Embed Size (px)
Citation preview


Tools for the Next Generation
Genomics has become Big Data.
The tools for viewing, analyzing it have not:
Utilities still perform single-pass alignments.
Viewers still use characters.
Alignment tools are still recursive.
This limits research and clinical analytics.

Sequence alignment is the core
Searching, comparing, aligning sequences.
Smith-Waterman algorithm is the standard.
Single pass: cannot fine-tune alignments.
Recursive: not suited to map-reduce / clouds.
Whole-gene: cannot align crossovers, CNV, reversals.
Fragment indexing only works for stable sequences.

Another approach: W-curve
Originally designed as a visualization technique.
Convert DNA sequence to geometry.
Visually compare relatively large sequences.
Geometry has richer content for computational approaches:
Parallel analysis.
Fuzzy math for comparison.

W-curve geometry
Each base is at the corner of a square.
All curves begin at ( 0, 0, 0 ).
Points are halfway to the next base's corner.
Z-axis advances in steps of one.
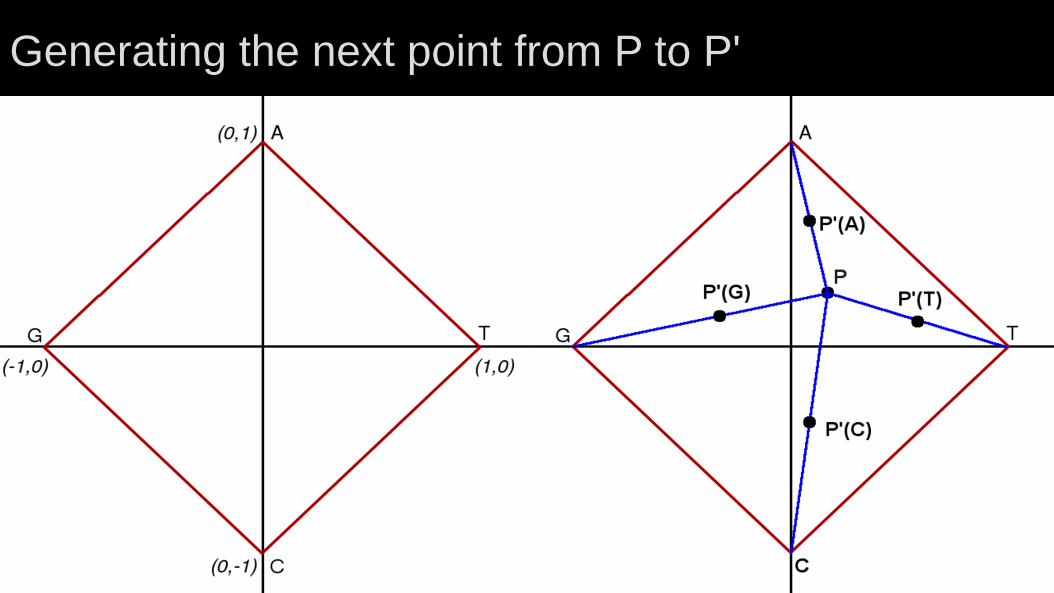
Generating the next point from P to P'

Starting a curve: CG...
From ( 0, 0, 0 )
Halfway to C ( 0.0, -0.5, 1 )
Halfway to G ( -0.5, -0.25, 2 )
First point is always 0.5 along an axis.

Visual analysis
The best pattern recognition engine known: Our Brians.
The W-curve's graphical pattern works well with people.
For example: Looking at wild, drug resistant, sample HIV.
Looking along the curve quickly shows differing sequences.

HIV-1 POL Fragment: Wild, 2 DR, Sample

Why it works
Autoregression is a central feature of the W-curve.
Curves converge quickly after a SNP or gap.
But remain apart long enough to see the difference.

Autoregression: SNP

Autogregression: GAP

Automating comparison
Geometry allows many approaches.
Simple one is aligning local points.
Use GIS:
Store the library as points.
Generate the sample as circles.
Find all of the points within a circle.
Cluster these to get the alignment.

Fuzzy Matching: Circles & Points.

Query your genes
Find all library points “near” each sample point.
Cluster them by z-offset between library, sample.
These are local alignments.
Local clusters be grouped to handle crossovers, gaps, insertions.

SQL is readable
Three values using postgis:
Library sequence.
Library base number.
Sample base offset.
select b.seq_id, b.base_no, a.base_no – b.base_no as 'delta',from sample a, library bwhere st_contains ( a.vertex, b.vertex )order by b.seq_id, b.delta, b.base_no;

Crossover:
Multiple library sequences are tiled onto the sample.
Adjacent when sorted by sample base.

Example: crossover gp120 ...1 6880 +931 6881 +931 6882 +93 ... 1 7665 +93 ...2 6307 -822 6308 -822 6310 -822 6311 -82 ... 2 7054 -82
Two library sequences: 1, 2.
Sort: ID, Offset, Base No.

Example: crossover gp120
1 6880 7665 +93
2 6307 7054 -82
Two sequences: 1, 2.
Sort: ID, Offset, Base No.
Collapse to fragments.

Example: crossover gp120
1 6973 7758 +93
2 6225 6972 -82
Two sequences: 1, 2.
Sort: ID, Offset, Base No.
Collapse to fragments.
Add offset to get sample bases.

Example: crossover gp1202 6225 6972 -821 6973 7758 +93
Two sequences: 1, 2.
Sort: ID, Offset, Base No.
Collapse to fragments.
Add offset to get sample bases.
Sort by sample base.

Example: crossover gp1202 6225 6972 -821 6973 7758 +93
Two sequences: 1, 2.
Sort: ID, Offset, Base No.
Collapse to fragments.
Add offset to get sample bases.
Sort by sample base.
Crossovers meet at 6972-3.
Similar algorithms for CNV, crossover chromosomes.

Multi-pass alignments
Messy genes: HIV-1, oconogenes.
Blast, Fasta, Clustal often spread the sample out too far.
Clustering provides tighter alignments.
Result can be re-filtered to remove extraneous data.
Scoring based on relevant portion of sequence.

Applying the cloud
The W-curve algorithm is suitable for map-reduce:
Individual triples are independent.
Clustering can be carried out in stages.
Each stage can accumulate clusters, fragments.
Work on large genes or databases can run in parallel.
Geometric data also suitable for machine-learning, fuzzy math.

Parallel execution: BLAST vs. W-curve
Parallel execution with BLAST requires fragmenting the sequence.
Separate outputs re-combined, usually manually.
Boundary issues at fragment boundaries.
W-curve alignment runs in parallel at all levels.
Samples do not have to be broken up.
No boundary issues.

Population, evolution, microbiome, plants:
Large-scale, often fuzzy matches.
Wide search in a large database.
Mix of large and small fragments.
Multiple alignments for each fragment.
All of these can be automated.

Example: Clustering HIV-1
Clades for HIV-1 use the entire gp120 sequence.
~80 bases out of 1500 make up CD4 binding site.
The rest is highly-variable.
BLAST & Clustal assume all variation is significant.
Result: The clades are based on 95% white noise.

Example: HIV-1
Classic HIV-1 antibody study.
Clades use entire gp120 sequence.
No diagonal: antibodies don't react with all of gp120.
Need to base clades on subset.

Applying the W-curve
First pass performs global alignments of gp120.
Candidate CD4 binding sites can be extracted.
Finer-grained alignment of 80+ bases in CD4 binding site.
Result is a set of clinically useful clades.

Other approaches
S-W cannot handle multi-base in FastQ data.
Has no way to deal with quality measures.
W-curve could generate multiple paths or a volume.
Comparison via fuzzy math with library sequences.
Result: Direct comparison of FastQ data.

Visualization
Interactive tools can help annotation, manipulation of sequences.
Tagging bases, shading common areas.
Selecting matching regions interactively for study.
Modifying difficult alignments by hand.
All of these can benefit from a graphical, visual approach.

Business Case
The W-curve algorithm is public.
Most of the automation is in filtering, however.
Different problems require special filters.
Job control, database optimization also matter.
Domain-specific knowledge is the real value added.
Custom filters, database generation & management.
Interactive user interface as either plugin or separate tool.

Contact
Steven Lembark
Workhorse [email protected]
+1 888 359 3508
Ed Bayham
EXPLOR [email protected]





























