
Remerciements
1
Université Paris Descartes
Ecole doctorale Biologie Sorbonne Paris Cité
Laboratoire : Origine et fonctions des cellules souches squelettiques au cours de la régénération osseuse UMR 1163 – Institut IMAGINE
Rôle du muscle au cours de la régénération osseuse: étude fonctionnelle de la contribution cellulaire et impact des traumatismes musculosquelettiques
Soutenue par Anaïs Julien
Thèse soumise et défendue en vue de l’obtention du diplôme de Docteur ès Science, spécialité Biologie Cellulaire
Le 30 Novembre 2018
Devant le jury composé de
Dr. Delphine DUPREZ Dr. Laurence VICO Dr. Lucie PEDUTO Dr. Frédéric RELAIX Dr. Céline COLNOT
Rapportrice Rapportrice Examinatrice Examinateur
Directrice de Thèse


Remerciements
3
A mon père, mon héros

Remerciements
4

Remerciements
5
« N’oublions pas que, lorsque l’on a découvert le radium, personne ne savait qu’il
pourrait être utilisé dans les hôpitaux. Les études étaient purement scientifiques, ce qui
prouve que le travail des chercheurs sert à quelque chose. Il faut faire des recherches
pour le plaisir de chercher, pour ce que la science offre de beau, en gardant à l’esprit
qu’une découverte scientifique peut, comme le radium, servir l’humanité. »
Marie Curie

Remerciements
6

Remerciements
7
Remerciements Avant tout, je voudrais avoir un mot pour mes deux professeurs de biologie du lycée, Mr.
Dimitri Garcia et Mr. Hervé Mortier qui m’ont transmis le gout de la biologie et le plaisir
d’apprendre. Avec eux à germer l’idée de faire une thèse jusqu’à ce que cela devienne le
but de mes études supérieures. J’arrive aujourd’hui à la fin de cette aventure et je
voudrais en profiter pour remercier toutes celles et ceux qui, de près ou de loin, en font
partie.
Je voudrais commencer par remercier très sincèrement les membres de mon jury,
Delphine Duprez, Laurence Vico, Lucie Peduto et Frédéric Relaix pour avoir accepté
d’évaluer mes travaux de thèse.
Céline, merci pour tout. Vous m’avez formé et tout appris. Je ne compte plus les heures
passées dans votre bureau à discuter des projets, à corriger mes écrits, à vérifier mes
diapositives ou à mettre sur pieds des expériences toujours plus ambitieuses. Vous avez
été la directrice de thèse idoine pour moi. Vous m’avez laissé assez d’indépendance pour
que je puisse m’amuser et avancer mais vous avez toujours été présente pour vous
assurer que je n’allais pas dans le mur. Votre patience à mon égard me surprend encore.
Votre confiance m’est aussi précieuse, et m’a aidée à m’épanouir. Pour tout ça, et bien
plus encore, merci du fond du cœur.
A mes petits Nains, Simon et Anuya ! Quel bonheur d’avoir passé cette année à vos côtés.
Anuya, tu sais tout le bien que je pense de toi. Je te remercie bien sûr pour ton aide
technique sans laquelle j’aurai certainement fusionner avec le microtome, mais encore
plus pour ta bonne humeur, ton sourire, ta patience et ta capacité à ne pas trop poser de
questions parfois (la boite de lames « à ranger » s’en souviens encore).
Simon, mon Nain ! Tu nous en auras fait des bêtises, pas besoin de les énumérer ça
risquerai d’être long… Mais franchement qu’est-‐ce que ça nous aura fait rire ! Tu es
maintenant la relève des petits Colnot. Je sais que la pression est grande, au vu des deux
exemples précédents mais tu devrais t’en sortir. Je suis heureuse de travailler avec toi
encore un peu et je sais que malgré tout ce que tu dis, c’est un plaisir partagé.

Remerciements
8
Malgré notre migration vers le 4eme étage, je n’oublie pas toutes les filles du labo 202 et
en particulier Laetitia. Merci d’avoir était là dans les coups de mous, pour faire la fête, en
culture et puis pour toutes nos discussions de fille !
Un grand merci aussi à l’équipe d’Agnès, à Brigitte, Juliette et Marcelo qui nous ont
accueilli au 4ème étage. On n’y a vraiment pas perdu au change et c’est un vrai plaisir de
travailler à vos côtés. Maurice a trouvé un vrai foyer à vos côtés, et lorsque Maurice va,
tout va ! Je n’oublie pas non plus les personnes du bureau 402b qui m’ont bien
gentiment accepté pendant l’écriture de ma thèse.
Merci aussi à l’équipe de Laurence, et en particulier à Maxence, Davide, Emilie et Ludo
dit Mister DB. On aura partagé de supers moments qui rendent l’expérience de thèse un
peu plus folle.
Ce projet n’aurait jamais pu être mené à son terme sans le travail du personnel de
l’animalerie, et en particulier à celui d’Emilie et de Crisitan. Cristian tu es un grand
professionnel et merci pour l’excellent travail que tu fournis au quotidien pour
surveiller nos petites souris.
Merci aussi à Mélinda, notre secrétaire sans qui l’administratif serait une vraie plaie.
Mon aventure de thèse est indissociable de mon aventure footballistique. J’ai vécu des
moments exceptionnels avec cette équipe, au PUC puis maintenant à Joinville. Le foot
fait partie intégrante de mon équilibre et m’a permis de mener cette thèse avec plus de
sérénité et de plaisir.
Merci à Stéphane, l’homme de l’ombre. Merci d’avoir toujours était là, discret mais
présent. Notre demi saison d’invincibilité est certainement l’un des meilleurs souvenirs
que je garde de ces 4 ans à tes côtés.
Merci à Claudia, le rouage indispensable à l’équilibre du groupe. M’entrainer avec toi est
toujours un vrai plaisir et discuter encore plus.
Et puis bien sûr merci à Julien ! Avant je tapais dans un ballon mais à tes côtés, j’ai appris
à jouer au football. C’est toujours un plaisir d’être sur le terrain à tes côtés. Ta
disponibilité pour les « à côté » font de toi une composante essentielle de nos vies.

Remerciements
9
Un grand merci aussi à Rudolphe, Valentin et Didjo pour votre présence et votre
soutient.
Et puis bien sûr, merci à toutes les filles avec qui j’ai partagé ces années. Quel plaisir
d’être à vos côtés, depuis 5 ans pour certaines et 6 pour Paga.
Au foot naissent de belles amitiés. Il suffit qu’une fille fasse le faire le taxi pour une autre,
et deux coéquipières se transforment en deux amies. Je pense que tu t’es reconnu Jo.
Merci pour tout, le taxi, nos discussions technico-‐tactiques, et puis tout le reste. Tu sais
tout le respect que j’ai pour toi. Et évidemment, merci à toi aussi Estelle. J’espère que tu
me pardonneras de t’avoir piqué ta femme après de multiples entrainements. J’aime tant
nos discussions, toujours le bon mot et le bon ton. Tu es souvent la voix de la sagesse et
c’est très appréciable. Merci les filles d’avoir toujours été présentes.
Je suis montée à Paris seule, débarquée de ma campagne avec pour information qu’il
fallait prendre direction nord/nord-‐est en sortant de la Gare de Lyon. Puis je suis entrée
au Magistère. Bien plus qu’une formation ou que des années fac, j’y ai trouvé une famille.
Alors merci à Chloé, Tomaso, Moc et tous les autres pour tous les moments passés
ensemble. Mais j’ai quand même une pensée plus particulière pour certains d’entre vous.
Lisa, nos discussions en cours de statistiques et tous les fous rires qui s’en sont suivis
sont gravés dans ma mémoire.
Christelle, entre sudistes nous avons su nous serrer les coudes dans cette jungle
parisienne. Enfin quelqu’un qui sait faire la bise !
Laure, mon petit rayon de soleil. Je suis très contente qu’on ait pu faire notre thèse au
même endroit !
Agathe, un tout un poème. Ta bonne humeur et ta gouaille apporte un peu de légèreté
dans mon quotidien. Je t’en remercie grandement.
Ludo ! Enfin quelqu’un qui apprécie le Seigneur des Anneaux et Harry Potter à leur juste
valeur ! Nos après-‐midi cinéma restent de super souvenirs !
Marie, merci d’avoir été là quasiment au quotidien. Nos repas chez Speedy ont contribué
à forger une belle amitié. Merci pour tout.
Amandine, la seule personne avec qui je peux parler plus de 5 heures d’affilée à
n’importe quelle heure du jour ou de la nuit sans problème. Tes réflexions alambiquées
et tes choix capillaires resteront toujours un mystère pour moi mais ne t’inquiète pas je
t’aime quand même.

Remerciements
10
J’ai fini le lycée il y a presque 10 ans maintenant, mais Alex et Nancy vous restez deux
fondamentaux de mon existence. Ma Coin-‐Coin, merci pour toutes les soirées passées à
papoter de tout et de rien quand je rentre chez moi. Merci aussi à Loic pour ta gentillesse
et ton humour. Vous formez une famille magnifique et savoir que j’en fais partie un petit
peu me touche.
Alex, que dire… Si j’avais à te définir, je dirais que tu es un peu mon phare dans la
tempête. Je sais que je peux compter sur toi et même quand on ne se voit qu’une fois par
an tu sais toujours quoi faire pour m’aider. On ne la fera peut-‐être jamais cette coloc’
mais finalement on reste amie et c’est bien ce qui compte.
Merci à ma poulette Béro et à mon poulet Rastouill préférés. Merci pour votre soutien,
vos visites parisiennes, nos soirées, et tous les souvenirs que l’on partage. Malgré mon
éloignement, vous ne m’avez pas oublié et ça fait très chaud au cœur.
Cette fin de thèse est intimement liée à mon aventure au sein de l’association YRII. J’y
suis rentrée un peu par hasard et c’est devenu une évidence. De discussions en projets
en passant par de bonnes parties de rigolades, ça a été un vrai bonheur d’évoluer à vos
côtés.
Claire, Clarisse, Hicham et Cyril je ne sais pas par où commencer… Vous avez été un de
mes piliers cette année et être à vos côtés est une bouffée d’oxygène. Merci de m’avoir
accepté parmi vous.
Hicham, merci pour ton hospitalité et ta générosité, les petits dej du dimanche, les
soirées et encore plus. Je n’oublierai jamais ton coup de fil au mois de novembre l’année
dernière. Je te l’ai déjà dit mais t’es un mec en or, ne change pas.
Clarisse, merci d’être toi. Toujours à 200 à l’heure, à faire dix mille trucs à la fois mais tu
sais aussi prendre le temps d’écouter et de discuter quand les autres en ont besoin. Et ça
fait du bien.
Cyril, mon partenaire de blagues. Je n’aurai jamais pensé trouver quelqu’un avec un tel
humour. On se marre bien ensemble et nos fous rires sont réguliers. Merci pour ça, parce
que c’est une des meilleures façons que j’ai trouvées pour décompresser.
Claire, c’était pas gagné mais à force de te côtoyer j’ai découvert une fille super derrière
sa carapace. J’aime ta franchise et ta droiture mais encore plus partager un moment avec
toi. T’es la première de la bande à partir et pour sûr tu vas me manquer.

Remerciements
11
Un grand merci à Oriane. Tu es bien plus que ma co-‐thésarde. On a partagé tellement de
choses en 4 ans que je ne pourrais pas faire la liste. Toujours là dans les coups durs, les
bons moments, toujours disponible et de bons conseils. Tu as été à la fois une collègue,
une amie, une grande sœur et un modèle. Nos fous rires, notre complicité et notre travail
à quatre mains restent des moments précieux.
Un grand merci aussi à toi Tonton pour nos repas au restaurant du samedi soir. Ne
t’inquiète pas Titou, je ne t’oublie pas. Tu es devenu un beau jeune homme, félicitations.
Merci à toi Olivier de nous avoir accueilli comme on est. C’est rassurant de savoir que
l’on peut compter sur toi.
Merci à toi Mamie. Voir la fierté dans tes yeux quand tu parles de moi m’est précieux. Je
sais que je ne suis pas la personne la plus disponible mais tes messages me font toujours
plaisir même si mes réponses peuvent être rares. J’espère que tu me pardonnes.
Ma ptite Choupinette. A la fois ma secrétaire, ma meilleure amie, ma colloc’ et ma
maman de substitution.
-‐ Qu’est-‐ce que je ferai sans toi ?
-‐ Ben la même chose mais sans moi !
Tu crois vraiment ce mensonge ? Parce que moi non. Sans toi je n’aurai jamais connu
Pompomville, le brie de Meaux, la vie des MAPK, le shaking body, les rochers à la noix de
coco, radio Latiiiiiina à 7h du mat’… Mais tout ça n’est qu’un prêté pour un rendu et je
suis fière de pouvoir dire que maintenant tu sais que l’on dit Luberon et non pas
Lubéron, que tu connais la règle du hors-‐jeu, que tu acceptes une autre lessive que
LeChat ou que tu manges du poisson. Et puis surtout sans nous, Pierre Hermès aurait fait
faillite ! Tout ça pour dire que l’on s’est bien trouvé que ces 5 ans à tes côtés resteront
une merveilleuse page de mon histoire. Merci pour tout, pour ton soutien, tes conseils, ta
bienveillance, ton calme et ta patience. Merci d’être toi. Parce que finalement, tout ça,
c’est toi, c’est moi, c’est nous.
Merci aussi à Evelyne, Philippe, Valérie, Eric et Lorette de m’avoir accueilli parmi vous.

Remerciements
12
Bien sûr pour finir, merci à vous Papa, Maman et Bobichou parce que cette thèse c’est
quand même aussi la vôtre.
Mon Mini-‐Moi, heureusement que je t’ai. Maman a bien fait de ne pas accepter ma
proposition quand tu es née. Je me sentirai bien seule sans ma petite sœur. Je suis
heureuse de voir ce qu’est notre relation, un soutien réciproque indéfectible. Merci pour
tes encouragements surtout dans les moments difficiles, ta disponibilité et puis aussi
pour tes connaissances en géographies qui me font toujours beaucoup rire. Tu peux être
fière de toi et de ce que tu deviens parce que moi je le suis.
Papa et Maman merci de m’avoir toujours poussé plus loin, de m’avoir donné des
valeurs et des repères qui ont fait ce que je suis aujourd’hui.
Maman, je pense que je ne serai jamais arrivé là sans toi. Merci pour toutes ces heures
passées au téléphone à me conseiller, me remonter le moral, m’aiguiller, tout
simplement à t’occuper de moi. Te savoir à mes côtés est une des choses les plus
précieuses que je possède.
Papa, tu as fait Bac -‐5. Et bien moi j’ai fait Bac +8, ça équilibre bien les choses non ? Merci
de m’avoir transmis ton gout du sport et ta curiosité. A force de regarder les émissions
sur le fonctionnement du stylo bille ou sur les constructions impossibles, je ne pouvais
que faire une thèse pour comprendre un peu mieux comment fonctionne le vivant. Tu
restes l’un des repères majeurs de mon existence. Si l’étoile du berger guide les marins
dans la nuit noire, tu guides mes pas sur le chemin de la vie. Je sais que tu es fier de moi
et sache que je le suis au moins autant de toi.

Remerciements
13

Remerciements
14

Role of skeletal muscle during bone repair: functional study of the cellular contribution and impact of musculoskeletal trauma
Tissue regeneration relies on stem cells that are activated, and then proliferate and
differentiate to repair the damaged or diseased tissue. Bone exhibits great capacities to
regenerate via the local recruitment of osteo-‐chondro-‐progenitors (OCPs) within bone
marrow and periosteum, the tissue lining the outer surface of bone. In the first part of
this thesis work, we show that periosteum contains skeletal stem cells (SSCs) and that
periosteal cells (PCs) have higher regenerative capacities than BMSCs. The regenerative
and self-‐renewing potential of PCs is dependent of the extracellular matrix protein
Periostin that contributes to the periosteal niche of SSCs.
Despite its great capacities of regeneration, bone fails to heal properly in 10% of bone
injuries and delayed healing is increased in patients with soft tissue damage in 46% of
cases. The role of skeletal muscle is well known in the orthopedic field but the cellular
and molecular mechanisms underlying bone-‐muscle crosstalk during bone repair are
poorly understood. In the second part of the thesis, we show that muscle satellite cells
are required for bone repair as source of growth factors and that skeletal muscle is a
source of OCPs during bone repair. In the third part of the thesis, we then characterized
the skeletal muscle derived cells using cell lineage tracing in genetic mouse models and
tissue grafting. We show that OCPs recruited from both skeletal muscle and periosteum
during bone repair are derived from the Prx1 lineage. FACS and molecular analyses
indicate that the Prx1-‐derived muscle cells are muscle interstitial cells distinct from
endothelial, hematopoietic and myogenic cells, but overlaps with the muscle
fibro/adipogenic progenitors population marked by Pdgfrα. To better characterize
bone-‐muscle crosstalk, we developed a new musculoskeletal trauma model. Under
ethical approval, tibial fractures were induced in adult mice with or without injury to
muscles surrounding the tibia. Histomorphometric analyses show that muscle injury
delays callus, cartilage and bone formation. This was accompanied by abnormal callus
organization with the presence of unresorbed cartilage and fibrosis leading to the
absence of bone bridging and non-‐union. Using grafting experiments, we show that the
contribution of skeletal muscle and periosteum to cartilage within the callus are
decreased in fractures with muscle injury. In the trauma environment, fibrosis within
the fracture callus is derived from the Prx1-‐derived muscle lineage but not from the

16
periosteum. Imatinib treatment, which targets PDGFRα, Bcr-‐Abl and c-‐Kit proteins,
decreases fibrosis and ameliorates bone repair in context of musculoskeletal trauma.
In conclusion, we identified an interstitial cell population within skeletal muscle that
contributes directly to cartilage and bone during fracture repair, and is the source of
fibrosis in a traumatic injury environment causing fracture non-‐union. This study
provides a cellular basis for delayed bone regeneration in severe musculoskeletal
injuries.
Keywords : muscle, bone, bone repair, muscle-‐bone interactions, lineage tracing, genetic of mouse model

17
Rôle du muscle au cours de la régénération osseuse: étude fonctionnelle de la contribution cellulaire et impact des traumatismes musculosqueletiques
La régénération tissulaire est basée sur l’activation, le recrutement et la différenciation
des cellules souches. Au cours de la régénération osseuse, les cellules stromales de la
moelle osseuse (CSMO) et les cellules du périoste (CP) sont deux sources d'ostéo-‐
chondro-‐progéniteurs (OCP). Dans la première partie de mon travail de thèse, nous
avons montré que le périoste contient des SSCs et que les CPs ont des capacités de
régénération plus élevées que les CSMOs. Le potentiel d'auto-‐renouvellement est
modulé par la niche des SSCs au sein du périoste et dépend de la protéine
extracellulaire, Périostine.
Malgré ses grandes capacités de régénération, l'os présente un retard de régénération
dans 10% des cas de lésions osseuses et cette proportion s’élève à 46% lors d’une
fracture avec atteinte des tissus environnants tels que le muscle. Le rôle du muscle
squelettique est bien connu dans le domaine orthopédique et il a été décrit comme une
source potentielle de cellules et de facteurs au cours de la réparation osseuse.
Cependant, les mécanismes cellulaires et moléculaires sous-‐jacents aux interactions os-‐
muscle au cours de la réparation osseuse sont mal compris. Dans la deuxième partie de
mon travail de thèse, nous avons montré que les cellules satellites musculaires sont
nécessaires à la réparation osseuse en tant que source de facteurs de croissance et que
le muscle est aussi une source d’OCPs pendant la régénération osseuse. La troisième
partie de mon travail de thèse a porté sur la caractérisation des cellules dérivées du
muscle squelettique en combinant des approches de lignage cellulaire et greffes de
tissus. Les OCPs sont activement recrutés du muscle squelettique pendant la réparation
osseuse et sont dérivés du lignage embryonnaire mésenchymateux Prx1, qui marque
également les CPs. Les analyses de cytométrie et moléculaires indiquent que les cellules
musculaires dérivées du lignage Prx1 sont des cellules interstitielles musculaires
distinctes des cellules endothéliales, hématopoïétiques et myogéniques, mais
chevauchant avec les progéniteurs fibro/adipogéniques musculaires marqués
par Pdgfrα. Pour mieux comprendre les interactions os-‐muscle au cours de la
régénération osseuse, nous avons développé un nouveau modèle de traumatisme
musculosquelettique. Après approbation éthique, des fractures du tibia ont été induites
chez des souris adultes avec ou sans blessure des muscles entourant le tibia. Les
analyses histomorphométriques montrent que les lésions musculaires retardent la

18
formation du cal, du cartilage et de l’os. Cela est accompagné d'une organisation
anormale du cal avec la présence de cartilage et de fibrose non résorbés conduisant à
l'absence de pontage osseux et à la non-‐consolidation de la fracture. Par des expériences
de greffes tissulaires, nous montrons que la contribution des muscles squelettiques et
du périoste au cartilage est diminuée dans le modèle de traumatisme
musculosquelettique. Dans l'environnement traumatique, le lignage Prx1 musculaire
forme le tissu fibrotique, contrairement au périoste. Dans le but de diminuer la fibrose,
nous avons utilisé l'Imatinib, qui cible les protéines PDGFRα, Bcr-‐Abl et c-‐Kit. Nous
montrons que l'Imatinib améliore la réparation osseuse dans un contexte de
traumatisme musculosquelettique.
En conclusion, nous avons identifié une population cellulaire interstitielle dans le
muscle squelettique qui contribue directement à la formation du cartilage et de l’os lors
de la régénération osseuse, mais qui est aussi la source de la fibrose dans un
environnement de lésion traumatique, provoquant une régénération imparfaite. Cette
étude fournit une base cellulaire et moléculaire pour traiter les déficits de régénération
osseuse dans les blessures musculosquelettiques sévères.
Mots clés : os, muscle, régénération osseuse, interactions os-‐muscle, lignage cellulaire,
génétique de la souris

Introduction
19
Table des matières Table des matières ...................................................................................................................... 19
Liste des illustrations ................................................................................................................. 21 Liste des abréviations ................................................................................................................. 23
Introduction ................................................................................................................................... 25 1. Le tissu osseux et la régénération osseuse ................................................................. 25 1.1 Structure du tissu osseux. ........................................................................................................................... 25 1.1.1. Les os longs ................................................................................................................................................................ 25 1.1.2. Les os courts et les os plats ................................................................................................................................. 26
1.2. Composition et fonctions du tissu osseux .......................................................................................... 27 1.3. Le développement osseux ......................................................................................................................... 27 1.3.1. L’ossification endochondrale des os longs ................................................................................................... 28 1.3.2. L’ossification intramembranaire ...................................................................................................................... 31
1.4. Croissance et maintien du tissu osseux ............................................................................................... 31 1.4.1. Croissance en longueur et en épaisseur ........................................................................................................ 31 1.4.2. Maintien homéostatique du tissu osseux ...................................................................................................... 32
1.5. La régénération osseuse après fracture .............................................................................................. 35 1.5.1. Les étapes de la régénération osseuse par voie endochondrale ........................................................ 35 1.5.2. Régulation moléculaire de la régénération osseuse ................................................................................ 38 1.5.3. Le rôle de l’environnement mécanique au cours de la régénération osseuse .............................. 39
1.6. Sources de cellules lors de la régénération osseuse ...................................................................... 40 1.6.1. Le concept de cellule souche mésenchymateuse ....................................................................................... 41 1.6.2. La moelle osseuse, une source minimale de cellules ............................................................................... 43 1.6.3. Le périoste, une source majeure de cellules ................................................................................................ 44 1.6.4. Le muscle, une source de cellules pour la réparation osseuse ? ......................................................... 45 1.6.5. Les cellules utilisées en thérapie humaine ................................................................................................... 45
2. Le muscle squelettique ...................................................................................................... 48 2.1. Les cellules myogéniques du muscle squelettiques ....................................................................... 48 2.1.1. Développement musculaire .............................................................................................................................. 49 2.1.1. Régénération musculaire ................................................................................................................................... 50
2.2. Les cellules non-‐myogénique du muscule squelettiques ............................................................. 52 2.2.1. Le système vasculaire musculaire ................................................................................................................... 53 2.2.1.a. Les vaisseaux ................................................................................................................................................... 53 2.2.1.b. Les péricytes .................................................................................................................................................... 53 2.2.1.c. Les mésoangioblastes ................................................................................................................................... 55
2.2.2. Le système fibro-‐mésenchymateux ................................................................................................................. 56 2.2.2.a. Les fibroblastes ............................................................................................................................................... 57 2.2.2.b. Les cellules mésenchymateuses musculaires / FAPs ..................................................................... 57 2.2.1.d. Les cellules intersticielles PW1+ (PICs) ............................................................................................... 59
3. Les interactions os-‐muscle ............................................................................................... 61 3.1. Interactions biomécaniques ..................................................................................................................... 61 3.2. Interactions moléculaires .......................................................................................................................... 63 3.3. Interactions os-‐muscle et ossification hétérotopique ................................................................... 65 3.4. Interactions os-‐muscle au cours de la régénération osseuse ..................................................... 66 3.5. Modèles murins d’étude du rôle du muscle dans la régénération osseuse .......................... 68
Objectifs de thèse ......................................................................................................................... 71
Article 1 ........................................................................................................................................... 73
Article 2 ......................................................................................................................................... 103 Article 3 (en cours de soumission) ....................................................................................... 121

Introduction
20
Discussion ..................................................................................................................................... 163
Références .................................................................................................................................... 171
Curriculum Vitae ........................................................................................................................ 211

Introduction
21
Liste des illustrations Figure 1 : Anatomie d’un os long Figure 2 : Schéma du développement osseux des os longs Figure 3 : Schéma de la croissance en longueur et en épaisseur des os longs Figure 4 : Schéma du remodelage osseux Figure 5 : Etapes de la régénération osseuse Figure 6 : Sources de cellules au cours de la régénération osseuse Figure 7 : Structure du muscle squelettique Figure 8 : Schéma du développement musculaire au cours de l’embryogenèse Figure 9 : Schéma de la régénération musculaire Figure 10 : Schéma du système périvasculaire musculaire Figure 11 : Schéma du système fibro-‐mésenchymateux musculaire Figure 12 : Schéma des interactions os-‐muscle

Introduction
22

Introduction
23
Liste des abréviations αSMA α Smooth Muscle Actin ADAM12 Désintégrine-‐métalloprotéinase 12 Agc Aggrécane Angpt Angiopoiétine AP Alkaline phosphatase BMP Bone Morphogenetic Protein cBMA concentrate of Bone Marrow Aspirate CCR C-‐C chemokine receptor CD29 Cluster de différentiation 29, Intergrine β1 Col Collagène COX2 Cyclooxygénase-‐2 CP Cellules du périoste CSF-‐1 Colony stimulator factor 1 CSH Cellules souches hématopoïétiques CSM Cellules souches mésenchymateuses CSMO Cellules souches de la moelle osseuse CSS Cellules souches squelettiques Cxcl12 C-‐X-‐C motif chemokine 12, stromal cell-‐derived factor 1 EDL Extensor digitus lengus FAP Progéniteurs fibro/adipogéniques FGF Fibroblast Growth Factor FGFR3 Fibroblast Growth Factor Receptor 3 FOP Fibrodysplasia Ossifians Progressiva FRM Facteurs de Régulation Myogéniques HGF Hepatocyte Growth Factor IGF Insulin Growth Factor IHH Indian Hedgehog Homolog IL Interleukine LeptR Leptin receptor M-‐CSF Macrophage Colony-‐Stimulating Factor MAB Mésoangioblastes MAPK Mitogen-‐Activated Protein Kinases MHC Myosin Heavy Chain Mmp Métalloprotéinase de la matrice MO Moelle Osseuse Mstn Myostatine Myf5 Myogenic factor 5 NG2 Neural/glial antigen 2 OCP Ostéochondroprogéniteurs OH Ossification hétérotopique Osx Osterix Pax3/7 Paired box 3/7 PDGF Platelet Derived Growth Factor PDGFRα Platelet Derived Growth Factor Receptor α

Introduction
24
PIC PW1 intersticial cells PMV Perte Musculaire Volumique Postn Périostine Prx1 Paired related homeobox 1 PTH Para-‐Thyroid Hormone PTHrP Para-‐Thyroid Hormone-‐related Protein PW1/Peg3 Paternally expressed 3 RANKL Receptor Activator of Nuclear Factor κ-‐B Ligand Runx Runt-‐related transciption factor Sca1 Stem cells antigen-‐1 SCDM Cellules Souches Dérivées du Muscle TA Tibialis anterior TGF β Transforming Growth Factor β

Introduction
25
Introduction
Le système musculosquelettique assure différentes fonctions essentielles du corps telles
que le mouvement, le stockage de minéraux ou la protection des organes internes. Les
deux principaux composants du système musculosquelettique sont les os et les muscles
striés squelettiques. Chez l’Homme, le squelette est composé de 206 os et le système
musculaire de 570 muscles striés[3],[4].
1. Le tissu osseux et la régénération osseuse
1.1 Structure du tissu osseux.
A l’âge adulte, les os du squelette (os sésamoïdes non compris) peuvent être divisés en
trois catégories anatomiques : les os longs, les os courts et les os plats.
1.1.1. Les os longs
Les os longs sont situés dans les membres supérieurs et inférieurs. Ils sont formés de deux
épiphyses situées aux deux extrémités et d’une diaphyse correspondant à la partie centrale de
l’os. Epiphyse et diaphyse sont séparées par la métaphyse qui contient la plaque de croissance
permettant la croissance en longueur des os longs. Les épiphyses sont composées d’os
spongieux (ou trabéculaire) contenant la moelle osseuse rouge et sont recouvertes par le
cartilage articulaire. Elles servent de site d’ancrage aux tendons et aux ligaments au niveau des
articulations. La diaphyse est constituée d’os compact (cortex) composé d’ostéons ou système
de Harvers (ostéocyte et canal neuro-‐vasculaire entouré d’os lamellaire). A l’intérieur de la
diaphyse se trouve la cavité médullaire occupée par la moelle osseuse[5]. Chez l’enfant, la cavité
médullaire des os longs est le siège majeur de l’hématopoïèse, alors que chez l’adulte,
l’hématopoïèse a lieu principalement au sein des os plats. La cavité de la moelle contient
différents types de cellules dont les cellules hématopoïétiques et les cellules stromales de la
moelle qui forment la niche des cellules souches hématopoïétiques (CSH)[6],[7]. La moelle
osseuse et le cortex sont en contact direct via l’endoste[8]. L’endoste est un tissu hétérogène
formé de cellules bordantes contenant des ostéoblastes, des ostéoclastes et des vaisseaux[9]. Sur
sa face externe, le cortex est recouvert d’une fine membrane appelée périoste qui sert d’ancrage
aux muscles adjacents et permet la croissance en épaisseur du cortex[10-‐12]. Le périoste est riche

Introduction
26
en neurofibres[13, 14], vaisseaux[15] et contient, entre autres, des cellules souches osseuses[16-‐18]
(Fig. 1).
1.1.2. Les os courts et les os plats
Les vertèbres, les os du coup du pied, les os du poignet sont des os courts alors que les
os du crâne, le sternum, les omoplates ou les os du bassin sont des os plats. Les os courts
et plats sont dépourvus de cavité médullaire et sont formés de deux couches d’os
compact. Entre ces deux couches, se trouve une quantité variable d’os spongieux qui
Figure 1: Anatomie d’un os long. (A) Structure générale d’un os long (B) Structure d’une épiphyse (C) Structure d’une diaphyse[5]

Introduction
27
contient la moelle rouge. Comme pour les os longs, le périoste tapisse la surface externe
de l’os compact. Les os plats ne contiennent pas de moelle jaune (formée principalement
d’adipocytes) mais uniquement de la moelle rouge et sont donc un site important de
formation des globules rouge chez l’adulte[19].
1.2. Composition et fonctions du tissu osseux
La matrice osseuse est composée de matière organique et de matière minérale. La
matrice organique est formée de collagène de type I (principalement), III et V, de
glycoaminoglycanes (décorine, biglycan), de glycoprotéines (ostéonectine,
thrombospondine, fibronectine), d’ostéocalcine et de protéines de la famille SIBLING[3].
Ces protéines de la matrice extracellulaire sont liées à des facteurs de croissance (TGF β,
IGF, FGF, PDGF) impliqués dans la formation osseuse et le maintien homéostatique du
tissu osseux[20]. La matière organique est associée aux cristaux d’hydroxyapatite de
formule Ca5(PO4)3(OH) qui forment la matière minérale[21].
De part leur composition, les os assurent des fonctions mécaniques, métaboliques et
hormonales. L’organisation lamellaire des fibrilles de collagène et des cristaux
d’hydroxyapatite confère à l’os une dureté et une résistance mécanique nécessaire au
maintien et à la mobilité du corps ainsi qu’à la protection d’organes vitaux tels que le
cerveau ou les organes internes[22]. Les os sont aussi la réserve principale de calcium et
de phosphate du corps. Ces deux ions jouent un rôle important dans le métabolisme et la
structure cellulaire, la régulation de certaines voies de signalisation[3, 23]. Enfin, le tissu
osseux remplit des fonctions endocrines, via la sécrétion de l’hormone ostéocalcine
notamment. L’ostéocalcine régule la sécrétion de l’insuline au niveau du pancréas,
promeut la production de testostérone dans les testicules, la sécrétion de l’interleukine
6 (IL-‐6) du muscle squelettique, et agit sur les fonctions cognitives du cerveau[24, 25] . Les
os sont aussi un site majeur d’hématopoïèse chez l’adulte.
1.3. Le développement osseux
Le développement osseux a lieu selon deux types d’ossification : ossification
endochondrale principalement pour les os longs et courts ou ossification
intramembranaire pour certains os du crâne par exemple[26]. Au cours de l’ossification

Introduction
28
endochondrale, chaque élément squelettique est composé dans un premier temps de
cartilage qui est ensuite ossifié. Au cours de l’ossification intramembranaire, chaque
élément squelettique se forme sans intermédiaire cartilagineux.
1.3.1. L’ossification endochondrale des os longs
L’ossification endochondrale des os longs est initiée par les condensations
mésenchymateuses de cellules mésodermiques dérivant du lignage Prx1 au niveau des
bourgeons de membres[27] (Fig.2A). L’expression des Bone Morphogenetic Proteins
(BMP) au sein des cellules Prx1+ permettent la croissance des condensations
mésenchymateuses [28, 29]. Les cellules à la périphérie des condensations s’allongent et
s’alignent pour former le périchondre, alors que les cellules au centre des condensations
qui expriment Sox9 se différencient en chondrocytes prolifératifs exprimant Col2A1[30]
(Fig.2B). Ces chondrocytes secrètent la matrice cartilagineuse riche en collagène de type
II et en protéoglycans. Ces chondrocytes se différencient ensuite en chondrocytes pré-‐
hypertrophiques puis hypertrophiques et sécrétent le collagène de type X (Fig.2C). Cette
étape est finement régulée par plusieurs voies de signalisation. Smad4, un effecteur de la
voie BMP, induit l’expression de Runx2 au sein des chondrocytes pré-‐hypertrophiques
ce qui permet la différenciation hypertrophique finale des chondrocytes[31, 32]. La
différenciation chondrocytaire est aussi régulée par la boucle de régulation IHH-‐PTHrP
(Indian hedgehog homolog -‐ Parathyroid hormone-‐related Protein). IHH est exprimé par
les chondrocytes pré-‐hypertrophiques et PTHrP par les cellules péri-‐articulaires. Les
chondrocytes pré-‐hypertrophiques expriment IHH qui diffuse jusqu’aux cellules péri-‐
articulaires. En réponse, ces cellules sécrètent PTHrP qui diffuse dans la plaque de
croissance et promeut la prolifération des chondrocytes. Dès que le niveau de PTHrP
diminue sous un seuil critique, les chondrocytes sortent du cycle cellulaire et entament
leur différenciation[33, 34]. Les voies BMP et IHH-‐PTHrP sont en partie régulées par la
signalisation FGF via FGFR3. En effet, l’absence de FGFR3 induit une augmentation de
l’expression de IHH et de BMP4. Dans le cas contraire où le récepteur FGFR3 est activé
de façon constitutive, l’expression de IHH et BMP4 est diminuée et la différenciation des
chondrocytes pré-‐hypertrophiques en chondrocytes hypertrophiques est bloquée[35].
Les chondrocytes hypertrophiques secrètent le facteur angiogénique Vascular
Endothelial Growth Factor (VEGF), qui permet l’invasion vasculaire (Fig.2D). L’invasion

Introduction
29
vasculaire permet la migration des cellules hématopoïétiques et des cellules
ostéogéniques Osterix+ (Osx+) au niveau du centre d’ossification primaire[36-‐38]. Sous
l’action de BMP2 et BMP4, les cellules Osx+ expriment Runx2 et se différencient en
ostéoblastes au sein du centre d’ossification primaire et au niveau du périchondre [39, 40].
Au sein des ostéoblastes Osx+, Smad4 interagit avec Runx2 et la voie canonique de Wnt
pour induire la formation de matrice osseuse[41]. Ainsi, les ostéoblastes secrètent du
collagène de type I, principal composant de la matrice osseuse. Une autre source
d’ostéoblastes provient de la transdifférenciation des chondrocytes hypertrophiques.
Les chondrocytes restants sont éliminés par apoptose[42-‐45]. Les ostéoblastes entourés de
matrice osseuse se différencient ensuite en ostéocytes. Les ostéoclastes qui dérivent des
monocytes résorbent de la matrice cartilagineuse via l’action de MMP9 notamment afin
de former la cavité médullaire[46, 47] (Fig.2E). Les centres d’ossifications secondaires se
développent en parallèle au sein des épiphyses après l’invasion vasculaire. Le cartilage
épiphysaire est ensuite résorbé et remplacé par de l’os spongieux contenant la moelle
osseuse [48] (Fig. 2F).
Des approches de lignage cellulaire ont permis de mieux caractériser l’origine des
progéniteurs squelettiques impliqués au cours du développement osseux. Des
transplantations de cartilage embryonnaire dans la capsule rénale ont montré l’absence
de recrutement systémique des ostéoblastes et leur provenance du périchondre[49]. Les
ostéoblastes du périchondre Osx+ migrent le long des vaisseaux vers le centre
d’ossification primaire pour former l’os[50]. Au stade post-‐natal, le lignage Osx+ forment
les ostéoblastes mais aussi lieu aux cellules stromales de la moelle, tout comme le
lignage Nestin+ [51][52]. Par ailleurs, une autre étude utilisant des transplantations dans la
capsule rénale, a montré qu’uniquement les os formés par ossification endochondrale
permettaient la formation de la niche des HSCs au sein de la cavité de la moelle
osseuse[53].

Introduction
30
Figure 2: Schéma représentatif du développement de l’os par voie endochondrale. Les cellules du bourgeon de membre forment les condensations mésenchymateuses composées de chondrocytes entourés par le périchondre. Les chondrocytes se différencient en chondrocytes hypertrophiques au centre du cartilage permettant l’invasion vasculaire qui amène des ostéoblastes et cellules hématopoïétiques pour former la cavité de la moelle osseuse. La matrice cartilagineuse est résorbée par les ostéoclastes et les ostéoblastes forment la matrice osseuse. La croissance osseuse est ensuite assurée par les plaques de croissances et les centres d’ossification secondaires[2].

Introduction
31
1.3.2. L’ossification intramembranaire
Les os plats du crâne sont issus des dérivés de crête neurale ou du mésoderme, et sont
formés via le processus d’ossification intramembranaire. Après l’étape de condensation,
les cellules mésenchymateuses expriment le marqueur ostéoblastique Osx, se
différencient en ostéoblastes et sécrètent le collagène de type I sans former de matrice
cartilagineuse. Les cellules à l’intérieur des condensations sont alors emprisonnées dans
la matrice osseuse et se différencient en ostéocytes après l’invasion vasculaire. Les
ostéoblastes situés à l’extérieur des condensations mésenchymateuses sécrètent de la
matrice osseuse qui forme l’os compact. Les cellules mésenchymateuses bordant cet os
compact donnent lieu au périoste et la moelle osseuse rouge infiltre l’os spongieux [54-‐56].
1.4. Croissance et maintien du tissu osseux
1.4.1. Croissance en longueur et en épaisseur La croissance osseuse en longueur est assurée par la plaque de croissance située à la
jonction entre l’épiphyse et la diaphyse[43]. La plaque de croissance est composée de
chondrocytes organisés « en colonne »[57]. Les chondrocytes situées sous l’os spongieux
de l’épiphyse sont indifférenciés et sont appelés chondrocytes de réserve. Les
chondrocytes de réserve expriment Sox9 induisant l’expression de gènes permettant la
production de matrice cartilagineuse tels que Col2a1 ou le protéoglycan aggrecan
(Agc)[58]. En proliférant, les chondrocytes s’organisent en colonne. L’inhibition de Sox9
corrélée à l’expression de Runx2 et Runx3 par les chondrocytes prolifératifs
permet l’expression du collagène de type X et la différenciation en chondrocytes
hypertrophiques[59]. Les chondrocytes hypertrophiques produisent la matrice
extracellulaire et des facteurs permettant sa minéralisation. L’arrêt de l’expression de
Col10 corrélé à l’expression de Runx2 et de la métalloprotéinase de la matrice 13
(Mmp13), initie la différenciation terminale des chondrocytes au niveau de la jonction
avec les trabécules osseux de la métaphyse[58, 60, 61]. Les mécanismes régulant la
transition cartilage-‐os ne sont pas entièrement élucidés. Les chondrocytes
hypertrophiques peuvent soit entrer en apoptose ou se transdifférencier en
ostéoblastes[45, 62, 63] (Fig. 3A). Les mécanismes moléculaires sont inconnus mais les

Introduction
32
facteurs impliqués dans le processus d’ossification endochondrale, tels que les BMPs,
Wnt, FGFs, TGF, pourraient aussi réguler spécifiquement cette étape.
La croissance en épaisseur de l’os se fait au niveau du périoste, le long de la diaphyse et
consiste en la formation de nouveaux ostéons. Les cellules ostéoblastiques du périoste
s’invaginent au niveau d’une artèriole. Les ostéoblastes secrètent de la matrice
extracellulaire de façon concentrique pour former de l’os lamellaire où ils s’auto-‐
emprisonnent au fur et à mesure. Cette apposition de matrice permet ainsi la croissance
en épaisseur du cortex (Fig 3B).
1.4.2. Maintien homéostatique du tissu osseux
Le tissu osseux enchaine continuellement des cycles de résorption et de formation
osseuse. La résorption osseuse est effectuée par les ostéoclastes et la formation osseuse
par les ostéoblastes. Ce processus, appelé remodelage osseux, est très finement régulé.
Dans les cas où la balance entre formation/résorption osseuse est déséquilibrée,
Figure 3: Schéma de la croissance osseuse. (A) Croissance en longueur. (B) Croissance en épaisseur des os.

Introduction
33
différentes pathologies telle que l’ostéoporose peuvent se développer[64, 65]. Le
remodelage osseux se déroule en cinq phases : l’initiation, la résorption, la transition, la
formation et la terminaison[1, 66]. La phase d’initiation débute par le recrutement de
cellules hématopoïétiques précurseurs d’ostéoclastes (pré-‐ostéoclastes) via la
circulation sanguine en réponse à une tension mécanique particulière ou à certaines
hormones (œstrogène, PTH par exemple) au site de remodelage. La sécrétion des
cytokines CSF-‐1 et RANKL par les ostéocytes permettent la prolifération des pré-‐
ostéoclastes et leur différenciation terminale en ostéoclastes[67-‐69]. Grâce à leur activité
protéolytique, les ostéoclastes résorbent la matrice osseuse via l’action des cystéines-‐
protéinases et des métalloprotéinases de la matrice[70]. Après résorption de la matrice,
les ostéomacs (macrophages résidents situés au niveau de l’endoste et/ou du périoste)
éliminent la matrice osseuse résorbée pour permettre la synthèse de la nouvelle matrice
par les ostéoblastes[71-‐73]. Les ostéoblastes sont ensuite activés et synthétisent la
nouvelle matrice osseuse. Cette phase de transition entre l’action des ostéoclastes et des
ostéoblastes n’est pas entièrement caractérisée, mais certaines protéines telles que
TGFβ ou IGF-‐1 sont considérés comme des « facteurs de couplages » agissant à la fois sur
les ostéoclastes pour diminuer leur action et sur les ostéoblastes pour favoriser la
synthèse de matrice osseuse[74, 75] [76]. Pendant la phase de formation, les pré-‐
ostéoblastes migrent au niveau des sites de résorption, se différencient en ostéoblastes
et sécrètent du collagène de type I et des protéoglycans[66]. Des cristaux
d’hydroxyapatite sont incorporés dans la matrice de collagène pour former la matrice
osseuse. Les ostéocytes sécrètent la Semaphorin3A (Sem3A), qui inhibe les ostéoclastes
et active les ostéoblastes[77]. Dès que la quantité d’os formé est équivalente à celle
résorbée, la phase de terminaison est initiée au cours de laquelle certains ostéoblastes
entrent en apoptose et d’autres se différencient en ostéocytes. La surface de l’os est
ensuite reconstituée. Le processus de remodelage est terminé[1, 66].

Introduction
34
Figure 4 : Schéma du cycle de remodellage osseux. Phase d’initiation : en réponse à un stimulus, des ostéoclastes sont recrutés et résorbent la matrice osseuse. Phase de transition : les facteurs de couplage (TGFβ, IGF-‐1) inhibent les ostéoclastes et activent les ostéoblastes. Phase de formation : les ostéoblastes sécrètent de la matrice osseuse. Adapté de [1]

Introduction
35
1.5. La régénération osseuse après fracture
1.5.1. Les étapes de la régénération osseuse par voie endochondrale
La régénération osseuse après fracture est un mécanisme très efficace qui permet à l’os
de retrouver sa forme et sa fonction d’origine. Il existe de nombreux points communs
entre le développement osseux et la régénération osseuse, comme les types cellulaires
intervenant dans ces deux processus (cellules mésenchymateuses, chondrocytes,
ostéoblastes et ostéoclastes), les voies de signalisation communes telles que BMP, FGF
ou TGF-‐β et les deux voies d’ossification : voie endochondrale ou voie
intramembranaire[78]. Cependant, la régénération osseuse est aussi régulée par
l’inflammation et les contraintes mécaniques qui n’interviennent pas ou différemment
au cours du développement osseux
Chez la souris, de nombreux modèles de réparation osseuse des os longs décrivent un
processus en quatre étapes faisant intervenir de façon coordonnée l’ossification
endochondrale et l’ossification intramembranaire : la phase inflammatoire (formation
de l’hématome, infiltration des cellules inflammatoires et recrutement des cellules
souches/progéniteurs osseux), la phase de cal mou ou fibro-‐cartilagineux
(vascularisation du site de fracture et formation du cartilage et de l’os), la phase de cal
dur ou osseux (résorption du cartilage et remplacement par le tissu osseux) et phase de
remodelage osseux (résorption de la matrice osseuse)[79, 80].
Après fracture, la première étape de la régénération osseuse consiste en la formation
d’un hématome due à la rupture des vaisseaux. La formation de l’hématome est une
étape cruciale et coïncide avec la sécrétion de facteurs pro-‐inflammatoires permettant le
recrutement des cellules inflammatoires et des ostéoclastes[81, 82]. Les cytokines M-‐CSF,
IL-‐1β, TNFα, IL-‐1 et IL-‐6 sont sécrétées[83, 84] et permettent la mobilisation des
neutrophiles[85]. Ces neutrophiles sécrètent à leur tour des cytokines telles que CCR2[86],
IL-‐6[87, 88] ou IL-‐17[89, 90] qui participent au recrutement des macrophages pro-‐
inflammatoires (M1) et des lymphocytes T. La sécrétion des interleukines IL-‐4 et IL-‐13
et l’invagination des neutrophiles par les macrophages M1 induit un changement
phénotypique des macrophages pro-‐inflammatoire M1 vers des macrophages anti-‐
inflammatoires M2[91, 92]. Ce changement phénotypique permet la diminution de la
sécrétion des molécules pro-‐inflammatoires et l’augmentation de la sécrétion des
molécules anti-‐inflammatoires telle que TGF-‐β, connue pour induire la chondrogenèse,

Introduction
36
le recrutement et la prolifération des précurseurs squelettiques[75, 91, 93, 94]. Les
macrophages sécrètent VEGF et PDGF-‐BB ce qui contribue à la revascularisation du cal [95, 96]. Le contrôle de la durée de la phase inflammatoire est fondamental pour une
régénération osseuse efficace. En effet, une phase inflammatoire prolongée (dans le cas
de maladies auto-‐immunes telles que le lupus ou le diabète) ou réduite conduit à une
régénération altérée pouvant aller jusqu’à l’absence de consolidation osseuse[97-‐101].
Durant la phase inflammatoire, les cellules immunitaires sécrètent des facteurs
trophiques tels que CXCL12 qui participent au recrutement et à l’établissement des
cellules mésenchymateuses CCR4+ au niveau du site de fracture[102-‐104].
L’activation des cellules souches osseuses a lieu dans les 3 premiers jours après fracture
conduisant à la formation du cal mou ou cal fibro-‐cartilagineux[97, 105]. L’expression de
facteurs de croissance tels que les BMP[106, 107], FGF[108, 109], VEGF[110, 111], IGF[112] induit la
différenciation de cellules souches osseuses en chondrocytes et ostéoblastes[113].
Pendant la deuxième étape de réparation, le cal mou est composé majoritairement de
cartilage au centre du cal où les contraintes mécaniques sont élevées et d’os en
périphérie où les contraintes mécaniques sont plus faibles. En périphérie du cal, les
cellules souches squelettiques se différencient directement en ostéoblastes et sécrètent
du collagène de type I pour former la matrice osseuse[114]. Au centre du cal, Les
progéniteurs squelettiques prolifèrent et se différencient en chondrocytes[115] qui
secrètent des protéines de la matrice cartilagineuse telles que le collagène de type II et
aggrecan. Les chondrocytes se différencient ensuite en chondrocytes hypertrophiques
qui sécrètent du collagène de type X et du VEGF permettant la revascularisation du cal.
Lors de la troisième étape, dite du « cal dur», le cartilage est activement résorbé via
l’action de Mmp9 ou Mmp13 pour être remplacé par l’os[116, 117]. De même que lors du
processus d’ossification endochondrale pendant l’embryogenèse, les chondrocytes
hypertrophiques peuvent être éliminés par apoptose ou se transdifférencier en
ostéoblastes. Il a cependant été observé que certains chondrocytes hypertrophiques
proches des vaisseaux se remettent à prolifèrer et ré-‐expriment des facteurs de cellules
souches comme Oct4, Nanog et Sox2 [118-‐120]. Ces chondrocytes pourraient ensuite se
différencier en ostéoblastes. Le cartilage est remplacé par l’os grâce à l’action des
ostéoblastes sécrétant le collagène de type I. Pendant la quatrième étape de
régénération osseuse, l’os spongieux subit un remodelage sous l’action des ostéoclastes
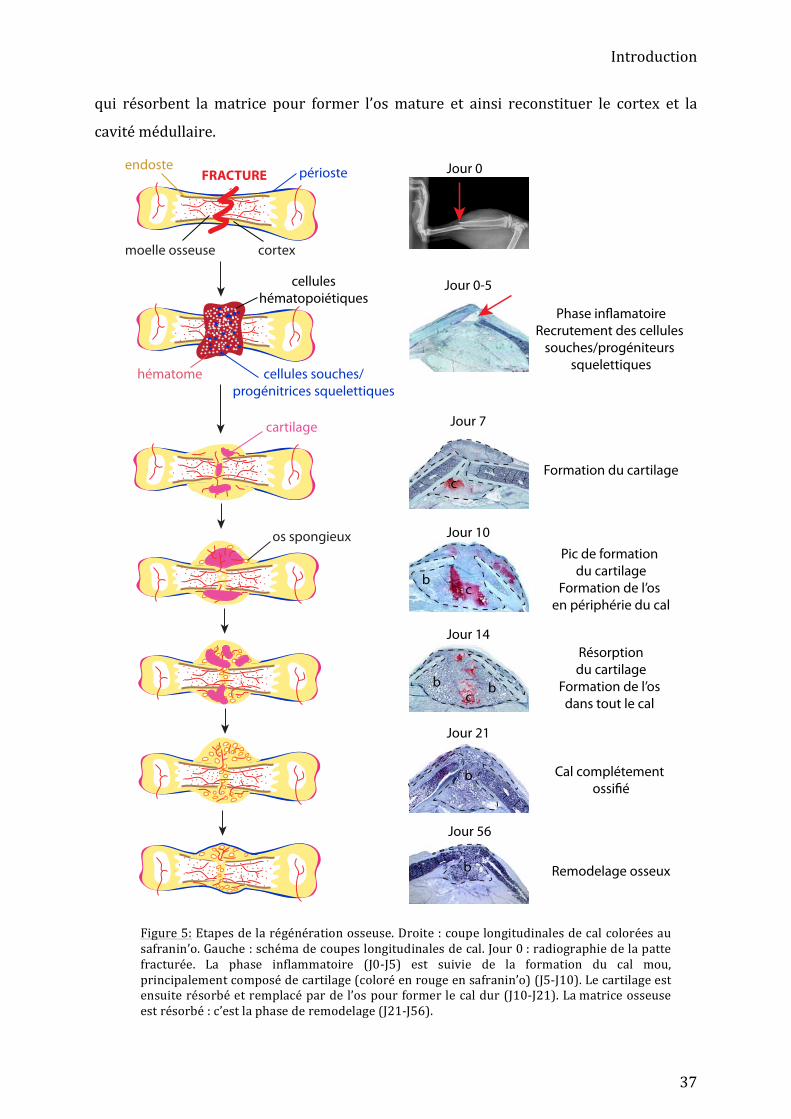
Introduction
37
qui résorbent la matrice pour former l’os mature et ainsi reconstituer le cortex et la
cavité médullaire.
Figure 5: Etapes de la régénération osseuse. Droite : coupe longitudinales de cal colorées au safranin’o. Gauche : schéma de coupes longitudinales de cal. Jour 0 : radiographie de la patte fracturée. La phase inflammatoire (J0-‐J5) est suivie de la formation du cal mou, principalement composé de cartilage (coloré en rouge en safranin’o) (J5-‐J10). Le cartilage est ensuite résorbé et remplacé par de l’os pour former le cal dur (J10-‐J21). La matrice osseuse est résorbé : c’est la phase de remodelage (J21-‐J56).
FRACTURE
celluleshématopoiétiques
hématome cellules souches/progénitrices squelettiques
périosteendoste
moelle osseuse cortex
cartilage
os spongieux
c
cb
cb b
b
b
Jour 0
Jour 0-5
Phase inflamatoireRecrutement des cellules
souches/progéniteurs squelettiques
Jour 7
Formation du cartilage
Pic de formation du cartilage
Formation de l’os en périphérie du cal
Jour 10
Jour 14Résorptiondu cartilage
Formation de l’os dans tout le cal
Cal complétement ossifié
Remodelage osseux
Jour 21
Jour 56

Introduction
38
1.5.2. Régulation moléculaire de la régénération osseuse
De nombreuses voies de signalisation dont BMP, Wnt, IHH-‐PTHrP et PTH sont
impliquées dans la régénération osseuse et participent à l’activation, au recrutement et à
la différenciation des cellules souches/progénitrices.
Du fait de leur potentiel ostéogénique important, les BMPs sont actuellement utilisés
cliniquement en orthopédie. Cependant, au vu de l’efficacité variable des traitements et
des effets secondaires observés, le rôle précis de ces molécules au cours de la
régénération osseuse notamment doit être mieux caractérisé. En ce sens, des approches
de délétion conditionnelle ont été développées afin de mieux comprendre leur rôle au
cours de la régénération osseuse. Les souris Prx1Cre/+ ;BMP7fl/fl et Prx1Cre/+ ;BMP4fl/fl ne
présentent aucune malformation osseuse ni retard de régénération osseuse. Cependant,
les souris Prx1Cre/+ ;BMP2fl/fl présentent une déficience sévère de régénération osseuse ce
qui démontre le rôle essentiel de BMP2 au cours de la régénération osseuse[106]. Des
approches de greffes de segments osseux combinées à l’inactivation de Bmp2 ont permis
de montrer que BMP2 est nécessaire à l’activation et à la différenciation des cellules du
périoste[121].
Contrairement au développement embryonnaire où le réseau de signalisation HH est
indispensable pour la différenciation chondrocytaire, au cours de la régénération
osseuse l’ablation de la voie de signalisation HH au sein des chondrocytes Col II+
n’affecte pas la régénération osseuse. Cependant, l’ablation de HH au sein des
ostéoblastes Col I+ induit un retard de régénération et un défaut de minéralisation[122].
Cela corrèle avec le fait que les cellules du périoste présente un défaut de différentiation
ostéogénique in vitro en absence d’HH[121].
La voie de signalisation PTH est aussi impliquée au cours de la régénération osseuse. Les
souris PTH KO présentent une régénération imparfaite avec une diminution du volume
du cal et du cartilage. De plus, les injections quotidiennes d’iPTH (intermitent PTH)
accélèrent la régénération osseuse, diminuent l’apoptose des ostéoblastes mais
augmentent leur maturation [123, 124]. De ce fait, PTH est utilisée comme traitement
contre l’ostéoporose afin d’augmenter la formation osseuse. L’administration d’iPTH
induit la diminution de l’expression de sclérostine, un antagoniste de la voie Wnt,
sécrétée par les ostéoblastes et les ostéocytes[125]. La voie Wnt est impliquée dans de
nombreux processus développementaux et de régénération. Dans le cas de la

Introduction
39
régénération osseuse, la suractivation de Wnt améliore la régénération alors que
l’inhibition de la voie altère sévèrement la régénération osseuse[126, 127]. Wnt pourrait
agir sur les cellules du périoste et régulerait leur différenciation en chondrocytes et
ostéoblastes[128].
1.5.3. Le rôle de l’environnement mécanique au cours de la régénération osseuse
La stimulation mécanique est un facteur important de l’homéostasie osseuse. cependant,
les résultats de l’effet de l’hypergravité sur le tissu osseux sont nuancés. Cet effet dépend
des os étudiés, de l’âge, du fond génétique des animaux et de l’intensité et de la durée de
l’hypergravité. Il semble que l’hypergravité chronique peut être bénéfique à une
intensité de 2g avec une augmentation de la densité osseuse et une diminution du
nombre d’ostéoclastes, mais elle peut être délétère à une intensité de 3g avec des effets
inverses à ceux observés à 2g [129-‐131]. La perte de masse osseuse due à l’absence de
stimulus mécanique est connue chez les spationautes, les personnes immobilisées ou
paralysées [132-‐134], et cette perte de masse osseuse est corrélée à une perte de masse
musculaire [135]. La microgravité induit une augmentation de l’activité ostéoclastique, la
mort des ostéocytes et affecte les capacités de différenciation des ostéoblastes [136, 137].
La stimulation mécanique du tissu osseux joue aussi un rôle au cours de l’exercice (voir
partie 3.1). Les effets de la gravité sur la régénération osseuse ne sont pas décrits dans la
littérature.
Au cours de la régénération osseuse, de nombreux modèles animaux illustrent l’effet de
l’environnement mécanique sur la nature du processus d’ossification. Une instabilité
mécanique favorise l’ossification par voie intramembranaire, alors que l’instabilité
favorise l’ossification par voie endochondrale[117, 138, 139]. Le rôle de l’environnement
mécanique est connu mais son effet est dépendant du type et de la position de la fracture [140, 141]. Chez l’homme, l’absence complète de stabilisation du site de fracture induit une
augmentation de la taille du cal [142, 143], une formation excessive de cartilage [144] et une
diminution de la vascularisation [145], qui augmentent significativement le risque de non-‐
consolidation. Cependant, la stimulation mécanique du site de fracture par l’activité
musculaire ou par ultrasons peut accélérer la régénération et améliorer la qualité du
tissu osseux néoformé[146, 147]. Les mécanismes d’action des stimuli mécaniques sur les
étapes de la régénération osseuse restent méconnus. Les stimuli mécaniques pourraient

Introduction
40
avoir un effet au cours de la phase inflammatoire [148], de l’invasion vasculaire [145], de la
différenciation des cellules progénitrices [113] ou du remodelage osseux [117].
1.6. Sources de cellules lors de la régénération osseuse
Si les étapes de la régénération osseuse sont bien connues, les cellules impliquées dans
la formation du cartilage et de l’os restent encore à mieux caractériser. Des expériences
de transplantations de populations de cellules souches hématopoïétiques (CSHs)
clonales ont montré que les CSHs peuvent se différencier en ostéoblastes[149].
Cependant, des analyses de lignage cellulaire utilisant la souris CD45Cre/+ ont montré que
le lignage hématopoïétique n’est pas une source d’ostéoblastes au cours de la
régénération osseuse[150]. Des expériences de parabiose ont suggéré un recrutement
systémique d’ostéoblastes lors de la régénération osseuse, mais ce recrutement semble
minimal[151, 152]. Cependant, le recrutement systémique permet l’apport de cellules
immunitaires et d’ostéoclastes, qui jouent un rôle important dans les étapes de la
réparation osseuse [153]. Les cellules souches/progénitrices squelettiques ont donc une
origine locale au cours de la régénération osseuse, dont trois sources principalement
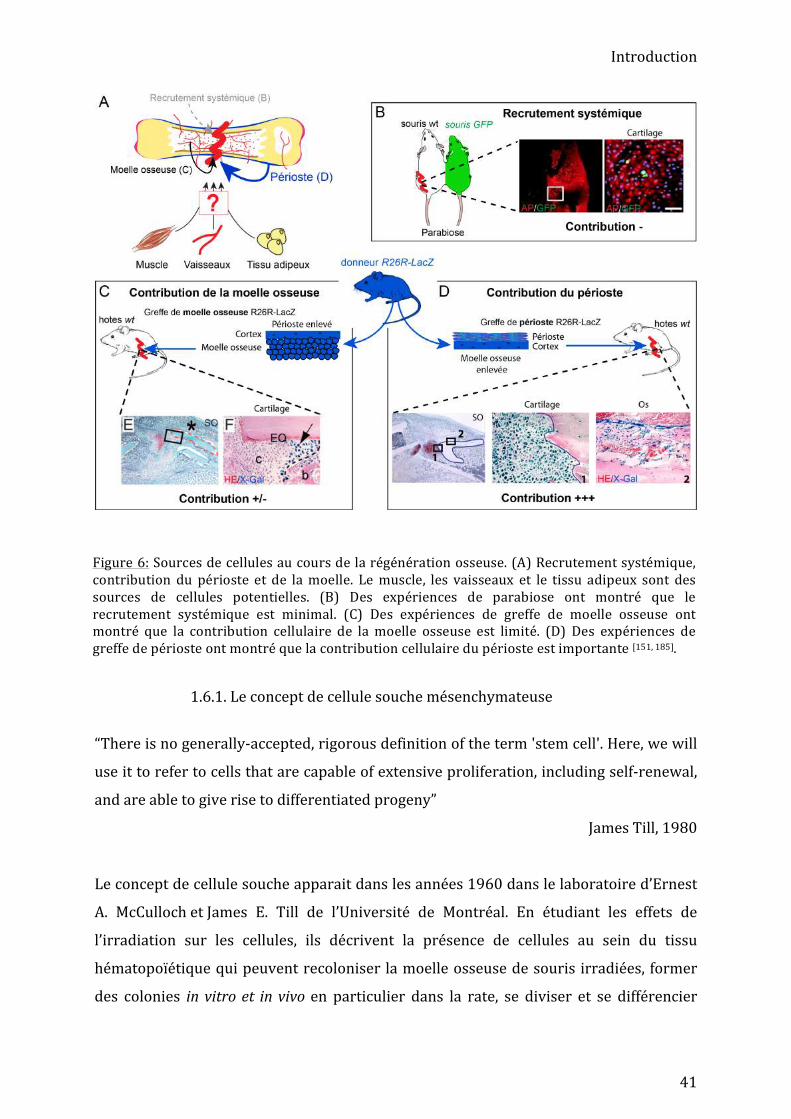
étudiés la moelle osseuse, le périoste et le muscle [154] (Fig. 6).
Introduction
41
1.6.1. Le concept de cellule souche mésenchymateuse
“There is no generally-‐accepted, rigorous definition of the term 'stem cell'. Here, we will
use it to refer to cells that are capable of extensive proliferation, including self-‐renewal,
and are able to give rise to differentiated progeny”
James Till, 1980
Le concept de cellule souche apparait dans les années 1960 dans le laboratoire d’Ernest
A. McCulloch et James E. Till de l’Université de Montréal. En étudiant les effets de
l’irradiation sur les cellules, ils décrivent la présence de cellules au sein du tissu
hématopoïétique qui peuvent recoloniser la moelle osseuse de souris irradiées, former
des colonies in vitro et in vivo en particulier dans la rate, se diviser et se différencier
Figure 6: Sources de cellules au cours de la régénération osseuse. (A) Recrutement systémique, contribution du périoste et de la moelle. Le muscle, les vaisseaux et le tissu adipeux sont des sources de cellules potentielles. (B) Des expériences de parabiose ont montré que le recrutement systémique est minimal. (C) Des expériences de greffe de moelle osseuse ont montré que la contribution cellulaire de la moelle osseuse est limité. (D) Des expériences de greffe de périoste ont montré que la contribution cellulaire du périoste est importante [151, 185].

Introduction
42
dans les lignages érythrocytaire, granulocytaire et mégacaryocytaire [155, 156]. Ils
émettent alors l’hypothèse que chaque colonie présente dans la rate a été formée à
partir d’une unique cellule de moelle osseuse précédemment injectée, et la valide par
des analyses génétiques[157]. Des cellules de la moelle osseuse sont donc capables de se
diviser à l’infini, de se différencier en différents lignages et de reformer des cellules
souches, à partir d’une cellule unique : le concept de cellule souche est né[158, 159].
Dans les années 1960-‐1970, Friedenstein AJ montre qu’une sous population de cellules
de la moelle osseuse a un potentiel ostéogénique[160]. Ces cellules sont facilement
différentiables des cellules hématopoïétiques de par leur rapide adhérence au plastique
et leur forme fuselée, et certaines d’entre elles peuvent former des colonies (CFU-‐F) in
vitro[161, 162]. Ces cellules sont alors nommées « osteogenic stem cells » ou « bone marrow
stromal cells »[161, 163]. Dans les années 1990, suite à des travaux de différenciation
adipogénique, ostéogénique et chondrogénique in vitro, ces cellules sont renommées
« mesenchymal stem cell » (cellules souches mésenchymateuses)[164-‐166].
Les cellules souches mésenchymateuses (CSMs) humaines sont définies comme des
cellules adhérentes au plastique, capables de se différencier in vitro en ostéoblastes,
adipocytes et chondrocytes et d’exprimer un panel de marqueurs de surface minimal :
négatifs (<2%) pour les marqueurs CD45, CD34, CD11b, CD19 et HLA-‐DR et positifs
(>95%) pour les marqueurs CD105, CD73 et CD90[167]. D’autres cellules de la moelle
dont les fibroblastes ou les CSHs sont capables d’adhérer au plastique. Ainsi, les études
faites in vitro à partir de moelle osseuse totale utilisent une population adhérente au
plastique hétérogène[168, 169]. Les marqueurs de surface ne sont pas exclusifs et d’autres
types cellulaires comme les péricytes notamment peuvent les exprimer[170].
Malgré ces critères utilisés pour définir les CSMs, leur terminologie est remise en cause.
Premièrement, le terme mésenchyme défini un tissu conjonctif embryonnaire qui dérive
du mésoderme puis de l’ectoderme et de la crête neurale[171]. Le mésenchyme ne désigne
donc pas un tissu présent à l’âge adulte. Deuxièmement, dans la majorité des études, le
caractère « souche » des CSMs est majoritairement évalué par des analyses clonales in
vitro. En absence d’expériences d’auto-‐renouvellement in vivo, leur caractère souche est
débattu dans la littérature mis à part dans la moelle osseuse où une population de
cellules CD146+ a été identifié comme étant capable de former de l’os et de reconstituer
la niche des cellules souches hématopoïétiques dans le cas de transplantations sous-‐
cutanées successives[172].

Introduction
43
Les cellules mésenchymateuses ont été isolées à partir de tous les tissus/organes[173].
Cependant, les propriétés et les fonctions de ces CSMs ne sont pas équivalentes pour
tous les tissus. Les cellules souches mésenchymateuses de la moelle osseuse (CSMOs)
sont capables de former du tissus adipeux et osseux in vivo et in vitro mais pas du
muscle squelettique alors que les CSMs dérivées du muscle squelettique ont été
rapportées comme ayant une capacité adipogénique et ostéogénique limitée mais
capables de former des myofibres in vitro[173, 174]. Ainsi, la terminologie des CSMs évolue
et il a été proposé de renommer ces cellules « cellules stromales mésenchymateuses» [164, 175-‐177].
1.6.2. La moelle osseuse, une source minimale de cellules
Chez l’adulte, la moelle osseuse est majoritairement composée d’adipocytes, de cellules
stromales et de cellules hématopoïétiques (CSHs, érythrocytes, leucocytes,
macrophages, ostéoclastes et neutrophiles)[178]. Les cellules stromales comprennent les
cellules souches mésenchymateuses (CSMs), des fibroblastes, des cellules endothéliales,
des péricytes, des cellules de Schwann (cellules gliales du système nerveux
périphérique) et des nerfs[179]. Le rôle principal des cellules stromales est de former la
niche des cellules souches hématopoïétiques pour soutenir l’hématopoïèse[180-‐182].
De par leur capacité à se différencier en ostéoblastes et chondrocytes et leur facilité
d’accès, les CSMs sont utilisées pour développer des approches de thérapie cellulaire
pour la régénération osseuse[183, 184]. Cependant des analyses de lignage cellulaire par
transplantation chez la souris ont montré que la contribution endogène des CSMs lors de
la régénération osseuse est minimale[185] (Fig.6C). Des approches de lignage cellulaire
utilisant le système Cre-‐Lox et les marqueurs αSMA, Gremlin 1, Leptin-‐Recepteur
(LeptR) et Mx1 ont été utilisés. Des chondrocytes et ostéoblastes issus de ces lignages
sont retrouvées au sein du cal[182, 186-‐188]. Cependant, ces lignages ne sont pas restreints
uniquement à la moelle osseuse[189, 190].
Les CSMs ont un rôle immunomodulateur et paracrine maintenant bien reconnu[191]. Des
greffes de CSMs au site de fracture induisent une diminution de l’expression de
cytokines pro-‐inflammatoires, démontrant leur rôle anti-‐inflammatoire au cours de la
régénération osseuse[192]. Les CSMs sécrètent des facteurs tels que VEGF, Cxcl12, CCL7
ou FGF essentiels au recrutement et à la différenciation des progéniteurs squelettiques

Introduction
44
en chondrocytes ou en ostéoblastes[193-‐195]. Le rôle indirect des CSMs est donc
fondamental lors de la régénération osseuse.
1.6.3. Le périoste, une source majeure de cellules
En 1757 Duhamel et Monceau décrivent pour la première fois une membrane entourant
le cortex capable de former de l’os[196]. En 1986, la description anatomique du périoste
est reportée[197]. Le périoste est constitué de deux couches : la couche externe en contact
avec le muscle appelée « couche fibreuse » et la couche interne en contact avec le cortex,
appelée « cambium layer ». La couche externe, composée de fibroblastes, est riche en
collagène et en fibres réticulaires conférant au périoste l’élasticité nécessaire lors des
mouvements. La couche interne du périoste est composée d’ostéoblastes et
d’ostéoprogéniteurs et est ancrée dans le cortex grâce à des fibres de collagène appelées
fibres de Sharpey. Le périoste est aussi riche en vaisseaux, en péricytes et en fibres
nerveuses[198, 199].
Dès le milieu du XIXème siècle, Dupuytren propose l’hypothèse selon laquelle le périoste
et la moelle osseuse sont deux sources de cellules lors de la formation du cartilage au
sein du cal[200]. En l’absence de marqueur spécifique des cellules du périoste, des greffes
tissulaires ont été réalisées pour évaluer le potentiel du périoste en tant que source de
cellules lors de la régénération osseuse. Des greffes segmentaires de fémur isolées de
souris Rosa26-‐LacZ transplantées dans un grand défaut osseux chez des souris hôtes
sauvages ont montré des cellules LacZ+ dérivant de la greffe dans le cartilage et l’os
néoformés. En absence de périoste au niveau de la greffe, la formation de cartilage et
d’os est réduite et la revascularisation du cal est compromise[201]. Dans une autre étude,
des greffes de périoste isolées de souris Rosa26-‐LacZ au site de fracture de souris ont
montré un grand nombre de chondrocytes et d’ostéoblastes LacZ+ dérivés de la greffe
de périoste dans le cal[185]. Le périoste est donc une source importante de cellules lors de
la régénération osseuse[2, 154] (Fig. 6C).

Introduction
45
1.6.4. Le muscle, une source de cellules pour la réparation osseuse ?
L’hypothèse de la contribution des tissus adjacents au site de fracture à la régénération
osseuse est acceptée étant donné la possibilité d’isoler des CSMs à partir de nombreux
tissus tels que le tissu adipeux et le muscle. De nombreuses études montrent la capacité
ostéogénique et chondrogénique des cellules musculaires. Stimulées in vitro avec
l’osteoactivin ou des BMPs, les cellules musculaires immortalisées C2C12 ou les cellules
satellites (cellules souches musculaires) sont capables de se différencier en
ostéoblastes[202, 203]. Une étude a montré que chez les souris MyoDCre/+ ;Z/AP+ dont le
lignage myogénique (fibres et cellules satellites) est marqué par l’expression de la
phosphatase alcaline, une contribution de ce lignage peut être détectée après fractures
stabilisées par une tige intra-‐médullaire et blessure du muscle et du périoste. Dans le cas
de fractures simples sans blessure des tissus adjacents, aucune contribution du muscle
au cal n’est observée (absence de cellules AP+ dans le cal) [204]. Une autre étude a
suggéré que l’action corrélée de Sox9 et Nkx3.2 régule négativement l’expression de
Pax3 dans les cellules musculaires pour permettre leur différenciation chondrogénique
en réponse à une fracture[205]. Des cellules du muscle, isolées à partir de muscles
adjacents à la fracture, présentent un potentiel ostéogénique in vitro plus élevés que les
CSMOs[206]. Des étapes de pré-‐plating permettent de sélectionner les cellules sur leur
habilité à adhérer au plastique. Les fibroblastes adhèrent en premier alors que les
cellules satellites et les myoblastes mettent plus de temps à adhérer[207]. Les cellules
isolées après six pré-‐plating et transfectées avec un adénovirus rh-‐BMP2 sont capables
de réparer un défaut de la calvaria, ce qui n’est pas le cas des cellules non-‐
transfectées[208]. Ainsi, si le rôle des cellules du tissu musculaire en tant que source de
cellules au cours de la régénération osseuse est suggéré, l’origine et le rôle exact des
cellules du muscle au cours de la régénération osseuse reste à définir[209, 210].
1.6.5. Les cellules utilisées en thérapie humaine
Même si le tissu osseux a une capacité de régénération élevée, 10% des fractures
simples et 40% des fractures complexes avec atteintes des vaisseaux et des tissus
adjacents, ne régénèrent pas correctement[211], [212]. Les traitements actuels des fractures
complexes (fractures poly-‐traumatiques, poly-‐fracture) est principalement chirurgical et
peut faire intervenir l’injection de facteurs de croissance (BMP, PDGF) au site de fracture

Introduction
46
ou les greffes osseuses de la crête iliaque[213-‐215]. Le cout de ces traitements[212, 216], la
douleur endurée par les patients, les risques d’effets secondaires (ossification ectopique
dans le cas de traitement local avec des BMPs)[217, 218] et de non-‐réparation[219]
démontrent que la prise en charge et les traitements des fractures ne sont pas optimaux.
Le « diamond concept » en ingénierie tissulaire combine l’utilisation de cellules
souches/progénitrices, de biomatériaux et de facteurs de croissance. L’ensemble est
transplanté au site de fracture tout en maitrisant sa stabilisation[220]. Si les résultats sont
encourageants, l’efficacité de cette méthode n’est toujours pas optimale et des effets
secondaires sont présents[221].
Du fait de son accessibilité, la moelle osseuse est actuellement la principale source de
cellules souches/progénitrices squelettiques utilisées en orthopédie. La moelle osseuse
est notamment utilisée en tant que greffe autologue. La technique de la membrane
induite avec greffe autologue de moelle osseuse et d’os spongieux de la crête est utilisée
pour le traitement des grands défauts osseux. L’efficacité de cette technique est
controversée et les risques de complications sont élevés. Cependant, la formation de la
membrane induite au site de résection promeut la différenciation ostéogénique des
cellules de la moelle osseuse via l’activation des réseaux de signalisation Smad et
MAPK[217, 222, 223]. De récentes approches utilisant du concentré de moelle osseuse
(concentrate of bone marrow aspirate, cBMA) ou des cellules de la moelle osseuse
amplifiées en culture visent à enrichir la population de cellules transplantées en cellules
souches [224, 225]. Lors de l’utilisation de cBMA, la moelle totale (issue le plus souvent de
la crête iliaque) est centrifugée et une partie des cellules hématopoïétiques est enlevée.
La suspension de cellules restantes contient des CSMOs, des cellules progénitrices ainsi
que des facteurs de croissance et des cytokines (TGFβ, BMPs, IL-‐1). Le cBMA est ensuite
mélangée à une matrice (gel de fibrine, matrice osseuse déminéralisée) et supplémenté
ou non par des BMPs exogènes. Les résultats sont prometteurs pour le traitement des
troubles articulaires et des fractures avec un temps de régénération diminué[222, 226].
Enfin, la Federal Drug Agency et l'European Medecines Agency ont accordé le grade
clinique aux CSMOs amplifiées in vitro en encadrant strictement leurs conditions de
culture[227, 228]. Les CSMOs sont extraites de la moelle osseuse puis purifiées et amplifiées
en culture pour être transplantées au moment de la chirurgie. De la même façon que

Introduction
47
pour le cBMA, les cellules sont généralement mélangées à une matrice et supplémentées
par des facteurs exogènes[229-‐231].
Ces méthodes rencontrent différentes limitations. La quantité de CSMOs récupérée
dépend de l’âge des patients. Plus l’âge est avancé, plus le nombre de CSMOs diminue.
L’amplification des cellules in vitro nécessite des contrôles phénotypiques et
génotypiques importants pour s’assurer que les cellules n’accumulent pas d’anomalies
génétiques pouvant aboutir au développement de pathologies comme le cancer. Enfin, la
nécessité d’utiliser des matériaux de soutien et de supplémenter les cellules à greffer en
facteurs de croissance peut entrainer des effets secondaires importants[232]. De plus, il
est possible que les cellules de la moelle osseuses aient une capacité à former de l’os et
du cartilage limitée et que leur rôle soit d’avantage trophique en participant à la
sécrétion de facteurs impliqués dans le recrutement de cellules progénitrices endogènes
ou dans l’inflammation[191]. Il est donc fondamental de mener des études permettant le
suivi sur le long terme de ces cellules afin de mieux comprendre leur rôle et ainsi de
pouvoir améliorer les traitements[233].
Le périoste étant une source majeure de cellules au cours du processus endogène de
régénération osseuse, l’utilisation de cellules du périoste est considérée mais pas encore
appliquée en clinique[234]. La présence d’un périoste intact pour une régénération
osseuse efficace est bien connue des orthopédistes mais du fait de son accessibilité
limitée, les cellules du périoste sont très peu utilisées en orthopédie[235]. Cependant, des
greffes de périoste ont été utilisées dans un essai clinique pour traiter des cas avancés
de parodontite chronique. Des greffes autologues de périoste ou de matrice protéique
ont été effectuées au site de blessure. Un an après, les patients traités avec les greffes de
périoste montrent une amélioration de la formation osseuse[236]. Des études ont été
menées pour évaluer le potentiel des greffes de périoste pour traiter des défauts osseux
de grande taille ou des défauts du cartilage[237].
Le muscle est utilisé cliniquement pour supporter la régénération osseuse en cas de
fracture complexe ou de grand défaut osseux. Il permet notamment d’améliorer la
vascularisation, de limiter les infections au niveau du site de fracture ou de sécréter des
facteur[238]. La capacité ostéogénique et chondrogénique du muscle squelettique est
décrite dans la littérature orthopédique dans le cas d’ossification ectopique ou dans la
fibrodysplasie ossifiante progressive. Cependant, le rôle du muscle squelettique en tant
que source de cellules au cours de la régénération osseuse est peu renseigné.
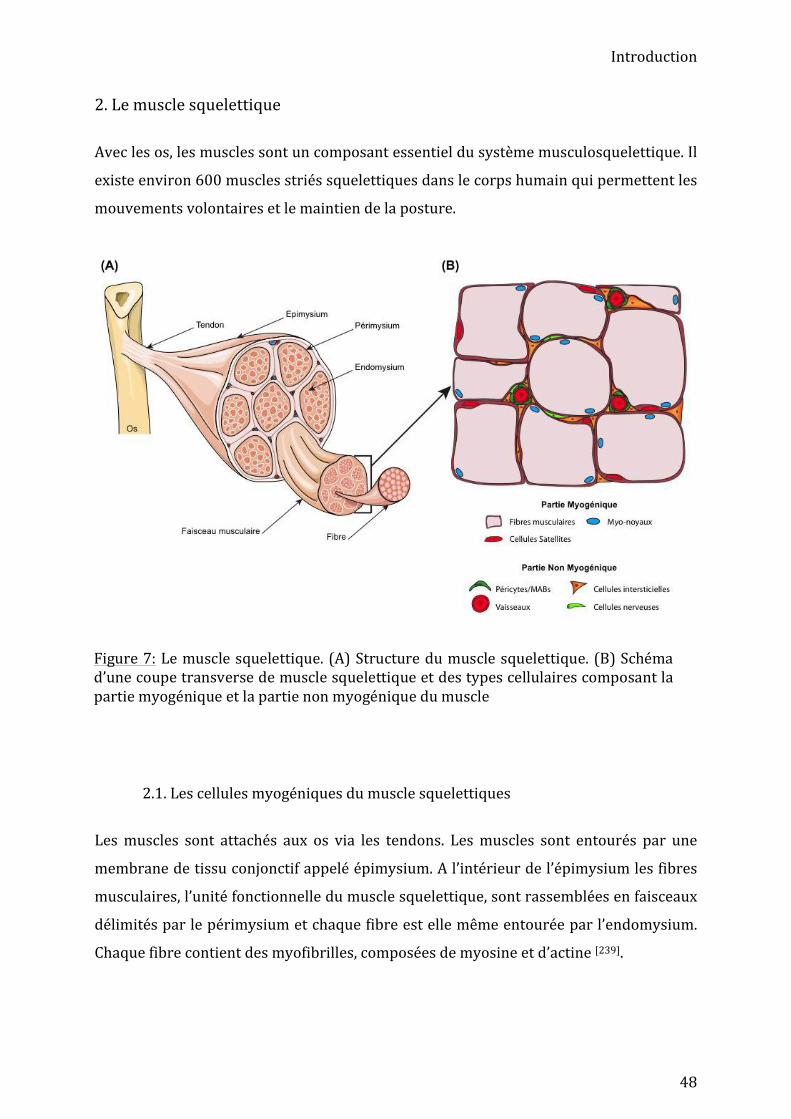
Introduction
48
2. Le muscle squelettique
Avec les os, les muscles sont un composant essentiel du système musculosquelettique. Il
existe environ 600 muscles striés squelettiques dans le corps humain qui permettent les
mouvements volontaires et le maintien de la posture.
2.1. Les cellules myogéniques du muscle squelettiques
Les muscles sont attachés aux os via les tendons. Les muscles sont entourés par une
membrane de tissu conjonctif appelé épimysium. A l’intérieur de l’épimysium les fibres
musculaires, l’unité fonctionnelle du muscle squelettique, sont rassemblées en faisceaux
délimités par le périmysium et chaque fibre est elle même entourée par l’endomysium.
Chaque fibre contient des myofibrilles, composées de myosine et d’actine [239].
Figure 7: Le muscle squelettique. (A) Structure du muscle squelettique. (B) Schéma d’une coupe transverse de muscle squelettique et des types cellulaires composant la partie myogénique et la partie non myogénique du muscle

Introduction
49
2.1.1. Développement musculaire Les cellules du muscle squelettique des membres est issu des somites et se met en place
en trois grandes étapes : la myogenèse primaire au stade embryonnaire, la myogenèse
secondaire au stade fœtal et le développement postnatal. L’étude de modèles murins a
permis de définir la cascade génétique impliquée dans la myogenèse dans les membres.
Lors de la myogenèse primaire, entre E11.5 et E14.5, les cellules mésodermiques des
somites Pax3+ qui expriment c-‐met/HGF, appelées myoblastes, délaminent et migrent au
niveau du bourgeon de membre[240]. Ces cellules prolifèrent et s’engagent dans la voie
myogénique sous l’effet des facteurs de régulation myogéniques (FRM) Myf5 et
MyoD[241]. Sous l’action de MRF4 et Myogenin, deux autres FRM, les myoblastes sortent
du cycle cellulaire et se différencient en myocytes[242]. Les myocytes fusionnent alors
entre eux et l’expression de la chaine lourde de la myosine (MHC) permet la formation
de fibres dites primaires[243]. Lors de la myogenèse secondaire, à partir de E14.5, les
cellules progénitrices myogéniques sont Pax3+/Pax7+ puis régulent négativement
l’expression de Pax3. Les cellules Pax7+ se différencient en myoblastes, migrent le long
des vaisseaux et fusionnent soit entres eux, soit avec les fibres primaires pour former les
fibres secondaires. Les fibres sont ensuite innervées et la lame basale est formée. Les
progéniteurs musculaires Pax7+ qui n’expriment pas les FRMs formeront le pool de
cellules satellites[244]. Le développement musculaire se poursuit après la naissance.
Pendant la période postnatale, les fibres subissent un processus d’hypertrophie mais
leur nombre reste constant. Les cellules satellites prolifèrent à un taux élevé, se
différencient en myoblastes sous l’action des FRMs et fusionnent avec les myofibres déjà
présentes. Dans le même temps, de nouvelles myofibrilles sont mises en place au sein
des myofibres, permettant au muscle d’achever sa formation[245]. Si les FRMs et les gènes
Pax3/Pax7 contrôlent la myogenèse, le réseau de signalisation complet de régulation
comprend de nombreux autres acteurs. Par exemple, la signalisation Notch au sein du
tissu musculaire est critique pour le maintien du pool de cellules souches. En effet, en
absence du ligand Notch, Delta 1, le pool de cellules satellites est réduit ce qui conduit à
une hypotrophie musculaire[246]. Les facteurs de croissance HGF, IGF et FGF sont
impliqués tout au long de la myogenèse : HGF permet la migration des myoblastes, IGF la
fusion des myoblastes et FGF est impliqué dans la prolifération des myoblastes et
réprime la différenciation. Enfin, myostatine (Mstn) régule l’hypertrophie des fibres
musculaires[245]. Le réseau de signalisation BMP joue aussi un rôle fondamental dans le
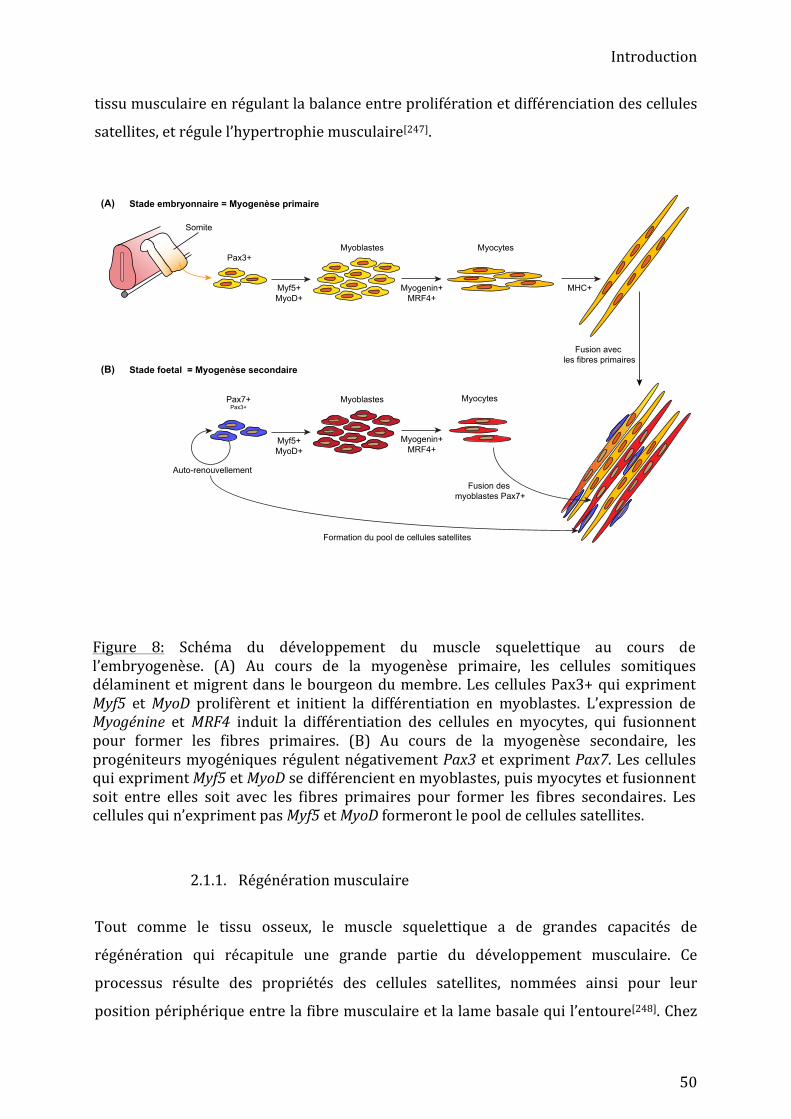
Introduction
50
tissu musculaire en régulant la balance entre prolifération et différenciation des cellules
satellites, et régule l’hypertrophie musculaire[247].
2.1.1. Régénération musculaire
Tout comme le tissu osseux, le muscle squelettique a de grandes capacités de
régénération qui récapitule une grande partie du développement musculaire. Ce
processus résulte des propriétés des cellules satellites, nommées ainsi pour leur
position périphérique entre la fibre musculaire et la lame basale qui l’entoure[248]. Chez
Figure 8: Schéma du développement du muscle squelettique au cours de l’embryogenèse. (A) Au cours de la myogenèse primaire, les cellules somitiques délaminent et migrent dans le bourgeon du membre. Les cellules Pax3+ qui expriment Myf5 et MyoD prolifèrent et initient la différentiation en myoblastes. L’expression de Myogénine et MRF4 induit la différentiation des cellules en myocytes, qui fusionnent pour former les fibres primaires. (B) Au cours de la myogenèse secondaire, les progéniteurs myogéniques régulent négativement Pax3 et expriment Pax7. Les cellules qui expriment Myf5 et MyoD se différencient en myoblastes, puis myocytes et fusionnent soit entre elles soit avec les fibres primaires pour former les fibres secondaires. Les cellules qui n’expriment pas Myf5 et MyoD formeront le pool de cellules satellites.
Pax3+
Somite
Myf5+
MyoD+
Myogenin+
MRF4+
MHC+
Stade embryonnaire = Myogenèse primaire
Pax7+Pax3+
Myf5+
MyoD+
Myogenin+
MRF4+
Fusion avec
les fibres primaires
Fusion des
myoblastes Pax7+
Stade foetal = Myogenèse secondaire
Auto-renouvellement
Formation du pool de cellules satellites
MyocytesMyoblastes
MyocytesMyoblastes
(A)
(B)

Introduction
51
l’adulte, les cellules satellites, qui représentent entre 2.5% et 6% du nombre de noyaux
dans un muscle sain, sont définies par l’expression du gène Pax7 et leur position
anatomique entre les fibres et la lame basale[249]. Le rôle central des cellules satellites au
cours de la régénération musculaire a été établi par des analyses de lignage cellulaire, de
déplétion et de transplantation[250-‐253]. En effet, les souris Pax7 KO présentent un retard
de croissance, une musculature deux fois moins importante que celle des souris
sauvages, mais une organisation normale du tissu musculaire. Cependant, après la
naissance, les souris Pax7 KO perdent progressivement leurs cellules satellites, ce qui
entraine un déficit sévère de régénération musculaire[254]. La déplétion des cellules
satellites Pax7+ chez les souris Pax7CreER/+; R26RDTA/+ au stade adulte entraine un échec
complet de régénération[250, 255].
Après une blessure, la régénération musculaire débute par une phase dégénérative,
caractérisée par la nécrose des fibres musculaires et l’infiltration de cellules
inflammatoires notamment de macrophages au niveau de la lésion musculaire[256]. Le
recrutement de macrophages et de neutrophiles permet le nettoyage du site de blessure.
La sécrétion de cytokines et de facteurs de croissance par les cellules hématopoïétiques
et les fibres en dégénérescence telles que IL5, IL6 ou HGF permet l’activation, le
recrutement et la prolifération des cellules satellites[257]. En parallèle, les cellules
satellites sont activées, prolifèrent et se différencient en myoblastes. Le contrôle du
devenir des cellules souches est effectué, entre autres, par les FRMs comme au cours du
développement. Ces FRMs agissent séquentiellement à chaque étape de la régénération
musculaire. Lors de la première étape l’expression de Myf5 induit l’activation des
cellules satellites, et la co-‐expression de Myf5 et MyoD permet la prolifération des
cellules satellites[258-‐260]. Par la suite, l’augmentation de l’expression de MyoD entraine
l'expression de p21, un inhibiteur du cycle cellulaire et parallèlement, une diminution de
l'activité des cdk (cyclin-‐dependent kinase), entraînant l'arrêt du cycle cellulaire et la
progression dans la différenciation[261]. Après la phase de prolifération, l'expression de
Myogénine est augmentée dans les cellules myogéniques et est associée au programme
de différenciation terminale. Ce dernier est complété par l'activation des protéines
spécifiques du muscle telles que la chaîne lourde de la MHC et la créatine kinase
musculaire. Les myocytes mononucléés fusionnent enfin pour former un syncytium
multinucléé (myotube) et, après maturation, une cellule musculaire contractile[262].

Introduction
52
D’autres populations cellulaires au sein du muscle, distinctes des cellules satellites ont
été décrites comme ayant une capacité myogénique : les cellules souches dérivées du
muscle (SCDM), les cellules progénitrices CD133+[263], les cellules interstitielles PW1+
(PICs)[264], les cellules Twist2+[265], les cellules endothéliales CD34+[266, 267] ou encore les
péricytes[268]. Cependant, ces marqueurs ne sont pas exclusifs des précurseurs
myogéniques[269, 270].
2.2. Les cellules non-‐myogénique du muscule squelettiques
Le tissu non-‐myogénique musculaire désigne toutes les cellules présentes au sein du
muscle excepté les cellules satellites, les myoblastes et les fibres musculaires (Fig. 7). Il
comprend les vaisseaux, les nerfs, les péricytes, les fibroblastes, les cellules
mésenchymateuses et les adipocytes. Souvent appelé tissu de soutien, le tissu non
myogénique contient des cellules remplissant des fonctions essentielles assurant
l’apport en oxygène et en nutriments (vaisseaux) , la sécrétion de facteurs ou de
protéines de la matrice extracellulaire (fibroblastes et cellules mésenchymateuses) ,
l’innervation des fibres essentielles aux mouvements (nerfs) ou une réserve d’énergie
(adipocytes) [271]. Ce tissu de soutien peut être divisé en trois catégories : le système
nerveux, le système vasculaire (vaisseaux, péricytes et mésoangioblastes) et le système
fibro-‐mésenchymateux.
Figure 9: Schéma de la régénération musculaire. Après une blessure musculaire, les cellules satellites sont activées, se multiplient et se différencient de façon asymétrique. Les cellules n’exprimant pas Myf5 et MyoD reconstituent le pool de cellules satellites, alors que celles exprimant Myf5 et MyoD se différencient en myoblastes puis myocytes. Les myocytes fusionnent pour reformer des fibres musculaires fonctionnelles.

Introduction
53
2.2.1. Le système vasculaire musculaire
2.2.1.a. Les vaisseaux
Les cellules endothéliales du muscle squelettiques dans le membre ont la même origine
mésodermique, le dermomyotome, que les précurseurs myogéniques[272]. Les
précurseurs endothéliaux, appelés angioblastes, migrent en premier au niveau du
bourgeon de membre, en amont des précurseurs myogéniques. Les angioblastes
sécrètent PDGFβ qui stimule la production de matrice extracellulaire, nécessaire à la
myogenèse[273]. La balance entre différenciation endothéliale et myogénique est
orchestrée par la répression mutuelle entre FoxC2 qui induit la différenciation
endothéliale et Pax3/7 qui induisent la différenciation myogénique. Les cellules pré-‐
endothéliales migrent au niveau du bourgeon de membre pour établir un pool de
cellules mononuclées qui se différencient en cellules endothéliales en parallèle de la
formation des myofibres[274, 275].
2.2.1.b. Les péricytes
Les péricytes sont observés pour la première fois en 1871 et définis deux ans plus tard
par le français Charles-‐Marie Benjamin Rouget comme des cellules contractiles
entourant les cellules endothéliales des petits vaisseaux[276]. Les péricytes sont
caractérisés par leur position anatomique et l’expression de différents marqueurs :
neural/glial antigen 2 (NG2), PDGFRβ ou phosphatase alkaline (AP)[268, 277, 278]. Ces
différents marqueurs semblent identifier différentes sous-‐populations de péricytes qui
se chevauchent potentiellement, mais il n’existe aucun marqueur spécifique des
péricytes qui permet leur identification stricte[279]. Les péricytes sont nécessaires à
l’homéostasie vasculaire et remplissent des fonctions essentielles comme la stabilisation
des vaisseaux[280], la régulation du flux sanguin[281-‐283] et l’établissement et la maturation
de la vascularisation au cours du développement[278, 284].
La mise en place des péricytes au cours du développement est organe/tissu
dépendant[285]. Les péricytes du membre dérivent du mésoderme, et plus
particulièrement de la splanctopleure et des somites[279, 286]. Chez les souris PDGFβ-‐
déficientes, les péricytes sont absents. Cependant, l’angiogenèse a lieu mais les vaisseaux
sont dilatés en l’absence de péricytes[278]. Les cellules endothéliales sécrètent le facteur

Introduction
54
de croissance PDGFβ qui induit le recrutement de cellules périvasculaires (péricytes,
mésoangioblastes)[287]. L’hypothèse d’une transition endothélio-‐mésenchymateuse de
cellules endothéliales sous le contrôle de Notch a été proposée, notamment dans le tissu
cardiaque[279, 288].
L’utilisation de modèles transgéniques a permis de mieux caractériser la population
péricytaire du muscle squelettique et ses rôles au sein du muscle squelettique. La souris
NG2Cre a permis de montrer que les péricytes NG2+ stimulent la croissance des
myofibres au stade post-‐natal, maintiennent la quiescence des cellules satellites via
l’action de IGF1 et angiopoietin 1 (ANGPT1)[289]. L’expression de NG2 et du filament
intermédiaire Nestin définie deux sous-‐populations péricytaires. Les péricytes de type I,
NG2+/Nestin+, forment du tissu fibrotique après blessure ou durant le vieillissement.
Les péricytes de types 2, NG2+/Nestin-‐, participent à l’angiogenèse normale et
tumorale[290-‐292] et sont les seuls à pouvoir se différencier en oligodendrocytes et en
cellules de Schwann, cellules nécessaires lors de l’innervation du muscle après
blessure[290, 293]. Les mécanismes régissant la différenciation de ces deux sous
populations restent mal compris. Cependant, il semble que la laminine inhibe la
prolifération et l’adipogenèse des péricytes de type 1 et favorise la myogenèse et la
prolifération des péricytes de types 2[294, 295].
L’étude de la souris APCreERT2 a montré que certains péricytes AP+ génèrent des cellules
satellites et des myofibres durant la croissance musculaire, contribuent activement à la
régénération musculaire et ont une capacité de différenciation myogénique in vitro[268,
296]. Il apparait aussi que les myoblastes Pax3+/Myf5+ ont la capacité à se différencier en
péricytes. En effet, sous l’action de Dll4 et PDGFRβ les myoblastes se différencient en
péricytes. A l’inverse, chez les souris MyoDCre ;RosaNCID où la voie Notch est activée dans
les cellules MyoD+, les gènes myogéniques sont régulés négativement, alors que les
gènes péricytaires sont activés [297].
Certains péricytes expriment aussi la désintégrine-‐métalloprotéinase 12 (ADAM12). Des
analyses de lignage ont montré que les cellules ADAM12+ participaient activement à la
formation de tissu fibrotique pendant la régénération musculaire[298].
Certains péricytes expriment aussi les marqueurs PDGFRα, Tie2 ou GLAST. Des analyses
de lignage utilisant ces marqueurs ont montré que les cellules PDGFRα+, Tie2+ ou
GLAST+ pouvaient former du cartilage et de l’os au cours de l’ossification hétérotopique.

Introduction
55
Ces données suggèrent donc que les péricytes auraient des capacités chondrogénique et
ostéogénique [299-‐302].
2.2.1.c. Les mésoangioblastes
Les mésoangioblastes (MAB) sont des cellules périvasculaires dérivées de l’aorte
dorsale, participant à la formation de différents tissus d’origine mésodermique (muscle
squelettique, cartilage, os), ayant une capacité clonogénique in vitro et exprimant les
marqueurs AP, CD34, Kit et Flk4[303]. Au cours du développement embryonnaire, la
compétition entre les BMPs sécrétés par les cellules endothéliales et Noggin (un
inhibiteur de la voie BMP) sécrété par les myofibres induit la différentiation des
progéniteurs périvasculaires en cellules périvasculaires ou en myotubes,
respectivement[304]. L’expression du gène Pax3 est indispensable aux MABs pour
s’engager dans la différenciation myogénique[305]. Au cours de la régénération
musculaire, l’expression du gène PW1/Peg3 permet la migration des MABs le long des
vaisseaux jusqu’au site de blessure et induit la différenciation myogénique des MABs [306]. Des analyses in vitro montrent que les MABs peuvent se différencier en adipocytes
en réponse à l’insuline et en ostéoblastes en réponse au BMP-‐2 ou TGFβ-‐1[307].
De par leur capacité myogénique, les MABs sont utilisés en thérapie cellulaire pour
traiter les dystrophies musculaires. Les MABs sont isolés à partir de culture d’explant de
vaisseaux issus d’embryon ou de souris jeunes[303, 308]. Des MABs infectés avec un
lentivirus exprimant α-‐sarcoglycan (α-‐SG) ont été injectés dans l’artère fémorale de
souris dystrophiques α-‐SG-‐/-‐. Quatre mois après le traitement, les muscles irrigués par
l’artère fémorale retrouvent une constitution et une fonction comparable aux muscles
wt[308]. Les MABs ont aussi été utilisés en thérapie cellulaire pour traiter des chiens
atteints de la dystrophie musculaire de Duchenne. Les MABs, transfectés par un
lentivirus exprimant la microdystrophynine humaine, ont été injectés dans l’artère
fémorale. Les chiens traités gardent une fonction musculaire normale, ce qui n’est pas le
cas des chiens contrôles qui décèdent au bout d’un an[309]. Ces études ont mené à la mise
en place d’un essai clinique chez l’Homme en 2011 (référence de l’essai clinique :
EudraCT #2011-‐000176-‐33). Afin de mieux comprendre les mécanismes d’intégration
et de régénération des MABs, deux études ont montré le rôle central des réseaux de
signalisation BMP et Notch au cours de la différentiation des MABs en myofibres in vitro

Introduction
56
et in vivo[310, 311]. L’expression de Delta-‐like ligand 1 (Dll1, activateur de la voie Notch) au
sein des MABs améliore leur capacité de différentiation myogénique et leur capacité
d’intégration, tout comme l’inhibition de la voie BMP par Noggin ou Dorsomorphine.
Malgré ces résultats, les fonctions physiologiques des MABs restent méconnues.
Les péricytes et les MABs sont des cellules périvasculaires qui présentent plusieurs
points communs tels que leur capacité de différenciation myogénique, adipogénique et
ostéogénique, leur localisation anatomique périvasculaire proche et l’expression de
certains marqueurs[296, 312, 313]. Il est donc possible que ces deux populations cellulaires
se chevauchent, du moins en partie (Figure 10).
2.2.2. Le système fibro-‐mésenchymateux
Le tissu conjonctif du muscle dans le membre provient des lames latérales du
mésoderme et sa mise en place au cours du développement est concomitante à la
myogenèse. Il existe peu de marqueurs permettant de décrire le développement et le
rôle du tissu conjonctif. Les marqueurs Tcf4 (transcription factor 4), Tbx5 (T-‐box
transcription factor), Osr1/2 (Odded-‐Skipped 1 et 2) et PDGFRα ont été étudiés mais ne
Figure 10: Les cellules périvasculaires : péricytes et mésoangioblastes. Les péricytes de type 1 sont NG2+/Nestin+ et peuvent être source de fibrose ou de tissu adipeux. Les péricytes de type 2 sont NG2+/Nestin-‐ et peuvent se différencier en cellules endothéliales, nerveuses et musculaires. Les mésoangioblastes (MAB) peuvent donner des chondrocytes, ostéoblastes, adipocytes et du muscle squelettique.

Introduction
57
permettent pas de définir des populations bien distinctes[314]. Même si le rôle du tissu
conjonctif reste à mieux caractériser, il est décrit comme étant un tissu de soutien
permettant une myogenèse normale.
2.2.2.a. Les fibroblastes
Les fibroblastes du muscle sont définis par leur localisation en périphérie des fibres, leur
forme fuselée et l’expression du collagène de type IV. Leur principale fonction
homéostasique est la production de matrice extracellulaire[315]. Tcf4 est exprimé dans le
tissu mésodermique des membres en développement [316]. La déplétion des cellules Tcf4
chez les souris Tcf4GFPCre+neo/+;R26RDTA/+ entraine une diminution du nombre de
myofibres au cours de la myogenèse chez la souris, démontrant que la présence des
fibroblastes Tcf4+ est essentielle à la myogenèse[317]. De plus, la déplétion des
fibroblastes Tcf4+ au cours de la régénération entraine une différenciation prématurée
des cellules satellites et la formation de myofibres de petite taille[318]. La déplétion des
cellules satellites chez les souris Pax7CreERT/+;R26RDTA/+ au cours de la régénération
musculaire cause une absence complète de régénération musculaire mais aussi une
production excessive de tissu fibrotique associée à une augmentation significative du
nombre de fibroblastes Tcf4+[319]. Ces résultats ont mis en évidence l’interaction des
cellules satellites et des fibroblastes Tcf4+ au cours de la régénération musculaire. Des
études in vitro sur des cellules humaines ont montré avec que la co-‐culture de
fibroblastes Tcf7L2+ (équivalent de Tcf4 chez l’Homme) et de progéniteurs
myogéniques stimulait la différenciation myogénique [320].
2.2.2.b. Les cellules mésenchymateuses musculaires / FAPs
L’étude de la souris Osr1Cre a permis de montrer que le lignage marqué par Osr1 au cours
du développement donne lieu à une sous population des cellules mésenchymateuses
musculaires à l’âge adulte, et que cette population est essentielle à la myogenèse[321].
Deux études distinctes mettent en évidence la présence d’une population de cellules
mésenchymateuses musculaires avec un rôle adipogénique et fibrogénique dépendant
de l’environnement tissulaire. Grâce à des analyses de cytométrie en flux, les
populations CD45-‐, CD31-‐, intégrineα7-‐, Sca1+, CD34+ d’une part et CD45-‐, CD31-‐,
SM/C2.6-‐, PDGFRα+ d’autre part ont été identifiées comme ayant un potentiel fibro-‐
adipogénique in vivo et in vitro et améliorant la myogenèse in vitro. Ces deux populations

Introduction
58
sont nommées progéniteurs fibro-‐adipogeniques (FAPs) et cellules mésenchymateuses,
respectivement[322]. Cette population mésenchymateuse PDGFRα+ est aussi présente
dans le tissu musculaire humain avec les mêmes potentiels fibro-‐adipogéniques in
vitro[323]. Les FAPs et les cellules mésenchymateuses PDGFRα+ ont donc des points
communs tels que leur capacité de différenciation in vitro et in vivo. Le rôle de ces
cellules a été beaucoup étudié dans le cadre de la régénération musculaire après une
blessure aigue ou dans le cadre de blessure chronique[324].
In vivo, le devenir des FAPs et des cellules mésenchymateuses est dépendant de la
nature de la blessure et de l’environnement musculaire. Après une blessure musculaire
au glycérol, les FAPs et les cellules mésenchymateuses PDGFRα+ forment des
adipocytes. Cependant, après une blessure à la toxine, les FAPs et les cellules
mésenchymateuses PDGFRα+ prolifèrent et forment le tissu fibrotique transitoire
observé au début de la régénération musculaire [325, 326]. Des expériences de co-‐culture
ont montré que les FAPs améliorent la différentiation musculaire des progéniteurs
myogéniques[324]. Cette étape de fibrose transitoire est essentielle pour une
régénération musculaire normale. En effet, chez des souris sauvages blessées par
injection de notexine, l’administration de Nilotinib, un inhibiteur de tyrosine kinase,
entraine une diminution du nombre de FAPs et une régénération musculaire
imparfaite[307]. La balance entre l’adipogenèse et la fibrogenèse est contrôlée par
différents mécanismes. Les éosinophiles recrutés au site de blessure sécrètent de l’IL4
nécessaire à la prolifération, à l’activation et à l’inhibition de l’adipogenèse des FAPs[327,
328]. Des expériences in vitro ont montré que les cellules satellites inhibent la
différenciation adipogénique des cellules mésenchymateuses[325]. La polyadénylation de
l’intron 16 du transcrit PDGFRα diminue l’activation des FAPs et la formation de tissu
fibrotique[329].
Contrairement aux blessures aigues, dans le modèle murin de la Dystrophie Musculaire
de Duchenne (mdx), la voie de signalisation PDGFRα est activée en continue avec pour
conséquence la formation de tissu fibrotique persistant[330, 331].
Les cellules marquées par PDGFRα et le réseau de signalisation PDGFRα sont donc une
cible de choix pour la thérapie. En effet, la fibrose chronique ou la formation de tissu
adipeux ectopique sont généralement corrélées à une régénération tissulaire imparfaite
observée notamment dans le cas de dystrophies musculaires. De nombreuses études ont
été menées dans le but de diminuer la formation de tissu fibrotique et d’améliorer la

Introduction
59
régénération myogénique. L’Imatinib, un inhibiteur des voies PDGFR, c-‐Kit et Abl utilisé
en clinique pour traiter les cancers de l’estomac, a été administré à des souris
dystrophiques mdx et les résultats montrent que l’Imatinib diminue la formation de
tissu fibrotique en diminuant notamment le nombre de cellules PDGFRα+[332, 333]. Par
ailleurs, l’inhibition de TGFβ-‐1 par du Nilotinib, induit l’apoptose des FAPs via
l’expression de TNFα chez les souris mdx, ce qui diminue la formation de tissu
fibrotique[210].
2.2.1.d. Les cellules intersticielles PW1+ (PICs)
En cherchant à établir les mécanismes de régulation de la myogenèse en amont de
l’action des MRF, le gène PW1/Peg3 (noté PW1)a été identifié comme étant fortement
exprimé au cours de la gastrulation puis à partir de E16.5 dans le muscle squelettique et
dans certaines populations cellulaires du système nerveux central[334]. Des analyses de
lignage cellulaire utilisant la souris PW1nLacZ exprimant LacZ sous contrôle du
promoteur PW1 montrent que les cellules PW1+ embryonnaires et du sac vitellin sont
capables de donner lieu à des cellules endothéliales progénitrices ou différenciées, in
vitro et in vivo[335, 336]. PW1 étant exprimé dans le muscle adulte[334], différentes études
ont été menées pour mieux comprendre le rôle des PICs. In vitro, les PICs ont une
capacité clonogénique élevée ce qui suggère que certaines cellules pourraient être des
cellules souches. In vivo, les PICs présentent une capacité de différentiation dans
différents lignages tels que le lignage myogénique, hépatique ou neuronal[337]. Au sein du
muscle squelettique, les PICs sont capables de former des myofibres et des adipocytes à
l’âge adulte. Si toutes les PICs expriment PW1, la différence entre les PICs
« myogéniques » et les PICs « adipocytaires » est basée sur l’expression de PDGFRα.
Uniquement les PICs exprimant PDGFRα forment des adipocytes. De plus, Pax7 est
requis pour la différentiation myogénique des PICs, qui sont distinctes des cellules
satellites[264, 338] (Figure 11).

Introduction
60
Figure 11: Schéma du système fibro-‐mésenchymateux musculaire. Les cellules intersticielles PW1+ (PICs) peuvent donner du tissu musculaire et adipeux. Les fibroblastes Tcf4+ peuvent former du tissu fibrotique. Les cellules mésenchymateuses et les progéniteurs fibro-‐adipogéniques (FAPs) peuvent former du tissu adipeux et fibrotique.
Cellule Intersticielle PW1+(PICs)
Tissu adipeux Muscle squelettique
Fibroblastes Tcf4 +
Tissu fibrotique
Cellule mésenchymateuse/Progéniteur fibro-adipogénique
Tissu adipeux Tissu fibrotique
Myofibres
Cellule satellite

Introduction
61
3. Les interactions os-‐muscle
L’os et le muscle squelettique sont deux tissus liés physiquement via le périoste et les
tendons, mécaniquement de par leur rôle locomoteur et moléculairement de par la
sécrétion par ces deux tissus de facteurs nécessaires à leur développement, leur
croissance, leur maintien homéostasique et leur régénération après blessure[339]. Au
cours du développement et de la croissance, ces deux tissus se forment en parallèle. La
présence d’un muscle normal et contractile est nécessaire au développement normal des
os. A l’âge adulte, les interactions os-‐muscle sont fondamentales pour le maintien
homéostasique de ces deux tissus. La perte de masse ou de fonction de l’un des deux
impacte directement la structure de l’autre. Ce mécanisme est encore plus visible au
cours du vieillissement ou de nombreuses maladies osseuses impactent négativement le
muscle et inversement[340].
3.1. Interactions biomécaniques
L’importance de la stimulation mécanique musculaire au cours du développement et de
l’homéostasie osseuse est bien connue[341].
Au cours du développement, chez les souris Myf5nlacZ/nlacZ:MyoD−/− où les muscles ne se
forment pas et chez les souris Pax3Sp/Sp où la formation des muscles est réduite, les os
longs sont plus courts, la taille des centres d’ossification primaire est réduite et les
articulations sont fusionnées[342, 343]. La paralysie musculaire durant le développement
osseux dérégule les centres d’ossifications primaires, conduisant à des structures
cartilagineuses et à des os plus courts. La paralysie induit l’arrêt de la prolifération des
chondrocytes au cours de la formation des arrêtes osseuses, ce qui corrèle avec la
diminution de la taille des centres d’ossification primaire des os longs[306, 344].
A l’âge adulte, la stimulation mécanique de l’os par le muscle est nécessaire pour
maintenir la masse osseuse. Dans le cas de situation de microgravité telle que les vols
spatiaux, la perte de masse osseuse est corrélée à la perte de masse musculaire[345-‐347].
La stimulation mécanique via l’exercice physique (course à pied, squats) combinée à un
régime alimentaire supplémenté en vitamine D permet de diminuer la perte de masse
osseuse et musculaire, ce qui suggère une interaction entre le tissu osseux et
musculaire[348]. La paralysie musculaire induite par l’injection de toxine botulique dans

Introduction
62
les muscles de la patte conduit à une résorption osseuse rapide qui affecte la forme du
tibia[349, 350]. Cependant, un effet direct de la toxine botulique sur l’os ne peut pas être
exclu. Le rôle des contractions musculaires a aussi été étudié grâce au modèle murin
(modèle mdg) dont les muscles ne se contractent pas. Chez les souris mdg, la paralysie
dérégule la balance entre ostéoblastes et ostéoclastes, ce qui, pour le tibia, conduit à une
forme altérée[351]. En réponse à la paralysie musculaire, l’ostéoclastogenèse est induite
via la surexpression de RANKL et NFATc1. L’inhibition de NFATc1 abolit
l’ostéoclastogenèse et la perte de volume osseux[352].
A l’opposé, l’activité physique est un stimulus de la formation osseuse. Dans un modèle
de rates ovariectomisées, la résorption osseuse est plus importante que la formation
osseuse, augmentant la fragilité osseuse. Cependant, si ces animaux sont soumis à une
activité physique, la balance résorption/formation osseuse est rétablie et la perte de
masse osseuse ralentie [353]. L’effet de l’activité physique pourrait être dû en partie par
une augmentation de la différenciation des cellules mésenchymateuses en ostéoblastes
plutôt qu’en adipocytes. La stimulation mécanique active l’expression de β-‐caténine qui
en conséquence réduit l’expression de facteurs pro-‐adipogénique (PPARγ et
adiponectine) et augmente l’expression de facteurs ostéogéniques (WIST, COX2)[344, 354,
355].
Chez l’Homme, la sarcopénie, généralement définie comme la perte élevée de masse
musculaire, et l’ostéoporose peuvent être liées[356]. Ces deux maladies présentent
plusieurs dérégulations moléculaires communes dont la diminution d’IGF-‐1 sérique et
l’augmentation de l’expression de cytokines pro-‐inflammatoire telles que TNF-‐α et IL-‐6.
IGF-‐1 et IL-‐6 sont notamment connues pour médier les interactions entre le muscle et
l’os (voir 3.2). IL-‐6 est surexprimé chez les patients atteints de sarcopénie et est connu
pour stimuler l’activité ostéoclastique[357, 358]. Cela pourrait donc induire un phénotype
ostéoporotique.
La diminution de l’activité physique est une composante majeure chez les patients
atteints de sarcopénie. Nous avons vu ci dessous que la stimulation mécanique de l’os
par le muscle est essentielle au maintien du tissu osseux. La perte d’activité physique est
donc concomitante du développement de l’ostéoporose[359]. Cela a notamment pour
conséquence l’augmentation du risque de fracture chez les patients atteints de
sarcopénie. Les mécanismes moléculaire et cellulaires sous-‐jacents restent mal connus.

Introduction
63
Si l’activité physique apparait comme une solution pour ralentir la sarcopénie et
l’ostéoporose, cela reste difficile à mettre en place[360, 361].
3.2. Interactions moléculaires
L’os et le muscle ont des fonctions endocrines reconnues et agissent de façon
systémique pour réguler diverses fonctions physiologiques[24]. Cependant, les
interactions moléculaires directes entre l’os et le muscle sont moins bien
caractérisées[362].
Les molécules étant spécifiquement sécrétées par le muscle, sont appelées myokines et
par l’os, ostéokines. Une des premières myokines identifiées est Mstn. Mstn est exprimée
majoritairement par les fibres musculaires mais les adipocytes peuvent aussi exprimer
Mstn [363, 364]. Mstn appartient à la super-‐famille TGFβ et régule négativement la masse
musculaire[363]. Les souris Mstn-‐/-‐ présentent une augmentation du volume osseux, ce qui
peut laisser suggérer que Mstn agit sur le tissu osseux[365, 366]. Des expériences in vitro
ont montré que la déplétion de Mstn augmente la capacité des CSMO à se différencier en
ostéoblastes[367]. De plus, Mstn inhibe la prolifération des chondrocytes et leur
différenciation en régulant négativement Sox9 in vitro[368]. Dans un modèle d’arthrite
rhumatoïde, Mstn est sur-‐exprimée ce qui a pour conséquence d’augmenter l’activité
ostéoclastique et donc la résorption osseuse et la dégénérescence articulaire.
L’inhibition de Mstn améliore le phénotype et diminue l’inflammation inhérente à
l’arthrite rhumatoïde[369]. La myokine Mstn agit donc sur différents composants de l’os :
les chondrocytes, les ostéoblastes et les ostéoclastes.
Certaines interleukines (IL-‐6, IL-‐7, IL-‐8, IL-‐15 notamment) sont considérées comme des
myokines du fait de leur expression élevée en réponse à la contraction musculaire, bien
qu’elles soient exprimées aussi par des cellules non myogéniques comme les cellules
immunitaires[370]. IL-‐6 est sécrétée par les myofibres lors de la contraction musculaire
mais aussi par les ostéoblastes et les ostéocytes. Le récepteur à l’IL-‐6 (IL-‐6R) est lui
exprimé par les cellules musculaires, les ostéoclastes et les ostéoblastes. IL-‐6 active les
voies JAK/STAT, AKT ou mTOR ce qui explique l’effet pléiotropique observée dans le
tissu osseux : IL-‐6 stimule l’ostéoclastogenèse de façon indirecte via la suractivation de
RANKL dans les ostéoblastes mais in vitro, IL-‐6 induit la différenciation ostéoblastique
des CSMOs[371-‐374].

Introduction
64
IGF-‐1 est un facteur de croissance principalement sécrété par le foie, qui sous l’action de
l’hormone de croissance, stimule la prolifération et la différenciation des
chondrocytes[375, 376]. Cependant, IGF-‐1 est aussi sécrété par les ostéocytes, les
ostéoblastes et les fibres musculaires au cours de l’exercice et est présent dans les lysats
musculaires totaux[377]. IGF-‐1 et le récepteur à IGF-‐1 (IGF-‐1R) sont abondamment
exprimés à la liaison os-‐muscle au niveau du périoste[378]. La protéine GRP94 est
nécessaire pour la production d’IGF-‐1 et d’IGF-‐2[379]. La déplétion spécifique de GRP94
dans les cellules musculaires induit une diminution de la quantité d’IGF-‐1 circulant qui
conduit à une petite taille avec une diminution de la densité osseuse[380]. Le rôle d’IGF-‐1
en tant que mécano senseur est connu et l’expression d’IGF-‐1 est corrélée à
l’augmentation de la densité osseuse après entrainement.
Malgré ces études, il est cependant difficile de savoir si les effets observés sont dus aux
interactions moléculaires directes entre l’os et le muscle ou à un effet systémique dû à la
sécrétion des interleukines et facteurs de croissance par différents tissus.
Les interactions os-‐muscle sont des interactions bidirectionnelles où des molécules
sécrétées par l’os peuvent impacter le muscle et inversement. Au cours de la formation
osseuse par ossification endochondrale, les chondrocytes sécrètent abondamment IHH
qui est nécessaire à leur différenciation[34]. Les souris IHH-‐/-‐ présentent une
augmentation drastique de l’apoptose chez les myoblastes en cours de différentiation.
IHH permet donc la survie des myoblastes et leur différentiation au cours de la
myogenèse. De plus, la surexpression de IHH induit une augmentation de la masse
musculaire sans affecter le tissu osseux. Le phénotype observé dans les myoblastes est
donc indépendant du phénotype osseux[381].
Les études démontrant les interactions strictes entre l’os et le muscle ont
majoritairement été réalisées in vitro. Des expériences de co-‐culture indirectes entre des
cellules myogéniques C2C12 et des ostéoblastes MC3T3-‐E1 montrent que les exosomes
des cellules myogéniques C2C12 promeuvent la différentiation ostéoblastiques[382]. La
culture et différentiation d’ostéoblastes MC3T3-‐E1 avec du milieu conditionné de C2C12
qui sur-‐expriment ou qui n’expriment pas certains facteurs (FAM5C, ostéoglycine) a
permis de montrer le rôle potentiel de ces molécules dans les interactions os-‐muscle[383-‐
385]. Des études in vivo seront nécessaires pour démontrer la relevance fonctionnelle de
ces observations.

Introduction
65
Le système Cre-‐lox peut être une solution pour étudier les interactions os-‐muscle
puisqu’il permet l’inactivation ou la surexpression de protéines au sein de lignages
cellulaires spécifiques. L’inactivation de Mbtps1 (membrane-‐bound transcription factor
peptidase, site 1) dans les ostéocytes via la souris Dmp1Cre induit l’expression des gènes
Pax7, Myog et MyoD chez les souris âgées, dont l’expression est normalement induite au
cours de la régénération musculaire. Aucun phénotype osseux n’est décrit, à part une
augmentation de 25% de la rigidité osseuse. L’expression de Mbtps1 dans les ostéocytes
participe donc à l’homéostasie musculaire[386, 387]. Ce type d’expérience permet
d’analyser directement les interactions os-‐muscle mais l’apparition d’un phénotype dans
le lignage où la déplétion a été faite peut induire des erreurs d’interprétation des
résultats.
3.3. Interactions os-‐muscle et ossification hétérotopique
Le rôle important des interactions entre l’os et le muscle au cours de la formation
osseuse est reconnu. De nombreuses études portent sur l’ossification hétérotopique
(OH). L’OH peut avoir deux origines distinctes. Une origine génétique dans le cas de la
Fibrodysplasie Ossifiante Progressive (FOP), causée par des mutations dans le récepteur
BMPRI et activant de façon constitutive la voie BMP[348]. Une origine traumatique où
après une fracture complexe combinée à un traumatisme musculaire ou après le
traitement d’une fracture par administration de BMP-‐2 ou BMP-‐7, de l’os ectopique est
formé [388-‐390]. L’origine cellulaire de l’OH reste mal comprise. Des études de lignage in
vivo ont pu exclure le lignage myogénique et le lignage hématopoïétique en tant que
contributeur cellulaire à la formation d’os ectopique. Les lignages Tie2 (lignage
endothélial), αSMA (lignage fibro-‐mésenchymateux) et GLAST (mésenchymateux)
contribuent directement à la formation des chondrocytes, des ostéoblastes et du tissu
fibroprolifératif de l’OH. Il n’est pas exclu que certains de ces lignages se chevauchent et
englobent aussi une partie du lignage péricytaire [301, 342, 391]. D’un point de vue
moléculaire, la sur-‐activation de la réponse inflammatoire corrèle avec le
déclenchement de l’ossification, puis l’activation de différents réseaux de signalisation
tels que les BMP, Wnt et Hif1α induisant la formation osseuse par ossification
endochondrale[210, 388].

Introduction
66
3.4. Interactions os-‐muscle au cours de la régénération osseuse
Les interactions entre l’os et le muscle sont fondamentales pour une régénération
osseuse efficace. En effet, alors que 5 à 10% des fractures simples présentent un retard
de régénération, ce chiffre augmente à 40% lorsque la fracture est combinée à une
atteinte vasculaire et à une blessure du muscle[211, 392]. En outre, plus la blessure
musculaire est importante, plus le risque de retard de régénération est élevé[393]. Afin
d’améliorer la régénération osseuse, des lambeaux de muscle sont utilisés pour couvrir
les fractures complexes ou les grands défauts osseux[394]. Ces lambeaux de muscle
peuvent avoir différentes fonctions : servir de bio-‐réacteur pour améliorer
l’implantation des cellules mésenchymateuses transplantées[395], améliorer la
vascularisation du site de fracture[396], pourvoir le site de fracture en facteurs de
croissance et cytokines (endogène ou exogène)[397] et limiter le risque d’infection[398].
L’utilisation de modèles murins a permis de mettre en évidence que la présence de
lambeau musculaire augmente directement la formation osseuse [399].
D’un point de vu mécanique, contrairement au développement où la paralysie
musculaire retarde la formation osseuse, dans la cas de la régénération osseuse, si les
deux fragments osseux restent alignés, la paralysie musculaire induite par l’injection de
toxine botulique permet de stabiliser le site de fracture et donc peut avoir des effets
bénéfiques[400]. Cependant, si le site de fracture n’est pas stabilisé et que les deux
fragments d’os forment un angle important alors l’activité musculaire est essentielle
pour réduire la fracture et induire le réalignement des fragments osseux[401, 402]. Chez
l’Homme, après fracture si les deux parties de l’os sont trop éloignées, la réduction de la
fracture par les muscles peut être insuffisante, et une intervention chirurgicale est
nécessaire.
Les interactions moléculaires os-‐muscle décrites au cours du développement jouent
aussi un rôle au cours de la régénération osseuse. Pendant la régénération osseuse, le
muscle peut aussi influencer la réponse inflammatoire. Dans le modèle murin mdx,
l’environnement inflammatoire est dérégulé dans le tissu musculaire et induit un retard
de régénération osseuse, restauré en partie par le traitement au PLX3397 qui inhibe les
monocytes [403, 404]. Après fracture, TNF-‐α et IL-‐6 sont sécrétées pendant la phase
inflammatoire et induisent la migration et la différentiation ostéogénique in vitro des
« cellules souches dérivées du muscle » SCDMs[206].

Introduction
67
Après fracture, Mstn est exprimée par les fibres musculaires mais aussi par les
chondrocytes dès jour 4 post-‐fracture[368]. L’administration de Mstn diminue la taille de
la cal, augmente la formation de tissu fibreux et donc retarde la régénération osseuse. De
plus, les souris Mstn-‐/-‐ présentent des volumes de cal et d’os et une rigidité du cal plus
élevée que les souris Mstn+/+. Mstn est donc impliqué dans la formation du cal et de l’os
au cours de la régénération osseuse[405, 406]. D’autres voies de signalisation telles BMP ou
IGF-‐1que pourraient être impliquées dans les interactions os-‐muscle au cours de la
régénération osseuse mais, à ce jour, il n’existe pas d’études démontrant leur rôle direct [209]. A l’échelle cellulaire, plusieurs études suggèrent de façon indirecte la contribution
cellulaire du muscle à la formation du cal et la contribution endogène n’est pas
démontrée[195].. Les SCDMs ne contribuent pas à la régénération osseuse mais
préalablement transfectées par un rétrovirus exprimant BMP4, les SCDMs sont capables
de contribuer à la régénération osseuse par voie endochondrale et intramembranaire [407]. Cependant, la sur-‐expression de BMP peut conduire à la formation d’os ectopique et
les cellules SCDMs sont peu caractérisées[208]. Les cellules αSMA+ sont présentes dans le
tissu interstitiel musculaire, dans le tissu osseux (ostéoblastes), et forment du cartilage
et de l’os au cours de la régénération osseuse par voie endochondrale[391, 408]. Par
définition, les cellules mésenchymateuses du muscle sont capables de se différencier en
ostéoblastes et chondrocytes in vitro. Des transplantations sous cutanées montrent la
capacité des cellules mésenchymateuses musculaires humaines à former de l’os in
vivo[302, 409]. Les cellules mésenchymateuses musculaires pourraient donc aussi
contribuer à la régénération osseuse [410].
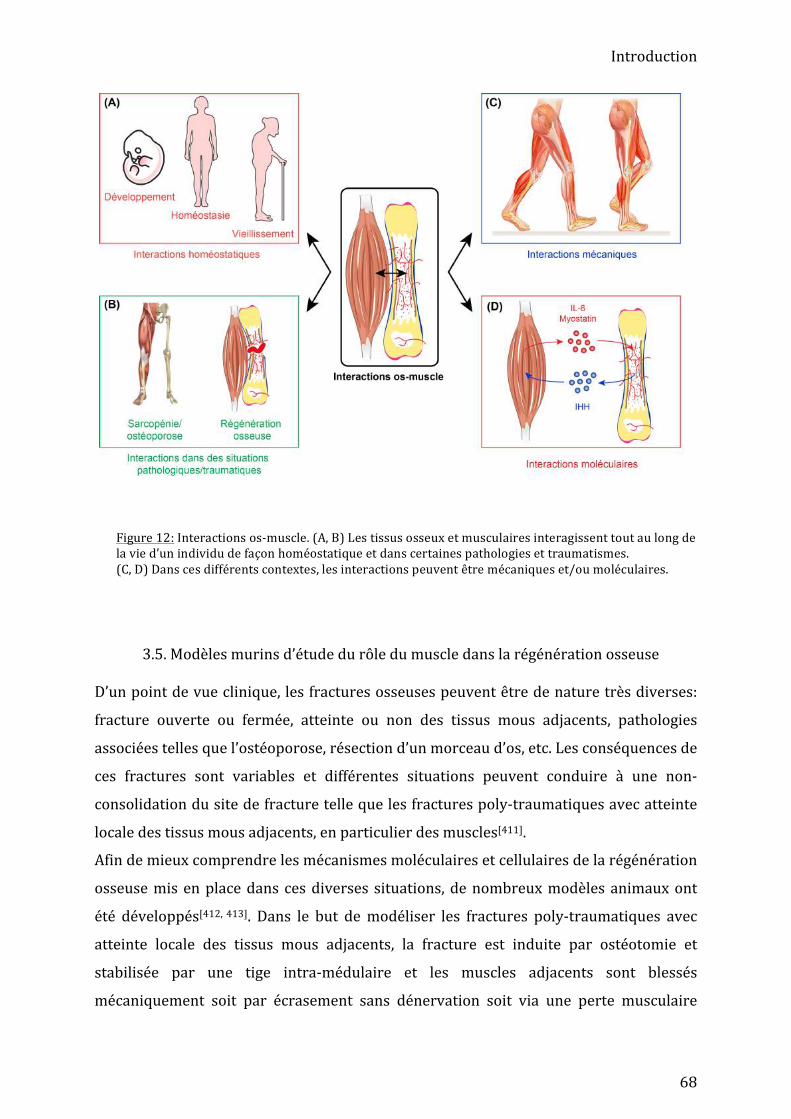
Introduction
68
3.5. Modèles murins d’étude du rôle du muscle dans la régénération osseuse D’un point de vue clinique, les fractures osseuses peuvent être de nature très diverses:
fracture ouverte ou fermée, atteinte ou non des tissus mous adjacents, pathologies
associées telles que l’ostéoporose, résection d’un morceau d’os, etc. Les conséquences de
ces fractures sont variables et différentes situations peuvent conduire à une non-‐
consolidation du site de fracture telle que les fractures poly-‐traumatiques avec atteinte
locale des tissus mous adjacents, en particulier des muscles[411].
Afin de mieux comprendre les mécanismes moléculaires et cellulaires de la régénération
osseuse mis en place dans ces diverses situations, de nombreux modèles animaux ont
été développés[412, 413]. Dans le but de modéliser les fractures poly-‐traumatiques avec
atteinte locale des tissus mous adjacents, la fracture est induite par ostéotomie et
stabilisée par une tige intra-‐médulaire et les muscles adjacents sont blessés
mécaniquement soit par écrasement sans dénervation soit via une perte musculaire
Figure 12: Interactions os-‐muscle. (A, B) Les tissus osseux et musculaires interagissent tout au long de la vie d’un individu de façon homéostatique et dans certaines pathologies et traumatismes. (C, D) Dans ces différents contextes, les interactions peuvent être mécaniques et/ou moléculaires.

Introduction
69
volumétrique (PMV) du muscle tibialis anterior (TA) [414, 415]. La blessure par écrasement
correspond à un écrasement des muscles du compartiment antérolatéral (TA, extensor
halucis longus, extensor digitorum lengus et peroneus tertius) de la jambe. La PMV
consiste à retirer un morceau de tissu musculaire du TA. Ces deux modèles ont pour
caractéristiques principales d’induire la rupture de la lame basale et la destruction des
cellules musculaires. Il en résulte une régénération musculaire imparfaite avec la
formation d’un tissu fibrotique persistant[416, 417]. Dans le cas de la blessure musculaire
par écrasement, la régénération osseuse par voie intramembranaire n’est pas impactée.
Cependant, la PMV induit la formation de cartilage au sein du cal et retarde la
régénération osseuse[414, 415].
L’utilisation de ces modèles permet de mieux comprendre l’impact d’une blessure du
muscle sur la régénération osseuse, mais les connaissances cellulaires et moléculaires
restent très sommaires.

Introduction
70

Objectifs de thèse
71
Objectifs de thèse
Les troubles musculosqueletiques représentent la deuxième cause d'invalidité au
monde. La prise en charge des patients et les traitements actuels restent complexes et
très couteux et la thérapie cellulaire est une perspective prometteuse. Les CSMs sont en
développement pour être utilisé en clinique mais d'autres sources de cellules (périoste,
muscle squelettique) pourraient être utilisées. Il est donc important de mieux
caractériser les différentes sources de cellules intervenant lors de la régénération
osseuse pour mieux comprendre leurs rôles au cours du processus endogène de
régénération osseuse, leurs déficiences dans les retards de régénération et améliorer la
prise en charge des patients atteints de troubles musculosquelettiques. Mon travail de
thèse a porté sur le rôle des cellules du périoste et principalement sur le rôle du muscle
et la caractérisation des ostéochondroprogéniteurs (OCP) provenant du muscle pendant
la régénération osseuse.
Première partie : Rôle du périoste dans la régénération osseuse et caractérisation des
cellules de périoste
Le périoste a été décrit comme une source majeure de cellules au cours de la
régénération osseuse. Cependant, les cellules du périoste sont très peu caractérisées.
Dans cette partie, j’ai participé à la caractérisation les cellules du périoste au niveau
cellulaire et moléculaire, et à la comparaison de leur potentiel de régénération par
rapport aux CSMOs (Article 1).
Deuxième partie : Rôle des cellules satellites, les cellules souches du muscle
squelettique, dans la régénération osseuse
Le rôle du muscle squelettique au cours de la régénération osseuse est admis mais la
contribution in vivo n’est pas connu. Dans cette deuxième partie, nous avons étudié le
rôle des cellules satellites au cours de la régénération osseuse. Nous avons montré que,
si des cellules souches musculaires étaient capables de former du cartilage au sein du
cal, la contribution cellulaire des cellules satellites est très faible. Cependant, les cellules
satellites secrètent des facteurs de croissance (BMP, IFG) nécessaires à la régénération
osseuse (Article 2).

Objectifs de thèse
72
Troisième partie : Rôle des traumatismes musculosquelettiques et impact sur les
osteochondroprogéniteurs issus du muscle dans la régénération osseuse
Dans le but de caractériser le rôle du muscle au cours de la régénération osseuse, nous
avons développé un nouveau modèle de blessure poly-‐traumatique combinant fracture
et blessure des muscles adjacents. Dans cette étude, nous avons utilisé ce modèle afin de
caractériser les cellules du muscle formant le cartilage et l’os dans le cal et leurs
déficiences dans les retards de régénération liées aux traumatismes (Article 3, en cours
de soumission).

Article 1, Duchamp et al, Nature Communications, 2018
73
Article 1
ARTICLE
Periosteum contains skeletal stem cells with highbone regenerative potential controlled by PeriostinOriane Duchamp de Lageneste1, Anaïs Julien1, Rana Abou-Khalil1, Giulia Frangi1, Caroline Carvalho1,Nicolas Cagnard2, Corinne Cordier3, Simon J. Conway4 & Céline Colnot 1
Bone regeneration relies on the activation of skeletal stem cells (SSCs) that still remain poorly
characterized. Here, we show that periosteum contains SSCs with high bone regenerative
potential compared to bone marrow stromal cells/skeletal stem cells (BMSCs) in mice.
Although periosteal cells (PCs) and BMSCs are derived from a common embryonic
mesenchymal lineage, postnatally PCs exhibit greater clonogenicity, growth and differentia-
tion capacity than BMSCs. During bone repair, PCs can efficiently contribute to cartilage and
bone, and integrate long-term after transplantation. Molecular profiling uncovers genes
encoding Periostin and other extracellular matrix molecules associated with the enhanced
response to injury of PCs. Periostin gene deletion impairs PC functions and fracture
consolidation. Periostin-deficient periosteum cannot reconstitute a pool of PCs after injury
demonstrating the presence of SSCs within periosteum and the requirement of Periostin in
maintaining this pool. Overall our results highlight the importance of analyzing periosteum
and PCs to understand bone phenotypes.
DOI: 10.1038/s41467-018-03124-z OPEN
1 INSERM UMR1163, Imagine Institute, Paris Descartes University, 75015 Paris, France. 2 Paris-Descartes Bioinformatics Platform, 75015 Paris, France.3 INSERM US24 - CNRS UMS3633 Cytometry Platform, Paris Descartes University, 75015 Paris, France. 4 Herman B. Wells Center for Pediatric Research,Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN 46202, USA. Correspondence and requests for materials should beaddressed to C.C. (email: [email protected])
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 1
1234
5678
90():,;

Article 1, Duchamp et al, Nature Communications, 2018
74
The skeleton is a central component of vertebrates’ body,providing structural support and protection for majororgans. The 206 bones constituting the human skeleton
store vital minerals, form muscle attachments, and comprise theniche for hematopoiesis. Bones are constantly challengedmechanically and can remodel or regenerate throughout life. Thedevelopment, growth, and regeneration of this essential organsystem rely on two robust ossification processes, intramem-branous ossification occurring by direct differentiation ofmesenchymal precursors into osteoblasts and endochondralossification marked by the formation of an intermediate cartilagetemplate1. Vascular invasion of this cartilage template drives thereplacement of cartilage by the bone marrow cavity and bone.During this crucial step of skeletal development, hematopoieticstem cells (HSCs) migrate into the developing bone to establishtheir niche within the marrow cavity. In parallel, bone-formingcells distribute in various bone compartments along the innersurface of bone (endosteum), metaphyseal trabeculae, and on theouter surface of the bone within the periosteum. It is wellestablished that these two processes of ossification can be reca-pitulated postnatally to very efficiently repair injured bones2–5.This reactivation of the skeletogenic program requires there-expression of key transcription factors and growth factorsregulating skeletal development. Yet the skeletal stem cells (SSCs)that permit this regenerative process and the mechanisms of stemcell activation in response to bone injury remain elusive.
Research on the biology of SSCs has mostly concentrated untilnow on the characterization of bone marrow stromal cells/skeletalstem cells (BMSCs), that form the niche for HSCs, regulate boneturnover, and show multipotency and self-renewal capacities aftersubcutaneous transplantation6–11. SSC populations are very het-erogeneous, making it a challenge to identify specific markers totrace these cells in vivo. Recent advances with genetic mousemodels have identified several markers to define various sub-populations of SSCs that appear during limb development andpost-natal growth, and play a role in bone maintenance andrepair12–22. However, these markers do not distinguish the tissueorigins of activated SSCs in response to bone injury. AlthoughBMSCs are largely used for enhancing bone repair through cell-based therapy, it has become clear that BMSCs are not the centralcellular component of endogenous skeletal repair. In contrast, theperiosteum is largely involved in bone strength maintenance andits preservation is crucial for normal bone repair23–31. Theperiosteum is a thin layer of vascularized tissue lining the bonesurface, supporting the tendon and muscle attachments, andhighly responsive to mechanical stress. Several studies haverevealed the periosteum as a major source of SSCs for bone repair,but this population has been largely overlooked until now30,32,33.We hypothesized that bone marrow and periosteum compriseSSC populations with distinct functions in bone biology andspecifically during endogenous bone repair.
Here we uncover common embryonic origins of BMSCs andperiosteal cells (PCs), but increased regenerative capacities andlong-term integration of PCs during bone regeneration in mice.Periosteum grafting shows that a pool of PCs is reconstituted andmaintained within periosteum in response to injury and can bere-activated after subsequent injuries revealing the presence ofSSCs within periosteum. Molecular profiling of PCs and BMSCsin response to injury identifies specific factors expressed in theextracellular matrix (ECM) of periosteum, including Periostin.Bone repair is compromised in Periostin KO mice due toimpaired periosteum and PC functions. Unlike wild-type peri-osteum, Periostin-deficient periosteum cannot reconstitute a poolof PCs and contribute to healing after successive bone injuriescausing severe repair defects. Periostin is, therefore, a keyregulator of SSCs in periosteum and their niche.
ResultsPCs and BMSCs share specific markers. In the absence of aunique marker to define SSCs, we used Prx1, a marker of themesenchymal lineage in developing limbs34,35. BMSCs wereobtained by flushing bone marrow of tibias and femurs followedby lineage depletion. Remaining long bones free of bone marrowwere placed in culture and PCs were let to grow out of the boneexplants (Fig. 1a and Supplementary Fig. 1a). In primary culturesof PCs and BMSCs isolated from Prx1-Cre;YFPfl/+ mice, thepopulations negative for hematopoietic and endothelial makersand double-positive for Sca1/CD29 and Sca1/CD10536 weremostly Prx1-derived YFP-positive (Fig. 1a–b and SupplementaryFig. 1b). The populations that were positive for hematopoieticand endothelial makers were mainly YFP-negative (Supplemen-tary Fig. 1c). By qRT-PCR, Prx1-sorted PCs from Prx1-Cre;mTmG mice overexpressed markers previously shown to definemouse BMSCs, such as PDGFRα37, Gremlin 119, Cxcl128,Nestin15, but not Leptin Receptor (Leptin R)20,21. Prx1-sorted PCsalso overexpressed the pericyte marker NG238 and did notoverexpress the fibroblast marker Vimentin compared to thePrx1-negative population39 (Fig. 1c and Supplementary Fig. 1d).Secondary colony forming efficiency assay (CFE) show higherclonogenicity of PCs compared to BMSCs (Fig. 1d). Cell-growthanalyses revealed higher cell growth of PCs compared withadherent bone marrow cells (aBM, prior to lineage depletion) andBMSCs (Fig. 1e). PCs can differentiate in osteogenic, adipogenic,and chondrogenic lineages in vitro with an increased potential forchondrogenesis compared to BMSCs and aBM (Fig. 1f).
Common embryonic origin of PCs and BMSCs. BMSCs formthe niche for HSCs and are established within the bone marrowcompartment when cartilage anlagen are vascularized during longbone development40–42. In mice, this step occurs from the develop-mental stage E14.5 during the formation of the primary ossificationcenter. Simultaneously, the periosteum forms and will include PCs.Whether BMSCs and PCs derive from the same pool of mesenchy-mal cells within each skeletal element or whether BMSCs can bebrought by blood vessels from another local or systemic source is stillnot well understood. To address this question, we performed renalcapsule transplantations for cell-lineage analyses. On one hand wetracked cells derived from transplanted cartilage elements that are notyet vascularized and do not yet comprise a bone marrow compart-ment. On the other hand, we tracked cells brought by the wild-typehost vasculature that support the vascularization of cartilage grafts inthe renal capsule43,44 (Fig. 2a). First we transplanted E14.5 Prx1-Cre;YFPfl/+ grafts into wild-type hosts. In fully developed bones 8 weekspost-transplantation, the PC and BMSC populations positive forSca1/CD29/CD105 coincided with the donor-derived YFP-positivepopulation marked by Prx1 (Fig. 2b and Supplementary Fig. 2a).Therefore, PCs and BMSCs were both derived from the transplantedcartilage element. Conversely, after transplantation of E14.5 wild-typefemoral cartilages into Prx1-Cre;YFPfl/+ hosts, PC and BMSCpopulations positive for Sca1/CD29 were YFP-negative, confirmingthat PCs and BMSCs are derived from the graft and not from thehost. These results show that both BMSCs and PCs are derived fromthe local Prx1-mesenchymal lineage forming each embryonic skeletalelement and are not brought by blood vessels during the establish-ment of the primary ossification center (Supplementary Fig. 2b, c).
Local recruitment of Prx1-derived cells during bone repair. ThePrx1-derived cells within adult bones have been shown toparticipate in bone repair34,45. We localized Prx1-derived cellswithin the intact periosteum and activated periosteum 3 dayspost-fracture in adult Prx1-Cre;mTmG mice in which Prx1-derived cells are GFP-positive and all other cells are Tomato-
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
2 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications
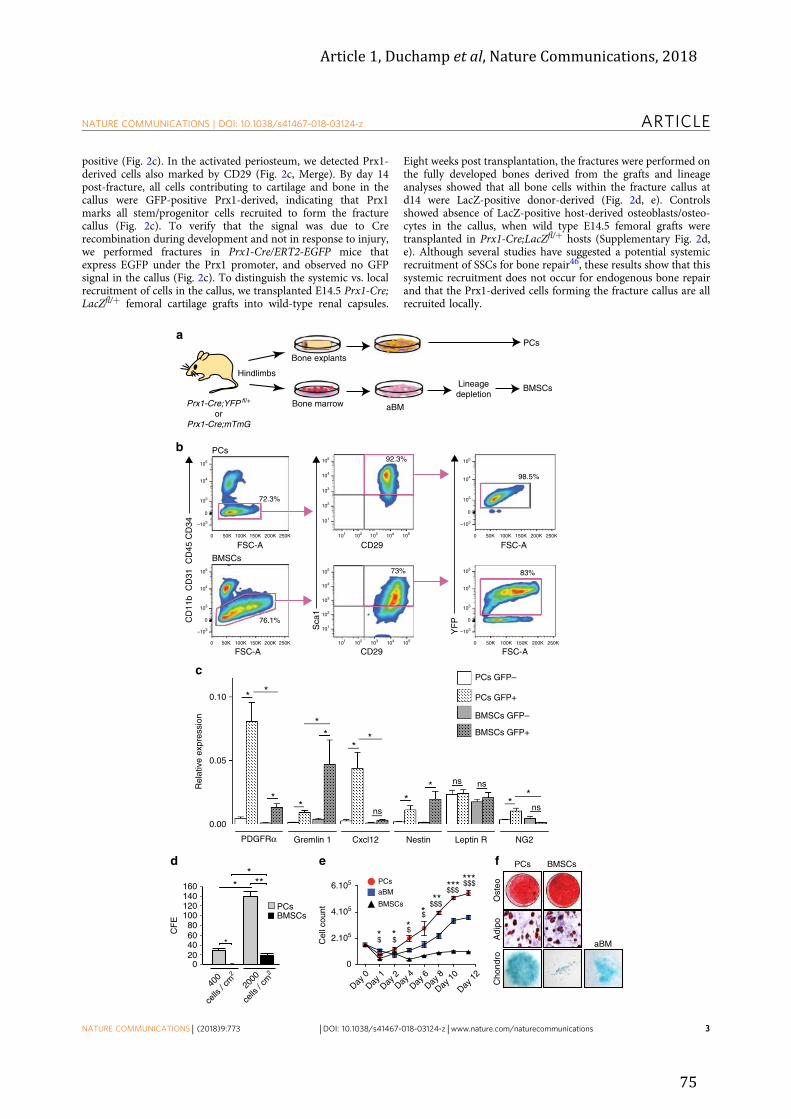
Article 1, Duchamp et al, Nature Communications, 2018
75
positive (Fig. 2c). In the activated periosteum, we detected Prx1-derived cells also marked by CD29 (Fig. 2c, Merge). By day 14post-fracture, all cells contributing to cartilage and bone in thecallus were GFP-positive Prx1-derived, indicating that Prx1marks all stem/progenitor cells recruited to form the fracturecallus (Fig. 2c). To verify that the signal was due to Crerecombination during development and not in response to injury,we performed fractures in Prx1-Cre/ERT2-EGFP mice thatexpress EGFP under the Prx1 promoter, and observed no GFPsignal in the callus (Fig. 2c). To distinguish the systemic vs. localrecruitment of cells in the callus, we transplanted E14.5 Prx1-Cre;LacZfl/+ femoral cartilage grafts into wild-type renal capsules.
Eight weeks post transplantation, the fractures were performed onthe fully developed bones derived from the grafts and lineageanalyses showed that all bone cells within the fracture callus atd14 were LacZ-positive donor-derived (Fig. 2d, e). Controlsshowed absence of LacZ-positive host-derived osteoblasts/osteo-cytes in the callus, when wild type E14.5 femoral grafts weretransplanted in Prx1-Cre;LacZfl/+ hosts (Supplementary Fig. 2d,e). Although several studies have suggested a potential systemicrecruitment of SSCs for bone repair46, these results show that thissystemic recruitment does not occur for endogenous bone repairand that the Prx1-derived cells forming the fracture callus are allrecruited locally.
Prx1-Cre;YFP fl/+
orPrx1-Cre;mTmG
Lineagedepletion
a
b
ed f
Adi
po
Hindlimbs
BMSCs
83%
Ost
eo
BMSCs PCs
Bone marrow
98.5%
CD
11b
CD
31 C
D45
CD
34
PCs
Bone explants
Sca
1
BMSCs
76.1%
YF
P
FSC-A
FSC-A FSC-A
FSC-ACD29
CD29
c
72.3%
PCs
Cho
ndro
Day 0
Day 1Day
2Day
4Day
6Day
8
Day 10
Day 12
0
2.105
Cel
l cou
nt
PCsaBM
BMSCs4.105
6.105
105
104
103
–103
0 50K 100K 150K 200K 250K
0
105
104
103
–103
0 50K 100K 150K 200K 250K
0
105
104
103
–103
0 50K 100K 150K 200K 250K
0
105
104
103
–103
0 50K 100K 150K 200K 250K
0
*$ **
***
******
$$
$$$$
$$$$$$
92.3%
73%
aBM
aBM
16014012010080604020
0
400
cells
/ cm2
CF
E
2000
cells
/ cm2
PCsBMSCs
*
* ***
0.00
0.05
0.10
Rel
ativ
e ex
pres
sion
PDGFRα Gremlin 1 Cxcl12 Nestin Leptin R NG2
PCs GFP–
PCs GFP+
BMSCs GFP–
BMSCs GFP+
*
*
*
*
**
**
ns*
**
*
ns ns
ns
105
104
103
102
101
101 102 103 104 105
105
104
103
102
101
101 102 103 104 105
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 3

Article 1, Duchamp et al, Nature Communications, 2018
76
Higher regenerative capacity of PCs compared to BMSCs. Todirectly compare the regenerative potential of PCs and BMSCs tobone repair in vivo, we transplanted GFP-labeled PCs and BMSCsin the fracture site of wild-type adult mice for in vivo lineagetracing (Fig. 3a). By day 7-post fracture (d7), transplanted PCswere found in the center of the callus and showed increasedcontribution to the callus compared to BMSCs (Fig. 3b andSupplementary Fig. 3a). Similar results were obtained for aBMand BMSC populations indicating that cell depletion of bonemarrow cells did not compromise their biological activity (Sup-plementary Fig. 3a). This increased contribution of PCs was notdue to changes in cell proliferation or cell death compared withBMSCs (Supplementary Fig. 3b and c). The majority of BMSCsstayed at the periphery of the callus and PCs integrated far intothe callus and cartilage by day 10 (Fig. 3b and SupplementaryFig. 3b, BMSCs right panel). Wound healing assays showed thatPCs migrated faster than BMSCs in vitro, which could at least inpart explain their higher regenerative potential in vivo (Supple-mentary Fig. 3d). Lineage analyses of PCs and BMSCs isolatedfrom Prx1-Cre;mTmG donors showed that PCs derived exclu-sively from the Prx1-mesenchymal lineage contributed to carti-lage and bone within the callus, while BMSCs had less potential toform cartilage and did not participate in forming new bone atlater stages (Fig. 3c).
Molecular profiling of the periosteum response to injury. Inorder to uncover the molecular signatures of PCs defining theirhigh regenerative potential compared to BMSCs, we performedmicroarray analyses of PCs and BMSCs isolated from un-injured(d0) and injured (day 3 post-fracture) tibias (Fig. 4a). Sampleclustering showed that all biological replicates clustered togetherand that PCs from un-injured bone (PCd0) are a distinct popu-lation compared to other groups in particular to BMSCd0. Afterfracture, PCd3 are closer to BMSCd3 (Fig. 4b). To identify genesets that distinguish PCs and BMSCs before or after injury, weperformed GSEA analyses comparing either PCd0 vs. BMSCd0 orPCd3 vs. BMSCd3. At both d0 and d3 post-fracture, PCs sharecommon GO categories such as “stemness”, “limb development”,and “ECM”. In contrary, BMSCs are enriched in GO categoriessuch as “downregulation of stemness”, “bone resorption”, and“immune and hematopoietic lineage” (Fig. 4c, d and Supple-mentary Fig. 4a, b). The number of differentially expressed genesin response to injury was greater in PCs compared to BMSCs(Fig. 4e). We then focused on genes specifically upregulated inPCs after fracture, but not in BMSCs and found 203 genesdefining the “periosteum response to injury” (PRI) gene set(Fig. 4f). GSEA analysis comparing PCs and BMSCs showed thatPRI genes are enriched in five different functions (Fig. 4g, in red).In order to find candidate genes that confer higher regenerative
capacities to PCs, we excluded the “stemness” GO categories andmerged “response to external stimulus” and “regulation ofexternal stimulus” into “external stimulus”. We intersected the“external stimulus”, “matrisome”, and “extracellular space” genesets to identify 9 candidate genes of interest (Fig. 4h). We focusedon Periostin (Postn) gene previously described as being specifi-cally expressed within periosteum47. Among the “Postn-linkedgenes” (complete list of Postn-linked genes in SupplementaryTable 3), 6 genes were found in common with the PRI gene setand belong to the matricellular protein and small leucin richproteoglycan families (Fig. 4i and Supplementary Table 3).Together, these findings reveal that PCs and BMSCs have distinctmolecular profiles and that PCs are more responsive to boneinjury. The molecular response to injury is marked in PCs withthe early upregulation of genes encoding ECM proteins, whichplay important roles in cell–matrix interactions and may be keyelements for SSC activation after bone injury.
Lack of Periostin impairs periosteum function and bonehealing. To elucidate the role of the matricellular proteinPeriostin in the periosteal response to injury, we analyzedPeriostin (Postn) expression by qRT-PCR and immuno-fluorescence. qRT-PCR analyses of PCs and BMSCs in un-injuredtibias and tibias 3 days post-injury confirmed the specific upre-gulation of Postn gene in PCs at day 3 compared to day 0 and toBMSCs (Fig. 5a). Periostin-positive cells were detected byimmunofluorescence in the cambial layer of the periosteum alongthe un-injured tibia (Fig. 5b). No expression of Periostin wasdetected in the uninjured and activated bone marrow andendosteum (Supplementary Fig. 5a). By qRT-PCR Postn wasupregulated in sorted GFP-positive PCs isolated from Prx1-Cre;mTmG mice (Fig. 5b). Three days after fracture, Periostin washighly expressed in the cambial cell layer of the activated peri-osteum also containing CD29 expressing cells. By day 14, Peri-ostin was expressed at the junction between late hypertrophiccartilage and bone, and by osteoblasts and osteocytes in the newbone matrix. By 28 days, Periostin was detected in the newlyformed periosteum at the periphery of the ossified callus (Fig. 5b).To functionally assess the role of Periostin during bone repair, weinduced tibial fractures in wild-type controls (WT) and PeriostinKO (KO) mice that have been reported to exhibit post-natalgrowth retardation and skeletal defects including reduced trabe-cular bone density in long bones48. Periostin KO mice exhibitimpaired bone regeneration marked by reduced callus size andbone volume throughout all stages of repair. Periostin KO micefailed to achieve maximum cartilage volume by day 10 followedby delayed cartilage resorption, leading to fibrosis and a non-union at day 28 (Fig. 5c, d). Periostin KO mice also displayedabnormal repair of unicortical bone defects that heal through
Fig. 1 FACS and in vitro analyses of PCs and BMSCs. a Experimental design of periosteal cells (PCs) and bone marrow stromal cells/skeletal stem cells(BMSCs) cultures from Prx1-Cre;YFPfl/+ or Prx1-Cre;mTmG mouse hindlimbs. Bone marrow cells were flushed from hindlimbs and plated to obtain adherentbone marrow cells (aBM). After expansion, lineage depletion was performed to isolate BMSCs with no further passage. The flushed bones were placed inculture to isolate in one step the PCs migrating out of the explants. b Flow cytometry analyses of PCs and BMSCs isolated from Prx1-Cre;YFPfl/+ mice. PCsand BMSCs negative for endothelial/hematopoietic markers (CD31, CD11b, CD34, and CD45) and double-positive for Sca1/CD29 are largely YFP+(derived from Prx1-mesenchymal lineage). c Quantitative RT-PCR analyses of FACS sorted GFP-positive and GFP-negative PCs and BMSCs isolated fromPrx1-Cre;mTmGmice. Results show overexpression of the markers PDGFRα, Gremlin1, Cxcl12, and Nestin and to a lesser extent NG2 in GFP-positive comparedto GFP-negative PCs, but not LeptinR. d CFE assays showing PCs forming colonies at cell density as low as 400 cells/cm2 14 days after plating and BMSCsat 2000 cells/cm2 14 days after plating. Colonies were stained with Giemsa blue and counted under microscope. e Cell-growth assay shows that PCs growfaster than adherent bone marrow cells (aBM) and BMSCs. The cells were plated at the same density (105 cells/dish) and counted every day during thefirst two days then every two days for 12 days (* represents the comparison between PCs and aBM, $ represents the comparison between PCs andBMSCs). f In vitro differentiation of PCs and BMSCs into osteogenic (3 weeks), adipogenic (3 weeks), and chondrogenic (2 weeks) lineages as shown byalizarin red S, Oil red O, and alcian blue staining, respectively. Due to the poor chondrogenic capacity of BMSCs, aBM were assessed for chondrogenesis.Statistical differences between the groups (n= 3 or 4 per group) were determined using Mann–Whitney test (*,$ p≤ 0.05, **,$$ p < 0.001,***,$$$ p < 0.0005). All data represent mean ± SD
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
4 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications

Article 1, Duchamp et al, Nature Communications, 2018
77
direct bone formation indicating that healing via both intra-membranous and endochondral ossification is affected in theabsence of Periostin (Supplementary Fig. 5b, c). To assess thespecific impact of Periostin gene invalidation on the periosteum,GFP-wild type or -Periostin KO periosteum grafts were trans-planted at the fracture site of wild-type mice (Fig. 6a). Periostin
KO periosteal grafts exhibit a decreased contribution to repair inthe wild-type environment compared to WT grafts, thus Periostinis essential for periosteum activation and contribution to bonerepair (Fig. 6b).
To determine if the defective periosteum response to fracturein Periostin KO mice was linked to impaired PCs, we isolated
b
a
c
E14.5 Prx1-Cre;LacZ fl/+ femur
FSC-A CD29 FSC-A
CD
11b
CD
31 C
D45
CD
34
YF
P
Sca
198.8%
Prx1-Cre;mTmG mice
d14
83.6%
PCs105
104
103
–103
0
50K 100K 150K 200K 250K0 50K 100K 150K 200K 250K0
FSC-ACD29FSC-A
8 weeks
Un-injured periosteum
Prx1-Cre;mTmG mice callus d14 post fracture
d
e
DAPI/GFP/Tomato DAPI/GFP/Tomato
cabm
m
cal
po
m
c
DAPI/GFP/Tomato 250 µm
250 µm1 mm
Lineagetracing
Activated periosteum d3 post fracture
58.8%
20 µm
DAPI/GFP/Tomato/CD29/Merge
c
m
po ]
TC
BMSCs
97.2%
49.2% 96.9%
BMSCs
PCs
E14.5 Prx1Cre; YFP fl /+ femur
8 weeks
Wild-typeRenal capsule
Wild-typeRenal capsule
Xgal/TRAP
Prx1-Cre/ERT2,-EGFP micecallus d14 post fracture
bm
cal
DAPI/GFP m
1 mm
105105
104
103
–103
0
104
103
102
101
101 102 103 104 105
101 102 103 104 105
105 105
104
103
102
101
105
104
103
–103
0
104
103
–103
0
50K 100K 150K 200K 250K0 50K 100K 150K 200K 250K0
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 5

Article 1, Duchamp et al, Nature Communications, 2018
78
activated PCs from wild-type and Periostin KO mice 3 days aftertibial fracture. The capacity of Periostin KO PCs to form cloneswas decreased compared to WT in CFE assays (Fig. 6c). PeriostinKO PCs had impaired osteogenesis and adipogenesis compared towild-type PCs in vitro, although chondrogenic potential was notaffected (Fig. 6d). By qRT-PCR the Periostin-linked genes,Lumican, Decorin, Osteoglycin, Thrombospondin 2,and Dermato-pontin upregulated in PCs in response to injury and/or encodingECM proteins (Fig. 4i and Supplementary Table 3) showeddecreased expression in Periostin KO PCs compared to wild type(Fig. 6e). Endoglin 1 expression was upregulated in Periostin KOPCs compared to wild type and the expression of Asporin,Fibrillin 1, Tenascin C, Biglycan, SPARC, and Col3a1 was notaffected (Fig. 6e). The expression of these genes in Periostin KOBMSCs compared to wild type was not affected (SupplementaryFig. 5d). These results indicate that Periostin KO PCs havedeficient stem cell properties in vitro and decreased expression ofother ECM proteins that are normally specifically upregulated inPCs in response to injury. We then assessed the ability of wild-type PCs to rescue impaired bone healing in Periostin KO mice(Fig. 6f). GFP-wild-type trans-planted PCs showed high expres-sion of Periostin and great capacity to integrate into cartilage(Fig. 6g). Bone volume was increased (WT in KO) compared tocontrols (KO in KO) and cartilage volume was decreasedindicating a partial rescue of the phenotype (Fig. 6h).
Periostin is required to maintain the pool of PCs. To assessmore directly the role of Periostin in regulating PCs and theirperiosteum niche, we used periosteum transplantation (Fig. 7a).We first assessed whether periosteum contains cells that were ableto repair bone and re-populate the periosteum after injury. Aftertransplantation of GFP periosteal grafts at the fracture site ofwild-type hosts (Fig. 7a)32, periosteum-derived GFP-positive cellslargely contributed to cartilage in the callus, and rare GFP-positive cells were localized in the newly formed periosteum byd28 (Fig. 7b). To evaluate the GFP-positive cells that persisted inthe callus, we isolated PCs and BMSCs from ossified calluses aftertransplantation. GFP-positive cells that were negative for hema-topoietic and endothelial marker, and positive for Sca1/CD29,were only detected in PCs, but not in BMSCs cultures (Fig. 7c).This indicated that PCs within GFP donor periosteum couldrepopulate the newly formed periosteum. Following a secondinjury, these rare GFP-positive PCs could be re-activated tocontribute to bone repair and were detected within cartilage andbone by day 7 (Fig. 7b). By day 28, rare GFP-positive cells wereagain detected in the new periosteum indicating the ability of PCsto re-populate the periosteum after the second injury (Fig. 7b). Ina third cycle of injury, these re-activated PCs could again
contribute to cartilage within the callus (Fig. 7b). Quantitativeanalyses revealed the ability of PCs to expand extensively fromperiosteum in the 3 cycles of injuries (Supplementary Fig. 6a).The contribution to repair of these rare PCs within the newperiosteum did not decrease between the second and third injurycycles, indicating that the contribution was not due to a popu-lation of progenitor cells that would be exhausted overtime(Supplementary Fig. 6a). When we performed the same experi-ment with Periostin KO grafts into wild-type hosts, the ability ofPCs to persist in the new periosteum after fracture and contributeto repair in a second injury cycle was abolished, leading todefective callus formation and fibrosis (Fig. 7d). Further, trans-plantation of Periostin KO grafts into a Periostin KO fracture siteamplified the bone healing defect as shown by the completeabsence of callus formation and bridging (Supplementary Fig. 6bas compared to the phenotype shown in Fig. 5d). Since we did notdetect decreased cell proliferation in the periosteum of PeriostinKO mice (Supplementary Fig. 6c), these results show that thePeriostin KO phenotype is not due to a deficient proliferation, butthe inability of PCs to maintain a pool of PCs in the periosteumand support bone healing in the absence of Periostin.
DiscussionBone regeneration is a well-orchestrated process allowing thebone to recover its proper shape and functions without the for-mation of scar tissue. Skeletal stem cells are activated in the earlysteps of bone regeneration and are the basis for this extraordinarycapacity of bone to regenerate, but their endogenous origins andthe mechanisms of activation are still poorly understood. Manystudies have focused on the characterization of BMSCs, which arecurrently used in cell-based therapy approaches in orthopedics. Inthis study, we have identified that PCs have an enhanced capacityfor cell growth and clonogenicity, as well as superior regenerativecapacities compared to BMSCs. In microarray analyses, PCs havethe key characteristics of SSCs, since they express “stemness” and“limb or skeletal system development” gene sets. In contrast,BMSCs are enriched in GO categories, such as “downregulationof stemness”, “bone resorption” and “immune and hematopoieticlineage”, suggesting that these cells play an indirect role duringbone repair. Previous reports suggested that endogenous BMSCsare restricted to the bone marrow compartment during bonerepair and indirectly stimulate healing via the secretion of growthfactors32,49. Their role in regulating hematopoiesis and boneresorption remains to be further addressed in the context of bonerepair. We show here that after transplantation at an injury site,BMSCs have reduced capacity to form cartilage and bone duringskeletal regeneration compared to PCs that show great engraft-ment capacity further revealing the stem cell properties of PCs.
Fig. 2 PCs and BMSCs derive from the Prx1-mesenchymal lineage. a Experimental design for renal capsule transplantations. Femoral cartilages beforevascular invasion were isolated from E14.5 Prx1-Cre;YFPfl/+ embryos and transplanted under the renal capsule of wild-type hosts. PCs and BMSCs wereisolated from mature skeletal elements 8 weeks post-transplantation as shown in Fig. 1a. b Flow cytometry analyses of PCs and BMSCs isolated from Prx1-Cre;YFPfl/+ mature skeletal elements grown under renal capsule. Both PCs and BMSCs that are negative for endothelial/hematopoietic markers (CD31,CD11b, CD34, and CD45) and positive for Sca1/CD29 are mostly YFP-positive (Prx1-donor-derived) (n= 3 per group). c Localization of Prx1-derived cellsin the periosteum and fracture callus of Prx1-Cre;mTmGmouse. The un-injured periosteum is derived from Prx1-lineage (GFP+). In the activated periosteumat day 3 post fracture, some Prx1-derived cells (GFP/pointed by green arrows) colocalize with CD29-positive cells (Merge CD29+ GFP+/pointed by whitearrows). In the fracture callus (d14), all chondrocytes and osteoblasts/osteocytes are derived from Prx1-lineage (GFP+). The fractures performed on Prx1-Cre/ERT2,-EGFP mice, where EGFP is expressed under the Prx1 promoter, show no GFP signal in the callus. d Experimental design for cell-lineage analysesof Prx1-derived cells during bone regeneration in renal capsule. Femoral cartilages were isolated from E14.5 Prx1-Cre+/−;LacZfl/+ embryos and transplantedunder the renal capsule of wild-type hosts. After 8 weeks, mature femurs underwent osteotomy and were collected at d14 post-fracture for cell-lineagetracing. e TC and Xgal/TRAP double-staining on longitudinal sections of Prx1-Cre+/−;LacZfl/+ fractured femurs in wild-type hosts (top) showing new bonewithin the callus entirely donor-derived, i.e., LacZ+ TRAP− (black arrowheads: osteocytes) and some osteoclasts (TRAP+ LacZ+ with endogenous beta-galactosidase activity). Scale bar: 0.5 mm. TC: Masson’s trichrome, TRAP: Tartrate resistant acid phosphatase, m: muscle, c: cortex, po: periosteum, bm:bone marrow, cal: callus, ca: cartilage, white dashed line: callus, orange lightning bolt: fracture, orange arrow: fracture site, black arrow: points to theperiosteum, black arrowhead: osteocytes in new bone
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
6 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications
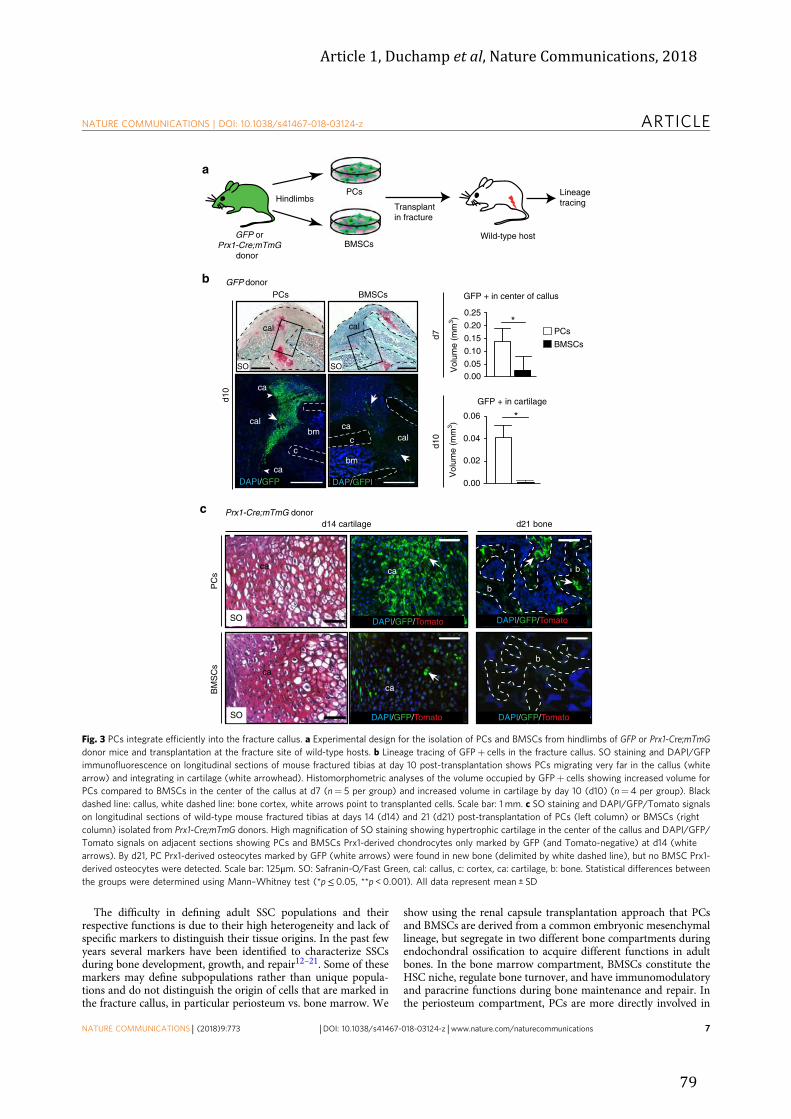
Article 1, Duchamp et al, Nature Communications, 2018
79
The difficulty in defining adult SSC populations and theirrespective functions is due to their high heterogeneity and lack ofspecific markers to distinguish their tissue origins. In the past fewyears several markers have been identified to characterize SSCsduring bone development, growth, and repair12–21. Some of thesemarkers may define subpopulations rather than unique popula-tions and do not distinguish the origin of cells that are marked inthe fracture callus, in particular periosteum vs. bone marrow. We
show using the renal capsule transplantation approach that PCsand BMSCs are derived from a common embryonic mesenchymallineage, but segregate in two different bone compartments duringendochondral ossification to acquire different functions in adultbones. In the bone marrow compartment, BMSCs constitute theHSC niche, regulate bone turnover, and have immunomodulatoryand paracrine functions during bone maintenance and repair. Inthe periosteum compartment, PCs are more directly involved in
Hindlimbs
BMSCs
a
b
PCs
GFP orPrx1-Cre;mTmG
donor
Transplantin fracture
Lineagetracing
Prx1-Cre;mTmG donor
GFP donor
PC
sB
MS
Cs
d14 cartilage d21 bone
DAPI/GFP/Tomato
ca b
b
ca
ca
PCs BMSCs
0.000.050.100.150.200.25
*
GFP + in center of callus
d7d1
0
GFP + in cartilage
0.00
0.02
0.04
0.06 *
c
SO
ca
SO
Vol
ume
(mm
3 )
SO
cal
DAPI/GFP/Tomato
PCs
d10
cal
c
caDAPI/GFP
bm
ca
Wild-type host
Vol
ume
(mm
3 )bm
calca
DAP/GFPI
c
BMSCs
cal
SO
b
DAPI/GFP/Tomato DAPI/GFP/Tomato
Fig. 3 PCs integrate efficiently into the fracture callus. a Experimental design for the isolation of PCs and BMSCs from hindlimbs of GFP or Prx1-Cre;mTmGdonor mice and transplantation at the fracture site of wild-type hosts. b Lineage tracing of GFP+ cells in the fracture callus. SO staining and DAPI/GFPimmunofluorescence on longitudinal sections of mouse fractured tibias at day 10 post-transplantation shows PCs migrating very far in the callus (whitearrow) and integrating in cartilage (white arrowhead). Histomorphometric analyses of the volume occupied by GFP+ cells showing increased volume forPCs compared to BMSCs in the center of the callus at d7 (n= 5 per group) and increased volume in cartilage by day 10 (d10) (n= 4 per group). Blackdashed line: callus, white dashed line: bone cortex, white arrows point to transplanted cells. Scale bar: 1 mm. c SO staining and DAPI/GFP/Tomato signalson longitudinal sections of wild-type mouse fractured tibias at days 14 (d14) and 21 (d21) post-transplantation of PCs (left column) or BMSCs (rightcolumn) isolated from Prx1-Cre;mTmG donors. High magnification of SO staining showing hypertrophic cartilage in the center of the callus and DAPI/GFP/Tomato signals on adjacent sections showing PCs and BMSCs Prx1-derived chondrocytes only marked by GFP (and Tomato-negative) at d14 (whitearrows). By d21, PC Prx1-derived osteocytes marked by GFP (white arrows) were found in new bone (delimited by white dashed line), but no BMSC Prx1-derived osteocytes were detected. Scale bar: 125μm. SO: Safranin-O/Fast Green, cal: callus, c: cortex, ca: cartilage, b: bone. Statistical differences betweenthe groups were determined using Mann–Whitney test (*p≤ 0.05, **p < 0.001). All data represent mean ± SD
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 7

Article 1, Duchamp et al, Nature Communications, 2018
80
bone repair by forming cartilage and bone in the callus, while therole of PCs in other periosteum functions remains to be furthercharacterized. The renal capsule model also provided strongevidence that systemic recruitment of cells during bone repair isnegligible.
A hallmark of adult stem cells is their ability to self renew afterinjury. Self-renewal capacity has been established for other adultstem cells after tissue injury to show their ability to maintain a
pool of stem cells within the same anatomical location50. Theability of SSCs to self renew has never been addressed in thecontext of bone repair until now. Sacchetti and collaboratorsreported that BMSCs can form de novo a bone marrow stromaorganizing a hematopoietic environment surrounded by bonetissue after subcutaneous transplantation11. This model of het-erotopic bone formation, however, does not reproduce the ade-quate environment to assess PC renewal after bone injury.
a b
c
Microarray analyses
d
Developmental cell growth
Limb development
Cell fate specification
Cell morphogenesis
Cell fate commitment
Stem cell population maintenance
Bone stem cell
Stemness
Extracellular matrix
Bone resorption
Osteoclast differentiation
Downregulation of stemness
Downregulation of stem cell markers
0.015 0.003 0.000 0.003 0.008
p-value
Enriched in BMSCd0
Enriched in PCd0
PCd0 vs. BMSCd0 PCd3 vs. BMSCd3
ECM Glycoproteins
Extracellular matrix
Regulation of mesenchymalcell proliferation
Stemness
Stem population maintenance
Regulation of stem cell proliferation
Endochondral bone morphogenesis
Skeletal system development
Skeletal system morphogenesis
Downregulation of stemness
Regulation of bone resorption
0.000 0.010 0.036
p-value
e
PCsd0vsd3
BMSCsd0vsd3
0
1000
2000
3000
Num
ber o
f diff
eren
tially
expr
esse
d pr
obes
PC d3
BMSC d3
PC d0
BMSC d0
PCd3 > BMSCd3
PCd3 > PCd0
2018 203
0.3 0.6 0.90.025 0.050p -value
0.25
0.50
0.75
1.00
FD
R
Stem cellMatrisome
Response to external stimulusExtracellular space
Regulation of external stimulus
MatrisomeExternal stimulus
Extracellularspace
820 3
9
2
2 6
Ccl8Dcn
Cxcl1Cxcl12
837
Periosteum response to injury
Periosteum response to injury
PC
d3
15P
C d
3 17
PC
d3
19P
C d
3 20
BM
SC
d3
19B
MS
C d
3 17
BM
SC
d3
20B
MS
C d
3 15
BM
SC
d0
11B
MS
C d
0 13
BM
SC
d0
7B
MS
C d
0 10
PC
d0
11P
C d
0 13
PC
d0
7P
C d
0 10
g h i
f
LumAspnDcn
Gas6IL6TNF
Col3a1Postn
Enriched in BMSCd3
Enriched in PCd3
PCd3 > BMSCd3PCd3 > PCd0
8212009197
69 16
POSTN-linkedgenes
62
Egln1Col3a1Postn
Thbs2Fbn1Dpt
TNCOgnBgn
SPARC
Tibia
Fig. 4Microarray analyses of PCs and BMSCs in response to fracture. a Experimental design for microarray analyses of PCs and BMSCs isolated from wild-type un-injured tibias (d0) and from tibias 3 days post fracture (d3) (n= 4 per group). b Hierarchical clustering of biological replicates. c, d GSEA analysesof PCd0 vs. BMSCd0 and PCd3 vs. BMSCd3, respectively. PCs are enriched in stem cell, developmental, skeletal, and extracellular matrix gene sets (red)compared to BMSCs at both d0 and d3 (blue). e Number of differentially expressed probes in PCs and BMSCs in response to fracture. f Venn diagramshowing the intersection of PCd3 vs. PCd0 and PCd3 vs. BMSCd3 representing the periosteum response to injury (PRI). g GSEA analysis of PRI genes. Red,blue, and gray boxes correspond to significant, interesting, and non-useful functions, respectively. Five significant functions are identified “response toexternal stimulus, “regulation of external stimulus”, “extracellular space”, “matrisome”, and “stem cell” (red). h The GSEA significantly enriched GOcategories “response to external stimulus” and “regulation of external stimulus” were merged into “external stimulus” and compared by Venn to the“extracellular space” and “matrisome” GO categories resulting in a list of 9 common genes. i Venn diagram shows the intersection of PRI and Postn-linkedgenes resulting in a list of 6 genes (Complete list of 93 Postn linked genes in Supplementary Table 3)
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
8 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications
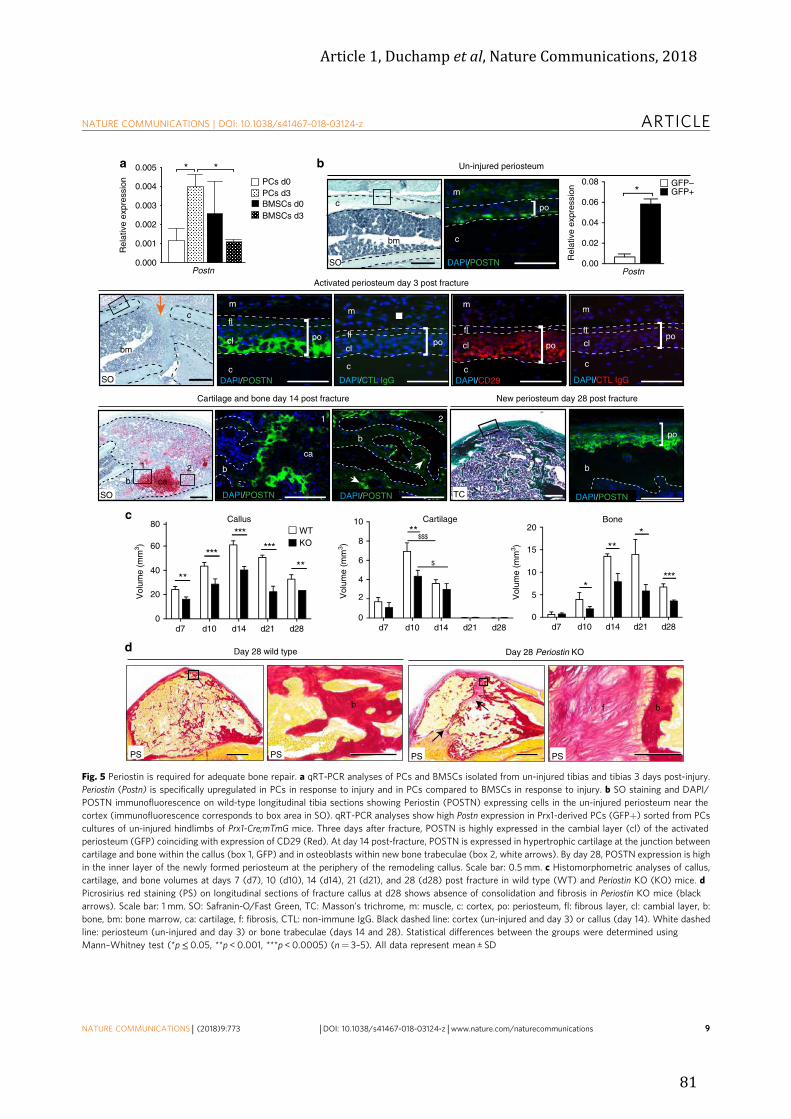
Article 1, Duchamp et al, Nature Communications, 2018
81
b
c
d
f bb
Day 28 wild type Day 28 Periostin KO
Un-injured periosteum
Activated periosteum day 3 post fracture
Cartilage and bone day 14 post fracture New periosteum day 28 post fracture
1
b
ca
1
b
po
po
c
m
fl
cl
DAPI/CD29
po
m
c
fl
1 2
cab
SO TC
0
20
40
60
80
****
***
*** ***
0
2
4
6
8
10**
d7 d10 d14 d21 d280
5
10
15
20
*
***
***
CallusWT
Cartilage Bone
b
2
SO
c
bm
c
bm
SO
PS
po
m
c
DAPI/POSTN
Vol
ume
(mm
3 )
Vol
ume
(mm
3 )
Vol
ume
(mm
3 )KO
d7 d10 d14 d21 d28d7 d10 d14 d21 d28
GFP–GFP+
Postn
DAPI/POSTN
DAPI/POSTN DAPI/POSTN
$$$
$
cl
fl
m
c
DAPI/CTL IgG
po cl
m
flcl
c
DAPI/CTL IgG
po
Rel
ativ
e ex
pres
sion
Rel
ativ
e ex
pres
sion
PCs d0PCs d3BMSCs d0BMSCs d3
Postn
a
0.000
0.001
0.002
0.003
0.004
0.005 **
DAPI/POSTN
PS PS PS
0.00
0.02
0.04
0.06
0.08*
Fig. 5 Periostin is required for adequate bone repair. a qRT-PCR analyses of PCs and BMSCs isolated from un-injured tibias and tibias 3 days post-injury.Periostin (Postn) is specifically upregulated in PCs in response to injury and in PCs compared to BMSCs in response to injury. b SO staining and DAPI/POSTN immunofluorescence on wild-type longitudinal tibia sections showing Periostin (POSTN) expressing cells in the un-injured periosteum near thecortex (immunofluorescence corresponds to box area in SO). qRT-PCR analyses show high Postn expression in Prx1-derived PCs (GFP+) sorted from PCscultures of un-injured hindlimbs of Prx1-Cre;mTmG mice. Three days after fracture, POSTN is highly expressed in the cambial layer (cl) of the activatedperiosteum (GFP) coinciding with expression of CD29 (Red). At day 14 post-fracture, POSTN is expressed in hypertrophic cartilage at the junction betweencartilage and bone within the callus (box 1, GFP) and in osteoblasts within new bone trabeculae (box 2, white arrows). By day 28, POSTN expression is highin the inner layer of the newly formed periosteum at the periphery of the remodeling callus. Scale bar: 0.5 mm. c Histomorphometric analyses of callus,cartilage, and bone volumes at days 7 (d7), 10 (d10), 14 (d14), 21 (d21), and 28 (d28) post fracture in wild type (WT) and Periostin KO (KO) mice. dPicrosirius red staining (PS) on longitudinal sections of fracture callus at d28 shows absence of consolidation and fibrosis in Periostin KO mice (blackarrows). Scale bar: 1 mm. SO: Safranin-O/Fast Green, TC: Masson’s trichrome, m: muscle, c: cortex, po: periosteum, fl: fibrous layer, cl: cambial layer, b:bone, bm: bone marrow, ca: cartilage, f: fibrosis, CTL: non-immune IgG. Black dashed line: cortex (un-injured and day 3) or callus (day 14). White dashedline: periosteum (un-injured and day 3) or bone trabeculae (days 14 and 28). Statistical differences between the groups were determined usingMann–Whitney test (*p≤ 0.05, **p < 0.001, ***p < 0.0005) (n= 3–5). All data represent mean ± SD
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 9

Article 1, Duchamp et al, Nature Communications, 2018
82
Therefore, we designed a strategy based on periosteal grafting andsubsequent injuries to specifically address the capacity of PCs tocontribute to repair and repopulate the periosteum cell com-partment after the repair process is completed. We show thatPCs, in addition to their enhanced capacity to regenerate bone
after transplantation, can maintain a pool of PCs within perios-teum after injury and are mobilized again after subsequentinjuries to repair bone. A similar approach based on single-muscle fiber transplantation was originally used to show extensiveexpansion to reform new muscle fibers in situ and self-renewal
FracturedGFP or Periostin KO-GFP
ActivatedPCs
Lineagetracing
d3
a
Transplantationin Periostin KO hosts
d14
d
b
g
Osteo
WT
c
f
0.00.10.20.30.4
*
0.00
0.05
0.10
0.15*
GFP+ in callus
GFP+ in cartilage
WT in WTKO in WT
KO
h
Transplantationin WT hosts
d14Lineage tracing
e
cal
graft
ca
DAPI/GFP
SOcal
graftca
WT in WT KO in WT
WT in KO
0
5
10
15
CF
E
*
WT KO
Vol
ume
(mm
3 )V
olum
e (m
m3 )
Vol
ume
(mm
3 )
Vol
ume
(mm
3 )
Vol
ume
(mm
3 )
Adipo Chondro
Callus CartilageBone
01020304050 ns
012345 *
5
10
15 *
0
KO in KOWT in KO
GFP or Periostin KO-GFP donor
Periosteumgraft
d14
Aspor
in
Fibrilli
n 1
Lumica
n
Decor
in
Osteog
lycin
Throm
bosp
ondin
2
c
cal
1
2
1
ca2
DAPI/GFP/POSTN/
Merge
Endog
lin 1
DAPI/GFP
SO
*
ns
ns
0.00
0.02
0.04
0.06
0.2
0.4
0.6
Rel
ativ
e ex
pres
sion
Rel
ativ
e ex
pres
sion
Dermato
ponti
n
* *
*
* *0.0
0.1
0.2
0.3
0.5
1.0
1.5
Tenas
cin C
Biglyc
an
SPARC
Col3a1
ns
ns
nsns
WTKO
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
10 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications

Article 1, Duchamp et al, Nature Communications, 2018
83
capacity of satellite cells51. This was not the case after trans-plantation of myoblasts, the muscle progenitors, which cannotcontribute to repair and self-renew52. The fact that a smallnumber of PCs within the initial periosteal graft can largelycontribute to cartilage, give rise to rare PCs within the newperiosteum and again largely contribute to cartilage in the nextinjury cycle indicates the presence of SSCs within the periosteum.Proliferation of progenitors could not provide a sufficient sourceof cells after three consecutive rounds of injury and repair, as theywould disappear overtime. Furthermore, we provide evidence thatthis capacity of PCs to re-integrate the newly formed periosteumand contribute to repair after a second injury is abolished in theabsence of Periostin without affecting the proliferation of PCs inresponse to injury. These data show that periosteum containsstem cells that can self renew during several injury cycles andPeriostin is required for this self-renewal capacity by regulatingthe periosteal niche of SSCs. More investigation will be requiredin the future to identify specific markers for the periosteal stemcell population and follow SSC activation and self-renewal at thesingle-cell level within periosteum in vivo. More data will also beneeded to compare the markers and tissue localization of PCs andBMSCs in mouse and human, as there are already known dif-ferences for BMSCs11,36,53.
An important question to elucidate is how SSCs are activatedin response to injury. Our microarray data highlight several ECMproteins that are upregulated in PCs at day 3 post fracture.Within these ECM proteins, we discovered Periostin, a matri-cellular protein regulating cell–cell and cell–matrix interactions.Periostin is highly expressed during development and in adulttissues submitted to mechanical stress, injury or other patholo-gical conditions54–56. Periostin plays a crucial role in inflamma-tory and tumor microenvironments57–60. In cancer, Periostincorrelates with bad prognosis61 and Periostin present in themetastatic niche supports cancer stem cell self-renewal andmetastatic colonization57. In response to bone injury, we showthat Periostin and other ECM proteins linked to Periostin areupregulated in PCs and Periostin is crucial for adequate bonerepair. In mice lacking Periostin, some of these ECM proteins aredownregulated in PCs, suggesting that Periostin and Periostin-associated ECM proteins all contribute to PC activation and nicheregulation in response to injury, allowing periosteal activation(Supplementary Fig. 7). Our results also re-enforce the impor-tance of a local periosteal response at the injury site to allow callusformation and bone repair. The local activation of PCs isnecessary for the bone repair process to occur and we show that alocal deficiency in this PC pool in Periostin KO periosteum issufficient to delay repair and induce non-union.
In conclusion, our results reveal the presence of SSCs withinperiosteum with higher regenerative potential compared to
BMSCs. Although PCs and BMSCs derive from commonmesenchymal progenitors during bone development and growth,the periosteum environment is essential to confer greater regen-erative properties to PCs. We show that PCs and their perios-teum niche are two key components that act locally to allow callusformation and bone bridging for fracture consolidation. Fur-thermore, PCs, and the ECM components that they produce,including Periostin, are essential for periosteum activation anddefine the enhanced regenerative potential of PCs. Together, theroles of PCs illustrated in this study will help refocus investigationon the periosteum to elucidate numerous bone phenotypesassociated with PCs rather than BMSCs defects. The skeletonpossesses high regenerative capacities, yet our understanding ofSSC origins, recruitment, and functions for the repair of thiscentral organ system will necessitate more investigation of theperiosteum microenvironment to find novel strategies to treatskeletal repair defects and bone diseases.
MethodsMice. C57BL/6ScNj, betaactin‐GFP (GFP), Prx1-Cre, Rosa-tdTomato-EGFP(mTmG), R26ReYFP, and R26ReLacZ transgenic and reporter mice were obtainedfrom Jackson Laboratory (Bar Harbor, ME). Prx1-Cre/ERT2-EGFP mice wereprovided by Dr. S. Murakami34. Periostin null mice from Simon J. Conwaylaboratory were crossed with the GFP mice for lineage tracing48. The mice werebred and genotyped in our laboratory. Five to eight-week-old mice were used forin vitro experiments and more than two-month-old for in vivo experiments. Allmouse primers for PCR genotyping (Supplementary Table 1) were purchased fromEurofins Scientific (Eurofins, Luxembourg). All procedures were approved by theParis Descartes University Ethical Committee. No specific randomization methodswere used for the study. However, experimental groups were homogeneous andcomposed of equivalent animals based on gender, age, and genotype. For eachexperimental group, the mice were from different litters and samples obtained frommultiple experiments (>2) to generate biological replicates.
Primary cultures of PCs and BMSCs. BMSCs and PCs were harvested from tibiasand femurs of un-injured mice (d0) or from tibias 3 days post fracture (d3). Themice were killed and their hindlimbs dissected. After removing the epiphyses,bones were flushed to isolate total bone marrow cells and aBM were expanded ingrowth media consisting of MEMα supplemented with 20% lot-selected non-heat-inactivated FBS, 1% penicillin-streptomycin (Life Technology, Carlsbad, Cali-fornia), and 10 ng/ml bFGF (R&D, Minneapolis, MN). When confluence wasreached, lineage depletion (CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G/C),7–4, and Ter-119 monoclonal antibodies, Miltenyi Biotec, San Diego, CA, ref. 130-090-858) was performed on aBM to obtain BMSCs that were directly used forin vitro and in vivo assays without further expansion. Although this step of lineagedepletion is not standard in the literature for bone marrow cells, we chose thisapproach to enrich BMSCs with skeletal progenitors in order to obtain a popula-tion more comparable to PCs for the purpose of this study. Primary PCs wereobtained by explant culture of the remaining flushed bones free of muscles andtendons. Explants were cultured in growth media and PCs migrated out of theexplanted within 3 days. After 2 weeks, the bones were removed and PCs weretrypsinized and directly used for in vitro and in vivo experiments without furtherexpansion. To determine if our method of explant culture was optimal to retrievePCs without contamination from the endosteum or the bone cortex, we performedthe same procedure by flushing the bone marrow preceded or followed by
Fig. 6 Impaired periosteum in Periostin KO mice. a Experimental design for the isolation of periosteum grafts from GFP or Periostin KO-GFP donors andtransplantation at the fracture site of wild type (WT) hosts for lineage tracing of periosteum-derived cells during bone repair. b SO staining and DAPI/GFPimmunofluorescence on longitudinal callus sections at day 14 reveals decreased contribution KO-GFP grafts (KO in WT) compared to GFP grafts (WT inWT) (arrowheads). Quantification of GFP signal shows decreased volume in callus and cartilage for KO-GFP grafts compared to GFP grafts. Scale bar: 1mm. c CFE assay on activated PCs isolated from WT and KO mice and plated at 400 cells/cm2 for 14 days. Colonies were stained with Giemsa blue andcounted. d In vitro differentiation assays of activated PCs isolated from WT and KO mice shows osteogenic differentiation (alizarin red S stain) at 2 weeksfor WT PCs, but not for KO PCs (left) and at 5 weeks for WT and KO PCs (right). Adipogenesis (Oil red O) is reduced in KO at 3 weeks andchondrogenesis (alcian blue stain) at 1 week is similar in WT and KO PCs. e Quantitative RT-PCR analyses of Periostin (Postn)-linked genes, some of themupregulated in PCs in response to injury and encoding ECM proteins (see Fig. 4i and Supplementary Table 3) inWT-PCs and KO PCs. f Experimental designfor the isolation of activated PCs from GFP or KO-GFP mice and transplantation at the fracture site of KO hosts (WT in KO and KO in KO, respectively). gPeriostin (POSTN/red) immunofluorescence on callus sections after transplantation of WT PCs (GFP/green) in KO hosts. PCs express POSTN when theyintegrate into the callus (Merge/Yellow, left and boxes 1 and 2) and stop expressing POSTN when they differentiate (box 2, white arrowhead pointing toPOSTN-negative and GFP-positive chondrocytes). Scale bar: 1 mm. h Histomorphometric analyses of callus, bone, and cartilage volumes at d14. Scale bar: 1mm. SO: Safranin-O, c: cortex, ca: cartilage, cal: callus, white dashed line: bone, black dashed line: callus, merge: GFP+ cells expressing POSTN. Statisticaldifferences between the groups were determined using Mann–Whitney test (*p < 0.05) (n= 3 or 4 per group). All data represent mean ± SD
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 11
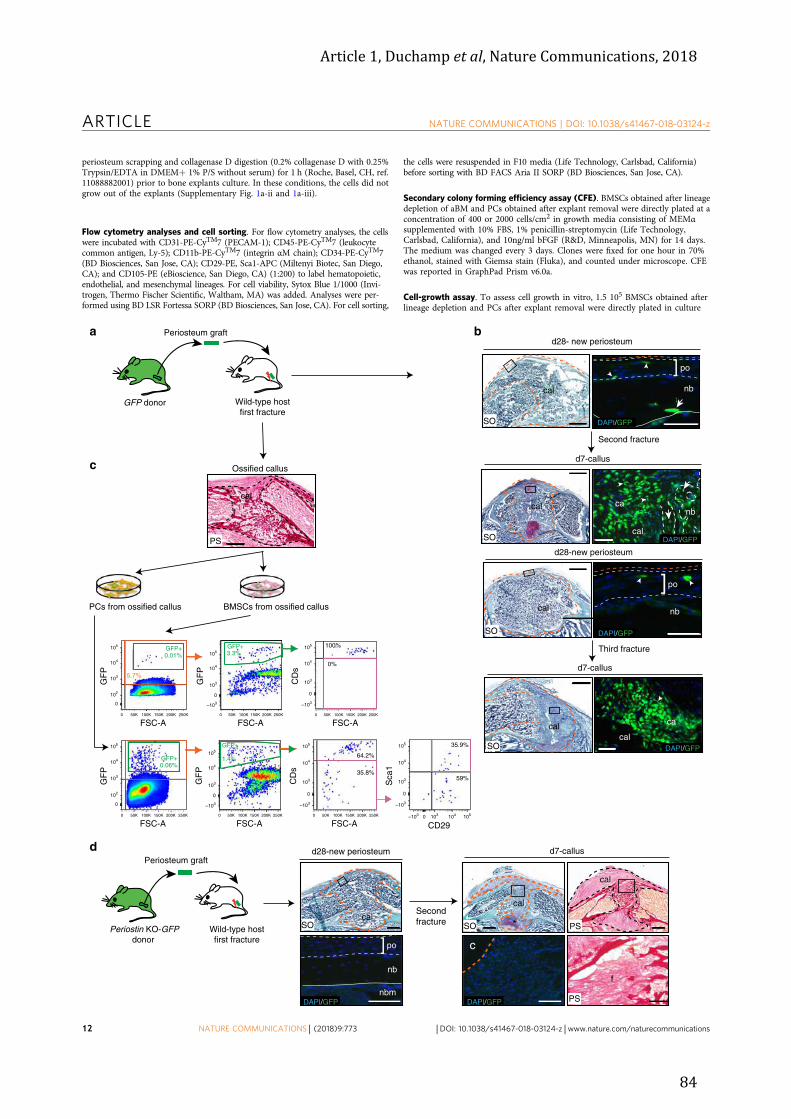
Article 1, Duchamp et al, Nature Communications, 2018
84
periosteum scrapping and collagenase D digestion (0.2% collagenase D with 0.25%Trypsin/EDTA in DMEM+ 1% P/S without serum) for 1 h (Roche, Basel, CH, ref.11088882001) prior to bone explants culture. In these conditions, the cells did notgrow out of the explants (Supplementary Fig. 1a-ii and 1a-iii).
Flow cytometry analyses and cell sorting. For flow cytometry analyses, the cellswere incubated with CD31-PE-CyTM7 (PECAM-1); CD45-PE-CyTM7 (leukocytecommon antigen, Ly-5); CD11b-PE-CyTM7 (integrin αM chain); CD34-PE-CyTM7(BD Biosciences, San Jose, CA); CD29-PE, Sca1-APC (Miltenyi Biotec, San Diego,CA); and CD105-PE (eBioscience, San Diego, CA) (1:200) to label hematopoietic,endothelial, and mesenchymal lineages. For cell viability, Sytox Blue 1/1000 (Invi-trogen, Thermo Fischer Scientific, Waltham, MA) was added. Analyses were per-formed using BD LSR Fortessa SORP (BD Biosciences, San Jose, CA). For cell sorting,
the cells were resuspended in F10 media (Life Technology, Carlsbad, California)before sorting with BD FACS Aria II SORP (BD Biosciences, San Jose, CA).
Secondary colony forming efficiency assay (CFE). BMSCs obtained after lineagedepletion of aBM and PCs obtained after explant removal were directly plated at aconcentration of 400 or 2000 cells/cm2 in growth media consisting of MEMαsupplemented with 10% FBS, 1% penicillin-streptomycin (Life Technology,Carlsbad, California), and 10ng/ml bFGF (R&D, Minneapolis, MN) for 14 days.The medium was changed every 3 days. Clones were fixed for one hour in 70%ethanol, stained with Giemsa stain (Fluka), and counted under microscope. CFEwas reported in GraphPad Prism v6.0a.
Cell-growth assay. To assess cell growth in vitro, 1.5 105 BMSCs obtained afterlineage depletion and PCs after explant removal were directly plated in culture
Second fracture
cal
SO
SO
cal
Third fracture
po
nb
DAPI/GFP
cal
cal
SO
cal
SOcal
ca
nb
po
a
GFP donor
Periosteum graft
Wild-type hostfirst fracture
b
c
PCs from ossified callus BMSCs from ossified callus
FSC-A
GF
P
FSC-A
GF
P
FSC-A FSC-A CD29
GFP+
5.7%
0.01%
35.8%
64.2%
0%
100%
35.9%
59%
GFP+0.06%
FSC-A
d28- new periosteum
d28-new periosteum
d7-callus
d7-callus
]
DAPI/GFP
]
DAPI/GFP
DAPI/GFP
GFP+3.3%
GFP+
1.4%
canb
Ossified callus
cal
PS
FSC-A
GF
P
GF
P
CD
sC
Ds
Sca
1
Wild-type hostfirst fracture
Secondfracture SOSO
Periosteum graftd28-new periosteum d7-callus
Periostin KO-GFPdonor
PS
PS
cal
cal
*
cal
fnb
] po
nbmDAPI/GFP DAPI/GFP
c
d
105
104
103
102
0
105
104
103
102
0
105
104
103
–103
0
105
104
103
–103
0
105
104
103
–103
0
50K 100K 150K 200K 250K0
50K 100K 150K 200K 250K0
50K 100K 150K 200K 250K0
50K 100K 150K 200K 250K0
105
104
103
–103
0
50K 100K 150K 200K 250K0
105
105
104
104
103
103
–103
–103
0
0
50K 100K 150K 200K 250K0
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
12 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications

Article 1, Duchamp et al, Nature Communications, 2018
85
dishes and cultured in growth medium. The cells were trypsinized for counting thefirst two days and then every two days during twelve days. Cell count was reportedin GraphPad Prism v6.0a.
In vitro osteogenic and adipogenic and chondrogenic differentiations. For eachdifferentiation protocol, BMSCs were used following lineage depletion and PCsfollowing explant removal without further passage. For osteogenic differentiation,the cells were plated at confluence in osteogenic medium containing MEMα with10% FBS supplemented with 0.1 μM dexamethasone, 0.2 mM L-ascorbic acid, and10 mM glycerol 2-phosphate disodium salt hydrate (Sigma, St. Louis, MO). Themedium was changed every three days during 2–5 weeks, and the cells were stainedwith 0.2% alizarin red S (Sigma, St. Louis, MO). For adipogenic differentiation, theconfluent cells were cultured with adipogenic medium containing MEMα with 10%FBS supplemented with 10 μg/ml insulin, 100 μM indomethacin, 0.5 mM 3-iso-butyl-1-methylxanthine, and 0.1 μM dexamethasone (Sigma, St. Louis, MO). Themedium was changed every 3 days during 3 weeks and the cells were stained withOil Red O solution (Sigma, St. Louis, MO). Nuclei were counterstained with Harrishematoxylin (DiaPath, Martinengo, Italy). Pictures of lipid droplets were takenunder light microscopy using Leica DM IRB light microscope and LASv4.3 software (Leica Microsystems Inc, Buffalo Grove, IL). For chondrogenic dif-ferentiation, the cells were resuspended at a concentration of 5.105 cells in 200 μl ofgrowth media and plated as micromass. After 2 h at 37 °C, the cells were coveredwith chondrogenic medium containing DMEM with 10% FBS supplemented with0.1 μM dexamethasone, 100 μg/ml sodium pyruvate, 40 μg/ml L-proline, 50 μg/mlL-ascorbic acid, 50 mg/ml ITS, and 10 ng/ml TGFβ1. The medium was changedevery 3 days during 1–2 weeks and the cells were stained with Alcian blue (Sigma,St. Louis, MO).
Wound healing assay. Wound healing assay was performed to assess migrationcapacity of cells in vitro. Forty-eight hours before the assay, BMSCs obtained afterlineage depletion and PCs after explant removal were directly plated in cultureinserts in μ-slide 8 well ibiTreat (Biovalley) and cultured in growth media. Beforestarting the assay, culture inserts were removed allowing a clear separation betweentwo migration fronts (wound). A volume of 10 μM of Cytosine β-D-arabinofuranoside hydrochloride (Sigma, St. Louis, MO) was added in the mediumto inhibit cell mitosis. Wound healing was recorded every 10 minutes over 50 to 72h using videomicroscopy (Nikon Eclipse Ti-E). Data were analyzed with ICYsoftware (bioimageanalysis.org) and reported in GraphPad Prism v6.0a.
Fractures and cell transplantations. Closed non-stabilized and open non-stabilized tibial fractures were performed in the mid-diaphysis under anesthesiaand analgesia2,32. For all surgeries, mice were anesthetized with an intraperitonealinjection of Médétomidine (1 mg/kg) and Kétamine (50 mg/ml) and received asubcutaneous injection of Buprenorphine (0,1 mg/kg) for analgesia. For closedfractures, the tibia was placed on the fracture jig and 460 g weight was droppedfrom 14 cm to create a closed, transverse fracture by three-point bending, whichwas confirmed by radiography. Opened non-stabilized tibial fractures were pro-duced by osteotomy. The anterior tibial surface was exposed by separating the bonefrom the surrounding muscles. Three holes were drilled in the tibial cortex using a0.4 mm drill bit and the bone was cut to create the fracture. After surgery, the micewere revived with a subcutaneous injection of Atipamezole (1 mg/kg) and wereallowed to move freely. The mice then received a second dose of analgesic 12–24 hafter surgery and subsequent doses as needed. For cell transplantations at thefracture site, 100,000 cells were embedded in a fibrin gel using a Tissucol® kit(Baxter, France TISSEEL, composed of human fibrinogen 15 mg/ml and thrombin9 mg/ml) and the cell pellet was transplanted at the time of fracture62.
Cortical bone defects. To assess the impact of Periostin deficiency on bone healingthrough intramembranous ossification, we performed unicortical bone defects(without breaking the bone) on wild-type and Periostin KO mice as previouslydescribed24. Briefly, after anesthesia and analgesia, the tibial surface was exposed,and a hole (1 mm in diameter) was drilled into one cortex without drilling into theopposite cortex. After surgery, the mice were revived as indicated above.
Periosteum grafting. Periosteum grafts isolated from the tibia GFP or Prx1-Cre;mTmG donor mice were transplanted at the site of open non-stabilized tibialfractures in 10-week-old wild-type or Periostin KO host mice32. For assessment oflong-term engraftment, successive fractures at one-month interval were performedat the site of initial bone graft in 5-week-old hosts and fracture calluses werecollected at days 7 and 28. For BMSCs and PCs cultures, day 14 ossified calluseswere retrieved by dissection. BMSCs were obtained by flushing the bone marrowcells from the fracture calluses followed by adherence and lineage depletion callusesas described above. PCs were cultured by explant cultures of remaining ossifiedparts of the fracture calluses. Fracture callus pieces were carefully cleaned andplaced in culture periosteum facing down. PCs and BMSCs were used for cellsorting and FACS analyses.
Renal capsule transplantation. Femoral grafts containing cartilage anlage sur-rounded by perichondrium were isolated from E14.5 donor embryos, transplantedin the renal capsule of adult host mice, and allowed to develop for 8 weeks to formfully mature bones43. PCs and BMSCs were isolated as described above from bonesderived from Prx1-Cre;YFPfl/+ donors transplanted in C57BL/6ScNj (wild type)hosts and analyzed via flow cytometry as indicated above. For lineage analysesduring bone repair, the bones derived from Prx1-Cre;LacZfl/+donors transplantedin wild-type hosts were fractured by osteotomy under anesthesia by exposing thekidney capsule and collected 7 or 14 days post fracture for lineage tracing usingXgal/TRAP staining on tissue sections as previously described63. In control sam-ples, the genotypes of the donor (wild type) and host (Prx1-Cre;YFPfl/+or Prx1-Cre;LacZfl/+) were reversed.
Histomorphometry and cell-lineage analyses. The mice were killed at specifiedtime points post fracture. Tibias were fixed in 4% paraformaldehyde, decalcified in19% EDTA, and processed for histomorphometric analyses of callus, cartilage, andbone on Safranin-O (SO) and Trichrome (TC) stained sections62,64. Picrosiriusstaining was performed on adjacent sections to visualize bone and fibrous tissue.For quantitative analyses of GFP-transplanted cells in fracture calluses, GFP signalwas analyzed on sections adjacent to Safranin-O and Trichrome using a ZeissImager D1 AX10 light microscope and ZEN software (Carl Zeiss MicroscopyGmbH, Gottinger, Germany).
Immunofluorescence and immunohistochemistry. For PCNA immuno-fluorescence, the sections were rehydrated, post fixed in 4% paraformaldehyde for10 min, treated with methanol for 10 min, permeabilized with 0.25% TritonX-100in PBS, and blocked with 5% Goat serum in 0.25% tritonX-100 in PBS for 15 min.The sections were then incubated with primary antibody rabbit anti mouse PCNA1:800 (Cell Signaling, Danvers, MA ref. 13110 s) or non-immune rabbit IgG asnegative control (Invitrogen, Thermo Fischer Scientific, Waltham, MA ref. 10500C) O/N at 4 °C. The sections were washed and incubated with secondary antibodyAlexa 546 goat anti rabbit 1:500 (Invitrogen, Thermo Fischer Scientific, Waltham,MA ref.11010) in 5% goat serum for one hour at RT, and mounted with Fluor-omount-G™ with DAPI (eBioscience, San Diego, CA).
For Cleaved Caspase 3 immunofluorescence, the sections were rehydrated, postfixed in 4% paraformaldehyde for 10 min, permeabilized with 0.25% TritonX-100in PBS, and blocked with 5% Goat serum in 0.25% tritonX-100 in PBS for onehour. The sections were then incubated with primary antibody rabbit anti mouse
Fig. 7 No reconstitution of the PC pool in Postn KO periosteum after fracture. a Experimental design for the isolation of periosteum graft from GFP donormice and transplantation at the fracture site of wild-type hosts. b SO staining and DAPI/GFP immunofluorescence on longitudinal sections of mousefractured tibias post transplantation with GFP periosteum graft. At d28 post fracture (d28-new periosteum), high magnification shows rare periosteum-derived GFP+ cells that integrate in the new bone to form osteocytes (white arrow) and in the new periosteum (white arrowheads). After a second fractureperformed at the level of the first callus, abundant periosteum-derived GFP+ cells are found in the callus and form cartilage (white arrowheads) and bone(white arrows) (d7-callus) and few GFP+ cells reintegrate the new periosteum at d28 (white arrowheads) (d28-new periosteum). After a third fracture,periosteum-derived GFP+ cells can again form cartilage efficiently in the callus by day 7 (white arrowhead) (day 7-callus). c Cell sorting and FACSanalyses on PCs and BMSCs isolated from ossified calluses (d14). PCs and BMSCs derived from the periosteum graft were detected based on theexpression of the GFP (0.06% and 0.01%, respectively). Cell sorting was performed to enrich the population in GFP+ cells (orange box) and FACSanalyses to assess the expression of hematopoietic-endothelial markers (CD11b, CD31, CD45, and CD34) and Sca1/CD29). In BMSCs cultures, GFP+ cellswere all positive for hematopoietic-endothelial markers (100%). For PC cultures, we detected a population that was negative for hematopoietic-endothelialmarkers (35.8%) and positive for Sca1/CD29 (35.9%) (n= 2 or 3). d Transplantation of Periostin KO grafts into wild-type hosts. No GFP+ cells aredetected in the new periosteum (d28–new periosteum), and no GFP+ chondrocytes contribute to the callus after a second injury. These Periostin KO graftsinduced fibrosis at the fracture site of wild-type hosts (d7–callus). SO: Safranin-O/Fast Green, PS: Picro Sirius, cal: callus, po: periosteum, nb: new bone, ca:cartilage, white dashed line: periosteum (d28) or new bone (d7), orange dashed line: callus, yellow line: periosteum transplant, asterisk: cartilage formationopposite to transplant (n= 4). Scale bar= 1 mm
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 13

Article 1, Duchamp et al, Nature Communications, 2018
86
28. O’Driscoll, S. W. & Fitzsimmons, J. S. The role of periosteum in cartilagerepair. Clin. Orthop. Relat. Res. 391, S190–S207 (2001).
29. Zhang, X., Awad, H. A., O’Keefe, R. J., Guldberg, R. E. & Schwarz, E. M. Aperspective: engineering periosteum for structural bone graft healing. Clin.Orthop. Relat. Res. 466, 1777–1787 (2008).
30. Zhang, X. et al. Periosteal progenitor cell fate in segmental cortical bone grafttransplantations: implications for functional tissue engineering. J. Bone Miner.Res. 20, 2124–2137 (2005).
31. Thabet, A. M. et al. Periosteal grafting for congenital pseudarthrosis of thetibia: a preliminary report. Clin. Orthop. Relat. Res. 466, 2981–2994 (2008).
32. Colnot, C. Skeletal cell fate decisions within periosteum and bone marrowduring bone regeneration. J. Bone Miner. Res. 24, 274–282 (2009).
33. van Gastel, N. et al. Engineering vascularized bone: osteogenic and proangiogenicpotential of murine periosteal cells. Stem Cells 30, 2460–2471 (2012).
34. Kawanami, A., Matsushita, T., Chan, Y. Y. & Murakami, S. Mice expressingGFP and CreER in osteochondro progenitor cells in the periosteum. Biochem.Biophys. Res. Commun. 386, 477–482 (2009).
35. Logan, M. et al. Expression of Cre Recombinase in the developing mouse limbbud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
36. Boxall, S. A. & Jones, E. Markers for characterization of bone marrowmultipotential stromal cells. Stem Cells Int. 2012, 975871 (2012).
37. Morikawa, S. et al. Prospective identification, isolation, and systemictransplantation of multipotent mesenchymal stem cells in murine bonemarrow. J. Exp. Med. 206, 2483–2496 (2009).
38. Crisan, M. et al. A perivascular origin for mesenchymal stem cells in multiplehuman organs. Cell Stem Cell 3, 301–313 (2008).
39. Chang, H. Y. et al. Diversity, topographic differentiation, and positional memoryin human fibroblasts. Proc. Natl Acad. Sci. USA 99, 12877–12882 (2002).
40. Kronenberg, H. M. Developmental regulation of the growth plate. Nature 423,332–336 (2003).
41. Bianco, P. & Robey, P. G. Skeletal stem cells. Development 142, 1023–1027 (2015).42. Bianco, P., Robey, P. G. & Simmons, P. J. Mesenchymal stem cells: revisiting
history, concepts, and assays. Cell Stem Cell 2, 313–319 (2008).43. Colnot, C., Lu, C., Hu, D. & Helms, J. A. Distinguishing the contributions of
the perichondrium, cartilage, and vascular endothelium to skeletaldevelopment. Dev. Biol. 269, 55–69 (2004).
44. Colnot, C., Romero, D.M., Huang, S. & Helms, J.A. Mechanisms of action ofdemineralized bone matrix in the repair of cortical bone defects. Clin. Orthop.Relat. Res. 435, 69–78 (2005).
45. Murao, H., Yamamoto, K., Matsuda, S. & Akiyama, H. Periosteal cells are a majorsource of soft callus in bone fracture. J. Bone Miner. Metab. 31, 390–398 (2013).
46. Kumagai, K., Vasanji, A., Drazba, J. A., Butler, R. S. & Muschler, G. F. Circulatingcells with osteogenic potential are physiologically mobilized into the fracturehealing site in the parabiotic mice model. J. Orthop. Res. 26, 165–175 (2008).
47. Horiuchi, K. et al. Identification and characterization of a novel protein,periostin, with restricted expression to periosteum and periodontal ligamentand increased expression by transforming growth factor beta. J. Bone Miner.Res. 14, 1239–1249 (1999).
48. Rios, H. et al. Periostin null mice exhibit dwarfism, incisor enamel defects, andan early-onset periodontal disease-like phenotype. Mol. Cell. Biol. 25,11131–11144 (2005).
49. Granero-Molto, F. et al. Regenerative effects of transplanted mesenchymalstem cells in fracture healing. Stem Cells 27, 1887–1898 (2009).
50. Kuang, S., Gillespie, M. A. & Rudnicki, M. A. Niche regulation of musclesatellite cell self-renewal and differentiation. Cell Stem Cell 2, 22–31 (2008).
51. Collins, C. A. & Partridge, T. A. Self-renewal of the adult skeletal musclesatellite cell. Cell Cycle 4, 1338–1341 (2005).
52. Sacco, A., Doyonnas, R., Kraft, P., Vitorovic, S. & Blau, H. M. Self-renewal andexpansion of single transplanted muscle stem cells. Nature 456, 502–506 (2008).
53. Dominici, M. et al. Minimal criteria for defining multipotent mesenchymalstromal cells. The international society for cellular therapy position statement.Cytotherapy 8, 315–317 (2006).
54. Bonnet, N. et al. The matricellular protein periostin is required for sostinhibition and the anabolic response to mechanical loading and physicalactivity. J. Biol. Chem. 284, 35939–35950 (2009).
55. Conway, S. J. et al. The role of periostin in tissue remodeling across health anddisease. Cell Mol. Life Sci. 71, 1279–1288 (2014).
56. Bonnet, N. et al. Periostin deficiency increases bone damage and impairsinjury response to fatigue loading in adult mice. PLoS ONE 8, e78347 (2013).
57. Malanchi, I. et al. Interactions between cancer stem cells and their nichegovern metastatic colonization. Nature 481, 85–89 (2011).
58. Shimazaki, M. et al. Periostin is essential for cardiac healing after acutemyocardial infarction. J. Exp. Med. 205, 295–303 (2008).
59. Sidhu, S. S. et al. Roles of epithelial cell-derived periostin in TGF-betaactivation, collagen production, and collagen gel elasticity in asthma. Proc.Natl Acad. Sci. USA 107, 14170–14175 (2010).
60. Zhou, H. M. et al. Spatiotemporal expression of periostin during skindevelopment and incisional wound healing: lessons for human fibrotic scarformation. J. Cell. Commun. Signal. 4, 99–107 (2010).
61. Hu, F., Wang, W., Zhou, H. C. & Shang, X. F. High expression of periostin isdramatically associated with metastatic potential and poor prognosis ofpatients with osteosarcoma. World J. Surg. Oncol. 12, 287 (2014).
62. Abou-Khalil, R. et al. Role of muscle stem cells during skeletal regeneration.Stem Cells 33, 1501–1511 (2015).
63. Colnot, C., Huang, S. & Helms, J. Analyzing the cellular contribution of bonemarrow to fracture healing using bone marrow transplantation in mice.Biochem. Biophys. Res. Commun. 350, 557–561 (2006).
64. Abou-Khalil, R. et al. Delayed bone regeneration is linked to chronicinflammation in murine muscular dystrophy. J. Bone Miner. Res. 29, 304–315(2014).
65. Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis anddisplay of genome-wide expression patterns. Proc. Natl Acad. Sci. USA 95,14863–14868 (1998).
66. Saeed, A. I. et al. TM4: a free, open-source system for microarray datamanagement and analysis. Biotechniques 34, 374–378 (2003).
67. Mootha, V. K. et al. PGC-1alpha-responsive genes involved in oxidativephosphorylation are coordinately downregulated in human diabetes. Nat.Genet. 34, 267–273 (2003).
68. Subramanian, A. et al. Gene set enrichment analysis: a knowledge-basedapproach for interpreting genome-wide expression profiles. Proc. Natl Acad.Sci. USA 102, 15545–15550 (2005).
69. Szklarczyk, D. et al. STRINGv10: protein–protein interaction networks,integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
AcknowledgementsWe thank E. Bonnelye, C. Bole-Feysot, M. Garfa-Traoré, N. Goudin, C. Lebreton, J.Megret, A. Rausell, C. Tarrin, C. de Ticornot, and F. Yang for advice and/or technicalassistance; R. Marcucio, F. Relaix, and S.S. Sidhu for reading the manuscript. This workwas supported by INSERM ATIP-Avenir, ANR-13-BSV1-001-01, FP7 Marie Curie IRG-268227, Osteosynthesis & Trauma Care Foundation, and NIAMS R01 AR057344 to C.Colnot.
Author contributionsC. Colnot supervised the project, designed and carried out experiments, analyzed thedata, and wrote the manuscript. O.D.L. designed and carried out experiments, analyzedthe data, and wrote the manuscript. R.A.K. designed, carried out experiments, andanalyzed the data. A.J., N.C., and G.F. carried out experiments and analyzed the data. C.Carvalho. and C. Cordier. provided advice and technical assistance. S.J.C. provided thePeriostin mouse strain. N.C. and S.J.C. reviewed the manuscript.
Additional informationSupplementary Information accompanies this paper at https://doi.org/10.1038/s41467-018-03124-z.
Competing interests: The authors declare no competing financial interests.
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims inpublished maps and institutional affiliations.
Open Access This article is licensed under a Creative CommonsAttribution 4.0 International License, which permits use, sharing,
adaptation, distribution and reproduction in any medium or format, as long as you giveappropriate credit to the original author(s) and the source, provide a link to the CreativeCommons license, and indicate if changes were made. The images or other third partymaterial in this article are included in the article’s Creative Commons license, unlessindicated otherwise in a credit line to the material. If material is not included in thearticle’s Creative Commons license and your intended use is not permitted by statutoryregulation or exceeds the permitted use, you will need to obtain permission directly fromthe copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
© The Author(s) 2018
NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z ARTICLE
NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications 15

Article 1, Duchamp et al, Nature Communications, 2018
87
Cleaved Caspase 3 1:200 (Cell Signaling, Danvers, MA ref. 9661) or non-immunerabbit IgG as negative control (Invitrogen, Thermo Fischer Scientific, Waltham,MA ref. 10500 C) O/N at 4 °C. The sections were washed and incubated withsecondary antibody Alexa 546 goat anti rabbit 1:800 (Invitrogen, Thermo FischerScientific, Waltham, MA ref.11010) in 5% goat serum for one hour at RT andmounted with Fluoromount-G™ with DAPI (eBioscience, San Diego, CA).
For Periostin immunofluorescence, the sections were rehydrated, blocked with5% donkey serum in PBS one hour at room temperature (RT), and incubated withprimary antibody goat anti mouse Periostin 1:400 (R&D, Minneapolis, MN ref.AF2955) or non-immune goat IgG as negative control (Life Technology, Carlsbad,California ref. 026202) overnight (O/N) at 4 °C. The sections were washed andincubated with secondary antibody Alexa 488 donkey anti goat or Alexa 546donkey anti goat 1:500 (Invitrogen, Thermo Fischer Scientific, Waltham, MA ref.A11055 or ref. A11056) for one hour at RT. Slides were mounted withFluoromount-G™ with DAPI (eBioscience, San Diego, CA).
For CD29 immunofluorescence, the sections were rehydrated, post fixed in 4%paraformaldehyde for 10 min, permeabilized with 0.25% TritonX-100 in PBS, andblocked with 5% donkey serum in PBS for 15 min. The sections were incubatedwith primary antibody goat anti mouse integrinβ1 5 μg/ml (R&D, Minneapolis,MN, ref. AF2405) or non-immune goat IgG as negative control (Life Technology,Carlsbad, California ref. 026202) O/N at 4 °C. The sections were washed andincubated with secondary antibody Alexa 546 donkey anti goat or Alexa 647donkey anti goat 1:500 (Invitrogen, Thermo Fischer Scientific, Waltham, MA) in5% donkey serum for one hour at RT and mounted with Fluoromount-G™ withDAPI (eBioscience, San Diego, CA).
For BrdU immunochemistry, the mice were beforehand injected with 50 mg/kgof BrdU (Sigma, St. Louis, MO ref. B5002) in 5% DMSO and their hindlimbs wereharvested three hours later and processed as previously described. The sectionswere dehydrated in ethanol baths and antigen retrieval was performed using 2 NHCl in 0.5% Triton-X100 for 30 min at RT. Endogenous peroxidase activity wasblocked using 3% H2O2 in PBS for 10 min. Sections were blocked in 5% goat serumin PBS for 1 h and incubated with primary antibody rat anti mouse BrdU 1:200(Abcam, Cambridge, UK ref. Ab6326) or no primary antibody as negative control,O/N at 4 °C. The sections were washed and incubated with secondary antibodybiotin goat anti rat 1:500 (Jackson ImmunoResearch, West Grove, PA ref.112066072) for 1 h at RT. After washing in PBS, the sections were incubated inStreptavidin-HRP 1:100 (BD Biosciences, San Jose, CA ref. 554066) for 30 min atRT. Finally, signal was revealed using DAKO kit (Agilent, Santa Clara, CA ref.K3467) and counterstained with 5% Methyl green. BrdU+ cells were countedunder microscope and reported in GraphPad Prism v6.0a.
RNA isolation and qRT-PCR. Total mRNA extraction from cells was performedusing RNeasy Plus Mini Kit (Qiagen, Germantown, MD) and following manu-facturer’s instructions. The concentration of extracted RNA was confirmed using aNanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE).All mouse primers (Supplementary Table 2) were purchased from Eurofins Sci-entific (Eurofins, Luxembourg). cDNA synthesis was performed using SuperscriptIII RT, RNaseOUT, Ribonuclease inhibitor, Oligo(dT)12–18, 10 mM dNTP mix,5X first-strand buffer, and 0.1 M DTT, following manufacturer’s instructions(Thermo Fischer Scientific, Waltham, MA). Real-time PCR was performed usingSYBR™ Green PCR Master Mix and detected using 7300 Real-Time PCR System(Thermo Fischer Scientific, Waltham, MA). Mouse GAPDH was used as an internalcontrol for all genes.
Microarray analyses. BMSCs and PCs were isolated and purified from uninjuredtibia (d0) (n= 4) and tibia 3 days post fracture (n= 4). The cells were harvestedand total RNA was extracted using Rneasy Plus mini Kit (Qiagen). RNA qualitywas assessed using Agilent Model 2100 Bioanalyzer (Agilent Technologies). Geneexpression analyses were performed using GeneChip Mouse 430 2.0 Array (Affy-metrix). Fluorescence data were imported into Affymetrix Expression Console andR Bioconductor analysis software. Data were normalized with RMA method,groups were compared by Student’s t-test and the results were filtered at p-value≤5% and fold change ≥1.2. Hierarchical clustering was performed using Multi-Experiment Viewer software (MeV)65,66. Gene Set Enrichment Analysis (GSEA)analysis was performed using all normalized probes on “curated gene set” and “GOgene set” collections of the Molecular Signatures Database v5.2 according to67,68.Postn-linked gene list was built using STRING database69 (http://string-db.org/cgi/input.pl?UserId=5bskVvnWAJdi&sessionId=hMz9XrOQGQ4P&input_page_show_search=on). We used the following parameters: active interaction sources: allchecked; minimum required interaction score: 0.4; maximum number of inter-actors to show: 1st shell: 100; and 2nd shell: 20. We obtained a list of 93 genes(Supplementary Table 3).
Statistical analyses. Statistical significance was determined with two-sidedMann–Whitney test and reported in GraphPad Prism v6.0a. P-values were deter-mined as follows: *,$p ≤ 0.05; **,$$p < 0.001; ***,$$$p < 0.0005. All samples wereincluded except for fractures that were proximal and/or distal or comminutedfractures. All analyses were performed using a blind numbering system.
Data availability. The microarray data have been deposited in the ArrayExpressdatabase under the accession number E-MTAB-6417. All other data are available inthe article and in the supplementary information files.
Received: 9 February 2017 Accepted: 19 January 2018
References1. Hall, B. K. Bones and Cartilage: Developmental and Evolutionary Skeletal
Biology. (Elsevier Academic Press, 2005).2. Colnot, C., Thompson, Z., Miclau, T., Werb, Z. & Helms, J. A. Altered fracture
repair in the absence of MMP9. Development 130, 4123–4133 (2003).3. Ferguson, C., Alpern, E., Miclau, T. & Helms, J. A. Does adult fracture repair
recapitulate embryonic skeletal formation? Mech. Dev. 87, 57–66 (1999).4. Vortkamp, A. et al. Recapitulation of signals regulating embryonic bone formation
during postnatal growth and in fracture repair. Mech. Dev. 71, 65–76 (1998).5. Yu, Y. Y., Lieu, S., Lu, C. & Colnot, C. Bone morphogenetic protein
2 stimulates endochondral ossification by regulating periosteal cell fate duringbone repair. Bone 47, 65–73 (2010).
6. Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cellniche. Nature 425, 841–846 (2003).
7. Zhang, J. et al. Identification of the haematopoietic stem cell niche and controlof the niche size. Nature 425, 836–841 (2003).
8. Greenbaum, A. et al. CXCL12 in early mesenchymal progenitors is requiredfor haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013).
9. Crane, J. L. & Cao, X. Bone marrow mesenchymal stem cells and TGF-betasignaling in bone remodeling. J. Clin. Invest. 124, 466–472 (2014).
10. Friedenstein, A. J., Chailakhyan, R. K. & Gerasimov, U. V. Bone marrowosteogenic stem cells: in vitro cultivation and transplantation in diffusionchambers. Cell. Tissue Kinet. 20, 263–272 (1987).
11. Sacchetti, B. et al. Self-renewing osteoprogenitors in bone marrow sinusoidscan organize a hematopoietic microenvironment. Cell 131, 324–336 (2007).
12. Chan, C. K. et al. Identification and specification of the mouse skeletal stemcell. Cell 160, 285–298 (2015).
13. Maes, C. et al. Osteoblast precursors, but not mature osteoblasts, move intodeveloping and fractured bones along with invading blood vessels. Dev. Cell.19, 329–344 (2010).
14. Marecic, O. et al. Identification and characterization of an injury-inducedskeletal progenitor. Proc. Natl Acad. Sci. USA 112, 9920–9925 (2015).
15. Mendez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form aunique bone marrow niche. Nature 466, 829–834 (2010).
16. Mizoguchi, T. et al. Osterix marks distinct waves of primitive and definitivestromal progenitors during bone marrow development. Dev. Cell. 29, 340–349(2014).
17. Ono, N. et al. Vasculature-associated cells expressing nestin in developingbones encompass early cells in the osteoblast and endothelial lineage. Dev.Cell. 29, 330–339 (2014).
18. Park, D. et al. Endogenous bone marrow MSCs are dynamic, fate-restrictedparticipants in bone maintenance and regeneration. Cell Stem Cell 10,259–272 (2012).
19. Worthley, D. L. et al. Gremlin 1 identifies a skeletal stem cell with bone,cartilage, and reticular stromal potential. Cell 160, 269–284 (2015).
20. Yue, R., Zhou, B. O., Shimada, I. S., Zhao, Z. & Morrison, S. J. Leptin receptorpromotes adipogenesis and reduces osteogenesis by regulating mesenchymalstromal cells in adult bone marrow. Cell Stem Cell 18, 782–796 (2016).
21. Zhou, B. O., Yue, R., Murphy, M. M., Peyer, J. G. & Morrison, S. J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source ofbone formed by adult bone marrow. Cell Stem Cell 15, 154–168 (2014).
22. Ono, N., Ono, W., Nagasawa, T. & Kronenberg, H. M. A subset ofchondrogenic cells provides early mesenchymal progenitors in growing bones.Nat. Cell Biol. 16, 1157–1167 (2014).
23. Colnot, C., Zhang, X. & Knothe Tate, M. L. Current insights on theregenerative potential of the periosteum: molecular, cellular, and endogenousengineering approaches. J. Orthop. Res. 30, 1869–1878 (2012).
24. Ferretti, C. & Mattioli-Belmonte, M. Periosteum derived stem cells forregenerative medicine proposals: boosting current knowledge. World J. StemCells 6, 266–277 (2014).
25. Roberts, S. J., van Gastel, N., Carmeliet, G. & Luyten, F. P. Uncovering theperiosteum for skeletal regeneration: the stem cell that lies beneath. Bone 70,10–18 (2015).
26. Doi, K. & Sakai, K. Vascularized periosteal bone graft from the supracondylarregion of the femur. Microsurgery 15, 305–315 (1994).
27. Fell, H. B. The osteogenic capacity in vitro of periosteum and endosteumisolated from the limb skeleton of fowl embryos and young chicks. J. Anat. 66,157–180 (1932). 111.
ARTICLE NATURE COMMUNICATIONS | DOI: 10.1038/s41467-018-03124-z
14 NATURE COMMUNICATIONS | �(2018)�9:773� | DOI: 10.1038/s41467-018-03124-z | www.nature.com/naturecommunications

Article 1, Duchamp et al, Nature Communications, 2018
88
1
Supplementary Information
Periosteum contains skeletal stem cells with high bone regenerative
potential controlled by Periostin
Duchamp de Lageneste et al.
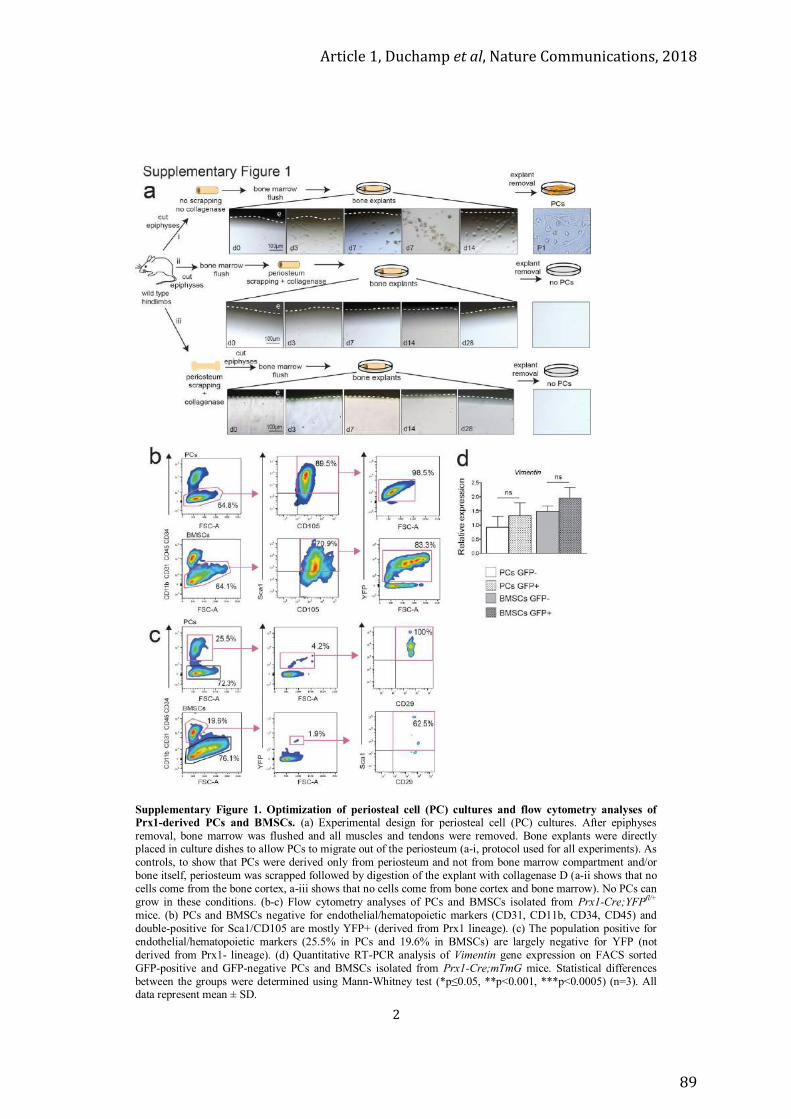
Article 1, Duchamp et al, Nature Communications, 2018
89
2
Supplementary Figure 1. Optimization of periosteal cell (PC) cultures and flow cytometry analyses of Prx1-derived PCs and BMSCs. (a) Experimental design for periosteal cell (PC) cultures. After epiphyses removal, bone marrow was flushed and all muscles and tendons were removed. Bone explants were directly placed in culture dishes to allow PCs to migrate out of the periosteum (a-i, protocol used for all experiments). As controls, to show that PCs were derived only from periosteum and not from bone marrow compartment and/or bone itself, periosteum was scrapped followed by digestion of the explant with collagenase D (a-ii shows that no cells come from the bone cortex, a-iii shows that no cells come from bone cortex and bone marrow). No PCs can grow in these conditions. (b-c) Flow cytometry analyses of PCs and BMSCs isolated from Prx1-Cre;YFPfl/+
mice. (b) PCs and BMSCs negative for endothelial/hematopoietic markers (CD31, CD11b, CD34, CD45) and double-positive for Sca1/CD105 are mostly YFP+ (derived from Prx1 lineage). (c) The population positive for endothelial/hematopoietic markers (25.5% in PCs and 19.6% in BMSCs) are largely negative for YFP (not derived from Prx1- lineage). (d) Quantitative RT-PCR analysis of Vimentin gene expression on FACS sorted GFP-positive and GFP-negative PCs and BMSCs isolated from Prx1-Cre;mTmG mice. Statistical differences between the groups were determined using Mann-Whitney test (*p≤0.05, **p<0.001, ***p<0.0005) (n=3). All data represent mean ± SD.
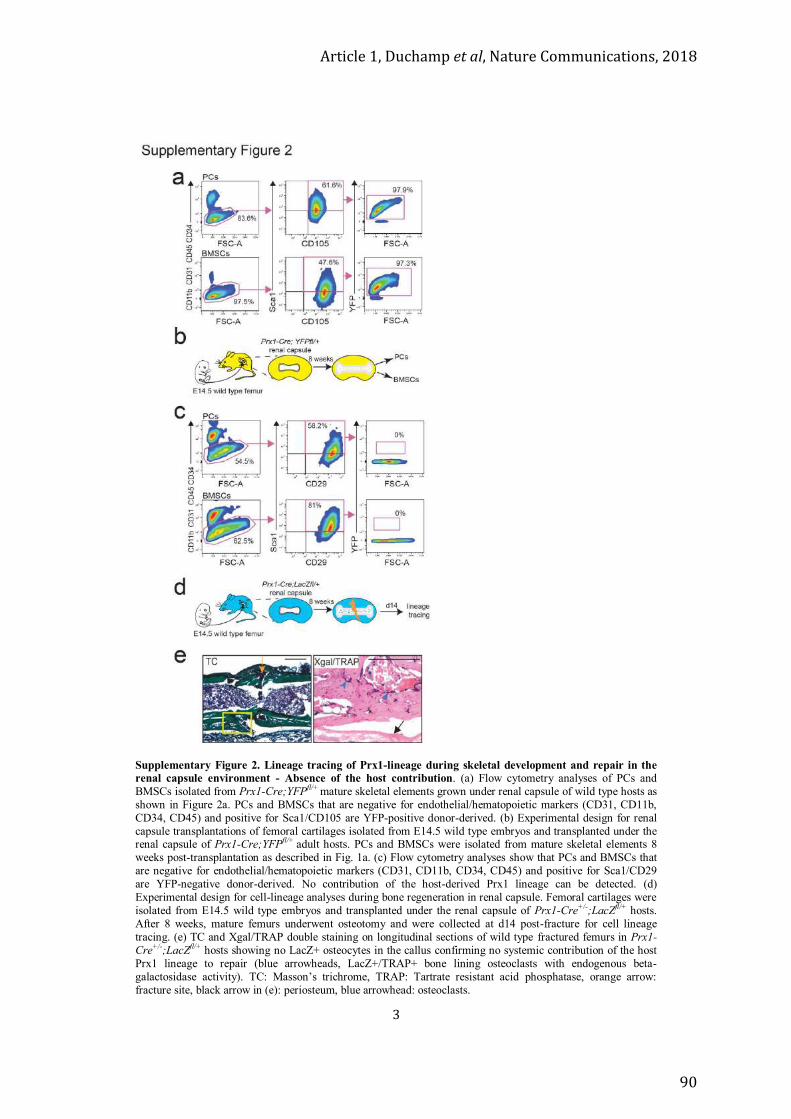
Article 1, Duchamp et al, Nature Communications, 2018
90
3
Supplementary Figure 2. Lineage tracing of Prx1-lineage during skeletal development and repair in the renal capsule environment - Absence of the host contribution. (a) Flow cytometry analyses of PCs and BMSCs isolated from Prx1-Cre;YFPfl/+ mature skeletal elements grown under renal capsule of wild type hosts as shown in Figure 2a. PCs and BMSCs that are negative for endothelial/hematopoietic markers (CD31, CD11b, CD34, CD45) and positive for Sca1/CD105 are YFP-positive donor-derived. (b) Experimental design for renal capsule transplantations of femoral cartilages isolated from E14.5 wild type embryos and transplanted under the renal capsule of Prx1-Cre;YFPfl/+ adult hosts. PCs and BMSCs were isolated from mature skeletal elements 8 weeks post-transplantation as described in Fig. 1a. (c) Flow cytometry analyses show that PCs and BMSCs that are negative for endothelial/hematopoietic markers (CD31, CD11b, CD34, CD45) and positive for Sca1/CD29 are YFP-negative donor-derived. No contribution of the host-derived Prx1 lineage can be detected. (d) Experimental design for cell-lineage analyses during bone regeneration in renal capsule. Femoral cartilages were isolated from E14.5 wild type embryos and transplanted under the renal capsule of Prx1-Cre+/-;LacZfl/+ hosts. After 8 weeks, mature femurs underwent osteotomy and were collected at d14 post-fracture for cell lineage tracing. (e) TC and Xgal/TRAP double staining on longitudinal sections of wild type fractured femurs in Prx1-Cre+/-;LacZfl/+ hosts showing no LacZ+ osteocytes in the callus confirming no systemic contribution of the host Prx1 lineage to repair (blue arrowheads, LacZ+/TRAP+ bone lining osteoclasts with endogenous beta-galactosidase activity). TC: Masson’s trichrome, TRAP: Tartrate resistant acid phosphatase, orange arrow: fracture site, black arrow in (e): periosteum, blue arrowhead: osteoclasts.
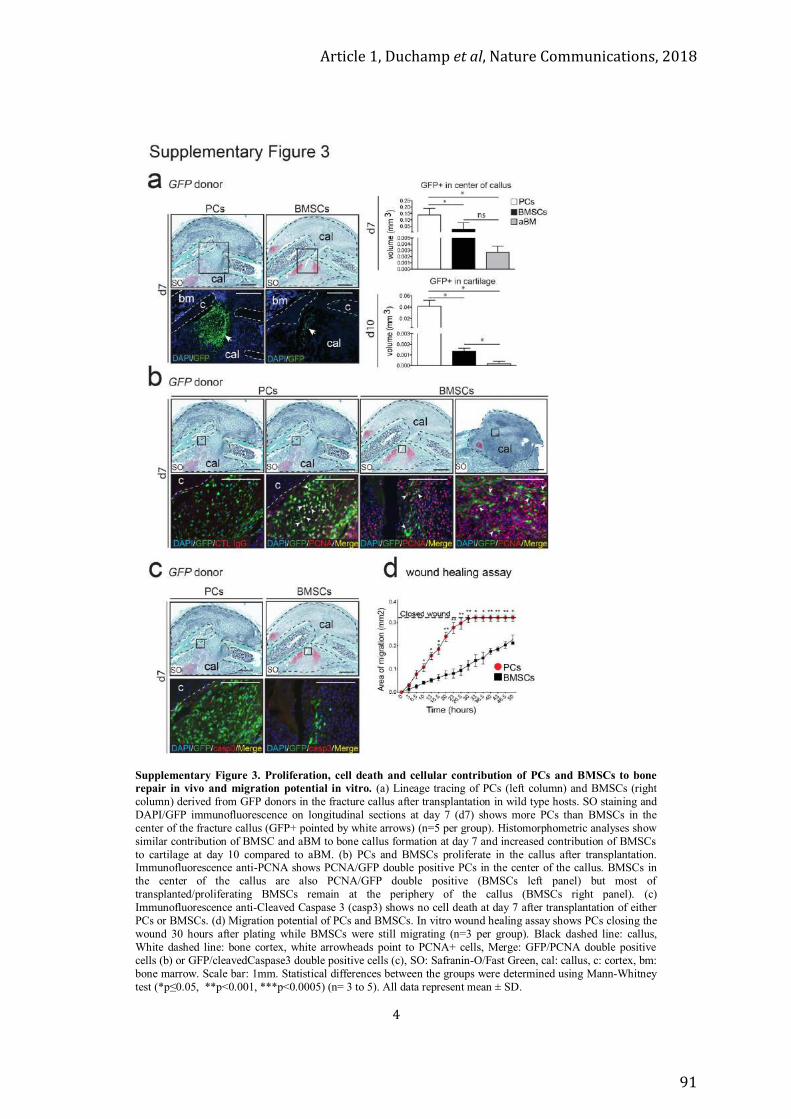
Article 1, Duchamp et al, Nature Communications, 2018
91
4
Supplementary Figure 3. Proliferation, cell death and cellular contribution of PCs and BMSCs to bone repair in vivo and migration potential in vitro. (a) Lineage tracing of PCs (left column) and BMSCs (right column) derived from GFP donors in the fracture callus after transplantation in wild type hosts. SO staining and DAPI/GFP immunofluorescence on longitudinal sections at day 7 (d7) shows more PCs than BMSCs in the center of the fracture callus (GFP+ pointed by white arrows) (n=5 per group). Histomorphometric analyses show similar contribution of BMSC and aBM to bone callus formation at day 7 and increased contribution of BMSCs to cartilage at day 10 compared to aBM. (b) PCs and BMSCs proliferate in the callus after transplantation. Immunofluorescence anti-PCNA shows PCNA/GFP double positive PCs in the center of the callus. BMSCs in the center of the callus are also PCNA/GFP double positive (BMSCs left panel) but most of transplanted/proliferating BMSCs remain at the periphery of the callus (BMSCs right panel). (c) Immunofluorescence anti-Cleaved Caspase 3 (casp3) shows no cell death at day 7 after transplantation of either PCs or BMSCs. (d) Migration potential of PCs and BMSCs. In vitro wound healing assay shows PCs closing the wound 30 hours after plating while BMSCs were still migrating (n=3 per group). Black dashed line: callus, White dashed line: bone cortex, white arrowheads point to PCNA+ cells, Merge: GFP/PCNA double positive cells (b) or GFP/cleavedCaspase3 double positive cells (c), SO: Safranin-O/Fast Green, cal: callus, c: cortex, bm: bone marrow. Scale bar: 1mm. Statistical differences between the groups were determined using Mann-Whitney test (*p≤0.05, **p<0.001, ***p<0.0005) (n= 3 to 5). All data represent mean ± SD.
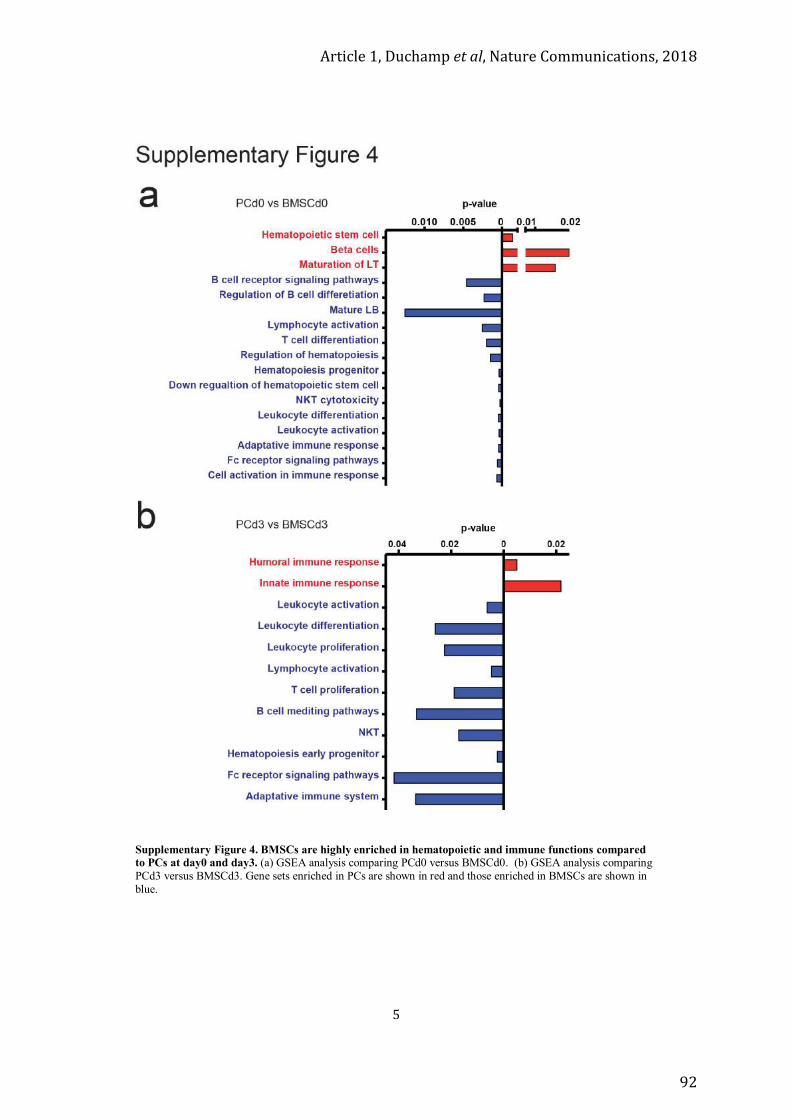
Article 1, Duchamp et al, Nature Communications, 2018
92
5
Supplementary Figure 4. BMSCs are highly enriched in hematopoietic and immune functions compared to PCs at day0 and day3. (a) GSEA analysis comparing PCd0 versus BMSCd0. (b) GSEA analysis comparing PCd3 versus BMSCd3. Gene sets enriched in PCs are shown in red and those enriched in BMSCs are shown in blue.
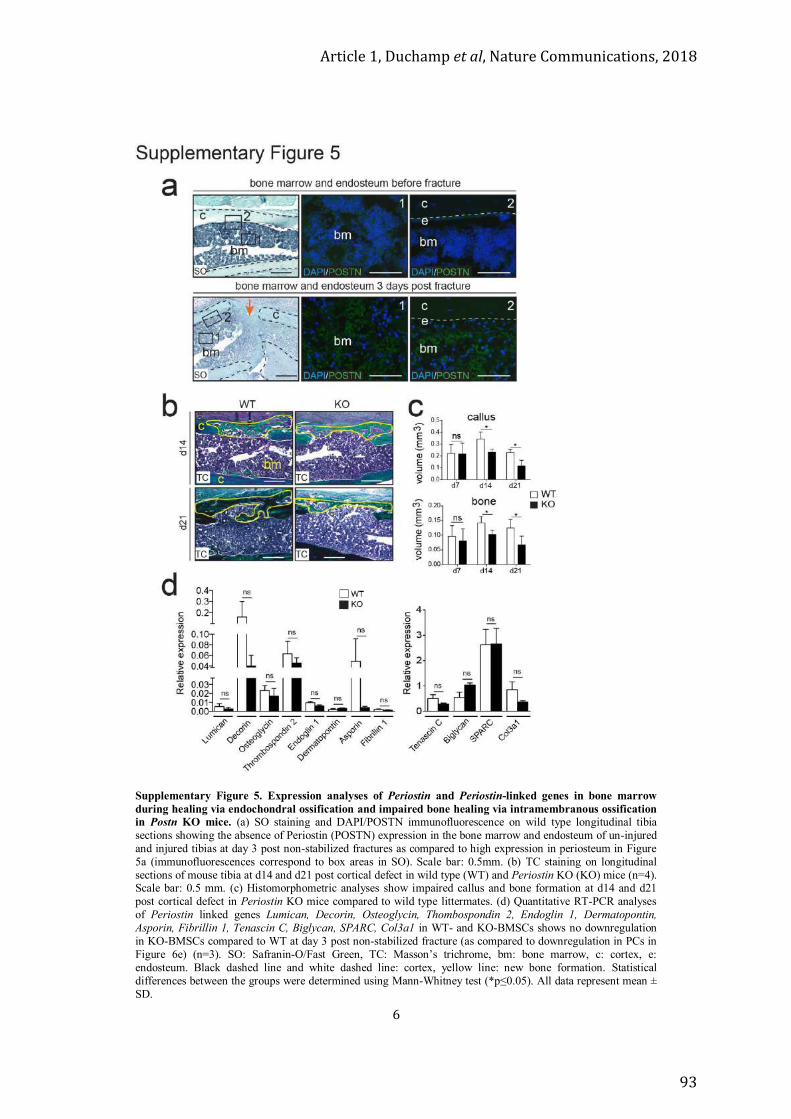
Article 1, Duchamp et al, Nature Communications, 2018
93
6
Supplementary Figure 5. Expression analyses of Periostin and Periostin-linked genes in bone marrow during healing via endochondral ossification and impaired bone healing via intramembranous ossification in Postn KO mice. (a) SO staining and DAPI/POSTN immunofluorescence on wild type longitudinal tibia sections showing the absence of Periostin (POSTN) expression in the bone marrow and endosteum of un-injured and injured tibias at day 3 post non-stabilized fractures as compared to high expression in periosteum in Figure 5a (immunofluorescences correspond to box areas in SO). Scale bar: 0.5mm. (b) TC staining on longitudinal sections of mouse tibia at d14 and d21 post cortical defect in wild type (WT) and Periostin KO (KO) mice (n=4). Scale bar: 0.5 mm. (c) Histomorphometric analyses show impaired callus and bone formation at d14 and d21 post cortical defect in Periostin KO mice compared to wild type littermates. (d) Quantitative RT-PCR analyses of Periostin linked genes Lumican, Decorin, Osteoglycin, Thombospondin 2, Endoglin 1, Dermatopontin, Asporin, Fibrillin 1, Tenascin C, Biglycan, SPARC, Col3a1 in WT- and KO-BMSCs shows no downregulation in KO-BMSCs compared to WT at day 3 post non-stabilized fracture (as compared to downregulation in PCs in Figure 6e) (n=3). SO: Safranin-O/Fast Green, TC: Masson’s trichrome, bm: bone marrow, c: cortex, e: endosteum. Black dashed line and white dashed line: cortex, yellow line: new bone formation. Statistical differences between the groups were determined using Mann-Whitney test (*p≤0.05). All data represent mean ± SD.

Article 1, Duchamp et al, Nature Communications, 2018
94
7
Supplementary Figure 6, related to Figure 7. The ability of periosteal cells to form cartilage and to colonize the new periosteal niche after periosteum transplantation is impaired in the absence of Periostin. (a) Quantitative analyses of GFP periosteal grafts from Figure 7a-c show the ability of periosteal cells derived from the GFP periosteal graft to expand extensively from periosteum in 3 rounds of injuries. The number of GFP+ cells is high in the cartilage in response to the fracture (d7) compared to the rare GFP+ cells found in the new periosteum at d28. (b) Transplantation of Periostin KO GFP graft into Periostin KO hosts shows that the absence of Periostin abolishes the ability of periosteal cells to re-populate the periosteal niche (d28-new periosteum, no GFP+ cells), and to form cartilage after a second injury (d7-callus, no GFP+ cells). As a consequence, the pseudarthrosis phenotype is observed as early as day 7 Periostin KO hosts causing a more severe bone repair defect compared to the first injury (d7, fibrosis showed by Picrosirius Staining, PS). (c) Immunohistochemistry anti-BrdU shows that cell proliferation is not affected in the periosteum in the absence of Periostin (black arrows: BrdU+ cells). SO: Safranin-O/Fast Green, cal: callus, po: periosteum, nb: new bone, m: muscle, white dashed line: periosteum, orange and black dashed lines: callus, black dashed line in (c): periosteum, n=2 to 4 per group. Scale bar= 1mm. Statistical differences between the groups were determined using Mann-Whitney test (*p≤0.05) (n=3). All data represent mean ± SD.
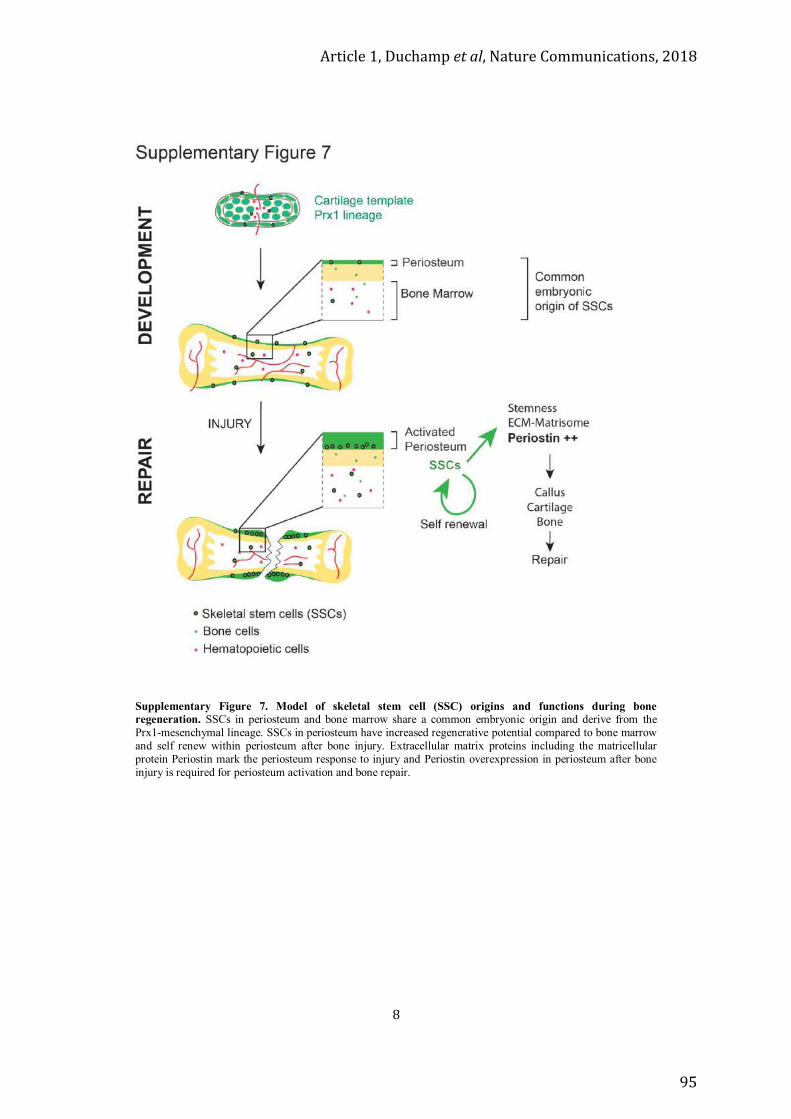
Article 1, Duchamp et al, Nature Communications, 2018
95
8
Supplementary Figure 7. Model of skeletal stem cell (SSC) origins and functions during bone regeneration. SSCs in periosteum and bone marrow share a common embryonic origin and derive from the Prx1-mesenchymal lineage. SSCs in periosteum have increased regenerative potential compared to bone marrow and self renew within periosteum after bone injury. Extracellular matrix proteins including the matricellular protein Periostin mark the periosteum response to injury and Periostin overexpression in periosteum after bone injury is required for periosteum activation and bone repair.

Article 1, Duchamp et al, Nature Communications, 2018
96
9
Supplementary Table 1. Primers for PCR genotyping name Primers
mouse Prx1-Cre 5’-CCTGGAAAATGCTTCTGTCCG-3’ 5’-CAGGGTGTTATAAGCAATCCC-3’
mouse mTmG 5’-CTCTGCTGCCTCCTGGCTTCT-3’ 5’-CGAGGCGGATCACAAGCAATA-3’ 5’-TCAATGGGCGGGGGTCGTT-3’
mouse R26ReYFP 5’-AAGACCGCGAAGAGTTTGTC-3’ 5’-GGAGCGGGAGAAATGGATATG-3’ 5’-AAAGTCGCTCTGAGTTGTTAT-3’
mouse R26ReLacZ 5'-AAAGTCGCTCTGAGTTGTTATCA-3' 5'- GTGGGAAGTCTTGTCCCTCC -3' 5'-CTTCCATTTGTCACGTCCTGC-3'
mouse Periostin 5’-AGTGTGCAGATGTTTGCTTG-3’ 5’-ACGAAATACAGTTTGGTAATCC-3’ 5’-CAGCGCATCGCCTTCTATCG-3’
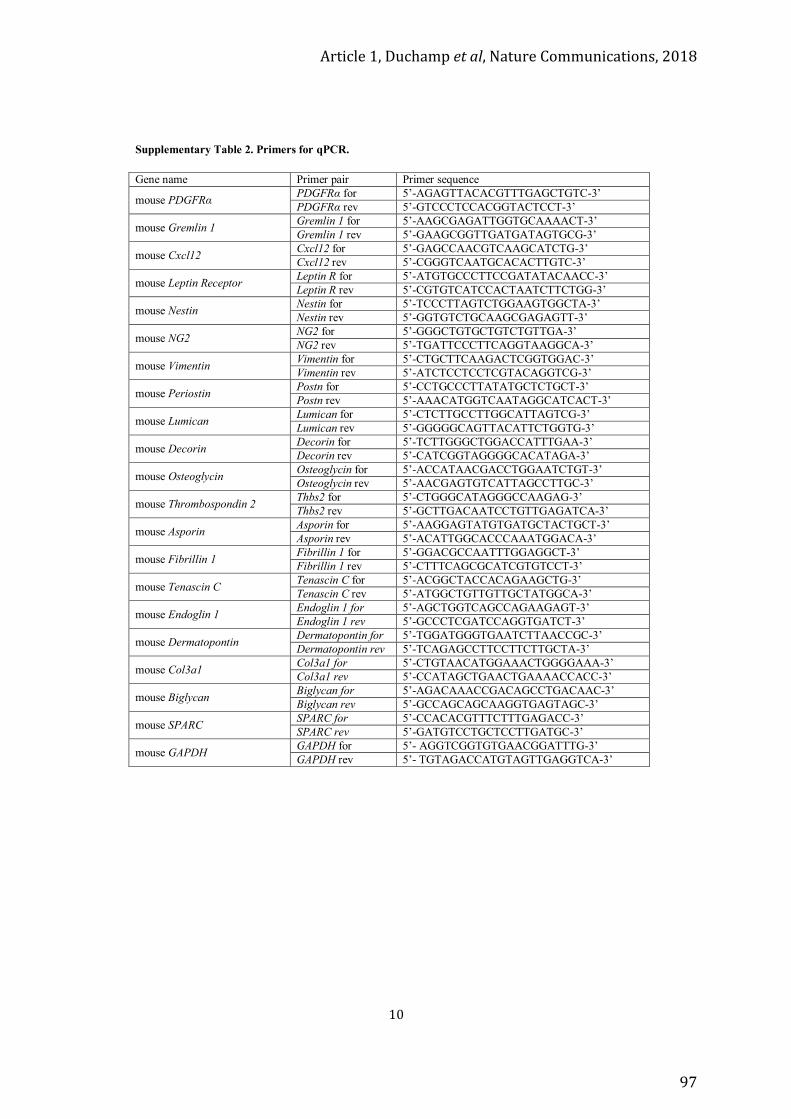
Article 1, Duchamp et al, Nature Communications, 2018
97
10
Supplementary Table 2. Primers for qPCR. Gene name Primer pair Primer sequence
mouse PDGFRα PDGFRα for 5’-AGAGTTACACGTTTGAGCTGTC-3’ PDGFRα rev 5’-GTCCCTCCACGGTACTCCT-3’
mouse Gremlin 1 Gremlin 1 for 5’-AAGCGAGATTGGTGCAAAACT-3’ Gremlin 1 rev 5’-GAAGCGGTTGATGATAGTGCG-3’
mouse Cxcl12 Cxcl12 for 5’-GAGCCAACGTCAAGCATCTG-3’ Cxcl12 rev 5’-CGGGTCAATGCACACTTGTC-3’
mouse Leptin Receptor Leptin R for 5’-ATGTGCCCTTCCGATATACAACC-3’ Leptin R rev 5’-CGTGTCATCCACTAATCTTCTGG-3’
mouse Nestin Nestin for 5’-TCCCTTAGTCTGGAAGTGGCTA-3’ Nestin rev 5’-GGTGTCTGCAAGCGAGAGTT-3’
mouse NG2 NG2 for 5’-GGGCTGTGCTGTCTGTTGA-3’ NG2 rev 5’-TGATTCCCTTCAGGTAAGGCA-3’
mouse Vimentin Vimentin for 5’-CTGCTTCAAGACTCGGTGGAC-3’ Vimentin rev 5’-ATCTCCTCCTCGTACAGGTCG-3’
mouse Periostin Postn for 5’-CCTGCCCTTATATGCTCTGCT-3’ Postn rev 5’-AAACATGGTCAATAGGCATCACT-3’
mouse Lumican Lumican for 5’-CTCTTGCCTTGGCATTAGTCG-3’ Lumican rev 5’-GGGGGCAGTTACATTCTGGTG-3’
mouse Decorin Decorin for 5’-TCTTGGGCTGGACCATTTGAA-3’ Decorin rev 5’-CATCGGTAGGGGCACATAGA-3’
mouse Osteoglycin Osteoglycin for 5’-ACCATAACGACCTGGAATCTGT-3’ Osteoglycin rev 5’-AACGAGTGTCATTAGCCTTGC-3’
mouse Thrombospondin 2 Thbs2 for 5’-CTGGGCATAGGGCCAAGAG-3’ Thbs2 rev 5’-GCTTGACAATCCTGTTGAGATCA-3’
mouse Asporin Asporin for 5’-AAGGAGTATGTGATGCTACTGCT-3’ Asporin rev 5’-ACATTGGCACCCAAATGGACA-3’
mouse Fibrillin 1 Fibrillin 1 for 5’-GGACGCCAATTTGGAGGCT-3’ Fibrillin 1 rev 5’-CTTTCAGCGCATCGTGTCCT-3’
mouse Tenascin C Tenascin C for 5’-ACGGCTACCACAGAAGCTG-3’ Tenascin C rev 5’-ATGGCTGTTGTTGCTATGGCA-3’
mouse Endoglin 1 Endoglin 1 for 5’-AGCTGGTCAGCCAGAAGAGT-3’ Endoglin 1 rev 5’-GCCCTCGATCCAGGTGATCT-3’
mouse Dermatopontin Dermatopontin for 5’-TGGATGGGTGAATCTTAACCGC-3’ Dermatopontin rev 5’-TCAGAGCCTTCCTTCTTGCTA-3’
mouse Col3a1 Col3a1 for 5’-CTGTAACATGGAAACTGGGGAAA-3’ Col3a1 rev 5’-CCATAGCTGAACTGAAAACCACC-3’
mouse Biglycan Biglycan for 5’-AGACAAACCGACAGCCTGACAAC-3’ Biglycan rev 5’-GCCAGCAGCAAGGTGAGTAGC-3’
mouse SPARC SPARC for 5’-CCACACGTTTCTTTGAGACC-3’ SPARC rev 5’-GATGTCCTGCTCCTTGATGC-3’
mouse GAPDH GAPDH for 5’- AGGTCGGTGTGAACGGATTTG-3’ GAPDH rev 5’- TGTAGACCATGTAGTTGAGGTCA-3’

Article 1, Duchamp et al, Nature Communications, 2018
98
11
Supplementary Table 3. List of 93 genes named “Postn linked genes”.
6 Genes from intersection of PCd3>PCd0 with Postn linked genes and with PCd3>BMSCd3
Postn periostin, osteoblast specific factor Aspn asporin
Col3a1 collagen, type III, alpha 1 Dcn decorin
Egln1 EGL nine homolog 1 (C, elegans) Lum lumican
16 Genes from intersection of PCd3>BMSCd3 with Postn linked genes
Bmpr1a bone morphogenetic protein receptor, type 1A Col12a1 collagen, type XII, alpha 1 Col1a1 collagen, type I, alpha 1 Col1a2 collagen, type I, alpha 2 Col5a1 collagen, type V, alpha 1 Col5a2 collagen, type V, alpha 2 Col6a3 collagen, type VI, alpha 3
Dpt dermatopontin Egfr epidermal growth factor receptor Fbn1 fibrillin 1
Mmp12 matrix metallopeptidase 12 Mmp2 matrix metallopeptidase 2 Mmp3 matrix metallopeptidase 3
Sparcl1 Sparc like 1 Tbx18 T-box18 Thbs2 thrombospondin 2
9 Genes from intersection of PCd3>PCd0 with Postn linked genes
Bcl2l11 BCL2-like 1 Erbb3 V-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian) Grb2 growth factor receptor bound protein 2 Gtf2f2 general transcription factor IIF, polypeptide 2 Itgb2 integrin beta 2 Itgb5 integrin beta 5
Lgals3 lectin, galactose binding, soluble 3 Runx3 Runt Related Transcription Factor 3 Tgfb1 transforming growth factor, beta 1
62 other Postn linked genes
Akt1 thymoma viral proto-oncogene 1
Bcar1 breast cancer anti-estrogen resistance 1 Bcl2l1 BCL2-like 1 Bgn biglycan
Bmp1 bone morphogenetic protein 1 Bmp2 bone morphogenetic protein 2 Bmp4 bone morphogenetic protein 4 Cbl Casitas B-lineage lymphoma
Cdh1 cadherin 1 Cdh11 cadherin 11 Cdx1 caudal type homeobox 1 Clca3 chloride channel calcium activated 3
Col14a1 collagen, type XIV, alpha 1 Col2a1 collagen, type II, alpha 1 Col4a1 collagen, type IV, alpha 1 Col6a1 collagen, type VI, alpha 1 Col6a2 collagen, type VI, alpha 2 Ctnnb1 catenin (cadherin associated protein), beta 1
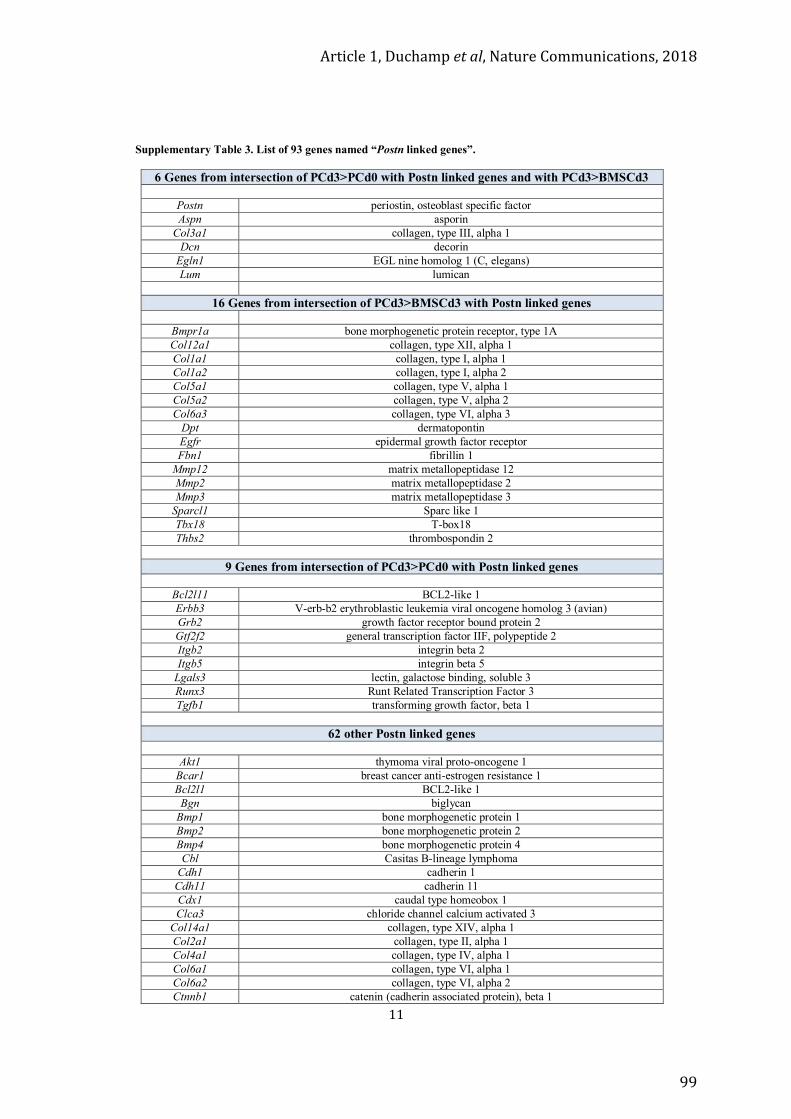
Article 1, Duchamp et al, Nature Communications, 2018
99
11
Supplementary Table 3. List of 93 genes named “Postn linked genes”.
6 Genes from intersection of PCd3>PCd0 with Postn linked genes and with PCd3>BMSCd3
Postn periostin, osteoblast specific factor Aspn asporin
Col3a1 collagen, type III, alpha 1 Dcn decorin
Egln1 EGL nine homolog 1 (C, elegans) Lum lumican
16 Genes from intersection of PCd3>BMSCd3 with Postn linked genes
Bmpr1a bone morphogenetic protein receptor, type 1A Col12a1 collagen, type XII, alpha 1 Col1a1 collagen, type I, alpha 1 Col1a2 collagen, type I, alpha 2 Col5a1 collagen, type V, alpha 1 Col5a2 collagen, type V, alpha 2 Col6a3 collagen, type VI, alpha 3
Dpt dermatopontin Egfr epidermal growth factor receptor Fbn1 fibrillin 1
Mmp12 matrix metallopeptidase 12 Mmp2 matrix metallopeptidase 2 Mmp3 matrix metallopeptidase 3
Sparcl1 Sparc like 1 Tbx18 T-box18 Thbs2 thrombospondin 2
9 Genes from intersection of PCd3>PCd0 with Postn linked genes
Bcl2l11 BCL2-like 1 Erbb3 V-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian) Grb2 growth factor receptor bound protein 2 Gtf2f2 general transcription factor IIF, polypeptide 2 Itgb2 integrin beta 2 Itgb5 integrin beta 5
Lgals3 lectin, galactose binding, soluble 3 Runx3 Runt Related Transcription Factor 3 Tgfb1 transforming growth factor, beta 1
62 other Postn linked genes
Akt1 thymoma viral proto-oncogene 1
Bcar1 breast cancer anti-estrogen resistance 1 Bcl2l1 BCL2-like 1 Bgn biglycan
Bmp1 bone morphogenetic protein 1 Bmp2 bone morphogenetic protein 2 Bmp4 bone morphogenetic protein 4 Cbl Casitas B-lineage lymphoma
Cdh1 cadherin 1 Cdh11 cadherin 11 Cdx1 caudal type homeobox 1 Clca3 chloride channel calcium activated 3
Col14a1 collagen, type XIV, alpha 1 Col2a1 collagen, type II, alpha 1 Col4a1 collagen, type IV, alpha 1 Col6a1 collagen, type VI, alpha 1 Col6a2 collagen, type VI, alpha 2 Ctnnb1 catenin (cadherin associated protein), beta 1

Article 1, Duchamp et al, Nature Communications, 2018
100
12
Ctnnd1 catenin (cadherin associated protein), beta 11 Egln2 EGL nine homolog 2 (C, elegans) Erbb2 V-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (avian) Erbb4 V-erb-b2 erythroblastic leukemia viral oncogene homolog 4 (avian) Fkbp4 FK506 binding protein 4 Fkbp5 FK506 binding protein 5 Fn1 fibronectin 1
Foxo1 forkhead box O1 Foxo3 forkhead box O3 Itga3 integrin alpha 3 Itgam integrin alpha M Itgax integrin alpha X Itgb1 integrin beta 1 Itgb3 integrin beta 3 Itgb6 integrin beta 6 Kdr kinase insert domain protein receptor
Mmp13 matrix metallopeptidase 13 Mmp1b matrix metallopeptidase 1b
Mtor mechanistic target of rapamycin (serine/threonine kinase) Ndufs2 NADH dehydrogenase (ubiquinone) Fe-S protein 2 Ndufs3 NADH dehydrogenase (ubiquinone) Fe-S protein 3 Ndufs7 NADH dehydrogenase (ubiquinone) Fe-S protein 7 Ndufv1 NADH dehydrogenase (ubiquinone) flavoprotein 1 Ndufv2 NADH dehydrogenase (ubiquinone) flavoprotein 2 Nos3 nitric oxide synthase 3, endothelial cell
Notch1 Notch gene homolog 1 (Drosophila) Nppa natriuretic peptide type A Ogn osteoglycin
Pdgfrab platelet derived growth factor receptor, beta polypeptide Ptk2 PTK2 protein tyrosine kinase 2 Pxn paxillin Rbpj recombination signal binding protein for immunoglobulin kappa J region
Serpinb2 serine (or cysteine) peptidase inhibitor, clade B, member 2 Rictor RPTOR Independent Companion Of MTOR Complex 2 Runx2 Runt Related Transcription Factor 2 Shc1 src homology 2 domain-containing transforming protein C1 Snai2 snail homolog 2 (Drosophila) Sost sclerostin
Sparc SPARC Tgfb2 transforming growth factor, beta 2 Tnc tenascin C
Twist1 twist homolog 1 (Drosophila) Vegfa vascular endothelial growth factor A Vegfc vascular endothelial growth factor C

Article 1, Duchamp et al, Nature Communications, 2018
101

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
102

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
103
Article 2
Role of Muscle Stem Cells During SkeletalRegenerationRANA ABOU–KHALIL,a FRANK YANG,b SHIRLEY LIEU,b ANAIS JULIEN,a JASELLE PERRY,b CATIA PEREIRA,aFREDERIC RELAIX,c THEODORE MICLAU,b RALPH MARCUCIO,b CELINE COLNOTaKey Words. Bone • Cre-loxP system • In vivo tracking • Satellite cells • Stem cell transplantation• CartilageABSTRACTAlthough the importance of muscle in skeletal regeneration is well recognized clinically, themechanisms by which muscle supports bone repair have remained elusive. Muscle flaps areoften used to cover the damaged bone after traumatic injury yet their contribution to bonehealing is not known. Here, we show that direct bone-muscle interactions are required for peri-osteum activation and callus formation, and that muscle grafts provide a source of stem cellsfor skeletal regeneration. We investigated the role of satellite cells, the muscle stem cells. Satel-lite cells loss in Pax72/2 mice and satellite cell ablation in Pax7CreERT2/1;DTAf/f mice impairedbone regeneration. Although satellite cells did not contribute as a large source of cells endoge-nously, they exhibited a potential to contribute to bone repair after transplantation. The frac-ture healing phenotype in Pax7CreERT2/1;DTAf/f mice was associated with decreased bonemorphogenetic proteins (BMPs), insulin-like growth factor 1, and fibroblast growth factor 2expression that are normally upregulated in response to fracture in satellite cells. ExogenousrhBMP2 improved bone healing in Pax7CreERT2/1;DTAf/f mice further supporting the role of satel-lite cells as a source of growth factors. These results provide the first functional evidence for adirect contribution of muscle to bone regeneration with important clinical implications as itmay impact the use of muscle flaps, muscle stem cells, and growth factors in orthopedic appli-cations. STEM CELLS 2015;33:1501–1511INTRODUCTION
Skeletal muscle and bone are closely linkedacross development, growth, and aging [1–3].Given their common mesodermal origin, it isnot surprising that the dependent associationof muscle and bone arises at the earlieststages of development. Muscle provides asource of mechanical stimuli for bone [4, 5].Muscle mass, bone mass, and strength arehighly correlated. Decreased muscle function,arising with disease or age, is clearly accompa-nied by diminished bone mass and morphol-ogy [1, 6, 7].
The functional interactions between mus-cle and bone are also critical during adult tis-sue regeneration. The clinical importance ofmuscle in fracture repair is clear as illustratedby the increased rate of delayed union andnonunion in patients with extreme traumaand soft tissue damage [8]. The lack of intactmuscle around the fracture site may hinderproper vascularization, release of osteogenicfactors, and/or recruitment of stem cells.Treatments for these fractures include antibi-otic therapy to prevent infection, fracture sta-bilization, bone grafting in case of critical size
defects, and soft tissue grafting such as fascio-cutaneous or muscle flaps [9]. Soft tissue cov-erage is important for protecting the woundor exposed tissues, reducing infection rate,and possibly increasing the blood supply [10].Muscle coverage of open fractures increasesthe rate of healing compared to fasciocutane-ous tissue [11, 12]. However, the specific rolesof muscle to support bone repair have notbeen elucidated at the cellular and molecularlevels.
Skeletal muscle and bone exhibit very effi-cient regenerative capacities supported byendogenous muscle stem cells, also called satel-lite cells, and skeletal stem cells, respectively. Inadult skeletal muscle, satellite cells are essentialfor the maintenance of muscle mass and formuscle regeneration [13, 14]. Pax3 and Pax7,two related paired-box transcription factors,mark the satellite cell population and areinvolved in the specification, maintenance ofskeletal muscle progenitors, and their engage-ment in the myogenic program during embryo-genesis [15–17]. While, Pax7 is considered asthe universal marker of adult satellite cells, Pax3expression is only confined to a subpopulation
aINSERM UMR1163,Universit!e Paris Descartes-Sorbonne Paris Cit!e, InstitutImagine, Hopital NeckerEnfants Malades, Paris,France; bDepartment ofOrthopaedic Surgery,University of California, SanFrancisco, San FranciscoGeneral Hospital,Orthopaedic TraumaInstitute, San Francisco,California, USA; cINSERM,U955, IMRB, UPEC, Cr!eteil,Paris, France
Correspondence: C!eline Colnot,Ph.D., INSERM UMR1163,Universit!e Paris Descartes-Sorbonne Paris Cit!e-InstitutImagine, Hopital Necker EnfantsMalades, 24 Boulevard duMontparnasse-75015 Paris,France. Telephone: 33-(0)1-42-75-42-33;e-mail: [email protected]
Received September 24, 2014;accepted for publicationDecember 7, 2014; firstpublished online in STEM CELLS
EXPRESS January 16, 2015.
VC AlphaMed Press1066-5099/2014/$30.00/0
http://dx.doi.org/10.1002/stem.1945
STEM CELLS 2015;33:1501–1511 www.StemCells.com VC AlphaMed Press 2015
REGENERATIVE MEDICINE

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
104
of satellite cells that express Pax7 after birth [13, 15–17].Whether muscle stem cells are involved in the muscle-boneinteractions in response to tissue injury is not known.
Following bone injury, skeletal stem cells are activatedduring the inflammatory phase of repair [18, 19]. We haveshown that skeletal muscle regulates inflammation duringbone regeneration [20]. Tumor necrosis factor-a can enhancebone healing and induce the differentiation of stromal cellspresent in muscle toward osteogenic and chondrogenic line-ages [21]. Although skeletal stem cells originate primarilyfrom the local periosteum [22–24], muscle-derived stem cellsmay also secondarily contribute to bone repair [25–27].Indeed, satellite cells can differentiate into osteoblasts andchondrocytes in vitro and in vivo [25, 28–30]. The recruitmentof skeletal stem cells is stimulated by growth factors includingbone morphogenetic proteins (BMPs) secreted at the fracturesite by inflammatory cells and bone cells [31–33]. Further-more, skeletal muscle produces numerous growth factors andcytokines such as insulin-like growth factor 1 (IGF1) and fibro-blast growth factor 2 (FGF2), two well-known osteogenic-related factors [5, 34]. Inversely, myostatin secreted by myo-fibers has negative effects on bone repair [5, 34, 35]. Theseobservations reveal important biochemical interactionsbetween muscle and bone, which have not been functionallyexamined so far.
Here, we provide functional evidence for the local cellularand molecular interactions between muscle and bone. Weconcentrate on the role of satellite cells that mediate thedirect muscle-bone interactions required for callus formation.We show that loss and ablation of satellite cells in miceimpair bone regeneration. We investigate the role of satellitecells as a source of cells and molecular signals, and show thatsatellite cells are activated in the muscle adjacent to the frac-tured bone leading to increased production of growth factorsthat are essential for bone regeneration. Hence, we establishthe cooperation of two adjacent tissues during musculoskel-etal regeneration, via the concomitant stimulation of stemcells within these tissues.MATERIALS AND METHODSAnimalsC57BL/6, Pax7CreERT2 beta-actin-GFP, Pax72/2, Pax3Cre, DTAf/f
(DTA5 diphtheria toxin fragment A), R26RLacZ, and R26R
eYFP
mice were obtained from Jackson Laboratory (Bar Harbor,ME) [17, 36, 37]. Pax7
CreERT2 were mated with DTAf/f,
R26RLacZ, and R26R
eYFP mice. Pax3Cre mice were bred with
R26RLacZ. Three-month-old male mice and age-matched wild-
type male littermates were used to conduct all experiments.To induce Cre recombinase activity, mice received intraperito-neal injections of Tamoxifen (Tmx) (Sigma, St. Louis, Missouri)at 3 mg/40 g b.wt. daily for 3 days, 1 week prior to fractureinjury. For satellite cell ablation in Pax7
CreERT2/1;DTA
f/f mice,due to lethality of mice [38], Tmx injections were adminis-trated 24 hours prior to fracture, immediately following frac-ture, and 24 hours later. In our hands, cre recombination andsatellite cell ablation efficiency was !80% (Supporting Infor-mation Fig. S1). All experiments were conducted according tothe Institutional Animal Care and Use Committee of Universityof California San Francisco and Paris Descartes University.
Closed Nonstabilized FracturesClosed standardized nonstabilized tibial fractures were createdunder anesthesia in the mid-diaphysis of the right tibia viathree-point bending as previously described [18, 20]. The tibiawas placed on the fracture jig, and a 500 g weight wasdropped from 3.5 cm to create the fracture. Mice wererevived and monitored closely until sacrifice.rhBMP2 TreatmentAt the time of the fracture, Tmx-induced Pax7
CreERT2/1;DTA
f/f
mice received a single injection of 10 mg of recombinanthuman BMP2 (rhBMP2) (Medtronic, Minneapolis, MN) inphosphate buffered saline (0.7 mg/ml) between the fracturedtibia ends using a syringe and 30-gauge needle (Hamilton,Reno, NV) [39].Open Nonstabilized FracturesOpen nonstabilized tibial fractures were produced by osteot-omy as previously described [20, 23]. The anterior tibial sur-face was exposed by separating the bone from thesurrounding muscles. Three holes were drilled in the tibialcortex using a 0.4 mm drill bit. Bone was cut until a fracturewas created. Prior to the open fracture, a 0.4 mm soft Milli-pore filter was placed at the periosteal surface of the boneand wrapped around the tibia at the level of the fracture cov-ering 3–4 mm distally and posteriorly. Precaution was takennot to damage the periosteum when separating periosteumand muscle using fine scissors and forceps during the proce-dure. The soft filter was placed on the posterior part of thetibia in-between muscle and bone. Following osteotomy, themuscles were sutured on the anterior part of the tibia inorder to hold the filter in place and to cover the entire sur-face of the tibia. In control samples, the muscle was sepa-rated from the muscle without disrupting the periosteum. Thesame procedure was followed except for the placement ofthe filter.Histological and Histomorphometric AnalysesHistomorphometric analyses were performed as previouslydescribed [20]. Briefly, mice were sacrificed following anesthe-sia and callus tissues were collected at days 5, 7, 14, and 21postfracture (n5 5 or 6 per group). Samples were fixed in 4%paraformaldehyde (PFA) overnight, decalcified in 19% EDTA(pH 7.4) for 14 days and, embedded in paraffin. Serial 10 mmlongitudinal paraffin sections were stained with Safranin-O/Fast Green to detect cartilage and modified Milligan’s Tri-chrome to detect bone. Images were captured using a LeicaDM 5000 B light microscope (Leica Microsystems GmbH, Ger-many) with an attached camera (Diagnostic Instruments, Inc.,Sterling Heights, MI), and Olympus CAST system (Olympus,Center Valley, PA). Images were analyzed with Adobe Photo-shop (Adobe, Inc., San Jose, CA) and Visiopharm (Visiopharm,Hørsholm, Denmark) to determine the component volumes ofbone and cartilage formation and reference volumes of callustissues as described in [20, 40, 41].Transplantation of Bone GraftsBone grafts, with or without muscle, were isolated from thetibia of Rosa26 donor mice that expressed LacZ ubiquitouslyas described in [23]. To follow cells derived from Rosa26
1502 Muscle Stem Cells in Bone Repair
VC AlphaMed Press 2015 STEM CELLS

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
105
muscle combined with the periosteum, muscle was leftattached to the periosteum. Bone grafts containing intact per-iosteum, with or without muscle, were placed in a tibial corti-cal bone defect adjacent to the fracture site of 3-month-oldrecipient C57BL/6 male mice [23].Muscle TransplantationA whole EDL (extensor digitorum longus) muscle was carefullyisolated from beta-actin-GFP donor mice, expressing green flu-orescent protein (GFP) ubiquitously, and transplanted into 3-month-old recipient C57BL/6 host male mice as described in[42]. Prior to open nonstabilized fracture injury, GFP-EDL mus-cle was transplanted adjacent to the tibia. The short proximaltendon of the graft was sutured to the tendon between thepatella and the knee of the host. The distal long tendon ofthe graft was sutured to the distal tendon of the host pero-neus muscle. Mice were revived and monitored closely untilsacrifice (n5 5).Myoblast CultureHind limb muscles were dissected from 2-month-old beta-
actin-GFP male mice and digested as previously described inBrack et al. [43]. Briefly, hind limb muscles were digestedwith 400 UI/ml collagenase type II (Life Technology, Carlsbad,CA) for 90 minutes at 37!C. Digested muscles were mechani-cally dissociated into single myofibers by repeated triturationusing Pasteur Pipette. Subsequently, myofiber fragments weredigested with 0.5 U/ml Dispase and 0.2% collagenase type II(Life Technologies, Carlsbad, CA) for 30 minutes at 37!C. Satel-lite cells were liberated from myofibers and plated into cul-ture dish in growth media (Hams F-10, 20% fetal bovineserum (FBS), 5 ng/ml bFGF). Primary myoblast cultures wereenriched by negatively selecting fibroblasts that attach to non-coated dishes. Only pure low-passage-myoblasts ("95%) wereused for transplantation.Fluorescent-Activated Cell Sorting of Satellite CellsMuscle stem cells (satellite cells) were freshly sorted fromTamoxifen-induced 2-month-old Pax7CreERT2/1;R26ReYFP/1 malemice. Briefly as described above, myogenic mononucleatedcells were enzymatically isolated from hind limb muscles. Cellswere incubated in Hams F-10 supplemented with 10% FBSand satellite cells were freshly sorted based on the expressionof YFP (yellow fluorescent protein). Flow cytometry cell sort-ing was performed using BD FACS Aria (Becton Dickinson[BD], Franklin Lakes, NJ) through Imagine Institute FlowCytometry Core Facility. Live cells were identified by negativestaining for Sytox Blue (1 mg/ml) (Invitrogen, Carlsbad, CA).Satellite cell sorting was optimized to achieve maximal cellpurity and viability. Only pure sorted satellite cells ("99%)were used for cell transplantation.Cell TransplantationAn open tibial fracture was performed as described above on 3-month-old recipient C57BL/6 host male mice. To transplant thecells to the fracture site, Tissucol kit (Baxter, France) was usedaccording to the manufacturer’s instructions and as previouslydescribed [44]. Myoblasts and freshly sorted muscle stem cells(105 cells) were embedded in highly and fast resorbable Tissu-col fibrin scaffold obtained by adding 15 ml of fibrinogen(30 mg/ml) followed by 15 ml of thrombin (18 mg/ml). After
gentle mixing, cells embedded into resorbable fibrin scaffoldwere transplanted into the fracture site of the C57BL/6 hostmice (n5 5). Mice were revived and monitored closely untilsacrifice.AntibodiesAffinity-purified rat anti-mouse CD31/PECAM antibody waspurchased from BD Pharmingen (San Diego, CA) to detectendothelial cells. Affinity-purified Chicken anti-GFP antibodywas purchased from Life Technologies to detect GFP or YFPproteins.ImmunofluorescenceFollowing transplantation of GFP-EDL, GFP myoblasts, andfreshly sorted Pax7 YFP1 satellite cells into the fracture site,fracture calluses were harvested. Samples were fixed in 4%paraformaldehyde overnight, decalcified in 19% EDTA (pH 7.4)for 14 days at 4!C and, subsequently, embedded for cryostatsectioning. Immunofluorescence was performed on slides pre-pared from sections located 300 mm apart throughout the cal-lus. Tissue sections for immunohistochemistry were fixed in4% PFA for 10 minutes, washed, permeabilized in 0.2% PBS,Triton X-100, and incubated with blocking solution (10% goatserum) for 30 minutes. Sections were stained in GFP antibodyovernight at 4!C and subsequently revealed with Alexa fluoro-phore conjugated chicken anti-IgG antibodies with DAPI for 1hour at room temperature.PECAM Immunohistochemistry and StereologicalAnalysesAnti-PECAM immunohistochemistry was performed on tissuesections located 300 mm apart throughout the callus as previ-ously described [20]. Briefly, after deparaffinization, sectionswere treated with 0.1% trypsin in PBS 13 for retrieval of anti-genicity. Endogenous peroxidase activity and nonspecific bind-ing sites were blocked by incubating sections in 0.3% H2O2 inphosphate buffer saline (PBS) 13 and 5% goat serum in PBS13 for 30 minutes and 90 minutes, respectively. Sectionswere then incubated with diluted primary antibody in 5%goat serum (1:100) at 4!C overnight. Sections were next incu-bated with diluted secondary antibody in 5% goat serum(1:250). Subsequently, sections were incubated with avidin/biotin enzyme complex (Vector Laboratories, Inc., Burlingame,CA) in PBS 13. Staining was detected using diaminobenzidine,and the tissue was counterstained with 0.02% Fast Green.Stereological analyses of endothelial cell surface density wereperformed as previously described [20].X-gal Staining and QuantificationBeta-galactosidase activity was detected by X-gal (5-bromo-4-chloro-3-indolyl-D-b-galactoside) staining as previouslydescribed [45]. Briefly, cryosections located 300 mm apartthroughout the callus were fixed in 0.2% glutaraldehyde solu-tion, washed three times for 15 minutes in a solution contain-ing 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NonidetP40 in PBS, and stained overnight in wash solution containing1 mg/ml X-gal, 2.1 mg/ml potassium ferrocyanide, 1.64 mg/ml potassium ferricyanide, and 20 mM Tris-HCl, pH 7.3, andlightly counterstained with eosin. To exclude X-Gal stainingdue to endogenous b-galactosidase activity in osteoclasts, weperformed double staining for b-galactosidase followed by
Abou-Khalil, Yang, Lieu et al. 1503
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
106
tartrate resistant acid phosphatase (TRAP) staining with a leu-kocyte acid phosphatase kit (Sigma, St. Louis, MO) [23]. Quan-tification of LacZ donor contribution was performed byfollowing the histomorphometric method described in Luet al. [40] to count X-gal-positive cells excluding the bonemarrow compartment (n5 5 or 6 per group).RNA Isolation and RTqPCRNonstabilized tibial fractures in Pax7
CreERT2/1;DTA
f/f and theirage-matched wild-type littermate were created as describedabove. Mice were sacrificed as described above at days 3 and5 postfracture. Following removal of surrounding skin, callustissues and all adjacent tissues located 0.5 cm distal and prox-imal to the callus boundaries were collected at day 5 post-fracture to analyze osteogenic and chondrogenic markers. Toassess the molecular contribution of muscle during boneregeneration, only the adjacent muscles surrounding calluswere collected after 3 days of bone regeneration. Total RNAwas extracted from muscles using Trizol reagent (Life Technol-ogies). Freshly fluorescence-activated cell-sorted (FACS) mus-cle stem cells were collected from muscle surrounding callusfrom uninjured or 3 days regenerating tibia of 3-month-oldPax7CreERT2/1;R26ReYFP/1mice. Total RNA was isolated fromFACS-sorted muscle stem cells using Qiagen Kit (Germantown,MD). The quantity of extracted RNA was confirmed using aNanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific,Wilmington, DE). Commercially available primers (SupportingInformation Table S1) were purchased from Qiagen (German-town, MD). cDNA synthesis was performed using an iScriptcDNA Synthesis Kit (Bio-Rad, Hercules, CA). Real-time PCR wasperformed using a QuantiTect SYBR Green PCR Kit (Qiagen)and detected using a CFX96 Touch Real-Time PCR DetectionSystem (Bio-Rad, Hercules, CA). GAPDH was used as an inter-nal control for all genes.Statistical AnalysesA minimum of five samples was used for each group. Statisti-cal significance was calculated with GraphPad Prism v6.0a.Student’s t test, one-way, and two-way ANOVA were used forstatistical analyses. In all experiments, p values <.05 wereconsidered significant.RESULTSTo functionally assess the role of skeletal muscle during boneregeneration, we blocked the physical contact between mus-cle and bone using an open tibial fracture model. At the timeof fracture, a Millipore filter was placed at the periosteal sur-face, preventing direct physical interactions between boneand muscle. Although callus size was comparable in the twogroups due to the presence of the filter and fibrous tissue(data not shown), the composition of the callus was signifi-cantly affected. At 7 and 10 days postfracture, relative carti-lage and relative bone volumes were significantly decreasedin the presence of the filter compared to calluses without fil-ter (Fig. 1A, 1B), indicating a delay in cartilage and bone dep-osition. We observed a deficient periosteal reaction in areaswhere the filter was in direct contact with the bone (Fig. 1C,asterisk). This was not due to periosteal damage or delayedangiogenesis as shown by the presence of blood vessels
stained with PECAM in areas where periosteal reaction wasimpaired (Fig. 1C). By day 10, periosteal reaction was eventu-ally noticeable but shifted in distal parts of the tibia, adjacentto the filter, while cartilage was mostly found at the level ofthe fracture site in the control group (Fig. 1C, arrows). Thiswas followed by a delay in cartilage resorption in the pres-ence of the filter as shown by an increase in the relative carti-lage volume by days 14 and 21 compared to controls (Fig.1C). Relative bone volume remained significantly lower in thepresence of the filter through day 21 indicating a sustaineddelay in bone deposition (Fig. 1B). Moreover, histological anal-yses showed that bone bridging was disrupted in areas wherethe filter was close to the fracture site (Fig. 1D).
Since direct interactions between muscle and bone appearnecessary for callus formation, we assessed the relative cellu-lar contribution of periosteum and muscle using bone graftingand genetic cell tracing [23]. As previously shown, Rosa26periosteal grafts transplanted into the fracture site of wild-type mice contribute locally to cartilage and bone within thecallus as illustrated by the presence of LacZ1 chondrocytesand osteoblasts within the callus (Fig. 2A). When muscle wasleft attached to the graft the percentage of LacZ1 graft con-tribution to the callus was significantly increased (Fig. 2B, 2C).To test the cellular contribution of muscle independent of theperiosteum, we transplanted an EDL muscle expressing GFPubiquitously adjacent to an open fracture of wild-type hostmice (Fig. 2D). At day 14, we observed GFP1 chondrocytes incartilage that stained for Safranin-O within the callus (Fig. 2E),indicating the muscle can provide a source of chondroprogeni-tors during bone regeneration.
To determine the role of satellite cells during boneregeneration, we assessed bone regeneration in Pax72/2
mice that exhibit progressive loss of satellite cells afterbirth [17]. Although the majority of Pax7-deficient mice dieat 2 weeks of age, the surviving Pax7-deficient mice weresmaller but exhibited normal tibia length by 3 months(Supporting Information Fig. S2) and survived until adult-hood [46, 47]. Pax7-deficient mice showed impaired boneregeneration as shown by the decrease in callus size, carti-lage, and bone volumes compared to wild-type mice (Fig.3A–3C). Impaired bone regeneration in Pax7
2/2 mice wasnot linked to a defect in angiogenesis as shown by theincrease in capillary surface compared to wild-type mice(Fig. 3F, 3G).
To confirm the importance of satellite cells during boneregeneration, satellite cells were ablated at the time of frac-ture after tamoxifen (Tmx) induction of Pax7CreERT2/1;DTAf/f
mice (Cre mice) and their Cre-negative control littermates(Ctrl) (Fig. 4A). In our experiments, Cre recombination andsatellite cell ablation efficiency was !80%, which is consistentwith previous reports [37]. By day 5 and day 7, callus size,cartilage, and bone volumes were significantly decreased inCre mice compared to Ctrl (Fig. 4B–4D). Long-term studieswere precluded due to the lethality of Pax7CreERT2/1;DTAf/f
mice 5–7 days after Tmx injections [38]. Histological analysesconfirmed the decrease in cartilage and bone matrix deposi-tion in Cre mice (Fig. 4E). This was correlated with adecreased expression of cartilage markers (collagen 2, col2
and collagen 10, col10) and bone markers (collagen 1, col1and osteocalcin, oc) (Fig. 4F). As observed in Pax72/2 mice, asignificant increase in capillary surface was observed in Cre
1504 Muscle Stem Cells in Bone Repair
VC AlphaMed Press 2015 STEM CELLS
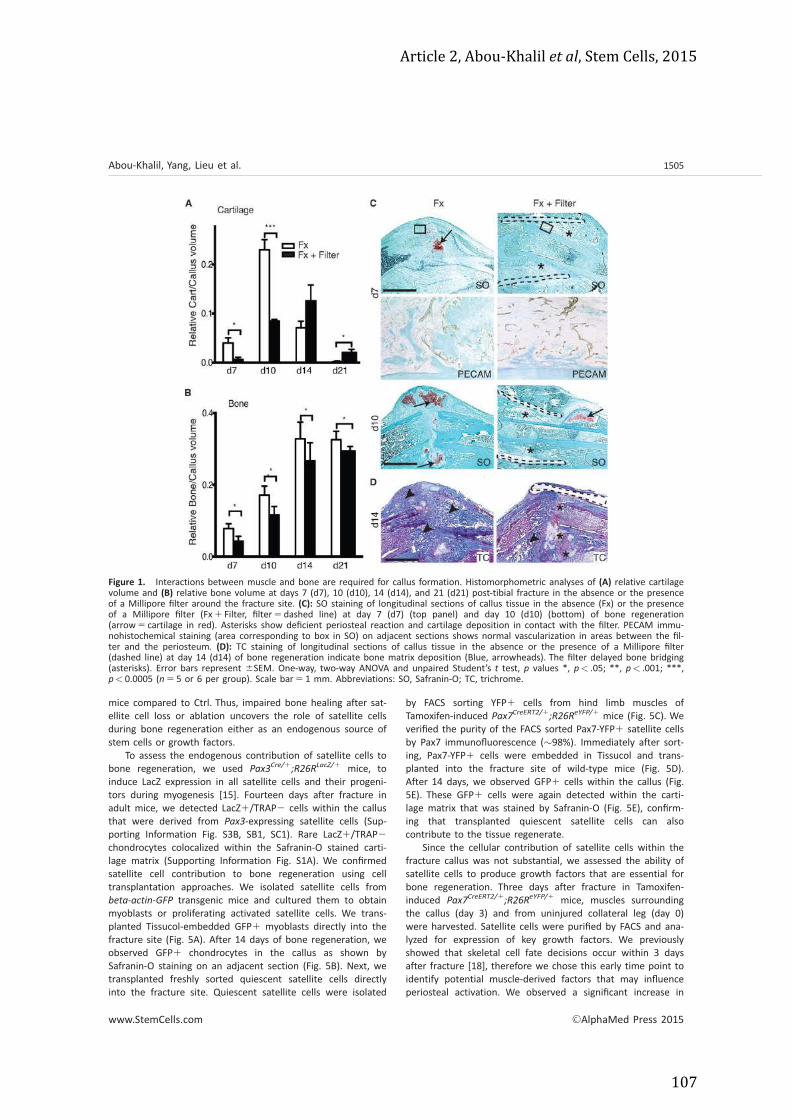
Article 2, Abou-‐Khalil et al, Stem Cells, 2015
107
mice compared to Ctrl. Thus, impaired bone healing after sat-ellite cell loss or ablation uncovers the role of satellite cellsduring bone regeneration either as an endogenous source ofstem cells or growth factors.
To assess the endogenous contribution of satellite cells tobone regeneration, we used Pax3
Cre/1;R26R
LacZ/1 mice, toinduce LacZ expression in all satellite cells and their progeni-tors during myogenesis [15]. Fourteen days after fracture inadult mice, we detected LacZ1/TRAP2 cells within the callusthat were derived from Pax3-expressing satellite cells (Sup-porting Information Fig. S3B, SB1, SC1). Rare LacZ1/TRAP2chondrocytes colocalized within the Safranin-O stained carti-lage matrix (Supporting Information Fig. S1A). We confirmedsatellite cell contribution to bone regeneration using celltransplantation approaches. We isolated satellite cells frombeta-actin-GFP transgenic mice and cultured them to obtainmyoblasts or proliferating activated satellite cells. We trans-planted Tissucol-embedded GFP1 myoblasts directly into thefracture site (Fig. 5A). After 14 days of bone regeneration, weobserved GFP1 chondrocytes in the callus as shown bySafranin-O staining on an adjacent section (Fig. 5B). Next, wetransplanted freshly sorted quiescent satellite cells directlyinto the fracture site. Quiescent satellite cells were isolated
by FACS sorting YFP1 cells from hind limb muscles ofTamoxifen-induced Pax7CreERT2/1;R26ReYFP/1 mice (Fig. 5C). Weverified the purity of the FACS sorted Pax7-YFP1 satellite cellsby Pax7 immunofluorescence (!98%). Immediately after sort-ing, Pax7-YFP1 cells were embedded in Tissucol and trans-planted into the fracture site of wild-type mice (Fig. 5D).After 14 days, we observed GFP1 cells within the callus (Fig.5E). These GFP1 cells were again detected within the carti-lage matrix that was stained by Safranin-O (Fig. 5E), confirm-ing that transplanted quiescent satellite cells can alsocontribute to the tissue regenerate.
Since the cellular contribution of satellite cells within thefracture callus was not substantial, we assessed the ability ofsatellite cells to produce growth factors that are essential forbone regeneration. Three days after fracture in Tamoxifen-induced Pax7
CreERT2/1;R26R
eYFP/1 mice, muscles surroundingthe callus (day 3) and from uninjured collateral leg (day 0)were harvested. Satellite cells were purified by FACS and ana-lyzed for expression of key growth factors. We previouslyshowed that skeletal cell fate decisions occur within 3 daysafter fracture [18], therefore we chose this early time point toidentify potential muscle-derived factors that may influenceperiosteal activation. We observed a significant increase in
Figure 1. Interactions between muscle and bone are required for callus formation. Histomorphometric analyses of (A) relative cartilagevolume and (B) relative bone volume at days 7 (d7), 10 (d10), 14 (d14), and 21 (d21) post-tibial fracture in the absence or the presenceof a Millipore filter around the fracture site. (C): SO staining of longitudinal sections of callus tissue in the absence (Fx) or the presenceof a Millipore filter (Fx1 Filter, filter5dashed line) at day 7 (d7) (top panel) and day 10 (d10) (bottom) of bone regeneration(arrow5 cartilage in red). Asterisks show deficient periosteal reaction and cartilage deposition in contact with the filter. PECAM immu-nohistochemical staining (area corresponding to box in SO) on adjacent sections shows normal vascularization in areas between the fil-ter and the periosteum. (D): TC staining of longitudinal sections of callus tissue in the absence or the presence of a Millipore filter(dashed line) at day 14 (d14) of bone regeneration indicate bone matrix deposition (Blue, arrowheads). The filter delayed bone bridging(asterisks). Error bars represent 6SEM. One-way, two-way ANOVA and unpaired Student’s t test, p values *, p< .05; **, p< .001; ***,p< 0.0005 (n5 5 or 6 per group). Scale bar5 1 mm. Abbreviations: SO, Safranin-O; TC, trichrome.
Abou-Khalil, Yang, Lieu et al. 1505
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
108
expression of bmp-2, 24, 26, and 27 in satellite cells after 3days of bone regeneration compared to quiescent satellitecells at day 0 (Fig. 6A). Expression of other growth factorssuch as Igf1 and Fgf2 was also increased in satellite cells atday 3 (Fig. 6A). We next determined the effect of satellite cellablation in Pax7
CreERT2/1;DTA
f/f (Cre) mice on the expressionof these growth factors during bone repair (Fig. 6B). Threedays after fracture, expression of bmp2, bmp4, bmp7 as wellas igf1 and fgf2 was significantly decreased in muscles sur-rounding the callus of Cre mice compared to Ctrl mice (Fig.6B). These results indicate that satellite cells express growthfactors, including BMPs, in response to injury and that thesemuscle-derived growth factors are significantly decreased inthe muscle surrounding the fracture callus following satellitecell ablation.
To determine whether impaired bone healing in Pax7-CreERT2/1
;DTAf/f mice is due to the decrease in muscle stem
cell-derived growth factors after satellite cell ablation, weinjected 10 mg of recombinant human BMP2 (rhBMP2)
directly to the fracture site in tamoxifen-treated Pax7-CreERT2/1
;DTAf/f mice (Fig. 6C). After 5 days of bone regenera-
tion, rhBMP2 treatment significantly increased callus size andboth cartilage and bone volumes compared to untreated con-trol mice (Ctrl) (Fig. 6C). Altogether our data provide strongfunctional evidence for the role of satellite cell-derived BMPduring bone regeneration.DISCUSSIONBone has a remarkable ability to regenerate following injury.However, in approximately 10% of all skeletal injuries boneregeneration is delayed or impaired, and there is even greaterrisk of delayed union or nonunion in patients with soft tissuedamage [8]. Muscle may be essential at several stages of thebone repair process. Our data provide a mechanism wherebythe muscle supports the normal process of bone healing by adirect interaction with the periosteum and by providing
Figure 2. Muscle improves the cellular contribution of periosteum during bone regeneration and directly contributes to cartilage. Bonegrafts (orange dotted lines, g: graft) with intact PO (A) or with intact periosteum and adjacent muscle (PO1muscle) (B) isolated fromRosa26 mice were grafted at the fracture site of wild-type mice. Sections through the fracture callus 10 days postfracture and bonegrafting stained with SO (top) and XGAL (bottom). (A): PO grafts gave rise to chondrocytes and osteoblasts/osteocytes (arrows) at theperiosteal surface. (B): PO combined with muscle increased the proportion of graft-derived cartilage and bone within the callus (arrows).(C): Stereological quantification of LacZ contribution within the callus. (D): EDL muscle isolated from beta-actin-GFP mice and trans-planted adjacent to the open tibial fracture of wild-type host mice (n5 5). (E): SO staining (left) of longitudinal sections of callus tissuesat day 14 (d14). Immunofluorescence of GFP (green) and DAPI (blue) (right) shows GFP1 cells within callus (delimited by a dotted line)on adjacent sections. Low magnification of GFP staining (top) corresponds to area in the black box in SO. White arrowhead points tothe GFP1 EDL graft. High magnifications of SO and GFP1 chondrocytes within cartilage (bottom) correspond to white box in GFP panel.Error bars represent 6SEM. Unpaired Student’s t test, p values **, p< .001 (n5 5 or 6 per group). Scale bar5 1 mm (A), 500 mm (E).Abbreviations: GFP, green fluorescent protein; PO, periosteum; SO, Safranin-O.
1506 Muscle Stem Cells in Bone Repair
VC AlphaMed Press 2015 STEM CELLS

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
109
osteogenic/chondrogenic factors through activation of satellitecells in the muscle adjacent to the fracture callus. Disruptingthese interactions may underlie the increased rate of nonun-ions in patients with significant soft-tissue injuries. Further-more, our results provide a mechanism by which musclegrafts covering soft tissue injuries stimulate healing [12].
The periosteum plays an indispensible role in bone regen-eration and is a major source of skeletal stem cells for carti-lage and bone formation [22, 23]. We used a model ofnonstabilized tibial fracture, which allowed us to amplify theendochondral ossification process and periosteal activation inorder to better represent the role of satellite cells and musclein fracture repair. We show that skeletal muscle adjacent tobone interacts with the periosteum and is essential for itsactivation in response to bone injury. Muscle obstruction,using a porous filter, impaired bone regeneration by inhibitingperiosteal activation in areas where direct muscle-periosteuminteractions were blocked, delaying chondrogenesis and osteo-genesis, and most importantly preventing bone bridging atlater time points. Using a periosteal graft model, we also
showed that muscle enhanced the periosteal contribution tobone regeneration confirming the importance of skeletal mus-cle during bone regeneration as a source of growth factorsand/or stem cells.
We established the cellular contribution of muscle duringbone regeneration by transplanting whole GFP-EDL muscleadjacent to a fracture site that gave rise to GFP1 chondro-cytes within the facture callus. We showed that satellite cellsplay a crucial role in bone regeneration as satellite cell loss inPax7
2/2 mice and satellite cell ablation in Pax7CreERT2/1
;DTAf/f
mice severely impaired bone regeneration. Interestingly, inboth the Pax7
2/2 and Pax7CreERT2/1
;DTAf/f mice, angiogenesis
was increased indicating that skeletal progenitors within bloodvessels did not compensate for the defect in bone regenera-tion [48]. Muscle-lineage analyses in Pax3
Cre/1;R26R
LacZ/1
mice revealed a contribution of satellite cells as an endoge-nous source of chondrocytes during bone regeneration. Localmuscle injury surrounding the callus activates satellites cellsin regenerating fibers. These activated satellite cells may bereleased to be integrated in the callus and become exposed
Figure 3. Loss of muscle stem cells impairs bone regeneration. Histomorphometric measurements of (A) total callus volume, (B) totalcartilage volume, and (C) total bone volume at days 7 (d7), 14 (d14), and 21 (d21) postfracture in control wild-type (Ctrl) and Pax7
2/2
mice. (D): SO staining of longitudinal sections of Ctrl and Pax72/2 callus tissues at day 7 (d7) (cartilage in red, callus outlined by a dot-
ted line). (E): TC staining of sections of Ctrl and Pax72/2 callus tissues at day 14 (d14) (bone matrix deposition in blue). (F): PECAM
immunostaining (arrowheads) of Ctrl and Pax72/2 fracture calluses after 7 days (d7) of bone regeneration (area corresponds to boxesin D). (G): Stereological quantification of blood vessels within the callus of Ctrl and Pax7
2/2 mice. Error bars represent 6SEM. One-way, two-way ANOVA and unpaired Student’s t test, p values *, p< .05; **, p< .001 (n5 5 or 6 per group). Scale bar5 1 mm. Abbrevi-ations: B, bone; Cg, cartilage; SO, Safranin-O; TC, trichrome.
Abou-Khalil, Yang, Lieu et al. 1507
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
110
osteogenic/chondrogenic factors through activation of satellitecells in the muscle adjacent to the fracture callus. Disruptingthese interactions may underlie the increased rate of nonun-ions in patients with significant soft-tissue injuries. Further-more, our results provide a mechanism by which musclegrafts covering soft tissue injuries stimulate healing [12].
The periosteum plays an indispensible role in bone regen-eration and is a major source of skeletal stem cells for carti-lage and bone formation [22, 23]. We used a model ofnonstabilized tibial fracture, which allowed us to amplify theendochondral ossification process and periosteal activation inorder to better represent the role of satellite cells and musclein fracture repair. We show that skeletal muscle adjacent tobone interacts with the periosteum and is essential for itsactivation in response to bone injury. Muscle obstruction,using a porous filter, impaired bone regeneration by inhibitingperiosteal activation in areas where direct muscle-periosteuminteractions were blocked, delaying chondrogenesis and osteo-genesis, and most importantly preventing bone bridging atlater time points. Using a periosteal graft model, we also
showed that muscle enhanced the periosteal contribution tobone regeneration confirming the importance of skeletal mus-cle during bone regeneration as a source of growth factorsand/or stem cells.
We established the cellular contribution of muscle duringbone regeneration by transplanting whole GFP-EDL muscleadjacent to a fracture site that gave rise to GFP1 chondro-cytes within the facture callus. We showed that satellite cellsplay a crucial role in bone regeneration as satellite cell loss inPax7
2/2 mice and satellite cell ablation in Pax7CreERT2/1
;DTAf/f
mice severely impaired bone regeneration. Interestingly, inboth the Pax7
2/2 and Pax7CreERT2/1
;DTAf/f mice, angiogenesis
was increased indicating that skeletal progenitors within bloodvessels did not compensate for the defect in bone regenera-tion [48]. Muscle-lineage analyses in Pax3
Cre/1;R26R
LacZ/1
mice revealed a contribution of satellite cells as an endoge-nous source of chondrocytes during bone regeneration. Localmuscle injury surrounding the callus activates satellites cellsin regenerating fibers. These activated satellite cells may bereleased to be integrated in the callus and become exposed
Figure 3. Loss of muscle stem cells impairs bone regeneration. Histomorphometric measurements of (A) total callus volume, (B) totalcartilage volume, and (C) total bone volume at days 7 (d7), 14 (d14), and 21 (d21) postfracture in control wild-type (Ctrl) and Pax7
2/2
mice. (D): SO staining of longitudinal sections of Ctrl and Pax72/2 callus tissues at day 7 (d7) (cartilage in red, callus outlined by a dot-
ted line). (E): TC staining of sections of Ctrl and Pax72/2 callus tissues at day 14 (d14) (bone matrix deposition in blue). (F): PECAM
immunostaining (arrowheads) of Ctrl and Pax72/2 fracture calluses after 7 days (d7) of bone regeneration (area corresponds to boxesin D). (G): Stereological quantification of blood vessels within the callus of Ctrl and Pax7
2/2 mice. Error bars represent 6SEM. One-way, two-way ANOVA and unpaired Student’s t test, p values *, p< .05; **, p< .001 (n5 5 or 6 per group). Scale bar5 1 mm. Abbrevi-ations: B, bone; Cg, cartilage; SO, Safranin-O; TC, trichrome.
Abou-Khalil, Yang, Lieu et al. 1507
www.StemCells.com VC AlphaMed Press 2015
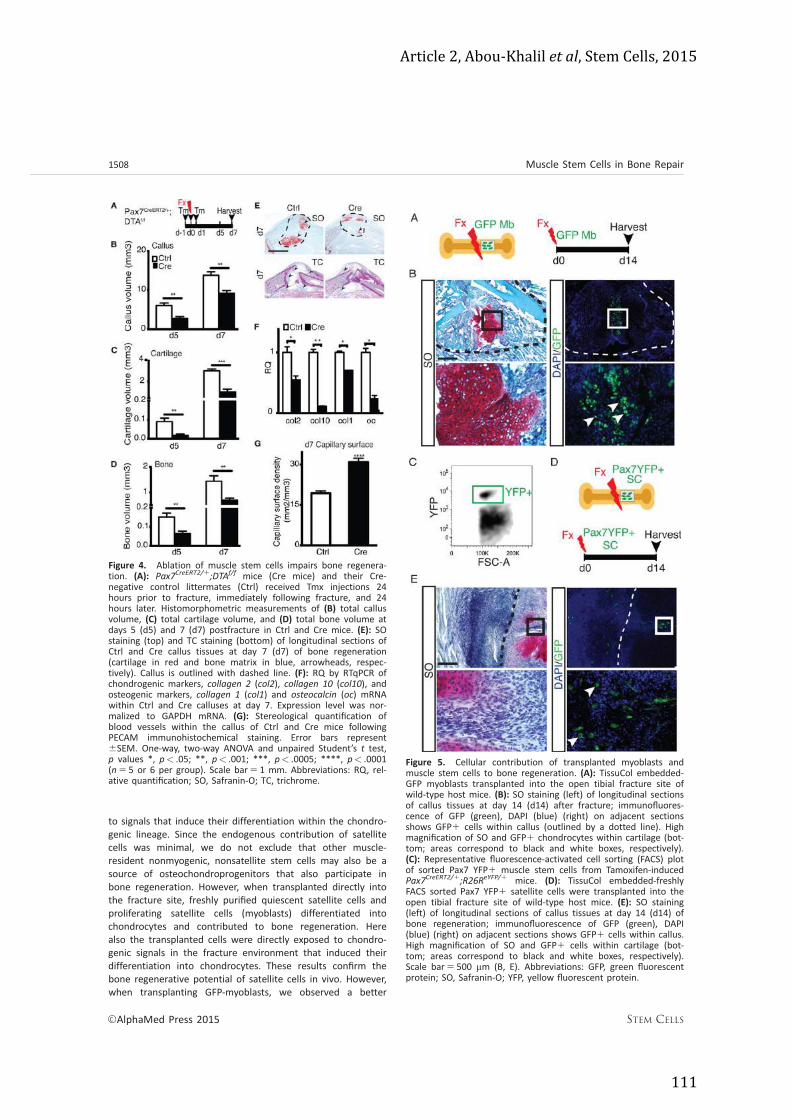
Article 2, Abou-‐Khalil et al, Stem Cells, 2015
111
to signals that induce their differentiation within the chondro-genic lineage. Since the endogenous contribution of satellitecells was minimal, we do not exclude that other muscle-resident nonmyogenic, nonsatellite stem cells may also be asource of osteochondroprogenitors that also participate inbone regeneration. However, when transplanted directly intothe fracture site, freshly purified quiescent satellite cells andproliferating satellite cells (myoblasts) differentiated intochondrocytes and contributed to bone regeneration. Herealso the transplanted cells were directly exposed to chondro-genic signals in the fracture environment that induced theirdifferentiation into chondrocytes. These results confirm thebone regenerative potential of satellite cells in vivo. However,when transplanting GFP-myoblasts, we observed a better
Figure 5. Cellular contribution of transplanted myoblasts andmuscle stem cells to bone regeneration. (A): TissuCol embedded-GFP myoblasts transplanted into the open tibial fracture site ofwild-type host mice. (B): SO staining (left) of longitudinal sectionsof callus tissues at day 14 (d14) after fracture; immunofluores-cence of GFP (green), DAPI (blue) (right) on adjacent sectionsshows GFP1 cells within callus (outlined by a dotted line). Highmagnification of SO and GFP1 chondrocytes within cartilage (bot-tom; areas correspond to black and white boxes, respectively).(C): Representative fluorescence-activated cell sorting (FACS) plotof sorted Pax7 YFP1 muscle stem cells from Tamoxifen-inducedPax7CreERT2/1;R26ReYFP/1 mice. (D): TissuCol embedded-freshlyFACS sorted Pax7 YFP1 satellite cells were transplanted into theopen tibial fracture site of wild-type host mice. (E): SO staining(left) of longitudinal sections of callus tissues at day 14 (d14) ofbone regeneration; immunofluorescence of GFP (green), DAPI(blue) (right) on adjacent sections shows GFP1 cells within callus.High magnification of SO and GFP1 cells within cartilage (bot-tom; areas correspond to black and white boxes, respectively).Scale bar5 500 mm (B, E). Abbreviations: GFP, green fluorescentprotein; SO, Safranin-O; YFP, yellow fluorescent protein.
Figure 4. Ablation of muscle stem cells impairs bone regenera-tion. (A): Pax7CreERT2/1;DTAf/f mice (Cre mice) and their Cre-negative control littermates (Ctrl) received Tmx injections 24hours prior to fracture, immediately following fracture, and 24hours later. Histomorphometric measurements of (B) total callusvolume, (C) total cartilage volume, and (D) total bone volume atdays 5 (d5) and 7 (d7) postfracture in Ctrl and Cre mice. (E): SOstaining (top) and TC staining (bottom) of longitudinal sections ofCtrl and Cre callus tissues at day 7 (d7) of bone regeneration(cartilage in red and bone matrix in blue, arrowheads, respec-tively). Callus is outlined with dashed line. (F): RQ by RTqPCR ofchondrogenic markers, collagen 2 (col2), collagen 10 (col10), andosteogenic markers, collagen 1 (col1) and osteocalcin (oc) mRNAwithin Ctrl and Cre calluses at day 7. Expression level was nor-malized to GAPDH mRNA. (G): Stereological quantification ofblood vessels within the callus of Ctrl and Cre mice followingPECAM immunohistochemical staining. Error bars represent6SEM. One-way, two-way ANOVA and unpaired Student’s t test,p values *, p< .05; **, p< .001; ***, p< .0005; ****, p< .0001(n5 5 or 6 per group). Scale bar5 1 mm. Abbreviations: RQ, rel-ative quantification; SO, Safranin-O; TC, trichrome.
1508 Muscle Stem Cells in Bone Repair
VC AlphaMed Press 2015 STEM CELLS

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
112
contribution to cartilage within the callus compared to sortedsatellite cells, suggesting that the GFP-myoblast cell popula-tion was maybe a heterogeneous cell population allowing abetter contribution to cartilage within the callus. Future invitro analyses may help further elucidate the cellular versusmolecular contributions of myogenic cells during bone repair.
More significantly, we show that satellite cells provide asource of growth factors during bone regeneration. The fac-tors regulating musculoskeletal interactions have not beenelucidated. BMPs may be produced by many cell types atthe fracture site including inflammatory cells, bone matrix,
and bone cells [33, 49]. We show that BMPS producedlocally by muscle stem cells are among the growth factorsprovided by muscle to support bone regeneration. Indeed,muscle stem cells expressed other growth factors such asIGF1 and FGF2 that are also crucial for bone regeneration[5, 34]. When treated with rhBMP2, the delayed bone regen-eration in Pax7
CreERT2/1;DTA
f/f mice was improved. Musclestem cells may play a role, in addition to other cell types,by regulating the BMP-dependent activation of skeletal stemcells within periosteum and callus formation [31, 39, 50],providing a functional explanation for the critical interactions
Figure 6. Molecular contribution of satellite cells to bone regeneration. (A): Pax7CreERT2/1;R26ReYFP/1 mice received Tamoxifen injec-tions daily for 3 days, 1 week prior to closed nonstabilized tibial fracture. Muscles surrounding callus were harvested and Pax7 YFP1satellite cells were freshly fluorescence-activated cell sorted (FACS) at d0 and d3. RQ by RTqPCR of bmp2, bmp4, bmp6, bmp7, igf1, fgf2mRNA, within freshly FACS sorted Pax7 YFP1 satellite cells at d0 (d0 SC) and d3 (d3 SC) of bone regeneration. Expression level was nor-malized to GAPDH mRNA. (B): Tamoxifen injections and closed-nonstabilized fracture of 3-month-old Pax7CreERT2/1;DTAf/f (Cre) mice andtheir matched control (Ctrl) littermates. Muscles surrounding callus were harvested at 3 days (d3). RQ by RTqPCR of bmp2, bmp4,bmp7, igf1, fgf2 mRNA within surrounding muscle at d3 of bone regeneration of Ctrl and Cre mice. Expression level was normalized toGAPDH mRNA. Error bars represent 6SEM. Unpaired Student’s t test, p values *, p< .05; **, p< .001 (n5 5 or 6 per group). (C):Tamoxifen injections and closed nonstabilized tibial fracture in 3-month-old Pax7CreERT2/1;DTAf/f (Cre) mice. Ten micrograms of rhBMP2in phosphate buffered saline was injected at the time of the fracture. Histomorphometric measurements of total callus volume (left),total cartilage volume (center), and total bone volume (right) at day 5 (d5) postfracture in Ctrl and BMP2-treated Cre mice. (D): Repre-sentative Safranin-O (top) and Trichrome (bottom) staining of callus sections of Ctrl and BMP2-treated Cre mice (arrowheads point toenhanced periosteal reaction and cartilage formation in treated mice). Error bars represent 6SEM. Unpaired Student’s t test, p values *,p< .05; **, p< .001 (n 55 or 6 per group). Scale bar5 1 mm. Abbreviations: RQ, relative quantification; SO, Safranin-O; TC, trichrome.
Abou-Khalil, Yang, Lieu et al. 1509
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
113
contribution to cartilage within the callus compared to sortedsatellite cells, suggesting that the GFP-myoblast cell popula-tion was maybe a heterogeneous cell population allowing abetter contribution to cartilage within the callus. Future invitro analyses may help further elucidate the cellular versusmolecular contributions of myogenic cells during bone repair.
More significantly, we show that satellite cells provide asource of growth factors during bone regeneration. The fac-tors regulating musculoskeletal interactions have not beenelucidated. BMPs may be produced by many cell types atthe fracture site including inflammatory cells, bone matrix,
and bone cells [33, 49]. We show that BMPS producedlocally by muscle stem cells are among the growth factorsprovided by muscle to support bone regeneration. Indeed,muscle stem cells expressed other growth factors such asIGF1 and FGF2 that are also crucial for bone regeneration[5, 34]. When treated with rhBMP2, the delayed bone regen-eration in Pax7
CreERT2/1;DTA
f/f mice was improved. Musclestem cells may play a role, in addition to other cell types,by regulating the BMP-dependent activation of skeletal stemcells within periosteum and callus formation [31, 39, 50],providing a functional explanation for the critical interactions
Figure 6. Molecular contribution of satellite cells to bone regeneration. (A): Pax7CreERT2/1;R26ReYFP/1 mice received Tamoxifen injec-tions daily for 3 days, 1 week prior to closed nonstabilized tibial fracture. Muscles surrounding callus were harvested and Pax7 YFP1satellite cells were freshly fluorescence-activated cell sorted (FACS) at d0 and d3. RQ by RTqPCR of bmp2, bmp4, bmp6, bmp7, igf1, fgf2mRNA, within freshly FACS sorted Pax7 YFP1 satellite cells at d0 (d0 SC) and d3 (d3 SC) of bone regeneration. Expression level was nor-malized to GAPDH mRNA. (B): Tamoxifen injections and closed-nonstabilized fracture of 3-month-old Pax7CreERT2/1;DTAf/f (Cre) mice andtheir matched control (Ctrl) littermates. Muscles surrounding callus were harvested at 3 days (d3). RQ by RTqPCR of bmp2, bmp4,bmp7, igf1, fgf2 mRNA within surrounding muscle at d3 of bone regeneration of Ctrl and Cre mice. Expression level was normalized toGAPDH mRNA. Error bars represent 6SEM. Unpaired Student’s t test, p values *, p< .05; **, p< .001 (n5 5 or 6 per group). (C):Tamoxifen injections and closed nonstabilized tibial fracture in 3-month-old Pax7CreERT2/1;DTAf/f (Cre) mice. Ten micrograms of rhBMP2in phosphate buffered saline was injected at the time of the fracture. Histomorphometric measurements of total callus volume (left),total cartilage volume (center), and total bone volume (right) at day 5 (d5) postfracture in Ctrl and BMP2-treated Cre mice. (D): Repre-sentative Safranin-O (top) and Trichrome (bottom) staining of callus sections of Ctrl and BMP2-treated Cre mice (arrowheads point toenhanced periosteal reaction and cartilage formation in treated mice). Error bars represent 6SEM. Unpaired Student’s t test, p values *,p< .05; **, p< .001 (n 55 or 6 per group). Scale bar5 1 mm. Abbreviations: RQ, relative quantification; SO, Safranin-O; TC, trichrome.
Abou-Khalil, Yang, Lieu et al. 1509
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
114
between muscle and bone that are required for periostealactivation. The role of muscle in periosteal activation via aBMP-dependent mechanism may be particularly relevant inthe context of endochondral ossification, as we previouslyshowed that cell fate within the periosteum was regulatedby BMP2 [39].CONCLUSIONSAltogether, our results elucidate the functional role of muscleduring bone regeneration, via the cellular and molecular con-tribution of satellite cells, the muscle stem cells, in the pro-cess of endochondral ossification. Muscle-derived growthfactors are primary actors in this context. Understanding themechanism by which skeletal muscle enhances bone regener-ation is crucial to define the causes for tissue repair dysfunc-tions after severe trauma, which often affects several tissues.By identifying the potential of satellite cells to provide asource of growth factors and skeletal stem cells in vivo, ourfindings may lead to direct clinical applications for the treat-ment of nonunion and for better understanding the bases ofmusculoskeletal repair defects associated with musculoskeletaldiseases and with musculoskeletal trauma. In future clinicalapplications, muscle flaps may not only help covering bonedefects and prevent infections, but also supporting bone heal-ing more directly. Indeed, muscle may provide a more effi-cient source of stem cells for cell therapies, as muscle stemcells may reveal superior in vivo regenerative capacities com-pared to bone marrow-derived mesenchymal stem cells usedwidely in tissue engineering approaches.
ACKNOWLEDGMENTSWe thank Caroline Carvalho for technical assistance and Cor-inne Cordier at the Imagine Institute flow cytometry corefacility. This work was supported by NIH-NIAMS R01AR057344, ANR-13-BSV1-001-01, INSERM ATIP-Avenir andSanofi R10071KS, FP7 Marie Curie IRG-268227, Osteosynthesisand Trauma Care Foundation 2011-CCSP, National ScienceFoundation Science Master’s Program Award (DGE-1011717),CIRM Bridges Master’s Training Grant (TBI-01194).AUTHOR CONTRIBUTIONSR.A.-K.: conception and design, collection and assembly ofdata, data analysis and interpretation, and manuscript writing;F.Y.: collection and assembly of data, data analysis and inter-pretation, and manuscript writing; S.L.: collection and assem-bly of data and data analysis; A.J.: collection and assembly ofdata and data analysis and interpretation; J.P. and C.P.: collec-tion and assembly of data; F.R.: provision of study material,data interpretation, and final approval of manuscript; T.M.and R.M.: financial support, data interpretation, and finalapproval of manuscript; C.C.: conception and design, financialsupport, collection and assembly of data, data analysis andinterpretation, manuscript writing, and final approval ofmanuscript. R.A.-K. and F.Y. contributed equally to this work.DISCLOSURE OF POTENTIAL CONFLICTS OF INTERESTThe authors indicate no potential conflicts of interest.REFERENCES1 Brotto M. Aging, sarcopenia and store-
operated calcium entry: A common link? CellCycle 2011;10:4201–4202.2 Sharir A, Stern T, Rot C et al. Muscleforce regulates bone shaping for optimalload-bearing capacity during embryogenesis.Development 2011;138:3247–3259.3 Karasik D. How pleiotropic genetics ofthe musculoskeletal system can informgenomics and phenomics of aging. Age(Dordr) 2011;33:49–62.4 Judex S, Rubin CT. Is bone formationinduced by high-frequency mechanical signalsmodulated by muscle activity? J MusculoskeletNeuronal Interact 2010;10:3–11.5 Hamrick MW. A role for myokines inmuscle-bone interactions. Exerc Sport Sci Rev2011;39:43–47.6 Brotto M. Lessons from the FNIH-NIA-FDA sarcopenia consensus summit. IBMSBonekey 2012;9.7 Hamrick MW, Ding KH, Pennington Cet al. Age-related loss of muscle mass andbone strength in mice is associated with adecline in physical activity and serum leptin.Bone 2006;39:845–853.8 Sen MK, Miclau T. Autologous iliac crestbone graft: Should it still be the gold stand-ard for treating nonunions? Injury 2007;38(suppl 1):S75–80.9 Yazar S, Lin CH, Lin YT et al. Outcomecomparison between free muscle and freefasciocutaneous flaps for reconstruction of
distal third and ankle traumatic open tibialfractures. Plast Reconstr Surg 2006;117:2468–2475; discussion 2476-2467.10 Ulusal AE, Lin CH, Lin YT et al. The useof free flaps in the management of type IIIBopen calcaneal fractures. Plast Reconstr Surg2008;121:2010–2019.11 Evans CH, Liu FJ, Glatt V et al. Use ofgenetically modified muscle and fat grafts torepair defects in bone and cartilage. Eur CellsMater 2009;18:96–111.12 Harry LE, Sandison A, Paleolog EM et al.Comparison of the healing of open tibialfractures covered with either muscle or fas-ciocutaneous tissue in a murine model.J Orthop Res 2008;26:1238–1244.13 Relaix F, Zammit PS. Satellite cells areessential for skeletal muscle regeneration:The cell on the edge returns centre stage.Development 2012;139:2845–2856.14 Murphy MM, Lawson JA, Mathew SJet al. Satellite cells, connective tissue fibro-blasts and their interactions are crucial formuscle regeneration. Development 2011;138:3625–3637.15 Relaix F, Rocancourt D, Mansouri Aet al. A Pax3/Pax7-dependent population ofskeletal muscle progenitor cells. Nature 2005;435:948–953.16 Relaix F, Rocancourt D, Mansouri Aet al. Divergent functions of murine Pax3and Pax7 in limb muscle development. GenesDev 2004;18:1088–1105.
17 Seale P, Sabourin LA, Girgis-Gabardo Aet al. Pax7 is required for the specification ofmyogenic satellite cells. Cell 2000;102:777–786.18 Colnot C, Thompson Z, Miclau T et al.Altered fracture repair in the absence ofMMP9. Development 2003;130:4123–4133.19 Wang X, Yu YY, Lieu S et al. MMP9 regu-lates the cellular response to inflammationafter skeletal injury. Bone 2013;52:111–119.20 Abou-Khalil R, Yang F, Mortreux M et al.Delayed bone regeneration is linked tochronic inflammation in murine musculardystrophy. J Bone Miner Res 2014;29:304–315.21 Glass GE, Chan JK, Freidin A et al. TNF-alpha promotes fracture repair by augment-ing the recruitment and differentiation ofmuscle-derived stromal cells. Proc Natl AcadSci USA 2011;108:1585–1590.22 Zhang X et al. Periosteal progenitor cellfate in segmental cortical bone graft trans-plantations: Implications for functional tissueengineering. J Bone Miner Res 2005;20:2124–2137.23 Colnot C. Skeletal cell fate decisionswithin periosteum and bone marrow duringbone regeneration. J Bone Miner Res 2009;24:274–282.24 Colnot C, Zhang X, Knothe Tate ML. Cur-rent insights on the regenerative potential ofthe periosteum: Molecular, cellular, andendogenous engineering approaches.J Orthop Res 2012;30:1869–1878.
1510 Muscle Stem Cells in Bone Repair
VC AlphaMed Press 2015 STEM CELLS

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
115
25 Liu R, Birke O, Morse A et al. Myogenicprogenitors contribute to open but notclosed fracture repair. BMC MusculoskeletDisord 2011;12:288.26 Wright V, Peng H, Usas A et al. BMP4-expressing muscle-derived stem cells differ-entiate into osteogenic lineage and improvebone healing in immunocompetent mice.Mol Ther 2002;6:169–178.27 Bosch P, Musgrave DS, Lee JY et al.Osteoprogenitor cells within skeletal muscle.J Orthop Res 2000;18:933–944.28 Asakura A, Komaki M, Rudnicki M. Mus-cle satellite cells are multipotential stem cellsthat exhibit myogenic, osteogenic, and adipo-genic differentiation. Differentiation 2001;68:245–253.29 Morrison JI, Loof S, He P et al. Salaman-der limb regeneration involves the activationof a multipotent skeletal muscle satellite cellpopulation. J Cell Biol 2006;172:433–440.30 Cairns DM, Liu R, Sen M et al. Interplayof Nkx3.2, Sox9 and Pax3 regulates chondro-genic differentiation of muscle progenitorcells. PLoS One 2012;7:e39642.31 Tsuji K, Bandyopadhyay A, Harfe BDet al. BMP2 activity, although dispensable forbone formation, is required for the initiationof fracture healing. Nat Genet 2006;38:1424–1429.32 Rosen V. Harnessing the parathyroidhormone, Wnt, and bone morphogeneticprotein signaling cascades for successfulbone tissue engineering. Tissue Eng Part BRev 2011;17:475–479.33 Yu YY, Lieu S, Lu C et al. Immunolocali-zation of BMPs, BMP antagonists, receptors,
and effectors during fracture repair. Bone2010;46:841–851.34 Hamrick MW, McNeil PL, Patterson SL.Role of muscle-derived growth factors inbone formation. J Musculoskelet NeuronalInteract 2010;10:64–70.35 Elkasrawy M, Immel D, Wen X et al.Immunolocalization of myostatin (GDF-8) fol-lowing musculoskeletal injury and the effectsof exogenous myostatin on muscle and bonehealing. J Histochem Cytochem 2012;60:22–30.36 Okabe M, Ikawa M, Kominami K et al.‘Green mice’ as a source of ubiquitous greencells. FEBS Lett 1997;407:313–319.37 Lepper C, Conway SJ, Fan CM. Adult sat-ellite cells and embryonic muscle progenitorshave distinct genetic requirements. Nature2009;460:627–631.38 Lepper C, Partridge TA, Fan CM. An abso-lute requirement for Pax7-positive satellitecells in acute injury-induced skeletal muscleregeneration. Development 2011;138:3639–3646.39 Yu YY, Lieu S, Lu C et al. Bone morpho-genetic protein 2 stimulates endochondralossification by regulating periosteal cell fateduring bone repair. Bone 2010;47:65–73.40 Lu C, Saless N, Hu D et al. Mechanicalstability affects angiogenesis during earlyfracture healing. J Orthop Trauma 2011;25:494–499.41 Howard V, Reed M. Unbiased Stereol-ogy, Three-dimensional Measurement inMicroscopy. NY: Bios Scientific PublishersLimited, 1998.
42 Grounds MD, Partridge TA. Isoenzymestudies of whole muscle grafts and move-ment of muscle precursor cells. Cell TissueRes 1983;230:677–688.43 Brack AS, Conboy MJ, Roy S et al.Increased Wnt signaling during aging altersmuscle stem cell fate and increases fibrosis.Science 2007;317:807–810.44 Bensaid W, Triffit JT, Blanchat C et al. Abiodegradable fibrin scaffold for mesenchy-mal stem cell transplantation. Biomaterials2003;24:2497–2502.45 Colnot C, Huang S, Helms J. Analyzingthe cellular contribution of bone marrow tofracture healing using bone marrow trans-plantation in mice. Biochem Biophys ResCommun 2006;350:557–561.46 Oustanina S, Hause G, Braun T. Pax7directs postnatal renewal and propagation ofmyogenic satellite cells but not their specifi-cation. EMBO J 2004;23:3430–3439.47 Kuang S, Charge SB, Seale P et al. Dis-tinct roles for Pax7 and Pax3 in adult regen-erative myogenesis. J Cell Biol 2006;172:103–113.48 Grcevic D, Pejda S, Matthews BG et al.In vivo fate mapping identifies mesenchymalprogenitor cells. Stem Cells 2012;30:187–196.49 Dimitriou R, Tsiridis E, Giannoudis PV.Current concepts of molecular aspects ofbone healing. Injury 2005;36:1392–1404.50 Lai X, Price C, Lu XL et al. Imaging andquantifying solute transport across perios-teum: Implications for muscle-bone crosstalk.Bone 2014;66:82–89.
See www.StemCells.com for supporting information available online.
Abou-Khalil, Yang, Lieu et al. 1511
www.StemCells.com VC AlphaMed Press 2015

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
116
Supplementary Figures
Role of muscle stem cells during skeletal regeneration
Abou-‐Khalil et al.
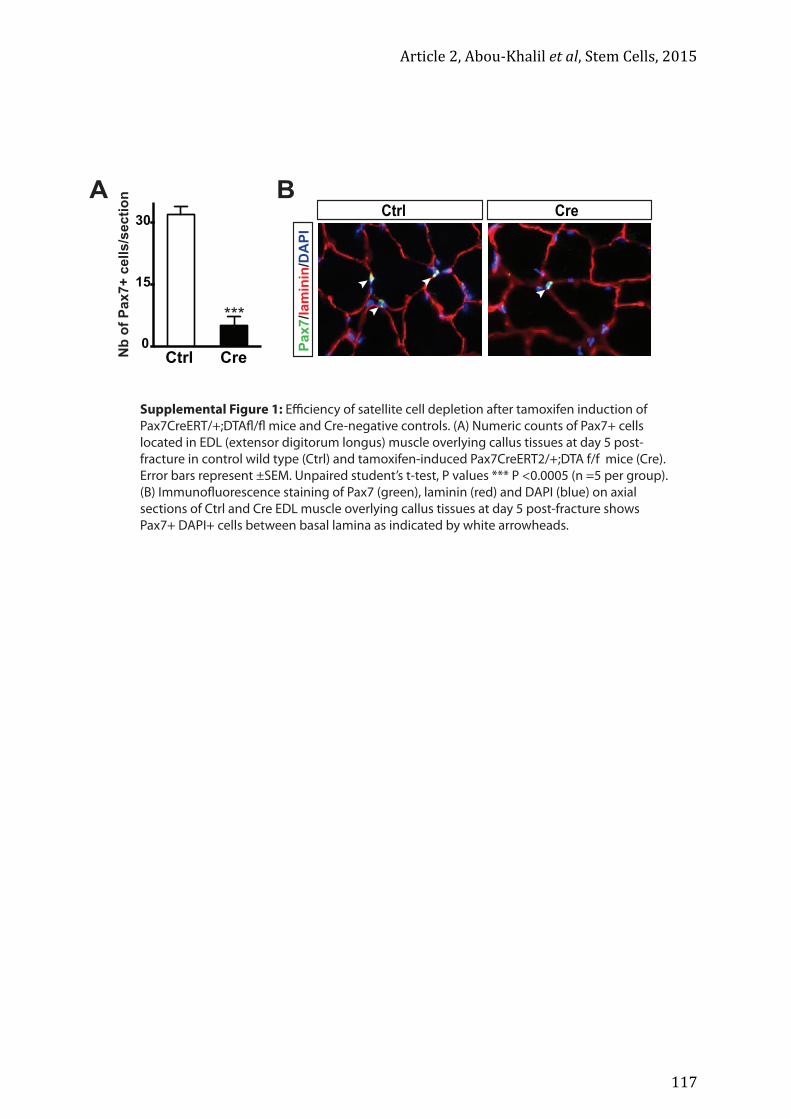
Article 2, Abou-‐Khalil et al, Stem Cells, 2015
117
BA
Ctrl Cre
Pax7
/lam
inin
/DA
PI
Ctrl Cre0
15
30
Nb
of P
ax7+
cel
ls/s
ectio
n
***
Supplemental Figure 1: Efficiency of satellite cell depletion after tamoxifen induction of Pax7CreERT/+;DTAfl/fl mice and Cre-negative controls. (A) Numeric counts of Pax7+ cells located in EDL (extensor digitorum longus) muscle overlying callus tissues at day 5 post-fracture in control wild type (Ctrl) and tamoxifen-induced Pax7CreERT2/+;DTA f/f mice (Cre). Error bars represent ±SEM. Unpaired student’s t-test, P values *** P <0.0005 (n =5 per group). (B) Immunofluorescence staining of Pax7 (green), laminin (red) and DAPI (blue) on axial sections of Ctrl and Cre EDL muscle overlying callus tissues at day 5 post-fracture shows Pax7+ DAPI+ cells between basal lamina as indicated by white arrowheads.

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
118

Article 2, Abou-‐Khalil et al, Stem Cells, 2015
119
Supplemental Figure 3: Endogenous contribution of muscle stem cells during bone regeneration
(A) Safranin-O (SO) staining of longitudinal sections of Pax3Cre/+;R26RLacZ/+ callus tissues at day 14 (d14)
of bone regeneration, dashed line delimitates callus area. (B) XGAL staining of adjacent sections of
Pax3Cre/+;R26RLacZ/+ callus tissues at day 14 (d14) of bone regeneration (area corresponds to dashed box
in SO staining) shows LacZ+ chondrocytes within the callus. (B.1) Representative LacZ+ chondrocytes within
the callus (area corresponds to Box 1 in XGAL staining). (B.2) Representative LacZ+ bone lining cells (area
corresponds to Box 2 in XGAL staining). (C) Representative TRAP staining on adjacent sections of
Pax3Cre/+;R26RLacZ/+ callus tissues at day 14 (d14) of bone regeneration. (C.1) LacZ+ chondrocytes within
callus are TRAP- (area corresponds to Box1 in TRAP staining), while LacZ+ osteoclasts (C.2) are TRAP+
(area corresponds to Box2 in TRAP staining). Mu: muscle; ca: callus; B: bone; Cg: cartilage. Scale bar: 1 mm (A)
XGAL
TRAP
A B
B.2
B.1
C.1
C.2
C
mu
ca
B
B
Cg
Cg
B
mu
ca
1
1
2
2
B

Article 3, Julien et al, En cours de soumission
120

Article 3, Julien et al, En cours de soumission
121
Article 3 (en cours de soumission) Muscle-‐derived profibrotic progenitors impair bone
healing in musculoskeletal trauma
Julien Anais1 , Kanagalingam Anuya1 , Megret Jérome2 , Relaix Frédéric3 and Colnot
Céline1,*
1 INSERM U1163, Imagine Institute, Paris Descartes University, 75015, Paris, France
2 INSERM US24 -‐ CNRS UMS3633 Cytometry Platform, Paris Descartes University,
75015, Paris, France
3 INSERM IMRB U955, Paris Est-‐Créteil University, 94000, Créteil, France
*Correspondence: [email protected]

Article 3, Julien et al, En cours de soumission
122

Article 3, Julien et al, En cours de soumission
123
ABSTRACT
Tissue regeneration relies on resident stem cells that are generally activated locally
within the injured tissue or organ. Failure to regenerate after injury can be associated
with abnormal stem cell function, inflammation and vascularization, as well as fibrous
tissue accumulation. The origin of fibrosis is a main concern in regenerative medicine
and chronic diseases as it interferes with tissue regeneration. In the musculoskeletal
system, the regenerative process depends on tissue-‐specific stem cells residing in
muscle and bone, i.e. satellite cells and periosteal cells respectively. Here we show that
bone injury leads to the recruitment of skeletal stem/progenitor cells not only from the
periosteum but also from adjacent skeletal muscles. Lineage tracing experiments reveal
that skeletal stem/progenitor cells within bone and muscle are derived from a common
mesenchymal lineage, marked by Prx1 and PDGFRα, and cooperate to repair bone in
adult. In a mouse model of bone fracture combined with muscle injury mimicking
musculoskeletal trauma in human, skeletal stem/progenitor cells within periosteum and
muscle are compromised leading to impaired bone healing. Absence of fracture
consolidation in this poly-‐trauma model is characterized by the accumulation of fibrous
tissue produced by muscle interstitial cells also derived from the Prx1 lineage and
overlapping with fibro-‐adipoprogenitors. Inhibition of PDGFRα signalling with
Imatinib® in this mouse model improves fibrotic remodelling and bone repair. In sum,
the results reveal a supporting role of skeletal muscle as a source of skeletal
stem/progenitor cells during bone repair and uncover skeletal muscle as the source of
fracture callus fibrosis in musculoskeletal trauma. This dual role of skeletal muscle in
bone regeneration suggests new pharmacological and cell-‐based approaches to treat
musculoskeletal trauma.

Article 3, Julien et al, En cours de soumission
124

Article 3, Julien et al, En cours de soumission
125
INTRODUCTION
The hallmark of tissue regeneration is the recruitment of tissue resident stem cells
concomitant with a controlled inflammatory response and transient fibrous tissue
deposition1,2. These initial steps of regeneration are then followed by new tissue
formation that necessitates a resolving phase of inflammation and removal of fibrotic
cells and associated extra-‐cellular matrix. This regenerative process can be
compromised in severe trauma and in pathological conditions such as chronic
inflammatory and fibrotic diseases including cardiovascular diseases, kidney diseases
and muscular dystrophy. Deregulated inflammation, unresorbed fibrous tissue together
with deficient stem cell function prevents functional recovery of the tissue3,4.
Bone regeneration is usually described as a scare-‐less and efficient regenerative process,
involving skeletal stem cells producing cartilage and bone that are then slowly
remodelled to reconstitute the initial shape and function of the injured bone. The origin
and role of fibrosis in this process is unknown although fibrous tissue has been reported
clinically in trauma patients5. Moreover, the nature of the skeletal stem cells and their
tissue origins is still not entirely elucidated as we lack tissue-‐specific markers. Although
bone marrow has long been studied as the source of skeletal progenitors for bone,
recent advances have shown that periosteum lying at the outer surface of bone is a
major source of skeletal stem/progenitors during endogenous repair6-‐10. Other reports
have also pointed at the contribution of surrounding tissues such as muscle11-‐14. It is
known clinically that an intact muscle around bone is required for bone repair as soft
tissue damage can severely impair bone healing, but the role of muscle in the context of
musculoskeletal trauma is not understood15-‐17.
Bone-‐muscle interactions are essential in bone physiology. Skeletal muscle is the source
of mechanical signals regulating bone development and contributing to bone loss
associated with aging18,19. Skeletal muscle is also the source of paracrine signals in bone
homeostasis and repair14,20. The presence of bone forming cells in muscle has also been
suspected since Urist first showed that bone formation can be induced within muscle21.
Given the close physical interactions between muscle and bone, traumatic bone injury
necessarily leads to concomitant injury to the adjacent skeletal muscle. Following

Article 3, Julien et al, En cours de soumission
126
muscle injury, muscle resident stem cells, the satellite cells, generate new muscle fibers
while reconstituting a pool of stem cells22. In parallel, a population of muscle interstitial
cells, the fibro/adipogenic progenitors, also expands, allowing satellite cell activation,
and then disappears to allow muscle regeneration to proceed23-‐25. Muscle regeneration
and the cross talk with the adjacent regenerating bone after musculoskeletal trauma
have never been investigated due, in part, to the lack of appropriate experimental
models of traumatic injury. In models of volumetric muscle loss, removal of muscle
tissue causes a complete deficit of muscle repair that normally relies on the presence of
satellite cell and basal lamina preventing the assessment of muscle cells in the bone
repair process26,27. Here, we report a new musculoskeletal trauma model, combining
fracture and mechanical injury to muscles surrounding the fracture site, in which muscle
and bone regeneration are impaired. Using cell lineage tracing in genetic mouse models
and tissue grafting, we show that osteochondroprogenitors are recruited from muscle
during bone repair in addition to cells recruited from the periosteum. In the trauma
environment, osteochondroprogenitors from muscle and periosteum are both
compromised and fibrogenic progenitors coming from muscle adjacent to the fracture
cause fibrosis accumulating in the callus. These skeletal muscle-‐derived
osteochondroprogenitors and profibrotic progenitors overlap with the muscle
fibro/adipogenic progenitor population marked by PDGFRa. Treatment with Imatinib
ameliorates fibrotic remodelling and bone repair after musculoskeletal trauma.

Article 3, Julien et al, En cours de soumission
127
RESULTS
Periosteum and muscle cooperate to repair bone but are compromised after
trauma
To assess the coordinated cellular and molecular events of muscle and bone healing, we
developed a clinically relevant mouse model of musculoskeletal trauma. In this model,
bone fracture is induced in adult mouse tibia and combined with mechanical injury to
surrounding skeletal muscles to mimic soft tissue trauma (Fig 1). While centronucleated
fibers are present in the entire regenerating muscle after CTX or BaCl2 injections28, the
mechanical injury leads to heterogeneous and delayed muscle regeneration as shown by
areas containing regenerating muscle fibers and areas containing fibrous tissue at days
14 and 30 post-‐injury (Fig. 1A). When combined with tibial fracture, injury to muscles
surrounding the tibia impairs bone healing marked by a delay in callus, cartilage and
bone formation by day 7 post-‐injury, followed by a delay in cartilage resorption and
bone remodelling (Fig. 1B). Histological analyses reveal a complete disorganization of
the callus with the presence of unresorbed cartilage islands, the presence of fibrosis and
an absence of bone bridging by day 21. By day 56, bone bridging was still not observed
confirming the fracture non-‐union (Fig. 1C). Bone healing was not impaired when only
the tibialis anterior (TA) muscle was injured indicating that the proximity of an intact
muscle is essential for bone repair and that a threshold of soft tissue trauma exists
above which bone healing cannot occur efficiently (Supplementary Fig 1).
To evaluate the impact of musculoskeletal trauma on cells residing within periosteum
and muscle, we traced GFP labelled periosteum-‐derived cells and Tomato labelled
muscle-‐derived cells after tissue grafting at the fracture site of wild type mice (Fig 2A).
In mice with fracture alone, concomitant recruitment of cells from muscle and
periosteum occurs in the callus with both GFP-‐positive and Tomato-‐positive cells found
in cartilage and bone (Fig. 2B-‐C). To confirm the physiological role of muscle, we
induced the fracture one month after muscle transplant to allow muscle graft
regeneration and observed again donor muscle-‐derived cells in cartilage and bone (Fig
2D-‐F). When muscle or periosteum was transplanted in the poly-‐trauma environment,
the cellular contribution to cartilage was decreased (Fig 2G-‐H). Further, muscle injury
did not impact healing via intramembranous ossification, which is correlated with the

Article 3, Julien et al, En cours de soumission
128
absence of cellular contribution of muscle in this process (Supplementary Fig 2). Muscle
and periosteum thus cooperate by providing a source of cells to support the process of
endochondral ossification, which is a predominant mode of tissue repair after fracture.
Combined trauma to muscle and bone reduces this coordinated contribution of muscle
and periosteum to bone healing leading the absence of fracture consolidation.
Periosteum and muscle cooperate to repair bone but are compromised after
trauma
To assess the coordinated cellular and molecular events of muscle and bone healing, we
developed a clinically relevant mouse model of musculoskeletal trauma. In this model,
bone fracture is induced in adult mouse tibia and combined with mechanical injury to
surrounding skeletal muscles to mimic soft tissue trauma (Fig 1). While centronucleated
fibers are present in the entire regenerating muscle after CTX or BaCl2 injections28, the
mechanical injury leads to heterogeneous and delayed muscle regeneration as shown by
areas containing regenerating muscle fibers and areas containing fibrous tissue at days
14 and 30 post-‐injury (Fig. 1A). When combined with tibial fracture, injury to muscles
surrounding the tibia impairs bone healing marked by a delay in callus, cartilage and
bone formation by day 7 post-‐injury, followed by a delay in cartilage resorption and
bone remodelling (Fig. 1B). Histological analyses reveal a complete disorganization of
the callus with the presence of unresorbed cartilage islands, the presence of fibrosis and
an absence of bone bridging by day 21. By day 56, bone bridging was still not observed
confirming the fracture non-‐union (Fig. 1C). Bone healing was not impaired when only
the tibialis anterior (TA) muscle was injured indicating that the proximity of an intact
muscle is essential for bone repair and that a threshold of soft tissue trauma exists
above which bone healing cannot occur efficiently (Supplementary Fig 1).
To evaluate the impact of musculoskeletal trauma on cells residing within periosteum
and muscle, we traced GFP labelled periosteum-‐derived cells and Tomato labelled
muscle-‐derived cells after tissue grafting at the fracture site of wild type mice (Fig 2A).
In mice with fracture alone, concomitant recruitment of cells from muscle and
periosteum occurs in the callus with both GFP-‐positive and Tomato-‐positive cells found
in cartilage and bone (Fig. 2B-‐C). To confirm the physiological role of muscle, we
induced the fracture one month after muscle transplant to allow muscle graft

Article 3, Julien et al, En cours de soumission
129
regeneration and observed again donor muscle-‐derived cells in cartilage and bone (Fig
2D-‐F). When muscle or periosteum was transplanted in the poly-‐trauma environment,
the cellular contribution to cartilage was decreased (Fig 2G-‐H). Further, muscle injury
did not impact healing via intramembranous ossification, which is correlated with the
absence of cellular contribution of muscle in this process (Supplementary Fig 2). Muscle
and periosteum thus cooperate by providing a source of cells to support the process of
endochondral ossification, which is a predominant mode of tissue repair after fracture.
Combined trauma to muscle and bone reduces this coordinated contribution of muscle
and periosteum to bone healing leading the absence of fracture consolidation.
Common mesenchymal lineages in muscle and periosteum repair bone
To characterize the muscle cell population recruited during bone repair, we performed
genetic lineage tracing using Prx1Cre;mTmG and PdgfrαCreERT;mTmG mouse lines. Prx1
and Pdgfra are markers of bone marrow stromal cells and periosteal cells10,29,30. Pdgfrα
has also been shown to label a population of mesenchymal progenitors in the muscle
interstitium called fibro/adipogenic progenitors (FAP)24. All skeletal stem/progenitors
giving rise to cartilage and bone are derived from the Prx1 lineage in the fracture callus
of Prx1Cre;mTmG mice (Fig 3A and 10) whereas 60% of cells were derived from the
Pdgfrα lineage in tamoxifen-‐induced PdgfrαCreERT2;mTmG mice (Fig. 3A). EDL and
periosteum grafts from Prx1Cre;mTmG donors transplanted at the fracture site of wild
type host give rise to chondrocytes and osteoblasts/osteocytes strictly derived from the
Prx1 lineage in the callus (Fig 3B-‐C). EDL and periosteum grafts from tamoxifen-‐induced
PdgfrαCreERT2;mTmG mice provide GFP-‐positive Pdgfrα-‐derived and Tomato-‐positive
non-‐Pdgfrα-‐derived chondrocytes and osteoblasts/osteocytes, indicating that PDGFRα
does not label all skeletal progenitors due to the 60% recombination efficiency of this
inducible Cre line (data not shown). To confirm that muscle tissue itself contains Prx1-‐
labelled skeletal stem/progenitors, we isolated muscle cells from Prx1Cre;mTmG mice
after removing fascia, tendon and fat. Transplanted Prx1-‐derived muscle cells integrate
into cartilage and bone while non-‐Prx1-‐derived cells did not (Fig 3D, E). Therefore,
periosteum and muscle contain Prx1-‐derived skeletal stem/progenitors that participate
in bone repair and that are also marked by Pdgfrα. We previously observed that freshly
isolated Pax7+ muscle stem cells can give rise to rare chondrocytes when transplanted

Article 3, Julien et al, En cours de soumission
130
at the fracture site14. However, the physiological contribution of muscle stem cells was
not detected when we performed fracture in Pax7CreERT2;mTmG mice or transplanted
Pax7CreERT2;mTmG EDL grafts at the fracture site of wild type mice, even when the
fracture was combined with muscle injury, thus excluding endogenous contribution
from the myogenic lineage itself (Supplementary Figure 3).
Prx1-‐derived muscle interstitial cells comprise osteo-‐chondrogenic and fibro-‐
adipogenic progenitors
In intact Prx1Cre;mTmG TA muscle, Prx1-‐derived GFP-‐labelled cells are localized in the
muscle interstitium next to capillaries and are distinct from CD31-‐positive endothelial
cells, αSMA-‐positive vascular smooth muscle cells. Prx1-‐derived muscle cells also
express the markers CD29 and PDGFRα, and some of them are positive for the pericyte
markers NG2 and PDGFRβ (Fig 4A). FACs analyses of freshly isolated cells from intact
muscles surrounding the tibia in Prx1Cre ;YFPfl/+ mice show that Prx1-‐derived cells
represent 32% of mononucleated cells within muscle and are CD45-‐CD11b-‐CD31-‐ (CDs-‐).
Some Prx1-‐derived muscle cells are CD34+ and α7integrin+ and the majority are
PDGFRα+, Sca1+ and CD29+ (Fig 4B). Prx1-‐derived muscle cells thus overlap with
osteochondroprogenitors (OCP) defined as CDs-‐Sca1+CD34-‐ cells and fibro-‐adipogenic
progenitors (FAP) defined as CDs-‐Sca1+CD34+ cells 10,25,31,32. Double positive
PDGFRα+/Prx1-‐derived YFP+ cells are found in both OCPs and FAPs populations (Fig
4C). RT-‐qPCR analysis of Prx1-‐derived YFP+ muscle cells at P1 show expression of CD34,
fibro-‐mesenchymal markers Cxcl12, Gremlin, Mx1, Pdgfrα, Nestin, Leptin Receptor, PW1,
Tcf4, αSMA and Vimentin, and pericyte markers NG2 and Pdfgrβ . We detected
expression of the tendon markers Tenomodulin (Tnmd) and Tenasin C (TnC) but not
Scleraxis (scx). The satellite cell marker Pax7 was not detected excluding any overlap
between the Prx1-‐derived lineage and the myogenic lineage (Fig. 4D). CFU-‐F assay
shows that Prx1-‐derived muscle cells exhibit higher clonogenicity than non-‐Prx1
derived muscle cells (Fig 4E). In vitro analyses further show that Prx1-‐derived muscle
cells can differentiate into osteogenic, adipogenic, chondrogenic and fibrogenic lineages
but fail to achieve myogenic differentiation (Fig 4F).

Article 3, Julien et al, En cours de soumission
131
Muscle is the source of callus fibrosis in musculoskeletal trauma
In response to a bone fracture, we observed expansion of the Prx1+ cell population
within muscle around the tibia at day 3 and a decrease by day 21 around the callus. In
the poly-‐trauma environment, the Prx1+ cell population expands largely throughout
injured muscles and persists within muscles surrounding the callus by day 21 (Fig 5A).
The expansion of the Prx1+ cell population coincides with the progression of callus
fibrosis, which is detected by day 7 after fracture and decreases rapidly during the
course of fracture repair but persists after musculoskeletal trauma (Fig 5B,
Supplementary Figure 4). The fibrous tissue in the fracture callus is also composed of
cells derived from the Prx1 lineage, and expresses the fibrotic markers Periostin and
PDGFRα (Fig 5C)33,34. Lineage analysis of muscle and periosteum-‐derived cells in the
poly-‐trauma environment showed that Prx1Cre;mTmG EDL grafts give rise to bone and
fibrous tissue in the callus of wild type hosts, while periosteum grafts only give rise to
bone by day 21(Fig 5D). To attenuate fibrosis, we treated mice with the clinically
approved tyrosine kinase inhibitor Imatinib that inhibits receptor phosphorylation
including PDGFRα35,36. Imatinib® treatment had no effect on bone repair at day 7 post-‐
fracture combined with muscle injury. However, by day 21 post-‐fracture combined with
muscle injury, mice injected with Imatinib® exhibit an improved bone repair phenotype
with decreased cartilage, bone and fibrosis volumes compared to control mice treated
with PBS (Fig. 5E, F). Prx1-‐derived cells within muscle surrounding the fracture site are
therefore the origin of callus fibrosis that can be targeted pharmacologically to improve
bone repair after musculoskeletal trauma.

Article 3, Julien et al, En cours de soumission
132

Article 3, Julien et al, En cours de soumission
133
DISCUSSION
The role of muscle during bone regeneration is well recognized clinically but the
underlying cellular and molecular mechanisms are poorly understood37. In the absence
of a relevant animal model to study muscle-‐bone interactions during skeletal
regeneration, we developed a model of bone fracture combined with a mechanical injury
of skeletal muscle. As observed in human, muscle injury in this model leads to fracture
non-‐union displayed by delayed callus formation, abnormal replacement of cartilage by
bone and absence of bone bridging. This phenotype is correlated with impaired
contribution to repair of periosteum-‐ and muscle-‐derived skeletal stem/progenitor cells
that cooperate to support endochondral ossification and fracture consolidation. In the
context of poly-‐trauma this coordinated regenerative process mediated by periosteum
and skeletal muscles is disrupted, and skeletal muscle adjacent to the fracture callus
becomes the source of persistent callus fibrosis compromising the repair process.
Although previous reports have suggested a potential contribution of muscle only after
periosteum stripping13, we clearly establish a direct role of muscle both in the
physiological response to bone facture and in trauma as a source of skeletal
stem/progenitor cells. The role of the myogenic lineage has been suggested previously
due to the ability of muscle stem cells to differentiate into osteoblasts and chondrocytes
in vitro and following in vivo transplantation14,38. Muscle contribution to fracture
healing has also been observed using the myogenic-‐specific Cre lines such as MyoD-‐Cre
and Pax3-‐Cre although the contribution was negligible in Pax3-‐Cre line13,14. Our results
based on the Prx1-‐Cre and Pax7-‐CreERT lines indicate that the endogenous contribution
of the myogenic lineage does not occur physiologically or in response to poly-‐trauma.
Instead, we identify a significant contribution from the interstitial compartment of the
muscle to bone regeneration.
The findings reveal that a unique characteristic of the skeletal regeneration process is
the multiple tissue origins of skeletal stem/progenitor cells residing in adjacent tissues
(bone marrow, periosteum, skeletal muscle) and recruited to form the fracture callus.
The Prx1-‐derived muscle cells share common features with periosteal cells (PCs) and
BMSCs such as their in vitro differentiation potential, bone repair contribution to
cartilage and bone, and marker profiles. These results suggest the therapeutic potential

Article 3, Julien et al, En cours de soumission
134
of muscle-‐derived cells for bone repair. Previous reports showed that non-‐skeletal
mesenchymal stromal cells (MSCs) expressing CD146 in human skeletal muscle do not
have chondro-‐osteogenic properties but instead myogenic potential in vitro31. Our
results show that Prx1-‐derived and PDGFRα-‐positive muscle interstitial cells
physiologically form cartilage and bone in the fracture callus and other studies have also
highlighted the involvement PDGFRα-‐positive muscle cells in heterotopic ossification39.
However, we never observed intramuscular ossification in the musculoskeletal trauma
model. Further, we show that the Prx1-‐derived muscle interstitial cells do not have
myogenic potential both in vitro and in vivo, and are derived from the same lineage as
BMSCs and PCs10. The bone regenerative potential of muscle interstitial cells is thus
defined by their embryonic origin, not by their tissue of origin. Currently, there are no
known markers available to isolate tissue-‐specific MSCs, discriminate their tissue
origins and track their fate in vivo 40-‐44. Some markers have been identified for BMSCs as
“skeletal stem cell (SSC) markers”40-‐48. However, these markers seem to define various
sub-‐populations and are also expressed in PCs10 and in muscle-‐derived MSCs as shown
in this study. Hence, our experimental model combining genetic lineage tracing and
tissue grafting is a powerful tool to differentiate the contribution of the various sources
of cells and their functions in musculoskeletal regeneration.
Although muscle interstitial cells contributing to bone repair share common
characteristics with bone marrow-‐derived and periosteum-‐derived cells, they also have
a fibrogenic potential not observed for periosteal cells and cause callus fibrosis in the
polytrauma environment. Fibrosis is a dynamic process that is common to many tissue
regeneration processes. Fibrotic tissue supports activation and proliferation of tissue
resident stem/progenitor cells and has to be remodelled to allow stem/progenitors cells
differentiate and achieve regeneration1,49. The dynamic of fibrosis has never been
studied in bone repair. We show that callus fibrosis follows the same pattern as in other
tissues and that impaired fibrotic remodelling leads to absence of bone repair. Targeting
fibrotic cells is essential to develop treatments modulating fibrotic remodelling but the
cellular origin of fibrotic cells remains elusive in many organs. We show that Prx1-‐
derived OCP but also fibrogenic cells within skeletal muscle are peri-‐vascular as
previously reported for cells causing fibrosis in other tissues, and they are distinct from
endothelial cells and myogenic cells50-‐54. The population of mesenchymal stromal

Article 3, Julien et al, En cours de soumission
135
cells/myofibroblasts responsible for fibrosis in various tissues such as kidney, lung,
heart, skin, skeletal muscle or bone marrow has been described as being highly
heterogeneous23,52,53,55-‐59. Over the past years several markers such as PDGFRα, Gli1 or
PDGFRβ have been used to identify pro-‐fibrogenic cells in various tissues1. In skeletal
muscle, fibro-‐adipogenic progenitors (FAPs) have been described as the major source of
pro-‐fibrotic cells24,25. Here, we show that FAP and osteo-‐chondrogenic progenitor (OCP)
populations within skeletal muscle overlap. This result uncovers new functions of FAPs
as a plastic population, which adapts its fate according to the environment.
This work thus highlights added complexity and cellular heterogeneity in
musculoskeletal regeneration, as it involves resident skeletal stem/progenitor cells
within bone as well as interstitial cells recruited from the adjacent muscle that can
support repair or have a negative impact when triggered down the fibrogenic pathway.
Several molecular therapies have been developed to treat fibrosis and many of them are
currently in clinics as Imatinib®, an inhibitor of PDGFR, Bcr-‐abl and c-‐kit signalling
pathways60,61 62. Due to the implication of PDGFRα-‐positive cells in skeletal muscle
fibrosis, dystrophic mice have been treated with Imatinib® and exhibit a decrease in
skeletal muscle fibrosis. In our model of musculoskeletal trauma, daily administration of
Imatinib® ameliorates the late stages of bone repair post-‐trauma but does not affect
early steps of regeneration. Imatinib® or other related drugs may offer new strategies in
orthopaedics to enhance bone regeneration.
In conclusion, our results brings new knowledge on the role muscle plays during bone
repair, and how soft tissues and specifically muscle-‐derived cells may be used or
targeted to improve bone repair in patients. We identify muscle interstitial cells marked
by Prx1-‐lineage and PDGFRα that participate directly in cartilage and bone formation
during fracture repair but fail to do so after traumatic injury to become the source of
fibrosis causing fracture non-‐union. This study provides a cellular basis for delayed bone
regeneration in trauma and suggests new cell-‐based and drug-‐based strategies to treat
patients affected by musculoskeletal injuries.

Article 3, Julien et al, En cours de soumission
136
METHODS
Mice
C57BL/6ScNj, beta-‐actin GFP, Prx1Cre/+, PdgfrαCreERT/+, Pax7CreERT2/+, Rosa-‐tdTomato-‐EGFP
(mTmG), RosaYFP and RosaDTA/+ mice were obtained from Jackson Laboratory (Bar
Harbor, ME). All primers for PCR genotyping were purchased from Eurofins Scientific,
France (Supplementary Table 1). All procedures were approved by the Paris Descartes
University Ethical Committee. For inducible Cre recombination, Tamoxifen (TMX, ref
T5648, Sigma) was prepared at a concentration of 10mg/mL diluted in corn oil and
heated at 60°C for 2h. TMX (300μL) was injected intraperitoneally 24h before fracture,
at the time of fracture and 24h post-‐fracture when using PdgfrαCreERT/+, and daily for
three consecutives day one week prior surgery when using Pax7CreERT2/+.
Tibial fractures and Imatinib treatment
For all surgical procedures, mice were anesthetized with an intraperitoneal injection of
Ketamine (50mg/mL) and Metedomidine (1mg/kg) and received a subcutaneous
injection of Buprenorphine (0.1mg/kg) for analgesia. Mice were revived with an
intraperitoneal injection of atipamezole (1mg/mL) and allowed to ambulate freely. Mice
received post-‐operative doses of analgesia and were monitored daily. As described
previously, open non-‐stabilized tibial fractures were produced by osteotomy in the mid-‐
diaphysis after exposing the tibial surface10. For Imatinib treatment, mice received daily
intraperitoneal injections of Imatinib® (50mg/kg/day, ref STI571, Selleckchem) or
vehicle (PBS) from the day of fracture until sacrifice.
Muscle injury
Under anaesthesia, a skin incision was made over the anterior-‐proximal surface of the
right tibia of ten to 12-‐week-‐old wild type mice. Muscles, including tibialis anterior (TA),
tibialis posterior, extensor digitorum longus (EDL), soleus, gastrocnemius muscles,
surrounding the tibia were compressed along their entire length using a hemostat in a
standardized and reproducible procedure. Each compression was applied for five
seconds. Mice were revived as described above and received soft food to facilitate
recovery.
EDL muscle and periosteum transplantation

Article 3, Julien et al, En cours de soumission
137
Donor mice were sacrificed by cervical dislocation. EDL-‐muscle was dissected from
tendon to tendon and transplanted adjacent to the fractured tibia as previously
described14. When fracture was induced one month after EDL muscle grafting, the tibia
was exposed as described above to perform osteotomy without affecting the grafted
muscle. Periosteal grafts were collected from donor mice and transplanted at the site of
fracture as previously described6,10.
Cells transplantation
Open fracture was performed as described above. 150 000 freshly sorted muscle cells
were embedded in TissuCol® kit TISSEEL (human fibrogen 15mg/mL and thrombin
9mg/mL, Baxter, France) and were transplanted at the fracture site as described in10,14.
Sample processing, histology and histomorphometry
Mice were sacrificed by cervical dislocation and fractured tibias were harvested at days
7, 14, 21, 28 or 56 post-‐surgery. Samples were fixed 24 hours in 4% PFA (ref 15714,
Euromedex) and decalcified in 19% EDTA (pH 7.4) (ref EU00084, Euromedex) for 21
days at 4°C. Samples were embedded in paraffin or cryopreserved to allow detection of
fluorescent reporters. For cryosections, samples were incubated in sucrose 30% at 4°C
over night, then embedded in OCT (ref F/62550-‐1, MMFrance) and stored at -‐80°C.
Serials sections were collected throughout the entire callus and histomorphometric
analysis performed on Safranin’o (SO), modified Massons’ Trichrome (TC) or Picrosirius
(PS) stained sections using a Zeiss Imager D1 AX10 light microscope and ZEN software
(Carl Zeiss Microscopy GmbH)63. Muscle tibialis anterior (TA) samples were harvested
at specific time point, fixed for 3 hours in PFA 4%, incubated in sucrose 30% for 2 hours
and embedded in OCT for cryosection.
Immunofluorescence
Anti-‐GFP (1/1000, ref ab13970 Abcam) and anti-‐Periostin (1/400, ref AF2955 R&D)
immunofluorescence was performed as described in10,14. For anti-‐αSMA
immunofluorescence, muscle cryosections were rehydrated in PBS for 5min, blocked in
goat anti-‐mouse IgG fragment diluted in PBS (1/100, ref 115-‐007-‐003, Jackson Immuno
Research) and incubated over night at 4°C with mouse anti-‐mouse αSMA (1/200, A2547
Sigma). Slides were rinsed for 3x5min in PBS 1x, incubated for 1hour with anti-‐mouse

Article 3, Julien et al, En cours de soumission
138
AF647 (1/1000, ref A21236 Life Technologies) and mounted with Fluoromount with
DAPI.
For anti-‐NG2, anti-‐PDGFRα and anti-‐CD31 immunofluorescences, muscle cryosections
were rehydrated in PBS for 5min, post-‐fixed in PFA 4% for 5min, rinsed 3x5min in 0.5%
PBST, blocked in 5% serum in 0.5%PBST and incubated over night at 4°C with primary
antibody: rabbit anti-‐NG2 (1/50 ref AB5320 Merck), goat anti-‐PDGFRα (1/200, ref
AF1062 R&D), goat anti-‐CD29 (1/100, ref AF2405 R&D) or rat anti-‐CD31 (1/50, ref
533370 Pharmigen). Slides were rinsed in PBS 3x5min and then incubated for 1h at
room temperature in goat anti-‐rabbit (1/250, ref 21245 Life Technologies), donkey anti-‐
goat (1/500, ref ab150135 Abcam) or goat anti-‐rat (1/50, ab6565 Abcam). Slides were
mounted with Fluoromount with DAPI (ref 495952, eBiosciences). For anti-‐CD29
immunofluorescence, muscle cryosections were rehydrated, post-‐fixed in PFA 4% for
10min, washed, permeabilized in PBS-‐Triton 0.25%, blocked in 1% BSA for 15min and
incubated with goat anti-‐mouse CD29 (1/50, ref 026202, R&D) overnight at at 4°C.
Slides were next rinsed and incubated in donkey anti-‐goat AF647 (1/500, ref ab150135
Abcam). Slides were mounted with Fluoromount with DAPI. Pictures were taken using a
Zeiss Imager D1 AX10 light microscope.
For anti-‐αSMA immunocytofluorescence, cells were fixed in PFA 4% for 15min, rinsed in
PBS, permeabilized in PBS-‐Triton 0.25%, blocked in 5% NGS, incubated with anti-‐αSMA-‐
Cy5 (ref AC12-‐0159-‐11, Clinisciences) for 1 hour and mounted with Fluoromount with
DAPI. Pictures were taken using a Zeiss Axio Vertical A1 light microscope.
Primary culture of muscle cells and periosteal cells
For primary culture of muscle cells, 10-‐12 weeks old mice were sacrificed by cervical
dislocation. Skin and fascia were removed. Tibialis anterior (TA), extensor digitus lengus
(EDL), plantaris and soleus muscles surrounding the tibia were dissected from tendon to
tendon. In a petri dish with 1mL of DMEM medium (ref 21063029, Invitrogen), tendon
and fat were removed and muscles were cut in small pieces using scissors. Muscles were
transferred in digesting medium: DMEM (ref 21063029, Invitrogen) with 1% Trypsin
(ref 210234, Roche) and 1% collagenase D (ref 11088866001, Roche) and incubated at
37°C for a minimum of 2 hours to digest all muscles. Every 20 min individualised cells
were removed and transferred into growth media on ice: αMEM (ref 32561029, Life
Technologies) with 1% penicillin-‐streptomycin (P/S) (ref 15140122, Life Technologies),

Article 3, Julien et al, En cours de soumission
139
20% lot-‐selected non-‐heat-‐inactivated foetal bovine serum (ref 10270106, Life
Technologies) and 10ng/ml bFGF (ref 3139-‐FB-‐025/CF, R&D) and digesting medium
was changed. Cells were then filtered through 100μm filters and 40μm filters. Cells were
centrifuged 10min at 1500 rpm and resuspended in 10ml of growth medium.
Primary culture of periosteal cells was performed as previously described10.
Briefly, 4-‐6 week old mice were sacrificed and flushed femurs and tibia were placed in
culture in growth media supplemented with FBS and bFGF as indicated above to allow
PCs to migrate out of the bone. When PCs reached confluence, bones were removed and
cells were trypsinised (ref 25200056, Life Technologies) and replated. PCs were used at
P1 for all experiments. In vitro adipogenesis, chondrogenesis, osteogenesis and CFU-‐F
assays were performed as previously described10. For myogenic differentiation, Prx1-‐
derived muscle cells were plated at 1000 cells per cm2 and induced with myogenic
medium containing F10 (ref 31550-‐02, Life Technologies), 2% horse serum (ref
26050088, Life Technologies) and 1% P/S for 3 days. For fibrogenic differentiation,
Prx1-‐derived muscle cells were grown until subconfluency and induced to fibrogenic
differentiation with DMEM high-‐glucose (ref 10566016, Life Technologies), 10% FBS,
1% P/S and TGF-‐β1 at 1ng/mL (ref T7039, Sigma).
Cell sorting and flow cytometry analyses
For cell sorting, cultured muscle cells and PCs at P1 were trypsinised and resuspended
in growth medium. Cells were centrifuged at 1500 rpm for 10min, resuspended in F10
media (ref 31550-‐023, Life Technologies) and filtered through 40μm filters. Freshly
digested cells were centrifuged after filtering through 40μm filters and resuspended in
F10 media. Sytox blue (1/1000, ref S34857, Thermofischer) was added just before
sorting. Cell sorting was performed on BD FACS Aria II SORP (BD Biosciences). For flow
cytometry analysis, 100 000 cells were incubated with CD31-‐PECy7 (PECAM-‐1, ref
561410 BD Biosciences); CD45-‐PECy7 (leukocyte common antigen, Ly-‐5, ref 552848 BD
Biosciences); CD11b-‐PECy7 (integrin αM chain, ref 552850 BD Biosciences); CD34-‐
AF700 (ref 560518 BD Biosciences); Sca1-‐APC (ref 130-‐102-‐343 Miltenyi Biotec).
Analyses were performed on BD LSR Fortessa SORP (BD Biosciences) and results
analysed using FlowJo, LLC software, version 10.2.

Article 3, Julien et al, En cours de soumission
140
RTqPCR analysis
Cells pellets were freezed at -‐80°C directly trypsinization. RNA extraction was
performed with RNAeasy Kit (ref 74134, Qiagen) following manufacture’s instructions.
Amount of RNA was quantified using NanoDrop 2000 UV-‐Vis Spectrophotometer
(Thermo Scientific). 500μg of RNA was used to synthetized cDNA. RNAs were mixed
with 1μL of oligo(dT)12-‐18 (ref 18418-‐012, Life Technologies) and 1μL 10mMdNTP Mix
(ref 18427-‐013, Life Technologies) and heated at 65°C for 5min and left on ice for 1min.
Next, 4μL 5X First-‐Strand buffer, 1μL 0.1M DTT, 1μL Superscript III RT® (ref 18080-‐
044, Life Technologies) and 1μL RNaseOUT® (ref 10777-‐019, Life Technologies) were
added and incubated at 50°C for 1h. The reaction was inactivated by heating at 70°C for
15min. qPCR mix was composed by 1μL of primers (see sup table), 4μL of RNAse free
H2O, 10μL of SYBR green Master Mix (ref 11744-‐100, Life Technologies) and 5μL of
cDNA and qPCR reactioin was performed using 7300 Real-‐Time PCR System
(Thermofischer Scientific). Mouse Gapdh was used as internal calibrator. qPCR analysis
was done following ΔΔCT methods as previously described10.
Statistical analyses
Statistical significance was determined with two-‐sided Mann-‐Whitney test and reported
in GraphPad Prism v6.0a. P-‐values were determined as follows: *p≤0.05; **p<0.01.
ACKNOWLEDGMENTS
We thank M. Garfa-‐Traoré, N. Goudin, C. Cordier, O. Duchamp de Lageneste and R. Prota
for advice and/or technical assistance. This work was supported by INSERM ATIP-‐
Avenir, FP7 Marie Curie IRG-‐268227 to C.C., ANR-‐13-‐BSV1-‐001-‐01 to C.C. and F.R. and
NIAMS R01 AR057344 and R01 AR072707 to C.C. and T. Miclau

Article 3, Julien et al, En cours de soumission
141
REFERENCES
1. Di Carlo, S.E. & Peduto, L. The perivascular origin of pathological fibroblasts. J Clin Invest 128, 54-‐63 (2018).
2. Pretheeban, T., Lemos, D.R., Paylor, B., Zhang, R.H. & Rossi, F.M. Role of stem/progenitor cells in reparative disorders. Fibrogenesis Tissue Repair 5, 20 (2012).
3. Lemos, D.R. & Duffield, J.S. Tissue-‐resident mesenchymal stromal cells: Implications for tissue-‐specific antifibrotic therapies. Sci Transl Med 10(2018).
4. El Agha, E., et al. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 21, 166-‐177 (2017).
5. Panteli, M., Pountos, I., Jones, E. & Giannoudis, P.V. Biological and molecular profile of fracture non-‐union tissue: current insights. J Cell Mol Med 19, 685-‐713 (2015).
6. Colnot, C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res 24, 274-‐282 (2009).
7. van Gastel, N., et al. Engineering vascularized bone: osteogenic and proangiogenic potential of murine periosteal cells. Stem Cells 30, 2460-‐2471 (2012).
8. Zhang, X., et al. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J Bone Miner Res 20, 2124-‐2137 (2005).
9. Debnath, S., et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133-‐139 (2018).
10. Duchamp de Lageneste, O., et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun 9, 773 (2018).
11. Harry, L.E., et al. Comparison of the healing of open tibial fractures covered with either muscle or fasciocutaneous tissue in a murine model. J Orthop Res 26, 1238-‐1244 (2008).
12. Glass, G.E., et al. TNF-‐alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-‐derived stromal cells. Proc Natl Acad Sci U S A 108, 1585-‐1590 (2011).
13. Liu, R., et al. Myogenic progenitors contribute to open but not closed fracture repair. BMC Musculoskelet Disord 12, 288 (2011).
14. Abou-‐Khalil, R., et al. Role of muscle stem cells during skeletal regeneration. Stem Cells 33, 1501-‐1511 (2015).
15. Willett, K., Al-‐Khateeb, H., Kotnis, R., Bouamra, O. & Lecky, F. Risk of mortality: the relationship with associated injuries and fracture treatment methods in patients with unilateral or bilateral femoral shaft fractures. J Trauma 69, 405-‐410 (2010).
16. Byrd, H.S., Cierny, G., 3rd & Tebbetts, J.B. The management of open tibial fractures with associated soft-‐tissue loss: external pin fixation with early flap coverage. Plastic and reconstructive surgery 68, 73-‐82 (1981).
17. Richards, R.R., McKee, M.D., Paitich, C.B., Anderson, G.I. & Bertoia, J.T. A comparison of the effects of skin coverage and muscle flap coverage on the early strength of union at the site of osteotomy after devascularization of a segment of canine tibia. J Bone Joint Surg Am 73, 1323-‐1330 (1991).

Article 3, Julien et al, En cours de soumission
142
18. Sharir, A., Stern, T., Rot, C., Shahar, R. & Zelzer, E. Muscle force regulates bone shaping for optimal load-‐bearing capacity during embryogenesis. Development 138, 3247-‐3259 (2011).
19. Shwartz, Y., Farkas, Z., Stern, T., Aszodi, A. & Zelzer, E. Muscle contraction controls skeletal morphogenesis through regulation of chondrocyte convergent extension. Dev Biol 370, 154-‐163 (2012).
20. Hamrick, M.W., McNeil, P.L. & Patterson, S.L. Role of muscle-‐derived growth factors in bone formation. J Musculoskelet Neuronal Interact 10, 64-‐70 (2010).
21. Urist, M.R., Silverman, B.F., Buring, K., Dubuc, F.L. & Rosenberg, J.M. The bone induction principle. Clin Orthop 53, 243-‐283 (1967).
22. Relaix, F. & Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139, 2845-‐2856 (2012).
23. Uezumi, A., et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 124, 3654-‐3664 (2011).
24. Uezumi, A., Fukada, S., Yamamoto, N., Takeda, S. & Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12, 143-‐152 (2010).
25. Joe, A.W., et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12, 153-‐163 (2010).
26. Willett, N.J., et al. Attenuated human bone morphogenetic protein-‐2-‐mediated bone regeneration in a rat model of composite bone and muscle injury. Tissue Eng Part C Methods 19, 316-‐325 (2013).
27. Hurtgen, B.J., et al. Autologous minced muscle grafts improve endogenous fracture healing and muscle strength after musculoskeletal trauma. Physiol Rep 5(2017).
28. Hardy, D., et al. Comparative Study of Injury Models for Studying Muscle Regeneration in Mice. PLoS One 11, e0147198 (2016).
29. Morikawa, S., et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206, 2483-‐2496 (2009).
30. Houlihan, D.D., et al. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-‐1 and PDGFR-‐alpha. Nat Protoc 7, 2103-‐2111 (2012).
31. Sacchetti, B., et al. No Identical "Mesenchymal Stem Cells" at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem cell reports 6, 897-‐913 (2016).
32. Boxall, S.A. & Jones, E. Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int 2012, 975871 (2012).
33. Ozdemir, C., et al. Periostin is temporally expressed as an extracellular matrix component in skeletal muscle regeneration and differentiation. Gene 553, 130-‐139 (2014).
34. Sohn, J., Lu, A., Tang, Y., Wang, B. & Huard, J. Activation of non-‐myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum Mol Genet 24, 3814-‐3829 (2015).
35. Huang, P., Zhao, X.S., Fields, M., Ransohoff, R.M. & Zhou, L. Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J 23, 2539-‐2548 (2009).

Article 3, Julien et al, En cours de soumission
143
36. Ito, T., et al. Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-‐marker expression in muscle mesenchymal progenitors. Neuromuscular disorders : NMD 23, 349-‐356 (2013).
37. Chan, J.K., Harry, L., Williams, G. & Nanchahal, J. Soft-‐tissue reconstruction of open fractures of the lower limb: muscle versus fasciocutaneous flaps. Plast Reconstr Surg 130, 284e-‐295e (2012).
38. Cairns, D.M., et al. Interplay of Nkx3.2, Sox9 and Pax3 regulates chondrogenic differentiation of muscle progenitor cells. PLoS One 7, e39642 (2012).
39. Lees-‐Shepard, J.B., et al. Activin-‐dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun 9, 471 (2018).
40. Chan, C.K., et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285-‐298 (2015).
41. Mendez-‐Ferrer, S., et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829-‐834 (2010).
42. Park, D., et al. Endogenous Bone Marrow MSCs Are Dynamic, Fate-‐Restricted Participants in Bone Maintenance and Regeneration. Cell Stem Cell 10, 259-‐272 (2012).
43. Worthley, D.L., et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160, 269-‐284 (2015).
44. Zhou, B.O., Yue, R., Murphy, M.M., Peyer, J.G. & Morrison, S.J. Leptin-‐receptor-‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15, 154-‐168 (2014).
45. Maes, C., et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Developmental cell 19, 329-‐344 (2010).
46. Mizoguchi, T., et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Developmental cell 29, 340-‐349 (2014).
47. Ono, N., et al. Vasculature-‐associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Developmental cell 29, 330-‐339 (2014).
48. Marecic, O., et al. Identification and characterization of an injury-‐induced skeletal progenitor. Proc Natl Acad Sci U S A 112, 9920-‐9925 (2015).
49. Judson, R.N., Zhang, R.H. & Rossi, F.M. Tissue-‐resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J 280, 4100-‐4108 (2013).
50. Birbrair, A., et al. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res 10, 67-‐84 (2013).
51. Birbrair, A., et al. Type-‐1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol 305, C1098-‐1113 (2013).
52. Dias, D.O., et al. Reducing Pericyte-‐Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell 173, 153-‐165 e122 (2018).
53. Kramann, R., et al. Perivascular Gli1+ progenitors are key contributors to injury-‐induced organ fibrosis. Cell Stem Cell 16, 51-‐66 (2015).
54. Soderblom, C., et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci 33, 13882-‐13887 (2013).

Article 3, Julien et al, En cours de soumission
144
55. Epperly, M.W., Guo, H., Gretton, J.E. & Greenberger, J.S. Bone marrow origin of myofibroblasts in irradiation pulmonary fibrosis. Am J Respir Cell Mol Biol 29, 213-‐224 (2003).
56. Kanisicak, O., et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7, 12260 (2016).
57. LeBleu, V.S., et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19, 1047-‐1053 (2013).
58. Rinkevich, Y., et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015).
59. Schneider, R.K., et al. Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 20, 785-‐800 e788 (2017).
60. Tampe, D. & Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat Rev Nephrol 10, 226-‐237 (2014).
61. Joensuu, H., Hohenberger, P. & Corless, C.L. Gastrointestinal stromal tumour. Lancet 382, 973-‐983 (2013).
62. Ardern-‐Holmes, S.L. & North, K.N. Treatment for plexiform neurofibromas in patients with NF1. Lancet Oncol 13, 1175-‐1176 (2012).
63. Abou-‐Khalil, R., et al. Delayed bone regeneration is linked to chronic inflammation in murine muscular dystrophy. J Bone Miner Res 29, 304-‐315 (2014).

Article 3, Julien et al, En cours de soumission
145

Article 3, Julien et al, En cours de soumission
146
Figure 1: Musculoskeletal trauma impairs bone healing. (A) Tibialis anterior muscle sections at 14 and 30 days post-‐injury (upper and lower
panel respectively) stained with Hematoxylin and Eosin (HE) and Picrosirius (PS). High
magnifications show centronucleated myofibers (arrowheads) in the regenerating area
(boxes 1,3) and centronucleated myofibers surrounded by fibrous tissue (asterisks) in
the fibrotic area (boxes 2,4). (B) Histomorphometric quantification of callus, cartilage
and bone volume in tibial fractures with or without muscle injury at days 7, 14, 21, 28
and 56 post-‐fracture. (C) Representative callus sections stained with Safranin-‐O (SO),
Trichrome (TC) and PS at days 21 and 56 showing fully ossified callus in fractures
without muscle injury (b, bone; boxes 1, 2). Fracture calluses with muscle injury exhibit
unresorbed cartilage (c), fibrous tissue (f, box 3) and absence of bone bridging (box 4,
orange arrowheads). Scale bars: low magnification of muscle and fracture calluses=
1mm; boxed areas=200μm. Statistical analyses were performed following Mann-‐
Whitney test (* p-‐value<0,05; ** p-‐value<0,01, n=5 per group). All data represent mean
± SD.

Article 3, Julien et al, En cours de soumission
147
B
Fracture Fracture with muscle injury
12
1 1
33
3
4
d14
d30
A
Cartilage8
d7 d14 d21 d28 d56
Volu
me
(mm
3 )
0
2
4
6
Bone
d7 d14 d21 d28 d560
5
10
15
Volu
me
(mm
3 )
Callus
0
20
40
60
d7 d14 d21 d28 d56
Volu
me
(mm
3 )
C
4
HE
d21
SO
SO
Regenerating area Fibrotic area
HE
2
4
Muscle injury
Fracture
Fracture
**
**
*
**
** *
*
d56
PS
PS
PS
2
PS
HE
HE PS
PS
TC
SO
PS
PS
PS
PS TC
TC
**
*
*
c
b
f
b
HE
HE
12
1 2
34
3 4Fracture with muscle injury
Figure 1

Article 3, Julien et al, En cours de soumission
148
Figure 2: Musculoskeletal trauma impacts muscle and periosteum contribution to
bone healing.
(A) Experimental design of combined GFP periosteum and mTmG EDL muscle
transplantation at the fracture site of wild type hosts. (B) Callus sections at 14 days post-‐
fracture stained with Safranin-‐O (SO, left). Adjacent sections mounted with DAPI (right)
show EDL muscle graft outside the callus (ca, limited by a yellow dotted line) and
muscle-‐derived cells within the callus (Tomato signal, red arrow) as well as periosteum
graft (PO graft, limited by a green dotted line) and periosteum-‐derived cells within the
callus (GFP signal, green arrow). (C) High magnifications of the callus showing cartilage
(left) and bone (right) stained with SO and TC (top panel), and derived from the EDL
graft (red, middle panel) or from the periosteum graft (green, bottom panel). Both
muscle and periosteum derived cells are found in cartilage (c) and bone (b, white
arrowhead). (D) Experimental design of GFP-‐EDL muscle graft transplanted next to un-‐
injured tibia of wild-‐type host, and tibial fracture induced after one month. (E) Callus
sections at 14 days post-‐fracture stained with SO (left, top) and mounted with DAPI
(right, top) showing GFP-‐EDL muscle graft outside the callus (limited by a yellow dotted
line) and muscle-‐derived cells in callus (GFP signal). (F) High magnification of cartilage
(box 1, c, left) and bone (box 2, right, white dotted line) containing GFP-‐positive EDL
muscle-‐derived chondrocytes and osteocytes respectively (white arrow). (G)
Experimental design of GFP-‐EDL muscle or GFP-‐periosteum graft transplanted at the
fracture site with or without muscle injury. (H) Histomorphometric analyses of the
percentage of GFP positive cartilage in the total GFP volume within callus showing
decreased contribution to cartilage of EDL and periosteum grafts in fracture with muscle
injury compared to fracture alone. c: cartilage, b: bone, bm: bone marrow. Scale bars:
B=500μm, C=100μm (scale bar for E and F?). Statistical analyses were performed
following Mann-‐Whitney test (* p-‐value<0,05; ** p-‐value<0,01, n=5 per groups). All data
represent mean ± SD.
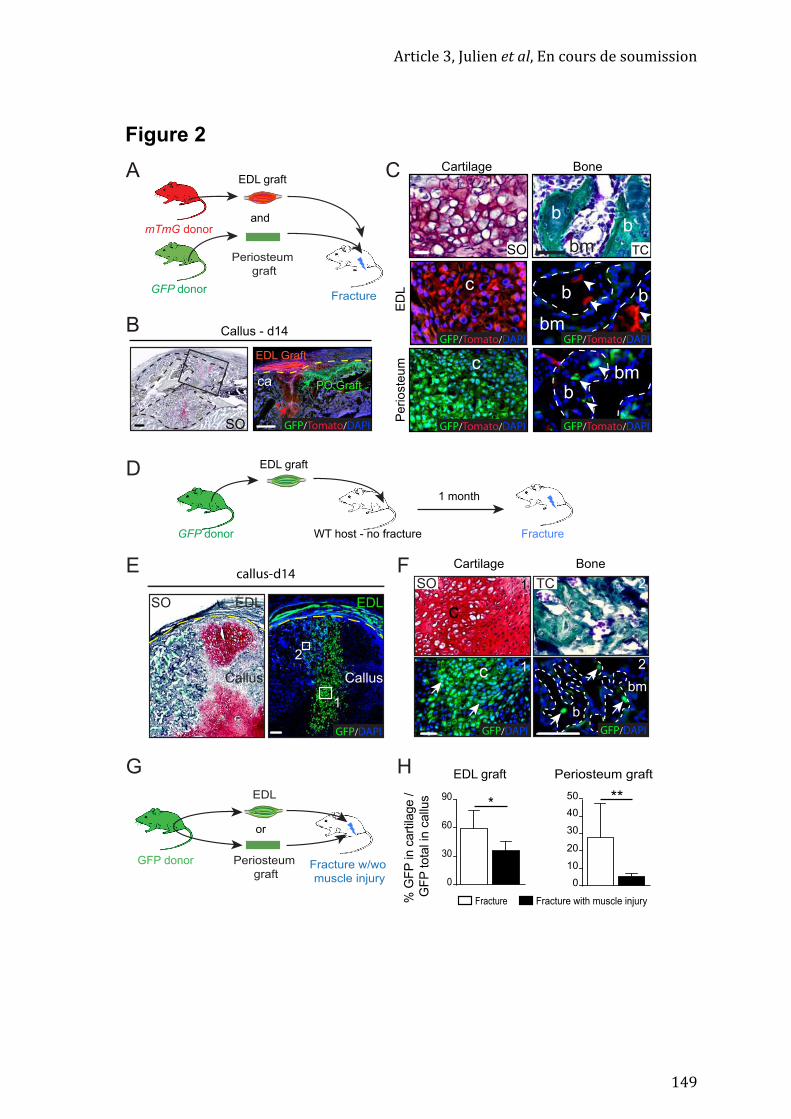
Article 3, Julien et al, En cours de soumission
149
ca
EDL Graft
PO Graft
G Periosteum graftEDL graft
Fracture Fracture with muscle injury% G
FP in
car
tilag
e /
GFP
tota
l in
callu
s
GFP donor Periosteum graft
Fracture w/wo muscle injury
or
Callus - d14
c
A Cartilage Bone
ED
LP
erio
steu
mb
b
SO
GFP/Tomato/DAPI
b bbm
bm
EDL
010
20
30
4050 **
0
30
60
90 *
cGFP/Tomato/DAPI
bbm TCSO
GFP/Tomato/DAPIGFP/Tomato/DAPI
GFP donor
Periosteum graft
EDL graft
and
Fracture
mTmG donor
C
E
D
callus-d14
cSO
1
1
2
GFP/DAPI GFP/DAPI
TC 2
EDL graft
WT host - no fracture
1 month
FractureGFP donor
EDL
Callus
1
2
GFP/DAPI
SO EDL
Callus
b
bm
GFP/Tomato/DAPI
Cartilage Bone
B
F
H
c
Figure 2

Article 3, Julien et al, En cours de soumission
150
Figure 3: Prx1 and PDGFRa mark muscle and periosteum-‐derived cells during
bone repair.
(A) Localization of Prx1-‐ and PDGFRα-‐derived cells in the fracture callus of
Prx1Cre;mTmG or tamoxifen-‐induced PDGFRαCreERT;mTmG mice respectively at day 14
post tibial fracture. Longitudinal callus sections were stained with Safranin-‐O (SO, left)
or mounted with DAPI (right) to visualize GFP and Tomato signals. High magnifications
show that in the calluses of Prx1Cre;mTmG mice all chondrocytes in cartilage (c) and
osteocytes in bone (b, white arrows) are Prx1-‐derived GFP-‐positive. In
PDGFRαCreERT;mTmG calluses, cartilage and bone contains GFP positive PDGFRα-‐
derived chondrocytes and osteocytes, as well as Tomato-‐positive cells that are not
PDGFRα-‐derived. (B) Experimental design of EDL muscle-‐ and periosteum-‐ grafts from
Prx1Cre;mTmG or tamoxifen-‐induced PDGFRαCreERT;mTmG mice transplanted at the
fracture site of wild type hosts. (C) Longitudinal callus sections stained with SO at day
14 post-‐fracture (left) and adjacent sections at high magnification show EDL-‐ (top) or
periosteum-‐derived (bottom) cells within the callus. Prx1Cre;mTmG EDL and periosteum
grafts (bottom) give rise exclusively to Prx1-‐derived GFP-‐positive chondrocytes (white
arrowheads) and osteocytes (white arrows) in the callus. PDGFRαCreERT;mTmG EDL
and periosteum grafts (right) give rise to GFP-‐positive PDGFRα-‐derived chondrocytes
(white arrowheads) and osteocytes (white arrows) but also to Tomato-‐positive PDGFRα-‐
derived chondrocytes (orange arrowhead). (D) Experimental design of muscle cells
isolation from hind limbs of Prx1Cre;mTmG donor mice and transplantation of sorted
Prx1-‐derived cells (GFP+) or non Prx1-‐derived cells (Tomato+) at the fracture site of
wild-‐type hosts. (E) Longitudinal callus sections at days 14 and 21 post-‐transplantation
stained with SO and Masson’s trichrome (TC) respectively. High magnifications of
adjacent sections show that Prx1-‐derived cells can form cartilage (c) by day 14 and bone
(b) and bone marrow (bm, orange arrowhead) by day 21 unlike non-‐Prx1 derived cells
that are not detected in the callus. Scale bars: SO=1mm, high magnification=50μm for
cartilage and 25μm for bone, n=3 per groups.

Article 3, Julien et al, En cours de soumission
151
A PdgfraCreERT;mT/mG
B
Prx1Cre;mT/mG
cartilage cartilagebone bone
ED
Lp
erio
ste
um
cartilage bone bone
C
Prx1Cre;mT/mG or
PdgfraCreERT;mT/mGdonor
wild type host
Periosteum
graft
EDL graft
or
GFP/Tomato/DAPI GFP/Tomato/DAPI GFP/Tomato/DAPI GFP/Tomato/DAPI
c
cb
bm bm
b
PdgfraCreERT;mT/mGPrx1Cre;mT/mG
SO
SO
Lineage
tracing
Prx1Cre;mT/mG Transplant at
the fracture siteCell sorting
FSC-A
Prx
1:G
FP
Muscle cell
digestion
DGFP+
cells
GFP-
cells
or
c
GFP/Tomato/DAPI
c
b
c
ca
muscle
GFP/Tomato/DAPI
GFP/Tomato/DAPIbm
b
bmb
GFP/Tomato/DAPI
GFP/Tomato/DAPI
cartilage
c
c
muscle
ca
GFP/Tomato/DAPI
GFP/Tomato/DAPIbm
b
b
bmGFP/Tomato/DAPI
GFP/Tomato/DAPI
cb
GFP/Tomato/DAPI
TCSO
Non-Prx1
derived cells
Prx1
derived cells
Prx1
derived cells
d14 post-transplantation d21 post-transplantation
GFP/Tomato/DAPIb
bm
E
SO
Fracture
Figure 3

Article 3, Julien et al, En cours de soumission
152
Figure 4: Characterization of Prx1-‐derived muscle cells recruited during bone
repair.
(A) Localization of Prx1-‐derived cells in adult skeletal muscle. Transverse sections of TA
muscle from Prx1Cre;mTmG mice stained with CD31, αSMA, PDGFRβ, NG2, CD29 or
PDGFRα (magenta). Nuclei are detected with DAPI. Prx1-‐derived cells are GFP-‐positive
and non Prx1-‐derived are Tomato-‐positive. (B, C) Flow cytometry analyses of freshly
isolated cells from muscles surrounding the tibia of Prx1Cre;YFPfl/+ adult mice. (B) Prx1-‐
derived YFP+ cells represent 32% of the total muscle cell population, are negative for
CD31, CD11b and CD45, and positive for CD34 (35,2%), α7integrin (11,1%), PDGFRα
(81%), Sca1 (77,5%) and CD29 (95,2%). Red curve represents experimental tube and
blue curve fluorescence minus one (FMO) control. (C) FAP and OCP populations contain
Prx1 derived cells (YFP+) and PDGFRα expressing cells (right panel). (D) RT-‐qPCR
analysis on Prx1-‐derived muscle YFP+ cells at P1 showed expression for CD34, Cxcl12,
Gremlin, Mx1, Pdgfrα, Nestin, Leptin Receptor, PW1, Tcf4, αSMA and Vimentin, NG2,
Pdfgrβ, Tensomodulin (Tnmd), Tenasin C (TnC) but not for Scleraxis (scx) and Pax7. (E)
Prx1-‐derived cells form more CFU-‐F and have a higher clonogenicity capacity than non-‐
Prx1-‐derived cells. (F) Osteogenic, adipogenic, chondrogenic, fibrogenic and myogenic in
vitro differentiation of Prx1-‐derived muscle cells. Prx1-‐derived muscle cells (P1)
undergo osteogenic, chondrogenic, adipogenic and fibrogenic differentiation but fail to
achieve myogenic differentiation. Statistical analyses were performed following Mann-‐
Whitney test (* p-‐value<0,05, n=3 per group). All data represent mean ± SD.
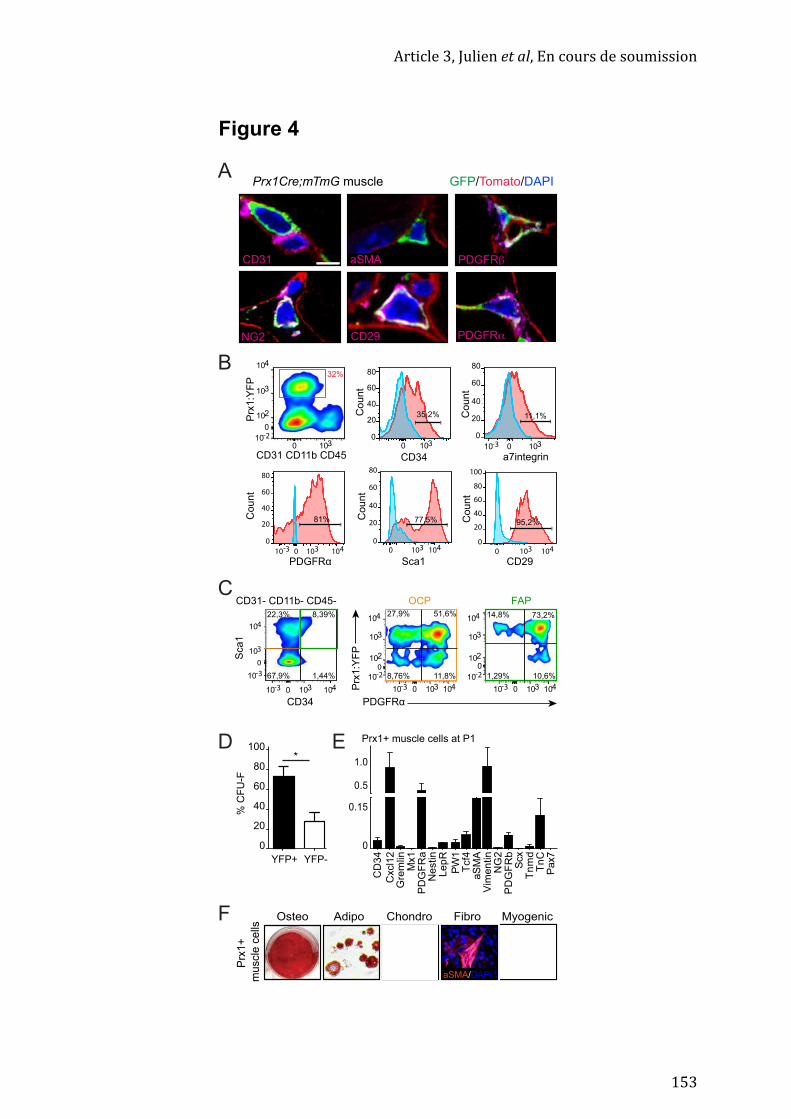
Article 3, Julien et al, En cours de soumission
153
A Prx1Cre;mTmG muscle
B
CD31 CD11b CD45
Prx
1:Y
FP
CD34
0
20
40
60
80
Sca1
77,5%
0 1030
20
40
60
80
PDGFRα
81%
10410
0 103
0
104
103
102
10
0 1030
20
40
60
80
35,2%
0 103 104
0 103
0
20
40
60
80
a7integrin
11,1%
10
0 1030
20
40
60
80
95,2%
104
100
CD29
Co
un
tC
ou
nt
Co
un
tC
ou
nt
Co
un
t
32%
CD31 aSMA
CD29NG2 PDGFRα
PDGFRβ
GFP/Tomato/DAPI
C
0
104
103
10
10 0 104103
1,44%
8,39%22,3%
67,9%
CD34
Sca
1
CD31- CD11b- CD45-
0
104
103
10
102
10 0 104103
51,6%27,9%
8,76% 11,8%
10 0 104103
104
103
10
102
0
10,6%1,29%
14,8% 73,2%
PDGFRα
Prx
1:Y
FP
Prx
1+
mu
scle
ce
lls
Osteo Adipo Chondro Myogenic
D
FibroF
CD
34
Cxcl1
2
Gre
mlin
Mx1
PD
GF
Ra
Ne
stin
Le
pR
PW
1
Tcf4
aS
MA
Vim
en
tin
NG
2
PD
GF
Rb
Scx
Tn
md
Tn
C
Pax7
0
0.15
0.5
1.0
aSMA/DAPI
20
40
60
80
100
*
0
YFP+ YFP-
% C
FU
-F
Prx1+ muscle cells at P1E
OCP FAP
Figure 4

Article 3, Julien et al, En cours de soumission
154
Figure 5: Callus fibrosis is produced by muscle Prx1-‐derived profibrotic
progenitors and can be targeted by Imatinib to improve bone healing.
(A) Transverse sections of intact tibia and muscle (d0) of Prx1Cre;mTmG mice, and days
3 and 21 post-‐fracture alone or fracture with muscle injury. Prx1+ derived cells (GFP+)
are detected at d3 within the muscle adjacent to the fracture site and throughout the
muscle after fracture combined with muscle injury. By day 21 post-‐surgery, Prx1+
derived cells are not observed in muscle surrounding the fracture callus but are
detected within the fibrous tissue thin muscle after fracture combined with muscle
injury. (B) Histomorphometric quantification of fibrosis volume in tibial fractures with
or without muscle injury at days 7, 14, 21, 28 and 56 post-‐fracture. (C) Upper panel:
longitudinal sections of fracture callus 21 days post-‐fracture with muscle injury of
Prx1Cre;mTmG mice stained with picrosirus (PS) showing fibrosis (black arrowhead, f,
boxed area) and adjacent sections showing Prx1-‐derived GFP+ cells (white arrowhead,
f) in fibrous tissue. Lower panel: PDGFRα (left) and Periostin (right)
immunofluorescence on callus section of fracture with muscle injury d21 post-‐fracture
of wild type mice show staining in fibrous tissue. (D) Upper panel: experimental design
of injured EDL muscle graft or periosteum graft from Prx1Cre;mTmG mice transplanted
at the fracture site of wild type hosts with muscle injury. Lower panel: longitudinal
callus sections stained with PS show fibrous tissue (f) within callus at day 21. Adjacent
sections show Prx1-‐derived muscle cells (GFP+) in fibrosis (f) and in bone (b) but no
Tomato+ cells (top panel). Periosteum derived GFP+ cells are detected in bone (b) but
not in fibrous tissue (asterisk) (E) Experimental design of Imatinib® treatment.
Fractures with muscle injury were performed on wild-‐type mice. Mice were daily treated
with Imatinib® (50mg/kg/day) or vehicle (PBS) from the time of surgery to harvesting
at days 7 or 21. (F) Histomorphometric analyses of total callus, cartilage, bone and
fibrosis volume of wild type mice with fracture combined to muscle injury daily treated
with Imatinib® (50mg/kg/day) or vehicle (PBS). c: cartilage, b: bone, f: fibrosis, m:
muscle. Scale bars: low magnification=1mm, high magnification=100μm. Statistical
analyses were performed following Mann-‐Whitney test (* p-‐value<0,05; ** p-‐value<0,01,
n=5 per group). All data represent mean ± SD.
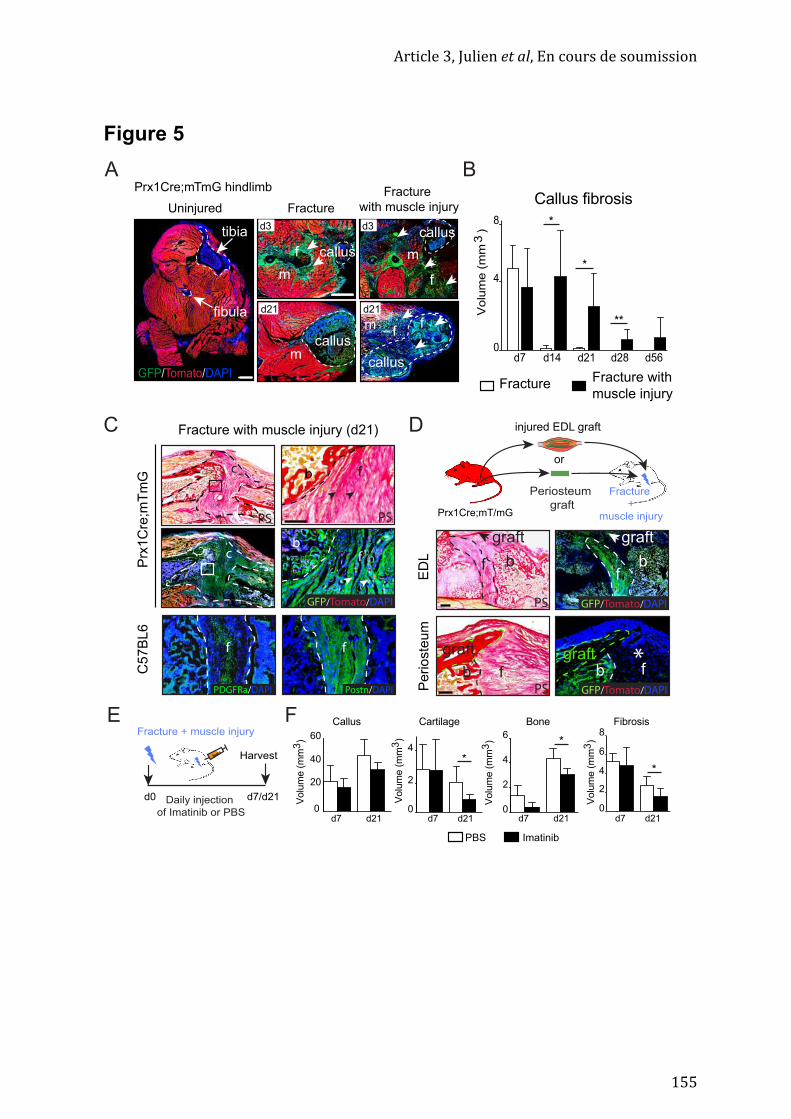
Article 3, Julien et al, En cours de soumission
155
Figure 5A
D
c
bf
Prx
1C
re;m
Tm
G
CE
DL
Pe
rio
ste
um
Prx1Cre;mT/mG
Periosteum
graft
injured EDL graft
or
Fracture
+
muscle injury
graft
b f*
f
graft
f f
f
graft
graft
b
GFP/Tomato/DAPI
GFP/Tomato/DAPI
PS
GFP/Tomato/DAPI
PS
b
Fracture with muscle injury (d21)
Postn/DAPIPDGFRa/DAPI PS
PS
f
E
b b
d7 d14 d21 d28 d56
*
*
**
0
4
8
Volu
me (
mm
3)
C5
7B
L6
FractureFracture with
muscle injury
c
*
PBS Imatinib
*
*
Volu
me (
mm
3)
Callus
0
20
40
60
Cartilage
0
2
4
0
2
4
Volu
me (
mm
3)
6
Volu
me (
mm
3)
Bone
0
2
4
6
8
Fibrosis
Volu
me (
mm
3)
d21d21d7 d7d21d21d7 d7
Uninjured
d3
Fracture
with muscle injury
callus
callustibia
fibula
d3
d21 d21
Fracture
Prx1Cre;mTmG hindlimb
GFP/Tomato/DAPIcallus
callus
f
m
m
m
m
Callus fibrosis
B
d0
Fracture + muscle injury
Daily injection
of Imatinib or PBS
Harvest
d7/d21
F
f
f
f
f

Article 3, Julien et al, En cours de soumission
156

Article 3, Julien et al, En cours de soumission
157
Supplementary Figures
Muscle-‐derived profibrotic progenitors impair bone
healing in musculoskeletal trauma
Julien A. et al.

Article 3, Julien et al, En cours de soumission
158
d7 d14 d21 d280
20
40
60
Callus volume (mm3)
*
*
0
2
4
6
8 **
*
****
****
0
5
10
15
*
**
*
No muscle injury Fracture and total muscle injuryFracture and TA muscle injury
Cartilage volume (mm3) Bone volume (mm3)
d7 d14 d21 d28 d7 d14 d21 d28
Supplementary Figure 1
Supplementary figure 1: Tibialis anterior (TA) muscle injury does not severely impact bone repair. Histomorphometric quantification of callus, cartilage and bone volumes in tibial fractures without muscle injury, with total muscle injury (as shown in Figure 1) or with TA muscle injury at days 7, 14, 21 and 28 post-fracture. Statistical analyses were performed following Mann-Whitney test (* p-value<0,05; ** p-value<0,01, n=5 per group). All data represent mean ± SD.

Article 3, Julien et al, En cours de soumission
159
A
No muscle injury Muscle injury
Cortical defect
no muscle injury muscle injury
0.0
0.4
d7 d21
0.00
0.05
0.10
d7 d21
Callus Bone
Volu
me (
mm
3)
d2
1
nsns ns ns
B
GFP donor
EDL graft
Cortical defect
GFP/DAPIcortex
EDLSO
Volu
me (
mm
3)
cortex
d7
TC TC
Supplementary figure 2: Bone repair of cortical defects via intramembranous ossification is not affected by muscle injury. (A) Upper panel: Representative longitudinal sections stained with Masson’s trichrome (TC) of mouse tibia at days 21 post cortical defect (black arrows) with or without muscle injury in wild type mice. Lower panel: Histomorphometric quantification of callus and bone volumes of cortical defect with or without muscle injury at day 7 and day 21 post-surgery shows no significant differences between the two groups. (B) Upper panel: experimental design of GFP -EDL muscle grafts transplanted next to a cortical defect. Lower panel: longitudinal sections of cortical defect with GFP-EDL-muscle graft stained with safranin’o (SO) and mounted with DAPI. No GFP+ muscle-derived cells are detected within the cortical defect. Scale bar = 500μm. Statistical analyses were performed following Mann-Whitney test (* p-value<0,05; ** p-value<0,01, n=4 per group). All data represent mean ± SD
Supplementary Figure 2

Article 3, Julien et al, En cours de soumission
160
EDL
callus
c
GFP/Tomato/DAPI GFP/Tomato/DAPI
SO
Pax7CreERT;mT/mGdonor
wild type host
EDL graft
c
c
Pax7CreERT;mT/mG miceA
B
C SO1
1
2
2
2
callus
cb
SO
*
Fracture
Fracture with muscle injury
callus
GFP/Tomato/DAPI
cb
GFP/Tomato/DAPI
GFP/Tomato/DAPI
SO
GFP/Tomato/DAPI
m
m
muscle
GFP/Tomato/DAPI
1
1 2
2
1
1
2
2 GFP/Tomato/DAPI
muscle
Supplementary figure 3: Pax7 does not mark muscle-derived cells during bone repair. (A) Localization of Pax7-derived cells in the fracture callus of tamoxifen-induced Pax7CreERT2;mTmG mice at day 14 post tibial fracture with or without muscle injury. Longitudinal callus sections stained with Safranin-O (SO, left) or mounted with DAPI (right) to visualize GFP and Tomato signals. High magnification shows that in the calluses of Pax7CreERT2;mTmG mice all chondrocytes in cartilage (c) and osteocytes in bone (b) are not Pax7-derived (Tomato-positive). In muscle (m) surrounding the fracture, new GFP+ Pax7-derived muscle fibers are not detected but around the fracture combined with muscle injury, all regenerating muscle fibers are GFP+. (B) Experimental design of EDL muscle grafts from Pax7CreERT2;mTmG mice transplanted adjacent to the fractured tibia of wild type hosts. (C) Longitudinal callus sections stained with SO at day 14 post-fracture (top) and adjacent section at high magnification shows EDL-derived cells (bottom) within the callus (delimited by a yellow line). Regenerating myofibers are visible within the EDL graft (GFP+, asterisk). Pax7CreERT2;mTmG EDL gives rise exclusively to non Pax7-derived chondrocytes (Tomato+) in the callus. Scale bar: SO=1mm, high magnification= 50μm.
Supplementary Figure 3
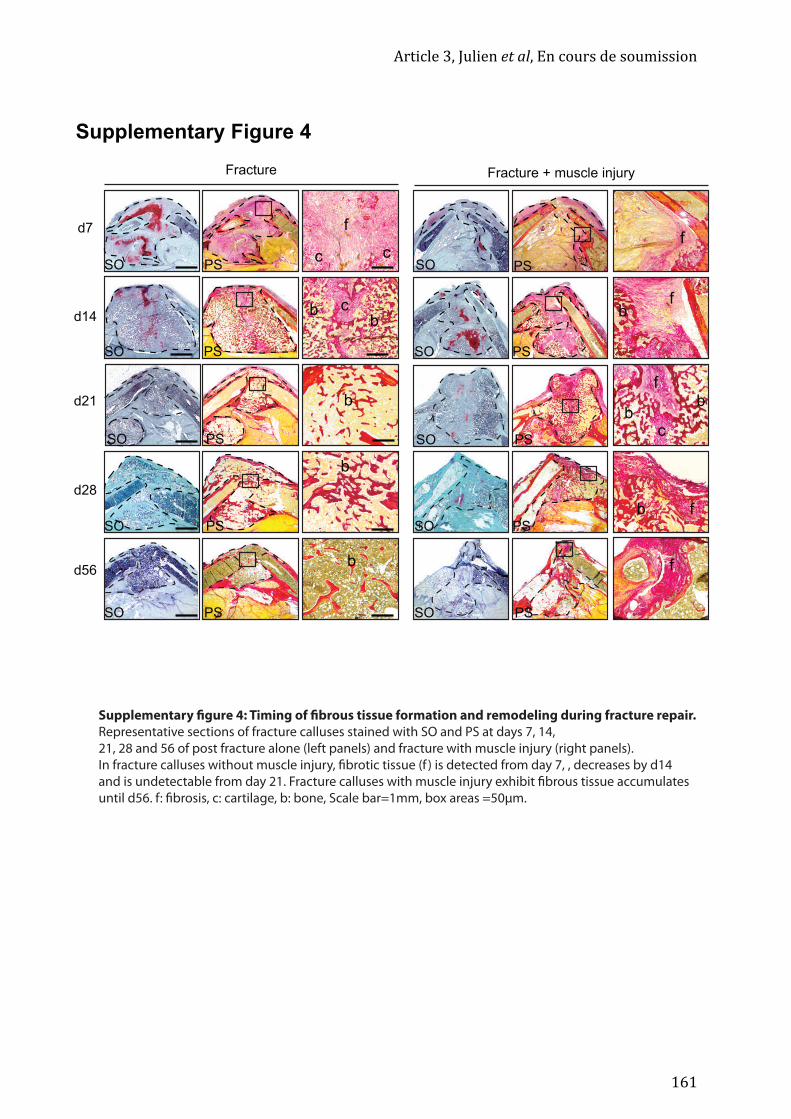
Article 3, Julien et al, En cours de soumission
161
Fracture
SO PS
b
d7
d14
d21
SO PS
SO PS
SO PS
SO PS
d28
d56
Fracture + muscle injury
SO PS
SO PS
SO PS
SO PS
SO PS
b
b
f
c c
cbb
f
f
f
f
f
cb
b
b
b
Supplementary figure 4: Timing of fibrous tissue formation and remodeling during fracture repair. Representative sections of fracture calluses stained with SO and PS at days 7, 14, 21, 28 and 56 of post fracture alone (left panels) and fracture with muscle injury (right panels). In fracture calluses without muscle injury, fibrotic tissue (f ) is detected from day 7, , decreases by d14 and is undetectable from day 21. Fracture calluses with muscle injury exhibit fibrous tissue accumulates until d56. f: fibrosis, c: cartilage, b: bone, Scale bar=1mm, box areas =50μm.
Supplementary Figure 4

Discussion
162

Discussion
163
Discussion
La régénération tissulaire dépend du recrutement, de l’activation et de la différenciation
de cellules souches. Déterminer la nature de l’origine des cellules souches est une
question fondamentale pour la compréhension des mécanismes de régénération
tissulaire. Les cellules impliquées dans la régénération osseuse proviennent de sources
tissulaires multiples. La recherche fondamentale et translationnelle porte
principalement sur les CSMOs. Cependant, nous avons montré dans la première partie
des résultats que le périoste contient des cellules osseuses dotées d’une capacité d’auto-‐
renouvellement et dont le potentiel de régénération est plus élevé que celui des CSMOs.
Dans la deuxième partie, avons montré que le muscle squelettique joue un rôle
important au cours de la régénération osseuse comme source de facteurs sécrétés, en
partie par les cellules souches musculaires , les cellules satellites, et comme source de
cellules participant directement à la formation du cartilage dans le cal. Dans la troisième
partie de ma thèse, nous avons caractérisé les OCPs provenant du muscle et l’impact
d’un traumatisme musculosquelettique sur les OCPs et le processus de régénération
osseuse. Nous avons développé un nouveau modèle de traumatisme
musculosquelettique combinant une fracture et une blessure mécanique du muscle qui
conduit à une absence de consolidation osseuse. Les OCPs provenant du muscle pendant
la régénération osseuse constituent une population de cellules interstitielles
musculaires dérivant du lignage mésenchymateux Prx1, exprimant PDGFRα un
marqueur des FAPs dans le muscle. Cependant, dans le cas d’une blessure traumatique,
les cellules dérivées du lignage Prx1 sont aussi source de fibrose contribuant
négativement au processus de régénération. Afin de diminuer l’accumulation de tissu
fibrotique, nous avons utilisé un inhibiteur de la voie PDGFR, l’Imatinib®. Le traitement
avec l’Imatinib® améliore la régénération osseuse après un traumatisme
musculosquelettique.

Discussion
164
Quelles sont les origines des cellules souches squelettiques activées en réponse à
une fracture ?
De nombreux tissus ont de grandes capacités de régénération et les mécanismes
cellulaires impliqués sont variés. La régénération musculaire est basée sur le
recrutement, l’activation et la différenciation des cellules souches spécifiques, les
cellules satellites[250, 251, 253]. La régénération des glomérules rénaux est permise grâce à
la transdifférenciation des cellules tubulaires épithéliales [418, 419]. Après une lésion
hépatique, les hépatocytes prolifèrent afin de restaurer la masse et la fonction hépatique [420, 421]. Ces différents mécanismes ne sont pas exclusifs et la régénération tissulaire
peut faire intervenir plusieurs d’entre eux. Dans le foie par exemple, les cellules ovales
sont décrites comme des cellules souches hépatiques potentielles et pourraient se
différencier directement en hépatocytes en parallèle de la prolifération des
hépatocytes[422]. Cependant, quelque soit le tissu, la question de l’origine des cellules
impliquées dans le processus de régénération est fondamentale. La contribution des
cellules résidentes au cours de la régénération tissulaire est bien décrite, mais la
contribution de cellules non-‐résidentes, recrutées à partir des tissus adjacents ou de
manière systémique via la vascularisation est peu caractérisée. La contribution de
cellules circulantes telles que les CSMOs ou les cellules CD34+ a été envisagée au cours
de la régénération de différents tissus [423-‐430] mais reste minoritaire comparée à la
contribution des cellules résidentes[431-‐433]. Néanmoins, ces cellules circulantes peuvent
participer à la régénération tissulaire via leur fusion avec les cellules résidentes et non
en tant que cellules souches/progénitrices au sens propre du terme[434-‐438]. Les cellules
circulantes ont aussi un rôle indirect nécessaire comme source de facteurs par exemple.
La contribution directe d’un tissu adjacent au tissu blessé n’est pas renseignée dans la
littérature. Cette question est pourtant essentielle puisque qu’un traumatisme impacte
généralement plusieurs tissus adjacents. Dans le cas de la régénération osseuse, les
travaux du laboratoire indiquent que le périoste est la source majeure d’OCPs alors que
les CSMOs agissent indirectement comme source de facteurs. La source majeure d’OCPs
semble donc être locale. L’étude menée pendant ma thèse apporte plusieurs éléments
concernant le rôle du muscle adjacent à l’os. Nous avons montré que le muscle
squelettique est une source importante de cellules squelettiques au cours de la
régénération osseuse. Si les cellules satellites contribuent principalement en tant que

Discussion
165
source de facteurs, les cellules interstitielles dérivant du lignage Prx1+ contribuent à la
régénération osseuse en formant des chondrocytes, des ostéoblastes et des ostéocytes.
Comment distinguer les cellules provenant de tissus adjacents pendant la
régénération osseuse ?
Les cellules du périoste qui contribuent à la régénération osseuse dérivent aussi du
lignage Prx1, comme les cellules du muscle squelettique. Actuellement, il n’existe aucun
marqueur tissu-‐spécifique permettant de distinguer les cellules provenant du périoste et
celles provenant du muscle squelettique[174]. Nous avons donc utilisé des approches de
greffes tissulaires de périoste et de muscle pour évaluer le potentiel de ces deux tissus
lors de la formation du cal. Cependant, la transplantation d’un muscle ou d’une portion
de périoste reflète une partie seulement de leur contribution et ne permet pas de
visualiser la contribution endogène totale et relative de ces deux tissus. Pour cela, nous
développons actuellement des approches de greffes segmentaires de tibias combinées à
du lignage génétique. Si la contribution des cellules du muscle Prx1 au cours de la
régénération osseuse est maintenant établie, il reste à savoir si elle est nécessaire à la
régénération osseuse et si leur absence altère ce processus.
Nos travaux montrent que les CSMOs, les PCs et les cellules du muscle qui contribuent à
la régénération osseuse ont de nombreux points communs. Dans le but de mieux
caractériser les CSMOs et d’identifier un marqueur des cellules souches squelettiques,
les marqueurs Mx1, Gremlin 1[186], LeptinR[187], Nestin[181, 439], PDGFRα[440], Prx1[182],
Cxcl12[441] et Osterix[51] chez la souris ou CD146 chez l’Homme[172] ont été identifiés
dans la moelle osseuse. Par des analyses moléculaires et de lignage cellulaire nous avons
montré que la plupart de ces marqueurs sont aussi exprimés dans les PCs et les cellules
interstitielles du muscle. Des analyses à l’échelle de la cellule unique pourraient être
utiles pour mieux comprendre l’architecture des populations de cellules impliquées lors
de la régénération osseuse et pour trouver un marqueur permettant de discriminer
l’origine tissulaire des cellules.

Discussion
166
Quels sont les mécanismes de recrutement des cellules souches en réponse à une
fracture ?
La compréhension des mécanismes d’activation et de recrutement des cellules souches
au cours d’un processus de régénération est fondamentale. L’analyse par microarray des
PCs et CSMOs avant fracture et 3 jours après fracture nous a permis d’identifier
Périostine (Postn) comme un facteur essentiel de la niche des cellules souches du
périoste. En absence de Postn, les cellules souches du périoste perdent leur potentiel
d’auto-‐renouvellement et leur capacité d’intégration à long terme. Nous cherchons
maintenant à comprendre les mécanismes d’activation des cellules recrutées du muscle
et leurs interactions avec les cellules de l’os au cours de la régénération osseuse. De
nombreuses voies de signalisation telles que Notch, IGF-‐1, HGF ou BMP ont été décrites
comme étant impliqués dans les mécanismes d’activation, de recrutement et de
différenciation des cellules souches au cours de la régénération[442-‐444]. La voie de
signalisation BMP est essentielle au cours du développement osseux et musculaire[29, 40,
445], lors de l’homéostasie tissulaire[247, 446] et au cours de la régénération[106, 447-‐449]. La
voie de signalisation BMP canonique est activée via la fixation des ligands BMP sur les
récepteurs de type 1 et 2, ce qui entraine leur dimérisation puis leur auto-‐
phosphorylation. Cela induit le recrutement du complexe de molécules effectrices
Smad1/5/8 qui sont à leur tour phosphorylées. La protéine Smad4 est alors recrutée par
le complexe pSmad1/5/8. Le complexe pSmad1/5/8-‐Smad4 est ensuite transloqué au
niveau du noyau pour agir au niveau des gènes cibles[450]. Les souris Prx1Cre/+;Bmp2fl/fl
présentent une régénération imparfaite avec une absence de formation du cal après
fracture[106]. BMP2 n’est pas nécessaire au maintien du pool de progéniteurs
squelettique au sein du périoste. Néanmoins, BMP2 est essentiel à l’activation et à la
différentiation chondrogénique des cellules du périoste lors de la régénération
osseuse[107, 113, 447]. Dans la deuxième partie des résultats, nous avons montré que les
cellules satellites des muscles entourant le tibia sécrètent des facteurs de croissance
dont les BMPs en réponse à la fracture. Les cellules satellites sont donc une source
supplémentaire de BMP pour la régénération osseuse. Au sein du tissu musculaire, au
cours de l’homéostasie tissulaire, les BMPs permettent l’expansion du pool de cellules
souches en retardant la différenciation myogénique. Une régulation négative de la voie
BMP conduit à une réduction de la taille des fibres régénérées de 40% et à une

Discussion
167
augmentation de la fibrose[449, 451]. Les BMPs ont aussi un rôle dans la régénération
musculaire. Les gènes Id sont connus pour être des cibles directes de la voie BMP et il a
été montré que chez les souris Id1/Id3 KO, la régénération musculaire est retardée[448].
Le rôle de la voie BMPs dans les interactions os-‐muscle reste encore à éclaircir. Afin
d’élucider les mécanismes d’activation des OCPs du muscle squelettique pendant la
régénération osseuse, nous travaillons sur un modèle murin de déplétion du récepteur 1
au BMP (BMPR1a) dans les cellules PDGFRα+. Pour cela, nous avons induit des fractures
sur les souris PDGFRαCreERT2/+ ;Alk3fl/fl et les souris contrôles PDGFRα+/+ ;Alk3fl/fl après
induction au tamoxifène. Les premiers résultats montrent un retard de régénération au
jour 14 post-‐fracture, ce qui suggère un rôle de la voie BMP au sein des cellules
PDGFRα+ au cours de la régénération osseuse. Ce phénotype reste à décrire plus en
détails, et une déplétion génétique combinée à des approches de transplantation
tissulaire sera nécessaire afin de distinguer le rôle de la voie BMPs au niveau des OCPs
du muscle et du périoste. Des études à grandes échelles seront aussi nécessaires afin
d’identifier de nouveaux facteurs intervenant dans l’activation des OCPs au cours de la
régénération osseuse.
Quelles sont les causes d’une régénération osseuse imparfaite et quel est le rôle
de la réponse fibrotique ?
Le processus de régénération tissulaire est généralement divisé en quatre étapes : la
formation de l’hématome, la phase inflammatoire, la phase proliférative et la phase de
remodelage[452-‐454]. Le timing de chaque phase est finement régulé et la dérégulation
d’une phase peut altérer la régénération. Les origines des déficiences de régénération
tissulaire peuvent être multiples mais le potentiel des cellules souches est rarement mis
en cause. Dans le cas du tissu osseux par exemple, un environnement mécanique
instable est une des causes de régénération imparfaite. En effet, l’instabilité induit une
accumulation de cartilage non résorbé qui peut avoir pour conséquence de
compromettre la formation du pont osseux[455, 456]. Cette question essentielle de la
transition cartilage-‐os n’a pas été développée au cours de mes travaux de thèse mais
notre modèle de traumatisme pourrait être un outil pour l’étudier.
Une autre cause de non-‐régénération peut-‐être une dérégulation de la phase
inflammatoire et par conséquence une mauvaise régulation des cellules du tissu de

Discussion
168
soutien et l’accumulation de tissu fibrotique[281, 457-‐459]. La fibrose est définie comme la
présence anormale de matrice extracellulaire (collagène notamment) en réponse à un
dommage aigue ou chronique, ce qui entraine un disfonctionnement du tissu lésé[460-‐462].
La fibrose est une problématique majeure dans le domaine de la régénération tissulaire.
De nombreuses études ont établi le lien entre l’inflammation et la formation de la
fibrose, renforçant un peu plus l’importance du contrôle des étapes précoces du
processus de régénération[463, 464]. Concernant le tissu osseux, notre laboratoire a
précédemment montré que chez les souris mdx, modèle murin de la dystrophie de
Duchenne, où la réponse inflammatoire est continue, la régénération osseuse est
retardée[404]. La déplétion des macrophages au moment de la fracture induit un retard
de régénération et la formation de tissu fibrotique au sein du cal[73]. Cox2 est un
médiateur de l’inflammation sécrété à la fois par les cellules immunitaires et par les
OCPs. Chez les souris Cox2-‐/-‐ la régénération osseuse est retardée et du tissu fibrotique
se forme au sein du cal. Des greffes segmentaires de tibias ont permis de mettre en
évidence le rôle primordial de Cox2 lors de l’activation des progéniteurs squelettiques
du périoste[465, 466]. Ces expériences mettent en évidence les interactions directes entre
le système immunitaire, les progéniteurs squelettiques et la formation de tissu
fibrotique[467]. Ces interactions sont retrouvées aussi au cours de la régénération
musculaire, notamment entre les cellules satellites et les macrophages[468-‐470] [463, 471, 472].
En utilisant des approches génétiques, il a été montré que le switch
macrophage/monocyte orchestre la régénération musculaire et la persistance des
macrophages pro-‐inflammatoires conduit à la formation aberrante de tissu
fibrotique[473]. Plusieurs cytokines et facteurs de croissance tels que IL-‐10 ou IGF-‐1 sont
sécrétés par les cellules immunitaires dont l’absence conduit à une régénération
anormale et à la formation de tissu fibrotique[474, 475]. L’origine des cellules formant le
tissu fibrotique a été décrite dans certains organes ou tissus comme le rein ou le muscle
squelettique, notamment grâce à des études de lignage utilisant différents marqueurs
tels que PDGFRα[326, 330, 476], Gli1[477] ou ADAM12[298]. Chez les souris dystrophiques, les
cellules PDGFRα+ s’accumulent et sont à l’origine de la fibrose[326, 330, 476, 478]. La voie de
signalisation TGF-‐β est suractivée, inhibant l’apoptose des FAPs et induisant la
formation de tissu fibrotique.
La formation de tissu fibrotique lors des étapes précoces reste cependant essentielle à la
régénération tissulaire. Dans le muscle squelettique, après une blessure aigue, les

Discussion
169
éosinophiles sécrètent de l’IL-‐4 et de l’IL-‐13 qui inhibent la différentiation adipogénique
des FAPs, pour promouvoir leur différentiation en fibroblastes et la sécrétion de matrice
extracellulaire [328]. La déplétion conditionnelle des fibroblastes Tcf4+ au cours de la
régénération musculaire entraine une régénération incomplète[318]. Des approches
génétiques ou des expériences de co-‐culture ont montré que les cellules interstitielles
musculaires (FAP, péricytes, CSMs) sont une source de facteurs (IGF-‐1, Angpt)
permettant la différentiation, prolifération et fusion des myoblastes[289, 322, 479, 480]. Les
cellules interstitielles musculaires, et plus particulièrement les FAPs et les cellules
PDGFRα+, ont donc un rôle ambivalent au cours de la régénération musculaire, à la fois
bénéfique lorsque leur présence est transitoire mais délétère si elle est persistante. La
formation de tissu fibrotique au début de la régénération tissulaire est donc décrite dans
de nombreux tissus. Si ce processus est bien renseigné dans le tissu musculaire, ce n’est
pas le cas dans l’os où l’origine cellulaire de la fibrose n’est pas caractérisée dans la
littérature. Mes travaux de thèse ont montré que la formation initiale de fibrose fait
partie du processus physiologique de réparation osseuse comme observé dans d’autres
tissus mais que la persistance de la fibrose après un poly-‐trauma est néfaste au
processus de régénération. Mes résultats éclairent aussi sur les origines tissulaires de la
fibrose. Dans le cas d’une fracture combinée à une blessure du muscle, le lignage
musculaire Prx1+ forme à la fois du cartilage et de l’os mais aussi du tissu fibrotique,
alors que le périoste ne contribue pas à la formation du tissu fibrotique non-‐résorbé.
Cette donnée ouvre la voie à de nouvelles perspectives thérapeutiques pour la prise en
charge des fractures complexes associées à un taux régénération imparfaite élevé. Au vu
de l’essor de la thérapie cellulaire, du potentiel de régénération des cellules du muscle et
de leur accessibilité, les cellules musculaires interstitielles apparaissent comme une
source potentielle de cellule pour le traitement des fractures. Cependant, du fait de leur
potentiel fibrogénique, des études complémentaires devront être menées afin de
comprendre comment la fibrogenèse est induite dans ces cellules. Au vu de tous ces
éléments, nous pouvons penser que, dans le modèle de blessure traumatique,
l’environnement inflammatoire est dérégulé et les cellules Prx1+ sont orientés vers la
voie fibrogénique.
De nombreuses molécules ont été développées dans le but de traiter la fibrose. Elles
ciblent différentes étapes du processus fibrotique comme la formation ou la maturation
des fibres de collagène, ou encore la prolifération ou la présence des fibrocytes[481, 482].

Discussion
170
Afin de traiter les dystrophies musculaires et de réduire la formation de tissu fibrotique,
différentes molécules ciblant la voie de signalisation PDGFRα telles que l’Imatinib® ou le
Nilotinib® par exemple ont été testées. Ces molécules sont utilisées cliniquement dans
d’autres pathologies, notamment pour traiter certains cancers[483, 484]. Le traitement de
souris mdx par les inhibiteurs de la voie de signalisation PDGFRα améliore le phénotype
dystrophique en inhibant l’action du TGF-‐β, un facteur pro-‐fibrogénique[210, 332, 333].
D’autres études démontrent le potentiel anti-‐fibrotique de l’Imatinib® dans des
blessures chroniques d’organes tels que le foie ou les poumons[281, 485, 486]. Cependant, il
n’existe aucune étude menée sur leur action sur le tissu osseux. Notre étude démontre
ainsi le potentiel thérapeutique de l’Imatinib® dans le traitement des fractures
complexes. Malgré ces résultats encourageants, certaines études démontrent la toxicité
de l’Imatinib®, notamment au niveau de la peau[487], du cœur où l’Imatinib® induit un
stress du réticulum endoplasmique et un dysfonctionnement de l’autophagie conduisant
à la mort par apoptose des cellules[488], et de l’os en stimulant la formation osseuse par
les ostéoblastes[489, 490]. Ces effets pléiotropiques peuvent s’expliquer par la non-‐
spécificité de l’Imatinib® qui a pour cible PDGFR, Bcr-‐abl ou c-‐Kit en particulier et par
l’expression pan-‐tissulaire de ces protéines. Des traitements locaux pourront donc être
envisagés.
Ces résultats apportent donc de nouvelles perspectives en thérapie cellulaire et/ou
pharmacologiques.

Références
171
Références
1. Negishi-‐Koga T, Takayanagi H: Bone cell communication factors and Semaphorins. Bonekey Rep 2012, 1:183.
2. Colnot C, Zhang X, Knothe Tate ML: Current insights on the regenerative potential of the periosteum: molecular, cellular, and endogenous engineering approaches. J Orthop Res 2012, 30(12):1869-‐1878.
3. Clarke B: Normal bone anatomy and physiology. Clin J Am Soc Nephrol 2008, 3 Suppl 3:S131-‐139.
4. Poole RM: The incredible machine; 1986.
5. Steele D: The Anatomy and Biology of the Human Skeleton; 1988.
6. Morrison SJ, Scadden DT: The bone marrow niche for haematopoietic stem cells. Nature 2014, 505(7483):327-‐334.
7. Mendelson A, Frenette PS: Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med 2014, 20(8):833-‐846.
8. Kiel MJ, Morrison SJ: Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol 2008, 8(4):290-‐301.
9. Deldar A, Lewis H, Weiss L: Bone lining cells and hematopoiesis: an electron microscopic study of canine bone marrow. Anat Rec 1985, 213(2):187-‐201.
10. Benjamin M, Toumi H, Ralphs JR, Bydder G, Best TM, Milz S: Where tendons and ligaments meet bone: attachment sites ('entheses') in relation to exercise and/or mechanical load. J Anat 2006, 208(4):471-‐490.
11. Blaisdell FE: The osteogenic function of the periosteum. Arch Surg 1925, 11(6):933-‐945.
12. Paulsen F, Tillmann B: Functional and clinical anatomy of the posterior insertion of the human vocal ligament. Eur Arch Otorhinolaryngol 1997, 254(9-‐10):442-‐448.
13. Gronblad M, Liesi P, Korkala O, Karaharju E, Polak J: Innervation of human bone periosteum by peptidergic nerves. Anat Rec 1984, 209(3):297-‐299.
14. Hohmann EL, Elde RP, Rysavy JA, Einzig S, Gebhard RL: Innervation of periosteum and bone by sympathetic vasoactive intestinal peptide-‐containing nerve fibers. Science 1986, 232(4752):868-‐871.
15. Chanavaz M: Anatomy and histophysiology of the periosteum: quantification of the periosteal blood supply to the adjacent bone with 85Sr and gamma spectrometry. J Oral Implantol 1995, 21(3):214-‐219.

Références
172
16. Ferretti C, Lucarini G, Andreoni C, Salvolini E, Bianchi N, Vozzi G, Gigante A, Mattioli-‐Belmonte M: Human Periosteal Derived Stem Cell Potential: The Impact of age. Stem Cell Rev 2015, 11(3):487-‐500.
17. Roberts SJ, van Gastel N, Carmeliet G, Luyten FP: Uncovering the periosteum for skeletal regeneration: the stem cell that lies beneath. Bone 2015, 70:10-‐18.
18. Duchamp de Lageneste O, Julien A, Abou-‐Khalil R, Frangi G, Carvalho C, Cagnard N, Cordier C, Conway SJ, Colnot C: Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun 2018, 9(1):773.
19. Gray H: Anatomy of Human body; 1858.
20. Bilezikian J, Raisz, LG., Martin, TJ.: Principles of Bone Biology, Third Edition: Academic Press; 2008.
21. D. CJ: Bones: Structure and mechanics: Princeton University Press; 2002.
22. Vaughan TJ, McCarthy CT, McNamara LM: A three-‐scale finite element investigation into the effects of tissue mineralisation and lamellar organisation in human cortical and trabecular bone. J Mech Behav Biomed Mater 2012, 12:50-‐62.
23. Penido M, Alon US: Phosphate homeostasis and its role in bone health. Pediatr Nephrol 2012, 27(11):2039-‐2048.
24. Karsenty G, Ferron M: The contribution of bone to whole-‐organism physiology. Nature 2012, 481(7381):314-‐320.
25. Obri A, Khrimian L, Karsenty G, Oury F: Osteocalcin in the brain: from embryonic development to age-‐related decline in cognition. Nat Rev Endocrinol 2018, 14(3):174-‐182.
26. Berendsen AD, Olsen BR: Bone development. Bone 2015, 80:14-‐18.
27. Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ: Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 2002, 33(2):77-‐80.
28. Lim J, Tu X, Choi K, Akiyama H, Mishina Y, Long F: BMP-‐Smad4 signaling is required for precartilaginous mesenchymal condensation independent of Sox9 in the mouse. Dev Biol 2015, 400(1):132-‐138.
29. Ovchinnikov DA, Selever J, Wang Y, Chen YT, Mishina Y, Martin JF, Behringer RR: BMP receptor type IA in limb bud mesenchyme regulates distal outgrowth and patterning. Dev Biol 2006, 295(1):103-‐115.
30. Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B: Sox9 is required for cartilage formation. Nat Genet 1999, 22(1):85-‐89.

Références
173
31. Retting KN, Song B, Yoon BS, Lyons KM: BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136(7):1093-‐1104.
32. Yan J, Li J, Hu J, Zhang L, Wei C, Sultana N, Cai X, Zhang W, Cai CL: Smad4 deficiency impairs chondrocyte hypertrophy via the Runx2 transcription factor in mouse skeletal development. J Biol Chem 2018, 293(24):9162-‐9175.
33. Lai LP, Mitchell J: Indian hedgehog: its roles and regulation in endochondral bone development. J Cell Biochem 2005, 96(6):1163-‐1173.
34. Yang J, Andre P, Ye L, Yang YZ: The Hedgehog signalling pathway in bone formation. Int J Oral Sci 2015, 7(2):73-‐79.
35. Naski MC, Colvin JS, Coffin JD, Ornitz DM: Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 1998, 125(24):4977-‐4988.
36. Berendsen AD, Olsen BR: How vascular endothelial growth factor-‐A (VEGF) regulates differentiation of mesenchymal stem cells. J Histochem Cytochem 2014, 62(2):103-‐108.
37. Colnot C: Cellular and molecular interactions regulating skeletogenesis. J Cell Biochem 2005, 95(4):688-‐697.
38. Duan X, Murata Y, Liu Y, Nicolae C, Olsen BR, Berendsen AD: Vegfa regulates perichondrial vascularity and osteoblast differentiation in bone development. Development 2015, 142(11):1984-‐1991.
39. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M et al: Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89(5):755-‐764.
40. Bandyopadhyay A, Tsuji K, Cox K, Harfe BD, Rosen V, Tabin CJ: Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet 2006, 2(12):e216.
41. Salazar VS, Zarkadis N, Huang L, Norris J, Grimston SK, Mbalaviele G, Civitelli R: Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-‐imperfecta-‐like phenotype. J Cell Sci 2013, 126(Pt 21):4974-‐4984.
42. Adams CS, Shapiro IM: The fate of the terminally differentiated chondrocyte: evidence for microenvironmental regulation of chondrocyte apoptosis. Crit Rev Oral Biol Med 2002, 13(6):465-‐473.
43. Kronenberg HM: Developmental regulation of the growth plate. Nature 2003, 423(6937):332-‐336.

Références
174
44. Yang L, Tsang KY, Tang HC, Chan D, Cheah KS: Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A 2014, 111(33):12097-‐12102.
45. Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B: Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet 2014, 10(12):e1004820.
46. Egawa S, Miura S, Yokoyama H, Endo T, Tamura K: Growth and differentiation of a long bone in limb development, repair and regeneration. Dev Growth Differ 2014, 56(5):410-‐424.
47. Touaitahuata H, Cres G, de Rossi S, Vives V, Blangy A: The mineral dissolution function of osteoclasts is dispensable for hypertrophic cartilage degradation during long bone development and growth. Dev Biol 2014, 393(1):57-‐70.
48. Wang Y, Menendez A, Fong C, ElAlieh HZ, Kubota T, Long R, Bikle DD: IGF-‐I Signaling in Osterix-‐Expressing Cells Regulates Secondary Ossification Center Formation, Growth Plate Maturation, and Metaphyseal Formation During Postnatal Bone Development. J Bone Miner Res 2015, 30(12):2239-‐2248.
49. Colnot C, Lu C, Hu D, Helms JA: Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol 2004, 269(1):55-‐69.
50. Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM: Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 2010, 19(2):329-‐344.
51. Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, Ono N, Kronenberg HM, Frenette PS: Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell 2014, 29(3):340-‐349.
52. Ono N, Ono W, Mizoguchi T, Nagasawa T, Frenette PS, Kronenberg HM: Vasculature-‐associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Dev Cell 2014, 29(3):330-‐339.
53. Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, Helms JA, Kuo CJ, Kraft DL, Weissman IL: Endochondral ossification is required for haematopoietic stem-‐cell niche formation. Nature 2009, 457(7228):490-‐494.
54. Franz-‐Odendaal TA: Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed) 2011, 16:2734-‐2746.

Références
175
55. Baek WY, Kim YJ, de Crombrugghe B, Kim JE: Osterix is required for cranial neural crest-‐derived craniofacial bone formation. Biochem Biophys Res Commun 2013, 432(1):188-‐192.
56. Duan X, Bradbury SR, Olsen BR, Berendsen AD: VEGF stimulates intramembranous bone formation during craniofacial skeletal development. Matrix Biol 2016, 52-‐54:127-‐140.
57. Wilsman NJ, Farnum CE, Leiferman EM, Fry M, Barreto C: Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J Orthop Res 1996, 14(6):927-‐936.
58. Lefebvre V, Smits P: Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today 2005, 75(3):200-‐212.
59. van der Eerden BC, Karperien M, Wit JM: Systemic and local regulation of the growth plate. Endocr Rev 2003, 24(6):782-‐801.
60. Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B: The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 2002, 16(21):2813-‐2828.
61. Mackie EJ, Tatarczuch L, Mirams M: The skeleton: a multi-‐functional complex organ: the growth plate chondrocyte and endochondral ossification. J Endocrinol 2011, 211(2):109-‐121.
62. Oshima Y, Akiyama T, Hikita A, Iwasawa M, Nagase Y, Nakamura M, Wakeyama H, Kawamura N, Ikeda T, Chung UI et al: Pivotal role of Bcl-‐2 family proteins in the regulation of chondrocyte apoptosis. J Biol Chem 2008, 283(39):26499-‐26508.
63. Tsang KY, Chan D, Cheah KS: Fate of growth plate hypertrophic chondrocytes: death or lineage extension? Dev Growth Differ 2015, 57(2):179-‐192.
64. Henriksen K, Bollerslev J, Everts V, Karsdal MA: Osteoclast activity and subtypes as a function of physiology and pathology-‐-‐implications for future treatments of osteoporosis. Endocr Rev 2011, 32(1):31-‐63.
65. Rachner TD, Khosla S, Hofbauer LC: Osteoporosis: now and the future. Lancet 2011, 377(9773):1276-‐1287.
66. Raggatt LJ, Partridge NC: Cellular and molecular mechanisms of bone remodeling. J Biol Chem 2010, 285(33):25103-‐25108.
67. Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-‐dos-‐Santos AJ, Van G, Itie A et al: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-‐node organogenesis. Nature 1999, 397(6717):315-‐323.

Références
176
68. Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-‐Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF et al: Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 2011, 17(10):1231-‐1234.
69. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA: Matrix-‐embedded cells control osteoclast formation. Nat Med 2011, 17(10):1235-‐1241.
70. Everts V, Delaisse JM, Korper W, Niehof A, Vaes G, Beertsen W: Degradation of collagen in the bone-‐resorbing compartment underlying the osteoclast involves both cysteine-‐proteinases and matrix metalloproteinases. J Cell Physiol 1992, 150(2):221-‐231.
71. Alexander KA, Raggatt LJ, Millard S, Batoon L, Chiu-‐Ku Wu A, Chang MK, Hume DA, Pettit AR: Resting and injury-‐induced inflamed periosteum contain multiple macrophage subsets that are located at sites of bone growth and regeneration. Immunol Cell Biol 2017, 95(1):7-‐16.
72. Cho SW, Soki FN, Koh AJ, Eber MR, Entezami P, Park SI, van Rooijen N, McCauley LK: Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc Natl Acad Sci U S A 2014, 111(4):1545-‐1550.
73. Vi L, Baht GS, Whetstone H, Ng A, Wei Q, Poon R, Mylvaganam S, Grynpas M, Alman BA: Macrophages promote osteoblastic differentiation in-‐vivo: implications in fracture repair and bone homeostasis. J Bone Miner Res 2015, 30(6):1090-‐1102.
74. Martin TJ, Sims NA: Osteoclast-‐derived activity in the coupling of bone formation to resorption. Trends Mol Med 2005, 11(2):76-‐81.
75. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J et al: TGF-‐beta1-‐induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 2009, 15(7):757-‐765.
76. Crane JL, Cao X: Bone marrow mesenchymal stem cells and TGF-‐beta signaling in bone remodeling. J Clin Invest 2014, 124(2):466-‐472.
77. Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H: Osteoprotection by semaphorin 3A. Nature 2012, 485(7396):69-‐74.
78. Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA: Fracture healing as a post-‐natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem 2003, 88(5):873-‐884.
79. Dimitriou R, Tsiridis E, Giannoudis PV: Current concepts of molecular aspects of bone healing. Injury 2005, 36(12):1392-‐1404.
80. Ferguson C, Alpern E, Miclau T, Helms JA: Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev 1999, 87(1-‐2):57-‐66.

Références
177
81. Kolar P, Schmidt-‐Bleek K, Schell H, Gaber T, Toben D, Schmidmaier G, Perka C, Buttgereit F, Duda GN: The early fracture hematoma and its potential role in fracture healing. Tissue Eng Part B Rev 2010, 16(4):427-‐434.
82. Schell H, Duda GN, Peters A, Tsitsilonis S, Johnson KA, Schmidt-‐Bleek K: The haematoma and its role in bone healing. J Exp Orthop 2017, 4(1):5.
83. Kon T, Cho TJ, Aizawa T, Yamazaki M, Nooh N, Graves D, Gerstenfeld LC, Einhorn TA: Expression of osteoprotegerin, receptor activator of NF-‐kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J Bone Miner Res 2001, 16(6):1004-‐1014.
84. Mountziaris PM, Mikos AG: Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng Part B Rev 2008, 14(2):179-‐186.
85. Kovtun A, Bergdolt S, Wiegner R, Radermacher P, Huber-‐Lang M, Ignatius A: The crucial role of neutrophil granulocytes in bone fracture healing. Eur Cell Mater 2016, 32:152-‐162.
86. Xing Z, Lu C, Hu D, Yu YY, Wang X, Colnot C, Nakamura M, Wu Y, Miclau T, Marcucio RS: Multiple roles for CCR2 during fracture healing. Dis Model Mech 2010, 3(7-‐8):451-‐458.
87. Wallace A, Cooney TE, Englund R, Lubahn JD: Effects of interleukin-‐6 ablation on fracture healing in mice. J Orthop Res 2011, 29(9):1437-‐1442.
88. Yang X, Ricciardi BF, Hernandez-‐Soria A, Shi Y, Pleshko Camacho N, Bostrom MP: Callus mineralization and maturation are delayed during fracture healing in interleukin-‐6 knockout mice. Bone 2007, 41(6):928-‐936.
89. Nam D, Mau E, Wang Y, Wright D, Silkstone D, Whetstone H, Whyne C, Alman B: T-‐lymphocytes enable osteoblast maturation via IL-‐17F during the early phase of fracture repair. PLoS One 2012, 7(6):e40044.
90. Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, Takayanagi H: IL-‐17-‐producing gammadelta T cells enhance bone regeneration. Nat Commun 2016, 7:10928.
91. Schlundt C, El Khassawna T, Serra A, Dienelt A, Wendler S, Schell H, van Rooijen N, Radbruch A, Lucius R, Hartmann S et al: Macrophages in bone fracture healing: Their essential role in endochondral ossification. Bone 2018, 106:78-‐89.
92. Wang J, Arase H: Regulation of immune responses by neutrophils. Ann N Y Acad Sci 2014, 1319:66-‐81.
93. Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL: Expression of a truncated, kinase-‐defective TGF-‐beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol 1997, 139(2):541-‐552.

Références
178
94. Wu M, Chen G, Li YP: TGF-‐beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 2016, 4:16009.
95. Gong L, Zhao Y, Zhang Y, Ruan Z: The Macrophage Polarization Regulates MSC Osteoblast Differentiation in vitro. Ann Clin Lab Sci 2016, 46(1):65-‐71.
96. Schmidt-‐Bleek K, Kwee BJ, Mooney DJ, Duda GN: Boon and Bane of Inflammation in Bone Tissue Regeneration and Its Link with Angiogenesis. Tissue Eng Part B Rev 2015, 21(4):354-‐364.
97. Claes L, Recknagel S, Ignatius A: Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol 2012, 8(3):133-‐143.
98. Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA: Impaired fracture healing in the absence of TNF-‐alpha signaling: the role of TNF-‐alpha in endochondral cartilage resorption. J Bone Miner Res 2003, 18(9):1584-‐1592.
99. Loi F, Cordova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB: Inflammation, fracture and bone repair. Bone 2016, 86:119-‐130.
100. Naik AA, Xie C, Zuscik MJ, Kingsley P, Schwarz EM, Awad H, Guldberg R, Drissi H, Puzas JE, Boyce B et al: Reduced COX-‐2 expression in aged mice is associated with impaired fracture healing. J Bone Miner Res 2009, 24(2):251-‐264.
101. Yamaguchi T, Takada Y, Maruyama K, Shimoda K, Arai Y, Nango N, Kosaki N, Takaishi H, Toyama Y, Matsuo K: Fra-‐1/AP-‐1 impairs inflammatory responses and chondrogenesis in fracture healing. J Bone Miner Res 2009, 24(12):2056-‐2065.
102. Otsuru S, Tamai K, Yamazaki T, Yoshikawa H, Kaneda Y: Circulating bone marrow-‐derived osteoblast progenitor cells are recruited to the bone-‐forming site by the CXCR4/stromal cell-‐derived factor-‐1 pathway. Stem Cells 2008, 26(1):223-‐234.
103. Toupadakis CA, Wong A, Genetos DC, Chung DJ, Murugesh D, Anderson MJ, Loots GG, Christiansen BA, Kapatkin AS, Yellowley CE: Long-‐term administration of AMD3100, an antagonist of SDF-‐1/CXCR4 signaling, alters fracture repair. J Orthop Res 2012, 30(11):1853-‐1859.
104. Yellowley C: CXCL12/CXCR4 signaling and other recruitment and homing pathways in fracture repair. Bonekey Rep 2013, 2:300.
105. Bielby R, Jones E, McGonagle D: The role of mesenchymal stem cells in maintenance and repair of bone. Injury 2007, 38 Suppl 1:S26-‐32.
106. Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V: BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet 2006, 38(12):1424-‐1429.

Références
179
107. Yu YY, Lieu S, Lu C, Miclau T, Marcucio RS, Colnot C: Immunolocalization of BMPs, BMP antagonists, receptors, and effectors during fracture repair. Bone 2010, 46(3):841-‐851.
108. Behr B, Leucht P, Longaker MT, Quarto N: Fgf-‐9 is required for angiogenesis and osteogenesis in long bone repair. Proc Natl Acad Sci U S A 2010, 107(26):11853-‐11858.
109. Behr B, Sorkin M, Manu A, Lehnhardt M, Longaker MT, Quarto N: Fgf-‐18 is required for osteogenesis but not angiogenesis during long bone repair. Tissue Eng Part A 2011, 17(15-‐16):2061-‐2069.
110. Hu K, Olsen BR: The roles of vascular endothelial growth factor in bone repair and regeneration. Bone 2016, 91:30-‐38.
111. Hu K, Olsen BR: Osteoblast-‐derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J Clin Invest 2016, 126(2):509-‐526.
112. Lau KW, Rundle CH, Zhou XD, Baylink DJ, Sheng MH: Conditional deletion of IGF-‐I in osteocytes unexpectedly accelerates bony union of the fracture gap in mice. Bone 2016, 92:18-‐28.
113. Yu YY, Lieu S, Lu C, Colnot C: Bone morphogenetic protein 2 stimulates endochondral ossification by regulating periosteal cell fate during bone repair. Bone 2010, 47(1):65-‐73.
114. Liu C, Carrera R, Flamini V, Kenny L, Cabahug-‐Zuckerman P, George BM, Hunter D, Liu B, Singh G, Leucht P et al: Effects of mechanical loading on cortical defect repair using a novel mechanobiological model of bone healing. Bone 2018, 108:145-‐155.
115. He X, Bougioukli S, Ortega B, Arevalo E, Lieberman JR, McMahon AP: Sox9 positive periosteal cells in fracture repair of the adult mammalian long bone. Bone 2017, 103:12-‐19.
116. Behonick DJ, Xing Z, Lieu S, Buckley JM, Lotz JC, Marcucio RS, Werb Z, Miclau T, Colnot C: Role of matrix metalloproteinase 13 in both endochondral and intramembranous ossification during skeletal regeneration. PLoS One 2007, 2(11):e1150.
117. Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA: Altered fracture repair in the absence of MMP9. Development 2003, 130(17):4123-‐4133.
118. Bahney CS, Hu DP, Taylor AJ, Ferro F, Britz HM, Hallgrimsson B, Johnstone B, Miclau T, Marcucio RS: Stem cell-‐derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res 2014, 29(5):1269-‐1282.
119. Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, Marcucio RS, Bahney CS: Cartilage to bone transformation during fracture healing is coordinated by

Références
180
the invading vasculature and induction of the core pluripotency genes. Development 2017, 144(2):221-‐234.
120. Park J, Gebhardt M, Golovchenko S, Perez-‐Branguli F, Hattori T, Hartmann C, Zhou X, deCrombrugghe B, Stock M, Schneider H et al: Dual pathways to endochondral osteoblasts: a novel chondrocyte-‐derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open 2015, 4(5):608-‐621.
121. Wang Q, Huang C, Xue M, Zhang X: Expression of endogenous BMP-‐2 in periosteal progenitor cells is essential for bone healing. Bone 2011, 48(3):524-‐532.
122. Baht GS, Silkstone D, Nadesan P, Whetstone H, Alman BA: Activation of hedgehog signaling during fracture repair enhances osteoblastic-‐dependent matrix formation. J Orthop Res 2014, 32(4):581-‐586.
123. Jilka RL, O'Brien CA, Ali AA, Roberson PK, Weinstein RS, Manolagas SC: Intermittent PTH stimulates periosteal bone formation by actions on post-‐mitotic preosteoblasts. Bone 2009, 44(2):275-‐286.
124. Ogita M, Rached MT, Dworakowski E, Bilezikian JP, Kousteni S: Differentiation and proliferation of periosteal osteoblast progenitors are differentially regulated by estrogens and intermittent parathyroid hormone administration. Endocrinology 2008, 149(11):5713-‐5723.
125. Bodine PV, Seestaller-‐Wehr L, Kharode YP, Bex FJ, Komm BS: Bone anabolic effects of parathyroid hormone are blunted by deletion of the Wnt antagonist secreted frizzled-‐related protein-‐1. J Cell Physiol 2007, 210(2):352-‐357.
126. Komatsu DE, Mary MN, Schroeder RJ, Robling AG, Turner CH, Warden SJ: Modulation of Wnt signaling influences fracture repair. J Orthop Res 2010, 28(7):928-‐936.
127. Secreto FJ, Hoeppner LH, Westendorf JJ: Wnt signaling during fracture repair. Curr Osteoporos Rep 2009, 7(2):64-‐69.
128. Ling L, Nurcombe V, Cool SM: Wnt signaling controls the fate of mesenchymal stem cells. Gene 2009, 433(1-‐2):1-‐7.
129. Gnyubkin V, Guignandon A, Laroche N, Vanden-‐Bossche A, Normand M, Lafage-‐Proust MH, Vico L: Effects of chronic hypergravity: from adaptive to deleterious responses in growing mouse skeleton. J Appl Physiol (1985) 2015, 119(8):908-‐917.
130. Iwaniec UT, Turner RT: Influence of body weight on bone mass, architecture and turnover. J Endocrinol 2016, 230(3):R115-‐130.
131. Wade CE: Responses across the gravity continuum: hypergravity to microgravity. Adv Space Biol Med 2005, 10:225-‐245.

Références
181
132. Edwards WB, Schnitzer TJ, Troy KL: The mechanical consequence of actual bone loss and simulated bone recovery in acute spinal cord injury. Bone 2014, 60:141-‐147.
133. Sievanen H: Immobilization and bone structure in humans. Arch Biochem Biophys 2010, 503(1):146-‐152.
134. Vico L, Collet P, Guignandon A, Lafage-‐Proust MH, Thomas T, Rehaillia M, Alexandre C: Effects of long-‐term microgravity exposure on cancellous and cortical weight-‐bearing bones of cosmonauts. Lancet 2000, 355(9215):1607-‐1611.
135. Lloyd SA, Lang CH, Zhang Y, Paul EM, Laufenberg LJ, Lewis GS, Donahue HJ: Interdependence of muscle atrophy and bone loss induced by mechanical unloading. J Bone Miner Res 2014, 29(5):1118-‐1130.
136. Gerbaix M, Gnyubkin V, Farlay D, Olivier C, Ammann P, Courbon G, Laroche N, Genthial R, Follet H, Peyrin F et al: One-‐month spaceflight compromises the bone microstructure, tissue-‐level mechanical properties, osteocyte survival and lacunae volume in mature mice skeletons. Sci Rep 2017, 7(1):2659.
137. Zayzafoon M, Gathings WE, McDonald JM: Modeled microgravity inhibits osteogenic differentiation of human mesenchymal stem cells and increases adipogenesis. Endocrinology 2004, 145(5):2421-‐2432.
138. Isaksson H, Comas O, van Donkelaar CC, Mediavilla J, Wilson W, Huiskes R, Ito K: Bone regeneration during distraction osteogenesis: mechano-‐regulation by shear strain and fluid velocity. J Biomech 2007, 40(9):2002-‐2011.
139. Miclau T, Lu C, Thompson Z, Choi P, Puttlitz C, Marcucio R, Helms JA: Effects of delayed stabilization on fracture healing. J Orthop Res 2007, 25(12):1552-‐1558.
140. Chao EY, Inoue N: Biophysical stimulation of bone fracture repair, regeneration and remodelling. Eur Cell Mater 2003, 6:72-‐84; discussion 84-‐75.
141. Perren SM: Evolution of the internal fixation of long bone fractures. The scientific basis of biological internal fixation: choosing a new balance between stability and biology. J Bone Joint Surg Br 2002, 84(8):1093-‐1110.
142. Grundnes O, Reikeras O: Effects of instability on bone healing. Femoral osteotomies studied in rats. Acta Orthop Scand 1993, 64(1):55-‐58.
143. Jagodzinski M, Krettek C: Effect of mechanical stability on fracture healing-‐-‐an update. Injury 2007, 38 Suppl 1:S3-‐10.
144. Histing T, Heerschop K, Klein M, Scheuer C, Stenger D, Holstein JH, Pohlemann T, Menger MD: Characterization of the healing process in non-‐stabilized and stabilized femur fractures in mice. Arch Orthop Trauma Surg 2016, 136(2):203-‐211.

Références
182
145. Lu C, Saless N, Hu D, Wang X, Xing Z, Hou H, Williams B, Swartz HM, Colnot C, Miclau T et al: Mechanical stability affects angiogenesis during early fracture healing. J Orthop Trauma 2011, 25(8):494-‐499.
146. Wang SJ, Lewallen DG, Bolander ME, Chao EY, Ilstrup DM, Greenleaf JF: Low intensity ultrasound treatment increases strength in a rat femoral fracture model. J Orthop Res 1994, 12(1):40-‐47.
147. Watanabe Y, Matsushita T, Bhandari M, Zdero R, Schemitsch EH: Ultrasound for fracture healing: current evidence. J Orthop Trauma 2010, 24 Suppl 1:S56-‐61.
148. Thompson Z, Miclau T, Hu D, Helms JA: A model for intramembranous ossification during fracture healing. J Orthop Res 2002, 20(5):1091-‐1098.
149. Mehrotra M, Williams CR, Ogawa M, LaRue AC: Hematopoietic stem cells give rise to osteo-‐chondrogenic cells. Blood Cells Mol Dis 2013, 50(1):41-‐49.
150. Otsuru S, Overholt KM, Olson TS, Hofmann TJ, Guess AJ, Velazquez VM, Kaito T, Dominici M, Horwitz EM: Hematopoietic derived cells do not contribute to osteogenesis as osteoblasts. Bone 2017, 94:1-‐9.
151. Kumagai K, Vasanji A, Drazba JA, Butler RS, Muschler GF: Circulating cells with osteogenic potential are physiologically mobilized into the fracture healing site in the parabiotic mice model. J Orthop Res 2008, 26(2):165-‐175.
152. Shirley D, Marsh D, Jordan G, McQuaid S, Li G: Systemic recruitment of osteoblastic cells in fracture healing. J Orthop Res 2005, 23(5):1013-‐1021.
153. Wang XX, Allen RJ, Jr., Tutela JP, Sailon A, Allori AC, Davidson EH, Paek GK, Saadeh PB, McCarthy JG, Warren SM: Progenitor cell mobilization enhances bone healing by means of improved neovascularization and osteogenesis. Plast Reconstr Surg 2011, 128(2):395-‐405.
154. Colnot C: Cell sources for bone tissue engineering: insights from basic science. Tissue Eng Part B Rev 2011, 17(6):449-‐457.
155. McCulloch EA, Till JE: The sensitivity of cells from normal mouse bone marrow to gamma radiation in vitro and in vivo. Radiat Res 1962, 16:822-‐832.
156. Till JE: Radiosensitivity and chromosome numbers in strain L mouse cells in tissue culture. Radiat Res 1961, 15:400-‐409.
157. Becker AJ, Mc CE, Till JE: Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963, 197:452-‐454.
158. Siminovitch L, McCulloch EA, Till JE: The Distribution of Colony-‐Forming Cells among Spleen Colonies. J Cell Comp Physiol 1963, 62:327-‐336.

Références
183
159. Till JE, McCulloch EA: Hemopoietic stem cell differentiation. Biochim Biophys Acta 1980, 605(4):431-‐459.
160. Friedenstein AJ, Piatetzky S, II, Petrakova KV: Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol 1966, 16(3):381-‐390.
161. Friedenstein AJ, Chailakhjan RK, Lalykina KS: The development of fibroblast colonies in monolayer cultures of guinea-‐pig bone marrow and spleen cells. Cell Tissue Kinet 1970, 3(4):393-‐403.
162. Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-‐Borok IV: Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation 1974, 17(4):331-‐340.
163. Owen M, Friedenstein AJ: Stromal stem cells: marrow-‐derived osteogenic precursors. Ciba Found Symp 1988, 136:42-‐60.
164. Bianco P, Robey PG, Simmons PJ: Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell 2008, 2(4):313-‐319.
165. Caplan AI: Mesenchymal stem cells. J Orthop Res 1991, 9(5):641-‐650.
166. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR: Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284(5411):143-‐147.
167. Dominici M, Le Blanc K, Mueller I, Slaper-‐Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D, Horwitz E: Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8(4):315-‐317.
168. Alt E, Yan Y, Gehmert S, Song YH, Altman A, Gehmert S, Vykoukal D, Bai X: Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-‐forming potential. Biol Cell 2011, 103(4):197-‐208.
169. Phinney DG, Kopen G, Isaacson RL, Prockop DJ: Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: variations in yield, growth, and differentiation. J Cell Biochem 1999, 72(4):570-‐585.
170. Traktuev DO, Merfeld-‐Clauss S, Li J, Kolonin M, Arap W, Pasqualini R, Johnstone BH, March KL: A population of multipotent CD34-‐positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ Res 2008, 102(1):77-‐85.
171. MacCord K: "Mesenchyme". In.: Embryo Project Encyclopedia; 2012.
172. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M et al: Self-‐renewing osteoprogenitors in bone

Références
184
marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131(2):324-‐336.
173. da Silva Meirelles L, Chagastelles PC, Nardi NB: Mesenchymal stem cells reside in virtually all post-‐natal organs and tissues. J Cell Sci 2006, 119(Pt 11):2204-‐2213.
174. Sacchetti B, Funari A, Remoli C, Giannicola G, Kogler G, Liedtke S, Cossu G, Serafini M, Sampaolesi M, Tagliafico E et al: No Identical "Mesenchymal Stem Cells" at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Reports 2016, 6(6):897-‐913.
175. Lindner U, Kramer J, Rohwedel J, Schlenke P: Mesenchymal Stem or Stromal Cells: Toward a Better Understanding of Their Biology? Transfus Med Hemother 2010, 37(2):75-‐83.
176. Prockop DJ: Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276(5309):71-‐74.
177. Caplan AI: Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl Med 2017, 6(6):1445-‐1451.
178. R. R: Rubin's Pathology: Clinicopathologic Foundations of Medicine; 2007.
179. Birbrair A, Frenette PS: Niche heterogeneity in the bone marrow. Ann N Y Acad Sci 2016, 1370(1):82-‐96.
180. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR et al: Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425(6960):841-‐846.
181. Ding L, Saunders TL, Enikolopov G, Morrison SJ: Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481(7382):457-‐462.
182. Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, Lin CP, Kronenberg HM, Scadden DT: Endogenous bone marrow MSCs are dynamic, fate-‐restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012, 10(3):259-‐272.
183. Arthur A, Zannettino A, Gronthos S: The therapeutic applications of multipotential mesenchymal/stromal stem cells in skeletal tissue repair. J Cell Physiol 2009, 218(2):237-‐245.
184. Turner L, Knoepfler P: Selling Stem Cells in the USA: Assessing the Direct-‐to-‐Consumer Industry. Cell Stem Cell 2016, 19(2):154-‐157.
185. Colnot C: Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res 2009, 24(2):274-‐282.

Références
185
186. Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y et al: Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160(1-‐2):269-‐284.
187. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ: Leptin-‐receptor-‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014, 15(2):154-‐168.
188. Grcevic D, Pejda S, Matthews BG, Repic D, Wang L, Li H, Kronenberg MS, Jiang X, Maye P, Adams DJ et al: In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells 2012, 30(2):187-‐196.
189. Deveza L, Ortinau L, Lei K, Park D: Comparative analysis of gene expression identifies distinct molecular signatures of bone marrow-‐ and periosteal-‐skeletal stem/progenitor cells. PLoS One 2018, 13(1):e0190909.
190. Kalajzic Z, Li H, Wang LP, Jiang X, Lamothe K, Adams DJ, Aguila HL, Rowe DW, Kalajzic I: Use of an alpha-‐smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone 2008, 43(3):501-‐510.
191. Caplan AI, Correa D: The MSC: an injury drugstore. Cell Stem Cell 2011, 9(1):11-‐15.
192. Granero-‐Molto F, Weis JA, Miga MI, Landis B, Myers TJ, O'Rear L, Longobardi L, Jansen ED, Mortlock DP, Spagnoli A: Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells 2009, 27(8):1887-‐1898.
193. Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T: Stromal cell-‐derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum 2009, 60(3):813-‐823.
194. Shinohara K, Greenfield S, Pan H, Vasanji A, Kumagai K, Midura RJ, Kiedrowski M, Penn MS, Muschler GF: Stromal cell-‐derived factor-‐1 and monocyte chemotactic protein-‐3 improve recruitment of osteogenic cells into sites of musculoskeletal repair. J Orthop Res 2011, 29(7):1064-‐1069.
195. Wang X, Wang Y, Gou W, Lu Q, Peng J, Lu S: Role of mesenchymal stem cells in bone regeneration and fracture repair: a review. Int Orthop 2013, 37(12):2491-‐2498.
196. D D: Lettre sur la formation des os dans les animaux, et du bois dans les arbres. Rec périod Obs Med Chir Pharm 1757, 7:153-‐160.
197. Tang XM, Chai BF: Ultrastructural investigation of osteogenic cells. Chin Med J (Engl) 1986, 99(12):950-‐956.

Références
186
198. Diaz-‐Flores L, Gutierrez R, Lopez-‐Alonso A, Gonzalez R, Varela H: Pericytes as a supplementary source of osteoblasts in periosteal osteogenesis. Clin Orthop Relat Res 1992(275):280-‐286.
199. Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O'Leary P et al: Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience 2002, 113(1):155-‐166.
200. C. D: On the injuries and diseases of bones: being selections from the collected edition of the clinical lectures of baron Dupuytren. London, Sydenham Society 1847.
201. Zhang X, Xie C, Lin AS, Ito H, Awad H, Lieberman JR, Rubery PT, Schwarz EM, O'Keefe RJ, Guldberg RE: Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J Bone Miner Res 2005, 20(12):2124-‐2137.
202. Asakura A, Komaki M, Rudnicki M: Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68(4-‐5):245-‐253.
203. Sondag GR, Salihoglu S, Lababidi SL, Crowder DC, Moussa FM, Abdelmagid SM, Safadi FF: Osteoactivin induces transdifferentiation of C2C12 myoblasts into osteoblasts. J Cell Physiol 2014, 229(7):955-‐966.
204. Liu R, Birke O, Morse A, Peacock L, Mikulec K, Little DG, Schindeler A: Myogenic progenitors contribute to open but not closed fracture repair. BMC Musculoskelet Disord 2011, 12:288.
205. Cairns DM, Liu R, Sen M, Canner JP, Schindeler A, Little DG, Zeng L: Interplay of Nkx3.2, Sox9 and Pax3 regulates chondrogenic differentiation of muscle progenitor cells. PLoS One 2012, 7(7):e39642.
206. Glass GE, Chan JK, Freidin A, Feldmann M, Horwood NJ, Nanchahal J: TNF-‐alpha promotes fracture repair by augmenting the recruitment and differentiation of muscle-‐derived stromal cells. Proc Natl Acad Sci U S A 2011, 108(4):1585-‐1590.
207. Rando TA, Blau HM: Primary mouse myoblast purification, characterization, and transplantation for cell-‐mediated gene therapy. J Cell Biol 1994, 125(6):1275-‐1287.
208. Lee JY, Qu-‐Petersen Z, Cao B, Kimura S, Jankowski R, Cummins J, Usas A, Gates C, Robbins P, Wernig A et al: Clonal isolation of muscle-‐derived cells capable of enhancing muscle regeneration and bone healing. J Cell Biol 2000, 150(5):1085-‐1100.
209. Davis KM, Griffin KS, Chu TG, Wenke JC, Corona BT, McKinley TO, Kacena MA: Muscle-‐bone interactions during fracture healing. J Musculoskelet Neuronal Interact 2015, 15(1):1-‐9.

Références
187
210. Lemos DR, Eisner C, Hopkins CI, Rossi FMV: Skeletal muscle-‐resident MSCs and bone formation. Bone 2015, 80:19-‐23.
211. Einhorn TA: Enhancement of fracture-‐healing. J Bone Joint Surg Am 1995, 77(6):940-‐956.
212. Hak DJ, Fitzpatrick D, Bishop JA, Marsh JL, Tilp S, Schnettler R, Simpson H, Alt V: Delayed union and nonunions: epidemiology, clinical issues, and financial aspects. Injury 2014, 45 Suppl 2:S3-‐7.
213. Axelrad TW, Kakar S, Einhorn TA: New technologies for the enhancement of skeletal repair. Injury 2007, 38 Suppl 1:S49-‐62.
214. Buza JA, 3rd, Einhorn T: Bone healing in 2016. Clin Cases Miner Bone Metab 2016, 13(2):101-‐105.
215. Mauffrey C, Barlow BT, Smith W: Management of segmental bone defects. J Am Acad Orthop Surg 2015, 23(3):143-‐153.
216. Dahabreh Z, Calori GM, Kanakaris NK, Nikolaou VS, Giannoudis PV: A cost analysis of treatment of tibial fracture nonunion by bone grafting or bone morphogenetic protein-‐7. Int Orthop 2009, 33(5):1407-‐1414.
217. Morelli I, Drago L, George DA, Gallazzi E, Scarponi S, Romano CL: Masquelet technique: myth or reality? A systematic review and meta-‐analysis. Injury 2016, 47 Suppl 6:S68-‐S76.
218. Wang EA, Rosen V, D'Alessandro JS, Bauduy M, Cordes P, Harada T, Israel DI, Hewick RM, Kerns KM, LaPan P et al: Recombinant human bone morphogenetic protein induces bone formation. Proc Natl Acad Sci U S A 1990, 87(6):2220-‐2224.
219. Bishop JA, Palanca AA, Bellino MJ, Lowenberg DW: Assessment of compromised fracture healing. J Am Acad Orthop Surg 2012, 20(5):273-‐282.
220. Giannoudis PV, Einhorn TA, Marsh D: Fracture healing: the diamond concept. Injury 2007, 38 Suppl 4:S3-‐6.
221. Giannoudis PV, Gudipati S, Harwood P, Kanakaris NK: Long bone non-‐unions treated with the diamond concept: a case series of 64 patients. Injury 2015, 46 Suppl 8:S48-‐54.
222. Giannoudis PV: Treatment of bone defects: Bone transport or the induced membrane technique? Injury 2016, 47(2):291-‐292.
223. Tang Q, Tong M, Zheng G, Shen L, Shang P, Liu H: Masquelet's induced membrane promotes the osteogenic differentiation of bone marrow mesenchymal stem cells by activating the Smad and MAPK pathways. Am J Transl Res 2018, 10(4):1211-‐1219.

Références
188
224. Auregan JC, Begue T, Rigoulot G, Glorion C, Pannier S: Success rate and risk factors of failure of the induced membrane technique in children: a systematic review. Injury 2016, 47 Suppl 6:S62-‐S67.
225. Im GI: Clinical use of stem cells in orthopaedics. Eur Cell Mater 2017, 33:183-‐196.
226. Chahla J, Mannava S, Cinque ME, Geeslin AG, Codina D, LaPrade RF: Bone Marrow Aspirate Concentrate Harvesting and Processing Technique. Arthrosc Tech 2017, 6(2):e441-‐e445.
227. Miller RP, Hanley PJ: Isolation and Manufacture of Clinical-‐Grade Bone Marrow-‐Derived Human Mesenchymal Stromal Cells. Methods Mol Biol 2016, 1416:301-‐312.
228. Robey PG, Kuznetsov SA, Ren J, Klein HG, Sabatino M, Stroncek DF: Generation of clinical grade human bone marrow stromal cells for use in bone regeneration. Bone 2015, 70:87-‐92.
229. Bara JJ, Richards RG, Alini M, Stoddart MJ: Concise review: Bone marrow-‐derived mesenchymal stem cells change phenotype following in vitro culture: implications for basic research and the clinic. Stem Cells 2014, 32(7):1713-‐1723.
230. Giannotti S, Trombi L, Bottai V, Ghilardi M, D'Alessandro D, Danti S, Dell'Osso G, Guido G, Petrini M: Use of autologous human mesenchymal stromal cell/fibrin clot constructs in upper limb non-‐unions: long-‐term assessment. PLoS One 2013, 8(8):e73893.
231. Quarto R, Mastrogiacomo M, Cancedda R, Kutepov SM, Mukhachev V, Lavroukov A, Kon E, Marcacci M: Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med 2001, 344(5):385-‐386.
232. James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, Ting K, Soo C: A Review of the Clinical Side Effects of Bone Morphogenetic Protein-‐2. Tissue Eng Part B Rev 2016, 22(4):284-‐297.
233. Derubeis AR, Cancedda R: Bone marrow stromal cells (BMSCs) in bone engineering: limitations and recent advances. Ann Biomed Eng 2004, 32(1):160-‐165.
234. Ball MD, Bonzani IC, Bovis MJ, Williams A, Stevens MM: Human periosteum is a source of cells for orthopaedic tissue engineering: a pilot study. Clin Orthop Relat Res 2011, 469(11):3085-‐3093.
235. Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L: Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 1994, 331(14):889-‐895.
236. Paolantonio M, Femminella B, Coppolino E, Sammartino G, D'Arcangelo C, Perfetti G, Perinetti G: Autogenous periosteal barrier membranes and bone grafts in

Références
189
the treatment of periodontal intrabony defects of single-‐rooted teeth: a 12-‐month reentry randomized controlled clinical trial. J Periodontol 2010, 81(11):1587-‐1595.
237. Harhaus L, Huang JJ, Kao SW, Wu YL, Mackert GA, Honer B, Cheng MH, Kneser U, Cheng CM: The vascularized periosteum flap as novel tissue engineering model for repair of cartilage defects. J Cell Mol Med 2015, 19(6):1273-‐1283.
238. Balogh ZJ, Reumann MK, Gruen RL, Mayer-‐Kuckuk P, Schuetz MA, Harris IA, Gabbe BJ, Bhandari M: Advances and future directions for management of trauma patients with musculoskeletal injuries. Lancet 2012, 380(9847):1109-‐1119.
239. Frontera WR, Ochala J: Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 2015, 96(3):183-‐195.
240. Dietrich S, Abou-‐Rebyeh F, Brohmann H, Bladt F, Sonnenberg-‐Riethmacher E, Yamaai T, Lumsden A, Brand-‐Saberi B, Birchmeier C: The role of SF/HGF and c-‐Met in the development of skeletal muscle. Development 1999, 126(8):1621-‐1629.
241. Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R: MyoD or Myf-‐5 is required for the formation of skeletal muscle. Cell 1993, 75(7):1351-‐1359.
242. Schafer K, Braun T: Early specification of limb muscle precursor cells by the homeobox gene Lbx1h. Nat Genet 1999, 23(2):213-‐216.
243. Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F: The formation of skeletal muscle: from somite to limb. J Anat 2003, 202(1):59-‐68.
244. Relaix F, Rocancourt D, Mansouri A, Buckingham M: A Pax3/Pax7-‐dependent population of skeletal muscle progenitor cells. Nature 2005, 435(7044):948-‐953.
245. Chal J, Pourquie O: Making muscle: skeletal myogenesis in vivo and in vitro. Development 2017, 144(12):2104-‐2122.
246. Schuster-‐Gossler K, Cordes R, Gossler A: Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc Natl Acad Sci U S A 2007, 104(2):537-‐542.
247. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A et al: BMP signaling controls muscle mass. Nat Genet 2013, 45(11):1309-‐1318.
248. Mauro A: Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 1961, 9:493-‐495.

Références
190
249. Seale P, Sabourin LA, Girgis-‐Gabardo A, Mansouri A, Gruss P, Rudnicki MA: Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102(6):777-‐786.
250. Lepper C, Partridge TA, Fan CM: An absolute requirement for Pax7-‐positive satellite cells in acute injury-‐induced skeletal muscle regeneration. Development 2011, 138(17):3639-‐3646.
251. Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-‐Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A: Pax7-‐expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011, 138(17):3647-‐3656.
252. Lepper C, Conway SJ, Fan CM: Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 2009, 460(7255):627-‐631.
253. Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M: Direct isolation of satellite cells for skeletal muscle regeneration. Science 2005, 309(5743):2064-‐2067.
254. von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA: Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A 2013, 110(41):16474-‐16479.
255. Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM: Self-‐renewal and expansion of single transplanted muscle stem cells. Nature 2008, 456(7221):502-‐506.
256. Chazaud B, Brigitte M, Yacoub-‐Youssef H, Arnold L, Gherardi R, Sonnet C, Lafuste P, Chretien F: Dual and beneficial roles of macrophages during skeletal muscle regeneration. Exerc Sport Sci Rev 2009, 37(1):18-‐22.
257. Serrano AL, Baeza-‐Raja B, Perdiguero E, Jardi M, Munoz-‐Canoves P: Interleukin-‐6 is an essential regulator of satellite cell-‐mediated skeletal muscle hypertrophy. Cell Metab 2008, 7(1):33-‐44.
258. Charge SB, Rudnicki MA: Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004, 84(1):209-‐238.
259. Kitzmann M, Carnac G, Vandromme M, Primig M, Lamb NJ, Fernandez A: The muscle regulatory factors MyoD and myf-‐5 undergo distinct cell cycle-‐specific expression in muscle cells. J Cell Biol 1998, 142(6):1447-‐1459.
260. Gunther S, Kim J, Kostin S, Lepper C, Fan CM, Braun T: Myf5-‐positive satellite cells contribute to Pax7-‐dependent long-‐term maintenance of adult muscle stem cells. Cell Stem Cell 2013, 13(5):590-‐601.
261. Chinzei N, Hayashi S, Ueha T, Fujishiro T, Kanzaki N, Hashimoto S, Sakata S, Kihara S, Haneda M, Sakai Y et al: P21 deficiency delays regeneration of skeletal muscular tissue. PLoS One 2015, 10(5):e0125765.

Références
191
262. Karalaki M, Fili S, Philippou A, Koutsilieris M: Muscle regeneration: cellular and molecular events. In Vivo 2009, 23(5):779-‐796.
263. Torrente Y, Belicchi M, Marchesi C, D'Antona G, Cogiamanian F, Pisati F, Gavina M, Giordano R, Tonlorenzi R, Fagiolari G et al: Autologous transplantation of muscle-‐derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant 2007, 16(6):563-‐577.
264. Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA: Identification and characterization of a non-‐satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol 2010, 12(3):257-‐266.
265. Liu N, Garry GA, Li S, Bezprozvannaya S, Sanchez-‐Ortiz E, Chen B, Shelton JM, Jaichander P, Bassel-‐Duby R, Olson EN: A Twist2-‐dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol 2017, 19(3):202-‐213.
266. Le Grand F, Auda-‐Boucher G, Levitsky D, Rouaud T, Fontaine-‐Perus J, Gardahaut MF: Endothelial cells within embryonic skeletal muscles: a potential source of myogenic progenitors. Exp Cell Res 2004, 301(2):232-‐241.
267. Tamaki T, Akatsuka A, Ando K, Nakamura Y, Matsuzawa H, Hotta T, Roy RR, Edgerton VR: Identification of myogenic-‐endothelial progenitor cells in the interstitial spaces of skeletal muscle. J Cell Biol 2002, 157(4):571-‐577.
268. Dellavalle A, Maroli G, Covarello D, Azzoni E, Innocenzi A, Perani L, Antonini S, Sambasivan R, Brunelli S, Tajbakhsh S et al: Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat Commun 2011, 2:499.
269. Sharabi AB, Aldrich M, Sosic D, Olson EN, Friedman AD, Lee SH, Chen SY: Twist-‐2 controls myeloid lineage development and function. PLoS Biol 2008, 6(12):e316.
270. Yong KS, Keng CT, Tan SQ, Loh E, Chang KT, Tan TC, Hong W, Chen Q: Human CD34(lo)CD133(lo) fetal liver cells support the expansion of human CD34(hi)CD133(hi) hematopoietic stem cells. Cell Mol Immunol 2016, 13(5):605-‐614.
271. Omelyanenko N.P. SLI, Mironov S., P.: Connective Tissue: Histophysiology, Biochemistry, Molecular Biology. 2017.
272. Buckingham M, Vincent SD: Distinct and dynamic myogenic populations in the vertebrate embryo. Curr Opin Genet Dev 2009, 19(5):444-‐453.
273. Tozer S, Bonnin MA, Relaix F, Di Savino S, Garcia-‐Villalba P, Coumailleau P, Duprez D: Involvement of vessels and PDGFB in muscle splitting during chick limb development. Development 2007, 134(14):2579-‐2591.

Références
192
274. Latroche C, Gitiaux C, Chretien F, Desguerre I, Mounier R, Chazaud B: Skeletal Muscle Microvasculature: A Highly Dynamic Lifeline. Physiology (Bethesda) 2015, 30(6):417-‐427.
275. Lagha M, Brunelli S, Messina G, Cumano A, Kume T, Relaix F, Buckingham ME: Pax3:Foxc2 reciprocal repression in the somite modulates muscular versus vascular cell fate choice in multipotent progenitors. Dev Cell 2009, 17(6):892-‐899.
276. C R: Mémoire sur le développement, la structures et les propriétés des capillaires sanguins et lymphatiques. Archs Physiol Norm Pathol 1873.
277. Ozerdem U, Grako KA, Dahlin-‐Huppe K, Monosov E, Stallcup WB: NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn 2001, 222(2):218-‐227.
278. Lindahl P, Johansson BR, Leveen P, Betsholtz C: Pericyte loss and microaneurysm formation in PDGF-‐B-‐deficient mice. Science 1997, 277(5323):242-‐245.
279. Cappellari O, Cossu G: Pericytes in development and pathology of skeletal muscle. Circ Res 2013, 113(3):341-‐347.
280. Diaz-‐Flores L, Gutierrez R, Varela H, Rancel N, Valladares F: Microvascular pericytes: a review of their morphological and functional characteristics. Histol Histopathol 1991, 6(2):269-‐286.
281. Abdul N, Dixon D, Walker A, Horabin J, Smith N, Weir DJ, Brewster NT, Deehan DJ, Mann DA, Borthwick LA: Fibrosis is a common outcome following total knee arthroplasty. Sci Rep 2015, 5:16469.
282. Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D: Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508(7494):55-‐60.
283. Pallone TL, Silldorff EP: Pericyte regulation of renal medullary blood flow. Exp Nephrol 2001, 9(3):165-‐170.
284. Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C: Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 2001, 153(3):543-‐553.
285. Bergers G, Song S: The role of pericytes in blood-‐vessel formation and maintenance. Neuro Oncol 2005, 7(4):452-‐464.
286. Kumar A, D'Souza SS, Moskvin OV, Toh H, Wang B, Zhang J, Swanson S, Guo LW, Thomson JA, Slukvin, II: Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep 2017, 19(9):1902-‐1916.

Références
193
287. Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C: Role of PDGF-‐B and PDGFR-‐beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126(14):3047-‐3055.
288. Timmerman LA, Grego-‐Bessa J, Raya A, Bertran E, Perez-‐Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-‐Belmonte JC et al: Notch promotes epithelial-‐mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004, 18(1):99-‐115.
289. Kostallari E, Baba-‐Amer Y, Alonso-‐Martin S, Ngoh P, Relaix F, Lafuste P, Gherardi RK: Pericytes in the myovascular niche promote post-‐natal myofiber growth and satellite cell quiescence. Development 2015, 142(7):1242-‐1253.
290. Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O: Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res 2013, 10(1):67-‐84.
291. Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O: Type-‐1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol 2013, 305(11):C1098-‐1113.
292. Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O: Type-‐2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 2014, 307(1):C25-‐38.
293. Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O: Skeletal muscle neural progenitor cells exhibit properties of NG2-‐glia. Exp Cell Res 2013, 319(1):45-‐63.
294. Gautam J, Nirwane A, Yao Y: Laminin differentially regulates the stemness of type I and type II pericytes. Stem Cell Res Ther 2017, 8(1):28.
295. Yao Y, Norris EH, Mason CE, Strickland S: Laminin regulates PDGFRbeta(+) cell stemness and muscle development. Nat Commun 2016, 7:11415.
296. Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R et al: Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol 2007, 9(3):255-‐267.
297. Cappellari O, Benedetti S, Innocenzi A, Tedesco FS, Moreno-‐Fortuny A, Ugarte G, Lampugnani MG, Messina G, Cossu G: Dll4 and PDGF-‐BB convert committed skeletal myoblasts to pericytes without erasing their myogenic memory. Dev Cell 2013, 24(6):586-‐599.
298. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L: Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med 2012, 18(8):1262-‐1270.

Références
194
299. Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O: Pericytes: multitasking cells in the regeneration of injured, diseased, and aged skeletal muscle. Front Aging Neurosci 2014, 6:245.
300. Kan L, Peng CY, McGuire TL, Kessler JA: Glast-‐expressing progenitor cells contribute to heterotopic ossification. Bone 2013, 53(1):194-‐203.
301. Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS: Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 2009, 91(3):652-‐663.
302. Oishi T, Uezumi A, Kanaji A, Yamamoto N, Yamaguchi A, Yamada H, Tsuchida K: Osteogenic differentiation capacity of human skeletal muscle-‐derived progenitor cells. PLoS One 2013, 8(2):e56641.
303. Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, Caprioli A, Sirabella D, Baiocchi M, De Maria R et al: The meso-‐angioblast: a multipotent, self-‐renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 2002, 129(11):2773-‐2783.
304. Ugarte G, Cappellari O, Perani L, Pistocchi A, Cossu G: Noggin recruits mesoderm progenitors from the dorsal aorta to a skeletal myogenic fate. Dev Biol 2012, 365(1):91-‐100.
305. Messina G, Sirabella D, Monteverde S, Galvez BG, Tonlorenzi R, Schnapp E, De Angelis L, Brunelli S, Relaix F, Buckingham M et al: Skeletal muscle differentiation of embryonic mesoangioblasts requires pax3 activity. Stem Cells 2009, 27(1):157-‐164.
306. Bonfanti C, Rossi G, Tedesco FS, Giannotta M, Benedetti S, Tonlorenzi R, Antonini S, Marazzi G, Dejana E, Sassoon D et al: PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence. Nat Commun 2015, 6:6364.
307. Tagliafico E, Brunelli S, Bergamaschi A, De Angelis L, Scardigli R, Galli D, Battini R, Bianco P, Ferrari S, Cossu G et al: TGFbeta/BMP activate the smooth muscle/bone differentiation programs in mesoangioblasts. J Cell Sci 2004, 117(Pt 19):4377-‐4388.
308. Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D'Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP et al: Cell therapy of alpha-‐sarcoglycan null dystrophic mice through intra-‐arterial delivery of mesoangioblasts. Science 2003, 301(5632):487-‐492.
309. Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I et al: Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444(7119):574-‐579.

Références
195
310. Costamagna D, Quattrocelli M, van Tienen F, Umans L, de Coo IF, Zwijsen A, Huylebroeck D, Sampaolesi M: Smad1/5/8 are myogenic regulators of murine and human mesoangioblasts. J Mol Cell Biol 2016, 8(1):73-‐87.
311. Quattrocelli M, Costamagna D, Giacomazzi G, Camps J, Sampaolesi M: Notch signaling regulates myogenic regenerative capacity of murine and human mesoangioblasts. Cell Death Dis 2014, 5:e1448.
312. Crisan M, Deasy B, Gavina M, Zheng B, Huard J, Lazzari L, Peault B: Purification and long-‐term culture of multipotent progenitor cells affiliated with the walls of human blood vessels: myoendothelial cells and pericytes. Methods Cell Biol 2008, 86:295-‐309.
313. Pierantozzi E, Vezzani B, Badin M, Curina C, Severi FM, Petraglia F, Randazzo D, Rossi D, Sorrentino V: Tissue-‐Specific Cultured Human Pericytes: Perivascular Cells from Smooth Muscle Tissue Have Restricted Mesodermal Differentiation Ability. Stem Cells Dev 2016, 25(9):674-‐686.
314. Nassari S, Duprez D, Fournier-‐Thibault C: Non-‐myogenic Contribution to Muscle Development and Homeostasis: The Role of Connective Tissues. Front Cell Dev Biol 2017, 5:22.
315. Chapman MA, Meza R, Lieber RL: Skeletal muscle fibroblasts in health and disease. Differentiation 2016, 92(3):108-‐115.
316. Kardon G, Harfe BD, Tabin CJ: A Tcf4-‐positive mesodermal population provides a prepattern for vertebrate limb muscle patterning. Dev Cell 2003, 5(6):937-‐944.
317. Mathew SJ, Hansen JM, Merrell AJ, Murphy MM, Lawson JA, Hutcheson DA, Hansen MS, Angus-‐Hill M, Kardon G: Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 2011, 138(2):371-‐384.
318. Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G: Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138(17):3625-‐3637.
319. Fry CS, Lee JD, Jackson JR, Kirby TJ, Stasko SA, Liu H, Dupont-‐Versteegden EE, McCarthy JJ, Peterson CA: Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J 2014, 28(4):1654-‐1665.
320. Mackey AL, Magnan M, Chazaud B, Kjaer M: Human skeletal muscle fibroblasts stimulate in vitro myogenesis and in vivo muscle regeneration. J Physiol 2017, 595(15):5115-‐5127.
321. Vallecillo-‐Garcia P, Orgeur M, Vom Hofe-‐Schneider S, Stumm J, Kappert V, Ibrahim DM, Borno ST, Hayashi S, Relaix F, Hildebrandt K et al: Odd skipped-‐related 1 identifies a population of embryonic fibro-‐adipogenic progenitors regulating myogenesis during limb development. Nat Commun 2017, 8(1):1218.

Références
196
322. Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM: Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 2010, 12(2):153-‐163.
323. Uezumi A, Fukada S, Yamamoto N, Ikemoto-‐Uezumi M, Nakatani M, Morita M, Yamaguchi A, Yamada H, Nishino I, Hamada Y et al: Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis 2014, 5:e1186.
324. Judson RN, Zhang RH, Rossi FM: Tissue-‐resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J 2013, 280(17):4100-‐4108.
325. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K: Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 2010, 12(2):143-‐152.
326. Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe-‐Suzuki Y et al: Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 2011, 124(Pt 21):3654-‐3664.
327. Dong Y, Silva KA, Dong Y, Zhang L: Glucocorticoids increase adipocytes in muscle by affecting IL-‐4 regulated FAP activity. FASEB J 2014, 28(9):4123-‐4132.
328. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A: Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153(2):376-‐388.
329. Mueller AA, van Velthoven CT, Fukumoto KD, Cheung TH, Rando TA: Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 2016, 540(7632):276-‐279.
330. Ieronimakis N, Hays A, Prasad A, Janebodin K, Duffield JS, Reyes M: PDGFRalpha signalling promotes fibrogenic responses in collagen-‐producing cells in Duchenne muscular dystrophy. J Pathol 2016, 240(4):410-‐424.
331. Sohn J, Lu A, Tang Y, Wang B, Huard J: Activation of non-‐myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum Mol Genet 2015, 24(13):3814-‐3829.
332. Huang P, Zhao XS, Fields M, Ransohoff RM, Zhou L: Imatinib attenuates skeletal muscle dystrophy in mdx mice. FASEB J 2009, 23(8):2539-‐2548.
333. Ito T, Ogawa R, Uezumi A, Ohtani T, Watanabe Y, Tsujikawa K, Miyagoe-‐Suzuki Y, Takeda S, Yamamoto H, Fukada S: Imatinib attenuates severe mouse dystrophy and inhibits proliferation and fibrosis-‐marker expression in muscle mesenchymal progenitors. Neuromuscul Disord 2013, 23(4):349-‐356.

Références
197
334. Relaix F, Weng X, Marazzi G, Yang E, Copeland N, Jenkins N, Spence SE, Sassoon D: Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Dev Biol 1996, 177(2):383-‐396.
335. Besson V, Smeriglio P, Wegener A, Relaix F, Nait Oumesmar B, Sassoon DA, Marazzi G: PW1 gene/paternally expressed gene 3 (PW1/Peg3) identifies multiple adult stem and progenitor cell populations. Proc Natl Acad Sci U S A 2011, 108(28):11470-‐11475.
336. Malinverno M, Corada M, Ferrarini L, Formicola L, Marazzi G, Sassoon D, Dejana E: Peg3/PW1 Is a Marker of a Subset of Vessel Associated Endothelial Progenitors. Stem Cells 2017, 35(5):1328-‐1340.
337. Cottle BJ, Lewis FC, Shone V, Ellison-‐Hughes GM: Skeletal muscle-‐derived interstitial progenitor cells (PICs) display stem cell properties, being clonogenic, self-‐renewing, and multi-‐potent in vitro and in vivo. Stem Cell Res Ther 2017, 8(1):158.
338. Pannerec A, Formicola L, Besson V, Marazzi G, Sassoon DA: Defining skeletal muscle resident progenitors and their cell fate potentials. Development 2013, 140(14):2879-‐2891.
339. Cianferotti L, Brandi ML: Muscle-‐bone interactions: basic and clinical aspects. Endocrine 2014, 45(2):165-‐177.
340. Bonewald LF, Kiel DP, Clemens TL, Esser K, Orwoll ES, O'Keefe RJ, Fielding RA: Forum on bone and skeletal muscle interactions: summary of the proceedings of an ASBMR workshop. J Bone Miner Res 2013, 28(9):1857-‐1865.
341. Mazess RB, Whedon GD: Immobilization and bone. Calcif Tissue Int 1983, 35(3):265-‐267.
342. Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, Rattenbach R, Relaix F, Maire P, Rountree RB et al: Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell 2009, 16(5):734-‐743.
343. Nowlan NC, Bourdon C, Dumas G, Tajbakhsh S, Prendergast PJ, Murphy P: Developing bones are differentially affected by compromised skeletal muscle formation. Bone 2010, 46(5):1275-‐1285.
344. Shwartz Y, Blitz E, Zelzer E: One load to rule them all: mechanical control of the musculoskeletal system in development and aging. Differentiation 2013, 86(3):104-‐111.
345. Gopalakrishnan R, Genc KO, Rice AJ, Lee SM, Evans HJ, Maender CC, Ilaslan H, Cavanagh PR: Muscle volume, strength, endurance, and exercise loads during 6-‐month missions in space. Aviat Space Environ Med 2010, 81(2):91-‐102.

Références
198
346. LeBlanc A, Schneider V, Shackelford L, West S, Oganov V, Bakulin A, Voronin L: Bone mineral and lean tissue loss after long duration space flight. J Musculoskelet Neuronal Interact 2000, 1(2):157-‐160.
347. Orwoll ES, Adler RA, Amin S, Binkley N, Lewiecki EM, Petak SM, Shapses SA, Sinaki M, Watts NB, Sibonga JD: Skeletal health in long-‐duration astronauts: nature, assessment, and management recommendations from the NASA Bone Summit. J Bone Miner Res 2013, 28(6):1243-‐1255.
348. Smith SM, Heer MA, Shackelford LC, Sibonga JD, Ploutz-‐Snyder L, Zwart SR: Benefits for bone from resistance exercise and nutrition in long-‐duration spaceflight: Evidence from biochemistry and densitometry. J Bone Miner Res 2012, 27(9):1896-‐1906.
349. Poliachik SL, Bain SD, Threet D, Huber P, Gross TS: Transient muscle paralysis disrupts bone homeostasis by rapid degradation of bone morphology. Bone 2010, 46(1):18-‐23.
350. Warner SE, Sanford DA, Becker BA, Bain SD, Srinivasan S, Gross TS: Botox induced muscle paralysis rapidly degrades bone. Bone 2006, 38(2):257-‐264.
351. Sharir A, Stern T, Rot C, Shahar R, Zelzer E: Muscle force regulates bone shaping for optimal load-‐bearing capacity during embryogenesis. Development 2011, 138(15):3247-‐3259.
352. Aliprantis AO, Stolina M, Kostenuik PJ, Poliachik SL, Warner SE, Bain SD, Gross TS: Transient muscle paralysis degrades bone via rapid osteoclastogenesis. FASEB J 2012, 26(3):1110-‐1118.
353. Westerlind KC, Wronski TJ, Ritman EL, Luo ZP, An KN, Bell NH, Turner RT: Estrogen regulates the rate of bone turnover but bone balance in ovariectomized rats is modulated by prevailing mechanical strain. Proc Natl Acad Sci U S A 1997, 94(8):4199-‐4204.
354. Case N, Xie Z, Sen B, Styner M, Zou M, O'Conor C, Horowitz M, Rubin J: Mechanical activation of beta-‐catenin regulates phenotype in adult murine marrow-‐derived mesenchymal stem cells. J Orthop Res 2010, 28(11):1531-‐1538.
355. David V, Martin A, Lafage-‐Proust MH, Malaval L, Peyroche S, Jones DB, Vico L, Guignandon A: Mechanical loading down-‐regulates peroxisome proliferator-‐activated receptor gamma in bone marrow stromal cells and favors osteoblastogenesis at the expense of adipogenesis. Endocrinology 2007, 148(5):2553-‐2562.
356. Miyakoshi N, Hongo M, Mizutani Y, Shimada Y: Prevalence of sarcopenia in Japanese women with osteopenia and osteoporosis. J Bone Miner Metab 2013, 31(5):556-‐561.

Références
199
357. Greenfield EM, Shaw SM, Gornik SA, Banks MA: Adenyl cyclase and interleukin 6 are downstream effectors of parathyroid hormone resulting in stimulation of bone resorption. J Clin Invest 1995, 96(3):1238-‐1244.
358. Joseph C, Kenny AM, Taxel P, Lorenzo JA, Duque G, Kuchel GA: Role of endocrine-‐immune dysregulation in osteoporosis, sarcopenia, frailty and fracture risk. Mol Aspects Med 2005, 26(3):181-‐201.
359. Ma HT, Griffith JF, Xu L, Leung PC: The functional muscle-‐bone unit in subjects of varying BMD. Osteoporos Int 2014, 25(3):999-‐1004.
360. Girgis CM: Integrated therapies for osteoporosis and sarcopenia: from signaling pathways to clinical trials. Calcif Tissue Int 2015, 96(3):243-‐255.
361. Tarantino U, Baldi J, Celi M, Rao C, Liuni FM, Iundusi R, Gasbarra E: Osteoporosis and sarcopenia: the connections. Aging Clin Exp Res 2013, 25 Suppl 1:S93-‐95.
362. Tagliaferri C, Wittrant Y, Davicco MJ, Walrand S, Coxam V: Muscle and bone, two interconnected tissues. Ageing Res Rev 2015, 21:55-‐70.
363. McPherron AC, Lawler AM, Lee SJ: Regulation of skeletal muscle mass in mice by a new TGF-‐beta superfamily member. Nature 1997, 387(6628):83-‐90.
364. Shan T, Liang X, Bi P, Kuang S: Myostatin knockout drives browning of white adipose tissue through activating the AMPK-‐PGC1alpha-‐Fndc5 pathway in muscle. FASEB J 2013, 27(5):1981-‐1989.
365. Hamrick MW: Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec A Discov Mol Cell Evol Biol 2003, 272(1):388-‐391.
366. Hamrick MW, McPherron AC, Lovejoy CO: Bone mineral content and density in the humerus of adult myostatin-‐deficient mice. Calcif Tissue Int 2002, 71(1):63-‐68.
367. Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, Kang B, Isales CM, Fulzele S, Wenger KH: Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-‐derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone 2007, 40(6):1544-‐1553.
368. Elkasrawy M, Fulzele S, Bowser M, Wenger K, Hamrick M: Myostatin (GDF-‐8) inhibits chondrogenesis and chondrocyte proliferation in vitro by suppressing Sox-‐9 expression. Growth Factors 2011, 29(6):253-‐262.
369. Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, Beckmann D, Paruzel P, Bertrand J, Redlich K et al: Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 2015, 21(9):1085-‐1090.
370. Brotto M, Johnson ML: Endocrine crosstalk between muscle and bone. Curr Osteoporos Rep 2014, 12(2):135-‐141.

Références
200
371. Bakker AD, Jaspers RT: IL-‐6 and IGF-‐1 Signaling Within and Between Muscle and Bone: How Important is the mTOR Pathway for Bone Metabolism? Curr Osteoporos Rep 2015, 13(3):131-‐139.
372. Erices A, Conget P, Rojas C, Minguell JJ: Gp130 activation by soluble interleukin-‐6 receptor/interleukin-‐6 enhances osteoblastic differentiation of human bone marrow-‐derived mesenchymal stem cells. Exp Cell Res 2002, 280(1):24-‐32.
373. Taguchi Y, Yamamoto M, Yamate T, Lin SC, Mocharla H, DeTogni P, Nakayama N, Boyce BF, Abe E, Manolagas SC: Interleukin-‐6-‐type cytokines stimulate mesenchymal progenitor differentiation toward the osteoblastic lineage. Proc Assoc Am Physicians 1998, 110(6):559-‐574.
374. Udagawa N, Takahashi N, Katagiri T, Tamura T, Wada S, Findlay DM, Martin TJ, Hirota H, Taga T, Kishimoto T et al: Interleukin (IL)-‐6 induction of osteoclast differentiation depends on IL-‐6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J Exp Med 1995, 182(5):1461-‐1468.
375. Nilsson A, Isgaard J, Lindahl A, Dahlstrom A, Skottner A, Isaksson OG: Regulation by growth hormone of number of chondrocytes containing IGF-‐I in rat growth plate. Science 1986, 233(4763):571-‐574.
376. Schoenle E, Zapf J, Humbel RE, Froesch ER: Insulin-‐like growth factor I stimulates growth in hypophysectomized rats. Nature 1982, 296(5854):252-‐253.
377. Bikle DD, Tahimic C, Chang W, Wang Y, Philippou A, Barton ER: Role of IGF-‐I signaling in muscle bone interactions. Bone 2015, 80:79-‐88.
378. Hamrick MW, McNeil PL, Patterson SL: Role of muscle-‐derived growth factors in bone formation. J Musculoskelet Neuronal Interact 2010, 10(1):64-‐70.
379. Ostrovsky O, Eletto D, Makarewich C, Barton ER, Argon Y: Glucose regulated protein 94 is required for muscle differentiation through its control of the autocrine production of insulin-‐like growth factors. Biochim Biophys Acta 2010, 1803(2):333-‐341.
380. Barton ER, Park S, James JK, Makarewich CA, Philippou A, Eletto D, Lei H, Brisson B, Ostrovsky O, Li Z et al: Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J 2012, 26(9):3691-‐3702.
381. Bren-‐Mattison Y, Hausburg M, Olwin BB: Growth of limb muscle is dependent on skeletal-‐derived Indian hedgehog. Dev Biol 2011, 356(2):486-‐495.
382. Xu Q, Cui Y, Luan J, Zhou X, Li H, Han J: Exosomes from C2C12 myoblasts enhance osteogenic differentiation of MC3T3-‐E1 pre-‐osteoblasts by delivering miR-‐27a-‐3p. Biochem Biophys Res Commun 2018, 498(1):32-‐37.

Références
201
383. Jahn K, Lara-‐Castillo N, Brotto L, Mo CL, Johnson ML, Brotto M, Bonewald LF: Skeletal muscle secreted factors prevent glucocorticoid-‐induced osteocyte apoptosis through activation of beta-‐catenin. Eur Cell Mater 2012, 24:197-‐209; discussion 209-‐110.
384. Tanaka K, Matsumoto E, Higashimaki Y, Katagiri T, Sugimoto T, Seino S, Kaji H: Role of osteoglycin in the linkage between muscle and bone. J Biol Chem 2012, 287(15):11616-‐11628.
385. Tanaka K, Matsumoto E, Higashimaki Y, Sugimoto T, Seino S, Kaji H: FAM5C is a soluble osteoblast differentiation factor linking muscle to bone. Biochem Biophys Res Commun 2012, 418(1):134-‐139.
386. Gorski JP, Huffman NT, Vallejo J, Brotto L, Chittur SV, Breggia A, Stern A, Huang J, Mo C, Seidah NG et al: Deletion of Mbtps1 (Pcsk8, S1p, Ski-‐1) Gene in Osteocytes Stimulates Soleus Muscle Regeneration and Increased Size and Contractile Force with Age. J Biol Chem 2016, 291(9):4308-‐4322.
387. Gorski JP, Price JL: Bone muscle crosstalk targets muscle regeneration pathway regulated by core circadian transcriptional repressors DEC1 and DEC2. Bonekey Rep 2016, 5:850.
388. Dey D, Wheatley BM, Cholok D, Agarwal S, Yu PB, Levi B, Davis TA: The traumatic bone: trauma-‐induced heterotopic ossification. Transl Res 2017, 186:95-‐111.
389. Jackson WM, Aragon AB, Onodera J, Koehler SM, Ji Y, Bulken-‐Hoover JD, Vogler JA, Tuan RS, Nesti LJ: Cytokine expression in muscle following traumatic injury. J Orthop Res 2011, 29(10):1613-‐1620.
390. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M et al: A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006, 38(5):525-‐527.
391. Matthews BG, Torreggiani E, Roeder E, Matic I, Grcevic D, Kalajzic I: Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone 2016, 84:69-‐77.
392. Sen MK, Miclau T: Autologous iliac crest bone graft: should it still be the gold standard for treating nonunions? Injury 2007, 38 Suppl 1:S75-‐80.
393. Gustilo RB, Mendoza RM, Williams DN: Problems in the management of type III (severe) open fractures: a new classification of type III open fractures. J Trauma 1984, 24(8):742-‐746.
394. Chou YC, Wu CC, Chan YS, Chang CH, Hsu YH, Huang YC: Medial gastrocnemius muscle flap for treating wound complications after double-‐plate fixation via two-‐incision approach for complex tibial plateau fractures. J Trauma 2010, 68(1):138-‐145.

Références
202
395. Al-‐Fotawei R, Ayoub AF, Heath N, Naudi KB, Tanner KE, Dalby MJ, McMahon J: Radiological assessment of bioengineered bone in a muscle flap for the reconstruction of critical-‐size mandibular defect. PLoS One 2014, 9(9):e107403.
396. Richards RR, McKee MD, Paitich CB, Anderson GI, Bertoia JT: A comparison of the effects of skin coverage and muscle flap coverage on the early strength of union at the site of osteotomy after devascularization of a segment of canine tibia. J Bone Joint Surg Am 1991, 73(9):1323-‐1330.
397. Liu F, Ferreira E, Porter RM, Glatt V, Schinhan M, Shen Z, Randolph MA, Kirker-‐Head CA, Wehling C, Vrahas MS et al: Rapid and reliable healing of critical size bone defects with genetically modified sheep muscle. Eur Cell Mater 2015, 30:118-‐130; discussion 130-‐111.
398. Chan JK, Harry L, Williams G, Nanchahal J: Soft-‐tissue reconstruction of open fractures of the lower limb: muscle versus fasciocutaneous flaps. Plast Reconstr Surg 2012, 130(2):284e-‐295e.
399. Harry LE, Sandison A, Paleolog EM, Hansen U, Pearse MF, Nanchahal J: Comparison of the healing of open tibial fractures covered with either muscle or fasciocutaneous tissue in a murine model. J Orthop Res 2008, 26(9):1238-‐1244.
400. Aydin A, Memisoglu K, Cengiz A, Atmaca H, Muezzinoglu B, Muezzinoglu US: Effects of botulinum toxin A on fracture healing in rats: an experimental study. J Orthop Sci 2012, 17(6):796-‐801.
401. Blecher R, Krief S, Galili T, Assaraf E, Stern T, Anekstein Y, Agar G, Zelzer E: The Proprioceptive System Regulates Morphologic Restoration of Fractured Bones. Cell Rep 2017, 20(8):1775-‐1783.
402. Rot C, Stern T, Blecher R, Friesem B, Zelzer E: A mechanical Jack-‐like Mechanism drives spontaneous fracture healing in neonatal mice. Dev Cell 2014, 31(2):159-‐170.
403. Abou-‐Khalil R, Colnot C: Cellular and molecular bases of skeletal regeneration: what can we learn from genetic mouse models? Bone 2014, 64:211-‐221.
404. Abou-‐Khalil R, Yang F, Mortreux M, Lieu S, Yu YY, Wurmser M, Pereira C, Relaix F, Miclau T, Marcucio RS et al: Delayed bone regeneration is linked to chronic inflammation in murine muscular dystrophy. J Bone Miner Res 2014, 29(2):304-‐315.
405. Elkasrawy M, Immel D, Wen X, Liu X, Liang LF, Hamrick MW: Immunolocalization of myostatin (GDF-‐8) following musculoskeletal injury and the effects of exogenous myostatin on muscle and bone healing. J Histochem Cytochem 2012, 60(1):22-‐30.

Références
203
406. Kellum E, Starr H, Arounleut P, Immel D, Fulzele S, Wenger K, Hamrick MW: Myostatin (GDF-‐8) deficiency increases fracture callus size, Sox-‐5 expression, and callus bone volume. Bone 2009, 44(1):17-‐23.
407. Gao X, Usas A, Proto JD, Lu A, Cummins JH, Proctor A, Chen CW, Huard J: Role of donor and host cells in muscle-‐derived stem cell-‐mediated bone repair: differentiation vs. paracrine effects. FASEB J 2014, 28(8):3792-‐3809.
408. Kinner B, Gerstenfeld LC, Einhorn TA, Spector M: Expression of smooth muscle actin in connective tissue cells participating in fracture healing in a murine model. Bone 2002, 30(5):738-‐745.
409. Mastrogiacomo M, Derubeis AR, Cancedda R: Bone and cartilage formation by skeletal muscle derived cells. J Cell Physiol 2005, 204(2):594-‐603.
410. Owston H, Giannoudis PV, Jones E: Do skeletal muscle MSCs in humans contribute to bone repair? A systematic review. Injury 2016, 47 Suppl 6:S3-‐S15.
411. Fong K, Truong V, Foote CJ, Petrisor B, Williams D, Ristevski B, Sprague S, Bhandari M: Predictors of nonunion and reoperation in patients with fractures of the tibia: an observational study. BMC Musculoskelet Disord 2013, 14:103.
412. M H-‐L: Mouse models in bone fracture healing research. Curr Mol Bio Rep 2016:101-‐111.
413. Mills LA, Simpson AH: In vivo models of bone repair. J Bone Joint Surg Br 2012, 94(7):865-‐874.
414. Hurtgen BJ, Ward CL, Garg K, Pollot BE, Goldman SM, McKinley TO, Wenke JC, Corona BT: Severe muscle trauma triggers heightened and prolonged local musculoskeletal inflammation and impairs adjacent tibia fracture healing. J Musculoskelet Neuronal Interact 2016, 16(2):122-‐134.
415. Utvag SE, Grundnes O, Rindal DB, Reikeras O: Influence of extensive muscle injury on fracture healing in rat tibia. J Orthop Trauma 2003, 17(6):430-‐435.
416. Czerwinska AM, Streminska W, Ciemerych MA, Grabowska I: Mouse gastrocnemius muscle regeneration after mechanical or cardiotoxin injury. Folia Histochem Cytobiol 2012, 50(1):144-‐153.
417. Mahdy MAA: Glycerol-‐induced injury as a new model of muscle regeneration. Cell Tissue Res 2018.
418. Chou YH, Pan SY, Yang CH, Lin SL: Stem cells and kidney regeneration. J Formos Med Assoc 2014, 113(4):201-‐209.
419. Yang HC, Liu SJ, Fogo AB: Kidney regeneration in mammals. Nephron Exp Nephrol 2014, 126(2):50.

Références
204
420. Fausto N, Campbell JS, Riehle KJ: Liver regeneration. Hepatology 2006, 43(2 Suppl 1):S45-‐53.
421. Maillet V, Boussetta N, Leclerc J, Fauveau V, Foretz M, Viollet B, Couty JP, Celton-‐Morizur S, Perret C, Desdouets C: LKB1 as a Gatekeeper of Hepatocyte Proliferation and Genomic Integrity during Liver Regeneration. Cell Rep 2018, 22(8):1994-‐2005.
422. Fausto N, Campbell JS: The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev 2003, 120(1):117-‐130.
423. Lemoli RM, Catani L, Talarico S, Loggi E, Gramenzi A, Baccarani U, Fogli M, Grazi GL, Aluigi M, Marzocchi G et al: Mobilization of bone marrow-‐derived hematopoietic and endothelial stem cells after orthotopic liver transplantation and liver resection. Stem Cells 2006, 24(12):2817-‐2825.
424. Matsumoto T, Kano K, Kondo D, Fukuda N, Iribe Y, Tanaka N, Matsubara Y, Sakuma T, Satomi A, Otaki M et al: Mature adipocyte-‐derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol 2008, 215(1):210-‐222.
425. Matsumoto T, Kuroda R, Mifune Y, Kawamoto A, Shoji T, Miwa M, Asahara T, Kurosaka M: Circulating endothelial/skeletal progenitor cells for bone regeneration and healing. Bone 2008, 43(3):434-‐439.
426. Poulsom R, Alison MR, Forbes SJ, Wright NA: Adult stem cell plasticity. J Pathol 2002, 197(4):441-‐456.
427. Camargo FD, Green R, Capetanaki Y, Jackson KA, Goodell MA: Single hematopoietic stem cells generate skeletal muscle through myeloid intermediates. Nat Med 2003, 9(12):1520-‐1527.
428. Ferrari G, Cusella-‐De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, Mavilio F: Muscle regeneration by bone marrow-‐derived myogenic progenitors. Science 1998, 279(5356):1528-‐1530.
429. Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ: Multi-‐organ, multi-‐lineage engraftment by a single bone marrow-‐derived stem cell. Cell 2001, 105(3):369-‐377.
430. LaBarge MA, Blau HM: Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell 2002, 111(4):589-‐601.
431. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP: Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005, 115(1):56-‐65.
432. Lin F, Moran A, Igarashi P: Intrarenal cells, not bone marrow-‐derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest 2005, 115(7):1756-‐1764.

Références
205
433. Matsumoto T, Kawamoto A, Kuroda R, Ishikawa M, Mifune Y, Iwasaki H, Miwa M, Horii M, Hayashi S, Oyamada A et al: Therapeutic potential of vasculogenesis and osteogenesis promoted by peripheral blood CD34-‐positive cells for functional bone healing. Am J Pathol 2006, 169(4):1440-‐1457.
434. Blau HM: A twist of fate. Nature 2002, 419(6906):437.
435. Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KB, Virag JI, Bartelmez SH, Poppa V et al: Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004, 428(6983):664-‐668.
436. Terada N, Hamazaki T, Oka M, Hoki M, Mastalerz DM, Nakano Y, Meyer EM, Morel L, Petersen BE, Scott EW: Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416(6880):542-‐545.
437. Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-‐Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M: Cell fusion is the principal source of bone-‐marrow-‐derived hepatocytes. Nature 2003, 422(6934):897-‐901.
438. Weimann JM, Charlton CA, Brazelton TR, Hackman RC, Blau HM: Contribution of transplanted bone marrow cells to Purkinje neurons in human adult brains. Proc Natl Acad Sci U S A 2003, 100(4):2088-‐2093.
439. Mendez-‐Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS: Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466(7308):829-‐834.
440. Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-‐Hara C, Nagoshi N, Sunabori T et al: Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 2009, 206(11):2483-‐2496.
441. Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC: CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-‐cell maintenance. Nature 2013, 495(7440):227-‐230.
442. Dumont NA, Wang YX, Rudnicki MA: Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142(9):1572-‐1581.
443. Zhang F, Zhang J, Li X, Li B, Tao K, Yue S: Notch signaling pathway regulates cell cycle in proliferating hepatocytes involved in liver regeneration. J Gastroenterol Hepatol 2018, 33(8):1538-‐1547.
444. Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL: Yap-‐dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526(7575):715-‐718.

Références
206
445. Stantzou A, Schirwis E, Swist S, Alonso-‐Martin S, Polydorou I, Zarrouki F, Mouisel E, Beley C, Julien A, Le Grand F et al: BMP signaling regulates satellite cell-‐dependent postnatal muscle growth. Development 2017, 144(15):2737-‐2747.
446. Sanchez-‐Duffhues G, Hiepen C, Knaus P, Ten Dijke P: Bone morphogenetic protein signaling in bone homeostasis. Bone 2015, 80:43-‐59.
447. Chappuis V, Gamer L, Cox K, Lowery JW, Bosshardt DD, Rosen V: Periosteal BMP2 activity drives bone graft healing. Bone 2012, 51(4):800-‐809.
448. Clever JL, Sakai Y, Wang RA, Schneider DB: Inefficient skeletal muscle repair in inhibitor of differentiation knockout mice suggests a crucial role for BMP signaling during adult muscle regeneration. Am J Physiol Cell Physiol 2010, 298(5):C1087-‐1099.
449. Friedrichs M, Wirsdoerfer F, Flohe SB, Schneider S, Wuelling M, Vortkamp A: BMP signaling balances proliferation and differentiation of muscle satellite cell descendants. BMC Cell Biol 2011, 12:26.
450. Salazar VS, Gamer LW, Rosen V: BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol 2016, 12(4):203-‐221.
451. Ono Y, Calhabeu F, Morgan JE, Katagiri T, Amthor H, Zammit PS: BMP signalling permits population expansion by preventing premature myogenic differentiation in muscle satellite cells. Cell Death Differ 2011, 18(2):222-‐234.
452. Falanga V: Wound healing and its impairment in the diabetic foot. Lancet 2005, 366(9498):1736-‐1743.
453. Pivonka P, Dunstan CR: Role of mathematical modeling in bone fracture healing. Bonekey Rep 2012, 1:221.
454. Tidball JG, Dorshkind K, Wehling-‐Henricks M: Shared signaling systems in myeloid cell-‐mediated muscle regeneration. Development 2014, 141(6):1184-‐1196.
455. Babhulkar S, Pande K, Babhulkar S: Nonunion of the diaphysis of long bones. Clin Orthop Relat Res 2005(431):50-‐56.
456. Gaston MS, Simpson AH: Inhibition of fracture healing. J Bone Joint Surg Br 2007, 89(12):1553-‐1560.
457. Bataller R, Brenner DA: Liver fibrosis. J Clin Invest 2005, 115(2):209-‐218.
458. Moyer AL, Wagner KR: Regeneration versus fibrosis in skeletal muscle. Curr Opin Rheumatol 2011, 23(6):568-‐573.
459. Porter JD, Khanna S, Kaminski HJ, Rao JS, Merriam AP, Richmonds CR, Leahy P, Li J, Guo W, Andrade FH: A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-‐deficient mdx mice. Hum Mol Genet 2002, 11(3):263-‐272.

Références
207
460. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-‐Piallat ML, Gabbiani G: The myofibroblast: one function, multiple origins. Am J Pathol 2007, 170(6):1807-‐1816.
461. Wynn TA: Cellular and molecular mechanisms of fibrosis. J Pathol 2008, 214(2):199-‐210.
462. Zeisberg M, Kalluri R: Cellular mechanisms of tissue fibrosis. 1. Common and organ-‐specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 2013, 304(3):C216-‐225.
463. Vannella KM, Wynn TA: Mechanisms of Organ Injury and Repair by Macrophages. Annu Rev Physiol 2017, 79:593-‐617.
464. Wynn TA, Vannella KM: Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44(3):450-‐462.
465. Xie C, Liang B, Xue M, Lin AS, Loiselle A, Schwarz EM, Guldberg RE, O'Keefe RJ, Zhang X: Rescue of impaired fracture healing in COX-‐2-‐/-‐ mice via activation of prostaglandin E2 receptor subtype 4. Am J Pathol 2009, 175(2):772-‐785.
466. Xie C, Ming X, Wang Q, Schwarz EM, Guldberg RE, O'Keefe RJ, Zhang X: COX-‐2 from the injury milieu is critical for the initiation of periosteal progenitor cell mediated bone healing. Bone 2008, 43(6):1075-‐1083.
467. Hadjiargyrou M, O'Keefe RJ: The convergence of fracture repair and stem cells: interplay of genes, aging, environmental factors and disease. J Bone Miner Res 2014, 29(11):2307-‐2322.
468. Bondesen BA, Mills ST, Kegley KM, Pavlath GK: The COX-‐2 pathway is essential during early stages of skeletal muscle regeneration. Am J Physiol Cell Physiol 2004, 287(2):C475-‐483.
469. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C et al: A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155(6):1282-‐1295.
470. Segawa M, Fukada S, Yamamoto Y, Yahagi H, Kanematsu M, Sato M, Ito T, Uezumi A, Hayashi S, Miyagoe-‐Suzuki Y et al: Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp Cell Res 2008, 314(17):3232-‐3244.
471. Kharraz Y, Guerra J, Mann CJ, Serrano AL, Munoz-‐Canoves P: Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm 2013, 2013:491497.
472. Tidball JG, Wehling-‐Henricks M: Shifts in macrophage cytokine production drive muscle fibrosis. Nat Med 2015, 21(7):665-‐666.

Références
208
473. Wang H, Melton DW, Porter L, Sarwar ZU, McManus LM, Shireman PK: Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am J Pathol 2014, 184(4):1167-‐1184.
474. Deng B, Wehling-‐Henricks M, Villalta SA, Wang Y, Tidball JG: IL-‐10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J Immunol 2012, 189(7):3669-‐3680.
475. Tonkin J, Temmerman L, Sampson RD, Gallego-‐Colon E, Barberi L, Bilbao D, Schneider MD, Musaro A, Rosenthal N: Monocyte/Macrophage-‐derived IGF-‐1 Orchestrates Murine Skeletal Muscle Regeneration and Modulates Autocrine Polarization. Mol Ther 2015, 23(7):1189-‐1200.
476. Uezumi A, Ikemoto-‐Uezumi M, Tsuchida K: Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front Physiol 2014, 5:68.
477. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD: Perivascular Gli1+ progenitors are key contributors to injury-‐induced organ fibrosis. Cell Stem Cell 2015, 16(1):51-‐66.
478. Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Munoz-‐Canoves P: Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 2011, 1(1):21.
479. Fiore D, Judson RN, Low M, Lee S, Zhang E, Hopkins C, Xu P, Lenzi A, Rossi FM, Lemos DR: Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res 2016, 17(1):161-‐169.
480. Kopinke D, Roberson EC, Reiter JF: Ciliary Hedgehog Signaling Restricts Injury-‐Induced Adipogenesis. Cell 2017, 170(2):340-‐351 e312.
481. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ: Diverse origins of the myofibroblast-‐implications for kidney fibrosis. Nat Rev Nephrol 2015, 11(4):233-‐244.
482. Tampe D, Zeisberg M: Potential approaches to reverse or repair renal fibrosis. Nat Rev Nephrol 2014, 10(4):226-‐237.
483. Aman J, van Bezu J, Damanafshan A, Huveneers S, Eringa EC, Vogel SM, Groeneveld AB, Vonk Noordegraaf A, van Hinsbergh VW, van Nieuw Amerongen GP: Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 2012, 126(23):2728-‐2738.
484. O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T et al: Imatinib compared with interferon and low-‐dose cytarabine for newly diagnosed chronic-‐phase chronic myeloid leukemia. N Engl J Med 2003, 348(11):994-‐1004.

Références
209
485. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB: Imatinib mesylate inhibits the profibrogenic activity of TGF-‐beta and prevents bleomycin-‐mediated lung fibrosis. J Clin Invest 2004, 114(9):1308-‐1316.
486. Rizzo AN, Sammani S, Esquinca AE, Jacobson JR, Garcia JG, Letsiou E, Dudek SM: Imatinib attenuates inflammation and vascular leak in a clinically relevant two-‐hit model of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2015, 309(11):L1294-‐1304.
487. Brouard M, Saurat JH: Cutaneous reactions to STI571. N Engl J Med 2001, 345(8):618-‐619.
488. Hu W, Lu S, McAlpine I, Jamieson JD, Lee DU, Marroquin LD, Heyen JR, Jessen BA: Mechanistic investigation of imatinib-‐induced cardiac toxicity and the involvement of c-‐Abl kinase. Toxicol Sci 2012, 129(1):188-‐199.
489. Berman E, Nicolaides M, Maki RG, Fleisher M, Chanel S, Scheu K, Wilson BA, Heller G, Sauter NP: Altered bone and mineral metabolism in patients receiving imatinib mesylate. N Engl J Med 2006, 354(19):2006-‐2013.
490. Mughal TI, Schrieber A: Principal long-‐term adverse effects of imatinib in patients with chronic myeloid leukemia in chronic phase. Biologics 2010, 4:315-‐323.

Références
210
Curriculum Vitae
211
Curriculum Vitae Anaïs JULIEN 27/04/1991
16 Avenue du fort +33 6 77 51 33 26 92120 Montrouge, France [email protected]
Formations**
2015% % Diplôme%universitaire%d’expérimentation%animale%niveau%1%9%Université%Paris%Descartes%% % % %% %%
201492018% Doctorat%en%Biologie%Cellulaire%9%Université%Paris%Descartes,%Paris,%France%201292014% Master%de%Génétique%9%Magistère%Européen%de%Génétique,%Université%Paris%Diderot,%Paris,%France%(Mention%Bien)%201192012% License%de%Génétique%9%Magistère%Européen%de%Génétique,%Université%Paris%Diderot,%Paris,%France%(Mention%Assez%Bien)%200992011% Classe%préparatoire%aux%grandes%écoles%section%BCPST,%Lycée%Thiers,%Marseille,%France%
*Expériences*professionnelles*
*
201492018%% Doctorat%à%l’Institut%IMAGINE,%Paris,%France.%PI:%Dr%COLNOT%Céline%Etudes' des' interactions' os.muscle' lors' de' la' régénération' osseuse.' Développement% d’un% modèle% de% blessure%musculosquelettique%et%caractérisation%des%cellules%interstitielles%du%muscle%squelettique%contribuant%à%la%régénération%osseuse%Rédaction%d’un%article%scientifique%en%cours%de%soumission%
%
2014% % Stage%de%Master%2%à%l’Institut%IMAGINE,%Paris,%France.%PI:%Dr%COLNOT%Céline%Etude'des'relations'os.muscle'pendant'la'régénération'osseuse'
'
2013% % Stage%de%Master%1%à%Cold%Spring%Harbor%Laboratory,%USA.%PI:%Dr%DUBNAU%Joshua%Etude'de'l’impact'de'la'mobilisation'du'rétrotransposon'Gypsy'dans'le'système'nerveux'
%
2012% % Stage%de%License%à%«%Umeå%Center%for%Molecular%Medecine%»,%Suède.%PI:%Dr%WILSON%Sara%Study'of'PUNC'gene,'potential'candidate'in'Netrin'1'pathway'in'neural'chick'development'
%
2009% Stage%volontaire%d’observation%à%l’Institut%de%Recherche%et%de%Développement%et%à%l’Université%de%Montpellier,%sous%la%direction%de%Dr.%Sylvie%Hurtrez9Bousses.%%%
Publications*scientifiques*
*
2018% Muscle8derived* profibrotic* progenitors* impair* bone* healing* in*musculoskeletal* trauma.% Julien* A,% Kanagalingam% A,%Megret%J,%Relaix%F%and%Colnot%C.%En%cours%de%soumission%Periosteum%contains%skeletal%stem%cells%with%high%bone%regenerative%potential%controlled%by%Periostin.%%Duchamp%de% Lageneste%O,% Julien*A,%Abou9Khalil% R,% Frangi%G,%Carvalho%C,%Cagnard%N,%Cordier%C,%Conway%SJ,% Colnot%C.%Nature%Communications.%2018%Feb%22;9(1):773.%
%
2017% BMP% signaling% regulates% satellite% cell9dependent% postnatal% muscle% growth.% Stantzou% A,% Schirwis% E,% Swist% S,% Alonso9Martin%S,%Polydorou% I,%Zarrouki%F,%Mouisel%E,%Beley%C,%Julien*A,%Le%Grand%F,%Garcia%L,%Colnot%C,%Birchmeier%C,%Braun%T,%Schuelke%M,%Relaix%F,%Amthor%H.%Development.%2017%Aug%1;144(15):273792747%
%
2015% Role%of%muscle%stem%cells%during%skeletal%regeneration.%Abou9Khalil%R,%Yang%F,%Lieu%S,%Julien*A,%Perry%J,%Pereira%C,%Relaix%F,%Miclau%T,%Marcucio%R,%Colnot%C.%Stem%Cells.%2015%May;33(5):1501911%
*Congrès*scientifiques*et*Prix*
*
2018% *% Julien*A,%Kanagalingam%A,%Duchamp%O,%Megret% J,%Relaix%F%and%Colnot%C.%Muscle' interstitial' cells' contribute' to'bone'repair' and' cause' fibrosis' in' musculoskeletal' trauma.% Congrès% annuel% de% l’American% Society% for% Bone% and% Mineral%Research%(ASBMR)'(Montréal)'.'Poster%*% Julien*A,%Kanagalingam%A,%Duchamp%O,%Megret% J,%Relaix%F%and%Colnot%C.%Muscle' interstitial' cells' contribute' to'bone'repair'and'cause'fibrosis' in'musculoskeletal'trauma.%Conférence%«%Exercise,% locomotion%and%musculoskeletal%system%»%(Lyon)%9%Pitch%poster%(Primé)%et%Poster%
% *%Julien*A,%Kanagalingam%A,%Megret%J,%Relaix%F%and%Colnot%C.%Muscle%interstitial%cells%contribute%to%bone%repair%and%cause%fibrosis%in%musculoskeletal%trauma.%Congrès%annuel%des%Jeunes%Chercheurs%de%l’Institut%IMAGINE%(Paris)%–%Présentation%orale%(1er%Prix)%
%
2016% *% Julien* A,% Alsonso9Martin% S,% Carvalho% C,% Relaix% F% and% Colnot% C.% Identification' of' a' novel' mesenchymal' stem' cell'population' from'muscle' that' contributes' to' bone' regeneration.% Congrès% annuel% des% Jeunes% Chercheurs% de% l’Institut%IMAGINE'(Paris)%9%Présentation%orale%(2eme%Prix)%
% *% Julien* A,% Alsonso9Martin% S,% Carvalho% C,% Relaix% F% and% Colnot% C.% Identification' of' a' novel' mesenchymal' stem' cell'population' from' muscle' that' contributes' to' bone' regeneration.' Présentation% de% poster% au% Journées% Françaises% de%Biologie%des%Tissus%Minéralisés%(JFBTM)'(Nancy)%9%Poster%(1er%Prix)%
%
2015% *%Julien*A,%Alsonso9Martin%S,%Carvalho%C,%Relaix%F%and%Colnot%C.%A'novel'model'of'muscle'injury'to'elucidate'muscle.bone'interaction'during'bone'repair.%Congrès%annuel%des%Jeunes%Chercheurs%de%l’Institut%IMAGINE%(Paris)%9%Présentation%orale%
% *%Julien*A,%Alsonso9Martin%S,%Carvalho%C,%Relaix%F%and%Colnot%C.%Muscle%injury%impairs%bone%regeneration%in%adult%mice.%Journées%Françaises%de%Biologie%des%Tissus%Minéralisés%(JFBTM)'(Clermon.Ferrand)%–%Présentation%Orale%
%Activités*et*loisirs*
%%
Activité%associative%Secrétaire%de%l’Association%des%Jeunes%Chercheurs%de%l’Institut%IMAGINE%(2018)%% %% % Directrice%du%comité%organisateur%du%Congrès%annuel%des%Jeunes%Chercheurs%de%l’Institut%IMAGINE%(2018)%%
Loisirs% % Football%(pratiqué%depuis%1998),%Cuisine%/%Gastronomie,%Lecture%
Recommended

















