Embed Size (px)
Citation preview

Haplotype resolved structural variation assembly with long reads
Mount Sinai: Ali Bashir, Oscar Rodriguez, Matthew Pendleton
Reed College: Anna Ritz, Alex Ledger
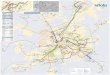
Summary Of 1kG/GIAB PacBio SV Calls
*Ran through a streamlined version of the SV pipeline may not be comparableOther Notes:- MEI calls use conservative parameters (likely undercalling insertions)- “Other” calls contain some improperly flagged insertions/deletions as well as complex events and inversions
Insertion Deletion Complex
Sample# of Calls
# of TR calls
# of Alu
#of L1
# of SVA
# of Calls
# of TR calls*
# of Alu # of L1
# of SVA
HG002 13471 5573 325 68 7 9639 6880 798 201 22 2493HG003* 12947 5133 411 74 5 9692 6776 411 74 5 2580HG004 12769 5066 475 160 96 9509 7233 971 282 33 2599
HG00512 9830 4164 366 75 67 7672 5781 768 275 23 2157HG00513 9761 4175 351 86 79 7791 5936 770 258 27 2314HG00514 1285 4866 212 42 3 9636 6770 767 222 26 2635HG00731 9874 4322 357 76 75 7678 5797 790 256 17 2174HG00732 11059 4884 400 85 85 8227 6274 813 271 24 2351HG00733
* 11769 5365 330 45 4 8848 6179 743 191 25 2313NA19238 7512 2999 280 72 59 6320 4765 628 237 12 1910NA19239 5909 2357 199 46 50 5061 3809 528 161 21 1468NA19240
* 13285 5185 345 78 7 9791 7596 911 275 23 2600

Haplotype Resolved Assemblies
• Two methods:– Falcon Unzip
• PacBio de novo diploid assembly method• Refer to Jason Chin’s talk!
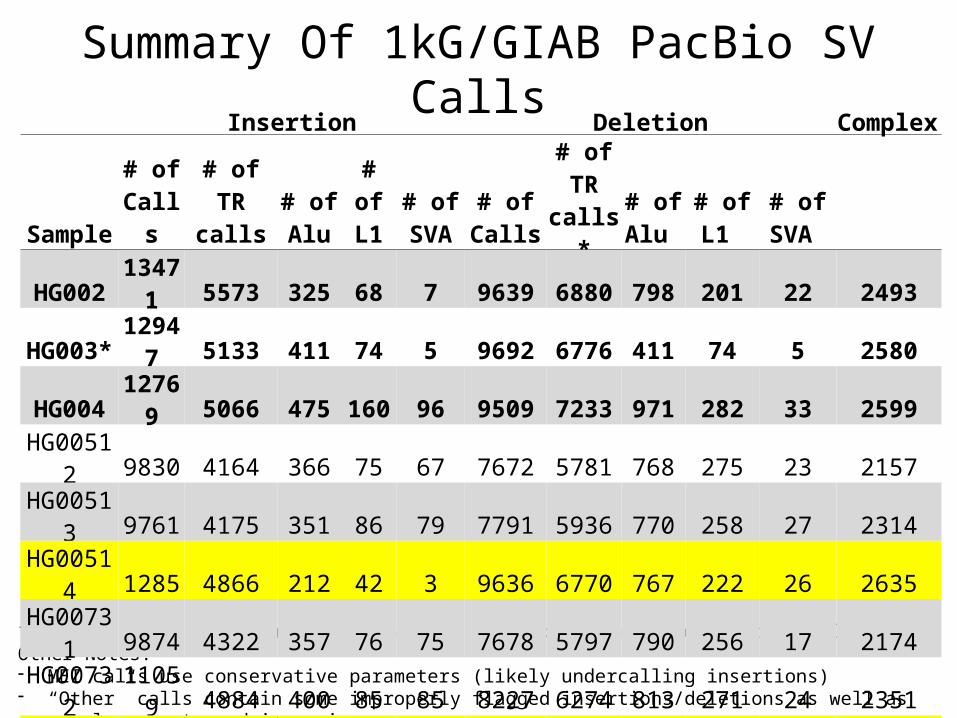
– 10X guided PacBio Assembly• Using long-range haplotypes from 10X to partition
PacBio reads for guided assembly
10X + PacBio
Genome
10X partitioned SNVs
Long reads

10X Partitioned PacBio Assembly
Select regions with greater than 4X
coverage
Assemble regions(currently leverages
MHAP/Canu/FALCON)
Contig created?Yes No
Split region using heterozygous SNPs
Create Local MSA between SNVs.
BLASR to map reads to contig and Quiver
for new contig
Map contig to reference
Updated MSPacMonstr SV pipeline
Realign via event specific alignment

Partitioned Haplotype Genome/Read Coverage
Percent of Genome Covered by >4 Reads # of Reads in each partition

How do things compare in the “callable regions” in all methods (partial results - deletions)
Many missed calls are a result of:1.) Slight discrepancies in the SV length cutoff (currently 50 bp)2.) Shifted Tandem Repeat Calls
Total Calls from Haplotype Approach- 18396Total Calls from FalconUnzip Assembly Approach- 28797

Assembly Calls “Missed” In Haplotype Calls
Partitioned Reads
Haplotype Contigs
Assembly Calls
Haplotype Calls

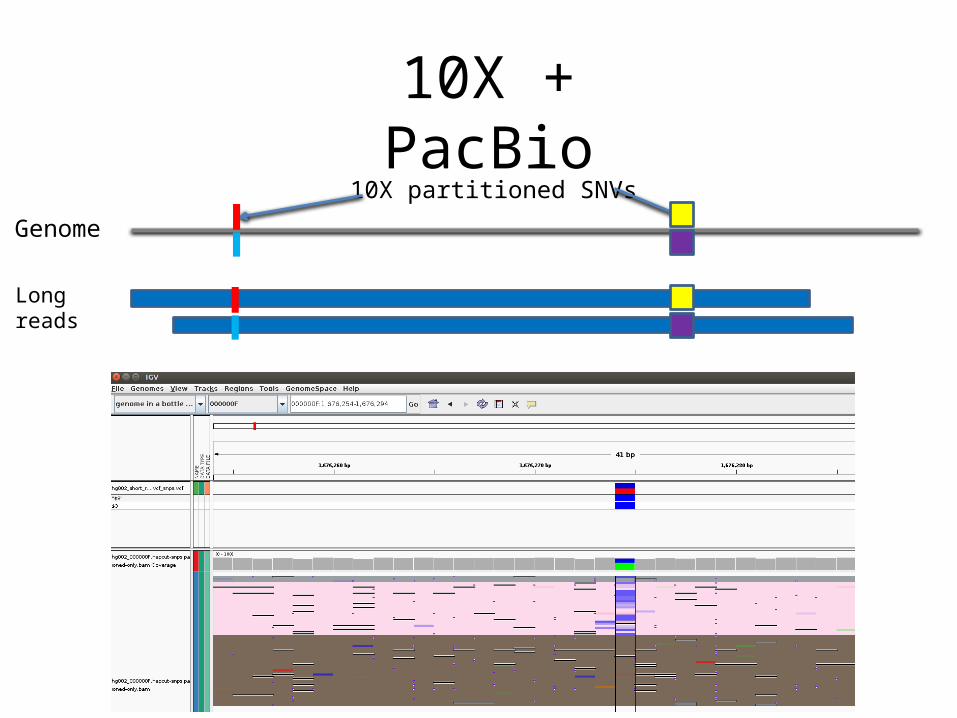
Haplotype Calls “Missed” In Assembly Calls
Partitioned Reads
Haplotype Contigs
Assembly Calls
Haplotype Calls
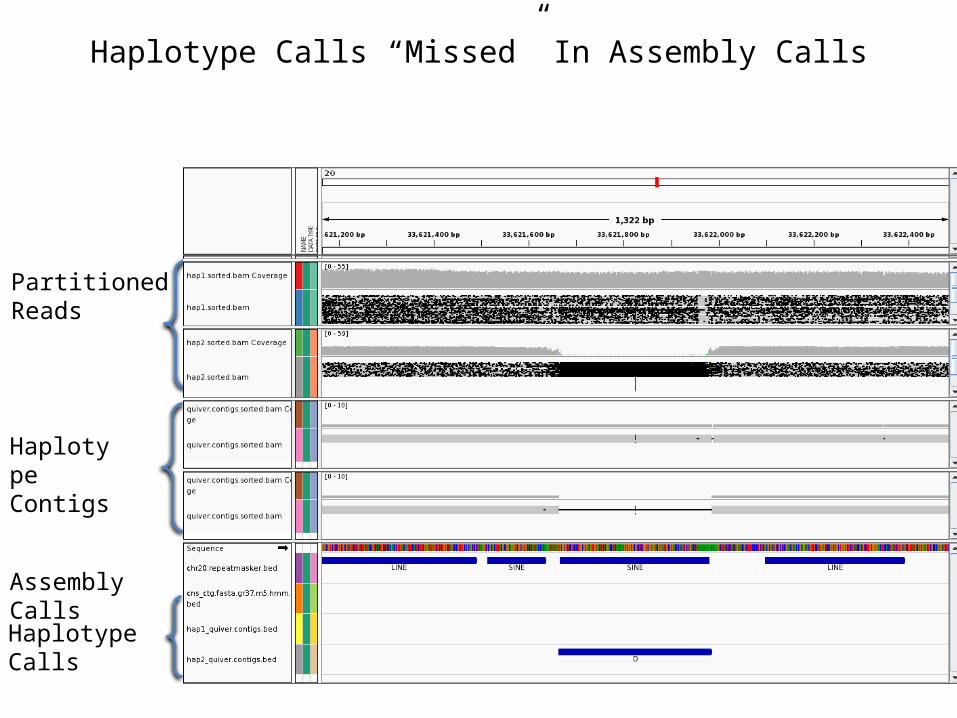
Haplotype Calls “Missed” In Assembly Calls
Partitioned Reads
Haplotype Contigs
Assembly Calls
Haplotype Calls
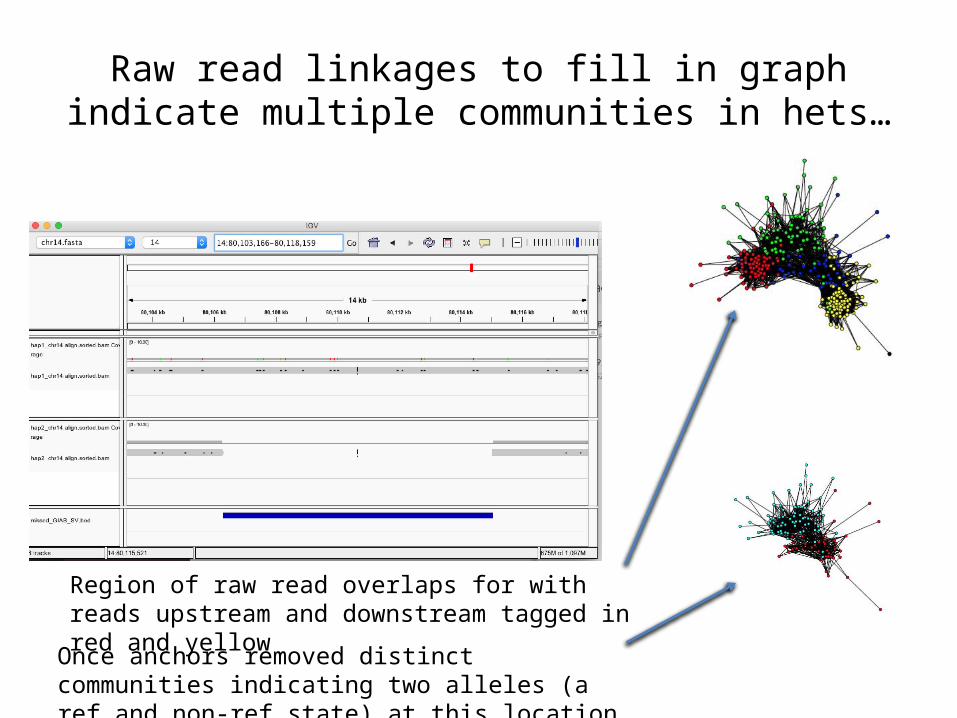
Raw read linkages to fill in graph indicate multiple communities in hets…
Once anchors removed distinct communities indicating two alleles (a ref and non-ref state) at this location
Region of raw read overlaps for with reads upstream and downstream tagged in red and yellow

And single communities in missed homozygous regions
Singe community once anchors removed
Region of raw read overlaps for with reads upstream and downstream tagged in red and yellow

Next steps- Low-coverage regions with MSA and other
strategies
- Filling in Reference Gaps with:- 1.) De Novo Assembly (from Falcon-Unzip)
- Reads not properly recruited because of ref divergence- 2.) Community detection algorithm (Ritz Lab at Reed)
- Trio phasing comparison via MSA

Acknowledgements
• Mount Sinai– Eric Schadt– Matt Pendleton– Ajay Ummat– Oscar Franzen– Gintaras Deikus– Robert Sebra– Oscar Rodriguez
• Reed College– Anna Ritz– Alex Ledger
• UCSF– Pui Kwok
• PacBio– Jason Chin
• 1000 Genomes SV Working Group
• UW – Mark Chaisson
• EMBL– Jan Korbel– Markus H.-Y.
Fritz– Tobias Rausch
• BioNano Genomics– Han Cao– Alex Hastie– Heng Dai– Andy Pang– Joyce Lee
• 10X Genomics– Patrick Marks– Deanna Church– Mike Schnall-Levin– Sofia
Kyriazopoulou-Panagiotopoulou

Intro to discussion
Merge deletions
within 1kb
Rank calls by
closeness of predicted
size to median size and select call in each region from best callset
Find calls supported
by 2+ technologies with size within 20%
Filter calls overlapping
seg dups, reference
N’s, or with call with
predicted size 2x larger
• Where are we currently lacking in integration?– Repeats– Multi-het events– Complex events
• How do we leverage our divergent calls?– Different precison of technologies– Using the richness of assembly

Repeats (like TRs) can shift boundaries for various callers

Example Workarounds
-Breakpoint calls (not assembled sequence):If L1 is within 10% of L2 size and L1 and L2 are
both in R we call it a “match”
- Assembly Calls Perform MSA to identify truly “homozygous”
sequences
L1
L2
Tandem Repeat
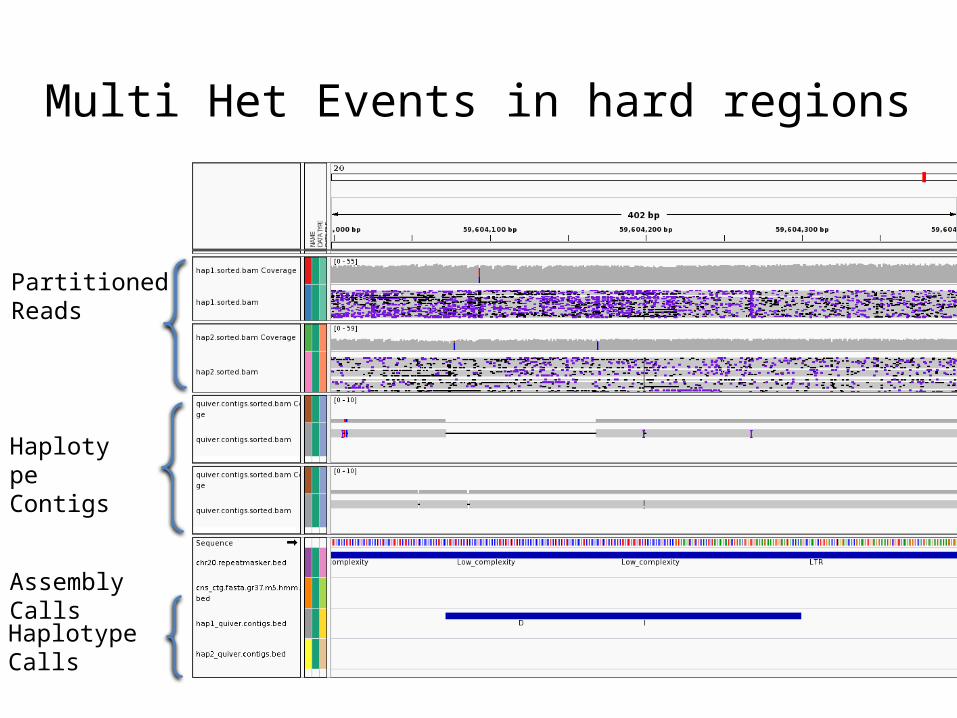
Multi Het Events in hard regions
Partitioned Reads
Haplotype Contigs
Assembly Calls
Haplotype Calls
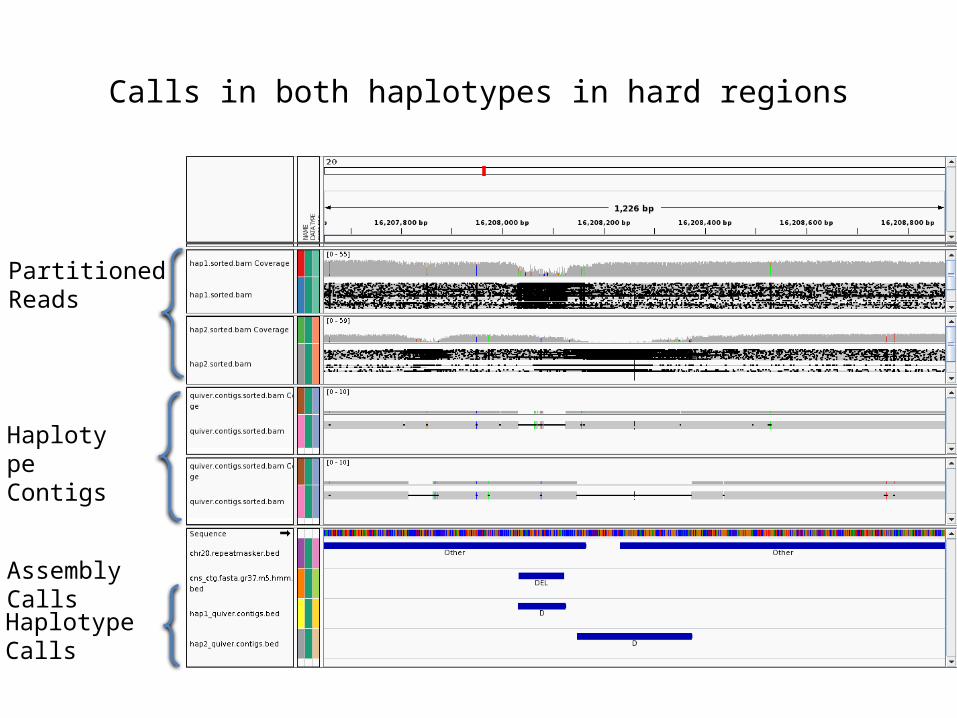
Calls in both haplotypes in hard regions
Partitioned Reads
Haplotype Contigs
Assembly Calls
Haplotype Calls
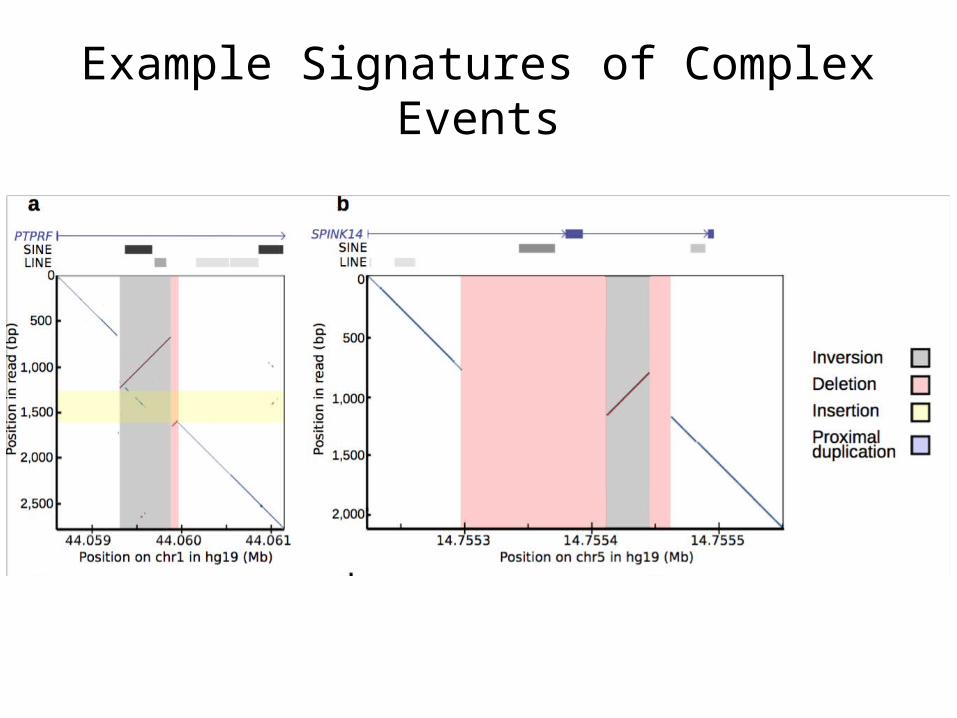
Example Signatures of Complex Events

Assembly provides detailed indications of quality
• Provides sequence of breakpoint• Potentially provides co-located events• Potentially provides information on accuracy of
the assembly in that region

MIS-ASSEMBLY
Slide from Jason Chin at the SMRT Informatics Workshop
Ctg 33 mapped to Chr14
Ctg 33
Ctg
33
map
ped
to C
hr1
Ctg 120
Mis-assembly point

Discussion
Merge deletions
within 1kb
Rank calls by
closeness of predicted
size to median size and select call in each region from best callset
Find calls supported
by 2+ technologies with size within 20%
Filter calls overlapping
seg dups, reference
N’s, or with call with
predicted size 2x larger

Things to resolve
Integration• How to compare events
with variable breakpoints across callsets?– Tandem repeats
• How to compare non-deletions?– Start with insertions?
• Distinguish precise breakpoints when possible
Typing• Leverage long-range
information to type with short reads?
• How to deal with imprecise breakpoints?
• At what point is something validated?– Potentially high-confidence
variants (or reference?)– Haplotype-separated