
Research Collection
Doctoral Thesis
Mitochondrial genomes of plant pathogenic fungi
Author(s): Torriani, Stefano F.F.
Publication Date: 2008
Permanent Link: https://doi.org/10.3929/ethz-a-005807786
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Diss. ETH No. 18123
MITOCHONDRIAL GENOMES OF PLANT PATHOGENIC FUNGI
A dissertation submitted to the
SWISS FEDERAL INSTITUTE OF TECHNOLOGY ZURICH For the degree of
DOCTOR OF SCIENCE
Presented by
STEFANO F. F. TORRIANI Dipl. Natw. ETH
Born 17 January 1980
Citizen of Switzerland
Accepted on recommendation of
Prof. Dr. Bruce A. McDonald, examiner
Prof. Dr. Alexander Widmer, co-examiner
2008


Table of contents
SUMMARY 1
RIASSUNTO 3
CHAPTER 1 5
General introduction
CHAPTER 2 25
The mitochondrial genome of Mycosphaerella graminicola
CHAPTER 3 51
The evolutionary history of Mycosphaerella spp. mitochondrial genome
CHAPTER 4 75
The mitochondrial genome of Phaeosphaeria nodorum
CHAPTER 5 123
QoI resistance evolution in European populations of Mycosphaerella graminicola
CHAPTER 6 143
Screening for QoI resistance in a global sample of Rhynchosporium secalis
CHAPTER 7 159
General conclusions
ACKNOWLEDGEMENTS 167
CURRICULUM VITAE 169
PUBBLICATIONS 169


To all those who manage to read from start to finish


1
Summary
The mitochondrial genomes of plant pathogenic fungi was the leitmotiv of this work that
1) completely annotated the mitochondrial genomes of two wheat-infecting pathogens,
Mycosphaerella graminicola and Phaeosphaeria nodorum, 2) inferred the mitochondrial
evolutionary history of M. graminicola and 3) studied the evolutionary mechanisms
influencing the occurrence and spread of strobilurin (QoI) fungicide resistances.
The two obtained mitochondrial genomes were the first analyzed from any species from
the class Dothideomycetes, including more than 6000 known species and some of the most
economically important plant pathogenic fungi. The fungal mitochondrial genomes displayed
some common characteristics interspecifically, including a standard set of genes involved in
oxidative phosphorylation, two genes for the large and the small ribosomal subunits and a set
of tRNAs genes, which often clustered in groups showing conservation in gene order
between organisms in the large Pezizomycotina subphylum of Ascomycota. The
mitochondrial tRNA gene order was proposed as an additional criterion in phylogenetic
analyses to distinguish between fungal classes. Another distinctive feature of fungal
mitochondrial genomes is the variable number of introns.
The evolutionary history of the M. graminicola mitochondrial genome was inferred using
four sequence loci, dating the divergence from the ancestral Mycosphaerella populations,
between 10,000 and 9,000 years ago, coinciding with the domestication of wheat and
essentially confirming the analysis based on nuclear markers. The insertion/deletion
polymorphisms distinguishing the mitochondrial haplotypes were characterized. A putative
gene predicted to encode protein acting as virulence factor proposed to increase the
pathogenicity to durum wheat (Triticum turgidum), was identified exclusively in
mitochondrial RFLP haplotype 4.
To provide better fungicide management strategies the evolutionary mechanisms driving
the emergence and spread of resistance need to be studied. QoIs target the protein encoded by
the mitochondrial gene cytochrome b. M. graminicola and the barley-infecting pathogen
Rhynchosporium secalis were therefore screened for QoI resistance. All 841 tested R. secalis
isolates from a global collection were completely sensitive with an estimated naïve resistance
frequency less than 0.12%. M. graminicola populations displayed high QoI resistance

2
frequencies in Europe, ranging from 30% to 100%. In M. graminicola, selection, parallel
genetic adaptation and migration were identified as the principal forces shaping the evolution
of fungicide resistances. The same evolutionary pattern of QoI resistance in M, graminicola
could be extended to other fungal pathosystems or fungicides having different modes of
action.

3
Riassunto
Il genoma mitocondriale dei funghi fitopatogeni è stato il filo conduttore di questo lavoro
durante il quale: 1) è stato annotato l’intero genoma mitocondriale di due patogeni del
frumento Mycosphaerella graminicola e Phaeosphaeria nodorum, 2) è stata dedotta la storia
evolutiva di M. graminicola, 3) sono stati investigati i meccanismi evolutivi responsabili
dello sviluppo e diffusione della resistenza ai fungicidi analoghi delle strobilurine.
I due genomi mitocondriali ottenuti sono i primi analizzati di specie appartenenti alla
classe Dothideomycetes, che comprende più di 6,000 specie e alcuni fra i funghi fitopatogeni
economicamente più dannosi. I genomi fungini mitocondriali mostrano alcune caratteristiche
comuni a livello interspecifico, come l’espressione di un gruppo di geni dedicati alla
fosforilazione ossidativa, due geni codificanti per altrettanti RNA ribosomiali (rRNA) e una
gamma di RNA di trasporto (tRNA), i quali sovente sono raggruppati in clusters e mostrano
una conservazione nell’ordine genico fra gli appartenenti alla suddivisione Pezizomycotina.
Questo lavoro propone di usare l’ordine genico dei tRNA mitocondriali quale criterio
aggiuntivo in analisi filogenetiche per differenziare classi fungine. Un numero variabile di
introni fra un organismo e l’altro è una caratteristica distintiva dei genomi mitocondriali dei
funghi.
La storia evolutiva del genoma mitocondriale di M. graminicola è stata dedotta usando
quattro loci genetici datando la separazione di questa specie dalle popolazioni ancestrali di
Mycosphaerella fra 10,000 e 9,000 anni fa, simultaneamente alla domesticazione del
frumento; ciò che conferma i risultati di una precedente analisi basata su loci nucleare. Le
inserzioni e delezioni di DNA, che distinguono gli aplotipi mitocondriali di M. graminicola,
sono state caratterizzate identificando un gene putativo (esclusivo dell’aplotipo 4) che si
suppone codifichi un fattore di virulenza in grado di aumentare la patogenicità verso il grano
duro (Triticum durum).
L’analisi dei meccanismi evolutivi alla base dello sviluppo e diffusione delle resistenze è
di fondamentale importanza per fornire migliori strategie di gestione dei fungicidi. M.
graminicola e Rhynchosporium secalis, un patogeno dell’orzo, sono stati dunque esaminati
alla ricerca d’isolati resistenti ai fungicidi analoghi delle strobilurine che agiscono sul

4
citocromo b, codificato da un gene mitocondriale. Tutti gli 841 isolati di R. secalis
provenienti dai cinque continenti sono risultati sensibili e la frequenza di resistenza stimata è
inferiore a 0.12%. Le popolazioni europee di M. graminicola invece mostrano frequenze di
resistenza variabili fra 30-100%. Dall’analisi di M. graminicola si deduce che selezione,
adattamento genetico parallelo e migrazione sono le principali forze che modellano
l’evoluzione delle resistenze. Questa interpretazione probabilmente può essere estesa ad altri
sistemi patologici fungini e a fungicidi aventi modi d’azione differenti da quello dei fungicidi
analoghi delle strobilurine.

CHAPTER 1
General introduction

Chapter 1 General introduction
6

Chapter 1 General introduction
7
Agriculture and pathogens - Past, Present and Future
About 12,000 years ago humanity began the slow development from a hunter-gatherer to
an agricultural society. Archeological and genetic findings support the origin of the
agriculture in the Fertile Crescent of the Middle East between 10,000 and 12,000 years ago
(Gopher et al. 2002; Salamini et al. 2002). This step, also known as the Neolithic revolution
(Childe 1953), was crucial for shaping the human society we know today. Without
agriculture the earth would be different, probably characterized by a reduced technology,
political organization, no or little urbanization, less pollution and more biodiversity. The wild
progenitors of the major Neolithic founder crops e.g. wheat (Triticum spp.), barley (Hordeum
vulgare) and pea (Pisum sativum) have been identified in the Middle East (Diamond 1997;
Haudry 1997). The continuous selection of desirable morphological traits has driven the slow
process of wild grass domestication, creating the crop species we know today (Sharma and
Waines 1980; Elias et al. 1996; Taenzler et al. 2002). The first crops were probably
composed of species mixtures, but over time Neolithic farmers of the Middle East preferred
planting crop species monocultures that subsequently spread to other regions in Europe and
Asia (Diamond 1997).
The invention of agriculture caused the simultaneous evolution of those pathogens
infecting the wild progenitors of the founder crops. With agriculture intensification the
landscape was modified, changing from a natural to an agricultural ecosystem, characterized
by greater environmental homogeneity, lower species diversity and higher density of more
host-specialized pathogens (Stukenbrock and McDonald 2008). The evolution of all three
pathogens studied in this work, (Mycospharella graminicola, Phaeosphaeria nodorum and
Rhynchosporium secalis) was shaped by human mediated environmental changes that created
agricultural ecosystems. Stukenbrock and McDonald (2008) identified the four more likely
scenarios for the evolutionary mechanisms by which plant pathogenic fungi have emerged in
agricultural ecosystem; 1) host-tracking, when pathogen and host coevolved during host
domestication (Stukenbrock et al. 2007), 2) host shifts and jumps, e.g. following the
introduction of new crops into new natural ecosystems or from a wild population (Couch et
al. 2005), 3) horizontal gene transfer, when a exogenous gene is introduced in a new genome

Chapter 1 General introduction
8
(Friesen et al. 2006), 4) interspecific hybridization, when a big part of an exogenous genome
is introduced into a new genome (Ioos et al. 2006).
As the beginning of agriculture influenced evolution of plant pathogenic fungi, it is
reasonable to postulate that present anthropogenic climate changes will affect future
evolution of the same organisms. A pioneering study, confirming that current
anthropogenically induced environmental changes influenced pathogen evolution, was
conducted using archived samples of wheat grain and leaves from the Broadbalk experiment
in Rothamsted (1844-2003). This study presented a strong correlation between changes in
atmospheric pollution and changes in ratio of plant pathogenic fungi M. graminicola and P.
nodorum (Bearchell et al. 2005).
Agriculture played, and will play in the future, a fundamental role in sustaining the
logarithmic increase of the global human population, which grew from 1.6 to 6.1 billion
inhabitants during the 20th century (Lutz and Qiang 2002) and will probably reach 9 billion
by 2050 (United Nation 2004). The Food and Agriculture Organization of the United Nations
(FAO newsroom 2008) identified the major long-term challenges that world agriculture is
facing; among them 1) land and water constraints, 2) low investments in rural infrastructure
and agricultural research 3) expensive agricultural inputs relative to farm-gate prices 4)
limited adaptation to climate changes. To feed the world population of 2050, the global food
production must nearly double. Since population growth will occur mostly in emerging
countries and in urban areas, the rural work force will need to be more productive and
therefore investments in agriculture renewal are needed. Another future problem for
agriculture has its roots in the limited availability of oil, whose price reached a new record of
147.27 US$ per barrel on 11 July 2008. A possible alternative to fossil fuels are the biofuels
that may provide up to 25% of the world’s energy needs over the next 20 years (FAO
newsroom 2006). As produced today biofuels offer new opportunities to farmers, especially
in tropical areas, but simultaneously competition for land between energy production and
nutritional purposes will cause future troubles.

Chapter 1 General introduction
9
Mitochondrial genome current view
The discovery of mitochondrion could not be credited to a single scientist, however the
first descriptions of this small rod-shaped organelle go back to 1840. The name mitochondria
was introduced by microbiologist Carl Benda (1857-1933) in 1898 from the Greek mitos
“thread” and khondrion “little granule”. Most knowledge about mitochondria arose during
research conducted over the last sixty years (Scheffler 2001). Especially from the sixties,
when DNA was discovered in mitochondria, sequences accumulate in databases
continuously.
At present, 1,486 mitochondrial (mt) genomes have been completely sequenced and
deposited in the NCBI database, 57 from fungi including 37 from Ascomycota. Fungal mt
genome sequencing is in its infancy, since only a few of the 70,000 described fungal species
have been studied (Paquin et al. 1997). Mt genomes have proven to be highly useful for
research dealing with evolutionary and systematic studies because of their uniparental
inheritance, near absence of genetic recombination and uniform background (Chen and
Hebert 1999). During evolution of mt genomes, several genes were lost or translocated to the
nucleus (Adams et al. 2000), with most mt proteins now encoded by nuclear genes and only
subsequently imported into mitochondrion by translocase complexes. Relatively few mt
proteins are synthesized directly within the organelle (Hartl et al. 1989; Brennicke et al.
1993). Mt genomes vary widely in size among fungi, ranging from approximately 18 to 109
kb (NCBI database). The main causes are differences in length and organization of intergenic
regions and invasion of some mt genes by mobile group I introns, containing homing
endonuclease genes of the LAGLIDADG family (Signorovitch et al. 2007). Fungal mtDNAs
are generally larger than animal mtDNAs and about an order of magnitude smaller than those
of plants (Burger et al. 2003). Other unique characteristics of mt genomes are 1) high A+T
content, 2) lack of methylation, 3) conservation in gene function, 4) high copy number 5)
alternative genetic code and 6) an independent rate of evolution relative to nuclear genomes
(Griffiths 1996; Campbell et al. 1999; Ballard and Whitlock 2004).

Chapter 1 General introduction
10
Chemical disease control vs mitochondrial genome
Chemical disease control was routinely applied from the 19th century, though some
chemicals, like brine arsenic and copper sulphate, were used even earlier in the treatment of
cereal seeds (Russell 2005). From 1824 sulphur was recommended to control powdery
mildew and other foliar pathogens (Robertson 1824). More chemicals were released by the
end of 19th century, including e.g. Bordeaux mixture and mercuric chloride. After the Second
World War, a period of big advances in chemical industry, chemical crop protection industry
exploded and from the 1960s a more focused R&D and a rapid market growth characterized
it (Russell 2005). Immediately, with the increased use and sometimes overuse of chemical
compounds, pathogens developed resistances and industry realized that the situation was
having a serious impact so that a major collaboration was established with the formation of
Fungicide Resistance Action Committee (FRAC) in the early 1980s. FRAC’s main duty is to
coordinate resistance management strategies (Russell 1995). In 2005 the total market value of
fungicides approached nine billions dollar (McDougall 2006).
Most of the known fungicides targeted nuclear genes, but in 1996 were introduced in
the market strobilurins fungicides (QoIs), inhibiting mt respiration in fungi. The discovery of
QoIs was inspired by a group of natural fungicidal derivates of ß-methoxyacrylic acid
produced by a range of Basidiomycete wood-rotting fungi (Bartlett et al. 2002). A single
amino acid substitution (G143A) in mt encoded protein cytochrome b, caused by a single
nucleotide polymorphism, is the major mechanism of QoI resistance in most plant pathogenic
fungi (Gisi et al. 2002). Other fungi like Alternaria solani (Pasche et al. 2005) or
Pyrenophora teres (Sierotzki et al. 2007) have an alternative mutation (F129L), conferring
only moderate resistance (FRAC International, Germany). Unknown mechanisms leading to
QoI resistance have been seen in Venturia inaequalis (Steinfeld et al. 2001) and Podosphaera
fusca (Fernández-Ortuño et al. 2008). Recently, it was postulated that for those fungi having
Type 1 intron located directly after G143, the mutation causing G143A change couldn’t
occur because it would affect the splicing process, producing a non-functional protein
(Grasso et al. 2006). Until now the evolutionary mechanisms behind QoI resistance were not
clear, for example was not sure if QoI resistance emerged only once in a distinct mt
haplotype, that subsequently spread to other locations by migration, or if it has been fixed

Chapter 1 General introduction
11
independently in different genetic and geographic backgrounds by parallel genetic adaptation.
Chen et al. (2007) proposed for the grape pathogen Plasmopara viticola the parallel
emergence of fungicide resistance, but no literature was found for cereal pathogens.
Sales of QoIs exceeded $600 million in 1999 (McDougall 2001). In 1998, only two years
after the launch, the first QoI resistance appeared in Blumeria graminis (Sierotzki et al.
2000). In the following years other pathogens became resistant e.g. Pseudoperonospora
cubensis in 1999 (Ishii et al. 2001), Pyricularia grisea in 2000 (Vincelli and Dixon 2002),
Mycosphaerella graminicola in 2001 (Fraaije et al. 2005). Since no cross-resistance exists
between QoIs and other widely used compounds, such as DeMethylation Inhibitors (DMI) or
chlorothalonil, disease control and postponement of resistance development could be
achieved by combining these chemicals in spray programs.
Mycosphaerella graminicola - The pathogen
Mycosphaerella graminicola (anamorph: Septoria tritici) causes Septoria tritici blotch of
wheat and other poaceous hosts worldwide (Eyal 1999). This disease is especially severe in
humid and temperate climates e.g. northwestern Europe, causing up to 40% economic loss
(Eyal 1981). The taxonomic placement of Mycosphaerella was until recently uncertain and it
was usually placed near Dothidea in the Dothideales (Kirk et al. 2001; Goodwin et al. 2004),
but recent analyses placed Mycosphaerella in the Capnodiales, a sister group to the
Dothideales and Myriangiales (Schoch et al. 2006). Stukenbrock et al. (2007), using nuclear
markers, fixed the divergence of M. graminicola from the ancestral populations infecting
uncultivated grasses about 10,500 years ago in the Fertile Crescent, proposing the
evolutionary scenario of the pathogen domestication along with the host. M. graminicola life
cycle presents both sexual and asexual stages, producing respectively ascospores potentially
dispersed over several kilometers (Sanderson 1972) and pycnidiospores, normally
disseminated by rain splash (Bannon and Cooke 1998). M. graminicola displays high nuclear
and low mt diversity (Zhan et al. 2003). High nuclear diversity is produced by combined
effect of large effective population sizes (Zhan and McDonald 2004), high gene flow (Boeger
et al. 1993; Zhan et al. 2003) and recurring sexual reproduction (Chen and McDonald 1996).
The low mt diversity was proposed to result from a selective sweep affecting only the mt

Chapter 1 General introduction
12
genome (Zhan et al. 2004). The best way to control Septoria tritici blotch is through
fungicides, like DMI or QoIs. DMI have been extensively used for over 20 years, displaying
wide variation in the baseline sensitivity (Gisi et al. 1997; Stergiopoulos et al. 2003) with a
slow but constant shift towards reduced sensitivity (Gisi et al. 2005; Mavroeidi and Shaw
2005). Genetic analyses of azole-resistant strains indicated a polygenic system for azole
resistance (De Waard 1994; Stergiopoulos et al. 2003). QoIs were introduced in 1996 and
developed quickly high levels of resistance in M. graminicola (Fraajie et al. 2005).
Phaeosphaeria nodorum - The pathogen
Phaeosphaeria (syn. Leptosphaeria) nodorum [anamorph: Stagonospora (syn. Septoria)
nodorum] causes the economically important disease Stagonospora nodorum leaf and glume
blotch disease on wheat. P. nodorum is the major foliar pathogen in Western Australia and
north central and north eastern North America (Solomon et al. 2006) as it was in Europe until
the 1970s before being replaced by M. graminicola (Bearchell et al. 2005). P. nodorum is a
filamentous fungus from the Dothiodeomycetes class that includes the causal pathogens of
many economically important plant diseases e.g. Leptosphaeria maculans, Cochliobolus
heterostrophus and Ascochyta blight of several legumes (Winka and Eriksson 1997). Many
plant pathogens of the Dothiomycetes produce host specific toxins (Wolpert et al. 2002). In
P. nodorum, proteinaceous host specific toxins were shown to be important virulence
determinants (Liu et al. 2004a and 2004b), like ToxA that has been horizontally transferred
into Pyrenophora tritici repentis, causing the emergence of a new disease of wheat called
yellow or tan spot in 1941 (Friesen et al. 2006). As M. graminicola, P. nodorum displays in
both sexual and asexual stages, with ascospores being blown over considerable distances and
pycnidiospores dispersed over short distances (Griffiths and Hann 1976; Brennan et al. 1985;
Arseniuk et al. 1998). The infection epidemiology is polycyclic with repeated cycles of both
sexual and asexual infection (Bathgate and Loughman 2001; McDonald unpublished). P.
nodorum is dispersed over long distances also by seed borne dispersal (Shah et al. 1995;
Bennett et al. 2005). Seed transmission is important but easily controlled by fungicides.
Epidemiology and population genetic evidence indicates that the pathogen undergoes sexual

Chapter 1 General introduction
13
reproduction in the field regularly and that, as M. graminicola, gene flow is global
(Stukenbrock et al. 2006). The migration among populations probably has not been
symmetrical, but rather reflects the historical movement of the host. P. nodorum is mainly
controlled, as M. graminicola, with DMI and QoIs. Cultural practices could reduce the
severity of the infections often in a minor way (Eyal 1999).
Rhynchosporium secalis - The pathogen
Rhynchosporium secalis causes a necrotic disease known as scald or leaf blotch on
barley, rye and various other poaceous hosts, especially on species of Agropyron, Bromus,
Hordeum and Lolium (Caldwell 1937; Shipton et al. 1974; Zaffarano et al. 2008). Scald is a
worldwide problem and in region with humid and cool temperature it can cause yield losses
of 35-40% (Shipton et al. 1974; Khan 1986). R. secalis produces two-celled conidia with a
typical beak (from here the name of the species, since beak in Greek is Rhyncos). No sexual
spore has been found for the genus Rhynchosporium, however high levels of genetic
variability similar to that of sexual reproducing fungi were identified (Goodwin et al. 1993;
McDonald et al. 1999; Salamati et al. 2000). The presence of both mating types at equal
frequency in the field is a further suggestion for the occurrence of a cryptic teleomorph
(Linde et al. 2003). R. secalis is mainly controlled through cultural practices like crop
rotation, treatment of seeds or resistant cultivars; fungicides remain a valid alternative. Many
resistance genes to barley scald were described (Webster and Jackson 1980; Reitan et al.
2002). Fungicides are largely applied in Europe for example by the end of the 1980s about
90% of winter barley crop was treated at least once (Oerke et al. 1994). Fungicides display
different efficacy ranging from high resistance frequencies for benzimidazol carbamates
(MBC) and DMI (Kendall et al. 1993; Locke and Phillips 1995) to the complete sensitivity to
QoIs (FRAC International, Germany). Barley and rye originated in the Fertile Crescent
approximately 10,000 to 12,000 years ago (Badr et al. 2000), but Zaffarano et al. (2006)
showed that the center of diversity did not coincide with center of origin of barley. Center of
pathogen diversity was found to be Scandinavia. This result was confirmed using the
avirulence gene NIP1 (Brunner et al. 2007). Zaffarano et al. (2008) confirmed the emergence

Chapter 1 General introduction
14
of R. secalis most likely between 1,200 and 3,600 years ago, following the introduction of
barley in to northern Europe through a host shift.
The present study - chapter by chapter
The present nearly four years study, was planned to provide new insights into the mt
genomes of plant pathogenic fungi. The mt genomes of P. nodorum (EU053989) and M.
graminicola (EU090238) were completely sequenced, annotated and released. This effort
represents about the 5% of all mtDNAs sequenced from ascomycetes, until 2008 globally.
The evolutionary history of the mt genome in Mycosphaerella populations infecting bread
wheat, durum wheat and wild grasses was elucidated. Finally the QoI resistance evolution
was studied in M. graminicola and R. secalis, showing that 1) in M. graminicola recurring
mutations introduced the QoI resistant allele into different genetic and geographic
backgrounds and that 2) R. secalis remains sensitive to QoIs with a naïve resistant allele
frequency predicted from a global collection of samples to be lower than 0.12%.
Chapter 1 gives an introduction about 1) history and evolution of agriculture, 2) present
knowledge of mt genomes, 3) correlation between mtDNAs and QoI resistance and 4)
general introduction about plant pathogenic fungi M. graminicola, P.nodorum and R. secalis.
Chapter 2 shows the intraspecific comparison and annotation of the completely
sequenced mt genome sequences from two isolates of M. graminicola and an estimation of
the intraspecific mt diversity based on three mt loci among 37 isolates sampled globally.
Chapter 3 infers the evolutionary history of the mt genome of Mycosphaerella
populations infecting Triticum durum, Triticum aestivum, Lolium multiflorum, Dactylis
glomerata and Agropyron repens and characterizes the insertions/deletions polymorphisms
associated with the main mtRFLP types, with particular attention to a 3 kb insertion found
exclusively in RFLP type 4, which was proposed to increase the parasitic fitness on T. durum.

Chapter 1 General introduction
15
Chapter 4 presents the P. nodorum genome sequence. My contribution to this
publication was limited to the characterization and annotation of mt genome.
Chapter 5 clarifies the evolutionary mechanisms behind the rapid emergence and spread
of QoI-resistant isolates of M. graminicola in Europe, by testing two hypothesis 1) QoI
resistance occurred only once and subsequently spread to other regions or 2) QoI resistance
occurred independently in several different genetic and geographic backgrounds.
Chapter 6 characterizes the mt gene cytochrome b of R. secalis, describes the
intraspecific and interspecific sequence diversity, presents the diagnostic tools needed to
detect the most common allele associated with QoI resistance and screens a global collection
of 841 isolates for QoI resistance.

Chapter 1 General introduction
16
References
Adams KL, Daley DO, Qiu YL, Whelan J, Palmer JD. 2000. Repeated, recent and diverse
transfer of a mitochondrial gene to the nucleus in flowering plants. Nature 408:354-357.
Arseniuk E, Góral T, Scharen AL. 1998. Seasonal pattern of spore dispersal of
Phaeosphaeria spp. and Stagonospora spp.. Plant Dis. 82:187-194.
Badr A, Müller K, Schäfer-Pregl R, Rabey HE, Effgen S, Ibrahim HH, Pozzi C, Rhde W,
Salamini F. 2000. On the origin and domestication history of barley (Hordeum vulgare).
Mol Biol Evol. 17:499-510.
Ballard JWO, Whitlock MC. 2004. The incomplete natural history of mitochondria. Mol
Ecol. 13:729-744.
Bannon FJ, Cooke BM. 1998. Studies on dispersal of Septoria tritici pycnidiospores in
wheat-clover intercrops. Plant Pathol. 47:49-56.
Bartlett DW, Clough JM, Godwin JR, Hall AA, Hamer M, Parr-Dobrzanski B. 2002. The
strobilurin fungicides. Pest Manag Sci. 58:649-662.
Bathgate JA, Loughman R. 2001. Ascospores are a source of inoculum of Phaeosphaeria
nodorum, P. avenaria f. sp. avenaria and Mycosphaerella graminicola in Western
Australia. Austral Plant Pathol. 30:317-322.
Bearchell SJ, Fraaije BA, Shaw MW, Fitt BDL. 2005. Wheat archive links long-term fungal
pathogen population dynamics to air pollution. PNAS 102:5438-5442.
Bennett RS, Milgroom MG, Bergstrom GC. 2005. Population structure of seedborne
Phaeosphaeria nodorum in New York wheat. Phytopathology 95:300-305.
Boeger JM, Chen RS, McDonald BA. 1993. Gene flow between geographic populations of
Mycosphaerella graminicola (anamorph Septoria tritici) detected with restriction
fragment length polymorphism markers. Phytopathology 83:1148-1154.
Brennan RM, Fitt BDL, Taylor GS, Colhoun J. 1985. Dispersal of Septoria nodorum
pycnidiospores by simulated raindrops in still air. Phytopathology 112:281-290.
Brennicke A, Grohmann L, Hiesel R, Knoop V, Schuster W. 1993. The mitochondrial
genome on its way to the nucleus: different stages of gene transfer in higher plants. FEBS
Lett. 325:140-145.

Chapter 1 General introduction
17
Brunner PC, Schurch S, McDonald BA. 2007. The origin and colonization history of the
barley scald pathogen Rhynchosporium secalis. J Evol Biol. 20:1311-1322.
Burger G, Gray MW, Lang BF. 2003. Mitochondrial genomes: anything goes. TRENDS
Genet. 19:709-716.
Caldwell RM. 1937. Rhynchosporium scald of barley, rye, and other grasses. J Agr Res.
55:175-197.
Campbell A, Mrazek J, Karlin S. 1999. Genome signature comparisons among prokaryote,
plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA 96:9184-9189.
Chen WJ, Delmotte F, Richard-Cervera S, Douence L, Greif C and Corio-Costet MF. 2007.
At least two origins of fungicide resistance in grapevine downy mildew populations. Appl
Environ Microb. 73:5162-5172.
Chen JZ, Hebert PDN. 1999. Intra-individual sequence diversity and hierarchical approach to
the study of mitochondrial DNA mutations. Mutat Res. 434:205-217.
Chen RS, McDonald BA. 1996. Sexual reproduction plays a major role in the genetic
structure of populations of the fungus Mycosphaerella graminicola. Genetics 142:1119-
1127.
Childe VG. 1953. New light on the most ancient Near East. New York: Praeger.
Couch BC, Fudal I, Lebrun MH, Tharreau D, Valent B, van Kim P, Kohn LM. 2005. Origins
of host-specific populations of the blast pathogen Magnaporthe oryzae in crop
domestication with subsequent expansion of pandemic clones on rice and weeds of rice.
Genetics 170:613-630.
De Waard MA. 1994. Resistance to fungicides which inhibit sterol 14-a-demethilation, an
historical perspective. In Heaney S, Slawson D, Hollomon DW, Smith M, Russell PE,
Parry DW. eds, Fungicide resistance. 3-10. BCPC Surrey, UK.
Diamond J. 1997. Guns, Germs, and Steel: the Fates of Human Societies. New York: Norton.
Elias EM, Steiger DK, Cantrell RG. 1996. Evaluation of lines derived from wild emmer
chromosome substitutions. II. Agronomic traits. Crop Sci. 36:228-233.
Eyal Z. 1981. Integrated control of Septoria diseases of wheat. Plant Dis. 65:763-768.
Eyal Z. 1999. The septoria tritici and stagonospora nodorum blotch disease of wheat. Eur J
Plant Pathol. 105:629-641.
FAO newsroom 2006. FAO sees major shift to bioenergy. 25 April 2006.

Chapter 1 General introduction
18
FAO newsroom 2008. Record harvest but troubles loom ahead. 6 November 2008.
Fernandez-Ortuno D, Torés JA, de Vicente A, Pérez-García A. 2008. Field resistance to QoI
fungicides in Podosphaera fusca is not supported by typical mutations in the
mitochondrial cytochrome b gene. Pest Manag Sci. 64:694-702.
Fraaije BA, Burnett FJ, Clark WS, Motteram J, Lucas JA. 2005. Resistance development to
QoI inhibitors in populations of Mycosphaerella graminicola in the UK. In: Dehne HW,
Gisi U, Kuck KH, Russell PE, Lyr H. eds, Modern Fungicides and Antifungal
Compounds IV. 63-71 BCPC, Alton, UK.
Friesen TL, Stukenbrock EH, Liu ZH, Meinhardt S, Ling H, Faris JD, Rasmussen JB,
Solomon PS, McDonald BA, Oliver RP. 2006. Emergence of a new disease as a result of
interspecific virulence gene transfer. Nat Genet. 38:953-956.
Gisi U, Hermann D, Ohl L, Steden C. 1997. Sensitivity profiles of Mycosphaerella
graminicola and Phytophthora infestans populations to different classes of fungicides.
Pestic Sci. 51:290-298.
Gisi U, Pavic L, Stanger C, Hugelshofer U, Sierotzki H. 2005. Dynamics of Mycosphaerella
graminicola populations in response to selection by different fungicides. In: Dehne HW,
Gisi U, Kuck KH, Russell PE, Lyr H. eds, Modern Fungicides and Antifungal
Compounds IV. 73-80 BCPC, Alton, UK.
Gisi U, Sierotzki H, Cook A, McCaffery A. 2002. Mechanisms influencing the evolution of
resistance to Qo inhibitor fungicides. Pest Manag Sci. 58:859-867.
Goodwin SB, Waalwijk C, Kema GHJ. 2004. Genetics and genomics of Mycosphaerella
graminicola: a model for the Dothideales. In: Arora DK, Khachatourians GG, eds,
Applied Mycology & Biotechnology. Volume 4. Fungal Genomics Elsevier Science B.V.,
Amsterdam, pp. 315-330.
Goodwin SB, Maroof MAS, Allard RW, Webster RK. 1993. Isozyme variation within and
among populations of Rhynchosporium secalis in Europe, Australia and the United States.
Mycol Res. 97:49-58.
Gopher A, Abbo S, Lev-Yadun ST. 2002. The ‘when’, the ‘where’ and the ‘why’ of the
Neolithic revolution in the Levant. Doc Praehist. 28:49-62.

Chapter 1 General introduction
19
Grasso V, Palermo S, Sierotzki H, Garibaldi A, Gisi U. 2006. Cytochrome b gene structure
and consequences for resistance to Qo inhibitor fungicides in plant pathogens. Pest
Manag Sci. 62:465-472.
Griffiths AJF. 1996. Mitochondrial inheritance in filamentous fungi. J Genet. 75:403-414.
Griffiths DC, Hann CAO. 1976. Dispersal of Septoria nodorum spores and spread of glume
blotch of wheat in the field. Transactions of the British Micological Society 67:413-418.
Hartl FU, Pfanner N, Nicholson DW, Neupert W. 1989. Mitochondrial protein import.
Biochim Biophys Acta 988:1-45.
Haudry A, Cenci A, Ravel C, Bataillon T, Brunel D, Poncet C, Hochu I, Poirier S, Santoni S,
Glémin S, David J. 2007. Grinding up wheat: a massive loss of nucleotide diversity since
domestication. Mol Biol Evol. 24:1506-1517.
Ioos R, Andrieux A, Marcais B, Frey P. 2006. Genetic characterization of the natural hybrid
species Phytophthora alni as inferred from nuclear and mitochondrial DNA analyses.
Fungal Genet Biol. 43:511-529.
Ishii H, Fraaije BA, Sugiyama T, Noguchi K, Nishimura K, Takeda T, Amano T, Hollomon
DW. 2001. Occurrence and molecular characterization of strobilurin resistance in
cucumber powdery mildew and downy mildew. Phytopathology 91:1166-1171.
Kendall SJ, Hollomon DW, Cooke LR, Jones DR. 1993. Changes in sensitivity to DMI
fungicides in Rhynchosporium secalis. Crop Prot. 12:357-362.
Khan TN. 1986. Effects of fungicide treatments on scald (Rhynchosporium secalis (Oud)
Davis) infection and yield of barley in western-Australia. Aus J Exp Agr. 26:231-235.
Kirk PM, Cannon PF, David JC, Stalpers JA. 2001. Ainsworth & Bisby´s Dictionary of the
Fungi. 9th eds, CAB International, Wallingford, UK.
Linde CC, Zala M, Ceccarelli S, McDonald BA. 2003. Further evidence for sexual
reproduction in Rhynchosporium secalis based on distribution and frequency of mating-
type alleles. Fungal Genet Biol. 40:115-125.
Liu ZH, Faris JD, Meinhardt SW, Ali S, Rasmussen JB, Friesen TL. 2004a. Genetic and
physical mapping of a gene conditioning sensitivity in wheat to a partially purified host-
selective toxin produced by Stagonospora nodorum. Phytopathology 94:1056-1060.

Chapter 1 General introduction
20
Liu ZH, Friesen TL, Rasmussen JB, Ali S, Meinhardt SW, Faris JD. 2004b. Quantitative trait
loci analysis and mapping of seedling resistance to Stagonospora nodorum leaf blotch in
wheat. Phytopathology 94:1061-1067.
Locke T, Phillips AN. 1995. The occurrence of carbendazim resistance in Rhyncosporium
secalis on winter barley in England and Wales in 1992 and 1993. Plant Pathol. 44:294-
300.
Lutz W, Qiang R. 2002. Determinants of human population growth. Phil Trans R Soc Lond
B. 357:1197-1210.
McDonald BA, Zhan J, Burdon JJ. 1999. Genetic structure of Rhynchosporium secalis in
Australia. Phytopathology 89:639-645.
McDougall P. 2001. Crop protection and agricultural biotechnology consultants, Report 249,
July 2001.
McDougall P. 2006. Phillips McDougall Agriservice Report. Pathhead, Midlothian, Scotland,
UK.
Oerke EC, Dehne HW, Schönbeck F, Weber A. 1994. Crop Production and Crop Protection.
297-364, Elsevier, Amsterdam, the Netherlands.
Paquin B, Laforest MJ, Forget L, Roewer I, Wang Z, Longcore J, Lang BF. 1997. The fungal
mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene
expression. Curr Genet. 31:380-395.
Pasche JS, Piche LM, Gudmestad NC. 2005. Effect of the F129L mutation in Alternaria
solani on fungicides affecting mitochondrial respiration. Plant Dis. 89:269-278.
Reitan L, Grønnerød S, Ristad TP, Salamati S, Skinnes H, Waugh R, Bjørnstad Å. 2002.
Characterization of resistance genes against scald (Rhynchosporium secalis (Oudem.) J.J.
Davis) in barley (Hordeum vulgare L.) lines from central Norway, by means of genetic
markers and pathotype tests. Euphytica 123:31-39.
Robertson J. 1824. Transaction of the London Horticultural Society 5:175.
Russell PE. 1995. Fungicide resistance: occurrence and management. J Agr Sci. 124:317-
323.
Russell PE. 2005. A century of fungicide evolution. J Agr Sci. 143:11-25.

Chapter 1 General introduction
21
Salamati S, Zhan J, Burdon JJ, McDonald BA. 2000. The genetic structure of field
populations of Rhynchosporium secalis from three continents suggests moderate gene
flow and regular recombination. Phytopathology 902:901-908.
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W. 2002. Genetics and
geography of wild cereal domestication in the Near East. Nat Rev Genet. 3:429-441.
Sanderson FR. 1972. A Mycosphaerella species as the ascogenous state of Septoria tritici
Rob. and Desm. New Zeal J Bot. 10:707-710.
Scheffler IE. 2001. A century of mitochondrial research: achievements and perspectives.
Mitochondrion 1:3-31.
Schoch CL, Shoemaker RA, Seifert KA, Hambleton S, Spatafora JW, Crous PW. 2006. A
multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia
98:1041-1052.
Shah DA, Bergstrom GC, Ueng PP. 1995. Initiation of Septoria nodorum blotch epidemics in
winter wheat by seedborne Stagonospora nodorum. Phytopathology 85:452.457.
Sharma HC, Waines JG. 1980. Inheritance of tough rachis in crosses of Triticum
monococcum and T. boeoticum. J Hered. 71:214-216.
Shipton WA, Boyd WJR, Ali SM. 1974. Scald of barley. Rev Plant Pathol. 53:840-861.
Sierotzki H, Frey R, Wullschleger J, Palermo S, Karlin S, Godwin J, Gisi U. 2007.
Cytochrome b gene sequence and structure of Pyrenophora teres and P. tritici-repentis
and implications for QoI resistance. Pest Manag Sci. 63:225-233.
Sierotzki H, Wullschleger J, Gisi U. 2000. Point mutation in cytochrome b gene conferring
resistance to strobilurin fungicides in Erysiphe graminis f. sp tritici field isolates. Pestic
Biochem and Phys. 68:107-112.
Signorovitch AY, Buss LW, Dellaporta SL. 2007. Comparative genomics of large
mitochondria in placozoans. PLOS Genetics 3:e13. Solomon PS, Lowe RGT, Tan K-C, Waters ODC, Oliver RP. 2006. Stagonospora nodorum:
cause of stagonospora nodorum blotch of wheat. Mol Plant Pathol. 7:147-156.
Steinfeld U, Sierotzki H, Parisi S, Poirey S, Gisi U. 2001. Sensitivity of mitochondrial
respiration to different inhibitors in Venturia inaequalis. Pest Manag Sci. 57:787-796.

Chapter 1 General introduction
22
Stergiopoulos I, van Nistelrooy JGM, Kema GHJ, De Waard MA. 2003. Multiple
mechanisms account for variation in base-line sensitivity to azole fungicides in field
isolates of Mycosphaerella graminicola. Pest Manag Sci. 59:1333-1343.
Stukenbrock EH, Banke S, Javan-Nikkhah M, McDonald BA. 2007. Origin and
domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric
speciation. Mol Biol Evol. 24:398-411.
Stukenbrock EH, Banke S, McDonald BA. 2006. Global migration patterns in the fungal
wheat pathogen Phaeosphaeria nodorum. Mol Ecol. 15:2895-2904.
Stukenbrock EH, McDonald BA. 2008 The origin of plant pathogens in agro-ecosystems.
Annu Rev Phytopathol. 46:75-100.
Taenzler B, Esposti RF, Vaccino P, Brandolini A, Effgen S, Heun M, Schäfer-Pregl R,
Borghi B, Salamini F. 2002. Molecular linkage map of Einkorn wheat: mapping of
storage-protein and soft-glume genes and bread-making quality QTLs. Genet Res Camb.
80:131-143.
United Nations (Population Division). 2004. World population to 2300. ST/ESA/SER.A/236,
New York, USA.
Vincelli P, Dixon E. 2002. Resistance to Q(o)I (strobilurin-like) fungicides in isolates of
Pyricularia grisea from perennial ryegrass. Plant Dis. 86:235-240.
Webster RK, Jackson LF. 1980. Sources of resistance in barley to Rhynchosporium secalis.
Plant Dis. 64:88-90.
Winka K, Eriksson OE. 1997. Supraordinal taxa of Ascomycota. Myconet. 1:1-16.
Wolpert TJ, Dunkle LD, Ciuffetti LM. 2002. Host-selective toxins and avirulence
determinants: What's in a name? Annu Rev Phytopathol 40:251-285.
Zaffarano PL, McDonald BA, Linde CC. 2008. Rapid speciation following recent host shifts
in the plant pathogenic fungus Rhynchosporium. Evolution 62:1418-1436.
Zaffarano PL, McDonald BA, Zala M, Linde CC. 2006. Global hierarchical gene diversity
analysis suggests the Fertile Crescent is not the center of origin of the barley scald
pathogen Rhynchosporium secalis. Phytopathology 96:941-950.
Zhan J, McDonald BA. 2004. The interaction among evolutionary forces in the pathogenic
fungus Mycosphaerella graminicola. Fungal Genet Biol. 41:590-599.

Chapter 1 General introduction
23
Zhan J, Pettway RE, McDonald BA. 2003. The global genetic structure of the wheat
pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low
mitochondrial diversity, regular recombination and gene flow. Fungal Genet Biol.
38:286-297.

Chapter 1 General introduction
24

CHAPTER 2
The mitochondrial gemome of Mycosphaerella graminicola
Stefano FF Torriani, Stephen B Goodwin, Gert HJ Kema, Jasmyn L Pangilinan, Bruce A.
McDonald. 2008. Intraspecific comparison and annotation of two complete mitochondrial
genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. Fungal
Genetics and Biology 45:628-637.

Chapter 2 mtDNA of M. graminicola
26

Chapter 2 mtDNA of M. graminicola
27
Abstract
The mitochondrial genomes of two isolates of the wheat pathogen Mycosphaerella
graminicola were sequenced completely and compared to identify polymorphic regions. This
organism is of interest because it is phylogenetically distant from other fungi with sequenced
mitochondrial genomes and it has shown discordant patterns of nuclear and mitochondrial
diversity. The mitochondrial genome of M. graminicola is a circular molecule of
approximately 43,960 bp containing the typical genes coding for 14 proteins related to
oxidative phosphorylation, one RNA polymerase, two rRNA genes and a set of 27 tRNAs.
The mitochondrial DNA of M. graminicola lacks the gene encoding the putative ribosomal
protein (rps5-like), commonly found in fungal mitochondrial genomes. Most of the tRNA
genes were clustered with a gene order conserved with many other ascomycetes. A sample of
thirty-five additional strains representing the known global mt diversity was partially
sequenced to measure overall mitochondrial variability within the species. Little variation
was found, confirming previous RFLP-based findings of low mitochondrial diversity. The
mitochondrial sequence of M. graminicola is the first reported from the family
Mycosphaerellaceae or the order Capnodiales. The sequence also provides a tool to better
understand the development of fungicide resistance and the conflicting pattern of high
nuclear and low mitochondrial diversity in global populations of this fungus.

Chapter 2 mtDNA of M. graminicola
28
Introduction
Mycosphaerella graminicola (anamorph Septoria tritici) is the causal agent of Septoria
tritici blotch of wheat and other poaceous hosts, and occurs worldwide across a wide range of
climates (Eyal 1999). The life cycle of M. graminicola includes both sexual and asexual
stages. The sexual stage permits genetic recombination and produces airborne ascospores
with the potential to be dispersed over several kilometers (Sanderson 1972), whereas the
asexual phase (S. tritici) produces pycnidiospores disseminated over limited distances from
plant to plant via rain splash (Bannon and Cooke 1998).
In most fungi studied to date there is concordance between genetic variation in the
mitochondrial (mt) and nuclear genomes (Zhan et al. 2004; Sommerhalder et al. 2007), with
some fungi having high levels of mt and nuclear diversity (Liu et al. 1996; Kudla et al. 2002)
and others having low mt and nuclear genetic variability (Kurdyla et al. 1995; Xia et al.
2000). However, the pattern of variability in M. graminicola is different; a comparison of
RFLP markers in nuclear and mt genomes showed a pattern of high nuclear and low mt
diversity in populations around the world (Zhan et al. 2003). Over 1300 nuclear RFLP
genotypes were found among 1673 isolates, with an average of 18 alleles per nuclear RFLP
locus. In contrast, only seven mtDNA haplotypes were found globally, with the two most
common representing 93% of the world population. The high nuclear diversity is thought to
be the consequence of high gene flow (Boeger et al. 1993), coupled with large effective
population sizes (Zhan and McDonald 2004) and recurring sexual reproduction (Chen and
McDonald 1996; Kema et al. 1996; Hunter et al. 1999). Zhan et al. (2004) suggested a
selective sweep to explain the low diversity found in the mtDNA. Selective sweeps may be
common in mt genomes because all of the genes are linked in one molecule so that selection
on one gene can affect the frequency of all genes through hitchhiking.
Mt genomes have proven to be highly useful for research in evolutionary biology and
systematics because of their uniparental inheritance, the near absence of genetic
recombination, and uniform genetic backgrounds (Chen and Hebert 1999). The evolution of
mtDNAs has been characterized by extensive loss and translocation of genes to the nucleus
(Adams et al. 2000) since their origin by endosymbiosis of a bacterial ancestor (John and
Whatley 1975). The result of this process is that most mt proteins are encoded by nuclear

Chapter 2 mtDNA of M. graminicola
29
genes whose products are imported into the mitochondrion by translocase complexes, leaving
relatively few mt proteins that are synthesized directly within the organelle (Hartl et al. 1989;
Brennicke et al. 1993).
Mt genomes are characterized by high A + T content, lack of methylation, conservation in
gene function, and high copy number (Campbell et al. 1999), and they can evolve at their
own rate relative to the nuclear genomes of the organisms in which they occur (Ballard and
Whitlock 2004). The size and topology of the mt genome, the number and nature of the
proteins it encodes, and even the genetic code itself can vary greatly among species (Gray et
al. 1999). Fungal mtDNAs are generally an order of magnitude smaller than those of plants
but larger than animal mtDNAs (Burger et al. 2003) and usually contain 14 genes encoding
hydrophobic subunits of respiratory chain complexes, as well as genes for the large (rnl) and
small (rns) ribosomal subunits and a set of tRNAs (Gray et al. 1999). The coding percent
ranges between 40 and 60% in the Pezizomycotina. Among fungi, mt genomes vary widely
in size, from approximately 18 to 109 kb (NCBI database). The variability of mt genome size
among species is strongly influenced by differences in length and organization of intergenic
regions, as well as by differences in intron content (from 0 to 30) and size (ranging from 0.15
and 4 kb). Burger et al. (2003) showed that there is no correlation between mtDNA size and
gene content.
The taxonomic placement of Mycosphaerella within the class Dothideomycetes until
recently was uncertain, and it usually was placed near Dothidea in the Dothideales (Kirk et
al. 2001; Goodwin et al. 2004). However, recent analyses of a multigene phylogeny showed
that Mycosphaerella belongs in the Capnodiales, a sister group to the Dothideales and
Myriangiales (Schoch et al. 2006). Though the mt genome of Stagonospora nodorum,
another member of the Dothideomycetes, was recently published (Hane et al. 2007), no mt
genomes have been published from Mycosphaerella or any species in the Capnodiales,
Dothideales, or Myriangiales.
The goals of this research were to obtain and annotate the first complete mitochondrial
genome sequence from the Mycosphaerella branch of the fungal evolutionary tree, and to test
a previous hypothesis of low mitochondrial diversity within global populations of M.
graminicola. Complete sequences of the mtDNA genomes from two isolates of M.
graminicola (one from North America and one from Europe) plus sequences at three

Chapter 2 mtDNA of M. graminicola
30
mitochondrial loci for 35 additional isolates representing most of the known global diversity
were compared, first to quantify the overall mtDNA sequence diversity in M. graminicola
and second to compare it with earlier findings of low diversity based on RFLP analysis. An
interspecific analysis of the tRNA genes flanking rnl of species in the Pezizomycotina
revealed a consensus in tRNA gene content and order.
Materials and methods
Fungal strains, DNA extraction, and library construction. Strain IPO323 was isolated
from a naturally infected leaf of the soft white wheat cultivar Arminda collected in Brabant,
the Netherlands during 1981 (Kema and Van Silfhout 1997). Fungal mycelia were produced
on liquid shake cultures, harvested, stored and prepared for DNA extraction as described in
Kema et al. (2002). Fungal spores and mycelia were ground with a Hybaid Ribolyser (model
FP120HY-230) for 10 s at 2,500 rpm with two tungsten carbide beads, and total genomic
DNA was extracted using the Promega Wizard Magnetic DNA Purification System for Food
as described by the manufacturer except with only 50 mg of lyophilised fungal material and
500 µl of lysis buffer. Plasmid libraries with insert sizes of 3 and 8 kb were created at the
U.S. Department of Energy’s Joint Genome Institute (JGI) and sequenced to 4× genomic
sequence coverage (~150,000 clones each).
Strain STBB1 was isolated from a wheat field 5 km southwest of College Station, Texas,
USA, during 1989. The entire mt genome of this isolate was purified from total DNA by
cesium chloride (CsCl) ultracentrifugation as described by Garber and Yoder (1983), with a
CsCl density of 1.6 g/ml. A library was constructed by digesting the purified mtDNA to
completion using the restriction enzyme HindIII, ligating the fragments into the plasmid
vector pUC18 and cloning in Escherichia coli strain DH5α.
DNA sequencing and assembly. Shotgun sequencing of the nuclear and mt genomes of
isolate IPO323 was through the Community Sequencing Program of the JGI
(www.jgi.doe.gov/CSP/) by analysis of libraries with insert sizes averaging 3, 8 and 40 kb.
The mt genome was assembled from ~7,680 sequencing reads from 10 plates of the 3-kb
library using phrap (http://www.phrap.org/) with its standard parameters. This corresponds to

Chapter 2 mtDNA of M. graminicola
31
roughly 5-6 Mb of sequence. Approximately 5.5% of the reads (~260 kb) represented
mtDNA so the initial sequence was assembled at a depth of about 6×. The average depth of
coverage for the entire project was 8.9× and was released publicly (http://genome.jgi-
psf.org/Mycgr1/Mycgr1.home.html) during November 2006.
The mtDNA library obtained from isolate STBB1 was sequenced using the BigDyeTM
Terminator v3.0 Cycle Sequencing kit and the primer walking strategy. The sequencing
reactions were in a total volume of 10 µl using 20-40 ng of plasmid DNA, 10 pmol of
primers and 2 µl of BigDye reaction mix, previously diluted 1:4. The cycling profile was 10 s
denaturation at 95°C, 5 s annealing at 50°C and 4 min extension at 60°C for 100 cycles. The
sequencing reactions were purified through Sephadex G-50 DNA Grade F (Amersham
Biosciences, Switzerland) before being loaded into an ABI 3100 automated sequencer
(Applied Biosystems). The sequences were aligned and analyzed with the Sequencher
version 4.2 software package (Gene Codes Corporation, Ann Arbor, MI) using the genetic
code of Pezizomycotina that diverged from the standard nuclear code for the codon TGA,
which was read as Trp and not as Stop. Sequencing of the isolate STBB1 mt library generated
approximately 75% of the entire mtDNA genome. Gaps in the STBB1 sequence were filled
by aligning the sequenced HindIII fragments to the complete mtDNA sequence of isolate
IPO323 and designing pairs of primers to amplify the missing regions in STBB1. The
amplicons were sequenced as described above to obtain the entire mt genome of isolate
STBB1.
Sequence annotation. The mtDNA sequence of M. graminicola was screened for
similarity with those from other organisms in the NCBI database using the BlastN tool.
Sequences showing matches with protein-coding genes of other organisms were subsequently
compared using the BlastX tool (Altschul et al. 1990). The mt sequences of strains IPO323
and STBB1 were aligned using the Sequencher program and screened manually for
polymorphisms including transitions, transversions, insertions and deletions (indels). The
genes coding for ribosomal RNAs were determined by comparison with sequences from
other fungi. The tRNAs were defined by tRNAscan-SE v1.21 (Lowe and Eddy 1997) and by
comparison with the NCBI database. Expression of mt genes was tested by blast searches
against databases of EST sequences (Kema et al. 2003; Soanes and Talbot 2006; Goodwin et

Chapter 2 mtDNA of M. graminicola
32
al. 2007). Repetitive elements, including minisatellites, simple-sequence repeats (SSRs) and
mononucleotide repeats were identified using the online program Perfect Microsatellite
Repeat Finder (http://sgdp.iop.kcl.ac.uk/nikammar/repeatfinder.html).
Intraspecific comparison and haplotype network. Three mtDNA loci, named Mg1,
Mg2 and Mg3, were used to assess the overall mt diversity within the species. These loci
were located in different regions of the mtDNA and were chosen because they displayed
different degrees of polymorphism in the comparison between STBB1 and IPO323. Mg1 was
located within orf1 and had no polymorphism. Mg2 included a portion of orf4 and had
several polymorphisms including single-nucleotide polymorphisms (SNPs), indels and
homopolymers of various lengths. Mg3 included the region with tRNA-Gly, tRNA-Asp, tRNA-
Ser and tRNA-Trp and had two polymorphic microsatellites and one homopolymer. Thirty-
five isolates (Table 1) belonging to four RFLP haplotypes (Zhan et al. 2003) and originating
from five continents were amplified using primers: Mg1F (5’-CCG GTC CCT CTA ATA
GTT CTG G-3’) and Mg1R (5’-TAA TCC GCC ATT ACT TCT CAG G-3’); Mg2F (5’-GGT
TCC AAT GGG TTT AAT GCT A-3’) and Mg2R (5’- TGG GTG TAG CTA GAA ACC
CTT C-3’); Mg3F (5’-AAG CTA CGC CTA TGG CTA ACA C-3’) and Mg3R (5’-AGG
TAA GAC GCA CGC ATT TC-3’). Each PCR reaction contained 5-10 ng of DNA in a 20-µl
reaction volume containing 10 pmol of each primer, 100 µM of each nucleotide, 2 µl of 10x
PCR buffer (1x PCR buffer: 10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl, 2 mM
MgSO4, 0.1% Triton X-100 [pH 8.8]) and 1 U of Taq DNA Polymerase (New England
Biolabs). The PCR amplifications were carried out under the following conditions: initial
denaturation at 96°C for 2 min, followed by 35 cycles of 96°C for 1 min, 56°C for 1 min, and
72°C for 1 min, with a final extension at 72°C for 5 min. The PCR products were sequenced
using the primers Mg1F, Mg2F and Mg3R, generating a total of 1,339 bp (338 bp for Mg1,
333 bp for Mg2 and 668 bp for Mg3). Sequencing reactions were performed as described
previously for STBB1. The program SNAP Workbench (Price and Carbone 2005) was used
to collapse the sequences into haplotypes and DnaSP (Rozas et al. 2003) was used to test for
recombination within and among the tested loci. The software package TCS version 1.21
(Clement et al. 2000) was used to infer intraspecific evolution of the M. graminicola mtDNA.
Chapter 2 mtDNA of M. graminicola
33
This program applies a statistical parsimony method to infer unrooted cladograms based on
Templeton’s 95% parsimony connection limit (Templeton et al. 1992).
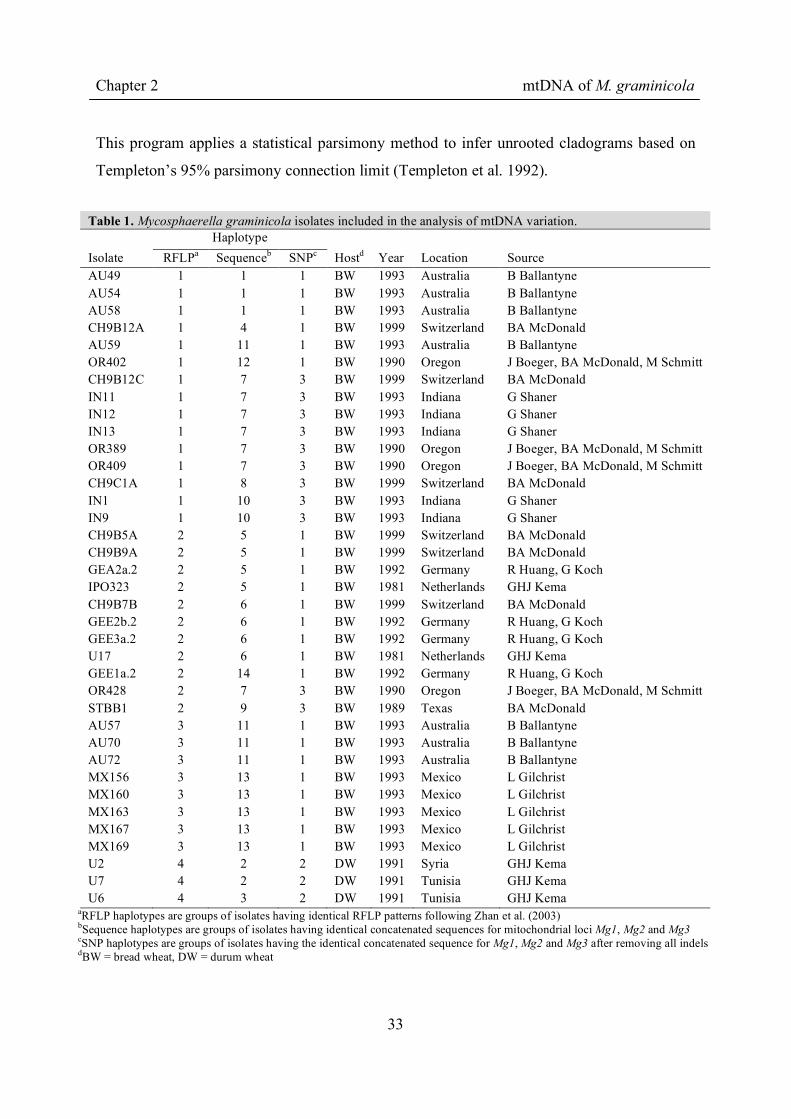
Table 1. Mycosphaerella graminicola isolates included in the analysis of mtDNA variation. Haplotype Isolate RFLPa Sequenceb SNPc Hostd Year Location Source AU49 1 1 1 BW 1993 Australia B Ballantyne AU54 1 1 1 BW 1993 Australia B Ballantyne AU58 1 1 1 BW 1993 Australia B Ballantyne CH9B12A 1 4 1 BW 1999 Switzerland BA McDonald AU59 1 11 1 BW 1993 Australia B Ballantyne OR402 1 12 1 BW 1990 Oregon J Boeger, BA McDonald, M Schmitt CH9B12C 1 7 3 BW 1999 Switzerland BA McDonald IN11 1 7 3 BW 1993 Indiana G Shaner IN12 1 7 3 BW 1993 Indiana G Shaner IN13 1 7 3 BW 1993 Indiana G Shaner OR389 1 7 3 BW 1990 Oregon J Boeger, BA McDonald, M Schmitt OR409 1 7 3 BW 1990 Oregon J Boeger, BA McDonald, M Schmitt CH9C1A 1 8 3 BW 1999 Switzerland BA McDonald IN1 1 10 3 BW 1993 Indiana G Shaner IN9 1 10 3 BW 1993 Indiana G Shaner CH9B5A 2 5 1 BW 1999 Switzerland BA McDonald CH9B9A 2 5 1 BW 1999 Switzerland BA McDonald GEA2a.2 2 5 1 BW 1992 Germany R Huang, G Koch IPO323 2 5 1 BW 1981 Netherlands GHJ Kema CH9B7B 2 6 1 BW 1999 Switzerland BA McDonald GEE2b.2 2 6 1 BW 1992 Germany R Huang, G Koch GEE3a.2 2 6 1 BW 1992 Germany R Huang, G Koch U17 2 6 1 BW 1981 Netherlands GHJ Kema GEE1a.2 2 14 1 BW 1992 Germany R Huang, G Koch OR428 2 7 3 BW 1990 Oregon J Boeger, BA McDonald, M Schmitt STBB1 2 9 3 BW 1989 Texas BA McDonald AU57 3 11 1 BW 1993 Australia B Ballantyne AU70 3 11 1 BW 1993 Australia B Ballantyne AU72 3 11 1 BW 1993 Australia B Ballantyne MX156 3 13 1 BW 1993 Mexico L Gilchrist MX160 3 13 1 BW 1993 Mexico L Gilchrist MX163 3 13 1 BW 1993 Mexico L Gilchrist MX167 3 13 1 BW 1993 Mexico L Gilchrist MX169 3 13 1 BW 1993 Mexico L Gilchrist U2 4 2 2 DW 1991 Syria GHJ Kema U7 4 2 2 DW 1991 Tunisia GHJ Kema U6 4 3 2 DW 1991 Tunisia GHJ Kema
aRFLP haplotypes are groups of isolates having identical RFLP patterns following Zhan et al. (2003) bSequence haplotypes are groups of isolates having identical concatenated sequences for mitochondrial loci Mg1, Mg2 and Mg3 cSNP haplotypes are groups of isolates having the identical concatenated sequence for Mg1, Mg2 and Mg3 after removing all indels dBW = bread wheat, DW = durum wheat

Chapter 2 mtDNA of M. graminicola
34
Results
Gene content and genome organization. The mt genome of M. graminicola is a circular
molecule of approximately 43,960 bp containing 15 protein-coding genes, the large (rnl) and
small (rns) ribosomal subunits, 27 tRNAs and eight putative open reading frames (orfs1-8) of
unknown function (Fig. 1; Table 2). The protein-coding genes included three ATP synthase
subunits (atp6, atp8 and atp9), the three cytochrome oxidase subunits I, II and III (cox1-3),
cytochrome b (cytb), seven nicotinamide adenine dinucleotide ubiquinone oxidoreductase
subunits (nad1-6, nad4L) and a DNA-directed RNA polymerase (RNA-Pol). These genes
were transcribed in two contiguous segments of opposite direction (Fig. 1).
A putative ribosomal protein (rps5-like) commonly found within rnl of ascomycetes was
missing (Fig. 1; Table 2). To test whether this gene could have been transferred to the nuclear
genome, blastp and tblastn searches were performed on the 8.9× draft genomic sequence of
M. graminicola. The blastp searches identified no matching proteins among the list of
annotated genes. However, the tblastn searches identified matches at better than e-5 on
scaffold 5 to rps5-like proteins from Phaeosphaeria nodorum (e-9) and Penicillium marneffei
(e-7), but not to those from the Sordariomycetes Hypocrea jecorina or Verticillium dahliae.
Therefore, this gene most likely occurs in the nuclear rather than the mitochondrial genome
of M. graminicola.
The eight putative orfs of unknown function are predicted to produce proteins containing
from 126 to 481 amino acids. The 3’ terminus of orf3 overlapped with orf2 for 52
nucleotides. The exact function of these putative proteins remains to be determined, but the
TMHMM2 method (Krogh et al. 2001) predicted orf2 to encode a non-membrane protein,
whereas the other orfs were predicted to encode proteins having from one (orf5 and orf8) to
ten (orf7) transmembrane domains. Expressed sequence tag (EST) databases (Kema et al.
2003; Soanes and Talbot 2006; Goodwin et al. 2007) provided evidence for the transcription
of orf5, orf6 and orf8.
Putative protein-coding genes covered 51.8% of the genome (including 15.9% composed
of putative orfs), while 4.5 and 11.5% corresponded to tRNA genes and both rnl and rns,
respectively. These values were similar to those reported for other ascomycetes (Table 2).
Overall, the M. graminicola mtDNA was 34.5% A, 33.5% T, 16.3% G and 15.7% C. MtDNA

Chapter 2 mtDNA of M. graminicola
35
AT-content was 68% with coding and non-coding parts of the genome having, on average,
the same AT-percentage.
Figure 1. Circular map of the mitochondrial genome of Mycosphaerella graminicola. Black blocks, grey blocks, hatched blocks and bars show, respectively, protein-coding, orfs, rRNA and tRNA genes. Arrows indicate the direction of transcription.
Chapter 2 mtDNA of M. graminicola
36
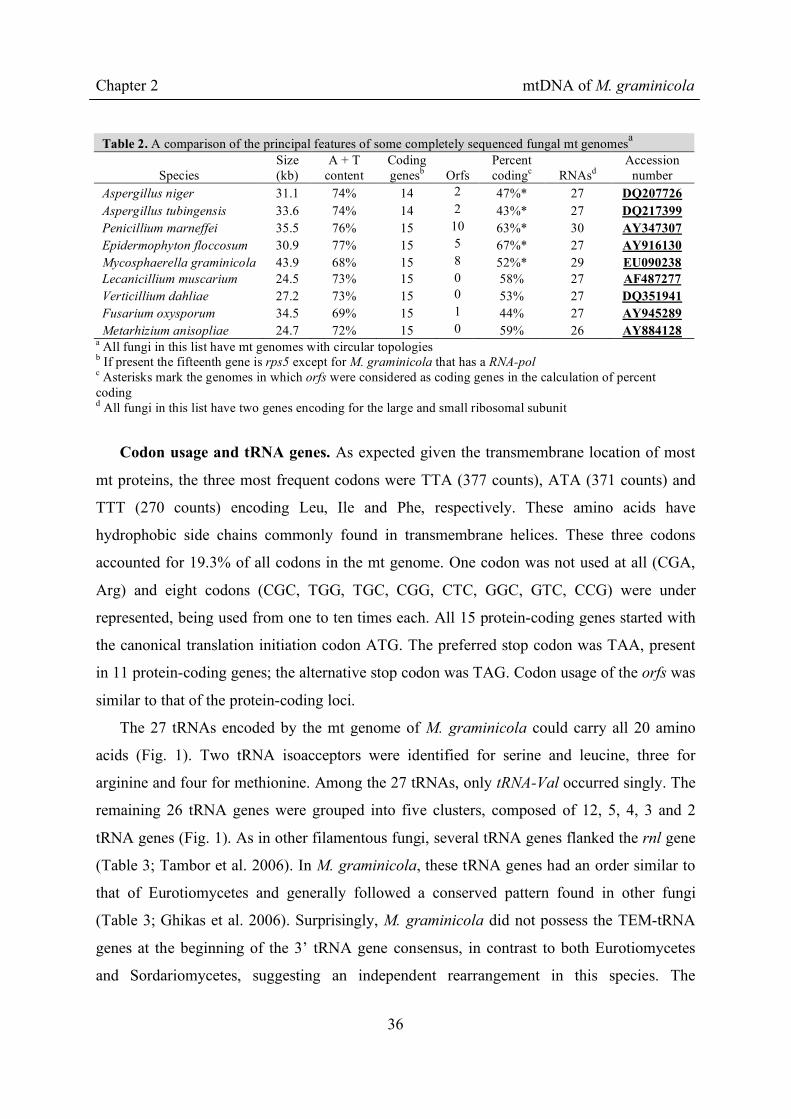
Table 2. A comparison of the principal features of some completely sequenced fungal mt genomesa
Species Size (kb)
A + T content
Coding genesb
Orfs
Percent codingc RNAsd
Accession number
Aspergillus niger 31.1 74% 14 2 47%* 27 DQ207726 Aspergillus tubingensis 33.6 74% 14 2 43%* 27 DQ217399 Penicillium marneffei 35.5 76% 15 10 63%* 30 AY347307 Epidermophyton floccosum 30.9 77% 15 5 67%* 27 AY916130 Mycosphaerella graminicola 43.9 68% 15 8 52%* 29 EU090238 Lecanicillium muscarium 24.5 73% 15 0 58% 27 AF487277 Verticillium dahliae 27.2 73% 15 0 53% 27 DQ351941 Fusarium oxysporum 34.5 69% 15 1 44% 27 AY945289 Metarhizium anisopliae 24.7 72% 15 0 59% 26 AY884128
a All fungi in this list have mt genomes with circular topologies b If present the fifteenth gene is rps5 except for M. graminicola that has a RNA-pol c Asterisks mark the genomes in which orfs were considered as coding genes in the calculation of percent coding d All fungi in this list have two genes encoding for the large and small ribosomal subunit
Codon usage and tRNA genes. As expected given the transmembrane location of most
mt proteins, the three most frequent codons were TTA (377 counts), ATA (371 counts) and
TTT (270 counts) encoding Leu, Ile and Phe, respectively. These amino acids have
hydrophobic side chains commonly found in transmembrane helices. These three codons
accounted for 19.3% of all codons in the mt genome. One codon was not used at all (CGA,
Arg) and eight codons (CGC, TGG, TGC, CGG, CTC, GGC, GTC, CCG) were under
represented, being used from one to ten times each. All 15 protein-coding genes started with
the canonical translation initiation codon ATG. The preferred stop codon was TAA, present
in 11 protein-coding genes; the alternative stop codon was TAG. Codon usage of the orfs was
similar to that of the protein-coding loci.
The 27 tRNAs encoded by the mt genome of M. graminicola could carry all 20 amino
acids (Fig. 1). Two tRNA isoacceptors were identified for serine and leucine, three for
arginine and four for methionine. Among the 27 tRNAs, only tRNA-Val occurred singly. The
remaining 26 tRNA genes were grouped into five clusters, composed of 12, 5, 4, 3 and 2
tRNA genes (Fig. 1). As in other filamentous fungi, several tRNA genes flanked the rnl gene
(Table 3; Tambor et al. 2006). In M. graminicola, these tRNA genes had an order similar to
that of Eurotiomycetes and generally followed a conserved pattern found in other fungi
(Table 3; Ghikas et al. 2006). Surprisingly, M. graminicola did not possess the TEM-tRNA
genes at the beginning of the 3’ tRNA gene consensus, in contrast to both Eurotiomycetes
and Sordariomycetes, suggesting an independent rearrangement in this species. The

Chapter 2 mtDNA of M. graminicola
37
secondary structures of tRNA-Phe and tRNA-Thr diverged from the expected cloverleaf form
as they contained nine instead of the canonical seven nucleotides in the anticodon loop
Figure 2. A linearized map showing polymorphisms between the mt genomes of Mycosphaerella graminicola isolates STBB1 and IPO323. Protein coding genes are presented as dark-grey blocks, orfs as light-grey blocks and ribosomal subunit as hatched blocks. The polymorphisms occurring between the mt genomes of isolates STBB1 and IPO323 are transversions (triangles), transitions (circles), microsatellites (plus signs), a frameshift (diamond), and a 17-bp-long indel (square). Arrows under genes indicate the direction of transcription and the small black dots close to the genes show mononucleotide repeats longer than 7 bases. The tRNAs are not presented to simplify the figure. Mg1, Mg2 and Mg3 were the three regions used to assess the total intraspecific mt diversity.
Repetitive elements and comparative genomics. One 27-mer minisatellite repeated
three times (located between the nad4 and nad4L genes), 186 SSRs and 51 mononucleotide
repeats larger than seven nucleotides (mainly located in non-coding regions), were found in
the mt genome of M. graminicola.
The total nucleotide diversity between IPO323 and STBB1 was 0.16%. The two M.
graminicola isolates differed by only 23 base substitutions, including fourteen transversions
and nine transitions. These changes represented 0.05% of the entire mt genome. Twenty-two
additional mutations were found between IPO323 and STBB1:18 were mononucleotide
repeats of different lengths (9 poly-A and 9 poly-T), two were tetra- (AAAT) or penta-
nucleotide microsatellite repeats (ATTTA), one was a frameshift mutation and the last was a
17-base deletion (Fig. 2). The nucleotide diversity among the global sample of 35 isolates
was 35% greater than that between only IPO323 and STBB1 for the same three
mitochondrial loci (Mg1, Mg2 and Mg3). Mg2 was the most variable locus, having 3 SNPs, a
17-bp indel, a polymorphic microsatellite with 2 alleles, and a mononucleotide repeat with 3
alleles. Mg1 had the fewest mutations, with two SNPs that were exclusive to isolates

Chapter 2 mtDNA of M. graminicola
38
collected from durum wheat (Triticum turgidum). Mg3 had a mononucleotide repeat with 2
alleles and 2 microsatellites, respectively with 2 and 4 alleles. All microsatellite alleles were
due to differences in the number of repeats. The concatenated sequences of Mg1, Mg2, and
Mg3 from all 37 isolates identified 14 haplotypes. If all mutations other than SNPs were
excluded from the analysis, only three haplotypes were found (Table 1). If the increase of
35% in nucleotide diversity detected for the Mg loci is extrapolated to the total genome, it
results in a value of 0.22% for mitochondrial nucleotide diversity in a global sample of 37
isolates representing most of the known mt variants. A haplotype network was inferred from
all three Mg loci using the concatenated alignments (Fig. 3).
The haplotype network did not show a clear pattern of geographical association; although
isolates from North America were in the top half and all of those at the bottom were from
Europe. Some frequent haplotypes such as H5, H6, and H7 included isolates of mixed origin,
while others (H1, H11 and H13) were geographically limited. The M. graminicola haplotypes
originating from durum wheat (Triticum turgidum ssp. durum) were distinguished from those
originating from bread wheat (T. aestivum) by three SNPs. The two sequenced haplotypes
(H5 and H9) represented different parts of the network. No evidence for recombination was
found in the mtDNA of M. graminicola using the DnaSP program.
Table 3. Comparison of tRNAa gene clusters flanking the rnl gene in several ascomycetesa
Species Class
5'-upstream regionb,c rnl 3'-downstream regionb,c Accession
number A. niger Eurotiomycetes KGDS1WIS2P rnl TEVM1M2L1AFL2QM3H DQ207726 A. tubingensis Eurotiomycetes KGDS1WIS2P rnl TEVM1M2L1AF2QLM3H DQ217399 P. marneffei Eurotiomycetes RKG1G2DS1WIS2P rnl TEVM1M2L1AFL2QM3H AY347307 E. floccosum Eurotiomycetes KGDS1IWS2P rnl TEVM1M2L1AFL2QM3H AY916130 M. graminicola Dothideomycetes GDS1WIS2P rnl M1L1EAFL2YQM2HRM3 EU090238 L. muscarium Sordariomycetes GVISW*P rnl TE1M1M2L1E2FKL2QHM3 AF487277 V. dahliae Sordariomycetes KGDS*VW*R*P1*P2 rnl TE1M1M2L1AFL2QHM3 DQ351941 F. oxysporum Sordariomycetes VISWP rnl TEM1M2L1AFKL2QHM3 AY945289 M. anisopliae Sordariomycetes YDS1N*G*LIS2W rnl TE M1M2L1AFKL2QHM3 AY884128
a The tRNA gene order of listed organisms is based on Genbank sequences b The underlined tRNA genes showed rearrangement if compared to the consensus (bold) c Capital letters refer to tRNA genes for: R=arginine, K=lysine, G=glycine, D=aspartic acid, S=serine,
W=tryptophan, I=isoleucine, P=proline, T=threonine, E=glutamic acid, V=valine, L=leucine, A=alanine, F=phenylalanine, Q=glutamine, H=histidine, Y=tyrosine, N=asparagine
* Asterisk indicates where functional genes interrupt the tRNA genes sequence 123 The numbers indicate the presence of more tRNA genes for the same amino acid in the consensus sequences

Chapter 2 mtDNA of M. graminicola
39
Discussion
The mt genomes of two strains of the plant pathogenic fungus M. graminicola originating
from different continents (Europe and North America) were sequenced completely, annotated
and compared to identify polymorphisms. Both isolates had mt genomes belonging to RFLP
haplotype 2 (Zhan et al. 2003; Table 1). The mtDNA of M. graminicola was circular and
A+T biased like those of most other fungi (Table 2).
These two M. graminicola sequences represent the first complete mt genomes of any
species in the genus Mycosphaerella or from the branch of the fungal evolutionary tree that
includes the Capnodiales, Dothideales, or Myriangiales (Schoch et al. 2006). Mycosphaerella
and its related asexual genera (e.g., Cercospora, Septoria) comprise one of the largest and
most economically important groups of pathogenic fungi (Goodwin et al. 2001) with several
thousand species infecting virtually every major family of plants (Corlett 1991). Species of
Mycosphaerella are not closely related to model fungi or those with completely sequenced mt
genomes, so represent a previously unsampled branch of the fungal evolutionary tree.
The mtDNA of M. graminicola contains genes for 14 inner mt membrane proteins
involved in electron transport and coupled oxidative phosphorylation, as well as rnl, rns and
RNA-Pol genes (Fig. 1). Except for presence of the RNA-Pol gene and absence of a gene
encoding a putative ribosomal protein (rps5-like), this is the standard set of mtDNA-encoded
genes found in other fungi. The rps5-like gene is found commonly in mt genomes of different
fungal species and it was postulated that mtDNA-encoded rps5 was present in the common
ancestor of fungal and animal mtDNAs (Bullerwell et al. 2000). As M. graminicola is one of
the few ascomycetes known to be lacking rps5, the absence of this gene could indicate an
independent loss in this species. A possible homolog of this gene was identified on scaffold 5
of the 8.9× draft genomic sequence of M. graminicola, so it may have been transferred to the
nuclear genome rather than having been lost.
Genes in the M. graminicola mtDNA had no introns, a finding that contrasts with other
fungal mtDNAs that possess large introns containing intron-encoded proteins, as found in
Podospora anserina (Cummings et al. 1990) and Penicillium marneffei (Woo et al. 2003).
Eight orfs, with no obvious homology to any other sequenced genes present in the GenBank
database, were found in the mt genome of M. graminicola. The functions of these putative

Chapter 2 mtDNA of M. graminicola
40
genes remain unclear, although some of them may represent highly diverged versions of
known mtDNA-encoded genes, no longer recognizable by identity searches (Gray et al.
1998). EST databases provided evidence for transcription of orf5, orf6 and orf8, indicating
that they may be expressed. Interestingly, these three orfs were the only ones of the eight that
were located adjacent to tRNA genes, so possibly they may be transcribed along with the
tRNAs but not translated.
All tRNA secondary structures had the expected cloverleaf form, but particularly
interesting were tRNA-Thr (UGU as anticodon) and tRNA-Phe (GAA as anticodon) because
they had nine nucleotides in the anticodon loop instead of the canonical seven. This rare
tRNA structure was described previously in Metarhizium anisopliae for tRNA-Thr and tRNA-
Glu (Ghikas et al. 2006), and in Verticillium dahliae for tRNA-Thr, tRNA-Glu, tRNA-Arg and
tRNA-Ser (Pantou et al. 2006).
Nuclear genomes, including that of M. graminicola (Goodwin et al. 2007), possess SSRs
that are known to be highly variable in terms of motif repeat number and distribution (Toth et
al. 2000; Katti et al. 2001;). This study presents a similar picture for the mt genome of M.
graminicola. SSRs and mononucleotide repeats may play a significant role in the regulation
and evolution of the entire molecule. In nuclear genomes it was demonstrated that these
highly variable tracts, if placed in promoter regions, could influence transcriptional activity
(Kashi et al. 1997) and could play an important role in creating and maintaining quantitative
genetic variation (Tautz et al. 1986; Kashi et al. 1997). In the mtDNA of M. graminicola,
mononucleotide repeats became less common in coding regions as their length increased.
Because most long mononucleotide repeats are located 5’-upstream of ATG start codons
(Fig. 2), we hypothesize that they might play a role in regulating transcription. These tracts
could be protein binding signals and, more precisely, upstream promoter elements, as
demonstrated previously in nuclear genomes (Kashi et al. 1997).
The intraspecific mt diversity was first assessed by comparing the total genome
sequences of two isolates (STBB1 and IPO323), giving a nucleotide diversity of 0.16%. In
order to assess species-wide variation, another 35 isolates were chosen, originating from five
continents and belonging to four of the seven known RFLP haplotypes (Table 1). Using these
additional isolates, the total mtDNA variation in M. graminicola was estimated to range from
0.16 to 0.22%, falling within the lower range of published intraspecific nucleotide diversities.

Chapter 2 mtDNA of M. graminicola
41
Figure 3. Phylogenetic relationships among Mycosphaerella graminicola mtDNA haplotypes inferred using a parsimony haplotype network. Haplotypes are presented as ovals with sizes proportional to the haplotype frequencies. Haplotypes containing isolates originating from durum wheat (Triticum turgidum ssp. durum) are grey. Open circles are hypothetical missing intermediate haplotypes. The completely sequenced haplotypes, H5 (IPO323) and H9 (STBB1), are indicated with bold text.

Chapter 2 mtDNA of M. graminicola
42
The nucleotide diversity would decrease to 0.12% if the 17-bp indel was excluded. This 17-
bp indel appears to be a recent mutation that occurred during the 1970s (Torriani SFF,
unpublished), suggesting that the M. graminicola mtDNA may be increasing in diversity
following the hypothesized selective sweep (Zhan et al. 2003). Other examples of low
intraspecific mtDNA nucleotide diversity based on complete mtDNA sequences were 0.2%
for the olive fly Bactrocera oleae (Nardi et al. 2003) and 0.36% for Drosophila simulans
(Ballard 2000).
These results support earlier findings of low mt diversity in M. graminicola obtained by
RFLP analysis (Zhan et al. 2003). While the haplotypic diversity based on sequences was
higher than that found using RFLPs, the total nucleotide diversity remains the lowest reported
to date in fungi. The greater number of haplotypes found through sequencing reflects the
higher resolution of this method, especially the ability to resolve small indels that are missed
by RFLP analysis (Fig. 3). In fact, if indels were removed from the sequence analysis and
only SNPs were considered, only three mt haplotypes were found, but they did not always
correspond with the RFLP data (Table 1). For example, isolates with RFLP haplotypes 1 and
2 were the most polymorphic and could have SNP haplotypes 1 or 3. Isolates with RFLP
haplotype 3 always had SNP haplotype 1. It was interesting that isolates of M. graminicola
adapted to durum wheat had unique RFLP and SNP haplotypes 4 and 2, respectively (Table
1). The nonrandom association between mitochondrial RFLP haplotypes and host species,
presumably caused by natural selection operating on the mt genome, was noted previously in
M. graminicola (Zhan et al. 2004) and other fungi (Gomes et al. 2000; Demanche et al.
2001). The intraspecific haplotype network (Fig. 3) that included all mutational events also
distinguished between haplotypes originating from bread wheat and durum wheat.
The contrasting genetic diversity among mt and nuclear genomes in M. graminicola
(Zhan et al. 2003 and 2004) raises intriguing questions about the mechanisms leading to this
phenomenon. At least two hypotheses can be proposed to account for the observed low levels
of mt variation, including a lower mutation rate in the mt genome or a selective sweep. A
comparison among the three yeast species Saccharomyces cerevisiae, Kluyveromyces lactis,
and Candida glabrata showed that the frequency of nucleotide changes is higher in nuclear
than in mt genomes (Clark-Walker 1991), which is the opposite of mammals where nuclear
genes evolve slower than mt genes (Saccone et al. 2000). On the other hand, the low level of

Chapter 2 mtDNA of M. graminicola
43
polymorphism in the M. graminicola mtDNAs may have been generated through fixation of
an advantageous mt mutation during a selective sweep. The selection of a favored mt
haplotype leading to low levels of polymorphism was suggested in the oomycete
Phytophthora infestans (Gavino and Fry 2002).
The analysis of intraspecific diversity in the largely conserved mt genome of M.
graminicola provides the basis for developing new tools essential to clarify the conflicting
patterns of nuclear and mt diversity and to understand its cause. For example, the
polymorphic microsatellites differed in numbers of 4- or 5-base repeats, making them
amenable to agarose gel assays. These polymorphisms have already been used to analyze
paternity in crosses (Ware 2006) and are now being used to analyze the evolution of
resistance to strobilurin fungicides in M. graminicola (Chapter 5).
Acknowledgments
We gratefully acknowledge Marcello Zala for technical support and Charles Crane for
bioinformatics analyses. DNA sequencing of isolate IPO323 was performed at the
Department of Energy's Joint Genome Institute through the Community Sequencing Program
(www.jgi.doe.gov/csp/). All sequencing data are public and may be accessed through
http://www.jgi.doe.gov/Mgraminicola. This project was supported by the Swiss National
Science Foundation (grant 3100A0-104145) and by USDA CRIS project 3602-22000-013-
00D.

Chapter 2 mtDNA of M. graminicola
44
References
Adams KL, Daley DO, Qiu YL, Whelan J, Palmer JD. 2000. Repeated, recent and diverse
transfer of a mitochondrial gene to the nucleus in flowering plants. Nature 408:354-357.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search
tool. J Mol Biol. 215:403-410.
Ballard JWO. 2000. Comparative genomics of mitochondrial DNA in Drosophila simulans. J
Mol Evol. 51:64-75.
Ballard JWO, Whitlock MC. 2004. The incomplete natural history of mitochondria. Mol
Ecol. 13:729-744.
Bannon FJ, Cooke BM. 1998. Studies on dispersal of Septoria tritici pycnidiospores in
wheat-clover intercrops. Plant Pathol. 47:49-56.
Boeger JM, Chen RS, McDonald BA. 1993. Gene flow between geographic populations of
Mycosphaerella graminicola (anamorph Septoria tritici) detected with restriction
fragment length polymorphism markers. Phytopathology 83:1148-1154.
Brennicke A, Grohmann L, Hiesel R, Knoop V, Schuster W. 1993. The mitochondrial
genome on its way to the nucleus: different stages of gene transfer in higher plants. FEBS
Lett. 325:140-145.
Bullerwell CE, Burger G, Lang F. 2000. A novel motif for identifying Rps3 homologs in
fungal mitochondrial genomes. Trends Biochem Sci. 25:363-365.
Burger G, Gray MW, Lang BF. 2003. Mitochondrial genomes: anything goes. TRENDS
Genet. 19:709-716.
Campbell A, Mrazek J, Karlin S. 1999. Genome signature comparisons among prokaryote,
plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA 96:9184-9189.
Chen RS, McDonald BA. 1996. Sexual reproduction plays a major role in the genetic
structure of populations of the fungus Mycosphaerella graminicola. Genetics 142:1119-
1127.
Chen JZ, Hebert PDN. 1999. Intra-individual sequence diversity and hierarchical approach to
the study of mitochondrial DNA mutations. Mutat Res. 434:205-217.
Clark-Walker GD. 1991. Contrasting mutation rates in mitochondrial and nuclear genes of
yeasts versus mammals. Curr Genet. 20:195-198.

Chapter 2 mtDNA of M. graminicola
45
Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene
genealogies. Mol Ecol. 9:1657-1659.
Corlett M. 1991. An annotated list of the published names in Mycosphaerella and Sphaerella.
Mycol Mem. 18:1-328.
Cummings DJ, McNally KL, Domenico JM, Matsuura ET. 1990. The complete DNA
sequence of the mitochondrial genome of Podospora anserina. Curr Genet. 17:375-402.
Demanche C, Berthelemy M, Petit T, Polack B, Wakefield AE, Dei-Cas E, Guillot J. 2001.
Phylogeny of Pneumocystis carinii from 18 primate species confirms host specificity and
suggests coevolution. J Clin Microbiol. 39:2126-2133.
Eyal Z. 1999. The Septoria/Stagonospora blotch disease of wheat: past, present and future.
In: van Ginkel M, McNab A, Krupinsky J, (Eds.), Septoria and Stagonospora Diseases of
Cereals: A Compilation Research, CIMMYT, Mexico. Eur J Plant Pathol. 105:629-641.
Garber RC, Yoder OC. 1983. Isolation of DNA from filamentous fungi and separation into
nuclear, mitochondrial, ribosomal, and plasmid components. Anal Biochem. 135:416-
422.
Gavino PD, Fry WE. 2002. Diversity in and evidence for selection on the mitochondrial
genome of Phytophthora infestans. Mycologia 94:781-793.
Ghikas DV, Kouvelis VN, Typas MA. 2006. The complete mitochondrial genome of the
entomopathogenic fungus Metarhizium anisopliae var. anisopliae: gene order and trn
gene clusters reveal a common evolutionary course for all Sordariomycetes, while
intergenic regions show variation. Arch Microbiol. 185:393-401.
Gomes EA, de Abreu LM, Borges AC, de Araujo EF. 2000. ITS sequences and mitochondrial
DNA polymorphism in Pisolithus isolates. Mycol Res. 104:911-918.
Goodwin SB, Dunkle LD, Zismann VL. 2001. Phylogenetic analysis of Cercospora and
Mycosphaerella based on the internal transcribed spacer region of ribosomal DNA.
Phytopathology 91:648-658.
Goodwin SB, van der Lee TAJ, Cavaletto JR, te Lintel-Hekkert B, Crane CF, Kema GHJ,
2007. Identification and genetic mapping of highly polymorphic microsatellite loci from
an EST database of the septoria tritici blotch pathogen Mycosphaerella graminicola.
Fungal Genet Biol. 44:398-414.

Chapter 2 mtDNA of M. graminicola
46
Goodwin SB, Waalwijk C, Kema GHJ. 2004. Genetics and genomics of Mycosphaerella
graminicola: a model for the Dothideales. In: Arora DK, Khachatourians GG (Eds.),
Applied Mycology & Biotechnology. Vol 4. Fungal Genomics Elsevier Science BV,
Amsterdam, pp. 315-330.
Gray MW, Burger G, Lang BF. 1999. Mitochondrial evolution. Science 283:1476-1481.
Gray MW, Lang BF, Cedergren R, Golding B, Lemieux C, Sankoff D, Turmel M, Brossard
N, Delage E, Littlejohn TG, Plante I, Rioux P, Saint-Louis D, Zhy Y, Burger G. 1998.
Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res.
26:865-887.
Hane J, Lowe R, Solomon P, Tan KC, Schoch CL, Spatafora JW, Crous PW, Kodira C,
Birren B, Torriani SSF, McDonald BA, Oliver RP. 2007. Dothideomycete-plant
interaction illuminated by genome sequencing and EST analysis of the wheat pathogen
Stagonospora nodorum. Plant Cell 19:3347-3368.
Hartl FU, Pfanner N, Nicholson DW, Neupert W. 1989. Mitochondrial protein import.
Biochim Biophys Acta 988:1-45.
Hunter T, Coker RR, Royle DJ. 1999. The teleomorph stage, Mycosphaerella graminicola, in
epidemics of septoria tritici blotch on winter wheat in the UK. Plant Pathol. 48:51-57.
John P, Whatley FR. 1975. Paracoccus denitrificans and the evolutionary origin of
mitochondria. Nature 254:495-498.
Kashi Y, King D, Soller M. 1997. Simple sequence repeats as a source of quantitative genetic
variation. Trends Genet. 13:74-78.
Katti MV, Ranjekar PK, Gupta VS. 2001. Differential distribution of simple sequence repeats
in eukaryotic genome sequences. Mol Biol Evol. 18:1161-1167.
Kema GHJ, Goodwin SB, Hamza S, Verstappen ECP, Cavaletto JR, Van der Lee TAJ,
Hagenaar-de Weerdt M, Bonants PJM, Waalwijk C. 2002. A combined AFLP and RAPD
genetic linkage map of Mycosphaerella graminicola, the septoria tritici leaf blotch
pathogen of wheat. Genetics 161:1497-1505.
Kema GHJ, van Silfhout CH. 1997. Genetic variation for the virulence and resistance in the
wheat Mycosphaerella graminicola pathosystem 3. Comparative seedling and adult plant
experiments. Phytopathology 87:266-272.

Chapter 2 mtDNA of M. graminicola
47
Kema GHJ, Verstappen ECP, Todorova M, Waalwijk C. 1996. Successful crosses and
molecular tetrad and progeny analyses demonstrate heterothallism in Mycosphaerella
graminicola. Curr Genet. 30:251-258.
Kema GHJ, Verstappen E, van der Lee T, Mendes O, Sandbrink H, Klein-Lankhorst R,
Zwiers L, Csukai M, Baker K, Waalwijk C. 2003. Gene hunting in Mycosphaerella
graminicola. Proceedings of the 22nd Fungal Genetics Conference, Asilomar, California,
USA Page 252.
Kirk PM, Cannon PF, David JC, Stalpers JA. 2001. Ainsworth & Bisby´s Dictionary of the
Fungi. 9th Ed., CAB International, Wallingford, UK.
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. 2001. Predicting transmembrane
protein topology with a hidden Markov model: Application to complete genomes. J Mol
Biol. 305:567-580.
Kudla J, Albertazzi FJ, Blazevic D, Hermann M, Bock R. 2002. Loss of the mitochondrial
cox2 intron 1 in a family of monocotyledonous plants and utilization of the mitochondrial
intron sequence for the construction of a nuclear intron. Mol Genet Genomics 267:223-
230.
Kurdyla TM, Guthrie PAI, McDonald BA, Appel DN. 1995. RFLPs in mitochondrial and
nuclear DNA indicate low levels of genetic diversity in the oak wilt pathogen
Ceratocystis fagacearum. Curr Genet. 27:373-378.
Lowe TM, Eddy SR. 1997. tRNAscan-SE: A program for improved detection of transfer
RNA genes in genomic sequence. Nucleic Acids Res. 25:955-964.
Liu YC, Cortesi P, Double ML, MacDonald WL, Milgroom MG. 1996. Diversity and
multilocus genetic structure in populations of Cryphonectria parasitica. Phytopathology
86:1344-1351.
Nardi F, Carapelli A, Dallai R, Frati F. 2003. The mitochondrial genome of the olive fly
Bactrocera oleae: two haplotypes from distant geographical locations. Insect Mol Biol.
12:605-611.
Pantou MP, Kouvelis VN, Typas MA. 2006. The complete mitochondrial genome of the
vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a
diagnostic tool for species identification. Curr Genet. 50:125-136.

Chapter 2 mtDNA of M. graminicola
48
Price EW, Carbone I. 2005. SNAP: workbench management tool for evolutionary population
genetic analysis. Bioinformatics 21:402-404.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. 2003. DnaSP, DNA polymorphism
analyses by the coalescent and other methods. Bioinformatics 19:2496-2497.
Saccone C, Gissi C, Lanave C, Larizza A, Pesole G, Reyes A. 2000. Evolution of the
mitochondrial genetic system: an overview. Gene 261:153-159.
Sanderson FR. 1972. A Mycosphaerella species as the ascogenous state of Septoria tritici
Rob. and Desm. New Zeal J Bot. 10:707-710.
Schoch CL, Shoemaker RA, Seifert KA, Hambleton S, Spatafora JW, Crous PW. 2006. A
multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia
98:1041-1052.
Soanes DM, Talbot NJ. 2006. Comparative genomic analysis of phytopathogenic fungi using
expressed sequence tag (EST) collections. Mol Plant Pathol. 7:61-70.
Sommerhalder RJ, McDonald BA, Zhan J. 2007. Concordant evolution of mitochondrial and
nuclear genomes in the wheat pathogen Phaeosphaeria nodorum. Fungal Genet Biol. 44:
764-772.
Tambor JHM, Guedes RF, Nobrega MP, Nobrega FG. 2006. The complete DNA sequence of
the mitochondrial genome of the dermatophyte fungus Epidermophyton floccosum. Curr
Genet. 49:302-308.
Tautz D, Trick M, Dover G. 1986. Cryptic simplicity in DNA is a major source of genetic
variation. Nature 322:652-656.
Templeton AR, Crandall KA, Sing CF. 1992. A cladistic analysis of the phenotypic
associations with haplotypes inferred from restriction endonuclease mapping and DNA
sequence data. III. Cladogram estimation. Genetics 132:619-633.
Toth G, Gaspari Z, Jurka J. 2000. Microsatellites in different eukaryotic genomes: survey and
analysis. Genome Res. 10:967-981.
Ware SB. 2006. Aspects of sexual reproduction in Mycosphaerella species on wheat and
barley: genetic studies on specificity, mapping, and fungicide resistance. Ph.D. thesis,
Wageningen University, the Netherlands.
Woo PCY, Zhen HJ, Cai JJ, Yu J, Lau SKP, Wang J, Teng JLL, Wong SSY, Tse RH, Chen
R, Yang HM, Liu B, Yuen KY. 2003. The mitochondrial genome of the thermal

Chapter 2 mtDNA of M. graminicola
49
dimorphic fungus Penicillium marneffei is more closely related to those of molds than
yeasts. FEBS Lett. 555:469-477.
Xia JQ, Correll JC, Lee FN, Ross WJ, Rhoads DD. 2000. Regional population diversity of
Pyricularia grisea in Arkansas and influence of host selection. Plant Dis. 84:877-884.
Zhan J, Kema GHJ, McDonald BA, 2004. Evidence for natural selection in the mitochondrial
genome of Mycosphaerella graminicola. Phytopathology 94:261-267.
Zhan J, McDonald BA. 2004. The interaction among evolutionary forces in the pathogenic
fungus Mycosphaerella graminicola. Fungal Genet Biol. 41:590-599.
Zhan J, Pettway RE, McDonald BA. 2003. The global genetic structure of the wheat
pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low
mitochondrial diversity, regular recombination and gene flow. Fungal Genet Biol.
38:286-297.

Chapter 2 mtDNA of M. graminicola
50

CHAPTER 3
The evolutionary history of Mycosphaerella spp. mitochondrial genome
Stefano FF Torriani, Patrick C Brunner, Bruce A. McDonald. 2009. Evolutionary history of
the mitochondrial genome in Mycosphaerella populations infecting bread wheat, durum
wheat, and wild grasses. To be submitted.

Chapter 3 M. graminicola mtDNA evolution
52

Chapter 3 M. graminicola mtDNA evolution
53
Abstract
Plant pathogens emerge in agro-ecosystems following different evolutionary mechanisms
over different time scales. Previous analysis based on nuclear loci indicated that
Mycosphaerella graminicola diverged from an ancestral population adapted to wild grasses
during the process of wheat domestication approximately 10,500 years ago. Here we present
a coalescence analysis based on four mitochondrial loci (total of 1338 bp) that dates the
origin of M. graminicola to approximately 10,000 years before present, further supporting the
hypothesis that Neolithic farmers shaped the evolution of the pathogen during the
development of wheat agriculture. The previously proposed non-random association between
mitochondrial genome haplotypes of M. graminicola and host-specific populations infecting
bread wheat (Triticum aestivum) or durum wheat (Triticum durum) was reconsidered after the
annotation of a 1935 bp putative gene exclusive to a mitochondrial DNA haplotype found at a
higher frequency on durum wheat. The putative protein was predicted to have ligase activity.
Ligases can act as virulence factors by manipulating host cellular pathways to overcome plant
innate immunity. This could explain the increased pathogenicity on durum wheat of those
mitochondrial haplotypes possessing this newly identified gene. Since this putative ligase
gene could not be detected in M. graminicola ancestors we propose that it was introduced
through horizontal gene transfer from a yet unknown donor organism.

Chapter 3 M. graminicola mtDNA evolution
54
Introduction
Archeological (Childe 1953; Moore et al. 2000; Gopher et al. 2002) and genetic findings
(Salamini et al. 2002) place the origins of agriculture in the Fertile Crescent of the Middle
East between 10,000 and 12,000 years ago. The wild progenitors of the major Neolithic
founder crops including wheat (Triticum spp.), barley (Hordeum vulgare) and pea (Pisum
sativum) have been identified in this region (Diamond 1997; Haudry et al. 2007). The
domestication of wild grasses was a slow process reflecting the gradual transition from a
hunter-gatherer to an agriculture-based society. The continuous selection of desired
morphological traits, like increased seed size, stiffness of the ear rachis and the ease of
release of the seed, altered the wild progenitors into the crop species known today (Sharma
and Waines 1980; Elias et al. 1996; Taenzler et al. 2002). Agricultural practices also
modified the physical and biotic environment by altering natural ecosystems characterized by
greater environmental heterogeneity, higher species diversity and lower host density into
agro-ecosystems with greater environmental homogeneity, lower species diversity and higher
host density (Stukenbrock and McDonald 2008). Natural ecosystems favor the evolution of
generalist pathogens able to infect several host species. In contrast, agro-ecosystems
characterized by extensive monoculture favor the evolution of host-specialized pathogens
that have higher virulence (Anderson and May 1982). Agriculture played a significant role in
selecting and spreading globally a number of pathogen species. At least four evolutionary
mechanisms have been proposed by which pathogens emerged in agro-ecosystems over
different time scales (Stukenbrock and McDonald 2008): 1) pathogen domestication along
with the host (e.g. Mycosphaerella graminicola; Stukenbrock et al. 2007), 2) host shifts and
host jumps (e.g. Magnaporthe oryzae; Couch et al. 2005), 3) horizontal gene transfer (e.g.
Pyrenophora tritici-repentis; Friesen et al. 2006), and 4) hybridization (e.g. Phytophthora
cambivora; Ioos et al. 2006). The impact of agricultural practices on the emergence of new
pathogens can be inferred by dating the divergence time between pathogens on cultivated
crops and their progenitors on wild plants. Up until now, very few studies have followed this
approach, with a focus on plant pathogenic fungi and always using nuclear markers (Couch et
al. 2005; Stukenbrock et al. 2007; Munkacsi et al. 2008; Zaffarano et al. 2008). These studies
confirmed that the origins of some fungal plant pathogens, including M. graminicola and M.

Chapter 3 M. graminicola mtDNA evolution
55
oryzae, coincided with the agricultural domestication of their host plant. M. graminicola
emerged as a new pathogen adapted to wheat from an ancestral population infecting wild
grasses in the Middle East during the process of wheat domestication around 10,500 years
ago (Stukenbrock et al. 2007). The new pathogen was first distributed throughout Europe,
during the spread of wheat cultivation approximately 5000-8000 years ago (McDonald et al.
1999; Banke et al. 2004). More recently (500 and 150 years ago, respectively), European
colonists introduced it together with its host into the Americas and Australia (Banke and
McDonald 2005).
M. graminicola is one of the most important foliar pathogens of wheat and occurs
globally across a wide range of climates (Polley and Thomas 1991; Eyal 1999). The life cycle
includes both sexual and asexual stages, with the sexual ascospores having the potential to be
blown over several kilometers (Sanderson 1972) and the asexual pycnidiospores
disseminated over limited distances, mainly via rain-splash (Bannon and Cooke 1998).
Disease management relies on breeding for resistance, fungicide applications and cultural
practices (Parker and Lovell 2001; Russell 2005; Loyce et al. 2008). M. graminicola is
characterized by high nuclear and low mitochondrial (mt) diversity in populations around the
world (Zhan et al. 2003). High gene flow (Boeger et al. 1993), large effective population
sizes (Zhan and McDonald 2004) and recurring sexual reproduction result in high observed
nuclear diversity (Chen and McDonald 1996; Hunter et al. 1999). In contrast, Zhan et al.
(2003) identified only seven mtRFLP haplotypes among a global collection of 1673 isolates,
with the two most frequent (type 1 and type 3) characterizing approximately 93% of the
isolates. Restriction mapping showed that differences among mtRFLP haplotypes were
mainly insertion or deletion polymorphisms (indels). MtRFLP type 1 differed from mtRFLP
type 2 by a 1.7 kb insertion, whereas mtRFLP type 4 differed from mtRFLP type 1 by a 3 kb
insertion within the 1.7 kb insertion (Zhan et al. 2004). Torriani et al. (2008) completely
sequenced two mtRFLP type 2 genomes and estimated, using thirty-five globally selected
samples from four different mtRFLP types, an intraspecific nucleotide diversity of 0.22% for
M. graminicola, which is one of the lowest values reported until now in any organism. A
selective sweep affecting only the mt genome was proposed to explain the high nuclear and
low mt diversity (Zhan et al. 2004). Previous studies revealed a nonrandom association
between both mtRFLP (Zhan et al. 2004) or mt sequence haplotypes (Torriani et al. 2008)

Chapter 3 M. graminicola mtDNA evolution
56
and host species of origin. MtRFLP type 4 isolates were collected only from durum wheat
(Triticum turgidum) and never detected on isolates from bread wheat (Triticum aestivum).
Because only minor genetic differences were found in the nuclear genome between
populations collected from durum- and bread wheat, it was hypothesized that natural
selection acted on host-specific (i.e. durum- wheat-adapted) mt haplotypes such as mtRFLP
type 4 (Zhan et al. 2004).
The first aim of this study was to infer the evolutionary history of M. graminicola using
sequence data from four mt loci. Pathogen populations closely related to M. graminicola,
collected from wild grasses at the center of origin (Stukenbrock et al. 2007), were added to
the analyses to obtain a more complete picture. The second goal was to characterize the indel
polymorphisms distinguishing the different mtRFLP types and in particular a 3 kb insertion
exclusive to mtRFLP haplotype 4 proposed to increase the parasitic fitness on durum wheat.
Materials and methods
Fungal Isolates. M. graminicola isolates collected from locations in Asia, Europe,
Africa, North and South America were obtained from infected leaves sampled from both
bread and durum wheat. Closely related fungal pathogens in the genus Mycosphaerella were
collected from uncultivated grasses of three different genera, Agropyron repens, Dactylis
glomerata and Lolium multiflorum in the northwestern Iranian province Ardabil.
Additionally, we included four isolates of Septoria passerinii, a closely related barley
pathogen collected from H. vulgare (Table 1). Pathogen isolation and DNA extraction were
performed as previously described (Linde et al. 2002; Zhan et al. 2003 and 2004). Most of the
populations were previously described using nuclear (McDonald et al. 1999; Linde et al.
2002; Banke et al. 2004; Stukenbrock et al. 2007) and mt markers (Zhan et al. 2003 and
2004; Torriani et al. 2008 and 2009).

Chapter 3 M. graminicola mtDNA evolution
57
Table 1. Pathogen populations included in this study
Pathogen Isolates Population Location Continent Host Plant Year Sampling strategy1 Collectors
Mycosphaerella Spp. 2 Iran 1 Ardabil Asia Lolium multiflorum 2004 Random Javan-Nikkhah M (S1 and S2) 2 Iran 2 Ardabil Asia Dactylis glomerata 2004 Random Javan-Nikkhah M 6 Iran 3 Ardabil Asia Lolium multiflorum 2004 Random Javan-Nikkhah M 4 Iran 4 Ardabil Asia Agropyron repens 2004 Random Javan-Nikkhah M 3 Iran 5 Ardabil Asia Lolium multiflorum 2004 Random Javan-Nikkhah M Mycosphaerella 20 Israel Sa'ad Asia Triticum aestivum 1992 Transect Yarden O graminicola 1 Syria Syria Asia Triticum turgidum 1995 Random Kema GHJ 18 Iran Mehran Asia Triticum aestivum 2001 Random Javan-Nikkhah M 8 England England Europe Triticum aestivum 1992 Transect Shaw C, Pjils C 9 Denmark Abed Europe Triticum aestivum 1994 Hierarchy Rasmussen M 8 Switzerland Hubersdorf Europe Triticum aestivum 1999 Hierarchy McDonald BA 6 United States 1 Oregon America Triticum aestivum 1990 Hierarchy Boeger J2
5 United States 2 Indiana America Triticum aestivum 1993 Hierarchy Shaner G 8 Uruguay Colonia America Triticum aestivum 1993 Hierarchy Diaz de Ackermann R 5 Algeria 1 Algeria Africa Triticum turgidum 1992-94 Random Kema GHJ 1 Algeria 2 Algeria Africa Triticum aestivum 1992-94 Random Kema GHJ 2 Tunisia 1 Tunisia Africa Triticum turgidum 1992-94 Random Kema GHJ 10 Tunisia 2 Tunisia Africa Triticum aestivum 2008 Mixed Boukef S 21 Tunisia 3 Tunisia Africa Triticum turgidum 2008 Mixed Boukef S Septoria passerinii 4 United States 3 N. Dakota America Hordeum vulgare 1995 Random Long D
1 Mixed sampling strategy included both isolates collected randomly and hierarchical. 2 and McDonald BA, Schmitt M
Polymerase Chain Reaction and Sequencing. Amplifications of the previously
described mtDNA loci Mg1, Mg2 and Mg3 (Torriani et al. 2008 and 2009) from
Mycosphaerella spp. isolates collected from uncultivated grasses were unsuccessful.
Therefore, other mtDNA regions were screened to develop four new mtDNA loci, Mg4, Mg5,
Mg6 and Mg7. Mg4 partially amplified nad5, Mg5 amplified the large ribosomal subunit
(rnl), while Mg6 and Mg7 were located 3’downstream of rnl in a region containing tRNA-
genes with a gene order found to be conserved interspecifically (Tambor et al. 2006; Torriani
et al. 2008). All isolates were amplified using the following primers: Mg4F (5’-AAC ACT
AAG TCT GTC ACG TAT CTG-3’) and Mg4R (5’-CCC TTC ATG ACA GGT TTC TAC
TC-3’); Mg5F (5’-TTA ATC TAT TCA GTT ATA ATT C-3’) and Mg5R (AGT CGT ACC
CTT CAT GAT TCC T-3’); Mg6F (5’-GAG CTA GCA CCG TTC TTA TAA ATG’-3) and
Mg6R (5’- AAA GAT CGG TGA GCC GTT AC-3’); Mg7F (5’-AAA CGG AGG TAA CGG
CTC AC-3’) and Mg7R (5’- GAT TAT CTT GTG GGC ATT GGT C-3’). PCR reactions
were carried out in 20 µl volumes containing 2 µl of 10x PCR buffer (1x PCR buffer: 10 mM
KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl, 2 mM MgSO4, 0.1% Triton X-100, [pH 8.8]), 0.1

Chapter 3 M. graminicola mtDNA evolution
58
mM of each dNTP, 10 pmol of each primer, 1 U of Taq DNA Polymerase (New England
Biolabs, England) and 4 µl of genomic DNA (5 to 10 ng final DNA concentration). Thermal
cycling conditions included initial denaturation at 96°C for 2 min followed by 35 cycles of
96°C for 1 min, 53°C for 90 sec, 72°C for 1 min. Finally, a 5 min PCR extension was carried
out at 72°C. The PCR products were sequenced using the primers Mg4R, Mg5F, Mg6F and
Mg7F. A total of 1338 bp was sequenced from each isolate, 483 bp for Mg4, 318 bp for Mg5,
156 bp for Mg6 and 381 bp for Mg7. Sequencing reactions were performed using the ABI
PRISM BigDye Terminator v3.0 ready reaction cycle sequencing kit (Applied Biosystems,
Foster City, CA). Sequencing reactions were in a final volume of 10 µl using 10 pmol of
primer, 2 µl BigDye reaction mix, previously diluted 1:4 and 4 µl of purified PCR products.
The cycling profile was: 10 sec denaturation at 95°C, 5 sec annealing at 50°C and 4 min
extension at 60°C for 100 cycles. All sequences were aligned and edited manually with
Sequencer 4.5 (Gene Codes Corporation, Ann Arbor, MI) using the Pezizomycotina genetic
code that diverged from the standard nuclear code for codon TGA, which was read as Trp
and not as Stop.
Mt RFLP polymorphism analysis. The M. graminicola mt genomic region including
the indel polymorphisms characterizing the different mtRFLP haplotypes was identified by
comparing Southern blots described in Zhan el al. (2003) and the recently released complete
mtDNA sequence (EU090238; Torriani et al. 2008). Primers Mgvar1F and Mgvar1R (Table
2) were used to amplify the mt indel polymorphic region (Fig. 1) from three mtRFLP types
(type 1, type 2 and type 4). MtRFLP types 1 and 3 were identical at this locus so only type 1
was analyzed. The reactions were in a total volume of 25 µl containing 2.5 µl of 10x long
PCR buffer with 15 mM MgCl2, 0.2 mM of each dNTP, 10 pmol of each primer, 1 U of long
PCR enzyme mix (containing a Taq DNA Polymerase and a second thermostable polymerase
that exhibits 3’-5’ exonuclease activity; Fermentas, Switzerland) and 5 µl of genomic DNA
(5 to 15 ng final DNA concentration). The thermal cycling conditions were: initial
denaturation at 94°C for 2 min followed by 10 cycles of 15 sec denaturation at 94°C, 30 sec
annealing at 60°C, 5 min extension at 68°C and 20 cycles with same profile but with 15 sec
more extension. Finally, a 10 min PCR extension was carried out at 68°C. The obtained PCR
amplicons were completely sequenced by primer walking using primers listed in Table 2. The

Chapter 3 M. graminicola mtDNA evolution
59
open reading frame unique to mtRFLP type 4 (mtRFLP4-ORF) was translated and screened
for similarity using the BlastP tool provided by the NCBI database (Altschul et al. 1990) and
tested for the presence of transmembrane domains with the TMHMM2 method (Krogh et al.
2001). The program ProtFun 2.2 (Jensen et al. 2002) was used to assess the putative function
of mtRFLP4-ORF encoded protein. ProtFun 2.2 produces ab initio prediction of protein
functions from amino acid sequences using a large number of other feature prediction
servers, most of them available online on the homepage of the Technical University of
Denmark (http://www.cbs.dtu.dk).
All isolates were tested for the presence/absence of mtRFLP4-ORF by multiplex PCR
using the Mg-loci settings described above and two pairs of primers, Mgvar6F and Mgvar6R
specific for mtRFLP4-ORF and Mg5F and Mg5R as a control for the method. The amplicons
were 397 bp and 562 bp, respectively.
Table 2. 5’-3’ sequence of primers used for sequencing the indels characterizing the mtDNA RFLP
types of Mycosphaerella graminicola.
Primer name Sequence (5'-3') Size (bp) RFLP-typesa
Mgvar1F ATACTAGCACTGGCTCCGTTTC 22 1, 2, 3, 4 Mgvar2F AACGCCCTTACTACAAGACGTG 22 1, 3, 4 Mgvar3F CCTAATTCCTTGTATTCCCTTTCC 24 4 Mgvar4F TGTATTACACTGAGATGCGTTGC 23 4 Mgvar5F AGGAGAACTTCGCAAGAATAGC 22 4 Mgvar6F TGACGTAATCAATCATCTGAGG 22 4 Mgvar1R ATACCCACACCTTTACCCAGAG 22 1, 2, 3, 4 Mgvar2R GGTGGTGCAGAATTTCCATTAG 22 1, 3, 4 Mgvar3R TGAGCAACATCTCTAGGACGAG 22 1, 3, 4 Mgvar4R TTCATCGCAAGAGATAGCAAAC 22 1, 3, 4 Mgvar5R AAGGTCCTCGACCAAACCTATC 22 4 Mgvar6R CAGGTGGGACAGTGTCATGTAG 22 4
a The mitochondrial RFLP-haplotypes that the primer can bind (only type 1-4 are considered)
Haplotype Network. To determine the phylogenetic relationships among the
Mycosphaerella populations collected from T. aestivum, T. durum, L. multiflorum, D.
glomerata and A. repens, the concatenated sequences of Mg4, Mg5, Mg6 and Mg7 were
analyzed using the program TCS 1.21 (Clement et al. 2000). All indels and infinite site
violations were removed prior to using the program SNAP Workbench (Price and Carbone
2005). TCS 1.21 infers an unrooted cladogram (Fig. 2) applying a statistical parsimony
method based on Templeton’s 95% parsimony connection limit (Templeton et al. 1992).

Chapter 3 M. graminicola mtDNA evolution
60
Coalescent analysis. The ancestral history of the mt genome in Mycosphaerella
populations infecting bread wheat, durum wheat and wild grasses was inferred by coalescent-
based genealogy using SNAP Workbench (Price and Carbone 2005). The concatenated (Mg4,
Mg5, Mg6 and Mg7) DNA sequences were collapsed into haplotypes using the program
SNAP map (Aylor et al. 2006) by excluding indels and removing infinite site violations. To
detect incompatibility among segregating sites, an incompatibility matrix was generated in
SNAP Clade and SNAP Matrix (Markwordt et al. 2004), and conflicting sites were removed
manually. The M. graminicola populations were considered as one panmictic population
because of the high gene flow operating between geographic populations in this species
(Boeger et al. 1993; Zhan et al. 2003). The coalescent analysis was run using four subdivided
lineages, including S. passerinii, M. graminicola and the Mycosphaerella populations S1 and
S2 adapted to uncultivated grasses, as described in Stukenbrock et al. (2007).
A migration matrix was generated with the program MIGRATE (Beerli and Felsenstein
2001) and used as the backward migration matrix for ancestral inference, using population
subdivision in the program GENETREE (Griffiths and Tavaré 1994) as implemented in the
SNAP workbench. The genealogy with the highest root probability was determined by
performing 500,000 simulations of the coalescent with five different starting random number
seeds. The tree with highest root probability was selected showing the distribution of
mutations along the branches of the pathogen populations (Fig. 3). To compare dates of
divergence with previously published data, the relative coalescence time scale was calibrated
at the separation of S. passerini from Mycosphaerella spp. by assuming a divergence time of
68,500 years bp, as reported in Stukenbrock et al. (2007) using nuclear markers. The nuclear
coalescence time was calculated using mutation rates from six loci and 360 individuals
(Stukenbrock et al. 2007).
Results
One hundred twenty-two M. graminicola isolates from four continents, collected from T.
aestivum (93) and T. durum (29), seventeen from Mycosphaerella spp. adapted to
uncultivated grasses L. multiflorum (11), D. glomerata (2) and A. repens (4) as well as four S.
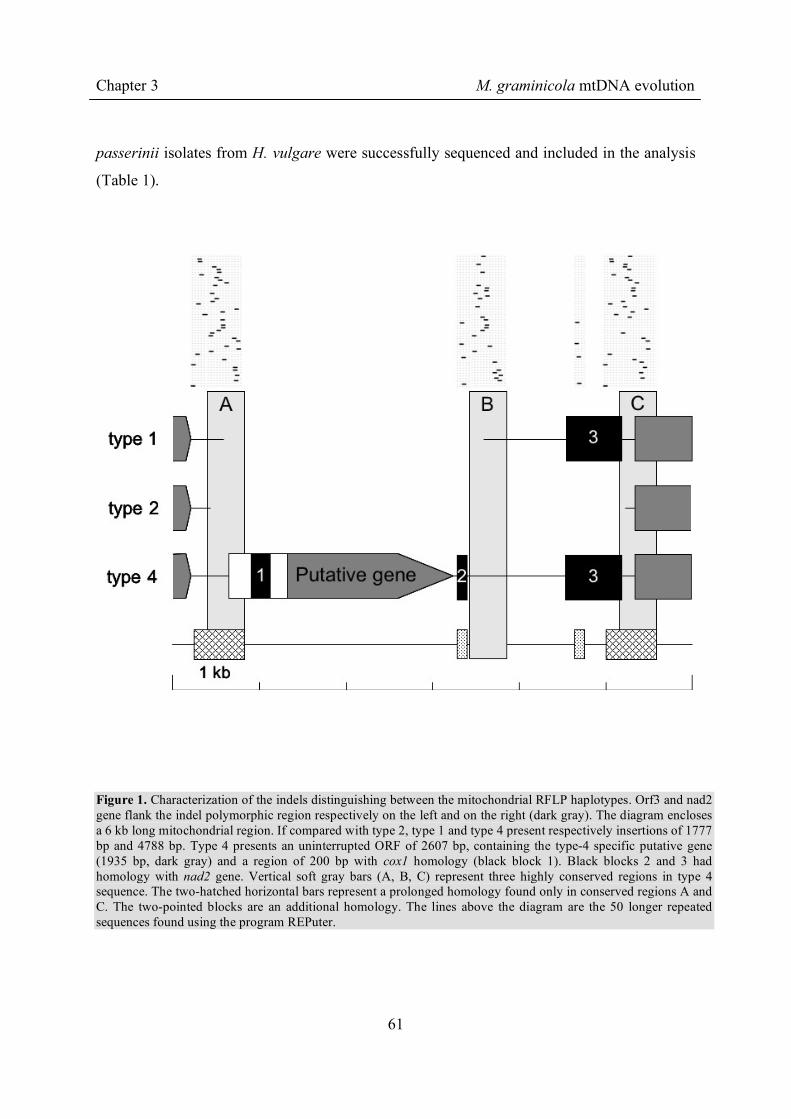
Chapter 3 M. graminicola mtDNA evolution
61
passerinii isolates from H. vulgare were successfully sequenced and included in the analysis
(Table 1).
Figure 1. Characterization of the indels distinguishing between the mitochondrial RFLP haplotypes. Orf3 and nad2 gene flank the indel polymorphic region respectively on the left and on the right (dark gray). The diagram encloses a 6 kb long mitochondrial region. If compared with type 2, type 1 and type 4 present respectively insertions of 1777 bp and 4788 bp. Type 4 presents an uninterrupted ORF of 2607 bp, containing the type-4 specific putative gene (1935 bp, dark gray) and a region of 200 bp with cox1 homology (black block 1). Black blocks 2 and 3 had homology with nad2 gene. Vertical soft gray bars (A, B, C) represent three highly conserved regions in type 4 sequence. The two-hatched horizontal bars represent a prolonged homology found only in conserved regions A and C. The two-pointed blocks are an additional homology. The lines above the diagram are the 50 longer repeated sequences found using the program REPuter.

Chapter 3 M. graminicola mtDNA evolution
62
Haplotype Network. The parsimony haplotype network was inferred from the
compatible sequence alignment of the four sequenced loci (Mg4-Mg7) excluding indels and
infinite violation sites. The 143 isolates collapsed into six distinct haplotypes, two for both M.
graminicola and S2 (S2a, S2b), and one each for S1 and S. passerinii (Fig. 2). Four major
evolutionary groups were identified; S. passerinii, M. graminicola, S1 and S2, consistent
with previous results based on nuclear sequences (Stukenbrock et al. 2007). The number of
mutational events between S. passerinii and S2 was 31, between S1 and M. graminicola nine
and between S2 and M. graminicola three. The most frequent M. graminicola haplotype,
representing more than 99% of all isolates, included samples collected from both durum and
bread wheat, with all isolates sampled from durum wheat collapsing into this haplotype. The
haplotype network showed that S1 could infect three different hosts (L. multiflorum, D.
glomerata and A. repens), whereas S2 was restricted to L. multiflorum (Fig. 2).
Figure 2. Parsimony haplotype network for the concatenated loci (Mg4, Mg5, Mg6 and Mg7). Haplotypes are presented as circle with sizes proportional to the haplotype frequencies; numbers in parenthesis refer to isolates enclosed in that haplotype. Different colors indicate the hosts from which the isolates were sampled. Dots are hypothetical missing intermediate haplotypes.

Chapter 3 M. graminicola mtDNA evolution
63
Coalescent analysis. The Program Genetree was used to provide coalescent-based
genealogies to differentiate between ancestral and descendent populations and to infer the
evolution of polymorphisms among haplotypes (Fig. 3). The analysis included 48 informative
polymorphic sites, which gave good resolution identifying the same six clades obtained in the
parsimony haplotype network (Fig. 2). A transversion from adenine to guanine separated S2
from S1 and, more recently, a transition from thymine to cytosine separated S1 from M.
graminicola (mutation numbers 25 and 1 respectively in Fig. 3). The S2 clade separated
earlier than S1 from M. graminicola. S1 was the clade that accumulated the most mutations
after the split from M. graminicola. S. passerinii separated at the deepest point in the
genealogy, suggesting a more ancient divergence from the other three Mycosphaerella clades.
One isolate from Denmark didn’t cluster with other M. graminicola isolates in the same
clade. After converting the coalescence time to real time by calibrating the separation
between S. passerini and Mycosphaerella spp. at 68,500 years ago (Stukenbrock et al. 2007),
we found that S2 diverged approximately 22,000 years ago and M. graminicola emerged as a
new pathogen about 10,000 years BP.
MtRFLP haplotype analysis. The indel polymorphic region of M. graminicola mtDNA
characterizing the different mtRFLP haplotypes (Zhan et al. 2003 and 2004) was amplified,
sequenced and analyzed for three different mtRFLP types, in order to include more than 90%
of the global mtRFLP haplotypic diversity (Zhan et al. 2003). The amplifications resulting
from mtRFLP types 1, 2 and 4 were respectively 2,495 bp, 719 bp and 5,506 bp in size.
MtRFLP types 1 had an insertion of 1,776 bp compared to type 2; type 4 differed from type 1
by a 3,011 bp insertion (Fig. 1). The MtRFLP type 4 carried a unique insertion containing a
2,607 bp ORF displaying two stretches of 175 bp and 200 bp, showing homology
respectively to nad2 (93% identity, e-value of 9e-66; black block 1 in Fig. 1) and cox1 (99%
identity, e-value of 6e-98; black block 2 in Fig. 1). The mtRFLP type 4 insertion also had 122
nucleotides with nad2 similarity (85% identity, e-value of 1e-30; black block 3 in Fig. 1),
repeated within a longer nad2-homologous stretch (639 bp, 93% identity and e-value of 0;
black block 4 in Fig. 1) present in both mtRFLP type 1 and type 4, but absent in mtRFLP
type 2 (Fig 1). Within the mtRFLP type 4 insertion a putative gene of 1,935 nucleotides was
identified, composed of 32% A, 33% T, 19% G and 16% C, with an AT-content of 65%. The

Chapter 3 M. graminicola mtDNA evolution
64
translation of the putative gene gave rise to a 644 amino acid protein with no specific match
against NCBI database sequences, but with a theoretical isoelectric point of 5.15 and an
estimated molecular weight of 74 KDa. The predicted protein displays three putative
transmembrane domains at positions 22-44, 29-81 and 97-119, five putative N-glycosylated
sites, one putative O-glycosylated site, 37 putative phosphorylation sites and it was
hypothesized to follow the secretory pathway. Based on the amino acid sequence, the protein
was predicted to have a ligase function.
A region approximately 435 bp long was found repeated three times (A-, B-, C-repeats;
soft grey bars Fig. 1) in mtRFLP type 4, always flanking a putative indel insertion/excision
site. MtRFLP types 1 and 2 had two and one copies of this repeat, respectively. The A-, B-
and C-repeats had a total nucleotide diversity of 16%. The A- and C-repeats showed a 153 bp
extended homology not found in the B-repeat (91% identity, e-value of 2e-50; hatched
horizontal bars Fig. 1).
All isolates were screened for the presence of mtRFLP4-ORF using a multiplex PCR
approach. MtRFLP4-ORF was not found in Mycosphaerella isolates sampled from the
uncultivated grasses (A. repens, D. glomerata, L. multiflorum), nor in S. passerinii collected
from H. vulgare. 29% of the tested M. graminicola isolates possessed the mtRFLP4-ORF,
including 93% of isolates sampled from durum wheat and 9% from bread wheat. All
mtRFLP4-ORF containing isolates from bread wheat were limited to a single North African
population (Tunisia 2, Table 1), where it occurred at a frequency of 80%. Samples containing
mtRFLP4-ORF were found in all populations of T. turgidum.

Chapter 3 M. graminicola mtDNA evolution
65
Figure 3. Coalescent-based genealogy of the combined mitochondrial loci Mg4, Mg5, Mg6, Mg7 showing the distribution of mutations in Mycospharella graminicola, Mycosphaerella S1 and S2 and Septoria passerinii. The split between M. graminicola and S1 was fixed at 10,500 (Stukenbrock et al. 2007) and used to convert the coalescence scale to real time. The number of isolates represented in each branch is shown below the tree. Time scale is given on the right from the past (top) to the present (bottom).

Chapter 3 M. graminicola mtDNA evolution
66
Discussion
Here we report the first analysis of the evolutionary history of the M. graminicola mt
genome. Earlier studies considered the low mtDNA variability and the nonrandom
association between mtRFLP haplotypes and host species (Zhan et al. 2003 and 2004;
Torriani et al. 2008). The obtained mt coalescence analyses provided an overview of the
divergence of Mycosphaerella populations since the split from S. passerinii, dated to
approximately 68,500 years ago. Mt coalescence supported nuclear analyses showing S.
passerinii branching at the deepest point of the coalescence, indicating that the separation
between the wheat pathogen M. graminicola and barley pathogen S. passerinii took place
before the domestication of their hosts. Mitochondrial and nuclear coalescence analyses
provided congruent results, indicating that both genomes shared the same evolutionary
history and reinforcing the hypothesis that M. graminicola emerged as a new pathogen during
the process of wheat domestication about 10,000 years ago in the Fertile Crescent from an
ancestral species able to infect several uncultivated hosts. Many archeological studies have
reported on the profound changes in Mesopotamia and other Neolithic cultures after the
beginning of agriculture and the related intensive selection and cultivation of selected crop
genotypes (Moore et al. 2000; Gopher et al. 2002). By selecting plants for agricultural
purposes Neolithic farmers not only altered the natural environment by creating agro-
ecosystems, they also changed host-pathogen interactions (Stukenbrock and McDonald
2008), shaping the evolution of pathogen populations and giving rise to “modern” plant
pathogen species, such as M. graminicola. Other pathogens thought to have been
domesticated along with their hosts are M. oryzae on rice (Couch et al. 2005), Phytophthora
infestans on potatoes (Gomez-Alpizar et al. 2007) and Ustilago maydis on maize (Munkacsi
et al. 2008).
For the first time we identified M. graminicola isolates from bread wheat belonging to
mtRFLP type 4. In previous surveys mtRFLP type 4 isolates were found only infecting
durum wheat (Zhan et al. 2004; Torriani et al. 2008). All mtRFLP type 4 isolates from bread
wheat belonged to the same Tunisian population. Tunisian cereal production is characterized
by a significantly higher production of durum wheat than bread wheat (Ben Salem et al.
1995). MtRFLP type 4 isolates could have increased in frequency through selection in durum

Chapter 3 M. graminicola mtDNA evolution
67
wheat fields, possibly due to higher pathogenicity on durum wheat conferred by mtRFLP4-
ORF, and subsequently spread into surrounding fields of bread wheat. Even though other
mtRFLP types may have higher fitness on bread wheat, the higher number of mtRFLP type 4
spores produced in the more common durum wheat fields may have allowed this haplotype to
become locally more frequent on bread wheat. Durum wheat has been grown on a large scale
for a long time in Tunisia. Since both wild relatives and domesticated wheat co-occur
(Medini et al. 2005), the introduction of the mtRFLP4-ORF into the mt genome of M.
graminicola could have taken place in this region. The coalescent analysis was not able to
distinguish mtRFLP4-ORF containing isolates from the others. Additionally, the mtRFLP4-
ORF was not found in S1 and S2, indicating the absence of this putative gene in M.
graminicola ancestors and suggesting a recent insertion event. Since horizontal gene transfer
(HGT; also known as lateral gene transfer) has already been demonstrated to promote the
development of new functions or increased virulence in recipient lineages (De la Cruz and
Davies 2000; Friesen et al. 2006; Keeling and Palmer 2008), we hypothesize that HGT may
have introduced mtRFLP4-ORF into M. graminicola mtDNA. HGTs often take place among
prokaryotes; leading to the acquisition of genes conferring antibiotic resistance (De la Cruz
and Davies 2000). Prokaryote to eukaryote transfers have also been found, e.g. an ancestor of
Saccharomyces cerevisiae received a gene encoding the enzyme dihydroorotate
dehydrogenase making the yeast facultatively anaerobic (Hall et al. 2005). Recently, Friesen
et al. (2006) demonstrated that a gene (ToxA) encoding a critical virulence factor was
laterally transferred from one fungal species to another, showing that HGT can occur
between fungi. HGT can introduce exogenous genetic material through symbiotic
relationship between prokaryotes and eukaryotes or by viral transfection (Gogarten 2003). A
third possible mechanism (specific to fungi) involves conidial anastomosis tubes forming
between two fungal species that regularly come into close contact (Friesen et al. 2006).
Because no similarity was found with any NCBI deposited sequences, a possible donor could
not be identified, even though the high A+T content of mtRFLP4-ORF (65%) suggests a mt
origin, since this is one of the distinctive feature of these genomes (Campbell et al. 1999).
The presence of the three 435 bp long repeats (A-, B-, C-repeat) flanking the
insertion/excision sites of indels, characterizing the different mtRFLP types, indicates that the
RFLP polymorphism at this locus was produced by two mutational events (Fig. 1).

Chapter 3 M. graminicola mtDNA evolution
68
MtRFLP4-ORF was predicted to have ligase function, a result that if confirmed would find
support in the literature. It was already noticed that different pathogen virulence factors
having ligase activity can enable pathogens to manipulate the proteasome-mediated
degradation of proteins in their hosts to promote disease by removing specific host proteins
(Huang et al. 2004, Angot et al. 2007; Speth et al. 2007). We postulate that the increased
pathogenicity of mtRFLP type 4 isolates to durum wheat could be due to the introgression of
the mtRFLP4-ORF that encodes a protein acting as a virulence factor.
Acknowledgments
We gratefully acknowledge Marcello Zala for technical support and Stephen Goodwin for
generously providing isolates from Septoria passerinii. This project was supported by the
Swiss National Science Foundation (grant 3100A0-104145). DNA sequencing was
conducted using the facilities of the Genetic Diversity Center at ETH Zurich.

Chapter 3 M. graminicola mtDNA evolution
69
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search
tool. J Mol Biol. 215:403-410.
Anderson RM, May RM. 1982. Coevolution of hosts and parasites. Parasitology 85:411-412.
Angot A, Vergunst A, Genin S, Peeters N. 2007. Exploitation of eukaryotic ubiquitin
signaling pathways by effectors translocated by bacterial type III and type IV secretion
systems. PLoS Pathogens 3:1-13.
Aylor DL, Price EW, Carbone I. 2006. SNAP: combine and map modules for multilocus
population genetic analysis. Bioinformatics 22:1399-1401.
Banke S, McDonald BA. 2005. Migration patterns among global populations of the
pathogenic fungus Mycosphaerella graminicola. Mol Ecol. 14:1881-1896.
Banke S, Peschon A, McDonald BA. 2004. Phylogenetic analysis of globally distributed
Mycosphaerella graminicola populations based on three DNA sequence loci. Fungal
Genet Biol. 41:226-238.
Bannon FJ, Cooke BM. 1998. Studies on dispersal of Septoria tritici pycnidiospores in
wheat-clover intercrops. Plant Pathol. 47:49-56.
Ben-Salem M, Daalouol A, Ayadi A. 1995. Le blé dur en Tunisie. Zaragoza (Spain):
CIHEAM-IAMZ p. 81-91.
Beerli P, Felsenstein J. 2001. Maximum likelihood estimation of a migration matrix and
effective population sizes in n subpopulations by using a coalescent approach. Proc Natl
Acad Sci USA 98:4563-4568.
Boeger JM, Chen RS, McDonald BA. 1993. Gene flow between geographic populations of
Mycosphaerella graminicola (anamorph Septoria tritici) detected with restriction
fragment length polymorphism markers. Phytopathology 83:1148-1154.
Campbell A, Mrazek J, Karlin S. 1999. Genome signature comparisons among prokaryote,
plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA 96:9184-9189.
Chen RS, McDonald BA. 1996. Sexual reproduction plays a major role in the genetic
structure of populations of the fungus Mycosphaerella graminicola. Genetics 142:1119-
1127.
Childe VG. 1953. New light on the most ancient Near East. New York: Praeger.

Chapter 3 M. graminicola mtDNA evolution
70
Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene
genealogies. Mol Ecol. 9:1657-1659.
Couch BC, Fudal I, Lebrun MH, Tharreau D, Valent B, et al. 2005. Origins of host-specific
populations of the blast pathogen Magnaporthe oryzae in crop domestication with
subsequent expansion of pandemic clones on rice and weeds of rice. Genetics 170:613-
630.
De la Cruz F, Davies J. 2000. Horizontal gene transfer and the origin of species: lesson from
bacteria. Trends Microbiol. 8:128-133.
Diamond J. 1997. Guns, Germs, and Steel: the Fates of Human Societies. New York: Norton
Elias EM, Steiger DK, Cantrell RG. 1996. Evaluation of lines derived from wild emmer
chromosome substitutions. II. Agronomic traits. Crop Sci. 36:228-233.
Emanuelsson O, Brunak S, von Heijne G, Nielsen H. 2007. Locating proteins in the cell
using TargetP, SignalP and related tools. Nat Protoc. 2:953-971.
Eyal Z. 1999. The Septoria/Stagonospora blotch disease of wheat: past, present and future.
In: van Ginkel M, McNab A, Krupinsky J. (Eds.), Septoria and Stagonospora Diseases of
Cereals: A Compilation Research, CIMMYT, Mexico. Eur J Plant Pathol. 105:629-641.
Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, Rasmussen JB, Solomon
PS, McDonald BA, Oliver RP. 2006. Emergence of a new disease as a result of
interspecific virulence gene transfer. Nat Genet. 38:953-956.
Gogarten JP. 2003. Gene transfer: gene swapping craze reaches eukaryotes. Curr Biol.
13:R53-R54.
Gomez-Alpizar L, Carbone I, Ristaino JB. 2007. An Andean origin of Phytophthora infestans
inferred from mitochondrial and nuclear gene genealogies. Proc Natl Acad Sci USA
104:3306-3311.
Gopher A, Abbo S, Lev-Yadun ST. 2002. The ‘when’, the ‘where’ and the ‘why’ of the
Neolithic revolution in the Levant. Doc Praehist. 28:49-62.
Griffiths RC, Tavaré S. 1994. Ancestral inference in population genetics. Stat Sci. 9:307-319.
Hall C, Brachat S, Dietrich FS. 2005. Contribution of horizontal gene transfer to the
evolution of Saccharomyces cerevisiae. Eukayot Cell 4:1102-1115.

Chapter 3 M. graminicola mtDNA evolution
71
Haudry A, Cenci A, Ravel C, Bataillon T, Brunel D, Poncet C, Hochu I, Poirier S, Santoni S,
Glémin S, David J. 2007. Grinding up wheat: a massive loss of nucleotide diversity since
domestication. Mol Biol Evol. 24:1506-1517.
Huang JN, Huang Q, Zhou X, Shen MM, Yen A, Yu SX, Dong GQ, Qu KB, Huang PY,
Anderson EM, Daniel-Issakani S, Buller RML Payan DG, Lu HH. 2004. The poxvirus
p28 virulence factor is a E3 ubiquiting ligase. J Biol Chem. 279:54110-54116.
Hunter T, Coker RR, Royle DJ. 1999. The teleomorph stage, Mycosphaerella graminicola, in
epidemics of septoria tritici blotch on winter wheat in the UK. Plant Pathol. 48:51-57.
Ioos R, Andrieux A, Marcais B, Frey P. 2006. Genetic characterization of the natural hybrid
species Phytophthora alni as inferred from nuclear and mitochondrial DNA analyses.
Fungal Genet. Biol. 43:511-529.
Jensen LJ, Gupta R, Blom N, Devos D, Tamames J, Kesmir C, Nielsen H, Staerfeldt HH,
Rapacki K, Workman C, Andersen CAF, Knudsen S, Krogh A, Valencia A, Brunak S.
2002. Ab initio prediction of human orphan pprotein function from post-translational
modifications and localization features. J Mol Biol. 319:1257-1265.
Keeling PJ, Palmer JD. 2008. Horizontal gene transfer in eukaryotic evolution. Nat Rev
Genet. 9:605-618.
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. 2001. Predicting transmembrane
protein topology with a hidden Markov model: Application to complete genomes. J Mol
Biol. 305:567-580.
Linde C, Zhan J, McDonald BA. 2002. Population structure of Mycosphaerella graminicola
from lesions to continents. Phytopathology 92:946-955.
Loyce C, Meynard JM, Bouchard C, Rolland B, Lonnet P, Bataillon P, Bernicot MH,
Bonnefoy M, Charrier X, Debote B, Demarquet T, Duperrier B, Félix I, Heddadj D,
Leblanc O, Leleu M, Mangin P, Méausoone M, Doussinault G. 2008. Interaction between
cultivar and crop management effects on winter wheat disease, lodging, and yield. Crop
Prot. 27:1131-1142.
Markwordt JD, Doshi R, Carbone I. 2004. SNAP clade and matrix. Distributed over the
internet, http://snap.cifr.ncsu.edu. Department of Plant Pathology, North Carolina
University.
McDonald BA, Zhan J, Yarden O, Hogan K, Garton J, Pettway RE. 1999. The population

Chapter 3 M. graminicola mtDNA evolution
72
genetics of Mycosphaerella graminicola and Phaeosphaeria nodorum. In: Lucas JA,
Bowyer P, Anderson HM, editors. Septoria on cereals: a study of Pathosystems.
Wallingford (UK): CABI. p. 44-69.
Medini M, Hamza S, Rebai A, Baum M. 2005. Analysis of genetic diversity in Tunisian
durum wheat cultivars and related wild species by SSR and AFLP markers. Genet Resour
Crop Ev. 52:21-31.
Moore AMT, Hillman GC, Legge AJ. 2000. Village on the Euphrates: from foraging to
farming at Abu Hureyra. New York: Oxford University Press.
Munkacsi AB, Stoxen S, May G. 2008. Ustilago maydis populations tracked maize through
domestication and cultivation in the Americas. Proc R Soc London Ser B. 275:1037-
1046.
Parker SR, Lovell DJ. 2001. Quantifying the benefits of seed treatment for foliar disease
control. 3rd Seed treatment symposium, Seed treatment: challenge and opportunities,
proceedings 181-188.
Polley RW, Thomas MR. 1991. Surveys of diseases of winter-wheat in England and Wales,
1976–1988. Ann Appl Biol. 119:1-20.
Price EW, Carbone I. 2005. SNAP: workbench management tool for evolutionary population
genetic analysis. Bioinformatics 21:402-404.
Russell PE. 2005. A century of fungicide evolution. J Agr Sci. 143:11-25.
Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W. 2002. Genetics and
geography of wild cereal domestication in the Near East. Nat Rev Genet. 3:429-441.
Sanderson FR. 1972. A Mycosphaerella species as the ascogenous state of Septoria tritici
Rob. and Desm. New Zeal J Bot. 10:707-710.
Sharma HC, Waines JG. 1980. Inheritance of tough rachis in crosses of Triticum
monococcum and T. boeoticum. J Hered. 71:214-216.
Speth EB, Lee YN, He SY. 2007. Pathogen virulence factors as molecular probes of basic
plat cellular functions. Curr Opin Plant Biol. 10:580-586.
Stukenbrock EH, Banke S, Javan-Nikkhah M, McDonald BA. 2007. Origin and
domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric
speciation. Mol Biol Evol. 24:398-411.
Stukenbrock EH, McDonald BA. 2008 The origin of plant pathogens in agro-ecosystems.

Chapter 3 M. graminicola mtDNA evolution
73
Annu Rev Phytopathol. 46:75-100.
Taenzler B, Esposti RF, Vaccino P, Brandolini A, Effgen S, Heun M, Schäfer-Pregl R,
Borghi B, Salamini F. 2002. Molecular linkage map of Einkorn wheat: mapping of
storage-protein and soft-glume genes and bread-making quality QTLs. Genet Res Camb.
80:131-143.
Tambor JHM, Guedes RF, Nobrega MP, Nobrega FG. 2006. The complete DNA sequence of
the mitochondrial genome of the dermatophyte fungus Epidermophyton floccosum. Curr
Genet. 49:302-308.
Templeton AR, Crandall KA, Sing CF. 1992. A cladistic analysis of the phenotypic
associations with haplotypes inferred from restriction endonuclease mapping and DNA
sequence data. III. Cladogram estimation. Genetics 132:619-633.
Torriani SFF, Brunner PC, McDonald BA, Sierotzki H. 2009. QoI resistance emerged
independently at least 4 times in European populations of Mycosphaerella graminicola.
Pest Manag Sci. 65:155-162.
Torriani SFF, Goodwin SB, Kema GHJ, Pangilinan JL, McDonald BA. 2008. Intraspecific
comparison and annotation of two complete mitochondrial genome sequences from the
plant pathogenic fungus Mycosphaerella graminicola. Fungal Genet Biol. 45:628-637.
Zaffarano PL, McDonald BA, Linde C. 2008. Rapid speciation following recent host shifts in
the plant pathogenic fungus Rhynchosporium. Evolution 62:1418-1436.
Zhan J, Kema GHJ, McDonald BA. 2004. Evidence for natural selection in the mitochondrial
genome of Mycosphaerella graminicola. Phytopathology 94:261-267.
Zhan J, McDonald BA. 2004. The interaction among evolutionary forces in the pathogenic
fungus Mycosphaerella graminicola. Fungal Genet Biol. 41:590-599.
Zhan J, Pettway RE, McDonald BA. 2003. The global genetic structure of the wheat
pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low
mitochondrial diversity, regular recombination and gene flow. Fungal Genet Biol.
38:286-297.

Chapter 3 M. graminicola mtDNA evolution
74

CHAPTER 4
The mitochondrial genome of Phaeosphaeria nodorum
Torriani’s contribution to the publication “Hane et al. (2007)” was the complete
annotation of the mitochondrial genome and related analysis.
James K Hane, Rohan GT Lowe, Peter S Solomon, Kar-Chun Tan, Conrad L Schoch, Joseph
W Spatafora, Pedro W Crous, Chinappa Kodira, Bruce W Birren, Stefano FF Torriani, Bruce
A McDonald, Richard P Oliver. 2007. Dothideomycete-Plant interactions illuminated by
genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. The
Plant Cell 19:3347-3368.

Chapter 4 mtDNA of P. nodorum
76

Chapter 4 mtDNA of P. nodorum
77
Abstract
Stagonospora nodorum is a major necrotrophic fungal pathogen of wheat (Triticum
aestivum), and a member of the Dothideomycetes, a large fungal taxon that include many
important plant pathogens affecting major crop plant families. Here, we report the acquisition
and initial analysis of a draft genome sequence for this fungus. The assembly comprises
37,164,227 bp of nuclear DNA contained in 107 scaffolds. The circular mitochondrial
genome comprises 49850 bp encoding 46 genes including four that are intron-encoded. The
nuclear genome assembly contains 26 classes of repetitive DNA comprising 4.5% of the
genome. Some of the repeats show evidence of repeat-induced point mutations consistent
with a frequent sexual cycle. ESTs and gene prediction models support a minimum of 10,762
nuclear genes. Extensive orthology was found between the polyketide synthase family in S.
nodorum and Cochliobolus heterostrophus, suggesting an ancient origin and conserved
functions for these genes. A striking feature of the gene catalogue was the large number of
genes predicted to encode secreted proteins; the majority has no meaningful similarity to any
other known genes. It is likely that genes for host-specific toxins, in addition to ToxA, will be
found amongst this group. ESTs obtained from axenic mycelium grown on oleate (chosen to
mimic early infection) and late-stage lesions sporulating on wheat leaves were obtained.
Statistical analysis shows that transcripts encoding proteins involved in protein synthesis and
in the production of extracellular proteases, cellulases and xylanases predominate in the
infection library. This suggests that the fungus is dependant on the degradation of wheat
macromolecular constituents to provide the carbon skeletons and energy for the synthesis of
proteins and other components destined for the developing pycnidiospores.

Chapter 4 mtDNA of P. nodorum
78
Introduction
Stagonospora (syn. Septoria) nodorum (Berk.) Castell. & Germano [teleomorph
Phaeosphaeria (syn. Leptosphaeria) nodorum (Müll.) Hedjar] is a major pathogen of wheat
(Triticum aestivum) in most wheat-growing areas of the world. It is the major cause of losses
due to foliar pathogens in Western Australia and north central and north eastern North
America (Solomon et al. 2006a) and until the 1970s, it was the major foliar necrotrophic
pathogen in Europe (Bearchell et al. 2005). Infection begins when spores (ascospores or
asexual pycnidiospores) land on leaf tissue (Bathgate and Loughman 2001; Solomon et al.
2006b). The spores rapidly germinate to produce hyphae which invade the leaf, utilising
hyphopodia to gain entry to epidermal cells or by growing directly through stomata (Solomon
et al. 2006b). The hyphae rapidly colonise the leaves and begin to produce pycnidia in 7-10
days. The infection has been divided into three metabolic phases (Solomon et al. 2003a). The
first phase is penetration of the host epidermis and is fuelled predominantly by lipid stores in
the spores (Solomon et al. 2004a); the second is proliferation throughout the interior of the
leaf, involves toxin release (Liu et al. 2006) and uses host-derived simple carbohydrate
sources (Solomon et al. 2004a); the final phase produces the new spores, but so far the
metabolic requirements for pycnidiation are unclear.
The infection epidemiology follows a polycyclic pattern with repeated cycles of both
asexual (Bathgate and Loughman 2001) and sexual (McDonald unpublished data) infection
throughout the growing season. New rounds of infection are initiated by rain-splash of
pycnidiospores and wind dispersal of ascospores. Eventually, wheat heads become infected,
causing the glume blotch symptom. In Mediterranean climates, the fungus over-summers in
senescent straw and stubble. Seed transmission can be important but is readily controlled by
fungicides. Infected stubble harbours the pseudothecia that produce airborne (Bathgate and
Loughman 2001), heterothallic (Bennett et al. 2003) ascospores to reinitiate the infection in
the following growing season. Epidemiological and population genetic evidence suggest the
fungus undergoes meiosis in the field regularly and that gene flow is global (Stukenbrock et
al. 2006). S. nodorum is a member of the Dothideomycetes class of filamentous fungi. This is
a newly recognised major class that broadly replaced the long-recognized loculoascomycetes

Chapter 4 mtDNA of P. nodorum
79
(Winka and Eriksson 1997) and includes the causal organisms of many economically
important plant diseases; notable examples are black leg (Leptosphaeria maculans), southern
corn leaf blight (Cochliobolus heterostrophus), barley net blotch (Pyrenophora teres), apple
scab (Venturia inaequalis), black sigatoka of banana (Mycosphaerella fijiensis), wheat leaf
blotch (M. graminicola), tomato leaf mould (Passalora [syn. Cladosporium] fulva) and
Ascochyta blight of many legume species (Ascochyta spp.). The taxon also includes large
numbers of saprobic species occurring on substrates ranging from dung to plant debris, a few
species associated with animals and several lichenized species (Del Prado et al. 2006).
Many plant pathogens in this group produce host-specific toxins (Wolpert et al. 2002).
These molecules interact with specific host factors to produce disease symptoms only in
selected genotypes of specific plant hosts. Well-known examples are found in Alternaria
Alternata, Cochliobolus heterostrophus and P. tritici-repentis, while species in the genera
Mycosphaerella, Corynespora and Stemphylium are also thought to produce host-specific
toxins (Agrios 2005). Proteinaceous host-specific toxins have recently been shown to be
important virulence determinants in S. nodorum (Liu et al. 2004a and 2004b) including one
whose gene is thought to have been interspecifically transferred to the wheat tan spot
pathogen, P. tritici-repentis (Friesen et al. 2006). Only one example of a host-specific toxin
has been identified outside of the Dothideomycetes (Wolpert et al. 2002). Non-host specific
toxins produced by species in this group include cercosporin (Cercospora) and solanopyrone
(Ascochyta) but are also produced by many other fungal taxa (Agrios 2005).
S. nodorum is an experimentally tractable organism, which is easily handled in defined
media, was one of the first fungal pathogens to be genetically manipulated (Cooley et al.
1988) and has been a model for fungicide development (Dancer et al. 1999). Molecular
analysis of pathogenicity determinants is aided by facile tools for gene ablation and rapid in
vitro phenotypic screens and thus far, a small number of genes required for pathogenicity
have been identified (Cutler et al. 1998; Cooley et al. 1999; Bailey et al. 2000; Bindschedler
et al. 2003; Solomon et al. 2003b, 2004a, 2004b; Solomon and Oliver 2004; Solomon et al.
2005 and 2006a). It has thus emerged as a model for dothideomycete pathology.
Whole genome sequences have been described for a handful of fungal saprobes and
pathogens (Galagan et al. 2003; Jones et al. 2004; Dean et al. 2005; Galagan et al. 2005;
Kamper et al. 2006). Here, we present an initial analysis of the genome sequence of S.

Chapter 4 mtDNA of P. nodorum
80
nodorum, the first dothideomycete genome sequence to be publicly released. Gene expression
studies using EST libraries from axenic mycelium and heavily infected wheat leaves
complement the genome sequence and provide the first broad-based analysis of the genomic
basis of infection by S. nodorum.
Materials and Methods
Fungal Strains. Stagonospora nodorum strain SN15 (Solomon et al. 2003b) was used for
both genome and EST libraries and has been deposited at the Fungal Genetics Stock Center.
The genome sequence was obtained as described (http://www.broad.mit.edu/annotation/
genome/ stagonospora_nodorum/Assembly.html). The sequences are available for download
from GenBank under accession number AAGI00000000.
Phylogenetic Analysis. A combined matrix of 41 taxa was generated from DNA
sequences obtained from two ribosomal (nuclear large and small subunit [nuc-LSU and nuc-
SSU]) and three protein genes (elongation factor 1 a [EF-1α] and the largest and second
largest subunits of RNA polymerase II [RPB1 and RPB2]). Data were obtained from the
Assembling the Fungal Tree of Life (AFTOL; www.aftol.org) and GenBank sequence
databases, which aligned in ClustalX (Thompson et al. 1997) and manually improved where
necessary. After introns and ambiguously aligned characters were excluded, 6694 bp were
used in the final analyses. In some cases, genes were missing (see Supplemental Table 3 and
Supplemental Data Set 3 online). The resulting data were combined and delimited into 11
partitions, including nuc-SSU, nuc-LSU, and the first, second, and third codon positions of
EF-1α, RPB1, and RPB2, with unique models applied to each partition. Metropolis coupled
Markov chain Monte Carlo analyses were conducted using MrBayes 3.1.2 (Huelsenbeck and
Ronquist 2001) with a six-parameter model of evolution (generalized time reversible)
(Rodriguez et al. 1990) and gamma distribution approximated with four categories and a
proportion of invariable sites. Trees were sampled every 100th generation for 5,000,000
generations. Three runs were completed to ensure that stationarity was reached, and 5000
trees were discarded as “burn in” for each. Posterior probabilities were determined by
calculating a 50% majority-rule consensus tree of 45,000 trees from a single run. Maximum

Chapter 4 mtDNA of P. nodorum
81
likelihood bootstrap proportions were calculated by doing 1000 replicates in RAxML-VI-
HPC (Stamatakis, 2006) with the GTRCAT model approximation and 25 rate categories with
the same data partitions as for the Bayesian runs.
Repetitive Elements. Repetitive elements were identified de novo using RepeatScout
v1.0.0 (Price et al. 2005) with a minimum threshold of 10 matches and a minimum repeat
length of 200 bp. Newly generated repeats were aligned to the genome assembly via
BLASTN v2.0 (Altschul et al. 1990). Hits were discarded if sequence identity fell below 35%
or alignment length was less than 200 bp. For each repeat, the number of filtered hits was
counted, and repeats were discarded if the number of hits to the assembly was less than 10.
Prototype repeat regions were defined by identification of sequence similarities among
themselves via BLASTN and deletion of redundant repeat sequence. Regions of the genome
assembly that matched to a repeat were aligned using MUSCLE v3.6 (Edgar 2004). To
identify repeat type and function, repeats were compared with the nonredundant sequence
database hosted by NCBI via BLASTN/BLASTX, and subrepeat regions within repeats were
also analyzed. Tandem repeats were identified using Tandem Repeat Finder (Benson 1999),
direct repeats were identified via MegaBLAST (Zhang et al. 2000), and inverted repeats were
identified using both MegaBLAST and eINVERTED (Rice et al. 2000).
Putative telomeric regions were predicted by considering scaffold ends containing
successive repetitive elements without interspersed predicted protein coding genes. The
occurrence of repeat classes within these regions was counted and compared with
occurrences throughout the genome. Where less than 85% of the repeats were found to be at
nongenic scaffold end regions, these classes were classified as telomeric repeats. The scaffold
ends were predicted to be physical telomeres if they contained three or more telomeric
repeats.
To analyze and compare the prevalence of RIP mutation, we developed a program to
compare RIP mutations between multiple sequences (Hane and Oliver, unpublished data).
RIPCAL compares the aligned sequences against a designated model sequence (in this case
the trimmed de novo repeats) and was configured to calculate RIP (Dean et al. 2005).

Chapter 4 mtDNA of P. nodorum
82
Gene Content. An automated genome annotation was initially created using the Calhoun
annotation system. A combination of gene prediction programs, FGENESH, FGENESH+,
and GENEID, and 317 manually curated transcripts were used. GENEWISE was used with
non-species specific parameters to predict genes from proteins identified by BLASTX. To
refine the initial genome annotation, a further 10,752 EST reads were obtained from an EST
library from oleate-grown mycelium and 10,751 from S. nodorum infected wheat. These EST
sequences were screened against a library of wheat ESTs, trimmed for vector sequence
manually, for poly(A) tail sequence using TrimEST (Rice et al. 2000), screened for unusable
sequences of poor quality, filtered for remaining sequence equal or bigger than 50 bp, and
aligned to the genome assembly using Sim4 (Florea et al. 1998). ESTs with multiple genomic
locations were assigned their optimum location based on percent identity, total alignment
length, and best location of the EST mated pair. Gene models were manually annotated
according to optimum EST alignments using Apollo (Lewis et al. 2002). Genes that were
fully supported by EST data were used to train the gene prediction program UNVEIL
(Majoros et al. 2003). Second-round gene annotations were created by combining gene
predictions with EST data, with EST data replacing predicted gene models, and UNVEIL
predictions preferred over first-round predictions. Genes with coding regions smaller than 50
amino acids were discarded. The numerical identifiers assigned during the first-round
predictions have been retained. New gene models were assigned loci from 20,000 onwards.
Updated loci (UNVEIL and EST supported) have been given the numerical suffix 2 (e.g.,
SNOG_16571.2). The 5354 unsupported genes have retained the numerical identifiers and
suffix 1. Where genic regions lack EST support, only the coding sequence is reported.
ESTs not aligned to the assembly by Sim4 were compared with the unassembled reads
via BLASTN. EST with matches to unassembled reads with an e-value of less than 1e-10 were
clustered into contigs using cap3 (Huang and Madan 1999). The assembled ESTs were tested
for single open reading frames using getORF (Rice et al. 2000), and possible genes were
determined by BLASTP (Altschul et al. 1990) to protein databases at NCBI. One new gene
(STAG_20208.1) was identified by this method. We have chosen not to alter genes where
gene models conflicted with homologs except where colinearity evidence confirms orthology
(Figure 4). Updated files of gene and protein sequences are available from R. Oliver

Chapter 4 mtDNA of P. nodorum
83
Proteins were assigned putative functional classes by searching for relevant PFAM
(Bateman et al. 1999) domains (Bateman et al. 2004) with HMMER v2.0 (Eddy, 1998) (see
Supplemental Data Set 2 and Supplemental Table 2 online). Putative gene ontology was
assigned via BLASTP with BLAST2GO (Conesa et al. 2005). Due to the relatively poor
identification of certain PKS and NRPS domains using resources like PFAM (Bateman et al.
2004) and CD-SEARCH (Marchler-Bauer and Bryant 2004; Marchler-Bauer et al. 2005),
PKS and NRPS domains and their modular organization were further elucidated with online
tools available at NRPS-PKS (Yadav et al. 2003; Ansari et al. 2004). Subcellular localization
and secretion was predicted via WoLF PSORT (Horton et al. 2007).
EST Library Construction. For the construction of the in planta cDNA library, wheat
(Triticum aestivum) cv Amery was infected with S. nodorum SN15 as a whole plant spray
and processed as a latent period assay (Solomon et al. 2006b). Necrotic tissue was excised
from the leaves at 10, 11, and 12 DAI for RNA isolation. RNA was isolated by the Trizol
method (Sigma-Aldrich).
Material for the oleate library was generated as follows: minimal medium(100 mL) +
sucrose (0.5% [w/v]) was inoculated with S. nodorum SN15 pycnidiospores (2.75 x 107) and
incubated at 22°C with shaking (130 rpm) for 4 d. Mycelia was harvested, washed, and added
to 100 mL of minimal media, including 0.05% (v/v) Tween 80 and 0.2% (w/v) oleate as the
carbon source. The culture was incubated as before for 30 h before being harvested, washed,
snap frozen in liquid nitrogen, and freeze-dried in a Maxi Dry Lyo (Heto Holten). mRNA
was extracted using the Messagemaker mRNA purification-cloning kit (Gibco/Invitrogen)
according to manufacturer’s instructions. A total of 2.3 mg of mRNA was used as template
for reverse transcription to cDNA. The in planta cDNA library was constructed using the
SMART cDNA library construction kit (Clontech), and the oleate library was constructed in
pSPORT1 (Gibco/Invitrogen). All manipulations were performed according to the
manufacturer’s instructions. Phage DNA was packaged using the GigapackIII Gold phage
packaging system (Stratagene) according to the manufacturer’s instructions. Phages were
grown, amplified, and mass-excised to bacterial clones according to the provided protocols.
The final libraries were both estimated to contain about 500,000 clones.

Chapter 4 mtDNA of P. nodorum
84
Quantitative PCR. Total RNA (1µg) was reverse transcribed to cDNA using iScript
reverse transcriptase premix (Bio-Rad) according to the manufacturer’s instructions. RNA
from three biological replicates was pooled for a single cDNA synthesis. cDNA reactions
were used as PCR template at 1:50 dilution for in vitro-grown SN15 samples and at 1:5
dilution for in planta-grown SN15 samples. Quantitative PCR reactions consisted of 10 µL of
iQ SYBR green supermix (Bio-Rad), forward and reverse primers (each at 1.2 µM), and 5 µL
of template DNA in a 20 µL reaction. Reactions were incubated in a Rotor-Gene 3000
thermocycler (Corbett Research). Cycling conditions were 3 min/95°C and then 35 cycles of
10s/95°C, 20s/57°C, and 20s/72°. Amplicon fluorescence from template of unknown
concentration was compared with that from genomic DNA standards of 25, 2.5, 0.25, and
0.025 ng/reaction. All reactions were performed with two technical replicates. Data were
analyzed using the Rotorgene software version 6.0 (Corbett Research). Primer sequences are
listed in Supplemental Table 4 online.
Accession Number. Sequence data from this article can be found in the GenBank/EMBL
data libraries under accession number AAGI00000000.
Results
Acquisition and analysis of the genome sequence. The genome sequence was obtained
using a whole-genome shotgun approach. Approximately 10x sequence coverage was
obtained as paired-end reads from plasmids of 4 kb and 10 kb plus 40-kb fosmids. Reads
were assembled using Arachne (Jaffe et al. 2003) forming 496 contigs totalling 37,071 kb.
The contig N50 was 179 kb, meaning an average base in the assembly lies within a contig of
at least 179 kb. Greater than 98% of the bases in the assembly have a consensus quality score
larger or equal of 40, corresponding to the standard error rate of fully finished sequence. The
contigs are connected in 109 scaffolds spanning 37,202 kb with a scaffold N50 of 1.05 Mb.
More than 50% of the genome is contained in the 13 largest scaffolds. Within the scaffolds,
only ~140 kb is estimated to lie within sequence gaps. Thus in terms of representation,
contiguity and sequence accuracy, the draft assembly is of high quality. The mitochondrial
genome comprises a circle of 49,850 bp (Genbank accession number EU053989). It replaces

Chapter 4 mtDNA of P. nodorum
85
two of the auto-assembled; 52 and 65. The nuclear genome is therefore assembled in 107
scaffolds of total length 37,164,227 bp.
General features of the assembly are described in Table 1. As detailed below, the
genomic sequencing was complemented by sequencing EST libraries. One library was
constructed from axenically-grown mycelium and after removing vector, polyA sequences,
poor quality sequences and sequences smaller than 100 bp, there were 7750 remaining ESTs.
Of these, 97.6% mapped to the genome assembly via Sim4 and 1.4% mapped to unassembled
reads. This indicates that the assembly has achieved good coverage of the genome. In all,
seven EST contigs and 13 singletons clustered to the unassembled reads.
Table 1. The assembly of the S. nodorum nuclear genome sequence Scaffold count 107 Scaffold median length/bp 32,376 Total/bp 37,164,227 Scaffold mean length/bp 347,329 Coverage ~10 x Gaps/number 387 G + C % 50.3 % Total length of gaps/bp 146,025 Scaffold minimum length/bp 2005 Max length of gap/bp 11,204 Scaffold maximum length/bp 2,531,949 Median length of gap/bp 325
Analysis of Repetitive elements. Repetitive elements were identified de novo, by
identifying sequence elements that existed in 10 or more copies, were greater than 200 bp,
and exhibited more than 65% sequence identity. The analysis revealed the presence of 25
repeat classes. Table 2 lists the general features of the repeats (see Supplemental Data Set 1
online). Only three, Molly, Pixie and Elsa, had been detected earlier (Genbank accession
numbers AJ277502, AJ277503, and AJ277966, respectively). Molly, Pixie, X15 and R37
show sequence characteristics of inverted terminal repeat-containing transposons, while Elsa,
R9 and X11 appear to be retrotransposons. Repetitive elements were found individually
throughout the genome but were often found in clusters spanning several kilobases.
Telomere-associated repeats were identified by searching for examples of the canonical
telomere repeat TTAGGG at the termini of auto-assembled scaffolds. Physically-linked
repetitive sequences were then analysed for association with the TTAGGG sequence repeats.
Between 19 and 38 copies of telomere-associated repeats were found in the assembly.

Chapter 4 mtDNA of P. nodorum
86
Repeat-induced point (RIP) mutation is a fungal-specific genome-cleansing process that
detects repeated DNA at meiosis and introduces C to T mutations into the copies (Cambareri
et al. 1989). Using the parameters defined for Magnaporthe grisea (Dean et al. 2005) we
identified RIP-like characteristics in several of the repeat classes (Table 2; Supplementary
Data Set 1 online). The transposons Molly and Elsa were the most clearly affected classes.
None of the telomere-associated repeats displayed RIP characteristics.
Mitochondrial Genome. The mitochondrial genome of S. nodorum assembled as a circular
molecule of approximately 49,850 bp, with an overall G + C content of 29.4%. It contains the
typical genes encoding 12 inner mitochondrial membrane proteins involved in electron
transport and coupled oxidative phosphorylation (nad1-6 and nad4L, cytb, cox1-3, atp6), the
5S ribosomal protein, three open reading frames (ORFs) of unknown function, and genes for
the large and small ribosomal RNAs (rnl and rns) (Fig. 1). The genes were coded on both
DNA strands. The 27 tRNAs can carry all 20 canonical amino acids. All tRNA secondary
structures showed the expected cloverleaf form, but tRNA-Thr and tRNA-Phe had 9
nucleotides in the anticodon loop instead of the typical seven, and tRNA-Arg2 had 11
nucleotides in its anticodon loop.
Gene content. The initial gene prediction process identified 16,957 gene models of
which 16,586 were located on the 107 nuclear scaffolds. A revised gene prediction
procedure, using new ESTs (see below) and 795 fully supported and manually annotated
gene models, identified 10,762 version 2 nuclear gene models. Of these, 617 genes
corresponded to a merging of version 1 genes; in 50 cases, three prior gene models were
merged and in one case, four genes were merged. ESTs that aligned to unassembled reads
identified one supported gene (SNOG_20000.2). A total of 5,354 version 1 gene models
were not supported but did not conflict with second-round predictions. We therefore conclude
that S. nodorum contains a minimum of 10,762 nuclear genes of which all but 125 are
supported by two gene prediction procedures and 2,696 are supported by direct experimental
evidence via EST alignment. These genes have the identification format SNOG_xxxxx.2,
were compared with the Genbank nonredundant protein NCBI database. Informative (not
hypothetical, predicted, putative or unknown) BLASTP (Altschul et al. 1990) hits with e-

Chapter 4 mtDNA of P. nodorum
87
values less than 1x10-6 were found for 7,116 genes (See Supplementary Data Set2 online). It
is estimated that at least 46.6% of the nuclear genome is transcribed and 38.8% is translated.
The 5,354 gene models without support from the reannotation have unaltered accession
numbers as SNOG_xxxxx.1 and are retained for further possible validation and analysis. As
952 of these unsupported gene models have BLASTP hits with e-values less than 1x10-6, we
predict that some will be validated as new evidence comes to hand.
Figure 1. The Structure of the Mitochondrial Genome. Black segments are exons, hatched segments are rRNA genes, and white segments are introns. The direction of transcription is indicated with the arrows.
Chapter 4 mtDNA of P. nodorum
88
Table 2. Features of repetitive element classes found in the nuclear genome
Repeat Name Class Count Full Length (bp)
Occupied (bp)
Percentage of Genome RIPa
Subtelomeric R22 Telomeric repeat 23 678 12565 0.03 NO X15 Telomeric repeat/transposon or remnant 37 6231 89126 0.24 NO X26 Telomeric repeat 38 4628 77020 0.21 NO X35 Telomeric repeat 19 1157 14393 0.04 NO X48 Telomeric repeat 22 265 5631 0.02 NO
Ribosomal Y1 rDNA repeat 113 9358 401890 1.08 NO
Other Elsa Transposon 17 5240 34287 0.09 YES Molly Transposon 40 1862 50453 0.14 YES Pixie Transposon 28 1845 39148 0.11 NO R10 Unknown 59 1241 44018 0.12 NO R25 Unknown 23 3320 44860 0.12 NO R31 Unknown 23 3031 40267 0.11 NO R37 Transposon or remnant 98 1603 104915 0.28 NO R38 Unknown 25 358 8556 0.02 NO R39 Unknown 29 2050 35758 0.10 NO R51 Unknown 39 833 25538 0.07 NO R8 Unknown 48 9143 275643 0.74 NO R9 Transposon or remnant 72 4108 163739 0.44 NO X0 Unknown 76 3862 145268 0.39 NO X11 Transposon or remnant 36 8555 128638 0.35 NO X12 Unknown 29 2263 25369 0.07 NO X23 Unknown 29 685 11679 0.03 NO X28 Unknown 30 1784 32002 0.09 NO X3 Unknown 213 9364 463438 1.24 NO X36 Unknown 10 512 5067 0.01 NO X96 Unknown 14 308 4321 0.01 NO SUM 4.52
a Evidence from the alignment that the repeat has been subjected to RIP mutation.

Chapter 4 mtDNA of P. nodorum
89
Table 3. Comparison of GO classification of unigene and transcript numbers between the planta and oleate library A. Biological Processes GO identifier Description Loci in planta oleate p-value1 Up-regulated in planta GO:0006412 Protein biosynthesis 128 710 325 5.97E-19 GO:0045493 Xylan catabolism 24 31 0 6.16E-09 GO:0006508 Proteolysis 78 110 39 8.29E-07 GO:0008643 Carbohydrate transport 58 22 0 1.26E-06 GO:0000272 Polysaccharide catabolism 12 24 1 4.29E-06 GO:0016068 Type I hypersensitivity 13 87 31 9.67E-06 GO:0030245 Cellulose catabolism 16 18 0 1.33E-05 GO:0005975 Carbohydrate metabolism 102 53 16 6.57E-05 GO:0042732 D-xylose metabolism 2 25 4 2.01E-04 GO:0009051 Pentose-phosphate shunt, oxidative branch 2 21 3 4.07E-04 Up-regulated oleate GO:0007582 Physiological process 24 8 48 1.04E-10 GO:0006629 Lipid metabolism 20 6 40 1.42E-09 GO:0006096 Glycolysis 21 42 92 5.93E-09 GO:0006108 Malate metabolism 4 12 45 5.48E-08 GO:0006099 Tricarboxylic acid cycle 20 50 85 4.11E-06 GO:0015986 ATP synthesis coupled proton transport 26 34 65 6.56E-06 GO:0006183 GTP biosynthesis 1 0 13 1.54E-05 GO:0006228 UTP biosynthesis 1 0 13 1.54E-05 GO:0006241 CTP biosynthesis 1 0 13 1.54E-05 GO:0006334 Nucleosome assembly 12 65 95 3.21E-05
B. Cellular Components GO identifier Description Loci in planta oleate p-value1 Up-regulated in planta GO:0005840 Ribosome 89 567 262 3.14E-15 GO:0015935 Small ribosomal subunit 11 93 33 4.86E-06 GO:0005576 Extracellular region 36 23 1 7.44E-06 GO:0005730 Nucleolus 6 23 2 4.15E-05 GO:0030529 Ribonucleoprotein complex 25 60 19 4.31E-05 GO:0030125 Clathrin vesicle coat 9 7 0 8.86E-03 GO:0016020 Membrane 338 127 122 1.07E-02 GO:0016021 Integral to membrane 401 243 213 1.38E-02 GO:0019867 Outer membrane 7 45 24 1.40E-02 GO:0005874 Microtubule 19 8 1 1.97E-02 Up-regulated oleate GO:0005829 Cytosol 34 19 50 1.01E-06 GO:0016469 Proton-transporting two-sector atpase complex 24 27 60 1.41E-06 GO:0000786 Nucleosome 11 65 95 3.21E-05 GO:0043234 Protein complex 33 15 29 1.23E-03 GO:0005739 Mitochondrion 102 138 147 1.62E-03 GO:0005634 Nucleus 288 183 186 1.90E-03 GO:0005777 Peroxisome 14 26 38 3.39E-03 GO:0005746 Mitochondrial electron transport chain 8 36 47 4.40E-03 GO:0045261 Proton-transporting ATP synthase complex, catalytic core F(1) 5 14 23 7.49E-03 GO:0005778 Peroxisomal membrane 3 1 7 8.63E-03

Chapter 4 mtDNA of P. nodorum
90
Table. 3 (continued) C. Molecular Functions GO Identifier Description Loci in planta oleate p-value1 Up-regulated in planta GO:0003735 Structural constituent of ribosome 111 709 328 2.09E-18 GO:0004553 Hydrolase activity, hydrolyzing O-glycosyl compounds 83 55 5 4.13E-10 GO:0004252 Serine-type endopeptidase activity 14 43 3 6.92E-09 GO:0005351 Sugar porter activity 64 22 0 1.26E-06 GO:0046556 Alpha-N-arabinofuranosidase activity 6 19 0 7.39E-06 GO:0030248 Cellulose binding 19 18 0 1.33E-05 GO:0004029 Aldehyde dehydrogenase (NAD) activity 1 15 0 7.85E-05 GO:0050661 NADP binding 6 23 3 1.60E-04 GO:0004185 serine carboxypeptidase activity 8 13 0 2.56E-04 GO:0004616 phosphogluconate dehydrogenase (decarboxylating) activity 4 22 3 2.56E-04 Up-regulated oleate GO:0004459 L-lactate dehydrogenase activity 2 5 44 2.07E-11 GO:0030060 L-malate dehydrogenase activity 2 5 44 2.07E-11 GO:0005498 Sterol carrier activity 3 7 46 1.02E-10 GO:0008415 Acyltransferase activity 17 4 34 4.64E-09 GO:0005506 Iron ion binding 91 89 128 3.72E-06 GO:0004550 Nucleoside diphosphate kinase activity 1 0 13 1.54E-05 GO:0005554 Molecular function unknown 49 40 65 7.95E-05 GO:0046933 Hydrogen-transporting ATP synthase activity, rotational mechanism 24 34 58 8.73E-05 GO:0046961 Hydrogen-transporting atpase activity, rotational mechanism 24 34 58 8.73E-05 GO:0050660 FAD binding 27 13 30 2.85E-04
1 Probability of significant difference between the libraries (Audic and Claverie 1997).
Table 4. Comparison of tRNA gene clusters flanking the rnl gene of the mitochondrial genome in several ascomycetes Organism 5-'upstream regionb rnl 3-'downstream regionb Accession number P. marneffei RKG1G2DS1WIS2P rnl TEVM1M2L1AFL2QM3H AY347307 A. niger KGDS1WIS2P rnl TEVM1M2L1AFL2QM3H DQ217399 M. graminicola GDS1WIS2P rnl M1L1EAFL2YQM2HRM3 EU090238 S. nodorum VKGDS1WIRS2P rnl T M1M2EAFLQHM3 EU053989 E. floccosum KGDSIWSP rnl TEVM1M2L1AFL2QM3H AY916130 H. jecorina ISWP rnl TEM1M2L1AFKL2QHM3 AF447590 P. anserina ISP rnl TEIM1L1AFL2QHM2 X55026
a The tRNA gene order of listed organisms is based on Genbank sequences b Capital letters refer to tRNA genes for: R=arginine, K=lysine, G=glycine, D=aspartic acid, S=serine,
W=tryptophan, I=isoleucine, P=proline, T=threonine, E=glutamic acid, V=valine, L=leucine, A=alanine, F=phenylalanine, Q=glutamine, H=histidine, Y=tyrosine.
1,2,3 The numbers indicate the presence of more tRNA genes for the same amino acid in the consensus sequence.

Chapter 4 mtDNA of P. nodorum
91
Gene expression during infection. Two EST libraries were constructed and analyzed as
part of this project. An in vitro library was constructed from axenic fungal mycelium
transferred to media with oleate as the sole carbon source; this is referred to as the oleate
library. An in planta library was made from bulked sporulating disease lesions on wheat 9, 10
and 11 days after infection (DAI). For both libraries 5,000 random clones were sequenced in
both directions. The oleate library was entirely fungal and hence particularly suited for
primary genome annotation purposes. The lipid growth media was chosen to replicate the
early stages of infection in which lipolysis, the glyoxalate cycle and gluconeogenesis are
thought to be critical metabolic requirements (Solomon et al. 2004a). The in planta library
was designed to reveal both plant and fungal genes expressed at a late stage of infection.
After trimming and removal of plant ESTs and alignment to the genome assembly, the in
planta library formed 1448 and the oleate library formed 1231 unigenes (See Supplement
Data Set 2 online). Only 427 of the unigenes were found in both libraries. Although this
represents 19% of the unigenes, it encompasses 46% of the ESTs, showing the genes
expressed uniquely in one library are of relatively low expression.
The unigenes obtained from in planta and oleate libraries were classified according to the
gene ontology (GO) categories. GO matches were obtained for 851 (59%) of the unigenes
found in the in planta and 736 (59%) of those found in the oleate library. Relative numbers of
ESTs and genes in different GO classes were compared (Table 3). GO categories were ranked
by statistical discrimination between the two libraries and the top 10 are shown for each of
biological process, cellular component and molecular function.
Coexpression of fungal gene clusters responsible for the synthesis of secondary
metabolites (Bok and Keller 2004) and pathogenicity (Kamper et al. 2006) has been
observed. We compared the number of ESTs found in the in planta and oleate libraries to
search for clusters of coexpressed genes. Using a window of 10 contiguous genes, we
scanned for regions with biased ratios of ESTs from either library. One putatively in planta-
induced region stood out. These six genes SNOG_16151.2 to SNOG_16157.2 were
exclusively in the in planta library with 4, 20, 0, 4 10 and 1 EST clones respectively. The
genes have best hits to a major falcilitatore superfamily transporter, a phytanoyl-CoA
dioxygenase, a CocE/NonD hydrolase, a salicylate hydroxylase and are next to a transcription
factor. Such a cluster may be involved in the degradation of phytols, phenylpropanoids and

Chapter 4 mtDNA of P. nodorum
92
catechols either for nutritional purposes providing trichloroacetic acid intermediates via the
beta-ketoadipate pathway or to detoxify wheat defence compound(s). It is intriguing that
overexpression of the thiolase in the beta-ketoadipate pathway in L. maculans markedly
reduced pathogenicity (Elliott and Howlett 2006). Experiments to test these ideas in S.
nodorum are in progress.
Discussion
As estimated genome size of S. nodorum is 37.2 Mbp, which is significantly larger than
the 28 to 32 Mbp previously estimated by pulsed-field gel analysis (Cooley and Caten 1991).
Electrophoretic karyotypes have proved to be unreliable estimators of total genome size in
many fungal species (Orbach et al. 1988 and 1989; Talbot et al. 1993). In the case of S.
nodorum, this discrepancy may be a consequence of comigration of chromosomal bands on
pulsed-field gels leading to an underestimation of genome size (Cooley and Caten 1991). The
electrophoretic karyotype was found to be highly variable between strains, even when
isolated from a single ascus, consistent with the generalisation that many fungal species have
plastic genome structures (Zolan 1995). The revised genome size estimate is comparable to
other sequenced filamentous fungi such as the rice pathogen Magnaporthe grisea, which is
currently estimated to be 41.6 Mbp and the non-pathogen Neurospora crassa now estimated
at 39.2 Mbp.
Phylogenetic analysis. The genome sequence has confirmed the phylogenetic placement
of S. nodorum in the class Dothideomycetes. Along with the Eurotiomycetes (containing
Aspergillus, and human pathogens such as Histoplasma), Lecanoromycetes (the majority of
the lichenized species), Leotiomycetes (containing numerous endophytes and the plant
pathogen Sclerotinia) and Sordariomycetes (with Neurospora, Magnaporthe and
Colletotrichum spp.) the Dothideomycetes is now recognised as a major clade of the
filamentous Ascomycota (James et al. 2006). Phylogenetic analyses of full fungal genomes
and large-scale taxon sampling agree with the placement of S. nodorum and point to the rapid
divergence of these major classes of ascomycetes (Robbertse et al. 2006; Spatafora et al.
2006). The tree in Fig. 2 is a focused sampling of the largest classes of the Ascomycota with
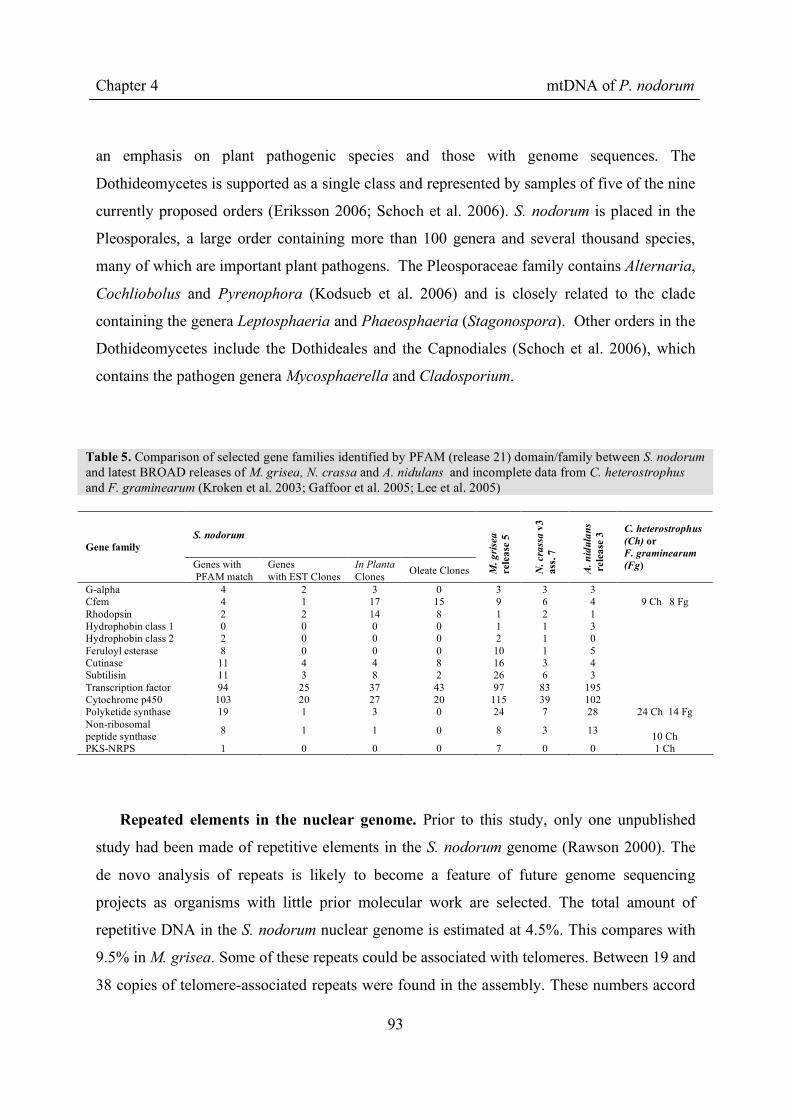
Chapter 4 mtDNA of P. nodorum
93
an emphasis on plant pathogenic species and those with genome sequences. The
Dothideomycetes is supported as a single class and represented by samples of five of the nine
currently proposed orders (Eriksson 2006; Schoch et al. 2006). S. nodorum is placed in the
Pleosporales, a large order containing more than 100 genera and several thousand species,
many of which are important plant pathogens. The Pleosporaceae family contains Alternaria,
Cochliobolus and Pyrenophora (Kodsueb et al. 2006) and is closely related to the clade
containing the genera Leptosphaeria and Phaeosphaeria (Stagonospora). Other orders in the
Dothideomycetes include the Dothideales and the Capnodiales (Schoch et al. 2006), which
contains the pathogen genera Mycosphaerella and Cladosporium.
Table 5. Comparison of selected gene families identified by PFAM (release 21) domain/family between S. nodorum and latest BROAD releases of M. grisea, N. crassa and A. nidulans and incomplete data from C. heterostrophus and F. graminearum (Kroken et al. 2003; Gaffoor et al. 2005; Lee et al. 2005)
S. nodorum Gene family
Genes with PFAM match
Genes with EST Clones
In Planta Clones Oleate Clones M
. gris
ea
rele
ase
5
N. c
rass
a v3
as
s. 7
A. n
idul
ans
rel
ease
3 C. heterostrophus
(Ch) or F. graminearum (Fg)
G-alpha 4 2 3 0 3 3 3 Cfem 4 1 17 15 9 6 4 9 Ch 8 Fg Rhodopsin 2 2 14 8 1 2 1 Hydrophobin class 1 0 0 0 0 1 1 3 Hydrophobin class 2 2 0 0 0 2 1 0 Feruloyl esterase 8 0 0 0 10 1 5 Cutinase 11 4 4 8 16 3 4 Subtilisin 11 3 8 2 26 6 3 Transcription factor 94 25 37 43 97 83 195 Cytochrome p450 103 20 27 20 115 39 102 Polyketide synthase 19 1 3 0 24 7 28 24 Ch 14 Fg Non-ribosomal peptide synthase 8 1 1 0 8 3 13 10 Ch PKS-NRPS 1 0 0 0 7 0 0 1 Ch
Repeated elements in the nuclear genome. Prior to this study, only one unpublished
study had been made of repetitive elements in the S. nodorum genome (Rawson 2000). The
de novo analysis of repeats is likely to become a feature of future genome sequencing
projects as organisms with little prior molecular work are selected. The total amount of
repetitive DNA in the S. nodorum nuclear genome is estimated at 4.5%. This compares with
9.5% in M. grisea. Some of these repeats could be associated with telomeres. Between 19 and
38 copies of telomere-associated repeats were found in the assembly. These numbers accord

Chapter 4 mtDNA of P. nodorum
94
well with the 14 to 19 chromosomes visualized by pulsed-field electrophoresis (Cooley and
Caten 1991). Studies of S. nodorum life cycle have indicated that it undergoes regular sexual
crossing, particularly in areas with Mediterranean-style climates with the associated need to
over-summer as ascospores (Bathgate and Loughman 2001; Stukenbrock et al. 2006). The
relatively low content of repetitive DNA is consistent with this observation. The prevalence
of clear cases of RIP further confirms that meiosis is a frequent event. RIP has been found to
varying degrees in other fungal genomes. It is notable that the closely related L. maculans has
previously been shown experimentally to exhibit RIP (Idnurm and Howlett 2003).
Mitochondrial Genome. A distinctive feature of fungal mitochondrial genomes is the
clustering of tRNA genes (Ghikas et al. 2006) and it is thought that both the tRNA gene
content and their placement will be conserved in fungi (Table 4). The 27 tRNA genes
clustered into five groups, with the two larger tRNA gene clusters flanking rnl, a pattern
common to other fungal mtDNAs (Tambor et al. 2006). The tRNA gene cluster 5’ to rnl had
GDS1WIS2P as a consensus with, its closest sequenced relative M. graminicola (Genbank
accession number EU090238), while the 3’-downstream consensus was EAFLQHM having
many tRNA genes in the same order found in other Ascomycetes. Variation in the order of
tRNAs, such as inversions in Epidermophyton floccosum (tRNA-Ile, tRNA-Tpr) or in
Podospora anserina and S. nodorum (tRNA-Met, tRNA-His), suggest that rearrangements
are common in fungal mtDNAs (Table 4).
Kouvelis et al. (2004) argued that gene pairs nad2-nad3, nad1-nad4, atp6-atp8, and cytb-
cox1 would remain joined in ascomycetes with some possible exceptions, as already detected
in M. graminicola that present only two of these gene pairs coupled. In S. nodorum, the atp8-
9 genes were not present and cytb-cox1 were uncoupled and, interestingly, the nad1 and nad4
genes were on sections of the mtDNA in an inverted orientation. Two orfs (orf1 and orf2) had
homologous sequences in the in planta EST library. While the M. graminicola mitochondrial
genome had almost no introns, four intron-encoded genes were found in S. nodorum, with
high homology to LAGLIDADG type endonucleases or GIY-YIG type nucleases (See
Supplemental Table 1 online). Intron-encoded proteins have also been reported in P. anserina
(Cummings et al. 1990) and Penicillium marneffei (Woo et al. 2003).
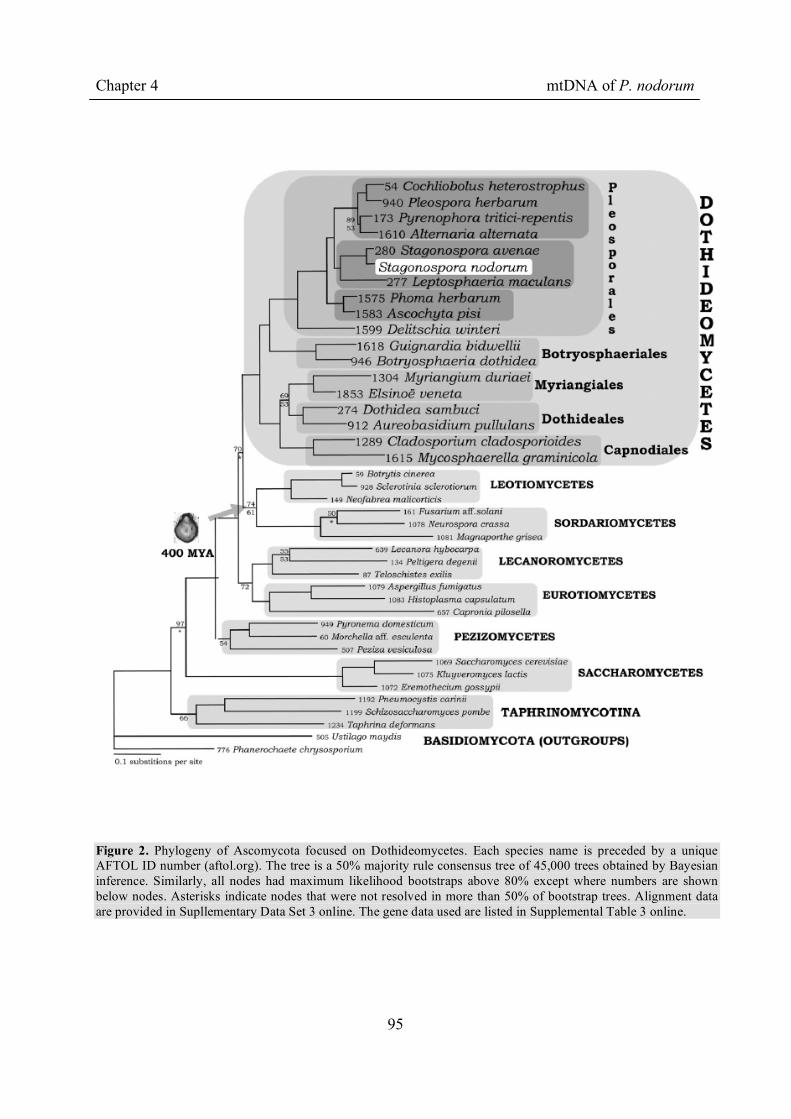
Chapter 4 mtDNA of P. nodorum
95
Figure 2. Phylogeny of Ascomycota focused on Dothideomycetes. Each species name is preceded by a unique AFTOL ID number (aftol.org). The tree is a 50% majority rule consensus tree of 45,000 trees obtained by Bayesian inference. Similarly, all nodes had maximum likelihood bootstraps above 80% except where numbers are shown below nodes. Asterisks indicate nodes that were not resolved in more than 50% of bootstrap trees. Alignment data are provided in Supllementary Data Set 3 online. The gene data used are listed in Supplemental Table 3 online.

Chapter 4 mtDNA of P. nodorum
96
Table 6. Potential orthologs to S. nodorum non-ribosomal peptide synthase genes
a Domain abbreviations; Adenylation A; Condensation C; Cyclisation Cy; Epimerisation E; Thiolation T; Thioesterase Te . Potential orthologs identified by best blastp hits to NRPS genes in C. heterostrophus (Lee et al. 2005) and by best informative hit to NR at NCBI of e-value < 1x10-10.
b The best hit of the identified gene in the S. nodorum gene set was observed reciprocally. c Percent similarity of ortholog pairs (Needleman and Wunsch 1970) via NEEDLE (Rice et al. 2000). d Domain structure and modular organisation for all sequences was determined via the online NRPS-PKS database (Ansari
et al. 2004). It should be noted that domain structure predictions may vary slightly from those stated in the original studies.
e Protein sequence for AbNPS2 currently available in supplementary appendix S2 (Kim et al. 2007).
Functional Analysis of Proteins. Our primary goal in obtaining the genome sequence
was to reveal genes likely to be involved in pathogenicity. Whilst some genes seem to be
specifically associated with pathogenicity in a single organism, other genes and gene families
have been generally associated with pathogenicity albeit they are also found in non-
pathogens (see http://www.phi-base.org/about.php; Baldwin et al. 2006). Some of the generic
functions are listed (Table 5; see Supplemental Data Set 2 and Supplemental Table 2 online).

Chapter 4 mtDNA of P. nodorum
97
EST support was found for 59 of the genes, with no statistically significant difference in the
numbers of ESTs in the in planta and oleate libraries.
Non-ribosomal peptide synthetases (NRPS) are modular genes that control the synthesis
of a diverse set of secondary metabolites including the dothideomycete host-specific toxins
AM-toxin, HC-toxin and victorin from Alternaria and Cochliobolus species (Wolpert et al.
2002). NRPS genes from C. heterostrophus have been analyzed in detail (Lee et al. 2005).
Comparison of both protein sequences and domain structure of the C. heterostrophus NRPS
genes (NPS1-11) with the eight identified in S. nodorum was undertaken (Table 6). Putative
orthology was determined by identifying reciprocal best hits amongst the gene sets. Three
pairs were identified. SNOG_02134.2 was linked to NPS2 and both appear to be orthologous
to the A. nidulans gene SidC (Eisendle et al. 2003), which is responsible for the synthesis of
ferricrocin, an intracellular iron storage and transport compound involved in protection
against iron toxicity (Eisendle et al. 2006). It is likely that SNOG_02134.2 and NPS2 have
similar roles. SNOG_14638.2 was linked to NPS6, a ubiquitous gene with a related role in
siderophore-mediated iron uptake and oxidative stress protection (Oide et al. 2006). Third,
SNOG_14834.2 appears to be directly related to NPS4, Psy1 from Alternaria brassicae
(Guillemette et al. 2004) and NPS2 from A. brassicicola (Kim et al. 2007). Ab NPS2 mutants
are reduced in virulence and the gene is predicted to encode a component of the conidial wall.
It is likely that SNOG_14834.2 will have a similar generic role. Reciprocal best-hit
relationships were observed between a further two SNOG NRPS genes and other fungal
genes; SNOG_09081.2 was related to PesA (Bailey et al. 1996) and SNOG_14908.2 to the
Salps2 gene from Hypocrea lixii (Vizcaino et al. 2006). These genes plus SNOG_14834.2,
SNOG_09488.2 and SNOG_01105.2 are all closely related to NPS4, suggesting this five-
gene subfamily is expanded in S. nodorum. Interestingly, a further seven NRPS genes from
C. heterostrophus (NPS3, 5, 8, 10, 11 and 12) have no obvious ortholog in S. nodorum,
suggesting expansion of this subfamily in the maize pathogen (Yoder and Turgeon 2001).
Intensive studies in C. heterostrophus showed that individual gene knockouts produced
altered phenotypes only for NPS6 (Lee et al. 2005) indicating redundancy of function. The
smaller complement of NRPS genes in M. grisea (eight) and S. nodorum indicate that these
organisms may be more fruitful models to study NRPS function. Furthermore, although
toxins have been implicated in the virulence of both pathogens, nonribosomal synthetases

Chapter 4 mtDNA of P. nodorum
98
cannot be expected to be a major source. No ESTs were found corresponding to these genes.
This may be due to their large size or to a generally low level of expression.Polyketide
synthases (PKS) are a second modular gene family strongly associated with pathogenicity
(Kroken et al. 2003; Gaffoor et al. 2005) being responsible for the production of T-toxin from
C. heterostrophus and PM-toxin from Mycosphaerella zeae-maydis (Yun et al. 1998; Baker
et al. 2006). S. nodorum is predicted to contain 19 PKS genes compared to the 24, 14 and 24
in the pathogens M. grisea, Fusarium graminearum and C. heterostrophus respectively, and
28 in the saprobe A. nidulans, but significantly more than the seven in N. crassa (Table 5).
Figure 3. Homology relationship of the S. nodorum secretome. The 1231 predicted extracellular proteins without GO annotation were compared with the latest releases of the U. maydis, N. crassa and M. grisea genomes. Counts are best hits of e-value < 1e-10 if predicted as extracellular by WolF PSORT.
Orthology relationships between the well-studied F. graminearum and C. heterostrophus
gene sets and S. nodorum are shown in Table 7. Reciprocal best Blast hits were observed
with eight genes from C. heterostrophus and five from F. graminearum. This close
relationship, particularly with C. heterostrophus, reflects the close phylogenetic relationship
and suggests an ancient origin for the majority of the PKS paralogs (Kroken et al. 2003).

Chapter 4 mtDNA of P. nodorum
99
SNOG_11981.2 is supported by five ESTs from the in planta library and none from the oleate
library, consistent with upregulation during infection and sporulation (no other PKSs have
EST support). This gene is orthologous to non-reducing clade 2 PKS genes that are
associated with 1,8-dihydroxynaphthalene melanin biosynthesis in Bipolaris oryzae and
many other pathogens (Kroken et al. 2003; Moriwaki et al. 2004; Amnuaykanjanasin et al.
2005). We showed previously that S. nodorum produces melanin from
dihydroxyphenlyalanine (Solomon et al. 2004b) for which a PKS would not be necessary.
This finding suggests that either S. nodorum may produce 1,8-dihydroxynaphthalene melanin
in addition to dihydroxyphenlyalanine melanin or that SNOG_11981.2 plays another role.
A single PKS-NRPS hybrid protein (SNOG_00308.2.) was identified in the genome of S.
nodorum as was found for F. graminearum (Gaffoor et al. 2005) and C. heterostrophus (Lee
et al. 2005). C. heterostrophus NPS7 and SNOG_00308.2 do not appear to be closely related
and it is likely that they evolved independently. In contrast, the hybrid protein of F.
graminearum (GzFUS1/FG12100) is closely related to SNOG_00308.2. Overall they are
54.5% similar and apart from an acyl binding domain found only in SNOG_00308.2 the
domain structures are identical. Gz FUS1 produces fusarin C, a mycotoxin (Gaffoor et al.
2005). The best hit (with 59.4% similarity) to SNOG_00308.2 in M. grisea is Ace1, a hybrid
PKS/NRPS that confers avirulence to M. grisea during rice (Oryza sativa) infection (Bohnert
et al. 2004). It will be intriguing to determine if the product of SNOG_00308.2 is required for
pathogenicity.
G-alpha proteins have been extensively studied in fungi (Lafon et al. 2006) and many are
required for pathogenicity. Distinct roles for three g-alpha genes have been revealed in M.
grisea, A. nidulans and N. crassa. It was therefore a surprise to find a fourth g-alpha gene in
the S. nodorum genome. This gene (SNOG_06158.2) was shown to be expressed in vitro and
in planta. It was investigated by gene disruption and its loss resulted in no discernable
phenotype (data not shown). A fourth g-alpha protein has been recently identified in both A.
oryzae (Lafon et al. 2006) and Ustilago maydis (Kamper et al. 2006), but neither shows
significant sequence similarity to that of S. nodorum. Indeed SNOG_06158.2 is most similar
to Gba3 amongst the U. maydis g-alphas and GpaB amongst the A. oryzae genes.

Chapter 4 mtDNA of P. nodorum
100
SN15 PKS
Domain/Module
Structure Best Hit Accession Organism Reciprocal SimilarityInferred Clade
Domain/Module
StructureInferred Function Ortholog
fg12100 F. graminearum No 21.60% KsAtAcp/KrAcp/Acp
PKS25 AAR90279 C. heterostrophus Yes 59.70% KsAtKrAcp
atX BAA20102 A. terreus Yes 69.00% KsAtKrAcp
F. graminearum No 42.10% KsAtErKrAcp
PKS3 AAR90258 C. heterostrophus Yes 45.80% KsAtErKrAcp
mlcB BAC20566 P. citrinum Yes 47.70% KsAtDhErKrAcp
F. graminearum Yes 38.90% KsAtAcp/ErKrAcp
PKS6 AAR90261 C. heterostrophus No 46.50% KsAtErKr
typeI PKS AAO62426 Phoma sp. C2932 No 49.80% KsAtErKrAcp
F. graminearum Yes 43.10% KsAtErKrAcp
PKS5 AAR90261 C. heterostrophus Yes 73.10% KsAtErKr
atl5 BAD83684 A. solani Yes 79.20% KsAtErKrAcp
F. graminearum No 31.30% KsAtErKrAcp
PKS14 AAR92221 G. moniliformis No 31.80% KsAtErKr
FUM1 AAD43562 G. moniliformis No 30.00% KsAtErKrAcp
F. graminearum No 36.20% KsAtAcp
PKS17 AAR90253 B. fuckeliana Yes 43.90% KsAtAcp
pksCT BAD44749 M. purpureus Yes 44.70% KsAtAcp
F. graminearum No 37.80% KsAtAcp/KrAcp/Acp
PKS16 AAR90270 C. heterostrophus Yes 67.90% KsAtKr
EqiS AAV66106 F. heterosporum Yes 39.80% KsAtKrAcp/Acp
F. graminearum No 40.30% KsAtAcp
PKS12 AAR90248 B. fuckeliana No 44.50% KsAtAcp/Acp
PKS AAS48892 N. haematococca No 47.80% KsAtAcp/Acp
F. graminearum Yes 41.20% KsAtAcp/Acp
PKS3 AAR92210 G. moniliformis No 49.30% KsAtAcp/Acp
PKS AAT69682 C. nicotianae No 60.70% KsAtAcp/Acp
F. graminearum No 58.90% KsAtErKrAcp
PKS11 AAR90266 C. heterostrophus Yes 56.10% KsAtErKrAcp
FUM1 AAD43562 G. moniliformis Yes 57.70% KsAtErKrAcp
F. graminearum No 43.20% KsAtErKrAcp
PKS8 AAR90244 B. fuckeliana Yes 59.70% KsAtErKrAcp
PKS2 ABB76806 C. heterostrophus Yes 44.90% KsAtErKrAcp
F. graminearum Yes 62.80% KsAtErKrAcp
PKS14 AAR90268 C. heterostrophus Yes 82.70% KsAtDHErKrAcp
FUM1 AAD43562 G. moniliformis No 53.90% KsAtErKrAcp
F. graminearum No 44.70% KsAtErKrAcp
PKS14 AAR92221 G. moniliformis Yes 48.70% KsAtErKr
PKSKA1 AAY32931 Xylaria sp. BCC 1067 No 47.30% KsAtErKrAcp
F. graminearum Yes 54.90% KsAtAcp
PKS18 AAR90272 C. heterostrophus Yes 85.70% KsAtAcp/Acp/Te
PKS1 BAD22832 B. oryzae Yes 88.20% KsAtAcp/Acp/Te
F. graminearum No 39.70% KsAtErKrAcp
PKS5 AAR92212 G. moniliformis No 38.20% KsAtAcp/ErKrAcp
alt5 BAD83684 A. solani No 37.80% KsAtErKrAcp
F. graminearum No 40.40% KsAtErKrAcp
PKS12 AAR92219 G. moniliformis No 45.10% KsAtAcp/Acp/ErKrAcp
FUM1 AAD43562 G. moniliformis No 39.10% KsAtErKrAcp
F. graminearum No 44.30% KsAtErKrAcp
PKS15 AAR90269 C. heterostrophus No 43.50% KsAtAcp/ErKrAcp
PKS BAC20566 P. citrinum No 39.10% KsAtDhErKrAcp
F. graminearum No 42.20% KsAtAcp
PKS1 BAD22832 B. oryzae No 41.30% KsAtAcp/Acp/Te
PKS14 AAR90250 B. fuckeliana No 67.60% KsAtAcp/Acp
F. graminearum No 43.90% KsAtErKrAcp
PKS10 AAR90246 B. fuckeliana Yes 68.80% KsAtErKrAcp
PKSKA1 AAY32931 Xylaria sp. BCC 1067 No 54.70% KsAtErKrAcp
Table 7. Potential Orthologs of S. nodorum PKS Genes
fungal 6MSAS
Synthesis melanin, autofusarin or
similar polyketide
SNOG_15965.2 KsAtErKrAcp
fg10548reduc PKS
clade IV Synthesis fumonisin
SNOG_15829.2 KsAtAcp
fg12040
non-reduc PKS
Synthesis fumonisin
SNOG_14927.2 KsAtDhKrAcp
fg01790reduc PKS
clade IV
Synthesis diketide moiety
compactin
SNOG_13032.2 KsAtAcpKr
fg01790reduc PKS
clade IV
Synthesis melanin
SNOG_12897.2 KsAtErKr
fg10548reduc PKS
clade I Synthesis alternapyrone
SNOG_11981.2 KsAtAcp/Acp/Acp/Te
fg12040non-reduc PKS
clade II
Synthesis fumonisin
SNOG_11272.2 KsKsAtErKrAcp
fg12055reduc PKS
clade IV
Synthesis zearalenone or similar
polyketide. Similar to HR type
PKS
SNOG_11076.2 KsAtDhErKrAcp
fg01790reduc PKS
clade IV
Synthesis fumonisin
SNOG_11066.2 KsKsAtErKrAcp
fg12055reduc PKS
clade III
High virulence; synthesis T-toxin;
zearalenone
SNOG_09623.2 KsAtAcp/ErKrAcp
fg01790reduc PKS
clade IV
Synthesis perithecial pigment,
autofusarin or similar polyketide
SNOG_08614.2 KsAtAcp/Acp/Te
fg12125non-reduc PKS
clade I
Synthesis perithecial pigment,
cercosporin or similar polyketide.
SNOG_08274.2 KsAcp/AtAcp/Acp
fg12040non-reduc PKS
clade II
Synthesis citrinin
SNOG_07866.2 KsAtKr
fg12100reduc PKS
clade II
Synthesis polyketide similar to
citrinin/lovastatin.
SNOG_06682.1 KsAtAcp
fg03964non-reduc PKS
clade III
Synthesis alternapyrone
SNOG_06676.2 KsAtErKr
fg01790reduc PKS
clade IV Synthesis fumonisin
SNOG_05791.2 KsAtErKrAcp
fg12109reduc PKS
clade I
Synthesis diketide moiety
compactin or similar polyketide.
SNOG_04868.2 KsAtErKrAcp
fg05794reduc PKS
clade I Synthesis squalestatin
SNOG_02561.2 KsAtErKrAcp
fg12109reduc PKS
clade I
SNOG_00477.2 KsAtKrAcp 6-methylsalicylic acid synthesis
Domain abbreviations; Acyl-carrier protein domain ACP; Acetyl transferase At; Dehytratase Dh; Enoyl Reductase Er; Keto Reductase Kr;
Keto Synthase Ks; Thioesterase Te. Potential orthologs to polyketide synthases C. heterostrophus and F. graminearum (Kroken et al.,
2003; Gaffoor et al., 2005) and by best informative hit to NR at NCBI of e-value < 1x10-10. Domain structure and modular organisation for
all sequences was determined via the online NRPS-PKS database (Ansari et al., 2004). It should be noted that domain structure predictions
may vary slightly from those stated in the original studies. Percent similarity of ortholog pairs was determined (Needleman and Wunsch,
1970) via NEEDLE (Rice et al., 2000). References for ortholog function: 6-methylsalicylic acid synthesis (Fujii et al. 1996); synthesis
diketide moiety compactin or similar polyketide (Abe et al. 2002a and 2002b); synthesis diketide (Abe et al. 2002a and 2002b); synthesis
squalestatin (Nicholson et al. 2001); synthesis alternapyrone (Fujii et al. 2005); synthesis fumonisin (Schimlzu et al. 2005; Proctor et al.
1999); Synthesis citrinin (Schimlzu et al. 2005); synthesis perithecial pigment, autofusarin or similar polyketide (Graziani et al. 2004;
Chung et al. 2003); high virulence, synthesis T-toxin, zearalenone (Gaffoor et al. 2005; Baker et al. 2006); Synthesis melanin (Moriwaki et
al. 2004).

Chapter 4 mtDNA of P. nodorum
101
G-alpha proteins transduce extracellular signals leading to infection-specific development
(Solomon et al. 2004b). Pth11 and ACI1 are two M. grisea genes encoding transmembrane
receptors that defined a new protein domain, CFEM (Kulkarni et al. 2003) with roles in g-
protein signalling. M. grisea has nine CFEM domain proteins and F. graminearum has eight.
Using a combination of domain searches and blast searches seeded with the M. grisea and F.
graminearum genes, we identified a total of six related genes (Table 8). Four of these have at
least a weak match to the CFEM domain but only three are predicted to be transmembrane
proteins. SNOG_09610.2 appears to be the ortholog of Pth11 and the F. graminearum gene
fg05821, suggesting these genes are ancient and conserved. Pth11 is required for appressorial
development and perception of suitable surfaces (DeZwaan et al. 1999). Neither S. nodorum
nor F. graminearum form classical appressoria (Solomon et al. 2006b) suggesting that the
genes have different functions in these species. SNOG_05942.2 is also closely related to
Pth11. Knockout strains for this gene were obtained but had no obvious phenotype (data not
shown). SNOG_03589.2 is also related to Pth11, and appears to be a transmembrane protein
but lacks the CFEM domain. Both SNOG_08876.2 and SNOG_15007.2 possess CFEM-
domains and a signal peptide but do not appear to be integral membrane proteins. Knockout
strains for SNOG_08876.2 were obtained but revealed no obvious phenotype (data not
shown). SNOG_15007.2 is similar to glycosylphosphatidylinositol-anchored CFEM-
containing proteins from three other fungal species. It appears to be heavily expressed both in
the oleate and in planta EST libraries with 17 and 27 ESTs respectively. A clear role for this
gene is as yet unknown. CFEM-domain proteins are thought to be involved in surface signal
perception and three candidates for this role have been identified. The paucity of such genes
in S. nodorum and lack of phenotype associated with deletion of two of them suggests that
this function may be covered by a different class of transmembrane receptors.
Hydrophobins are small, secreted proteins with eight Cys residues in a conserved pattern
that coat the fungal mycelium and spore (Wessels 1994). Class 2 hydrophobins are restricted
to ascomycetes, whereas class 1 hydrophobins are also found in other fungal divisions
(Linder et al. 2005). Two class 2 and no class 1 hydrophobin genes were found in the S.
nodorum genome (See Supplemental Figure 1 online). This is the first example of an
ascomycete genome with only class 2 hydrophobin genes. It is interesting to note that
although a range of Neurospora species all contained a gene orthologous to the class 1

Chapter 4 mtDNA of P. nodorum
102
hydrophobin EAS gene from N. crassa, they were expressed significantly only in species that
produced aerial conidia (Winefield et al. 2007). S. nodorum produces pycnidiospores in a
gelatinous cirrhus adapted for rain-dispersal (Solomon et al. 2006b). It may be that the
absence of aerial mitosporulation negates the need for class 1 hydrophobins.
Two rhodopsin-like genes were found in the genome, one of which is similar to the
bacteriorhodopsin-like gene found in L. maculans that is a proton pump (Sumii et al. 2005).
The likely physiological roles of these genes are currently under investigation.
Table 8. Potential orthologs of S. nodorum CFEM proteins identified by matches to PFAM domain PF05730 and best hit to M. grisea and F. graminearum CFEM proteins
a In addition to HMMER matches to PFAM accession PF05730 (using gathering cutoffs), CFEM domain containing proteins from Magnaporthe grisea (BROAD release 5: MGG_01149.5, MGG_01872.5, MGG_05531.5, MGG_05871.5, MGG_06724.5, MGG_06755.5, MGG_07005.5, MGG_09570.5, MGG_10473.5) and Fusarium graminearum (FGDB: fg00588, fg02077, fg02155, fg02374, fg03897, fg05175, fg05821, fg08554) were used as seeds to identify putative CFEM proteins by their best blastp match with an e-value < 1x10-10 to S. nodorum. b,c Cellular location was determined with WoLF PSORT (Horton et al. 2007) and presence of transmembrane spanning regions (7tm)
were determined by consensus between TMHMM (Krogh et al., 2001), TMPRED (Hofmann and Stoffel, 1993) and Phobius (Kall et al., 2004). Best hits of Stagonospora CFEM proteins to the non-redundant protein database at NCBI was determined by ‘informative’ blastp hits of e-value < 10-10 (hits excluding seed and self hits, which are not hypothetical or unknown and preferentially yield useful functional information).

Chapter 4 mtDNA of P. nodorum
103
The interaction between a pathogen and its host is to a large extent orchestrated by the
proteins that are secreted or localized to the cell wall or cell membrane. Pathogens such as M.
grisea, which kill and degrade host tissues, have been shown to secrete large numbers of
degradative enzymes (Dean et al. 2005). More surprisingly, the biotrophic pathogen U.
maydis was also found to secrete many proteins; many of the genes are clustered,
coexpressed and required for normal pathogenesis but are of unknown molecular functions
(Kamper et al. 2006). We have therefore analyzed the putative proteome of S. nodorum for
potentially secreted proteins and compared them with these pathogens and the saprobe N.
crassa. A total of 1782 proteins were predicted to be extracellular based on predictions using
WoLFPSORT; a further 1760 were predicted to be plasma membrane locatede (See
Supplemental Data Set 2 online). GO annotations were assigned via Blast2GO for 551 of the
putatively extracellular proteins (Table 9). They are dominated by carbohydrate and protein
degradation enzymes as would be expected and which is consistent with the EST analysis
(see below). The role of many fungal extracellular proteins is currently unknown. Among the
S. nodorum predicted extracellular proteins, 1231 had no GO annotation. Of these, only 410
had significant matches to putatively extracellular proteins from U. maydis, M. grisea, or N.
crassa (Figure 3). More of these 410 genes had homologs in M. grisea (13 + 91 = 104) than
in N. crassa (3 + 39 = 42). As M. grisea and N. crassa are phylogenetically equidistant from
S. nodorum, this suggests both that the pathogens secrete more proteins than N. crassa and
that the genes are related. Expanding this generalization will require genome analysis of a
saprobic dothideomycete. A further 251 genes (89 + 162) had homologs in both M. grisea
and N. crassa. Another 118 S. nodorum putatively secreted genes of unknown function had
significant similarity to genes encoding the U. maydis secretome, including 13 that uniquely
hit the biotrophic basidiomycete. We found no obvious patterns of clustering of any of these
secreted genes. The most striking finding was that 821 genes had no significant similarity to
predicted extracellular proteins of any of these fully sequenced genomes. These large
numbers of putatively secreted proteins of mostly unknown functions, whose genes appear to
be rapidly evolving, points to a hitherto unsuspected complexity and subtlety in the
interaction between the pathogen and its environments.
One newly recognized aspect of S. nodorum is the production of secreted proteinaceous
toxins. SNOG_16571.2 encodes a host-specific protein toxin called ToxA that determines the

Chapter 4 mtDNA of P. nodorum
104
interaction with the dominant wheat disease susceptibility gene Tsn1 (Friesen et al. 2006; Liu
et al. 2006). Population genetic evidence suggests that this gene was interspecifically
transferred to the wheat tan spot pathogen, P. tritici-repentis, prior to 1941, thereby
converting a minor into a major pathogen. An RGD motif that is involved in import into
susceptible plant cells is required for activity (Meinhardt et al. 2002; Manning et al. 2004;
Manning and Ciuffetti 2005). Biochemical and genetic evidence suggests that S. nodorum
isolates contain several other proteinaceous toxins (Liu et al. 2004a and 2004b). ToxA is a
13.2-kD protein, and current research indicates that the other toxins are also small proteins.
Among the predicted extracellular proteins, 840 are smaller than 30 kD and 26 have an RGD
motif (see Supplemental Data Set 2 online). Analysis of these toxin candidates is underway.
Table 9. The most abundant gene ontologies associated with the predicted secretome

Chapter 4 mtDNA of P. nodorum
105
Gene Expression during Infection. When analyzed by biological process and molecular
function, the in planta EST library was dominated by genes involved in the biosynthesis of
proteins and in the degradation and use of extracellular proteins and carbohydrates,
specifically cellulose and arabino-xylan (Table 3; see Supplemental Table 5 online). The
secretion of proteases (Bindschedler et al. 2003), xylanases, and cellulases suggests that the
fungus catabolizes plant proteins and carbohydrates for energy in planta at this late stage of
infection. Studies in a range of pathogens suggest that early infection is characterized by the
catabolism of internal lipid stores and that mid stages are characterized by the use of external
sugars and amino acids (Solomon et al. 2003a). These studies of late infection suggest that
polymerized substrates are used after the more easily available substrates are exhausted and
provide a novel perspective to in planta nutrition. The abundance of transcripts linked to
protein synthesis indicates that this process is very active during this stage of infection and
may be related to the massive secretion of degradative enzymes and to the synthesis of
proteins needed for sporulation. A similar picture was observed when the genes were
analyzed by cellular component (Table 3) with gene products destined for the ribosome
dominating. Genes whose products are targeted to the nucleolus and cytoskeleton illustrate
the importance of nuclear division and cytokinesis at this stage of infection as many new cell
types are elaborated in the developing pycnidium. Thirty-six gene products targeted to the
extracellular region include a protease, an a-glucuronidase, and various glucanases consistent
with the picture of polymerized substrate breakdown.
Late-stage infection has rarely been examined in plant-fungal interactions, and it is
therefore noteworthy that upregulation of transcripts involved in protein synthesis was also
observed in late-stage infections of wheat by M. graminicola (Keon et al. 2005). The high
rate of protein synthesis may be required for pycnidial biogenesis and may also represent an
attractive target for fungicidal intervention.
The oleate EST library was dominated by genes involved in lipid and malate metabolism,
the trichloroacetic acid cycle, and electron transport. To determine whether the ratio of ESTs
in the in planta and oleate libraries was indicative of early- and late stage infection, transcript
levels from eight candidate genes were determined by a quantitative PCR. The genes were
chosen from those found exclusively in the in planta library. Transcripts were quantified in
cDNA pools made from RNA isolated from in vitro and in planta growth of SN15 at early

Chapter 4 mtDNA of P. nodorum
106
and late infection time points. The in vitro cultures sampled at 4 DAI did not contain any
pycnidia, whereas cultures sampled at 18 DAI were heavily sporulating with many pycnidia.
To isolate RNA from both early and late infections of wheat, an SN15 infection latent period
assay was used to provide in planta infection transcripts (Solomon et al. 2003b). Lesions
were excised after 8 and 12 DAI, representing early and late infection stages. The three genes
most upregulated during growth in planta (SNOG_00557.2, SNOG_03877.2, and
SNOG_16499.2) all had less than 20 in planta ESTs, indicating that the EST frequencies
were useful predictors of gene expression at these levels. SNOG_00557.2 was the gene with
the highest peak expression level in planta and the largest difference between in vitro and in
planta conditions. As the gene is predicted to be an arabinofuranosidase, this reaffirmed the
important role of carbohydrolases during colonization of the host tissue. Overall, the four
tissue types represented clear states of nonsporulation and sporulation during both in vitro
and in planta growth. All the genes were found to be expressed in the 12-DAI infected
sample, and this was the highest level observed in all but one case (see Supplemental Figure
2 online). On the other hand, moderate to high levels of gene expression were found in the in
vitro samples from six of the eight genes. Attempts to use axenic samples to mimic infection
has a long history (Coleman et al. 1997; Talbot et al. 1997; Solomon et al. 2003a; Trail et al.
2003; Thomma et al. 2006). Our data indicate that oleate feeding is not a significantly closer
model for infection than starvation has proved to be.
Genome Architecture. Colinearity of gene order is a notable feature of closely related
plant and animal genomes but is represented in fungi by a complex pattern of dispersed
colinearity, such as observed between Aspergillus spp (Galagan et al. 2005). This low degree
of organizational conservation can be attributed to the 200 million year separation of these
species and the rarity of meiotic events that would tend to maintain chromosomal integrity. S.
nodorum is the first dothideomycete to have its sequence publicly released; thus, the closest
species for which whole-genome comparisons could be made are the Aspergilli and N.
crassa, which are separated from S. nodorum by approximately 400 million years of
evolution. Thus far, we have been unable to detect significant regions of colinearity with
other sequenced genomes, but it is possible that more sensitive methods and the release of

Chapter 4 mtDNA of P. nodorum
107
more closely related genome sequences will succeed in revealing patterns of chromosomal
level evolution.
Figure 4. Collinearity between S. nodorum SN15 and L. maculans identified by tBLASTX. (A) Mating type locus. (B) A 38-kb region of L. maculans.. Colours indicate an orthologous relationship between genes, whereas black indicates no relationship. Numbers in red give coordinates on the S. nodorum scaffold.
At a smaller scale, the mating type loci of L. maculans, C. heterostrophus, A. alternata,
and M. graminicola have been previously analyzed (Cozijnsen and Howlett 2003). Apart
from Mat1-1 and orf1, the only shared genes were a DNA ligase gene found in L. maculans
and M. graminicola and Gap1 found in L. maculans and C. heterostrophus. The colinearity
between L. maculans and S. nodorum is more complete. Fig. 4A shows the relationship of
gene order and orientation between these sibling species. It is clear that gene order and
orientation are conserved, though the intergenic regions have no discernable relationships.
This confirms the close phylogenetic relationship between these species, with S. nodorum
closest to L. maculans and more distantly related to C. heterostrophus.
There is tantalizing evidence of residual colinearity of the genes present in a contiguous
38-kb region of L. maculans DNA (Idnurm et al. 2003). Six of the nine L. maculans genes

Chapter 4 mtDNA of P. nodorum
108
have orthologs within a 200-kb region of S. nodorum DNA. Gene orientation is retained, but
they are interspersed with about 60 other predicted genes (Fig. 4b).
Figure 5. Organization of quinate cluster from severeal sequenced fungal genomes. Numbers under the S.
nodorum genes refer to the SNOG identifiers. The S. nodorum genes are located on scaffold 19 and 12,
respectively

Chapter 4 mtDNA of P. nodorum
109
A different pattern of colinearity is apparent in the quinate gene cluster. The quinate
genes regulate the catabolism of quinate and have a wide distribution in filamentous fungi
(Giles et al. 1991). The cluster comprises seven coregulated genes. In some cases, portions of
the cluster are repeated at other genomic locations. In S. nodorum, the main cluster is located
on scaffold 19 with two genes on scaffold 12. Fig. 5 shows the relationship of gene content
and order in six fungal species. As the species for comparison span 400 million years of
evolution, it is apparent that clustering of these genes per se is highly conserved. However,
the gene order and orientation shows no conservation apart from the constant juxtaposition of
homologs of Qa-1S and Qa1-F and of Qa-X and Qa-2. Even in these cases, though, some are
5’ to 5’ and others are 3’ to 3’. It appears, therefore, that there must be selection for retention
of clustering of these seven genes, even if the exact orientation may not be so important. It
may be that such proximity clustering is important in that it enables gene regulation by
chromatin remodelling mediated by a gene such as LaeA (Bok and Keller 2004).
SNOG_11365.2 is an apparent ortholog of LaeA.
Fungi have a truly ancient history, many times longer than that of animals and flowering
plants (Padovan et al. 2005). The extent of genetic diversity is correspondingly large.
Phylogenetic analysis of fungi has been hampered by a paucity of reliable morphological
indicators. As a consequence, phylogenetic reconstruction of the fungi has been particularly
unstable until the widespread introduction of multigene-based DNA sequence comparisons.
This study confirms the overall monophyletic characters of the Dothideomycetes and the
Pleosporales taxa. These groups contain many thousands of species and are notable for their
content of major plant pathogens infecting many important plant families. The origins of this
class, likely more than 400 million years ago, is considerably older than their plant hosts. It is
particularly interesting to note that all the fungal plant pathogens that are well established as
host-specific toxin producers are in this class (Stagonospora, Cochliobolus, Pyrenophora, M.
zeae-maydis and Alternaria). Taken as a whole, the pathogens in this class are mainly
described as necrotrophic, while a few are debatably described as biotrophic (Venturia and C.
fulvum) or hemibiotrophic (L. maculans, M. graminicola, and M. fijiensis). None of the
species in this class are obligate pathogens, and none possess classical haustoria (Oliver and
Ipcho 2004); furthermore, many other species are nonpathogenic. The genome sequence of S.
nodorum represents an important point of comparison from which to derive hypotheses about

Chapter 4 mtDNA of P. nodorum
110
the genetic basis of pathogenicity in these organisms. Genome sequences of several of these
species are in progress (Goodwin 2004), including A. brassicicola, M. fijiensis, M.
graminicola, L. maculans, and P. tritici-repentis, or have been completed but not released (C.
heterostrophus; Catlett et al. 2003). Three of these species, S. nodorum, M. graminicola, and
P. tritici-repentis, are wheat pathogens. The possibility of multiple pairwise comparisons of
gene content between phylogenetically and ecologically close species promises to be a
powerful method to derive workably small lists of candidate effector genes controlling
pathogenicity, host specificity, and life cycle characters and thus provide ideas for the
generation of novel crop protection strategies. The genome sequence provides the tools for
global transcriptome analysis, thereby identifying the genes expressed during different phases
of infection. This study has also highlighted secreted proteins, which appear to be markedly
more numerous than in nonpathogens but which have predominately mysterious roles.
Finally, the genome sequence is an essential prerequisite for the critical analysis of
hypotheses of interspecific gene transfer. This has already been identified in pathogens in
general (Richards et al. 2006) and S. nodorum in particular (Friesen et al. 2006; Stukenbrock
and McDonald 2007) and may emerge as a major feature in the evolution of these organisms.

Chapter 4 mtDNA of P. nodorum
111
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. Aligned Kyte and Doolittle Hydrophobicity Plots.
Supplemental Figure 2. Quantitative PCR Analysis of Gene Expression from in Planta-
Associated Genes.
Supplemental Table 1. Mitochondrial Genes.
Supplemental Table 2. Summary of PFAM Domains Used for Identification.
Supplemental Table 3. List of Species Used in This Study.
Supplemental Table 4. PCR Primers for Abundant in Planta-Associated Genes.
Supplemental Table 5. EST Data for Abundant in Planta-Associated Genes.
Supplemental Data Set 1. Repetitive Elements: Telomeres, Subrepeats, and Similarity
Scores.
Supplemental Data Set 2. Gene Summary.
Supplemental Data Set 3. Alignments for Fig. 1 as a Noninterleaved Nexus Formatted Text
File.
Acknowledgements
Funding was provided by the Australian Grains Research and Development Corporation
(UMU14; we particularly thank James Fortune, John Sandow, and John Lovett for their
support of this project), by the Swiss Federal Institute of Technology Zurich, by the Swiss
National Science Foundation (Grant 3100A0-104145), by MIT, and by the National Science
Foundation (DEB-0228725; Assembling the Fungal Tree of Life). We thank Soeren
Rasmussen for help with bioinformatics, Robert Lee for the oleate EST library, and Tim
Friesen (USDA) for comments on the manuscript. We dedicate this article to the recently
retired Chris Caten who has inspired our work on S. nodorum.

Chapter 4 mtDNA of P. nodorum
112
References
Abe Y, Suzuki T, Mizuno T, Ono C, Iwamoto K, Hosobuchi M, Yoshikawa H. 2002b. Effect
of increased dosage of the ML-236B (compactin) biosynthetic gene cluster on ML-236B
production in Penicillium citrinum. Mol Genet Genomics 268:130-137.
Abe Y, Suzuki T, Ono C, Iwamoto K, Hosobuchi M, Yoshikawa H. 2002a. Molecular
cloning and characterization of an ML-236B (compactin) biosynthetic gene cluster in
Penicillium citrinum. Mol Genet Genomics 267:636-646.
Agrios G. 2005. Plant Pathology. Burlington, MA: Elsevier Academic Press.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search
tool. J Mol Biol. 215:403-410.
Amnuaykanjanasin A, Punya J, Paungmoung P, Rungrod A, Tachaleat A, Pongpatta-
nakitshote S, Cheevadhanarak S, Tanticharoen M. 2005. Diversity of type I polyketide
synthase genes in the wood-decay fungus Xylaria sp. BCC 1067. FEMS Microbiol Lett.
251:125-136.
Ansari MZ, Yadav G, Gokhale RS, Mohanty D. 2004. NRPS-PKS: A knowledge-based
resource for analysis of NRPS/PKS megasynthases. Nucleic Acids Res. 32:W405-413.
Audic S, Claverie JM. 1997. The significance of digital gene expression profiles. Genome
Res. 7:986-995.
Bailey AM, Kershaw MJ, Hunt BA, Paterson IC, Charnley AK, Reynolds SE, Clarkson JM.
1996. Cloning and sequence analysis of an intron-containing domain from a peptide
synthetase-encoding gene of the entomopathogenic fungus Metarhizium anisopliae. Gene
173:195-197.
Baker SE, Kroken S, Inderbitzin P, Asvarak T, Li BY, Shi L, Yoder OC, Turgeon BG. 2006.
Two polyketide synthaseencoding genes are required for biosynthesis of the polyketide
virulence factor, T-toxin, by Cochliobolus heterostrophus. Mol Plant Microbe Interact.
19:139-149.
Baldwin TK, Winnenburg R, Urban M, Rawlings C, Koehler J, Hammond-Kosack KE. 2006.
The pathogen-host interactions database (PHI-base) provides insights into generic and
novel themes of pathogenicity. Mol Plant Microbe Interact. 19:1451-1462.

Chapter 4 mtDNA of P. nodorum
113
Bateman A, Birney E, Durbin R, Eddy SR, Finn RD, Sonnhammer EL. 1999. Pfam 3.1: 1313
multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids
Res. 27:260-262.
Bateman A, et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32: D138-
D141. Bathgate JA, Loughman R. 2001. Ascospores are a source of inoculum of
Phaeosphaeria nodorum, P. avenaria f. sp. avenaria and Mycosphaerella graminicola in
Western Australia. Australas Plant Pathol. 30:317-322.
Bearchell SJ, Fraaije BA, Shaw MW, Fitt BDL. 2005. Wheat archive links long-term fungal
pathogen population dynamics to air pollution. Proc Natl Acad Sci USA 102:5438-5442.
Bennett RS, Yun SH, Lee TY, Turgeon BG, Arseniuk E, Cunfer BM, Bergstrom GC. 2003.
Identity and conservation of mating type genes in geographically diverse isolates of
Phaeosphaeria nodorum. Fungal Genet Biol. 40:25-37.
Benson G. 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic
Acids Res. 27:573-580.
Bindschedler LV, Sanchez P, Dunn S, Mikan J, Thangavelu M, Clarkson JM, Cooper RM.
2003. Deletion of the SNP1 trypsin protease from Stagonospora nodorum reveals another
major protease expressed during infection. Fungal Genet Biol. 38:43-53.
Bohnert HU, Fudal I, Dioh W, Tharreau D, Notteghem JL, Lebrun MH. 2004. A putative
polyketide synthase/peptide synthetase from Magnaporthe grisea signals pathogen attack
to resistant rice. Plant Cell 16:2499-2513.
Bok JW, Keller NP. 2004. LaeA, a regulator of secondary metabolism in Aspergillus spp.
Eukaryot Cell 3:527-535.
Cambareri E, Jensen B, Schabtach E, Selker E. 1989. Repeat-induced G-C to A-T mutations
in Neurospora. Science 244:1571-1575.
Catlett NL, Yoder OC, Turgeon BG. 2003. Whole-genome analysis of two-component signal
transduction genes in fungal pathogens. Eukaryot Cell 2:1151-1161.
Chung KR, Ehrenshaft M, Wetzel DK, Daub ME. 2003. Cercosporin-deficient mutants by
plasmid tagging in the asexual fungus Cercospora nicotianae. Mol Genet Genomics
270:103-113.

Chapter 4 mtDNA of P. nodorum
114
Coleman M, Henricot B, Arnau J, Oliver RP. 1997. Starvation induced genes of the tomato
pathogen Cladosporium fulvum are also induced during growth in planta. Mol Plant
Microbe Interact. 10:1106-1109.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. 2005. Blast2GO: A
universal tool for annotation, visualization and analysis in functional genomics research.
Bioinformatics 21:3674-3676.
Cooley R, Shaw R, Franklin F, Caten C. 1988. Transformation of the phytopathogenic fungus
Septoria nodorum to hygromycin B resistance. Curr Genet. 13:383-386.
Cooley RN, Caten CE. 1991. Variation in electrophoretic karyotype between strains of
Septoria nodorum. Mol Gen Genet. 228:17-23.
Cozijnsen AJ, Howlett BJ. 2003. Characterisation of the mating-type locus of the plant
pathogenic ascomycete Leptosphaeria maculans. Curr Genet. 43:351-357.
Cummings D, McNally K, Domenico J, Matsuura E. 1990. The complete DNA sequence of
the mitochondrial genome of Podospora anserina. Curr Genet. 17:375-402.
Dancer J, Daniels A, Cooley N, Foster S. 1999. Septoria tritici and Stagonospora nodorum as
model pathogens for fungicide discovery. In Septoria on Cereals: A Study of
Pathosystems, Lucas JA, Bowyer P, Anderson HM, eds (New York: ABI Publishing), pp.
316-331.
Dean RA et al. 2005. The genome sequence of the rice blast fungus Magnaporthe grisea.
Nature 434:980-986.
Del Prado R, Schmitt I, Kautz S, Palice Z, Luecking R, Lumbsch HT. 2006. Molecular data
place Trypetheliaceae in Dothideomycetes. Mycol Res. 110:511-520.
DeZwaan TM, Carroll AM, Valent B, Sweigard JA. 1999. Magnaporthe grisea pth11p is a
novel plasma membrane protein that mediates appressorium differentiation in response to
inductive substrate cues. Plant Cell 11:2013-2030.
Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755-763.
Edgar RC. 2004. MUSCLE: A multiple sequence alignment method with reduced time and
space complexity. BMC Bioinformatics 5:113.
Eisendle M, Oberegger H, Zadra I, Haas H. 2003. The siderophore system is essential for
viability of Aspergillus nidulans: Functional analysis of two genes encoding l-ornithine N

Chapter 4 mtDNA of P. nodorum
115
5-monooxygenase (sidA) and a non-ribosomal peptide synthetase (sidC). Mol Microbiol.
49:359-375.
Eisendle M, Schrettl M, Kragl C, Müller D, Illmer P, Haas H. 2006. The intracellular
siderophore ferricrocin is involved in iron storage, oxidative-stress resistance,
germination, and sexual development in Aspergillus nidulans. Eukaryot Cell 5:1596-
1603.
Elliott CE, Howlett BJ. 2006. Overexpression of a 3-ketoacyl-CoA thiolase in Leptosphaeria
maculans causes reduced pathogenicity on Brassica napus. Mol Plant Microbe Interact.
19:588-596.
Eriksson OE. 2006. Outline of Ascomycota. Myconet 12:1-82.
Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. 1998. A computer program for aligning
a cDNA sequence with a genomic DNA sequence. Genome Res. 8:967-974.
Friesen TL, Stukenbrock EH, Liu ZH, Meinhardt S, Ling H, Faris JD, Rasmussen JB,
Solomon PS, McDonald BA, Oliver RP. 2006. Emergence of a new disease as a result of
interspecific virulence gene transfer. Nat Genet. 38:953-956.
Fujii I, Ono Y, Tada H, Gomi K, Ebizuka Y, Sankawa U. 1996. Cloning of the polyketide
synthase gene atX from Aspergillus terreus and its identification as the 6-methylsalicylic
acid synthase gene by heterologous expression. Mol Gen Genet. 253:1-10.
Fujii I, Yoshida N, Shimomaki S, Oikawa H, Ebizuka Y. 2005. An iterative type I polyketide
synthase PKSN catalyzes synthesis of the decaketide alternapyrone with regio-specific
octamethylation. Chem Biol. 12:1301-1309.
Gaffoor I, Brown D, Plattner R, Proctor R, Qi W, Trail F. 2005. Functional analysis of the
polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph
Fusarium graminearum). Eukaryot Cell 4:1926-1933.
Galagan JE et al. 2003. The genome sequence of the filamentous fungus Neurospora crassa.
Nature 422:859-868.
Galagan JE et al. 2005. Sequencing of Aspergillus nidulans and comparative analysis with A.
fumigatus and A. oryzae. Nature 438:1105-1115.
Ghikas DV, Kouvelis VN, Typas MA. 2006. The complete mitochondrial genome of the
entomopathogenic fungus Metarhizium anisopliae var. anisopliae: Gene order and trn

Chapter 4 mtDNA of P. nodorum
116
gene clusters reveal a common evolutionary course for all Sordariomycetes, while
intergenic regions show variation. Arch Microbiol. 185:393-401.
Giles NH, Geever RF, Asch DK, Avalos J, Case ME. 1991. Organization and regulation of
the qa (quinic acid) genes in Neurospora crassa and other fungi. J Hered. 82:1-7.
Goodwin SB. 2004. Minimum phylogenetic coverage: An additional criterion to guide the
selection of microbial pathogens for initial genomic sequencing efforts. Phytopathology
94:800-804.
Graziani S, Vasnier C, Daboussi MJ. 2004. Novel polyketide synthase from Nectria
haematococca. Appl Environ Microbiol. 70:2984-2988.
Guillemette T, Sellam A, Simoneau P. 2004. Analysis of a nonribosomal peptide synthetase
gene from Alternaria brassicae and flanking genomic sequences. Curr Genet. 45:214-
224.
Hofmann K, Stoffel W. 1993. TMbase - A database of membrane spanning proteins
segments. Biol Chem Hoppe Seyler 374:166.
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. 2007.
WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 35:W585-587.
Huang X, Madan A. 1999. CAP3: A DNA sequence assembly program. Genome Res. 9:868-
877.
Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees.
Bioinformatics 17:754-755.
Idnurm A, Howlett BJ. 2003. Analysis of loss of pathogenicity mutants reveals that repeat-
induced point mutations can occur in the Dothideomycete Leptosphaeria maculans.
Fungal Genet Biol. 39:31-37.
Idnurm A, Taylor JL, Pedras MSC, Howlett BJ. 2003. Small scale functional genomics of the
blackleg fungus, Leptosphaeria maculans: Analysis of a 38 kb region. Australas Plant
Pathol. 32:511-519.
Jaffe DB, Butler J, Gnerre S, Maucelli E, Lindblad-Toh K, Mesirov JP, Zody MC, Lander
ES. 2003. Whole genome sequence assembly for mammalian genomes: Arachne 2.
Genome Res. 13:91-96.
James T, et al. 2006. Reconstructing the early evolution of fungi using a six-gene phylogeny.
Nature 443:818-822.

Chapter 4 mtDNA of P. nodorum
117
Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G,
Thorstenson YR, Agabian N, Magee PT, Davis RW, Scherer S. 2004. The diploid
genome sequence of Candida albicans. Proc Natl Acad Sci USA 101:7329-7334.
Kall L, Krogh A, Sonnhammer EL. 2004. A combined transmembrane topology and signal
peptide prediction method. J Mol Biol. 338:1027-1036.
Kamper J et al. 2006. Insights from the genome of the biotrophic fungal plant pathogen
Ustilago maydis. Nature 444:97-101.
Keon J, Antoniw J, Rudd J, Skinner W, Hargreaves J, Hammond-Kosack K. 2005. Analysis
of expressed sequence tags from the wheat leaf blotch pathogen Mycosphaerella
graminicola (anamorph Septoria tritici). Fungal Genet Biol. 42:376-389.
Kim KH, Cho Y, La Rota M, Cramer RA Jr, Lawrence CB. 2007. Functional analysis of the
Alternaria brassicicola nonribosomal peptide synthetase gene AbNPS2 reveals a role in
conidial cell wall construction. Mol Plant Pathol. 8:23-29.
Kodsue, R, Dhanasekaran V, Aptroot A, Lumyong P, McKenzie EHC, Hyde KD, Jeewon R.
2006. The family Pleosporaceae: Intergeneric relationships and phylogenetic perspectives
based on sequence analyses of partial 28S rDNA. Mycologia 98:571-583.
Kouvelis V, Ghikas D, Typas M. 2004. The analysis of the complete mitochondrial genome
of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum
common gene organization in mtDNAs of Sordariomycetes: Phylogenetic implications.
Fungal Genet Biol. 41:930-940.
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane
protein topology with a hidden Markov model: Application to complete genomes. J Mol
Biol. 305 567-580.
Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. 2003. Phylogenomic analysis of
type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad
Sci USA 100:15670-15675.
Kulkarni RD, Kelkar HS, Dean RA. 2003. An eight-cysteinecontaining CFEM domain
unique to a group of fungal membrane proteins. Trends Biochem Sci. 28:118-121.
Lafon A, Han KH, Seo JA, Yu JH, d’Enfert C. 2006. G-protein and cAMP-mediated
signaling in aspergilli: A genomic perspective. Fungal Genet Biol. 43:490-502.

Chapter 4 mtDNA of P. nodorum
118
Lee BN, Kroken S, Chou DYT, Robbertse B, Yoder OC, Turgeon BG. 2005. Functional
analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a
factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot Cell
4:545-555.
Lewis SE, et al. 2002. Apollo: A sequence annotation editor. Genome Biol. 3:82.
Linder MB, Szilvay GR, Nakari-Setala T, Penttila ME. 2005. Hydrophobins: The protein-
amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 29:877-896.
Liu Z, Friesen T, Ling H, Meinhardt S, Oliver R, Rasmussen J, Faris J. 2006. The Tsn1-
ToxA interaction in the wheat- Stagonospora nodorum pathosystem parallels that of the
wheat-tan spot system. Genome 49:1265-1273.
Liu ZH, Faris JD, Meinhardt SW, Ali S, Rasmussen JB, Friesen TL. 2004a. Genetic and
physical mapping of a gene conditioning sensitivity in wheat to a partially purified host-
selective toxin produced by Stagonospora nodorum. Phytopathology 94:1056-1060.
Liu ZH, Friesen TL, Rasmussen JB, Ali S, Meinhardt SW, Faris JD. 2004b. Quantitative trait
loci analysis and mapping of seedling resistance to Stagonospora nodorum leaf blotch in
wheat. Phytopathology 94:1061-1067.
Lowe TM, Eddy SR. 1997. tRNAscan-SE: A program for improved detection of transfer
RNA genes in genomic sequences. Nucleic Acids Res. 25:955-964.
Majoros WH, Pertea M, Antonescu C, Salzberg SL. 2003. GlimmerM, Exonomy and Unveil:
Three ab initio eukaryotic genefinders. Nucleic Acids Res. 31:3601-3604.
Manning VA, Andrie RM, Trippe AF, Ciuffetti LM. 2004. Ptr ToxA requires multiple motifs
for complete activity. Mol Plant Microbe Interact. 17:491-501.
Manning VA, Ciuffetti LM. 2005. Localization of Ptr ToxA produced by Pyrenophora tritici-
repentis reveals protein import into wheat mesophyll cells. Plant Cell 17:3203-3212.
Marchler-Bauer A, et al. 2005. CDD: A Conserved Domain Database for protein
classification. Nucleic Acids Res. 33:D192-D196.
Marchler-Bauer A, Bryant SH. 2004. CD-Search: Protein domain annotations on the fly.
Nucleic Acids Res. 32:W327-331.
Meinhardt SW, Cheng WJ, Kwon CY, Donohue CM, Rasmussen JB. 2002. Role of the
arginyl-glycyl-aspartic motif in the action of Ptr ToxA produced by Pyrenophora tritici-
repentis. Plant Physiol. 130:1545-1551.

Chapter 4 mtDNA of P. nodorum
119
Moriwaki A, Kihara J, Kobayashi T, Tokunaga T, Arase S, Honda Y. 2004. Insertional
mutagenesis and characterization of a polyketide synthase gene (PKS1) required for
melanin biosynthesis in Bipolaris oryzae. FEMS Microbiol Lett. 238:1-8.
Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities
in the amino acid sequence of two proteins. J Mol Biol. 48:443-453.
Nicholson TP, Rudd BA, Dawson M, Lazarus CM, Simpson TJ, Cox RJ. 2001. Design and
utility of oligonucleotide gene probes for fungal polyketide synthases. Chem Biol. 8:157-
178.
Oide S, Moeder W, Krasnoff S, Gibson D, Haas H, Yoshioka K, Turgeon BG. 2006. NPS6,
encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron
metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant
Cell 18:2836-2853.
Oliver RP, Ipcho SVS. 2004. Arabidopsis pathology breathes new life into the necrotrophs
vs. biotrophs classification of fungal pathogens. Mol Plant Pathol. 5:347-352.
Orbach M. 1989. Electrophoretic characterisation of the Magnaporthe grisea genome. Fungal
Genet Newsl. 14:14.
Orbach M, Vollrath D, Davis R, Yanofsky C. 1988. An electrophoretic karyotype for
Neurospora crassa. Mol Cell Biol. 8:1469-1473.
Padovan AC, Sanson GF, Brunstein A, Briones MR. 2005. Fungi evolution revisited:
Application of the penalized likelihood method to a bayesian fungal phylogeny provides a
new perspective on phylogenetic relationships and divergence dates of ascomycota
groups. J Mol Evol. 60:726-735.
Peng T, Shubitz L, Simons J, Perrill R, Orsborn KI, Galgiani JN. 2002. Localization within a
proline-rich antigen (Ag2/PRA) of protective antigenicity against infection with
Coccidioides immitis in mice. Infect Immun. 70:3330-3335.
Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat families in large
genomes. Bioinformatics 21(Suppl 1):i351-i358.
Proctor RH, Desjardins AE, Plattner RD, Hohn TM. 1999. A polyketide synthase gene
required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating
population A. Fungal Genet Biol. 27:100-112.

Chapter 4 mtDNA of P. nodorum
120
Rawson JM. 2000. Transposable Elements in the Phytopathogenic Fungus Stagonospora
nodorum. PhD dissertation, UK University of Birmingham.
Rice P, Longden I, Bleasby A. 2000. EMBOSS: The European Molecular Biology Open
Software Suite. Trends Genet. 16:276-277.
Richards TA, Dacks JB, Jenkinson JM, Thornton CR, Talbot NJ. 2006. Evolution of
filamentous plant pathogens: Gene exchange across eukaryotic kingdoms. Curr Biol.
16:1857-1864.
Robbertse B, Reeves JB, Schoch CL, Spatafora JW. 2006. A phylogenomic analysis of the
Ascomycota. Fungal Genet Biol. 43:715-725.
Rodriguez F, Oliver JF, Martin A, Medina JR. 1990. The general stochastic model of
nucleotide substitution. J Theor Biol. 142:485-501.
Schoch CL, Shoemaker RA, Seifert KA, Hambleton S, Spatafora JW, Crous PW. 2006. A
multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia
98:1041-1052.
Shimizu T, Kinoshita H, Ishihara S, Sakai K, Nagai S, Nihira T. 2005. Polyketide synthase
gene responsible for citrinin biosynthesis in Monascus purpureus. Appl Environ
Microbiol. 71:3453-3457.
Solomon PS, Tan KC, Oliver RP. 2003a. The nutrient supply of pathogenic fungi; a fertile
field for study. Mol Plant Pathol. 4:203-210.
Solomon PS, Thomas SW, Spanu P, Oliver RP. 2003b. The utilisation of di/tripeptides by
Stagonospora nodorum is dispensable for wheat infection. Physiol Mol Plant Pathol.
63:191-199.
Solomon PS, Lee RC, Wilson TJG, Oliver RP. 2004a. Pathogenicity of Stagonospora
nodorum requires malate synthase. Mol Microbiol. 53:1065-1073.
Solomon PS, Lowe RGT, Tan KC, Waters ODC, Oliver RP. 2006a. Stagonospora nodorum:
cause of stagonospora nodorum blotch of wheat. Mol Plant Pathol. 7:147-156.
Solomon PS, Tan KC, Sanchez P, Cooper RM, Oliver RP. 2004b. The disruption of a G
alpha subunit sheds new light on the pathogenicity of Stagonospora nodorum on wheat.
Mol Plant Microbe Interact. 17:456-466.

Chapter 4 mtDNA of P. nodorum
121
Solomon PS, Waters OD, Simmonds J, Cooper RM, Oliver RP. 2005. The Mak2 MAP kinase
signal transduction pathway is required for pathogenicity in Stagonospora nodorum. Curr
Genet. 48:60-68.
Solomon PS, Wilson TJG, Rybak K, Parker K, Lowe RGT, Oliver RP. 2006b. Structural
characterisation of the interaction between Triticum aestivum and the dothideomycete
pathogen Stagonospora nodorum. Eur J Plant Pathol. 114:275-282.
Spatafora JW, et al. 2006. A five-gene phylogenetic analysis of the Pezizomycotina.
Mycologia 98:1018-1028.
Stamatakis A. 2006. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses
with thousands of taxa and mixed models. Bioinformatics 22:2688-2690.
Stukenbrock EH, Banke S, McDonald BA. 2006. Global migration patterns in the fungal
wheat pathogen Phaeosphaeria nodorum. Mol Ecol. 15:2895-2904.
Stukenbrock EH, McDonald BA. 2007. Geographic variation and positive diversifying
selection in the host specific toxin SnToxA. Mol Plant Pathol. 8:321-322.
Sumii M, Furutani Y, Waschuk SA, Brown LS, Kandori H. 2005. Strongly hydrogen-bonded
water molecule present near the retinal chromophore of Leptosphaeria rhodopsin, the
bacteriorhodopsinlike proton pump from a eukaryote. Biochemistry 44:15159-15166.
Talbot N, Salch Y, Ma M, Hamer J. 1993. Karyotypic variation within clonal lineages of the
rice blast fungus, Magnaporthe grisea. Appl Environ Microbiol. 59:585-593.
Talbot NJ, McCafferty HRK, Ma M, Moore K, Hamer JE. 1997. Nitrogen starvation of the
rice blast fungus Magnaporthe grisea may act as an environmental cue for disease
symptom expression. Physiol Mol Plant Pathol. 50:179-195.
Tambor J, Guedes R, Nobrega M, Nobrega F. 2006. The complete DNA sequence of the
mitochondrial genome of the dermatophyte fungus Epidermophyton floccosum. Curr
Genet. 49:302-308.
Thomma B, Bolton MD, Clergeot PH, De Wit P. 2006. Nitrogen controls in planta expression
of Cladosporium fulvum Avr9 but no other effector genes. Mol Plant Pathol. 7:125-130.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X
windows interface: Flexible strategies for multiple sequence alignment aided by quality
analysis tools. Nucleic Acids Res. 25:4876-4882.

Chapter 4 mtDNA of P. nodorum
122
Trail F, Xu JR, San Miguel P, Halgren RG, Kistler HC. 2003. Analysis of expressed
sequence tags from Gibberella zeae (anamorph Fusarium graminearum). Fungal Genet
Biol. 38:187-197.
Vizcaino JA, Cardoza RE, Dubost L, Bodo B, Gutierrez S, Monte E. 2006. Detection of
peptaibols and partial cloning of a putative peptaibol synthetase gene from T. harzianum
CECT 2413. Folia Microbiol. (Praha) 51:114-120.
Wessels JGH. 1994. Developmental regulation of fungal cell wall formation. Annu Rev
Phytopathol. 32:413-437.
Winefield RD, Hilario E, Beever RE, Haverkamp RG, Templeton MD. 2007. Hydrophobin
genes and their expression in conidial and aconidial Neurospora species. Fungal Genet
Biol. 44:250-257.
Winka K, Eriksson OE. 1997. Supraordinal taxa of Ascomycota. Mol Phylogenet Evol. 1:1-
16.
Wolpert TJ, Dunkle LD, Ciuffetti LM. 2002. Host-selective toxins and avirulence
determinants: What’s in a name? Annu Rev Phytopathol. 40:251-285.
Woo P, et al. 2003. The mitochondrial genome of the thermal dimorphic fungus Penicillium
marneffei is more closely related to those of molds than yeasts. FEBS Lett. 555:469-477.
Yadav G, Gokhale RS, Mohanty D. 2003. SEARCHPKS: A program for detection and
analysis of polyketide synthase domains. Nucleic Acids Res. 31:3654-3658.
Yoder OC, Turgeon BG. 2001. Fungal genomics and pathogenicity. Curr Opin Plant Biol.
4:315-321.
Yun SH, Turgeon BG, Yoder OC. 1998. REMI-induced mutants of Mycosphaerella zeae-
maydis lacking the polyketide PMtoxin are deficient in pathogenesis to corn. Physiol Mol
Plant Pathol. 52:53-66.
Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA
sequences. J Comput Biol. 7:203-214.
Zolan ME. 1995. Chromosome-length polymorphism in fungi. Microbiol Rev. 59:686-698.

CHAPTER 5
QoI resistance evolution in European populations of M. graminicola
Stefano FF Torriani, Patrick C Brunner, Bruce A. McDonald, Helge Sierotzki. 2009. QoI
resistance emerged independently at least four times in European populations of
Mycosphaerella graminicola. Pest Management Science 65:155-162

Chapter 5 QoI vs M. graminicola
124

Chapter 5 QoI vs M. graminicola
125
Abstract
QoI fungicides or quinone outside inhibitors (also called “strobilurins”) have been widely
used to control agriculturally important fungal pathogens since their introduction in 1996.
Strobilurins block the respiration pathway by inhibiting the cytochrome bc1 complex in
mitochondria. Several plant pathogenic fungi have developed field resistance. The first QoI
resistance in Mycosphaerella graminicola was detected retrospectively in UK in 2001 at a
low frequency in QoI treated plots. During the following seasons resistance reached high
frequencies across northern Europe. The aim of this study was to identify the main
evolutionary forces driving the rapid emergence and spread of QoI resistance in M.
graminicola populations. G143A mutation causing QoI resistance was first detected during
2002 in all tested populations and in eight distinct mtDNA sequence haplotypes. By 2004, 24
different mtDNA haplotypes contained the G143A mutation. Phylogenetic analysis showed
that strobilurin resistance was acquired independently through at least four recurrent
mutations at the same site of cytochrome b. Estimates of directional migration rates showed
that the majority of gene flow in Europe had occurred in a west-to-east direction. This study
demonstrated that recurring mutations independently introduced the QoI resistance allele into
different genetic and geographic backgrounds. The resistant haplotypes then increased in
frequency due to the strong fungicide selection and spread eastward through wind dispersal
of ascospores.

Chapter 5 QoI vs M. graminicola
126
Introduction
Mycosphaerella graminicola (anamorph: Septoria tritici) is the causal agent of Septoria
tritici leaf blotch, one of the most important foliar diseases of wheat in many parts of the
world (Hardwick et al. 2001; Bearchell et al. 2005). M. graminicola is a haploid, heterothallic
ascomycete, characterized by both sexual and asexual reproduction. Sexual spores play an
important role as the primary source of inoculum initiating an epidemic (Shaw et al. 1989;
Hunter et al. 1999; Zhan et al. 2001) but they also contribute to secondary infection and can
be dispersed over tens of kilometers (Sanderson 1972). The asexual pycnidiospores are
disseminated over limited distances from plant to plant via rain splash and they contribute
only to the secondary infection (Bannon et al. 1998).
M. graminicola was controlled mainly by demethylation-inhibiting (DMI) fungicides
until the introduction of strobilurin-based fungicides in 1996. This new class of fungicides
provided a superior disease control and additional favorable effects on the physiology of the
plant (Fraaije et al. 2003). QoI fungicides act by inhibiting the Qo site of the cytochrome bc1
complex in the mitochondria, thereby blocking the electron transfer process in the respiration
chain and causing an energy deficiency due to a lack of adenosine triphosphate (ATP)
(Bartlett et al. 2002). In most pathogens, including M. graminicola, resistance towards
strobilurins is conferred by a single nucleotide change in the mitochondrial cytochrome b
gene (cyt b), leading to an amino acid change at codon 143 from glycine to alanine (G143A)
(Gisi et al. 2002). In some fungi, such as Alternaria solani (Pasche et al. 2005) or
Pyrenophora teres (Sierotzki et al. 2007) another mutation in cyt b (F129L) was identified,
however this mutation results in a less resistant phenotype compared to the G143A mutation
(Gisi et al. 2002; Kim et al. 2003). Recently it was postulated that for fungi such as A. solani
and P. teres that possess an intron placed directly after G143, the mutation leading to the
G143A change cannot occur, because this mutation would strongly affect the splicing
process, causing a deficient cytochrome b (Grasso et al. 2006). Other unknown mechanisms
leading to resistance to QoI’s were reported in Venturia inaequalis (Steinfeld et al. 2001) and
Podosphaera fusca (Fernández-Ortuño et al. 2008).
The first QoI resistant isolates of M. graminicola were detected retrospectively in the UK
in 2001 at low frequency in QoI treated plots (Fraaije et al. 2005a) and subsequently in 2002

Chapter 5 QoI vs M. graminicola
127
in five European countries (Gisi et al. 2005). During 2003 and 2004 the frequency of resistant
isolates increased rapidly in northern Europe. The resistance is now widespread across the
entire UK (Fraaije et al. 2005a), whereas in France and Germany resistance levels are higher
in the north than in the south (Gisi et al. 2005). The north-south gradient in resistance
distribution is thought to be due to differences in the intensity of QoI use because of lower
disease pressure in southern regions.
The aim of this study was to clarify the evolutionary mechanisms behind the emergence
and spread of QoI resistant isolates in M. graminicola. The main hypotheses tested were:
1. The G143A mutation occurred only once or very few times, potentially in only one
mtDNA haplotype and/or one region, and was subsequently distributed to other regions
by migration.
2. The G143A mutation occurred independently in several different mtDNA haplotypes
and/or geographic regions.
Parallel genetic adaptation to drugs and pesticides was until now widely studied in
animals for insecticides (Andreev et al. 1999; Nardi et al. 2006) and nematicides (Elard et al.
1996), in plants for herbicides (Bernasconi et al. 1995) and in bacteria for antibiotics (Barlow
and Hall 2003; Hall et al. 2004). However, very little is presently known about the processes
driving the development of fungicide resistance alleles in plant pathogens. The unique
features of mitochondrial genomes, including their uniparental inheritance, the near absence
of genetic recombination, their small size and fixed complement of genes enabled us to
clearly differentiate between multiple or single occurrences and origins of fungicide
resistance in M. graminicola. To accomplish this, we sequenced three loci of the mtDNA
genome and assessed the QoI resistance/sensitivity in each isolate by identifying the
presence/absence of the G143A mutation.
Materials and methods
Fungal isolates. Isolates from geographically distinct European populations of M.
graminicola were obtained from infected leaves collected from locations in Germany (GER),

Chapter 5 QoI vs M. graminicola
128
England (UK), Denmark (DN), Ireland (IR) and France (FR). To address the temporal
dimension of evolution of QoI resistance, isolates from the same regions -whenever possible-
included samples collected before the use of QoI (pre-QoI) and samples collected after the
first occurrence of QoI resistant isolates (post-QoI). In total, 181 M. graminicola isolates
obtained from infected leaf samples by the methods previously described (McDonald et al.
1994; Linde et al. 2002) were used in this study (Table 1). The pre-QoI isolates from 1992
and 1994 were part of a previously described worldwide collection of M. graminicola
(McDonald et al. 1999; Linde et al. 2002; Zhan et al. 2003; Banke and McDonald 2005). The
post-QoI isolates from 2002 were supplied by Syngenta Switzerland (Gisi et al. 2005) and the
German isolates from 2004 were collected close to Dedelow in the north-eastern German
state of Brandenburg (Ger-D) and close to Quarnbek in the north-western German state of
Schleswig-Holstein (GER-Q) (Brunner et al. 2008).
PCR-RFLP to detect the QoI resistant allele. All 181 isolates were assayed using a
PCR-RFLP approach to detect the QoI-resistant A143 allele. Using the recently published
mitochondrial genome of M. graminicola (EU090238; Torriani et al. 2008), we designed two
primers to partially amplify cytb. The total PCR reaction volume was 20 µl per well,
containing 10 pmol of each primer (MgcytbF 5’-TCG TTA CTG GTG TTA CAC TTG C-3’
and MgcytbR 5’-GCC ATA ACA TAA TTC TCG CTG TCA CC -3’), 100 µM of each
nucleotide, 2 µl of 10x PCR buffer (1x PCR buffer: 10 mM KCl, 10 mM (NH4)2SO4, 20 mM
Tris-HCl, 2 mM MgSO4, 0.1% Triton X-100, [pH 8.8]), 1 U of Taq DNA Polymerase (New
England Biolabs, England) and 4 µl of M. graminicola genomic DNA (5 to 10 ng final DNA
concentration). Thermal cycling conditions were as follows: initial hold at 96°C for 2 min, 35
cycles of 96°C for 1 min, 50°C for 1 min, 72°C for 1 min. The final products were incubated
for 5 min at 72°C. A portion of each PCR product (10 µl) was digested using 1U Fnu4HI
(New England BioLabs, England) for 4 h at 37°C. Digests were visualized on a 1% agarose
gel with 1x Tris-borate-EDTA (TBE) buffer (Fig. 1).
Sequencing and sequence analysis. The mtDNA loci Mg1, Mg2 and Mg3 (Torriani et al.
2008) were amplified and sequenced for all 181 isolates. Sequences were aligned and edited

Chapter 5 QoI vs M. graminicola
129
manually using Sequencher 4.5 (Gene Code Corporation, Ann Arbor, MI, USA). The
concatenated nucleotide sequences were then collapsed into haplotypes recording insertions
and deletions (indels) with the program SNAP Workbench (Table 1 and Fig. 2; Price and
Carbone 2005). DnaSP was used to test for recombination within and among the tested loci
(Rozas et al. 2003).
Data analysis. We used the Kishino-Hasegawa test (KH test; Kishino and Hasegawa
1989) as implemented in PAUP* 4.0b.10 (Swofford 2002) to test different hypotheses of
resistance appearance by comparing tree topologies using the full mtDNA data set (all codon
positions). We compared the unconstrained tree obtained by Maximum Likelihood (ML) and
Maximum Parsimony (MP) analyses, respectively, to two alternative hypotheses of haplotype
genealogy; i) resistant (R) haplotypes were assumed to be monophyletic, ii) sensitive (S)
haplotypes were assumed to be monophyletic. Results and probability values (two-tailed test)
were obtained through 1000 nonparametric bootstrap replicates and the “full optimization”
criterion (Table 2).
Classic estimates of migration rates assume that populations have reached equilibrium
between drift and migration and are of equal size. However, the historical time frame we are
investigating is likely too short for haplotypes drawn from populations to have sorted into
distinct lineages (Avise 2001) and hence, these populations are probably not in equilibrium.
Thus, we applied the likelihood-based coalescent method implemented in MIGRATE version
2.1.3 (Beerli and Felsenstein 2001), which does not rely on the assumption of populations in
equilibrium or equal population size, and therefore provides a biologically more realistic
approach. Furthermore, MIGRATE estimates asymmetric/directional gene flow among
populations, making this approach particularly useful in epidemiological settings. Following
the authors’ recommendations, we made a first MIGRATE run with the default values using
FST to find the starting parameters for the four tested populations (UK-1994, DN-1994,
GER-Q-2004 and GER-D). Parameters obtained from this initial run were used to re-run the
program with the following Markov chain settings; short chains 10, long chains 3, averaging
over 3 replicates and a 4-chain heating scheme.
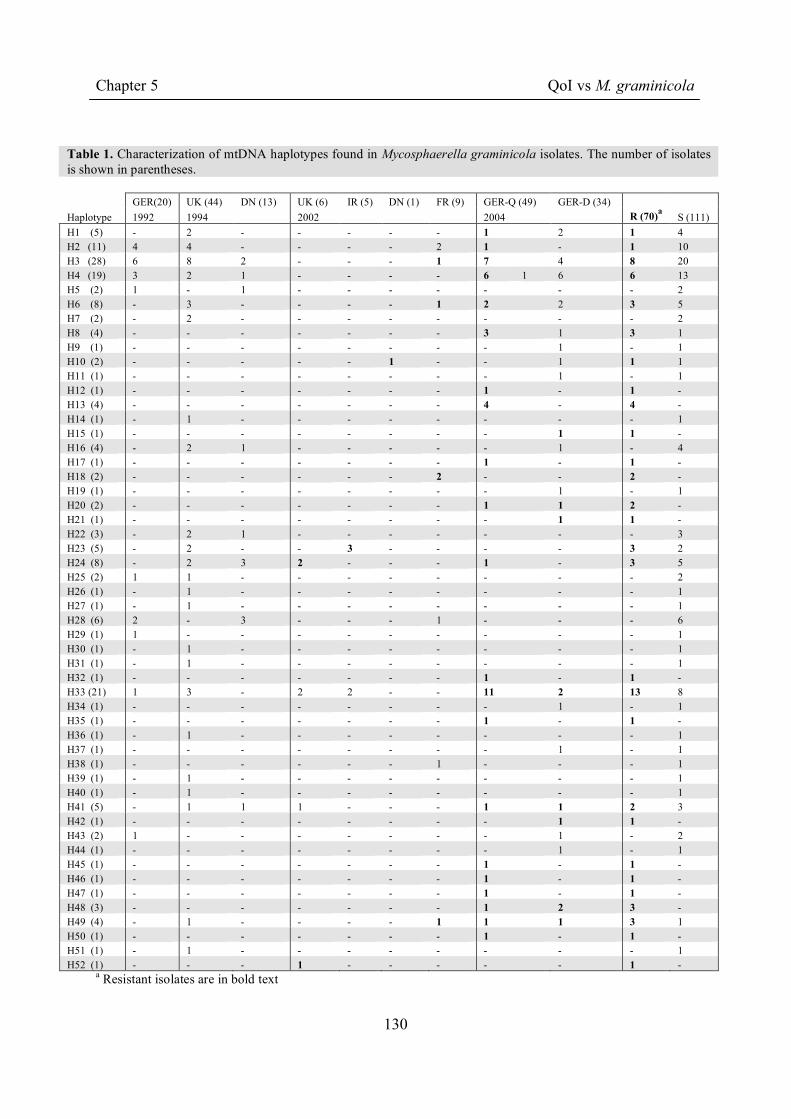
Chapter 5 QoI vs M. graminicola
130
Table 1. Characterization of mtDNA haplotypes found in Mycosphaerella graminicola isolates. The number of isolates is shown in parentheses. GER(20) UK (44) DN (13) UK (6) IR (5) DN (1) FR (9) GER-Q (49) GER-D (34) Haplotype 1992 1994 2002 2004 R (70)a S (111) H1 (5) - 2 - - - - - 1 2 1 4 H2 (11) 4 4 - - - - 2 1 - 1 10 H3 (28) 6 8 2 - - - 1 7 4 8 20 H4 (19) 3 2 1 - - - - 6 1 6 6 13 H5 (2) 1 - 1 - - - - - - - 2 H6 (8) - 3 - - - - 1 2 2 3 5 H7 (2) - 2 - - - - - - - - 2 H8 (4) - - - - - - - 3 1 3 1 H9 (1) - - - - - - - - 1 - 1 H10 (2) - - - - - 1 - - 1 1 1 H11 (1) - - - - - - - - 1 - 1 H12 (1) - - - - - - - 1 - 1 - H13 (4) - - - - - - - 4 - 4 - H14 (1) - 1 - - - - - - - - 1 H15 (1) - - - - - - - - 1 1 - H16 (4) - 2 1 - - - - - 1 - 4 H17 (1) - - - - - - - 1 - 1 - H18 (2) - - - - - - 2 - - 2 - H19 (1) - - - - - - - - 1 - 1 H20 (2) - - - - - - - 1 1 2 - H21 (1) - - - - - - - - 1 1 - H22 (3) - 2 1 - - - - - - - 3 H23 (5) - 2 - - 3 - - - - 3 2 H24 (8) - 2 3 2 - - - 1 - 3 5 H25 (2) 1 1 - - - - - - - - 2 H26 (1) - 1 - - - - - - - - 1 H27 (1) - 1 - - - - - - - - 1 H28 (6) 2 - 3 - - - 1 - - - 6 H29 (1) 1 - - - - - - - - - 1 H30 (1) - 1 - - - - - - - - 1 H31 (1) - 1 - - - - - - - - 1 H32 (1) - - - - - - - 1 - 1 - H33 (21) 1 3 - 2 2 - - 11 2 13 8 H34 (1) - - - - - - - - 1 - 1 H35 (1) - - - - - - - 1 - 1 - H36 (1) - 1 - - - - - - - - 1 H37 (1) - - - - - - - - 1 - 1 H38 (1) - - - - - - 1 - - - 1 H39 (1) - 1 - - - - - - - - 1 H40 (1) - 1 - - - - - - - - 1 H41 (5) - 1 1 1 - - - 1 1 2 3 H42 (1) - - - - - - - - 1 1 - H43 (2) 1 - - - - - - - 1 - 2 H44 (1) - - - - - - - - 1 - 1 H45 (1) - - - - - - - 1 - 1 - H46 (1) - - - - - - - 1 - 1 - H47 (1) - - - - - - - 1 - 1 - H48 (3) - - - - - - - 1 2 3 - H49 (4) - 1 - - - - 1 1 1 3 1 H50 (1) - - - - - - - 1 - 1 - H51 (1) - 1 - - - - - - - - 1 H52 (1) - - - 1 - - - - - 1 -
a Resistant isolates are in bold text

Chapter 5 QoI vs M. graminicola
131
Results
A total of 181 strains were analyzed, including 111 sensitive and 70 resistant isolates (Table
1). The first QoI resistant isolates were found in 2002 in four sampled locations. All 77 pre-
QoI samples were sensitive and 67% of post-QoI isolates were resistant. In 2004, GER-Q
had only one sensitive isolate while GER-D had 71% sensitive isolates (Table 1, Fig. 3a).
The concatenated sequences of the 181 analyzed isolates collapsed into 52 nucleotide
haplotypes, designated as H1 to H52 (Table 1 and Fig. 4), of which 29 included a single
isolate and ten included at least five isolates (Table 1; Fig. 2). The ten most frequent
haplotypes represented 64% of all isolates, including 69% of the pre- and 54% of the post-
QoI isolates. The remaining 42 “rare” haplotypes had on average 1.55 isolates per haplotype.
The program DNAsp did not detect any recombination events within or between the tested
mitochondrial loci Mg1, Mg2 and Mg3.
Nine of the ten most frequent haplotypes acquired the resistance mutation independently;
with only haplotype H28 having all sensitive isolates (Fig. 2). Twelve mtDNA sequence
haplotypes had both resistant and sensitive strains, 24 haplotypes were exclusively sensitive
and 16 were exclusively resistant. Resistant isolates were first detected in 2002 in all tested
geographical populations and belonged to 8 distinct mtDNA sequence haplotypes out of 13
total mtDNA haplotypes detected in that year. In 2002, none of the resistant sequence
haplotypes were found in more than one population. Two years later, 24 sequence haplotypes
out of the 33 detected were resistant and four of these haplotypes were found previously in
2002. We used the KH-tests to statistically assess different hypotheses of haplotype evolution
by comparing tree topologies. In both the ML and MP topologies the unconstrained trees
performed significantly better then the two alternative hypotheses, where trees with R-
haplotypes or S-haplotypes were considered monophyletic, respectively (Table 2). The
unconstrained ML tree formed an unrooted cladogram and identified four major clades, all of
which consisted of a mixture of R, S and mixed (RS) haplotypes (Fig. 4).
Estimates of directional migration rates indicated that the majority of gene flow in Europe
had occurred in a west-to-east direction (Table 3, Fig. 3b). For example, estimates of gene
flow were high (Nm = 86) from the UK to GER-Q, whereas there was no or little detectable

Chapter 5 QoI vs M. graminicola
132
gene flow in the opposite direction. Similarly, gene flow was 10 times higher from GER-Q
into GER-D than in the opposite direction.
Table 2. Kishino-Hasegawa tests to statistically assess different hypotheses of haplotype evolution by comparing tree topologies as implemented in PAUP. The unconstrained tree topologies obtained by Maximum Likelihood and Maximum Parsimony analyses, respectively, were compared to two alternative hypotheses of haplotype genealogy; i) resistant (R) haplotypes were assumed to be monophyletic, ii) sensitive (S) haplotypes were assumed to be monophyletic. Probabilities (P) of obtaining better trees were assessed using two-tailed tests, the full optimization criterion and 1000 bootstrap replicates. Tree Tree score Probability Maximum Likelihood -ln L -ln L diff P unconstrained 1042.71823 best R-constrained 1072.48505 29.76681 0.016* S-constrained 1072.48506 29.76683 0.016* Maximum Parsimony Length Length diff unconstrained 1158 best R-constrained 1207 49 < 0.001* S-constrained 1216 58 < 0.001* * P < 0.05
Figure 1. PCR-RFLP analysis of the cytb-locus for detection of the A143 strobilurin-resistant allele. a) The G143A mutation causes a second Fnu4HI restriction site in cyt b, conferring resistance to strobilurin. Molecular sizes are indicated in each fragment. b) An example of the PCR-RFLP results for two sensitive (S) and two resistant (R) isolates. The first lane is a 100-bp size ladder (M). The size of the digested products is shown.
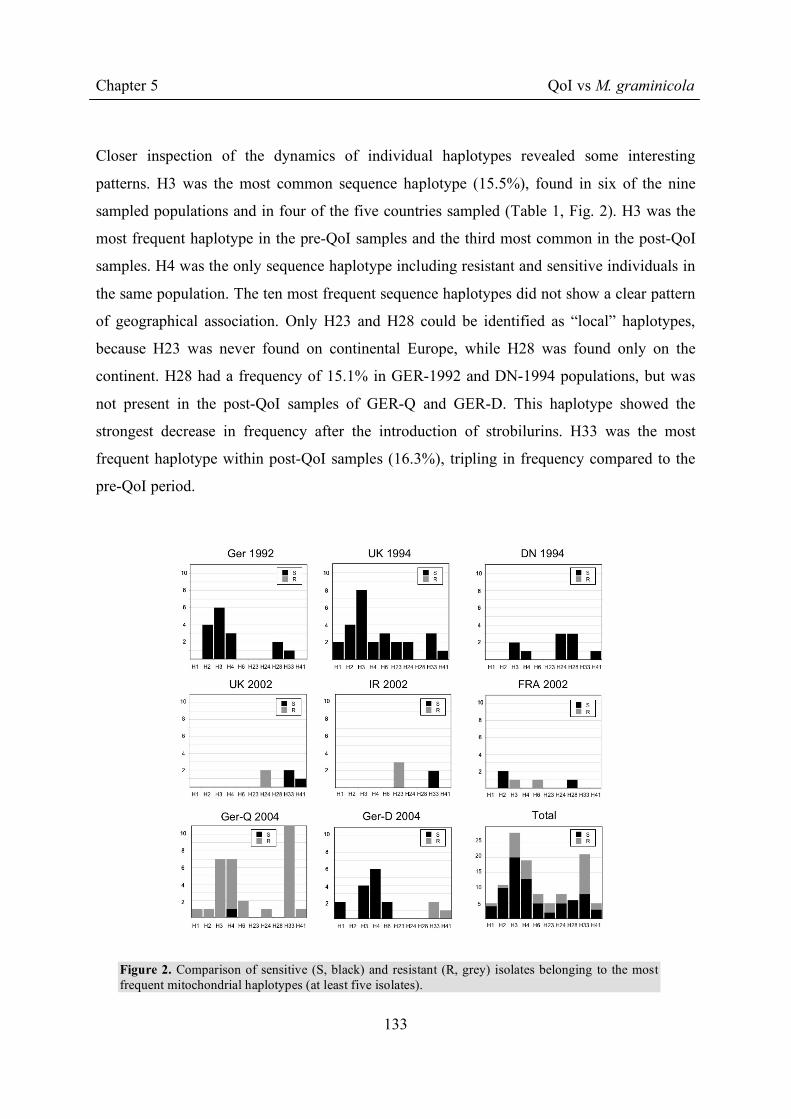
Chapter 5 QoI vs M. graminicola
133
Closer inspection of the dynamics of individual haplotypes revealed some interesting
patterns. H3 was the most common sequence haplotype (15.5%), found in six of the nine
sampled populations and in four of the five countries sampled (Table 1, Fig. 2). H3 was the
most frequent haplotype in the pre-QoI samples and the third most common in the post-QoI
samples. H4 was the only sequence haplotype including resistant and sensitive individuals in
the same population. The ten most frequent sequence haplotypes did not show a clear pattern
of geographical association. Only H23 and H28 could be identified as “local” haplotypes,
because H23 was never found on continental Europe, while H28 was found only on the
continent. H28 had a frequency of 15.1% in GER-1992 and DN-1994 populations, but was
not present in the post-QoI samples of GER-Q and GER-D. This haplotype showed the
strongest decrease in frequency after the introduction of strobilurins. H33 was the most
frequent haplotype within post-QoI samples (16.3%), tripling in frequency compared to the
pre-QoI period.
Figure 2. Comparison of sensitive (S, black) and resistant (R, grey) isolates belonging to the most frequent mitochondrial haplotypes (at least five isolates).

Chapter 5 QoI vs M. graminicola
134
Figure 3. a) Geographical locations of the six sampled post-QoI Mycosphaerella graminicola populations. The total number of isolates assayed for each population is indicated in the circles, which also show the frequencies of sensitive (black) and resistant (grey) strains. All pre-QoI populations were completely sensitive to strobilurins b) Migration patterns of Mycosphaerella graminicola among populations. Arrows sizes are proportional to the migration rate. Absence of an arrow indicates no or little detected migration (Nm < 0.5).

Chapter 5 QoI vs M. graminicola
135
Discussion
The main result of this study was to unequivocally demonstrate that QoI resistant M.
graminicola isolates arose independently in different genetic and geographic backgrounds,
falsifying the hypothesis that a single mutant haplotype emerged and subsequently spread
across other regions in Europe. This finding indicates that recurring mutations introduced the
resistance allele into several distinct mtDNA haplotypes, which then increased in frequency
due to the strong fungicide selection, and spread eastward through wind dispersal of
ascospores.
Since the introduction of QoI fungicides into the cereal market in 1996, QoI fungicide
resistant isolates were detected in field populations of a wide range of pathogen species,
including Blumeria graminis f. sp. tritici and hordei (Sierotzki et al. 2000) and Pyrenophora
tritici-repentis (Sierotzki et al 2007). Resistance in M. graminicola emerged simultaneously
in several locations during 2002, with a rapid increase in the frequency of resistant isolates
during the two following seasons, reaching especially high levels in Northern Europe (Gisi et
al 2005), where the G143A resistance allele was found in more than half of the identified
mtDNA sequence haplotypes (Table 1, Fig. 4). The hypothesis that the mutations to
resistance emerged independently in different genetic backgrounds is clearly supported by the
unconstrained ML tree (Fig. 4), which shows that in M. graminicola the A143-allele was
acquired through at least four distinct mutational events. Similar findings for the parallel
emergence of fungicide resistance were identified for the grape pathogen Plasmopara viticola
(Chen et al. 2007). This is the first time that intraspecific parallel evolution of fungicide
resistance has been demonstrated for a cereal pathogen and it confirms previous results from
P. viticola suggesting that “de novo” appearance of a resistance allele in distinct genetic
backgrounds may be common for plant pathogenic fungi. The interspecific parallel evolution
of resistance alleles was previously shown across a wide range of plant pathogens (Grasso et
al 2006).
No mutants were found when screening a worldwide collection of 1000 pre-QoI field
isolates of M. graminicola for the presence of the A143 resistance allele. Assuming a
binomial distribution for the mutation, the 95% confidence interval for the frequency of the
G143A mutation in the global population ranged from 0 to 0.003 (McDonald, unpublished).

Chapter 5 QoI vs M. graminicola
136
Figure 4. Unconstrained Maximum Likelihood tree obtained for all resistant (R, dark-grey hatched), susceptible (S, not hatched) and mixed (RS, soft-grey) mtDNA haplotypes.

Chapter 5 QoI vs M. graminicola
137
By 2002, eight mtDNA sequence haplotypes acquired the resistance allele. None of these
resistance haplotypes were present in more than one population, indicating that 2002 was the
starting point for the subsequent rapid emergence of resistant haplotypes. Published data
shows that the 143A allele was present at low frequency in QoI treated plots in the UK in
2001 (Fraaije et al. 2005a). The mutation was therefore present at detectable levels prior to
2002 but became much more common in 2002, a year of high Septoria disease pressure in
NW Europe. While recurring mutations were important for driving the evolution of
strobilurin resistance in M. graminicola, the subsequent role of natural or anthropogenic gene
flow should not be underestimated. In fact, directional migration rates clearly showed the
importance of gene flow in moving mtDNA haplotypes from Western to Eastern Europe
(Table 3, Fig. 3b). The airborne ascospores have the potential to be dispersed over many
kilometers (Fraaije et al. 2005b) following the prevailing wind directions across Europe. The
same patterns of M. graminicola migration in Europe were found using the nuclear-encoded
Cyp51 locus (Brunner et al. 2008).
The unique properties of mtDNA allowed us to unequivocally demonstrate the
intraspecific parallel evolution of QoI resistance. The lack of recombination found in the
mtDNA of M. graminicola using the present and previous dataset (Torriani et al. 2008)
means that the G143A mutation had to emerge independently in different genetic
backgrounds, instead of being shuffled into different backgrounds through sexual
recombination. Most modern pesticides, like DMI fungicides, have as their target nuclear-
encoded proteins whose genes are subject to recombination (Brunner et al. 2008). For these
resistances, recombination makes it difficult to determine how many times the mutation to
resistance occurred because the same mutation can be recombined into many different
genetic backgrounds. The results arising from this study provide a better insight into the
processes driving the development of fungicide resistance alleles in fungal populations. This
study confirms the evidence for the independent multiple acquisition of the resistant allele to
a fungicide at the intraspecific level in plant pathogenic fungi, and may provide new
perspectives regarding the development of appropriate management strategies.

Chapter 5 QoI vs M. graminicola
138
Table 3. Pairwise likelihood estimates of directional migration rates between major geographical regions (95% confidence intervals in parenthesis). Donor populations are shown on the left and receiving populations are given along the top. Migration rates are expressed as Nm, the number of immigrants per generation.
UK DN GER-Q GER-D UK 2.33 (0.54 – 5.74) 86.84 (57.67 – 121.55) 0.01(0.00 – 0.01) DN 0.46 (0.06 – 0.84) 16.11 (7.03 – 29.94) 5.52 (0.69 – 9.71)
GER-Q 0.25 (0.00 – 0.51) 4.21 (0.65 – 7.12) 37.82 (25.71 – 45.36) GER-D 0.04 (0.00 – 0.10) 0.05 (0.00 – 0.08) 3.58 (0.45 – 5.43)
Acknowledgements
This project was supported by the Swiss National Science Foundation (grant 3100A0-
104145). The ETH Genetic Diversity Center provided facilities for collecting all DNA
sequence data. We thank Andreas von Tiedemann and Christine Schäfer (Georg-August-
University of Göttingen) for generously providing isolates from recent German populations.

Chapter 5 QoI vs M. graminicola
139
References
Andreev D, Kreitman M, Phillips TW, Beeman RW, French-Constant RH. 1999. Multiple
origins of cyclodiene insecticide resistance in Tribolium castaneum (Coleoptera:
Tenebrionidae). J Mol Evol. 48:615-624.
Avise JC. 1994. Molecular Markers, Natural History and Evolution. Chapman and Hall, 511
pp, New York, USA.
Banke S, McDonald BA. 2005. Migration patterns among global populations of the
pathogenic fungus Mycosphaerella graminicola. Mol Ecol. 14:1881-1896.
Bannon FJ, Cooke BM. 1998. Studies on dispersal of Septoria tritici pycnidiospores in
wheat-clover intercrops. Plant Pathol. 47:49-56.
Bartlett DW, Clough JM, Godwin JR, Hall AA, Hamer M, Parr-Dobrzanski B. 2002. The
strobilurin fungicides. Pest Manag Sci. 58:649-662.
Barlow M, Hall BG. 2003. Experimental prediction of the natural evolution of antibiotic
resistance. Genetics 163:1237-1241.
Bearchell SJ, Fraaije BA, Shaw MW, Fitt BDL. 2005. Wheat archive links long-term fungal
pathogen population dynamics to air pollution. Proc Natl Acad Sci USA 102:5438-5442.
Beerli P, Felsenstein J. 2001. Maximum likelihood estimation of a migration matrix and
effective population sizes in n subpopulations by using a coalescent approach. Proc Natl
Acad Sci USA 98:4563-4568.
Bernasconi P, Woodworth AR, Rosen BA, Subramanian MV, Siehl DL.1995. A naturally-
occurring point mutation confers broad range tolerance to herbicides that target
acetolactate synthase. J Biol Chem. 270:17381-17385.
Brunner PC, Stefanato FL, McDonald BA. 2008. Evolution of the CYP51 gene in
Mycosphaerella graminicola: evidence for intragenic recombination and selective
replacement. Mol Plant Pathol. 9:305-316.
Chen WJ, Delmotte F, Richard-Cervera S, Douence L, Greif C, Corio-Costet MF. 2007. At
least two origins of fungicide resistance in grapevine downy mildew populations. Appl
Environ Microb. 73:5162-5172.

Chapter 5 QoI vs M. graminicola
140
Elard L, Comes AM, Humbert JF. 1996. Sequences of beta-tubulin cDNA from
benzimidazole-susceptible and -resistant strains of Teladorsagia circumcincta, a
nematode parasite of small ruminants. Mol Biochem Parasitol. 79:249-253.
Fernández-Ortuño D, Torés JA, de Vicente A, Pérez-García A. 2008. Field resistance to QoI
fungicides in Podosphaera fusca is not supported by typical mutations in the
mitochondrial cytochrome b gene. Pest Manag Sci. 64:694-702.
Fraaije BA, Burnett FJ, Clark WS, Motteram J, Lucas JA. 2005. Resistance development to
QoI inhibitors in populations of Mycosphaerella graminicola in the UK. In: Dehne HW,
Gisi U, Kuck KH, Russell PE, Lyr H, eds, Modern Fungicides and Antifungal
Compounds IV. pp 63-71 BCPC, Alton, UK.
Fraaije BA, Cools HJ, Fountaine J, Lovell DJ, Motteram J, West JS, Lucas JA. 2005. Role of
ascospores in further spread of QoI-resistant cytochrome b alleles (G143A) in field
populations of Mycosphaerella graminicola. Phytopathology 95:933-941.
Fraaije BA, Lucas JA, Clark WS, Burnett FJ. 2003 QoI resistance development in
populations of cereal pathogens in the UK. In: The BCPC International Congress-Crop
Science & Technology, pp. 689-694.
Gisi U, Pavic L, Stanger C, Hugelshofer U, Sierotzki H. 2005. Dynamics of Mycosphaerella
graminicola populations in response to selection by different fungicides. In Dehne HW,
Gisi U, Kuck KH, Russell PE and Lyr H, eds., Modern Fungicides and Antifungal
Compounds IV, pp 73-80, BCPC, Alton, UK.
Gisi U, Sierotzki H, Cook A, McCaffery A. 2002. Mechanisms influencing the evolution of
resistance to Qo inhibitor fungicides. Pest Manag Sci. 58:859-867.
Grasso V, Palermo S, Sierotzki H, Garibaldi A, Gisi U. 2006. Cytochrome b gene structure
and consequences for resistance to Qo inhibitor fungicides in plant pathogens. Pest
Manag Sci. 62:465-472.
Hall BG, Salipante SJ, Barlow M. 2004. Independent origins of subgroup B1 + B2 and
subgroup B3 metallo-beta-lactamase. J Mol Evol. 59:133-141.
Hardwick NV, Jones DR, Slough JE. 2001. Factors affecting diseases in winter wheat in
England and Wales, 1989-98. Plant Pathol. 50:453-432.
Hunter T, Coker RR, Royle DJ. 1999. The teleomorph stage, Mycosphaerella graminicola, in
epidemics of septoria tritici blotch on winter wheat in the UK. Plant Pathol. 48:51-57.

Chapter 5 QoI vs M. graminicola
141
Kim YS, Dixon EW, Vincelli P, Farman ML. 2003. Field resistance to strobilurin (QoI)
fungicides in Pyricularia grisea caused by mutations in the mitochondrial cytochrome b
gene. Phytopathology 93:891-900. Kishino H, Hasegawa M. 1989. Evaluation of the maximum likelihood estimate of the
evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J Mol Evol. 29:170-179.
Linde C, Zhan J, McDonald BA. 2002. Population structure of Mycosphaerella graminicola
from lesions to continents. Phytopathology 92:946-955.
McDonald BA, Miles J, Nelson LR, Pettway RE. 1994. Genetic variability in nuclear-DNA
in field populations of Stagonospora nodorum. Phytopathology 84:250-255.
McDonald BA, Zhan J, Yarden O et al. 1999. The population genetics of Mycosphaerella
graminicola and Phaeosphaeria nodorum. In Lucas JA, Bowyer P, Anderson HM, eds.,
Septoria on Cereals: A Study of Pathosystems, pp 44-69, CABI, Wallingford, UK.
Nardi F, Carapelli A, Vontas JG, Dallai R, Roderick GK, Frati F. 2006. Geographical
distribution and evolutionary history of organophosphate-resistant ACE alleles in the
olive fly (Bactrocera oleae). Insect Biochem Mol Biol. 36:593-602.
Pasche JS, Piche LM, Gudmestad NC. 2005. Effect of the F129L mutation in Alternaria
solani on fungicides affecting mitochondrial respiration. Plant Dis. 89:269-278.
Price EW and Carbone I. 2005. SNAP: workbench management tool for evolutionary
population genetic analysis. Bioinformatics 21:402-404.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. 2003. DnaSP, DNA polymorphism
analyses by the coalescent and other methods. Bioinformatics 19:2496-2497.
Sanderson FR. 1972. Mycosphaerella graminicola (Fuckel) Sanderson comb. nov., the
ascogenous state of Septoria tritici Rob. and Desm. New Zeal J Bot. 14:359-360.
Shaw MW, Royle DJ. 1989. Airborne inoculum as a major source of Septoria tritici
(Mycosphaerella graminicola) infections in winter wheat crops in the UK. Plant Pathol.
38:35-43.
Sierotzki H, Frey R, Wullschleger J, Palermo S, Karlin S, Godwin J, Gisi U. 2007.
Cytochrome b gene sequence and structure of Pyrenophora teres and P. tritici-repentis
and implications for QoI resistance. Pest Manag Sci. 63:225-233.

Chapter 5 QoI vs M. graminicola
142
Sierotzki H, Wullschleger J, Gisi U. 2000. Point-mutation in cytochrome b gene conferring
resistance to strobilurin fungicides in Erysiphe graminis f. sp. tritici field isolates. Pest
Biochem Phys. 68:107-112.
Steinfeld U, Sierotzki H, Parisi S, Poirey S, Gisi U. 2001. Sensitivity of mitochondrial
respiration to different inhibitors in Venturia inaequalis. Pest Manag Sci. 57:787-796.
Swofford DL. 2002. PAUP*. Phylogenetic Analysis Using Parsimony (and other methods),
Version 4. Sinauer Associates, Sunderland, MA.
Torriani SFF, Goodwin SB, Kema GHJ, Pangilinan JL, McDonald BA. 2008. Intraspecific
comparison and annotation of two complete mitochondrial genome sequences from the
plant pathogenic fungus Mycosphaerella graminicola. Fungal Genet Biol. 45:628-637.
Zhan J, Mundt CC, McDonald BA. 2001 Using restriction fragment length polymorphisms to
assess temporal variation and estimate the number of ascospores that initiate epidemics in
field populations of Mycosphaerella graminicola. Phytopathology 91:1011-1017.
Zhan J, Pettway RE, McDonald BA. 2003. The global genetic structure of the wheat
pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low
mitochondrial diversity, regular recombination, and gene flow. Fungal Genet Biol.
38:286-297.

CHAPTER 6
Screening for QoI resistance in a global sample of Rhynchosporium secalis
Stefano FF Torriani, Celeste C Linde, Bruce A. McDonald. 2009. Lack of G143A QoI
resistance allele and sequence conservation for the mitochondrial cytochrome b gene in a
global sample of Rhynchosporium secalis. Australasian Plant Pathology 38:202-207.

Chapter 6 QoI vs R. secalis
144

Chapter 6 QoI vs R. secalis
145
Abstract
Barley scald caused by Rhynchosporium secalis is often controlled by fungicides, most
recently by those in the strobilurin-based (QoI) class. Since the launch of QoIs in 1996 a
range of important plant pathogens including Blumeria graminis, Mycosphaerella fijiensis
and Plasmopara viticola, developed resistance. Present monitoring data indicate that R.
secalis populations remain sensitive. The primary molecular mechanism of QoI resistance in
several fungi is a point mutation at codon 143 in the mitochondrial encoded cytochrome b
gene (cytb), which causes the substitution of glycine by alanine (G143A). We characterized
the cytb gene of R. secalis, assessed the intraspecific and interspecific sequence diversity,
developed a PCR-RFLP diagnostic tool to detect the most common allele associated with QoI
resistance, screened a global collection of 841 R. secalis isolates for this allele and tested a
representative sample of isolates for QoI resistance in vitro. The results indicated a high
degree of conservation for the cytb gene at both intra- and interspecific levels and complete
QoI sensitivity in all R. secalis populations tested.

Chapter 6 QoI vs R. secalis
146
Introduction
Rhynchosporium secalis causes an important disease of barley known as leaf blotch or
scald. Scald occurs wherever barley is grown and can cause yield losses of 35% or more
(Shipton et al. 1974; Khan 1986). In addition to cultivated barley (Hordeum vulgare), R.
secalis infects rye and several wild grass species (Caldwell 1937), although the species on rye
is most likely a different species (Zaffarano et al. 2006 and 2008). R. secalis overwinters
mainly on infected leaves and crop residues which can serve as sources of primary inoculum
(Caldwell 1937; Skoropad 1959; Evans 1969; Ayesu-offei and Carter 1974). The pathogen
can also infect seed, although seed-borne inoculum may be only a minor source if the seed
are treated. Moist conditions and temperatures between 15-20°C favor host infection and
disease development, with typical symptoms produced within approximately 14 days
(Caldwell 1937; Skoropad 1957; Shipton et al. 1974).
Fungicides are used routinely in Europe to control scald and other principal fungal
diseases of barley. For example, by the end of the 1980s 90% of the winter barley crop was
treated at least once with fungicides (Oerke et al. 1994). Fungicides exhibited varying
efficacy against scald and resistance has developed to most fungicides (Cooke et al. 2004).
However, some of the newest generation of active ingredients, particularly QoI chemistry
types, remain effective. The QoI fungicides have as their specific target the quinol-oxidizing
(Qo) site of the mitochondrial cytb protein. The QoI fungicides act by blocking electron
transport, thereby inhibiting respiration (Bartlett et al. 2002). A point mutation at codon 143
in the cytb gene leading to an amino-acid change from glycine to alanine (G143A) has been
shown to be the major mechanism of resistance to QoI fungicides in Blumeria graminis,
Mycosphaerella fijiensis, Plasmopara viticola and other plant pathogenic fungi (Gisi et al.
2000; Sierotzki et al. 2000a and 2000b; Bartlett et al. 2002). Recently another mutation
(F129L), conferring only moderate resistance, was identified in other fungi, including
Alternaria solani (Pasche et al. 2005) and Pyrenophora teres (Sierotzki et al. 2007). Based on
current knowledge, G143A and F129L mutations display different resistance factors (RF =
Effective dose 50 [resistant strain] / Effective dose 50 [sensitive wild-type strain]) ranging
from 5 to 15 for F129L and usually greater than 100 for G143A (FRAC International,
Germany). The amino acid sequence of the cytochrome b protein is highly conserved both

Chapter 6 QoI vs R. secalis
147
within and between species, but some synonymous mutations in nucleotide sequences have
been found within species (Yokoyama et al. 2001). Generally the only non-synonymous
mutations identified within species were those conferring QoI resistance, but some
exceptions have been reported (Kim et al. 2003).
The QoI fungicides were introduced in 1996 and the first resistance appeared in B.
graminis in 1998 (Sierotzki et al. 2000b), followed by Pseudoperonospora cubensis in 1999
(Ishii et al. 2001) and Pyricularia grisea in 2000 (Vincelli and Dixon 2002). More recently
QoI resistant isolates have been detected in several other plant pathogens, including P.
viticola on grape, Sphaerotheca fuliginea on cucumber, M. fijiensis on banana, Venturia,
inaequalis on apple and Mycosphaerella graminicola on wheat (Ma et al. 2003; Fraaije et al.
2005). In the case of P. grisea, analysis of 58 isolates from nine different states in the USA
indicated that the cytb mutations encoding resistance were present in the fungal population
before the QoI fungicides were introduced (Kim et al. 2003). The sampling strategy in this
case was not appropriate to determine the frequency of the resistant mutations in the naïve
populations but Fraaije et al. (2002) showed that the frequency of resistant strains of B.
graminis could increase from 2% to 58% in a single season following three applications of
the fungicide. Thus even very low frequencies of resistant cytb mutations in naïve
populations could lead to a rapid loss in effectiveness of the QoI fungicides. Independent
multiple origins of the G143A resistance allele were recently documented in two plant
pathogenic fungi (Chen et al. 2007; Torriani et al. 2009).
Grasso et al. (2006) predicted that the presence of a Type 1 intron located directly after
the codon for glycine at position 143 would prevent the emergence of the G143A mutation by
blocking proper splicing of the intron. Until now the QoI fungicides have been effective
against scald and no resistance has been reported in the field, but it is not known if the lack of
resistance is due to the presence of a Type 1 intron after codon 143. If the intron is not
present, we consider it most likely that resistance will appear first in areas where QoIs are
used most intensively. If resistance appears, a molecular diagnostic tool will be useful to
detect and monitor the development and spread of QoI resistance. In anticipation of this
need, we set out to develop a diagnostic tool for the QoI resistance allele that is most likely to
emerge.

Chapter 6 QoI vs R. secalis
148
With the aim of developing a method for detection of the G143A mutation in R. secalis,
the objectives of this study were 1) to characterize the cytb gene and determine if it carries an
intron directly after the glycine codon at position 143, 2) to determine how much diversity
exists in the cytb gene in a selected worldwide collection, 3) to use the developed PCR-RFLP
method to search for the G143A mutation in R. secalis populations from around the world
and 4) to test a representative sample of isolates for QoI resistance in vitro.
Materials and methods
Fungal isolation and DNA extraction. The isolations of R. secalis analyzed in this
project were made from plant leaves as described by McDonald et al. (1999). DNA was
extracted following the protocol described by McDonald et al. (1999) and Linde et al. (2003).
Cytochrome b gene sequencing, characterization and intra/interspecific diversity.
The cytb gene of R. secalis was identified using a BLASTN search (Basic Local Alignment
Search Tool Nucleotide; NCBI, Bethesda, MD) of the sequences arising from a R. secalis
mitochondrial DNA library developed in our laboratory.
The entire cytb gene of 40 selected R. secalis isolates (Table 1) was PCR-amplified using
the primers RscytbF (5’-ACC AAC CAC AGC GAC TTA GAT AG-3’) and RscytbR (5’-
TGG GAT AGC AGT ATC CAC ACC-3’). Each PCR mixture contained 5-10 ng of DNA in
a 20 µL reaction volume containing 10 pmol of each primer, 100 µM of each nucleotide, 2
µL of 10× PCR buffer (1× PCR buffer: 10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl, 2
mM MgSO4, 0.1% Triton X-100, [pH 8.8]), and 1 U of Taq DNA Polymerase (New England
Biolabs, England). The PCR amplifications were carried out under the following conditions:
initial denaturation at 96°C for 2 min, followed by 35 cycles of 96°C for 1 min, 55°C for 1
min, and 72°C for 1 min, with a final extension at 72°C for 5 min. PCR products were
subjected to three separate sequencing reactions using the primers RscytbF, RscytbR and
RscytbstrR (5’-GCA GGT GGA GTT TGC ATA GG-3’), providing a sequence of 1631
nucleotides (FJ346339). Sequencing reactions were performed using the ABI PRISM BigDye
Terminator v3.0 ready reaction cycle sequencing kit (Applied Biosystems, Foster City, CA).
The sequencing PCRs were in a total volume of 10 µL using 20 to 40 ng DNA, 10 pmol of

Chapter 6 QoI vs R. secalis
149
primer and 2 µL BigDye reaction mix diluted 1:4. The cycling profile was: 10 sec
denaturation at 95°C, 5 sec annealing at 50°C and 4 min extension at 60°C for 100 cycles.
The PCR products were purified after the sequencing reaction to remove buffer salts and
unincorporated ddNTPs through Sephadex G-50 DNA Grade F (Amersham Biosciences,
Switzerland). The cleaned PCR products were then loaded into a 3100 ABI automated
sequencer. The sequences were visualized and analyzed with the Sequencher version 4.2
software package (Gene Codes Corporation, Ann Arbor, MI) using the genetic code of
Pezizomycotina that diverges from the standard nuclear code for the codon TGA, which was
read as Trp and not as Stop. The complete cytb sequences were translated and tested for
similarities with other organisms present in the GenBank database using BLASTP (Fig. 1).
Figure 1. Amino-acid sequence of Rhynchosporium secalis cytb protein deduced from the obtained DNA sequence. The amino-acid sequences from Venturia inaequalis (AF004559.1), Mycosphaerella graminicola (AY247413.1) and Aspergillus niger (D63375.1) are also shown. The conserved amino acids in all presented organisms are marked in light grey; dark gray denotes amino acids conserved in at least two organisms. The glycine at position 143, if replaced by alanine, is expected to confer resistance to strobilurin.

Chapter 6 QoI vs R. secalis
150
Detection of QoI resistance allele G143A using PCR-RFLP. A worldwide collection of
841 R. secalis isolates (Table 1) was screened for the G143A mutation known to confer the
highest QoI resistance in most fungal pathogens (Gisi et al. 2000; Sierotzki et al. 2000a and
2000b; Bartlett et al. 2002). The isolates were from field populations collected over the last
20 years and most of them were described earlier (McDonald et al. 1989 and 1999; Salamati
et al. 2000; Linde et al. 2003; Schürch et al. 2004, Zaffarano et al. 2006). For the screening,
specific primers for the R. secalis cytb were designed: RscytbstrF (5’-TGC ATT ACA ACC
CTA GTA TAT TAG-3’) and RscytbstrR. The PCR reactions and the thermal cycling
conditions were as described above except that the annealing temperature was decreased to
50°C. A portion of each PCR product (10 µL) was digested using 1U Fnu4HI (New England
BioLabs, England) for 4 h at 37°C. Digests were visualized on a 1% agarose gel with 1x tris-
borate-EDTA (TBE) buffer (Fig. 2). As a control we used M. graminicola isolates known to
be QoI-sensitive or QoI-resistant amplified using the MgcytbF and MgcytbR primers
described in Torriani et al. (2009).
Table 1. Origin of the 851 isolates of Rhynchosporium secalis used in this study
Continent (isolates)
Origin Years of collection
Sequenced isolates1
PCR-RFLP isolates
Sensitivity-test isolates1
Source or reference
Europe (n= 464) Switzerland 1999, 2000, 2001, 2003
2 97 - Linde et al. 2003, Schürch et al. 2004
Russia 2003 3 14 - L. Lebedeva England 1997 1 18 1 Schürch et al. 2004, J. Speakman France 1997 - 10 - Schürch et al. 2004, J. Speakman Turkey 1997 - 5 - Schürch et al. 2004, J. Speakman Germany 1997 1 61 1 Schürch et al. 2004, J. Speakman Sweden 1996 - 7 - Schürch et al. 2004, S. Salamati Finland 1996 2 67 2 Linde et al. 2003, Salamati et al. 2000,
Schürch et al. 2004 Norway 1996 2 185 2 Linde et al. 2003, Salamati et al. 2000,
Schürch et al. 2004 Africa (n=113) Tunisia 2003 2 - 2 A. Yahyaoyi South Africa 2002 2 59 2 Linde et al. 2003 Eritrea 2002 2 21 - A. Yahyaoyi Ethiopia 2001 2 31 2 Linde et al. 2003, Schürch et al. 2004 Asia (n=134) Azerbaijan 2003 2 - 2 A. Yahyaoyi Kazakhstan 2003 2 - 2 A. Yahyaoyi Syria 2002, 2003 2 62 2 M. Abang Jordan 2002 2 68 - Schürch et al. 2004 America (n=43) Argentina 2003 - 3 - B. Perez USA 1988 5 40 5 Linde et al. 2003, McDonald et al. 1989,
Salamati et al. 2000, Schürch et al. 2004 Oceania (n=97) New Zealand 2004 4 - 3 M. Cromey Australia 1996, 1997 4 93 1 Linde et al. 2003, McDonald et al.1999,
Salamati et al. 2000, Schürch et al. 2004 Total 40 841 27
1 The cytochrome b sequenced isolates and those tested for fungicide resistance were the same.

Chapter 6 QoI vs R. secalis
151
In vitro QoI sensitivity test. A representative sample of 27 isolates (Table 1), chosen
from the 40 used to assess the intraspecific diversity of cytb, was tested for QoI sensitivity in
vitro. Azoxystrobin was dissolved in methanol and adjusted to a concentration of 20 mg/mL.
The sensitivity of each isolate was determined by mycelial growth on potato dextrose agar
(PDA) amended with 0, 1, 10, or 100 µg/mL azoxystrobin and 0.5 mM salicylhydroxamic
acid (SHAM). Azoxystrobin and SHAM were added to autoclaved PDA cooled to 55°C. The
concentration of methanol was 0.5% (v/v) in all tests. PDA was also amended with methanol
to test for inhibitory effects of methanol. Conidia were suspended in water at 106 conidia/mL
and 5 µL aliquots of the conidial suspension were placed onto PDA in Petri dishes. Three
different isolates were tested simultaneously in each Petri dish, with three inoculations per
isolate. Each Petri dish assay was replicated three times. Following incubation at 18°C for
two weeks, mycelial growth was assessed visually and compared with the unamended PDA
and PDA amended with methanol controls (Fig. 3).
Figure 2. PCR-RFLP analysis of cytb-locus for detection of QoI resistance allele. a) The G143A mutation resulted in creation of a second Fnu4HI restriction site in cytb-locus for both R. secalis and M. graminicola. Molecular sizes are indicated within each fragment. b) An example of PCR amplification with the primers cytbstrF and cytbstrR from a QoI sensitive (s) isolate of R. secalis either undigested (RS s) or digested (RS s,d). As controls, a QoI sensitive (MG s) and a resistant (MG r) isolate of M. graminicola were amplified with the same PCR conditions using the primers MgcytbF and MgcytbR and digested (MG s,d; MG r,d). First and last lanes are a 100 bp size ladder. Arrows show the size of the bands of R. secalis (left) and from M. graminicola (right).

Chapter 6 QoI vs R. secalis
152
Results
Cytochrome b gene sequencing, characterization and intra/interspecific diversity. The
entire cytb gene (FJ346339) was sequenced for 40 isolates from five continents (Table 1) to
investigate the intraspecific diversity. No polymorphisms were detected, indicating a high
degree of conservation globally. Cytb gene is 1191 base pairs long, lacks introns, and is
predicted to encode a protein of 396 amino acids (Fig. 2; FJ346339). The organism with the
highest similarity was V. inaequalis with 89% identity and an e-value of zero (Table 2 and
Fig. 1). In general, organisms with the highest similarities were Ascomycetes, but some
similarity was also found with Basidiomycetes (Table 2).
PCR-RFLP analysis. A total of 841 field isolates (Table 1) were screened for the G143A
mutation using the primers RscytbstrF and RscytbstrR to amplify the cytb-locus surrounding
the G143A mutation site. The amplified PCR products were 652 bp in size and when digested
with the restriction enzyme Fnu4HI produced two fragments which were 418 and 234 bp
long (Fig. 2). A mutation that generated a second Fnu4HI restriction site (…GGNGC… to
…GCNGC…) in the cytb locus of R. secalis would provide evidence for the presence of the
QoI resistant allele as already shown for M. graminicola (Fig. 2; Torriani et al. 2009). All
isolates had the wild type allele, indicating sensitivity to the QoI fungicide.
Table 2. Fungi having significant alignments with cytochrome b of Rhynchosporium secalis Organism Taxa Accession number Similarity (%) E-value Venturia inaequalis Ascomycota O48334 89 0 Mycosphaerella graminicola Ascomycota YP_001648755 88 0 Aspergillus niger Ascomycota Q33798 83 0 Mycosphaerella fijiensis Ascomycota AAK27475 87 0 Penicillium marneffei Ascomycota Q6V9D6 85 0 Magnaporthe grisea Ascomycota AAO91629 82 0 Fusarium oxysporum Ascomycota AAW67488 82 0 Trichophyton rubrum Ascomycota Q9ZZ40 80 0 Blumeria graminis Ascomycota AAK26621 87 8e-173 Schizophyllum commune Basidiomycota Q94ZJ2 68 4e-149 Mycena viridimarginata Basidiomycota Q36445 64 2e-146

Chapter 6 QoI vs R. secalis
153
In vitro QoI sensitivity test. All 27 R. secalis isolates showed complete sensitivity to
azoxystrobin with no mycelial growth detected in PDA amended with azoxystrobin at 1, 10
or 100 ppm (data not presented).
Discussion
We conducted this research prior to the first report of QoI resistance in R. secalis,
anticipating that diagnostic tools and prior knowledge of the diversity found in the cytb gene
would be useful for monitoring the development and movement of fungicide-resistant
populations.
Figure 3. Example of the in vitro azoxystrobin sensitivity test for three of the twenty-seven tested Rhynchosporium secalis isolates.
The cytb of R. secalis is composed of 1191 nucleotides, has no introns, and encodes a
protein of 396 amino acids (Fig. 1; FJ346339). Our interest in this gene arises from the
finding that the cytb protein is the specific target for QoI fungicides. The replacement of
glycine by alanine at amino acid position 143 (G143A) is the major mechanism of resistance
for pathogenic fungi, as already demonstrated for V. inaequalis (Zheng et al. 2000), M.
fijiensis (Sierotzki et al. 2000a), B. graminis (Sierotzki et al. 2000b) and several other fungi.
The lack of an intron after codon 143 in R. secalis indicates that the G143A mutation will not
be lethal if it emerges (Grasso et al. 2006).

Chapter 6 QoI vs R. secalis
154
No polymorphisms were detected in the complete cytb DNA sequence in a global
collection of 40 isolates, indicating a high degree of conservation for this gene. The absence
of mutations for cytb is consistent with strong directional selection acting on this gene,
reflecting the importance of this protein in cellular respiration. The conservation seen in the
amino acid sequence within species is reflected at the interspecific level, with little variation
found between different fungal species. This contrasts with the nucleotide sequence variation
associated with the presence of different numbers of introns between species. Intron variation
in cytb reported thus far in other fungi range from no introns for M. graminicola (Torriani et
al. 2008) to the six introns in V. inaequalis (Zheng and Köller 1997).
A worldwide collection of 841 field isolates of R. secalis was screened for the presence of
the G143A mutation to determine the frequency of QoI resistant allele. No mutants were
found among the 841 isolates, indicating that the G143A mutation occurred at a frequency
lower than 0.0012 in the global population. The F129L mutation which confers only
moderate resistance in A. solani (Pasche et al. 2005), was not detected in the 40 isolates
sequenced in this study. The sensitivity test conducted on fungicide-amended PDA was
consistent with the molecular screens showing a complete sensitivity of R. secalis isolates to
QoI. These results indicate that resistant mutants do not exist at a detectable frequency in R.
secalis populations around the world, but selection could rapidly increase the frequency of
resistance when it emerges as Gisi et al. (2005) demonstrated for M. graminicola.
If resistance to QoI emerges, a molecular method will be useful to detect the resistant
allele. In anticipation of this need, the G143A resistance allele of R. secalis can be easily
detected through the PCR-RFLP method described here, using RscytbstrF and RscytbstrR as
primers and Fnu4HI as a restriction enzyme.
Acknowledgements
We thank M. Zala for his technical assistance and P. Ceresini for critical reading of the
manuscript. This project was supported by the Swiss National Science Foundation (grant
3100A0-104145). DNA sequence data were collected using facilities provided by the ETH
Genetic Diversity Center.

Chapter 6 QoI vs R. secalis
155
References
Ayesu-offei EN, Carter MV. 1974. Epidemiology of leaf scald of barley. Aust J Agr Res. 22:
383-390.
Bartlett DW, Clough JM, Godwin JR, Hall AA, Hamer M, Parr-Dobrzanski B. 2002. The
strobilurin fungicides. Pest Manag Sci. 58:647-662.
Caldwell RM. 1937. Rhynchosporium scald of barley, rye, and other grasses. J Agr Res.
55:175-197.
Chen WJ, Delmotte F, Richard-Cervera S, Douence L, Greif C, Corio-Costet MF. 2007. At
least two origins of fungicide resistance in grapevine downy mildew populations. Appl
Environ Microb. 73:5162-5172.
Cooke LR, Locke T, Lockley KD, Phillips A, Sadiq MDS, Coll R, Black L, Taggart PJ,
Mercer PC. 2004. The effect of fungicide programmes based on epoxiconazole on the
control and DMI sensitivity of Rhynchosporium secalis in winter barley. Crop Prot.
23:393-406.
Evans SG. 1969. Observations on development of leaf blotch and net blotch of barley from
barley debris, 1968. Plant Pathol. 18:116-118.
Fraaije BA, Burnett FJ, Clark WS, Motteram J, Lucas JA. 2005. Resistance development to
QoI inhibitors in populations of Mycosphaerella graminicola in the UK. In: Dehne HW,
Gisi U, Kuck KH, Russell PE and H. Lyr eds, Modern Fungicides and Antifungal
Compounds IV. 63-71 BCPC, Alton, UK
Fraaije BA, Butters JA, Coelho JM, Jones DR, Hollomon DW. 2002. Following the
dynamics of strobilurin resistance in Blumeria graminis f.sp tritici using quantitative
allele-specific real-time PCR measurements with the fluorescent dye SYBR Green. Plant
Pathol. 51:45-54.
Gisi U, Chin KM, Knapova G, Farber RK, Mohr U, Parisi S, Sierotzki H, Steinfeld U. 2000.
Recent developments in elucidating modes of resistance to phenylamide, DMI and
strobilurin fungicides. Crop Prot. 19:863-872.
Gisi U, Pavic L, Stanger C, Hugelshofer U, Sierotzki H. 2005. Dynamics of Mycosphaerella
graminicola populations in response to selection by different fungicides. In HW Dehne,

Chapter 6 QoI vs R. secalis
156
U Gisi, KH Kuck, PE Russell and H Lyr, eds., Modern Fungicides and Antifungal
Compounds IV, pp 73-80, BCPC, Alton, UK.
Grasso V, Palermo S, Sierotzki H, Garibaldi A, Gisi U. 2006. Cytochrome b gene structure
and consequences for resistance to Qo inhibitor fungicides in plant pathogens. Pest
Manag Sci. 62:465-472.
Ishii H, Fraaije BA, Sugiyama T, Noguchi K, Nishimura K, Takeda T, Amano T, Hollomon
DW. 2001. Occurrence and molecular characterization of strobilurin resistance in
cucumber powdery mildew and downy mildew. Phytopathology 91:1166-1171.
Khan TN. 1986. Effects of fungicide treatments on scald (Rhynchosporium secalis (Oud)
Davis) infection and yield of barley in western-Australia. Austr J Exp Agr. 26:231-235.
Kim YS, Dixon EW, Vincelli P, Farman ML. 2003. Field resistance to strobilurin (Q(o)I)
fungicides in Pyricularia grisea caused by mutations in the mitochondrial cytochrome b
gene. Phytopathology 93:891-900.
Linde CC, Zala M, Ceccarelli S, McDonald BA. 2003. Further evidence for sexual
reproduction in Rhynchosporium secalis based on distribution and frequency of mating-
type alleles. Fungal Genet Biol. 40:115-125.
Ma ZH, Felts D, Michailides TJ. 2003. Resistance to azoxystrobin in Alternaria isolates from
pistachio in California. Pest Biochem Phys. 77:66-74.
McDonald BA, McDermott JM, Allard RW, Webster RK. 1989. Coevolution of host and
pathogen populations in the Hordeum vulgare-Rhynchosporium secalis pathosystem. P
Natl Acad Sci USA 86:3924-3927.
McDonald BA, Zhan J, Burdon JJ. 1999. Genetic structure of Rhynchosporium secalis in
Australia. Phytopathology 89:639-645.
Oerke EC, Dehne HW, Schönbeck F, Weber A. 1994. Crop Production and Crop Prot.,
Elsevier, Amsterdam, pp. 297-364.
Pasche JS, Piche LM, Gudmestad NC. 2005. Effect of the F129L mutation in Alternaria
solani on fungicides affecting mitochondrial respiration. Plant Dis. 89:269-278.
Salamati S, Zhan J, Burdon JJ, McDonald BA. 2000. The genetic structure of field
populations of Rhynchosporium secalis from three continents suggests moderate gene
flow and regular recombination. Phytopathology 902:901-908.

Chapter 6 QoI vs R. secalis
157
Schürch S, Linde CC, Knogge W, Jackson LF, McDonald BA. 2004. Molecular population
genetic analysis differentiates two virulence mechanisms of the fungal avirulence gene
NIP1. Mol Plant Microbe In.10:1114-1125.
Shipton WA, Boyd WJR, Ali SM. 1974. Scald of barley. Rev Plant Pathol. 53:840-861.
Sierotzki H, Parisi S, Steinfeld U, Tenzer I, Poirey S, Gisi U. 2000a. Mode of resistance to
respiration inhibitors at the cytochrome bc(1) enzyme complex of Mycosphaerella
fijiensis field isolates. Pest Manag Sci. 56:833-841.
Sierotzki H, Wullschleger J, Gisi U (2000b) Point mutation in cytochrome b gene conferring
resistance to strobilurin fungicides in Erysiphe graminis f. sp tritici field isolates. Pest
Biochem Phys. 68:107-112.
Sierotzki H, Frey R, Wullschleger J, Palermo S, Karlin S, Godwin J, Gisi U. 2007.
Cytochrome b gene sequence and structure of Pyrenophora teres and P. tritici-repentis
and implications for QoI resistance. Pest Manag Sci. 63:225-233.
Skoropad WP. 1957. Temperature and humidity relationships in securing infection of barley
with Rhynchosporium secalis. Phytopathology 47:32-33.
Skoropad WP. 1959. Seed and seedling infection of barley by Rhynchosporium secalis.
Phytopathology 49:623-626.
Torriani SFF, Brunner PC, McDonald BA, Sierotzki H. 2009. QoI resistance emerged
independently at least 4 times in European populations of Mycosphaerella graminicola.
Pest Manag Sci. 65:155-162.
Torriani SFF, Goodwin SB, Kema GHJ, Pangilinan JL, McDonald BA. 2008. Intraspecific
comparison and annotation of two complete mitochondrial genome sequences from the
plant pathogenic fungus Mycosphaerella graminicola. Fungal Genet Biol. 45:628-637.
Vincelli P, Dixon E. 2002. Resistance to Q(o)I (strobilurin-like) fungicides in isolates of
Pyricularia grisea from perennial ryegrass. Plant Dis. 86:235-240.
Yokoyama K, Wang L, Miyaji M, Nishimura K. 2001. Identification, classification and
phylogeny of the Aspergillus section Nigri inferred from mitochondrial cytochrome b
gene. FEMS Microbiol lett. 200:241-246.
Zaffarano PL, McDonald BA, Zala M, Linde CC. 2006. Global hierarchical gene diversity
analysis suggests the Fertile Crescent is not the center of origin of the barley scald
pathogen Rhynchosporium secalis. Phytopathology 96:941-950.

Chapter 6 QoI vs R. secalis
158
Zaffarano PL, McDonald BA, Linde CC. 2008. Rapid speciation following recent host shifts
in the plant pathogenic fungus Rhynchosporium. Evolution 62:941-950.
Zheng DS, Köller W. 1997. Characterization of the mitochondrial cytochrome b gene from
Venturia inaequalis. Curr Genet. 32:661-666.
Zheng DS, Olaya G, Köller W. 2000. Characterization of laboratory mutants of Venturia
inaequalis resistant to the strobilurin-related fungicide kresoxim-methyl. Curr Genet.
38:148-155.

CHAPTER 7
General conclusions

Chapter 7 General conclusions
160

Chapter 7 General conclusions
161
The mitochondrial genomes of Mycosphaerella graminicola and Phaeosphaeria nodorum
represent the first completely annotated mitochondrial genomes of any species from the class
Dothideomycetes (Schoch et al. 2006), that includes economically important plant pathogenic
fungi affecting a variety of hosts, including Cochliobolus heterostrophus (southern blight on
corn), Leptosphaeria maculans (black leg on brassica crop), Pyrenophora tritici repentis (tan
spot on wheat), Venturia inaequalis (apple scab) and Mycosphaerella fijiensis (black
Sigatoka on bananas). In total the class Dothideomycetes comprises more than 6000 known
species. Only 57 complete fungal mitochondrial genome sequences are deposited in the
NCBI database today. The principal reason for the limited mitochondrial genome sequences
available was the expense of sequencing, but new low cost sequencing technologies, e.g.
pyrosequencing (Ronaghi et al. 1998) have increased the rate of new sequences releases. As
chapters 2 and 3 show, fungal mitochondrial genomes display some common features
interspecifically, including the expression of a standard set of genes involved in oxidative
phosphorylation, two genes encoding the large and the small ribosomal subunit (rnl and rns)
and a set of tRNA genes, often clustered in groups showing conservation in the gene order
(Ghikas et al. 2006). Although the number of completely sequenced fungal mtDNAs is
limited, it is clear that the tRNA gene clusters flanking rnl display a conserved gene order
among the classes Dothideomycetes, Eurotiomycetes and Sordariomycetes, forming the large
Pezizomycotina subphylum (with more than 32,000 described species; Kirk et al. 2001) of
Ascomycota. This mitochondrial genomic region could be used as an additional criterion in
phylogenetic analyses to distinguish between fungal classes. The evolutionary history is
recorded in the tRNA clusters as rearrangement in gene order; for example 1)
Sordariomycetes, in the tRNA genes located 3’-downstream rnl, had an inversion between
tRNA-Met and tRNA-His not found in the Eurotiomycetes or 2) Dothideomycetes, in the same
region, displayed the insertion of a tRNA-Glu not present neither in the Eurotiomycetes nor in
the Sordariomycetes. These tRNA gene rearrangements could also be reflected at order level
since M. graminicola underwent the loss of three tRNA genes if compared to P. nodorum and
all other fungal species.
A distinctive feature among fungal mitochondrial genome is that the intron content varies
widely between species, ranging from no or few introns like in M. graminicola and P.
nodorum to a substantial intron invasion as found in Leptosphaeria maculans (unpublished).

Chapter 7 General conclusions
162
If we take as example cytb, probably the best studied fungal mitochondrial gene because it
encodes the target of QoI fungicides, we find that M. graminicola and Rhynchosporium
secalis have an uninterrupted gene, P. nodorum displays one intron and L. maculans has six
introns and a total size 8.4x and 4.4x bigger than respectively M. graminicola and R. secalis.
Most mitochondrial introns are group I introns and most of the mitochondrial group I intron
open reading frames (ORFs) fall into two of the four main families of homing endonucleases
(Galburt and Stottard 2002; Lang et al. 2007), encoding LAGLIDADG and GIY-YIG as their
conserved sequence motifs (Belfort and Perlman 1995). Group I introns are commonly found
in fungal mitochondrial genes but are absent from most animal and protist mtDNA genomes
(Haugen et al. 2005). Intraspecifically it seems these introns are unevenly distributed among
the genes, with preferences for cox1, cytb and rnl (Lang et al. 2007). Some group I introns are
mobile and proliferate through intron homing, a mechanism able to move introns into
previously intronless genes (Lang. et al. 2007). Since introns are highly divergent sequences,
it is unlikely that they will remain conserved between taxa that separated millions of years
ago. A more likely explanation for the distribution of group I intron across the tree of life is
horizontal transfer (Dujon 1989). Future studies will be needed to clarify 1) why M.
graminicola is intronless, 2) if this is a common feature of all Mycospharellaceae, including
M. fijiensis and the S1/S2 populations from uncultivated hosts, 3) what would occur if a
group I intron will be introduced into the mtDNA and 4) if the larger number of group I
introns in L. maculans reflects an older invasion or if there are species where intron homing
is favored.
The evolutionary history of the mitochondrial genome inferred from mitochondrial
sequences dated the divergence of M. graminicola from the ancestral Mycosphaerella
populations, adapted to uncultivated grasses, to between 10,200 and 9,000 years ago,
confirming the coalescent analysis conducted using nuclear genes (Stukenbrock et al. 2007).
The epochal shift from a hunter-gatherer to an agricultural society left archeological traces,
telling of the profound changes occurring in Mesopotamia about 10,000 to 12,000 years ago
following cultivation of the first crops (Moore et al. 2000; Gopher et al. 2002). The evolution
of M. graminicola was therefore human-mediated through the beginning of agriculture, since
this pathogen was domesticated along with wheat. Analogously, the industrial revolution
brought changes to society as profound as the invention of agriculture 10,000 years ago. It is,

Chapter 7 General conclusions
163
therefore, not surprising that the first traces of evolutionary change thought to be a
consequence of the industrial revolution begin to be discovered today; some examples
include 1) the effect of air pollution on long term fungal pathogen population dynamics
(Bearchell et al. 2005), 2) the extinction/appearance of sensitive or resistant strains after
fungicide applications (chapter 5 and 3) the shift northwards of plant pathogens well adapted
in the South in response to global warming (La Porta et al. 2008).
The ability of horizontal gene transfer to increase virulence in recipient lineages is
documented in the literature, as recently was found for Pyrenophora tritici repentis that
received ToxA gene from P. nodorum (Friesen et al. 2006). Analogously this work suggests
the introgression of an exogenous putative gene in the mitochondrial RFLP haplotype 4 of M.
graminicola (chapter 3). The putative open reading frame (ORF) found in type 4, was absent
both intraspecifically, in the other mitochondrial haplotypes, and interspecifically in
Mycosphaerella spp. collected from uncultivated hosts. This study postulates that a horizontal
gene transfer introduced into M. graminicola the RFLP4-ORF that encodes for a protein,
acting as virulence factor. The RFLP4-ORF identification strengthens previous data, showing
nonrandom associations between mitochondrial haplotypes and infected hosts (Zhan et al.
2004). Future research will need to investigate the functions of the putative gene, when and if
it is transcribed and try to genetically silence it in mitochondrial type 4 and/or introduce it
into other mitochondrial types to test the effect on pathogenicity in durum wheat (Triticum
durum) and bread wheat (T. aestivum).
The importance of monitoring programs and studies trying to understand the evolutionary
mechanisms involved in the development of fungicide resistance, is well described in the
introductory sentence found on the homepage of Fungicide Resistance Action Committee
(FRAC): “Fungicides have become an integral part of efficient food production. The loss of a
fungicide to agriculture through resistance is a problem that affects us all”. In fact, the
development of a fungicide resistance causes critical yield losses, reduces profit and
decreases food production. Depending upon the fungicide mode of action, a pathogen can
evolve resistance gradually or suddenly. A decreased sensitivity can either be disruptive, like
found for QoIs, or gradual as found for the DMIs (Gisi et al. 2005). In the disruptive
mechanism the change in sensitivity can be rapid and in most cases is mediated by a single
amino acid substitution in the target protein, like the G143A mutations that confer QoI

Chapter 7 General conclusions
164
resistance. The continuous mechanism results in a stepwise reduction of sensitivity because
the nature of resistance is polygenic. M. graminicola, for example, evolved QoI resistance
suddenly in 2001, only five years after the QoI was first released on the market (Fraaije et al.
2005). As this work showed (chapter 5), the mutation causing G143A occurred several times
in different genetic and geographic backgrounds. The DMIs have been used for more than
twenty years and are still effective, although some reduction in efficacy has been detected
(Cools et al. 2005). When released, QoIs were supposed (wrongly) to display low risk to
develop resistance although the disruptive resistance mechanism because targeting the
product of a mitochondrial gene. The latest fungicide resistance management strategies
suggest using products in mixtures or in alternating spray programs to decrease the likelihood
that a pathogen develops resistance, since any isolates resistant to one fungicide could be
negatively selected by the second. As presented in this work a strong fungicide selection can
introduce the resistance allele independently several times through parallel evolution,
therefore a reduction of the fungicide amount in the environment has to be achieved to
decrease the selection pressure. An important piece of the fungicide resistance management
puzzle is based on effective monitoring to know the spread of the resistance and to evaluate
the effect of the different treatments and management strategies.
This PhD thesis brought new insights to better understand mitochondrial genetics and
evolution of some of the most economically damaging plant pathogenic fungi. These studies
have for the first time described the mitochondrial genomes from species belonging to the
Dothideomycetes class, providing the basis for further studies connected to this important
branch of the Ascomycetes. A more applied result from this work deals with fungicide
resistance management, identifying selection, parallel genetic adaptation and migration as the
principal evolutionary forces involved in the emergence and spread of fungicide resistance.

Chapter 7 General conclusions
165
References
Bearchell SJ, Fraaije BA, Shaw MW, Fitt BDL. 2005. Wheat archive links long-term fungal
pathogen population dynamics to air pollution. PNAS 102:5438-5442.
Belfort M, Perlman PS. 1995. Mechanisms of intron mobility. J Biol Chem. 270:30237-
30240.
Cools HJ, Fraaije BA, Lucas JA. 2005. Molecular examination of Septoria tritici isolates
with reduced sensitivities to triazoles. In Dehne HW, Gisi U, Kuck KH, Russell PE and
Lyr H, eds., Modern Fungicides and Antifungal Compounds IV, pp 103-114. BCPC
Alton, UK.
Dujon B. 1989. Group I introns as mobile genetic elements: facts and mechanistic
speculations - a review. Gene. 82:91-114.
Galburt EA, Stoddard BL. 2002. Catalytic mechanisms of restriction and homing
endonucleases. Biochemistry 41:13851-13860.
Ghikas DV, Kouvelis VN, Typas MA. 2006. The complete mitochondrial genome of the
entomopathogenic fungus Metarhizium anisopliae var. anisopliae: gene order and trn
gene clusters reveal a common evolutionary course for all Sordariomycetes, while
intergenic regions show variation. Arch Microbiol 185:393-401.
Gisi U, Pavic L, Stanger C, Hugelshofer U and Sierotzki H. 2005. Dynamics of
Mycosphaerella graminicola populations in response to selection by different fungicides.
In Dehne HW, Gisi U, Kuck KH, Russell PE and Lyr H, eds., Modern Fungicides and
Antifungal Compounds IV, pp 73-80, BCPC, Alton, UK.
Gopher A, Abbo S, Lev-Yadun ST. 2002. The ‘when’, the ‘where’ and the ‘why’ of the
Neolithic revolution in the Levant. Doc Praehist. 28:49-62.
Haugen p, Simon DM, Bhattacharya D. 2005. The natural history of group I introns Trends
Genet. 21:111-119.
Kirk PM, Cannon PF, David JC, Stalpers JA. 2001. Ainsworth and Bisby’s Dictionary of the
Fungi, 9th ed. CAB International, Wallingford, UK.
La Porta N, Capretti P, Thomsen IM, Kasanen R, Hietala AM, Von Weissenberg K. 2008.
Forest pathogens with higher damage potential due to climate change in Europe. Can J
Plant Pathol. 30:177-195.

Chapter 7 General conclusions
166
Lang BF, Laforest MJ, Burger G. 2007. Mitochondrial introns: a critical view. Trends Genet.
23:119-125.
Moore AMT, Hillman GC, Legge AJ. 2000. Village on the Euphrates: from foraging to
farming at Abu Hureyra. Oxford Univ. Press, New York, USA.
Ronaghi M, Uhlén M, Nyrén P. 1998. A sequencing method based on real-time
pyrophospate. Science 281:363-365.
Schoch CL, Shoemaker RA, Seifert KA, Hambleton S, Spatafora JW, Crous PW. 2006. A
multigene phylogeny of the Dothideomycetes using four nuclear loci. Mycologia
98:1041-1052.
Stukenbrock EH, Banke S, Javan-Nikkhah M, McDonald BA. 2007. Origin and
domestication of the fungal wheat pathogen Mycosphaerella graminicola via sympatric
speciation. Mol Biol Evol. 24:398-411.
Zhan J, McDonald BA. 2004. The interaction among evolutionary forces in the pathogenic
fungus Mycosphaerella graminicola. Fungal Genet Biol. 41:590-599.

Acknowledgements
167
Acknowledgments
I am grateful to many people for their support during this period. Especially thanks to:
Bruce McDonald for giving me the opportunity of this thesis
Patrick Brunner and Soren Banke for supervising me
Alex Widmer for being my co-examiner
Mamma Mia (non il musical) per tutto quello che mi ha dato
Marcello Zala, per quello che mi hai insegnato, dentro e fuori il labo
Paolo, per i peperoncini, i ricordi del Kerkis, ma soprattutto per avermi tenuto alto morale
A-(floor) people. Joana, Megan, Eva, Rubik, Pascal, Francesca, Carlotta
All others from the phytopathology group, Giovanni, Pato, Cate, Rex, Gob, Rochi, Frep,
Fabi, Patrice, Andi, Miske, Thalia, J(u)ana, Paulo, Danilo, Gogo, Cesare, Ueli, Uli, Gabri,
Thomas, Pierre, Moni, Celeste, Yvonne, Tina and those I forgot, because it is 5.30 am
I miei amici senza i quali la vita sarebbe noiosa
Teo e Geo (i coinqui), Alan, Dada, Olmo, Sandro, Vano, Guaina, Meui, Babs, Bastian, Lucio
(e tutto il Brè), Jamal, Tognone, Sam (il), Sam (la), Aida, Peda, Glauco, Puzzle, Daniel,
Stella, Nick R, Tania, Vero, Anna, Annina, Meda, Chiara, Mattia, Alice, Naty, The
Pussywarmers, Nick e Jenny, Crimy, Fedi, Ronzi, Fabbio, KiKa, Claudio, Alessio, Yvonne,
Imo, Canguri, Manu, Frige, Piffa, Zan, Teresa, Laura, Deon, Luli, Pisu e Rosy e tutti gli
altri…
Raoul, 3 anni non son bastati a capire l’ora della colazione, ma l’12.34 non si tocca. GRAZIE
Questa tesi è stata portata a termine nonostante
Medo, Eli, Aldo e Mitch
GRAZIE A TUTTI

Acknowledgements
168

Curriculum vitae
169
Curriculum Vitae
Stefano Torriani, born in Sorengo (TI), Switzerland, 17th January 1980
2005-2008 PhD student, Institute for Integrative Biology, Phytopathology Group, Federal Institute of Technology, ETH-Zurich, Switzerland. Project title: Mitochondrial genomes of plant pathogenic fungi. Supervised by Prof. Bruce A. McDonald and Dr. Patrick C. Brunner.
1999-2004 Diploma in Natural Sciences (Biology), ETH Zurich: Dipl. Natw. ETH 1995-1999 Liceo Lugano 1 (TI), scientific direction (Maturität Typus B) 1991-1995 Scuole Medie Istituto Elvetico, Lugano (TI) 1986-1991 Scuole Elementari Istituto Elvetico, Lugano (TI)
Pubblications • Stefano FF Torriani, Stephen B Goodwin, Gert HJ Kema, Jasmyn L Pangilinan, Bruce A.
McDonald. 2008. Intraspecific comparison and annotation of two complete mitochondrial
genome sequences from the plant pathogenic fungus Mycosphaerella graminicola.
Fungal Genetics and Biology 45:628-637.
• Stefano FF Torriani, Patrick C Brunner, Bruce A. McDonald, Helge Sierotzki. 2009. QoI
resistance emerged independently at least 4 times in European populations of
Mycosphaerella graminicola. Pest Management Science 65:155-162
• Stefano FF Torriani, Celeste C Linde, Bruce A. McDonald. 2009 Lack of G143A QoI
resistance allele and sequence conservation for the mitochondrial cytochrome b gene in a
global sample of Rhynchosporium secalis. Australasian Plant Pathology 38:202-207
• James K Hane, Rohan GT Lowe, Peter S Solomon, Kar-Chun Tan, Conrad L Schoch,
Joseph W Spatafora, Pedro W Crous, Chinappa Kodira, Bruce W Birren, Stefano FF

Curriculum vitae
170
Torriani, Bruce A McDonald, Richard P Oliver. 2007. Dothideomycete-Plant interactions
illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora
nodorum. The Plant Cell 19:3347-3368.
• Jiasui S Zhan, Stefano FF Torriani, Bruce A McDonald. 2007. Significant difference in
pathogenicyty between MAT1-1 and MAT1-2 isolates in the wheat pathogen
Mycosphaerella graminicola. Fungal Genetics and Biology 44:339-346.
Recommended

















