
Campione:
In provette con
EDTA
…E ogni tipo di tessuto con cellule nucleate

Sources of Biological Evidence
Blood
Semen
Saliva
Urine
Hair
Teeth
Bone
Tissue

Tracce biologiche:
Formazioni ungueali
Tracce biologiche:
Formazioni pilifere
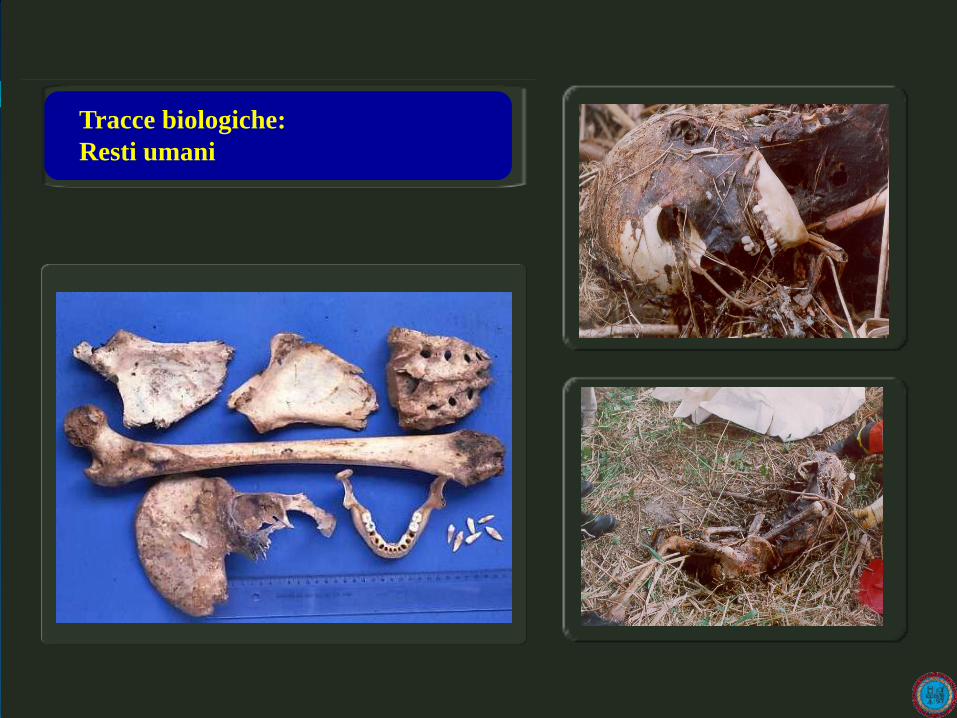
Tracce biologiche:
Resti umani

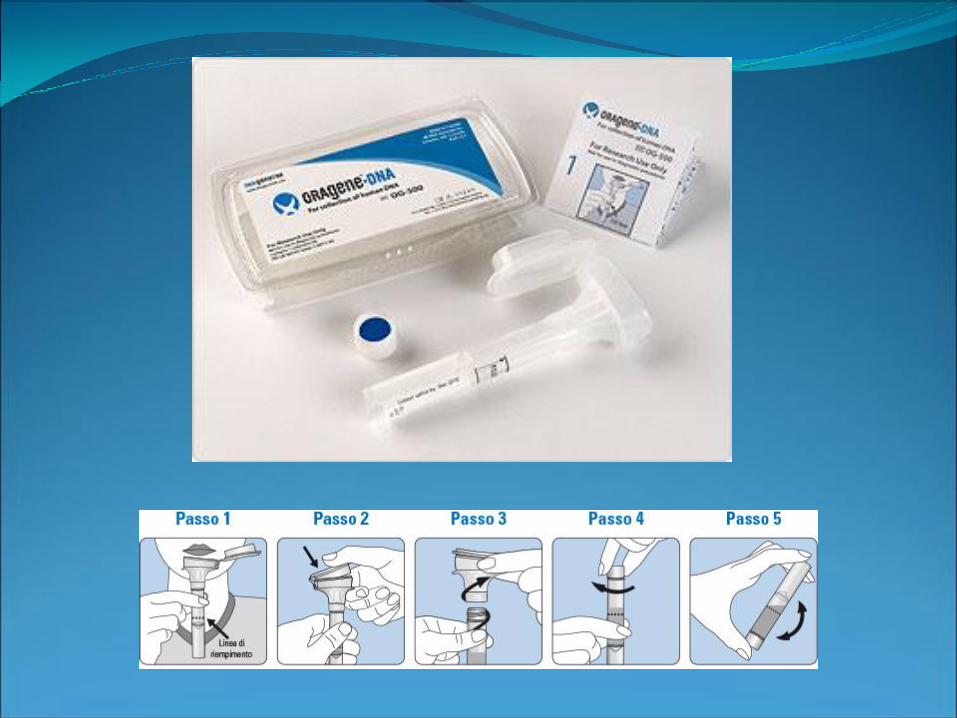
Prelievo buccale
oral swab

Raccolta di saliva
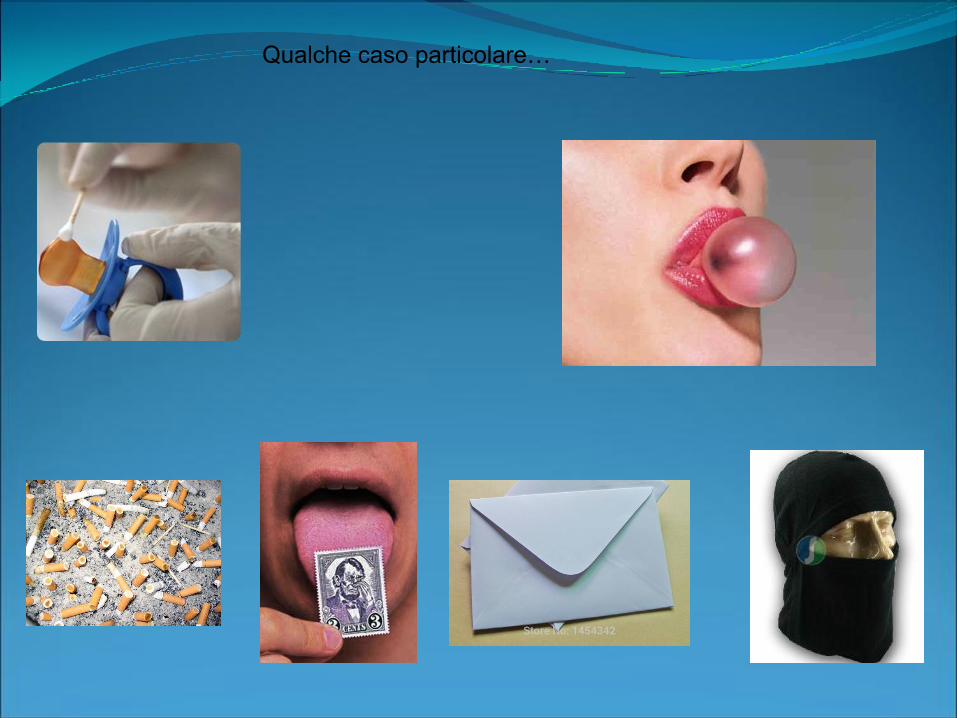
Qualche caso particolare…

Conservazione e trasporto dei campioni
Le tracce biologiche si conservano meglio se essiccate e poste in ambiente freddo; condizioni che riducono il tasso di crescita batterica e la degradazione del DNA.
I campioni devono essere sigillati e trasportati con cura
I campioni vanno conservati a +4°C (breve tempo), a – 20°C (lungo tempo) oppure a – 80 °C.
Eventuale conservazione in etanolo

Estrazione del DNA
• Metodo ideale
- elevata purezza
- elevato recupero
- sicuro: no reagenti pericolosi
- economico
- rapida esecuzione
• Scelta del metodo
- tipo di campione
- grado di purificazione richiesto
- tecnica impiegata per l’identificazione
- tempo richiesto
- numero di campioni da processare

Azione chelante
Lisi delle membrane
Rimozione delle proteine
Precipitazione del DNA
Solubilizzazione del DNA

Azione chelante
Soluzioni acquose contenti tensioattivi, agenti
chelanti, che bloccano l’attività della DNAasi,
generalmente sequestrando i cationi (in alcuni casi
determinano anche la lisi cellulare)
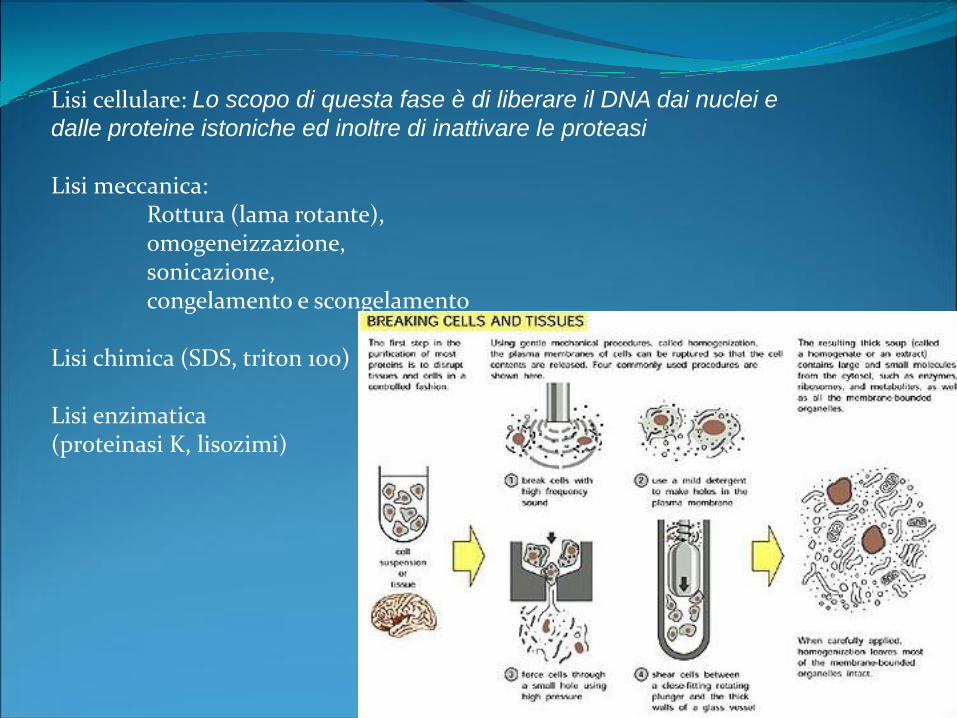
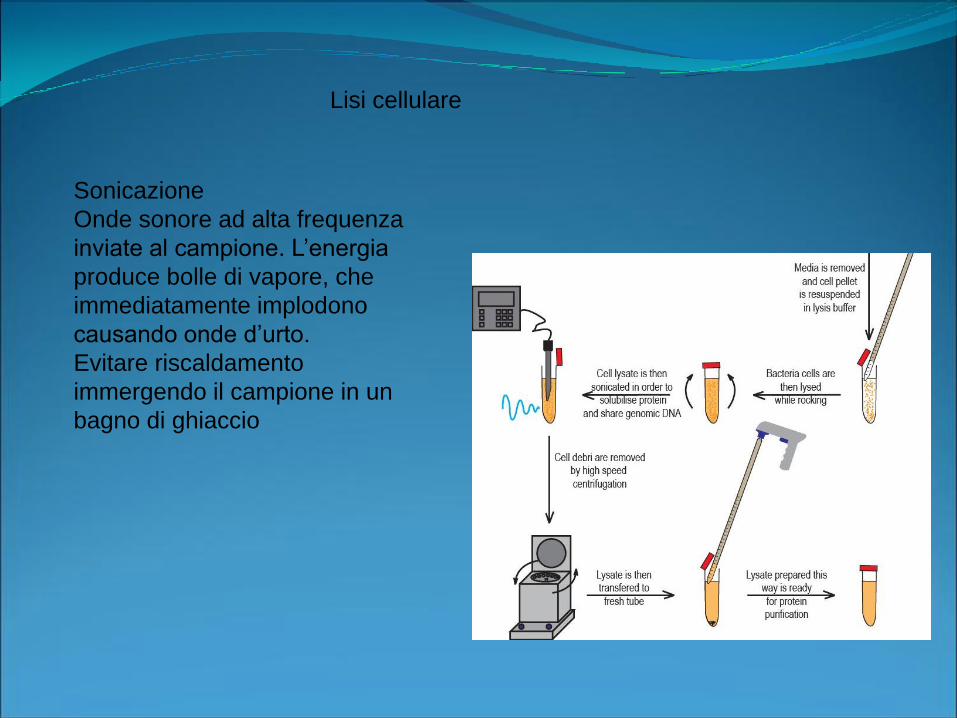
Lisi cellulare: Lo scopo di questa fase è di liberare il DNA dai nuclei e
dalle proteine istoniche ed inoltre di inattivare le proteasi
Lisi meccanica:Rottura (lama rotante), omogeneizzazione, sonicazione, congelamento e scongelamento
Lisi chimica (SDS, triton 100)
Lisi enzimatica (proteinasi K, lisozimi)
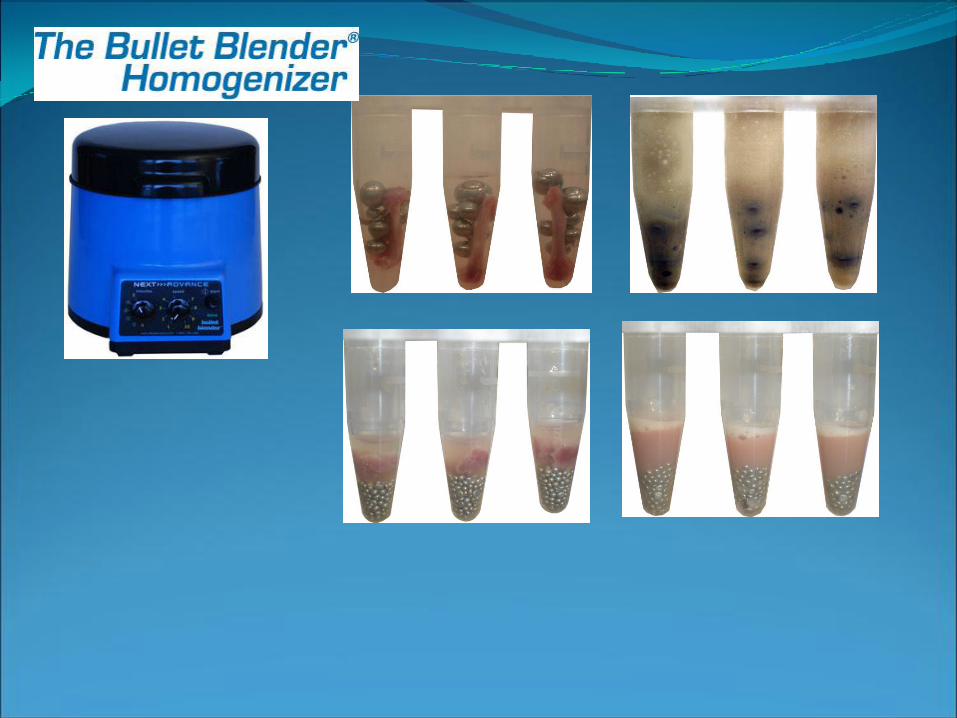
Lisi cellulare
Sonicazione
Onde sonore ad alta frequenza
inviate al campione. L’energia
produce bolle di vapore, che
immediatamente implodono
causando onde d’urto.
Evitare riscaldamento
immergendo il campione in un
bagno di ghiaccio

Lisi cellulare
Congelamento e scongelamento
Ingrossamento e rottura delle cellule. Necessari
diversi cicli

Lisi cellulare
Mortaio e pestello, previo congelamento con azoto liquido. X
cellule vegetali
Additivi e facilitatori della lisi.: con buffer ipotonico o lisozima (x
digerire la componente polissaccarida)
Lisi chimica:
Detergenti x lisi: teewn 20, sds, triton 100…

Lisi cellulare
Lisi enzimatica
Utilizzo di lisozimi, proteasi, cellulasi, …
Costoso e non sempre riproducibile
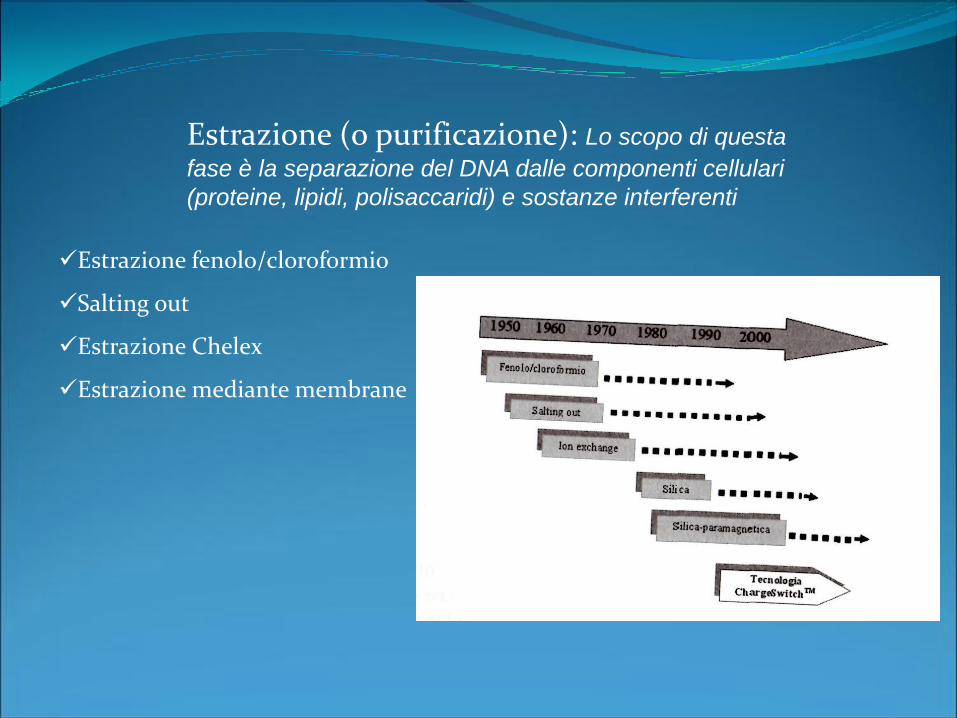
Estrazione fenolo/cloroformio
Salting out
Estrazione Chelex
Estrazione mediante membrane
Estrazione (o purificazione): Lo scopo di questa
fase è la separazione del DNA dalle componenti cellulari
(proteine, lipidi, polisaccaridi) e sostanze interferenti

Purificazione DNA con fenolo-cloroformio
Metodo di estrazione classico basato sulla diversa
solubilità delle molecole in soluzione (DNA/contaminanti)
tra due fasi non miscibili
- Lisi campione
- Tampone contenente SDS, EDTA, proteinasi K (500
μL)
- Estrazione DNA
- Trattamento 1:1 con fenolo-cloroformio alcool
isoamilico pH 8.0
(a pH 7.5-8.0 il DNA è nella fase acquosa; a pH 5.0-6.0 il
DNA è nella fase organica)
-Dopo Centrifugazione: 3 fasi:
- fase acquosa contenente DNA
- interfaccia contenente proteine denaturate
- fase organica contenente i lipidi
- Prelevare la fase acquosa superiore
Procedura a resa elevata, economica, tempi lunghi,
sostanze pericolose

Salting out (estrazione salina)
Le proteine risultano meno solubili in alte concentrazione di Sali (generalmente il NaCl)
In soluzione acquosa le proteine assumono una configurazione ripiegata con gli aminoacidi idrofobici all’interno e idrofilici all’esterno. In concentrazioni saline molto elevate , alcune molecole di acqua vengono attratte dagli ioni del sale, diminuendo così il numero di molecole di acqua disponibili per interagire con le proteine. Maggiore interazione proteina-proteina: le proteine così precipitano formando interazioni idrofobiche fra di loro.
Procedura rapida, sicura e poco costosa.

Estrazione Chelex
Il Chelex è una resina a scambio ionico in grado di legare gli ioni bivalenti come il magnesio rendendolo quindi indisponibile per il funzionamento delle nucleasi.
Produce DNA a singolo filamento. Non adatta per il DNA antico
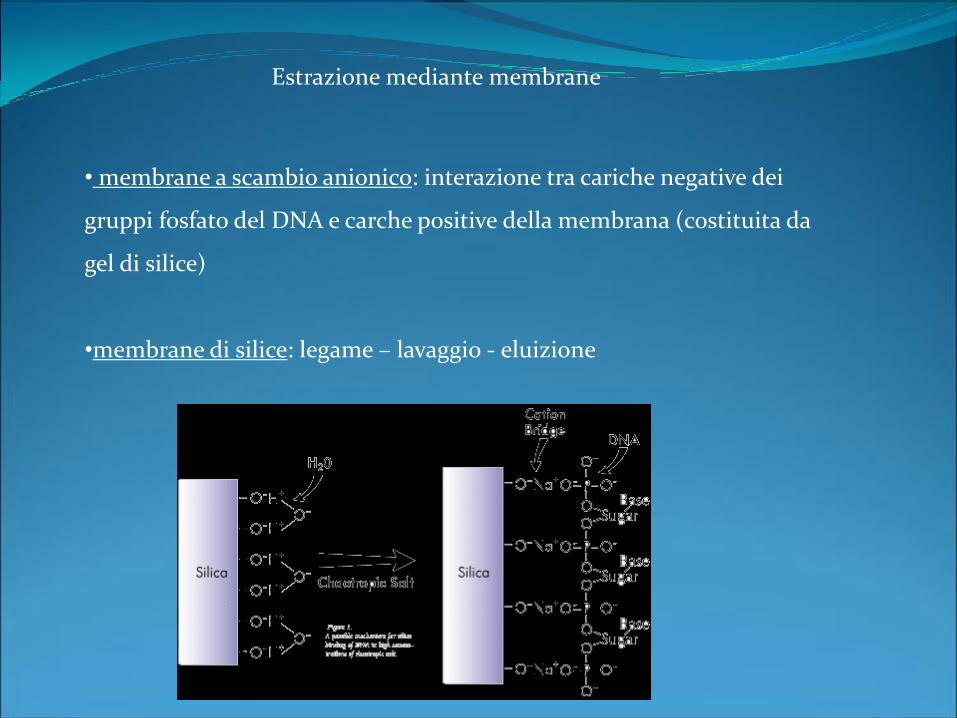
Estrazione mediante membrane
• membrane a scambio anionico: interazione tra cariche negative dei
gruppi fosfato del DNA e carche positive della membrana (costituita da
gel di silice)
•membrane di silice: legame – lavaggio - eluizione

Purificazione DNA su gel di silice
Il metodo è basato sulla proprietà degli a. nucleici
di adsorbirsi alla silice, a differenza delle proteine
e di altri composti organici
- Lisi campione:- con tampone contenente detergenti, proteinasi K, sali a 56°C per
10 min.
- Adsorbimento DNA al gel di silice contenuto in
una colonnina- in presenza di etanolo e sali cautropici il DNA si lega alla silice
- lavaggi con soluzioni contenenti alcool in grado di rimuovere
le proteine denaturate e gli altri componenti cellulari e lasciare
il DNA legato alla silice
- Eluizione DNA: con H2O o soluzioni a bassa concentrazione
salina
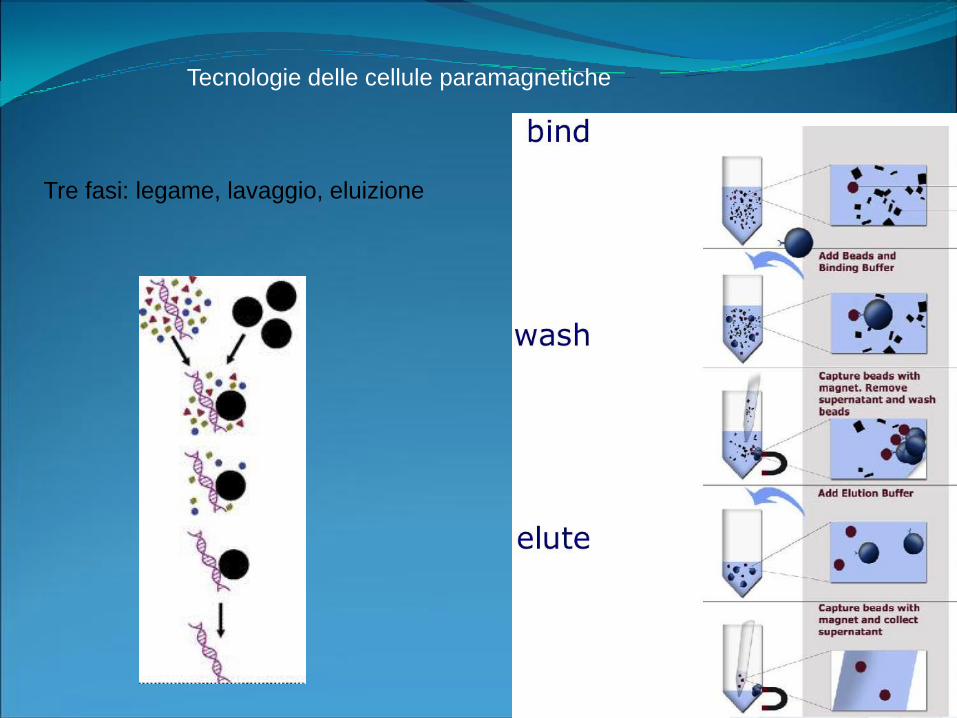
Tecnologie delle cellule paramagnetiche
Tre fasi: legame, lavaggio, eluizione

Estrazione Charge-Switch
Utilizzo di particelle paramagnetiche,
rivestite da molecole in grado di
cambiare la carica in base al pH del
mezzo
(la carica positiva interagisce con il
DNA e non con le proteine)
Metodo rapido. DNA con elevato
grado di purezza.

La resa e la purezza, intese sia come presenza in soluzione
dell’acido nucleico in esame sia come assenza di sostanze
contaminanti che, legandosi ai reagenti in soluzione, potrebbero
modificare i risultati dell’analisi.
Resa: quantità di acido nucleico che è possibile ottenere
Purezza: esprime quanto l’acido nucleico si trovi in presenza o
meno di contaminanti e in particolare si utilizza il rapporto di
assorbimento 260/280 nm.
Resa e purezza dell’estrazione

CampioniSpazzolino
tradizionale
Spazzolino in
etanoloListerine saliva
1 3,66 4,22 8,00 44,00
2 21,20 26,80 8,70 96,80
3 3,30 9,46 3,40 29,60
4 6,14 22,80 56,20 66,00
5 6,98 25,20 12,00 50,40
6 6,20 14,80 29,80 58,20
7 26,00 17,18 18,68 102,00
8 8,20 24,60 5,74 49,20
9 22,20 4,70 102,00 80,00
10 9,90 11,28 9,36 7,42
media 11,38 16,10 25,39 58,36
D.S. 8,42 8,54 31,22 29,22

Quantificazione DNA
Spettrofotometro:
Determina la quantità di luce ultravioletta assorbita dal campione in
esame.
2 differenti lunghezze d’onda: 260 e 280 nm.
lettura a 260: concentrazione di acido nucleico presente nel
campione.
lettura a 280 nm: proteine presenti nel campione
OD260/OD280: purezza dell’acido nucleico.
Purezza ottimale:OD260/OD280 intorno a 1,8, valori superiori
rivelano una presenza eccessiva di RNA. Se invece sono presenti
contaminazioni proteiche o residui di fenolo la OD260/OD280 avrà
un valore significativamente minore e non sarà possibile una
quantizzatine accurata del DNA.
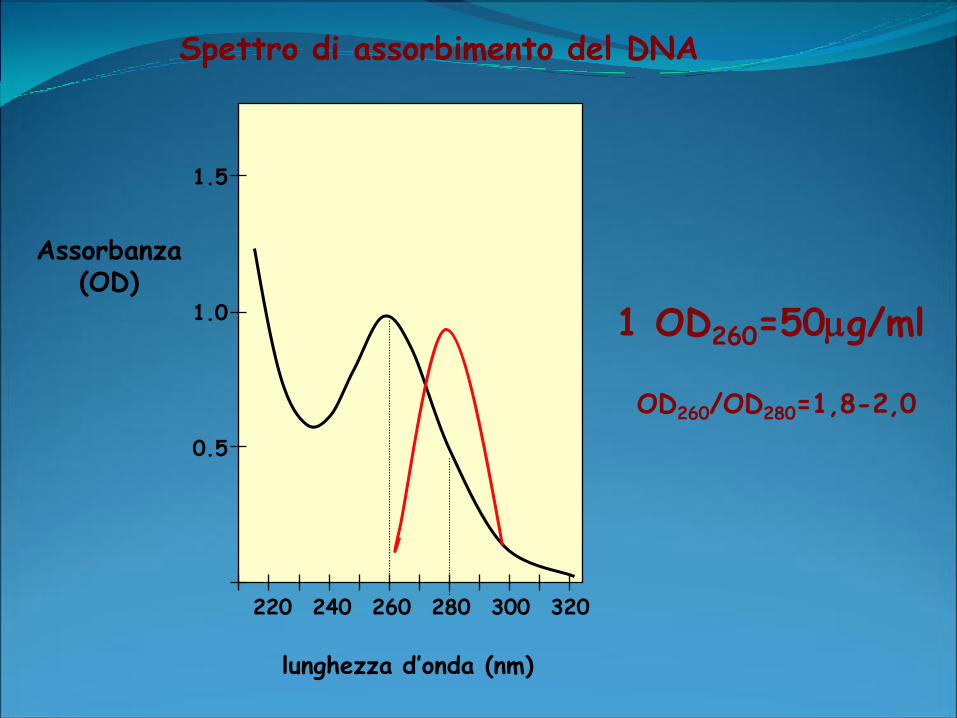
Spettro di assorbimento del DNA
220 240 260 280 300 320
0.5
1.0
1.5
lunghezza d’onda (nm)
Assorbanza(OD)
1 OD260=50g/ml
OD260/OD280=1,8-2,0
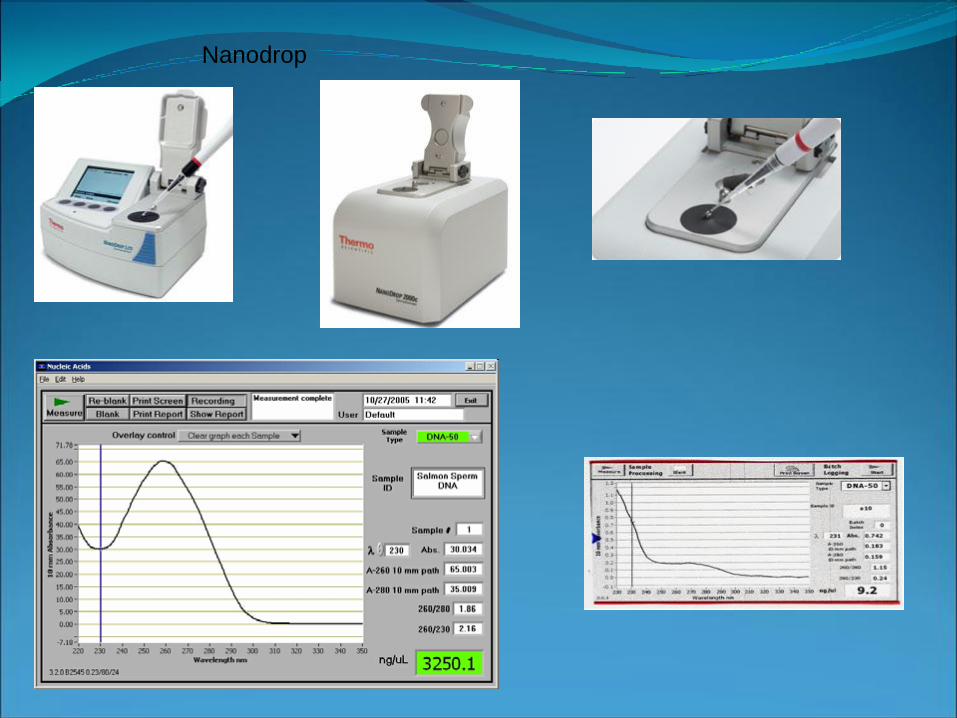
Nanodrop

Fluorimetro
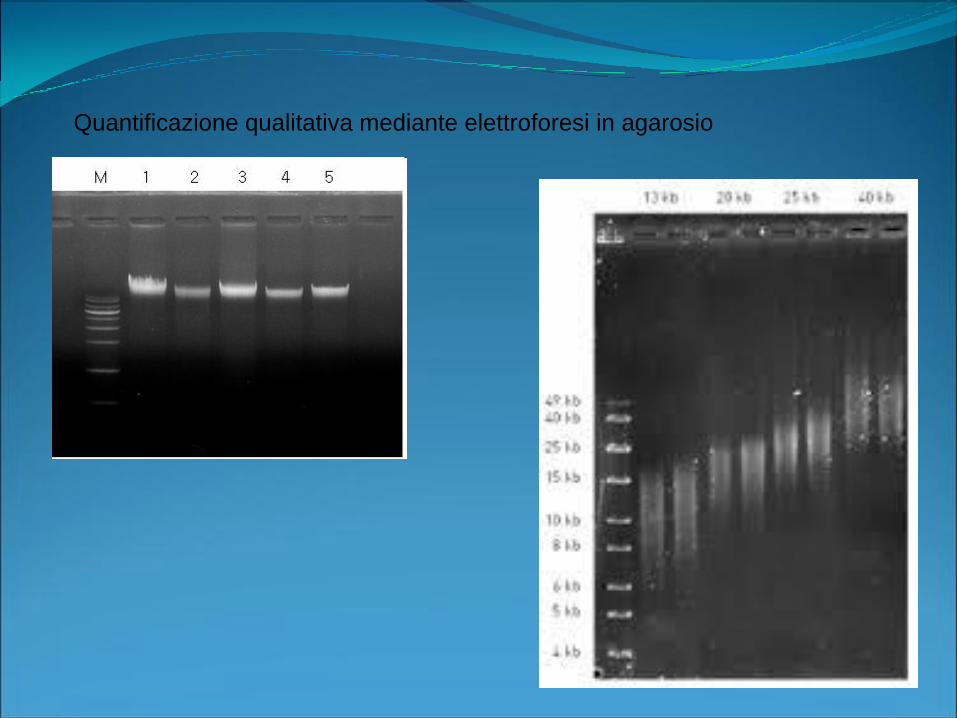
Quantificazione qualitativa mediante elettroforesi in agarosio

PCR
Mix:
DNA
dNTPs
Buffer+Mg
Taq
Primers

Numero di cicli123456789
101112131415161718192021222324252627282930
Numero di molecole di amplificati
248
163264
128256512
1.0242.0484.0968.192
16.38432.76865.536
131.072262.144524.288
1.048.5762.097.1524.194.3048.388.608
16.777.21635.544.43267.777.216
134.217.728268.435.456536.870.912
1.073.741.724
Resa teorica di una reazione di PCR a
partire da una singola copia di DNA
Y= N2n
Y= numero molecole di DNA
amplificato
N= numero molecole di DNA
di partenza
n= numero dei cicli di PCR

Log [DNA]
n° cicli
Fase Geometrica
Plateau
Fase Lineare
LA REAZIONE DI PCR

Competizione tra il prodotto dei cicli precedenti e i primers per l'ibridazione
Inattivazione termica dell’enzima
Riduzione del rapporto molare tra le concentrazioni della DNA-polimerasi e del DNA
Riduzione progressiva dell’efficienza di denaturazione e/o di ibridazione
Distruzione degli amplificati per l’attività esonucleasica 5’>3’ della polimerasi
Accumulo di pirofosfati (inibitori della polimerasi)
Progressiva diminuzione della concentrazione di uno o più componenti necessari alla reazione (?)
CAUSE DELL’EFFETTO PLATEAU
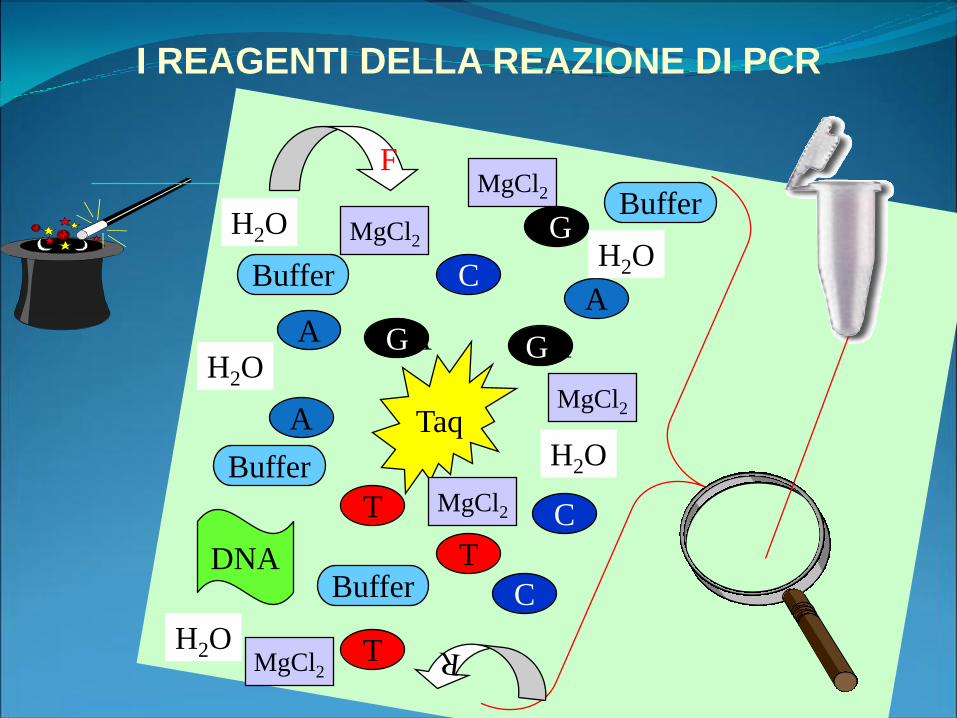
I REAGENTI DELLA REAZIONE DI PCR
A
GA
C
C
GA
G
A
C
T
T
A
T
Taq
DNA
MgCl2
MgCl2
MgCl2
MgCl2
MgCl2
Buffer
Buffer
Buffer
Buffer
F
H2OH2O
H2O
H2O
H2O

DNA TARGET:
Il DNA umano è costituito da circa 3.3 x 109 bp, (1bp= 650 dalton)
Il DNA contenuto in una cellula pesa circa 6.6 x10-6 ug
1 ug di DNA genomico corrisponde a circa 5 x 10-7 pmoli
Circa 150.000 cellule umane contengono 1 ug di DNA
3 pg di DNA genomico contengono 1 copia di ogni gene umano
QUANTITA’ OTTIMALE
0.1 a 1-2 ug di DNA genomico
Per reazione di PCR

Troppo DNA
Troppo RNA
Presenza di emoglobina
Eparina
Ioni metallici
SDS
Composti Aromatici
Etanolo
Urea
Detergenti
Ecc.
Inibitori della PCR
DNA TARGET

Estrema cura nella scelta della regione da amplificare
Estrema purezza
Lunghezza: 20-30 paia di basi (non meno di 16 bp)
Equilibrato rapporto AT/GC
Tm comprese tra 45-68° C
Tm simili (+/- 2 C)
OLIGONUCLEOTIDI O PRIMERS
SPECIFICI DEGENERATI UNIVERSALI
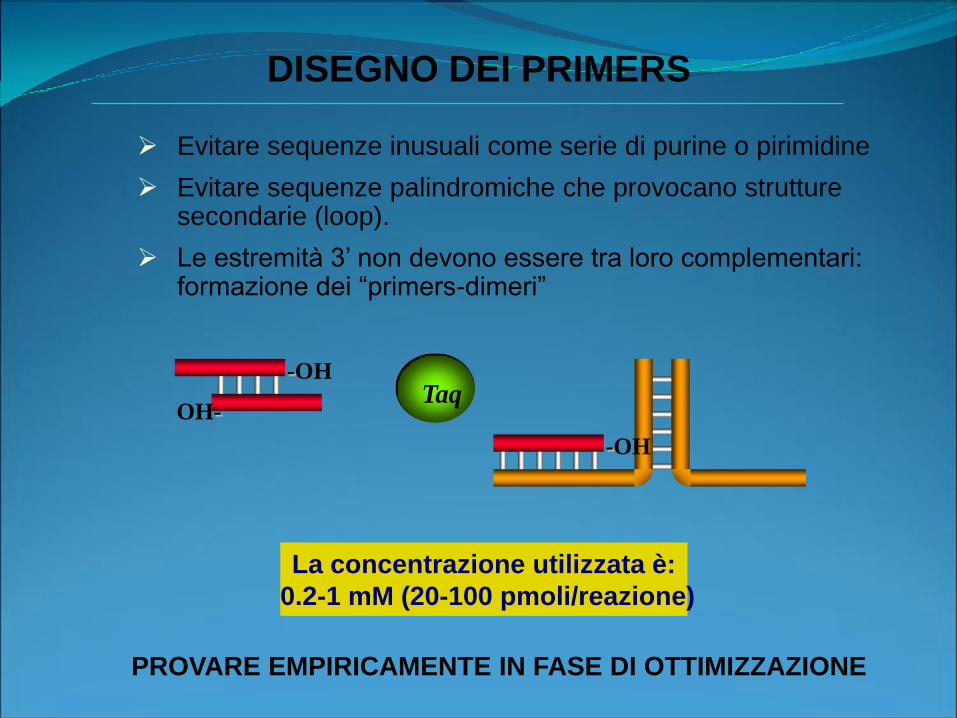
Evitare sequenze inusuali come serie di purine o pirimidine
Evitare sequenze palindromiche che provocano strutture secondarie (loop).
Le estremità 3’ non devono essere tra loro complementari: formazione dei “primers-dimeri”
-OH
OH-
-OH
Taq
DISEGNO DEI PRIMERS
PROVARE EMPIRICAMENTE IN FASE DI OTTIMIZZAZIONE
La concentrazione utilizzata è:
0.2-1 mM (20-100 pmoli/reazione)

Disegno dei primers
Per il disegno dei primer e delle coppie di primer spesso vengono
utilizzati dei programmi appositi accessibili gratuitamente via web.
Un elenco (non esaustivo!) di programmi utilizzabili come servizio
web è il seguente:
Primer3 URL: http://biotools.umassmed.edu/bioapps/primer3_
www.cgi oppure http://frodo.wi.mit.edu/
GeneFisher URL: http://bibiserv.techfak.uni-bielefeld.de/
genefisher2/
Oligonucleotide calculator URL: http://www.basic.northwe
stern.edu/biotools/oligocalc.html
Coppie di primer possono comunque anche essere disegnate
a mano.

La lunghezza ottimale di un primer dipende sia dal suo contenuto in
A+T, sufficientemente basso da poter avere Tm (e quindi Ta)
superiori a 50°C,
la probabilità di trovare una singola base (A, G, C o T) in una
sequenza casuale di DNA è ¼ (cioè 4-1); su un dinucleotide sarà il
prodotto delle singole probabilità cioè 4-1 X 4-1 = 4-2 . Per un
oligonucleotide di 16 basi sarà perciò 4-16 (=1/4 294 967 296) che
corrisponde alla dimensione di un genoma di un eucariote
complesso medio (come Homo sapiens) ed è 1000 volte più grande
del genoma di E. coli. Di conseguenza un oligonucleotide con
almeno 17 o più basi sarà estremamente specifico e primer di
questa lunghezza sono comunemente utilizzati
Il limite superiore per la lunghezza di un primer è dettato dalla sua
Tm.

TAMPONE DI REAZIONE
controllo pH (attività Taq polimerasi)
controllo forza ionica ….(termostabilità Taq polimerasi)
Cosolventi: diminuiscono la temperatura di denaturazione e di annealing:
DMSO, formamide, glicerolo
Standard PCR
1x Taq Buffer
10mM Tris-HCl: pH 8.3 a t.a.
50mM KCl
1.5mM MgCl2 (!!!!)

Necessario per l’attività enzimatica
Stabilizza l’ibridazione del DNA
Sottratto alla reazione da diversi fattori: DNA,
EDTA, supporto in vetro (In Situ), dNTPs.
Legato dai dNTP (gruppi fosfato)
Concentrazione ottimale: dNTP+ primers+ DNA
CONCENTRAZIONE DI MgCl2
La concentrazione utilizzata è:
0.5-5 Mm (generalmente 1.5 mM)

[Mg++] mM
CONCENTRAZIONE DI MgCl2
amplificato
specifico
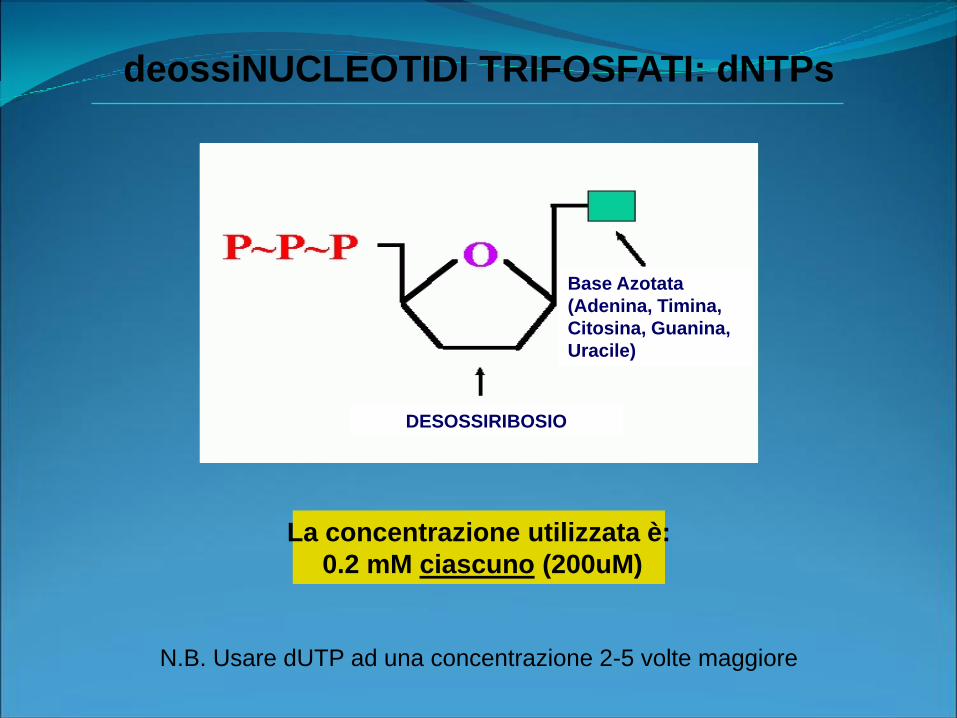
deossiNUCLEOTIDI TRIFOSFATI: dNTPs
Base Azotata
(Adenina, Timina,
Citosina, Guanina,
Uracile)
DESOSSIRIBOSIO
La concentrazione utilizzata è:
0.2 mM ciascuno (200uM)
N.B. Usare dUTP ad una concentrazione 2-5 volte maggiore

attività
3’ esonucleasica
attività
catalitica
attività
5’ esonucleasica
DNA POLIMERASI (Taq polimerasi)

LE CONDIZIONI DI REAZIONE

Temperatura e tempo di annealing
La temperatura di annealing (Ta) dei primer dipende dal loro contenuto in
G+C e dalla loro lunghezza e quindi dalla temperatura di fusione tra
primer e la sua elica complementare sul DNA stampo.
Considerando primer di lunghezza media di 20 basi una
formula empirica spesso utilizzata per il calcolo della Tm è la seguente:
Tm = [4(G + C) + 2(A + T)] °C.
Nel caso che i due primer abbiano Tm diverse generalmente si considera
quello con la Tm più bassa.
Solitamente si utilizza come temperatura di annealing la Tm-5 °C
anche se spesso l’utilizzo diretto della stessa Tm può portare ad avere
ottime rese nella reazione di PCR.

1) Ta costante durante i cicli;
2) Ta che diminuisce ciclo dopo ciclo (touch-down)
La strategia di reazione touch-down permette di rendere i
primi cicli di PCR
estremamente “stringenti”, cioè tali da promuovere
l’amplificazione solo di frammenti specifici rendendo instabili
eventuali annealing dei primer a sequenze di DNA non
perfettamente complementari.
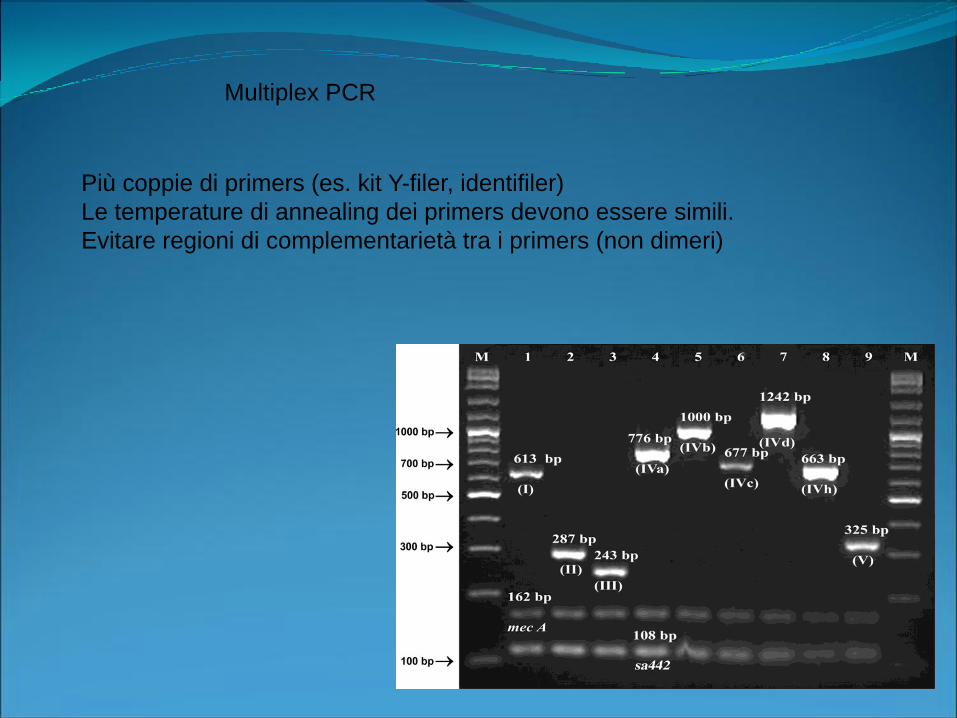
Multiplex PCR
Più coppie di primers (es. kit Y-filer, identifiler)
Le temperature di annealing dei primers devono essere simili.
Evitare regioni di complementarietà tra i primers (non dimeri)

Effetto del cloruro di magnesio (MgCl2) e della temperatura
di annealing (Ta) sulla specificità di una reazione di PCR.
Le frecce in alto e in basso indicano rispettivamente un aumento
o una diminuzione della Ta e del MgCl2.

Scelta dei primers
Condizioni di reazione
ContaminazioneSpecificità
Sensibilità
Scelta dei primers
Eterogeneità genomica
Presenza di inibitori
Tipo di campione
Efficienza della reazione
Riproducibilità
Standardizzazione delle fasi del metodo:
preparazione del campione
protocollo di PCR
sistema di rivelazione
PRINCIPALI FATTORI CHE INFLUENZANO LA
RESA DELLA PCR

Elettroforesi di acidi nucleici
•Gel di agarosio
•Gel di acrilammide


Come si prepara un gel d’agarosio
Alla soluzione d’agarosio
si aggiunge Etidio Bromuro
(EtBr)
Si scioglie l’agarosio in
polvere in una soluzione
tampone a 100°C
100V 0,2A
tampone
elettroforetico
generatore di corrente
Polo +
Polo -
gel di agarosio 1.2%
scatola elettroforetica
ELETTROFORESI IN GEL DI AGAROSIO

100V 0,2A
Polo +
Polo -
Loading dye aiuta il caricamento del campione nel pozzetto del gel. Contiene glicerolo, Blu di
bromofenolo e Blu di xilencianolo che migrano nel gel a velocità diversa.
ELETTROFORESI IN GEL DI AGAROSIO
100V 0,2A
Polo +
Polo -
ON
Loading dye aiuta il caricamento del campione nel pozzetto del gel. Contiene glicerolo, Blu di
bromofenolo e Blu di xilencianolo che migrano nel gel a velocità diversa.
ELETTROFORESI IN GEL DI AGAROSIO

100V 0,2A
Polo +
Polo -
ON
Loading dye aiuta il caricamento del campione nel pozzetto del gel. Contiene glicerolo, Blu di
bromofenolo e Blu di xilencianolo che migrano nel gel a velocità diversa.
ELETTROFORESI IN GEL DI AGAROSIO

Etidio Bromuro (EtBr):
colorante fluorescente
assorbe la luce UV a 300 nm
dando colore giallo-arancio
E’ utile sia per visualizzare,
sia per quantificare il
campione: l’intensità della
fluorescenza è, infatti,
proporzionale alla quantità
del campione.
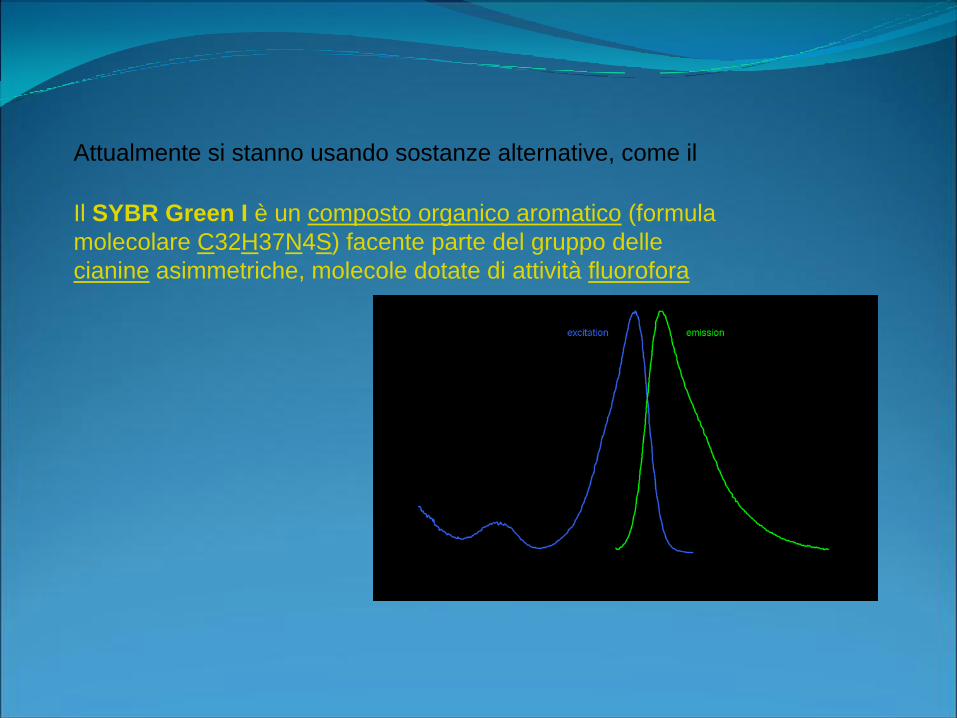
Attualmente si stanno usando sostanze alternative, come il
Il SYBR Green I è un composto organico aromatico (formula
molecolare C32H37N4S) facente parte del gruppo delle
cianine asimmetriche, molecole dotate di attività fluorofora
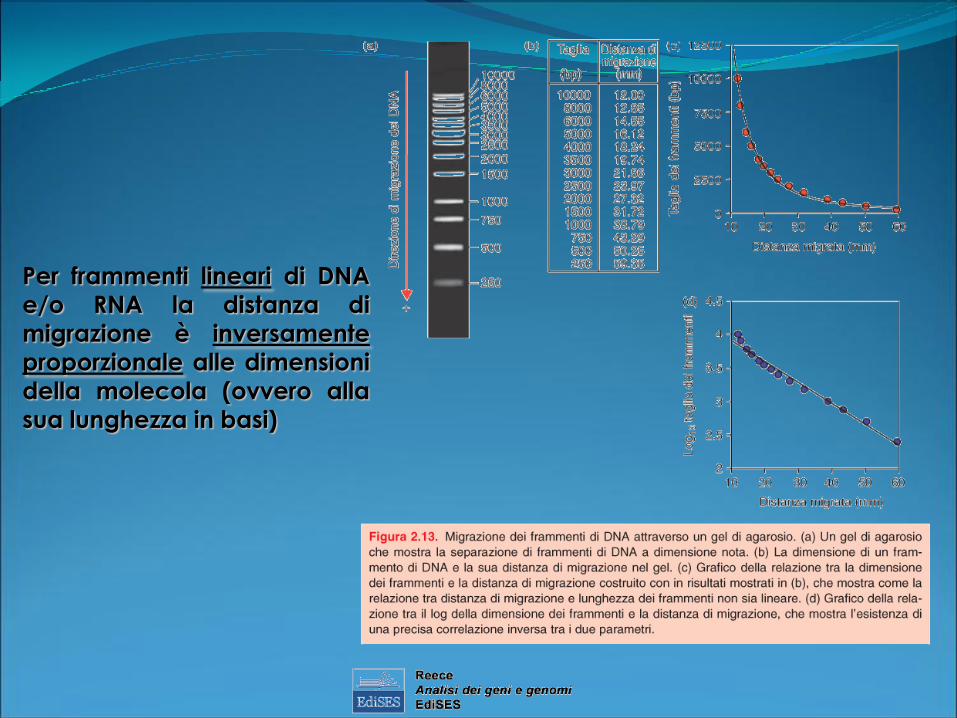
Per frammenti lineari di DNA
e/o RNA la distanza di
migrazione è inversamente
proporzionale alle dimensioni
della molecola (ovvero alla
sua lunghezza in basi)
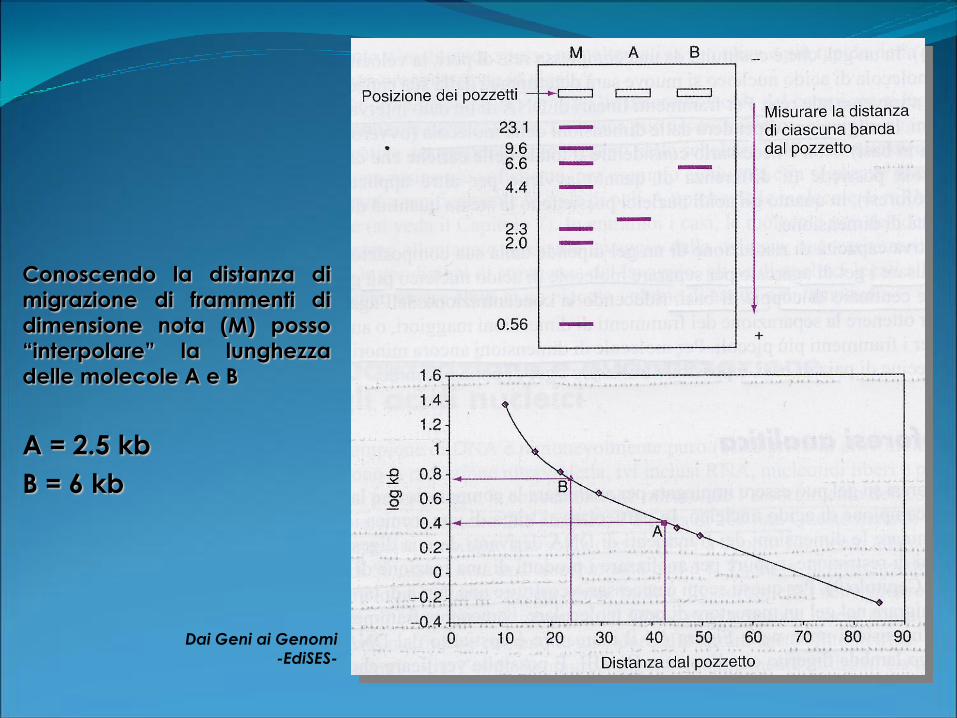
Conoscendo la distanza di
migrazione di frammenti di
dimensione nota (M) posso
“interpolare” la lunghezzadelle molecole A e B
Dai Geni ai Genomi
-EdiSES-
A = 2.5 kb
B = 6 kb

GEL DI AGAROSIO
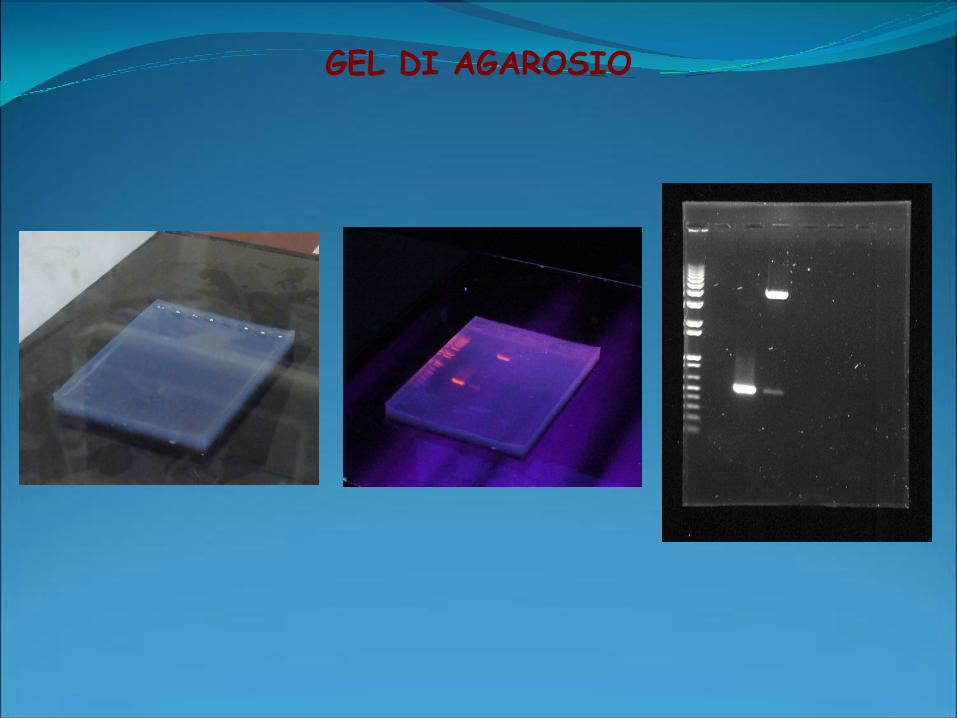
GEL DI AGAROSIO
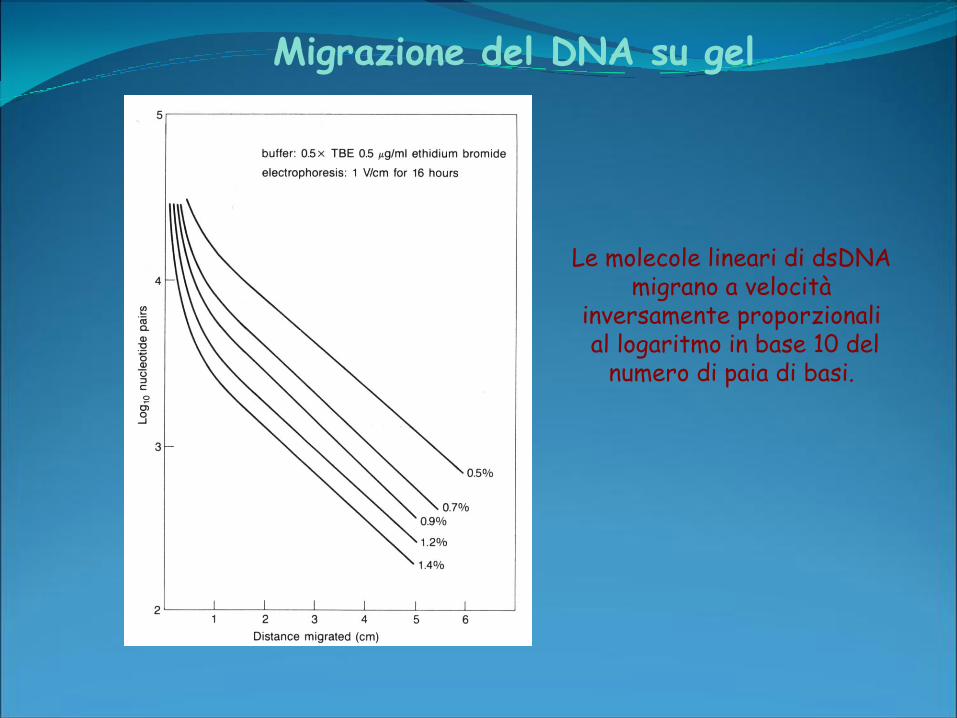
Migrazione del DNA su gel
Le molecole lineari di dsDNA migrano a velocità
inversamente proporzionalial logaritmo in base 10 del
numero di paia di basi.

Fattori che influenzano la migrazione
• Peso molecolare
•Concentrazione di agarosio
•Conformazione DNA
•Voltaggio applicato
• Intercalanti
•Tampone di elettroforesi

Visualizzazione del DNA

Marcatori di peso molecolare
HindIII

Separazione di frammenti minori di 1 kb
Permette di separare frammenti che differiscono di
una sola base (determinazione della sequenza del
DNA)
ELETTROFORESI IN GEL DI POLIACRILAMMIDE (PAGE)

Formazione di un gel di poliacrilammide

acrilamide/bis acrilamide 29:1 (6-15%)+ 7M Urea
Spessore del gel
0,4-1,5 mm
Il gel è composto da
acrilamide e bis acrilamide in
un rapporto caratteristico 29:1
che determina lo spessore dei
pori. Esso inoltre contiene urea
che funziona da denaturante.
ELETTROFORESI IN GEL DI POLIACRILAMMIDE (PAGE)
Risoluzione: 20 - 1000 nt

Polo +
Polo -
1000V 22 mA
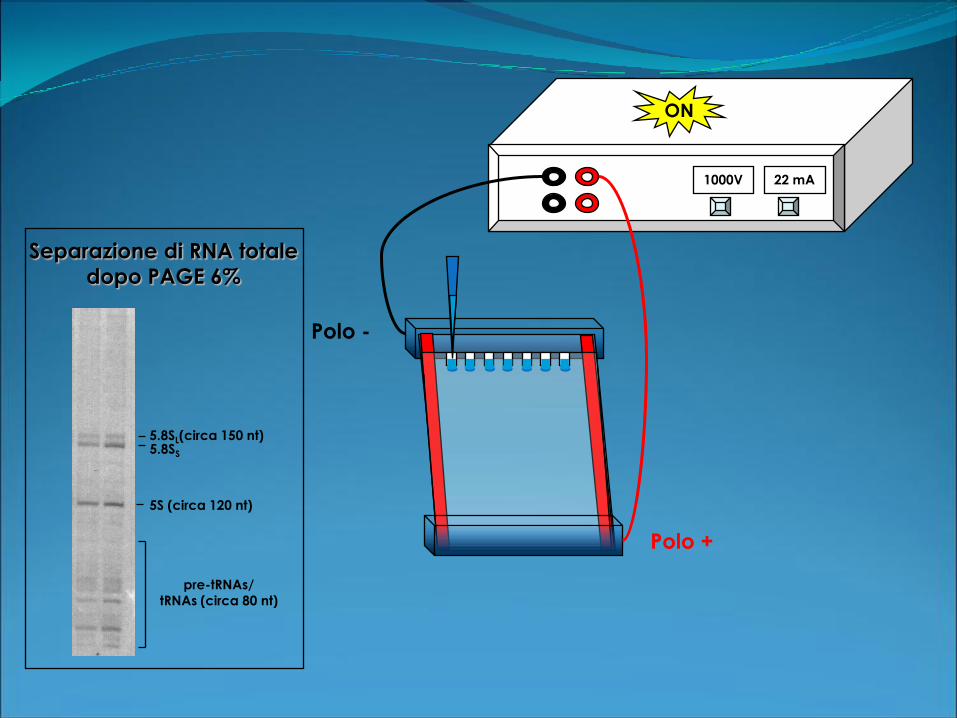
1000V 22 mA
ON
Polo +
Polo -
5.8SL(circa 150 nt)
5S (circa 120 nt)
pre-tRNAs/
tRNAs (circa 80 nt)
5.8SS
Separazione di RNA totale
dopo PAGE 6%
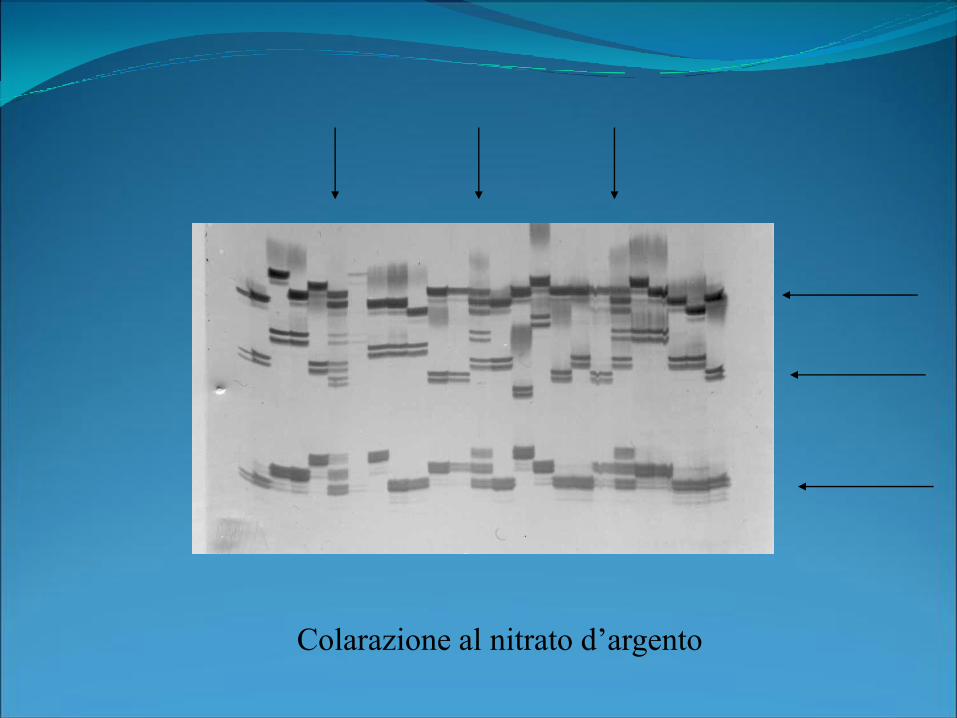
Colarazione al nitrato d’argento

Metodi di sequenziamento
• Sanger (enzimatico)
• Maxam e Gilbert (chimico)
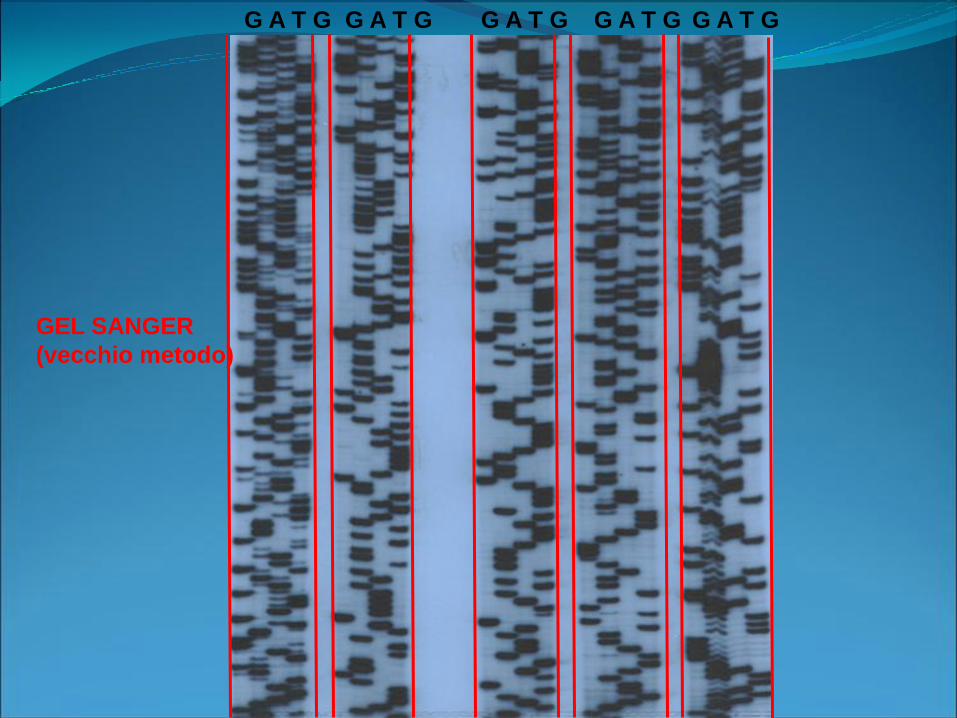
GEL SANGER
(vecchio metodo)
G A T G G A T G G A T G G A T G G A T G
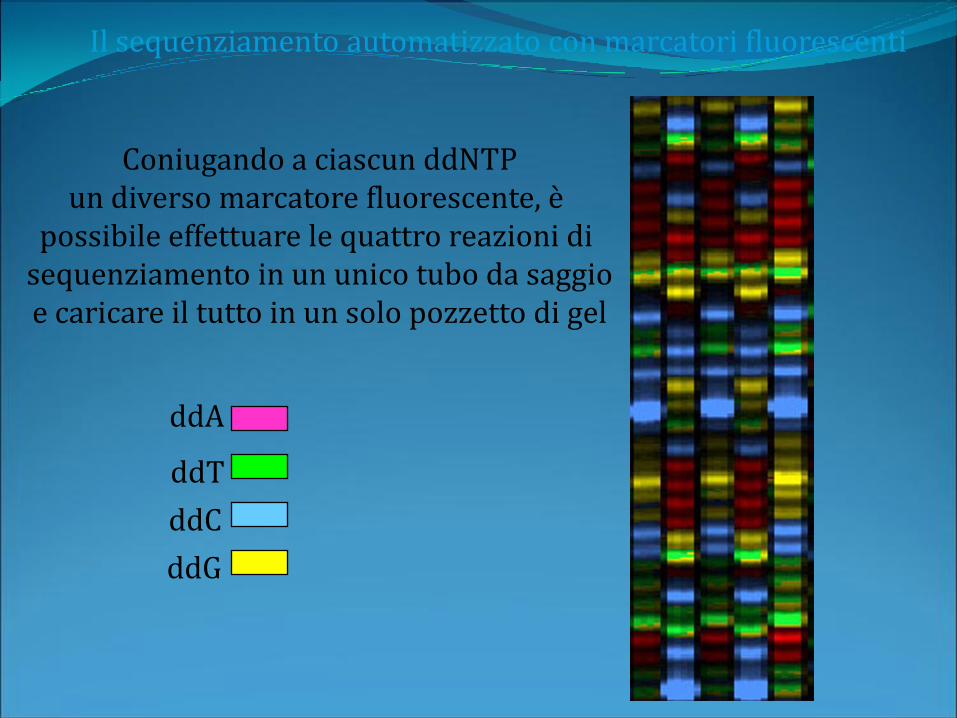
Il sequenziamento automatizzato con marcatori fluorescenti
Coniugando a ciascun ddNTPun diverso marcatore fluorescente, è
possibile effettuare le quattro reazioni di sequenziamento in un unico tubo da saggioe caricare il tutto in un solo pozzetto di gel
ddA
ddC
ddT
ddG

SEQUENZIATORE AUTOMATICO
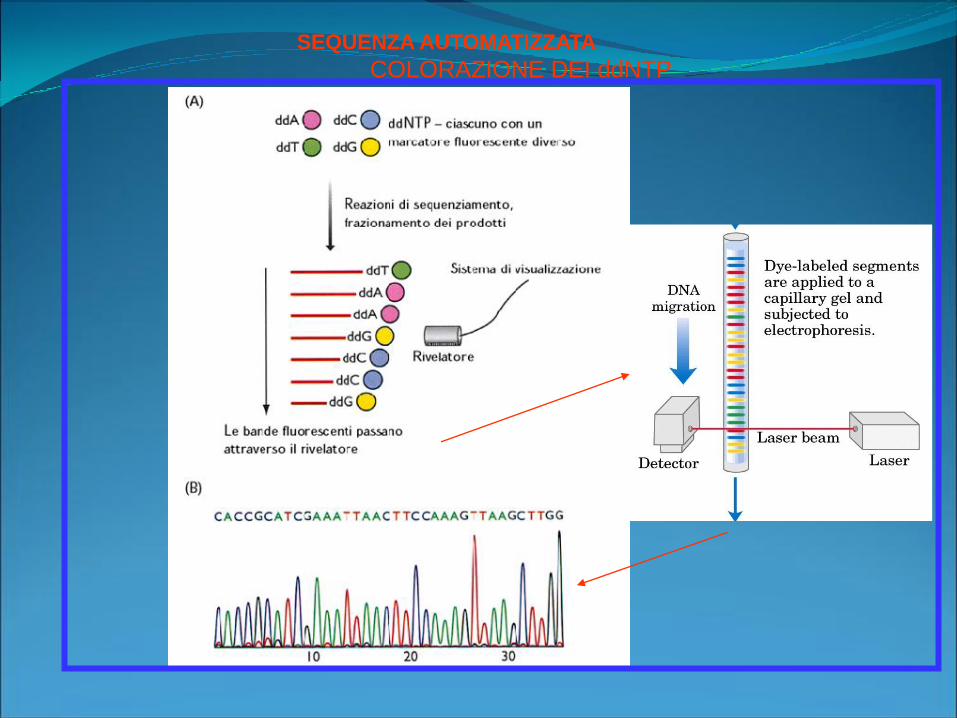
SEQUENZA AUTOMATIZZATA
COLORAZIONE DEI ddNTP

Throughput in 24 hours
3100 3700Sequencing
Std run 176 samples 768 samples
(800bp) (1000bp)
Rapid run 368 samples 1152 samples
(550bp) (700bp)
GenScan 736 samples 3456 samples
3100
3700
GA AT TCCCCT
10
GCAGGC GTGG
20
C TGCAGC CTG
30
GT TAT GA TTA
40
C TGT T A TGT T
50
GCTACTACT G
60
CTGACAA TGC
70
TGC TGCTGC T
80
TCTCCTCAC T
90
GTCTCCACT T
100
CCT TGAACA A
110
TGC GCC GTCA
120
TGC T TCT T TT
130
GCCTCC
CCGC T
140
GC TCCA GA AA
150
GC TA GGCC GC
160
A GAT CA GA AC
170
CA CCACA GT C
180
A A TA T CA C CA
190
C CT T CCT CT T
200
A TA GA T TC GG
210
A A T CT CA T GA
220
TA GGG GC TCA
230
GC C TC T GT GC
240
GA G T GGA GA G
250
AA GT T T
T GCA G
260
GC GA G C T GA G
270
GA GCA A T T GC
280
A GG T GA T A T G
290
A T GT GC TC GG
300
C T CA A GA A GC
310
GGGC C C GG AG
320
A G GA A GA A GT
330
C GT GC C GGG G
340
C TA A T T A T T G
350
GCA A A A CGA G
360
C T CT T GT T G
GT
370
A A A CA T T GA T
380
C CA A C T GG A A
390
T GT CA C TA A T
400
G G C GA A T CA A
410
T A T T C CA T A A
420
G G CA T GA T G G
430
T T GC T CA G A G
440
G CA G G A G A A G
450
A G CA A C GA A T
460
A C GA T C C TA T
470
AA A A G A T AA A
480
A
480
A CA T A A A TA A
490
A C A GT C T T GA
500
T T A T A T T C T G
510
G GT A T TA A A G
520
C CA CA A T CA G
530
A A CAA A TA TA
540
T GC T T T GTA T
550
C T T T T C T T GC
560
C T T CT T CA T T
570
A C C AA C T G C T
580
T C C GC GGC CA
590
C A T
Model 3700
BC 1.1.0.0
3100data.ab1.ab1
bdt
Lane 13
Signal G:3083 A:2887 T:2508 C:1336
DT3700POP6{BD}v0_3.mob
Points 3292 to 17806 Base 1: 3292
Page 1 of 2
Wed, Jan 19, 2000 5:10 PM
Tue, Aug 17, 1999 9:47 PM
Spacing: 14.21
T TA A GA G A
600
AC T T GT G GTA
610
A G ATAA GA A G
620
ATA T T TT A T T
630
C GT T CT GC T G
640
A C TT GC T GGA
650
T GT C GGG AA A
660
TA T T CT GCA T
670
TT GA T AAGAG
680
GCGGTTAA TT
690
GCA GATAT AA
700
T T GGTA GTGA
710
A AAGGGTC GT
720
TGCT
TATGGTC
730
ACCGT GAAGC
740
GA GT CAGCAG
750
CACAAGAATG
760
TGTGCCGTTC
770
T A GTTAT AT G
780
G TTGA TAT GG
790
AACCT GNTTA
800
GTC GG TTAAG
810
GNANAGATCT
820
A CCAAACA C C
830
TGCG GACTGA
840
TG AGATTA TT
850
TTTACTA TCT
860
A TTTGGG AAC
870
C
870
T A CGG CCT AA
880
CANCTCATGT
890
AT C TNNTNCG
900
TCGAA GGTGG
910
GTGTCNTGAN
920
AANCCT AA AC
930
GGNCCGAA GG
940
AACCTTGGGG
950
GCCCGACCCA
960
A NTTT GGGGC
970
A C ANNCAGGC
980
CTA TCGA CCC
990
G AAGGAAAGG
1000
C GCNGGCN
NCN
1010
CCTNCGCT TG
1020
CNTTNG GGGG
1030
G GCNNT GNA C
1040
CT T TNANNNC
1050
CCT T T CCG CG
1060
T T A A CGCA C N
1070
NNNNN NNNNN
1080
NNNNCAA NNN
1090
NNNNNNNNNN
1100
NNNNNNNNNN
1110
NNNNNNNNNN
1120
NNNNNNNNNN
1130
NN
NNNNNNNNN
1140
NNNNNNNNNN
1150
NNNNNNNNNN
1160
NNNNNNNNNN
1170
NNNNNNNNNN
1180
NNNNNNNNNN
1190
NNNNNNNNNN
1200
NNNNNNNNNN
1210
NNNNNNNNNN
1220
NNNNNNNNNN
1230
NNNNNNNNNN
1240
NNNN
Model 3700
BC 1.1.0.0
3100data.ab1.ab1
bdt
Lane 13
Signal G:3083 A:2887 T:2508 C:1336
DT3700POP6{BD}v0_3.mob
Points 3292 to 17806 Base 1: 3292
Page 2 of 2
Wed, Jan 19, 2000 5:10 PM
Tue, Aug 17, 1999 9:47 PM
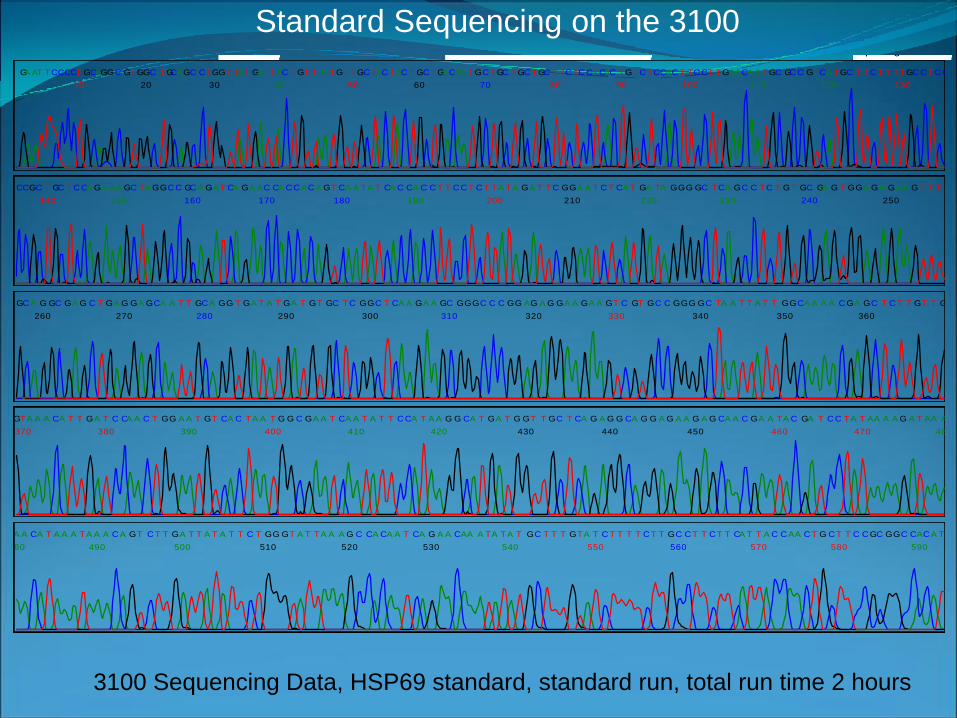
Spacing: 14.213100 Sequencing Data, HSP69 standard, standard run, total run time 2 hours
Standard Sequencing on the 3100

Possibili Cause Possibili Soluzioni
• Il primer non trova sito di
annealing
• Cambiare primer
• Il DNA presenta contaminazioni di
vario tipo• Ripreparare il DNA
• La quantità di DNA e' insufficiente • Aumentare la quantità di DNA
• La quantià di primer è insufficiente • Aumentare la quantità di primer
• Il primer è stato disegnato male, es.
temperatura di melting troppo bassa • Ridisegnare il primer
Reazione fallita: non presenta
picchi definiti e ha un alto
rumore di fondo

Possibili Cause Possibili Soluzioni
Campione che presenta rumore di
fondo sufficientemente alto da
causare ambiguità (N) nel
riconoscimento dei picchi da parte
del software
• La quantità di DNA e' insufficiente e il
segnale troppo debole
• Aumentare la quantità di DNA
• Il DNA presenta contaminazioni che
inibiscono la reazione
• Purificare meglio il DNA
• Ci sono altri templati contaminanti • Ripreparare il DNA
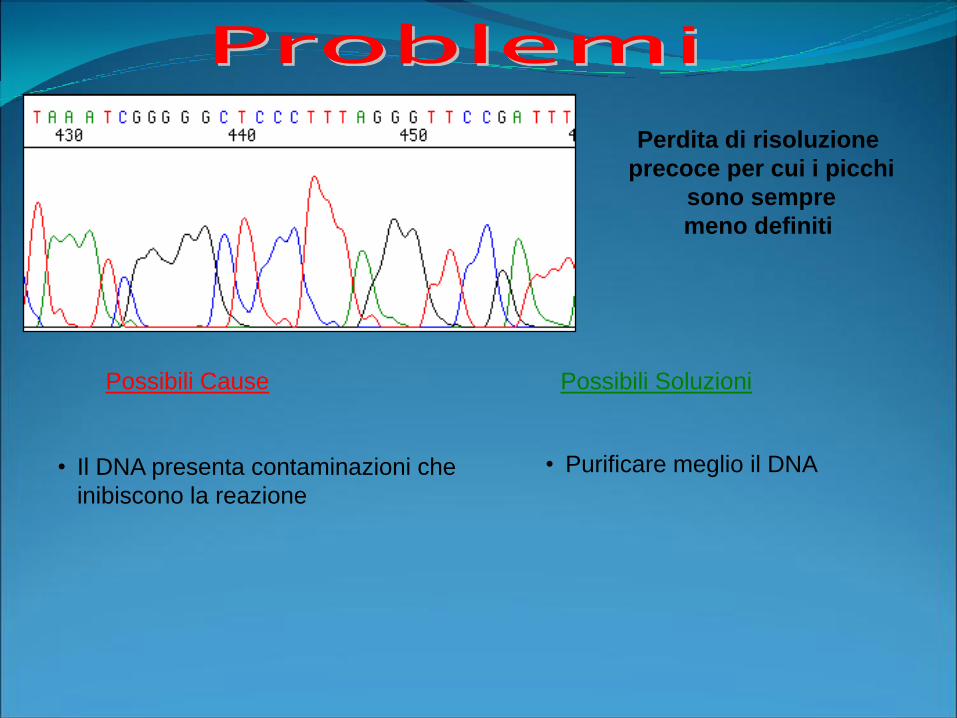
Possibili Cause Possibili Soluzioni
Perdita di risoluzione
precoce per cui i picchi
sono sempre
meno definiti
• Il DNA presenta contaminazioni che
inibiscono la reazione
• Purificare meglio il DNA

Possibili Cause Possibili Soluzioni
• Clone o PCR multiple con stesso
tratto iniziale, es. vettore • Ripreparare DNA in modo da avere
un tipo di templato unico
• Siti multipli di attacco del primer sul DNA • Cambiare primer
• Mutazione frame shift
• Slittamento dopo una regione omopolimerica
Sequenza
doppia
dopo un
tratto
buono
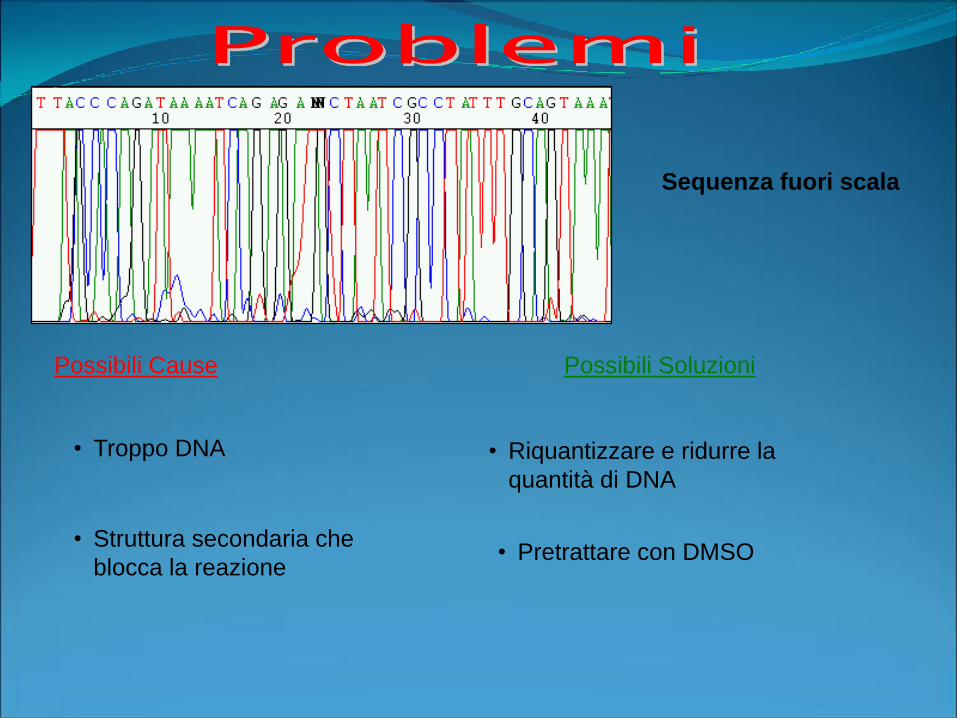
Possibili Cause Possibili Soluzioni
Sequenza fuori scala
• Troppo DNA • Riquantizzare e ridurre la
quantità di DNA
• Struttura secondaria che
blocca la reazione• Pretrattare con DMSO
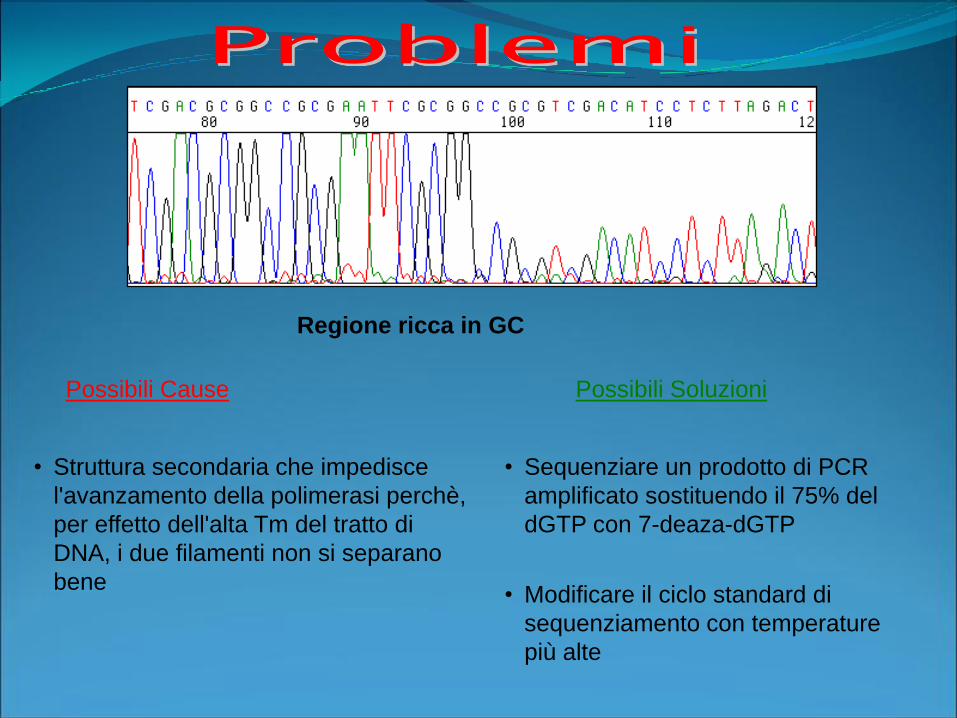
Possibili Cause Possibili Soluzioni
Regione ricca in GC
• Struttura secondaria che impedisce
l'avanzamento della polimerasi perchè,
per effetto dell'alta Tm del tratto di
DNA, i due filamenti non si separano
bene
• Sequenziare un prodotto di PCR
amplificato sostituendo il 75% del
dGTP con 7-deaza-dGTP
• Modificare il ciclo standard di
sequenziamento con temperature
più alte
Recommended

















