Tobacco Ech Virus - Virus de la gravure du tabacTGN Trans Golgi Network - Réseau trans-golgienTM TransMembranaire ou Domaine TransMembranaireTSPO TranSlocation PrOtein - protéine de translocationTspO Tryptophan rich sensory prOteinTTN TriakontaTétraNeuropeptideVDAC Voltage Dependent Anionic Channel - canal anionique voltage-dépendantWB Western BlotZPP Protoporphyrine zinc


Introduction


Chapitre1
Les protéines TSPO
Sommaire1.1 La famille TSPO : des protéines membranaires anciennes et conservées
à travers l’évolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.2 La TSPO animale : TranSlocator PrOtein 18 kDa . . . . . . . . . . . . 12
1.2.1 Historique et découverte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
1.2.2 Localisation et distribution . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
1.2.3 Information génétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.2.4 Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
1.2.5 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
1.2.6 Adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
1.2.7 Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
1.3 La TSPO2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 521.3.1 Localisation cellulaire et subcellulaire . . . . . . . . . . . . . . . . . . . . . 54
1.3.2 Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
1.3.3 Fonction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
1.3.4 Structure et adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
1.4 La TSPO végétale : l’AtTSPO . . . . . . . . . . . . . . . . . . . . . . . . . 571.4.1 Structure génétique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
1.4.2 Expression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
1.4.3 Biodégradation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
1.4.4 Localisation de l’AtTSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
1.4.5 Adressage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
1.4.6 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
1.4.7 La TSPO chez la mousse Physcomitrella patens . . . . . . . . . . . . . . . . 68
1.5 La TSPO bactérienne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 691.5.1 Information génétique et localisation cellulaire . . . . . . . . . . . . . . . . 69
1.5.2 Fonctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
1.5.3 Structures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
1.6 Objectifs de la thèse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82
1.1 La famille TSPO : des protéines membranaires anciennes etconservées à travers l’évolution
La TSPO est présente dans presque tous les organismes des 3 grands domaines du vivant :les Bacteria, les Eukarya et les Archaea, comme l’illustre la Fig. 1.1. Les quelques exceptions

8 Chapitre 1. Les protéines TSPO
concernent des levures comme Saccharomyces cerevisiae [Riond et al., 1991] où la TSPO estabsente. Les animaux et plantes possèdent même potentiellement 2 protéines TSPO, dénomméesTSPO1 et TSPO2, voire 3 dans le cas de la mousse [Frank et al., 2007]. Nous reviendrons plustard sur le cas de la TSPO2 et, jusqu’à la Sec. 1.3, TSPO fera référence uniquement à la TSPO1.
Figure 1.1: Arbre phylogénétique de la famille des protéines TSPO au sein des 3domaines du vivant : Bacteria, Archae et Eukarya [Fan et al., 2012].
La Fig. 1.2 montre un alignement de séquences d’acides aminés (a.a) de quelques protéinesTSPO issues de différentes espèces de chaque règne. Le gène Tspo code pour une protéine à5 domaines transmembranaires (TM) en hélice α (Fig. 1.2). De manière générale on constateque les protéines TSPO partagent une homologie de séquence considérable. Le niveau globald’identité entre les TSPO mammalienne et bactérienne est de ∼30% [Yeliseev et al., 1997] et ilest de ∼25% entre les TSPO végétale et mammalienne [Guillaumot et al., 2009a]. Un tableaurécapitulant les pourcentages d’identité entre les séquences de différents homologues TSPO estdonné en Annexe A.
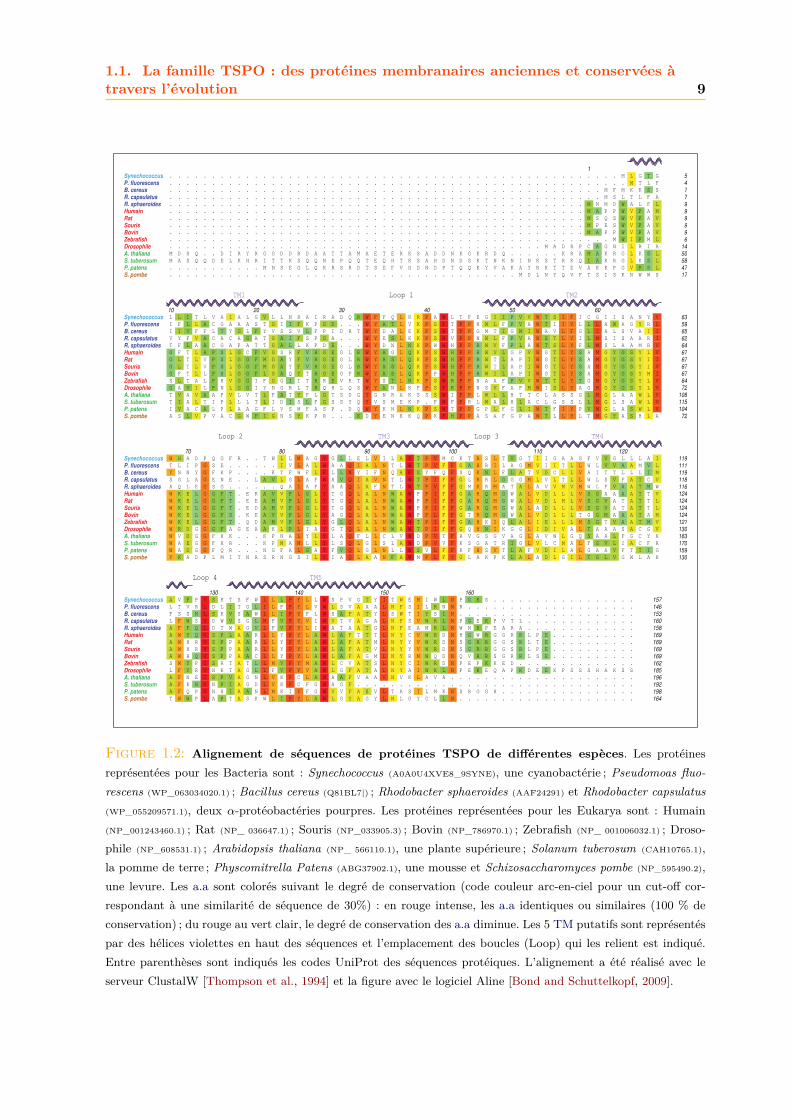
1.1. La famille TSPO : des protéines membranaires anciennes et conservées àtravers l’évolution 9
R. sphaeroides
B. cereusR. capsulatus
HumainRatSourisBovinZebrafishDrosophileA. thaliana
S. pombe
S. tuberosum
P. fluorescens
P. patens
Synechococcus
R. sphaeroides
B. cereusR. capsulatus
HumainRatSourisBovinZebrafishDrosophileA. thaliana
S. pombe
S. tuberosum
P. fluorescens
P. patens
Synechococcus
R. sphaeroides
B. cereusR. capsulatus
HumainRatSourisBovinZebrafishDrosophileA. thaliana
S. pombe
S. tuberosum
P. fluorescens
P. patens
Synechococcus
R. sphaeroides
B. cereusR. capsulatus
HumainRatSourisBovinZebrafishDrosophileA. thaliana
S. pombe
S. tuberosum
P. fluorescens
P. patens
Synechococcus
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M N M D W A L F L 9
T F L A A C G A P A T T G A L L K P D E . . . W Y D N L N K P W W N P P R W V F P L A W T S L Y F L M S L A A M R V 64
A Q L E G S G . . . . . . Q A L A F Y A A Q L A F N T L W T P V F F G M K R M A T A L A V V M V M W L F V A A T M W 116
A F F Q L D T W A G V L F V P Y L I W A T A A T G L N F E A M R L N W N R P E A R A . . . . . . . . . . . . . 158
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M F M K K S S 7
I I V F F L T Y G L F Y V S S V L F P I D R T W Y D A L E K P S W T P P G M T I G M I W A V L F G L I A L S V A I I 65
Y N N Y G F K P . . . . K T F W F L F L L N Y I F N Q A F S Y F Q F S Q K N L F L A T V D C L L V A I T T L L L I M 119
F S S N L S K V S A W L L I P Y F L W S A F A T Y L S W T I Y S I N . . . . . . . . . . . . . . . . . . . . . 153
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M S L T L F A 7
V Y F V A C A C A G A T G A I F S P G A . . . W Y E S L K K P S W V P P N W L F P V A W S T L Y I L M S I S A A R I 62
S G L A G E N E . . L A V L G L A F W A V Q I A V N T L W T P I F F G L R R L G G G M L V L T L L W L S V F A T C V 118
L F W S V D W V S G L M F V P Y V I W V T V A G A L N F S V W R L N P G E K P V T L . . . . . . . . . . . . . 160
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M A P P W V P A M 9
G F T L A P S L G C F V G S R F V H G E G L R W Y A G L Q K P S W H P P H W V L G P V W G T L Y S A M G Y G S Y L V 67
W K E L G G F T . E K A V V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L V D L L L V S G A A A A T T V 124
A W Y Q V S P L A A R L L Y P Y L A W L A F T T T L N Y C V W R D N H G W R G G R R L P E . . . . . . . . . . 169
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M S Q S W V P A V 9
G L T L V P S L G G F M G A Y F V R G E G L R W Y A S L Q K P S W H P P R W T L A P I W G T L Y S A M G Y G S Y I I 67
W K E L G G F T . E E A M V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L V D L M L V S G V A T A T T L 124
A W H R V S P P A A R L L Y P Y L A W L A F A T M L N Y Y V W R D N S G R R G G S R L T E . . . . . . . . . . 169
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M P E S W V P A V 9
G L T L V P S L G G F M G A Y F V R G E G L R W Y A S L Q K P S W H P P R W T L A P I W G T L Y S A M G Y G S Y I V 67
W K E L G G F T . E D A M V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L A D L L L V S G V A T A T T L 124
A W H R V S P P A A R L L Y P Y L A W L A F A T V L N Y Y V W R D N S G R R G G S R L P E . . . . . . . . . . 169
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M A P P W V P A V 9
G F T L L P S L G G F L G A Q Y T R G E G F R W Y A S L Q K P P W H P P R W I L A P I W G T L Y S A M G Y G S Y M I 67
W K E L G G F S . K E A V V P L G L Y A G Q L A L N W A W P P L F F G T R Q M G W A L V D L L L T G G M A A A T A M 124
A W H Q V S P P A A C L L Y P Y L A W L A F A G M L N Y R M W Q D N Q V R R S G R R L S E . . . . . . . . . . 169
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M W I P M L 6
S L T A L P H V G G I F G G I I T R R E V K T W Y S T L N K P S W R P P N A A F P V V W T T L Y T G M G Y G S Y L V 64
W K E L G G F T . Q D A M V P L G L Y G L Q L A L N W A W T P I F F G A H K I Q L A L I E L L L M S G T V A A T M V 121
S W Y P I S R T A T L L M V P Y M A W L C V A T S L N Y C I W R D N P E P K K E D . . . . . . . . . . . . . . 162
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M A D R P C A G N I L R I A 14
G A V I L P N L G G I Y N G R L T R Q H L Q S W Y A N L K F P S F K P P N S V F A P M W I S L Y A G M G Y G S Y L V 72
W R D G G G F A G E A A K L P L I A Y G T Q L A L N W A W T P I F F G Q H N I K G G L I D I V A L T A A A S A C G V 130
L F Y R V N K T A G L L F V P Y V A W L G F A T A L N Y A I W K L N P E K E Q A P K D E E K P S S S H A K S S 185
M D S Q . . D I R Y R G G D D R D A A T T A M A E T E R K S A D D N K G K R D Q . . . . . . K R A M A K R G L K S L 50
T V A V A A P V L V T L F A T Y F L G T S D G Y G N R A K S S S W I P P L W L L H T T C L A S S G L M G L A A W L V 108
W V D G G F H K . . . K P N A L Y L Y L A Q F L L C L V W D P V T F R V G S G V A G L A V W L G Q S A A L F G C Y K 163
A F N E I S P V A G N L V K P C L A W A A F V A A V N V K L A V A . . . . . . . . . . . . . . . . . . . . . . 196
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M D L N Y Q V F T S I S K N W W S 17
A S L V P V A C G W F I G N S Y K P R . . . K D Y E N K K Q P K F H P P A S A F G P A W T L L Y L T M G Y A S H L A 72
Y K A D P L M I T N A S R N G S I L Y I A Q L A A N F A W M P L F Y G L A K P K L A L A D L G I L T G L V G W L A K 130
T W W P L A P T A S K W L I P Y L A W L G Y A G Y L N L G Y C L L N . . . . . . . . . . . . . . . . . . . . . 164
M A S Q Q D E L K H R I T T K S Q Q N E P Q Q T E Q H T K S A H D N D S K T N K N I N K S T R K Q I A K R G L K S L 58
T I A L T I P L L L T L I D I S L F G S S Y Q Y V S M E K P . F W F P R L W A L H L A C L G S S L L M G L S A W L V 115
W A E G G F H R . . . K P M A M L L Y L S Q L G L S L A W D P V V F K S G A T R I G L V L C M A L F G V L I A C F R 170
A F K N V N P I A G D L V K P C F G W A G F . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M T L F 4
I F L L A C G A A A S T G I I F K P G D . . . W Y A T L V K P S F T P P N W L F P V A W T I I Y L L L A W A G Y R L 59
T L I P G S E . . . . . . I V L A L W A A Q I A L N T L W T P V F F G A H R I L A G M V I I T L L W L V V A A M V L 111
L T V R L D L I T G L I L F P Y L V W L S V A A A L N F S I L R N N R . . . . . . . . . . . . . . . . . . . . 146
. . . . . . . . . . . M N S E G L Q K R S R D T S E F V H D N D P T Q Q K Y V A K A Y R K T T E V A K K P G V P S L 47
I V A C A L P L A A G F L V S M F A S P . D Q W Y K N L N K P S W T P P G P L F G L I W T F I Y P V M G L A S W L V 104
W A D G G F Q R . . . N G F A L G A Y F V Q L G L N L L W S V L F F K F H S V T L A F V D I L A L G A A V F T T I G 159
A F Q P V N H I A A N L M K I Y F G W V V F A S V L T A S I L M K N S R G G H . . . . . . . . . . . . . . . . 198
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M L G T G 5
L L I T L V A I A L G V L L N R A I R A D Q R W F F Q L R R P A W L T F E G I I P V V W T S I F I C G I I S A N Y V 63
W H A D P Q G F R . . T W L L M A G Y G L L E L V I L A Y T P V M C K T R S L T V G T I I G A A G F V V G L L L A I 119
A V F P V S K T S F W L L L P F L L W S P V G T Y I T W E M I W L N P G K S . . . . . . . . . . . . . . . . . 157
1
10 20 30 40 50 60
70 80 90 100 110 120
130 140 150 160
TM1 TM2
TM3 TM4
TM5Loop 4
Loop 3Loop 2
Loop 1
Figure 1.2: Alignement de séquences de protéines TSPO de différentes espèces. Les protéinesreprésentées pour les Bacteria sont : Synechococcus (A0A0U4XVE8_9SYNE), une cyanobactérie ; Pseudomoas fluo-rescens (WP_063034020.1) ; Bacillus cereus (Q81BL7|) ; Rhodobacter sphaeroides (AAF24291) et Rhodobacter capsulatus(WP_055209571.1), deux α-protéobactéries pourpres. Les protéines représentées pour les Eukarya sont : Humain(NP_001243460.1) ; Rat (NP_ 036647.1) ; Souris (NP_033905.3) ; Bovin (NP_786970.1) ; Zebrafish (NP_ 001006032.1) ; Droso-phile (NP_608531.1) ; Arabidopsis thaliana (NP_ 566110.1), une plante supérieure ; Solanum tuberosum (CAH10765.1),la pomme de terre ; Physcomitrella Patens (ABG37902.1), une mousse et Schizosaccharomyces pombe (NP_595490.2),une levure. Les a.a sont colorés suivant le degré de conservation (code couleur arc-en-ciel pour un cut-off cor-respondant à une similarité de séquence de 30%) : en rouge intense, les a.a identiques ou similaires (100 % deconservation) ; du rouge au vert clair, le degré de conservation des a.a diminue. Les 5 TM putatifs sont représentéspar des hélices violettes en haut des séquences et l’emplacement des boucles (Loop) qui les relient est indiqué.Entre parenthèses sont indiqués les codes UniProt des séquences protéiques. L’alignement a été réalisé avec leserveur ClustalW [Thompson et al., 1994] et la figure avec le logiciel Aline [Bond and Schuttelkopf, 2009].

10 Chapitre 1. Les protéines TSPO
Selon la théorie de l’endosymbiose [Margulis, 1996], les eucaryotes ont pour origine une cellule primitive ayant"engloutie" une série de bactéries plus petites, lesquelles auraient fourni à la cellule hôte des organites tels quela mitochondrie, le noyau et le chloroplaste. Le 1er évènement serait l’internalisation d’une Archaea dans uneBacteria, ce qui aurait donné le noyau, amorçant ainsi la lignée des Eukarya. Puis d’autres internalisationsauraient suivi : la mitochondrie dériverait d’une endosymbiose avec une α-protéobactérie pourpre (bactériesautotrophes a et photosynthétiques), permettant à la cellule eucaryote d’utiliser l’oxygène nouvellement apparudans l’environnement comme source d’énergie ; le chloroplaste serait apparu plus tardivement et dériverait d’unorganisme apparenté aux cyanobactéries (bactéries réalisant une photosynthèse oxygénique), permettant ainsiaux cellules d’utiliser l’énergie lumineuse [Govindjee and Shevela, 2011].
a. L’autotrophie est la production, par un organisme vivant, de matière organique par réduction de matièreinorganique. Ce mode de nutrition caractérise les végétaux chlorophylliens (verts), les cyanobactéries, et lesbactéries sulfureuses. Les matières inorganiques utilisées sont généralement le carbone ou l’azote.
Encadré 1 (La théorie de l’endosymbiose).
Ces protéines possèdent des localisations différentes (qui seront discutées ultérieurement)ce qui peut nous renseigner sur leur lien : membrane plasmique pour la TSPO bactérienne,membrane mitochondriale pour la TSPO animal et membrane du reticulum endoplasmique(RE) et de l’appareil de Golgi (AG) pour la TSPO des végétaux supérieurs.
La protéine TSPO animale mitochondriale est plus proche de la protéine bactérienne (p. ex.Rhodobacter capsulatus, Rhodobacter sphaeroides, Bacillus cereus) qu’elle ne l’est de la TSPOappartenant aux végétaux supérieurs (p. ex. Arabidospis Thaliana, Solanum tuberosum) [Lin-demann et al., 2004]. Or les α-protéobactéries, telle R. capsulatus, sont à l’origine des mito-chondries selon la théorie endosymbiotique, Encadré 1, (alors que les chloroplastes des plantesont pour origine une cyanobactérie 1) ainsi la TSPO animale descendrait de ces bactéries. La
1. Les cyanobactéries sont des bactéries procaryotes coloniales. Elles réalisent la photosynthèse oxygénique etpeuvent donc transformer l’énergie solaire en énergie chimique utilisable par la cellule en fixant CO2 et en libérantdu O2. Apparues il y a environ 3,8 milliards d’années, elles ont contribué à l’expansion des formes actuelles de

1.1. La famille TSPO : des protéines membranaires anciennes et conservées àtravers l’évolution 11
protéine TSPO végétale appartenant aux plantes dites inférieures - comprenant les mousses(Physcomitrella patens), les lichens, les champignons 2 (p. ex. Schizosaccharomyces pombe) etles algues - est plus proche de la TSPO mitochondriale animale (p. ex. humain, souris, rat,bovin), qu’elle ne l’est de la protéine bactérienne. La présence d’homologues de la TSPO a éga-lement été rapportée chez les cyanobactéries (Synechoccus), pressenties pour être les ancêtres desplastes 3 végétaux [Yeliseev et al., 1997]. Pourtant, la protéine TSPO des cyanobactéries présenteseulement entre 7 et 17% d’a.a identiques avec la TSPO des végétaux supérieurs. Les végétauxdits supérieurs et inférieures pourraient donc aussi descendre du groupe des α-protéobactéries[Lindemann et al., 2004] ; par la suite la TSPO des végétaux supérieurs se serait plus éloignéede ses homologues des végétaux inférieurs et animales.
Les protéines TSPO appartiennent donc à une famille de protéines ancienne et hautementconservée à travers l’évolution. La TSPO est ainsi retrouvée chez les animaux, végétaux, champi-gnons, archae et bactéries. Cela laisse suggérer qu’au cours de l’évolution, il y ait pu y avoir uneconservation de fonction fondamentale pour la cellule, en plus d’une conservation de la struc-ture de cette protéine qui est, comme nous allons le voir dans la suite de cette introduction, uneprotéine énigmatique aux multiples facettes.
La TSPO a été initialement découverte chez les mammifères en 1977 [Braestrup and Squires,1977] et ce n’est que bien plus tard, en 1995, que n’a été découvert son homologue chez la bactérieRhodobacter sphaeroides [Yeliseev and Kaplan, 1995], puis presque 10 ans après chez la planteArabidopsis thaliana [Lindemann et al., 2004].
Les données concernant TSPO sont donc très nombreuses chez les mammifères pour desraisons historiques relatives à sa découverte mais aussi pour des raisons d’ordre médical, unesurproduction de TSPO ayant été fréquemment associée à de nombreuses pathologies chez l’êtrehumain.
L’introduction qui suit vise d’une part à être une revue des données concernant les relationsstructure-fonction de la protéine TSPO animale, principalement, de la TSPO2, des TSPO vé-gétale et bactérienne ; et d’autre part à faire transparaître le contexte dans lequel s’est situémon travail de thèse puisque le point de vue qui dominait sur la TSPO depuis près de 25 ans acomplétement été remis en question ces 2 dernières années.
vie sur Terre par leur production d’O2 par photosynthèse2. Jusqu’au milieu du XXe siècle, les naturalistes classèrent les champignons parmi les plantes. En 1969 Robert
H. Whittaker les individualise enfin dans un règne particulier, les Fungi3. Le plaste est un organite présent dans les cellules des eucaryotes chlorophylliens (algues et plantes). Il
possède un ADN propre, il est dit semi-autonome. Suivant la cellule, les plastes peuvent se spécialiser pouraccomplir certaines fonctions. Ainsi, p. ex., les chloroplastes sont le siège de la photosynthèse, les amyloplastessont spécialisés dans le stockage d’amidon et les chromoplastes contiennent des pigments comme la caroténoïde.

12 Chapitre 1. Les protéines TSPO
1.2 La TSPO animale : TranSlocator PrOtein 18 kDa
1.2.1 Historique et découverte
Les Benzodiazépines (BZ) sont des molécules qui agissent sur le système nerveux central (SNC ; il est constituédu cerveau et de la moelle épinière) et qui sont utilisées en médecine pour leurs propriétés amnésiantes, anxioly-tiques, sédatives et hypnotiques, myorelaxantes et anticonvulsivantes. Dans le SNC, ces molécules se fixent surle récepteur central aux BZ (CBR - Central BZ Receptor) [Braestrup and Squires, 1977, Mohler and Okada,1977]. Le CBR fait partie d’un complexe macromoléculaire, GABAA, qui contient aussi le récepteur à l’acideγ-aminobutyrique (GABA ; un composé chimique naturel qui inhibe le système nerveux) et un canal chlorure[DeLorey and Olsen, 1992]. En se fixant sur le CBR, les BZ augmentent l’affinité du complexe pour le GABAet augmente son effet d’inhibition [DeLorey and Olsen, 1992].
Encadré 2 (Les Benzodiazépines).
La TSPO a été appelée initialement et pendant très longtemps PBR pour Peripheral-typeBenzodiazepine Receptor (Récepteur Périphérique aux BZ). En effet, en 1977, en cherchantdes sites de liaisons aux BZ, Encadré. 2, Braestrup and Squires observent la fixation dudiazépam (une BZ plus connue sous le nom de ValiumR©) dans le rein du rat et dans d’autrestissus périphériques (foie et poumon) avec une affinité similaire à celle du CBR. Cettefixation hors du SNC a donc valu à la protéine impliquée le nom de PBR pour la différencierdu CBR [Verma and Snyder, 1989]. Par la suite, le PBR s’est révélé être présent dansla plupart des tissus du rat [Anholt et al., 1985, Gavish et al., 1999], mais aussi SNC àsavoir dans les cellules gliales 4[Richards and Mohler, 1984, Benavides et al., 1990, Marangoset al., 1982], mais avec une expression variable selon les tissus [Bribes et al., 2004]. Enplus de leur distribution tissulaire, le PBR et le CBR se distinguent par leurs propriétéspharmacologiques, physiologiques et structurales [Gavish et al., 1999, Casellas et al., 2002].
La présence ubiquitaire du PBR et non uniquement périphérique, la mise au point de ligandspharmacologiques n’appartenant pas à la famille des BZ, la caractérisation d’une fonction detransport de molécules et non de récepteur ont conduit en 2006 [Papadopoulos et al., 2006a]à une modification du nom de la protéine en TSPO pour TranSlocase PrOtein, reflétant sonrôle supposé de transporteur décrit en détail plus loin (Sec. 1.2.5). Cette nouvelle appellationa permis aussi de faire un lien avec son homologue bactérien de R. sphaeroides nommé TspOpour "tryptophan-rich sensory protein" [Yeliseev and Kaplan, 1999, 2000] en lien avec sa fonctionsupposée que nous aborderons Sec. 1.5.2.
4. Les cellules gliales forment l’environnement des neurones. Elles assurent le maintien de l’homéostasie,produisent la myéline et jouent un rôle de soutien et de protection du tissu nerveux en apportant les nutrimentset l’oxygène, en éliminant les cellules mortes et en combattant les pathogènes. Il existe 4 variétés de cellulesgliales : les astrocytes, les oligodendrocytes, les cellules épendymaires et les cellules microgliales ou microglie.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 13
1.2.2 Localisation et distribution
1.2.2.1 Distribution dans les tissus
La TSPO est retrouvée dans tout le corps mais son taux d’expression varie considérablementen fonction des tissus [Anholt et al., 1985, Woods and Williams, 1996, Zisterer and Williams,1997, Austin et al., 2013, Gavish et al., 1999, Giatzakis and Papadopoulos, 2004] :
• Forte expression de la TSPO (8 - 20 pmol/mg) : tissus stéroïdogéniques 5 et les tissus desécrétion c.-à-d. glandes surrénales, glande pinéale (petite glande endocrine du cerveaudes vertébrés), glandes salivaires, épithélium olfactif, testicules, ovaires, utérus et placenta[Austin et al., 2013, Bribes et al., 2004, Ostuni et al., 2008].• Expression intermédiaire de la TSPO (5 - 8 pmol/mg) : reins et cœur [Austin et al., 2013,Gavish et al., 1999].
• Faible expression de la TSPO (10 - 70 fmol/mg) : foie et cerveau [Austin et al., 2013,Gavish et al., 1999, Batarseh et al., 2010].
Au caractère ubiquitaire de la protéine s’ajoute une hétérogénéité de son expression au seinmême d’un organe. Dans les glandes surréales, la partie médullaire est visiblement dépourvuede TSPO contrairement au cortex où la TSPO est fortement exprimée [Anholt et al., 1986]. Dela même manière, dans les testicules la TSPO est essentiellement localisée dans les cellules deLeydig 6 et au niveau de l’anse de Henlé 7 [De Souza et al., 1985]. Dans les cellules sanguines,l’expression de la TSPO est plus élevée dans les monocytes 8 et les granulocytes neutrophiles 9
et plus faible dans les plaquettes et les érythrocytes (globules rouges) [Canat et al., 1993]. Dansle SNC, la TSPO n’est pas présente dans les neurones [Marangos et al., 1982, Anholt et al.,1986] mais dans les membranes nucléaires des cellules gliales et plus particulièrement dans lesastrocytes 10 et la microglie 11 [Kuhlmann and Guilarte, 2000].
5. Les hormones stéroïdiennes sont des stéroïdes (un groupe de lipides dont font parti les stérols et les vitaminesD) se comportant comme des hormones. On peut les regrouper en cinq catégories selon leurs récepteurs : lesglucocorticoïdes, les minéralocorticoïdes, les androgènes, les œstrogènes et les progestatifs.
6. Les cellules de Leydig contrôlent le développement et le maintien des caractères sexuels primaires et secon-daires, et jouent un rôle dans le fonctionnement de l’appareil génital masculin et le comportement sexuel.
7. L’anse de Henlé est le tube en forme de U responsable du transport de l’urine dans le rein.8. Les monocytes sont des cellules immunitaires, ce sont les plus grands de nos globules blancs (leucocytes).9. Les granulocytes neutrophiles sont des cellules sanguines appartenant à la lignée des leucocytes, ils ont
donc un rôle dans le système immunitaire.10. Les astrocytes sont des cellules en forme d’étoile impliquées de manière essentielle dans l’élaboration des
connections neuronales, le contrôle de la neurotransmission, l’approvisionnement en nutriments et le métabolismeénergétique du système nerveux ainsi que la génération de neurones dans le cerveau adulte.11. La microglie est une population de cellules gliales constituée de macrophages résidents du cerveau et de la
moelle épinière. Ces cellules sont capables de changer de forme très rapidement pour migrer d’un endroit à unautre, formant ainsi la principale défense immunitaire active du système nerveux central. Elle auraient aussi unrôle physiologique dans la neurogenèse et le fonctionnement des synapses.

14 Chapitre 1. Les protéines TSPO
1.2.2.2 Localisation subcellulaire
L’utilisation du ligand pharmacologique le plus affin sous sa forme radiomarquée, le [3H]-PK11195 (cf. § 1.2.4.1), sur des glandes surrénales de rat a permis de montrer que la TSPOest majoritairement localisée dans la membrane mitochondriale externe (MME) [Anholt et al.,1986, O’Beirne et al., 1990]. Par la suite, cette localisation subcellulaire a été confirmée par desétudes, avec ce même ligand ou le [3H]-Ro5-4864 (cf. § 1.2.4.1), sur de nombreux tissus tels queles testicules, le poumon, le rein, le coeur, le foie et le muscle squelettique [Antkiewicz-Michaluket al., 1988a,b, Mukherjee and Das, 1989]. Plus précisément, la TSPO se situe dans la MME auniveau des sites de contact entre les membranes externe et interne [O’Beirne et al., 1990, Cultyet al., 1999].
Bien que la TSPO soit principalement localisée dans la MME, elle a aussi été trouvée margi-nalement dans les membranes de plusieurs cellules (peut-être à cause d’une contamination parles mitochondries) :
• dans la membrane plasmique d’érythrocytes humaines, qui sont dépourvues de mitochon-dries et de noyaux [Olson et al., 1988] ;• dans la membrane plasmique de tissus périphériques tels que les tissus musculaires ; [Dobleet al., 1985], hépatiques [O’Beirne et al., 1990, Woods and Williams, 1996] et les cellulesde Leydig de rat [Garnier et al., 1993], le cortex surrénale de souris [Oke et al., 1992] ;• dans la membrane nucléaire de cellules : tumorales mammaires [Hardwick et al., 1999],hépatiques [Corsi et al., 2005] et gliales du SNC [Kuhlmann and Guilarte, 2000].
1.2.3 Information génétique
La TSPO mitochondriale est une protéine de 169 a.a soit ∼18 kDa codée par un gènenucléaire. Ce gène Tspo de 11-kbp est retrouvé en un seul exemplaire dans le génome humain oùil est localisé sur le locus 22q13.31 du chromosome 22 [Riond et al., 1991, Chang et al., 1992].
L’ADNc 12 de Tspo a été cloné à partir de multiples espèces dont l’Homme [Riond et al.,1991, Lin et al., 1993], la vache [Parola et al., 1991], le cochon, le rat [Sprengel et al., 1989,Krueger et al., 1990, Casalotti et al., 1992] et la souris [Garnier et al., 1994b]. Le clonageet la caractérisation du gène Tspo ont montré qu’il est composé de 4 exons 13, avec un largeintron, contenant des séquences répétitives, qui sépare le 1er et le second exon. Le 1er exon,des parties du 2e exon et la moitié du 4e exon ne seraient pas traduits [Gavish et al., 1999,Batarseh and Papadopoulos, 2010]. Le domaine de l’exon 2 a été décrit comme lié à la liaisonde l’isoquioline PK11195 et en partie à la liaison des BZ spécifiques de la TSPO [Farges et al.,1994, Papadopoulos et al., 1997].
Le promoteur du gène Tspo humain ne possède pas de boîte TATA, qui est une séquenced’ADN qui sert, en partie, de lieu de reconnaissance pour l’ARN polymérase chez les eucaryotes,
12. L’ADN complémentaire (ou ADNc), est la copie en ADN d’un ARN messager. Ainsi il représente, sousforme d’un simple brin, la partie codante de la région du génome ayant été transcrit en cet ARNm.13. L’exon est la partie codante de l’ADN au sein d’un gène et un intron est la partie non codante d’un gène.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 15
mais contient 5 sites putatifs (boite GC) de liaison aux facteurs de transcription (FT) Sp.Or, cette architecture du promoteur est communément associée aux "gènes de ménage" (ouhousekeeping gene ; gènes constitutifs qui codent principalement pour des protéines essentiellesaux fonctions cellulaires de base) [Batarseh and Papadopoulos, 2010]. De plus, une architecturesimilaire est retrouvée dans les promoteurs de plusieurs gènes impliqués dans la régulation de laprolifération cellulaire et la cancérogenèse [DiMario, 2002]. Le promoteur du gène Tspo murinpossède aussi des sites de liaison putatifs supplémentaires pour des FT (AP1, Ets, Myc, SOX,SRY) impliqués dans des fonctions telles que la régulation du développement embryonnaire,l’apoptose et la prolifération et différentiation cellulaire [Giatzakis and Papadopoulos, 2004]. Deplus, le gène Tspo est sous le contrôle de voies de transduction du signal (PKcε, AP1, ERK1/2)sensibles aux dérivés réactifs de l’oxygène (ROS - Reactive Oxygen Species). Ces voies régulentde nombreux processus biologiques tels que l’adaptation métabolique, la différenciation et laprolifération cellulaire [Batarseh et al., 2010].
La présence de tous ces éléments laisse déjà suggérer un rôle de la TSPO dans ces fonctions.
1.2.4 Ligands
L’expression variable de la TSPO selon les tissus ainsi que les différences observées dans lesétudes de localisation membranaire (plasmique, mitochondriale, nucléaire) nous amène à nousquestionner sur la fonction et le rôle de la protéine selon le tissu considéré. La découverte de laTSPO comme récepteur à de multiples ligands spécifiques très différents les uns des autres, luia valu d’être impliquée dans de très nombreuses fonctions physiologiques. Le Tab. 1.1 donne unaperçu de ces fonctions avant qu’elles ne soient décrites en détails plus loin dans la Sec.1.2.5.Cette section vise à présenter les principaux ligands de la TSPO, dont une partie est issue dela caractérisation pharmacologique intense dont la TSPO a fait l’objet depuis plus de 25 ans.
1.2.4.1 Les ligands synthétiques
La découverte des propriétés de liaison des BZ à la TSPO et de son lien avec de nombreuxtroubles neurologiques et psychiatriques ont fait de la protéine une cible de choix des phar-macologistes. En effet, des études ont montré une diminution du niveau de la TSPO dans lesplaquettes et les lymphocytes de patients suicidaires, schizophrènes, présentant des troubles del’anxiété ou souffrant de stress post-traumatique [Arbo et al., 2015]. De plus, une augmentationde la TSPO a été détectée dans le cerveau de patients dépressifs sévères [Setiawan et al., 2015].C’est pourquoi le développement de molécules pour le traitement de ces pathologies a été etest toujours très actif [Scarf et al., 2012]. Ainsi, de très nombreuses molécules spécifiques de laTSPO et ayant une forte affinité ont été synthétisées durant ces 25 dernières années. Ces ligandsregroupent plusieurs familles de molécules, comme les benzodiazépines et les isoquinolines, dontle point commun est de comporter des assemblages polycycliques. [Le Fur et al., 1983a,b, Anziniet al., 1996, Cinone et al., 2000, Cappelli et al., 2008, Scarf et al., 2012].
16 Chapitre 1. Les protéines TSPO
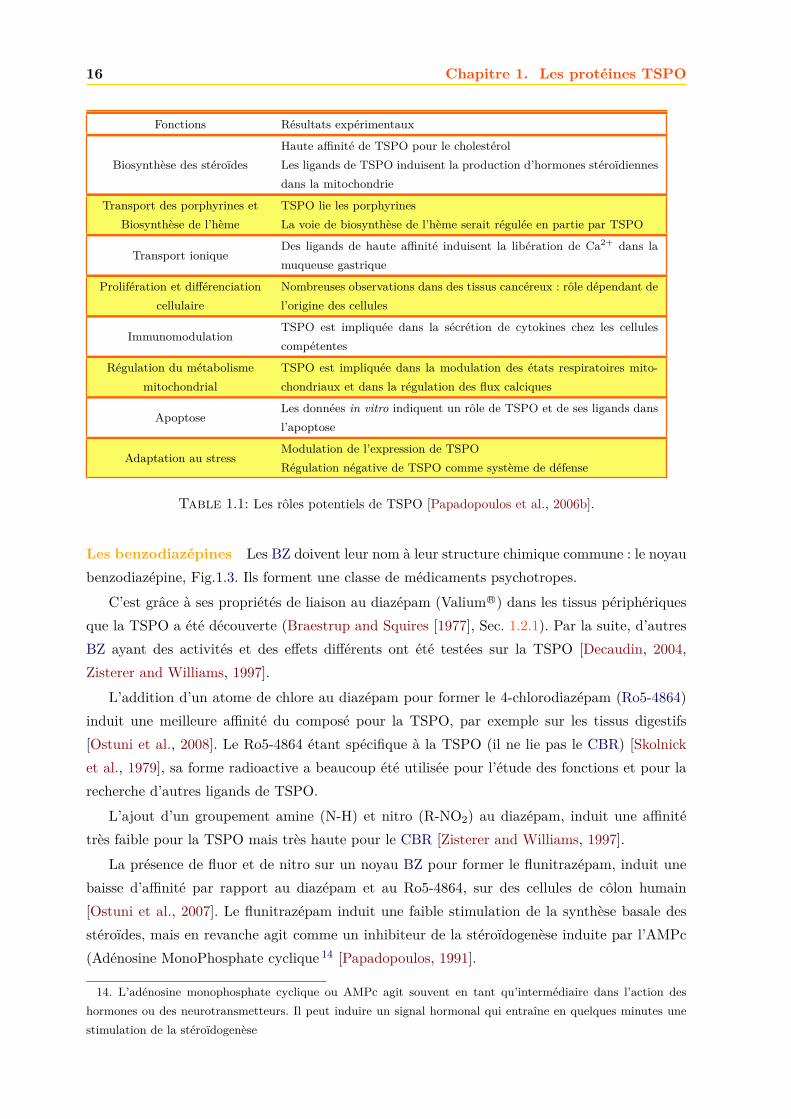
Fonctions Résultats expérimentaux
Biosynthèse des stéroïdesHaute affinité de TSPO pour le cholestérolLes ligands de TSPO induisent la production d’hormones stéroïdiennesdans la mitochondrie
Transport des porphyrines etBiosynthèse de l’hème
TSPO lie les porphyrinesLa voie de biosynthèse de l’hème serait régulée en partie par TSPO
Transport ioniqueDes ligands de haute affinité induisent la libération de Ca2+ dans lamuqueuse gastrique
Prolifération et différenciationcellulaire
Nombreuses observations dans des tissus cancéreux : rôle dépendant del’origine des cellules
ImmunomodulationTSPO est impliquée dans la sécrétion de cytokines chez les cellulescompétentes
Régulation du métabolismemitochondrial
TSPO est impliquée dans la modulation des états respiratoires mito-chondriaux et dans la régulation des flux calciques
ApoptoseLes données in vitro indiquent un rôle de TSPO et de ses ligands dansl’apoptose
Adaptation au stressModulation de l’expression de TSPORégulation négative de TSPO comme système de défense
Table 1.1: Les rôles potentiels de TSPO [Papadopoulos et al., 2006b].
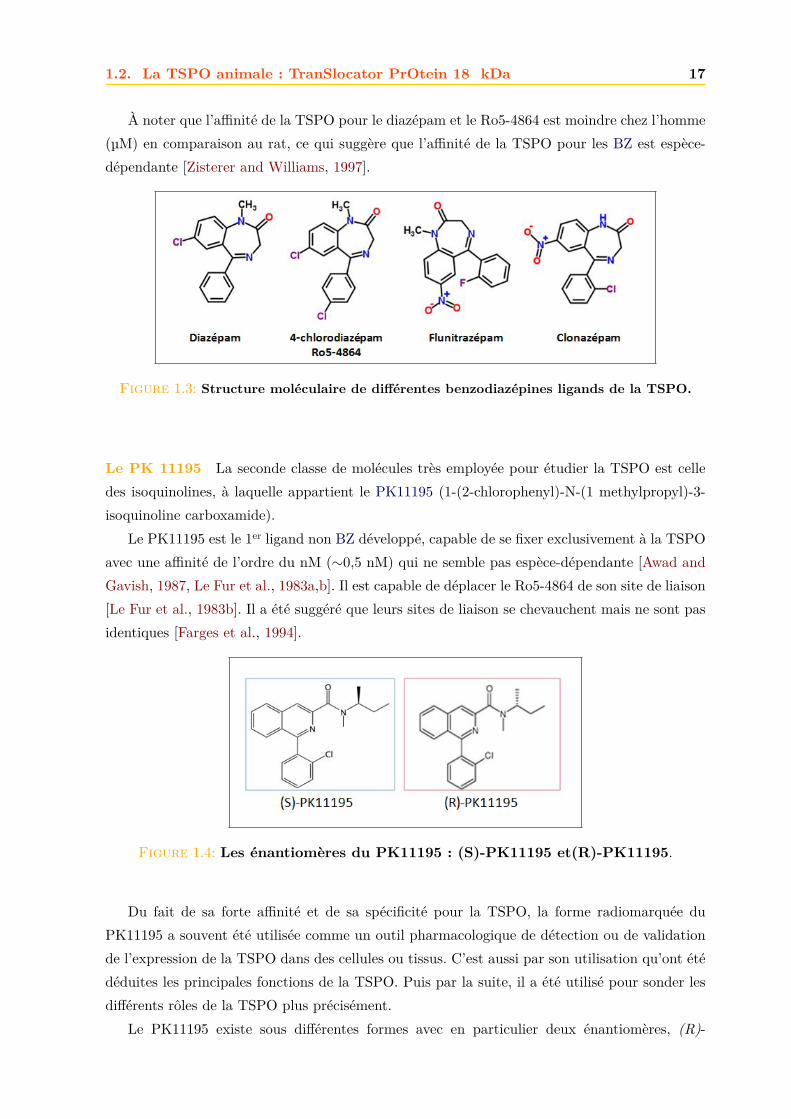
Les benzodiazépines Les BZ doivent leur nom à leur structure chimique commune : le noyaubenzodiazépine, Fig.1.3. Ils forment une classe de médicaments psychotropes.
C’est grâce à ses propriétés de liaison au diazépam (ValiumR©) dans les tissus périphériquesque la TSPO a été découverte (Braestrup and Squires [1977], Sec. 1.2.1). Par la suite, d’autresBZ ayant des activités et des effets différents ont été testées sur la TSPO [Decaudin, 2004,Zisterer and Williams, 1997].
L’addition d’un atome de chlore au diazépam pour former le 4-chlorodiazépam (Ro5-4864)induit une meilleure affinité du composé pour la TSPO, par exemple sur les tissus digestifs[Ostuni et al., 2008]. Le Ro5-4864 étant spécifique à la TSPO (il ne lie pas le CBR) [Skolnicket al., 1979], sa forme radioactive a beaucoup été utilisée pour l’étude des fonctions et pour larecherche d’autres ligands de TSPO.
L’ajout d’un groupement amine (N-H) et nitro (R-NO2) au diazépam, induit une affinitétrès faible pour la TSPO mais très haute pour le CBR [Zisterer and Williams, 1997].
La présence de fluor et de nitro sur un noyau BZ pour former le flunitrazépam, induit unebaisse d’affinité par rapport au diazépam et au Ro5-4864, sur des cellules de côlon humain[Ostuni et al., 2007]. Le flunitrazépam induit une faible stimulation de la synthèse basale desstéroïdes, mais en revanche agit comme un inhibiteur de la stéroïdogenèse induite par l’AMPc(Adénosine MonoPhosphate cyclique 14 [Papadopoulos, 1991].
14. L’adénosine monophosphate cyclique ou AMPc agit souvent en tant qu’intermédiaire dans l’action deshormones ou des neurotransmetteurs. Il peut induire un signal hormonal qui entraîne en quelques minutes unestimulation de la stéroïdogenèse
1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 17
À noter que l’affinité de la TSPO pour le diazépam et le Ro5-4864 est moindre chez l’homme(µM) en comparaison au rat, ce qui suggère que l’affinité de la TSPO pour les BZ est espèce-dépendante [Zisterer and Williams, 1997].
Figure 1.3: Structure moléculaire de différentes benzodiazépines ligands de la TSPO.
Le PK 11195 La seconde classe de molécules très employée pour étudier la TSPO est celledes isoquinolines, à laquelle appartient le PK11195 (1-(2-chlorophenyl)-N-(1 methylpropyl)-3-isoquinoline carboxamide).
Le PK11195 est le 1er ligand non BZ développé, capable de se fixer exclusivement à la TSPOavec une affinité de l’ordre du nM (∼0,5 nM) qui ne semble pas espèce-dépendante [Awad andGavish, 1987, Le Fur et al., 1983a,b]. Il est capable de déplacer le Ro5-4864 de son site de liaison[Le Fur et al., 1983b]. Il a été suggéré que leurs sites de liaison se chevauchent mais ne sont pasidentiques [Farges et al., 1994].
Figure 1.4: Les énantiomères du PK11195 : (S)-PK11195 et(R)-PK11195.
Du fait de sa forte affinité et de sa spécificité pour la TSPO, la forme radiomarquée duPK11195 a souvent été utilisée comme un outil pharmacologique de détection ou de validationde l’expression de la TSPO dans des cellules ou tissus. C’est aussi par son utilisation qu’ont étédéduites les principales fonctions de la TSPO. Puis par la suite, il a été utilisé pour sonder lesdifférents rôles de la TSPO plus précisément.
Le PK11195 existe sous différentes formes avec en particulier deux énantiomères, (R)-

18 Chapitre 1. Les protéines TSPO
PK11195 et (S)-PK11195, Fig. 1.4, présentant des affinités très différentes. En effet, d’aprèsDubroeucq et al. [1983], Shah et al. [1994], le (S)-PK11195 aurait une affinité 2 fois moindreque le (R)-PK11195 sur plusieurs tissus de rat. La prise en compte de cette différence a eu un rôlecrucial dans la détermination de la structure atomique de la TSPO de souris par RMN puisqueseul un énantiomère, le (R)-PK11195 possédant une affinité meilleure, a permis la stabilisationde la structure 3D [Jaremko et al., 2014], nous y reviendrons dans la Sec.1.2.7.
Autres ligands Le PK11195, surtout le (R)-PK 11195, marqué au 11C est utilisé dans lestechniques d’imageries in vivo notamment pour la tomographie par émission de positrons (TEP)[Banati et al., 1999, Schuitemaker et al., 2013]. Mais les problèmes de spécificité et de solubilitéont conduit à développer de nouvelles molécules comme outils d’imagerie [Chauveau et al., 2008].Parmi ces nouveaux ligands synthétiques récemment mis en évidence, il y a le le PBR28, Fig. 1.5.Il s’agit d’un composé aryloxyanilide, qui présente une affinité pour la TSPO comparable au(R)-PK 11195 mais dont la forme radiomarquée présente une meilleure spécificité que le (R)-PK11195 [Harberts et al., 2013, Owen et al., 2014].
Au développement, par les pharmaciens, de ligands de la TSPO pour l’imagerie médicales’ajoute le développement de ligands pour le traitement des troubles psychiatriques. Parmi cesligands de la TSPO on peut citer (Fig. 1.5) :
· les benzoxazolones et leurs dérivés [Fukaya et al., 2013], qui sont conçus à partir del’ouverture du cycle diazepine du Ro5-4864· le XBD 173 ou emapunil [N-benzyl-N-ethyl-2-(7-methyl-8-oxo-2-phenylpurin-9-yl)acetamide] [Owen et al., 2011]. Il possède l’avantage d’éviter les effets secondaires desBZ [Rupprecht et al., 2009]· l’etifoxine, qui appartient au benzoaxine, une famille proche des BZ et ayant des effetssemblables mais structurellement différente. Lui ausi possède l’avantage d’éviter les effetssecondaires des BZ [Nguyen et al., 2006]· le ZBD-2 [N-benzyl-N-ethyl-2-(7,8-dihydro-7-benzyl-8-oxo-2-phenyl-9H-purin-9-yl) aceta-mide]. En plus d’un effet anxiolytique, il permettrait de réguler la douleur via une pro-duction de neurosteroïdes [Wang and Tonggu, 2015]· le YL-IPA08 [N-ethyl-N-(2-pyridinylmethyl)-2-(3,4-ichlorophenyl)-7-methyl imidazo [1,2-a] pyridine-3-acetamide hydrochloride]. Il a un effet contre les troubles liés à un stresspost-traumatique via la synthèse de alloprégnénolone.[Zhang et al., 2014]
Nous verrons par la suite, dans le §1.2.5 qui s’intéresse aux fonctions de la TSPO, que cesligands synthétiques ont encore d’autres effets qui intéressent le corps médical.
1.2.4.2 Les ligands naturels
Au départ, la recherche des ligands naturels a été réalisée en incubant des extraits mem-branaires riches en protéine TSPO ou des mitochondries avec les ligands pharmacologiquesradioactifs caractérisés, présentés ci-dessus (p. ex. [3H]PK11195, [3H]Ro5-4864, [3H]diazepam),afin d’identifier les molécules naturelles capables de déplacer la liaison du traceur radioactif.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 19
Figure 1.5: Structure moléculaire de différents ligands de TSPO dévellopés pourune possible utilisation dans l’imagerie médical et le traitements des troubles psy-chiatriques.
Le DBI - Diazepam Binding Inhibitor Le DBI (Diazepam Binding Inhibitor) a été ini-tialement identifié comme ligand endogène des récepteurs des BZ (PBR et CBR), grâce à sespropriétés d’inhibition de la liaison du diazépam dans les cerveaux de rat [Guidotti et al., 1983]et d’humain [Ferrero et al., 1986]. Il présente une affinité de l’ordre du nM pour la TSPO [Costaand Guidotti, 1991]. Ce polypeptide de 86 a.a (9 kDa) est le précurseur commun des endozépines- ligands des récepteurs des BZ [Guidotti et al., 1983] - et il génère, par clivage protéolytique(au niveau de résidus lysines isolés), un certain nombre de fragments biologiquement actifsdont les plus connus sont : l’OctaDécaNeuropeptide (ODN ou DBI33−50) qui agit sur le CBR[Bormann and Clapham, 1985] et le TriakontaTétraNeuropeptide (TTN ou DBI17−50) qui agitsélectivement sur la TSPO [Berkovich et al., 1990, Gavish and Weizman, 1997].
Le gène du DBI est largement exprimé, tant au niveau du système nerveux central que dansla plupart des organes périphériques [Gray et al., 1986]. Le DBI et ses peptides apparentés sont,en particulier, très abondants dans les organes stéroïdogéniques [Alho et al., 1990, Duparc et al.,2003].
Les résultats d’un certain nombre de travaux réalisés sur différents modèles animaux ontpermis de montrer que le DBI et ses peptides dérivés sont impliqués dans la stéroïdogenèse :
• Les études menées sur différents modèles de cellules stéroïdogéniques d’animaux montrentque le DBI active le transport du cholestérol et stimule la biosynthèse des hormonesstéroïdes (testostérone et prégnénolone) et des neurostéroïdes [Papadopoulos et al., 1992,Garnier et al., 1993, Lesouhaitier et al., 2000, Do-Rego et al., 2001].• La suppression du DBI par un oligonucléotide antisens 15, entraine une inhibition de laproduction de stéroïdes [Garnier et al., 1994a, Boujrad et al., 1993].
• Le TTN stimule la formation de prégnénolone mitochondriale et déplace la liaison desligands de la TSPO dans des extraits rénaux, testiculaires et de cerveau [Papadopou-los et al., 1992, Papadopoulos, 1991]. De plus, l’administration de TTN provoque uneaugmentation dose dépendante de la sécrétion de la testostérone au niveau de fragments
15. Un oligonucléotide anti-sens est un fragment d’ARN, généralement synthétique, qui peut se lier spécifique-ment à un ARN messager (ARNm) naturel : sa séquence nucléotidique est complémentaire de celle de l’ARNmqu’il cible. Sa liaison bloque la synthèse de la protéine codée par cet ARNm

20 Chapitre 1. Les protéines TSPO
testiculaires humains perfusés, tandis que l’ODN n’a pas d’effet [Duparc et al., 2003].
Le DBI est donc important pour le maintien de la stéroïdogenèse constitutive et la régulationhormonale.
Figure 1.6: Superposition des structures cristallographique et RMN du DBI (ou Acyl-CoA Binding protein - ACBP). En couleur, la structure cristallographique du DBI humain (fichierPDB 2FJ9). En gris, la structure RMN du DBI bovin (fichier PDB 2ABD). Les DBIs humain et bovinpartagent ∼95 % d’identité. Les résidus indiqués correspondent aux sites de coupures donnant naissanceaux fragments actifs ODN (33-50) et TTN(17-50). Image réalisée avec le programme PyMOL.
La structure du DBI déterminée par RMN [Andersen and Poulsen, 1993] et par cristallo-graphie [Taskinen et al., 2007] révèle un assemblage de 4 hélices, Fig. 1.6. Le peptide TTNactif sur la TSPO correspond à une région contenant une seule hélice, mais les a.a réellementimpliqués dans l’interaction avec la TSPO ne sont pas caractérisés. Des études bioinformatiquesont permis de suggérer la participation des résidus M46, L47 et F49 dans la partie C-terminaledu peptide dans l’interaction avec la TSPO [Cinone et al., 2000]. En absence d’une structure 3Dde la TSPO, ces études ont été menées par une méthode appelée 3D QSAR (three-dimensionalQuantitative Structure Activity Relationship), qui se base sur une comparaison des structureset des caractéristiques physico-chimiques des ligands de TSPO pour obtenir des informationssur la nature des interactions entre ligands/TSPO.
Le Cholestérol Le cholestérol est un lipide de la famille des stérols. C’est une moléculehydrophobe à l’exception de son groupe hydroxyle en position 3 β qui lui donne son carac-tère amphiphile. Il est formé d’un noyau tétracyclique stérol rigide, substitué par une chaîneiso-octyle flexible en position 17, Fig. 1.7. Sa structure 3D plane lui permet une interaction hy-drophobe très favorable avec les phospholipides au sein de la bicouche lipidique de la membrane,en rendant cette dernière moins déformable (plus rigide 16 et moins perméable).
Le cholestérol est absent des bactéries et des plantes mais il est le composant majeur des
16. Par contre, à basse température, le cholestérol retarde la transition gel/liquide des lipides, en les empêchantd’interagir entre eux par interaction de Van der Waals lorsqu’ils se rapprochent les uns des autres : à bassetempérature, le cholestérol a un effet plutôt fluidifiant

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 21
Figure 1.7: Éléments structuraux du cholestérol.
membranes cellulaires eucaryotes, il représente 20 à 25% des lipides totaux de la cellule. Ce-pendant, dans les membranes cellulaires, le cholestérol est en proportion variable, significativedans la membrane plasmique et le RE, pratiquement nulle dans la MME. Il a par ailleurs uneinfluence sur les nombreuses protéines présentes dans les membranes : il module l’activité deces protéines soit de manière indirecte en influençant la dynamique membranaire [Lee, 2004],soit de manière spécifique en interagissant directement avec ces protéines et en modifiant leurstructure tridimensionnelle [Addona et al., 1998]. Il a également été décrit, qu’il pouvait modulerl’oligomérisation fonctionnelle des protéines via sa dimérisation [Hanson et al., 2008].
La TSPO a été proposée avoir un rôle dans la translocation du cholestérol avec lequel elleprésente une affinité de ∼5 nM [Lacapere et al., 2001]. Son interaction avec la TSPO se faitsur un site impliquant des résidus situés du côté C-terminal, appelé CRAC pour Cholesterol-Recognition Amino Acid Consensus [Li and Papadopoulos, 1998].
Le cholestérol est le précurseur de plusieurs molécules dans les cellules qui l’utilisent pour laformation de dérivés dont les hormones stéroïdiennes (cortisol, cortisone, et aldostérone dans lessurrénales, mais aussi oestrogènes et testostérone dans les gonades). Dans le foie, le cholestérol,provenant des tissus périphériques, est métabolisé en acides biliaires sécrétés vers l’intestin. Dansla peau, le 7-déshydrocholestérol, un précurseur du cholestérol , est également le précurseur dela vitamine D3 qui intervient dans la calcification des os.
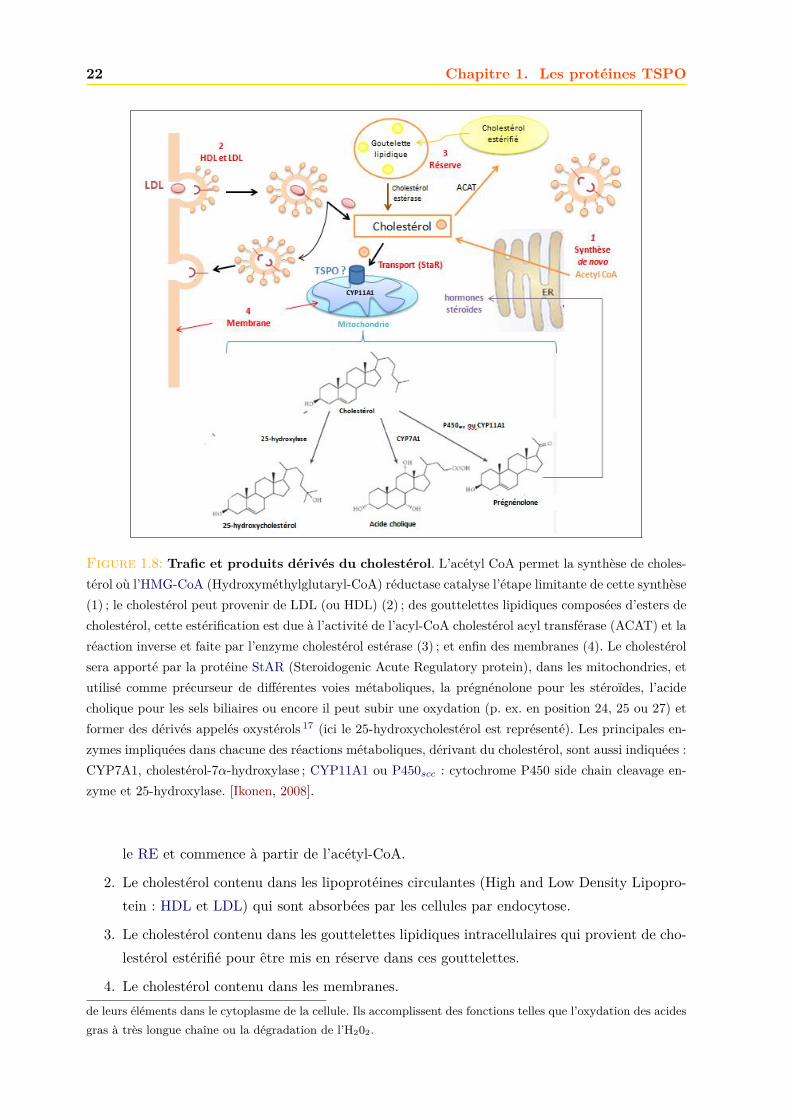
Il existe quatre sources de cholestérol dans les cellules [Miller, 2007, Ikonen, 2008], Fig.1.8 :
1. Le cholestérol synthétisé in cellulo représente un peu moins de la moitié du cholestéroldans le corps. La biosynthèse dans le foie représente ∼10% et dans les intestins ∼15% dela quantité produite par jour. La voie de biosynthèse du cholestérol comprend les enzymesqui se trouvent dans le cytoplasme, le RE et les peroxysomes 18. Sa synthèse se fait dans
18. Les peroxysomes sont des vésicules entourées d’une membrane simple de type bicouche lipidique. Ils nepossèdent ni gène, ni machinerie capable d’assurer la traduction et vont donc être obligés d’incorporer l’ensemble
22 Chapitre 1. Les protéines TSPO
Figure 1.8: Trafic et produits dérivés du cholestérol. L’acétyl CoA permet la synthèse de choles-térol où l’HMG-CoA (Hydroxyméthylglutaryl-CoA) réductase catalyse l’étape limitante de cette synthèse(1) ; le cholestérol peut provenir de LDL (ou HDL) (2) ; des gouttelettes lipidiques composées d’esters decholestérol, cette estérification est due à l’activité de l’acyl-CoA cholestérol acyl transférase (ACAT) et laréaction inverse et faite par l’enzyme cholestérol estérase (3) ; et enfin des membranes (4). Le cholestérolsera apporté par la protéine StAR (Steroidogenic Acute Regulatory protein), dans les mitochondries, etutilisé comme précurseur de différentes voies métaboliques, la prégnénolone pour les stéroïdes, l’acidecholique pour les sels biliaires ou encore il peut subir une oxydation (p. ex. en position 24, 25 ou 27) etformer des dérivés appelés oxystérols 17 (ici le 25-hydroxycholestérol est représenté). Les principales en-zymes impliquées dans chacune des réactions métaboliques, dérivant du cholestérol, sont aussi indiquées :CYP7A1, cholestérol-7α-hydroxylase ; CYP11A1 ou P450scc : cytochrome P450 side chain cleavage en-zyme et 25-hydroxylase. [Ikonen, 2008].
le RE et commence à partir de l’acétyl-CoA.
2. Le cholestérol contenu dans les lipoprotéines circulantes (High and Low Density Lipopro-tein : HDL et LDL) qui sont absorbées par les cellules par endocytose.
3. Le cholestérol contenu dans les gouttelettes lipidiques intracellulaires qui provient de cho-lestérol estérifié pour être mis en réserve dans ces gouttelettes.
4. Le cholestérol contenu dans les membranes.de leurs éléments dans le cytoplasme de la cellule. Ils accomplissent des fonctions telles que l’oxydation des acidesgras à très longue chaîne ou la dégradation de l’H202.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 23
Les porphyrines Les porphyrines sont des molécules à structure cyclique (cycle tétrapyrole),essentielles aux organismes vivants et qui participent à de nombreuses fonctions comme letransport et le stockage de l’oxygène (hémoglobine, myoglobines, hémoprotéines), l’oxydationde substrats (cytochromes, hémoprotéines), la photosynthèse chez les plantes et les bactériesphotosynthétiques (chlorophylle), la synthèse de neuromédiateurs (Vitamine B12), ou encore laméthanogenèse chez les bactéries (coenzyme F430), Fig.1.9.
La conjugaison des doubles liaisons de ces molécules en fait des composés très colorés (ab-sorbance maximale vers 405 nm - bande de Soret), souvent photosensibles, et leur confère despropriétés de fluorescence (vers 620 nm pour une excitation à 400 nm).
Figure 1.9: La protoporphyrine IX (PPIX), son précurseur le coproporphyrinogèneIII et des produits dérivés chélatés biologiquement actifs. La ZPP (Protoporphyrinezinc) est un composé trouvé dans le sang, et plus précisément dans les globules rouges quandla production de l’hème est inhibée par une intoxication par le plomb (saturnisme) et/ou unecarence en fer (anémie).
La voie de biosynthèse des porphyrines comporte une voie principale dont le produit finalest la protoporphyrine IX (PPIX), principale porphyrine chez les mammifères. Cette voie estexclusivement endogène et s’effectue en plusieurs étapes, alternativement dans la mitochondrieet dans le cytosol notamment les dernières réactions de cette biosynthèse font intervenir uneétape de traversée de la membrane mitochondriale [Krieg et al., 2000]. Ensuite, les porphyrinespeuvent fixer des métaux par réaction avec les sels métalliques correspondants, chez l’animal ledérivé métallique porphyrique le plus important est l’hème, Fig. 1.10.
La liaison de molécules issues d’extraits sanguins a permis d’identifier les porphy-rines en tant que ligands naturels de TSPO. Ainsi TSPO présente une affinité impor-tante, de l’ordre du nM, pour la PPIX et l’hème [O’Beirne et al., 1990, Verma et al.,1987], et moindre pour le coproporphyrinogène III, un précurseur des porphyrines qui
18. Les oxystérols sont des molécules produites par oxydation ou hydroxylation du cholestérol. Ce sont descomposés ayant un rôle important en physiologie et en physiopathologie chez les mammifères. Certains sontconsidérés comme des molécules de signalisation pouvant avoir une action régulatrice dans plusieurs métabolismescomme la biosynthèse du cholestérol, des hormones stéroïdiennes et des acides biliaires. D’autres oxystérols,par contre, sont considérés comme des composés toxiques pouvant induire des perturbations structurales etmétaboliques au sein des cellules pouvant induire des pathologies. Enfin, plusieurs oxystérols semblent égalementcapables de perturber des mécanismes biologiques comme le processus inflammatoire.[Souidi et al., 2004]

24 Chapitre 1. Les protéines TSPO
Figure 1.10: Schéma de la biosynthèse de l’hème dans la mitochondrie. La synthèsede l’hème a lieu principalement dans la moelle osseuse (85%) et le foie. Au niveau cellulaireinterviennent au moins 8 enzymes (indiquées en bleu) différentes ayant des localisations cyto-plasmique et mitochondriale. Le 1ercycle cycle tétrapyrole, l’uroporphyrinogène III, est forméen 3 étapes à partir de l’acide δ-aminolévulinique (ALA). L’uroporphyrinogène III est convertien hème en 4 étapes à partir des intermédiaires : coproporphyrinogène III, protoporphyrinogèneIX et protoporphyrine IX (PPIX). L’hème sert ensuite de co-facteur aux hémo-protéines : hé-moglobines, myoglobine, catalases, peroxidases, cytochromes et à des hémo-protéines senseursà gaz comme O2 ou NO.
est produit dans le cytosol et transloqué dans la mitochondrie où il est converti enPPIX [Taketani et al., 1995b], Fig.1.10. La substitution du fer de l’hème par un zinc ré-duit de 8 à 10 fois l’affinité pour la TSPO, de même le remplacement du fer par del’étain ou du cobalt conduit à des produits 1000 fois moins affins [Gavish et al., 1999].Le site de liaison de ces porphyrines sur la TSPO semble être le même que celui du ligandpharmacologique PK11195 [Li et al., 2015], nous y reviendrons plus tard.
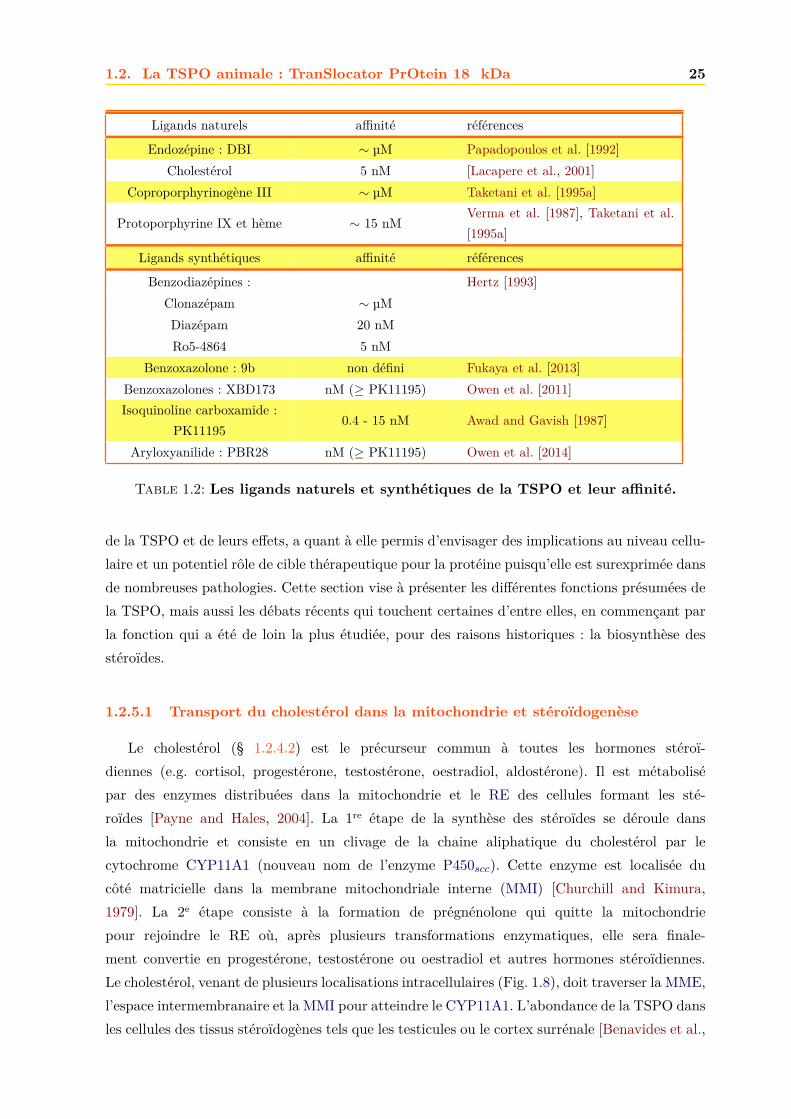
Le Tab. 1.2 résume les principaux ligands de la TSPO et leur affinité typique observée carpour certains ligands ces affinités peuvent variées fortement d’un tissu à l’autre.
1.2.5 Fonctions
Les liens établis entre les différents ligands endogènes (DBI, porphyrines, cholestérol) et lesdifférentes localisations membranaires et tissulaires de la TSPO ont laissé suggérer la participa-tion de la protéine à plusieurs fonctions. La découverte de nombreux ligands pharmacologiques
1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 25
Ligands naturels affinité références
Endozépine : DBI ∼ µM Papadopoulos et al. [1992]Cholestérol 5 nM [Lacapere et al., 2001]
Coproporphyrinogène III ∼ µM Taketani et al. [1995a]
Protoporphyrine IX et hème ∼ 15 nMVerma et al. [1987], Taketani et al.[1995a]
Ligands synthétiques affinité références
Benzodiazépines : Hertz [1993]Clonazépam ∼ µMDiazépam 20 nMRo5-4864 5 nM
Benzoxazolone : 9b non défini Fukaya et al. [2013]Benzoxazolones : XBD173 nM (≥ PK11195) Owen et al. [2011]Isoquinoline carboxamide :
PK111950.4 - 15 nM Awad and Gavish [1987]
Aryloxyanilide : PBR28 nM (≥ PK11195) Owen et al. [2014]
Table 1.2: Les ligands naturels et synthétiques de la TSPO et leur affinité.
de la TSPO et de leurs effets, a quant à elle permis d’envisager des implications au niveau cellu-laire et un potentiel rôle de cible thérapeutique pour la protéine puisqu’elle est surexprimée dansde nombreuses pathologies. Cette section vise à présenter les différentes fonctions présumées dela TSPO, mais aussi les débats récents qui touchent certaines d’entre elles, en commençant parla fonction qui a été de loin la plus étudiée, pour des raisons historiques : la biosynthèse desstéroïdes.
1.2.5.1 Transport du cholestérol dans la mitochondrie et stéroïdogenèse
Le cholestérol (§ 1.2.4.2) est le précurseur commun à toutes les hormones stéroï-diennes (e.g. cortisol, progestérone, testostérone, oestradiol, aldostérone). Il est métabolisépar des enzymes distribuées dans la mitochondrie et le RE des cellules formant les sté-roïdes [Payne and Hales, 2004]. La 1re étape de la synthèse des stéroïdes se déroule dansla mitochondrie et consiste en un clivage de la chaine aliphatique du cholestérol par lecytochrome CYP11A1 (nouveau nom de l’enzyme P450scc). Cette enzyme est localisée ducôté matricielle dans la membrane mitochondriale interne (MMI) [Churchill and Kimura,1979]. La 2e étape consiste à la formation de prégnénolone qui quitte la mitochondriepour rejoindre le RE où, après plusieurs transformations enzymatiques, elle sera finale-ment convertie en progestérone, testostérone ou oestradiol et autres hormones stéroïdiennes.Le cholestérol, venant de plusieurs localisations intracellulaires (Fig. 1.8), doit traverser la MME,l’espace intermembranaire et la MMI pour atteindre le CYP11A1. L’abondance de la TSPO dansles cellules des tissus stéroïdogènes tels que les testicules ou le cortex surrénale [Benavides et al.,

26 Chapitre 1. Les protéines TSPO
1983, De Souza et al., 1985], sa localisation dans la MME et la capacité de ses ligands à stimulerla stéroïdogenèse, a conduit à supposer que TSPO pouvait être impliquée dans la synthèse deshormones stéroïdes et dans le mécanisme de translocation du cholestérol à travers la MME. Plusrécemment, il a été montré que la protéine ATAD3A (ATPase family AAA Domain-containingprotein 3), dans la MMI, faisait le lien avec le CYP11A1 [Rone et al., 2012].
La 1re preuve associant TSPO à la stéroïdogenèse est l’observation d’une stimulation de labiosynthèse des stéroïdes dans les cellules stéroïdogéniques traitées avec les ligands synthétiquesde la TSPO (diazepam, Ro5-4864, PK11195) et son ligand endogène DBI. [Besman et al., 1989,Mukhin et al., 1989, Yanagibashi et al., 1989, Papadopoulos et al., 1990, Krueger et al., 1990,Papadopoulos, 1991, Papadopoulos et al., 1992, Boujrad et al., 1994, Papadopoulos et al., 1997].
Par la suite, d’autres études spécifiques sur la TSPO ont renforcé cette idée en montrant :
• Une inhibition de la synthèse des stéroïdes dans des cellules tumorales de Leydig, suite àl’inhibition de l’expression de TSPO [Boujrad et al., 1993, Papadopoulos et al., 1997], puisune restauration de cette synthèse suite à une transfection des cellules avec un vecteurcontenant l’ADNc de la TSPO[Papadopoulos et al., 1997].
• De nombreuses hormones, dont l’oestradiol et l’aldostérone, régule la TSPO [Batarseh andPapadopoulos, 2010, Basile et al., 1988, Mazurika et al., 2009] et leur présence apparaitnécessaire à l’expression constitutive : une diminution de la TSPO est observée dans lestissus stéroïdogènes suite à l’ablation de l’hypophyse, mais pas dans le cas d’une ablationdes glandes surrénales ou d’une castration [Anholt et al., 1985, Veenman and Gavish,2012] - or les hormones stéroïdes sont principalement produites par nos gonades, sur ordrede l’hypothalamus et de l’hypophyse - une sur-exposition aux hormones peut diminuer lesniveaux de la TSPO [Batarseh and Papadopoulos, 2010, Mazurika et al., 2009]. Il y auraitdonc une régulation hormonale de l’expression et de la fonction de la protéine.• La TSPO lie le cholestérol avec une affinité de l’ordre du nM [Lacapere et al., 2001].
Des études plus récentes ont avancé que la TSPO faisait partie de plusieurs complexesprotéiques, appelés le "transduceosome" [Liu et al., 2006] et le "metabolon" [Rone et al., 2012]par lesquels se fait le transfert du cholestérol du cytosol vers la matrice mitochondriale, Fig. 1.11.Le transduceosome initie le transport du cholestérol par stimulation hormonale, il est composé deprotéines cytosoliques et de la MME : PKA-RI, TSPO, VDAC, StAR, ACBD3 [Papadopouloset al., 2015]. Le metabolon est un complexe de 800 kDa composé de protéines de la MME,de l’espace intermembranaire et de la MMI - TSPO, VDAC, ATAD3A et CYP11A1 - et agitcomme intermédiaire entre l’import du cholestérol et sa conversion en prégnénolone. Un modèlede transport du cholestérol a été proposé, Fig. 1.11 : différentes composantes cytosoliques dutransduceosome tels que l’ACBD3, PKA-RI ou StAR lieraient et apporteraient le cholestérol àla TSPO jusqu’au complexe protéique formé par TSPO et VDAC.
À la suite de la liaison d’un ligand spécifique (p.ex. le DBI) sur la TSPO, celle-ci assure latranslocation du cholestérol à travers la MME. Puis, le transfert du cholestérol à travers l’espaceintermembranaire est assuré par l’ATAD3A dont le N-terminal est ancré dans MME, ce qui lui

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 27
Figure 1.11: Schéma des différentes protéines formant le transduceosome et le metabolon,complexes impliqués dans la régulation de la stéroïdogenèse [Papadopoulos et al., 2015]. Le"Transduceosome" est constitué de protéines cytosoliques et des protéines de la MME, il est indiqué parun encadré gris. Le "Metabolon" est un complexe protéique constitué des protéines de la MME, de lamatrice mitochondriale et de la MMI, il est indiqué par un encadré rouge. Dans les tissus stéroïdogéniques,le Transduceosome est activé en réponse à une stimulation hormonale permettant d’activer une voie designalisation AMPcdépendante qui va activer la protéine PKA-RI (PKA-RIα : cAMP-dependent proteinkinase regulatory subunit Ialpha). Cette protéine interagit avec ACBD3 (Acyl-Coenzyme A BindingDomain containing 3 protein) et permet la phosphorylation de la protéine StAR (Steroidogenic Acuteregulatory protein). Sous cette forme phosphorylée, la protéine StAR possédant un domaine C-terminalva s’ancrer dans la MME où il interagit avec les protéines du Metabolon telles que la TSPO et VDAC(Voltage-Dependent Anion Channel). Le cholestérol apporté par StAR peut être transporté à travers laMME par la TSPO à la suite d’une liaison avec un ligand (p. ex. DBI), puis à travers la matrice parl’ATAD3A (ATPase family AAA Domain-containing protein 3) qui fait la jonction entre la MME et laMMI. Finalement, le cholestérol est dirigé vers le cytochrome CYP11A1, dans la matrice mitochondriale,qui va le convertir en prégnénolone, précurseur des stéroïdes. Comment se fait la sortie hors de lamitochondrie de la prégnénolone ? . la TSPO y participe t-elle ? (flèche bleue). Cela reste à découvrir.Figure adaptée de [Papadopoulos et al., 2015]

28 Chapitre 1. Les protéines TSPO
permet de faire un pont entre la MME et la MMI où le cholestérol sera catalysé en prégnénolonepar le CYP11A1 [Liu et al., 2006, Rone et al., 2012, Papadopoulos et al., 2015]. Cette dernièresort de la mitochondrie par un mécanisme inconnu et est convertie en progestérone, testostéroneet oestradiol dans le RE.
Il a été proposé que la TSPO permet la translocation du cholestérol, en se comportant soitcomme un échangeur, soit comme un canal [Lacapère and Papadopoulos, 2003]. Dans le casd’un canal, le transport de la molécule à travers la membrane se ferait suivant leur gradient dediffusion. Dans le cas d’un transport par échange, l’hypothèse de fonctionnement propose que :dans un 1er temps la liaison du cholestérol suivi par la liaison d’un ligand activateur (PK 11195 ouDBI, le ligand endogène). La liaison de ce dernier conduit à un changement de conformation dela TSPO permettant au cholestérol de traverser la membrane. Une fois le cholestérol libéré, uneautre molécule située au niveau de l’espace intermembranaire mitochondrial (prégnénolone parexemple) se lierait à son tour à la TSPO. La dissociation du PK 11195 induirait un changementconformationnel qui permettrait la translocation de ce produit vers l’extérieur permettant declore un cycle de transport [Lacapère and Papadopoulos, 2003].
D’autre part, il a aussi été proposé que la TSPO pourrait former des polymères, régulant letransport de cholestérol et la vitesse de formation des stéroïdes [Delavoie et al., 2003, Rone et al.,2012]. Ces polymères de TSPO ont été montrés se former en présence de ROS [Delavoie et al.,2003]. Les auteurs proposent que la TSPO monomérique peut lier le cholestérol mais ne pas letransporter à travers la membrane mitochondriale, seuls les polymères de TSPO le pourraient.Ainsi à la suite d’une stimulation hormonale (les hormones étant à l’origine de la productionde ROS), la proportion de polymères de TSPO augmenterait, et sous l’activation d’un ligandpermettrait d’assurer la translocation du cholestérol à travers la membrane mitochondriale.Cependant, ce que les auteurs suggèrent n’est pas clair et plutôt confus. En effet, les auteursmontrent aussi que l’hormone progestérone, inhibe la liaison du cholestérol et des ligands parles polymères.
Une tentative pour générer des souris Knock-out (KO ; cf. Encadré 3 et Annexe B) a montréentraîner une létalité embryonnaire précoce sans que les auteurs puissent en expliquer la raison[Papadopoulos et al., 1997]. Dès lors, la TSPO a été considérée comme un élément indispensableaux organismes et indispensable à la biosynthèse des stéroïdes.
Remise en cause de la fonction Le caractère indispensable de la TSPO dans le dévelop-pement embryonnaire et la stéroïdogenèse à récemment était remis en cause.
1. En 2014, Morohaku et al. ont généré des souris TSPO Knock-out KO conditionnel (cf.Encadré 3 et Annexe B) spécifique aux cellules testiculaires de Leydig. La même année,Banati et al., Tu et al. ont généré des souris TSPO KO global ciblant les exons 2 et 3 (cf.Sec 1.2.3) de la TSPO. Chez ces animaux, aucune présence de la protéine n’a été observée,et aucun effet sur les niveaux des stéroïdes et sur la fertilité n’a été observé. Les auteursont alors conclu que la TSPO n’était ni essentielle, ni impliquée dans la biosynthèse des

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 29
La création d’une souris Knock-out (KO), dont un gène connu a été inactivé, permet d’évaluer les conséquencede la perte de la fonction chez la souris adulte ou d’évaluer, si elle est létale à quelle stade du développement ladéficience apparait. Le KO d’un gène consiste en son inactivation par sa délétion ou sa mutation et se base surla technique de recombinaison homologue.Le Knock-out conditionnel permet l’invalidation d’un gène dans un tissus ou à partir d’un stade de déve-loppement particulier. Il est utile lorsqu’un gène est exprimé dans plusieurs tissu ou à différents stades dedéveloppement. La technique la plus souvent utilisée est celle de la recombinaison Cre-lox.Le Knock-down d’un gène consiste en son inactivation partielle par diminution de son expression. Pour qu’ungène puisse s’exprimer et produire ses effets son ARN messager doit d’abord être traduit en protéine. Latechnique du Knock-down consiste donc à utiliser des ARN dits "interférents" (ARNi), ayant la même séquenceque l’ARNm du gène à inactiver, pour spécifiquement les détruire ou inhiber le processus de traduction de laprotéine. Cette méthode permet d’inactiver un gène mais de manière plus subtile que le Knock-out décrit plushaut. Ces techniques sont expliquées plus en détails en Annexe B.
Encadré 3 (Knock-out global, conditionnel et Knock Down).
stéroïdes et le transport du cholestérol, auquel cas des effets auraient été observés sur lestesticules et les surrénales.
2. En contradiction avec ces découvertes, en 2015, Fan et al. observent une forte mortalitéembryonnaire de leurs souris KO TSPO conditionnel spécifique aux gonades et ciblantles exons 2 et 3 de la TSPO. De plus, des souris KO TSPO conditionnel spécifique auxgonades et aux surrénales (conduit par un promoteur différent des précédentes souris KOutilisées dans l’étude) ne montrent pas de mortalité au stade embryonnaire mais une pertede leur capacité à former de la corticostérone, une hormone secrétée par le cortex de laglande surrénale à partir du cholestérol, en réponse à un traitement hormonal.
Ces résultats suggèrent un rôle important de TSPO dans le développement embryonnaire etdans la stéroïdogenèse induite par les hormones [Fan et al., 2015].
Les résultats des KO doivent être interprétés avec précautions : il existe une variabilité dansles résultats dépendant en grande partie de la souche à partir de laquelle les cellules souchesembryonnaires (SE) sont dérivées. Les schémas de sélection influent sur le processus de sélectionet les taux de mortalité sont rarement signalés. De plus, la perte d’un gène peut être compensépar un autre amenant ainsi à des phénotypes normaux ou peu altérés. L’équipe de Papadopouloset al. [2015] proposent que la létalité rapportée dans leur article de 1997 [Papadopoulos et al.,1997], lors de la délétion du gène Tspo entier de 11 kb dans les cellules SE, serait due à la présencede 2 gènes qui recouvrent partiellement le locus Tspo et dont l’expression est importante pourla survie.
Néanmoins, ces résultats soulèvent des questions sur la fonction exacte de la TSPO dans lastéroïdogenèse. Bien qu’aucun effet physiologique de la délétion du gène Tspo sur la stéroïdoge-nèse gonadique et surrénale n’ait été observé. Il sera intéressant d’étudier les effets des ligands

30 Chapitre 1. Les protéines TSPO
TSPO sur les niveaux de stéroïdes chez ces animaux génétiquement modifiés.De plus, il semble y avoir des redondances fonctionnelles pour les protéines mitochondriales,
à cause de leur importance dans la cellule et dans la physiologie animale. En effet, les sourisVDAC KO sont viables et affichent peu de changements phénotypiques manifestes [Messinaet al., 2012, Raghavan et al., 2012]. Ainsi, malgré l’importance apparente de VDAC, d’autresprotéines semblent reprendre ses fonctions après sa suppression. Il en est de même pour ANT(Adenine nucleotide translocator) qui conduit les échanges ATP-ADP [Graham et al., 1997] quisont essentielles à la vie de la cellules. Peut-être est-ce aussi le cas pour la TSPO? Ou alorssi la TSPO n’est pas requise pour la biosynthèse des stéroïdes quelle est sa fonction dans lacellule ? Ces observations appellent à d’autres études pour identifier les mécanismes cellulaireset moléculaires conduisant à l’importation du cholestérol dans la synthèse de stéroïdes.
L’expression de la TSPO dans de nombreux types de cellules qui ne produisent pas destéroïdes, dans les tissus comme les poumons, les reins, le foie, la rate et la moelle osseuse et lesintestins a laissé suggérer d’autres fonctions pour la TSPO tant au niveau mitochondrial quecellulaire.
1.2.5.2 La régulation de la biosynthèse et la biodégradation des porphyrines
Figure 1.12: Schéma illustrant l’implication hypothétique de la TSPO dans la bio-synthèse de l’hème.
Chez le rat, des taux d’expression élevés de la TSPO ont été détectés dans la moelle osseuse[Anholt et al., 1985], qui est le lieu de fabrication majeure des cellules sanguines telles queles érythrocytes qui transportent l’oxygène via l’hémoglobine. Une localisation au niveau deplusieurs cellules du système sanguin a également été montrée [Canat et al., 1993]. Commedécrit précedemment (§ 1.2.4.2), des expériences in vitro (sur des cellules et avec la TSPO

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 31
recombinante) ont montré que la TSPO pouvait lier plusieurs types de porphyrines telles quela coproporphyrine III, la protoporphyrine IX (PPIX) et l’hème (une porphyrine avec un ionfer) [Verma et al., 1987, Taketani et al., 1995b, Wendler et al., 2003]. Une fixation in vivo de laPPIX sur la TSPO chez le rat a même été observée par imagerie [Ozaki et al., 2010].
Ces données ont posé la question d’une implication possible de la TSPO dans la biosynthèsedes hèmes, et plus particulièrement du rôle de la TSPO dans la translocation des porphyrines,intermédiaires de la biosynthèse des hèmes, à travers la MME, Fig.1.12. D’autres observationsont montré que l’induction de la TSPO coïncidait avec l’induction d’enzymes de la biosynthèse del’hème et que la conversion du coproporphyrinogène III - précurseur produit dans le cytosol puistransloqué dans la mitochondre - en protoporphyrinogène IX diminuée en présence de ligandsBZs [Taketani et al., 1994, 1995b]. Enfin, un Knock-Down de la TSPO résulte en l’accumulationde PPIX exogènes dans la mitochondrie [Zeno et al., 2012].
Bien que la liaison des porphyrines telles que l’hème, la coproporphyrine III et la PPIX surla TSPO a bien été mise en évidence dans plusieurs espèces et plusieurs tissus, son implicationdans la biosynthèse de l’hème et le mécanisme de translocation à travers les membranes desmitochondries n’est pas élucidé. A quel moment la TSPO pourrait-elle intervenir dans le méta-bolisme de l’hème, Fig.1.12 ? La TSPO intervient-elle seule ou en formant des complexes ? Nouspouvons faire supposer que la TSPO peut être impliquée dans :
• la translocation de l’acide aminolévulinique (ALA) de la matrice mitochondriale vers lecytosol où il est transformé après plusieurs réactions en coproporphyrinogène III ;• la translocation du coproporphyrinogène III du cytosol vers l’espace intermembranaire mi-tochondrial où il est transformé en protoporphyrinogène IX, qui à son tour est transforméen PPIX et en hème après fixation d’un atome de fer ;• la sortie de l’hème de la mitochondrie pour les protéines à hème non mitochondriales,comme l’hémoglobine, la myoglobine et les oxydases ;• la sortie de la PPIX de la mitochondrie en cas d’hyperproduction p. ex.
La TSPO pourrait aussi participer à la dégradation des porphyrines comme il l’a été montrépour la TSPO de plante et la TSPO bactérienne (Cf. Sec. 1.4.6 et Sec. 1.5.2) et ainsi participerà l’homéostasie REDOX, cela est présenté dans la suite du manuscrit.
Remise en cause de la fonction Dans leur récente étude, Zhao et al. [2015] proclame quela TSPO n’est pas essentielle dans la biosynthèse de l’hème. En effet dans le but d’étudier lesliens supposés entre la TSPO et les pophyrines, les auteurs ont généré des souris TSPO KOglobal, des cellules TSPO KO de différents tissus et des cellules cancéreuses TSPO KO. Leursrésultats montrent que la déficience en TSPO n’a pas d’effet sur : l’erythropoïseProcessus dematuration des globules rouges., la biosynthèse de l’hème, la bioconversion de l’ALA en PPIXet la mort cellulaire induite par la phototoxicité de la PPIX . Les seuls effets observés sontsur l’homéostasie énergétique mitochondriale dans les fibroblastes 19 : il y a une diminution du
19. Les fibroblastes sont les cellules résidentes du derme.

32 Chapitre 1. Les protéines TSPO
taux de consommation d’oxygène et du potentiel de la membrane mitochondriale indicant unemodification du métabolisme cellulaire mais sans impact sur la biosynthèse des porphyrines.
Là encore des mécanismes de compensation pourraient être à l’origine de la non observationd’effets dramatiques de la délétion du gène Tspo. Chez les plantes et les bactéries, il a étésuggéré que la TSPO avait un rôle en lien avec les porphyrines. Chez les plantes la TSPO n’estpas essentielle (KO viable) [Guillaumot et al., 2009a] alors que chez les bactéries cela dépendde la souche : la délétion du gène Tspo est viable chez Rhodobacter sphaeroides [Yeliseev andKaplan, 1999] et Synorhizobium meliloti [Davey and de Bruijn, 2000] mais est toxique chez P.fluorescens Pf0-1 (thèse de Leneveu-Jenvrin [2014]).
1.2.5.3 Le transport d’ions et de nucléotides, le PMPT, l’apoptose et les cancers
Il a été montré que la TSPO est impliquée dans le transport d’ions (Ca2+ ou Cl−) dans lesastrocytes [Gandolfo et al., 2001], le rein [Basile et al., 1988] et la muqueuse gastrique [Ostuniet al., 2004]. Le mécanisme proposé est que l’activation de la TSPO par ses ligands provoque uneaugmentation du Ca2+ cytosolique (sortie de Ca2+ de la mitochondrie), induisant une activationde canaux présents dans les membranes plasmiques et conduisant à la sécrétion du chlore parles cellules [Basile et al., 1988, Ostuni et al., 2004, Gandolfo et al., 2001].
La perméabilité mitochondriale transitoire réfère à l’ouverture d’un pore non spécifique, le PMPT, dans la MMIconduit au passage d’ions et de petites molécules < 1.5 kDa. L’ouverture du PMPT est traditionnellement liéeà un dysfonctionnement mitochondrial suite à un stress : dépolarisation mitochondriale, arrêt de la synthèsede l’ATP, libération de Ca2+, déplétion de pyridine nucléotide (NAD et NADP), inhibition de la respiration,gonflement osmotique de la matrice qui peut conduire à son tour à la rupture de la MME et à la libérationdes protéines pro-apoptotiques tel que le cytochrome c contenu dans les crêtes de la mitochondrie. Ces effetsnéfastes, qui peuvent mener à l’apoptose - ou mort cellulaire programmée, processus par lequel des cellulesdéclenchent leur autodestruction en réponse à un signal - ne sont visibles que pour des ouvertures du PMPT delongue durée, tandis que les ouvertures à court terme peuvent être impliquées dans la régulation physiologiquede Ca2+ et dans l’homéostasie des ROS. [Bernardi and Di Lisa, 2015]. En condition normale, un recyclagepermanent des mitochondries a lieu via la mitophagie a, les événements de fusion/fission et la biogénèse. Demême, en condition normal, l’apoptose permet l’élimination des cellules anormales. Les 1res études suggéraientl’implication du VDAC, de l’ANT et de la TSPO dans ce pore. Mais des récentes études montrent qu’il seraitformé par un dimère d’ATP synthase F0F1 [Giorgio et al., 2013].
a. La mitophagie est la dégradation sélective des mitochondries par autophagie. Elle se produit souventlorsque les mitochondries sont défectueuses suite à des dommages ou un stress.
Encadré 4 (Le pore mitochondrial de perméabilité transitoire (PMPT)).
Des études de solubilisation et de co-immunoprécipitation de protéines mitochondriales derein de rat ont montré que la TSPO peut s’associer à 2 protéines : la porine de la MME VDACet l’ANT [McEnery et al., 1992].
Ces 2 protéines, qui interviennent dans les flux ioniques et les transports de nucléotides

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 33
dépendants du potentiel de membrane, ont été longtemps pressenties pour être des constituantsdu pore mitochondrial de perméabilité transitoire (PMPT), Encadré. 4, mais sa compositionexacte n’est pas encore clairement élucidée [Bernardi, 2013, Bernardi and Di Lisa, 2015]. CePMPT est impliqué dans certains évènements qui induisent l’apoptose, cf. Encadré.4. En raisondes relations existant entre la TSPO, le VDAC et l’ANT, il a été suggéré que la TSPO fassepartie du PMPT [Rone et al., 2009a,b]. En effet, Il a été montré que les ligands PK1195,Ro5-4864 et PPIX induisaient l’ouverture l’ouverture du PMPT [Kinnally et al., 1993, Chelliet al., 2001, Azarashvili et al., 2007]. Alors que l’utilisation d’un AC anti-TSPO peut bloquerl’ouverture du PMPT. De plus, l’activation de TSPO, suite à une stimulation avec du chlorurede cobalt (CoCl2), un agent mimant l’hypoxie (générant des ROS), engendre la mort cellulairepar apoptose [Zeno et al., 2009], cela laisse encore une fois envisager un lien entre TSPO etPMPT.
Remise en cause de la fonction L’implication de la TSPO dans le PMPT a donc été denombreuses fois évoquée par plusieurs équipes, mais on peut noter qu’un même groupe a donné2 avis opposés sur son implication à quelques années d’intervalle [Sileikyte et al., 2011, 2014].La remise en cause vient de l’utilisation, par cette équipe, de souris TSPO KO conditionnelspécifique au coeur ou foie qui suggère que les effets attribués à la TSPO ou à ses ligandsnaturels ou artificiels sur l’activité du PMPT pourraient être indirects [Sileikyte et al., 2014].En effet, ces auteurs montrent que la TSPO ne joue pas de rôle dans la régulation ou la structuredu PMPT. L’activation du PMPT par les ligands TSPO, à des concentrations élevées (∼ µM),se ferait par des mécanismes indépendants de TSPO, lesquels cela reste à découvrir [Selvarajand Stocco, 2015, Bernardi, 2013, Bernardi and Di Lisa, 2015]. Cette idée est appuyée par lefait qu’il a été découvert assez récemment, que l’ATP synthase FOF1 faisait partie du PMPT[Giorgio et al., 2013] et que le PK11195, encore une fois à des concentrations élevées, peutinteragir avec lui et l’inhiber [Johnson et al., 2006, Cleary et al., 2007, Seneviratne et al., 2012].D’autres part, le PK11195 pourrait réguler le signalement Ca2+ intracellulaire via des effets surla protéine pro-apoptotique Bcl-2 et ceci permettrait de rendre compte de son rôle dans la mortcellulaire par apoptose [Campanella et al., 2008].
Régulation du métabolisme cellulaire lors de la prolifération cellulaire en particulierdans les cancers. La prolifération cellulaire et le cancer ont un lien avec le contrôle del’apoptose : une défectuosité du processus d’apoptose représente une des causes majeures dudéveloppement et de la progression du cancer. De même, la modification de la régulation decertains processus cellulaires, y compris ceux qui sont associés aux mitochondries et au transportdu cholestérol (les cellules tumorales présentent une accumulation du cholestérol [Yoshioka et al.,2000]), est en partie responsable de la prolifération effrénée des cellules cancéreuses.
Une surexpression de TSPO ou une augmentation de la liaison des ligands de TSPO ontété observées dans de nombreux tissus cancéreux - du sein, des ovaires, du colon, du foie, dela prostate, dans les tumeurs gliales du cerveau - et son expression semble corrélée au degré

34 Chapitre 1. Les protéines TSPO
de malignité de la tumeur. Ces données suggèrent un rôle de la TSPO dans la cancérogenèse[Batarseh and Papadopoulos, 2010, Miettinen et al., 1995, Corsi et al., 2008, Venturini et al.,1998, Hardwick et al., 1999]. Il a été proposé que la TSPO joue un rôle dans cette prolifération,notamment à cause de l’effet anti-prolifératif observé de la part des ligands TSPO dans un largeéventail de type de cellules cancéreuses [Austin et al., 2013]. Par exemple :
• Dans le cancer du sein humain, de nombreux ligands pharmacologiques de la TSPOcontrôlent la prolifération cellulaire [Galiegue et al., 2004, Han et al., 2003, Hardwicket al., 1999, Papadopoulos et al., 2000]. De plus, un Knock-down de la TSPO diminue laprolifération cellulaire in vitro et la croissance tumorale in vivo.• Dans le cancer du cerveau (neuroblastome), le PK11195 induit l’apoptose et une diminu-tion de la prolifération des cellules tumorales [Mendonca-Torres and Roberts, 2013].
Les mécanismes conduisant à la surproduction de la TSPO dans les cancers ne sont pasencore élucidés : s’agit-il d’une augmentation du nombre de mitochondries dans les cellules oudu nombre de TSPO par mitochondrie ? [Venneti et al., 2006] Le rôle spécifique de la TSPOdans les cellules cancéreuses n’est pas clair non plus. Les ligands TSPO ont-ils un effet sur lescellules cancéreuses via l’apoptose ? Dans ce cas, les mécanismes impliqués sont-ils dépendantsou indépendants de la TSPO?
Une hypothèse émise est que la TSPO pourrait être impliquée dans l’accumulation du choles-térol dans les cellules tumorales, et ainsi influencer le degré de fluidité des membranes, activantainsi des mécanismes de signalisation impliqués dans la prolifération cellulaire [Singleton andBourguignon, 2004]. En attendant, cela fait de la TSPO une cible thérapeutique potentielle enutilisant ses ligands artificiels tels que le PK 11195 ou le Ro5-4864 [Austin et al., 2013]. De plus,l’utilisation de ces ligands radiomarqués permet l’imagerie des cancers et de leurs évolutions.
1.2.5.4 L’homéostatie énergétique mitochondriale et l’homéostatie REDOX/ROS(cf. Encadré 5)
La respiration mitochondriale Des études anciennes ont suggéré un rôle de la TSPO dansle métabolisme énergétique mitochondriale. Dans les mitochondries de cellules de rein, du foie,des glandes surrénales et du cerveau de rongeurs [Hirsch et al., 1988, Larcher et al., 1989],un effet inhibiteur des ligands TSPO sur la consommation d’O2 a laissé suggérer une possibleinteraction de la protéine avec les enzymes de la chaîne respiratoire [Veenman and Gavish,2006]. La présence de la TSPO dans les mitochondries a fait l’objet de nombreuses études surles interactions de la TSPO avec la chaine respiratoire, source de production de ROS. Maisaucune n’a permis de conclure de comment la TSPO est impliquée dans la respiration cellulaire[Gavish et al., 1999].
Plus récemment, [Zhao et al., 2015] ont montré sur des cellules de fibroblastes TSPO KO,une diminution de la consommation en O2, des niveaux d’ATP et du potentiel membranaireindiquant un changement dans l’homéostasie énergétique de la cellule, qui pourrait influencerdes événements et des réponses cellulaires. Une diminution des niveaux d’ATP a également été

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 35
L’homéostasie énergétique se définit comme le maintien de l’équilibre entre les apports et les dépenses d’énergie. Ilnécessite une coordination entre l’activité cellulaire, la disponibilité des nutriments et la régulation des processusde production énergétique. La respiration mitochondriale est un ensemble de réactions qui permette de produirel’énergie nécessaire au fonctionnement de la cellule sous forme d’ATP : le glucose, grâce à la glycolyse, au cycle deKrebs et à la chaîne respiratoire, permet la production d’ATP ; de même, la dégradation des acides gras selon unensemble de réactions d’oxydations permet d’obtenir l’énergie nécessaire à leur survie. Puisque la mitochondrieest le lieu central de consommation de l’oxygène durant la phosphorylation oxydative, la formation de ROS par lachaîne respiratoire mitochondriale est normale et fait partie intégrante de l’homéostasie énergétique et REDOX a
de la cellule. Les ROS ainsi qu’un potentiel REDOX équilibré sont déterminants pour assurer correctement lesfonctions moléculaires de la cellule comme l’organisation des mécanismes de défense et de signalisation cellulairequi régulent la vie cellulaire (croissance, prolifération et différentiation) en général. Cependant, si l’homéostasieREDOX de la cellule est affectée par une élévation trop marquée des ROS, on parle alors de stress oxydant,des dommages peuvent être causés aux structures biologiques par ces agents oxydants et des concentrationsanormales de ROS dans les mitochondries peuvent amorcer l’apoptose. L’inflammation est étroitement liée à unétat d’oxydo-réduction particulier comportant une production de ROS, mais là encore une production prolongéede ces molécules devient la source de nombreuses pathologies, humaines en particulier, notamment de maladiesneurodégénératives et de cancers.
a. L’homéostatie REDOX est l’équilibre entre les oxydants et les réducteurs. Le déséquilibre est la plupartdu temps causé par un excès d’oxydants et/ou un manque de réducteurs.
Encadré 5 (Homéostasie énergétique, REDOX et stress oxydant dans les mitochondries).
observée sur les cellules microgliales KO TSPO [Banati et al., 2014].
Comme nous l’avons vu précédemment, Morohaku et al. [2014], Tu et al. [2014] (mêmeéquipe), ont réussi à générer les 1res souris KO TSPO. Le dernier KO de la TSPO, réalisépar cette équipe concerne les cellules stéroïdogéniques de Leydig [Tu et al., 2016]. Les auteursobservent sur ces cellules

36 Chapitre 1. Les protéines TSPO
• un changement dans le métabolisme énergétique mitochondriale de ces cellules : une pro-duction de l’énergie à partir des acides gras plutôt que du glucose (cf. Encadré 5), associéeà une augmentation significative de l’oxydation des acides gras.• une augmentation de la production de ROS et une régulation positive des gènes impliquésdans le oxydation des acides gras .
Par contre certains résultats sont en contradictions avec les KO réalisés par Banati et al. [2014]et par cette même équipe [Zhao et al., 2015] sur d’autres types de cellules : la migroglie [Banatiet al., 2014] et les fibroblastes [Zhao et al., 2015]. Ils n’observent ni changement dans la consom-mation d’O2, ni dans le potentiel membranaire mitochondrial. Les auteurs suggèrent alors quecette différence est due aux types de cellules utilisés : la microglie et les fibroblastes ne sontpas des cellules où le métabolisme lipidique est important contrairement aux cellules stéréogé-niques qui sont capables d’un métabolisme lipidique actif permetant de maintenir leur niveaude consommation oxygènique. Cette étude est la première qui lie TSPO et acides gras. Ellemontre que la TSPO affecte l’homéostasie énergétique mitochondriale à travers une modulationde l’oxydation des acides gras.
La régulation des ROS La TSPO pourrait avoir un rôle de régulation du stress oxydatif.D’une part, plusieurs études montrent qu’une surexpression de la TSPO atténue la produc-
tion de ROS [Carayon et al., 1996, de Tassigny et al., 2013, Veenman et al., 2008] :
• Dans les cellules des tissus hématopoïétiques 20, une surexpression de la TSPO dans lescellules résistantes au peroxyde d’hydrogène suggère un rôle possible de la protéine dansun mécanisme antioxydant qui pourrait impliquer les porphyrines [Carayon et al., 1996]ou la régulation de la superoxyde dismutase, une protéine anti-oxydante [Wright andReichenbecher, 1999].
• Sur des cellules de glioblastome (tumeur cérébrale), exposées à de la PPIX, la TSPOen présence de ROS, prévient l’accumulation intracellulaire de PPIX [Zeno et al., 2012].Ceci suggére un rôle anti-ROS de la TSPO via une dégradation de l’excès de PPIX [Zenoet al., 2012], qui est comme nous le verrons à la Sec.1.5.2.2 une capacité qu’ont les TSPObactériennes.• Dans les couches superficielles de l’épiderme, la surexpression de la TSPO induit uneprotection transitoire contre les ROS générés par une exposition aux UV [Stoebner et al.,2001].• La surexpression de la TSPO inhibe la production mitochondriale de ROS dans des cellulesendothéliales 21 [Joo et al., 2015].
D’autre part, plusieurs études montrent qu’une surexpression de la TSPO engendre l’effetinverse [Gatliff et al., 2014, Gatliff and Campanella, 2015, Klubo-Gwiezdzinska et al., 2012] :
• Une surproduction de la TSPO engendre une production de ROS, qui est inhibée après unKnock-down de la protéine [Gatliff et al., 2014, Gatliff and Campanella, 2015]. De plus, les
20. Les cellules hématopoïétiques sont à l’origine des différentes cellules du sang.21. Les cellules endothéliales tapissent la face interne des vaisseaux.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 37
auteurs observent que la TSPO (en collaboration avec VDAC), via cette augmentation desROS, inhibe la mitophagie en empêchant l’ubiquitination des protéines 22. Ils en concluentque la TSPO et VDAC pourraient, via les ROS être des régulateurs du contrôle de laqualité des mitochondries.• Tout récemment, Guilarte et al. [2016] montrent que, dans la microglie activée 23 l’inter-action entre la TSPO et la NADPH oxydase (NOX2) - un complexe enzymatique qu’onretrouve dans les phagocytes, producteur de ROS - est liée à une production de ROS.Cette dernière induit une réponse antioxydante dont le but pourrait être de maintenirl’homéostasie REDOX dans les cellules gliales afin de prolonger leur durée de vie lorsd’un stress. Mais lorsque la microglie est chroniquement activée, ce qui se produit sou-vent dans les processus de neuroinflammation chronique (p. ex. maladies dégénératives),l’équilibre de l’interaction TSPO-NOX2 est rompu. Les mécanismes de défense ne sontalors plus en mesure de régler le niveau de la production de ROS, lesquels créeraient alorsdes dommages à la microglie et aux neurones environnants [Guilarte et al., 2016].
Toutes ces données indiquent que la TSPO est liée à la régulation des l’homéostatisie RE-DOX/ROS, qui à l’air d’être une fonction dépendante des tissus [Gatliff and Campanella, 2016].Cette idée est renforcée par le fait que des FT qui opèrent an aval d’une voie sensible aux ROScontrôlent la transcription du gène Tspo [Gatliff et al., 2014, Gatliff and Campanella, 2015].
On peut aussi rappeler qu’il a été montré que la présence de ROS peut induire la formationde polymères (dimère, tetramère et octamère) covalents de TSPO [Delavoie et al., 2003]. LaTSPO pourrait donc jouer son rôle de régulateur de ROS par polymérisation.
La production des ROS est étroitement liée aux processus d’inflammation, l’étude de Gui-larte et al. [2016], en donne un bon aperçu. Ainsi, les observations rapportées ci-dessus, concer-nant le rôle de la TSPO dans l’homéostatie des ROS, peuvent être reliées aux données qui sontprésentées dans le paragraphe qui suit sur le rôle de la TSPO dans le processus d’inflammation.Le détermination du rôle de la TSPO dans le processus de d’inflammation est un défi importantpuisque c’est de cette implication que la TSPO tire une grande partie de son potentiel de ciblethérapeutique. Il en est de même dans le cas des cancers où la TSPO fait office de cible pourleur diagnostic et leur traitement. Dans le cas des cancers l’inflammation chronique peut êtreégalement un facteur à risque.
1.2.5.5 L’inflammation
L’inflammation hors du SNC L’inflammation est la réponse des tissus vivants, vascularisés,à une agression : infection, trauma, brûlure, allergie, nécrose, cellules cancéreuses, etc. Les cel-lules de l’inflammation incluent notamment, les lymphocytes, les phagocytes et les fibroblastes.Ces cellules, une fois activées, libèrent des composés médiateurs de l’inflammation : histamine,cytokines (p. ex. TNF-α), ROS, etc. Les conséquences fonctionnelles de cette activation sont
22. Les protéines destinées à être dégrader sont marquées par des ubiquitines.23. L’activation de la microglie est abordée dans la section qui suit : Encadré 6

38 Chapitre 1. Les protéines TSPO
l’élimination du pathogène (p. ex. par phagocytose) et/ou la réparation de la lésion.Il a été montré que la TSPO et ses ligands étaient impliqués dans la réponse inflammatoire,
par exemple dans l’ischémie de re-perfusion 24[Faure et al., 2002, Favreau et al., 2009].TSPO est également surexprimée au stade précoce de l’inflammation dans les maladies
inflammatoires de l’intestin telles que la maladie de Crohn ou la rectocolite hémorragique. Deplus, des ligands de TSPO améliorent la réparation de l’intestin. La protéine peut donc servircomme marqueur des processus de réparation et cela laisse envisager des approches pour letraitement de ces maladies [Ostuni et al., 2010b].
Durant un stress inflammatoire, la libération de TNF-α et de ROS peuvent déstabiliserl’ultrastucture mitochondriale et induire la mort cellulaire par apoptose. La TSPO aurait unrôle dans le maintient de cette architecture mitochondriale et le contrôle de la production deROS [Issop et al., 2016].
L’inflammation dans le SNC : les neuropathologies. Dans un cerveau sain, l’expressionde la TSPO est faible et limitée aux cellules gliales (astrocytes et microglie) [Chen and Guilarte,2008, Cosenza-Nashat et al., 2009, Papadopoulos et al., 2006c, Veenman and Gavish, 2000].Cependant, dans un cerveau endommagé, son expression augmente considérablement dans lescellules gliales activées de la zone de la lésion ou de l’inflammation [Chen et al., 2004, Chen andGuilarte, 2006, Kuhlmann and Guilarte, 2000].
En plus de cela, plusieurs données suggèrent que la TSPO pourrait participer à la régulationde la neuroinflammation :
• Les ligands TSPO - PK11195 [Veiga et al., 2005, 2007], XBD173 [Karlstetter et al., 2014],Ro5-4864 [Veiga et al., 2005, Ryu et al., 2005] et endozépines (DBI, TTN ; cf. Sec.1.2.4.2)[Wang et al., 2014] - diminuent la neuroinflammation : diminution du niveau de microglieactivée, de la production de cytokines pro-inflammatoires, de la production de ROS etrégulation de l’activité des macrophages• simultanément à la TSPO, le DBI (cf. Sec.1.2.4.2) est lui aussi régulé positivement dansles astrocytes et les cellules de Müller activés 25 [Wang et al., 2014] ;
• un Knock-down de la TSPO dans des cellules microgliales activées augmente l’inflamma-tion [Bae et al., 2014].
Une hypothèse proposée est que la TSPO pourrait être surexprimée pour réguler, avec sesligands, l’activation de la microglie en participant à la communication entre les astrocytes et lamicroglie (Cf. Encadré 6) [Wang et al., 2014, Bae et al., 2014] : la TSPO pourrait être médiateurd’un signal via p. ex. les ROS ou l’expression et la sécrétion de TNF-α.
Pour compléter cette idée, on peut la mettre en relation avec les travaux de Guilarte et al.[2016], Choi et al. [2011], cité précédemment, qui montrent une interaction entre NOX-2 etTSPO pour produire des ROS dans le but de générer une réponse antioxydante. Ces donnés
24. L’ischémie de re-perfusion est une lésion du tissu causée par le retour soudain du sang dans le tissu aprèsune période de manque d’oxygéné (ischémie).25. Les cellules de Müller sont des cellules gliales de la rétine.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 39
L’activation des cellules microgliales (ou microglie) et des astrocytes est une réponse caractéristique du SNCface à une inflammation du tissu nerveux.
(1) Hypoxie, traumatisme, infection, AVC, auto-immunité, peuvent être parmi les facteurs déclenchant l’acti-vation des astrocytes et de la microglie. Cette activation mène à des changements morpholoqiques (2) et à lasécrétion de molécules mediatrices de l’inflammation (p. ex. cytokines, glutamate, ATP, NO, ROS). (3) Lesastrocytes et la microglie activés présentent des fonctions homéostatiques telles que la diminution du stress oxy-datif, la libération de médiateurs anti-inflammatoires (comme les cytokines interleukines). De plus la microglieactivée en tant que macrophage, peut libérer des agents pro-inflammatoire (p. ex. TNF-α) pour stimuler lesfonctions immunes, et peuvent utiliser la phagocyte pour éliminer des éléments étrangers. (4) L’inactivationnormale de la microglie activée est en partie médiée par les astrocytes qui : inhibent la phagocytose microgliale,diminuent la production du TNF-α par la microglie et les ROS et libérent des molécules anti-inflammatoires. Engénérale, ce processsus inflammatoire est une réponse protectrice permettant de détruire et éliminer les agentsnuisibles et les tissus endommagés bénéficiant ainsi la réparation des tissus. (5)-(6) Une activation chroniquedes astrocytes et de la microglie ou des astrocytes non contrôlées sont, entre autre, associés à une libérationexcessive de molécules pro-inflammatoires, un stress oxydatif (génération de ROS) et un déséquilibre ionique.(7) Cet ensemble de phénomènes a de nombreux effets néfastes, y compris l’épileptogenèse, les convulsions, leslésions neuronales qui peuvent mener à la mort des neurones. Ces effets néfastes peuvent maintenir l’activationpathologique chronique des astrocytes et de la microglie. La neuroinflammation chronique peut aboutir à desmaladies neurodégénératives ou des désordres psychiatriques. Schéma adapté de [Kosonowska et al., 2015].
Encadré 6 (Le processus de neuroinflammation).

40 Chapitre 1. Les protéines TSPO
suggèrent donc un rôle de régulateur dans l’activation de la microglie par la TSPO en lien avecune régulation du stress oxydatif, Fig 1.13a.
La neuroinflammation a un rôle clé dans le processus de guérison des lésions du cerveau,mais une activation chronique peut devenir dangereuse pour les cellules neuronales et constituéun facteur important dans les processus de neurodégénération, Encadré. 6.
Les maladies neurodégénératives La neuroinflammation est donc une composantecommune des neuropathologies telles que les maladies neurodérégénératives et les lésions trau-matiques du cerveau et elle pourrait aussi jouer un rôle dans les troubles psychiatriques [Reuset al., 2015].
De nombreuses études montrent une sur-expression de la TSPO, dans des modèles d’animauxet des tissus postmortem humains présentant des neuropathologies telles que la sclérose enplaque, la maladie d’Alzheimer, la maladie d’Huntington, la maladie de Parkison, l’épilepsie,l’encéphalite, Les gliomes, les accidents vasculaires cérébraux [Venneti et al., 2006, Chen andGuilarte, 2008, Papadopoulos et al., 2006c, Veenman and Gavish, 2000].
De nombreux ligands TSPO ont montrés un effet sur ces maladies neurodégénératives. Parexemple :
• chez la souris, la combinaison du PK11195 et du Ro5-4864 réduit les niveaux de β-amyloïdes, typiques de la maladie d’Alzheimer [Barron et al., 2013] ;• l’etifoxine (une benzoxazine, cf. Sec. 1.2.4.1), dans des modèles de souris de sclérose en
plaque [Daugherty et al., 2013] et le SSR180575 (7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide) dans des modèles de souris de neuro-pathie diabétique 26 montrent des effets neuroprotecteurs [Ferzaz et al., 2002].
Comment ces ligands exercent cet effet neuroprotecteur ? Comme les effets des ligands TSPO,dans les modèles animaliers de troubles psychiatrique et neurodégénératifs, sont souvent associésavec une augmentation locale des niveaux de neurostéroïdes, ces derniers pourraient apporterune réponse.
En effet, certains neurostéroïdes (progestérone, allopregnénolone) exercent un effet neuropro-tectant (réduction de la perte des neurones, amélioration de la récupération comportementale etcognitive) dans un grand nombre de maladies neurodégénératives et de troubles psychiatriques[Charalampopoulos et al., 2008, Bristot et al., 2014, Melcangi et al., 2014, Schule et al., 2014].Or, les ligands pharmacologiques de la TSPO - PK11195, Ro5-4864, XBD173, SSR180575, Eti-foxine, etc. - stimulent la formation de neurostéroïdes [COSTA et al., 2006, Papadopoulos et al.,1992, Rupprecht et al., 2010]. Ces neurostéroïdes sont synthétisés par les neurones et les cellulesgliales [Baulieu, 1997]. Ainsi, une hypothèse est que les ligands TSPO pourraient exercer leursactions neurothérapeutiques en partie en induisant la synthèse des neurostéroïdes dans les cel-lules gliales. [Serra et al., 1999, Verleye et al., 2005, Daugherty et al., 2013, Rupprecht et al.,2009, Giatti et al., 2009, Zhang et al., 2014]. Cependant, il n’y a pas de preuves directes d’un lien
26. La neuropathie diabétique est une complication du diabète qui consiste en une atteinte des nerfs.
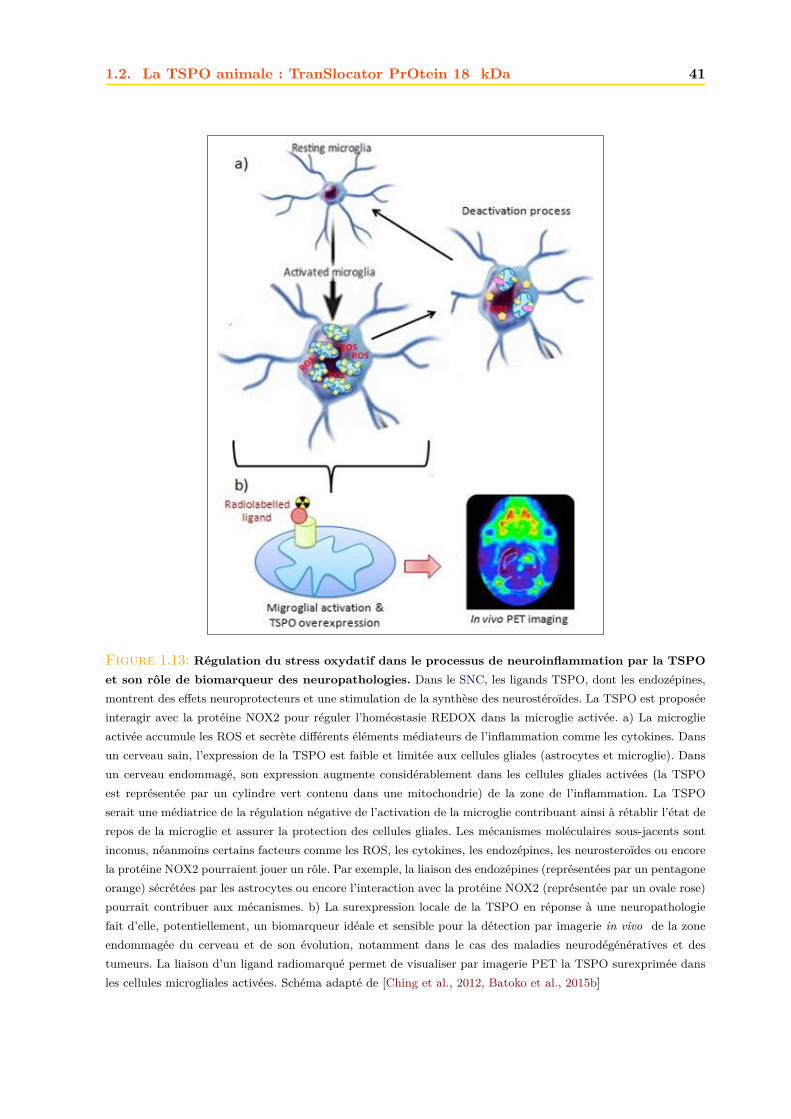
1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 41
Figure 1.13: Régulation du stress oxydatif dans le processus de neuroinflammation par la TSPOet son rôle de biomarqueur des neuropathologies. Dans le SNC, les ligands TSPO, dont les endozépines,montrent des effets neuroprotecteurs et une stimulation de la synthèse des neurostéroïdes. La TSPO est proposéeinteragir avec la protéine NOX2 pour réguler l’homéostasie REDOX dans la microglie activée. a) La microglieactivée accumule les ROS et secrète différents éléments médiateurs de l’inflammation comme les cytokines. Dansun cerveau sain, l’expression de la TSPO est faible et limitée aux cellules gliales (astrocytes et microglie). Dansun cerveau endommagé, son expression augmente considérablement dans les cellules gliales activées (la TSPOest représentée par un cylindre vert contenu dans une mitochondrie) de la zone de l’inflammation. La TSPOserait une médiatrice de la régulation négative de l’activation de la microglie contribuant ainsi à rétablir l’état derepos de la microglie et assurer la protection des cellules gliales. Les mécanismes moléculaires sous-jacents sontinconus, néanmoins certains facteurs comme les ROS, les cytokines, les endozépines, les neurosteroïdes ou encorela protéine NOX2 pourraient jouer un rôle. Par exemple, la liaison des endozépines (représentées par un pentagoneorange) sécrétées par les astrocytes ou encore l’interaction avec la protéine NOX2 (représentée par un ovale rose)pourrait contribuer aux mécanismes. b) La surexpression locale de la TSPO en réponse à une neuropathologiefait d’elle, potentiellement, un biomarqueur idéale et sensible pour la détection par imagerie in vivo de la zoneendommagée du cerveau et de son évolution, notamment dans le cas des maladies neurodégénératives et destumeurs. La liaison d’un ligand radiomarqué permet de visualiser par imagerie PET la TSPO surexprimée dansles cellules microgliales activées. Schéma adapté de [Ching et al., 2012, Batoko et al., 2015b]

42 Chapitre 1. Les protéines TSPO
entre l’augmentation des neurostéroïdes et l’action des ligands. On ne sait pas si l’augmentationde la neurostéroïdogène par les ligands de la TSPO pourrait être contrôlée par des mécanismesdépendants ou indépendants de la TSPO [Tu et al., 2014].
Les lésions du cerveau Dans les pathologies liées aux lésions du cerveau, la surexpressionde la TSPO est aussi directement corrélée avec le degré des dommages. Ainsi, la TSPO peutêtre utilisée, via l’utilisation de ces ligands radiomarqués, comme biomarqueur de l’état et laprogression des lésions traumatiques [Papadopoulos and Lecanu, 2009]. De plus, plusieurs étudesont montré les effets neuroprotecteurs des ligands de la TSPO dans des modèles expérimentalsde lésion du cerveau : par exemple Soustiel et al. [2008], Veiga et al. [2005] montrent qu’uneadministration de Ro5-4864 augmente le nombre de neurones survivants et préserve le réseauneuronal. De plus, le PK11195 tout comme le Ro5-4864 permettent la régénération des nerfsdans des modèles animaliers de lésion du nerf sciatique [Lacor et al., 1999, Leonelli et al., 2005,Mills et al., 2005, 2008].
Les mécanismes et les implications physiologiques de comment et pourquoi la TSPO répondà une lésion du cerveau sont inconnus.
Toutes ces études sur la TSPO en lien avec la neuroinflammation (activation de la micro-glie, maladies neurodégénératives, lésions du cerveau) ont aboutit à l’utilisation de la TSPOcomme biomarqueur des pathologies actives du cerveau. L’utilisation des ligands radiomarquésde la TSPO permet la visualisation des cellules gliales activées dans lesquelles la TSPO estsurexprimée, Fig1.13b. Ceci fournit une excellente approche pour :
• la détection des maladies du cerveau ;• l’étude de la progression de la neuroinflammation et de la neurodégénérescence ;• l’étude de la progression tumorale ;• et le suivi des effets des stratégies thérapeutiques.
1.2.6 Adressage
Comme la plupart des PMb, la TSPO est génétiquement codée dans le noyau, translatéedans le cytosol et importée dans la mitochondrie. Contrairement aux PMb de la matrice ou de lamembrane interne de la mitochondrie, les protéines de la MME n’ont pas de peptide signal pourl’adressage à la mitochondrie. Elles sont adressées à la MME via des séquences d’a.a internes,cf. Enc.7.
À ce jour ces signaux d’adressage à la MME n’ont pas été identifiés ou prédits pour les PMbà plusieurs TM et le signal ou les signaux dans la TSPO sont inconnus. Cependant, Rone et al.[2009b] ont identifié, par mutations, 2 zones nécessaires pour l’adressage, l’importation et lafonction de la TSPO :
• Un motif de Schellman allant des a.a 103 à 108, qui fournit l’orientation structurale néces-saire pour l’importation et l’insertion de la TSPO. Ce motif, souvent retrouvé terminantune Hα, est stabilisé par des interactions hydrophobes entre les a.a autour d’un résidu

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 43
Contrairement à l’intégration co-translationnelle des PMb dans la membrane du RE, il a été proposé que laplupart des PMb mitochondriales soient complètement synthétisées comme des protéines précurseurs, des pré-protéines, par des ribosomes libres dans le cytosol, puis transférées dans la mitochondrie par un mécanismepostranslationnel. La pré-protéine est dans une conformation non repliée et est associée avec des protéineschaperonnes (Hsc70 et Hsp90) qui la maintienne dans une conformation permettant sa translocation. Quel estle signal qui mène à la liaison des protéines chaperonne à la pré-protéine ? Cela n’est pas clair.Les pré-protéines peuvent être dotées d’une extension N-terminale de 20 à 50 (caractérisée par une tendanceà se replier en une Hα amphipatique avec une face hydrophobe et l’autre chargée négativement), appelée pré-séquence ou peptide signal, qui permet leur adressage à la surface de la mitochondrie et leur transport et leur trivers les différents compartiments mitochondriales. Cependant, de nombreuses pré-protéines ne possèdent pas depeptide signal et à la place possèdent un signal ou des signaux d’adressage interne dans leur séquence. En effet,les PMb de la matrice ou de la MMI sont généralement dotées de ce peptide signal contrairement aux protéinesde la MME. La nature de ces signaux internes reste assez vague.Il a été montré que le complexe TOM (Translocase of the Outer Membrane) facilite l’importation de la plupartdes PMb mitochondriales. TOM20 et TOM70 sont les 2 principaux récepteurs de ce complexe qui reconnaissentla pré-protéine. Le domaine cytosolique de TOM70 possède des motifs qui reconnaissent préférentiellement lespré-protéines hydrophobes qui ont une séquence signale interne. Ces motifs non seulement reconnaissent la pré-protéine mais aussi interagissent avec les chaperonnes, principalement Hsp70 et Hsp90 chez les animaux. Cecomplexe utilise l’ATP pour transporter et insérer la pré-protéine et la libérer des chaperonnes. TOM20 est lerécepteur des peptides signals, il interagit avec la face hydrophobe de cette pré-séquence.Ainsi ce complexe TOM est le candidat tout désigné pour faire partie de la machinerie qui décode les signauxd’adressage des protéines de la membrane externe et qui contrôle leur insertion dans la membrane. Cependant, onne sait pas grand chose des mécanismes par lesquels ces protéines sont reconnues à la surface de la mitochondrieet sont insérées dans la membrane externe. [Akopian et al., 2013, Rapaport, 2003, Walther and Rapaport, 2009]
Encadré 7 (L’adressage des protéines membranaires (PMb) dans la MME).
glycine en position i+4 (en position 106 chez la TSPO) [Viguera and Serrano, 1995]. Dansla TSPO, ce motif se situe dans la boucle cytosolique reliant la TM3 et TM4 (cf. Sec.1.2.7,Fig.4.12). L’élimination des a.a 103 à 108, de même que la mutation ponctuelle G106A -qui perturbe le motif de Schellman en réduisant la flexibilité - donnent lieu à des agrégatsde GFP-TSPO dans les zones adjacentes à la mitochondrie.• Le domaine C-terminal et le motif CRAC requis pour l’insertion dans la MME.
La TSPO est adressée à la MME via des interactions avec des chaperonnes cytosoliques (Hsc70et Hsp90), Fig. 1.14. À la MME, la TSPO interagit avec TOM70 et elle est libérée des chape-ronnes d’une manière dépendante de l’ATP. Son adressage et insertion dépendent de la structurede la TSPO (domaine C-terminale et le motif de Schellman) mais ne nécessite pas les autrescomposants du complexe TOM. Son insertion dans la MME est assistée par la protéine Metaxin1 (une PMb de la MME suggérée être impliquée dans l’importation des protéines dans la mi-tochondrie). À ce moment la TSPO est probablement sous sa forme repliée. L’insertion dans laMME pourrait être accélérée par un traitement au cAMP (cyclic Adenosine MonoPhosphate,un second messager qui mène à une augmentation de la synthèse lipidique, protéique et de la

44 Chapitre 1. Les protéines TSPO
phosphorylation protéique). Une fois l’importation terminée, la TSPO pourrait s’associer avecd’autres protéines de la MME, comme VDAC, pour former des complexes.
Figure 1.14: Mécanismes d’importation de la TSPO dans la MME. La TSPO est adressée à laMME à travers des interactions avec les chaperonnes cytosoliques, Hsp70 et Hsp90. À la MME, TSPOinteragit avec TOM70 et est libérée des chaperonnes d’une manière dépendante de l’ATP. L’insertionde la TSPO dépend de Metaxin 1 et de sa structure, où à la fois le domaine C-terminal et le motif deSchellman motif (a.a 103-108) sont nécessaire pour former un complexe de 66 kDa. Une fois l’importationachevée, la TSPO peut s’associer avec d’autres protéines de la MME comme VDAC et ANT. D’aprèsRone et al. [2009b].
1.2.7 Structure
La résolution d’une structure d’une PMb est toujours un défi, Encadré. 8. Au début de mathèse, aucune structure atomique à haute résolution de la TSPO n’existait, bien qu’une grandevariété de techniques avaient été utilisées pour acquérir des données structurales à partir de laprotéine recombinante.
La TSPO est composée de 169 a.a, ce qui représente environ 18 kDa, dont environ 7.7% d’a.aaromatiques, en particulier elle possède 12 tryptophanes (Trp). C’est pourquoi son homologuebactérien fut nommé "Tryptophane rich Sensory PrOtein".
La structure primaire de cette protéine est fortement conservée chez les mammifères, il y a∼80% d’identité entre les séquences humaine, bovine, murine et de rat [Culty et al., 1999]. L’ana-lyse des différentes séquences en acides aminés suggérait la présence de 5 domaines hydrophobes[Fan et al., 2012].
Les 1ers travaux de topologie par marquage des épitopes ont caractérisé la nature transmem-branaire de ces fragments avec la partie N-terminale dirigée vers l’espace intermembranaire et

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 45
Alors que les PMb sont la cible de plus de 50% des médicaments et qu’elles représentent 20 à 40% des protéinescodées par les génomes, leur étude accuse un large retard par rapport à celle des protéines solubles (moins de7% des structures déposés à la PDB (Protein Data Bank) sont des structures de PMb. Cette différence provientd’une part des difficultés à produire des PMb pures et stables en quantités nécessaires à la détermination desstructures 3D et d’autre part des difficultés à les cristalliser. Même s’il est possible de surexprimer chez la bactérieune PM, leur extraction et leur isolement nécessitent l’utilisation de détergents a, afin d’une part de respecterleur nature mixte hydrophile/hydrophobe et d’autre part de conserver leur état natif. Le grand dilemme estque la présence de détergent nécessaire à la solubilisation des PMb, rend ces dernières le plus souvent instablesconformationnellement, partiellement ou totalement dépliées ou encore plus ou moins rapidement inactivées.D’où la nécessité de reproduire un environnement mimétique proche de leur environnement natif membranairepour que la protéine adopte un repliement tertiaire stable permettant l’acquisition de données de bonne qualité.Le choix de l’environnement mimétique membranaire est donc une étape cruciale et, étant donné qu’il n’existepas de guide universel, une étude au cas par cas est nécessaire. Il existe différents environnements tels que : lesmicelles de détergent ou les bicelles lipides/détergent ; les amphipols qui permettent de substituer le détergentpar un polymère ; les nanodisques dans lesquels une protéine appelée MSP (Membrane Scaffold Protein) s’enrouleautour du noyau hydrophobe de lipides contenant la PM ; les lipodisques formées par des phospholipides et lepolymère SMA (anhydride maléique stérique) ; et les liposomes, vésicules sphériques de phospholipides avec unc ?ur aqueux. Une fois la PMb stabilisée dans un environnement, elle peut être étudiée par différentes approchesexpérimentales.
a. Molécules amphiphiles qui se substituent aux lipides et s’adsorbent sur la surface transmembranaire (trèsfortement hydrophobe) des PMb
Encadré 8 (Biologie structurale des protéines membranaires).
la partie C-terminale cytoplasmique [Joseph-Liauzun et al., 1998].
L’analyse de la structure secondaire de la TSPO entière, en solution, par dichroïsme circulaire(DC) montra un repliement majoritaire en Hα [Jamin and Lacapère, 2008, Murail et al., 2008].Des travaux RMN sur 5 peptides synthétiques correspondants aux 5 domaines hydrophobes ont

46 Chapitre 1. Les protéines TSPO
confirmé qu’ils adoptaient une structure en hélice α (Hα) [Murail et al., 2008].On peut noter que cette structuration dépend de la présence du ligand . En effet, Les spectres
RMN 2D HSQC 27 montrent que la structure tertiaire de la TSPO est flexible mais l’introductiondu ligand PK11195 entraîne une stabilisation d’une structure tertiaire (ou conformation) [Murailet al., 2008].
En 2010, des cristaux bidimensionnels d’un homologue bactérien de la TSPO (Rhodobactersphaeroides) ont été observés par cryomicroscopie électronique et ont permis d’obtenir un modèle3D à faible résolution (10Å) [Korkhov et al., 2010]. Dans ce modèle, la TSPO s’associe en dimèreet on peut observer dans chacun des monomères un faisceau de 5 Hα.
En mars 2014, la stabilisation structurale par le (R)-PK11195 (§ 1.2.4.1) en RMN du liquide,a permis la résolution de la 1re structure atomique de la TSPO (de souris ; mTSPO) en micellesde détergent DPC [Jaremko et al., 2014].
Cette structure 3D de la mTSPO en complexe avec le PK11195 confirme que la TSPO estcomposée de 5 Hα transmembranaires (TM1 à TM5), qui lorsque l’on regarde depuis le cytosoldans le sens des aiguilles d’une montre, forment un faisceau compact selon l’ordre : TM1-TM2-TM5-TM4-TM3, Fig.1.15a. La mTSPO contient un grand nombre de résidus prolines.La TM1 est perturbée par la Pro15, alors que les TM2 et TM3 sont perturbées par un couplede prolines : Pro44-Pro45 et Pro96-Pro97 respectivement. La TM5 possède une Pro139 qui lacoude vers l’espace intermembranaire. Or, les prolines sont des a.a qui, de part leur géométrieparticulière, tendent à interrompre les structures secondaires des protéines telles que les Hα etles feuillets β. Les boucles reliant les hélices transmembranaires sont courtes, à l’exception decelle reliant la TM1 et TM2 (résidus Gly28 à Pro45) . Les 7 résidus de Glu29 à Ala35 se replienten une petite hélice α (Hα′) qui est inclinée le long de l’axe de la TM1 et qui positionne laboucle TM1-TM2 proche de la TM5, Fig1.15a. La TSPO contient également un grand nombrede résidus Trp (12 au total) parmi eux, 4 sont très conservés : le Trp33 localisé dans l’Hα′ dans laboucle TM1-TM2, le Trp53 dans la TM2, le Trp126 localisé à la fin de la TM4 et le Trp143 dansla TM5. Les Trp53 et Trp143 sont proches et contribuent à l’interface TM2/TM5. Ces mêmesTrp pointent vers l’intérieur de la protéine et créent plusieurs contacts avec des résidus voisinsce qui pourrait expliquer que les TM2, TM4 et TM5 soient les plus stables et les plus conservéesà travers l’évolution.
Le C-terminal est hautement chargé positivement et dynamique tout comme l’est le N-terminal. Ils n’ont pas de structure stable.
Liaison du PK11195 En accord avec la prédiction de stoechiométrie de 1 :1 pour la liaisondes ligands [Lacapere et al., 2001], un seul PK11195 est observé sur la protéine. Le ligand vientse lier dans une poche formée par les 5 hélices transmembranaires dans la partie supérieurecytosolique du faisceau hélical. Cette cavité hydrophobe est recouverte par la longue boucle qui
27. En RMN du liquide, le spectre HSQC constitue une empreinte de la protéine d’étude et de son état derepliement. L’observation d’un nombre de tâche au moins égale au nombre d’a.a de la protéine, est un signe quela protéine adopte une conformation unique stable
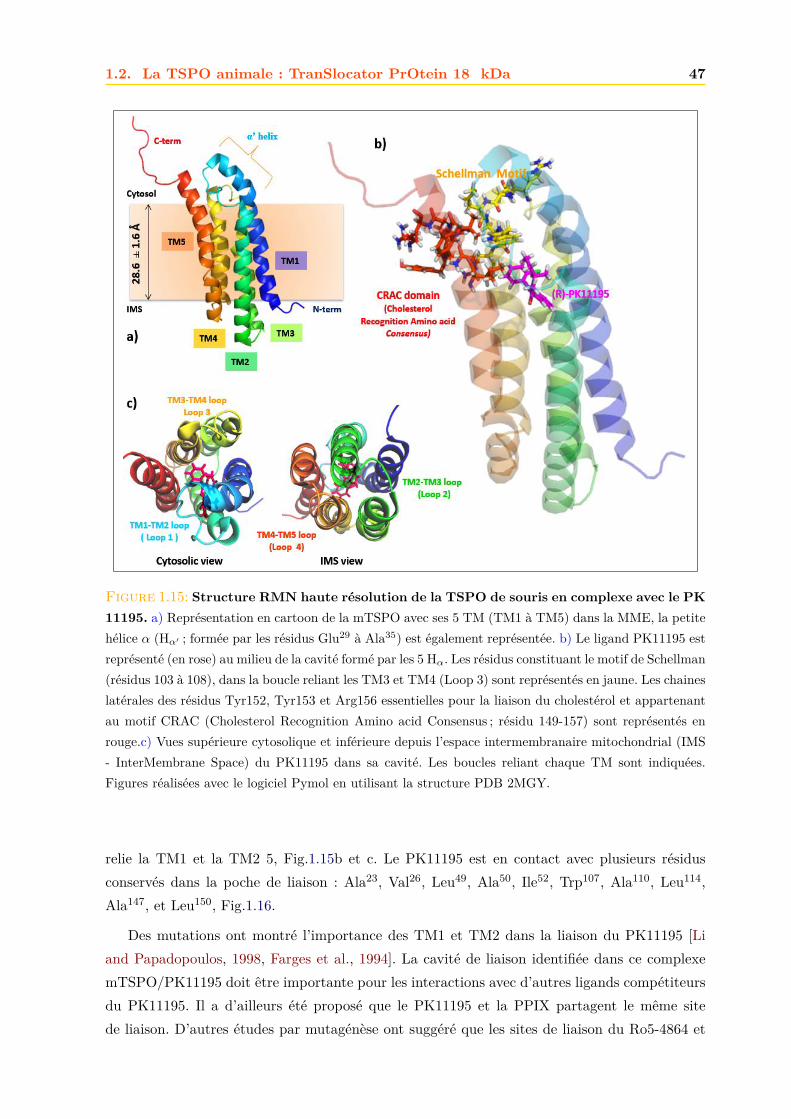
1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 47
Figure 1.15: Structure RMN haute résolution de la TSPO de souris en complexe avec le PK11195. a) Représentation en cartoon de la mTSPO avec ses 5 TM (TM1 à TM5) dans la MME, la petitehélice α (Hα′ ; formée par les résidus Glu29 à Ala35) est également représentée. b) Le ligand PK11195 estreprésenté (en rose) au milieu de la cavité formé par les 5 Hα. Les résidus constituant le motif de Schellman(résidus 103 à 108), dans la boucle reliant les TM3 et TM4 (Loop 3) sont représentés en jaune. Les chaineslatérales des résidus Tyr152, Tyr153 et Arg156 essentielles pour la liaison du cholestérol et appartenantau motif CRAC (Cholesterol Recognition Amino acid Consensus ; résidu 149-157) sont représentés enrouge.c) Vues supérieure cytosolique et inférieure depuis l’espace intermembranaire mitochondrial (IMS- InterMembrane Space) du PK11195 dans sa cavité. Les boucles reliant chaque TM sont indiquées.Figures réalisées avec le logiciel Pymol en utilisant la structure PDB 2MGY.
relie la TM1 et la TM2 5, Fig.1.15b et c. Le PK11195 est en contact avec plusieurs résidusconservés dans la poche de liaison : Ala23, Val26, Leu49, Ala50, Ile52, Trp107, Ala110, Leu114,Ala147, et Leu150, Fig.1.16.
Des mutations ont montré l’importance des TM1 et TM2 dans la liaison du PK11195 [Liand Papadopoulos, 1998, Farges et al., 1994]. La cavité de liaison identifiée dans ce complexemTSPO/PK11195 doit être importante pour les interactions avec d’autres ligands compétiteursdu PK11195. Il a d’ailleurs été proposé que le PK11195 et la PPIX partagent le même sitede liaison. D’autres études par mutagénèse ont suggéré que les sites de liaison du Ro5-4864 et

48 Chapitre 1. Les protéines TSPO
Figure 1.16: Le site de liaison du PK11195. Vue sélective de la poche de liaison du PK11195 àpartir de la stucture de la mTSPO en complexe avec ce ligand. Les résidus directement en contact avec le(R)-Pk11195 sont indiqués. Les résidus montrés importants dans la liaison du Ro5-4864, par mutagénèseset analyses bioinformatiques, sont encadrés en violet [Farges et al., 1994, Li et al., 2013]. Figure adaptéede Jaremko et al. [2015b].
du PK11195 se chevauchent[Farges et al., 1994]. Ces mêmes études ont montré que les résidusArg24, Glu29, Leu31, Arg32, Leu37, Lys39, Pro40, Ser41, Trp42, Trp107, Val154 et Trp161 sontimportants dans la liaison du RO5-4864. Comme attendu, les résidus impliqués dans la liaisondu PK11195 n’interfèrent pas avec le site de liaison supposé du cholestérol (CRAC). Il y a doncbien 2 sites de liaisons distincts pour le PK11195 et le cholestérol.
Figure 1.17: Représentation du passage du cholestérol dans le canal formé par les 5 Hα
de la TSPO. En blanc : les hélices α ; en jaune : le cholestérol. La vue de côté montre la liaison ducholestérol au domaine CRAC. D’après Rupprecht et al. [2010]

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 49
Figure 1.18: Vue inférieure depuis l’espace intermembranaire mitochondrial (IMS)de le structure RMN de la mTSPO avec les résidus constituant le motif CRAC(Cholesterol Recognition Amino acid Consensus [L/V-x1−5-Y-x1−5-R/K] ; résidus149-157). Ces résidus sont représentés en orange et rouge. Les chaines latérales des résidusTyr152, Tyr153 et Arg156 essentielles pour la liaison du cholestérol sont représentées en rouge.Le PK 11195 est représenté en magenta au centre de la cavité.
Liaison du Cholestérol Dans le modèle cristallographique bactérien proposé par Korkhovet al. [2010], il est proposé que la TSPO sous forme monomérique pourrait former un tunnelintérieur hydrophobe bordé par le motif CRAC [L/V-x1−5-Y-x1−5-R/K] pour la liaison et latranslocation du cholestérol [Rupprecht et al., 2010], Fig.1.17. Il a alors été proposé que ledomaine CRAC, situé dans le domaine C-terminal, était localisé le long de TM5 (résidu 149 à157 - VLNYYVWRD ; [Li and Papadopoulos, 1998]). Ce domaine serait au sein d’une crevasseformée par des résidus aromatiques qui enserrent le noyau stérol alors que le groupe hydroxylserait chapeauté par une arginine, et que la chaine alkyle ferait face à des résidus hydrophobescomme la valine [Jamin et al., 2005]. Cependant, dans la structure RMN, on ne voit pas un teltunnel hydrophobe qui pourrait permettre la translocation du cholestérol du côté cytosoliquevers le la matrice mitochondriale. De plus, les chaines latérales des résidus Tyr152, Tyr153, etArg156 essentielles pour la liaison du cholestérol [Li and Papadopoulos, 1998, Jamin et al.,2005] sont localisées à l’extérieur de la structure et pointent vers l’environnement membranaire,Fig 1.18. La grande question est alors comment pourrait se faire le transport du cholestérol sitelle est une fonction de la protéine ? Plusieurs auteurs ont suggéré qu’étant donné la capacitéconnue du cholestérol à se dimériser, celle-ci pourrait mener à l’oligomérisation de la TSPO cequi permettrait d’assurer la translocation du cholestérol à travers la membrane mitochondrialeentre 2 monomères (certains transporteurs ayant été montrés ne pouvant fonctionner qu’à l’étatdimérique) [Jaremko et al., 2014, Li et al., 2013]. Cependant, la TSPO de mammifère n’a étémontrée former des dimères que sous certaines conditions qui ne sont pas liées à la dimérisation
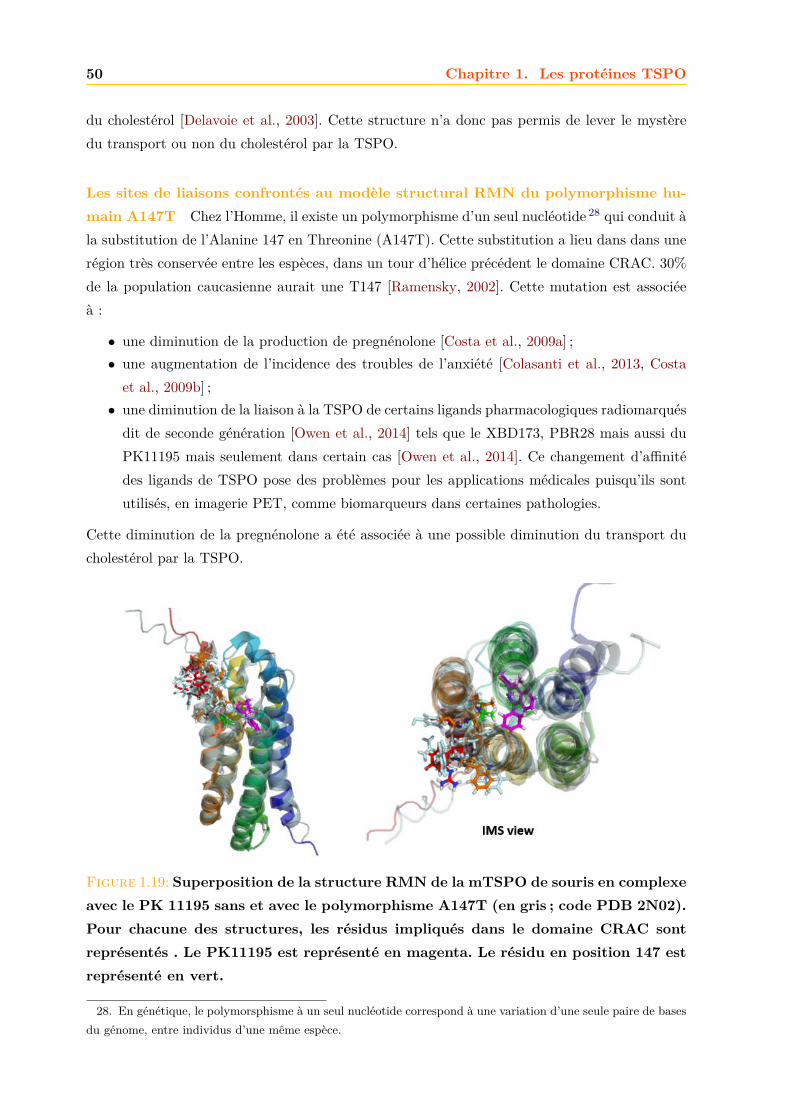
50 Chapitre 1. Les protéines TSPO
du cholestérol [Delavoie et al., 2003]. Cette structure n’a donc pas permis de lever le mystèredu transport ou non du cholestérol par la TSPO.
Les sites de liaisons confrontés au modèle structural RMN du polymorphisme hu-main A147T Chez l’Homme, il existe un polymorphisme d’un seul nucléotide 28 qui conduit àla substitution de l’Alanine 147 en Threonine (A147T). Cette substitution a lieu dans dans unerégion très conservée entre les espèces, dans un tour d’hélice précédent le domaine CRAC. 30%de la population caucasienne aurait une T147 [Ramensky, 2002]. Cette mutation est associéeà :
• une diminution de la production de pregnénolone [Costa et al., 2009a] ;• une augmentation de l’incidence des troubles de l’anxiété [Colasanti et al., 2013, Costaet al., 2009b] ;• une diminution de la liaison à la TSPO de certains ligands pharmacologiques radiomarquésdit de seconde génération [Owen et al., 2014] tels que le XBD173, PBR28 mais aussi duPK11195 mais seulement dans certain cas [Owen et al., 2014]. Ce changement d’affinitédes ligands de TSPO pose des problèmes pour les applications médicales puisqu’ils sontutilisés, en imagerie PET, comme biomarqueurs dans certaines pathologies.
Cette diminution de la pregnénolone a été associée à une possible diminution du transport ducholestérol par la TSPO.
Figure 1.19: Superposition de la structure RMN de la mTSPO de souris en complexeavec le PK 11195 sans et avec le polymorphisme A147T (en gris ; code PDB 2N02).Pour chacune des structures, les résidus impliqués dans le domaine CRAC sontreprésentés . Le PK11195 est représenté en magenta. Le résidu en position 147 estreprésenté en vert.
28. En génétique, le polymorsphisme à un seul nucléotide correspond à une variation d’une seule paire de basesdu génome, entre individus d’une même espèce.

1.2. La TSPO animale : TranSlocator PrOtein 18 kDa 51
La question se pose alors : comment une mutation qui est loin du domaine de liaison duPK11195, peut-elle modifier la liaison/le transport du cholestérol ? Dans le but de répondreà cette question, un an après la résolution du complexe mTSPO/PK11195 en DPC, la mêmeéquipe a déterminé la structure d’un mutant mTSPO présentant ce polymorphosmime A147T[Jaremko et al., 2015b]. Encore une fois cette structure a été résolue en DPC et en présence duligand (R)-PK11195. En effet, leur mutant A147T lie le (R)-PK11195 avec une affinité similaireà la protéine sauvage. Leur structure montre que la substitution A147T résulte seulement en unchangement conformationnelle local autour du site de mutation. Mais la mutation n’influencepas l’intégrité du domaine CRAC. Cette structure n’a donc pas permis de savoir pourquoi cepolymorphisme diminue l’affinité de certains ligands, mais pas du (R)-PK11195. Elle n’a pas nonplus permis de déterminer quel est son lien avec la diminution de la production de pregnénoloneprobablement due à une diminution du transport du cholestérol.
À ce jour, il n’y pas de structure de la TSPO avec et sans cholestérol et c’est probablementce qui manque pour pouvoir lever le voile sur la fonction qui lie la TSPO au cholestérol. Celane pourra se faire en détergent puisque la protéine ne lie pas le cholestérol dans ces conditions.L’étude de la TSPO2, une isoforme de la TSPO découverte chez les mammifères et les oiseaux,pourrait contribuer à mieux comprendre la protéine. En effet, cette protéine possède la capacitéde liaison du cholestérol mais présente une très faible affinité pour le PK11195 pourtant le ligandle plus affin de la TSPO mitochondriale.

52 Chapitre 1. Les protéines TSPO
1.3 La TSPO2
En 2009, l’équipe de Fan et al. découvre un nouveau membre de la famille TSPO : laprotéine TSPO2. D’après les auteurs, le gène Tspo2 ne serait présent que chez les oiseaux etles mammifères. Il émergerait d’un événement de duplication ancien du gène Tspo1, avant ladivergence entre la lignée des Aves (les oiseaux) et des Mammifères, il y a environ 300 millionsd’années. Cependant d’après les arbres phylogénétiques consensuels, Fig.1.20, on devrait aussiretrouver le gène Tspo2 chez les reptiles et les tortues. Et effectivement depuis, le séquençagede quelques espèces de reptiles et de tortues montre la présence d’un gène Tspo2. Mais pourle moment aucunes données sur l’expression et la fonctionnalité dans ces espèces n’ont étéreportées.
Chez l’Homme et la souris, Tspo2 se trouve sur le chromosome 6 et 17, respectivement. Lesgènes Tspo2 et Tspo1 auraient vus leurs fonctions évoluer durant l’évolution de la famille degène Tspo [Fan et al., 2009], il s’agit donc de 2 gènes paralogues.
Cette protéine partage 35% d’identité avec la TSPO1. Un alignement des séquences deTSPO1 et TSPO2 de plusieurs espèces est présenté Fig. 1.21. Pour éviter toute confusion leterme TSPO1 sera à présent employé, pour se référer à la protéine TSPO mitochondriale 18kDa qui fut l’objet de la partie précédente.
Figure 1.20: Arbre phylogénétique consensuelle des tétrapodes. La séparation Mammi-fère/Aves a eu lieu il y a environ 300 millions d’années. http://www.onezoom.org/index.htm.
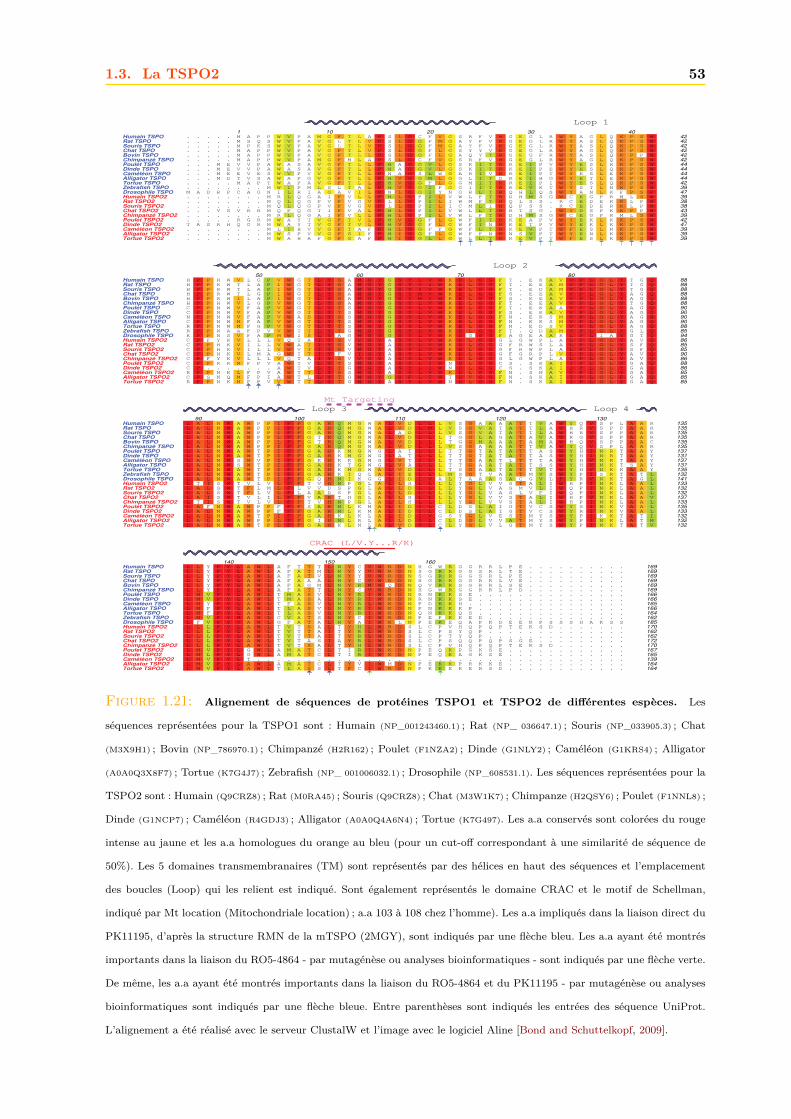
1.3. La TSPO2 53
Humain TSPORat TSPOSouris TSPO
Bovin TSPO
Zebrafish TSPODrosophile TSPO
Souris TSPO2
Humain TSPO2Rat TSPO2
Dinde TSPO2Poulet TSPO2
Alligator TSPO2Caméléon TSPO2
Chimpanzé TSPO2Chat TSPO2
Poulet TSPODinde TSPO
Chat TSPO
Chimpanzé TSPO
Caméléon TSPOAlligator TSPO
Tortue TSPO2
Tortue TSPO
Humain TSPORat TSPOSouris TSPO
Bovin TSPO
Zebrafish TSPODrosophile TSPO
Souris TSPO2
Humain TSPO2Rat TSPO2
Dinde TSPO2Poulet TSPO2
Alligator TSPO2Caméléon TSPO2
Chimpanzé TSPO2Chat TSPO2
Poulet TSPODinde TSPO
Chat TSPO
Chimpanzé TSPO
Caméléon TSPOAlligator TSPO
Tortue TSPO2
Tortue TSPO
Humain TSPORat TSPOSouris TSPO
Bovin TSPO
Zebrafish TSPODrosophile TSPO
Souris TSPO2
Humain TSPO2Rat TSPO2
Dinde TSPO2Poulet TSPO2
Alligator TSPO2Caméléon TSPO2
Chimpanzé TSPO2Chat TSPO2
Poulet TSPODinde TSPO
Chat TSPO
Chimpanzé TSPO
Caméléon TSPOAlligator TSPO
Tortue TSPO2
Tortue TSPO
Humain TSPORat TSPOSouris TSPO
Bovin TSPO
Zebrafish TSPODrosophile TSPO
Souris TSPO2
Humain TSPO2Rat TSPO2
Dinde TSPO2Poulet TSPO2
Alligator TSPO2Caméléon TSPO2
Chimpanzé TSPO2Chat TSPO2
Poulet TSPODinde TSPO
Chat TSPO
Chimpanzé TSPO
Caméléon TSPOAlligator TSPO
Tortue TSPO2
Tortue TSPO
. . . . . M A P P W V P A M G F T L A P S L G C F V G S R F V H G E G L R W Y A G L Q K P S W
H P P H W V L G P V W G T L Y S A M G Y G S Y L V W K E L G G F T . E K A V V P L G L Y T G Q
L A L N W A W P P I F F G A R Q M G W A L V D L L L V S G A A A A T T V A W Y Q V S P L A A R
L L Y P Y L A W L A F T T T L N Y C V W R D N H G W R G G R R L P E . . . . . . . . . .
. . . . . M S Q S W V P A V G L T L V P S L G G F M G A Y F V R G E G L R W Y A S L Q K P S W
H P P R W T L A P I W G T L Y S A M G Y G S Y I I W K E L G G F T . E E A M V P L G L Y T G Q
L A L N W A W P P I F F G A R Q M G W A L V D L M L V S G V A T A T T L A W H R V S P P A A R
L L Y P Y L A W L A F A T M L N Y Y V W R D N S G R R G G S R L T E . . . . . . . . . .
. . . . . M P E S W V P A V G L T L V P S L G G F M G A Y F V R G E G L R W Y A S L Q K P S W
H P P R W T L A P I W G T L Y S A M G Y G S Y I V W K E L G G F T . E D A M V P L G L Y T G Q
L A L N W A W P P I F F G A R Q M G W A L A D L L L V S G V A T A T T L A W H R V S P P A A R
L L Y P Y L A W L A F A T V L N Y Y V W R D N S G R R G G S R L P E . . . . . . . . . .
. . . . . M A P P W V P A V G F T L L P S L G G F L G A Q Y T R G E G F R W Y A S L Q K P P W
H P P R W I L A P I W G T L Y S A M G Y G S Y M I W K E L G G F S . K E A V V P L G L Y A G Q
L A L N W A W P P L F F G T R Q M G W A L V D L L L T G G M A A A T A M A W H Q V S P P A A C
L L Y P Y L A W L A F A G M L N Y R M W Q D N Q V R R S G R R L S E . . . . . . . . . .
. . . . . . . . M W I P M L S L T A L P H V G G I F G G I I T R R E V K T W Y S T L N K P S W
R P P N A A F P V V W T T L Y T G M G Y G S Y L V W K E L G G F T . Q D A M V P L G L Y G L Q
L A L N W A W T P I F F G A H K I Q L A L I E L L L M S G T V A A T M V S W Y P I S R T A T L
L M V P Y M A W L C V A T S L N Y C I W R D N P E P K K E D . . . . . . . . . . . . . .
M A D R P C A G N I L R I A G A V I L P N L G G I Y N G R L T R Q H L Q S W Y A N L K F P S F
K P P N S V F A P M W I S L Y A G M G Y G S Y L V W R D G G G F A G E A A K L P L I A Y G T Q
L A L N W A W T P I F F G Q H N I K G G L I D I V A L T A A A S A C G V L F Y R V N K T A G L
L F V P Y V A W L G F A T A L N Y A I W K L N P E K E Q A P K D E E K P S S S H A K S S
. . . . . . . . M Q L Q G P V F V G V P L L G P I L I C M L I H Q P S S . R C E D E R K L P W
C P P H K V I L L V W V T I Y S V M G Y A S Y L V W K E L G G G F R W P L A L P L G L Y S F Q
L A L S W T F L V L F L A A D S P G L A L L D L L L L Y G L V A S L V F I W Q P I N K L A A L
L L L P Y L A W L T V T T A I T Y R L W R D S L C P T Y Q P . . . . . . . . . . . . . .
. . . . . . . . M R L Q G A I F V L L P H L G P I L V W L F T R D H M S G W C E G P R M L S W
C P F Y K V L L L V Q T A I Y S V V G Y A S Y L V W K D L G G G L G W P L A L P L G L Y A V Q
L T I S W T V L V L F F T V H N P G L A L L H L L L L Y G L V V S T A L I W H P I N K L A A L
L L L P Y L A W L T V T S A L T Y H L W R D S L C P V H Q P Q P T E K S D . . . . . . .
. . . . . . . . M Q L Q G P V F V G V P L L G P I L I W M F T H Q L S S . R C E D E K K L P W
C P P H K V I L L V W A T I Y S V M G Y A S Y L V W K D L G G G F R W S L A L P L G L Y S F Q
L A L S W T F L M L F L V V D S P G L A L L D L L L L Y G L V A S M V L I W Q P I N K L A A L
L L L P Y L A W L T V T T A I T Y R L W R D S L C P S Y Q P . . . . . . . . . . . . . .
T A S R H Q G R M W A Y T V G F T V L P H V G G F L G W F I N R K E T P V W Y E K L K K P S W
C P . . . . . . . A W T V L Y T G M G Y A S Y L I W N D L G G C S . S K A I I P L G L Y G A Q
L A L N W A W P P F F F G A R N L K M A L I D I L C L D S L A I G T V C S W Y R I N K V A A L
L M L P Y L G W L A M A T C L T I R I W K D N P E Q K A G K S E . . . . . . . . . . . .
. . . . . R G R M W A Y T V G F T V L P H V G G F L G W F I T R K E A P V W Y E K L K K P S W
C P P R K M F P V A W T V L Y T S M G Y A S Y L I W N D L G G C S . S K A I I P L G L Y G A Q
L A F N W A W P P F F F S A R N L K M A L I D I L C L D S L A I G T V C S W Y S I N K V A A L
L M V P Y L G W L A M A T C L T I R I W K D N P E Q K P G K S E . . . . . . . . . . . .
. . . . . . . . M W S P V V G F S I F P H I G G F L G W F F N R N S V P T W Y E N L K K P S W
C P S N Q M F P I A W T L L Y T G M G Y G S F L I W T D V G G F N . S K A I I P L G L F G A Q
L A L N W A W P P L F F G I H N L R L A L L D I L C L D G L V V A T M Y S W Y P I N K L A T M
L M V P Y L A W L A M A T C L T Y V I W M D N P E K K P R K K E . . . . . . . . . . . .
. . . . . . . . M L I H V V G F T A F P H L G G F F G W F L T R K E V P T W F E S L N K P S W
R P P N K L F P V A W T T L Y S S M G Y A S Y L V W K D L G G F N . S N A V V P L G L Y G A Q
L A L N W A W T P I F F G A H K L K L A L I D I V G S Y G L V E S T M Y S W Y P I K K T A T I
L M V P Y L T . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . M R L Q G A I F V L L P H L G P I L V W L F T R D H M S G W C E G P R M L S W
C P F Y K V L L L V Q T A I Y S V V G Y A S Y L V W K D L G G G L G W P L A L P L G L Y A V Q
L T I S W T V L V L F F T V H N P G L A L L H L L L L Y G L V V S T A L I W H P I N K L A A L
L L L P Y L A W L T V T S A L T Y H L W R D S L C P V H Q P Q P T E K S D . . . . . . .
. . . V S V R R M Q P Q G T I F V A L P H L G P I L V W L L T R R T S G . W Y D S P K K P P W
C P P H K V L M A G W I T I Y F V I G Y A S Y L V W K D L G G G F G R P L V L P L G L Y A V Q
L A I S W T V L I L F F V A R T H G L A L L H L L L L Y G L V V S T A L I W H P V N K L A A V
L L L P Y L A W L T V T A S I A Y R L W R D S L C P S Q Q P Q P S G E . . . . . . . . .
. . . M E V V P A W A S A V G F T L L P H A G G V L G S K I T R R E I P V W Y E S L R K P S W
C P P N W V F A P V W G T L Y T S M G Y G S Y L V W K E L G G F T . E K A A V P L G L Y A G Q
L A L N W A W T P I F F G A H K M G W G L A T L L L T T G T A T A T T A S W Y H I N R T A A Y
L M V P Y L A W L T M A S V L N Y R I W K D N R N K K S E . . . . . . . . . . . . . . .
. . . M E V V P A W A S A V G F T L L P H A G G V L G S K I T R K E I P E W Y E S L Q K P S W
C P P N W V F A P V W G T L Y T S M G Y G S Y L V W K E L G G F S . E K A V V P L G L Y A G Q
L A L N W A W T P I F F G A H K M G W G L A T L L L T T G T A T A T T A S W Y H I N R T A A Y
L M V P Y L A W L T M A S A L N Y R I W K D N R N K K S E . . . . . . . . . . . . . . .
. . . . . M A P P W V P A V G F T L V P S L G G F L G S Y Y V R G E G L R W Y A G L Q K P S W
H P P R W T L G P I W G T L Y S A M G Y G S Y M V W K E L G G F S . E E A V V P L G L Y A G Q
L A L N W A W P P L F F G T R Q M G W A L V D L L L T G G L A G A T A V A W R G V S P P A A R
L L Y P Y L A W L A F A A A L N Y C V W R D N H G R R G G R R L V E . . . . . . . . . .
. . . . . M A P P W V P A M G F M L A P S L G C F V G S R F V H G E G L R W Y A G L Q K P S W
H P P H W V L G P V W G T L Y S A M G Y G S Y L V W K E L G G F T . E E A V V P L G L Y T G Q
L A L N W A W P P I F F G A R Q M G W A L V D L L L V S G A A A A T T V A W Y Q V S P L A A R
L L Y P Y L A W L A F A T T L N Y C V W R D N H G W R G G R R L P D . . . . . . . . . .
. . . M E E V E S W V P V V G F T L L P N A G S L W G A R I V K R E I P T W Y K S L E K P S W
N P P N W V F A P V W A S L Y S S M G Y G S Y L V W K E L G G F N . E K S M V P L G L Y A G N
L A L N W S W V P I F F G K H K K G W G L V T I L L T T G A A T L T T T A W Y H V N K T A A Y
L M Y P Y L A W L T F A S V L N Y R L W K D N P D K K H . . . . . . . . . . . . . . . .
. . . M D T V S A W A P G V G F T L L P H V G G M L G G L I T D R E I H G W Y E Y L E K P S W
C P P N W M F A P V W G T L Y T S M G Y G S Y L V W K E L G G F N . K K S M V P L G L Y A G Q
L A L N W S W T P I F F G A H K T G W G L V A L L L T T G A A T A T T S S W Y H V N K T S A Y
L M F P Y L A W L T L A S V L N Y R I W K D N P N K K K P . . . . . . . . . . . . . . .
. . . . . . . . M W A H A F G F S A F P H I G G L L G W L L T R K E V P T W F E S L K K P S W
R P P N K M F P V V W T T L Y T G M G Y A S Y L V W N D L G G F N . S K A I I P L G L Y G A Q
L A L N W A W T P L F F G A H K L N L A L V D V L C L Y G L V L G T M Y S W Y P I N K T A T V
L M V P Y L A W L T L A S S L T F C I W R D N P K K E K E R S D . . . . . . . . . . . .
. . . . . M A P T W A P A V G F T L L P H V G G F L G G M I T K R E I P I W Y E S L Q K P S W
R P P N W M F G P V W G T L Y T S M G Y G S Y L V W K E L G G F N . E D S V V P L G L Y A G Q
L A L N W A W T P I F F G A H K M G W A L V D L L L T S G A A T A T T V T W Y H V N K K A A Y
L M F P Y L A W L T L A S V L N Y R I W K D N Q N K K Q S . . . . . . . . . . . . . . .
1 10 20 30 40
50 60 70 80
90 100 110 120 130
140 150 160
42
88
135
169
42
88
135
169
42
88
135
169
42
88
135
169
39
85
132
162
47
94
141
185
38
85
132
162
39
86
133
170
38
85
132
162
47
86
133
165
42
88
135
167
39
85
132
164
39
85
132
139
39
86
133
170
43
90
137
172
44
90
137
166
44
90
137
166
42
88
135
169
42
88
135
169
44
90
137
165
44
90
137
166
39
85
132
164
42
88
135
164
Loop 2
Loop 1
Loop 4Loop 3
Mt Targeting
CRAC (L/V.Y...R/K)
Figure 1.21: Alignement de séquences de protéines TSPO1 et TSPO2 de différentes espèces. Les
séquences représentées pour la TSPO1 sont : Humain (NP_001243460.1) ; Rat (NP_ 036647.1) ; Souris (NP_033905.3) ; Chat
(M3X9H1) ; Bovin (NP_786970.1) ; Chimpanzé (H2R162) ; Poulet (F1NZA2) ; Dinde (G1NLY2) ; Caméléon (G1KRS4) ; Alligator
(A0A0Q3X8F7) ; Tortue (K7G4J7) ; Zebrafish (NP_ 001006032.1) ; Drosophile (NP_608531.1). Les séquences représentées pour la
TSPO2 sont : Humain (Q9CRZ8) ; Rat (M0RA45) ; Souris (Q9CRZ8) ; Chat (M3W1K7) ; Chimpanze (H2QSY6) ; Poulet (F1NNL8) ;
Dinde (G1NCP7) ; Caméléon (R4GDJ3) ; Alligator (A0A0Q4A6N4) ; Tortue (K7G497). Les a.a conservés sont colorées du rouge
intense au jaune et les a.a homologues du orange au bleu (pour un cut-off correspondant à une similarité de séquence de
50%). Les 5 domaines transmembranaires (TM) sont représentés par des hélices en haut des séquences et l’emplacement
des boucles (Loop) qui les relient est indiqué. Sont également représentés le domaine CRAC et le motif de Schellman,
indiqué par Mt location (Mitochondriale location) ; a.a 103 à 108 chez l’homme). Les a.a impliqués dans la liaison direct du
PK11195, d’après la structure RMN de la mTSPO (2MGY), sont indiqués par une flèche bleu. Les a.a ayant été montrés
importants dans la liaison du RO5-4864 - par mutagénèse ou analyses bioinformatiques - sont indiqués par une flèche verte.
De même, les a.a ayant été montrés importants dans la liaison du RO5-4864 et du PK11195 - par mutagénèse ou analyses
bioinformatiques sont indiqués par une flèche bleue. Entre parenthèses sont indiqués les entrées des séquence UniProt.
L’alignement a été réalisé avec le serveur ClustalW et l’image avec le logiciel Aline [Bond and Schuttelkopf, 2009].

54 Chapitre 1. Les protéines TSPO
1.3.1 Localisation cellulaire et subcellulaire
Les études par microscopie de fluorescence, avec une protéine TSPO2 (humaine ou murine)fusionné à une GFP, montre une localisation majoritaire de la protéine dans les Mb du RE etdu noyau, alors que rappelons le la TSPO1 est majoritairement mitochondriale. On retrouvela TSPO2 dans les tissus hématopoiétiques (moelle osseuse et le tissu lymphoïde : thymus,ganglions, rate, foie etc.) et dans les cellules érythroïdes [Fan et al., 2009, Nakazawa et al.,2009].
1.3.2 Ligands
Outre sa différence de localisation avec la TSPO1, la TSPO2 a perdu la capacité de liaisondes ligands spécifiques de la TSPO1. En effet, les TSPO2 humaine et murine ne montrent pas deliaison spécifique pour le PK11195. Cela est confirmé par l’analyse des séquences (Fig. 1.21) quimontrent qu’un grand nombre de résidus impliqués dans la liaison du PK11195, chez la TSPO1,ne sont pas conservés chez la TSPO2. Cependant, la TSPO2 conserve la capacité de liaison ducholestérol. En effet, le motif CRAC de liaison du cholestérol est présent en son C-terminal etson élimination entraine une diminution de la liaison du cholestérol sur la protéine [Fan et al.,2009].
1.3.3 Fonction
La redistribution du cholestérol intracellulaire est une étape importante durant l’érythro-poïèse, qui est le processus de maturation des GR, Encadré 9. La distribution de la TSPO2 dansles cellules érythrocytes et sa liaison au cholestérol ont laissé penser à un lien entre la protéineet la synthèse, l’import et/ou le trafic du cholestérol durant l’érythropoïèse.
La 1re étude qui évoque cette possible fonction pour la TSPO2 a été réalisée par Nakazawaet al.. Les auteurs ont identifiés, chez l’embryon de poulet, une protéine TSPO2, nomméePBRL. Le gène Tspo2 apparait être uniquement exprimé durant l’érytropoïèse pimitive (c.-à-d. étapes allant des cellules souches pluripotentes jusq’à la formation des réticulocytes, cf.Encadré. 9) et cette expression est étroitement corrélée avec celle des gènes de l’hémoglobine.De plus, l’inhibtion pharmacologique du gène Tspo2 conduit à une diminution du niveau deprotéines globines (protéines qui constituent la molécule d’hémoglobine) durant l’érythropoïèse.Ces données suggèrent un rôle de la TSPO2 dans l’érythropoïèse, peut-être via la régulation dela concentration en hèmes cytoplasmiques si un parallèle avec la TSPO1 est fait.
Une seconde étude, réalisée par Fan et al., montre que bien que la TSPO2 soit présentedans toutes les cellules érythroïdes, elle n’est que faiblement exprimée durant les 1res étapes del’érythropoïèse (c.-à-d. dans les étapes allant des cellules souches à la formation des proérythro-blastes) mais est hautement exprimée durant les dernières étapes du processus de maturation(c.-à-d. érythroblastes et reticulocytes). De plus, l’expression de Tspo2 est fortement corrélée àla régulation négative des enzymes impliquées dans la biosynthèse du cholestérol. Ces données

1.3. La TSPO2 55
Les hématies ou globules rouges (GR) du sang contiennent l’hémoglobine, qui permet de fixer l’oxygène auniveau des poumons et le transporter vers les tissus. L’érythropoïèse est le processus par lequel l’organismeassure la production des GR. Le siège de l’érytropoïèse varie en fonction de l’âge :
— Chez l’embryon humain l’érythropoïèse a lieu dans la rate à partir de 3 mois puis à partir du 5e moisl’érythropoïèse se situe dans la moelle osseuse.
— Chez l’enfant la moelle de tous les os est le siège d’une érythropoïèse.
— Chez l’adulte l’érythropoïèse s’effectue dans la moelle osseuse des os plats et courts, essentiellement lesos de la tête, du tronc et de la partie proximale des membres.
Les différentes étapes de l’érytropoïèse sont présentées sur la figure ci-dessous
L’étape terminale de différenciation des érythrocytes nécessite une bonne synthèse de l’hème pour assurer lademande des érythroblastes matures qui ont besoin de fer et d’hème pour former de l’hémoglobine.
Encadré 9 (L’érythropoïèse).
montrent que la TSPO2 pourrait jouer un rôle dans le trafic intracellulaire du cholestérol. Eneffet, les érythrocytes ont besoin de diminuer la fluidité de leur membrane pour l’expulsion dunoyau et ainsi assurer la fin du processus de maturation. Ainsi, lors des dernières étapes de ma-turation des érythrocytes, la TSPO2 re-distribuerait le cholestérol libre pour assurer le maintiende l’équilibre entre les réserves de cholestérol libre et les exigences de la membrane cellulaire desérythroblastes pour leur intégrité et leur fluidité. Des changements dans cet équilibre conduisentà une concentration en cholestérol élevée dans les cellules précurseurs provoquant une rétentionnucléaire qui inhibe la progression du processus de différenciation, Fig. 1.22. Cette idée estrenforcée par le fait que la délétion du domaine CRAC de la TSPO2, entraîne une distributioninégale dramatique du cholestérol dans les Mb du RE et les Mb nucléaires.

56 Chapitre 1. Les protéines TSPO
Figure 1.22: Fonction proposée pour la TSPO2 dans le processus d’érytropoïèse.
1.3.4 Structure et adressage
Tout comme la TSPO1, l’analyse de la séquence de la TSPO2 montre 5 domaines TM,Fig.1.21.
Les caractéristiques structurales qui mènent à une différence de localisation de la TSPO2par rapport à la TSPO1 reste inconnues. Néanmoins, une comparaison des séquences entre laTSPO1 et TSPO2 montre une divergence à l’intérieur des TM3 et TM4. [Rone et al., 2009b] ontmontré que le second domaine (entre les TM3 et TM4) cytoplasmique est en partie responsablede l’insertion de la TSPO1 dans la mitochondrie. Le domaine correspondant dans la TSPO2n’apparait donc pas être capable d’adresser la TSPO2 à la mitochondrie. Ainsi la protéine auraitplutôt tendance à être retenue dans les membranes du RE et du noyau [Fan et al., 2012]. Il estaussi possible que cet adressage de la TSPO2 soit contrôlé par une séquence hydrophobe interneà la protéine.

1.4. La TSPO végétale : l’AtTSPO 57
1.4 La TSPO végétale : l’AtTSPO
Une autre protéine TSPO, possède tout comme la TSPO2, une localisation dans les mem-branes du RE. Il s’agit de l’homologue de la TSPO1 (TSPO mitochondriale 18kDa) chez lesvégétaux. La découverte de la TSPO végétale remonte aux années 2000 : Corsi et al. [2004],Lindemann et al. [2004], Frank et al. [2007], Guillaumot et al. [2009a,b]. Le modèle le plusutilisé pour étudier la TSPO chez les végétaux étant Arabidopsis thaliana, cette section porteraessentiellement sur les études menées sur cette protéine AtTSPO.
1.4.1 Structure génétique
Chez A.thaliana, un seul gène (AtTspo) code pour la protéine AtTSPO, il se situe sur le locusAt2g47770. Ce gène code pour une protéine de 196 a.a (∼ 21 kDa) qui possède une extensionen N-terminal (∼ 40 a.a). Cette extension, de composition variable selon les plantes, n’est pasretrouvée chez les TSPO bactérienne et mammalienne [Lindemann et al., 2004, Guillaumotet al., 2009a], Fig.1.23 et Fig. 1.2. Cette séquence pourrait être impliquée dans l’adressage de laprotéine, nous y reviendrons plus loin. De plus la protéine végétale possède un C-terminal pluscourt que ces homologues.
AtTspo possède 3 codons ATG possibles d’initiation à la traduction lesquels peuvent coderpour la méthionine initiale en position M1, M21 et M42, soit 3 versions de différentes longueursde la protéine AtTSPO : 196, 176 et 155 a.a respectivement Fig.1.24. À noter, que la délétiondu gène est viable pour la plante [Vanhee et al., 2011a].
L’AtTSPO possède 25% d’identité avec la TSPO mitochondriale de mammifère et 15% avecla TSPO bactérienne. Il y a un grand degré de diversité dans les séquences des homologues desTSPO végétales (Cf. Annexe A et Fig.1.2 de le Sec.1.1). Les alignements entre les séquences del’AtTSPO, des homologues d’autres végétaux et la TSPO mitochondriale humaine montrent quele domaine CRAC de liaison du cholestérol n’est pas conservé chez la TSPO végétale, Fig. 1.23.La liaison de la protéine avec des stérols végétaux n’a pas encore été explorée.
1.4.2 Expression
Sous des conditions normales de croissance, l’AtTSPO est fortement exprimée dans les tissuspauvres en eau, tels que les graines sèches et les grains de pollens, et son expression augmenteavec la maturation des graines puis diminue avec leur germination Vanhee et al. [2011b].
Par contre, la protéine est quasiment indétectable dans les tissus végétatifs (racine, tige,feuille) [Guillaumot et al., 2009a]. Néanmoins, son expression dans ces tissus peut être induitetransitoirement par un stress osmotique ou salin, une déficience en magnésium ou par traitementà l’hormone de stress ABA ( Acide Abscissique), voir Annexe C "Les végétaux face au stressabiotiques". La protéine apparait donc être une protéine de réponse au stress (Annexe.C).[Guillaumot et al., 2009a, Vanhee et al., 2011b, Kreps et al., 2002, Seki et al., 2002].
Paradoxalement, les plantes ou les cellules végétales transgéniques surexprimant constituti-
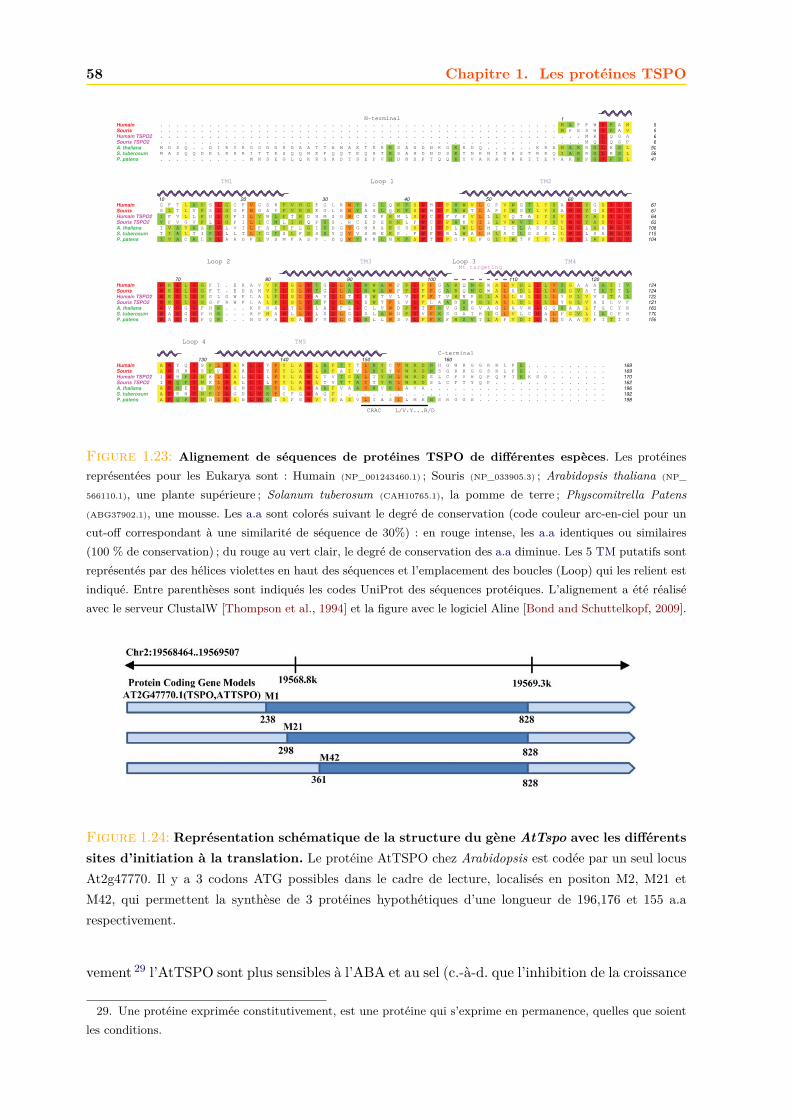
58 Chapitre 1. Les protéines TSPO
HumainSouris
A. thalianaS. tuberosumP. patens
Souris TSPO2Humain TSPO2
HumainSouris
A. thalianaS. tuberosumP. patens
Souris TSPO2Humain TSPO2
HumainSouris
A. thalianaS. tuberosumP. patens
Souris TSPO2Humain TSPO2
HumainSouris
A. thalianaS. tuberosumP. patens
Souris TSPO2Humain TSPO2
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . P P A 9
F T C S R G L G G P G A 67
. E K G P W A A T 124
R Y T C H W G G R L P . . . . . . . . . . 169
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . P E S A 9
T V Y G L T A P G A 67
. E D G P W A A T T 124
H R P R Y V Y S G G S L P . . . . . . . . . . 169
M D S Q . . D I R Y R G G D D R D A A T T A M A E T E R S A D D N K G R D Q . . . . . . K R A R K 50
T A L V T F T F L T D G G N R A S S I L H T T C A S S G L A 108
V G F H . . . K P N A Y L A F L C L V D T V G G V A G A V W G Q A A F G C Y K 163
E G N C A V A V A V A . . . . . . . . . . . . . . . . . . . . . . 196
M A S Q Q D E L K H R I T T K S Q Q N E P Q Q T E Q H T S A H D N D S T N K N I N K S T R K Q R K 58
T T I L L T D S F S Y Q V S M E P . F F R L A H A C G S S L L S A 115
A G F H . . . K P A L L S G S L D V S G A T R G L C A F I C F R 170
K G D C F G A G . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192
. . . . . . . . . . . M N S E G L Q K R S R D T S E F V D N D P T Q Q Y V A K A Y R K T T E V A K P 47
C L A A G F V S M F A S P . D Q K N T G P F G F P L 104
A G F Q . . . N G F A A F G L L V F T F A G A A F T G 159
H N I F G V V T A S L M K S R G G H . . . . . . . . . . . . . . . . 198
6
63
121
162
6
64
122
170
W
G S F G E R A Q H G
F T A V G Q A
Y Q S G R R
W
G S G F G A E R A S Q H G
F T A M G Q
S A G R R R
A
S
N
N T
V S I
A
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M Q Q G P
F V G V L P I C L Q P S . R C E D E L P C K L
G F R W P L A F S T L V L A D P Y A S L V F
I K L L T V T S L C P T Y Q P . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M R Q G A
F V L H P I W T D H M S G C E G P M L C F Y K L Q T A
G L G W P L A A V T S T V L V T V N P H Y V S A
I H K L L T V T S L C P V H Q P Q P T K S D . . . . . . .
M A V P M
L A P L G V F V H G W Y L K P S W P P H W V L V W T L Y S M G Y S Y L V
W K E L G G V P L G L Y T Q L A L N W A W P I F F A R M G A L V D L L L V S G A T V
A W V P L A A L L P Y L A W L A F T T L N Y V W R D N E
M V P V
L L P L G M F V R G W Y L K P S W P P R W L I W T L Y S M G Y S Y I V
W K E L G G V P L G L Y T Q L A L N W A W P I F F A R M G A L D L L L V S G V A T L
A W V P A A L L P Y L A W L A F T L N Y V W R D N E
K K M A K G L S L
V A V A P V L Y G S Y K S W P P W L L L L M G A W L V
W D G K L L Y Q L W P V F R S L L S L
A F N I P V A L V K P L A W A F A V N K L
K K I A K G L S L
I A L P L L I I L G S Y K W P W L L L L M G W L V
W E G R M M L Y Q L L A W P V F K I L V M L G V L A
A F V N P I A L V K P W F
H K K G V P S L
I V A A P L L W Y L K P S W P P L L I W I Y V M G A S W L V
W D G R L G Y Q L L N W L F F K H S V L A V D I L L V T
A F Q P V N I A A L M K Y W V F S L I N
L
V P L G L I M I H S R K W P P H V I L V W V T I Y S V M G Y A S Y L V
W K E L G G L P L G L Y S Q L A L W F L F A S G L A L L D L L L L G L V
W Q P I N L A A L L P Y L A W L T T A I Y R L W R D
L
I L P L G L V L F R W R S W P V L L V I Y S V V G Y A S Y L V
W K D L G G L P L G L Y Q L I W L F F H G L A L L L L L L G L V T L
W P I N L A A L L P Y L A W L T S A L Y H L W R D E
1
10 20 30 40 50 60
70 80 90 100 110 120
130 140 150 160
TM1 TM2
TM3 TM4
TM5Loop 4
Loop 3Loop 2
Loop 1
Mt targeting
L/V.Y...R/DCRAC
C-terminal
N-terminal
Figure 1.23: Alignement de séquences de protéines TSPO de différentes espèces. Les protéinesreprésentées pour les Eukarya sont : Humain (NP_001243460.1) ; Souris (NP_033905.3) ; Arabidopsis thaliana (NP_
566110.1), une plante supérieure ; Solanum tuberosum (CAH10765.1), la pomme de terre ; Physcomitrella Patens(ABG37902.1), une mousse. Les a.a sont colorés suivant le degré de conservation (code couleur arc-en-ciel pour uncut-off correspondant à une similarité de séquence de 30%) : en rouge intense, les a.a identiques ou similaires(100 % de conservation) ; du rouge au vert clair, le degré de conservation des a.a diminue. Les 5 TM putatifs sontreprésentés par des hélices violettes en haut des séquences et l’emplacement des boucles (Loop) qui les relient estindiqué. Entre parenthèses sont indiqués les codes UniProt des séquences protéiques. L’alignement a été réaliséavec le serveur ClustalW [Thompson et al., 1994] et la figure avec le logiciel Aline [Bond and Schuttelkopf, 2009].
Figure 1.24: Représentation schématique de la structure du gène AtTspo avec les différentssites d’initiation à la translation. Le protéine AtTSPO chez Arabidopsis est codée par un seul locusAt2g47770. Il y a 3 codons ATG possibles dans le cadre de lecture, localisés en positon M2, M21 etM42, qui permettent la synthèse de 3 protéines hypothétiques d’une longueur de 196,176 et 155 a.arespectivement.
vement 29 l’AtTSPO sont plus sensibles à l’ABA et au sel (c.-à-d. que l’inhibition de la croissance
29. Une protéine exprimée constitutivement, est une protéine qui s’exprime en permanence, quelles que soientles conditions.

1.4. La TSPO végétale : l’AtTSPO 59
induite par ABA ou NaCl est plus forte pour ces plantes). De plus, les cellules exposées à lalumière sont moins verdoyantes. La surexpression d’ AtTSPO peut donc être préjudiciable pourles cellules végétales.
Ces observations mènent à l’hypothèse que : lors d’un stress, l’AtTSPO ne serait nécessaireà la cellule que de manière transitoire pour amplifier la perception et/ou la signalisation dé-pendante de l’ABA, qui comme présenté en Annexe C, est une hormone synthétisée en grandequantité en période de stress pour permettre l’expression de gènes impliqués dans la protectionde la cellule. L’hormone pourrait par la suite participer à la régulation négative de la protéine.
L’analyse du promoteur du gène AtTspo, Fig.1.25, a permis l’identification de motifs (ouélément de réponse 30) incluant :
• DRE drought responsive element : élément de réponse à la sécheresse ;• ABRE et ABA responsive element ;• IBOX et GBOX éléments de réponse à la lumière ;• et PRE élément de régulation de la PP9-Mg.
Figure 1.25: Représentation schématique de la région (environ 400 pb) en amont deAt2g47770. Les différents motifs constituant cette région sont : drought-responsive element (DRE) ;abscisic acid-responsive element (ABRE), light-responsive elements IBOX et GBOX, et le magnesium-protoporphyrin IX regulatory element (PRE). D’après [Guillaumot et al., 2009a]
La présence de ces éléments de réponse renforce l’hypothèse que l’expression d’AtTSPO estdépendante de la production de l’hormone de stress ABA et d’autres stimuli environnementauxet ceci probablement via la liaison de ces éléments par des facteurs de transcription (FT) telsque AREB1, AREB2 ou ABF3 [Guillaumot et al., 2009a,b, Yoshida et al., 2010]. L’expressionde la TSPO chez les plantes apparait donc être régulée de manière complexe (régulation au ni-veau transcriptionnelle, post transcriptionnelle Balsemao-Pires et al. [2011]). Ceci est en accordavec le fait que même s’il a été montrée que la délétion d’At2g47770 est viable et sans chan-
30. La fonction des éléments de réponse est de permettre la régulation de l’expression des gènes par des facteursde transcription (FT) spécifiques. Ces FT vont se lier aux gènes et coordonner le recrutement du complexede l’ARN polymérase permettant la transcription du gène en ARN messager, afin de synthétiser les protéinescorrespondantes. Un élément de réponse peut être également répresseur inhibant alors l’expression du gène.

60 Chapitre 1. Les protéines TSPO
gement flagrant sur le phénotype, la sur-expression d’AtTSPO peut être préjudiciables commenous l’avons vu précédemment (augmentation de la sensibilité et diminution de la verdoyance)[Guillaumot et al., 2009a, Vanhee et al., 2011c].
1.4.3 Biodégradation
Les tétrapyrroles a sont des molécules que l’on retrouve chez tous les organismes du règne vivant et qui possèdentun large éventail de propriétés chimiques telles que l’absorption de la lumière, le transfert d’électrons et la liaisonde l’oxygène. Ce sont les composants essentiels de nombreux processus biologiques fondamentaux tels que laphotosynthèse ou la respiration.Chez les plantes, on retrouve 4 types de tétrapyrroles qui dérivent d’une voie de biosynthèse commune et chaquetype possède des protéines de liaison à différentes localisations :
— La chlorophylle qui intervient dans la photosynthèse ; on la retrouve dans la membrane thylakoïde deschloroplastes.
— L’hème ; on le retrouve dans le cytosol et dans la mitochondrie où il joue un rôle essentiel dans la respi-ration. Il permet la formation de nombreuses hémoprotéines tels que l’leghémoglobine b, les peroxydases,ou les cytochromes.
— Le sirohème ; on le retrouve dans les plastes où il est contenu dans la nitrite réductase et la sulfiteréductase, il contrôle le transfert d’électrons du centre de ces enzymes pour la réduction des nitrites oudes sulfites.
— La pytochromobiline ; on la retrouve dans le cytosol où la famille des phytochromes photorecepteursl’utilisent pour percevoir la lumière rouge et la lumière rouge lointaine, mais sous activation par la lumièreles phytochromes deviennent localisés dans le noyau.
Malgré les différentes localisations des protéines liant les tétrapyrroles, ces derniers sont tous synthétisés dans
les plastes (exception des 2 dernières étapes de la synthèse de l’hème qui peuvent se faire à la fois dans la
mitochondrie et le chloroplaste ; on peut rappeler que chez les animaux, les tétrapyrroles sont synthétisés dans
la mitochondrie). Par contre, les co-facteurs des tétrapyrroles sont éparpillés, en particulier les hémoprotéines
sont retrouvées partout dans la cellule. Cependant, il a été proposé, qu’une fraction de protoporphyrinogène IX
est aussi transférée des plastes jusqu’à la mitochondrie où elle est convertie en protohème par la protoporphy-
rinogène oxidase et la ferrochélatase. Le 1er précurseur pour tous les tétrapyrroles est l’ALA. [Mochizuki et al.,
2010, Czarnecki and Grimm, 2012]. Les propriétés chimiques des tétrapyrroles peuvent aussi causer de sévères
dommages photo-oxydatifs et la mort de la cellule sous certaines conditions. En effet, l’excitation non régulée
par la lumière des porphyrines (les tétrapyrroles cycliques) peut mener à la génération de radicaux libres et de
ROS. C’est pourquoi, le métabolisme et le transport des tétrapyrroles requièrent une régulation précise.
a. Les tétrapyrroles sont des molécules constituées de 4 cycles de pyrrole reliés entre par des groupes méthines.b. La leghémoglobine est une hémoprotéine fixatrice d’O2 présente chez les plantes légumineuses et qui a une
structure très proche de l’hémoglobine.
Encadré 10 (La biosynthèse des tétrapyrroles chez les végétaux).
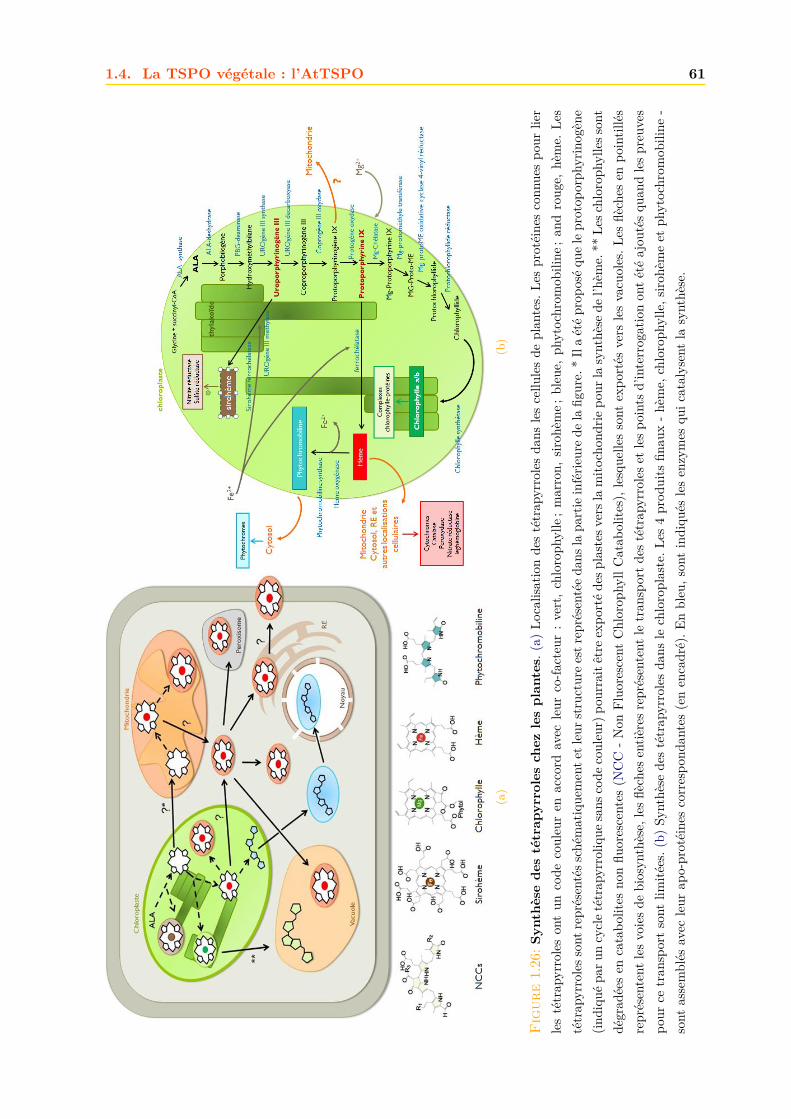
1.4. La TSPO végétale : l’AtTSPO 61
(a)
(b)
Fig
ure1.26
:Syn
thèsede
stétrap
yrroleschez
lesplan
tes.
(a)Lo
calisationde
stétrap
yrrolesda
nslescellu
lesde
plan
tes.
Lesprotéinesconn
uespo
urlie
rlestétrap
yrroleson
tun
code
couleuren
accord
avec
leur
co-fa
cteu
r:v
ert,
chloroph
ylle;m
arron,
siroh
ème;
bleue,
phytochrom
obiline
;and
roug
e,hème.
Les
tétrap
yrroless
ontr
eprésentés
schématiquemente
tleu
rstruc
ture
estr
eprésentée
dans
lapa
rtieinférie
urede
lafig
ure.*Ilaétéprop
oséqu
eleprotop
orph
yrinog
ène
(indiqu
épar
uncyclet
étrapy
rrolique
sans
code
couleu
r)po
urraitêtre
expo
rtéde
splastes
vers
lamito
chon
driepo
urla
synthèse
del’h
ème.**
Lesc
hlorop
hylle
sson
tdégrad
éesen
catabo
lites
nonflu
orescentes
(NCC
-Non
Fluo
rescentChlorop
hyllCatab
olite
s),lesqu
ellessont
expo
rtés
vers
lesvacuoles.L
esflèches
enpo
intillés
représentent
lesv
oies
debiosyn
thèse,
lesfl
èchese
ntièresr
eprésententletran
sportd
estétrap
yrrolese
tles
points
d’interrog
ationon
tété
ajou
tésq
uand
lesp
reuv
espo
urce
tran
sportsont
limité
es.(b)
Synthèse
destétrap
yrrolesda
nsle
chloroplaste.L
es4prod
uits
finau
x-h
ème,
chloroph
ylle,siro
hèmeet
phytochrom
obiline
-sont
assemblés
avec
leur
apo-protéinescorrespo
ndan
tes(enen
cadré).E
nbleu
,son
tindiqu
éslesen
zymes
quic
atalysentla
synthèse.

62 Chapitre 1. Les protéines TSPO
Guillaumot et al. [2009a,b], Vanhee et al. [2011a,c] ont montré que dans les tissus végéta-tifs, l’expression d’AtTSPO induite par l’ABA est transitoire et la dégradation de l’AtTSPO,exprimée constitutivement ou induite, est augmentée par stimulation de la biosynthèse des té-trapyrroles (en nourrissant les cellules à l’ALA). Ceci laisse suggérer que la régulation négativepost-translationnelle de l’AtTSPO pourrait dépendre du métabolisme des tétrapyrroles, Enca-dré 10 et Fig. 1.26. Cette hypothèse est renforcée par le fait qu’AtTSPO lie la PPIX in vitroet l’hème in vitro et in vivo et que cette liaison est nécessaire à sa dégradation. De plus, ladégradation d’AtTSPO se ferait via une voie active dépendante de l’autophagie. Cela a été misen évidence, entre autre, par le fait que la dégradation de la protéine est inhibée dans les mu-tants déficients en atg5 une protéine nécessaire à l’autophagie. En plus de la liaison à l’hème, laliaison de l’"ubiquitine-like" protéine ATG8 impliquée dans l’autophagie, serait nécessaire pourla dégradation de la protéine.
1.4.4 Localisation de l’AtTSPO
Les 1res études réalisées sur la TSPO de plusieurs plantes montraient majoritairement unelocalisation mitochondriale de la protéine, rappelant la localisation de la TSPO mitochondrialede mammifère :
• Corsi et al. [2004] rapportent, par immuoréaction avec un AC anti-mTSPO, la présenced’une protéine TSPO de 30-36 kDa, dans les fractions nucléaires des tissus méristéma-riques 31 de Solanum tuberosum (pomme de terre)• Lindemann et al. [2004] détectent par immunomarquage avec un sérum anti-mTSPO, une
location de la protéine dans la membrane externe des plastes et surtout des mitochondriesde D. lanata. De plus, des protéines mitochondriales d’ A. thaliana et de D. lanata furentreconnues par un AC dirigé contre un peptide déduit de la séquence d’AtTSPO. Lesauteurs rapportent également une liaison des ligands de la TSPO mammalienne (PK11195,RO5-4864) aux fractions mitochondriales de différentes espèces de plantes et à l’AtTSPOrecombinante, supposée, qu’ils ont réussi à exprimer en très faible quantité.• Finalement, Frank et al. [2007] détectent une localisation mitochondriale de la protéinePpTSPO1 (Physcomitrella paten) fusionnée au N-terminal de la protéine fluorescente GFP(PpSPO1-GFP), lorsque celle-ci est surexprimée temporairement dans les protoplastes dela moussePhyscomitrella patens.
Par contre, les études plus récentes montrent une localisation tout autre pour l’AtTSPO. Ellen’est ni localisée dans la membrane plasmique, ni dans la mitochondrie, ni dans le chloroplastemais dans les organites impliqués dans le voie de sécrétion constitutive intracellulaire, c.-à-d.principalement au niveau des membranes du RE et de l’appareil de Golgi (AG) :
• Guillaumot et al. [2009a] utilisent des cellules A. thaliana surexprimant l’AtTSPO pourexaminer sa localisation. Les expériences par western blot, en utilisant un AC anti-
31. Le méristème est un amas de cellules indifférenciées capables de se diviser (mitoses) puis de se différencieren acquérant une structure et une fonction

1.4. La TSPO végétale : l’AtTSPO 63
AtTSPO, montre que la protéine est co-localisée avec Atsec22, une protéine caractéristiquede l’AG et du RE. De plus, la protéine de fusion YFP-AtTSPO fluorescente est retrouvéeégalement dans le RE et l’AG et non dans la mitochondrie.
• Vanhee et al. [2011b] montrent par des expériences d’immunofluorescence, que dans lescellules d’A. thaliana, l’AtTSPO est co-localisée avec AtArf1, une autre protéine carac-téristique de l’AG. De plus, là encore, l’utilisation de la fluorescence confirme la nonlocalisation d’YFP-AtTSPO dans la mitochondrie et sa présence dans la voie de sécré-tion. D’après les auteurs, AtTSPO serait transportée du RE vers l’AG dans lequel elles’accumulerait. Cependant, un fait intéressant est que l’expression hétérologue d’AtTSPOchez la levure, Saccharomyces cerevisiae, donne une protéine localisée dans les mitochon-dries et interagissant avec la porine VDAC, ce qui rappelle fortement la localisation de laprotéine TSPO mitochondriale mammalienne 18 kDa.• Finalement, Hachez et al. [2014] montrent que la protéine interagit physiquement avecl’aquaporine PIP2 ;7. Cette dernière est adressée à la membrane plasmique et est impliquéedans le transport de l’eau dans la membrane de l’AG et du RE.
La localisation d’AtTSPO apparait donc controversée. Ces résultats pourrait suggèrer quel’AtTSPO possède différentes localisation selon le contexte cellulaire et génétique (AtTSPOpossède 3 codons d’initiation possibles, §. 1.4.1). balsemao2011, par des techniques de marquagefluorescent, a examiné la localisation subcellualire de la protéine entière fusionnée à une GFP(M1AtTSPO-GFP) et des protéines tronquées en position M21 et M42 (M21AtTSPO-GFP etM42AtTSPO-GFP respectivement). Les données obtenues montraient : une localisation de laprotéine entière (M1AtTSPO-GFP) dans le RE de la plante ; et une localisation des protéinestronquées (M21AtTSPO-GFP et M42AtTSPO-GFP) dans les mitochondries. À cela s’ajoute,l’observation d’une délocalisation de la protéine entière après un traitement salin : M1TSPO-GFP est localisée dans les plastes. Cette étude laissa donc suggérer une importance du codond’initiation dans la localisation d’AtTSPO, ainsi qu’un possible rôle du N-terminal et du stressdans l’adressage. Cependant, en discutant avec le Prof. Henri Batoko (Université Catholique deLouvain), qui travaille depuis de nombreuses années sur la protéine, nous avons appris que cetteétude n’était pas fiable. En effet, il existerait des erreurs méthodologiques et d’interprétationsdans les fusions fluorescentes que les auteurs ont réalisé notamment en ce qui concerne latopologie de la protéine résultante qui serait adressée aux plastes. Le Prof. H. Batoko n’ajamais obtenu une protéine fonctionnelle en ajoutant une étiquette quelle qu’elle soit en C-terminal d’AtTSPO. Celui-ci nous a donc confirmé une localisation de l’AtTSPO limitée à lavoie de sécrétion (AG et RE) de la cellule.
1.4.5 Adressage
L’AtTSPO est adressée à la membrane du RE et de l’AG contrairement à la TSPO1 quiest adressée à la membrane mitochondriale. Quelle pourrait être l’origine de cette différence delocalisation ?
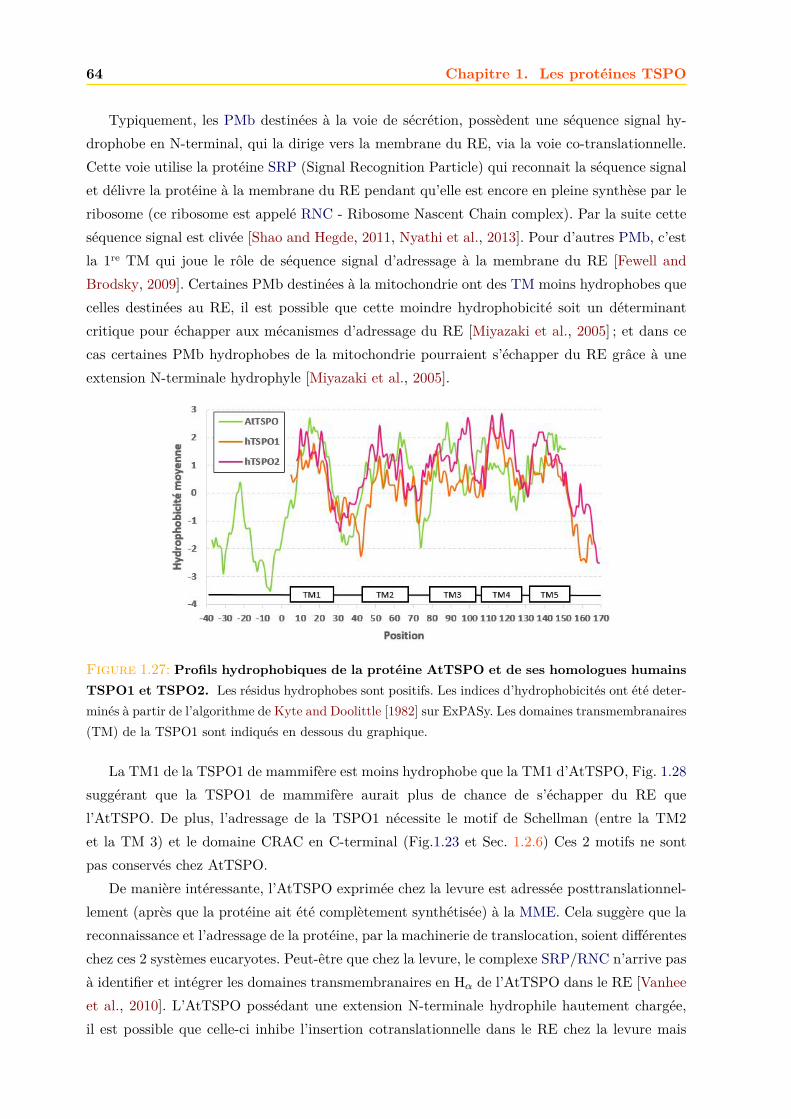
64 Chapitre 1. Les protéines TSPO
Typiquement, les PMb destinées à la voie de sécrétion, possèdent une séquence signal hy-drophobe en N-terminal, qui la dirige vers la membrane du RE, via la voie co-translationnelle.Cette voie utilise la protéine SRP (Signal Recognition Particle) qui reconnait la séquence signalet délivre la protéine à la membrane du RE pendant qu’elle est encore en pleine synthèse par leribosome (ce ribosome est appelé RNC - Ribosome Nascent Chain complex). Par la suite cetteséquence signal est clivée [Shao and Hegde, 2011, Nyathi et al., 2013]. Pour d’autres PMb, c’estla 1re TM qui joue le rôle de séquence signal d’adressage à la membrane du RE [Fewell andBrodsky, 2009]. Certaines PMb destinées à la mitochondrie ont des TM moins hydrophobes quecelles destinées au RE, il est possible que cette moindre hydrophobicité soit un déterminantcritique pour échapper aux mécanismes d’adressage du RE [Miyazaki et al., 2005] ; et dans cecas certaines PMb hydrophobes de la mitochondrie pourraient s’échapper du RE grâce à uneextension N-terminale hydrophyle [Miyazaki et al., 2005].
Figure 1.27: Profils hydrophobiques de la protéine AtTSPO et de ses homologues humainsTSPO1 et TSPO2. Les résidus hydrophobes sont positifs. Les indices d’hydrophobicités ont été deter-minés à partir de l’algorithme de Kyte and Doolittle [1982] sur ExPASy. Les domaines transmembranaires(TM) de la TSPO1 sont indiqués en dessous du graphique.
La TM1 de la TSPO1 de mammifère est moins hydrophobe que la TM1 d’AtTSPO, Fig. 1.28suggérant que la TSPO1 de mammifère aurait plus de chance de s’échapper du RE quel’AtTSPO. De plus, l’adressage de la TSPO1 nécessite le motif de Schellman (entre la TM2et la TM 3) et le domaine CRAC en C-terminal (Fig.1.23 et Sec. 1.2.6) Ces 2 motifs ne sontpas conservés chez AtTSPO.
De manière intéressante, l’AtTSPO exprimée chez la levure est adressée posttranslationnel-lement (après que la protéine ait été complètement synthétisée) à la MME. Cela suggère que lareconnaissance et l’adressage de la protéine, par la machinerie de translocation, soient différenteschez ces 2 systèmes eucaryotes. Peut-être que chez la levure, le complexe SRP/RNC n’arrive pasà identifier et intégrer les domaines transmembranaires en Hα de l’AtTSPO dans le RE [Vanheeet al., 2010]. L’AtTSPO possédant une extension N-terminale hydrophile hautement chargée,il est possible que celle-ci inhibe l’insertion cotranslationnelle dans le RE chez la levure mais

1.4. La TSPO végétale : l’AtTSPO 65
pas chez la plante, comme il l’a été montré pour la protéine ABCme dans l’étude de Miyazakiet al. [2005] [Vanhee et al., 2010]. Comme décrit pour la TSPO1, l’AtTSPO pourrait alors êtreinsérée dans la MME via la voie dépendante de TOM70 [Vanhee et al., 2010]. L’AtTSPO par-tage la même localisation subcellulaire que la TSPO2, cette dernière est aussi plus hydrophobede manière générale que la TSPO1, Fig. 1.28. Cependant, la TSPO de plante n’apparait pasplus proche de la TSPO2 qu’elle ne l’est de la TSPO1, Fig. 1.23. On ne sait pas si la TSPO2conserve sa localisation dans le RE lorsqu’elle est exprimée chez la levure.
1.4.6 Fonctions
1.4.6.1 AtTPSPO, ABA et réponse au stress
L’AtTSPO est rapidement régulée négativement après son induction et il a été montré qu’elleest transcriptionnellement régulée par les FT bZip de type AREB1, AREB2, AREB3, qui sontimpliqués dans la signalisation dépendante de l’ABA [Vanhee et al., 2011c]. Il est connu quel’enzyme 8’-hydroxylase catalyse la 1re étape de la dégradation oxydative de l’ABA [Krochkoet al., 1998]. Il a alors été proposé qu’indirectement l’AtTSPO pouvait réguler l’activité decette enzyme et ainsi participer à l’homéostatie de l’ABA [Vanhee and Batoko, 2011]. L’AtTSPOserait donc une protéine importante dans l’adaptation au stress de la plante et pourrait moduler,au moins en partie, la perception/signalisation dépendante de l’ABA durant le stress. L’ABAparticiperait ensuite à la régulation négative post-translationnelle de la protéine et l’AtTSPOparticiperait également à l’homéostatie de l’ABA. [Guillaumot et al., 2009b, Vanhee et al.,2011c, Vanhee and Batoko, 2011]. C’est pourquoi, en partie, la cellule végétale aurait besoind’une voie efficace et hautement régulée pour n’induire la protéine que transitoirement durantle stress. Puis, elle la dégraderait car à long terme, la protéine entrainerait des interférencesdans la signalisation et l’homéostatie de l’ABA.
1.4.6.2 AtTSPO, stress oxydatif et homéostasie des porphyrines
Les stress abiotiques ou un traitement par l’ABA génèrent un stress oxydatif chez les végé-taux [Xiong and Zhu, 2003], Encadré 11.
L’induction par l’ABA de l’expression d’AtTSPO est transitoire et s’accompagne simulta-nément d’une augmentation du niveau d’hèmes libres, elle aussi transitoire [Guillaumot et al.,2009a]. De plus, l’augmentation de l’ABA intracellulaire inhibe l’expression des protéines liantla chlorophylle [Guillaumot et al., 2009a]. Ainsi, l’augmentation de l’ABA, lors d’un stress, ré-duirait le flux de PPIX vers la branche chlorophylienne privilégiant ainsi la branche de l’hème(Fig. 1.26). Cette augmentation de la production des hèmes serait nécessaire pour protéger lecellule des dommages du stress : une partie des hèmes libres synthétisés peut quitter les plastespour être incorporée dans les enzymes "piégeurs" de ROS, et l’autre partie peut être dégradéedans les plastes par l’hème oxygènase pour produire la biliverdine IXα, un puissant antioxydant.
Cependant, un excès d’hèmes/porphyrines libres génère aussi la production de ROS. Or, des

66 Chapitre 1. Les protéines TSPO
La production des ROS est normale pour tous les organismes, il s’agit d’une conséquence inévitable de la pho-tosynthèse oxygénique et des conditions de stress environnementaux. La cellule dispose d’un système complexede détoxification contre les ROS, comprenant une série d’enzymes et de molécules antioxydantes (superoxidedismutase, catalase, ascorbate peroxidase et glutathione réductase).Le stress oxydatif survient lorsqu’il y a un déséquilibre entre la production de ROS et la capacité de la cellule àles contourner ou les détoxifier. La conséquence de ce stress est l’apparition de dégâts souvent irréversibles pourla cellule. C’est pourquoi, un contrôle efficace de ce stress est crucial pour la survie de la plante.Comme mentionné précédemment, les porphyrines peuvent être des molécules potentiellement toxiques pourla cellule. Particulièrement, les hèmes libres du cytosol et la protoporphyrine IX (PPIX) qui peuvent générerspontanément des ROS en présence de lumière.Cependant, quand l’hème est lié à un "piégeur" de ROS comme la catalase ou la peroxydase, il joue un rôleessentiel dans la détoxification des ROS. De plus, la dégradation de l’hème, par l’hème oxygènase, donne labilliverdine-IXα, un puissant antioxydant [Otterbein et al., 2003, Balestrasse et al., 2005]. Ainsi, encore une fois,un contrôle précis des porphyrines libres dans la cellule est nécessaire pour sa survie.
Encadré 11 (Le stress oxydatif chez les végétaux).
lignées cellulaires transgéniques d’Arabidospis surexprimant l’AtTSPO et dont la synthèse destétrapyrroles a été stimulée, ont un niveau réduit de porphyrines libres. L’AtTSPO serait doncchargée de lier ces hèmes/porphyrines pour les dégrader via une voie d’autophagie spécifique(§.1.4.3). La protéine aurait donc un rôle de "piégeur de porphyrines". La question de commentet où l’ATSPO acquière l’hème reste à être élucidée.
Par contre, des lignées cellulaires transgéniques d’Arabidospis sur-exprimant l’AtTSPO ac-cumulent plus de ROS que les cellules WT en présence ou en absence d’ABA. Ainsi, encore unefois, la protéine AtTSPO est nécessaire mais que de manière transitoire, pour limiter l’accumu-lation, dans la cellule, d’hèmes libres et d’autres pophyrines générateurs de ROS. Car à plus longterme, le piégeage continu des porphyrines par la protéine va devenir toxique pour la cellule, enprivant les apo-hémoprotéines d’acquérir leur hème et particulièrement les hémoprotéines anti-oxydantes impliquées dans l’élimination des ROS. De plus, comme vu précédemment, l’AtSPOindirectement régule l’homéostasie de l’ABA via l’enzyme 8’-hydroxylase or cette dernière aégalement besoin de la liaison à l’hème pour être active.
Les ROS ne sont pas toujours toxique pour la cellule, car il s’agit également des molécules designalisation (messagers secondaires), à une concentration appropriée ils sont un facteur postitifdans la réponse au stress. Pour son bon fonctionnement, la cellule végétale doit donc régulerfinement l’homéostasie des ROS en utilisant les connexions complexes qu’il y a entre l’AtTSPO,les porphyrines et l’ABA.
1.4.6.3 La régulation des aquaporines par l’AtTSPO
Lors d’un stress lié à la sécheresse, la plante doit limiter ses sorties d’eau pour assurersa survie. Le flux d’eau à travers les membranes des cellules, pour réguler la croissance et

1.4. La TSPO végétale : l’AtTSPO 67
Figure 1.28: Modèle hypothétique de la fonction de régulation du stress oxydatif parl’AtTSPO. Dans les plantes, un stress abiotique induit une augmentation des niveaux de l’hormone destress ABA ainsi qu’un stress oxydatif (ROS). Pendant ce stress, le signalement dépendant de l’ABAinduit transitoirement la surexpression de l’AtTSPO, dans les membranes du RE et de l’AG, de mêmequ’une augmentation de la biosynthèse des hèmes, dans les chloroplastes, pour faire face aux besoins enhèmes des hémoprotéines "piégeurs" de ROS. L’AtTSPO agit également comme un "piégeur" de ROSen faisant face aux possibles excès d’hèmes ou de porphyrines libres cytosoliques qui pourraient causerdes dommages oxydatifs à la cellule. Pour cela, l’AtTSPO lie l’hème et interagit avec la protéine ATG8,via un motif AIM conservé (121LYLYL125), pour être dégradée (avec son hème) par autophagie. Cettedégradation permet aussi de réguler négativement le niveau d’AtTSPO dans la cellule, puisqu’une surex-pression continue de la protéine est toxique pour la cellule et induit un stress oxydatif en interférant avecl’activité des hémoprotéines impliquées dans l’élimination des ROS. L’AtTSPO agit aussi indirectementcomme régulateur négatif de l’ABA, via la régulation de l’activité de l’enzyme de dégradation de celle-ci,la 8’-hydroxylase, qui a besoin de la liaison de l’hème pour être active. Figure adaptée de [Vanhee et al.,2011c, Vanhee and Batoko, 2011]
la transpiration, est modulé par les aquaporines présentes dans la membrane plasmique. Larégulation négative des aquaporines, dans le but de limiter le transport de l’eau entre les celluleset la deshydratation des cellules, apparait donc jouer un grand rôle dans la réponse de la planteface à ce type de stress.
Pour réduire le niveau à la membrane plasmique d’une de ces aquaporines, la PIP2 ;7,l’AtTSPO pourrait interagir avec celle-ci lorsqu’elle est en pleine translation vers la membraneplasmique. Le complexe TSPO-PIP2 ;7 perait alors être dégradé par la voie de l’autophagie[Hachez et al., 2014].

68 Chapitre 1. Les protéines TSPO
1.4.7 La TSPO chez la mousse Physcomitrella patens
Chez la mousse Physcomitrella patens, 3 gènes codent pour 5 protéines homologue à laTSPO1 mamalienne. L’une d’entre elle, PpTSPO1, fusionnée à une GFP, a été détectée dansdes organelles ressemblant aux mitochondries [Frank et al., 2007]. Depuis aucune autre étudesn’a été réalisées pour confirmer ou non cette localisation et on ne sait pas si toutes ces protéinesTSPO de P. patens ont la même localisation sub-cellulaire.
La PpTSPO1 est régulée positivement au niveau transcriptionnel par un stress salin. Toutcomme chez A. thaliana, la délétion du gène codant pour cette protéine est viable chez cetteespèce. Cependant, un tel mutant est caractérisé par une accumulation d’hème et de PPIX[Frank et al., 2007]. Ceci suggère donc, tout comme pour l’AtTSPO, un rôle de la PpTSPO1dans le métabolisme (dégradation) des porphyrines. De plus, ce mutant est plus sensible austress salin et produit 2.6 fois plus ROS que la mousse sauvage [Frank et al., 2007]. Ceci laissepenser que la PpTSPO1 est impliquée dans l’homéostasie des ROS chez cette espèce [Lehtonenet al., 2012]. Ce mutant est également plus sensible à une infection fongique ou bactérienne,suggérant une implication de TSPO dans les mécanismes de défense de la plante [Lehtonenet al., 2012].

1.5. La TSPO bactérienne 69
1.5 La TSPO bactérienne
L’existence de la 1re protéine bactérienne homologue à la TSPO mitochondriale 18kDa(TSPO1) fût rapportée en 1995 par Yeliseev and Kaplan [1995] chez la bactérie Rhodobactersphaeroides. Elle fut dénommée TspO pour Tryptophan rich sensory protein (le O majusculereflétant sont rôle de senseur d’oxygène). C’est sur cette bactérie que les principaux travauxdécrivant les relations structure-fonction ont été effectués.
La bactérie R. sphaeroides R. sphaeroides est une bactérie pourpre photosynthétique ap-partenant à la subdivision des α-protéobactéries. Cette bactérie, pour assurer les besoins éner-gétiques nécessaires à sa croissance, peut pratiquer une respiration aérobie (utilisation d’O2) ouanaérobie, c.-à-d. qu’en réponse à une diminution des niveaux d’O2, elle peut passer à un modede croissance photosynthétique ou encore mettre en place un métabolisme fermentatif. Ainsi,en aérobiose, les systèmes photosynthétiques (SP) sont pratiquement inexistants mais quand lateneur en O2 diminue, le SP membranaire, qui est localisé dans la membrane interne, est activé.Celui-ci permet la capture de l’énergie lumineuse et sa convertion en énergie chimique grâceaux caroténoïdes et aux bactériochlorophylles (Bchl) [Oh and Kaplan, 2001, Zeilstra-Ryalls andKaplan, 1998], Encadré. 12.
1.5.1 Information génétique et localisation cellulaire
Chez R. sphaeroides, le gène Tspo, de 480 nucléotides, code pour une protéine RsTSPO de148 a.a soit ∼17 kDa. Tspo est localisé au sein du cluster Crt impliqué dans la voie de biosynthèsedes caroténoïdes [Yeliseev and Kaplan, 1995]. Cependant la délétion du gène Tspo n’a aucuneincidence sur cette voie métabolique [Yeliseev and Kaplan, 1995]. La protéine TspO est localiséedans la membrane externe de la bactérie, la partie N-terminale apparait du coté extracellulaire,alors que la partie C-terminale est tournée vers l’espace périplasmique. La protéine RsTSPOprésente 32% d’identité avec la protéine TSPO mitochondriale humaine 18 kDa (hTSPO1)[Yeliseev and Kaplan, 1995].
1.5.2 Fonctions
1.5.2.1 RsTSPO et la photosynthèse
Yeliseev and Kaplan, Zeng and Kaplan ont montré que la RsTspO était principalementproduite en condition de stress anoxique (diminution du taux d’O2) ou lumineux (absence delumière). De plus, en mimant les conditions observées lors du passage du métabolique aérobie à laphotosynthèse - passage de forte vers une faible teneur en O2 - ils ont observé, chez un mutant dugène Tspo, une forte synthèse des pigments photosynthétiques (Bchl) en comparaison du WT.Cependant, cette augmentation n’est que transitoire. Il a alors été proposé que la RsTSPOpuisse agir comme un régulateur négatif, mais seulement provisoirement, des gènes impliquésdans la biosynthèse des pigments photosynthétiques en réponse à une diminution de la tension

70 Chapitre 1. Les protéines TSPO
en oxygène [Yeliseev and Kaplan, 1995]. C’est pourquoi la TSPO bactérienne a été dénomméeTryptophan-rich sensory protein.
Lorsque la teneur en O2 diminue (dans le noir ou en présence de lumière), il y a induction des systèmesphotosynthétiques (SP) localisés dans la membrane cytoplasmique de R. sphaeroides.Le SP, comprenant 2 complexes photo-collecteurs et un centre réactionnelle pour le transfert d’électrons via desréactions d’oxydo-réduction, est composé en partie par les caroténoïdes et Bchl (une porphyrine), qui permettentde capter l’énergie lumineuse et de la convertir en énergie chimique. Alors que les Bchl sont plus particulièrementimpliquées dans la capture de l’énergie lumineuse, les caroténoïdes ont d’avantage pour fonction de maintenirla structure de la membrane et de la protéger contre les dommages photo-oxydatifs. Dans le noir, en conditionsanaérobie, la bactérie pratique une respiration en utilisant le dimethyl sulfoxide (DMSO) ou l’amine-N-oxidecomme accepteur d’électrons à la place de l’oxygène. Le centre réactionnel est alors nécessaire dans ce type derespiration [Zannoni, 1995].La production du SP membranaire est sous le contrôle de 3 systèmes de régulation principaux : FnrL, qui est unrégulateur global intervenant en anaérobiose ; le système à 2 composants PrrB/A et le système répresseur/anti-répresseur PpsR/AppA. Ces systèmes régulent l’expression des gènes dont les produits sont impliqués dans labiosynthèse du SP en réponse aux changements de tension en O2.La lumière et l’oxygène sont perçus et intégrés par le système represseur/anti-répresseur PpsR/AppA, danslequel PpSR est un répresseur des gènes photosynthétiques et AppA un anti répresseur sensible à la lumièreet l’oxygène. Ainsi, en conditions lumineuse et aérobie, PpsR réprime les gènes photosynthétiques alors qu’encondition anaérobie, la formation du complexe AppA/PpsR permet l’expression de ces gènes et donc la formationdu SP. Il a été montré qu’AppA et PpsR sont des hémoprotéines. L’activité de PpsR serait donc contrôlée àla fois par AppA et la liaison de l’hème [Yin et al., 2012, 2013]. Cette liaison pourrait alors stimuler l’activitéantirépresseur de AppA pour permettre la redirection de la synthèse des tétrapyrroles des hèmes vers la branchedes Bchl. Ceci permettrait alors une synthèse maximale des SP.
Par ailleurs, les bactéries photosynthétiques doivent coordonner la synthèse des PPIX, hèmes et des Bchl, puisqueleur surproduction est toxique : la combinaison O2/photosynthèse générant un stress oxydatif.
Encadré 12 (R. sphaeroides et l’anaérobie).
Il a été proposé une régulation transitoire par la RsTSPO, via la régulation du flux d’inter-médiaires des porphyrines, du système répresseur/anti-répresseur PpsR/AppA (Encadré. 12),

1.5. La TSPO bactérienne 71
lequel réprime les gènes photosynthétiques en conditions lumineuse et aérobie [Yeliseev and Ka-plan, 1995]. Précisément, cette régulation permettrait de moduler l’activité de l’hémoprotéineanti-répresseur AppA. En particulier, il a été envisagé que la RsTSPO était impliquée dans leflux de copropophyrine III, un intermédiaire de la synthèse de la PPIX, de l’hème et de la Bchl[Oh and Kaplan, 2004, Zeilstra-Ryalls and Kaplan, 1998, Zeng and Kaplan, 2001]. Cependantla coproporphyrine III est toujours excrétée par R. sphaeroides en absence de la RsTSPO, sug-gérant que ce transport n’est pas TSPO-dépendant. Il se pourrait alors que la protéine régulel’activité de AppA à travers la séquestration ou la dégradation de l’hème ou de l’un de cesintermédiaires comme la PPIX [Batoko et al., 2015a].
La régulation négative transitoire des gènes photosynthétiques ainsi observée lors du passageen condition anaérobie, pourrait permettre d’assurer que l’augmentation de la synthèse des SP,nécessaires à la respiration de la bactérie (qui se fait en privilégiant la branche des BChl plutôtque celle des hèmes), ne se produise pas avant qu’il y ait suffisamment d’hèmes disponibles pourtraiter la fonction essentielle de transfert d’électrons : les électrons issus de la photosynthèsedoivent faire la navette vers le PS via des cytochromes contenant des hèmes.
1.5.2.2 La dégradation des porphyrines
Ginter et al. [2013], Guo et al. [2015] ont montré que les TSPO de différentes bactéries (Chlo-robium tepidum, Archaeoglobus fulgidus, Rhodobacter sphaeroides, Bacillus cereus) catalysent ladégradation de la PPIX et ceci d’une manière dépendante de l’oxygène et de la lumière (encondition aérobie). Cette dégradation s’accompagne d’une diminution des ROS produit par laphoto-oxydation de la PPIX. En condition anaérobie, la dégradation de la PPIX est moindre.La photodégradation de la PPIX par la TSPO bactérienne donne un composé inconnu maissemblable à la biliverdine, il a été nommé Bilindigin.
À noter que la hTSPO1, possédant la Thr147 du polymorphysme A147T, et la hTSPO2 ontété testées mais ne montrent aucun produit de dégradation de la PPIX [Guo et al., 2015]. LahTSPO1 (ou une autre TSPO mammalienne) n’a quant à elle jamais été testée.
La TSPO pourrait catalyser à la fois l’oxydation vinyl/formyl et aussi le clivage du pontméthyne reliant les noyaux pyrroles porteurs de groupement vinyle. Cette activité enzymatiquede dégradation est partiellement inhibée par le PK11195 et nécessite 2 Trp en position 51 et138 (chez la BcTSPO). Le Trp138 est relativement bien conservé à travers les protéines TSPO,contrairement au Trp51 qui n’est pas conservé chez les plantes et chez certaines TSPO2 animales.Il a été proposé que ces 2 résidus interagissent avec la PPIX via le pont méthyne clivable et legroupement vinyle.
Les tétrapyrroles libres, comme la PPIX, sont hautement contrôlés par la cellule puisque leurphoto-excitation génère des ROS. Ainsi, cette dégradation de la PPIX par la TSPO pourraitprotéger les bactéries de l’effet oxydatif de l’accumulation des porphyrines [Ginter et al., 2013,Guo et al., 2015]. De plus, la TSPO pourrait aussi promouvoir la neutralisation des ROS viace composé inconnu, issu de la dégradation de la PPIX, semblable à l’antioxydant biliverdine,
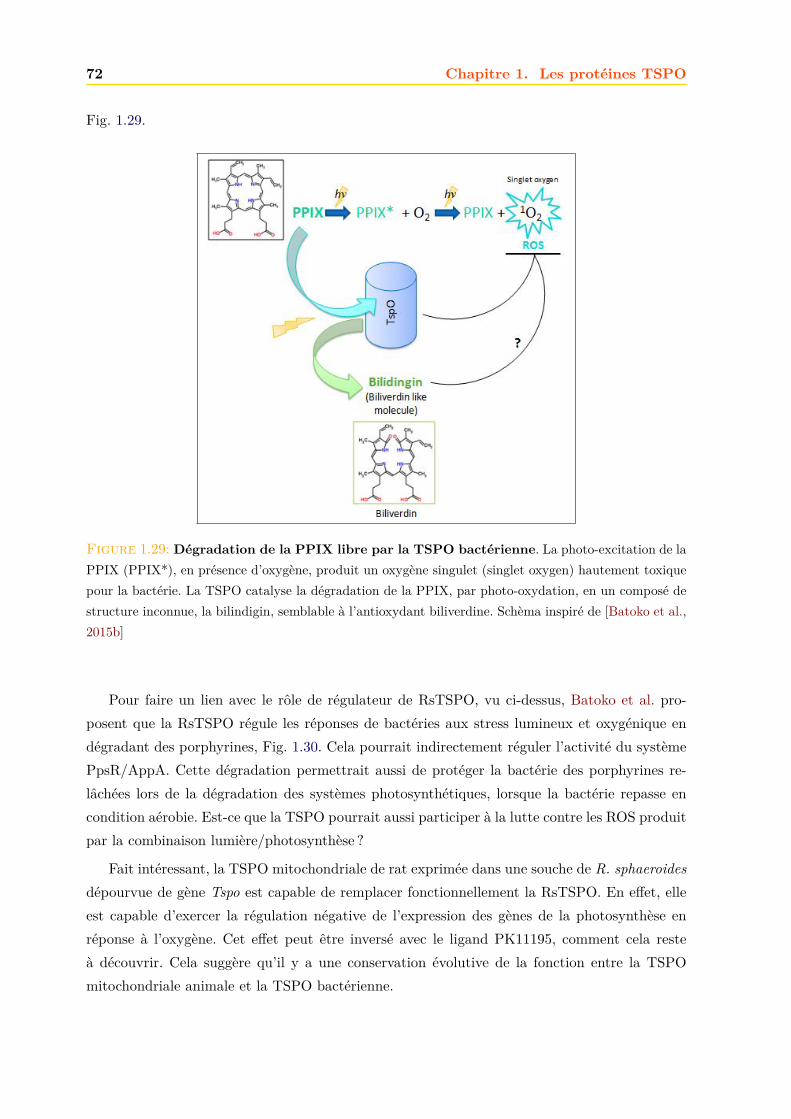
72 Chapitre 1. Les protéines TSPO
Fig. 1.29.
Figure 1.29: Dégradation de la PPIX libre par la TSPO bactérienne. La photo-excitation de laPPIX (PPIX*), en présence d’oxygène, produit un oxygène singulet (singlet oxygen) hautement toxiquepour la bactérie. La TSPO catalyse la dégradation de la PPIX, par photo-oxydation, en un composé destructure inconnue, la bilindigin, semblable à l’antioxydant biliverdine. Schèma inspiré de [Batoko et al.,2015b]
Pour faire un lien avec le rôle de régulateur de RsTSPO, vu ci-dessus, Batoko et al. pro-posent que la RsTSPO régule les réponses de bactéries aux stress lumineux et oxygénique endégradant des porphyrines, Fig. 1.30. Cela pourrait indirectement réguler l’activité du systèmePpsR/AppA. Cette dégradation permettrait aussi de protéger la bactérie des porphyrines re-lâchées lors de la dégradation des systèmes photosynthétiques, lorsque la bactérie repasse encondition aérobie. Est-ce que la TSPO pourrait aussi participer à la lutte contre les ROS produitpar la combinaison lumière/photosynthèse ?
Fait intéressant, la TSPO mitochondriale de rat exprimée dans une souche de R. sphaeroidesdépourvue de gène Tspo est capable de remplacer fonctionnellement la RsTSPO. En effet, elleest capable d’exercer la régulation négative de l’expression des gènes de la photosynthèse enréponse à l’oxygène. Cet effet peut être inversé avec le ligand PK11195, comment cela resteà découvrir. Cela suggère qu’il y a une conservation évolutive de la fonction entre la TSPOmitochondriale animale et la TSPO bactérienne.
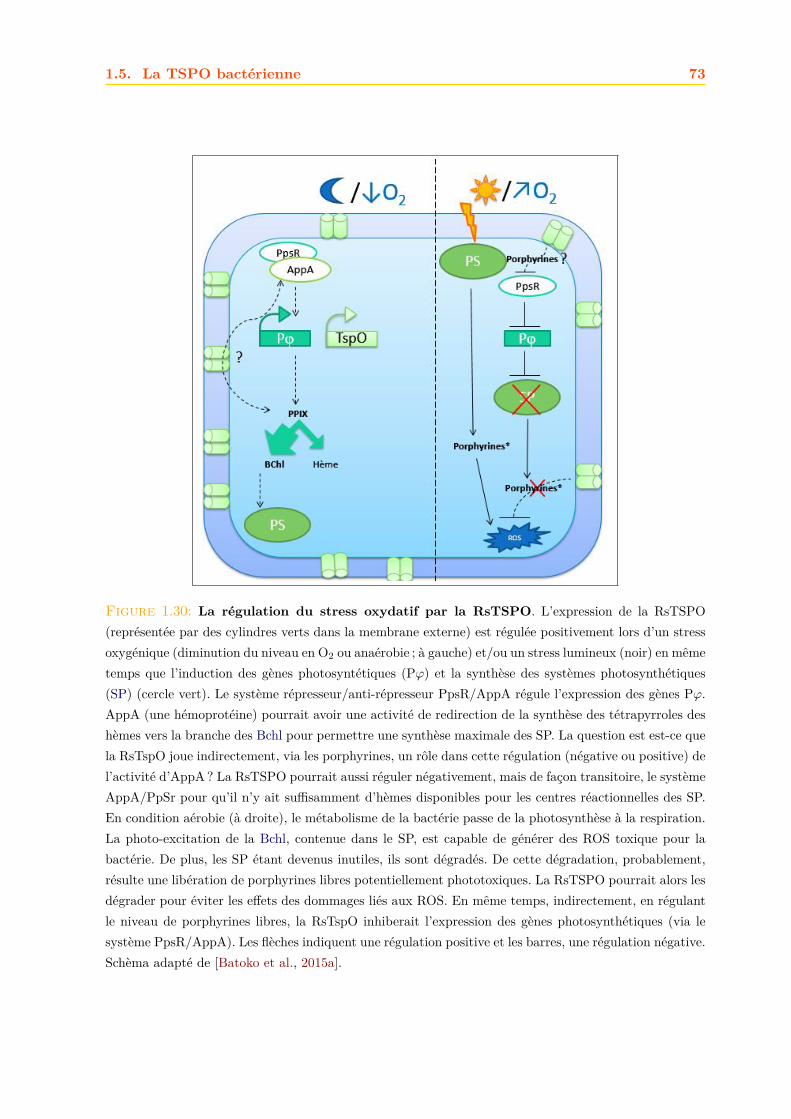
1.5. La TSPO bactérienne 73
Figure 1.30: La régulation du stress oxydatif par la RsTSPO. L’expression de la RsTSPO(représentée par des cylindres verts dans la membrane externe) est régulée positivement lors d’un stressoxygénique (diminution du niveau en O2 ou anaérobie ; à gauche) et/ou un stress lumineux (noir) en mêmetemps que l’induction des gènes photosyntétiques (Pϕ) et la synthèse des systèmes photosynthétiques(SP) (cercle vert). Le système répresseur/anti-répresseur PpsR/AppA régule l’expression des gènes Pϕ.AppA (une hémoprotéine) pourrait avoir une activité de redirection de la synthèse des tétrapyrroles deshèmes vers la branche des Bchl pour permettre une synthèse maximale des SP. La question est est-ce quela RsTspO joue indirectement, via les porphyrines, un rôle dans cette régulation (négative ou positive) del’activité d’AppA? La RsTSPO pourrait aussi réguler négativement, mais de façon transitoire, le systèmeAppA/PpSr pour qu’il n’y ait suffisamment d’hèmes disponibles pour les centres réactionnelles des SP.En condition aérobie (à droite), le métabolisme de la bactérie passe de la photosynthèse à la respiration.La photo-excitation de la Bchl, contenue dans le SP, est capable de générer des ROS toxique pour labactérie. De plus, les SP étant devenus inutiles, ils sont dégradés. De cette dégradation, probablement,résulte une libération de porphyrines libres potentiellement phototoxiques. La RsTSPO pourrait alors lesdégrader pour éviter les effets des dommages liés aux ROS. En même temps, indirectement, en régulantle niveau de porphyrines libres, la RsTspO inhiberait l’expression des gènes photosynthétiques (via lesystème PpsR/AppA). Les flèches indiquent une régulation positive et les barres, une régulation négative.Schèma adapté de [Batoko et al., 2015a].

74 Chapitre 1. Les protéines TSPO
1.5.2.3 Fonctions chez d’autres Bactéries
Chez Sinorhizobium meliloti, une bactérie pourpre tout commeR. sphaeroides, la SmTSPOcontrôle avec FixL un locus nommé ndi pour "nutrient deprivation induced locus". L’expres-sion de ce locus est induite en réponse à des stress environnementaux, comme la carence ennutriments, la diminution de la tension en O2 [Davey and de Bruijn, 2000]. FixL est un senseurspécifique de O2. Cette protéine comprend un domaine de fixation à la membrane cytoplas-mique, un domaine PAS contenant un site de fixation pour l’hème, capable de percevoir lapression partielle en oxygène. FixL est nécessaire à l’induction de l’expression du locus ndi enréponse à une faible tension en oxygène. FixL et la SmTSPO pourraient contrôler la biosynthèsedes porphyrines en réponse à un faible niveau d’oxygène. La régulation exercée par la SmTSPOsur l’expression de ndi apparait être épistatique sur FixL : en effet, aucune expression de ndin’a pu être mise en évidence dans un mutant délété du gène Tspo, même si FixL est présent[Davey and de Bruijn, 2000].
Chez la cyanobactérie Fremyella diplosiphon, la FdTSPO est régulée positivement en réponseà une déficience en nutriment et aux stress. De plus, une délétion du gène Tspo entraîne unediminution du niveau des ROS lors d’un stress salin ou osmotique, alors que lors d’un stressoxydatif ces mêmes mutants montrent une augmentation des niveaux des ROS. La FdTSPOparticiperait donc aux processus de régulation liés aux stress.
1.5.3 Structures
Ces 2 dernières années, à la résolution de la structure de la mTSPO par RMN s’est ajoutéela détermination des structures cristallographiques de 2 TSPO bactériennes, celles de Rhodo-bacter sphaeroïdes RsTspO et de Bacillus cereus BcTspO. Ces 2 structures bactériennes offrentl’opportunité de pouvoir faire des comparaisons entre bactéries et mammifères.
1.5.3.1 Structures cristallographiques de la BcTSPO
Guo et al. ont produit plusieurs TSPO recombinantes, exprimées chez E. coli, de plusieurs or-ganismes bactériens et vertébrés mais apparemment seule la TSPO de Bacillus cereus (BcTSPO)a bien voulu cristalliser . La BcTspO recombinante extraite en détergent existe sous 3 états d’oli-gomérisation différents : monomérique, dimérique et polymérique. Les structures cristallinesobtenues sont :
• 2 Apo BcTspO en phase lipidique cubique 32 (LCP) cristallisées à partir de la fractioncontenant un mélange de protéine sous forme monomérique et dimérique. La maille cris-talline obtenue est, dans les 2 cas, monomérique. Une structure a été obtenue à hauterésolution, 1.6 Å (4RYO), et l’autre à 2 Å.
32. La phase lipidique cubique (LCP) est une bicouche lipidique en trois dimensions pénétrée par un systèmeinterconnecté de canaux aqueux. La monooléine (un monoglycéride d’acide oléique) est le lipide de choix pourformer des LCP. Les PMb sont incorporées dans la LCP où ils diffusent en 3D. Le grand avantage de la LCP estque la protéine reste dans un environnement plus natif.

1.5. La TSPO bactérienne 75
• Une Apo BcTspO (4RYJ) en micelles de détergent cristallisée à partir de la fractioncontenant la protéine sous forme oligomérique. La maille cristalline obtenue est dimériqueet la structure a une résolution de 4.1 Å.• Un complexe BcTspO-PK11195 en LCP (4RYI). La maille cristalline obtenue est sousforme dimérique et la structure a une résolution de 3.5 Å. Il n’est pas précisé à partir dequelle fraction a été obtenue ce cristal.
Chacune des 4 structures cristallisées montrent 5 hélices α transmembranaires organisées,lorsque l’on regarde du C-terminal, dans le sens des aiguilles d’une montre selon l’ordre : TM1-TM2-TM5-TM4-TM3. Une longue boucle, dans laquelle on observe une petite hélice α (α’) reliela TM1 et la TM2 et recouvre la poche C-terminale, Fig. 1.31.a.
Figure 1.31: Structures cristallographiques de la BcTSPO avec et sans la présence duligand PK11195. a) Vue supérieure périplasmique de la BcTSPO (4RYO), en LCP, avec ces 5 TM(TM1 à TM5). b) Représentation du complexe dimère de BcTSPO/PK11195 (4RIY ; le PK11195 estreprésenté en rose). c) Vue sélective de la poche de liaison du PK11195. Les résidus indiqués ont étédécrits comme étant en contact de Van der Waals avec le ligand [Guo et al., 2015]. d) Superpositiondes structures de la BsTspO en détergent (4RYJ, en gris) et en LCP (colorée). La superposition a étéréalisée en isolant le monomère de la structure dimérique de la BcTSPO en détergent. e) Superpositiondes structures, en LCP, de l’Apo BsTSPO (colorée) et de la BcTSPO en complexe avec le PK11195(gris). La superposition a été réalisée en isolant un monomère de la structure dimérique de la BcTSPOen complexe avec le ligand. Figures réalisées avec le logiciel Pymol en utilisant la structure PDB 4RYO,4RYI et 4RYJ.
L’interface des associations dimériques est constituée par des résidus de la TM1 et de laTM3, Fig. 1.31.b. Les monomères de l’apo BcTSPO (monomère isolé du dimère) en LCP et enmicelles de détergent sont semblables, Fig. 1.31.d. Ils ne différent que légèrement dans la bouclereliant la TM1 et la TM2.

76 Chapitre 1. Les protéines TSPO
Le PK11195 se lie dans une cavité formée par les 5 hélices transmembranaires. Les résidusindiqués, selon les auteurs, comme étant en contact de Van der Walls sont représentés Fig. 1.31.c.Finalement, ce qui est très surprenant avec ces structures, c’est que les conformations desprotéines avec et sans ligand sont identiques 1.31.e.
1.5.3.2 Structures cristallographiques de la RsTSPO
La RsTSPO recombinante exprimée chez E. coli, extraite et purifiée en détergent est sousforme dimérique [Li et al., 2015]. À partir de cette fraction Li et al. ont obtenu la structurecristallographique de la RsTSPO, en LCP, à 2.5 Å de résolution. La maille cristalline obtenueest un dimère. Cette structure montre 5 hélices α transmembranaires organisées, lorsque l’onregarde du côté C-terminal, dans le sens des aiguilles d’une montre, selon l’ordre : TM1-TM2-TM5-TM4-TM3. Cependant, la structure n’a pas entièrement était résolue, il manque pourchacun des monomères, formant l’association dimérique, la boucle 1 reliant la TM1 et la TM2.
Les auteurs ont tenté de nombreuses mutations pour arriver à obtenir une structure cristallo-graphique de la protéine. La protéine porteuse de la mutation A139T (mimant le polymorphismehumain A147T, cf. §.1.2.7) a permis d’obtenir, toujours à partir de la protéine se trouvant sousforme dimérique en détergent, 2 structures cristallographiques en LCP : une structure RsTSPOA139T haute résolution (4UC1) à 1.8 Å dont la maille cristalline est un dimère, Fig. 1.32.a ;et une structure RsTSPO A139T (4UC2) à 2.4 Å dont la maille cristalline est un trimère,Fig. 1.32.c.
Pour chacune des 3 structures obtenues, le dimère est formé de monomères compactes dontl’interface est composé de 37 résidus issus des TM3 (16 a.a), TM1 (12 a.a) et TM4 (3 a.a),(Fig. 1.32.c). Cette interface est très étroite et ne permet vraisemblablement pas le transport demolécules. Le dimère observé dans ces cristaux semble être l’unité structurale et fonctionnellede la RsTSPO [Li et al., 2015].
Les ligands PK11195, PPIX et cholestérol, bien que présentant une affinité moindre (del’ordre du µM) pour la RsTSPO que pour la hTSPO (nM), la RsTSPO les lie toujours. Lamutation A139T diminue l’affinité (de 4 à 5 fois) de la BcTSPO pour ces ligands [Li et al.,2015]. Les différences structurales observées entre le WT et le mutant A139T sont en accordavec ces différences d’affinités des ligands. En effet, des différences majeures se produisent dansles TM2 et TM5 et dans la boucle reliant TM1 et TM2 : la TM2 bascule de 7.7˚ vers la TM5alors que la TM5 devient moins coudée résultant une association plus proches des TM2 et TM5.Ces changements s’accompagnent d’un repositionnement des chaines latérales des résidus F144
et L142 dans la TM5 qui sont 2 résidus du domaine CRAC, Fig. 1.32.b.L’addition de cholestérol ou de PK11195 au milieu de cristallisation n’a pas permis leur
résolution. Cependant, dans un des monomères de l’association trimérique du mutant RsTSPOA139T, les auteurs ont eu la surprise de trouver une porphyrine (PPIX) co-purifiée avec laprotéine, Fig. 1.32.c. De façon aussi surprenante, les structures de la protéine avec et sansPPIX se superposent relativement bien (à l’exception de la boucle 1) et aucun changement de

1.5. La TSPO bactérienne 77
Figure 1.32: Structures cristallographiques de la RsTspO WT et du mutant A139T avecet sans la PPIX. a) Représentation en cartoon du dimère RsTSPO, en LCP, présentant la mutationA139T (4UC2). b) Superposition de la structure WT (beige) et A139T (vert). La superposition a étéréalisée en isolant le monomère de la structure dimérique du mutant A139T. La structure monomérique(4UC3) de la BcTSPO WT n’est pas complètement résolue. c) En haut, représentation du trimère, enLCP, de RsTspO A139T (4UC1) dont l’un des monomères contient une PPIX (en rose). En bas, vuesupérieure périplasmique de ce trimère. d) Superposition des structures, en LCP, de l’Apo RsTSPO (engris) et du monomère, isolé du trimère, en complexe avec la PPIX (coloré). Figures réalisées avec lelogiciel Pymol en utilisant la structure PDB 4UC1, 4UC2 et 4UC3.
conformation n’est observé, Fig. 1.32.d.
1.5.3.3 Comparaison des structures bactériennes
Bien que les structures cristallographiques viennent de 2 bactéries différentes, une gramnégative (R. spaheroides) et une gram positive (B. cereus), et qu’elles ne partagent que 23%d’identité de séquence, elles sont remarquablement similaires, avec ou sans ligand ! Fig. 1.33.Les hélices transmembranaires se superposent très bien et la longue boucle reliant la TM1 et laTM2 adopte une structure très similaire avec une courte hélice α′ au milieu de la boucle.
La seule différence notable est l’organisation des dimères qui ne présente pas la même inter-face : TM1 et TM2 pour la BcTSPO ; TM1 et TM3 pour la RsTSPO, Fig. 1.31.b, Fig. 1.32.cet Fig. 1.34. Cette différence soulève les questions suivantes : est-ce que l’interface du dimèreest conservée ? est-ce qu’il a une signification fonctionnelle pertinente ? L’observation d’unestructure dimérique de la RsTSPO extraite et purifiée en détergent ainsi que dans les diffé-rents cristaux souligne probablement l’importance du dimère. Alors que le fait de retrouverla BcTSPO sous forme monomérique en solution pourrait signifier que les cristaux dimériquesobtenus résultent d’un effet d’empilement du cristal.

78 Chapitre 1. Les protéines TSPO
Figure 1.33: Superpositions des structures cristallographiques de la BcTSPO et du mutantRsTSPO A139T. a) Superposition des structures monomériques de l’APO BcTSPO (4RYO, coloré) etde l’APO RsTSPO A139T (4UC1, beige). b) Superposition des structures monomériques de la BcTSPOen complexe avec le PK11195 (4RYI, coloré) et de la RsTSPO A139T en complexe avec la PPIX (4UC2,beige). Les superpositions ont été réalisées en isolant un monomère de la structure dimérique et trimériquede la BcTSPO et de son mutant A139T, respectivement. Figures réalisées avec le logiciel Pymol.
Figure 1.34: Alignements des séquences des TSPO humaine, murine, de rat, de R. sphae-roides et de B. cereus montrant la différence des interfaces des dimères chez les 2 bactéries.Les a.a impliqués dans l’interface du dimère de la RsTSPO sont indiqués par des triangles cyans et ceuxde l’interface du dimère de la BcTSPO sont indiqués par des ronds cyans. D’après [Li et al., 2016].
1.5.3.4 Comparaison des structures bactériennes et mammalienne
Dans toutes les structures résolues, le monomère de TSPO adopte un faisceau de 5 Hαtransmembranaire (TM). Cette similarité souligne une forte conservation du repliement desprotéines TSPO durant l’évolution.
La structure RMN de la mTSPO1 en DPC résolue par Jaremko et al. montre des différencesnotables avec les structures cristallographiques bactériennes.
Premièrement, la mTSPO est instable (multiples conformations) et peu structurée en ab-sence de ligand, et seule la présence en large excès du PK11195 peut stabiliser une conformation.Ceci contraste remarquablement avec l’apparente similitude des structures cristallographiquesbactériennes aussi bien avec et sans ligand. Jaremko et al. [2015b] proposent que la flexibilitéapparente de la mTSPO Apo illustre une plasticité conformationnelle native de la protéine. Li
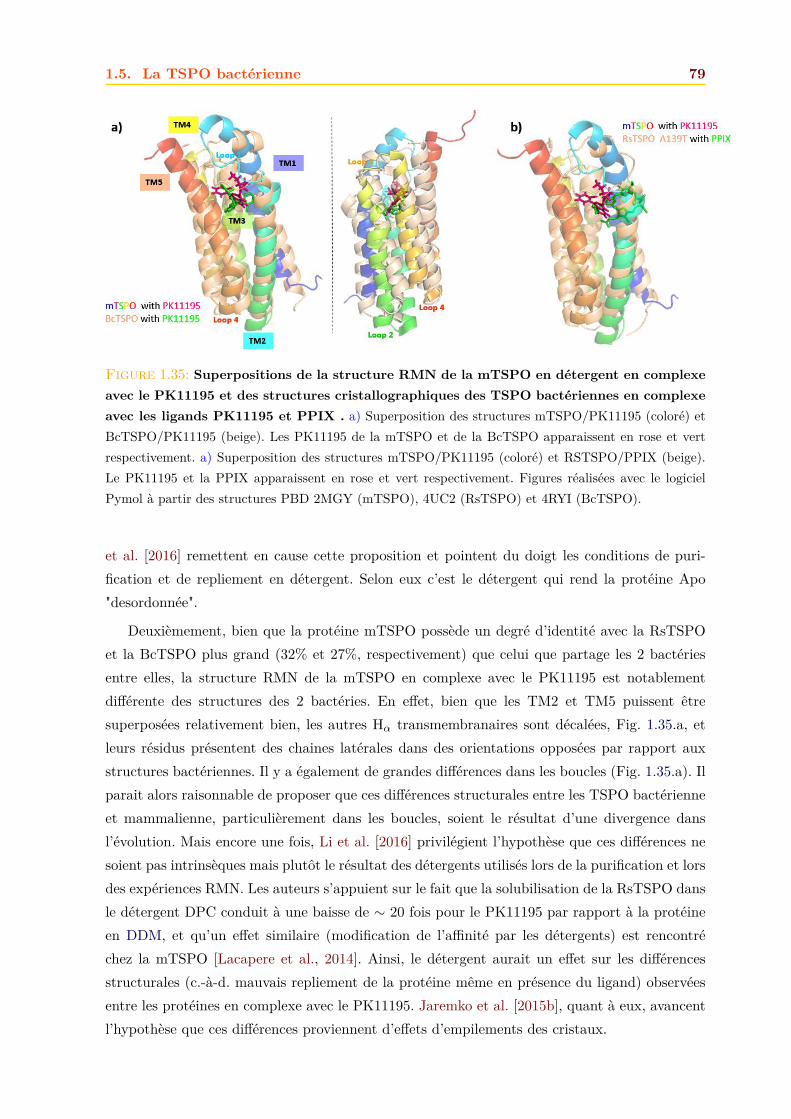
1.5. La TSPO bactérienne 79
Figure 1.35: Superpositions de la structure RMN de la mTSPO en détergent en complexeavec le PK11195 et des structures cristallographiques des TSPO bactériennes en complexeavec les ligands PK11195 et PPIX . a) Superposition des structures mTSPO/PK11195 (coloré) etBcTSPO/PK11195 (beige). Les PK11195 de la mTSPO et de la BcTSPO apparaissent en rose et vertrespectivement. a) Superposition des structures mTSPO/PK11195 (coloré) et RSTSPO/PPIX (beige).Le PK11195 et la PPIX apparaissent en rose et vert respectivement. Figures réalisées avec le logicielPymol à partir des structures PBD 2MGY (mTSPO), 4UC2 (RsTSPO) et 4RYI (BcTSPO).
et al. [2016] remettent en cause cette proposition et pointent du doigt les conditions de puri-fication et de repliement en détergent. Selon eux c’est le détergent qui rend la protéine Apo"desordonnée".
Deuxièmement, bien que la protéine mTSPO possède un degré d’identité avec la RsTSPOet la BcTSPO plus grand (32% et 27%, respectivement) que celui que partage les 2 bactériesentre elles, la structure RMN de la mTSPO en complexe avec le PK11195 est notablementdifférente des structures des 2 bactéries. En effet, bien que les TM2 et TM5 puissent êtresuperposées relativement bien, les autres Hα transmembranaires sont décalées, Fig. 1.35.a, etleurs résidus présentent des chaines latérales dans des orientations opposées par rapport auxstructures bactériennes. Il y a également de grandes différences dans les boucles (Fig. 1.35.a). Ilparait alors raisonnable de proposer que ces différences structurales entre les TSPO bactérienneet mammalienne, particulièrement dans les boucles, soient le résultat d’une divergence dansl’évolution. Mais encore une fois, Li et al. [2016] privilégient l’hypothèse que ces différences nesoient pas intrinsèques mais plutôt le résultat des détergents utilisés lors de la purification et lorsdes expériences RMN. Les auteurs s’appuient sur le fait que la solubilisation de la RsTSPO dansle détergent DPC conduit à une baisse de ∼ 20 fois pour le PK11195 par rapport à la protéineen DDM, et qu’un effet similaire (modification de l’affinité par les détergents) est rencontréchez la mTSPO [Lacapere et al., 2014]. Ainsi, le détergent aurait un effet sur les différencesstructurales (c.-à-d. mauvais repliement de la protéine même en présence du ligand) observéesentre les protéines en complexe avec le PK11195. Jaremko et al. [2015b], quant à eux, avancentl’hypothèse que ces différences proviennent d’effets d’empilements des cristaux.

80 Chapitre 1. Les protéines TSPO
À noter, que la structure récemment résolue du mutant mTSPO contenant le polymorphismehumain A147T, montre moins d’altérations de la structure comparée au WT, en comparaison dece qui a été observé dans la structure du même mutant créé chez RsTSPO (A139T). Encore unefois, Jaremko2015 avancent des effets d’empilements des cristaux pour expliquer ces différenceset Li et al. l’effet du détergent sur le repliement. De plus Li et al. argumentent que chacunede leurs structures qui résultent pourtant de formes cristallines différentes (c.-à-d. avec unarrangement unique et des contacts intermoléculaires différents) mènent toutes globalement àla même structure 3D.
Finalement, une autre différence est l’état oligomérique des différentes structures résolues :dimérique pour la RsTSPO ; monomérique et dimérique pour la BcTSPO ; et monomérique pourla mTSPO. Le dimère de BcTSPO présente une interface complètement différente de celle deRsTSPO, comme vu précédemment.
R. sphaeroidesB. cereus
HumainSouris
R. sphaeroidesB. cereus
HumainSouris
R. sphaeroidesB. cereus
HumainSouris
M N M D W A L F L T F L A A C G A P A T T G A L L K P D E . . . W Y D N L N K P W W N P P R W V F P L A W T S L Y F 55
L M S L A A M R V A Q L E G S G . . . . . Q A L A F Y A A Q L A F N T L W T P V F F G M K R M A T A L A V V M V M W 108
L F V A A T M W A F F Q L D T W A G V L F V P Y L I W A T A A T G L N F E A M R L N W N R P E A R A . . . 158
. . M F M K K S S I I V F F L T Y G L F Y V S S V L F P I D R T W Y D A L E K P S W T P P G M T I G M I W A V L F G 56
L I A L S V A I I Y N N Y G F K P . . . K T F W F L F L L N Y I F N Q A F S Y F Q F S Q K N L F L A T V D C L L V A 111
I T T L L L I M F S S N L S K V S A W L L I P Y F L W S A F A T Y L S W T I Y S I N . . . . . . . . . . . 153
M A P P W V P A M G F T L A P S L G C F V G S R F V H G E G L R W Y A G L Q K P S W H P P H W V L G P V W G T L Y S 58
A M G Y G S Y L V W K E L G G F T E K A V V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L V D L L L V S 116
G A A A A T T V A W Y Q V S P L A A R L L Y P Y L A W L A F T T T L N Y C V W R D N H G W R G G R R L P E 169
M P E S W V P A V G L T L V P S L G G F M G A Y F V R G E G L R W Y A S L Q K P S W H P P R W T L A P I W G T L Y S 58
A M G Y G S Y I V W K E L G G F T E D A M V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L A D L L L V S 116
G V A T A T T L A W H R V S P P A A R L L Y P Y L A W L A F A T V L N Y Y V W R D N S G R R G G S R L P E 169
1 10 20 30 40 50
60 70 80 90 100 110
120 130 140 150 160
TM3
TM4 TM5Loop 4
Loop 3Loop 2
N-terminal
C-terminal
TM1 TM2
CRAC L/V.Y...R/DLAF Enhancement motif
A139T in BacteriaA147T in human
Figure 1.36: Alignements des séquences des TSPO humaine, murine, de rat, de R. sphae-roides et de B. cereus montrant les différents a.a impliqués dans la liaison du PK11195. Lesa.a en interaction avec le PK11195 chez la mTSPO et la BcTSPO sont indiqués par des triangles et desflèches roses, respectivement.
Comparaison des sites de liaisons des ligands Une molécule ressemblant à une PPIXa été résolue dans la structure cristallographique de RsTSPO. Dans chacune des 3 structures(mTSPO-PK11195, BcSTPO-PK11195 et RsTSPO-PPIX), le ligand se lie dans la même cavitécentrale mais avec des positons différentes, Fig. 1.35. En effet, le PK11195 se lie au milieu de lacavité, interagissant avec des résidus des 5 TM, alors que la PPIX est insérée plus profondémentdans la cavité dans une région entre la TM1 et la TM2, Fig. 1.35. Cette cavité centrale jouedonc un rôle important dans la liaison des ligands.
Néanmoins, les résidus impliqués dans la liaison du PK11195 restent à être confirmés. Eneffet, le PK11195 interagit avec une orientation et un ensemble de résidus différents dans lastructure cristallographique de la BcTSPO en comparaison à la structure RMN de la mTSPO,Fig. 1.36. De plus, la résolution de la structure de la BcTSPO étant relativement faible (3.5 Å)la détermination de la position précise du ligand est quelque peu ambigüe chez cette dernière[Li et al., 2016].

1.5. La TSPO bactérienne 81
La RsTSPO bien que possédant le motif CRAC de liaison au cholestérol possède une affinité 1000 fois plus faiblepour ce dernier que son homologue mammalien.Le mutant RsTSPO A139T (mimant le polymorphsime humain A147T) qui montre une diminution de l’affinitépour le cholestérol par rapport au WT. En 2015, Li et al. identifie chez ce mutant un motif d’augmentation deliaison du cholestérol, le motif hydrophobe LAF. Ces 3 résidus, adjacents au motif CRAC et précédant donc lesite A147T, sont hautement variables chez la bactérie mais très conservés chez les mammifères. Le mutant A139TRsTSPO avec ses 3 résidus A136-T137-A138 remplacés par la séquence humaine LAF montre une augmentationétonnante de l’affinité pour le cholestérol (le Kd passe de 20 nM à 80 µM) qui devient similaire à l’affinitéreportée pour la TSPO de mammifère [Li et al., 2015] . Cela suggère un rôle important du motif LAF dansl’affinité nM pour le cholestérol observée chez la TSPO de mammifère.
Encadré 13 (Le motif LAF).
La liaison du cholestérol Les TSPO des différentes espèces montrent toutes le motif CRACexposé à la membrane plutôt que formant une partie d’un tunnel à l’intérieur de la protéine, oud’un tunnel à l’interface des dimères de TSPO. De plus, il n’y a aucun résidu du motif CRACou du motif LAF, Encadré 13, présent à l’interface des dimères.
Li et al. [2016] propose un modèle de liaison de cholestérol à partir de la structure cristallo-graphique de la RsTSPO. Celui-ci est présenté Fig. 1.37. Cependant, l’orientation du cholestéroldans ce modèle est inversée par rapport à ce qui a été prédit par d’autres analyses (c.-à-d. noyaustérol associé au motif CRAC et chaine alkyl vers le motif LAF [Jamin et al., 2005]) [Li et al.,2015]. La chaine est orientée vers l’extérieur du plan membranaire et plus proche du motifCRAC que du motif LAF. Les auteurs proposent donc une voie externe pour le transport ducholestérol. Ce transport nécessiterait un dimère de dimère de TSPO, ou la TSPO en complexeavec un autre partenaire (VDAC) et dans ce cas le transport se réalise à l’interface entre les 2protéines.
Figure 1.37: Site de liaison du cholestérolle plus favorable prédit pour la RsTSPO.Le cholestérol en noir traverse les TM5 et TM4et il est localisé au voisinage du motif LAF etCRAC. Ce site de liaison est parallèle au sitede liaison d’une monooléine trouvée à la mêmeposition dans différents cristaux de la RsTSPO(4UC1, 4UC2, and 5DUO). Les chaines laté-rales de certains résidus constituant le motif LAF(ATA chez RsTSPO) et CRAC sont représentées(A136, T137, et A138 colorés en orange et L142,F144, et R148 colorés en jaune) [Li et al., 2016].

82 Chapitre 1. Les protéines TSPO
1.6 Objectifs de la thèse
Au départ, mon projet de thèse était de résoudre une structure 3D par RMN de la mTSPO1(TSPO mitochondriale 18 kDa murine), en détergent, permettant d’ouvrir la voie aux étudesfonctionnelles de la protéine.
À mon arrivé au laboratoire, la production de la protéine recombinante mTSPO1 double-ment marquée 15N,13C, en grande quantité et pure pour les études structurales en RMN duliquide était au point. Il avait déjà été montré que l’ajout du ligand spécifique de la TSPO, lePK 11195, permettait de stabiliser la mTSPO1 (0.1 mM) dans le DPC donnant des spectresRMN 2D avec une bonne dispersion des pics [Murail et al., 2008]. Toutefois, à forte concentra-tion (1 mM), condition nécessaire pour acquérir des spectres 3D indispensables à l’attributionséquentielle des résidus et à la résolution d’une structure 3D, il apparaissait une instabilité dela protéine et du ligand. Cette instabilité était probablement due à la co-existence de plusieursconformations de la protéine. Mon 1er objectif était donc de déterminer des conditions de sta-bilisation conformationnelle de la mTSPO1 (choix du détergent, ajout de lipides, ligands). Larésolution, en mars 2014, de la structure de la mTSPO1 en DPC en complexe avec PK11195,par une équipe allemande, a mis un frein à ces études en RMN du liquide.
La maîtrise par le laboratoire de la réincorporation de la mTSPO1 dans des liposomespermettait d’envisager des études de la protéine dans un environnement plus proche de sonenvironnement natif et plus fonctionnel, révélé par une liaison de haute affinité du PK1115.Mon 2e objectif était alors d’optimiser les conditions de reconstitution pour entamer les étudespar RMN du solide à l’angle magique. De telles études ayant été récemment développées avecsuccès pour une d’autre classe de PM [Das et al., 2012, Marassi et al., 2011, Park et al., 2012].
La découverte, en 2009, d’une isoforme de la TSPO, chez les animaux et les oiseaux, pré-sentant une forte homologie de séquence mais des fonctions et des localisations membranairesdifférentes, nous ont poussé à l’étudier. Mon 3e objectif a été de surexprimer l’isoforme TSPO2humaine, sous forme recombinante, pour entamer les relations structure-fonction de cette pro-téine.
Les TSPO formant une famille très conservée cela suggère une fonction commune entreanimaux, bactéries et plantes, qui aurait été conservée au cours de l’évolution. Cette hypothèsenous a conduits à nous intéresser à différentes espèces. La résolution des structures atomiquesdes bactéries, en 2015, nous a amené à étudier la TSPO de plante, pour laquelle il n’y avait pasde données structurales. La TSPO de plante se différencie, principalement, de ses homologuespar la présence d’une extension N-terminale. Mon 4e objectif a alors été de surexprimer le Nterd’AtTSPO, d’en déterminer sa structure et ses relations structure/fonction.

Matériels et Méthodes


Chapitre2
Matériels et Méthodes
Sommaire2.1 Expression et Production des TSPO . . . . . . . . . . . . . . . . . . . . . 86
2.1.1 Les souches bacteriennes d’Escherichia coli . . . . . . . . . . . . . . . . . . 86
2.1.2 Les vecteurs d’expression . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
2.1.3 Les milieux de cultures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
2.1.4 Transformation des bactéries en bactéries compétentes . . . . . . . . . . . . 91
2.1.5 Transgénèse des bactéries . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
2.1.6 Amplification et purification des plasmides . . . . . . . . . . . . . . . . . . 92
2.1.7 Culture des bactéries . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
2.1.8 Production acellualire ou cell free . . . . . . . . . . . . . . . . . . . . . . . . 97
2.2 Purification des TSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
2.2.1 Les détergents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
2.2.2 Extraction des protéines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
2.2.3 Purification sur Résine Ni-NTA . . . . . . . . . . . . . . . . . . . . . . . . . 104
2.2.4 Purification du peptide Nter d’AtTSPO . . . . . . . . . . . . . . . . . . . . 106
2.3 Caractérisation des TSPO . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
2.3.1 Gel SDS-PAGE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 108
2.3.2 Western Blot . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
2.3.3 Spectrométrie de masse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
2.3.4 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
2.3.5 Fluorescence intrinsèque . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
2.3.6 Liaisons des Ligands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
2.3.7 Mesure de la liaison des ligands par séparation des ligands libres et liés surRésine Ni-NTA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
2.3.8 Liaison de ligands radioactifs . . . . . . . . . . . . . . . . . . . . . . . . . . 120
2.3.9 Les lipides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
2.3.10 Reconstitution de la mTSPO1 dans des protéoliposomes . . . . . . . . . . . 122
2.4 La RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
2.4.1 Principe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
2.4.2 La relaxation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 126
2.4.3 Le signal RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
2.4.4 La RMN du liquide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
2.4.5 La RMN du solide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 134

86 Chapitre 2. Matériels et Méthodes
2.1 Expression et Production des TSPO
2.1.1 Les souches bacteriennes d’Escherichia coli
Voici une description des différents souches bactériennes d’Escherichia coli utilisées lors decette étude. Les souches sont conservées à −80 ◦C dans une solution contenant 15 % de glycérol.
DH5 α La souche E. coli DH5 α est couramment utilisée pour le clonage 1 de fragmentsd’ADN et la préparation des plasmides.
Pour augmenter la stabilité des insertions plasmidiques, cette bactérie possède le gène recA1muté. Cette mutation permet d’éliminer la recombinaison homologue qui pourrait causer unréarrangement du plasmide ou sa délétion. Elle possède également une mutation sur le gèneendA1 qui inactive l’endonucléase I qui dégraderait l’ADN plasmidique. Cette souche a étéutilisée pour le clonage et l’amplification des différents plasmides codant pour les protéinesTSPO. Elle n’est pas utilisée pour l’expression des protéines, puisqu’elle contient des protéasesqui dégraderaient les protéines exprimées.
BL21(DE3) La souche E. coli BL21 (DE3) constitue un système de choix pour la surexpres-sion de gènes et la production de protéines recombinantes.
Elle porte une mutation dans les gènes ompT et lon qui empêche l’expression des protéasescodées par ces gènes, qui si elles étaient présentes dégraderaient la protéine recombinante.
Ces bactéries ont, dans leur génome, le gène qui code pour l’ARN polymérase T7 (insertchromosomique "DE3" issu du phage T7), ce qui permet la surproduction de protéines dontles gènes sont sous le contrôle du promoteur T7. Ce dernier est, lui même sous le contrôledu promoteur lactose lacUV5 2 inductible par l’IPTG (Isopropyl-β-D-1-thiogalactopyranoside).Ainsi, en absence d’IPTG, la transcription du gène d’intérêt et de l’ARN polymérase T7 estréprimée grâce à la fixation du répresseur LacI sur l’opérateur lactose lacO. L’ajout d’IPTGinduit la transcription de l’ARN polymérase T7 à partir du promoteur lacUV5 par l’ARNpolymérase d’E. coli. L’ARN polymérase T7 transcrit ensuite le gène d’intérêt à partir dupromoteur T7, Fig. 2.1.
À noter, que les bactéries E. coli possèdent un opéron lactose, lui aussi sous le contrôledu promoteur-opérateur lactose. Cette opéron est nécessaire au transport et au métabolismedu lactose. Il est composé de trois gènes structurels : lacZ, lacY et lacA. lacZ code pour laβ-galactosidase qui hydrolyse le lactose en glucose et galactose ; lacY code la β-galactosideperméase ; et lacA code la β-thiogalactoside acétyltransférase.
1. Le clonage moléculaire consiste à insérer un fragment d’ADN dans un vecteur approprié comme p. ex. unplasmide (molécule d’ADN double brin circulaire distinct de l’ADN chromosomique de la cellule.). Le nouveauplasmide ainsi créé sera ensuite introduit dans une cellule hôte.
2. Le promoteur lacUV5 est une variante du promoteur lac de la souche sauvage d’E. coli. Il contient unemutation impliquant une paire de base lui permettant de recruter l’ARN polymérase plus efficacement.
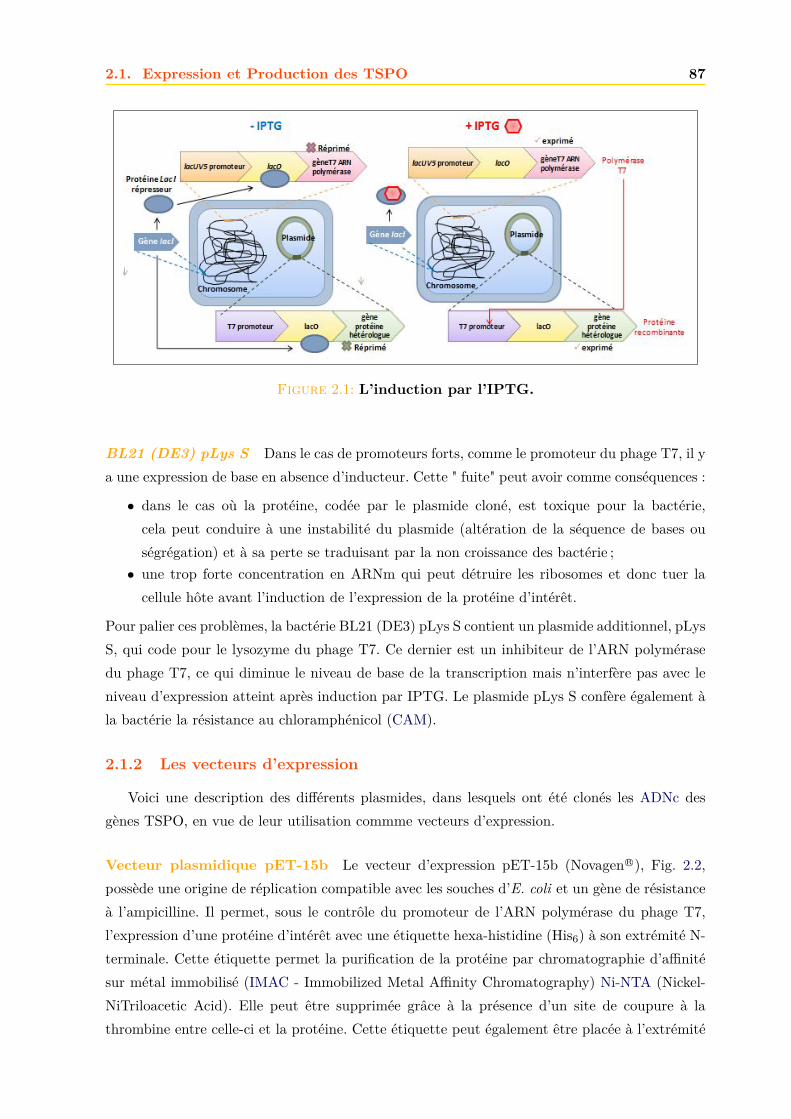
2.1. Expression et Production des TSPO 87
Figure 2.1: L’induction par l’IPTG.
BL21 (DE3) pLys S Dans le cas de promoteurs forts, comme le promoteur du phage T7, il ya une expression de base en absence d’inducteur. Cette " fuite" peut avoir comme conséquences :
• dans le cas où la protéine, codée par le plasmide cloné, est toxique pour la bactérie,cela peut conduire à une instabilité du plasmide (altération de la séquence de bases ouségrégation) et à sa perte se traduisant par la non croissance des bactérie ;• une trop forte concentration en ARNm qui peut détruire les ribosomes et donc tuer lacellule hôte avant l’induction de l’expression de la protéine d’intérêt.
Pour palier ces problèmes, la bactérie BL21 (DE3) pLys S contient un plasmide additionnel, pLysS, qui code pour le lysozyme du phage T7. Ce dernier est un inhibiteur de l’ARN polymérasedu phage T7, ce qui diminue le niveau de base de la transcription mais n’interfère pas avec leniveau d’expression atteint après induction par IPTG. Le plasmide pLys S confère également àla bactérie la résistance au chloramphénicol (CAM).
2.1.2 Les vecteurs d’expression
Voici une description des différents plasmides, dans lesquels ont été clonés les ADNc desgènes TSPO, en vue de leur utilisation commme vecteurs d’expression.
Vecteur plasmidique pET-15b Le vecteur d’expression pET-15b (NovagenR©), Fig. 2.2,possède une origine de réplication compatible avec les souches d’E. coli et un gène de résistanceà l’ampicilline. Il permet, sous le contrôle du promoteur de l’ARN polymérase du phage T7,l’expression d’une protéine d’intérêt avec une étiquette hexa-histidine (His6) à son extrémité N-terminale. Cette étiquette permet la purification de la protéine par chromatographie d’affinitésur métal immobilisé (IMAC - Immobilized Metal Affinity Chromatography) Ni-NTA (Nickel-NiTriloacetic Acid). Elle peut être supprimée grâce à la présence d’un site de coupure à lathrombine entre celle-ci et la protéine. Cette étiquette peut également être placée à l’extrémité
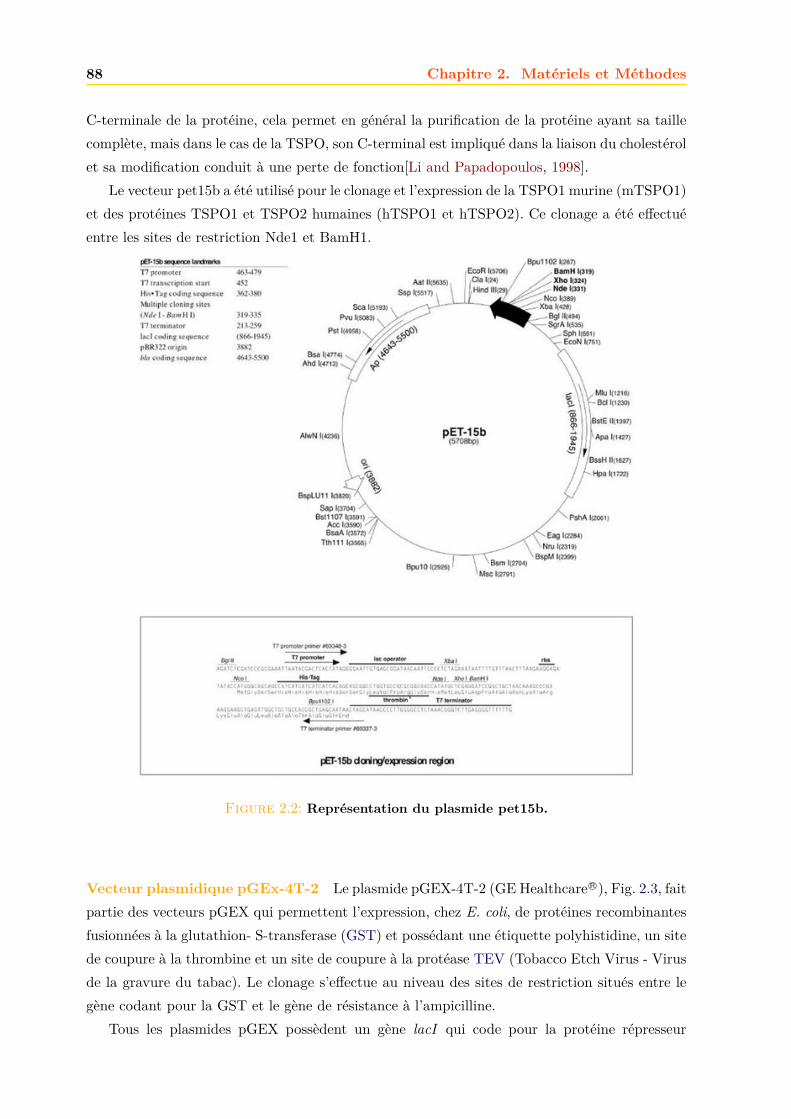
88 Chapitre 2. Matériels et Méthodes
C-terminale de la protéine, cela permet en général la purification de la protéine ayant sa taillecomplète, mais dans le cas de la TSPO, son C-terminal est impliqué dans la liaison du cholestérolet sa modification conduit à une perte de fonction[Li and Papadopoulos, 1998].
Le vecteur pet15b a été utilisé pour le clonage et l’expression de la TSPO1 murine (mTSPO1)et des protéines TSPO1 et TSPO2 humaines (hTSPO1 et hTSPO2). Ce clonage a été effectuéentre les sites de restriction Nde1 et BamH1.
Figure 2.2: Représentation du plasmide pet15b.
Vecteur plasmidique pGEx-4T-2 Le plasmide pGEX-4T-2 (GE HealthcareR©), Fig. 2.3, faitpartie des vecteurs pGEX qui permettent l’expression, chez E. coli, de protéines recombinantesfusionnées à la glutathion- S-transferase (GST) et possédant une étiquette polyhistidine, un sitede coupure à la thrombine et un site de coupure à la protéase TEV (Tobacco Etch Virus - Virusde la gravure du tabac). Le clonage s’effectue au niveau des sites de restriction situés entre legène codant pour la GST et le gène de résistance à l’ampicilline.
Tous les plasmides pGEX possèdent un gène lacI qui code pour la protéine répresseur

2.1. Expression et Production des TSPO 89
LacI qui se lie à l’opérateur tac, ne permettant pas l’expression de la protéine d’intérêt avantl’induction par IPTG. L’ajout d’IPTG induit l’expression du promoteur tac, ce qui permet latranscription du gène gst suivi du gène d’intérêt. Bien que les vecteurs pGEX ne sont pas sousle contrôle du promoteur T7, ils peuvent être utilisés chez la bactérie E. coli BL21 (DE3) carle promoteur tac est sous le contrôle de l’ARN polymèrase de l’hôte E. coli.
Figure 2.3: Représentation du plasmide pGEX-4T-2.
2.1.3 Les milieux de cultures
Le milieu SOC Le milieu SOC (Super Optimal Broth) est un milieu riche utilisé pour laculture des bactéries lors de la transgénèse des bactéries compétentes. Il permet leur régénérationaprès le choc thermique. Sa composition est donnée dans le Tab. 2.1.
Le milieu LB Le milieu LB (Lysogeny Broth, Bertani, G.) a été utilisé pour cultiver lesdifférentes souches bactériennes citées précédemment. Pour 1L d’eau distillée, il est composéde : 10 g de tryptone, 5 g d’extrait de levure et 10 g de NaCl ; le tout est ajusté à pH 7.2 avecdu NaOH. Il est stérilisé à l’autoclave avant chaque culture bactérienne.
Le milieu M9 Le milieu de culture minimum M9 ne contient pas de sources de carbone etd’azote nécessaires à la synthèse des a.a des protéines par les bactéries. Il permet alors, parajout de source nutritionnelle isotopique - ammonium 15NH4Cl, [13C]-glucose ou deutérium2H - d’obtenir la surexpression d’une protéine marquée. Ce milieu contient aussi les élémentsnutritifs de base (sodium, magnésium, chlore, calcium et phosphore) nécessaires à la croissance
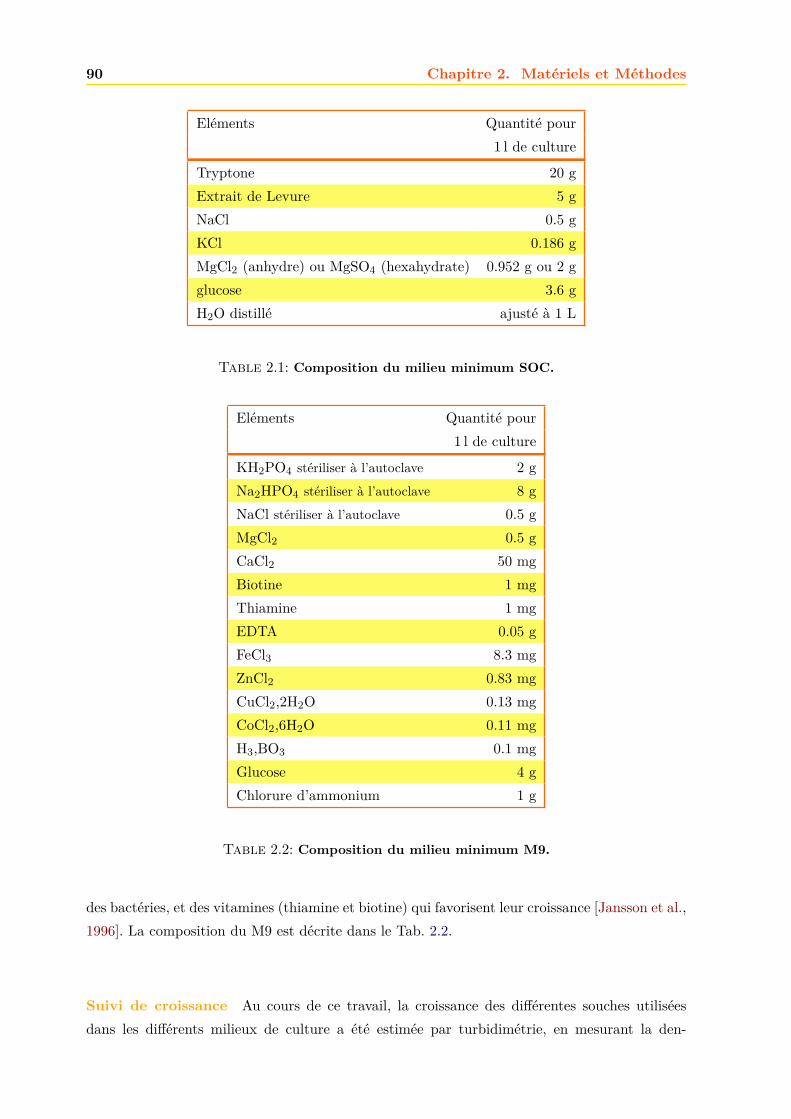
90 Chapitre 2. Matériels et Méthodes
Eléments Quantité pour1 l de culture
Tryptone 20 gExtrait de Levure 5 gNaCl 0.5 gKCl 0.186 gMgCl2 (anhydre) ou MgSO4 (hexahydrate) 0.952 g ou 2 gglucose 3.6 gH2O distillé ajusté à 1 L
Table 2.1: Composition du milieu minimum SOC.
Eléments Quantité pour1 l de culture
KH2PO4 stériliser à l’autoclave 2 gNa2HPO4 stériliser à l’autoclave 8 gNaCl stériliser à l’autoclave 0.5 gMgCl2 0.5 gCaCl2 50 mgBiotine 1 mgThiamine 1 mgEDTA 0.05 gFeCl3 8.3 mgZnCl2 0.83 mgCuCl2,2H2O 0.13 mgCoCl2,6H2O 0.11 mgH3,BO3 0.1 mgGlucose 4 gChlorure d’ammonium 1 g
Table 2.2: Composition du milieu minimum M9.
des bactéries, et des vitamines (thiamine et biotine) qui favorisent leur croissance [Jansson et al.,1996]. La composition du M9 est décrite dans le Tab. 2.2.
Suivi de croissance Au cours de ce travail, la croissance des différentes souches utiliséesdans les différents milieux de culture a été estimée par turbidimétrie, en mesurant la den-

2.1. Expression et Production des TSPO 91
sité optique (DO) à 600 nm 3 par spectrophotométrie à l’aide du spectrophotomètre OD600DiluPhotometerTM (IMPLEN).
2.1.4 Transformation des bactéries en bactéries compétentes
Afin de permettre l’entrée dans la bactérie du plasmide contenant la séquence d’ADN codantpour la protéine d’intérêt, celle-ci est rendue compétente c.-à-d. qu’on la rend perméable. Voiciles différentes étapes de cette transformation :
1. Des bactéries (BL21 (DE3), BL21 (DE3) pLys S ou DH5α) sont mises en culture pendantune nuit à 37 ◦C sous agitation à 200 rpm (rotations par minute) dans 5ml de LB stérile.
2. Le lendemain, cette pré-culture est transférée dans un erlen de 500 ml stérile contenant100ml de milieu LB stérile sans antibiotique. La solution est incubée à 37 ◦C sous agitationà 200 rpm jusqu’à obtenir une DO600 comprise entre 0,4 et 0,6, c.-à-d. jusqu’à leur phase decroissance exponentielle, phase durant laquelle les bactéries sont en division ce qui assured’avoir les membranes étirées au maximum et donc une paroi bactérienne plus fragile.
3. Une fois cette DO atteinte, la culture est mise dans la glace pendant 30 min pour arrêterla prolifération bactérienne.
4. puis la solution est centrifugée à 1200 × g pendant 10 min à 4 ◦C pour éliminer le LB.
5. Les culots bactériens sont repris dans 10 ml d’une solution de CaCl2 0.1 M stérile. Lamembrane bactérienne est notamment constituée d’une double couche de phospholipideschargés négativement par leur phosphate. Cette membrane est donc altérée en présenced’une solution concentrée en ions Ca2+, lequel permet de former des nanopores réversiblespar lesquels l’ADN pourra pénétrer par la suite.
6. Une dernière centrifugation est réalisée et les culots bactériens obtenus sont resuspendusdans 800 µl de CaCl2 0.1 M stérile et froid.
7. Les bactéries sont gardées sur glace pendant 10 min et sont maintenant compétenteset prêtes à l’emploi. Elles peuvent être conservées à -80 ◦C avec 15 % glycérol pendantplusieurs mois.
2.1.5 Transgénèse des bactéries
Une fois les bactéries rendues compétentes, le plasmide peut être introduit, c’est la transgé-nèse. Toutes les opérations qui suivent ont été effectuées sur glace afin de ne pas endommagerles bactéries :
1. À partir d’une solution à 10 ng/µl de plasmide, on mélange 100 µl de bactéries compétentesavec 1 à 5 µl de solution plasmidique.
3. Cette longueur d’onde de 600 nm est choisie car peut de choses absorbent à ce λ par contre la diffusion estforte.

92 Chapitre 2. Matériels et Méthodes
2. On incube sur glace pendant 30 min pour fixer l’ADN sur les membranes bactériennespuis les bactéries subissent pendant 45 sec un choc thermique à 42 ◦C afin d’élargir lesnanopores créés dans les membranes et ainsi permettre l’entrée du plasmide.
3. Les bactéries sont ensuite remises sur la glace pendant 2 min pour refermer les pores dela paroi.
4. 900 µl de milieu de culture SOC autoclavé sont ajoutés pour laisser croître les bactéries1h à 37 ◦C sous une agitation de 200 rpm.
5. 20 µl de solution bactérienne sont déposées et étalées sur une gélose LB-agar à 50 µg/mld’ampiciline. Puisque tous les plasmides utilisés lors de cette étude, possède un gène derésistance à cette antibiotique, seules les bactéries ayant incorporées le plasmide survi-vront.
6. Les géloses sont incubées toute la nuit à 37 ◦C. Les colonies bactériennes ainsi obtenuessont prêtes à être cultivées (pré-culture) en vue de la production de protéine recombinanteou d’une amplification du plasmide incorporé ou pour réaliser des stocks.
7. La pré-culture peut être réalisée sur la nuit, en incubant une colonie dans 5 ml de milieuLB complémentés avec les antibiotiques adéquates (Amp 50 µg/ml et�ou CAM 30 µg/ml.Ces bactéries peuvent alors être prélevées pour lancer une culture à plus grand volume ouelles peuvent être aliquotées et congelées pour réaliser un stock utilisable plus tard.
2.1.6 Amplification et purification des plasmides
Il est important de disposer d’une importante quantité de plasmides de qualité pour entre-prendre la surexpression d’une protéine. Au départ la construction plasmidique codant pour laprotéine d’intérêt est souvent initialement acheter en faible quantité, il est donc nécessaire del’amplifier pour pouvoir créer un stock qui servira aux différentes expériences (transgénèse dedifférentes souches bactériennes, séquençage, cell free, etc.).
Cette amplification du plasmide est réalisée à partir des bactéries DH5α qui sont particu-lièrement recommandées pour cet usage. 600 µl de bactéries DH5α ont donc été transforméesavec le plasmide d’intérêt et incubées dans 100 ml de LB (Amp 50mg/ml) sur la nuit à 37 ◦Cet à 200 rpm. Ensuite, pour extraire et purifier le plasmide, le kit de purification "QIAGENR©
Plasmid Maxi Kit" a été utilisé. En voici le protocole :
1. La solution bactérienne (500 ml) cultivée sur la nuit est centrifugée à 6000 × g, pendant15 min à 4 ◦C.
2. Le culot bactérien est resuspendu dans 10 ml de tampon de resuspension (toutes lescompositions des tampons sont données Tab. 2.3).
3. On ajoute 10 ml de tampon de lyse et on laisse incuber 5 min à Tamb.
4. 10 ml de tampon de neutralisation sont ajoutés et la solution est incubée sur glace pendant20 min.

2.1. Expression et Production des TSPO 93
Tampon Composition
Resuspension 50 mM Tris-Cl pH 8.0 ; 10 mM EDTA ; ajout de 100 µl RNase ALyse 200 mM NaOH, 1% SDS w
v
Neutralisation 3.0 M d’acétate de potassium pH 5.5Tampon d’équilibrage
QBT750 mM NaCl ; 50 mM MOPS pH 7.0 ; 15% isopropanol vv ; 0.15%Triton R© X-100 v
v
Lavage 1.0 M NaCl ; 50 mM MOPS pH 7.0 ; 15% isopropanol vvÉlution 1.25 M NaCl ; 50 mM Tris·Cl pH 8.5 ; 15% isopropanol vv
TE (Tris EDTA) 10 mM Tris-Cl pH 8.0 ; 1 mM EDTATBE (Tris Borate EDTA)
5X0.44 M Tris-base ; 0.44 M Acide Borique ; 0.1 M EDTA pH 8
Table 2.3: Composition des tampons du Kit "QIAGEN R© Plasmid Maxi Kit" .
5. La solution est centrifugée à 20000 × g pendant 30 min à 4 ◦C. Cette centrifugation permetde séparer l’ADN génomique, les débris cellulaire, le KDS (produit insoluble issu de l’ajoutd’acétate de potassium au SDS) et les protéines de l’ADN plasmidique contenues dans lesurnageant.
6. La colonne QIAGEN, possédant une résine échangeuse d’ions, est équilibrée avec 10 mlde tampon d’équilibrage puis elle est chargée avec le surnageant, contenant le plasmide,qui entre dans la résine par gravité.
7. La colonne est lavée avec 2 × 30 ml de tampon de lavage puis l’ADN est élué avec 15 mlde tampon d’élution.
8. L’ADN est précipité par ajout de 10.5 ml d’isopropanol, le tout est centrifuqé à 15000 ×g pendant 30 min à 4 ◦C.
9. Le culot obtenu et qui contient l’ADN est lavé avec 5 ml d’éthanol 70 % et de nouveaucentrifugé à 15000 × g pendant 10 min.
10. Le culot est séché à l’air libre pendant 5-10 min puis l’ADN plasmidique redissout dansun volume approprié (50 à 100 µl) de tampon TE.
Contrôle de la qualité du plasmide Pour vérifier l’intégrité et déterminer la quantité deplasmides recombinants ainsi amplifiés et purifiés par la méthode décrites ci-dessus, 3 techniquesont été utilisées.
Le séquençage 5 à 20 ng de plasmide ont été envoyés à séquencer au service de séquençage,par la méthode Sanger, proposé par Beckman Coulter Genomics. Cela nous permet de vérifierque la séquence est bien celle attendue.

94 Chapitre 2. Matériels et Méthodes
Le gel d’agarose L’électrophorèse D’ADN sur gel d’agarose est une technique qui permetde séparer et d’identifier des fragments d’ADN, afin d’en déterminer la taille. Elle est basée sur laséparation des acides nucléiques, via les mailles formées par l’agarose, en fonction de leur taille etde leur conformation spatiale. En effet, plus une molécule d’ADN est grosse, plus elle sera ralentiedans les mailles du gel et migrera donc moins loin qu’une molécule plus petite. Cependant, laconformation spatiale de l’ADN joue également un rôle important dans la migration. Pour unemolécule d’ADN de même taille, un ADN relâché va migrer moins loin qu’un ADN linéarisé,qui lui-même va migrer moins loin qu’un ADN surenroulé, Fig. 2.4. La taille des fragments qu’ilest possible de séparer est comprise entre 0,2 et 50 kb.
Les fragments d’ADN sont facilement détectés sur le gel grâce à un colorant fluorescent. Onpeut ainsi visualiser en lumière UV des quantités très faibles d’ADN (de l’ordre de 5-10 ng parbande). L’électrophorèse en gel d’agarose est donc une technique très sensible ; elle est de plusrapide et simple à mettre en oeuvre. La séparation est possible car à pH basique, l’ADN (oul’ARN) est chargé négativement de par les charges portées par ses groupements phosphates.Par conséquent, sous l’impulsion d’un champ électrique (de l’ordre de 100 V), l’ADN va migrervers l’anode en fonction de sa taille.
Figure 2.4: Illustration des 3 conformationsde l’ADN plasmidique visibles sur un geld’agarose. La migration d’un plasmide dépend àla fois de sa taille et de sa structure. Une moléculed’ADN sous la forme enroulée migre plus vite qu’unemolécule d’ADN de même taille sous forme linéaireou relâchée. Il est impossible de déterminer la tailled’une molécule enroulée, la taille correcte est donnéepar la bande de l’ADN linéaire.
Voici le protocole de réalisation du gel d’agarose et de sa révélation :
1. 0.8 g d’agarose sont mélangés à 100 ml de tampon TBE (la proportion d’agarose dépendde la taille des molécules d’ADN à séparer).
2. La solution d’agarose 0.8 % wv est chauffée au micro-onde pour dissoudre l’agarose jusqu’à
obtention d’un mélange transparent et homogène.
3. Quand le flacon devient saisissable à la main, le gel est coulé sur un support, prévu à ceteffet (∼ 0.5 cm d’épaisseur), muni d’un peigne permettant de marquer l’empreinte despuits de dépôt de l’ADN.
4. Tout de suite après, le colorant de visualisation de l’ADN (SYBRR© safe DNA gel stain dela marque INVITROGENTM est ajouté : ∼3 µl pour 30 ml.
5. Le gel est laissé polymériser pendant 30 min, on enlève le peigne et on dépose le gel dansla cuve de migration. Le gel est alors recouvert de tampon TBE 0.5X (Tab. 2.3).

2.1. Expression et Production des TSPO 95
6. Les échantillons d’ADN, préalablement mélangés à du colorant bleu (Blue/orange 6XPROMEGATM , p. ex. 3µl d’ADN pour 3 µl de bleu, sont déposés dans les puits - il faut∼ 50 ng d’ADN par puits.
7. L’un des puits est rempli avec 3-4 µl de marqueur de poids moléculaire (INVITROGENTM
Tracket, 1kb DNA ladder 0.1 µl. On laisse migrer le gel ∼ 20 min à 135V.
8. Finalement, le gel est révélé par exposition à une lampe UV pendant quelques minutes.
Quantification fluorimétrique de l’ADN double brin La technique la plus com-munément utilisée pour déterminer la concentration de l’ADN plasmidique est la mesure del’absorbance à 260 nm (ou densité optique - DO260) et sa pureté est caractérisée par le rapportDO260 (acides nucléiques)/DO280 (protéines contaminantes) ; une préparation d’ADN est ditepure si elle présente un rapport le plus près possible de 2. Cependant, l’inconvénient de cettetechnique est qu’elle ne fait pas de distinction entre ADN (double ou simple brin), ARN, nu-cléotides libres, protéines et sels, lesquels absorbent tous à 260 nm. Pour y remédier, nous avonsmesuré la concentration de nos plasmides avec l’appareil QubitR©. Il permet une quantificationfluorimétrique de l’ADN double brin et il est bien plus sensible que la quantification baséesur l’absorbance UV. Cet appareil utilise une sonde fluorescente spécifique qui émet seulementquand elle est liée à l’ADN double brin pour des concentrations allant de 100 pg/µl à 1000 ng/µl.
La mesure est faite en incubant 1-20 µl d’échantillon dans un tube avec 180-199µl de solutioncontenant le réactif fluorescent, pendant 2 min à Tamb ; et en insérant les tubes dans l’appareilqui donne alors la concentration en ADN double brin de l’échantillon.
2.1.7 Culture des bactéries
Deux cultures différentes ont été utilisées dans cette étude pour produire les protéines :
• Une culture dans un milieu riche (LB) pour une large production dans le but de caractériserbiochimiquement les protéines TSPO recombinantes.• Une culture dans un milieu minimum (M9) pour une production de protéines marquées
15N ou 15N,13C (pour les études RMN). Ces cultures ont concerné la protéine mTSPO1(ou hTSPO1) et le peptide N-terminal (Nter) de l’AtTSPO. Toutes les cultures ont étéréalisées à 37 ◦C dans l’incubateur Innova 44 Incubator Shaker, sous agitation à 200 rpm.
Culture en milieu riche LB Les méthodes présentées ici sont données pour un volume deculture de 2L :
1. des bactéries E. coli (souche BL21 (DE3) ou pLys S) transformées par le plasmide d’ex-pression, codant pour la protéine TSPO de mammifère (mTSPO1, hTSPO1 ou hTSPO2)ou le peptide Nter de l’AtTSPO (TSPO d’A. thaliana) fusionné à la GST, sont mises enpré-culture. Celle-ci est réalisée pendant la nuit, dans 150 ml de milieu LB contenant lesantibiotiques adéquates, à savoir :
• 50 µg/ml d’ampicilline pour les bactéries BL21 (DE3) ;

96 Chapitre 2. Matériels et Méthodes
• 50 µg/ml d’ampiciline et 30µg/ml de CAM pour les bactéries BL21 (DE3) pLys S.
Cette pré-culture permet de relancer la croissance des bactéries conservées en présencede glycérol (15 à 40%) à -80 ◦C. La mesure de la DO600 permet de vérifier la croissancebactérienne.
2. Après une nuit, les 150 ml de cette pré-culture sont dilués dans 2L de LB, contenantles antibiotiques comme décrit précédemment pour la pré-culture, de manière à obtenirune culture dont la DO600 initiale est de ∼ 0.2. Les 2L de culture sont répartis dans6 Erlenmeyers de 5L pour une meilleure oxygénation sous agitation. Cette culture estsuivie jusqu’à atteindre une DO600 de ∼ 0.6 c.-à-d. au début de la phase de croissanceexponentielle de la bactérie, valeur à laquelle la synthèse de la protéine ou du peptide estinduite par ajout d’IPTG à une concentration finale de 1 mM.
3. Après 4h (pour le Nter d’AtTSPO) ou plus (l’induction sur la nuit est possible pour lesprotéines TSPO1), les milieux bactériens sont centrifugés (Beckman, rotor JLA 10-900)pendant 20 min à 6700 × g et à 4 ◦C dans des bouteilles de 1L.
4. Les culots de bactéries peuvent être conservés à -20 ◦C ou repris chacun par 30 ml de tam-pon Hepes 50 mM pH 7.8, NaCl 150 mM. Les suspensions obtenues peuvent être utiliséesdirectement pour la purification ou conservées à -20 ◦C pour un traitement ultérieur.
Marquage totale : culture en milieu M9 Le protocole s’inspire de la méthode de Marley[Marley et al., 2001]
1. Une pré-culture de 150 ml et une culture de 2L en milieu LB sont réalisées comme décritci-dessus et la culture est suivie jusqu’à atteindre une DO600 de ∼3-3.5.
2. La culture est centrifugée (Beckman, rotor JLA 10-900) pendant 20 min à 6700 × g et4 ◦C dans des bouteilles de 1L.
3. Les culots sont lavés par 30 ml de M9 et repris dans 2L de M9 complémenté avec :
• 1 g/l de chlorure d’ammonium marqué au 15N comme source d’azote et 10 g/l deglucose, pour une protéine marquée seulement au 15N.
• 1 g/l de chlorure d’ammonium marqué au 15N et 4 g/l de glucose marqué au 13C,pour une protéine doublement marquée 15N,13C. À noter que les protocoles de lalittérature suggèrent jusqu’à 20 g de glucose et 1 à 2 g d’ammonium par litre deculture (concentration finale des 2 composés de l’ordre de 10 mM). Cependant, lecoût élevé du glucose marqué 13C, nous conduit à réduire sa concentration à 4 g/l.
4. Les 2L de culture M9 sont répartis dans 6 Erlenmeyers de 5L pour une bonne oxygénationsous agitation et lorsque les bactéries recommencent à pousser (∼ 30 à 45 min après ledébut de la culture en M9), la production de la protéine ou du peptide Nter est induitepar ajout d’IPTG à 1 mM finale.
5. Après 4 h d’induction, pour le peptide Nter d’AtTSPO, ou plus pour les protéines TSPO,les milieux bactériens sont centrifugés dans des bouteilles de 1L. Cette centrifugation(Beckman, rotor JLA 10-900) est réalisée à 6700 × g pendant 20 min à 4 ◦C.

2.1. Expression et Production des TSPO 97
6. Les culots sont lavés et peuvent être conservés à −20 ◦C ou repris chacun par :
• 30 ml de tampon Hepes 50 mM pH 7.8, NaCl 150 mM (tampon A) pour les protéinesTSPO.• 40-50 ml de tampon Hepes 50 mM pH 7.8, NaCl 500 mM (tampon A’), pour lepeptide Nter d’AtTSPO. Une telle concentration en sel est utilisée pour tenter dedissocier l’ADN qui pourrait boucher la colonne lors de la purification.
7. Les milieux bactériens sont centrifugés (Beckman, rotor JA 25.5), dans des tubes de 30ml, à 6700 × g pendant 20 min à 4 ◦C.
8. Les culots sont lavés et repris par 20-30 ml de tampon A, dans le cas des protéines TSPOet par 15 ml de tampon A’ dans le cas du peptide Nter d’AtTSPO ;
9. Une pastille d’inhibiteur de protéases (cOmplete ULTRA Tablets, Mini, EDTA-free,ROCHE) est ajoutée.
Suivi de l’induction Pour confirmer la sur-expression des protéines et suivre le niveau d’ex-pression, des aliquotes de solution bactérienne ont été prélevés avant induction et à différentstemps après l’induction lors de la culture. Ces aliquotes de 1 ml, normalisés à une DO de 1, sontcentrifugés pendant 10 min à 4000 × g, puis les culots bactériens sont repris dans un tamponcontenant du SDS (1%) pour lyser les bactéries. 20 à 30 µl sont déposés sur un gel SDS-PAGEpolyacrylamide 12%.
2.1.8 Production acellualire ou cell free
Les expériences de cell free (ou production acellulaire) ont été réalisées en collaboration avecMarie-France Giraud à l’Institut de Biochimie et Génétique Cellulaires (IBGC) de Bordeauxpour produire la protéine hTSPO2.
Ce système de production in vitro est très attractif car il permet de contourner certainesdifficultés techniques telles que l’insertion dans la membrane ou la toxicité des protéines mem-branaires surexprimées. La synthèse de la protéine se faisant dans des lysats cellulaires plutôtque dans des cellules cultivées, on appelle cette méthode le cell free. La production des pro-téines en cell free peut être réalisée avec différentes sortes et espèces d’extrait cellulaire et cetteapproche présente plusieurs avantages par rapport aux méthodes in vivo. Cependant c’est uneméthode très couteuse.
Le cell free nécessite 2 composants de base (Fig. 2.5) :
• L’ADN codant pour la protéine d’intérêt.• Une solution de réaction contenant la machinerie moléculaire nécessaire à la transcriptionet à la translation. Les extraits cellulaires issus d’E. coli fournissent toutes ou la plupartde ces molécules dont : l’ARN polymerase pour la transcription par les ARNm, les ri-bosomes pour la translation polypeptidique, l’ARNt et les acides aminés, les co-facteursenzymatiques et la source d’énergie (ATP).

98 Chapitre 2. Matériels et Méthodes
Ces composés sont distribués dans 2 solutions l’une appelée "Reaction Mix" (mélange réaction-nel) qui contient les composants acteurs de la réaction de synthèse, et l’autre "Feeding Mix"(mélange d’alimentation) qui contient les ressources (substrat) de faible PM nécessaires à laréaction de synthèse de la protéine.
Figure 2.5: Principe du cell free.
La méthode du cell free nécessite plusieurs préparations :
1. Il faut avoir une quantité suffisante du plasmide codant pour la protéine d’intérêt : environ15 µg de plasmide pour une expérience de 1 ml de mélange réactionnel. Il faut donc aupréalable amplifier l’ADN par clonage bactérien puis le purifier comme il l’a été décritauparavant.
2. Il faut préparer les extraits cellulaires (ici S30) : fermentation des bactéries E. coli, récu-pération des culots bactériens et lavages, lyse des bactéries, élimination des membranespar centrifugation et dialyse contre un tampon.
3. Il faut ensuite préparer les différents mélanges : le mélange "Master Mix", Tab. 2.4 ; lemélange réactionnel "Reaction Mix" ; et enfin le mélange "Feeding Mix", Tab. 2.5. Lesvolumes donnés dans ces tableaux sont pour une expérience avec un volume de mélangeréactionnel de 1 ml.
Réaliser la réaction de synthèse de la protéine dans un seul compartiment contenant tous lescomposants de la réaction cell free ne permet pas d’obtenir un rendement d’expression protéiquetrès élevé. C’est pourquoi, la réaction de synthèse se fait en utilisant un système d’échange encontinu (CECF - continous exchange cell free). Ce système consiste à créer 2 compartiments,séparés par une membrane semi-perméable (membrane de dialyse) ayant une limite de 10 kDa.L’un des compartiments contient le mélange réactionnel (Reaction Mix) avec donc les moléculesde haut PM : c’est dans ce compartiment que se réalise la transcription et la translation, etc’est aussi dans ce compartiment que la protéine est retenue. L’autre compartiment contient le"Feeding" mélange fournissant ainsi un réservoir de précurseurs de faible PM. C’est aussi dans
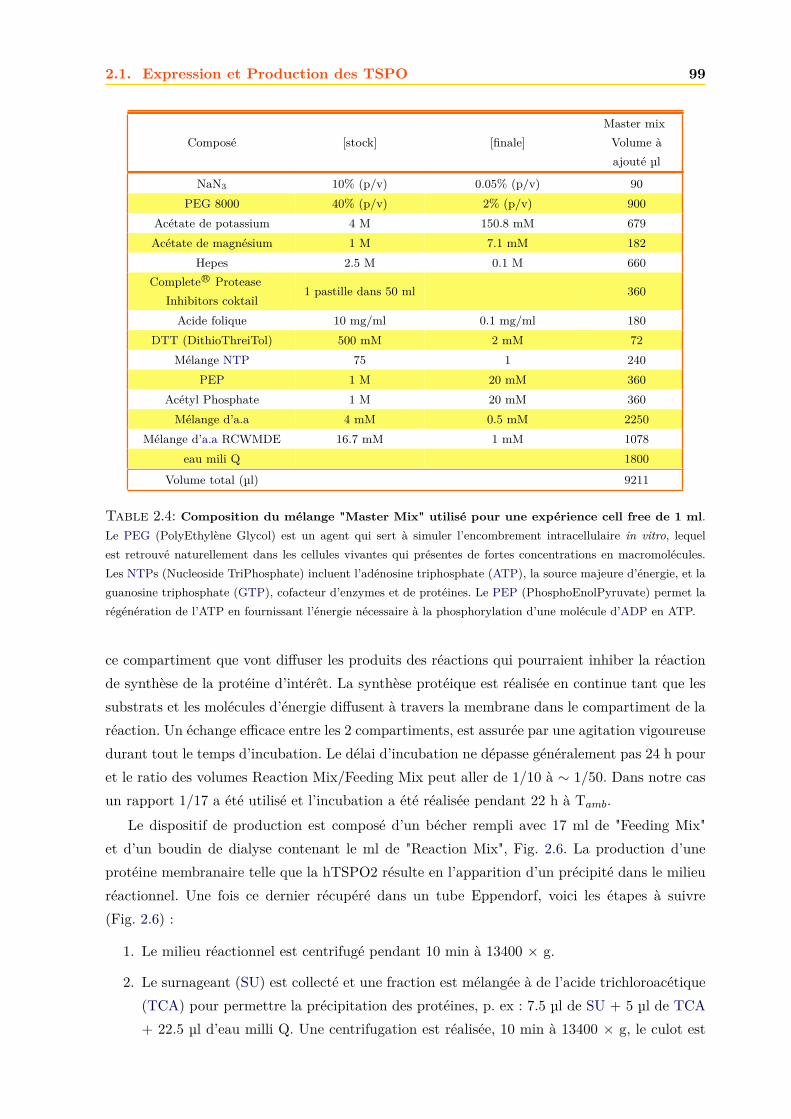
2.1. Expression et Production des TSPO 99
Composé [stock] [finale]Master mixVolume àajouté µl
NaN3 10% (p/v) 0.05% (p/v) 90PEG 8000 40% (p/v) 2% (p/v) 900
Acétate de potassium 4 M 150.8 mM 679Acétate de magnésium 1 M 7.1 mM 182
Hepes 2.5 M 0.1 M 660CompleteR© Protease
Inhibitors coktail1 pastille dans 50 ml 360
Acide folique 10 mg/ml 0.1 mg/ml 180DTT (DithioThreiTol) 500 mM 2 mM 72
Mélange NTP 75 1 240PEP 1 M 20 mM 360
Acétyl Phosphate 1 M 20 mM 360Mélange d’a.a 4 mM 0.5 mM 2250
Mélange d’a.a RCWMDE 16.7 mM 1 mM 1078eau mili Q 1800
Volume total (µl) 9211
Table 2.4: Composition du mélange "Master Mix" utilisé pour une expérience cell free de 1 ml.Le PEG (PolyEthylène Glycol) est un agent qui sert à simuler l’encombrement intracellulaire in vitro, lequelest retrouvé naturellement dans les cellules vivantes qui présentes de fortes concentrations en macromolécules.Les NTPs (Nucleoside TriPhosphate) incluent l’adénosine triphosphate (ATP), la source majeure d’énergie, et laguanosine triphosphate (GTP), cofacteur d’enzymes et de protéines. Le PEP (PhosphoEnolPyruvate) permet larégénération de l’ATP en fournissant l’énergie nécessaire à la phosphorylation d’une molécule d’ADP en ATP.
ce compartiment que vont diffuser les produits des réactions qui pourraient inhiber la réactionde synthèse de la protéine d’intérêt. La synthèse protéique est réalisée en continue tant que lessubstrats et les molécules d’énergie diffusent à travers la membrane dans le compartiment de laréaction. Un échange efficace entre les 2 compartiments, est assurée par une agitation vigoureusedurant tout le temps d’incubation. Le délai d’incubation ne dépasse généralement pas 24 h pouret le ratio des volumes Reaction Mix/Feeding Mix peut aller de 1/10 à ∼ 1/50. Dans notre casun rapport 1/17 a été utilisé et l’incubation a été réalisée pendant 22 h à Tamb.
Le dispositif de production est composé d’un bécher rempli avec 17 ml de "Feeding Mix"et d’un boudin de dialyse contenant le ml de "Reaction Mix", Fig. 2.6. La production d’uneprotéine membranaire telle que la hTSPO2 résulte en l’apparition d’un précipité dans le milieuréactionnel. Une fois ce dernier récupéré dans un tube Eppendorf, voici les étapes à suivre(Fig. 2.6) :
1. Le milieu réactionnel est centrifugé pendant 10 min à 13400 × g.
2. Le surnageant (SU) est collecté et une fraction est mélangée à de l’acide trichloroacétique(TCA) pour permettre la précipitation des protéines, p. ex : 7.5 µl de SU + 5 µl de TCA+ 22.5 µl d’eau milli Q. Une centrifugation est réalisée, 10 min à 13400 × g, le culot est
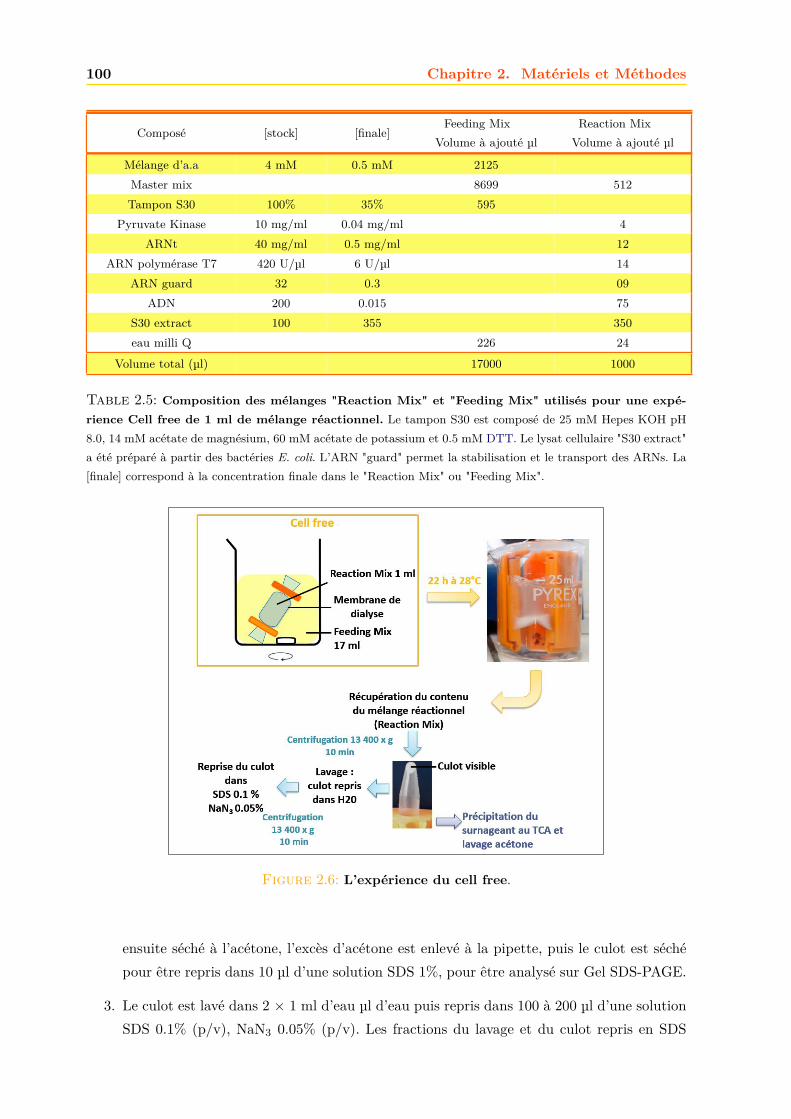
100 Chapitre 2. Matériels et Méthodes
Composé [stock] [finale]Feeding Mix
Volume à ajouté µlReaction Mix
Volume à ajouté µl
Mélange d’a.a 4 mM 0.5 mM 2125Master mix 8699 512Tampon S30 100% 35% 595
Pyruvate Kinase 10 mg/ml 0.04 mg/ml 4ARNt 40 mg/ml 0.5 mg/ml 12
ARN polymérase T7 420 U/µl 6 U/µl 14ARN guard 32 0.3 09
ADN 200 0.015 75S30 extract 100 355 350eau milli Q 226 24
Volume total (µl) 17000 1000
Table 2.5: Composition des mélanges "Reaction Mix" et "Feeding Mix" utilisés pour une expé-rience Cell free de 1 ml de mélange réactionnel. Le tampon S30 est composé de 25 mM Hepes KOH pH8.0, 14 mM acétate de magnésium, 60 mM acétate de potassium et 0.5 mM DTT. Le lysat cellulaire "S30 extract"a été préparé à partir des bactéries E. coli. L’ARN "guard" permet la stabilisation et le transport des ARNs. La[finale] correspond à la concentration finale dans le "Reaction Mix" ou "Feeding Mix".
Figure 2.6: L’expérience du cell free.
ensuite séché à l’acétone, l’excès d’acétone est enlevé à la pipette, puis le culot est séchépour être repris dans 10 µl d’une solution SDS 1%, pour être analysé sur Gel SDS-PAGE.
3. Le culot est lavé dans 2 × 1 ml d’eau µl d’eau puis repris dans 100 à 200 µl d’une solutionSDS 0.1% (p/v), NaN3 0.05% (p/v). Les fractions du lavage et du culot repris en SDS

2.2. Purification des TSPO 101
sont conservées pour être analysées sur gel SDS-PAGE.
4. Finalement, le culot resuspendu peut être purifié sur résine Ni-NTA.
2.2 Purification des TSPO
2.2.1 Les détergents
Les détergents qui ont été utilisés lors de cette étude, pour solubiliser la protéine membra-naire TSPO, et leurs caractéristiques sont présentés dans le Tab. 2.6. Les plus utilisés furentle SDS (Sodium Dodécyl Sulfate) et le DPC (Dodécyl PhosphoCholine). Chaque détergent secaractérise par une concentration micellaire critique (cmc), concentration au-dessus de laquelledes micelles se forment spontanément (c’est dans ces micelles que les PMb vont s’insérer).

102 Chapitre 2. Matériels et Méthodes
Détergents
long
ueur
dela
chaîne
Naturede
latête
PMg.mol
−1
cmcen
mM
(mg/
ml)à
25◦ C
dans
l’eau
form
ulechim
ique
Sodium
Dod
écyl
Sulfa
te-S
DS
12:0
anioniqu
e28
8.4
4-8(∼
1-2.3)
DoD
écyl
Phosph
oCho
line-D
PC12
:0Zw
itterioniqu
e35
1.4
1.1(∼
0.4)
n-Dod
ecyl-β-D
-Maltosid
e-
DDM
12:0
nonioniqu
e51
0.6
0.17
(∼0.08
)
1,2-DiH
eptano
yl-sn-glycero-3-
Phosph
oCho
line-
DHPC
6:0
Zwitt
erioniqu
e45
3.50
715
(∼6.8)
Tab
le2.6:
Structureet
caractéristiqu
esde
sdé
tergents
(Sigma-Aldrich
TM)utiliséspo
url’é
tude
desTSP
O.

2.2. Purification des TSPO 103
2.2.2 Extraction des protéines
2.2.2.1 Extraction de la mTSPO1
La mTSPO1 (ou la hTSPO1) est extraite d’E.coli où elle est en majeure partie localiséedans les corps d’inclusion bactériens (seule une faible fraction est membranaire) par lyse auxultrasons, avec l’appareil Vibra-CellTM 4. Cette lyse se fait sur glace (car l’échauffement induitpar les ultrasons peut être délétère pour la protéine) pendant 1 min à une puissance de 40%et une sonde de 3 mm. Une fois la lyse des bactéries effectuée, voici les étapes à réaliser pourrécupérer la protéine en détergent :
1. Une centrifugation dans des tubes de 30 ml à 13776 × g pendant 20 min et à Tamb permetde récupérer les corps d’inclusion (CI) dans le culot.
2. La mTSPO1 étant une protéine membranaire, l’ajout d’un détergent qui mime l’actiondes phospholipides autour des zones hydrophobes de la protéine est primordial pour lamaintenir soluble. Le SDS a été choisi pour extraire la mTSPO1 des CI et la maintenirsoluble. Elle est extraite des CI par ajout de 19 ml de tampon A et de 1 ml de SDS à20% (p/v, poids/volume). On obtient ainsi une solution à 1% en SDS soit 10 mg/ml (∼10 cmc). À partir de là, le SDS devenant solide à 4 ◦C toutes les étapes se font à Tamb.
3. Une dernière centrifugation de 30 min à 48400×g (Beckman, rotor JA 20) permet derécupérer les CI sous forme de culot alors que la protéine solubilisée en SDS est dans lesurnageant.
4. Finalement, de la benzonase (1 µl à 250U/µl) est ajoutée afin de cliver les ADNs et lesARNs présents. Du NaN3 à 1 mM finale est ajouté pour éviter une contamination par desbactéries.
L’extraction de la protéine des CI pourrait être obtenue par l’action d’autres agents commele chlorure de guanidinium (GdmCl) ou l’urée [Palmer and Wingfield, 2012]. Cependant, lasolubilisation des CI par le GdmCl ou l’urée est, en général, suivie d’une étape de renaturation,ce qui aurait conduit à rajouter une étape pour les substituer par un détergent afin de maintenirla protéine soluble.
Le cas de la protéine hTSPO2 étant plus complexe il sera présenté dans la partie "Résultatset Discussions".
2.2.2.2 Extraction du peptide Nter de l’AtTSPO fusionné à la GST
La protéine de fusion GST-Nter est extraite d’E.coli, où elle est localisée dans le cytosol, parlyse aux ultrasons pendant 1 min sur glace. Celle-ci est réalisée comme décrit ci-dessus pour lamTSPO1.
4. La lyse peut aussi se faire par des méthodes mécaniques en utilisant par exemple des billes de verre ou unepresse Aminco-French.

104 Chapitre 2. Matériels et Méthodes
Une centrifugation à 13776 × g pendant 20 min à Tamb permet de séparer les CI, qui seretrouvent dans le culot, de la protéine de fusion qui est dans le surnageant. 1µl de benzonase(Semba Biosciences ; 250U/µl est ajoutée au surnageant afin de cliver les ADN et les ARNprésents. Une pastille d’anti-protéase est ajoutée ainsi que du NaN3 à 1 mM finale pour éviterune contamination bactérienne.
2.2.3 Purification sur Résine Ni-NTA
Les protéines recombinantes ont été purifiées par chromatographie d’affinité sur une colonnede résine Ni-NTA (Nickel-NiTriloacetic Acid) agarose (QiagenR© ; capacité de liaison jusqu’à 50mg de protéine/ml ) en utilisant la présence de l’étiquette His6 dans les cas des TSPO1 et dela TSPO2, et de l’étiquette His10 dans le cas du de la protéine de fusion GST-Nter, Fig. 2.7.Une 1re étape, la charge, permet de fixer la protéine sur la résine. Puis, une 2e étape, le lavage,permet d’éliminer les contaminants (acides nucléiques, protéines). Enfin, une 3e étape, l’élution,permet de décrocher la protéine de la résine, par ajout d’imidazole qui entre en compétitionavec l’histidine car c’est un analogue structural.
Figure 2.7: Principe de la purification sur Ré-sine Ni-NTA . L’ion Nickel Ni2+ possède 6 élec-trons de valence. L’acide nitrilotriacétique (NTA)forme 4 liaisons covalentes de coordination avec 4des électrons du Ni2+. Le Ni2+ une fois complexé,gardent 2 sites accessibles qui peuvent être occu-pés par des les histidines de l’étiquette His6, p. ex.,d’une protéine ou d’un peptide. En absence d’histi-dines ces 2 sites peuvent être occupés par des molé-cules d’eau .
2.2.3.1 Minipurification
Pour estimer rapidement la quantité de protéine présente dans la solution à purifier et ajusterle volume de résine à utiliser, une purification d’une petite quantité est réalisée avant de procéderà la purification totale. Cette "minipurification" a été utilisée dans le cas de la purification dela TSPO1. Elle est réalisé en suivant ces étapes :
1. Dans un tube Eppendorf, 1 ml de la solution issue des CI solubilisés est incubé avec 100 µlde résine Ni-NTA, préalablement lavée et équilibrée avec le tampon de charge A, pendant15 min à Tamb sous agitation.
2. La résine est récupérée par centrifugation et est lavée avec 3 × 1 ml de tampon de lavage,50 mM Hepes, 5 mM Imidazole, 150 mM NaCl contenant 1% de SDS (p/v).
3. La protéine fixée sur la résine est éluée dans 3 × 1 ml de tampon d’élution, 50 mM Hepes,250 mM Imidazole, 150 mM NaCl contenant 1% de SDS (p/v).

2.2. Purification des TSPO 105
La concentration de la protéine présente dans les fractions d’élution a été estimée par la mesurede la DO à 280 nm en utilisant l’ε de la TSPO comme décrit dans le paragraphe ci-dessous.
2.2.3.2 Méthode générale de mesure de la concentration en protéine
Toutes les quantités de protéines ont été calculées à partir de la DO à 280 nm. Celle-ci estmesurée sur les spectres d’absorption enregistrés entre 240 et 320/350 nm sur un spectrophoto-mètre UV-Visible (UNICAM UV300).
Le calcul se fait en utilisant la loi de Beer Lambert : I = I0eεlC ; avec ε le coefficient d’ex-tinction de la protéine en ml.mg−1.cm−1 ; l le trajet optique en cm et C la concentration enprotéine. En pratique, cela revient à faire C = DO280
εlC .Les coefficients d’extinction utilisés sont :
• 3.88 ml.mg−1.cm−1 pour la mTSPO1(hTSPO1) ;• 2.88 ml.mg−1.cm−1 pour la hTSPO2 ;• 1.57 ml.mg−1.cm−1 pour la protéine GST, 0.27 ml.mg−1.cm−1 pour le peptide Nter
d’AtTSPO soit 1.84 ml.mg−1.cm−1 pour la protéine de fusion Nter-GST.
2.2.3.3 Purification totale de la mTSPO1
1. Jusqu’à ∼2 ml de résine Ni-NTA (selon la quantité de protéines estimé avec la minipurifi-caiton ou avec l’expérience) sont déposés sur une colonne Biorad (environ 2 ml de volumede lit et 10 ml de réservoir sur 1 cm de large).
2. La résine est ensuite lavée et équilibrée par 5 ml de tampon A : Hepes 50 mM, NaCl 500mM, pH 7.8.
3. La solution contenant les protéines extraites des CI solubilisés en SDS est chargée surla résine. Ce qui passe à travers la colonne - le non fixé (NF) - est récupéré dans uncontenant.
4. Pour éliminer les protéines fixées de manière non spécifique, la résine est lavée par 4 ×le volume de résine avec le tampon A puis avec 8 volumes de tampon A contenant 5mM d’imidazole. Au cours de cette étape, on peut effectuer l’échange de détergents sinécessaire.
5. L’élution de la protéine recombinante est réalisée avec un tampon d’élution qui contient250 mM d’imidazole et du détergent.
Toutes les étapes (charge, lavages et élution) sont analysées en enregistrant les spectres d’ab-sorption (240-320 nm) et en mesurant les valeurs à 280 nm de toutes les fractions collectées.
L’échange de détergent L’échange de détergent est réalisé au cours de l’étape de lavagelors de la purification sur la colonne Ni-NTA. Une fois la TSPO1 en SDS (SDS qui nous aservi à solubiliser les CI) fixée sur la résine NTA-Ni), la substitution du détergent est réaliséepar lavage avec 2 volumes de colonne contenant une concentration du nouveau détergent 2

106 Chapitre 2. Matériels et Méthodes
fois plus concentrée que celle du SDS. Cette étape est suivie par une autre étape de lavage (4volumes de colonne) qui permet l’ajustement de la concentration du détergent de substitutionà la concentration finale désirée. Finalement, la TSPO1 est éluée avec le tampon d’élutioncontenant de l’imidazole et le détergent de substitution. Lors de cette étude c’est surtout laDPC qui a servi de détergent de substitution.
2.2.4 Purification du peptide Nter d’AtTSPO
Purification de la protéine de fusion sur résine Ni-NTA Cette purification est réaliséde la même manière que pour la mTSPO1 sur résine Ni-NTA.
1. La solution contenant les protéines de fusion est chargée sur la résine (1.2-1.4 ml).
2. Puis, la résine est lavée par 2 ml de tampon A’ (Hepes 50 mM pH 7.8, NaCl 500 mM).
3. La résine est de nouveau lavée mais cette fois avec des concentrations croissantes d’imida-zole de 10 à 50 mM par volume de 2 ml. Ce gradient permet d’éliminer le plus efficacementpossible les contaminants sans décrocher la protéine de fusion.
4. L’élution de la protéine est réalisée par 5 × 1 ml de tampon d’élution qui contient 250mM d’imidazole.
Toutes les étapes (charge, lavages et élution) sont analysées en enregistrant les spectres d’ab-sorption (240-320 nm) et en mesurant les valeurs à 280 nm de toutes les fractions collectées.
Clivage de la protéine de fusion par digestion par la protéase TEV Les fractionsd’élution après avoir été regroupées sont dialysées sur la nuit à Tamb, pour éliminer l’imida-zole, contre un tampon de dialyse NaPO4 10 mM pH 7.8 NaN3 1 mM contenant une pastilled’antiprotéase. Le pH a été choisi pour permettre une activité optimale de la protéase TEV.Le tampon phosphate, quant à lui, est choisi pour, par la suite, faciliter les études en spectro-métrie de masse et en RMN. En effet, une trop forte concentration en sel rend : en RMN, leréglage de l’accord de la sonde (matching et tuning) difficile et augmente la durée de l’impulsion90˚ du proton, tous ces effets combinés peuvent entraîner une baisse du rapport S/N ; et enspectrométrie de masse, une ionisation inefficace de l’échantillon.
L’enzyme TEV est une protéase du virus de la mosaïque du tabac (TEV - Tobacco etchvirus) qui reconnaît la séquence d’a.a Glu-Asn-Leu-Tyr-Phe-Gln-Glu. L’enzyme coupe entre laglutamine (Gln) et la glycine (Gly), Fig. 2.8. Dans une protéine de fusion, cette séquence d’a.aest placée entre celle de la protéine d’intérêt et la GST afin d’éliminer cette dernière. Vu la raretéde cette séquence, les chances que la protéine soit clivée à un autre endroit dans sa séquencesont relativement faibles.
Des aliquotes de 20 µl, à la concentration de 0.2µg/µl, de protéase TEV possédant uneétiquette histidine, nous ont généreusement été fournies par Mr Henri Batoko. La TEV estajoutée avec la protéine de fusion dans un contenant en verre avec un agitateur magnétiqueou dans un tube Eppendorf sous agitation en fonction du volume à digérer. La digestion estréalisée à Tamb.

2.2. Purification des TSPO 107
Dans la littérature, pour un ratio de 1 µg de TEV pour 50 à 100 µg de protéines, la digestionse fait généralement pendant : 4 à 6h à 30 ◦C ou sur la nuit à 4 ◦C ou à Tamb. Ne disposant pasde quantité de TEV suffisante nous avons travaillé à plus faible ratio (de l’ordre du millième) etsur des temps de digestion beaucoup plus longs (> 16h). Ensuite, des fractions avant et aprèsdigestion sont déposées sur gel SDS-PAGE acrylamide 12% pour vérifier que le clivage de laprotéine de fusion GST-Nter est total.
Figure 2.8: Schèma représentant la coupure de la protéine de fusion GST-Nter par laprotéase TEV. La TEV reconnaît une séquence spécifique et coupe entre la glutamine (Gln) et laglycine (Gly) de cette séquence.
Purification du Nter d’AtTSPO sur résine Ni-NTA Après élimination de l’étiquetteGST du peptide Nter. Celui-ci est purifié une dernière fois sur résine Ni-NTA pour éliminer laGST libre et les protéines contaminantes.
L’étape de charge permet de fixer la GST et la TEV sur la résine car toutes les 2 possèdentune étiquette polyhistidine. Puis, les étape de lavages permettent d’éliminer les contaminants(acides nucléiques, protéines) et de récupérer le peptide.
1. ∼ 1 ml final de résine Ni-NTA sont déposés sur une colonne Biorad.
2. La résine est équilibrée par 5 ml de tampon NaPO4 10 mM pH 7.8.
3. L’étape de charge permet de fixer la GST libre et la TEV sur la résine. Ce qui passe àtravers la colonne - le non fixé (NF) - est récupéré dans un tube. Il contient les protéinescontaminantes et une très faible fraction de peptide Nter. En effet, le volume mort de lacolonne fait que le peptide sort lors des étapes de lavages.
4. La résine est donc lavée avec du tampon NaPO4 10 mM pH 6.5 (ce pH est plus adéquatepour la RMN) par 3 × 2 ml.
5. L’élution par 200 mM d’imidazole n’est pas nécessairement utile puisqu’elle permet justede détacher la GST de la résine.

108 Chapitre 2. Matériels et Méthodes
2.3 Caractérisation des TSPO
2.3.1 Gel SDS-PAGE
Une fois la protéine ou le peptide purifié, il est possible d’évaluer son degré de pureté enfaisant un gel d’électrophorèse SDS-PAGE.
L’électrophorèse SDS-PAGE (Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis- électrophorèse en gel de polyacrylamide contenant du dodécysulfate de sodium) est une tech-nique consistant à faire migrer des protéines dans un gel, sous l’influence d’un champ électrique,permettant ainsi leur séparation (via les mailles formées par l’acrylamide) selon leur chargeélectrique et pour des charges identiques, selon leur poids moléculaire PM.
Dans le cas du SDS-PAGE, la séparation est réalisée en conditions dissociantes en raison del’action de 2 composés :
• Le SDS : il possède une longue queue hydrocarbonée hydrophobe et une extrémité chargéenégativement. Il se lie aux régions hydrophobes des protéines et leur confère une chargenette négative. Cela signifie que seul le PM apparent (et non réel) des protéines sera lefacteur de leur séparation.• Le β-mercaptoéthanol : il réduit les ponts disulfures des protéines. Cela permet de séparerles différents polypeptides reliés par de tels ponts au sein d’une même protéine.
Lors de cette étude, nous avons utilisés à la fois des gel polyacrylamide pré-coulés 12% ou4-20% (Mini-PROTEANR© TGXTM Precast Protein Gels, 10-well, 30 µl) et des gels 12% coulésmanuellement. La préparation de ces derniers est décrit ci-dessous.
Préparation du gel La grosseur des mailles du gel est fonction de la concentration d’acry-lamide et du taux de bis-acrylamide. Plus la concentration est élevée, plus les mailles serontpetites et les molécules les mieux séparées seront celles de faible PM. Le choix du % d’acry-lamide du gel dépend de la taille de la protéine d’intérêt. Un gel a 12% d’acrylamide permetune gamme de séparation en taille de 10 à 200 kDa. Le gel polyacrylamide est composé de2 parties : une partie inférieure (gel de séparation) destinée à séparer les protéines selon leurmasse moléculaire, Tab. 2.7, et une partie supérieure (gel de stacking) destinée à concentrer lesprotéines, Tab. 2.8.
Une fois ces solutions préparées voici les étapes à suivre (appareil BioRad-mini protean IIavec des gels de 7,2 cm × 8,6 cm × 1 mm d’épaisseur) :
1. Le gel de séparation est coulé entre des plaques de verre fixées sur un support. Du butanolest ajouté à sa surface pour ne pas que le gel séche.
2. Une fois le gel de séparation polymérisé (∼ 45min), le butanol est enlevé, et la surface dugel est lavé à l’eau.
3. Le gel de stacking est coulé et un peigne est enchâssé entre les plaques.
4. Après polymérisation du gel, le peigne est retiré formant ainsi des puits. La taille et lenombre des dents des peignes sont variables ce qui permet de déposer des volumes allant

2.3. Caractérisation des TSPO 109
Éléments Quantité pour 10 ml
eau distillée 3.35 ml1.5 M Tris-HCL, pH 8.8 2.5 ml20 % (p/v) SDS 50 µlSolution 30% Acrylamide/Bis Solution 37.5 :1 4 ml10% ammonium persulfate fraichement préparé 50 µlTEMED 5 µl
Table 2.7: Composition du gel de séparation. La matrice est créée par la copolymérisationd’acrylamide et de bis-acrylamide. Le TEMED et le persulfate d’ammonium sont les catalyseurs de laréaction de polymérisation, il est donc nécessaire de les mettre en dernier.
Éléments Quantité pour 5 ml
Solution 30% Acrylamide/Bis Solution 37.5 :1 3.7 ml1M Tris pH 6.8 1.25 ml20 % (p/v) SDS 25 µl10% ammonium persulfate fraichement préparé 25 µlTEMED 5 µl
Table 2.8: Composition du gel de stacking.
de 20 µl à 200 µl d’échantillon de protéines à séparer.
5. Les gels sont placés dans une cuve d’électrophorèse contenant un tampon de migration,Tab 2.9.
6. Les échantillons protéiques sont mélangés dans un rapport 4/1 (v/v) avec du tampon dedissociation (5X), Tab. 2.10. P. ex, 60 µl d’échantillon et 15 µl de tampon de dissociation ;
7. Les protéines (de 100 à 0,1 ng de protéines/puit) sont déposées dans les puits du gel, àl’aide d’une seringue Hamilton, de même qu’un marqueur de PM (∼ 4-5 µl) qui sont desprotéines standards de masses molaires connues. Le gel est soumis à un courant électriqueà voltage constante (150 V, courant variable maximum) pendant ∼1h permettant leurmigration dans le gel.
8. Après migration, le gel est démoulé. Les protéines sont fixées dans le gel par une solutionqui contient du méthanol et de l’acide acétique.
9. Les protéines sont révélées par une coloration, p. ex. avec le bleu de Coomassie (R250) oule nitrate d’argent (plus sensible).
Coloration au Bleu de Coomassie La coloration au bleu de Coomassie est la méthodela plus communément utilisée pour révéler les bandes protéiques du gel. Pour qu’une bandesoit clairement visible, avec cette méthode, il faut qu’elle contienne 20-50 ng de protéines. Les

110 Chapitre 2. Matériels et Méthodes
Éléments Quantité pour 1L
1 M Tris-HCl pH 8.4 25 mM (3 g)glycine 200 mM (15 g)20 % (p/v) SDS 5 ml (1 g)
Table 2.9: Composition du tampon de migration (1X).
Éléments Quantité pour 5 ml
0.5 M Tris pH 6.8 1 mlglycerol 80% 1.25 ml20 % (p/v) SDS 250 µlβ-mercaptoéthanol 50 µlBleu de Coomassie R250 2.5 mgeau milli Q 2.45 ml
Table 2.10: Composition du tampon de dissociation (5X).
gels sont immergés dans une solution de coloration contenant du bleu de Coomassie, Tab. 2.11,pendant au moins 1h. L’excès de colorant est éliminé et le gel est décoloré dans la solution dedécoloration, Tab. 2.11, jusqu’à révélation de la migration des protéines.
Coloration au nitrate d’argent La coloration des gels au nitrate d’argent est une méthodeplus sensible que la coloration au bleu de Coomassie. Elle permet de détecter des protéines àpartir de 0,1 ng par bande. Cependant, elle est beaucoup plus longue et contraignante à mettreen oeuvre. Lors du protocole toutes les étapes se font sous agitation et avec de l’eau milli Q.
1. Après migration, les protéines présentes dans le gel de polyacrylamide sont fixées pendant30 min au minimum à l’aide d’une solution de fixation contenant : 50% (v/v) d’éthanol,5% (v/v) d’acide acétique et 45% (v/v) d’eau .
2. Les gels sont placés pendant 15 min dans la solution de fixation diluée au 10e.
3. Les gels sont lavés dans 3 × 5 min dans un bain d’eau.
4. Pour être sensibilisés, ils sont placés pendant 1 min dans un bain de thiosulfate de sodium0,02% (p/v). Cette étape de sensibilisation est arrêtée par 3 lavages de 30 s dans l’eau
Solution de coloration Solution de décoloration
2.5% (p/v) Coomassie (Brilliant Blue R250 BIO-RAD)40% (v/v) éthanol 40% (v/v) éthanol10% (v/v) acide acétique 10% (v/v) acide acétique60% (v/v) eau 60% (v/v) eau
Table 2.11: Composition des solutions utilisées lors de la coloration d’un gel SDS-PAGEau bleu de Coomassie.

2.3. Caractérisation des TSPO 111
(pour réduire le bruit de fond).
5. La réaction a l’argent est faite en immergeant les gels, pendant 20 min, dans une solutioncontenant pour 50 ml : 2 mg (soit 0.2% p/v) de nitrate d’argent et 37.5 µl de formaldéhyde37%. L’arrêt de la réaction est faite en lavant les gels dans l’eau 2 × 30 secondes.
6. Les gels sont placés dans solution de développement, composée pour 50 ml de solutionde : 3 g de carbonate de sodium, 37 µl de formaldéhyde 37% et 1 ml de thiosulfate desodium 2%. La durée de la révélation peut varier de quelques secondes à une vingtaine demin selon la vitesse d’apparition des bandes.
7. Finalement, la révélation est arrêtée par une solution d’acide acétique 5% (v/v) pendant∼5 min (jusqu’à ce que la coloration convienne).
2.3.2 Western Blot
Figure 2.9: Les différentes étapes du Western Blot. Sources : https://www.licor.com/
et https://www.moleculardevices.com.
Un western blot, ou immunoblot, est une méthode de biologie moléculaire qui permet ladétection de protéines spécifiques sur une membrane, grâce à l’utilisation d’anticorps (AC) 5
Cette technique a beaucoup été utilisée pour la recherche de la hTSPO2. Les WBs ont étéréalisés à l’institut national de la transfusion sanguine (INTS) en collaboration avec ClaudeHattab et du Pr. Mariano Ostuni.
5. Un anticorps est une protéine complexe utilisée par le système immunitaire pour détecter les protéines. Ilspeuvent y parvenir grâce à zone spécifique qui ne reconnaît qu’une partie très précise de la protéine (ou antigène),l’épitope

112 Chapitre 2. Matériels et Méthodes
Tampon de transfert SDS-PAGE (1X) Quantité pour 1L
1 M Tris-HCl pH 8.4 25 mM (3 g)glycine 200 mM (15 g)éthanol 200 ml
Table 2.12: Composition des tampons utilisés lors d’un Western Blot.
Le même protocole initial de l’électrophorèse SDS-PAGE a été utilisé pour le WB saufqu’après l’électrophorèse, les protéines ont été électrotransférées sur une membrane de nitrocel-lulose à l’aide du système de transfert "X-Cell Blot Module" (Biorad). Le principe consiste àrecouvrir le gel de la membrane et d’appliquer un courant électrique entraînant le transfert desprotéines du gel sur la membrane. La fixation des protéines à la membrane se fait grâce à desinteractions hydrophobes et ioniques entre la membrane et les protéines. Pour se faire :
1. L’appareil de transfert et des éponges sont lavés avec de l’eau juste avant usage, puis sontimmergés dans du tampon de transfert, Tab. 2.12.
2. Les membranes de transfert et du papier buvard Whatman sont découpés de manière àêtre un peu plus large que le gel SDS-PAGE. Puis ils sont immergés dans dans du tamponde transfert.
3. Le gel SDS-PAGE est démonté, puis le tout est installé dans le module de transfert se-lon l’ordre suivant : 2 éponges/2 papiers buvards/gel/papier buvard/membrane/3 papiersbuvards/2 éponges.
4. Le module est introduit dans le réservoir de l’appareil de transfert et les 2 sont remplis detampon de transfert.
5. Le transfert a été effectué sous voltage constant 3 V, 250 W, pendant ∼ 1h30 ;
À la fin du transfert, la détection de la protéine d’intérêt est réalisée avec l’utilisation d’unAC spécifique, dit primaire, qui se lient à « sa » bande de protéines, avant d’être reconnu parun AC secondaire. Ce dernier est conjugué à une molécule pour la coloration.
Les AC sans liaison spécifique sont éliminés par lavage à l’aide d’un tampon contenant undétergent.
1. La membrane est récupérée avec une pince, déposée sur du papier serin puis égouttée.
2. La membrane est immergée, pendant 1h et sous agitation à 37 ◦C, dans la solution desaturation TBS 6-Tween 0.5% lait 5% : 20 mM Tris-HCl, pH 7.5, 0.15 M NaCl, 0.5% Tween-20 (un détergent ; Sigma-Aldrich) et lait 5% (p/v) (écrémé RégilaitR©. Cette étape appelée"blocage de la membrane" est indispensable pour limiter les interactions non spécifiquesultérieures entre les AC et la membrane. Les protéines contenues dans le lait se lient àla membrane dans tous les sites non-occupés par la protéine-cible. Ainsi, lorsque les ACsont appliqués lors de l’étape suivante, ils ne peuvent (idéalement) plus s’attacher à la
6. TBS signifie Tris Buffer Saline.

2.3. Caractérisation des TSPO 113
membrane que sur les sites de liaison de la protéine-cible. Cela réduit le "bruit de fond"dans le produit final du transfert de western et donne des résultats plus clairs, éliminantles faux positifs.
3. La membrane est incubée, pendant 1h et sous agitation à 37 ◦C (ou à 4 ◦C sur la nuit), dans10 ml de solution de blocage à laquelle est ajouté l’AC primaire à la dilution appropriée :
• Pour la détection de la hTSPO2 : 25 µl pour une dilution au 1/400. Le terme AC n’estpas vraiment correct dans ce cas, puisque qu’il s’agit d’un sérum 7 d’immunisationde lapin qui a été utilisé. Celui-ci a été fait en injectant un peptide de 13 a.a, cor-respondant au C-terminal de la hTSPO2. Il contient donc à la fois l’AC anti-TSPO2mais aussi tous les autres AC développés par le lapin. Avant injection du peptide, unsérum pré-immunisation du lapin a été prélevé et il est utilisé comme contrôle dansles WB ;• Pour la détection de l’étiquette His6 : 2 µl pour une dilution au 1/5000. L’AC utiliséest un AC monoclonal 8 commercial (6*His, His-Tag Antibody ProteinTechTM).
4. La membrane est ensuite lavée, 3× 10 min, sous agitation à Tamb, avec du tampon TBS-0.5% Tween-20 : 25 mM Tris, ph 7.4, 140 mM NaCl, 3 mM KCl, 0.5% Tween
5. La membrane est de nouveau saturée pendant 15 min à 37 ◦C, sous agitation, avec letampon de saturation ;
6. La membrane est incubée pendant 1 h à Tamb et sous agitation dans 10 ml de tampon desaturation auquel a été ajouté l’AC secondaire conjugué à l’HRP (Horseradish Peroxidase- peroxydase de raifort ; il catalyse l’oxydation du luminol en présence de peroxyde d’hy-drogène et cette réaction produit une lumière) : 2.5 µl pour une dilution au 1/ 4000. L’ACutilisé est un AC commercial anti-lapin couplé à l’HRP (Goat Anti-Rabbit (GAR)-HRP,Paris Biotech) ;
7. La membrane est lavée de nouveau 2 × 10 min fois avec du TBS 0,5% Tween et 1 × 10avec du TBS sans Tween ;
8. Enfin, la membrane est séchée sur du papier absorbant et révélée par autoradiographiesur film à l’aide du kit Enhanced Chemiluminiscence (ECL, Perkin Elmer), de films MSbiofilms (KODAK) et du développeur Curix 60 developer (Agfa Healthcare) :
• La solution de l’ECL est préparée en mélangeant 5 ml de chacun des réactifs A etB contenant : un substrat luminescent de la péroxydase (luminol), un catalyseur(péroxyde d’hydrogène) ainsi qu’un amplificateur chimique (phénol) de la réactiond’oxydation.• La membrane est incubée dans 4 ml de cette solution pendant 2 min sous agitation.La péroxydase en marquant l’AC secondaire oxyde donc le luminol en présence du
7. Le sérum sanguin est le partie liquide du sang contenant les AC.8. Les anticorps monoclonaux sont des anticorps reconnaissant le même épitope car ils sont issus d’une seule
lignée de plasmocytes, provenant d’une seule cellule

114 Chapitre 2. Matériels et Méthodes
péroxyde d’hydrogène. Cette réaction aboutit à l’émission d’une lumière (chimiolu-minescence).
• La membrane est séchée sur un papier absorbant puis placée entre 2 feuilles deplastique transparent (film saran). Elle est alors placée dans une cassette pour filmradiographique.• La membrane est exposée en chambre noire (utilisant des lumières rouges) : le filmest placé sur le dessus de la membrane, la cassette est refermée. Sous l’exposition dela lumière due à la réaction, une image des AC liés à la membrane est créée sur lefilm (bandes sombres). L’exposition peut durée de 1 min à 1 h selon l’intensité dusignal attendue.• Le film est retiré de la cassette puis développé immédiatement après à l’aide du dé-veloppeur. Selon l’intensité du signal obtenue, d’autres films peuvent être réalisésen variant la durée d’exposition pour optimiser les résultats. En effet,l’émission lu-mineuse continue 24 heures en décroissant au fil du temps. Il est donc possible sinécessaire de refaire une ou plusieurs autres expositions avec de nouveaux films.
Le Dot Blot Le Dot blot est une technique similaire au Western blot sauf que les échantillonsne sont pas séparés par gel d’électrophorèse mais directement déposés sur la membrane en nitro-cellulose. C’est une technique beaucoup plus rapide et moins complexe que le WB. Cependant,elle n’offre aucune information sur la taille de la protéine-cible. Elle permet de confirmer laprésence ou l’absence d’une protéine grâce à l’utilisation d’un AC avec une spécificité élevéepour détecter la protéine cible. Cette technique a été utilisé pour confirmer la présence du Nterde l’AtTSPO purifié.
Lors de ce protocole, la membrane utilisée est une membrane en PVDB (PolyVinyliDeneFluoride) qui offre une meilleure rétention des protéines adsorbées qu’une membrane en nitro-cellulose en raison de sa plus grande hydrophobicité.
1. La membrane est lavée à l’éthanol 100% (elle devient semi-transparente), 3 fois à l’eau etséchée sur un papier absorbant. La membrane PVBD étant très hydrophobe, les solutionsaqueuse utilisés lors du protocole ne pourraient pas pénétrer dans la membrane sansce lavage à l’éthanol. Ce dernier permet donc d’activer la membrane en l"hydratant" etaméliore ainsi la liaison des protéines.
2. Les échantillons (de 1 à 5 µg de protéines totales par point) sont déposés sur la membrane.Puis elle est laissée sécher à l’air.
3. La membrane est bloquée, pendant 30 min à 37 ◦C ou 1h à Tamb sous agitation, par untampon de saturation TBS-SDS 0.05% lait 5% : 50 mM Tris pH 7.6, 150 mM NaCl, SDS0.05% (p/v) et lait écrémé (Régilait) 5% (p/v) ;
4. la membrane est ensuite incubée, pendant 1 h sous agitation à 37 ◦C (ou à 4 ◦C sur lanuit) dans 15 ml de solution TBS-SDS 0.05% lait 2.5% à laquelle est ajouté l’AC primaireanti-Nter, à une dilution appropriée : 5 µl pour une dilution au 1/3000.

2.3. Caractérisation des TSPO 115
5. La membrane est lavée 3 × 10 min par un tampon TBS-SDS 0.05%.
6. La membrane est de nouveau bloquée, pendant 10 min sous agitation à Tamb par untampon de saturation TBS-SDS 0.05% lait écrémé 10% (p/v).
7. La membrane est incubée, pendant 45 min sous agitation à Tamb, dans 15 ml de solutionTBS-SDS 0.05% lait 2.5% à laquelle est ajouté l’AC secondaire, à une dilution appropriée :3 µl pour une dilution au 1/5000. L’AC secondaire utilisé est un AC commercial anti-lapin couplé à la phosphatase alcaline (Goat anti-Rabbit IgG 9- whole molecule - AlkalinePhosphatase, SIGMA).
8. La membrane est lavée 3 × min dans le tampon TBS-SDS %.
9. Finalement, la révélation se fait par ajout de NBT (Nitro-Blue Tetrazonium) et du BCIP(BromoChlorylIndoloPhosphate) : 1 pastille (BCIP/NBT Sigma Fast) pour 10 ml. LeBCIP est un substrat de la phosphatase alcaline qui libère une fonction réductrice indoleinstable qui fait se lier 2 molécules du réactif et réduit le NBT en provoquant un précipitéviolet. La présence de la protéine-cible se traduit donc par l’apparition du colorationviolette sur la membrane.
2.3.3 Spectrométrie de masse
Figure 2.10: Principe d’un spectrométre de masse.
La spectrométrie de masse est une technique d’analyse qui permet la détermination desmasses moléculaires des composés analysés ainsi que leur identification. Elle est fondée sur laséparation d’ions en phase gazeuse en fonction de leur rapport masse/charge (m/z), Fig. 2.10.
Toutes les expériences de spectrométrie de masse ont été effectuées sur un instrument de typeMALDI-TOF. C’est un spectromètre de masse couplant une source d’ionisation laser assistéepar une matrice (MALDI, Matrix-Assisted Laser Desorption/Ionisation) et un analyseur detemps de vol (TOF, Time-Of-Flight mass spectrometry).
2.3.3.1 Préparation des échantillons
Les échantillons de la protéine ou du peptide purifiés (environ 1 µl) ont été mélangés àune matrice (1 µl d’alpha-cyano-4-hydroxy-cinnamic acid - HCCA). Ce mélange a été déposé
9. Les IgGs sont des glycoprotéines consituant une classe d’anticorps. L’IgG est le principal type d’AC trouvédans le sang. L’AC anti-lapin IgG reconnait les chaines longues et courtes de toutes les sous-classes d’AC primaires

116 Chapitre 2. Matériels et Méthodes
sur une cible et après séchage un laser ionise les molécules et les désorbent de cette matrice.Dans le cas des protéines TSPO purifiées en SDS, du fait de l’interférence de celui-ci (détergentionique), le SDS est complémenté avec du DDM (détergent non anionique) par dilution. Desstandards de PM connus sont déposés sur la cible pour déterminer correctement le PM desprotéines d’intérêts.
Dans le cas de la mTSPO1, nous avons testé plusieurs échantillons (stock rapporté à ∼ 1mg/ml) dilués à différents rapport (1/10, 1/100, 1/1000) dans une solution de DDM à 0.1%finale (soit 10 cmc ; 1 mg/ml). Il s’avère que, la mTSPO1 apparait surtout sous forme dichargéeet le meilleur rapport signal/bruit est obtenu pour une protéine diluée au 1/100.
Les échantillons de protéines isolées sur gel SDS-PAGE sont préparés comme décrit ci-dessous :
1. Tout d’abord, les bandes du gel SDS-page d’intérêts sont découpées au scalpel. Chaquebande excisée est déposée dans un tube Eppendorf.
2. Les bandes sont découpées en morceaux, dans les tubes Eppendorf, pour optimiser lapénétration de la trypsine dans les mailles du gel. Ces morceaux ne doivent pas être troppetits pour ne pas les aspirer dans le cône de la pipette lors des diverses étapes de ladigestion.
3. Un 1er lavage est effectué pour éliminer le bleu de Coomassie et enlever le SDS qui in-hibe l’activité de la trypsine. Ce lavage est réalisé sous agitation, en ajoutant 200 µl d’unmélange H2O/acétonitrile (ACN) (9/1) pendant 2×15 min. Le mélange H2O/ACN estensuite enlevé à la pipette.
4. 200µl d’ACN pure sont ajoutés pour sécher le gel pendant 2×15 min. Les fragments de gelsse déshydratent, ceci permettra par la suite au DTT (DiThioThréitol) de mieux pénétrerdans les mailles du gel. L’ACN est retiré à la pipette.
5. On ajoute 30 µl d’un mélange DTT 10 mM et bicarbonate d’ammonium NH4HCO3 100mM pH 8. On chauffe au bain marie à 56 ◦C pendant 45 min. Cette opération permet laréduction des ponts disulfures des protéines. Le mélange DTT/NH4HCO3 est enlevé à lapipette.
6. On ajoute 30µl d’IAA (IodoAcétAmide) à 10 mg/ml (10 mg de poudre dans 1 ml deNH4HCO3 100 mM pH 8). On incube pendant 30 min dans l’obscurité. Ceci permetl’alkylation des cystéines pour éviter qu’elles ne reforment des ponts disulfures. L’IAA estéliminé à la pipette.
7. Afin d’éliminer les traces de DTT et d’IAA, les bandes sont lavées 2×15 min avec 100 µlH2O/ACN (9/1), sous agitation. Puis 1×10 min avec de l’ACN. Les fragments de gel sedéshydratent et forment une boule compacte, ce qui va favoriser ensuite l’entrée de latrypsine par réhydratation des morceaux de gel. l’ACN est retiré à la pipette.
8. 30 µl d’une solution de trypsine (0.04 ng/µl, Gold Mass Spec PROMEGA) dans du tamponNH4HCO3 50 mM pH 8 contenant du DDM 0.1% final sont ajoutés à chaque échantillon.

2.3. Caractérisation des TSPO 117
L’ajout de DDM est nécessaire pour obtenir une digestion de la TSPO. Les bandes sontincubées dans cette solution pendant 45 min. Il est nécessaire à cette étape de se placerdans la glace pourlimiter la réaction d’autolyse de la trypsine.
9. Le tampon trypsique est élimé à la pipette et 30 µl de tampon NH4HCO3 50 mM pH 8DDM 0.1% final sont ajoutés et incubés toute la nuit au froid ;
10. Finalement, 20 à 30 µl sont prélevés et déposés dans l’injecteur du spectromètre orbitrap.
2.3.4 Dichroïsme circulaire
Pour juger de l’état de repliement des TSPO ou du peptide Nter d’AtTSPO, la spectroscopiepar dichroïsme circulaire (DC) est particulièrement appropriée.
La technique repose sur la capacité qu’ont les structures optiquement actives d’absorber defaçon inégale la lumière polarisée circulairement à droite de la lumière polarisée circulairement àgauche. L’absorption préférentielle de l’une de ces deux polarisations résultera en une déviationde la résultante qui formera une ellipse au lieu d’une lumière polarisé plane, Fig. 2.11a. Pour lesprotéines, l’absorption de la lumière polarisée entre 160 nm et 260 nm est principalement dueà la présence de liaisons peptiques, pour lesquelles on observe les transitions n → Π∗ vers 220nm et Π → Π∗ vers 190 nm. La mesure de la déviation θ à plusieurs λ permet des signaturescaractéristiques pour chaque type d’élément de structure secondaire (hélices, feuillets, etc.). Unexemple est donné en Fig.2.11b pour les 4 structures secondaires trouvées le plus souvent dansles protéines.
La mesure est généralement reportée en ellipticité molaire par résidu [θ] endeg.cm2.dmol−1.res−1.
[θ]λ = 100× θλc× l × n
Avec θ, l’ellipticité (˚), c la concentration (mol/l), l la longueur de la cuve (cm) et n le nombrede liaisons peptiques.
La spectroscopie DC permet ainsi à partir du spectre de la protéine et d’une base de donnéesde référence, de déterminer le % de structures secondaires existant dans la protéine en solution.Cela permet de qualifier la qualité du repliement des échantillons de protéine en amont des étudesstructurales. La proportion des différentes structures secondaires est estimée par déconvolutionpar les logiciels CDPRO et CDfriend. Les spectres de DC ont été mesurés avec un spectromètreJASCO J-80 équipé d’un portoir de cuvette thermostatée par un thermorégulateur à effet Peltier.Pour chaque échantillon, un spectre a été mesuré entre 185 et 270 nm à Tamb avec une cuvede 200 µl de chemin optique 1 mm, un pas de 0,2 nm, une vitesse de scan de 20 nm/min, untemps de réponse de 1 s et les spectres moyennés sur 5 acquisitions. Le spectre du tampon depréparation de l’échantillon a été soustrait de chaque spectre de protéine.
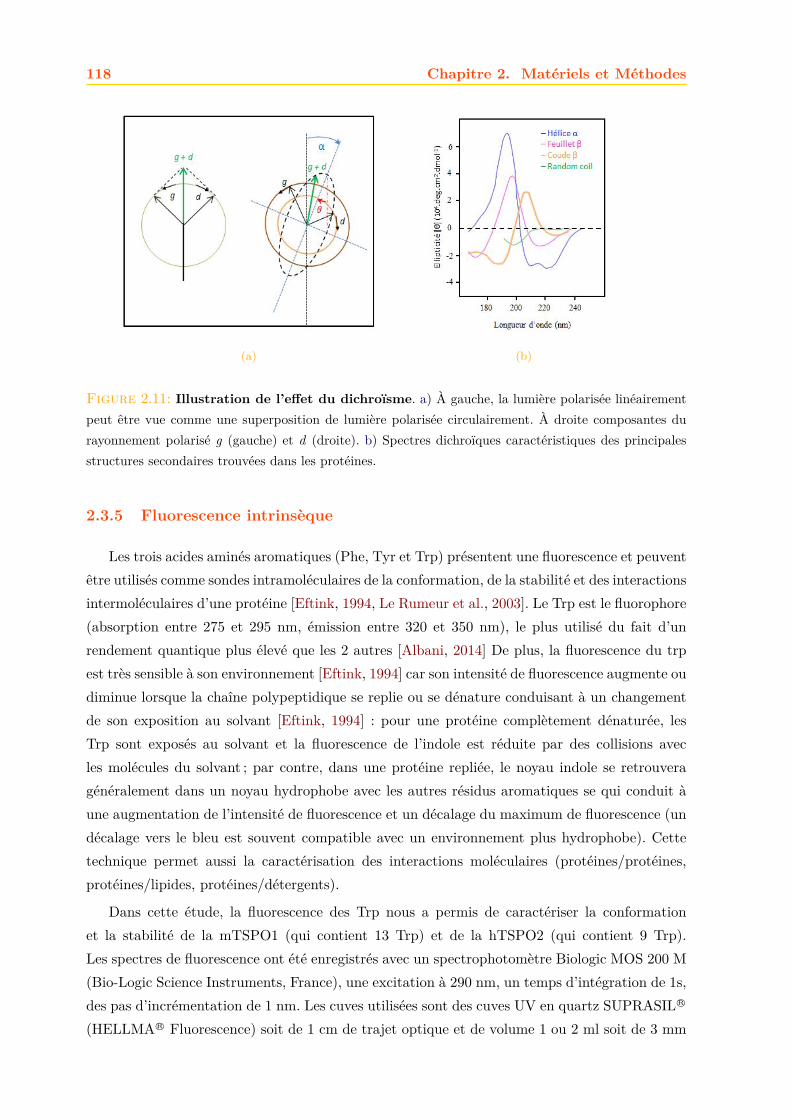
118 Chapitre 2. Matériels et Méthodes
(a) (b)
Figure 2.11: Illustration de l’effet du dichroïsme. a) À gauche, la lumière polarisée linéairementpeut être vue comme une superposition de lumière polarisée circulairement. À droite composantes durayonnement polarisé g (gauche) et d (droite). b) Spectres dichroïques caractéristiques des principalesstructures secondaires trouvées dans les protéines.
2.3.5 Fluorescence intrinsèque
Les trois acides aminés aromatiques (Phe, Tyr et Trp) présentent une fluorescence et peuventêtre utilisés comme sondes intramoléculaires de la conformation, de la stabilité et des interactionsintermoléculaires d’une protéine [Eftink, 1994, Le Rumeur et al., 2003]. Le Trp est le fluorophore(absorption entre 275 et 295 nm, émission entre 320 et 350 nm), le plus utilisé du fait d’unrendement quantique plus élevé que les 2 autres [Albani, 2014] De plus, la fluorescence du trpest très sensible à son environnement [Eftink, 1994] car son intensité de fluorescence augmente oudiminue lorsque la chaîne polypeptidique se replie ou se dénature conduisant à un changementde son exposition au solvant [Eftink, 1994] : pour une protéine complètement dénaturée, lesTrp sont exposés au solvant et la fluorescence de l’indole est réduite par des collisions avecles molécules du solvant ; par contre, dans une protéine repliée, le noyau indole se retrouveragénéralement dans un noyau hydrophobe avec les autres résidus aromatiques se qui conduit àune augmentation de l’intensité de fluorescence et un décalage du maximum de fluorescence (undécalage vers le bleu est souvent compatible avec un environnement plus hydrophobe). Cettetechnique permet aussi la caractérisation des interactions moléculaires (protéines/protéines,protéines/lipides, protéines/détergents).
Dans cette étude, la fluorescence des Trp nous a permis de caractériser la conformationet la stabilité de la mTSPO1 (qui contient 13 Trp) et de la hTSPO2 (qui contient 9 Trp).Les spectres de fluorescence ont été enregistrés avec un spectrophotomètre Biologic MOS 200 M(Bio-Logic Science Instruments, France), une excitation à 290 nm, un temps d’intégration de 1s,des pas d’incrémentation de 1 nm. Les cuves utilisées sont des cuves UV en quartz SUPRASILR©
(HELLMAR© Fluorescence) soit de 1 cm de trajet optique et de volume 1 ou 2 ml soit de 3 mm

2.3. Caractérisation des TSPO 119
de trajet optique et de volume 80 µl.
2.3.6 Liaisons des Ligands
L’interaction non covalente entre une protéine P et de son ligand L donne lieu à un complexeréversible :
P + L� PL
L’objectif principal en pratique de ces études de relations ligand/protéine est la détermina-tion de la capacité de fixation, appelée affinité, du ligand pour la protéine. Elle est caractériséepar la concentration du ligand occupant 50% des sites de liaisons de la protéine. Cette concen-tration est appelée constante de dissociation Kd et elle peut être calculée par le rapport entreles concentrations à l’équilibre des formes libres des 2 espèces et de la forme complexée :
Kd = [L][P ][LP ]
Plus cette constante est petite, plus l’affinité entre les 2 molécules est forte.
2.3.7 Mesure de la liaison des ligands par séparation des ligands libres etliés sur Résine Ni-NTA.
Figure 2.12: Illustration de la méthode de mesure de liaison des ligands (PK11195 etPPIX) par séparation des ligands libres et liés sur Résine Ni-NTA.
Comme toutes les chromatographies, la chromatographie d’affinité permet de séparer lescomposés d’un mélange. Nous avons utilisé cette capacité de séparation pour mesurer la liaisonde ligand sur la mTSPO1 en la fixant par son étiquette His6 sur la résine Ni-NTA. Nous avonsainsi séparé la protéine ayant un ligand lié et le ligand libre. La mTSPO1 a été mise en présencede son ligand dans un tube eppendorf puis incubée à Tamb avec la résine Ni-NTA. Après unesimple centrifugation de 1 min à 2000 × g, la mTSPO1 liée à la résine se retrouve au fond du tubeet le ligand libre dans le surnageant, Fig. 2.12. L’enregistrement des spectres de fluorescence auxλ d’émission et excitation de la mTSPO1, 330 et 290 nm respectivement, et aux λd’ d’émissionet excitation de la PPIX 405 et 620 nm respectivement, permet de suivre la fixation de lamTSPO1 et liaison de ses ligands.

120 Chapitre 2. Matériels et Méthodes
2.3.8 Liaison de ligands radioactifs
Pour la mTSPO1 reconstituée en protéoliposomes (le protocole de reconstitution sera pré-sentée plus loin), nous avons réalisé des mesures de liaison avec le PK11195 radioactif. Pource faire, les protéoliposomes sont incubés dans des solutions de concentrations croissantes de[3H]-PK11195 radioactif et filtrés sur un filtre Whatman GF/F . Cette filtration permettant deséparer le PK 11195 lié du libre. Les filtres sont ensuite mis en présence de 3 ml de liquide descintillation et la radioactivité est mesurée (WALLAC 1409 liquid scintillation counter ; chaquemesure 3 fois afin de faire des moyennes). Pour déterminer le PK11195 lié spécifiquement etnon spécifiquement, lors de l’incubation, 2 expériences sont effectuées en parallèle : avec ou sansexcès de ligand non radioactif (ligand froid). En absence de ligand froid, les sites de liaisonsspécifiques sur la mTSPO1 sont occupés par le ligand radioactif, Fig. 2.13 à droite. Au contraire,en excès de PK11195 froid, ils sont occupés par ce dernier, Fig. 2.13 à gauche. Ainsi il est pos-sible de calculer la liaison spécifique puisque l’expérience sans excès de PK 11195 froid, nousdonne la liaison totale ("LTot=Lspé+Lnonspé") et celle en excès de PK11195 froid nous donnela liaison non spécifique. Une simulation du tracé du PK11195 lié à la TSPO1 en fonction duPK11195 total par la courbe y = x×Bmax
x+Kd, permet d’obtenir la constante de dissociation Kd
et la capacité maximale de liaison Bmax (en nmol/mg de protéines).
Figure 2.13: Principe de la filtration avec le ligand radioactif (rouge) et ligand froid (bleu).
2.3.9 Les lipides
Il existe plusieurs types de lipides naturels avec des chaines aliphatiques plus ou moinsinsaturées et différents types de têtes polaires. Les lipides utilisés dans ce travail sont présentésdans le Tab. 2.13.
2.3.9.1 Préparation des lipides pour les protéoliposomes
Voici comment a été préparé le mélange DMPC/DMPE utilisé pour former les liposomesdans lesquels va venir s’incoporer la mTSPO1 :

2.3. Caractérisation des TSPO 121
Lipideslongueurde lachaîne
Nature de latête
PM Tm ◦C Structure
1,2-DiMyristoyl-sn-glycero-3-PhosphoCholine
DMPC14 :0 Zwitterionique 678 24
1,2-DiMyristoyl-sn-glycero-3-
PhosphoEthanolamineDMPE
14 :0 Zwitterionique 635.8 50
1,2-DiMyristoyl-sn-glycero-3-
PhosphorylGlycerolDMPG
14 :0 Anionique 688.85 23
Table 2.13: Structure et caractéristiques des lipides (Avanti PolarTM) utilisés lors de cetteétude.
1. La poudre de DMPC est mélangée à la poudre de DMPE à un ratio DMPC/DMPE de9/1 (p/p) (p. ex. 30 mg de DMPE et 270 mg de DMPC).
2. Les phospholipides sont dissous dans quelques ml de chloroforme puis la solution estdéposée dans un ballon en verre. Le solvant est évaporé en utilisant un évaporateur rotatif(BuchiTM). Après évaporation du chloroforme, on obtient un mince film de lipides sur laparoi du ballon.
3. Un tampon aqueux est ajouté et le film est repris en vortexant jusqu’à solubilisation totaledes lipides.
4. Le mélange est congelé dans l’azote liquide et lyophilisé sur la nuit pour enlever les der-nières traces de solvant.
5. Finalement, les liposomes de DMPC/DMPE sont obtenus en ajoutant un tampon aqueux,à un volume calculé pour obtenir la concentration souhaitée (p. ex. 3 ou 6 ml pour obtenirune solution stock à 100 ou 50 mg/ml, respectivement, à partir de la pesé de 30 mg deDMPE et 270 mg de DMPC). La solution est vortexée pour solubiliser tous les lipidespuis mise dans un bain sonicateur pour obtenir des liposomes unilamellaires.
2.3.9.2 Préparation des Bicelles
Les bicelles dont des disques formés par une bicouche plane de phospholipides à longuechaine (p. ex. DMPC ou DMPG) dont les côtés sont stabilisés par des phospholipides à courtes

122 Chapitre 2. Matériels et Méthodes
chaînes (DHPC) 10, Fig. 2.14.
Figure 2.14: Modèle de bicelleDMPC/DHPC. La morphologie des bi-celles varie en fonction de la température, de laconcentration totale en lipide et du rapport q :rapport de la concentration des lipides sur celledu détergent.
Pour l’étude de Nter d’AtTSPO Lors de l’étude du peptide Nter d’AtTSPO 2 types debicelles ont été réalisées : DMPC/DHPC (1/2 mole/mole) et DMPG/DHPC (1/2 mole/mole).Pour des bicelles à 50 mM de DHPC et 25 mM, voici la préparation :
1. la poudre de DHPC est pesée, ∼23 mg/ml, et dissoute (vortex) dans un tampon NaPO4
10 mM pH 6.1-6.2 ;
2. la poudre de DMPC (DMPG) est pesée, ∼17 mg/ml (17.22 mg/ml), et la solution deDHPC lui est ajoutée ;
3. le mélange est vortexé pendant 1 min ;
4. des cycles chaud/froid sont réalisés (∼3 fois) pour former des bicelles homogènes : lemélange est incubé dans un bain à 37 ◦C et congelé dans de l’azote liquide.
Des micelles de PI(4,5)P2 (1,2-dioleoyl-sn-glycero-3-[phosphatidylinositol-4,5-bisphosphate]Avanti PolarTM) ont également été préparées pour l’étude du Nter. La poudre de PI(4,5)P2a été dissoute dans un tampon NaPO4 10 mM pH 6.1-6.2 à la concentration désirée puis lasolution soniquée.
Pour l’étude de la mTSPO1 Les échantillons de bicelles ont été préparés par l’ajout deDHPC à 1M à la protéine reconstituée dans les liposomes de DMPC/DMPE (9 :1 p/p).
2.3.10 Reconstitution de la mTSPO1 dans des protéoliposomes
2.3.10.1 Principe de la reconstitution
La réincorporation des PMb purifiées en détergent dans une membrane lipidique sous formede protéoliposomes est une technique qui permet d’étudier leur fonction et leur structure dansun système plus proche de leur membrane native. Cela est important puisque de nombreusesPMb n’expriment leur activité seulement lorsqu’elles sont correctement repliées et insérées dansce type de bicouche lipidique synthétique[Wang and Tonggu, 2015, Rigaud, 2002].
La procédure générale d’incorporation implique différentes étapes :
10. Les chaines courtes de la DHPC agissent comme un détergent doux. Elle possède une cmc et peut doncformer des micelles c’est pourquoi ses caractéristiques sont présentés dans la Sec.2.2.1 "Les détergents".

2.3. Caractérisation des TSPO 123
1. le mélange de la protéine solubilisée en détergent avec des lipides pour former un complexeternaire (protéine/lipide/détergent) ;
2. l’échange du détergent autour de la protéine par les lipides ;
3. la formation de la vésicule par élimination du détergent.
Plusieurs protocoles de reconstitution de PMb dans des liposomes existent [Rigaud andLevy, 2003]. Les techniques couramment utilisées pour l’élimination du détergent sont la dilution[Dolder et al., 1996], la dialyse [Jap et al., 1992] et l’absorption sélective du détergent sur desBiobeads BB (billes hydrophobes poreuses en polystyrène) [Rigaud et al., 1997]. La méthoded’élimination de détergent utilisée dans notre cas a été la méthode d’adsorption sur des billesde polystyrène Biobeads (Bio-Beads SM2, Bio-Rad) [Rigaud et al., 1997, Lacapere et al., 2001].
Plusieurs paramètres sont à prendre en compte pour obtenir une bonne réincoporation dela protéine dans les liposomes : le choix des lipides, le rapport lipide/protéine (LPR - LipidProtein Ration) et le taux d’élimination du détergent. Dans le cas de la mTSPO1, des étudesprécédentes [Lacapere et al., 2001, Ostuni et al., 2010b, Teboul et al., 2012, Lacapere et al.,2014] ont montrées que :
• des protéoliposomes constitués d’un mélange DMPC/DMPE (9/1 poids/poids) permettentla liaison de ligands par la mTSPO1 ;• qu’un LPR au dessus de 0.5 mène à la formation de protéoliposomes ;• l’absorption du détergent par les BioBeads permet le contrôle du taux d’élimination dudétergent et l’obtention de protéoliposomes en quelques heures.
2.3.10.2 Suivi de la reconstitution
La reconstitution peut ensuite être suivie par plusieurs méthodes :
• la turbidimétrie, la formation des protéoliposomes au fur et à mesure de l’élimination dudétergent crée une modification de la turbidité observable par une augmentation de laDO350nm et DO550nm (λ auquel les liposomes diffusent sans interférence avec le spectrede la mTSPO1) ;• la fluorescence intrinsèque de la mTSPO1, le Trp possédant une fluorescence intrinsèquedépendante de son environnement local ;• la DLS, les protéoliposomes formés étant des objets plus grands que les complexes ternairesprotéine-détergent-lipide ;• et la microscopie électronique.
Absorbance, Florescence Les mesures d’absorbance/turbidimètrie et les spectres d’absorp-tion ont été réalisées sur :
• Pour une reconstitution en partant d’une protéine à 0.1-0.2 mg/ml, avec des cuves de 1cm de trajet optique et de volume 1 ou 2 ml.• Pour une reconstitution en partant d’une protéine à 1-2 mg/ml, le spectrophotomètreUV-Vis NanoDrop 2000 (Thermo Scientific) a été utilisé (la mesure nécessite seulement le

124 Chapitre 2. Matériels et Méthodes
dépôt d’une goute de 1 à 2 µl d’échantillon) et la fuorescence a été mesurée avec des cuvesde 3 mm de trajet optique et de volume 80microl.
La Diffusion Dynamique de Lumière (DLS) Les mesures de la taille des objets présentsen solution ont été obtenues avec l’appareil Protein Solutions DynaPro-99 (yatt Technology,France).
La microscopie électronique La ME permet de suivre et de contrôler la réincorporation dela protéine en observant la formation des protéoliposomes. Les échantillons (∼ 10µl) sont déposéssur des grilles de cuivre (400 mesh) lavées, préalablement rendues hydrophiles et recouvertes d’unfilm de carbone. La coloration négative de l’échantillon a été obtenue en utilisant de l’acétated’uranyle (UA ; 2% p/p), lequel fait apparaître les bords des vésicules de protéoliposomes enblanc (absence de colorant) et le centre des vésicules en noir (accumulation de colorant - lefaisceau d’électron est arrêté ou fortement dévié par les atomes lourds d’uranyle). L’observationa été réalisée grâce à un microscope électronique à transmission JEOL JEM-100CX. Différentschamps ont été observés à plusieurs grandissements permettant de contrôler l’homogénéité dela solution et de mesurer la taille des protéoliposomes reconstitués.
Pour la cryoME, un microscope électronique à transmission JEOL JEM-2100 a été utilisé.Les échantillons (∼ 2-4 µl ont été déposés sur des grilles recouvert d’une fine couche de carbonetroué (Quantifoil grids, EMS France) et rapidement congelés en étant plongés dans un puitd’éthane liquide à l’aide d’un système de guillautine. L’excès d’éthane liquide est éliminé avecdu papier filtre, l’échantillon est placé sur un porte échantillon qui est sous atmosphère d’azoteet rapidement observé pour ne pas qu’il ne se réchauffe.
2.3.10.3 La reconstitution en pratique
De manière pratique, la mTSPO1 purifiée en SDS est diluée dans un tampon (Hepes NaOH10 mM ph 7.8 SDS 2 cmc (soit 2 mg/ml) ou NaPO4 10 mM pH 6.2-6.5 pour les études RMN),à une concentration finale de l’ordre de :
• soit à 0.1 mg/ml - cette concentration permet de réaliser les mesures d’absorbance et defluorescence sans risquer la saturation du signal. C’est typiquement la concentration quia été utilisée en routine depuis de nombreuses années dans le laboratoire.• soit à une concentration beaucoup plus élevée 1-2 mg/ml - cette concentration permet detravailler avec des plus petits volumes et a été testée pour la reconstitution des protéoli-posomes destinés à l’étude en RMN du solide. Cependant, elle nécessite l’utilisation d’unnanodrop pour la mesure de la concentration en protéine ou de se restreindre à l’enregis-trement du spectre dans la région ou il n’y pas de signal de la protéine. La reconstitutiondans ces conditions est beaucoup plus difficile à contrôler.
Le mélange de lipides DMPC/DMPE (9/1 p/p) est ajouté de manière à obtenir le rapportlipide-protéine (LPR) souhaité. Pour la RMN du solide un LPR de 1 a d’abord été utilisé puis un
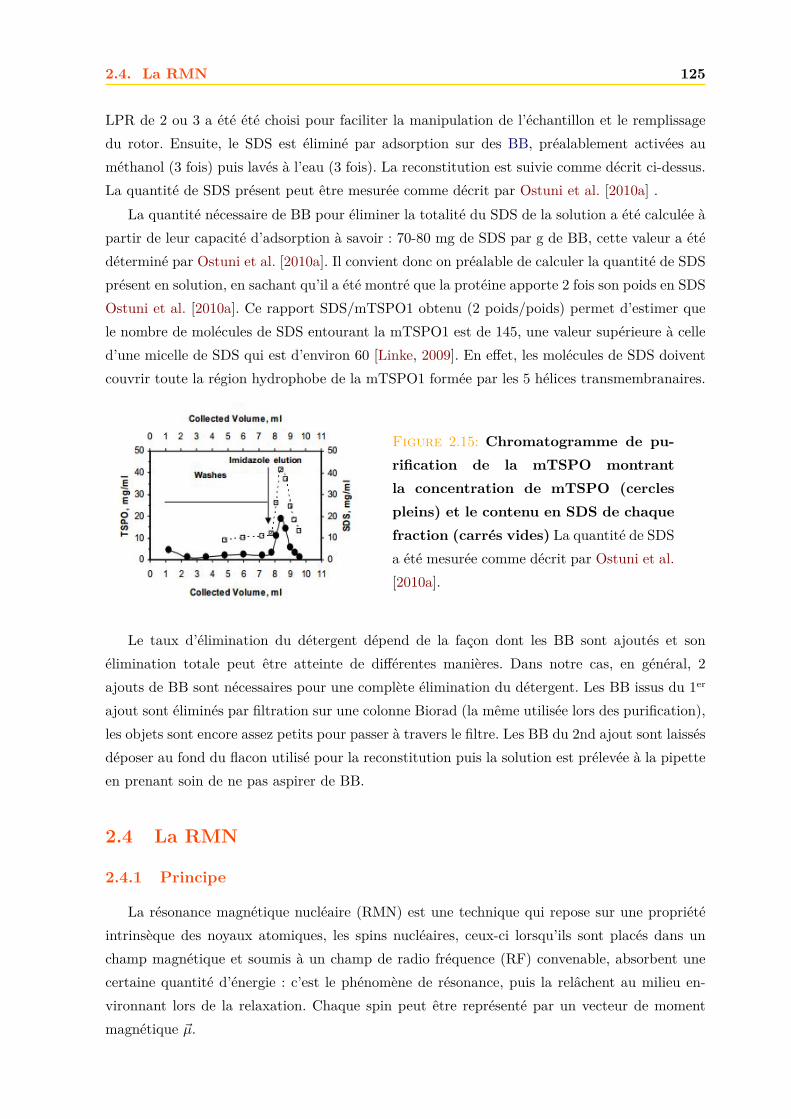
2.4. La RMN 125
LPR de 2 ou 3 a été été choisi pour faciliter la manipulation de l’échantillon et le remplissagedu rotor. Ensuite, le SDS est éliminé par adsorption sur des BB, préalablement activées auméthanol (3 fois) puis lavés à l’eau (3 fois). La reconstitution est suivie comme décrit ci-dessus.La quantité de SDS présent peut être mesurée comme décrit par Ostuni et al. [2010a] .
La quantité nécessaire de BB pour éliminer la totalité du SDS de la solution a été calculée àpartir de leur capacité d’adsorption à savoir : 70-80 mg de SDS par g de BB, cette valeur a étédéterminé par Ostuni et al. [2010a]. Il convient donc on préalable de calculer la quantité de SDSprésent en solution, en sachant qu’il a été montré que la protéine apporte 2 fois son poids en SDSOstuni et al. [2010a]. Ce rapport SDS/mTSPO1 obtenu (2 poids/poids) permet d’estimer quele nombre de molécules de SDS entourant la mTSPO1 est de 145, une valeur supérieure à celled’une micelle de SDS qui est d’environ 60 [Linke, 2009]. En effet, les molécules de SDS doiventcouvrir toute la région hydrophobe de la mTSPO1 formée par les 5 hélices transmembranaires.
Figure 2.15: Chromatogramme de pu-rification de la mTSPO montrantla concentration de mTSPO (cerclespleins) et le contenu en SDS de chaquefraction (carrés vides) La quantité de SDSa été mesurée comme décrit par Ostuni et al.[2010a].
Le taux d’élimination du détergent dépend de la façon dont les BB sont ajoutés et sonélimination totale peut être atteinte de différentes manières. Dans notre cas, en général, 2ajouts de BB sont nécessaires pour une complète élimination du détergent. Les BB issus du 1er
ajout sont éliminés par filtration sur une colonne Biorad (la même utilisée lors des purification),les objets sont encore assez petits pour passer à travers le filtre. Les BB du 2nd ajout sont laissésdéposer au fond du flacon utilisé pour la reconstitution puis la solution est prélevée à la pipetteen prenant soin de ne pas aspirer de BB.
2.4 La RMN
2.4.1 Principe
La résonance magnétique nucléaire (RMN) est une technique qui repose sur une propriétéintrinsèque des noyaux atomiques, les spins nucléaires, ceux-ci lorsqu’ils sont placés dans unchamp magnétique et soumis à un champ de radio fréquence (RF) convenable, absorbent unecertaine quantité d’énergie : c’est le phénomène de résonance, puis la relâchent au milieu en-vironnant lors de la relaxation. Chaque spin peut être représenté par un vecteur de momentmagnétique ~µ.

126 Chapitre 2. Matériels et Méthodes
Figure 2.16: Illustration de l’effet Zee-man. Différence d’énergie des états de spind’un noyau induite par un champ magnétiqueB0.
Soumis à un champ magnétique B0, le moment ~µ interagit avec B0 et décrit un mouvementde précession autour de B0, avec une fréquence ν0 appelée fréquence de Larmor. Soumis à cechamp B0 la projection du moment magnétique ( ~µ = γ~~I ; avec γ le rapport gyromagnétique dunoyau et ~ la constante de Planck/ 2π) d’un noyau de spin I non nul prendra 2I+1 orientationspar rapport à la direction de ce champ. cet effet est appelé effet Zeeman . Dans le cas des noyauxde spin I = 1/2 (p. ex. 1H, 13C, 15N, 19F, 31P) 2 états, correspondant au nombre quantiquem, sont autorisés : l’état α de basse énergie (m = 1/2) lorsque le spin nucléaire est dans lamême direction que le champ appliqué et l’état β de haute énergie (m= -1/2), lorsque le spinnucléaire a une orientation antiparallèle au champ appliqué. La différence d’énergie sera alors∆E = −mγ~B0=hν0, Fig. 2.16.
L’équation de Boltzmann Nα
Nβ= e
∆EkT définit le rapport du nombre de spins dans l’état α
(Nα) et l’étatβ (Nβ) (avec k constante de Boltzmann et T la température).Dans le cadre de la RMN par transformée de Fourier, l’échantillon est introduit dans un
champ magnétique constant et soumis simultanément à des impulsions RF (Radiofréquence)qui basculent l’aimantation perpendiculairement à B0 (plan xOy). En effet, la fréquence doitêtre proche de la fréquence de résonance, ν0 de l’atome observé, pour que l’absorption de l’énergieassociée permette une transition de l’état α vers l’état β. Ensuite, le champ RF est coupé lesnoyaux excités reviennent à l’état d’équilibre par le processus de "relaxation".
La mesure, en fonction du temps, de la décroissance de l’aimantation dans le plan xOy,fournit un signal de RMN, appelé signal de précession libre ou "Free Induction Decay" (FID).La transformée de Fourier (TF) de ce FID permet d’obtenir le spectre RMN de la moléculequi se caractérise par la présence de différentes raies dont la position et la forme dépendent desinteractions subies par les noyaux.
2.4.2 La relaxation
La relaxation est le processus par lequel l’aimantation, qui est la résultante des spins del’échantillon, après avoir été basculée dans le plan transversal (xOy), revient progressivement àl’équilibre avec l’environnement c-à-d. parallèle à B0. La vitesse de relaxation est influencée parles propriétés physiques de la molécule et l’échantillon, de sorte que l’ étude de ce phénomènede relaxation peut p. ex. donner des informations sur les mouvements internes de la molécule.
Il existe 2 types de relaxations, Fig. 2.17 :

2.4. La RMN 127
• La relaxation longitudinale ou spin - réseau caractérisée par le temps T1, qui correspondau retour à l’équilibre de l’aimantation vers sa valeur d’équilibre selon l’axe Oz. Dans ceprocessus, le système de spins échange de l’énergie avec le réseau sous forme de chaleurdissipée à travers l’échantillon. En règle générale, cela se passe selon un processus mono-exponentiel. Dans les liquides, les temps de relaxation longitudinale T1 des protons sonttypiquement de l’ordre de quelques centaines de ms à quelques s. La relaxation longitudi-nale est liée à l’environnement moléculaire du noyau.• La relaxation transversale ou spin-spin caractérisée par le temps T2, qui correspond au re-tour à l’équilibre de l’aimantation dans le plan perpendiculaire à B0. Il y a échange d’éner-gie entre les spins nucléaires. Le plus souvent l’aimantation transversale décroît monoexponentiellement. Les temps de relaxation transversale T2, qui couvrent une gamme devaleurs extrêmement larges. Dans les solides, ils peuvent être plus courts que la ms, alorsque, dans les liquides, ils sont proches des T1 et sont donc de l’ordre de quelques centainesde ms à quelques s. À une faible valeur de T2, il faut associer un FID court dans le temps,c-à-d. un signal large dépendant des fréquences.
Figure 2.17: Les relaxations longitudinale T1 et transversale T2
2.4.3 Le signal RMN
L’observable fondamentale de la RMN à TF est le FID (Free Induction Decay) qui résultede la relaxation de tous les noyaux présents dans l’échantillon. Le FID est donc la somme desFID de chaque noyau qui résonne à une fréquence caractéristique. Ce FID est décrit par unecourbe exponentielle décroissante : I(t)=e(iωt)e(−R2×t)=e(iωt)e−t/T2 ; avec I(t) l’intensité de laFID au temps t. Une fois acquise, la TF du domaine temporel au domaine fréquentielle permetd’observer le déplacement chimique, l’intensité et la largeur de raie de chaque résonance.
Déplacement chimique La valeur du déplacement chimique (δ) traduit l’effet de l’envi-ronnement électronique autour du noyau. Ce dernier produit un champ local qui s’oppose ous’ajoute à B0 selon la position du noyau observé par rapport au nuage électronique. Cet effetest pris en compte sous la forme d’une constante d’écran σ : ν = ν0(1− σ).
Les noyaux de même espèce, mais situés dans des environnements chimiques différents,présentent ainsi des fréquences de résonance différentes, d’où le nom de "déplacement chimique"
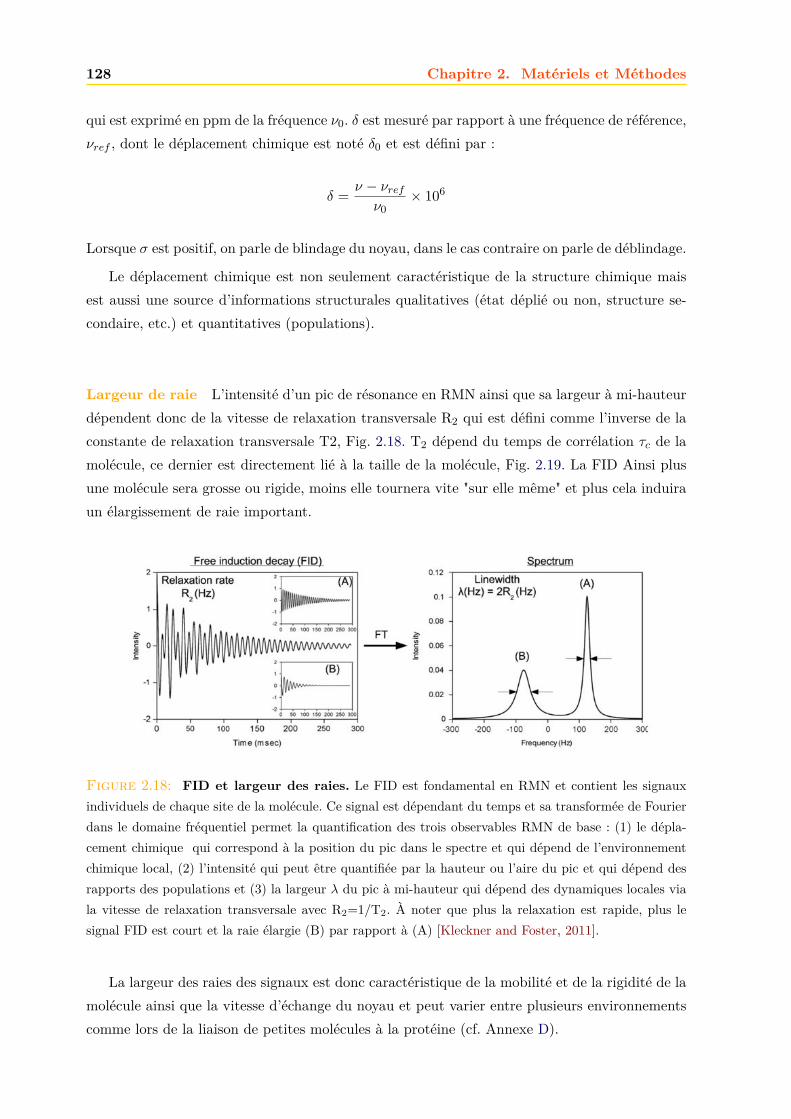
128 Chapitre 2. Matériels et Méthodes
qui est exprimé en ppm de la fréquence ν0. δ est mesuré par rapport à une fréquence de référence,νref , dont le déplacement chimique est noté δ0 et est défini par :
δ = ν − νrefν0
× 106
Lorsque σ est positif, on parle de blindage du noyau, dans le cas contraire on parle de déblindage.
Le déplacement chimique est non seulement caractéristique de la structure chimique maisest aussi une source d’informations structurales qualitatives (état déplié ou non, structure se-condaire, etc.) et quantitatives (populations).
Largeur de raie L’intensité d’un pic de résonance en RMN ainsi que sa largeur à mi-hauteurdépendent donc de la vitesse de relaxation transversale R2 qui est défini comme l’inverse de laconstante de relaxation transversale T2, Fig. 2.18. T2 dépend du temps de corrélation τc de lamolécule, ce dernier est directement lié à la taille de la molécule, Fig. 2.19. La FID Ainsi plusune molécule sera grosse ou rigide, moins elle tournera vite "sur elle même" et plus cela induiraun élargissement de raie important.
Figure 2.18: FID et largeur des raies. Le FID est fondamental en RMN et contient les signauxindividuels de chaque site de la molécule. Ce signal est dépendant du temps et sa transformée de Fourierdans le domaine fréquentiel permet la quantification des trois observables RMN de base : (1) le dépla-cement chimique qui correspond à la position du pic dans le spectre et qui dépend de l’environnementchimique local, (2) l’intensité qui peut être quantifiée par la hauteur ou l’aire du pic et qui dépend desrapports des populations et (3) la largeur λ du pic à mi-hauteur qui dépend des dynamiques locales viala vitesse de relaxation transversale avec R2=1/T2. À noter que plus la relaxation est rapide, plus lesignal FID est court et la raie élargie (B) par rapport à (A) [Kleckner and Foster, 2011].
La largeur des raies des signaux est donc caractéristique de la mobilité et de la rigidité de lamolécule ainsi que la vitesse d’échange du noyau et peut varier entre plusieurs environnementscomme lors de la liaison de petites molécules à la protéine (cf. Annexe D).

2.4. La RMN 129
Figure 2.19: Comportement de T1 et T2
en fonction du temps de corrélation τc
défini comme le temps moyen que met lamolécule pour tourner de 1 radian.
2.4.4 La RMN du liquide
Toutes les expériences RMN du liquide ont été réalisées au laboratoire avec un spectomètreRMN 500 MHz équipé d’une cryosonde (Bruker) en collaboration avec le Pr. Olivier Lequin.
2.4.4.1 Spectre RMN homonucléaire 1D 1H d’une protéine
Le spectre RMN d’une molécule est caractérisé par des propriétés particulières permettantd’obtenir des informations sur l’environnement des noyaux étudiés. P. ex., pour un atome consi-déré, son δ est caractéristique du groupement chimique auquel il appartient et si plusieurs atomesrésonnent à la même fréquence, l’aire du pic de résonance correspondant est proportionnelle aunombre de ces atomes.
Le spectre 1D 1H d’une protéine est souvent très complexe puisqu’il contient la superposi-tion des signaux des différents protons des résidus. Les protons appartenant à un groupementfonctionnel particulier auront tendance à résonner dans une gamme de fréquence caractéris-tique de ce groupement, comme illustré sur la Fig. 2.20. Cependant, des variations du δ sontobservées pour la plupart des noyaux des a.a dues au repliement adopté par la protéine. Cettedispersion des signaux est appelée dispersion spectrale et peut être utilisée comme indicateurde la formation de structure secondaire et tertiaire. De plus, les changements structuraux lorsde liaisons protéine-ligand peuvent être caractérisés par l’étude de l’évolution du spectre RMNde la protéine lors de la liaison à ce ligand.
2.4.4.2 Spectre RMN hétéronucléaire 2D d’une protéine
Même si les spectres RMN 1D permettent d’obtenir des informations sur la protéine, leuranalyse reste complexe à cause des superpositions des résonances. De plus, elle ne permet pasde donner la moindre indication de structure tertiaire. Le passage à 2 dimensions permet nonseulement de mieux séparer les informations mais aussi d’établir des corrélations entre ces infor-mations : entre les couplages et les déplacements chimiques ou entre les spectres obtenus pourdifférents noyaux p. ex. Pour cela le spectre proton est édité dans une dimension supplémentairequi correspond soit à la fréquence de résonance de l’azote (spectres 1H,15N HSQC), soit à celledu carbone (spectres 1H,13C HSQC).

130 Chapitre 2. Matériels et Méthodes
Figure 2.20: Gammes de dépla-cement chimique des 1H repor-tées sur le spectre RMN 1D 1Hde la mTSPO1. Figure adaptée de[Galvagnion, 2011].
Dans une expérience RMN 1D la phase d’acquisition a lieu juste après la séquence d’impul-sions et l’intensité des signaux des protons est tracée en fonction d’un axe de fréquence. Dansune expérience 2D, l’étape d’acquisition est séparée de la phase d’excitation par des stadesintermédiaires appelés évolution et le mélange. Le spectre 2D est alors obtenu par acquisitionsuccessive d’une série de spectres 1D suivant 4 étapes :
1. La 1re période, période de préparation, permet l’excitation du système de spins par une ouplusieurs impulsions RF et la relaxation des spins entre chaque acquisition.
2. Ensuite, les spins évoluent en fonction de leur fréquence de résonance pendant la périoded’évolution dont la durée t1 est incrémentée régulièrement c.-à-d. que la 1re fois la valeur de t1est proche de zéro et le premier spectre est acquis. La 2e fois, t1 est augmentée par ∆t ("dwelltime") et un autre spectre est acquis. Ceci est répétée jusqu’à ce qu’il y ait une "collection"suffisante de FID pour l’analyse par TF 2D.
3. La période de mélange permet un échange d’aimantation (transfert de polarisation), par unenouvelle application d’une ou plusieurs impulsions, entre 2 populations de spins qui sontcorrélées. Ce transfert de polarisation peut se faire de différentes manières :
• Par couplage scalaire scalaire : il s’agit du couplage indirect qui a pour origine les inter-actions entre spins nucléaires et spins électroniques des liaisons chimiques. Leur portéeest limitée par le nombre de liaisons chimiques qui séparent les noyaux.• Par relaxation croisée qui se base sur le couplage dipolaire. Le couplage dipolaire est uncouplage direct à travers l’espace entre spins nucléaires. Sa portée dépend de la distanced qui sépare les deux noyaux couplés. Le couplage dipolaire est invisible en RMN duliquide à cause du mouvement brownien. C’est un phénomène de relaxation croisée quiva pouvoir transférer l’aimantation entre les 2 noyaux couplés. Lorsque la relaxationd’un noyau est dépendante de l’état des noyaux voisins cela constitue le phénomène de

2.4. La RMN 131
relaxation croisée qui se manifeste sous la forme de l’effet Overhauser nucléaire (nOe).Cet effet varie selon une loi en 1/d6 .
4. Finalement la période de détection permet l’enregistrement du signal (FID) pendant un tempst2. Ce signal dépend de l’évolution du système de spins pendant t1 et de la nature de la périodede mélange.
2.4.4.3 Le spectre HSQC - Heteronuclear Single Quantum Coherence
La principale expérience 2D RMN utilisé lors de l’étude la mTSPO1 ou du Nter d’AtTSPOest l’expérience 2D 2D 1H,15N HSQC Celle ci permet de corréler le déplacement chimique del’hétéronoyau 15N au(x) déplacement(s) chimique(s) du (des) 1H auquel il est lié. La polarisationest transférée du noyau 1H au noyau 15N qui lui est directement lié via le couplage scalaire 1JNH .Puis, le déplacement chimique du noyau 15N évolue pendant une période t1 et la polarisationest re-transférée vers le noyau 1H pour la détection pendant une période t2.
(a)(b)
Figure 2.21: Expérience HSQC. a) Représentation du transfert d’aimantation dans un acide aminélors de la séquence HSQC. b) Schéma de la séquence d’impulsions HSQC : la période de préparationest constituée d’une période de retour à l’équilibre de l’aimantation et d’un motif INEPT (InsensitiveNuclei Enhanced by Polarization Transfer) de durée = τ (1/4JNH) qui transfère l’aimantation des 1Hvers les 15N auxquels ils sont directement liés. La période d’évolution, de durée t1, est un écho de spin quipermet que seuls les déplacements chimiques 15N s’expriment pendant le temps t1 mais aussi que l’effetdes couplages 15N,1H soit refocalisé. La période de mélange est un motif INEPT INVERSE, il permet detransférer l’aimantation des 15N vers les 1H qui leur sont couplés. Enfin, durant la période de détection,le signal RMN 1H est détecté en présence d’un découplage 15N et donc, durant t2, les couplages 1H,15Nsont éliminés du spectre.
L’HQSC est généralement la 1re expérience hétéronucléaire réalisée lors de l’étude d’uneprotéine. Elle constitue une empreinte de la protéine et permet de se rendre compte de l’étatde son repliement. Elle permet donc d’évaluer si d’autres expériences RMN sont susceptibles defonctionner. Dans une protéine, chaque résidu est caractérisé par la liaison N-H de sa fonctionamide, à l’exception des prolines (dont l’azote ne porte aucun hydrogène) et du résidu N-terminal(l’hydrogène de la fonction amine étant labile). Chacune des liaisons N-H du squelette d’une

132 Chapitre 2. Matériels et Méthodes
protéine enrichie en 15N donne lieu à un pic de corrélation sur le spectre 1H,15N HSQC . Lesliaisons Nε-Hε des chaines latérales des Trp et les liaisons Nδ-Hδ/Nε-Hε des chaines latéralesdes Asn/Gln respectivement peuvent aussi être visibles sur le spectre.
2.4.4.4 L’attribution des résonances
Pour pouvoir réaliser l’attribution des δ, les expériences 3D sont indispensables. Le spectre2D hétéro nucléaire (1H,15N HSQC) est édité dans une troisième dimension qui correspond à lafréquence de résonance de l’atome 13C.
L’attribution séquentielle des résonances 1HN, 13Cα, 13Cβ et 15NH de la chaine principaledu peptide Nter d’AtTSPO a été réalisée à partir de l’analyse du spectre 2D 1H-15N HSQCet des spectres 3D HNCACB, CBCA(CO)HN, HN(CA)CO et HNCO et en suivant la mé-thode décrite ci-dessous (http://nmr.chem.uu.nl/~abonvin/tutorials/Assignment-Data/
assignment.html).Tout d’abord, pour chaque pic de corrélation NH du spectre 2D 1H-15N HSQC, c.-à-d. pour
chaque résidu i, on compare les plans 1H-13C des spectres 3D HNCACB et CBCA(CO)HN auniveau du δ(15NH(i)) dans la dimension 15N (Fig. 2.22.a, b,c, d et e) : les pics de corrélationsituées sur les plans 1H-13C du spectre HNCACB ont pour ordonnées le δ du 13Cα(i) , 13Cα(i−1),13Cβ(i) et 13Cβ(i−1) (Fig. 2.22.d ; les pics du résidu i apparaissent plus intensément que ceuxdu i-1) ; ceux situées sur les plans 1H-13C du spectre CBCA(CO)HN ont pour ordonnée le δdu 13Cα(i−1) et 113Cβ(i−1), uniquement (Fig. 2.22.e). Cette différence permet ainsi de dissocierle couple δ(13Cα(i))/δ(13Cβ(i)) du couple δ(13Cα(i−1))/ δ(13Cβ(i−1)). Les δ des 13Cα et 13Cβétant spécifiques pour chacun des 20 a.a (une base de données comprenant les statistiques desδ calculées les 20 a.a existe - http://www.bmrb.wisc.edu/ref_info/statful.htm) le systèmede spin du résidu i et du résidu i-1 peut être déterminé.
L’attribution des δ des 13CO est possible par analyse des spectres 3D HNCO et HN(CA)CO(Fig. 2.22.f et g) à partir de l’attribution des δ des 1HN et 15NH (Figure 2.17A). Pour unrésidu i, on se place sur le plan 1H-13C de l’expérience HNCO au niveau du δ( 15NH(i)) dans ladimension 15N (Fig. 2.22.g) ; le pic de corrélation situé sur ce plan correspond au groupementCO du résidu i-1 et a pour abscisse et ordonnée δ( 1HN(i)) et δ( 13CO(i-1)), respectivement.L’expérience HN(CA)CO (Fig. 2.22.f) nous donne pour un plan donné, les CO du résidu i eti-1 permettant de faire les liens séquentiels.
2.4.4.5 Préparation des échantillons pour les expériences en RMN du liquide
La sensibilité d’une expérience RMN, c.-à-d. le rapport signal/bruit (S/B), suit la relation :S/B ∝ nγe
√γ3dB
30t ; avec n le nombre de noyaux observés, γe le rapport gyromagnétique du
noyau excité, γd celui du noyau détécté, B0 le champ magnétique et t le temps d’acqusition.Le S/B dépendra donc des noyaux étudiés et de la concentration de l’échantillon Pour uneexpérience RMN 1H, une concentration de 50 µM en protéine permet une acquisition d’unspectre de bonne qualité en quelques min. Pour les expériences multidimentionnelles (1H, 13C,

2.4. La RMN 133
Figure 2.22: Méthodologie d’attribution séquentielle des δ des 1HN, 13Cα, 13Cβ et 15NHdu squelette peptique. Les noyaux indiqués en rouge et bleu sont ceux dont le δ est mesuré ; lesnoyaux indiqués en bleu clair et vert sont impliqués dans les transferts d’aimantation mais leur δ n’estpas mesuré.
15N une concentration de quelques centaines de µM est nécessaire pour pouvoir obtenir unspectre avec une bonne sensibilité (une telle concentration permet d’obtenir un spectre HSQC1H-15N en moins de 30 min). Si la quantité de matière est divisée par 2, le temps d’acquisitiondes données devra être multiplié par 4 pour obtenir un spectre équivalent. Un volume de 500 µlest nécessaire pour les tubes RMN standards de 5 mm de diamètre et 10% de D2O sont ajoutéspour le "lock" (stabilisation du champ magnétique).
Préparation des solutions de PPIX pour la RMN La PPIX tout comme l’hème, depar leur structure ont une tendance à l’agrégation en solution aqueuse. Cette agrégation ouempilement ("stacking") est le résultat d’interactions intermoléculaires π-π (interactions noncovalentes entre les noyaux aromatiques). Elle dépend du pH et de la force ionique de la solution[Scolaro et al., 2002]. La PPIX est sous forme monomérique à un pH compris entre 0 et 3 ;dimérique à un pH > 8 et les plus grands agrégats sont retrouvé à un pH compris entre 3 et7. Cependant en RMN le pH idéal est autour de 6, lequel permet un échange lent des protonsN-H avec le solvant . La solution stock de PPIX est donc préparée en ajoutant la molécule sousforme de poudre à une solution NaOH 1M.

134 Chapitre 2. Matériels et Méthodes
Préparation des échantillons de mTSPO1 en présence des ligands hydrophobes pourla RMN Les molécules hydrophobes comme le PK11195, sont conservés dans de l’éthanol(EtOH) pour permettre leur solubilisation : p. ex. le PK11195 ne présente une solubilité que del’ordre de 300 µM dans l’eau alors qu’elle est de 50 mM dans l’EtOH. Lors de la préparationde nos échantillons pour la RMN nous préparons des films de PK 11195 (ou d’autres ligandshydrophobes), à la concentration désirée, par évaporation de la solution alcoolique, dans untube Eppendorf, par un flux d’air comprimé ou d’azote. Ensuite, la protéine en détergent estajoutée dans le tube contenant le film. Le tube est vortexé vigoureusement pour reprendre lefilm.
2.4.5 La RMN du solide
2.4.5.1 Les interactions en RMN du solide
L’intérêt des biologistes pour la RMN en phase solide prit beaucoup plus de temps à sedévelopper. En effet, s’il est courant d’obtenir des spectres très bien résolus en RMN en phaseliquide, ceci est loin d’être le cas en phase solide : en liquide la largeur des signaux est de l’ordredu Hz alors qu’en solide elle peut facilement excéder des dizaines de kHz ou même plusieursMHz. La raison en est simple : en RMN du liquide le mouvement brownien des moléculespermet de moyenner à zéro les interactions anisotropes (dépendantes de l’orientation de lamolécule dans son milieu) ; seules persistent les interactions isotropes, telles le couplage scalaire,et le déplacement chimique. Les raies obtenues sur un spectre de RMN en phase liquide sontdonc plutôt fines. Dans le cas de la RMN du solide, il n’y a pas ou peu de mouvements dansl’échantillon solide. Les interactions anisotropes ne sont donc pas moyennées et dominent lespectre de RMN, qui possède alors des raies très larges. Par conséquent, il fallut tout d’aborddisséquer les interactions anisotropes présentes en RMN du solide pour mettre au point destechniques permettant de réduire la largeur des raies.
Les améliorations méthodologiques, combinées au développement de séquences d’impulsionsspécifiques à la RMN en phase solide ainsi que les progrès dans la préparation des échantillons,conduisirent à la première détermination structurale tridimensionnelle de protéine la SH3 (7kDa) par cette technique en 2002 [Castellani et al., 2002]
Dans la matière condensée, l’effet Zeeman d’un noyau est perturbé par la proximité spatialedes électrons et des autres noyaux qui l’entourent. Comparativement à l’effet Zeeman dominant,ces interactions sont considérées comme des perturbations. En solide, ces interactions sont :l’interaction dipolaire, quadrupolaire, scalaire et de déplacement chimique, Fig.2.23. Voici unebrève description de ces perturbations et de leur contribution en RMN du solide par rapport àla RMN du liquide .
l’interaction de déplacement chimique Comme nous l’avons vu dans la section précé-dente, le déplacement chimique (δ) dépend de l’environnement électronique du noyau. Toutécart d’une distribution sphérique des électrons autour du noyau donne une contribution ani-
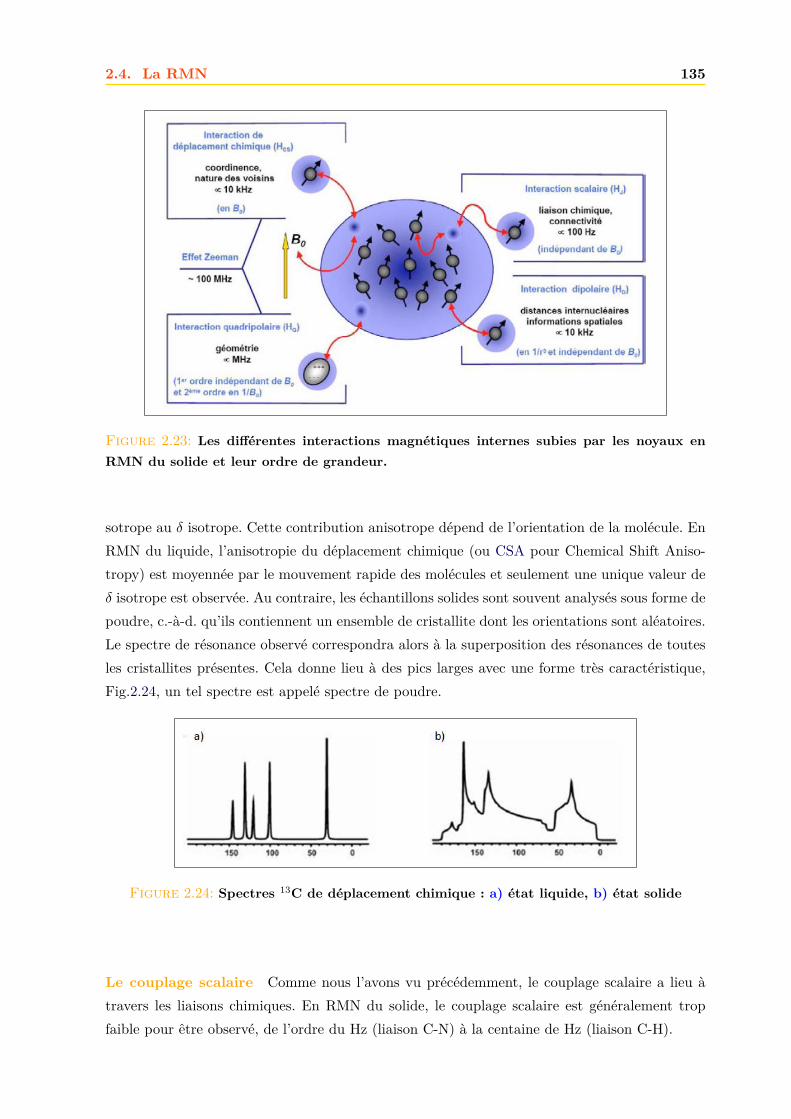
2.4. La RMN 135
Figure 2.23: Les différentes interactions magnétiques internes subies par les noyaux enRMN du solide et leur ordre de grandeur.
sotrope au δ isotrope. Cette contribution anisotrope dépend de l’orientation de la molécule. EnRMN du liquide, l’anisotropie du déplacement chimique (ou CSA pour Chemical Shift Aniso-tropy) est moyennée par le mouvement rapide des molécules et seulement une unique valeur deδ isotrope est observée. Au contraire, les échantillons solides sont souvent analysés sous forme depoudre, c.-à-d. qu’ils contiennent un ensemble de cristallite dont les orientations sont aléatoires.Le spectre de résonance observé correspondra alors à la superposition des résonances de toutesles cristallites présentes. Cela donne lieu à des pics larges avec une forme très caractéristique,Fig.2.24, un tel spectre est appelé spectre de poudre.
Figure 2.24: Spectres 13C de déplacement chimique : a) état liquide, b) état solide
Le couplage scalaire Comme nous l’avons vu précédemment, le couplage scalaire a lieu àtravers les liaisons chimiques. En RMN du solide, le couplage scalaire est généralement tropfaible pour être observé, de l’ordre du Hz (liaison C-N) à la centaine de Hz (liaison C-H).
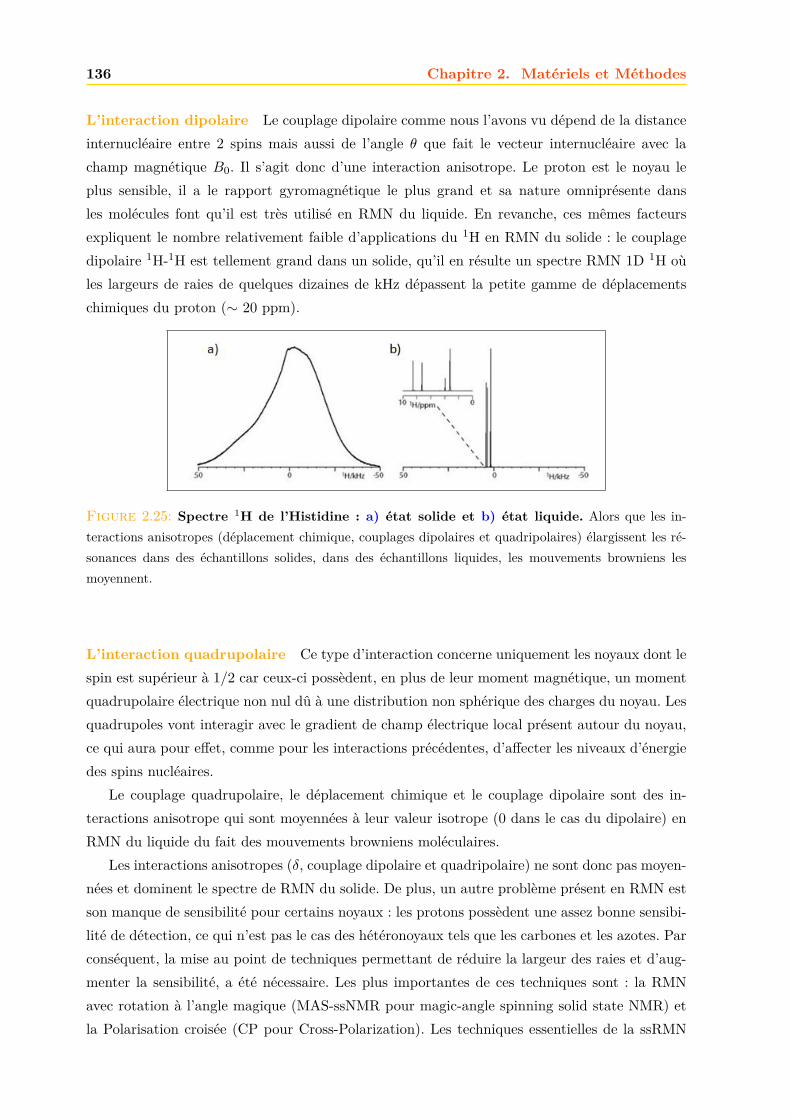
136 Chapitre 2. Matériels et Méthodes
L’interaction dipolaire Le couplage dipolaire comme nous l’avons vu dépend de la distanceinternucléaire entre 2 spins mais aussi de l’angle θ que fait le vecteur internucléaire avec lachamp magnétique B0. Il s’agit donc d’une interaction anisotrope. Le proton est le noyau leplus sensible, il a le rapport gyromagnétique le plus grand et sa nature omniprésente dansles molécules font qu’il est très utilisé en RMN du liquide. En revanche, ces mêmes facteursexpliquent le nombre relativement faible d’applications du 1H en RMN du solide : le couplagedipolaire 1H-1H est tellement grand dans un solide, qu’il en résulte un spectre RMN 1D 1H oùles largeurs de raies de quelques dizaines de kHz dépassent la petite gamme de déplacementschimiques du proton (∼ 20 ppm).
Figure 2.25: Spectre 1H de l’Histidine : a) état solide et b) état liquide. Alors que les in-teractions anisotropes (déplacement chimique, couplages dipolaires et quadripolaires) élargissent les ré-sonances dans des échantillons solides, dans des échantillons liquides, les mouvements browniens lesmoyennent.
L’interaction quadrupolaire Ce type d’interaction concerne uniquement les noyaux dont lespin est supérieur à 1/2 car ceux-ci possèdent, en plus de leur moment magnétique, un momentquadrupolaire électrique non nul dû à une distribution non sphérique des charges du noyau. Lesquadrupoles vont interagir avec le gradient de champ électrique local présent autour du noyau,ce qui aura pour effet, comme pour les interactions précédentes, d’affecter les niveaux d’énergiedes spins nucléaires.
Le couplage quadrupolaire, le déplacement chimique et le couplage dipolaire sont des in-teractions anisotrope qui sont moyennées à leur valeur isotrope (0 dans le cas du dipolaire) enRMN du liquide du fait des mouvements browniens moléculaires.
Les interactions anisotropes (δ, couplage dipolaire et quadripolaire) ne sont donc pas moyen-nées et dominent le spectre de RMN du solide. De plus, un autre problème présent en RMN estson manque de sensibilité pour certains noyaux : les protons possèdent une assez bonne sensibi-lité de détection, ce qui n’est pas le cas des hétéronoyaux tels que les carbones et les azotes. Parconséquent, la mise au point de techniques permettant de réduire la largeur des raies et d’aug-menter la sensibilité, a été nécessaire. Les plus importantes de ces techniques sont : la RMNavec rotation à l’angle magique (MAS-ssNMR pour magic-angle spinning solid state NMR) etla Polarisation croisée (CP pour Cross-Polarization). Les techniques essentielles de la ssRMN
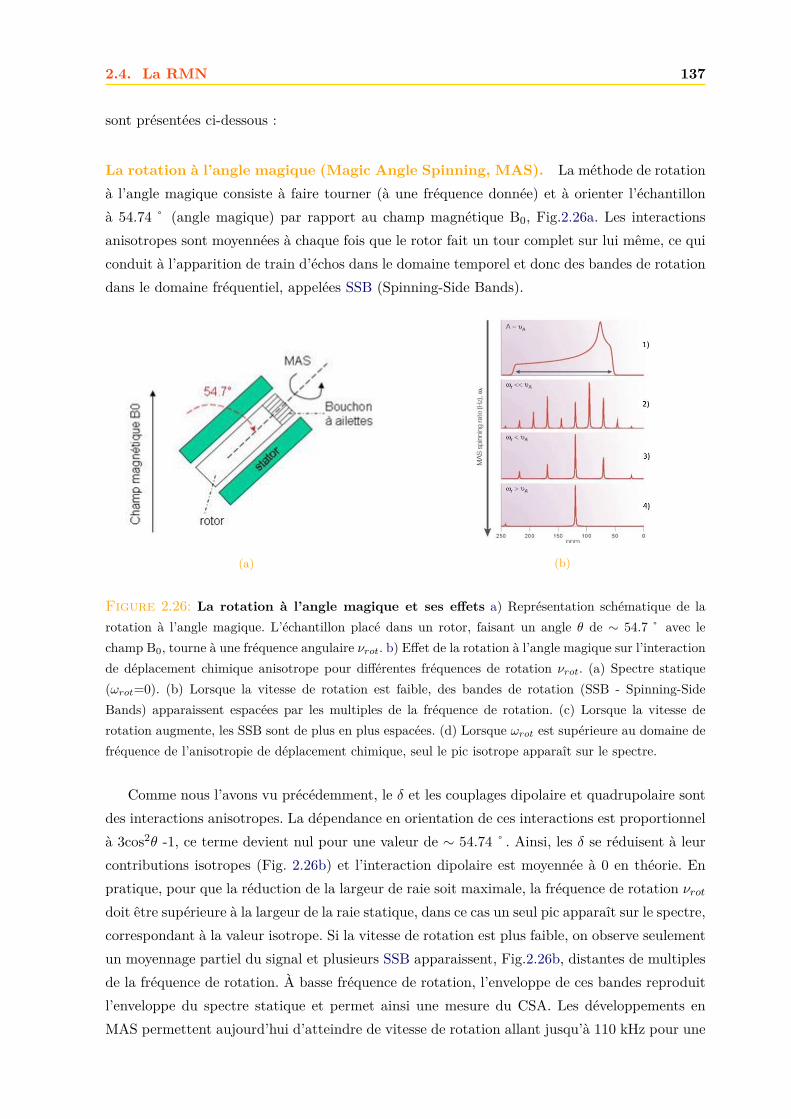
2.4. La RMN 137
sont présentées ci-dessous :
La rotation à l’angle magique (Magic Angle Spinning, MAS). La méthode de rotationà l’angle magique consiste à faire tourner (à une fréquence donnée) et à orienter l’échantillonà 54.74˚ (angle magique) par rapport au champ magnétique B0, Fig.2.26a. Les interactionsanisotropes sont moyennées à chaque fois que le rotor fait un tour complet sur lui même, ce quiconduit à l’apparition de train d’échos dans le domaine temporel et donc des bandes de rotationdans le domaine fréquentiel, appelées SSB (Spinning-Side Bands).
(a) (b)
Figure 2.26: La rotation à l’angle magique et ses effets a) Représentation schématique de larotation à l’angle magique. L’échantillon placé dans un rotor, faisant un angle θ de ∼ 54.7˚ avec lechamp B0, tourne à une fréquence angulaire νrot. b) Effet de la rotation à l’angle magique sur l’interactionde déplacement chimique anisotrope pour différentes fréquences de rotation νrot. (a) Spectre statique(ωrot=0). (b) Lorsque la vitesse de rotation est faible, des bandes de rotation (SSB - Spinning-SideBands) apparaissent espacées par les multiples de la fréquence de rotation. (c) Lorsque la vitesse derotation augmente, les SSB sont de plus en plus espacées. (d) Lorsque ωrot est supérieure au domaine defréquence de l’anisotropie de déplacement chimique, seul le pic isotrope apparaît sur le spectre.
Comme nous l’avons vu précédemment, le δ et les couplages dipolaire et quadrupolaire sontdes interactions anisotropes. La dépendance en orientation de ces interactions est proportionnelà 3cos2θ -1, ce terme devient nul pour une valeur de ∼ 54.74˚. Ainsi, les δ se réduisent à leurcontributions isotropes (Fig. 2.26b) et l’interaction dipolaire est moyennée à 0 en théorie. Enpratique, pour que la réduction de la largeur de raie soit maximale, la fréquence de rotation νrotdoit être supérieure à la largeur de la raie statique, dans ce cas un seul pic apparaît sur le spectre,correspondant à la valeur isotrope. Si la vitesse de rotation est plus faible, on observe seulementun moyennage partiel du signal et plusieurs SSB apparaissent, Fig.2.26b, distantes de multiplesde la fréquence de rotation. À basse fréquence de rotation, l’enveloppe de ces bandes reproduitl’enveloppe du spectre statique et permet ainsi une mesure du CSA. Les développements enMAS permettent aujourd’hui d’atteindre de vitesse de rotation allant jusqu’à 110 kHz pour une

138 Chapitre 2. Matériels et Méthodes
sonde conçue pour des rotors 0.7 mm. De telles vitesse ne peuvent être atteindre qu’avec desrotors de petits volume ce qui limite considérablement la quantité de matériel utilisé et doncla sensibilité, ce qui peut être très pénalisant pour certains échantillons biologiques. Pour detels systèmes, cette stratégie peut être gagnante si la perte de sensibilité due à la réduction duvolume est compensée par le gain en sensibilité induit par l’augmentation de la fréquence derotation. Le couplage dipolaire homonucléaire pour 1H est de ∼ 50 kHz, la vitesse de rotationn’est pas encore assez grande pour le moyenner. Au contraire, pour le 13C, le couplage dipolairehomonucléaire 13C-13C est faible à cause de la distance entre les noyaux 13C et de leur γ pluspetit.
La polarisation croisée (Cross polarization - CP). L’interaction dipolaire hétéronu-cléaire est source de perte de résolution et on cherche le plus souvent à l’éliminer. Cependant,elle peut être utilisée avantageusement dans une expérience de polarisation croisée qui, combi-née à la MAS, est appelée CP-MAS. C’est une technique permettant un gain en sensibilité enmesurant le signal d’un noyau peu sensible (typiquement 13C ou 15N) par l’intermédiaire d’unnoyau abondant voisin (typiquement 1H). Il s’agit d’exciter le noyau abondant et de faire untransfert de polarisation, via le couplage dipolaire, vers le noyau "dilué" que l’on veut observer,Fig. 2.27.
Figure 2.27: Séquence d’impulsions pour l’expérience 13C CP-MAS. P3 : durée de l’impulsion90˚ 1H à la puissance PL2 ; P15 : temps de contact aux puissance PL1 pour le 13C et SP0 pour le 1H ;P31 : durée du pulse du découplage à la puissance PL13.
Ce transfert de polarisation est d’autant plus efficace que les distances sont courtes et quela liaison est rigide entre les deux noyaux. Elle permet non seulement un gain en sensibilitéimportant, mais aussi en temps expérimental car les temps de répétition exploité est celui dunoyau abondant. En MAS, pour un transfert efficace entre 2 spins 1/2, I et S, il faut satisfairela condition de Hartmann-Hahn à savoir : ωI = ωS ± nωr soit γIBS = γSBI ± nωr avec BI etBS les valeurs des champs RF appliqués aux fréquences respectives de I et S, n un entier etωr la fréquence de rotation de l’échantillon à l’angle magique. Les spins sont alors soumis, dansleurs référentiels respectifs, au même champ effectif. Le gain en sensibilité est égale à γI
γS.

2.4. La RMN 139
Dans le cas d’un transfert de polarisation du 1H vers le 13C, le rapport signal sur bruit estmultiplié par 4 ( γ1H
γ13C= 4). De plus, le temps de relaxation longitudinale (T1) du 1H étant très
inférieure à celui du 13C, ceci implique que les expériences de CP sont plus courtes que cellesd’excitation directe des 13C.
Figure 2.28: Condition de Hartmann-Hahn pour la polarisation croisée.
Le découplage hétéronucléaire Le couplage dipolaire hétéronucléaire avec les protons voi-sins est généralement l’interaction qui contribue majoritairement à la largeur de raie observéepour un spin dilué comme le 13C. La rotation à l’angle magique, en intervenant sur la partiespatiale des interactions, permet de moduler les effets de leur anisotropie. Cependant, lorsqueles interactions sont fortes, elles ne peuvent pas être complètement moyennées par le MAS. Onpeut alors utiliser un découplage hétéronucléaire qui, lui, va agir sur les interactions. Les typesde découplage les plus utilisés sont le découplage par onde continue (CW) et les découplagesmulti-impulsionnels, p. ex. TPPM, SPINAL, XiX.
L’expérience PDSD La PDSD (Proton-Driven Spin Diffusion) est l’expérience standarden ssNMR et peut être considérée comme l’équivalent à l’état solide de l’HSQC (l’impulsion90˚ initial de la séquence d’impulsions HSQC en RMN du liquide est ici remplacée par uneCP). C’est la 1re expérience qu’il faut enregistrer sur son échantillon de protéine pour évaluerla qualité générale des spectres qu’il sera possible d’obtenir avec cet échantillon. Elle consisteen un transfert de polarisation des noyaux 1H au 13C. De là, la polarisation est transférée àd’autres noyaux 13C qui sont proches dans l’espace, Fig. 2.29a. Comme son nom le suggère, lesprotons sont impliqués dans ce transfert (bien que cela ne soit pas spécifiquement indiqué surle schéma).
Sur le spectre apparaitra un pic croisé pour tous les atomes 13C qui sont corrélés les unsaux autres jusqu’à une certaine distance. Cela signifie qu’avec des temps de mélange courttous les noyaux 13C au sein d’un même résidu seront couplés les uns aux autres (corrélationsintrarésiduelles). En augmentant la durée du temps de mélange les corrélations à plus longueéchelle apparaitront, c.-à-d. des corrélations entre les résidus.
Expériences NCA et NCO Lors de l’étude de la mTSPO1 reconstituée en liposomes, 2expériences ont été réalisées pour étudier la faisabilité de l’attribution des résonances de la

140 Chapitre 2. Matériels et Méthodes
(a) (b)
Figure 2.29: L’expérience PDSD a) Transferts de polarisations entre les noyaux. Les noyaux indiquésen rouge sont ceux dont le δ est mesuré ; les noyaux indiqués en bleu sont impliqués dans les transfertsde polarisation mais leur δ n’est pas mesuré. b) Séquence d’impulsions. La CP permet le transfert depolarisation/aimantation vers les noyaux X qui sont alors excités. Pendant le temps d’évolution (t1)le δ évolue sous les effets des interactions. Cette période prend fin avec une impulsion 90˚ qui metl’aimantation le long de l’axe z. Le temps de mélange (mixing) permet aux noyaux X de communiquerà travers les protons par diffusion de spin. Puis, une autre impulsion 90˚ bascule l’aimantation dans leplan transverse et le FID est acquis. Des découplages sont appliqués pendant t2 et l’enregistrement dusignal pour éliminer les interactions dipolaires 1H-X.
protéine. Ces expériences, NCA et NCO, se basent sur un double transfert de polarisation,Fig. 2.30a. Dans ces expériences, la magnétisation est transférée des 1H au 15N via la CP puissélectivement sur les 13Cα ou sur les 13CO en utilisant une CP spécifique. L’expérience NCAfournit des corrélations intramoléculaires (Ni,Ci) et l’expérience NCO permet d’obtenir les liensséquentielles entre le Ni et le COi−1.
2.4.5.2 Remplissage du rotor
Pour la RMN du solide, les protéoliposomes doivent être concentrés par centrifugation. Ils’agit d’une étape critique, en effet le remplissage du rotor (de 2.5, 3.2 ou 4 mm de diamètre)avec un maximum d’échantillon reconstitué fait parti des difficultés rencontrées lorsque l’onsouhaite faire de la RMN du solide avec des PMb. Celui-ci s’est révélé être un véritable challengelors de cette thèse. Différents protocoles sont décrits dans la littérature et tous se basent surl’obtention d’un culot de protéoliposomes par centrifugation. Plusieurs dispositifs sont utiliséspour introduire le culot dans le rotor :
— Introduction manuelle du culot dans le rotor en utilisant la pointe d’une spatule puis lacentrifugation du rotor, placé dans un tube de 1.5 ml, à 10 000 × g pendant 1 min à Tamb.Cette opération est répétée jusqu’à ce que ∼ 30 mg d’échantillon soit introduits dans lerotor [Abdine et al., 2012].

2.4. La RMN 141
(a) (b)
(c)
Figure 2.30: Transferts de polarisations entre les noyaux dans les expériences a) NCA et b)NCO et c) séquence d’impulsions NCA/NCO. Une CP permet le transfert de magnétisation des1H au 15N. Après un temps d’évolution t1, la magnétisation est transférée des 15N au 13Cα ou 13CO enutilisant une CP spécifique. Pour ce faire la fréquence de la porteuse 15N est mise proche des fréquencesdes 13Cα ou des 13CO durant la période de transfert ou bien entre les 2 pour détecter les 2 régions à lafois. Finalement le FID est acquis dans la dimension 13C.
— Assemblage "fait maison" de tubes et cônes de pipette, Fig.2.31a [Das et al., 2013].
— Un dispositif de centrifugation "fait maison", Fig 2.31b [Bockmann et al., 2009].
Lors de ma thèse plusieurs méthodes ont été testées pour réussir à remplir correctement unrotor (ie : que 100 % de la protéine reconstituée soit introduit dans le rotor MAS). Cette étapes’est révélée être une étape critique pour l’étude de la protéine en ssNMR.
Quoi qu’il en soit, que l’on parte d’un grand ou d’un petit volume de reconstitution, au finalla façon la plus simple de remplir le rotor est d’obtenir le culot dans un tube Eppendorff de 1.5ml.
Dans notre cas, la nature gluante des protéoliposomes a rendu presque impossible le pipet-tage ou l’utilisation d’une spatule, comme décrit par [Abdine et al., 2012], pour le transfertdu culot dans le rotor MAS sans que l’on perde une grande partie de notre échantillon quis’effrite en petits morceaux qui restent collés sur les parois du tube ou de la pipette. Nousavons construit différents dispositifs, en s’inspirant de la méthode de remplissage présentée parDas et al. [2013]. Ne disposant pas des mêmes outils que les auteurs, il n’a pas été possible dereproduire à l’identique leur méthode. En particulier nous ne disposons pas au laboratoire desmêmes tubes de centrifugation et des centrifugeuses qui vont avec.
La stratégie de remplissage utilisée lors de cette étude est présentée Fig.2.33. Lors des pre-
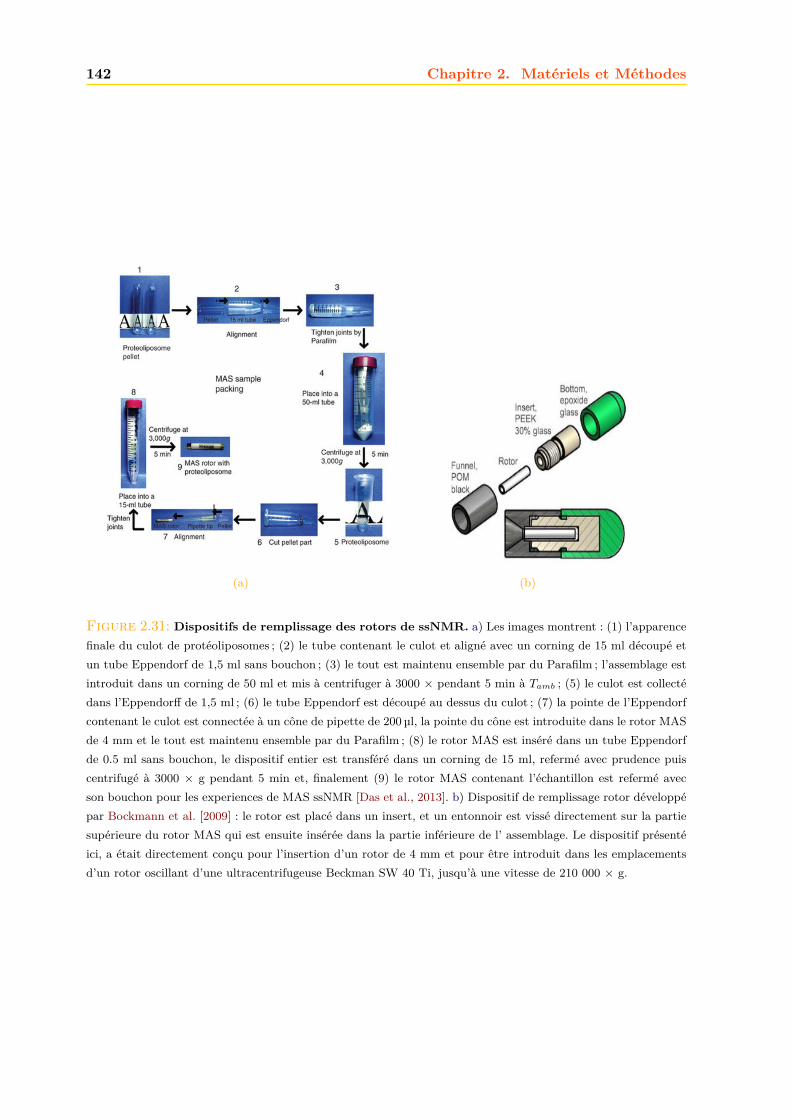
142 Chapitre 2. Matériels et Méthodes
(a) (b)
Figure 2.31: Dispositifs de remplissage des rotors de ssNMR. a) Les images montrent : (1) l’apparencefinale du culot de protéoliposomes ; (2) le tube contenant le culot et aligné avec un corning de 15 ml découpé etun tube Eppendorf de 1,5 ml sans bouchon ; (3) le tout est maintenu ensemble par du Parafilm ; l’assemblage estintroduit dans un corning de 50 ml et mis à centrifuger à 3000 × pendant 5 min à Tamb ; (5) le culot est collectédans l’Eppendorff de 1,5 ml ; (6) le tube Eppendorf est découpé au dessus du culot ; (7) la pointe de l’Eppendorfcontenant le culot est connectée à un cône de pipette de 200 µl, la pointe du cône est introduite dans le rotor MASde 4 mm et le tout est maintenu ensemble par du Parafilm ; (8) le rotor MAS est inséré dans un tube Eppendorfde 0.5 ml sans bouchon, le dispositif entier est transféré dans un corning de 15 ml, refermé avec prudence puiscentrifugé à 3000 × g pendant 5 min et, finalement (9) le rotor MAS contenant l’échantillon est refermé avecson bouchon pour les experiences de MAS ssNMR [Das et al., 2013]. b) Dispositif de remplissage rotor développépar Bockmann et al. [2009] : le rotor est placé dans un insert, et un entonnoir est vissé directement sur la partiesupérieure du rotor MAS qui est ensuite insérée dans la partie inférieure de l’ assemblage. Le dispositif présentéici, a était directement conçu pour l’insertion d’un rotor de 4 mm et pour être introduit dans les emplacementsd’un rotor oscillant d’une ultracentrifugeuse Beckman SW 40 Ti, jusqu’à une vitesse de 210 000 × g.

2.4. La RMN 143
mières reconstitutions, nous partions d’un grand volume 40-50 ml à 0.1-0.2 mg/ml de protéine,ce qui nous permettais de suivre aisément la fluorescence et l’absorbance avec les appareils dis-ponibles au laboratoire en utilisant des cuves de 1 cm de chemin optique et une contenance de 2ou 1 ml. Un tel volume à cette concentration permettait d’espérer de remplir théoriquement lerotor de ∼5-10 mg de protéine reconstituée. Puis, une fois la reconstitution terminée, la solutionde protéoliposomes est centrifugée, le culots est repris dans un volume manipulable de tampon,puis la solution est re-centrifugée dans un tube Eppendorf de 1.5 ml. L’extrémité de ce tubecontenant le culot est découpée avec un cutter et insérée sur un cône de pipette de 1 ml, luiaussi légèrement découpé à son extrémité pour s’ajuster à l’Eppendorf, le tout est maintenu parun parafilm. Une autre option est d’insérer un cône de 2 ml (dans ce cas le cône est légèrementà l’intérieur de l’Eppendorf). Dans tous les cas les 2 types de cônes s’ajustent à l’intérieur (undécoupage peut être nécessaire) du rotor ssNMR de 4 mm. L’ensemble du dispositif fait maisonest placé dans un corning de 15 ml puis centrifugé. Le problème de ce dispositif est que de nom-breuses fuites se produisent. De plus la résistance des tubes ne permet pas de les centrifugertrès rapidement (vitesse maximale de 3000 à 20000 × g selon la marque).
La quantification de l’efficacité du remplissage du rotor par cette technique est très difficile.Une tentative en pipetant directement dans le rotor s’est avéré peu efficace. Nous avons essayéd’estimer la quantité de lipides dans un rotor HRMAS (High Resolution Magic Angle Spinning)par RMN du proton, puis d’utiliser le LPR connu pour remonter à la concentration en protéineà l’intérieur du rotor.
Une autre technique de remplissage a été testée en s’inspirant de Bockmann et al. [2009].En effet, nous avons obtenu par Luminita Duma, avec qui nous avons colaborée sur cetet étude,un dispositif de remplissage de rotor par centrifugation comme celui utilisé par Bockmann et al.[2009]. Il s’agissait d’un dispositif destiné à un rotor 3 mm, il a été nécessaire de l’envoyer àl’atelier de l’Université pour le rendre utilisable avec nos rotors de 4 mm et les tubes de notreultra-centrifugeuse. L’utilisation de ce dispositif 11 a permis de rendre le protocole de remplissagede rotor beaucoup plus simple et d’assurer un meilleur transfert du culot de protéoliposomesde l’échantillon à l’intérieur du rotor. La Fig.2.35 illustre le protocole de remplissage avec cenouveau dispositif.
2.4.5.3 La RMN du solide en pratique
Réglages et optimisations Avant de pouvoir commencer à acquérir les spectres ssNMR surnotre échantillon de protéolipomes de mTSPO1 de nombreux réglages et optimisations ont dûêtre réalisés.
1. Tout d’abord, le réglage de l’angle magique sur un échantillon de KBr. Celui ci est choisicar le 79Br a une fréquence de résonance très proche de celle du 13C et en tant que noyauquadrupolaire il est très sensible aux défauts de réglage de l’angle magique.
11. D’ailleurs un dispositif similaire est depuis peu commercialiser.
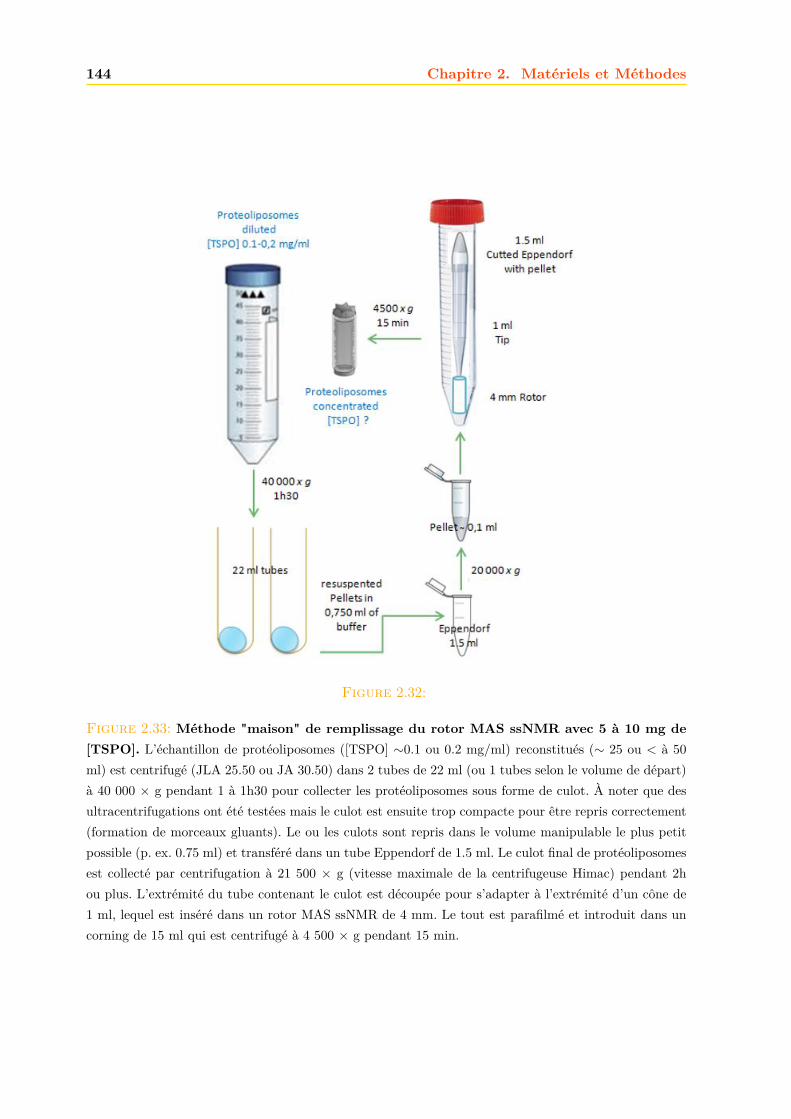
144 Chapitre 2. Matériels et Méthodes
Figure 2.32:
Figure 2.33: Méthode "maison" de remplissage du rotor MAS ssNMR avec 5 à 10 mg de[TSPO]. L’échantillon de protéoliposomes ([TSPO] ∼0.1 ou 0.2 mg/ml) reconstitués (∼ 25 ou < à 50ml) est centrifugé (JLA 25.50 ou JA 30.50) dans 2 tubes de 22 ml (ou 1 tubes selon le volume de départ)à 40 000 × g pendant 1 à 1h30 pour collecter les protéoliposomes sous forme de culot. À noter que desultracentrifugations ont été testées mais le culot est ensuite trop compacte pour être repris correctement(formation de morceaux gluants). Le ou les culots sont repris dans le volume manipulable le plus petitpossible (p. ex. 0.75 ml) et transféré dans un tube Eppendorf de 1.5 ml. Le culot final de protéoliposomesest collecté par centrifugation à 21 500 × g (vitesse maximale de la centrifugeuse Himac) pendant 2hou plus. L’extrémité du tube contenant le culot est découpée pour s’adapter à l’extrémité d’un cône de1 ml, lequel est inséré dans un rotor MAS ssNMR de 4 mm. Le tout est parafilmé et introduit dans uncorning de 15 ml qui est centrifugé à 4 500 × g pendant 15 min.

2.4. La RMN 145
Figure 2.34:
Figure 2.35: Autre méthode de remplissage du rotor MAS ssNMR avec un nouveau dispo-sitif. L’échantillon de protéoliposomes ([TSPO] ∼2 ou 3 mg/ml) reconstitués (∼ 4-5 ml) est centrifugé(HIMAC) dans un tube Eppendorf de 1.5 ml à 21 500 × g toute la nuit pour collecter les protéoliposomessous forme de culot. Puis le tube contenant le culot de protéoliposomes est plongé dans de l’azote liquidepour être congelé. Le culot congelé se décolle ensuite facilement du tube et il est rapidement inséré dansle dispositif de remplissage du rotor avec une spatule. Ce dispositif est mis dans un corning de 15 ml etcentrifugé à 4 500 × g pendant 15 min ou plus à 20 ◦C pour faire "fondre" et descendre le culot dans lerotor 4 mm ssNMR.
2. Ensuite le réglage de la CP entre 1H et 13C. Les impulsions doivent être calibrées pourla CP, pour cela un échantillon d’adamantane doublement marqué est utilisé comme ré-férence, cela se fait en plusieurs étapes :
(a) Calibration de l’impulsion 1H 90˚ (P3 et PL2).
(b) Calibration de l’impulsion 13C 90˚ (P1 et PL11).
(c) ajustement de la puissance du 13C pour satisfaire la condition de Hartmann-Hahn(HH). Pour atteindre la condition de HH, les champs RF des 1H et des 13C doiventdifférer par la fréquence de rotation. La condition HH nécessaire pour la CP esttrès sensible. Cette sensibilité signifie que même des erreurs de réglages mineures ouune instabilité instrumentale peut conduire à une perte dramatique du signal. Unefaçon de surmonter cette sensibilité est d’utiliser une rampe d’impulsion montantesur le noyau 1H. Ainsi la puissance correspondante à la condition de HH est répartiesur une plus grande zone et devient insensible aux petites erreurs et aux différentesvitesses de rotation.
3. Il faut ensuite procéder au réglage de la CP entre 1H et 15N. Cela se fait comme décritci-dessus pour la CP 1H et 13C sauf que l’échantillon de référence utilisé est un échantillonde glycine doublement marqué.
4. Pour les expériences NCA et NCO , il est nécessaire d’optimiser toutes les durées etles puissances des impulsions des 2 CPs (CP 1H-15N et CP 15N-13C). Pour ce faire, unéchantillon de cristaux de la protéine GB1 est utilisé.
Les expériences ssRMN ont été réalisées avec Luminata DUMA (Université de Compiègne),Piotr Tekely et Rudra Purusottam (ENS). Pour les expériences ssRMN 1H un spectromètre

146 Chapitre 2. Matériels et Méthodes
"narrow-bore" 400 MHz Bruker AVANCE DRX avec une sonde 4 mm HR-MAS double résonance(1H,X) a été utilisé. Pour les expériences ssRMN 13C,15N ont été utilisés : un spectromètre"wide-bore" 400 MHz Bruker AVANCE DRX avec une sonde 4 mm CP-MAS triple résonance(1H,X,Y) ; et un spectromètre "narrow-bore" 500 MHz Bruker AVANCE III avec une sonde 4mm CP-MAS double résonance (1H,X).

Résultats et discussions


Chapitre3
La mTSPO1
Sommaire3.1 Production de la mTSPO1 recombinante . . . . . . . . . . . . . . . . . . 149
3.1.1 Expression dans E.coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 150
3.1.2 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
3.2 Caractérisation de la mTSPO1 recombinante en détergent . . . . . . . 157
3.2.1 Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
3.2.2 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 158
3.2.3 Repliement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 159
3.2.4 RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
3.3 Effets des ligands sur la mTSPO1 recombinante . . . . . . . . . . . . . . 162
3.3.1 Fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 163
3.3.2 Séparation des ligands libres et liés . . . . . . . . . . . . . . . . . . . . . . . 164
3.3.3 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
3.3.4 RMN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
3.3.5 Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
3.4 La mTSPO1 en RMN du Solide . . . . . . . . . . . . . . . . . . . . . . . . 172
3.4.1 Reconstitution de la mTSPO1 dans des liposomes . . . . . . . . . . . . . . 172
3.4.2 Remplissage du rotor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177
3.4.3 Expériences CP-MAS 1D 13C . . . . . . . . . . . . . . . . . . . . . . . . . . 182
3.4.4 Expériences 13C-13C PDSD . . . . . . . . . . . . . . . . . . . . . . . . . . . 189
3.4.5 Expériences 2D 15N-13C NCO et NCA . . . . . . . . . . . . . . . . . . . . . 190
3.4.6 Conclusions, discussions et perspectives . . . . . . . . . . . . . . . . . . . . 191
3.1 Production de la mTSPO1 recombinante
Les études structurales des PMb nécessitent une grande quantité de protéines (notammentmarquées) purifiées et concentrées. Cependant, la mTSPO1 est naturellement peu abondantedans les cellules, afin de palier ce problème, elle a été surexprimée sous la forme d’une protéinerecombinante en bactérie, puis purifiée en détergent sur colonne d’affinité grâce à son étiquettepoly-histidine [Li and Papadopoulos, 1998]. Le protocole est décrit de manière détaillé dans laSec. 2.1.7 du M&M. La mise au point du protocole d’expression et de purification de la protéine,non marquée ou marquée 15N,13C, pour les études structurales, a été réalisée lors des travauxde thèse de Soria Harbi Iatmanen [Harbi Iatmanen, 2014].
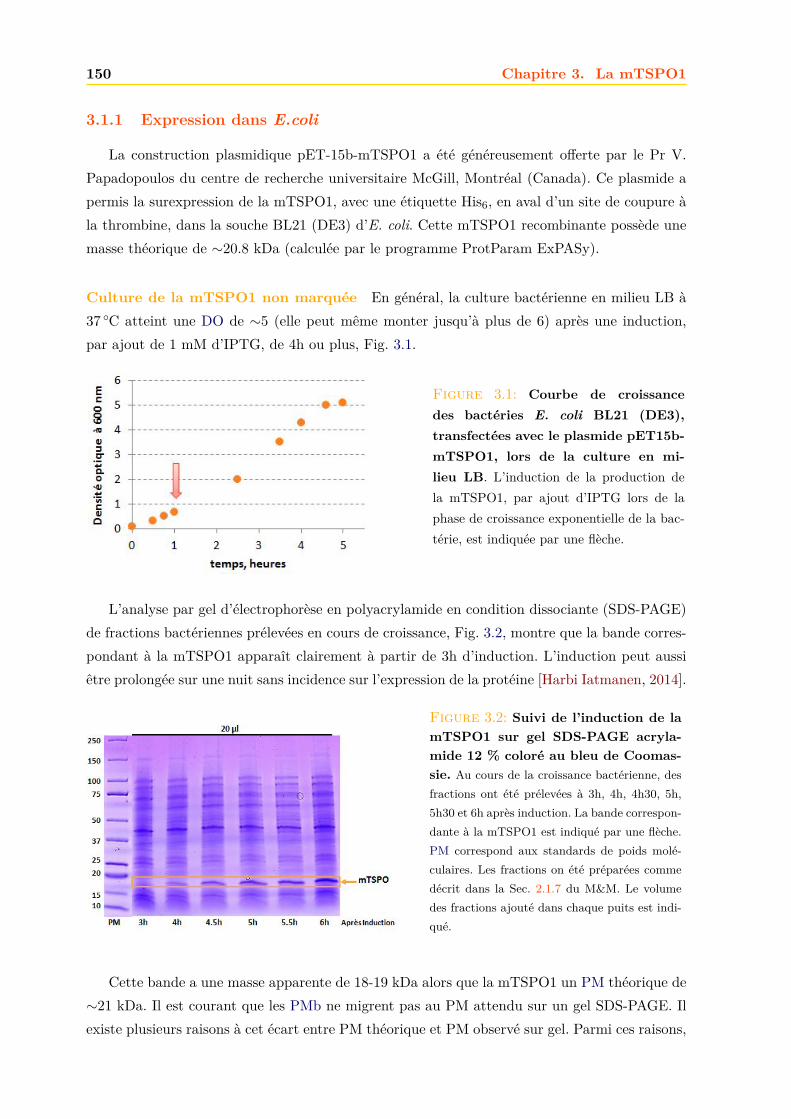
150 Chapitre 3. La mTSPO1
3.1.1 Expression dans E.coli
La construction plasmidique pET-15b-mTSPO1 a été généreusement offerte par le Pr V.Papadopoulos du centre de recherche universitaire McGill, Montréal (Canada). Ce plasmide apermis la surexpression de la mTSPO1, avec une étiquette His6, en aval d’un site de coupure àla thrombine, dans la souche BL21 (DE3) d’E. coli. Cette mTSPO1 recombinante possède unemasse théorique de ∼20.8 kDa (calculée par le programme ProtParam ExPASy).
Culture de la mTSPO1 non marquée En général, la culture bactérienne en milieu LB à37 ◦C atteint une DO de ∼5 (elle peut même monter jusqu’à plus de 6) après une induction,par ajout de 1 mM d’IPTG, de 4h ou plus, Fig. 3.1.
Figure 3.1: Courbe de croissancedes bactéries E. coli BL21 (DE3),transfectées avec le plasmide pET15b-mTSPO1, lors de la culture en mi-lieu LB. L’induction de la production dela mTSPO1, par ajout d’IPTG lors de laphase de croissance exponentielle de la bac-térie, est indiquée par une flèche.
L’analyse par gel d’électrophorèse en polyacrylamide en condition dissociante (SDS-PAGE)de fractions bactériennes prélevées en cours de croissance, Fig. 3.2, montre que la bande corres-pondant à la mTSPO1 apparaît clairement à partir de 3h d’induction. L’induction peut aussiêtre prolongée sur une nuit sans incidence sur l’expression de la protéine [Harbi Iatmanen, 2014].
Figure 3.2: Suivi de l’induction de lamTSPO1 sur gel SDS-PAGE acryla-mide 12 % coloré au bleu de Coomas-sie. Au cours de la croissance bactérienne, desfractions ont été prélevées à 3h, 4h, 4h30, 5h,5h30 et 6h après induction. La bande correspon-dante à la mTSPO1 est indiqué par une flèche.PM correspond aux standards de poids molé-culaires. Les fractions on été préparées commedécrit dans la Sec. 2.1.7 du M&M. Le volumedes fractions ajouté dans chaque puits est indi-qué.
Cette bande a une masse apparente de 18-19 kDa alors que la mTSPO1 un PM théorique de∼21 kDa. Il est courant que les PMb ne migrent pas au PM attendu sur un gel SDS-PAGE. Ilexiste plusieurs raisons à cet écart entre PM théorique et PM observé sur gel. Parmi ces raisons,
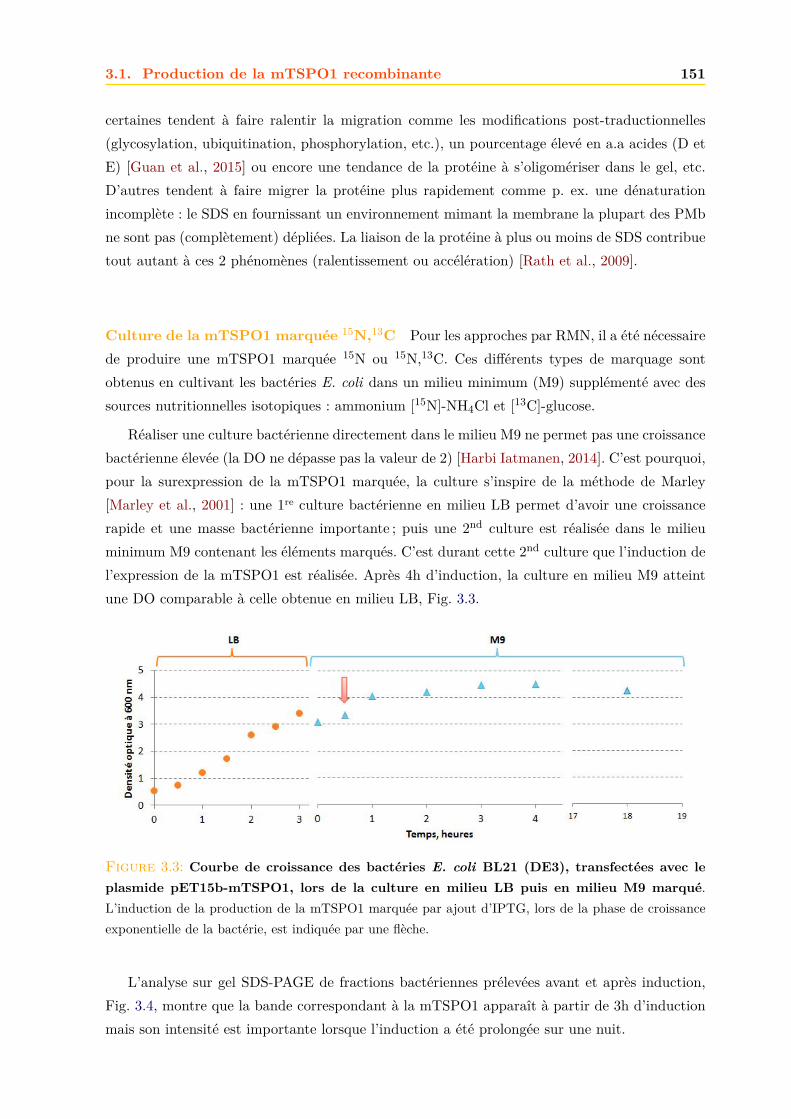
3.1. Production de la mTSPO1 recombinante 151
certaines tendent à faire ralentir la migration comme les modifications post-traductionnelles(glycosylation, ubiquitination, phosphorylation, etc.), un pourcentage élevé en a.a acides (D etE) [Guan et al., 2015] ou encore une tendance de la protéine à s’oligomériser dans le gel, etc.D’autres tendent à faire migrer la protéine plus rapidement comme p. ex. une dénaturationincomplète : le SDS en fournissant un environnement mimant la membrane la plupart des PMbne sont pas (complètement) dépliées. La liaison de la protéine à plus ou moins de SDS contribuetout autant à ces 2 phénomènes (ralentissement ou accélération) [Rath et al., 2009].
Culture de la mTSPO1 marquée 15N,13C Pour les approches par RMN, il a été nécessairede produire une mTSPO1 marquée 15N ou 15N,13C. Ces différents types de marquage sontobtenus en cultivant les bactéries E. coli dans un milieu minimum (M9) supplémenté avec dessources nutritionnelles isotopiques : ammonium [15N]-NH4Cl et [13C]-glucose.
Réaliser une culture bactérienne directement dans le milieu M9 ne permet pas une croissancebactérienne élevée (la DO ne dépasse pas la valeur de 2) [Harbi Iatmanen, 2014]. C’est pourquoi,pour la surexpression de la mTSPO1 marquée, la culture s’inspire de la méthode de Marley[Marley et al., 2001] : une 1re culture bactérienne en milieu LB permet d’avoir une croissancerapide et une masse bactérienne importante ; puis une 2nd culture est réalisée dans le milieuminimum M9 contenant les éléments marqués. C’est durant cette 2nd culture que l’induction del’expression de la mTSPO1 est réalisée. Après 4h d’induction, la culture en milieu M9 atteintune DO comparable à celle obtenue en milieu LB, Fig. 3.3.
Figure 3.3: Courbe de croissance des bactéries E. coli BL21 (DE3), transfectées avec leplasmide pET15b-mTSPO1, lors de la culture en milieu LB puis en milieu M9 marqué.L’induction de la production de la mTSPO1 marquée par ajout d’IPTG, lors de la phase de croissanceexponentielle de la bactérie, est indiquée par une flèche.
L’analyse sur gel SDS-PAGE de fractions bactériennes prélevées avant et après induction,Fig. 3.4, montre que la bande correspondant à la mTSPO1 apparaît à partir de 3h d’inductionmais son intensité est importante lorsque l’induction a été prolongée sur une nuit.

152 Chapitre 3. La mTSPO1
Figure 3.4: Suivi de l’induction de lamTSPO1 15N,13C sur gel SDS-PAGE acryla-mide 12 % coloré au bleu de Coomassie. Aucours de la croissance bactérienne, des fractions ontété prélevées, avant et après induction (à 0, 1, 2, 3 et18h). La bande correspondante à la mTSPO1 mar-quée est indiquée par une flèche. Une fraction demTSPO1 non marquée et purifiée a été égalementdéposé comme marqueur. PM, correspond aux stan-dards de poids moléculaires. Le volume des fractionsajouté dans chaque puits est indiqué.
3.1.2 Purification
3.1.2.1 Extraction des corps d’inclusion
La surproduction de la mTSPO1 dans les bactéries E. coli a pour conséquence son incorpo-ration majoritaire dans les corps d’inclusion (CI) [Robert and Lacapere, 2010].
Un taux d’expression élevé favorise les interactions hydrophobes intermoléculaires par rap-port aux interactions internes, et empêche souvent le repliement correct des protéines recombi-nantes qui finissent dans des agrégats insolubles, les CI [Georgiou and Valax, 1999]. Cet état aété longtemps considéré comme un inconvénient pour la purification de protéines recombinantes.
Toutefois, cette accumulation dans les CI présente un avantage pratique. Tout d’abord,la protéine peut être récupérée simplement par des séries de centrifugations et de lavages duculot. Ainsi, l’isolement de la protéine dans les CI peut constituer en elle-même une étape depurification [Marston and Hartley, 1990]. Ensuite la protéine est protégée contre la protéolyse.
La mTSPO1 a été extraite des CI par ajout de SDS - (Sodium Dodécyl Sulfate). Ce détergentfort, en fournissant un environnement hydrophobe semblable à celui présent dans les membranes,permet de maintenir la protéine soluble.
3.1.2.2 Chromatographie d’affinité Ni-NTA
Après extraction des CI, la mTSPO1, grâce à son étiquette His6 est purifiée manuellementpar chromatographie d’affinité sur résine Ni-NTA.
Minipurification Avant de procéder à la purification totale, une minipurification a été réa-lisée comme décrit Sec.2.2.3.1 du M&M . Celle-ci permet d’une part d’estimer la quantité demTSPO1 présente dans la solution à purifier et d’autre part d’ajuster le volume de résine à uti-liser pour ne pas perdre de protéine et pour obtenir des fractions collectées les plus concentréespossibles (la résine que nous utilisions ayant une capacité de ∼50 mg de protéines/ml).
Les différentes fractions obtenues, lors de cette minipurification, ont été analysées par spec-trométrie d’absorption (spectre entre 240 et 320/350 nm), Fig. 3.5a. Les 1res fractions collectées
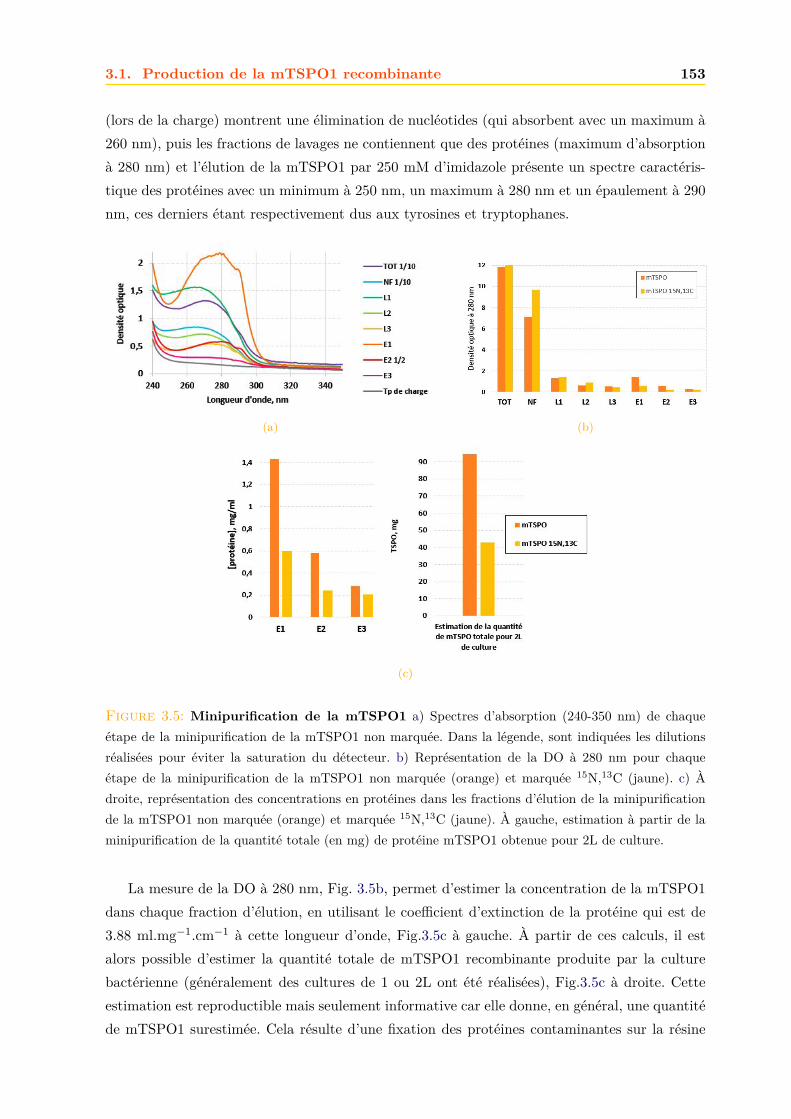
3.1. Production de la mTSPO1 recombinante 153
(lors de la charge) montrent une élimination de nucléotides (qui absorbent avec un maximum à260 nm), puis les fractions de lavages ne contiennent que des protéines (maximum d’absorptionà 280 nm) et l’élution de la mTSPO1 par 250 mM d’imidazole présente un spectre caractéris-tique des protéines avec un minimum à 250 nm, un maximum à 280 nm et un épaulement à 290nm, ces derniers étant respectivement dus aux tyrosines et tryptophanes.
(a) (b)
(c)
Figure 3.5: Minipurification de la mTSPO1 a) Spectres d’absorption (240-350 nm) de chaqueétape de la minipurification de la mTSPO1 non marquée. Dans la légende, sont indiquées les dilutionsréalisées pour éviter la saturation du détecteur. b) Représentation de la DO à 280 nm pour chaqueétape de la minipurification de la mTSPO1 non marquée (orange) et marquée 15N,13C (jaune). c) Àdroite, représentation des concentrations en protéines dans les fractions d’élution de la minipurificationde la mTSPO1 non marquée (orange) et marquée 15N,13C (jaune). À gauche, estimation à partir de laminipurification de la quantité totale (en mg) de protéine mTSPO1 obtenue pour 2L de culture.
La mesure de la DO à 280 nm, Fig. 3.5b, permet d’estimer la concentration de la mTSPO1dans chaque fraction d’élution, en utilisant le coefficient d’extinction de la protéine qui est de3.88 ml.mg−1.cm−1 à cette longueur d’onde, Fig.3.5c à gauche. À partir de ces calculs, il estalors possible d’estimer la quantité totale de mTSPO1 recombinante produite par la culturebactérienne (généralement des cultures de 1 ou 2L ont été réalisées), Fig.3.5c à droite. Cetteestimation est reproductible mais seulement informative car elle donne, en général, une quantitéde mTSPO1 surestimée. Cela résulte d’une fixation des protéines contaminantes sur la résine

154 Chapitre 3. La mTSPO1
plus importante que lors de la purification totale. Ces protéines contaminantes contribuantalors, tout comme la mTSPO1, au maximum à 280 nm. Expérimentalement, on a observéque la quantité de mTSPO1, après la purification totale, est au moins 2 fois plus faible quel’estimation faite à partir de la minipurification.
Sur la Fig.3.5, sont représentées à la fois les données de la minipurification de la mTSPO1marquée 15N,13C et non marquée. Une comparaison de ces données montre que pour le mêmevolume de culture, la production de la mTSPO1 marquée est environ 2 fois moindre que laproduction de la mTSPO1 non marquée.
Purification de la mTSPO1 non marquée Après la minipurification et une fois le volumede résine ajusté, le mélange issu de la solubilisation des CI est chargé sur la colonne de purifi-cation (Sec. 2.2.3.3 du M&M) et, comme pour la minipurification, le spectre d’absorption desdifférentes fractions est enregistré, Fig. 3.6a .
Pour contrôler l’efficacité de la purification les fractions collectées sont déposées sur gel SDS-PAGE. Les bandes protéiques présentes sont révélées par coloration au bleu de Coomassie ou àl’argent. Le gel de la Fig.3.6c nous montre la présence d’une bande correspondant à la mTSPO1dans les fractions d’élution.
Tout comme pour la minipurification, la quantité de protéine présente dans chaque fractioncollectée a été calculée à partir de la mesure de la DO280, Fig. 3.6b, et en utilisant le coefficientd’extinction de la mTSPO1. Ceci permet de tracer le chromatogramme de purification de laprotéine, Fig. 3.6d.
En fonction de la pureté et de la quantité de protéines, on sélectionne les fractions qui vontêtre gardées et regroupées pour être dialysées contre des tampons adéquats (sans imidazole,ni NaCl p. ex.). Le stock de mTSPO1 recombinante ainsi réalisé est conservé soit à 4 ◦C pourun usage rapide ou à -20 ◦C pour une conservation plus longue. La mTSPO1 purifiée en SDSest stable (pas de dégradation) plusieurs semaines à 4 ◦C et plusieurs années congelée à -20 ◦C[Harbi Iatmanen, 2014].
Au final, cette méthode de production nous a permis d’obtenir :
• une quantité de mTSPO1 recombinante autour de 40 mg/L de culture ;
• des stocks de protéines recombinantes concentrées : p. ex., à l’issue de la purification de lamTSPO1 présentée Fig. 3.6, le regroupement des fractions d’élution les plus concentrées(pic maximum à 16 mg/ml), nous a permis de former un stock de 1.2 ml à ∼12.5 mg/mlde protéines.
Cette concentration ainsi que la stabilité de la protéine sont tout à fait satisfaisantes et per-mettent de réaliser les différents tests biochimiques et biophysiques de caractérisation de laprotéine.
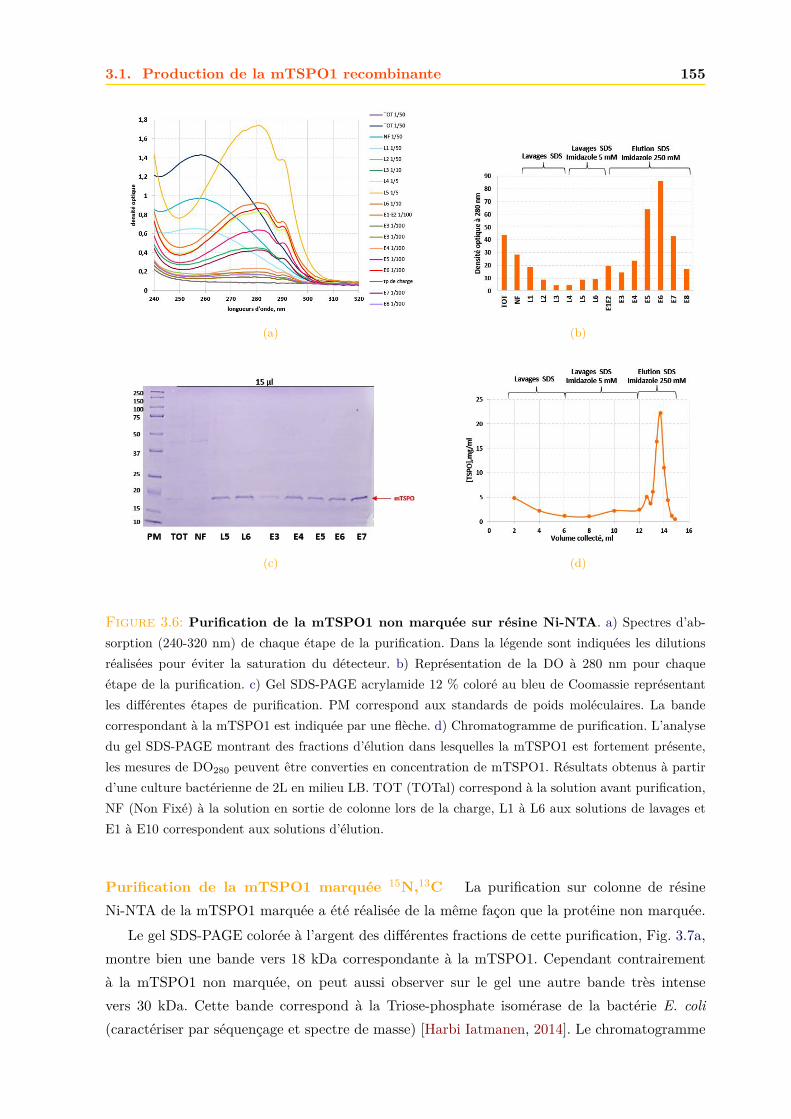
3.1. Production de la mTSPO1 recombinante 155
(a) (b)
(c) (d)
Figure 3.6: Purification de la mTSPO1 non marquée sur résine Ni-NTA. a) Spectres d’ab-sorption (240-320 nm) de chaque étape de la purification. Dans la légende sont indiquées les dilutionsréalisées pour éviter la saturation du détecteur. b) Représentation de la DO à 280 nm pour chaqueétape de la purification. c) Gel SDS-PAGE acrylamide 12 % coloré au bleu de Coomassie représentantles différentes étapes de purification. PM correspond aux standards de poids moléculaires. La bandecorrespondant à la mTSPO1 est indiquée par une flèche. d) Chromatogramme de purification. L’analysedu gel SDS-PAGE montrant des fractions d’élution dans lesquelles la mTSPO1 est fortement présente,les mesures de DO280 peuvent être converties en concentration de mTSPO1. Résultats obtenus à partird’une culture bactérienne de 2L en milieu LB. TOT (TOTal) correspond à la solution avant purification,NF (Non Fixé) à la solution en sortie de colonne lors de la charge, L1 à L6 aux solutions de lavages etE1 à E10 correspondent aux solutions d’élution.
Purification de la mTSPO1 marquée 15N,13C La purification sur colonne de résineNi-NTA de la mTSPO1 marquée a été réalisée de la même façon que la protéine non marquée.
Le gel SDS-PAGE colorée à l’argent des différentes fractions de cette purification, Fig. 3.7a,montre bien une bande vers 18 kDa correspondante à la mTSPO1. Cependant contrairementà la mTSPO1 non marquée, on peut aussi observer sur le gel une autre bande très intensevers 30 kDa. Cette bande correspond à la Triose-phosphate isomérase de la bactérie E. coli(caractériser par séquençage et spectre de masse) [Harbi Iatmanen, 2014]. Le chromatogramme
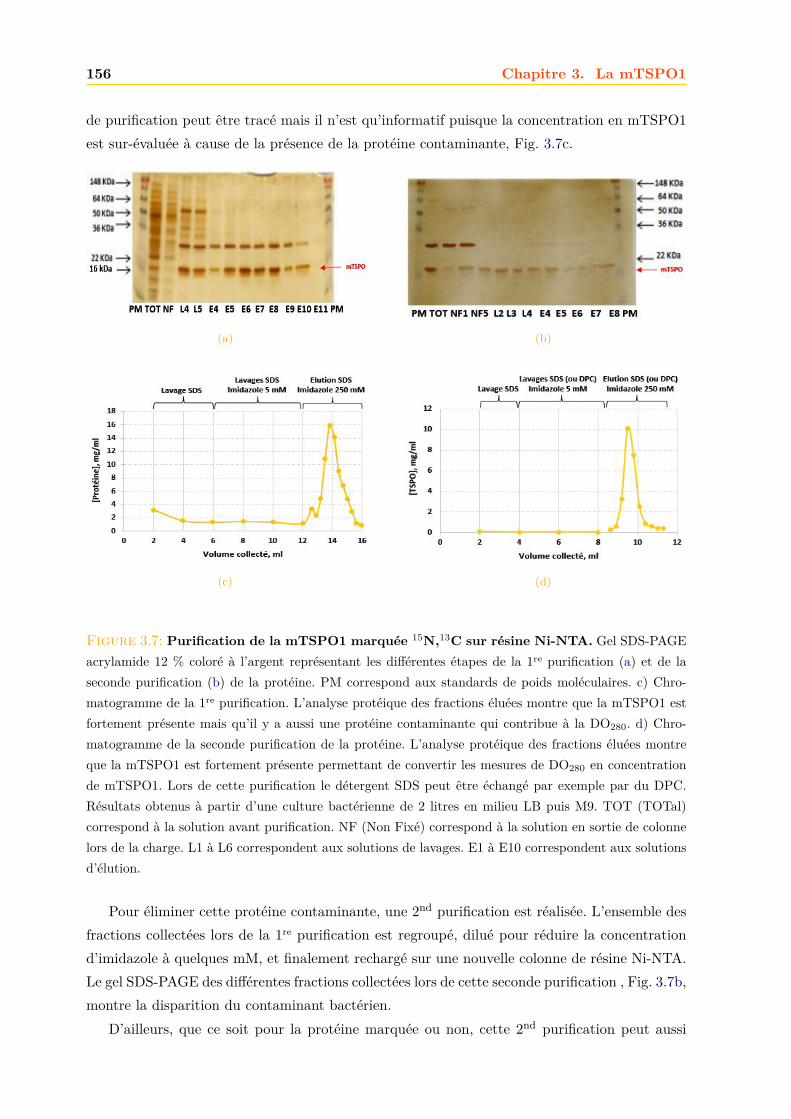
156 Chapitre 3. La mTSPO1
de purification peut être tracé mais il n’est qu’informatif puisque la concentration en mTSPO1est sur-évaluée à cause de la présence de la protéine contaminante, Fig. 3.7c.
(a) (b)
(c) (d)
Figure 3.7: Purification de la mTSPO1 marquée 15N,13C sur résine Ni-NTA. Gel SDS-PAGEacrylamide 12 % coloré à l’argent représentant les différentes étapes de la 1re purification (a) et de laseconde purification (b) de la protéine. PM correspond aux standards de poids moléculaires. c) Chro-matogramme de la 1re purification. L’analyse protéique des fractions éluées montre que la mTSPO1 estfortement présente mais qu’il y a aussi une protéine contaminante qui contribue à la DO280. d) Chro-matogramme de la seconde purification de la protéine. L’analyse protéique des fractions éluées montreque la mTSPO1 est fortement présente permettant de convertir les mesures de DO280 en concentrationde mTSPO1. Lors de cette purification le détergent SDS peut être échangé par exemple par du DPC.Résultats obtenus à partir d’une culture bactérienne de 2 litres en milieu LB puis M9. TOT (TOTal)correspond à la solution avant purification. NF (Non Fixé) correspond à la solution en sortie de colonnelors de la charge. L1 à L6 correspondent aux solutions de lavages. E1 à E10 correspondent aux solutionsd’élution.
Pour éliminer cette protéine contaminante, une 2nd purification est réalisée. L’ensemble desfractions collectées lors de la 1re purification est regroupé, dilué pour réduire la concentrationd’imidazole à quelques mM, et finalement rechargé sur une nouvelle colonne de résine Ni-NTA.Le gel SDS-PAGE des différentes fractions collectées lors de cette seconde purification , Fig. 3.7b,montre la disparition du contaminant bactérien.
D’ailleurs, que ce soit pour la protéine marquée ou non, cette 2nd purification peut aussi

3.2. Caractérisation de la mTSPO1 recombinante en détergent 157
permettre l’échange du SDS par un autre détergent (§ 2.2.3.3 du M&M). Cependant cet échangene peut se faire avec tous les détergents [Robert and Lacapere, 2010]. P. ex, le Triton X100 oul’octylglucoside ne permettent pas l’élution de la protéine alors que le DPC (Dodécyl Phospho-Choline) ou le DDM (Dodecylmaltoside ) en présence d’un peu de SDS le permettent [Harbi Iat-manen, 2014].
Après cette une 2nd purification, le chromatogramme de purification peut alors être tracé,Fig.3.7d, pour sélectionner les fractions les plus concentrées (la substitution du SDS par duDPC donne le même type de chromatogramme). Ces dernières sont dialysées, pour éliminerl’imidazole et les sels, dans un tampon adéquat pour les études en RMN (tampon NaPO4 10mM, pH 6.1-6.5, SDS ou DPC).
Au final, cette méthode de production nous a permis d’obtenir une protéine TSPO double-ment marquée 15N,13C en quantité et pureté suffisantes pour entreprendre les études structuralesde la protéine par RMN. Typiquement, on obtient une quantité de mTSPO1 marquée autour de15-20 mg/L de culture. À l’issu des 2 purifications successives, un stock d’∼1.2 ml à 7 mg/ml demTSPO1 recombinante 15N,13C est obtenu (pic maximum à 10 mg/ml sur le chromatogrammede la Fig.3.7d).
La spectrométrie de masse MALDI-TOF a permis de confirmer la production de la mTSPO1[Lacapere et al., 2014, Sagan and Bolbach, 2009]. La masse monochargée mesurée est de 20870Da, ce qui correspond bien à la mTSPO1 augmentée de son étiquette His6, mais pour laquellela méthionine (Met) en N-terminal a été perdue comme cela a déjà été décrit pour d’autresprotéines recombinantes [Fernandez-San Millan et al., 2007].
3.2 Caractérisation de la mTSPO1 recombinante en détergent
Le choix du détergent est une étape importante pour l’étude d’une PMb car celui-ci doitpréserver au mieux le repliement natif de la protéine. La mTSPO1 peut être purifiée en présencede différents détergents comme le SDS ou le DPC. Dans une 1re étape nous avons caractérisé lamTSPO1 dans ces deux environnements.
3.2.1 Fluorescence
La TSPO étant une protéine riche en tryptophane (Trp), nous avons donc utilisé sa fluores-cence intrinsèque pour la caractériser.
Les mesures de fluorescence intrinsèque de la mTSPO1 purifiée en micelles de SDS(mTSPO1/SDS ; SDS 2 cmc - 2 mg/ml), montre qu’un ajout de DPC (10 cmc - 4 mg/ml)entraîne une augmentation de ∼100% de la fluorescence, Fig. 3.8a. De même, la mTSPO1purifiée en micelles de DPC (mTSPO1/DPC ; 5 cmc - 2 mg/ml), montre une diminution de l’in-tensité de fluorescence de ∼100%, lorsque que du SDS est ajouté (5 cmc - 5 mg/ml), Fig. 3.8b.Ces changements de fluorescence sont donc complètement réversibles par ajout du détergent enquantité suffisante.
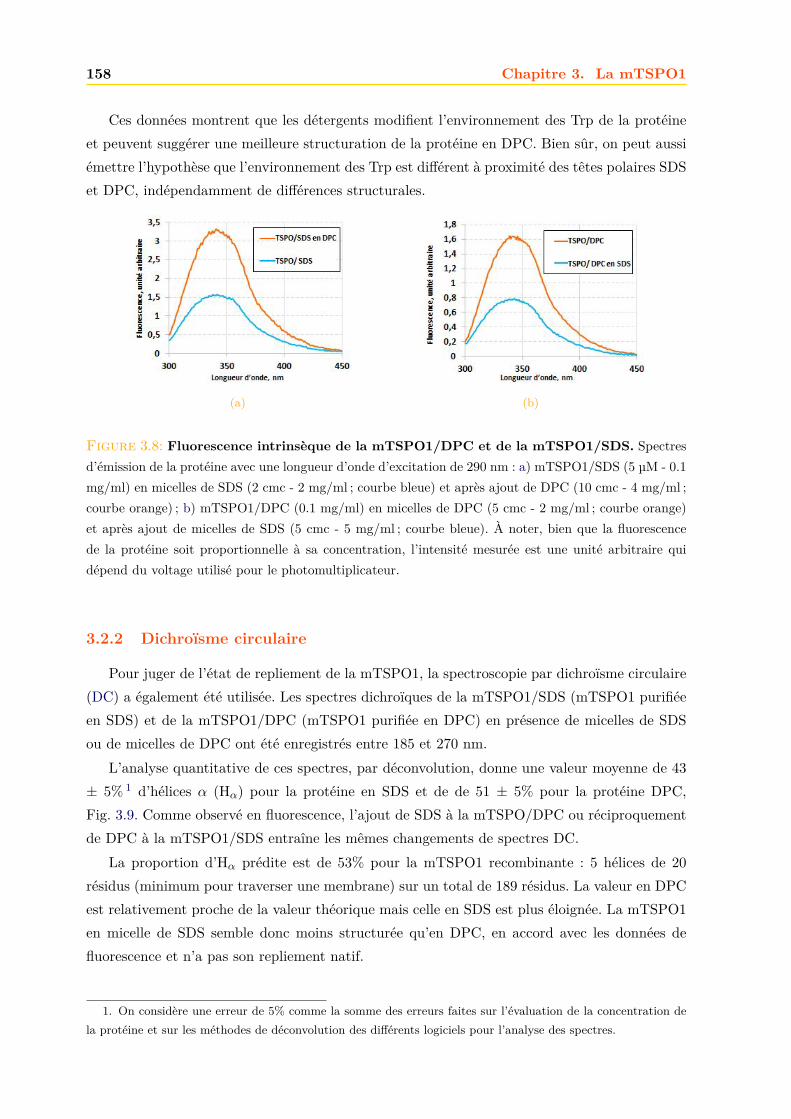
158 Chapitre 3. La mTSPO1
Ces données montrent que les détergents modifient l’environnement des Trp de la protéineet peuvent suggérer une meilleure structuration de la protéine en DPC. Bien sûr, on peut aussiémettre l’hypothèse que l’environnement des Trp est différent à proximité des têtes polaires SDSet DPC, indépendamment de différences structurales.
(a) (b)
Figure 3.8: Fluorescence intrinsèque de la mTSPO1/DPC et de la mTSPO1/SDS. Spectresd’émission de la protéine avec une longueur d’onde d’excitation de 290 nm : a) mTSPO1/SDS (5 µM - 0.1mg/ml) en micelles de SDS (2 cmc - 2 mg/ml ; courbe bleue) et après ajout de DPC (10 cmc - 4 mg/ml ;courbe orange) ; b) mTSPO1/DPC (0.1 mg/ml) en micelles de DPC (5 cmc - 2 mg/ml ; courbe orange)et après ajout de micelles de SDS (5 cmc - 5 mg/ml ; courbe bleue). À noter, bien que la fluorescencede la protéine soit proportionnelle à sa concentration, l’intensité mesurée est une unité arbitraire quidépend du voltage utilisé pour le photomultiplicateur.
3.2.2 Dichroïsme circulaire
Pour juger de l’état de repliement de la mTSPO1, la spectroscopie par dichroïsme circulaire(DC) a également été utilisée. Les spectres dichroïques de la mTSPO1/SDS (mTSPO1 purifiéeen SDS) et de la mTSPO1/DPC (mTSPO1 purifiée en DPC) en présence de micelles de SDSou de micelles de DPC ont été enregistrés entre 185 et 270 nm.
L’analyse quantitative de ces spectres, par déconvolution, donne une valeur moyenne de 43± 5% 1 d’hélices α (Hα) pour la protéine en SDS et de de 51 ± 5% pour la protéine DPC,Fig. 3.9. Comme observé en fluorescence, l’ajout de SDS à la mTSPO/DPC ou réciproquementde DPC à la mTSPO1/SDS entraîne les mêmes changements de spectres DC.
La proportion d’Hα prédite est de 53% pour la mTSPO1 recombinante : 5 hélices de 20résidus (minimum pour traverser une membrane) sur un total de 189 résidus. La valeur en DPCest relativement proche de la valeur théorique mais celle en SDS est plus éloignée. La mTSPO1en micelle de SDS semble donc moins structurée qu’en DPC, en accord avec les données defluorescence et n’a pas son repliement natif.
1. On considère une erreur de 5% comme la somme des erreurs faites sur l’évaluation de la concentration dela protéine et sur les méthodes de déconvolution des différents logiciels pour l’analyse des spectres.
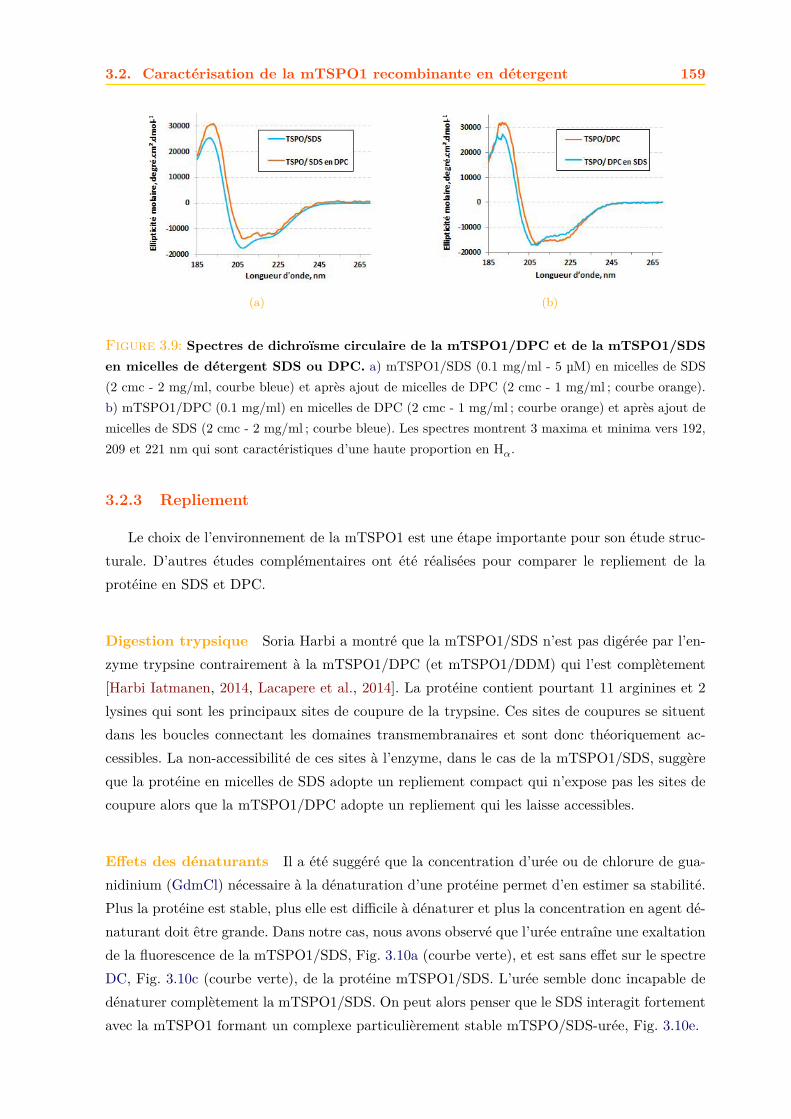
3.2. Caractérisation de la mTSPO1 recombinante en détergent 159
(a) (b)
Figure 3.9: Spectres de dichroïsme circulaire de la mTSPO1/DPC et de la mTSPO1/SDSen micelles de détergent SDS ou DPC. a) mTSPO1/SDS (0.1 mg/ml - 5 µM) en micelles de SDS(2 cmc - 2 mg/ml, courbe bleue) et après ajout de micelles de DPC (2 cmc - 1 mg/ml ; courbe orange).b) mTSPO1/DPC (0.1 mg/ml) en micelles de DPC (2 cmc - 1 mg/ml ; courbe orange) et après ajout demicelles de SDS (2 cmc - 2 mg/ml ; courbe bleue). Les spectres montrent 3 maxima et minima vers 192,209 et 221 nm qui sont caractéristiques d’une haute proportion en Hα.
3.2.3 Repliement
Le choix de l’environnement de la mTSPO1 est une étape importante pour son étude struc-turale. D’autres études complémentaires ont été réalisées pour comparer le repliement de laprotéine en SDS et DPC.
Digestion trypsique Soria Harbi a montré que la mTSPO1/SDS n’est pas digérée par l’en-zyme trypsine contrairement à la mTSPO1/DPC (et mTSPO1/DDM) qui l’est complètement[Harbi Iatmanen, 2014, Lacapere et al., 2014]. La protéine contient pourtant 11 arginines et 2lysines qui sont les principaux sites de coupure de la trypsine. Ces sites de coupures se situentdans les boucles connectant les domaines transmembranaires et sont donc théoriquement ac-cessibles. La non-accessibilité de ces sites à l’enzyme, dans le cas de la mTSPO1/SDS, suggèreque la protéine en micelles de SDS adopte un repliement compact qui n’expose pas les sites decoupure alors que la mTSPO1/DPC adopte un repliement qui les laisse accessibles.
Effets des dénaturants Il a été suggéré que la concentration d’urée ou de chlorure de gua-nidinium (GdmCl) nécessaire à la dénaturation d’une protéine permet d’en estimer sa stabilité.Plus la protéine est stable, plus elle est difficile à dénaturer et plus la concentration en agent dé-naturant doit être grande. Dans notre cas, nous avons observé que l’urée entraîne une exaltationde la fluorescence de la mTSPO1/SDS, Fig. 3.10a (courbe verte), et est sans effet sur le spectreDC, Fig. 3.10c (courbe verte), de la protéine mTSPO1/SDS. L’urée semble donc incapable dedénaturer complètement la mTSPO1/SDS. On peut alors penser que le SDS interagit fortementavec la mTSPO1 formant un complexe particulièrement stable mTSPO/SDS-urée, Fig. 3.10e.
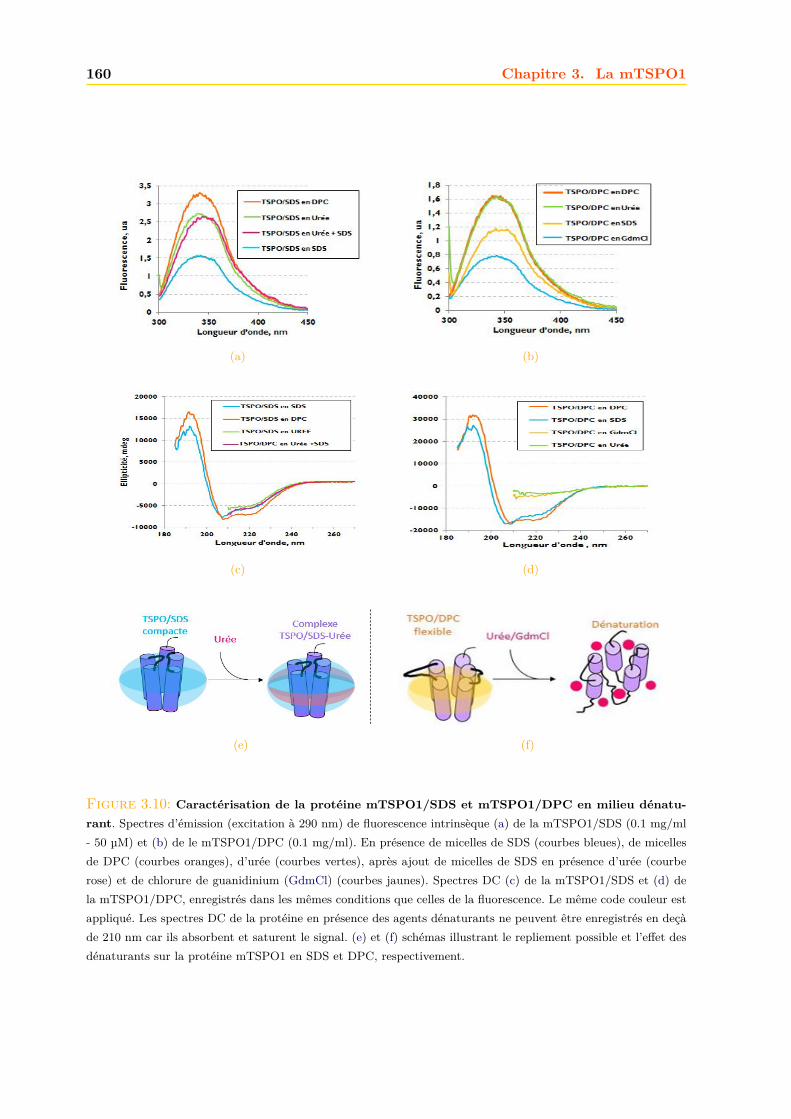
160 Chapitre 3. La mTSPO1
(a) (b)
(c) (d)
(e) (f)
Figure 3.10: Caractérisation de la protéine mTSPO1/SDS et mTSPO1/DPC en milieu dénatu-rant. Spectres d’émission (excitation à 290 nm) de fluorescence intrinsèque (a) de la mTSPO1/SDS (0.1 mg/ml- 50 µM) et (b) de le mTSPO1/DPC (0.1 mg/ml). En présence de micelles de SDS (courbes bleues), de micellesde DPC (courbes oranges), d’urée (courbes vertes), après ajout de micelles de SDS en présence d’urée (courberose) et de chlorure de guanidinium (GdmCl) (courbes jaunes). Spectres DC (c) de la mTSPO1/SDS et (d) dela mTSPO1/DPC, enregistrés dans les mêmes conditions que celles de la fluorescence. Le même code couleur estappliqué. Les spectres DC de la protéine en présence des agents dénaturants ne peuvent être enregistrés en deçàde 210 nm car ils absorbent et saturent le signal. (e) et (f) schémas illustrant le repliement possible et l’effet desdénaturants sur la protéine mTSPO1 en SDS et DPC, respectivement.
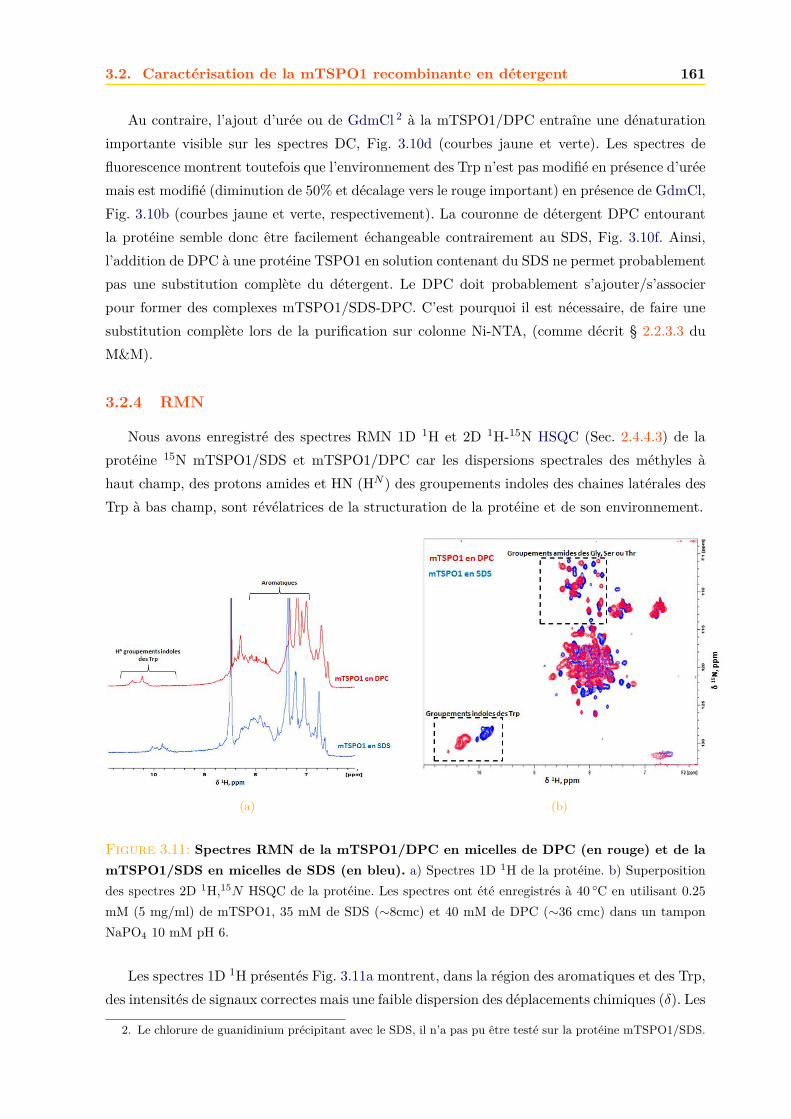
3.2. Caractérisation de la mTSPO1 recombinante en détergent 161
Au contraire, l’ajout d’urée ou de GdmCl 2 à la mTSPO1/DPC entraîne une dénaturationimportante visible sur les spectres DC, Fig. 3.10d (courbes jaune et verte). Les spectres defluorescence montrent toutefois que l’environnement des Trp n’est pas modifié en présence d’uréemais est modifié (diminution de 50% et décalage vers le rouge important) en présence de GdmCl,Fig. 3.10b (courbes jaune et verte, respectivement). La couronne de détergent DPC entourantla protéine semble donc être facilement échangeable contrairement au SDS, Fig. 3.10f. Ainsi,l’addition de DPC à une protéine TSPO1 en solution contenant du SDS ne permet probablementpas une substitution complète du détergent. Le DPC doit probablement s’ajouter/s’associerpour former des complexes mTSPO1/SDS-DPC. C’est pourquoi il est nécessaire, de faire unesubstitution complète lors de la purification sur colonne Ni-NTA, (comme décrit § 2.2.3.3 duM&M).
3.2.4 RMN
Nous avons enregistré des spectres RMN 1D 1H et 2D 1H-15N HSQC (Sec. 2.4.4.3) de laprotéine 15N mTSPO1/SDS et mTSPO1/DPC car les dispersions spectrales des méthyles àhaut champ, des protons amides et HN (HN ) des groupements indoles des chaines latérales desTrp à bas champ, sont révélatrices de la structuration de la protéine et de son environnement.
(a) (b)
Figure 3.11: Spectres RMN de la mTSPO1/DPC en micelles de DPC (en rouge) et de lamTSPO1/SDS en micelles de SDS (en bleu). a) Spectres 1D 1H de la protéine. b) Superpositiondes spectres 2D 1H,15N HSQC de la protéine. Les spectres ont été enregistrés à 40 ◦C en utilisant 0.25mM (5 mg/ml) de mTSPO1, 35 mM de SDS (∼8cmc) et 40 mM de DPC (∼36 cmc) dans un tamponNaPO4 10 mM pH 6.
Les spectres 1D 1H présentés Fig. 3.11a montrent, dans la région des aromatiques et des Trp,des intensités de signaux correctes mais une faible dispersion des déplacements chimiques (δ). Les
2. Le chlorure de guanidinium précipitant avec le SDS, il n’a pas pu être testé sur la protéine mTSPO1/SDS.

162 Chapitre 3. La mTSPO1
chaines latérales des Trp de la mTSPO1/SDS en micelles de SDS (2 cmc) apparaissent entre 9.6et 10.1 ppm tandis que ceux de la mTSPO1/DPC en micelles de DPC (5 cmc) apparaissent entre10 et 10.5 ppm. L’ajout de DPC en quantité suffisante (> à 5 cmc) à la protéine mTSPO1/SDS,en solution de micelles de SDS, déplace le massif des Trp vers les δ observées pour la TSPO/DPC(données non présentées). Ceci montre que les détergents après purification de la mTSPO1peuvent se substituer bien que cette substitution ne soit pas complète en solution.
La superposition des spectres 2D 1H,15N HSQC de la protéine TSPO/SDS en solution demicelles de SDS et celle de TSPO/DPC en solution de micelles de DPC, Fig. 3.11b, montrent queles 2 spectres ont une dispersion spectrale et une homogénéité des largeurs des pics semblables.Certaines régions du spectre de la protéine ne sont pas affectées (superposition des pics) parle changement de détergent ; les pics de corrélations subissent juste un léger décalage de leur δreflétant le changement de l’environnement : les détergents SDS et DPC ayant différentes têtespolaires, l’interaction avec un même résidu sera différente. Comme observé sur les spectres 1D1H, les Trp de la protéine se montrent les plus "sensibles" aux changements de l’environnement :le massif des Trp subit un décalage important en fonction de la tête polaire du détergent enaccord avec la localisation des Trp aux interfaces lipide/eau [de Jesus and Allen, 2013].
Par contre, dans certaines parties du spectre, en particulier dans la région des groupementsamides du squelette peptidique des Gly, Ser et Thr, les pics des groupements HN de la protéinesont bien différenciés entre les détergents. En plus de l’effet du changement d’environnement celapeut refléter des changements structuraux dans la protéine. La qualité des spectres ne permetpas de dire si le SDS ou le DPC conduit à un meilleur repliement de la protéine. Toutefois, au vudes données présentées ci-dessus (fluorescence, CD, digestion trypsique, effets des dénaturants),il est certain que la mTSPO1 n’est pas dans sa conformation fonctionnelle native et le DPCs’avère un meilleur agent mimétique de son environnement membranaire natif.
Dans les 2 cas, les spectres montrent de nombreuses superpositions des résonances et unmauvais étalement dans la région des Trp. La mTSPO1 possède 12 Trp, 12 pics de corrélationsdevraient donc apparaître dans cette région mais ils n’y sont pas. Cela suggère l’existence dedifférentes conformations de la mTSPO1 en échange intermédiaire à l’échelle des déplacementschimiques de la RMN (cf. Annexe D "L’échange chimique en RMN"). La structure tertiaire de lamTSPO1 à la fois en SDS et en DPC semble donc peu stable et l’attribution des résonances dansces 2 conditions est bien trop complexe, une telle instabilité rendant impossible l’acquisition desspectres 3D nécessaires pour l’attribution (cf. Sec. 2.4.4.4).
3.3 Effets des ligands sur la mTSPO1 recombinante
Les mesures de liaison d’un ligand sont des bons contrôles pour vérifier si la protéine estfonctionnelle. La TSPO1 possède de nombreux ligands pharmacologiques de classes chimiquesdifférentes (PK11195, Ro5-4864, etc.) ainsi que quelques ligands naturels (PPIX, cholestérol).
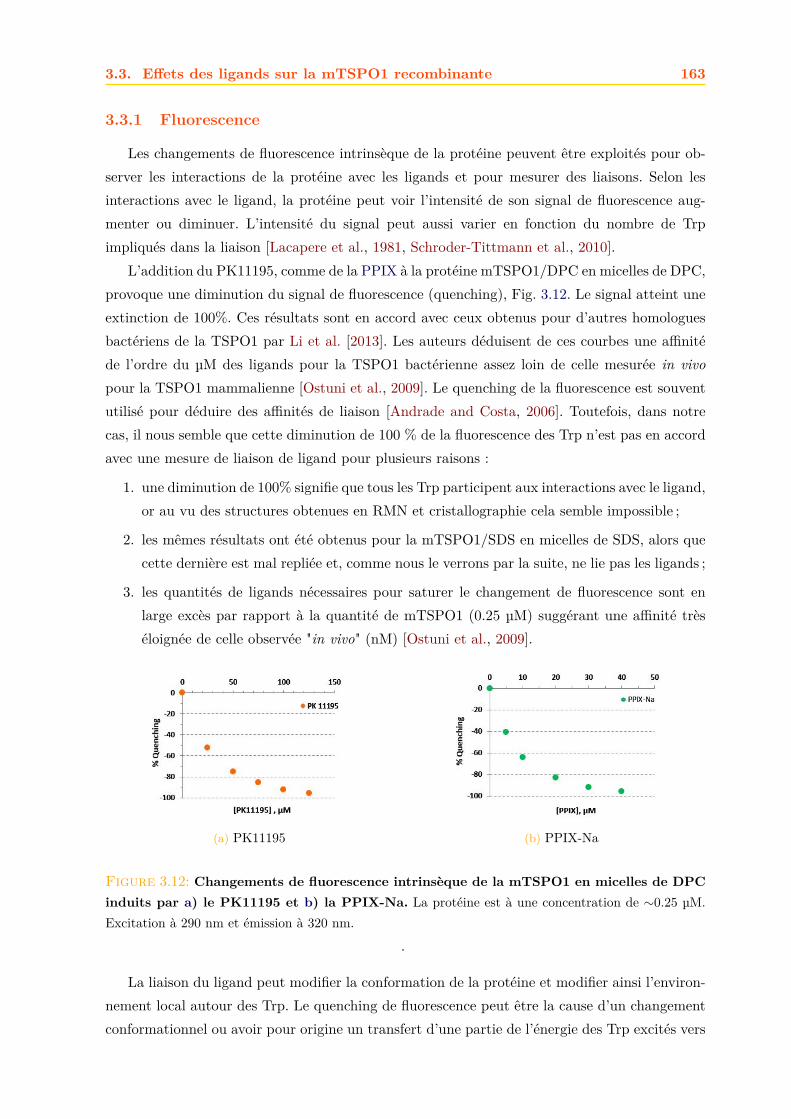
3.3. Effets des ligands sur la mTSPO1 recombinante 163
3.3.1 Fluorescence
Les changements de fluorescence intrinsèque de la protéine peuvent être exploités pour ob-server les interactions de la protéine avec les ligands et pour mesurer des liaisons. Selon lesinteractions avec le ligand, la protéine peut voir l’intensité de son signal de fluorescence aug-menter ou diminuer. L’intensité du signal peut aussi varier en fonction du nombre de Trpimpliqués dans la liaison [Lacapere et al., 1981, Schroder-Tittmann et al., 2010].
L’addition du PK11195, comme de la PPIX à la protéine mTSPO1/DPC en micelles de DPC,provoque une diminution du signal de fluorescence (quenching), Fig. 3.12. Le signal atteint uneextinction de 100%. Ces résultats sont en accord avec ceux obtenus pour d’autres homologuesbactériens de la TSPO1 par Li et al. [2013]. Les auteurs déduisent de ces courbes une affinitéde l’ordre du µM des ligands pour la TSPO1 bactérienne assez loin de celle mesurée in vivopour la TSPO1 mammalienne [Ostuni et al., 2009]. Le quenching de la fluorescence est souventutilisé pour déduire des affinités de liaison [Andrade and Costa, 2006]. Toutefois, dans notrecas, il nous semble que cette diminution de 100 % de la fluorescence des Trp n’est pas en accordavec une mesure de liaison de ligand pour plusieurs raisons :
1. une diminution de 100% signifie que tous les Trp participent aux interactions avec le ligand,or au vu des structures obtenues en RMN et cristallographie cela semble impossible ;
2. les mêmes résultats ont été obtenus pour la mTSPO1/SDS en micelles de SDS, alors quecette dernière est mal repliée et, comme nous le verrons par la suite, ne lie pas les ligands ;
3. les quantités de ligands nécessaires pour saturer le changement de fluorescence sont enlarge excès par rapport à la quantité de mTSPO1 (0.25 µM) suggérant une affinité trèséloignée de celle observée "in vivo" (nM) [Ostuni et al., 2009].
(a) PK11195 (b) PPIX-Na
Figure 3.12: Changements de fluorescence intrinsèque de la mTSPO1 en micelles de DPCinduits par a) le PK11195 et b) la PPIX-Na. La protéine est à une concentration de ∼0.25 µM.Excitation à 290 nm et émission à 320 nm.
.
La liaison du ligand peut modifier la conformation de la protéine et modifier ainsi l’environ-nement local autour des Trp. Le quenching de fluorescence peut être la cause d’un changementconformationnel ou avoir pour origine un transfert d’une partie de l’énergie des Trp excités vers
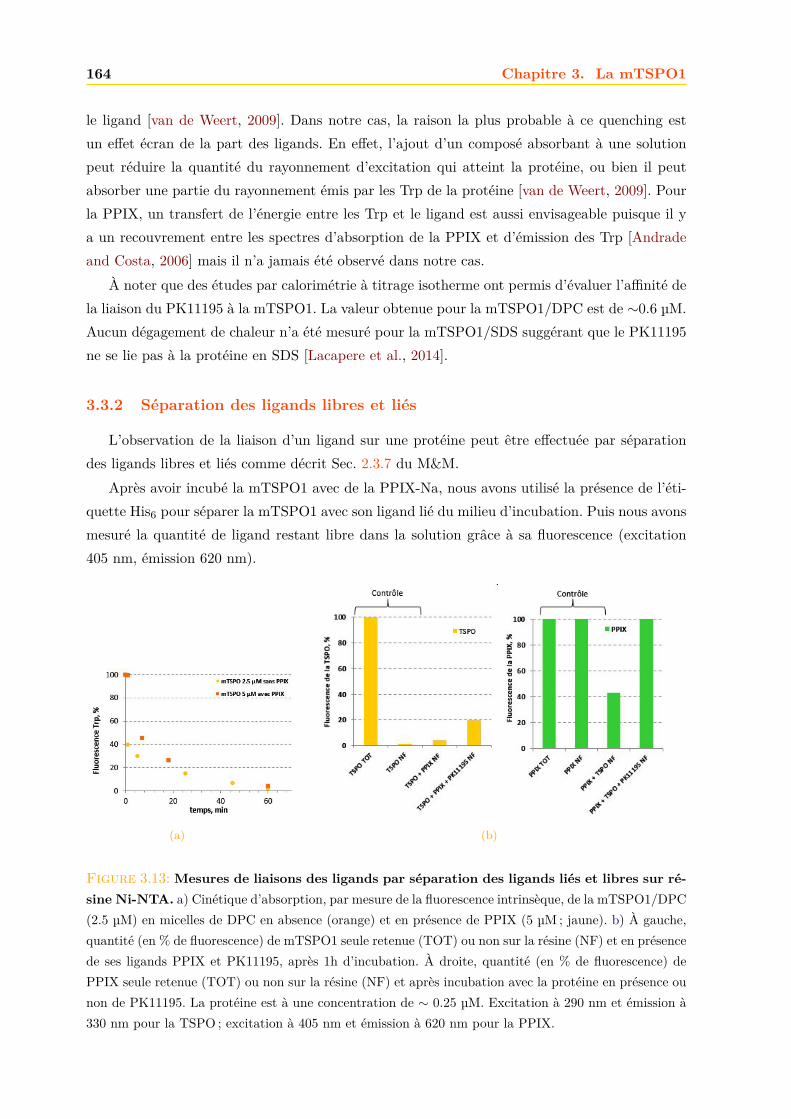
164 Chapitre 3. La mTSPO1
le ligand [van de Weert, 2009]. Dans notre cas, la raison la plus probable à ce quenching estun effet écran de la part des ligands. En effet, l’ajout d’un composé absorbant à une solutionpeut réduire la quantité du rayonnement d’excitation qui atteint la protéine, ou bien il peutabsorber une partie du rayonnement émis par les Trp de la protéine [van de Weert, 2009]. Pourla PPIX, un transfert de l’énergie entre les Trp et le ligand est aussi envisageable puisque il ya un recouvrement entre les spectres d’absorption de la PPIX et d’émission des Trp [Andradeand Costa, 2006] mais il n’a jamais été observé dans notre cas.
À noter que des études par calorimétrie à titrage isotherme ont permis d’évaluer l’affinité dela liaison du PK11195 à la mTSPO1. La valeur obtenue pour la mTSPO1/DPC est de ∼0.6 µM.Aucun dégagement de chaleur n’a été mesuré pour la mTSPO1/SDS suggérant que le PK11195ne se lie pas à la protéine en SDS [Lacapere et al., 2014].
3.3.2 Séparation des ligands libres et liés
L’observation de la liaison d’un ligand sur une protéine peut être effectuée par séparationdes ligands libres et liés comme décrit Sec. 2.3.7 du M&M.
Après avoir incubé la mTSPO1 avec de la PPIX-Na, nous avons utilisé la présence de l’éti-quette His6 pour séparer la mTSPO1 avec son ligand lié du milieu d’incubation. Puis nous avonsmesuré la quantité de ligand restant libre dans la solution grâce à sa fluorescence (excitation405 nm, émission 620 nm).
(a) (b)
Figure 3.13: Mesures de liaisons des ligands par séparation des ligands liés et libres sur ré-sine Ni-NTA. a) Cinétique d’absorption, par mesure de la fluorescence intrinsèque, de la mTSPO1/DPC(2.5 µM) en micelles de DPC en absence (orange) et en présence de PPIX (5 µM; jaune). b) À gauche,quantité (en % de fluorescence) de mTSPO1 seule retenue (TOT) ou non sur la résine (NF) et en présencede ses ligands PPIX et PK11195, après 1h d’incubation. À droite, quantité (en % de fluorescence) dePPIX seule retenue (TOT) ou non sur la résine (NF) et après incubation avec la protéine en présence ounon de PK11195. La protéine est à une concentration de ∼ 0.25 µM. Excitation à 290 nm et émission à330 nm pour la TSPO ; excitation à 405 nm et émission à 620 nm pour la PPIX.
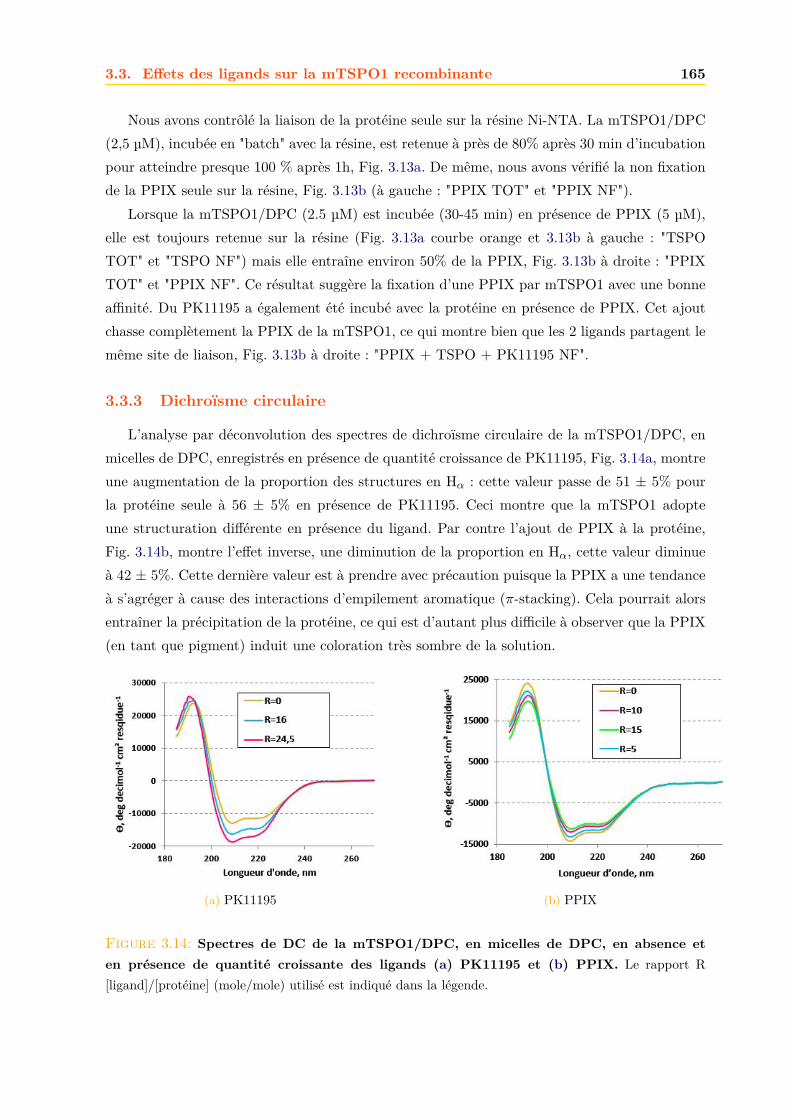
3.3. Effets des ligands sur la mTSPO1 recombinante 165
Nous avons contrôlé la liaison de la protéine seule sur la résine Ni-NTA. La mTSPO1/DPC(2,5 µM), incubée en "batch" avec la résine, est retenue à près de 80% après 30 min d’incubationpour atteindre presque 100 % après 1h, Fig. 3.13a. De même, nous avons vérifié la non fixationde la PPIX seule sur la résine, Fig. 3.13b (à gauche : "PPIX TOT" et "PPIX NF").
Lorsque la mTSPO1/DPC (2.5 µM) est incubée (30-45 min) en présence de PPIX (5 µM),elle est toujours retenue sur la résine (Fig. 3.13a courbe orange et 3.13b à gauche : "TSPOTOT" et "TSPO NF") mais elle entraîne environ 50% de la PPIX, Fig. 3.13b à droite : "PPIXTOT" et "PPIX NF". Ce résultat suggère la fixation d’une PPIX par mTSPO1 avec une bonneaffinité. Du PK11195 a également été incubé avec la protéine en présence de PPIX. Cet ajoutchasse complètement la PPIX de la mTSPO1, ce qui montre bien que les 2 ligands partagent lemême site de liaison, Fig. 3.13b à droite : "PPIX + TSPO + PK11195 NF".
3.3.3 Dichroïsme circulaire
L’analyse par déconvolution des spectres de dichroïsme circulaire de la mTSPO1/DPC, enmicelles de DPC, enregistrés en présence de quantité croissance de PK11195, Fig. 3.14a, montreune augmentation de la proportion des structures en Hα : cette valeur passe de 51 ± 5% pourla protéine seule à 56 ± 5% en présence de PK11195. Ceci montre que la mTSPO1 adopteune structuration différente en présence du ligand. Par contre l’ajout de PPIX à la protéine,Fig. 3.14b, montre l’effet inverse, une diminution de la proportion en Hα, cette valeur diminueà 42 ± 5%. Cette dernière valeur est à prendre avec précaution puisque la PPIX a une tendanceà s’agréger à cause des interactions d’empilement aromatique (π-stacking). Cela pourrait alorsentraîner la précipitation de la protéine, ce qui est d’autant plus difficile à observer que la PPIX(en tant que pigment) induit une coloration très sombre de la solution.
(a) PK11195 (b) PPIX
Figure 3.14: Spectres de DC de la mTSPO1/DPC, en micelles de DPC, en absence eten présence de quantité croissante des ligands (a) PK11195 et (b) PPIX. Le rapport R[ligand]/[protéine] (mole/mole) utilisé est indiqué dans la légende.
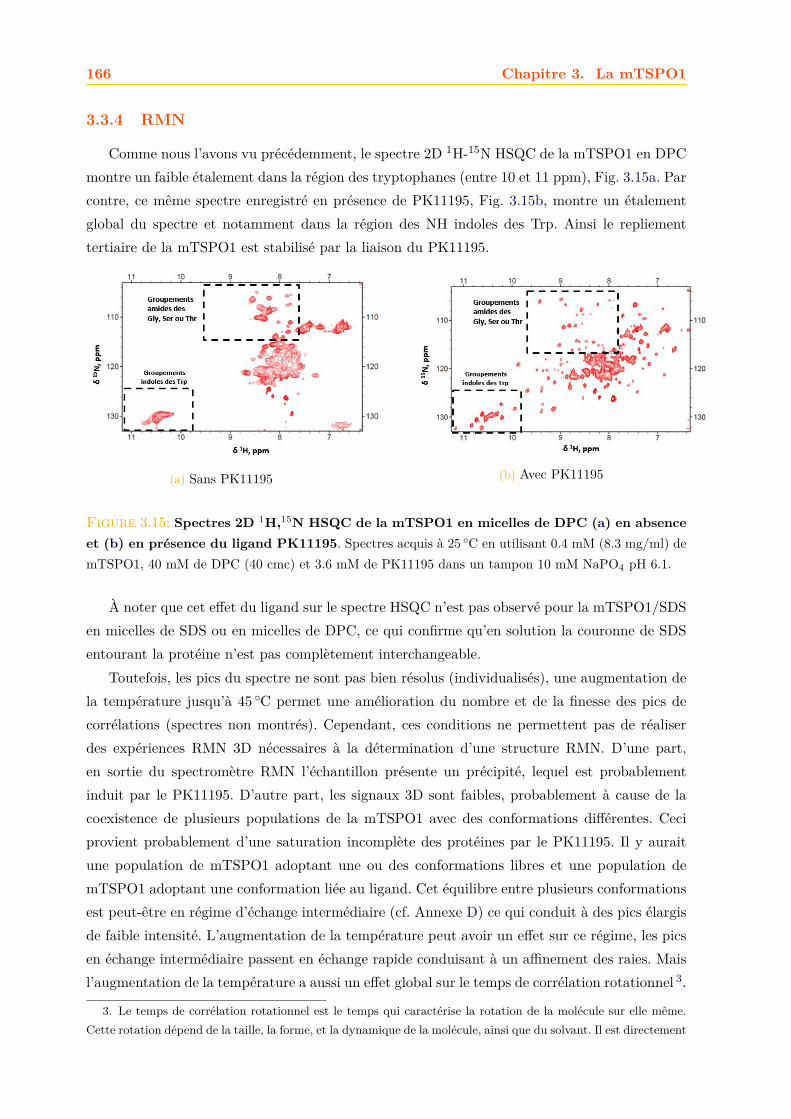
166 Chapitre 3. La mTSPO1
3.3.4 RMN
Comme nous l’avons vu précédemment, le spectre 2D 1H-15N HSQC de la mTSPO1 en DPCmontre un faible étalement dans la région des tryptophanes (entre 10 et 11 ppm), Fig. 3.15a. Parcontre, ce même spectre enregistré en présence de PK11195, Fig. 3.15b, montre un étalementglobal du spectre et notamment dans la région des NH indoles des Trp. Ainsi le repliementtertiaire de la mTSPO1 est stabilisé par la liaison du PK11195.
(a) Sans PK11195 (b) Avec PK11195
Figure 3.15: Spectres 2D 1H,15N HSQC de la mTSPO1 en micelles de DPC (a) en absenceet (b) en présence du ligand PK11195. Spectres acquis à 25 ◦C en utilisant 0.4 mM (8.3 mg/ml) demTSPO1, 40 mM de DPC (40 cmc) et 3.6 mM de PK11195 dans un tampon 10 mM NaPO4 pH 6.1.
À noter que cet effet du ligand sur le spectre HSQC n’est pas observé pour la mTSPO1/SDSen micelles de SDS ou en micelles de DPC, ce qui confirme qu’en solution la couronne de SDSentourant la protéine n’est pas complètement interchangeable.
Toutefois, les pics du spectre ne sont pas bien résolus (individualisés), une augmentation dela température jusqu’à 45 ◦C permet une amélioration du nombre et de la finesse des pics decorrélations (spectres non montrés). Cependant, ces conditions ne permettent pas de réaliserdes expériences RMN 3D nécessaires à la détermination d’une structure RMN. D’une part,en sortie du spectromètre RMN l’échantillon présente un précipité, lequel est probablementinduit par le PK11195. D’autre part, les signaux 3D sont faibles, probablement à cause de lacoexistence de plusieurs populations de la mTSPO1 avec des conformations différentes. Ceciprovient probablement d’une saturation incomplète des protéines par le PK11195. Il y auraitune population de mTSPO1 adoptant une ou des conformations libres et une population demTSPO1 adoptant une conformation liée au ligand. Cet équilibre entre plusieurs conformationsest peut-être en régime d’échange intermédiaire (cf. Annexe D) ce qui conduit à des pics élargisde faible intensité. L’augmentation de la température peut avoir un effet sur ce régime, les picsen échange intermédiaire passent en échange rapide conduisant à un affinement des raies. Maisl’augmentation de la température a aussi un effet global sur le temps de corrélation rotationnel 3.
3. Le temps de corrélation rotationnel est le temps qui caractérise la rotation de la molécule sur elle même.Cette rotation dépend de la taille, la forme, et la dynamique de la molécule, ainsi que du solvant. Il est directement

3.3. Effets des ligands sur la mTSPO1 recombinante 167
Une solution qui paraît logique pour obtenir la saturation des sites de liaison des protéinespar le PK11195 est d’augmenter sa concentration. Or en augmentant le rapport PK11195 surmTSPO1, on provoque la formation d’un précipité de PK11195 qui est une molécule très hy-drophobe. En effet, ce composé peu soluble (jusqu’à 300 µM en tampon phosphate) complexifiegrandement la tâche puisque même solubilisé en détergent comme le DPC (il faut environ5 moles de DPC pour solubiliser une mole de PK11195 dans des gammes de concentrationsfaibles), il précipite à forte concentration [Lacapere et al., 2014, Harbi Iatmanen, 2014]. Unschéma illustrant ce phénomène est proposé Fig. 3.16.
Figure 3.16: Schèma illustrant l’équilibre entre les populations de mTSPO1 libres et liéesau PK11195 ainsi que la précipitation du PK11195 à forte concentration. La mTSPO1 purifiéeen DPC, mise dans un tampon contenant des micelles de DPC, est entourée d’une couronne de DPC etcoexiste avec des micelles de détergent libre. Le PK11195, qui est peu soluble, va se positionner sur lesite de liaison de la protéine, dans les micelles de DPC mais aussi dans la couronne de DPC entourant laprotéine. Il coexiste alors un équilibre entre la population de mTSPO1 libres et la population de mTSPO1complexées au PK11195. Lorsque la concentration de PK11195 est augmentée pour obtenir la saturationdes sites de liaisons, et ainsi obtenir une population de protéines uniquement complexées, les micellesde DPC se saturent également. Cet équilibre entre saturation de la mTSPO1 et les micelles est instablecar une saturation des micelles de détergent conduit à l’exclusion du PK11195 et à sa précipitation sousforme d’agrégats.
La saturation de la mTSPO1 par le PK11195 en présence de DPC n’a jamais pu êtreoptimisée, la recherche de condition modifiant l’affinité de la protéine pour le ligand et l’ajoutde lipides n’ont jamais pu aboutir à un échantillon pour lequel l’ensemble de la population demTSPO1 adopte une conformation unique et stable. Néanmoins, Jaremko et al. ont réussi àsurmonter ce problème en utilisant un énantiomère du PK11195 (sous sa forme (R)) au lieudu mélange racémique (Sec. 1.2.4.1 de l’introduction). La forme (R) possède une affinité 2 foisplus grande pour la mTSPO1 que le mélange racémique dans les conditions de la RMN. Celasuggère que la forme S contribue le plus fortement à la précipitation du mélange racémique.
lié au volume et au poids moléculaire de la protéine. À haute température ce temps est plus court qu’à bassetempérature, conduisant souvent à une amélioration de la largeur des raies
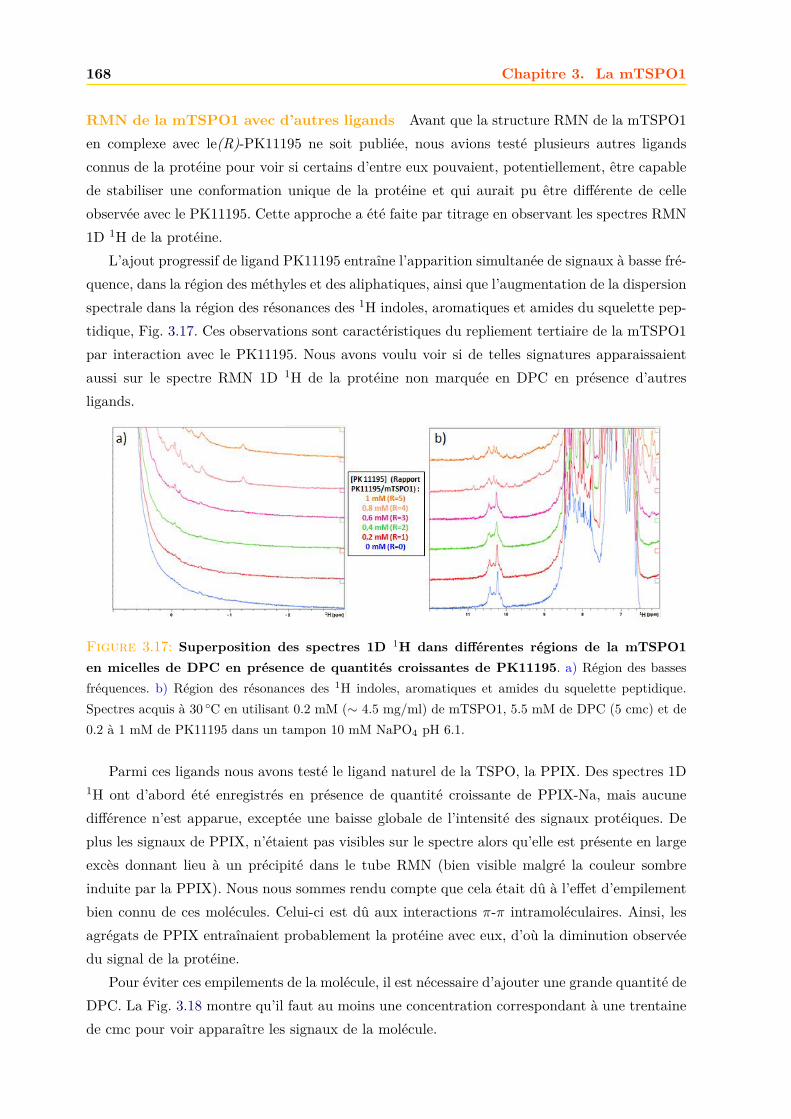
168 Chapitre 3. La mTSPO1
RMN de la mTSPO1 avec d’autres ligands Avant que la structure RMN de la mTSPO1en complexe avec le(R)-PK11195 ne soit publiée, nous avions testé plusieurs autres ligandsconnus de la protéine pour voir si certains d’entre eux pouvaient, potentiellement, être capablede stabiliser une conformation unique de la protéine et qui aurait pu être différente de celleobservée avec le PK11195. Cette approche a été faite par titrage en observant les spectres RMN1D 1H de la protéine.
L’ajout progressif de ligand PK11195 entraîne l’apparition simultanée de signaux à basse fré-quence, dans la région des méthyles et des aliphatiques, ainsi que l’augmentation de la dispersionspectrale dans la région des résonances des 1H indoles, aromatiques et amides du squelette pep-tidique, Fig. 3.17. Ces observations sont caractéristiques du repliement tertiaire de la mTSPO1par interaction avec le PK11195. Nous avons voulu voir si de telles signatures apparaissaientaussi sur le spectre RMN 1D 1H de la protéine non marquée en DPC en présence d’autresligands.
Figure 3.17: Superposition des spectres 1D 1H dans différentes régions de la mTSPO1en micelles de DPC en présence de quantités croissantes de PK11195. a) Région des bassesfréquences. b) Région des résonances des 1H indoles, aromatiques et amides du squelette peptidique.Spectres acquis à 30 ◦C en utilisant 0.2 mM (∼ 4.5 mg/ml) de mTSPO1, 5.5 mM de DPC (5 cmc) et de0.2 à 1 mM de PK11195 dans un tampon 10 mM NaPO4 pH 6.1.
Parmi ces ligands nous avons testé le ligand naturel de la TSPO, la PPIX. Des spectres 1D1H ont d’abord été enregistrés en présence de quantité croissante de PPIX-Na, mais aucunedifférence n’est apparue, exceptée une baisse globale de l’intensité des signaux protéiques. Deplus les signaux de PPIX, n’étaient pas visibles sur le spectre alors qu’elle est présente en largeexcès donnant lieu à un précipité dans le tube RMN (bien visible malgré la couleur sombreinduite par la PPIX). Nous nous sommes rendu compte que cela était dû à l’effet d’empilementbien connu de ces molécules. Celui-ci est dû aux interactions π-π intramoléculaires. Ainsi, lesagrégats de PPIX entraînaient probablement la protéine avec eux, d’où la diminution observéedu signal de la protéine.
Pour éviter ces empilements de la molécule, il est nécessaire d’ajouter une grande quantité deDPC. La Fig. 3.18 montre qu’il faut au moins une concentration correspondant à une trentainede cmc pour voir apparaître les signaux de la molécule.
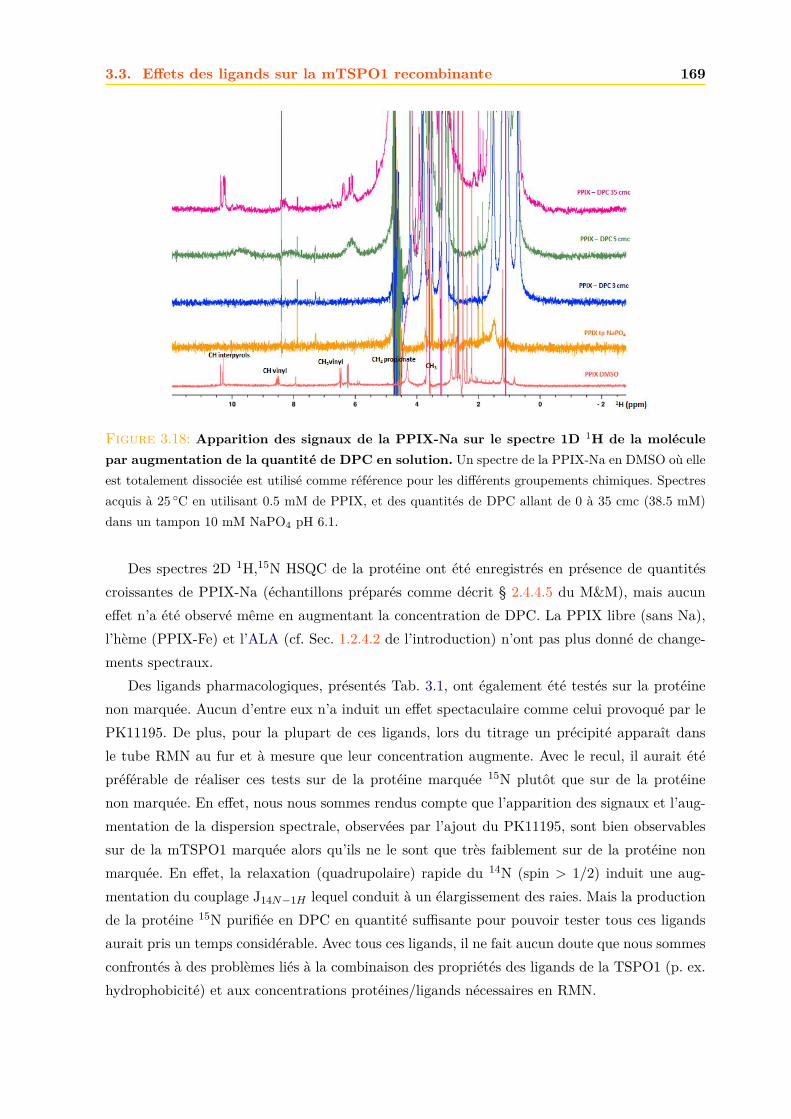
3.3. Effets des ligands sur la mTSPO1 recombinante 169
Figure 3.18: Apparition des signaux de la PPIX-Na sur le spectre 1D 1H de la moléculepar augmentation de la quantité de DPC en solution. Un spectre de la PPIX-Na en DMSO où elleest totalement dissociée est utilisé comme référence pour les différents groupements chimiques. Spectresacquis à 25 ◦C en utilisant 0.5 mM de PPIX, et des quantités de DPC allant de 0 à 35 cmc (38.5 mM)dans un tampon 10 mM NaPO4 pH 6.1.
Des spectres 2D 1H,15N HSQC de la protéine ont été enregistrés en présence de quantitéscroissantes de PPIX-Na (échantillons préparés comme décrit § 2.4.4.5 du M&M), mais aucuneffet n’a été observé même en augmentant la concentration de DPC. La PPIX libre (sans Na),l’hème (PPIX-Fe) et l’ALA (cf. Sec. 1.2.4.2 de l’introduction) n’ont pas plus donné de change-ments spectraux.
Des ligands pharmacologiques, présentés Tab. 3.1, ont également été testés sur la protéinenon marquée. Aucun d’entre eux n’a induit un effet spectaculaire comme celui provoqué par lePK11195. De plus, pour la plupart de ces ligands, lors du titrage un précipité apparaît dansle tube RMN au fur et à mesure que leur concentration augmente. Avec le recul, il aurait étépréférable de réaliser ces tests sur de la protéine marquée 15N plutôt que sur de la protéinenon marquée. En effet, nous nous sommes rendus compte que l’apparition des signaux et l’aug-mentation de la dispersion spectrale, observées par l’ajout du PK11195, sont bien observablessur de la mTSPO1 marquée alors qu’ils ne le sont que très faiblement sur de la protéine nonmarquée. En effet, la relaxation (quadrupolaire) rapide du 14N (spin > 1/2) induit une aug-mentation du couplage J14N−1H lequel conduit à un élargissement des raies. Mais la productionde la protéine 15N purifiée en DPC en quantité suffisante pour pouvoir tester tous ces ligandsaurait pris un temps considérable. Avec tous ces ligands, il ne fait aucun doute que nous sommesconfrontés à des problèmes liés à la combinaison des propriétés des ligands de la TSPO1 (p. ex.hydrophobicité) et aux concentrations protéines/ligands nécessaires en RMN.
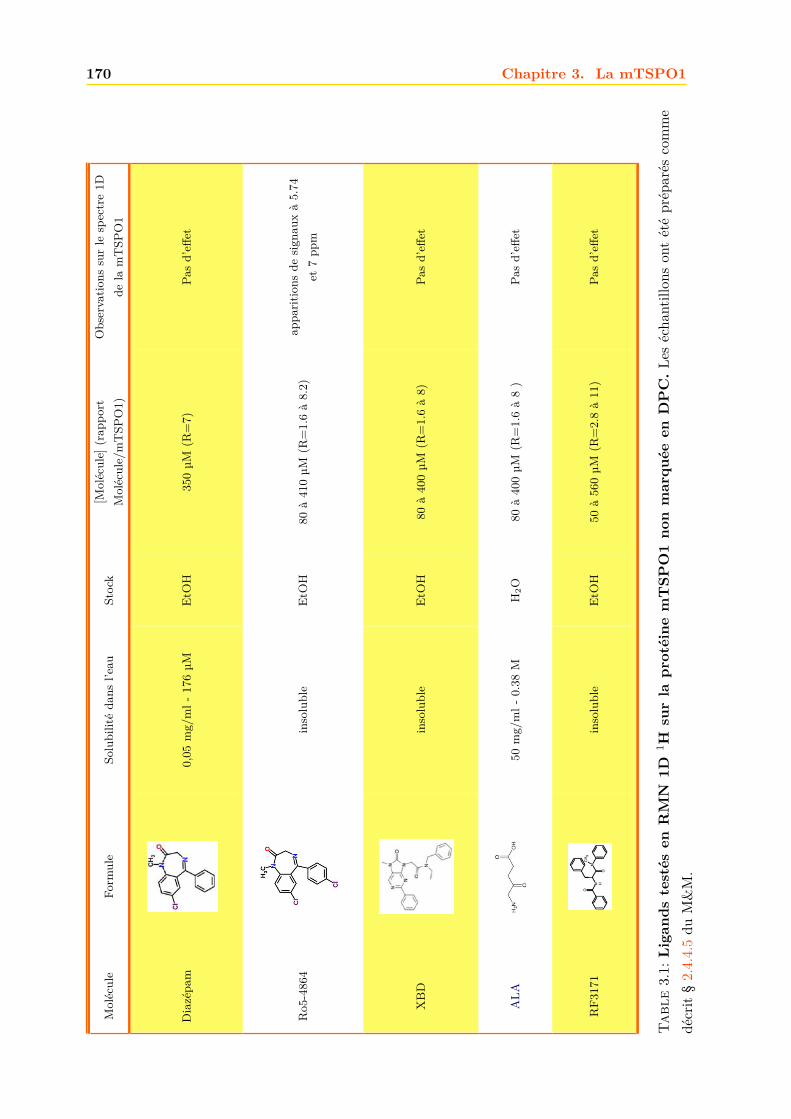
170 Chapitre 3. La mTSPO1
Molécule
Form
ule
Solubilitéda
nsl’e
auStock
[Molécule]
(rap
port
Molécule/mTSP
O1)
Observ atio
nssurle
spectre1D
dela
mTSP
O1
Diazépa
m0,05
mg/
ml-
176
µMEt
OH
350
µM(R
=7)
Pasd’eff
et
Ro5
-486
4insoluble
EtOH
80à41
0µM
(R=1.6à8.2)
appa
ritions
desig
naux
à5.74
et7pp
m
XBD
insoluble
EtOH
80à40
0µM
(R=1.6à8)
Pasd’eff
et
ALA
50mg/
ml-
0.38
MH
2O
80à40
0µM
(R=1.6à8)
Pasd’eff
et
RF3
171
insoluble
EtOH
50à56
0µM
(R=2.8à11
)Pa
sd’eff
et
Tab
le3.1:Ligand
stestés
enRMN
1D1 H
surla
protéine
mTSP
O1no
nmarqu
éeen
DPC.L
esécha
ntillon
son
tétépréparés
comme
décrit§2.4.4.5du
M&M.

3.3. Effets des ligands sur la mTSPO1 recombinante 171
3.3.5 Conclusions
La mTSPO1 a été surexprimée dans E. coli puis extraite des corps d’inclusion par SDSet purifiée en présence de détergent (SDS ou DPC) grâce à son étiquette His6 sur résine Ni-NTA. Ce protocole nous a permis d’obtenir une mTSPO1 recombinante, marquée ou non, enquantité et pureté satisfaisantes pour son étude structurale en particulier en RMN. Les donnéesobtenues montrent que la mTSPO1 purifiée en SDS n’est pas repliée correctement et ne permetpas la liaison des ligands. La mTSPO1 en DPC semble mieux repliée et permet la liaisons desligands. La détermination d’une structure atomique d’une protéine membranaire est une étapeindispensable pour mieux comprendre son fonctionnement. Par RMN, cela nécessite l’acquisitiond’une série de spectres 3D qui ne peuvent être obtenus qu’avec une protéine "stable". Les donnéesque nous avions au début de ma thèse montraient que la mTSPO1 était difficilement stabilisableen solution en présence de détergent DPC. La présence du ligand spécifique, le PK11195, montraune modification spectaculaire des spectres 2D 1H,15N HSQC en solution [Murail et al., 2008],mais ne semblait pas, à forte concentration, stabiliser suffisamment la protéine pour acquérirles spectres 3D. Dans ces conditions, l’affinité de la mTSPO1 pour son ligand était de l’ordredu µM. Ainsi la quantité de PK11195 à ajouter pour saturer la protéine dans les conditionsde la RMN (mTSPO1 de l’ordre du mM) demandait d’ajouter de fortes quantités de PK11195en prenant en compte sa faible solubilité et son insertion dans les micelles de détergent. Latentative de stabilisation de la protéine avec d’autres ligands (PPIX, Ro5-4864, etc.) nous aconfronté aux mêmes problèmes que ceux rencontrés avec le PK11195 lors l’étude de la protéineen RMN.
Face à aux difficultés rencontrées pour stabiliser la protéine en RMN du liquide, en pa-rallèle nous avions débuté l’étude structurale par RMN du solide de la protéine reconstituéesen protéoliposomes. Puis les résolutions des structures par Jaremko2014,Li2014,Guo2014 et lesinterrogations qu’elles ont amené, nous ont conforté dans l’idée que cette étude en RMN dusolide pouvait permettre une avancée dans la compréhension de la protéine.

172 Chapitre 3. La mTSPO1
3.4 La mTSPO1 en RMN du Solide
3.4.1 Reconstitution de la mTSPO1 dans des liposomes
Dans le but de fournir un environnement plus proche de sa membrane mitochondriale na-turelle, nous avons réintroduit la mTSPO1 dans la bicouche lipidique (cf Sec.4.12 du M&M).Brièvement, cette reconstitution a été obtenue en mélangeant la mTSPO1 purifiée en SDS avecdes lipides (DMPC/DMPE, rapport 9/1 p/p) dans un tampon contenant suffisamment de SDSpour solubiliser à la fois la protéine et les lipides. Puis, le détergent est éliminé par ajouts deBB [Lacapere et al., 2001], Fig. 3.19a, pour permettre la formation des protéoliposomes.
Différents rapports lipide/protéine (LPR) ont été utilisés lors de l’étude en RMN du solide(ssNMR) : LPR allant de 1 (soit 30 molécules de lipides/molécule de mTSPO1) à 10.
Le suivi de la reconstitution peut se faire par la combinaison de plusieurs approches : fluo-rescence intrinsèque, turbidimétrie (mesure de l’absorbance/DO à 550 nm), microscopie électro-nique ou cryomicroscopie et diffusion dynamique de lumière DLS [Ostuni et al., 2010b, Lacapereet al., 2014] :
1. L’augmentation de la fluorescence intrinsèque de la mTSPO1, Fig 3.19b en rose qui in-dique un changement de l’environnement des Trp. Ceci pourrait révéler un changement deconformation de la protéine lorsqu’elle s’entoure d’une membrane lipidique et l’adoptiond’un repliement plus proche de son repliement natif. Cette augmentation s’accompagned’un décalage vers le bleu (blueshift) compatible avec un environnement des Trp plushydrophobe [Uversky and Longhi, 2010, Maestro and Sanz, 2005], Fig. 3.19c.
2. L’augmentation de la turbidimétrie de la solution à 350 nm et 550 nm indique la formationde protéoliposomes qui diffusent la lumière. Alors que les complexes mTSPO1/SDS, petitsobjets, ne la diffusent quasiment pas. Fig. 3.19b en vert et orange : p. ex. la DO à 550 nmpasse d’une valeur de ∼0,1 à une valeur > 1 où visuellement la solution apparaît trouble.
3. L’évolution de la taille et de la forme des objets présents en solution est observable direc-tement par ME et cryoME, Fig. 3.19d, Mais peut être limitée par le champ d’observation.
4. La taille des objets ou des populations est mesurable par DLS.
La combinaison des données de DLS et de microscopie électrique donne des protéoliposomesune taille de ∼ 200 nm, pas forcément homogène, (3.19d, image c)) pour les protéoliposomes.On peut noter que, très souvent, le temps d’enlever les BB du second ajout, les protéoliposomesforment des agrégats ou fusionnent. Cela est visible par microscopie (image 4 de la Fig. 3.19d),par DLS (population avec des tailles » 200 nm), par une légère diminution de la fluorescence(∼ 15% par rapport à la valeur maximale) et par une grande valeur des DO à 350 et 550 nm(3) et 4) sur le graphique Fig. 3.19b).
On peut noter que le changement d’environnement des Trp révélé par les données defluorescence intrinsèque est également observable par RMN. Les spectres 1D 1H-RMN de lamTSPO1/DPC en présence de DMPC/DMPE montrent un décalage du déplacement chimiquedu massif des groupements NH indoles des Trp reflétant leur changement d’environnement,
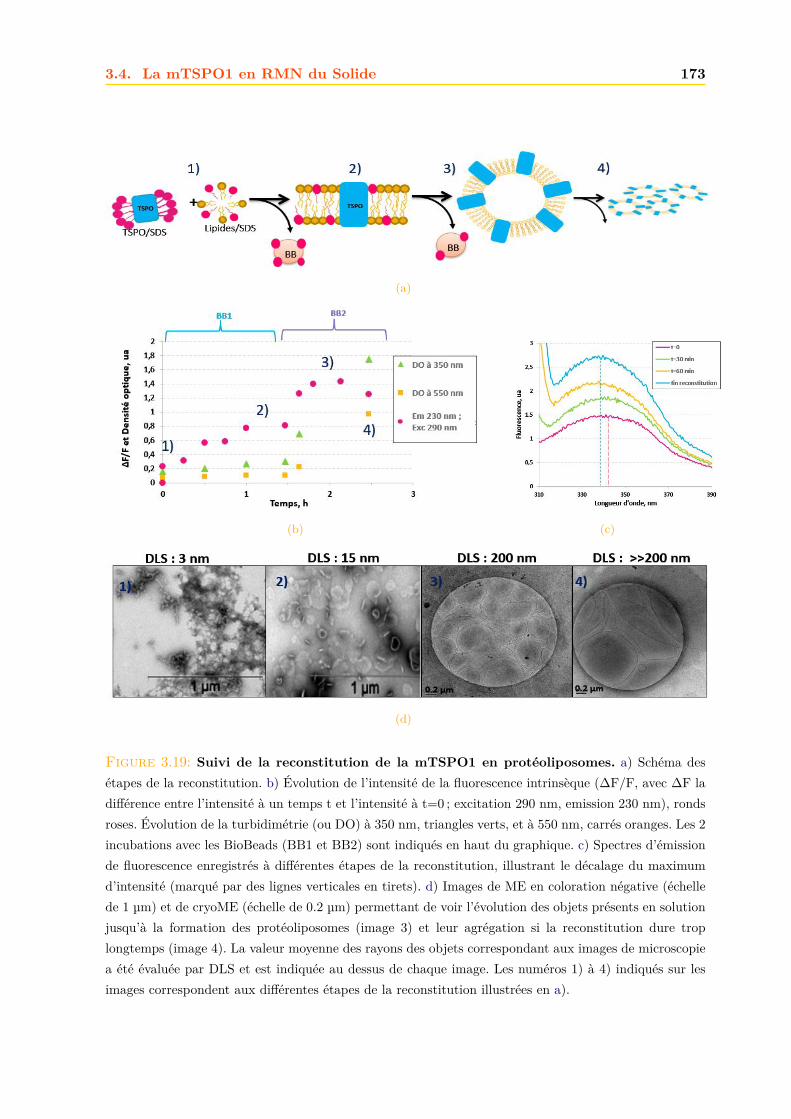
3.4. La mTSPO1 en RMN du Solide 173
(a)
(b) (c)
(d)
Figure 3.19: Suivi de la reconstitution de la mTSPO1 en protéoliposomes. a) Schéma desétapes de la reconstitution. b) Évolution de l’intensité de la fluorescence intrinsèque (∆F/F, avec ∆F ladifférence entre l’intensité à un temps t et l’intensité à t=0 ; excitation 290 nm, emission 230 nm), rondsroses. Évolution de la turbidimétrie (ou DO) à 350 nm, triangles verts, et à 550 nm, carrés oranges. Les 2incubations avec les BioBeads (BB1 et BB2) sont indiqués en haut du graphique. c) Spectres d’émissionde fluorescence enregistrés à différentes étapes de la reconstitution, illustrant le décalage du maximumd’intensité (marqué par des lignes verticales en tirets). d) Images de ME en coloration négative (échellede 1 µm) et de cryoME (échelle de 0.2 µm) permettant de voir l’évolution des objets présents en solutionjusqu’à la formation des protéoliposomes (image 3) et leur agrégation si la reconstitution dure troplongtemps (image 4). La valeur moyenne des rayons des objets correspondant aux images de microscopiea été évaluée par DLS et est indiquée au dessus de chaque image. Les numéros 1) à 4) indiqués sur lesimages correspondent aux différentes étapes de la reconstitution illustrées en a).
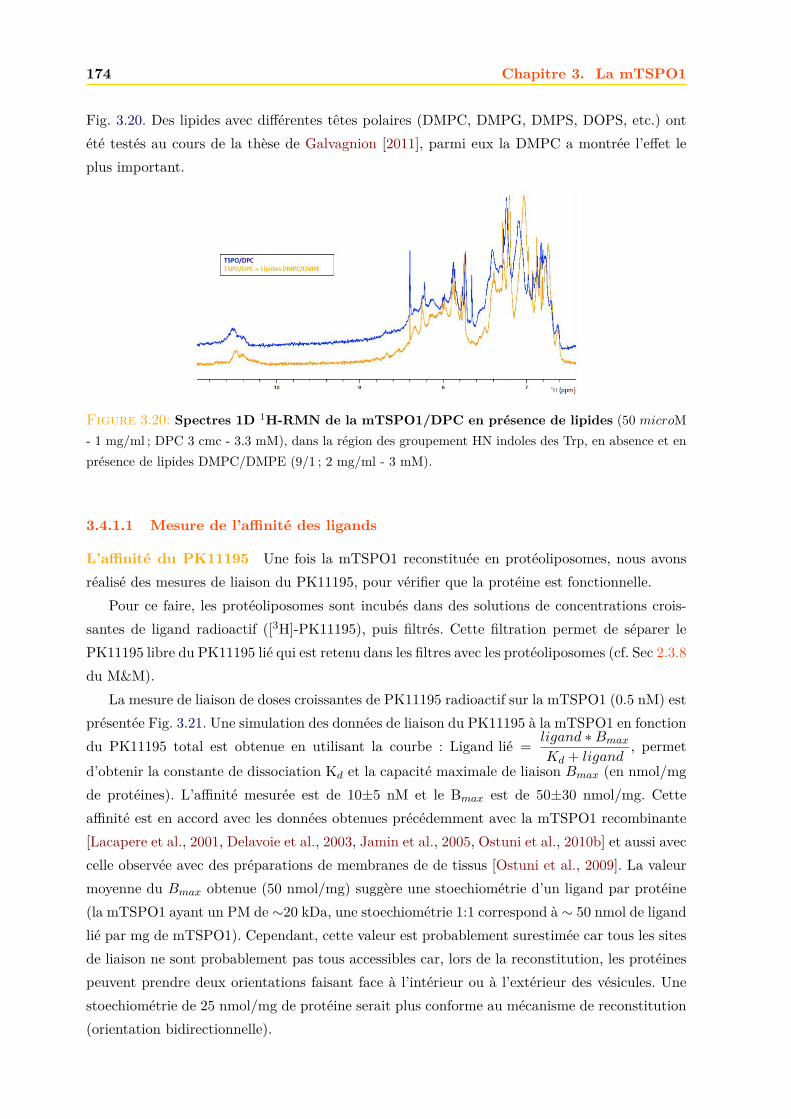
174 Chapitre 3. La mTSPO1
Fig. 3.20. Des lipides avec différentes têtes polaires (DMPC, DMPG, DMPS, DOPS, etc.) ontété testés au cours de la thèse de Galvagnion [2011], parmi eux la DMPC a montrée l’effet leplus important.
Figure 3.20: Spectres 1D 1H-RMN de la mTSPO1/DPC en présence de lipides (50 microM- 1 mg/ml ; DPC 3 cmc - 3.3 mM), dans la région des groupement HN indoles des Trp, en absence et enprésence de lipides DMPC/DMPE (9/1 ; 2 mg/ml - 3 mM).
3.4.1.1 Mesure de l’affinité des ligands
L’affinité du PK11195 Une fois la mTSPO1 reconstituée en protéoliposomes, nous avonsréalisé des mesures de liaison du PK11195, pour vérifier que la protéine est fonctionnelle.
Pour ce faire, les protéoliposomes sont incubés dans des solutions de concentrations crois-santes de ligand radioactif ([3H]-PK11195), puis filtrés. Cette filtration permet de séparer lePK11195 libre du PK11195 lié qui est retenu dans les filtres avec les protéoliposomes (cf. Sec 2.3.8du M&M).
La mesure de liaison de doses croissantes de PK11195 radioactif sur la mTSPO1 (0.5 nM) estprésentée Fig. 3.21. Une simulation des données de liaison du PK11195 à la mTSPO1 en fonctiondu PK11195 total est obtenue en utilisant la courbe : Ligand lié = ligand ∗Bmax
Kd + ligand, permet
d’obtenir la constante de dissociation Kd et la capacité maximale de liaison Bmax (en nmol/mgde protéines). L’affinité mesurée est de 10±5 nM et le Bmax est de 50±30 nmol/mg. Cetteaffinité est en accord avec les données obtenues précédemment avec la mTSPO1 recombinante[Lacapere et al., 2001, Delavoie et al., 2003, Jamin et al., 2005, Ostuni et al., 2010b] et aussi aveccelle observée avec des préparations de membranes de de tissus [Ostuni et al., 2009]. La valeurmoyenne du Bmax obtenue (50 nmol/mg) suggère une stoechiométrie d’un ligand par protéine(la mTSPO1 ayant un PM de ∼20 kDa, une stoechiométrie 1:1 correspond à ∼ 50 nmol de ligandlié par mg de mTSPO1). Cependant, cette valeur est probablement surestimée car tous les sitesde liaison ne sont probablement pas tous accessibles car, lors de la reconstitution, les protéinespeuvent prendre deux orientations faisant face à l’intérieur ou à l’extérieur des vésicules. Unestoechiométrie de 25 nmol/mg de protéine serait plus conforme au mécanisme de reconstitution(orientation bidirectionnelle).
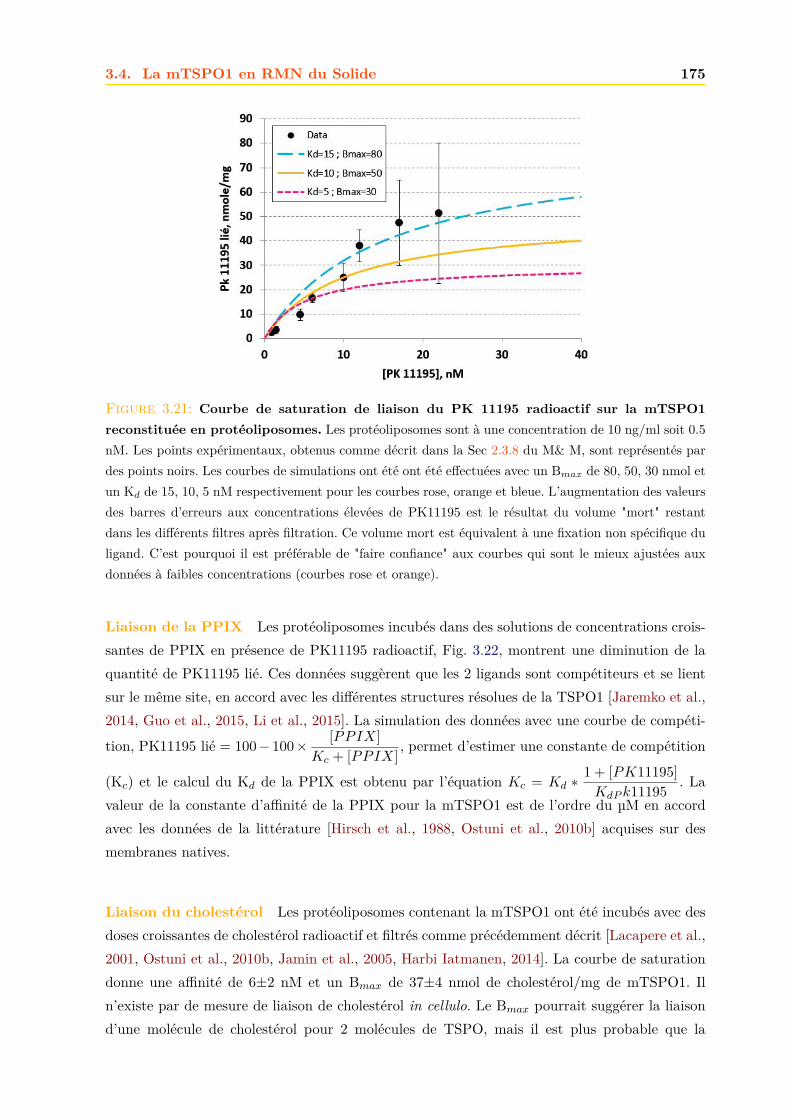
3.4. La mTSPO1 en RMN du Solide 175
Figure 3.21: Courbe de saturation de liaison du PK 11195 radioactif sur la mTSPO1reconstituée en protéoliposomes. Les protéoliposomes sont à une concentration de 10 ng/ml soit 0.5nM. Les points expérimentaux, obtenus comme décrit dans la Sec 2.3.8 du M& M, sont représentés pardes points noirs. Les courbes de simulations ont été ont été effectuées avec un Bmax de 80, 50, 30 nmol etun Kd de 15, 10, 5 nM respectivement pour les courbes rose, orange et bleue. L’augmentation des valeursdes barres d’erreurs aux concentrations élevées de PK11195 est le résultat du volume "mort" restantdans les différents filtres après filtration. Ce volume mort est équivalent à une fixation non spécifique duligand. C’est pourquoi il est préférable de "faire confiance" aux courbes qui sont le mieux ajustées auxdonnées à faibles concentrations (courbes rose et orange).
Liaison de la PPIX Les protéoliposomes incubés dans des solutions de concentrations crois-santes de PPIX en présence de PK11195 radioactif, Fig. 3.22, montrent une diminution de laquantité de PK11195 lié. Ces données suggèrent que les 2 ligands sont compétiteurs et se lientsur le même site, en accord avec les différentes structures résolues de la TSPO1 [Jaremko et al.,2014, Guo et al., 2015, Li et al., 2015]. La simulation des données avec une courbe de compéti-
tion, PK11195 lié = 100− 100× [PPIX]Kc + [PPIX] , permet d’estimer une constante de compétition
(Kc) et le calcul du Kd de la PPIX est obtenu par l’équation Kc = Kd ∗1 + [PK11195]KdPk11195 . La
valeur de la constante d’affinité de la PPIX pour la mTSPO1 est de l’ordre du µM en accordavec les données de la littérature [Hirsch et al., 1988, Ostuni et al., 2010b] acquises sur desmembranes natives.
Liaison du cholestérol Les protéoliposomes contenant la mTSPO1 ont été incubés avec desdoses croissantes de cholestérol radioactif et filtrés comme précédemment décrit [Lacapere et al.,2001, Ostuni et al., 2010b, Jamin et al., 2005, Harbi Iatmanen, 2014]. La courbe de saturationdonne une affinité de 6±2 nM et un Bmax de 37±4 nmol de cholestérol/mg de mTSPO1. Iln’existe par de mesure de liaison de cholestérol in cellulo. Le Bmax pourrait suggérer la liaisond’une molécule de cholestérol pour 2 molécules de TSPO, mais il est plus probable que la
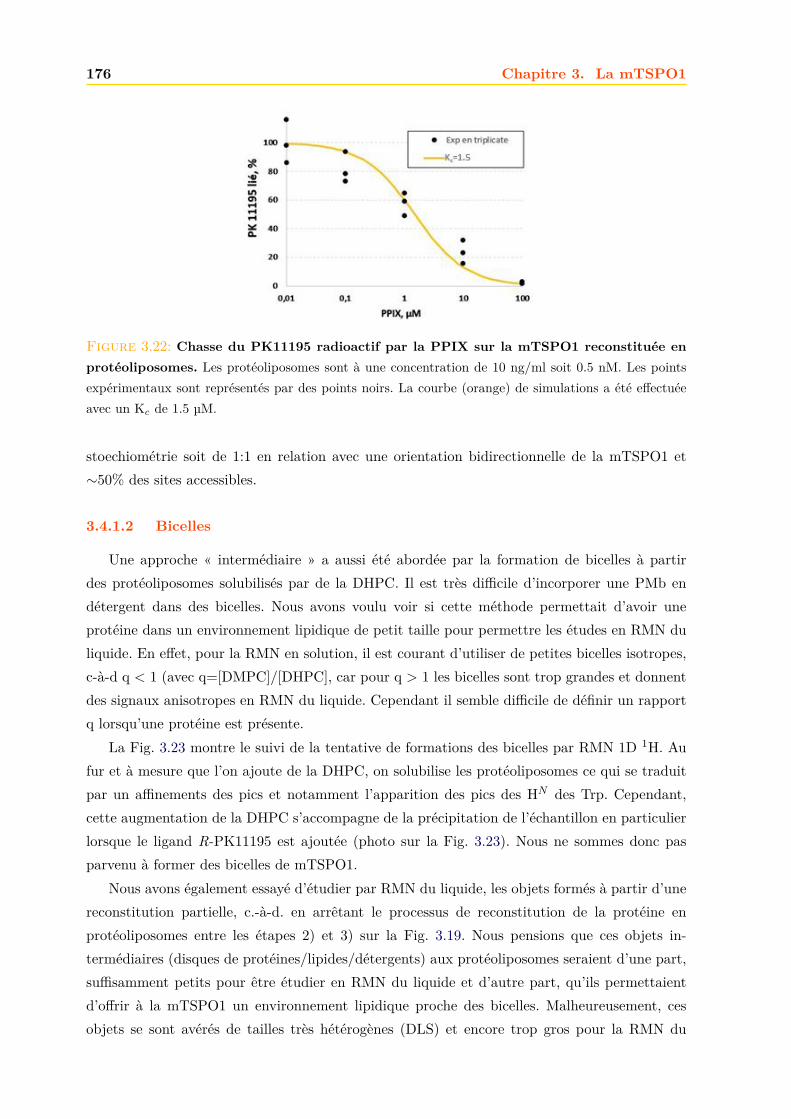
176 Chapitre 3. La mTSPO1
Figure 3.22: Chasse du PK11195 radioactif par la PPIX sur la mTSPO1 reconstituée enprotéoliposomes. Les protéoliposomes sont à une concentration de 10 ng/ml soit 0.5 nM. Les pointsexpérimentaux sont représentés par des points noirs. La courbe (orange) de simulations a été effectuéeavec un Kc de 1.5 µM.
stoechiométrie soit de 1:1 en relation avec une orientation bidirectionnelle de la mTSPO1 et∼50% des sites accessibles.
3.4.1.2 Bicelles
Une approche « intermédiaire » a aussi été abordée par la formation de bicelles à partirdes protéoliposomes solubilisés par de la DHPC. Il est très difficile d’incorporer une PMb endétergent dans des bicelles. Nous avons voulu voir si cette méthode permettait d’avoir uneprotéine dans un environnement lipidique de petit taille pour permettre les études en RMN duliquide. En effet, pour la RMN en solution, il est courant d’utiliser de petites bicelles isotropes,c-à-d q < 1 (avec q=[DMPC]/[DHPC], car pour q > 1 les bicelles sont trop grandes et donnentdes signaux anisotropes en RMN du liquide. Cependant il semble difficile de définir un rapportq lorsqu’une protéine est présente.
La Fig. 3.23 montre le suivi de la tentative de formations des bicelles par RMN 1D 1H. Aufur et à mesure que l’on ajoute de la DHPC, on solubilise les protéoliposomes ce qui se traduitpar un affinements des pics et notamment l’apparition des pics des HN des Trp. Cependant,cette augmentation de la DHPC s’accompagne de la précipitation de l’échantillon en particulierlorsque le ligand R-PK11195 est ajoutée (photo sur la Fig. 3.23). Nous ne sommes donc pasparvenu à former des bicelles de mTSPO1.
Nous avons également essayé d’étudier par RMN du liquide, les objets formés à partir d’unereconstitution partielle, c.-à-d. en arrêtant le processus de reconstitution de la protéine enprotéoliposomes entre les étapes 2) et 3) sur la Fig. 3.19. Nous pensions que ces objets in-termédiaires (disques de protéines/lipides/détergents) aux protéoliposomes seraient d’une part,suffisamment petits pour être étudier en RMN du liquide et d’autre part, qu’ils permettaientd’offrir à la mTSPO1 un environnement lipidique proche des bicelles. Malheureusement, cesobjets se sont avérés de tailles très hétérogènes (DLS) et encore trop gros pour la RMN du
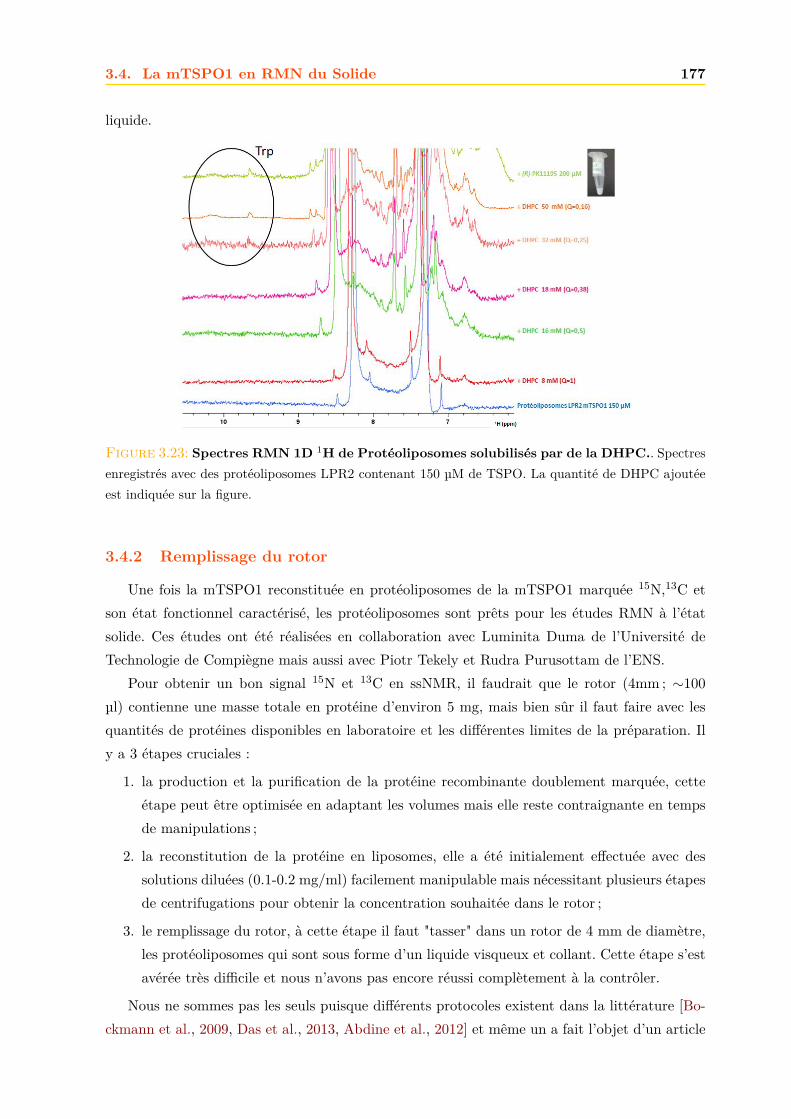
3.4. La mTSPO1 en RMN du Solide 177
liquide.
Figure 3.23: Spectres RMN 1D 1H de Protéoliposomes solubilisés par de la DHPC.. Spectresenregistrés avec des protéoliposomes LPR2 contenant 150 µM de TSPO. La quantité de DHPC ajoutéeest indiquée sur la figure.
3.4.2 Remplissage du rotor
Une fois la mTSPO1 reconstituée en protéoliposomes de la mTSPO1 marquée 15N,13C etson état fonctionnel caractérisé, les protéoliposomes sont prêts pour les études RMN à l’étatsolide. Ces études ont été réalisées en collaboration avec Luminita Duma de l’Université deTechnologie de Compiègne mais aussi avec Piotr Tekely et Rudra Purusottam de l’ENS.
Pour obtenir un bon signal 15N et 13C en ssNMR, il faudrait que le rotor (4mm ; ∼100µl) contienne une masse totale en protéine d’environ 5 mg, mais bien sûr il faut faire avec lesquantités de protéines disponibles en laboratoire et les différentes limites de la préparation. Ily a 3 étapes cruciales :
1. la production et la purification de la protéine recombinante doublement marquée, cetteétape peut être optimisée en adaptant les volumes mais elle reste contraignante en tempsde manipulations ;
2. la reconstitution de la protéine en liposomes, elle a été initialement effectuée avec dessolutions diluées (0.1-0.2 mg/ml) facilement manipulable mais nécessitant plusieurs étapesde centrifugations pour obtenir la concentration souhaitée dans le rotor ;
3. le remplissage du rotor, à cette étape il faut "tasser" dans un rotor de 4 mm de diamètre,les protéoliposomes qui sont sous forme d’un liquide visqueux et collant. Cette étape s’estavérée très difficile et nous n’avons pas encore réussi complètement à la contrôler.
Nous ne sommes pas les seuls puisque différents protocoles existent dans la littérature [Bo-ckmann et al., 2009, Das et al., 2013, Abdine et al., 2012] et même un a fait l’objet d’un article

178 Chapitre 3. La mTSPO1
Nature protocols Das et al. [2013]. Tous commencent par un culot de protéoliposomes par cen-trifugation mais divergent sur la manière de remplir le rotor. Nous en avons essayé plusieurs etnous avons construit différents dispositifs "maisons", en s’inspirant de la méthode de remplissageprésentée par Das et al. [2013], et en l’adaptant à nos outils comme décrit Sec.2.4.5.2 du M&M.
Après la reconstitution, plusieurs étapes de centrifugation ont été réalisées, nous avons me-suré par fluorescence et absorbance le contenu des surnageants pour s’assurer que tous lesprotéoliposomes étaient bien dans le culot. Toutefois, lors des resuspensions il est très difficilede savoir si tout le matériel a bien été repris. Les solutions devenant de plus en plus visqueuses,le pipettage d’un petit volume pour mesurer la quantité de protéines (par absorbance en diluantet en solubilisant dans un tampon contenant du SDS) s’est avéré faussé car il est difficile depipetter un volume précis et homogène.
Ainsi, la concentration du matériel transféré dans le rotor a été difficilement contrôlable.Nous avons essayé de limiter le nombre de centrifugations en faisant des reconstitutions avecdes solutions concentrées (1-2 mg/ml en protéine). Cependant, le remplissage du rotor lui mêmes’est avéré difficile à contrôler. L’utilisation d’un nouveau dispositif, présenté Fig. 2.31 de laSec.2.4.5.2 du M&M, a permis d’améliorer cette étape de remplissage. Toutefois, l’ensembledes 2 étapes de concentrations des protéoliposomes et de remplissage du rotor n’est pas encorecomplètement optimisé.
La quantification de ce que nous avions dans notre rotor c.-à-d. l’efficacité du remplissagedu rotor, est très difficile. Une 1 tentative en pipettant pour mesurer la quantité de protéinesdirectement dans le rotor s’est avérée peu efficace. Une 2e tentative a consisté à essayer d’estimerla quantité de lipides contenus dans le rotor en enregistrant des spectres 1D 1H des lipides, puisconnaissant le LPR auquel les protéoliposomes ont été reconstitués de remonter à la concen-tration en protéine à l’intérieur du rotor. Concrètement, dans un 1er temps, nous avons doncrempli, à la pipette, plusieurs rotors HR-MAS avec différentes concentrations de liposomes deDMPC. Le remplissage des rotors ne posent pas de problème dans ces conditions car les lipidessont très fluides. Pour chacun de ces rotors, le spectre 1D 1H a été enregistré, Fig. 3.24a. Les2 pics les plus intenses apparaissent à 1.3 et 3.3 ppm et correspondent d’une part, aux protonsdes carbones 4 à 13 de la chaine aliphatique et d’autre part, aux protons des carbones γ de latête polaire. Le tracé de l’intensité de ces 2 pics en fonction de la concentration en DMPC peutalors être utilisé pour calibrer le contenu en lipides des rotors, Fig. 3.24b.
Dans un second temps, nous avons enregistré le spectre 1H d’un rotor rempli de protéolipo-somes de mTSPO1 à LPR 2, pour lequel en théorie il devrait y avoir 6 mg total de protéines et12 mg de lipides dans les 100 µl d’échantillon, pour en estimer le contenu en lipides, Fig. 3.25a.Malheureusement, les pics (à 1.3 et 3.3 ppm) des lipides de cet échantillon sont très faibles eten reportant leur intensité sur les courbes de calibrations, Fig. 3.25b nous obtenons une esti-mation de seulement 21 mg/ml de DMPC (en théorie nous aurions du avoir 120 mg/ml) soitune quantité 2.1 mg de lipides totales dans le rotor, soit une quantité de mTSPO1 de ∼ 1 mgtotale à la place des 6 mg attendus. Bien que la mTSPO1 soit reconstituée dans un mélange
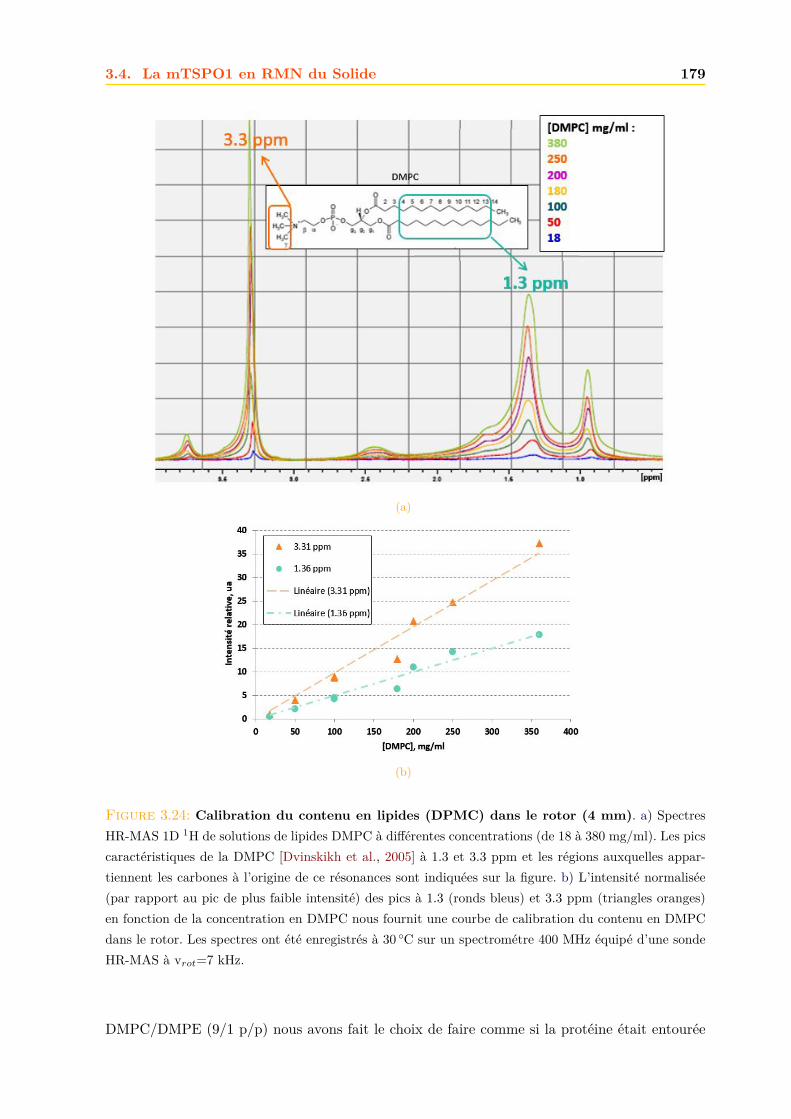
3.4. La mTSPO1 en RMN du Solide 179
(a)
(b)
Figure 3.24: Calibration du contenu en lipides (DPMC) dans le rotor (4 mm). a) SpectresHR-MAS 1D 1H de solutions de lipides DMPC à différentes concentrations (de 18 à 380 mg/ml). Les picscaractéristiques de la DMPC [Dvinskikh et al., 2005] à 1.3 et 3.3 ppm et les régions auxquelles appar-tiennent les carbones à l’origine de ce résonances sont indiquées sur la figure. b) L’intensité normalisée(par rapport au pic de plus faible intensité) des pics à 1.3 (ronds bleus) et 3.3 ppm (triangles oranges)en fonction de la concentration en DMPC nous fournit une courbe de calibration du contenu en DMPCdans le rotor. Les spectres ont été enregistrés à 30 ◦C sur un spectrométre 400 MHz équipé d’une sondeHR-MAS à vrot=7 kHz.
DMPC/DMPE (9/1 p/p) nous avons fait le choix de faire comme si la protéine était entourée
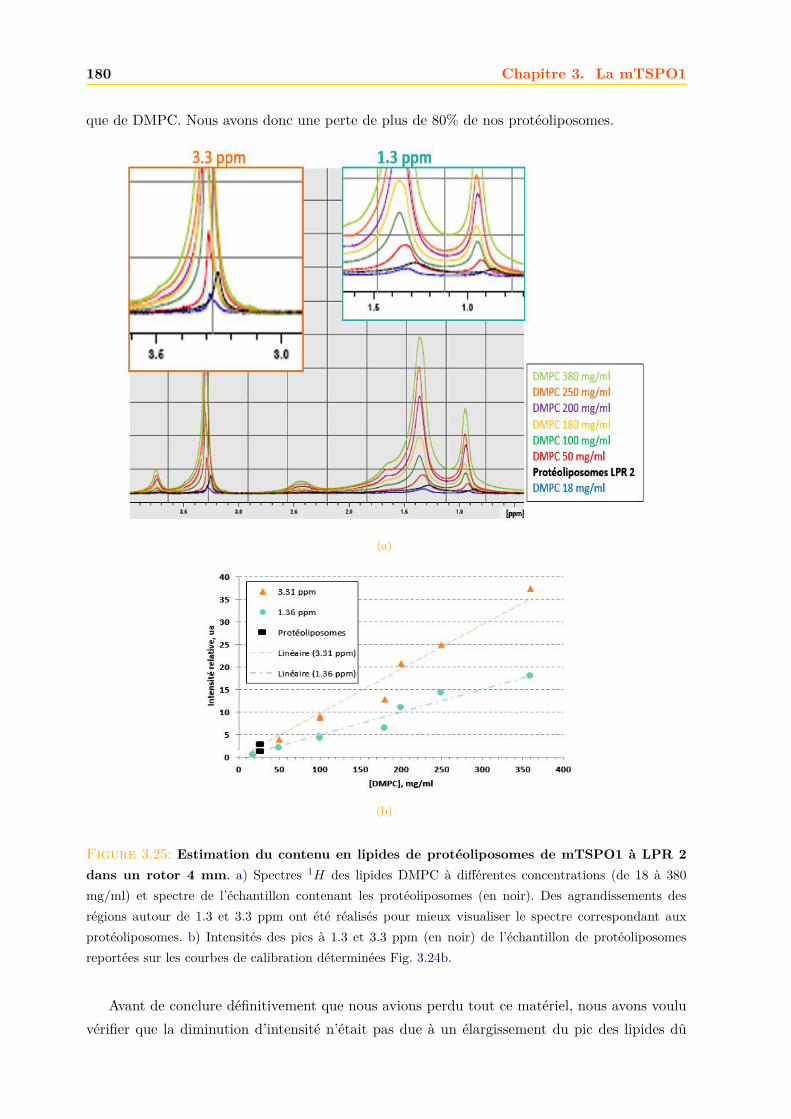
180 Chapitre 3. La mTSPO1
que de DMPC. Nous avons donc une perte de plus de 80% de nos protéoliposomes.
(a)
(b)
Figure 3.25: Estimation du contenu en lipides de protéoliposomes de mTSPO1 à LPR 2dans un rotor 4 mm. a) Spectres 1H des lipides DMPC à différentes concentrations (de 18 à 380mg/ml) et spectre de l’échantillon contenant les protéoliposomes (en noir). Des agrandissements desrégions autour de 1.3 et 3.3 ppm ont été réalisés pour mieux visualiser le spectre correspondant auxprotéoliposomes. b) Intensités des pics à 1.3 et 3.3 ppm (en noir) de l’échantillon de protéoliposomesreportées sur les courbes de calibration déterminées Fig. 3.24b.
Avant de conclure définitivement que nous avions perdu tout ce matériel, nous avons vouluvérifier que la diminution d’intensité n’était pas due à un élargissement du pic des lipides dû

3.4. La mTSPO1 en RMN du Solide 181
(a)
(b)
(c)
Figure 3.26: Spectres CP-MAS 13C : a) protéoliposomes avec un LPR 10, b) liposomes purs(DMPC/DMPE 9/1) c) soustraction des spectres a) et b). Les déplacements chimiques caractéristiquesdes différents groupes sont indiqués. Spectres enregistrés à 25 ◦C avec une vrot de 7 kHz avec découplagehétéronucléaire.
à la présence des protéines. En effet, nous observons un décalage vers les basses fréquencesdes 2 pics majoritaires de nos lipides dans l’échantillon de protéoliposomes, montrant que laprotéine modifie l’environnement des lipides. Il est donc ainsi fort probable que la présence dela protéine cause un élargissement des pics de lipides, ce qui nous donne une sur-évaluation dela perte. Dans le but de déterminer comment la protéine affecte le spectre HR-MAS 1D 1Hlipidique, nous avons essayé de préparer plusieurs échantillons de protéoliposomes à différentsLPR en gardant la quantité de lipides constante. Malheureusement, il fut impossible de remplirles rotors de la même façon, chaque LPR ne présentant pas le même "comportement". P. ex, unéchantillon LPR 10 est facilement manipulable alors qu’un échantillon LPR 1 est très collant.Les données ne purent donc être interprétées.
Une autre approche pour rendre le remplissage du rotor plus facile pourrait être d’utiliser desLPR plus élevés et de profiter de la meilleure fluidité du système. Cependant, dans ces conditionsles signaux des lipides deviennent importants et peuvent masquer ceux de la protéine. En effet,Le spectre CP-MAS 13C d’un échantillon de protéoliposomes de mTSPO1 à un LPR de 10 (p/p),Fig. 3.26a, montre la présence de pics très intenses (entre 10 et 80 ppm), provenant des 13Cen abondance naturelle des lipides. Un spectre de liposomes pures (DMPC/DMPE) enregistré,

182 Chapitre 3. La mTSPO1
Fig. 3.26b, confirme que ces signaux proviennent des lipides. Le spectre de la Fig. 3.26c montrela soustraction des deux 1er spectres, après ajustement des intensités, et donne le spectre dela mTSPO1 seule. Ce spectre présente des pics larges qui résultent essentiellement d’un grandnombre des signaux proches.
Dans la suite de nos expériences, nous avons cherché un compromis et travaillé avec unéchantillon de protéoliposomes reconstitué à LPR 3 (0.2 mg/ml initial), le remplissage a étéeffectué par centrifugations successives (Fig. 2.33 du M&M) et la quantité de mTSPO1 dans lerotor de 4 mm a été estimée à 3 mg alors qu’elle aurait due être de 9 mg.
3.4.3 Expériences CP-MAS 1D 13C
Dans les solides, les interactions dipolaires sont dominantes et donc privilégiées pour lesexpériences de transfert car plus efficaces. Pour tout système en général et les lipides, on peutrigidifier le système en baisant la température.
L’état physique des lipides dans la bicouche des membranes change de façon très importante sous l’effetdes variations de température. Ces changements correspondent à des transitions de phases. Une transi-tion de phase correspond à un changement dans l’organisation et la mobilité des chaines d’hydrocarburesà l’intérieur de la bicouche. En dessous de la température de transition, les lipides sont en phase gel : leslipides se trouvent dans un état ordonné, leurs chaines hydrocarbonées sont en configuration all-trans- les chaines sont rigides parallèles les unes aux autres - Fig. 3.27a. Au dessus de la température detransition, les lipides sont en phase cristal-liquide : l’augmentation de la température induit de plus enplus de mouvements de rotation autour des liaisons C-C, les lipides se retrouvent alors dans un état plusdésordonné, leurs chaines hydrocarbonées contiennent un grand nombre de conformères gauches - uneou plusieurs des liaisons C-C des chaines acyles sont tordues - on parle alors de lipides en conformationall-gauche, Fig. 3.27b. La température de transition (Tm) dépend de la longueur, de la nature de latête polaire et du degré de d’insaturation des chaines acyles : les températures de transition faibles sontassociées à des chaines plus courtes et des degrés d’insaturation plus élevés. P. ex, la Tm de la DMPCest de 24˚, DPPC 42 ◦C, DOPC −22 ◦C, DMPE 49 ◦C. Les spectres 13C ssNMR des phospholipidessont sensibles aux mouvements de la bicouche lipidique présents en phase cristal-liquide et permettentdonc d’étudier la dynamique des différentes régions (tête polaire, squelette glycérol et chaine acyle) desmolécules de lipides [Cornell, 1980].
Encadré 14 (Transition de phase des lipides).
Température Les structures et les dynamiques des PMb sont fortement affectées par lespropriétés des lipides qui les entourent, Encadré 14. Nous nous sommes donc posés la questionde l’effet de la température sur le spectre CP-MAS 1D 13C notamment à cause de la températurede transition des lipides (pour la DMPC la température de transition Tm est de 23 ◦C).
Dans un 1er temps, des spectres CP-MAS 1D 13C ont été enregistrés sur des protéolipomes

3.4. La mTSPO1 en RMN du Solide 183
(a) (b)
Figure 3.27: Illustration desconformations all-trans et all-gauche sur la structure d’unglycérophospholipides. a : confor-mation all-trans. b : lipide avec desconformations trans et gauche,les conformations all-gauche sontindiquées par des flèches. Source :http://nptel.ac.in/courses/
102103044/module2/lec10/7.
html.
DMPC/DMPE (9/1 p/p) de mTSPO1 entre 284 et 313 K (température du thermocouple).Sur ces spectres présentés Fig. 3.28a on voit clairement que le pic attribué aux conforma-tions all-trans à ∼33 ppm se déplace vers ∼31 ppm avec l’augmentation de la température. Cedéplacement est attribuable à une conversion des conformations all-trans en all-gauche. À latempérature de transition on voit la présence simultanée des pics des conformations all-trans etall-gauche qui montre la co-existence des 2 populations. Ces résultats sont en accord avec ceuxobtenus sur un échantillon de liposomes de DMPC pures et dont la transition gel/cristal-liquidea été analysée en détail dans Purusottam et al. [2015], Fig. 3.29.
Le suivi de l’intensité des résonances des lipides all-gauche et all-trans en fonction de la tem-pérature, sur les spectres CP-MAS 1D 13C des protéoliposomes, permet d’estimer la températurede transition des lipides de l’échantillon, Fig. 3.28b. Pour notre échantillon de protéopliposomesla Tm des lipides est estimée à ∼ 293 K (20 ◦C), une valeur inférieure à celle de la DMPCpure (24 ◦C). Au vu du mélange de lipides utilisé (DMPC/DMPE, 9/1 p/p) et de la Tm de laDMPE (49 ◦C), on pourrait s’attendre à une température de transition légèrement plus élevéeque celle de la DMPC pure (24 ◦C). Iglic [2011] reporte que les interactions protéine-lipide ont,en général, pour effet de diminuer la température de transition à cause d’une stabilisation deslipides par les protéines. Ainsi, l’observation de cette diminution de la température de transitionde phase serait due à la présence de la mTSPO1 dans la bicouche lipidique à notre LPR de 3.En réalité, la température de l’échantillon est plus grande que la température indiquée par lethermocouple à cause de la friction induite par la rotation à l’angle magique. Cet échauffementdépend du type de sonde et de la fréquence de rotation. Pour un rotor 4 mm aux fréquencesde rotation utilisées, cet échauffement ne devrait pas dépasser 10 ◦C. Ainsi, notre approche dedétermination de la Tm est sujette aux erreurs introduites par l’échauffement dû à la frictionet à la contribution des signaux protéiques à l’intensité des résonances des lipides.
La présence de ce signal qui reflète la dynamique (ordre-désordre) et la conformation de lachaîne acyle des phospholipides, se révèle donc être une empreinte simple de la transition de
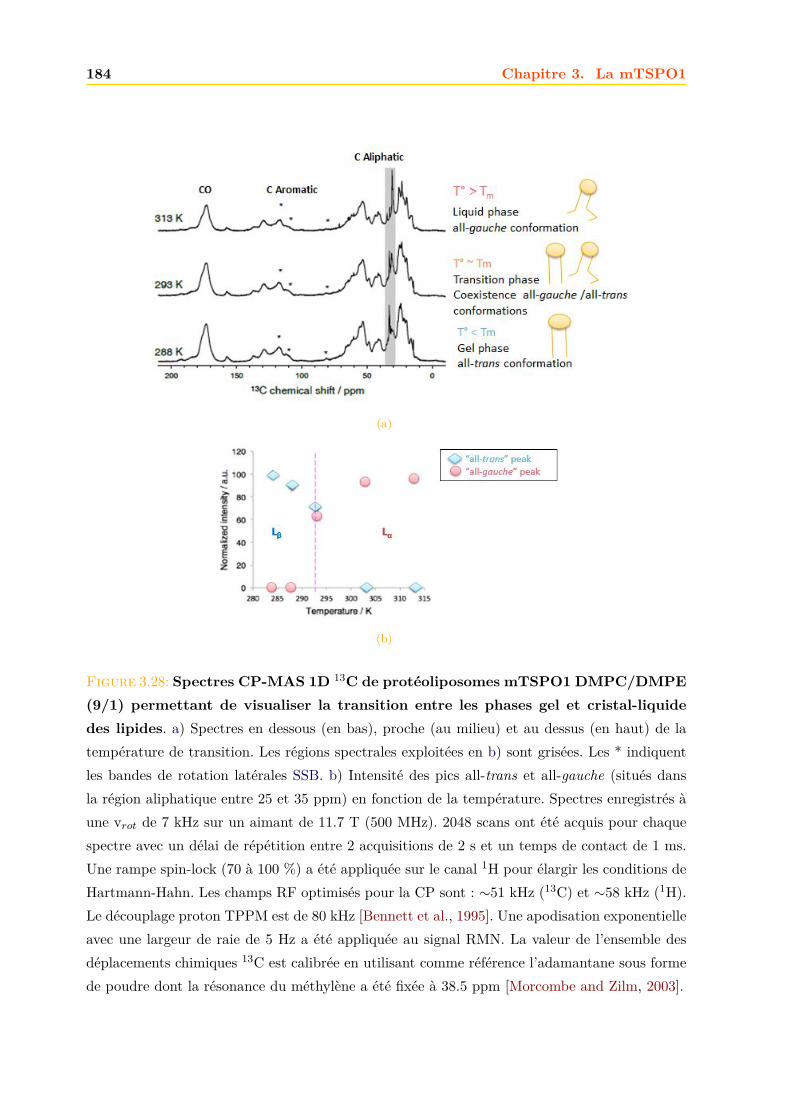
184 Chapitre 3. La mTSPO1
(a)
(b)
Figure 3.28: Spectres CP-MAS 1D 13C de protéoliposomes mTSPO1 DMPC/DMPE(9/1) permettant de visualiser la transition entre les phases gel et cristal-liquidedes lipides. a) Spectres en dessous (en bas), proche (au milieu) et au dessus (en haut) de latempérature de transition. Les régions spectrales exploitées en b) sont grisées. Les * indiquentles bandes de rotation latérales SSB. b) Intensité des pics all-trans et all-gauche (situés dansla région aliphatique entre 25 et 35 ppm) en fonction de la température. Spectres enregistrés àune vrot de 7 kHz sur un aimant de 11.7 T (500 MHz). 2048 scans ont été acquis pour chaquespectre avec un délai de répétition entre 2 acquisitions de 2 s et un temps de contact de 1 ms.Une rampe spin-lock (70 à 100 %) a été appliquée sur le canal 1H pour élargir les conditions deHartmann-Hahn. Les champs RF optimisés pour la CP sont : ∼51 kHz (13C) et ∼58 kHz (1H).Le découplage proton TPPM est de 80 kHz [Bennett et al., 1995]. Une apodisation exponentielleavec une largeur de raie de 5 Hz a été appliquée au signal RMN. La valeur de l’ensemble desdéplacements chimiques 13C est calibrée en utilisant comme référence l’adamantane sous formede poudre dont la résonance du méthylène a été fixée à 38.5 ppm [Morcombe and Zilm, 2003].
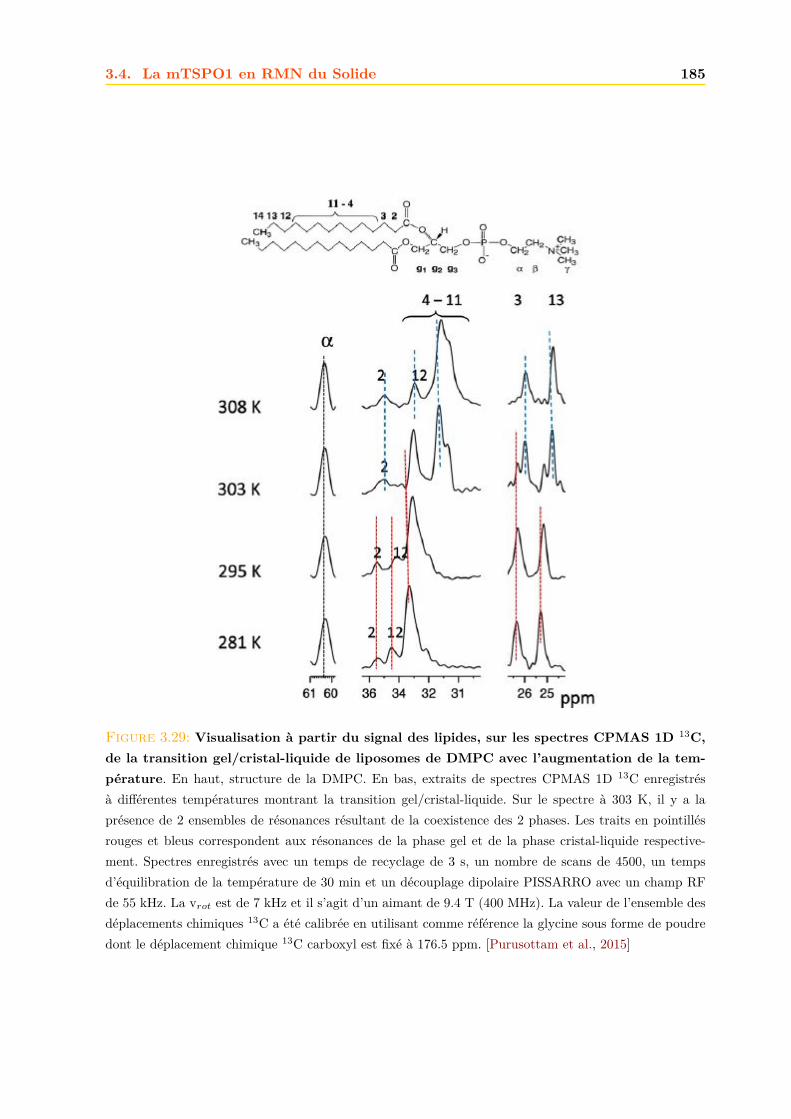
3.4. La mTSPO1 en RMN du Solide 185
Figure 3.29: Visualisation à partir du signal des lipides, sur les spectres CPMAS 1D 13C,de la transition gel/cristal-liquide de liposomes de DMPC avec l’augmentation de la tem-pérature. En haut, structure de la DMPC. En bas, extraits de spectres CPMAS 1D 13C enregistrésà différentes températures montrant la transition gel/cristal-liquide. Sur le spectre à 303 K, il y a laprésence de 2 ensembles de résonances résultant de la coexistence des 2 phases. Les traits en pointillésrouges et bleus correspondent aux résonances de la phase gel et de la phase cristal-liquide respective-ment. Spectres enregistrés avec un temps de recyclage de 3 s, un nombre de scans de 4500, un tempsd’équilibration de la température de 30 min et un découplage dipolaire PISSARRO avec un champ RFde 55 kHz. La vrot est de 7 kHz et il s’agit d’un aimant de 9.4 T (400 MHz). La valeur de l’ensemble desdéplacements chimiques 13C a été calibrée en utilisant comme référence la glycine sous forme de poudredont le déplacement chimique 13C carboxyl est fixé à 176.5 ppm. [Purusottam et al., 2015]

186 Chapitre 3. La mTSPO1
phase gel/cristal-liquide des phospholipides des liposomes. Il peut être exploité pour estimer latempérature de l’échantillon et s’assurer p. ex., que les phospholipides sont bien en phase cristal-liquide ce qui permet la rotation uniaxiale rapide de la protéine dans la bicouche, cette conditionétant nécessaire pour utiliser les techniques de ssNMR présentés dans Marassi et al. [2011], Daset al. [2012], Park et al. [2012, 2010], Das et al. [2015], Opella [2013]. La méthode communémentutilisée pour s’assurer de cette condition utilise les SSB (Spinning Side Band), elle est présentéeFig. 3.30a. Cette figure montre les spectres ssNMR, dans la gamme des déplacements chimiquesdes carbonyles (spectre 13CO-CSA), enregistrés a une température en dessous (10 ◦C) et audessus (25 ◦C) de la température de transition gel/cristal-liquide de la bicouche de phospholipideDMPC/DMPE. Dans les conditions d’une rotation à l’angle magique relativement faible, 3 kHz,le spectre obtenue à 25 ◦C, où la bicouche est en phase cristal-liquide, ne présente pas de SSB.Cela prouve que la mTSPO1 subit une diffusion rotationnelle dans la bicouche suffisammentrapide pour moyenner le spectre, Fig. 3.30b. En comparaison, le spectre 13CO-CSA obtenu à10 ◦C (Fig. 3.30a), où la bicouche lipidique est en phase gel, présente une famille de SSB. Celamontre qu’à cette température, la diffusion rotationnelle n’est pas suffisante pour moyennerl’anisotropie de déplacement chimique.
Cette diffusion rotationnelle peut fournir par la suite les bases pour la méthode de déter-mination structurale développée dans les articles Marassi et al. [2011], Das et al. [2012], Parket al. [2012, 2010], Das et al. [2015], Opella [2013]. En effet, bien que leurs spectres initiauxsoient acquis sur une protéine qui ne subit pas une diffusion rotationnelle rapide, par la suite,ils tirent avantage de cette rotation rapide des PMbs (p. ex. la protéine GPCR) autour de lanormale à la bicouche lipidique pour obtenir de nombreuses informations sur la protéine. Cesinformations sont équivalentes à celles disponibles en RMN du solide avec des échantillons sta-tiques et orientés lesquels, laquelle fournie des contraintes de distances et angulaires pour lescalculs de la structure tridimentionnelle.
L’inspection du δ des pics des 13C du fragment central des chaines acyles des lipides, enabondance naturelle, se révèle donc être une bonne alternative pour le contrôle de cette condition(diffusion rotationnelle de la protéine) ou pour . Cela permet de ne pas avoir recours à l’étudeplus exigeante des SSB uniquement visibles sur des spectres enregistrés à faible νrot [Purusottamet al., 2015].
Étude de la mobilité de l’échantillon de protéoliposomes Le transfert de polarisationest l’expérience clé pour la détection des noyaux peu sensibles par RMN. Le transfert dans lesliquides est souvent réalisé à travers le couplage scalaire J via l’expérience INEPT, alors quedans les solides le couplage dipolaire est utilisé avec l’expérience de polarisation croisée (CP -Cross Polarisation). Les membranes lipidiques ont la particularité de se comporter soit commedes liquides soit comme des solides, selon la température utilisée. La combinaison des différentesméthodes de transfert - CP, excitation directe et INEPT - permet d’obtenir des informationssur la mobilité des lipides et de la protéine [Eddy et al., 2015]. Différents spectres RMN 1D 13C

3.4. La mTSPO1 en RMN du Solide 187
(a) (b)
Figure 3.30: Les effets de la diffusion rotationnelle de la protéine. a) Spectres 13CO-CSAenregistrés à vrot de 3 kHz des proteoliposomes à 10 ◦C (283 K) et 25 ◦C (298 K). Les SSB (SpinningSide Band) correspondent aux bandes de rotations [Purusottam et al., 2015]. b) Représentation d’unePMb en diffusion rotationnelle, par rapport à la normale à la bicouche lipidique (flèches oranges), dansun échantillon en rotation à l’angle magique par rapport au champ magnétique (flèche bleu) [Das et al.,2015]. Encart bleu, représentation du modèle de mosaïque fluide de la membrane biologique [Singer andNicolson, 1972]. Les flèches indiquent la diffusion rotationnelle de la PMb autour de la normale à labicouche [Opella, 2013].
de notre échantillon de protéoliposomes mTSPO1 (LPR 3), dans les conditions de rotation àl’angle magique ont été enregistrés à faible température (284 K - 10 ◦C) :
1. Dans l’expérience CP-MAS le transfert de polarisation entre les 1H et 13C se fait viales couplages dipolaires, c.-à-d. à travers l’espace, ainsi on ne voit que les signaux desmolécules rigides. Le spectre CP-MAS 1D 13C présenté Fig.3.31 (au milieu), révèle lesfragments rigides des lipides et de la protéine.
2. Contrairement à l’expérience CP-MAS, dans l’expérience INEPT refocalisée la polarisa-tion est transférée entre les 1H et 13C via les couplages scalaires J, c-à-d. via les liaisons. Onne voit alors que les signaux des molécules mobiles. Le spectre INEPT 1D-13C, présentéFig.3.31 (en haut), révèle qu’à 284 K, il y a toujours des lipides mobiles avec un pics très fin(∼ 52 ppm) correspondant au CH3 des têtes cholines de la DMPC. Les "signaux INEPT"sont plus faibles que l’intensité des "signaux CP-MAS", indiquant qu’aucun mouvementtrès rapide n’est observé.
3. L’expérience d’excitation directe, quant à elle, est une sorte de combinaison d’une expé-rience INEPT et CP-MAS puisqu’elle détecte le système entier, c.-à-d. qu’on voit à la foisles parties rigides et mobiles, Fig. 3.31 (en bas).
Le spectre 15N CP-MAS a également été enregistré dans les mêmes conditions de tempéra-ture, Fig. 3.32. Ce spectre montre les résonances 15N du squelette et des chaînes latérales de laprotéine.
Ces expériences montrent qu’à une température inférieure à la Tm des lipides, les protéoli-
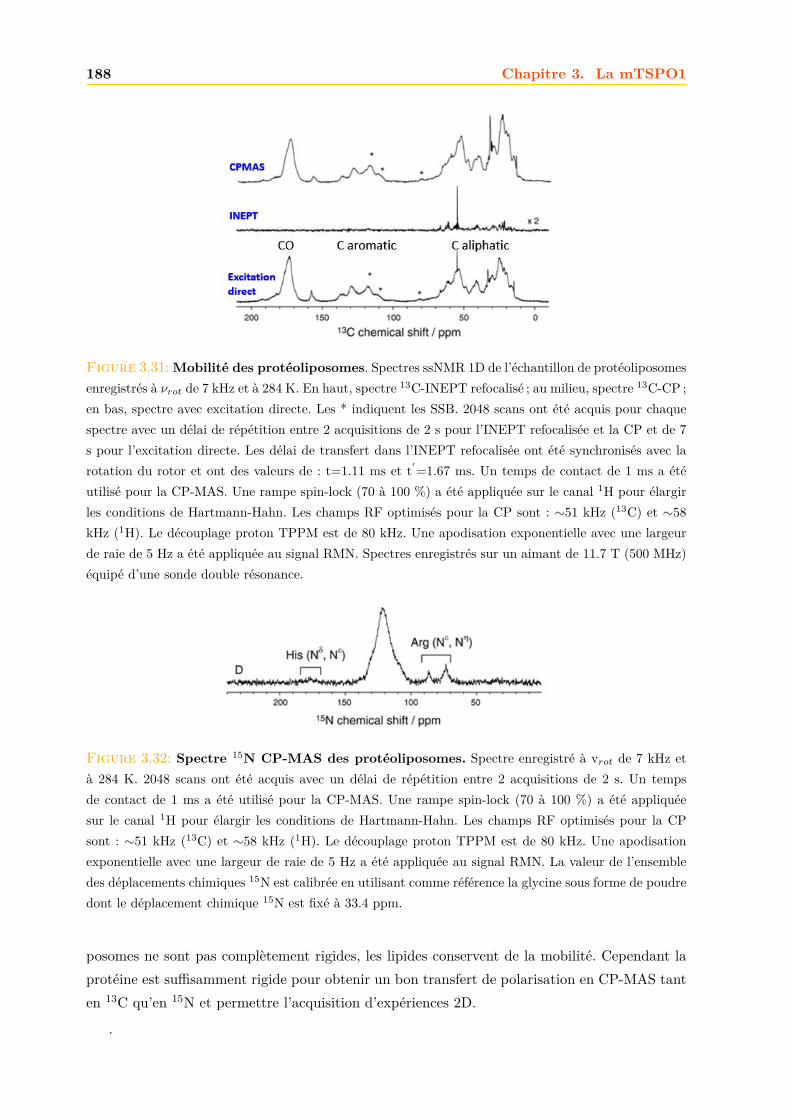
188 Chapitre 3. La mTSPO1
Figure 3.31:Mobilité des protéoliposomes. Spectres ssNMR 1D de l’échantillon de protéoliposomesenregistrés à νrot de 7 kHz et à 284 K. En haut, spectre 13C-INEPT refocalisé ; au milieu, spectre 13C-CP ;en bas, spectre avec excitation directe. Les * indiquent les SSB. 2048 scans ont été acquis pour chaquespectre avec un délai de répétition entre 2 acquisitions de 2 s pour l’INEPT refocalisée et la CP et de 7s pour l’excitation directe. Les délai de transfert dans l’INEPT refocalisée ont été synchronisés avec larotation du rotor et ont des valeurs de : t=1.11 ms et t′=1.67 ms. Un temps de contact de 1 ms a étéutilisé pour la CP-MAS. Une rampe spin-lock (70 à 100 %) a été appliquée sur le canal 1H pour élargirles conditions de Hartmann-Hahn. Les champs RF optimisés pour la CP sont : ∼51 kHz (13C) et ∼58kHz (1H). Le découplage proton TPPM est de 80 kHz. Une apodisation exponentielle avec une largeurde raie de 5 Hz a été appliquée au signal RMN. Spectres enregistrés sur un aimant de 11.7 T (500 MHz)équipé d’une sonde double résonance.
Figure 3.32: Spectre 15N CP-MAS des protéoliposomes. Spectre enregistré à vrot de 7 kHz età 284 K. 2048 scans ont été acquis avec un délai de répétition entre 2 acquisitions de 2 s. Un tempsde contact de 1 ms a été utilisé pour la CP-MAS. Une rampe spin-lock (70 à 100 %) a été appliquéesur le canal 1H pour élargir les conditions de Hartmann-Hahn. Les champs RF optimisés pour la CPsont : ∼51 kHz (13C) et ∼58 kHz (1H). Le découplage proton TPPM est de 80 kHz. Une apodisationexponentielle avec une largeur de raie de 5 Hz a été appliquée au signal RMN. La valeur de l’ensembledes déplacements chimiques 15N est calibrée en utilisant comme référence la glycine sous forme de poudredont le déplacement chimique 15N est fixé à 33.4 ppm.
posomes ne sont pas complètement rigides, les lipides conservent de la mobilité. Cependant laprotéine est suffisamment rigide pour obtenir un bon transfert de polarisation en CP-MAS tanten 13C qu’en 15N et permettre l’acquisition d’expériences 2D.
.
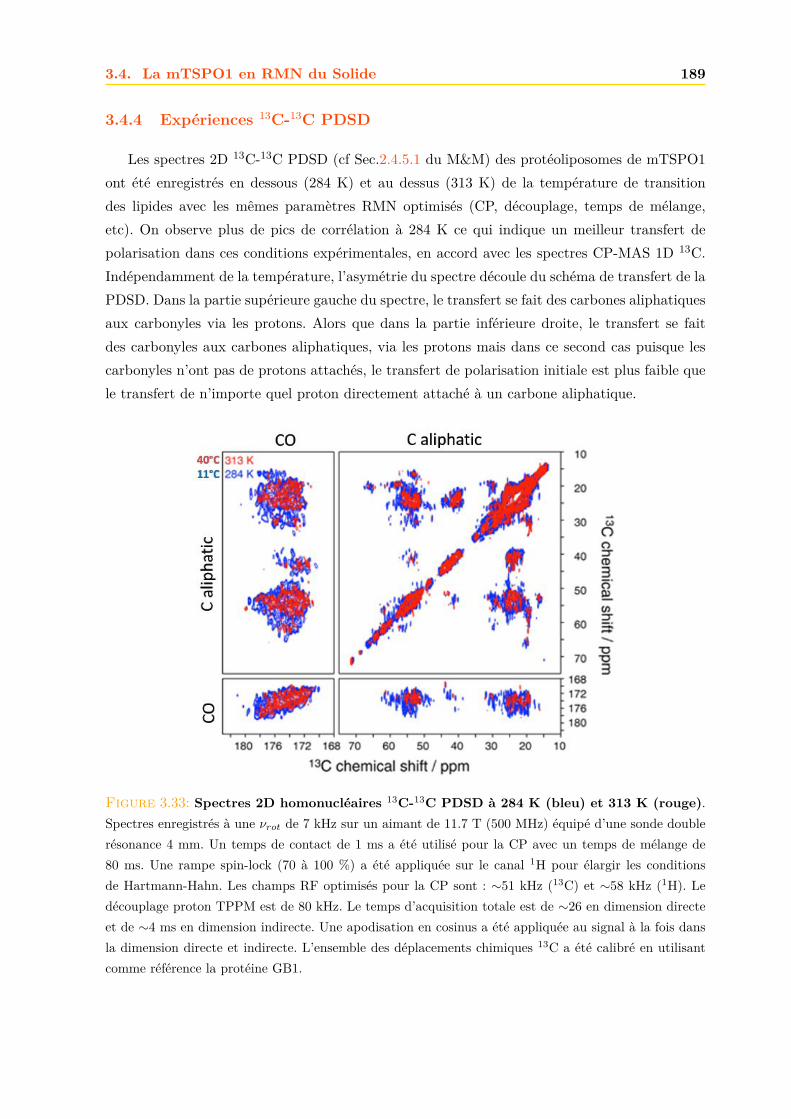
3.4. La mTSPO1 en RMN du Solide 189
3.4.4 Expériences 13C-13C PDSD
Les spectres 2D 13C-13C PDSD (cf Sec.2.4.5.1 du M&M) des protéoliposomes de mTSPO1ont été enregistrés en dessous (284 K) et au dessus (313 K) de la température de transitiondes lipides avec les mêmes paramètres RMN optimisés (CP, découplage, temps de mélange,etc). On observe plus de pics de corrélation à 284 K ce qui indique un meilleur transfert depolarisation dans ces conditions expérimentales, en accord avec les spectres CP-MAS 1D 13C.Indépendamment de la température, l’asymétrie du spectre découle du schéma de transfert de laPDSD. Dans la partie supérieure gauche du spectre, le transfert se fait des carbones aliphatiquesaux carbonyles via les protons. Alors que dans la partie inférieure droite, le transfert se faitdes carbonyles aux carbones aliphatiques, via les protons mais dans ce second cas puisque lescarbonyles n’ont pas de protons attachés, le transfert de polarisation initiale est plus faible quele transfert de n’importe quel proton directement attaché à un carbone aliphatique.
Figure 3.33: Spectres 2D homonucléaires 13C-13C PDSD à 284 K (bleu) et 313 K (rouge).Spectres enregistrés à une νrot de 7 kHz sur un aimant de 11.7 T (500 MHz) équipé d’une sonde doublerésonance 4 mm. Un temps de contact de 1 ms a été utilisé pour la CP avec un temps de mélange de80 ms. Une rampe spin-lock (70 à 100 %) a été appliquée sur le canal 1H pour élargir les conditionsde Hartmann-Hahn. Les champs RF optimisés pour la CP sont : ∼51 kHz (13C) et ∼58 kHz (1H). Ledécouplage proton TPPM est de 80 kHz. Le temps d’acquisition totale est de ∼26 en dimension directeet de ∼4 ms en dimension indirecte. Une apodisation en cosinus a été appliquée au signal à la fois dansla dimension directe et indirecte. L’ensemble des déplacements chimiques 13C a été calibré en utilisantcomme référence la protéine GB1.
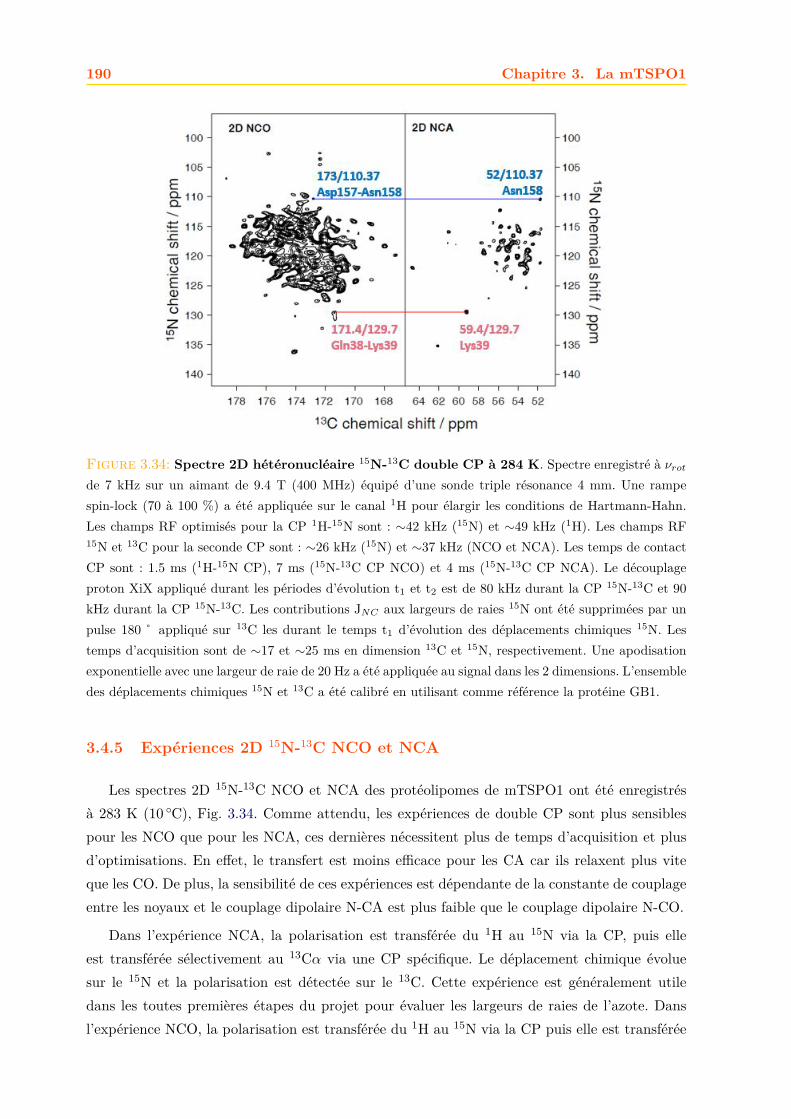
190 Chapitre 3. La mTSPO1
Figure 3.34: Spectre 2D hétéronucléaire 15N-13C double CP à 284 K. Spectre enregistré à νrotde 7 kHz sur un aimant de 9.4 T (400 MHz) équipé d’une sonde triple résonance 4 mm. Une rampespin-lock (70 à 100 %) a été appliquée sur le canal 1H pour élargir les conditions de Hartmann-Hahn.Les champs RF optimisés pour la CP 1H-15N sont : ∼42 kHz (15N) et ∼49 kHz (1H). Les champs RF15N et 13C pour la seconde CP sont : ∼26 kHz (15N) et ∼37 kHz (NCO et NCA). Les temps de contactCP sont : 1.5 ms (1H-15N CP), 7 ms (15N-13C CP NCO) et 4 ms (15N-13C CP NCA). Le découplageproton XiX appliqué durant les périodes d’évolution t1 et t2 est de 80 kHz durant la CP 15N-13C et 90kHz durant la CP 15N-13C. Les contributions JNC aux largeurs de raies 15N ont été supprimées par unpulse 180˚ appliqué sur 13C les durant le temps t1 d’évolution des déplacements chimiques 15N. Lestemps d’acquisition sont de ∼17 et ∼25 ms en dimension 13C et 15N, respectivement. Une apodisationexponentielle avec une largeur de raie de 20 Hz a été appliquée au signal dans les 2 dimensions. L’ensembledes déplacements chimiques 15N et 13C a été calibré en utilisant comme référence la protéine GB1.
3.4.5 Expériences 2D 15N-13C NCO et NCA
Les spectres 2D 15N-13C NCO et NCA des protéolipomes de mTSPO1 ont été enregistrésà 283 K (10 ◦C), Fig. 3.34. Comme attendu, les expériences de double CP sont plus sensiblespour les NCO que pour les NCA, ces dernières nécessitent plus de temps d’acquisition et plusd’optimisations. En effet, le transfert est moins efficace pour les CA car ils relaxent plus viteque les CO. De plus, la sensibilité de ces expériences est dépendante de la constante de couplageentre les noyaux et le couplage dipolaire N-CA est plus faible que le couplage dipolaire N-CO.
Dans l’expérience NCA, la polarisation est transférée du 1H au 15N via la CP, puis elleest transférée sélectivement au 13Cα via une CP spécifique. Le déplacement chimique évoluesur le 15N et la polarisation est détectée sur le 13C. Cette expérience est généralement utiledans les toutes premières étapes du projet pour évaluer les largeurs de raies de l’azote. Dansl’expérience NCO, la polarisation est transférée du 1H au 15N via la CP puis elle est transférée

3.4. La mTSPO1 en RMN du Solide 191
sélectivement au 13CO via une CP spécifique. Le déplacement chimique évolue sur le 15N etla polarisation est détectée sur le 13C. Les spectres NCO et NCA permettent d’extraire lesconnections inter-résiduelle Ni-COi−1 et intra-résiduelle Ni-CAi.
Par exemple, 2 tâches isolées, en bleue et rouge sur la Fig. 3.34, ont été identifiées au mêmedéplacement chimique 15N dans chacun des spectres NCA et NCO. La 1re tentative d’attributiondu résidu i peut être faite en utilisant les coordonnées (Ni-CAi) de son pic de corrélation surle spectre NCA et le programme PLUQ [Fritzsching et al., 2013]. Ce programme utilise unegrande base de données de déplacements chimiques des noyaux 15N et 13C issus de la BMRB(Biological Magnetic Resonance Bank) pour prédire rapidement, à partir des coordonnées dupic de corrélation, le type d’a.a et la structure secondaire à laquelle il est probable d’appartenir.Ensuite une fois le type d’a.a déterminé pour le résidu i, ses coordonnées sur le spectre NCO etles déplacements chimiques des autres 13C sur la PDSD sont utilisés pour valider l’attributionproposée. La 1re tentative d’attribution du résidu i-1 est obtenue à partir des ses coordonnéessur le spectre CO et de sa confrontation à la séquence en acide aminé de la protéine. Ensuite,les corrélations entre ce CO et les carbones aliphatiques donnent accès au type de résidu quipeut être validé par le programme PLUQ.
3.4.6 Conclusions, discussions et perspectives
Etre capable d’étudier une PMb dans un environnement proche de celui de la bicouche lipi-dique devrait permettre que la protéine soit dans une conformation biologiquement pertinente,qui est, autant que possible celle présente dans la cellule vivante. Nos résultats montrent quela détermination de la structure atomique de la mTSPO1 dans un tel environnement sembleaccessible par RMN du solide.
Toutefois nos échantillons ne sont pas encore suffisamment concentrés pour donner des si-gnaux permettant une attribution complète des déplacements chimiques. En effet, les nom-breuses autres expériences 3D (triple CP ; NCACX, NCOCX, CHHC, NHHC) nécessaires pourobtenir l’attribution séquentielle nécessiteront des acquisitions plus longues que les expériences1D et 2D faites jusqu’à présent [Wiegand et al., 2016]. L’étape de remplissage avec ses problèmesde pertes, reste donc encore à optimiser et il sera nécessaire de procéder à des reconstitutionsavec une plus grande quantité (15-20 mg) de protéines au départ, par rapport à ce que nousfaisions d’habitude (5 à 10 mg).
La faible résolution des pics va nécessiter l’utilisation de spectromètre à plus haut champstypes 800 ou 900 MHz pour pouvoir l’augmenter. De tels spectromètres ne possèdent souvent pasde sonde adaptée aux rotors 4 mm que nous avons eu l’habitude d’utiliser lors de cette étude. Eneffet, pour ce type d’appareil il s’agit souvent de sonde 3.2 ou 1.3 mm (c.-à-d. rotors de diamètrede 3.2 et 1.3 mm, respectivement). Encore une fois, la concentration des protéoliposomes etle remplissage de tels rotors restera à résoudre. Néanmoins, ces rotors plus petits présententl’avantage de pouvoir tourner plus vite à l’angle magique permettant d’augmenter encore plusla résolution spectrale.

192 Chapitre 3. La mTSPO1
Effet de la Température La température se révèle un paramètre important pour l’efficacitédu transfert de polarisation dans la CP. Nos expériences PDSD ont pu être améliorées endiminuant la température à 11 ◦C (température inférieure à la température de transition deslipides).À cette basse température, il y a nettement plus de pics de corrélations sur le spectre(orange vs rouge, Fig. 3.35) et c’est dans ces conditions que nous avons enregistré les spectresNCO et NCA.
Nos toutes dernières expériences montrent que nous pouvons encore améliorer nos spectresen diminuant d’avantage la température. La Fig. 3.35b, montre clairement une augmentationde l’intensité des pics de résonances lorsque la température est abaissée à -30 ◦C. Les pics desaromatiques (vers 120-130 ppm) deviennent clairement visibles. De plus, l’effet de la diminutionde la température permet une augmentation impressionnante du nombre de corrélations dans laPDSD (Fig. 3.35, bleu). Cette augmentation à la fois de l’intensité et du nombre de corrélationsindique une augmentation de l’efficacité de transfert de la CP à -30 ◦C par rapport à 11 et 40 ◦C.Cela suggère qu’il existe une rigidification de la protéine indépendante des lipides (T < Tm)qui pourrait être liée à une "congélation" de l’eau. Il se peut que cette congélation de l’eau dansnotre échantillon conduise à une réduction de la mobilité des protéines et des lipides commecela l’a été montré pour d’autres protéines [Perry et al., 2002, Kloepper et al., 2007].
Le fait qu’il soit nécessaire de descendre à une telle température pour rigidier la protéinepourrait suggérer que la protéine dans sa bicouche lipidique à Tamb ou physiologique (37 ◦Cest dynamique, elle subit des mouvements dans la membrane et des réarrangements confor-mationnels locaux. L’échantillon présente une inhomogénéité conformationnelle (la TSPO estdans plusieurs états qui coexistent dans la membrane) ce qui induit plusieurs signaux de chaquecarbone et donc un élargissement des raies. Cela signifie que les données de RMN du liquideoù la mTSPO1 n’est pas stable conformationnellement sans ligand reflètent une réalité de laprotéine puisque cette instabilité est observée à la fois en RMN du liquide en détergent et enRMN du solide en membrane.
Le gain de sensibilité obtenu avec cette diminution en température peut s’avérer bénéfiquepour l’enregistrement des spectres double et triple CP nécessaires à l’attribution des résonancespour remonter par la suite à une structure 3D d’une conformation unique et stable de la protéine.Cependant, les spectres 13C-13C restent très encombrés et il est fort probable qu’un passage pasun marquage sélectif de certains a.a soit nécessaire pour faciliter l’attribution.
Effet du ligand Nous avons effectué des expériences préliminaires pour tenter de voir l’effetdu PK11195 sur le spectre de la mTSPO1. Des protéoliposomes ont été incubés avec du PK11195en excès pour saturer les sites de liaisons. Le tout a été centrifugé (Beckmann, 1h, 200000 × g)et le culot repris dans un petit volume de surnageant pour remplir le rotor. Nous nous sommesassurés par des tests de liaison de ligand radioactif que le fait de concentrer les protéolipomesavec le PK11195, permettait de conserver la saturation des sites de la mTSPO1.
La Fig. 3.36 montre les spectres de corrélation homonucléaire 13C-13C des protéoliposomes,
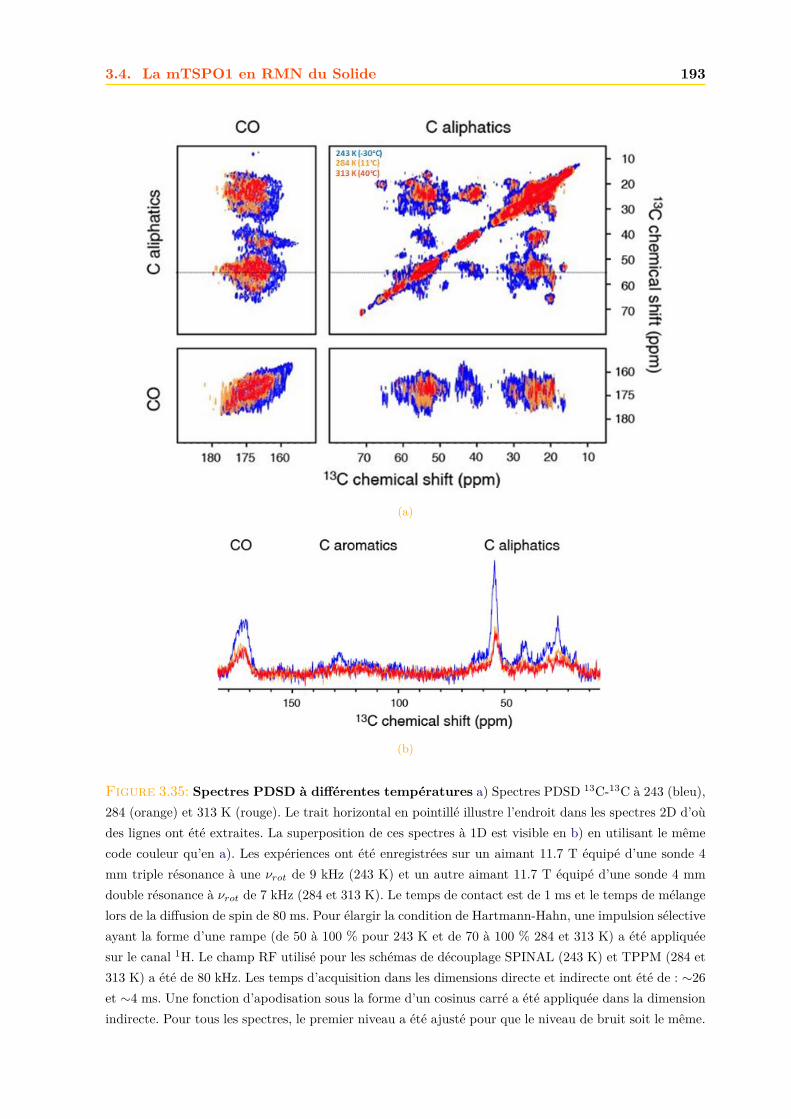
3.4. La mTSPO1 en RMN du Solide 193
(a)
(b)
Figure 3.35: Spectres PDSD à différentes températures a) Spectres PDSD 13C-13C à 243 (bleu),284 (orange) et 313 K (rouge). Le trait horizontal en pointillé illustre l’endroit dans les spectres 2D d’oùdes lignes ont été extraites. La superposition de ces spectres à 1D est visible en b) en utilisant le mêmecode couleur qu’en a). Les expériences ont été enregistrées sur un aimant 11.7 T équipé d’une sonde 4mm triple résonance à une νrot de 9 kHz (243 K) et un autre aimant 11.7 T équipé d’une sonde 4 mmdouble résonance à νrot de 7 kHz (284 et 313 K). Le temps de contact est de 1 ms et le temps de mélangelors de la diffusion de spin de 80 ms. Pour élargir la condition de Hartmann-Hahn, une impulsion sélectiveayant la forme d’une rampe (de 50 à 100 % pour 243 K et de 70 à 100 % 284 et 313 K) a été appliquéesur le canal 1H. Le champ RF utilisé pour les schémas de découplage SPINAL (243 K) et TPPM (284 et313 K) a été de 80 kHz. Les temps d’acquisition dans les dimensions directe et indirecte ont été de : ∼26et ∼4 ms. Une fonction d’apodisation sous la forme d’un cosinus carré a été appliquée dans la dimensionindirecte. Pour tous les spectres, le premier niveau a été ajusté pour que le niveau de bruit soit le même.
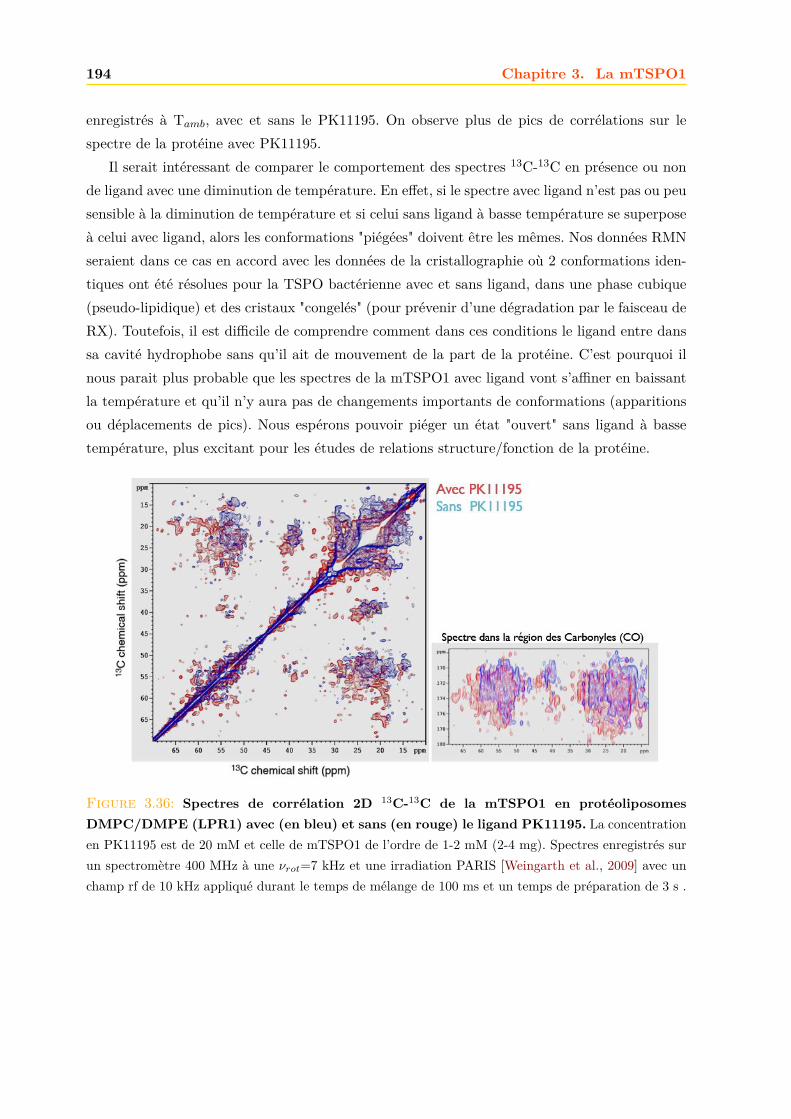
194 Chapitre 3. La mTSPO1
enregistrés à Tamb, avec et sans le PK11195. On observe plus de pics de corrélations sur lespectre de la protéine avec PK11195.
Il serait intéressant de comparer le comportement des spectres 13C-13C en présence ou nonde ligand avec une diminution de température. En effet, si le spectre avec ligand n’est pas ou peusensible à la diminution de température et si celui sans ligand à basse température se superposeà celui avec ligand, alors les conformations "piégées" doivent être les mêmes. Nos données RMNseraient dans ce cas en accord avec les données de la cristallographie où 2 conformations iden-tiques ont été résolues pour la TSPO bactérienne avec et sans ligand, dans une phase cubique(pseudo-lipidique) et des cristaux "congelés" (pour prévenir d’une dégradation par le faisceau deRX). Toutefois, il est difficile de comprendre comment dans ces conditions le ligand entre danssa cavité hydrophobe sans qu’il ait de mouvement de la part de la protéine. C’est pourquoi ilnous parait plus probable que les spectres de la mTSPO1 avec ligand vont s’affiner en baissantla température et qu’il n’y aura pas de changements importants de conformations (apparitionsou déplacements de pics). Nous espérons pouvoir piéger un état "ouvert" sans ligand à bassetempérature, plus excitant pour les études de relations structure/fonction de la protéine.
Figure 3.36: Spectres de corrélation 2D 13C-13C de la mTSPO1 en protéoliposomesDMPC/DMPE (LPR1) avec (en bleu) et sans (en rouge) le ligand PK11195. La concentrationen PK11195 est de 20 mM et celle de mTSPO1 de l’ordre de 1-2 mM (2-4 mg). Spectres enregistrés surun spectromètre 400 MHz à une νrot=7 kHz et une irradiation PARIS [Weingarth et al., 2009] avec unchamp rf de 10 kHz appliqué durant le temps de mélange de 100 ms et un temps de préparation de 3 s .

Chapitre4
La hTSPO2
Sommaire4.1 Expression et Production de la hTSPO2 dans E. coli . . . . . . . . . . . 195
4.1.1 Intégrité et qualité du plasmide pET15b-hTSPO2 . . . . . . . . . . . . . . 195
4.1.2 Expression dans les bactéries BL21 (DE3) pLys S . . . . . . . . . . . . . . . 197
4.1.3 Expression dans les bactéries BL21 (DE3) classiques . . . . . . . . . . . . . 206
4.1.4 Optimisations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
4.1.5 Conclusions, discussions et perspectives . . . . . . . . . . . . . . . . . . . . 209
4.2 Production de la protéine hTSPO2 en système acellulaire ou Cell Free 213
4.2.1 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
4.2.2 Caractérisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
4.3 Conclusions et Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . 218
Chez les mammifères, la protéine membranaire TSPO 18 kDa existe sous 2 isoformes :TSPO1 et TSPO2. Ces 2 protéines sont suggérées être impliquées dans des fonctions différenteset sont localisées dans des membranes différentes : TSPO1 dans la MME et TSPO2 dans lamembrane plasmique (MP) des globules rouges (GR).
À l’heure actuelle il y a très peu de données sur la TSPO2 qui fut découverte en 2009.La détermination de sa structure est une étape importante pour comprendre aussi bien safonction que celle de la TSPO1. Tout comme la TSPO1, l’abondance naturelle de la TSPO2n’est pas suffisante pour entreprendre les études structurales. Nous avons donc tenté de mettreau point la production de la TSPO2 humaine (hTSPO2) recombinante chez la bactérie et sapurification, en se basant sur les méthodes employées pour produire et purifier les isorformesmTSPO1 et hTSPO1 [Robert and Lacapere, 2010], ces dernières partageant 80% d’identité.Les isoformes TSPO (mTSPO1/mTSPO2 et hTSPO1/hTSPO2) partagent ∼ 35% d’identité et47% de similarité, Fig. 4.1. Chez ces 2 isoformes, le domaine CRAC de liaison du cholestérol estconservé mais ni le C-terminal ni le motif d’adressage caractérisé chez la mTSPO1 [Rone et al.,2009b] ne le sont, tout comme la haute affinité pour le PK11195 [Fan et al., 2009, 2012].
4.1 Expression et Production de la hTSPO2 dans E. coli
4.1.1 Intégrité et qualité du plasmide pET15b-hTSPO2
La construction plasmidique pET15b-hTSPO2 a été réalisée par la société ProteoGenix.Nous leur avons demandé la construction de ce plasmide, en leur fournissant la séquence de lahTSPO2 et en leur spécifiant le site de clonage désiré - entre les sites de restriction NdeI/BamHI- NdeI étant juste en aval de l’étiquette histidine et du site de clivage à la thrombine.

196 Chapitre 4. La hTSPO2
Humain TSPO
Souris TSPO
Souris TSPO2
Humain TSPO2
Humain TSPO
Souris TSPO
Souris TSPO2
Humain TSPO2
Humain TSPO
Souris TSPO
Souris TSPO2
Humain TSPO2
A P P A C S R H W G 58
. K P W 115
A Q S P R Y T C H G W R . . . 169
P E S L A Y S H W T A 58
. D P W A 115
T R S P P R Y V Y S G S . . . 169
. . . Q Q G P V V G L I C Q P S S . R D E R P V 54
V G F R W L F S T V L A D S P 112
Y V S L V F I Q T T S T Y Q . . . . . . . 162
. . . R Q G A I V V W D H S G P R M F Y Q T A 55
V G L G W L A V T S T V V T V P H 113
Y V V S A I T T S V H Q Q P T K S D 170
1 10 20 30 40 50
60 70 80 90 100 110
120 130 140 150 160
Y P P
G V
Y
G Y P P
G M
A R
I L E
V
H I T M E
V
M W P A M G T S F G G E G L R A G L Q G
A F T E A G N A P G M
S A A A T A A F N N R G G R L P
M W P A V G T S F G G E G L R A L Q G
A F T E A G N A P G M
S A A T A H A F N N R G G R L P
M P M C L C K L L
A P A L L
A P N K L L A R L C P P
M P L C G L C K L L
A P A L L
H P N K L L A H L C P P
V F L P L G V F V H W K S W P P H V L V W T L Y S
M G Y S Y L V W K E L G G V P L G L Y T Q L A L W W I F F A R Q G A L V D L L L V
G T V W V L A A L L P Y L A W L T T L Y V W R D E
V L V P L G M F V R W K S W P P R L I W T L Y S
M G Y S Y I V W K E L G G V P L G L Y T Q L A L W W I F F A R Q G A L D L L L V
G V T L W V A A L L P Y L A W L T L Y V W R D E
L F V P L G L I H K W P P H V I V W T I Y S
M G Y S Y L V W K E L G G L P L G L Y S Q L A L W F L F A G A L L D L L L L
G L W I L A A L L P Y L A W L T T I Y L W R D
L F L L P L G L F R W S W P V L V I Y S
V G Y S Y L V W K D L G G L P L G L Y Q L I W L F F H N G A L L L L L L
G L T L W I L A A L L P Y L A W L T S L Y L W R D E
Loop 2
Loop 1
Loop 4
Loop 3
Mt Targeting TSPO1
N-terminal
C-terminal
CRAC Cholesterol Binding
Figure 4.1: Alignement de séquences des protéines TSPO1 et TSPO2 murines et humaines.Les séquences représentées pour la TSPO1 sont : Humain (NP_001243460.1 ; polymorphisme A147T) et Souris(NP_033905.3). Les séquences représentées pour la TSPO2 sont : Humain (Q9CRZ8) et Souris (Q9CRZ8). Lesa.a conservés sont colorées du rouge intense au jaune et les a.a homologues du orange au bleu (pour un cut-offcorrespondant à une similarité de séquence de 50%). Les 5 domaines transmembranaires (TM) sont représentés pardes hélices en haut des séquences et l’emplacement des boucles (Loop) qui les relient est indiqué. Sont égalementreprésentés le domaine CRAC [L/V-x1−5-Y-x1−5-R/D] et le motif de Schellman, correspondant à la séquenced’insertion chez la TSPO1, est indiqué par "Mt location" (Mitochondriale location ; a.a 103 à 108 chez l’homme).Les a.a impliqués dans la liaison direct du PK11195, d’après la structure RMN de la mTSPO1 (2MGY), sontindiqués par une flèche bleue. Les a.a ayant été montrés importants dans la liaison du RO5-4864 - par mutagénèseou analyses bioinformatiques - sont indiqués par une flèche verte. De même, les a.a ayant été montrés importantsdans la liaison du RO5-4864 et du PK11195 - par mutagénèse ou analyses sont indiqués par une flèche bleue.Entre parenthèses sont indiqués les entrées des séquences UniProt. L’alignement a été réalisé avec le serveurClustalW et l’image avec le logiciel Aline [Bond and Schuttelkopf, 2009].
Ce plasmide code pour une protéine hTSPO2, avec une étiquette hexahistidine, de 190 a.asoit un PM théorique de ∼21.2 kDa (calcul fait avec le programme ProtParam ExPASy). Laprotéine non recombinante, c.-à-d. sans l’étiquette His6 et le site de coupure à la thrombine,possède 170 a.a soit un PM de ∼19 kDa.
Avant de commencer l’expression de la hTSPO2, des bactéries DH5α d’E. coli ont été trans-féctées (100 µl de bactéries transféctées avec 50 ng de plasmide), multipliées (500 ml) et lyséespour avoir un stock d’ADN plasmidique (comme décrit Sec.2.1.5 du M&M). Celui-ci a étépurifiée et nous avons contrôler son intégrité et sa qualité (cf. Sec. 2.1.6 du M&M).
La DO260 est utilisée pour déterminer la concentration de l’ADN, et une valeur autour de 1.8pour le ratio DO260/DO280 nous donne une indication sur la pureté de l’ADN. Cependant, cettevaleur ne permet pas de savoir si celui-ci est intègre puisqu’il ne fait pas de distinction entreADN double brin et ADN simple brin, ou nucléotides libres qui résulteraient d’une dégradationdu plasmide.
La combinaison du séquençage, d’un gel d’agarose et de l’utilisation de l’appareil fluorimé-trique Qubit c©, utilisant une sonde fluorescente spécifique qui émet seulement quand elle est liéeà l’ADN double brin, nous a assuré définitivement de la qualité du plasmide :
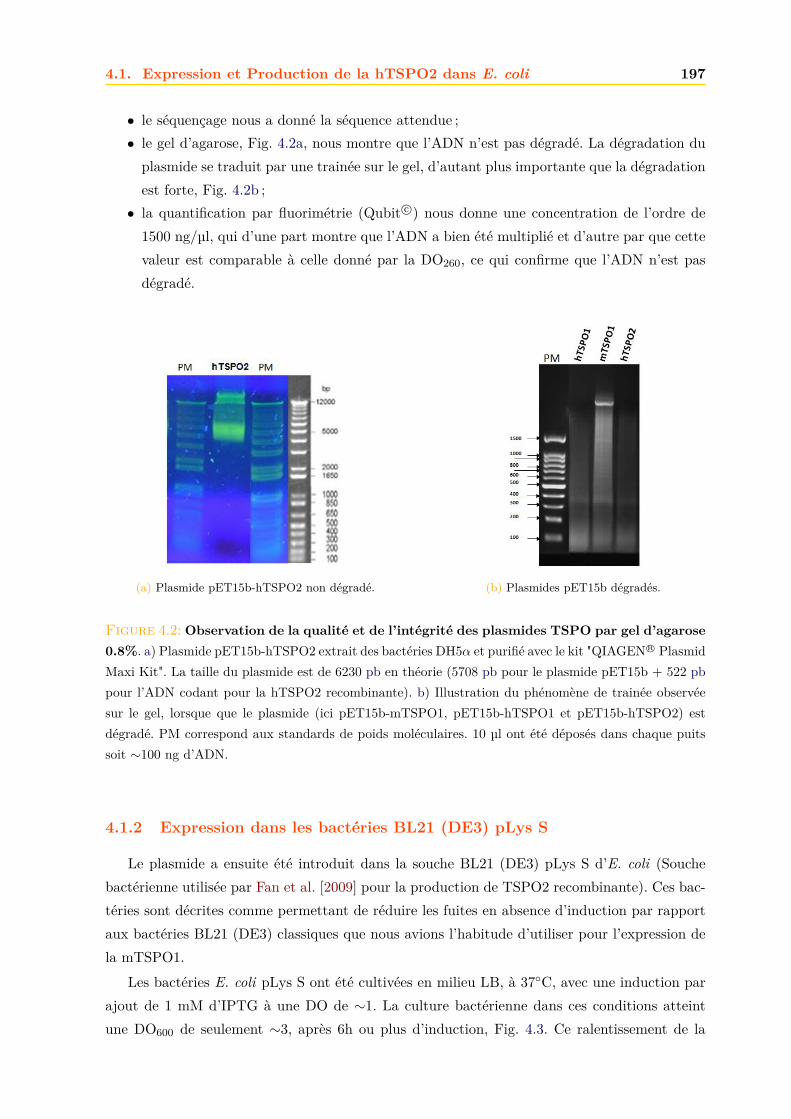
4.1. Expression et Production de la hTSPO2 dans E. coli 197
• le séquençage nous a donné la séquence attendue ;• le gel d’agarose, Fig. 4.2a, nous montre que l’ADN n’est pas dégradé. La dégradation duplasmide se traduit par une trainée sur le gel, d’autant plus importante que la dégradationest forte, Fig. 4.2b ;
• la quantification par fluorimétrie (Qubit c©) nous donne une concentration de l’ordre de1500 ng/µl, qui d’une part montre que l’ADN a bien été multiplié et d’autre par que cettevaleur est comparable à celle donné par la DO260, ce qui confirme que l’ADN n’est pasdégradé.
(a) Plasmide pET15b-hTSPO2 non dégradé. (b) Plasmides pET15b dégradés.
Figure 4.2: Observation de la qualité et de l’intégrité des plasmides TSPO par gel d’agarose0.8%. a) Plasmide pET15b-hTSPO2 extrait des bactéries DH5α et purifié avec le kit "QIAGENR© PlasmidMaxi Kit". La taille du plasmide est de 6230 pb en théorie (5708 pb pour le plasmide pET15b + 522 pbpour l’ADN codant pour la hTSPO2 recombinante). b) Illustration du phénomène de trainée observéesur le gel, lorsque que le plasmide (ici pET15b-mTSPO1, pET15b-hTSPO1 et pET15b-hTSPO2) estdégradé. PM correspond aux standards de poids moléculaires. 10 µl ont été déposés dans chaque puitssoit ∼100 ng d’ADN.
4.1.2 Expression dans les bactéries BL21 (DE3) pLys S
Le plasmide a ensuite été introduit dans la souche BL21 (DE3) pLys S d’E. coli (Souchebactérienne utilisée par Fan et al. [2009] pour la production de TSPO2 recombinante). Ces bac-téries sont décrites comme permettant de réduire les fuites en absence d’induction par rapportaux bactéries BL21 (DE3) classiques que nous avions l’habitude d’utiliser pour l’expression dela mTSPO1.
Les bactéries E. coli pLys S ont été cultivées en milieu LB, à 37◦C, avec une induction parajout de 1 mM d’IPTG à une DO de ∼1. La culture bactérienne dans ces conditions atteintune DO600 de seulement ∼3, après 6h ou plus d’induction, Fig. 4.3. Ce ralentissement de la
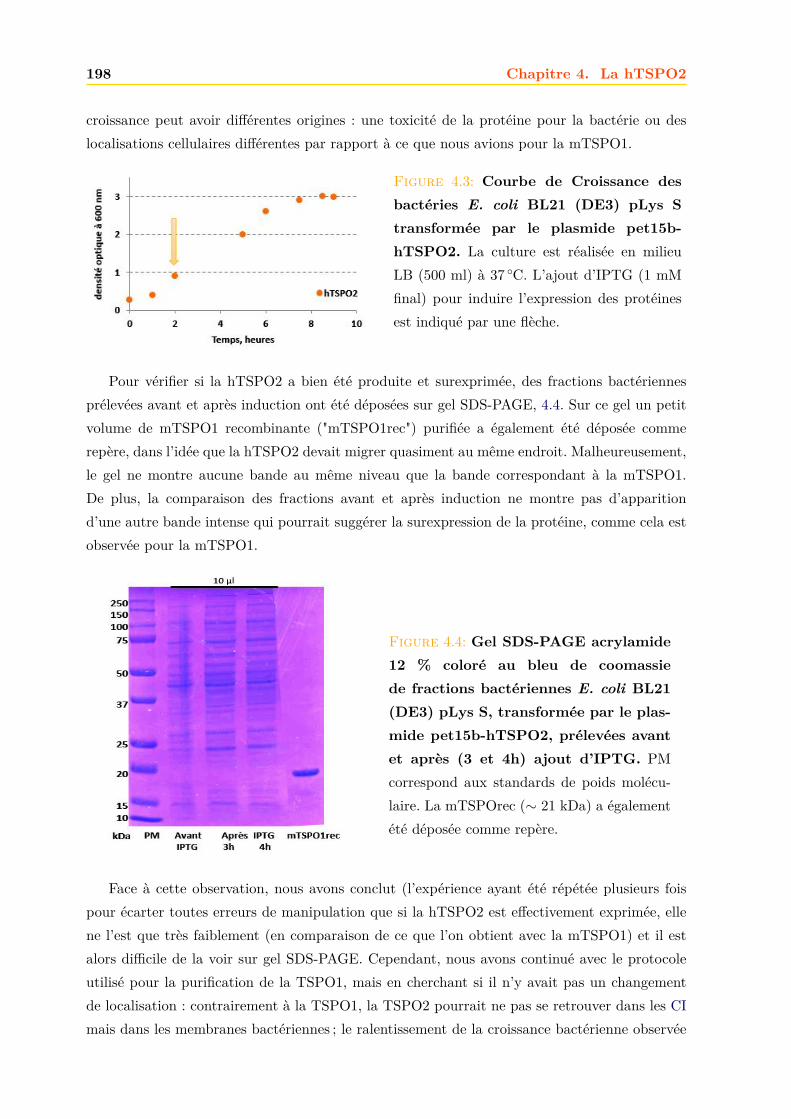
198 Chapitre 4. La hTSPO2
croissance peut avoir différentes origines : une toxicité de la protéine pour la bactérie ou deslocalisations cellulaires différentes par rapport à ce que nous avions pour la mTSPO1.
Figure 4.3: Courbe de Croissance desbactéries E. coli BL21 (DE3) pLys Stransformée par le plasmide pet15b-hTSPO2. La culture est réalisée en milieuLB (500 ml) à 37 ◦C. L’ajout d’IPTG (1 mMfinal) pour induire l’expression des protéinesest indiqué par une flèche.
Pour vérifier si la hTSPO2 a bien été produite et surexprimée, des fractions bactériennesprélevées avant et après induction ont été déposées sur gel SDS-PAGE, 4.4. Sur ce gel un petitvolume de mTSPO1 recombinante ("mTSPO1rec") purifiée a également été déposée commerepère, dans l’idée que la hTSPO2 devait migrer quasiment au même endroit. Malheureusement,le gel ne montre aucune bande au même niveau que la bande correspondant à la mTSPO1.De plus, la comparaison des fractions avant et après induction ne montre pas d’apparitiond’une autre bande intense qui pourrait suggérer la surexpression de la protéine, comme cela estobservée pour la mTSPO1.
Figure 4.4: Gel SDS-PAGE acrylamide12 % coloré au bleu de coomassiede fractions bactériennes E. coli BL21(DE3) pLys S, transformée par le plas-mide pet15b-hTSPO2, prélevées avantet après (3 et 4h) ajout d’IPTG. PMcorrespond aux standards de poids molécu-laire. La mTSPOrec (∼ 21 kDa) a égalementété déposée comme repère.
Face à cette observation, nous avons conclut (l’expérience ayant été répétée plusieurs foispour écarter toutes erreurs de manipulation que si la hTSPO2 est effectivement exprimée, ellene l’est que très faiblement (en comparaison de ce que l’on obtient avec la mTSPO1) et il estalors difficile de la voir sur gel SDS-PAGE. Cependant, nous avons continué avec le protocoleutilisé pour la purification de la TSPO1, mais en cherchant si il n’y avait pas un changementde localisation : contrairement à la TSPO1, la TSPO2 pourrait ne pas se retrouver dans les CImais dans les membranes bactériennes ; le ralentissement de la croissance bactérienne observée

4.1. Expression et Production de la hTSPO2 dans E. coli 199
pourrait être dû alors au fait que la machinerie bactérienne est énormément sollicitée pourintégrer la protéine recombinante dans ses membranes.
Figure 4.5: Schéma illustrant les étapes de traitement des bactéries : (1) culture ;(2) séparation du cytosol (Cyt) (contenant les protéines solubles, l’ADN/ARN et les débrismembranaires) des corps (CI) insolubles ; (3) la TSPO2 est-elle dans les membranes obtenuespar centrifugation de la fraction cytosolique, (4) ou peut-elle être extraite des CI solubilisés enSDS 1% ou (6) ?
À la suite de la culture (étape (1) sur la Fig. 4.5), les bactéries ont été lysées et les corpsd’inclusions (CI) séparés, par centrifugation, du cytosol (Cyt) contenant les protéines solubles,l’ADN/ARN et les membranes bactériennes (Mb ; étape (2) sur la Fig. 4.5). Ensuite, Ces Mbont été isolées du reste du cytosol par centrifugation pendant, 1h à 50000×g et 4 ◦C (étape (3)sur la Fig. 4.5). Les CI ont été solubilisés en SDS pour en extraire les protéines (étape (4) sur laFig. 4.5). Après ces différentes étapes, nous avons pu procéder à la purification de ces fractions.
4.1.2.1 Purification des corps d’inclusion
Après l’extraction des protéines des CI par solubilisation avec du SDS 1%, la purificationsur colonne de résine Ni-NTA (1 ml) a été réalisée. Les spectres d’absorption de chacune desétapes de la purification (charge, lavages et élution) ont été enregistrés entre 240 et 350 nm,Fig. 4.6a.
Au fur et à mesure des lavages (L1 à L5), l’absorption à 260 nm (caractéristique des acidesnucléiques) diminue au profit d’une augmentation de celle à 280 nm, avec l’apparition d’unépaulement à 290 nm et d’un minimun à 250 nm lesquels sont caractéristiques des protéines,Fig. 4.6a.
Les fractions (E1 à E6) de l’élution par 200 mM d’imidazole donnent des spectres d’absorp-tion correspondant à des protéines. Nous avons alors pensé que cela pouvait être des spectres dela hTSPO2. Cependant, on peut noter que l’absorbance, entre 310 et 350 nm, dans ces fractionsd’élution ne retombe pas au niveau de la ligne de base. Ceci suggère la présence de d’objets,diffusant à ces longueurs d’onde, comme des membranes.
La représentation des DO280, Fig. 4.6b, montre une forte concentration en protéines pour lafraction E3.
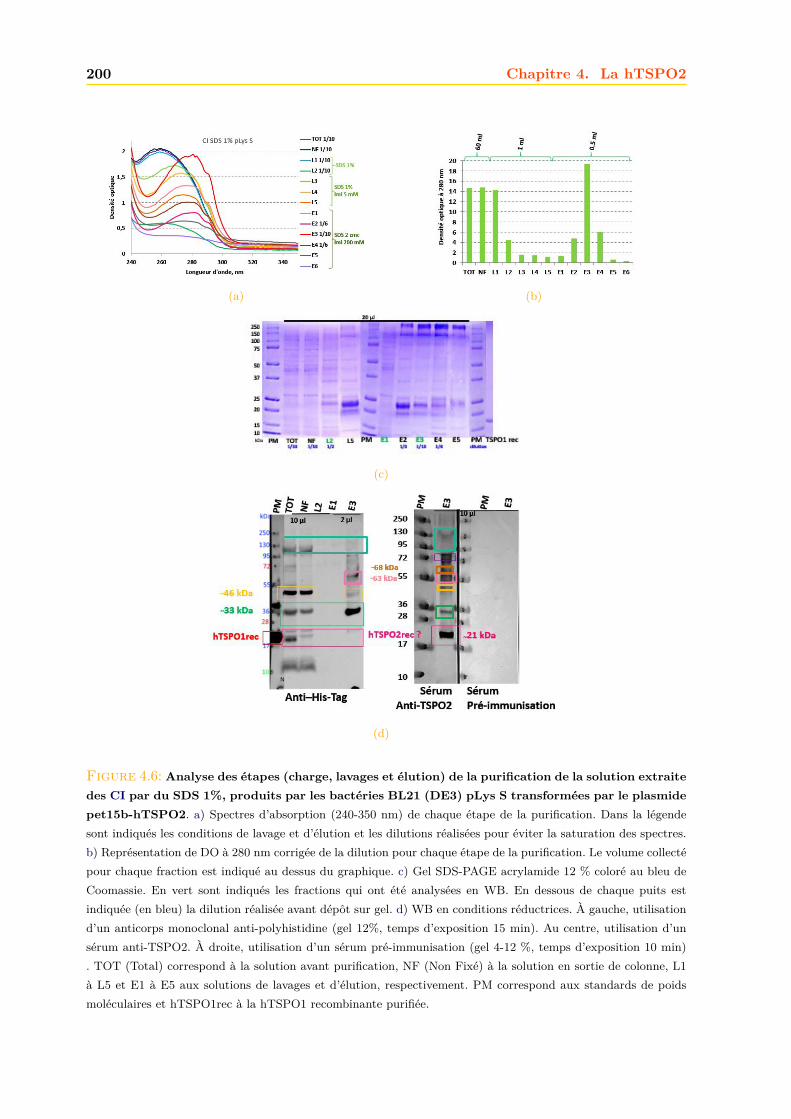
200 Chapitre 4. La hTSPO2
(a) (b)
(c)
(d)
Figure 4.6: Analyse des étapes (charge, lavages et élution) de la purification de la solution extraitedes CI par du SDS 1%, produits par les bactéries BL21 (DE3) pLys S transformées par le plasmidepet15b-hTSPO2. a) Spectres d’absorption (240-350 nm) de chaque étape de la purification. Dans la légendesont indiqués les conditions de lavage et d’élution et les dilutions réalisées pour éviter la saturation des spectres.b) Représentation de DO à 280 nm corrigée de la dilution pour chaque étape de la purification. Le volume collectépour chaque fraction est indiqué au dessus du graphique. c) Gel SDS-PAGE acrylamide 12 % coloré au bleu deCoomassie. En vert sont indiqués les fractions qui ont été analysées en WB. En dessous de chaque puits estindiquée (en bleu) la dilution réalisée avant dépôt sur gel. d) WB en conditions réductrices. À gauche, utilisationd’un anticorps monoclonal anti-polyhistidine (gel 12%, temps d’exposition 15 min). Au centre, utilisation d’unsérum anti-TSPO2. À droite, utilisation d’un sérum pré-immunisation (gel 4-12 %, temps d’exposition 10 min). TOT (Total) correspond à la solution avant purification, NF (Non Fixé) à la solution en sortie de colonne, L1à L5 et E1 à E5 aux solutions de lavages et d’élution, respectivement. PM correspond aux standards de poidsmoléculaires et hTSPO1rec à la hTSPO1 recombinante purifiée.

4.1. Expression et Production de la hTSPO2 dans E. coli 201
Analyse sur Gels SDS-PAGE et en WB Le contenu protéique de ces fractions collectéesa été analysé sur gels SDS-PAGE colorés au bleu de Coomassie, Fig. 4.6c. Puis, certaines deces mêmes fractions (TOT, NF, L2, E1, E3) ont été analysées par Western-Blot (WB) pourrechercher spécifiquement la hTSPO2. Un AC dirigé contre l’étiquette polyhistidine (AC anti-His-tag) a tout d’abord été utilisé pour détecter l’étiquette his6 de la hTSPO2, Fig. 4.6d àgauche. Ensuite, un sérum anti-hTSPO2 a été utilisé, sur la fraction d’élution, pour détecterspécifiquement la TSPO2, Fig. 4.6d au centre. Ce sérum a été obtenu par immunisation d’unlapin avec, comme antigène, un peptide correspondant aux 13 derniers a.a de la hTSPO2.
Ce sérum contient les AC dirigés contre la TSPO2 mais peut aussi contenir d’autres ACqui peuvent être présent en raison de l’histoire immunologique de l’animal. En conséquence,nous avons utilisé comme contrôle un sérum pré-immunisation 1 (Fig. 4.6d, à droite). Le WBen présence de ce dernier ne révèle aucune bande.
La présence de hTSPO2 serait confirmée de manière certaine par la détection d’une bandeintense dans l’élution, à un PM apparent compatible avec le PM de la hTSPO2, à la fois surle WB anti-His-tag et sur le WB anti-TSPO2. Des petits volumes de mTSPO1 recombinante(mTSPO1rec) et de hTSPO1 rcombinante (hTSPO1rec) ont été déposés comme repère pourfaciliter l’éventuelle détection de la bande correspondante à la hTSPO2.
Sur le gel SDS-PAGE (Fig. 4.6c), les fractions correspondant au Total (TOT), avant puri-fication, et au non fixé (NF) ne nous ont pas permis de constater la disparition d’une bandepouvant correspondre à la fixation de la hTSPO2 sur la résine. Dans l’élution, aucune banden’apparait clairement à la même hauteur que la mTSPO1rec. Cependant plusieurs bandes in-tenses apparaissent entre 20 et 28 kDa, nous avons alors pensé que peut-être l’une d’entreelle correspondait à la hTSPO2 (21.2 kDa), mais on constate également la présence d’autresprotéines contaminantes entre 100 et 250 kDa.
Ensuite les WB (anti-His-tag et anti-TSPO2) révèlent la présence :
• Une bande intense à un PM apparent de ∼ 44 kDa (encadré jaune), sur l’anti-His-tag quicorrespond à une protéine non retenue sur la colonne. Cette même bande n’est retrouvéque faiblement dans la fraction E3 et sur le WB anti-TSPO2.• Une série de bandes plus ou moins intenses, dans la fraction d’élution E3, à des PMsapparents de ∼33 kDa (bande très intense sur l’anti-His-tag et bien visible sur l’anti-TSPO2 ; encadré vert), ∼63 kDa (encadré rose) ∼68 kDa (encadré marron) et ∼72 kDa(encadré violet).• Une bande très intense sur l’anti-TSPO2 (E3) à un PM de ∼ 21 kDa (encadré rose), unpeu au dessus de la bande correspondant à la hTSPO1rec (encadré rouge), soit le PM dela hTSPO2. Cependant cette bande n’est clairement visible que dans la fraction TOT surle WB anti-His-tag.
1. Le sérum pré-immunisation a été extrait de l’animal avant l’injection de l’antigène, il contient donc tousles AC générés par l’animal mais pas ceux dirigés contre la protéine d’intérêt. Ainsi, la comparaison de ces 2 WBpermet d’identifier des bandes qui pourraient être non spécifiques à la hTSPO2

202 Chapitre 4. La hTSPO2
De ces gels, nous avons conclu qu’au vu de son intensité et de son PM apparent, la bande à21 ∼kDa sur le WB anti-TSPO2 (encadré rose) pouvait correspondre à la hTSPO2. De plus, lesautres bandes présentes dans la fraction d’élution à la fois en anti-his-tag et anti-TSPO2, (∼33kda, ∼63 kDa ?) pouvaient laisser imaginer la présence de polymères de hTSPO2 déjà décritpar l’équipe du Pr. Mariano Ostuni de l’Institut National de Transfusion sanguine (INTS), aveclequel nous avons collaboré sur ce projet, de même qu’avec Claude Chattab pour la réalisationdes WB. L’équipe travaille sur les isoformes TSPO1 et TSPO2 dans les globules rouges (GR) etelle retrouve majoritairement la hTSPO2 native (19 kDa) sous forme polymérique : l’analyse delysat de fantômes de GR (Ghost RB ; un GR dépourvu d’hémoglobine) par WB avec un sérumanti-hTSPO2, montre une hTSPO2 qui apparaît à des PM apparents de ∼ 36, 54 and 72 kDasuggérant une hTSPO2 sous forme dimérique, trimérique et tétradrimérique respectivement(thèse de Irène Marginedas actuellement en cours).
Cependant, le gel SDS-PAGE (Fig. 4.6c) montre que nos fraction d’élution ne sont pas pures.Ceci est confirmé par le grand nombre de bandes observés par les WBs certaines pourrait êtrespécifiques de la hTSPO2 en complexe avec d’autres protéines. D’autres sont non spécifiques etpeuvent être due à la présence de réactions parasites des AC secondaire ou primaire avec desprotéines contaminantes.
4.1.2.2 Purification du cytosol
Après purification des CI, nous avons procédé à la purification, sur résine Ni-NTA, de lasolution cytosolique totale et de la solution cytosolique sans Mb (séparées par centrifugation àforte vitesse). Pour permettre la solubilisation d’éventuelles PMb, comme la hTSPO2, du SDSà 1% final a été ajouté dans chacune des solutions.
Pour chaque étape de la purification (charge, lavages et élution), les spectres d’absorption ontété enregistrées entre 240 et 320 nm. On observe pour la solution cytosolique totale, l’élutionde protéines dans les fractions E3 et E4, Fig. 4.6a, avec une ligne de base (entre 300 et 350nm) qui ne retourne pas au niveau de celle du tampon. Nous n’avons pas d’explication de cephénomène, nous avons testé sans effet l’ajout de SDS supplémentaire ou une centrifugation quiauraient dû éliminer une présence de Mb.
Les DO280 sont représentés Fig. 4.7b pour les 2 solutions cytosoliques (totale et sans MB).Leur comparaison avec la solution extraite des CI solubilisés en SDS, montre un plus fortcontenu en protéine dans les fractions d’élution issues des CI que dans les fractions cytosoliques.Logiquement, la comparaison des fractions "avec et sans Mb" montre que l’isolement des Mbs’accompagne d’une diminution de la quantité totale en protéines du cytosol.
Analyse sur Gels SDS-PAGE et en WB Le contenu protéique des fractions collectéeslors de la purification du cytosol a été analysé sur gels SDS-PAGE colorés au bleu de Coomas-sie, Fig. 4.7c. Sur ces gels a été déposé, à titre de comparaison, des fractions provenant de lapurification des extraits issus des CI solubilisés. Comme précédemment, un petit volume de
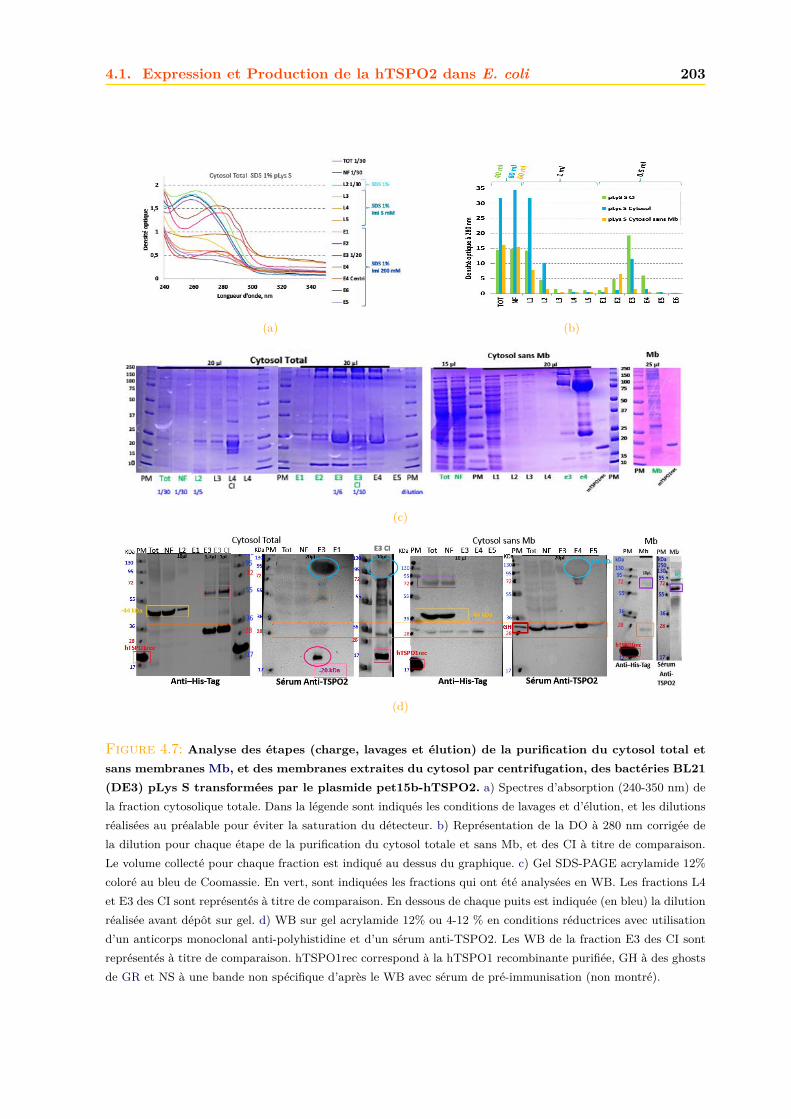
4.1. Expression et Production de la hTSPO2 dans E. coli 203
(a) (b)
(c)
(d)
Figure 4.7: Analyse des étapes (charge, lavages et élution) de la purification du cytosol total etsans membranes Mb, et des membranes extraites du cytosol par centrifugation, des bactéries BL21(DE3) pLys S transformées par le plasmide pet15b-hTSPO2. a) Spectres d’absorption (240-350 nm) dela fraction cytosolique totale. Dans la légende sont indiqués les conditions de lavages et d’élution, et les dilutionsréalisées au préalable pour éviter la saturation du détecteur. b) Représentation de la DO à 280 nm corrigée dela dilution pour chaque étape de la purification du cytosol totale et sans Mb, et des CI à titre de comparaison.Le volume collecté pour chaque fraction est indiqué au dessus du graphique. c) Gel SDS-PAGE acrylamide 12%coloré au bleu de Coomassie. En vert, sont indiquées les fractions qui ont été analysées en WB. Les fractions L4et E3 des CI sont représentés à titre de comparaison. En dessous de chaque puits est indiquée (en bleu) la dilutionréalisée avant dépôt sur gel. d) WB sur gel acrylamide 12% ou 4-12 % en conditions réductrices avec utilisationd’un anticorps monoclonal anti-polyhistidine et d’un sérum anti-TSPO2. Les WB de la fraction E3 des CI sontreprésentés à titre de comparaison. hTSPO1rec correspond à la hTSPO1 recombinante purifiée, GH à des ghostsde GR et NS à une bande non spécifique d’après le WB avec sérum de pré-immunisation (non montré).

204 Chapitre 4. La hTSPO2
mTSPO1rec a été déposé comme repère. L’analyse des fractions "Cytosol Total" montre la pré-sence de nombreuses protéines que nous n’observons pas sur le gel "Cytosol sans Mb". Toutefois,on ne retrouve pas les protéines manquantes sur le gel "Mb". Il est encore une fois il est difficilede distinguer une bande qui pourrait correspondre à la hTSPO2.
Afin de mieux identifier si la hTSPO2 est présente, certaines de ces mêmes fractions ont étéanalysées par Western-Blot (AC anti-His-tag et sérum anti-TSPO2 ), Fig. 4.7d.
Sur les WBs anti-His-tag, de la protéine hTSPO1 recombinante (encadré rouge clair) aété déposée comme repère. Toutefois aucune bande n’apparait à ce PM dans les différentespurifications (Cytosol tot et sans Mb).
Sur le WB anti-tspo2 "Cytosol sans Mb", des fantômes de GR (GH, encadré rouge sombre)ont été déposés. Ces fantômes contiennent de la hTSPO2 native sous forme dimérique. LeWB des fractions "Cytosol dans Mb" montre la présence d’une bande (encadré orange), aumême niveau que celle correspondant à ces fantômes, à la fois présente dans le WB anti-TSPO2(bande très intense) et dans l’anti-His-tag, Fig. 4.7c au centre. Cette bande est également visiblefaiblement, dans la fraction E3 "Cytosol totale", sur le WB anti-TSPO2 ; et elle est apparaitintensément dans le WB anti-His-tag, Fig. 4.7c à gauche. Il est aussi possible que cette bandesoit présente dans la fraction membranaire, encadré orange sur la Fig. 4.7c à droite. De plus,toujours dans cette fraction E3 "Cytosol + Mb" une bande intense apparait à un PM compatibleavec celui de la hTSPO2 (∼ 20 kDa, encadré magenta). L’absence de cette bande de la fraction"cytosol sans Mb" laisse suggérer que la protéine est présente dans les membranes, cependantla bande n’est pas observée dans le WB (Fig. 4.7d à droite) de la fraction membranaire.
De ces observations, nous avons conclu que nous avions probablement de la hTSPO2 sousforme dimérique et monomérique dans le cytosol de la bactérie. Cependant nous sommes confron-tés à un problème de pureté.
Caractérisation par spectrométrie de masse Nous avons essayé d’identifier une bandepouvant correspondre à la hTSPO2 sur nos gels SDS-PAGE par une analyse par ORBITRAPdes peptides obtenus après digestion trypsique de certaines bandes du gel SDS-PAGE. Ainsi,nous avons choisi de digérer les 4 bandes les plus intenses (PM ∼20-25, ∼25, ∼40 et ∼100kDa) qui apparaissaient sur du gel "Cytosol sans MB" (gel qui venait d’être réalisé) et les plusréactives à l’anti-TSPO2. Il est à noter qu’au moment de cette expérience nous ne disposionspas de tous les WB et notamment ceux suggérant la présence d’un monomère de hTSPO2 (endessous du PM 20 kDa).
Figure 4.8: Identification des protéinescorrespondantes aux bandes encadrées surle gel SDS-PAGE de la purification de lasolution cytosolique sans Mb.

4.1. Expression et Production de la hTSPO2 dans E. coli 205
Cette analyse ne nous a pas permis d’identifier la hTSPO2 mais nous a permis de mieuxcaractériser et de comprendre l’origine de la contamination protéique de nos purifications, enrévélant la présence de :
• la FKPP-type peptidyl-propyl cis-trans isomérase SlyD : 20.853 kDa ; bande 3 Fig. 4.8,• la Polyaline alinopropyltransférase : 32.3 kDa ; bande 2 sur la Fig. 4.8,• le facteur d’élongation tu 2 : 43.314 kDa ; bande 4 sur la Fig. 4.8. Les bandes observées∼ 44 kDa (encadré orange) sur nos différents WB sont propablement dues à une réactionparasite de cette protéine avec nos AC utilisés .• la β-galactosidase : 116 kDa ; bande 1 sur la Fig. 4.8. Cette protéine intervient dansle métabolisme du galactose et des sphingolipides. Elle est sous le contrôle de l’opéronlactose 2 (cf. § 2.1.1). Or, l’IPTG a la capacité de se lier et d’inhiber le répresseur lactose,induisant ainsi la transcription du gène codant pour la protéine. C’est cette protéine quenous retrouvons et qui parasite la plupart de nos WBs (bande encadré en cyan), quece soit lors de la purification de la solution extraite des CI solubilisés ou des solutionscytosoliques totales et sans Mb.
De nombreux travaux [Bolanos-Garcia and Davies, 2006, Parsy et al., 2007, Bartlow et al.,2011] ont montré que de nombreuses protéines bactériennes "natives" de E. coli se fixent surla colonne de résine Ni-NTA. En effet, au moins 3 histidines spatialement proches dans l’espacesuffisent à permettre la fixation d’une protéine sur la résine. L’abondance de ces protéines, dontla majorité sont des protéines de réponse au stress [Bolanos-Garcia and Davies, 2006], entreen compétition avec la protéine recombinante. Ceci est d’autant plus vrai lorsque les protéinesrecombinantes ne sont pas exprimées en grande quantité [Bolanos-Garcia and Davies, 2006]comme on le voit clairement pour les purifications de la hTSPO2.
Bien que nous soyons convaincu que la hTSPO2 soit exprimée et soit présente sous formemonomérique et oligomérique nous sommes confrontés à un gros problème de pureté. En par-ticulier, l’abondance de la β-galactosidase (formant une "patate" sur nos gels SDS-PAGE etWBs), dont l’expression est induite par ajout d’IPTG, nous a fait nous demander si l’utilisationdes bactéries E. coli pLys S était le meilleur choix. Dans une documentation commerciale deThermofisher il est montré une surexpression de la β-galactosidase dans les bactéries pLys Spar rapport aux bactéries "classiques". Nous avons donc décider de retenter l’expression de laprotéine à partir de bactéries BL21 (DE3) classique que nous avions l’habitude d’utiliser pourla surexpression de la protéine mTSPO1 (ou de la hTSPO1).
2. L’opéron lactose, ou opéron lac est un opéron nécessaire au transport et au métabolisme du lactose chezEscherichia coli.
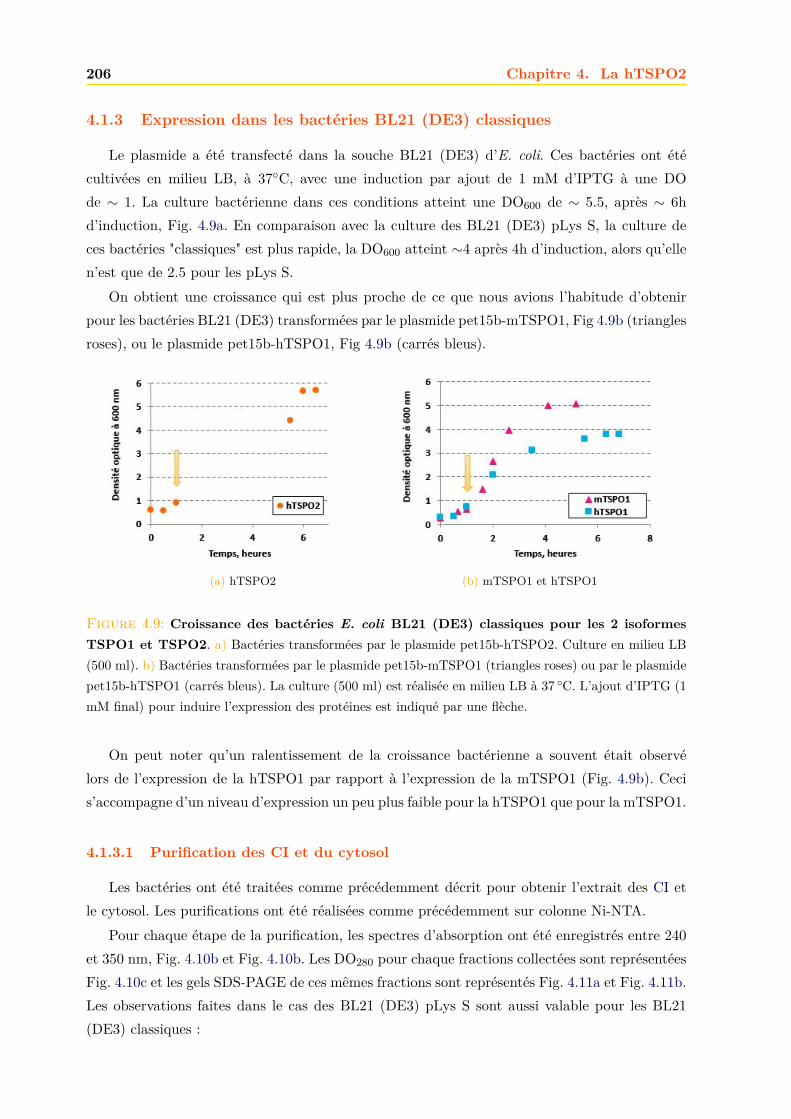
206 Chapitre 4. La hTSPO2
4.1.3 Expression dans les bactéries BL21 (DE3) classiques
Le plasmide a été transfecté dans la souche BL21 (DE3) d’E. coli. Ces bactéries ont étécultivées en milieu LB, à 37◦C, avec une induction par ajout de 1 mM d’IPTG à une DOde ∼ 1. La culture bactérienne dans ces conditions atteint une DO600 de ∼ 5.5, après ∼ 6hd’induction, Fig. 4.9a. En comparaison avec la culture des BL21 (DE3) pLys S, la culture deces bactéries "classiques" est plus rapide, la DO600 atteint ∼4 après 4h d’induction, alors qu’ellen’est que de 2.5 pour les pLys S.
On obtient une croissance qui est plus proche de ce que nous avions l’habitude d’obtenirpour les bactéries BL21 (DE3) transformées par le plasmide pet15b-mTSPO1, Fig 4.9b (trianglesroses), ou le plasmide pet15b-hTSPO1, Fig 4.9b (carrés bleus).
(a) hTSPO2 (b) mTSPO1 et hTSPO1
Figure 4.9: Croissance des bactéries E. coli BL21 (DE3) classiques pour les 2 isoformesTSPO1 et TSPO2. a) Bactéries transformées par le plasmide pet15b-hTSPO2. Culture en milieu LB(500 ml). b) Bactéries transformées par le plasmide pet15b-mTSPO1 (triangles roses) ou par le plasmidepet15b-hTSPO1 (carrés bleus). La culture (500 ml) est réalisée en milieu LB à 37 ◦C. L’ajout d’IPTG (1mM final) pour induire l’expression des protéines est indiqué par une flèche.
On peut noter qu’un ralentissement de la croissance bactérienne a souvent était observélors de l’expression de la hTSPO1 par rapport à l’expression de la mTSPO1 (Fig. 4.9b). Cecis’accompagne d’un niveau d’expression un peu plus faible pour la hTSPO1 que pour la mTSPO1.
4.1.3.1 Purification des CI et du cytosol
Les bactéries ont été traitées comme précédemment décrit pour obtenir l’extrait des CI etle cytosol. Les purifications ont été réalisées comme précédemment sur colonne Ni-NTA.
Pour chaque étape de la purification, les spectres d’absorption ont été enregistrés entre 240et 350 nm, Fig. 4.10b et Fig. 4.10b. Les DO280 pour chaque fractions collectées sont représentéesFig. 4.10c et les gels SDS-PAGE de ces mêmes fractions sont représentés Fig. 4.11a et Fig. 4.11b.Les observations faites dans le cas des BL21 (DE3) pLys S sont aussi valable pour les BL21(DE3) classiques :
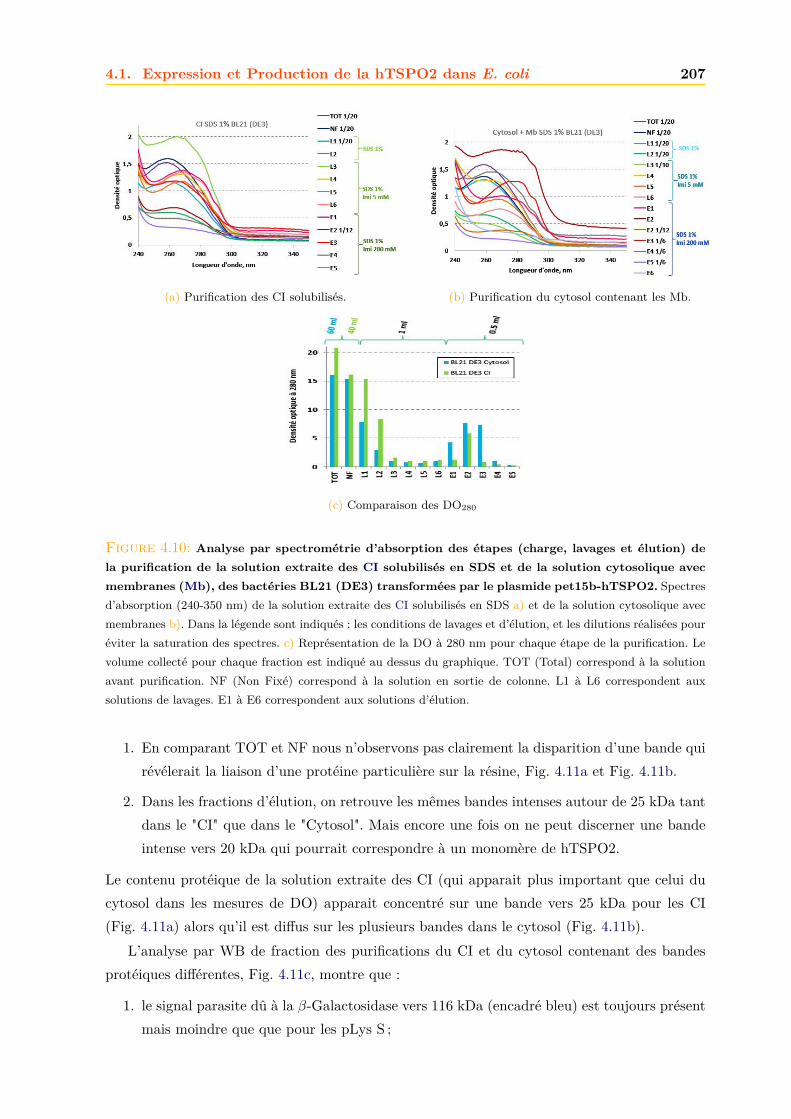
4.1. Expression et Production de la hTSPO2 dans E. coli 207
(a) Purification des CI solubilisés. (b) Purification du cytosol contenant les Mb.
(c) Comparaison des DO280
Figure 4.10: Analyse par spectrométrie d’absorption des étapes (charge, lavages et élution) dela purification de la solution extraite des CI solubilisés en SDS et de la solution cytosolique avecmembranes (Mb), des bactéries BL21 (DE3) transformées par le plasmide pet15b-hTSPO2. Spectresd’absorption (240-350 nm) de la solution extraite des CI solubilisés en SDS a) et de la solution cytosolique avecmembranes b). Dans la légende sont indiqués : les conditions de lavages et d’élution, et les dilutions réalisées pouréviter la saturation des spectres. c) Représentation de la DO à 280 nm pour chaque étape de la purification. Levolume collecté pour chaque fraction est indiqué au dessus du graphique. TOT (Total) correspond à la solutionavant purification. NF (Non Fixé) correspond à la solution en sortie de colonne. L1 à L6 correspondent auxsolutions de lavages. E1 à E6 correspondent aux solutions d’élution.
1. En comparant TOT et NF nous n’observons pas clairement la disparition d’une bande quirévélerait la liaison d’une protéine particulière sur la résine, Fig. 4.11a et Fig. 4.11b.
2. Dans les fractions d’élution, on retrouve les mêmes bandes intenses autour de 25 kDa tantdans le "CI" que dans le "Cytosol". Mais encore une fois on ne peut discerner une bandeintense vers 20 kDa qui pourrait correspondre à un monomère de hTSPO2.
Le contenu protéique de la solution extraite des CI (qui apparait plus important que celui ducytosol dans les mesures de DO) apparait concentré sur une bande vers 25 kDa pour les CI(Fig. 4.11a) alors qu’il est diffus sur les plusieurs bandes dans le cytosol (Fig. 4.11b).
L’analyse par WB de fraction des purifications du CI et du cytosol contenant des bandesprotéiques différentes, Fig. 4.11c, montre que :
1. le signal parasite dû à la β-Galactosidase vers 116 kDa (encadré bleu) est toujours présentmais moindre que que pour les pLys S ;
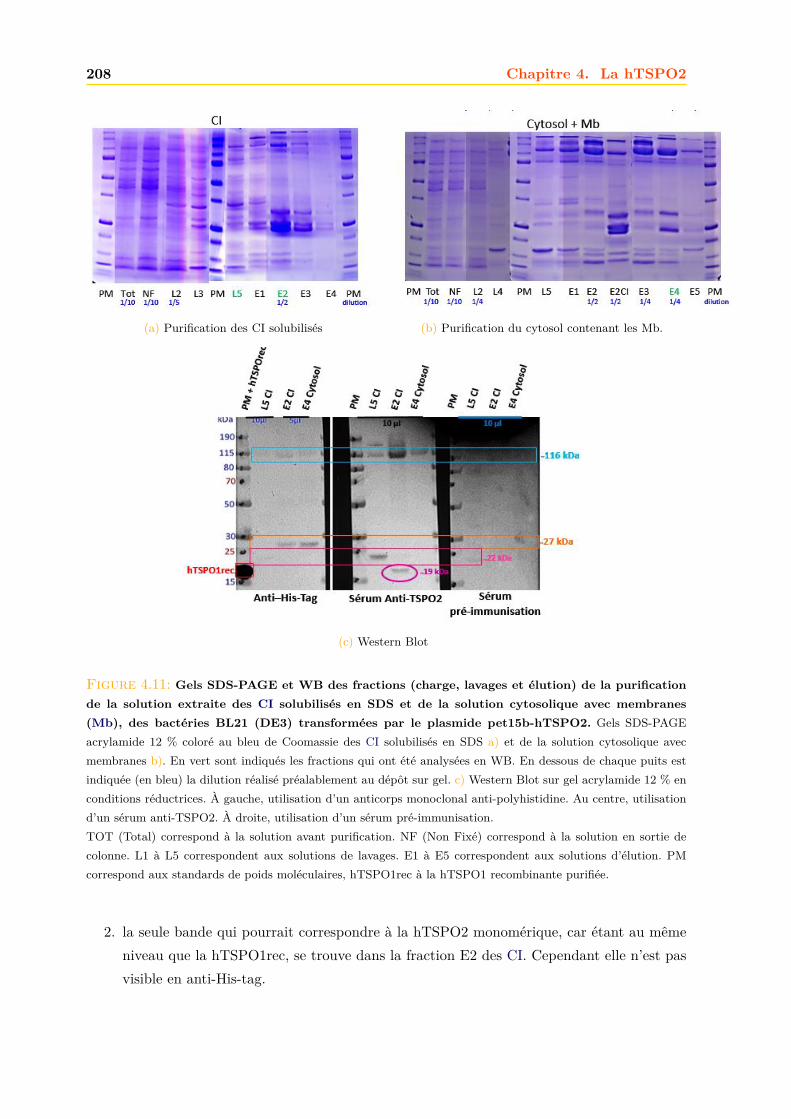
208 Chapitre 4. La hTSPO2
(a) Purification des CI solubilisés (b) Purification du cytosol contenant les Mb.
(c) Western Blot
Figure 4.11: Gels SDS-PAGE et WB des fractions (charge, lavages et élution) de la purificationde la solution extraite des CI solubilisés en SDS et de la solution cytosolique avec membranes(Mb), des bactéries BL21 (DE3) transformées par le plasmide pet15b-hTSPO2. Gels SDS-PAGEacrylamide 12 % coloré au bleu de Coomassie des CI solubilisés en SDS a) et de la solution cytosolique avecmembranes b). En vert sont indiqués les fractions qui ont été analysées en WB. En dessous de chaque puits estindiquée (en bleu) la dilution réalisé préalablement au dépôt sur gel. c) Western Blot sur gel acrylamide 12 % enconditions réductrices. À gauche, utilisation d’un anticorps monoclonal anti-polyhistidine. Au centre, utilisationd’un sérum anti-TSPO2. À droite, utilisation d’un sérum pré-immunisation.TOT (Total) correspond à la solution avant purification. NF (Non Fixé) correspond à la solution en sortie decolonne. L1 à L5 correspondent aux solutions de lavages. E1 à E5 correspondent aux solutions d’élution. PMcorrespond aux standards de poids moléculaires, hTSPO1rec à la hTSPO1 recombinante purifiée.
2. la seule bande qui pourrait correspondre à la hTSPO2 monomérique, car étant au mêmeniveau que la hTSPO1rec, se trouve dans la fraction E2 des CI. Cependant elle n’est pasvisible en anti-His-tag.

4.1. Expression et Production de la hTSPO2 dans E. coli 209
4.1.4 Optimisations
Nous avons tenté plusieurs optimisations dans le but de réussir à sur-exprimée la hTSPO2.
1. Nous avons fait plusieurs transfections de plasmide dans plusieurs lots de bactéries (BL21(DE3) classique ou pLys S) commerciales, disponibles au laboratoire, pour obtenir descolonies fraîches et relancer des cultures. Mais cela n’a rien donné.
2. Nous avons testé la souche bactérienne RosettaTM (DE3) d’E. coli sans succès.
3. Nous avons testé en parallèle, à partir d’une même préculture, plusieurs température deculture : 37 ◦C, 27 ◦C, 17 ◦C. Mais l’analyse par gels SDS-PAGE et par WB des fractionsbacteriennes avant et après induction par 1 mM d’IPTG, ne montre pas de sur-expressionde la protéine.
4. Nous avons testé l’effet de la rifampicine. En effet, de nombreuses études ont reportél’effet bénéfique, sur le rendement de production de protéines recombinantes, de l’ajoutde rifampicine, ∼45 min, après l’induction par ITPG [Kuderova et al., 1999, Tabor andRichardson, 1985, Lee et al., 1995]. La rifampicine est un antibiotique qui inhibe l’ARNpolymèrase bactérien mais pas celui du bacteriophage T7. Ainsi, elle permet d’inhiberla production additionnelle des protéines hôtes d’E. coli mais pas celle de la protéinerecombinante sous le contrôle du promoteur T7. Un délai entre l’induction par IPTG etl’ajout de rifampicine est nécessaire pour permettre à la bactérie de produire assez depolymèrase T7. Nous avons donc lancé plusieurs cultures en parallèle avec ajout, après45 min d’induction, de 0, 10, 100 et 200 µg/ml final de rifampicine. Cependant, unecomparaison des fractions bactériennes sur gels SDS-PAGE, avant et après induction, nemontre aucun changement probant. La présence de rifamipicine ne semble pas affecter leniveau d’expression des protéines hôtes.
4.1.5 Conclusions, discussions et perspectives
L’analyse des Western Blot nous suggère une expression de la hTSPO2 sous forme mo-nomérique dans les CI pour les bactéries BL21 (DE3) classiques et sous forme dimérique etmonomérique, à la fois dans les CI et la partie cytosolique soluble (sans membrane), pour lesbactéries BL21 (DE3) pLys S. Cette dernière localisation semble difficile à imaginer pour uneprotéine membranaire. Une hypothèse pourrait être que la centrifugation réalisée pour isolerles Mb (1h à 50000×g) du reste du contenu cytosolique n’a pas était suffisante en vitesse ouen temps. Cependant, la localisation dans la fraction soluble reste possible. Il a été décrit dansl’étude statistique réalisée par [Korepanova et al., 2009], que bien que la plupart des PMb soientexprimées sous forme insoluble dans les CI et dans les Mb, un certain pourcentage (∼ 10%) estretrouvé dans la fraction cytosolique soluble en même temps que dans les CI et les membranes.
De plus, une autre explication, à cette différence de localisation entre BL21 (DE3) classiqueset pLys S pourrait être révéler par différents "comportements" de ces 2 types de bactéries. Leurcroissance est différente. Les bactéries pLys S sont beaucoup plus fragiles que les classiques :

210 Chapitre 4. La hTSPO2
après la 1re centrifugation qui permet de collecter les culots bactériens après la culture, leurreprise conduit à une solution visqueuse, typique de la présence d’ADN, suggérant que les bac-téries étaient déjà lysées avant même la sonication. Les CI des bactéries pLys S sont minusculeset mous alors que ceux de BL21 classiques sont gros et compacts. Ainsi, la présence d’unehTSPO2 dans la fraction cytosolique provient peut être d’une "fuite" de la protéine hors des CIoù elle se trouvait initialement.
Dans tous les cas nous ne sommes par parvenus à l’objectif qui était de sur-exprimée laprotéine hTSPO2 dans les bactéries E.coli et de la purifier à partir d’un protocole qui pourtantfonctionne très bien pour son isorforme TSPO1.
La seule étude qui présente un protocole de production de la (h,m)TSPO2 recombinante, avecune étiquette His6 en N-terminal, est celle de [Fan et al., 2009]. Ces protéines ont été obtenues partransformation de bactéries Bl21 (DE3) pLys S avec le vecteur d’expression pET-28c. Ce vecteurplasmidique permet une configuration His-Tag/Trombine/T7-Tag en N-terminal en plus d’uneétiquette polyhistidines optionnelle en C-terminal. Le "T7-Tag" est un épitope de 11 résiduscodé par le gène 10 du bacteriophage T7. Il est utilisé pour la purification ou la détectionpar immunodétection de la protéine par un AC monoclonal anti-T7-Tag. L’ADN codant pourla protéine TSPO2 a été cloné entre les sites de restriction NdeI/Xho1. La production estinduite par 1 mM d’IPTG à 30 ◦C. Dans ces conditions, l’analyse par WB des lysats cellulairesprésentée dans cet article révèle une bande unique à un PM apparent de 20 kDa, correspondantà la mTSPO2 recombinante. Cette bande n’est pas présente dans les CI mais dans la fractioncytosolique (il n’est pas précisé si elle est retrouvée dans les Mb ou la fraction cytosoliquesoluble). Le WB n’étant pas montré en entier (seulement entre 80 et 10 kDa) et bien que lesauteurs disent avoir réalisé des WB anti-His-tag et anti-TSPO2, il n’est pas précisé lequel des2 est présenté sur la figure. Les gels SDS-PAGE qu’ils ont réalisé ne sont pas présentés. Il estalors difficile pour nous de comparer cette étude avec nos expériences, puisque nous ne pouvonspas juger des quantités et de l’état de pureté qu’ils ont obtenu pour leurs fractions contenant lahTSPO2. En effet, une production de protéine recombinante "relativement faible" peut suffirepour mener des expériences de biochimie fonctionnelle, telles que celles présentées dans l’article,contrairement aux études de biologie structurale pour lesquelles nous avons besoin d’une grandequantité de protéine recombinante pure.
D’autres optimisations pourraient être envisagées pour permettre la sur-expression de lahTSPO2 recombinante au laboratoire :
1. Un changement de vecteur plasmidique. P. ex, le gène d’intérêt peut être cloné dans unsystème PGEX qui permet la production d’une protéine recombinante fusionnée à la GST.Cette étiquette GST permet une augmentation de l’expression du gène d’intérêt et uneaugmentation de la solubilité de la protéine de fusion [Gopal and Kumar, 2013].
2. Un changement de souche bactérienne d’E. coli : BL21 Star qui permet une augmenta-tion de la stabilité des ARNm ; C41 (DE3) conçue pour permettre la sur-expression desprotéines "toxiques" ou C43 (DE3) conçue pour l’expression des protéines globulaires et

4.1. Expression et Production de la hTSPO2 dans E. coli 211
membranaires [Dumon-Seignovert et al., 2004, Hattab et al., 2014] ;
3. La transfection des bactéries avec une plus faible quantité de plasmide pour éviter uneéventuelle instabilité (mutation, digestion, agrégation) de celui-ci qui peut causer sa perte[Palomares et al., 2004].
4. Un changement des paramètres de culture : concentration d’IPTG, pH, ajout de com-posants chimiques permettant l’expression de protéines chaperonnes, etc. [Hattab et al.,2014, Gopal and Kumar, 2013].
5. Une purification par gel filtration, permettant la séparation des protéines selon leur taille,pourrait être réalisée afin de séparer la hTSPO2 de ses contaminants.
6. Un changement de système d’expression : système eucaryote (p. ex les levure ou cellulesd’insectes et de mammifères) ou système acellulaire, aussi appelé "cell-free". C’est cettedernière technique que nous avons choisi d’utiliser pour tenter de sur-exprimée notreprotéine hTSPO2. Cela fera l’objet de la dernière section.
Humain TSPO
Humain TSPO2
Humain TSPO
Humain TSPO2
Humain TSPO
Humain TSPO2
M A P P W P A M G T A S C F G S R V G E G L R Y A L Q P H H W G P G 58
A G F T . E K A V T G N A W P P G M W 115
S A A A A T A Y Q S P R Y A F T N C N H G W R G G R R L P . . . 169
. . . M R Q G A I V L H P I V W L T D H M S G C E P R M L C F Y K L L Q T A 55
V A G L G W P L A A V T S T V L V T V P L H 113
Y L V V S A I H P N K L L T V A T H S L C P V H Q P Q P T K S D 170
1 10 20 30 40 50
60 70 80 90 100 110
120 130 140 150 160
K P W T
A A D
V F L P L G V F H W G S W P V L V L Y S
M G Y S Y L V W K E L G G V P L G L Y Q L L W I F F R Q G A L V L L L V
G T V W V L A A L L P Y L A W L T T L Y V W R D E
L F L P L G L F R W G S W P V L V I Y S
V G Y S Y L V W K D L G G L P L G L Y Q L I W L F F H N G A L L L L L L
G T L W I L A A L L P Y L A W L T S L Y L W R D E
Loop 2
Loop 1
Loop 4
Loop 3
Mt Targeting TSPO1
N-terminal
C-terminal
CRAC Cholesterol Binding
(a) Alignement de séquences des protéines hTSPO1 et hTSPO2.
(b) Composition en acides aminés des isoformes hu-maines TSPO1 et TSPO2. Données obtenues à partirl’outil ProtParam.
(c) Comparaison des profils hydrophobiques des sé-quences des isoformes humaines TSPO1 et TSPO2.Les résidus hydrophobes sont positifs. Les domainestransmembranaires sont indiqués. Figure issue de [Fanet al., 2009].
Figure 4.12: Diffèrence entre les 2 isoformes humaines TSPO1 et TSPO2

212 Chapitre 4. La hTSPO2
Finalement, la question se pose de savoir pourquoi nous ne parvenons pas à sur-exprimeren bactérie la hTSPO2, alors que cela se fait très bien pour son isoforme TSPO1. Est-ce unproblème de séquence, de repliement ou de translation ?
1. Cela pourrait être due à des différences structurales liées à des variations de séquencesou d’hydrophobicité entre les protéines, Fig.4.12a et Fig.4.12b. Il a été décrit qu’une fortehydrophobicité des PMb peut altérer l’expression dans un système bactérien [Price et al.,2011]. D’un point de vue des séquences, le C-terminal, la boucle 1 et la boucle 2 ne sont pasconservés entre les 2 isoformes. Une comparaison du profil d’hydrophobicité de la TSPO1et TSP2 humaine montre une grande similarité à la fois dans les régions transmembrainescytosoliques, Fig. 4.12c. La boucle 1 apparait cependant plus hydrophobe chez la TSPO2.Il est difficile à dire si cela peut être à l’origine des problèmes rencontrés pour produire lahTSPO2 recombinante. La hTSPO2 possède 2 cystéines dans la boucle 1 qui pourraientpeut être former un ponts disulfure qui pourrait interferer avec son expression [Rosanoand Ceccarelli, 2014].
2. On sait que in vivo les 2 protéines ne possèdent pas la même localisation subcellulaire- mitochondriale pour la TSPO1, endoplasmique et nucléaire pour la TSPO2 - mais au-cune des protéines ne possèdent de peptide signal. Pour la TSPO1 le domaine C-terminalainsi que le motif de Schellman dans la boucle 3 ont été montrés avoir un rôle importantdans son adressage et son insertion dans la MME [Rone et al., 2009b]. Par contre pourla TSPO2 il n’y a pour le moment pas de signal de localisation connu. Peut être que lamachinerie d’E. coli n’est pas adaptée pour assister l’adressage, l’insertion et le repliementde la TSPO2 dans le membrane, comme cela est le cas pour d’autres PMb [Wagner et al.,2007, Klepsch et al., 2011, Ojemalm et al., 2013, Bowie, 2005]. P. ex, il a été démontréque la production de PMb hétérologue peut conduire à la saturation de la machineriede biogenèse d’E. coli, ce qui affecte négativement les capacités de la bactérie (mauvaisrepliement/aggregation des protéines dans le cytoplasme, troubles de la respiration, pro-duction d’ATP inefficace) [Klepsch et al., 2011]. Certaines PM nécessitent des chaperonnesspécifiques à leur localisation, pour leur repliement correct [Kota and Ljungdahl, 2005], ilest alors possible qu’elles soient absentes chez l’hôte utilisé.
3. Il se pourrait que la vitesse de décodage du cadre de lecture (c.-à-d. le temps mis par unribosome pour traduire un ARNm) ne soit pas le même pour nos 2 protéines à cause dedifférence dans leur séquence des bases. [Chu et al., 2014] reporte qu’il existe des codons"rapides" et des codons "lents" lesquels mènent à une libération lente du codon start parles ribosomes ce qui ne permet pas un démarrage efficace de la traduction.

4.2. Production de la protéine hTSPO2 en système acellulaire ou Cell Free 213
4.2 Production de la protéine hTSPO2 en système acellulaireou Cell Free
Ne parvenant pas à sur-exprimer en grande quantité et à purifier la hTSPO2 recombinantechez la bactérie E .coli nous avons fait le choix de passer à un autre système de production :le système acellulaire ou cell-free. Il a pour principe l’utilisation d’extrait de la machinerietranscriptionnelle et traductionnelle, p. ex. de la bactérie E. coli, complété par des a.a (marquésou non) et des molécules d’ATP, entre autres, pour produire une protéine spécifique à partird’un plasmide. Cette approche par cell free a été réalisée en collaboration avec Marie-FranceGiraud à L’IBG de Bordeaux.
Le protocole de production de la protéine en cell free est décrit Sec. 2.1.8 du M&M. Briè-vement, le plasmide pet15b-htTSPO2 (∼15 µg) a été ajouté un 1 ml de mélange réactionnelpuis la réaction de synthèse s’est faite en utilisant un système d’échange en continu. Celui-ciest constitué d’un compartiment de réaction contenant la machinerie cellulaire et l’informationgénétique, et d’un compartiment d’alimentation contenant les cofacteurs, a.a, tampons et autrescomposants . Une membrane de dialyse séparant les deux compartiments favorise la sortie desmolécules « déchets » et l’entrée de molécules nécessaires à la réaction, grâce au gradient deconcentration résultant de l’activité de synthèse.
Après incubation pendant 22 h à Tamb, un précipité est visible dans le compartiment dumilieu réactionnel comme illustré Fig. 4.13a. En effet puisqu’aucun environnement hydrophoben’est fourni, la protéine membranaire précipite après translation.
Après récupération de la solution réactionnelle contenant le précipité, celle-ci est centrifugée.Le culot et le surnageant obtenus sont traités comme décrit Fig. 4.13a. Le culot est lavé à l’eaupuis repris dans 200 µl de tampons SDS 1% pour permettre la solubilisation de la hTSPO2. Pourvérifier que toute la protéine a bien été concentré dans le culot, une petite fraction du surnageant(SU) est traité au TCA pour permettre la précipitation d’éventuelles protéines présentes. Cettefraction est également reprise dans un tampon SDS 1% pour être analysée sur gel. La Fig. 4.13bmontre le gel SDS-PAGE coloré au bleu de Coomassie sur lequel a été déposée une fraction dechaque étape du traitement. Dans la fraction "culot", on observe une bande au PM attendu pourla hTSPO2. Cette bande n’est pas retrouvée pas dans les fractions "lavage" et "SU". Il sembledonc que la protéine hTSPO2 a bien été produite mais de nombreuses protéines contaminantessont encore présentes, le culot solubilisé a donc été purifié sur résine NI-NTA avant d’être analysépar WB.
4.2.1 Purification
Le culot solubilisé, contenant la protéine, a été dilué 10 fois dans un tampon SDS 0.5%Imidazole 2 mM pH 7.8 avant d’être purifié sur résine Ni-NTA (0.5 ml). À chaque étape (charge,lavages et élution) un spectre d’absorbance a été enregistré, Fig. 4.14a. Les fractions d’élutionmontrent un spectre caractéristique de protéines. À partir des mesures des DO à 280 nm,

214 Chapitre 4. La hTSPO2
(a)
(b)
Figure 4.13: Production de la hTSPO2 par la méthode du cell free en mode précipité.a) Schéma illustrant les étapes du processus. b) Gel SDS-PAGE acrylamide 12 % coloré au bleu deCoomassie des différentes fractions issues de la centrifugation du milieu réactionnel pour séparer culotet surnageant SU). Les numéros de (1) à (3) attribués aux différentes fractions déposées sur le gelcorrespondent aux différentes étapes du traitement du culot et du SU illustrées en a). PM correspondaux standards de poids moléculaires et MPS1 à une protéine kinase produite en routine en cell free aulaboratoire.
Fig. 4.14b, la quantité de hTSPO2 présente dans les fractions éluées a été estimée à partirde l’ε de celle-ci, Fig. 4.14b (données encadrées). . La fraction d’élution E2 (1 ml) est la plusconcentrée et la quantité en hTSPO2 est estimée au maximum à ∼ 0.1 mg.
L’analyse sur WB d’un volume de la solution du culot resuspendu en tampon SDS et de lafraction E2 a permis de confirmer la présence de la hTSPO2, Fig.4.14c : une bande intense aumême PM que la hTSPO1 recombinante apparait à la fois sur le WB "anti-His-Tag" et sur leWB "anti-TSPO2".
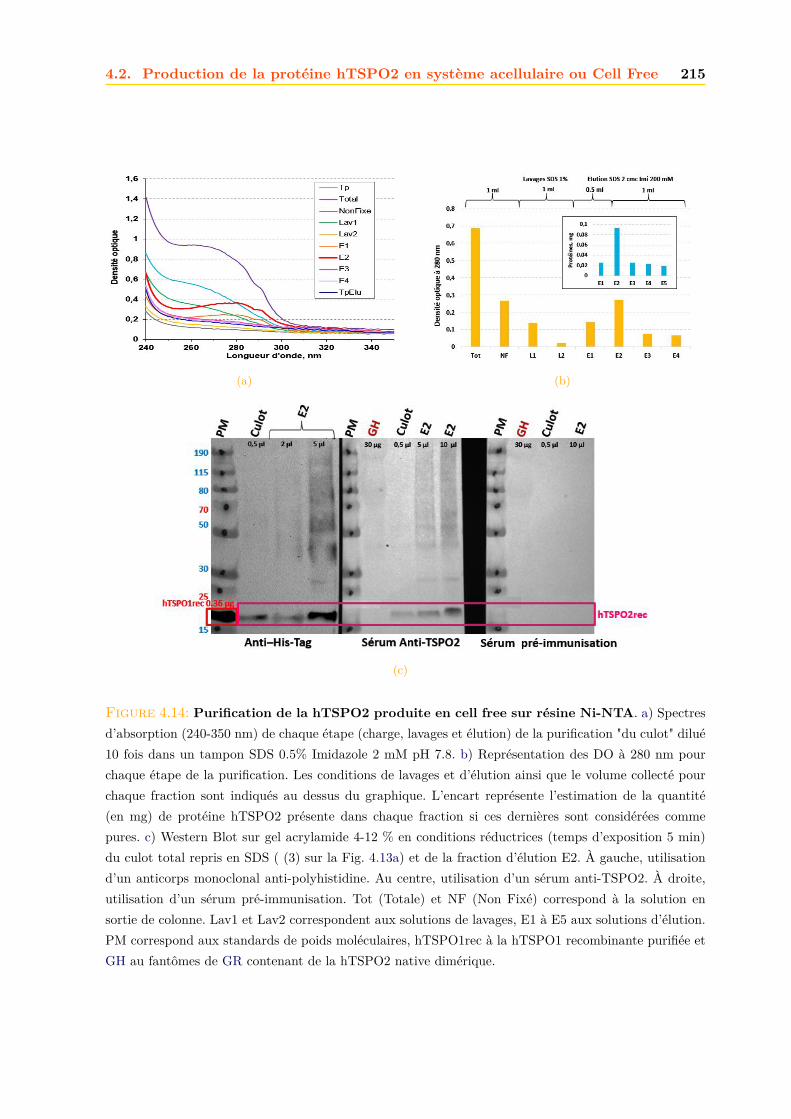
4.2. Production de la protéine hTSPO2 en système acellulaire ou Cell Free 215
(a) (b)
(c)
Figure 4.14: Purification de la hTSPO2 produite en cell free sur résine Ni-NTA. a) Spectresd’absorption (240-350 nm) de chaque étape (charge, lavages et élution) de la purification "du culot" dilué10 fois dans un tampon SDS 0.5% Imidazole 2 mM pH 7.8. b) Représentation des DO à 280 nm pourchaque étape de la purification. Les conditions de lavages et d’élution ainsi que le volume collecté pourchaque fraction sont indiqués au dessus du graphique. L’encart représente l’estimation de la quantité(en mg) de protéine hTSPO2 présente dans chaque fraction si ces dernières sont considérées commepures. c) Western Blot sur gel acrylamide 4-12 % en conditions réductrices (temps d’exposition 5 min)du culot total repris en SDS ( (3) sur la Fig. 4.13a) et de la fraction d’élution E2. À gauche, utilisationd’un anticorps monoclonal anti-polyhistidine. Au centre, utilisation d’un sérum anti-TSPO2. À droite,utilisation d’un sérum pré-immunisation. Tot (Totale) et NF (Non Fixé) correspond à la solution ensortie de colonne. Lav1 et Lav2 correspondent aux solutions de lavages, E1 à E5 aux solutions d’élution.PM correspond aux standards de poids moléculaires, hTSPO1rec à la hTSPO1 recombinante purifiée etGH au fantômes de GR contenant de la hTSPO2 native dimérique.
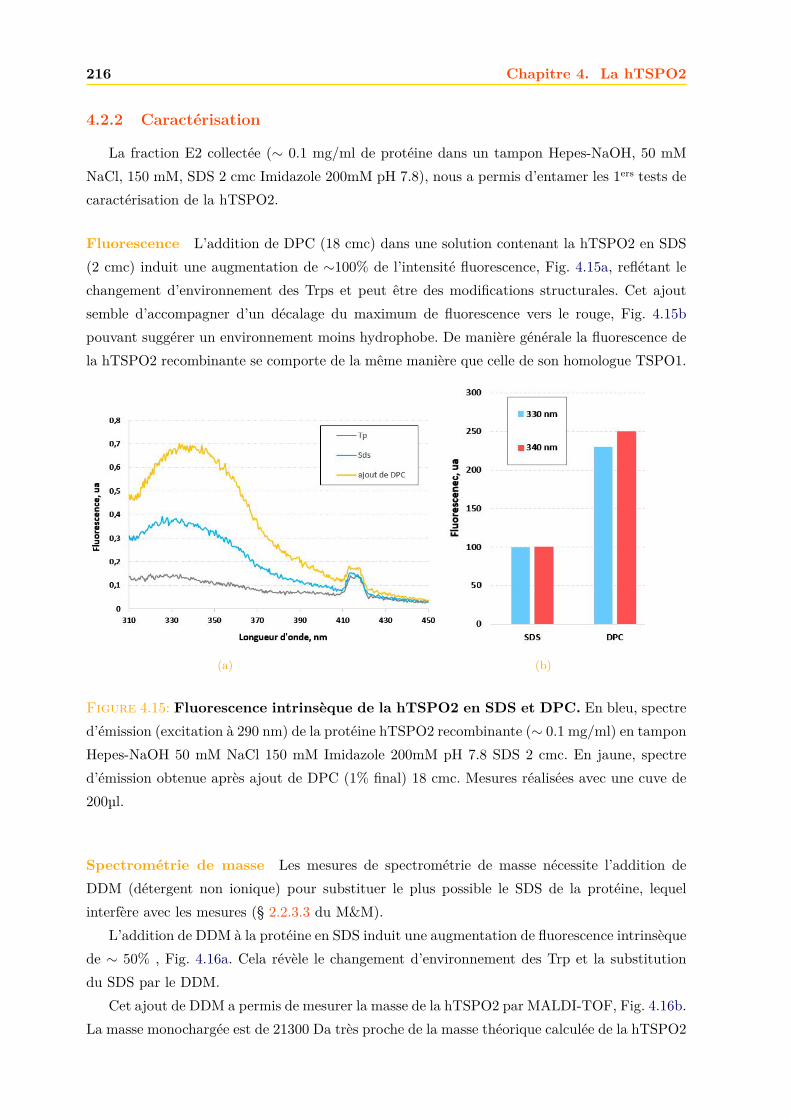
216 Chapitre 4. La hTSPO2
4.2.2 Caractérisation
La fraction E2 collectée (∼ 0.1 mg/ml de protéine dans un tampon Hepes-NaOH, 50 mMNaCl, 150 mM, SDS 2 cmc Imidazole 200mM pH 7.8), nous a permis d’entamer les 1ers tests decaractérisation de la hTSPO2.
Fluorescence L’addition de DPC (18 cmc) dans une solution contenant la hTSPO2 en SDS(2 cmc) induit une augmentation de ∼100% de l’intensité fluorescence, Fig. 4.15a, reflétant lechangement d’environnement des Trps et peut être des modifications structurales. Cet ajoutsemble d’accompagner d’un décalage du maximum de fluorescence vers le rouge, Fig. 4.15bpouvant suggérer un environnement moins hydrophobe. De manière générale la fluorescence dela hTSPO2 recombinante se comporte de la même manière que celle de son homologue TSPO1.
(a) (b)
Figure 4.15: Fluorescence intrinsèque de la hTSPO2 en SDS et DPC. En bleu, spectred’émission (excitation à 290 nm) de la protéine hTSPO2 recombinante (∼ 0.1 mg/ml) en tamponHepes-NaOH 50 mM NaCl 150 mM Imidazole 200mM pH 7.8 SDS 2 cmc. En jaune, spectred’émission obtenue après ajout de DPC (1% final) 18 cmc. Mesures réalisées avec une cuve de200µl.
Spectrométrie de masse Les mesures de spectrométrie de masse nécessite l’addition deDDM (détergent non ionique) pour substituer le plus possible le SDS de la protéine, lequelinterfère avec les mesures (§ 2.2.3.3 du M&M).
L’addition de DDM à la protéine en SDS induit une augmentation de fluorescence intrinsèquede ∼ 50% , Fig. 4.16a. Cela révèle le changement d’environnement des Trp et la substitutiondu SDS par le DDM.
Cet ajout de DDM a permis de mesurer la masse de la hTSPO2 par MALDI-TOF, Fig. 4.16b.La masse monochargée est de 21300 Da très proche de la masse théorique calculée de la hTSPO2
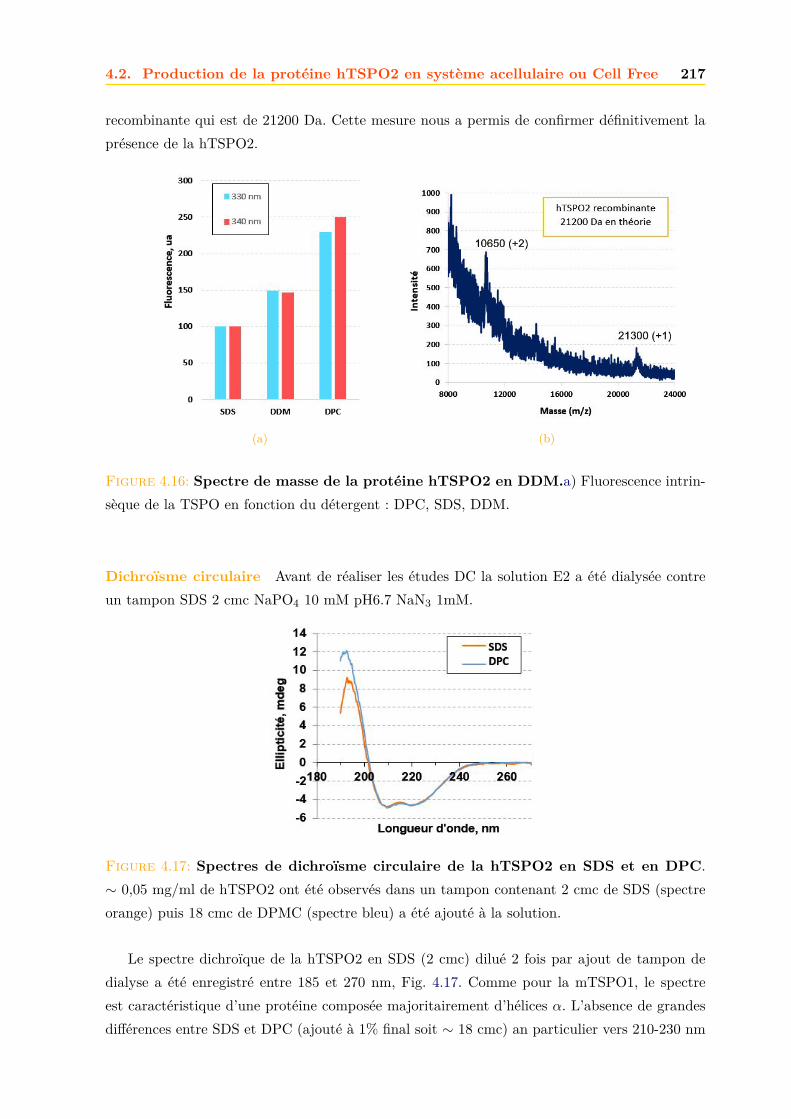
4.2. Production de la protéine hTSPO2 en système acellulaire ou Cell Free 217
recombinante qui est de 21200 Da. Cette mesure nous a permis de confirmer définitivement laprésence de la hTSPO2.
(a) (b)
Figure 4.16: Spectre de masse de la protéine hTSPO2 en DDM.a) Fluorescence intrin-sèque de la TSPO en fonction du détergent : DPC, SDS, DDM.
Dichroïsme circulaire Avant de réaliser les études DC la solution E2 a été dialysée contreun tampon SDS 2 cmc NaPO4 10 mM pH6.7 NaN3 1mM.
Figure 4.17: Spectres de dichroïsme circulaire de la hTSPO2 en SDS et en DPC.∼ 0,05 mg/ml de hTSPO2 ont été observés dans un tampon contenant 2 cmc de SDS (spectreorange) puis 18 cmc de DPMC (spectre bleu) a été ajouté à la solution.
Le spectre dichroïque de la hTSPO2 en SDS (2 cmc) dilué 2 fois par ajout de tampon dedialyse a été enregistré entre 185 et 270 nm, Fig. 4.17. Comme pour la mTSPO1, le spectreest caractéristique d’une protéine composée majoritairement d’hélices α. L’absence de grandesdifférences entre SDS et DPC (ajouté à 1% final soit ∼ 18 cmc) an particulier vers 210-230 nm

218 Chapitre 4. La hTSPO2
peut être due à une structuration déjà relativement bonne de la hTSPO2 en SDS par rapportà la mTSPO1 dont le signal est beaucoup plus dissymétrique dans cette région.
4.3 Conclusions et Perspectives
La méthode du cell free nous a permis de produire une hTSPO2 recombinante que nous avonspu purifier pour entreprendre les premiers tests de caractérisation. Cependant la quantité deprotéine obtenue (de l’ordre de la centaine de µ) ne permet pas encore d’envisager la productionde la protéine marquée 15N,13C nécessaire à son étude par RMN. Des optimisations sont donc àréaliser pour augmenter le rendement. Pour cela il est envisagé de changer de vecteur plasmidiqueet de cloner l’ADN codant pour la hTSPO2 dans un vecteur pIVEX. Ce vecteur a été conçuet optimisé pour l’expression des protéines avec une étiquette polyhistidines en cell free avec lamachinerie d’E. coli [Berrier et al., 2004, Roge and Betton, 2005].
Si ce changement de vecteur permet d’obtenir un rendement satisfaisant, il sera alors possiblede passer à la production de la hTSPO2 marquée, qui ne pose en théorie aucune difficulté (le seulpoint négatif est le coût) puisqu’il suffit d’ajouter les a.a marqués au milieu réactionnel. De plus,le cell free permet de procéder au marquage sélectif de la protéine en ajoutant spécifiquementle ou les a.a marqué(s) désiré(s), ce qui permet de simplifier les spectres RMN qui sont souventsurchargés pour une protéine uniformément marquée. Ce marquage sélectif est souvent pluscomplexe lorsque l’on produit la protéine en bactérie. En effet, l’ajout d’a.a marqués dans unmilieu minimum contenant une source de carbone non marqué nécessite des souches bactériennesauxotrophes afin d’éviter les problèmes de redistribution des isotopes marqués ou "scrambling"intervenant dans la biosynthèse des a.a.
Dans cette dernière étude en cell free nous avons produit la protéine en mode précipité ce-pendant cette méthode est aussi adaptée pour produire la protéine membranaire en détergentou encore directement dans un environnement lipidique, tels que des liposomes ou des nano-disques, en absence de détergent. Cela permet d’obtenir une protéine dans un environnementproche sa membrane native dans lequel elle est le plus souvent correctement repliée, stable,fonctionnelle et prête pour les études en RMN du solide. La description détaillée d’une telleprodédure dans le cas du canal bactérien macano-sensible Mscl est donnée dans Abdine et al.[2012]. Une perspective à long terme est donc de pourvoir appliquer ce même protocole à lahTSPO2 pour résoudre sa structure 3D dans une bicouche lipidique.

Chapitre5
Le peptide Nter d’AtTSPO
Sommaire5.1 Un nouveau rôle pour l’ATtspo ? . . . . . . . . . . . . . . . . . . . . . . . 2195.2 Production du Nter marqué 15N,13C . . . . . . . . . . . . . . . . . . . . 223
5.2.1 Expression dans E. coli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2235.2.2 Purification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2235.2.3 Confirmation de la présence du peptide Nter . . . . . . . . . . . . . . . . . 228
5.3 Caractérisation du peptide . . . . . . . . . . . . . . . . . . . . . . . . . . . 2295.3.1 Dichroïsme circulaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2305.3.2 Résolution de la structure du peptide par RMN . . . . . . . . . . . . . . . . 2305.3.3 Interaction du peptide avec le calcium . . . . . . . . . . . . . . . . . . . . . 233
5.4 Interaction du peptide avec les lipides . . . . . . . . . . . . . . . . . . . . 2355.4.1 Bicelles de DMPG/DHPC . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2355.4.2 Micelles de PI(4,5)P2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 239
5.5 Conclusions, perspectives et discussions . . . . . . . . . . . . . . . . . . . 244
5.1 Un nouveau rôle pour l’ATtspo ?
Comme nous l’avons vu dans l’introduction générale, la TSPO végétale se caractérise par uneextension N-terminale (Nter) qu’on ne retrouve ni chez la TSPO mammalienne ni chez la TSPObactérienne, Fig. 5.1. Les questions qui se posent alors sont : pourquoi l’apparition du Nter chezla plante durant l’évolution ? Quel est son rôle ? Une première hypothèse était que le Nter selonsa longueur avait un rôle dans l’adressage de la protéine dans différentes organelles, d’après lestravaux de Balsemao-Pires et al. [2011] sur l’AtTSPO. Mais nos récentes discussions avec lePr Henri Batoko nous laissent penser que cette hypothèse est réfutable puisque son équipe n’ajamais détecté d’AtTSPO que ce soit dans les fractions mitochondriales ou de plastes mais dansle RE et l’AG.
Le Pr. H. Batoko et son équipe ont montré par des expériences de biochimie que l’AtTSPOinteragit avec différents lipides et en particulier avec les phosphatidylinositol phosphates (PIP,données non publiées). Ils ont également montré que ces interactions sont perdues après unedélétion du Nter de la protéine. Le Pr. H. Batoko nous a aussi décrit que d’autres expériences(non publiées) sur S. pombe laissent suggérer que la TSPO est nécessaire pour la distributiondes PIP dans les membranes.
Toutes ces informations laissent donc supposer un rôle de l’AtTSPO dans la distribution desPIP et l’homéostasie des lipides dans les membranes, qui lui serait conféré par son extensionNter. Ce rôle pourrait avoir une portée plus grande puisque les PIP possèdent un rôle essentieldans la vie de la cellule et notamment au niveau de la signalisation.

220 Chapitre 5. Le peptide Nter d’AtTSPO
HumainA. thaliana
Humain TSPO2R. sphaeroîdes|
HumainA. thaliana
Humain TSPO2R. sphaeroîdes|
HumainA. thaliana
Humain TSPO2R. sphaeroîdes|
HumainA. thaliana
Humain TSPO2R. sphaeroîdes|
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M A P P W V P A M G F T L A P S L 17
G C F V G S R F V H G E G L R W Y A G L Q K P S W H P P H W V L G P V W G T L Y S A M G Y G S Y L V W K E L G G F T 75
. E K A V V P L G L Y T G Q L A L N W A W P P I F F G A R Q M G W A L V D L L L V S G A A A A T T V A W Y Q V S P L 132
A A R L L Y P Y L A W L A F T T T L N Y C V W R D N H G W R G G R R L P E . . . 169
M D S Q D I R Y R G G D D R D A A T T A M A E T E R K S A D D N K G K R D Q K R A M A K R G L K S L T V A V A A P V 58
L V T L F A T Y F L G T S D G Y G N R A K S S S W I P P L W L L H T T C L A S S G L M G L A A W L V W V D G G F H K 116
. . . K P N A L Y L Y L A Q F L L C L V W D P V T F R V G S G V A G L A V W L G Q S A A L F G C Y K A F N E I S P V 171
A G N L V K P C L A W A A F V A A V N V K L A V A . . . . . . . . . . . . . . . 196
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M R L Q G A I F V L L P H L 14
G P I L V W L F T R D H M S G W C E G P R M L S W C P F Y K V L L L V Q T A I Y S V V G Y A S Y L V W K D L G G G L 72
G W P L A L P L G L Y A V Q L T I S W T V L V L F F T V H N P G L A L L H L L L L Y G L V V S T A L I W H P I N K L 130
A A L L L L P Y L A W L T V T S A L T Y H L W R D S L C P V H Q P Q P T E K S D 170
1 10
20 30 40 50 60 70
80 90 100 110 120 130
140 150 160
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . M N M D W A L F L T F L A A 14
C G A P A T T G A L L K P D E W Y D N L N K P W W N P P R W V F P L A W T S L Y F L M S L A A M R V A Q L E G S G . 71
. . . . . Q A L A F Y A A Q L A F N T L W T P V F F G M K R M A T A L A V V M V M W L F V A A T M W A F F Q L D T W 124
A G V L F V P Y L I W A T A A T G L N F E A M R L N W N R P E A R A . . . . . . 158
TM1 TM2
TM3 TM4
TM5
C-terminal
N-terminal
Loop 1
Loop 3 Loop 4
Loop 2
CRAC L/V.Y...R/D
Figure 5.1: Nter d’AtTSPO. Séquence de la protéine entière AtTSPO de 196 a.a soit ∼ 21kDa. En vert sont représentés les 49 a.a du peptide constituant la partie N-terminale étudiéelors de cette étude. Sont également représentées les séquences des TSPO1 et TSPO2 humaineset de la bactéries R. sphaeroides.
Les phosphatidylinositol phosphates Les lipides majeures des membranes eucaryotessont les glycérophospholipides : phosphatidylcholine (>50% des phospholipides de la plupartdes membranes eucaryotes), phosphatidyléthanolamine phosphatidylsérine, phosphatidylinosi-tol (PI) et l’acide phosphatidique. Le RE est le principal site de synthèse des lipides, lesquelssont ensuite rapidement transportés aux autres organelles (mitochondrie, membrane plasmique,peroxysome, chloroplaste, etc) [van Meer et al., 2008]. Il y a différents moyens pour les lipidesd’être transportés :
• les vésicules ;• les protéines solubles ;• "flip flop" à travers la bicouche lipidique ;• et les mouvements aux sites de contact.
Le trafic lipidique entre les différentes organelles chez les cellules des plantes est très similairede celui retrouvé chez les cellules animales [Hurlock et al., 2014].
Dans le cas des PIP, leur précurseur (PI) est synthétisé dans le RE puis il est ensuite délivréaux autres Mb par transport vésiculaire ou par des protéines appelées PhosphatIdylinositolTransfer Proteins (PITP)
Les phosphatidylinositol phosphates sont des lipides anioniques qui bien que représentantmoins de 1% des lipides totaux des Mb ont un rôle indispensable dans de nombreux processuscellulaires tels que le contrôle du trafic vésiculaire et les événements de signalisation [De Matteisand Godi, 2004, McLaughlin and Murray, 2005, Lemmon, 2008, Balla et al., 2009] :
• les PIP sont connus comme étant la source de seconds messagers (inositol 1,4,5-triphosphate, diacylglycérol) impliqués dans la transduction des signaux provenant des

5.1. Un nouveau rôle pour l’ATtspo ? 221
récepteurs de la surface cellulaire ;• ils sont des activateurs de nombreux canaux ioniques et d’enzymes ;• ils sont impliqués dans le transport vésiculaire des protéines entre les différents comparti-ments membranaires 1 cellulaires, incluant les mécanismes d’exocytose et d’endocytose ;• ils modulent la distribution et le métabolisme des lipides à travers leur relation étroiteavec les protéines de transfert, participant ainsi à l’homéostasie des membranes ;• bien qu’en très faible quantité dans la Mb, ils sont des sites d’ancrage spécifiques auxdifférents compartiments membranaires de la cellule, et contribuent ainsi à leur identité :les compartiments membranaires sont enrichis ou dépourvues de PIP spécifiques, ce quidonne une composition unique pour chaque compartiment. Il a été proposé que cetteorganisation s’apparente à un "code", qui peut être lu par des protéines qui interagissentspécifiquement avec ces PIP [Kutateladze, 2010]. Récemment, Simon et al. ont montréque le "code PIP" n’est pas suffisant pour expliquer la sélection des protéines liant leslipides parmi les différents compartiments membranaires de la cellule chez la plante. LesPIP sont des lipides négativement chargés et cette propriété physique pourrait contribuerà l’identité des Mb en régulant leurs charges de surface. P. ex, la membrane plasmique aune signature électrostatique spécifique contrôlée par le PI4P et ce champ électrostatiquecontrôle la localisation, le recrutement et la fonction de plusieurs protéines agissant enaval de récepteurs kinase ou impliquées dans certaines voies de signalisation hormonale[Simon et al., 2013].
Il existe 7 différentes espèces de PIP, qui sont issues de la phosphorylation du phosphatidy-linositol (PI) à différentes positions (3, 4 et/ou 5) de l’anneau inositol, Fig. 5.2 :
• 3 phosphatidylinositol monophosphates : PI3P, PI4P et PI5P ;• 3 phosphatidylinositol biphosphate : PI(3,4)P2, PI(3,5)P2 et PI(4,5)P2 ;• et 1 phosphatidylinositol triphosphate : PI(3,4,5)P3.
Le contrôle de leur fonction se fait par cette phosphorylation/déphosphorylation par des kinaseset phosphatases spécifiques des différentes membranes intracellulaires [Lemmon, 2008].
Dans les cellules épithéliales des racines d’Arabidopsis, Simon et al. retrouvent, Fig. 5.3 :
• le PI(4,5)P2 localisé strictement dans la membrane plasmique et non dans les comparti-ments endomembranaires (noyau, RE, AG, lysosomes, vésicules, endosomes) ;• le PI3P localisé dans les endosomes tardifs (late)/pré-vacuolaires (PVC) et de façonmoindre dans les tonoplastes (membranes des vacuoles) ;• le PI4P localisé dans la membrane plasmique et dans l’AG, les endosomes précoces (early)du réseau trans-golgien (Trans Golgi Network ou TGN ; un réseau de canalicules) et lesendosomes recyclés.
1. Les cellules eucaryotes sont organisées en compartiments limités par une membrane et fonctionnellementdistincts. Ces compartiments membranaires comprennent les différentes organites de la cellule - le noyau, lesmitochondries, l’AG, le RE, les peroxysomes, les lysosomes - et les vésicules qui servent de transporteurs entreles organites.

222 Chapitre 5. Le peptide Nter d’AtTSPO
Figure 5.2: Les phosphatidylinositol phosphates - PIP. Les phosphatidylinositols sont des phos-pholipides composés d’un squelette diacylglycérol hydrophobe (R) lié a un anneau inositol hydrophilevia un liaison phosphodiester. Ils peuvent être phosphorylés (en rouge) à différentes positions sur leuranneau inositol pour produire 7 espèces de PIPs différentes. Les PIP présents chez les plantes sont enca-drés en vert. Le schéma du métabolisme des PIP chez les plantes est encadré en orange. Figure adaptéede http://www.illuminatedcell.com/Phosphoinositides.html.
Figure 5.3: Localisations des PI3P, PI4P and PI(4,5)P2 dans la cellule épithélialed’Arabidopsis. Le gradient d’intensité de la localisation dans les compartiments intracellulaires estreprésenté par la largeur du triangle et un dégradé de couleur. Le tonoplaste est le terme désignant lamembrane des vacuoles. La différence entre les endosomes précoces "early" et tardifs "late" est que cesderniers sont plus acides et possèdent des hydrolases acides. Figure adaptée de [Simon et al., 2013].
Dans le but d’explorer le rôle du Nter d’AtTSPO, nous avons donc décidé en collaboration

5.2. Production du Nter marqué 15N,13C 223
avec le Pr. H. Batoko, de le surexprimer, d’étudier sa structure par RMN et son comportementen présence de lipides et de PIP.
5.2 Production du Nter marqué 15N,13C
5.2.1 Expression dans E. coli
Les bactéries BL21 d’E.coli transformées avec le plasmide pGEX4-T2 contenant le Nterd’AtTSPO (non conservé chez les mammifères et les bactéries, Fig.5.1) ont été généreusementoffertes par le Pr. H. Batoko de l’université catholique de Louvain (Belgique), avec lequel nousavons travaillé sur ce projet. Ce plasmide nous a permis la surexpression du peptide correspon-dant au 49 1ers a.a de la partie N-terminale de la protéine AtTSPO, fusionnée à une protéineGST pour une meilleure solubilisation et purification du peptide, Fig. 5.4. Après quelques étapesde mise au point en milieu riche LB nous avons travaillé en milieu M9 pour obtenir une protéinemarquée. Dans les conditions d’une induction de ∼4 h à 37 ◦C et après ajout de 1 mM d’IPTG,la culture bactérienne en milieu M9, contenant les sources isotopiques marquées 15N,13C, atteintune DO de ∼ 5, Fig.5.5a.
L’analyse sur gel SDS-PAGE de fractions bactériennes prélevées avant et après inductionmontre bien l’apparition d’une bande intense au PM de la protéine de fusion GST, Fig. 5.5b.
Figure 5.4: Représentation schématique de la protéine de fusion GST-Nter surexpriméepar les bactéries E. coli. La protéine GST a un PM de ∼ 26 kDa. La GST est liée en C-terminal àun site de coupure à la thrombine, une étiquette de 10 histidines, un site de coupure à la protéase TEV(Tobacco Etch Virus) et finalement au peptide Nter d’AtTSPO. Le peptide Nter a un PM de ∼ 5.3 kDaet la construction entière de la protéine de fusion a un PM de ∼ 35 kDa. Le slash entre les résidus Glnet Gly dans le site de coupure à la TEV représente l’endroit dans la séquence où la protéase va cliver.
5.2.2 Purification
Le protocole de purification du peptide Nter est présenté Sec.2.2.4 du M&M.
Purification de la protéine de fusion GST-Nter sur résine Ni-NTA Les culots bacté-riens ont été repris dans un tampon hautement salin pour dissocier les ADN présents qui, lorsdes ers essais avaient bouchés notre colonne de purification.
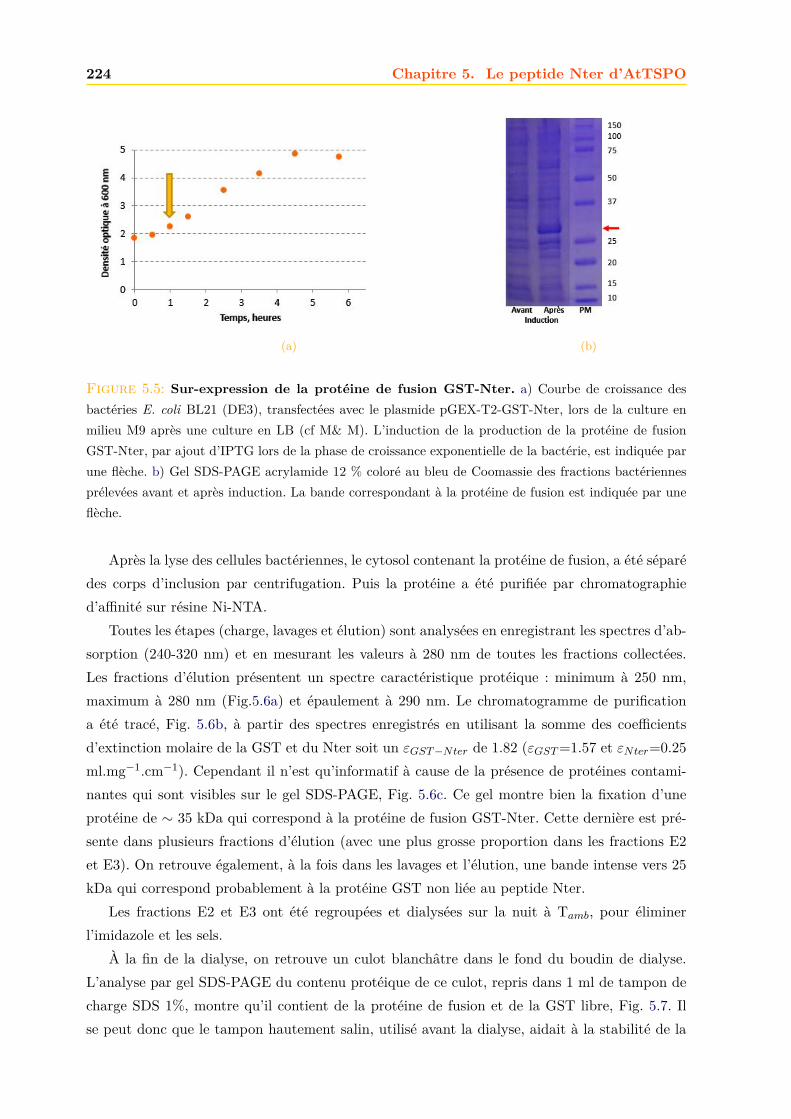
224 Chapitre 5. Le peptide Nter d’AtTSPO
(a) (b)
Figure 5.5: Sur-expression de la protéine de fusion GST-Nter. a) Courbe de croissance desbactéries E. coli BL21 (DE3), transfectées avec le plasmide pGEX-T2-GST-Nter, lors de la culture enmilieu M9 après une culture en LB (cf M& M). L’induction de la production de la protéine de fusionGST-Nter, par ajout d’IPTG lors de la phase de croissance exponentielle de la bactérie, est indiquée parune flèche. b) Gel SDS-PAGE acrylamide 12 % coloré au bleu de Coomassie des fractions bactériennesprélevées avant et après induction. La bande correspondant à la protéine de fusion est indiquée par uneflèche.
Après la lyse des cellules bactériennes, le cytosol contenant la protéine de fusion, a été séparédes corps d’inclusion par centrifugation. Puis la protéine a été purifiée par chromatographied’affinité sur résine Ni-NTA.
Toutes les étapes (charge, lavages et élution) sont analysées en enregistrant les spectres d’ab-sorption (240-320 nm) et en mesurant les valeurs à 280 nm de toutes les fractions collectées.Les fractions d’élution présentent un spectre caractéristique protéique : minimum à 250 nm,maximum à 280 nm (Fig.5.6a) et épaulement à 290 nm. Le chromatogramme de purificationa été tracé, Fig. 5.6b, à partir des spectres enregistrés en utilisant la somme des coefficientsd’extinction molaire de la GST et du Nter soit un εGST−Nter de 1.82 (εGST=1.57 et εNter=0.25ml.mg−1.cm−1). Cependant il n’est qu’informatif à cause de la présence de protéines contami-nantes qui sont visibles sur le gel SDS-PAGE, Fig. 5.6c. Ce gel montre bien la fixation d’uneprotéine de ∼ 35 kDa qui correspond à la protéine de fusion GST-Nter. Cette dernière est pré-sente dans plusieurs fractions d’élution (avec une plus grosse proportion dans les fractions E2et E3). On retrouve également, à la fois dans les lavages et l’élution, une bande intense vers 25kDa qui correspond probablement à la protéine GST non liée au peptide Nter.
Les fractions E2 et E3 ont été regroupées et dialysées sur la nuit à Tamb, pour éliminerl’imidazole et les sels.
À la fin de la dialyse, on retrouve un culot blanchâtre dans le fond du boudin de dialyse.L’analyse par gel SDS-PAGE du contenu protéique de ce culot, repris dans 1 ml de tampon decharge SDS 1%, montre qu’il contient de la protéine de fusion et de la GST libre, Fig. 5.7. Ilse peut donc que le tampon hautement salin, utilisé avant la dialyse, aidait à la stabilité de la
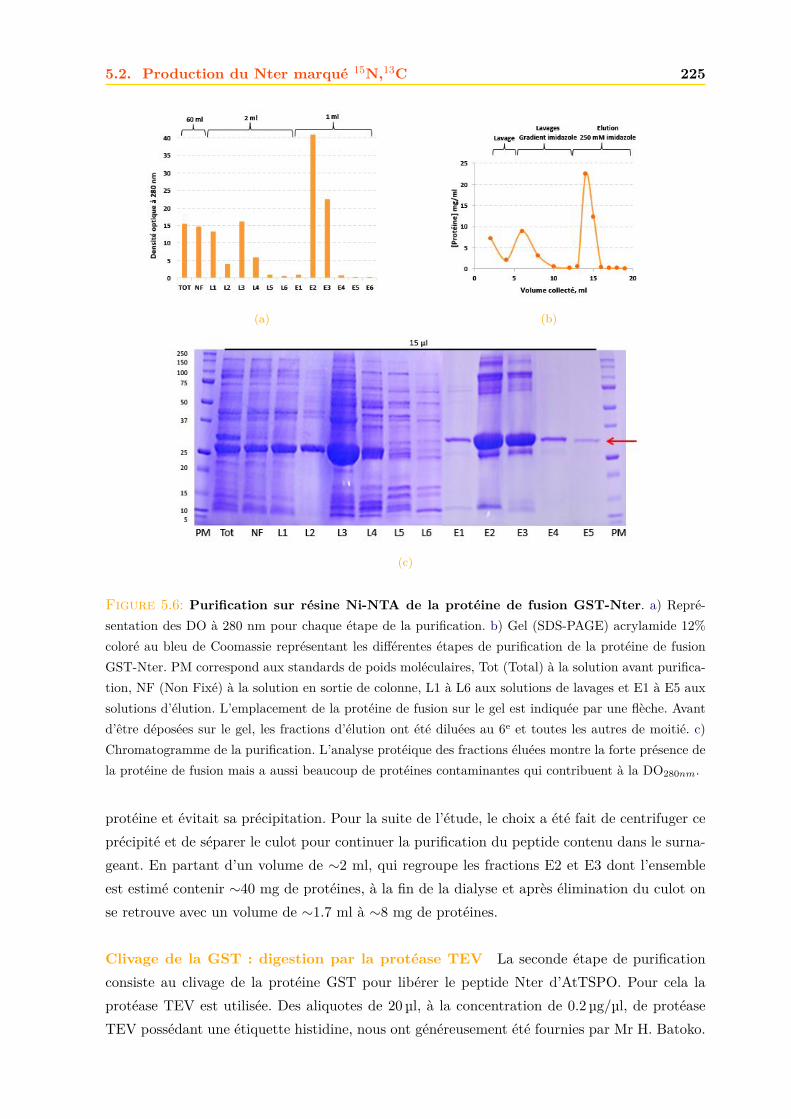
5.2. Production du Nter marqué 15N,13C 225
(a) (b)
(c)
Figure 5.6: Purification sur résine Ni-NTA de la protéine de fusion GST-Nter. a) Repré-sentation des DO à 280 nm pour chaque étape de la purification. b) Gel (SDS-PAGE) acrylamide 12%coloré au bleu de Coomassie représentant les différentes étapes de purification de la protéine de fusionGST-Nter. PM correspond aux standards de poids moléculaires, Tot (Total) à la solution avant purifica-tion, NF (Non Fixé) à la solution en sortie de colonne, L1 à L6 aux solutions de lavages et E1 à E5 auxsolutions d’élution. L’emplacement de la protéine de fusion sur le gel est indiquée par une flèche. Avantd’être déposées sur le gel, les fractions d’élution ont été diluées au 6e et toutes les autres de moitié. c)Chromatogramme de la purification. L’analyse protéique des fractions éluées montre la forte présence dela protéine de fusion mais a aussi beaucoup de protéines contaminantes qui contribuent à la DO280nm.
protéine et évitait sa précipitation. Pour la suite de l’étude, le choix a été fait de centrifuger ceprécipité et de séparer le culot pour continuer la purification du peptide contenu dans le surna-geant. En partant d’un volume de ∼2 ml, qui regroupe les fractions E2 et E3 dont l’ensembleest estimé contenir ∼40 mg de protéines, à la fin de la dialyse et après élimination du culot onse retrouve avec un volume de ∼1.7 ml à ∼8 mg de protéines.
Clivage de la GST : digestion par la protéase TEV La seconde étape de purificationconsiste au clivage de la protéine GST pour libérer le peptide Nter d’AtTSPO. Pour cela laprotéase TEV est utilisée. Des aliquotes de 20 µl, à la concentration de 0.2µg/µl, de protéaseTEV possédant une étiquette histidine, nous ont généreusement été fournies par Mr H. Batoko.
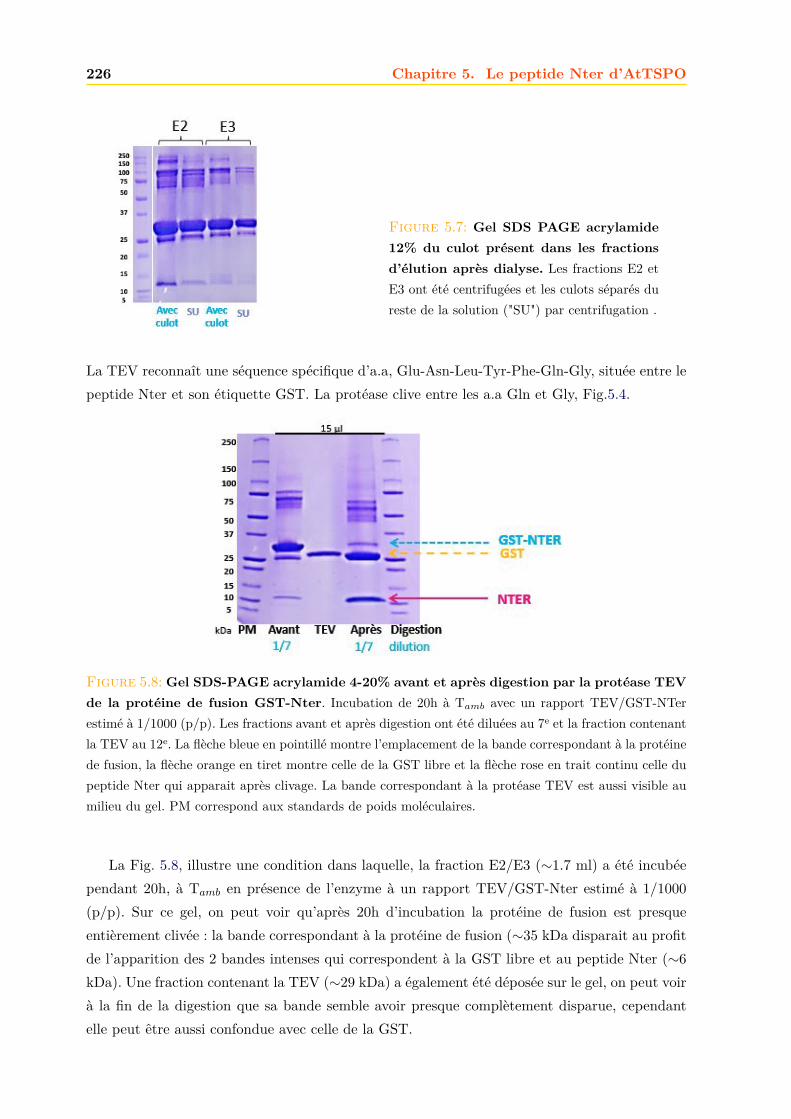
226 Chapitre 5. Le peptide Nter d’AtTSPO
Figure 5.7: Gel SDS PAGE acrylamide12% du culot présent dans les fractionsd’élution après dialyse. Les fractions E2 etE3 ont été centrifugées et les culots séparés dureste de la solution ("SU") par centrifugation .
La TEV reconnaît une séquence spécifique d’a.a, Glu-Asn-Leu-Tyr-Phe-Gln-Gly, située entre lepeptide Nter et son étiquette GST. La protéase clive entre les a.a Gln et Gly, Fig.5.4.
Figure 5.8: Gel SDS-PAGE acrylamide 4-20% avant et après digestion par la protéase TEVde la protéine de fusion GST-Nter. Incubation de 20h à Tamb avec un rapport TEV/GST-NTerestimé à 1/1000 (p/p). Les fractions avant et après digestion ont été diluées au 7e et la fraction contenantla TEV au 12e. La flèche bleue en pointillé montre l’emplacement de la bande correspondant à la protéinede fusion, la flèche orange en tiret montre celle de la GST libre et la flèche rose en trait continu celle dupeptide Nter qui apparait après clivage. La bande correspondant à la protéase TEV est aussi visible aumilieu du gel. PM correspond aux standards de poids moléculaires.
La Fig. 5.8, illustre une condition dans laquelle, la fraction E2/E3 (∼1.7 ml) a été incubéependant 20h, à Tamb en présence de l’enzyme à un rapport TEV/GST-Nter estimé à 1/1000(p/p). Sur ce gel, on peut voir qu’après 20h d’incubation la protéine de fusion est presqueentièrement clivée : la bande correspondant à la protéine de fusion (∼35 kDa disparait au profitde l’apparition des 2 bandes intenses qui correspondent à la GST libre et au peptide Nter (∼6kDa). Une fraction contenant la TEV (∼29 kDa) a également été déposée sur le gel, on peut voirà la fin de la digestion que sa bande semble avoir presque complètement disparue, cependantelle peut être aussi confondue avec celle de la GST.
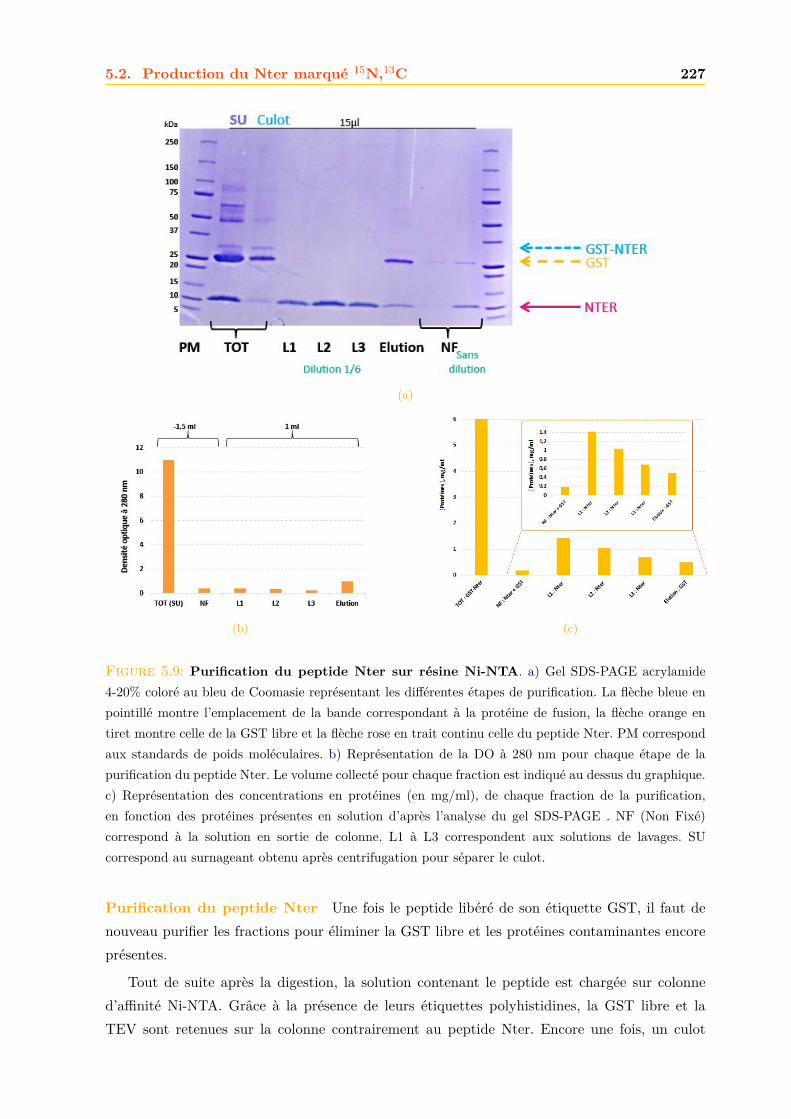
5.2. Production du Nter marqué 15N,13C 227
(a)
(b) (c)
Figure 5.9: Purification du peptide Nter sur résine Ni-NTA. a) Gel SDS-PAGE acrylamide4-20% coloré au bleu de Coomasie représentant les différentes étapes de purification. La flèche bleue enpointillé montre l’emplacement de la bande correspondant à la protéine de fusion, la flèche orange entiret montre celle de la GST libre et la flèche rose en trait continu celle du peptide Nter. PM correspondaux standards de poids moléculaires. b) Représentation de la DO à 280 nm pour chaque étape de lapurification du peptide Nter. Le volume collecté pour chaque fraction est indiqué au dessus du graphique.c) Représentation des concentrations en protéines (en mg/ml), de chaque fraction de la purification,en fonction des protéines présentes en solution d’après l’analyse du gel SDS-PAGE . NF (Non Fixé)correspond à la solution en sortie de colonne. L1 à L3 correspondent aux solutions de lavages. SUcorrespond au surnageant obtenu après centrifugation pour séparer le culot.
Purification du peptide Nter Une fois le peptide libéré de son étiquette GST, il faut denouveau purifier les fractions pour éliminer la GST libre et les protéines contaminantes encoreprésentes.
Tout de suite après la digestion, la solution contenant le peptide est chargée sur colonned’affinité Ni-NTA. Grâce à la présence de leurs étiquettes polyhistidines, la GST libre et laTEV sont retenues sur la colonne contrairement au peptide Nter. Encore une fois, un culot

228 Chapitre 5. Le peptide Nter d’AtTSPO
contenant de la GST en majorité, s’est reformé après la digestion (Fig. 5.9a, "SU" et "NF"). Leculot a été séparé par centrifugation avant que la solution ne soit déposée sur la colonne.
L’analyse des fractions sur gel SDS-PAGE, Fig. 5.9a, montre que la fraction non fixée (NF)contient les protéines contaminantes et une très faible fraction de peptide Nter. En effet, levolume mort de la colonne correspond à peu près au volume du dépôt et le peptide ne sortque lors des étapes de lavages après que la 1re fraction déposée est chassée le volume mort. Lafraction éluée contient la GST qui a été décrochée par l’imidazole.
La mesure des DO à 280 nm, Fig. 5.9b, permet d’estimer la quantité de protéines présentedans chaque fraction. Pour les fractions TOT et NF, le gel SDS-PAGE montrant la présence àla fois de la GST et du Nter, l’ε utilisé est la somme des ε de la protéine et du peptide. Pour lesfractions de lavages, le gel ne montrant que la présence du Nter, seul l’ε de celui-ci a été utilisé.Pour la fraction d’élution seul celui de la GST a été utilisé.
Dans ces conditions de purification, la fraction la plus concentrée en protéine est L1 (1 ml)dont la quantité en peptide a été estimée à ∼ 1.3 mg (220 µM), Fig. 5.9c. Les fractions L2 etL3, ont été estimées, quant à elles, à ∼ 1 mg (180 µM) et ∼ 0.8 mg (140 µM), respectivement.Cependant l’ε de la protéine étant petit (0.25), le signal en spectrométrie UV du peptide estfaible. La moindre présence de GST (ou d’autres protéines contaminantes absorbant à 280 nm),même si elle n’est pas visible sur le gel, peut contribuer à la DO à 280 nm. La quantité de Nterest alors probablement sur-estimée.
5.2.3 Confirmation de la présence du peptide Nter
Dot Blot Le technique du dot-blot, consistant à déposer les fractions protéiques sur unemembrane de nitrocellulose sous forme de points puis à effectuer les étapes d’un western blotclassique (§ 2.3.2 du M&M), nous a permis de confirmer la présence du peptide dans nos fractionsde lavages, Fig 5.10. L’AC primaire utilisé est un AC dirigé contre le Nter d’AtTSPO (fournipar le Pr H. Batoko).
Figure 5.10: Confirmation de la présence du peptide N-ter par Dot Blot. Les fractions col-lectées lors de la purification et déposées sur la membrane sont la solution en sortie de colonne NF (NonFixé) et les solutions de lavages L1 à L3. L’AC primaire utilisé est un AC dirigé spécifiquement contrele Nter.
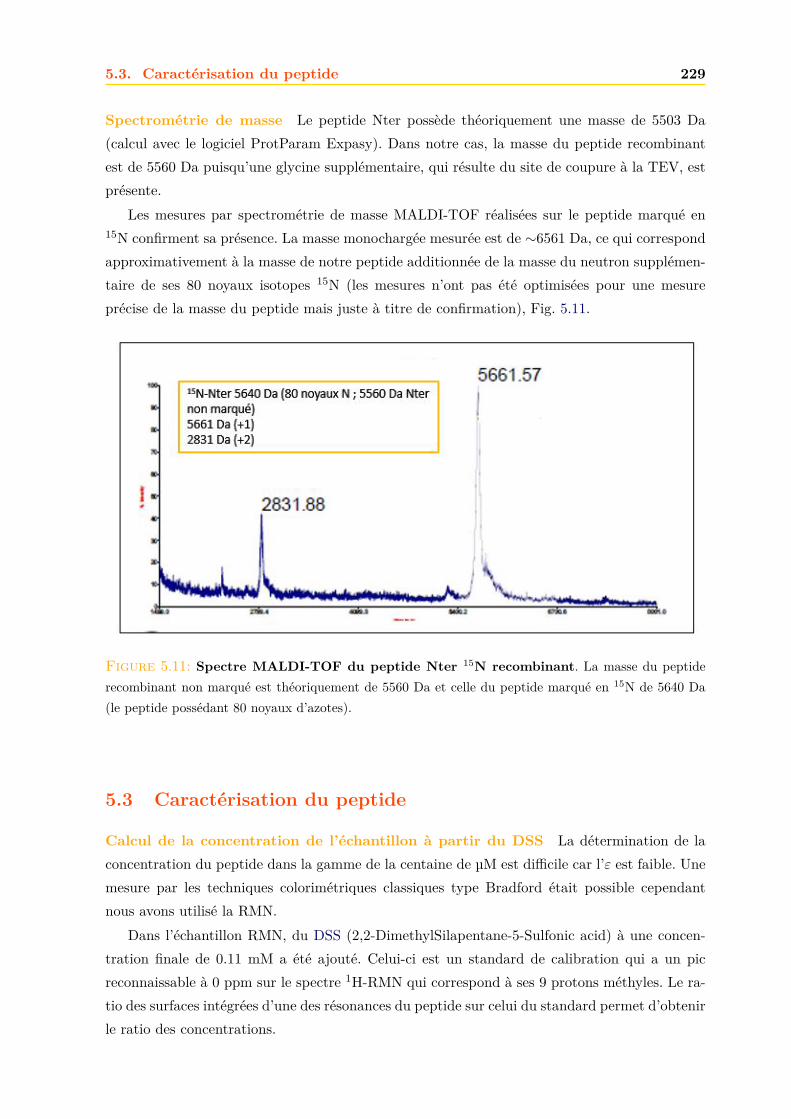
5.3. Caractérisation du peptide 229
Spectrométrie de masse Le peptide Nter possède théoriquement une masse de 5503 Da(calcul avec le logiciel ProtParam Expasy). Dans notre cas, la masse du peptide recombinantest de 5560 Da puisqu’une glycine supplémentaire, qui résulte du site de coupure à la TEV, estprésente.
Les mesures par spectrométrie de masse MALDI-TOF réalisées sur le peptide marqué en15N confirment sa présence. La masse monochargée mesurée est de ∼6561 Da, ce qui correspondapproximativement à la masse de notre peptide additionnée de la masse du neutron supplémen-taire de ses 80 noyaux isotopes 15N (les mesures n’ont pas été optimisées pour une mesureprécise de la masse du peptide mais juste à titre de confirmation), Fig. 5.11.
Figure 5.11: Spectre MALDI-TOF du peptide Nter 15N recombinant. La masse du peptiderecombinant non marqué est théoriquement de 5560 Da et celle du peptide marqué en 15N de 5640 Da(le peptide possédant 80 noyaux d’azotes).
5.3 Caractérisation du peptide
Calcul de la concentration de l’échantillon à partir du DSS La détermination de laconcentration du peptide dans la gamme de la centaine de µM est difficile car l’ε est faible. Unemesure par les techniques colorimétriques classiques type Bradford était possible cependantnous avons utilisé la RMN.
Dans l’échantillon RMN, du DSS (2,2-DimethylSilapentane-5-Sulfonic acid) à une concen-tration finale de 0.11 mM a été ajouté. Celui-ci est un standard de calibration qui a un picreconnaissable à 0 ppm sur le spectre 1H-RMN qui correspond à ses 9 protons méthyles. Le ra-tio des surfaces intégrées d’une des résonances du peptide sur celui du standard permet d’obtenirle ratio des concentrations.
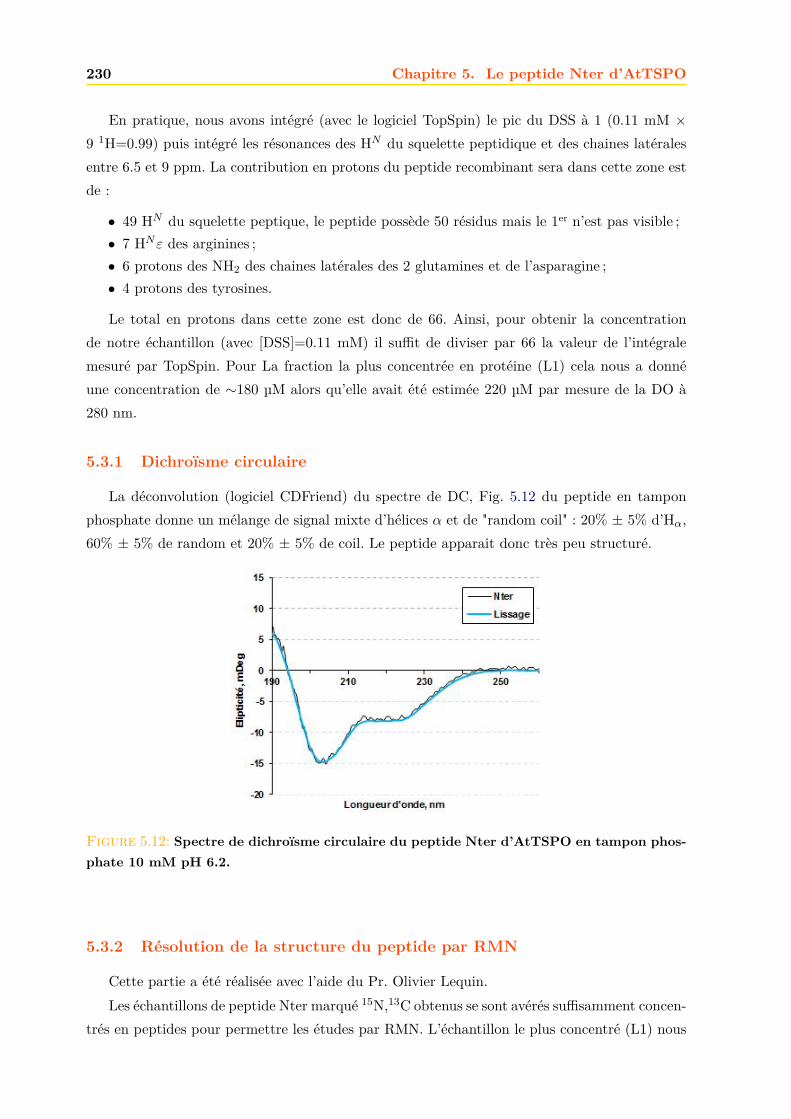
230 Chapitre 5. Le peptide Nter d’AtTSPO
En pratique, nous avons intégré (avec le logiciel TopSpin) le pic du DSS à 1 (0.11 mM ×9 1H=0.99) puis intégré les résonances des HN du squelette peptidique et des chaines latéralesentre 6.5 et 9 ppm. La contribution en protons du peptide recombinant sera dans cette zone estde :
• 49 HN du squelette peptique, le peptide possède 50 résidus mais le 1er n’est pas visible ;• 7 HNε des arginines ;• 6 protons des NH2 des chaines latérales des 2 glutamines et de l’asparagine ;• 4 protons des tyrosines.
Le total en protons dans cette zone est donc de 66. Ainsi, pour obtenir la concentrationde notre échantillon (avec [DSS]=0.11 mM) il suffit de diviser par 66 la valeur de l’intégralemesuré par TopSpin. Pour La fraction la plus concentrée en protéine (L1) cela nous a donnéune concentration de ∼180 µM alors qu’elle avait été estimée 220 µM par mesure de la DO à280 nm.
5.3.1 Dichroïsme circulaire
La déconvolution (logiciel CDFriend) du spectre de DC, Fig. 5.12 du peptide en tamponphosphate donne un mélange de signal mixte d’hélices α et de "random coil" : 20% ± 5% d’Hα,60% ± 5% de random et 20% ± 5% de coil. Le peptide apparait donc très peu structuré.
Figure 5.12: Spectre de dichroïsme circulaire du peptide Nter d’AtTSPO en tampon phos-phate 10 mM pH 6.2.
5.3.2 Résolution de la structure du peptide par RMN
Cette partie a été réalisée avec l’aide du Pr. Olivier Lequin.Les échantillons de peptide Nter marqué 15N,13C obtenus se sont avérés suffisamment concen-
trés en peptides pour permettre les études par RMN. L’échantillon le plus concentré (L1) nous

5.3. Caractérisation du peptide 231
a permis l’acquisition de spectres de bonne qualité en termes de dispersion spectrale et d’ho-mogénéité des largeurs de raies. De plus, l’échantillon s’est révélé être stable pendant le tempsnécessaire à l’acquisition de données RMN 3D permettant l’attribution des signaux des différentsrésidus du peptide sur le spectre 2D 1H,15N HSQC, Fig. 5.13.
Dans un 1er temps, les résonances des atomes du squelette peptidique (13Cα, 13Cβ, 15NHet 1HN) ont été attribuées par analyse des spectres 3D HNCACB, CBCA(CO)NH, HNCO etHN(CA)CO, et du spectre 2D 1H,15N HSQC du peptide en tampon phosphate 10 mM, pH 6.2(la méthodologie d’attribution est présentée Sec. 2.4.4.4 du M&M).
Les résidus utilisés comme points de départs dans l’attribution séquentielle sont des résidusdont le déplacement chimique (δ) du 13Cα, 13Cβ, 15NH et 1HN est particulier. Il s’agit toutparticulièrement :
• des glycines (Gly) dont le δ du 15NH est ∼ 110 ppm et qui ne possèdent pas de 13Cβ etdont les 13Cα sont blindés (δ faible),• des thréonines (Thr) et des sérines (Ser) dont les δ des 13Cβ est au voisinage et au-delàde 60 ppm ;• et des alanines (Ala) dont le δ du 13Cβ est en dessous de 20 ppm.
Ainsi les signaux des Ser (S4, S29 et S50) ; des Thr (T19, T20 et T25) ; et des Gly (G11, G12,G35 et G47) et des résidus voisins (i-1 et i+1) ont ainsi pu être rapidement attribués.
Ensuite le processus d’attribution est semblable à un puzzle avec certaine partie plus dureque d’autre, p. ex. il a été difficile d’attribuer les signaux des "motifs" de résidus se répétant dansla séquence : AMA (2 fois présents), les arginines et les lysines des motifs KR (3 fois présents).De plus les signaux de leur groupement amide sont proches sur le spectre HSQC et leur 13Cαet 13Cβ résonnent dans la même gamme de fréquence sur les spectres 3D. Les expériencesHNCACB, CBCA(CO)NH, nous ont permis d’attribuer la plupart des résonances du squelettepeptique, puis les expériences HNCO et HN(CA)CO ont permis de lever l’ambigüité sur lesparties un peu plus complexes.
En complément des expériences citées ci-dessus, une expérience 2D CBCA(CO)NH optimiséepour détecter les groupements CO-NH2 a permis d’attribuer les signaux des chaines latéralesdes résidus Asn et Gln visibles entre 6.8 et 7.6 ppm en dimension 1H sur le spectre HSQC.
Analyse des déplacements chimiques Le déplacement chimique (δ) des noyaux de lachaine principale d’un résidu est influencé par plusieurs facteurs dont la structure secondairedans laquelle celui-ci est engagé. Ainsi, à partir des δ des noyaux 13CO, 13Cα, 13Cβ, 1Hα, 1HN et15N , il est possible d’évaluer les éléments de structure secondaire de la protéine ou du peptide. Laméthode consiste à calculer la différence (∆δ) entre le déplacement chimique expérimental (δobs)et une valeur de référence (δcoil) correspondant au même noyau pour le même acide aminé dansune conformation aléatoire "Random coil". La valeur obtenue est appelée déplacement chimiquesecondaire (CSD ou Chemical Shift Deviation) [Wishart and Sykes, 1994a,b]. Les CSD n’ontpas la même signification selon la nature du noyau. Ainsi pour le proton 1H, une succession
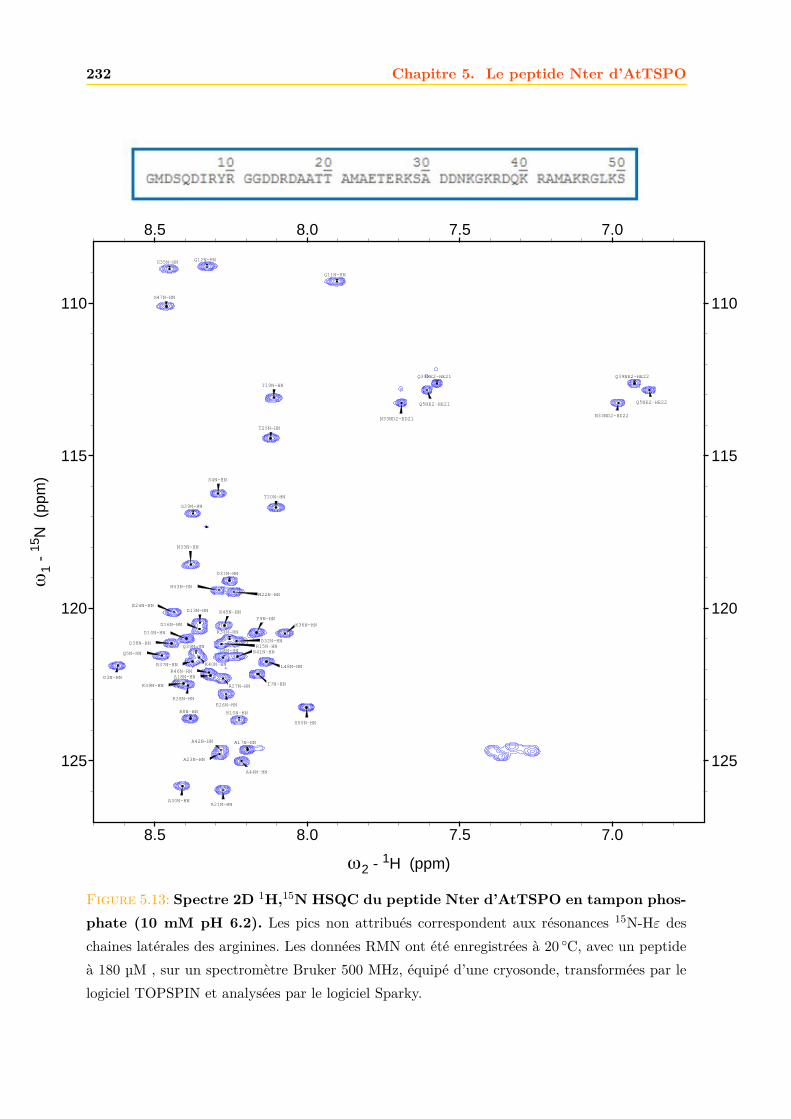
232 Chapitre 5. Le peptide Nter d’AtTSPO
8.5
8.5
8.0
8.0
7.5
7.5
7.0
7.0
ω2 - 1H (ppm)
125 125
120 120
115 115
110 110
ω1
- 15N
(pp
m)
A21N-HNA30N-HN
A44N-HN
A23N-HN
A17N-HN
R10N-HNR8N-HN
S50N-HN
E26N-HN
K49N-HN R27N-HN
A18N-HNI7N-HN
D3N-HN
R37N-HN L48N-HNK40N-HN
D6N-HN R41N-HNQ5N-HNQ39N-HN R15N-HN
D38N-HN
K34N-HN
D32N-HN
D14N-HN
K36N-HNY9N-HN
D16N-HN
K45N-HND13N-HNE24N-HN
M22N-HN
M43N-HN
D31N-HN
A42N-HN
R46N-HN
K28N-HN
G11N-HN
N33N-HN
S29N-HN
S4N-HN
T20N-HN
T25N-HN
T19N-HN
G12N-HNG35N-HN
G47N-HN
N33ND2-HD21
Q5NE2-HE21
Q39NE2-HE21 Q39NE2-HE22
Q5NE2-HE22
N33ND2-HD22
Figure 5.13: Spectre 2D 1H,15N HSQC du peptide Nter d’AtTSPO en tampon phos-phate (10 mM pH 6.2). Les pics non attribués correspondent aux résonances 15N-Hε deschaines latérales des arginines. Les données RMN ont été enregistrées à 20 ◦C, avec un peptideà 180 µM , sur un spectromètre Bruker 500 MHz, équipé d’une cryosonde, transformées par lelogiciel TOPSPIN et analysées par le logiciel Sparky.

5.3. Caractérisation du peptide 233
d’indices supérieurs à 0.1 ppm en valeur absolue reflète une structure secondaire stable et lesigne en précise le type : positif pour un feuillet β et négatif pour une hélice α. Dans le casdes 13Cα, il s’agit du contraire, une valeur ∆δCα positive indique une structure en hélice α,et une valeur négative indique une structure en feuillet β. Une succession de valeurs de ∼ ± 1ppm peut être interprétée comme une structure secondaire complètement formée, et des valeursplus faibles indiquent une structure partiellement formée, c.-à-d. une propension à adopter unestructure secondaire.
Les protéines désordonnées ayant souvent des δ très proches des valeurs "random coil",Marsh et al. [2006] ont amélioré la méthode de Wishart and Sykes dans le but de détecter lesstructures secondaires "transitoires" de ces protéines. En effet, ces protéines, bien que dans unétat désordonné, possèdent des quantités significatives de structures non aléatoires. Les auteurscombinent les CSD de différents noyaux - 13Cα, 13Cβ, 1Hα, 1HN et 15N (en pratique, tous nesont pas nécessaires seuls quelques uns sont utilisés pour le calcul) - dans une seule valeur pourobtenir une proportion de structure secondaire définit par une "score SSP (Secondary StructurePropensity)" pour chaque résidu. La contribution des différents δ est pondérée par sa sensibilitéaux structures secondaires α et β. Ce score représente donc la fraction de structure α ou β.Pour un résidu donnée, un score SSP de 1 ou -1 reflète une structure entièrement en hélice αou en feuillet β, respectivement, Fig. 5.14a. Tandis qu’un score de 0,5 indique qu’il y a 50% deconformères dans l’ensemble désordonné qui sont en hélice α à cette position. Une succession descore SSP de +1 ou -1 indique un élément de structure secondaire stable entièrement en héliceα ou en feuillet β, respectivement [Marsh et al., 2006].
Les auteurs ont incorporé cette nouvelle méthode dans un programme SSP que nous avonsutilisé pour le peptide Nter, de même que la méthode CSD. Les résultats sont présentés Fig.5.14bet Fig.5.14c. Ces figures nous montrent que 2 segments du Nter semblent avoir une petitepropension à faire de l’hélice α : une région centrée sur l’alanine 21 (résidus 16-27) et une autrerégion centrée sur le résidu aspartate 38 (résidus 31-45). Moins de 25% des conforméres del’ensemble des résidus présents dans ces 2 régions sont en structure α (Fig.5.14b). Globalementle peptide entier apparait donc très flexible et peu ordonnée, en accord avec les données dedichroïsme circulaire qui indique la présence de ∼ 20% d’Hα
5.3.3 Interaction du peptide avec le calcium
Le peptide Nter possédant beaucoup de charges dans sa séquence, nous avons voulu voir sile calcium pouvait induire la formation de structures secondaires stables.
En effet, certaines protéines après liaison à l’ion calcium subissent des changements confor-mationnels avec la formation de structures secondaires et tertiaires stables : c’est le cas de laprotéine T1SS (Type I Secretion System) qui est désordonnée et qui se replie en structure pa-rallèle β-roll après liaison du Ca2+ sur un motif riche en glycine et aspartate. D’autres protéinescontiennent un motif hélice-boucle-hélice dans lequel le calcium vient se lier aux chaines latéralesdes a.a chargés négativement (aspartate et glutamate) et à une glycine, créant d’importantes
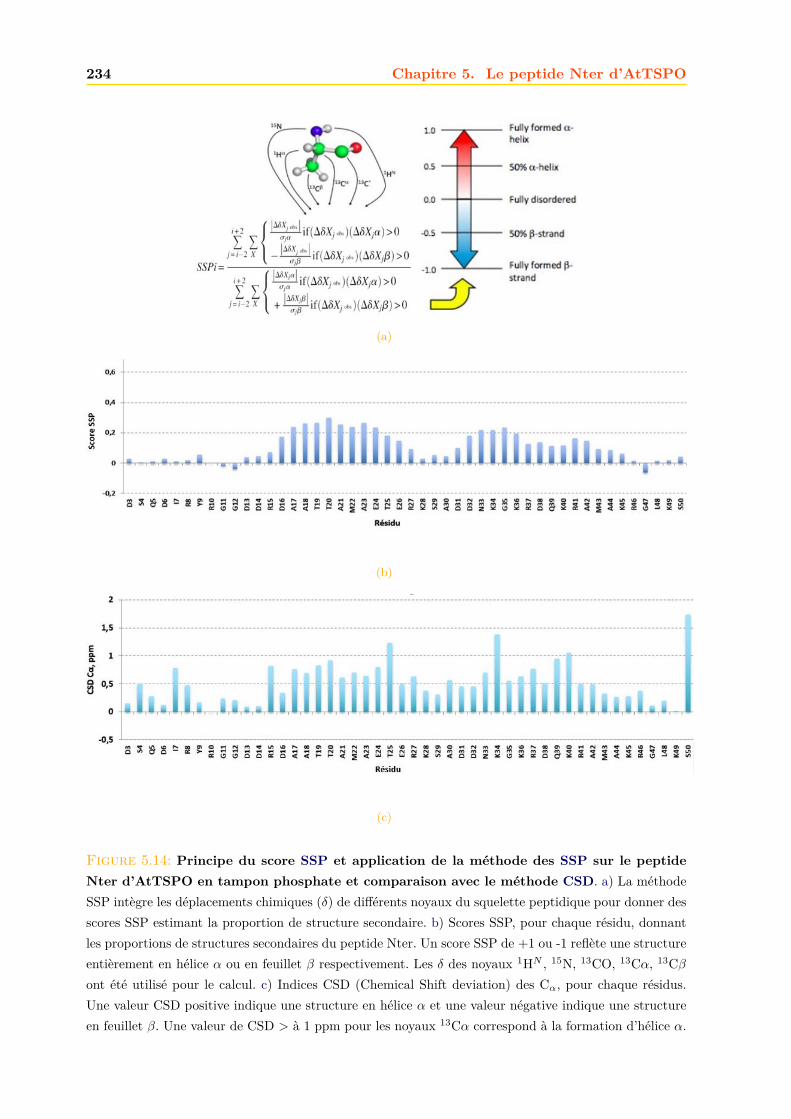
234 Chapitre 5. Le peptide Nter d’AtTSPO
(a)
(b)
(c)
Figure 5.14: Principe du score SSP et application de la méthode des SSP sur le peptideNter d’AtTSPO en tampon phosphate et comparaison avec le méthode CSD. a) La méthodeSSP intègre les déplacements chimiques (δ) de différents noyaux du squelette peptidique pour donner desscores SSP estimant la proportion de structure secondaire. b) Scores SSP, pour chaque résidu, donnantles proportions de structures secondaires du peptide Nter. Un score SSP de +1 ou -1 reflète une structureentièrement en hélice α ou en feuillet β respectivement. Les δ des noyaux 1HN , 15N, 13CO, 13Cα, 13Cβont été utilisé pour le calcul. c) Indices CSD (Chemical Shift deviation) des Cα, pour chaque résidus.Une valeur CSD positive indique une structure en hélice α et une valeur négative indique une structureen feuillet β. Une valeur de CSD > à 1 ppm pour les noyaux 13Cα correspond à la formation d’hélice α.

5.4. Interaction du peptide avec les lipides 235
modifications structurales (p. ex. chez la protéine CBP3 de Dictyostelium discoideum dont lerôle est inconnu [Chenal et al., 2008, O’Brien et al., 2015, Mishig-Ochiriin et al., 2005]).
Nous avons réalisé une titration du peptide Nter (75 µM) par le calcium (de 1 à 3 mM) etchaque étape du titrage a été suivie par l’acquisition d’un spectre 2D 1H,15N HSQC. Aucun effetn’a été observé sur le spectre du peptide (spectres non montrés) suggérant aucune interactiondu Nter d’AtTSPO avec le calcium.
5.4 Interaction du peptide avec les lipides
Pour déterminer si le peptide Nter interagit avec les lipides et gagne en structuration (héliceα), nous avons préparé des bicelles de phospholipides comme décrit Sec.2.3.9.2 du M&M.
Ce modèle membranaire a souvent été utilisé pour étudier les interactions peptides (ouprotéines)/lipides [Whiles et al., 2002, Biverstahl et al., 2004, Nowicka et al., 2016, Anderssonet al., 2004, Perez et al., 2013]. Dans notre cas nous avons testé 2 types de bicelles : des bicellesDMPG/DHPC chargées négativement et des bicelles neutres (zwitterioniques) DMPC/DHPC,Fig. 5.15.
Figure 5.15:Représentation schématique de la structure et de la charge des bicelles utiliséeslors de cette étude. [Perez et al., 2013].
5.4.1 Bicelles de DMPG/DHPC
Pour commencer, le peptide ayant une charge positive, nous avons testé le lipide anioniqueDMPG avec des bicelles DMPG/DHPC . Le spectre 2D 1H-15N HSQC a été enregistré enprésence 75 µM de peptide (concentration déduite à partir du DSS) et de (25 mM/12.5 mM)de DMPG/DHPC.
La superposition des spectres 2D 1H,15N HSQC obtenus avec et sans bicelles est présentéeFig. 5.16. On observe le décalage de nombreux pics en présence de bicelles par rapport au spectreen absence, ce qui suggère une interaction du peptide avec les bicelles de DMPG/DHPC.
Pour s’assurer qu’il s’agit bien d’une interaction avec la DMPG, nous avons effectué uncontrôle avec des bicelles neutres (zwitterioniques) DPMC/DHPC. Les spectre 2D 1H-15N HSQC
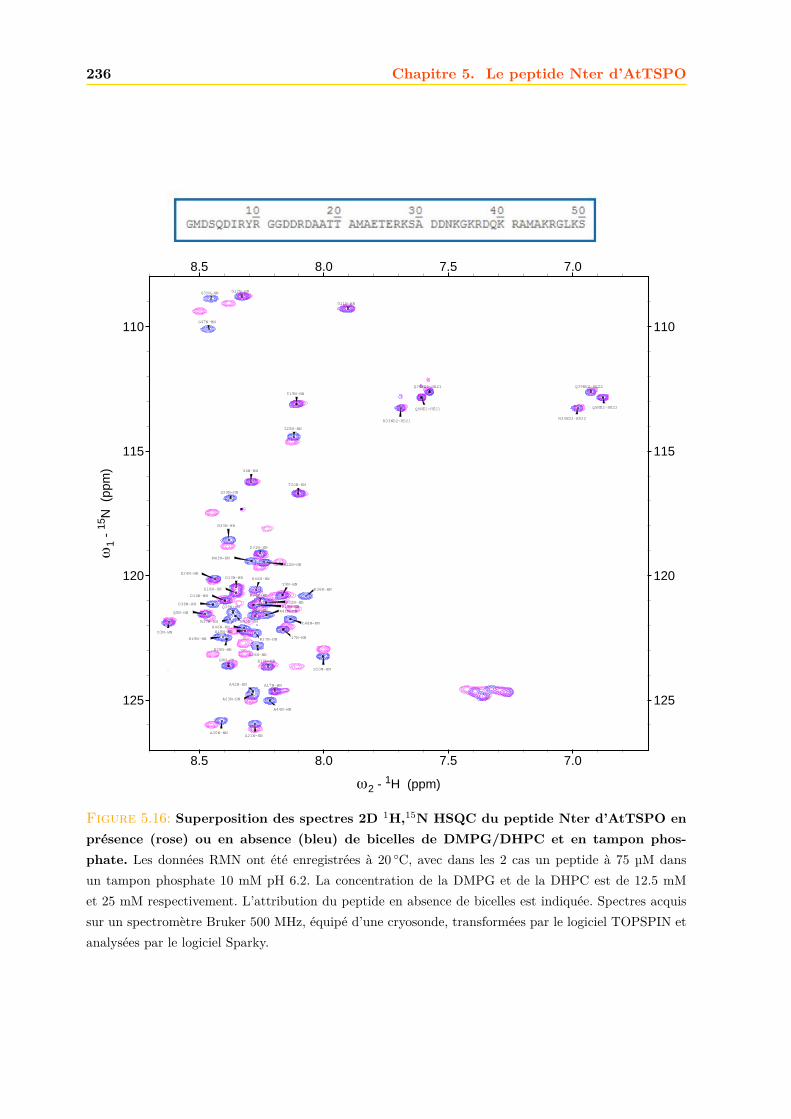
236 Chapitre 5. Le peptide Nter d’AtTSPO
8.5
8.5
8.0
8.0
7.5
7.5
7.0
7.0
ω2 - 1H (ppm)
125 125
120 120
115 115
110 110
ω1
- 15N
(pp
m)
A21N-HNA30N-HN
A44N-HN
A23N-HN
A17N-HN
R10N-HNR8N-HN
S50N-HN
E26N-HN
K49N-HN R27N-HN
A18N-HNI7N-HN
D3N-HN
R37N-HN L48N-HNK40N-HN
D6N-HN R41N-HNQ5N-HNQ39N-HN R15N-HN
D38N-HN
K34N-HN
D32N-HN
D14N-HN
K36N-HNY9N-HN
D16N-HN
K45N-HND13N-HNE24N-HN
M22N-HN
M43N-HN
D31N-HN
A42N-HN
R46N-HN
K28N-HN
G11N-HN
N33N-HN
S29N-HN
S4N-HN
T20N-HN
T25N-HN
T19N-HN
G12N-HNG35N-HN
G47N-HN
N33ND2-HD21
Q5NE2-HE21
Q39NE2-HE21 Q39NE2-HE22
Q5NE2-HE22
N33ND2-HD22
Figure 5.16: Superposition des spectres 2D 1H,15N HSQC du peptide Nter d’AtTSPO enprésence (rose) ou en absence (bleu) de bicelles de DMPG/DHPC et en tampon phos-phate. Les données RMN ont été enregistrées à 20 ◦C, avec dans les 2 cas un peptide à 75 µM dansun tampon phosphate 10 mM pH 6.2. La concentration de la DMPG et de la DHPC est de 12.5 mMet 25 mM respectivement. L’attribution du peptide en absence de bicelles est indiquée. Spectres acquissur un spectromètre Bruker 500 MHz, équipé d’une cryosonde, transformées par le logiciel TOPSPIN etanalysées par le logiciel Sparky.
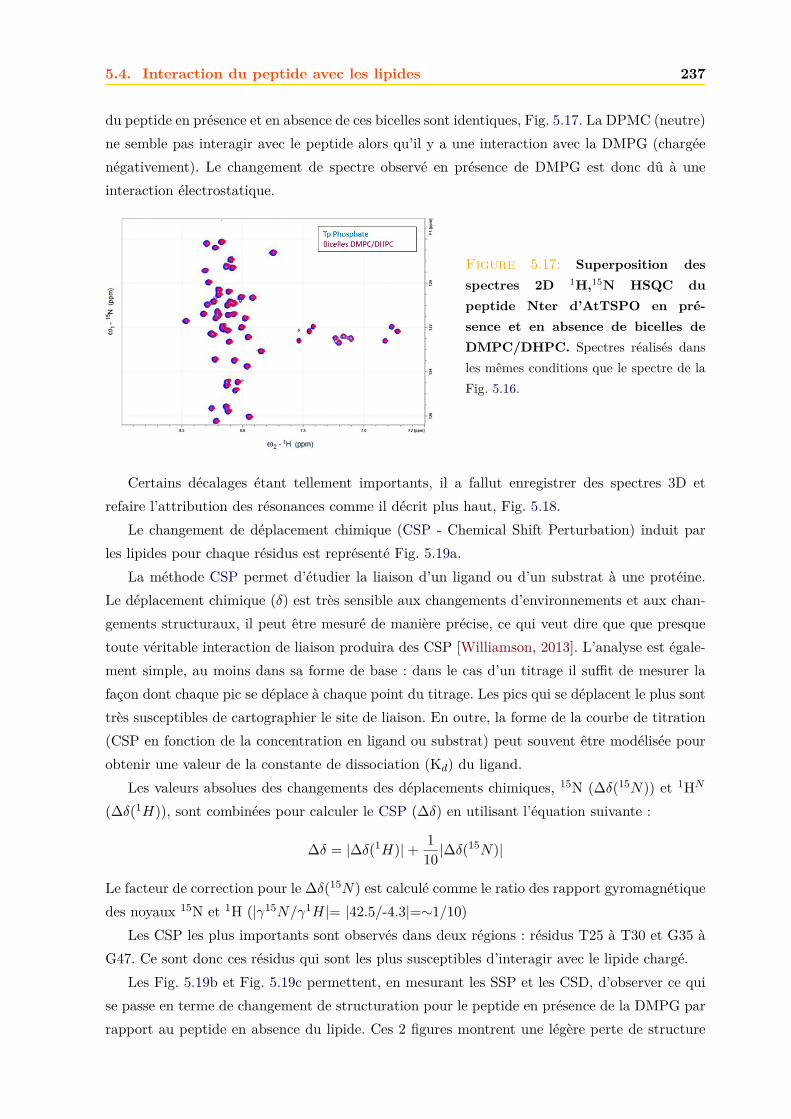
5.4. Interaction du peptide avec les lipides 237
du peptide en présence et en absence de ces bicelles sont identiques, Fig. 5.17. La DPMC (neutre)ne semble pas interagir avec le peptide alors qu’il y a une interaction avec la DMPG (chargéenégativement). Le changement de spectre observé en présence de DMPG est donc dû à uneinteraction électrostatique.
Figure 5.17: Superposition desspectres 2D 1H,15N HSQC dupeptide Nter d’AtTSPO en pré-sence et en absence de bicelles deDMPC/DHPC. Spectres réalisés dansles mêmes conditions que le spectre de laFig. 5.16.
Certains décalages étant tellement importants, il a fallut enregistrer des spectres 3D etrefaire l’attribution des résonances comme il décrit plus haut, Fig. 5.18.
Le changement de déplacement chimique (CSP - Chemical Shift Perturbation) induit parles lipides pour chaque résidus est représenté Fig. 5.19a.
La méthode CSP permet d’étudier la liaison d’un ligand ou d’un substrat à une protéine.Le déplacement chimique (δ) est très sensible aux changements d’environnements et aux chan-gements structuraux, il peut être mesuré de manière précise, ce qui veut dire que que presquetoute véritable interaction de liaison produira des CSP [Williamson, 2013]. L’analyse est égale-ment simple, au moins dans sa forme de base : dans le cas d’un titrage il suffit de mesurer lafaçon dont chaque pic se déplace à chaque point du titrage. Les pics qui se déplacent le plus sonttrès susceptibles de cartographier le site de liaison. En outre, la forme de la courbe de titration(CSP en fonction de la concentration en ligand ou substrat) peut souvent être modélisée pourobtenir une valeur de la constante de dissociation (Kd) du ligand.
Les valeurs absolues des changements des déplacements chimiques, 15N (∆δ(15N)) et 1HN
(∆δ(1H)), sont combinées pour calculer le CSP (∆δ) en utilisant l’équation suivante :
∆δ = |∆δ(1H)|+ 110 |∆δ(
15N)|
Le facteur de correction pour le ∆δ(15N) est calculé comme le ratio des rapport gyromagnétiquedes noyaux 15N et 1H (|γ15N/γ1H|= |42.5/-4.3|=∼1/10)
Les CSP les plus importants sont observés dans deux régions : résidus T25 à T30 et G35 àG47. Ce sont donc ces résidus qui sont les plus susceptibles d’interagir avec le lipide chargé.
Les Fig. 5.19b et Fig. 5.19c permettent, en mesurant les SSP et les CSD, d’observer ce quise passe en terme de changement de structuration pour le peptide en présence de la DMPG parrapport au peptide en absence du lipide. Ces 2 figures montrent une légère perte de structure
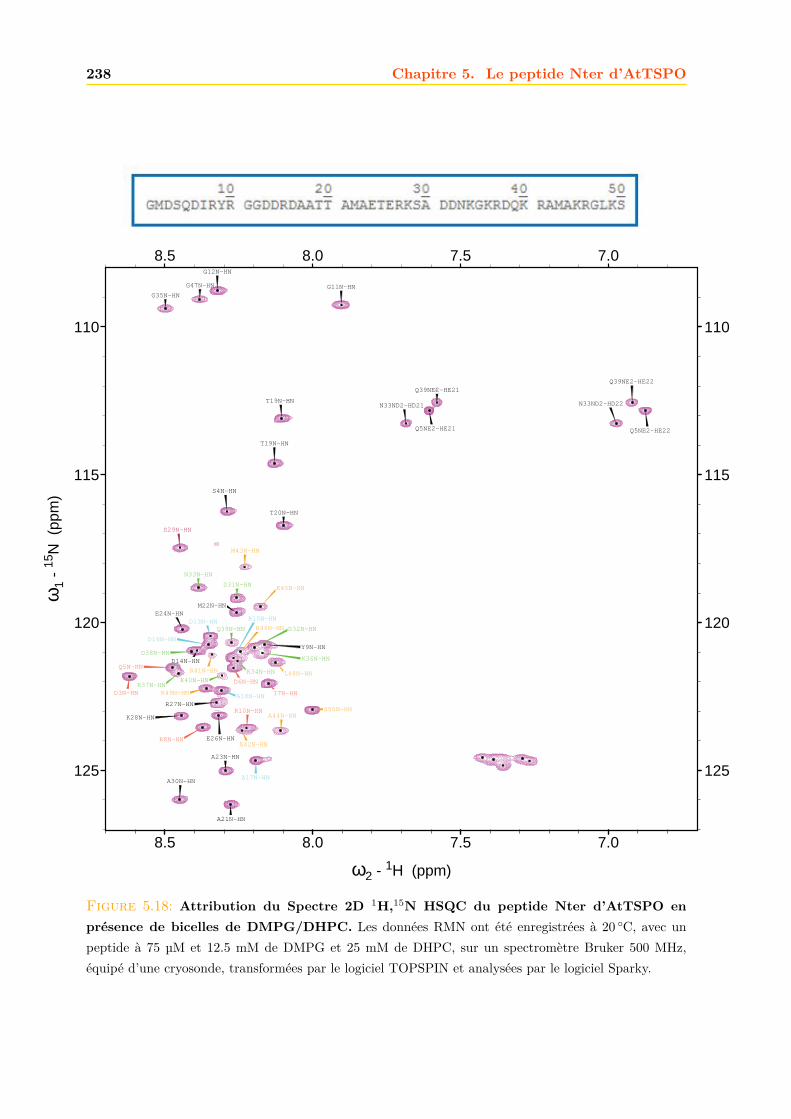
238 Chapitre 5. Le peptide Nter d’AtTSPO
8.5
8.5
8.0
8.0
7.5
7.5
7.0
7.0
ω2 - 1H (ppm)
125 125
120 120
115 115
110 110
ω1
- 15
N (
ppm
)
R10N-HN
D3N-HN
D6N-HN
Q5N-HN
S4N-HN
G35N-HN
G47N-HN
G12N-HN
G11N-HN
T19N-HN
T19N-HN
Q39NE2-HE21
Q5NE2-HE21
N33ND2-HD21 N33ND2-HD22
Q39NE2-HE22
Q5NE2-HE22
T20N-HN
S29N-HN
M43N-HN
N33N-HN
D31N-HN K45N-HN
M22N-HNE24N-HN
D13N-HN
D14N-HND38N-HN
D16N-HN
R41N-HN
R37N-HN
Q39N-HN
K40N-HN
R15N-HN
R46N-HN D32N-HN
Y9N-HN
L48N-HN
K36N-HN
K34N-HN
I7N-HN
S50N-HNA44N-HN
K49N-HN A18N-HNR27N-HN
K28N-HN
R8N-HN E26N-HN
A23N-HN
A17N-HNA30N-HN
A21N-HN
A42N-HN
Figure 5.18: Attribution du Spectre 2D 1H,15N HSQC du peptide Nter d’AtTSPO enprésence de bicelles de DMPG/DHPC. Les données RMN ont été enregistrées à 20 ◦C, avec unpeptide à 75 µM et 12.5 mM de DMPG et 25 mM de DHPC, sur un spectromètre Bruker 500 MHz,équipé d’une cryosonde, transformées par le logiciel TOPSPIN et analysées par le logiciel Sparky.
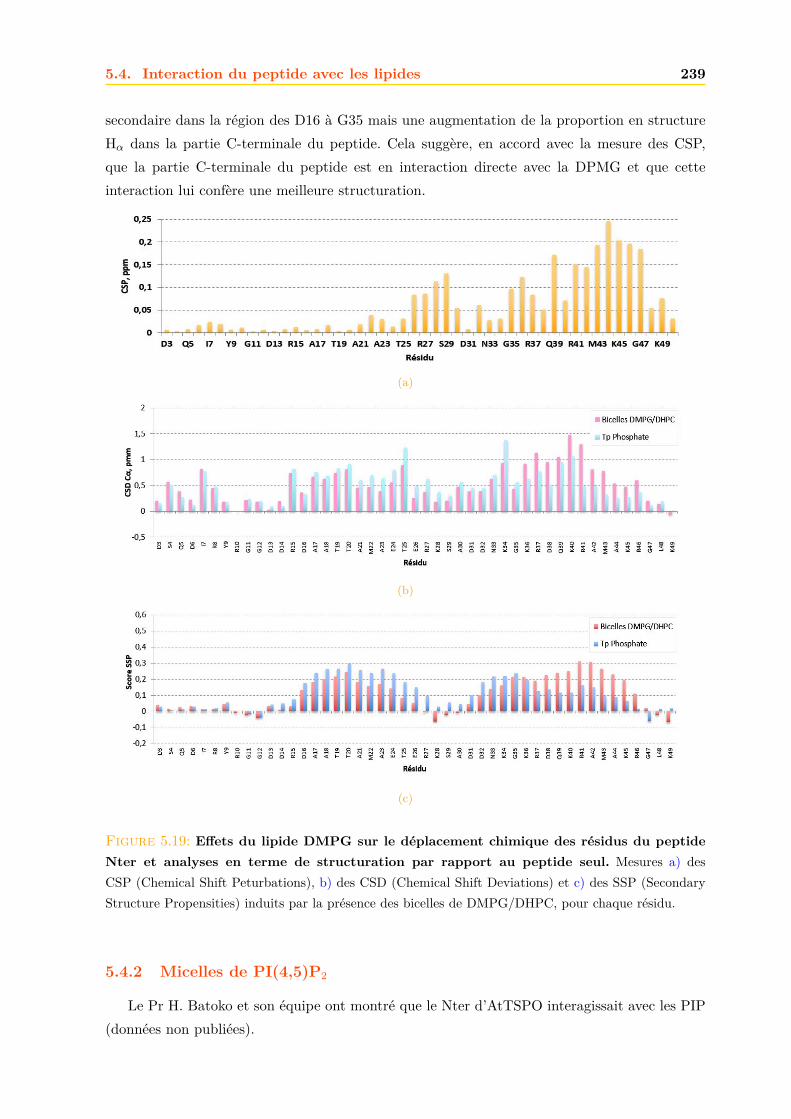
5.4. Interaction du peptide avec les lipides 239
secondaire dans la région des D16 à G35 mais une augmentation de la proportion en structureHα dans la partie C-terminale du peptide. Cela suggère, en accord avec la mesure des CSP,que la partie C-terminale du peptide est en interaction directe avec la DPMG et que cetteinteraction lui confère une meilleure structuration.
(a)
(b)
(c)
Figure 5.19: Effets du lipide DMPG sur le déplacement chimique des résidus du peptideNter et analyses en terme de structuration par rapport au peptide seul. Mesures a) desCSP (Chemical Shift Peturbations), b) des CSD (Chemical Shift Deviations) et c) des SSP (SecondaryStructure Propensities) induits par la présence des bicelles de DMPG/DHPC, pour chaque résidu.
5.4.2 Micelles de PI(4,5)P2
Le Pr H. Batoko et son équipe ont montré que le Nter d’AtTSPO interagissait avec les PIP(données non publiées).
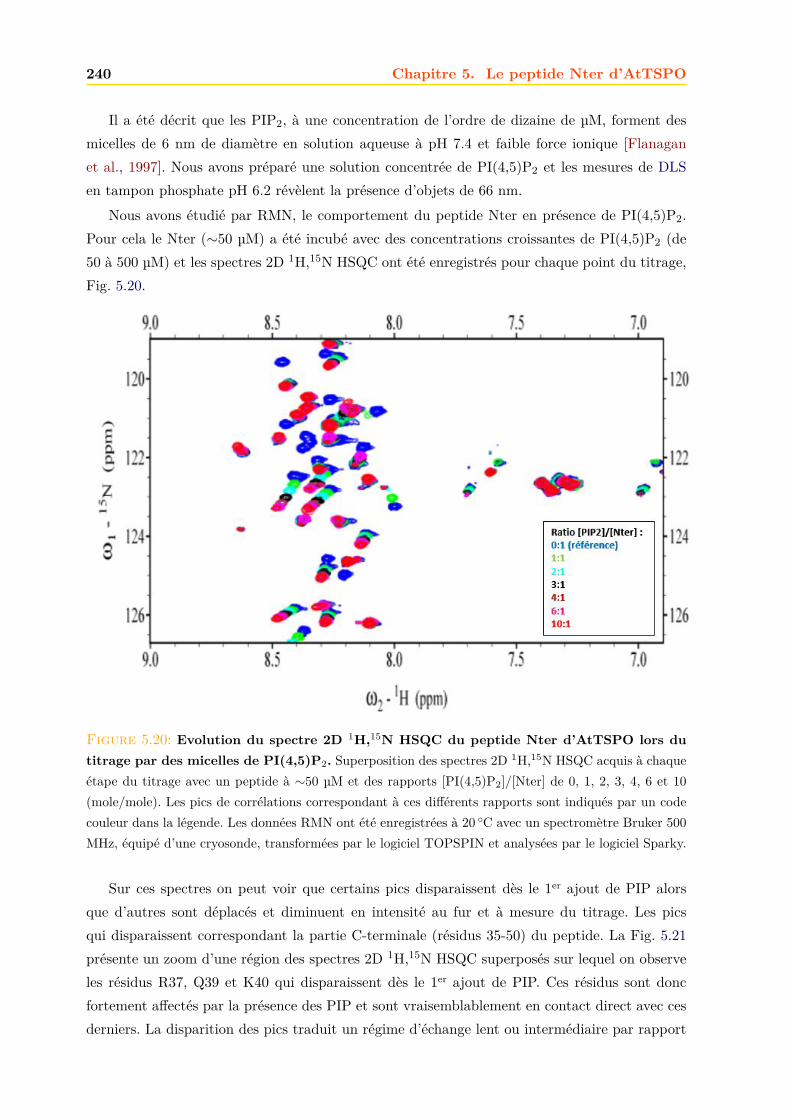
240 Chapitre 5. Le peptide Nter d’AtTSPO
Il a été décrit que les PIP2, à une concentration de l’ordre de dizaine de µM, forment desmicelles de 6 nm de diamètre en solution aqueuse à pH 7.4 et faible force ionique [Flanaganet al., 1997]. Nous avons préparé une solution concentrée de PI(4,5)P2 et les mesures de DLSen tampon phosphate pH 6.2 révèlent la présence d’objets de 66 nm.
Nous avons étudié par RMN, le comportement du peptide Nter en présence de PI(4,5)P2.Pour cela le Nter (∼50 µM) a été incubé avec des concentrations croissantes de PI(4,5)P2 (de50 à 500 µM) et les spectres 2D 1H,15N HSQC ont été enregistrés pour chaque point du titrage,Fig. 5.20.
Figure 5.20: Evolution du spectre 2D 1H,15N HSQC du peptide Nter d’AtTSPO lors dutitrage par des micelles de PI(4,5)P2. Superposition des spectres 2D 1H,15N HSQC acquis à chaqueétape du titrage avec un peptide à ∼50 µM et des rapports [PI(4,5)P2]/[Nter] de 0, 1, 2, 3, 4, 6 et 10(mole/mole). Les pics de corrélations correspondant à ces différents rapports sont indiqués par un codecouleur dans la légende. Les données RMN ont été enregistrées à 20 ◦C avec un spectromètre Bruker 500MHz, équipé d’une cryosonde, transformées par le logiciel TOPSPIN et analysées par le logiciel Sparky.
Sur ces spectres on peut voir que certains pics disparaissent dès le 1er ajout de PIP alorsque d’autres sont déplacés et diminuent en intensité au fur et à mesure du titrage. Les picsqui disparaissent correspondant la partie C-terminale (résidus 35-50) du peptide. La Fig. 5.21présente un zoom d’une région des spectres 2D 1H,15N HSQC superposés sur lequel on observeles résidus R37, Q39 et K40 qui disparaissent dès le 1er ajout de PIP. Ces résidus sont doncfortement affectés par la présence des PIP et sont vraisemblablement en contact direct avec cesderniers. La disparition des pics traduit un régime d’échange lent ou intermédiaire par rapport
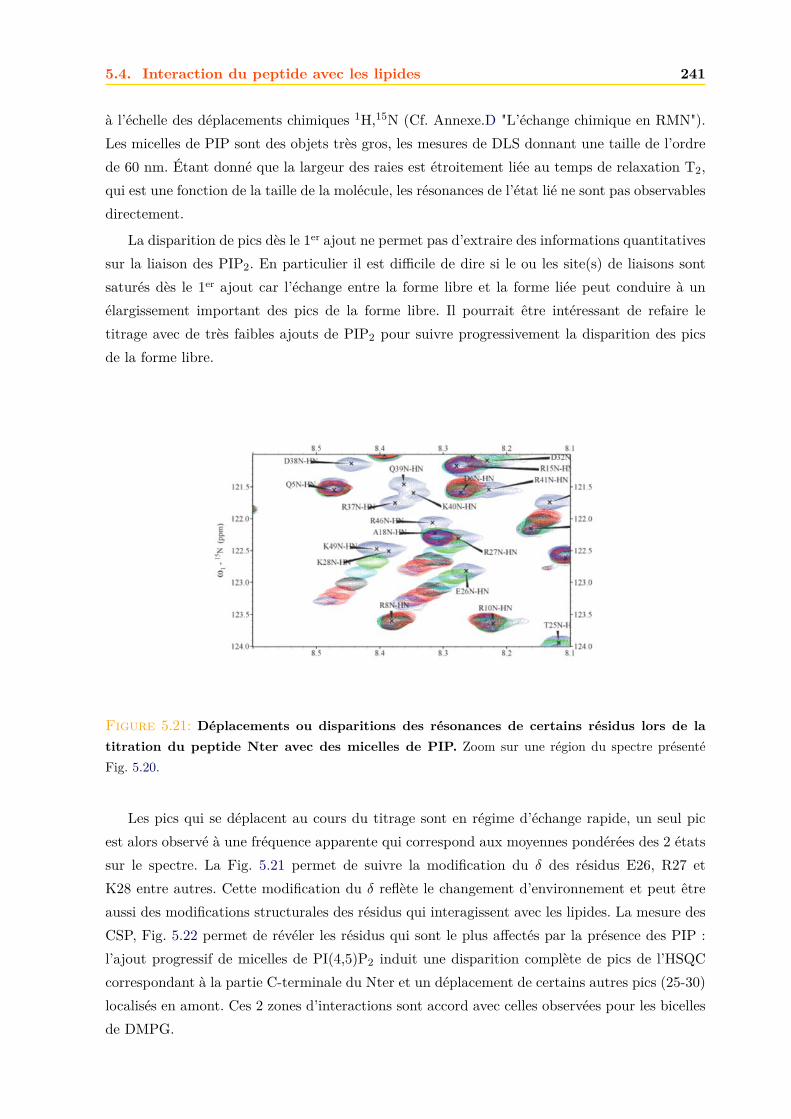
5.4. Interaction du peptide avec les lipides 241
à l’échelle des déplacements chimiques 1H,15N (Cf. Annexe.D "L’échange chimique en RMN").Les micelles de PIP sont des objets très gros, les mesures de DLS donnant une taille de l’ordrede 60 nm. Étant donné que la largeur des raies est étroitement liée au temps de relaxation T2,qui est une fonction de la taille de la molécule, les résonances de l’état lié ne sont pas observablesdirectement.
La disparition de pics dès le 1er ajout ne permet pas d’extraire des informations quantitativessur la liaison des PIP2. En particulier il est difficile de dire si le ou les site(s) de liaisons sontsaturés dès le 1er ajout car l’échange entre la forme libre et la forme liée peut conduire à unélargissement important des pics de la forme libre. Il pourrait être intéressant de refaire letitrage avec de très faibles ajouts de PIP2 pour suivre progressivement la disparition des picsde la forme libre.
Figure 5.21: Déplacements ou disparitions des résonances de certains résidus lors de latitration du peptide Nter avec des micelles de PIP. Zoom sur une région du spectre présentéFig. 5.20.
Les pics qui se déplacent au cours du titrage sont en régime d’échange rapide, un seul picest alors observé à une fréquence apparente qui correspond aux moyennes pondérées des 2 étatssur le spectre. La Fig. 5.21 permet de suivre la modification du δ des résidus E26, R27 etK28 entre autres. Cette modification du δ reflète le changement d’environnement et peut êtreaussi des modifications structurales des résidus qui interagissent avec les lipides. La mesure desCSP, Fig. 5.22 permet de révéler les résidus qui sont le plus affectés par la présence des PIP :l’ajout progressif de micelles de PI(4,5)P2 induit une disparition complète de pics de l’HSQCcorrespondant à la partie C-terminale du Nter et un déplacement de certains autres pics (25-30)localisés en amont. Ces 2 zones d’interactions sont accord avec celles observées pour les bicellesde DMPG.
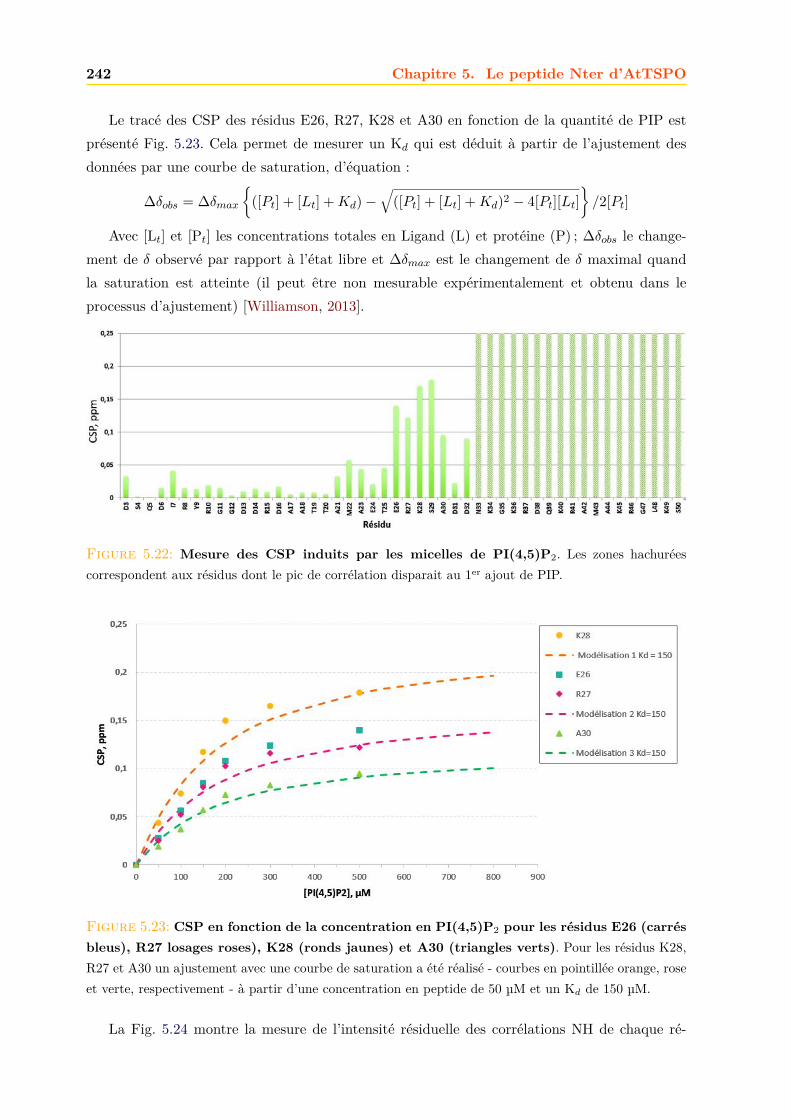
242 Chapitre 5. Le peptide Nter d’AtTSPO
Le tracé des CSP des résidus E26, R27, K28 et A30 en fonction de la quantité de PIP estprésenté Fig. 5.23. Cela permet de mesurer un Kd qui est déduit à partir de l’ajustement desdonnées par une courbe de saturation, d’équation :
∆δobs = ∆δmax{
([Pt] + [Lt] +Kd)−√
([Pt] + [Lt] +Kd)2 − 4[Pt][Lt]}/2[Pt]
Avec [Lt] et [Pt] les concentrations totales en Ligand (L) et protéine (P) ; ∆δobs le change-ment de δ observé par rapport à l’état libre et ∆δmax est le changement de δ maximal quandla saturation est atteinte (il peut être non mesurable expérimentalement et obtenu dans leprocessus d’ajustement) [Williamson, 2013].
Figure 5.22: Mesure des CSP induits par les micelles de PI(4,5)P2. Les zones hachuréescorrespondent aux résidus dont le pic de corrélation disparait au 1er ajout de PIP.
Figure 5.23: CSP en fonction de la concentration en PI(4,5)P2 pour les résidus E26 (carrésbleus), R27 losages roses), K28 (ronds jaunes) et A30 (triangles verts). Pour les résidus K28,R27 et A30 un ajustement avec une courbe de saturation a été réalisé - courbes en pointillée orange, roseet verte, respectivement - à partir d’une concentration en peptide de 50 µM et un Kd de 150 µM.
La Fig. 5.24 montre la mesure de l’intensité résiduelle des corrélations NH de chaque ré-
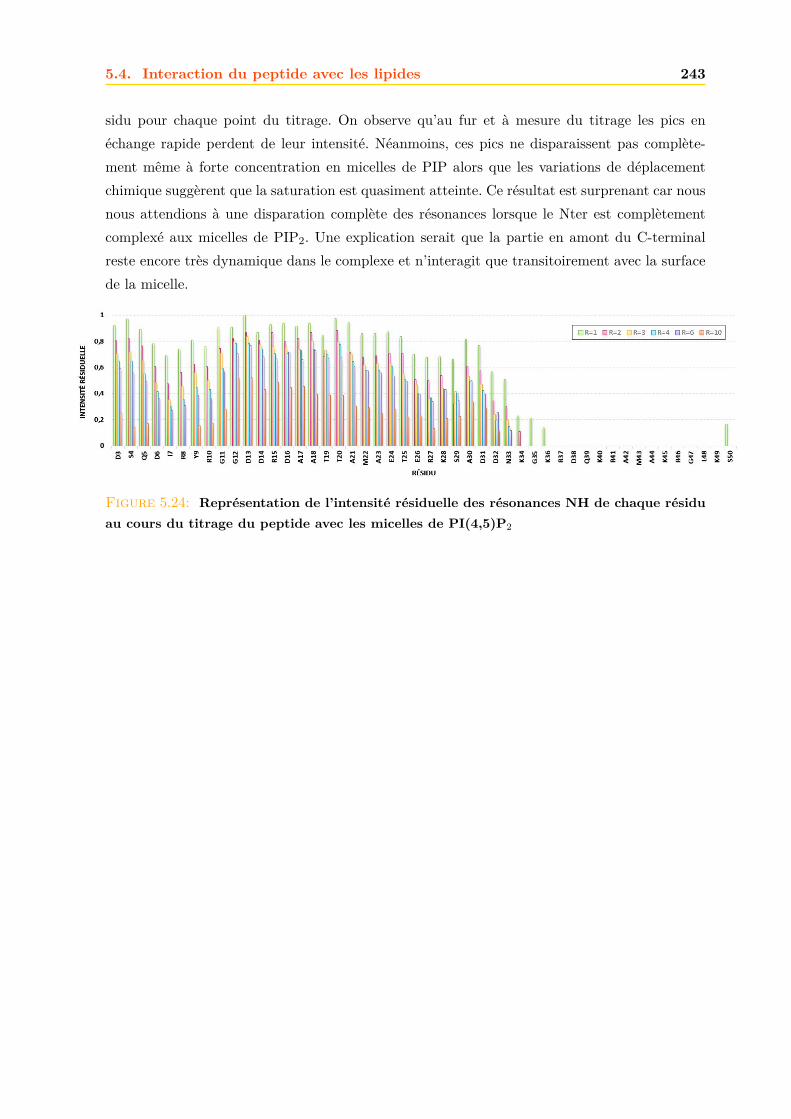
5.4. Interaction du peptide avec les lipides 243
sidu pour chaque point du titrage. On observe qu’au fur et à mesure du titrage les pics enéchange rapide perdent de leur intensité. Néanmoins, ces pics ne disparaissent pas complète-ment même à forte concentration en micelles de PIP alors que les variations de déplacementchimique suggèrent que la saturation est quasiment atteinte. Ce résultat est surprenant car nousnous attendions à une disparation complète des résonances lorsque le Nter est complètementcomplexé aux micelles de PIP2. Une explication serait que la partie en amont du C-terminalreste encore très dynamique dans le complexe et n’interagit que transitoirement avec la surfacede la micelle.
Figure 5.24: Représentation de l’intensité résiduelle des résonances NH de chaque résiduau cours du titrage du peptide avec les micelles de PI(4,5)P2

244 Chapitre 5. Le peptide Nter d’AtTSPO
5.5 Conclusions, perspectives et discussions
En conclusion, nous avons réussi à surexprimer le peptide recombinant N-terminald’AtTSPO et obtenu des quantités suffisantes, pures de peptide marquée pour entreprendredes études RMN malgré un problème de précipitation au cours de la purification.
La qualité des spectres RMN nous a permis de faire l’attribution des résonances de chaquepic et l’analyse des déplacements chimiques nous a montré que le peptide était très flexible ettrès peu structuré. Seules 2 régions montrent une propension à former des structures secondairesen hélice α.
Les expériences 2D ou 3D NOESY (Nuclear Overhauser SpectroscopY) reposent sur lamesure des NOE (Effet Nucléaire Overhauser) qui fournissent des informations sur les distancesentre atomes voisins. Ces distances, à moins de 5 Å, peuvent être intra-résidu ou inter-résidus(séquentielle, moyenne portée, longue portée). L’analyse des déplacements chimiques montrantque le peptide est très flexible et très peu structuré, les NOE ne seront que peu nombreux apriori. Cependant, même en faible nombre, il serait intéressant de les mesurer car ils pourraitnous renseigner d’avantage sur d’éventuelle structuration locale telle que des hélices α ou desstructures de type coude.
Dans le but d’explorer l’hypothèse que l’AtTSPO, grâce à son extension N-terminale, pouvaitavoir un rôle dans le transfert des lipides et notamment des PIP, entre les membranes, auniveau des sites de contacts, nous avons étudié le comportement du peptide Nter en présencede différents lipides : DMPC, DMPG et PI(4,5)P2.
Le spectre HSQC du peptide n’est pas affecté en présence de bicelles neutres deDMPC/DHPC contrairement au spectre en présence de bicelles anioniques DMPG/DHPC àdes ratios similaires. Cela montre que le peptide interagit avec les lipides et qu’il s’agit d’uneinteraction électrostatique : les charges négatives des phosphates et les charges positives desrésidus (K et R). De plus, ces interactions permettent une meilleure structuration de la partieC-terminale du peptide.
Le spectre HSQC du peptide en présence de micelles de PI(4,5)P2 montre 2 régimesd’échange : des pics en échange lent et/ou intermédiaire, correspondant aux résidus de la partieCter et des pics en échange rapide correspondant aux résidus en amont du Cter. Les résidusde la partie Cter se trouvent probablement en contact direct avec les PIP alors que les résidusen amont restent très flexibles et interagissent moins fortement avec les PIP, nous pourrionsreprésenter cela comme présenté Fig. 5.25 . Cependant, bien qu’ayant pu calculer une constantede dissociation, il est difficile de définir combien il y a de sites de liaisons par peptide. Desexpériences supplémentaires de mesures des vitesses de relaxation sont à envisager pour obtenirplus d’informations à ce sujet.
Pour nous rapprocher aux plus des conditions retrouvées dans les membranes, à savoirquelques % de PIP nous envisageons de faire des bicelles DMPC/DHPC enrichies en PI(4,5)P2.Une 1re tentative a été réalisée avec l’ajout de 1 et 2 % de PI(4,5)P2 mais cet ajout n’a montréaucun effet. Un % plus élevé serait donc à tester. Nous avons également dans l’idée de tester

5.5. Conclusions, perspectives et discussions 245
Figure 5.25: Schéma représentant les interactions du peptide Nter d’AtTSPO à lasurface des micelles de PI(4,5)P2. Le peptide Nter, qui possède une charge glo-bale positive, apparait intrinsèquement désordonné mais possède des éléments destructures secondaires "transitoires " en hélices α (non représentés). Les micelles dePI(4,5)P2 étant chargées négativement, l’interaction entre le peptide se ferait viades interactions électrostatiques. Cependant différent régimes d’échanges existent. Le peptide"lié" existe sous forme d’un large ensemble d’état où chaque résidu peut être ou ne pas être encontact direct avec les PIP. À titre d’exemple, le cercle rose représente un résidu de la partieC-terminale du peptide en échange lent qui est soit en contact direct/lié avec les PIP soit libre,et pour lequel 3 autres configurations sont illustrés avec le cercle bleu, représentant un résiduen amont de cette partie C-terminale, qui lui est en échange rapide et très flexible.
d’autres PIP spécifiques de certains organelles comme le PI3P (localisé majoritairement dansles endosomes et la membrane des vacuoles) ou encore la PI4P (localisé dans la membraneplasmique, l’AG et le réseau trans-golgien). Cela permettrait de voir si l’interaction du Nteravec ces PIP est différente de celle observé avec le PI(4,5)P2. En particulier, voir si certainsinduisent une meilleure structuration du peptide.
Dans la littérature, il a été décrit différentes interactions des PIP avec des protéines : lesprotéines qui reconnaissent les PIP et celles qui les transfèrent. Si l’AtTSPO transfert effecti-vement les PIP, il peut être intéressant d’étudier ses protéines pour voir si des points communsde dégagent.
Les protéines reconnaissants les PIP Typiquement, la liaison des protéines aux PIPimplique des interactions électrostatiques avec les charges négatives du ou des phosphate(s) dunoyau inositol. Dans certains cas (protéines solubles), des a.a hydrophobes adjacents renforcent
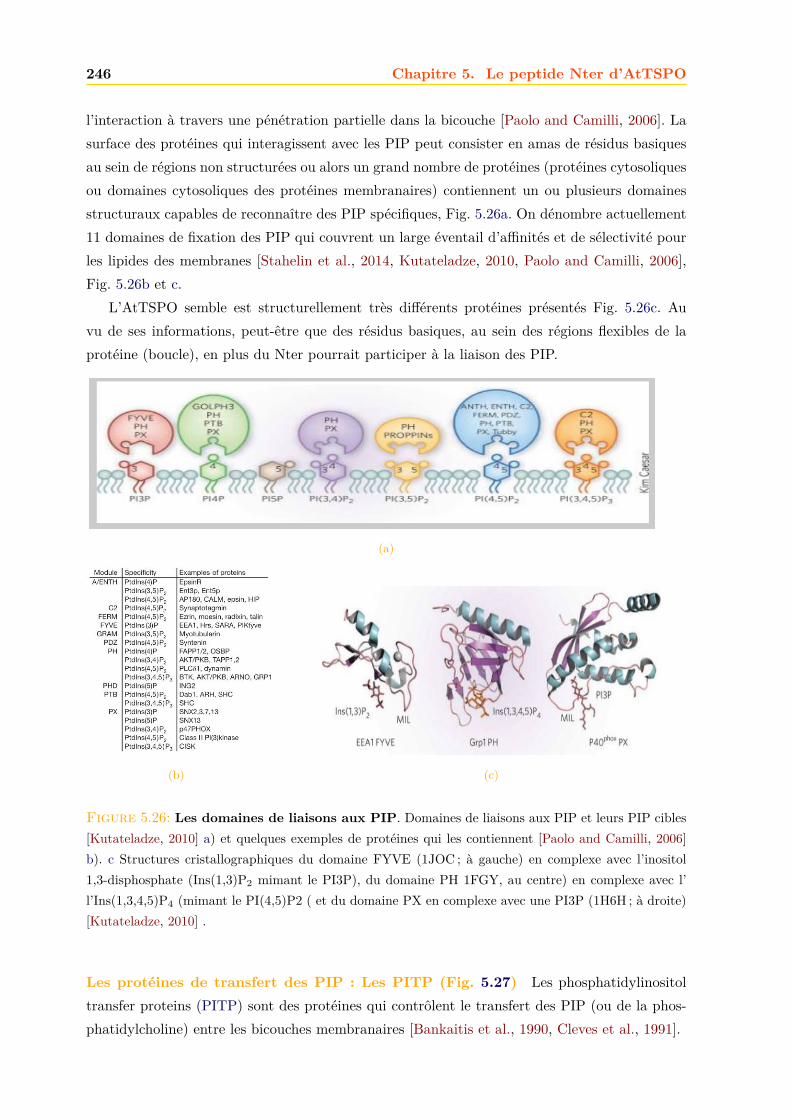
246 Chapitre 5. Le peptide Nter d’AtTSPO
l’interaction à travers une pénétration partielle dans la bicouche [Paolo and Camilli, 2006]. Lasurface des protéines qui interagissent avec les PIP peut consister en amas de résidus basiquesau sein de régions non structurées ou alors un grand nombre de protéines (protéines cytosoliquesou domaines cytosoliques des protéines membranaires) contiennent un ou plusieurs domainesstructuraux capables de reconnaître des PIP spécifiques, Fig. 5.26a. On dénombre actuellement11 domaines de fixation des PIP qui couvrent un large éventail d’affinités et de sélectivité pourles lipides des membranes [Stahelin et al., 2014, Kutateladze, 2010, Paolo and Camilli, 2006],Fig. 5.26b et c.
L’AtTSPO semble est structurellement très différents protéines présentés Fig. 5.26c. Auvu de ses informations, peut-être que des résidus basiques, au sein des régions flexibles de laprotéine (boucle), en plus du Nter pourrait participer à la liaison des PIP.
(a)
(b) (c)
Figure 5.26: Les domaines de liaisons aux PIP. Domaines de liaisons aux PIP et leurs PIP cibles[Kutateladze, 2010] a) et quelques exemples de protéines qui les contiennent [Paolo and Camilli, 2006]b). c Structures cristallographiques du domaine FYVE (1JOC ; à gauche) en complexe avec l’inositol1,3-disphosphate (Ins(1,3)P2 mimant le PI3P), du domaine PH 1FGY, au centre) en complexe avec l’l’Ins(1,3,4,5)P4 (mimant le PI(4,5)P2 ( et du domaine PX en complexe avec une PI3P (1H6H ; à droite)[Kutateladze, 2010] .
Les protéines de transfert des PIP : Les PITP (Fig. 5.27) Les phosphatidylinositoltransfer proteins (PITP) sont des protéines qui contrôlent le transfert des PIP (ou de la phos-phatidylcholine) entre les bicouches membranaires [Bankaitis et al., 1990, Cleves et al., 1991].

5.5. Conclusions, perspectives et discussions 247
Les PITP ont un PM de ∼ 35 kDa et possèdent un haut niveau de conservation chezles eucaryotes [Routt and Bankaitis, 2004, Phillips et al., 2006]. Ils peuvent être divisés en 2branches basées sur leur homologie de séquence :
1. les PITP des levures et des plantes qui sont hautement similaires au niveau de leur sé-quence [Bankaitis et al., 1989, 1990] ;
2. et les PITP des animaux (mammifères, amphibiens, insectes) qui sont structurellementhomologues [Bankaitis et al., 1989, Dickeson et al., 1989].
Cependant, il n’y a absolument pas de similarité de séquence entre les 2 groupes. En fait, lesPITP des levures et des plantes partagent une homologie avec la protéine mammalienne deliaison du rétinaldéhyde [Salama et al., 1990] et les protéines de liaisons des phosphoinositidesdes plantes supérieures [Kapranov et al., 2001, Kearns et al., 1998]. Néanmoins, malgré l’absencede similarité entre les séquences des PITP de mammifères et de levures/plantes, elles présententdes activités identiques in vitro [Routt and Bankaitis, 2004, Phillips et al., 2006].
Chez la levure, la PITP majeure est Sec14p, qui est impliquée dans le trafic membranairedans le compartiment du réseau trans-golgien [Cleves et al., 1991]. Alors que la levure possède 6protéines homologues à Sec14, les eucarytotes supérieurs en ont beaucoup plus, p. ex. l’Hommeet la souris en ont plus de 23 [Bankaitis et al., 2007]. Chez Arabidopsis il y a 31 homologues deSec14p qui sont appelés SFH pour "Sec Fourteen Homologs" qui sont divisés en 2 sous-famillesmajeures :
1. La sous-famille "Sec14-nodulin" (qui contient un domaine noduline 2 en C-terminal). Cedomaine contient des motifs similaires aux motifs polybasiques qui lient les PIP, menantà l’hypothèse que les protéines "Sec14-nodulin" peuvent stimuler la synthèse des PIPainsi que les organiser et les séquestrer, ce qui serait idéal pour des protéines régulant ladistribution spatiale et temporelle des PIP [Thole and Nielsen, 2008].
2. La sous famille des protéines "Sec14-GOLD" qui contiennent un domaine GOLD en C-terminal (GOLgi Dynamic domain). Le domaine GOLD a d’abord été identifié dans lesprotéines cargo impliquées dans le transport vésiculaire [Thole and Nielsen, 2008].
Cette classe de protéine, Fig. 5.28, semble fonctionner sous forme d’une cavité qui lie le PIPpour le transporter d’une membrane à l’autre. Ce qui parait loin de ce que l’on peut imaginerpour l’AtTSPO insérée dans une membrane.
Les protéines de transfert des PIP aux sites de contact Les sites de contacts membra-naires (SCM) sont de petits intervalles cytosoliques (10-30 nm) entre le RE et les membranesdes autres organelles cellulaires tels que la mitochondrie, l’AG, la membrane plasmique, lesendosomes, les gouttelettes lipidiques [Selitrennik and Lev, 2016], Fig. 5.29a. Ces sites très dy-namiques qui permettent le transport des ions calcium et différents métabolites, ont un rôle
2. La noduline est une protéine qui est synthétisée dans les nodules de certaines plantes en réponse à uneinfection par un microorganisme fixateur d’azote, et qui est impliquée dans le développement et le fonctionnementde ces nodules.

248 Chapitre 5. Le peptide Nter d’AtTSPO
Figure 5.27: Les PITP se lient aux Phos-phatidylinositols (PI) et phosphatidylcho-lines (PC) et les transfèrent entre les com-partiments membranaires (p. ex. le RE ou lamembrane plasmique). Les PITP sont toujoursoccupées par une seule molécule soit de PI soit dePC. L’affinité des PITP pour le PI est 16 fois plusgrande que pour la PC et dépend de la distributionrelative du PI et de la PC. [Hsuan and Cockcroft,2001]
(a) (b)
Figure 5.28: Structures des PITP a) Structure d’une PITP de plante (SFH1), ressemblant à la protéineSec14p de levure, liant une PIP (bleu) et une PC (rouge) à 2 sites distincts. Les PITP de levure et de plantehomologues à la Sec14-p adoptent un repliement unique et compacte, qui dans le cas de Sec14-p elle même estcomposé de 12 hélices α, 6 brins β et 8 hélices3−10. Ces PITP lieraient leur substrat d’une façon à ce que la têtepolaire du phospholipide est orientée vers le solvant et la chaine acyl enfouie à l’intérieur de la protéine. Pour celaSec14p ne contient qu’une seule poche hydrophobe qui ne peut accommoder qu’une molécule de phosholipide.La tête inositol est stabilisée par un réseau de liaisons hydrogènes [Grabon et al., 2015, Routt and Bankaitis,2004]. b) Structure de la PITP de mammifère (PITPα) Les PITP doivent exister avec 2 conformations : uneconformation "ouverte" (1KCM ; à gauche) et une conformation "fermée" (1T27 ; à droite) dans laquelle le lipide(ici la PC) est enfoui à l’intérieur de la structure au centre des 8 brins β et des 3 hélices α. La boucle permettantl’échange du lipide ainsi que la partie C-terminale de la protéine agissent comme un couvercle au dessus de lacavité [Selitrennik and Lev, 2016].
dans la signalisation et sont souvent stabilisés par des complexes d’attache [Helle et al., 2013].De plus en plus de preuves montrent que les SCM permettent le transport des lipides entres lesbicouches grâce à des protéines qui extraient le lipide d’une membrane d’un des organelles pourle délivrer à une autre, Fig. 5.29b. De nombreuses études montrent que les PITP pourraientfaire partie de ses protéines et faciliteraient alors le transport des PIP entre les différents com-partiments membranaires pour permettre une régulation rapide de la composition lipidique desmembranes [Selitrennik and Lev, 2016, Yu et al., 2016].
L’ensemble des données disponibles de la littérature, sur les protéines qui lient les PIP etsur les PITP, concerne plutôt les protéines solubles avec une cavité de liaison mais il existeune autre famille de PITP beaucoup moins étudiée qui possède un domaine d’ancrage aux

5.5. Conclusions, perspectives et discussions 249
membranes (Fig. 5.29b en orange) et qui contient des helices transmembranaires [Wong andLevine, 2016]. Ces protéines agissent aux SCM. L’AtTSPO fait peut-être partie de cet classe dePIPT. Le rôle de l’AtTSPO reste encore à définir et en particulier le mécanisme d’interactionavec les membranes et les PIP. Est-ce que l’AtTSPO agit aux sites de contacts comme certainesPITP, p. ex. à l’interface entre RE-AG, RE-plaste ou RE-mitochondrie ? L’AtTSPO assure-t-ellela distribution des lipides et en particulier des PIP entre les différentes organelles assurant ainsil’homéostasie lipidique des membranes ? L’AtTSPO participe-t-elle indirectement à la régulationde différentes voies de signalisation en transférant les PIP ? C’est en tout cas à ces questionsqu’il serait important de pouvoir répondre.
(a) (b)
Figure 5.29: a) Localisations des sites de contacts membranaires : A. Réticulum endoplasmique (ER)- membrane plasmique(PM) ; B. ER - Mitochondrie (Mito) ; C. ER - endosome précoce (LE)/coprsmultivésiculaires (MVB) et ER - lysosome ; D. ER - Appareil de Golgi ; E. ER - chloroplastes (chloro) ;F. ER - peroxisome (Pex) ; G. ER - goutelettes lipidiques (LD) ; H. sites de contact entre les membranesinterne et externe de la mitochondrie et des chloroplastes. [Toulmay and Prinz, 2011] b) Modèle demécanismes de transport non vésiculaires aux niveaux des sites de contacts. Les lipides (en rouges)peuvent être échangés rapidement entre les membranes via des protéines membranaires, des protéinessolubles de transfert ou via des tunnels hydrophobes. [Beh and Quon, 2016].
En attentant, la mise en évidence par RMN de l’interaction du Nter d’AtTSPO avec leslipides DMPG et PIP est un premier pas vers ce qui pourrait être une fonction encore méconnuede la protéine TSPO chez la plante.


Conclusions et Perspectives


Chapitre6
Conclusions et Perspectives
La détermination de la structure des protéines membranaires est une étape clé dans l’étudedes relations structure/fonction de ces protéines. Leur faible abondance naturelle nécessiteleur surexpression. Au cours de ce travail, j’ai pu expérimenter la difficulté de cette approche.Tout d’abord, lorsque j’ai voulu appliquer à l’isoforme hTSPO2 le même protocole d’expressiondans les bactéries et de purification que celui optimisé pour la mTSPO1, j’ai été confrontée àd’énormes difficultés. Il a fallu développer une autre approche par voie acellulaire pour obtenir laprotéine. Ensuite, lorsque j’ai voulu produire l’AtTSPO qui possède une extension N-terminale,d’une cinquante de résidus supplémentaires par rapport à la mTSPO1 ou à la hTSPO2, l’ad-dition d’une protéine de fusion (GST) censé faciliter son expression s’est avéré toxique pour labactérie. Cependant, l’expression du Nter seul, couplé à cette même protéine de fusion, s’estavérée efficace.
L’obtention de la mTSPO1 recombinante marquée 15N,13C en quantité et pureté suffisantes,m’a permis de continuer son étude structurale déjà entamée au laboratoire. Les études en RMNdu liquide de la protéine en détergent (DPC), se sont avérées difficiles à cause de l’instabilité dela mTSPO1 en absence de ligand. La stabilisation d’une conformation unique en présence duligand PK11195 (le plus affin) n’a pas pu être obtenue aux fortes concentrations nécessaires enRMN (mM). En effet, le PK11195, molécule très hydrophobe, induit la formation d’un précipité(protéine/ligand) à fortes concentrations. Cette difficulté a pu être surmontée par Jaremko et al.en 2014, avec l’utilisation de l’enantiomère (R)-PK11195, conduisant à la première résolutiond’une structure d’un membre de la famille TSPO.
Les structures de la mTSPO1 ainsi que celles de deux de ses homologues bactériens (BcTSPOet RsTSPO), obtenues par cristallographie [Guo et al., 2015, Li et al., 2015], se sont révéléesêtre des informations importantes mais elles n’ont pas permis d’élucider le ou les mécanisme(s)de fonctionnement de la protéine. En particulier, de savoir s’il s’agit ou non d’une protéine detransport. Ces structures se sont également révélées très surprenantes, puisque que les formesbactériennes ne montrent aucun changement de conformation de la protéine avec et sans li-gand. De plus, les structures bactériennes apparaissent stables sans ligand alors que les étudespar RMN du liquide montrent une mTSPO1 instable sans ligand. Cela a amené à une petite"confrontation" de la part des auteurs Jaremko et al. et Li et al. dans laquelle chacun défend satechnique et pointe du doigt les défauts de l’autre !
Pour y voir plus clair, il apparaît donc important d’obtenir une structure de la mTSPO1sans ligand que se soit par RMN ou cristallographie. Pour le moment les essais de cristallisationde la mTSPO1 n’ont jamais permis d’obtenir des cristaux exploitables pour déterminer unestructure atomique [Lacapere et al., 2014]. Néanmoins en septembre 2015, Jaremko et al. ontréussi à attribuer des séquences spécifiques de la protéine sans ligand. Pour ce faire, ils ontréalisé une variété d’expériences multidimensionnelles et utilisé l’attribution de la mTSPO1 en

254 Chapitre 6. Conclusions et Perspectives
complexe avec le (R)-PK11195. Ils ont également tenu compte du fait que les déplacementschimiques des Cα et des Cβ dépendent principalement des structures secondaires et sont moinsaffectés par la structure tertiaire. De cette façon, ils ont attribué spécifiquement 87 résidus (sur169) de la protéine. Ces résidus ne sont pas distribués uniformément sur toute la séquence maispermettent d’élucider partiellement la structure secondaire de la mTSPO1 en absence de sonligand de haute affinité. Elles permettent aussi de déterminer qu’elles sont les parties de laprotéine qui sont le plus fortement affectées par les changements conformationnels, Fig. 6.1.
(a)
(b)
Figure 6.1: Structure secondaire de la mTSPO1 e détergent et sans ligand vue par la RMNdu liquide. a) Schéma des résidus de la mTSPO1 détectés dans les expériences RMN de Jaremko et al.[2015b]. b) Contenu en hélices de la mTSPO1 déterminé à partir des différences entre les déplacementschimiques (δ) observées et les valeurs des δ en conformation aléatoire "random coil" pour la mTSPO sansligand (noir) et en complexe avec le (R)-PK11195 (gris). Plus la valeur est proche de 1 plus la structureen hélice est stable. En haut sont représentés schématiquement les régions avec une structure en Hαstable (mTSPO1/PK11195) ou instable. Les régions en blanc subissent d’importants élargissements deraies dus aux échanges conformationnels, les régions hachées sont désordonnées. [Jaremko et al., 2015b].
Dans cette étude, les auteurs montrent que la boucle 1 reliant les TM1 et TM2, laquelleest proche de la poche de liaison du PK11195, garde sa conformation en hélice (hélice α’)malgré l’absence de ligand. Bien que la mTSPO1 garde un haut contenu en Hα dans unevariété de détergents, sa structure tertiaire n’apparait pas stable. En absence de PK11195,la mTSPO1 semble adopter de multiples conformations en échange. D’après les auteurs, devastes mouvements, sur des échelles de temps allant de la pico-seconde à la micro-seconde, se

255
produisent le long de la structure primaire de la protéine. Ceci mène à une perte de stabilité desinteractions tertiaires et à des dépliements locaux des hélices transmembranaires au voisinagedu site de liaison du ligand. Les auteurs suggèrent que la nature flexible de la mTSPO1 souligneune plasticité conformationnelle de la protéine comme on peut la rencontrer chez des protéinesmembranaires telles que les GPCR (les récepteurs couplés aux protéines G) qui interagissentavec une grande diversité de ligands par l’intermédiaire de leurs domaines extracellulaires et/oudans la partie extracellulaire de leurs segments TM [Deupi and Kobilka, 2007]. Ces récepteursprocèdent à des réarrangements conformationnels de leurs segments TM à proximité des sites deliaison pour s’ajuster aux caractéristiques structurales de leurs ligands. Néanmoins pour Li et al.cet "état désordonné" serait dû aux détergents. La détermination d’une structure de la mTSPO1(avec et sans ligand) dans son milieu naturel en présence de lipides permettrait de trancher laquestion et peut-être d’accéder à certaines étapes de son mécanisme de fonctionnement, commel’activation par les ligands.
L’approche par RMN du solide, de la protéine reconstituée dans un environnement plusproche de sa membrane native est tout à fait possible au vu des résultats obtenues lors de cettethèse. En effet, les spectres 2D 13C-13C ainsi que NCA et NCO de la mTSPO1 sans ligandlaissent envisager qu’il serait possible d’obtenir une structure à plus ou moins long terme. Deplus, les spectres RMN du solide des protéliposomes de mTSPO1 en présence et en absence deligand PK11195 montrent des différences nettes suggérant des changements de conformation.Toutefois, le fait que l’ajout de PK11195 induise plus de pics de corrélation dans le spectre2D 13C-13C pourrait suggérer juste une structuration et non un changement de conformation,comme observé en RMN du liquide avec la protéine en détergent. Néanmoins, les données deRMN du solide suggère une plus forte structuration de la protéine dans un environnementlipidique qu’en présence de détergent.
Du point de vue de la fonction la plus étudiée de la TSPO1 animale, la fixation et le transportdu cholestérol, il n’existe pas de données structurales. Il pourrait être intéressant de faire desétudes par RMN du solide de protéoliposomes contenant du cholestérol puisqu’il a été montréque la mTSPO1 reconstituée en présence de DMPC/DMPE lie le cholestérol avec une affiniténM [Lacapere et al., 2001]. Une comparaison de la mTSPO1 avec son isoforme hTSPO2 seraitégalement intéressante car la première est décrite comme transportant le cholestérol avec uneactivation par le ligand Pk11195 alors que la la seconde lie le cholestérol de la même manièremais présente une très faible affinité pour le PK11195. Cependant, cette fonction est controverséechez les animaux et ne peut pas être décrite chez les bactéries qui n’ont pas de cholestérol.
Nos travaux sur le Nter de l’AtTSPO suggèrent une nouvelle fonction pour les TSPO deplante, directement liée à cette extension d’une cinquantaine de résidus absente chez les animauxet les bactéries. En effet, le changement structural du Nter d’AtTSPO en présence de lipideschargés dont les PIP, pourrait être révélateur d’une implication de la protéine dans le trafic etl’homéostasie lipidique. Ceci pourrait avoir lieu en particulier au niveau des sites de contactsdes différents compartiments membranaires. Toutefois, le rôle des domaines transmembranaires

256 Chapitre 6. Conclusions et Perspectives
de l’AtTSPO reste à définir. La structure globale de l’AtTSPO avec son Nter reste aussi àdéterminer. Ce nouveau rôle, chez les plantes, pourrait s’ajouter à celui déjà reporté dans labiodégradation d’hèmes et de porphyrines par autophagie du complexe AtTSPO/Hème [Vanheeet al., 2011c]. Ce mécanisme n’est observé qu’en condition d’un stress abiotique puisque laprotéine AtTSPO n’est exprimée que dans ces conditions [Guillaumot et al., 2009a].
Figure 6.2: Les TSPO forme une fa-mille conservée et ancienne de protéinesqui, semblent être impliquées en appa-rence dans une multitude de fonctionsà travers les espèces dans les 3 grandsrègnes du vivant. Des endosymbioses au-raient permis le transfert de la protéineet des ses fonctions entre les bactéries etles eucaryotes. Ainsi, des bactéries aux hu-mains, il pourrait y avoir eu une conser-vation d’un rôle physiologique commun àtoute les TSPO en rapport avec la régula-tion du stress cellulaire et de l’homéosta-sie redox par le métabolisme tétrapyrroleavec des mécanismes différents d’une es-pèce à l’autre. En plus de cette fonctioncommune, chaque espèce aurait acquisedes fonctions spécifiques au cours de leurévolution. Adapté de [Li et al., 2016, Veen-man et al., 2016, Batoko et al., 2015b].
Les études des TSPO bactérienne, végétale et animale révèlent que toutes ses protéines ontdes liens avec le stress et les porphyrines, Fig. 6.2. En effet, les différentes TSPO sont induitesen conditions de stress : chez les bactéries lors d’un changement de lumière ou du niveau d’O2 ;chez les plantes lors d’un stress abiotique et chez les animaux lors de l’inflammation. D’autrespart, les porphyrines ont souvent été citées comme ayant un rôle dans la régulation du stressoxydatif. Toutes les TSPO lient les porphyrines, en particulier la PPIX et l’hème, in vivo et invitro avec une affinité de l’ordre du nM [Veenman et al., 2016]. Le lien entre stress et porphyrineschez les bactéries a été décrit plusieurs fois : chezR.sphaeroides, la RhTSPO est proposée régulerl’expression des gènes photosynthétiques via les porphyrines [Yeliseev and Kaplan, 1999]. ChezB. cereus, la BsTSPO catalyse la dégradation de la PPIX qui pourrait générer des ROS toxiques[Guo et al., 2015] confirmant les résultats obtenues avec plusieurs autres bactéries [Ginter et al.,2013]. Un rôle similaire a été proposé dans les cellules de mammifères (glioblastomes) ou desKO de TSPO génèrent une accumulation de PPIX et une augmentation des ROS [Zeno et al.,2012]. Guilarte et al. [2016] propose que lors de l’inflammation de la microglie chez les mam-mifères, la TSPO mitochondriale, aux niveaux des sites de contact, permettrait la sortie des

257
porphyrines vers la protéine NOX2 (une hémoprotéine) du RE pour induire la génération deROS et maintenir l’homéostasie REDOX.
Les résultats obtenus sur les TSPO bactériennes suggèrent que c’est à travers ces espècesqu’il faut peut-être se tourner pour découvrir cette fonction commune à toutes les TSPO quiaurait été conservée au cours de l’évolution, Fig. 6.2. Dans les cristaux de RsTSPO obtenus parLi et al. une PPIX est liée dans un seul des monomères du trimère, alors que dans les cristauxde BcTSPO obtenus par Guo et al. aucune PPIX n’est observée. Pourquoi retrouve-t-on laPPIX seulement dans l’un des monomères d’un des cristaux ? Est-ce dû à la cristallisation ?Nous pensons avoir une part de la réponse car les premières études réalisées au laboratoire(en collaboration avec Hugues Nury et Florine Dupeux à l’IBS Grenoble) montrent la présenced’un tétrapyrrole lors de la purification de la BcTSPO recombinante à partir de bactéries E.coli. Le tétrapyrolle disparaitrait complètement lors de la cristallisation de la BcTSPO alorsqu’il pourrait rester partiellement pour la RsTSPO. Nous envisageons des études de RMN de laBcTSPO avec et sans PPIX pour voir si par cette approche il existe ou non deux conformationsdistinctes. Puis, nous souhaiterions caractériser les différents intermédiaires du processus dedégradation de la PPIX/hème conduisant à la formation du composé inconnu, appelée bilindiginressemblant à la biliverdine, Fig. 6.3.
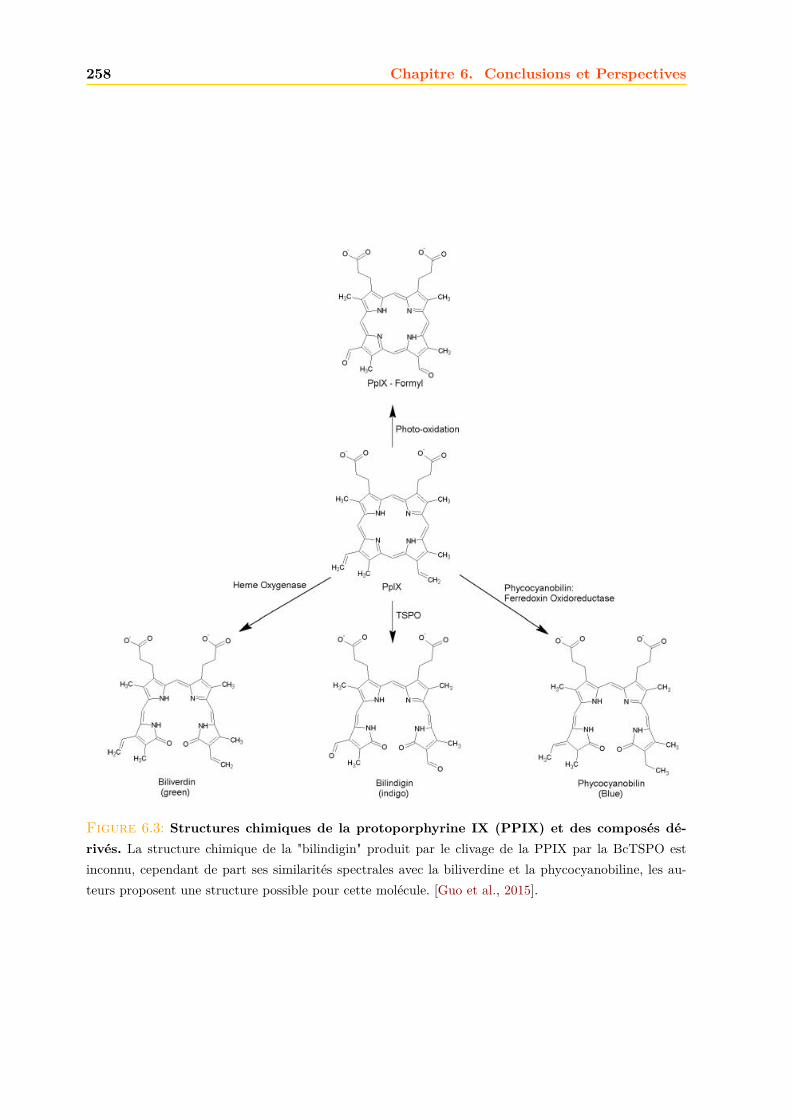
258 Chapitre 6. Conclusions et Perspectives
Figure 6.3: Structures chimiques de la protoporphyrine IX (PPIX) et des composés dé-rivés. La structure chimique de la "bilindigin" produit par le clivage de la PPIX par la BcTSPO estinconnu, cependant de part ses similarités spectrales avec la biliverdine et la phycocyanobiline, les au-teurs proposent une structure possible pour cette molécule. [Guo et al., 2015].

Annexe

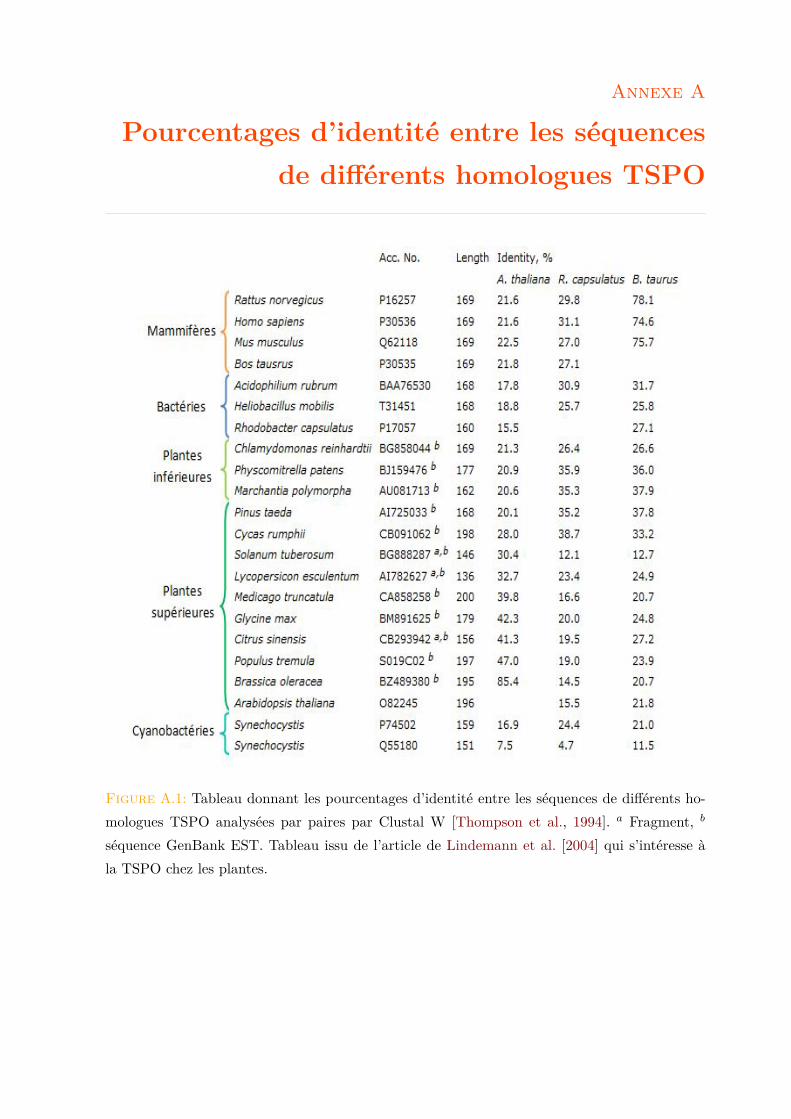
Annexe A
Pourcentages d’identité entre les séquencesde différents homologues TSPO
Figure A.1: Tableau donnant les pourcentages d’identité entre les séquences de différents ho-mologues TSPO analysées par paires par Clustal W [Thompson et al., 1994]. a Fragment, b
séquence GenBank EST. Tableau issu de l’article de Lindemann et al. [2004] qui s’intéresse àla TSPO chez les plantes.


Annexe B
Knock-out global, conditionnel etKnock-down
B.1 Le Knock-out global
La création d’une souris Knock-out (KO), dont un gène connu a été inactivé, permet d’éva-luer les conséquences de la perte de la fonction chez la souris adulte ou d’évaluer, si elle estlétale à quelle stade du développement la déficience apparait. Le KO d’un gène consiste enson inactivation par sa délétion ou sa mutation et se base sur la technique de recombinaisonhomologue. Pour créer une souris KO, Il est nécessaire de disposer du gène à invalider et desséquences le bordant dans le génome avant de pouvoir procéder à son invalidation. Ensuite leschercheurs procèdent à plusieurs étapes :
1. Un morceau d’ADN synthétique, appelé vecteur de ciblage, est réalisé pour remplacerle gène à invalider (gène A), p. ex. : le plasmide portant le gène lac Z (résistant à unantibiotique) qui est entouré des mêmes séquences qui bordent le gène A dans le génome.Ce vecteur est introduit dans des cellules souches embryonnaires SE, lesquelles serontcapables de se différencier en presque tout type de cellule adulte, cela signifie que si ungène est éliminé dans une cellule SE, les effets peuvent être observés dans tous les tissusd’une souris adulte.

264 Annexe B. Knock-out global, conditionnel et Knock-down
2. Dans quelques cellules SE, un évènement de recombinaison homologue a lieu : grâce à unereconnaissance des séquences communes entre le vecteur et le génome de la cellule, unecopie du gène A est remplacée par le vecteur de ciblage.
3. Les cellules sont cultivées sur un milieu possédant l’antibiotique. Ainsi les quelques cellulesSE qui ont réalisé la recombinaison homologue sont sélectionnées et multipliées. Une desdeux copies du gène est ainsi invalidée dans leur génome.
4. L’injection de cellules ES recombinées dans de très jeunes embryons de souris (ces celluless’intègrent à l’embryon) permet l’obtention d’individus mosaïques dérivant pour partie decellules non modifiées, et pour une autre partie de cellules recombinées.
5. Le croisement d’une souris mosaïque, dont les cellules sexuelles dérivent des cellules ESrecombinées (ces souris sont choisies car elles seules sont capables de transmettre le gèneinvalidé à leur descendance) avec une souris sauvage ne possédant pas cette mutationpermet d’obtenir, pour 50% de la descendance, des souris hétérozygotes possédant unecopie du gène sauvage et une copie du gène invalidé.
6. Le croisement des hétérozygotes obtenus précédemment permet l’obtention (dans la pro-portion de 1/4 de la descendance) d’individus homozygotes : ce sont les souris "KO", chezqui le gène a été invalidé.
(Source : http://planet-vie.ens.fr/content/invalidation-gene-knock-out)

B.2. Le Knock-out conditionnel 265
B.2 Le Knock-out conditionnel
Le Knock-out conditionnel permet l’invalidation d’un gène dans un tissu ou à partir d’unstade de développement particulier. Il est utile lorsqu’un gène est exprimé dans plusieurs tissusou à différents stades de développement. La technique la plus souvent utilisée est celle de larecombinaison Cre-lox. La protéine Cre est une recombinase, qui reconnaît dans un segmentd’ADN, une séquence de 34 pb appelée loxP. Lorsque 2 sites loxP sont orientés dans le mêmesens, la Cre recombinase induit la recombinaison entre les 2 sites loxP et ainsi induit la délétionde la séquence d’ADN située entre les 2 sites. À l’issue de cette recombinaison l’allèle mutéporte un site loxP a priori neutre. Le principe du KO conditionnel est de développer dansun 1er temps des souris porteuses d’un allèle dans lequel 2 sites loxP encadrent une partieessentielle du gène d’intérêt sans pour autant perturber son fonctionnement. Ces souris sontensuite croisées avec une souris transgénique qui exprime la Cre dans un tissu spécifique choisi,grâce à l’emploi de séquences régulatrices appropriées. Chez les souris double-transgéniquesissues de ce croisement, la recombinaison entraîne l’excision des séquences situées entre les sitesloxP et donc l’inactivation du gène d’intérêt dans le tissu choisi. Cette stratégie permet decontourner le problème de la létalité embryonnaire, mais aussi d’examiner les effets de cettemutation dans n’importe quel tissu.
B.3 Le Knock-down
Le knock-down d’un gène consiste en son inactivation partielle par diminution de son expres-sion. Pour qu’un gène puisse s’exprimer et produire ses effets son ARN messager doit d’abordêtre traduit en protéine. La technique du knock-down consiste donc à utiliser des ARN dits"interférents" (ARNi), ayant la même séquence que l’ARNm du gène à inactiver, pour spécifi-quement les détruire ou inhiber le processus de traduction de la protéine. Cette méthode permetd’inactiver un gène mais de manière plus subtile que le knock-out.


Annexe C
Les végétaux face au stress abiotiques
Il existe plusieurs types de stress abiotiques, qui peuvent affecter la croissance, le développement oula productivité d’une plante :
• température : trop chaud ou au contraire trop froid pouvant causer le gel• eau : déficit, excès• radiations : dans le visible, l’UV ou les radiations ionisantes• chimiques : sels, métaux lourds ou pesticides• nutritionnel : carence ou excès
De plus, ces stress abiotiques provoquent un stress secondaire dû à l’accumulation d’espèces réactives del’oxygène (ROS - Reactive oxygen species) : c’est le stress oxydatif.
Pour faire face à ces différents stress, la plante va mettre en place des mécanismes de tolérance. Cesmécanismes auront pour but d’éviter soit une perte d’homéostasie, et donc une fragilisation des mem-branes et des protéines, soit des déformations élastiques ou plastiques. Aussi, en réponse à la sécheresse,la salinité ou le froid, le ralentissement de la croissance observé permet à la plante de diminuer les effetsdu stress, p. ex en utilisant les réserves glucidiques pour la protection cellulaire ou en réduisant la surfaceexposée au stress.
Les mécanismes mis en place pour lutter contre ces stress impliquent parfois les mêmes voies designalisation, conduisant aux mêmes stratégies de protection de la cellule. En effet, le rétablissement del’équilibre osmotique, la protection des membranes et des protéines et la détoxification du milieu sontsouvent les objectifs des stratégies de tolérance. Un schéma simplifié de la réponse générale (moléculaireet biochimique) des plantes face aux stress abiotiques est présenté Fig.C.1. Bien sûr, la réponse est enréalité bien plus complexe car chaque stress abiotique aboutit à des stratégies adaptatives spécifiques.Cette réponse "générique" peut de décomposer en 4 étapes :
1. Perception du stress : le stress est perçu par des récepteurs/senseurs membranaires qui initientdes signaux. Les phytocytochromes, les histidine-kinases (HK, ex : AHK1 chez A.thaliana), leskinases RLK (receptor-like kinases) ou encore les récepteurs couplés aux protéines G (RCPG) fontparties de ces récepteurs/senseurs membranaires.
2. Transduction du signal : les signaux sont transduits via de nombreuses voies de signalisa-tion (il existe une voie de transduction dépendante de l’ABA - Encadré 15 - et d’autres ABA-indépendantes) par l’intermédiaire de messagers secondaires tels que les transducteurs du signal,l’inositol phosphatase, les ROS ou le Ca2+. Par ailleurs, l’augmentation du Ca2+ cytosolique estanalysée par des senseurs de calcium qui provoquent une cascade de phosphorylation de protéines(MAPK - Mitogen-actived Protein Kinase 1, CDPK - Calcium-Dependent Protein Kinase 2, SOS -Salt Overly Sensitive 3) qui permettent la progression du signal de stress et donnent lieu à l’acti-vation de FT impliqués dans la réponse au stress .
1. La cascade MAPK est composée de différentes protéines kinases qui s’activent entre elles d’une manièreséquentielle via la phosphorylation, pour transmettre le signal de stress et finalement activer des FT.
2. Les protéines CDPKs pourraient percevoir les changements intracellulaires de concentration de Ca2+ et lestraduire en une cascade de phosphorylations spécifiques pour activer des FT.
3. Le stress salin initie un signal Ca2+ qui active des complexes de protéines Kinases SOS, puis la phospho-rylation de protéines SOS va mener à l’activation de TFs, qui permettra finalement d’aboutir à la stimulation oula suppression de l’activité de transporteurs impliqués dans l’homéostasie ionique.

268 Annexe C. Les végétaux face au stress abiotiques
3. Signalisation hormonale : la perception du stress donne lieu en parallèle à la production d’hor-mones comme l’ABA, l’acide salycilique et l’éthylène. Ces hormones conduisent à la mise en placede mécanismes de tolérance par des voies identiques ou parallèles à celles décrites ci-dessus. P. ex,l’ABA module le degré d’ouverture des stomates 4.
Chez les plantes, l’ABA (acide abscissique - C15H20O4) est une phytohormone concentrée au niveau du pa-renchyme des racines et des feuilles matures, à l’intérieur des plastes. Cette molécule est d’origine lipidique(sesquiterpénoïde) et elle est capable de traverser les membranes. Cette hormone intervient dans des méca-nismes tels que la dormance et la maturation des graines, l’inhibition de la germination ou encore la lutte contredifférents stress abiotiques.
Encadré 15 (L’hormone ABA).
4. Réponses aux stress. La réponse de la plante peut être de 2 types :
• Précoces : activités enzymatiques, activation de transporteurs et de canaux, synthèse enquelques minutes de FTs de microARN. P. ex, en cas de sécheresse, la plante voudra limitersa perte d’eau notamment, par la fermeture de ses stomates. Ceci, est rendu possible grâceà l’ABA qui en se fixant à un récepteur spécifique, déclenche une voie de signalisation,par l’intermédiaire du Ca2+, qui régulent des canaux ioniques provoquant la fermeture desstomates. En temps que réponse précoce, la plante peut aussi modifier sa balance ionique endétournant son métabolisme. Pour cela, elle va synthétiser des petites molécules organiquesosmoprotectantes, les osmolytes, tels que des a.a (proline, valine, et isoleucine), des sucres(glucose, fructose, mannitol, inositol, sorbitol, etc.) ou la glycine betaïne. Ces osmolytespermettent un ajustement osmotique (notamment en augmentant la pression osmotiquedans la cellule afin d’éviter la fuite de l’eau) et une stabilisation des membranes et de laconformation des protéines.
• Tardives : Les FT activés par le stress - p. ex les leucines Zipper de la famille bZip pourla voie de signalisation dépendante de l’ABA - reconnaissent des motifs (appelés élémentsde réponse) de façon spécifique dans les promoteurs des gènes. Parmi ces motifs, il y a lesmotifs ABRE (ABA Responsive Element) pour la transduction dépendante de l’ABA, etles éléments de réponse à la déshydratation (DRE) pour la transduction indépendante àcette hormone. Cette reconnaissance induit les gènes permettant d’exprimer des gènes co-dant pour des protéines qui vont agir dans la protection directe ou indirecte (rétablissementde l’équilibre osmotique, protection des membranes et des protéines, détoxification par éli-mination des ROS, etc.). Ces protéines vont agir en synergie pour avoir la réponse la plusadaptée possible et ainsi assurer la survie de la cellule.
4. Les stomates sont des micropores, situés à la surface des feuilles et des tiges à travers lesquels s’opèrentdes échanges gazeux avec l’atmosphère et par lesquels s’échappe l’eau de transpiration des plantes.
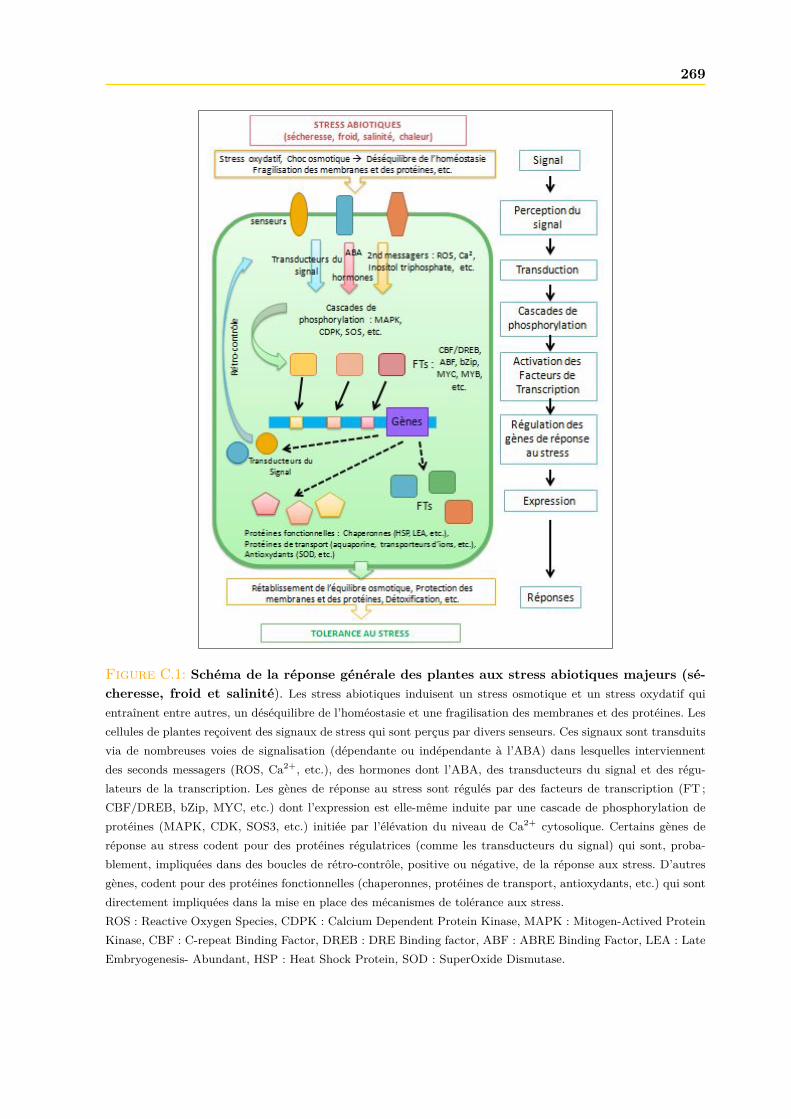
269
Figure C.1: Schéma de la réponse générale des plantes aux stress abiotiques majeurs (sé-cheresse, froid et salinité). Les stress abiotiques induisent un stress osmotique et un stress oxydatif quientraînent entre autres, un déséquilibre de l’homéostasie et une fragilisation des membranes et des protéines. Lescellules de plantes reçoivent des signaux de stress qui sont perçus par divers senseurs. Ces signaux sont transduitsvia de nombreuses voies de signalisation (dépendante ou indépendante à l’ABA) dans lesquelles interviennentdes seconds messagers (ROS, Ca2+, etc.), des hormones dont l’ABA, des transducteurs du signal et des régu-lateurs de la transcription. Les gènes de réponse au stress sont régulés par des facteurs de transcription (FT ;CBF/DREB, bZip, MYC, etc.) dont l’expression est elle-même induite par une cascade de phosphorylation deprotéines (MAPK, CDK, SOS3, etc.) initiée par l’élévation du niveau de Ca2+ cytosolique. Certains gènes deréponse au stress codent pour des protéines régulatrices (comme les transducteurs du signal) qui sont, proba-blement, impliquées dans des boucles de rétro-contrôle, positive ou négative, de la réponse aux stress. D’autresgènes, codent pour des protéines fonctionnelles (chaperonnes, protéines de transport, antioxydants, etc.) qui sontdirectement impliquées dans la mise en place des mécanismes de tolérance aux stress.ROS : Reactive Oxygen Species, CDPK : Calcium Dependent Protein Kinase, MAPK : Mitogen-Actived ProteinKinase, CBF : C-repeat Binding Factor, DREB : DRE Binding factor, ABF : ABRE Binding Factor, LEA : LateEmbryogenesis- Abundant, HSP : Heat Shock Protein, SOD : SuperOxide Dismutase.


Annexe D
L’échange chimique en RMN
L’échange chimique en RMN se réfère à tout processus, dans la gamme de la µs-ms, danslequel le noyau échange entre 2 (ou plusieurs) environnements ce qui provoque une modificationde ces paramètres RMN : déplacement chimique (δ), couplage scalaire, couplage dipolaire etrelaxation.
Cet échange chimique correspond en général, dans le cas d’une protéine, à des mouvementsinternes comme un changement de conformation, ou à une interaction avec une autre moléculecomme lors de la formation d’un complexe protéine/ligand.
Si on prend l’exemple des résonances d’un noyau en échange entre deux états A et B quiapparaissent sur le spectre à des fréquences νA et νB respectivement, ces états pourront êtrecaractéristiques de la fixation réversible d’un ligand ou d’un échange conformationnel. L’effetsur les spectres va dépendre, Fig. D.1.a :
— des populations de chaque état (pA et pB) ; du taux d’échange kex (τex) défini comme lasomme des constantes de dissociation et d’association kon+koff , kex quantifie le nombremoyen d’événements d’échange stochastique par unité de temps et est donc exprimée ens−1 ;
— et de ∆ν défini comme la différence des déplacements chimiques |∆νA-∆νB|. [Klecknerand Foster, 2011].
De plus, cette échange sera caractérisée par un régime : rapide, intermédiaire ou lent à l’échellede temps des déplacements chimiques, Fig. D.1.b :
— Lorsque l’échange entre les états A et B est relativement lent à l’échelle des fréquences(kex � ∆ν), on observe alors 2 pics sur le spectre RMN aux fréquences νA et νB. Outre leurδ, chaque pic se distingue par son intensité et sa largeur de raie. Ces 2 derniers paramètresdépendent des populations et de la constante de relaxation T2 (laquelle dépend de lataille de la molécule) de chaque état. P.ex. lors d’expériences de titrage d’une protéineavec un partenaire, dans ce contexte de régime d’échange lent, l’intensité des pics estproportionnelle aux populations des états A (libre) et B (lié) de la protéine au cours dutitrage, Fig. D.1.d. Le pic minoritaire peut alors devenir indétectable. De plus cette baissed’intensité peut être accentuée par la taille de l’état lié : si le complexe protéine/partenaireformé est grand cela affecte T2 ce qui produit un élargissement de raie supplémentaire.La largeur des raies est donné par : 1
πT2+ k
π— Lorsque la vitesse d’échange augmente, le changement va commencer à se produire à
l’échelle des déplacements chimiques (kex ∼ ∆ν) , on parle d’échange intermédiaire. Unemolécule sera alors dans un état A au début de l’expérience, mais le temps que la séquenced’impulsions prend fin la molécule sera dans l’état B. Un ou deux pics élargis sont observés.L ’élargissement des raies observé est dû à l’interférence de l’interconversion entre l’étatA et B.
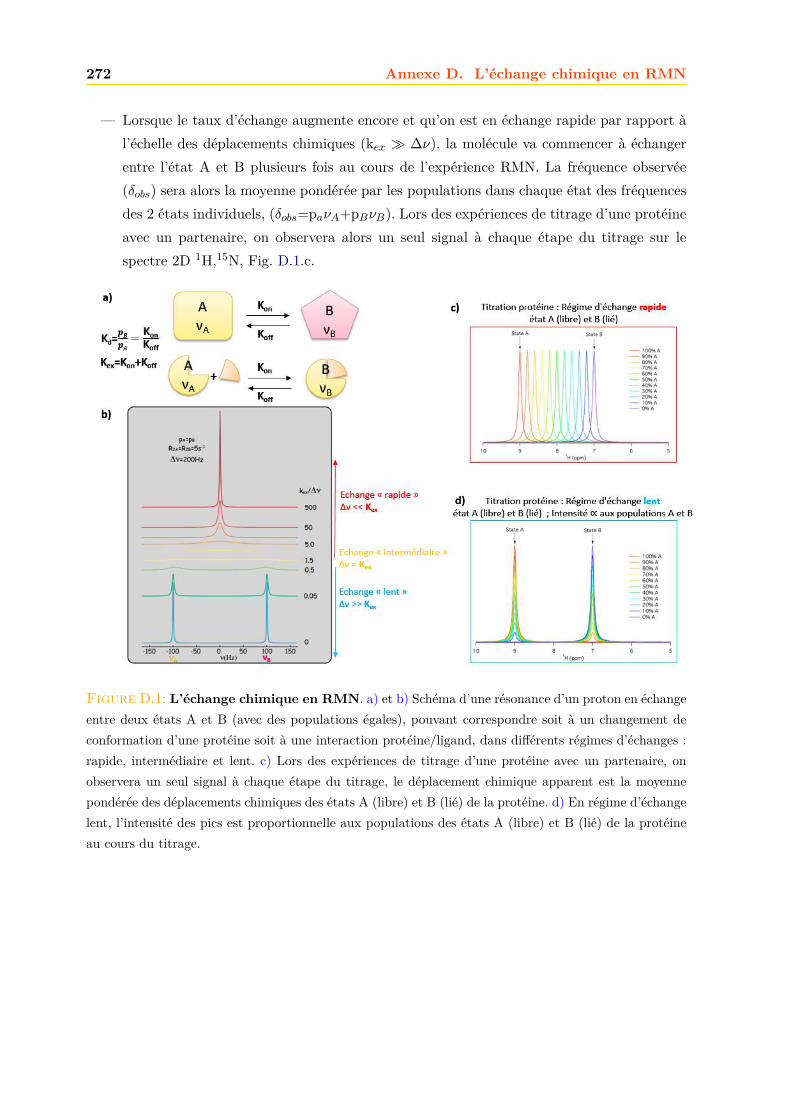
272 Annexe D. L’échange chimique en RMN
— Lorsque le taux d’échange augmente encore et qu’on est en échange rapide par rapport àl’échelle des déplacements chimiques (kex � ∆ν), la molécule va commencer à échangerentre l’état A et B plusieurs fois au cours de l’expérience RMN. La fréquence observée(δobs) sera alors la moyenne pondérée par les populations dans chaque état des fréquencesdes 2 états individuels, (δobs=paνA+pBνB). Lors des expériences de titrage d’une protéineavec un partenaire, on observera alors un seul signal à chaque étape du titrage sur lespectre 2D 1H,15N, Fig. D.1.c.
Figure D.1: L’échange chimique en RMN. a) et b) Schéma d’une résonance d’un proton en échangeentre deux états A et B (avec des populations égales), pouvant correspondre soit à un changement deconformation d’une protéine soit à une interaction protéine/ligand, dans différents régimes d’échanges :rapide, intermédiaire et lent. c) Lors des expériences de titrage d’une protéine avec un partenaire, onobservera un seul signal à chaque étape du titrage, le déplacement chimique apparent est la moyennepondérée des déplacements chimiques des états A (libre) et B (lié) de la protéine. d) En régime d’échangelent, l’intensité des pics est proportionnelle aux populations des états A (libre) et B (lié) de la protéineau cours du titrage.

Annexe E
Articles

274 Annexe E. Articles
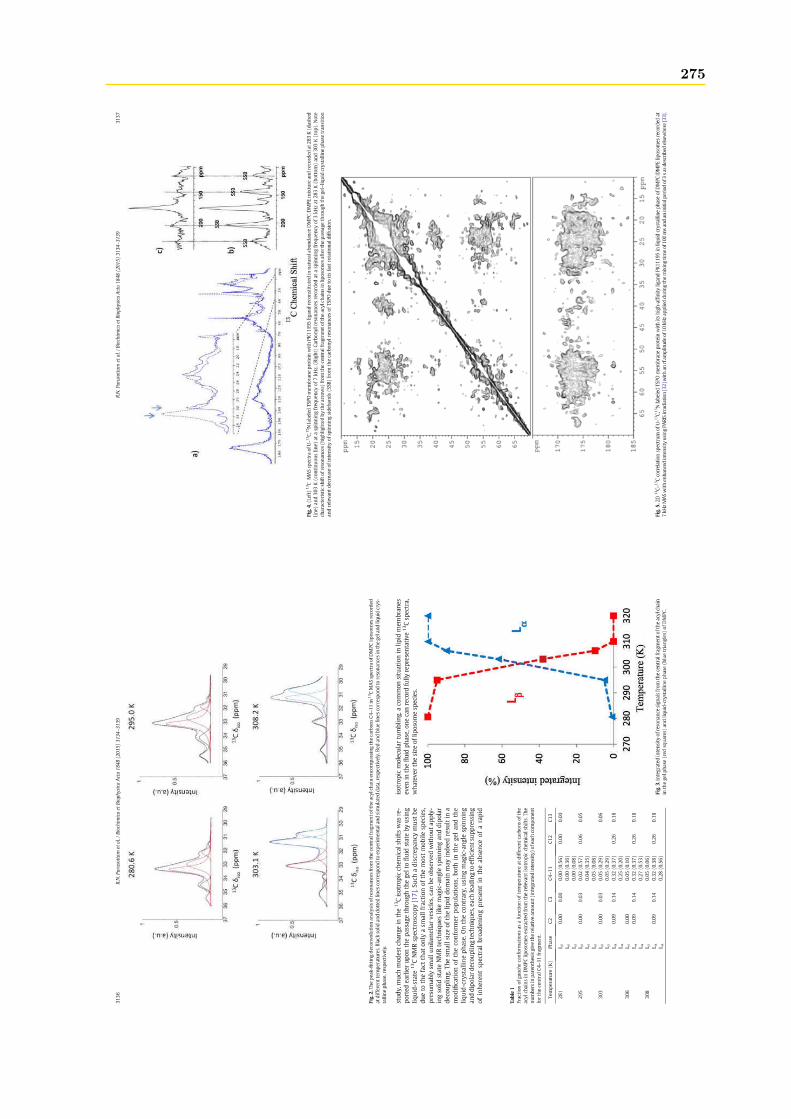
275

276 Annexe E. Articles

277

278 Annexe E. Articles

279

280 Annexe E. Articles

281

282 Annexe E. Articles

283

284 Annexe E. Articles
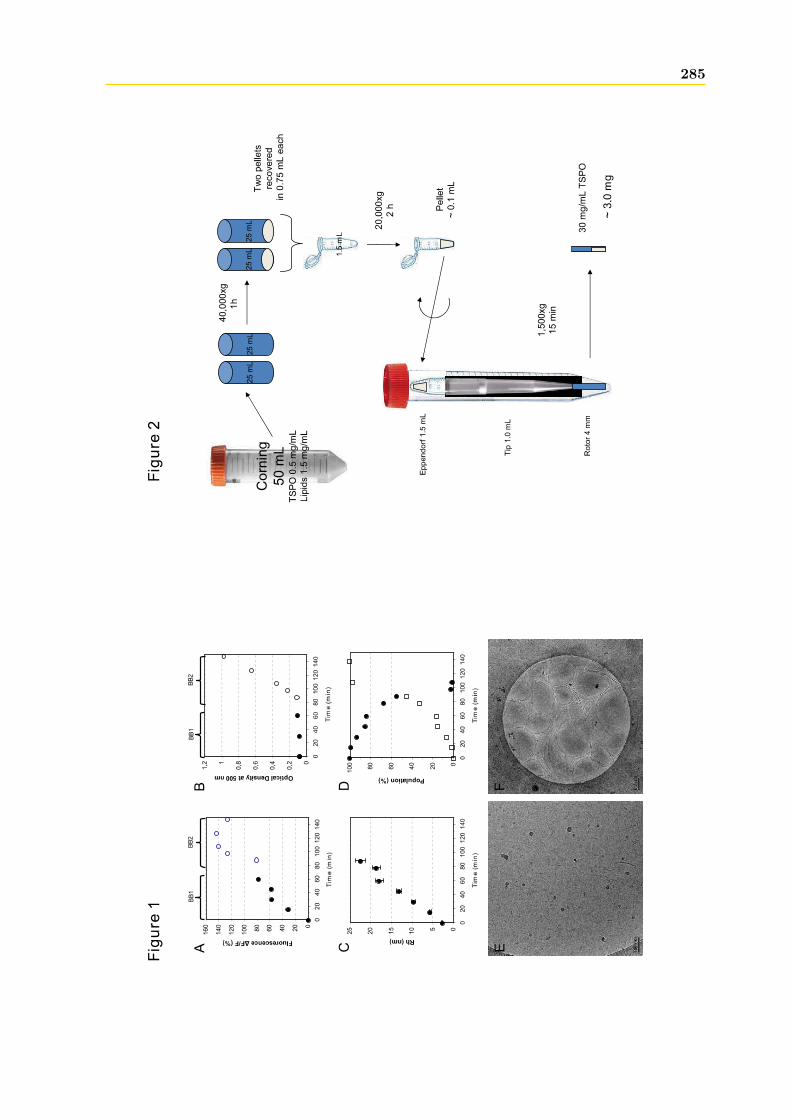
285
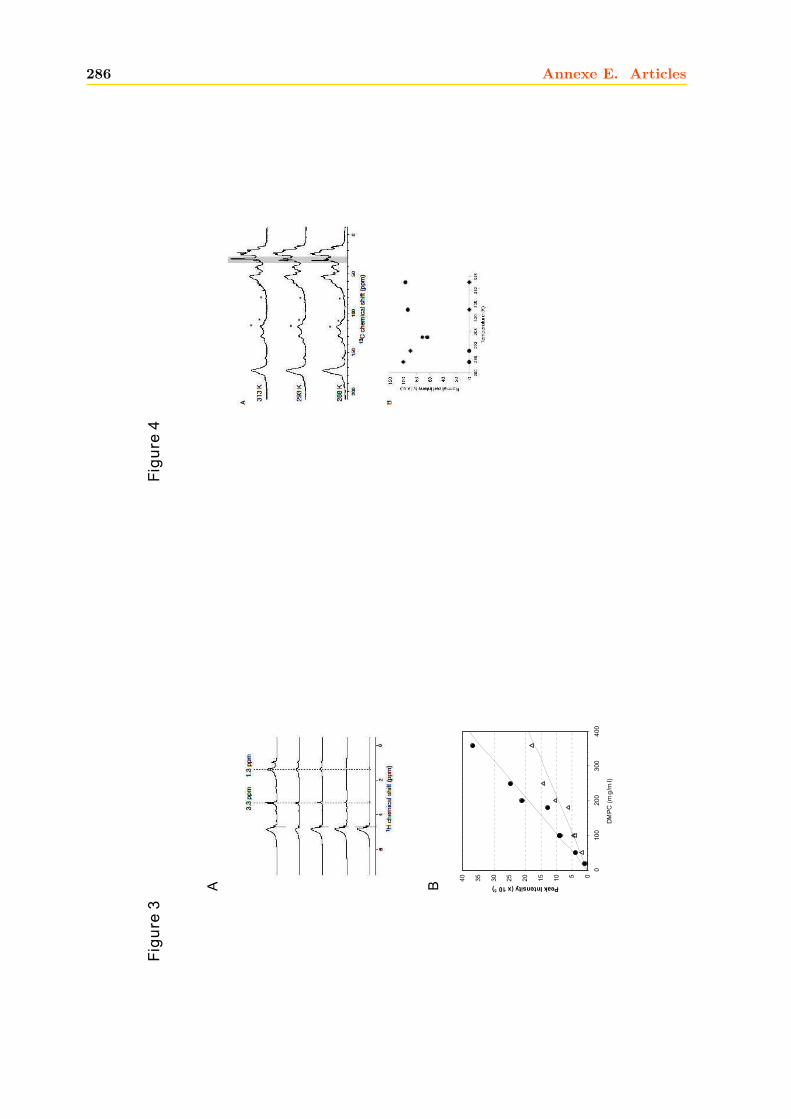
286 Annexe E. Articles

287
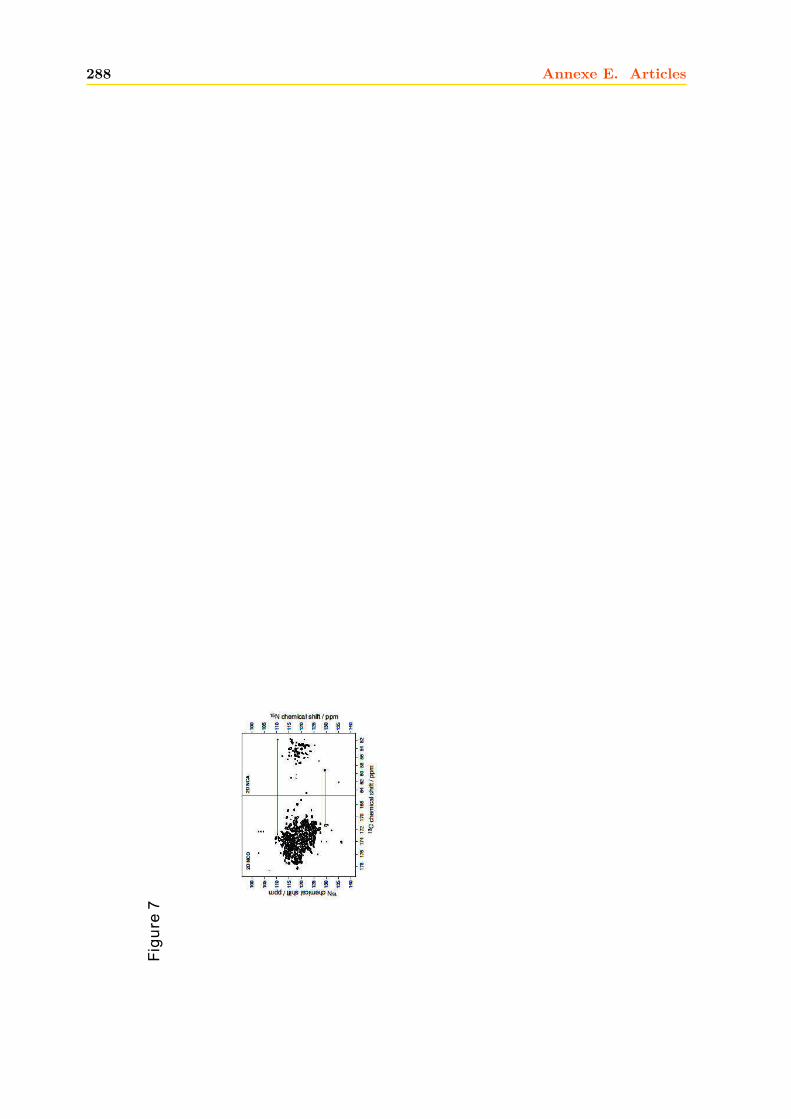
288 Annexe E. Articles

289

290 Annexe E. Articles

291
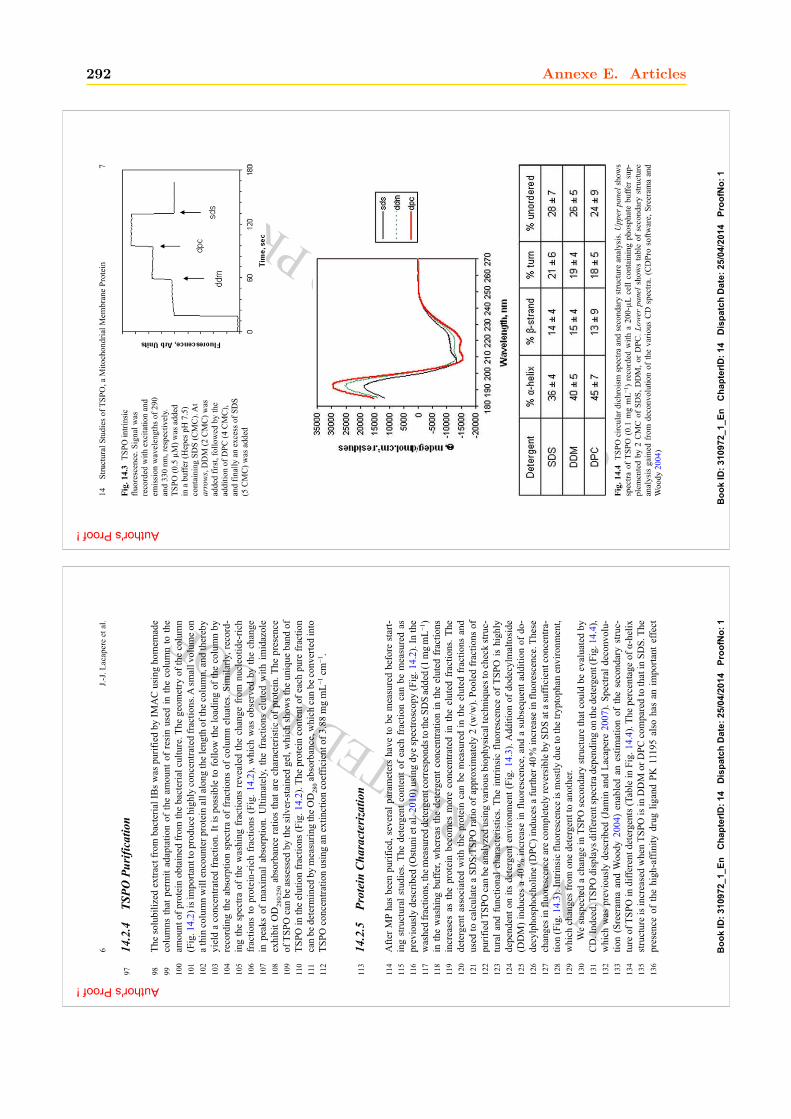
292 Annexe E. Articles
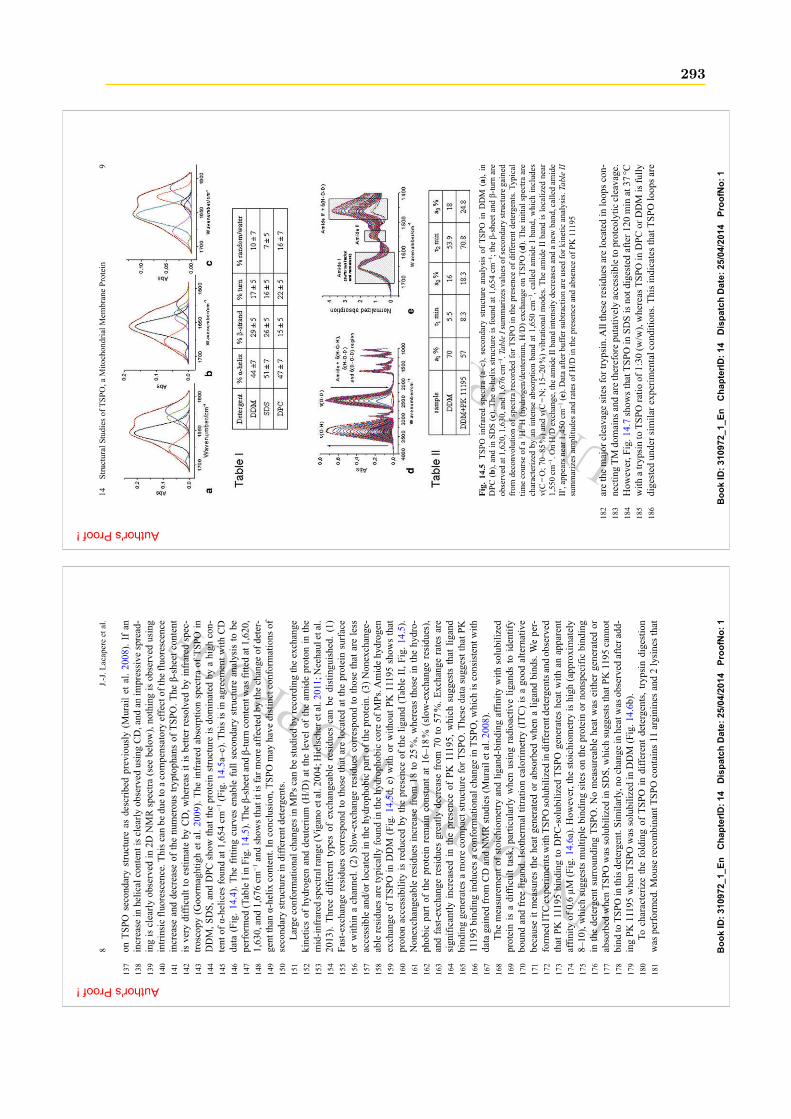
293
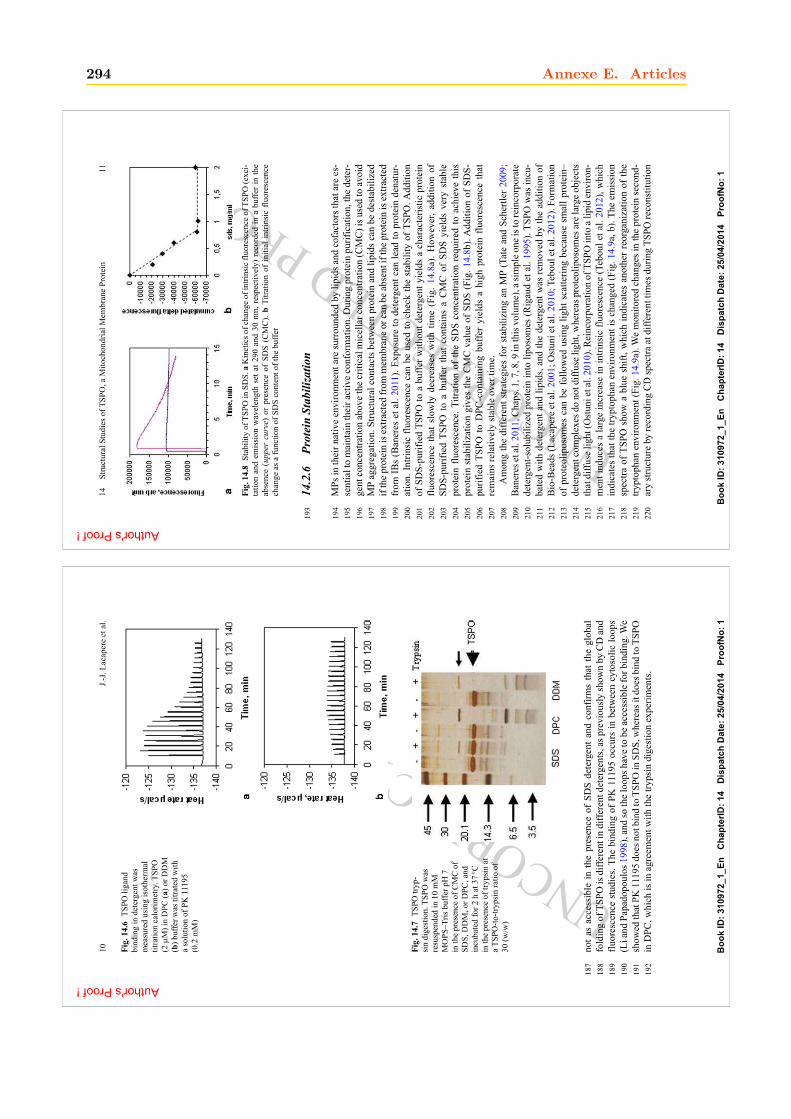
294 Annexe E. Articles
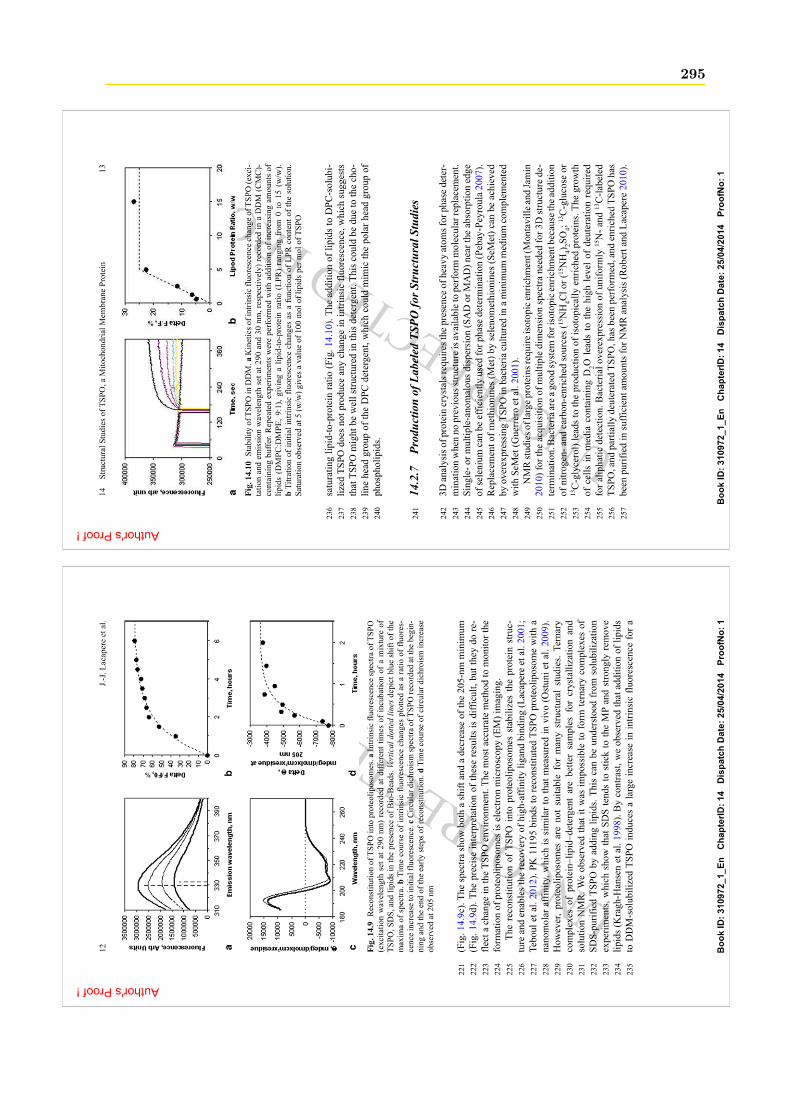
295

296 Annexe E. Articles

297

298 Annexe E. Articles
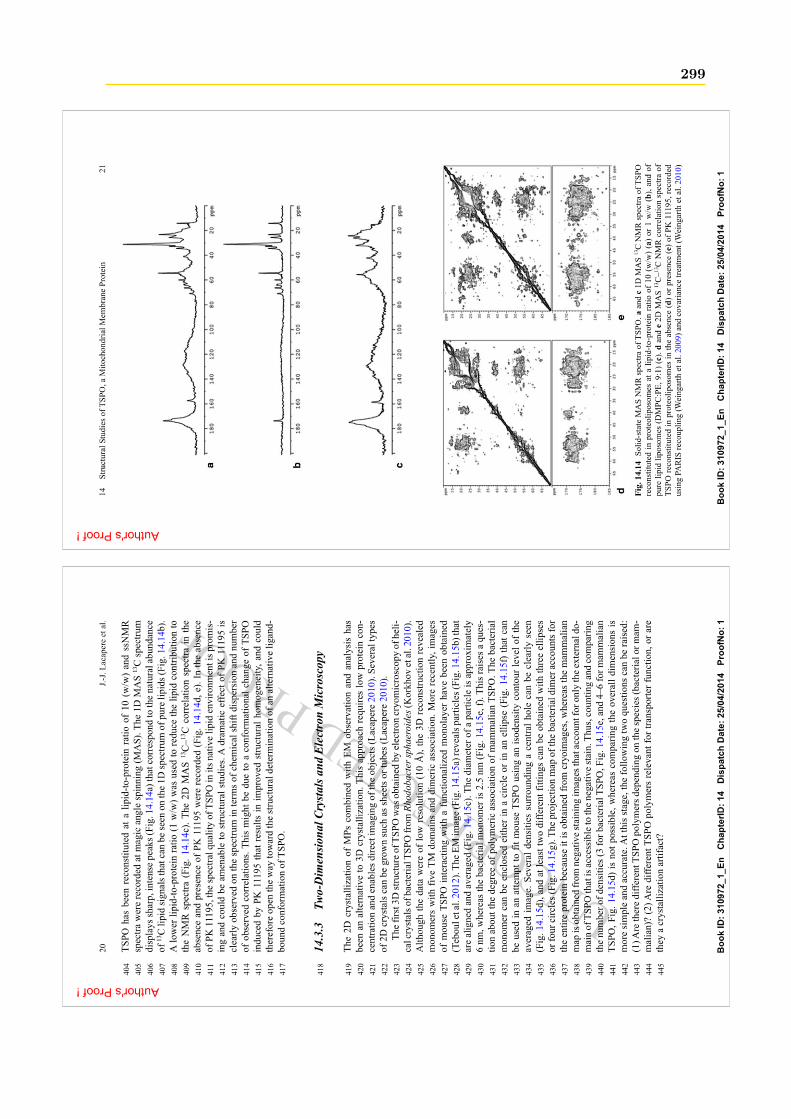
299

300 Annexe E. Articles

301
302 Annexe E. Articles

303


305

306 Annexe E. Articles

307

308 Annexe E. Articles

309
310 Annexe E. Articles
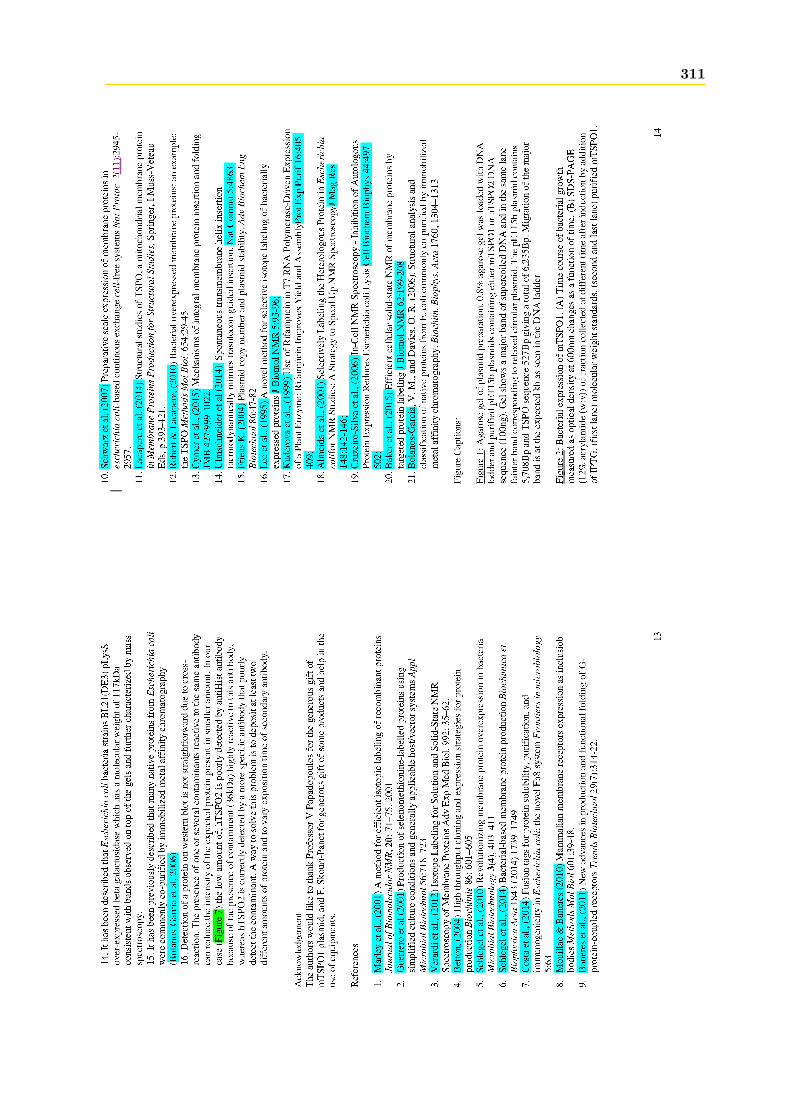
311

312 Annexe E. Articles
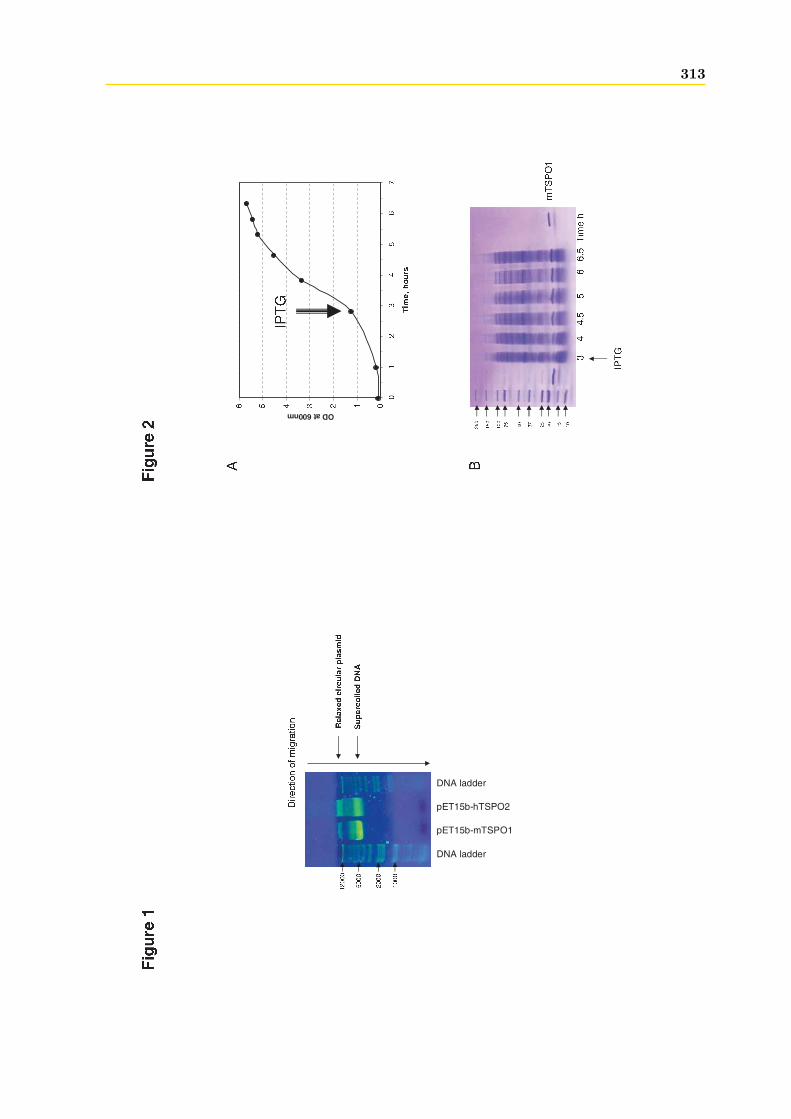
313
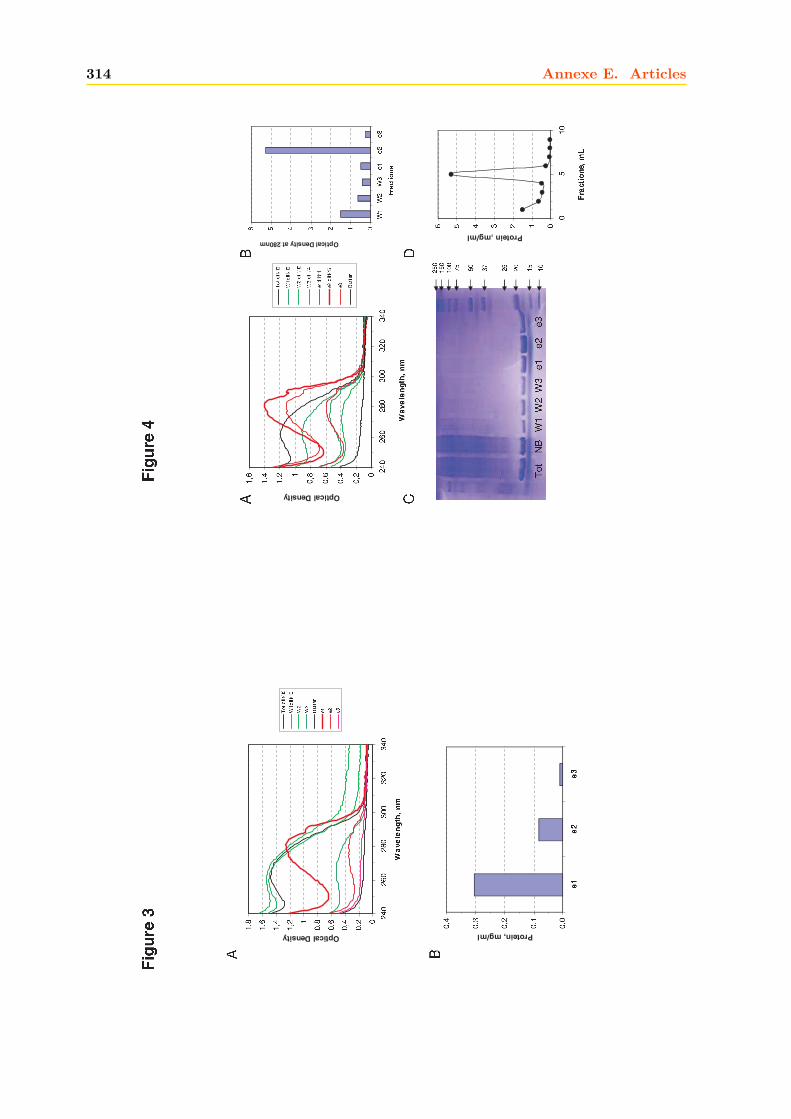
314 Annexe E. Articles
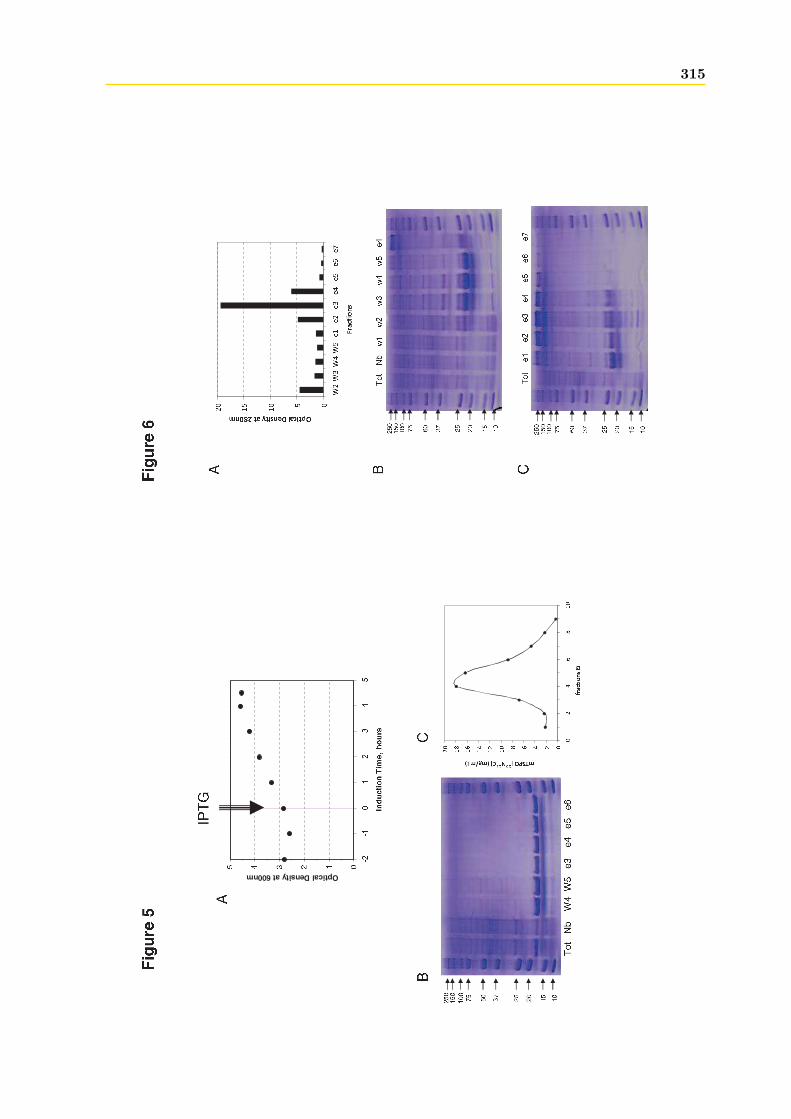
315
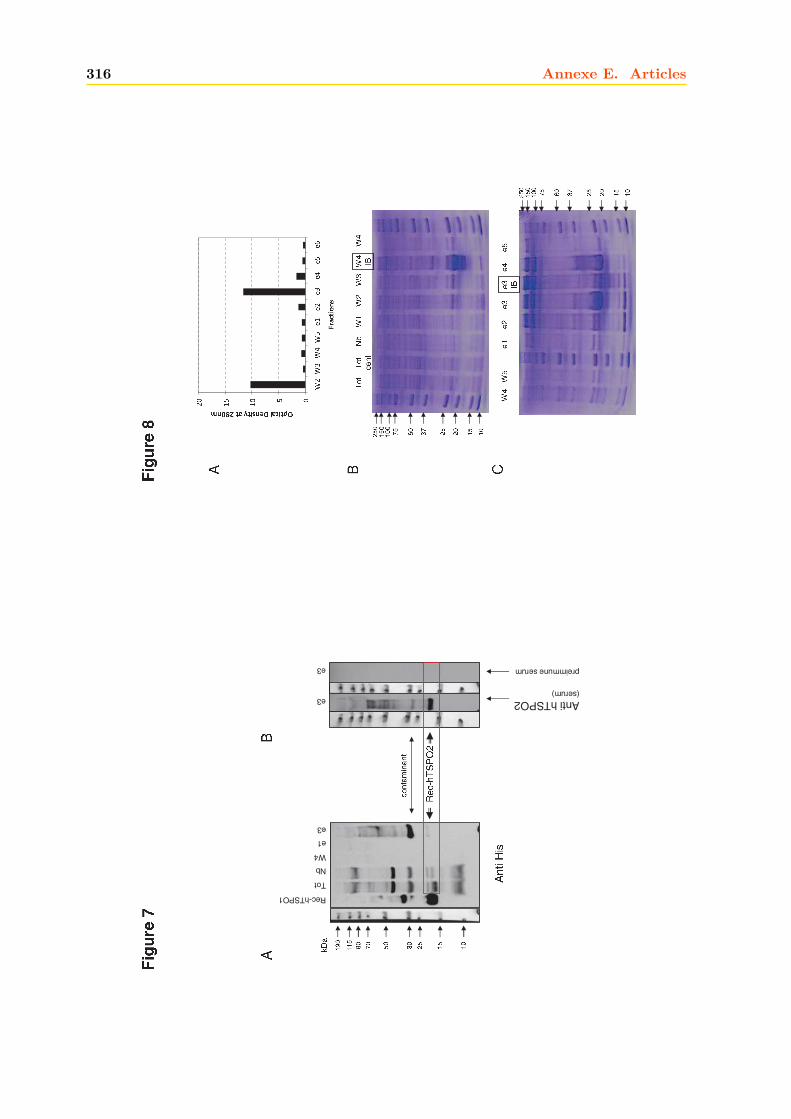
316 Annexe E. Articles
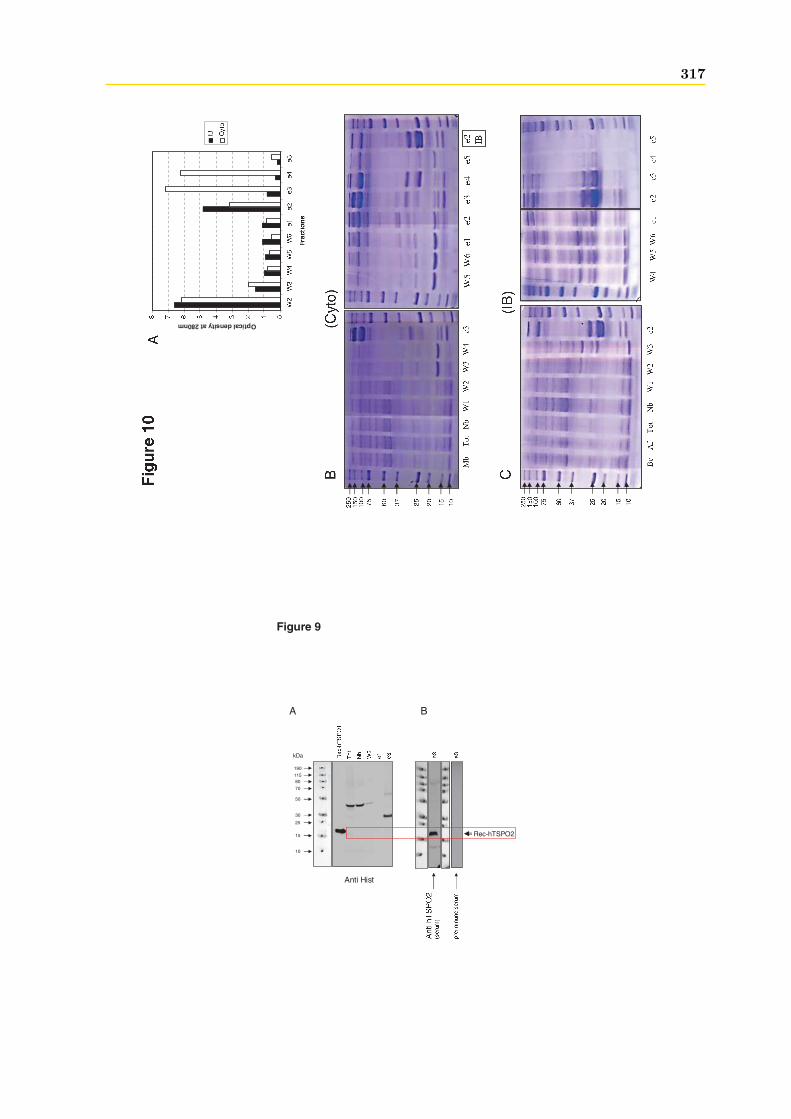
317
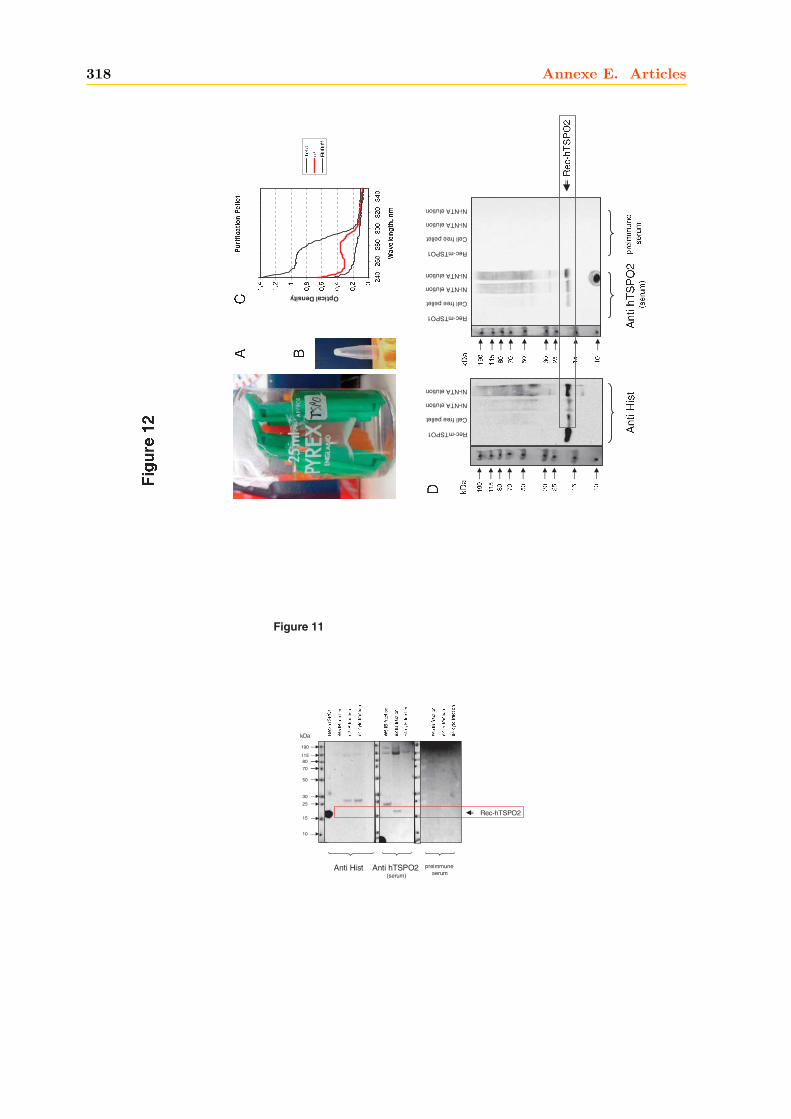
318 Annexe E. Articles

Bibliographie
A. Abdine, K. H. Park, and D. E. Warschawski. Cell-free membrane protein expression for solid-stateNMR. Methods Mol. Biol., 831 :85–109, 2012. (Cité en pages 140, 141, 177 et 218.)
G. H. Addona, H. Sandermann, M. A. Kloczewiak, S. S. Husain, and K. W. Miller. Where does cholesterolact during activation of the nicotinic acetylcholine receptor ? Biochim. Biophys. Acta, 1370(2) :299–309, Mar 1998. (Cité en page 21.)
D. Akopian, K. Shen, X. Zhang, and S. O. Shan. Signal recognition particle : an essential protein-targetingmachine. Annu. Rev. Biochem., 82 :693–721, 2013. (Cité en page 43.)
J. R. Albani. Origin of tryptophan fluorescence lifetimes. Part 2 : fluorescence lifetimes origin of trypto-phan in proteins. J Fluoresc, 24(1) :105–117, Jan 2014. (Cité en page 118.)
H. Alho, P. Bovolin, and E. Slobodyansky. Diazepam binding inhibitor (DBI) processing : immunohis-tochemical studies in the rat brain. Neurochem. Res., 15(2) :209–216, Feb 1990. (Cité en page 19.)
K. V. Andersen and F. M. Poulsen. The three-dimensional structure of acyl-coenzyme A binding proteinfrom bovine liver : structural refinement using heteronuclear multidimensional NMR spectroscopy. J.Biomol. NMR, 3(3) :271–284, May 1993. (Cité en page 20.)
A. Andersson, J. Almqvist, F. Hagn, and L. Maler. Diffusion and dynamics of penetratin in differentmembrane mimicking media. Biochim. Biophys. Acta, 1661(1) :18–25, Feb 2004. (Cité en page 235.)
Suzana M. Andrade and Sílvia M. B. Costa. Spectroscopic studies of water-soluble porphyrins withprotein encapsulated in bis(2-ethylhexyl)sulfosuccinate (AOT) reverse micelles : Aggregation versuscomplexation. Chemistry - A European Journal, 12(4) :1046–1057, jan 2006. (Cité en pages 163et 164.)
R. R. Anholt, E. B. De Souza, M. L. Oster-Granite, and S. H. Snyder. Peripheral-type benzodiazepinereceptors : autoradiographic localization in whole-body sections of neonatal rats. J. Pharmacol. Exp.Ther., 233(2) :517–526, May 1985. (Cité en pages 12, 13, 26 et 30.)
R. R. Anholt, P. L. Pedersen, E. B. De Souza, and S. H. Snyder. The peripheral-type benzodiazepinereceptor. Localization to the mitochondrial outer membrane. J. Biol. Chem., 261(2) :576–583, Jan1986. (Cité en pages 13 et 14.)
L. Antkiewicz-Michaluk, A. Guidotti, and K. E. Krueger. Molecular characterization and mitochondrialdensity of a recognition site for peripheral-type benzodiazepine ligands. Mol. Pharmacol., 34(3) :272–278, Sep 1988a. (Cité en page 14.)
L. Antkiewicz-Michaluk, A. G. Mukhin, A. Guidotti, and K. E. Krueger. Purification and characterizationof a protein associated with peripheral-type benzodiazepine binding sites. J. Biol. Chem., 263(33) :17317–17321, Nov 1988b. (Cité en page 14.)
M. Anzini, A. Cappelli, S. Vomero, G. Giorgi, T. Langer, G. Bruni, M. R. Romeo, and A. S. Basile.Molecular basis of peripheral vs central benzodiazepine receptor selectivity in a new class of peripheralbenzodiazepine receptor ligands related to alpidem. J. Med. Chem., 39(21) :4275–4284, Oct 1996. (Citéen page 15.)
B. D. Arbo, F. Benetti, L. M. Garcia-Segura, and M. F. Ribeiro. Therapeutic actions of translocator pro-tein (18 kDa) ligands in experimental models of psychiatric disorders and neurodegenerative diseases.J. Steroid Biochem. Mol. Biol., 154 :68–74, Nov 2015. (Cité en page 15.)

320 Bibliographie
C. J. Austin, J. Kahlert, M. Kassiou, and L. M. Rendina. The translocator protein (TSPO) : a noveltarget for cancer chemotherapy. Int. J. Biochem. Cell Biol., 45(7) :1212–1216, Jul 2013. (Cité enpages 13 et 34.)
M. Awad and M. Gavish. Binding of [3H]Ro 5-4864 and [3H]PK 11195 to cerebral cortex and peripheraltissues of various species : species differences and heterogeneity in peripheral benzodiazepine bindingsites. J. Neurochem., 49(5) :1407–1414, Nov 1987. (Cité en pages 17 et 25.)
T. Azarashvili, D. Grachev, O. Krestinina, Y. Evtodienko, I. Yurkov, V. Papadopoulos, and G. Rei-ser. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeabilitytransition pore opening in rat brain mitochondria. Cell Calcium, 42(1) :27–39, Jul 2007. (Cité enpage 33.)
Keun-Ryung Bae, Hyun-Jung Shim, Deebika Balu, Sang Ryong Kim, and Seong-Woon Yu. Translocatorprotein 18 kDa negatively regulates inflammation in microglia. Journal of Neuroimmune Pharmaco-logy, 9(3) :424–437, apr 2014. (Cité en page 38.)
K. B. Balestrasse, G. O. Noriega, A. Batlle, and M. L. Tomaro. Involvement of heme oxygenase asantioxidant defense in soybean nodules. Free Radic. Res., 39(2) :145–151, Feb 2005. (Cité en page 66.)
T. Balla, Z. Szentpetery, and Y. J. Kim. Phosphoinositide signaling : new tools and insights. Physiology(Bethesda), 24 :231–244, Aug 2009. (Cité en page 220.)
E. Balsemao-Pires, Y. Jaillais, B. J. Olson, L. R. Andrade, J. G. Umen, J. Chory, and G. Sachetto-Martins. The Arabidopsis translocator protein (AtTSPO) is regulated at multiple levels in responseto salt stress and perturbations in tetrapyrrole metabolism. BMC Plant Biol., 11 :108, 2011. (Cité enpages 59 et 219.)
R. B. Banati, G. W. Goerres, R. Myers, R. N. Gunn, F. E. Turkheimer, G. W. Kreutzberg, D. J.Brooks, T. Jones, and J. S. Duncan. [11C](R)-PK11195 positron emission tomography imaging ofactivated microglia in vivo in Rasmussen’s encephalitis. Neurology, 53(9) :2199–2203, Dec 1999. (Citéen page 18.)
R. B. Banati, R. J. Middleton, R. Chan, C. R. Hatty, W. W. Kam, C. Quin, M. B. Graeber, A. Parmar,D. Zahra, P. Callaghan, S. Fok, N. R. Howell, M. Gregoire, A. Szabo, T. Pham, E. Davis, and G. J. Liu.Positron emission tomography and functional characterization of a complete PBR/TSPO knockout.Nat Commun, 5 :5452, 2014. (Cité en pages 28, 35 et 36.)
V. A. Bankaitis, D. E. Malehorn, S. D. Emr, and R. Greene. The Saccharomyces cerevisiae SEC14 geneencodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgicomplex. J. Cell Biol., 108(4) :1271–1281, Apr 1989. (Cité en page 247.)
V. A. Bankaitis, J. R. Aitken, A. E. Cleves, and W. Dowhan. An essential role for a phospholipid transferprotein in yeast Golgi function. Nature, 347(6293) :561–562, Oct 1990. (Cité en pages 246 et 247.)
V. A. Bankaitis, P. Vincent, M. Merkulova, K. Tyeryar, and Y. Liu. Phosphatidylinositol transfer proteinsand functional specification of lipid signaling pools. Adv. Enzyme Regul., 47 :27–40, 2007. (Cité enpage 247.)
A. M. Barron, L. M. Garcia-Segura, D. Caruso, A. Jayaraman, J. W. Lee, R. C. Melcangi, and C. J.Pike. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J.Neurosci., 33(20) :8891–8897, May 2013. (Cité en page 40.)
Patrick Bartlow, Guy T. Uechi, John J. Cardamone, Tamanna Sultana, McKinzie Fruchtl, Robert R.Beitle, and Mohammad M. Ataai. Identification of native escherichia coli BL21 (DE3) proteins thatbind to immobilized metal affinity chromatography under high imidazole conditions and use of 2d-

Bibliographie 321
DIGE to evaluate contamination pools with respect to recombinant protein expression level. ProteinExpression and Purification, 78(2) :216–224, aug 2011. (Cité en page 205.)
A. S. Basile, H. W. Lueddens, and P. Skolnick. Regulation of renal peripheral benzodiazepine receptorsby anion transport inhibitors. Life Sci., 42(6) :715–726, 1988. (Cité en pages 26 et 32.)
A. Batarseh and V. Papadopoulos. Regulation of translocator protein 18 kDa (TSPO) expression inhealth and disease states. Mol. Cell. Endocrinol., 327(1-2) :1–12, Oct 2010. (Cité en pages 14, 15, 26et 34.)
A. Batarseh, J. Li, and V. Papadopoulos. Protein kinase C epsilon regulation of translocator protein(18 kDa) Tspo gene expression is mediated through a MAPK pathway targeting STAT3 and c-Juntranscription factors. Biochemistry, 49(23) :4766–4778, Jun 2010. (Cité en pages 13 et 15.)
H. Batoko, V. Veljanovski, and P. Jurkiewicz. Enigmatic Translocator protein (TSPO) and cellular stressregulation. Trends Biochem. Sci., 40(9) :497–503, Sep 2015a. (Cité en pages 71 et 73.)
H. Batoko, V. Veljanovski, and P. Jurkiewicz. Enigmatic Translocator protein (TSPO) and cellular stressregulation. Trends Biochem. Sci., 40(9) :497–503, Sep 2015b. (Cité en pages 41, 72 et 256.)
E. E. Baulieu. Neurosteroids : of the nervous system, by the nervous system, for the nervous system.Recent Prog. Horm. Res., 52 :1–32, 1997. (Cité en page 40.)
Christopher Beh and Evan Quon. Membrane contact sites : Complex zones for membrane associationand lipid exchange. LPI, page 55, feb 2016. (Cité en page 249.)
J. Benavides, N. Croci, and M. Strolin Benedetti. Inhibition by aminoacetonitrile and propargylamine ofglycine cleavage system from rat brain and liver mitochondria. Biochem. Pharmacol., 32(2) :287–291,Jan 1983. (Cité en page 25.)
J. Benavides, A. Dubois, B. Gotti, F. Bourdiol, and B. Scatton. Cellular distribution of omega 3 (per-ipheral type benzodiazepine) binding sites in the normal and ischaemic rat brain : an autoradiographicstudy with the photoaffinity ligand [3H]PK 14105. Neurosci. Lett., 114(1) :32–38, Jun 1990. (Cité enpage 12.)
Andrew E. Bennett, Chad M. Rienstra, Michèle Auger, K. V. Lakshmi, and Robert G. Griffin. Hete-ronuclear decoupling in rotating solids. The Journal of Chemical Physics, 103(16), 1995. (Cité enpage 184.)
A. Berkovich, P. McPhie, M. Campagnone, A. Guidotti, and P. Hensley. A natural processing pro-duct of rat diazepam binding inhibitor, triakontatetraneuropeptide (diazepam binding inhibitor 17-50)contains an alpha-helix, which allows discrimination between benzodiazepine binding site subtypes.Mol. Pharmacol., 37(2) :164–172, Feb 1990. (Cité en page 19.)
P. Bernardi. The mitochondrial permeability transition pore : a mystery solved ? Front Physiol, 4 :95,2013. (Cité en page 33.)
P. Bernardi and F. Di Lisa. The mitochondrial permeability transition pore : molecular nature and roleas a target in cardioprotection. J. Mol. Cell. Cardiol., 78 :100–106, Jan 2015. (Cité en pages 32 et 33.)
C. Berrier, K. H. Park, S. Abes, A. Bibonne, J. M. Betton, and A. Ghazi. Cell-free synthesis of afunctional ion channel in the absence of a membrane and in the presence of detergent. Biochemistry,43(39) :12585–12591, Oct 2004. (Cité en page 218.)
M. J. Besman, K. Yanagibashi, T. D. Lee, M. Kawamura, P. F. Hall, and J. E. Shively. Identification ofdes-(Gly-Ile)-endozepine as an effector of corticotropin-dependent adrenal steroidogenesis : stimulationof cholesterol delivery is mediated by the peripheral benzodiazepine receptor. Proc. Natl. Acad. Sci.U.S.A., 86(13) :4897–4901, Jul 1989. (Cité en page 26.)

322 Bibliographie
H. Biverstahl, A. Andersson, A. Graslund, and L. Maler. NMR solution structure and membrane interac-tion of the N-terminal sequence (1-30) of the bovine prion protein. Biochemistry, 43(47) :14940–14947,Nov 2004. (Cité en page 235.)
A. Bockmann, C. Gardiennet, R. Verel, A. Hunkeler, A. Loquet, G. Pintacuda, L. Emsley, B. H. Meier,and A. Lesage. Characterization of different water pools in solid-state NMR protein samples. J.Biomol. NMR, 45(3) :319–327, Nov 2009. (Cité en pages 141, 142, 143 et 177.)
V. M. Bolanos-Garcia and O. R. Davies. Structural analysis and classification of native proteins fromE. coli commonly co-purified by immobilised metal affinity chromatography. Biochim. Biophys. Acta,1760(9) :1304–1313, Sep 2006. (Cité en page 205.)
C. S. Bond and A. W. Schuttelkopf. ALINE : a WYSIWYG protein-sequence alignment editor forpublication-quality alignments. Acta Crystallogr. D Biol. Crystallogr., 65(Pt 5) :510–512, May 2009.(Cité en pages 9, 53, 58 et 196.)
J. Bormann and D. E. Clapham. gamma-Aminobutyric acid receptor channels in adrenal chromaffincells : a patch-clamp study. Proc. Natl. Acad. Sci. U.S.A., 82(7) :2168–2172, Apr 1985. (Cité enpage 19.)
N. Boujrad, J. R. Hudson, and V. Papadopoulos. Inhibition of hormone-stimulated steroidogenesis incultured Leydig tumor cells by a cholesterol-linked phosphorothioate oligodeoxynucleotide antisenseto diazepam-binding inhibitor. Proc. Natl. Acad. Sci. U.S.A., 90(12) :5728–5731, Jun 1993. (Cité enpages 19 et 26.)
N. Boujrad, J. L. Gaillard, M. Garnier, and V. Papadopoulos. Acute action of choriogonadotropinon Leydig tumor cells : induction of a higher affinity benzodiazepine-binding site related to steroidbiosynthesis. Endocrinology, 135(4) :1576–1583, Oct 1994. (Cité en page 26.)
J. U. Bowie. Solving the membrane protein folding problem. Nature, 438(7068) :581–589, Dec 2005.(Cité en page 212.)
C. Braestrup and R. F. Squires. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc. Natl. Acad. Sci. U.S.A., 74(9) :3805–3809, Sep 1977. (Cité enpages 11, 12 et 16.)
E. Bribes, D. Carriere, C. Goubet, S. Galiegue, P. Casellas, and J. Simony-Lafontaine. Immunohis-tochemical assessment of the peripheral benzodiazepine receptor in human tissues. J. Histochem.Cytochem., 52(1) :19–28, Jan 2004. (Cité en pages 12 et 13.)
G. Bristot, B. Ascoli, C. Gubert, B. Panizzutti, F. Kapczinski, and A. R. Rosa. Progesterone and itsmetabolites as therapeutic targets in psychiatric disorders. Expert Opin. Ther. Targets, 18(6) :679–690,Jun 2014. (Cité en page 40.)
M. Campanella, G. Szabadkai, and R. Rizzuto. Modulation of intracellular Ca2+ signalling in HeLa cellsby the apoptotic cell death enhancer PK11195. Biochem. Pharmacol., 76(11) :1628–1636, Dec 2008.(Cité en page 33.)
X. Canat, P. Carayon, M. Bouaboula, D. Cahard, D. Shire, C. Roque, G. Le Fur, and P. Casellas.Distribution profile and properties of peripheral-type benzodiazepine receptors on human hemopoieticcells. Life Sci., 52(1) :107–118, 1993. (Cité en pages 13 et 30.)
A. Cappelli, G. Giuliani, S. Valenti, M. Anzini, S. Vomero, G. Giorgi, C. Sogliano, E. Maciocco, G. Biggio,and A. Concas. Synthesis and structure-activity relationship studies in peripheral benzodiazepinereceptor ligands related to alpidem. Bioorg. Med. Chem., 16(6) :3428–3437, Mar 2008. (Cité enpage 15.)

Bibliographie 323
P. Carayon, M. Portier, D. Dussossoy, A. Bord, G. Petitpretre, X. Canat, G. Le Fur, and P. Casellas.Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells againstoxygen radical damage. Blood, 87(8) :3170–3178, Apr 1996. (Cité en page 36.)
S. O. Casalotti, G. Pelaia, A. G. Yakovlev, T. Csikos, D. R. Grayson, and K. E. Krueger. Structure ofthe rat gene encoding the mitochondrial benzodiazepine receptor. Gene, 121(2) :377–382, Nov 1992.(Cité en page 14.)
P. Casellas, S. Galiegue, and A. S. Basile. Peripheral benzodiazepine receptors and mitochondrial func-tion. Neurochem. Int., 40(6) :475–486, May 2002. (Cité en page 12.)
F. Castellani, B. van Rossum, A. Diehl, M. Schubert, K. Rehbein, and H. Oschkinat. Structure of aprotein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature, 420(6911) :98–102,Nov 2002. (Cité en page 134.)
Y. J. Chang, R. T. McCabe, H. Rennert, M. L. Budarf, R. Sayegh, B. S. Emanuel, P. Skolnick, and J. F.Strauss. The human "peripheral-type" benzodiazepine receptor : regional mapping of the gene andcharacterization of the receptor expressed from cDNA. DNA Cell Biol., 11(6) :471–480, 1992. (Citéen page 14.)
I. Charalampopoulos, E. Remboutsika, A. N. Margioris, and A. Gravanis. Neurosteroids as modulatorsof neurogenesis and neuronal survival. Trends Endocrinol. Metab., 19(8) :300–307, Oct 2008. (Cité enpage 40.)
F. Chauveau, H. Boutin, N. Van Camp, F. Dolle, and B. Tavitian. Nuclear imaging of neuroinflammation :a comprehensive review of [11C]PK11195 challengers. Eur. J. Nucl. Med. Mol. Imaging, 35(12) :2304–2319, Dec 2008. (Cité en page 18.)
B. Chelli, A. Falleni, F. Salvetti, V. Gremigni, A. Lucacchini, and C. Martini. Peripheral-type ben-zodiazepine receptor ligands : mitochondrial permeability transition induction in rat cardiac tissue.Biochem. Pharmacol., 61(6) :695–705, Mar 2001. (Cité en page 33.)
M. K. Chen and T. R. Guilarte. Imaging the peripheral benzodiazepine receptor response in centralnervous system demyelination and remyelination. Toxicol. Sci., 91(2) :532–539, Jun 2006. (Cité enpage 38.)
M. K. Chen and T. R. Guilarte. Translocator protein 18 kDa (TSPO) : molecular sensor of brain injuryand repair. Pharmacol. Ther., 118(1) :1–17, Apr 2008. (Cité en pages 38 et 40.)
M. K. Chen, K. Baidoo, T. Verina, and T. R. Guilarte. Peripheral benzodiazepine receptor imaging inCNS demyelination : functional implications of anatomical and cellular localization. Brain, 127(Pt 6) :1379–1392, Jun 2004. (Cité en page 38.)
A. Chenal, J.I. Guijarro, B. Raynal, M. Delepierre, and D. Ladant. RTX calcium binding motifs areintrinsically disordered in the absence of calcium : IMPLICATION FOR PROTEIN SECRETION.Journal of Biological Chemistry, 284(3) :1781–1789, nov 2008. (Cité en page 235.)
A. S. Ching, B. Kuhnast, A. Damont, D. Roeda, B. Tavitian, and F. Dolle. Current paradigm of the18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation andneurodegenerative diseases. Insights Imaging, 3(1) :111–119, Feb 2012. (Cité en page 41.)
J. Choi, M. Ifuku, M. Noda, and T. R. Guilarte. Translocator protein (18 kDa)/peripheral benzodiazepinereceptor specific ligands induce microglia functions consistent with an activated state. Glia, 59(2) :219–230, Feb 2011. (Cité en page 38.)
D. Chu, E. Kazana, N. Bellanger, T. Singh, M. F. Tuite, and T. von der Haar. Translation elongationcan control translation initiation on eukaryotic mRNAs. EMBO J., 33(1) :21–34, Jan 2014. (Cité enpage 212.)

324 Bibliographie
P. F. Churchill and T. Kimura. Topological studies of cytochromes P-450scc and P-45011 beta in bovineadrenocortical inner mitochondrial membranes. Effects of controlled tryptic digestion. J. Biol. Chem.,254(20) :10443–10448, Oct 1979. (Cité en page 25.)
N. Cinone, H. D. Hotje, and A. Carotti. Development of a unique 3D interaction model of endogenousand synthetic peripheral benzodiazepine receptor ligands. J. Comput. Aided Mol. Des., 14(8) :753–768,Nov 2000. (Cité en pages 15 et 20.)
J. Cleary, K. M. Johnson, A. W. Opipari, and G. D. Glick. Inhibition of the mitochondrial F1F0-ATPaseby ligands of the peripheral benzodiazepine receptor. Bioorg. Med. Chem. Lett., 17(6) :1667–1670, Mar2007. (Cité en page 33.)
A. E. Cleves, T. P. McGee, E. A. Whitters, K. M. Champion, J. R. Aitken, W. Dowhan, M. Goebl,and V. A. Bankaitis. Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass therequirement for an essential phospholipid transfer protein. Cell, 64(4) :789–800, Feb 1991. (Cité enpages 246 et 247.)
A. Colasanti, D. R. Owen, D. Grozeva, E. A. Rabiner, P. M. Matthews, N. Craddock, and A. H. Young.Bipolar Disorder is associated with the rs6971 polymorphism in the gene encoding 18 kDa TranslocatorProtein (TSPO). Psychoneuroendocrinology, 38(11) :2826–2829, Nov 2013. (Cité en page 50.)
B.A. Cornell. The dynamics of the carbonyl groups in phospholipid bilayers from a study of their 13cchemical shift anisotropy. Chemical Physics Letters, 72(3) :462–465, jun 1980. (Cité en page 182.)
L. Corsi, R. Avallone, E. Geminiani, F. Cosenza, I. Venturini, and M. Baraldi. Peripheral benzodiazepinereceptors in potatoes (Solanum tuberosum). Biochem. Biophys. Res. Commun., 313(1) :62–66, Jan2004. (Cité en pages 57 et 62.)
L. Corsi, E. Geminiani, R. Avallone, and M. Baraldi. Nuclear location-dependent role of peripheralbenzodiazepine receptor (PBR) in hepatic tumoral cell lines proliferation. Life Sci., 76(22) :2523–2533, Apr 2005. (Cité en page 14.)
L. Corsi, E. Geminiani, and M. Baraldi. Peripheral benzodiazepine receptor (PBR) new insight in cellproliferation and cell differentiation review. Curr Clin Pharmacol, 3(1) :38–45, Jan 2008. (Cité enpage 34.)
M. Cosenza-Nashat, M. L. Zhao, H. S. Suh, J. Morgan, R. Natividad, S. Morgello, and S. C. Lee.Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based onimmunohistochemical localization in abnormal human brain. Neuropathol. Appl. Neurobiol., 35(3) :306–328, Jun 2009. (Cité en page 38.)
B. Costa, S. Pini, P. Gabelloni, E. Da Pozzo, M. Abelli, L. Lari, M. Preve, A. Lucacchini, G. B. Cassano,and C. Martini. The spontaneous Ala147Thr amino acid substitution within the translocator proteininfluences pregnenolone production in lymphomonocytes of healthy individuals. Endocrinology, 150(12) :5438–5445, Dec 2009a. (Cité en page 50.)
B. Costa, S. Pini, C. Martini, M. Abelli, P. Gabelloni, S. Landi, M. Muti, C. Gesi, L. Lari, A. Cardini,S. Galderisi, A. Mucci, A. Lucacchini, and G. B. Cassano. Ala147Thr substitution in translocatorprotein is associated with adult separation anxiety in patients with depression. Psychiatr. Genet., 19(2) :110–111, Apr 2009b. (Cité en page 50.)
E. Costa and A. Guidotti. Diazepam binding inhibitor (DBI) : a peptide with multiple biological actions.Life Sci., 49(5) :325–344, 1991. (Cité en page 19.)
E. COSTA, D. L. CHENEY, D. R. GRAYSON, A. KORNEYEV, P. LONGONE, L. PANI, E. RO-MEO, E. ZIVKOVICH, and A. GUIDOTTI. Pharmacology of neurosteroid biosynthesis. role of the

Bibliographie 325
mitochondrial DBI receptor (MDR) complex. Annals of the New York Academy of Sciences, 746(1) :223–242, dec 2006. (Cité en page 40.)
M. Culty, H. Li, N. Boujrad, H. Amri, B. Vidic, J. M. Bernassau, J. L. Reversat, and V. Papadopoulos. Invitro studies on the role of the peripheral-type benzodiazepine receptor in steroidogenesis. J. SteroidBiochem. Mol. Biol., 69(1-6) :123–130, 1999. (Cité en pages 14 et 44.)
O. Czarnecki and B. Grimm. Post-translational control of tetrapyrrole biosynthesis in plants, algae, andcyanobacteria. J. Exp. Bot., 63(4) :1675–1687, Feb 2012. (Cité en page 60.)
Bibhuti B. Das, Henry J. Nothnagel, George J. Lu, Woo Sung Son, Ye Tian, Francesca M. Marassi, andStanley J. Opella. Structure determination of a membrane protein in proteoliposomes. J. Am. Chem.Soc., 134(4) :2047–2056, feb 2012. (Cité en pages 82 et 186.)
Bibhuti B. Das, Sang Ho Park, and Stanley J. Opella. Membrane protein structure from rotationaldiffusion. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1848(1) :229–245, jan 2015. (Citéen pages 186 et 187.)
N. Das, D. T. Murray, and T. A. Cross. Lipid bilayer preparations of membrane proteins for orientedand magic-angle spinning solid-state NMR samples. Nat Protoc, 8(11) :2256–2270, Nov 2013. (Citéen pages 141, 142, 177 et 178.)
Daniel J. Daugherty, Vimal Selvaraj, Olga V. Chechneva, Xiao-Bo Liu, David E. Pleasure, and WenbinDeng. A TSPO ligand is protective in a mouse model of multiple sclerosis. EMBO Molecular Medicine,5(6) :891–903, may 2013. (Cité en page 40.)
M. E. Davey and F. J. de Bruijn. A homologue of the tryptophan-rich sensory protein TspO and FixLregulate a novel nutrient deprivation-induced Sinorhizobium meliloti locus. Appl. Environ. Microbiol.,66(12) :5353–5359, Dec 2000. (Cité en pages 32 et 74.)
A. J. de Jesus and T. W. Allen. The role of tryptophan side chains in membrane protein anchoring andhydrophobic mismatch. Biochim. Biophys. Acta, 1828(2) :864–876, Feb 2013. (Cité en page 162.)
M. A. De Matteis and A. Godi. PI-loting membrane traffic. Nat. Cell Biol., 6(6) :487–492, Jun 2004.(Cité en page 220.)
E. B. De Souza, R. R. Anholt, K. M. Murphy, S. H. Snyder, and M. J. Kuhar. Peripheral-type benzo-diazepine receptors in endocrine organs : autoradiographic localization in rat pituitary, adrenal, andtestis. Endocrinology, 116(2) :567–573, Feb 1985. (Cité en pages 13 et 26.)
A. d. de Tassigny, R. Assaly, S. Schaller, R. M. Pruss, A. Berdeaux, and D. Morin. Mitochondrialtranslocator protein (TSPO) ligands prevent doxorubicin-induced mechanical dysfunction and celldeath in isolated cardiomyocytes. Mitochondrion, 13(6) :688–697, Nov 2013. (Cité en page 36.)
D. Decaudin. Peripheral benzodiazepine receptor and its clinical targeting. Anticancer Drugs, 15(8) :737–745, Sep 2004. (Cité en page 16.)
Franck Delavoie, Hua Li, Matthew Hardwick, Jean-Claude Robert, Christoforos Giatzakis, Gabriel Pé-ranzi, Zhi-Xing Yao, Jean Maccario, Jean-Jacques Lacapère, and Vassilios Papadopoulos. In vivoand in vitro peripheral-type benzodiazepine receptor polymerization : functional significance in drugligand and cholesterol binding †. Biochemistry, 42(15) :4506–4519, apr 2003. (Cité en pages 28, 37,50 et 174.)
T. M. DeLorey and R. W. Olsen. Gamma-aminobutyric acidA receptor structure and function. J. Biol.Chem., 267(24) :16747–16750, Aug 1992. (Cité en page 12.)
X. Deupi and B. Kobilka. Activation of G protein-coupled receptors. Adv. Protein Chem., 74 :137–166,2007. (Cité en page 255.)

326 Bibliographie
S. K. Dickeson, C. N. Lim, G. T. Schuyler, T. P. Dalton, G. M. Helmkamp, and L. R. Yarbrough.Isolation and sequence of cDNA clones encoding rat phosphatidylinositol transfer protein. J. Biol.Chem., 264(28) :16557–16564, Oct 1989. (Cité en page 247.)
J. X. DiMario. Activation and repression of growth factor receptor gene transcription (Review). Int. J.Mol. Med., 10(1) :65–71, Jul 2002. (Cité en page 15.)
J. L. Do-Rego, A. G. Mensah-Nyagan, D. Beaujean, J. Leprince, M. C. Tonon, V. Luu-The, G. Pelletier,and H. Vaudry. The octadecaneuropeptide ODN stimulates neurosteroid biosynthesis through acti-vation of central-type benzodiazepine receptors. J. Neurochem., 76(1) :128–138, Jan 2001. (Cité enpage 19.)
A. Doble, J. Benavides, O. Ferris, P. Bertrand, J. Menager, N. Vaucher, M. C. Burgevin, A. Uzan,C. Gueremy, and G. Le Fur. Dihydropyridine and peripheral type benzodiazepine binding sites :subcellular distribution and molecular size determination. Eur. J. Pharmacol., 119(3) :153–167, Dec1985. (Cité en page 14.)
M. Dolder, A. Engel, and M. Zulauf. The micelle to vesicle transition of lipids and detergents in thepresence of a membrane protein : towards a rationale for 2D crystallization. FEBS Lett., 382(1-2) :203–208, Mar 1996. (Cité en page 123.)
M.C. Dubroeucq, C.L.A. Renault, and F.G.R. Le. Nouveaux dérivés d’arène et d’hétéroarènecarboxa-mides, leur procédé de préparation et médicaments les contenant, November 16 1983. EP Patent App.EP19,830,400,749. (Cité en page 18.)
L. Dumon-Seignovert, G. Cariot, and L. Vuillard. The toxicity of recombinant proteins in Escherichiacoli : a comparison of overexpression in BL21(DE3), C41(DE3), and C43(DE3). Protein Expr. Purif.,37(1) :203–206, Sep 2004. (Cité en page 211.)
C. Duparc, H. Lefebvre, M. C. Tonon, H. Vaudry, and J. M. Kuhn. Characterization of endozepines inthe human testicular tissue : effect of triakontatetraneuropeptide on testosterone secretion. J. Clin.Endocrinol. Metab., 88(11) :5521–5528, Nov 2003. (Cité en pages 19 et 20.)
S. V. Dvinskikh, V. Castro, and D. Sandstrom. Probing segmental order in lipid bilayers at variablehydration levels by amplitude- and phase-modulated cross-polarization NMR. Phys Chem Chem Phys,7(18) :3255–3257, Sep 2005. (Cité en page 179.)
M. T. Eddy, Y. Su, R. Silvers, L. Andreas, L. Clark, G. Wagner, G. Pintacuda, L. Emsley, and R. G.Griffin. Lipid bilayer-bound conformation of an integral membrane beta barrel protein by multidi-mensional MAS NMR. J. Biomol. NMR, 61(3-4) :299–310, Apr 2015. (Cité en page 186.)
M. R. Eftink. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J.,66(2 Pt 1) :482–501, Feb 1994. (Cité en page 118.)
J. Fan, M. B. Rone, and V. Papadopoulos. Translocator protein 2 is involved in cholesterol redistributionduring erythropoiesis. Journal of Biological Chemistry, 284(44) :30484–30497, sep 2009. (Cité enpages 52, 54, 195, 197, 210 et 211.)
J. Fan, P. Lindemann, M. G. Feuilloley, and V. Papadopoulos. Structural and functional evolution ofthe translocator protein (18 kDa). Curr. Mol. Med., 12(4) :369–386, May 2012. (Cité en pages 8, 44,56 et 195.)
J. Fan, E. Campioli, A. Midzak, M. Culty, and V. Papadopoulos. Conditional steroidogenic cell-targeteddeletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc.Natl. Acad. Sci. U.S.A., 112(23) :7261–7266, Jun 2015. (Cité en page 29.)
R. Farges, E. Joseph-Liauzun, D. Shire, D. Caput, G. Le Fur, and P. Ferrara. Site-directed mutagenesis

Bibliographie 327
of the peripheral benzodiazepine receptor : identification of amino acids implicated in the binding siteof Ro5-4864. Mol. Pharmacol., 46(6) :1160–1167, Dec 1994. (Cité en pages 14, 17, 47 et 48.)
J. P. Faure, T. Hauet, Z. Han, J. M. Goujon, I. Petit, G. Mauco, M. Eugene, M. Carretier, and V. Papa-dopoulos. Polyethylene glycol reduces early and long-term cold ischemia-reperfusion and renal medullainjury. J. Pharmacol. Exp. Ther., 302(3) :861–870, Sep 2002. (Cité en page 38.)
F. Favreau, L. Rossard, K. Zhang, T. Desurmont, E. Manguy, A. Belliard, S. Fabre, J. Liu, Z. Han,R. Thuillier, V. Papadopoulos, and T. Hauet. Expression and modulation of translocator protein andits partners by hypoxia reoxygenation or ischemia and reperfusion in porcine renal models. Am. J.Physiol. Renal Physiol., 297(1) :F177–190, Jul 2009. (Cité en page 38.)
A. Fernandez-San Millan, I. Farran, A. Molina, A. M. Mingo-Castel, and J. Veramendi. Expression ofrecombinant proteins lacking methionine as N-terminal amino acid in plastids : human serum albuminas a case study. J. Biotechnol., 127(4) :593–604, Jan 2007. (Cité en page 157.)
P. Ferrero, M. R. Santi, B. Conti-Tronconi, E. Costa, and A. Guidotti. Study of an octadecaneuropeptidederived from diazepam binding inhibitor (DBI) : biological activity and presence in rat brain. Proc.Natl. Acad. Sci. U.S.A., 83(3) :827–831, Feb 1986. (Cité en page 19.)
B. Ferzaz, E. Brault, G. Bourliaud, J. P. Robert, G. Poughon, Y. Claustre, F. Marguet, P. Liere, M. Schu-macher, J. P. Nowicki, J. Fournier, B. Marabout, M. Sevrin, P. George, P. Soubrie, J. Benavides,and B. Scatton. SSR180575 (7-chloro-N,N,5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5-b]indole-1-acetamide), a peripheral benzodiazepine receptor ligand, promotes neuronal survival andrepair. J. Pharmacol. Exp. Ther., 301(3) :1067–1078, Jun 2002. (Cité en page 40.)
Sheara W. Fewell and Jeffrey L. Brodsky. Trafficking Inside Cells : Pathways, Mechanisms and Regu-lation, chapter Entry into the Endoplasmic Reticulum : Protein Translocation, Folding and QualityControl, pages 119–142. Springer New York, New York, NY, 2009. ISBN 978-0-387-93877-6. (Cité enpage 64.)
L.A. Flanagan, C.C. Cunningham, J. Chen, G.D. Prestwich, K.S. Kosik, and P.A. Janmey. The structureof divalent cation-induced aggregates of PIP2 and their alteration by gelsolin and tau. BiophysicalJournal, 73(3) :1440–1447, sep 1997. (Cité en page 240.)
W. Frank, K. M. Baar, E. Qudeimat, M. Woriedh, A. Alawady, D. Ratnadewi, L. Gremillon, B. Grimm,and R. Reski. A mitochondrial protein homologous to the mammalian peripheral-type benzodiazepinereceptor is essential for stress adaptation in plants. Plant J., 51(6) :1004–1018, Sep 2007. (Cité enpages 8, 57, 62 et 68.)
K. J. Fritzsching, Y. Yang, K. Schmidt-Rohr, and M. Hong. Practical use of chemical shift databases forprotein solid-state NMR : 2D chemical shift maps and amino-acid assignment with secondary-structureinformation. J. Biomol. NMR, 56(2) :155–167, Jun 2013. (Cité en page 191.)
T. Fukaya, T. Ishiyama, S. Baba, and S. Masumoto. Identification of a novel benzoxazolone derivativeas a selective, orally active 18 kDa translocator protein (TSPO) ligand. J. Med. Chem., 56(20) :8191–8195, Oct 2013. (Cité en pages 18 et 25.)
S. Galiegue, P. Casellas, A. Kramar, N. Tinel, and J. Simony-Lafontaine. Immunohistochemical assess-ment of the peripheral benzodiazepine receptor in breast cancer and its relationship with survival.Clin. Cancer Res., 10(6) :2058–2064, Mar 2004. (Cité en page 34.)
Céline Galvagnion. Etude structurale de la protéine de translocation et de ses domaines hélice-boucle-hélice transmembranaires par RMN en solution. PhD thesis, Université Pierre et Marie Curie, 2011.(Cité en pages 130 et 174.)

328 Bibliographie
P. Gandolfo, E. Louiset, C. Patte, J. Leprince, O. Masmoudi, M. Malagon, F. Gracia-Navarro, H. Vaudry,and M. C. Tonon. The triakontatetraneuropeptide TTN increases [CA2+]i in rat astrocytes throughactivation of peripheral-type benzodiazepine receptors. Glia, 35(2) :90–100, Aug 2001. (Cité enpage 32.)
M. Garnier, N. Boujrad, B. O. Oke, A. S. Brown, J. Riond, P. Ferrara, M. Shoyab, C. A. Suarez-Quian,and V. Papadopoulos. Diazepam binding inhibitor is a paracrine/autocrine regulator of Leydig cellproliferation and steroidogenesis : action via peripheral-type benzodiazepine receptor and independentmechanisms. Endocrinology, 132(1) :444–458, Jan 1993. (Cité en pages 14 et 19.)
M. Garnier, N. Boujrad, S. O. Ogwuegbu, J. R. Hudson, and V. Papadopoulos. The polypeptidediazepam-binding inhibitor and a higher affinity mitochondrial peripheral-type benzodiazepine re-ceptor sustain constitutive steroidogenesis in the R2C Leydig tumor cell line. J. Biol. Chem., 269(35) :22105–22112, Sep 1994a. (Cité en page 19.)
M. Garnier, A. B. Dimchev, N. Boujrad, J. M. Price, N. A. Musto, and V. Papadopoulos. In vitroreconstitution of a functional peripheral-type benzodiazepine receptor from mouse Leydig tumor cells.Mol. Pharmacol., 45(2) :201–211, Feb 1994b. (Cité en page 14.)
J. Gatliff and M. Campanella. TSPO is a REDOX regulator of cell mitophagy. Biochem. Soc. Trans.,43(4) :543–552, Aug 2015. (Cité en pages 36 et 37.)
J. Gatliff and M. Campanella. TSPO : kaleidoscopic 18-kDa amid biochemical pharmacology, controland targeting of mitochondria. Biochem. J., 473(2) :107–121, Jan 2016. (Cité en page 37.)
J. Gatliff, D. East, J. Crosby, R. Abeti, R. Harvey, W. Craigen, P. Parker, and M. Campanella. TSPOinteracts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Au-tophagy, 10(12) :2279–2296, 2014. (Cité en pages 36 et 37.)
M. Gavish and R. Weizman. Role of peripheral-type benzodiazepine receptors in steroidogenesis. ClinNeuropharmacol, 20(6) :473–481, Dec 1997. (Cité en page 19.)
M. Gavish, I. Bachman, R. Shoukrun, Y. Katz, L. Veenman, G. Weisinger, and A. Weizman. Enigma ofthe peripheral benzodiazepine receptor. Pharmacol. Rev., 51(4) :629–650, Dec 1999. (Cité en pages 12,13, 14, 24 et 34.)
G. Georgiou and P. Valax. Isolating inclusion bodies from bacteria. Meth. Enzymol., 309 :48–58, 1999.(Cité en page 152.)
S. Giatti, M. Pesaresi, G. Cavaletti, R. Bianchi, V. Carozzi, R. Lombardi, O. Maschi, G. Lauria, L. M.Garcia-Segura, D. Caruso, and R. C. Melcangi. Neuroprotective effects of a ligand of translocatorprotein-18 kDa (Ro5-4864) in experimental diabetic neuropathy. Neuroscience, 164(2) :520–529, Dec2009. (Cité en page 40.)
C. Giatzakis and V. Papadopoulos. Differential utilization of the promoter of peripheral-type benzodia-zepine receptor by steroidogenic versus nonsteroidogenic cell lines and the role of Sp1 and Sp3 in theregulation of basal activity. Endocrinology, 145(3) :1113–1123, Mar 2004. (Cité en pages 13 et 15.)
Christopher Ginter, Irene Kiburu, and Olga Boudker. Chemical catalysis by the translocator protein (18kDa). Biochemistry, 52(21) :3609–3611, may 2013. (Cité en pages 71 et 256.)
V. Giorgio, S. von Stockum, M. Antoniel, A. Fabbro, F. Fogolari, M. Forte, G. D. Glick, V. Petronilli,M. Zoratti, I. Szabo, G. Lippe, and P. Bernardi. Dimers of mitochondrial ATP synthase form thepermeability transition pore. Proc. Natl. Acad. Sci. U.S.A., 110(15) :5887–5892, Apr 2013. (Cité enpages 32 et 33.)
Gopal Jee Gopal and Awanish Kumar. Strategies for the production of recombinant protein in escherichiacoli. The Protein Journal, 32(6) :419–425, jul 2013. (Cité en pages 210 et 211.)

Bibliographie 329
Govindjee and Dmitriy Shevela. Adventures with cyanobacteria : a personal perspective. Frontiers inPlant Science, 2(28), 2011. ISSN 1664-462X. (Cité en page 10.)
Aby Grabon, Danish Khan, and Vytas A. Bankaitis. Phosphatidylinositol transfer proteins and instruc-tive regulation of lipid kinase biology. Biochimica et Biophysica Acta (BBA) - Molecular and CellBiology of Lipids, 1851(6) :724–735, jun 2015. (Cité en page 248.)
B. H. Graham, K. G. Waymire, B. Cottrell, I. A. Trounce, G. R. MacGregor, and D. C. Wallace. Amouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in theheart/muscle isoform of the adenine nucleotide translocator. Nat. Genet., 16(3) :226–234, Jul 1997.(Cité en page 30.)
P. W. Gray, D. Glaister, P. H. Seeburg, A. Guidotti, and E. Costa. Cloning and expression of cDNAfor human diazepam binding inhibitor, a natural ligand of an allosteric regulatory site of the gamma-aminobutyric acid type A receptor. Proc. Natl. Acad. Sci. U.S.A., 83(19) :7547–7551, Oct 1986. (Citéen page 19.)
Y. Guan, Q. Zhu, D. Huang, S. Zhao, L. Jan Lo, and J. Peng. An equation to estimate the differencebetween theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. SciRep, 5 :13370, 2015. (Cité en page 151.)
A. Guidotti, C. M. Forchetti, M. G. Corda, D. Konkel, C. D. Bennett, and E. Costa. Isolation, cha-racterization, and purification to homogeneity of an endogenous polypeptide with agonistic actionon benzodiazepine receptors. Proc. Natl. Acad. Sci. U.S.A., 80(11) :3531–3535, Jun 1983. (Cité enpage 19.)
T. R. Guilarte, M. K. Loth, and S. R. Guariglia. TSPO Finds NOX2 in Microglia for Redox Homeostasis.Trends Pharmacol. Sci., Mar 2016. (Cité en pages 37, 38 et 256.)
D. Guillaumot, S. Guillon, T. Deplanque, C. Vanhee, C. Gumy, D. Masquelier, P. Morsomme, andH. Batoko. The Arabidopsis TSPO-related protein is a stress and abscisic acid-regulated, endoplasmicreticulum-Golgi-localized membrane protein. Plant J., 60(2) :242–256, Oct 2009a. (Cité en pages 8,32, 57, 59, 60, 62, 65 et 256.)
D. Guillaumot, S. Guillon, P. Morsomme, and H. Batoko. ABA, porphyrins and plant TSPO-relatedprotein. Plant Signal Behav, 4(11) :1087–1090, Nov 2009b. (Cité en pages 57, 59, 62 et 65.)
Y. Guo, R. C. Kalathur, Q. Liu, B. Kloss, R. Bruni, C. Ginter, E. Kloppmann, B. Rost, and W. A.Hendrickson. Structure and activity of tryptophan-rich TSPO proteins. Science, 347(6221) :551–555,jan 2015. (Cité en pages 71, 74, 75, 175, 253, 256, 257 et 258.)
C. Hachez, V. Veljanovski, H. Reinhardt, D. Guillaumot, C. Vanhee, F. Chaumont, and H. Batoko.The Arabidopsis abiotic stress-induced TSPO-related protein reduces cell-surface expression of theaquaporin PIP2 ;7 through protein-protein interactions and autophagic degradation. Plant Cell, 26(12) :4974–4990, Dec 2014. (Cité en pages 63 et 67.)
Z. Han, R. S. Slack, W. Li, and V. Papadopoulos. Expression of peripheral benzodiazepine receptor (PBR)in human tumors : relationship to breast, colorectal, and prostate tumor progression. J. Recept. SignalTransduct. Res., 23(2-3) :225–238, 2003. (Cité en page 34.)
M. A. Hanson, V. Cherezov, M. T. Griffith, C. B. Roth, V. P. Jaakola, E. Y. Chien, J. Velasquez,P. Kuhn, and R. C. Stevens. A specific cholesterol binding site is established by the 2.8 A structureof the human beta2-adrenergic receptor. Structure, 16(6) :897–905, Jun 2008. (Cité en page 21.)
E. Harberts, D. Datta, S. Chen, J. E. Wohler, U. Oh, and S. Jacobson. Translocator protein 18 kDa(TSPO) expression in multiple sclerosis patients. J Neuroimmune Pharmacol, 8(1) :51–57, Mar 2013.(Cité en page 18.)

330 Bibliographie
Soria Harbi Iatmanen. Production et caractérisation structurale et fonctionnelle de la protéine membra-naire recombinante TSPO. PhD thesis, Faculté de Médecine X Bichat, 2014. (Cité en pages 149, 150,151, 154, 155, 157, 159, 167 et 175.)
M. Hardwick, D. Fertikh, M. Culty, H. Li, B. Vidic, and V. Papadopoulos. Peripheral-type benzodiazepinereceptor (PBR) in human breast cancer : correlation of breast cancer cell aggressive phenotype withPBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport ofcholesterol. Cancer Res., 59(4) :831–842, Feb 1999. (Cité en pages 14 et 34.)
Georges Hattab, Annabelle Y. T. Suisse, Oana Ilioaia, Marina Casiraghi, Manuela Dezi, Xavier L. War-net, Dror E. Warschawski, Karine Moncoq, Manuela Zoonens, and Bruno Miroux. Membrane proteinproduction in escherichia coli : Overview and protocols. In Membrane Proteins Production for Struc-tural Analysis, pages 87–106. Springer Science Business Media, 2014. (Cité en page 211.)
Sebastian C.J. Helle, Gil Kanfer, Katja Kolar, Alexander Lang, Agnès H. Michel, and Benoît Korn-mann. Organization and function of membrane contact sites. Biochimica et Biophysica Acta (BBA)- Molecular Cell Research, 1833(11) :2526–2541, nov 2013. (Cité en page 248.)
L Hertz. Binding characteristics of the receptor and coupling to transport proteins. Academic Press,London, 1993. (Cité en page 25.)
R. Hirsch, M. Eckhaus, H. Auchincloss, D. H. Sachs, and J. A. Bluestone. Effects of in vivo administrationof anti-T3 monoclonal antibody on T cell function in mice. I. Immunosuppression of transplantationresponses. J. Immunol., 140(11) :3766–3772, Jun 1988. (Cité en pages 34 et 175.)
J. Hsuan and S. Cockcroft. The PITP family of phosphatidylinositol transfer proteins. Genome Biol., 2(9) :REVIEWS3011, 2001. (Cité en page 248.)
Anna K. Hurlock, Rebecca L. Roston, Kun Wang, and Christoph Benning. Lipid trafficking in plantcells. Traffic, 15(9) :915–932, jul 2014. (Cité en page 220.)
Ales Iglic, editor. Advances in Planar Lipid Bilayers and Liposomes, Volume 14. Academic Press, 2011.ISBN 0123877202. (Cité en page 183.)
Elina Ikonen. Cellular cholesterol trafficking and compartmentalization. Nature Reviews Molecular CellBiology, 9(2) :125–138, feb 2008. (Cité en pages 21 et 22.)
L. Issop, M. A. Ostuni, S. Lee, M. Laforge, G. Peranzi, P. Rustin, J. F. Benoist, J. Estaquier, V. Papado-poulos, and J. J. Lacapere. Translocator Protein-Mediated Stabilization of Mitochondrial Architectureduring Inflammation Stress in Colonic Cells. PLoS ONE, 11(4) :e0152919, 2016. (Cité en page 38.)
N. Jamin, J. M. Neumann, M. A. Ostuni, T. K. Vu, Z. X. Yao, S. Murail, J. C. Robert, C. Giatzakis,V. Papadopoulos, and J. J. Lacapere. Characterization of the cholesterol recognition amino acidconsensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol., 19(3) :588–594,Mar 2005. (Cité en pages 49, 81, 174 et 175.)
Nadège Jamin and Jean-Jacques Lacapère. Circular Dichroism : Folding and Conformational Changes ofMembrane Proteins, pages 241–258. Wiley-VCH Verlag GmbH Co. KGaA, 2008. ISBN 9783527621224.(Cité en page 45.)
M. Jansson, Y. C. Li, L. Jendeberg, S. Anderson, G. T. Montelione, and B. Nilsson. High-level productionof uniformly 15n- and 13c-enriched fusion proteins in Escherichia coli. J. Biomol. NMR, 7(2) :131–141,Mar 1996. (Cité en page 90.)
B. K. Jap, M. Zulauf, T. Scheybani, A. Hefti, W. Baumeister, U. Aebi, and A. Engel. 2D crystallization :from art to science. Ultramicroscopy, 46(1-4) :45–84, Oct 1992. (Cité en page 123.)
L. Jaremko, M. Jaremko, K. Giller, S. Becker, and M. Zweckstetter. Structure of the mitochondrial

Bibliographie 331
translocator protein in complex with a diagnostic ligand. Science, 343(6177) :1363–1366, Mar 2014.(Cité en pages 18, 46, 49, 78, 167, 175 et 253.)
Łukasz Jaremko, Mariusz Jaremko, Karin Giller, Stefan Becker, and Markus Zweckstetter. Conforma-tional flexibility in the transmembrane protein TSPO. Chemistry - A European Journal, 21(46) :16555–16563, sep 2015a. (Cité en page 253.)
Mariusz Jaremko, Łukasz Jaremko, Karin Giller, Stefan Becker, and Markus Zweckstetter. Structuralintegrity of the a147t polymorph of mammalian TSPO. ChemBioChem, 16(10) :1483–1489, jun 2015b.(Cité en pages 48, 51, 78, 79 et 254.)
K. M. Johnson, J. Cleary, C. A. Fierke, A. W. Opipari, and G. D. Glick. Mechanistic basis for therapeutictargeting of the mitochondrial F1F0-ATPase. ACS Chem. Biol., 1(5) :304–308, Jun 2006. (Cité enpage 33.)
H. K. Joo, Y. R. Lee, G. Kang, S. Choi, C. S. Kim, S. Ryoo, J. B. Park, and B. H. Jeon. The 18-kDa Translocator Protein Inhibits Vascular Cell Adhesion Molecule-1 Expression via Inhibition ofMitochondrial Reactive Oxygen Species. Mol. Cells, 38(12) :1064–1070, Dec 2015. (Cité en page 36.)
E. Joseph-Liauzun, P. Delmas, D. Shire, and P. Ferrara. Topological analysis of the peripheral benzodia-zepine receptor in yeast mitochondrial membranes supports a five-transmembrane structure. J. Biol.Chem., 273(4) :2146–2152, Jan 1998. (Cité en page 45.)
P. Kapranov, S. M. Routt, V. A. Bankaitis, F. J. de Bruijn, and K. Szczyglowski. Nodule-specificregulation of phosphatidylinositol transfer protein expression in Lotus japonicus. Plant Cell, 13(6) :1369–1382, Jun 2001. (Cité en page 247.)
Marcus Karlstetter, Caroline Nothdurfter, Alexander Aslanidis, Katharina Moeller, Felicitas Horn, Re-becca Scholz, Harald Neumann, Bernhard H F Weber, Rainer Rupprecht, and Thomas Langmann.Translocator protein (18 kDa) (TSPO) is expressed in reactive retinal microglia and modulates micro-glial inflammation and phagocytosis. Journal of Neuroinflammation, 11(1) :3, 2014. (Cité en page 38.)
M. A. Kearns, D. E. Monks, M. Fang, M. P. Rivas, P. D. Courtney, J. Chen, G. D. Prestwich, A. B.Theibert, R. E. Dewey, and V. A. Bankaitis. Novel developmentally regulated phosphoinositide bindingproteins from soybean whose expression bypasses the requirement for an essential phosphatidylinositoltransfer protein in yeast. EMBO J., 17(14) :4004–4017, Jul 1998. (Cité en page 247.)
K. W. Kinnally, D. B. Zorov, Y. N. Antonenko, S. H. Snyder, M. W. McEnery, and H. Tedeschi. Mi-tochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions ofligands. Proc. Natl. Acad. Sci. U.S.A., 90(4) :1374–1378, Feb 1993. (Cité en page 33.)
I. R. Kleckner and M. P. Foster. An introduction to NMR-based approaches for measuring proteindynamics. Biochim. Biophys. Acta, 1814(8) :942–968, Aug 2011. (Cité en pages 128 et 271.)
M. M. Klepsch, J. O. Persson, and J. W. de Gier. Consequences of the overexpression of a eukaryoticmembrane protein, the human KDEL receptor, in Escherichia coli. J. Mol. Biol., 407(4) :532–542,Apr 2011. (Cité en page 212.)
K. D. Kloepper, D. H. Zhou, Y. Li, K. A. Winter, J. M. George, and C. M. Rienstra. Temperature-dependent sensitivity enhancement of solid-state NMR spectra of alpha-synuclein fibrils. J. Biomol.NMR, 39(3) :197–211, Nov 2007. (Cité en page 192.)
J. Klubo-Gwiezdzinska, K. Jensen, A. Bauer, A. Patel, J. Costello, K. D. Burman, L. Wartofsky, M. J.Hardwick, and V. V. Vasko. The expression of translocator protein in human thyroid cancer and itsrole in the response of thyroid cancer cells to oxidative stress. J. Endocrinol., 214(2) :207–216, Aug2012. (Cité en page 36.)

332 Bibliographie
Alla Korepanova, Fei P. Gao, Yuanzi Hua, Huajun Qin, Robert K. Nakamoto, and Timothy A. Cross.Cloning and expression of multiple integral membrane proteins from mycobacterium tuberculosis inescherichia coli. Protein Science, 14(1) :148–158, jan 2009. (Cité en page 209.)
V. M. Korkhov, C. Sachse, J. M. Short, and C. G. Tate. Three-dimensional structure of TspO by electroncryomicroscopy of helical crystals. Structure, 18(6) :677–687, Jun 2010. (Cité en pages 46 et 49.)
E. Kosonowska, K. Janeczko, and Z. Setkowicz. Inflammation induced at different developmental stagesaffects differently the range of microglial reactivity and the course of seizures evoked in the adult rat.Epilepsy Behav, 49 :66–70, Aug 2015. (Cité en page 39.)
J. Kota and P. O. Ljungdahl. Specialized membrane-localized chaperones prevent aggregation of polytopicproteins in the ER. J. Cell Biol., 168(1) :79–88, Jan 2005. (Cité en page 212.)
J. A. Kreps, Y. Wu, H. S. Chang, T. Zhu, X. Wang, and J. F. Harper. Transcriptome changes forArabidopsis in response to salt, osmotic, and cold stress. Plant Physiol., 130(4) :2129–2141, Dec 2002.(Cité en page 57.)
R. C. Krieg, S. Fickweiler, O. S. Wolfbeis, and R. Knuechel. Cell-type specific protoporphyrin IXmetabolism in human bladder cancer in vitro. Photochem. Photobiol., 72(2) :226–233, Aug 2000.(Cité en page 23.)
J. E. Krochko, G. D. Abrams, M. K. Loewen, S. R. Abrams, and A. J. Cutler. (+)-Abscisic acid 8’-hydroxylase is a cytochrome P450 monooxygenase. Plant Physiol., 118(3) :849–860, Nov 1998. (Citéen page 65.)
K. E. Krueger, A. G. Mukhin, L. Antkiewicz-Michaluk, M. R. Santi, D. R. Grayson, A. Guidotti, R. Spren-gel, P. Werner, and P. H. Seeburg. Purification, cloning, and expression of a peripheral-type benzo-diazepine receptor. Adv. Biochem. Psychopharmacol., 46 :1–13, 1990. (Cité en pages 14 et 26.)
A. Kuderova, E. Nanak, M. Truksa, and B. Brzobohaty. Use of rifampicin in T7 RNA polymerase-drivenexpression of a plant enzyme : rifampicin improves yield and assembly. Protein Expr. Purif., 16(3) :405–409, Aug 1999. (Cité en page 209.)
A. C. Kuhlmann and T. R. Guilarte. Cellular and subcellular localization of peripheral benzodiazepinereceptors after trimethyltin neurotoxicity. J. Neurochem., 74(4) :1694–1704, Apr 2000. (Cité enpages 13, 14 et 38.)
T. G. Kutateladze. Translation of the phosphoinositide code by PI effectors. Nat. Chem. Biol., 6(7) :507–513, Jul 2010. (Cité en pages 221 et 246.)
J. Kyte and R. F. Doolittle. A simple method for displaying the hydropathic character of a protein. J.Mol. Biol., 157(1) :105–132, May 1982. (Cité en page 64.)
J. J. Lacapere, M. P. Gingold, P. Champeil, and F. Guillain. Sarcoplasmic reticulum ATPase phos-phorylation from inorganic phosphate in the absence of a calcium gradient. Steady state and kineticfluorescence studies. J. Biol. Chem., 256(5) :2302–2306, Mar 1981. (Cité en page 163.)
J. J. Lacapere, F. Delavoie, H. Li, G. Peranzi, J. Maccario, V. Papadopoulos, and B. Vidic. Structuraland functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res.Commun., 284(2) :536–541, Jun 2001. (Cité en pages 21, 25, 26, 46, 123, 172, 174, 175 et 255.)
Jean-Jacques Lacapère and Vassilios Papadopoulos. Peripheral-type benzodiazepine receptor : structureand function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids, 68(7-8) :569–585, sep 2003. (Cité en page 28.)
Jean-Jacques Lacapere, Soria Iatmanen-Harbi, Lucile Senicourt, Olivier Lequin, Piotr Tekely, Rudra N.Purusottam, Petra Hellwig, Sebastien Kriegel, Stephanie Ravaud, Céline Juillan-Binard, Eva PebayPeyroula, and Vassilios Papadopoulos. Structural studies of TSPO, a mitochondrial membrane protein.

Bibliographie 333
In Membrane Proteins Production for Structural Analysis, pages 393–421. Springer Science BusinessMedia, 2014. (Cité en pages 79, 123, 157, 159, 164, 167, 172 et 253.)
P. Lacor, P. Gandolfo, M. C. Tonon, E. Brault, I. Dalibert, M. Schumacher, J. Benavides, and B. Ferzaz.Regulation of the expression of peripheral benzodiazepine receptors and their endogenous ligandsduring rat sciatic nerve degeneration and regeneration : a role for PBR in neurosteroidogenesis. BrainRes., 815(1) :70–80, Jan 1999. (Cité en page 42.)
J. C. Larcher, J. L. Vayssiere, F. J. Le Marquer, L. R. Cordeau, P. E. Keane, A. Bachy, F. Gros, andB. P. Croizat. Effects of peripheral benzodiazepines upon the O2 consumption of neuroblastoma cells.Eur. J. Pharmacol., 161(2-3) :197–202, Feb 1989. (Cité en page 34.)
G. Le Fur, F. Guilloux, P. Rufat, J. Benavides, A. Uzan, C. Renault, M. C. Dubroeucq, and C. Gue-remy. Peripheral benzodiazepine binding sites : effect of PK 11195, 1-(2-chlorophenyl)-N-methyl-(1-methylpropyl)-3 isoquinolinecarboxamide. II. In vivo studies. Life Sci., 32(16) :1849–1856, Apr 1983a.(Cité en pages 15 et 17.)
G. Le Fur, N. Vaucher, M. L. Perrier, A. Flamier, J. Benavides, C. Renault, M. C. Dubroeucq, C. Gue-remy, and A. Uzan. Differentiation between two ligands for peripheral benzodiazepine binding sites,[3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci., 33(5) :449–457, Aug 1983b.(Cité en pages 15 et 17.)
E. Le Rumeur, Y. Fichou, S. Pottier, F. Gaboriau, C. Rondeau-Mouro, M. Vincent, J. Gallay, andA. Bondon. Interaction of dystrophin rod domain with membrane phospholipids. Evidence of a closeproximity between tryptophan residues and lipids. J. Biol. Chem., 278(8) :5993–6001, Feb 2003. (Citéen page 118.)
A. G. Lee. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta, 1666(1-2) :62–87, Nov 2004. (Cité en page 21.)
Karen M. Lee, Elliot J. Androphy, and James D. Baleja. A novel method for selective isotope labeling ofbacterially expressed proteins. Journal of Biomolecular NMR, 5(1) :93–96, 1995. (Cité en page 209.)
Mikko T. Lehtonen, Eeva M. Marttinen, Motomu Akita, and Jari P.T. Valkonen. Fungi infecting culti-vated moss can also cause diseases in crop plants. Ann Appl Biol, 160(3) :298–307, mar 2012. (Citéen page 68.)
M. A. Lemmon. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol., 9(2) :99–111, Feb 2008. (Cité en pages 220 et 221.)
Charlène Leneveu-Jenvrin. Virulence et adaptation à l’environnement de souches de Pseudomonas fluo-rescens et Pseudomonas mosselii ; Contribution à l’étude des conditions d’expression et des fonctionsde la protéine TSPO chez P. fluorescens. PhD thesis, Université de Rouen, 2014. (Cité en page 32.)
E. Leonelli, J. G. Yague, M. Ballabio, I. Azcoitia, V. Magnaghi, M. Schumacher, L. M. Garcia-Segura,and R. C. Melcangi. Ro5-4864, a synthetic ligand of peripheral benzodiazepine receptor, reducesaging-associated myelin degeneration in the sciatic nerve of male rats. Mech. Ageing Dev., 126(11) :1159–1163, Nov 2005. (Cité en page 42.)
O. Lesouhaitier, M. K. Kodjo, F. Cartier, V. Contesse, L. Yon, C. Delarue, and H. Vaudry. The effectof the endozepine triakontatetraneuropeptide on corticosteroid secretion by the frog adrenal glandis mediated by activation of adenylyl cyclase and calcium influx through T-type calcium channels.Endocrinology, 141(1) :197–207, Jan 2000. (Cité en page 19.)
F. Li, J. Liu, Y. Zheng, R. M. Garavito, and S. Ferguson-Miller. Crystal structures of translocatorprotein (TSPO) and mutant mimic of a human polymorphism. Science, 347(6221) :555–558, jan 2015.(Cité en pages 24, 76, 81, 175, 253, 255 et 257.)

334 Bibliographie
Fei Li, Yan Xia, Jens Meiler, and Shelagh Ferguson-Miller. Characterization and modeling of the oli-gomeric state and ligand binding behavior of purified translocator protein 18 kDa from rhodobactersphaeroides. Biochemistry, 52(34) :5884–5899, aug 2013. (Cité en pages 48, 49 et 163.)
Fei Li, Jian Liu, Nan Liu, Leslie A. Kuhn, R. Michael Garavito, and Shelagh Ferguson-Miller. Translo-cator protein 18 kDa (TSPO) : An old protein with new functions ? Biochemistry, 55(20) :2821–2831,may 2016. (Cité en pages 78, 79, 80, 81 et 256.)
H. Li and V. Papadopoulos. Peripheral-type benzodiazepine receptor function in cholesterol transport.Identification of a putative cholesterol recognition/interaction amino acid sequence and consensuspattern. Endocrinology, 139(12) :4991–4997, Dec 1998. (Cité en pages 21, 47, 49, 88 et 149.)
D. Lin, Y. J. Chang, J. F. Strauss, and W. L. Miller. The human peripheral benzodiazepine receptorgene : cloning and characterization of alternative splicing in normal tissues and in a patient withcongenital lipoid adrenal hyperplasia. Genomics, 18(3) :643–650, Dec 1993. (Cité en page 14.)
P. Lindemann, A. Koch, B. Degenhardt, G. Hause, B. Grimm, and V. Papadopoulos. A novel Arabidopsisthaliana protein is a functional peripheral-type benzodiazepine receptor. Plant Cell Physiol., 45(6) :723–733, Jun 2004. (Cité en pages 10, 11, 57, 62 et 261.)
D. Linke. Detergents : an overview. Meth. Enzymol., 463 :603–617, 2009. (Cité en page 125.)
J. Liu, M. B. Rone, and V. Papadopoulos. Protein-protein interactions mediate mitochondrial cholesteroltransport and steroid biosynthesis. J. Biol. Chem., 281(50) :38879–38893, Dec 2006. (Cité en pages 26et 28.)
Beatriz Maestro and Jesús M. Sanz. Accumulation of partly folded states in the equilibrium unfoldingof the pneumococcal choline-binding module c-LytA. Biochem. J., 387(2) :479–488, apr 2005. (Citéen page 172.)
P. J. Marangos, J. Patel, J. P. Boulenger, and R. Clark-Rosenberg. Characterization of peripheral-typebenzodiazepine binding sites in brain using [3H]Ro 5-4864. Mol. Pharmacol., 22(1) :26–32, Jul 1982.(Cité en pages 12 et 13.)
Francesca M. Marassi, Bibhuti B. Das, George J. Lu, Henry J. Nothnagel, Sang Ho Park, Woo Sung Son,Ye Tian, and Stanley J. Opella. Structure determination of membrane proteins in five easy pieces.Methods, 55(4) :363–369, dec 2011. (Cité en pages 82 et 186.)
L. Margulis. Archaeal-eubacterial mergers in the origin of Eukarya : phylogenetic classification of life.Proc. Natl. Acad. Sci. U.S.A., 93(3) :1071–1076, Feb 1996. (Cité en page 10.)
J. Marley, M. Lu, and C. Bracken. A method for efficient isotopic labeling of recombinant proteins. J.Biomol. NMR, 20(1) :71–75, May 2001. (Cité en pages 96 et 151.)
Joseph A. Marsh, Vinay K. Singh, Zongchao Jia, and Julie D. Forman-Kay. Sensitivity of secondarystructure propensities to sequence differences between - and -synuclein : Implications for fibrillation.Protein Sci., 15(12) :2795–2804, dec 2006. (Cité en page 233.)
F. A. Marston and D. L. Hartley. Solubilization of protein aggregates. Meth. Enzymol., 182 :264–276,1990. (Cité en page 152.)
C. Mazurika, L. Veenman, R. Weizman, M. Bidder, S. Leschiner, I. Golani, I. Spanier, G. Weisinger, andM. Gavish. Estradiol modulates uterine 18 kDa translocator protein gene expression in uterus andkidney of rats. Mol. Cell. Endocrinol., 307(1-2) :43–49, Aug 2009. (Cité en page 26.)
M. W. McEnery, A. M. Snowman, R. R. Trifiletti, and S. H. Snyder. Isolation of the mitochondrial benzo-diazepine receptor : association with the voltage-dependent anion channel and the adenine nucleotidecarrier. Proc. Natl. Acad. Sci. U.S.A., 89(8) :3170–3174, Apr 1992. (Cité en page 32.)

Bibliographie 335
S. McLaughlin and D. Murray. Plasma membrane phosphoinositide organization by protein electrostatics.Nature, 438(7068) :605–611, Dec 2005. (Cité en page 220.)
R. C. Melcangi, S. Giatti, D. Calabrese, M. Pesaresi, G. Cermenati, N. Mitro, B. Viviani, L. M. Garcia-Segura, and D. Caruso. Levels and actions of progesterone and its metabolites in the nervous systemduring physiological and pathological conditions. Prog. Neurobiol., 113 :56–69, Feb 2014. (Cité enpage 40.)
M. C. Mendonca-Torres and S. S. Roberts. The translocator protein (TSPO) ligand PK11195 inducesapoptosis and cell cycle arrest and sensitizes to chemotherapy treatment in pre- and post-relapseneuroblastoma cell lines. Cancer Biol. Ther., 14(4) :319–326, Apr 2013. (Cité en page 34.)
A. Messina, S. Reina, F. Guarino, and V. De Pinto. VDAC isoforms in mammals. Biochim. Biophys.Acta, 1818(6) :1466–1476, Jun 2012. (Cité en page 30.)
H. Miettinen, J. Kononen, H. Haapasalo, P. Helen, P. Sallinen, T. Harjuntausta, H. Helin, and H. Alho.Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human as-trocytomas : relationship to cell proliferation. Cancer Res., 55(12) :2691–2695, Jun 1995. (Cité enpage 34.)
W. L. Miller. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol trans-porter. Biochim. Biophys. Acta, 1771(6) :663–676, Jun 2007. (Cité en page 21.)
C. Mills, M. Makwana, A. Wallace, S. Benn, H. Schmidt, I. Tegeder, M. Costigan, R. H. Brown, G. Rai-vich, and C. J. Woolf. Ro5-4864 promotes neonatal motor neuron survival and nerve regeneration inadult rats. Eur. J. Neurosci., 27(4) :937–946, Feb 2008. (Cité en page 42.)
C. D. Mills, J. L. Bitler, and C. J. Woolf. Role of the peripheral benzodiazepine receptor in sensoryneuron regeneration. Mol. Cell. Neurosci., 30(2) :228–237, Oct 2005. (Cité en page 42.)
Tsogbadrakh Mishig-Ochiriin, Chang-Hun Lee, Sun-Young Jeong, Beom-Jun Kim, Chang-Hoon Choi,Hyung-Soon Yim, and Sa-Ouk Kang. Calcium-induced conformational changes of the recombinantCBP3 protein from dictyostelium discoideum. Biochimica et Biophysica Acta (BBA) - Proteins andProteomics, 1748(2) :157–164, may 2005. (Cité en page 235.)
E. Miyazaki, Y. Kida, K. Mihara, and M. Sakaguchi. Switching the sorting mode of membrane proteinsfrom cotranslational endoplasmic reticulum targeting to posttranslational mitochondrial import. Mol.Biol. Cell, 16(4) :1788–1799, Apr 2005. (Cité en pages 64 et 65.)
N. Mochizuki, R. Tanaka, B. Grimm, T. Masuda, M. Moulin, A. G. Smith, A. Tanaka, and M. J. Terry.The cell biology of tetrapyrroles : a life and death struggle. Trends Plant Sci., 15(9) :488–498, Sep2010. (Cité en page 60.)
H. Mohler and T. Okada. Benzodiazepine receptor : demonstration in the central nervous system.Science, 198(4319) :849–851, Nov 1977. (Cité en page 12.)
C. R. Morcombe and K. W. Zilm. Chemical shift referencing in MAS solid state NMR. J. Magn. Reson.,162(2) :479–486, Jun 2003. (Cité en page 184.)
Kanako Morohaku, Susanne H. Pelton, Daniel J. Daugherty, W. Ronald Butler, Wenbin Deng, and VimalSelvaraj. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormonebiosynthesis. Endocrinology, 155(1) :89–97, 1 2014. ISSN 0013-7227. (Cité en pages 28 et 35.)
S. Mukherjee and S. K. Das. Subcellular distribution of "peripheral type" binding sites for [3H]Ro5-4864in guinea pig lung. Localization to the mitochondrial inner membrane. J. Biol. Chem., 264(28) :16713–16718, Oct 1989. (Cité en page 14.)
A. G. Mukhin, V. Papadopoulos, E. Costa, and K. E. Krueger. Mitochondrial benzodiazepine receptors

336 Bibliographie
regulate steroid biosynthesis. Proc. Natl. Acad. Sci. U.S.A., 86(24) :9813–9816, Dec 1989. (Cité enpage 26.)
S. Murail, J. C. Robert, Y. M. Coic, J. M. Neumann, M. A. Ostuni, Z. X. Yao, V. Papadopoulos,N. Jamin, and J. J. Lacapere. Secondary and tertiary structures of the transmembrane domains of thetranslocator protein TSPO determined by NMR. Stabilization of the TSPO tertiary fold upon ligandbinding. Biochim. Biophys. Acta, 1778(6) :1375–1381, Jun 2008. (Cité en pages 45, 46, 82 et 171.)
F. Nakazawa, C. Alev, M. Shin, Y. Nakaya, L. M. Jakt, and G. Sheng. PBRL, a putative peripheralbenzodiazepine receptor, in primitive erythropoiesis. Gene Expr. Patterns, 9(2) :114–121, Feb 2009.(Cité en page 54.)
N. Nguyen, E. Fakra, V. Pradel, E. Jouve, C. Alquier, M. E. Le Guern, J. Micallef, and O. Blin. Efficacy ofetifoxine compared to lorazepam monotherapy in the treatment of patients with adjustment disorderswith anxiety : a double-blind controlled study in general practice. Hum Psychopharmacol, 21(3) :139–149, Apr 2006. (Cité en page 18.)
U. Nowicka, M. Hoffman, L. Randles, X. Shi, L. Khavrutskii, K. Stefanisko, N. I. Tarasova, K. H. Darwin,and K. J. Walters. Mycobacterium tuberculosis copper-regulated protein SocB is an intrinsicallydisordered protein that folds upon interaction with a synthetic phospholipid bilayer. Proteins, 84(2) :193–200, Feb 2016. (Cité en page 235.)
Y. Nyathi, B. M. Wilkinson, and M. R. Pool. Co-translational targeting and translocation of proteinsto the endoplasmic reticulum. Biochim. Biophys. Acta, 1833(11) :2392–2402, Nov 2013. (Cité enpage 64.)
G. B. O’Beirne, M. J. Woods, and D. C. Williams. Two subcellular locations for peripheral-type ben-zodiazepine acceptors in rat liver. Eur. J. Biochem., 188(1) :131–138, Feb 1990. (Cité en pages 14et 23.)
Darragh P. O’Brien, Belen Hernandez, Dominique Durand, Véronique Hourdel, Ana-Cristina Sotomayor-Pérez, Patrice Vachette, Mahmoud Ghomi, Julia Chamot-Rooke, Daniel Ladant, Sébastien Brier, andAlexandre Chenal. Structural models of intrinsically disordered and calcium-bound folded states of aprotein adapted for secretion. Sci. Rep., 5 :14223, sep 2015. (Cité en page 235.)
J. I. Oh and S. Kaplan. Generalized approach to the regulation and integration of gene expression. Mol.Microbiol., 39(5) :1116–1123, Mar 2001. (Cité en page 69.)
Jeong-Il Oh and Samuel Kaplan. Generalized approach to the regulation and integration of gene expres-sion. Molecular Microbiology, 39(5) :1116–1123, feb 2004. (Cité en page 71.)
K. Ojemalm, S. C. Botelho, C. Studle, and G. von Heijne. Quantitative analysis of SecYEG-mediatedinsertion of transmembrane α − helicesintothebacterialinnermembrane. J. Mol. Biol., 425(15) :2813−−2822, Aug2013.(Citéenpage 212.)
B. O. Oke, C. A. Suarez-Quian, J. Riond, P. Ferrara, and V. Papadopoulos. Cell surface localizationof the peripheral-type benzodiazepine receptor (PBR) in adrenal cortex. Mol. Cell. Endocrinol., 87(1-3) :1–6, Sep 1992. (Cité en page 14.)
J. M. Olson, B. J. Ciliax, W. R. Mancini, and A. B. Young. Presence of peripheral-type benzodiazepinebinding sites on human erythrocyte membranes. Eur. J. Pharmacol., 152(1-2) :47–53, Jul 1988. (Citéen page 14.)
Stanley J. Opella. Structure determination of membrane proteins in their native phospholipid bilayerenvironment by rotationally aligned solid-state NMR spectroscopy. Accounts of Chemical Research,46(9) :2145–2153, sep 2013. (Cité en pages 186 et 187.)

Bibliographie 337
M. A. Ostuni, K. Marazova, G. Peranzi, B. Vidic, V. Papadopoulos, R. Ducroc, and J. J. Lacapere.Functional characterization and expression of PBR in rat gastric mucosa : stimulation of chloridesecretion by PBR ligands. Am. J. Physiol. Gastrointest. Liver Physiol., 286(6) :G1069–1080, Jun2004. (Cité en page 32.)
M. A. Ostuni, R. Ducroc, G. Peranzi, M. C. Tonon, V. Papadopoulos, and J. J. Lacapere. Translocatorprotein (18 kDa) ligand PK 11195 induces transient mitochondrial Ca2+ release leading to transepi-thelial Cl- secretion in HT-29 human colon cancer cells. Biol. Cell, 99(11) :639–647, Nov 2007. (Citéen page 16.)
M. A. Ostuni, O. R. Tumilasci, G. Peranzi, E. M. Cardoso, L. N. Contreras, A. L. Arregger, V. Papado-poulos, and J. J. Lacapere. Effect of translocator protein (18 kDa)-ligand binding on neurotransmitter-induced salivary secretion in rat submandibular glands. Biol. Cell, 100(7) :427–439, Jul 2008. (Citéen pages 13 et 16.)
M. A. Ostuni, G. Peranzi, R. A. Ducroc, M. Fasseu, B. Vidic, J. Dumont, V. Papadopoulos, and J. J.Lacapere. Distribution, pharmacological characterization and function of the 18 kDa translocatorprotein in rat small intestine. Biol. Cell, 101(10) :573–586, Oct 2009. (Cité en pages 163 et 174.)
M. A. Ostuni, S. Iatmanen, D. Teboul, J. C. Robert, and J. J. Lacapere. Characterization of membraneprotein preparations : measurement of detergent content and ligand binding after proteoliposomesreconstitution. Methods Mol. Biol., 654 :3–18, 2010a. (Cité en page 125.)
M. A. Ostuni, L. Issop, G. Peranzi, F. Walker, M. Fasseu, C. Elbim, V. Papadopoulos, and J. J. Laca-pere. Overexpression of translocator protein in inflammatory bowel disease : potential diagnostic andtreatment value. Inflamm. Bowel Dis., 16(9) :1476–1487, Sep 2010b. (Cité en pages 38, 123, 172, 174et 175.)
L. E. Otterbein, M. P. Soares, K. Yamashita, and F. H. Bach. Heme oxygenase-1 : unleashing theprotective properties of heme. Trends Immunol., 24(8) :449–455, Aug 2003. (Cité en page 66.)
D. R. Owen, A. J. Lewis, R. Reynolds, R. Rupprecht, D. Eser, M. R. Wilkins, I. Bennacef, D. J. Nutt, andC. A. Parker. Variation in binding affinity of the novel anxiolytic XBD173 for the 18 kDa translocatorprotein in human brain. Synapse, 65(3) :257–259, Mar 2011. (Cité en pages 18 et 25.)
D. R. Owen, Q. Guo, N. J. Kalk, A. Colasanti, D. Kalogiannopoulou, R. Dimber, Y. L. Lewis, V. Libri,J. Barletta, J. Ramada-Magalhaes, A. Kamalakaran, D. J. Nutt, J. Passchier, P. M. Matthews, R. N.Gunn, and E. A. Rabiner. Determination of [(11)C]PBR28 binding potential in vivo : a first humanTSPO blocking study. J. Cereb. Blood Flow Metab., 34(6) :989–994, Jun 2014. (Cité en pages 18, 25et 50.)
Harushige Ozaki, Sami S. Zoghbi, Jinsoo Hong, Ajay Verma, Victor W. Pike, Robert B. Innis, andMasahiro Fujita. In vivo binding of protoporphyrin IX to rat translocator protein imaged with positronemission tomography. Synapse, 64(8) :649–653, apr 2010. (Cité en page 31.)
I. Palmer and P. T. Wingfield. Preparation and extraction of insoluble (inclusion-body) proteins fromEscherichia coli. Curr Protoc Protein Sci, Chapter 6 :Unit6.3, Nov 2012. (Cité en page 103.)
Laura A. Palomares, Sandino Estrada-Moncada, and Octavio T. Ramírez. Recombinant Gene Expres-sion : Reviews and Protocols, chapter Production of Recombinant Proteins, pages 15–51. HumanaPress, Totowa, NJ, 2004. ISBN 978-1-59259-774-1. (Cité en page 211.)
Gilbert Di Paolo and Pietro De Camilli. Phosphoinositides in cell regulation and membrane dynamics.Nature, 443(7112) :651–657, oct 2006. (Cité en page 246.)
V. Papadopoulos. Identification and purification of a human Sertoli cell-secreted protein (hSCSP-80)

338 Bibliographie
stimulating Leydig cell steroid biosynthesis. J. Clin. Endocrinol. Metab., 72(6) :1332–1339, Jun 1991.(Cité en pages 16, 19 et 26.)
V. Papadopoulos and L. Lecanu. Translocator protein (18 kDa) TSPO : an emerging therapeutic targetin neurotrauma. Exp. Neurol., 219(1) :53–57, Sep 2009. (Cité en page 42.)
V. Papadopoulos, A. G. Mukhin, E. Costa, and K. E. Krueger. The peripheral-type benzodiazepinereceptor is functionally linked to Leydig cell steroidogenesis. J. Biol. Chem., 265(7) :3772–3779, Mar1990. (Cité en page 26.)
V. Papadopoulos, P. Guarneri, K. E. Kreuger, A. Guidotti, and E. Costa. Pregnenolone biosynthesis inC6-2B glioma cell mitochondria : regulation by a mitochondrial diazepam binding inhibitor receptor.Proc. Natl. Acad. Sci. U.S.A., 89(11) :5113–5117, Jun 1992. (Cité en pages 19, 25, 26 et 40.)
V. Papadopoulos, H. Amri, N. Boujrad, C. Cascio, M. Culty, M. Garnier, M. Hardwick, H. Li, B. Vidic,A. S. Brown, J. L. Reversa, J. M. Bernassau, and K. Drieu. Peripheral benzodiazepine receptor incholesterol transport and steroidogenesis. Steroids, 62(1) :21–28, Jan 1997. (Cité en pages 14, 26, 28et 29.)
V. Papadopoulos, A. Kapsis, H. Li, H. Amri, M. Hardwick, M. Culty, P. G. Kasprzyk, M. Carlson,J. P. Moreau, and K. Drieu. Drug-induced inhibition of the peripheral-type benzodiazepine receptorexpression and cell proliferation in human breast cancer cells. Anticancer Res., 20(5A) :2835–2847,2000. (Cité en page 34.)
V. Papadopoulos, M. Baraldi, T. R. Guilarte, T. B. Knudsen, J. J. Lacapere, P. Lindemann, M. D.Norenberg, D. Nutt, A. Weizman, M. R. Zhang, and M. Gavish. Translocator protein (18kDa) : newnomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecularfunction. Trends Pharmacol. Sci., 27(8) :402–409, Aug 2006a. (Cité en page 12.)
V. Papadopoulos, M. Baraldi, T. R. Guilarte, T. B. Knudsen, J. J. Lacapere, P. Lindemann, M. D.Norenberg, D. Nutt, A. Weizman, M. R. Zhang, and M. Gavish. Translocator protein (18kDa) : newnomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecularfunction. Trends Pharmacol. Sci., 27(8) :402–409, Aug 2006b. (Cité en page 16.)
V. Papadopoulos, L. Lecanu, R. C. Brown, Z. Han, and Z. X. Yao. Peripheral-type benzodiazepinereceptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience, 138(3) :749–756, 2006c. (Cité en pages 38 et 40.)
V. Papadopoulos, Y. Aghazadeh, J. Fan, E. Campioli, B. Zirkin, and A. Midzak. Translocator protein-mediated pharmacology of cholesterol transport and steroidogenesis. Mol. Cell. Endocrinol., 408 :90–98, Jun 2015. (Cité en pages 26, 27, 28 et 29.)
Sang Ho Park, Bibhuti B. Das, Anna A. De Angelis, Mario Scrima, and Stanley J. Opella. Mechanically,magnetically, and “rotationally aligned” membrane proteins in phospholipid bilayers give equivalentangular constraints for NMR structure determination. The Journal of Physical Chemistry B, 114(44) :13995–14003, nov 2010. (Cité en page 186.)
Sang Ho Park, Bibhuti B. Das, Fabio Casagrande, Ye Tian, Henry J. Nothnagel, Mignon Chu, HansKiefer, Klaus Maier, Anna A. De Angelis, Francesca M. Marassi, and Stanley J. Opella. Structure ofthe chemokine receptor CXCR1 in phospholipid bilayers. Nature, oct 2012. (Cité en pages 82 et 186.)
A. L. Parola, D. G. Stump, D. J. Pepperl, K. E. Krueger, J. W. Regan, and H. E. Laird. Cloning andexpression of a pharmacologically unique bovine peripheral-type benzodiazepine receptor isoquinolinebinding protein. J. Biol. Chem., 266(21) :14082–14087, Jul 1991. (Cité en page 14.)
Céline B. Parsy, Caroline J. Chapman, Antony C. Barnes, John F. Robertson, and Andrea Murray.Two-step method to isolate target recombinant protein from co-purified bacterial contaminant SlyD

Bibliographie 339
after immobilised metal affinity chromatography. Journal of Chromatography B, 853(1-2) :314–319,jun 2007. (Cité en page 205.)
A. H. Payne and D. B. Hales. Overview of steroidogenic enzymes in the pathway from cholesterol toactive steroid hormones. Endocr. Rev., 25(6) :947–970, Dec 2004. (Cité en page 25.)
Y. Perez, M. Maffei, A. Igea, I. Amata, M. Gairi, A. R. Nebreda, P. Bernado, and M. Pons. Lipid bindingby the Unique and SH3 domains of c-Src suggests a new regulatory mechanism. Sci Rep, 3 :1295, 2013.(Cité en page 235.)
Ashlee Perry, Michael P. Stypa, Brandon K. Tenn, and Kristin K. Kumashiro. Solid-state 13c NMRreveals effects of temperature and hydrationon elastin. Biophysical Journal, 82(2) :1086–1095, feb2002. (Cité en page 192.)
Scott E. Phillips, Patrick Vincent, Kellie E. Rizzieri, Gabriel Schaaf, Vytas A. Bankaitis, and Eric A.Gaucher. The diverse biological functions of phosphatidylinositol transfer proteins in eukaryotes.Critical Reviews in Biochemistry and Molecular Biology, 41(1) :21–49, jan 2006. (Cité en page 247.)
W Nicholson Price, Samuel K Handelman, John K Everett, Saichiu N Tong, Ana Bracic, Jon D Luff,Victor Naumov, Thomas Acton, Philip Manor, Rong Xiao, Burkhard Rost, Gaetano T Montelione,and John F Hunt. Large-scale experimental studies show unexpected amino acid effects on proteinexpression and solubility in vivo in e. coli. Microb Inform Exp, 1(1) :6, 2011. (Cité en page 212.)
Rudra.N. Purusottam, Lucile Sénicourt, Jean-Jacques Lacapère, and Piotr Tekely. Probing the gel toliquid-crystalline phase transition and relevant conformation changes in liposomes by 13c magic-anglespinning NMR spectroscopy. Biochimica et Biophysica Acta (BBA) - Biomembranes, 1848(12) :3134–3139, dec 2015. (Cité en pages 183, 185, 186 et 187.)
A. Raghavan, T. Sheiko, B. H. Graham, and W. J. Craigen. Voltage-dependant anion channels : novelinsights into isoform function through genetic models. Biochim. Biophys. Acta, 1818(6) :1477–1485,Jun 2012. (Cité en page 30.)
V. Ramensky. Human non-synonymous SNPs : server and survey. Nucleic Acids Research, 30(17) :3894–3900, sep 2002. (Cité en page 50.)
D. Rapaport. Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins.EMBO Rep., 4(10) :948–952, Oct 2003. (Cité en page 43.)
Arianna Rath, Mira Glibowicka, Vincent G. Nadeau, Gong Chen, and Charles M. Deber. Detergentbinding explains anomalous SDS-PAGE migration of membrane proteins. Proceedings of the NationalAcademy of Sciences, 106(6) :1760–1765, jan 2009. (Cité en page 151.)
G. Z. Reus, G. R. Fries, L. Stertz, M. Badawy, I. C. Passos, T. Barichello, F. Kapczinski, and J. Quevedo.The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders.Neuroscience, 300 :141–154, Aug 2015. (Cité en page 40.)
J. G. Richards and H. Mohler. Benzodiazepine receptors. Neuropharmacology, 23(2B) :233–242, Feb1984. (Cité en page 12.)
J. L. Rigaud. Membrane proteins : functional and structural studies using reconstituted proteoliposomesand 2-D crystals. Braz. J. Med. Biol. Res., 35(7) :753–766, Jul 2002. (Cité en page 122.)
J. L. Rigaud and D. Levy. Reconstitution of membrane proteins into liposomes. Meth. Enzymol., 372 :65–86, 2003. (Cité en page 123.)
J. L. Rigaud, G. Mosser, J. J. Lacapere, A. Olofsson, D. Levy, and J. L. Ranck. Bio-Beads : an efficientstrategy for two-dimensional crystallization of membrane proteins. J. Struct. Biol., 118(3) :226–235,Apr 1997. (Cité en page 123.)

340 Bibliographie
J. Riond, P. Leplatois, P. Laurent, G. Le Fur, D. Caput, G. Loison, and P. Ferrara. Expression andpharmacological characterization of the human peripheral-type benzodiazepine receptor in yeast. Eur.J. Pharmacol., 208(4) :307–312, Dec 1991. (Cité en pages 8 et 14.)
J. C. Robert and J. J. Lacapere. Bacterial overexpressed membrane proteins : an example : the TSPO.Methods Mol. Biol., 654 :29–45, 2010. (Cité en pages 152, 157 et 195.)
J. Roge and J. M. Betton. Use of pIVEX plasmids for protein overproduction in Escherichia coli. Microb.Cell Fact., 4 :18, Jun 2005. (Cité en page 218.)
M. B. Rone, J. Fan, and V. Papadopoulos. Cholesterol transport in steroid biosynthesis : role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta, 1791(7) :646–658, Jul2009a. (Cité en page 33.)
M. B. Rone, A. S. Midzak, L. Issop, G. Rammouz, S. Jagannathan, J. Fan, X. Ye, J. Blonder, T. Veenstra,and V. Papadopoulos. Identification of a dynamic mitochondrial protein complex driving cholesterolimport, trafficking, and metabolism to steroid hormones. Mol. Endocrinol., 26(11) :1868–1882, Nov2012. (Cité en pages 26 et 28.)
Malena B. Rone, Jun Liu, Josip Blonder, Xiaoying Ye, Timothy D. Veenstra, Jason C. Young, andVassilios Papadopoulos. Targeting and insertion of the cholesterol-binding translocator protein intothe outer mitochondrial membrane. Biochemistry, 48(29) :6909–6920, jul 2009b. (Cité en pages 33,42, 44, 56, 195 et 212.)
Germán L. Rosano and Eduardo A. Ceccarelli. Recombinant protein expression in escherichia coli :advances and challenges. Front. Microbiol., 5, apr 2014. (Cité en page 212.)
S. M. Routt and V. A. Bankaitis. Biological functions of phosphatidylinositol transfer proteins. Biochem.Cell Biol., 82(1) :254–262, Feb 2004. (Cité en pages 247 et 248.)
R. Rupprecht, G. Rammes, D. Eser, T. C. Baghai, C. Schule, C. Nothdurfter, T. Troxler, C. Gentsch,H. O. Kalkman, F. Chaperon, V. Uzunov, K. H. McAllister, V. Bertaina-Anglade, C. D. La Rochelle,D. Tuerck, A. Floesser, B. Kiese, M. Schumacher, R. Landgraf, F. Holsboer, and K. Kucher. Translo-cator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science, 325(5939) :490–493, Jul 2009. (Cité en pages 18 et 40.)
Rainer Rupprecht, Vassilios Papadopoulos, Gerhard Rammes, Thomas C. Baghai, Jinjiang Fan, NagarajuAkula, Ghislaine Groyer, David Adams, and Michael Schumacher. Translocator protein (18 kDa)(TSPO) as a therapeutic target for neurological and psychiatric disorders. Nature Reviews DrugDiscovery, 9(12) :971–988, dec 2010. (Cité en pages 40, 48 et 49.)
Jae K. Ryu, Hyun B. Choi, and James G. McLarnon. Peripheral benzodiazepine receptor ligand PK11195reduces microglial activation and neuronal death in quinolinic acid-injected rat striatum. Neurobiologyof Disease, 20(2) :550–561, nov 2005. (Cité en page 38.)
S. Sagan and G. Bolbach. Analyse des protéines membranaires par spectrométrie de masse : toujours unamour impossible ? Spectra Analyse, 270 :19–21, 2009. (Cité en page 157.)
S. R. Salama, A. E. Cleves, D. E. Malehorn, E. A. Whitters, and V. A. Bankaitis. Cloning and charac-terization of Kluyveromyces lactis SEC14, a gene whose product stimulates Golgi secretory functionin Saccharomyces cerevisiae. J. Bacteriol., 172(8) :4510–4521, Aug 1990. (Cité en page 247.)
A. M. Scarf, C. Luus, E. Da Pozzo, S. Selleri, C. Guarino, C. Martini, L. M. Ittner, and M. Kassiou.Evidence for complex binding profiles and species differences at the translocator protein (TSPO) (18kDa). Curr. Mol. Med., 12(4) :488–493, May 2012. (Cité en page 15.)
K. Schroder-Tittmann, E. Bosse-Doenecke, S. Reedtz-Runge, C. Ihling, A. Sinz, K. Tittmann, and R. Ru-

Bibliographie 341
dolph. Recombinant expression, in vitro refolding, and biophysical characterization of the humanglucagon-like peptide-1 receptor. Biochemistry, 49(36) :7956–7965, Sep 2010. (Cité en page 163.)
A. Schuitemaker, M. A. Kropholler, R. Boellaard, W. M. van der Flier, R. W. Kloet, T. F. van der Doef,D. L. Knol, A. D. Windhorst, G. Luurtsema, F. Barkhof, C. Jonker, A. A. Lammertsma, P. Scheltens,and B. N. van Berckel. Microglial activation in Alzheimer’s disease : an (R)-11-C]PK11195 positronemission tomography study. Neurobiol. Aging, 34(1) :128–136, Jan 2013. (Cité en page 18.)
C. Schule, C. Nothdurfter, and R. Rupprecht. The role of allopregnanolone in depression and anxiety.Prog. Neurobiol., 113 :79–87, Feb 2014. (Cité en page 40.)
Luigi Monsù Scolaro, Mariangela Castriciano, Andrea Romeo, Salvatore Patanè, Eugenio Cefalì, andMaria Allegrini. Aggregation behavior of protoporphyrin IX in aqueous solutions : clear evidenceof vesicle formation. The Journal of Physical Chemistry B, 106(10) :2453–2459, mar 2002. (Cité enpage 133.)
M. Seki, M. Narusaka, J. Ishida, T. Nanjo, M. Fujita, Y. Oono, A. Kamiya, M. Nakajima, A. Enju, T. Sa-kurai, M. Satou, K. Akiyama, T. Taji, K. Yamaguchi-Shinozaki, P. Carninci, J. Kawai, Y. Hayashizaki,and K. Shinozaki. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, coldand high-salinity stresses using a full-length cDNA microarray. Plant J., 31(3) :279–292, Aug 2002.(Cité en page 57.)
M. Selitrennik and S. Lev. The role of phosphatidylinositol-transfer proteins at membrane contact sites.Biochemical Society Transactions, 44(2) :419–424, apr 2016. (Cité en pages 247 et 248.)
V. Selvaraj and D. M. Stocco. The changing landscape in translocator protein (TSPO) function. TrendsEndocrinol. Metab., 26(7) :341–348, Jul 2015. (Cité en page 33.)
M. S. Seneviratne, D. Faccenda, V. De Biase, and M. Campanella. PK11195 inhibits mitophagy targetingthe F1Fo-ATPsynthase in Bcl-2 knock-down cells. Curr. Mol. Med., 12(4) :476–482, May 2012. (Citéen page 33.)
M. Serra, P. Madau, M. F. Chessa, M. Caddeo, E. Sanna, G. Trapani, M. Franco, G. Liso, R. H. Purdy,M. L. Barbaccia, and G. Biggio. 2-Phenyl-imidazo[1,2-a]pyridine derivatives as ligands for peripheralbenzodiazepine receptors : stimulation of neurosteroid synthesis and anticonflict action in rats. Br. J.Pharmacol., 127(1) :177–187, May 1999. (Cité en page 40.)
E. Setiawan, A. A. Wilson, R. Mizrahi, P. M. Rusjan, L. Miler, G. Rajkowska, I. Suridjan, J. L. Kennedy,P. V. Rekkas, S. Houle, and J. H. Meyer. Role of translocator protein density, a marker of neuroin-flammation, in the brain during major depressive episodes. JAMA Psychiatry, 72(3) :268–275, Mar2015. (Cité en page 15.)
F. Shah, S. P. Hume, V. W. Pike, S. Ashworth, and J. McDermott. Synthesis of the enantiomers of[N-methyl-11C]PK 11195 and comparison of their behaviours as radioligands for PK binding sites inrats. Nucl. Med. Biol., 21(4) :573–581, May 1994. (Cité en page 18.)
S. Shao and R. S. Hegde. Membrane protein insertion at the endoplasmic reticulum. Annu. Rev. CellDev. Biol., 27 :25–56, 2011. (Cité en page 64.)
J. Sileikyte, V. Petronilli, A. Zulian, F. Dabbeni-Sala, G. Tognon, P. Nikolov, P. Bernardi, and F. Ric-chelli. Regulation of the inner membrane mitochondrial permeability transition by the outer mem-brane translocator protein (peripheral benzodiazepine receptor). J. Biol. Chem., 286(2) :1046–1053,Jan 2011. (Cité en page 33.)
J. Sileikyte, E. Blachly-Dyson, R. Sewell, A. Carpi, R. Menabo, F. Di Lisa, F. Ricchelli, P. Bernardi,and M. Forte. Regulation of the mitochondrial permeability transition pore by the outer membrane

342 Bibliographie
does not involve the peripheral benzodiazepine receptor (Translocator Protein of 18 kDa (TSPO)). J.Biol. Chem., 289(20) :13769–13781, May 2014. (Cité en page 33.)
Mathilde Laetitia Audrey Simon, Matthieu Pierre Platre, Sonia Assil, Ringo van Wijk, William YaweiChen, Joanne Chory, Marlène Dreux, Teun Munnik, and Yvon Jaillais. A multi-colour/multi-affinitymarker set to visualize phosphoinositide dynamics in arabidopsis. The Plant Journal, 77(2) :322–337,dec 2013. (Cité en pages 221 et 222.)
S. J. Singer and G. L. Nicolson. The fluid mosaic model of the structure of cell membranes. Science, 175(4023) :720–731, Feb 1972. (Cité en page 187.)
P. A. Singleton and L. Y. Bourguignon. CD44 interaction with ankyrin and IP3 receptor in lipid raftspromotes hyaluronan-mediated Ca2+ signaling leading to nitric oxide production and endothelial celladhesion and proliferation. Exp. Cell Res., 295(1) :102–118, Apr 2004. (Cité en page 34.)
P. Skolnick, P. J. Marangos, P. Syapin, F. K. Goodwin, and S. M. Paul. CNS benzodiazepine receptors :physiological studies and putative endogenous ligands. Pharmacol. Biochem. Behav., 10(5) :815–823,May 1979. (Cité en page 16.)
M. Souidi, S. Dubrac, M. Parquet, D. H. Volle, J. M. Lobaccaro, D. Mathe, O. Combes, P. Scanff,C. Lutton, and J. Aigueperse. [Oxysterols : metabolism, biological role and associated diseases].Gastroenterol. Clin. Biol., 28(3) :279–293, Mar 2004. (Cité en page 23.)
J. F. Soustiel, M. Zaaroor, E. Vlodavsky, L. Veenman, A. Weizman, and M. Gavish. Neuroprotectiveeffect of Ro5-4864 following brain injury. Exp. Neurol., 214(2) :201–208, Dec 2008. (Cité en page 42.)
R. Sprengel, P. Werner, P. H. Seeburg, A. G. Mukhin, M. R. Santi, D. R. Grayson, A. Guidotti, andK. E. Krueger. Molecular cloning and expression of cDNA encoding a peripheral-type benzodiazepinereceptor. J. Biol. Chem., 264(34) :20415–20421, Dec 1989. (Cité en page 14.)
Robert V. Stahelin, Jordan L. Scott, and Cary T. Frick. Cellular and molecular interactions of phos-phoinositides and peripheral proteins. Chemistry and Physics of Lipids, 182 :3–18, sep 2014. (Cité enpage 246.)
P. E. Stoebner, P. Carayon, P. Casellas, M. Portier, T. Lavabre-Bertrand, P. Cuq, J. P. Cano, J. Mey-nadier, and L. Meunier. Transient protection by peripheral benzodiazepine receptors during the earlyevents of ultraviolet light-induced apoptosis. Cell Death Differ., 8(7) :747–753, Jul 2001. (Cité enpage 36.)
S. Tabor and C. C. Richardson. A bacteriophage T7 RNA polymerase/promoter system for controlledexclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A., 82(4) :1074–1078, Feb 1985. (Citéen page 209.)
S. Taketani, H. Kohno, M. Okuda, T. Furukawa, and R. Tokunaga. Induction of peripheral-type ben-zodiazepine receptors during differentiation of mouse erythroleukemia cells. A possible involvement ofthese receptors in heme biosynthesis. J. Biol. Chem., 269(10) :7527–7531, Mar 1994. (Cité en page 31.)
S. Taketani, H. Kohno, T. Furukawa, and R. Tokunaga. Involvement of peripheral-type benzodiazepinereceptors in the intracellular transport of heme and porphyrins. J. Biochem., 117(4) :875–880, Apr1995a. (Cité en page 25.)
S. Taketani, H. Kohno, T. Furukawa, and R. Tokunaga. Involvement of peripheral-type benzodiazepinereceptors in the intracellular transport of heme and porphyrins. J. Biochem., 117(4) :875–880, Apr1995b. (Cité en pages 24 et 31.)
J. P. Taskinen, D. M. van Aalten, J. Knudsen, and R. K. Wierenga. High resolution crystal structuresof unliganded and liganded human liver ACBP reveal a new mode of binding for the acyl-CoA ligand.Proteins, 66(1) :229–238, Jan 2007. (Cité en page 20.)

Bibliographie 343
D. Teboul, S. Beaufils, J. C. Taveau, S. Iatmanen-Harbi, A. Renault, C. Venien-Bryan, V. Vie, and J. J.Lacapere. Mouse TSPO in a lipid environment interacting with a functionalized monolayer. Biochim.Biophys. Acta, 1818(11) :2791–2800, Nov 2012. (Cité en page 123.)
Julie M Thole and Erik Nielsen. Phosphoinositides in plants : novel functions in membrane trafficking.Current Opinion in Plant Biology, 11(6) :620–631, dec 2008. (Cité en page 247.)
J. D. Thompson, D. G. Higgins, and T. J. Gibson. CLUSTAL W : improving the sensitivity of progressivemultiple sequence alignment through sequence weighting, position-specific gap penalties and weightmatrix choice. Nucleic Acids Res., 22(22) :4673–4680, Nov 1994. (Cité en pages 9, 58 et 261.)
Alexandre Toulmay and William A Prinz. Lipid transfer and signaling at organelle contact sites : thetip of the iceberg. Current Opinion in Cell Biology, 23(4) :458–463, aug 2011. (Cité en page 249.)
L. N. Tu, K. Morohaku, P. R. Manna, S. H. Pelton, W. R. Butler, D. M. Stocco, and V. Selvaraj.Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with noeffects on steroid hormone biosynthesis. J. Biol. Chem., 289(40) :27444–27454, Oct 2014. (Cité enpages 28, 35 et 42.)
L. N. Tu, A. H. Zhao, M. Hussein, D. M. Stocco, and V. Selvaraj. Translocator Protein (TSPO) AffectsMitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology, 157(3) :1110–1121, Mar2016. (Cité en page 35.)
Vladimir N. Uversky and Sonia Longhi, editors. Instrumental Analysis of Intrinsically Disordered Pro-teins. Wiley-Blackwell, mar 2010. (Cité en page 172.)
Marco van de Weert. Fluorescence quenching to study protein-ligand binding : Common errors. JFluoresc, 20(2) :625–629, dec 2009. (Cité en page 164.)
Gerrit van Meer, Dennis R. Voelker, and Gerald W. Feigenson. Membrane lipids : where they are andhow they behave. Nature Reviews Molecular Cell Biology, 9(2) :112–124, feb 2008. (Cité en page 220.)
C. Vanhee and H. Batoko. Arabidopsis TSPO and porphyrins metabolism : a transient signaling connec-tion ? Plant Signal Behav, 6(9) :1383–1385, Sep 2011. (Cité en pages 65 et 67.)
C. Vanhee, S. Guillon, D. Masquelier, H. Degand, M. Deleu, P. Morsomme, and H. Batoko. A TSPO-related protein localizes to the early secretory pathway in arabidopsis, but is targeted to mitochondriawhen expressed in yeast. Journal of Experimental Botany, 62(2) :497–508, sep 2010. (Cité en pages 64et 65.)
C. Vanhee, S. Guillon, D. Masquelier, H. Degand, M. Deleu, P. Morsomme, and H. Batoko. A TSPO-related protein localizes to the early secretory pathway in Arabidopsis, but is targeted to mitochondriawhen expressed in yeast. J. Exp. Bot., 62(2) :497–508, Jan 2011a. (Cité en pages 57 et 62.)
C. Vanhee, S. Guillon, D. Masquelier, H. Degand, M. Deleu, P. Morsomme, and H. Batoko. A TSPO-related protein localizes to the early secretory pathway in Arabidopsis, but is targeted to mitochondriawhen expressed in yeast. J. Exp. Bot., 62(2) :497–508, Jan 2011b. (Cité en pages 57 et 63.)
C. Vanhee, G. Zapotoczny, D. Masquelier, M. Ghislain, and H. Batoko. The Arabidopsis multistressregulator TSPO is a heme binding membrane protein and a potential scavenger of porphyrins viaan autophagy-dependent degradation mechanism. Plant Cell, 23(2) :785–805, Feb 2011c. (Cité enpages 60, 62, 65, 67 et 256.)
L. Veenman and M. Gavish. The peripheral-type benzodiazepine receptor and the cardiovascular system.Implications for drug development. Pharmacol. Ther., 110(3) :503–524, Jun 2006. (Cité en page 34.)
L. Veenman and M. Gavish. The role of 18 kDa mitochondrial translocator protein (TSPO) in program-med cell death, and effects of steroids on TSPO expression. Curr. Mol. Med., 12(4) :398–412, May2012. (Cité en page 26.)

344 Bibliographie
L. Veenman, Y. Shandalov, and M. Gavish. VDAC activation by the 18 kDa translocator protein (TSPO),implications for apoptosis. J. Bioenerg. Biomembr., 40(3) :199–205, Jun 2008. (Cité en page 36.)
L. Veenman, A. Vainshtein, N. Yasin, M. Azrad, and M. Gavish. Tetrapyrroles as Endogenous TSPOLigands in Eukaryotes and Prokaryotes : Comparisons with Synthetic Ligands. Int J Mol Sci, 17(6),2016. (Cité en page 256.)
Leo Veenman and Moshe Gavish. Peripheral-type benzodiazepine receptors : Their implication in braindisease. Drug Dev. Res., 50(3-4) :355–370, 2000. (Cité en pages 38 et 40.)
S. Veiga, I. Azcoitia, and L. M. Garcia-Segura. Ro5-4864, a peripheral benzodiazepine receptor ligand,reduces reactive gliosis and protects hippocampal hilar neurons from kainic acid excitotoxicity. J.Neurosci. Res., 80(1) :129–137, Apr 2005. (Cité en pages 38 et 42.)
Sergio Veiga, Paloma Carrero, Olga Pernia, Iñigo Azcoitia, and Luis Miguel Garcia-Segura. Translocatorprotein (18 kDa) is involved in the regulation of reactive gliosis. Glia, 55(14) :1426–1436, 2007. (Citéen page 38.)
S. Venneti, B. J. Lopresti, and C. A. Wiley. The peripheral benzodiazepine receptor (Translocatorprotein 18kDa) in microglia : from pathology to imaging. Prog. Neurobiol., 80(6) :308–322, Dec 2006.(Cité en pages 34 et 40.)
I. Venturini, M. L. Zeneroli, L. Corsi, R. Avallone, F. Farina, H. Alho, C. Baraldi, C. Ferrarese, N. Pe-cora, M. Frigo, G. Ardizzone, A. Arrigo, R. Pellicci, and M. Baraldi. Up-regulation of peripheralbenzodiazepine receptor system in hepatocellular carcinoma. Life Sci., 63(14) :1269–1280, 1998. (Citéen page 34.)
M. Verleye, Y. Akwa, P. Liere, N. Ladurelle, A. Pianos, B. Eychenne, M. Schumacher, and J. M. Gillardin.The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroidlevels in rat brain. Pharmacol. Biochem. Behav., 82(4) :712–720, Dec 2005. (Cité en page 40.)
A. Verma and S. H. Snyder. Peripheral type benzodiazepine receptors. Annu. Rev. Pharmacol. Toxicol.,29 :307–322, 1989. (Cité en page 12.)
A. Verma, J. S. Nye, and S. H. Snyder. Porphyrins are endogenous ligands for the mitochondrial(peripheral-type) benzodiazepine receptor. Proc. Natl. Acad. Sci. U.S.A., 84(8) :2256–2260, Apr 1987.(Cité en pages 23, 25 et 31.)
A. R. Viguera and L. Serrano. Experimental analysis of the Schellman motif. J. Mol. Biol., 251(1) :150–160, Aug 1995. (Cité en page 43.)
S. Wagner, L. Baars, A. J. Ytterberg, A. Klussmeier, C. S. Wagner, O. Nord, P. A. Nygren, K. J. vanWijk, and J. W. de Gier. Consequences of membrane protein overexpression in Escherichia coli. Mol.Cell Proteomics, 6(9) :1527–1550, Sep 2007. (Cité en page 212.)
D. M. Walther and D. Rapaport. Biogenesis of mitochondrial outer membrane proteins. Biochim.Biophys. Acta, 1793(1) :42–51, Jan 2009. (Cité en page 43.)
LiGuo Wang and LiGe Tonggu. Membrane protein reconstitution for functional and structural studies.Science China Life Sciences, 58(1) :66–74, jan 2015. (Cité en pages 18 et 122.)
M. Wang, X. Wang, L. Zhao, W. Ma, I. R. Rodriguez, R. N. Fariss, and W. T. Wong. Macroglia-microglia interactions via TSPO signaling regulates microglial activation in the mouse retina. Journalof Neuroscience, 34(10) :3793–3806, mar 2014. (Cité en page 38.)
Markus Weingarth, Dan E. Demco, Geoffrey Bodenhausen, and Piotr Tekely. Improved magnetizationtransfer in solid-state NMR with fast magic angle spinning. Chemical Physics Letters, 469(4-6) :342–348, feb 2009. (Cité en page 194.)

Bibliographie 345
G. Wendler, P. Lindemann, J. J. Lacapere, and V. Papadopoulos. Protoporphyrin IX binding andtransport by recombinant mouse PBR. Biochem. Biophys. Res. Commun., 311(4) :847–852, Nov 2003.(Cité en page 31.)
J. A. Whiles, R. Deems, R. R. Vold, and E. A. Dennis. Bicelles in structure-function studies of membrane-associated proteins. Bioorg. Chem., 30(6) :431–442, Dec 2002. (Cité en page 235.)
T. Wiegand, C. Gardiennet, F. Ravotti, A. Bazin, B. Kunert, D. Lacabanne, R. Cadalbert, P. Guntert,L. Terradot, A. Bockmann, and B. H. Meier. Solid-state NMR sequential assignments of the N-terminaldomain of HpDnaB helicase. Biomol NMR Assign, 10(1) :13–23, Apr 2016. (Cité en page 191.)
M. P. Williamson. Using chemical shift perturbation to characterise ligand binding. Prog Nucl MagnReson Spectrosc, 73 :1–16, Aug 2013. (Cité en pages 237 et 242.)
D. S. Wishart and B. D. Sykes. The 13C chemical-shift index : a simple method for the identificationof protein secondary structure using 13C chemical-shift data. J. Biomol. NMR, 4(2) :171–180, Mar1994a. (Cité en pages 231 et 233.)
David S Wishart and Brian D Sykes. [12] chemical shifts as a tool for structure determination. InMethods in Enzymology, pages 363–392. Elsevier BV, 1994b. (Cité en page 231.)
L. H. Wong and T. P. Levine. Lipid transfer proteins do their thing anchored at membrane contactsitesâ ? but what is their thing ? Biochem. Soc. Trans., 44(2) :517–527, Apr 2016. (Cité en page 249.)
M. J. Woods and D. C. Williams. Multiple forms and locations for the peripheral-type benzodiazepinereceptor. Biochem. Pharmacol., 52(12) :1805–1814, Dec 1996. (Cité en pages 13 et 14.)
G. Wright and V. Reichenbecher. The effects of superoxide and the peripheral benzodiazepine receptorligands on the mitochondrial processing of manganese-dependent superoxide dismutase. Exp. CellRes., 246(2) :443–450, Feb 1999. (Cité en page 36.)
L. Xiong and J. K. Zhu. Regulation of abscisic acid biosynthesis. Plant Physiol., 133(1) :29–36, Sep2003. (Cité en page 65.)
K. Yanagibashi, Y. Ohno, N. Nakamichi, T. Matsui, K. Hayashida, M. Takamura, K. Yamada, S. Tou, andM. Kawamura. Peripheral-type benzodiazepine receptors are involved in the regulation of cholesterolside chain cleavage in adrenocortical mitochondria. J. Biochem., 106(6) :1026–1029, Dec 1989. (Citéen page 26.)
A. A. Yeliseev and S. Kaplan. A sensory transducer homologous to the mammalian peripheral-type benzo-diazepine receptor regulates photosynthetic membrane complex formation in Rhodobacter sphaeroides2.4.1. J. Biol. Chem., 270(36) :21167–21175, Sep 1995. (Cité en pages 11, 69, 70 et 71.)
A. A. Yeliseev and S. Kaplan. A novel mechanism for the regulation of photosynthesis gene expressionby the TspO outer membrane protein of Rhodobacter sphaeroides 2.4.1. J. Biol. Chem., 274(30) :21234–21243, Jul 1999. (Cité en pages 12, 32 et 256.)
A. A. Yeliseev and S. Kaplan. TspO of rhodobacter sphaeroides. A structural and functional modelfor the mammalian peripheral benzodiazepine receptor. J. Biol. Chem., 275(8) :5657–5667, Feb 2000.(Cité en page 12.)
A. A. Yeliseev, K. E. Krueger, and S. Kaplan. A mammalian mitochondrial drug receptor functions asa bacterial "oxygen" sensor. Proc. Natl. Acad. Sci. U.S.A., 94(10) :5101–5106, May 1997. (Cité enpages 8 et 11.)
L. Yin, V. Dragnea, and C. E. Bauer. PpsR, a regulator of heme and bacteriochlorophyll biosynthesis,is a heme-sensing protein. J. Biol. Chem., 287(17) :13850–13858, Apr 2012. (Cité en page 70.)

346 Bibliographie
L. Yin, V. Dragnea, G. Feldman, L. A. Hammad, J. A. Karty, C. E. Dann, and C. E. Bauer. Redox andlight control the heme-sensing activity of AppA. MBio, 4(5) :e00563–00513, 2013. (Cité en page 70.)
T. Yoshida, Y. Fujita, H. Sayama, S. Kidokoro, K. Maruyama, J. Mizoi, K. Shinozaki, and K. Yamaguchi-Shinozaki. AREB1, AREB2, and ABF3 are master transcription factors that cooperatively regulateABRE-dependent ABA signaling involved in drought stress tolerance and require ABA for full acti-vation. Plant J., 61(4) :672–685, Feb 2010. (Cité en page 59.)
Y. Yoshioka, J. Sasaki, M. Yamamoto, K. Saitoh, S. Nakaya, and M. Kubokawa. Quantitation by (1)H-NMR of dolichol, cholesterol and choline-containing lipids in extracts of normal and phathologicalthyroid tissue. NMR Biomed, 13(7) :377–383, Nov 2000. (Cité en page 33.)
H. Yu, Y. Liu, D. R. Gulbranson, A. Paine, S. S. Rathore, and J. Shen. Extended synaptotagmins areCa2+-dependent lipid transfer proteins at membrane contact sites. Proc. Natl. Acad. Sci. U.S.A., 113(16) :4362–4367, Apr 2016. (Cité en page 248.)
Davide Zannoni. Aerobic and anaerobic electron transport chains in anoxygenic phototrophic bacteria.In Advances in Photosynthesis and Respiration, pages 949–971. Springer Science Business Media,1995. (Cité en page 70.)
J. H. Zeilstra-Ryalls and S. Kaplan. Role of the fnrL gene in photosystem gene expression and photo-synthetic growth of Rhodobacter sphaeroides 2.4.1. J. Bacteriol., 180(6) :1496–1503, Mar 1998. (Citéen pages 69 et 71.)
X. Zeng and S. Kaplan. TspO as a modulator of the repressor/antirepressor (PpsR/AppA) regulatorysystem in rhodobacter sphaeroides 2.4.1. Journal of Bacteriology, 183(21) :6355–6364, nov 2001. (Citéen pages 69 et 71.)
S. Zeno, M. Zaaroor, S. Leschiner, L. Veenman, and M. Gavish. CoCl(2) induces apoptosis via the 18kDa translocator protein in U118MG human glioblastoma cells. Biochemistry, 48(21) :4652–4661, Jun2009. (Cité en page 33.)
S. Zeno, L. Veenman, Y. Katz, J. Bode, M. Gavish, and M. Zaaroor. The 18 kDa mitochondrial trans-locator protein (TSPO) prevents accumulation of protoporphyrin IX. Involvement of reactive oxygenspecies (ROS). Curr. Mol. Med., 12(4) :494–501, May 2012. (Cité en pages 31, 36 et 256.)
Li-Ming Zhang, Zhi-Kun Qiu, Nan Zhao, Hong-Xia Chen, Yan-Qin Liu, Jiang-Ping Xu, You-Zhi Zhang,Ri-Fang Yang, and Yun-Feng Li. Anxiolytic-like effects of YL-IPA08, a potent ligand for the translo-cator protein (18 kDa) in animal models of post-traumatic stress disorder. The International Journalof Neuropsychopharmacology, 17(10) :1659–1669, apr 2014. (Cité en pages 18 et 40.)
Amy H. Zhao, Lan N. Tu, Chinatsu Mukai, Madhu P. Sirivelu, Viju V. Pillai, Kanako Morohaku, RoyCohen, and Vimal Selvaraj. Mitochondrial translocator protein (TSPO) function is not essential forheme biosynthesis. Journal of Biological Chemistry, 291(4) :1591–1603, dec 2015. (Cité en pages 31,34 et 36.)
D. M. Zisterer and D. C. Williams. Peripheral-type benzodiazepine receptors. Gen. Pharmacol., 29(3) :305–314, Sep 1997. (Cité en pages 13, 16 et 17.)

Bibliographie 347

Résumé :Les TSPO forment une famille ancienne et hautement conservée à travers l’évolution de protéines à
5 domaines transmembranaires, que l’on retrouve aussi bien chez les animaux que les plantes, ou encoreles bactéries.
La TSPO animale (ou TSPO1), la plus étudiée des TSPO à ce jour, se trouve majoritairement dansles tissus stéroïdiens où sa fonction précise est controversée. Chez certaines espèces animales, il existeune isoforme, la TSPO2, localisée dans la membrane plasmique des globules rouges alors que la TSPO1est mitochondriale. La fonction de la TSPO2 n’est pas bien caractérisée. La TSPO végétale, localiséedans le réticulum endoplasmique, possède une extension N-terminale que n’ont pas les TSPO animale etbactérienne. Elle semble être impliquée dans la régulation du stress, tout comme la TSPO bactérienne.
Les études structure/fonction des différentes TSPO réalisées dans ce travail ont nécessité leur pro-duction par voie recombinante car elles sont naturellement peu abondantes.
Nous avons produit la TSPO1 murine marquée 15N,13C par surexpression dans la bactérie E. coli. Puisla protéine purifiée en détergent (SDS et DPC) a été étudiée par différentes techniques (CD, fluorescence,RMN). L’ajout du ligand spécifique de la TSPO1, le PK11195, stabilise une conformation en DPC cequi a permis en 2014 la résolution de sa structure, par RMN du liquide, par une équipe allemande. Afind’étudier la TSPO1 dans un environnement plus proche de sa membrane native, nous l’avons reconstituéedans des liposomes DMPC/DMPE et étudiée par RMN du solide. Les 1ers résultats sont encourageantset ouvrent une nouvelle approche expérimentale pour la détermination de sa structure en présence et,plus particulièrement, en absence de ligand.
La surexpression de la TSPO2 humaine dans la bactérie E. coli s’est avérée difficile et nous avonsdû la réalisée par système acellulaire (cell-free). Les quantités obtenues par cette méthode permettentd’envisager le développement futur des études des relations structure/fonction.
La production et la purification du Nter de la TSPO d’A. thaliana marqué 15N,13C ont permis ladétermination de sa structure par RMN du liquide. Son interaction avec des lipides chargés mise enévidence par les études RMN, suggère une nouvelle fonction de l’AtTSPO dans le trafic lipidique.
Mots clés : TSPO1, TSPO2, AtTSPO1, Protéine Membranaire, Structure, RMN
Abstract :TSPO are five-transmembrane domain proteins that form a protein family highly conserved throu-
ghout evolution and that are found in animals as well as in plants and bacteria.Animal TSPO (referred to as TSPO1), the most studied TSPO, is highly expressed in tissues involved
in steroid biosynthesis where its precise role remains controversial. In some animal species the presenceof a less characterized TSPO isoform, TSPO2, has been reported. TSPO2 was found to be located inthe plasma membrane of red blood cells whereas TSPO1 is located in mitochondrial outer membrane.Plant TSPO, which is located in the endoplasmic reticulum, possesses an N-terminal extension that isabsent in bacterial and animal TSPO. This TSPO, along with the bacterial TSPO, seems to be involvedin stress regulation.
The structural and functional studies of TSPO proteins conducted in this work required their pro-duction through recombinant expression because they are naturally non-abundant proteins.
We made use of E. coli to produce the recombinant 15N,13C-labelled mouse TSPO1. Then the proteinpurified in detergent was studied through several methods (CD, fluorescence, NMR).
High-affinity binding of PK11195 to TSPO1 stabilizes a conformation in DPC, which made possiblethe structure determination of the protein in solution by NMR by a German team. We have incorporatedTSPO1 into DMPC/DPME liposomes in order to provide a native-like environment and we then studiedit by solid-state NMR. Preliminary results are encouraging and open up a new approach for TSPO1structure determination in presence or in absence of ligand.
Human TSPO2 overexpression in E. coli proved to be difficult and we therefore use the cell-freemethod. The amounts we obtained by this method allows us to consider future developments of structure-function relationship studies.
Production and purification of 13C,15N labelled N-terminal of A. thaliana TSPO have made it possibleto determine its structure by liquid state NMR. Interaction of this peptide with charged lipids revealedby NMR, suggests a new fonction of AtTSPO in lipid trafficking.
Keywords : TSPO1, TSPO2, AtTSPO1, Membrane Protein, Structure, NMR.






























