
`
Tim Mercer Genome In A Bottle - Sept 16th
Representing the human genome with synthetic spike-in controls.
DISCLAIMER: The Garvan Institute of Medical Research has filed patentapplications on some techniques described in this study.
Tim Mercer Garvan Institute for Medical Research
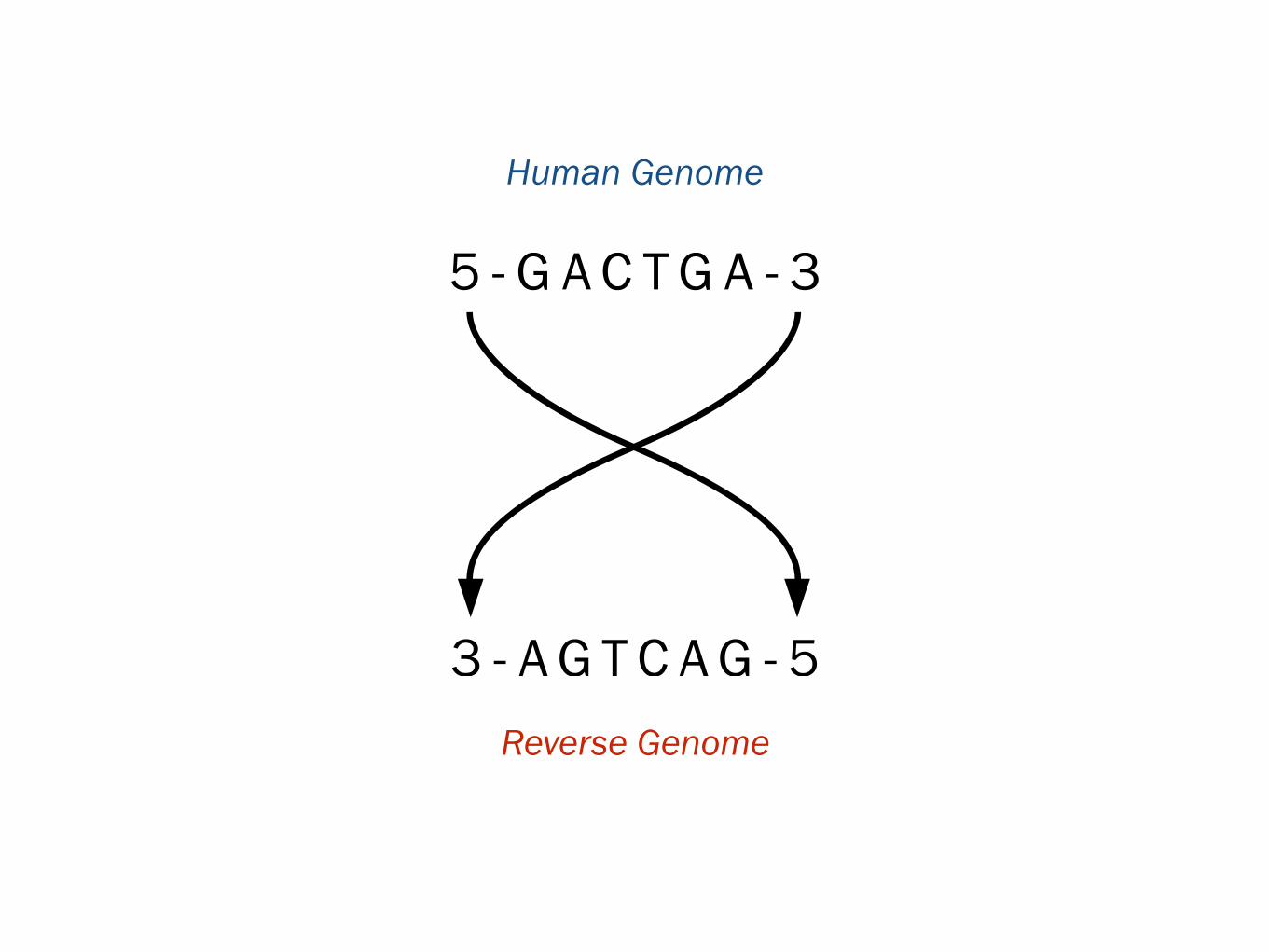
Human Genome
Reverse Genome
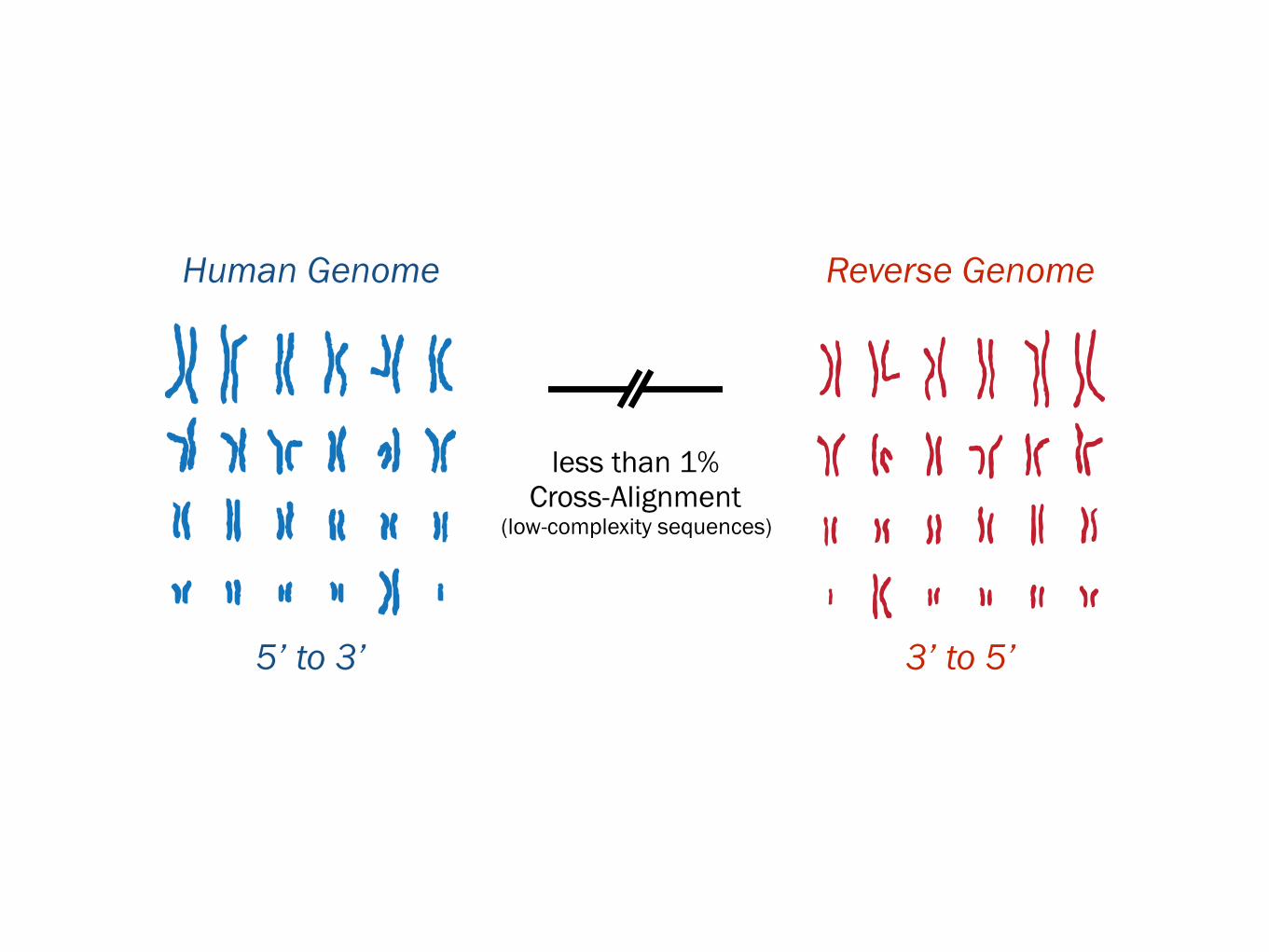
Human Genome
5’ to 3’Synthetic Genome
3’ to 5’Human Genome Reverse Genome
5’ to 3’ 3’ to 5’
less than 1% Cross-Alignment
(low-complexity sequences)
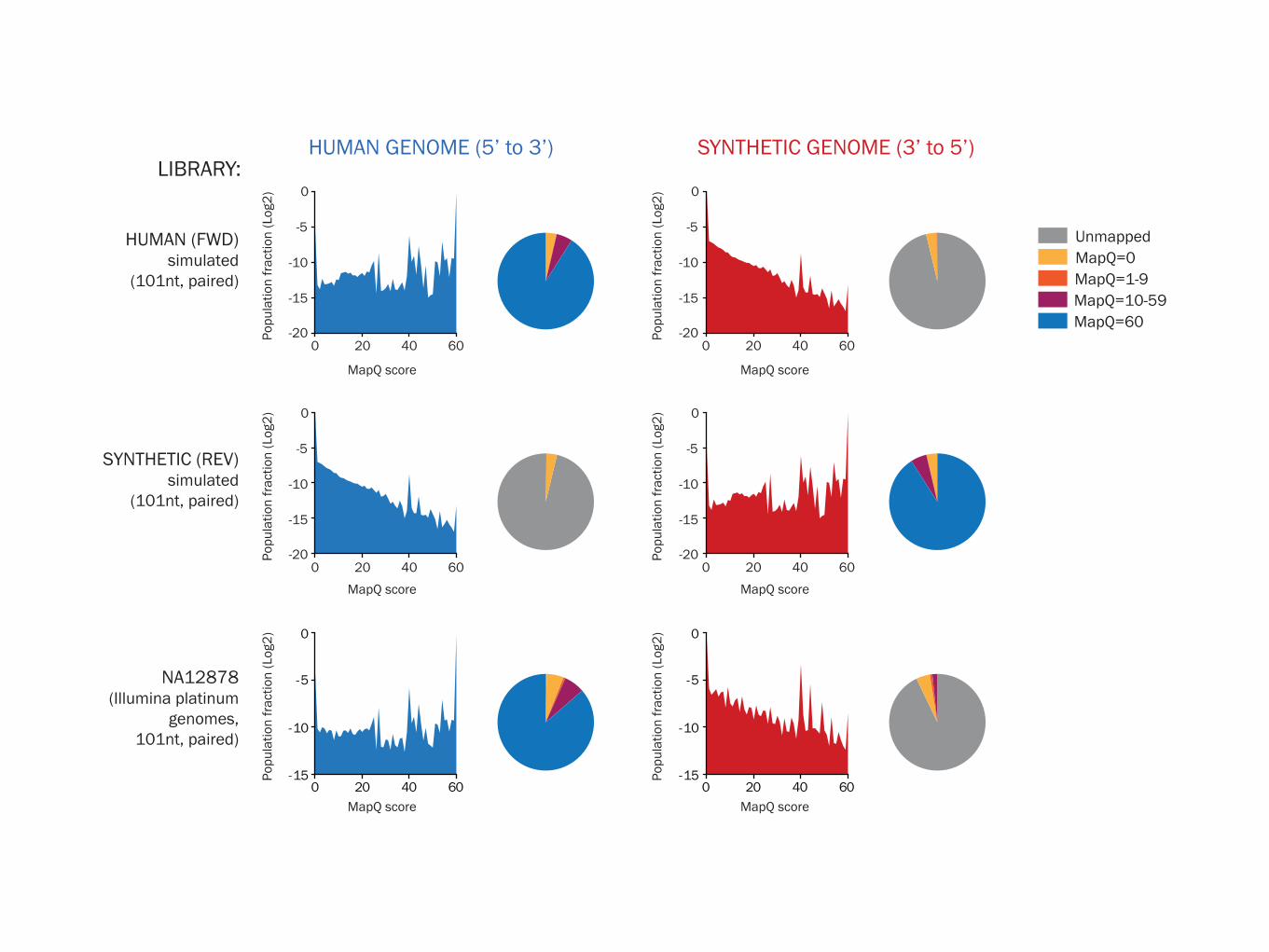
HUMAN (FWD)simulated
(101nt, paired)
SYNTHETIC (REV) simulated
(101nt, paired)
NA12878(Illumina platinum
genomes,101nt, paired)
-20
-15
-10
-5
0
Popu
latio
n fra
ctio
n (L
og2)
Popu
latio
n fra
ctio
n (L
og2)
Popu
latio
n fra
ctio
n (L
og2)
Popu
latio
n fra
ctio
n (L
og2)
UnmappedMapQ=0MapQ=1-9MapQ=10-59MapQ=60
0 20 40 60-15
-10
-5
0
Popu
latio
n fra
ctio
n (L
og2)
MapQ score
0 20 40 60
MapQ score
-20
-15
-10
-5
0
0 20 40 60
-20
-15
-10
-5
0
0 20 40 60MapQ score
-20
-15
-10
-5
0
Popu
latio
n fra
ctio
n (L
og2)
0 20 40 60MapQ score
0 20 40 60
-10
-5
0
MapQ score
-15
MapQ score
HUMAN GENOME (5’ to 3’) SYNTHETIC GENOME (3’ to 5’)LIBRARY:
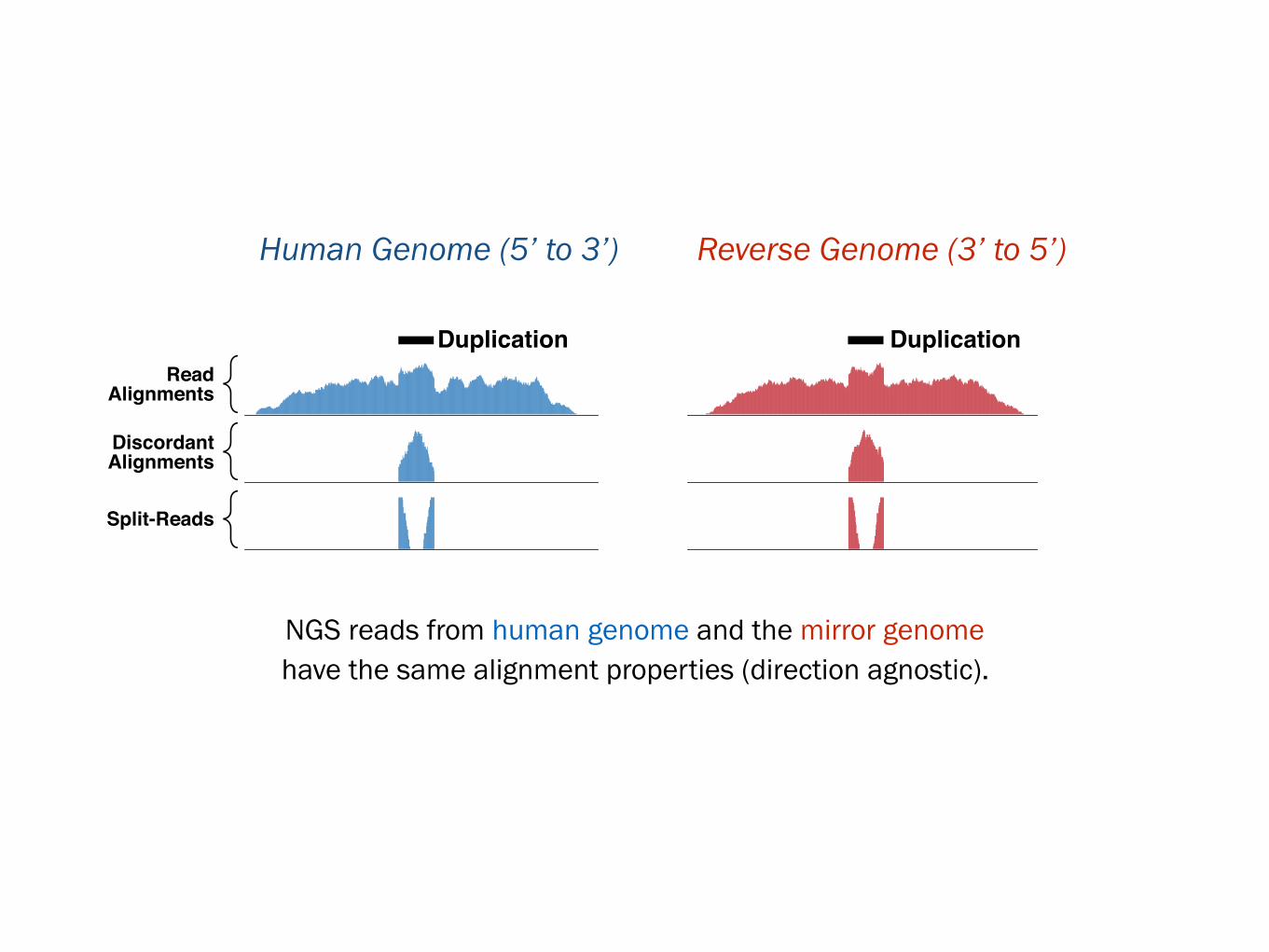
Read
Alignments
Split-Reads
Discordant
Alignments
Duplication Duplication
Human Genome
5’ to 3’Synthetic Genome
3’ to 5’
NGS reads from human genome and the mirror genome have the same alignment properties (direction agnostic).
Human Genome (5’ to 3’) Reverse Genome (3’ to 5’)
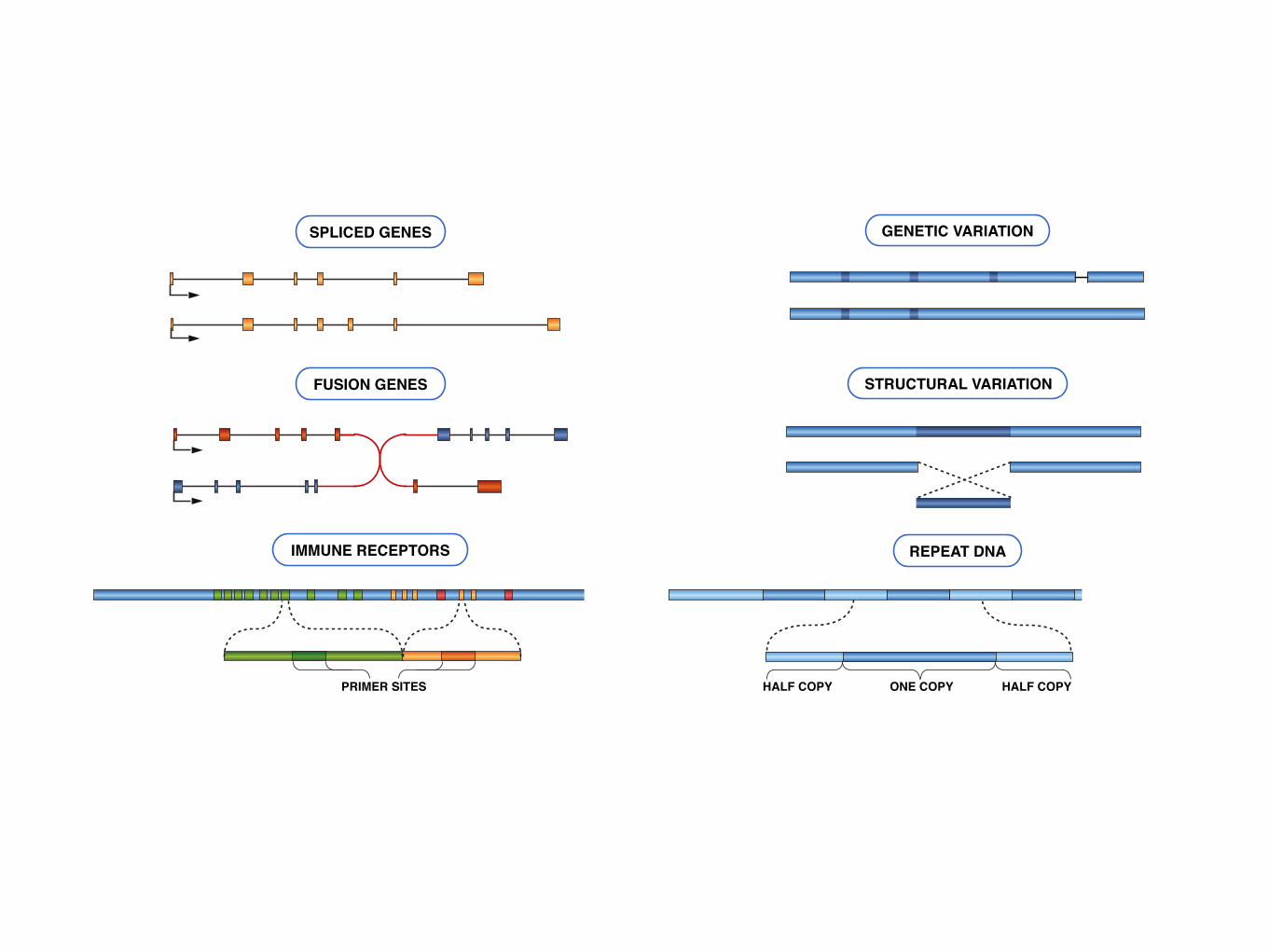
SPLICED GENES
FUSION GENES
IMMUNE RECEPTORS
GENETIC VARIATION
PRIMER SITES
STRUCTURAL VARIATION
REPEAT DNA
ONE COPYHALF COPY HALF COPY
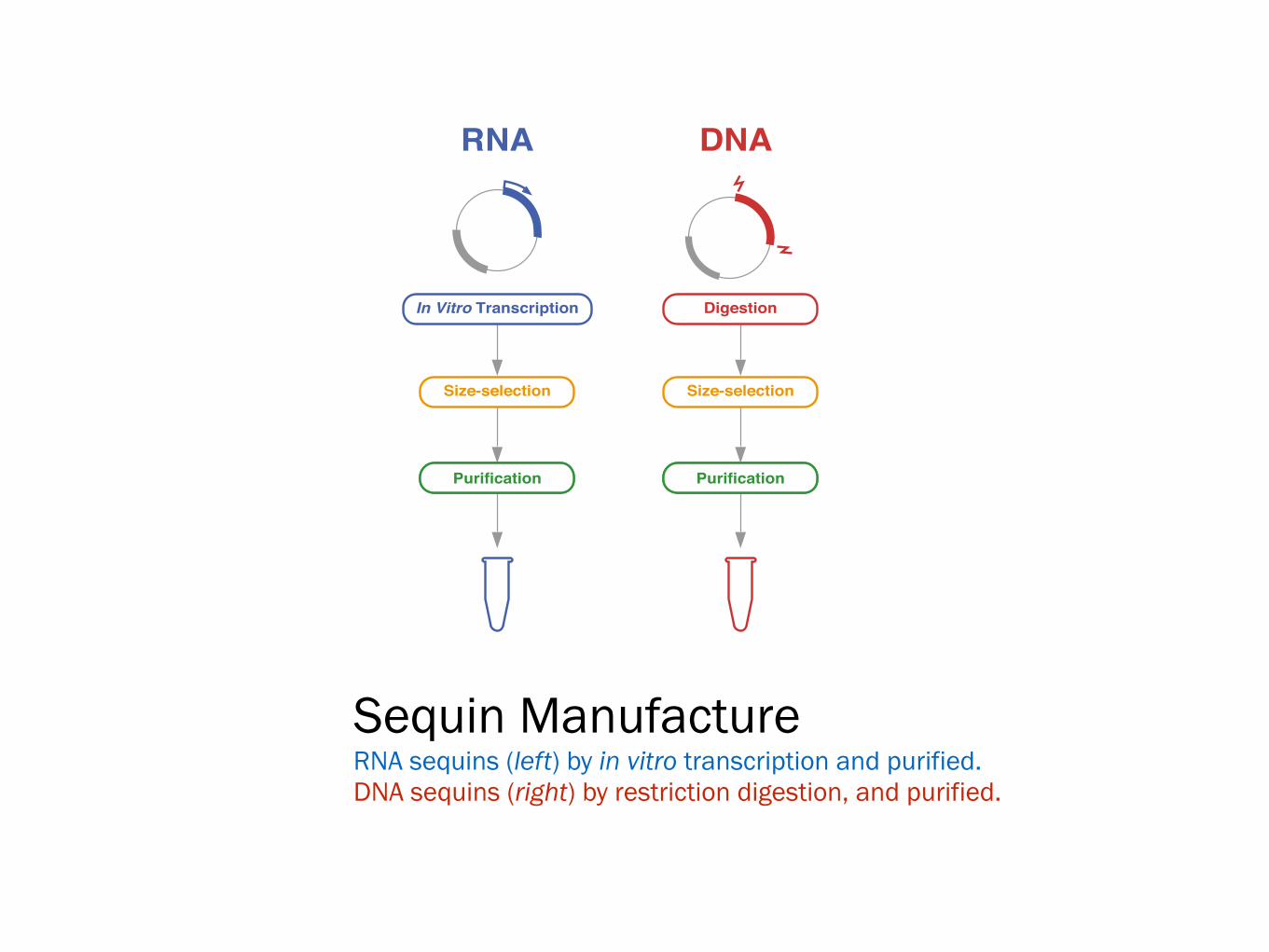
RNA DNA
Size-selection
Purification
In Vitro Transcription Digestion
Size-selection
Purification
Sequin Manufacture RNA sequins (left) by in vitro transcription and purified. DNA sequins (right) by restriction digestion, and purified.

Expected Abundance
Ob
serv
ed A
bun
da
nce
Expected Abundance
Ob
serv
ed A
bun
da
nce
Mix A
Mix B
Mixture A Mixture B Fold-difference
Variable Sequins(Measure differences between samples)
Constant Sequins(Normalise between samples)
Mixtures Individual (RNA or DNA) sequins are combined to emulate quantitive features (eg. gene expression, splicing, allele frequency) and establish internal reference ladders.
Expected Abundance
Observ
ed
Ab
un
da
nce
Observ
ed
Ab
un
da
nce
Expected AbundanceMix A
Mix B
Mixture A Mixture B Fold-difference
Variable Sequins(Measure differences between samples)
Mix A Mix B Fold-Change
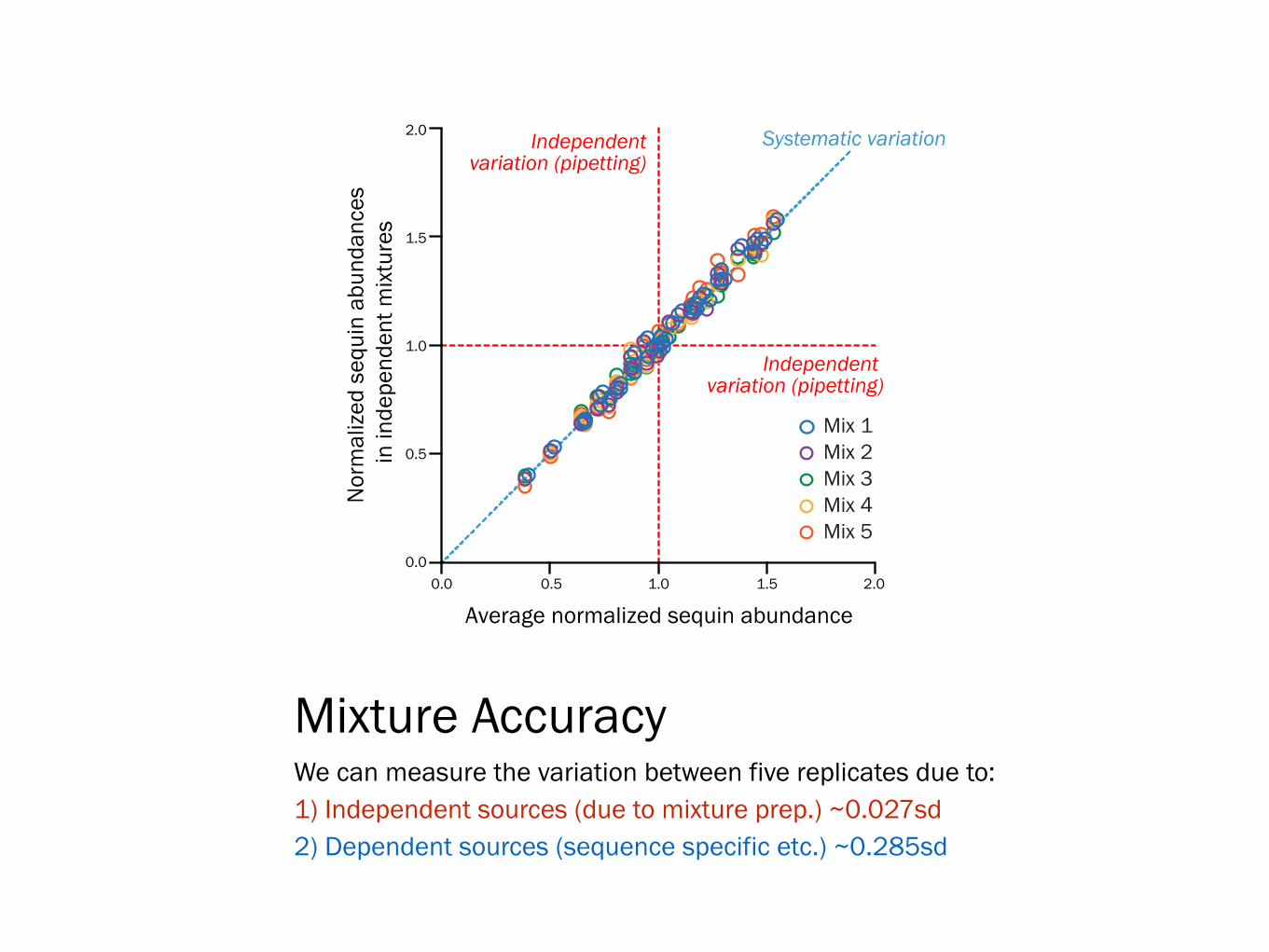
Mixture Accuracy We can measure the variation between five replicates due to: 1) Independent sources (due to mixture prep.) ~0.027sd 2) Dependent sources (sequence specific etc.) ~0.285sd
0.0 0.5 1.0 1.5 2.00.0
0.5
1.0
1.5
2.0
Average normalized sequin abundance
Nor
mal
ized
seq
uin
abun
danc
es in
inde
pend
ent m
ixtu
res
Mix 2Mix 3Mix 4Mix 5
Systematic variationIndependentvariation (pipetting)
Independent variation (pipetting)
Mix 1

Sequins are added to a RNA/DNA sampleat a fractional concentrations (typically 2-3%).
The combined sample is then sequenced, with a proportional fraction of the reads derived from sequins in the final library.

To distinguish reads in the library that derive from sequins, we align the library to a combined index comprising both the human genome (hg38) and also the mirror genome.
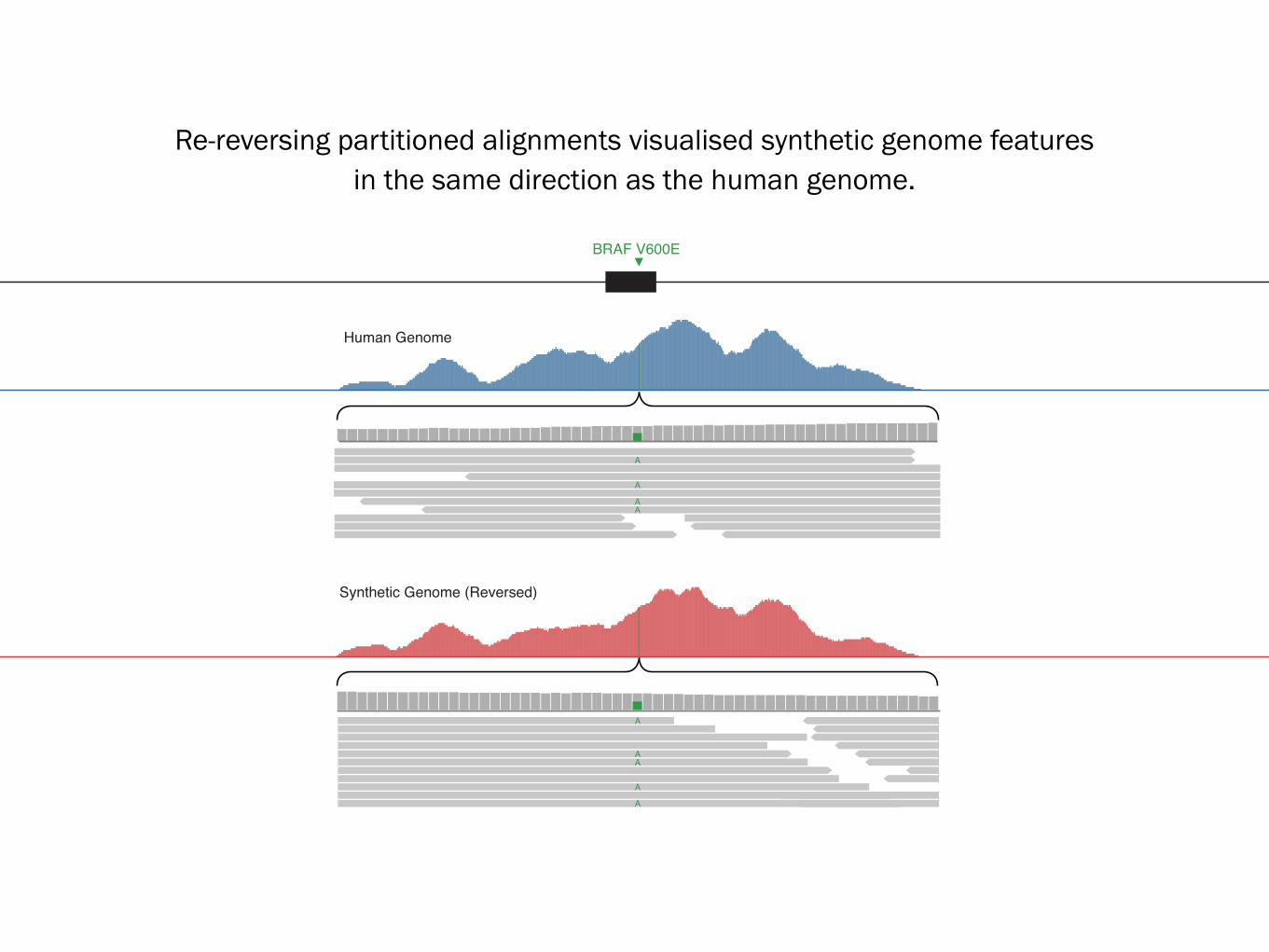
Human Genome
Synthetic Genome (Reversed)
A
A
A
A
BRAF V600E
A
A
A
A
A
Re-reversing partitioned alignments visualised synthetic genome features in the same direction as the human genome.


GENETIC VARIANTS

$
!$
Sequence Read
Coverage
Alignments
Identified Variants
Homozygous Variation
Heterozygous Variation InDels
Sequin A
Sequin B
in silico ChromosomeVariant A
Variant B
Homozygous Variation
Heterozygous Variation
Sequin B
Sequin A
Manufacture sequins
Combine with genome DNA
for sequencing and analysis

1kb length240 SNPs/Indels sampled from dbSNP(Deveson et al., Nature Methods 2016)
v2 (available shortly)
1.8kb length99 SNVs/Indels sampled from NA12878 high confidence (v2)99 SNVs/Indels in difficult regions(high & low GC, mono/di/tri nucleotide repeats)heterozygous / homozygous as per annotation
v1
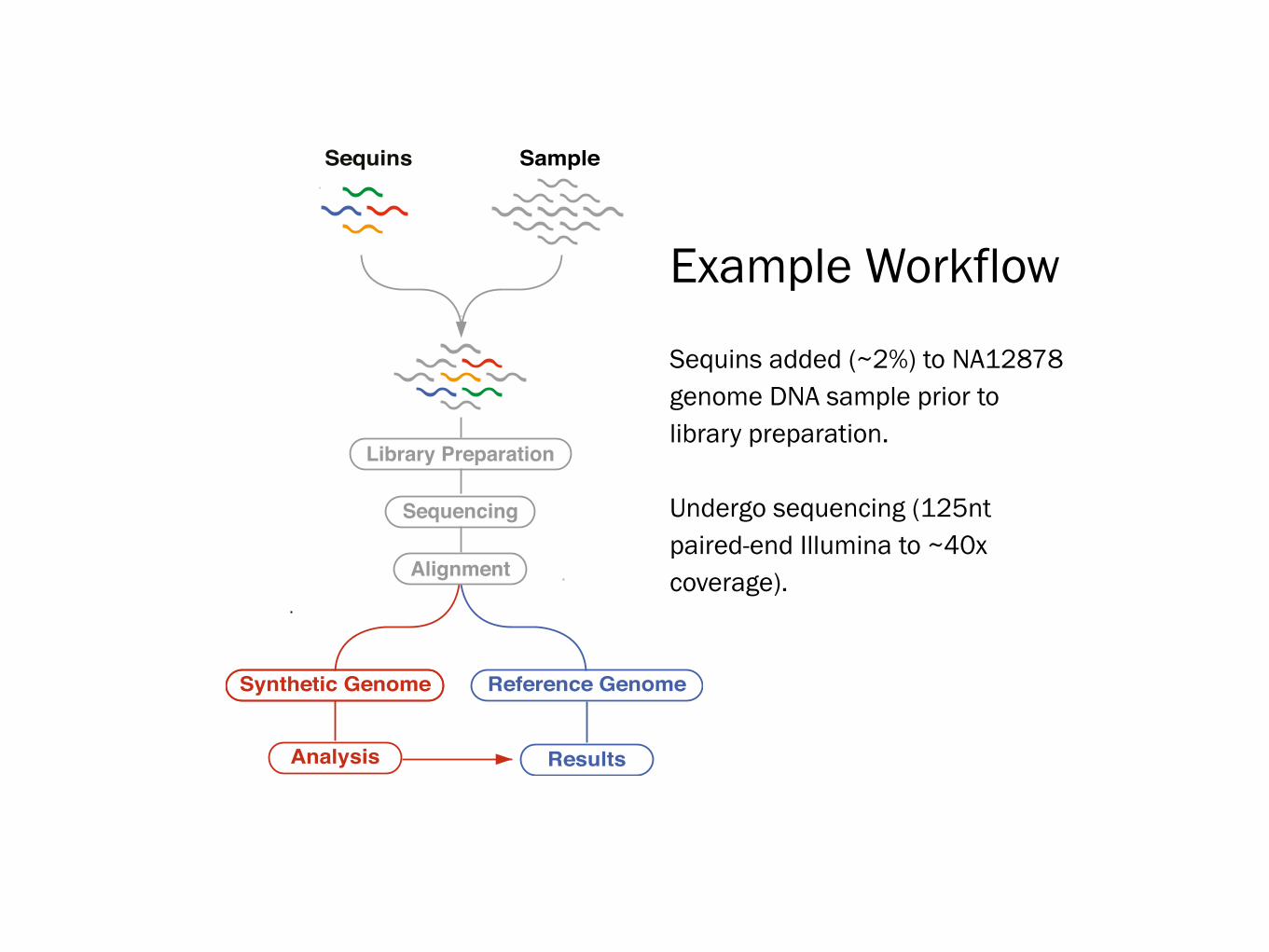
Library Preparation
Reference GenomeSynthetic Genome
SampleSequins
Sequencing
Alignment
Analysis Results
Example Workflow
Sequins added (~2%) to NA12878 genome DNA sample prior to library preparation.
Undergo sequencing (125nt paired-end Illumina to ~40x coverage).
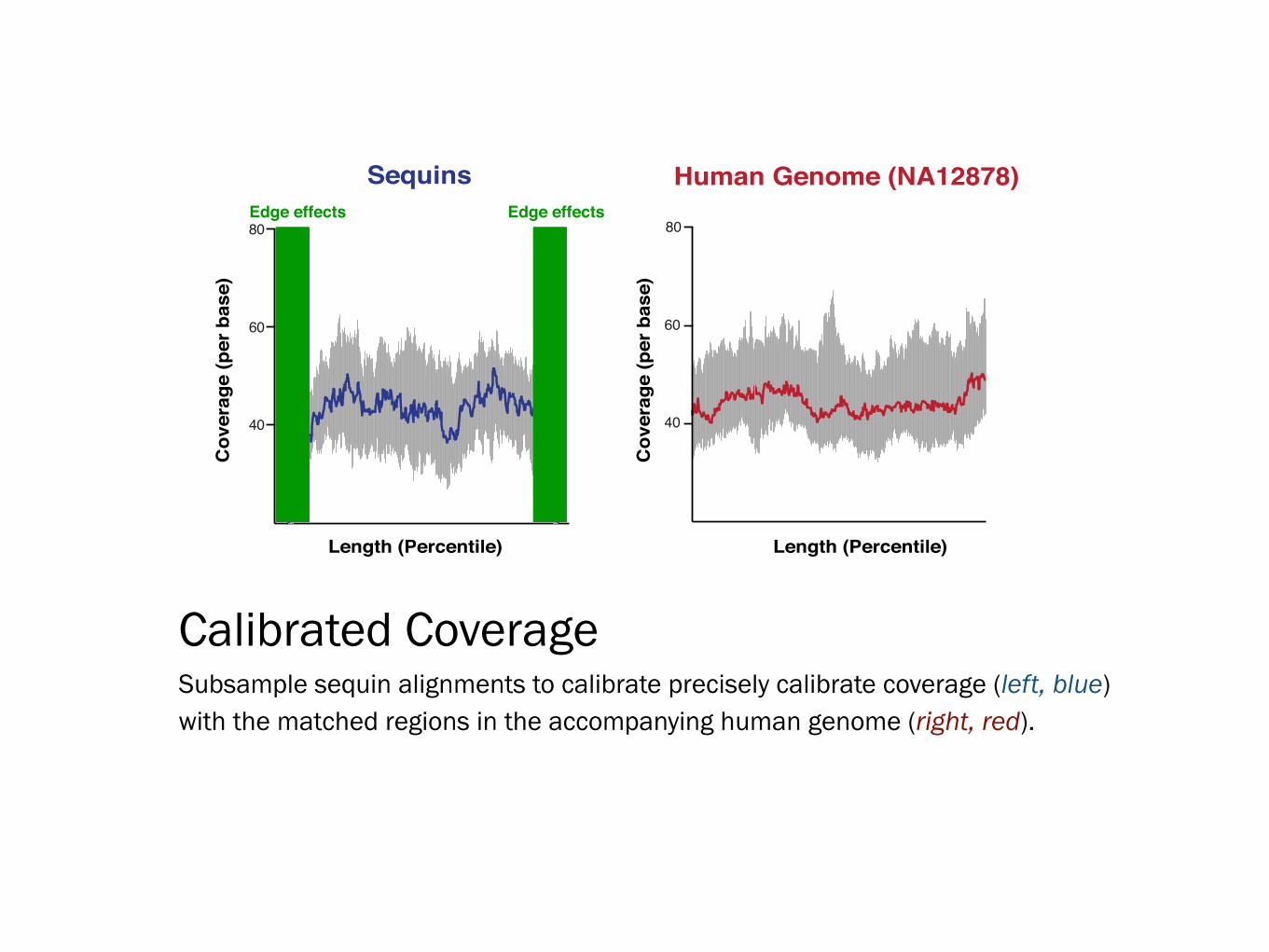
Calibrated Coverage Subsample sequin alignments to calibrate precisely calibrate coverage (left, blue) with the matched regions in the accompanying human genome (right, red).
40
60
80
40
60
80
Length (Percentile) Length (Percentile)
Edge effects Edge effectsC
ov
era
ge
(p
er b
as
e)
Co
ve
ra
ge
(p
er b
as
e)
Sequins Human Genome (NA12878)
0 25 500
50
100
0 25 500
50
100
Median Per-Base CoverageMedian Per-Base CoverageSe
nsiti
vity
(%)
Sens
itivi
ty (%
)
Sequins
NA12878
""
"
"
""
"
"
!
!!!
!
!
! #
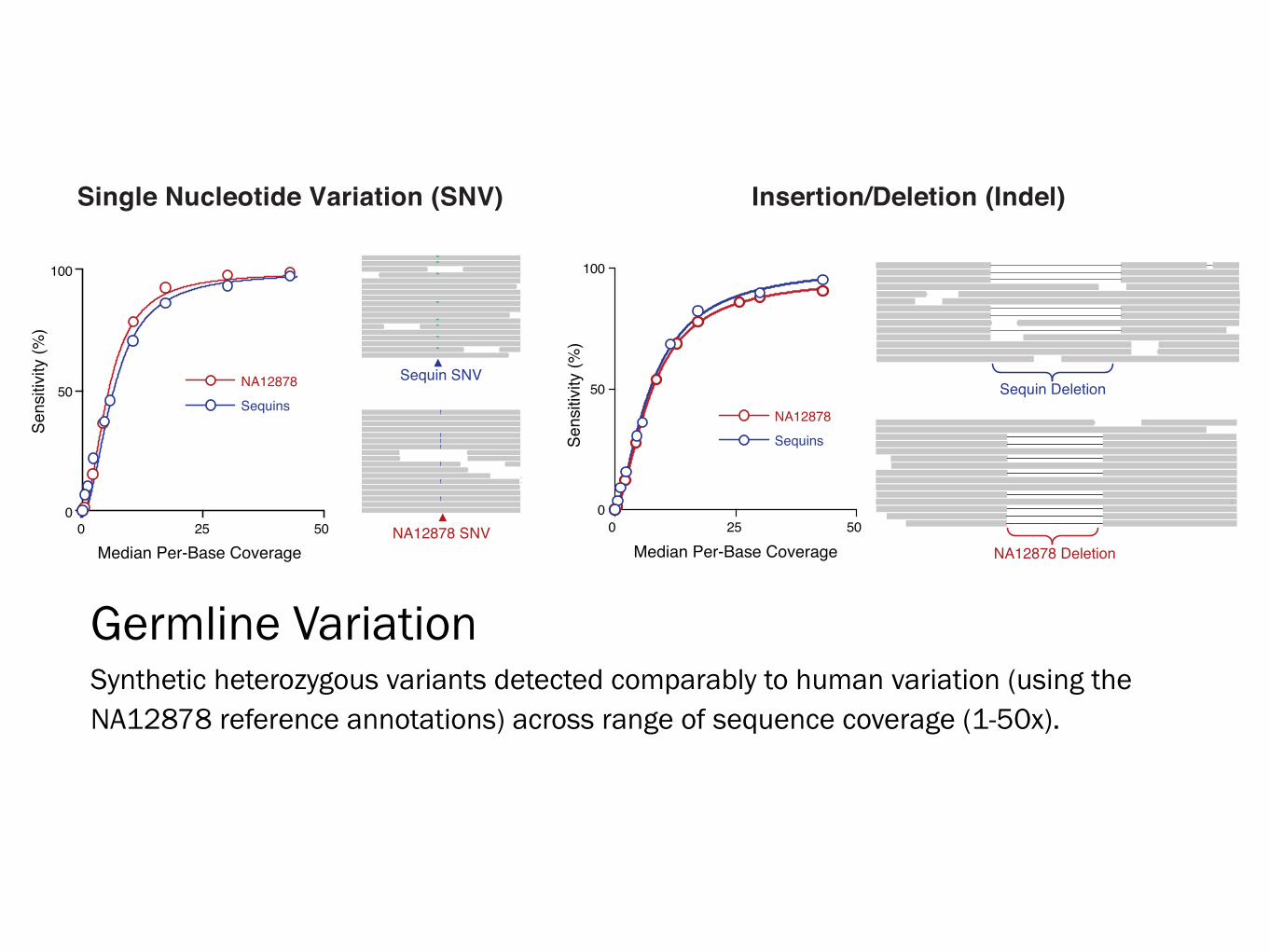
Sequin SNVSequin Deletion
NA12878 SNVNA12878 Deletion
Single Nucleotide Variation (SNV) Insertion/Deletion (Indel)
Sequins
NA12878
Germline Variation Synthetic heterozygous variants detected comparably to human variation (using the NA12878 reference annotations) across range of sequence coverage (1-50x).

Somatic Mutations By titrating ‘variant’ sequins relative to ‘reference’ sequins, we can establish the range of somatic mutation frequencies observed in complex tumour sub-populations.
Sequin Frequencies
FOXP 1/FLT3
FLT3/IDH1
CXCL17
TP53
IDH2/RUNX1
Griffith et. al., Cell Systerms 2015
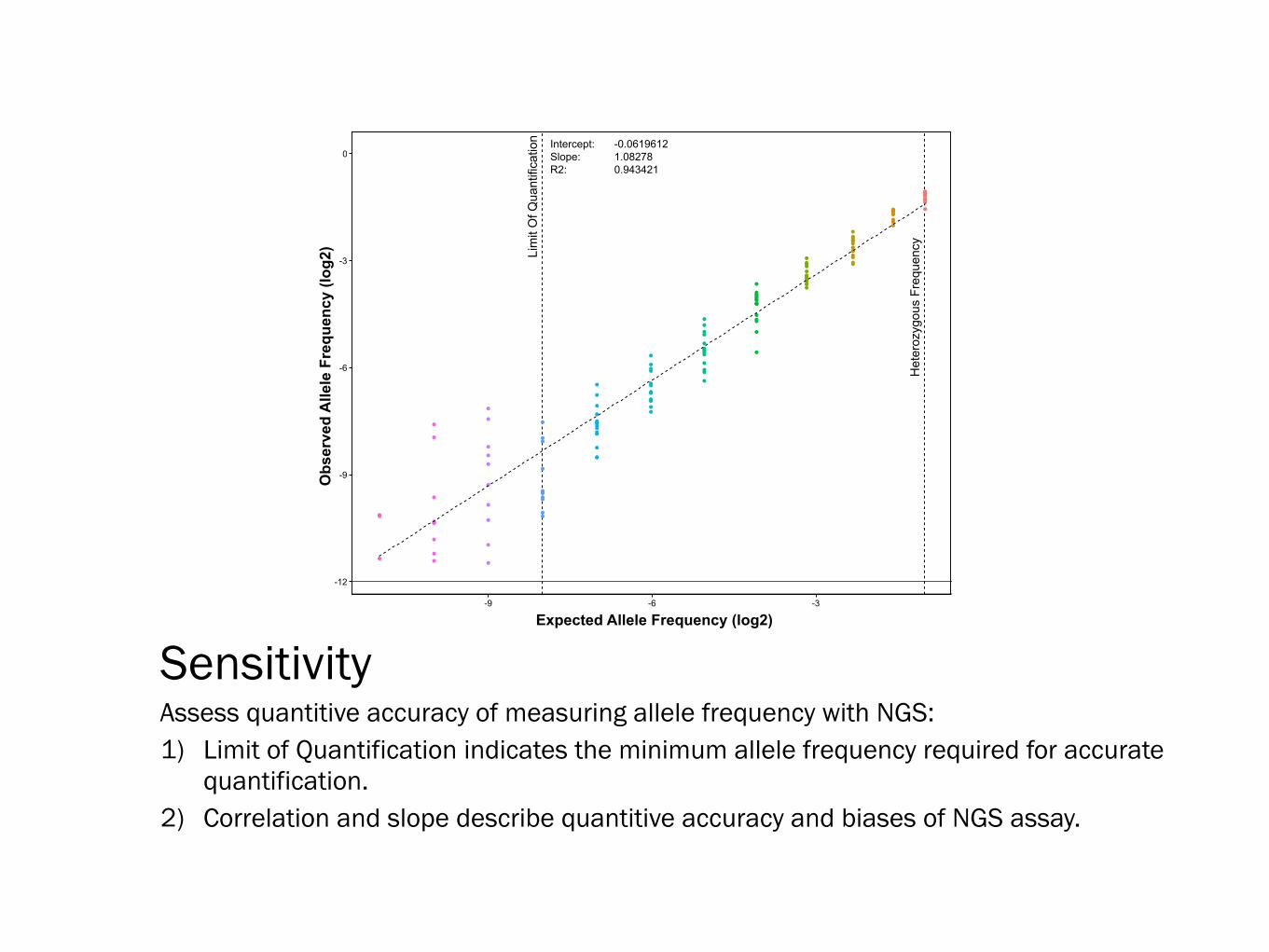
Sensitivity Assess quantitive accuracy of measuring allele frequency with NGS: 1) Limit of Quantification indicates the minimum allele frequency required for accurate
quantification. 2) Correlation and slope describe quantitive accuracy and biases of NGS assay.
Het
eroz
ygou
s Fr
eque
ncy
-12
-9
-6
-3
0
-9 -6 -3
Expected Allele Frequency (log2)
Ob
se
rv
ed
All
ele
Fre
qu
en
cy
(lo
g2
) Lim
it O
f Qua
ntifi
catio
n Intercept: -0.0619612Slope: 1.08278R2: 0.943421
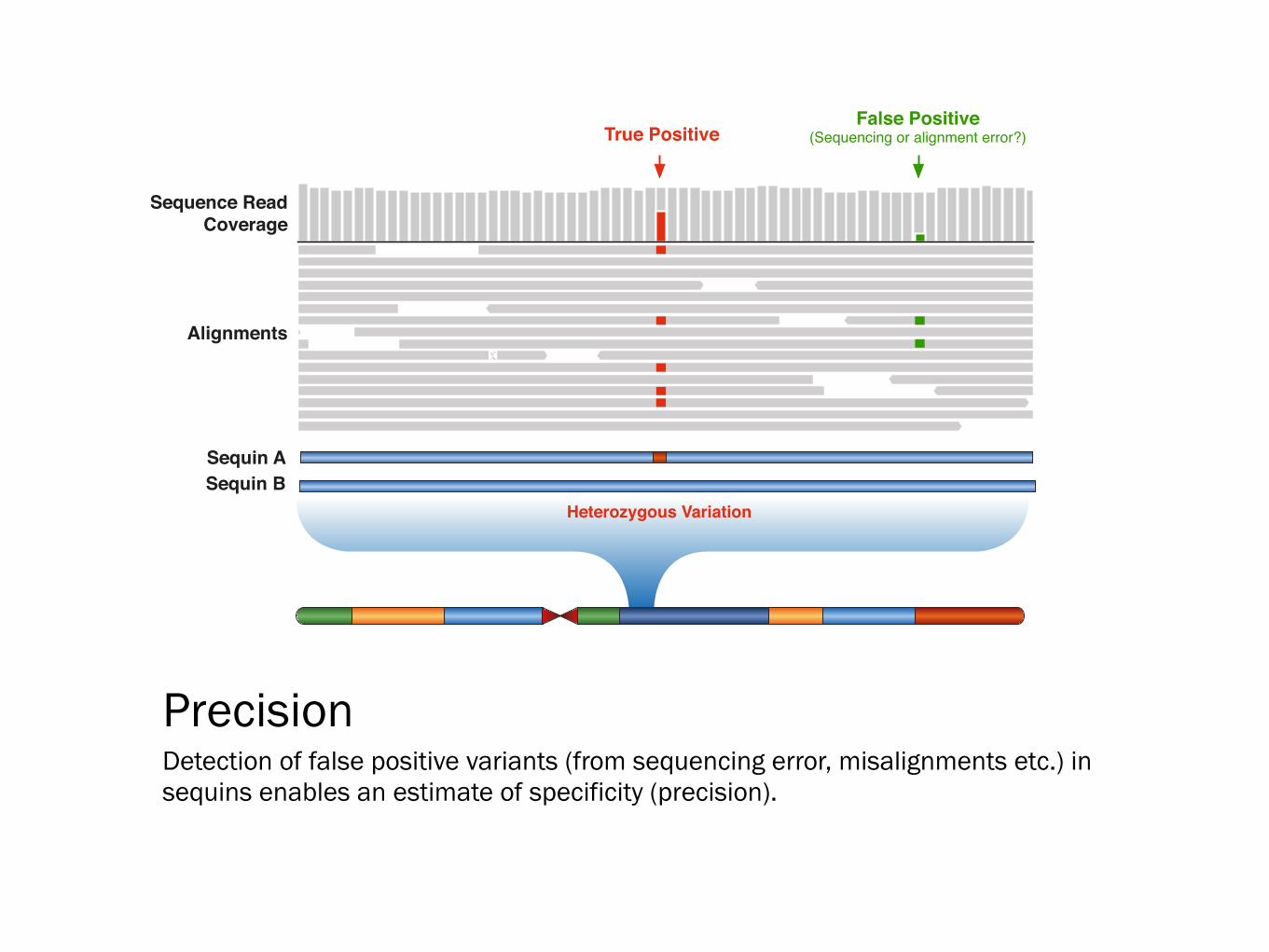
Precision Detection of false positive variants (from sequencing error, misalignments etc.) in sequins enables an estimate of specificity (precision).
$
!$
Sequence Read
Coverage
Alignments
Heterozygous Variation
Sequin A
Sequin B
True PositiveFalse Positive
(Sequencing or alignment error?)
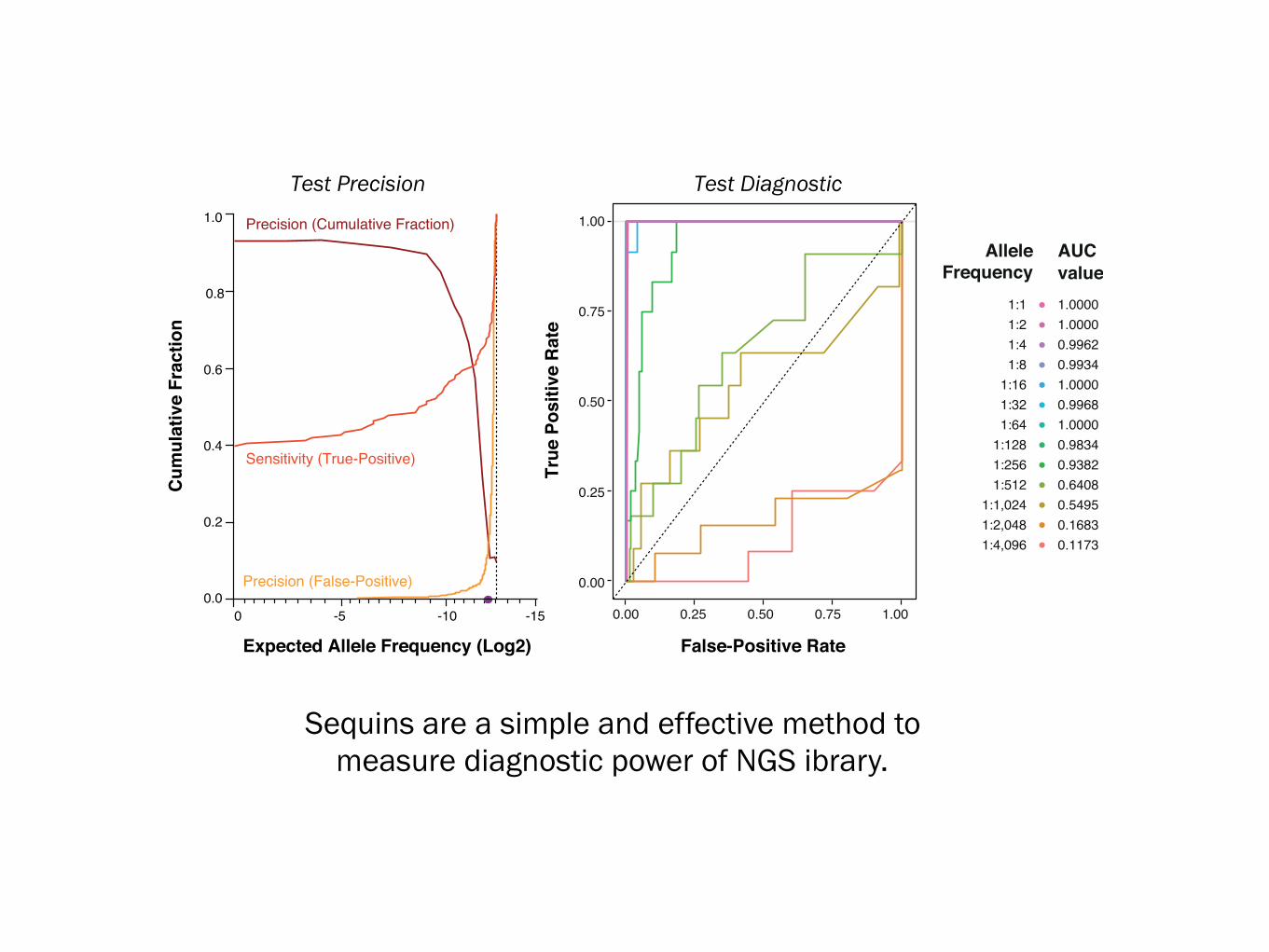
Sequins are a simple and effective method tomeasure diagnostic power of NGS ibrary.
0 -5 -10 -15
1:11:21:41:8
1:161:321:64
1:1281:2561:512
1:1,0241:2,0481:4,096
AlleleFrequency
0.00 0.25 0.50 0.75 1.00
0.00
0.25
0.50
0.75
1.00
0.0
0.2
0.4
0.6
0.8
1.0
1.00001.00000.99620.99341.00000.99681.00000.98340.93820.64080.54950.16830.1173
AUCvalue
Precision (Cumulative Fraction)
Sensitivity (True-Positive)
Precision (False-Positive)
Cum
ulat
ive
Frac
tion
False-Positive Rate
True
Pos
itive
Rat
e
Expected Allele Frequency (Log2)
Test Precision Test Diagnostic
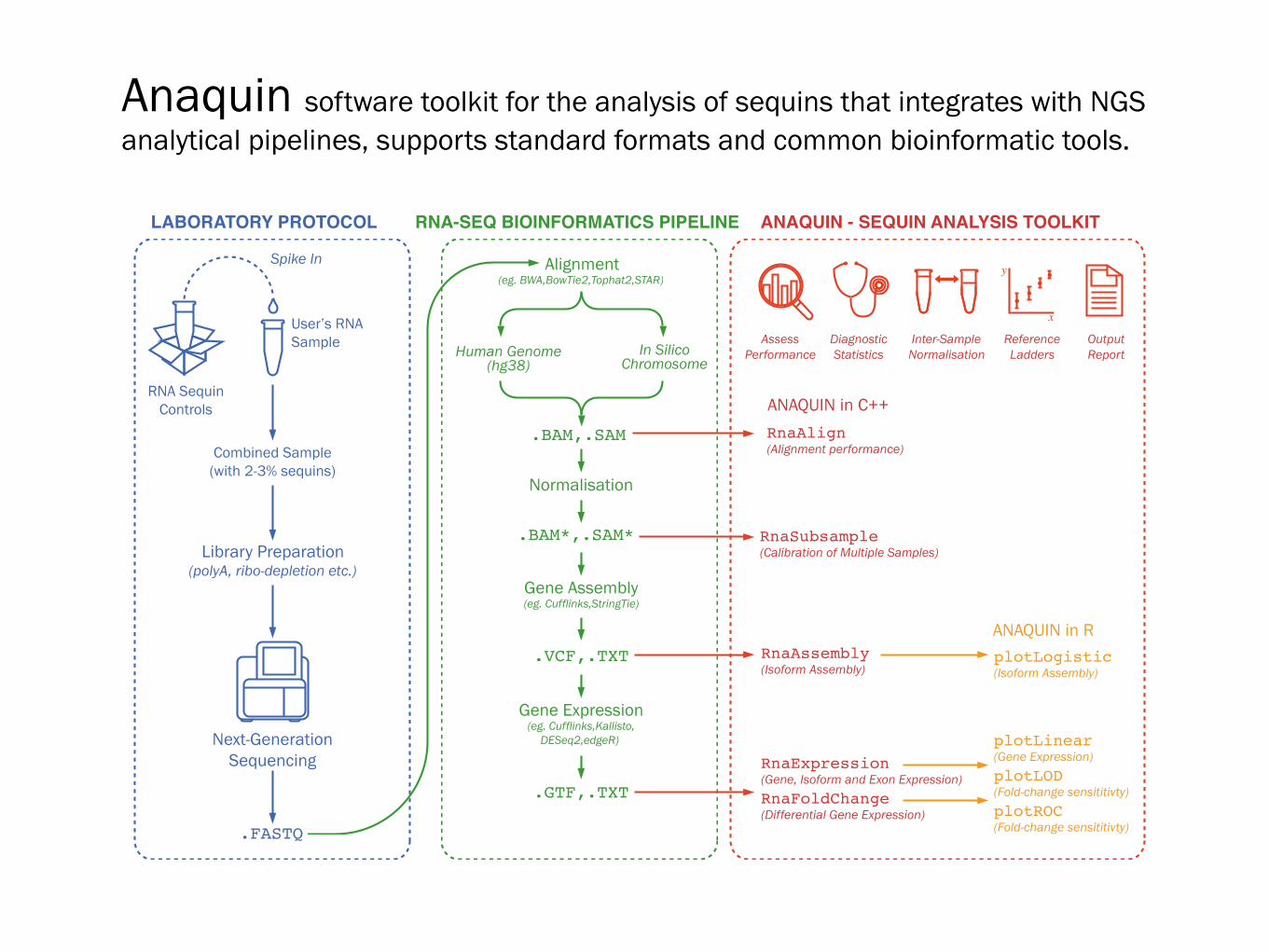
RnaAlign
(Alignment performance)
RnaExpression
(Gene, Isoform and Exon Expression)RnaFoldChange
(Differential Gene Expression)
plotLinear
(Gene Expression)plotLOD
(Fold-change sensititivty)plotROC
(Fold-change sensititivty)
RnaSubsample
(Calibration of Multiple Samples)
RnaAssembly
(Isoform Assembly)plotLogistic
(Isoform Assembly)
Library Preparation(polyA, ribo-depletion etc.)
Next-GenerationSequencing
User’s RNASample
RNA SequinControls
Combined Sample(with 2-3% sequins)
Spike In
.FASTQ
ANAQUIN in C++
ANAQUIN in R
LABORATORY PROTOCOL
Alignment(eg. BWA,BowTie2,Tophat2,STAR)
Gene Assembly(eg. Cufflinks,StringTie)
Normalisation
Gene Expression(eg. Cufflinks,Kallisto,
DESeq2,edgeR)
.BAM,.SAM
.BAM*,.SAM*
.VCF,.TXT
.GTF,.TXT
RNA-SEQ BIOINFORMATICS PIPELINE
Human Genome(hg38)
In SilicoChromosome
x
y
DiagnosticStatistics
Inter-SampleNormalisation
ReferenceLadders
OutputReport
AssessPerformance
ANAQUIN - SEQUIN ANALYSIS TOOLKIT
Anaquin software toolkit for the analysis of sequins that integrates with NGS analytical pipelines, supports standard formats and common bioinformatic tools.

SEQUINS ARE FREE FOR NON-PROFIT RESEARCH, request an aliquot from www.sequin.xyz

Acknowledgments: Ted Wong
Jim Blackburn Ira Deveson
Bindu Kanakamedala Simon Hardwick
Wendy Chen James Ferguson
John Mattick Katrina Frankcombe
Peter Whitfield
Further Reading ‘Representing genetic variation with synthetic DNA standards.’
by Deveson et al., (2016) Nature Methods
‘Spliced synthetic genes as internal controls in RNA sequencing experiments’ by Hardwick et al., (2016) Nature Methods
Recommended

















