Embed Size (px)
Citation preview

Chapter One - Introduction
2
1.1- Background In our previous work (1) different phases of (vanadium- phosphorus-
oxygen) (VPO) catalyst were prepared, characterized and evaluated by
catalytic test in order to produce Maleic anhydride (MA) via selective
oxidation of n-butane.
Maleic anhydride (MA), and its derivatives malic acid and fumaric
acid, are produced with a worldwide capacity of about (2.8 X 10 6) metric
tons per year. While the market demand for (MA), increased from
(0.616 X 10 6) metric tons in 2000 to almost (1.27 X 10 6) metric tons in
2004(2-4).
These multifunctional chemical intermediates have found applications
in almost any field of industrial chemistry. The principal use of (MA) is in
manufacturing of unsaturated polyester resins (UPR) ( 63 %), lubricating
oil additives (10 %) , copolymers (9 %) ,alkenyl succinic anhydride (5 %) ,
malic acid (3 %) ,fumaric acid (2 %) agricultural chemicals (1%),
miscellaneous, including reactive plasticizers , sulfosuccinic acid esters ,
and alkyd resins (% 7). Furthermore, due to its double bond and anhydride
function, (MA) is a versatile intermediate for the production of co-
polymers of (MA), for example, ethylene glycol and vinyl monomer.
Recently, potential new uses of (MA) have been found in its conversion to
(1-4) butanediol and the manufacturing of tetrahydrofuran (THF) and
butyrolactone via hydrogenation (5).
Maleic anhydride (MA) and the two di-acid isomers were first prepared
in the 1830's, but it took about 100 years before commercial manufacture
was performed in 1933. The National Aniline and Chemical Company Inc.
started a process for the production of (MA) based on benzene oxidation
using a vanadium oxide catalyst. Smaller amounts of maleic acid were also
formed as by product in the production of phthalic anhydride. The use of

Chapter One - Introduction
3
benzene as a feedstock for the production of (MA) was dominant in the
world until the late 1980's (6).
Currently, worldwide production of (MA) is based on the major feed
stocks benzene, butenes and n-butane (1-3,6-14). Most of the capacity is
produced via fixed–bed oxidation of benzene, though benzene is being
displaced by butane as a feed stock (all production in United States is
butane based) (1-3,6-14), because butane is a lower cost and environmentally
more desirable raw material and because butane oxidation produces a
clean product stream, forming mainly (MA) and carbon oxides as shown
below:
C4H10 + 3.5 O2 C4H2O3 + 4 H2O ∆ H = - 1236 KJ/ mole (1.1)
C4H10 + 4.5 O2 4 CO + 5 H2O ∆ H = - 1521 KJ/ mole (1.2)
C4H10 + 6.5 O2 4 CO2 + 5 H2O ∆ H = - 2656 KJ/ mole (1.3)
It is obvious that CO and CO2 are thermodynamically more favored
products. Only kinetic control by a catalyst will enhance the formation of
(MA). In practice, the process is operating at a yield of approximately
(60%) to the desired product. CO and CO2 are the sole carbon containing
by–products in a ratio of about unity. Suppression of the unselective and
very exothermic oxidation to carbon oxides requires sufficient heat transfer
capacities of the reactor. Nonetheless, hot spots are frequently met in (MA)
production plants.
Processes for the production of MA From n-Butane
In general, three different types of process can be distinguished in
commercial production of (MA) from n-butane; fixed-bed processes

Chapter One - Introduction
4
(Scheme 1.1), fluidized–bed processes (Scheme 1.2) and the re-circulating-
solids process (Scheme 1.3).
The fixed-bed reactor consists of a number of tubes that are packed
with coarse catalyst bodies. The reactants flow through these tubes. As a
result of the obstruction of the gas flow by the catalyst bodies, a pressure
drop across the bed is exhibited. Therefore, pressure has to be applied at
the inlet to ensure an adequate flow rate. The magnitude of the pressure
drop is depending on the flow rate, the length of the catalyst bed and the
size to the catalyst bodies. Since the selective oxidation of n-butane to
(MA) is highly exothermic, fixed-bed reactors must containing a facility to
remove the reaction heat. This can be done in various ways: the bed can be
split into different sections, with provision for cooling the gas between the
sections ,or using a reactor containing a large number of tubes, along
which a cooling gas or the liquid is recirculated. However, hot spots can
occur easily in fixed-bed reactors. These can be prevented by using larger
catalyst bodies, a less active catalyst, or by dilution of the catalyst with an
inert solid (support). In view of the explosion limits and the flammability
of mixtures of n-butane and air, only low concentrations of n- butane can
be applied (2 - 4 %).Furthermore, the gases must be mixed and pre-heated
before entering the reaction zone. In a fixed-bed reactor ,the concentration
of n-butane will decrease when moving to the end of the tube .To maintain
a sufficiently high selectivity at the exit of the reactor a less active catalyst
is installed at the entrance and a very active catalyst at the end of the
reactor (4,5).
A single passed fixed - bed reactor was used in our previous work (1) ,
(1 m long and 0.019 m in diameter) mounted in four heating zones vertical
furnace. The flow of n-butane was controlled before entering the reactor
using gasometer. The total gas flow after leaving the reactor was also
measured and fixed at industrial conditions (space velocity 2000 h -1).The

Chapter One - Introduction
5
gas mixture (1.5 Vol. % n-butane in air ) was preheated before passing on
the catalyst using inert alumina granules in the upper half of the reactor .
The lower half of the reactor (0.1 L) was filled with (2.3 mm) granules by
(4) temperature indicators controllers (TIC).Maleic produced was passed
through scrubber and was dissolved in water to produce Maleic acid .this
acid was titrated with (0.01 N) NaOH using phenolphthalein as indicator.
Scheme 1.1: Huntsman fixed-bed reactor for MA production (4).
In a fluidized-bed process, reaction gases flow upward through a bed
of catalyst particles .When the force of the gas flow on the catalyst bed is
equal to the weight of the bed, the catalyst bed expands significantly and
the catalyst bodies are brought in continuous motion. Because of this
motion ,better heat transfer characteristics are established and ,hence, hot
spots cannot occur in a fluidized-bed reactor comprise the fact that reaction
gases can be used without pre-mixing and pre-heating before entering the
reactor. Furthermore, higher n-butane concentrations can be used due to a
decreased explosion risk compared to the fixed-bed process (4). The flow
diagram for fluidized-bed (Scheme 1.2) process is comparable to that of
the fixed-bed process (Scheme 1.1).Several different ways have been

Chapter One - Introduction
6
developed to produce bulk (VPO) catalyst particles of the right size and the
attrition resistance for fluidized-bed purposes (16) .When the catalyst bodies
are too small, they will be blown out of the reactor .Large bodies, on the
other hand, call for extremely high linear gas flow rates in the reactor,
therefore, the particle size of fluidized-bed catalysts usually ranges from
(10-150 microns). Development of fluid-bed butane system has been
reported (17,18).These systems offer the advantage of operation at higher
butane concentration (up to 4%) and thus lower original costs. However,
less selectivity at the higher butane loadings and the intermediate problems
with attrition of (VPO) materials need to be overcome before this process
becomes economically preferred (17,18).
Scheme 1.2: ALMA Fluidized bed reactor for MA (4).
DuPont commercially operates the third type of process in their
recirculation-solids reactor (17,18).In this case, oxidation of n-butane and
regeneration of the catalyst are carried out in two separate reaction zones
(Scheme 1.3) .The selectivity to (MA) is increased, because the oxidation
of n-butane is carried out in absence of oxygen (19,20) . In the first step, n-
butane reacts with lattice oxygen from the catalyst. In this stage, the

Chapter One - Introduction
7
catalyst is reduced by n-butane resulting in the selective formation of MA,
which is removed in a stripper. Regeneration of the reduced catalyst with
oxygen takes place in the second reactor zone. In principle, the oxidation
state of the catalyst can be controlled optimally in this way (17,18).This
process operates at lower conversion per pass and with higher selectivities
than normally encountered in fixed or fluid bed systems. It also uses a
unique attrition resistance system (19-21).
Scheme 1.3: DUPONT re-circulating – solids process for the production of THF
from n-butane via MA (4).
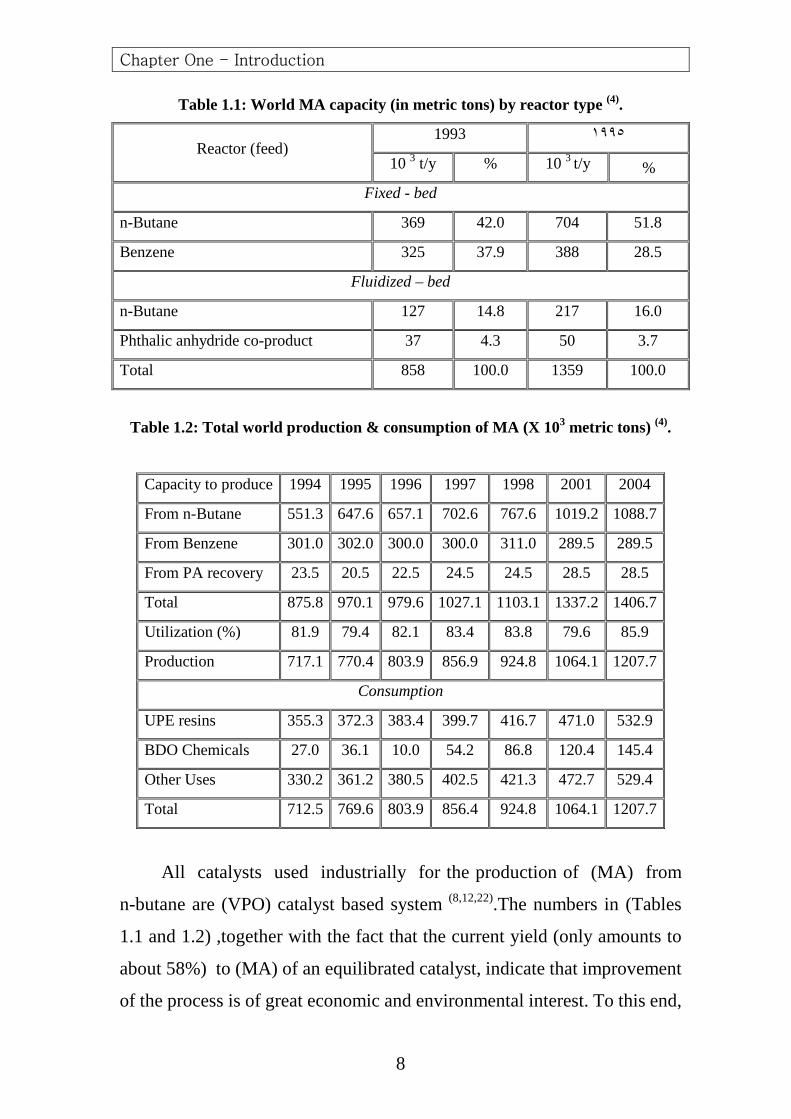
Table 1.1 shows the 1993 and 1995 world-wide (MA) production
capacity for the different processes (4).As can be seen from the table, both
fixed-bed and fluidized-bed butane based processes have been growing at
the expense of benzene-based processes. Furthermore, it seems that
production of (MA) with fluidized –bed technology would not surpass
production with fixed-bed technology which was still operated at higher
yields to (MA). Table 1.2 represents the long – term (MA) production –
consumption balance (4) .The average of the annual demand growth which
was estimated to be about (4%). By the year 2004, total world –wide
consumption will be increased by about (76.5%) as compared to 1998.
Chapter One - Introduction
8
Table 1.1: World MA capacity (in metric tons) by reactor type (4).
Reactor (feed) 1993 ١٩٩٥
10 3 t/y % 10 3 t/y %
Fixed - bed
n-Butane 369 42.0 704 51.8
Benzene 325 37.9 388 28.5
Fluidized – bed
n-Butane 127 14.8 217 16.0
Phthalic anhydride co-product 37 4.3 50 3.7
Total 858 100.0 1359 100.0
Table 1.2: Total world production & consumption of MA (X 103 metric tons) (4).
Capacity to produce 1994 1995 1996 1997 1998 2001 2004
From n-Butane 551.3 647.6 657.1 702.6 767.6 1019.2 1088.7
From Benzene 301.0 302.0 300.0 300.0 311.0 289.5 289.5
From PA recovery 23.5 20.5 22.5 24.5 24.5 28.5 28.5
Total 875.8 970.1 979.6 1027.1 1103.1 1337.2 1406.7
Utilization (%) 81.9 79.4 82.1 83.4 83.8 79.6 85.9
Production 717.1 770.4 803.9 856.9 924.8 1064.1 1207.7
Consumption
UPE resins 355.3 372.3 383.4 399.7 416.7 471.0 532.9
BDO Chemicals 27.0 36.1 10.0 54.2 86.8 120.4 145.4
Other Uses 330.2 361.2 380.5 402.5 421.3 472.7 529.4
Total 712.5 769.6 803.9 856.4 924.8 1064.1 1207.7
All catalysts used industrially for the production of (MA) from
n-butane are (VPO) catalyst based system (8,12,22).The numbers in (Tables
1.1 and 1.2) ,together with the fact that the current yield (only amounts to
about 58%) to (MA) of an equilibrated catalyst, indicate that improvement
of the process is of great economic and environmental interest. To this end,

Chapter One - Introduction
9
several developments can be considered. First, the bulk (VPO) catalyst
should exhibit a higher attrition resistance (mechanical strength) in order to
be more suitable for fluidized-bed process. Secondly, the activation period
for the catalyst should be shortened. This will result in an earlier
achievement of optimum performance. However, the catalyst formulation
could be changed; resulting in better properties and an improved catalytic
performance. Furthermore, there is also a comprehensive demand for a
cheaper and more reproducible preparation procedure for the currently
applied (VPO) catalyst. In any case, these improvements can never be
achieved without thoroughly investigations of the catalytic and structural
properties of the active (VPO) phase.
1.2- Scope of the Literature Survey
The n-butane to (MA) reaction is a fascinating complex system. This
catalytic system performance a (14-electron) oxidation involving the
abstraction of (8 hydrogen atoms) and insertion of (3 oxygen atoms) as
described in (eq. 1.4) (21).
CH3
CH3
+ 3.5 O2
O
O
O + 4 H2O ... (1.4)
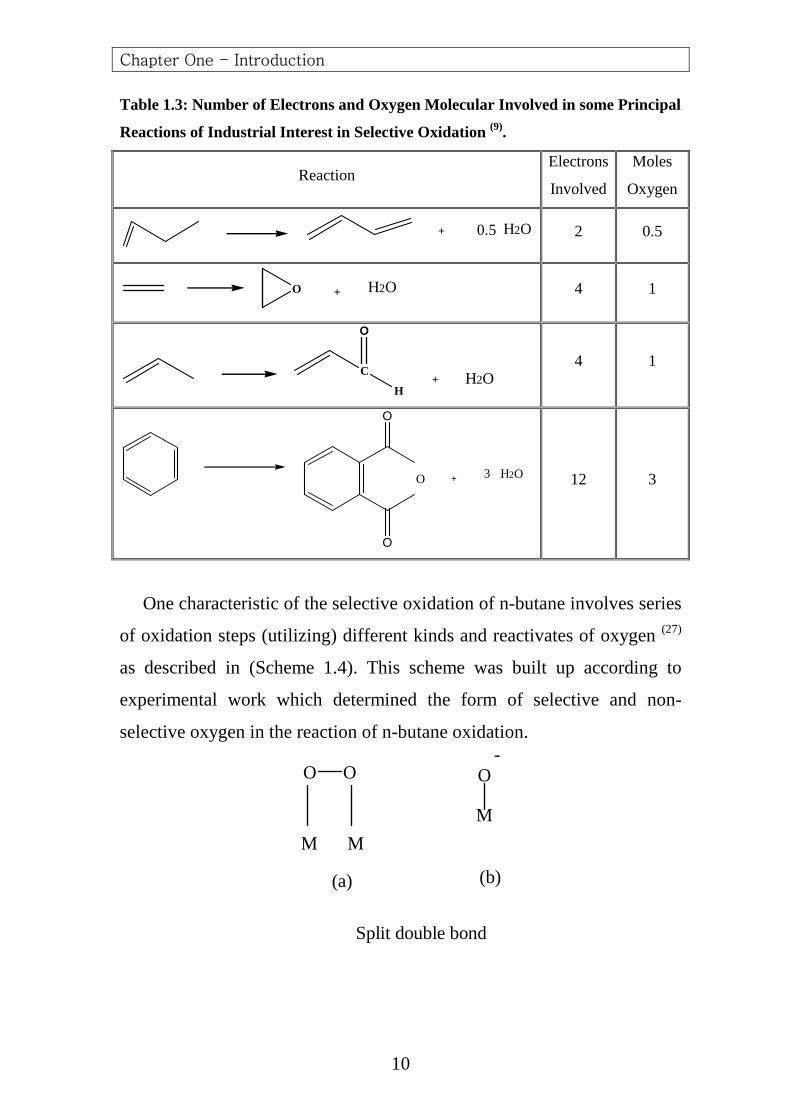
Compared to other industrially practiced hydrocarbon selective
oxidation reactions (23-26), it is the most complex one (Table 1.3). It is the
only example of an industrially practiced selective oxidation reaction
involving alkane activation. Knowledge gained through study and
understanding of this system may contribute to advances in alkane
activation in general.
Chapter One - Introduction
10
Table 1.3: Number of Electrons and Oxygen Molecular Involved in some Principal
Reactions of Industrial Interest in Selective Oxidation (9).
Reaction Electrons
Involved
Moles
Oxygen
+ 0.5 H2O
2 0.5
O + H2O
4 1
C
O
H+ H2O
4 1
O
+ 3 H2O
O
O
12 3
One characteristic of the selective oxidation of n-butane involves series
of oxidation steps (utilizing) different kinds and reactivates of oxygen (27)
as described in (Scheme 1.4). This scheme was built up according to
experimental work which determined the form of selective and non-
selective oxygen in the reaction of n-butane oxidation.
O O
M M
(a)
M
O-
(b)
Split double bond

Chapter One - Introduction
11
O M O
O
O
O O
O M O
(c) (d) (e)
O M O
OO
Radical Pi adsorbed
Scheme 1.4: Kinds of oxygen adsorbed species (28)
1.3 -Structure of the catalyst
1.3.1- Crystalline (VPO) phases
The (VPO) system is characterized by the facile formation of a number
of crystalline phases of vanadium (III), vanadium (IV) and of vanadium
(V).In the more interesting (P/V) ratio near (1.0), the different crystalline
(α, β) (29-35), polymorphic (α1, α11, γ, δ) ( 29,35-37) or hydrated phases of
(VOPO4) (34-41) have been extensively studied. In general, the vanadyl
orthophosphate crystal structure consists of (VO6) and (PO4) groups
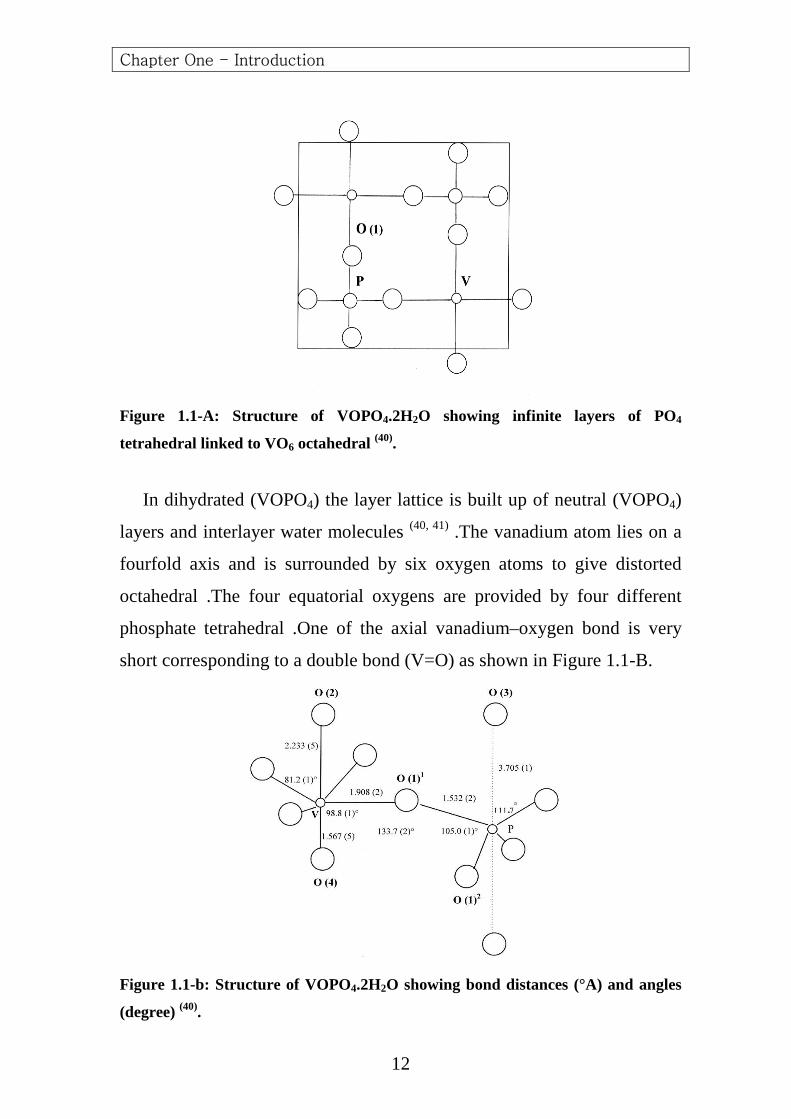
arranged in layers [VOPO4] ∞, held together by long (V-O) bonds or by
hydrogen bonds (Figure 1.1-A). The layered structure leads to rich
intercalation chemistry, with formation of layered solids consisting of
alternating inorganic (42-48) and organic (49-56) layer or the formation of
solvated inorganic intercalation compounds (43).
Chapter One - Introduction
12
Figure 1.1-A: Structure of VOPO4.2H2O showing infinite layers of PO4
tetrahedral linked to VO6 octahedral (40).
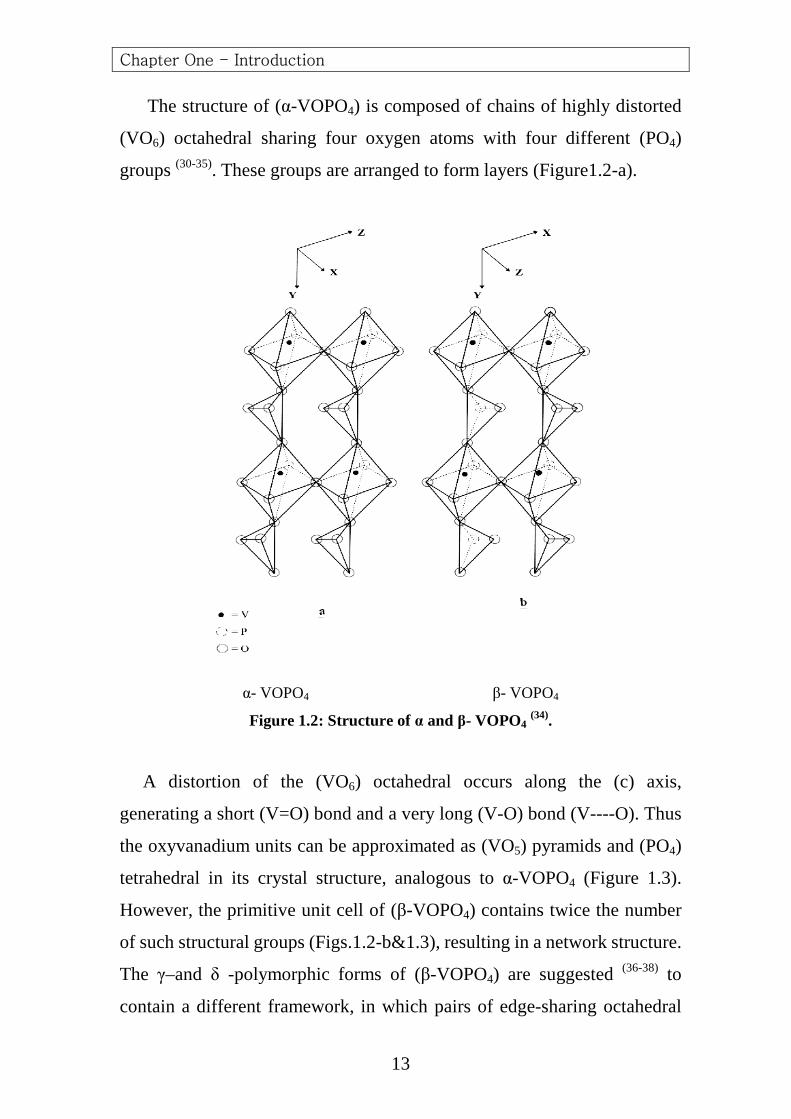
In dihydrated (VOPO4) the layer lattice is built up of neutral (VOPO4)
layers and interlayer water molecules (40, 41) .The vanadium atom lies on a
fourfold axis and is surrounded by six oxygen atoms to give distorted
octahedral .The four equatorial oxygens are provided by four different
phosphate tetrahedral .One of the axial vanadium–oxygen bond is very
short corresponding to a double bond (V=O) as shown in Figure 1.1-B.
Figure 1.1-b: Structure of VOPO4.2H2O showing bond distances (°A) and angles
(degree) (40).
Chapter One - Introduction
13
The structure of (α-VOPO4) is composed of chains of highly distorted
(VO6) octahedral sharing four oxygen atoms with four different (PO4)
groups (30-35). These groups are arranged to form layers (Figure1.2-a).
α- VOPO4 β- VOPO4
Figure 1.2: Structure of α and β- VOPO4 (34).
A distortion of the (VO6) octahedral occurs along the (c) axis,
generating a short (V=O) bond and a very long (V-O) bond (V----O). Thus
the oxyvanadium units can be approximated as (VO5) pyramids and (PO4)
tetrahedral in its crystal structure, analogous to α-VOPO4 (Figure 1.3).
However, the primitive unit cell of (β-VOPO4) contains twice the number
of such structural groups (Figs.1.2-b&1.3), resulting in a network structure.
The γ–and δ -polymorphic forms of (β-VOPO4) are suggested (36-38) to
contain a different framework, in which pairs of edge-sharing octahedral

Chapter One - Introduction
14
with trans vanadyl oxygens are alternately unshared or shared with
phosphate tetrahedral. Such pairs do not exist in the (α- and β-VOPO4)
forms. During thermal treatment at high temperature the consecutive
transition has been observed (36).
δ- VOPO4 γ-VOPO4 β-VOPO4 …. 1.5
α-VOPO4
β-VOPO4
Figure 1.3: Comparison between structure of α and β-VOPO4 (34).
Chapter One - Introduction
15
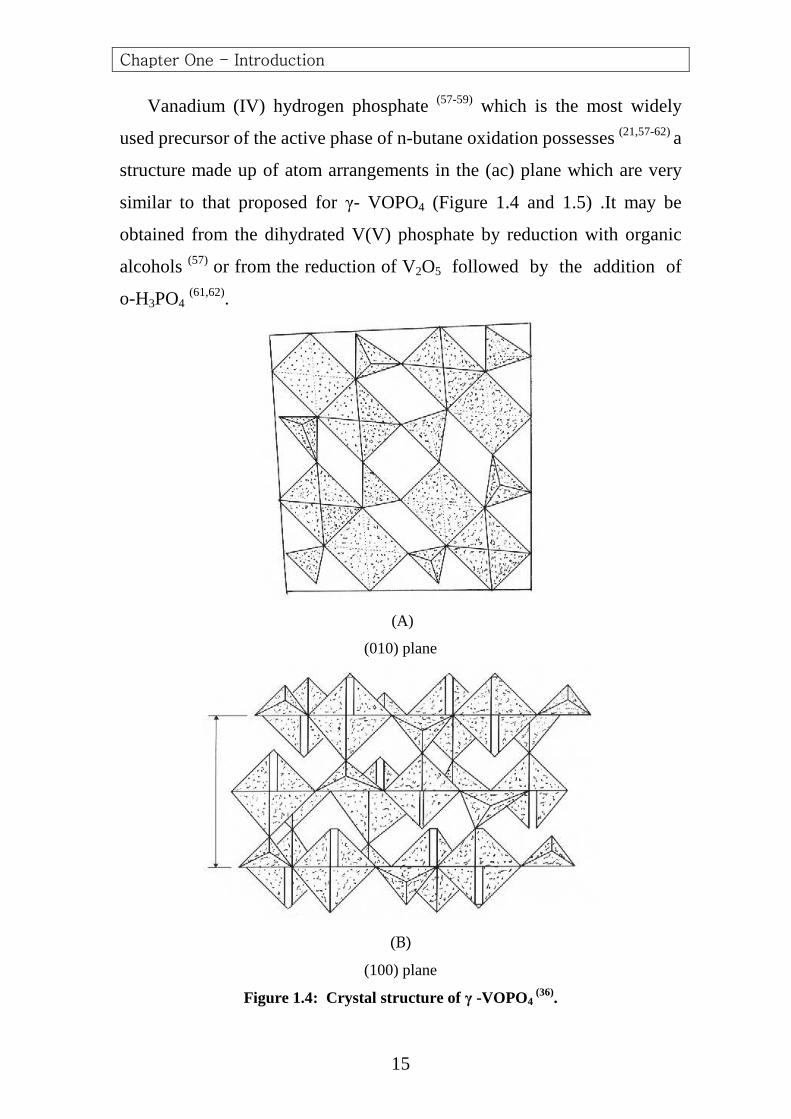
Vanadium (IV) hydrogen phosphate (57-59) which is the most widely
used precursor of the active phase of n-butane oxidation possesses (21,57-62) a
structure made up of atom arrangements in the (ac) plane which are very
similar to that proposed for γ- VOPO4 (Figure 1.4 and 1.5) .It may be
obtained from the dihydrated V(V) phosphate by reduction with organic
alcohols (57) or from the reduction of V2O5 followed by the addition of
o-H3PO4 (61,62).
(A)
(010) plane
(B)
(100) plane
Figure 1.4: Crystal structure of γ -VOPO4 (36).

Chapter One - Introduction
16
Figure 1.5: Crystal structure of VOHPO4.0.5 H2O (010) plane (59).
Both the hemi hydrate and tetrahydrate of vanadium hydrogen
phosphate are known (58) and their structures are closely related, showing a
layered structure and pairs of face sharing vanadium (IV) octahedral
hydrogen phosphate (OH) groups are directed into the interlayer space.
The coordination geometry around both vanadium and phosphorus is
similar in the two structures (Figure 1.5 and 1.6) .The possible formation
of the tri-hydrate VOHPO4 has also been reported (63).
Figure 1.6: View of the infinite double chains of (VO6) octahedra and (PO4)
tetrhedra running along the (100) direction in VOHPO4.4H2O (75).

Chapter One - Introduction
17
The intercalation compounds of vanadium hydrogen phosphate with
organic molecules or inorganic cations and anions such as (V n+) of HPO42-
are reported (23-33,36,37,63-72) .This intercalation chemistry is of particular
importance in the description of the structurally and catalytically related
chemistry of VOHPO4.0.5 H2O and of its derived phases.
Two main effects observed in the preparation of (VOHPO4.0.5 H2O)
may be, in fact, strictly connected to intercalation, properties. First, non
stoichiometry is easily accommodated as evidenced by the preparation of
compounds with (0.9-1.2) P: V ratio without any apparent modification of
structural and morphological properties (62).Second, the preparation
conditions have a pronounced effect on the microstructure, i.e., on the
morphology, solid - state reactivity, and the presence of disorder in the
stacking fold of crystalline planes of its layered structure.
In fact, reduction of the starting V (V) compound may be realized (62) by
using, for example, aqueous HCl or isobutyl alcohol. In both cases, almost
pure vanadyl hydrogen phosphate is obtained, but with different
microstructure (61,62,73). The layers of vanadyl hydrogen phosphate (010)
plane (Figure 1.5) are interconnected in tri dimensional structure by weak
hydrogen bonding of phosphates and of water molecules. The organic
alcohol competes with this effect, reducing the bonding between the planes
and allowing the formation of crystals in which these (010) planes are
predominantly exposed (plate like morphology) (21,56,59,62,74) .This effect, in
addition to the increase in surface area modifies the surface properties due
to a change in the relative ratio of crystalline planes at the surface. The
alcohol also can remain partially intercalated between layers (62) .This
effect induces some local modification of the vanadyl hydrogen phosphate
structure, which can strongly modify its solid-state reactivity (61,73).
Ball et al.(75) proposed that the known mono hydrogen phosphate phases
can be classified in three different structural types, namely type I,

Chapter One - Introduction
18
(VOHPO4.0.5H2O) ;type II (α-VOHPO4.2H2O); type III, (VOHPO4.4H2O)
and (β-VOHPO4.2H2O) .practically ,the β-dihydrate appears as
intermediate in the thermal treatment of the tetrahydrate from both
thermogravimetric and thermodiffractometric experiments .Moreover , the
structure of (β-VOHPO4.2H2O) is closely related with that of
(VOHPO4.4H2O) (Figure 1.6, 1.7-a and 1.7-b). Both compounds present a
similar arrangement of (VO6) octahedra and (PO4) tetrahedral forming
alternating chains which lie along the (c) direction in the β-dihydrate and
along the a direction in the tetrahydrate and along the (a) direction in the
tetrahydrate. The coordination geometry around both vanadium and
phosphorus atom is similar in both structures. Each phosphate group
contains three oxygen atoms (shared with three different vanadium atoms)
and a hydroxyl group. The (V) atoms show very similar coordination
polyhedra, having a water molecule trans-coordinated to the axial (V=O)
group and a second coordinated water molecule in the equatorial plane of
the (VO6) octahedra. The similarity of these structures suggests that the
dehydration of the tetrahydrate into the dihydrate may precede
topotacitically. However, several ways for the reorganization of the infinite
double chains that lie parallel to the (a) axis (6.379 °A ) of the tetrahydrate
may be imagined to give the interconnected single chains running in the (c)
direction (12.623 °A = 2 X 6.379 °A) of the β-dihydrate . All the possible
models that imagined need to break some bonds (at least 2 or 4 per unit
cell), to rotate some polyhedra and to reconstruct the bonds.
Figure 1.7- a: Projection of the structure of β-VOHPO4. 2H2O along (100) (75).

Chapter One - Introduction
19
Figure 1.7-b: Projection of the structure of β-VOHPO4. 2H2O along (010) (75).
The transformation of V (IV) and V (V) phases into vanadyl
pyrophosphate is an important step in forming active catalysts, by thermal
treatment (Calcinations) at 400 °C, the VOHPO4.0.5 H2O dehydrates to
(VO)2P2O7 .Alternatively, the vanadyl pyrophosphate may be obtained by
reduction from VOPO4 or by thermal treatment of different crystalline or
amorphous (V-P-O-X) phases (where X indicates a generic thermally
decomposable anion or cation such as (C2O4 2- or NH4
+ ) (63, 76).
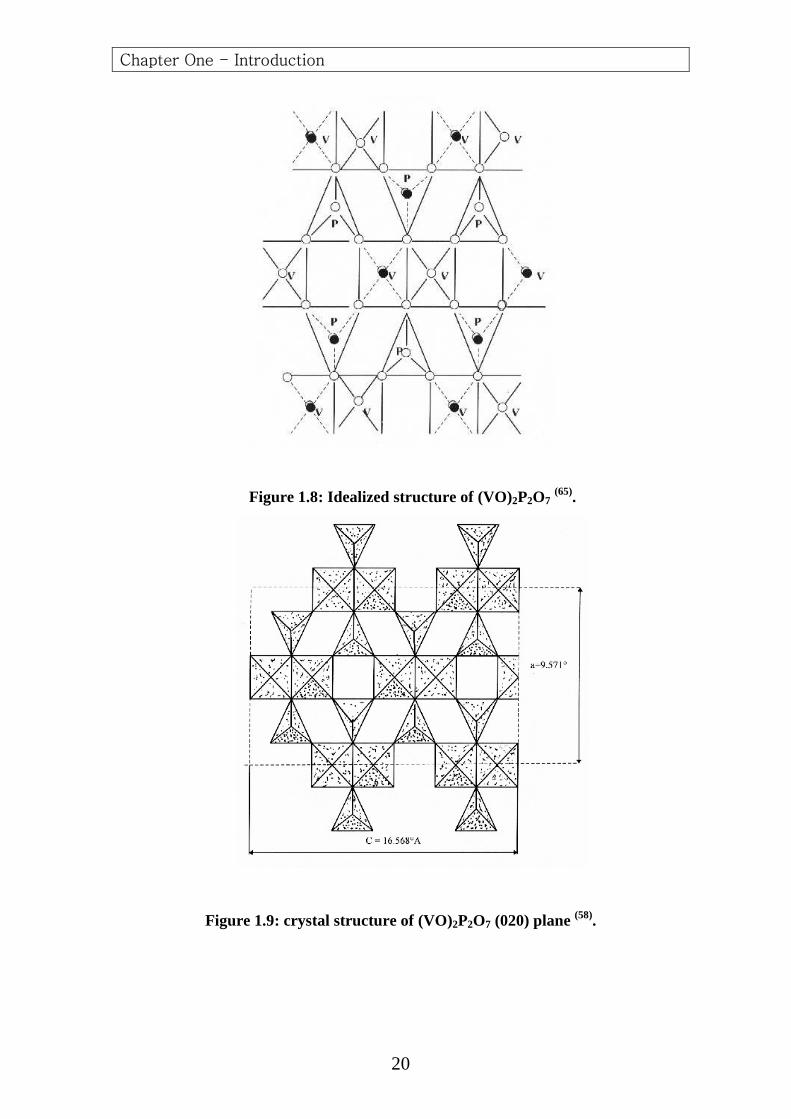
The vanadyl pyrophosphate is built of chains of (V) polyhedral linked
by pyrophosphate groups (60,77) .The (V) atoms are linked through the
oxygen atoms of the vanadyl in (V-O-V) chains in the (c) direction, and
the (V) atom octahedra are linked in pairs through a common edge,
forming double chains in this direction. The vanadyl groups in the paired
vanadium octahedra are oriented trans to one another (Figs. 1.8 and 1.9).
Chapter One - Introduction
20
Figure 1.8: Idealized structure of (VO)2P2O7 (65).
Figure 1.9: crystal structure of (VO)2P2O7 (020) plane (58).

Chapter One - Introduction
21
The unit cell of (VO)2P2O7 is orthorhombic (77,78) and is topological
similar in its (bc plane) to that of (VOHPO4.0.5H2O) in the (ab)
plane (56,60).The change from face-shared to edge-shard vanadium
octahedral in converting the hemihydrate to the pyrophosphate results in a
small expansion of one axis, but the other in plane dimension shows little
change according the pseudomorphic relations between the two crystalline
structures (59) as shown in (Figure1.10) this indicates a topotactic
mechanism of transformation, and this result has been confirmed with
Scanning Electron Microscopy. (21,56,59,62) .The layer spacing decreases from
(5.69°A) in the hemihydrate to (3.91°A) in the pyrophosphate. This is
consistent with removing the water molecules shared by the vanadium
pairs and filling the resulting vanadium coordination site with the oxygen
atoms of vanadyl groups from the layers above. This transformation
requires only very small displacements of the atoms. Importantly, since the
conversion of the hemihydrate to pyrophosphate can take place without
breaking any (V-O-P) bonds, the structural order/disorder and morphology
of the precursor phase are maintained during the transformation to vanadyl
pyrophosphate. This means that it is possible to control some of the
structural /morphological characteristics of vanadyl pyrophosphate by
controlling the specific nature of the vanadyl hydrogen phosphate
hemihydrate precursor phase (62,73). Furthermore the terminal vanadyl
oxygen atoms in the face – shared octahedral pairs of vanadyl hydrogen
phosphate have a syn arrangement; while in vanadyl pyrophosphate they
are in anti positions. These arrangements in the layer stacking direction
result in the initial formation of (VO)2P2O7 crystalline with many defects
(36,37,65,79). Alternatively, Bordes and Courtine (70,74) discussed the possible
presence of disorder in the crystalline structure of (VO)2P2O7 in terms of
different crystalline phases (β- and γ- VO)2P2O7) in which former
possesses a network structure versus a layered structure for the γ- phase.
Chapter One - Introduction
22
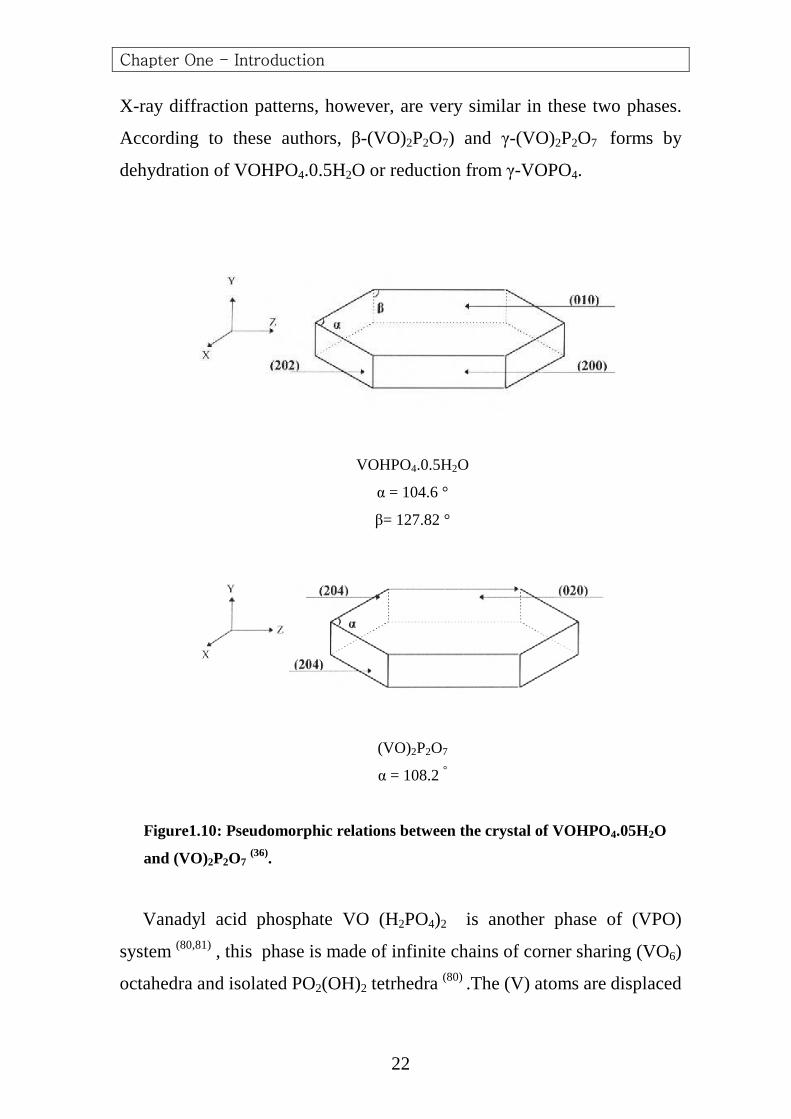
X-ray diffraction patterns, however, are very similar in these two phases.
According to these authors, β-(VO)2P2O7) and γ-(VO)2P2O7 forms by
dehydration of VOHPO4.0.5H2O or reduction from γ-VOPO4.
VOHPO4.0.5H2O
α = 104.6 °
β= 127.82 °
(VO)2P2O7
α = 108.2 °
Figure1.10: Pseudomorphic relations between the crystal of VOHPO4.05H2O
and (VO)2P2O7 (36).
Vanadyl acid phosphate VO (H2PO4)2 is another phase of (VPO)
system (80,81) , this phase is made of infinite chains of corner sharing (VO6)
octahedra and isolated PO2(OH)2 tetrhedra (80) .The (V) atoms are displaced

Chapter One - Introduction
23
(0.364) °A from the equatorial plane along the fourfold axis. As a result,
one short bond (1.600 °A) which characterizes the vanadyl (VO2+) ion,
forms in almost regular oxygen octahedra .The bond between the (V atom)
and the oxygen in the trans position is significantly longer(2.382 °A).
Furthermore ,the equatorial planes of the octahedra are alternatively
rotated of ( +18° and - 18°) around the chain axis (Fig. 1.11-a ).Phosphate
tetrahedra act as bidentate bridges via oxygen atoms for (V) atoms
belonging to adjacent chains, (OH) corners established a contact between
tetrahedra to form hydrogen bonds (Figs. 1.11- a &1.11-b ).
(A)
Figure 1.11-a: Crystal structure of VO (H2PO4)2 along XY axis (80).

Chapter One - Introduction
24
(B)
Figure 1.11-b: Crystal structure of VO (H2PO4)2 along XZ (80).
1.3.2 - Active phase in n-Butane Selective Conversion to MA
Because the (VPO) system is characterized by the facile formation of a
number of crystalline phases, the structure of the active phase must be
discussed in term of factors such as, oxidation state, (P/V) ratio, and
crystal phase transformations under reaction atmosphere.
The various crystal phases can interconvert as a function of the reducing or
oxidizing properties of the reactants, the time on stream, and the reaction
temperature (29,37,61,82,83).
The orthophosphate (VOPO4) phases are transformed to (VO)2P2O7 by
reaction with the hydrocarbon mixture. In this reduction process single
(VO6) octahedral form pairs by loss of oxide anions. However, the
different (VOPO4) phases previously discussed possess different
reducibility to vanadyl pyrophosphate, depending on the structure or
morphology (36).

Chapter One - Introduction
25
The Complex solid-state chemistry of (VPO) system has led to some
confusion and contradictions in the literature concerning the nature of the
active phase in the n-butane oxidation and the identification of the active
site involved in the different steps of the reaction.
Bordes and Courtine (37) suggested that the active sites in n-butane
oxidation to (MA) were associated with coherent interfaces between slabs
of (100) VOPO4 and of (010) (VO)2P2O7 along the (100) and (201) planes,
respectively. On the contrary Volta et al. (84,85) believed that the active sites
are not associated with interfacial effects between two crystalline phases.
On the basis of comparison between X-ray diffraction, they suggested that
the active phase for selective oxidation of n-butane consists of a mixture of
well-crystallized (VO)2P2O7 (V4+) and an amorphous surface (VPO) phase
of (V5+) involving many corner-sharing VO6 octahedra. This amorphous
phase may be interpreted as a precursor of (β-VOPO4), which forms at
higher reaction temperatures.
Hodnett and Delmon (67-69) used a prereduction treatment with hydrogen
in order to improve selectivity to maleic anhydride and suggest that the
best catalyst consists of an oxidized surface layer built upon a reduced core
of a V (IV) phase. The selectivity is not related to the presence of a
specific well-crystallized phase, but only to the distribution of vanadium
oxidation states between the bulk and the surface.
The active catalyst must possess an optimal [V (IV)/V (V)] ratio for
selectivity in n-butane oxidation according to Zozhigalov et al., (86) from
studies of V-P compounds with variable V (IV) /V (V) ratios. These
compounds preserve the layer structure of the initial (α-V5+OPO4)
compound. Optimal performance properties are associated with catalysts
containing [4-9 V (V) ions per V (IV) ion]. n-butane oxidation and (MA)
take place at the expense of the catalyst surface oxygen and are
accompanied by its reduction to (VO)2P2O7. On (VO)2P2O7 however, the

Chapter One - Introduction
26
rate of reduction of the catalyst is lower than that of butane to (MA)
reaction, and therefore these authors conclude that the V(V) phase is
reduced and that the V(IV) phase is oxidized under the dynamic conditions
of catalytic reaction (86).
Weing and Schrader (74) claimed that only (VO)2P2O7 is the active and
selective phase in n-butane oxidation to (MA). A slight excess of catalyst
phosphorus (P/ V=1.1 catalysts) is necessary to stabilize the active phase
The excess phosphorus creates a distortion of (P2O7)4- crystal
environment. The α-VOPO4 is considered by these authors and others (21)
as active, but non selective phase.
Many authors proposed that the vanadyl pyrophosphate (VO)2P2O7 as
the active phase (1,9,21,56,62,73,77,80,87) .Trifiro et al. (62,88-90) attributed the
activity of the catalyst to the V(IV) phase (the vanadyl pyrophosphate),
whereas the selectivity to maleic anhydride(MA) was connected to the
presence of a very limited and controlled amount of V (V) sites .
The same discordance as to the nature of the active phase is present in
the literature concerning the optimal (P/V) ratio of the catalysts, even
though there is a general agreement that phosphorus stabilizes the (+4)
valance state of vanadium and limits its oxidation (8,74,86,90).
Garbassi et al. (91) have found that the specific conversion of
n-butane increases by an order of magnitude for a (P/V) ratio just
exceeding unity, but extended X-ray absorption measurements do not show
any structural effect of the phosphorus.
The (P/V) ratio is a key parameter in determining catalyst selectivity
and activity according to Weing and Schrader (74). Selectivity for (MA)
increase with catalyst phosphorus loading, whereas specific activity of
both selective and nonselective oxidation decreases on increase of
phosphorus content in the range (0.9-1.2) P/V range. Best catalytic

Chapter One - Introduction
27
performances are exhibited with a catalyst with (P/V = 1.1) according to
Buchanan and Sunderesan (92).
Similar results were observed by Pepera et al. (93). According to Bosh
and co-workers (94) who studied the selective oxidation of n-butane to (MA)
under oxygen deficient over (VPO) catalyst, they found that the selectivity
was strongly influenced by the actual surface V (V)/V (IV) ratio).
Garbassi et al. (91) found a value of (P/V) surface ratio in the
(2.0-2.8) range for P/V bulk ratio in the (1.0-1.4) range.
Hodnett and Delmon (67-69) reported that the surface (P/V) ratio is (1.0)
for bulk stoichiometric (P/V) values of 1.0 or higher. They concluded
therefore that the reactivity of near-surface layers is hardly affected by the
(P/V) ratio, but bulk reactivity is drastically curtailed.
Selectivity to (MA) from n-butane maximizes for (P/V) =1.0 according
to Ai (70), who associated this catalytic effect with the presence of strong-
acid sites to activate n-butane. Finally, an optimal value of (P/V) around
(1.0) was suggested by Trifiro et al. (73,88-90) and Contractor et al. (21).
The active sites has been proposed as being the (200) plane of
(VO)2P2O7 crystallites that preferentially expose the (200) plane have been
found to exhibit higher selectivities for n-butane oxidation (95).
Transformation Electron Microscopy has shown that catalyst selectivity
correlated with the disappearance of amorphous platelets (96). Seven
different active sites have been suggested for the (200) plane of (VO)2P2O7
catalysts as shown in (Figure 1.12) whereas:
a- Bronsted acid sites, probably –POH groups.
b- Lewis acid sites, probably V IV and V V.
c- One electron redox couples, (VV / V IV), (V IV / VIII ).
d- Two electron redox couples (V V / VIII ).
e- Bridging oxygen, V-O-V, or V-O-P.
f- Terminal oxygen VV= O.

Chapter One - Introduction
28
g- Activated molecular oxygen peroxo and superoxo species (96).
Figure.1.12: Termination of the (200) plane of (VO)2P2O7 and proposed active
sites for oxidation (s1) = Lewis acid, (s2) = Lewis acid site, (s3) = terminal
oxygen, (s4) = bridging oxygen, (s5) = superoxo and peroxo site, (s6) = V v/ viv
redox couple (97).
1.4- Kinetics of n-Butane Oxidation
A central question in analyzing the problem of bridging the gap
between surface science and applied catalysis approaches is the
verification of the possibility of describing the macro kinetic behavior
using rate equations and constants derived from the analysis of the kinetics
of the single elementary steps. Impressive results have been obtained in
this direction (98) .The fitting of macro kinetic data on the basis of a
microkinetic model is usually considered the best demonstration of the
applicability of the suggested reaction mechanism under real working
conditions (99). Is it this true also for more complex and multifunctional
reactions? In order to replay to this question it is good to briefly recall the
principles of the Langmuir description of the catalytic reaction on an

Chapter One - Introduction
29
"ideal" surface, because this is the basis of most of the macrokinetic
models for deriving the reaction rates over catalytic surfaces. On the ideal
Langmuir surface there is one type only of active sites, the energetic of
chemisorption is independent on the coverage, and there is no interaction
between substrate (Figure 1.13-a). Real surfaces are far from the ideal
Langmuir surface, as schematically represented in (Figure 1.13- b), due to
the presence of surface heterogeneities the simplest is the presence of
steps and kinks, but as mentioned In the introduction the situation is far
more complex on oxides) which imply a distribution in the energetic of
interaction, and the presence of interaction between the adsorbates .A
chemical bond of a molecule with a surface site implies the donation of
electrons to or from the surface with thus a modification of the catalyst
conduction band, band gap, etc.; therefore, the energetic of interaction of
molecule with the surface is not independent from the surface coverage,
and the adsorption of " spectator" species, i. e. species which do not play
a direct role in the reaction mechanism, should be also considered. This
problem is even more accentuated in selective oxidation reactions where a
multielectron transfer occurs, as mentioned before.
It may be thus expected that kinetic models based on Langmuir
approach do not correctly fit macrokinetic data, but on the contrary it is a
common experience that these models are well suited to describe the
macrokinetic behavior. How can this dilemma be solved? One possible
interpretation, which is schematically shown in (Figure 1.13-c), is that
during catalytic reaction the largest part of the catalyst surface is covered
by "strongly chemisorbed" species which are characterized species
characterized by a surface lifetimes much longer than of reaction
intermediates and therefore reactant or intermediate seen a local situation
similar to "ideal" surface. This pointed out that the concept of " clean"
catalyst surface, i. e. of reaction of a molecule at single specific active sites

Chapter One - Introduction
30
without considering the modification of the surface reactivity induced by
the presence of other co-adsorbates (reactants, intermediates, " spectator"
species), may not lead to correct description of the " real" working catalyst
surface and reaction mechanism, especially when complex, multi steps
reactions (selective oxidation reactions, for example) are considered.

Chapter One - Introduction
31
A- A simple model of the ideal Langmuir surface
B- A simple model of real surface with kinks and steps, surface interaction between
adsorbates and electron donation of a chemisorbed molecule to catalyst conduction
band (strongly chemisorbed species)
C-Working catalyst surface with the largest part of the surface occupied from strongly
chemisorbed (reactant intermediate)
Figure 1.13- Model of the ideal Langmuir surface (99).

Chapter One - Introduction
32
Several studies have been conducted in the eighties to derive kinetic
expressions to describe the reaction sequence for n-butane oxidation to
Maleic Anhydride in steady- state conditions on (VPO) system (86,87,91,100,101).
Escardino et al. (100) studied the kinetic of n-butane oxidation in fluidized -
bed reactor over (VPO) catalyst with (P/V= 0.8) at (676-753) K.
A triangular reaction network was proposed as shown in Scheme 1.5.
CH3
CH3
O
O
O
CO , CO2
r1
r2 r3
Scheme1.5: A triangular reaction network on n-butane to MA (100).
Maleic Anhydride (MA) and Carbon oxides (COx) were formed directly
from n-butane (at rates r1 and r2 respectively), and MA was also oxidized
to carbon oxides (at rate r3). At n-butane concentrations typical of
industrial reactors, the rate of n-butane oxidation was controlled by the
reaction between n-butane gas and surface oxygen.

Chapter One - Introduction
33
Wohlfahrt and Hofmann (101) investigated n-butane oxidation kinetics
over a wide range of n-butane and oxygen concentration at (719-777K)
over (VPO) catalyst.
Centi, et al. (87) used a very active catalyst prepared in an organic
medium which permitted low reaction temperatures (573-613K) to be
employed. Under these conditions it was found that the rate of reaction of
n-butane to carbon dioxide did not depend on the hydrocarbon
concentration, but only on the concentration of oxygen.
The order of reaction maintained in the literature depended on the
method of analysis of available data from experimental results of
hydrocarbon depletion and oxygen partial pressure. The comparison of
these kinetics results stressed the importance of surface catalyst behavior
(selectivity to MA) as well as the role of the redox properties of vanadium
in determining the activity of the catalyst (87,101).
1.5- Surface Modifications by Interaction of n-Butane with
Catalyst Surface
Centi et al. (87) used fresh catalyst for their kinetics study and suggested
that, a critical factor governing the selectivity at very high butane
conversion is the instability of the formed (MA) in the back end of the
catalytic bed. It is thus worthwhile to analyze the variations in the catalyst
surface as a function of position in the catalytic bed (86). These experiments
showed formation of V (V) at the end of catalyst bed which was attributed
to formation the more oxidizing atmosphere present.
Trifiro et al. (89) studied the oxidation of n-butane at low and high
hydrocarbon concentrations on vanadium (IV)-phosphorus (1:1) mixed
oxide in relation to the surface modifications induced by the reaction
medium. The results showed the presence, in the catalyst, of high amounts

Chapter One - Introduction
34
of V (III) together with V (IV) and the absence of V (V), whereas lower
amounts of V (III) together with both V (IV) and V (V) are formed, when
maleic anhydride is formed. It is suggested that two redox couples operate
in (MA) synthesis:
a- V (IV)-V (III) in the synthesis of olefins from n-butane.
b- V (V)-V (IV) in the synthesis of (MA) from the olefins is formed.
Mori and co-workers (102) studied the oxidation of n-butane on various
unsupported or supported V(V) oxides, they confirmed that (V(V)=O)
species are very active in total combustion of butane, these experiments
results indicate the important relationship between catalytic behavior and
the surface modification induced by the medium itself. The redox
properties of the fed influence the surface oxidation state of the catalyst,
which in turn profoundly affected the nature of the products formed in the
reaction.
Centi and Perathoner (99) referred to (VO)2P2O7 as the active phase in
the industrial catalysts for the selective oxidation of n- butane to (MA).
This catalyst is very selective in n-butane oxidation, but when propane is
fed Instead of n-butane, only carbon oxides and traces of other products
(propene mainly) are detected.
A first question is thus why the decrease in length of the carbon chain
produces a so drastic change in the selectivity on the same catalyst? The
answer can be that from n-butane a stable product against consecutive
oxidation (MA) forms, but not in the case of propane oxidation. However,
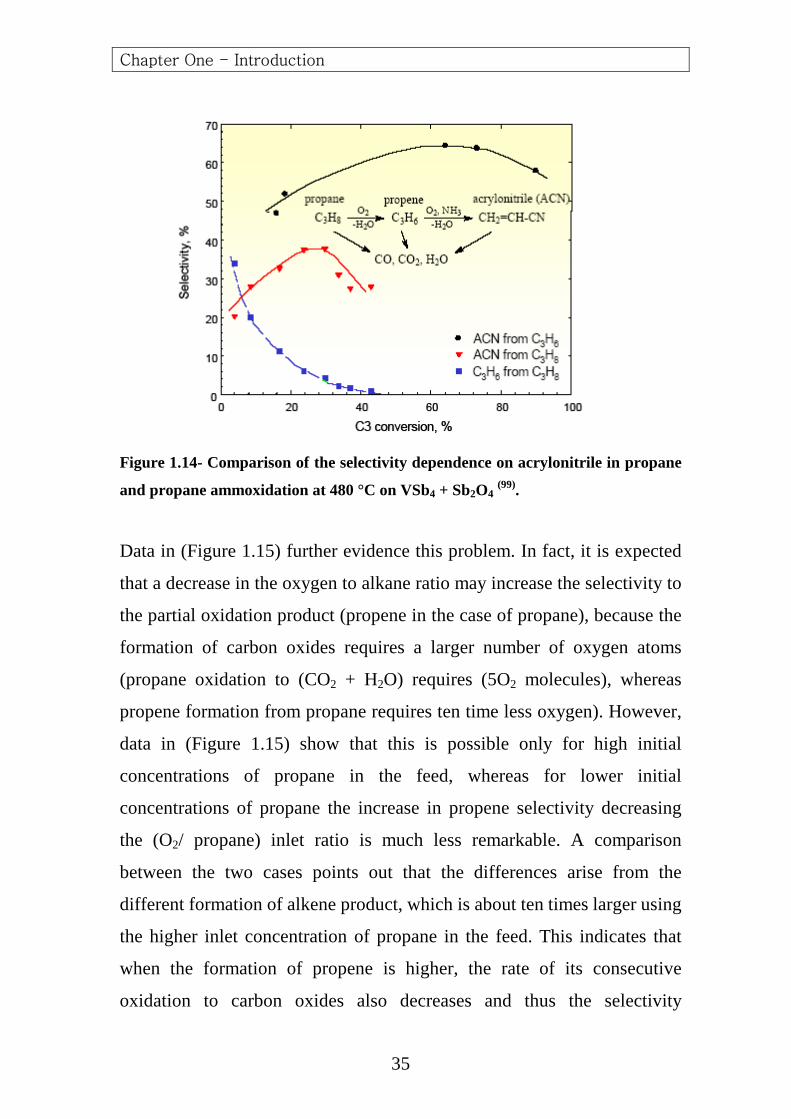
this answer responds to only part of the problem. The data reported in
(Figure 1.14) have already pointed that the sensitivity of the reaction
product against consecutive oxidation is not the only factor that determines
the rate of consecutive oxidation to carbon oxides.
Chapter One - Introduction
35
Figure 1.14- Comparison of the selectivity dependence on acrylonitrile in propane
and propane ammoxidation at 480 °C on VSb4 + Sb2O4 (99).
Data in (Figure 1.15) further evidence this problem. In fact, it is expected
that a decrease in the oxygen to alkane ratio may increase the selectivity to
the partial oxidation product (propene in the case of propane), because the
formation of carbon oxides requires a larger number of oxygen atoms
(propane oxidation to (CO2 + H2O) requires (5O2 molecules), whereas
propene formation from propane requires ten time less oxygen). However,
data in (Figure 1.15) show that this is possible only for high initial
concentrations of propane in the feed, whereas for lower initial
concentrations of propane the increase in propene selectivity decreasing
the (O2/ propane) inlet ratio is much less remarkable. A comparison
between the two cases points out that the differences arise from the
different formation of alkene product, which is about ten times larger using
the higher inlet concentration of propane in the feed. This indicates that
when the formation of propene is higher, the rate of its consecutive
oxidation to carbon oxides also decreases and thus the selectivity
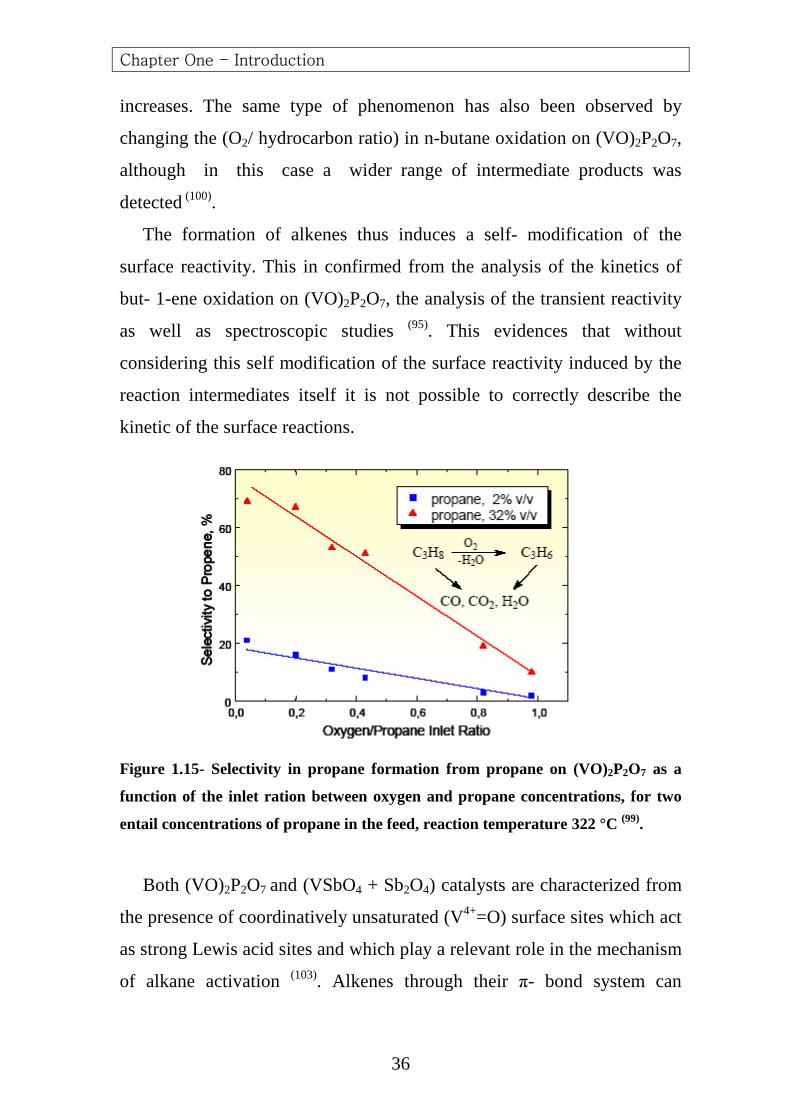
Chapter One - Introduction
36
increases. The same type of phenomenon has also been observed by
changing the (O2/ hydrocarbon ratio) in n-butane oxidation on (VO)2P2O7,
although in this case a wider range of intermediate products was
detected (100).
The formation of alkenes thus induces a self- modification of the
surface reactivity. This in confirmed from the analysis of the kinetics of
but- 1-ene oxidation on (VO)2P2O7, the analysis of the transient reactivity
as well as spectroscopic studies (95). This evidences that without
considering this self modification of the surface reactivity induced by the
reaction intermediates itself it is not possible to correctly describe the
kinetic of the surface reactions.
Figure 1.15- Selectivity in propane formation from propane on (VO)2P2O7 as a
function of the inlet ration between oxygen and propane concentrations, for two
entail concentrations of propane in the feed, reaction temperature 322 °C (99).
Both (VO)2P2O7 and (VSbO4 + Sb2O4) catalysts are characterized from
the presence of coordinatively unsaturated (V4+=O) surface sites which act
as strong Lewis acid sites and which play a relevant role in the mechanism
of alkane activation (103). Alkenes through their π- bond system can

Chapter One - Introduction
37
chemisorb on these sites forming relatively stable chemisorbed species,
although they may be considered "spectator" species, because they are not
directly involved in the mechanism of further selective oxidation of these
alkene intermediates. Oxygen also strongly chemisorbs on the surface
Lewis acid sites forming thermal stable species (they desorb above 450-
500°C), but which play a relevant role in the mechanism of oxidation.
When the surface concentration of the intermediate alkenes in alkane
oxidation is as high as to limit the amount of chemisorbed oxygen, due to
this competitive chemisorption, it is thus possible to control the population
of oxygen adspecies by this mechanism. This explains the considerable
promotion of selectivity to Partial oxidation products by increasing alkane
inlet concentration (Figure 1.15) and, on the other hand, explains also the
apparent contradiction of the different rate of acrylonitrile consecutive
oxidation when forms from propene instead of that from propane
(Figure 1.14).
Although propene forms from propane as a reaction intermediate, its
concentration is clearly higher when it is fed directly and thus its effect in
limiting the concentration of Surface oxygen species is present even for
higher conversions of the hydrocarbon. Therefore, the maximum in the
formation of acrylonitrile is observed at higher hydrocarbon conversions
when the alkene is fed instead of the alkane on the same catalyst
(Figure 1.14).
In the oxidation of n-butane ،partial oxidation product (butenes and
butadiene) are observed at low oxygen concentrations when a very limited
number of vanadium (V) species are present on the surface of the catalyst.
The change of valance state of vanadium on the catalytic surface upon a
balance of three factors:
a- The redox potential of the feed.
b- The rate of oxidation of the catalyst at the temperature of reaction.
Chapter One - Introduction
38
c- The rate of reduction of the catalyst at the temperature of reaction.
1.6- Relationship between Redox Properties & Catalytic
Behavior
As pointed out in the section on catalyst structure ( section 1.3), the
binary (VPO) system was rather complicated because of the great variety
of observed phases and the difficulties encountered in the development of
successful and reproducible syntheses of pure single phases stable under
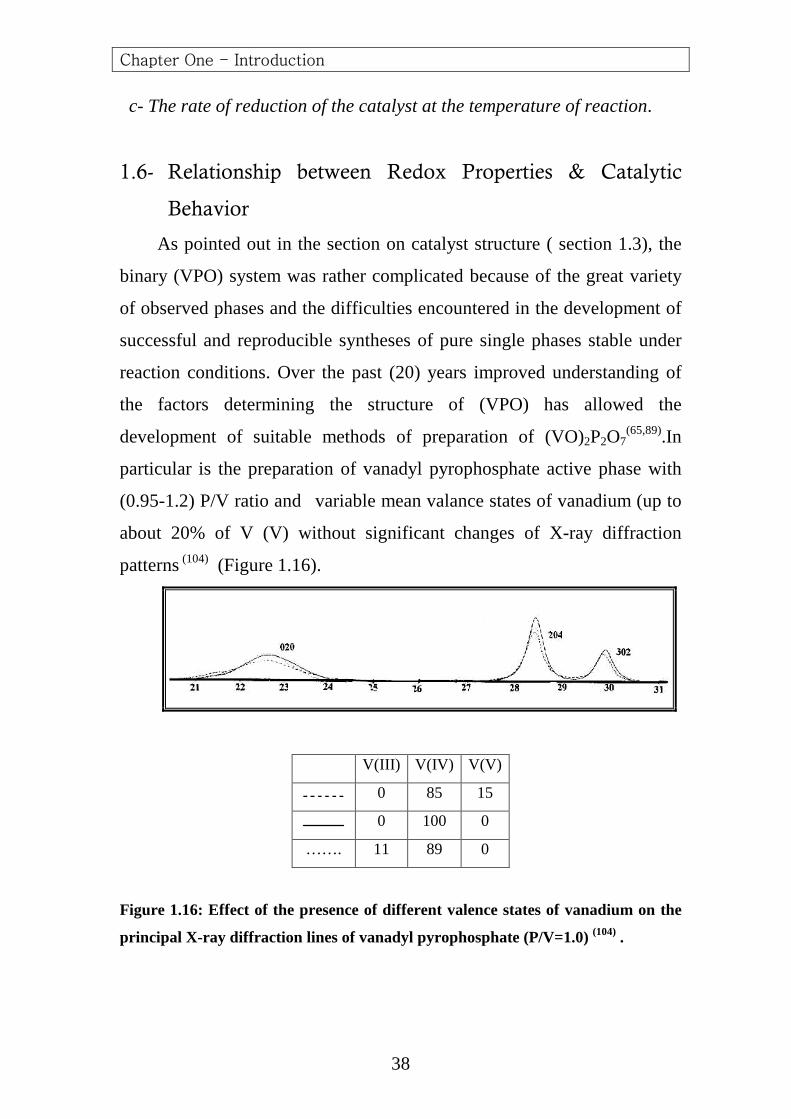
reaction conditions. Over the past (20) years improved understanding of
the factors determining the structure of (VPO) has allowed the
development of suitable methods of preparation of (VO)2P2O7(65,89).In
particular is the preparation of vanadyl pyrophosphate active phase with
(0.95-1.2) P/V ratio and variable mean valance states of vanadium (up to
about 20% of V (V) without significant changes of X-ray diffraction
patterns (104) (Figure 1.16).
V(III) V(IV) V(V)
0 85 15
0 100 0
……. 11 89 0
Figure 1.16: Effect of the presence of different valence states of vanadium on the
principal X-ray diffraction lines of vanadyl pyroph osphate (P/V=1.0) (104) .

Chapter One - Introduction
39
The variation of the content of (P) in the composition modifies the
catalytic properties in n-butane oxidation (86) and the redox properties of the
catalysts (104), a slight deficiency of phosphorus, does not change the rate of
V (IV) oxidation to V (V). On the other hand, an excess of phosphorus
only slightly influences the rate of oxidation, but strongly affects the rate
of reduction. This effect is in part attributed to a decrease of the number of
the active site, but also reflects a particular kind of interaction of
phosphorus upon the vanadium ions (61,104). In order to correlate the
observed variations in the redox properties with the catalytic behavior in n-
butane oxidation on the fresh catalyst used by these authors, it is necessary
to distinguish between tests at low conversion and high conversion.
At low conversion, the catalysts with (P/V ratio = 0.95 and 1.01) show
the same activity, selectivity, and kinetic behavior (86). However, at high
conversion of n-butane (80%) the catalyst deficient in phosphorus and with
the observed higher rate of vanadium oxidation forms primarily carbon
oxides. The catalyst with more phosphorus with respect to the
stoichiometric ratio of (1.0) are less active and less selective (90), but do not
show the strong decline of (MA) selectivity at the highest conversion.
It is reasonable to correlate these catalytic effects with the redox
properties of the catalyst (105). The strong increase of the rate of vanadium
oxidation in the phosphorus-deficient catalyst leads to an enhancement of
the rate of consecutive oxidation of ( MA) (effect on the selectivity),
whereas the decreased rate of reduction of vanadium in the catalyst with
higher (P) content than a stoichiometric one leads to the reduced rate of
hydrocarbon depletion (effect on the activity)
Nakamura et al. (105) also suggested that some V (V) ions were
necessary for (MA) synthesis, but the optimum mean of valance state of
catalyst is close to four. Similar conclusions were presented by Weing and
Schrader (74). They reported catalyst phosphorus loading is a key parameter

Chapter One - Introduction
40
in determining catalyst selectivity and activity. An increase of (P) content
in the (0.9-1.2 P/V) ratio decreases specific activity in n-butane depletion
and increases the selectivity to (MA). The same selectivity was reported by
Pytnitskaya et al. (71), Buchanan and Sundarsan (92), also found a correlation
in vanadium phosphorus oxides between the concentration of V (IV) ions
in the discharged catalyst and the activity in n-butane oxidation.
The results of surface modifications induced by the interaction of C-4
hydrocarbons as well as the results of (P/V) ratio effect on redox properties
and on (selectivity/activity) are interpreted on the basis of two different
types of reaction in going from the alkane to (MA): a first step is oxidative
dehydrogenation up to adsorbed butadiene and second step is further
oxidation of this intermediate. The first step is suggested to be controlled
by the rate of V (IV) reduction, whereas the second step is controlled by
the amount of V (V) available. However, when the content of V (V) is too
high, the oxidation proceeds further to carbon oxides.
These data suggest a strong relationship between selectivity to (MA) at
high conversion and rate of formation of V (V) as reported by Trifiro and
co-worker (62) (Figure 1.17).
(A)
Conversion
3 >2 >1 (in disorder arrangement) (62)
Figure 1.17-a: Maleic Anhydride yield VS conversion at 300°C in n-butane
oxidation for different catalyst with P/V= 1.0, feed = 0.6% n-butane, 12% O2 (62).
MA
%
Chapter One - Introduction
41
(B)
Time (minutes)
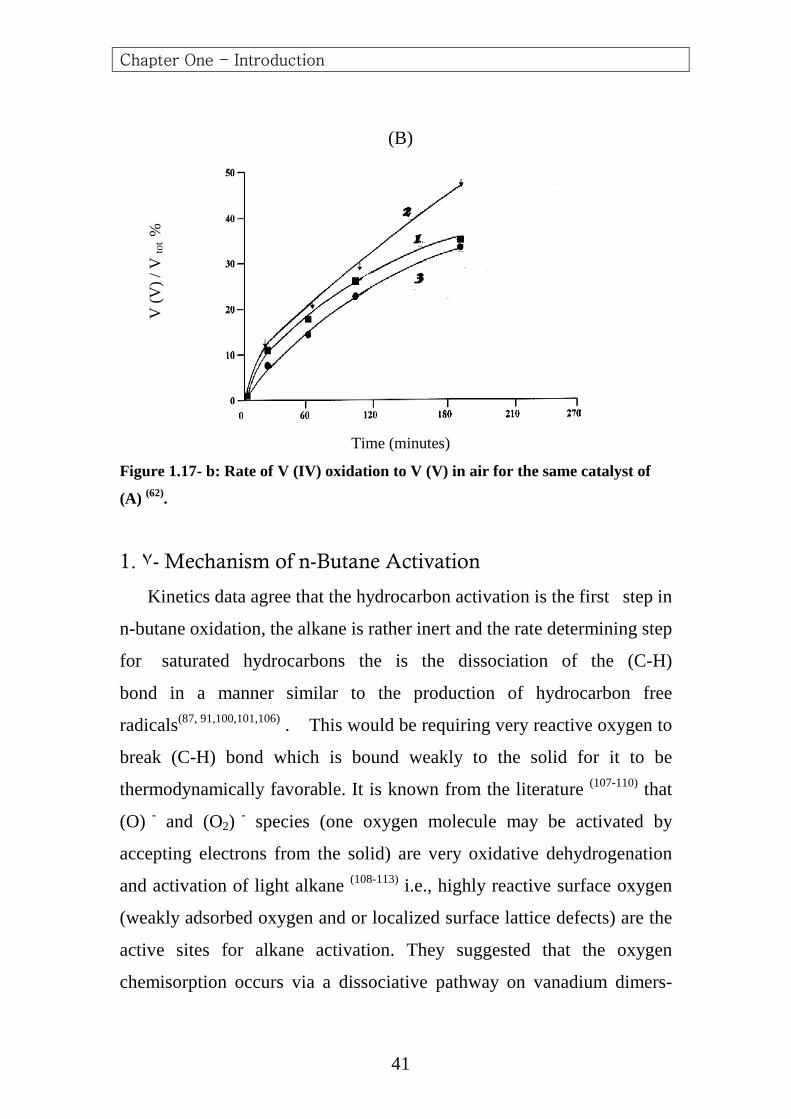
Figure 1.17- b: Rate of V (IV) oxidation to V (V) in air for the same catalyst of
(A) (62).
1. ٧ - Mechanism of n-Butane Activation
Kinetics data agree that the hydrocarbon activation is the first step in
n-butane oxidation, the alkane is rather inert and the rate determining step
for saturated hydrocarbons the is the dissociation of the (C-H)
bond in a manner similar to the production of hydrocarbon free
radicals(87, 91,100,101,106) . This would be requiring very reactive oxygen to
break (C-H) bond which is bound weakly to the solid for it to be
thermodynamically favorable. It is known from the literature (107-110) that
(O) - and (O2) - species (one oxygen molecule may be activated by
accepting electrons from the solid) are very oxidative dehydrogenation
and activation of light alkane (108-113) i.e., highly reactive surface oxygen
(weakly adsorbed oxygen and or localized surface lattice defects) are the
active sites for alkane activation. They suggested that the oxygen
chemisorption occurs via a dissociative pathway on vanadium dimers-
V (
V)
/ V to
t %

Chapter One - Introduction
42
leading to a (V (V)-O*) type surface species capable of activating the
alkane.
Due to the topotactic transformation that occurs without the breaking
of (VPO) connections and due to the higher electronegativity of (P) with
respect to (V), the presence of defects in the structure of vanadyl
pyrophosphate is reflected on the surface in an enhancement of Lewis
acidity of surface unsaturated vanadium ions (Figure 1.18). Strong acid
sites on (VO)2P2O7 were observed also by Puttock and Rochester (111,112) by
infrared spectroscopy.
(A)
A- Medium –strong Lewis acid sites, well crystalline structure
(B)
B- Very strong Lewis acid sites, disordered crystalline structure
Figure 1.18: Model of Lewis acid sites in (VO)2P2O7 with different degrees of
disorder in the stacking fold of (020) planes (62).

Chapter One - Introduction
43
All above observations may be summarized in the following model:
The presence of defects in the crystalline structure creates strain in the
V-P-O bonds and would create a surface-activated reactive couple.
V O Pδ δ+
− − − − −−
→
……….. (1.5)
This reactive couple was proposed by Centi et al. (73) as being
responsible for the concerted mechanism of the removal of two hydrogen
atoms, according to the (Figure 1.19-a, 1.19-b and Figure 1.20) which show
the redox cycle proposed to this mechanism of activation of n-butane.The
model is based on the idea that (V (IV)–oxygen) are the active sites of butane
dehydrogenation (62,99).
Munson and et al. (113) had developed an experimental protocol to study
the mechanism of this reaction in which 13C-isotopically labeled n-butane
is flowed over a catalyst bed and the reaction products are analyzed using 13C NMR spectroscopy. This strongly suggests that the total oxidation of
n-butane on (VPO) catalysts involves the oxidation and abstraction of the
two methyl groups of n-butane, and the two methylene groups of n-butane
form ethylene. An organometallic mechanism is proposed to explain these
results.
In conclusion, it is necessary to try to develop new models of the
surface reactivity which include new evidence on aspects such as (i) the
role of chemisorbed species on the surface reactivity, (ii) the presence of
multiple pathways of reaction (iii) the dynamics of catalyst reconstruction
(iv) the mobility of surface adspecies, etc. The consideration of all these
possible effects in analyzing the surface reactivity will make possible the
design of new catalysts as well as the understanding of surface
reactivity at oxide surfaces (99).
Chapter One - Introduction
44
HC
CMe
Me
H
V O
V
O
OP
H
(A) A- Detail of the concerted mechanism of (2H) abstraction.
VV
O
O
O
CCH
CH3
H3C
H
H
H
V VO
O
O
O
O
O
O
H
HO
-H2O
VV
O
O O
O
O2
O
O
O
O
3+
O
CH
HC
H3C
CH3
4+
(B) B- Redox Cycle involved in the mechanism of activation.
Figure 1.19: Proposed mechanism of n-butane activation on VO)2P2O7(73).
Chapter One - Introduction
45
O O
R
O O
+
O
O OO
OO O
COOHHOOC
HOOC
O O
O O
CH3COOH
O
CHO
O O
COX
Furan
MA
Dihydrofuran
Butadiene
n-Butane
Methyl vinyl Ketone
But-3-ene-oxide
Crotonaldehyde
Maleic acid
Butirrolactone
But-1-ene-3-oxide
2-Butoxide
2-Butanone
Butenes
H2O
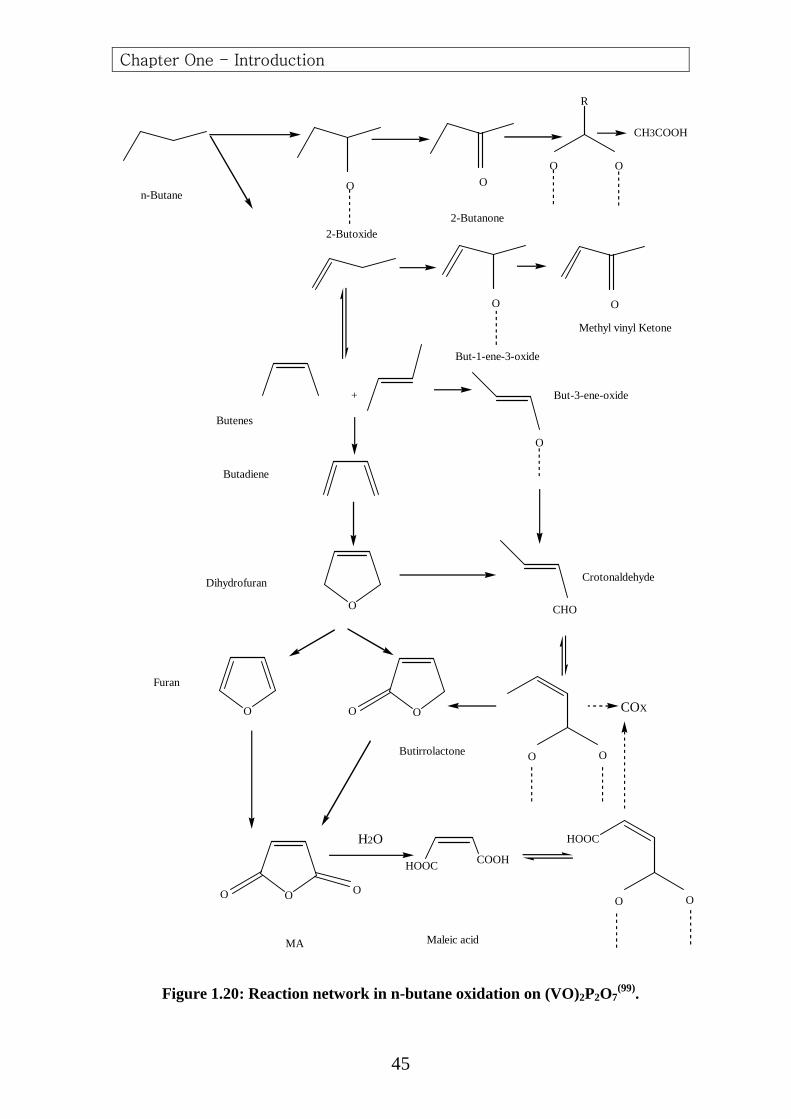
Figure 1.20: Reaction network in n-butane oxidation on (VO)2P2O7(99).

Chapter One - Introduction
46
1.8 - Role of Adsorbed & Lattice Oxygen Species
In the selective oxidation of n-butane to (MA) on vanadyl
pyrophosphate (VO)2P2O7 catalysts, many results have been reported on
the respective roles of lattice and gas-phase oxygen in the formation of
partial and complete oxidation products (113).
Three types of lattice oxygen species with different reactivites are
present at the surface of vanadyl pyrophosphate (Figure 1.21) (24).
OVO
VV
P
O
VV
(a) (b) (c)
Figure 1.21: The Three Types of Oxygen Species (24).
The participation of lattice oxygen is a general characteristic of metal
oxide catalyzed reaction in selective hydrocarbon oxidation (114-117).
Pepera et al. (93) concluded that the lattice oxygen ions located in the
top few surface layers are responsible for the oxidation of n-butane to
(MA, CO and CO2). Another authors (106 , 117, 118) obtained experimental
evidence to support this conclusion. Abon and co-workers (119) claimed on
the basis of isotopic labeling results that only lattice oxygen is active for
the formation of (MA) and other products. The circulating fluidized-bed
riser reactor technology for (MA) production described in the literature

Chapter One - Introduction
47
(18,21,119) is based upon the fact that, under anaerobic conditions, the lattice
oxygen of (VPO) can selectively oxidize butane to (MA) (18,21,119).
This problem becomes more complex when oxygen is co-fed with
butane under steady state reaction conditions employed in industrial fixed-
bed reactor processes. In addition to the conventional Mars-Van Krevelen
mechanism, where lattice oxygen is the active agent for butane oxidation,
gas-phase oxygen, surface lattice oxygen and / or activated chemisorbed
oxygen have all been proposed as important oxidants in the formation of
Maleic anhydride or in the formation of unselective products, (CO and
CO2)(96,107,120-122). For example, Trifiro et al. (95) proposed that adsorbed
oxygen is responsible for selective oxidation and that it is involved in the
oxygen insertion steps required for the formation of (MA). Ebner and co-
workers (95,106) concluded that adsorbed oxygen is selective only in the
(MA) formation step. They proposed that two types of oxygen are involved
in butane oxidation: surface lattice oxygen that is responsible for ring
closure, and activated chemisorbed oxygen, (O*), that is involved in the
further step of (MA) formation. Rodemerck et al. (121) reported that
adsorbed oxygen is active but not selective, i. e.; it can only produce
(CO2). In contrast to all of these studies, Zazhigalov et al. (122) concluded
that (MA) formation over (VO)2P2O7 is mainly due to gas- phase oxygen.
The controversy in the literature about the roles of lattice and
chemisorbed oxygen calls for further clarification of the puzzle. Wang and
co-workers (123) used a novel microbalance reactor to carry out kinetic
analyses of butane oxidation by (VPO) catalysts and of the oxidation of
partially reduced (VPO) with oxygen .These authors conclude from there
experiments that both lattice oxygen and adsorbed oxygen on (VPO)
catalyst can selectively oxidize butane to (MA). Under aerobic conditions,
the oxidation of butane by adsorbed oxygen species is much faster than
by lattice oxygen.

Chapter One - Introduction
48
The first attempt to rationalize the problem of types of oxygen species
involved in the mechanism of C-4 alkene transformation to (MA) was
reported by Weiss et al. (124) , vanadium-oxygen double bonds (V=O) were
suggested to be the active sites both in:
The oxydehydrogenation step from butenes to butadiene, the
consecutive steps of oxygen insertion to (MA).The first step involves a
homolytic (C-H) dissociation of the adsorbed olefine and formation of the
allyl radical coordinated to a metallic transition ion as a surface π-allyl
complex. The allyl carbcation obtained can form the diene by loss of a
proton through reaction with a nucleophilic agent. The allyl ester that
forms can undergo a rapid reversible rearrangement in which each end of
the carbon of the skeleton is alternatively bonded to the lattice oxygen.
This lattice oxygen possesses a weak electrophilic reactivity. On the
contrary, electrophilic oxygen must be involved in the attack on the diene.
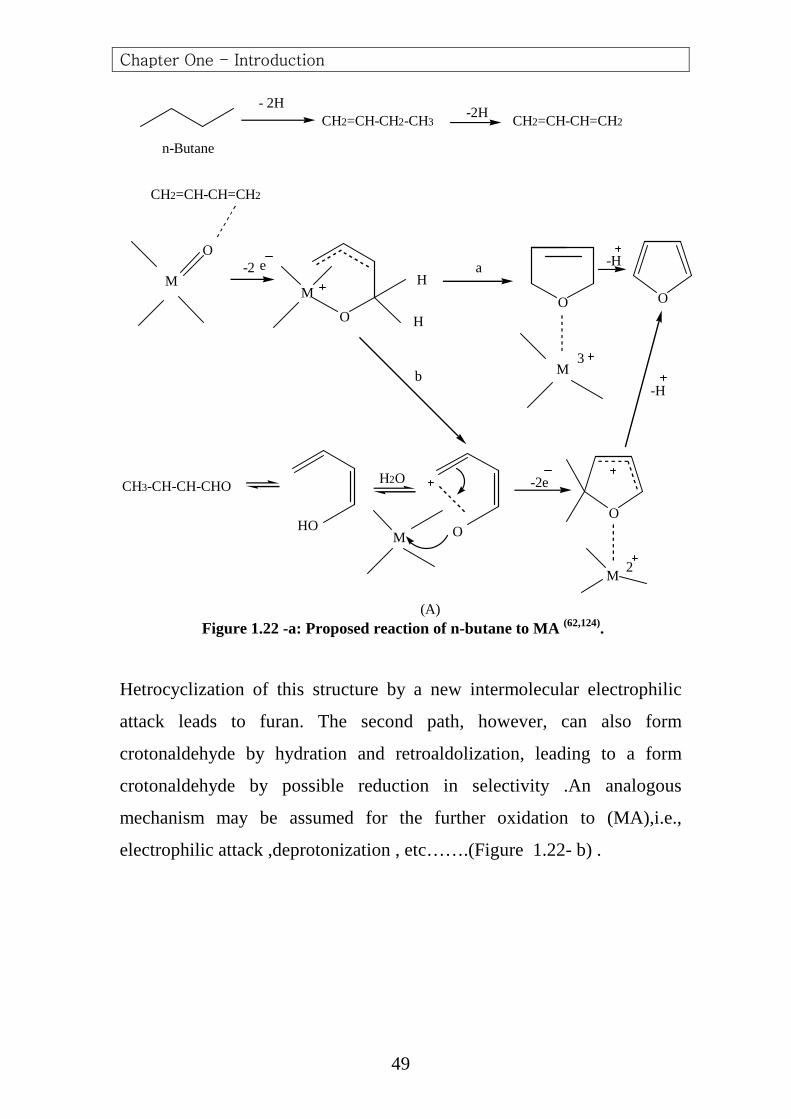
The proposed mechanism is shown in (Figure 1.22-a). The product of
attack on the diene can occur according to two-selective pathways, the first
one involves 1, 4- cyclization to 2, 5-dihydrofuran which is easily
dehydrogenated to furan; the second pathway is deprotonization of the
allyl carbcation to a dienolate structure.
Chapter One - Introduction
49
n-Butane
- 2HCH2=CH-CH2-CH3 CH2=CH-CH=CH2
CH2=CH-CH=CH2
O
M
O
MH
HO
M3
CH3-CH-CH-CHO
HO OM
O
M2
-2H
-2 e a
O
b
H2O -2e
-H
-H
(A) Figure 1.22 -a: Proposed reaction of n-butane to MA (62,124).
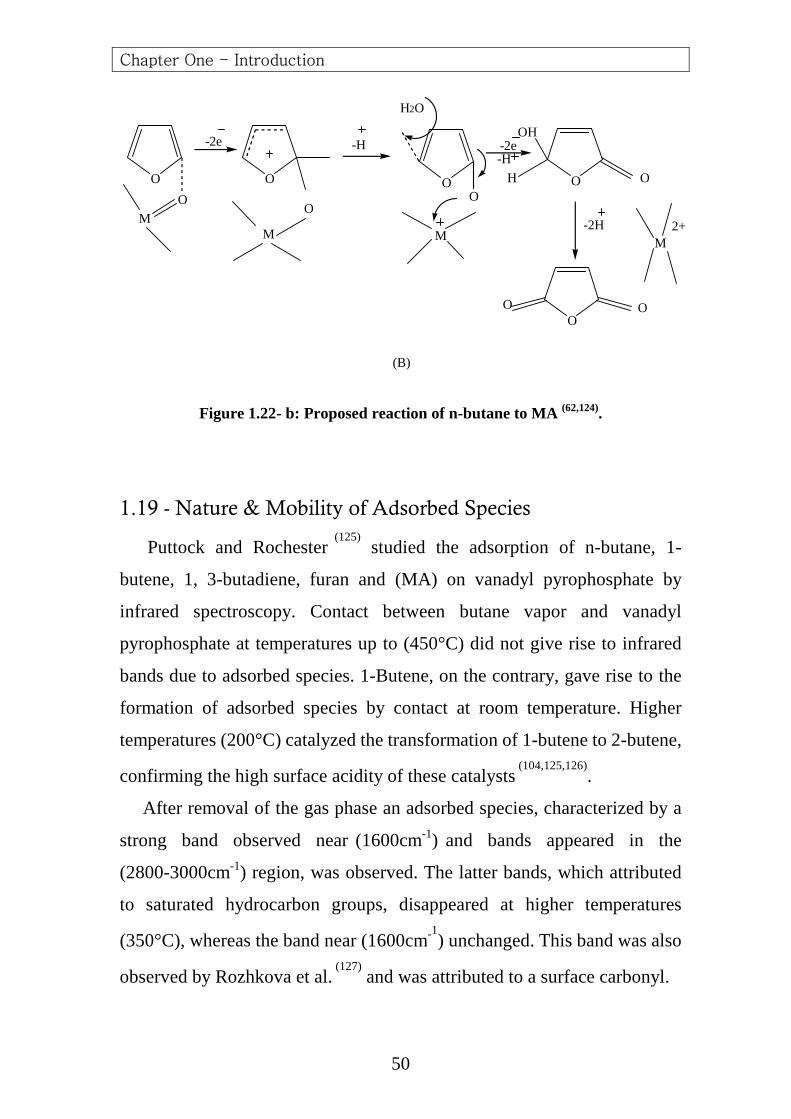
Hetrocyclization of this structure by a new intermolecular electrophilic
attack leads to furan. The second path, however, can also form
crotonaldehyde by hydration and retroaldolization, leading to a form
crotonaldehyde by possible reduction in selectivity .An analogous
mechanism may be assumed for the further oxidation to (MA),i.e.,
electrophilic attack ,deprotonization , etc…….(Figure 1.22- b) .
Chapter One - Introduction
50
O
-2e
O
M
O
O
M
-H
O
H2O
O
M
-2e-H
O
OH
OH
-2H
OOO
M2+
(B)
Figure 1.22- b: Proposed reaction of n-butane to MA (62,124).
1.19 - Nature & Mobility of Adsorbed Species
Puttock and Rochester (125)
studied the adsorption of n-butane, 1-
butene, 1, 3-butadiene, furan and (MA) on vanadyl pyrophosphate by
infrared spectroscopy. Contact between butane vapor and vanadyl
pyrophosphate at temperatures up to (450°C) did not give rise to infrared
bands due to adsorbed species. 1-Butene, on the contrary, gave rise to the
formation of adsorbed species by contact at room temperature. Higher
temperatures (200°C) catalyzed the transformation of 1-butene to 2-butene,
confirming the high surface acidity of these catalysts (104,125,126)
.
After removal of the gas phase an adsorbed species, characterized by a
strong band observed near (1600cm-1) and bands appeared in the
(2800-3000cm-1) region, was observed. The latter bands, which attributed
to saturated hydrocarbon groups, disappeared at higher temperatures
(350°C), whereas the band near (1600cm-1) unchanged. This band was also
observed by Rozhkova et al. (127)
and was attributed to a surface carbonyl.

Chapter One - Introduction
51
A similar spectrum of adsorbed species was found in the interaction of
furan with the catalyst. The primary interaction of furan and vanadyl
pyrophosphate was suggested to involve coordination between the oxygen
atoms of associatively adsorbed furan molecules and coordinatively
unsaturated exposed vanadium cations with Lewis acid properties. Furan
showed a slight oxidation to maleic anhydride in the absence of oxygen but
the presence of oxygen promoted maleic anhydride was primarily non
dissociatively adsorbed on (VO)2P
2O
7, but two further bands at (1560 and
1450cm-1), attributed to carboxylate anions, suggested a partial oxidation
of the adsorbed (MA).
Weing and Schrader (74)
studied by infrared spectroscopy the n-butane
interaction with (VPO) catalysts with variable (P/V= 0.9, 1.0 and 1.1)
using an in situ FTIR cell. These authors presented evidence for the
presence of reactant (n-butane), partially oxidized product (MA),
combustion products (CO, CO2, and H2O) and reactive surface species
(Maleic acid and olefins) on the vanadyl pyrophosphate. Another
observations reported by Pepera and co-workers (93)
agree with the
hypothesis that the catalytic process in this complex reaction involves the
shuttling of hydrogen a way from and oxygen toward the intermediate
adsorbed on the surface, and the intermediate forms a stable surface
species (which does not adsorbed or react) to (MA). This mechanism is a
key to the selectivity and the absence of intermediate products in the
reaction of n-butane conversion to (MA).
1.10- Promoted (VPO) Catalysts
Numerous attempts have been made to synthesize improved (VPO)
catalysts by adding various amounts of other metallic elements. Although

Chapter One - Introduction
52
reports of some of these attempts have appeared in literature (128-146), the
vast majority were found in the form of patents (64,128-130,133-136)
.
The effect of these promoters was studied by many authors; Ai (131,132)
studied the effect of alkali metal addition on activity and selectivity. It was
reported that the addition of less than (10% Li) showed no remarkable
effect, Ai also studied the effect of the methods of preparing V2O
5-P
2O
5-
ZrO2 catalysts on their activity and selectivity in the oxidation of n-butane
by changing the procedures of ZrO2addition. Ai found that the best
performance was obtained with the catalyst prepared by adding
simultaneously two solutions of ethyleneglycol in which ZnOCl2 and o-
H3PO
4 have been dissolved to a powder of the precursor of the (V
2O
5-
P2O
5) catalyst prepared in an organic medium. The presence of the vanadyl
pyrophosphate crystalline phase was detected in all of the catalysts. (131,132)
The addition of suitable activators to (VPO) catalyst was reported to
give an improved yield of (MA) by several patent claims (6,133-136)
using a
catalyst containing (P-V-Mo) to oxidation n-butane to (MA), this catalyst
gave a maximum (MA) yield of (88%).
Zazhigalov and co-workers (128,137) studied the effect of added alkali and
alkaline-earth metals (Li, Na, K, Mg, Ca or Ba) on the properties of (VPO)
catalysts. The results showed that the addition of alkali and alkaline earth
metals to these catalysts produced two effects, on the one hand, there was
an increase in the content of oxygen on the surface and hence an increase
in the number of acid centers, on the other hand, the presence of these
promoters increase the (P/V) ratio on the surface of the catalyst and this
explains their stabilizing effect. It should be noted that the greatest increase
in the surface content of phosphorus was observed when (Li) is added, and
(Li) was considered to be one of the best stabilizing additives

Chapter One - Introduction
53
A new catalyst belonging to the (V-P-Mo-O) system selective in the
mild oxidation of n-butane to (MA) were prepared and characterized by
Courtine et al. (139)
These attempts led to the conclusion that (Mo)could be
substituted up to (7%) in (VOPO4) phases. Thermal analysis of the
hydrated precursor, XRD and IR spectroscopies of both hydrated and
anhydrous solid phases obtained showed that the solid solutions
isostructural with (VOPO4.2H2O) and (α-VOPO4) receptively could be
formulated.
Zazhigalov and co-workers (140) studied the properties of cobalt-promot
(VO) ٢ P2O7 in the oxidation of n-butane. This study focused on the
influence of cobalt additives on the composition of the vanadium-
containing catalyst, and on cobalt's other properties which were important
for the production of industrial (VPO) catalysts. Fresh catalysts were
composed of (VOHPO4.0.5H2O) phase. After reaction the catalysts
contained (VO)2P2O7. Cobalt was uniformly distributed in the pellets. Its
presence increased the content of phosphorus at the surface, which
modified the surface acidity and in turn improved the selectivity for n-
butane oxidation. No changes of the profile of phosphorus with depth were
observed, even after (500h) on stream, the surface composition of the
catalyst remained unchanged. Cobalt stabilized the catalyst performance
by forming cobalt phosphate which reduced phosphorus losses, improves
its catalytic properties and prolonged its lifetime.
A series of (VPO) catalysts with different (P/V) ratios and with or
without (indium and TEOS) additives have been characterized by
controlled-environment (XRD, ICP, ESCA, TEM, SEM, BET) and
chemical titration used for n-butane oxidation to (MA) (141) .The best
catalyst contained slight excess (P /V) with indium and tetra ethyl ortho-
silicate (TEOS) promoters. It was found that excess P increased the

Chapter One - Introduction
54
resistance of catalyst precursors toward oxidation and resulted in (VPO)
catalyst with a large exposed platelet face of layer morphology. The
promoters reduce the thickness of the platelet face of layer morphology.
The promoters reduce the thickness of the platelet and facilitate the
oxidation of the precursor which contains disordered (VOHPO4.0.5HO)
.The combination of the promoters and excess P results in a (VPO) catalyst
with appropriate oxidizability and morphology and gave high yields of
MA.
Guliants et al. (142) investigated the oxidation of n-butane to (MA)
over a model (Nb, Si, Ti, V and Zr) promoted bulk (VPO) and supported
vanadia catalyst , The promoters were concentrated in the surface region of
the bulk (VPO) catalysts .For the supported vanadia catalyst ,the vanadia
phase was present as a two-dimensional metal oxide over layer on the
different oxide supports (TiO2,ZrO2,Nb2O5,Al2O3 and SiO2) .No
correlation was found between the electronegativity of the promoter or
oxide support cation and the catalytic properties. Both promoted bulk
(VPO) and supported vanadia catalysts containing surface niobia species
were the most active and selective to (MA). These data suggested that the
activation of n-butane on both the bulk and supported vanadia catalysts
probably required both surface redox and acid sites, and that the acidity
also played an important role in controlling further kinetic steps of n-
butane oxidation.
The presence of (Na) in technical grade (V2O5) leads to solid solution
formation, this phenomenon lead to prepare VOPO4.2H2O with a new (P/V
=1.1). (1)
Sajip and co-workers (143) described and discussed the effect of Co and
Fe doping on (VPO) catalyst, prepared by organic method (with
isobutanol). At low levels, both Co and Fe dopants significantly enhanced
the selectivity and the intrinsic activity to MA. A combination of powder

Chapter One - Introduction
55
X-ray diffraction, P NMR spin-echo mapping spectroscopy and
transmission electron microscopy ,together with catalysts tests data, was
utilized to analyze the origin of the effects of Co doping. Co appears to be
essentially insoluble in crystalline (VO)2P2O7 and was preferentially
distributed in and stabilized an amorphous (VPO) material .It is suggested
that the origin of the promotional effect of (Co) was associated with its
interaction with the disordered (VPO) phase. The same techniques have
been used to analyze the Fe-doped catalyst, but at present it is not possible
to be definitive concerning the specific location of the Fe-dopant within
the phases present .Previous studies have indicated that (Fe) can form a
solid solution within (VO)2P2O7 and therefore it is probable that the (Fe)
amorphous vanadium phosphate phases are formed in the catalyst system.
Other authors (144) studied the effect of (Bi) on (VPO) catalyst, they
found that the incorporation of (Bi) into the (VPO) lattice lowered the
overall vanadium oxidation state from (4.24 to 4.08) .It also lowered both
the peak maximum temperature for the desorption of oxygen from the
lattice from (1001 K) (undoped) to (964 K) with shoulder at (912 K). The
total oxygen desorbed from the Bi-doped catalyst was only one-fourth that
of the undoped catalyst, while the amount of oxygen removed by TPR was
roughly the same for both catalysts. These results suggested that in
anaerobic oxidation, the Bi-doped catalyst will have roughly the same
activity as in undoped catalyst in C-4 hydrocarbon oxidation but would
have a higher selectivity to products such as olefins and (MA).
The barothermal and mechanochemical treatment of (VPO-Bi)
precursor was investigated (145), the barothermal treatment led to increase of
the relative ratio (001) plane of precursor without changes of the phase
composition. The (P/V) surface ratio increased more than two times and
the phosphorus surplus forms the islands on catalyst surface which
decrease the available active surface fragments. The change of Bronsted /

Chapter One - Introduction
56
Lewis acidity ratio of surface as result of treatment also was observed. The
activity of the catalyst in n-butane oxidation less change up to several
value of P/V ratio and decreased with its growth. The selectivity to (MA)
increase (more than 10% mol. %) practically in all interval of the (P/V)
ratio changes. Good correlation was observed between the selectivity to
Maleic Anhydride (MA) and Bronsted acidity of the catalyst.
The mechanochemical treatment of precursor showed an enhancement of
n-butane conversion and an improvement in (MA) selectivity and yield.
The sample milled in water exhibited a rise in conversion to (91%) but
selectivity increased only some percents. The maximal increase of the
selectivity after treatment in ethanol (more than 15 mol. %) and growth of
activity (but only 5-6 %) was observed. The correlation between the
changes of selectivity and Bronsted acidity was established (145).
1.11- Preparation of the catalysts
In accordance with the growing importance of the C-4 partial oxidation
route to (MA), numerous patents (16,19,20,146,147) have been awarded in the
past years for process which involved the preparation of (VPO) catalyst.
Various routes are available to prepare Vanadyl Hydrogen Phosphate
Hemihydrate VOHPO4.05H2O precursor, as outlined in Figure 1.23:
V2O5 + H3PO4 + i-BuOH
Organic Route
V2O5 + NH2OHHCl
VOHPO4.0.5H2O
(VO)2P2O7
Aqueous Route
+ H3PO4
Figure 1.23: Synthesis routes for vanadyl pyrophosphate catalysts (97).
The aqueous route entails the reduction ( V5+ ) by NH2OH , HCl or

Chapter One - Introduction
57
other reducing agents followed by the addition of H3PO4, VOHPO4.0.5H2O
is recovered by crystallization or evaporation.
The organic route involves the reduction of V2O5 by an organic solvent
(an alcohol), followed by the reaction with o-H3PO4 and recovery of the solid.
After recovery of the precursor, it is washed to remove trace amounts of
water soluble (V5+) compound and then calcined in nitrogen at (773) K
followed by final activation in air or n-butane/air at (673) K. It is critical that
calcination and activation be carefully controlled or the catalyst can be over
oxidized resulting in the formation of VOPO4 which is non selective
catalyst for n-butane (97).
The synthesis route and the reaction conditions during synthesis, affect the
morphology of and ultimately the catalyst performance. The precursor
prepared by the aqueous synthesis route is generally more crystalline than the
catalyst prepared by the organic route (95) .The organic synthesis route results
in platelet crystalline morphology; the size of the platelets and the way they
pack are determined by the choice of organic solvent. Isobutanol produces
rosette morphology where the platelets agglomerate. With sec. butyl or t-butyl
alcohol well formed platelets forms that do not agglomerate (148). The crystal
ordering of the precursor is also affected by the solvent. Large alcoholic
molecules, such as benzyl alcohol, appear to produced platelets with stacking
faults, deduced from the broadening of the (200) reflection in the XRD (149).
Stacking fault defects in the precursor have been correlated with improved
catalyst performance (148).
In recent years Hutchings and co-workers (149) prepared the precursor
by using V2O4 as a starting material with water as solvent. They also used
V2O4 with either H3PO4 99% or H4P2O7; reacted in autoclave at 145 ˚C to
form well crystalline VOHPO4.0.5H2O.The surface area of the precursor is
significantly enhanced when water is added as a solvent. On activation in
(n-butane / air) the catalyst surface area is increased from (4 m2/gm) for the

Chapter One - Introduction
58
precursor to (10-13 m2/gm). The selectivity to (MA) observed to be very
similar to other non- promoted (VPO) catalyst reported in the litterateur.
Although this study considered to unlikely that V2O4 can be used as a
starting material for the synthesis of commercial vanadium phosphate
catalyst, this study does show that relatively high area catalysts can be
achieved using water as solvent.
A dihydrate phases of (VPO) catalyst VOPO4.2H2O was prepared for
the first time in 1965 by Ladwig (33) by suspend V2O5 with rapped stirring
in distilled water and H3PO4 with (P/V = 7.3).The mixture was refluxed for
24 hours. The resulting precipitate was filtered, washed with distilled water
until a red color appeared then it was washed for several times with
acetone and left to dry.
No sign in the litterateur to prepare this phase with (P/V ratio =1.1).In
2000, several attempts were unsuccessfully exerted to prepare this phase in
the above ratio starting from V2O5 produced by Fluka and BDH company,
but all of these trials were not successful, on the other hand this phase was
successfully prepared starting from technical grade V2O5 with shorter
reaction time (16h), due to the intercalation effect of (Na ion) which led to
form solid solution of this phase. (1)
A new method through intercalation of VOPO4.2H2O crystallites in
primary alcohol (2- propanol or 1-butanol), followed by reduction with the
alcohol, have been investigated for the preparation of catalyst precursor.
Lamellar compounds were formed consisting of (V 4+, P +5 and alkyl
group) (150).
According to Bordes and Courtine (29), the calcination of VOPO4.2H2O
at 700 °C leads to obtain α-VOPO4.
Recently Bartley et al. (151) prepare vanadium (V) phosphate using
solvent-free method, by reacting (V2O5) with (H3PO4) in the absence of
water at 150 °C ,the resulting material was designed as anhydrous VOPO4.

Chapter One - Introduction
59
This material is readily hydrate to formVOPO4.2H2O. Activation of this
phase with n-butane/air leads to formation of (α1-VOPO4), while the
reaction of anhydrous (VOPO4) with alcohol leads to the exclusive
formation of VO (H2PO4)2.
Finally, there are three important parameters affect in determining
the final characteristics and activity of these catalysts which can be
summarized as:
a- The reducing agent and solvent (8, 70, 61,152-157).
b- The (P/ V) ratio
(8,146,148,155).
c- Activation conditions (29, 71)
.

Chapter One - Introduction
60
Conclusion of Bulk (VPO) catalyst
Oxidation of n-butane on the unsupported or bulk (VPO) catalysts is
the only known commercial process for an alkane oxidation.
A number of V (IV) and V (V) phosphates exist in the (VPO) system
and the correlation of catalytic performance with crystalline structure has
been reviewed (95,158).
Vanadyl (IV) pyrophosphate, (VO)2P2O7, has been identified as
critical for active and selective industrial catalysts (158). Some argue that the
(V5+/V4+) dimeric species in the top most oxidized layer of (VO)2P2O7 are
the active sites (8,159) , while others believe that the active sites lie within
the micro domains of crystalline vanadyl (V) orthophosphates, (β -, αII- ,
γ-, and δ-VOPO4), or (vanadyl (IV) meta phosphate) VO(PO3)2, formed
on the (100) faces of vanadyl pyrophosphate under the catalytic reaction
conditions (106). A recent kinetic study demonstrated that the best bulk
(VPO) catalysts contained only crystalline vanadyl (IV) pyrophosphate
after reaching the steady state (142). The vanadyl (IV) pyrophosphate phase
displayed preferential exposure of the (100) planes which contain vanadyl
dimers associated with active sites for n-butane oxidation (142). A high-
resolution Electron Microscopy study demonstrated that the surface (100)
planes in the fresh catalysts are covered with (1.5 nm) amorphous layered
which completely disappears within (23 days) of n-butane oxidation (96).
The kinetic studies of the bulk (VPO) catalysts further demonstrated the
similarity between the bulk (VPO) catalysts and supported catalytic
systems.
The catalytic activity of the bulk (VPO) catalysts was confined to a
very thin surface region of the (100) crystalline planes of vanadyl (IV)
pyrophosphate (1-2 atomic layers) (92).These findings suggested that the
crystalline vanadyl (IV) pyrophosphate phase in the bulk (VPO) catalysts
functioned as a support for the active surface. Such (VO)2P2O7 support

Chapter One - Introduction
61
stabilized some specific surface termination of the (100) planes with
(V4+/V5+) species and phosphate groups without contributing its lattice
oxygen to n-butane oxidation. Unfortunately, various spectroscopic
techniques, such as (Raman, ESR, UV-Vis, IR and EXAFS/XANES), are
unable to provide information about the molecular structure of the surface
present in the bulk (VPO) catalysts because of the much stronger signals
from the catalyst bulk than the catalyst surface. Limitations of both the
bulk and surface characterization techniques currently available, coupled
with the complex solid-state chemistry of vanadium phosphates, have led
to considerable confusion and contradictions in the literature concerning
the identity of the active sites involved in different steps of n-butane
oxidation to (MA) over the bulk (VPO) catalysts.
1.12- Supported (VPO) Catalyst:
Supported metal oxide catalysts consist of an active metal oxide
component, e.g. vanadia, deposited at the surface of an oxide support, such
as SiO2, TiO2 and Al2O3.
Currently, considerable interest is devoted to study selective oxidation
of alkanes on supported vanadia catalysts. The selective oxidation of
alkanes is highly desirable due to their potentially low environmental
impact and the relatively low cost of raw materials. Some typical systems
being studied are partial oxidation of n-butane to (MA) on supported
(VPO) catalysts (105,160) and oxidative dehydrogenation of n-butane (161).
Many fundamentals questions still remain unanswered about supported
vanadia catalysts despite the importance of this catalytic system.
This section concerns with structure-property relationships in C4-
hydrocarbon oxidation on supported vanadia catalysts.

Chapter One - Introduction
62
1.12.1-Molecular Structure of Vanadia Species in Fresh Catalysts
Structure of supported vanadia catalysts is a strong function of the
surface coverage. Surface coverage of vanadia over layer supported on
metal oxides has been determined experimentally using several
spectroscopic techniques, such as (Raman, IR, XPS, 51V NMR and UV-
Vis), as well as temperature-programmed reduction (TPR) and redox
reactions. Among these techniques, Raman spectroscopy is particularly
discriminating between the surface vanadia species and the crystalline
(V2O5). Characteristic Raman bands of polymeric vanadia species
corresponding to the (V-O-V) stretches, appear below (1000 cm-1) at high
coverages, while the isolated vanadate species present at sub monolayer
coverages produce only one Raman band of the terminal (V=O) bond at
(1020-1040cm-1). The finger print Raman features allow detecting
monolayer formation immediately prior to appearance of crystalline
(V2O5). Based on Raman spectroscopy measurements, monolayer surface
coverage on a number of oxide supports (Al2O3, TiO2, ZrO2, Nb2O5 and
CeO2) was found to be (7-8 VOx/nm2) (162) where nm is nanometer.
Vanadia supported on silica exhibited unusually low monolayer surface
coverage of only (0.7VOx/nm2). Lower density and reactivity of the
hydroxyls explain such low monolayer surface coverage on the silica
surface, which anchor vanadia species to the support. However,
commercial grade supports, such as pigment grade Titania, typically
contain monolayer quantities of (P, Na, K, Ca, etc.), which interact with
surface vanadia to form an amorphous phase (163). In such systems, the
surface vanadia titrates both the oxide support and the surface impurities,
delaying formation of crystalline V2O5 until several vanadia monolayers
are formed. Thus, the observed "monolayer'' vanadia coverage in such
impurity-containing systems can be (2-4) times greater than that found
above for the well defined supported catalysts.

Chapter One - Introduction
63
A number of synthesis methods were used to prepare supported
vanadia catalysts, non aqueous impregnation with vanadium alkoxides (169,170), such as incipient impregnation of support powder like (TiO2) with
aqueous solution of NH4VO3 in oxalic acid (164). Aqueous impregnation
with vanadium oxalate is favored in commercial preparation due to its high
solubility in water and the absence of undesirable volatile organic solvents.
Under ambient conditions the supported catalysts contain multilayers of
adsorbed water. Under such conditions the bridging (V-O-support) bonds
are hydrolyzed and the hydrated vanadia species are dissolved in the thin
aqueous layer.
A new generation of supports based on ceramics with high thermal
conductivity and without oxygen atoms were prepared and tested to
disperse the active (VO)2P2O7 phase (165). The best example is a new
relatively large specific surface area (β-SiC < 20 m2/g) prepared via the
shape memory synthesis. The (β-SiC-supported VPO) approved to be a
much better catalyst in terms of (MA) selectivity and yield than its bulk
equivalent, in fixed-bed configuration of reactor. The ability of this
ceramic to well disperse (VPO) was explained by the presence of an inter
phase (a glue) containing the (V, P, Si, C and O) elements, which did not
interfere with the formation of the active and selective (VPO) phase from
the (VOHPO4.0.5H2O).The real nature of this inter phase still needs to be
clarified. The high (MA) selectivity obtained on (VPO/β-SiC) was
attributed to both the absence of microporosity on the support and more
importantly, to the heat sink role of this support avoiding the formation of
hot spots on the catalytic sites and protecting the product, MA ,from
further oxidation into CO and CO2.
The uses of other exotic heat conductive materials as support such as
(Si3N4) and (BN) ,proves the validity of the concept of heat transfer from

Chapter One - Introduction
64
the surface to the bulk of the catalyst, without taking into account the
macroscopic transfer of heat outside the reactor (165).
It was finally demonstrated, from an industrial point of view ,that the
presence of the (β-SiC) support could provide high volume yield of (MA/
volume of catalyst) in a fixed-bed reactor because it was possible to work
with relatively high concentrations of n-butane ,contrary to what is
normally possible with the conventional bulk catalyst .In addition because
of the good dispersion, a large fraction of the expensive unused internal
part of the bulk active phase was replaced by cheap and light (β-SiC)
support .The optimal amount of active phase was found at (30wt %) of the
total composite. Finally, also demonstrated was the protective role of the
support on the thermal stability of the reactive phase which was much
more resistant to overheating accidents than the bulk material.
This new concept of heat control is currently being studied and used
for other similar problems where nonoxidic conductive materials are
potentially the answer to the difficulties that have been encountered in
many applications.
1.12.2- Molecular structure in dehydrated state
The moisture adsorbed in supported vanadia catalysts under ambient
conditions may be removed by heating supported catalysts in non reducing
oxygen containing atmosphere at elevated temperature, (573-973 K). Such
treatment also helps maintain the (5+) oxidation state of the surface
vanadia species. At temperatures below (473 K), the Raman stretch of the
terminal (V=O) bond occurring at (1020-1040 cm-1) is shifted significantly
downward due to extensive hydration of the surface vanadia. At
temperatures above (473 K) a small amount of moisture hydrogen bonded
to surface vanadia results in a few cm-1 downward shift of the Raman

Chapter One - Introduction
65
(V=O) band. At these temperatures (18O) labeling experiments showed that
the reversibly adsorbed moisture is also capable of rapidly undergoing
(18O) exchange with the terminal (V=O) bonds.
The structural studies suggested that the dehydrated vanadia species
on (Al2O3,TiO2, ZrO2, Nb2O5 and CeO2) was predominantly present as
isolated and polymeric tetrahedral VO4 units (Figure 1.24).The (18O)
labeling experiments of supported (VOx/ Al2O3, TiO2, ZrO2) showed that
these surface species possessed only one (terminal V = O) bond and three
bridging (Vـ�O-support) bonds for the isolated species, while the polymeric
surface species has only one (Vـ�O-support) bond per vanadium atom ,the
other two are being bridging V-O-V bonds (Figure 1.24). (166)
Spectroscopic techniques (X-ray absorption, Raman and UV-Vis.)
were utilized to monitor the effect of dehydration for vanadia supported on
(SiO2, Al2O3, TiO2, ZrO2 and HfO2) prepared with (VOx) surface densities
ranging from (0.46- 11.1 VOx/nm2) (166). UV-Vis. and near –edge spectra
indicate that supported (VOx) species exist in larger domains and have
higher – coordinate centers when hydrated. Dehydration consistently leads
to a breakup of these domains. Raman spectra suggest that (V=O) bonds in
mono and polyvanadate species are most susceptible to hydration. On all
supports, dehydration leads to the development of monovanadate species.
Polyvanadate on (TiO2, ZrO2, and HfO2) also undergo structural changes
when hydrated. Interpretation of this is more difficult because of the broad
Raman bands which allow only qualitative assignments. Support material
plays an important role in determining the extent of hydration, but this role
might arise from the ability to support polymeric vanadia species.

Chapter One - Introduction
66
Figure 1.24: Molecular structure of:
(A) Dehydrated isolated
(B) Polymerized vanadia species on oxide supports (166).
1.12.3- Structural changes during Hydrocarbon Oxidation
Wachs et al (161) investigated the oxidation of n-butane to (MA) over a
series of model-supported vanadia catalysts where the vanadia phase was
present as a two- dimensional metal oxide overlayer on the different oxide
supports ( TiO2, ZrO2, CeO2, Nb2O5, Al2O3, and SiO2). No correlation was
found between the properties of the terminal (V = O) bond and the butane
oxidation turnover frequency (TOF) which is mean the number molecules
converted per vanadium per second , during in situ Raman spectroscopy
study. Furthermore, neither the n-butane oxidation (TOF) nor (MA)
selectivity was related to the extent of reduction of the surface vanadia
species. The n-butane oxidation (TOF) was essentially independent of the
surface vanadia coverage, suggesting that the n-butane activation requires
only one surface vanadia site. The (MA) TOF, however, increased by a
factor of (2–3) as the surface vanadia coverage was increased to monolayer
coverage. The higher (MA) (TOF) at near monolayer coverages suggests
that a pair of adjacent vanadia sites may efficiently oxidize n-butane to
Maleic Anhydride. Other factors may also play a contributing role such as
(increase in surface Bronsted acidity and decrease in the number of

Chapter One - Introduction
67
exposed support cation sites. Varying the specific oxide support changed
the n- butane oxidation (TOF) by 50:
Ti > Ce > Zr ~ Nb > Al > Si
As well as the (MA) selectivity.
The (MA) selectivity closely followed the Lewis acid strength of the
oxide support cations:
Al > Nb > Ti > Si > Zr > Ce
The addition of acidic surface metal oxides (W, Nb, and P) to the surface
vanadia layer was found to have a beneficial effect on the n- butane
oxidation (TOF) and the (MA). The creation of bridging (V–O–P) bonds
had an especially positive effect on the (MA) selectivity .
1.12.4–Structure-Activity Relationships in Oxidation of
C4-hydrocarbons
Several studies on C4-hydrocarbon oxidation have been reported in the
literature (102,106,160).
Nakamura et al. (105) studied oxidation of 1-butene on alumina-
supported catalysts and reported reasonably high selectivities to (MA). The
results of their study showed that supported (VPO) catalysts could
selectively oxidize C4- hydrocarbons to (MA). They concluded that the
activity and selectivity of their supported catalysts for the oxidation of 1-
butene to (MA) were closely related to the oxidation state and the degree
of aggregation of vanadium ions. They implicated the (V4+/V5+) redox
mechanism of oxidation in which (V = O) bonds played an important role.
Mori et al. (102) studied oxidation of n-butane on titania-supported
vanadia catalysts at high temperature (693-763 K) and high n-butane
conversion. Such severe experimental conditions led to an over oxidation
of n-butane to (COx) and, likely, to combustion of the partial oxidation
products, including (MA). They found that the reaction rate under such

Chapter One - Introduction
68
conditions was proportional to the amount of (V 5+= O) species in the
catalyst and concluded that the reaction proceeded via the reduction-
oxidation or Mars-van Krevelen mechanism according to which the
surface (V = O) species contained the active oxygen.
Busca et al. (167) employed titania-supported vanadia catalysts in n-
butane oxidation and observed over oxidation of the hydrocarbon on very
active high surface area catalyst (117m2/g) at (723 K). When they
supported (10 wt% V2O5) on the low surface area Titania (18.4 m2/g),
vanadia formed micro crystals which were detrimental to selective
oxidation and led to n-butane combustion.
Owens and Kung (160) studied n-butane dehydrogenation on silica-
supported vanadia catalysts at (793) K. They observed that the isolated
vanadia species present at low surface coverage (0.53 and 0.58 wt% V)
were responsible for the high total dehydrogenation selectivity (1-butene,
cis-/trans-2-butene, and 1, 3-butadiene as shown in (Figure 1.25) (160).
1 2 3 4
Figure 1.25: Oxidation of butane on dispersed vanadia species (160).
The presence of the crystalline V2O5 species at high surface coverage
(998, 703, 526, 480, 404, 304 and 284 cm-1) Raman bands for the (6.4 wt%
V) catalyst contributed to the production of total oxidation products.
C4H9
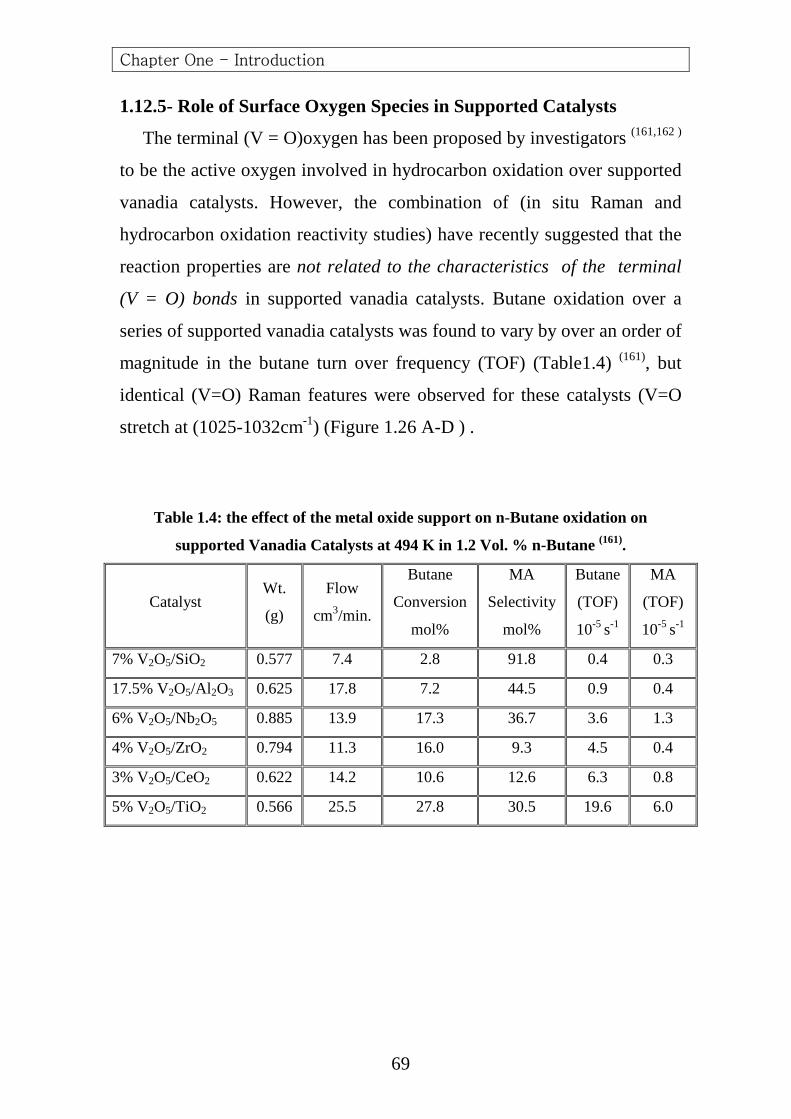
Chapter One - Introduction
69
1.12.5- Role of Surface Oxygen Species in Supported Catalysts
The terminal (V = O)oxygen has been proposed by investigators (161,162 )
to be the active oxygen involved in hydrocarbon oxidation over supported
vanadia catalysts. However, the combination of (in situ Raman and
hydrocarbon oxidation reactivity studies) have recently suggested that the
reaction properties are not related to the characteristics of the terminal
(V = O) bonds in supported vanadia catalysts. Butane oxidation over a
series of supported vanadia catalysts was found to vary by over an order of
magnitude in the butane turn over frequency (TOF) (Table1.4) (161), but
identical (V=O) Raman features were observed for these catalysts (V=O
stretch at (1025-1032cm-1) (Figure 1.26 A-D ) .
Table 1.4: the effect of the metal oxide support on n-Butane oxidation on
supported Vanadia Catalysts at 494 K in 1.2 Vol. % n-Butane (161).
Catalyst Wt.
(g)
Flow
cm3/min.
Butane
Conversion
mol%
MA
Selectivity
mol%
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
7% V2O5/SiO2 0.577 7.4 2.8 91.8 0.4 0.3
17.5% V2O5/Al 2O3 0.625 17.8 7.2 44.5 0.9 0.4
6% V2O5/Nb2O5 0.885 13.9 17.3 36.7 3.6 1.3
4% V2O5/ZrO2 0.794 11.3 16.0 9.3 4.5 0.4
3% V2O5/CeO2 0.622 14.2 10.6 12.6 6.3 0.8
5% V2O5/TiO2 0.566 25.5 27.8 30.5 19.6 6.0
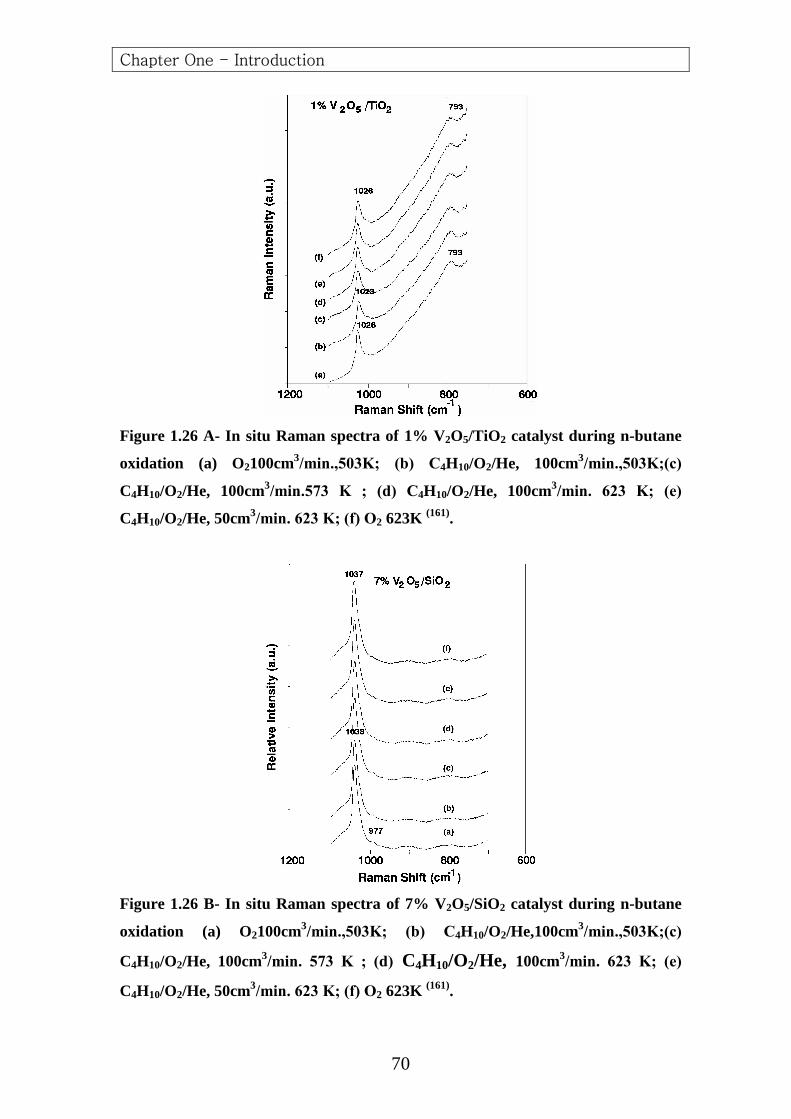
Chapter One - Introduction
70
Figure 1.26 A- In situ Raman spectra of 1% V2O5/TiO 2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He, 100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min.573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161).
Figure 1.26 B- In situ Raman spectra of 7% V2O5/SiO2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He,100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161).
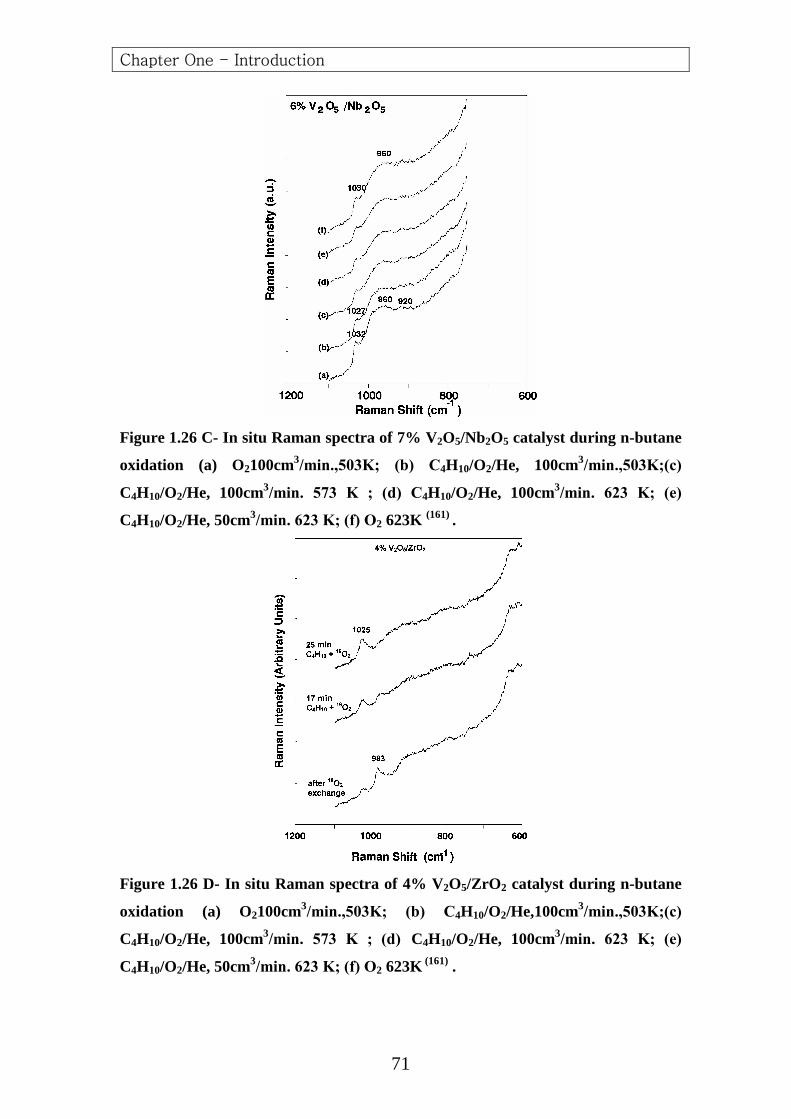
Chapter One - Introduction
71
Figure 1.26 C- In situ Raman spectra of 7% V2O5/Nb2O5 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He, 100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161) .
Figure 1.26 D- In situ Raman spectra of 4% V2O5/ZrO 2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He,100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161) .
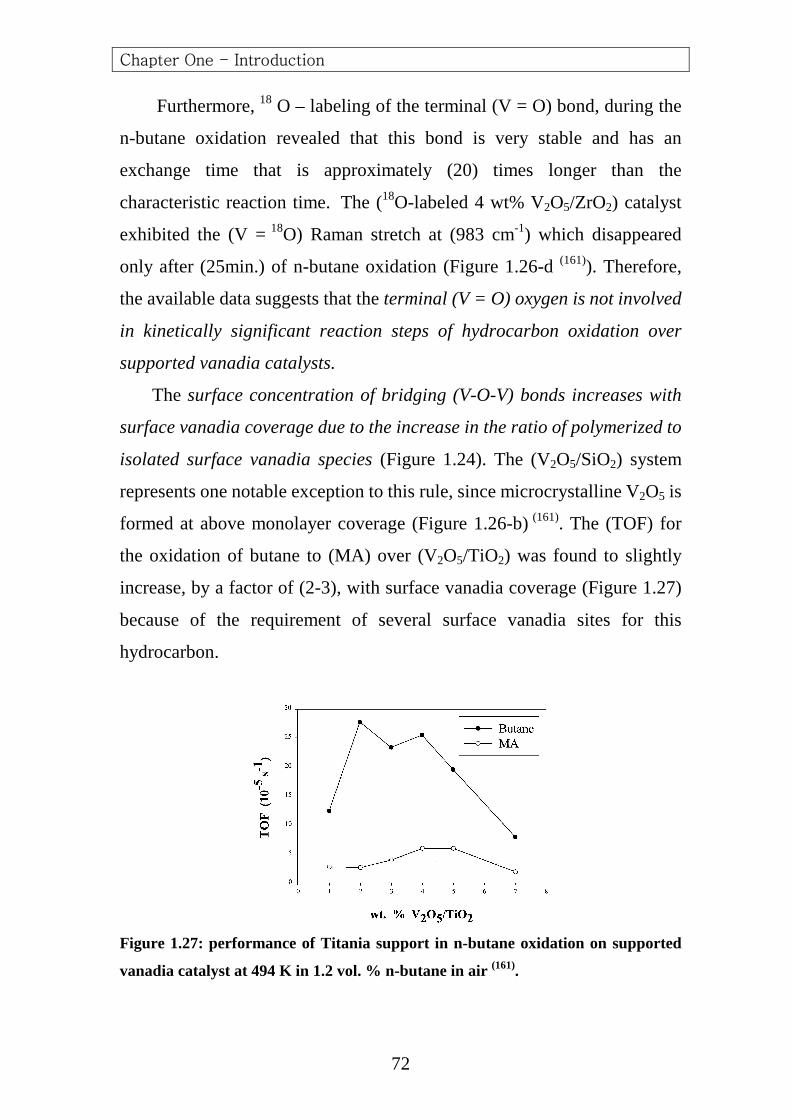
Chapter One - Introduction
72
Furthermore, 18 O – labeling of the terminal (V = O) bond, during the
n-butane oxidation revealed that this bond is very stable and has an
exchange time that is approximately (20) times longer than the
characteristic reaction time. The (18O-labeled 4 wt% V2O5/ZrO2) catalyst
exhibited the (V = 18O) Raman stretch at (983 cm-1) which disappeared
only after (25min.) of n-butane oxidation (Figure 1.26-d (161)). Therefore,
the available data suggests that the terminal (V = O) oxygen is not involved
in kinetically significant reaction steps of hydrocarbon oxidation over
supported vanadia catalysts.
The surface concentration of bridging (V-O-V) bonds increases with
surface vanadia coverage due to the increase in the ratio of polymerized to
isolated surface vanadia species (Figure 1.24). The (V2O5/SiO2) system
represents one notable exception to this rule, since microcrystalline V2O5 is
formed at above monolayer coverage (Figure 1.26-b) (161). The (TOF) for
the oxidation of butane to (MA) over (V2O5/TiO2) was found to slightly
increase, by a factor of (2-3), with surface vanadia coverage (Figure 1.27)
because of the requirement of several surface vanadia sites for this
hydrocarbon.
Figure 1.27: performance of Titania support in n-butane oxidation on supported
vanadia catalyst at 494 K in 1.2 vol. % n-butane in air (161).
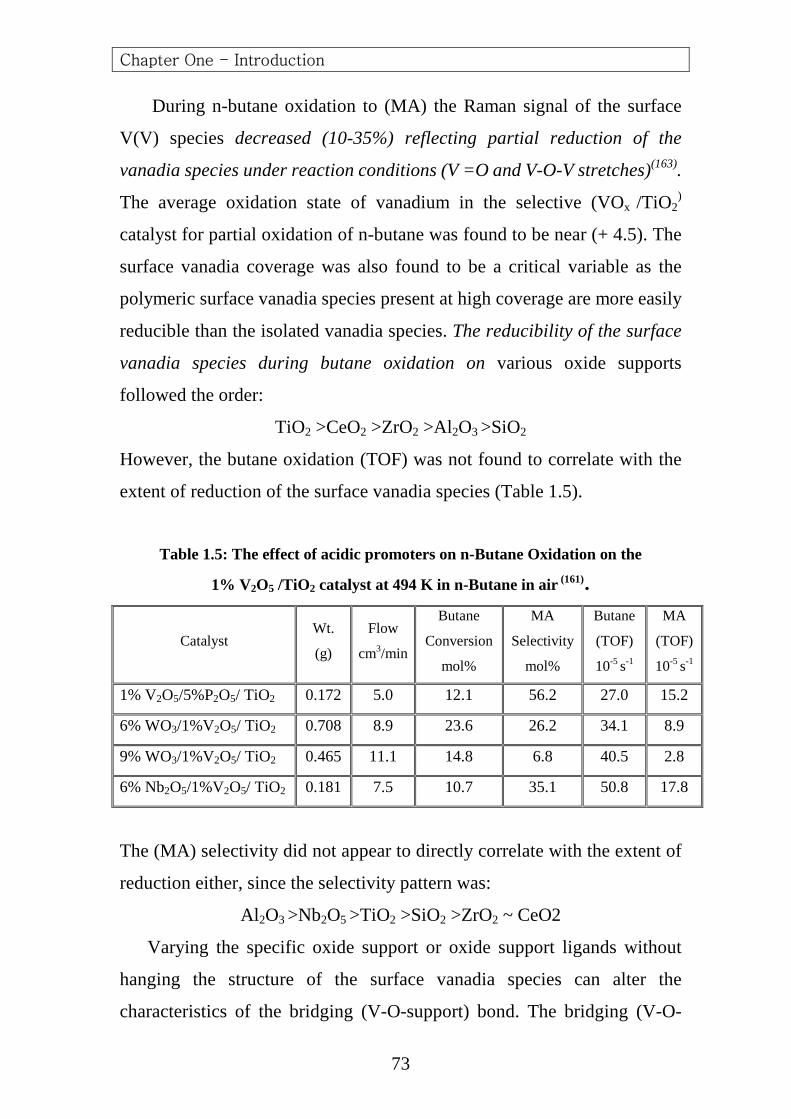
Chapter One - Introduction
73
During n-butane oxidation to (MA) the Raman signal of the surface
V(V) species decreased (10-35%) reflecting partial reduction of the
vanadia species under reaction conditions (V =O and V-O-V stretches)(163).
The average oxidation state of vanadium in the selective (VOx /TiO2)
catalyst for partial oxidation of n-butane was found to be near (+ 4.5). The
surface vanadia coverage was also found to be a critical variable as the
polymeric surface vanadia species present at high coverage are more easily
reducible than the isolated vanadia species. The reducibility of the surface
vanadia species during butane oxidation on various oxide supports
followed the order:
TiO2 >CeO2 >ZrO2 >Al2O3 >SiO2
However, the butane oxidation (TOF) was not found to correlate with the
extent of reduction of the surface vanadia species (Table 1.5).
Table 1.5: The effect of acidic promoters on n-Butane Oxidation on the
1% V2O5 /TiO2 catalyst at 494 K in n-Butane in air (161).
Catalyst Wt.
(g)
Flow
cm3/min
Butane
Conversion
mol%
MA
Selectivity
mol%
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
1% V2O5/5%P2O5/ TiO2 0.172 5.0 12.1 56.2 27.0 15.2
6% WO3/1%V2O5/ TiO2 0.708 8.9 23.6 26.2 34.1 8.9
9% WO3/1%V2O5/ TiO2 0.465 11.1 14.8 6.8 40.5 2.8
6% Nb2O5/1%V2O5/ TiO2 0.181 7.5 10.7 35.1 50.8 17.8
The (MA) selectivity did not appear to directly correlate with the extent of
reduction either, since the selectivity pattern was:
Al 2O3 >Nb2O5 >TiO2 >SiO2 >ZrO2 ~ CeO2
Varying the specific oxide support or oxide support ligands without
hanging the structure of the surface vanadia species can alter the
characteristics of the bridging (V-O-support) bond. The bridging (V-O-

Chapter One - Introduction
74
support) bond appears to be associated with the critical oxygen required
for hydrocarbon oxidation reactions since changing the specific oxide
support dramatically affects the (TOF) approximately two orders of
magnitude for n-butane oxidation to (MA) (Table1.4). The general trend
appears to be:
CeO2 >ZrO2 ~TiO2 >Nb2O5 >Al2O3 >SiO2
This suggests that bridging oxygens in the (V-O-support bonds) that are
more electronegative or basic, corresponding to oxide support cations with
lower electronegativity, are associated with the critical oxygen required
for hydrocarbon oxidation reactions over supported vanadia catalysts. The
formation of the (V-O-P) bond has a particularly positive effect on the
butane oxidation (TOF) and (MA) selectivity (Table 1.6). The
(1%V2O5/5% P2O5/TiO2) catalyst displayed the highest selectivity to (MA)
among all systems studied (Table 1.5). At (494 K in 1.2 vol. % n-butane in
air) and (12.1 mol% n-butane conversion), the selectivity to (MA) reached
(56.2 mol %). Such catalytic behavior is consistent with the above
observation that (bridging V-O) support bonds are critical in the oxidation
of n-butane to (MA).
The insight into the number of critical surface vanadia sites required in
hydrocarbon oxidation reactions can be gained by examination of the
variation of the (TOF) with surface vanadia coverage. Those reactions that
require only one surface site will exhibit a (TOF) that is independent of
surface vanadia coverage .The oxidation of butane to (MA) over titania-
supported vanadia catalysts (Table 1.6 and Figure 1.27) exhibited an
increase in (TOF) with surface vanadia coverage. This may reflect the
requirement of multiple surface vanadia sites or the influence of other
factors, such as surface acidity influence of (bridging V-O-V bonds),
structural changes, etc...
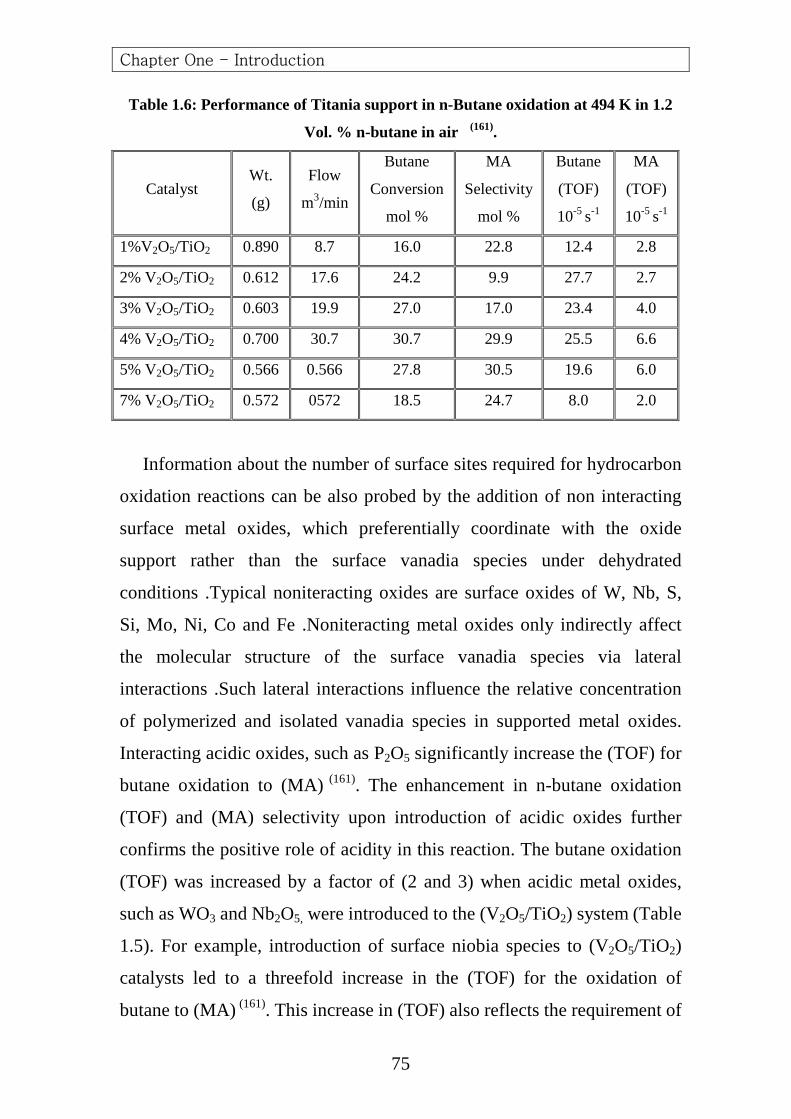
Chapter One - Introduction
75
Table 1.6: Performance of Titania support in n-Butane oxidation at 494 K in 1.2
Vol. % n-butane in air (161).
Catalyst Wt.
(g)
Flow
m3/min
Butane
Conversion
mol %
MA
Selectivity
mol %
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
1%V2O5/TiO2 0.890 8.7 16.0 22.8 12.4 2.8
2% V2O5/TiO2 0.612 17.6 24.2 9.9 27.7 2.7
3% V2O5/TiO2 0.603 19.9 27.0 17.0 23.4 4.0
4% V2O5/TiO2 0.700 30.7 30.7 29.9 25.5 6.6
5% V2O5/TiO2 0.566 0.566 27.8 30.5 19.6 6.0
7% V2O5/TiO2 0.572 0572 18.5 24.7 8.0 2.0
Information about the number of surface sites required for hydrocarbon
oxidation reactions can be also probed by the addition of non interacting
surface metal oxides, which preferentially coordinate with the oxide
support rather than the surface vanadia species under dehydrated
conditions .Typical noniteracting oxides are surface oxides of W, Nb, S,
Si, Mo, Ni, Co and Fe .Noniteracting metal oxides only indirectly affect
the molecular structure of the surface vanadia species via lateral
interactions .Such lateral interactions influence the relative concentration
of polymerized and isolated vanadia species in supported metal oxides.
Interacting acidic oxides, such as P2O5 significantly increase the (TOF) for
butane oxidation to (MA) (161). The enhancement in n-butane oxidation
(TOF) and (MA) selectivity upon introduction of acidic oxides further
confirms the positive role of acidity in this reaction. The butane oxidation
(TOF) was increased by a factor of (2 and 3) when acidic metal oxides,
such as WO3 and Nb2O5, were introduced to the (V2O5/TiO2) system (Table
1.5). For example, introduction of surface niobia species to (V2O5/TiO2)
catalysts led to a threefold increase in the (TOF) for the oxidation of
butane to (MA) (161). This increase in (TOF) also reflects the requirement of

Chapter One - Introduction
76
multiple surface metal oxide sites for this hydrocarbon oxidation. In
contrast to non interacting additives that mainly affect oxidation reactions
requiring multiple surface sites, interacting additives affect all hydrocarbon
oxidation reactions since they directly alter the structure and reactivity of
the surface vanadia sites. Interacting metal oxides, especially basic oxides,
retard the reduction of surface vanadia species. Consequently, the (TOF)
for all hydrocarbon oxidation reactions are lowered when basic additives
are introduced (163,164).
1.12.6 - Role of Acidity of Supported Vanadia Catalysts
The oxide supports possess only surface Lewis acid sites, with the
exception of silica where no Lewis acid sites exist on the surface. The
relative Lewis acid strength of these sites follows the order
Al 2O3 >Nb2O5 >TiO2 >ZrO2
In contrast to the oxide supports, unsupported V2O5 crystalline powders
displays both surface Bronsted and Lewis acid sites (167) and an increase in
the number of surface Bronsted acid sites.
Supported (VPO) catalysts typically exhibit surface enrichment in
phosphate similar to the conventional unsupported or bulk (VPO)
catalysts (106,156,161). In these catalysts the concentration of the surface
Bronsted acid sites increases with the surface (P/V) ratio. The surface
enrichment in phosphorus stabilizes the V (IV) oxidation state, which
results in decreased activity in n-butane oxidation. On the other hand, the
increased surface Bronsted acidity facilitates desorption of partially
oxidized products with acidic properties, such as (MA) leading to
significant improvement in (MA) selectivity (162).According to Grasselli st
al. (168), slight excess phosphate (P/V = 1.1-1.2) forms a protective
pyrophosphate "fence" around active surface sites at the surface preventing
overoxidation of the reactive intermediates by the active oxygen diffusing

Chapter One - Introduction
77
fast at the surface. However, the changes in the activity and selectivity of
the (VPO) catalysts at much higher phosphorus enrichment (P/V ratio near
2.0) were associated with formation of microcrystalline VO (PO3)2 (169).
Reports in the literature have demonstrated that the VO (PO3)2phase is
inactive in n-butane oxidation (169). Therefore, the catalytic behavior of the
VO (PO3)2 system is explained by the presence of other (VPO) impurity
phases, such as (VO)2P2O7 and various (VOPO4) phases (169). The oxidation
of n-butane on well-defined supported vanadia catalysts demonstrated that
the substitution of (V-O-Ti) for less reducible and more acidic (V-O-P)
bonds has a positive effect on the n-butane oxidation (TOF) and (MA)
selectivity . This observation indicates that the oxidation of n-butane
to (MA) requires both a redox site and some acidic functionality (170 ) .
1.12.7: The Role of Support
We can conclude that although supports are essentially diluents, they
play important multifunctional roles which can be summarized as
following:
1- Economic, to reduce cost by extending expensive phase in catalyst.
2- Mechanical, to give mechanical strength, to optimize bulk density, to
prove a heat sink or a heat source, and to dilute the overactive phase.
3- Geometric, to increase the surface area, to optimize the porosity of a
catalyst, to optimize crystal and particle size, to allow the catalyst
particles adopt the most favorable configuration of a catalyst.
4- Chemical, to react with the catalyst either to improve specific activity
or to minimize sintering and to accept or donate chemical entities,
porosity via a spillover mechanism.
5- Deactivation, to stabilize the catalyst against sintering and to minimize
poisoning.

Chapter One - Introduction
78
Conclusions
Significant progress had been achieved in recent years in studying
molecular structures of the surface vanadia species present in supported
metal oxide catalysts. The detailed structural information on well-defined
systems provided a foundation for developing structure-reactivity
relationships required to molecularly engineer supported vanadia catalysts
for oxidation reactions. The nature of the metal oxide support was found to
play a crucial role in defining catalytic properties of vanadia monolayer in
n-butane oxidation to (MA). The terminal vanadyl oxygen does not appear
to critically influence the reactivity properties of the surface vanadia
species during hydrocarbon oxidation reactions. The bridging (V-O-V)
oxygen plays only a minor role in enhancing the n-butane oxidation (TOF),
primarily due to the preference of multiple active sites in this oxidation
reaction. The bridging (V-O-support oxygen), however, appears to be the
most critical oxygen since its properties can change the (TOF) for
hydrocarbon oxidation reactions by as much as four orders of magnitude.
The specific phase of the oxide support as well as the specific preparation
method does not appear to influence the molecular structure or reactivity
of the surface vanadia species. The number of surface vanadia sites
required for a hydrocarbon oxidation reaction is dependent on the specific
reactant molecule. Oxidation reactions requiring only one surface vanadia
site are generally not sensitive to the surface vanadia coverage and the
presence of non interacting metal oxides. Oxidation reactions requiring
multiple surface vanadia sites are very sensitive to surface vanadia
coverage and the presence of non interacting metal oxides. However,
interacting metal oxides influence all hydrocarbon oxidation reactions
since they modify the surface vanadia sites. Acidic and basic metal oxides
also influence the selectivity of hydrocarbon oxidation reactions, but the
effect appears to be reaction-specific.

Chapter One - Introduction
79
Unlike bulk (VPO) catalysts, detailed surface structural information on
a molecular level can be obtained from model supported vanadia catalysts
containing two-dimensional over layers of surface vanadia species (172).
Raman spectroscopy provides direct fundamental surface information
about:
1- The ratio of isolated and polymerized surface vanadia species
(Fig.1.24).
2- Terminal (V=O) and bridging (V-O-V) bonds.
3- Extent of reduction of the surface vanadia species during catalysis.
4- Influence of the oxide support ligands.
5- Influence of (acidic/basic) metal oxide additives (promoters/poisons).
6- Participation of specific (V- O) bonds in catalysis (with the aid of
oxygen- 18O labeled isotope experiments.
Thus, the in situ Raman studies during oxidation of reactions over model
supported vanadia catalysts can provide new insights into the surface
properties of oxide catalysts which are not attainable with bulk metal oxide
catalysts.
The studies of the model supported catalysts provided several important
insights about the origins of the catalytic activity of the (VPO) system that
were not possible with the bulk (VPO) catalysts. Firstly, it was shown that
the (V=O) oxygen was not involved in n-butane oxidation to (MA) and
that the (V-O-support) bonds contained active oxygen for n-butane
oxidation. The (V-O-P) bonds were particularly beneficial for both the
activity and selectivity of the supported vanadia catalysts (Table 1.6 (163)),
suggesting that this oxygen species may be responsible for selective
oxidation of n-butane. Secondly, it was demonstrated that the oxidation of
n-butane to (MA) could occur at a single vanadia site, although adjacent
sites were more efficient (Figure 1.27).

Chapter One - Introduction
80
The results of n-butane oxidation on titania-supported vanadia
catalysts suggested that isolated surface vanadia species are capable of
n-butane oxidation to (MA), although multiple vanadia sites are more
efficient in this oxidation. Microcrystalline vanadia was found to be
detrimental for the process of (MA) formation. The kinetic studies of the
supported vanadia system provided more direct evidence that the multiple
vanadia sites were better at oxidizing n-butane to (MA).
The selectivity of the supported catalysts to (MA) is correlated with
the Lewis acid strength of the metal oxide promoters. Especially high
selectivity to (MA) was found when the (V-O-P) bonds formed after
addition of phosphorus oxide in accordance with previous observations in
supported and unsupported bulk (VPO) catalysts. These findings indicate
that the supported vanadia catalysts represent a suitable model system
capable of providing insights into the mechanism of n-butane oxidation on
bulk (VPO) catalysts.
Lastly, the use of acidic additives with supported (VPO) catalysts
further demonstrated their similarity to the bulk (VPO) system. Moreover,
it shed some light on the possible role of promoters that improve the
performance of the bulk (VPO) catalysts. The acidic additives used in the
supported vanadia catalysts enhance those catalytic oxidations which
require a combination of a redox and acidic site for selective catalysis. The
acidic additives promoted both the rate of n-butane oxidation and the (MA)
formation on supported vanadia catalysts (Table 1.6).
The supported (VPO) system proved to be a good model for the bulk,
i .e. unsupported (VPO) system. The in situ Raman studies of the
supported vanadia catalysts provided several important insights into the
nature of the selective n-butane oxidation that were not possible with
unsupported vanadium-phosphorus-oxygen (VPO) catalysts due to the
experimental limitations. The many discovered similarities between the

Chapter One - Introduction
81
bulk and supported (VPO) systems have important implications for the
design of new selective (VPO) catalysts. The nature of the oxide support,
the surface coverage of the active vanadia species and the acidity of metal
oxide additive are the most important determinants of the catalytic activity
and selectivity in n-butane oxidation. These structure-reactivity
relationships pave the way for molecular engineering of selective active
sites for hydrocarbon oxidation in supported (VPO) catalysts.
The aim of work
The aim of our previous work was precisely to use technical grade
vanadium pentoxide locally produced to prepare a catalyst to convert
n-butane to MA.
Our aim in this work is to study the effect of Titania (anatase) on the
physical, chemical and structural properties of our previous already
tested catalyst in order to prepare an industrial catalyst to increase the
feasibility of vanadium pentoxide production in our country and supply
further prospects to use it.

Chapter Two - Experimental
83
2.1: Samples Analysis
Ten techniques are used for analysis were employed in this work.
Eight of them were carried in international laboratories in four countries
(United States of America, Germany, United Arab Emirates, and
Ukraine) and two of them were performed locally as shown below:
1-The determination of vanadium, nickel, iron, sodium, calcium,
potassium, magnesium, molybdenum, silicon and aluminum were
carried out by atomic absorption spectrophotometer Model AA-680
Shimadzu, vanadium, sodium, silicon, aluminum, molybdenum and
calcium were determined in an N2O- acetylene flame at (318.4) nm,
(589.0) nm, (251.6) nm, (309.3) nm, (313.3) nm, and (422.7) nm
Principle lines. Nickel, iron, potassium, magnesium were determined in
Air- Acetylene flame at (232.0) nm, (248.3) nm, (766.5) nm, and
(285.2) nm Principle lines respectively. This measurement was carried
out in Ibn Sena State Company-Baghdad- Iraq.
2-The qualitative identification of the samples were performed on
STOE STADIP X-ray diffractometer with Cu Kα-radiation in Fritz-
Haber Institute, Berlin, Germany.
3-Thermogravimetric analysis (TG) and differential thermo-
gravimetric analysis (DTA) were carried out by MOM, Hungary, Q-
derivatograph , system F.Paulik, J.Paulik, L.Erday , in flowing air at
(10°C/min.). Determinations were carried in Institute for Sorption and
Problems of Endoecology National Academy of Sciences of Ukraine,
Ukraine

Chapter Two - Experimental
84
4- Fourier Transform Infra - Red analysis using Fluka, technical
gradeV2O5, and prepared phases were run using KBr disk by FT-IR-
8300 Fourier transform infrared spectrophotometer, Shimadzu between
4000-400 cm-1 in Al-Nahrain University-Collage of Science-Chemistry
Department- Baghdad – Iraq.
5- Laser Raman spectra were obtained in Jovin Yvon – Horiba Lab
Raman equipped with a CCD detector and with three different
excitation sources having wavelengths using (He-Ne laser) to give
632.8 nm (Red laser) and 514 nm (green laser). Backscattering
geometry of the micro-Raman technique (X 50) was used to record
spectra of the powder samples. Typical laser powers ranged from 3.1 to
5.0 mW was used; integration time was around 30 s for each spectrum;
and up to 10 Raman spectra were averaged for each sample. The
analysis was carried out in School of Chemical Engineering and
Materials Science- University of Oklahoma-U.S.A.
6- Specific Surface Area was determined by method of argon
temperature desorption on measured by GASOCHROM-1 equipment
(Mosneftekip).The mixture containing 8 vol. % of argon in helium (Ar
in He) was used. The standard – Al2O3 was with SSA = 4.2 m2/g (BET
measurement). The obtained value of specific surface area was the
average value from 5 measurements for one sample. The determination
error is 4-5 %.The specific surface area was calculated according to the
following equation:
SSA (sample) = (SAr sample/SAr standard) X SSA st X (g st / g sample)

Chapter Two - Experimental
85
This analysis was performed in Institute for Sorption and Problems of
Endoecology National Academy of Sciences of Ukraine, Ukraine.
7- The composition and the oxidation state of the surface of prepared
samples were obtained by X- Ray Photo Electron Spectroscopy (XPS).
Data were recorded on a physical electronics PHI 5800 ESCA system
with a background pressure of an approximately 2.0 X 10-9 Torr. The
electron take- off angle was 45° with respect to the sample surface. An
800-µm spot size and 23 eV pass energy were typically used for the
analysis. The binding energies were corrected by reference to the
carbon 1s line C/s line at 284.8 eV for hydrocarbon. A nonlinear
Shirley-type background was used for the area analysis of each peak.
The fitting of the XPS spectra was carried out with asymmetric peaks,
using the MulitPak software from Physical Electronics in School of
Chemical Engineering and Materials Science - University of Oklahoma-
U.S.A.
8- The surface acidic properties of the samples were studied by
adsorption of pyridine (py) and 2, 6 Dimethyl pyridine (dmpy). Using
(CHROM-5) chromatograph with flame ionization detector (FID) was
used. The sample (0.5 ml with specified weight) was loaded in
chromatographic column (short column with l=15 cm).The Argon
(carrier gas) was bubbled in special glass pot with “py” or “dmpy” and
was introduced by a valve pulses on column at (T=100 °C).The
difference between initial peak without sample and that passed through
sample of (“py”) or (“dmpy”) was determining the amount of adsorbed
in case in pulses the outlet peak was equal to initial peak the full

Chapter Two - Experimental
86
quantity of adsorbed base was determined .It was known that “py”
adsorbed on Lewis (LA) and Bronsted (BA) acid centers (total acidity
but “dmpy” adsorbed only on (BA) centers. The amount of Lewis acid
centers could be determined by difference (A py – A dmpy). This
measurement was carried out in Institute for Sorption and Problems of
Endoecology National Academy of Sciences of Ukraine.
9- The morphology of the surface of the prepared samples was
examined by Scanning Electron micrographs (SEM) using Scanning
Electron Microscope type JEOL HSM- 6400 (0.2- 45 KV accelerating
voltage , X 10- 300000 magnification ), which is a high resolution
Scanning Electron Microscope with a modern digital image processing
systems. The samples were coated with a thin layer of gold using
coating unit to prevent electrostatic charging on the surface of the
sample. This test was made by School of Mechanical Engineering -
American University of Sharjah – U.A.E.
10- A Hitachi S-4700 scanning Electron Microscope (SEM) equipped
with operating at 25 Kv on energy dispersive X-ray (EDX)
spectrometer (YAG) was used for examination of surface morphology
and composition, in Institute for Sorption and Problems of Endoecology
National Academy of Sciences of Ukraine, Ukraine.

Chapter Two - Experimental
٨٧
2.2 - Chemicals:
The chemicals used in this work are listed in Table 2.1 below
Table 2.1: Chemicals used in this work
No. Chemical Materials Molecular formula Purity % Co.
1 Acetone (CH3)2 CO 99 Merck
2 Benzyl alcohol C6H5 CH2OH 99 BDH
3 Diphenyl amine (C6H5)2 NH 99 Fluka
4 Isobutyl alcohol C4H10OH 99 Fluka
5 Iron (II) ammonium sulfate Fe (SO4)2 (NH4)2. 6H2O 99 Fluka
6 Oxalic acid C2O4H2 >99.99 BDH
7 o-Phosphoric acid O-H3PO4 89-92 BDH
8 Potassium bromide KBr 99 Merck
9 Potassium permanganate KMnO4 99 BDH
10 Sulfuric acid H2SO4 >95.97 Fluka
11 Titanium dioxide (anatase) TiO2 > 99 Fluka
12 Vanadium pentoxide V2O5 >99.99 Fluka
13 Vanadium pentoxide V2O5 (Technical Grade) 93.87
Ibn-Sena
State Co.
Iraq

Chapter Two - Experimental
٨٨
2.3: Preparation:
Two preparation routs are used in this work in order to prepare catalyst
precursor phases (Scheme 2 – 1). The (P/V) ratio used in all preparation
routes is mole ratio.
2.3.1: Route (A):
Route (A) is designated to prepare supported and unsupported of two
phases: intermediate phase (VOPO4.2H2O P/V = 1.1) and its derivative
(VO (H2PO4)2 P/V = 1.1).The intermediate phase was prepared and
isolated in order to study the correlation between these two phases as
separate phase. The reduction step is carried out after the preparation of
intermediate
2.3.1.1: Intermediate phase (VOPO4.2H2O P/V = 1.1)
2.3.1.1.1: Sample No.1 – un (unsupported)
According to the method which had been conducted for the first time in
our previous work (1), the quantity of (24) g of locally produced (technical
grade) V2O5 was suspended by rapid stirring in (96) ml distilled Water and
(17) ml of O- H3PO4 in order to obtain a molar ratio (P/V = 1.1). The
mixture was refluxed for 16 hrs. The color of mixture changed form brown
to yellow during (30) min. The resulting yellow precipitate was filtered ,
washed with distill water until a red color appeared in washing water, then
washed with acetone for several times and left to dry.
Several attempts were unsuccessfully exerted to prepare this phase with
molar ratio (P/V =1.1) from vanadium pentoxide (laboratory grade)

Chapter Two - Experimental
٨٩
produced by both Fluka and BDH Companies; the reaction did not proceed
even after 48 hours of continuous reflux.
2.3.1.1.2: Sample No. 1 – supp (supported)
Technical grade V2O5 (24 g) was mixed and ground with (1.92) g of
Titania (anatase) in order to obtain (TiO2 /V2O5 = 8% weight ratio). The
same procedure (as described in above section 2.3.1.1.1) was conducted to
prepare this supported phase.
2.3.1.2: Derivative (VO (H2PO4)2)
2.3.1.2.1: Sample No.3 – un (un supported)
To a suspension of (VOPO4.2H2O P/V = 1.1), 24 ml of benzyl alcohol
and (48) ml isobutanol were added and refluxed with stirring for 7 hrs.
During this time the color was changed from yellow to dark green. The
sample was filtered and dried at 125 °C for overnight.
2.3.1.2.2: Sample No. 3 – supp (supported Sample)
Technical grade V2O5 (24 g) was mixed and ground with (1.92) g of
Titania (anatase) in order to obtain (TiO2 /V2O5 = 8% weight ratio).
The same method which described above was conducted to prepare
supported sample with longer time reaction (13) hrs. The resulting
precursor was light green.

Chapter Two - Experimental
٩٠
2.3.2: Route B (VOHPO4.0.5H2O)
The reduction of V (V) step has taken place at the beginning of this
preparation route.
2.3.2.1: Sample No. 2 (Reference Sample)
According to Trifiro (171), V2O5 (Fluka) (24 g) was suspended by rapid
stirring with a mixture of (24 ml) benzyl alcohol and (48) ml isobutyl
alcohol.
The vanadium oxide-alcohol mixture was refluxed for 3 hours at 120
°C. The color of the mixture changed from brown to black. The apparently
reduced vanadium suspension was cooled to 40 °C prior to the addition of
22 ml O- H3PO4 in order to obtain a molar ratio for ( P/V = 1.1) . The
suspension was again heated to 120°C and refluxed for additional 3 hrs.
During this time reduced vanadium (IV) - phosphate was formed as
indicated by a change in color from black to blue. The resulting precursor
was separated by filtration and dried at 125°C for overnight. The
preparation of this sample shown in the Scheme 2-1.
2.3.2.2: Sample No. 2- un (unsupported)
The method described above (Section 2.3.2.1) was followed to prepare this
sample from technical grade V2O5. A green precursor was obtained.

Chapter Two - Experimental
٩١
2.3.2.3: Sample No. 2- supp (Supported)
V2O5 (24) g of technical grade, was mixed and ground with (1.92) g of
Titania (anatase) in order to obtain (TiO2 /V2O5 = 8% weight ratio). The
same procedure was conducted in order to prepare the supported phase (as
described in above section 2.3.2.1).

Chapter One - Introduction
2
1.1- Background In our previous work (1) different phases of (vanadium- phosphorus-
oxygen) (VPO) catalyst were prepared, characterized and evaluated by
catalytic test in order to produce Maleic anhydride (MA) via selective
oxidation of n-butane.
Maleic anhydride (MA), and its derivatives malic acid and fumaric
acid, are produced with a worldwide capacity of about (2.8 X 10 6) metric
tons per year. While the market demand for (MA), increased from
(0.616 X 10 6) metric tons in 2000 to almost (1.27 X 10 6) metric tons in
2004(2-4).
These multifunctional chemical intermediates have found applications
in almost any field of industrial chemistry. The principal use of (MA) is in
manufacturing of unsaturated polyester resins (UPR) ( 63 %), lubricating
oil additives (10 %) , copolymers (9 %) ,alkenyl succinic anhydride (5 %) ,
malic acid (3 %) ,fumaric acid (2 %) agricultural chemicals (1%),
miscellaneous, including reactive plasticizers , sulfosuccinic acid esters ,
and alkyd resins (% 7). Furthermore, due to its double bond and anhydride
function, (MA) is a versatile intermediate for the production of co-
polymers of (MA), for example, ethylene glycol and vinyl monomer.
Recently, potential new uses of (MA) have been found in its conversion to
(1-4) butanediol and the manufacturing of tetrahydrofuran (THF) and
butyrolactone via hydrogenation (5).
Maleic anhydride (MA) and the two di-acid isomers were first prepared
in the 1830's, but it took about 100 years before commercial manufacture
was performed in 1933. The National Aniline and Chemical Company Inc.
started a process for the production of (MA) based on benzene oxidation
using a vanadium oxide catalyst. Smaller amounts of maleic acid were also
formed as by product in the production of phthalic anhydride. The use of

Chapter One - Introduction
3
benzene as a feedstock for the production of (MA) was dominant in the
world until the late 1980's (6).
Currently, worldwide production of (MA) is based on the major feed
stocks benzene, butenes and n-butane (1-3,6-14). Most of the capacity is
produced via fixed–bed oxidation of benzene, though benzene is being
displaced by butane as a feed stock (all production in United States is
butane based) (1-3,6-14), because butane is a lower cost and environmentally
more desirable raw material and because butane oxidation produces a
clean product stream, forming mainly (MA) and carbon oxides as shown
below:
C4H10 + 3.5 O2 C4H2O3 + 4 H2O ∆ H = - 1236 KJ/ mole (1.1)
C4H10 + 4.5 O2 4 CO + 5 H2O ∆ H = - 1521 KJ/ mole (1.2)
C4H10 + 6.5 O2 4 CO2 + 5 H2O ∆ H = - 2656 KJ/ mole (1.3)
It is obvious that CO and CO2 are thermodynamically more favored
products. Only kinetic control by a catalyst will enhance the formation of
(MA). In practice, the process is operating at a yield of approximately
(60%) to the desired product. CO and CO2 are the sole carbon containing
by–products in a ratio of about unity. Suppression of the unselective and
very exothermic oxidation to carbon oxides requires sufficient heat transfer
capacities of the reactor. Nonetheless, hot spots are frequently met in (MA)
production plants.
Processes for the production of MA From n-Butane
In general, three different types of process can be distinguished in
commercial production of (MA) from n-butane; fixed-bed processes

Chapter One - Introduction
4
(Scheme 1.1), fluidized–bed processes (Scheme 1.2) and the re-circulating-
solids process (Scheme 1.3).
The fixed-bed reactor consists of a number of tubes that are packed
with coarse catalyst bodies. The reactants flow through these tubes. As a
result of the obstruction of the gas flow by the catalyst bodies, a pressure
drop across the bed is exhibited. Therefore, pressure has to be applied at
the inlet to ensure an adequate flow rate. The magnitude of the pressure
drop is depending on the flow rate, the length of the catalyst bed and the
size to the catalyst bodies. Since the selective oxidation of n-butane to
(MA) is highly exothermic, fixed-bed reactors must containing a facility to
remove the reaction heat. This can be done in various ways: the bed can be
split into different sections, with provision for cooling the gas between the
sections ,or using a reactor containing a large number of tubes, along
which a cooling gas or the liquid is recirculated. However, hot spots can
occur easily in fixed-bed reactors. These can be prevented by using larger
catalyst bodies, a less active catalyst, or by dilution of the catalyst with an
inert solid (support). In view of the explosion limits and the flammability
of mixtures of n-butane and air, only low concentrations of n- butane can
be applied (2 - 4 %).Furthermore, the gases must be mixed and pre-heated
before entering the reaction zone. In a fixed-bed reactor ,the concentration
of n-butane will decrease when moving to the end of the tube .To maintain
a sufficiently high selectivity at the exit of the reactor a less active catalyst
is installed at the entrance and a very active catalyst at the end of the
reactor (4,5).
A single passed fixed - bed reactor was used in our previous work (1) ,
(1 m long and 0.019 m in diameter) mounted in four heating zones vertical
furnace. The flow of n-butane was controlled before entering the reactor
using gasometer. The total gas flow after leaving the reactor was also
measured and fixed at industrial conditions (space velocity 2000 h -1).The

Chapter One - Introduction
5
gas mixture (1.5 Vol. % n-butane in air ) was preheated before passing on
the catalyst using inert alumina granules in the upper half of the reactor .
The lower half of the reactor (0.1 L) was filled with (2.3 mm) granules by
(4) temperature indicators controllers (TIC).Maleic produced was passed
through scrubber and was dissolved in water to produce Maleic acid .this
acid was titrated with (0.01 N) NaOH using phenolphthalein as indicator.
Scheme 1.1: Huntsman fixed-bed reactor for MA production (4).
In a fluidized-bed process, reaction gases flow upward through a bed
of catalyst particles .When the force of the gas flow on the catalyst bed is
equal to the weight of the bed, the catalyst bed expands significantly and
the catalyst bodies are brought in continuous motion. Because of this
motion ,better heat transfer characteristics are established and ,hence, hot
spots cannot occur in a fluidized-bed reactor comprise the fact that reaction
gases can be used without pre-mixing and pre-heating before entering the
reactor. Furthermore, higher n-butane concentrations can be used due to a
decreased explosion risk compared to the fixed-bed process (4). The flow
diagram for fluidized-bed (Scheme 1.2) process is comparable to that of
the fixed-bed process (Scheme 1.1).Several different ways have been

Chapter One - Introduction
6
developed to produce bulk (VPO) catalyst particles of the right size and the
attrition resistance for fluidized-bed purposes (16) .When the catalyst bodies
are too small, they will be blown out of the reactor .Large bodies, on the
other hand, call for extremely high linear gas flow rates in the reactor,
therefore, the particle size of fluidized-bed catalysts usually ranges from
(10-150 microns). Development of fluid-bed butane system has been
reported (17,18).These systems offer the advantage of operation at higher
butane concentration (up to 4%) and thus lower original costs. However,
less selectivity at the higher butane loadings and the intermediate problems
with attrition of (VPO) materials need to be overcome before this process
becomes economically preferred (17,18).
Scheme 1.2: ALMA Fluidized bed reactor for MA (4).
DuPont commercially operates the third type of process in their
recirculation-solids reactor (17,18).In this case, oxidation of n-butane and
regeneration of the catalyst are carried out in two separate reaction zones
(Scheme 1.3) .The selectivity to (MA) is increased, because the oxidation
of n-butane is carried out in absence of oxygen (19,20) . In the first step, n-
butane reacts with lattice oxygen from the catalyst. In this stage, the

Chapter One - Introduction
7
catalyst is reduced by n-butane resulting in the selective formation of MA,
which is removed in a stripper. Regeneration of the reduced catalyst with
oxygen takes place in the second reactor zone. In principle, the oxidation
state of the catalyst can be controlled optimally in this way (17,18).This
process operates at lower conversion per pass and with higher selectivities
than normally encountered in fixed or fluid bed systems. It also uses a
unique attrition resistance system (19-21).
Scheme 1.3: DUPONT re-circulating – solids process for the production of THF
from n-butane via MA (4).
Table 1.1 shows the 1993 and 1995 world-wide (MA) production
capacity for the different processes (4).As can be seen from the table, both
fixed-bed and fluidized-bed butane based processes have been growing at
the expense of benzene-based processes. Furthermore, it seems that
production of (MA) with fluidized –bed technology would not surpass
production with fixed-bed technology which was still operated at higher
yields to (MA). Table 1.2 represents the long – term (MA) production –
consumption balance (4) .The average of the annual demand growth which
was estimated to be about (4%). By the year 2004, total world –wide
consumption will be increased by about (76.5%) as compared to 1998.

Chapter One - Introduction
8
Table 1.1: World MA capacity (in metric tons) by reactor type (4).
Reactor (feed) 1993 ١٩٩٥
10 3 t/y % 10 3 t/y %
Fixed - bed
n-Butane 369 42.0 704 51.8
Benzene 325 37.9 388 28.5
Fluidized – bed
n-Butane 127 14.8 217 16.0
Phthalic anhydride co-product 37 4.3 50 3.7
Total 858 100.0 1359 100.0
Table 1.2: Total world production & consumption of MA (X 103 metric tons) (4).
Capacity to produce 1994 1995 1996 1997 1998 2001 2004
From n-Butane 551.3 647.6 657.1 702.6 767.6 1019.2 1088.7
From Benzene 301.0 302.0 300.0 300.0 311.0 289.5 289.5
From PA recovery 23.5 20.5 22.5 24.5 24.5 28.5 28.5
Total 875.8 970.1 979.6 1027.1 1103.1 1337.2 1406.7
Utilization (%) 81.9 79.4 82.1 83.4 83.8 79.6 85.9
Production 717.1 770.4 803.9 856.9 924.8 1064.1 1207.7
Consumption
UPE resins 355.3 372.3 383.4 399.7 416.7 471.0 532.9
BDO Chemicals 27.0 36.1 10.0 54.2 86.8 120.4 145.4
Other Uses 330.2 361.2 380.5 402.5 421.3 472.7 529.4
Total 712.5 769.6 803.9 856.4 924.8 1064.1 1207.7
All catalysts used industrially for the production of (MA) from
n-butane are (VPO) catalyst based system (8,12,22).The numbers in (Tables
1.1 and 1.2) ,together with the fact that the current yield (only amounts to
about 58%) to (MA) of an equilibrated catalyst, indicate that improvement
of the process is of great economic and environmental interest. To this end,

Chapter One - Introduction
9
several developments can be considered. First, the bulk (VPO) catalyst
should exhibit a higher attrition resistance (mechanical strength) in order to
be more suitable for fluidized-bed process. Secondly, the activation period
for the catalyst should be shortened. This will result in an earlier
achievement of optimum performance. However, the catalyst formulation
could be changed; resulting in better properties and an improved catalytic
performance. Furthermore, there is also a comprehensive demand for a
cheaper and more reproducible preparation procedure for the currently
applied (VPO) catalyst. In any case, these improvements can never be
achieved without thoroughly investigations of the catalytic and structural
properties of the active (VPO) phase.
1.2- Scope of the Literature Survey
The n-butane to (MA) reaction is a fascinating complex system. This
catalytic system performance a (14-electron) oxidation involving the
abstraction of (8 hydrogen atoms) and insertion of (3 oxygen atoms) as
described in (eq. 1.4) (21).
CH3
CH3
+ 3.5 O2
O
O
O + 4 H2O ... (1.4)
Compared to other industrially practiced hydrocarbon selective
oxidation reactions (23-26), it is the most complex one (Table 1.3). It is the
only example of an industrially practiced selective oxidation reaction
involving alkane activation. Knowledge gained through study and
understanding of this system may contribute to advances in alkane
activation in general.
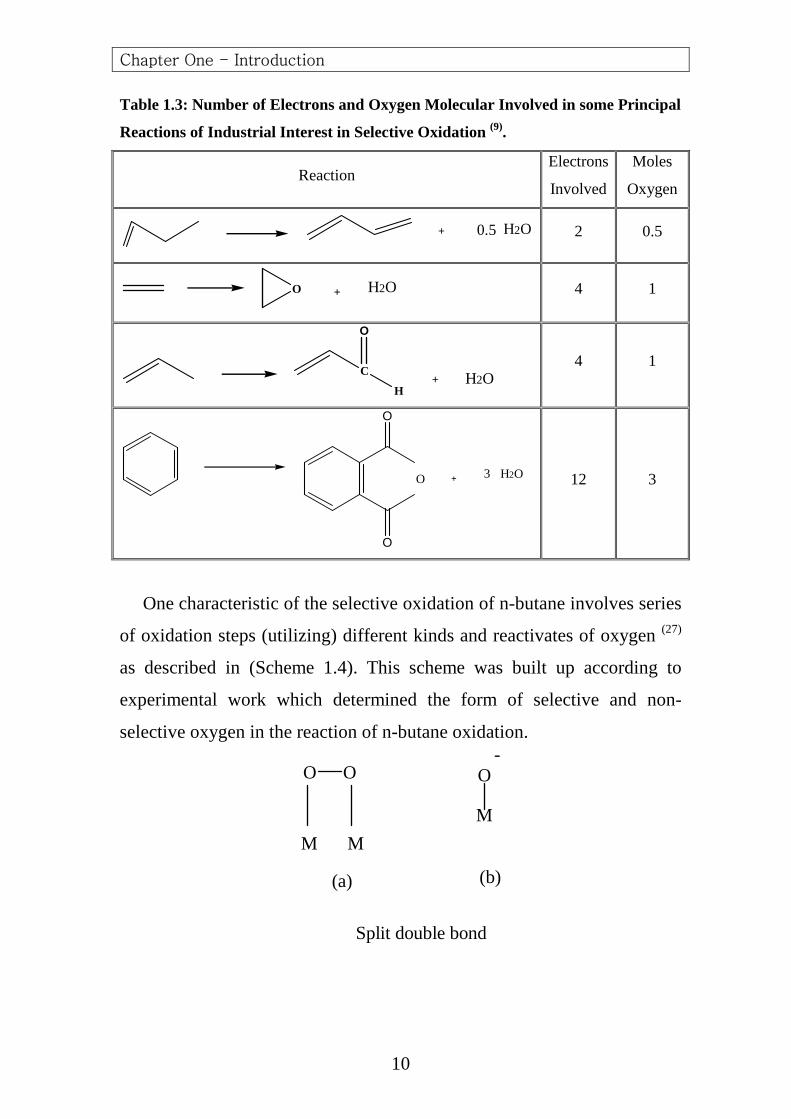
Chapter One - Introduction
10
Table 1.3: Number of Electrons and Oxygen Molecular Involved in some Principal
Reactions of Industrial Interest in Selective Oxidation (9).
Reaction Electrons
Involved
Moles
Oxygen
+ 0.5 H2O
2 0.5
O + H2O
4 1
C
O
H+ H2O
4 1
O
+ 3 H2O
O
O
12 3
One characteristic of the selective oxidation of n-butane involves series
of oxidation steps (utilizing) different kinds and reactivates of oxygen (27)
as described in (Scheme 1.4). This scheme was built up according to
experimental work which determined the form of selective and non-
selective oxygen in the reaction of n-butane oxidation.
O O
M M
(a)
M
O-
(b)
Split double bond

Chapter One - Introduction
11
O M O
O
O
O O
O M O
(c) (d) (e)
O M O
OO
Radical Pi adsorbed
Scheme 1.4: Kinds of oxygen adsorbed species (28)
1.3 -Structure of the catalyst
1.3.1- Crystalline (VPO) phases
The (VPO) system is characterized by the facile formation of a number
of crystalline phases of vanadium (III), vanadium (IV) and of vanadium
(V).In the more interesting (P/V) ratio near (1.0), the different crystalline
(α, β) (29-35), polymorphic (α1, α11, γ, δ) ( 29,35-37) or hydrated phases of
(VOPO4) (34-41) have been extensively studied. In general, the vanadyl
orthophosphate crystal structure consists of (VO6) and (PO4) groups
arranged in layers [VOPO4] ∞, held together by long (V-O) bonds or by
hydrogen bonds (Figure 1.1-A). The layered structure leads to rich
intercalation chemistry, with formation of layered solids consisting of
alternating inorganic (42-48) and organic (49-56) layer or the formation of
solvated inorganic intercalation compounds (43).

Chapter One - Introduction
12
Figure 1.1-A: Structure of VOPO4.2H2O showing infinite layers of PO4
tetrahedral linked to VO6 octahedral (40).
In dihydrated (VOPO4) the layer lattice is built up of neutral (VOPO4)
layers and interlayer water molecules (40, 41) .The vanadium atom lies on a
fourfold axis and is surrounded by six oxygen atoms to give distorted
octahedral .The four equatorial oxygens are provided by four different
phosphate tetrahedral .One of the axial vanadium–oxygen bond is very
short corresponding to a double bond (V=O) as shown in Figure 1.1-B.
Figure 1.1-b: Structure of VOPO4.2H2O showing bond distances (°A) and angles
(degree) (40).
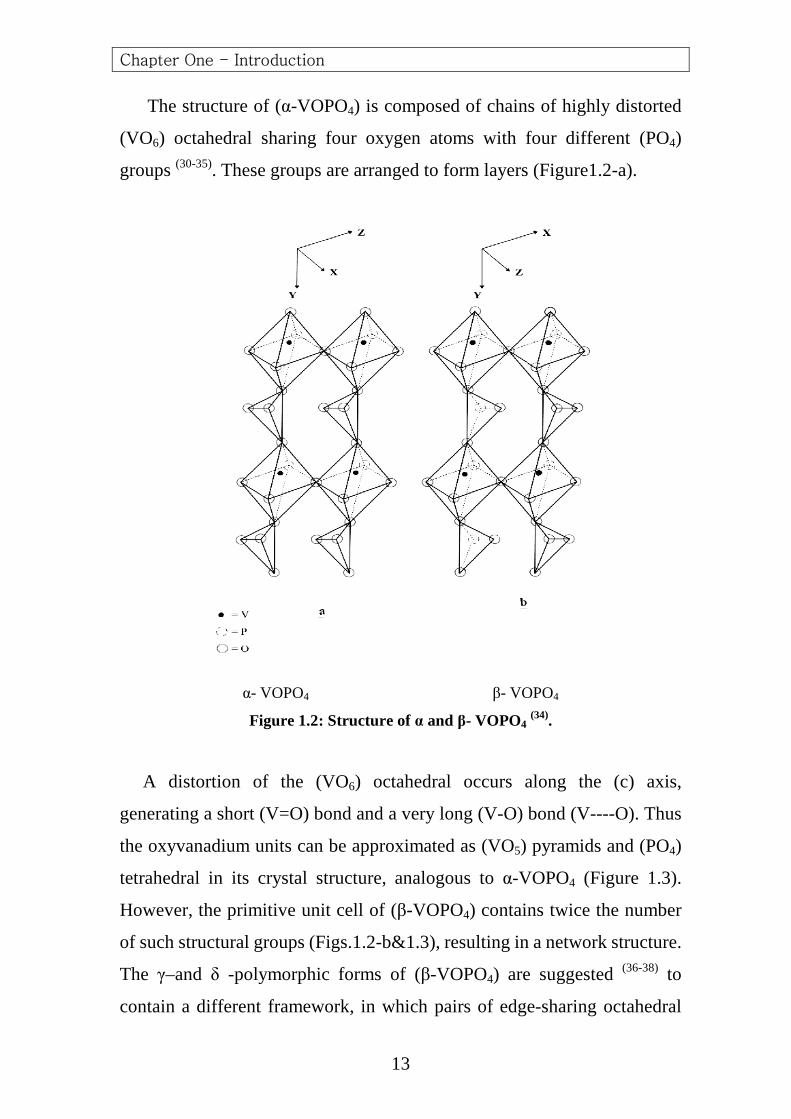
Chapter One - Introduction
13
The structure of (α-VOPO4) is composed of chains of highly distorted
(VO6) octahedral sharing four oxygen atoms with four different (PO4)
groups (30-35). These groups are arranged to form layers (Figure1.2-a).
α- VOPO4 β- VOPO4
Figure 1.2: Structure of α and β- VOPO4 (34).
A distortion of the (VO6) octahedral occurs along the (c) axis,
generating a short (V=O) bond and a very long (V-O) bond (V----O). Thus
the oxyvanadium units can be approximated as (VO5) pyramids and (PO4)
tetrahedral in its crystal structure, analogous to α-VOPO4 (Figure 1.3).
However, the primitive unit cell of (β-VOPO4) contains twice the number
of such structural groups (Figs.1.2-b&1.3), resulting in a network structure.
The γ–and δ -polymorphic forms of (β-VOPO4) are suggested (36-38) to
contain a different framework, in which pairs of edge-sharing octahedral

Chapter One - Introduction
14
with trans vanadyl oxygens are alternately unshared or shared with
phosphate tetrahedral. Such pairs do not exist in the (α- and β-VOPO4)
forms. During thermal treatment at high temperature the consecutive
transition has been observed (36).
δ- VOPO4 γ-VOPO4 β-VOPO4 …. 1.5
α-VOPO4
β-VOPO4
Figure 1.3: Comparison between structure of α and β-VOPO4 (34).

Chapter One - Introduction
15
Vanadium (IV) hydrogen phosphate (57-59) which is the most widely
used precursor of the active phase of n-butane oxidation possesses (21,57-62) a
structure made up of atom arrangements in the (ac) plane which are very
similar to that proposed for γ- VOPO4 (Figure 1.4 and 1.5) .It may be
obtained from the dihydrated V(V) phosphate by reduction with organic
alcohols (57) or from the reduction of V2O5 followed by the addition of
o-H3PO4 (61,62).
(A)
(010) plane
(B)
(100) plane
Figure 1.4: Crystal structure of γ -VOPO4 (36).
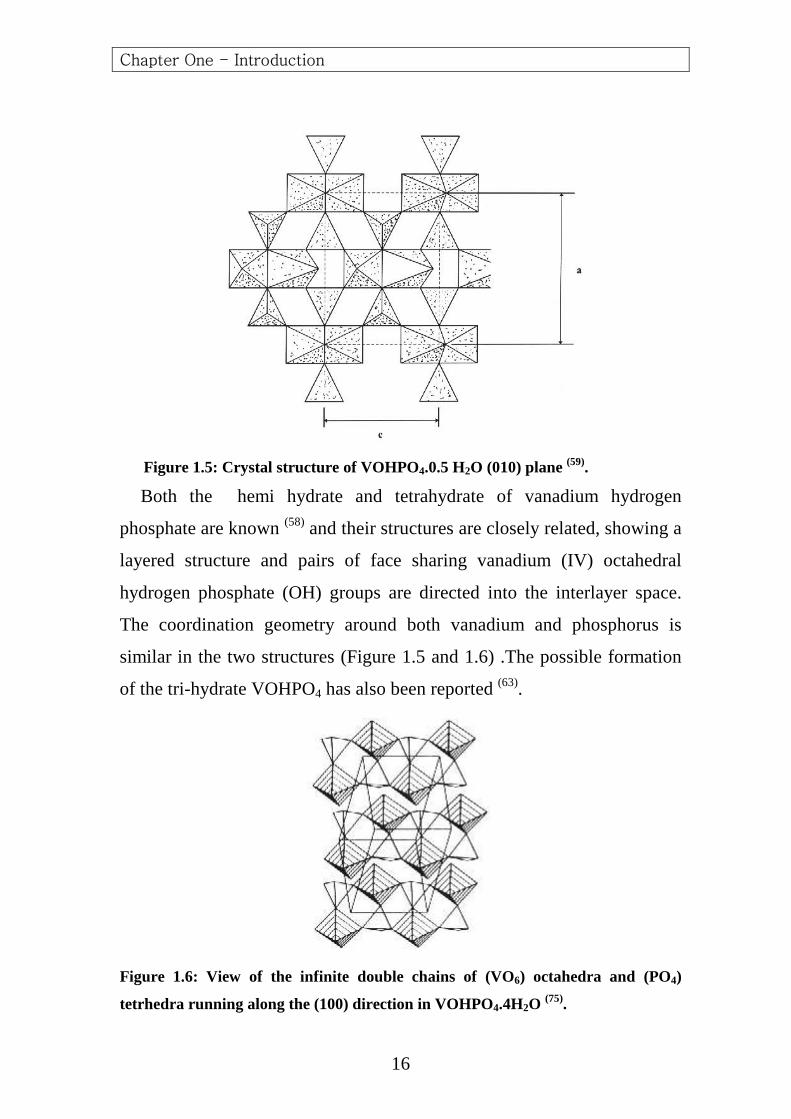
Chapter One - Introduction
16
Figure 1.5: Crystal structure of VOHPO4.0.5 H2O (010) plane (59).
Both the hemi hydrate and tetrahydrate of vanadium hydrogen
phosphate are known (58) and their structures are closely related, showing a
layered structure and pairs of face sharing vanadium (IV) octahedral
hydrogen phosphate (OH) groups are directed into the interlayer space.
The coordination geometry around both vanadium and phosphorus is
similar in the two structures (Figure 1.5 and 1.6) .The possible formation
of the tri-hydrate VOHPO4 has also been reported (63).
Figure 1.6: View of the infinite double chains of (VO6) octahedra and (PO4)
tetrhedra running along the (100) direction in VOHPO4.4H2O (75).

Chapter One - Introduction
17
The intercalation compounds of vanadium hydrogen phosphate with
organic molecules or inorganic cations and anions such as (V n+) of HPO42-
are reported (23-33,36,37,63-72) .This intercalation chemistry is of particular
importance in the description of the structurally and catalytically related
chemistry of VOHPO4.0.5 H2O and of its derived phases.
Two main effects observed in the preparation of (VOHPO4.0.5 H2O)
may be, in fact, strictly connected to intercalation, properties. First, non
stoichiometry is easily accommodated as evidenced by the preparation of
compounds with (0.9-1.2) P: V ratio without any apparent modification of
structural and morphological properties (62).Second, the preparation
conditions have a pronounced effect on the microstructure, i.e., on the
morphology, solid - state reactivity, and the presence of disorder in the
stacking fold of crystalline planes of its layered structure.
In fact, reduction of the starting V (V) compound may be realized (62) by
using, for example, aqueous HCl or isobutyl alcohol. In both cases, almost
pure vanadyl hydrogen phosphate is obtained, but with different
microstructure (61,62,73). The layers of vanadyl hydrogen phosphate (010)
plane (Figure 1.5) are interconnected in tri dimensional structure by weak
hydrogen bonding of phosphates and of water molecules. The organic
alcohol competes with this effect, reducing the bonding between the planes
and allowing the formation of crystals in which these (010) planes are
predominantly exposed (plate like morphology) (21,56,59,62,74) .This effect, in
addition to the increase in surface area modifies the surface properties due
to a change in the relative ratio of crystalline planes at the surface. The
alcohol also can remain partially intercalated between layers (62) .This
effect induces some local modification of the vanadyl hydrogen phosphate
structure, which can strongly modify its solid-state reactivity (61,73).
Ball et al.(75) proposed that the known mono hydrogen phosphate phases
can be classified in three different structural types, namely type I,

Chapter One - Introduction
18
(VOHPO4.0.5H2O) ;type II (α-VOHPO4.2H2O); type III, (VOHPO4.4H2O)
and (β-VOHPO4.2H2O) .practically ,the β-dihydrate appears as
intermediate in the thermal treatment of the tetrahydrate from both
thermogravimetric and thermodiffractometric experiments .Moreover , the
structure of (β-VOHPO4.2H2O) is closely related with that of
(VOHPO4.4H2O) (Figure 1.6, 1.7-a and 1.7-b). Both compounds present a
similar arrangement of (VO6) octahedra and (PO4) tetrahedral forming
alternating chains which lie along the (c) direction in the β-dihydrate and
along the a direction in the tetrahydrate and along the (a) direction in the
tetrahydrate. The coordination geometry around both vanadium and
phosphorus atom is similar in both structures. Each phosphate group
contains three oxygen atoms (shared with three different vanadium atoms)
and a hydroxyl group. The (V) atoms show very similar coordination
polyhedra, having a water molecule trans-coordinated to the axial (V=O)
group and a second coordinated water molecule in the equatorial plane of
the (VO6) octahedra. The similarity of these structures suggests that the
dehydration of the tetrahydrate into the dihydrate may precede
topotacitically. However, several ways for the reorganization of the infinite
double chains that lie parallel to the (a) axis (6.379 °A ) of the tetrahydrate
may be imagined to give the interconnected single chains running in the (c)
direction (12.623 °A = 2 X 6.379 °A) of the β-dihydrate . All the possible
models that imagined need to break some bonds (at least 2 or 4 per unit
cell), to rotate some polyhedra and to reconstruct the bonds.
Figure 1.7- a: Projection of the structure of β-VOHPO4. 2H2O along (100) (75).

Chapter One - Introduction
19
Figure 1.7-b: Projection of the structure of β-VOHPO4. 2H2O along (010) (75).
The transformation of V (IV) and V (V) phases into vanadyl
pyrophosphate is an important step in forming active catalysts, by thermal
treatment (Calcinations) at 400 °C, the VOHPO4.0.5 H2O dehydrates to
(VO)2P2O7 .Alternatively, the vanadyl pyrophosphate may be obtained by
reduction from VOPO4 or by thermal treatment of different crystalline or
amorphous (V-P-O-X) phases (where X indicates a generic thermally
decomposable anion or cation such as (C2O4 2- or NH4
+ ) (63, 76).
The vanadyl pyrophosphate is built of chains of (V) polyhedral linked
by pyrophosphate groups (60,77) .The (V) atoms are linked through the
oxygen atoms of the vanadyl in (V-O-V) chains in the (c) direction, and
the (V) atom octahedra are linked in pairs through a common edge,
forming double chains in this direction. The vanadyl groups in the paired
vanadium octahedra are oriented trans to one another (Figs. 1.8 and 1.9).
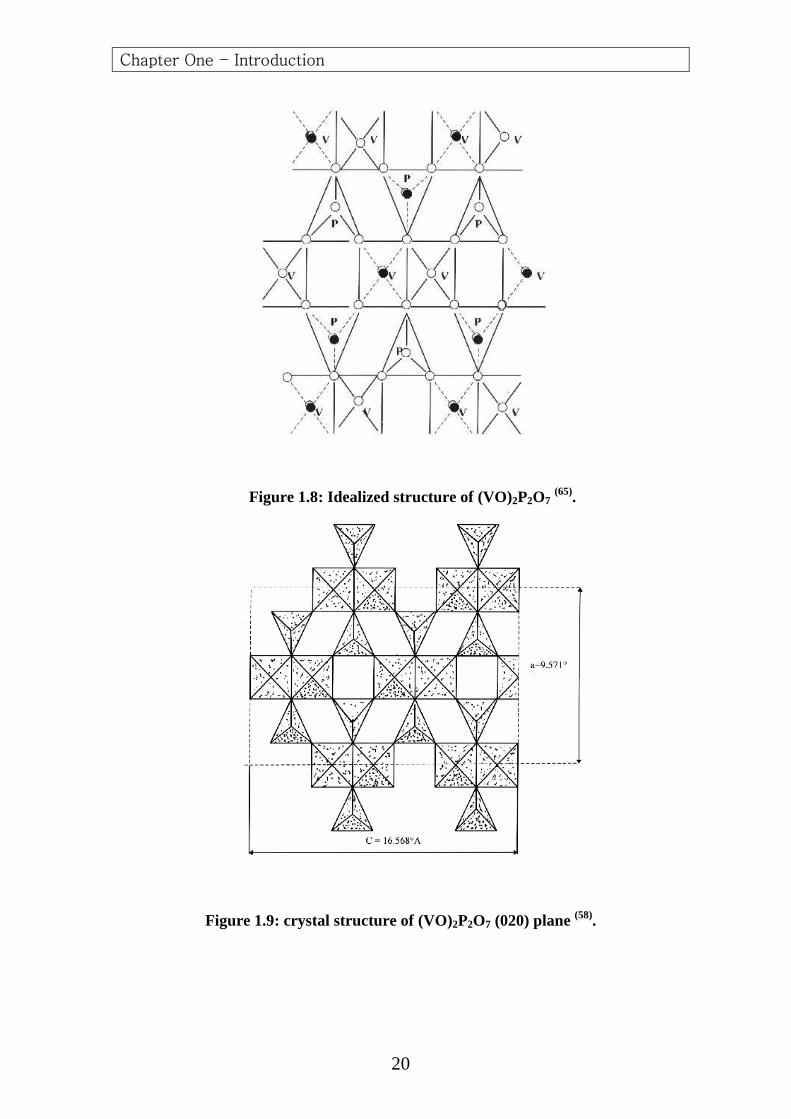
Chapter One - Introduction
20
Figure 1.8: Idealized structure of (VO)2P2O7 (65).
Figure 1.9: crystal structure of (VO)2P2O7 (020) plane (58).

Chapter One - Introduction
21
The unit cell of (VO)2P2O7 is orthorhombic (77,78) and is topological
similar in its (bc plane) to that of (VOHPO4.0.5H2O) in the (ab)
plane (56,60).The change from face-shared to edge-shard vanadium
octahedral in converting the hemihydrate to the pyrophosphate results in a
small expansion of one axis, but the other in plane dimension shows little
change according the pseudomorphic relations between the two crystalline
structures (59) as shown in (Figure1.10) this indicates a topotactic
mechanism of transformation, and this result has been confirmed with
Scanning Electron Microscopy. (21,56,59,62) .The layer spacing decreases from
(5.69°A) in the hemihydrate to (3.91°A) in the pyrophosphate. This is
consistent with removing the water molecules shared by the vanadium
pairs and filling the resulting vanadium coordination site with the oxygen
atoms of vanadyl groups from the layers above. This transformation
requires only very small displacements of the atoms. Importantly, since the
conversion of the hemihydrate to pyrophosphate can take place without
breaking any (V-O-P) bonds, the structural order/disorder and morphology
of the precursor phase are maintained during the transformation to vanadyl
pyrophosphate. This means that it is possible to control some of the
structural /morphological characteristics of vanadyl pyrophosphate by
controlling the specific nature of the vanadyl hydrogen phosphate
hemihydrate precursor phase (62,73). Furthermore the terminal vanadyl
oxygen atoms in the face – shared octahedral pairs of vanadyl hydrogen
phosphate have a syn arrangement; while in vanadyl pyrophosphate they
are in anti positions. These arrangements in the layer stacking direction
result in the initial formation of (VO)2P2O7 crystalline with many defects
(36,37,65,79). Alternatively, Bordes and Courtine (70,74) discussed the possible
presence of disorder in the crystalline structure of (VO)2P2O7 in terms of
different crystalline phases (β- and γ- VO)2P2O7) in which former
possesses a network structure versus a layered structure for the γ- phase.
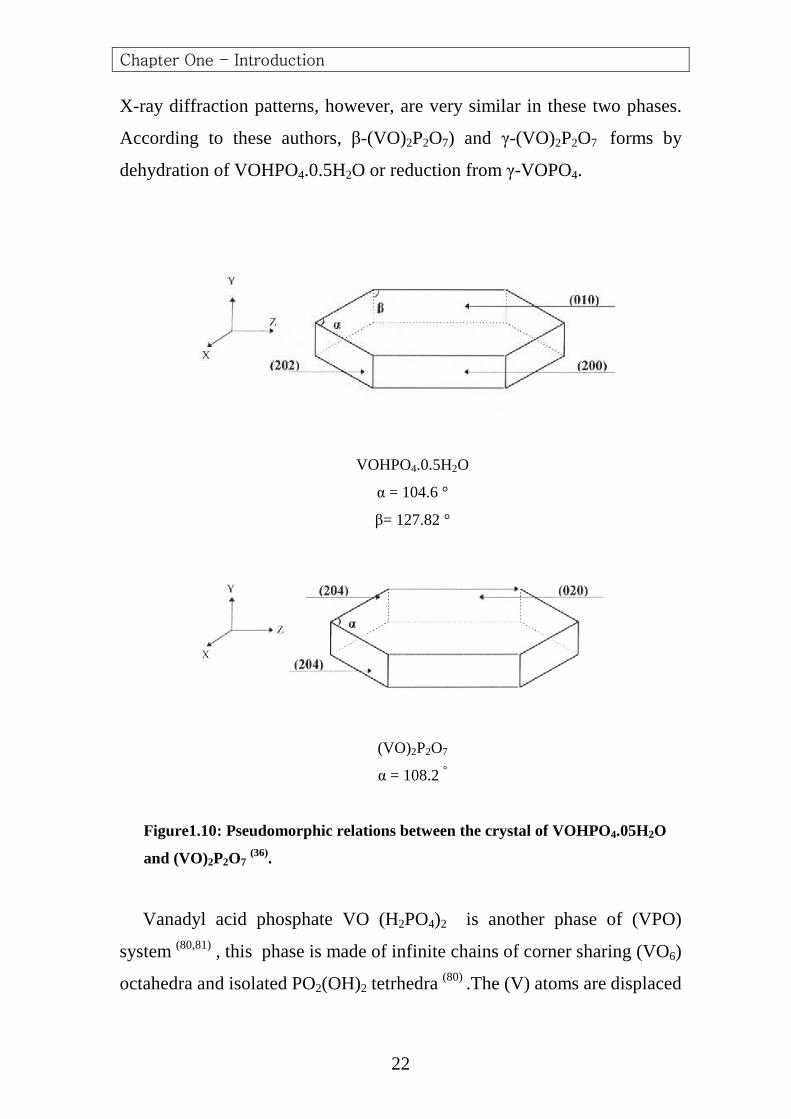
Chapter One - Introduction
22
X-ray diffraction patterns, however, are very similar in these two phases.
According to these authors, β-(VO)2P2O7) and γ-(VO)2P2O7 forms by
dehydration of VOHPO4.0.5H2O or reduction from γ-VOPO4.
VOHPO4.0.5H2O
α = 104.6 °
β= 127.82 °
(VO)2P2O7
α = 108.2 °
Figure1.10: Pseudomorphic relations between the crystal of VOHPO4.05H2O
and (VO)2P2O7 (36).
Vanadyl acid phosphate VO (H2PO4)2 is another phase of (VPO)
system (80,81) , this phase is made of infinite chains of corner sharing (VO6)
octahedra and isolated PO2(OH)2 tetrhedra (80) .The (V) atoms are displaced

Chapter One - Introduction
23
(0.364) °A from the equatorial plane along the fourfold axis. As a result,
one short bond (1.600 °A) which characterizes the vanadyl (VO2+) ion,
forms in almost regular oxygen octahedra .The bond between the (V atom)
and the oxygen in the trans position is significantly longer(2.382 °A).
Furthermore ,the equatorial planes of the octahedra are alternatively
rotated of ( +18° and - 18°) around the chain axis (Fig. 1.11-a ).Phosphate
tetrahedra act as bidentate bridges via oxygen atoms for (V) atoms
belonging to adjacent chains, (OH) corners established a contact between
tetrahedra to form hydrogen bonds (Figs. 1.11- a &1.11-b ).
(A)
Figure 1.11-a: Crystal structure of VO (H2PO4)2 along XY axis (80).

Chapter One - Introduction
24
(B)
Figure 1.11-b: Crystal structure of VO (H2PO4)2 along XZ (80).
1.3.2 - Active phase in n-Butane Selective Conversion to MA
Because the (VPO) system is characterized by the facile formation of a
number of crystalline phases, the structure of the active phase must be
discussed in term of factors such as, oxidation state, (P/V) ratio, and
crystal phase transformations under reaction atmosphere.
The various crystal phases can interconvert as a function of the reducing or
oxidizing properties of the reactants, the time on stream, and the reaction
temperature (29,37,61,82,83).
The orthophosphate (VOPO4) phases are transformed to (VO)2P2O7 by
reaction with the hydrocarbon mixture. In this reduction process single
(VO6) octahedral form pairs by loss of oxide anions. However, the
different (VOPO4) phases previously discussed possess different
reducibility to vanadyl pyrophosphate, depending on the structure or
morphology (36).

Chapter One - Introduction
25
The Complex solid-state chemistry of (VPO) system has led to some
confusion and contradictions in the literature concerning the nature of the
active phase in the n-butane oxidation and the identification of the active
site involved in the different steps of the reaction.
Bordes and Courtine (37) suggested that the active sites in n-butane
oxidation to (MA) were associated with coherent interfaces between slabs
of (100) VOPO4 and of (010) (VO)2P2O7 along the (100) and (201) planes,
respectively. On the contrary Volta et al. (84,85) believed that the active sites
are not associated with interfacial effects between two crystalline phases.
On the basis of comparison between X-ray diffraction, they suggested that
the active phase for selective oxidation of n-butane consists of a mixture of
well-crystallized (VO)2P2O7 (V4+) and an amorphous surface (VPO) phase
of (V5+) involving many corner-sharing VO6 octahedra. This amorphous
phase may be interpreted as a precursor of (β-VOPO4), which forms at
higher reaction temperatures.
Hodnett and Delmon (67-69) used a prereduction treatment with hydrogen
in order to improve selectivity to maleic anhydride and suggest that the
best catalyst consists of an oxidized surface layer built upon a reduced core
of a V (IV) phase. The selectivity is not related to the presence of a
specific well-crystallized phase, but only to the distribution of vanadium
oxidation states between the bulk and the surface.
The active catalyst must possess an optimal [V (IV)/V (V)] ratio for
selectivity in n-butane oxidation according to Zozhigalov et al., (86) from
studies of V-P compounds with variable V (IV) /V (V) ratios. These
compounds preserve the layer structure of the initial (α-V5+OPO4)
compound. Optimal performance properties are associated with catalysts
containing [4-9 V (V) ions per V (IV) ion]. n-butane oxidation and (MA)
take place at the expense of the catalyst surface oxygen and are
accompanied by its reduction to (VO)2P2O7. On (VO)2P2O7 however, the

Chapter One - Introduction
26
rate of reduction of the catalyst is lower than that of butane to (MA)
reaction, and therefore these authors conclude that the V(V) phase is
reduced and that the V(IV) phase is oxidized under the dynamic conditions
of catalytic reaction (86).
Weing and Schrader (74) claimed that only (VO)2P2O7 is the active and
selective phase in n-butane oxidation to (MA). A slight excess of catalyst
phosphorus (P/ V=1.1 catalysts) is necessary to stabilize the active phase
The excess phosphorus creates a distortion of (P2O7)4- crystal
environment. The α-VOPO4 is considered by these authors and others (21)
as active, but non selective phase.
Many authors proposed that the vanadyl pyrophosphate (VO)2P2O7 as
the active phase (1,9,21,56,62,73,77,80,87) .Trifiro et al. (62,88-90) attributed the
activity of the catalyst to the V(IV) phase (the vanadyl pyrophosphate),
whereas the selectivity to maleic anhydride(MA) was connected to the
presence of a very limited and controlled amount of V (V) sites .
The same discordance as to the nature of the active phase is present in
the literature concerning the optimal (P/V) ratio of the catalysts, even
though there is a general agreement that phosphorus stabilizes the (+4)
valance state of vanadium and limits its oxidation (8,74,86,90).
Garbassi et al. (91) have found that the specific conversion of
n-butane increases by an order of magnitude for a (P/V) ratio just
exceeding unity, but extended X-ray absorption measurements do not show
any structural effect of the phosphorus.
The (P/V) ratio is a key parameter in determining catalyst selectivity
and activity according to Weing and Schrader (74). Selectivity for (MA)
increase with catalyst phosphorus loading, whereas specific activity of
both selective and nonselective oxidation decreases on increase of
phosphorus content in the range (0.9-1.2) P/V range. Best catalytic

Chapter One - Introduction
27
performances are exhibited with a catalyst with (P/V = 1.1) according to
Buchanan and Sunderesan (92).
Similar results were observed by Pepera et al. (93). According to Bosh
and co-workers (94) who studied the selective oxidation of n-butane to (MA)
under oxygen deficient over (VPO) catalyst, they found that the selectivity
was strongly influenced by the actual surface V (V)/V (IV) ratio).
Garbassi et al. (91) found a value of (P/V) surface ratio in the
(2.0-2.8) range for P/V bulk ratio in the (1.0-1.4) range.
Hodnett and Delmon (67-69) reported that the surface (P/V) ratio is (1.0)
for bulk stoichiometric (P/V) values of 1.0 or higher. They concluded
therefore that the reactivity of near-surface layers is hardly affected by the
(P/V) ratio, but bulk reactivity is drastically curtailed.
Selectivity to (MA) from n-butane maximizes for (P/V) =1.0 according
to Ai (70), who associated this catalytic effect with the presence of strong-
acid sites to activate n-butane. Finally, an optimal value of (P/V) around
(1.0) was suggested by Trifiro et al. (73,88-90) and Contractor et al. (21).
The active sites has been proposed as being the (200) plane of
(VO)2P2O7 crystallites that preferentially expose the (200) plane have been
found to exhibit higher selectivities for n-butane oxidation (95).
Transformation Electron Microscopy has shown that catalyst selectivity
correlated with the disappearance of amorphous platelets (96). Seven
different active sites have been suggested for the (200) plane of (VO)2P2O7
catalysts as shown in (Figure 1.12) whereas:
a- Bronsted acid sites, probably –POH groups.
b- Lewis acid sites, probably V IV and V V.
c- One electron redox couples, (VV / V IV), (V IV / VIII ).
d- Two electron redox couples (V V / VIII ).
e- Bridging oxygen, V-O-V, or V-O-P.
f- Terminal oxygen VV= O.

Chapter One - Introduction
28
g- Activated molecular oxygen peroxo and superoxo species (96).
Figure.1.12: Termination of the (200) plane of (VO)2P2O7 and proposed active
sites for oxidation (s1) = Lewis acid, (s2) = Lewis acid site, (s3) = terminal
oxygen, (s4) = bridging oxygen, (s5) = superoxo and peroxo site, (s6) = V v/ viv
redox couple (97).
1.4- Kinetics of n-Butane Oxidation
A central question in analyzing the problem of bridging the gap
between surface science and applied catalysis approaches is the
verification of the possibility of describing the macro kinetic behavior
using rate equations and constants derived from the analysis of the kinetics
of the single elementary steps. Impressive results have been obtained in
this direction (98) .The fitting of macro kinetic data on the basis of a
microkinetic model is usually considered the best demonstration of the
applicability of the suggested reaction mechanism under real working
conditions (99). Is it this true also for more complex and multifunctional
reactions? In order to replay to this question it is good to briefly recall the
principles of the Langmuir description of the catalytic reaction on an

Chapter One - Introduction
29
"ideal" surface, because this is the basis of most of the macrokinetic
models for deriving the reaction rates over catalytic surfaces. On the ideal
Langmuir surface there is one type only of active sites, the energetic of
chemisorption is independent on the coverage, and there is no interaction
between substrate (Figure 1.13-a). Real surfaces are far from the ideal
Langmuir surface, as schematically represented in (Figure 1.13- b), due to
the presence of surface heterogeneities the simplest is the presence of
steps and kinks, but as mentioned In the introduction the situation is far
more complex on oxides) which imply a distribution in the energetic of
interaction, and the presence of interaction between the adsorbates .A
chemical bond of a molecule with a surface site implies the donation of
electrons to or from the surface with thus a modification of the catalyst
conduction band, band gap, etc.; therefore, the energetic of interaction of
molecule with the surface is not independent from the surface coverage,
and the adsorption of " spectator" species, i. e. species which do not play
a direct role in the reaction mechanism, should be also considered. This
problem is even more accentuated in selective oxidation reactions where a
multielectron transfer occurs, as mentioned before.
It may be thus expected that kinetic models based on Langmuir
approach do not correctly fit macrokinetic data, but on the contrary it is a
common experience that these models are well suited to describe the
macrokinetic behavior. How can this dilemma be solved? One possible
interpretation, which is schematically shown in (Figure 1.13-c), is that
during catalytic reaction the largest part of the catalyst surface is covered
by "strongly chemisorbed" species which are characterized species
characterized by a surface lifetimes much longer than of reaction
intermediates and therefore reactant or intermediate seen a local situation
similar to "ideal" surface. This pointed out that the concept of " clean"
catalyst surface, i. e. of reaction of a molecule at single specific active sites

Chapter One - Introduction
30
without considering the modification of the surface reactivity induced by
the presence of other co-adsorbates (reactants, intermediates, " spectator"
species), may not lead to correct description of the " real" working catalyst
surface and reaction mechanism, especially when complex, multi steps
reactions (selective oxidation reactions, for example) are considered.

Chapter One - Introduction
31
A- A simple model of the ideal Langmuir surface
B- A simple model of real surface with kinks and steps, surface interaction between
adsorbates and electron donation of a chemisorbed molecule to catalyst conduction
band (strongly chemisorbed species)
C-Working catalyst surface with the largest part of the surface occupied from strongly
chemisorbed (reactant intermediate)
Figure 1.13- Model of the ideal Langmuir surface (99).

Chapter One - Introduction
32
Several studies have been conducted in the eighties to derive kinetic
expressions to describe the reaction sequence for n-butane oxidation to
Maleic Anhydride in steady- state conditions on (VPO) system (86,87,91,100,101).
Escardino et al. (100) studied the kinetic of n-butane oxidation in fluidized -
bed reactor over (VPO) catalyst with (P/V= 0.8) at (676-753) K.
A triangular reaction network was proposed as shown in Scheme 1.5.
CH3
CH3
O
O
O
CO , CO2
r1
r2 r3
Scheme1.5: A triangular reaction network on n-butane to MA (100).
Maleic Anhydride (MA) and Carbon oxides (COx) were formed directly
from n-butane (at rates r1 and r2 respectively), and MA was also oxidized
to carbon oxides (at rate r3). At n-butane concentrations typical of
industrial reactors, the rate of n-butane oxidation was controlled by the
reaction between n-butane gas and surface oxygen.

Chapter One - Introduction
33
Wohlfahrt and Hofmann (101) investigated n-butane oxidation kinetics
over a wide range of n-butane and oxygen concentration at (719-777K)
over (VPO) catalyst.
Centi, et al. (87) used a very active catalyst prepared in an organic
medium which permitted low reaction temperatures (573-613K) to be
employed. Under these conditions it was found that the rate of reaction of
n-butane to carbon dioxide did not depend on the hydrocarbon
concentration, but only on the concentration of oxygen.
The order of reaction maintained in the literature depended on the
method of analysis of available data from experimental results of
hydrocarbon depletion and oxygen partial pressure. The comparison of
these kinetics results stressed the importance of surface catalyst behavior
(selectivity to MA) as well as the role of the redox properties of vanadium
in determining the activity of the catalyst (87,101).
1.5- Surface Modifications by Interaction of n-Butane with
Catalyst Surface
Centi et al. (87) used fresh catalyst for their kinetics study and suggested
that, a critical factor governing the selectivity at very high butane
conversion is the instability of the formed (MA) in the back end of the
catalytic bed. It is thus worthwhile to analyze the variations in the catalyst
surface as a function of position in the catalytic bed (86). These experiments
showed formation of V (V) at the end of catalyst bed which was attributed
to formation the more oxidizing atmosphere present.
Trifiro et al. (89) studied the oxidation of n-butane at low and high
hydrocarbon concentrations on vanadium (IV)-phosphorus (1:1) mixed
oxide in relation to the surface modifications induced by the reaction
medium. The results showed the presence, in the catalyst, of high amounts

Chapter One - Introduction
34
of V (III) together with V (IV) and the absence of V (V), whereas lower
amounts of V (III) together with both V (IV) and V (V) are formed, when
maleic anhydride is formed. It is suggested that two redox couples operate
in (MA) synthesis:
a- V (IV)-V (III) in the synthesis of olefins from n-butane.
b- V (V)-V (IV) in the synthesis of (MA) from the olefins is formed.
Mori and co-workers (102) studied the oxidation of n-butane on various
unsupported or supported V(V) oxides, they confirmed that (V(V)=O)
species are very active in total combustion of butane, these experiments
results indicate the important relationship between catalytic behavior and
the surface modification induced by the medium itself. The redox
properties of the fed influence the surface oxidation state of the catalyst,
which in turn profoundly affected the nature of the products formed in the
reaction.
Centi and Perathoner (99) referred to (VO)2P2O7 as the active phase in
the industrial catalysts for the selective oxidation of n- butane to (MA).
This catalyst is very selective in n-butane oxidation, but when propane is
fed Instead of n-butane, only carbon oxides and traces of other products
(propene mainly) are detected.
A first question is thus why the decrease in length of the carbon chain
produces a so drastic change in the selectivity on the same catalyst? The
answer can be that from n-butane a stable product against consecutive
oxidation (MA) forms, but not in the case of propane oxidation. However,
this answer responds to only part of the problem. The data reported in
(Figure 1.14) have already pointed that the sensitivity of the reaction
product against consecutive oxidation is not the only factor that determines
the rate of consecutive oxidation to carbon oxides.

Chapter One - Introduction
35
Figure 1.14- Comparison of the selectivity dependence on acrylonitrile in propane
and propane ammoxidation at 480 °C on VSb4 + Sb2O4 (99).
Data in (Figure 1.15) further evidence this problem. In fact, it is expected
that a decrease in the oxygen to alkane ratio may increase the selectivity to
the partial oxidation product (propene in the case of propane), because the
formation of carbon oxides requires a larger number of oxygen atoms
(propane oxidation to (CO2 + H2O) requires (5O2 molecules), whereas
propene formation from propane requires ten time less oxygen). However,
data in (Figure 1.15) show that this is possible only for high initial
concentrations of propane in the feed, whereas for lower initial
concentrations of propane the increase in propene selectivity decreasing
the (O2/ propane) inlet ratio is much less remarkable. A comparison
between the two cases points out that the differences arise from the
different formation of alkene product, which is about ten times larger using
the higher inlet concentration of propane in the feed. This indicates that
when the formation of propene is higher, the rate of its consecutive
oxidation to carbon oxides also decreases and thus the selectivity
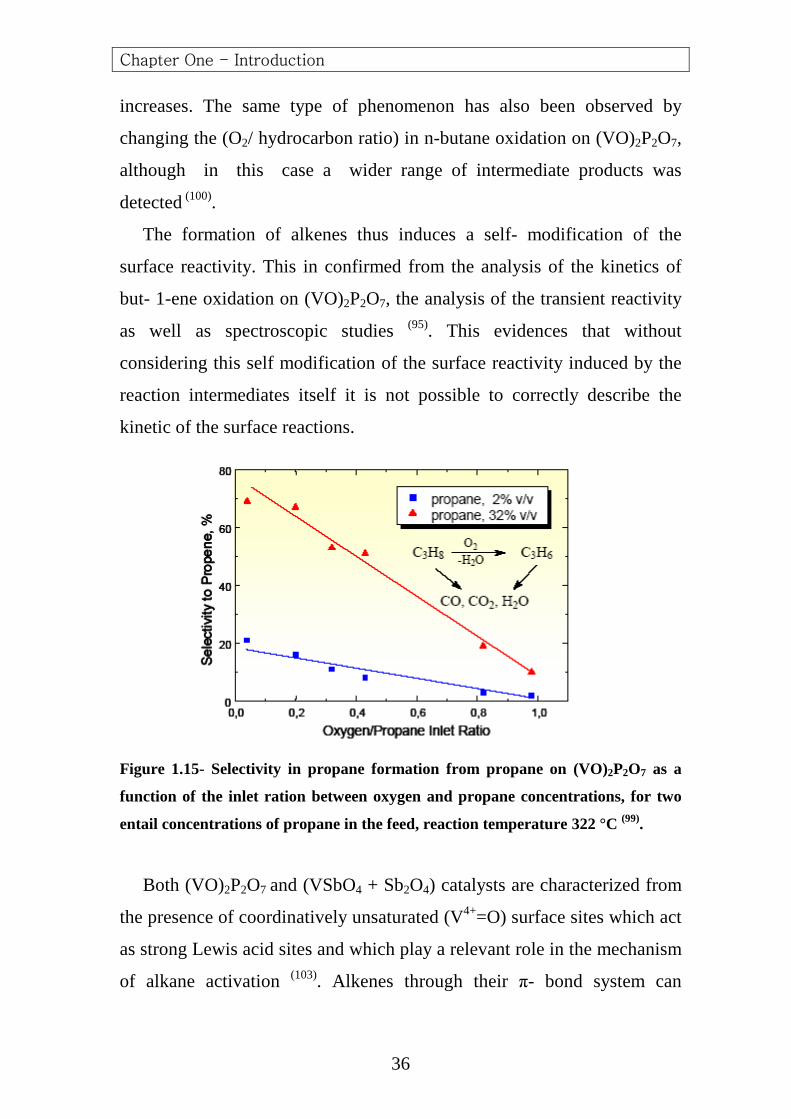
Chapter One - Introduction
36
increases. The same type of phenomenon has also been observed by
changing the (O2/ hydrocarbon ratio) in n-butane oxidation on (VO)2P2O7,
although in this case a wider range of intermediate products was
detected (100).
The formation of alkenes thus induces a self- modification of the
surface reactivity. This in confirmed from the analysis of the kinetics of
but- 1-ene oxidation on (VO)2P2O7, the analysis of the transient reactivity
as well as spectroscopic studies (95). This evidences that without
considering this self modification of the surface reactivity induced by the
reaction intermediates itself it is not possible to correctly describe the
kinetic of the surface reactions.
Figure 1.15- Selectivity in propane formation from propane on (VO)2P2O7 as a
function of the inlet ration between oxygen and propane concentrations, for two
entail concentrations of propane in the feed, reaction temperature 322 °C (99).
Both (VO)2P2O7 and (VSbO4 + Sb2O4) catalysts are characterized from
the presence of coordinatively unsaturated (V4+=O) surface sites which act
as strong Lewis acid sites and which play a relevant role in the mechanism
of alkane activation (103). Alkenes through their π- bond system can

Chapter One - Introduction
37
chemisorb on these sites forming relatively stable chemisorbed species,
although they may be considered "spectator" species, because they are not
directly involved in the mechanism of further selective oxidation of these
alkene intermediates. Oxygen also strongly chemisorbs on the surface
Lewis acid sites forming thermal stable species (they desorb above 450-
500°C), but which play a relevant role in the mechanism of oxidation.
When the surface concentration of the intermediate alkenes in alkane
oxidation is as high as to limit the amount of chemisorbed oxygen, due to
this competitive chemisorption, it is thus possible to control the population
of oxygen adspecies by this mechanism. This explains the considerable
promotion of selectivity to Partial oxidation products by increasing alkane
inlet concentration (Figure 1.15) and, on the other hand, explains also the
apparent contradiction of the different rate of acrylonitrile consecutive
oxidation when forms from propene instead of that from propane
(Figure 1.14).
Although propene forms from propane as a reaction intermediate, its
concentration is clearly higher when it is fed directly and thus its effect in
limiting the concentration of Surface oxygen species is present even for
higher conversions of the hydrocarbon. Therefore, the maximum in the
formation of acrylonitrile is observed at higher hydrocarbon conversions
when the alkene is fed instead of the alkane on the same catalyst
(Figure 1.14).
In the oxidation of n-butane ،partial oxidation product (butenes and
butadiene) are observed at low oxygen concentrations when a very limited
number of vanadium (V) species are present on the surface of the catalyst.
The change of valance state of vanadium on the catalytic surface upon a
balance of three factors:
a- The redox potential of the feed.
b- The rate of oxidation of the catalyst at the temperature of reaction.
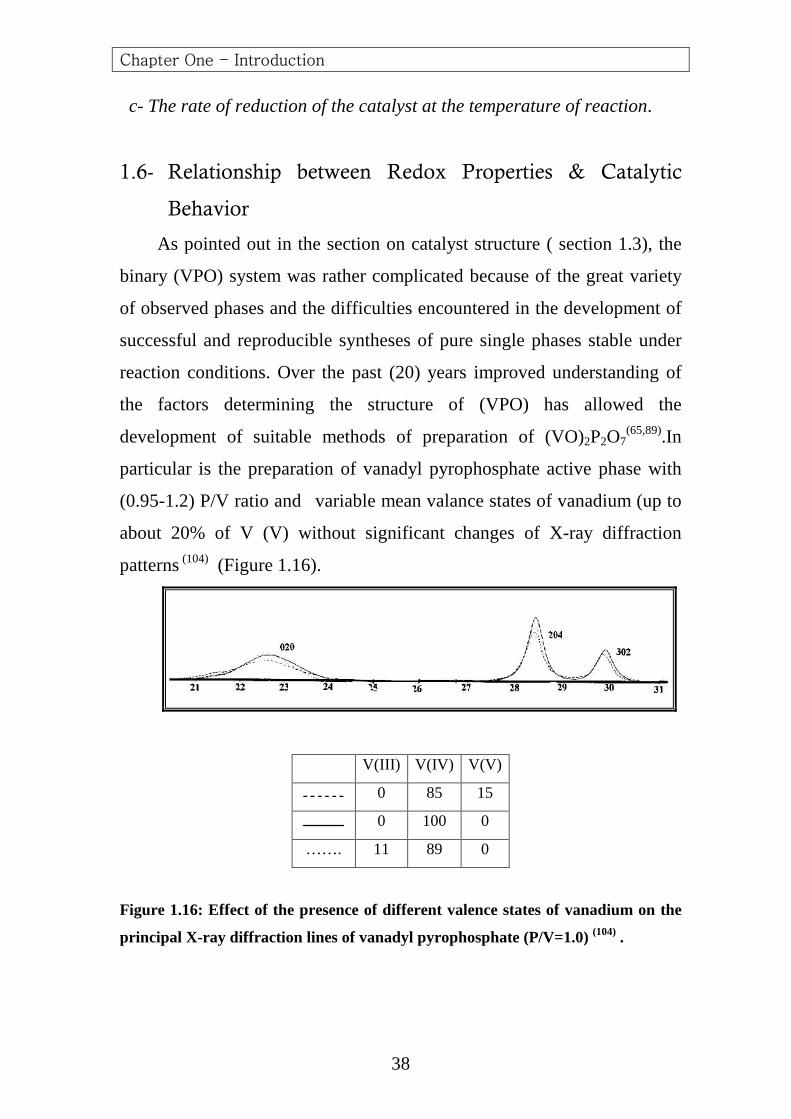
Chapter One - Introduction
38
c- The rate of reduction of the catalyst at the temperature of reaction.
1.6- Relationship between Redox Properties & Catalytic
Behavior
As pointed out in the section on catalyst structure ( section 1.3), the
binary (VPO) system was rather complicated because of the great variety
of observed phases and the difficulties encountered in the development of
successful and reproducible syntheses of pure single phases stable under
reaction conditions. Over the past (20) years improved understanding of
the factors determining the structure of (VPO) has allowed the
development of suitable methods of preparation of (VO)2P2O7(65,89).In
particular is the preparation of vanadyl pyrophosphate active phase with
(0.95-1.2) P/V ratio and variable mean valance states of vanadium (up to
about 20% of V (V) without significant changes of X-ray diffraction
patterns (104) (Figure 1.16).
V(III) V(IV) V(V)
0 85 15
0 100 0
……. 11 89 0
Figure 1.16: Effect of the presence of different valence states of vanadium on the
principal X-ray diffraction lines of vanadyl pyroph osphate (P/V=1.0) (104) .

Chapter One - Introduction
39
The variation of the content of (P) in the composition modifies the
catalytic properties in n-butane oxidation (86) and the redox properties of the
catalysts (104), a slight deficiency of phosphorus, does not change the rate of
V (IV) oxidation to V (V). On the other hand, an excess of phosphorus
only slightly influences the rate of oxidation, but strongly affects the rate
of reduction. This effect is in part attributed to a decrease of the number of
the active site, but also reflects a particular kind of interaction of
phosphorus upon the vanadium ions (61,104). In order to correlate the
observed variations in the redox properties with the catalytic behavior in n-
butane oxidation on the fresh catalyst used by these authors, it is necessary
to distinguish between tests at low conversion and high conversion.
At low conversion, the catalysts with (P/V ratio = 0.95 and 1.01) show
the same activity, selectivity, and kinetic behavior (86). However, at high
conversion of n-butane (80%) the catalyst deficient in phosphorus and with
the observed higher rate of vanadium oxidation forms primarily carbon
oxides. The catalyst with more phosphorus with respect to the
stoichiometric ratio of (1.0) are less active and less selective (90), but do not
show the strong decline of (MA) selectivity at the highest conversion.
It is reasonable to correlate these catalytic effects with the redox
properties of the catalyst (105). The strong increase of the rate of vanadium
oxidation in the phosphorus-deficient catalyst leads to an enhancement of
the rate of consecutive oxidation of ( MA) (effect on the selectivity),
whereas the decreased rate of reduction of vanadium in the catalyst with
higher (P) content than a stoichiometric one leads to the reduced rate of
hydrocarbon depletion (effect on the activity)
Nakamura et al. (105) also suggested that some V (V) ions were
necessary for (MA) synthesis, but the optimum mean of valance state of
catalyst is close to four. Similar conclusions were presented by Weing and
Schrader (74). They reported catalyst phosphorus loading is a key parameter
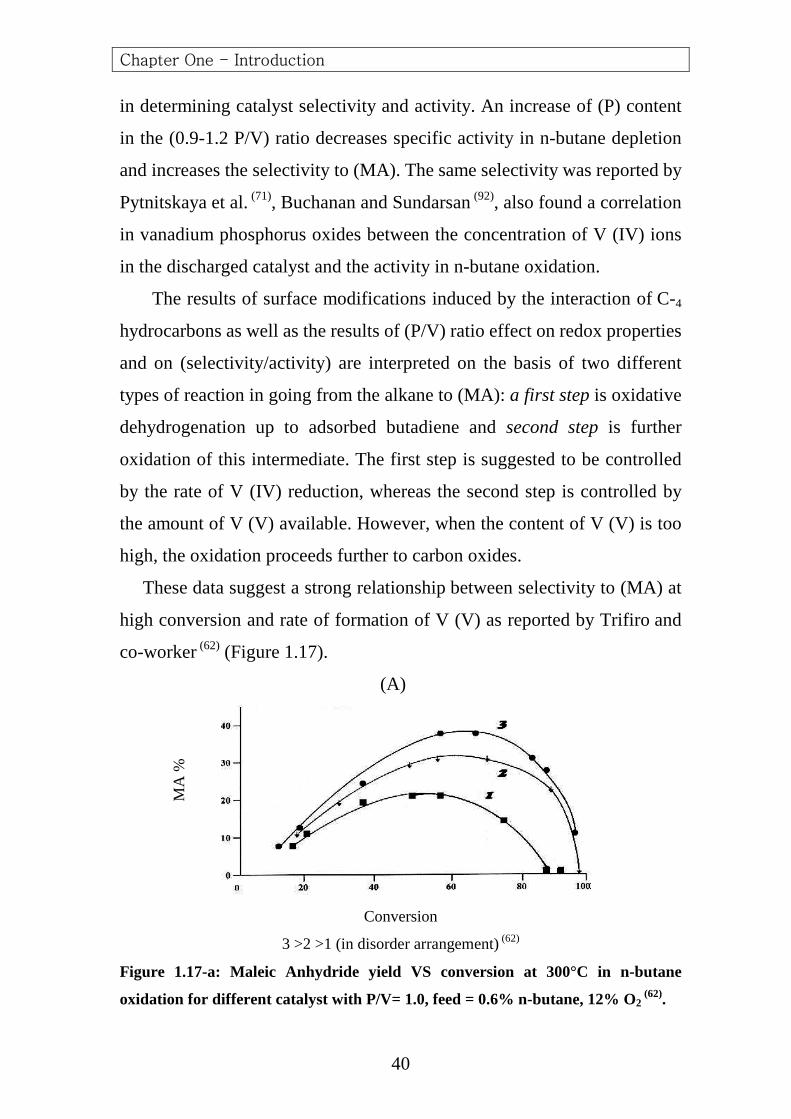
Chapter One - Introduction
40
in determining catalyst selectivity and activity. An increase of (P) content
in the (0.9-1.2 P/V) ratio decreases specific activity in n-butane depletion
and increases the selectivity to (MA). The same selectivity was reported by
Pytnitskaya et al. (71), Buchanan and Sundarsan (92), also found a correlation
in vanadium phosphorus oxides between the concentration of V (IV) ions
in the discharged catalyst and the activity in n-butane oxidation.
The results of surface modifications induced by the interaction of C-4
hydrocarbons as well as the results of (P/V) ratio effect on redox properties
and on (selectivity/activity) are interpreted on the basis of two different
types of reaction in going from the alkane to (MA): a first step is oxidative
dehydrogenation up to adsorbed butadiene and second step is further
oxidation of this intermediate. The first step is suggested to be controlled
by the rate of V (IV) reduction, whereas the second step is controlled by
the amount of V (V) available. However, when the content of V (V) is too
high, the oxidation proceeds further to carbon oxides.
These data suggest a strong relationship between selectivity to (MA) at
high conversion and rate of formation of V (V) as reported by Trifiro and
co-worker (62) (Figure 1.17).
(A)
Conversion
3 >2 >1 (in disorder arrangement) (62)
Figure 1.17-a: Maleic Anhydride yield VS conversion at 300°C in n-butane
oxidation for different catalyst with P/V= 1.0, feed = 0.6% n-butane, 12% O2 (62).
MA
%
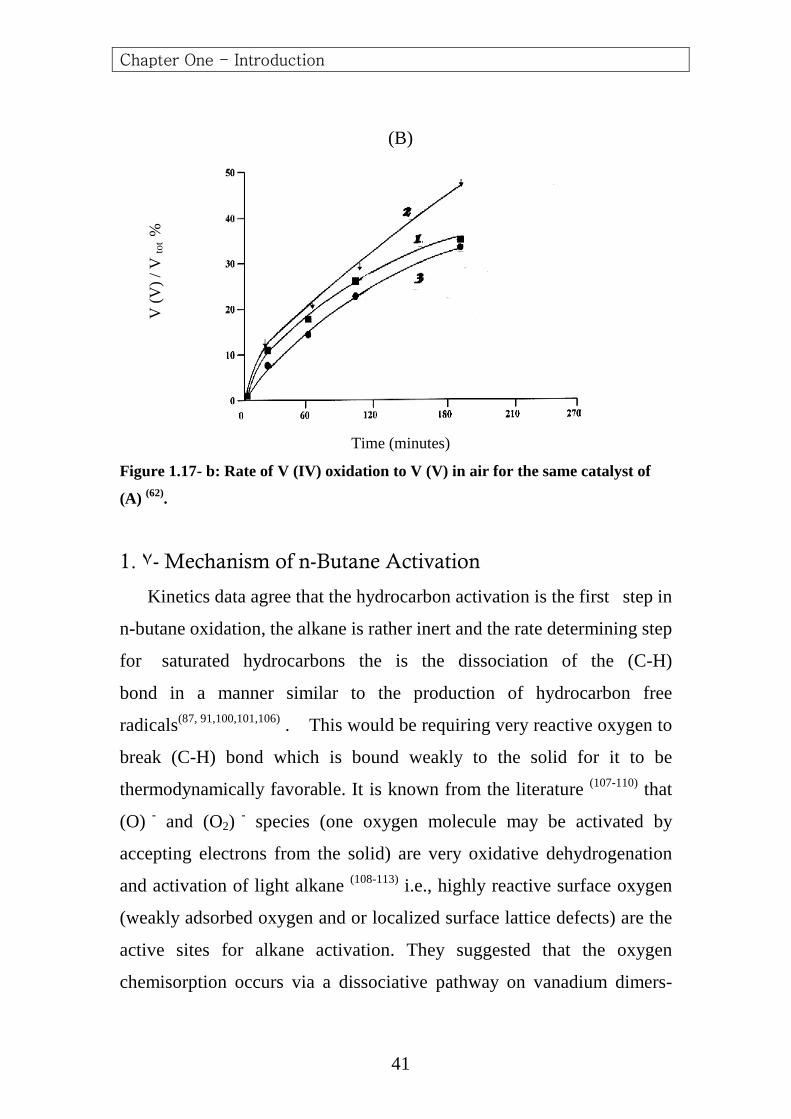
Chapter One - Introduction
41
(B)
Time (minutes)
Figure 1.17- b: Rate of V (IV) oxidation to V (V) in air for the same catalyst of
(A) (62).
1. ٧ - Mechanism of n-Butane Activation
Kinetics data agree that the hydrocarbon activation is the first step in
n-butane oxidation, the alkane is rather inert and the rate determining step
for saturated hydrocarbons the is the dissociation of the (C-H)
bond in a manner similar to the production of hydrocarbon free
radicals(87, 91,100,101,106) . This would be requiring very reactive oxygen to
break (C-H) bond which is bound weakly to the solid for it to be
thermodynamically favorable. It is known from the literature (107-110) that
(O) - and (O2) - species (one oxygen molecule may be activated by
accepting electrons from the solid) are very oxidative dehydrogenation
and activation of light alkane (108-113) i.e., highly reactive surface oxygen
(weakly adsorbed oxygen and or localized surface lattice defects) are the
active sites for alkane activation. They suggested that the oxygen
chemisorption occurs via a dissociative pathway on vanadium dimers-
V (
V)
/ V to
t %

Chapter One - Introduction
42
leading to a (V (V)-O*) type surface species capable of activating the
alkane.
Due to the topotactic transformation that occurs without the breaking
of (VPO) connections and due to the higher electronegativity of (P) with
respect to (V), the presence of defects in the structure of vanadyl
pyrophosphate is reflected on the surface in an enhancement of Lewis
acidity of surface unsaturated vanadium ions (Figure 1.18). Strong acid
sites on (VO)2P2O7 were observed also by Puttock and Rochester (111,112) by
infrared spectroscopy.
(A)
A- Medium –strong Lewis acid sites, well crystalline structure
(B)
B- Very strong Lewis acid sites, disordered crystalline structure
Figure 1.18: Model of Lewis acid sites in (VO)2P2O7 with different degrees of
disorder in the stacking fold of (020) planes (62).

Chapter One - Introduction
43
All above observations may be summarized in the following model:
The presence of defects in the crystalline structure creates strain in the
V-P-O bonds and would create a surface-activated reactive couple.
V O Pδ δ+
− − − − −−
→
……….. (1.5)
This reactive couple was proposed by Centi et al. (73) as being
responsible for the concerted mechanism of the removal of two hydrogen
atoms, according to the (Figure 1.19-a, 1.19-b and Figure 1.20) which show
the redox cycle proposed to this mechanism of activation of n-butane.The
model is based on the idea that (V (IV)–oxygen) are the active sites of butane
dehydrogenation (62,99).
Munson and et al. (113) had developed an experimental protocol to study
the mechanism of this reaction in which 13C-isotopically labeled n-butane
is flowed over a catalyst bed and the reaction products are analyzed using 13C NMR spectroscopy. This strongly suggests that the total oxidation of
n-butane on (VPO) catalysts involves the oxidation and abstraction of the
two methyl groups of n-butane, and the two methylene groups of n-butane
form ethylene. An organometallic mechanism is proposed to explain these
results.
In conclusion, it is necessary to try to develop new models of the
surface reactivity which include new evidence on aspects such as (i) the
role of chemisorbed species on the surface reactivity, (ii) the presence of
multiple pathways of reaction (iii) the dynamics of catalyst reconstruction
(iv) the mobility of surface adspecies, etc. The consideration of all these
possible effects in analyzing the surface reactivity will make possible the
design of new catalysts as well as the understanding of surface
reactivity at oxide surfaces (99).
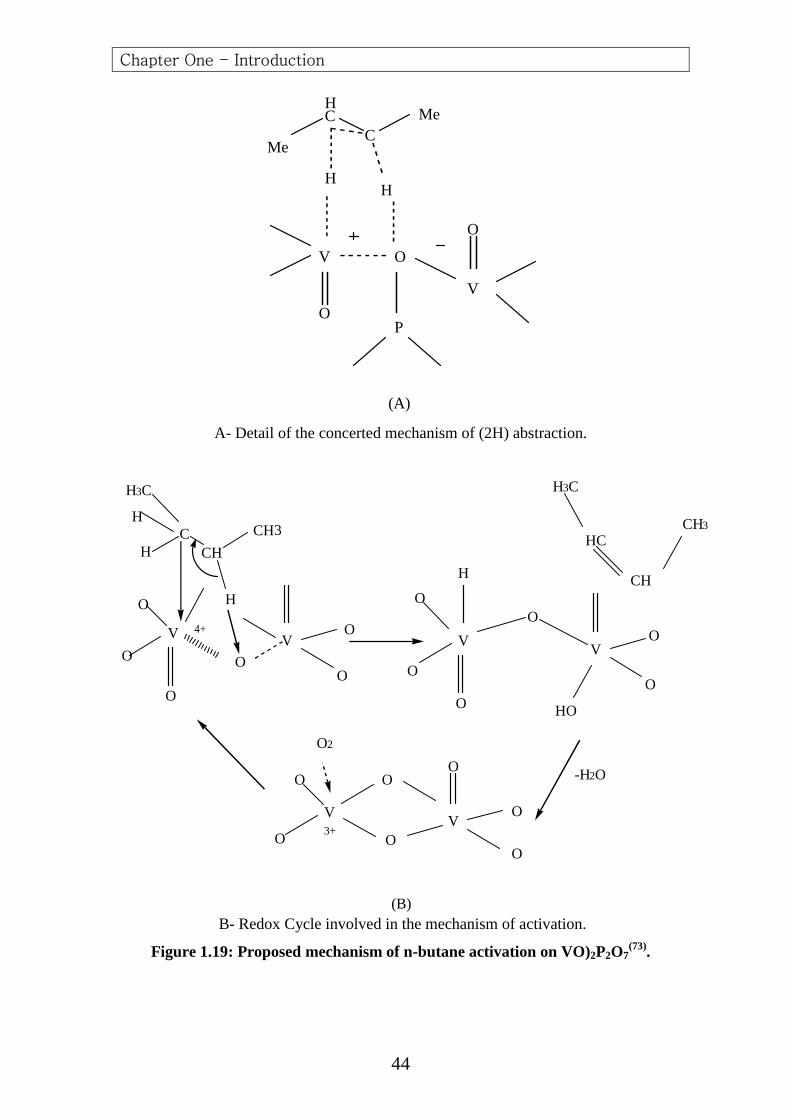
Chapter One - Introduction
44
HC
CMe
Me
H
V O
V
O
OP
H
(A) A- Detail of the concerted mechanism of (2H) abstraction.
VV
O
O
O
CCH
CH3
H3C
H
H
H
V VO
O
O
O
O
O
O
H
HO
-H2O
VV
O
O O
O
O2
O
O
O
O
3+
O
CH
HC
H3C
CH3
4+
(B) B- Redox Cycle involved in the mechanism of activation.
Figure 1.19: Proposed mechanism of n-butane activation on VO)2P2O7(73).
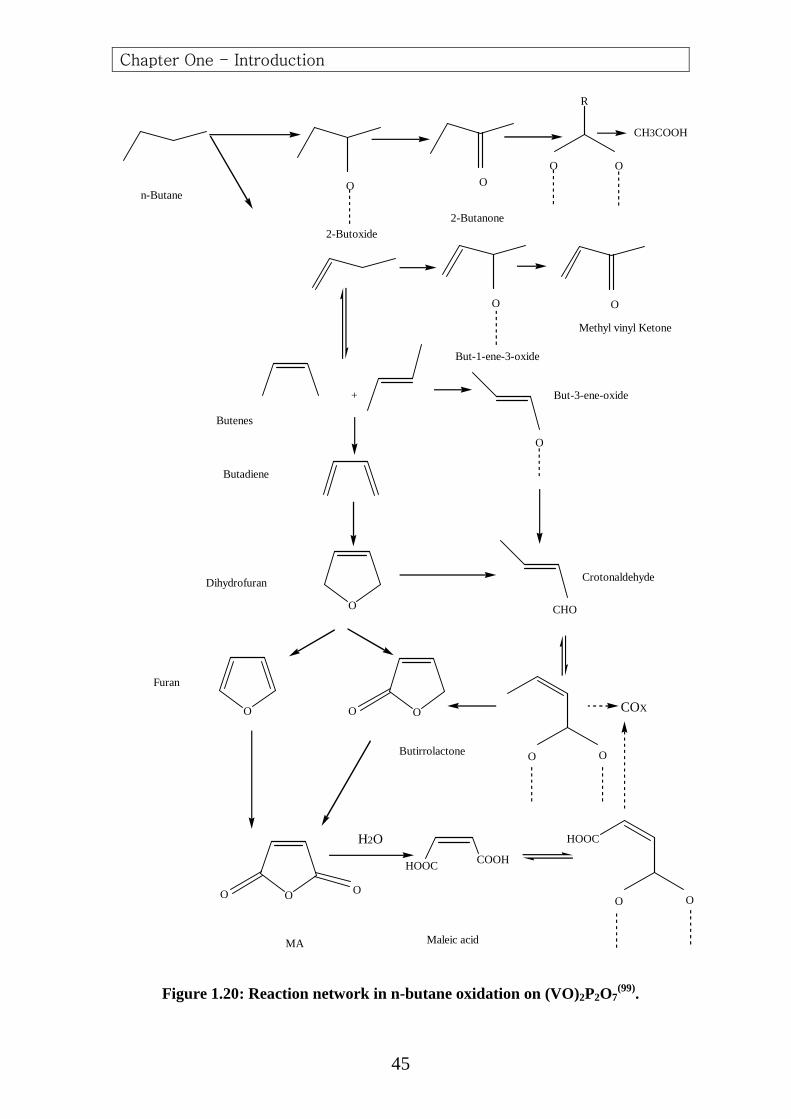
Chapter One - Introduction
45
O O
R
O O
+
O
O OO
OO O
COOHHOOC
HOOC
O O
O O
CH3COOH
O
CHO
O O
COX
Furan
MA
Dihydrofuran
Butadiene
n-Butane
Methyl vinyl Ketone
But-3-ene-oxide
Crotonaldehyde
Maleic acid
Butirrolactone
But-1-ene-3-oxide
2-Butoxide
2-Butanone
Butenes
H2O
Figure 1.20: Reaction network in n-butane oxidation on (VO)2P2O7(99).

Chapter One - Introduction
46
1.8 - Role of Adsorbed & Lattice Oxygen Species
In the selective oxidation of n-butane to (MA) on vanadyl
pyrophosphate (VO)2P2O7 catalysts, many results have been reported on
the respective roles of lattice and gas-phase oxygen in the formation of
partial and complete oxidation products (113).
Three types of lattice oxygen species with different reactivites are
present at the surface of vanadyl pyrophosphate (Figure 1.21) (24).
OVO
VV
P
O
VV
(a) (b) (c)
Figure 1.21: The Three Types of Oxygen Species (24).
The participation of lattice oxygen is a general characteristic of metal
oxide catalyzed reaction in selective hydrocarbon oxidation (114-117).
Pepera et al. (93) concluded that the lattice oxygen ions located in the
top few surface layers are responsible for the oxidation of n-butane to
(MA, CO and CO2). Another authors (106 , 117, 118) obtained experimental
evidence to support this conclusion. Abon and co-workers (119) claimed on
the basis of isotopic labeling results that only lattice oxygen is active for
the formation of (MA) and other products. The circulating fluidized-bed
riser reactor technology for (MA) production described in the literature

Chapter One - Introduction
47
(18,21,119) is based upon the fact that, under anaerobic conditions, the lattice
oxygen of (VPO) can selectively oxidize butane to (MA) (18,21,119).
This problem becomes more complex when oxygen is co-fed with
butane under steady state reaction conditions employed in industrial fixed-
bed reactor processes. In addition to the conventional Mars-Van Krevelen
mechanism, where lattice oxygen is the active agent for butane oxidation,
gas-phase oxygen, surface lattice oxygen and / or activated chemisorbed
oxygen have all been proposed as important oxidants in the formation of
Maleic anhydride or in the formation of unselective products, (CO and
CO2)(96,107,120-122). For example, Trifiro et al. (95) proposed that adsorbed
oxygen is responsible for selective oxidation and that it is involved in the
oxygen insertion steps required for the formation of (MA). Ebner and co-
workers (95,106) concluded that adsorbed oxygen is selective only in the
(MA) formation step. They proposed that two types of oxygen are involved
in butane oxidation: surface lattice oxygen that is responsible for ring
closure, and activated chemisorbed oxygen, (O*), that is involved in the
further step of (MA) formation. Rodemerck et al. (121) reported that
adsorbed oxygen is active but not selective, i. e.; it can only produce
(CO2). In contrast to all of these studies, Zazhigalov et al. (122) concluded
that (MA) formation over (VO)2P2O7 is mainly due to gas- phase oxygen.
The controversy in the literature about the roles of lattice and
chemisorbed oxygen calls for further clarification of the puzzle. Wang and
co-workers (123) used a novel microbalance reactor to carry out kinetic
analyses of butane oxidation by (VPO) catalysts and of the oxidation of
partially reduced (VPO) with oxygen .These authors conclude from there
experiments that both lattice oxygen and adsorbed oxygen on (VPO)
catalyst can selectively oxidize butane to (MA). Under aerobic conditions,
the oxidation of butane by adsorbed oxygen species is much faster than
by lattice oxygen.

Chapter One - Introduction
48
The first attempt to rationalize the problem of types of oxygen species
involved in the mechanism of C-4 alkene transformation to (MA) was
reported by Weiss et al. (124) , vanadium-oxygen double bonds (V=O) were
suggested to be the active sites both in:
The oxydehydrogenation step from butenes to butadiene, the
consecutive steps of oxygen insertion to (MA).The first step involves a
homolytic (C-H) dissociation of the adsorbed olefine and formation of the
allyl radical coordinated to a metallic transition ion as a surface π-allyl
complex. The allyl carbcation obtained can form the diene by loss of a
proton through reaction with a nucleophilic agent. The allyl ester that
forms can undergo a rapid reversible rearrangement in which each end of
the carbon of the skeleton is alternatively bonded to the lattice oxygen.
This lattice oxygen possesses a weak electrophilic reactivity. On the
contrary, electrophilic oxygen must be involved in the attack on the diene.
The proposed mechanism is shown in (Figure 1.22-a). The product of
attack on the diene can occur according to two-selective pathways, the first
one involves 1, 4- cyclization to 2, 5-dihydrofuran which is easily
dehydrogenated to furan; the second pathway is deprotonization of the
allyl carbcation to a dienolate structure.
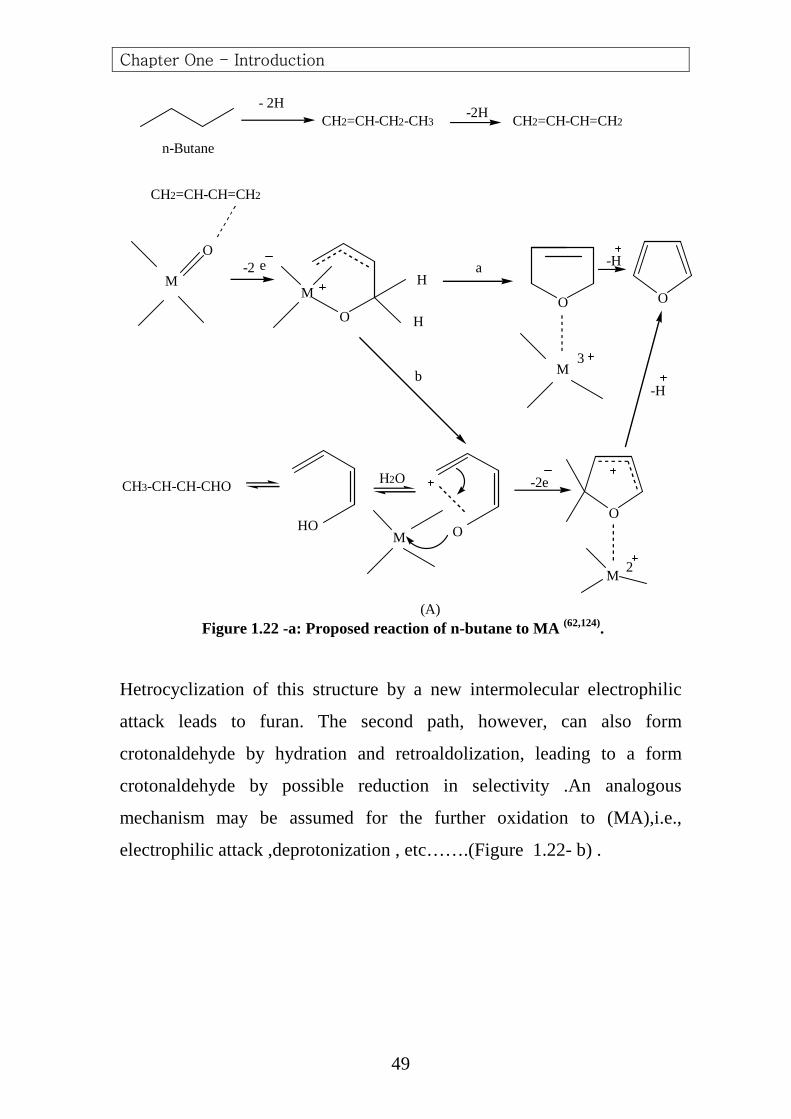
Chapter One - Introduction
49
n-Butane
- 2HCH2=CH-CH2-CH3 CH2=CH-CH=CH2
CH2=CH-CH=CH2
O
M
O
MH
HO
M3
CH3-CH-CH-CHO
HO OM
O
M2
-2H
-2 e a
O
b
H2O -2e
-H
-H
(A) Figure 1.22 -a: Proposed reaction of n-butane to MA (62,124).
Hetrocyclization of this structure by a new intermolecular electrophilic
attack leads to furan. The second path, however, can also form
crotonaldehyde by hydration and retroaldolization, leading to a form
crotonaldehyde by possible reduction in selectivity .An analogous
mechanism may be assumed for the further oxidation to (MA),i.e.,
electrophilic attack ,deprotonization , etc…….(Figure 1.22- b) .
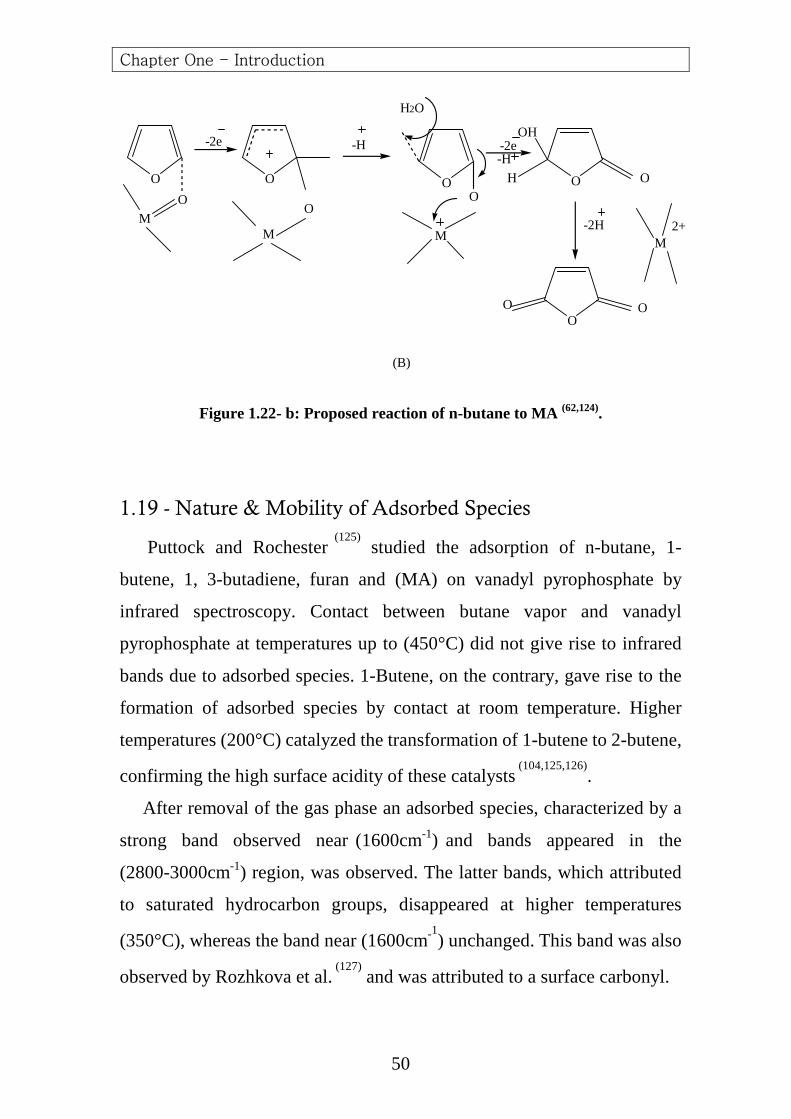
Chapter One - Introduction
50
O
-2e
O
M
O
O
M
-H
O
H2O
O
M
-2e-H
O
OH
OH
-2H
OOO
M2+
(B)
Figure 1.22- b: Proposed reaction of n-butane to MA (62,124).
1.19 - Nature & Mobility of Adsorbed Species
Puttock and Rochester (125)
studied the adsorption of n-butane, 1-
butene, 1, 3-butadiene, furan and (MA) on vanadyl pyrophosphate by
infrared spectroscopy. Contact between butane vapor and vanadyl
pyrophosphate at temperatures up to (450°C) did not give rise to infrared
bands due to adsorbed species. 1-Butene, on the contrary, gave rise to the
formation of adsorbed species by contact at room temperature. Higher
temperatures (200°C) catalyzed the transformation of 1-butene to 2-butene,
confirming the high surface acidity of these catalysts (104,125,126)
.
After removal of the gas phase an adsorbed species, characterized by a
strong band observed near (1600cm-1) and bands appeared in the
(2800-3000cm-1) region, was observed. The latter bands, which attributed
to saturated hydrocarbon groups, disappeared at higher temperatures
(350°C), whereas the band near (1600cm-1) unchanged. This band was also
observed by Rozhkova et al. (127)
and was attributed to a surface carbonyl.

Chapter One - Introduction
51
A similar spectrum of adsorbed species was found in the interaction of
furan with the catalyst. The primary interaction of furan and vanadyl
pyrophosphate was suggested to involve coordination between the oxygen
atoms of associatively adsorbed furan molecules and coordinatively
unsaturated exposed vanadium cations with Lewis acid properties. Furan
showed a slight oxidation to maleic anhydride in the absence of oxygen but
the presence of oxygen promoted maleic anhydride was primarily non
dissociatively adsorbed on (VO)2P
2O
7, but two further bands at (1560 and
1450cm-1), attributed to carboxylate anions, suggested a partial oxidation
of the adsorbed (MA).
Weing and Schrader (74)
studied by infrared spectroscopy the n-butane
interaction with (VPO) catalysts with variable (P/V= 0.9, 1.0 and 1.1)
using an in situ FTIR cell. These authors presented evidence for the
presence of reactant (n-butane), partially oxidized product (MA),
combustion products (CO, CO2, and H2O) and reactive surface species
(Maleic acid and olefins) on the vanadyl pyrophosphate. Another
observations reported by Pepera and co-workers (93)
agree with the
hypothesis that the catalytic process in this complex reaction involves the
shuttling of hydrogen a way from and oxygen toward the intermediate
adsorbed on the surface, and the intermediate forms a stable surface
species (which does not adsorbed or react) to (MA). This mechanism is a
key to the selectivity and the absence of intermediate products in the
reaction of n-butane conversion to (MA).
1.10- Promoted (VPO) Catalysts
Numerous attempts have been made to synthesize improved (VPO)
catalysts by adding various amounts of other metallic elements. Although

Chapter One - Introduction
52
reports of some of these attempts have appeared in literature (128-146), the
vast majority were found in the form of patents (64,128-130,133-136)
.
The effect of these promoters was studied by many authors; Ai (131,132)
studied the effect of alkali metal addition on activity and selectivity. It was
reported that the addition of less than (10% Li) showed no remarkable
effect, Ai also studied the effect of the methods of preparing V2O
5-P
2O
5-
ZrO2 catalysts on their activity and selectivity in the oxidation of n-butane
by changing the procedures of ZrO2addition. Ai found that the best
performance was obtained with the catalyst prepared by adding
simultaneously two solutions of ethyleneglycol in which ZnOCl2 and o-
H3PO
4 have been dissolved to a powder of the precursor of the (V
2O
5-
P2O
5) catalyst prepared in an organic medium. The presence of the vanadyl
pyrophosphate crystalline phase was detected in all of the catalysts. (131,132)
The addition of suitable activators to (VPO) catalyst was reported to
give an improved yield of (MA) by several patent claims (6,133-136)
using a
catalyst containing (P-V-Mo) to oxidation n-butane to (MA), this catalyst
gave a maximum (MA) yield of (88%).
Zazhigalov and co-workers (128,137) studied the effect of added alkali and
alkaline-earth metals (Li, Na, K, Mg, Ca or Ba) on the properties of (VPO)
catalysts. The results showed that the addition of alkali and alkaline earth
metals to these catalysts produced two effects, on the one hand, there was
an increase in the content of oxygen on the surface and hence an increase
in the number of acid centers, on the other hand, the presence of these
promoters increase the (P/V) ratio on the surface of the catalyst and this
explains their stabilizing effect. It should be noted that the greatest increase
in the surface content of phosphorus was observed when (Li) is added, and
(Li) was considered to be one of the best stabilizing additives

Chapter One - Introduction
53
A new catalyst belonging to the (V-P-Mo-O) system selective in the
mild oxidation of n-butane to (MA) were prepared and characterized by
Courtine et al. (139)
These attempts led to the conclusion that (Mo)could be
substituted up to (7%) in (VOPO4) phases. Thermal analysis of the
hydrated precursor, XRD and IR spectroscopies of both hydrated and
anhydrous solid phases obtained showed that the solid solutions
isostructural with (VOPO4.2H2O) and (α-VOPO4) receptively could be
formulated.
Zazhigalov and co-workers (140) studied the properties of cobalt-promot
(VO) ٢ P2O7 in the oxidation of n-butane. This study focused on the
influence of cobalt additives on the composition of the vanadium-
containing catalyst, and on cobalt's other properties which were important
for the production of industrial (VPO) catalysts. Fresh catalysts were
composed of (VOHPO4.0.5H2O) phase. After reaction the catalysts
contained (VO)2P2O7. Cobalt was uniformly distributed in the pellets. Its
presence increased the content of phosphorus at the surface, which
modified the surface acidity and in turn improved the selectivity for n-
butane oxidation. No changes of the profile of phosphorus with depth were
observed, even after (500h) on stream, the surface composition of the
catalyst remained unchanged. Cobalt stabilized the catalyst performance
by forming cobalt phosphate which reduced phosphorus losses, improves
its catalytic properties and prolonged its lifetime.
A series of (VPO) catalysts with different (P/V) ratios and with or
without (indium and TEOS) additives have been characterized by
controlled-environment (XRD, ICP, ESCA, TEM, SEM, BET) and
chemical titration used for n-butane oxidation to (MA) (141) .The best
catalyst contained slight excess (P /V) with indium and tetra ethyl ortho-
silicate (TEOS) promoters. It was found that excess P increased the

Chapter One - Introduction
54
resistance of catalyst precursors toward oxidation and resulted in (VPO)
catalyst with a large exposed platelet face of layer morphology. The
promoters reduce the thickness of the platelet face of layer morphology.
The promoters reduce the thickness of the platelet and facilitate the
oxidation of the precursor which contains disordered (VOHPO4.0.5HO)
.The combination of the promoters and excess P results in a (VPO) catalyst
with appropriate oxidizability and morphology and gave high yields of
MA.
Guliants et al. (142) investigated the oxidation of n-butane to (MA)
over a model (Nb, Si, Ti, V and Zr) promoted bulk (VPO) and supported
vanadia catalyst , The promoters were concentrated in the surface region of
the bulk (VPO) catalysts .For the supported vanadia catalyst ,the vanadia
phase was present as a two-dimensional metal oxide over layer on the
different oxide supports (TiO2,ZrO2,Nb2O5,Al2O3 and SiO2) .No
correlation was found between the electronegativity of the promoter or
oxide support cation and the catalytic properties. Both promoted bulk
(VPO) and supported vanadia catalysts containing surface niobia species
were the most active and selective to (MA). These data suggested that the
activation of n-butane on both the bulk and supported vanadia catalysts
probably required both surface redox and acid sites, and that the acidity
also played an important role in controlling further kinetic steps of n-
butane oxidation.
The presence of (Na) in technical grade (V2O5) leads to solid solution
formation, this phenomenon lead to prepare VOPO4.2H2O with a new (P/V
=1.1). (1)
Sajip and co-workers (143) described and discussed the effect of Co and
Fe doping on (VPO) catalyst, prepared by organic method (with
isobutanol). At low levels, both Co and Fe dopants significantly enhanced
the selectivity and the intrinsic activity to MA. A combination of powder

Chapter One - Introduction
55
X-ray diffraction, P NMR spin-echo mapping spectroscopy and
transmission electron microscopy ,together with catalysts tests data, was
utilized to analyze the origin of the effects of Co doping. Co appears to be
essentially insoluble in crystalline (VO)2P2O7 and was preferentially
distributed in and stabilized an amorphous (VPO) material .It is suggested
that the origin of the promotional effect of (Co) was associated with its
interaction with the disordered (VPO) phase. The same techniques have
been used to analyze the Fe-doped catalyst, but at present it is not possible
to be definitive concerning the specific location of the Fe-dopant within
the phases present .Previous studies have indicated that (Fe) can form a
solid solution within (VO)2P2O7 and therefore it is probable that the (Fe)
amorphous vanadium phosphate phases are formed in the catalyst system.
Other authors (144) studied the effect of (Bi) on (VPO) catalyst, they
found that the incorporation of (Bi) into the (VPO) lattice lowered the
overall vanadium oxidation state from (4.24 to 4.08) .It also lowered both
the peak maximum temperature for the desorption of oxygen from the
lattice from (1001 K) (undoped) to (964 K) with shoulder at (912 K). The
total oxygen desorbed from the Bi-doped catalyst was only one-fourth that
of the undoped catalyst, while the amount of oxygen removed by TPR was
roughly the same for both catalysts. These results suggested that in
anaerobic oxidation, the Bi-doped catalyst will have roughly the same
activity as in undoped catalyst in C-4 hydrocarbon oxidation but would
have a higher selectivity to products such as olefins and (MA).
The barothermal and mechanochemical treatment of (VPO-Bi)
precursor was investigated (145), the barothermal treatment led to increase of
the relative ratio (001) plane of precursor without changes of the phase
composition. The (P/V) surface ratio increased more than two times and
the phosphorus surplus forms the islands on catalyst surface which
decrease the available active surface fragments. The change of Bronsted /

Chapter One - Introduction
56
Lewis acidity ratio of surface as result of treatment also was observed. The
activity of the catalyst in n-butane oxidation less change up to several
value of P/V ratio and decreased with its growth. The selectivity to (MA)
increase (more than 10% mol. %) practically in all interval of the (P/V)
ratio changes. Good correlation was observed between the selectivity to
Maleic Anhydride (MA) and Bronsted acidity of the catalyst.
The mechanochemical treatment of precursor showed an enhancement of
n-butane conversion and an improvement in (MA) selectivity and yield.
The sample milled in water exhibited a rise in conversion to (91%) but
selectivity increased only some percents. The maximal increase of the
selectivity after treatment in ethanol (more than 15 mol. %) and growth of
activity (but only 5-6 %) was observed. The correlation between the
changes of selectivity and Bronsted acidity was established (145).
1.11- Preparation of the catalysts
In accordance with the growing importance of the C-4 partial oxidation
route to (MA), numerous patents (16,19,20,146,147) have been awarded in the
past years for process which involved the preparation of (VPO) catalyst.
Various routes are available to prepare Vanadyl Hydrogen Phosphate
Hemihydrate VOHPO4.05H2O precursor, as outlined in Figure 1.23:
V2O5 + H3PO4 + i-BuOH
Organic Route
V2O5 + NH2OHHCl
VOHPO4.0.5H2O
(VO)2P2O7
Aqueous Route
+ H3PO4
Figure 1.23: Synthesis routes for vanadyl pyrophosphate catalysts (97).
The aqueous route entails the reduction ( V5+ ) by NH2OH , HCl or

Chapter One - Introduction
57
other reducing agents followed by the addition of H3PO4, VOHPO4.0.5H2O
is recovered by crystallization or evaporation.
The organic route involves the reduction of V2O5 by an organic solvent
(an alcohol), followed by the reaction with o-H3PO4 and recovery of the solid.
After recovery of the precursor, it is washed to remove trace amounts of
water soluble (V5+) compound and then calcined in nitrogen at (773) K
followed by final activation in air or n-butane/air at (673) K. It is critical that
calcination and activation be carefully controlled or the catalyst can be over
oxidized resulting in the formation of VOPO4 which is non selective
catalyst for n-butane (97).
The synthesis route and the reaction conditions during synthesis, affect the
morphology of and ultimately the catalyst performance. The precursor
prepared by the aqueous synthesis route is generally more crystalline than the
catalyst prepared by the organic route (95) .The organic synthesis route results
in platelet crystalline morphology; the size of the platelets and the way they
pack are determined by the choice of organic solvent. Isobutanol produces
rosette morphology where the platelets agglomerate. With sec. butyl or t-butyl
alcohol well formed platelets forms that do not agglomerate (148). The crystal
ordering of the precursor is also affected by the solvent. Large alcoholic
molecules, such as benzyl alcohol, appear to produced platelets with stacking
faults, deduced from the broadening of the (200) reflection in the XRD (149).
Stacking fault defects in the precursor have been correlated with improved
catalyst performance (148).
In recent years Hutchings and co-workers (149) prepared the precursor
by using V2O4 as a starting material with water as solvent. They also used
V2O4 with either H3PO4 99% or H4P2O7; reacted in autoclave at 145 ˚C to
form well crystalline VOHPO4.0.5H2O.The surface area of the precursor is
significantly enhanced when water is added as a solvent. On activation in
(n-butane / air) the catalyst surface area is increased from (4 m2/gm) for the

Chapter One - Introduction
58
precursor to (10-13 m2/gm). The selectivity to (MA) observed to be very
similar to other non- promoted (VPO) catalyst reported in the litterateur.
Although this study considered to unlikely that V2O4 can be used as a
starting material for the synthesis of commercial vanadium phosphate
catalyst, this study does show that relatively high area catalysts can be
achieved using water as solvent.
A dihydrate phases of (VPO) catalyst VOPO4.2H2O was prepared for
the first time in 1965 by Ladwig (33) by suspend V2O5 with rapped stirring
in distilled water and H3PO4 with (P/V = 7.3).The mixture was refluxed for
24 hours. The resulting precipitate was filtered, washed with distilled water
until a red color appeared then it was washed for several times with
acetone and left to dry.
No sign in the litterateur to prepare this phase with (P/V ratio =1.1).In
2000, several attempts were unsuccessfully exerted to prepare this phase in
the above ratio starting from V2O5 produced by Fluka and BDH company,
but all of these trials were not successful, on the other hand this phase was
successfully prepared starting from technical grade V2O5 with shorter
reaction time (16h), due to the intercalation effect of (Na ion) which led to
form solid solution of this phase. (1)
A new method through intercalation of VOPO4.2H2O crystallites in
primary alcohol (2- propanol or 1-butanol), followed by reduction with the
alcohol, have been investigated for the preparation of catalyst precursor.
Lamellar compounds were formed consisting of (V 4+, P +5 and alkyl
group) (150).
According to Bordes and Courtine (29), the calcination of VOPO4.2H2O
at 700 °C leads to obtain α-VOPO4.
Recently Bartley et al. (151) prepare vanadium (V) phosphate using
solvent-free method, by reacting (V2O5) with (H3PO4) in the absence of
water at 150 °C ,the resulting material was designed as anhydrous VOPO4.

Chapter One - Introduction
59
This material is readily hydrate to formVOPO4.2H2O. Activation of this
phase with n-butane/air leads to formation of (α1-VOPO4), while the
reaction of anhydrous (VOPO4) with alcohol leads to the exclusive
formation of VO (H2PO4)2.
Finally, there are three important parameters affect in determining
the final characteristics and activity of these catalysts which can be
summarized as:
a- The reducing agent and solvent (8, 70, 61,152-157).
b- The (P/ V) ratio
(8,146,148,155).
c- Activation conditions (29, 71)
.

Chapter One - Introduction
60
Conclusion of Bulk (VPO) catalyst
Oxidation of n-butane on the unsupported or bulk (VPO) catalysts is
the only known commercial process for an alkane oxidation.
A number of V (IV) and V (V) phosphates exist in the (VPO) system
and the correlation of catalytic performance with crystalline structure has
been reviewed (95,158).
Vanadyl (IV) pyrophosphate, (VO)2P2O7, has been identified as
critical for active and selective industrial catalysts (158). Some argue that the
(V5+/V4+) dimeric species in the top most oxidized layer of (VO)2P2O7 are
the active sites (8,159) , while others believe that the active sites lie within
the micro domains of crystalline vanadyl (V) orthophosphates, (β -, αII- ,
γ-, and δ-VOPO4), or (vanadyl (IV) meta phosphate) VO(PO3)2, formed
on the (100) faces of vanadyl pyrophosphate under the catalytic reaction
conditions (106). A recent kinetic study demonstrated that the best bulk
(VPO) catalysts contained only crystalline vanadyl (IV) pyrophosphate
after reaching the steady state (142). The vanadyl (IV) pyrophosphate phase
displayed preferential exposure of the (100) planes which contain vanadyl
dimers associated with active sites for n-butane oxidation (142). A high-
resolution Electron Microscopy study demonstrated that the surface (100)
planes in the fresh catalysts are covered with (1.5 nm) amorphous layered
which completely disappears within (23 days) of n-butane oxidation (96).
The kinetic studies of the bulk (VPO) catalysts further demonstrated the
similarity between the bulk (VPO) catalysts and supported catalytic
systems.
The catalytic activity of the bulk (VPO) catalysts was confined to a
very thin surface region of the (100) crystalline planes of vanadyl (IV)
pyrophosphate (1-2 atomic layers) (92).These findings suggested that the
crystalline vanadyl (IV) pyrophosphate phase in the bulk (VPO) catalysts
functioned as a support for the active surface. Such (VO)2P2O7 support

Chapter One - Introduction
61
stabilized some specific surface termination of the (100) planes with
(V4+/V5+) species and phosphate groups without contributing its lattice
oxygen to n-butane oxidation. Unfortunately, various spectroscopic
techniques, such as (Raman, ESR, UV-Vis, IR and EXAFS/XANES), are
unable to provide information about the molecular structure of the surface
present in the bulk (VPO) catalysts because of the much stronger signals
from the catalyst bulk than the catalyst surface. Limitations of both the
bulk and surface characterization techniques currently available, coupled
with the complex solid-state chemistry of vanadium phosphates, have led
to considerable confusion and contradictions in the literature concerning
the identity of the active sites involved in different steps of n-butane
oxidation to (MA) over the bulk (VPO) catalysts.
1.12- Supported (VPO) Catalyst:
Supported metal oxide catalysts consist of an active metal oxide
component, e.g. vanadia, deposited at the surface of an oxide support, such
as SiO2, TiO2 and Al2O3.
Currently, considerable interest is devoted to study selective oxidation
of alkanes on supported vanadia catalysts. The selective oxidation of
alkanes is highly desirable due to their potentially low environmental
impact and the relatively low cost of raw materials. Some typical systems
being studied are partial oxidation of n-butane to (MA) on supported
(VPO) catalysts (105,160) and oxidative dehydrogenation of n-butane (161).
Many fundamentals questions still remain unanswered about supported
vanadia catalysts despite the importance of this catalytic system.
This section concerns with structure-property relationships in C4-
hydrocarbon oxidation on supported vanadia catalysts.

Chapter One - Introduction
62
1.12.1-Molecular Structure of Vanadia Species in Fresh Catalysts
Structure of supported vanadia catalysts is a strong function of the
surface coverage. Surface coverage of vanadia over layer supported on
metal oxides has been determined experimentally using several
spectroscopic techniques, such as (Raman, IR, XPS, 51V NMR and UV-
Vis), as well as temperature-programmed reduction (TPR) and redox
reactions. Among these techniques, Raman spectroscopy is particularly
discriminating between the surface vanadia species and the crystalline
(V2O5). Characteristic Raman bands of polymeric vanadia species
corresponding to the (V-O-V) stretches, appear below (1000 cm-1) at high
coverages, while the isolated vanadate species present at sub monolayer
coverages produce only one Raman band of the terminal (V=O) bond at
(1020-1040cm-1). The finger print Raman features allow detecting
monolayer formation immediately prior to appearance of crystalline
(V2O5). Based on Raman spectroscopy measurements, monolayer surface
coverage on a number of oxide supports (Al2O3, TiO2, ZrO2, Nb2O5 and
CeO2) was found to be (7-8 VOx/nm2) (162) where nm is nanometer.
Vanadia supported on silica exhibited unusually low monolayer surface
coverage of only (0.7VOx/nm2). Lower density and reactivity of the
hydroxyls explain such low monolayer surface coverage on the silica
surface, which anchor vanadia species to the support. However,
commercial grade supports, such as pigment grade Titania, typically
contain monolayer quantities of (P, Na, K, Ca, etc.), which interact with
surface vanadia to form an amorphous phase (163). In such systems, the
surface vanadia titrates both the oxide support and the surface impurities,
delaying formation of crystalline V2O5 until several vanadia monolayers
are formed. Thus, the observed "monolayer'' vanadia coverage in such
impurity-containing systems can be (2-4) times greater than that found
above for the well defined supported catalysts.

Chapter One - Introduction
63
A number of synthesis methods were used to prepare supported
vanadia catalysts, non aqueous impregnation with vanadium alkoxides (169,170), such as incipient impregnation of support powder like (TiO2) with
aqueous solution of NH4VO3 in oxalic acid (164). Aqueous impregnation
with vanadium oxalate is favored in commercial preparation due to its high
solubility in water and the absence of undesirable volatile organic solvents.
Under ambient conditions the supported catalysts contain multilayers of
adsorbed water. Under such conditions the bridging (V-O-support) bonds
are hydrolyzed and the hydrated vanadia species are dissolved in the thin
aqueous layer.
A new generation of supports based on ceramics with high thermal
conductivity and without oxygen atoms were prepared and tested to
disperse the active (VO)2P2O7 phase (165). The best example is a new
relatively large specific surface area (β-SiC < 20 m2/g) prepared via the
shape memory synthesis. The (β-SiC-supported VPO) approved to be a
much better catalyst in terms of (MA) selectivity and yield than its bulk
equivalent, in fixed-bed configuration of reactor. The ability of this
ceramic to well disperse (VPO) was explained by the presence of an inter
phase (a glue) containing the (V, P, Si, C and O) elements, which did not
interfere with the formation of the active and selective (VPO) phase from
the (VOHPO4.0.5H2O).The real nature of this inter phase still needs to be
clarified. The high (MA) selectivity obtained on (VPO/β-SiC) was
attributed to both the absence of microporosity on the support and more
importantly, to the heat sink role of this support avoiding the formation of
hot spots on the catalytic sites and protecting the product, MA ,from
further oxidation into CO and CO2.
The uses of other exotic heat conductive materials as support such as
(Si3N4) and (BN) ,proves the validity of the concept of heat transfer from

Chapter One - Introduction
64
the surface to the bulk of the catalyst, without taking into account the
macroscopic transfer of heat outside the reactor (165).
It was finally demonstrated, from an industrial point of view ,that the
presence of the (β-SiC) support could provide high volume yield of (MA/
volume of catalyst) in a fixed-bed reactor because it was possible to work
with relatively high concentrations of n-butane ,contrary to what is
normally possible with the conventional bulk catalyst .In addition because
of the good dispersion, a large fraction of the expensive unused internal
part of the bulk active phase was replaced by cheap and light (β-SiC)
support .The optimal amount of active phase was found at (30wt %) of the
total composite. Finally, also demonstrated was the protective role of the
support on the thermal stability of the reactive phase which was much
more resistant to overheating accidents than the bulk material.
This new concept of heat control is currently being studied and used
for other similar problems where nonoxidic conductive materials are
potentially the answer to the difficulties that have been encountered in
many applications.
1.12.2- Molecular structure in dehydrated state
The moisture adsorbed in supported vanadia catalysts under ambient
conditions may be removed by heating supported catalysts in non reducing
oxygen containing atmosphere at elevated temperature, (573-973 K). Such
treatment also helps maintain the (5+) oxidation state of the surface
vanadia species. At temperatures below (473 K), the Raman stretch of the
terminal (V=O) bond occurring at (1020-1040 cm-1) is shifted significantly
downward due to extensive hydration of the surface vanadia. At
temperatures above (473 K) a small amount of moisture hydrogen bonded
to surface vanadia results in a few cm-1 downward shift of the Raman

Chapter One - Introduction
65
(V=O) band. At these temperatures (18O) labeling experiments showed that
the reversibly adsorbed moisture is also capable of rapidly undergoing
(18O) exchange with the terminal (V=O) bonds.
The structural studies suggested that the dehydrated vanadia species
on (Al2O3,TiO2, ZrO2, Nb2O5 and CeO2) was predominantly present as
isolated and polymeric tetrahedral VO4 units (Figure 1.24).The (18O)
labeling experiments of supported (VOx/ Al2O3, TiO2, ZrO2) showed that
these surface species possessed only one (terminal V = O) bond and three
bridging (Vـ�O-support) bonds for the isolated species, while the polymeric
surface species has only one (Vـ�O-support) bond per vanadium atom ,the
other two are being bridging V-O-V bonds (Figure 1.24). (166)
Spectroscopic techniques (X-ray absorption, Raman and UV-Vis.)
were utilized to monitor the effect of dehydration for vanadia supported on
(SiO2, Al2O3, TiO2, ZrO2 and HfO2) prepared with (VOx) surface densities
ranging from (0.46- 11.1 VOx/nm2) (166). UV-Vis. and near –edge spectra
indicate that supported (VOx) species exist in larger domains and have
higher – coordinate centers when hydrated. Dehydration consistently leads
to a breakup of these domains. Raman spectra suggest that (V=O) bonds in
mono and polyvanadate species are most susceptible to hydration. On all
supports, dehydration leads to the development of monovanadate species.
Polyvanadate on (TiO2, ZrO2, and HfO2) also undergo structural changes
when hydrated. Interpretation of this is more difficult because of the broad
Raman bands which allow only qualitative assignments. Support material
plays an important role in determining the extent of hydration, but this role
might arise from the ability to support polymeric vanadia species.

Chapter One - Introduction
66
Figure 1.24: Molecular structure of:
(A) Dehydrated isolated
(B) Polymerized vanadia species on oxide supports (166).
1.12.3- Structural changes during Hydrocarbon Oxidation
Wachs et al (161) investigated the oxidation of n-butane to (MA) over a
series of model-supported vanadia catalysts where the vanadia phase was
present as a two- dimensional metal oxide overlayer on the different oxide
supports ( TiO2, ZrO2, CeO2, Nb2O5, Al2O3, and SiO2). No correlation was
found between the properties of the terminal (V = O) bond and the butane
oxidation turnover frequency (TOF) which is mean the number molecules
converted per vanadium per second , during in situ Raman spectroscopy
study. Furthermore, neither the n-butane oxidation (TOF) nor (MA)
selectivity was related to the extent of reduction of the surface vanadia
species. The n-butane oxidation (TOF) was essentially independent of the
surface vanadia coverage, suggesting that the n-butane activation requires
only one surface vanadia site. The (MA) TOF, however, increased by a
factor of (2–3) as the surface vanadia coverage was increased to monolayer
coverage. The higher (MA) (TOF) at near monolayer coverages suggests
that a pair of adjacent vanadia sites may efficiently oxidize n-butane to
Maleic Anhydride. Other factors may also play a contributing role such as
(increase in surface Bronsted acidity and decrease in the number of

Chapter One - Introduction
67
exposed support cation sites. Varying the specific oxide support changed
the n- butane oxidation (TOF) by 50:
Ti > Ce > Zr ~ Nb > Al > Si
As well as the (MA) selectivity.
The (MA) selectivity closely followed the Lewis acid strength of the
oxide support cations:
Al > Nb > Ti > Si > Zr > Ce
The addition of acidic surface metal oxides (W, Nb, and P) to the surface
vanadia layer was found to have a beneficial effect on the n- butane
oxidation (TOF) and the (MA). The creation of bridging (V–O–P) bonds
had an especially positive effect on the (MA) selectivity .
1.12.4–Structure-Activity Relationships in Oxidation of
C4-hydrocarbons
Several studies on C4-hydrocarbon oxidation have been reported in the
literature (102,106,160).
Nakamura et al. (105) studied oxidation of 1-butene on alumina-
supported catalysts and reported reasonably high selectivities to (MA). The
results of their study showed that supported (VPO) catalysts could
selectively oxidize C4- hydrocarbons to (MA). They concluded that the
activity and selectivity of their supported catalysts for the oxidation of 1-
butene to (MA) were closely related to the oxidation state and the degree
of aggregation of vanadium ions. They implicated the (V4+/V5+) redox
mechanism of oxidation in which (V = O) bonds played an important role.
Mori et al. (102) studied oxidation of n-butane on titania-supported
vanadia catalysts at high temperature (693-763 K) and high n-butane
conversion. Such severe experimental conditions led to an over oxidation
of n-butane to (COx) and, likely, to combustion of the partial oxidation
products, including (MA). They found that the reaction rate under such

Chapter One - Introduction
68
conditions was proportional to the amount of (V 5+= O) species in the
catalyst and concluded that the reaction proceeded via the reduction-
oxidation or Mars-van Krevelen mechanism according to which the
surface (V = O) species contained the active oxygen.
Busca et al. (167) employed titania-supported vanadia catalysts in n-
butane oxidation and observed over oxidation of the hydrocarbon on very
active high surface area catalyst (117m2/g) at (723 K). When they
supported (10 wt% V2O5) on the low surface area Titania (18.4 m2/g),
vanadia formed micro crystals which were detrimental to selective
oxidation and led to n-butane combustion.
Owens and Kung (160) studied n-butane dehydrogenation on silica-
supported vanadia catalysts at (793) K. They observed that the isolated
vanadia species present at low surface coverage (0.53 and 0.58 wt% V)
were responsible for the high total dehydrogenation selectivity (1-butene,
cis-/trans-2-butene, and 1, 3-butadiene as shown in (Figure 1.25) (160).
1 2 3 4
Figure 1.25: Oxidation of butane on dispersed vanadia species (160).
The presence of the crystalline V2O5 species at high surface coverage
(998, 703, 526, 480, 404, 304 and 284 cm-1) Raman bands for the (6.4 wt%
V) catalyst contributed to the production of total oxidation products.
C4H9
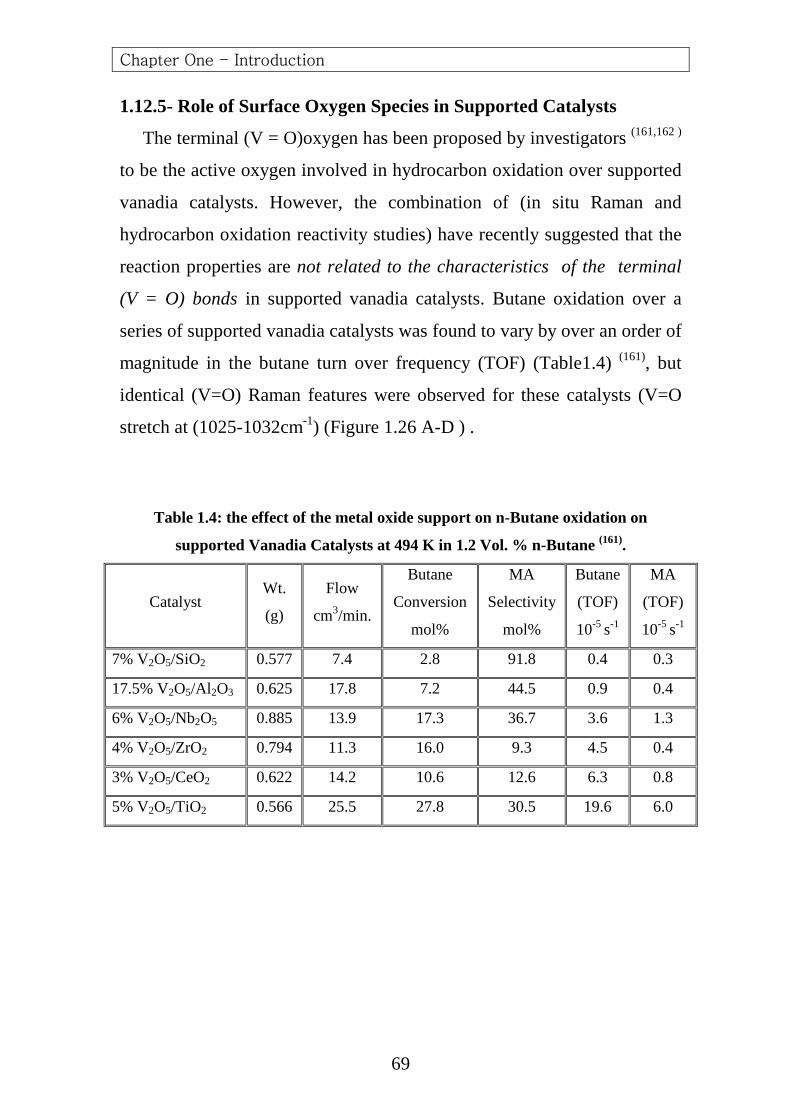
Chapter One - Introduction
69
1.12.5- Role of Surface Oxygen Species in Supported Catalysts
The terminal (V = O)oxygen has been proposed by investigators (161,162 )
to be the active oxygen involved in hydrocarbon oxidation over supported
vanadia catalysts. However, the combination of (in situ Raman and
hydrocarbon oxidation reactivity studies) have recently suggested that the
reaction properties are not related to the characteristics of the terminal
(V = O) bonds in supported vanadia catalysts. Butane oxidation over a
series of supported vanadia catalysts was found to vary by over an order of
magnitude in the butane turn over frequency (TOF) (Table1.4) (161), but
identical (V=O) Raman features were observed for these catalysts (V=O
stretch at (1025-1032cm-1) (Figure 1.26 A-D ) .
Table 1.4: the effect of the metal oxide support on n-Butane oxidation on
supported Vanadia Catalysts at 494 K in 1.2 Vol. % n-Butane (161).
Catalyst Wt.
(g)
Flow
cm3/min.
Butane
Conversion
mol%
MA
Selectivity
mol%
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
7% V2O5/SiO2 0.577 7.4 2.8 91.8 0.4 0.3
17.5% V2O5/Al 2O3 0.625 17.8 7.2 44.5 0.9 0.4
6% V2O5/Nb2O5 0.885 13.9 17.3 36.7 3.6 1.3
4% V2O5/ZrO2 0.794 11.3 16.0 9.3 4.5 0.4
3% V2O5/CeO2 0.622 14.2 10.6 12.6 6.3 0.8
5% V2O5/TiO2 0.566 25.5 27.8 30.5 19.6 6.0
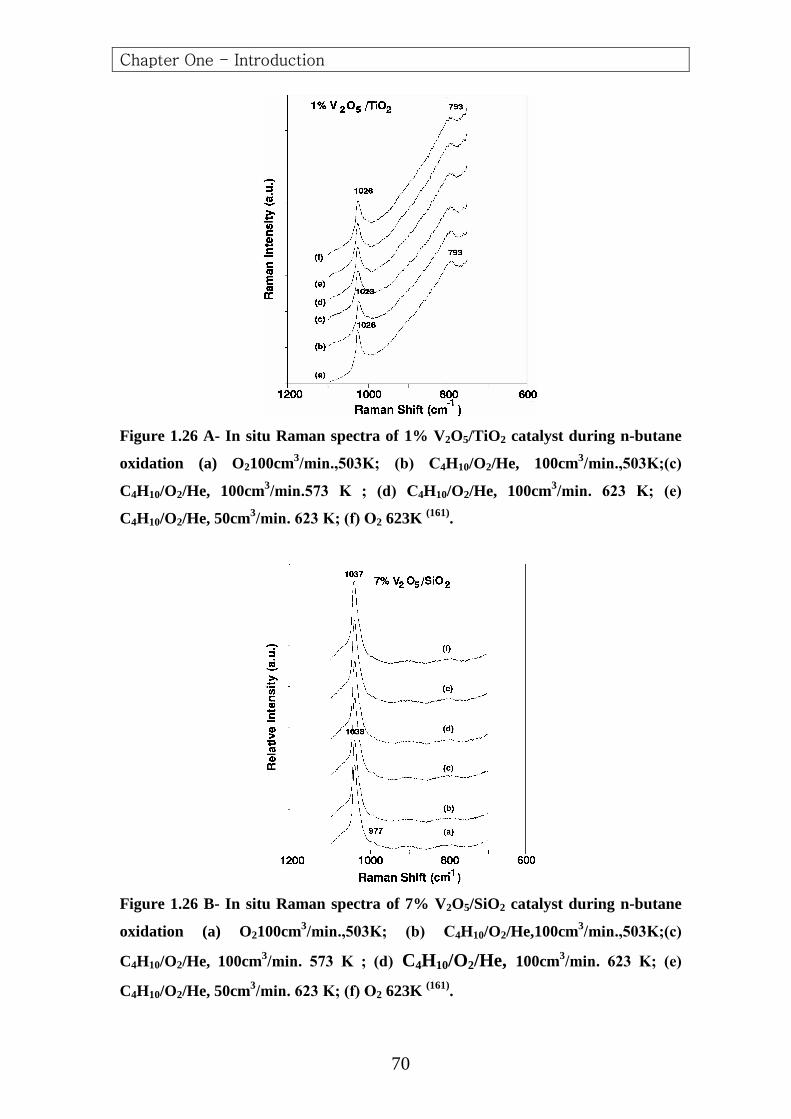
Chapter One - Introduction
70
Figure 1.26 A- In situ Raman spectra of 1% V2O5/TiO 2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He, 100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min.573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161).
Figure 1.26 B- In situ Raman spectra of 7% V2O5/SiO2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He,100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161).
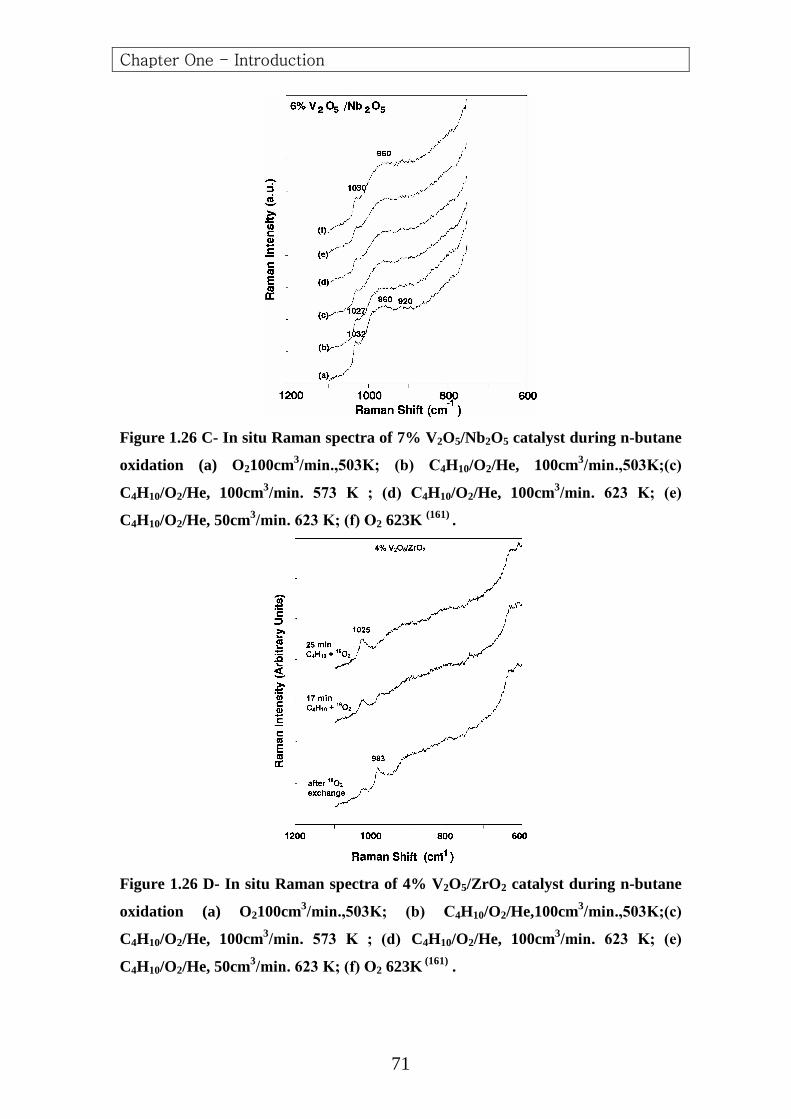
Chapter One - Introduction
71
Figure 1.26 C- In situ Raman spectra of 7% V2O5/Nb2O5 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He, 100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161) .
Figure 1.26 D- In situ Raman spectra of 4% V2O5/ZrO 2 catalyst during n-butane
oxidation (a) O2100cm3/min.,503K; (b) C4H10/O2/He,100cm3
/min.,503K;(c)
C4H10/O2/He, 100cm3/min. 573 K ; (d) C4H10/O2/He, 100cm3
/min. 623 K; (e)
C4H10/O2/He, 50cm3/min. 623 K; (f) O2 623K (161) .
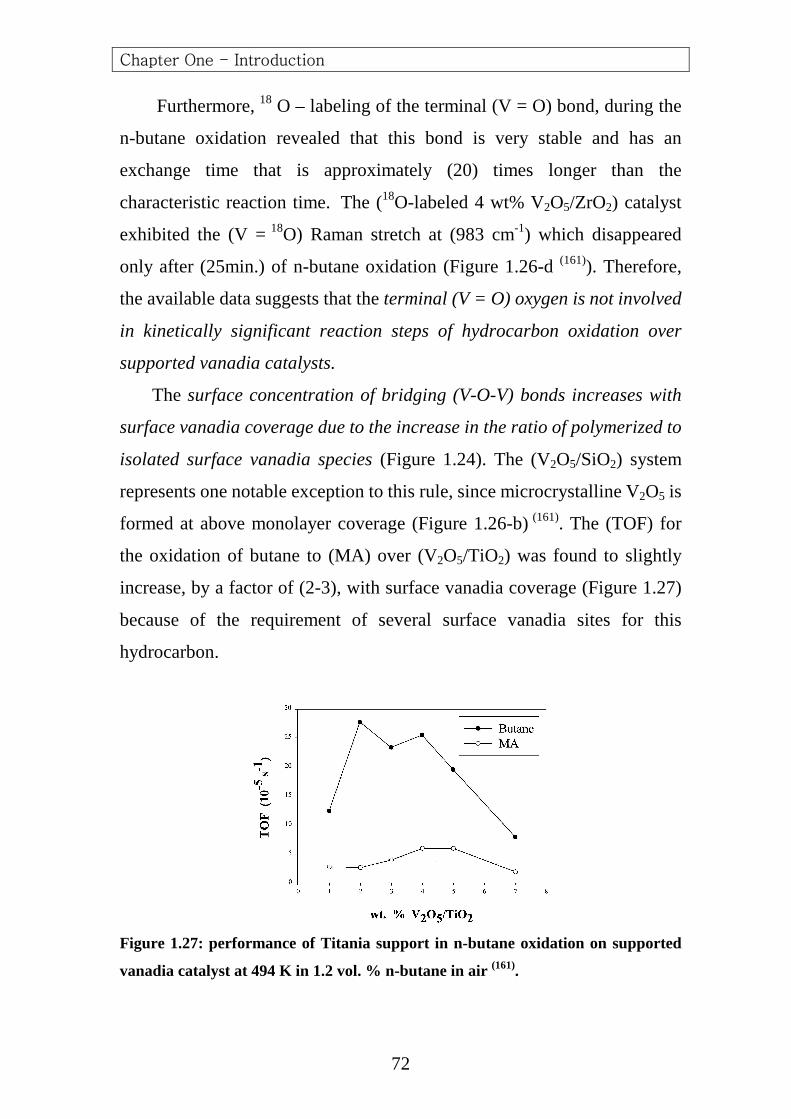
Chapter One - Introduction
72
Furthermore, 18 O – labeling of the terminal (V = O) bond, during the
n-butane oxidation revealed that this bond is very stable and has an
exchange time that is approximately (20) times longer than the
characteristic reaction time. The (18O-labeled 4 wt% V2O5/ZrO2) catalyst
exhibited the (V = 18O) Raman stretch at (983 cm-1) which disappeared
only after (25min.) of n-butane oxidation (Figure 1.26-d (161)). Therefore,
the available data suggests that the terminal (V = O) oxygen is not involved
in kinetically significant reaction steps of hydrocarbon oxidation over
supported vanadia catalysts.
The surface concentration of bridging (V-O-V) bonds increases with
surface vanadia coverage due to the increase in the ratio of polymerized to
isolated surface vanadia species (Figure 1.24). The (V2O5/SiO2) system
represents one notable exception to this rule, since microcrystalline V2O5 is
formed at above monolayer coverage (Figure 1.26-b) (161). The (TOF) for
the oxidation of butane to (MA) over (V2O5/TiO2) was found to slightly
increase, by a factor of (2-3), with surface vanadia coverage (Figure 1.27)
because of the requirement of several surface vanadia sites for this
hydrocarbon.
Figure 1.27: performance of Titania support in n-butane oxidation on supported
vanadia catalyst at 494 K in 1.2 vol. % n-butane in air (161).
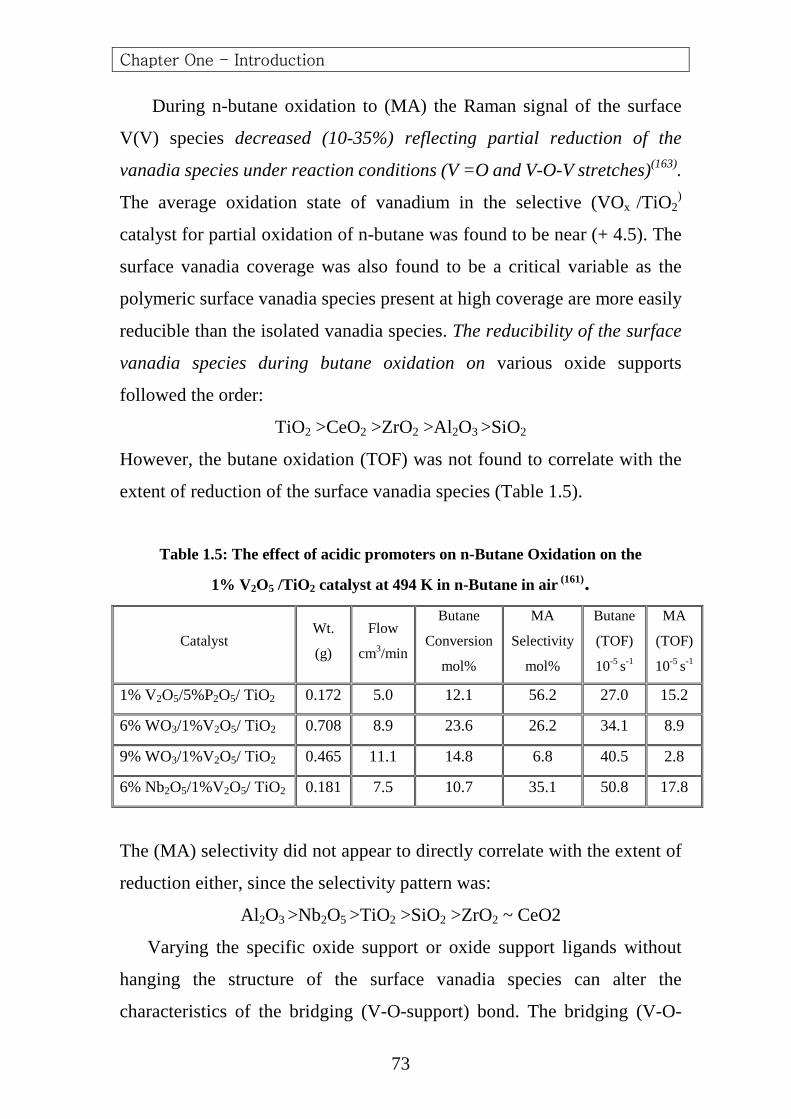
Chapter One - Introduction
73
During n-butane oxidation to (MA) the Raman signal of the surface
V(V) species decreased (10-35%) reflecting partial reduction of the
vanadia species under reaction conditions (V =O and V-O-V stretches)(163).
The average oxidation state of vanadium in the selective (VOx /TiO2)
catalyst for partial oxidation of n-butane was found to be near (+ 4.5). The
surface vanadia coverage was also found to be a critical variable as the
polymeric surface vanadia species present at high coverage are more easily
reducible than the isolated vanadia species. The reducibility of the surface
vanadia species during butane oxidation on various oxide supports
followed the order:
TiO2 >CeO2 >ZrO2 >Al2O3 >SiO2
However, the butane oxidation (TOF) was not found to correlate with the
extent of reduction of the surface vanadia species (Table 1.5).
Table 1.5: The effect of acidic promoters on n-Butane Oxidation on the
1% V2O5 /TiO2 catalyst at 494 K in n-Butane in air (161).
Catalyst Wt.
(g)
Flow
cm3/min
Butane
Conversion
mol%
MA
Selectivity
mol%
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
1% V2O5/5%P2O5/ TiO2 0.172 5.0 12.1 56.2 27.0 15.2
6% WO3/1%V2O5/ TiO2 0.708 8.9 23.6 26.2 34.1 8.9
9% WO3/1%V2O5/ TiO2 0.465 11.1 14.8 6.8 40.5 2.8
6% Nb2O5/1%V2O5/ TiO2 0.181 7.5 10.7 35.1 50.8 17.8
The (MA) selectivity did not appear to directly correlate with the extent of
reduction either, since the selectivity pattern was:
Al 2O3 >Nb2O5 >TiO2 >SiO2 >ZrO2 ~ CeO2
Varying the specific oxide support or oxide support ligands without
hanging the structure of the surface vanadia species can alter the
characteristics of the bridging (V-O-support) bond. The bridging (V-O-

Chapter One - Introduction
74
support) bond appears to be associated with the critical oxygen required
for hydrocarbon oxidation reactions since changing the specific oxide
support dramatically affects the (TOF) approximately two orders of
magnitude for n-butane oxidation to (MA) (Table1.4). The general trend
appears to be:
CeO2 >ZrO2 ~TiO2 >Nb2O5 >Al2O3 >SiO2
This suggests that bridging oxygens in the (V-O-support bonds) that are
more electronegative or basic, corresponding to oxide support cations with
lower electronegativity, are associated with the critical oxygen required
for hydrocarbon oxidation reactions over supported vanadia catalysts. The
formation of the (V-O-P) bond has a particularly positive effect on the
butane oxidation (TOF) and (MA) selectivity (Table 1.6). The
(1%V2O5/5% P2O5/TiO2) catalyst displayed the highest selectivity to (MA)
among all systems studied (Table 1.5). At (494 K in 1.2 vol. % n-butane in
air) and (12.1 mol% n-butane conversion), the selectivity to (MA) reached
(56.2 mol %). Such catalytic behavior is consistent with the above
observation that (bridging V-O) support bonds are critical in the oxidation
of n-butane to (MA).
The insight into the number of critical surface vanadia sites required in
hydrocarbon oxidation reactions can be gained by examination of the
variation of the (TOF) with surface vanadia coverage. Those reactions that
require only one surface site will exhibit a (TOF) that is independent of
surface vanadia coverage .The oxidation of butane to (MA) over titania-
supported vanadia catalysts (Table 1.6 and Figure 1.27) exhibited an
increase in (TOF) with surface vanadia coverage. This may reflect the
requirement of multiple surface vanadia sites or the influence of other
factors, such as surface acidity influence of (bridging V-O-V bonds),
structural changes, etc...
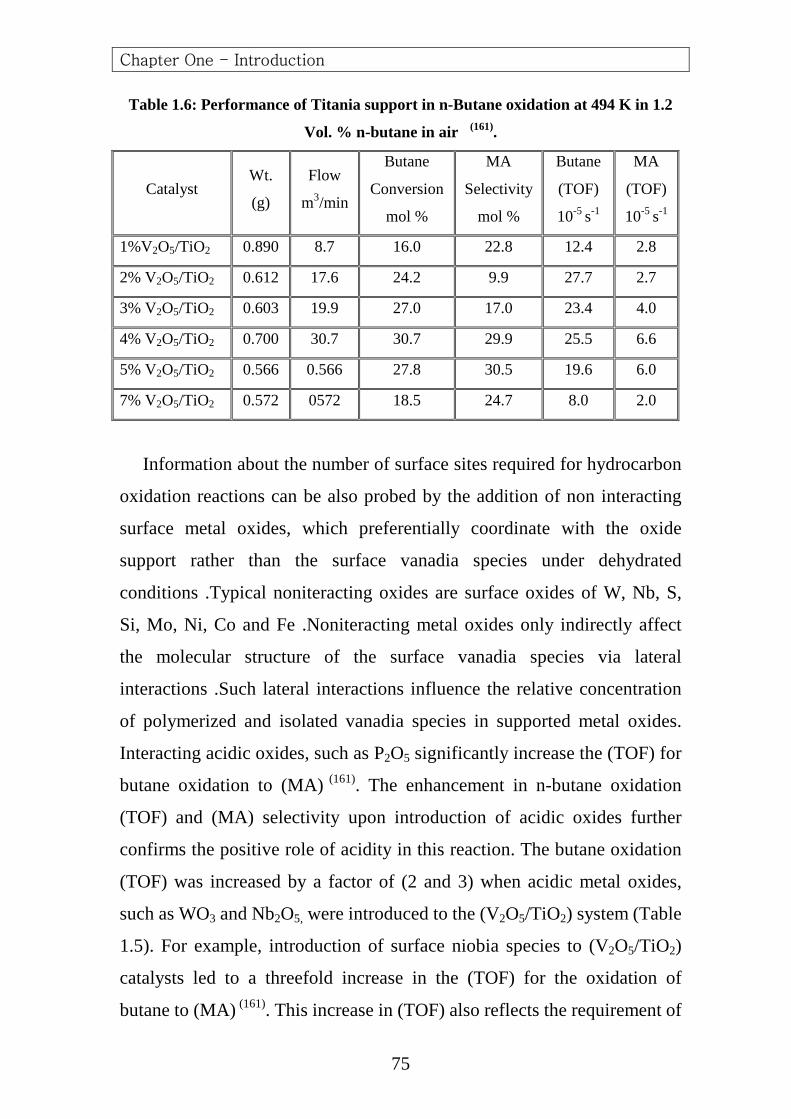
Chapter One - Introduction
75
Table 1.6: Performance of Titania support in n-Butane oxidation at 494 K in 1.2
Vol. % n-butane in air (161).
Catalyst Wt.
(g)
Flow
m3/min
Butane
Conversion
mol %
MA
Selectivity
mol %
Butane
(TOF)
10-5 s-1
MA
(TOF)
10-5 s-1
1%V2O5/TiO2 0.890 8.7 16.0 22.8 12.4 2.8
2% V2O5/TiO2 0.612 17.6 24.2 9.9 27.7 2.7
3% V2O5/TiO2 0.603 19.9 27.0 17.0 23.4 4.0
4% V2O5/TiO2 0.700 30.7 30.7 29.9 25.5 6.6
5% V2O5/TiO2 0.566 0.566 27.8 30.5 19.6 6.0
7% V2O5/TiO2 0.572 0572 18.5 24.7 8.0 2.0
Information about the number of surface sites required for hydrocarbon
oxidation reactions can be also probed by the addition of non interacting
surface metal oxides, which preferentially coordinate with the oxide
support rather than the surface vanadia species under dehydrated
conditions .Typical noniteracting oxides are surface oxides of W, Nb, S,
Si, Mo, Ni, Co and Fe .Noniteracting metal oxides only indirectly affect
the molecular structure of the surface vanadia species via lateral
interactions .Such lateral interactions influence the relative concentration
of polymerized and isolated vanadia species in supported metal oxides.
Interacting acidic oxides, such as P2O5 significantly increase the (TOF) for
butane oxidation to (MA) (161). The enhancement in n-butane oxidation
(TOF) and (MA) selectivity upon introduction of acidic oxides further
confirms the positive role of acidity in this reaction. The butane oxidation
(TOF) was increased by a factor of (2 and 3) when acidic metal oxides,
such as WO3 and Nb2O5, were introduced to the (V2O5/TiO2) system (Table
1.5). For example, introduction of surface niobia species to (V2O5/TiO2)
catalysts led to a threefold increase in the (TOF) for the oxidation of
butane to (MA) (161). This increase in (TOF) also reflects the requirement of

Chapter One - Introduction
76
multiple surface metal oxide sites for this hydrocarbon oxidation. In
contrast to non interacting additives that mainly affect oxidation reactions
requiring multiple surface sites, interacting additives affect all hydrocarbon
oxidation reactions since they directly alter the structure and reactivity of
the surface vanadia sites. Interacting metal oxides, especially basic oxides,
retard the reduction of surface vanadia species. Consequently, the (TOF)
for all hydrocarbon oxidation reactions are lowered when basic additives
are introduced (163,164).
1.12.6 - Role of Acidity of Supported Vanadia Catalysts
The oxide supports possess only surface Lewis acid sites, with the
exception of silica where no Lewis acid sites exist on the surface. The
relative Lewis acid strength of these sites follows the order
Al 2O3 >Nb2O5 >TiO2 >ZrO2
In contrast to the oxide supports, unsupported V2O5 crystalline powders
displays both surface Bronsted and Lewis acid sites (167) and an increase in
the number of surface Bronsted acid sites.
Supported (VPO) catalysts typically exhibit surface enrichment in
phosphate similar to the conventional unsupported or bulk (VPO)
catalysts (106,156,161). In these catalysts the concentration of the surface
Bronsted acid sites increases with the surface (P/V) ratio. The surface
enrichment in phosphorus stabilizes the V (IV) oxidation state, which
results in decreased activity in n-butane oxidation. On the other hand, the
increased surface Bronsted acidity facilitates desorption of partially
oxidized products with acidic properties, such as (MA) leading to
significant improvement in (MA) selectivity (162).According to Grasselli st
al. (168), slight excess phosphate (P/V = 1.1-1.2) forms a protective
pyrophosphate "fence" around active surface sites at the surface preventing
overoxidation of the reactive intermediates by the active oxygen diffusing

Chapter One - Introduction
77
fast at the surface. However, the changes in the activity and selectivity of
the (VPO) catalysts at much higher phosphorus enrichment (P/V ratio near
2.0) were associated with formation of microcrystalline VO (PO3)2 (169).
Reports in the literature have demonstrated that the VO (PO3)2phase is
inactive in n-butane oxidation (169). Therefore, the catalytic behavior of the
VO (PO3)2 system is explained by the presence of other (VPO) impurity
phases, such as (VO)2P2O7 and various (VOPO4) phases (169). The oxidation
of n-butane on well-defined supported vanadia catalysts demonstrated that
the substitution of (V-O-Ti) for less reducible and more acidic (V-O-P)
bonds has a positive effect on the n-butane oxidation (TOF) and (MA)
selectivity . This observation indicates that the oxidation of n-butane
to (MA) requires both a redox site and some acidic functionality (170 ) .
1.12.7: The Role of Support
We can conclude that although supports are essentially diluents, they
play important multifunctional roles which can be summarized as
following:
1- Economic, to reduce cost by extending expensive phase in catalyst.
2- Mechanical, to give mechanical strength, to optimize bulk density, to
prove a heat sink or a heat source, and to dilute the overactive phase.
3- Geometric, to increase the surface area, to optimize the porosity of a
catalyst, to optimize crystal and particle size, to allow the catalyst
particles adopt the most favorable configuration of a catalyst.
4- Chemical, to react with the catalyst either to improve specific activity
or to minimize sintering and to accept or donate chemical entities,
porosity via a spillover mechanism.
5- Deactivation, to stabilize the catalyst against sintering and to minimize
poisoning.

Chapter One - Introduction
78
Conclusions
Significant progress had been achieved in recent years in studying
molecular structures of the surface vanadia species present in supported
metal oxide catalysts. The detailed structural information on well-defined
systems provided a foundation for developing structure-reactivity
relationships required to molecularly engineer supported vanadia catalysts
for oxidation reactions. The nature of the metal oxide support was found to
play a crucial role in defining catalytic properties of vanadia monolayer in
n-butane oxidation to (MA). The terminal vanadyl oxygen does not appear
to critically influence the reactivity properties of the surface vanadia
species during hydrocarbon oxidation reactions. The bridging (V-O-V)
oxygen plays only a minor role in enhancing the n-butane oxidation (TOF),
primarily due to the preference of multiple active sites in this oxidation
reaction. The bridging (V-O-support oxygen), however, appears to be the
most critical oxygen since its properties can change the (TOF) for
hydrocarbon oxidation reactions by as much as four orders of magnitude.
The specific phase of the oxide support as well as the specific preparation
method does not appear to influence the molecular structure or reactivity
of the surface vanadia species. The number of surface vanadia sites
required for a hydrocarbon oxidation reaction is dependent on the specific
reactant molecule. Oxidation reactions requiring only one surface vanadia
site are generally not sensitive to the surface vanadia coverage and the
presence of non interacting metal oxides. Oxidation reactions requiring
multiple surface vanadia sites are very sensitive to surface vanadia
coverage and the presence of non interacting metal oxides. However,
interacting metal oxides influence all hydrocarbon oxidation reactions
since they modify the surface vanadia sites. Acidic and basic metal oxides
also influence the selectivity of hydrocarbon oxidation reactions, but the
effect appears to be reaction-specific.

Chapter One - Introduction
79
Unlike bulk (VPO) catalysts, detailed surface structural information on
a molecular level can be obtained from model supported vanadia catalysts
containing two-dimensional over layers of surface vanadia species (172).
Raman spectroscopy provides direct fundamental surface information
about:
1- The ratio of isolated and polymerized surface vanadia species
(Fig.1.24).
2- Terminal (V=O) and bridging (V-O-V) bonds.
3- Extent of reduction of the surface vanadia species during catalysis.
4- Influence of the oxide support ligands.
5- Influence of (acidic/basic) metal oxide additives (promoters/poisons).
6- Participation of specific (V- O) bonds in catalysis (with the aid of
oxygen- 18O labeled isotope experiments.
Thus, the in situ Raman studies during oxidation of reactions over model
supported vanadia catalysts can provide new insights into the surface
properties of oxide catalysts which are not attainable with bulk metal oxide
catalysts.
The studies of the model supported catalysts provided several important
insights about the origins of the catalytic activity of the (VPO) system that
were not possible with the bulk (VPO) catalysts. Firstly, it was shown that
the (V=O) oxygen was not involved in n-butane oxidation to (MA) and
that the (V-O-support) bonds contained active oxygen for n-butane
oxidation. The (V-O-P) bonds were particularly beneficial for both the
activity and selectivity of the supported vanadia catalysts (Table 1.6 (163)),
suggesting that this oxygen species may be responsible for selective
oxidation of n-butane. Secondly, it was demonstrated that the oxidation of
n-butane to (MA) could occur at a single vanadia site, although adjacent
sites were more efficient (Figure 1.27).

Chapter One - Introduction
80
The results of n-butane oxidation on titania-supported vanadia
catalysts suggested that isolated surface vanadia species are capable of
n-butane oxidation to (MA), although multiple vanadia sites are more
efficient in this oxidation. Microcrystalline vanadia was found to be
detrimental for the process of (MA) formation. The kinetic studies of the
supported vanadia system provided more direct evidence that the multiple
vanadia sites were better at oxidizing n-butane to (MA).
The selectivity of the supported catalysts to (MA) is correlated with
the Lewis acid strength of the metal oxide promoters. Especially high
selectivity to (MA) was found when the (V-O-P) bonds formed after
addition of phosphorus oxide in accordance with previous observations in
supported and unsupported bulk (VPO) catalysts. These findings indicate
that the supported vanadia catalysts represent a suitable model system
capable of providing insights into the mechanism of n-butane oxidation on
bulk (VPO) catalysts.
Lastly, the use of acidic additives with supported (VPO) catalysts
further demonstrated their similarity to the bulk (VPO) system. Moreover,
it shed some light on the possible role of promoters that improve the
performance of the bulk (VPO) catalysts. The acidic additives used in the
supported vanadia catalysts enhance those catalytic oxidations which
require a combination of a redox and acidic site for selective catalysis. The
acidic additives promoted both the rate of n-butane oxidation and the (MA)
formation on supported vanadia catalysts (Table 1.6).
The supported (VPO) system proved to be a good model for the bulk,
i .e. unsupported (VPO) system. The in situ Raman studies of the
supported vanadia catalysts provided several important insights into the
nature of the selective n-butane oxidation that were not possible with
unsupported vanadium-phosphorus-oxygen (VPO) catalysts due to the
experimental limitations. The many discovered similarities between the

Chapter One - Introduction
81
bulk and supported (VPO) systems have important implications for the
design of new selective (VPO) catalysts. The nature of the oxide support,
the surface coverage of the active vanadia species and the acidity of metal
oxide additive are the most important determinants of the catalytic activity
and selectivity in n-butane oxidation. These structure-reactivity
relationships pave the way for molecular engineering of selective active
sites for hydrocarbon oxidation in supported (VPO) catalysts.
The aim of work
The aim of our previous work was precisely to use technical grade
vanadium pentoxide locally produced to prepare a catalyst to convert
n-butane to MA.
Our aim in this work is to study the effect of Titania (anatase) on the
physical, chemical and structural properties of our previous already
tested catalyst in order to prepare an industrial catalyst to increase the
feasibility of vanadium pentoxide production in our country and supply
further prospects to use it.

Chapter Two - Experimental Part
٩٣
2.4: REDOX TITRATION
2.4.1: Redox Titration experiments
The redox titration for the technical grade V2O5 and prepared catalytic
phases was carried out as described below:
A- Preparation of the samples for Redox Titration
According to Nakamura (105), (0.1) g of each solid sample was dissolved
in (17 ml) o-H3PO4 and boiled until a clear solution was obtained for the
unsupported phases and a dissolved supported samples. This solution was
added to a solution containing (10) ml H2SO4 which was diluted with
(250) ml distilled water. This solution was divided into two parts (100 ml)
for each part.
B- Method of titration
After standardization of potassium permanganate KMnO4 solution with
oxalic acid (0.05N), one of the above two parts of solution was titrated
with Potassium Permanganate KMnO4 to the end point. Then the
titration was continued with (0.05N) iron (II) ammonium sulphate
Fe (SO4)2(NH4)2.6H2O using diphenyl amine as indicator.
The other part of solution in section (A) was titrated with (0.05N) of iron
(II) ammonium sulphate Fe (SO4)2(NH
4)2.6H
2O after the addition of one
drop of diphenyl amine as indicator.

Chapter Two - Experimental Part
٩٤
C- Method of Calculation
According to Buchanan (92) the average oxidation number of Vanadium
was calculated according to this equation:
OHNHSOFeofVol
mlKMnOofVolnV 26.)4()4(.
)(.5
22
4−= .............. 2. 1
The amount of V (III), V (IV) and V (V) was calculated according to
the following equations:-
T1 = V (IV) + 2V (III) ………………………… (2.2)
T2 = V (V) + V (IV) + V (III) ………………... (2.3)
T3 = V (V) …………………………………. (2.4)
Where:
T1 = Vol. of KMnO4 added to the first part of solution represents
the amount of V (IV) and V (III).
T2 = Vol. of Fe (SO4)2(NH4)2.6H2O added to the first part
solution represents the amount of V (V), V (IV) and V (III).
T3 = Vol. of Fe (SO4)2(NH4)2.6H2O added to the second part of
solution represents the amount of V (V).

Chapter Two - Experimental Part
٩٥
2.4.2: Redox Titration of reaction evolution:
A redox titration was conducted for supported VO (H2PO4)2 during the
reaction evolution (i.e. during the preparation of the phase), with interval
of reaction time of one hour, up to 7 hours. The investigated samples were
filtered, dried at 125 °C and then treated in same method described in
section 2.4.1.

Chapter Three
Results and Discussion

Part One
Results

Chapter Three – Part One – Results
٩٨
3.1: Results
The results obtained from using the techniques mentioned in chapter
two are highlighted in this section in reserving the sequences of analysis in
order to follow systematic and logic presentation of data, this approach can
facilitate results prospective prior to effective and global discussion.
3.1.1: The Elemental Analysis of technical grade V2O5
Table 3.1 represents the elemental analysis of technical grade V2O5 for
vanadium and impurities percentage which carried out by atomic
absorption.
It is obviously from the table that the higher concentration of the
impurities is represented by Sodium.
Table 3.1: Elemental Analysis of technical grade V2O5 used in this work.
V% Na% Al% Si% Ca% Fe% Mg% K% Ni% Mo% Remarks
52.520 0.770 0.170 0.060 0.160 0.060 0.050 0.041 0.006 0.017 Sample *
52.520 0.780 0.170 0.070 0.170 0.070 0.050 0.039 0.008 0.017 General
Sample**
* Sample: is the main source of material used in this research
**General Sample: random sample from V2O5 production line inventory

Chapter Three – Part One – Results
٩٩
3.1.2: Identification of prepared Phases by XRD
The X- ray diffraction pattern of VOHPO4.0.5H2O, VHPO4.H2O/ TiO2,
VO (H2PO4)2 and VO (H2PO4)2 /TiO2 (samples No.2-un, 2-supp, 3-un and
3-supp) are shown in Figure 3.1. The initial sample was used for the
calibration of the X- ray Diffractometer this sample is known (VPO)
system catalyst which is used for Propane Oxidation. The identification of
these phases is given in (Table 3.5), while the X-ray diffraction data of the
above samples (unsupported, supported and reported) are given in tables
3.2, 3.3and 3.4 respectively.
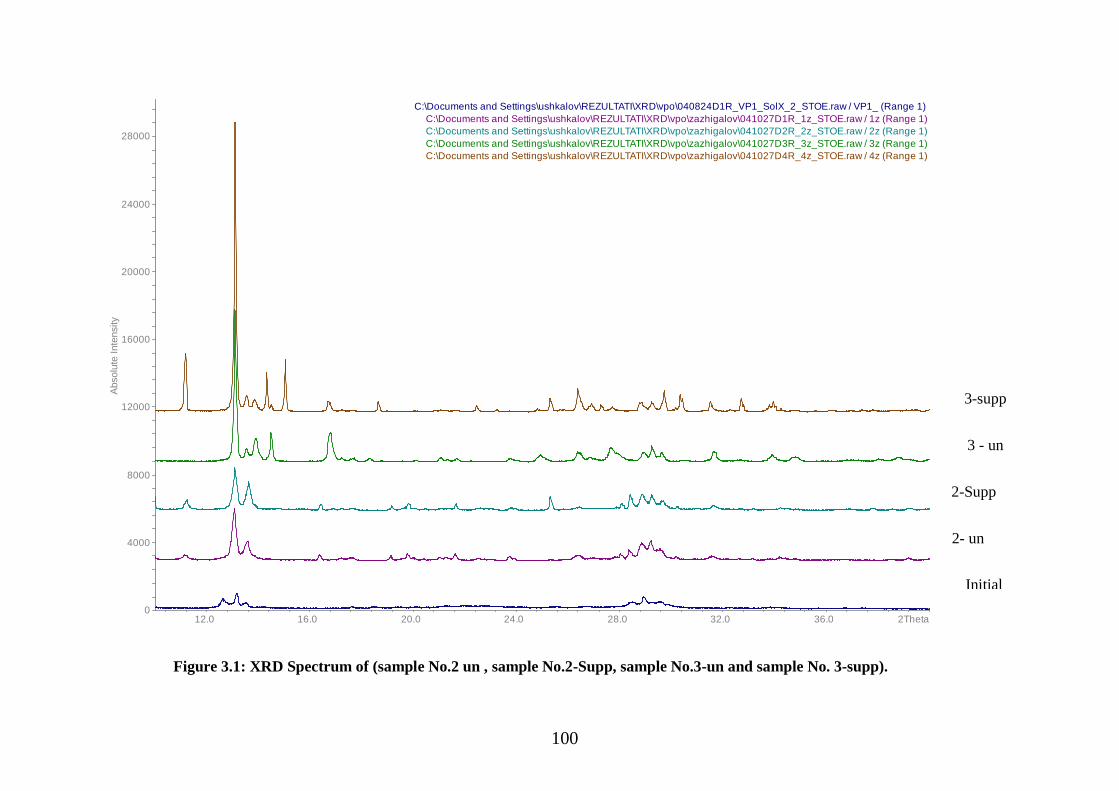
100
2Theta12.0 16.0 20.0 24.0 28.0 32.0 36.0
0
4000
8000
12000
16000
20000
24000
28000
Abs
olut
e In
tens
ityC:\Documents and Settings\ushkalov\REZULTATI\XRD\vpo\040824D1R_VP1_SolX_2_STOE.raw / VP1_ (Range 1)
C:\Documents and Settings\ushkalov\REZULTATI\XRD\vpo\zazhigalov\041027D1R_1z_STOE.raw / 1z (Range 1)C:\Documents and Settings\ushkalov\REZULTATI\XRD\vpo\zazhigalov\041027D2R_2z_STOE.raw / 2z (Range 1)C:\Documents and Settings\ushkalov\REZULTATI\XRD\vpo\zazhigalov\041027D3R_3z_STOE.raw / 3z (Range 1)C:\Documents and Settings\ushkalov\REZULTATI\XRD\vpo\zazhigalov\041027D4R_4z_STOE.raw / 4z (Range 1)
Initial
2- un
2-Supp
3 - un
Figure 3.1: XRD Spectrum of (sample No.2 un , sample No.2-Supp, sample No.3-un and sample No. 3-supp).
3-supp
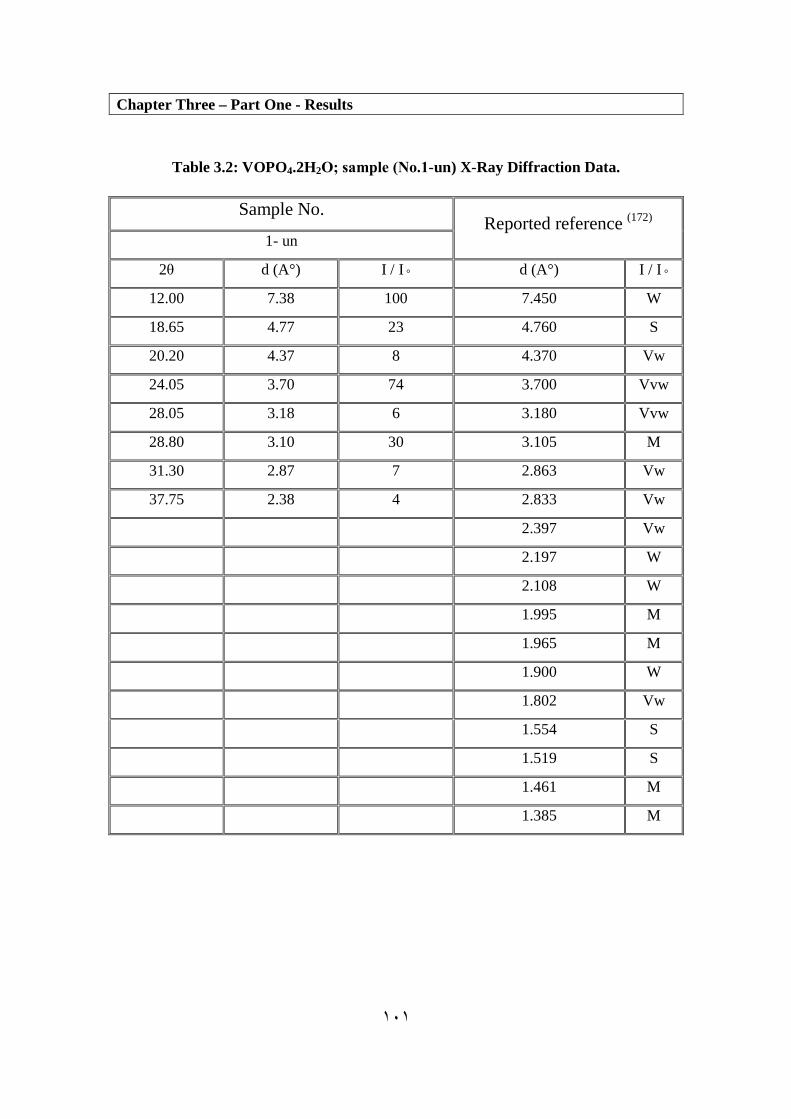
Chapter Three – Part One - Results
١٠١
Table 3.2: VOPO4.2H2O; sample (No.1-un) X-Ray Diffraction Data.
Sample No.
Reported reference (172) 1- un
2θ d (A°) I / I ° d (A°) I / I °
12.00 7.38 100 7.450 W
18.65 4.77 23 4.760 S
20.20 4.37 8 4.370 Vw
24.05 3.70 74 3.700 Vvw
28.05 3.18 6 3.180 Vvw
28.80 3.10 30 3.105 M
31.30 2.87 7 2.863 Vw
37.75 2.38 4 2.833 Vw
2.397 Vw
2.197 W
2.108 W
1.995 M
1.965 M
1.900 W
1.802 Vw
1.554 S
1.519 S
1.461 M
1.385 M
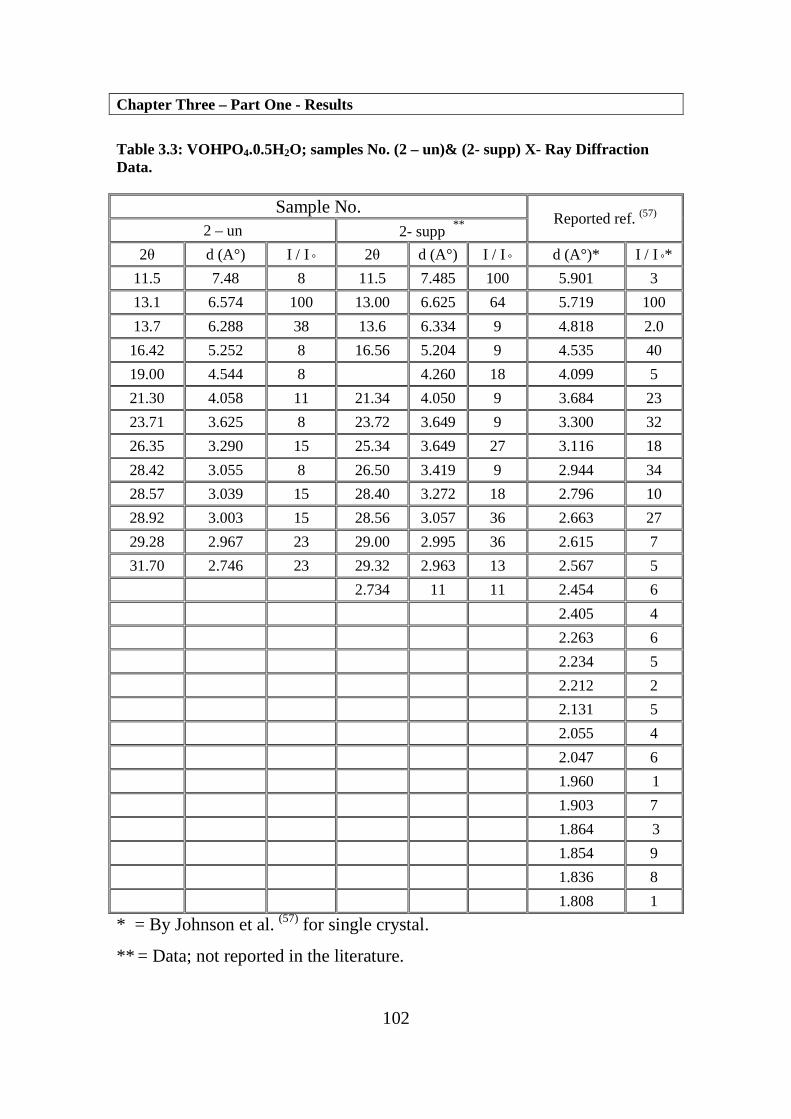
Chapter Three – Part One - Results
102
Table 3.3: VOHPO4.0.5H2O; samples No. (2 – un)& (2- supp) X- Ray Diffraction Data.
Sample No. Reported ref. (57)
2 – un 2- supp **
2θ d (A°) I / I ° 2θ d (A°) I / I ° d (A°)* I / I °*
11.5 7.48 8 11.5 7.485 100 5.901 3
13.1 6.574 100 13.00 6.625 64 5.719 100
13.7 6.288 38 13.6 6.334 9 4.818 2.0
16.42 5.252 8 16.56 5.204 9 4.535 40
19.00 4.544 8 4.260 18 4.099 5
21.30 4.058 11 21.34 4.050 9 3.684 23
23.71 3.625 8 23.72 3.649 9 3.300 32
26.35 3.290 15 25.34 3.649 27 3.116 18
28.42 3.055 8 26.50 3.419 9 2.944 34
28.57 3.039 15 28.40 3.272 18 2.796 10
28.92 3.003 15 28.56 3.057 36 2.663 27
29.28 2.967 23 29.00 2.995 36 2.615 7
31.70 2.746 23 29.32 2.963 13 2.567 5
2.734 11 11 2.454 6
2.405 4
2.263 6
2.234 5
2.212 2
2.131 5
2.055 4
2.047 6
1.960 1
1.903 7
1.864 3
1.854 9
1.836 8
1.808 1
* = By Johnson et al. (57) for single crystal.
** = Data; not reported in the literature.

Chapter three – Part One - Results
103
Table 3.4: VO (H2PO4) 2, samples (No.3 – un & 3- supp) X- Ray Diffraction Data.
Sample No. Reported
reference (172) Reported
reference ( 80) 3- un 3- supp ** 2θ d (A°) I / I ° 2θ d (A°) I / I ° d (A°) I / I ° d (A°) I / I °
11.50 7.485 18 13.28 6.483 100 6.34 M 6.327 100 13.28 6.486 100 13.72 6.279 7 3.99 S 4.482 4 13.72 6.279 3 14.28 6.031 10 3.586 S 3.988 12 14.28 6.031 2 14.57 5.914 17 3.378 M 3.637 1 14.71 5.858 13 16.57 5.204 20 3.173 F 3.581 29 15.28 5.641 2 21.42 4.035 2 2.983 M 3.375 8 16.57 5.204 3 24.90 3.478 7 2.837 F 3.167 75 18.64 4.630 1 26.71 3.246 8 2.483 M 2.977 5 21.42 4.034 3 29.00 2.995 10 2.375 W 2.835 45 22.57 3.832 7 29.57 2.938 5 2.312 Vw 2.480 9 23.00 3.761 2 30.00 2.969 7 2.215 Vw 2.371 4 25.42 3.407 1 31.71 2.744 2 2.098 M 2.213 1 26.78 3.230 3 33.85 2.576 7 2.007 W 2.111 5 27.42 3.163 5 1.995 W 2.096 19 27.57 3.147 5 1.955 Vvw 2.003 9 29.00 2.995 3 1.945 Vw 1.954 2 29.57 2.938 3 1.902 W 1.945 1 30.14 2.884 3 1.867 Vw 1.900 1 30.71 2.832 1 1.785 W 1.887 3 31.71 2.742 1 1.758 W 1.758 6 32.71 2.663 1 1.688 W - - 33.85 2.575 1 1.683 M 34.71 2.514 4 1.584 W
** = Data; are not reported in the literature.

Chapter Three – Part One - Results
104
Table 3.5: Identification of prepared phases by XRD
Sample No. Identified Phases
2 –un VOHPO4.0.5H2O▼+ VOHPO4.H2O ■ + VO(H2PO3)2
■
2 – supp VOHPO4.H2O ▼ + VO(H2PO4)2■ +TiO2 (a)
3 – un VO (H2PO4)2 ▼ + VO(H2PO3)2
■
3 – supp VO(H2PO4)2 ▼ + VO(H2PO3)2
■ VH2P3O10.2H2O ■ + TiO2 (a)
▼ = Main phase. ■ = Trace amount.
a = Anatase.
3.1.3: Thermal Analysis:
The results of Thermogravimetric and differential thermal analysis for the
supported and unsupported catalytic phases are presented in (Figs. 3.2- 3.5)
and (Tables 3.6 and 3.7.
Reported date in the literatures is numerous and some of them are in
contradictory due to the analysis environments like inert atmosphere, air,
nitrogen, oxygen, helium and argon. Our results are performed in air and are
in accordance with related reported data and the rate of heating (36, 57, 81, 80-83).
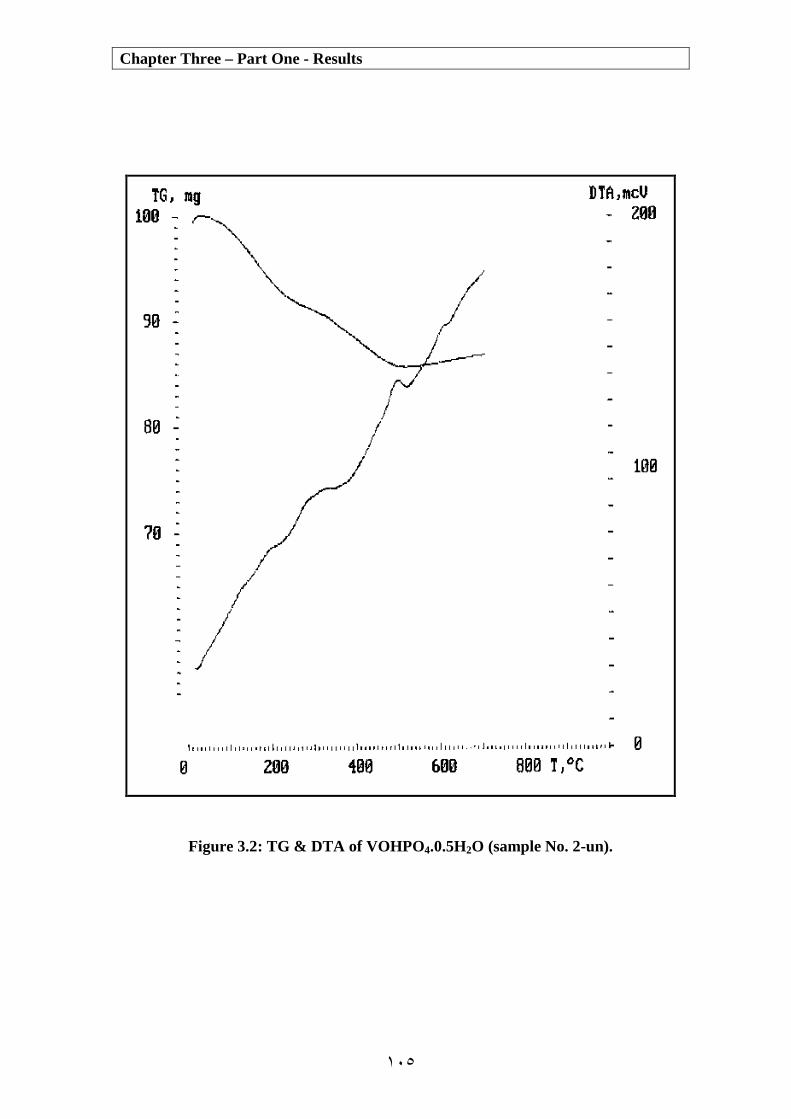
Chapter Three – Part One - Results
١٠٥
Figure 3.2: TG & DTA of VOHPO 4.0.5H2O (sample No. 2-un).
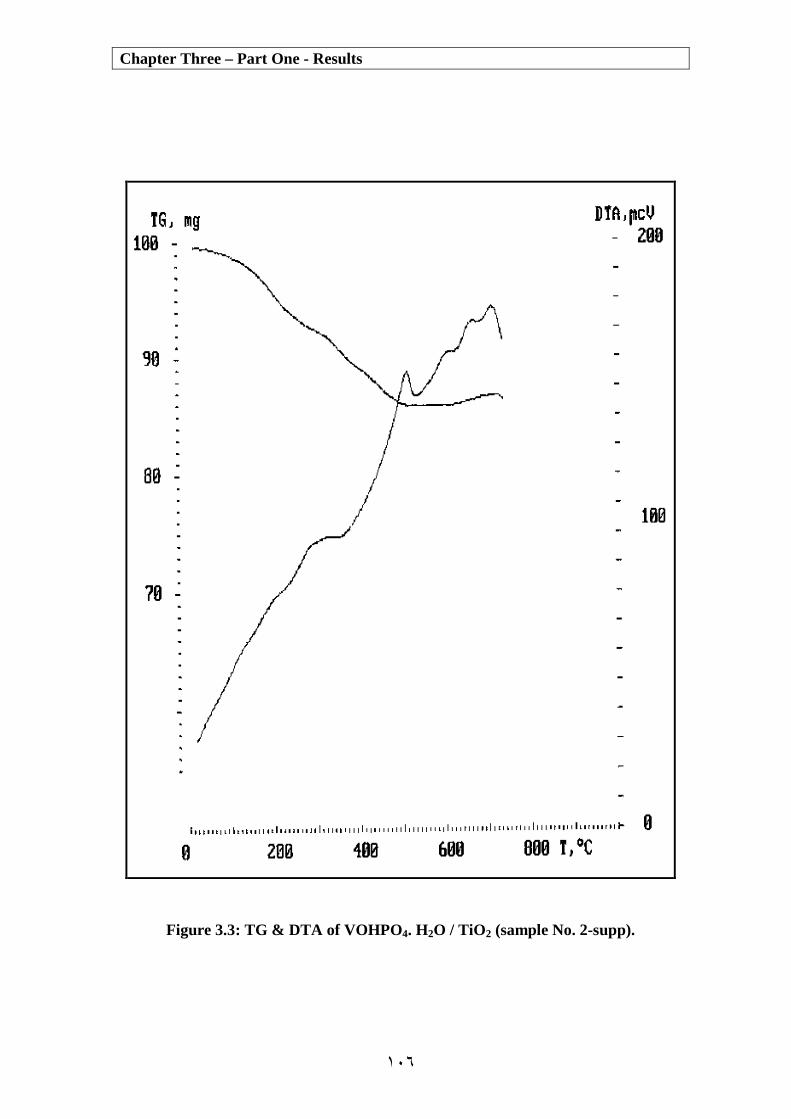
Chapter Three – Part One - Results
١٠٦
Figure 3.3: TG & DTA of VOHPO 4. H2O / TiO2 (sample No. 2-supp).
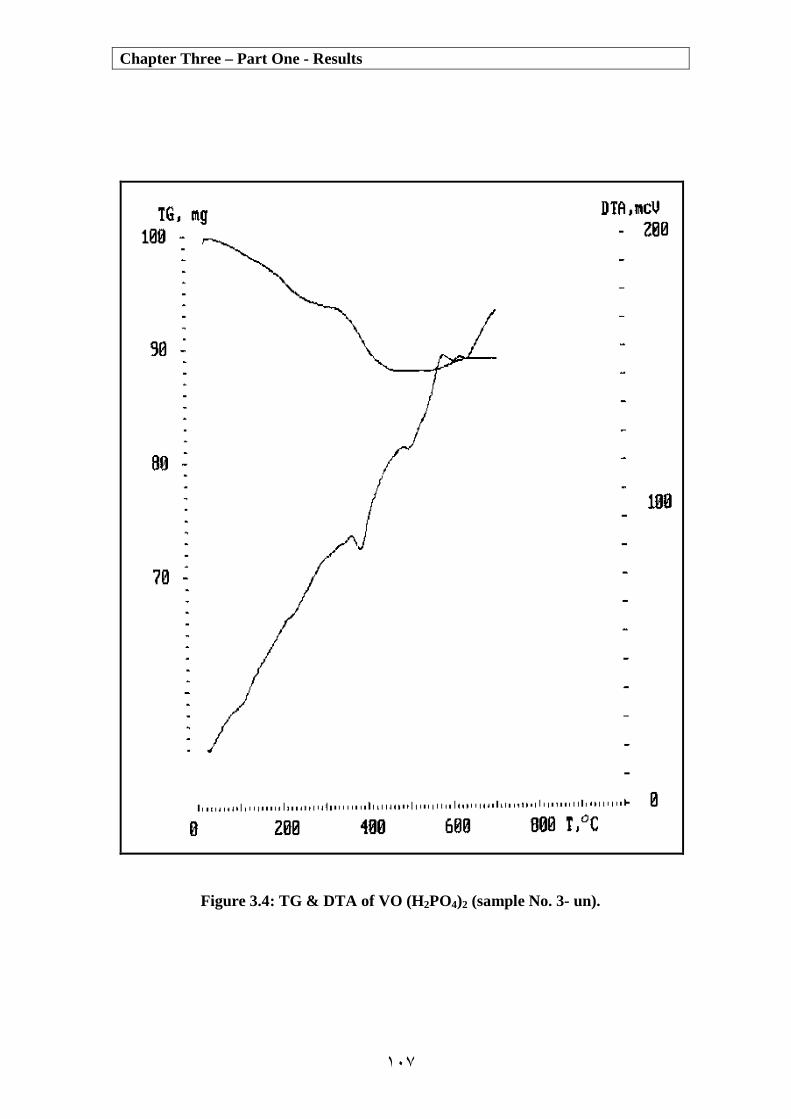
Chapter Three – Part One - Results
١٠٧
Figure 3.4: TG & DTA of VO (H 2PO4)2 (sample No. 3- un).
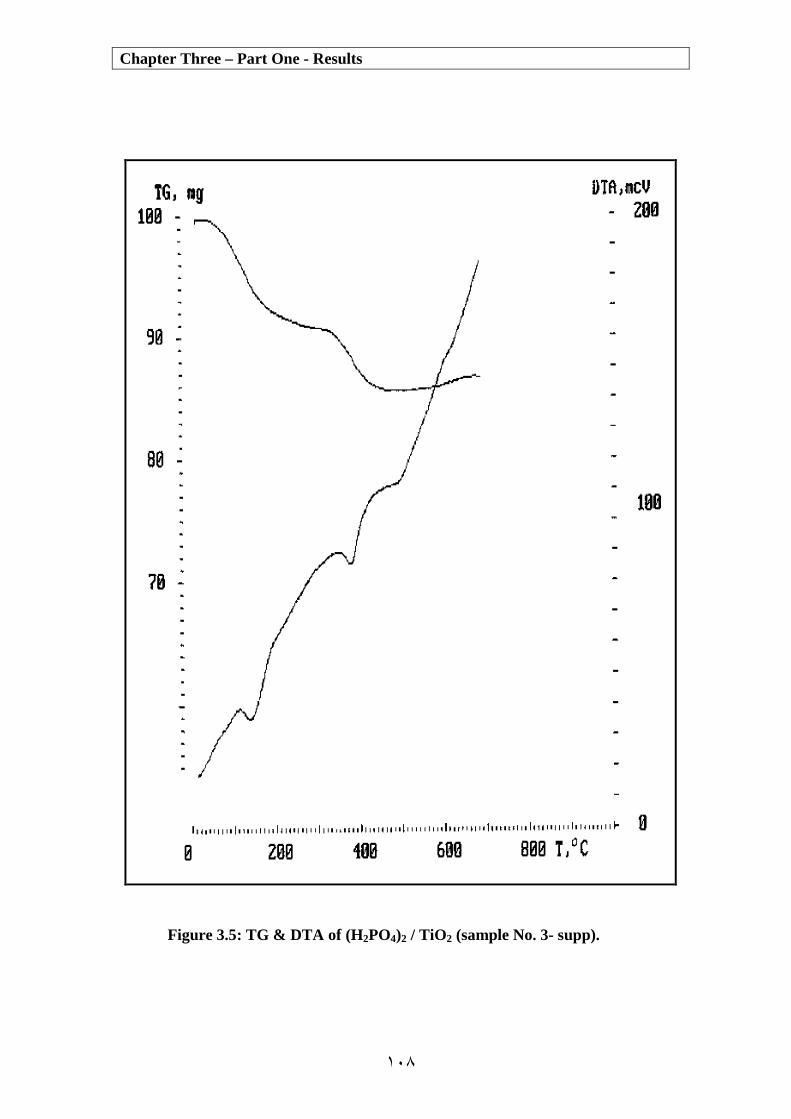
Chapter Three – Part One - Results
١٠٨
Figure 3.5: TG & DTA of (H 2PO4)2 / TiO2 (sample No. 3- supp).
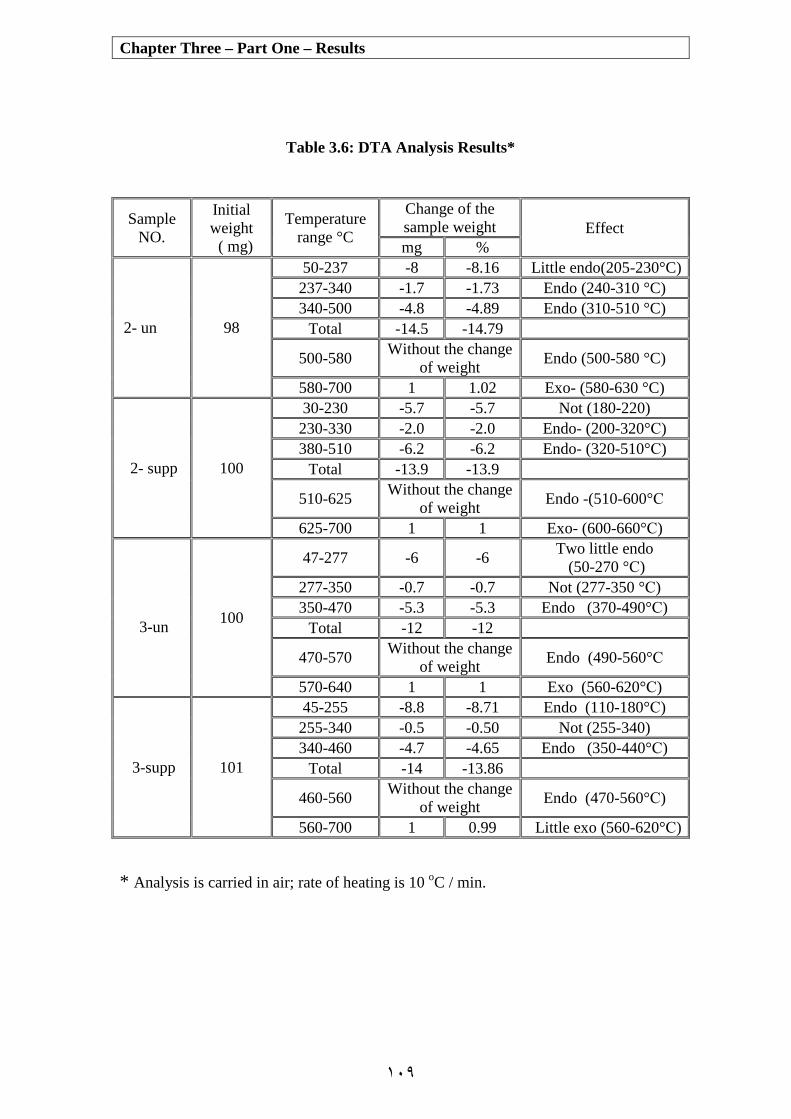
Chapter Three – Part One – Results
١٠٩
Table 3.6: DTA Analysis Results*
Sample NO.
Initial weight ( mg)
Temperature range °C
Change of the sample weight Effect mg %
2- un 98
50-237 -8 -8.16 Little endo(205-230°C) 237-340 -1.7 -1.73 Endo (240-310 °C) 340-500 -4.8 -4.89 Endo (310-510 °C)
Total -14.5 -14.79
500-580 Without the change
of weight Endo (500-580 °C)
580-700 1 1.02 Exo- (580-630 °C)
2- supp 100
30-230 -5.7 -5.7 Not (180-220) 230-330 -2.0 -2.0 Endo- (200-320°С) 380-510 -6.2 -6.2 Endo- (320-510°С)
Total -13.9 -13.9
510-625 Without the change
of weight Endo -(510-600°С
625-700 1 1 Exo- (600-660°С)
3-un
100
47-277 -6 -6 Two little endo (50-270 °С)
277-350 -0.7 -0.7 Not (277-350 °С) 350-470 -5.3 -5.3 Endo (370-490°С)
Total -12 -12
470-570 Without the change
of weight Endo (490-560°С
570-640 1 1 Exo (560-620°С)
3-supp 101
45-255 -8.8 -8.71 Endo (110-180°С) 255-340 -0.5 -0.50 Not (255-340) 340-460 -4.7 -4.65 Endo (350-440°С)
Total -14 -13.86
460-560 Without the change
of weight Endo (470-560°С)
560-700 1 0.99 Little exo (560-620°С)
* Analysis is carried in air; rate of heating is 10 oC / min.
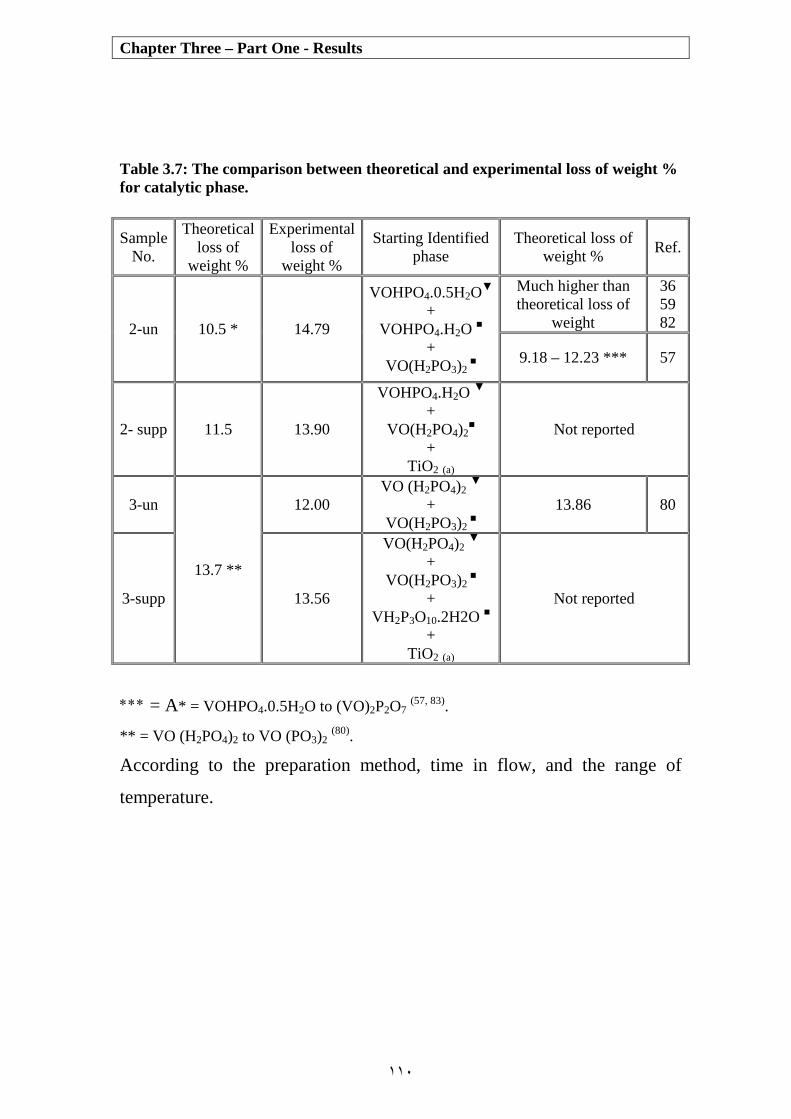
Chapter Three – Part One - Results
١١٠
Table 3.7: The comparison between theoretical and experimental loss of weight % for catalytic phase.
Sample No.
Theoretical loss of
weight %
Experimental loss of
weight %
Starting Identified phase
Theoretical loss of weight %
Ref.
2-un 10.5 * 14.79
VOHPO4.0.5H2O▼
+ VOHPO4.H2O ■
+ VO(H2PO3)2
■
Much higher than theoretical loss of
weight
36 59 82
9.18 – 12.23 *** 57
2- supp 11.5 13.90
VOHPO4.H2O ▼
+ VO(H2PO4)2
■ +
TiO2 (a)
Not reported
3-un
13.7 **
12.00 VO (H2PO4)2
▼ +
VO(H2PO3)2 ■
13.86 80
3-supp 13.56
VO(H2PO4)2 ▼
+ VO(H2PO3)2
■ +
VH2P3O10.2H2O ■
+ TiO2 (a)
Not reported
*** = A* = VOHPO4.0.5H2O to (VO)2P2O7 (57, 83).
** = VO (H 2PO4)2 to VO (PO3)2 (80).
According to the preparation method, time in flow, and the range of
temperature.

Chapter Three – Part One - Results
١١١
3.1.3- Fourier Transform Infrared (FTIR) Spectrum
Figures 3.6 and 3.7 show the FTIR spectrum of laboratory grade
(Fluka) and technical grade V2O5.The observed bands frequencies and
their vibrational assignments for V2O5 are shown in (Table 3.8).
Figures 3.8 and 3.9 represent the FTIR spectrum of laboratory grade
(Fluka) and technical grade V2O5 supported on Titania (anatase), all bands
were listed in (Table 3.9).
The spectrum of VOPO4.2H2O sample (No.1-un and 1-supp) is given in
Figures 3.10 and 3.11.The phase’s bands, for the above samples are
reported in (Table 3.10).
Figures 3.12, 3.13 and 3.14 can show the FTIR spectra recorded for
VOHPO4.0.5H2O and VOHPO4.H2O/TiO2 phase, reference sample
(sample No.2 , No. 2- un and sample No.2-supp) the bands positions are
tabulated in (Table 3.11).
The FTIR of spectrum of VO (H2PO4)2, samples (No. 3- un and No. 3-
supp) are shown in (Figures 3.15 & 3.16) respectively. The positions and
the assignment of the FTIR bands for this phase are given in (Table 3. 12).
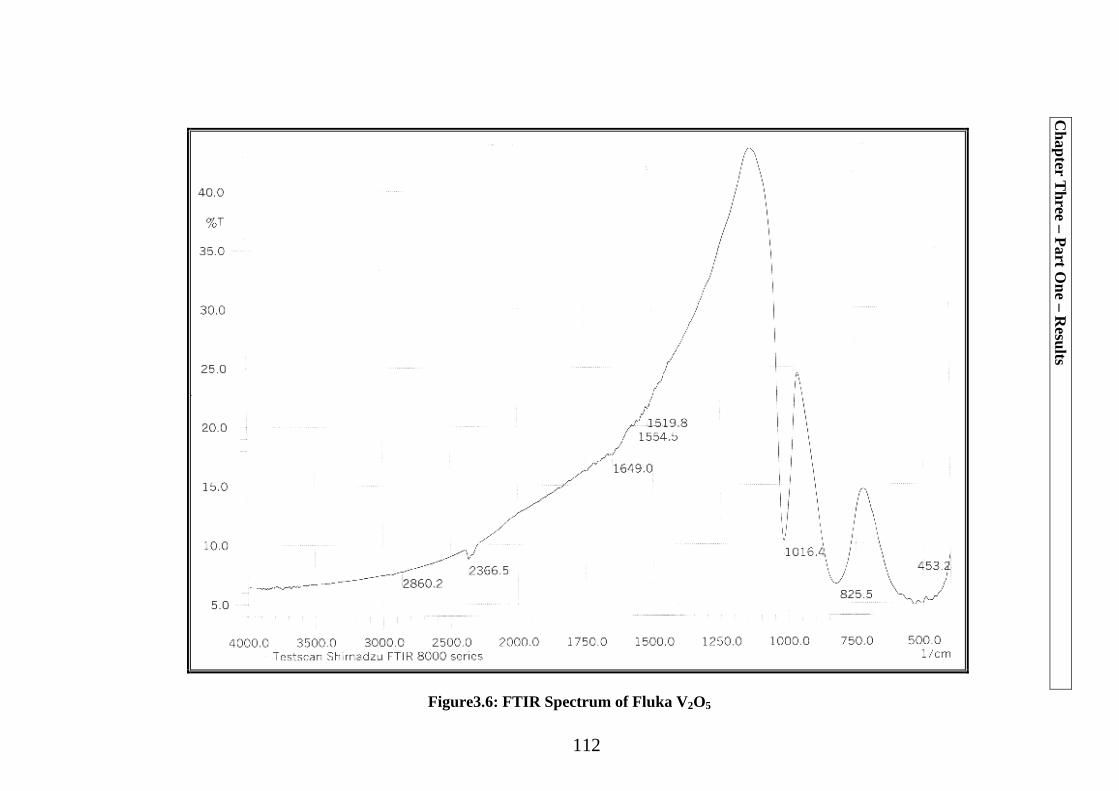
Chapter T
hree – Part O
ne – Results
Figure3.6: FTIR Spectrum of Fluka V2O5
112
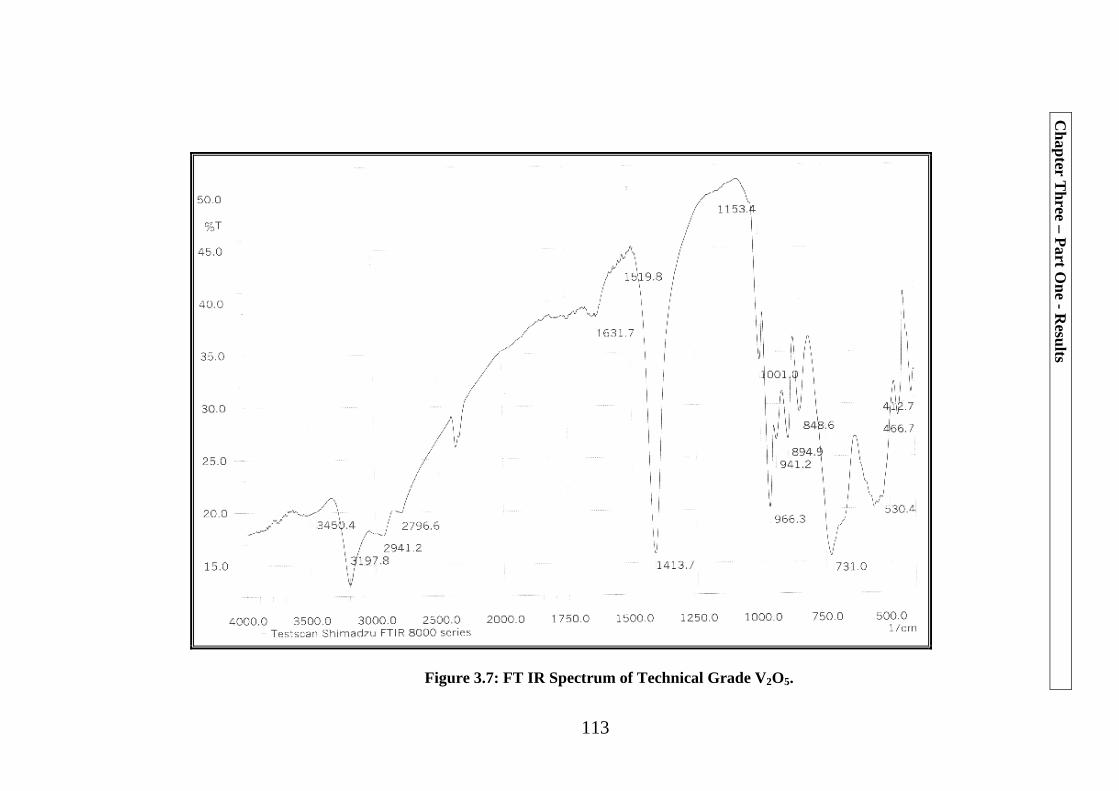
Chapter T
hree – Part O
ne - Results
Figure 3.7: FT IR Spectrum of Technical Grade V2O5.
113

Chapter Three – Part One – Results
114
Table 3.8: FT-IR Data of V2O5
Band Frequencies (cm -1) Attribution Reported Data Ref.
Fluka V2O5 T.G. V2O5
N.B 3450 (Sh) ν O-H (H2O) 3400 172
N.B 3198 (M) δ N-H ( NH4)+
3400-3180 172 3300-3030 174
N.B 2941(Sh) δ O-H (H2O)
3020-2270 172 N.B 2797 (Vw) δ O-H (H2O)
2366 (W) 2380 (w) δ O-H (H2O)
1649 (Vw) 1632 (Vw) δ O-H (H2O) 1640 62
1554 (Vw) N.B δ O-H (H2O) 1590 - 1500 172
1520 (Vw) 1520 (Vw) δ O-H (H2O) 1590 - 1500 172
N.B 1413 (Vs) δ N-H ( NH4)+
1300 - 1430 172 Near 1429 174 Near 1450 189
1016 (Vs) 1001 (M) δ V=O * 1024 1018
1050-800
176 189
N.B 966 (S) ν V=O 172 175 176
N.B 941 (W) δ M=O * 1000-850 176 Near 1000 189
N.B 895 (M) δ M=O * 1000-850 176 Near 1000 189
825 (Vs) 849 (S) ν V-O-V
818 - 850
172 175 176
N.B 731 (S) δ N-H 850-750 176
540 530 (W) δ V - O 520 - 540 172
453 467 (M) δ V - O 470 172
N.B 413 (W) δ V - O 416 62
*M = Na, Si, Fe, Ti ……etc
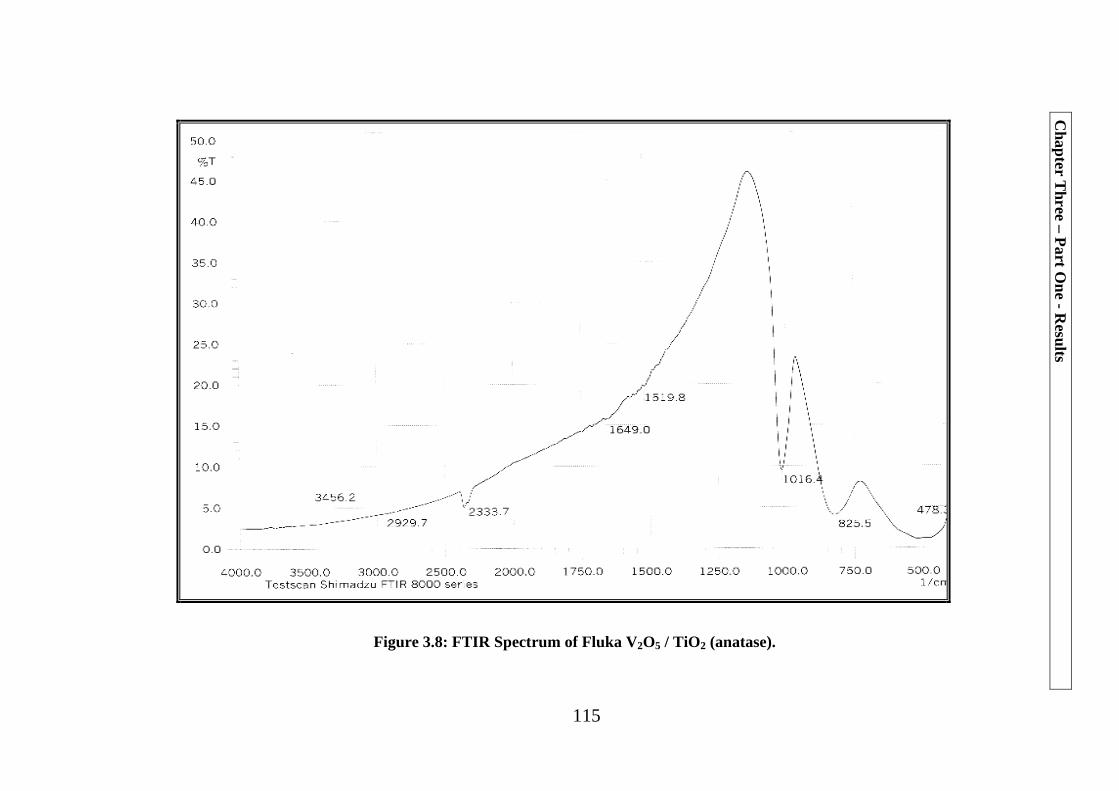
Chapter T
hree – Part O
ne - Results
Figure 3.8: FTIR Spectrum of Fluka V2O5 / TiO2 (anatase).
115

Chapter T
hree – Part O
ne – Results
Figure 3.9: FTIR Spectrum of Technical Grade V2O5 / TiO2 (anatase).
116
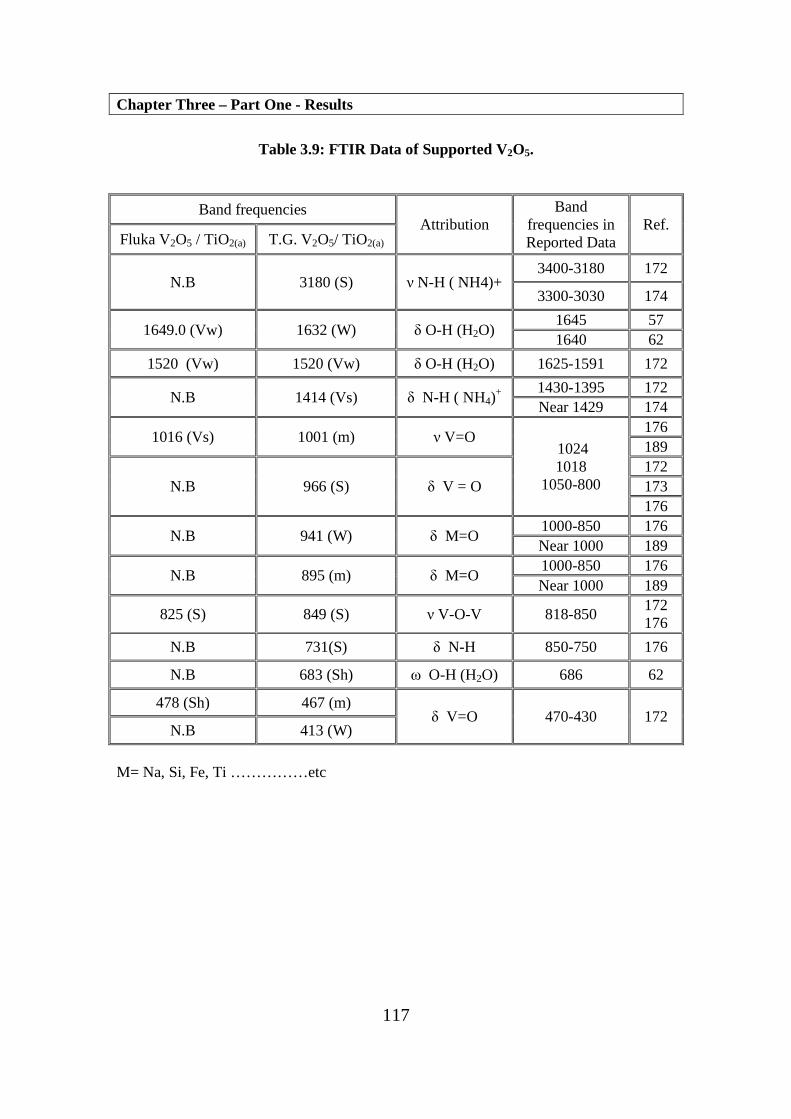
Chapter Three – Part One - Results
117
Table 3.9: FTIR Data of Supported V2O5.
Band frequencies Attribution
Band frequencies in Reported Data
Ref. Fluka V2O5 / TiO2(a) T.G. V2O5/ TiO2(a)
N.B 3180 (S) ν N-H ( NH4)+ 3400-3180 172
3300-3030 174
1649.0 (Vw) 1632 (W) δ O-H (H2O) 1645 57 1640 62
1520 (Vw) 1520 (Vw) δ O-H (H2O) 1625-1591 172
N.B 1414 (Vs) δ N-H ( NH4)+
1430-1395 172 Near 1429 174
1016 (Vs) 1001 (m) ν V=O 1024 1018
1050-800
176 189
N.B 966 (S) δ V = O 172 173 176
N.B 941 (W) δ M=O 1000-850 176 Near 1000 189
N.B 895 (m) δ M=O 1000-850 176 Near 1000 189
825 (S) 849 (S) ν V-O-V 818-850 172 176
N.B 731(S) δ N-H 850-750 176
N.B 683 (Sh) ω O-H (H2O) 686 62
478 (Sh) 467 (m) δ V=O 470-430 172
N.B 413 (W)
M= Na, Si, Fe, Ti ……………etc
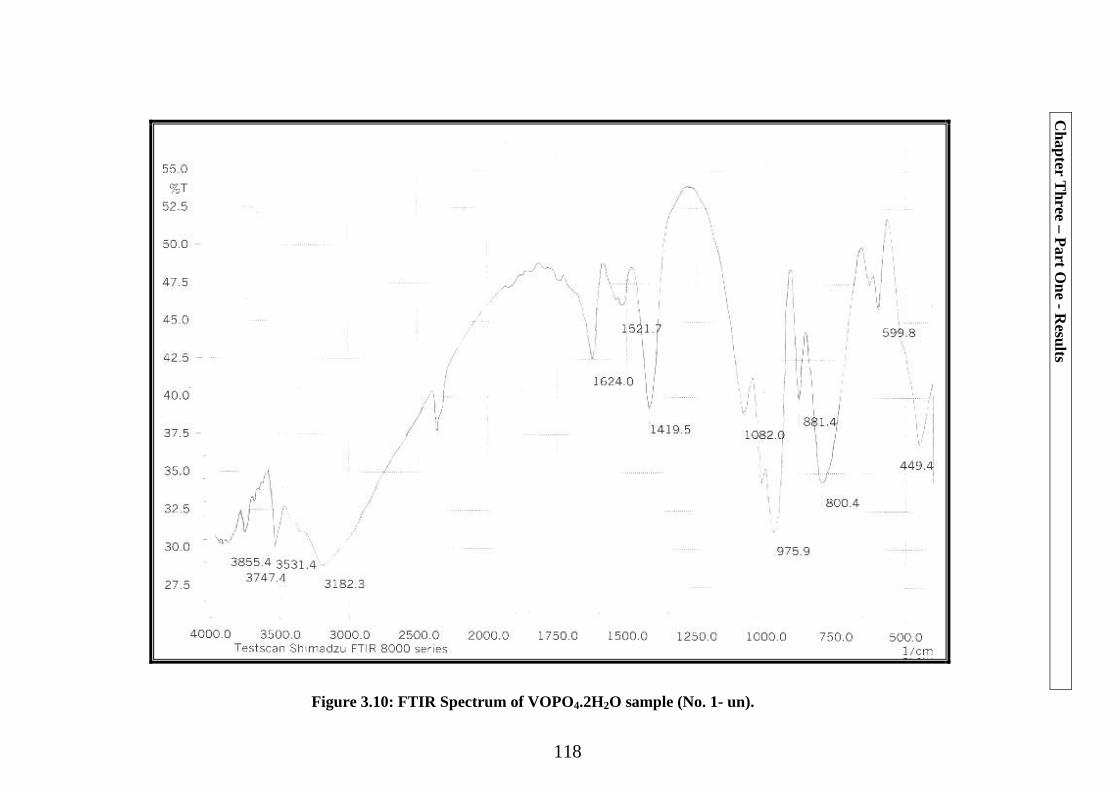
Chapter T
hree – Part O
ne - Results
Figure 3.10: FTIR Spectrum of VOPO4.2H2O sample (No. 1- un).
118
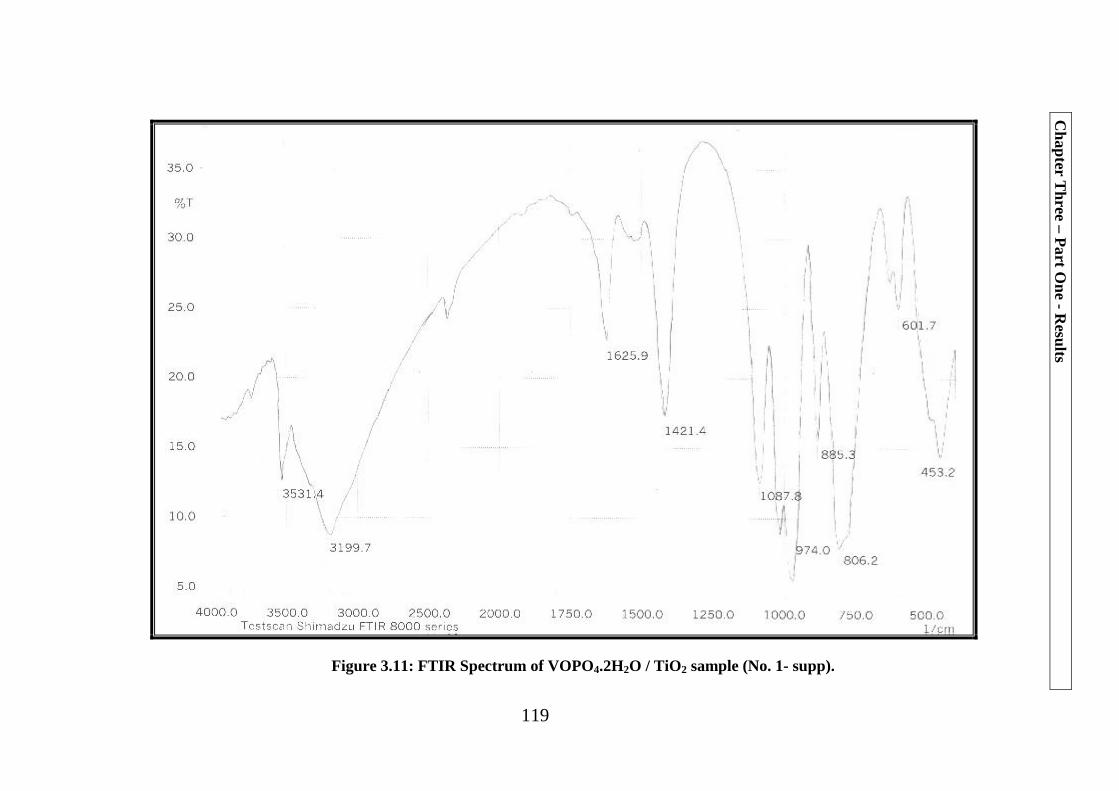
Chapter T
hree – Part O
ne - Results
Figure 3.11: FTIR Spectrum of VOPO4.2H2O / TiO2 sample (No. 1- supp).
119
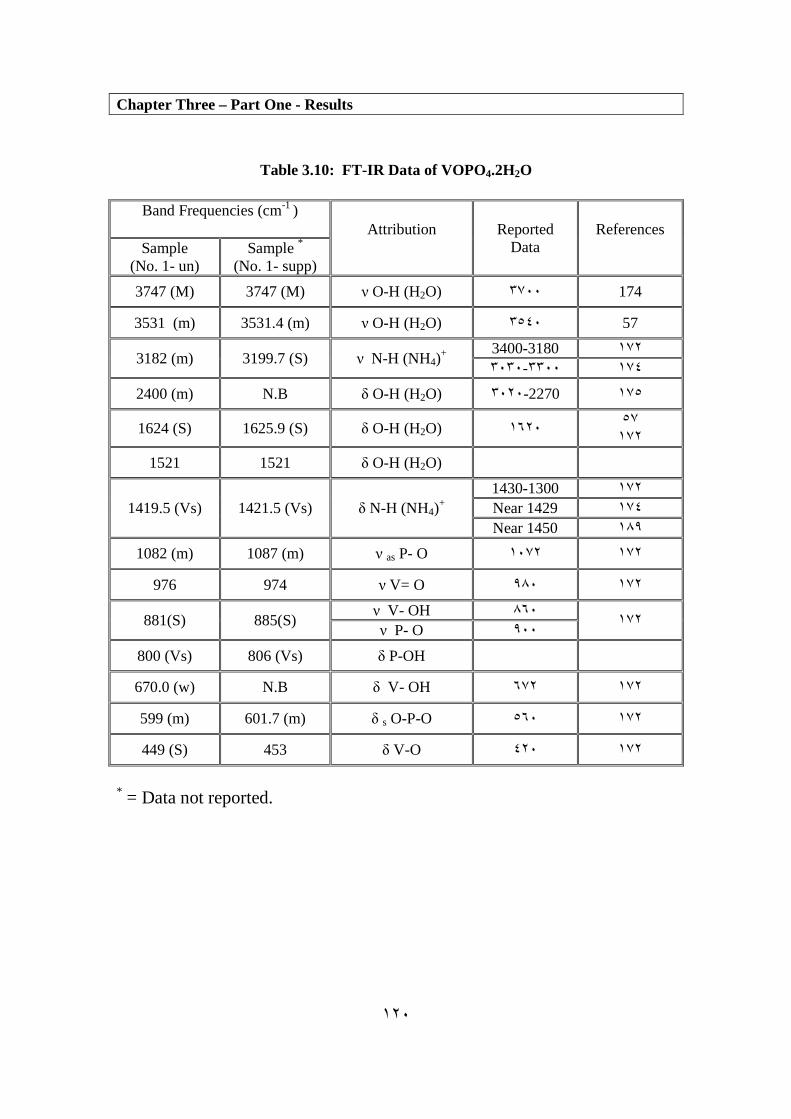
Chapter Three – Part One - Results
١٢٠
Table 3.10: FT-IR Data of VOPO4.2H2O
Band Frequencies (cm-1 )
Attribution
Reported Data
References Sample
(No. 1- un) Sample *
(No. 1- supp)
3747 (M) 3747 (M) ν O-H (H2O) ٣٧٠٠ 174
3531 (m) 3531.4 (m) ν O-H (H2O) ٣٥٤٠ 57
3182 (m) 3199.7 (S) ν N-H (NH4)+
3400-3180 ١٧٢ ٣٠٣٠- ٣٣٠٠ ١٧٤
2400 (m) N.B δ O-H (H2O) ٣٠٢٠-2270 ١٧٥
1624 (S) 1625.9 (S) δ O-H (H2O) ١٦٢٠ ٥٧ ١٧٢
1521 1521 δ O-H (H2O)
1419.5 (Vs) 1421.5 (Vs) δ N-H (NH4)+
1430-1300 ١٧٢ Near 1429 ١٧٤ Near 1450 ١٨٩
1082 (m) 1087 (m) ν as P- O ١٧٢ ١٠٧٢
976 974 ν V= O ١٧٢ ٩٨٠
881(S) 885(S) ν V- OH ٨٦٠
١٧٢ ν P- O ٩٠٠
800 (Vs) 806 (Vs) δ P-OH
670.0 (w) N.B δ V- OH ١٧٢ ٦٧٢
599 (m) 601.7 (m) δ s O-P-O ١٧٢ ٥٦٠
449 (S) 453 δ V-O ١٧٢ ٤٢٠
* = Data not reported.
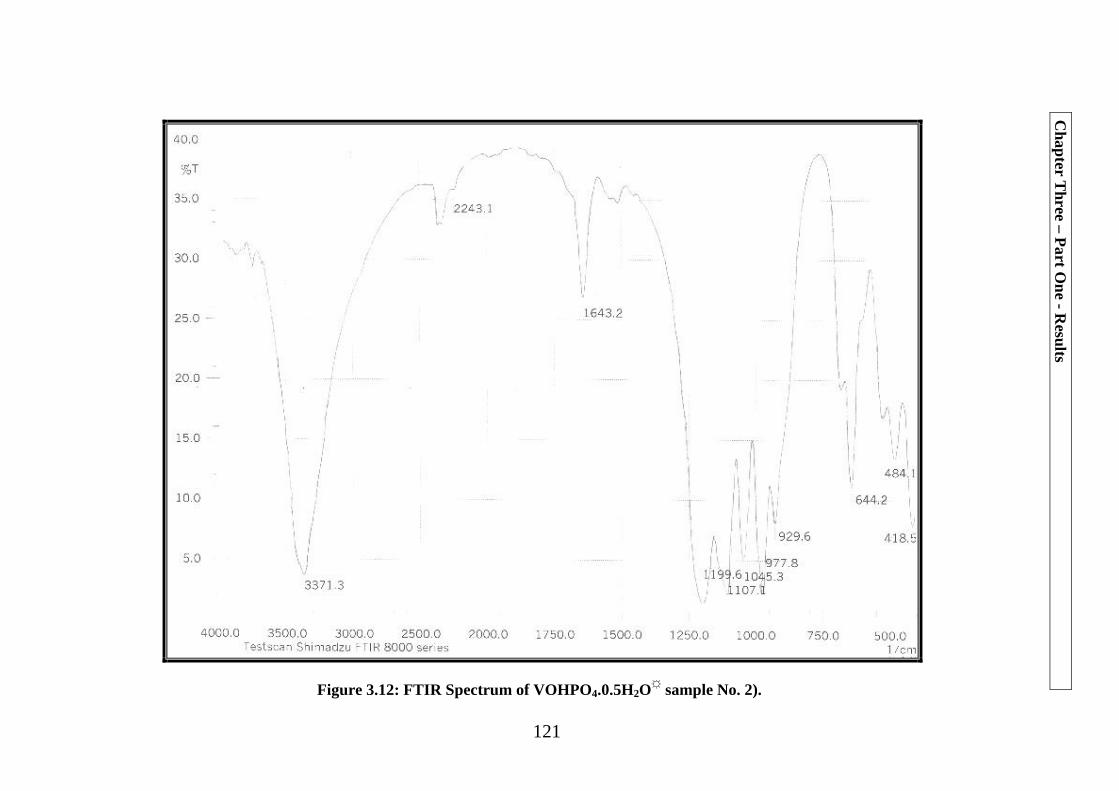
Chapter T
hree – Part O
ne - Results
Figure 3.12: FTIR Spectrum of VOHPO4.0.5H2O☼ sample No. 2).
121
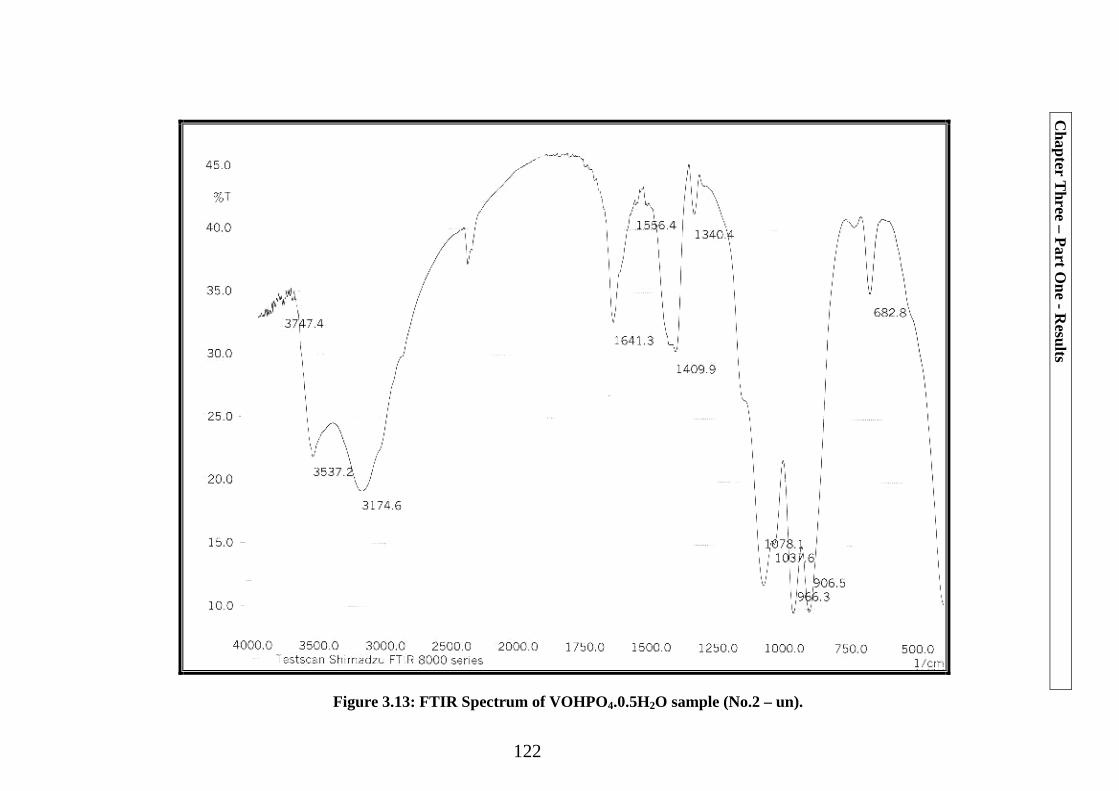
Chapter T
hree – Part O
ne - Results
Figure 3.13: FTIR Spectrum of VOHPO4.0.5H2O sample (No.2 – un).
122
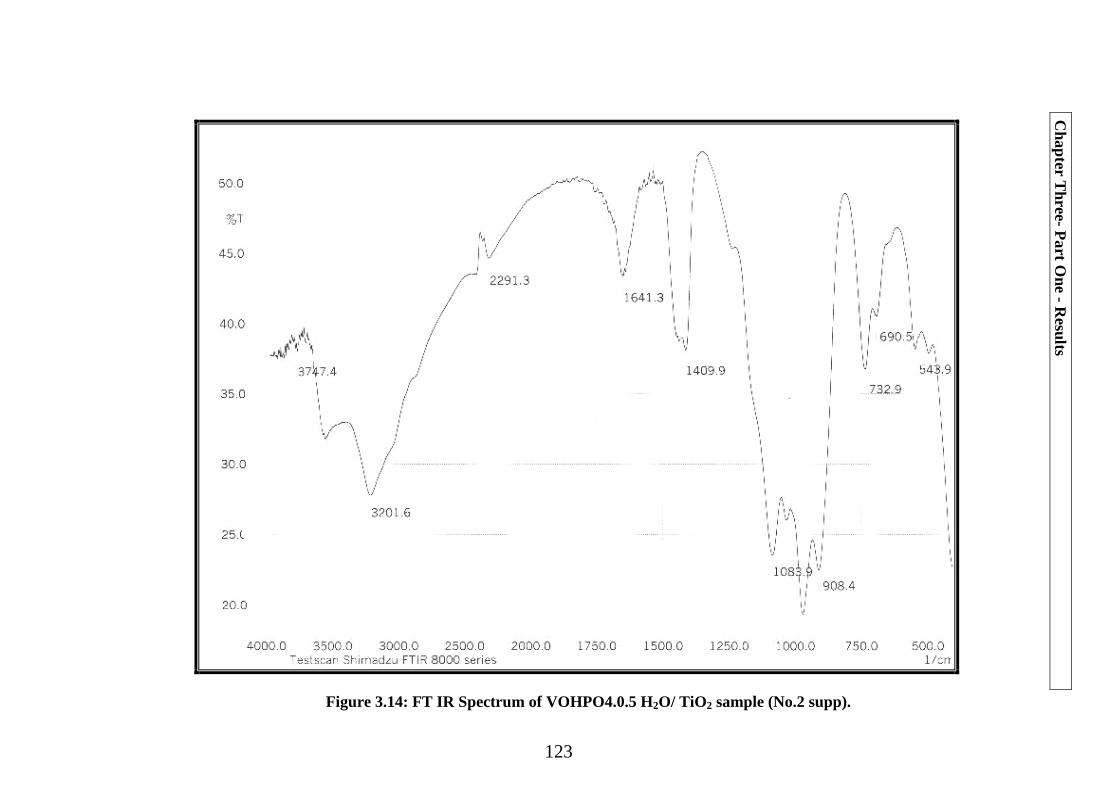
Chapter T
hree- Part O
ne - Results
Figure 3.14: FT IR Spectrum of VOHPO4.0.5 H2O/ TiO2 sample (No.2 supp).
123
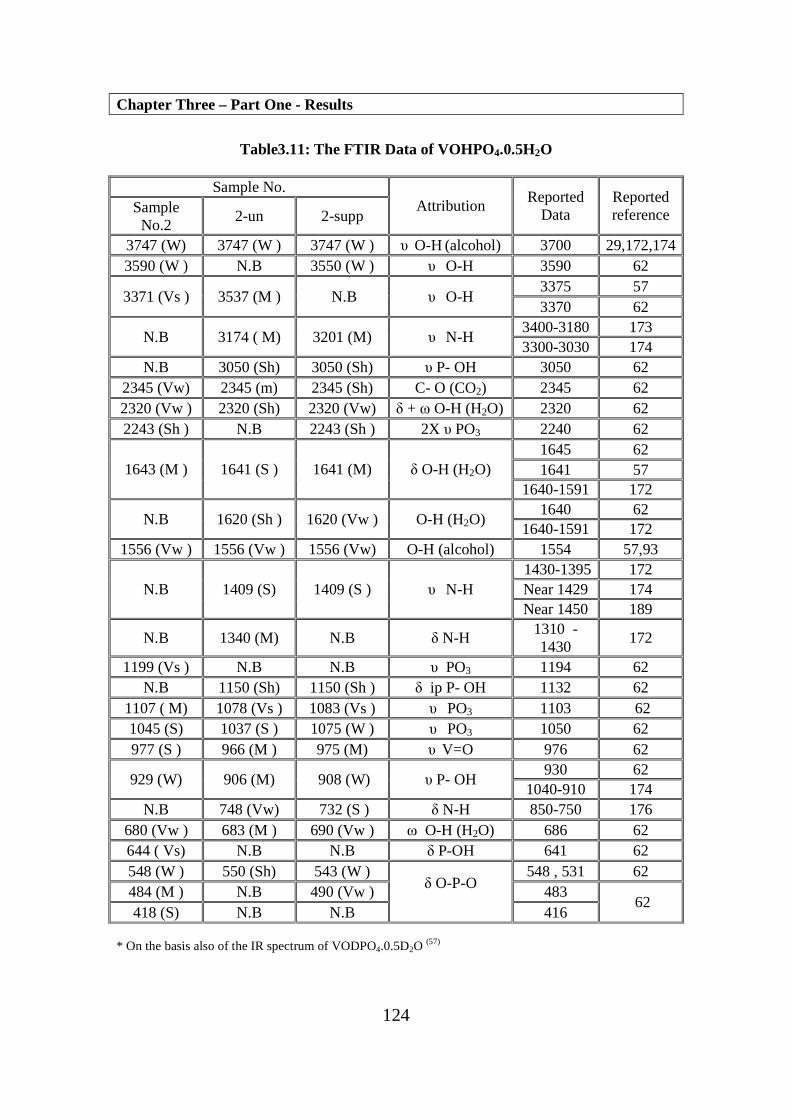
Chapter Three – Part One - Results
124
Table3.11: The FTIR Data of VOHPO4.0.5H2O
Sample No. Attribution
Reported Data
Reported reference Sample
No.2 2-un 2-supp
3747 (W) 3747 (W ) 3747 (W ) υ O-H (alcohol) 3700 29,172,174 3590 (W ) N.B 3550 (W ) υ O-H 3590 62
3371 (Vs ) 3537 (M ) N.B υ O-H 3375 57 3370 62
N.B 3174 ( M) 3201 (M) υ N-H 3400-3180 173 3300-3030 174
N.B 3050 (Sh) 3050 (Sh) υ P- OH 3050 62 2345 (Vw) 2345 (m) 2345 (Sh) C- O (CO2) 2345 62 2320 (Vw ) 2320 (Sh) 2320 (Vw) δ + ω O-H (H2O) 2320 62 2243 (Sh ) N.B 2243 (Sh ) 2X υ PO3 2240 62
1643 (M ) 1641 (S ) 1641 (M) δ O-H (H2O) 1645 62 1641 57
1640-1591 172
N.B 1620 (Sh ) 1620 (Vw ) O-H (H2O) 1640 62
1640-1591 172 1556 (Vw ) 1556 (Vw ) 1556 (Vw) O-H (alcohol) 1554 57,93
N.B 1409 (S) 1409 (S ) υ N-H 1430-1395 172 Near 1429 174 Near 1450 189
N.B 1340 (M) N.B δ N-H 1310 - 1430
172
1199 (Vs ) N.B N.B υ PO3 1194 62 N.B 1150 (Sh) 1150 (Sh ) δ ip P- OH 1132 62
1107 ( M) 1078 (Vs ) 1083 (Vs ) υ PO3 1103 62 1045 (S) 1037 (S ) 1075 (W ) υ PO3 1050 62 977 (S ) 966 (M ) 975 (M) υ V=O 976 62
929 (W) 906 (M) 908 (W) υ P- OH 930 62
1040-910 174 N.B 748 (Vw) 732 (S ) δ N-H 850-750 176
680 (Vw ) 683 (M ) 690 (Vw ) ω O-H (H2O) 686 62 644 ( Vs) N.B N.B δ P-OH 641 62 548 (W ) 550 (Sh) 543 (W )
δ O-P-O
548 , 531 62 484 (M ) N.B 490 (Vw ) 483
62 418 (S) N.B N.B 416
* On the basis also of the IR spectrum of VODPO4.0.5D2O (57)
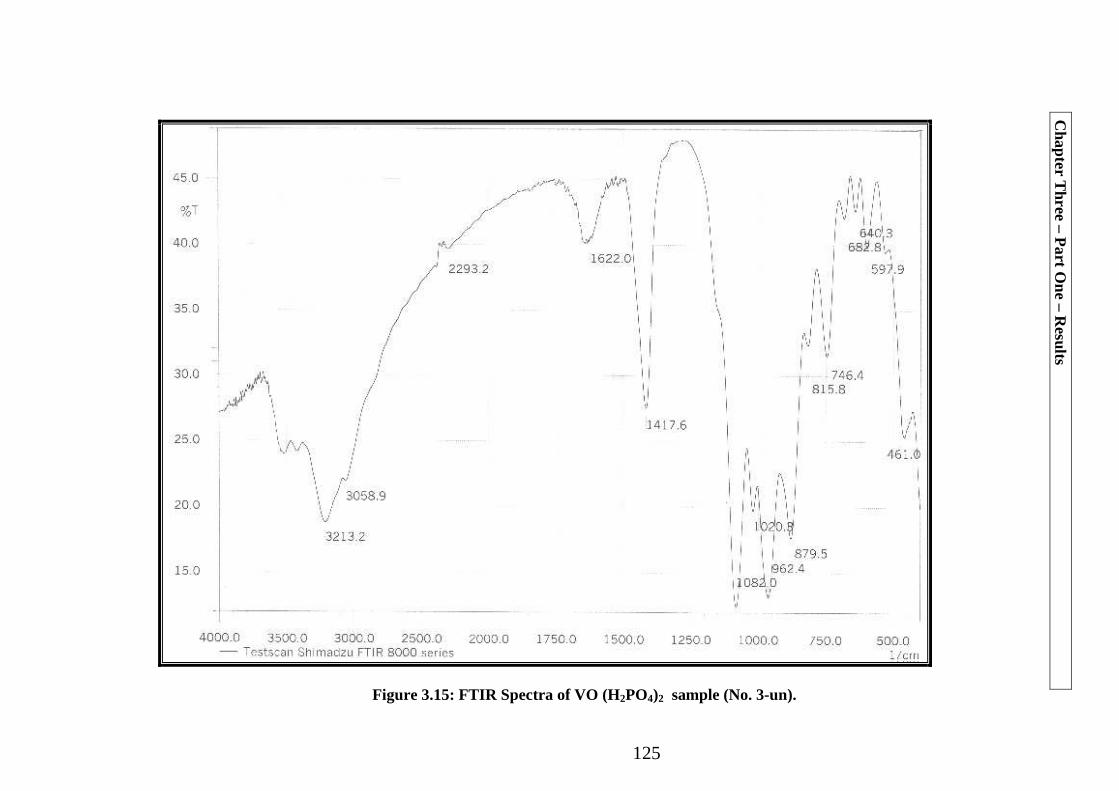
Chapter T
hree – Part O
ne – Results
Figure 3.15: FTIR Spectra of VO (H2PO4)2 sample (No. 3-un).
125
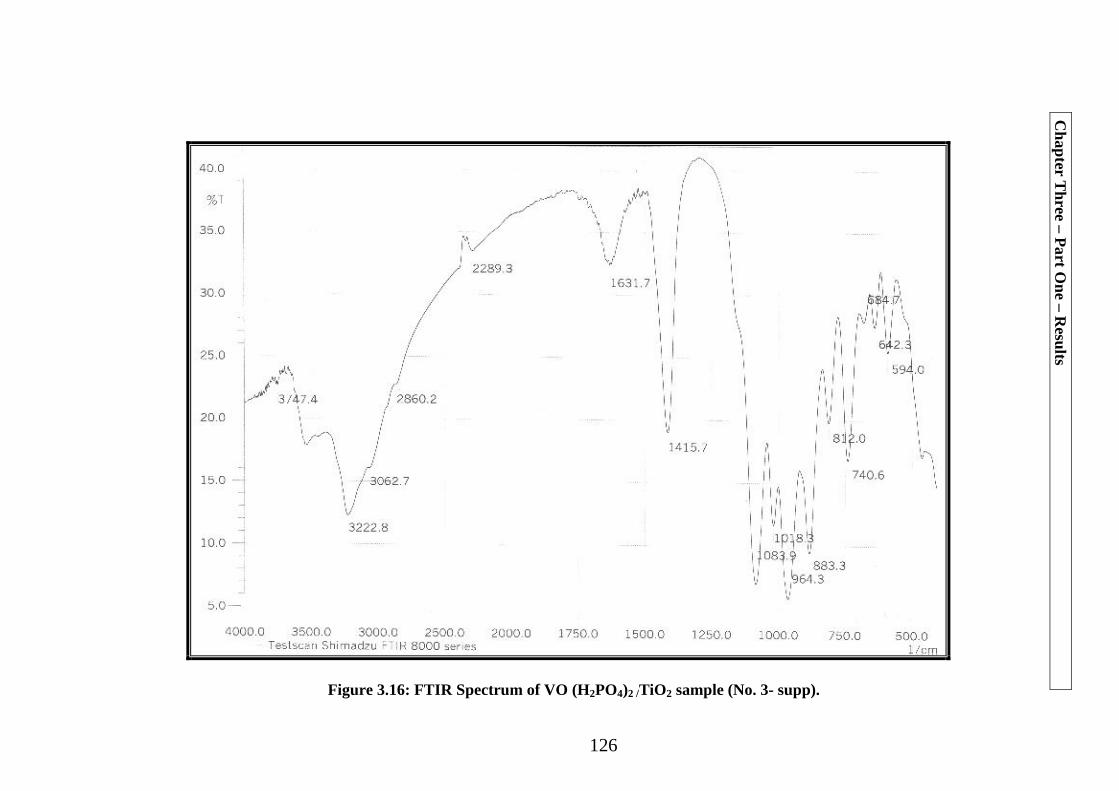
Chapter T
hree – Part O
ne – Results
Figure 3.16: FTIR Spectrum of VO (H2PO4)2 /TiO 2 sample (No. 3- supp).
126

Chapter Three – Part One - Results
127
Table3.12: The FT-IR Data of VO (H2PO4)2
Sample No. Attribution
Reported Data
Reference 3-un 3-supp
N.B 3747(Vw) υ O-H 3700 57,91,173
3550 (W ) 3550 (m) υ O-H (H2O) N.B 172
3400 (W) 3400 (Sh) υ O-H (H2O) 3400 172 υ P-OH 3420 80
3213 (M) 3223 (M) υ N-H (NH4) +
3400-3180 172 3300-3030 174
N.B N.B
υ O-H 3020-2270 172 N.B 2860 (Sh)
2293 (Vw) 2289 (W)
1622 (M) 1632 (M) δ O-H 1620 172
1418 (Vs) 1416 (Vs) υ N-H (NH4) +
1430-1395 172 Near 1429 174 Near 1450 189
1340 (W) N.B δ N-H
1300 172
δ ip P- OH 80
N.B N.B υ O-P-O 1175 172 1185 80
N.B N.B υ O-P-O 1155 172
1082 (Vs) 1084 (M) υ O-P-O 1100 172 1110 80
1020 (W) 1019 (M) υ V = O 1015 172
962 (M) 964 (S) υ P- O 930 172 υ V = O 935 80
N.B N.B υ V-OH 910 172
879 (M) 883 (M) υ V-OH 890 172 υ P-OH 885 80
816 (M) 812 (M) υ P-OH 800 172
746 (S) 741 (S) δ N-H 850-750 176
684 (W) 685 W) ω O-H (H2O) 686 62
640 642 (M)
δ O- P- O
645
172
N.B N.B 610
598 (M) 594 (M) 600
N.B N.B δ O- P- O
+ δ V- O
495
461 (Vw) 450 (Vw) 470 -430

Chapter Three – Part One – Results
١٢٨
3.1.5- Laser Raman Spectroscopy
Laser Raman spectroscopy analysis was carried out using red and green
laser for VOHPO4.0.5H2O and VO (H2PO4)2. Raman bands were shown in
Figures 3.17-A and 3.17-B. The obtained data were tabulated in (Tables
3.16 & 3.17) respectively.
In red laser (excitation line = 632.8 nm), all samples show the main
bands correspond to V= O, P-O-P and crystalline V2O5.
Different bands were obtained in green laser (excitation line = 514 nm)
some bands were appeared and others were disappeared as shown in the
below tables.
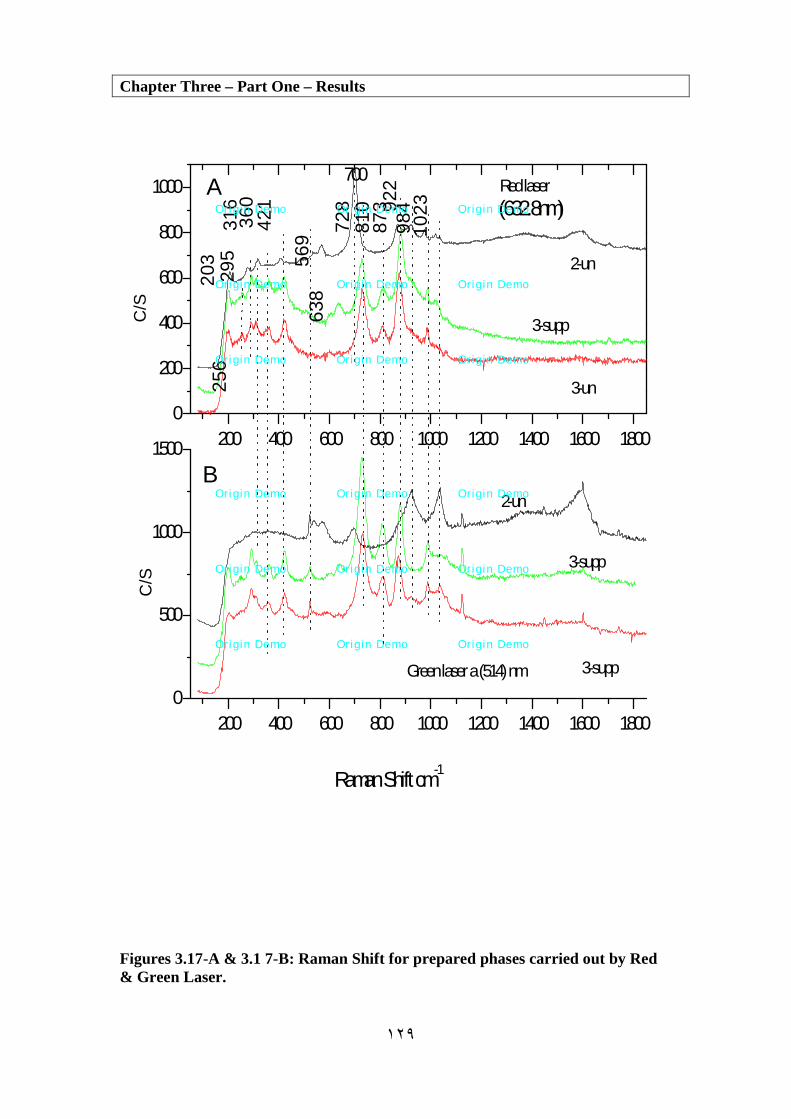
Chapter Three – Part One – Results
١٢٩
Figures 3.17-A & 3.1 7-B: Raman Shift for prepared phases carried out by Red & Green Laser.
200 400 600 800 1000 1200 1400 1600 18000
500
1000
1500 200 400 600 800 1000 1200 1400 1600 18000
200
400
600
800
1000
3-supp
2-un
Green laser a (514) nm
Raman Shift cm-1
3-supp
BOrigin Demo Origin Demo Origin Demo
Origin Demo Origin Demo Origin Demo
Origin Demo Origin Demo Origin Demo
C/S
C
/S
700
728
810
873
984
421
360
316
203
1023
569
638
922
295
256
2-un
3-supp
3-un
Red laserA(632.8nm)Origin Demo Origin Demo Origin Demo
Origin Demo Origin Demo Origin Demo
Origin Demo Origin Demo Origin Demo
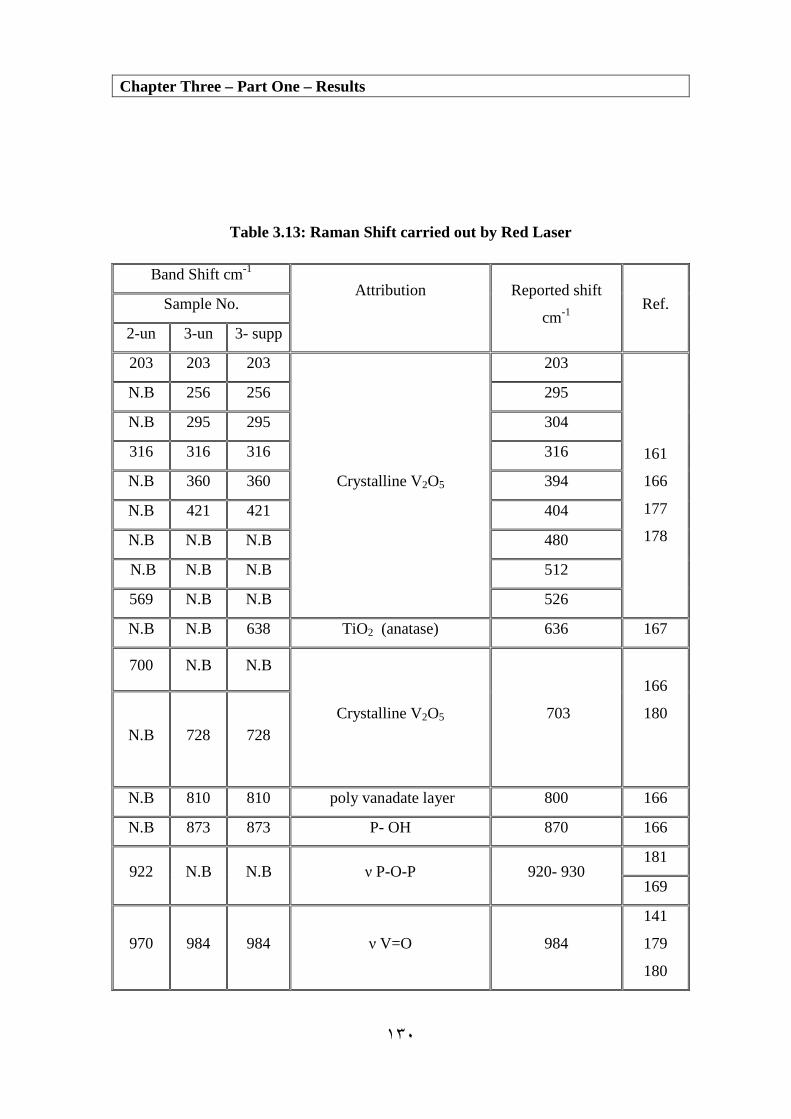
Chapter Three – Part One – Results
١٣٠
Table 3.13: Raman Shift carried out by Red Laser
Band Shift cm-1 Attribution
Reported shift
cm-1 Ref. Sample No.
2-un 3-un 3- supp
203 203 203
Crystalline V2O5
203
161
166
177
178
N.B 256 256 295
N.B 295 295 304
316 316 316 316
N.B 360 360 394
N.B 421 421 404
N.B N.B N.B 480
N.B N.B N.B 512
569 N.B N.B 526
N.B N.B 638 TiO2 (anatase) 636 167
700 N.B N.B
Crystalline V2O5
703
166
180
N.B 728 728
N.B 810 810 poly vanadate layer 800 166
N.B 873 873 P- OH 870 166
922 N.B N.B ν P-O-P 920- 930 181
169
970 984 984 ν V=O 984
141
179
180
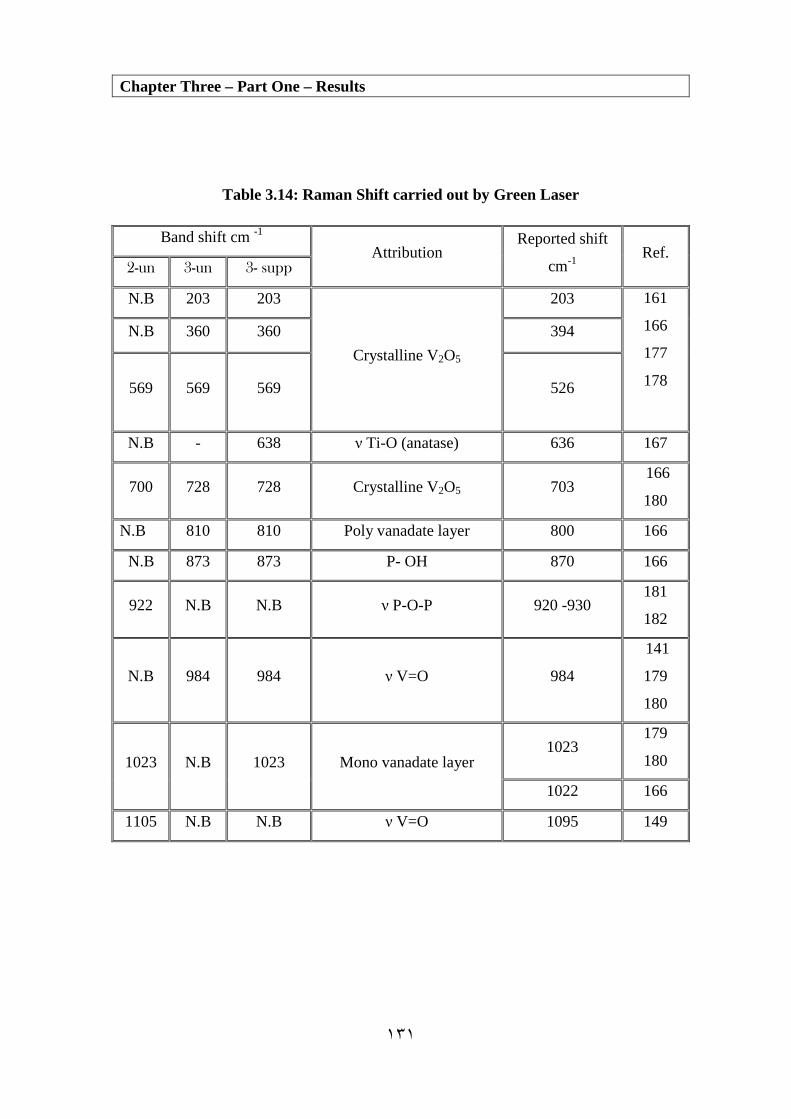
Chapter Three – Part One – Results
١٣١
Table 3.14: Raman Shift carried out by Green Laser
Band shift cm -1 Attribution
Reported shift
cm-1 Ref.
2-un 3-un 3- supp
N.B 203 203
Crystalline V2O5
203 161
166
177
178
N.B 360 360 394
569 569 569 526
N.B - 638 ν Ti-O (anatase) 636 167
700 728 728 Crystalline V2O5 703 166
180
N.B 810 810 Poly vanadate layer 800 166
N.B 873 873 P- OH 870 166
922 N.B N.B ν P-O-P 920 -930 181
182
N.B 984 984 ν V=O 984
141
179
180
1023 N.B 1023 Mono vanadate layer 1023
179
180
1022 166
1105 N.B N.B ν V=O 1095 149
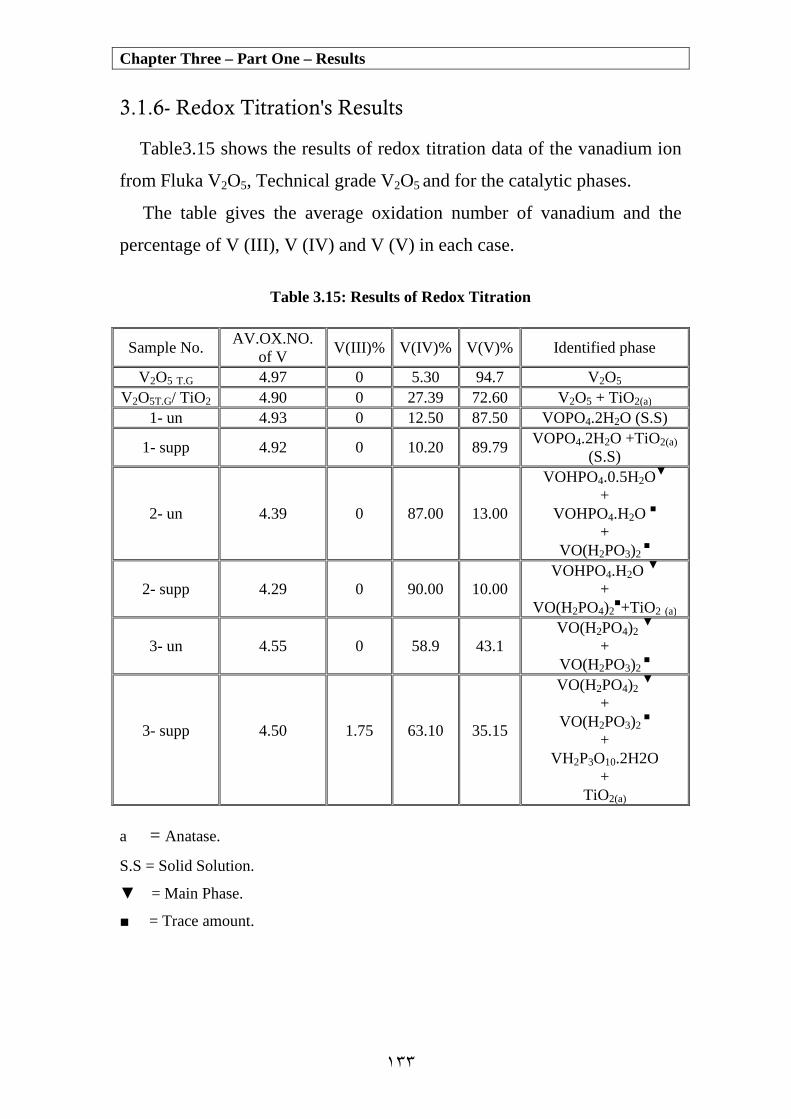
Chapter Three – Part One – Results
١٣٣
3.1.6- Redox Titration's Results
Table3.15 shows the results of redox titration data of the vanadium ion
from Fluka V2O5, Technical grade V2O5 and for the catalytic phases.
The table gives the average oxidation number of vanadium and the
percentage of V (III), V (IV) and V (V) in each case.
Table 3.15: Results of Redox Titration
Sample No. AV.OX.NO.
of V V(III)% V(IV)% V(V)% Identified phase
V2O5 T.G 4.97 0 5.30 94.7 V2O5 V2O5T.G/ TiO2 4.90 0 27.39 72.60 V2O5 + TiO2(a)
1- un 4.93 0 12.50 87.50 VOPO4.2H2O (S.S)
1- supp 4.92 0 10.20 89.79 VOPO4.2H2O +TiO2(a)
(S.S)
2- un 4.39 0 87.00 13.00
VOHPO4.0.5H2O▼
+ VOHPO4.H2O ■
+ VO(H2PO3)2
■
2- supp 4.29 0 90.00 10.00 VOHPO4.H2O ▼
+ VO(H2PO4)2
■+TiO2 (a)
3- un 4.55 0 58.9 43.1 VO(H2PO4)2
▼ +
VO(H2PO3)2 ■
3- supp
4.50
1.75
63.10
35.15
VO(H2PO4)2 ▼
+ VO(H2PO3)2
■ +
VH2P3O10.2H2O +
TiO2(a) a = Anatase.
S.S = Solid Solution.
▼ = Main Phase.
■ = Trace amount.
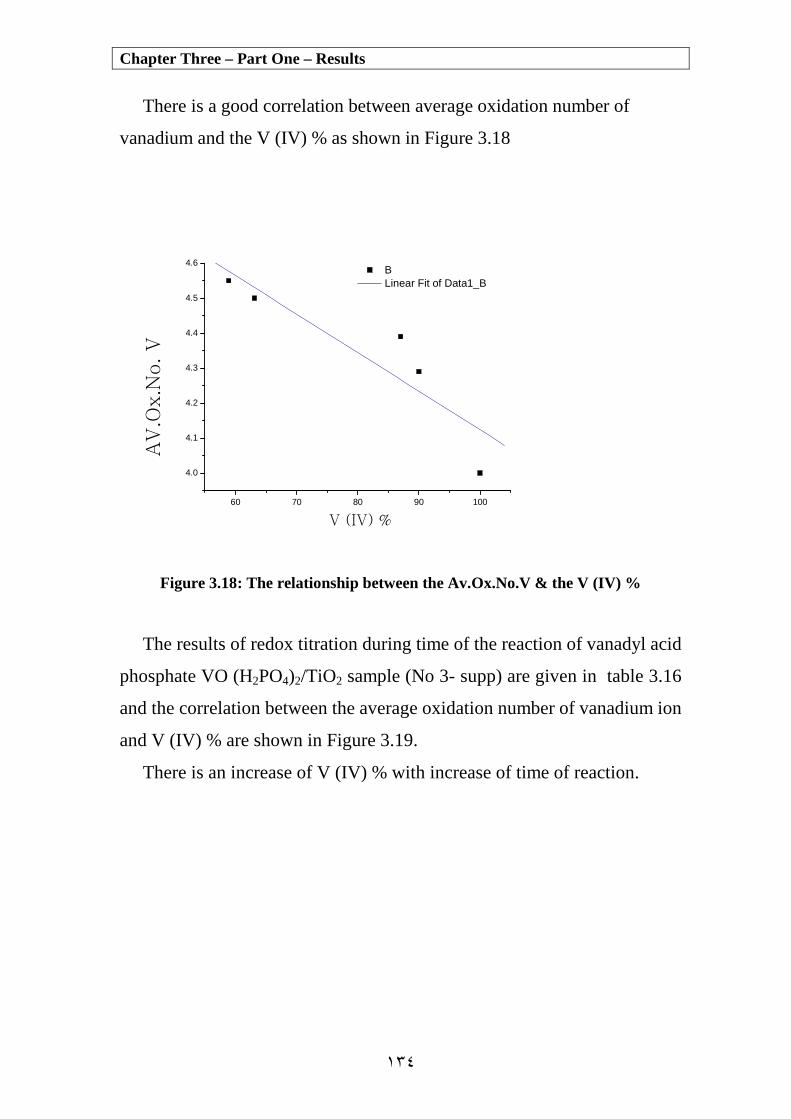
Chapter Three – Part One – Results
١٣٤
There is a good correlation between average oxidation number of
vanadium and the V (IV) % as shown in Figure 3.18
60 70 80 90 100
4.0
4.1
4.2
4.3
4.4
4.5
4.6 B Linear Fit of Data1_B
AV
.Ox.N
o. V
V (IV) %
Figure 3.18: The relationship between the Av.Ox.No.V & the V (IV) %
The results of redox titration during time of the reaction of vanadyl acid
phosphate VO (H2PO4)2/TiO2 sample (No 3- supp) are given in table 3.16
and the correlation between the average oxidation number of vanadium ion
and V (IV) % are shown in Figure 3.19.
There is an increase of V (IV) % with increase of time of reaction.
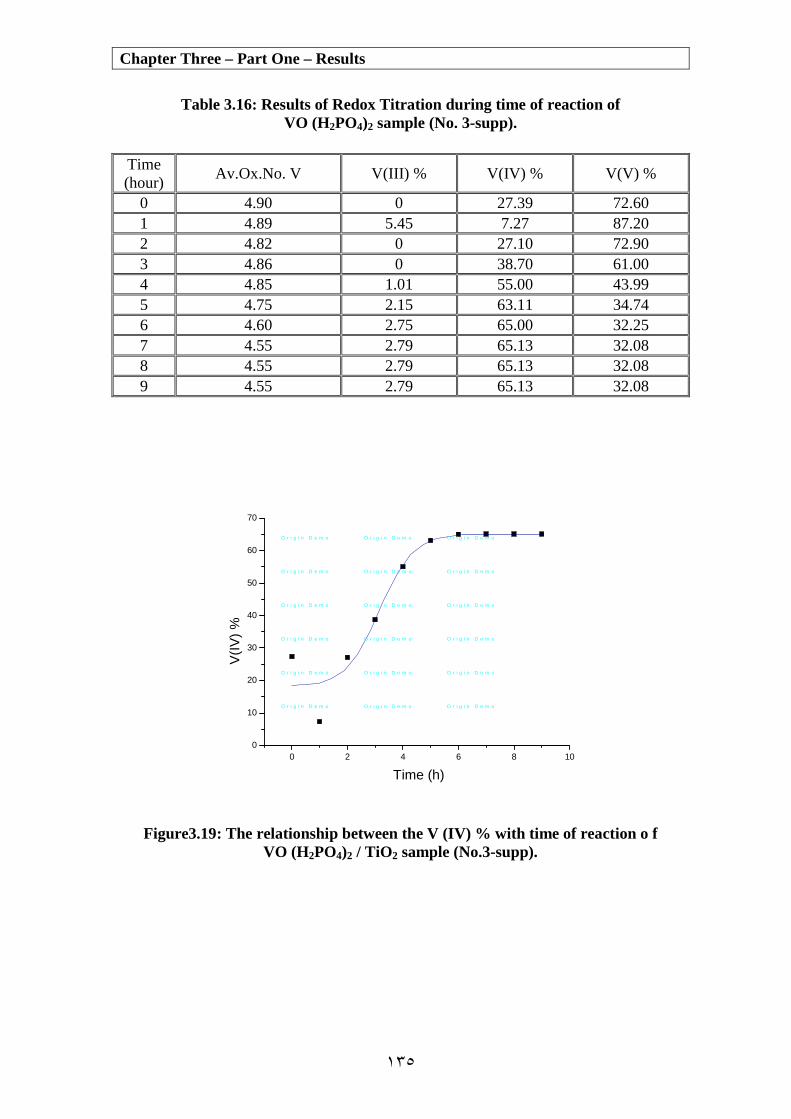
Chapter Three – Part One – Results
١٣٥
Table 3.16: Results of Redox Titration during time of reaction of VO (H2PO4)2 sample (No. 3-supp).
Time (hour)
Av.Ox.No. V V(III) % V(IV) % V(V) %
0 4.90 0 27.39 72.60 1 4.89 5.45 7.27 87.20 2 4.82 0 27.10 72.90 3 4.86 0 38.70 61.00 4 4.85 1.01 55.00 43.99 5 4.75 2.15 63.11 34.74 6 4.60 2.75 65.00 32.25 7 4.55 2.79 65.13 32.08 8 4.55 2.79 65.13 32.08 9 4.55 2.79 65.13 32.08
0 2 4 6 8 100
10
20
30
40
50
60
70
V(I
V)
%
Time (h)
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
O r i g i n D e m o O r i g i n D e m o O r i g i n D e m o
Figure3.19: The relationship between the V (IV) % with time of reaction o f VO (H2PO4)2 / TiO2 sample (No.3-supp).
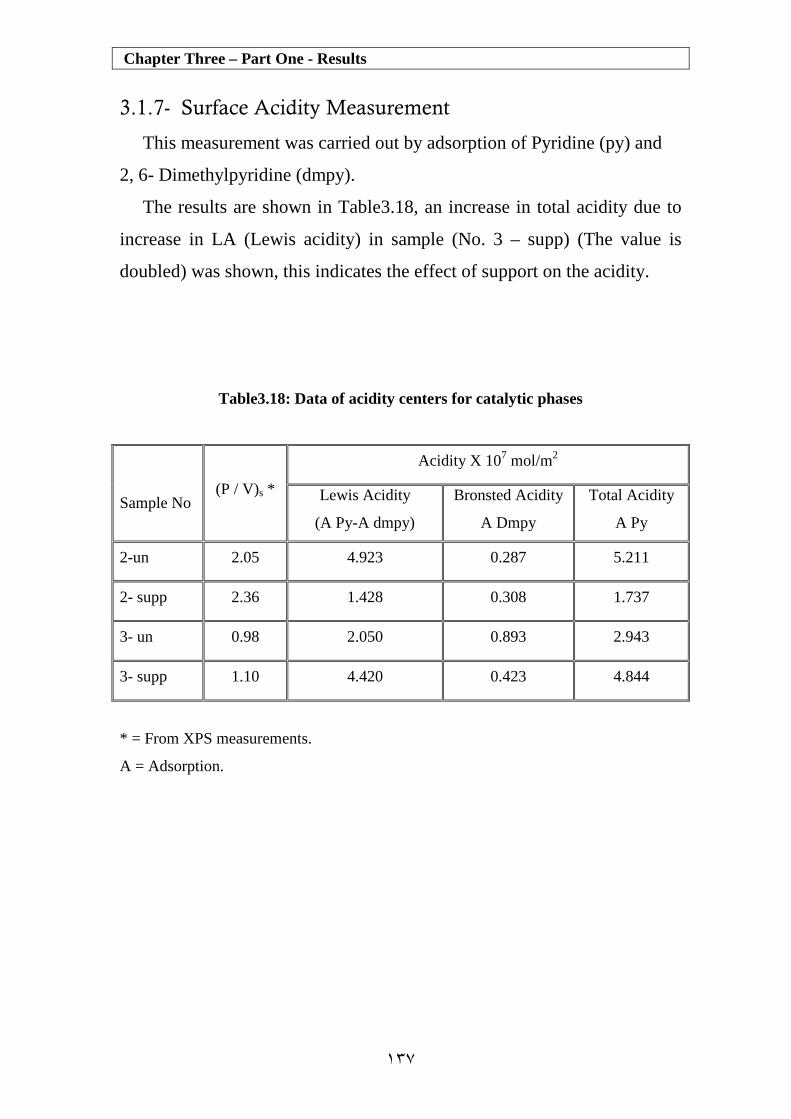
Chapter Three – Part One - Results
١٣٧
3.1.7- Surface Acidity Measurement
This measurement was carried out by adsorption of Pyridine (py) and
2, 6- Dimethylpyridine (dmpy).
The results are shown in Table3.18, an increase in total acidity due to
increase in LA (Lewis acidity) in sample (No. 3 – supp) (The value is
doubled) was shown, this indicates the effect of support on the acidity.
Table3.18: Data of acidity centers for catalytic phases
Sample No (P / V)s *
Acidity X 107 mol/m2
Lewis Acidity
(A Py-A dmpy)
Bronsted Acidity
A Dmpy
Total Acidity
A Py
2-un 2.05 4.923 0.287 5.211
2- supp 2.36 1.428 0.308 1.737
3- un 0.98 2.050 0.893 2.943
3- supp 1.10 4.420 0.423 4.844
* = From XPS measurements.
A = Adsorption.
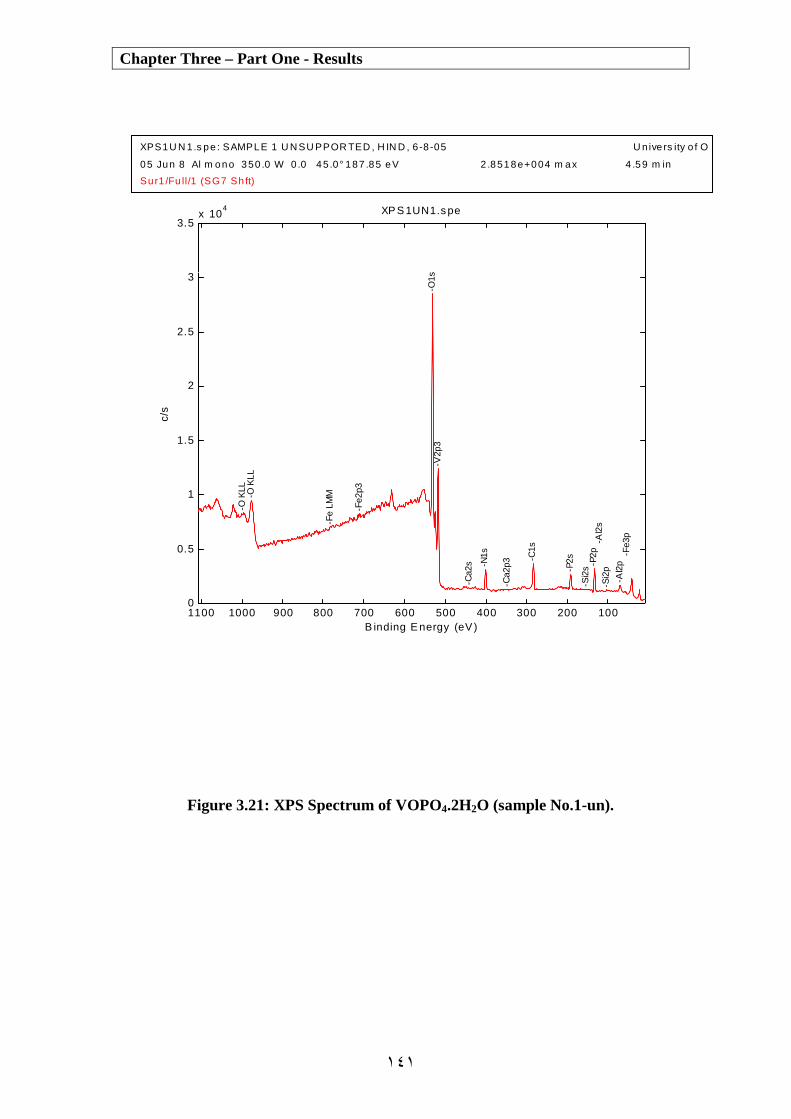
Chapter Three – Part One - Results
١٤١
XPS1U N 1.s pe : SAMPLE 1 U N SU PPOR TED , H IN D , 6-8-05 U nive rs ity o f O
05 Jun 8 Al m ono 350 .0 W 0 .0 45 .0° 187 .85 eV 2 .8518e+004 m ax 4 .59 m in
Sur1 /Fu ll/1 (SG7 Shft)
100200300400500600700800900100011000
0.5
1
1.5
2
2.5
3
3.5x 10
4 XP S 1UN1.spe
B inding E nergy (eV)
c/s
-O
KLL
-O
KLL
-O
1s
-N
1s
-C
1s
-P2
s
-P2
p
-V
2p3
-S
i2s
-S
i2p
-A
l2s
-A
l2p
-Fe
LM
M
-Fe
3p
-Fe
2p3
-C
a2s
-C
a2p3
Figure 3.21: XPS Spectrum of VOPO4.2H2O (sample No.1-un).
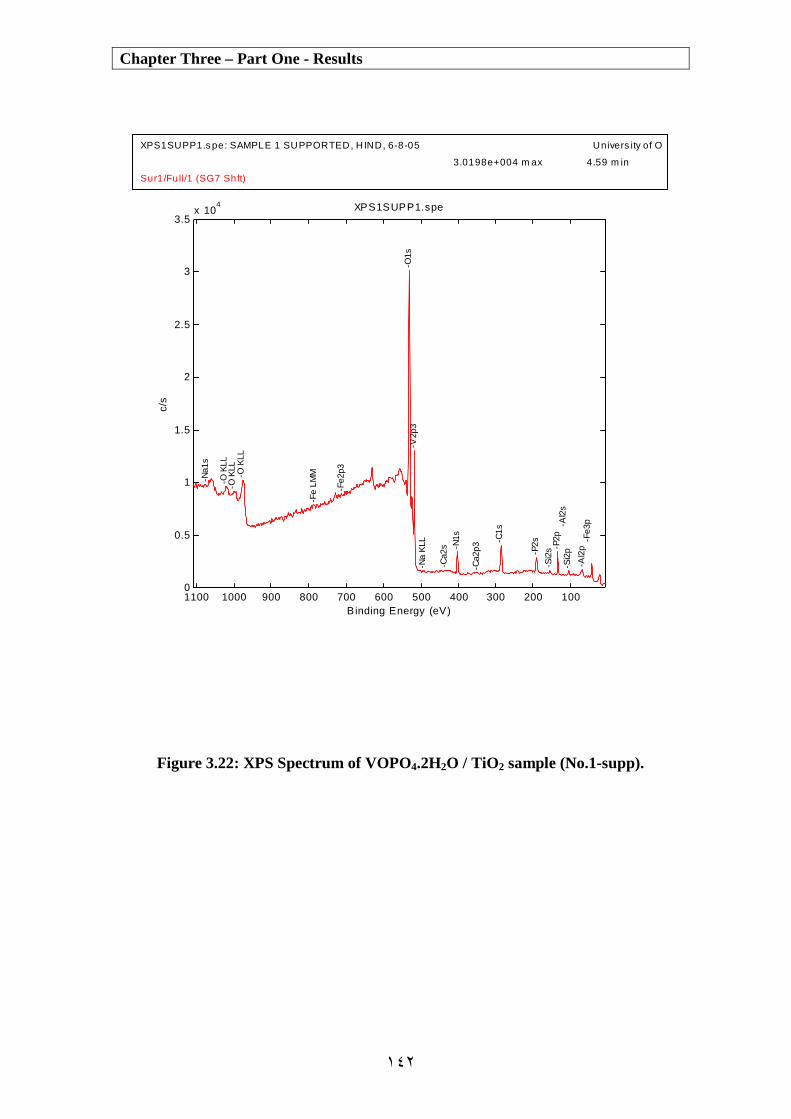
Chapter Three – Part One - Results
١٤٢
XPS1SUPP1.s pe: SAMPLE 1 SUPPORTED, HIND, 6-8-05 Univers ity of O
3.0198e+004 m ax 4.59 m in
Sur1/Full/1 (SG7 Shft)
100200300400500600700800900100011000
0.5
1
1.5
2
2.5
3
3.5x 10
4 XPS1SUPP1.spe
Binding Energy (eV)
c/s
-O
KLL
-O
KLL
-O
KLL
-O
1s
-N
1s -C
1s
-P2
s
-S
i2s -
P2p
-S
i2p
-N
a K
LL
-N
a1s
-C
a2s
-C
a2p3
-V
2p3
-A
l2s
-A
l2p
-Fe
LM
M
-Fe
3p
-Fe
2p3
Figure 3.22: XPS Spectrum of VOPO4.2H2O / TiO2 sample (No.1-supp).
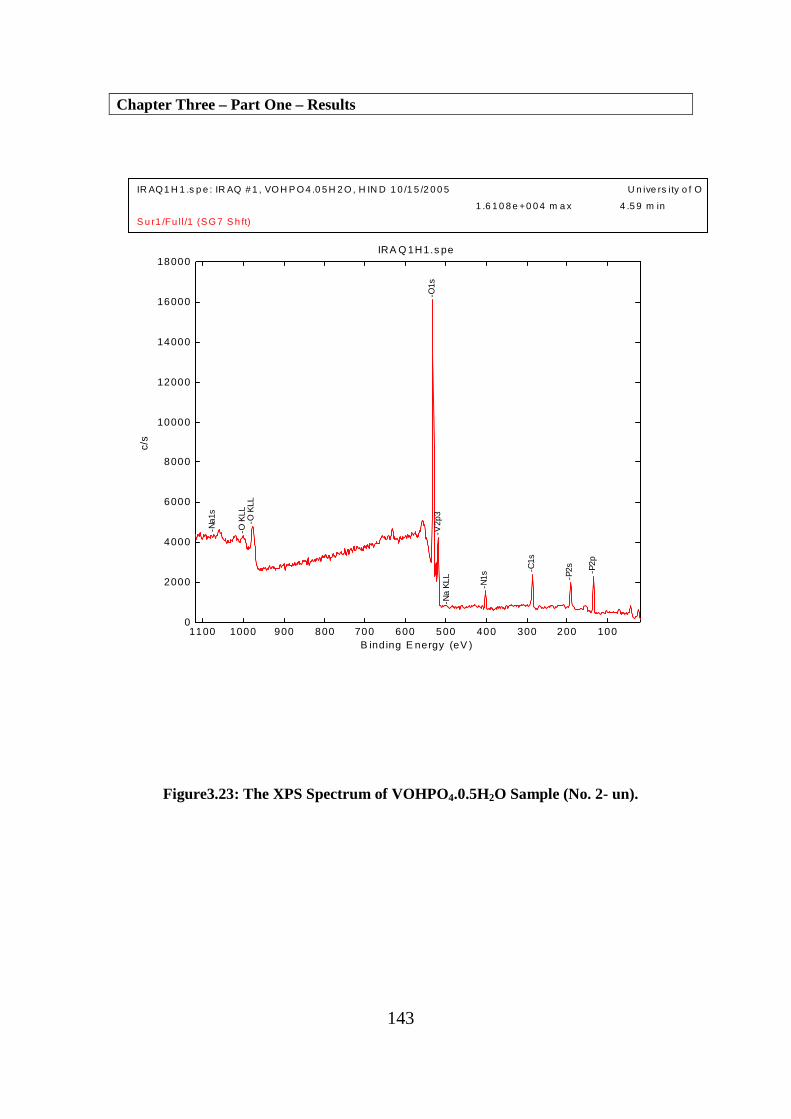
Chapter Three – Part One – Results
143
IR AQ1 H 1 .s p e : IR AQ # 1 , VO H P O 4 .0 5 H 2 O , H IN D 1 0 /1 5 /2 0 0 5 U n ive rs ity o f O
1 .6 1 0 8 e +0 0 4 m a x 4 .5 9 m in
S u r1 /Fu ll /1 (S G 7 S h ft)
100200300400500600700800900100011000
2000
4000
6000
8000
10000
12000
14000
16000
18000IRA Q 1H1.s pe
B ind ing E nergy (eV )
c/s
-O
KLL
-O
KLL
-O
1s
-N
1s
-C
1s
-P2
s
-P2
p
-V
2p3
-N
a K
LL
-N
a1s
Figure3.23: The XPS Spectrum of VOHPO4.0.5H2O Sample (No. 2- un).
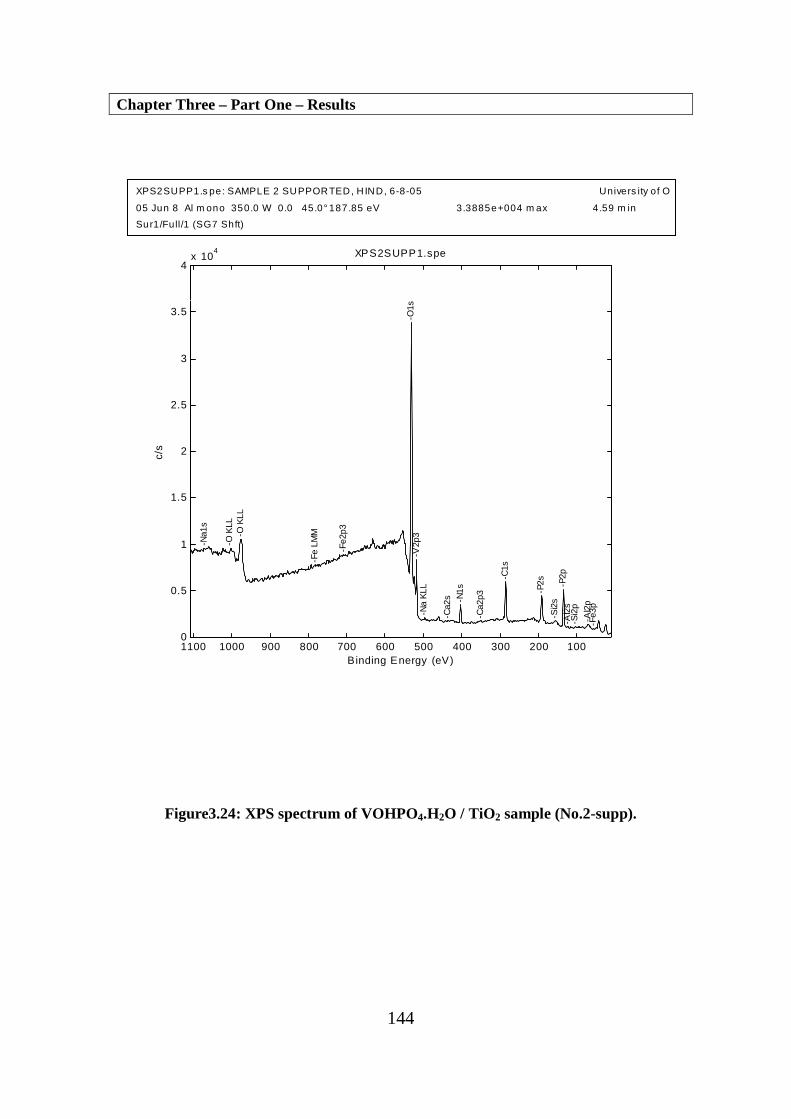
Chapter Three – Part One – Results
144
XPS2SUPP1.s pe: SAMPLE 2 SUPPORTED, H IND, 6-8-05 Univers ity of O
05 Jun 8 Al m ono 350.0 W 0.0 45.0° 187.85 eV 3.3885e+004 m ax 4.59 m in
Sur1/Full/1 (SG7 Shft)
100200300400500600700800900100011000
0.5
1
1.5
2
2.5
3
3.5
4x 10
4 XPS2SUPP1.spe
Binding Energy (eV)
c/s
-O
KLL
-O
KLL
-O
1s
-N
1s
-C
1s
-P2
s
-P2
p
-N
a K
LL
-N
a1s
-C
a2s
-C
a2p3
-V
2p3
-Fe
LM
M
-Fe
3p
-Fe
2p3
-A
l2s
-A
l2p
-S
i2s
-S
i2p
Figure3.24: XPS spectrum of VOHPO4.H2O / TiO2 sample (No.2-supp).
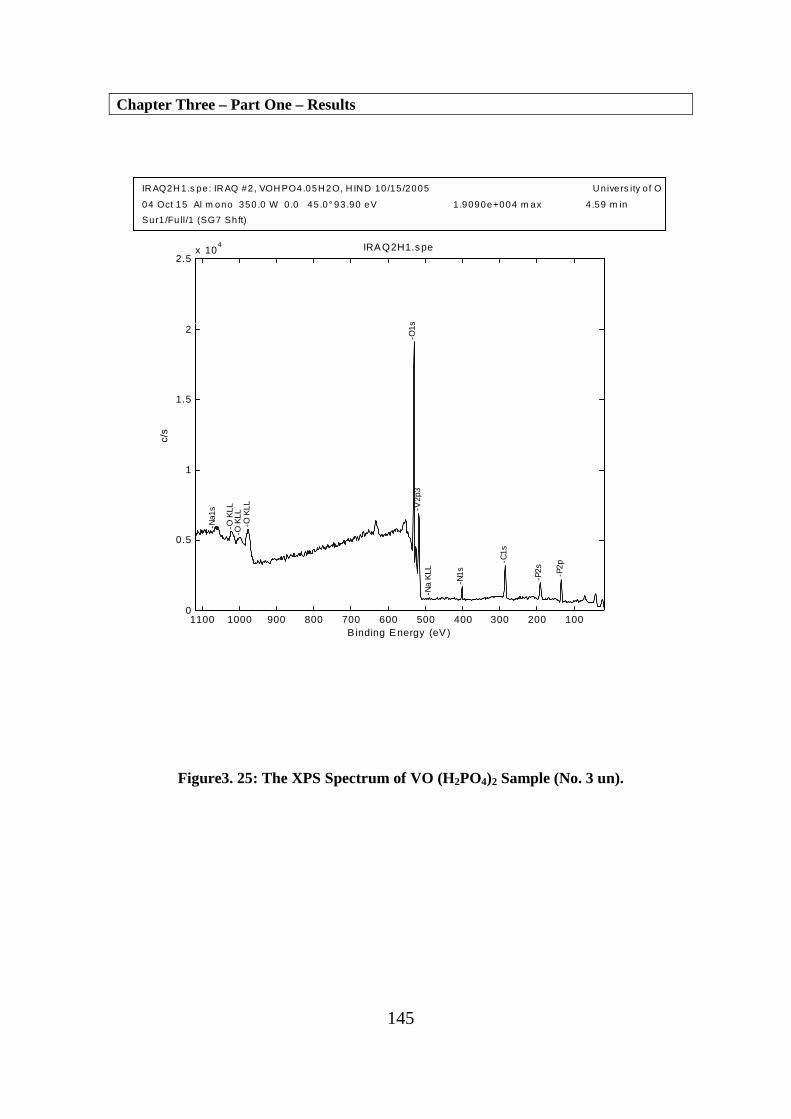
Chapter Three – Part One – Results
145
IR AQ2H 1.s pe: IR AQ #2, VOH PO4.05H 2O, H IN D 10/15 /2005 U n ivers ity o f O
04 Oct 15 Al m ono 350.0 W 0 .0 45 .0° 93 .90 eV 1.9090e+004 m ax 4.59 m in
Sur1 /Fu ll/1 (SG7 Shft)
100200300400500600700800900100011000
0.5
1
1.5
2
2.5x 10
4 IRAQ2H1.spe
B inding Energy (eV)
c/s
-O
KLL
-O
KLL
-O
KLL
-O
1s
-N
1s
-C
1s
-P2
s
-P2
p
-V
2p3
-N
a K
LL
-N
a1s
Figure3. 25: The XPS Spectrum of VO (H2PO4)2 Sample (No. 3 un).
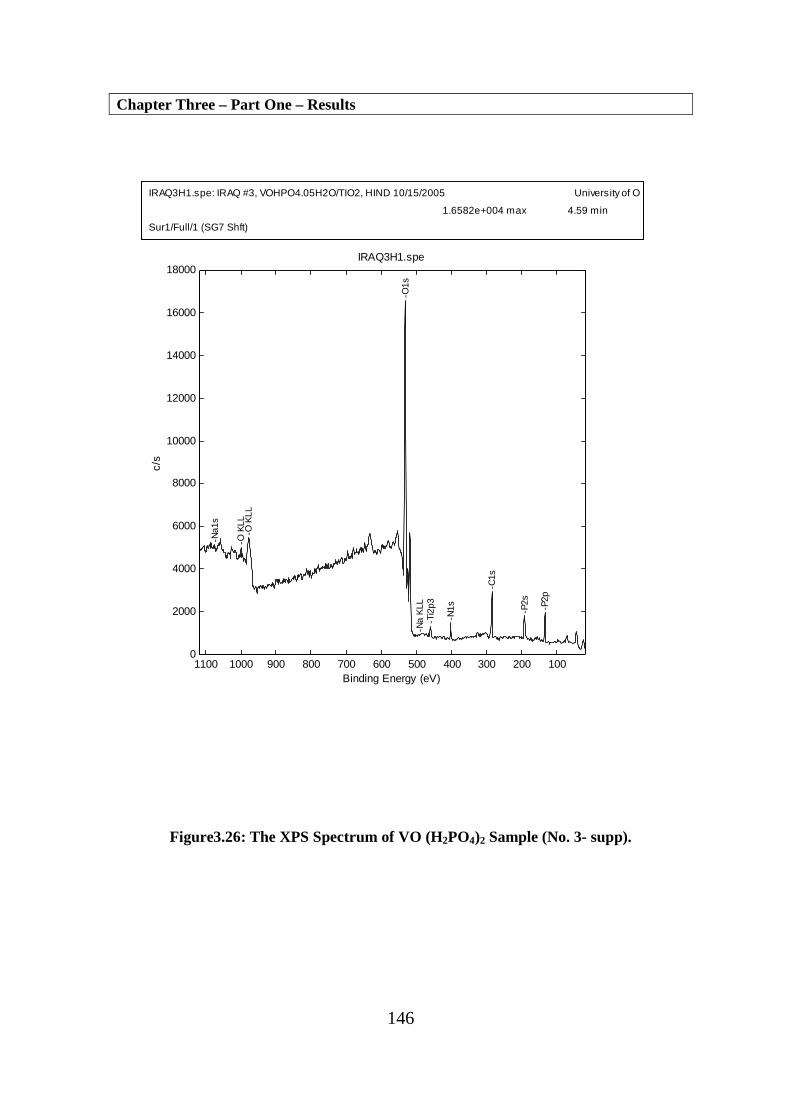
Chapter Three – Part One – Results
146
IRAQ3H1.spe: IRAQ #3, VOHPO4.05H2O/TIO2, HIND 10/15/2005 University of O
1.6582e+004 max 4.59 min
Sur1/Full/1 (SG7 Shft)
100200300400500600700800900100011000
2000
4000
6000
8000
10000
12000
14000
16000
18000IRAQ3H1.spe
Binding Energy (eV)
c/s
-O
KLL
-O
KLL
-O
1s
-Ti
2p3
-N
1s
-C
1s
-P2
s
-P2
p
-N
a K
LL
-N
a1s
Figure3.26: The XPS Spectrum of VO (H2PO4)2 Sample (No. 3- supp).
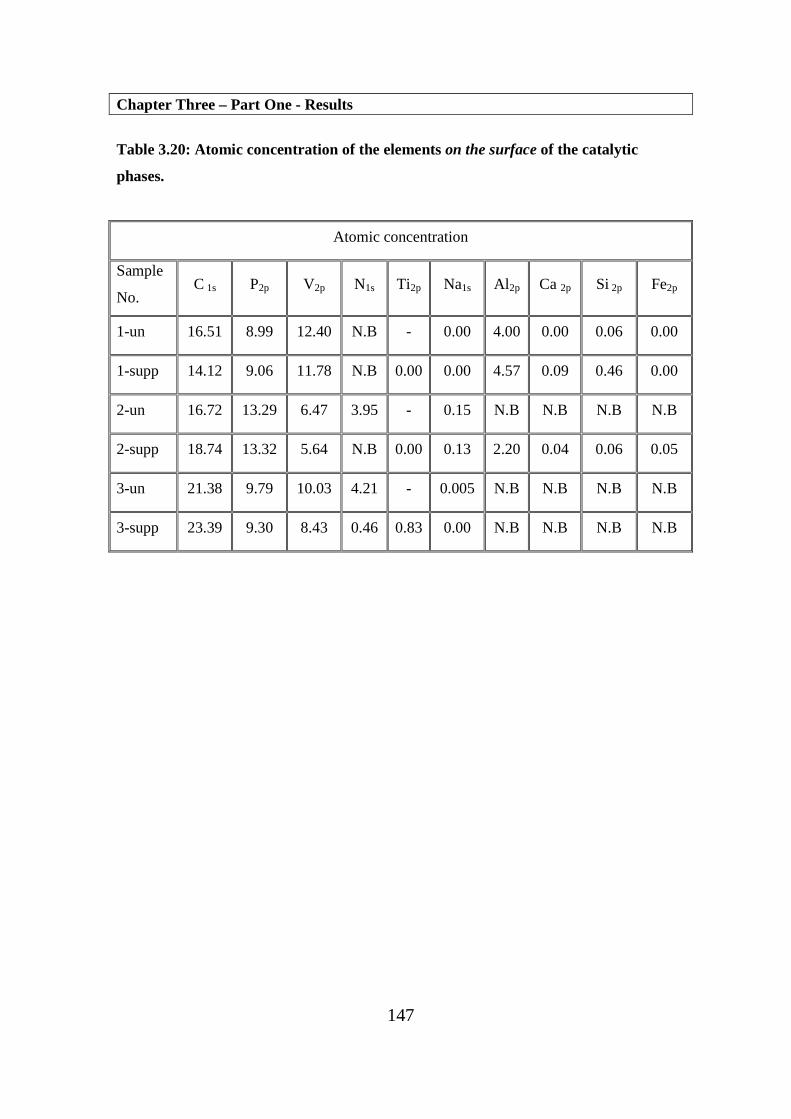
Chapter Three – Part One - Results
147
Table 3.20: Atomic concentration of the elements on the surface of the catalytic
phases.
Atomic concentration
Sample
No. C 1s P2p V2p N1s Ti2p Na1s Al2p Ca 2p Si 2p Fe2p
1-un 16.51 8.99 12.40 N.B - 0.00 4.00 0.00 0.06 0.00
1-supp 14.12 9.06 11.78 N.B 0.00 0.00 4.57 0.09 0.46 0.00
2-un 16.72 13.29 6.47 3.95 - 0.15 N.B N.B N.B N.B
2-supp 18.74 13.32 5.64 N.B 0.00 0.13 2.20 0.04 0.06 0.05
3-un 21.38 9.79 10.03 4.21 - 0.005 N.B N.B N.B N.B
3-supp 23.39 9.30 8.43 0.46 0.83 0.00 N.B N.B N.B N.B
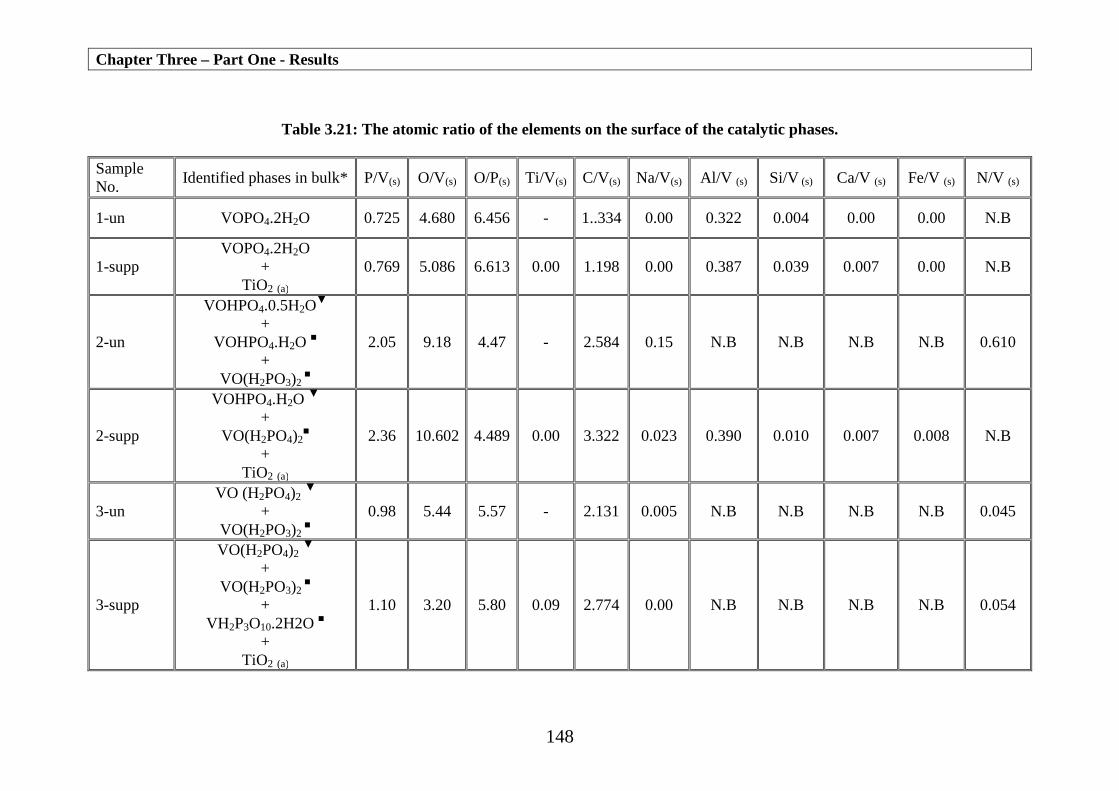
Chapter Three – Part One - Results
148
Table 3.21: The atomic ratio of the elements on the surface of the catalytic phases.
Sample No.
Identified phases in bulk* P/V(s) O/V(s) O/P(s) Ti/V(s) C/V(s) Na/V(s) Al/V (s) Si/V (s) Ca/V (s) Fe/V (s) N/V (s)
1-un VOPO4.2H2O 0.725 4.680 6.456 - 1..334 0.00 0.322 0.004 0.00 0.00 N.B
1-supp VOPO4.2H2O
+ TiO2 (a)
0.769 5.086 6.613 0.00 1.198 0.00 0.387 0.039 0.007 0.00 N.B
2-un
VOHPO4.0.5H2O▼
+ VOHPO4.H2O ■
+ VO(H2PO3)2
■
2.05 9.18 4.47 - 2.584 0.15 N.B N.B N.B N.B 0.610
2-supp
VOHPO4.H2O ▼
+ VO(H2PO4)2
■ +
TiO2 (a)
2.36 10.602 4.489 0.00 3.322 0.023 0.390 0.010 0.007 0.008 N.B
3-un VO (H2PO4)2
▼ +
VO(H2PO3)2 ■
0.98 5.44 5.57 - 2.131 0.005 N.B N.B N.B N.B 0.045
3-supp
VO(H2PO4)2 ▼
+ VO(H2PO3)2
■ +
VH2P3O10.2H2O ■ +
TiO2 (a)
1.10 3.20 5.80 0.09 2.774 0.00 N.B N.B N.B N.B 0.054

Chapter Three – Part One - Results
١٤٩
3.1.10- Scanning Electron Microscope (SEM) Analysis
Scanning electron images clearly show that the morphology of
VOPO4.2H2O with designed (P/V = 1.1) (Fig. 3.27 A and B ) is dominated
by small polygene-like particles with few small particles, while the
supported phase (Figure 3.28 A-D) is compose of larger particles.
The morphology of VOHPO4.0.5H2O (Figure 3.29 A-D) consist of
small lamellar morphology .It is obviously that using the organic medium
as reducing agent leads to obtain fine particles.
Scanning Electron Micrographs, shown in (Figure 3.30 and 3.31) for
VO(H2PO4)2 confirm the similarity in crystalline size between the
unsupported ( Fig. 3.30) and supported ( Fig. 3.31) phases, both of them
composed of well – formed plate- like crystals.
Figure 3.32 is representing the comparison of unsupported with
supported phases

Chapter Three - Part One - Results
١٥٠
(A) X 1000 (B) X 2500
Figure3.27: SEM Microstructure of VOPO4.2H2O (sample No. 1- un).
(A) X 740 (B) X 2219
(C) X 2969 (D) X 3699
Figure3.28: SEM Microstructure of VOPO4.2H2O / TiO2 (sample No. 1- supp).

Chapter Three – Part One – Results
١٥١
(A) X 400 (B) X 1000
(C) X 5000 (D) X 10000
Figure 3.29: SEM Microstructure of VOHPO4.0.5H2O (sample (No. 2- un).

Chapter Three – Part One - Results
152
(A) X 740 (B) X 1400
(C) X 2219 (D) X 2969
(E) 2959X (F) X 3693
Figure 3.30: SEM; Microstructure of VO (H2PO4)2 (sample (No. 3- un).

Chapter Three – Part One – Results
153
(A) X 740 (B) X 2219
(C) X 2953 (D) X 3693
Figure 3.31: SEM; Microstructure of VO (H2PO4)2/TiO2 (sample No. 3- supp).

Chapter Three – Part One - Results
154
(A) X 740 (un) (B) X 740 (supp)
(A) X 2962 (un) (B) X 2962 (supp)
(A) X 3693 (un) (B) X 3693 (supp)
Figure 3.32: A comparison between the morphology of unsupported & supported VO (H2PO4)2 (sample No.3-un and 3-supp).

Chapter Three – Part One - Results
١٥٥
3.1.11- Energy Dispersive X- Ray (EDX) Analysis
Besides the Scanning Electron Microscopy (SEM) for the information
of morphology, the energy dispersive X-Ray analysis (EDX) for
qualitative and quantities elemental analysis were applied in this study,
using the same instrument, in analyzing the surface layer up to 1 µm.
The qualitative information for each sample is given in the tables which
are appending to the figures.
The surface of VOHPO4.0.5H2O sample (No.2- un) is shown in
(Figures 3.33 and 3.34); this sample has a plate- like morphology. All
selected spots of this specimen indicate the presence of (O, V, and P) and
indicate the shell (K, L, M….etc) of these atoms. The weight ratio (P/V)
on the surface varied between (1.032 and 1.708) while atomic ratio for the
same components are ranged between (1.700- 2.811) .knowing that the
absolute designed atomic ratio for all prepared phases was (1.8).
The supported phase sample (No.2- supp) has also a plate –like
morphology with small rhombus of Titania (anatase) (figure 3.35). The
weight ratio of (P/V) is between (1.632 – 1.887) .The minimum atomic
ratio is (2.684) while the maximum value is (3.108).
For VO (H2PO4)2 sample (No.3- un), it has blocky and plate-like
morphology with regular face (Figures 3.36-3.38).
Finally VO (H2PO4)2/ TiO2, sample (No.3- supp), has flat plate with
different shape dispersed of Titania particles as appeared in (figures
3.39- 3.41).
The elemental analyses on the surface of above samples are listed in
(Table 3.22).

Chapter Three – Part One - Results
156
Element Wt % At %
O K 47.15 68.14
P K 26.91 20.09
V K 25.94 11.77
Total 100 100
Figure 3.33: The EDX results of VOHPO4.0.5H2O (sample No.2- un).
Element Wt % At %
O K 58.31 76.86
P K 22.05 15.01
V K 19.64 8.13
Total 100 100
Element Wt % At %
O K 64.64 81.18
P K 19.14 12.42
V K 16.22 6.4
Total 100 100
O K 52.92 72.56
P K 25.79 18.27
V K 21.29 9.17
Total 100 100
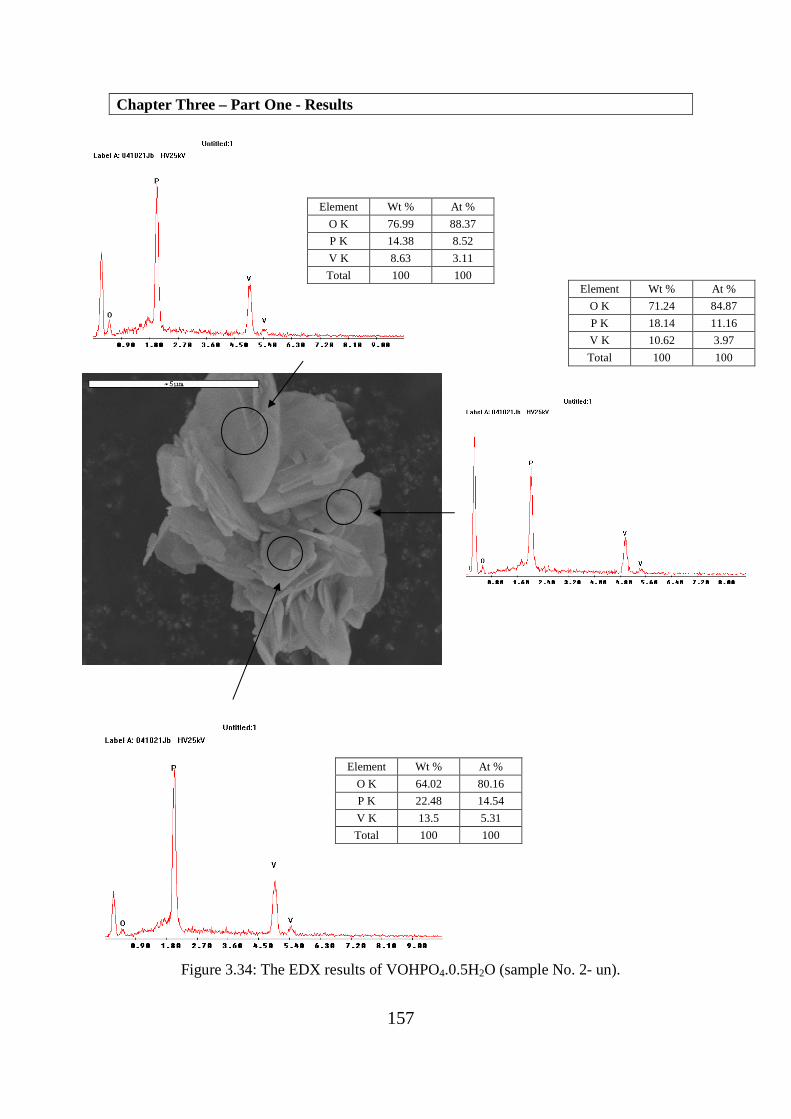
Chapter Three – Part One - Results
157
Figure 3.34: The EDX results of VOHPO4.0.5H2O (sample No. 2- un).
Element Wt % At %
O K 76.99 88.37
P K 14.38 8.52
V K 8.63 3.11
Total 100 100
Element Wt % At %
O K 64.02 80.16
P K 22.48 14.54
V K 13.5 5.31
Total 100 100
Element Wt % At %
O K 71.24 84.87
P K 18.14 11.16
V K 10.62 3.97
Total 100 100
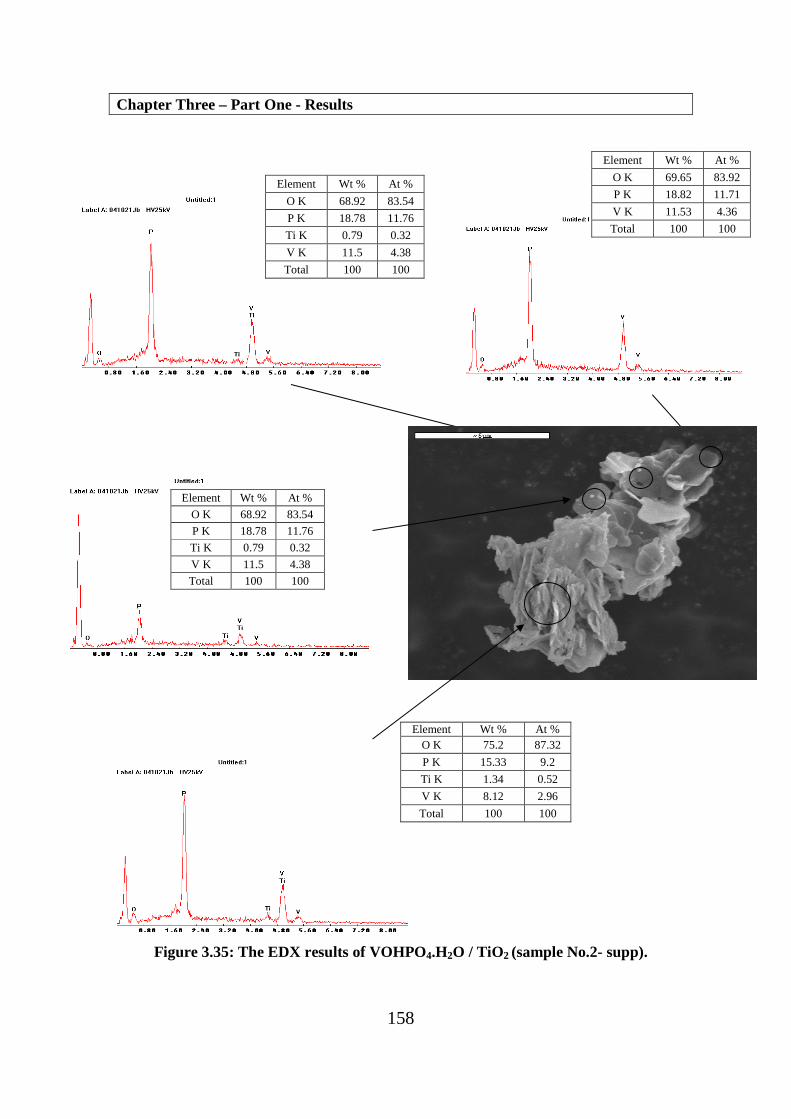
Chapter Three – Part One - Results
158
Figure 3.35: The EDX results of VOHPO4.H2O / TiO2 (sample No.2- supp).
Element Wt % At %
O K 69.65 83.92
P K 18.82 11.71
V K 11.53 4.36
Total 100 100
Element Wt % At %
O K 68.92 83.54
P K 18.78 11.76
Ti K 0.79 0.32
V K 11.5 4.38
Total 100 100
Element Wt % At %
O K 68.92 83.54
P K 18.78 11.76
Ti K 0.79 0.32
V K 11.5 4.38
Total 100 100
Element Wt % At % O K 75.2 87.32
P K 15.33 9.2
Ti K 1.34 0.52
V K 8.12 2.96
Total 100 100
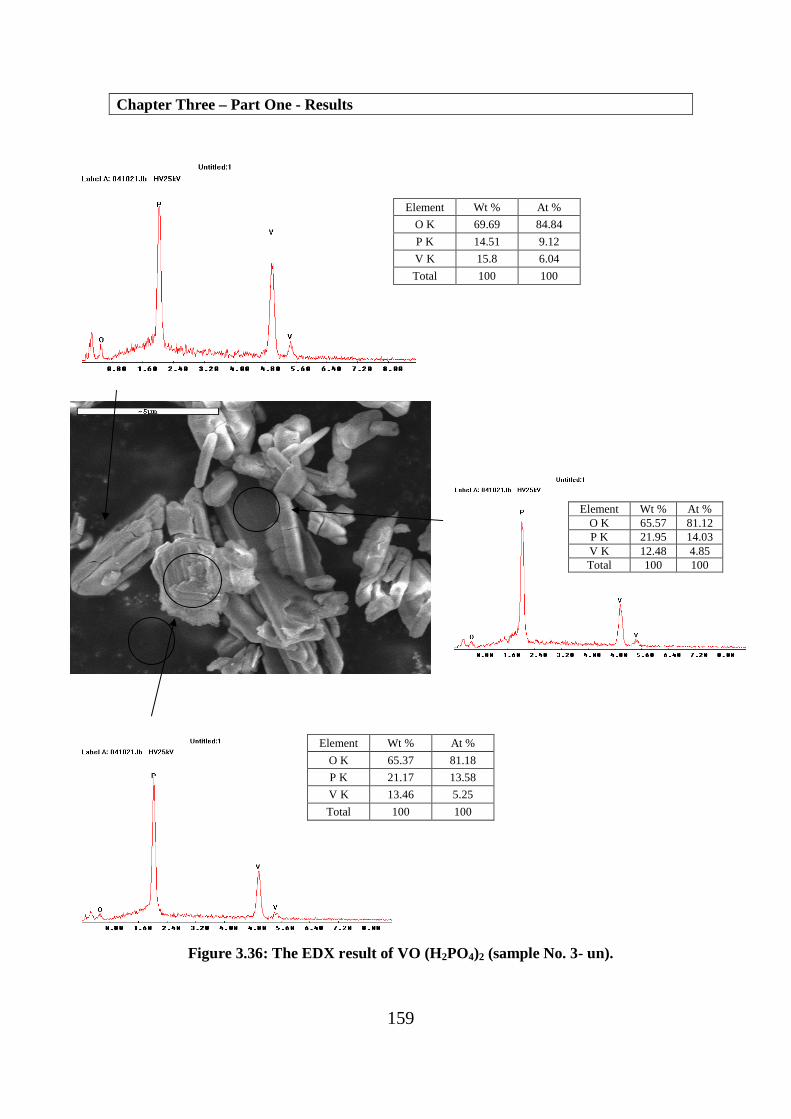
Chapter Three – Part One - Results
159
Figure 3.36: The EDX result of VO (H2PO4)2 (sample No. 3- un).
Element Wt % At %
O K 69.69 84.84
P K 14.51 9.12
V K 15.8 6.04
Total 100 100
Element Wt % At % O K 65.57 81.12 P K 21.95 14.03 V K 12.48 4.85 Total 100 100
Element Wt % At %
O K 65.37 81.18
P K 21.17 13.58
V K 13.46 5.25
Total 100 100
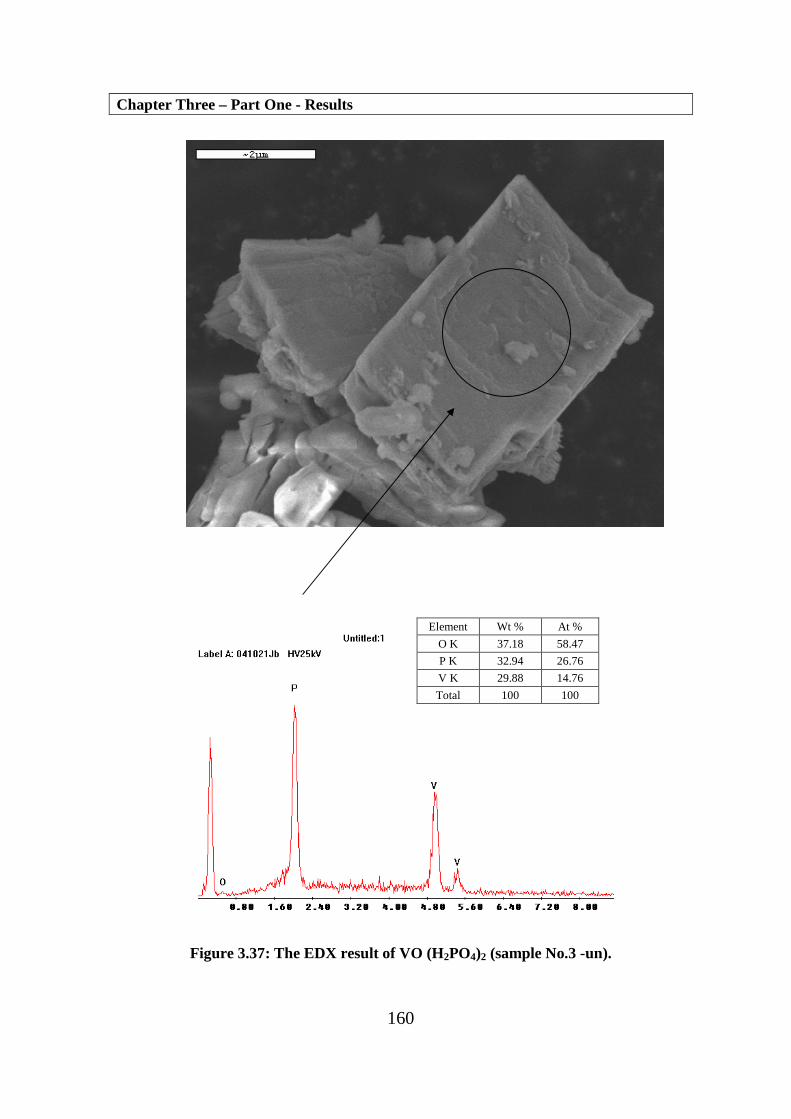
Chapter Three – Part One - Results
160
Figure 3.37: The EDX result of VO (H2PO4)2 (sample No.3 -un).
Element Wt % At %
O K 37.18 58.47
P K 32.94 26.76
V K 29.88 14.76
Total 100 100

Chapter Three – Part One - Results
161
Figure 3.38: The EDX result of VO (H2PO4)2 (sample No. 3- un).
Element Wt % At %
O K 65.66 81.17
P K 21.93 14.01
V K 12.41 4.82
Total 100 100
Element Wt % At %
O K 73.62 87.08
P K 13.02 7.96
V K 13.36 4.96
Total 100 100
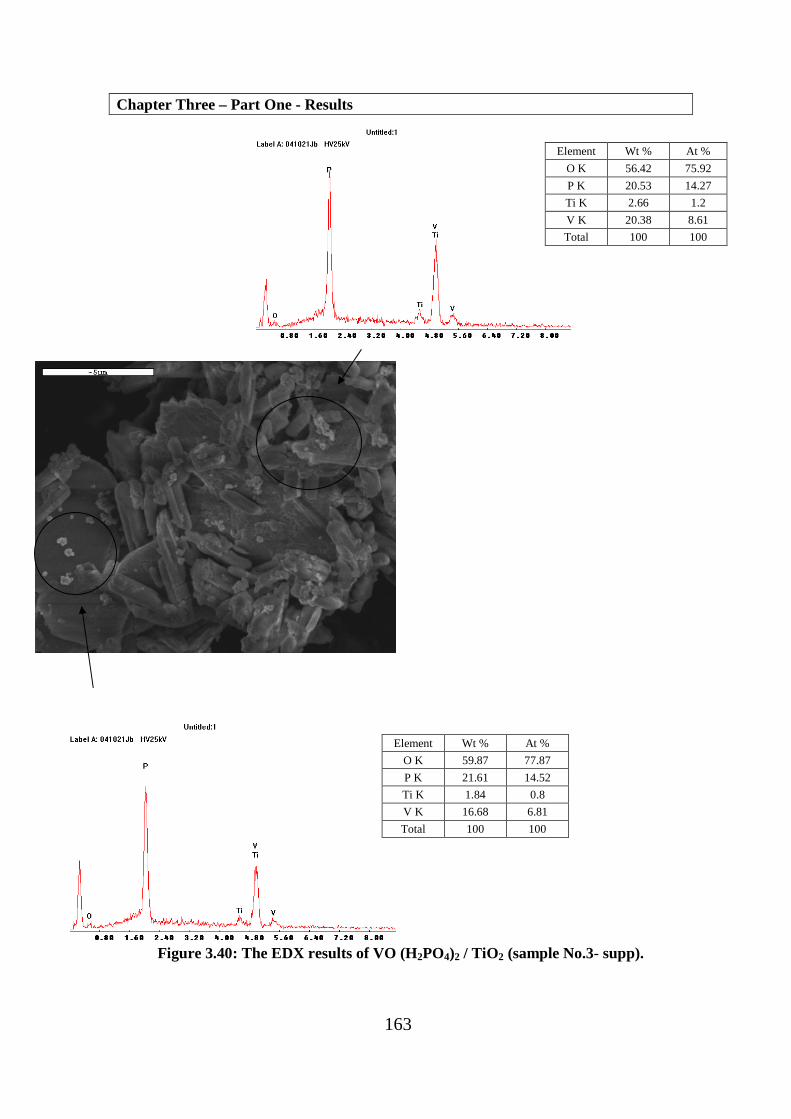
Chapter Three – Part One - Results
163
Figure 3.40: The EDX results of VO (H2PO4)2 / TiO2 (sample No.3- supp).
Element Wt % At %
O K 56.42 75.92
P K 20.53 14.27
Ti K 2.66 1.2
V K 20.38 8.61
Total 100 100
Element Wt % At %
O K 59.87 77.87
P K 21.61 14.52
Ti K 1.84 0.8
V K 16.68 6.81
Total 100 100

Chapter Three – Part One - Results
164
Figure 3.41: The EDX results of VO (H2PO4)2 / TiO2 (sample No.3- supp).
Element Wt % At %
O K 74.24 87.65
P K 11.45 6.98
Ti K 2.62 1.03
V K 11.7 4.34
Total 100 100

Chapter Three – Part Two - Discussion
١٦٧
3.2.1- Effects of Preparation method
Unlike route (A), the essential steps of the preparation of the precursor
vanadyl hydrogen phosphate hemihydrate (VOHPO4.0.5H2O) are listed
below for rout (B):
1- Reduction of V2O5 to (VO 2+) species under reflux in an organic
medium using (benzyl alcohol and isobutanol as a reducing agent.
2- Reaction with o- H3PO4 to form the VOHPO4 .0.5H2O precursor
phase.
3- Separation of this phase by vacuum filtration.
4- Drying overnight at 125°C.
While, the preparation of route (A) depends on the reaction with
Phosphoric acid to produce VOPO4.2H2O before the reduction step with
the mixture of (benzyl alcohol and isobutanol); In some instances already
prepared VOPO4.2H2O (sample No.1un and 1-supp) or a suspension of this
phase was reacted in the manner described to yield and hold the (P/V) ratio
constant at near 1.1
The selection of these routs is related to our previous work (1), since the
product of route (A) (new innovation) exhibited good catalytic activity and
the product of route (B) are recognized as official catalyst for n-butane
oxidation in the literature (21, 57 - 62) . The addition of support material
(TiO2) anatase to the materials of both routes and determine its effects on
their products, is the core of this study
Several effects were observed in the preparation steps of catalytic
precursor phases it may be summarized by these notes:
1 -In rout (A) Scheme 2.1, depending on the observed change in product
color, there is an increase of five hours of required reaction time for
VOPO4.2H2O / TiO2 preparation (16 h for unsupported sample and 21h for
supported one), and there is an increase of 13 hours of required reaction

Chapter Three – Part Two - Discussion
١٦٨
time necessary to prepare VO (H2PO4)2/TiO2 (21 hours for unsupported
VO (H2PO4)2 and 34 hours for supported phase).
In these two cases (supported and unsupported) the final main phases
are the same (vanadyl acid phosphate), but the only difference is in the
accompanying trace phase which is VO (H2PO3)2 .There is also an
additional phase which is VOH2P3O10.2H2O (where V Is in (3+) oxidation
state), exists in the supported phases .These anomaly phenomena can be
attributed to several factors:
A- The dispersion of variant solid quantity in fixed volume quantity of
liquid.
B- The intercalation of TiO2 within the structure of VOPO4.2H2O i.e.
TiO2 intercalate it self within the lamellar layers of VOPO4.2H2O,
prevents the process of organic material intercalation
C- In general the formation of VO (H2PO4)2 was due to conversion of a
mixture of alcoholic reducing agent (isobutanol and benzyl alcohol) to
the corresponding aldehyde and ketone. According to Bartley (81) who
found that the presence of such mixture lead to the formation of
benzoic acid and benzaldehyde which also behave as reducing agent
in place of alcohol, would result in different oxidation products. They
proposed that the involvement of an aldehyde or ketone as a reducing
agent is via the enol tautomer. For most aldehydes and ketones only a
small proportion of the enol tautomer is present. But in the presence of
a Bronsted acid, the equilibrium is more favorable and more enol is
formed. As demonstrated that the ketone and aldehyde operates as
reducing agent in the enol form, VOPO4.2H2O is reacted with
aldehyde and such reaction led to obtain vanadyl acid phosphate
VO (H2PO4)2.

Chapter Three – Part Two - Discussion
١٦٩
The preparation method in route A (this work) is a new method to prepare
vanadyl acid phosphate VO (H2PO4)2, which was previously prepared by
others in different methods (80- 82).
2- In route (B) Scheme 2.1, depending on the observed change in
product color, the reaction produced hydrogen phosphate hemihydrate
(VOHPO4.0.5H2O) in unsupported phase, and produced vanadyl hydrogen
phosphate mono hydrate (VOHPO4.H2O) in supported phase .The effect of
support is directed toward the addition of one single water molecule in the
final product instead of hemihydrate normally expected in this case.
When supporting on TiO2 ,the effect of formation of VOHPO4.H2O
instead of VOHPO4.0.5H2O is related to the competition between H2O
molecules and organic reducing agents, during the reaction, to be
intercalated in the layers formed (in suspended solid already formed,
containing TiO2).Although the X-ray diffraction pattern of these two
phases are identical (Fig.3.1 and Table 3.3) (with slight shift when
containing TiO2) but in thermogravimetric measurement there is a
significant loss of weight ) is observed of unsupported phase (Table 3.7) .
The extra loss of weight indicated that the presence of organic species is
accumulated in such extend in VOHPO4.0.5H2O phase, that they are being
easily expulsed ,this explanation is fortified by the data presented in (Table
3.6) for these two supported and unsupported phases in the range of (50-
230°) C .the loss of weight in the unsupported phase is related to extra
amount of organic molecules intercalated between layers ,there is no such
study of thermogravimetric analysis in the literature concerning the
presence of TiO2 in prepared phases.
The huge departure of organic molecules from unsupported phase is
observed in our previous work (1) even under investigation of this phase in
situ by Scanning Electron Microscope (SEM).The change of the structure

Chapter Three – Part Two - Discussion
١٧٠
is obvious during the inspection due to the evolved heat from the contact
of electronic beam and the sample.
Table 3.5, in rout (B) highlighted the formation of vanadyl acid
phosphate VO (H2PO4)2 in trace quantity accompanying the formation of
VOHPO4.H2O / TiO2.
Table 3.5 reveals that VOHPO4.0.5H2O which is formed without
support contains traces of other phases of (VPO) system. The explanation
of the presence of such phases is given by Bartley (81), as stated before, it is
due to the reaction of the products (aldehyde and ketone) involved from
alcohol oxidation in the precense of small amount of o- H3PO4.The organic
phase (route B) permit the reaction between (benzyl alcohol + isobutanol)
to obtain the reduced phase V2O4 once the H3PO4 is added, the majority of
V2O4 is converted to VOHPO4.0.5H2O in huge quantity. Some traces of
resultant product of alcohol oxidation which present in reaction
environment will convert small quantity of V2O4 to other phases of (VPO)
system. The presence of TiO2 is selective to produce vanadyl acid
phosphate VO (H2PO4)2. In contrary in (route A) the phosphoric acid is
added just in the beginning of the reaction. The small quantity of
phosphoric acid arises in the suspension media direct the reaction quickly
(when adding alcohol as reducing agent) toward the formation of vanadyl
acid phosphate VO (H2PO4)2.
The (P/V) ratio in VOHPO4.0.5H2O is higher than designed ratio
(1.8 atomic ratio) because of presence more than one phase, impurities,
and support which have high electronegativity which led to increase
(P/V) ratio (148 ) .
3.2.2- Promoters, impurities, and Solid Solution effects
The presence of support (TiO2) in 8 % wt percent in the crystal phase
network structure led to cancel the combined effects of promoters (or

Chapter Three – Part Two - Discussion
١٧١
impurities) which already exist in the unsupported phases; like
intercalation effect. Unlike the intercalation, the solid solution effect is still
present. The selection of this support and its percentage is suggested by
published literature (183-186)
The XRD and Raman spectra of the bulk (VPO) precursors which
demonstrate the presence of only vanadyl hydrogen phosphate
hemihydrate VOHPO4.0.5H2O, vanadyl hydrogen phosphate monohydrate
VOHPO4.H2O and vanadyl acid phosphate VO (H2PO4)2 .These bulk
characterization techniques did not detect promoter phases in the bulk
(VPO) since these techniques are blind for traces of impurities (116,187).
The intercalation of the promoters into the layered structure of the
VOHPO4.0.5H2O precursor and the cleavage of its (010) plane should
result in an increase of the surface area and solid solution
formation (1, 172).
Furthermore, the promoter elements in the most active and selective
(Na promoted catalyst) were concentrated in the surface region despite the
low promoter content, which probably indicates higher distribution of the
Na promoter in the (VPO) surface matrix. Therefore, it appears that the
promoter elements may partially form a solid solution in the bulk in the
absence of the support.
However, this process is primarily limited to the surface region and
affects the catalytic properties of the bulk (VPO) catalysts (172).
Thus the obtained results from this work determine the pathway for the
increase of the activity and selectivity of the catalyst for n-butane
oxidation. The increase of activity could be obtained by the introduction in
base (VPO) composition of the element donor, which increases the
effective negative charge on oxygen of catalyst surface, Ti
(electronegativity = 1.6) belong to this type of additives. However, the
content of other additives in the catalysts has been not significant. The

Chapter Three – Part Two - Discussion
١٧٢
introduction of the additives which increase the surface acidity favors to an
increases of the selectivity to products of partial n-butane oxidation, in this
case the influence of additives origin on acidity type did not determine
(and as a result it was very difficult to predict ) the group effective
promoters, which increase the selectivity to definite anhydride.
3.2.3 -Thermal Behaviors (DTA and TGA)
Generally, in all cases; the thermal behaviors included four
distinguished combined elements which correspond to (a) the loss of
adsorbed organic molecules which attributed to the residue of benzyl
alcohol and isobutanol, (b) decomposition of intercalated species, (c) the
loss of water molecules and (d) finally the stage of topotactic
transformation of the precursor to (VO)2P2O7 in route (B) and to
VO (PO3)2 in route (A).
In vanadyl hydrogen phosphate hemihydrate VOHPO4.0.5H2O (sample
(No.2-un), four endothermic phenomena were occurred (Figure 3.2). The
first one is a little endothermic, and it was occurred between (205- 230 °C)
with a loss of weight equal to 8.16 %. The second endothermic
phenomenon happened between (240- 310) °C with a loss of weight 1.73
%.Third endothermic peak is extended from (310 to 510 °C) with loss of
weight 4.90 %. The last one is laid between (500- 580) °C without any
change in the weight. These endothermic peaks followed by an exothermic
peak between (580- 630) °C with gain of weight.
The total loss of weight was (14.79 %), this value is greater than
theoretical loss (10.52%) to obtain vanadyl pyrophosphate, and this
difference could be attributed to intercalated species and to the presence of
inorganic impurities in technical grade V2O5.

Chapter Three – Part Two - Discussion
١٧٣
The vanadyl hydrogen phosphate monohydrate VOHPO4.H2O sample
(No. 2- supp) has different behavior, it has five stages the first one is
neither endothermic, nor exothermic which occurred between (180 – 220)
°C with loss of weight (5.7 %), followed by three endothermic phenomena:
the first is laid between (200- 320) °C occupied with loss in weight
equaled to 2 %. The second endothermic step happened in temperatures
rang between (320 – 510) °C with loss of weight 6.2 %. The last one was
occurred in rang between 510 and 600 °C with no change in final weight.
The total loss was 13.9%.
The gain in weight in the last exothermic peak between (600- 660) °C
was seen .It is obviously that the decomposition of supported sample was
extending to a higher range of temperatures which reflects the grater
resistance of such sample to the heat treatment.
Vanadyl acid phosphate VO (H2PO4)2 sample (No.3- un) shows another
different behavior (Figure 3.4), It started with two little endothermic peak
extended between (80 to 110) °C for the first and between (180 to 245) °C
for the second with loss 6 %; then another loss (0.7 %) was happened in
the range of temperature (277- 350) °C by neither endo nor exothermic
changes. The forth loss (5.3 %) was occurred in (370- 490) °C range, by an
endothermic phenomena. An additional endothermic peak was laid
between (490 – 560) °C without any change in the weight. Finally a little
exothermic peak was laid between (560 – 620) °C with again in weight
(0.99 %).
The theoretical loss of this phase is (13.7 %) (80), which equaled to loss
of two water molecule. The difference in the loss weight between our
sample and the literature could be explained by the presence of trace
amount of other species.
The supported sample showed a different behavior; it dehydrate in the
first endo stage between (110 -180 0 °C with loss of weight 8.71%

Chapter Three – Part Two - Discussion
١٧٤
followed by (neither endo or exothermic) at (255- 340) °C, the loss in this
stage was (0.5 %). The third stage was endothermic stage (350 – 440 °C)
occupied with loss of weight (4.65 %). in the range (470 – 560 °C) the last
endothermic peak was seen it without any change in the weight and finally
the last stage; which was a little exothermic between ( 560 and 620)°C
with gain in weight (0.99 %).The total loss in weight for this sample is
(13.8 %), this result is in agreement with the literature for our product (81) .
The presence of TiO2 in these sample and its behavior affect the presence
of other species, this confirm our suggestion above related to the
competition of intercalated species
The gain in weight in all samples at higher temperature with an
exothermic peak is presumably due to reoxidation of the samples that is
partially reduced on pyrolesis of the adsorbed or intercalated organics (57).
From above presentation ; two ranges of temperature have to be
considered .The first ,from room temperature up to 340 °C corresponds to
the departure of adsorbed species ,and the second stage ,from (340 to
510)°C, to the transformation of the solid .while the first range is
approximately always the same ,with a mass loss occurring in the two
steps at around (50 and 237) °C (Table 3.6) ,the second range differs from
one precursor to another .The loss which may be attributed to the departure
of physisorbed (mixture of isobutanol and benzyl alcohol ) and water are
reported in (Table 3.6) . These results showed that the dopant have a great
effect on the physisorption of water (172). This agrees with our results when
considering the effect of TiO2 on the selective adsorption of both water and
organic molecule to produce vanadyl hydrogen phosphate monohydrate
VOHPO4.H2O.
The topotactic mechanism of dehydration of vanadyl hydrogen
phosphate monohydrate VOHPO4.H2O, vanadyl hydrogen phosphate
hemihydrate VOHPO4.0.5H2O and vanadyl acid phosphate VO (H2PO4)2

Chapter Three – Part Two - Discussion
١٧٥
to (VO)2P2O7 and VO (PO3)2 has been described (57,81,174). The
VOHPO4.0.5H2O retains its water of hydration up to 350 °C. The
departure of these molecules results in vacancy formation at the apex of
vanadium oxygen octahedral. In this stage an electronic rearrangement
proceed with respect to the promoters or TiO2 which corresponds to the
formation of VOHPO4 layers .These layers will join expelling the
constitutional water molecule to form (P2O7) group form (2 HPO4) groups.
This second water departure was observed for VOHPO4.0.5H2O at 430 °C.
Doping with a low percentage 8 % of Titania delays the temperature of
dehydration. Doping with Titania at low percentage also promotes
vanadium oxidation this can be explained by better dispersion of Titania as
evidenced by energy depressive x-ray EDX result (Table 3.22).
The promoting effect of Titania on the oxidation state of vanadium
which increases V (V)/ V (IV) balance on the n-butane / air atmosphere is
highly important to explain the catalytic activity of this dopant in n-butane
oxidation to (MA). This is made possible by the formation of very limited
solid solution of type ((VO) 1-x Ti x). 2 P2O7) (186). This conform the
specificity of this support added in such amount as concluded
previously(183-187).
3.2.4 - FTIR and Raman structural properties
In FTIR spectra, there are large differences between laboratory grade
V2O5 (Fluka) and technical grade V2O5 (Fig. 3.6, Fig. 3.7 and table 3.8).
The FTIR spectra of the derived phases from the technical grade are also
different (Figures 3.10-3.16) these differences are due to:
a- The impurities of original material (Table 3.1).
b- The additional material used in the preparation methods.

Chapter Three – Part Two - Discussion
١٧٦
c- The formation of other phases as previously shown in XRD (section
3.1.2).
Due to the complexity of these above three effects and due to the
absence of the request of single pure phase capable to affect the activity of
the catalyst. These effects are governed by the concentration and quality of
these impurities added, although they are very little. Each element of these
impurities repeats the same effects with the main phases exist.
In addition the presence of TiO2 (8%) which is remarkable percentage
that could cause additional relational effects, now and then; they are
difficult to be clarified by using spectrum of FTIR .The matter is still open
to use several other techniques which can be applied or in accordance with
the main idea exposed.
The effect of these impurities especially (Na) was studied in our
previous work (1), the presence of this element leads to formation a solid
solution and this phenomenon leads, in X-ray, to shift the (2 θ) in a
comparison with the standard samples. The formation of solid solution still
exists in our present work; it is obvious from the comparison of the d-
spacing value which is reported in the literature (table 3.2, 3.3 and 3.4). It
is worthy to say that the effect of impurities in XRD spectrum is differing
from the effect on FTIR.
It is obviously that the technical grade (V2O5) contains amount of
ammonium meta vanadate (NH4VO3) which was indicated according to the
literatures (172,174) by the presence of band at 3197 cm-1 and band
between 1300 and 1430 cm-1 which refer to the stretching frequency of
N-H bond of (NH4) +.
It is clear that technical grade have several different bands which are
not similar to those which shown in Fluka V2O5, these bands are refer to
impurities which already exist in technical grade V2O5 (section 3.1.1).and
it also contained shifted bands in comparison with Fluka V2O5.

Chapter Three – Part Two - Discussion
١٧٧
The obtained data of FTIR of VOPO4.2H2O are in good agreement with
Bordes et al. (29,172).
The spectra of the precursor vanadyl hydrogen phosphate hemihydrate
VOHPO4 0.5H2O prepared in route (B) (Figures 3.13 and 3.14 and table
3.11) characterized by bands near (3174) cm-1 and (3201) cm-1 which
attributed to (υ N-H bond) however the standard sample (prepared from
pure Fluka V2O5. can't exhibit these bands (Figure 3.12) .This indicates the
presence of ammonium metavanadate since the frequency of N-H bond
belongs to ammonium group which is certainly intercalated between
lamellar layers. XPS results also indicates the presence of Nitrogen atom
(fig 3.21-3.24) which is related to of nitrogenous trace compound
This phase exhibited a band centered at (1645) cm -1, such frequency is
typical of water coordination ((in plane) deformation) in agreement with
the crystal structure.
Two bands are appeared at (1409) cm-1 (strong) and (1340) cm-1 in
the measured unsupported sample (No.2-un) the last one does not appear in
the supported sample ,the modification of the structure could lead to the
vanishing of one band of N-H bond .
Some bands (1199, 1107, 1045 and 929) cm-1 appeared in the spectrum
of standard sample and also appear in (1078, 1037and 906 cm-1) for
unsupported sample (No.2-un.) and near (1083, 1075 and 908 cm-1) for
supported sample No.2-supp).All these bands belong to the υ P-O bond
which is being extended between (1200- 900) cm-1 (57).
Meanwhile the υ V=O appeared at frequencies (975, 966, 972) cm-1 in
spectrum of sample (No.2) [reference sample of vanadyl hydrogen
phosphate hemihydrate prepared from V2O5 Fluka], sample No.2-un and
sample No.2-supp) respectively, This band normally appear at 1016 cm-1
in pure Fluka V2O5 and at 966 cm-1 in technical grade V2O5.

Chapter Three – Part Two - Discussion
١٧٨
The spectrum of this phase is also remarked by presence of a week band
centered at (748) cm-1 in unsupported sample (No.2-un), and strong band in
the region of (732) cm-1 in supported sample (No.2-supp) (which exist
originally in the technical grade V2O5 belongs to general bond (M = O)
which refers to the presence of impurities present in technical grade V2O5
(Table 3.1).
The spectrum of vanadyl acid phosphate VO (H2PO4)2 is characterized
by a week band near (3430) cm-1 which corresponding to (υ P-OH) bond
this is in agreement with Villeneuve (80).
The band of N-H bond appears near (3213 and 1418) cm-1 for
unsupported phase and near (3222.8 and 1415.8) cm-1 in the supported
phase, the positions of υ V= O is in good agreement with Bordes et.al.
(1015) cm-1 (172), (1020.3, 1018.9) cm-1 for unsupported and supported
phase respectively
The literature mentioned several bands corresponding to (υ P-O) cm-1
near (1185, 1110 and 910) cm-1 (81,175) in our case only one band appeared
near (1082) cm-1 for unsupported and (1084) cm-1 for supported phase.
This phenomenon indicates the presence of the deformation.
There are two bands centered at (879.5, and 815.5) cm-1 and (838.3 and
812) cm-1 in unsupported and supported phases respectively, these bands
are attributed to δ P-OH and / or δ V-OH. These results are in accordance
with published literature (80,172).
In conclusion the effect of support and intercalation between phases and
the effect of ammonium metavanadate are summarized in the following
points:
1- Slightly shifted bands are resultant of support effect presence.
2- All prepared phases are deformed.
3- The effect of Ammonium ion result from original ammonium

Chapter Three – Part Two - Discussion
١٧٩
metavanadate is clear in the spectrum of technical grade V2O5 and its
derived phases.
4- The impurities existent in technical grade V2O5 has certain influence
on the spectrum of the derived phases.
The dependence only on FTIR technique in explanation of the
unknown bands lead to some risk without the use of XRD, Raman
technique, and XPS in order to identify the prepared phases.
The data [table (3.13, 3.14) Fig (3.17, 3.18)] which rose from laser
Raman spectroscopy is approximated with the literature. Raman
spectroscopy is a useful technique for the identification of a wide range of
substances , solids, liquids, and gases. It is a straightforward, non-
destructive technique requiring no sample preparation. Raman
spectroscopy involves illuminating a sample with monochromatic light and
using a spectrometer to examine light scattered by the sample. Due to the
nature of this technique which is based on regrouping the similar and
different effects on the same bonds behind one single band obtained.
From our Data offered by University of Oklahoma, there are two
different types of vanadate layer, the first at (1023) cm -1 which pointed out
to monovanadate layer, this band are shown in both vanadyl hydrogen
phosphate hemihydrate phase, and vanadyl acid phosphate phase
(supported and unsupported). The second type is a polyvanadate layer at
(810) cm-1, which appeared only in VO (H2PO4)2 (supported and
unsupported).
Generally the Raman spectra of catalysts (81) have a strong broad band
with Raman shift of (935) cm -1, which is characteristic of VO (H2PO4)2,
this band is absent in our data. However, other bands for the same phase
(575, and 900) cm -1 are also missing from our spectra. Iglesia (166) suggest
that the characteristic band of hydrated (5VOx/TiO2) produces too poorly
resolved bands centered at about (800 and 900) cm-1, indicating the

Chapter Three – Part Two - Discussion
١٨٠
presence of polyvanadate species. Raman intensity in the region of (800)
cm-1 may also be due to second- order scattering from TiO2. Upon
dehydration, the broad polyvanadate bands remain, but the band centered
near (900) cm-1 becomes less intense. The appearance of a band at (1022)
cm-1 indicates the formation of monovanadate species. Four bands at (149,
395, 511, and 639) cm-1 are attributable to anatase TiO2, do not change
throughout hydration- dehydration cycles. These suggestions are in
accordance with our results above.
Since the green laser is higher energy than red laser, the distribution of
sodium and oxygen in the space of V2O5 crystalline network structure in
the moment of photon applying will cause the arises of atoms space
distribution more than in red laser , the matter which can be explained
significantly in the disappearance of number of bands (cystallinaty bands)
and the appearance of other bands which are related to vanadium and
phosphors these bands nominated as previously indicated as polyvanadate
layers
The appearance of V=O bond near (984) cm-1 for VO (H2PO4)2 and
near (970) cm-1 in VOHPO4.0.5H2O in week state (in red laser) and at
(1590) cm-1 for VOHPO4.0.5H2O (in green laser) is noted in relation to the
literature which gives higher intensity for this bands. This phenomena
indicate that Raman shows regroup several effects of impurities and
collect these effects in single (may be less sharp) bands. When using green
laser and its higher energy the masked effect (can not be seen with red
laser) would be highlighted. This supports the crystalline structure and
reflected to single sharp band.
Based on a combination of the powder XRD and Raman spectroscopy
studies, it can be concluded that the precursor phases of catalyst comprise
disordered materials.

Chapter Three – Part Two - Discussion
١٨١
2.3.5 - Bulk Oxidation States (Redox Titration)
Our results in section (3.1.6) are in agreement with the literature (68, 92,141)
which indicate that the best catalyst is the one which has an oxidation
number varied between (4.3- 4.6).
It is very clear that the support TiO2 has a positive effect toward the
average oxidation number of vanadium ion and the V (IV) % content i.e. it
decrease the average oxidation number (approached to +4) and increase the
percentage content of V (IV) .
It is clear from the results of redox titration of reaction evolution that
the time of reaction for VO (H2PO4)2 syntheses sample (No.3-supp) which
is (13h for reduction step) is enough for preparation reaction time
2.3.6- Effect of support on the specific surface area
No changes in the values of specific surface area (10 m2 /gm) are
observed in VOHPO4.0.5H2O (unsupported and supported) (Table 3.17A );
this is probably due to the presence of sodium ion (Na) in this phase.
According to Riva et al. (190) the presence of this ion with vanadium
pentoxide (V2O5) leads to the formation of new bond (Na- V); and the
presence of such species decreases the specific surface area. It also may be
seen according to Volta et al.(173) that whereas any increase with the
amount of dopant, at the same time, the surface areas of the solid will
decrease. The effect of support here is to equalize the effect of sodium.
For VO (H2PO4)2 phase, where (Na ion) was dissolved while
preparation (i.e. separated from the suspended solid) in water, which used
in the first stage of the preparation (Scheme 2.1), this lead to increase the
specific surface area for the supported phase (Table 3.17-A).No figures are
reported in open literatures about the supported phase surface area. The

Chapter Three – Part Two - Discussion
١٨٢
result given by this work of supported vanadyl acid phosphate surface area
(12 m2 / gm) is considered as the highest surface area found in the
literature for this phase, the increase of specific surface area has (with
other combined factors ) a great influence on catalytic activity
However, the specific surface area of VOPO4.2H2O/TiO2 as separated
phase (sample No.1- supp) is high (16 m2 /gm) in comparison with the
unsupported sample (11 m2 /gm) (sample No.1-un); In this case, there is
good indication, here, for the effect of support and, the effect washing out
the Sodium ion and the effect of obscenity of adsorbed species (which thy
will be added later on to the reduced phases when using reducing agents).
These results were given by method of (Argon Temperature
Desorption) offered by the National Academy of Science in Ukraine. This
method is unlike BET, did not permit to determine the pore shape or other
characteristics of morphology. It is well known that the pore shape for
unsupported sample by using BET method is cylindrical with two opened
ends. (1)
2.3.7 Acidity Effect
According to Zazhigalove (145) the Lewis centers can effect on the
activity of the catalyst i.e. higher conversion of n-butane to product.
However, the Bronsted centers (BA) can contribute more precisely on the
selective production of Maleic Anhydride (MA).
The acidic properties of phases were defined by adsorption of pyridine
(py) and 2, 6-dimethylpyridine (dmpy) in chromatic variant. This method
permits to determine the total acidity and amount of Bronsted and Lewis
acidity centers.
In comparison between the two types of phases selected in this work
route (A) and route (B), we can remake the jump of Lewis acidity centers

Chapter Three – Part Two - Discussion
١٨٣
(LA) value in the route (A) for supported phase (Table 3. 18) .This is good
indicator and presumption of the catalytic activity while the decrease in
Bronsted acidity centers (BA) will probably affect the selectivity.
The increase in activity (due to the increase in Lewis acidity (LA)
centers) is attributed to the deformation of crystalline network according to
Busca (189). These deformations play great role in the renewal of the
surface while the reaction proceeds, more surfaces is exposed and hence
more reaction could be occurred. i.e., the more surface exposed more
pyridine could be adsorbed and the more acidity exhibition.
According to Busca (189) who discussed this matter thoroughly. These
results can be confirmed by using FT-IR in such distinguished bands of
absorption.
2.3.8 - Surface Chemistry by XPS analysis
X-ray photoelectron spectroscopy (XPS), a surfaces nondestructive
measurement technique, is well suited to study a surface chemistry by
using appropriate depth profiling methods, can yield useful "chemical
depth profiles" by virtue of the strong "chemical shift" in the binding
energy of core – electron levels due to chemical bonding . XPS has been
applied to many different areas of applied and basic research.
Several important results were obtained from XPS measurement offered
by Oklahoma University in this work, maintained in section (3.1.9):
1- Identification of elements already exists in the surface of precursors
like (V, P, O, Ti, and Na). (Table 3.19)
2- Identification of several types of Oxygen species on the surface in
coordination with other element in situ (Fig 3.22 – 3.24).
3- Determination of oxidation state of vanadium ion on the surface (table
3.19). According to Watches (162), Jerry Li (180), Ruitenbeek (186), and Moser

Chapter Three – Part Two - Discussion
١٨٤
and Schrader (188), this value indicates that the average oxidation state of
vanadium on the surface, for all measured samples, is (5+).
4- Determination of atomic concentration of each element on the surface
(Table 3.20). This can lead to the determination of surface P/V ratio. The
surface enrichment in phosphorus which was determined by XPS
measurement; has been also reported on (VPO) catalysts (95).This surface
excess of Phosphorus and Oxygen has been tentatively explained by the
presence of other phases like VO (H2PO3)2 and VH2P3O10.2H2O.
XPS data appeared to be inadequate because surface and bulk
contributions are averaged in the spectra .Furthermore, the atomic
sensitivity factors for vanadium and phosphorus have proven to be not
satisfactory to enable one to quantify the XPS results. However, with
experiments at different incident angles will be able to establish that
surface phosphate enrichment which has occurred in all unsupported
(VPO) (186) .
Our XPS results exhibit no trace of Vanadium (IV) on the surface in
contradictory with the results of Bartley (81) for the catalyst derived from
Vanadyl Acid Phosphate VO (H2PO4)2, who suggest that although the
surface comprises Vanadium predominately in the (V) oxidation state, he
can not completely roll out a contribution from a small surface
concentration of V (IV).
The percentage of Na ion (as well other impurities)is varied between
the measured samples due to the preparation method, it is higher in
VOHPO4.0.5H2O (sample no.2-un) in relation to VO (H2PO4)2 sample
(no.3- un) .It is obvious that Na ion is dissolved in water which used
during preparation of the later sample. The presence of Titania leads to
neglected role of Na ion on the surface (sample no.3-supp).

Chapter Three – Part Two - Discussion
١٨٥
2.3.9- Morphology measurements by SEM
There are several parameters affecting on the morphology of the
prepared phases: P/V ratio, preparation method, and reducing agent.
It is worthy to note that the same phase prepared from technical grade of
V2O5 but with (P/V = 7.3), has a plate-like crystallites (1), which means that
the preparation method and the (P/V ratio) affect on the morphology of the
catalytic phases.
In conclusion there is no effect (in specified picture magnification) is
observed on the morphology for support (Titania anatase) on the
morphology of prepared phases (Figure 3.32).
Liskowski (191) has reported blocky morphology with regular sided of
long crystalline ,the ratio of the sides being 1:1: 2 regardless of the
dimension of crystal .However it is clear that there are not single crystal
phase since the surfaces are marked with many indentations. In contras ,
Vanadyl Acid Phosphate VO (H2PO4)2 prepared by Hutchings (149) using
aldehydes and ketones as reducing agents had needle shaped crystal which
at higher magnification did not reveal the presence of surface indentations.
Hence the solvent used to prepare VO (H2PO4)2 can control the
morphology of the crystallite. Our crystallites morphology which obtained
by alternative method of preparation and also the vanadyl hydrogen
phosphate hemihydrate VOHPO4.0.5H2O obtained by this work the using a
mixture of (benzyl alcohol and isobutanol) were not similar to crystallites
prepared by using aldehyde and ketone prepared by Hutchings (149) .
Scanning Electron images clearly show that the morphology of
VOPO4.2H2O (Figure 3.28) is dominated by large flat platelets with small
particles on the surface these results are in agreement with Bordes (172) and
Liskowski (191).

Chapter Three – Part Two - Discussion
١٨٦
2.3.10 – Identification of elemental composition by EDX
Analysis
EDX analysis stands for energy dispersive X-ray analysis .it is a
technique used for identifying the elemental composition of the specimen
or an area of interested thereof.
The distribution of the atoms on the surface are varied between the
selected spots, this point out the presence of more than one phase on the
surface in each prepared samples.
The inspection of the selected spot leads to conclude that the atoms were
randomly distributed. This observation was concluded from the
examination of closely different spots. This technique, however, has
failed in identification of impurities (192).
The presence of Titania as support facilitates the redistribution of
vanadium on the surface to be more homogenous in sample (No.2-supp),
this opinion must be taken with precaution when Titania percentage in the
selected spot is increased as it led to decrease the vanadium ratio.
The relationship between EDX and SEM:
It is obviously from these images that there are differences in the
crystal type which selected from different spots, but it is difficult to specify
that such differences were related to different phases, this technique is
incapable to identify the phases, so the SEM technique could be helpful
when using X-ray in determining different positions of atoms in crystallites
The distribution of Titania in VO (H2PO4)2 sample No.3-supp is better than
VOHPO4.0.5H2O
One of the uses of EDX is to identify the distribution and coordination
of atoms on the surface in depth of 1 µm i.e. the distribution of atoms
related to the identified phases on the bulk.

Chapter Three – Part Two - Discussion
١٨٧
This can be achieved by a hypothetic method which links the
distribution of atoms on the surface with these on the bulk by a percentage
of related atoms. The method of calculation consists of determining the
average value of P/V, V/O and P/O ratio for each sample and a adopting a
model which can exhibit the fractionate value for each atom. The results
show that the assumed phases on the surface could be something like:
V 1-x P 1-y O 1-z
This assumed method of calculation actually neglects the presence of
water molecule (OH group) and the presence of impurities in the proposed
structure and on the surface .However, for the supported phase the
distribution of titania is non- uniform and arbitrary depending on the
position of examination spot, so can be assumed as adjacent atom not
linked to the above proposed surface structure.
It is obvious that all samples have a higher (P/V) ratio. The increase in
this ratio is attributed to presence of different trace identified phases (95);
the presence of such phases is a normal phenomenon in (VPO) system (171).
2.3.11- Resumption of the catalytic activity
The unsupported phase vanadyl acid phosphate VO (H2PO4)2 had
been examined in terms of catalytic activity in our previous work in
February 2000 (1).
The addition of TiO2 (anatase) to this phase is mainly to determine the
chemical and physical effects (Structural and superficial) on this phase.

Chapter Three – Part Two - Discussion
١٨٨
From above points of discussion of these effects, presumption could be
conducted and catalytic properties could be assumed.
The reactivity of Vanadyl Acid Phosphate VO (H2PO4)2 was
determined in literature by Bartley (81) in June 2000. A detailed study of
n-butane oxidation over (VPO) catalyst derived from in situ activation of
Vanadyl Acid Phosphate VO (H2PO4)2 is described thoroughly .This
study suggests by using detailed powder X-ray diffraction and Laser
Raman Spectroscopy that , the catalyst derived from vanadyl acid
phosphate VO (H2PO4)2 is highly disordered , containing some
transformed (intermediate species) .
However, our method of preparation is quite alternative method to
prepare this phase in regard to the work of Hutching. The above points of
discussion also conclude the formation of disordered material.
According to these common points between these two comparable lines
of work, we can assume certain extra activity and selectivity due to the
presence of support TiO2 (anatase) well dispersed on the surface of the
catalyst as it was indicted by EDX (2.3.10).

Conclusions &
Future Prospects

١٩٠
Conclusions
According to this study we can conclude the following:
1- The possibility of preparing of Vanadyl Acid Phosphate VO (H2PO4)2
from technical grade Vanadium pentoxide, and with designed molar
ratio (P/V= 1.1), as an alternative method never mentioned in the
literature.
2- Trace amounts of co-exist phases are accompanying the main
prepared phase whatever the origin of V2O5 is.
3- The addition of (TiO2 anatase) could change the resultant phases
prepared in route (B), since a monohydrate composition
(VOHPO4.H2O) is obtained, instead of Vanadyl Hydrogen Phosphate
Hemihydrate (VOHPO4.0.5H2O) normally prepared without the use
of support .
4- Vanadyl Acid Phosphate VO (H2PO4)2 is always the same phase
prepared with or without the addition of TiO2. No effect of support is
observed on the preparation of this phase by route (A).
5- The effect of support on the thermal behaviors is obvious on the
prepared phases by both methods (A & B).
6- The presence of wide shift in the bands positions in the FTIR. These
shifts reflect the deformation of the angles and lengths of bonds in all
derived phases from these in technical grade V2O5.
7- The presence of mono vanadate layer in all prepared phases. However
the poly vanadate layers are identified only in the prepared Vanadyl
acid phosphate. This phenomenon is very useful to support the
employment of this phase, as a catalyst in the reaction of n-butane to
Maleic Anhydride. It may indicate a presence of catalytic activity.
8- The oxidation state of vanadium ion changes toward (4+) In the

١٩١
presence of the support.
9- There is a significant increase in specific surface area when adding
support to of intermediate VOPO4.2H2O preparation.
10- A specific surface area values of VO (H2PO4)2 and VO (H2PO4)2 /
TiO2 (10, 11 m2/g) are measured in this work for the first time.
11- The results of (XPS) point out the presence of structural elements
(V, P, O) and detectable percentage of inorganic impurities on the
surface of the prepared phases. They also imply the presence of
Nitrogen (N 1s) which could be attributed to Ammonium
metavanadate (NH4VO3).
12- Presence of several oxygen species in the surface of the prepared
phases.
13- The (P/V) atomic ratio on the surface of both Vanadyl Acid
Phosphate and Vanadyl Hydrogen Phosphate Hemihydrate in the
presence of support is near (1).
14- The Lewis acidity centers value of VO (H2PO4)2 in the presence of
support is doubled; a fact that reflects the good catalytic activity.
15- All phases have a lamellar morphology. Using a mixture of (Benzyl
Alcohol and Isobutanol) led to obtain a fine crystal in Vanadyl
Hydrogen Phosphate Hemihydrate VOHPO4.0.5H2O.
16- Presence of TiO2 in selected spots goes down to the depth of 1 µm
from the surface.
17- The intercalation of organic and /or inorganic species between the
lamellar layers of the catalyst precursor, thus, resulting in low
temperature dehydration transformation to the active phase
(VO)2P2O7 .All the distortion phenomena accompanying this
transformation are due to either the reaction of lattice oxygen with
organic solvents intercalated to produce vacancies, or the

١٩٢
incorporation of inorganic impurities inside the lattice structure
modification intramolcular bonds to produce solid solution.
18- The ability of solid solution formation phenomenon while preparing
the intermediate phase and the derived supported phase in the method
of preparation.
19- Many signs in these conclusions lead to predict a higher catalytic
activity for Vanadyl Acid Phosphate VO (H2PO4)2 prepared in new
method. This work is in accordance with the reported catalytic
activity in the literature.

١٩٣
Future Prospects
1- Studying the effect of catalytic test parameters like flow rate, contact
time, reaction temperature, air/butane mixing ratio on the conversion
and selectivity of n- butane reaction using supported Vanadyl Acid
Phosphate VO (H2PO4)2 in order to perform a feasibility study.
2- Determining the catalyst life time and studying the catalyst
regeneration.
3- Studying the poisoning and sintering of the catalyst.
4- Determining of crystallographic parameters including Millar Indices of
the phases prepared in this work.
5- Drawing a map of the distribution of atoms on the surface of precursor
and the derived catalyst. And localize the exact active spots
responsible for higher activity and selectivity in n –butane reaction to
produce Maleic anhydride.
6- Studying the catalytic reactor design required to support the indicated
chemical, physical, and engineering parameters to develop economical
industrial process.

References
١٩٥
References 1. Al-TA'EE, H.A. M.Sc Thesis, University of Baghdad, Iraq, 2000.
3. Malow, M. Hydrocarbon Process, 1980, 11, 149.
2. Ponder, T. Hydrocarbon Process, 1992,3,33.
4. Felthouse. T.R.; Burnet, J.C.; Michael, S.F; Mummy, M.J; Kuo, Y.
http://jws-edck.interscience.wiley.com.:809/kirk_articles _fs.html.Jan 2001.
5. http://www.the-innovation–group.com/ChemProfiles/MaleicAnhydride,
htm Oct.2003.
6. Robinson, W.D .Ency. Chem. Tech. 1979, 14, 770.
7. John, J.M.; William, A.C. Ency. Chem. Proc. Des. 1988,29,35.
8. Hodnett, B.K. Catal. Rev. Sci. Eng. 1985, 27, 373.
9. Wohlfahrt, k.; Emig, G. Hydrocarbon Process. 1980, 6, 83.
10. Budi, F.; Neri, A.; Stefani, G. Hydrocarbon Process. 1982, 1, 159.
11. Ponder .T. Hydrocarbon Process. 1979, 11, 88.
12. Arnold, S.C.; Sucire, G.D.; Verde, V.; Neri, L. Hydrocarbon Process.1985,
9,123.
13. Varma, R.A.; Saraf, D.A. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 7.
14. Obenaus, F. Droste, W. Ulman's Ency. Ind. Chem. 1985, A-4, 486.
15. Palmer, D.A.; Holzhauer, J.K. U.S. Patent 1982,4 342 699,.
16. McDermott, J.X. U.S. Patent 1979, 4 151 116,.
17. Wellauer , T.P.; Gresswell, D.L. ; Newson , E.J. Chem. Eng. Sci. 1986, 41,
765
18. Heylin, M. Chem. Eng. News 1987, 2, 24.
19. Contractor, R.M. U.S. Patent 1991, 5 021 588.
20. Contractor, R.M. U.S. Patent 1987, 4 668 802.
21. Contractor, R.M.; Bergna, H.E.; Horowitz, H.S. ; Blackstone , C.M ;
Malon, B. ; Torardi , C.C.; Griffiths ; B.; Chowdhry , U.; Sleight, A. Catal.
Today, 1987, 1, 49.

References
١٩٦
22. Hucknall, D.J. Selective Oxidation of Hydrocarbon, Academic Press.
London, 1974.
23. Dadyburjor, D.B.; Jewar, S.S.; Ruckenstein, R. Catal. Rev. Sci. Eng. 1979,
19, 293.
24. Centi, G.; Trifiro, F. Appl. Catal. 1984, 12, 1.
25. Kung, H.H. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25,171.
26. Grasselli, R.K.; Burrington, J.D.; Brazdil, J.F. Faraday Discuss. Chem. Soc.
1981, 72, 203.
27. Centi, G.; Trifiro, F.; Vaccari, A.; Fajonk, G.M.; Teichner, S.J. Bull. Soc.
Chim. Fr. 1981, 7-8, 290.
28. Trimm, D.L. Design of Industrial Catalysis, Elsevier, 1980, 81.
29. Bordes, E.; Courtine, P.J. Catal. 1979, 57, 235.
30. Jordan, B.; Calvo, C. Can. J. Chem. 1973, 51, 2621.
31. Jordan, B.; Calvo, C. Acta Crystallogr. Sect. B. 1979, B 32, 2899.
32. Bahargava, R.N.; Condrate, R.A. Appl. Spectrosc. 1977, 31, 230.
33. Ladwig, G.Z. Anorg. Allg. Chem. 1965, 338, 266.
34. Bordes, E. ; Courtine. P.; Pannetier, G. Ann. Chim. (Paris) 1973, 8,105.
35. Ballutaud. D.; Bordes, E.; Courtine. P.; Mater. Res. Bull. 1982, 17, 519.
36. Bordes, E.; Courtine. P.; Johnson, J.W.; Raminsona, A. Mater. Sci.
Monogr. 1985, 28, 887.
37. Bordes, E.; Courtine. P.; J. Chem. Soc. Chem. Commun. 1985, 294.
38. Tim, S.C. ; Baugherg, J.T. ;Harrison, W.T. ; Dussack ,L.L. ; Jacobson ,
A.J.; Johnson , J.W. Bordes, E. Ionics 1996, 84, 219.
39. Techez, M.; Theobald, F.; Bordes, E. J. Solid State .Chem.1981, 40, 280.
40. Tietiz, H.R. Aust. J. Chem. 1981, 34, 2035.
41. Chernorukov, N.G.; Egorov, N.P.; Korshunov, I.A. Russ. J. Inorg. Chem.
(Engl. Transl.) 1978, 23, 1479.
42. Johnson, J.W.; Jacobson, A.J.; Brody, J.F.; Rich, S.M. Inorg. Chem. 1982,

References
١٩٧
21, 3820.
43. Jacobson, A.J.; Johnson, J.W.; Brody, J.F.; Scanlon, J.C.; Lewansowski,
J.T.Inorg. Chem. 1985, 24, 1782.
44. Johnson, J.W.; Johnston , D.C. ; King , H.E.; Halbert, T.R,; Brody, J.F.;
Goshorn, D.P. Inorg. Chem. 1988, 27, 1646.
45. Huan, G.; Johnson, D.P. J. Solid State. Chem. 1991, 93, 514.
46. Voughey, J.R.; Harrison, W.T.A.; Jacobson, A.J.; Inorg. Chem. 1994, 33,
2481.
47. Harrison , W.A. ; Lim , S.C,; Voughey, J. T. ;Jacobson, A.J.; Goshorn,
D.P.; Johnson, J.W.; . J. Solid State. Chem. 1994, 113, 444.
48. Harrison , W.A. ; Voughey, J. T. ; Jacobson, A.J.; Goshorn, D.P.; Johnson,
J.W.; J. Solid State. Chem. 1995, 116, 77.
49. Harrison , W.A. ; Lim, S.C. ; Dussack, A.L. ; Jacobson, A.J.; Goshorn,
D.P.; Johnson, J.W.; J. Solid State. Chem. 1995, 118, 241.
50. Johnson, J.W.; Brody, J.F.; Alexander, R.M. Chem. Mater. 1990, 2, 198.
51. Huan, G.; Jacobson, A.J.; Johnson, J.W.; Gorcoran, E.W.Chem. Mater.
1990, 2, 91.
52. Huan, G.; Johnson, J.W.; Jacobson, A.J. Chem. Mater. 1990, 2, 719.
53. Huan, G.; Johnson, J.W.; Jacobson, A.J. J. Solid State. Chem. 1990, 89,
220.
54. Huan, G.; Johnson, J.W.; Jacobson, A.J. J. Solid State. Chem. 1991, 91,
385.
55. Huan, G.; Jacobson, A.J.; Johnson, J.W.; Goshorn, D.P.; Chem. Mater.
1992, 4, 631.
56. Huan, G.; Johnson, J.W.; Brody, J.F.; Goshorn, D.P.; Mater. Chem. Phys.
1993, 35, 199.
57. Johnson, J.W.; Jacobson, A.J.; Brody, J.F. J. Am. Chem. Soc. 1984, 106,
8123.

References
١٩٨
58. Leonowicz, M.E.; Johnson, J.W.; Brody, J.F.; Shannon, H.F.; New Sam, J.
M. J. Solid State. Chem. 1985, 56, 370.
59. Bordes, E.; Courtine. P.; J. Solid State. Chem. 1984, 55, 270.
60. Torardi, C.C.; Calabress, J.C. Inorg. Chem. 1984,23,1308.
61. Centi, G.; Trifiro, F.; Poli, G. Appl. Catal. 1985, 19, 225.
62. Busca, G.; Cavani, F.; Centi, G.; Trifiro, F. J. Catal. 1986,99,400.
63. Garbassi, F.; Bart, J.C.; Monitino, F. Petrini, Appl. Catal. 1985, 16, 271.
64. Udovich, C.A.; Edwards, R.C.; U.S. Patent 1986, 4 564 688.
65. Cavani, F.; Centi, G.; Trifiro, F.J. Chem. Soc. Chem. Commun. 1985, 492.
66. Martini, G.; Morselli, L.; Riva, A; Trifiro, F. React. Kinet. Catal. Lette.
1978, 8, 431.
67. Hodnett, B.K.; Delmon. B. J. Catal. 1984, 88, 43.
68. Hodnett, B.K.; Delmon. B. Ind. Eng. Chem. Fundam. 1984, 23, 465.
69. Hodnett, B.K.; Delmon. B. Appl. Catal. 1985, 15, 141.
70. Ai, M. J. Catal. 1986, 100, 336.
71. Pyatnitskaya, A.I.; Komashko, G.A.; Zazhigalov, V.A.; Gorokhovatskii,
Ya. B. React. Kinet. Catal. Lette. 1977, 6, 341.
72. Zhang, Y.; Martin, A.; Wolf, G.; Robs, U.; Worzala, S. Luck, H. B. Chem.
Mater. 1996, 8, 1135.
73. Trifiro, F.; Busca, G.; Centi, G.; Appl. Catal. 1986, 25, 265.
74. Weing, R.W.; Schrader, G.L. Ind. Eng. Chem. Fundam. 1986. 25, 612.
75. Ball, A.; Ferey, G.; Amoros, P.; Villeneuve, G. J. Solid State. Chem. 1989,
79, 169.
76. Shimoda, T.; Okuhara, T.; Misono, M. Bull. Chem. Soc. Jpn. 1985, 58,
2163.
77. Gorbunova, Yu. E.; Linde, S.A. Sov. Phys. Dokl. 1979, 24, 138.
78. Johnson, D.C.; Johnson, J.W.; J. Chem. Soc. Chem. Commun. 1985, 1720.
79. Johnson, J.W.; Johnson, D.C.; Jacobson, A.J. Preprints 4th.International

References
١٩٩
Symposium on the Scientific Vases for the Preparation of Heterogeneous
Catalysts (Louvain – Ia- Neuve, Belgium, Sep. 1986, P 85).
80. Villeneuve, G.; Erragh, A.; Beltran, D.; Drillon, M.; Hagenmuller, P. Matt.
Res. Bull, 1986, 21, 62.
81. Bartly , J.K.; Rhodes, C. ; Kiely ,C.J.; Carley, A.F.; Hutchings , G.J. Phys.
Chem. Chem. Phys. 2000, 2, 4999.
82. Cavani, F; Centi, G.; Trifiro, F.; Poli, G.J. Thermal. Anal. 1985, 30, 1241.
83. Cavani, F; Centi, G.; Riva, A. Trifiro, F. Catal. Today, 1987, 1, 17.
84. Volta, J.C. ; Bergeret, G.; Brover, J.P.; David , M.; Gallezot, P.; Hecquet,
G.J. Chem. Soc. Chem. Commun. 1986, 825.
85. Volta, J.C.; Bergeret, G.; David, M.; Broyer, J.P.; Hecquet, G. Catal.
Today, 1987, 1, 37.
86. Zazhigalov, V.; Pyatnitskaya, A.I.; Bechetokova, I.V.; Komashkoo, G.A.;
Ladwig, G.; Belousov, V.M. React. Kinet. Catal. Lette. 1983, 23, 119.
87. Centi, G.; Trifiro, F.; Fornasari, G. Ind. Eng. Chem. Prod. Res. Dev. 1985,
24, 32.
88. Trifiro, F.; Cavani, F; Centi, G. Appl. Catal. 1985, 15, 151.
89. Trifiro, F.; Morselli, L.; Urban, L. J. Catal. 1982, 75, 112.
90. Trifiro, F.; Centi, G.; Fornasari, G. J. Catal. 1984, 89, 44.
91. Garbassi, F.; Bart, J.C.; Tassinari, R.; Vlaic, G.; Lagard, P. J. Catal. 1986,
98, 317.
92. Buchanan, J.S.; Sundaresan, S. Appl. Catal. 1986, 26, 211.
93. Pepera , M. A.; Callahan, J.L.; Desmond, M.J. ; Milberger, E.C. ; Blum,
P.R.; Bremer, N.J. J. Am. Chem. Soc. 1985, 107, 4883.
94. Bosch, H.; Bruggink, A.A.; Ross. J.R. Appl. Catal. 1987, 31, 323.
95. Centi, G.; Trifiro, F.; Abner , J.R.; Franchetti , V.M. Chem. Rev. 1988,
88,55.
96. Guliants, V.V.; Benziger, J.B.; Sundaresan, S.; Wachse, I.E. Catal. Lette.

References
٢٠٠
1995, 32,379.
97. Benziger, J.B.; Guliants, V.V.; Sundaresan, S. Catal. Today. 1997, 33, 49.
98. Topso, H. Studies in Surface Science and Catal. 2000, 103, 1.
99. Centi, G.; Perathoner, S. Int. J. Mol. Sci. 2001, 2,183.
100. Escardino, A.; Sola, C.; Ruiz, F. An. Quim. 1973, 69, 1157, Summarized in
Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 7.
101. Wohlfahrt, K.; Hofmann, H. Chem. Ing. Tech. 1980, 52,811.
102 Mori, M.; Miyamoto, A.; Murakami, Y. J. Phys. Chem. 1985, 89, 4265.
103. Busca, G. Centi, G.; Trifiro, F. J. Am. Chem. Soc. 1985, 107, 7757.
104. Cavani, F.; Centi, G.; Manenti, I.; Trifiro, F. Ind. Eng. Chem. Prod. Res.
Dev. 1985, 24, 221.
105. Nakamura, M.; Kawi, K.; Fujiwara, Y. J .Catal. 1974, 34, 345.
106. Gleaves, J.T.; Ebner, J.R.; Kuechler .T.C. Catal. Rev. Sci. Eng. 1988, 30,
49.
107. Yang, T.J.; Lunsford, J.H. J. Catal. 1980, 63, 506.
108. Che, M.; Tench, A. J. Adv. Catal. 1982, 31, 77.
109. Nechiporuk, P.P.; Mishchenko, Yu. A.; Avetisov, A.K.; Dulin, D.A.;
Alinovski, I.O.; Gel'bashtein, A.I. Kinet. Catal. 1986, 27, 1304.
110. Khan, M. M.; Somorjai, G. A. J. Catal. 1985, 91, 263.
111. Puttock, S.J.; Rochester, C.H .J. Chem. Soc. Faraday Trans 1,
1986,82,2773.
112. Puttock, S.J.; Rochester, C.H .J. Chem. Soc. Faraday Trans 1,
1986,82,3013.
113. Munson, E.; Chen, B.; J. Am. Chem. Soc. 2002,124, 1638.
114. Ueda, W; Moro-Oka, Y.; Ikawa, T. J. Chem. Soc. Faraday Trans. 1, 1982,
78, 495.
115. Glaeser, L. C; Brazdil, J.F.; Hazle, M.A.; Mehicic, M.; Grasselli, R.K. J.
Chem. Soc. Faraday Trans. 1, 1982, 78, 495.

References
٢٠١
116. Askander, J. Ph.D Thesis, University of Technology of Compiegne, France,
1982.
117. Taufiq- Yap, Y.H.; Sakakini, B.H.; Waugh, K.C. Catal. Lette. 1997, 48,
105.
118. Beg, S.K.; Rao, M.S.; Delichere, P. Catal.Today 1991, 30, 1819.
119. Abon, M.; Bere, K.E.; Delichere, P. Catal. Today 1997, 33, 15.
120. Contractor, R.M.; Horowitz, H.S.; Sisler, G.M.; Bordes, E.; Catal. Today
1997, 37, 51.
121. Rodemerk, B.; Kubias, H.; Zanthoff, H.W.; Baerns, M. Appl. Catal. 1997,
53,203.
122. Zazhigalov, V.A.; Zaitsev, P.Yu. Belousov, N.; Wyustnek, W.H. React.
Kinet. Catal. Lette. 1984, 24, 375.
123. Wang, D.; Barteau, M.A. Catal. Lette. 2003, 90, 7.
124. Weiss, F.; Marion, J.; Metzger, J.; Coginon, J.M. Kinet. Catal. 1973, 14, 32.
125. Puttock, S.J.; Rochester, C.H. J. Chem. Soc. Faraday Trans. 1, 1986, 82,
3023.
126. Busca, G.; Centi, G.; Trifiro, F.; Lorenzelli, V. J. Phys. Chem. 1986, 90,
1337.
127. Rozhkova, E.V.; Gerei, S.V.; Gorokhovatskii, Ya. B. Kinet. Catal. 1975,
15, 618.
128. Baron, B.J.U.S. Patent 1981, 4 251 390.
129. Schineder, R.A. U.S. Patent 1975, 3 864 280.
130. Schineder, R.A. U.S. Patent 1977 , 4 043 943.
131. Ai. M. Chem. Abstr. 1971, 75, 117918 V.
132. Ai. M. J. Appl. Catal. 1986, 28, 223.
133. Lemanski , M . F .; Bremer, N . J.; Milberger, E.C. U. S. Patent 1980,
4 293 498.
134. Udovich, C.A. Bertolacini, R.J. U.S. Patent 1982,4 328 126.

References
٢٠٢
135. Ebner, J.R.; Gleaves, J.T. U.S. Patent 1986, 4 626 412.
136. Edwards, R.C.; Meyers, B.L. U.S. Patent 1987,4 652 543.
137. Zazhigalov, V.A Kinet. Catal. 1986, 23, 364.
138. Zazhigalov, V.A; Konovalova, N.D.; Stock, E. Kinet. Catal. 1988, 29,364
139. Raminsona, A.; Bordes, E.; Courtine, P. J.Solid State Chem. 1987, 68, 1.
140. Zazhigalov, V.A; Haber, J.; Stoch ,J.; Pyatnitzkaya ,A.I.; Komashko, G.A.;
Belousov, V.M. Appl. Catal. 1993, 96,135.
141. Cheng, W. Appl. Catal. 1996, 147, 55.
142. Guliants, V.V.; Benziger, J.B.; Sundaresan, S.; Wachse, I.E.; Jehng, M.
Catal. Lette. 1996, 28,275.
143. Sajip, S.; Bartley, J.K.; Burrows, A.; Sananés – Schulz, M.; Tuel ,A.; Volta,
J.C. ; Kiely, C.J.; Hutching, G.J. New J. Chem. 2001, 25,125.
144. Taufiq- Yap, Y.H.; Tan, K.C.; Waugh,K.C.; Hussein, M.Z. ; Ramil, I. ;
Abdul Rahman, M.B. Catal. Lette. 2003,98,87.
145. Zazhigalov, V.A; Yaremenko, V.E.; Ayub, I.; Su, D.; Willinger, ;
Kharlamov.; Ushkalov.; Schlogle, R.DGMK- Conference , Munich 2004.
146. Stefani, G. Fontana, P. U.S. Patent 1978, 4 085 122.
147. Katsumoto, K.; Marquis, M. U.S. Patent 1979, 4 132 670.
148. Horowitz, H.S.; Blackstone, C.M.; Sleight, A.W.; Tufer, G. Appl. Catal.
1988, 38,193.
149. Bartley, J.K.; Sanchez, J.A.L.; Hutching, G.J. Catal. Today 2003, 81, 197.
150. Kamiya, Y.; Ueki. S.; Hiysohi, N.; Yamamoto, N.; Okuhara, T. Catal.
Today 2003, 78, 281.
151. Bartley, J.K.; Kiely, C.J.; Wells, R.P.K.; Hutching, G.J. Catal. Lette. 2001,
72, 99.
152. Buchanan, J.S.; Apostolakis, J.; Sundaresan, S. Appl. Catal. 1985, 19, 65.
153. Centi, G.; Trifiro, F. Catal. Rev. Sci. Eng. 1986, 28,165.
154. Martine, G.; Trifiro, F.; Vaccari, A.J. Phys. Chem.1982, 86, 1573.

References
٢٠٣
155. Brkic, D.; Trifiro, F. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 333.
156. Varma, R.L.; Saraf, D.M. J. Catal. 1978, 55,361.
157. Cavani, F.; Centi, G.; Trifiro, F. Ind. Eng. Chem. Prod. Res. Dev. 1983, 22,
570.
158. Centi, G. Catal. Today 1993, 16, 5.
159. Hodnett, B.K. Catal. Today 1987, 1, 477.
160. Owens, L.; Kung, H.H. J. Catal. 1993, 144, 202.
161. Guliants, V.V. Catal. Today 1999, 51, 255.
162. Watchs , I.E.; Jehng, J.M.; Deo, G.; Weckhuysen, B.M.; Guliants, V.V.;
Benziger, J.B. ; Sundaresan, S.J. Catal. 1997, 170, 75.
163. Deo, G.; Watchs, I.E. J. Catal. 1994, 146, 323.
164. Deo, G.; Watchs, I.E. J. Catal. 1994, 146, 335.
165. Ledoux, M.J.; Crouzet, C.; Pham- Huu, C.; Turines, V.; Kourtakis, K.;
Mills, P.L.; Lerou, J. Catal. 2001, 203, 495.
166. Olthof, B.; Khodakov, A.; Bell, A.T.; Iglesia, E. J. Phys. Chem. B 2000,
104, 1516.
167. Busca, G.; Lorenzelli, V.J. Mol. Catal. 1989, 50, 231.
168. Agaskar, P.A.; DeCaul, L.; Grasslli, R.K. 1994, 23, 339.
169. Guliants, V.V.; Benziger, J.B.; Sundaresan, S.; Wachse, I.E.; Jehang, J.M.
Catal. Today. 1996, 28,275.
170. Zazhigalov, V.A; Haber, J.; Stoch ,J.; Bacherikova, I.V.; Komashko , G.A.;
Pyatnitzkaya ,A.I.; Appl. Catal. 1996, 134,225.
171. Centi, G.; Nieto, J.L.; Ungarelli, F.; Trifiro, F. Catal. Lette. 1990, 4,309.
172. Bordes, E. Thesis, University of Technology of Compiegne, France, 1980.
173. Martin, V.; Miller, J.M.; Volta, J.C.J. Thermal Anal. 1998, 86, 731.
174. Silverstin, M.R; Webster, X.F. Spectrometric Identification of Organic
Compounds, John Wiley and Sons Inc. 1998.
175. Machei, T.; Ruiz, P.; Delmon, B. J. J. Chem. Faraday Trans 1990, 86, 731.

References
٢٠٤
176. Nakamoto, K. Infra- Red and Raman Spectra of Inorganic and Coordination
Compounds, P.B John Wiley and Sons Inc. 5 th.1997.
177. Scharf, U.; Schneider, M.; Baiker, A.; Wokaun, A.J. Catal. 1994, 149, 344.
178. Abelo, Li; Husson, E.; Replin, Y.; Lucazau. U.G.; Spectrochim. Acta. 1983,
39,641
179. Went, G.T.; Oyama, St.; Bell, A. J. Phys. Chem. 1990, 94, 240.
180. Dr. Jerry, Li, University of Oklahoma, U.S.A.; Personal Communication
2005.
181. Abdelouahab, F.B.; Olier, R.; Guilhanme, N.; Lefebrre, F.; Volta, J.C.J.
Catal. 1992, 134, 151.
182. Overbeek, R.A.; Warringa, P.A.; Crombag, M.J.D.; Visser L.M.; Van
Dellen, A.J.; Geus, J.W. Appl. Catal. 1996, 135, 209.
183. Overbeek, R.A.; Pekelharing, A.R.C.J.; Van Dellen, A.J.; Geus, J.W. Appl.
Catal. 1996, 135, 231.
184. Ruitenbeek, M.; Van Dellen, A.J.; Koningsberger D.C.; Geus, J.W. Studies
in Surface Science and Catalysis 1998, 118, 549.
185. Ruitenbeek, M.; Van Dellen, A.J.; Barbon, B.; Van Faassen, E.E.;
Koningsberger D.C.; Geus, J.W. Catal. Lette. 1998, 55,133.
186. Ruitenbeek, M. Ph.D Thesis, University of Utrecht, Holland, 1999.
187. Askander, J. DEA Report, University of Technology of Compiegne, France,
1978.
188. Moser, T.P.; Schrader, G.L. J. Catal. 1985, 92, 216.
189. Busca, G.; Lavalley, J. Spectrochim Acta.1986, 42A, 443.
190. Riva, V.; Martítn, C. J. Catal. 1988, 114, 473.
191. Liskowski, A.; Su, D.C.; Schlogl, R.; Bartly, J.K.; Hutchings, G.H; Macros.
Micronal 2003, 9, 316.
192. www.semiconfast.com/edx.htm. June 2005.

References

.ھــــ�� ��ـــ�هللا �ـــ�� �ــ�ــ� ا�ـــ��ـــ� : ط�� ا����راه
�� : ���ان ا�ط�و�ـ�����ـ ) ا��"�ز ("! ��ات ا����ـ�( ا)ـ'��ـ ص ا%ــ�$�ــ وا���ــ ).2%�6ر أو�%4ـ�3 –�2�د�0م ( �ـ/�م ا-ـ�ا,ـ+ ا�ـ%���ة
. ٢٠٠٥/ ٨/ ٣: "�ر90 ا���78
.٣٧د – ٣٥ز – ٨٥٣م –�ــ� ا�!,ـــ�A– Bــ@�اد : ا%?3 ���ان
D"�Eا B8٥٥٩٢٠٤٠: ر
٠٧٩٠١٧٣٠٧٥٥: رB8 ا�4ال
�� [email protected]: ا���0 ا�?��و

Effects of Titania (Anatase) on
Structural & Superficial Properties
of
(VPO) Catalytic System
A thesis
Submitted to the College of Science
Al-Nahrain University
In partial fulfillment of the requirements for the
Degree of Doctor of Philosophy of Science in
Chemistry
By
Hind Abdullah Ali Hammo AL-TA'EE
B.Sc. Baghdad University 1987
M.Sc. Baghdad University 2000
June 2005

Special Dedication
To
My country, My Family,
And To My supervisors

III
We certify that this thesis was prepared under our supervision at Al-
Nahrain University / Collage of Science as a partial fulfillment of the
requirement for the degree of Ph.D in Chemistry.
Signature : Signature :
Name : Dr. Jalal A. Askander Name : Dr. Shahbaz A. Maki Address : ALFRED ENGINEERING Address : Chemistry Department Date : / /2005 Date : / /2005
Supervisor Supervisor
In view of the available recommendations, I forward this thesis for
debate by the examination committee.
Signature : Name : Dr. Shahbaz A. Maki
Address : Chairman of Chemistry Department Date : / /2005

IV
We certify that we have read this thesis and as examining
committee examined the student (Hind A. Ali AL-TA'EE) in its
content and that in our opinion it is adequate for the partial fulfillment
of the requirement for the degree of Ph.D in chemistry, with ( ) .
Signature : Signature :
Name : Dr. Moayyed G .Jalhoom Name : Prof. Falih H. Musa Title :Chief of Scientific Researchers Title : Professor
(Member)
(Member)
Signature
:
Signature
:
Name :Dr. Samir M. Jasim Name : Dr. Ramzi R. Ali Al-Ani
Title : Assistant Professor Title : Assistant Professor
(Member)
(Member)
Signature:
Signature:
Name: Dr. Jalal A. Askander Name: Dr .Shahbaz A. Maki Title: Senior Scientific Researcher Title: Assistant Professor
(Supervisor) (Supervisor)
Approved by the council of Collage of Science
Signature : Name : Laith A. Al-Ani
Address : Dean of the College of Science – Al-Nahrain University Date :
Signature :
Name : Prof. Saadoon A. Isa
Title : Vice president of Scientific affairs – Al-Nahrain University
(Chairman)

V
Acknowledgements
I would like to express my deep appreciation and sincere gratitude to my supervisor
Dr. Jalal A. Askander for his suggestions of this work, for his valuable guidance
constructive criticisms, and encouragement during the development of this work and in
the preparation of this thesis.
I wish to express my gratitude and thanks to Dr. Shahbaz A. Maki for his assistance
and supervision during this work and during the preparation of this thesis.
I also would like to thanks Dr. Ameer A. Ameer for his supervision assistance at the
first period of this work.
My thanks go to the Chairman and the staff of the department of Chemistry, College
of Science (Al- Nahrain University) for their encouragement. My deep thank to Dr.
Sawsan Harith for her explanations for FTIR results. My thanks to Mrs. Nasreen and
Mrs. Yassmen for their help in performed FTIR analysis
Deep thanks to Prof. Valery Zazhigalov the head department of heterogeneous
catalysis at The National Academy of Science of Ukraine for his help by making several
analysis (DTA, SSA, and Acidity miscreants, EDX and his generosity by sending the
samples to Germany in order to perform XRD there), I also would like to appreciate his
help by sending some of his published papers.
I would like to express my deep thanks to Prof. Danielle Resasco and Dr. Jerry Li at
University of Oklahoma – U.S.A, for there help in making XPS and Raman analysis and
sending some important papers.
My deep thanks for Mr. Dahfar S. Al-Ani and Dr. Hani Al-Kadi and Dr. Hussein Al-
Hindawi at American University of Sharjah for there help in making SEM analysis.
I would like to thanks Prof. E. Bordes (CNRS laboratories –University of Lille France)
and Prof. Jozef Kowalewski (Stockholm University - Sweden) for their supports and
invitations to perform part of this work in their laboratories in future
My appreciates to Prof. G. Hutchings (Chairman of Chemistry Department at
Cardiff University- England), Dr. M. Ruitenbeek (DSM Centre – Holland) and Prof.
Sleight, A. (U.S.A), Prof. V. V. Guliants (Cincinnati University – U.S.A), and Prof. G.
Clarizia (research Institute on Membrane Technology, ITM - CNR ,former IRMERC-CNR-
Italy) for their assistance by sending many useful published papers.
Finally my deep thanks and appreciate to everyone who helps me in this work.
Hind

VI
Symbols and Abbreviations
VPO = Vanadium – Phosphorus – Oxygen. MA = Maleic Anhydride. XRD = X-ray diffraction. ICP = Inductively Coupled Plasma. ESCA = Electron Spectroscopy Chemical Analysis. SEM = Scanning Electron Microscope. BET = Bruner – Emit – Teller. TEOS = Tetra ethyl o- silicate. EXAFS = Energy X-ray absorption fine structure. TOF = Turnover frequency = number of molecules
converted per Vanadium per second. ▼ = Main phase. ■ = Trace amount. SSA = Specific surface area. LA = Lewis acidity centers. BA = Bronsted acidity centers. XPS = X-ray photoelectron spectroscopy. Py = Pyridine. Dmpy = 2, 6- Dimethyl Pyridine. un = Unsupported. supp = Supported. A = Anatase. TG = Thermogravimetric analysis. DTA = Differential thermogravimetric analysis. Endo = Endothermic. Exo = Exothermic. T.G = Technical grade. S.S = Solid solution. AV.OX.No.Of V = Average oxidation number of Vanadium ion. eV = Electron volt. (s) = On surface. EDX = Energy dispersive X-ray analysis.

XI
Contents Page No. Certificate III Acknowledgement V Symbols and Abbreviations VI Summary VII Contents XI List of tables XIII List of figures XV List of scheme XV
VIII Chapter One
Introduction 1.1 Background 2 1.1.1 Processes for the production of MA from n-butane 3 1.2 Scope of the literature survey 9 1.3 Structure of the catalyst 11 1.3.1 Crystalline (VPO) phases 11 1.3.2 Active phase of n–butane selective conversion of MA 24 1.4 Kinetic of n-butane oxidation 28 1.5 Surface modifications by interaction of n-butane with
catalyst surface 33
1.6 Relationship between redox properties of catalytic behavior 38 1.7 Mechanism of n-butane activation 41 1.8 Role of adsorbed and lattice oxygen species 46 1.9 Nature and mobility of adsorbed species 50 1.10 Promoted (VPO) catalyst 51 1.11 Preparation of the catalyst 56 Conclusion of bulk (VPO) catalyst 60 1.12 Supported (VPO) catalyst 61 1. 12.1 Molecular structure of Vanadia species in fresh catalyst
molecular 62
1.12.2 Molecular structure in dehydrated state 64 1.12.3 Structural changes during hydrocarbon oxidation 66 1.12.4 Structure–activity relationships in oxidation of C-4
hydrocarbon 67
1.12.5 Role of surface oxygen species in supported catalysts 69 1.12.6 Role of acidity of supported vanadia catalyst 76 1.12.7 Role of Support 77

XII
Contents Page No.
Conclusions 78 Aim of work 81
Chapter two Experimental Part
2.1 Samples analysis 83 2.2 Chemicals 87 2.3 Preparation 88 2.3.1 Route A 88 2.3.2 Route B (VOHPO4.0.5H2O) 90 2.4 Redox titration 93
Chapter three Results and Discussion Part One ( The results)
3.1 Results 98 3.1.1 The elemental analysis of technical grade V2O5 98 3.1.2 Identification of prepared phases by XRD 199 3.1.3 Thermal analysis 104 3.1.4 Fourier Transform Infra-Red (FT-IR) spectrum 111 3.1.5 Laser Raman Spectroscopy 128 3.1.6 Redox titration 132 3.1.7 Specific surface area measurement 135 3.1.8 Surface acidity measurement 136 3.1.9 X-ray photoelectron spectroscopy (XPS) analysis 138 3.1.10 Scanning Electron Microscopes (SEM) analysis 149 3.1.11 Energy dispersive X-ray (EDX) analysis 155
Part Two (Discussion) 3.2.1 Effect of preparation method 167 3.2.2 Promoters, impurities, and Solid Solution effects 170 3.2.3 Thermal behaviors (DTA and (TG) 172 3.2.4 FT-IR and Raman structural properties 175 3.2.5 Bulk oxidation state (Redox titration) 181 3.2.6 Effect of support on the specific surface area 181 3.2.7 Acidity effect 182 3.2.8 Surface chemistry by XPS analysis 183 3.2.9 Morphology measurement by SEM 185 3.2.10 Identify the elemental composition by using EDX 196 3.2.11 Resumption of the catalytic activity 197 Conclusions 190 Future prospects 193 References 195

XIII

XII
List of Tables Fig. No. Title Page No. 1.1 World MA capacity (in metric tons) by reactor type. 6 1.2 Total world production and consumption of MA (X10 3
metric tons). 6
1.3 Number of electrons and oxygen molecules involved in some principle reactions of industrial intersect in selective oxidation
8
1.4 The effect of metal oxide support in n-butane oxidation on supported Vanadia catalysts at 494 K in 1.2 Vol.% n-butane.
58
1.5 Performance of titania support in n-butane oxidation at 479 K in 1.2 Vol.% n-butane in air.
63
1.6 The effect of acidic promoters on n-butane oxidation on the 1% V2O5 /TiO2 catalyst at 497 K in n-butane in air.
63
2.1 Chemical used in this work. 73 3.1 Elemental analysis of technical grade V2O5 used in this
work 82
3.2 VOPO4.2H2O (sample No.1-un) X-ray diffraction data. 84 3.3 VOHPO4.0.5H2O and VOHPO4.H2O samples (No.2-un and
2-supp) X-ray diffraction data. 85
3.4 VO (H2PO4)2 (sample No.3-un and 3-supp) X-ray diffraction data.
86
3.5 Identification of prepared phases by XRD. 87 3.6 DTA analysis results of precursors. 92 3.7 The comparison between theoretical and experimental loss of
weight % for catalytic phase. 93
3.8 Fourier Transform Infra-Red data of V2O5. 97 3.9 Fourier Transform Infra-Red data of supported V2O5. 100 3.10 Fourier Transform Infra-Red data of VOPO4.2H2O. 103 3.11 Fourier Transform Infra-Red data of VOHPO4.0.5H2O and
VOHPO4.H2O. 107
3.12 Fourier Transform Infra-Red data of VO (H2PO4)2. 110 3.13 Raman shift carried out by Red laser. 113 3.14 Raman shift carried out by Green laser. 114 3.15 Results of redox titration 115 3.16 Results of redox titration during time of reaction for
supported VO (H2PO4)2. 116
3.17-A Specific surface area for the catalytic phases. 118 3.17-B Specific surface area for VOPO4.2H2O 119 3.18 Data of acidity centers on the surface of catalytic phases 120 3.19 The binding energy of elements on the surface of catalytic
phases. 122
3.20 Atomic concentration of the elements on the surface of the catalytic phases.
129
3.21 The atomic ratio of the elements on the surface of the catalytic phases.
130
3.22 EDX results of the catalytic phases. 147

XIII
List of Figures Figure No. Title Page No.
1.1-A Structure of VOPO4.2H2O showing infinite layers of PO4 tetrahedral linked to VO6 octahedral
10
1.1-B Structure of VOPO4.2H2O showing bond distance °(A) and angles (degree)
11
1.2 Structure of α- and β- VOPO4 12 1.3 Comparison between structure of α- and β- VOPO4 13
1.4 Crystal structure γ-VOPO4
A- 010 plane B- 100 plane
14
1.5 Crystal structure of VOHPO4.0.5H2O (010) plane 15
1.6 View of the infinite double chains of (VO6) octahedra and (PO4) tetrahedra running along (100) direction in VOHPO4.0.5H2O
15
1.7-A Projection of the structure of β- VOHPO4.2H2O along (100)
17
1.7-B Projection of the structure of β- VOHPO4.2H2O along (010)
17
1.8 Idealized structure of (VO)2P2O7 (020) pane 18 1.9 Crystal structure of (VO)2P2O7 (020) plane. 18
1.10 Pseudomorphizm relations between the crystal structure of VOHPO4.0.5H2O and (VO)2P2O7.
20
1.11 Crystal structure of VO (H2PO4)2 21
1.12 Termination of the (200) plane of (VO)2P2O7 and proposed active sites for oxidation
24
1.13 Models of the ideal Langmuir surface 26
1.14 Comparison of the selectivity dependence on acrylonitrile in propane and propane ammoxidation at 480°C on VSb4 + Sb2O4.
29
1.15
Selectivity in propane formation from propane on (VO)2P2O7 as a function of the inlet between oxygen and propane concentration for two entail concentration of propane in the feed reaction temperature 322 °C
30
1.16 Effect of the presence of different valance states of Vanadium on the principle X-ray diffraction lines of Vanadyl pyrophosphate (P/V = 1.0)
32
1.17-A Maleic Anhydride yield Vs conversion at 300 °C in n-butane oxidation for different catalysts with (P/V = 1.0) feed: 0.6 n-butane, 12 % O2.
33
1.17-B Rate of V (IV) oxidation to V (V) in air for the same catalysts (A).
34
1.18 Model of Lewis acid sites (VO)2P2O7 with different degrees of disorder in the stacking fold of (020) planes.
38
1.19 Proposed mechanism of n-butane activation on (VO)2P2O7
39
1.20 Reaction network in n-butane oxidation on (VO)2P2O7 40
Figure No. Title Page No.

XIV
1.21 The three types of oxygen species 41 1.22-A Proposed reaction of n-butane to MA 43 1.22-B Proposed reaction of n-butane to MA 49 1.23 Synthesis routes for Vanadyl Pyrophosphate catalysts 51
1.24 Molecular structure of dehydrated isolated and polymerized vanadia species on oxide supports.
54
1.25 Oxidation of n-butane on dispersed Vanadia species. 57
1.26-A In situ Raman spectra of 1% V2O5 / TiO2 catalysts during n-butane oxidation.
60
1.26-B In situ Raman spectra of 7% V2O5/SiO2 catalysts during n-butane oxidation.
60
1.26-C In situ Raman spectra of 7% V2O5/Nb2O5 catalysts during n-butane oxidation.
61
1.26-D In situ Raman spectra of 7% V2O5/ZrO2 catalysts during n-butane oxidation.
61
1.27 Performance of Titania support in n-butane oxidation on supported Vanadia catalysts at 494 K in 1.2 Vol. % n-butane in air.
63
3.1 XRD spectrum of samples (2-un, 2-supp,3-un and 3-supp).
83
3.2 TG and DTA of VOHPO4.0.5H2O (sample No.2-un). 88 3.3 TG and DTA of VOHPO4. H2O (sample No.2-supp). 89 3.4 TG and DTA of VO (H2PO4)2 (sample No.3-un). 90 3.5 TG and DTA of VO (H2PO4)2 / TiO2 (sample No.3-supp). 91 3.6 FT-IR spectrum of Fluka V2O5. 95 3.7 FT-IR spectrum of technical grade V2O5 95 3.8 FT-IR spectrum of Fluka V2O5 / TiO2. 98 3.9 FT-IR spectrum of technical grade V2O5 / TiO2. 99 3.10 FT-IR spectrum of VOPO4.2H2O (sample No.1-un). 101
3.11 FT-IR spectrum of VOPO4.2H2O/ TiO2 (sample No.1-supp).
102
3.12 FT-IR spectrum of VOHPO4.0.5H2O ◊. 104
3.13 FT-IR spectrum of VOHPO4.0.5H2O (sample No.2-un) 105
3.14 FT-IR spectrum of VOHPO4.H2O/TiO2 (sample No.2-supp)
106
3.15 FT-IR spectrum of VO (H2PO4)2 (sample No.3-un). 108
3.16 FT-IR spectrum of VO (H2PO4)2/TiO2 (sample No.3-supp).
109
3.17A,3.17B Raman shift for prepared phases carried out by Red Laser and Green Laser.
112
3.18 The relationship between the AV.OX.No.of V ion and V (IV) %
116
3.19 The relation between the V (IV) percent with time. 117
3.20 The binding energy spectrum of the elements on the surface.
121
3.21 The XPS spectrum of VOPO4.2H2O (sample No.1-un). 123
3.22 The XPS spectrum of VOPO4.2H2O/TiO2 (sample No.1-supp).
124
Figure No. Title Page No. 3.23 The XPS spectrum of VOHPO4.0.5H2O (sample No.2-un) 125

XV
3.24 The XPS spectrum of VOHPO4.H2O/ TiO2 (sample No.2-supp)
126
3.25 The XPS spectrum of VO (H2PO4)2 (sample No.3-un). 127
3.26 The XPS spectrum of VO (H2PO4)2/TiO2 (sample No.3-supp).
128
3.27 SEM; Microstructure of VOPO4.2H2O (sample No.1-un). 132
3.28 SEM; Microstructure of VOPO4.2H2O/TiO2 (sample
No.1-supp). 132
3.29 SEM ; Microstructure of VOHPO4.0.5H2O (sample No.2-
un) 133
3.30 SEM; Microstructure of VO (H2PO4)2 (sample No.3-un). 134
3.31 SEM; Microstructure of VO (H2PO4)2/TiO2 (sample No.3-
supp). 135
3.32 A comparison between the morphology of unsupported
and supported 136
3.33 The EDX result of VOHPO4.0.5H2O (sample No.2-un) 138 3.34 The EDX result of VOHPO4.0.5H2O (sample No.2-un) 139
3.35 The EDX result of VOHPO4. H2O/ TiO2 (sample No.2-supp)
140
3.36 The EDX result of VO (H2PO4)2 (sample No.3-un). 141 3.37 The EDX result of VO (H2PO4)2 (sample No.3-un). 142 3.38 The EDX result of VO (H2PO4)2 (sample No.3-un). 143
3.39 The EDX result of VO (H2PO4)2/TiO2 (sample No.3-
supp). 144
3.40 The EDX result of VO (H2PO4)2/TiO2 (sample No.3-
supp). 145
3.41 The EDX result of VO (H2PO4)2/TiO2 (sample No.3-
supp). 146

XVI
List of Schemes Scheme No. Title Page No.
1.1 Huntsman fixed-bed reactor for MA production.
4
1.2 ALMA Fluidized-bed reactor for MA. 5
1.3 DUPANT recalculating – solid process for the production of THF from n-butane via MA.
5
1.4 Kinds of oxygen adsorbed species 9
1.5 A triangular reaction network on n-butane to MA.
27
2.1 Phase's preparation diagram from technical grade V2O5.
77

VII
Summary
The catalytic activity of Vanadyl Acid Phosphate towards the reaction
of n-butane oxidation to Maleic Anhydride ( MA) was achieved in our
previous work .So it is useful to study the effect of addition on (TiO2)
anatase as a support on structural properties and on its surface chemistry.
This phase is prepared in alternative method never maintained in the
literature. Route (A) depends on the reaction of the technical grade (V2O5)
with (o-H3PO4 ) in order to prepare the intermediate phase (VOPO4.2H2O)
which is then being reduced by using a mixture of ( Benzyl Alcohol and
Isobutanol) to reach the assigned phase VO (H2PO4)2 . While the
traditional route (B) depends on the reduction of Vanadium pentoxide
using alcohol mixture and then adding phosphoric acid to the reduced
phase.
The structural properties and surface chemistry of Vanadyl Acid
Phosphate and the intermediate phase prepared by route (A) and Vanadyl
Hydrogen Phosphate Hemihydrate prepared by route (B) are studied by
having confidence in number of physical and chemical analysis performed
in international known laboratories in U.S.A, Germany, Republic of
Ukraine and U.A.E. by using special techniques devoted in catalytic
structural and surfaces studies.
The identification of prepared phases is carried out by using X- ray
diffraction (XRD) in Fritz – Haber institute. The results of phase
identification show the co- existence of traces of other phases like VO
(H2PO3)2 with the dominated main phase prepared by rout (A). These
results also indicate that never reach very pure prepared phase which is

VIII
known phenomena in the field of preparation of (VPO) system. It was
appeared through these identification examination that the addition of TiO2
anatase could affect the resultant phases prepared in rout (B), since a
monohydrate composition (VOHPO4.H2O) is obtained in sated of Vanadyl
Hydrogen Phosphate Hemihydrate (VOHPO4.0.5H2O) normally prepared
without the use of support . No effect of support is observed on the main
prepared phase by rout (A).
The differential thermogravimetric behaviors for the supported and
unsupported precursors are studied.. The percentage of total loss of weight
for Vanadyl Acid Phosphate VO (H2PO4)2 / TiO2 is determined for the first
time.
The comparison of FT- IR spectrum of prepared supported and
unsupported phases with laboratory grade and technical grade of
Vanadium pentoxide are achieved. The obtained results emphasis on the
presence of wide shift in the band positions, these shifts reflects the
deformation of the angles and length of bonds in these phases. It is well
known that FT-IR is not adequate technique to study solid state structural
properties. However laser Raman spectroscopy could be helpful in this
aspect. Numbers of samples using this technique were carried out in
Oklahoma University laboratories in U.S.A. The presence of (TiO2
anatase) is accorded in the structure and layer of mono vanadate layer are
identified in all phases prepared, however the poly vanadate layer are
identified only in the prepared Vanadyl acid phosphate, this phenomenon
is very useful to support the employment of this phase, as a catalyst in the
reaction of n-butane to Maleic Anhydride and it is very good sign of
expected catalytic activity.
Vanadium ion oxidation stare and the percentage distribution for V (5+)
, V (4+) and V (3+) oxidation state are studied for supported and
unsupported phases by using redox titration technique , the results indicate

IX
that in the presence of the support the vanadium ion oxidation state
decreases toward (4+) oxidation state.
Desorption at Argon temperature technique was used for specific
surface area determination. The results state that there are significant
increases in specific surface area when adding support to intermediate
VOPO4.2H2O preparation. A specific surface area value for the first time is
assigned for the phase prepared by rout (A).
The X- ray photoelectron spectroscopy (XPS) technique was used on
samples in University of Oklahoma – U.S.A in order to study the surface
chemistry of the precursor VOHPO4.0.5H2O , VO (H2PO4)2 and
VOPO4.2H2O , the results point out the presence of structural elements (V,
P ,O), and detectable percentage of inorganic impurities on the surface and
also imply the presence of Nitrogen (N 1s) which is attributed to
Ammonium metavanadate, This compound was used as intermediate in
technical grade vanadium pentoxide preparation. The results also suggest
the presence of several oxygen species on the phases prepared. By using
this technique which can offer the (P/ V) atomic ratio, the prediction of
catalytic activity of supported phase can be assumed.
The Lewis and Bronsted acidity centers on the surface of the samples
were effectuated in National Academy of Science laboratories in republic
of Ukraine by using pyridine and 2, 6- Dimethyl pyridine adsorption. The
results indicated the doubled value of Lewis acidity centers in the
presence of support in phase VO (H2PO4)2. This is another signal to
predict activity as maintained in the literatures.
The investigation of surface of prepared phases samples were
performed in the American University of Sharjah. The results indicate
obtaining of lamellar crystalline characterized which is identical to these
proposed by literature.

X
Finally the combination between energy dispersive X-ray (EDX)
technique with Scanning Electron Microscope (SEM) technique was
carried out on samples in National Academy of Science laboratories in
republic of Ukraine in order to reach the elementally atomic percentage
down to depth of 1 µm from the surface. The results indicate the presence
of TiO2 in selected spots.
In general this work emphasize on the intercalation phenomenon
between structure layers and solid solution formation through out several
analysis which are made. This work can predict the catalytic activity of
Vanadyl Acid Phosphate VO (H2PO4)2 prepared phase enhancement by the
presence of TiO2 as support in the n-butane oxidation to Maleic Anhydride

ىـعل) ازـاتـنأ( الـتـيـتـانـيـايرات ـتأث
ةـيـة والسطحـبيـص التركيـالخصائ
) أوكسجين –فسفور –ناديوم ڤ( العوامل المـساعـدة لنظام
ةــالـرس
نـريـهـة النـعـامـج –وم ـلـعـة الـيـلـى كـة الـدمـقـم
اءـيـمـيـفي الك ةـسفـفل توراهـدك ةـيل درجـبات نـطلـزئي لمتـال جـكأستكم
من قبل
يـائـو الطـمـي حـلـداهللا عـد عبـنـه
١٩٨٧ –جامعة بغداد –بكلوريوس
٢٠٠٠ -جامعة بغداد –ماجستير
٢٠٠٥ زيرانح ـه١٤٢٦ جمادي االولى

ي ومـا ـبـرَ ِ◌ أمـر مـنْ وحُ ل الـرُ ح قـُـوُ الـرٌ ِ◌ عـن كَ ونَ سئلُ يَ وَ ﴿ ﴾ االقـليال ً ِ العلمَ◌ م منأوتيتُ
صدق اهللا العظيم )سورة االسراء ( ٨٤ا���

VII
Summary
This work is a continuous of our previous work in M.Sc study. Our
previous work was concerned with the use of locally produced V2O5
(technical grade) in the preparation of catalyst successfully employed in
the oxidation of n-butane to Malice Anhydride. The catalyst was
evaluated using geomechanic system (France) specially built for catalytic
test and modified to our proposes. The results had indicated to a functional
catalyst. The catalytic properties were attributed to several factors (highly
disordered, P/V around 1.1, adequate specific surface area, preparation
method and other factors) (1).
Since the catalytic activity of Vanadyl Acid Phosphate towards the
reaction of n-butane oxidation to maleic anhydride (MA) was achieved in
our previous work. It is useful to study the effect of addition (TiO2) anatase
as a support on structural properties and surface chemistry of the catalyst.
This precursor is prepared in an alternative method never mentioned in the
literature. Route (A) depends on the reaction of the technical grade (V2O5)
with (o- H3PO4 ) in order to prepare the intermediate phase (VOPO4.2H2O)
which is then reduced by a mixture of (benzyl alcohol and isobutanol) to
reach the assigned phase VO (H2PO4)2 . While the traditional route (B)
depends on the reduction of vanadium pentoxide by alcohol mixture and
then adding phosphoric acid to the reduced phase.
The structural properties and surface chemistry of vanadyl acid
phosphate and the intermediate phase prepared by route (A) and vanadyl
hydrogen phosphate hemihydrate prepared by route (B) are studied after
carrying out physical and chemical analysis in international known
laboratories in U.S.A, Germany, Republic of Ukraine and U.A.E. by using
special techniques devoted in catalytic structural and surfaces studies.

VIII
The identification of the prepared phases was carried out by using X-
ray diffraction (XRD). The results of phases identification show the co-
existence of traces of other phases like VO (H2PO3)2 with the dominated
main phase prepared by rout (A). These results also indicate that a very
pure phase was never reached phase which is known phenomenon in the
field of preparation of (VPO) system. It was found through these
identification examination that the addition of TiO2 (anatase) could affect
the resultant phases prepared in rout (B), since a monohydrate composition
(VOHPO4.H2O) is obtained instead of Vanadyl Hydrogen Phosphate
Hemihydrate (VOHPO4.0.5H2O) normally prepared without the use of
support . No effect of support is observed on the main phase prepared by
route (A).
The differential thermogravimetric behaviors for the precursors were
studied. This study highlights only the supported and unsupported
precursors in order to assign the effect of TiO2 (anatase) on the thermal
behavior, two effects were observed; the first one is increase the
percentage of total loss of weight for supported Vanadyl Acid Phosphate
(13.86 %), the second effect is the types of stage which in the precursor
started it dehydration; the unsupported phase started the dehydration in (2
endothermic peak between 50-27º C) while the supported phase started its
dehydration in one endothermic peak between ( 110-180 º C ). The total
loss in weight of supported VO (H2PO4)2 is reported here for the first time.
The comparison of FTIR spectrum of prepared supported and
unsupported phases with laboratory grade and technical grade of vanadium
pentoxide are achieved. The obtained results emphasis on the presence of
wide shifts in the band positions. These shifts reflect the deformation of
the angles and lengths of bonds in these phases. Laser Raman spectroscopy
could be helpful in this aspect. Structural properties were further
confirmed by studying the effect of presence of TiO2 (anatase) is accorded

IX
in the structure, and layers of mono vanadate layer are identified in all
prepared phases. However the poly vanadate layers are identified only in
the prepared Vanadyl acid phosphate. This phenomenon is very useful to
support the employment of this phase, as a catalyst in the conversion of n-
butane to maleic anhydride and it is very good sign of expected catalytic
activity.
The oxidation state of Vanadium ion and the percentage distribution for
V (5+), V (4+) and V (3+) in the bulk of precursors are studied for
supported and unsupported phases by using redox titration technique. The
results indicated that in the presence of the support the vanadium ion
oxidation states change towards (4+) oxidation state.
Desorption at Argon temperature technique was used for specific
surface area determination. The results state that there are significant
increases in the specific surface area when adding support to the
intermediate phase VOPO4.2H2O (16 m2.g -1) and the result show an
increase in this value for supported VO (H2PO4)2 (11 m2. g -1). A specific
surface area value for supported phase is reported here for the first time.
The X- ray photoelectron spectroscopy (XPS) technique was used in
order to study the surface chemistry of the two precursors
(VOHPO4.0.5H2O), and VO (H2PO4)2 and the intermediate phase
VOPO4.2H2O. The results pointed out the presence of structural elements
(V, P ,O), and detectable percentage of inorganic impurities on the surface
and also imply the presence of Nitrogen (N 1s) which is attributed to
ammonium metavanadate, This compound was used as intermediate in
technical grade vanadium pentoxide preparation. The results also suggest
the presence of several oxygen species on the prepared phases. The results
also indicate that the average oxidation state for prepared precursors on the
surface is (5 +). By using this technique which can offer the (P/ V) atomic

X
ratio, the prediction of catalytic activity of supported phase can be
assumed.
The Lewis and Bronsted acidity centers on the surface of the samples
were effectuated by adsorption of Pyridine and 2, 6- Dimethyl pyridine
adsorption. The results indicated the doubled value of Lewis acidity
centers in the presence of the support in VO (H2PO4)2 phase (2.050 X107
mol. m2) for unsupported sample and (4.420050 X107mol.m2) for
supported sample). This is another signal to predict the activity as
mentioned in the literature.
The investigation of the surface of the prepared phase's samples has
indicated the formation of lamellar crystalline character which is identical
to those proposed by literature.
Finally, the combination between energy dispersive X-ray (EDX)
techniques with Scanning Electron Microscope (SEM) technique was
carried out on samples in order to reach the elementally atomic percentage
down to depth of 1 µm from the surface. The results indicated the presence
of TiO2 in selected spots.
In general this work emphasize on the intercalation phenomenon
between structure layers and solid solution formation through out several
analysis which are made. This work can predict enhancement of Vanadyl
Acid Phosphate VO (H2PO4)2 prepared phase by the presence of TiO2 as a
support in the n-butane oxidation to Maleic Anhydride.

الصــــــــــةــــالخ
هــتم عملنــا الســابق باســتخدام أ. بق فــي دراســة الماجســتيراالســ لبحثنــاهــذا البحــث هــو اســتمرار
مســاعد اســتخدم بنجــاح فــي عامــلالمنــتج محليــا فــي تحضــير ، خــامس اوكســيد الفنــاديوم الصــناعي
اعد باسـتخدام منظومـة فرنسـية تـم تقيـيم العامـل المسـ. تان الطبيعي الى انهدريد المالييـكبيو أكسدة ال
اشـارت .بنيت لغرض تقييم العوامل المساعدة وقد تم تحويرهـا الغـراض فحـص هـذا العامـل المسـاعد
. ي الــى انهدريــد المالييــكعــالنتــائج الــى فعاليــة العامــل المســاعد المحضــر فــي تحويــل البيوتــان الطبي
ديوم العــالي ، نســبة الفســفور الــى الفنــالالنتظــام ا( الــى عــدة عوامــل منهــا ةعزيــت الفعاليــة التحفيزيــ
).، المساحة السطحية المناسبة ، طريقة التحضير وعوامل أخرى١,١المساوية الى
اكسـدة تفاعـل تجـاها ) Vanadyl Acid Phosphate(بعـد تاكيـد الفعاليـة التحفيزيـة للطـور
ثير اضـافة الطـور السـاند ثـاني من المفيد دراسة تـأ صبحأ ،الطبيعي الى انهدريد المالييك البيوتان
امـل عللعلـى الخصـائص التركيبيـة وكيميـاء السـطح ) TiO2 Anatase ( أوكسـيد التيتـانيوم
) أ ( يعتمــد الطريــق اذ ، االدبيــاتبطريقــة لــم يســبق االشــارة اليهــا فــي حضــر هــذا الطــور. المحفــز
لتحضــير الطـور الوســطي يكعلـى تفاعـل خــامس اوكسـيد الفنــاديوم الصـناعي مـع حــامض الفوسـفور
(VOPO4.2H2O) وتانول ـيـــــول البنزايــــل وااليزوبـن كحـــــج مـــــدام مزيـــــخـــــالــــذي يجــــري اختزالــــه باست
، بينمــا تعتمــد الطريقــة التقليديــة) Vanadyl Acid Phosphate( وصــوال الــى الطــور المعــين
ت واضــافة حــامض بواســطة مــزيج الكحــوال الفنــاديوم دعلــى اختــزال خــامس اوكســي ))ب (الطريــق (
. الفسفوريك على الطور المختزل
))ا ( الطريق (ور الوسطي ـوالط ورـذا الطـهـح لـاء السطـيـدراسة الخواص التركيبية وكيم تتم
ويـة اميياسـتنادا الـى عـدد مـن التحاليـل الفيزياويـة والك) )ب(الطريق ( بموجبايضا طور المحضر لوا
ميـة المعروفـة فـي الواليـات المتحـدة والمانيـا وأوكرانيـا واالمـارات لت العافي عدد مـن المختبـرا المنجزة
اذ ، وباســــتخدام تقنيـــات متخصصــــة فــــي دراســـات الســــطوح والعوامــــل المســــاعدة ، العربيـــة المتحــــدة
ـــود االشـــعة الســـينية للمس ـــة حي ــــشخصـــت االطـــوار المحضـــرة باســـتخدام تقني X-ray ). احيقـ
Diffraction )ن ـار مــود اثــوجـ ترافق ))ا ( الطريق (في وارـلالط ئج التشخيصرت نتاـأظهلقد و
كمـا . (Main Phase) ي الغالـبــــمـع الطـور الرئيس) VO (H2PO3)2)( اطوار اخرى مثل
ير االطـوار ـروفة فـي مجـال تحضــمعـ ةرـ ي وهـي ظاهــور نقــول الى طــكن الوصـارت الى انه اليمـأش
المـادة اضـافة كمـا تبـين مـن خـالل هـذا الفحـص ان ،)اوكسـجين –ر فسفو –فناديوم ( ام ـظنفي
اذ) ب (علـى نـواتج التفاعـل للطريـق اثـر قـد ) TiO2 Anatase التيتانيـا (Support) اندةـــالس
ة تحتــوي علــى ـبـــكير ت الحصــول علــىحيــث تــم انــه قــاد الــى عــدم الحصــول علــى الطــور المطلــوب
في . (VOHPO4.0.5H2O) بدال من نصف جزيئة VOHPO4.H2O)( دةـاء واحـزيئة مـج

حيـث تـم )) أ( الطريـق ( اندة على الطور الرئيسي المحضر مـنــــحين لم يؤثر وجود المادة الس
.كطور رئيسي في كلتا الحالتين ) VO(H2PO4)2 ( الحصول على
و تناولت الدراسة تسليط ، ) Precursors( وك الحراري التفاضلي لالطوار البادئةلدرس الس
) Support(لتحديد تأثير المادة الساندة )المسندة وغير المسندة( بادئة فقط لالضوء على االطوار ا
وقد اشارت النتائج الى ارتفاع نسبة الفقـدان فـي الـوزن فـي حالـة االطـوار . اوك الحراري لهلعلى الس
ــــــث بلغــــــت المســــــندة فــــــي حــــــين بلغــــــت TiO2 /(VO(H2PO4)2 للطــــــور % ١٣,٨٦(حي
كـذلك اشـارت النتـائج الـى ان فقـدان المـاء لهـذا الطـور فـي .للطور غير المسـند ) % ١٢,٠٠(النسبة
في المدى endothermic)(حالة كونه غير مسند في المرحلة االولى تتم بقمتين ماصتين للحرارة
اعثــة للحــرارة فــي المــدى فــي حــين ان الطــور المســند يبـدأ بقمــة واحــدة ب) درجـة مئويــة ٢٧٠- ٥٠(
TiO2 /VO( للطــور الــوزن دانـقـــوية لفـة المئـــتــم تحديــد النسبــ لقــد .)درجــة مئويــة ١٨٠-١١٠(
(H2PO4)2 ( بالطريق المحضر )الول مرة) ا.
Fourier Transform Infrared spectroscopy) الحمـراء اطيـاف االشـعة تحـت درست
FTIR) مقارنتهــا مــع خــامس أوكســيد توتمــ ) المســندة وغيــر المســندة( لكافــة االطــوار المحضــرة
مـع ذلـك المشـتق القياسـي ) VOHPO4.0.5H2O(وكـذلك تمـت مقارنـة الطـور ،الفنـاديوم النقـي
دلــت النتــائج علــى وجــود انحــراف كبيــر فــي مواقــع الحــزم و . الصــناعي مــن خــامس اوكســيد الفنــاديوم
فـي) deformation( بقية االطـوار وهـذا مؤشـر علـى وجـود التشـوه علىوانسحب هذا االنحراف
.هذه االطوارفي زوايا واطوال االواصر
( Solid state) التعويل على هذه التقنية فـي دراسـة المـادة الصـلبة ه اليمكنمن المعروف ان
، )Laser Raman Spectroscopy(ةيـــــلـــــذا تـــــم االســـــتعانة بتقنيـــــة مطيـــــاف رامـــــان الليزر
رف علـى ـعــتـكمـا تـم ال ) TiO2 Anatase( التيتانيـاجـود مـن خـالل هـذه التقنيـة تـم التحقـق مـن و
وتبـين مـن خـالل Poly) ( أو متعـددة )(Mono ت منفـردةـما أذا كانــات الفناديـت فيــة طبقــنوعيـ
ود ـت علـى وجــفيما دل ،ميع االطوار المحضرة بالطريقتينـجردة متكونة في ـفـنـالفحص ان الطبقة الم
جــدا حيــث تــدعم ةظــاهرة مفيــد وهــي ) VO (H2PO4)2( ي الطــور ـددة فـــتعـــردة والمـنفـــة المـالطبقــ
مهمـة اشـارة يوهـ ،الـى المالييـك انهدريـدسـدة البيوتـان الطبيعـي أك عـلهذا الطور في تفال نااستخدام
.له على الفعالية المتوقعة
لحـاالت االكسـدة حددت حالة االكسدة اليون الفناديوم وكذلك النسبة المئوية اليونـات الفنـاديوم
سندة باستخدام تسحيح للالطوار المسندة وغير الم (bulk)في الكتلة الخماسية والرباعية والثالثية
حالـة االكسـدة اليــون تحـول واشـارت النتـائج الــى ، (Redox Titration) أختـزال –االكسـدة
. لجميع االطوار المحضرة بوجود المادة الساندة+ ) ٤( الفناديوم باتجاه حالة االكسدة الرباعية

Desorption at Argon( االركــــون حــــرارة عنــــد درجــــة االمتــــزاز أســــتخدمت طريقــــة
temperature ( لقيـــاس المســـاحة الســـطحية للطـــور ) (VOHPO4.0.5H2O . وقـــد اشـــارت
ـــى حـــدوث ارتفـــاع ملحـــوظ فـــي المســـاحة الســـطحية للطـــو ـــائج ال (VOPO4.2H2O)ر الوســـطيالنت
للطـور غيـر الواحـد متـر مربـع للغـرام ١١( حيث ارتفعـت مـن )Support (بوجود المادة الساندة
لمســــاحة اكــــذلك تــــم الول مــــرة تحديــــد قيمة ) متــــر مربــــع للغــــرام للطــــور المســــند ١٦ المســــند الــــى
متــر مربــع للغــرام ١٠(حيــث كانــت )VO (H2PO4)2 ) (ا (ضــر بالطريقــةحالســطحية للطــور الم
. )للطور المسند الواحد متر مربع للغرام ١١للطور غير المسند وارتفعت الى
) VO (H2PO4)2و VOHPO4.0.5H2O) (لبادئـــة كيميـــاء الســـطح لالطـــوار ا تدرســـ
X-ray photoelectron)باســــــتخدام تقنيــــــة ) VOPO4.2H2O (وللطــــــور الوســــــطي
spectroscopy XPS) الـــى وجـــود اشـــارت النتـــائج لقـــد . ألطيـــاف االشـــعة الســـينية الضـــوئية
رات الشـوائب ذنسـبة البـاس بهـا مـن علـى السـطح والـى وجـود (P,V,O)عناصر التركيبة البلورية
وهـي التـي يحتويهـا علـى سـطوح االطـوار المحضـرة ) Fe, Ca , Na, Al , Si( مثـل الالعضـوية
ان مصـدر النتـروجين علـى .وجـود النتـروجين الـى كمـا اشـارت ،) (V2O5خامس أوكسيد الفنـاديوم
Ammonium metavanadate)( االمونيــوم فنــاداتميتا ود الــىـيعــ ة ـقاســـوار المـســطوح االطــ
NH4VO3 واكــدت وجــود .الصــناعي خــامس أوكســيد الفنــاديوم هــو طــور وســطي فــي انتــاج و
على تأكيد حصـول ظـاهرة المحلـول ايضادلت هذه التقنية و فصائل مختلفة اليونات االوكسجين،
اشــارت النتــائج . فــي بعــض االطــوار بســبب التيتــانيوم والصــوديوم (solid solution) الصــلب
الل ـن خــكمـا تـم مـ+) ٥(الـة االكسـدة اليـون الفنـاديوم فـي االطـار المحضـرة هـو كذلك الـن معـدل ح
فـــي وهـــي مرتفعـــة (P/V ratio) ب الذريـــة للفســـفور والفنـــاديومـســــهـــذه التقنيـــة التوصـــل الـــى الن
VOحيـــث بلغـــت هـــذه النســـبة فـــي الطـــور االطـــوار المســـندة عنهـــا فـــي االطـــوار غيـــر المســـندة
(H2PO4)2 / TiO2 )تقائيــة العامــل ننســبة مهمــة فــي الــتحكم بفعاليــة واهــذه ال تعــدو ، )١,١٠
. المساعد
ام طريقــــة دـتخـــــباس نمــــاذجللقيســــت حامضــــية مراكــــز لــــويس وبرونشــــد علــــى ســــطوح االطــــوار
Dimethyl -2,6) )وثنــائي مثيــل البــردين -٦،٢(و ) (Pyridineامدصــاص البــردين
Pyridine) د تضـاعفت بوجـود المـادة السـاندة ة حامضـية مراكـز لـويس قـقيمـ ان واظهرت النتائج)
Support ( في الطور) VO (H2PO4)2 ( حيث بلغـت) X 10 7 مـول للمتـر المربـع ٢,٠٥٠
للطـور المسـند )مول للمتر المربع الواحد 4.420X 107( في حين بلغت للطورغير المسند )الواحد
.لك المشار اليها في ادبيات الموضوعوهذه اشارة اخرى الى فعالية افضل من ت ،
Scanning Electron)ةـيــــنـقـدام تـخـــتـضرة باسـوح االطـــوار المحــــطـــس ةـاينــــمع تـتمــ
Microscope SEM) ودلـت النتـائج علـى الحصـول علـى سـطوح ، المجهـر االلكترونـي الماسـح

لضـمان فـي ادبيـات الموضـوع،مشـابهة لمـا موجـود وهي صـفة ا،له ) Lamellar( مستوية رقائقية
. فعالية أفضل للعامل المساعد
نيـة مـع تق ) Energy Dispersive X-ray( وأخيـرا تـم موالفـة تقنيـة تشـتت االشـعة السـينية
للوصـول الـى ) (Scanning Electron Microscope SEMالمجهـر االلكترونـي الماسـح
واشـارت تلـك النتـائج بوضـوح ، نمـاذج عـدة فـي عن السطحµ 1) ( لسمكالنسب الذرية للعناصر
.افة في البقع المختارة سالى وجود التيتانيوم على هذه الم
بــين الطبقــات الوكســيد التيتــانيوم (Intercalation) لقــد أكــد هــذا البحــث وجــود ظــاهرة اقحــام
اليــــل تحمــــن خــــالل ال . (Solid solution) حصــــول ظــــاهرة المحلــــول الصــــلب و ،البلوريــــة
.ذا البحثـي هـها فـم اجرائـعددة التي تـالمت
التنبــؤ بفعاليــة ،) Vanadyl Acid Phosphate( ة التــي حضــر فيهــا الطــور ـبالطريقــ كنـمـــي
سيؤدي الى تحسين الصـفات (Support)ان استخدام المادة الساندة كما ،مساعد عامل ك له جيدة
. صناعيا الييكانهدريد الم النتاجالطبيعي اكسدة البيوتان تفاعل فيالتحفيزية له





























